ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к гидрохлоридной соли, цитратной соли, фосфатной соли или сульфатной соли соединения 1, кристаллическим формам вышеуказанных солей и способу их получения. Настоящее изобретение также относится к их применению в получении лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] Согласно исследованию Международной организации здравоохранения (WHO) церебральный инсульт стал второй по распространенности причиной смертности после ишемической болезни сердца; церебральный инсульт также с высокой степенью вероятности приводит к пороку развития и инвалидности. Это серьезно влияет на качество жизни пациентов и их семьи. Следовательно, необходимо найти способ улучшения состояния здоровья пациентов, перенесших инсульт, и восстановления функционирования их организма и трудоспособности, чтобы они могли иметь лучшее качество жизни и хороший прогноз. Снижение бремени является целесообразным не только для отдельных индивидуумов, но также для всего общества.
[0003] Винпоцетин, который представлен формулой B-I, представляет собой индольный алкалоид, экстрагированный из растения барвинка малого. Винпоцетин является сильно жирорастворимым и может легко проходить через гематоэнцефалический барьер, так что он присутствует в высокой концентрации в тканях головного мозга и обладает хорошей эффективностью. Винпоцетин разработан венгерской компанией Gedeon Richter Со. в 1978 году. Он имеет более чем 30-летнюю историю в Европе. Его применяют главным образом для ослабления симптомов, вызванных, среди прочего, последствиями церебрального инфаркта, последствиями внутримозгового кровоизлияния и церебрального атеросклероза. С тех пор, как винпоцетин впервые появился на рынке, он считается общепринятым лекарственным препаратом для лечения сердечно-сосудистых и цереброваскулярных заболеваний. Недавно было обнаружено, что винпоцетин может оказывать улучшение в отношении возрастного нарушения памяти и умственной деятельности здоровых людей. Кроме того, обнаружено, что винпоцетин также может оказывать действие, направленное на лечение, среди прочего, спутанности сознания, синдрома дефицита внимания, раздражительности, зрительных и слуховых расстройств и перепадов настроения. Кроме того, в соответствии с данными клинических испытаний в отношении более 67% припадков частота припадков в значительной степени снижалась или их вообще не было. Он производит значительный терапевтический эффект в отношении генерализованных тонико-клонических припадков.
[0004] Частота возникновения церебрального инсульта и уровень инвалидизации вследствие его являются очень высокими в Китае, что стало огромной нагрузкой для китайской системы медицины. Винпоцетин широко применяется в Китае для лечения церебрального инсульта и других связанных заболеваний; это основной способ лечения для улучшения прогноза при церебральном инсульте. Однако терапевтический эффект винпоцетина все еще является сомнительным, а биодоступность таблеток винпоцетина крайне низкая.
