ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Неприменима.
УРОВЕНЬ ТЕХНИКИ
STING (стимулятор генов интерферона) является сигнальной молекулой врожденного ответа на дцДНК в цитозоле. Имеются сообщения о делеции STING, наблюдаемой при некоторых видах рака. Кроме того, также сообщалось о нарушении регуляции передачи сигналов STING при раковых заболеваниях у человека, таких как меланома (Xia T, et al., «Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis» Cancer Res. 2016) и рак толстой кишки. (Xia T, et al., «Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis» Cell Rep. 2016; 14:282-97). Примечательным является тот факт, что в этих исследованиях результаты геномного анализа показали, что причиной потери экспрессии STING является не делеция или мутация гена, а эпигенетические изменения (Xia, Cancer Res., 2016; Xia, Cell Rep., 2016). Активность STING по защите от рака также подтверждена данными, полученными в исследованиях на мышах. У мышей с нокаутом по STING наблюдали нарушение контроля опухоли (Woo SR, et al. «STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors» Immunity 2014;41:830-42).
Кроме того, роль STING в защите онтогенеза была продемонстрирована на нескольких спонтанных моделях мышей, включая глиому (Ohkuri T, et al., «Protective role of STING against gliomagenesis: Rational use of STING agonist in anti-glioma immunotherapy» Oncoimmunology. 2015; 4:e999523) и рак толстой кишки (Zhu Q, et al., «Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation» J. Immunol. 2014; 193:4779-82). Этот противоопухолевый эффект может быть связан со способностью STING противодействовать чрезмерной активации NF-kB и STAT3 (Woo 2014; Chandra D, et al. «STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer» Cancer Immunol Res. 2014; 2:901-10; Corrales L, et al., «Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity» Cell Rep. 2015; 11:1018-30; Curran E, et al. «STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia» Cell Rep. 2016; 15:2357-66; Tang CH, et al. «Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells» Cancer Res. 2016; 76:2137-52). Эта противоопухолевая активность, вероятно, связана с нарушением сосудистой сети опухоли и последующей индукцией адаптивного иммунного ответа (Corrales L, et al., «The host STING pathway at the interface of cancer and immunity» J. Clin. Invest. 2016; 126:2404-11). Соответственно, прямая или внутриопухолевая стимуляция STING в микроокружении опухоли агонистом может представлять собой новый подход к лечению рака.
Кроме того, рак мочевого пузыря является четвертым и одиннадцатым по распространенности раком у мужчин и женщин, соответственно, и отличается уникальным путем введения лекарственного средства пациентам с раком на ранней стадии (Kamat et al., «What is new in non-muscle-invasive bladder cancer in 2016?» Turk J Urol 2017; 43(1):9-13). В 2018 году в США было выявлено примерно 81190 новых случаев рака мочевого пузыря, и ожидалось примерно 17240 смертельных случаев от этого заболевания (веб-сайт Американского онкологического общества). Назначение БЦЖ (Bacillus Calmette-Gueri) является терапией первой линии при немышечноинвазивном раке мочевого пузыря с высоким риском (NMIBC). К сожалению, до 40% пациентов не реагируют на лечение БЦЖ, и многим из этих пациентов необходимо пройти процедуру радикальной цистэктомии для полного удаления мочевого пузыря (Zlotta, A.R., et al., «The management of BCG failure in non-muscle-invasive bladder cancer: an update,» Can Urol Assoc J 2009; 3(Suppl4): S199-205). В результате этого качество жизни пациентов после операции ухудшается. Следовательно, существует серьезная неудовлетворенная медицинская потребность в эффективных альтернативных методах лечения рака мочевого пузыря, позволяющих избежать или отсрочить радикальную цистэктомию.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у нуждающегося в лечении пациента, включающий введение пациенту Соединения 1 или его фармацевтически приемлемой соли:
 (Соединение 1).
(Соединение 1).
В некоторых вариантах осуществления изобретения фармацевтически приемлемая соль, вводимая в соответствии с представленными в настоящем описании вариантами осуществления, представляет собой соль диаммония или соль триэтиламина (TEA). В дополнительных вариантах осуществления описано лечение рака мочевого пузыря путем введения пациенту, нуждающемуся в лечении рака мочевого пузыря, фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель.
В дополнительных вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у нуждающегося в лечении пациента, включающий введение пациенту Соединения 2 или его фармацевтически приемлемой соли:
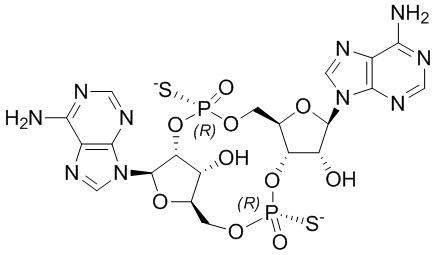 Соединение 2.
Соединение 2.
В дополнительных вариантах осуществления изобретения предлагается применение Соединения 1, Соединения 2 или их фармацевтически приемлемой соли, раскрытых в настоящем описании, для приготовления фармацевтической композиции для лечения рака мочевого пузыря.
В вариантах осуществления изобретения предлагается применение соединения 1, соединения 2, их фармацевтически приемлемой соли или фармацевтической композиции, содержащей соединение 1, соединение 2 или их фармацевтически приемлемую соль, при лечении рака мочевого пузыря.
В вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря, включающий идентификацию индивидуума, у которого рак мочевого пузыря поддается лечению с помощью соединения 1, соединения 2, их фармацевтически приемлемой соли или фармацевтической композиции, содержащей соединение 1, соединение 2 или их фармацевтически приемлемую соль; и введение указанному индивиду терапевтически эффективного количества соединения 1, соединения 2, фармацевтически приемлемой соли или фармацевтической композиции, с помощью которых рак мочевого пузыря идентифицирован как поддающийся лечению.
В некоторых вариантах осуществления изобретения индивидуума идентифицируют как имеющего рак мочевого пузыря, поддающийся лечению с помощью Соединения 1, Соединения 2, их фармацевтически приемлемой соли или фармацевтической композиции, содержащей Соединение 1, Соединение 2 или их фармацевтически приемлемую соль, по наличию у указанного индивидуума вариантного аллеля REF STING.
В некоторых вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у пациента, имеющего аллель REF STING, включающий введение указанному пациенту Соединения 1, Соединения 2, их фармацевтически приемлемой соли или фармацевтической композиции, содержащей Соединение 1, Соединение 2 или их фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретения предлагается способ лечения рака у пациента, имеющего аллель WT STING, включающий введение указанному пациенту Соединения 1, Соединения 2, их фармацевтически приемлемой соли или фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретения предлагается способ лечения рака у пациента, имеющего аллель AQ STING, включающий введение указанному пациенту Соединения 1, Соединения 2, фармацевтически приемлемой соли Соединения 1, Соединения 2 или фармацевтической композиции, содержащей Соединение 1, Соединение 2 или их фармацевтически приемлемую соль.
В некоторых осуществления изобретения предлагается способ лечения рака у пациента, имеющего аллель HAQ STING, включающий введение указанному пациенту Соединения 1, Соединения 2, фармацевтически приемлемой соли Соединения 1, Соединения 2 или фармацевтической композиции, содержащей Соединение 1, Соединение 2 или их фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретения Соединение 1 представлено в виде свободной кислоты. В некоторых вариантах осуществления изобретения соединение представлено в виде соли NH4 или соли триэтиламина (TEA).
В вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества Соединения 1 или его фармацевтически приемлемой соли, как указано выше.
В вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у пациента, включающий введение указанному пациенту Соединения 1, Соединения 2 или их фармацевтически приемлемой соли, или фармацевтической композиции, раскрытых в настоящем описании. Рак мочевого пузыря, лечение которого раскрыто в настоящем описании, может представлять собой уротелиальную карциному.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 показан синтез Соединения 1a. Этот синтез также описан в заявке на патент США № 15/898,533, поданной 17 февраля 2018 г. и включенной в настоящее описание посредством ссылки.
На фиг. 2А и фиг. 2B показан альтернативный синтез Соединения 1 и Соединения 1a. Этот альтернативный синтез также показан на фиг. 2C - фиг. 2E.
На фиг. 3 показана 1Н ЯМР спектрограмма Соединения 1.
На фиг. 4A, фиг. 4B и фиг. 4C показаны результаты анализа методом рентгеновской кристаллографии (фиг. ORTEP), соответственно, асимметричного кристалла Соединения 1, первой молекулы асимметричного кристалла и второй молекулы асимметричного кристалла.
На фиг. 5 показано изображение рентгеновской кристаллической структуры человеческого WT STING в комплексе с соединением 1.
На фиг. 6 показан C-концевой домен человеческого REF STING в комплексе с соединением 1.
На фиг. 7 показаны объемы опухолей, количественно определенные методом МРТ, в дни 20-21.
На фиг. 8 показаны кривые выживаемости для всех групп лечения рака мочевого пузыря, описанных в Примере 4.
На фиг. 9 показана карта вектора экспрессии для WT STING (pLenti-WT human STING-Puro).
Фиг. 10 и фиг. 11 относятся к примеру 108 и представляют лечебную активность соединения 1a на модели двойной опухоли CT26.
Фиг. 12 относится к примеру 109 и представляет график объема опухолей, которые были подвергнуты лечению, и кривую выживаемости.
Фиг. 13 относится к примеру 110 представляет график объема опухолей, которые были подвергнуты лечению, и кривую выживаемости.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Предлагается соединение (Соединение 1) и его фармацевтически приемлемые соли, а также фармацевтические композиции, которые могут быть полезными при лечении рака мочевого пузыря. Соединение 2 или его фармацевтически приемлемая соль, а также фармацевтические композиции, включающие Соединение 2 или его фармацевтически приемлемую соль, также могут быть полезными при лечении рака мочевого пузыря. Соединения могут активировать стимулятор генов интерферона (STING).
Соединение 1 представляет собой:
.
Соединение 2 представляет собой:
.
В некоторых вариантах осуществления Соединение 1 представлено в виде свободной кислоты. В некоторых вариантах осуществления соединение представлено, например, в виде соли NH4. Ссылка на «Соединение 1а» указывает на диаммониевую соль Соединения 1.
В вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества Соединения 1, Соединения 2 или их фармацевтически приемлемой соли.
Также предлагаются фармацевтические композиции для лечения рака мочевого пузыря, включающие соединение, раскрытое в настоящем описании, или его фармацевтически приемлемую соль, а также фармацевтически приемлемый наполнитель. Приведенные в настоящем описании варианты осуществления изобретения могут быть использованы для лечения рака мочевого пузыря или для приготовления лекарственных средств, полезных для лечения рака мочевого пузыря.
Специалистам в данной области известно, что заместители, связанные с атомами фосфора (P1, P2), имеющие как одинарные, так и двойные связи, могут быть подвержены таутомеризации. Например, таутомеризация соединения может происходить в условиях равновесия. Один из примеров показан ниже:
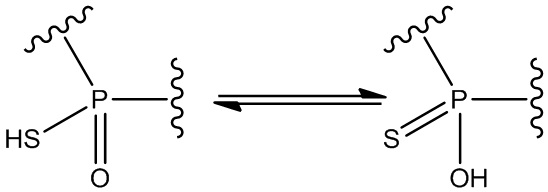 .
.
Такие таутомеры следует рассматривать как входящие в объем формулы изобретения. Структурное представление любого таутомера заданного соединения будет представлять одно и то же соединение.
Методы лечения
В вариантах осуществления изобретения предоставляется способ лечения рака мочевого пузыря у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества Соединения 1 или его фармацевтически приемлемой соли.
В некоторых вариантах осуществления Соединение 1 представлено в виде свободной кислоты или ее фармацевтически приемлемой соли. В некоторых вариантах осуществления вводимое соединение представлено в виде соли NH4, свободной кислоты или ее фармацевтически приемлемой соли. В некоторых вариантах осуществления соединение представлено в виде соли NH4.
В некоторых вариантах осуществления изобретения предлагается способ лечения рака мочевого пузыря, включающий введение нуждающемуся в лечении пациенту терапевтически эффективного количества агониста STING. В таких вариантах осуществления введение необязательно может быть внутрипузырным введением. Примеры потенциальных агонистов STING, которые можно использовать при лечении рака мочевого пузыря и которые необязательно могут вводиться внутрипузырно, включают, без ограничения, соединения, представленные в заявке на патент США 15/898,533, поданной 17 февраля 2018; заявке PCT PCT/US2018/018556, поданной 17 февраля 2018; заявке PCT PCT/US2018/018561, поданной 17 февраля 2018; US 2014/0205653 A1; US 2014/0329889 A1; US 2014/0341976 A1; US 2007/0149462 A1; WO 2018/098203 A1; WO 2018/198076 A1; WO 2018/198084 A1; WO 2018/140831 A2; US 2014/0341976 A1; WO 2015/185565 A1; US 7,709,458 B2; US 7,592,326; US 7,569,555 B2; US 2014/0205653 A1; US 2014/0341976 A1; US 2015/0056224 A1; US 2016/0362441 A1; US 2017/0158724 A1; US 2017/044206 A1; US 5,547,941; US 7,569,555 B2; US 7,592,326 B2; US 7,709,458 B2; US 9,549,944 B2; WO 2009/133560 A1; WO 2015/074145 A1; WO 2015/077354 A1; WO 2015/185565 A1; WO 2016/100261 A1; WO 2016/120305 A1;WO 2016/145102 A1; WO 2017/027645 A1; WO 2017/027646 A1; WO 2017/075477 A1; WO 2019/043634 A2; WO 2019/046496 A1; WO 2019/046498 A1; WO 2019/046500 A1; WO 2019/046511 A1; WO 2017/093933 A1;WO 2017/123657 A1; WO 2017/175156 A1; EP 1740,192 B1; CN 102199183 A; Fu, J., et al., «STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade,» Sci. Translational Med., 7(283):283ra52, (April 15, 2015); Corrales, L. et al., «Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity,» Cell Reports, 11:1018-1030 (2015); и Lioux, T. et al., «Design, Synthesis, and Biological Evaluation of Novel Cyclic Adenosine-Inosine Monophosphate (cAIMP) Analogs That Activate Stimulator of Interferon Genes (STING),» J. Med. Chem., 59:10253-10267 (2016). Все указанные документы, включая упомянутые в них соединения, включены в настоящее описание посредством ссылки; если какой-либо компонент любого из этих документов противоречит или иным образом несовместим с настоящим описанием, то настоящее описание является приоритетным.
Дозировка
Оптимальная доза для лечения рака мочевого пузыря может быть определена эмпирически для каждого индивидуума с помощью известных методов и будет зависеть от множества факторов, включая активность агентов; возраст, массу тела, общее состояние здоровья, пол и диету человека; время и способ введения; и другие лекарственные средства, которые принимает человек. Оптимальные дозы могут быть установлены путем обычного тестирования и с помощью обычных процедур, хорошо известных в данной области. Указанные выше соединения можно вводить любым подходящим способом.
«Фармацевтически приемлемая соль» в контексте настоящего описания относится к солям, полученным в результате реакции присоединения между кислотой или основанием и соединений по настоящему изобретению. Фармацевтически приемлемая соль представляет собой любую соль, которая сохраняет активность исходного соединения и не оказывает никакого чрезмерно вредного или нежелательного воздействия на организм субъекта, которому она вводится, и в контексте, в котором она вводится. Фармацевтически приемлемые соли включают, без ограничения, комплексы металлов и соли как неорганических, так и карбоновых кислот. Фармацевтически приемлемые соли также включают соли металлов, таких как алюминий, кальций, железо, магний, марганец, и комплексные соли. Кроме того, фармацевтически приемлемые соли включают, без ограничения, кислые соли, такие как соли уксусной, аспарагиновой, алкилсульфоновой, арилсульфоновой, аксетиловой, бензолсульфоновой, бензойной, бикарбонатной, бисерной, дивинной, масляной кислоты, эдетат кальция, камзиловой, угольной, хлорбензойной, лимонной, эдетовой, 1,2-этандисульфоновой (edisylic), лаурилсерной (estolic), этансульфоновой (esyl), этансульфиновой (esylic), муравьиновой, фумаровой, глюкогептоновой (gluceptic), глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексаминовой, гексилрезорциновой, гидрабаминовой, бромистоводородной, соляной кислоты, гидрохлорида, йодистоводородной, гидроксинафтоевой, изэтиновой, молочной, лактобионовой, малеиновой, яблочной, малоновой, миндальной, метансульфоновой, метилазотной, метилсерной, муциновой, муконовой, напсиловой, азотной, щавелевой, п-нитрометансульфоновой, памоиновой, пантотеновой, фосфорной, моногидрофосфорной, дигидрофосфорной, фталевой, полигалактуроновой, пропионовой, салициловой, стеариновой, янтарной, сульфаниловой, сульфоновой, серной, дубильной, винной, хлортеофиллиновой (teoclic), толуолсульфоновой кислоты и т.п. Также могут быть получены соли натрия и соли калия.
Вариантами осуществления могут быть соли диаммония. Фармацевтически приемлемые соли могут быть производными аминокислот, включая, помимо прочего, цистеин. Способы получения соединений в виде солей известны специалистам в данной области (см., например, Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66:1, 1977).
«Эффективное количество» или «терапевтически эффективное количество» терапевтического агента представляет собой количество, достаточное для получения наблюдаемого терапевтического улучшения по сравнению с раком мочевого пузыря у субъекта или пациента, который не проходил лечение.
Активные агенты, раскрытые в настоящем описании, могут быть объединены с фармацевтически приемлемым носителем с получением фармацевтических составов на их основе. Конкретный выбор носителя и состава будет зависеть от конкретного пути введения, для которого предназначена композиция.
В контексте настоящего описания термин «фармацевтически приемлемый носитель» относится к нетоксичному носителю, адъюванту или несущей среде, не нарушающей фармакологическую активность соединения, с которым она образует состав. Фармацевтически приемлемые носители, адъюванты или носители, которые можно использовать в композициях по настоящему изобретению, включают, без ограничения, сорбиновую кислоту, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий-карбоксиметилцеллюлозу, полиакрилаты, воски, полиэтиленгликоль и ланолин.
Композиции по настоящему изобретению могут быть подходящими для парентерального, перорального введения, введения в виде ингаляционного спрея, местного, ректального, назального, трансбуккального, внутрипузырного, вагинального введения или введения с помощью имплантированного резервуара и т.д. В некоторых вариантах осуществления состав содержит ингредиенты, которые получены из природных или неприродных источников. В некоторых вариантах осуществления состав или носитель могут быть предоставлены в стерильной форме. Неограничивающие примеры стерильного носителя включают воду, не содержащую эндотоксинов, или апирогенную воду. Композиции можно вводить внутрипузырно.
В контексте настоящего описания термин «парентеральный» включает подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, внутригрудинные, интратекальные, внутрипеченочные, внутриочаговые и внутричерепные инъекции или инфузии. В конкретных вариантах осуществления соединения вводят внутривенно, перорально, подкожно или внутримышечно. Стерильные инъекционные формы композиций по настоящему изобретению могут представлять собой водную или масляную суспензию. Эти суспензии могут быть составлены согласно методам, известным в данной области, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор или суспензию в нетоксичном разбавителе или растворителе, пригодном для парентерального введения. К приемлемым носителям и растворителям, которые можно использовать, относятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. В некоторых вариантах осуществления композиции вводят внутрипузырно.
Для этой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- или диглицериды. Для приготовления инъекционных препаратов также применимы жирные кислоты и их глицеридные производные полезны, а также натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в полиоксиэтилированных вариантах. Эти масляные растворы или суспензии также могут содержать разбавитель или диспергирующий агент на основе длинноцепочечного спирта, такой как карбоксиметилцеллюлоза или аналогичные диспергирующие агенты, которые обычно используются в приготовлении фармацевтически приемлемых дозированных форм, включая эмульсии и суспензии. Для приготовления состава также могут быть использованы другие обычно используемые поверхностно-активные вещества, такие как Твины (Tween), Спаны (Span) и другие эмульгирующие агенты, которые обычно используются при производстве фармацевтически приемлемых твердых, жидких или других лекарственных форм.
Для перорального введения соединение или соль могут быть представлены в приемлемой пероральной лекарственной форме, включая, без ограничения, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Также могут быть добавлены лубриканты, такие как стеарат магния. Для перорального введения в форме капсул подходящие разбавители включают лактозу и сушеный кукурузный крахмал. Если для перорального применения требуются водные суспензии, активный ингредиент можно комбинировать с эмульгирующими и суспендирующими агентами. При необходимости также могут быть добавлены определенные подсластители, ароматизаторы или красители. Также могут быть добавлены консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают, без ограничения, различные антибактериальные и противогрибковые агенты, такие как растворители, например этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, соли четвертичного аммония и парабены (такие как метилпарабен, этилпарабен, пропилпарабен и др.).
Под «немедленным высвобождением» подразумевают обычное высвобождение, при котором высвобождение лекарственного вещества начинается сразу после введения. Используемый в настоящем описании термин «немедленное высвобождение» включает лекарственные формы, которые обеспечивают растворение лекарственного вещества в содержимом желудочно-кишечного тракта без задержки или продления процесса растворения или абсорбции лекарственного вещества. Целью является быстрое высвобождение лекарственного вещества после введения, например, с возможностью высвобождения по меньшей мере 80% лекарственного вещества в течение приблизительно 30 минут после начала растворения в тесте на растворение.
«Замедленное высвобождение» или «пролонгированное высвобождение» относится к лекарственным формам, характеристики которых по высвобождению лекарственного вещества выбирают в зависимости от времени и/или местоположения для достижения терапевтических целей или преимуществ, которые не достигаются обычными лекарственными формами, такими как раствор или лекарственная форма с немедленным высвобождением.
Термин «устойчивый» означает, что для данного активного агента достигнут уровень в плазме, который поддерживается последующими дозами активного агента на этом же уровне или на уровне, который выше минимального эффективного терапевтического уровня и ниже минимального токсичного уровня в плазме для данного активного агента.
Термин «диапазон доз», используемый в настоящем описании, относится к верхнему и нижнему пределу приемлемого изменения количества указанного агента. Обычно пациентам, проходящим лечение, может вводиться доза агента в любом количестве в указанном диапазоне.
Термин «лечить» используется в настоящем описании для обозначения облегчения, уменьшения или ослабления по меньшей мере одного симптома заболевания у субъекта. Например, в отношении рака мочевого пузыря термин «лечить» может означать купирование, отсрочку начала (т.е. период до клинического проявления заболевания или симптома заболевания) и/или снижение риска развития или ухудшения симптома рака мочевого пузыря. Термин «защищать» используется в настоящем описании для обозначения профилактики с целью задержки или лечения, или того и другого в зависимости от ситуации, начала развития, продолжения или ухудшения симптомов рака мочевого пузыря у субъекта.
Термин «субъект» или «пациент» включает животных, которые способны страдать от рака мочевого пузыря или болеть им. Примеры субъектов или пациентов включают млекопитающих, например людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных животных, не относящихся к человеку. В некоторых вариантах осуществления субъект представляет собой человека, например человека, страдающего от рака мочевого пузыря, подверженного риску заболевания или потенциально способного страдать от него.
Термин «примерно» или «приблизительно» обычно означает в пределах 20%, более предпочтительно в пределах 10% и наиболее предпочтительно в пределах 5% заданного значения или диапазона. В качестве альтернативы, особенно в биологических системах, термин «примерно» означает приблизительно в пределах логарифма (т.е. порядка величины), предпочтительно в пределах двукратной величины от заданного значения.
Использование терминов в единственном числе в контексте описания изобретения (особенно в контексте приведенной ниже формулы изобретения) следует толковать как охватывающее также и множественное число, если только из контекста в явном виде не следует иное или иное в явном виде не противоречит контексту. Термины «содержащий», «имеющий», «включающий» и «состоящий из» следует толковать как неограниченные термины (т.е. означающие «включая, без ограничения»), если не указано иное. Перечисление диапазонов значений в настоящем описании писпользуется исключительно с целью сокращенной ссылки на каждое отдельное значение, попадающее в указанный диапазон, если в настоящем описании не указано иное, и каждое отдельное значение включено в описание, как если бы оно было указано отдельно в настоящем описании.
Типичные нарушения пролиферации клеток, которые можно лечить с помощью одного или более раскрытых в настоящем описании соединений, включают, без ограничения, рак, предраковую стадию или предраковое состояние и метастатические поражения в ткани и органах тела. Нарушения пролиферации клеток могут включать гиперплазию, метаплазию и дисплазию.
Соединение, раскрытое в настоящем описании, или его фармацевтически приемлемая соль могут использоваться для лечения или профилактики нарушения пролиферации клеток, или для лечения или профилактики рака у субъекта с повышенным риском развития рака по сравнению с популяцией в целом, или для применения для идентификации подходящих кандидатов для таких целей.
Фармацевтические составы и способы введения
Предлагаются фармацевтические составы, содержащие Соединение 1 или его фармацевтически приемлемую соль, для лечения рака мочевого пузыря. Фармацевтические составы могут дополнительно содержать носитель или наполнитель, стабилизатор, ароматизатор и/или краситель.
Соединение 1 или его фармацевтически приемлемую соль можно вводить различными путями, известными специалистам в данной области. Пути введения включают пероральное введение и внутрипузырное введение. В некоторых вариантах осуществления фармацевтический состав, содержащий соединение или его фармацевтически приемлемую соль, можно принимать перорально в форме жидкости, сиропа, таблетки, капсулы, порошка, капсулы с покрытыми частицами, жевательной таблетки или растворимого диска. Альтернативно фармацевтические составы по настоящему изобретению можно вводить внутривенно или трансдермально. Дополнительные пути введения известны специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, Gennaro A. R., Ed., 20.sup.th Edition, Mack Publishing Co., Easton, Pa).
В некоторых вариантах осуществления составы с соединением или фармацевтически приемлемой соли готовят в виде пасты, желе или суспензии. Например, лекарственное вещество растворяют, захватывают или суспендируют в форме частиц лекарственного вещества, микрокапсулированных частиц или полимерных частиц лекарственного вещества в гелеобразном растворе или полутвердом виде. Преимущество перорального желеобразного состава состоит в том, что его легче вводить пациентам, которым трудно глотать таблетки, капсулы или пилюли. В некоторых вариантах осуществления соединение тщательно перемешивают и суспендируют в подходящей среде с образованием пасты или геля. В случае перорального введения необязательно могут быть добавлены дополнительные агенты для придания аромата. Арахисовое масло или альгинат, приправленный малиной и подсластителем, являются примерами многих подходящих агентов, маскирующих вкус. В различных вариантах осуществления составы в виде пасты или желе также могут быть приготовлены с подходящими связующими или вспомогательными веществами, известными в данной области для местного применения.
Способы приготовления составов с замедленным высвобождением в форме таблеток, капсул или пилюль известны в данной области. В некоторых вариантах осуществления состав с замедленным высвобождением получают путем покрытия активного ингредиента лекарственного средства полимером, предпочтительно нерастворимым в воде полимером. Примером нерастворимого в воде полимера, используемого в области фармацевтики в качестве покрывающего агента с замедленным высвобождением, является агент для энтеросолюбильного покрытия или покрывающий агент, защищающий от воздействия желудочного сока. Нерастворимый в воде полимер может включать, например, этилцеллюлозу, очищенный шеллак, белый шеллак, сополимер аминоалкилметакрилата RS, фталат гидроксипропилметилцеллюлозы, ацетат-сукцинат гидроксипропилметилцеллюлозы, карбоксиметилэтилцеллюлозу, ацетат-фталат целлюлозу, сополимер L метакриловой кислоты, сополимер LD метакриловой кислоты, сополимер S метакриловой кислоты, сополимер E аминоалкилметакрилата или поливинилацеталь диэтиламиноацетата.
