[001] По настоящей заявке испрашивается преимущество приоритета в соответствии с китайской заявкой № 202011521011.6, поданной 21 декабря 2020 года.
ОБЛАСТЬ ТЕХНИКИ
[002] Настоящее изобретение относится к солевой форме соединения изохинолинонового типа в качестве ингибитора ROCK и способу ее получения, а также включает применение солевой формы в получении лекарственного средства для лечения глаукомы или глазной гипертензии.
УРОВЕНЬ ТЕХНИКИ
[003] Rho-ассоциированная киназа (ROCK), серин/треониновая протеинкиназа, представляет собой нижележащую эффекторную молекулу RHO и экспрессируется повсеместно в организме человека. ROCK вовлечена в регуляцию легкой цепи миозина (ЛЦМ). Подходит для лечения вазодилатации. Также ROCK может воздействовать на трабекулярные клетки пути оттока, уменьшая сопротивление оттоку водянистой влаги за счет расслабления трабекулярных клеток. Недавние исследования показывают, что ингибиторы ROCK также могут способствовать восстановлению эндотелиальных клеток роговицы и предотвращать фиброз, что имеет большие перспективы применения.
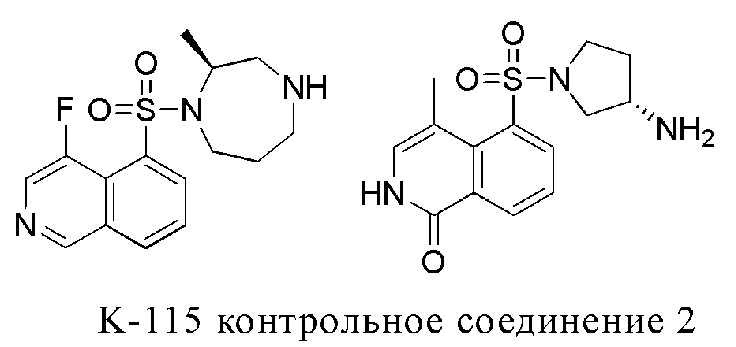
[004] Изохинолинсульфонамидные соединения представляют собой важный класс ингибиторов ROCK. Фасудил и K-115 (WO2006057397A1), которые были выпущены в настоящее время, оба представляют собой изохинолинсульфонамидные соединения. Фасудил, как новый лекарственный препарат с обширным фармакологическим действием, является ингибитором RHO-киназы, который расширяет кровеносные сосуды за счет повышения активности фосфатазы легких цепей миозина, снижает напряжение эндотелиальных клеток, улучшает микроциркуляцию в тканях головного мозга, не вызывая и не усугубляя внутримозговое «обкрадывание». И в то же время он может противодействовать факторам воспаления, защищать нервы, противостоять апоптозу и способствовать регенерации нервов. Тем не менее одобрение K-115 имеет очень широкое потенциальное применение, включая глаукому, глазную гипертензию, осложнения диабетического поражения сетчатки, возрастную макулодистрофию, повреждение роговицы, восстановление после хирургического лечения катаракты и глаукомы и т.д., и может быть дополнительно расширено до лекарственных препаратов системного действия.
[005] В WO2007026664A1 сообщается о ряде соединений с ингибирующим действием в отношении ROCK, таких как контрольное соединение 2, которые обладают хорошей ферментативной активностью, но нуждаются в улучшении с точки зрения мембранной проницаемости, фармакокинетики, лекарственной ориентированности и других аспектов. В настоящем изобретении сообщается о классе подобных соединений, которые были структурно модифицированы и имеют значительно улучшенные свойства в этом отношении.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
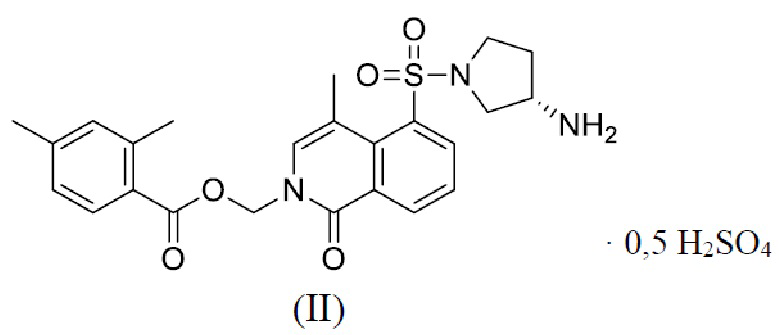
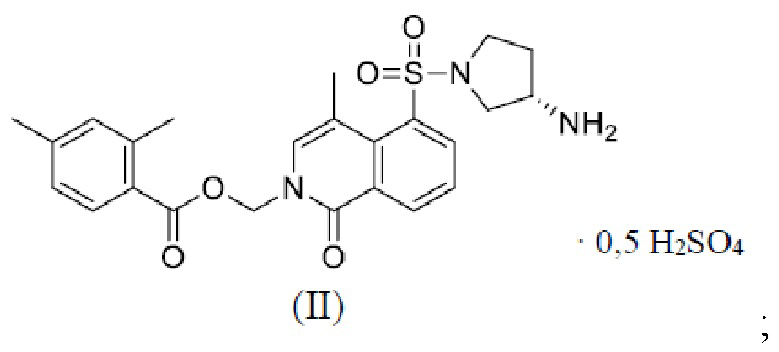
[006] В настоящем изобретении предложено соединение формулы (II),
[007] В настоящем изобретении также предложена кристаллическая форма А соединения формулы (II), причем порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 6,33±0,20°, 10,62±0,20° и 13,11±0,20°,
[008] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы А содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 6,55°± 0,20°, 10,62±0,20°, 12,57±0,20° и 13,11±0,20°.
[009] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы А содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 10,62±0,20°, 12,57±0,20°, 13,11±0,20°, 17,85±0,20°, 18,51±0,20° и 20,99±0,20°.
[0010] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы А содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 6,55±0,20°, 10,62±0,20°, 12,57±0,20°, 13,11±0,20°, 14,20±0,20°, 16,37±0,20°, 17,85±0,20°, 18,51±0,20°, 19,56±0,20°, 20,99±0,20°, 25,53±0,20° и 26,35±0,20°.
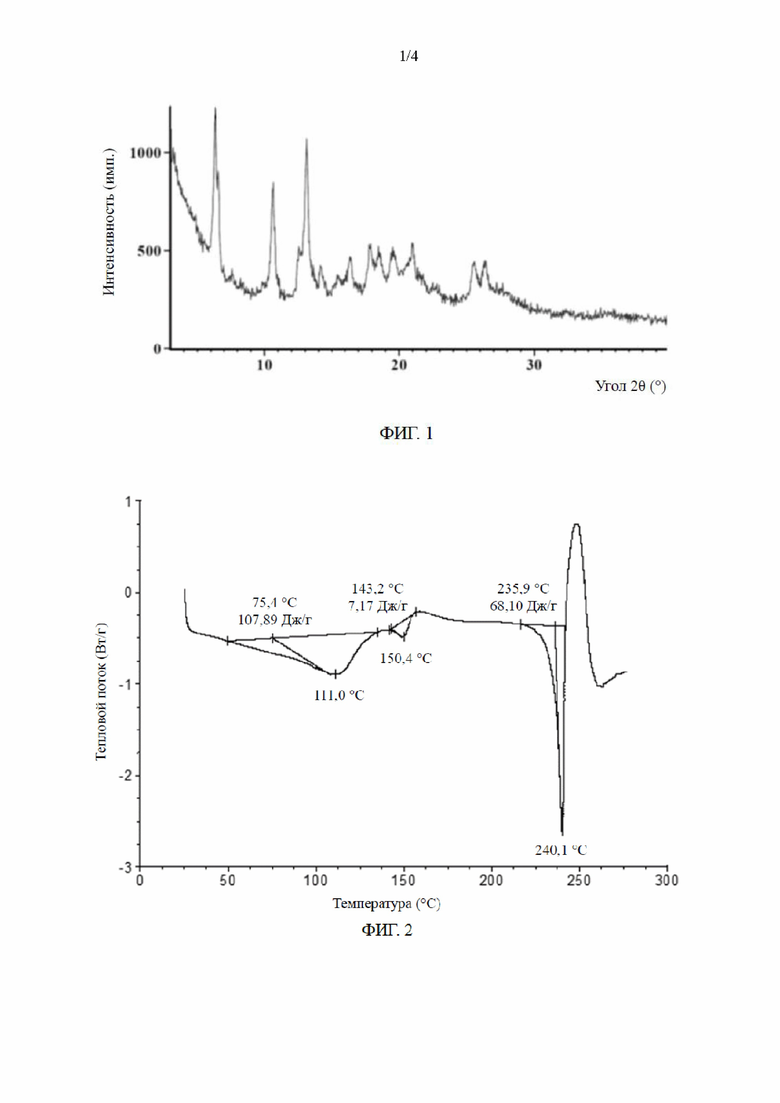
[0011] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы А по сути является такой, как показано на ФИГ. 1.
[0012] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы А являются такими, как показано в табл. 1.
[0013] Таблица 1: Данные анализа порошковой рентгеновской дифрактограммыкристаллической формы А соединения формулы (II)
(Å)
(импульсы)
(%)
(Å)
(импульсы)
(%)
[0014] В некоторых вариантах осуществления настоящего изобретения порошковую рентгеновскую дифрактограмму кристаллической формы А получают с использованием источника облучения Cu-Kα.
[0015] В некоторых вариантах осуществления настоящего изобретения кривая дифференциальной сканирующей калориметрии кристаллической формы А содержит эндотермический пикс начальным значением при 235,9±3,0°C.
[0016] В некоторых вариантах осуществления настоящего изобретения кривая дифференциальной сканирующей калориметрии кристаллической формы А по сути является такой, как показано на ФИГ. 2.