[0005] Эпилепсия является синдромом хронического рецидивирующего транзиторного нарушения функции головного мозга и характеризуется аномальным разрядом нейронов в головном мозге, что приводит к риску возникновения рецидивирующих припадков. Эпилепсия является распространенным заболеванием нервной системы. По распространенности она уступает лишь церебральному инсульту. Количество пациентов с эпилепсией в Китае является высоким, и при этом большинство пациентов - это молодежь и дети возрастом до 20 лет. Уровень инвалидизации и уровень смертности при данном заболевании также являются высокими, что стало проблемой, вызывающей беспокойство всего общества. Винпоцетин характеризуется различными показателями степени эффективности у более 67% пациентов с эпилепсией, особенно в случаях генерализованных тонико-клонических припадков.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0006] Настоящее изобретение предусматривает гидрохлоридную соль, цитратную соль, фосфатную соль или сульфатную соль соединения 1:
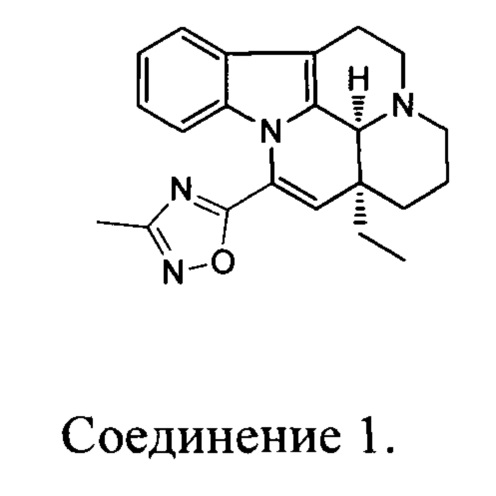
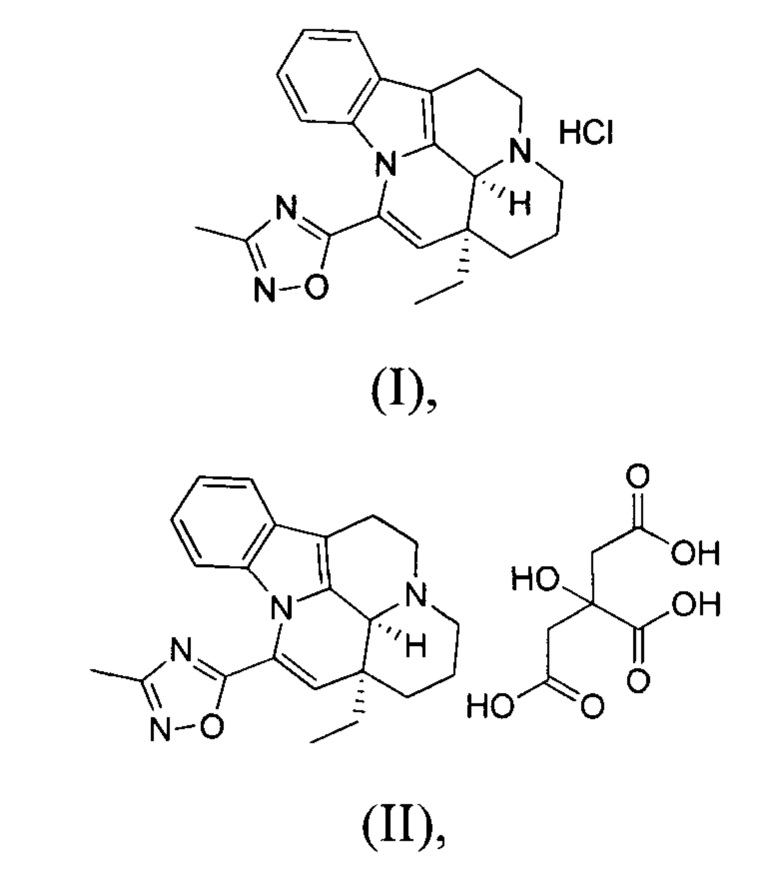
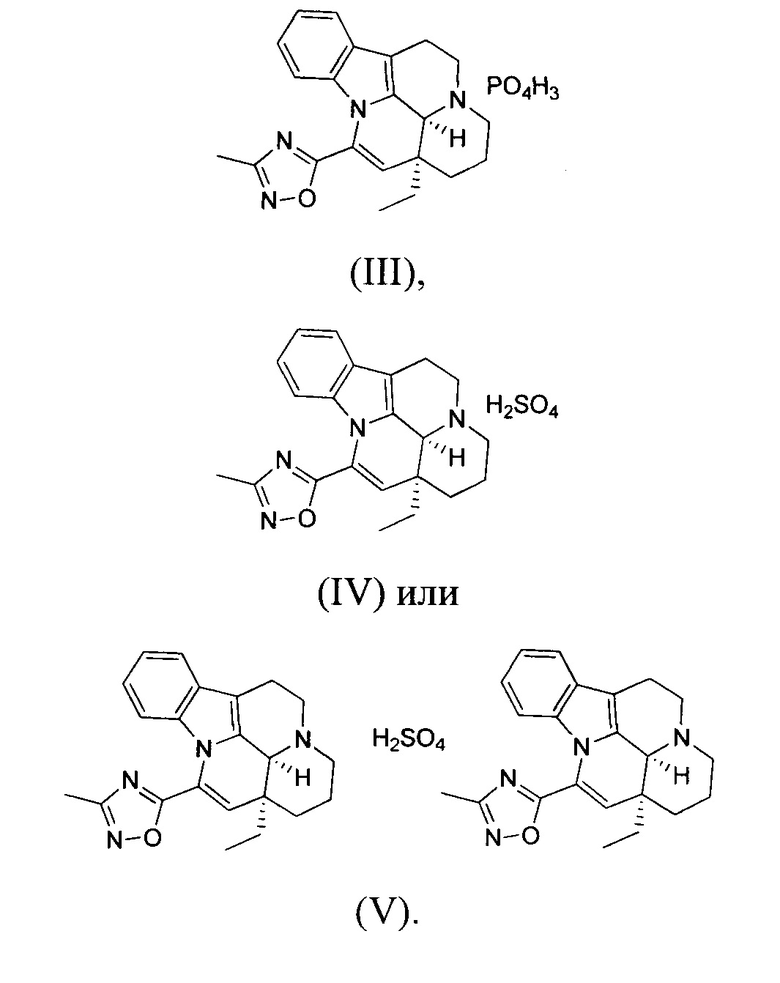
[0007] В некоторых вариантах осуществления настоящего изобретения гидрохлоридная соль, цитратная соль, фосфатная соль или сульфатная соль вышеуказанного соединения 1 выбрана из:
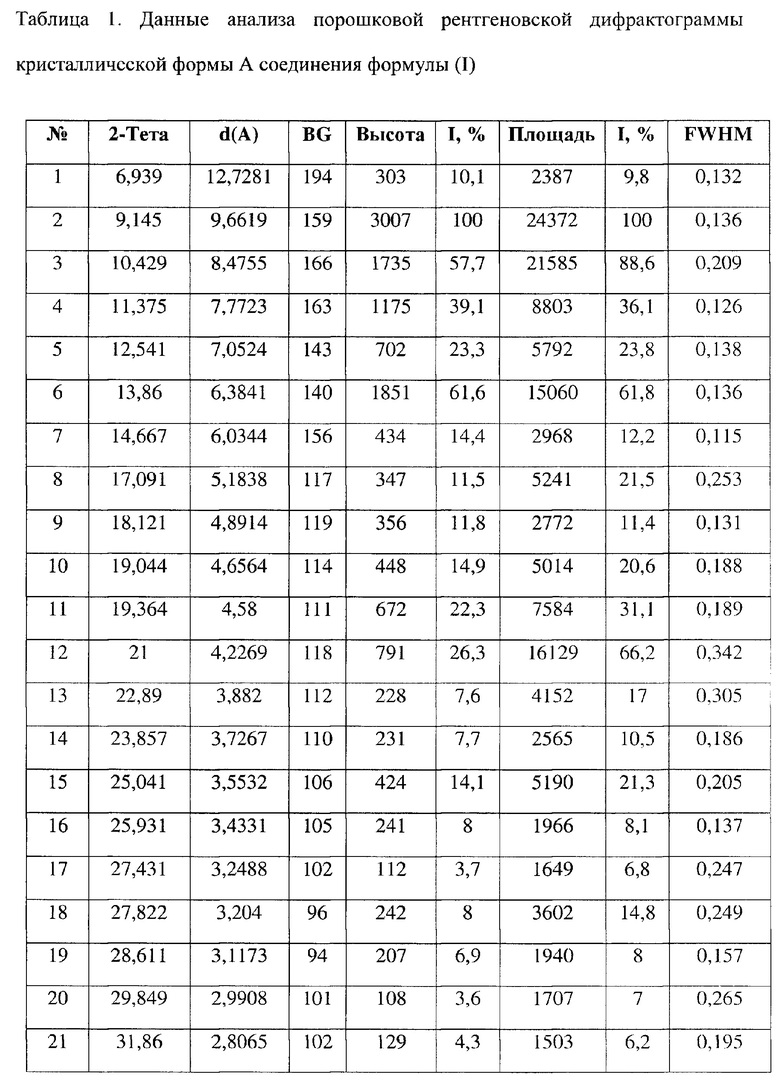
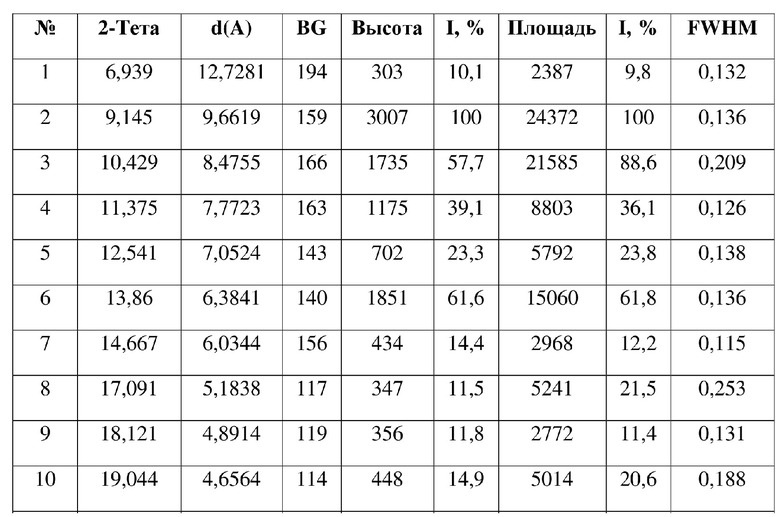
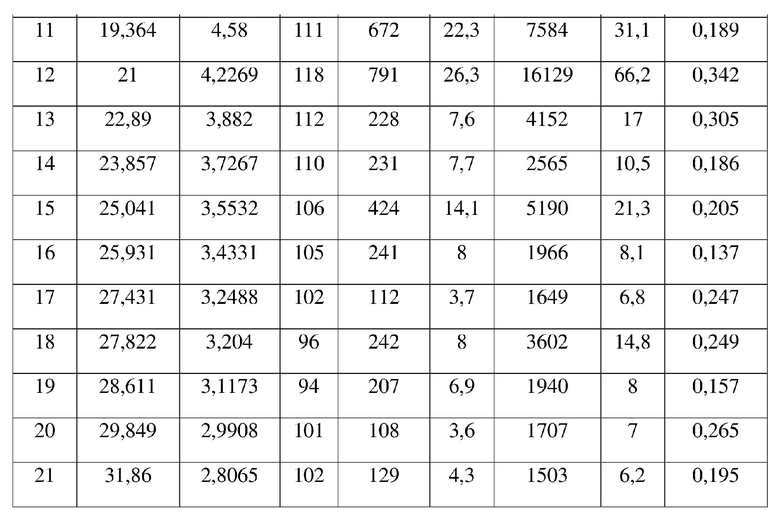
[0008] Настоящее изобретение также предусматривает кристаллическую форму А соединения формулы (I), где порошковая рентгеновская дифрактограмма кристаллической формы А содержит характеристические дифракционные пики при углах 2θ, составляющих 9,14±0,2°, 10,43±0,2°, 11,38±0,2°, 12,54±0,2°, 13,86±0,2°, 19,04±0,2°, 19,36±0,2°, 21,00±0,2°.
[0009] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы А соединения формулы (I) являются такими, как представлены в таблице 1.
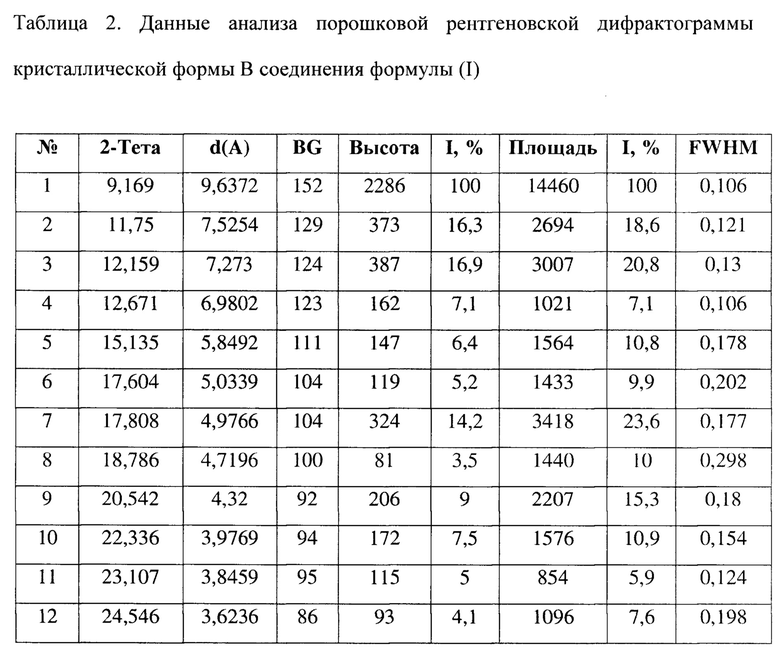
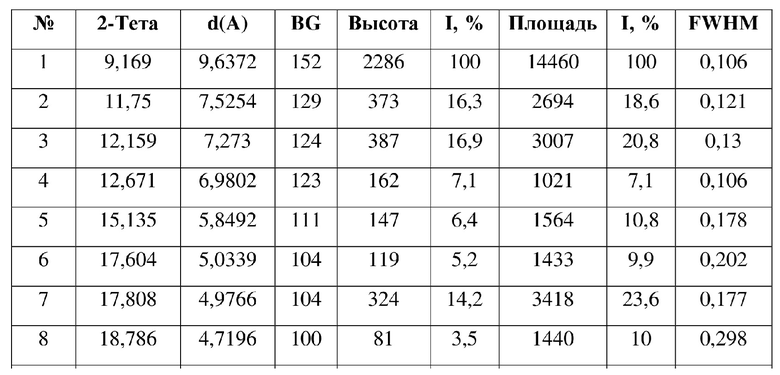
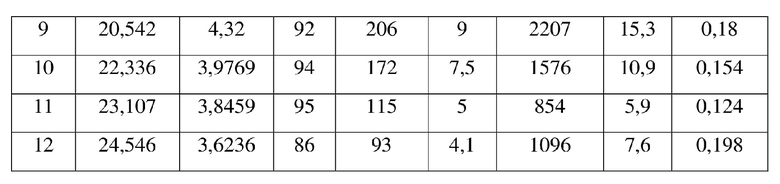
[0010] Настоящее изобретение также предусматривает кристаллическую форму В соединения формулы (I), где порошковая рентгеновская дифрактограмма кристаллической формы В содержит характеристические дифракционные пики при углах 2θ, составляющих 9,17±0,2°, 11,75±0,2°, 12,16±0,2°, 12,67±0,2°, 15,14±0,2°, 17,81±0,2°, 20,54±0,2°, 22,34±0,2°.
[0011] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы В соединения формулы (I), описанной выше, являются такими, как представлены в таблице 2.