Тип, степень замещения и молекулярная масса нерастворимых в воде полимеров могут зависеть от растворимости активного ингредиента в воде или спирте, требуемой скорости замедленного высвобождения и т.п. Нерастворимые в воде полимеры можно использовать по отдельности или в комбинации. Также могут быть включены гидрогенизированное масло, стеариновая кислота или цетанол в качестве вспомогательного покрывающего агента и триглицерид со средней длиной цепи, триацетин, триэтилцитрат или цетанол в качестве пластификатора.
В некоторых вариантах осуществления состав с замедленным высвобождением представляет собой матричную таблетку или гранулу. Активный ингредиент может быть покрыт полимерами 3-х разных типов. Эти полимеры трех разных типа могут включать: 1) нерастворимый в воде полимер, такой как этилцеллюлоза; 2) рН-независимый гелеобразующий полимер, такой как гидроксипропилметилцеллюлоза; и 3) рН-зависимый гелеобразующий полимер, такой как альгинат натрия. Эти полимеры трех разных типов могут быть использованы вместе для снижения скорости высвобождения лекарственных веществ.
Лекарственные формы: свойства высвобождения
Составы с замедленным высвобождением могут обеспечивать длительный эффект. Однако действие и/или биодоступность активного ингредиента может меняться в зависимости от нескольких факторов, таких как, например, окно абсорбции, носители или наполнители, используемые в составе, способ доставки состава и/или время прохождения активного ингредиента через желудочно-кишечный тракт пациента.
Терапевтическое средство может содержать по меньшей мере одну часть с замедленным высвобождением для выполнения функции замедленного высвобождения и одну часть с немедленным высвобождением для выполнения функции немедленного высвобождения. В некоторых вариантах осуществления, когда терапевтическое средство представляет собой единичную дозу, указанная доза может быть в форме таблеток, сформированных из смеси гранул с замедленным высвобождением, составляющих часть с замедленным высвобождением, и гранул с немедленным высвобождением, составляющих часть с немедленным высвобождением, препарата в виде капсулы, полученного путем заполнения капсулы гранулами с замедленным высвобождением и гранулами с немедленным высвобождением, или таблетки с прессованным покрытием, у которой внешний слой, составляющий часть с немедленным высвобождением, сформирован на внутреннем ядре, составляющем часть с замедленным высвобождением. Однако вышеупомянутые варианты осуществления не ограничены.
Более того, отсутствуют конкретные ограничения на состояние содержимого лекарственного вещества в композиции, или в части с немедленным высвобождением, или в части с замедленным высвобождением; соединение может быть равномерно диспергировано в композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может содержаться только в одной части композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может быть распределено таким образом, чтобы создавался градиент концентрации.
Часть с замедленным высвобождением в композиции по настоящему изобретению может содержать по меньшей мере одно pH-независимое полимерное вещество или pH-зависимое полимерное вещество для регулирования высвобождения лекарственного вещества.
pH-независимое полимерное вещество, используемое в настоящем изобретении, может включать полимерное вещество, заряд которого практически не изменяется при значениях pH, обычных для желудочно-кишечного тракта, в частности при pH от 1 до 8. Это означает, например, что полимерное вещество не имеет функциональных групп, заряд которых изменяется в зависимости от pH, таких как основные функциональные группы, например аминогруппы, или кислотные функциональные группы, например группы карбоновых кислот. Следует о™етить, что pH-независимое полимерное вещество может быть включено для придания композиции по настоящему изобретению функции замедленного высвобождения, но также может быть включено с другой целью. Более того, pH-независимое полимерное вещество, используемое в настоящем изобретении, может быть нерастворимым в воде или может набухать в воде или растворяться в воде с образованием геля.
Примеры нерастворимых в воде pH-независимых полимерных веществ включают, без ограничения, простые эфиры целлюлозы, сложные эфиры целлюлозы и сополимеры метакриловой кислоты и акриловой кислоты (торговое название Eudragit, производства Rohm GmbH & Co. KG, Darmstadt, Germany). Примеры включают, без ограничения, алкиловые эфиры целлюлозы, такие как этилцеллюлоза (торговое название Ethocel, производства Dow Chemical Company, USA), этилметилцеллюлоза, этилпропилцеллюлоза или изопропилцеллюлоза и бутилцеллюлоза, аралкиловые эфиры целлюлозы, такие как бензилцеллюлоза, простые эфиры цианоалкил целлюлозы, такие как цианоэтилцеллюлоза, сложные эфиры целлюлозы и органических кислот, такие как бутират-ацетат целлюлоза, ацетат целлюлоза, пропионат целлюлоза или бутират целлюлоза и пропионат-ацетат целлюлоза, сополимеры этилакрилата-метилметакрилата (торговое название Eudragit NE, производства Rohm GmbH & Co. KG, Darmstadt, Germany) и сополимер RS аминоалкилметакрилата (торговые названия Eudragit RL, Eudragit RS). Отсутствуют какие-либо особые ограничения на средний диаметр частиц нерастворимого в воде полимера, используемого в настоящем изобретении, но обычно чем ниже средний диаметр частиц, тем лучше рабочие характеристики, при этом средний диаметр частиц предпочтительно составляет от 0,1 до 100 мкм, более предпочтительно от 1 до 50 мкм, особенно предпочтительно от 3 до 15 мкм, наиболее предпочтительно от 5 до 15 мкм. Кроме того, примеры водорастворимых или набухающих в воде pH-независимых полимерных веществ включают, без ограничения, полиэтиленоксид (торговое название Polyox, производства Dow Chemical Company, молекулярная масса от 100000 до 7000000), низкозамещенную гидроксипропилцеллюлозу (торговое название L-HPC, производства Shin-Etsu Chemical, Japan), гидроксипропилцеллюлозу (торговое название HPC, производства Nippon Soda, Co., Ltd, Japan), гидроксипропилметилцеллюлозу (торговые названия Metolose 60SH, 65SH, 90SH, производства Shin-Etsu Chemical, Japan) и метилцеллюлозу (торговое название Metolose SM, производства Shin-Etsu Chemical, Japan).
В некоторых вариантах осуществления композиция может содержать одно pH-независимое полимерное вещество или может содержать несколько pH-независимых полимерных веществ. pH-независимое полимерное вещество, когда используется в раскрытых вариантах осуществления, может быть нерастворимым в воде полимерным веществом, более предпочтительно этилцеллюлозой, сополимером этилакрилата и метилметакрилата (торговое название Eudragit NE) или сополимером RS аминоалкилметакрилата (торговое название Eudragit RL, Eudragit RS). Особенно предпочтительным является, по меньшей мере, одно из следующих веществ: этилцеллюлоза и сополимер RS аминоалкилметакрилата. Наиболее предпочтительной является этилцеллюлоза. Отсутствуют конкретные ограничения на количество pH-независимого полимерного вещества в композиции; его количество может быть подобрано соответствующим образом в зависимости от цели, такой как контроль замедленного высвобождения лекарственного средства.
pH-зависимое полимерное вещество, которое можно использовать в представленных в настоящем описании вариантах осуществления, может быть полимерным веществом, заряд которого изменяется при значениях pH, обычных для желудочно-кишечного тракта, в частности при pH от 1 до 8. Это означает, например, полимерное вещество, содержащее функциональные группы, заряд которых изменяется в зависимости от pH, такие как основные функциональные группы, например аминогруппы, или кислотные функциональные группы, например группы карбоновых кислот. pH-зависимые функциональные группы pH-зависимого полимерного вещества предпочтительно представляют собой кислотные функциональные группы, причем pH-зависимое полимерное вещество наиболее предпочтительно содержит группы карбоновой кислоты.
pH-зависимое полимерное вещество, используемое в настоящем изобретении, может быть нерастворимым в воде или может набухать в воде или растворяться в воде с образованием геля. Примеры pH-зависимых полимерных веществ, используемых в настоящем изобретении, включают, без ограничения, энтеросолюбильные полимерные вещества. Примеры энтеросолюбильных полимерных веществ включают, без ограничения, сополимеры метакриловой кислоты и метилметакрилата (Eudragit L100, Eudragit S100, производства Rohm GmbH & Co. KG, Darmstadt, Germany), сополимеры метакриловой кислоты и этилакрилата (Eudragit L100-55, Eudragit L30D-55, производства Rohm GmbH & Co. KG, Darmstadt, Germany), фталат гидроксипропилметилцеллюлозы (HP-55, HP-50, производства Shin-Etsu Chemical, Япония), сукцинат-ацетат гидроксипропилметилцеллюлозу (AQOAT, производства Shin-Etsu Chemical, Japan), карбоксиметилэтилцеллюлозу (CMEC, производства Freund Corporation, Japan) и фталат-ацетат целлюлозу.
Примеры pH-зависимых полимерных веществ, которые набухают в воде или растворяются в воде с образованием геля, включают, без ограничения, альгиновую кислоту, пектин, карбоксивиниловый полимер и карбоксиметилцеллюлозу. В настоящем изобретении композиция может содержать одно pH-зависимое полимерное вещество или может содержать несколько pH-зависимых полимерных веществ. pH-зависимое полимерное вещество, используемое в настоящем изобретении, предпочтительно представляет собой энтеросолюбильное полимерное вещество, более предпочтительно сополимер метакриловой кислоты и этилакрилата, сополимер метакриловой кислоты и метилметакрилата, фталат гидроксипропилметилцеллюлозы или сукцинат-ацетат гидроксипропилметилцеллюлозу, особенно предпочтительным является сополимер метакриловой кислоты кислоты и этилакрилата.
Когда в процессе производства композиции по настоящему изобретению используется pH-зависимое полимерное вещество, то коммерчески доступный продукт может быть использован непосредственно в виде порошка или гранул или в виде суспензии, в которой pH-зависимое полимерное вещество уже диспергировано в растворителе, или такой коммерчески доступный продукт может быть диспергирован в воде или органическом растворителе для его использования. Чем меньше диаметр частиц pH-зависимого полимерного вещества, тем лучше рабочие характеристики, при этом pH-зависимое полимерное вещество предпочтительно находится в виде порошка. В случае сополимера метакриловой кислоты и этилакрилата примером является Eudragit L100-55. Отсутствуют конкретные ограничения на средний диаметр частиц pH-зависимого полимерного вещества, используемого в настоящем изобретении, но средний диаметр частиц предпочтительно составляет от 0,05 до 100 мкм, более предпочтительно от 0,05 до 70 мкм, наиболее предпочтительно от 0,05 до 50 мкм. Более того, отсутствуют конкретные ограничения на количество pH-зависимого полимерного вещества; например, в случае энтеросолюбильного полимерного вещества количество обычно составляет от 0,1 до 90 мас. частей, предпочтительно от 1 до 70 мас. частей, более предпочтительно от 5 до 60 мас. частей, особенно предпочтительно от 10 до 50 мас. частей в расчете на 100 мас. частей композиции.
Терапевтическое средство согласно представленным вариантам осуществления необязательно может дополнительно содержать любую из различных добавок, например любой фармакологически приемлемый носитель, такой как разбавитель, лубрикант, связующее вещество и разрыхлитель, а также любой консервант, краситель, подсластитель, пластификатор, вещество для пленочного покрытия и так далее. Примеры разбавителей включают, без ограничения, лактозу, маннит, двухосновный фосфат кальция, крахмал, прежелатинизированный крахмал, кристаллическую целлюлозу, неуплотненный оксид кремния, синтетический алюмосиликат, метасиликат алюмината магния и т.п. Примеры лубрикантов включают, без ограничения, стеарат магния, стеарат кальция, тальк, стеарилфумарат натрия и т.п. Примеры связующих веществ включают, без ограничения, гидроксипропилцеллюлозу, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон и т.п. Примеры разрыхлителей включают, без ограничения, карбоксиметилцеллюлозу, кальций-карбоксиметилцеллюлозу, натрий-кроскармеллозу, карбоксиметилкрахмал натрия, низкозамещенную гидроксипропил целлюлозу и т.п.
Примеры консервантов включают, без ограничения, сложные эфиры параоксибензойной кислоты, хлорбутанол, бензиловый спирт, фенилэтиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту и т.п. Предпочтительные примеры красителей включают, без ограничения, водонерастворимые красочные пигменты, природные пигменты (например, β-каротин, хлорофилл, красный оксид железа), желтый оксид железа, красный оксид железа, черный оксид железа и т.п. Предпочтительные примеры подсластителей включают, без ограничения, сахарин натрия, глицирризат калия, аспартам, стевию и т.п. Примеры пластификаторов включают, без ограничения, сложные эфиры глицерина и жирных кислот, триэтилцитрат, пропиленгликоль, полиэтиленгликоль и т.п. Примеры агентов для пленочного покрытия включают, без ограничения, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу и т.п.
Методы изготовления
Для получения представленных вариантов осуществления можно использовать один традиционный способ или комбинацию традиционных способов. Например, при производстве гранул, содержащих лекарственное вещество, в виде части с замедленным высвобождением или части с немедленным высвобождением, гранулирование является основной операцией, но ее можно комбинировать с другими операциями, такими как смешивание, сушка, просеивание и классификация. В качестве метода грануляции можно использовать, например, метод влажной грануляции, при котором к порошку добавляют связующее вещество и растворитель и осуществляют грануляцию, метод сухой грануляции, при котором порошок прессуют и выполняют грануляцию, метод грануляции расплава, при котором добавляют связующее вещество, которое плавится при нагревании, выполняют нагревание и грануляцию, и т.п.
Кроме того, в зависимости от способа грануляции можно использовать соответствующий рабочий метод, например метод грануляции при перемешивании с использованием планетарного миксера, шнекового миксера и т.п., метод грануляции при высокоскоростном перемешивании с использованием миксера Henschel, миксера Super и т.п., метод экструдирования и грануляции с использованием цилиндрического гранулятора, роторного гранулятора, шнекового экструдирующего гранулятора, пеллетного гранулятора или т.п., метод влажной грануляции с высоким усилием сдвига, метод грануляции в псевдоожиженном слое, метод грануляции с последующим сжатием, метод дробления и грануляции, или метод грануляции распылением. После грануляции можно выполнить сушку с помощью сушилки, методом псевдоожиженного слоя и т.п., измельчение и просеивание для получения гранул или мелких гранул, предназначенных для использования. Кроме того, в процессе приготовления композиции по настоящему изобретению можно использовать растворитель для грануляции. Отсутствуют какие-либо особые ограничения, применительно к растворителю для грануляции, который может представлять собой воду или любой из различных органических растворителей, например воду, низший спирт, такой как метанол или этанол, кетон, такой как ацетон или метилэтилкетон, метиленхлорид или их смеси.
Для вариантов осуществления с замедленным высвобождением гранулы получают путем совместного перемешивания, по меньшей мере одного лекарственного вещества и по меньшей мере одного вещества, выбранного из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, необязательно добавления разбавителя и связующего вещества, с последующей грануляцией с получением гранулированного материала. Полученный гранулированный материал сушат с помощью лотковой сушилки, сушилки с псевдоожиженным слоем и т.п., и просеивают, используя мельницу или осциллятор, получая таким образом гранулы с замедленным высвобождением. В качестве альтернативы, в способе производства гранул с замедленным высвобождением по настоящему изобретению можно добавить по меньшей мере одно лекарственное вещество, по меньшей мере одно вещество, выбранное из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, и необязательно разбавителя и связующего вещества, и используя сухой уплотнитель, такой как роликовый уплотнитель или поршневая машина для таблетирования, выполнить прессование при перемешивании, а затем грануляцию путем измельчения до подходящего размера. Гранулированный материал, полученный с помощью такого гранулятора, можно использовать в виде гранул или мелких гранул в соответствии с настоящим изобретением или можно дополнительно измельчить с помощью силовой мельницы, валкового гранулятора, скоростной мельницы и т.п. и просеять с получением гранул с замедленным высвобождением. Следует обратить внимание на то, что гранулы с немедленным высвобождением могут быть изготовлены таким же способом, что и гранулы с замедленным высвобождением.
Формованный прессованием продукт может быть изготовлен в виде части с замедленным высвобождением, которая содержит лекарственное вещество, или части с немедленным высвобождением, или в виде композиции, раскрытой в настоящем описании, с помощью одного традиционного метода или комбинации традиционных методов. Например, используя по меньшей мере одно лекарственное вещество, по меньшей мере, одно вещество, выбранное из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, разбавитель, такой как маннит или лактоза, связующее вещество, такое как поливинилпирролидон или кристаллическая целлюлоза, разрыхлитель, такой как кармеллоза натрия или кросповидон, и лубрикант, такой как стеарат магния или тальк, выполняют таблетирование традиционным методом, с помощью которого можно получить прессованный продукт. В этом случае таблетирование является основной операцией в способе производства прессованного продукта, но его можно комбинировать с другими операциями, такими как перемешивание, сушка, формирование сахарного покрытия и нанесение покрытия.
Примеры способа таблетирования включают, без ограничения, прямое прессование, при котором по меньшей мере одно лекарственное вещество и фармакологически приемлемые добавки смешивают вместе, а затем смесь непосредственно прессуют в таблетки с помощью машины для таблетирования, и сухое прессование гранул или влажное прессование гранул, при котором гранулы с замедленным высвобождением или гранулы с немедленным высвобождением по настоящему изобретению подвергают прессованию после необязательного добавления лубриканта или разрыхлителя. Отсутствуют какие-либо особые ограничения на машину для таблетирования, используемую при прессовании; например, можно использовать машину для таблетирования с одним пуансоном, роторную машину для таблетирования или машину для таблетирования с нанесением прессованного покрытия.
Содержащие лекарственное вещество гранулы с замедленным высвобождением или гранулы с немедленным высвобождением, или формованный прессованием продукт в соответствии с раскрытыми вариантами осуществления могут использоваться в естественном виде в форме гранул или таблетки в качестве композиции, но также могут быть подвергнуты дальнейшей обработке для получения композиции. Например, на формованный прессованием продукт или гранулы можно нанести пленочное покрытие, используя материал-основу для пленочного покрытия, такой как этилцеллюлоза, казеин, метилцеллюлоза, гидроксипропилметилцеллюлоза, сополимер L метакриловой кислоты, фталат-ацетат целлюлозу, шеллак и т.п., или сахарное покрытие, используя жидкость для сахарного покрытия, содержащую сахарозу, сахарный спирт, порошок гуммиарабика, тальк или т.п., с получением таблеток с пленочным покрытием или таблеток с сахарным покрытием. Одним из растворителей в этом методе нанесения покрытия может быть очищенная вода, но также можно использовать органический растворитель, такой как спирт, кетон, простой эфир или хлорированный углеводород, или их смесь. Например, в качестве органического растворителя можно использовать этанол, ацетон, метиленхлорид и т.п. Кроме того, в качестве устройства для нанесения покрытия можно использовать устройство, обычно используемое для нанесения покрытий при производстве лекарственных средств, например, включая устройство для нанесения покрытия распылением, в котором нанесение покрытия происходит путем распыления жидкости для покрытия или т.п., и роторный гранулятор с псевдоожиженным слоем для нанесения слоя покрытия.
В случае изготовления препаратов в виде капсул капсулы можно изготавливать путем заполнения твердых желатиновых капсул или капсул из ГПМЦ гранулами с замедленным высвобождением или гранулами с немедленным высвобождением, как указано выше, или мини-таблетками с помощью автоматической машины для заполнения капсул. В качестве альтернативы, в случае препаратов, вводимых из расчета на ампулу, или в случае сухого сиропа, который при приеме используется в смеси с водой или аналогичной жидкостью, гранулы с замедленным высвобождением или гранулы с немедленным высвобождением, как указано выше, могут быть смешаны с загустителем или диспергирующим агентом для диспергирования этих гранул, после чего смесь преобразуют в гранулы или таблетки. Кроме того, жидкость или желе можно приготовить с использованием воды и веществ, выбранных из диспергирующих агентов, эмульгаторов, загустителей, консервантов, регуляторов pH, подсластителей, ароматизаторов, отдушек и т.д. Однако, что касается других способов изготовления, их применение также не ограничено.
Для более полного понимания раскрытых в настоящем описании вариантов осуществления, ниже приведены примеры. Следует понимать, что эти примеры предназначены только для иллюстративных целей и не должны рассматриваться как ограничивающие изобретение.
ПРИМЕРЫ
В примерах могут использоваться следующие сокращения:
All: аллил
DMT: 4,4'-диметокситритил
(DMTO-:
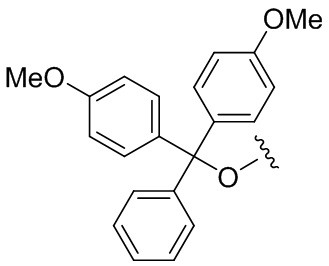 )
)
Bz: бензоил
Основание Хунига: i-Pr2NEt (диизопропилэтиламин)
АллилOH: аллиловый спирт
OAll: -OCH2CHCH2
ACN: ацетонитрил
All: -CH2CHCH2
2-нитроBnBr: 2-нитробензилбромид
Bz: бензоил
i-Pr: изопропил
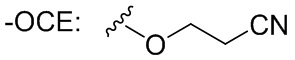
CE: цианоэтил
()
DEAD: диэтилазодикарбоксилат
DIAD: диизопропилазодикарбоксилат
DCM: дихлорметан
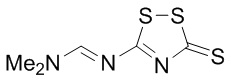
DDTT: N, N-диметил-N'-(3-тиоксо-3H-1,2,4-дитиазол-5-ил)формамид
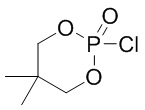
DMOCP: 2-хлор-5,5-диметил-1,3,2-диоксафосфинана 2-оксид
TBS: t-бутилдиметилсилил
3H-бензо[c][1,2]дитиол-3-он:

Пример 1 - Синтез Соединения 1a
Полная схема синтеза приведена на фиг. 1.
Этап A

К смеси (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ил(2-цианоэтил)диизопропилфосфорамидита (соединение 100) (смесь диастереомеров фосфора; 80,0 г, 91,332 ммоль, 1 экв., каталог ChemGenes Corporation, # ANP-9151), аллилового спирта (9,63 мл, 142 ммоль) и трифенилфосфина (38,3 г, 146 ммоль, 1,60 экв.) в THF (1,1 л) добавляли DEAD (40 мас.% раствор в толуоле; 54,2 мл, 137 ммоль, 1,5 экв.) при температуре окружающей среды (rt). Перемешивание продолжали при температуре окружающей среды, и реакцию контролировали с помощью ЖХ/МС. По завершении (19 ч) смесь концентрировали в вакууме (35°C), и полученную смесь очищали колоночной хроматографией на силикагеле (800 г x2 колонки, от 40 до 60% EtOAc в н-гептане, забуференном 0,5% триэтиламином) с получением Соединения 101 в виде белой пены (выход - 84,2 г в виде смеси диастереомеров фосфора).
1H ЯМР (3:2 смесь диастереомеров фосфора 400 МГц, CDCl3) δ 1,14-1,21 (м, 12H) 2,40 (т, J=6,2 Гц, 1,2H) 2,59 (т, J=6,2 Гц, 0,8H) 3,27 (д, J=8,6 Гц, 1H) 3,52-3,66 (м, 5H) 3,78 (с, 2,4H) 3,79 (с, 3,6H) 4,28-4,34 (м, 1H) 4,84-4,96 (м, 0,4 H) 4,99 (д, J=5,5 Гц, 2H) 4,95-5,10 (м, 0,6H) 5,05 (д, J=10,9 Гц, 1H) 5,22 (уш.д, J=17,6 Гц, 1H) 5,64 (уш.д, J=53,2 Гц, 0,6H) 5,70 (уш.д, J=51,6 Гц, 0,4H) 5,96-6,75 (м, 1H) 6,20 (д, J=16,0 Гц, 0,6H) 6,24 (д, J=17,2 Гц, 0,4H) 6,74-6,79 (м, 4H) 7,02-7,06 (м, 2H) 7,17-7,24 (м, 8H) 7,32-7,34 (м, 2H) 7,41-7,44 (м, 2H) 8,11 (с, 1H) 8,52 (с, 0,4H) 8,54 (с, 0,6H).
Этап В
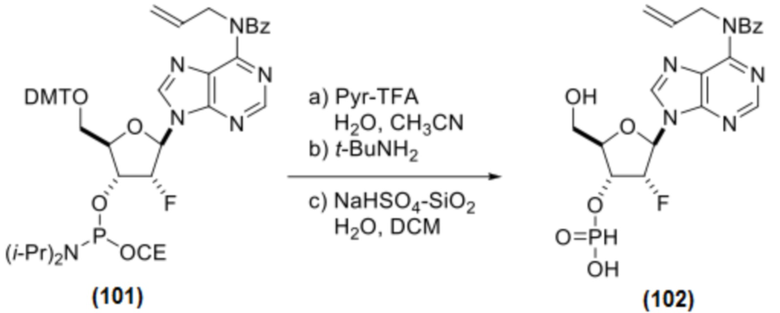
К раствору Соединения 101 (3,00 г, 3,28 ммоль, 1 экв.) в ацетонитриле (30 мл) добавляли воду (0,118 мл, 6,55 ммоль, 2,0 экв.) и соль трифторацетат пиридина (0,759 г, 3,93 ммоль, 1,2 экв.). После перемешивания при температуре окружающей среды в течение 1 минуты добавляли трет-бутиламин (14,5 г, 21,0 мл, 0,20 моль, 60 экв.). После полного отщепления цианоэтильной группы (контроль с помощью ЖХ/МС) реакционную смесь концентрировали в вакууме и дважды подвергали азеотропной перегонке с ацетонитрилом. Неочищенную смесь растворяли в DCM (45,0 мл) и обрабатывали водой (0,118 мл, 6,55 ммоль, 2,0 экв.) и NaHSO4-SiO2 (1,18 г, 6,55 ммоль, 2 экв.) при температуре окружающей среды. После полного отщепления DMT-группы (контроль с помощью ЖХ/МС, приблизительно 1 час) реакционную смесь фильтровали и дважды промывали смесью DCM/MeOH (9/1, 20 мл). Объединенные фильтраты концентрировали в вакууме и обрабатывали 1:1 смесью н-гептан/толуол (~30 мл). Верхний слой удаляли декантированием. Эту же операцию повторяли еще раз со смесью н-гептан/толуол (1/1, 30 мл), и нижний слой дважды подвергали азеотропной перегонке с ацетонитрилом с получением соединения 102 (предполагаемый теоретический выход 100%). Продукт использовали на следующем этапе без дополнительной очистки.
Этап С

К смеси Соединения 102 (1,56 г, 3,27 ммоль, 1 экв.) и Соединения 101 (3,00 г, 3,28 ммоль, 1 экв.) в ацетонитриле (30 мл) добавляли соль трифторацетат пиридина (азеотропно высушенную с пиридином; 0,760 г, 3,94%, ммоль, 1,25 экв.). Через 5 минут добавляли DDTT (0,840 г, 4,09 ммоль, 1,30 экв., каталог ChemGenes Corporation, № RN-1588), и после полной сульфуризации (контроль с помощью ЖХ/МС) реакционную смесь концентрировали в вакууме. Остаток растворяли в DCM (30 мл) и обрабатывали водой (0,57 мл, 32 ммоль, 10 экв.) и 6% дихлоруксусной кислотой (1,56 мл, 18,9 ммоль, 6,0 экв.) в DCM (30 мл). Через 20 минут реакцию останавливали добавлением пиридина (20 мл) и концентрировали в вакууме. Остаток подвергали азеотропной перегонке с пиридином, получая соединение 103 (3,22 г, предполагаемый теоретический выход 100%). Продукт использовали на следующем этапе без дополнительной очистки.