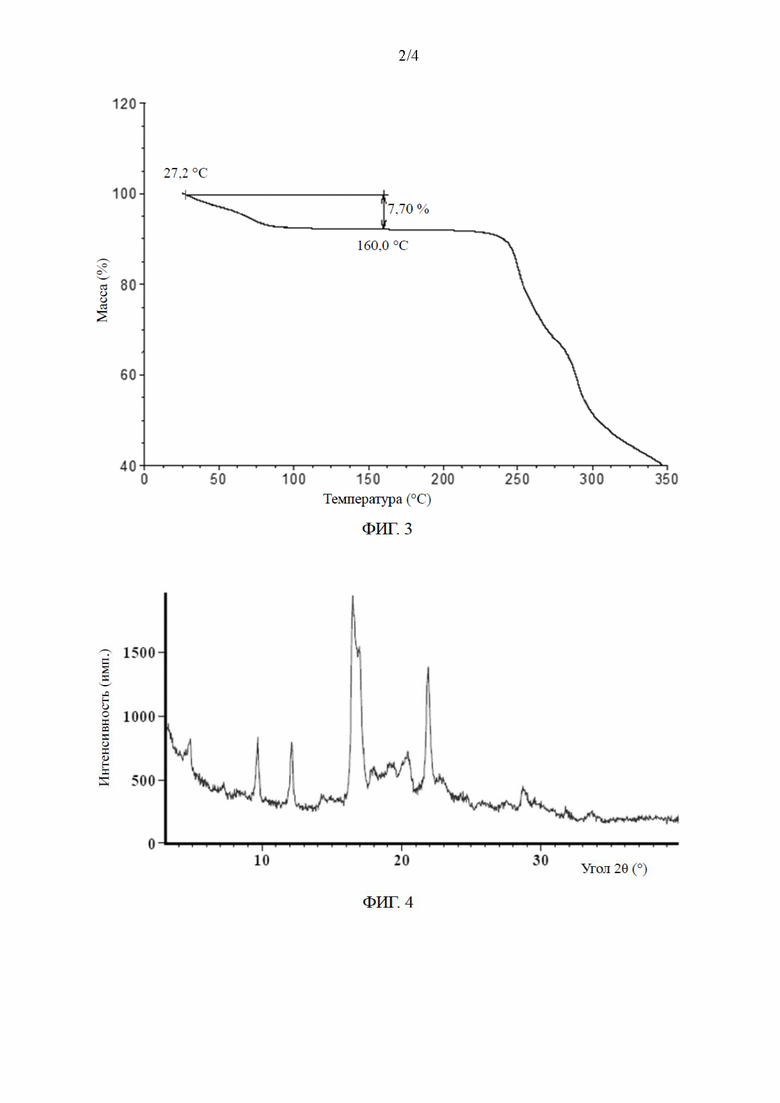
[0017] В некоторых вариантах осуществления настоящего изобретения кривая термогравиметрического анализа кристаллической формы А показывает потерю массы до 7,70% при 160,0±3,0°C.
[0018] В некоторых вариантах осуществления настоящего изобретения кривая термогравиметрического анализа кристаллической формы А по сути является такой, как показано на ФИГ. 3.
[0019] В настоящем изобретении также предложена кристаллическая форма Б соединения формулы (II), причем порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 16,48±0,20°, 16,95±0,20° и 21,87±0,20°,
[0020] В настоящем изобретении также предложена кристаллическая форма Б соединения формулы (II), причем порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 9,69±0,20°, 12,12±0,20° и 21,87±0,20°,
[0021] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20° и 21,87 ±0,20°.
[0022] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 9,69±0,20°, 12,12±0,20°, 16,48±0,20°, 16,95±0,20° и 21,87±0,20°.
[0023] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 9,69±0,20°, 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20°, 19,23±0,20°, 20,37±0,20° и 21,87±0,20°.
[0024] В настоящем изобретении предложена кристаллическая форма Б соединения формулы (II), причем порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 16,48±0,20°, 16,95±0,20°, и/или 21,87±0,20°, и/или 12,12±0,20°, и/или 17,94±0,20°, и/или 9,69±0,20°, и/или 20,37±0,20°, и/или 21,87±0,20°, и/или 4,80±0,20°, и/или 14,61±0,20°, и/или 19,23±0,20°, и/или 27,53±0,20°, и/или 28,72±0,20°, и/или 33,61±0,20°.
[0025] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 4,80±0,20°, 9,69±0,20°, 12,12±0,20°, 14,61±0,20°, 16,48± 0,20°, 16,95±0,20°, 17,94±0,20°, 19,23±0,20°, 20,37±0,20°, 21,87±0,20°, 27,53±0,20°, 28,72±0,20° и 33,61±0,20°.
[0026] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 4,80±0,20°, 9,69±0,20°, 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20°, 19,23±0,20°, 20,37±0,20° и 21,87±0,20°.
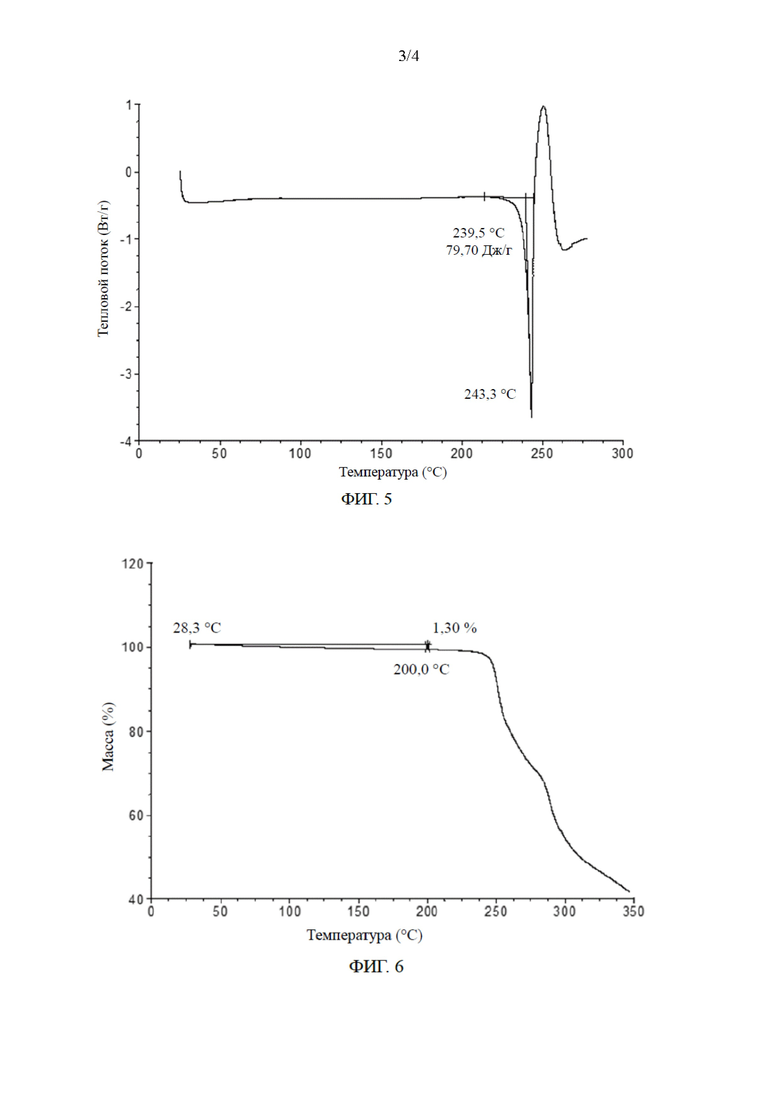
[0027] В некоторых вариантах осуществления настоящего изобретения порошковая рентгеновская дифрактограмма кристаллической формы Б по сути является такой, как показано на ФИГ. 4.
[0028] В некоторых вариантах осуществления настоящего изобретения данные анализа порошковой рентгеновской дифрактограммы кристаллической формы Б являются такими, как показано в табл. 2.
[0029] Таблица 2: Данные анализа порошковой рентгеновской дифрактограммы кристаллической формы Б соединения формулы (II)
(Å)
(импульсы)
(%)
(Å)
(импульсы)
(%)
[0030] В некоторых вариантах осуществления настоящего изобретения порошковую рентгеновскую дифрактограмму кристаллической формы Б получают с использованием источника облучения Cu-Kα.
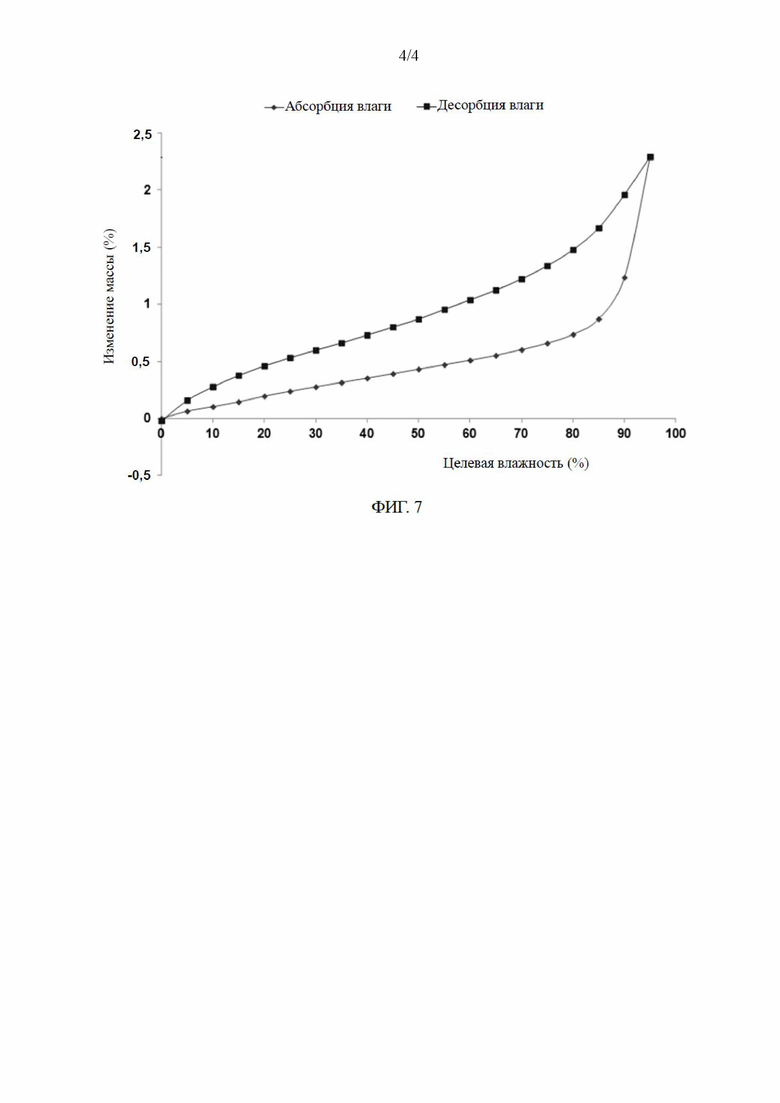
[0031] В некоторых вариантах осуществления настоящего изобретения кривая дифференциальной сканирующей калориметрии кристаллической формы Б содержит эндотермический пик с начальным значением при 239,5±3,0°C.