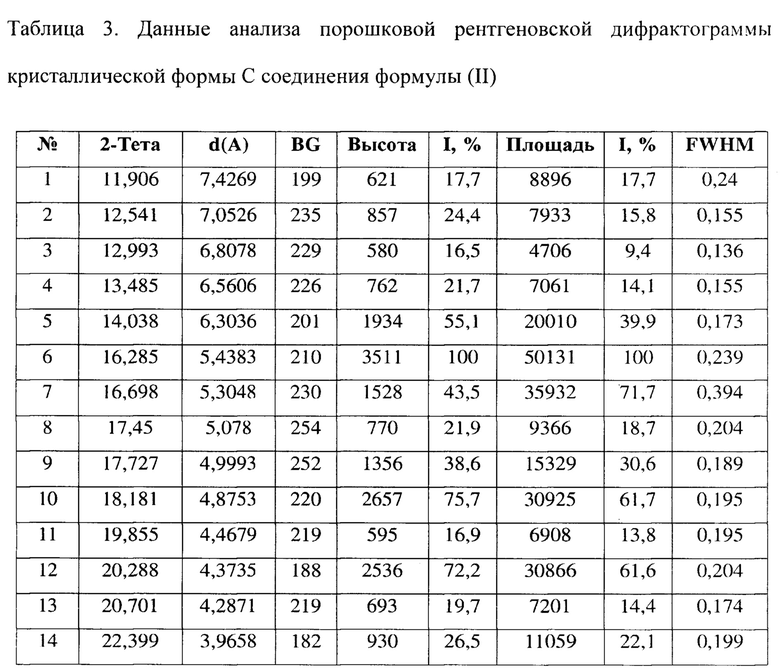
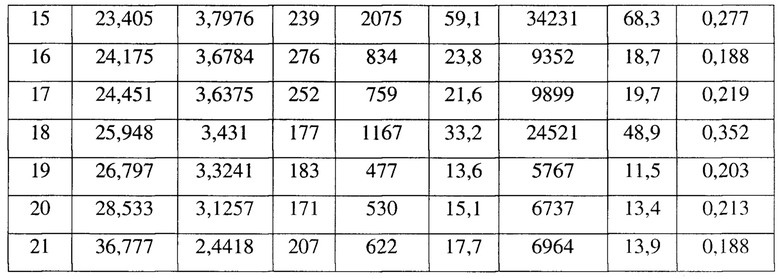
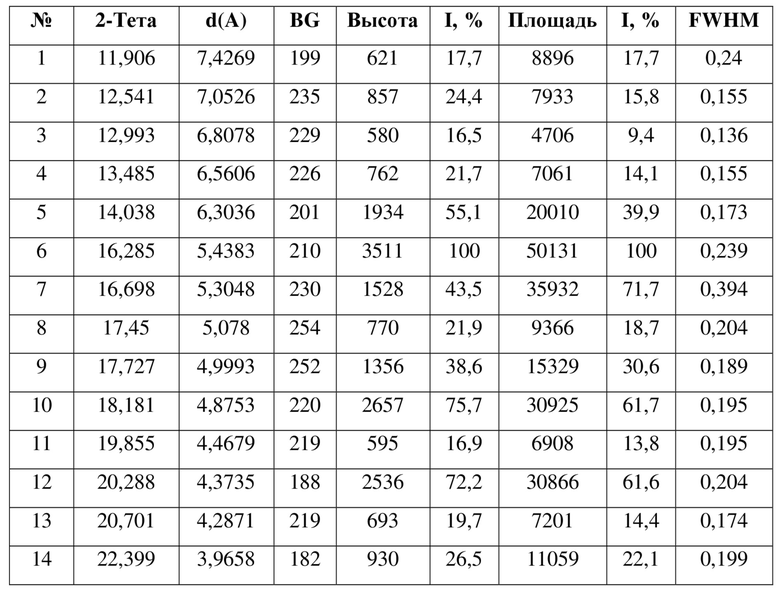
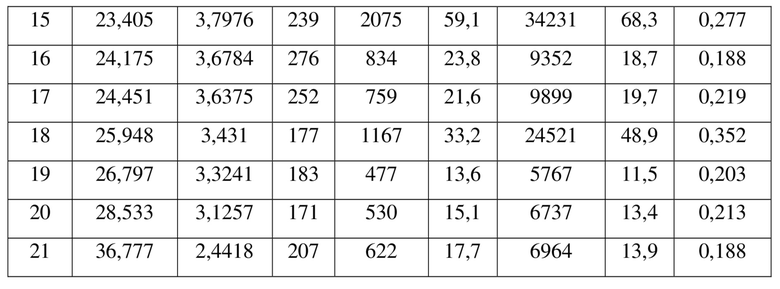
[0012] Настоящее изобретение также предусматривает кристаллическую форму С соединения формулы (II), где порошковая рентгеновская дифрактограмма кристаллической формы С содержит характеристические дифракционные пики при углах 2θ, составляющих 14,04±0,2°, 16,28±0,2°, 16,70±0,2°, 17,73±0,2°, 18,18±0,2°, 20,29±0,2°, 23,40±0,2°, 25,95±0,2°.
[0013] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы С соединения формулы (II) являются такими, как представлены в таблице 3.
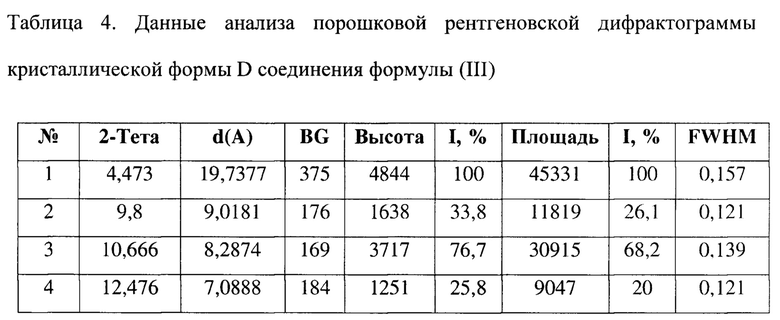
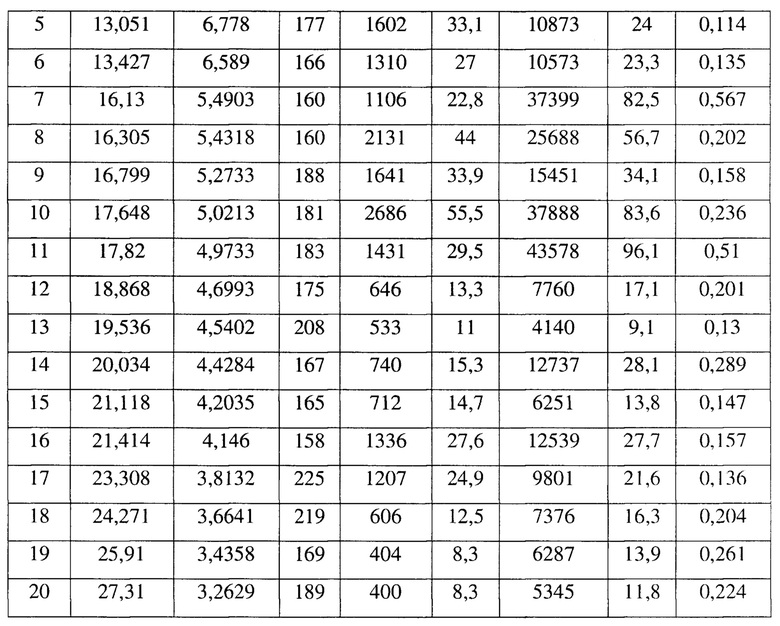
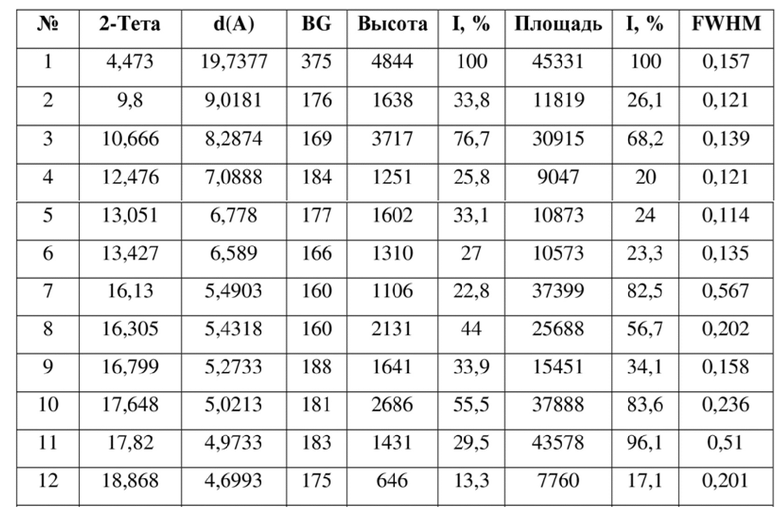
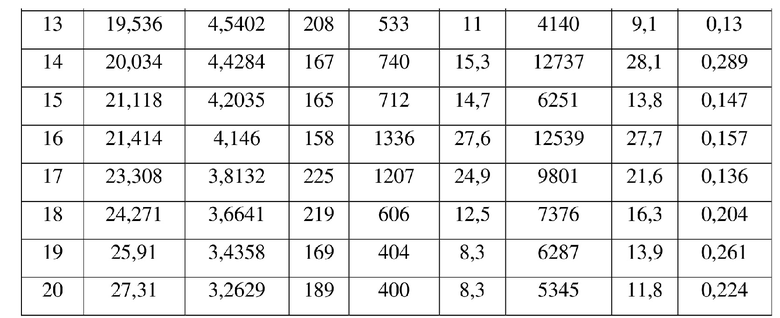
[0014] Настоящее изобретение также предусматривает кристаллическую форму D соединения формулы (III), где порошковая рентгеновская дифрактограмма кристаллической формы D содержит характеристические дифракционные пики при углах 2θ, составляющих 4,47±0,2°, 9,80±0,2°, 10,67±0,2°, 13,05±0,2°, 16,30±0,2°, 16,80±0,2°, 17,65±0,2°, 17,82±0,2°.
[0015] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы D соединения формулы (III) являются такими, как представлены в таблице 4.
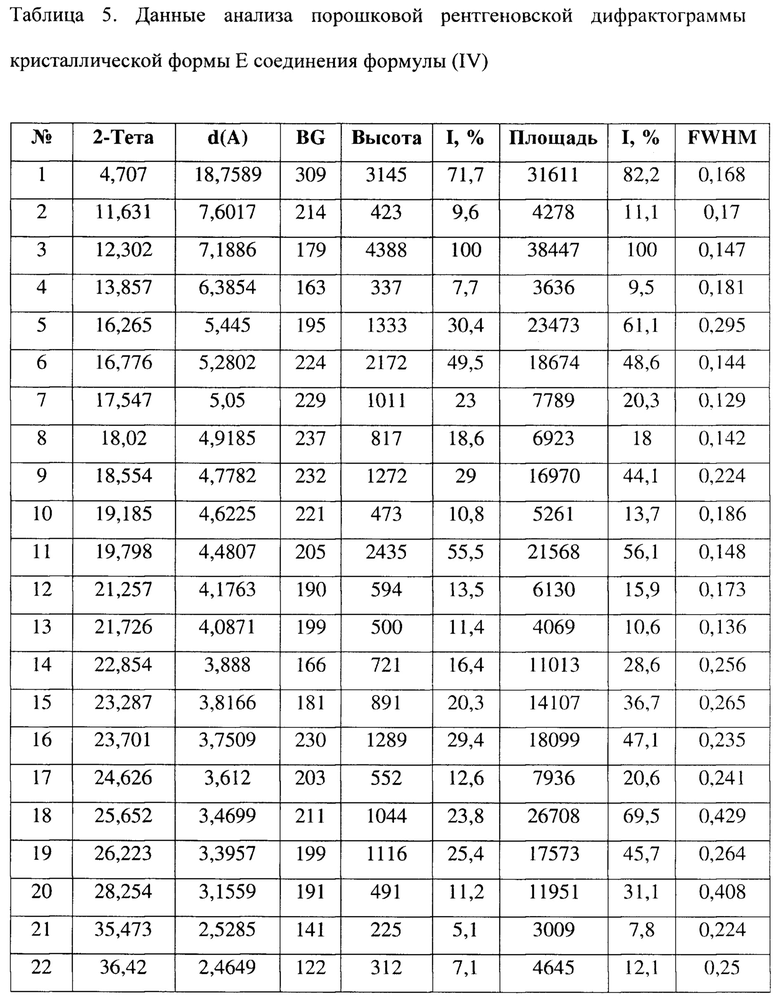
[0016] Настоящее изобретение также предусматривает кристаллическую форму Е соединения формулы (IV), где порошковая рентгеновская дифрактограмма кристаллической формы Е содержит характеристические дифракционные пики при углах 2θ, составляющих 4,71±0,2°, 12,30±0,2°, 16,26±0,2°, 16,78±0,2°, 19,80±0,2°, 23,70±0,2°, 25,65±0,2°, 26,22±0,2°.