Этап D
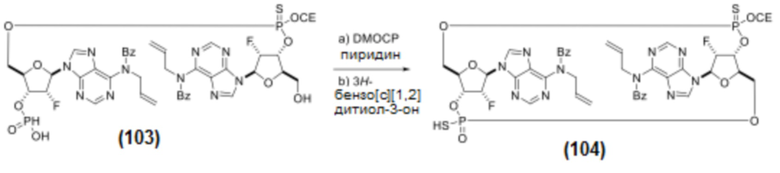
К раствору Соединения 103 (3,22 г, 3,15 ммоль, 1 экв.) в пиридине (100 мл) добавляли DMOCP (1,45 г, 7,88 ммоль, 2,50 экв.) при температуре окружающей среды. После полной макроциклизации (контроль с помощью ЖХ/МС) добавляли воду (1,7 мл, 94,5 ммоль, х10 раз относительно DMOCP), а затем 3H-бензо[c][1,2]дитиол-3-он (0,795 г, 4,73 ммоль, 1,5 экв.). После полной сульфуризации (примерно 40 минут) реакционную смесь частично концентрировали в вакууме примерно до 15 мл и выливали в смесь насыщенного водного NaHCO3 (50 мл) и воды (30 мл). После 10 мин перемешивания при температуре окружающей среды смесь экстрагировали смесью EtOAc/MTBE (1:1) (60 мл х3 раза). Органические слои объединяли, промывали рассолом (25 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (0-20% MeOH в DCM) с получением Соединения 104 (3,31 г, 3,20 ммоль, предполагаемый теоретический выход 100%) в виде коричневого масла. Продукт использовали на следующем этапе без дополнительной очистки.
Этап E
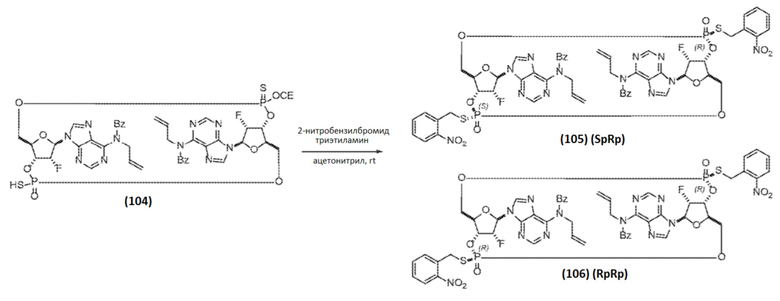
К раствору Соединения 104 (3,31 г, 3,20 ммоль, 1 экв.) в ацетонитриле (66,2 мл) добавляли 2-нитробензилбромид (2,42 г, 11,2 ммоль, 3,50 экв.) и триэтиламин (1,78 мл, 12,8 ммоль, 4,00 экв.). После завершения реакции (контроль с помощью ЖХ/МС, приблизительно 20 часов при температуре окружающей среды) реакционную смесь концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (60% этилацетат/н-гептан до 100% этилацетата) с получением 0,568 г продукта в виде смеси диастереомеров фосфора. Разделение диастереомеров препаративной ВЭЖХ дало Соединение 105 (SR-изомер; 0,225 г, 0,180 ммоль, общий выход 5,6% от Соединения 101) и Соединение 106 (RR-изомер; 0,187 г, 0,149 ммоль, общий выход 4,7% от Соединения 1).
Соединение 105 (SpRp) 1H ЯМР (400 МГц, CDCl3) δ=8,63 (с, 1H), δ=8,61 (с, 1H), 8,04-8,00 (м, 2H), 7,99 (с, 1H), 7,90 (с, 1H), 7,65-7,44 (м, 8H), 7,40-7,31 (м, 4H), 7,25-7,21 (м, 4H), 6,15-5,89 (м, 5H), 5,61 (дд, J=52,0, 5,1 Гц, 1H), 5,55 (ддд, J=51,2, 4,7, 2,7 Гц, 1H) 5,51-5,42 (м, 1H), 5,31-5,22 (м, 2H), 5,11 (дд, J=3,9, 9,8 Гц, 2H), 5,04-4,95 (м, 4H), 4,55-4,37 (м, 7H), 4,29-4,12 (м, 3H)
Соединение 106 (RpRp) 1H ЯМР (400 МГц, CDCl3) δ=8,65 (с, 2H), 8,06 (дд, J=1,4, 8,0 Гц, 2H), 7,98 (с, 2H), 7,57-7,52 (м, 6H), 7,47-7,32 (м, 6H), 7,25-7,21 (м, 4H), 6,15 (д, J=18,7 Гц, 2H), 6,09-5,99 (м, 2H), 5,82-5,76 (м, 2H), 5,60 (дд, J=51,8, 4,9 Гц, 2H), 5,27 (дд, J=1,2, 17,2 Гц, 2H), 5,12 (дд, J=1,0, 10,4 Гц, 2H), 5,06-4,96 (м, 4H), 4,55-4,40 (м, 4H), 4,36-4,24 (м, 4H), 4,21-4,02 (м, 2H)
Условия проведения препаративной ВЭЖХ:
Этап F

К нагретому (90°C) раствору Соединения 105 (519 мг, 0,414 ммоль, 1 экв.) в толуоле (519 мл) добавляли катализатор Ховейда-Граббса™ 2-го поколения ((1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенилметилен)рутений; производства SIGMA-ALDRITCH®, Кат. № 569755; CAS 301224-40-8; 91 мг, 0,15 ммоль, 0,35 экв.) и хинон (0,102 мл, 1,243 ммоль, 3,0 экв.). Смесь кипятили с обратным холодильником, и за ходом реакции следили с помощью ЖХ/МС. Через 3 часа дополнительно добавляли катализатор (91 мг, 0,15 ммоль, 0,35 экв.), и реакцию продолжали еще 3 часа. После охлаждения смесь обрабатывали DMSO (0,59 мл, 8,3 ммоль, 20 экв.) при температуре окружающей среды в течение 15 часов, концентрировали в вакууме и очищали колоночной хроматографией на силикагеле (SiO2 25 г, 66% этилацетат в н-гептане до 100% этилацетата) с получением Соединения 107 (200 мг, 0,163 ммоль, выход 39%) в виде коричневой сухой пены.
1H ЯМР (400 МГц, CDCl3) δ=8,19 (с, 1H), 8,12 (дд, J=7,8 Гц, 1,9 Гц, 1H), 8,10 (с, 1H), 8,02 (д, J=8,2 Гц, 1H), 7,89 (с, 1H), 7,63 (уш.д, J=7,0 Гц, 1H), 7,53-7,41 (м, 10H), 7,35-7,30 (м, 2H), 7,25-7,20 (м, 4H), 6,23 (д, J=17,6 Гц, 1H), 6,14 (д, J=18.8 Гц, 1H), 5,86-5,75 (м, 1H), 5,75 (дт, J=15,3, 5,0 Гц, 1H), 5,67 (дт, J=15,3, 4,7 Гц, 1H), 5,60 (дд, J=52,0, 3,9 Гц, 1H), 5,48 (дд, J=50,4, 3,9 Гц, 1H), 5,50-5,39 (м, 1H), 4,91-4,64 (м, 4H), 4,57-4,25 (м, 9H), 4,15 (д, J=7,03 Гц, 1H), 4,11 (д, J=7,03 Гц, 1H)
Этап G
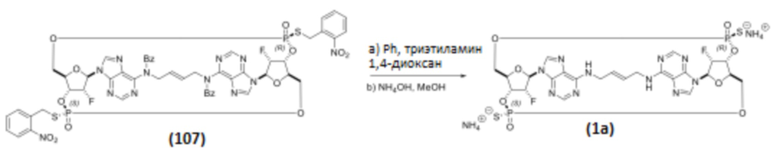
К раствору Соединения 107 (88 мг, 0,072 ммоль, 1 экв.) в 1,4-диоксане (1,76 мл) добавляли тиофенол (0,88 мл, 8,55 ммоль, 119 экв.) и триэтиламин (0,88 мл, 6,31 ммоль, 88 экв.). Полученную смесь перемешивали при температуре окружающей среды. После завершения реакции (контроль с помощью ЖХ/МС, 13 часов) добавляли метанол (5,28 мл) и 28% гидроксид аммония (3,52 мл), и полученную смесь нагревали до 50°C. После завершения реакции (контроль с помощью ЖХ/МС, 5 часов) смесь охлаждали до температуры окружающей среды, и полученную коричневатую суспензию фильтровали и промывали водой (15 мл). Фильтрат снова фильтровали для удаления дополнительных твердых частиц. Конечный фильтрат дважды экстрагировали 1:1 смесью толуола с гептаном (30 мл). Водный слой концентрировали в вакууме и затем ресуспендировали в воде (6 мл). Полученное твердое вещество фильтровали, и фильтрат подвергали препаративной ВЭЖХ с получением диаммониевой соли Соединения 1 (также называемого Соединением 1a) (39 мг, 0,050 ммоль, выход 70%) в виде белого твердого вещества.
Соединение 1a (SpRp, транс) 1H ЯМР (400 МГц, CD3OD) δ=9,05 (с, 1H), 8,33 (с, 1H), 8,25 (с, 1H), 8,12 (с, 1H), 6,34 (уш.с, 2H), 5,88 (уш.с, 2H), 5,66 (уш.д, J=51,6 Гц, 1H), 5,59 (уш.д, J=52,2 Гц, 1H) 5,01 (уш.с, 2H), 4,68-4,34 (м, 6H), 4,07-3,82 (м, 2H), 3,79-3,55 (м, 2H); 31P ЯМР (162 МГц, CD3OD) δ=55,48 (с, 1P), 55,16 (с, 1P).
Соединение 1a. Условия проведения препаративной ВЭЖХ:
Пример 1.1 - Альтернативный синтез Соединения 1a
Альтернативный путь синтеза Соединения 1a представлен на фиг.2А и фиг.2B, а также на фиг.2C и описан ниже.
Этап 1
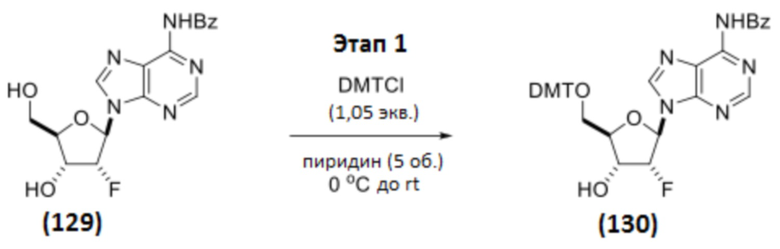
Соединение 129 (570 г, 1,53 моль, 1 мас.%, 1 объем, 1 экв.) растворяли в пиридине (2,85 л, 35,2 моль, 4,89 мас.%, 5,0 объемов, 23 экв.). Смесь охлаждали до 2,6°C и обрабатывали 4,4’-диметокситритилхлоридом (DMTCl; 543 г, 1,60 моль, 0,953 мас.%, 1,05 экв.). Смесь перемешивали при 0-5°C в течение 2 ч, а затем давали нагреться до температуры окружающей среды. Реакцию контролировали с помощью ЖХ/МС, и полное превращение подтверждали после перемешивания в течение ночи. Реакционную смесь охлаждали до температуры ниже 5°C и гасили обработкой MeOH (124 мл, 3,05 моль, 0,172 мас.%, 0,217 об., 2,0 экв.) в течение 15 минут. Смесь упаривали совместно с толуолом (2,00 л, 3,04 мас.%, 3,51 об.) в вакууме, а затем разбавляли смесью EtOAc (2,850 л, 4,5 мас.%, 5,0 об.) и н-гептана (2,85 л, 3,42 мас.%, 5,0 об.). Органический слой промывали насыщенным NaHCO3 (9 мас.% раствора в воде; 2,0 л, 3,5 об.). Дополнительный EtOAc (2,85 л, 4,5 мас.%, 5,0 об.) добавляли для полного растворения сырого продукта. После перемешивания в течение 5 минут два слоя разделяли. Органический слой промывали водой (2,0 л, 3,5 мас.%, 3,5 об.). Твердое вещество начинало медленно осаждаться из органического слоя. Водный слой отделяли. Затем органический слой концентрировали до приблизительно 1 объема. Неочищенный продукт суспендировали в смеси н-гептана (2,00 л, 2,40 мас.%, 3,51 об.) и толуола (0,50 л, 0,76 мас.%, 0,88 об.). После перемешивания в течение 15 минут бледно-желтое твердое вещество собирали вакуумной фильтрацией. Осадок на фильтре последовательно промывали: (1) смесью н-гептана (0,60 л, 0,72 мас.%, 1,05 об.) и толуола (0,30 л, 0,46 мас.%, 0,53 об.), и затем (2) н-гептаном (3,00 л, 3,6 мас.%, 5,26 об.). Твердое вещество сушили без нагревания в течение 30 минут и затем переносили на поддоны для сушки при 50°C в вакуумной печи в течение ночи с получением Соединения 130 в виде бледно-желтого твердого вещества (996,7 г, 1,47 моль, 1,75 мас.%, выход 97%).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,99 (с, 1H), 8,76 (с, 1H), 8,21 (с, 1H), 8,04-8,00 (м, 2H), 7,64-7,59 (м, 1H), 7,57-7,50 (м, 2H), 7,41-7,36 (м, 2H), 7,32-7,15 (м, 7H), 6,83-6,76 (м, 4H), 6,31 (дд, J=2,5, 17,0 Гц, 1H), 5,68 (ддд, J=2,3, 4,7, 52,7 Гц, 1H), 4,88-4,77 (м, 1H), 4,26-4,21 (м, 1H), 3,77 (с, 6H), 3,57 (дд, J=3,1, 10,9 Гц, 1H), 3,43 (дд, J=4,1, 10,7 Гц, 1H), 2,60 (уш.с, 1H)
Этап 1’
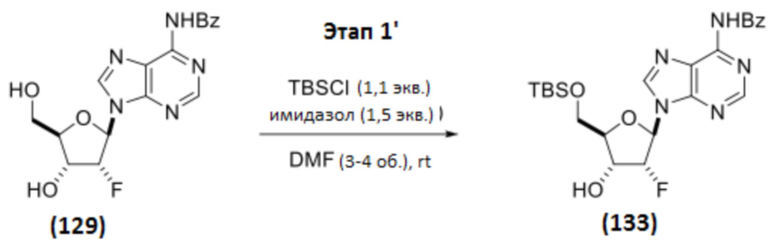
Соединение 129 (430 г, 1,15 моль, 1 мас.%, 1 об., 1 экв.) и имидазол (118 г, 1,73 моль, 0,274 мас.%, 1,50 экв.) растворяли в DMF (1,72 л, 3,78 мас.%, 4,0 об.), и полученную смесь охлаждали до 5°C. Добавляли TBS-Cl (191 г, 1,27 моль, 0,444 мас.%, 1,10 экв.). Смесь перемешивали при 0-11°C в течение 2 часов, оставляли медленно нагреваться до температуры окружающей среды (за ходом следили с помощью ЖХ/МС). Реакцию завершали через 6 часов после добавления TBS-Cl, оставляли перемешиваться при температуре окружающей среды в течение дополнительных 20 часов. Смесь охлаждали до 2°C и обрабатывали метанолом (93 мл, 74 г, 2,3 моль, 0,17 мас.%, 0,22 мас.%, 2,0 экв.) в течение 10 минут. Реакционную смесь разбавляли смесью MTBE (1,72 л, 1,23 кг, 2,96 мас.%, 4,0 об.) и EtOAc (1,72 л, 1,55 кг, 3,60 мас.%, 4,0 об.), и затем насыщенным NH4Cl (28 мас.% раствор в воде; 2,15 л, 5,0 об.). Твердое вещество начинало медленно выпадать из раствора. Смесь оставляли нагреваться до 24°C, и к ней добавляли воду (1,08 л, 1,08 кг, 2,5 мас.%, 2,5 об.) (внутренняя температура=22°C). Из смеси начинали выпадать новые твердые частицы. К смеси дополнительно добавляли воду (1,08 л, 1,08 кг, 2,5 мас.%, 2,5 об.) и MTBE (1,40 л, 1,04 кг, 2,4 мас.%, 3,3 об.). Грязно-белое твердое вещество собирали вакуумной фильтрацией. Реактор промывали водой (320 мл, 0,74 об.), а затем MTBE (1,80 л, 1,33 кг, 3,10 мас.%, 4,19 об.) для переноса любого оставшегося твердого вещества на фильтр. Осадок на фильтре последовательно промывали: (1) водой (1,80 л, 1,80 кг, 4,2 мас.%, 4,2 об.), (2) водой (1,80 л, 1,80 кг, 4,2 мас.%, 4,2 об.), (3) смесью MTBE (0,90 л, 0,67 кг, 1,5 мас.%, 2,1 об.) и н-гептана (0,90 л, 0,62 кг, 1,4 мас.%, 2,1 об.), (4) смесью MTBE (0,90 л, 0,67 кг, 1,5 мас.%, 2,1 об.) и н-гептана (0,90 л, 0,62 кг, 1,4 мас.%, 2,1 об.). Извлеченное твердое вещество сушили в вакууме при 40°C в течение 2 дней с получением Соединения 133 в виде белого твердого вещества (483 г, 0,991 моль, 1,12 мас.%, выход 86%).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,97 (с, 1H), 8,82 (с, 1H), 8,36 (с, 1H), 8,04-8,00 (м, 2H), 7,64-7,58 (м, 1H), 7,56-7,51 (м, 2H), 6,40 (дд, J=2,3, 16,0 Гц, 1H), 5,45 (ддд, J=2,7, 4,3, 53,1 Гц, 1H), 4,75-4,66 (м, 1H), 4,22-4,17 (м, 1H), 4,07 (дд, J=2,3, 11,7 Гц, 1H), 3,91 (дд, J=2,7, 11,7 Гц, 1H), 2,38 (дд, J=2,7, 7,0 Гц, 1H), 0,92 (с, 9H), 0,11 (с, 3H), 0,11 (с, 3H).
Этап 2
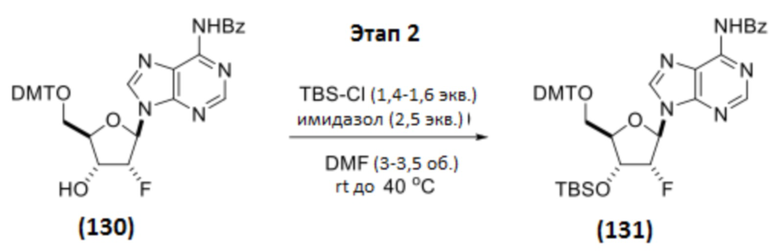
Соединение 130 (993 г, 1,47 моль, 1 мас.%, 1 об., 1 экв.) и имидазол (150 г, 2,20 моль, 0,151 мас.%, 1,5 экв.) растворяли в DMF (3,48 л, 3,28 кг, 3,3 мас.%, 3,5 об.), и смесь охлаждали до 5°C. Добавляли TBS-Cl (244 г, 1,62 моль, 0,245 мас.%, 1,10 экв.). Реакционную смесь перемешивали при 0-5°C в течение 2 часов, оставляли медленно нагреваться до температуры окружающей среды и контролировали с помощью ЖХ/МС. Через 17 часов дополнительно добавляли имидазол (100 г, 1,47 моль, 0,10 мас.%, 1,0 экв.) и TBS-Cl (111 г, 735 ммоль, 0,112 мас.%, 0,50 экв.) и перемешивание продолжали при температуре окружающей среды в течение 2 часов и при 35°C в течение 2 часов. Полученную смесь охлаждали до 13,6°C и обрабатывали МеОН (119 мл, 2,94 моль, 2 экв.) в течение 10 минут. В отдельный реактор добавляли лед (5 кг, 5 мас.%) и насыщенный NH4Cl (28 мас.% раствор в воде; 5,0 л, 5 об.). Реакционную смесь добавляли к смеси лед/NH4Cl. Из раствора сразу начало выпадать грязно-белое твердое вещество. К смеси дополнительно добавляли 2 кг льда (2 кг, 2 мас.%) и воду (3,0 л, 3 об.). Реакционную колбу ополаскивали водой (0,50 л, 0,5 об.), и к смеси добавляли ополаскиватель. К смеси добавляли н-гептан (2,00 л, 2 об.), и перемешивание продолжали в течение 10 минут. Грязно-белое твердое вещество собирали вакуумной фильтрацией. Осадок на фильтре промывали: (1) водой (4,0 л, 4,0 об.), (2) водой (4,0 л, 4,0 об.), (3) н-гептаном (4,0 л, 4,0 об.), (4) н-гептаном (4,0 л, 4,0 об.). Извлеченное твердое вещество сушили в вакууме при 45°C в течение 4 дней с получением Соединения 131 в виде грязно-белого твердого вещества (1,095 кг, 1,39 моль, 1,10 мас.%, выход 94%).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=9,09 (с, 1H), 8,78 (с, 1H), 8,28 (с, 1H), 8,02 (д, J=7,4 Гц, 2H), 7,63-7,59 (м, 1H), 7,55-7,50 (м, 2H), 7,37 (д, J=7,1 Гц, 2H), 7,29-7,17 (м, 7H), 6,79 (д, J=7,9 Гц, 4H), 6,29 (дд, J=2,9, 16,2 Гц, 1H), 5,60 (ддд, J=2,7, 3,9, 53,1 Гц, 1H), 4,78 (ддд, J=4,7, 6,4, 15,8 Гц, 1H), 4,26-4,22 (м, 1H), 3,77 (с, 6H), 3,58 (дд, J=3,1, 10,9 Гц, 1H), 3,26 (дд, J=3,7, 10,7 Гц, 1H), 0,85 (с, 9H), 0,10 (с, 3H), 0,02 (с, 3H)
Этап 3

Соединение 131 (1000 г, 1,27 моль, 1 мас.%, 1 об., 1 экв.) и транс-2-бутен-1,4-диол (геометрию олефина подтверждали 1H-ЯМР; 335 г, 3,80 моль, 0,335 мас.%, 3,0 экв.) дважды подвергали азеотропной перегонке с THF (3,0 л, 3,0 об.). Остаток растворяли в смеси THF (10 л, 10 об.) и толуола (15 л, 15 об.). Добавляли трифенилфосфин (432 г, 1,65 моль, 0,432 мас.%, 1,3 экв.) и затем реакционную смесь охлаждали до -5°C. Медленно в течение 20 минут добавляли DIAD (0,320 л, 1,65 моль, 333 г, 0,333 мас.%, 0,320 об., 1,3 экв.), поддерживая Т-внутреннюю ниже 5°C. Реакцию перемешивали при 0-5°C в течение 1 ч и контролировали с помощью ЖХ/МС. Ледяную баню убирали, и смеси давали нагреться до комнатной температуры. После перемешивания в течение ночи (17 ч) добавляли трифенилфосфин (83 г, 0,32 моль, 0,083 мас.%, 0,25 экв.) и DIAD (62 мл, 0,32 моль, 64 г, 0,064 мас.%, 0,062 об., 0,25 экв.). Еще через 1 час при комнатной температуре реакционную смесь разбавляли MTBE (10 л, 10 об.), дважды промывали полунасыщенным NaCl (18 мас.% раствор в воде; 2×4 л) и концентрировали в вакууме до густого масла. Смесь повторно растворяли в смеси МТВЕ (4,00 л, 4 об.) и н-гептана (0,50 л, 0,5 об.), и затем охлаждали до 0°C. К раствору добавляли затравку оксида трифенилфосфина. Из раствора начинали медленно выпадать твердые частицы, и раствор перемешивали в течение ночи. Белое твердое вещество собирали вакуумной фильтрацией и промывали MTBE (2 л, 2 об.) с выделением 540 г оксида трифенилфосфина. Фильтрат концентрировали и очищали через Biotage 150L KP-Sil (SiO2 5 кг; предварительно пропитанный 1% TEA в Hep/etOAc; элюенты: гептан/EtOAc (48 л 33% EtOAc с 1% TEA, 24 л 50% EtOAc с 1% TEA, 24 л 66% EtOAc с 1% TEA) → 100% EtOAc с 1% TEA). За колонкой следили с помощью ТСХ (2:1 EtOAc/н-гептан). Фракции чистого продукта объединяли и концентрировали под вакуумом с получением соединения 132 в виде бледно-белой твердой пены (634 г, содержавшие 14 мас.% побочного продукта, полученного из DIAD, нетто 545 г, 0,63 моль, скорректированный выход 50%). Фракции смеси объединяли и концентрировали под вакуумом с получением твердой бледно-желтой пены (750 г), которую подвергали повторной очистке через Biotage 150M HP-Sphere (2,5 кг SiO2; предварительно пропитаный 1% TEA в Hep/EtOAc; загруженный образец с элюентами толуола: Hep/EtOAc/1% TEA (12 л 50% EtOAc с 1% TEA, 16 л 66% EtOAc с 1% TEA) → EtOAc с 1% TEA). За колонкой следили с помощью ТСХ (2/1/0,03 EtOAc/н-гептан/TEA). Фракции чистого продукта объединяли и концентрировали под вакуумом с получением дополнительного соединения 132 в виде бледно-белой твердой пены (206 г, 0,24 моль, выход 18%).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,58 (с, 1H), 8,10 (с, 1H), 7,43-7,37 (м, 2H), 7,32-7,28 (м, 2H), 7,24-7,15 (м, 8H), 7,03-6,98 (м, 2H), 6,78-6,73 (м, 4H), 6,18 (дд, J=2,7, 17,2 Гц, 1H), 5,88 (тд, J=5,5, 15,6 Гц, 1H), 5,77 (тд, J=5,1, 15,6 Гц, 1H), 5,60 (ддд, J=2,7, 4,3, 53,1 Гц, 1H), 5,03-4,96 (м, 2H), 4,91 (ддд, J=4,5, 6,6, 16,6 Гц, 1H), 4,18-4,14 (м, 1H), 3,88-3,82 (м, 2H), 3,78 (с, 6H), 3,52 (дд, J=2,7, 10,9 Гц, 1H), 3,14 (дд, J=3,5, 10,9 Гц, 1H), 0,85 (с, 9H), 0,10 (с, 3H), 0,01 (с, 3H).