[0032] В некоторых вариантах осуществления настоящего изобретения кривая дифференциальной сканирующей калориметрии кристаллической формы Б по сути является такой, как показано на ФИГ. 5.
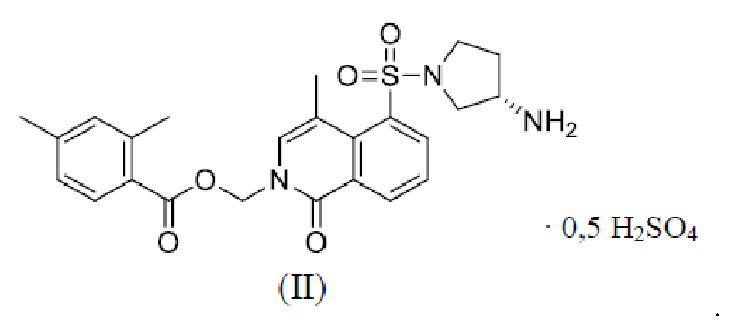
[0033] В некоторых вариантах осуществления настоящего изобретения кривая термогравиметрического анализа кристаллической формы Б показывает потерю массы до 1,30% при 200,0±3,0°C.
[0034] В некоторых вариантах осуществления настоящего изобретения кривая термогравиметрического анализа кристаллической формы Б по сути является такой, как показано на ФИГ. 6.
[0035] В настоящем изобретении дополнительнопредложен способ получения кристаллической формы Б соединения формулы (II). Способ получения включает:
(а) добавление кристаллической формы А соединения формулы (II) в растворитель с образованием суспензии; и
(б) перемешивание суспензии при 50°C в течение 3 ч, фильтрацию и сушку.
[0036] Указанный растворитель выбран из группы, состоящей из изопропанола, тетрагидрофурана, ацетонитрила, 2-бутанона и этилацетата.
[0037] В некоторых вариантах осуществления настоящего изобретения предложено применение соединения формулы (II), кристаллической формы А и кристаллической формы Б в получении лекарственных препаратов, связанных с ингибиторами ROCK.
[0038] В некоторых вариантах осуществления настоящего изобретения предложено применение соединения формулы (II), кристаллической формы А и кристаллической формы Б в получении лекарственного средства для лечения глаукомы или глазной гипертензии.
[0039] Технические эффекты
[0040] Соединение формулы (I) значительно повышает экспозиционное количество активного препарата и значительно повышает максимальную концентрацию лекарственного препарата в плазме крови Cмакс и время действия. В модели острой интраокулярной гипертензии соединение формулы (I) демонстрирует хорошие эффекты снижения глазного давления при различных тестируемых дозах и характеризуется определенной корреляцией дозы, а амплитуда снижения внутриглазного давления и продолжительность действия лучше, чем у K-115. Соединение формулы (I) обладает превосходной эффективностью (наивысший эффект снижения внутриглазного давления и наиболее продолжительное время действия). Соединение формулы (I) характеризуется высокой системной безопасностью.
[0041] Определение и пояснение
[0042] Если не указано иное, следующие используемые в настоящем документе термины и фразы подразумеваютследующие значения. Конкретную фразу или термин не следует считать неопределенными или неясными в отсутствие конкретного определения, а следует понимать в соответствии с их обычным значением. Когда в настоящем документе встречается торговое наименование, подразумевается, что оно относится к продукту или его активному ингредиенту, соответствующему торговому наименованию.
[0043] Промежуточное соединение по настоящему изобретению может быть получено различными способами синтеза, хорошо известными специалистам в данной области техники, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, полученные путем их комбинирования с другими способами химического синтеза, и эквивалентными замещающими способами, хорошо известными специалистам в данной области техники. Предпочтительные варианты осуществления включают без ограничения примеры по настоящему изобретению.
[0044] Химические реакции в конкретных вариантах осуществления по настоящему изобретению осуществляются в подходящих растворителях, а растворители должны быть подходящими для химических изменений по настоящему изобретению, а также необходимых для этого реагентов и материалов. Для получения соединения по настоящему изобретению, специалистам в данной области техники иногда необходимо модифицировать или выбрать шаги синтеза или реакционные процедуры на основе существующих вариантов осуществления.
[0045] Структура соединения по настоящему изобретению может быть подтверждена стандартными методами, хорошо известными специалистам в данной области техники; и если в настоящем изобретении задействована абсолютная конфигурация соединения, абсолютная конфигурация может быть проверена с помощью стандартных технических средств в данной области техники. Например, используется метод рентгеновской дифракции по отдельному кристаллу (РДОК), в котором данные интенсивности дифракции собираются для культивируемого отдельного кристалла с использованием дифрактометра Bruker D8 Venture с облучением CuKα в качестве источника света, и режим сканирования: ϕ/сканирование; и после сбора соответствующих данных используется прямой метод (Shelxs97) для анализа кристаллической структуры, который может подтвердить абсолютную конфигурацию.
[0046] Настоящее изобретение конкретно описано ниже с помощью примеров, но эти примеры не предназначены для каких-либо ограничений настоящего изобретения.
[0047] Все растворители, используемые в настоящем изобретении, коммерчески доступны и могут использоваться без дополнительной очистки.
[0048] В настоящем изобретении используются следующие сокращения: : к.т. представляет собой комнатную температуру; ТГФ представляет собой тетрагидрофуран; NMP представляет собой N-метил-2-пирролидон; MeSO3H представляет собой метансульфоновую кислоту; ДМЭ представляет собой диметиловый эфир этиленгликоля; ДХМ представляет собой дихлорметан; Xphos представляет собой 2-дициклогексилфосфино-2’,4,’6’-триизопропилбифенил; EtOAc представляет собой этилацетат; MeOH представляет собой метанол; ацетон представляет собой 2-пропанон; 2-Me-ТГФ представляет собой 2-метилтетрагидрофуран; и ИПС представляет собой изопропанол.
[0049] Соединения названы в соответствии с общепринятыми принципами именования в данной области техники или с использованием программного обеспечения ChemDraw®, а коммерчески доступные соединения используются под названиями из каталога поставщиков.
[0050] Метод порошковой рентгеновской дифракции (ПРД)
[0051] Модель прибора: порошковый рентгеновский дифрактометр PANalytical X’pert3
[0052] Метод испытаний: приблизительно 10 мг образца для ПРД-детектирования.
[0053] Подробные параметры ПРД следующие:
[0054] источник облучения: Cu, Kα1=1,540598 Å; Cu, Kα2=1,544426 Å
[0055] напряжение трубки: 40 кВ; ток трубки: 40 мА
[0056] диапазон сканирования: 3-40 град
[0057] ширина шага сканирования: 0,0263 град
[0058] продолжительность шага сканирования: 46,665 секунд.
[0059] Метод дифференциальной сканирующей калориметрии (ДСК)
[0060] Модель прибора: дифференциальный сканирующий калориметр TA2500
[0061] Метод испытаний: образец (приблизительно 1-5 мг) для испытаний помещали в алюминиевый диск ДСК, причем алюминиевый диск был закрыт крышкой и не имел перфорированных отверстий; и образец нагревали от 25°C (комнатная температура) при скорости нагрева 10°C/мин при 50 мл/мин N2 до разложения образца.
[0062] Метод термического гравиметрического анализа (ТГА)
[0063] Модель прибора: термогравиметрический анализатор TAQ5000
[0064] Метод испытаний: образец (приблизительно 1-5 мг) для испытаний помещали в алюминиевый диск ТГА, причем алюминиевый диск был открыт; и образец нагревали от комнатной температуры до 350°C при скорости нагрева 10°C/мин при 10-25 мл/мин N2.
[0065] Метод динамической сорбции паров (ДСП)
[0066] Модель прибора: прибор для собственной динамической сорбции паров SMS DVS
[0067] Эксперимент в отношении динамической сорбции паров состоит из адсорбции и десорбции. Обычно считается, что при установленной относительной влажности адсорбция или десорбция образца влагой при этой относительной влажности считается достигшей равновесия, когда масса образца достигает dm/dt ≤ 0,01%.
[0068] Температура испытания образца: T=25°C
[0069] Время установления равновесия: dm/dt: 0,01%/мин
[0070] Диапазон изменения относительной влажности: 0%-95%-0%; RH (%)
[0071] Тестовое изменение влажности на каждом шаге: 5%
[0072] Оценка гигроскопичности классифицируется следующим образом:
[0073] Примечание: ΔW% указывает на гигроскопический прирост массы тестируемого образца при 25±1°C и 80±2% RH.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0074] ФИГ. 1 представляет собой диаграмму ПРД кристаллической формы А соединения формулы (II) при облучении Cu-Kα;
[0075] ФИГ. 2 представляет собой диаграмму ДСК кристаллической формы А соединения формулы (II);
[0076] ФИГ. 3 представляет собой диаграмму ТГА кристаллической формы А соединения формулы (II);
[0077] ФИГ. 4 представляет собой диаграмму ПРД кристаллической формы Б соединения формулы (II) при облучении Cu-Kα;
[0078] ФИГ. 5 представляет собой диаграмму ДСК кристаллической формы Б соединения формулы (II);
[0079] ФИГ. 6 представляет собой диаграмму ТГА кристаллической формы Б соединения формулы (II); и
[0080] ФИГ. 7 представляет собой диаграмму ДСП кристаллической формы Б соединения формулы (II).
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0081] Для лучшего понимания содержания настоящего изобретения дополнительное описание сделано в сочетании с конкретными примерами, которые не следует рассматривать как ограничения содержания настоящего изобретения.