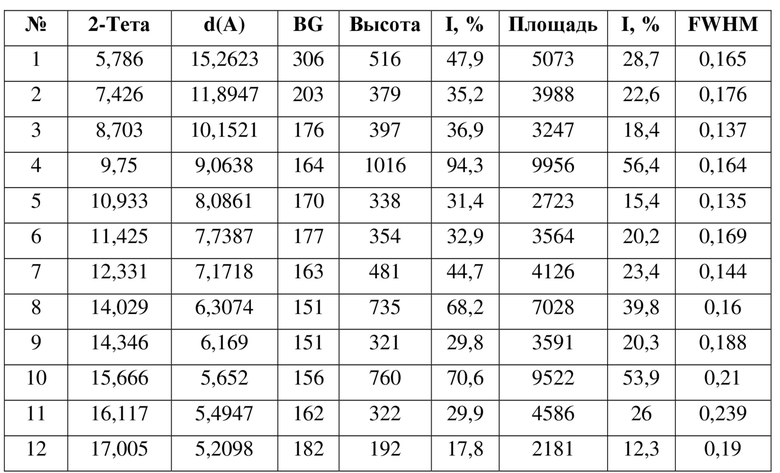
[0017] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы Е соединения формулы (IV) являются такими, как представлены в таблице 5.
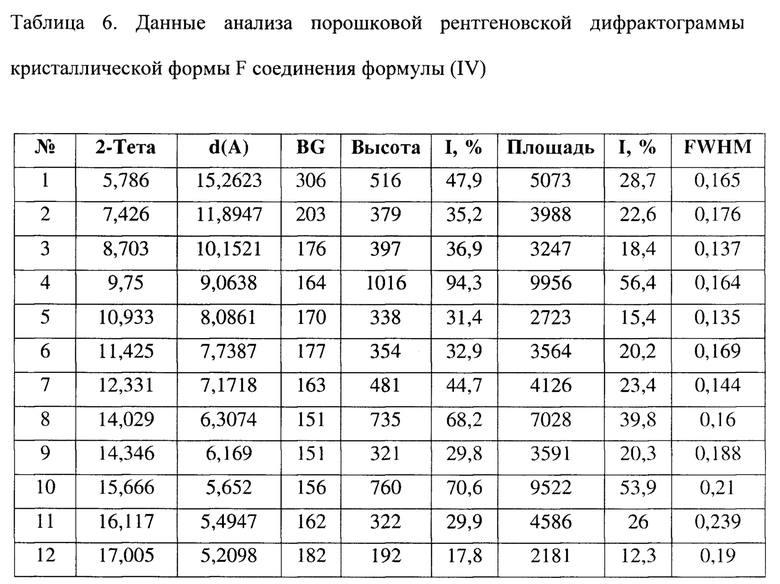
[0018] Настоящее изобретение также предусматривает кристаллическую форму F соединения формулы (IV), где порошковая рентгеновская дифрактограмма кристаллической формы F содержит характеристические дифракционные пики при углах 2θ, составляющих 5,79±0,2°, 9,75±0,2°, 14,03±0,2°, 15,67±0,2°, 17,46±0,2°, 18,86±0,2°, 20,42±0,2°, 20,99±0,2°.
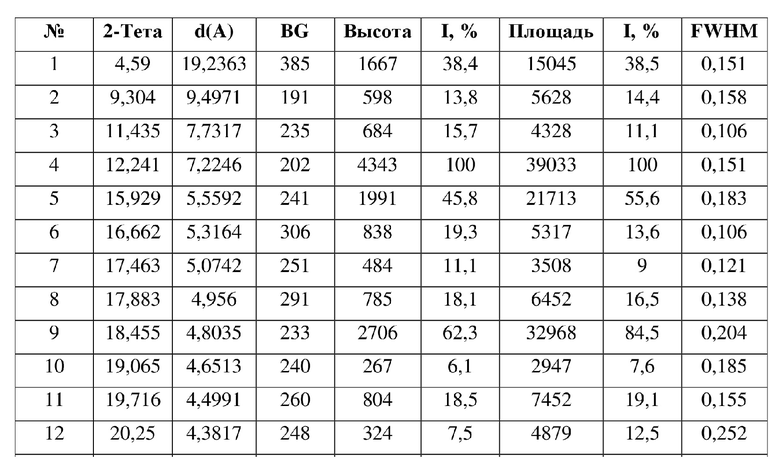
[0019] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы F соединения формулы (IV) являются такими, как представлены в таблице 6.
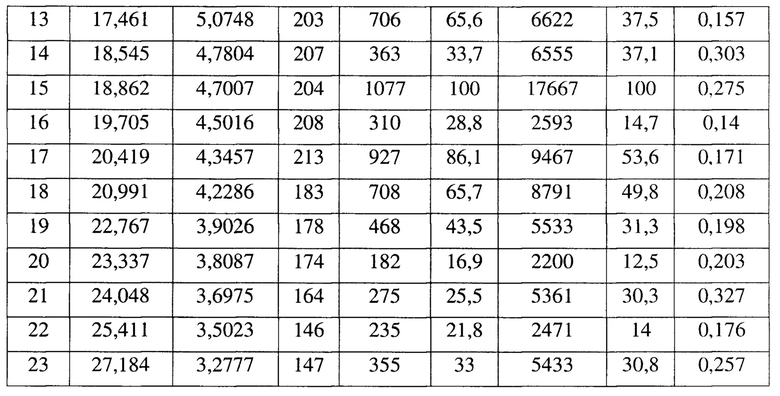
[0020] Настоящее изобретение также предусматривает кристаллическую форму G соединения формулы (V), где порошковая рентгеновская дифрактограмма кристаллической формы G содержит характеристические дифракционные пики при углах 2θ, составляющих 4,59±0,2°, 12,24±0,2°, 15,93±0,2°, 16,66±0,2°, 18,46±0,2°, 19,72±0,2°, 22,10±0,2°, 23,56±0,2°.
[0021] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы G соединения формулы (V) являются такими, как представлены в таблице 7.
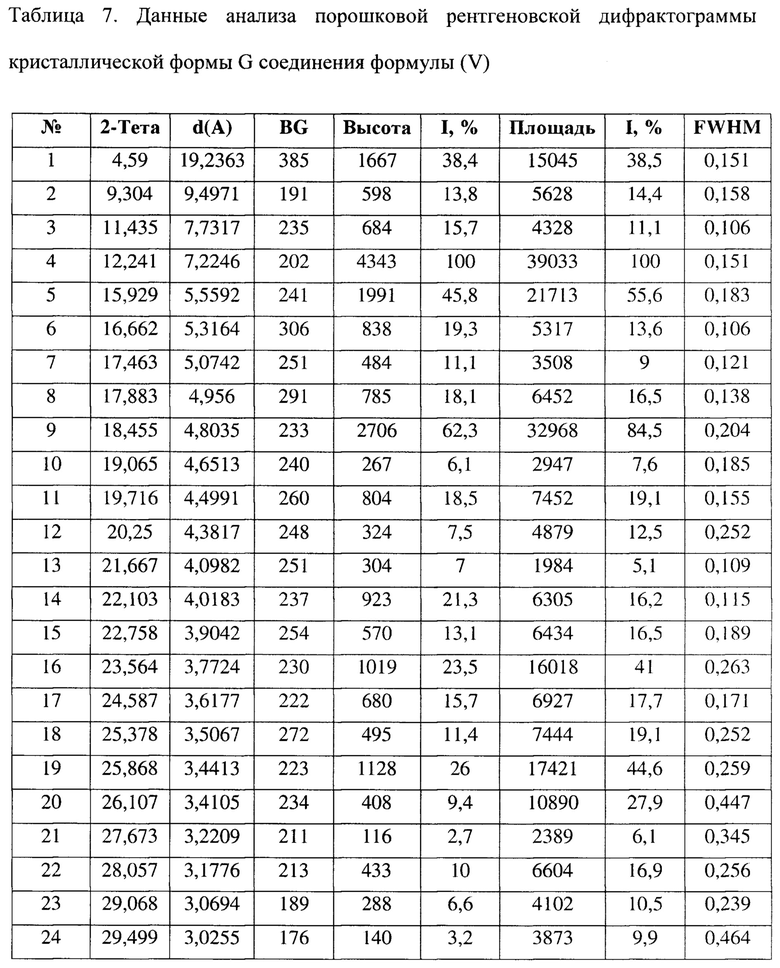
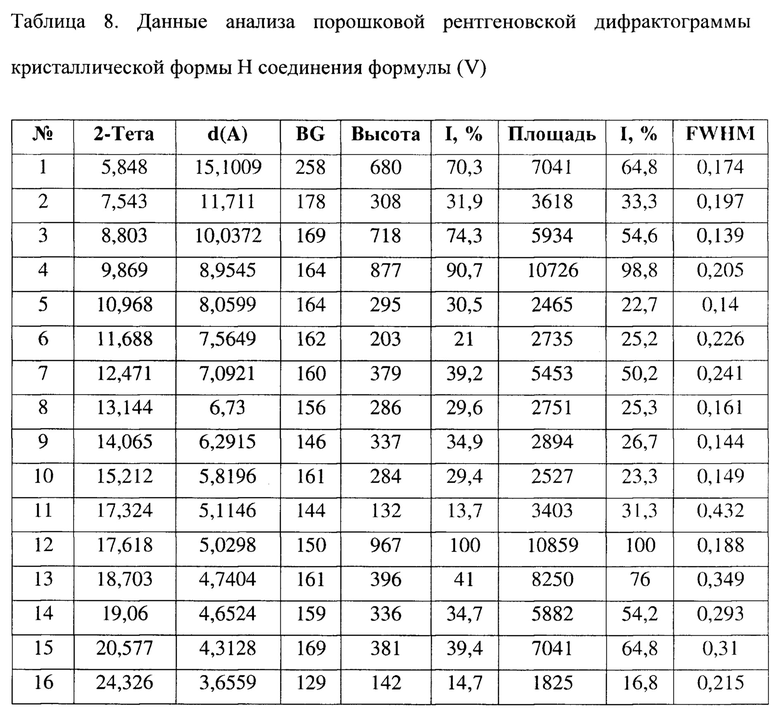
[0022] Настоящее изобретение также предусматривает кристаллическую форму Н соединения формулы (V), где порошковая рентгеновская дифрактограмма кристаллической формы Н содержит характеристические дифракционные пики при углах 2θ, составляющих 5,85±0,2°, 8,80±0,2°, 9,87±0,2°, 12,47±0,2°, 14,06±0,2°, 17,62±0,2°, 18,70±0,2°, 20,58±0,2°.