Этап 4

Соединение 132 (800 г, 0,930 моль, 1 мас.%, 1 об., 1 экв.) и соединение 133 (522 г, 1,07 моль, 0,652 мас.%, 1,15 экв.) азеотропно сушили с ТГФ (2×3 л, 2×3,8 объема) и повторно растворяли в THF (9,60 л, 8,45 кг, 12,0 об.) при комнатной температуре. Добавляли трифенилфосфин (317 г, 1,21 моль, 0,396 мас.%, 1,30 экв.), и смесь охлаждали ниже -5°C. DIAD (226 мл, 1,16 моль, 235 г, 0,294 мас.%, 0,283 об., 1,25 экв.) добавляли при Т-внутренний при температуре ниже 7°C. Реакционной смеси давали медленно нагреться до комнатной температуры. Реакцию контролировали с помощью ЖХ/МС. Через 21 час реакционную смесь концентрировали в вакууме до густого масла, подвергали азеотропной перегонке с н-гептаном (2,00, 1,37 кг, 1,71 мас.%, 2,50 об.) и затем повторно растворяли в смеси MTBE (2,40 л, 1,78 кг, 2,2 мас.%, 3,0 об.) и н-гептана (800 мл, 547 г, 0,68 мас.%, 1,0 об.). В раствор добавляли затравку оксида трифенилфосфина и охлаждали до 5°C, разбавляли н-гептаном (400 мл, 274 г, 0,34 мас.%, 0,50 об.), и перемешивали при 5°C в течение 30 минут. Белый твердый осадок собирали вакуумной фильтрацией и промывали 2:1 (об./об.) смесью МТВЕ и н-гептана (1,8 л) с получением оксида трифенилфозохина (455 г). Фильтрат концентрировали под вакуумом и очищали через Biotage 150L KP-Sil (SiO2 5 кг; предварительно пропитанный 1% TEA; образец загружали путем растворения в элюентах толуола: 9:1 гептан/EtOAc (16 л) и 15 TEA, 3,6:1 (46 л), 2:1 (20 л) и 1% TEA, 1:1 (30 л) и 1% TEA, и 100% EtOAc (16 л) и 1% TEA). Объединенные фракции чистого продукта концентрировали под вакуумом с получением Соединения 134 в виде твердой пены грязно-белого цвета (662,2 г). Фракции смеси объединяли и концентрировали под вакуумом (480 г). Белое нерастворимое твердое вещество, образовавшееся при разбавлении толуолом (300 мл) перед загрузкой в Biotage 150L, удаляли вакуумной фильтрацией. Растворимый в толуоле материал очищали с помощью Biotage 150M HP-Sphere (SiO2 2,5 кг (предварительно обработанный 1% TEA); загрузка образца с толуолом; элюенты: 2:1 гептан/EtOAc (26 л) с 1% TEA, 1:1 (25 л) с 1% TEA, 1:4 (34 л) с 1% TEA). Колонку контролировали с помощью ТСХ (гептан/EtOAc, 1:1). Объединенные фракции чистого продукта концентрировали под вакуумом с получением дополнительного соединения 134 в виде твердой пены грязно-белого цвета (165,5 г. Общий вес: 662,2+165,5 г=827,7 г, 930 ммоль, 1,03 мас.%, выход 67%).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.47 (с, 1H), 8,39 (с, 1H), 8,20 (с, 1H), 8,01 (с, 1H), 7,38-7,31 (м, 5H), 7,27-7,19 (м, 6H), 7,14-7,06 (м, 3H), 6,93-6,87 (м, 2H), 6,76 (д, J=8,6 Гц, 4H), 6,26 (дд, J=2,0, 16,0 Гц, 1H), 6,15 (дд, J=2,7, 17,2 Гц, 1H), 5,86 (дд, J=4,7, 15,2 Гц, 1H), 5,80 (дд, J=4,7, 15,2 Гц, 1H), 5,51 (ддд, J=2,7, 4,3, 52,8 Гц, 1H), 5,31 (ддд, J=2,0, 4,3, 52,8 Гц, 1H), 4,87 (д, J=4,7 Гц, 2H), 4,85-4,81 (м, 1H), 4,79 (д, J=4,3 Гц, 2H), 4,71-4,59 (м, 1H), 4,20-4,13 (м, 2H), 4,06 (дд, J=2,7, 11,3 Гц, 1H), 3,90 (дд, J=2,7, 11,7 Гц, 1H), 3,77 (с, 6H), 3,52 (дд, J=3,1, 10,9 Гц, 1H), 3,18 (дд, J=3,9, 10,9 Гц, 1H), 0,92 (с, 9H), 0,84 (с, 9H), 0,10 (с, 3H), 0,09 (с, 6H), 0,07 (с, 3H)
Этап 5-6
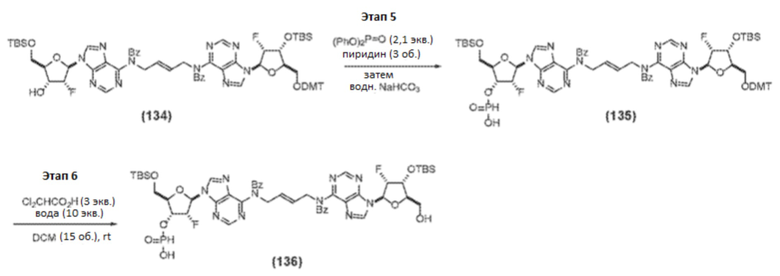
К раствору Соединения 134 (410,7 г, 309 ммоль, 1 мас.%, 1 об., 1 экв.) в пиридине (1,23 л, 1,21 кг, 15,2 моль, 2,9 мас.%, 3,0 об., 49 экв.) добавляли дифенилфосфит (90 мл, 109 г, 0,46 моль, 0,26 мас.%, 0,22 об., 1,5 экв.). Реакцию перемешивали при комнатной температуре и контролировали с помощью ЖХ/МС. Через 2 часа (конверсия 80%) добавляли дополнительный дифенилфосфит (29,9 мл, 36,2 г, 155 ммоль, 0,088 мас.%, 0,073 об., 0,50 экв.). Еще через 1 час добавляли дополнительный дифенилфосфит (6,0 мл, 7,2 г, 31 ммоль, 0,018 мас.%, 0,015 об., 0,10 экв.), и реакцию продолжали еще 0,5 часа (конверсия 98%). Реакционную смесь добавляли к смеси насыщенного NaHCO3 (9 мас.% раствор в воде; 2,1 л, 5 об.) и воды (1,0 л, мл, 2,5 об.), поддерживая Т-внутреннюю от 4,7 до 12°С. Реактор промывали небольшим объемом EtOAc. Перемешивание продолжали при комнатной температуре в течение 30 минут и контролировали реакцию с помощью ЖХ/МС (конверсия 100%). Реакционную смесь дважды экстрагировали 1:1 смесью EtOAc и MTBE (2×8,2 л, 2×20 об.). Объединенные органические слои промывали водой (4,1 л, 10 об.), концентрировали в вакууме и подвергали азетропной перегонке с толуолом (3×4,1 л, 3×10 об.; непрерывная подача) для удаления пиридина с получением Соединения 135 (осталось 0,55 экв. пиридина).
Этап 6. Неочищенное соединение 135 растворяли в дихлорметане (3,08 л, 4,07 кг, 9,9 мас.%, 7,5 об.) при температуре окружающей среды. Добавляли воду (55,7 мл, 0,136 об., 10 экв.) и затем раствор дихлоруксусной кислоты (77 мл, 120 г, 0,93 моль, 0,29 мас.%, 0,19 об., 3,0 экв.) в DCM (3,08 л, 7,5 об.) при поддержании внутренней T ниже 25°C. (Превращение в оранжевый раствор). Через 30 мин добавляли триэтилсилан (Et3SiH; 494 мл, 359 г, 3,09 моль, 0,875 мас.%, 1,20 об., 10,0 экв.) (Т-внутренняя изменилась с 18,2°C до 17°C) и перемешивание продолжали в течение 20 минут. Добавляли триэтиламин (431 мл, 313 г, 3,09 моль, 0,762 мас.%, 1,05 об., 10,0 экв.) (Т-внутренняя изменилась с 17,8°C до 22°C). Смесь концентрировали до 1,55 кг (3,8 мас.%), повторно растворяли в EtOAc (6,2 л, 5,5 кг, 14 мас.%, 15 об.), последовательно промывали: (1) водой (1,0 л, 2,5 об.) и насыщенным NaHCO3 (9 мас.% раствор в воде, 0,82 л, 2,0 об.). Раствор неочищенного продукта EtOAc хранили при -20°C в течение ночи; 0,82 л, 2,0 об.), и на следующий день раствор концентрировали в вакууме при 25°C. Полученную таким образом неочищенную смесь (654 г) растирали с: (1) н-гептаном (3,01 л, 7,5 об.), (2) смесью н-гептана (2,46 л, 6,0 об.) и толуола (0,82 л, 2,0 об.). Часть раствора (супернатант) сливали, а твердое вещество, оставшееся на дне, растворяли в ацетонитриле (4,1 л, 10 об.). Смесь концентрировали в вакууме при 25°C и дважды подвергали азеотропной перегонке с ацетонитрилом, получая соединение 136. Продукт использовали на следующем этапе без очистки (предполагаемый теоретический выход 100%).
Этап 7
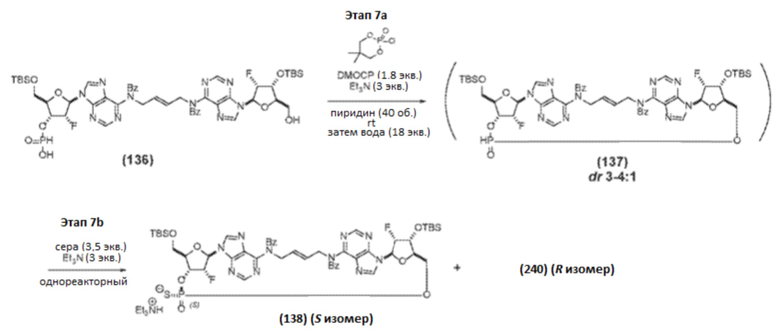
Этап 7а. Соединение 136 (337 г, 309 ммоль, 1 мас.%, 1 об., 1 экв.) растворяли в безводном пиридине (13,5 л, 13,2 кг, 39 мас.%, 40 об.) при комнатной температуре. Добавляли триэтиламин (129 мл, 927 ммоль, 94 г, 0,28 мас.%, 0,38 об., 3,0 экв.) и затем 2-хлор-5,5-диметил-1,3,2-диоксафосфинан 2-оксид (DMOCP; 103 г, 556 ммоль, 0,31 мас.%, 1,80 экв.). Полученную смесь перемешивали при температуре окружающей среды в течение 30 минут и контролировали с помощью ЖХ/МС (100% конверсия) с получением Соединения 137.
Этап 7b. К указанной выше смеси Соединения 137 добавляли ТЕА (129 мл, 927 ммоль, 94 г, 0,28 мас.%, 0,38 об., 3,0 экв.), воду (100 мл, 5,56 моль, 0,30 мас.%, 0,30 мас.%, 18 экв.) и серу (34,7 г, 1,08 моль, 0,10 мас.%, 3,5 экв.). Через 90 минут (100% конверсия) добавляли NaHCO3 (9 мас.% раствор в воде; 3,37 л, 10 об.), поддерживая Т-внутреннюю ниже 30°C (от 16,6°C до 27°C). Полученную смесь фильтровали для удаления солей. Фильтрат концентрировали под вакуумом, разбавляли МТВЕ (5,1 л, 15 об.) и дважды промывали NaCl (30 мас.% раствор в воде; 2×1,35 л, 2×4 об.). Нерастворимые твердые вещества отфильтровывали, фильтрат концентрировали в вакууме и подвергали азеотропной перегонке с толуолом (4,0 л, 12 об.). Полученное твердое вещество удаляли фильтрацией, а неочищенную смесь растворяли в толуоле и очищали через Biotage 150L KP-Sil (SiO2 5 кг; предварительно обработанный Hep/EtOAc/TEA (1,5/1,5/0,03 CV); элюировали: EtOAc/TEA (3/0,03 CV), EtOAc/MeOH/TEA (4/0,2/0,04 CV), EtOAC/MeOH/TEA (2/0,2/0,02 CV) колонку контролировали с помощью ТСХ (EtOAC/MeOH/TEA=9/1/0,1). Фракции, содержащие изомер Sp, объединяли и концентрировали в вакууме с получением Соединения 138 в виде твердой пены светло-розового цвета (Sp-изомер; 154 г, 128 ммоль, 0,46 мас.%, выход 41,3%). Фракции, содержащие изомер Rp, объединяли и концентрировали в вакууме с получением соединения 240 в виде твердой пены светло-розового цвета (Rp-изомер; 64 г, 53 ммоль, 0,19 мас.%, выход 17%).
Соединение 138 (Sp-изомер):
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,51 (с, 1H), 8,50 (с, 1H), 8,22 (с, 1H), 8,14 (с, 1H), 7,49-7,44 (м, 2H), 7,38-7,27 (м, 4H), 7,25-7,21 (м, 2H), 7,14 (т, J=7,1 Гц, 2H), 6,44 (дд, J=2,5, 13,9 Гц, 1H), 6,18 (д, J=15,2 Гц, 1H), 5,78 (тд, J=6,3, 15,6 Гц, 1H), 5,69 (тд, J=4,7, 15,6 Гц, 1H), 5,56 (дд, J=3,9, 50,8 Гц, 1H), 5,20-5,06 (м, 1H), 4,95-4,79 (м, 4H), 4,69 (дд, J=4,3, 16,0 Гц, 1H), 4,54-4,38 (м, 3H), 4,35 (д, J=5,5 Гц, 1H), 4,32-4,29 (м, 1H), 4,05 (дд, J=1,6, 11,7 Гц, 1H), 3,91 (дд, J=3,1, 11,7 Гц, 1H), 3,14-3,06 (м, 6H), 1,30 (т, J=7,4 Гц, 9H), 0,91 (с, 9H), 0,90 (с, 9H), 0,12 (с, 3H), 0,08 (с, 3H), 0,06 (с, 3H), 0,05 (с, 3H)
Соединение 240 (Rp-изомер):
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,54 (с, 1H), 8,38 (с, 1H), 8,33 (с, 1H), 8,01 (с, 1H), 7,39-7,09 (м, 10H), 6,39 (дд, J=2,3, 14,1 Гц, 1H), 6,13 (д, J=17,2 Гц, 1H), 5,72 (д, J=3,1 Гц, 2H), 5,68 (дд, J=4,3, 51,2 Гц, 1H), 5,43-5,29 (м, 1H), 5,10-4,96 (м, 3H), 4,90-4,83 (м, 2H), 4,78-4,72 (м, 1H), 4,52 (ддд, J=3,9, 6,6, 17,2 Гц, 1H), 4,44-4,35 (м, 2H), 4,31-4,26 (м, 1H), 4,20-4,12 (м, 2H), 3,87 (дд, J=3,5, 11,7 Гц, 1H), 3,79-3,77 (м, 1H), 3,15-3,09 (м, 6H), 1,33 (т, J=7,4 Гц, 9H), 0,94 (с, 9H), 0,89 (с, 9H), 0,13 (с, 3H), 0,12 (с, 3H), 0,10 (с, 3H), 0,09 (с, 3H)
Этап 8
Соединение 138 (221 г, 183 ммоль, 1 мас.%, 1 об., 1 экв.) растворяли в смеси пиридина (530 мл, 6,56 моль, 519 г, 2,3 мас.%, 2,4 об.) и TEA (2,65 л, 19,0 моль, 1,93 кг, 8,7 мас.%, 12 об., 104 экв.). Добавляли тригидрофторид триэтиламина (264 мл, 1,62 моль, 262 г, 1,2 мас.%, 1,2 об., 8,9 экв. в виде комплекса, 27 экв. HF) и смесь перемешивали при комнатной температуре, контролируя при этом степень конверсии с помощью ЖХ/МС. Через 3 часа (конверсия 97%) добавляли метокситриметилсилан (™SOMe; 1,40 л, 10,2 моль, 1,06 кг, 4,8 мас.%, 6,3 об., 55 экв.), и перемешивание продолжали в течение 30 минут. Реактор покрывался липким твердым веществом. Часть раствора (супернатант) сливали. Твердое вещество дважды растирали с толуолом (2×2,2 л, 2×10 об.; супернатант сливали). Неочищенное твердое вещество, оставшееся в реакторе, растворяли в дихлорметане (2,2 л, 10 об.) и промывали NH4Cl (28 мас.% раствор в воде; 2,2 л, 10 об.). Водный слой снова экстрагировали дихлорметаном (2,2 л, 10 об.). Объединенные органические слои промывали смесью NaCl (36 мас.% раствор в воде; 1,1 л, 5 об.) и воды (1,1 л, 5 об.), и затем концентрировали под вакуумом с получением Соединения 139 в виде желто-коричневой сухой пены (152 г, 155 ммоль, 0,70 мас.%, выход 85%). Неочищенный продукт использовали на следующем этапе без очистки.
Этап 9
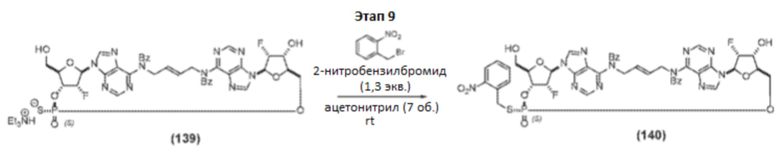
Соединение 139 (150 г, 153 ммоль, 1 мас.%, 1 об., 1 экв.) подвергали азеотропной перегонке с ацетонитрилом (4 л, 27 об.), и затем повторно растворяли в ацетонитриле (1,05 л, 0,83 кг, 5,5 мас.%, 7,0 об.) при rt (комнатной температуре). 2-Нитробензилбромид (44,4 г, 205 ммоль, 0,30 мас.%, 1,34 экв.) добавляли при комнатной температуре, и реакцию контролировали с помощью ЖХ/МС. Через 23 часа (100% конверсия) добавляли EtOAc (1,50 л, 10 об.), NH4Cl (28 мас.% раствор в воде; 300 мл, 2 об.), и воду (300 мл, 2 об.) (pH=6), и полученную смесь частично концентрировали под вакуумом при 25°C до 1,11 кг. Добавляли EtOAc (2,25 л, 15 об.), и смесь перемешивали в течение 5 минут. Два слоя разделяли. Водный слой экстрагировали этилацетатом (750 мл, 5 об.). Объединенные органические слои последовательно промывали: (1) смесью NaCl (36 мас.% раствор в воде; 300 мл, 2 об.) и воды (300 мл, 2 об.) и (2) водой (600 мл, 4 об.). Затем органический слой концентрировали под вакуумом и подвергали азеотропной перегонке с н-гептаном (1,50 л, 10 объемов). К неочищенному твердому веществу добавляли MTBE (0,95 л, 6,3 об.), и смесь нагревали при 40°C. Смесь разбавляли EtOAc (300 мл, 2 об.) и медленно охлаждали до 0°C. Плотному твердому веществу давали осесть, и супернатант откачивали через трубку с фильтрующей фриттой. Твердое вещество дважды промывали МТВЕ (2х300 мл, 2х2 объема; супернатант каждый раз откачивали через трубку с фильтрующей фриттой) и сушили в вакууме при 40°C в течение ночи с получением Соединения 140 в виде бледно-желтого твердого вещества (156 г). Фильтрат концентрировали в вакууме, получая коричневое масло (17,8 г), которое очищали через Biotage Snap-Ultra 340 г (элюенты: от 0 до 5% MeOH в EtOAc) с получением дополнительного соединения 140 в виде бледно-желтого твердого вещества (5,8 г). Общий выход: 156 г+5,8 г=161,8 г (нетто 152 ммоль, чистота 95%, выход 99%)
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,46 (с, 1H), 8,15 (с, 1H), 8,10 (с, 1H), 8,09-8,06 (м, 1H), 7,89 (с, 1H), 7,54-7,51 (м, 1H), 7,49-7,45 (м, 4H), 7,37-7,28 (м, 3H), 7,24-7,19 (м, 3H), 7,16-7,11 (м, 2H), 6,22 (д, J=16,8 Гц, 1H), 6,14 (дд, J=2,7, 17,2 Гц, 1H), 5,83-5,61 (м, 3H), 5,60-5,48 (м, 1H), 5,07 (дд, J=3,5, 51,6 Гц, 1H), 5,06-4,96 (м, 1H), 4,79 (дд, J=4,9, 15,8 Гц, 1H), 4,69 (д, J=5,9 Гц, 2H), 4,67-4,56 (м, 1H), 4,48-4,40 (м, 3H), 4,37-4,30 (м, 1H), 4,27 (д, J=5,9 Гц, 2H), 4,19-4,13 (м, 1H), 3,93-3,85 (м, 1H), 3,85-3,78 (м, 1H)
Этап 10-11
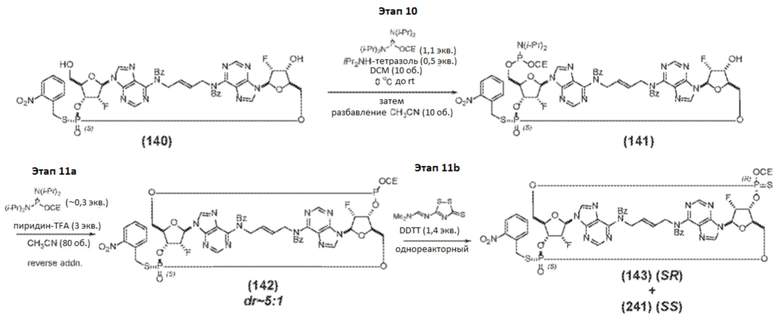
Этап 10. Соединение 140 (чистота 95%, нетто 73,2 г, 72,3 ммоль, 1 мас.%, 1 об., 1 экв.) и 2-цианоэтил N, N, N',N'-тетраизопропилфосфородиамидит (25,3 мл, 79,5 ммоль, 0,33 мас.% 0,35 об., 1,10 экв.) подвергали азеотропной перегонке с безводным ацетонитрилом три раза (3×2 л), повторно растворяли в дихлорметане (0,73 л, 10 об.) и охлаждали до 0-5°C. Добавляли тетразолид диизопропиламмония (6,19 г, 36,1 ммоль, 0,085 мас.%, 0,50 экв.). Полученную реакционную смесь перемешивали при 0°C в течение 10 часов, нагревали до 10°C в течение 2 часов, выдерживали при 10°C в течение 10 часов и нагревали до комнатной температуры в течение 2 часов. За реакцией следили с помощью ЖХ/МС и TLC (EtOAc с 0,5% TEA). Через 18 часов добавляли безводный ацетонитрил (0,73 л, 10 об.), и смесь хранили при -20°C в течение 3 дней.
Этап 11a. Смесь, полученную на этапе 10, нагревали до температуры окружающей среды и добавляли через капельную воронку порциями (100 мл каждые 30 минут, в течение 9 часов) к смеси соли трифторацетата пиридина (предварительно дважды подвергнутой азетропиной перегонке с пиридином; 41,9 г, 217 ммоль, 0,57 мас.%, 3,0 экв.) и ацетонитрила (5,85 л, 80 об.). Реакцию контролировали с помощью ЖХ/МС. Через 13 часов добавляли раствор 2-цианоэтил N, N, N’,N’-тетраизопропилфосфородиамидита (5,8 мл, 18 ммоль, 0,25 экв.) в ацетонитриле (24 мл) в течение 4 часов. Количество дополнительного реагента определяли на основе переаминирования Соединения 140 (~30% по данным ЖХ/МС). Более высокую степень конверсии диола наблюдали через 6 часов.
Этап 11b. Добавляли (диметиламинометилиден)амино)-3H-1,2,4-дитиазолин-3-тион (DDTT; 20,8 г, 101 ммоль, 0,28 мас.%, 1,4 экв.), и перемешивание продолжали в течение 1 часа. Реакционную смесь частично концентрировали до ~800 мл и разбавляли MTBE (1,46 л, 20 об.), NaHCO3 (9 мас.% раствор в воде; 1,1 л, 15 об.) и водой (0,37 л, 5 об.), pH=8. Слои разделяли, и водный слой экстрагировали смесью МТВЕ (1,46 л, 20 об.) и EtOAc (1,10 л, 15 об.). Объединенные органические слои дважды промывали 30% водным NaCl (2×0,73 л, 2×10 об.), концентрировали в вакууме при 35°C и подвергали азеотропной перегонке с толуолом (1,46 л, 20 об.). Результаты ЖХ/МС и ТСХ (EtOAc) показали, что соотношение соединения 143 (SpRp, желаемое) к соединению 241 (SpSp) составляет 5:1.
Неочищенный продукт очищали через Biotage 150M KP-Sil, (SiO2 2,5 кг; элюенты: EtOAc/Hep: 2:1 (4 CV), 3:1 (2,5 CV), 4:1 (2,5 CV), 100% EA. (3 CV), 5-10% MeOH в EA 4 CV) с получением Соединения 143 (36 г, 31,5 ммоль, выход 44%).
Соединение 143 (SpRp): 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,59 (с, 1H), 8,10 (с, 1H), 8,03-7,99 (м, 1H), 7,91 (с, 1H), 7,56-7,53 (м, 2H), 7,49-7,40 (м, 5H), 7,35-7,28 (м, 2H), 7,24-7,16 (м, 4H), 6,92 (с, 1H), 6,29 (д, J=14,9 Гц, 1H), 6,08 (д, J=20,7 Гц, 1H), 5,97-5,83 (м, 1H), 5,76 (тд, J=4,7, 15,6 Гц, 1H), 5,61-5,51 (м, 2H), 5,40 (д, J=4,3 Гц, 1H), 5,29-5,17 (м, 1H), 4,91 (дд, J=7,4, 14,9 Гц, 1H), 4,86-4,75 (м, 3H), 4,63 (дд, J=3,7, 9,2 Гц, 1H), 4,58-4,43 (м, 5H), 4,34-4,19 (м, 4H), 2,79 (тд, J=5,9, 16,8 Гц, 1H), 2,66 (тд, J=6,3, 16,8 Гц, 1H).
Соединение 241 (SpSp) 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8,11 (с, 1H), 8,03 (д, J=8,2 Гц, 1H), 7,94 (с, 1H), 7,90 (с, 1H), 7,61 (с, 1H), 7,56-7,40 (м, 7H), 7,33-7,28 (м, 2H), 7,23-7,17 (м, 4H), 6,22 (д, J=17,6 Гц, 1H), 6,15 (д, J=18,8 Гц, 1H), 5,85 (дд, J=3,5, 51,2 Гц, 1H), 5,75-5,45 (м, 5H), 4,95-4,23 (м, 14H), 2,82 (т, J=6,1 Гц, 2H).
Этап 12
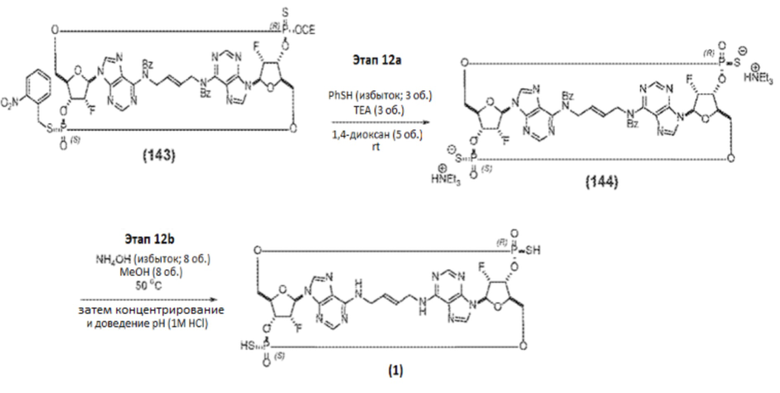
Соединение 143 (71,6 г, 62,6 ммоль, 1 мас.%, 1 об., 1 экв.) растворяли в 1,4-диоксане (0,43 л, 6 об.). Добавляли тиофенол (215 мл, 2,09 моль, 230 г, 3,2 мас.%, 3 объема, >30 экв.), и затем триэтиламин (215 мл, 1,54 моль, 156 г, 2,2 мас.%, 3 об.). Наблюдали некоторый экзотермический эффект (T-внутренняя увеличилась на ~7°C), поэтому для охлаждения и поддержания T-внутренней ниже 27°C использовали баню вода/лед. Реакцию контролировали с помощью ЖХ/МС. Через 2 часа добавляли MeOH (0,57 л, 8 об.) и NH4OH (28 мас.%; 15 моль, 0,57 л, 8 об., >200 экв.). Полученную смесь нагревали при 50°C в течение 5 часов, охлаждали до комнатной температуры и перемешивали в течение ночи. Через 14 часов добавляли воду (0,72 л, 10 об.) (выпадение твердого вещества не наблюдали), и смесь трижды экстрагировали смесью н-гептана и толуола (3×0,86 л, 3×12) 1:1 (об./об.), и затем толуолом (0,57 л, 8 об.). Водный слой концентрировали в вакууме при 40-50°C и разбавляли водой (1,07 л, 15 об.). Полученную суспензию оставляли на ночь при комнатной температуре. Полученное твердое вещество отфильтровывали, промывая водой (0,36 л, 5 об.). Фильтрат все еще оставался мутным, и его фильтровали через целит и фильтр Куно. Мутность все еще присутствовала. В течение 1 часа добавляли HCl (1,0 М раствор в воде; 132 мл, 132 ммоль, 2,1 экв.), и проверяли pH (pH<2). Перемешивание продолжали при комнатной температуре в течение 1 ч, и смесь фильтровали. Осадок на фильтре промывали водой (8×0,20 л), сушили в вакуумной печи при 35°C в течение 2 дней и без нагревания в течение 1 дня с получением Соединения 1 в виде бледно-оранжевого твердого вещества (44,88 г, 60,1 ммоль, 0,63 мас.%, выход 96%).