[0082] Промежуточное соединение 1-4 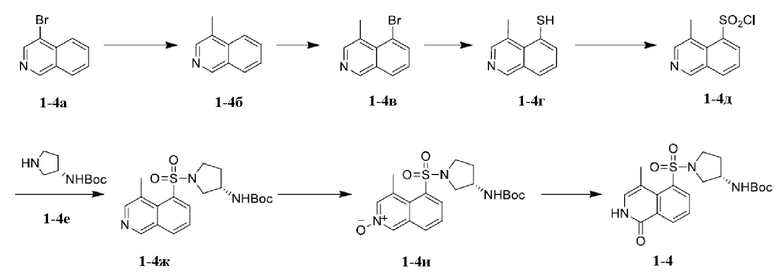
[0083] Шаг 1:
[0084] При контролируемой внутренней температуре от 25 до 35°C толуол (24 л), соединение 1-4а (4000 г), карбонат натрия (6120 г), метилбороновую кислоту (3465 г), 2-дициклогексилфосфино-2',6'-диметоксибифенил (98,66 г) и трис(дибензилиденацетон)дипалладий (88,03 г) последовательно добавляли в реактор при перемешивании, реактор трижды подвергали вытеснению азотом, внутреннюю температуру повышали до 90°C и перемешивали смесь при 90 до 100°C в течение 13 ч. В реактор добавляли воду (4 л) для растворения твердого вещества, реакционный раствор охлаждали до комнатной температуры и фильтровали с диатомитом, а осадок на фильтре промывали метил-трет-бутиловым эфиром (4 л). Промывочный раствор и фильтрат объединяли, доводили концентрированной хлористоводородной кислотой (8 л) до pH = 3 и отстаивали жидкость для расслаивания. Водную фазу экстрагировали метил-трет-бутиловым эфиром (5 л), и водную фазу доводили водным раствором гидроксида натрия до pH=9 и дважды экстрагировали этилацетатом (12 л); и объединенную органическую фазу концентрировали под вакуумом до тех пор, пока масса больше не уменьшалась, с получением соединения 1-4б.
[0085] МС-ИЭР (M+H) +: расчетное значение 144, и измеренное значение 144.
[0086] Шаг 2:
[0087] Поддерживая температуру в реакторе (50 л) ниже 30°С, в реактор медленно добавляли концентрированную серную кислоту (13,36 л). Температуру в реакторе контролировали так, чтобы она была ниже 50°C, при которой соединение 1-4б (2500 г) медленно добавляли в реактор с помощью воронки постоянного перепада давления. Температуру в реакторе поддерживали на уровне от -10 до 0°C, при которой в реактор порциями медленно добавляли N-бромсукцинимид (3070,25 г), и реакционный раствор перемешивали в течение 13 ч при температуре от -10 до 8°C. Внутреннюю температуру поддерживали на уровне ниже 50°C, и реакционный раствор медленно выливали в ледяную воду (8 л). Внутреннюю температуру поддерживали на уровне ниже 50°C, и реакционный раствор доводили до pH = 9 путем медленного добавления по каплям водного раствора гидроксида натрия. Добавляли этилацетат (10 л) и реакционный раствор перемешивали в течение 10 мин. Реакционный раствор фильтровали, и осадок на фильтре промывали этилацетатом (12,5 л). Водную фазу экстрагировали этилацетатом (10 л×3), и объединенную органическую фазу концентрировали при пониженном давлении при 45°C. К концентрированному осадку для растирания добавляли н-гептан (10 л) с последующей фильтрацией с отсасыванием с получением коричнево-красного твердого вещества. Твердое вещество сушили в вакуумном сушильном шкафу (40°C) с получением соединения 1-4в.
[0088] МС-ИЭР (M+H)+: расчетные значения 222, 224, и измеренные значения 222, 224.
[0089] Шаг 3:
[0090] В реактор (50 л) добавляли растворитель N, N-диметилацетамид (10 л) и при перемешивании добавляли соединение 1-4в (1003,65 г). Внутреннюю температуру поддерживали на уровне ниже 55°C, при которой в реактор (50 л) порциями медленно добавляли триметоксид натрия (1276,97 г); и реакционную смесь перемешивали при 50°C в течение 30 мин. Температуру реактора повышали до 120°C, в это время внутренняя температура составляла 109°C, и в этих условиях в реакторе осуществляли перемешивание в течение 12 ч. Температуру реактора доводили до 50°C так, чтобы температура реакционного раствора снижалась до 40-50°C; реакционный раствор фильтровали с диатомитом и промывали этилацетатом (10 л); и фильтрат добавляли с водой (10 л) и экстрагировали этилацетатом (5 л×2) с получением водной фазы. Водную фазу доводили до pH = 7 концентрированной хлористоводородной кислотой и экстрагировали этилацетатом (5 л×2); и органические фазы объединяли, промывали водой (10 л×3) и концентрировали при пониженном давлении до тех пор, пока масса больше не изменялась, с получением соединения 1-4г.
[0091] МС-ИЭР (M+H)+: расчетное значение 176, и измеренное значение 176.
[0092] Шаг 4:
[0093] В очищенный 50-л высоко- и низкотемпературный реактор добавляли дихлорметан (8 л) и перемешивали. В реактор добавляли соединение 1-4г (1474,02 г), и температуру масляной бани регулировали таким образом, чтобы температура в реакторе поддерживалась на уровне от 0 до 10°C. В реактор порциями медленно добавляли концентрированную хлористоводородную кислоту (4,38 л), и внутреннюю температуру поддерживали на уровне от 5 до 15°C. После завершения добавления концентрированной хлористоводородной кислоты температуру в реакторе снижали до -5°C. В реактор порциями медленно добавляли по каплям водный раствор гипохлорита натрия (21,35 л), и весь процесс контролировали при температуре от 0 до 10°C. Реакционный раствор фильтровали, а осадок на фильтре промывали метил-трет-бутиловым эфиром (2 л×2) и затем собирали. Собранный осадок на фильтре помещали в сушильный шкаф и сушили с получением соединения 1-4д.
[0094] МС-ИЭР (M+H)+:расчетное значение 242, и измеренное значение 242.
[0095] Шаг 5:
[0096] В 50-л реактор добавляли дихлорметан (10 л) и перемешивали; внутреннюю температуру поддерживали на уровне от 0 до 10°C, и в реактор добавляли соединение 1-4д (1462,35 г). Внутреннюю температуру поддерживали на уровне от 0 до 10°C и в реакционный раствор медленно добавляли по каплям N, N-диизопропилэтиламин (934,63 г); и внутреннюю температуру поддерживали на уровне от 0 до 10°C, и в реакционный раствор порциями медленно добавляли соединение 1-4е (1230,24 г). После завершения добавления температуру повышали до 10-20°C и смесь дополнительно перемешивали в течение 0,5 ч. Органическую фазу промывали насыщенным водным раствором хлорида аммония (3 л×3), и объединенный насыщенный водный раствор хлорида аммония экстрагировали дихлорметаном (3 л). Органические фазы по отдельности концентрировали при пониженном давлении до тех пор, пока масса больше не изменялась, а затем сушили при помощи вакуумного сушильного шкафа с получением соединения 1-4ж.
[0097] МС-ИЭР (M+H)+:расчетное значение 392, и измеренное значение 392.
[0098] Шаг 6:
[0099] В очищенную 5-л трехгорлую колбу добавляли 3,1 л безводного дихлорметана и перемешивали. В реакционную колбу добавляли взвешенное соединение 1-4ж (451,34 г); и после растворения твердого вещества помещали реакционную колбу в баню с ледяной водой так, чтобы внутренняя температура реакционной системы снижалась до 0°C. В 5-л реакционную колбу порциями медленно добавляли взвешенную м-хлорпербензойную кислоту (429,81 г), и температуру поддерживали при температуре от 0 до 10°C в течение всего процесса. После завершения добавления м-хлорпербензойной кислоты баню с ледяной водой реакционной колбы удаляли и заменяли масляной баней, и внутреннюю температуру реакционной колбы стабилизировали при температуре 20-25°C и перемешивали в течение 12 ч. Реакционную колбу помещали в баню с ледяной водой и перемешивали в течение 1 ч и фильтровали; осадок на фильтре ополаскивали дихлорметаном (400 мл×2); к фильтрату добавляли по каплям 10% водный раствор тиосульфата натрия при температуре от 25 до 30°C до тех пор, пока калий йодид-крахмальная индикаторная полоска не изменяла цвет на синий; и в систему добавляли по каплям насыщенный водный раствор бикарбоната натрия до pH = 7. Смеси давали отстояться для расслаивания, органическую фазу сохраняли, а водную фазу однократно экстрагировали 3 л дихлорметана. Органические фазы объединяли, промывали 1% водным раствором бикарбоната натрия (4 л×10), сушили безводным сульфатом натрия и фильтровали; и фильтрат концентрировали при пониженном давлении. Твердое вещество, полученное концентрированием, сушили в сушильном шкафу с получением соединения 1-4и.
[01] МС-ИЭР (M+H)+:расчетное значение 408, и измеренное значение 408.
[02] Шаг 7:
[03] В 5-л трехгорлую колбу добавляли соединение 1-4и (301,42 г) и тетрагидрофуран (1,5 л) и хорошо перемешивали, и добавляли по каплям триэтиламин (206 мл) при внутренней температуре 10-20°C и хорошо перемешивали. Добавляли по каплям ангидрид трифторуксусной кислоты (206 мл) при внутренней температуре от 20 до 30°C и осуществляли реакцию в течение 0,5 ч при температуре от 20 до 30°C. Добавляли реакционный раствор с водой (1,5 л) и хорошо перемешивали, и реакционный раствор становился мутным. Добавляли твердый гидроксид натрия (600 г) при внутренней температуре от 40 до 50°C и перемешивали в течение 0,5 ч, и большое количество твердого вещества выпадало в осадок; и добавляли 1,5 л метил-трет-бутилового эфира и перемешивали в течение 0,5 ч. Реакционный раствор отделяли и верхнюю органическую фазу фильтровали с получением твердого вещества. Твердое вещество растирали с метил-трет-бутиловым эфиром/этилацетатом (4,4 л, 10: 1) с получением взвеси, которую затем фильтровали с получением белого твердого вещества. Белое твердое вещество сушили в сушильном шкафу с получением неочищенного продукта соединения 1-4 (371,52 г). В 50-л реактор добавляли тетрагидрофуран (7 л), внутреннюю температуру поддерживали на уровне от 20 до 25°C, и неочищенный продукт соединения 1-4 (1713,19 г, объединенный с другими партиями) добавляли и перемешивали при внутренней температуре от 20 до 25°C в течение 0,5 ч, затем добавляли 8,6 л воды, перемешивали при температуре от 20 до 25°C в течение 0,5 ч, добавляли твердый гидроксид натрия (800 г) и перемешивали при внутренней температуре от 20 до 25°C в течение 0,5 ч. Добавляли метил-трет-бутиловый эфир (8,6 л) и перемешивали при внутренней температуре от 20 до 25°C в течение 0,5 ч. Раствор разделяли, верхнюю органическую фазу фильтровали, твердое вещество, полученное при фильтрации, сохраняли, а твердое вещество сушили в сушильном шкафу до постоянной массы с получением соединения 1-4.