[0023] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы Н соединения формулы (V) являются такими, как представлены в таблице 8.
[0024] Настоящее изобретение также предусматривает способ получения вышеуказанных кристаллических форм, включающий приведение свободного основания в контакт с кислотой, промывание и высушивание.
[0025] Настоящее изобретение также предусматривает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения, описанного выше, или кристаллической(-их) формы(форм), описанной(-ых) выше, в качестве активного ингредиента и фармацевтически приемлемый носитель.
[0026] Настоящее изобретение также предусматривает применение соединения, описанного выше, в изготовлении лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии.
[0027] Настоящее изобретение также предусматривает применение кристаллической формы, описанной выше, в получении лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии.
[0028] Настоящее изобретение также предусматривает применение фармацевтической композиции, описанной выше, в получении лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии.
Технический эффект
[0029] Кристаллическая форма А и кристаллическая форма В соединения формулы (I), кристаллическая форма С соединения формулы (II), кристаллическая форма D соединения формулы (III), кристаллическая форма Е и кристаллическая форма F соединения формулы (IV), кристаллическая форма G и кристаллическая форма Н соединения формулы (V), предусмотренные в настоящем изобретении, характеризуются свойствами стабильности, хорошей растворимостью и хорошей гигроскопичностью. Такие кристаллические формы имеют хорошие перспективы в развитии фармацевтической промышленности.
[0030] Способ получения каждой кристаллической формы по настоящему изобретению является простым; не требуются жесткие условия и высокотоксичные растворители. Полученные кристаллические формы характеризуются высокой чистотой и хорошим выходом; они подходят для расширения масштаба производства до промышленного.
Определение
[0031] Если не определено иное, то используемые в данном документе термины и фразы имеют указанное ниже значение. Если определенные термин или фраза конкретно не заданы, то такие термин или фраза не должны считаться неопределенными. Скорее термины применяются в рамках их общепринятых значений. Используемые в данном документе торговые названия предназначены для обозначения соответствующих коммерческих продуктов или их активных ингредиентов.
[0032] Соединения по настоящему изобретению можно получить с помощью различных способов синтеза, хорошо известных специалистам в данной области, в том числе с помощью вариантов осуществления, описанных ниже, вариантов осуществления, объединяющих в себе варианты осуществления, описанные ниже, и другие способы синтеза, и эквивалентных альтернатив, известных специалистам в данной области. Предпочтительные варианты осуществления включают без ограничения такие варианты осуществления по настоящему изобретению.
[0033] Применяемые в настоящем изобретении растворители являются коммерчески доступными. Используемые в данном документе сокращения представляют собой следующее: водн. означает водный; HATU означает O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC означает N-(3-диметиламинопропил)-N'-этилкарбодиимид; м-СРВА означает 3-хлорпероксибензойную кислоту; экв. означает эквивалент; CDI означает карбонилдиимидазол; DCM означает дихлорметан; РЕ означает петролейный эфир; DIAD означает диизопропилазодикарбоксилат; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; МеОН означает метанол; CBz означает бензилоксикарбонил, который представляет собой защитную группу для аминогруппы; Boc означает трет-бутилкарбонил, который представляет собой защитную группу для аминогруппы; HOAc означает уксусную кислоту; NaCNBH3 означает цианоборгидрид натрия; к.т. означает комнатную температуру; O/N означает в течение ночи; THF означает тетрагидрофуран; Вос2О означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SOCl2 означает тионилхлорид; CS2 означает сероуглерод; TsOH означает п-толуолсульфоновую кислоту; NFSI означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS означает 1-хлорпирролидин-2,5-дион; н-Bu4NF означает тетрабутиламмония фторид; iPrOH означает 2-пропанол; т.пл. означает точку плавления; LDA означает диизопропиламид лития; CDCl3 означает дейтерированный хлороформ; ЕА означает этилацетат; MeOD означает дейтерированный метанол; IPA означает изопропанол; PDE означает фосфодиэстеразу; AMP означает аденозинмонофосфат; GMP означает гуанозинмонофосфат.
[0034] Применяемые в настоящем изобретении растворители являются коммерчески доступными. Коммерчески доступные соединения указаны вместе с названиями по каталогу, предоставленными поставщиками.
[0035] Способ порошковой рентгеновской дифракции соответствует следующему.
[0036] Оборудование: рентгеновский дифрактометр Bruker D8 ADVANCE; мишень: Cu: K-Alpha; длина волны λ=1,54179 Å; напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 4-40°; скорость вращения образца: 15 об./мин.; скорость сканирования: 10°/ мин.
ПОДРОБНОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0037] Настоящее изобретение подробно описано ниже с помощью вариантов осуществления, но данные варианты осуществления не предназначены для ограничения настоящего изобретения. В данном документе было подробно описано настоящее изобретение, также в данном документе раскрыты варианты осуществления настоящего изобретения, при этом специалисту в данной области будет очевидно, что в вариантах осуществления настоящего изобретения могут быть сделаны различные модификации и изменения без отклонения от сущности и объема настоящего изобретения.
ЭТАЛОННЫЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 1. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ 1
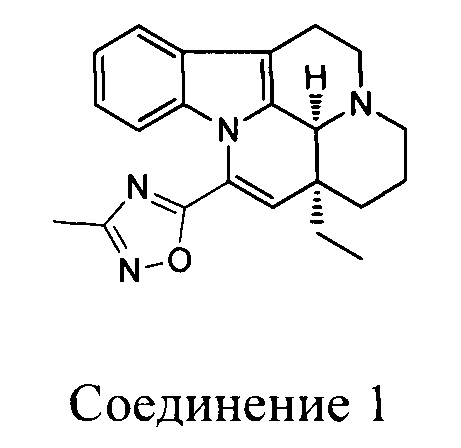
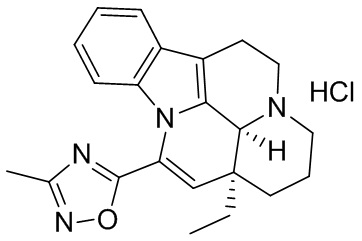
[0038] К раствору (41S,13аS)-13а-этил-2,3,41,5,6,13а-гексагидро-1Н-индоло[3,2,1-de]пиридо[3,2,1-ij][1,5]нафтиридин-12-карбоновой кислоты (14 г, 43,4 ммоль), 1-гидроксибензотриазола (300 мг, 2,17 ммоль) и триэтиламина (31 мл, 217 ммоль) в N,N-диметилформамиде (200 мл) добавляли тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (14,6 г, 45,6 ммоль) и гидрохлорид N-гидроксиацетамидина (5,28 г, 47,8 ммоль) в указанном порядке и реакционную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли солевой раствор, затем полученную смесь фильтровали и фильтрат разбавляли водой и экстрагировали с помощью дихлорметана. Экстрагированное вещество высушивали над безводным сульфатом натрия, выпаривали низкокипящие компоненты. Оставшийся неочищенный продукт в N,N-диметилформамиде непосредственно нагревали под действием микроволнового излучения до 160°С и подвергали реакции в течение 50 мин. Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии с использованием щелочи с получением целевого соединения (4,0 г, выход: 25%).
[0039] 1Н ЯМР (CDCl3, 400 МГц) δppm 7,46 (d, J=6,8 Гц, 1H), 7,13-7,06 (m, 2Н), 6,73 (d, J=8,0 Гц, 1H), 6,08 (s, 1H), 4,23 (s, 1H), 3,38-3,34 (m, 2Н), 3,29-3,28 (m, 2Н), 2,65-2,63 (m, 2Н), 2,55-2,51 (m, 1Н), 2,51 (s, 3Н), 1,97-1,92 (m, 2Н), 1,59-1,55 (m, 2Н), 1,45-1,41 (m, 1H), 1,11-1,10 (m, 1H), 1,00 (t, J=7,2 Гц, 3Н).
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 1. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ ФОРМУЛЫ (I) И ЕГО КРИСТАЛЛИЧЕСКИХ ФОРМ
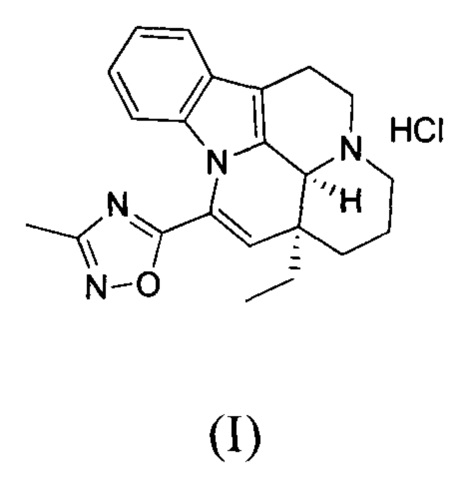
[0040] Получение соединения формулы (I)
[0041] Соединение 1 (15,00 г, 41,61 ммоль, 1,00 экв.) помещали в трехгорлую колбу объемом 500 мл. Добавляли 150 мл этилацетата и 15 мл дихлорметана, реакционную систему заменяли три раза азотом. К реакционной жидкости добавляли по каплям 1 н. HCl/EA (60 мл). Реакционную смесь перемешивали при 25°С в течение 30 минут, что приводило к появлению большого количества белого твердого вещества; белое твердое вещество затем фильтровали. Фильтрационный осадок промывали однократно с помощью 50 мл этилацетата. Высушивали фильтрационный осадок с получением белого продукта (15,00 г, 37,79 ммоль, 90,82%). 1Н ЯМР (400 МГц, CDCl3)=7,52 (dd, J=1,8 Гц, 1Н), 7,24-7,22 (m, 2H), 6,78 (dd, J=4,0 Гц, 1H), 6,14 (s, 1H), 4,78 (s, 1H), 3,83-3,66 (m, 2H), 3,32-3,01 (m, 4H), 2,55 (s, 3H), 2,33-2,25 (m, 3H), 1,81-1,68 (m, 1H), 1,28-1,27 (m, 1H), 1,12 (t, J=8,0 Гц, 3Н).