Этап 13
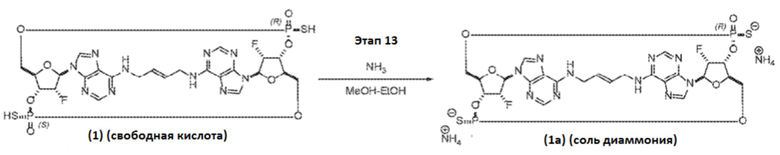
К Соединению 1 в форме свободной кислоты (22,42 г, 30,03 ммоль, 1 мас.%, 1 об., 1 экв.) добавляли аммиак (2,0 М раствор в МеОН; 220 мл, 440 ммоль, 10 об., 15 экв.). Добавляли EtOH (55 мл, 2,5 об.), и полученный раствор фильтровали через фильтр Куно (0,45 микрон; PTFE), промывая 1:1 смесью (об./об.) MeOH и EtOH (90 мл, 4 об.). Фильтрат концентрировали в вакууме при 30°C, получая грязно-белое твердое вещество, которое сушили при комнатной температуре в течение ночи, измельчали шпателем (легко крошилось) и сушили в вакууме при комнатной температуре. Затем выделенное твердое вещество суспендировали в толуоле (250 мл) и перемешивали при комнатной температуре в течение 30 минут. После этого твердое вещество собирали фильтрацией в вакууме и дважды промывали толуолом (2×50 мл). Затем твердое вещество сушили в вакууме в вакуумном сушильном шкафу с получением 22,4 г соединения 1a (диаммониевой соли соединения 1).
Перекристаллизация: Соединение 1a (22,14 г, 28,36 ммоль, 1 мас.%, 1 об., 1 экв.) растворяли в смеси воды (664 мл, 30 об.) и гидроксида аммония (28 мас.%; 2,5 мл, 18 ммоль, 0,63 экв.) (pH=9-10) и трижды экстрагировали толуолом (3×300 мл, 3×14 об.), трижды EtOAc (3×200 мл, 3×9 об.) и трижды толуолом (3×300 мл, 3х14 об.). Полученный водный слой обрабатывали HCl (1,0 М раствор в воде; 90 мл, 90 ммоль, 3,2 экв.) в течение 3,5 часов (pH≤2). Смесь перемешивали в течение 30 минут, и затем твердый осадок собирали вакуумной фильтрацией. Осадок на фильтре трижды промывали водой (3×200 мл, 3×9 об.) и сушили в вакууме в течение ночи. К твердому веществу добавляли аммиак (2,0 М раствор в МеОН; 250 мл, 500 ммоль, 17,6 экв.) и этанол (100 мл), и полученную смесь концентрировали в вакууме до появления кристаллов (~100 мл), после чего процесс концентрации останавливали, и смесь перемешивали в течение 20 минут. Добавляли этанол (45 мл), и смесь частично концентрировали (удаляли 45 мл). Эту операцию повторяли еще два раза, после чего смесь охлаждали до 0°C и перемешивали в течение 3,5 часов. Белое твердое вещество собирали фильтрованием в вакууме и промывали холодным этанолом (20 мл), и затем этилацетатом (2х50 мл). Белое твердое вещество сушили в вакууме при комнатной температуре в течение 3 дней с получением Соединения 1a в виде белого твердого вещества (16,6 г, 21,3 ммоль, 0,75 мас.%, выход 75%). Фильтрат концентрировали в вакууме и сушили в вакууме при комнатной температуре в течение 3 дней с получением соединения 1a в виде твердого вещества грязно-белого цвета (4,16 г, 5,3 ммоль, выход 18%).
Пример 1.2-1H ЯМР анализ соединения 1
1H ЯМР спектрограмма соединения 1a приведена на фиг.3. Полученный спектр был следующим:
1H ЯМР спектр (400 МГц, DMSO-d6, δH 2,49 ppm, 80°C)
δ (ppm): 3,05-3,13 (4H, м), 3,70 (1H, дд, J=13, 5 Гц), 3,78 (1H, дд, J=12, 4 Гц), 4,21-4,24 (2H, м), 4,28 (1H, м), 4,38 (1H, м), 4,53-4,68 (2H, м), 5,22 (1H, м), 5,76 (2H, с), 5,78 (1H, м), 6,26 (1H, м), 6,29 (1H, м), 8,13 (1H, с), 8,14 (1H, с), 8,36 (1H, уш.с), 8,59 (1H, уш.с).
Пример 1.3 - Рентгеноструктурный анализ Соединения 1
Примерно 2 мг соединения 1 растворяли в 600 мкл воды. 120 мкл этого раствора помещали в другой стеклянный флакон, и затем этот флакон хранили в закрытом контейнере с 3 мл MeCN при комнатной температуре в течение 1 недели. Использовали метод подготовки проб методом диффузии паров H2O/MeCN.
Бесцветный монокристалл блочной (ячеистой) структуры (0,1×0,1×0,1 мм), обнаруженный в растворе для кристаллизации, диспергировали в жидкости Parabar 10312 и устанавливали на устройстве Dual-Thickness MicroMounts™ (MiTeGen). Дифракционные данные собирали при -160°C с помощью XtaLAB PRO P200 MM007HF (Rigaku) методом осцилляций по оси ω с использованием монохроматизированного излучения Cu-Kα с применением зеркал с многослойным покрытием.
На фиг. 4A показано полученное с помощью программы ORTEP изображение молекул Соединения 1 в виде асимметричной единицы вместе с неупорядоченными молекулами воды. На фиг. 4B показана кристаллическая структура одной из молекул Соединения 1, приведенной на фиг. 4А. На фиг. 4C показана кристаллическая структура другой молекулы Соединения 1, показанной на фиг. 4А.
Кристаллическая структура Соединения 1 была получена с конечным R-фактором, равным 0,1354. Flack-параметр был почти нулевым (0,083 (17)), что указывает на то, что абсолютная конфигурация Соединения 1 представляет собой (R, S). Анализ кристаллической структуры также показал, что в большом канале соединения 1 присутствовало много молекул воды, что указывает на то, что молекулы воды могут легко выскальзывать из канала. Анализ также подтвердил, что конформации обеих кристаллографически независимых молекул асимметричной единицы являются практически одинаковыми.
Дополнительные параметры рентгеноструктурного анализа приведены ниже:
Пример 2. Полученная с помощью рентгеноструктурного анализа структура, подтверждающая комплекс с WT STING
Чтобы лучше понять механизм, с помощью которого предлагаемые новые соединения связываются с мишенями, с помощью рентгеноструктурного анализа определяли кристаллическую структуру WT STING в комплексе с соединениями.
А. Экспрессия и очистка С-концевого домена WT STING (остатки 155-341)
Последовательность ДНК, кодирующую человеческий белок WT STING от аминокислоты 155 до аминокислоты 341 (SEQ ID NO: 4), клонировали в вектор pET21b, после метки His-TEV-Sumo на N-конце (SEQ ID NO: 5). Последовательность pET21b депонирована в addgene и доступна по адресу: addgene.org/vector-database/2550/; эта последовательность включена в настоящее описание посредством ссылки.
Клетки E. coli BL21 (DE3)-Codon Plus трансформировали этой плазмидой, и экспрессию рекомбинантного белка индуцировали 0,1 мМ изопропил-β-D-1-тиогалактопиранозидом (IPTG). Белок очищали от растворимой фракции клеточного лизата с помощью аффинной хроматографии на Ni-NTA колонке. Метку His-TEV-Sumo удаляли с помощью протеазы Sumo и отделяли от WT STING 155-341 без метки, используя вторую колонку Ni-NTA для аффинной хроматографии. Белок дополнительно очищали с помощью анионообменной и эксклюзионной хроматографии и хранили в буфере, содержащем 20 мМ Трис-HCl pH 7,5 и 150 мМ NaCl, при концентрации 35 мг/мл.
В. Кристаллизация и определение структуры С-концевого домена WT STING в комплексе с соединением 1
Для совместной кристаллизации WT STING 155-341 с соединением 1 белок WT STING разбавляли до 10 мг/мл буфером для хранения (20 мМ Трис-HCl pH 7,5 и 150 мМ NaCl) и смешивали с соединением 1 (100 мМ маточный раствор в DMSO) в молярном соотношении 1:5. Смесь инкубировали в течение 4 часов при 4°C и центрифугировали при 13000 об/мин в течение 20 минут перед кристаллизацией. Устанавливали сетчатые лотки для кристаллизации с помощью метода диффузии паров в варианте висячей капли при 18°C. Кристаллы выращивали путем смешивания 1 мкл раствора WT STING/Соединение 1 с равным объемом раствора для лунок, содержащего 100 мМ HEPES (pH=7,5), 200 мМ CaCl2, и, когда кристаллы мгновенно замораживали в жидком азоте, то в качестве криозащитного агента использовали 15% (мас./об.) ПЭГ 8000, 20% (мас./об.) ПЭГ 400. Наборы данных дифракции собирали с помощью детектора Pilatus на канале луча SSRF BL19U1 и обрабатывали с помощью HKL3000 и программы SCALEPACK2MTZ в программном пакете CCP4.
Структуру WT STING 155-341, связанного с Соединением 1, определяли путем молекулярного замещения с помощью программы PHASER (Maximum Likelihood Molecular Replacement) с PDB ID 4F9E в качестве начальной поисковой модели. Присутствие Соединения 1 между областями контакта димера WT STING подтверждали на карте разности Fo-Fc, рассчитанной с модельными фазами. Строили модель и дополняли вручную с помощью программы Coot и дорабатывали с помощью программы Refmac5 в программном пакете CCP4. Окончательная уточненная структура представлена с разрешением 2,38 Å в пространственной группе P212121 с элементарной ячейкой, измеренной при a=33,820, b=78,110, c=132,212, α=90,00, β=90,00, γ=90,00. В каждой асимметричной единице идентифицировали две копии WT STING 155-341, связывающиеся с одной молекулой соединения 1 в области контакта димера.
C. Взаимодействие соединения 1 с WT STING, наблюдаемое в кристаллической структуре, полученной с помощью рентгеноструктурного анализа
На фиг. 5 показано полученное с помощью рентгеноструктурного анализа изображение кристаллической структуры STING WT человека в комплексе с соединением 1. Исследовали полученную с помощью рентгеноструктурного анализа кристаллическую структуру WT STING человека в комплексе с соединением 1, которое совместно кристаллизовали из образца соединения 1a. Соединение связывается с областью контакта кармана, образованного димером белка WT STING. Две грани аденинового основания соединения находятся в π-π стэкинг-взаимодействии с Tyr240 и гуанидиновой группой Arg238, соответственно. Транс-олефиновый линкер находится в ван-дер-ваальсовом взаимодействии с алифатической частью боковой цепи Arg 238. Заместитель фтора в положении C2’-рибозной группы соединения находится в гидрофобной полости, определяемой Thr263, Pro264 и Tyr163. Отрицательно заряженная тиофосфатная группа соединения образует солевой мостик с Arg238 и водородные связи с Ser162 и Thr267, соответственно. Кроме того, тиофосфатная группа также находится в электростатическом взаимодействии с гуанидиновой группой Arg232. Область LID петли WT STING, состоящая из 226-243 остатков, обернута вокруг двух основных групп и транс-олефинового линкера.
Пример 3. Определение кристаллической структуры REF STING в комплексе с Соединением 1 с помощью рентгеноструктурного анализа
А. Экспрессия и очистка С-концевого домена REF STING (остатки 155-341, SEQ ID NO: 6)
Последовательность ДНК, кодирующую человеческий белок REF STING от аминокислоты 155 до аминокислоты 341 (SEQ ID NO: 6), клонировали в вектор pET21b после метки His-TEV-Sumo на N-конце (SEQ ID NO: 7). Последовательность pET21b депонирована в addgene и доступна по адресу: addgene.org/vector-database/2550/; эта последовательность включена в настоящее описание посредством ссылки.
Клетки E. coli BL21 (DE3)-Plus Codon трансформировали этой плазмидой, и экспрессию рекомбинантного белка индуцировали 0,1 мМ изопропил-β-D-1-тиогалактопиранозидом (IPTG). Белок очищали от растворимой фракции клеточного лизата с помощью аффинной хроматографии на колонке Ni-NTA. His-TEV-Sumo метку удаляли с помощью протеазы Sumo и отделяли от не содержащего метку REF STING_155-341 с помощью второй колонки Ni-NTA для аффинной хроматографии. Белок дополнительно очищали с помощью анионообменной и эксклюзионной хроматографии и хранили в буфере, содержащем 20 мМ Трис-HCl pH 7,5 и 150 мМ NaCl, при концентрации 24 мг/мл.
В. Кристаллизация и определение структуры С-концевого домена REF STING в комплексе с соединением 1
Для совместной кристаллизации REF STING_155-341 с Соединением 1, белок REF STING разбавляли до 10 мг/мл буфером для хранения (20 мМ Трис-HCl pH 7,5 и 150 мМ NaCl) и смешивали с Соединением 1 (100 мМ маточный раствор в DMSO) в мольном соотношении 1:5. Смесь инкубировали в течение 4 часов при 4°C и центрифугировали при 13000 об/мин в течение 20 минут перед кристаллизацией. Устанавливали сетчатые лотки для кристаллизации с помощью метода диффузии паров в варианте висячей капли при 18°C. Кристаллы выращивали путем смешивания 1 мкл раствора REF STING/Соединение 1 с равным объемом раствора для лунок, содержащего 100 мМ HEPES (pH=7,5), 200 мМ CaCl2, и, когда кристаллы мгновенно замораживали в жидком азоте, в качестве криозащитного реагента использовали 15% (мас./об.) ПЭГ 8000, 20% (мас./об.) ПЭГ 400. Наборы данных дифракции собирали с помощью детектора Pilatus на канале луча SSRF BL18U1 и обрабатывали с помощью HKL3000 и программы SCALEPACK2MTZ в программном пакете CCP4. Эта структура показана на фиг. 6.
Структуру REF STING 155-341, связанного с Соединением 1, определяли путем молекулярного замещения с помощью программы PHASER (Maximum Likelihood Molecular Replacement), используя ранее определенную структуру WT STING 155-341 (как описано выше) в качестве начальной поисковой модели. Присутствие Соединения 1 между областями контакта димера WT STING подтверждали на карте разности Fo-Fc, рассчитанной с модельными фазами. Строили модель и дополняли вручную с помощью программы Coot и дорабатывали с помощью программы Refmac5 в программном пакете CCP4. Окончательная уточненная структура представлена с разрешением 2,76 Å в пространственной группе P212121 с элементарной ячейкой, измеренной при a=33,733, b=77,831, c=131,689, α=90,00, β=90,00, γ=90,00. В каждой асимметричной единице идентифицировали две копии REF STING 155-341, связывающиеся с одной молекулой Соединения 1 в области контакта димера.
С. Взаимодействие соединения 1 с REF STING, наблюдаемое в кристаллической структуре, полученной с помощью рентгеноструктурного анализа
На фиг. 6 показана полученная с помощью рентгеноструктурного анализа кристаллическая структура REF STING человека в комплексе с Соединением 1, которое было совместно кристаллизовано из образца Соединения 1a. Соединение связывается с областью контакта кармана, образованного димером белка STING. Две грани аденинового основания соединения находятся в π-π стэкинг-взаимодействии с Tyr240 и гуанидиновой группой Arg238, соответственно. Транс-олефиновый линкер находится в ван-дер-ваальсовом взаимодействии с алифатической частью боковой цепи Arg238, в то время как гуанидиновая часть боковой цепи Arg238 находится в π-π-стэкинг-взаимодействии с находящейся снаружи имидазольной группой боковой цепи His232. Олефиновый линкер находится в контакте с взаимодействующей парой боковых цепей Arg238 и His232. Заместитель фтора в положении C2’-рибозной группы соединения расположен в гидрофобной полости, определяемой Thr263, Pro264 и Tyr163. Отрицательно заряженная тиофосфатная группа соединения образует солевой мостик с Arg238 и водородные связи с Ser162 и Thr267, соответственно. Область петли LID REF STING, состоящая из остатков с 226 по 243, обернута вокруг двух групп оснований и транс-олефинового линкера.
Пример 4 - Оценка соединения 1а in vivo на модели рака мочевого пузыря у мышей при внутрипузырном введении
Ортотопическая модель рака мочевого пузыря MBT-2 у мышей была создана и охарактеризована Ли и др. (Lee et al., «Tumor Establishment Features of Orthotopic Murine Bladder Cancer Models,» Urological Oncology 2012; 53:396-400) в 2012 году, и было показано, что эта модель имеет гистологию рака мочевого пузыря, аналогичную человеческому. Следовательно, для оценки активности Соединения 1 против рака мочевого пузыря была выбрана эта модель. Для создания модели применяли протокол с процедурой предварительной обработки с помощью HCl, описанный Lee, et al. Вкратце, 30 мкл 0,1 н. раствора HCl вводили в мочевой пузырь мыши через катетер, и оставляли этот раствор HCl в мочевом пузыре на 15 секунд. Затем раствор HCl заменяли 30 мкл 0,1 н. раствором NaOH с последующей стадией промывки 1х PBS (pH 7,4). Процедура последующей имплантации опухолевых клеток была изменена по сравнению с опубликованной. Вкратце, после стадии промывки опухолевые клетки MBT-2 (2×106 клеток в 50 мкл среды RPMI1640) вводили в мочевой пузырь и оставляли на 45 минут. В конце этого 45-минутного периода мочевой пузырь опорожняли.
Через три дня после имплантации опухолевых клеток мышей лечили разными дозами соединения 1a, БЦЖ (OncoTICE®, Merck Canada Inc.), мышиным анти-PD-1 антителом (клон № RMP1-14, Bioxcell) и комбинацией соединения 1a с анти-PD-1 антителом по схеме, приведенной в таблице 1. Соединение 1a и БЦЖ вводили внутрипузырно, тогда как анти-PD1 антитело вводили внутрибрюшинной инъекцией.
В каждый день дозирования БЦЖ разводили в 0,9% NaCl (Lavoisier, France) до конечной концентрации 16,875 мг/мл для введения дозы. Любой оставшийся дозируемый раствор после использования уничтожали. Маточный раствора анти-PD-1 антитела разбавляли 1x PBS (Lonza, France) для получения маточного раствора 1 мг/мл, использованного для введения дозы. Для Соединения 1a сначала готовили 10 мг/мл маточный раствор, растворяя высушенный порошок в 1x PBS. Затем получали различные концентрации Соединения 1a, включая 5 мг/мл (400 мкг/группа мышей), 2,5 мг/мл (200 мкг/группа мышей) и 1,25 мг/мл (100 мкг/группа мышей) путем дальнейшего разбавления маточного раствора в 1х PBS.
Таблица 1. Терапевтические агенты и схемы дозирования
1. IVe, внутривенное введение
2. IP, внутрибрюшинная инъекция
С 20 по 21 день рост опухоли в мочевом пузыре количественно оценивали с помощью МРТ (магнитно-резонансной томографии), и размеры опухолей у всех мышей отображали в виде графика, как показано на фиг. 7. На фиг. 7 показано количественное определение объема опухоли с помощью МРТ с 20 по 21 день у всех исследуемых животных. Как показано на фиг. 7, все группы, обработанные соединением 1a (группы с 4 по 7), показали гораздо меньшие объемы опухоли, чем группа носителя (группа 1), группа БЦЖ (группа 2) и группа анти-PD1 антитела (группа 3).
Соединение 1a также показало статистически значимое улучшение выживаемости по сравнению с животными, которым вводили носитель, как показано на фиг. 8.
Как БЦЖ, так и анти-PD1 антитело не продемонстрировали значительную противораковую активность в этой ортотопической модели рака мочевого пузыря мыши, что позволяет предположить, что используемая модель представляет собой модель резистентности/рефрактерности к БЦЖ/PD1 антителам.
Пример 103. Репортерный анализ активности конструкции агониста HAQ STING
Для определения EC50 использовали клетки THP1-Dual™ (InvivoGen, кат. № thpd-nfis). Клетки THP1 Dual™ были охарактеризованы поставщиком Invivogen (Insight 201402-1) как носители генотипа HAQ STING. Клетки выращивали и поддерживали в условиях, рекомендованных производителем. Для определения ЕС50 осуществляли индукцию пути фактора регуляции интерферона (IRF), описанную в руководстве производителя. Вкратце, клетки засевали и обрабатывали различными концентрациями соединения в течение 20 часов в условиях инкубации при 37°C, 5% CO2. Клетки ресуспендировали и добавляли раствор QUANTI-Luc™ (кат. №: rep-qlc1). Полученное световое излучение измеряли люминометром (Envision, Perkin Elmer). Полученные сигналы наносили на график и рассчитывали ЕС50 с помощью программы GraphPad Prism7.
Значения ЕС50 человеческого STING (мкМ) для Соединения 1a представлены ниже в Таблице 2.
Пример 104 - Репортерный анализ специфических вариантов STING
STING человека имеет 4 основных варианта, включая варианты WT, HAQ, REF и AQ. REF-STING, также обозначаемый как R232H, например, встречается примерно у 14% человеческой популяции. По сравнению с аллелем дикого типа, R232H имеет пониженный ответ на циклические динуклеотиды бактерий и метазоа. Подробная информация об этих 4 основных вариантах, а также о других редких вариантах приедена у Yi G, et al., «Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides» PLoS One 2013; 8:e77846. Репортерные клеточные линии, специфичные к вариантам STING, создавали с использованием клеток THP1-Dual™ KO-STING (InvivoGen, кат № thpd-kostg) и трех векторов экспрессии белка-варианта STING. Карта вектора экспрессии для WT STING показана на фиг. 9. Для двух других векторов экспрессии в этом векторе использовали разные последовательности вариантов STING, при этом WT STING заменяли на соответствующую нуклеотидную последовательность.
Готовили векторы, экспрессирующие варианты STING, WT-STING, REF-STING и AQ-STING, которые стабильно трансфицировали в клетки THP1-Dual™ KO-STING для использования в репортерном анализе специфических вариантов STING, WT-STING, REF-STING и AQ-STING, соответственно. Значения ЕС50 определяли, как описано выше в Примере 103 относительно репортерного анализа активности конструкции агониста HAQ STING. Результаты показаны ниже в Таблице 2. Последовательности ДНК, использованные для этих вариантов STING, приведены в SEQ ID NO: 1 (нуклеотидная последовательность человеческого WT STING), SEQ ID NO: 2 (нуклеотидная последовательность человеческого REF STING) и SEQ ID NO: 3 (нуклеотидная последовательность человеческого AQ STING).
Человеческий WT STING:
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccggtgaccgggctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccggacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga (SEQ ID NO: 1).
Человеческий REF STING:
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccggtgaccatgctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccggacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga (SEQ ID NO: 2)
Человеческий AQ STING:
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccgctgaccgagctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccagacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga (SEQ ID NO: 3)
Пример 105 - Репортерный анализ активности агониста мышиного STING
Клетки ISG RAW-Lucia™ (InvivoGen, кат. № rawl-isg) использовали для репортерного анализа агониста мышиного STING. Значения ЕС50 определяли, как описано выше в Примере 103, в репортерном анализе активности агониста HAQ STING. Результаты показаны ниже в таблице 2.
Пример 106. Анализ методом дифференциальной сканирующей флуориметрии (DSF).
Анализ DSF использовали для измерения физического взаимодействия между соединениями и рекомбинантным белком STING. Усеченный рекомбинантный белок STING (а.к. 155-341) (SEQ ID NO: 4) экспрессировали в E. coli и выделяли для анализа, как описано ниже. Матрицу для анализа готовили в 384-луночных планшетах с конечным объемом 10 мкл на лунку, состоящей из 1 мкМ рекомбинантного белка STING (а.к. 155-341) (SEQ ID NO: 4), 100 мМ PBS (pH 7,4), дополненный 100 мМ KCl, 5х оранжевый краситель SYPRO и 50 мкМ соединения (конечная концентрация DMSO 0-1%). Анализы выполняли в системе ПЦР в реальном времени QuantStudio 12K Flex с использованием градиента температуры от 25°C до 95°C со скоростью 0,05°C/мин и фильтров возбуждения и испускания 470 и 586 нм, соответственно. По производным кривым флуоресценции, полученным с помощью программного обеспечения Applied Biosystems® Protein Thermal Shift (версия алгоритма 1.3), вычисляли температуру плавления (Tm) несвязанного и связанного с лигандом рекомбинантного белка STING и разницу температур плавления (dTm D).
Обычно считается, что соединения со значениями ΔTm больше 0 физически взаимодействуют с тестируемым белком, при этом значение ΔTm находится в прямой зависимости со сродством связывания соединения. В данном случае соединение 1a показало ΔTm 17,6 (таблица 2), что указывает на физическое взаимодействие с белком STING.
Таблица 2. Характеристика соединения 1a in vitro
Пример 107 - Анализ стимуляции PBMC человека ex vivo
Человеческую кровь, полученную от 5 здоровых доноров, собирали в 10,0 мл пробирки с гепарином натрия BD Vacutainer (кат. № 367874). Выделение мононуклеарных клеток периферической крови (PBMC) выполняли в 50 мл пробирках SIGMA ACCUSPIN (кат. № A2055) и SIGMA ACCUSPIN System-HISTOPAQUE-1077 (кат. № A7054) согласно протоколу производителя. Слой PBMC собирали и промывали 1x фосфатно-солевым буфером (PBS), как предложено Sigma. Подсчитывали РВМС и, наконец, суспендировали при концентрации 1×106/мл в RPMI (Corning кат. № 10-041-CV) с добавлением 10% фетальной бычьей сыворотки (FBS) (Gibco кат. № 20140.79). Один мл клеток (1×106) переносили в 5 мл полипропиленовую пробирку с круглым дном Falcon (кат. № 352063) и стимулировали различными концентрациями (0, 0,1, 1, 10 мкМ) в течение 24 часов в инкубаторе с 5% CO2 при 37°C.