[04] МС-ИЭР (M+H)+: расчетное значение 408, и измеренное значение 408.
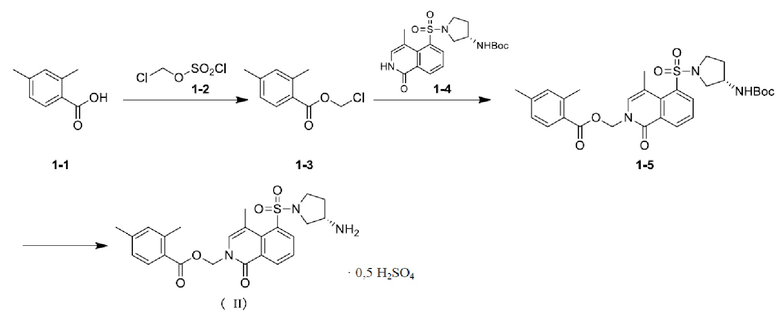
[05] Пример 1:Получение кристаллической формы А соединения формулы (II)
[06] Шаг 1:
[07] При перемешивании и защите азотом в реактор последовательно добавляли дихлорметан (8,6 л), воду (8,6 л) и исходное вещество 1-1 (575,34 г). Температуру в реакторе поддерживали на уровне от 10 до 15°C, и в реактор последовательно добавляли бикарбонат натрия (1289,36 г), гидросульфат тетрабутиламмония (133,87 г) и исходное вещество 1-2 (760,25 г), и перемешивали в течение 2 часов при внутренней температуре от 10 до 15°C. Реакционному раствору давали отстояться для расслаивания, и органическую фазу концентрировали при пониженном давлении до тех пор, пока фракции не вытекали. Концентрированный раствор переносили в трехгорлую колбу и перемешивали при внутренней температуре от 60 до 70°C в течение 12 ч, а затем охлаждали естественным путем до комнатной температуры с получением неочищенного продукта. Неочищенный продукт быстро фильтровали с силикагелем и ополаскивали дихлорметаном до тех пор, пока не оставалось остатка продукта. Элюат концентрировали до приблизительно 1,2 л при пониженном давлении, а затем промывали 1% водным раствором бикарбоната натрия (3 л); органическую фазу сушили с безводным сульфатом натрия (500 г), фильтровали, а затем концентрировали при пониженном давлении до тех пор, пока фракции не вытекали, с получением промежуточного соединения 1-3, которое непосредственно использовали на следующем шаге без очистки.
[08] МС-ИЭР (M+H)+: расчетное значение 199, и измеренное значение 199.
[09] 1H ЯМР (400 МГц, CD3Cl) δ 7,81 - 7,79 (д, J = 8 Гц, 1H), 7,01 - 6,95 (м, 2H), 5,84 (с, 2H), 2,52 (с, 3H), 2,27 (с, 3H).
[010] Шаг 2:
[011] В реактор добавляли 2-метилтетрагидрофуран (9702 мл) и перемешивали, и внутреннюю температуру поддерживали на уровне от 10 до 20°C; и затем последовательно добавляли соединение 1-4 (1078,26 г), карбонат цезия (1017,63 г) и промежуточное соединение 1-3 (631,71 г). Температуру в реакторе повышали до 57-63°C, и смесь перемешивали в течение 2 ч и 40 мин до остановки реакции. Температуру в реакторе понижали до 15-25°C, а затем добавляли реакционный раствор с водой (10,78 л), перемешивали и оставляли отстаиваться для расслаивания; полученную в результате водную фазу дважды экстрагировали 2-метилтетрагидрофураном (5390 мл×2); и объединенную органическую фазу концентрировали при пониженном давлении до приблизительно 2,2 л. Добавляли концентрированный раствор с ацетоном (1078 мл) и н-гептаном (2156 мл) и дополнительно концентрировали до тех пор, пока фракции не вытекали, с получением рыхлого твердого вещества. Внутреннюю температуру поддерживали на уровне от 10 до 30°C, и твердое вещество растирали и перемешивали три раза со смешанным растворителем (11,0495 л, ацетон: н-гептан = 1:40), фильтровали и ополаскивали н-гептаном (1078 мл). Осадок на фильтре сушили под вакуумом до постоянной массы с получением промежуточного соединения 1-5.
[012] МС-ИЭР (M+H)+: расчетное значение 570, и измеренное значение 570.
[013] 1H ЯМР (400 МГц, CD3Cl) δ 8,86 - 8,78 (м, 1H), 8,13 - 8,04 (м, 1H), 7,93 - 7,86 (м, 1H), 7,58 - 7,49 (м, 1H), 7,39 - 7,32 (м, 1H), 7,09 - 6,99 (м, 2H), 6,14 (с, 2H), 4,86 - 4,76 (м, 1H), 4,42 - 4,30 (м, 1H), 3,73 - 3,61 (м, 2H), 3,57 - 3,47 (м, 1H), 3,46 - 3,37 (м, 1H), 2,76 - 2,67 (м, 3H), 2,59 (с, 3H), 2,39 - 2,29 (м, 4H), 2,09 - 1,97 (м, 1H), 1,46 (с, 9H).
[014] Шаг 3:
[015] В 5-л трехгорлую колбу с внутренней температурой, поддерживаемой на уровне от 20 до 30°C, последовательно добавляли безводный тетрагидрофуран (4,24 л) и концентрированную серную кислоту (712,43 г) и хорошо перемешивали для последующего использования. В реактор с внутренней температурой, поддерживаемой на уровне от 20 до 30°C, последовательно добавляли промежуточное соединение 1-5 (1062,15 г) и тетрагидрофуран (4,24 л) и хорошо перемешивали. Затем в систему добавляли по каплям предварительно приготовленный раствор серной кислоты в тетрагидрофуране, затем реакционную смесь перемешивали в течение 4 ч, поддерживая внутреннюю температуру на уровне от 35 до 45°C, и в системе постепенно выпадало большое количество белого осадка. В реактор медленно добавляли метил-трет-бутиловый эфир (8,48 л) и перемешивали в течение 0,5 ч, поддерживая внутреннюю температуру на уровне от 20 до 30°C; реакционный раствор фильтровали, а отфильтрованное твердое вещество последовательно растирали со смешанным растворителем (метил-трет-бутиловый эфир/тетрагидрофуран = 10,6 л/10,6 л) и метил-трет-бутиловым эфиром (21,2 л) при температуре от 20 до 30°C в течение 0,5 ч, а затем последовательно пять раз растирали с водой (21,2 л×5) до тех пор, пока pH фильтрата не составлял приблизительно 7. Внутреннюю температуру поддерживали на уровне от 20 до 30°C, и твердое вещество последовательно растирали в метил-трет-бутиловом эфире (15,9 л) и смешанном растворителе (метил-трет-бутиловый эфир/тетрагидрофуран = 10,6 л/5,3 л), а затем фильтровали, ополаскивали и сушили в вакууме при 45°C или ниже с получением кристаллической формы А соединения формулы (II). Содержание сульфатного радикала составляло 9,17%, что выявляли при помощи ионной хроматографии. Следовательно, был сделан вывод, что в соединении формулы (II) также содержалось 0,5 сульфатной соли.
[016] МС-ИЭР (M+H)+: расчетное значение 470, и измеренное значение 470. 1H ЯМР (400 МГц, DMSO-d6) δ 8,64 - 8,58 (м, 1H), 8,26 (д, J = 8 Гц, 1H), 7,74 - 7,62 (м, 3H), 7,16 - 7,11 (м, 1H), 7,11 - 7,05 (м, 1H), 6,08 (с, 2H), 3,94 - 3,89 (м, 1H), 3,70 - 3,55 (м, 2H), 3,55 - 3,44 (м, 1H), 3,43 - 3,27 (м, 1H), 2,59 (с, 3H), 2,48 - 2,45 (м, 3H), 2,35 - 2,29 (м, 1H), 2,29 - 2,24 (м, 3H), 2,16 - 1,90 (м, 1H).
[017] Характеристика кристаллической формы А соединения формулы (II):
[018] (1) анализ порошковой рентгеновской дифракции: результаты испытаний показаны на ФИГ. 1 и в табл. 1;
[019] (2) анализ дифференциальной сканирующей калориметрии: результаты испытаний показаны на ФИГ. 2; и
[020] (3) оценка методом термического гравиметрического анализа: результаты испытаний показаны на ФИГ. 3.