[0042] Получение кристаллической формы А
[0043] Примерно 50 мг соединения формулы (I) добавляли к метанолу (1,5 мл). Суспензию перемешивали при 40°С в течение трех дней. Оставшееся твердое вещество центрифугировали (10 мин. при 14000 об./мин.) для отделения и высушивали в течение ночи в вакуумной печи при 40°С с получением кристаллической формы А.
[0044] Получение кристаллической формы В
[0045] Кристаллическую форму В получали с применением способа получения кристаллической формы А, за исключением того, что метанол заменяли ацетоном для получения кристаллической формы В.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 2. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ ФОРМУЛЫ (II) И ЕГО КРИСТАЛЛИЧЕСКОЙ ФОРМЫ С
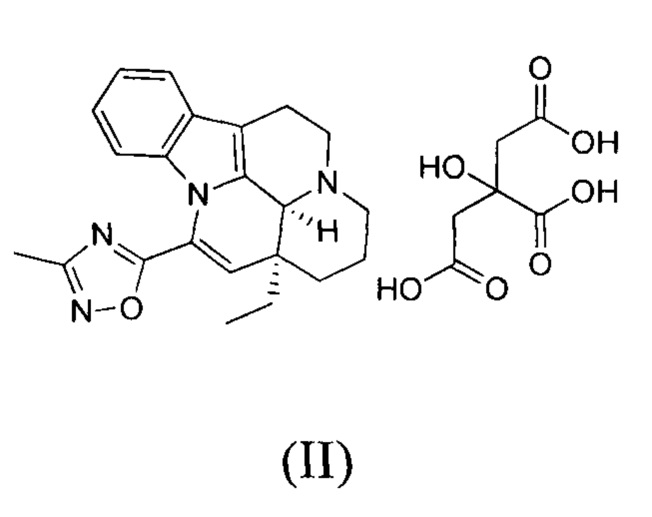
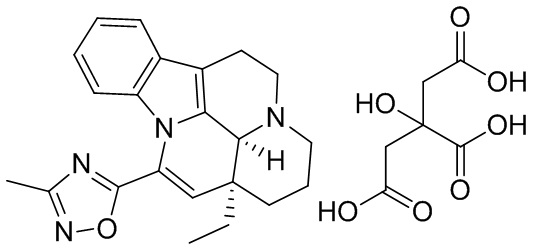
[0046] В трехгорлую колбу объемом 100 мл добавляли соединение 1 (2,00 г, 5,55 ммоль, 1,00 экв.) и лимонную кислоту (1,17 г, 6,11 ммоль, 1,10 экв.); также добавляли 30 мл этанола и реакционную систему заменяли три раза азотом. Температуру реакционной смеси повышали до 85-95°С. Когда внутренняя температура достигала 45-60°С, реакционный раствор становился прозрачным. Когда внутренняя температура достигала 60°С или выше, начинала проявляться мутность. Температуру реакционной смеси поддерживали на уровне 85-95°С и реакционную смесь перемешивали в течение 30 минут, что приводило к появлению большого количества белого твердого вещества. Нагревание прекращали и понижали внутреннюю температуру до 20-30°С с последующим фильтрованием. Фильтрационный осадок промывали однократно с помощью 200 мл этанола. Фильтрационный осадок высушивали с получением белого продукта (2,50 г, 4,56 ммоль, 82,21%), который представлял собой кристаллическую форму С.
[0047] 1Н ЯМР (400 МГц, MeOD) ppm 7,50-7,70 (m, 1Н), 7,06-7,30 (m, 2H), 6,64-6,84 (m, 1H), 6,21 (s, 1H), 3,69-3,76 (m, 2H), 2,94-3,13 (m, 2H), 2,87 (dd, J=15,56, 1,00 Гц, 2H), 2,77 (d, J=15,31 Гц, 2H), 2,52 (s, 3Н), 1,89-2,15 (m, 3Н), 1,11 (t, J=7,40 Гц, 3Н).
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 3. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ ФОРМУЛЫ (III) И ЕГО КРИСТАЛЛИЧЕСКОЙ ФОРМЫ D
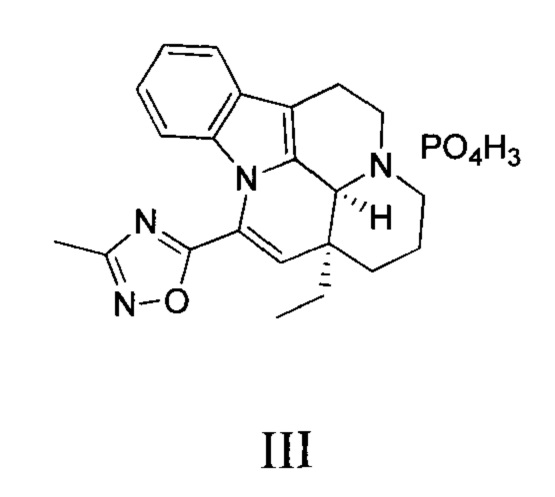
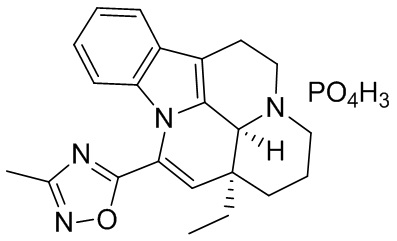
[0048] Получение соединения формулы (III)
[0049] Помещали соединение 1 (1,00 г, 2,77 ммоль, 1,00 экв.) в трехгорлую колбу объемом 100 мл; также добавляли 15 мл этанола и реакционную систему заменяли три раза азотом. В реакционную жидкость добавляли по каплям фосфорную кислоту (319,36 мг, 2,77 ммоль, 1,00 экв.). Температуру реакционной смеси повышали до 60°С, реакционную смесь поддерживали при температуре 60°С и перемешивали в течение 30 минут, что приводило к появлению большого количества белого твердого вещества. Нагревание прекращали; когда внутренняя температура понижалась до 20-30°С, реакционную смесь фильтровали. Фильтрационный осадок промывали однократно с помощью 20 мл этанола. Фильтрационный осадок высушивали с получением белого продукта (1,20 г, 2,61 ммоль, 94,30%). 1Н ЯМР (400 МГц, MeOD) = 7,57 (d, J=6,8 Гц, 1Н), 7,17 (t, J=6,0 Гц, 2H), 6,72 (d, J=7,5 Гц, 1H), 6,20 (s, 1H), 3,75 (d, J=6,3 Гц, 2H), 3,23 (d, J=15,1 Гц, 2H), 3,14-2,96 (m, 2H), 2,50 (s, 3Н), 2,14-1,96 (m, 3Н), 1,83-1,64 (m, 2H), 1,28-1,21 (m, 1H), 1,10 (t, J=7,3 Гц, 3Н).
[0050] Получение кристаллической формы D
[0051] Примерно 30 мг соединения формулы (III) добавляли к этанолу (0,5 мл) и перемешивали при 40°С в течение трех дней. Оставшееся соединение центрифугировали (10 мин. при 14000 об./мин.) для отделения и высушивали в течение ночи в вакуумной печи при 40°С с получением сухого твердого вещества, которое представляло собой кристаллическую форму D.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 4. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ ФОРМУЛЫ (IV) И ЕГО КРИСТАЛЛИЧЕСКИХ ФОРМ
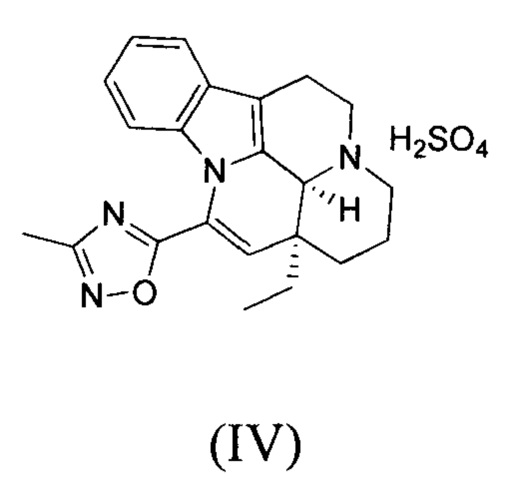
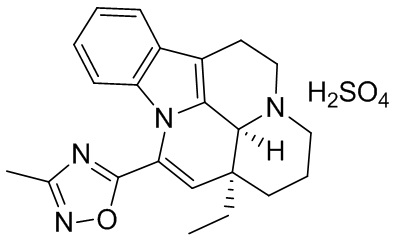
[0052] Получение соединения формулы (IV)
[0053] Соединение 1 (1,00 г, 2,77 ммоль, 1,00 экв.) добавляли в трехгорлую колбу объемом 100 мл, также добавляли 15 мл этилацетата и 3 мл дихлорметана; реакционную систему заменяли три раза азотом. В реакционную жидкость добавляли по каплям 1 мл серной кислоты (272,10 мг, 2,77 ммоль, 1,00 экв.), разбавленной водой. Температуру реакционной смеси поддерживали на уровне 25°С и осуществляли перемешивание в течение 30 минут, что приводило к появлению большого количества белого твердого вещества. Белое твердое вещество фильтровали, фильтрационный осадок промывали однократно с помощью 10 мл этилацетата. Фильтрационный осадок высушивали с получением белого продукта (1,10 г, 2,40 ммоль, 86,61%). 1Н ЯМР (400 МГц, MeOD) = 7,65-7,53 (m, 1Н), 7,25-7,12 (m, 2H), 6,79-6,68 (m, 1H), 6,19 (s, 1H), 5,02 (s, 1H), 3,90-3,74 (m, 2H), 3,34 (d, J=12,3 Гц, 1H), 3,26-3,04 (m, 3H), 2,48 (s, 3H), 2,06-1,89 (m, 3H), 1,81-1,67 (m, 2H), 1,29-1,16 (m, 1H), 1,10 (t, J=7,4 Гц, 3Н).