Через 24 часа после инкубации пробирки центрифугировали при 1400 об/мин в течение 5 минут, и супернатанты собирали. Супернатант хранили при -80°C до последующего измерения IFNβ. Измерение IFNβ выполняли с использованием базового набора IFN-β человека (Meso Scale Diagnostics, кат. № K151ADA) и использовали протокол, предоставленный производителем. IFN-бета оценивали путем считывания аналитического планшета на MESO SECTOR Imager 2400 с помощью программы MSD Discovery Workbench 4.0. Анализ белка IFNβ выполняли через 24 часа. Результаты показали, что соединение 1a может индуцировать продуцирование первичного белка IFNβ человека в РВМС дозозависимым образом.
Результаты, представленные в таблице 3, отражают среднее значение измерений, проведенных у пяти разных доноров.
Таблица 3 Анализ стимуляции человеческих РВМС ex vivo
Для количественного определения мРНК IFNβ выделяли общую РНК с помощью набора RNeasy Mini Kit (Qiagen, Germany) в соответствии с протоколом производителя. Количественное определение мРНК IFNβ выполняли с помощью количественной ПЦР. Вкратце, общую РНК (от 400 до 1000 нг) преобразовали в кДНК в реакционном объеме 60 мкл с помощью SuperScript VILO MasterMix (Life Technologies, USA). Полученные кДНК (10 нг) впоследствии амплифицировали с помощью анализов экспрессии TaqMan от Applied Biosystems с использованием РНК-специфических праймеров для IFNB1 (Hs01077958_s1) и GAPDH (Hs99999905_m1). Анализ количественной ПЦР выполняли с помощью TaqMan Fast Advanced Master Mix (Life Technologies, США) в системе ПЦР в реальном времени Applied Biosystems Quantstudio 12K Flex с начальным 2-минутным шагом при 50°C, затем при 95°C в течение 2 сек, и далее следовали 40 циклов при 95°C продолжительностью 1 сек и 60°C в течение 20 сек. Относительную экспрессию гена рассчитывали после нормализации относительно эталонного гена GAPDH с помощью метода 2-δδCT. Расчеты выполняли с помощью программного обеспечения Applied Biosystems Quantstudio 12K Flex v1.2.2. Кратные изменения мРНК IFNβ по сравнению с образцами, обработанными носителем, суммированы в таблице 4. Результаты показали, что соединение 1a может индуцировать мРНК IFNβ в первичных PBMC в зависимости от дозы и времени. В таблице 4 показано среднее значение, рассчитанное для пяти разных доноров.
Таблица 4 - Анализ 3-часовой и 24-часовой стимуляции человеческих PBMC (мРНК) ex vivo
Пример 108. Противораковый эффект Соединения 1a на модели двойной опухоли CT26.
Соединение 1a тестировали на противораковую активность на модели двойной опухоли CT26, которая представляет собой модель рака толстой кишки у мышей. Каждой самке мыши Balb/cJ в возрасте 5-6 недель (Jackson Labs, Bar Harbor, Maine) подкожно имплантировали опухолевые клетки CT26 с обеих сторон, по 105 клеток с каждой стороны. В исследовании А лечение начинали через 5 дней (1,25 мг/кг, 2,5 мг/кг и 5 мг/кг) после имплантации опухоли, когда средний размер опухоли достигал приблизительно 100 мм3. В исследовании B лечение начинали через 8 дней (0,6 мг/кг и 10 мг/кг) после имплантации опухоли, когда средний размер опухоли достигал приблизительно 120 мм3. Схема лечения представлена в таблицах 5 и 6.
Таблица 5. Схема дозирования в исследовании A
*I.T. - внутрь опухоли
Таблица 6. Схема дозирования в исследовании В
*I.T. - внутрь опухоли
Все мыши в этом исследовании имели две подкожные опухоли CT26. «Обработанная опухоль» указывает на опухоль с прямым введением соединения, тогда как «необработанная опухоль» указывает на опухоль без прямого введения соединения. Объем опухоли отслеживали на протяжении всего эксперимента. Объем опухоли измеряли два раза в неделю после начала лечения. Опухолевую нагрузку рассчитывали на основе измерений, выполненных штангенциркулем, по формуле для объема вытянутого эллипсоида (LxW2)/2, где L и W - соответствующие ортогональные измерения длины и ширины (мм).
Соединение 1a показало высокую лечебную активность в модели двойной опухоли CT26 (фиг.10 и фиг.11). Даже при самой низкой дозе, испытанной в исследовании, уровень излечения обработанных опухолей составил 20% (фиг. 8, доза 0,6 мг/кг). В то же время, к концу исследования самая высокая доза позволила излечить 100% животных от этой опухоли (10 мг/кг). Дозозависимый противоопухолевый эффект наблюдали также для необработанных опухолей. Группа с максимальной дозой (10 мг/кг) показала 80% лечебный эффект; все более низкие дозы также показали активность ингибирования роста опухоли. Следовательно, соединение 1a продемонстрировало терапевтическое окно в пределах от 0,6 мг/кг до 10 мг/кг, при этом противоопухолевая активность оказалась не только локальной, но и системной, о чем свидетельствовало наличие эффектов в дистальном участке опухоли, не подвергнутом инъекции. В качестве заключения, эти результаты показывают, что местное введение Соединения 1а может индуцировать как местную, так и системную (скрытую) противораковую активность.
Пример 109. Противораковый эффект Соединения 1a на модели метастазов в печень CT26
Соединение 1a тестировали на противораковую активность на модели метастазов в печень CT26. Анестезированным самкам мышей BALB/cJ в возрасте 5-6 недель (Jackson Labs, Harbor, Maine) имплантировали в селезенку опухолевые клетки CT26, экспрессирующие люциферазу (5×105 клеток на мышь). Последующий 10-минутный период ожидания обеспечил циркуляцию опухолевых клеток в печени животных. Затем селезенки удаляли, животных зашивали, и давали им возможность восстановиться. Через три дня опухолевые клетки CT26 (105 клеток на мышь) снова имплантировали, на этот раз подкожно (п/к) под область правой передней конечности, чтобы обеспечить развитие опухолевой массы для введения соединения. Через девять дней после инъекции внутрь селезенки однократно вводили соединение внутрь подкожной опухоли (10 мг/кг).
Местный противораковый эффект соединения измеряли по его влиянию на подкожную опухоль, в то время как скрытый эффект соединения оценивали по общей выживаемости обработанных мышей по сравнению с контрольными мышами, получавшими носитель, основываясь на пагубном эффекте растущей массы опухоли в печени каждой мыши. Соединение 1a показало как сильную активность в отношении локальных п/к опухолей, так и лечебную системную активность у 9 из 10 обработанных животных (фиг. 12). Эти результаты показывают, что местное введение соединения 1а может вызывать как местную, так и системную (абсорбционную) противораковую активность, включая глубокое поражение, например, в печени.
Пример 110. Противораковый эффект Соединения 1a на ортотопической модели мозга GL261
Соединение 1a тестировали на противораковую активность на ортотопической модели мозга GL261. GL261 представляет собой линию клеток глиомы мыши. Клетки мышиной глиомы GL261, экспрессирующие люциферазу (2×104 клеток/мышь), имплантировали внутричерепно самкам мышей-альбиносов B6 в возрасте 5-6 недель (Jackson Labs, Bar Harbor, Maine). Спустя три-четыре дня клетки GL261 имплантировали подкожно (106 клеток/мышь) под область правой передней конечности, чтобы обеспечить развитие опухолевой массы для введения соединения. Через десять дней после имплантации внутричерепных опухолевых клеток однократно вводили соединение внутрь подкожной опухоли (10 мг/кг). Локальный противораковый эффект соединения измеряли по его влиянию на подкожную опухоль, в то время как скрытый эффект соединения оценивали по общей выживаемости обработанных мышей по сравнению с контрольными мышами, получавшими носитель, основываясь на пагубном эффекте растущей массы опухоли в мозгу каждой мыши. Соединение 1а показало как сильную активность в отношении локальных подкожных опухолей, так и терапевтическую системную активность у 5 из 8 обработанных животных (фиг. 13). Эти результаты показывают, что местное введение Соединения 1a может вызывать как локальную, так и системную (абсорбционную) противораковую активность, включая глубокое поражение, например, в головном мозге.
Пример 111 - Сравнения
Значения ЕС50 вычисляли для анализов человеческого STING: WT STING, HAQ STING, AQ STING и REF STING, при непосредственном сравнении, используя соединение 1a по изобретению, природный лиганд STING (2'3'cGAMP) и предполагаемый агонист STING - ML RR-S2 CDA, описанные Corrales, et al. в работе «Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity,» Cell Reports (2015) 11:1018-1030, которая включена в настоящее описание посредством ссылки. Анализы выполняли, как описано в приведенных выше примерах. Следует обратить внимание на то, что указанные в таблице 7 результаты анализов ограничены анализами, выполненными путем непосредственного сравнения, и могут не отражать усредненные значения, определенные в большем количестве испытаний, как указано выше или где-либо еще. Также следует обратить внимание на то, что «2’3’cGAMP» - это то же самое, что «ML cGAMP», как указано в публикации Cell Reports.
Таблица 7
(мкМ)
В таблице 7 также приведены константы диссоциации (связывания) (Kd) для связывания человеческого STING дикого типа с каждым из трех тестируемых соединений, измеренные методом изотермической титрационной калориметрии (ITC). ITC - это метод микрокалориметического титрования, который позволяет измерить термодинамические свойства, связанные с межмолекулярными взаимодействиями. Исходя из результатов, полученных в этих тестах, соединение 1a, по-видимому, образует самую прочную связь с WT STING по сравнению с другими протестированными соединениями.
Материалы
Рекомбинантный человеческий белок STING дикого типа (аа, 139-379, H232R) получали в результате экспрессии в E.coli конструкции, кодирующей цитозольный домен человеческого STING WT, содержащий аминокислоты 139-379.
Реагенты
Источники реагентов, использованных в этом исследовании, приведены ниже:
Приготовление буфера для белков
Белок STING хранили при -60°C в аликвотах по 90 мкл и 100 мкл каждая в концентрации 3,0 мг/мл и 20 мг/мл, соответственно, в PBS, pH 7,5, содержащем 5% глицерин. В день анализа аликвоты белка размораживали, разбавляли до 400 мкл и заменяли буфер на PBS, используя центробежный фильтр Amicon Ultra (отсечка 10 кДа, 0,5 мл), центрифугируя минимум четыре раза по 10 мин со скоростью 14000 g в микроцентрифуге Eppendorf, и затем окончательно разбавляли до 20-30 мкМ (в зависимости от эксперимента), используя 1X PBS. Концентрацию белка определяли с помощью спектрофотометра Nanodrop 2000 с коэффициентом экстинкции белка 22140 (М-1см-1).
Приготовление образцов
В 1,5 мл микроцентрифужные пробирки вносили по 200 мкл 1 мМ маточных растворов Соединения 1a, 2’3’cGAMP и ML RR-S2CDA. Перед каждым экспериментом образцы разбавляли до концентраций от 200 мкМ до 500 мкМ (в зависимости от эксперимента).
Методы
Анализы выполняли на приборе Affinity ITC (TA Instruments № 609003.901), оснащенном приспособлением для очистки ITC (TA Instruments № 601800.901). Раствор белка STING (приблизительно 400 мкл, содержащие от 20 до 30 мкМ белка STING) пипетировали в 185 мкл лунку калориметра, допуская некоторое приемлемое избыточное количество. Контрольная лунка содержала эквивалентное количество воды Milli-Q. Инкубацию выполняли при 25°C с 20-ю инъекциями по 2,5 мкл соединений в концентрации от 100 до 300 мкМ. Для получения термограмм, состоящих из нескольких пиков выделяемого тепла (мккал/сек), представляющих скорость нагревания при каждой инъекции, использовали управляющее программное обеспечение ITC Run версии 3.3.0.0 (TA Instruments). Для корректировки базовой линии, корректировки тепла от пустого образца или разбавленного образца (при насыщении) и для интеграции уровней тепловых пиков с получением графических значений «Q» использовали программное обеспечение для анализа Nano Analyze версии 3.70 (TA Instruments). Полученные изотермы подгоняли с помощью независимой модели для получения термодинамических параметров.
Получали и регистрировали значения Kd и n (молярные отношения в точках перегиба кривой). Оптимальные условия для концентраций белка и лиганда получали из предварительных экспериментов.
Полученные результаты
Получали термограммы скорости нагревания и из них получали изотермы связывания тестируемых соединений с рекомбинантным человеческим STING дикого типа (а.к. 139-379, H232R). Связывание каждого соединения со STING было эндотермическим, на что указывает отрицательная скорость нагревания с экзотермической (положительное направление) теплотой разбавления (наблюдаемой после насыщения соединения белком). Было показано, что 2’,3’cGAMP вызывает аналогичный эндотермический ответ на различные варианты STING. Соединение 1a обеспечивает самое низкое значение Kd=0,04 мкМ, затем следует 2’3’cGAMP с Kd=0,07 мкМ и затем - ML RR-S2 CDA с Kd=0,40 мкМ. Все соединения показали значения n, близкие к 0,5, что позволяет предположить, что белок STING присутствует в виде димера и связывается при соотношении 1 моль соединения на 2 моль STING.
Пример 112. Идентификация потенциальных метаболитов
Для оценки образования основных метаболитов соединение 1а инкубировали в гепатоцитах мыши CD-1, крысы Sprague Dawley, собаки породы бигль, обезьяны Cynomolgus и человека.
Материалы
Криоконсервированные объединенные гепатоциты приобретали у компаний ThermoFisher Scientific (Waltham, MA), Xenotech, LLC (Kansas City, KS) и In Vitro ADMET Laboratories (Columbia, MD), а соответствующие среды приобретали у компаний In Vitro ADMET Laboratories (Columbia, MD) и Life Technologies (Carlsbad, CA). Окрашивающий раствор AOPI и фосфатный буфер приобретали у компаний Corning Life Sciences (Tewksbury, MA) и Nexcelom Bioscience (Lawrence, MA), соответственно. Все химические вещества, реагенты и растворители, использованные в анализе, имели либо аналитическую чистоту, либо степень чистоты для ВЭЖХ.
Схема и процедуры эксперимента
Инкубация гепатоцитов
Соединение 1a взвешивали и растворяли в воде для ВЭЖХ, содержащей 0,12% PBS с муравьиной кислотой, до концентрации 1020 ммоль/л. Затем раствор разбавляли отдельно в 2,5 раза до 4 ммоль/л, и затем дополнительно разбавляли в 21000 раз, используя среду E Вильямса, содержащую 0,1% человеческий сывороточный альбумин и 2 ммоль/л L-глутамина, с получением маточного раствора с концентрацией 20 мкмоль/л.
Перед инкубацией криоконсервированные гепатоциты размораживали на водяной бане при 37°C. Содержимое одной пробирки с криоконсервированными гепатоцитами добавляли в каждую коническую пробирку объемом 50 мл с криоконсервированной средой для восстановления гепатоцитов (UCRM), приобретенной у компании In Vitro ADMET Laboratories (Colambia, MD). Клетки центрифугировали на центрифуге Beckman (Brea, CA) с ротором GH 3.8 при 740 об/мин в течение 10 минут при температуре 4°C. Супернатант удаляли, и клетки повторно суспендировали для подсчета в среде для посева. После ресуспендирования клеток в среде для посева 20 мкл суспензии переносили и смешивали с 20 мкл окрашивающего раствора AOPI. Раствор осторожно перемешивали, и клетки подсчитывали с помощью целлометра (Nexcelom, Lawrence, MA). После подсчета клетки повторно суспендировали при концентрации 1 или 2 миллиона жизнеспособных клеток/мл в среде E Вильямса, содержащей 2 ммоль/л L-глутамина (pH 7,4).
Суспензию гепатоцитов (50 мкл/лунку) добавляли в 48-луночный планшет. Для начала реакции добавляли пятьдесят микролитров рабочего маточного раствора, содержащего соединение 1a (20 мкмоль/л). Планшет помещали в инкубатор для культуры тканей (5% CO2/95% влажность и 37°C), и реакцию останавливали добавлением 200 мкл стоп-раствора, состоящего из смеси 100% метанол/ацетонитрил (1/1, об./об.) с 2010 нг/мл фуросемида и 0,2 мкмоль/л (R)-пропранолола, через 5, 30, 60, 120, 180 и 240 минут. Смесь центрифугировали и фильтровали, и супернатант собирали для анализа. Конечные концентрации криоконсервированных гепатоцитов составляли 1×106 клеток/мл. Конечная инкубационная концентрация Соединения 1a составляла 10 мкмоль/л.
Условия ЖХ-МС/МС для идентификации метаболитов
Система ЖХ-МС/МС состояла из ВЭЖХ-системы Shimadzu и гибридного квадрупольного масс-спектрометра AB-SCIEX Triple TOF 5600 TOF (Framingham, MA). Система ВЭЖХ Shimadzu (Kyoto, Japan) состоит из модуля коммуникационной шины (CBM-20A), автоматического пробоотборника (SIL-30AC) с прикрепленным устройством для смены штативов (Rack Changer II), двух насосов (LC-30AD) и нагревателя колонки (СТО-30А). Масс-спектрометр калибровали, используя растворы для отрицательной и положительной калибровки AB-SCIEX APCI (Framingham, MA). Образцы, полученные в результате инкубации с гепатоцитами, анализировали как в отрицательном, так и в положительном режиме сканирования. Ниже приведено краткое описание основных аналитических методов и настроек приборов. Изменение настроек спектрометра зависело от типа аналита.
Условия ЖХ-МС/МС:
B: метанол/вода=95/5 (об./об.) с 5 мМ ацетата аммония
Параметры установки:
Параметры установки
DP -100,000 или 100,000
Диапазон сканирования: 120,0-1000,0
Параметры установки
CES 5,000
DP -100,000 или 100,000
Диапазон сканирования: 100,0-1000,0
Анализ данных по активности
Данные масс-спектрометрии получали с помощью программного обеспечения AB-Sciex Analyst TF (версия 1.5.1; Framingham, MA). Хроматограммы и спектры получали с помощью программного обеспечения AB-Sciex PeakView (версия 2.2.0.1; Framingham, MA). Сравнение относительных площадей пиков хроматограмм экстрагированных ионов основано на ±0,0002 Да ожидаемого точного отношения массы к заряду (m/z) для каждого представляющего интерес аналита.
Полученные результаты
После инкубации с гепатоцитами метаболит не обнаруживался. В представленных аналитических условиях Соединение 1а показало время удерживания, равное приблизительно 7,8 минут. В режиме отрицательного сканирования соединение 1a показало депротонированный молекулярный ион m/z 745 (C24H25F2N10O8P2S2-) и дважды депротонированный молекулярный ион m/z 372 (C24H24F2N10O8P2S22-). Основные наблюдаемые ионы МС/МС продукта имели m/z 533 (C19H19FN10O4PS-) и m/z 186 (C9H8N5-). В режиме положительного сканирования Соединение 1a имело протонированный молекулярный ион m/z 747 (C24H27F2N10O8P2S2+) и основные ионы МС/МС продуктов мели m/z 651 (C24H26F2N10O6PS+), m/z 252 (C10H11FN5O2+) и m/z 188 (C9H10N5+). Данные МС и МС/МС подтверждают структуру Соединения 1a.
Соединение 1a было стабильным при инкубации с гепатоцитами мыши, крысы, собаки, обезьяны и человека. В этом исследовании не было выявлено обнаруживаемого метаболита соединения 1a. В образцах, полученных в результате инкубации с гепатоцитами, детектируемым было только само соединение 1a, что подтверждено фрагментами тандемной масс-спектрометрии (MS/MS).
Все документы, на которые в настоящем описании имеются ссылки, включены в него посредством ссылки, и, если какой-либо включенный документ противоречит представленному описанию, то настоящее описание имеет преимущественную силу. Специалисты в данной области поймут, что в материал, представленный в настоящем описании, могут быть внесены различные изменения и модификации, и что такой материал охвачен объемом и сущностью изобретения.
СПИСКИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
SEQ ID NO: 1 (человеческий STING дикого типа):
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccggtgaccgggctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccggacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga
SEQ ID NO: 2 (человеческий REF STING):
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccggtgaccatgctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccggacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga
SEQ ID NO: 3 (человеческий AQ STING):
atgccccactccagcctgcatccatccatcccgtgtcccaggggtcacggggcccagaaggcagccttggttctgctgagtgcctgcctggtgaccctttgggggctaggagagccaccagagcacactctccggtacctggtgctccacctagcctccctgcagctgggactgctgttaaacggggtctgcagcctggctgaggagctgcgccacatccactccaggtaccggggcagctactggaggactgtgcgggcctgcctgggctgccccctccgccgtggggccctgttgctgctgtccatctatttctactactccctcccaaatgcggtcggcccgcccttcacttggatgcttgccctcctgggcctctcgcaggcactgaacatcctcctgggcctcaagggcctggccccagctgagatctctgcagtgtgtgaaaaagggaatttcaacgtggcccatgggctggcatggtcatattacatcggatatctgcggctgatcctgccagagctccaggcccggattcgaacttacaatcagcattacaacaacctgctacggggtgcagtgagccagcggctgtatattctcctcccattggactgtggggtgcctgataacctgagtatggctgaccccaacattcgcttcctggataaactgccccagcagaccgctgaccgagctggcatcaaggatcgggtttacagcaacagcatctatgagcttctggagaacgggcagcgggcgggcacctgtgtcctggagtacgccacccccttgcagactttgtttgccatgtcacaatacagtcaagctggctttagccgggaggataggcttgagcaggccaaactcttctgccagacacttgaggacatcctggcagatgcccctgagtctcagaacaactgccgcctcattgcctaccaggaacctgcagatgacagcagcttctcgctgtcccaggaggttctccggcacctgcggcaggaggaaaaggaagaggttactgtgggcagcttgaagacctcagcggtgcccagtacctccacgatgtcccaagagcctgagctcctcatcagtggaatggaaaagcccctccctctccgcacggatttctcttga
SEQ ID NO: 4 (остатки 155-341 WT STING):
VAHGLAWSYYIGYLRLILPELQARIRTYNQHYNNLLRGAVSQRLYILLPLDCGVPDNLSMADPNIRFLDKLPQQTGDRAGIKDRVYSNSIYELLENGQRAGTCVLEYATPLQTLFAMSQYSQAGFSREDRLEQAKLFCRTLEDILADAPESQNNCRLIAYQEPADDSSFSLSQEVLRHLRQEEKEEV
SEQ ID NO: 5 (His-TEV-Sumo-WT STING 155-341)
MHHHHHHSSGVDLGTENLYFQSNAMSDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGKEMDSLRFLYDGIRIQADQTPEDLDMEDNDIIEAHREQIGGGSVAHGLAWSYYIGYLRLILPELQARIRTYNQHYNNLLRGAVSQRLYILLPLDCGVPDNLSMADPNIRFLDKLPQQTGDRAGIKDRVYSNSIYELLENGQRAGTCVLEYATPLQTLFAMSQYSQAGFSREDRLEQAKLFCRTLEDILADAPESQNNCRLIAYQEPADDSSFSLSQEVLRHLRQEEKEEV
SEQ ID NO: 6 (остатки 155-341 REF STING):
VAHGLAWSYYIGYLRLILPELQARIRTYNQHYNNLLRGAVSQRLYILLPLDCGVPDNLSMADPNIRFLDKLPQQTGDHAGIKDRVYSNSIYELLENGQRAGTCVLEYATPLQTLFAMSQYSQAGFSREDRLEQAKLFCRTLEDILADAPESQNNCRLIAYQEPADDSSFSLSQEVLRHLRQEEKEEV
SEQ ID NO: 7 (His-TEV-Sumo-REF STING 155-341)
MHHHHHHSSGVDLGTENLYFQSNAMSDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGKEMDSLRFLYDGIRIQADQTPEDLDMEDNDIIEAHREQIGGGSVAHGLAWSYYIGYLRLILPELQARIRTYNQHYNNLLRGAVSQRLYILLPLDCGVPDNLSMADPNIRFLDKLPQQTGDHAGIKDRVYSNSIYELLENGQRAGTCVLEYATPLQTLFAMSQYSQAGFSREDRLEQAKLFCRTLEDILADAPESQNNCRLIAYQEPADDSSFSLSQEVLRHLRQEEKEEV
--->
СПИСКИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Eisai R&D Management Co., Ltd.