[021] Пример 2: Получение соединения формулы (I)
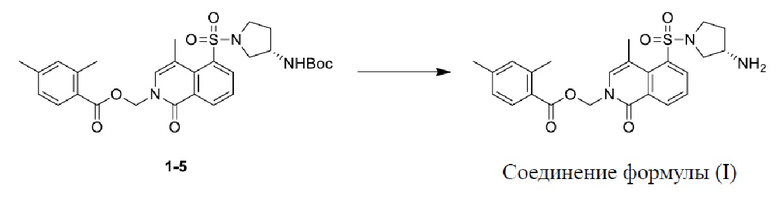
[022] Соединение 1-5 (2,2 г, 3,84 ммоль) растворяли в этилацетате (35 мл), и к реакционному раствору добавляли раствор водорода и этилацетата (4 М, 20 мл) и перемешивали при 15°C в течение 12 ч. Добавляли реакционный раствор с насыщенным водным раствором бикарбоната натрия для доведения pH до 8 и экстрагировали этилацетатом (60 мл × 2); органическую фазу сушили с безводным сульфатом натрия (5 г), фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта; и неочищенный продукт очищали при помощи высокоэффективной жидкостной хроматографии (нейтральная система) с получением соединения формулы (I). МС-ИЭР [M+H]+: расчетное значение 470, и измеренное значение 470. 1H ЯМР (400 МГц, CD3OD) δ = 8,71 (дд, J = 1,4, 8,0 Гц, 1H), 8,24 (дд, J = 1,5, 7,8 Гц, 1H), 7,81 (д, J = 7,9 Гц, 1H), 7,64 (т, J = 7,9 Гц, 1H), 7,54 (д, J = 0,9 Гц, 1H), 7,09 (с, 1H), 7,05 (д, J = 7,9 Гц, 1H), 6,14 (с, 2H), 3,77 - 3,62 (м, 3H), 3,59 - 3,50 (м, 1H), 3,20 (дд, J = 4,9, 9,5 Гц, 1H), 2,70 (д, J = 0,9 Гц, 3H), 2,53 (с, 3H), 2,32 (с, 3H), 2,30 - 2,23 (м, 1H), 1,96 - 1,86 (м, 1H).
[023] Пример 3: Получение кристаллической формы Б соединения формулы (II)
[024] Соответствующее количество кристаллической формы А соединения формулы (II) взвешивали и помещали в сосуд для отбора проб, и добавляли определенный объем растворителя в табл. 3 для получения суспензий или растворов различных отдельных растворителей. После непрерывного перемешивания суспензии при 50°C в течение 3 ч, образец фильтровали и отфильтрованное твердое вещество сушили в вакууме в вакуумном сушильном шкафу при 45°C для удаления остаточного растворителя.
[025] Таблица 3: Получение кристаллической формы Б соединения формулы (II)
[026] Характеристика кристаллической формы Б соединения формулы (II):
[027] (1) анализ порошковой рентгеновской дифракции: результаты испытаний показаны на ФИГ. 4 и в табл. 2;
[028] (2) анализ дифференциальной сканирующей калориметрии: результаты испытаний показаны на ФИГ. 5; и
[029] (3) оценка методом термического гравиметрического анализа: результаты испытаний показаны на ФИГ. 6.
[030] Пример 4: Исследование гигроскопичности кристаллической формы Б соединения формулы (II)
[031] Экспериментальные материалы:
[032] прибор для собственной динамической сорбции паров SMS DVS
[033] Экспериментальный метод:
[034] для испытания 10-15 мг кристаллической формы Б соединения формулы (II) помещали в лоток для проб для ДСП.
[035] Экспериментальные результаты:
[036] диаграмма ДСП кристаллической формы Б соединения формулы (II) показана на ФИГ. 7, где ΔW=0,736%.
[037] Экспериментальный вывод:
[038] кристаллическая форма Б соединения формулы (II) характеризовалась гигроскопическим приростом массы 0,736% при 25°C и 80% RH, демонстрируя незначительную гигроскопичность.
[039] Пример 5: Исследование стабильности в твердом состоянии кристаллической формы Б соединения формулы (II)
[040] Кристаллическую форму Б соединения формулы (II) помещали при 40°C/75%RH (запечатывали двухслойным пакетом из ПЭНП и дополнительно запечатывали пакетом из алюминиевой фольги со швами по периметру) на 6 месяцев, и помещали при 25°C/60%RH (запечатывали двухслойным пакетом из ПЭНП и дополнительно запечатывали пакетом из алюминиевой фольги со швами по периметру) на 12 месяцев, соответственно. Кристаллическую форму испытывали отдельно в каждой точке отбора проб для определения стабильности кристаллической формы образца.
[041] Таблица 4: Экспериментальные результаты исследования стабильности в твердом состоянии кристаллической формы Б соединения формулы (II)
[042] Вывод: кристаллическая форма Б соединения формулы (II) характеризовалась хорошей стабильностью при ускоренных и длительных испытаниях.
[043] Эксперименты по биологическому тестированию
[044] Экспериментальный пример 1: Фармакокинетический тест в водянистой влаге
[045] Экспериментальная цель:
[046] Соединение представляло собой молекулу пролекарства, содержащую сложноэфирную функциональную группу, которая может гидролизоваться до молекулы активного препарата (исходная субстанция препарата) под действием обильного количества сложноэфирной гидролазы в глазной ткани при введении с глазными каплями. В этом эксперименте определяли скорость образования активных фармацевтических ингредиентов in vivo и экспозиционное количество активных фармацевтических ингредиентов.
[047] Экспериментальные материалы:
[048] Самцы новозеландских белых кроликов в возрасте от 3 до 6 месяцев, массой от 2,0 до 5,0 кг, приобретенные у Pizhou Oriental Breeding Co., Ltd.
[049] Приготовление образца глазных капель:
[050] Используемый растворитель представлял собой 1,2% гидроксипропилметилцеллюлозу E5/20,5% полоксамер P407/1,6% полоксамер P188.
[051] Экспериментальная операция:
[052] Дозировка глазных капель составляла 0,5 мг/глаз, вводили в оба глаза. Образцы водянистой влаги собирали через 0,25 ч, 0,5 ч, 2 ч, 4 ч, 8 ч, и 24 ч после введения. Осуществляли количественное определение всех образцов при помощи жидкостной хроматографии в сочетании с масс-спектрометрией-масс-спектрометрии в водянистой влаге экспериментальных животных. Измеренные значения концентрации рассчитывали при помощи некомпартментной модели WinNonlin в соответствии с данными о зависимости концентрации водянистой влаги от времени, такими как время полужизни, максимальная концентрация, время достижения максимальной концентрации, экспозиционное количество в единицу времени в водянистой влаге и т.д.
[053] Таблица 5: Результаты фармакокинетического теста в водянистой влаге новозеландских кроликов
(ч)
(ч)
(нМ.ч)
[054] "--": не выявлено.
[055] Вывод: результаты показали, что в водянистой влаге в основном обнаруживались активные метаболиты после гидролиза сложных эфиров вместо испытываемых соединений (молекул пролекарства); соединение формулы (I) значительно повышало экспозиционное количество активного препарата и максимальную концентрацию, а время действия лекарственных препаратов в крови значительно продлялось.
[056] Экспериментальный пример 2: Тест на снижение внутриглазного давления у новозеландских кроликов, страдающих острой внутриглазной гипертензией
[057] Экспериментальная цель:
[058] Острую внутриглазную гипертензию индуцировали у кроликов путем инъекции вязкоэластичного агента в переднюю камеру, и проверяли влияние соединения формулы (I) на снижение глазного давления путем введения глазных капель в различных концентрациях.
[059] Экспериментальные материалы:
[060] Самцы новозеландских белых кроликов в возрасте от 97 до 127 суток, массой от 2,5 до 3,4 кг, приобретенные у Pizhou Oriental Breeding Co., Ltd.
[061] Экспериментальная операция:
[062] 50 самцов новозеландских белых кроликов случайным образом разделяли на 5 групп в зависимости от массы тела, 10 кроликов/группа. В переднюю камеру правого глаза животных 1-5 групп вводили медицинский гель гиалуроната натрия в виде однократной дозы, 100 мкл/глаз, для индуцирования глазной гипертензии у животных. Через 5-15 мин после формирования в правый глаз вводили по каплям растворитель, К-115, и исследуемый образец (соединения формулы (I) различных концентраций), а в левый глаз вводили по каплям растворитель, 50 мкл/глаз; и внутриглазное давление у животных измеряли в обоих глазах до введения, а также через 2, 4, 6, 8 и 10 ч после введения, соответственно. Экспериментальные результаты показаны в табл. 6.
[063] Таблица 6: Результаты измерения внутриглазного давления (среднее ±СПС) в обоих глазах до и после моделирования и введения животным в группах
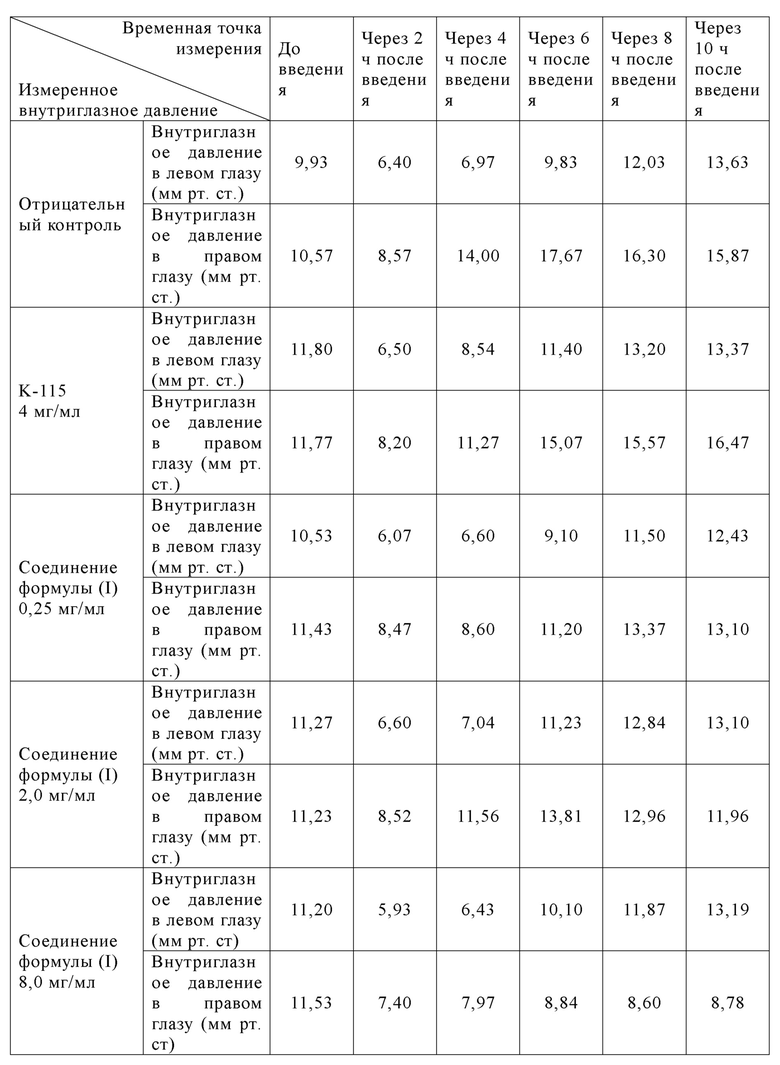
[064] Вывод: в модели острой внутриглазной гипертензии соединение формулы (I) продемонстрировало хороший эффект снижения внутриглазного давления при различных тестируемых дозах, и в то же время характеризовалось определенной корреляцией дозы, а амплитуда снижения внутриглазного давления и продолжительный эффект были лучше, чем у K-115.