[0054] Получение кристаллической формы Е
[0055] Приблизительно 30 мг соединения формулы (IV) добавляли к растворителю IPA:H2O = 1:9 (0,5 мл) и перемешивали при 40°С в течение трех дней. Оставшееся соединение центрифугировали (10 мин. при 14000 об./мин.) для отделения и высушивали в течение ночи в вакуумной печи при 40°С с получением сухого твердого вещества, которое представляло собой кристаллическую форму Е.
[0056] Получение кристаллической формы F
[0057] Кристаллическую форму F получали с применением способа получения кристаллической формы Е, за исключением того, что растворитель IPA:H2O = 1:9 заменяли этанолом для получения кристаллической формы Е.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ 5. ПОЛУЧЕНИЕ СОЕДИНЕНИЯ ФОРМУЛЫ (V) И ЕГО КРИСТАЛЛИЧЕСКИХ ФОРМ
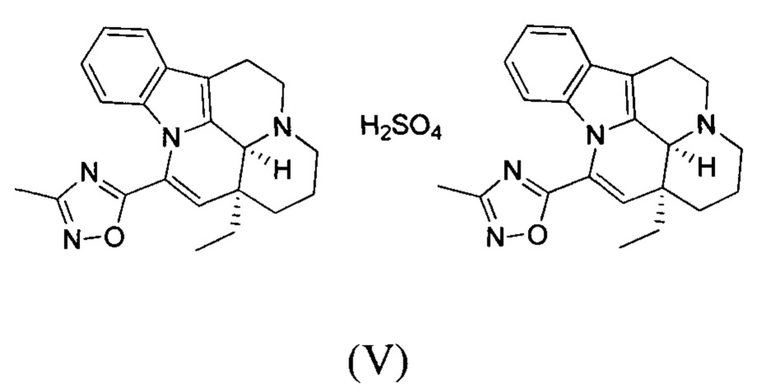
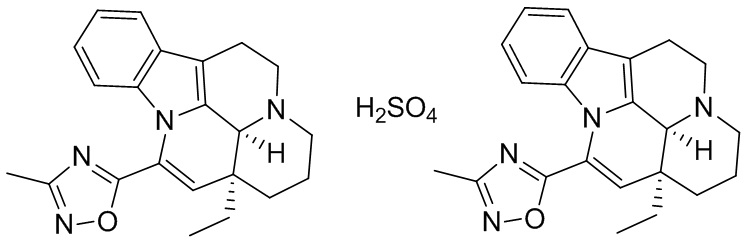
[0058] Получение соединения формулы (V)
[0059] Соединение 1 (1,00 г, 2,77 ммоль, 1,00 экв.) добавляли в трехгорлую колбу объемом 100 мл, также добавляли 15 мл этилацетата и 3 мл дихлорметана; реакционную систему заменяли три раза азотом. В реакционную жидкость добавляли по каплям 1 мл серной кислоты (135,84 мг, 1,39 ммоль, 0,50 экв.), разбавленной водой. Температуру реакционной смеси поддерживали на уровне 25°С и реакционную смесь перемешивали в течение 30 минут, что приводило к появлению большого количества белого твердого вещества. Белое твердое вещество фильтровали, фильтрационный осадок промывали однократно с помощью 10 мл этилацетата. Фильтрационный осадок высушивали с получением белого продукта (500,00 мг, 1,09 ммоль, 39,37%). 1Н ЯМР (400 МГц, MeOD) = 7,59 (dd, J=2,4, 4,6 Гц, 1Н), 7,27-7,08 (m, 2H), 6,73 (d, J=7,5 Гц, 1H), 6,29-6,13 (m, 1H), 5,06 (d, J=14,8 Гц, 1H), 3,93-3,73 (m, 2H), 3,47-3,31 (m, 1H), 3,28-3,02 (m, 3H), 2,53-2,41 (m, 3H), 2,11-1,88 (m, 3H), 1,77 (d, J=4,8 Гц, 2H), 1,25 (d, J=10,3 Гц, 1H), 1,15-1,02 (m, 3H).
[0060] Получение кристаллической формы G
[0061] Примерно 30 мг соединения формулы (V) добавляли к растворителю (0,5 мл). Реакционную смесь перемешивали при 40°С в течение трех дней. Проводили центрифугирование (10 мин. при 14000 об./мин.) для отделения оставшегося соединения в виде твердого вещества; выделенное соединение высушивали под вакуумом при 40°С в течение ночи. Получали сухое твердое вещество, которое представляло собой кристаллическую форму G.
[0062] Получение кристаллической формы Н
[0063] Кристаллическую форму Н получали с применением способа получения кристаллической формы G, за исключением того, что растворитель IPA:H2O = 1:9 заменяли EtOAc для получения кристаллической формы Н.
[0064] Экспериментальный пример 1: выявление in vitro фосфодиэстеразы (PDE)
[0065] Принцип эксперимента
[0066] В анализе измеряли активность фермента PDE1A на основе выявления флуоресцентной поляризации при выработке AMP/GMP. Принцип реакции заключался в связывании AMP/GMP с его антителом, при этом замещая AMP/GMP с меткой AlexaFluor 633.
[0067] Экспериментальные реагенты:
[0068] Реакционный буфер: 10 мМ Трис-HCl, рН=7,5, 5 мМ хлорид магния, 0,01% Brij 35, 1 мМ DTT и 1% DMSO;
[0069] ферментный субстрат: 1 М сАМР или cGMP (Са2+-кальмодулин в качестве кофактора PDE1A);
[0070] реагенты для выявления:
[0071] антитело к AMP2/GMP2 Transcreener ®;
[0072] AMP2/GMP2 с маркером AlexaFluor 633.
[0073] Экспериментальные процедуры и способы
[0074] 1. Фермент человека, подлежащий тестированию (приобретенный от SignalChem), и субстрат разбавляли свежеприготовленным реакционным буфером.
[0075] 2. Раствор фермента (концентрация 3 пМ) добавляли в лунки реакционного планшета.
[0076] 3. Применяли Echo 550 для добавления ряда растворов соединения в 100% DMSO в лунки реакционного планшета при необходимых концентрациях; указанные лунки реакционного планшета содержали раствор фермента. Реакционный планшет затем инкубировали при комнатной температуре в течение 10 минут.
[0077] 4. Субстратный раствор добавляли в лунки реакционного планшета, содержащие фермент и раствор соединения, для инициирования реакции.
[0078] 5. Реакционный планшет инкубировали в течение 1 часа при комнатной температуре со встряхиванием.
[0079] 6. Добавляли смесь для выявления (стоп-буфер с антителом и меченым веществом) для остановки ферментативной реакции; реакционный планшет инкубировали в течение 90 минут со встряхиванием.
[0080] 7. Применяли следующие устройства для выявления: EnVision (PerkinElmer), Cy5FP Ex FP 620, Em S-pol 688/P-pol 688, D658fp/D688 с зеркалом FP; флуоресцентную поляризацию выявляли с применением Ex/Em 620/688.
[0081] Анализ данных.
[0082] Ферментативную активность, соответствующую сигналу FP, определяли с помощью стандартной кривой AMP/GMP с применением DMSO в качестве контроля в таблице Excel. Ферментативную активность преобразовывали в концентрацию продукта в нМ. Анализ проводили с применением GraphPad Prism и рассчитывали значения IC50.
[0083] Результаты эксперимента представлены в таблице 1.

Настоящее изобретение относится к гидрохлоридной соли, цитратной соли, фосфатной соли или сульфатной соли соединения 1, а также к кристаллическим формам вышеуказанных солей и способу их получения. Соединение по изобретению предназначено для получения лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии. 11 н. и 9 з.п. ф-лы, 9 табл.
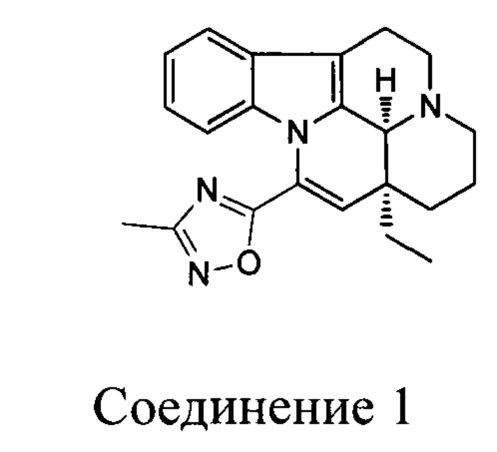
1. Гидрохлоридная соль, цитратная соль, фосфатная соль или сульфатная соль соединения 1:
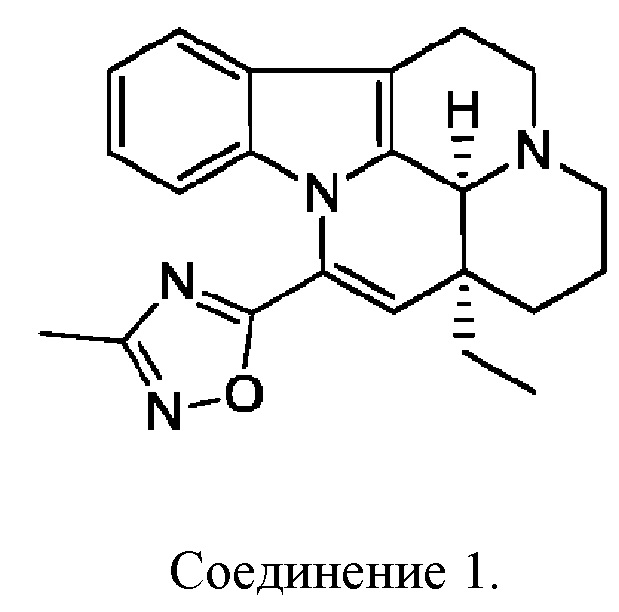
2. Гидрохлоридная соль, цитратная соль, фосфатная соль или сульфатная соль соединения 1 по п. 1, выбранная из:
(I),
(II),
(III),
(IV) или
(V).