Kim, Dae-Shik
<120> СПОСОБЫ ЛЕЧЕНИЯ РАКА МОЧЕВОГО ПУЗЫРЯ
<130> 0080171-000500
<160> 7
<170> PatentIn version 3.5
<210> 1
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 1
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg cgccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccggt gaccgggctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccgga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag 1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag 1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttga 1140
<210> 2
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 2
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg cgccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccggt gaccatgctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccgga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag 1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag 1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttga 1140
<210> 3
<211> 1140
<212> ДНК
<213> Homo sapiens
<400> 3
atgccccact ccagcctgca tccatccatc ccgtgtccca ggggtcacgg ggcccagaag 60
gcagccttgg ttctgctgag tgcctgcctg gtgacccttt gggggctagg agagccacca 120
gagcacactc tccggtacct ggtgctccac ctagcctccc tgcagctggg actgctgtta 180
aacggggtct gcagcctggc tgaggagctg cgccacatcc actccaggta ccggggcagc 240
tactggagga ctgtgcgggc ctgcctgggc tgccccctcc gccgtggggc cctgttgctg 300
ctgtccatct atttctacta ctccctccca aatgcggtcg gcccgccctt cacttggatg 360
cttgccctcc tgggcctctc gcaggcactg aacatcctcc tgggcctcaa gggcctggcc 420
ccagctgaga tctctgcagt gtgtgaaaaa gggaatttca acgtggccca tgggctggca 480
tggtcatatt acatcggata tctgcggctg atcctgccag agctccaggc ccggattcga 540
acttacaatc agcattacaa caacctgcta cggggtgcag tgagccagcg gctgtatatt 600
ctcctcccat tggactgtgg ggtgcctgat aacctgagta tggctgaccc caacattcgc 660
ttcctggata aactgcccca gcagaccgct gaccgagctg gcatcaagga tcgggtttac 720
agcaacagca tctatgagct tctggagaac gggcagcggg cgggcacctg tgtcctggag 780
tacgccaccc ccttgcagac tttgtttgcc atgtcacaat acagtcaagc tggctttagc 840
cgggaggata ggcttgagca ggccaaactc ttctgccaga cacttgagga catcctggca 900
gatgcccctg agtctcagaa caactgccgc ctcattgcct accaggaacc tgcagatgac 960
agcagcttct cgctgtccca ggaggttctc cggcacctgc ggcaggagga aaaggaagag 1020
gttactgtgg gcagcttgaa gacctcagcg gtgcccagta cctccacgat gtcccaagag 1080
cctgagctcc tcatcagtgg aatggaaaag cccctccctc tccgcacgga tttctcttga 1140
<210> 4
<211> 187
<212> Белок
<213> Homo sapiens
<400> 4
Val Ala His Gly Leu Ala Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu
1 5 10 15
Ile Leu Pro Glu Leu Gln Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr
20 25 30
Asn Asn Leu Leu Arg Gly Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu
35 40 45
Pro Leu Asp Cys Gly Val Pro Asp Asn Leu Ser Met Ala Asp Pro Asn
50 55 60
Ile Arg Phe Leu Asp Lys Leu Pro Gln Gln Thr Gly Asp Arg Ala Gly
65 70 75 80
Ile Lys Asp Arg Val Tyr Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn
85 90 95
Gly Gln Arg Ala Gly Thr Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln
100 105 110
Thr Leu Phe Ala Met Ser Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu
115 120 125
Asp Arg Leu Glu Gln Ala Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile
130 135 140
Leu Ala Asp Ala Pro Glu Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr
145 150 155 160
Gln Glu Pro Ala Asp Asp Ser Ser Phe Ser Leu Ser Gln Glu Val Leu
165 170 175
Arg His Leu Arg Gln Glu Glu Lys Glu Glu Val
180 185
<210> 5
<211> 311
<212> Белок
<213> Искусственная последовательность
<220>
<223> Конструкция His-TEV-Sumo-WT STING
<400> 5
Met His His His His His His Ser Ser Gly Val Asp Leu Gly Thr Glu
1 5 10 15
Asn Leu Tyr Phe Gln Ser Asn Ala Met Ser Asp Ser Glu Val Asn Gln
20 25 30
Glu Ala Lys Pro Glu Val Lys Pro Glu Val Lys Pro Glu Thr His Ile
35 40 45
Asn Leu Lys Val Ser Asp Gly Ser Ser Glu Ile Phe Phe Lys Ile Lys
50 55 60
Lys Thr Thr Pro Leu Arg Arg Leu Met Glu Ala Phe Ala Lys Arg Gln
65 70 75 80
Gly Lys Glu Met Asp Ser Leu Arg Phe Leu Tyr Asp Gly Ile Arg Ile
85 90 95
Gln Ala Asp Gln Thr Pro Glu Asp Leu Asp Met Glu Asp Asn Asp Ile
100 105 110
Ile Glu Ala His Arg Glu Gln Ile Gly Gly Gly Ser Val Ala His Gly
115 120 125
Leu Ala Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu
130 135 140
Leu Gln Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu
145 150 155 160
Arg Gly Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys
165 170 175
Gly Val Pro Asp Asn Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu
180 185 190
Asp Lys Leu Pro Gln Gln Thr Gly Asp Arg Ala Gly Ile Lys Asp Arg
195 200 205
Val Tyr Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala
210 215 220
Gly Thr Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala
225 230 235 240
Met Ser Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu
245 250 255
Gln Ala Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala
260 265 270
Pro Glu Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala
275 280 285
Asp Asp Ser Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg
290 295 300
Gln Glu Glu Lys Glu Glu Val
305 310
<210> 6
<211> 187
<212> Белок
<213> Homo sapiens
<400> 6
Val Ala His Gly Leu Ala Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu
1 5 10 15
Ile Leu Pro Glu Leu Gln Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr
20 25 30
Asn Asn Leu Leu Arg Gly Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu
35 40 45
Pro Leu Asp Cys Gly Val Pro Asp Asn Leu Ser Met Ala Asp Pro Asn
50 55 60
Ile Arg Phe Leu Asp Lys Leu Pro Gln Gln Thr Gly Asp His Ala Gly
65 70 75 80
Ile Lys Asp Arg Val Tyr Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn
85 90 95
Gly Gln Arg Ala Gly Thr Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln
100 105 110
Thr Leu Phe Ala Met Ser Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu
115 120 125
Asp Arg Leu Glu Gln Ala Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile
130 135 140
Leu Ala Asp Ala Pro Glu Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr
145 150 155 160
Gln Glu Pro Ala Asp Asp Ser Ser Phe Ser Leu Ser Gln Glu Val Leu
165 170 175
Arg His Leu Arg Gln Glu Glu Lys Glu Glu Val
180 185
<210> 7
<211> 311
<212> Белок
<213> Искусственная последовательность
<220>
<223> Конструкция His-TEV-Sumo-REF STING 155-341
<400> 7
Met His His His His His His Ser Ser Gly Val Asp Leu Gly Thr Glu
1 5 10 15
Asn Leu Tyr Phe Gln Ser Asn Ala Met Ser Asp Ser Glu Val Asn Gln
20 25 30
Glu Ala Lys Pro Glu Val Lys Pro Glu Val Lys Pro Glu Thr His Ile
35 40 45
Asn Leu Lys Val Ser Asp Gly Ser Ser Glu Ile Phe Phe Lys Ile Lys
50 55 60
Lys Thr Thr Pro Leu Arg Arg Leu Met Glu Ala Phe Ala Lys Arg Gln
65 70 75 80
Gly Lys Glu Met Asp Ser Leu Arg Phe Leu Tyr Asp Gly Ile Arg Ile
85 90 95
Gln Ala Asp Gln Thr Pro Glu Asp Leu Asp Met Glu Asp Asn Asp Ile
100 105 110
Ile Glu Ala His Arg Glu Gln Ile Gly Gly Gly Ser Val Ala His Gly
115 120 125
Leu Ala Trp Ser Tyr Tyr Ile Gly Tyr Leu Arg Leu Ile Leu Pro Glu
130 135 140
Leu Gln Ala Arg Ile Arg Thr Tyr Asn Gln His Tyr Asn Asn Leu Leu
145 150 155 160
Arg Gly Ala Val Ser Gln Arg Leu Tyr Ile Leu Leu Pro Leu Asp Cys