[065] Экспериментальный пример 3: Тест на снижение внутриглазного давления и офтальмологическую токсичность у новозеландских кроликов с нормальным внутриглазным давлением путем повторного введения глазных капель в течение 14 суток
[066] Экспериментальная цель:
[067] Эффект снижения внутриглазного давления и потенциальную офтальмологическую токсичность соединения формулы (I) проверяли у кроликов с нормальным внутриглазным давлением путем повторного введения глазных капель в течение 14 суток.
[068] Экспериментальные материалы:
[069] Самцы новозеландских белых кроликов в возрасте от 97 до 127 суток, массой от 2,6 до 3,5 кг, приобретенные у Pizhou Oriental Breeding Co., Ltd.
[070] Экспериментальная операция I:
[071] Самцов новозеландских белых кроликов случайным образом разделяли на 7 групп в зависимости от массы тела, 6 кроликов/группа. Животным в 1-7 группах вводили по каплям растворитель/контроль/исследуемый образец в оба глаза в объеме введения 50 мкл/глаз, один раз в сутки в течение 14 суток подряд, и сутки первого введения записывали как 1 сутки. Внутриглазное давление у животных измеряли до введения, а также через 1, 2, 4, 6, 8 и 10 ч после введения, соответственно. На 2-14 сутки внутриглазное давление у животных в группе введения K-115 измеряли через 1 ч после суточного введения; и внутриглазное давление у животных в остальных группах измеряли через 4 ч после суточного введения. Экспериментальные результаты показаны в табл. 7, 8 и 9:
[072] Таблица 7: Изменения внутриглазного давления (среднее ±СПС) у животных в соответствующих группах в обоих глазах до и после введения в 1 сутки 
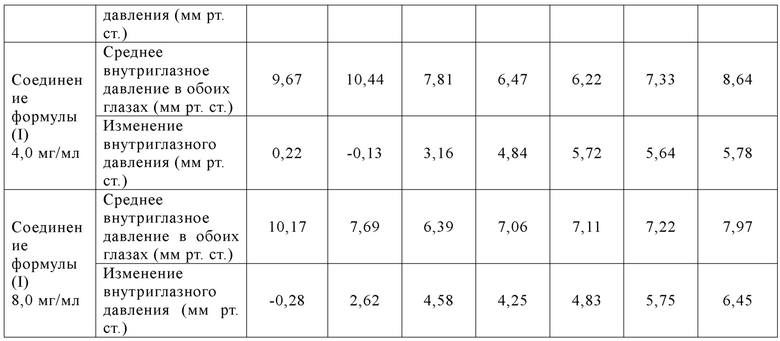
Таблица 8: Изменения внутриглазного давления (среднее ±СПС) у животных в соответствующих группах в обоих глазах до и после повторного введения со 2 по 7 сутки
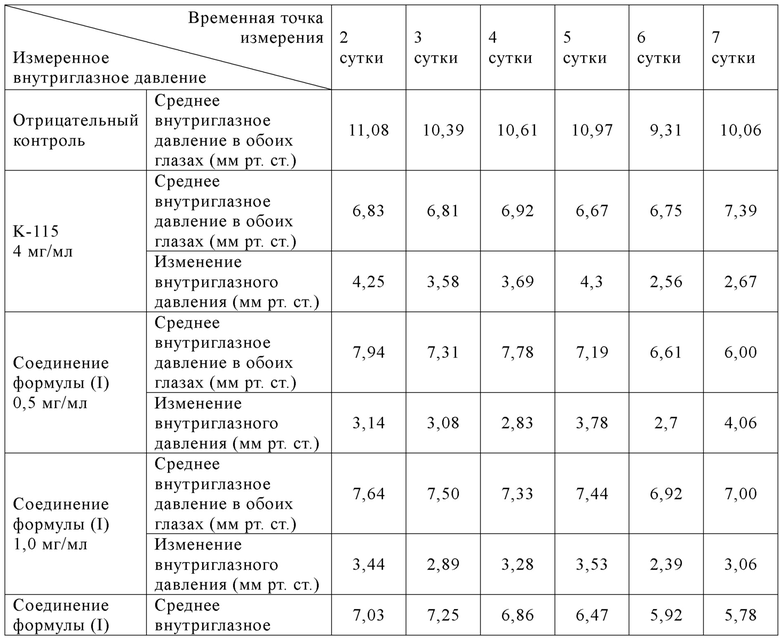
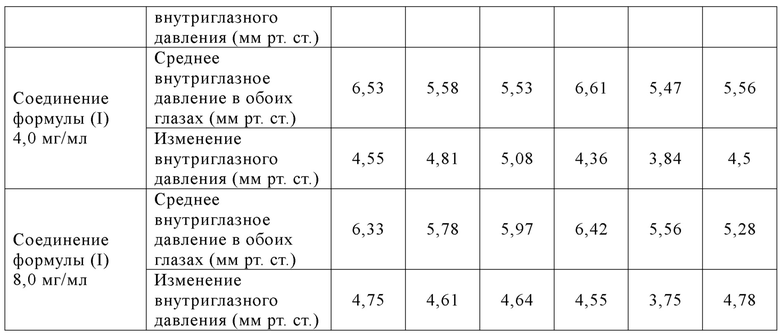
Таблица 9: Изменения внутриглазного давления (среднее ±СПС) у животных в соответствующих группах в обоих глазах до и после повторного введения с 8 по 14 сутки

[073] Вывод: однократное введение соединения формулы (I) продемонстрировало лучшую эффективность (наивысший эффект снижения внутриглазного давления и наиболее продолжительное время действия) при всех испытанных дозах (0,5-8,0 мг/мл), что было значительно лучше, чем у K-115. В случае 14 суток последовательного введения соединение формулы (I) могло стабильно поддерживать эффект значительного снижения внутриглазного давления при дозе 0,5 мг/мл, что все еще было значительно лучше, чем у К-115 при оценке максимального (Cмакс) эффекта снижения внутриглазного давления.
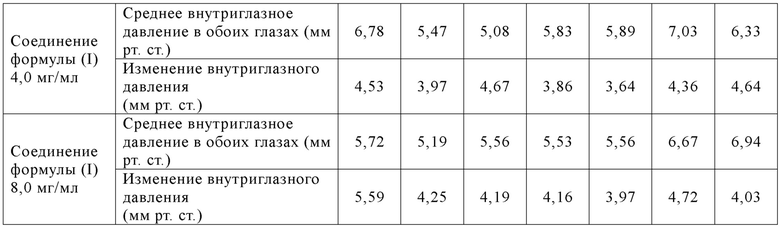
[074] Экспериментальная операция II:
[075] 42 самцов новозеландских белых кроликов случайным образом группировали в 7 групп в зависимости от массы тела, 6 кроликов/группа. Среди животных в 1-7 группах в левый глаз вводили по каплям физиологический раствор, а в правый глаз вводили по каплям растворитель/контроль/исследуемый образец в объеме введения 50 мкл/глаз соответственно, один раз в сутки в течение 14 суток подряд, и сутки первого введения записывали как 1 сутки. Внутриглазное давление у животных измеряли до введения в 1 сутки, а также через 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 10 ч после введения в 1 сутки (таблица 10).
[076] Животных подвергали определению реакции на раздражение глаз и обнаружению флуоресцеина натрия (оценивали в соответствии с критериями подсчета баллов) в обоих глазах при помощи ручной щелевой лампы перед началом теста (сутки -2/сутки -1), перед суточным введением (1 сутки - 14 сутки) и через 1 ч, 2 ч, 4 ч, 24 ч, 48 ч и 72 ч после последнего введения (14 сутки).
[077] Таблица 10: Изменения внутриглазного давления (среднее ±СПС) у животных в соответствующих группах в обоих глазах до и после введения в 1 сутки
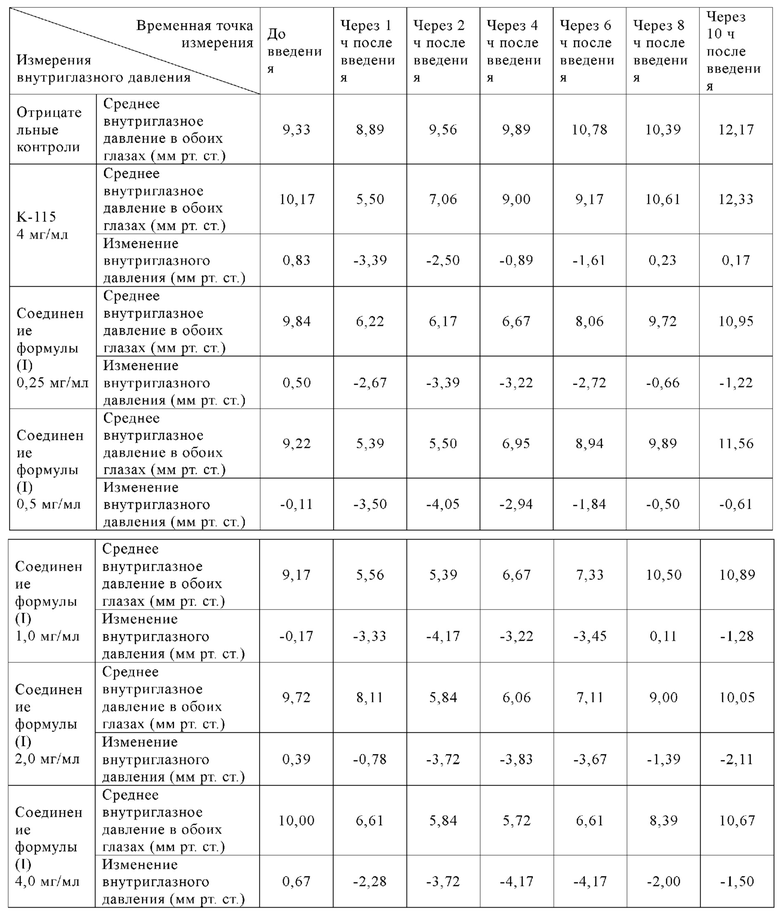
[078] Вывод: однократное введение соединения формулы (I) продемонстрировало лучшую эффективность (наивысший эффект снижения внутриглазного давления и наиболее продолжительное время действия) при всех испытанных дозах (0,25-4,0 мг/мл), что было значительно лучше, чем у K-115.