3. Кристаллическая форма A соединения формулы (I) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы A содержит характеристические дифракционные пики при углах 2Θ, составляющих 9,14±0,2°, 10,43±0,2°, 11,38±0,2°, 12,54±0,2°, 13,86±0,2°, 19,04±0,2°, 19,36±0,2°, 21,00±0,2°.
4. Кристаллическая форма A соединения формулы (I) по п. 3, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
5. Кристаллическая форма B соединения формулы (I) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы B содержит характеристические дифракционные пики при углах 2Θ, составляющих 9,17±0,2°, 11,75±0,2°, 12,16±0,2°, 12,67±0,2°, 15,14±0,2°, 17,81±0,2°, 20,54±0,2°, 22,34±0,2°.
6. Кристаллическая форма B соединения формулы (I) по п. 5, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
7. Кристаллическая форма C соединения формулы (II) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы C содержит характеристические дифракционные пики при углах 2Θ, составляющих 14,04±0,2°, 16,28±0,2°, 16,70±0,2°, 17,73±0,2°, 18,18±0,2°, 20,29±0,2°, 23,40±0,2°, 25,95±0,2°.
8. Кристаллическая форма C соединения формулы (II) по п. 7, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
9. Кристаллическая форма D соединения формулы (III) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы D содержит характеристические дифракционные пики при углах 2Θ, составляющих 4,47±0,2°, 9,80±0,2°, 10,67±0,2°, 13,05±0,2°, 16,30±0,2°, 16,80±0,2°, 17,65±0,2°, 17,82±0,2°.
10. Кристаллическая форма D соединения формулы (III) по п. 9, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
11. Кристаллическая форма E соединения формулы (IV) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы E содержит характеристические дифракционные пики при углах 2Θ, составляющих 4,71±0,2°, 12,30±0,2°, 16,26±0,2°, 16,78±0,2°, 19,80±0,2°, 23,70±0,2°, 25,65±0,2°, 26,22±0,2°.
12. Кристаллическая форма E соединения формулы (IV) по п. 11, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
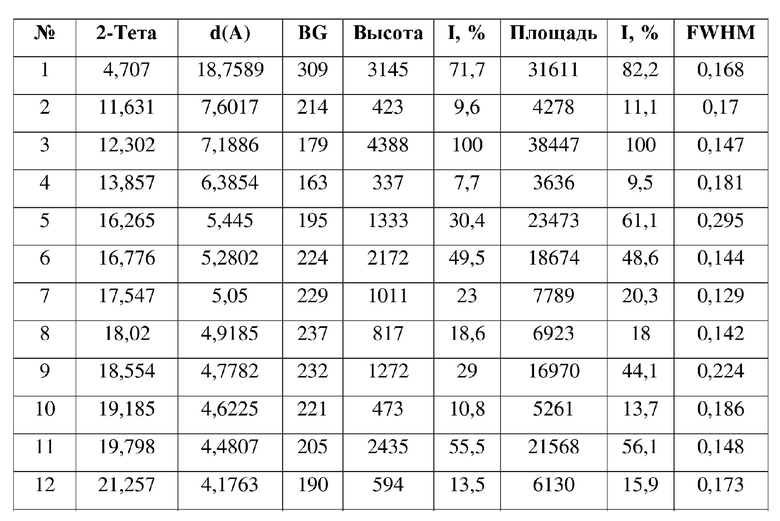

13. Кристаллическая форма F соединения формулы (IV) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы F содержит характеристические дифракционные пики при углах 2Θ, составляющих 5,79±0,2°, 9,75±0,2°, 14,03±0,2°, 15,67±0,2°, 17,46±0,2°, 18,86±0,2°, 20,42±0,2°, 20,99±0,2°.
14. Кристаллическая форма F соединения формулы (IV) по п. 13, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:

15. Кристаллическая форма G соединения формулы (V) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы G содержит характеристические дифракционные пики при углах 2Θ, составляющих 4,59±0,2°, 12,24±0,2°, 15,93±0,2°, 16,66±0,2°, 18,46±0,2°, 19,72±0,2°, 22,10±0,2°, 23,56±0,2°.
16. Кристаллическая форма G соединения формулы (V) по п. 15, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
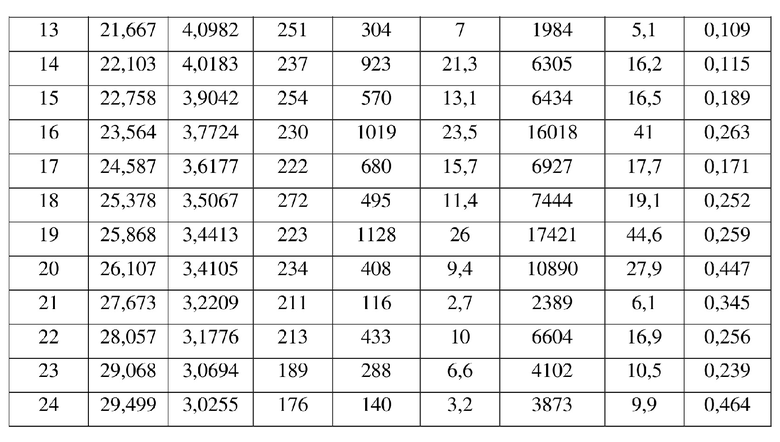
17. Кристаллическая форма H соединения формулы (V) по п. 2, где порошковая рентгеновская дифрактограмма кристаллической формы H содержит характеристические дифракционные пики при углах 2Θ, составляющих 5,85±0,2°, 8,80±0,2°, 9,87±0,2°, 12,47±0,2°, 14,06±0,2°, 17,62±0,2°, 18,70±0,2°, 20,58±0,2°.
18. Кристаллическая форма H соединения формулы (V) по п. 17, где данные анализа ее порошковой рентгеновской дифрактограммы представлены в таблице:
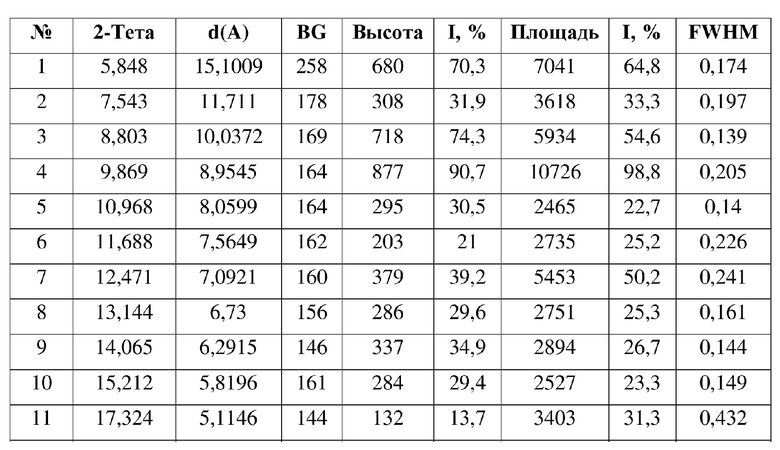
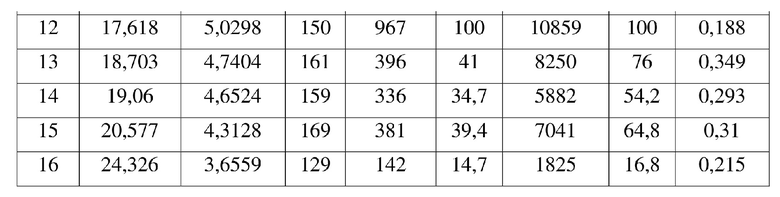
19. Способ получения кристаллической формы по любому из пп. 3-18,
где кристаллическую форму А получают следующим образом:
50 мг соединения формулы (I) добавляют к 1,5 мл метанола с образованием реакционной смеси; полученную реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы А;
где кристаллическую форму В получают следующим образом:
50 мг соединения формулы (I) добавляют к 1,5 мл ацетона с образованием реакционной смеси; полученную реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы В;
где кристаллическую форму С получают следующим образом:
1,00 экв. соединения 1 и 1,10 экв. лимонной кислоты добавляют к 30 мл этанола с образованием реакционной смеси; реакционную смесь перемешивают при 85-95°C; понижают температуру реакционной смеси до 20-30°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы С;
где кристаллическую форму D получают следующим образом:
30 мг соединения формулы (III) добавляют к 0,5 мл этанола с образованием реакционной смеси; реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы D;
где кристаллическую форму Е получают следующим образом:
30 мг соединения формулы (IV) добавляют к 0,5 мл растворителя IPA:H2O = 1:9 с образованием реакционной смеси; реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы Е;
где кристаллическую форму F получают следующим образом:
30 мг соединения формулы (IV) добавляют к 0,5 мл этанола с образованием реакционной смеси; реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы F;
где кристаллическую форму G получают следующим образом:
30 мг соединения формулы (V) добавляют к 0,5 мл растворителя IPA:H2O = 1:9 с образованием реакционной смеси; реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы G;
где кристаллическую форму H получают следующим образом:
30 мг соединения формулы (V) добавляют к 0,5 мл EtOAc с образованием реакционной смеси; реакционную смесь перемешивают при 40°C; отделяют и высушивают твердое вещество в реакционной смеси с получением кристаллической формы H.
20. Применение соединения по п. 1 или кристаллической формы по любому из пп. 3-7 в получении лекарственного препарата, предназначенного для лечения церебрального инсульта или эпилепсии.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Adam Vas, Balazs Gulyas: "Eburnamine derivatives and the Brain", MEDICINAL RESEARCH REVIEWS, 2005, v.25, no.6, p.737-757 | |||
| ДИАЗАБЕНЗОФТОРАНТРЕНОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2697513C2 |
| Способ получения производных эбурнамина | 1972 |
|
SU438183A1 |
| US 4749707 A, 07.06.1988 | |||
| Питательный кран для вагонных резервуаров воздушных тормозов | 1921 |
|
SU189A1 |