165 170 175
Gly Val Pro Asp Asn Leu Ser Met Ala Asp Pro Asn Ile Arg Phe Leu
180 185 190
Asp Lys Leu Pro Gln Gln Thr Gly Asp His Ala Gly Ile Lys Asp Arg
195 200 205
Val Tyr Ser Asn Ser Ile Tyr Glu Leu Leu Glu Asn Gly Gln Arg Ala
210 215 220
Gly Thr Cys Val Leu Glu Tyr Ala Thr Pro Leu Gln Thr Leu Phe Ala
225 230 235 240
Met Ser Gln Tyr Ser Gln Ala Gly Phe Ser Arg Glu Asp Arg Leu Glu
245 250 255
Gln Ala Lys Leu Phe Cys Arg Thr Leu Glu Asp Ile Leu Ala Asp Ala
260 265 270
Pro Glu Ser Gln Asn Asn Cys Arg Leu Ile Ala Tyr Gln Glu Pro Ala
275 280 285
Asp Asp Ser Ser Phe Ser Leu Ser Gln Glu Val Leu Arg His Leu Arg
290 295 300
Gln Glu Glu Lys Glu Glu Val
305 310
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ АНТИТЕЛО-STING АГОНИСТ И ИХ ПРИМЕНЕНИЕ В ИММУНОТЕРАПИИ | 2020 |
|
RU2826228C2 |
| ПРОИЗВОДНЫЕ 3-(5-ГИДРОКСИ-1-ОКСОИЗОИНДОЛИН-2-ИЛ) ПИПЕРИДИН-2,6-ДИОНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ С "ЦИНКОВЫМИ ПАЛЬЦАМИ" 2 (IKZF2) СЕМЕЙСТВА IKAROS | 2019 |
|
RU2797559C2 |
| ПРОИЗВОДНЫЕ 3-(5-АМИНО-1-ОКСОИЗОИНДОЛИН-2-ИЛ)ПИПЕРИДИН-2,6-ДИОНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ С "ЦИНКОВЫМИ ПАЛЬЦАМИ" 2 СЕМЕЙСТВА IKAROS (IKZF2) | 2019 |
|
RU2815714C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ МОЧЕВИНЫ И КОМПОЗИЦИИ НА ИХ ОСНОВЕ В КАЧЕСТВЕ ИНГИБИТОРОВ АТФАЗЫ SMARCA2/BRM | 2019 |
|
RU2798462C2 |
| КОНЬЮГАТ АНТИТЕЛА К КЛАУДИНУ И ЛЕКАРСТВЕННОГО СРЕДСТВА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2826119C1 |
| ЛИНКЕРЫ НА ОСНОВЕ СУЛЬФОМАЛЕИМИДА И СООТВЕТСТВУЮЩИЕ КОНЪЮГАТЫ | 2019 |
|
RU2815199C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2020 |
|
RU2787992C1 |
| ПРОИЗВОДНЫЕ МАЙТАНЗИНОИДА С САМОРАСЩЕПЛЯЮЩИМИСЯ ПЕПТИДНЫМИ ЛИНКЕРАМИ И ИХ КОНЪЮГАТЫ | 2018 |
|
RU2765098C2 |
| КОНЪЮГАТ АНТИТЕЛА К TROP-2 И АНАЛОГА ЭКЗАТЕКАНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2830167C1 |
| ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА | 2020 |
|
RU2813356C2 |
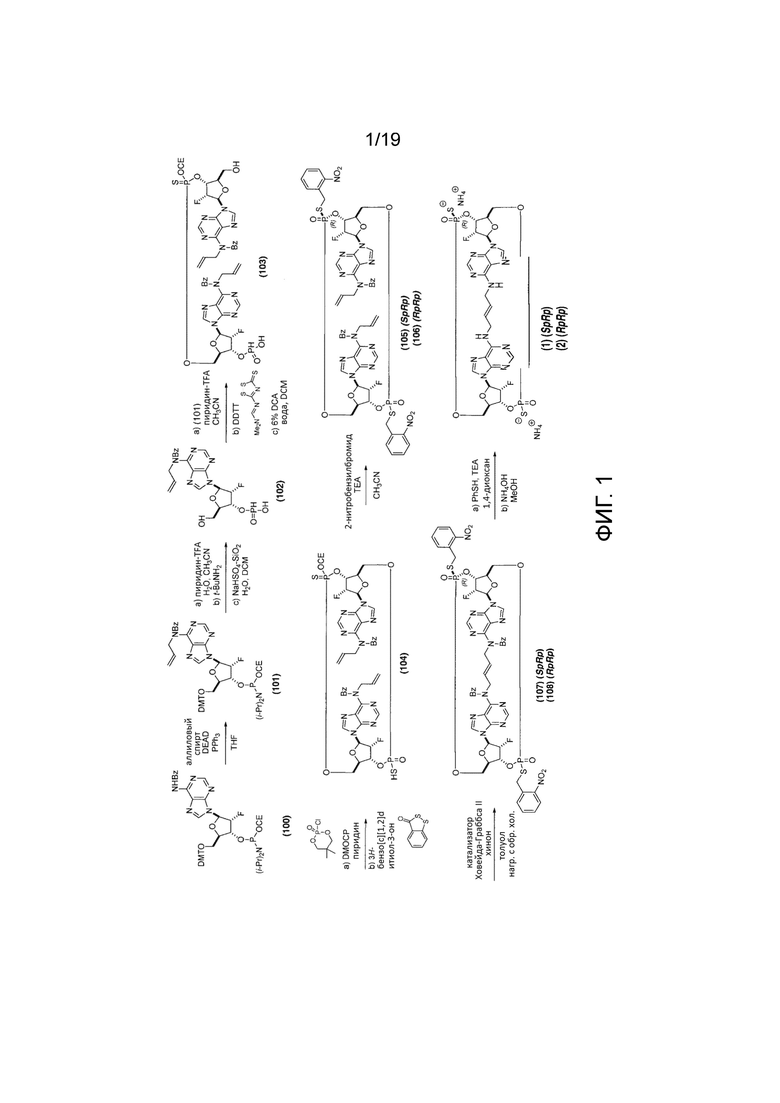
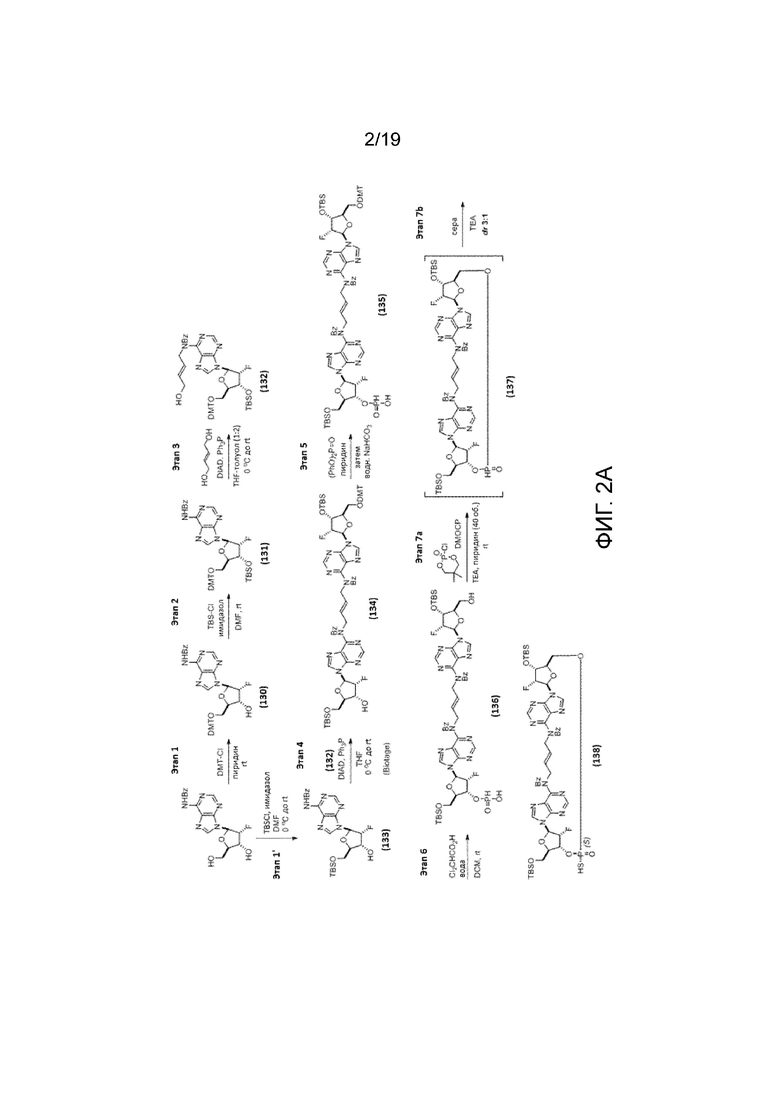
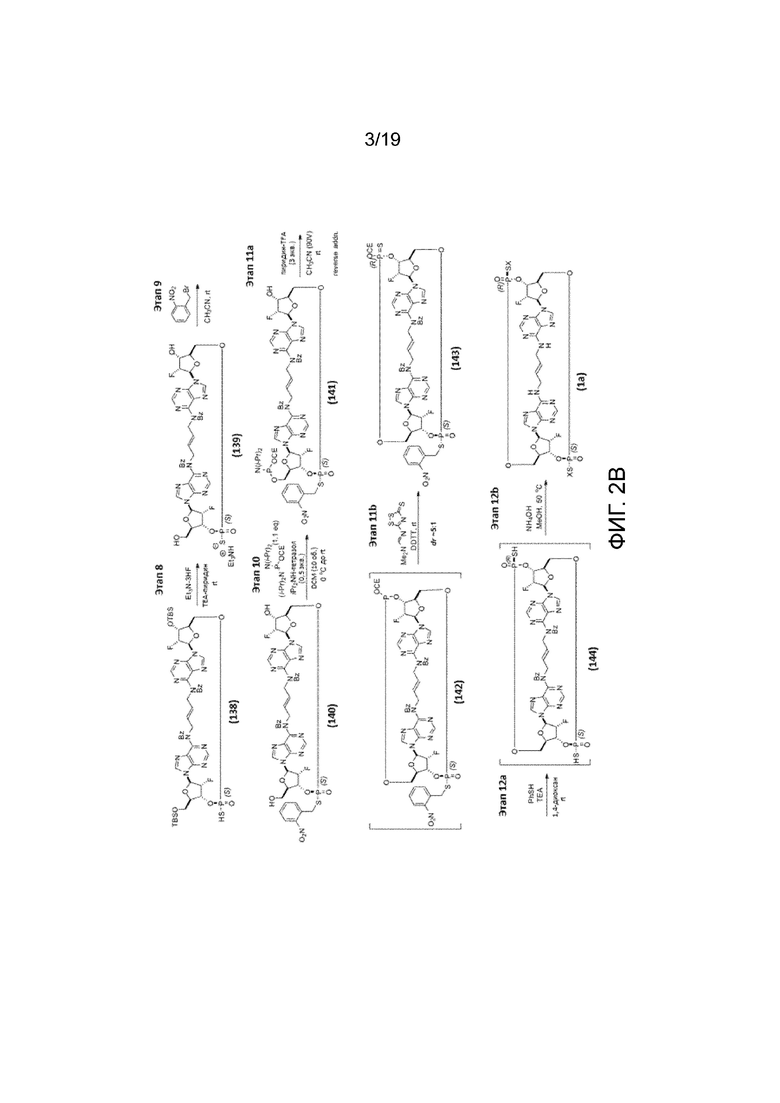

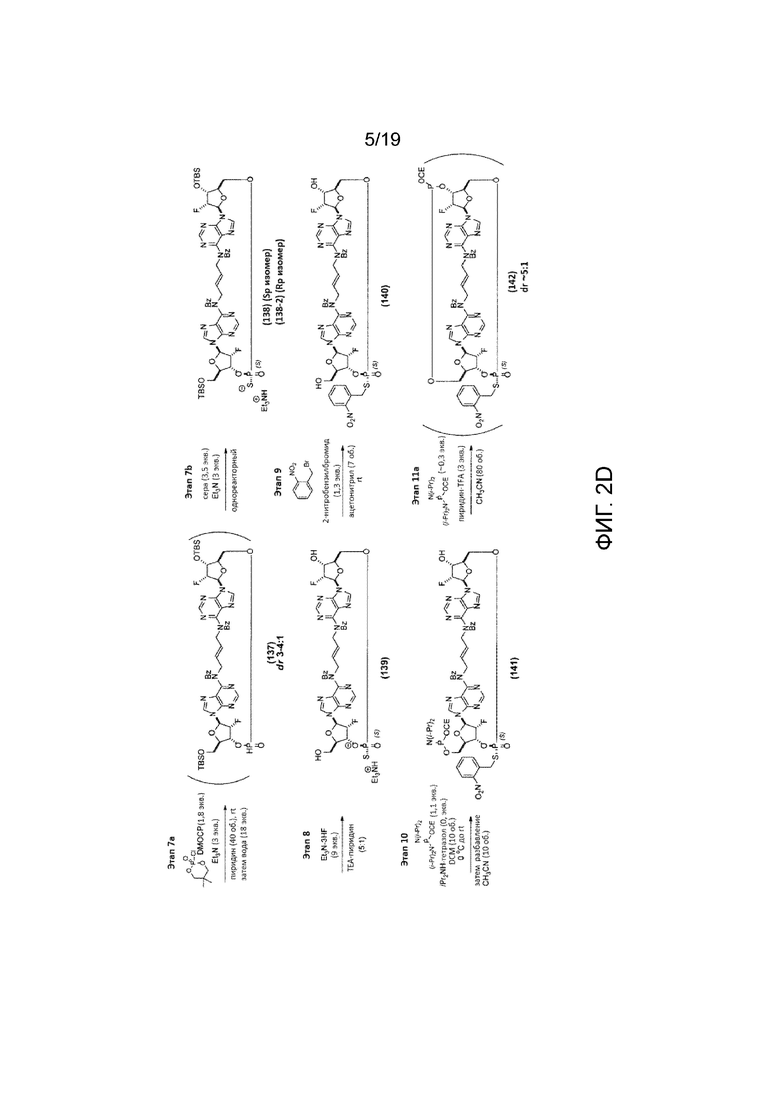
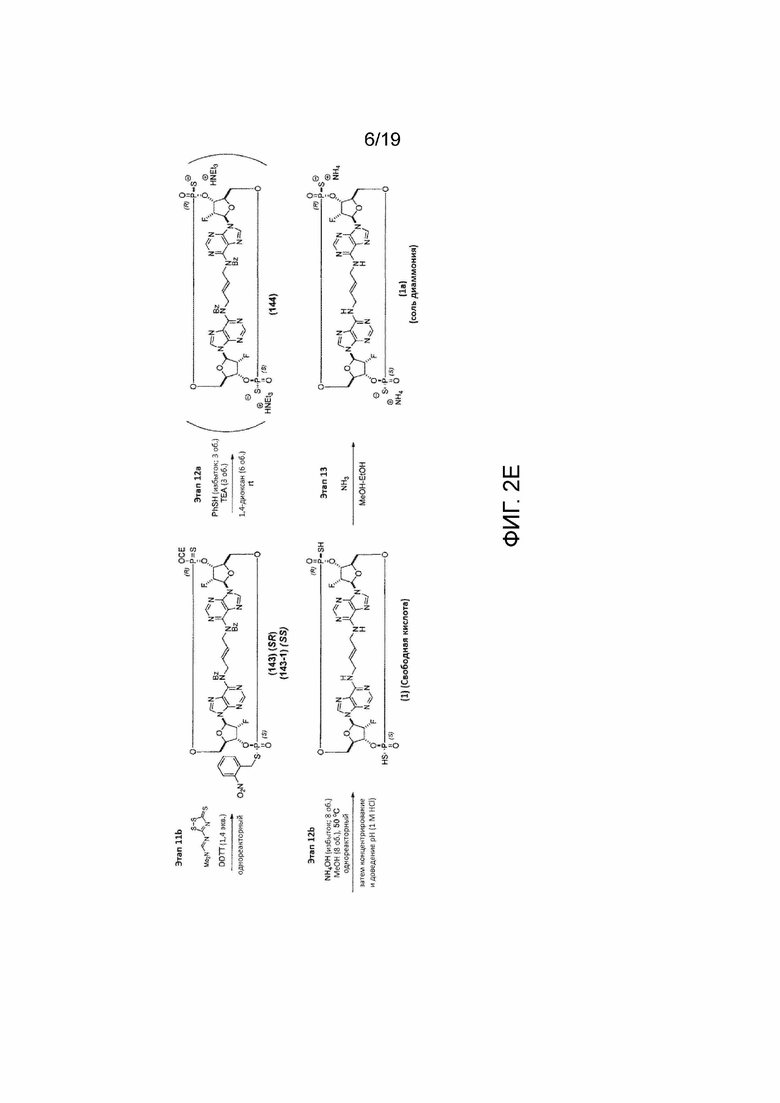
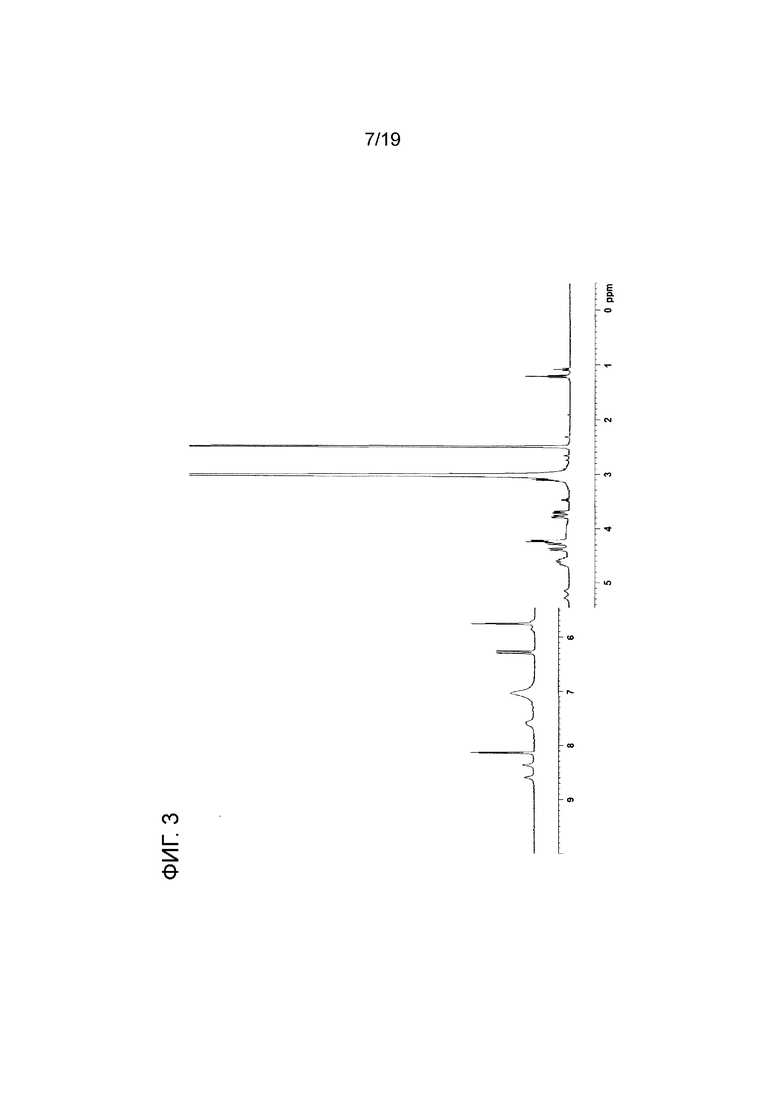
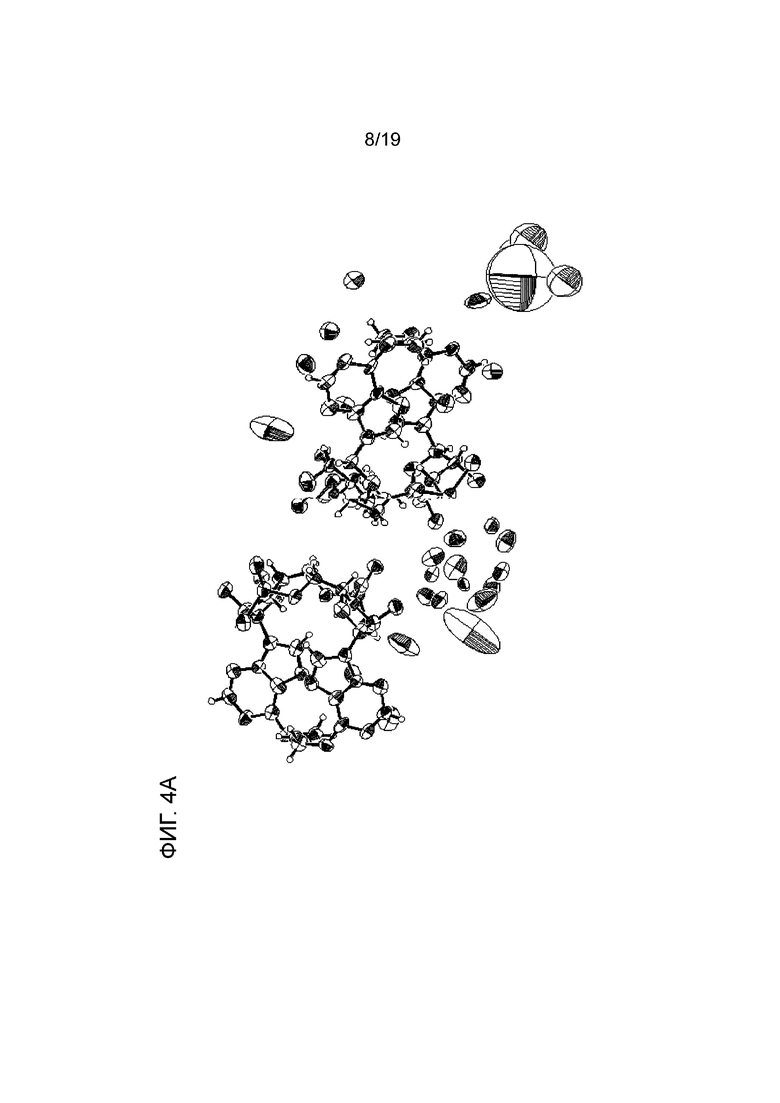
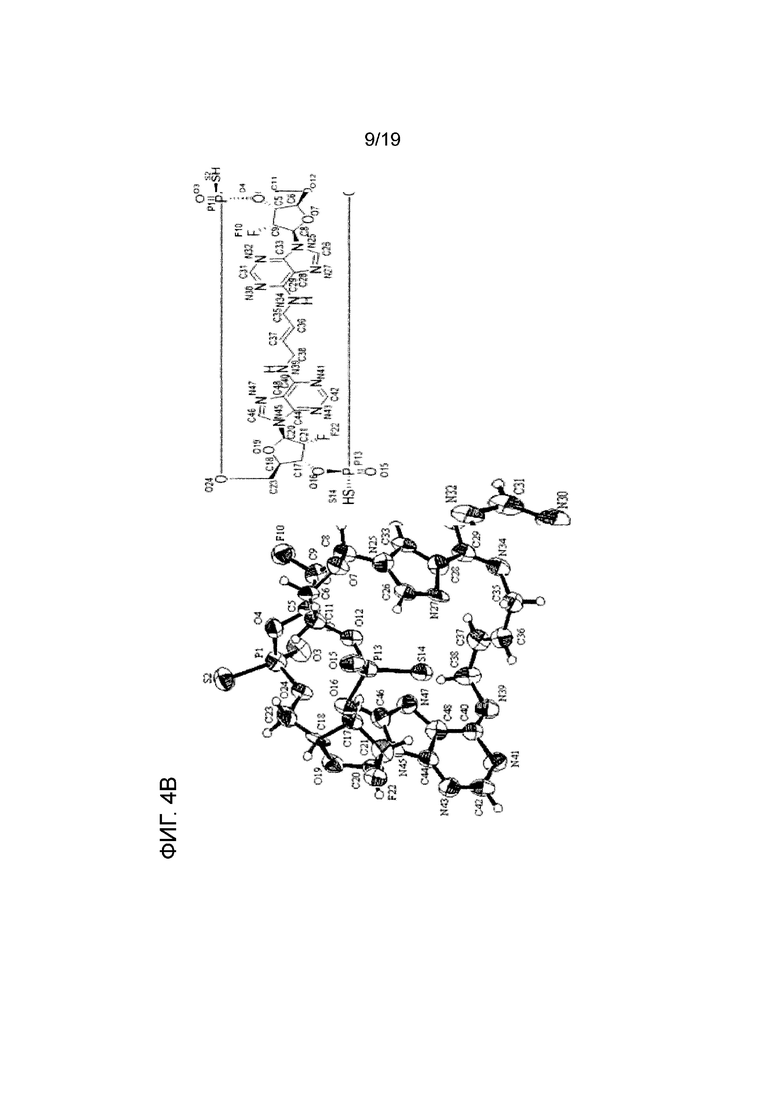
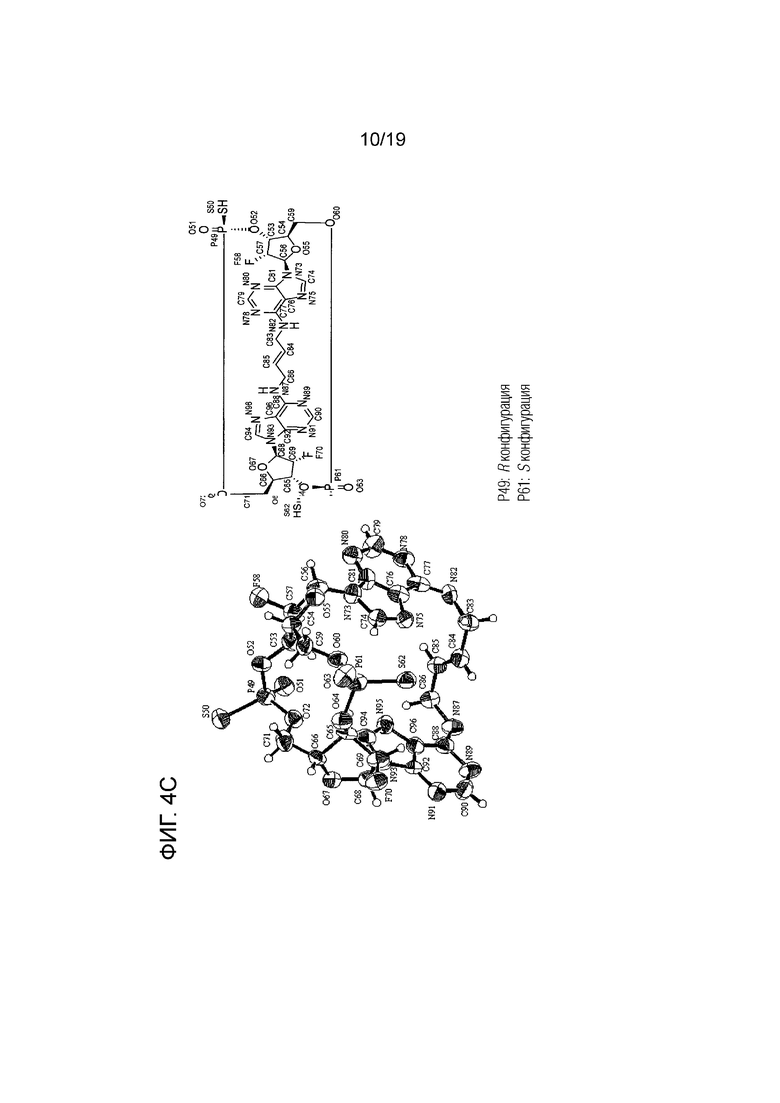
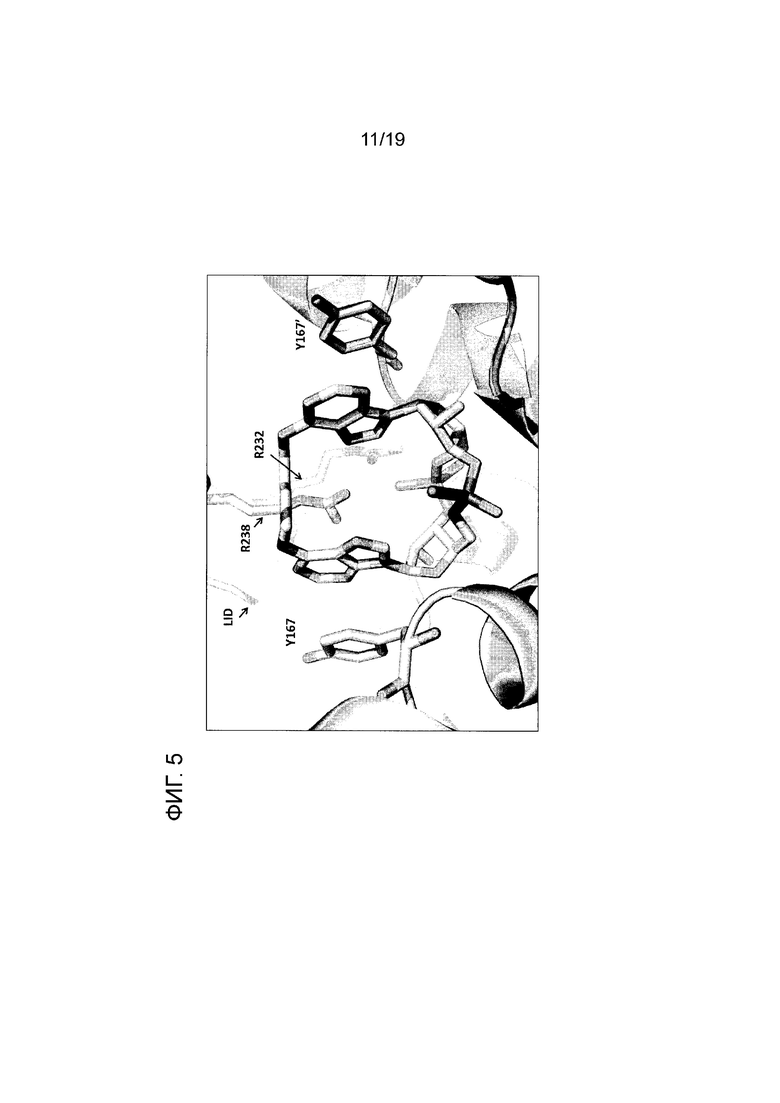


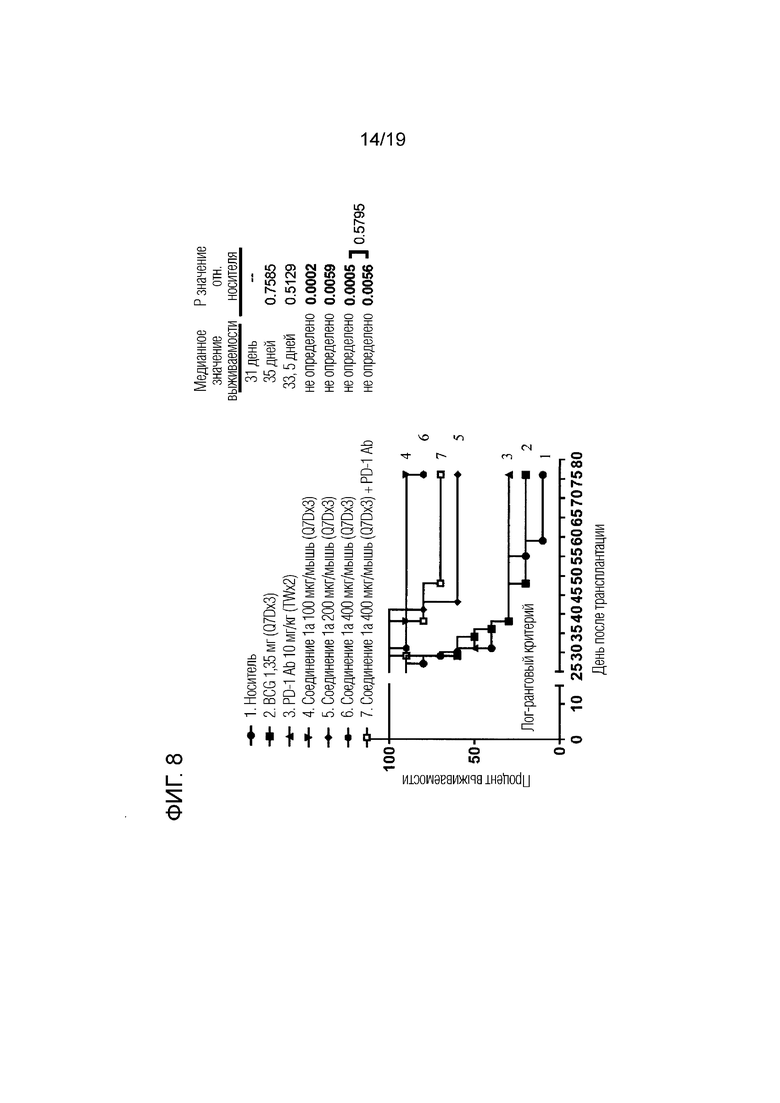
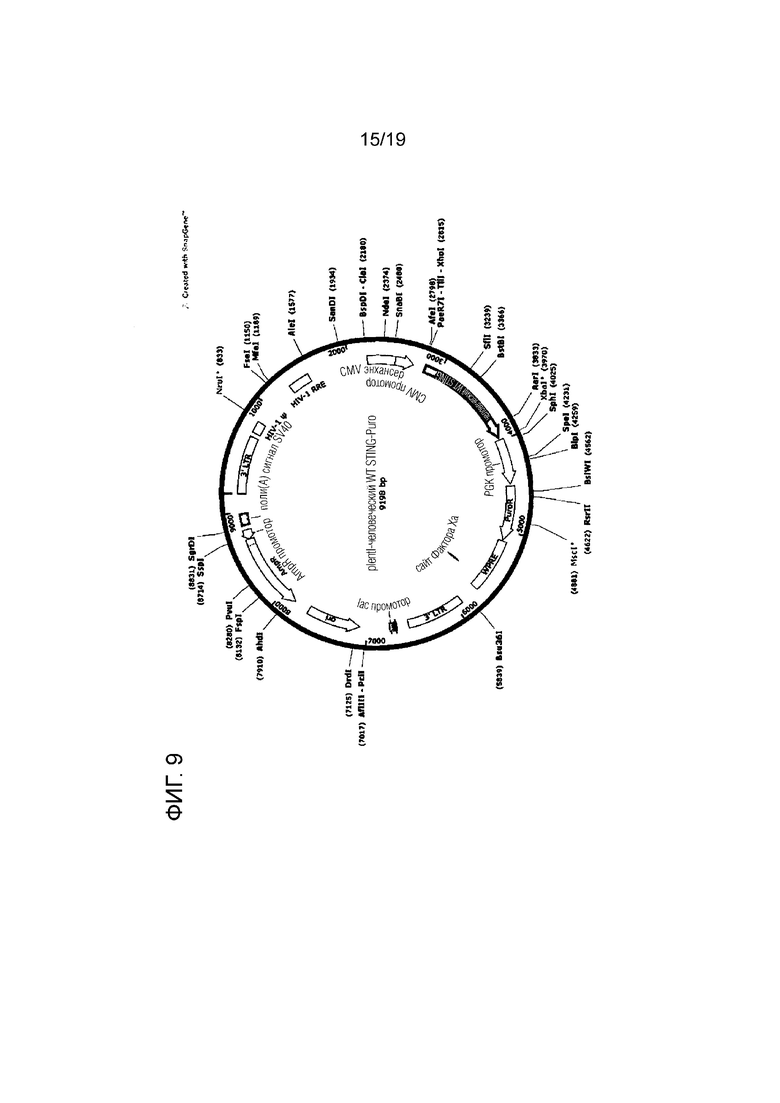

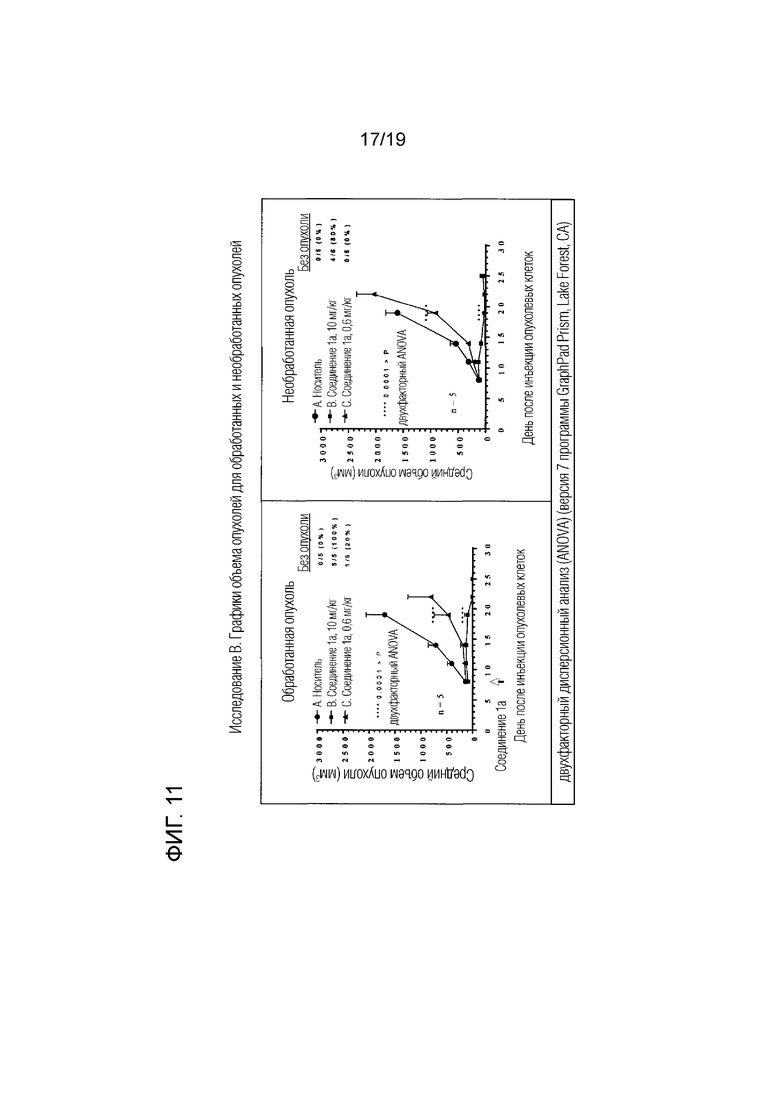
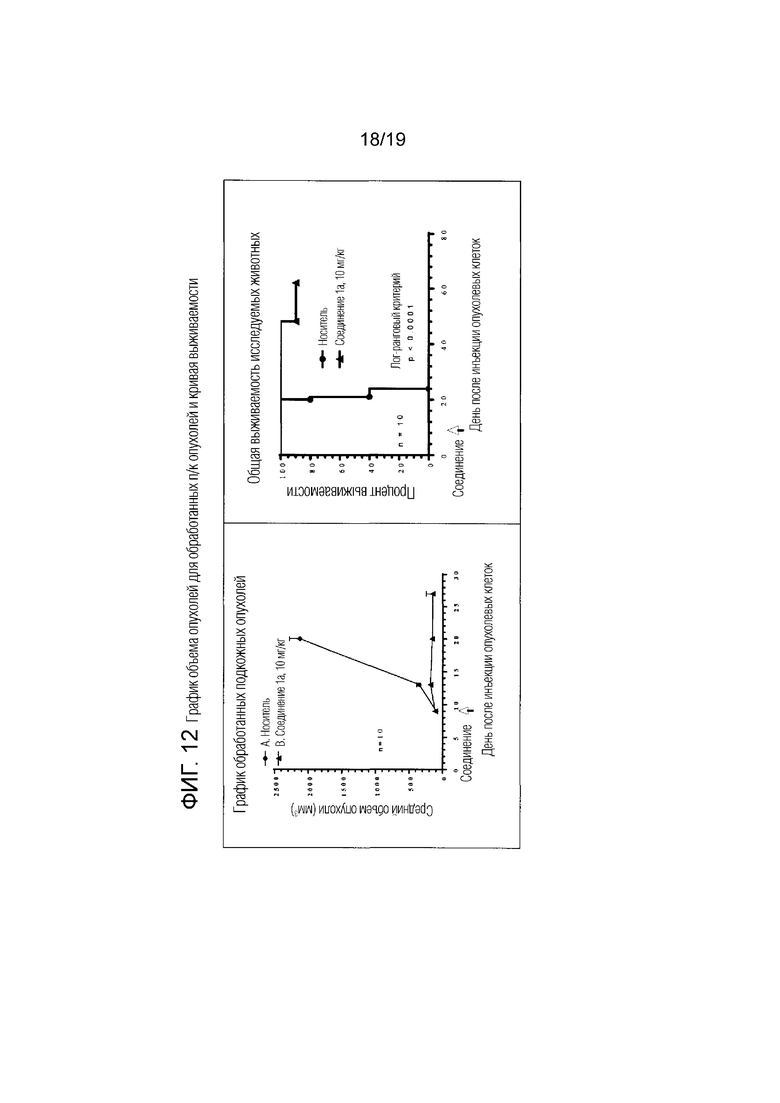
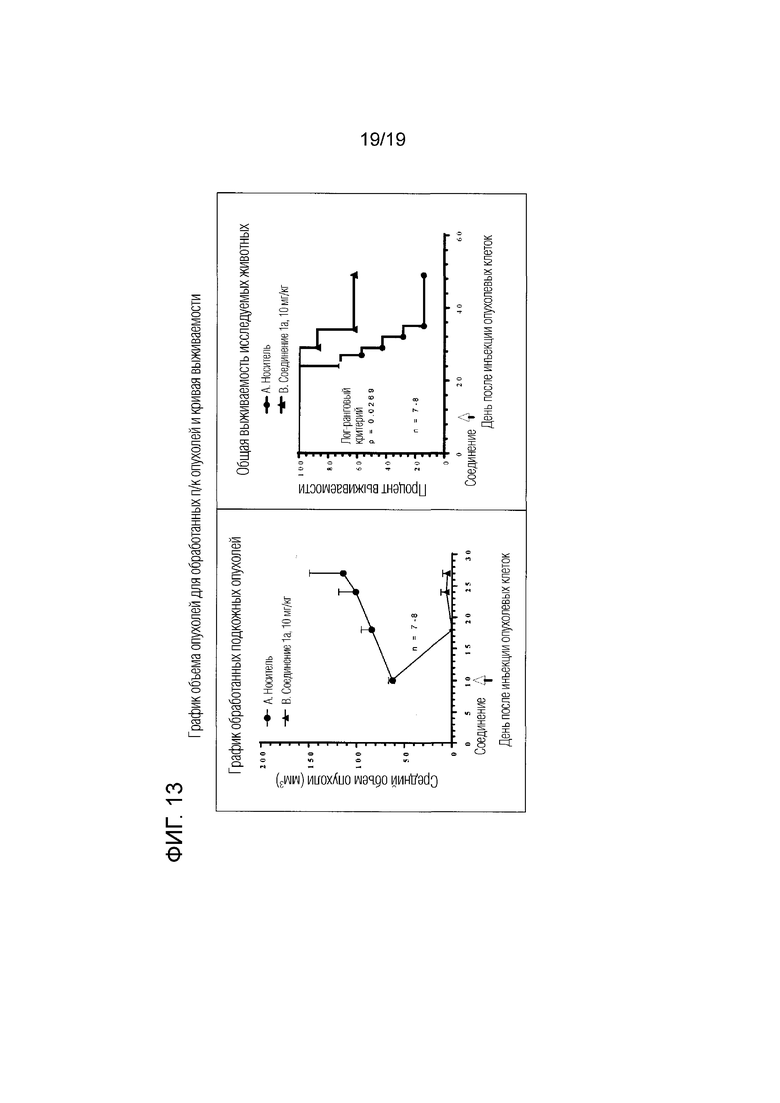
Изобретение относится к способу лечения рака мочевого пузыря, в том числе имеющего аллель REF STING, аллель WT STING, аллель AQ STING или аллель HAQ STING, включающий введение нуждающемуся в лечении пациенту эффективного количества соединения 1 или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль, а также к применению соединения 1 или его фармацевтически приемлемой соли для приготовления фармацевтической композиции для лечения рака мочевого пузыря. Технический результат: лечение рака мочевого пузыря, позволяющее избежать или отсрочить радикальную цистэктомию. 10 н. и 7 з.п. ф-лы, 13 ил., 7 табл., 112 пр.
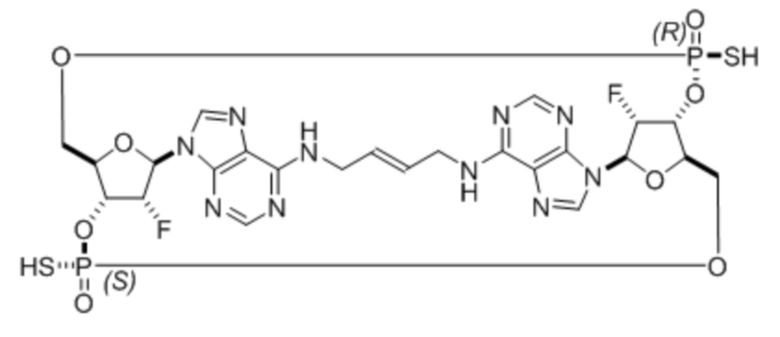
(Соединение 1)
1. Способ лечения рака мочевого пузыря, включающий введение нуждающемуся в лечении пациенту эффективного количества соединения 1 или его фармацевтически приемлемой соли:
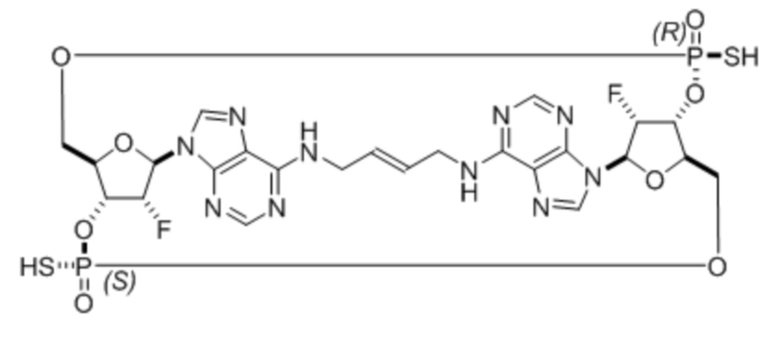
(Соединение 1).
2. Способ по п.1, в котором указанная фармацевтически приемлемая соль представляет собой соль диаммония.
3. Способ лечения рака мочевого пузыря, включающий введение нуждающемуся в лечении пациенту эффективного количества фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль:
(Соединение 1)
и фармацевтически приемлемый наполнитель.
4. Способ лечения рака мочевого пузыря, включающий:
идентификацию индивидуума, у которого рак мочевого пузыря поддается лечению с помощью соединения
(Соединение 1),
фармацевтически приемлемой соли соединения 1 или фармацевтической композиции, содержащей соединение 1 или фармацевтически приемлемую соль соединения 1; и
введение указанному индивидууму эффективного количества этого соединения, фармацевтически приемлемой соли или фармацевтической композиции, с помощью которых рак мочевого пузыря был идентифицирован как поддающийся лечению.
5. Способ по п.4, в котором указанный индивидуум идентифицирован как имеющий рак мочевого пузыря, поддающийся лечению с помощью Соединения 1, фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1, по наличию у пациента аллеля варианта REF STING.
6. Способ лечения рака мочевого пузыря у пациента, имеющего аллель REF STING, включающий введение указанному пациенту эффективного количества соединения
(Соединение 1),
фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1.
7. Способ лечения рака мочевого пузыря у пациента, имеющего аллель WT STING, включающий введение указанному пациенту эффективного количества соединения
(Соединение 1),
фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1.
8. Способ лечения рака мочевого пузыря у пациента, имеющего аллель AQ STING, включающий введение указанному пациенту эффективного количества соединения
(Соединение 1),
фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1.
9. Способ лечения рака мочевого пузыря у пациента, имеющего аллель HAQ STING, включающий введение указанному пациенту эффективного количества соединения
(Соединение 1),
фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1.
10. Способ по любому из пп.1-9, где введение Соединения 1, фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1, выполняют внутрипузырно.
11. Способ по любому из пп.1-9, где рак мочевого пузыря представляет собой немышечно-инвазивный рак мочевого пузыря.
12. Способ по любому из пп.1-11, где введение Соединения 1, фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей любое из вышеперечисленного, выполняют внутрипузырно.
13. Применение соединения 1 или его фармацевтически приемлемой соли для приготовления фармацевтической композиции для лечения рака мочевого пузыря:
(Соединение 1).
14. Применение Соединения 1, его фармацевтически приемлемой соли или фармацевтической композиции, содержащей Соединение 1 или его фармацевтически приемлемую соль, при лечении рака мочевого пузыря:
(Соединение 1).
15. Применение по п.13 или 14, где введение Соединения 1, фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей Соединение 1 или фармацевтически приемлемую соль Соединения 1, выполняют внутрипузырно.
16. Применение по п.13 или 14, где рак мочевого пузыря представляет собой немышечно-инвазивный рак мочевого пузыря.
17. Применение по любому из пп.13-16, где введение Соединения 1, фармацевтически приемлемой соли Соединения 1 или фармацевтической композиции, содержащей любое из вышеперечисленного, выполняют внутрипузырно.
| WO 2018152450 A1, 23.08.2018 | |||
| WO 2016120305 A1, 04.08.2016 | |||
| WO 2017123657 A1, 20.07.2017 | |||
| ПОДВИЖНОЙ ВОЗВЫШЕННЫЙ КОНТРРЕЛЬС ТУПОЙ КРЕСТОВИНЫ | 1928 |
|
SU29856A1 |