[079] Животных подвергали определению реакции на раздражение глаз в обоих глазах при помощи ручной щелевой лампы на сутки -2 - сутки -1 перед началом теста, перед первым введением ежесуточно (1 сутки - 14 сутки) и через 1 ч, 2 ч, 4 ч, 24 ч, 48 ч и 72 ч после последнего введения. Критерии подсчета баллов были следующими:
[080] Оценка реакции на раздражение глаз: максимальное количество баллов в отношении роговицы, радужной оболочки, конъюнктивы, отека и секреции суммировали, чтобы получить общую оценку симптомов раздражения глаз в каждой временной точке для каждого глаза животного. На основании оценки симптомов раздражения глаз рассчитывали среднюю оценку каждой группы животных в каждой временной точке наблюдения и определяли степень раздражения глаз каждой группы животных в каждой временной точке в соответствии со следующей таблицей.
[081] Критерии оценки раздражения глаз:
[082] Обнаружение флуоресцеина натрия: после определения реакции на раздражение глаз для каждого глаза обнаружение флуоресцеина натрия осуществляли при помощи ручной щелевой лампы, и критерии подсчета баллов были следующими:
[083] Экспериментальные результаты были следующими:
[084] В соответствии с критериями оценки раздражения глаз общий показатель реакции на раздражение глаз в каждой временной точке в каждой группе составлял менее 3, и все они были классифицированы как не вызывающие раздражения в соответствии с критериями.
[085] Во время теста все показатели теста с флуоресцеином натрия для глаз животных в каждой группе, обработанной физиологическим раствором, растворителем, К-115 и соединением формулы (I), составляли менее 1. Животные в каждой группе имели окрашивание с показателем флуоресцентного окрашивания роговицы 1 при каждой обработке и в отдельной временной точке, что считалось физиологическим окрашиванием. Во всех группах не было повреждений эпителия роговицы в любой временной точке.
[086] Вывод: в условиях данного теста К-115 добавляли в глаза по каплям в концентрации 4 мг/мл в течение 14 суток подряд по 50 мкл/глаз/сутки, не выявляя раздражения. Соединение формулы (I) добавляли в глаза по каплям в концентрации от 0,25 до 4 мг/мл в течение 14 суток подряд по 50 мкл/глаз/сутки, не выявляя раздражения.
[087] Экспериментальный пример 4: Токсикокинетический тест
[088] Экспериментальная цель:
[089] Скорость образования активных фармацевтических ингредиентов в плазме крови и экспозиционное количество активных фармацевтических ингредиентов определяли через 14 суток.
[090] Экспериментальные материалы:
[091] Самцы новозеландских белых кроликов в возрасте от 3 до 6 месяцев, массой от 2,0 до 5,0 кг, приобретенные у Pizhou Oriental Breeding Co., Ltd.
[092] Экспериментальная операция:
[093] Через 14 суток последовательного введения образцы крови собирали через 0 часов (до введения) и через 0,5, 1, 2, 4, 8 и 24 часа после введения в группе соединений формулы (I) (8,0 мг/мл) на 14-15 сутки. Приблизительно 0,8 мл цельной крови брали из ушной артерии или подкожной вены задней конечности (или другого подходящего участка) животного, подвергнутого токсикокинетическому тесту, и помещали в промаркированную пробирку для сбора крови с дикалиевой солью ЭДТА (K2EDTA) в качестве антикоагулянта. Плазму получали центрифугированием при 3000 об/мин и температуре от 2°С до 8°С в течение 10 мин в течение 60 мин после забора крови. Во всех образцах количественно определяли содержание вводимого соединения в плазме крови экспериментального животного при помощи технологии сочетания жидкостной хроматографии и масс-спектрометрии.
[094] Таблица 11: Результаты испытаний активных соединений в плазме крови новозеландских кроликов через 14 суток последовательного введения
[095] Примечание: BQL указано ниже предела обнаружения.
[096] Вывод: при высокой дозе 8 мг/мл концентрация метаболита соединения формулы (I) составляла 0,934 нг/мл через 4 ч после введения и была ниже предела обнаружения через 8 ч после введения, что свидетельствует о высокой системной безопасности.
Изобретение относится к соединению формулы (II) и его кристаллической форме А, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 6,55±0,20°, 10,62±0,20°, 12,57±0,20° и 13,11±0,20°, и кристаллической форме Б, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20° и 21,87±0,20°. Также изобретение относится к способу получения кристаллической формы Б соединения формулы (II), включающему (а) добавление кристаллической формы А соединения формулы (II) в растворитель с образованием суспензии; и (б) перемешивание суспензии при 50°C в течение 3 ч, фильтрацию и сушку, причем растворитель выбран из группы, состоящей из изопропанола, тетрагидрофурана, ацетонитрила, 2-бутанона и этилацетата. Соединение формулы (II) по изобретению применяют при получении лекарственного средства для лечения глаукомы или глазной гипертензии. 5 н. и 12 з.п. ф-лы, 7 ил., 11 табл., 9 пр.

1. Соединение формулы (II),
2. Кристаллическая форма А соединения формулы (II), в которой порошковая рентгеновская дифрактограмма кристаллической формы А содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 6,55±0,20°, 10,62±0,20°, 12,57±0,20° и 13,11±0,20°,
3. Кристаллическая форма А по п. 2, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 10,62±0,20°, 12,57±0,20°, 13,11±0,20°, 17,85±0,20°, 18,51±0,20° и 20,99±0,20°.
4. Кристаллическая форма А по п. 2, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0,20°, 6,33±0,20°, 6,55±0,20°, 10,62±0,20°, 12,57±0,20°, 13,11±0,20°, 14,20±0,20°, 16,37±0,20°, 17,85±0,20°, 18,51±0,20°, 19,56±0,20°, 20,99±0,20°, 25,53±0,20° и 26,35±0,20°.
5. Кристаллическая форма А по п. 3, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 3,30°, 6,33°, 6,55°, 10,62°, 12,57°, 13,11°, 14,20°, 16,37°, 17,85°, 18,51°, 19,56°, 20,99°, 25,53° и 26,35°.
6. Кристаллическая форма А по п. 5, в которой данные анализа порошковой рентгеновской дифрактограммы п указаны ниже:
(Å)
(%)
7. Кристаллическая форма А по любому из пп. 2-6, в которой кривая дифференциальной сканирующей калориметрии кристаллической формы А содержит эндотермический пик с начальным значением при 235,9 °C±3,0°C.
8. Кристаллическая форма А по любому из пп. 2-6, в которой кривая термогравиметрического анализа кристаллической формы А показывает потерю массы до 7,70% при 160,0±3,0°C.
9. Кристаллическая форма Б соединения формулы (II), в которой порошковая рентгеновская дифрактограмма кристаллической формы Б содержит характеристические дифракционные пики при следующих углах 2θ: 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20° и 21,87±0,20°,
10. Кристаллическая форма Б по п. 9, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 9,69±0,20°, 12,12±0,20°, 16,48±0,20°, 16,95±0,20° и 21,87 ±0,20°.
11. Кристаллическая форма Б по п. 10, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 9,69±0,20°, 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20°, 19,23±0,20°, 20,37±0,20° и 21,87±0,20°.
12. Кристаллическая форма Б по п. 11, в которой порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих углах 2θ: 4,80±0,20°, 9,69±0,20°, 12,12±0,20°, 14,61±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20°, 19,23±0,20°, 20,37±0,20°, 21,87±0,20°, 27,53±0,20°, 28,72±0,20° и 33,61±0,20°.
13. Кристаллическая форма Б по п. 12, в которой данные анализа порошковой рентгеновской дифрактограммы указаны ниже:
14. Кристаллическая форма Б по любому из пп. 9-13, в которой кривая дифференциальной сканирующей калориметрии кристаллической формы Б содержит эндотермический пик с начальным значением при 239,5±3,0°C.
15. Кристаллическая форма Б по любому из пп. 9-13, в которой кривая термогравиметрического анализа кристаллической формы Б показывает потерю массы до 1,30% при 200,0±3,0°C.
16. Способ получения кристаллической формы Б соединения формулы (II), включающий:
(а) добавление кристаллической формы А соединения формулы (II) в растворитель с образованием суспензии; и
(б) перемешивание суспензии при 50°C в течение 3 ч, фильтрацию и сушку,
причем кристаллическая форма A соединения формулы (II) или кристаллическая форма Б соединения формулы (II) имеет следующую структуру
порошковая рентгеновская дифрактограмма кристаллической формы А соединения формулы (II) содержит характеристические дифракционные пики при следующих углах 2θ: 3,30±0.20°, 6,33±0,20°, 6,55±0,20°, 10,62±0,20°, 12,57±0,20° и 13,11±0.20°;
порошковая рентгеновская дифрактограмма кристаллической формы Б соединения формулы (II) содержит характеристические дифракционные пики при следующих углах 2θ: 12,12±0,20°, 16,48±0,20°, 16,95±0,20°, 17,94±0,20° и 21,87±0,20°;
причем растворитель выбран из группы, состоящей из изопропанола, тетрагидрофурана, ацетонитрила, 2-бутанона и этилацетата.
17. Применение соединения по п. 1 в получении лекарственного средства для лечения глаукомы или глазной гипертензии.
| CN 101622243 B, 04.12.2013 | |||
| CN 105085478 A, 25.11.2015 | |||
| СУЛЬФОНАМИДНОЕ СОЕДИНЕНИЕ | 2006 |
|
RU2376300C1 |
| Электрический звуковой сигнальный прибор | 1927 |
|
SU11283A1 |
