ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям 3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона или их фармацевтически приемлемым солям, которые действуют как ингибиторы МЕК и могут применяться для лечения аномального роста клеток, такого как рак, у пациентов. Настоящее изобретение также относится к фармацевтическим композициям, содержащим эти соединения, и к способам применения этих соединений и композиций при лечении аномального роста клеток, такого как рак, у субъекта, нуждающегося в этом. Настоящее изобретение также относится к твердым формам 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, к фармацевтическим композициям, содержащим эти твердые формы, и к способам применения этих твердых форм и их композиций при лечении аномального роста клеток, такого как рак, у субъекта, нуждающегося в этом.
УРОВЕНЬ ТЕХНИКИ
Киназа МЕК (киназа митоген-активируемой протеинкиназы (МАРКК)) является важным компонентом пути выживания клеток Ras-RAF-MEK-ERK. Путь Ras активируется путем связывания факторов роста, цитокинов и гормонов с родственными им рецепторами. Однако в раковых клетках этот путь конститутивно активируется и приводит к увеличению выживаемости раковых клеток, пролиферации клеток, ангиогенезу и метастазированию. Опухоли, демонстрирующие конститутивную активацию этого пути, включают, но этим не ограничивая, опухоли толстой кишки, поджелудочной железы, молочной железы, головного мозга, яичников, легких и кожи. Активация Ras (обусловленная передачей сигналов в восходящем направлении или в результате активации точечных мутаций в онкогене Ras) приводит к фосфорилированию и активации киназы Raf, которая, в свою очередь, фосфорилирует и активирует МЕК1 и МЕК2 (называемые также МАРКК1 и МАРКК2). МЕК1 и МЕК2 представляют собой киназы двойной специфичности, которые активируют ERK1 и ERK2 путем фосфорилирования и активации киназы ERK1/2 (называемой также МАР-киназой), которая дополнительно фосфорилирует и регулирует функцию таких белков, как Mcl-1, Bim и Bad, играющих активную роль в выживании клеток и апоптозе. Следовательно, активация этого опосредованного фосфорилированием каскада реакций приводит к усилению клеточной пролиферации, повышению выживаемости клеток и снижению гибели клеток, что необходимо для инициации и поддержания онкогенного фенотипа. Известно, что ингибирование этого пути, в частности, ингибирование активности МЕК, позволяет достигать положительного эффекта при лечении гиперпролиферативных заболеваний. Ингибиторы МЕК проявляли различную степень активности в нескольких случаях, в том числе при меланоме с мутацией BRAF V600, меланоме с мутацией NRAS, серозном раке яичников с низкой степенью злокачественности, плексиформных нейрофибромах, раке щитовидной железы и глиомах с низкой степенью злокачественности, с более ограниченными ответами при KRAS-мутантном раке поджелудочной железы или раке легких.
Известно, что типы рака, в случае которых часто происходит метастазирование в головной мозг, например, меланома и немелкоклеточный рак легких, несут изменения, активирующие путь МАРК, такие как мутация BRAF V600E и KRAS G12 (Cancer Genome Atlas N., Cell 2015; 161: 1681 - 96). Несмотря на то, что активирующие мутации могут возникать на разных уровнях канонического пути, тем не менее, все они требуют передачи сигнала через митогенную/внеклеточную сигнал-регулируемую киназу (МЕК) для увеличения пролиферации и выживаемости (Schubbert S, Shannon К, Bollag G., Nat Rev Cancer. 2007; 7:295-308). Общая активация пути МАРК при злокачественных новообразованиях, а также центральное расположение МЕК и в нисходящем направлении, вызывают потенциальный интерес к применению ингибиторов МЕК для лечения внутричерепных опухолей.
Гематоэнцефалический интерфейс включает эндотелий церебральных микрососудов, образующий гематоэнцефалический барьер (ВВВ), и эпителий хориоидальных сплетений, образующий гематоликворный барьер (BCSFB). Гематоэнцефалический барьер (ВВВ) представляет собой высокоселективный физический, транспортный и метаболический барьер, отделяющий центральную нервную систему (CNS) от крови. ВВВ может препятствовать проникновению конкретных лекарственных средств в ткани головного мозга и, следовательно, является ограничивающим фактором при доставке многих периферически вводимых средств в CNS. Эффективность действия многих молекулярно-направленных средств при опухолях центральной нервной системы ограничена проникновением через гематоэнцефалический барьер (ВВВ), который состоит из монослоя эндотелиальных клеток, соединенных плотными контактами, которые служат физическим барьером, защищающим мозг. Кроме того, эти эндотелиальные клетки экспрессируют множество эффлюксных переносчиков, включающих Р-гликопротеин (Р-gp) и белок резистентности рака молочной железы (BCRP), которые, как известно, выводят из головного мозга многие противораковые средства (Ohtsuki and Terasaki, 2007, Pharm Res 24:1745-1758; Agarwal et al., 2011, Pharm Res 24:1745-1758). Подобно гематоэнцефалическому барьеру, гематоликворный барьер предотвращает проникновение большинства переносимых с кровью веществ в головной мозг, при этом обеспечивая селективное проникновение конкретных веществ в головной мозг и способствуя выведению мозговых метаболитов и продуктов метаболизма головного мозга в кровь.
Поэтому, все еще сохраняется потребность в создании лекарственных препаратов для лечения опухолей, опосредованных МЕК, в том числе лекарственных препаратов, которые способны проникать через ВВВ и/или BCSFB и воздействовать на опухоли в CNS.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В изобретении предлагаются, помимо всего прочего, соединения формулы I и формулы II и их фармацевтически приемлемые соли. Такие соединения могут ингибировать активность МЕК, воздействуя, вследствие этого, на биологические функции, и могут применяться для лечения субъекта, имеющего МЕК-ассоциированную опухоль. Кроме того, в изобретении предлагаются твердые формы 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Также предлагаются фармацевтические композиции и лекарственные препараты, включающие соединения любой из описанных в изобретении формул и их фармацевтически приемлемые соли, которые могут применяться для лечения субъекта, имеющего МЕК-ассоциированную опухоль, в форме монотерапии или в комбинации с дополнительными противоопухолевыми терапиями. Кроме того, в изобретении предлагаются способы получения соединений, их фармацевтически приемлемых солей и фармацевтических композиций любой из описанных в изобретении формул и их фармацевтически приемлемых солей, и способы их применения. Это краткое изложение сущности изобретения предложено для ознакомления в упрощенной форме с выбором концепций, которые далее описаны более подробно. Предполагается, что это краткое изложение не определяет ключевые признаки или основные признаки заявленного предмета изобретения, и также предполагается, что оно не используется отдельно в качестве вспомогательного средства при определении объема заявленного предмета изобретения.
В соответствии с вариантом осуществления изобретения, предлагается соединение формулы I
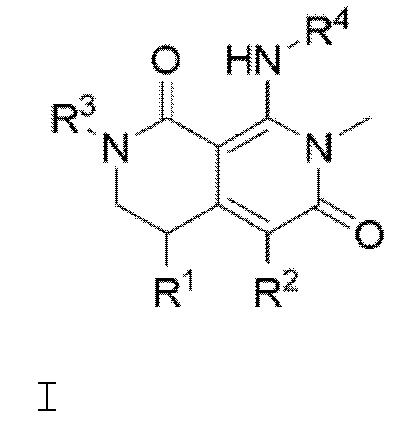
или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
Кроме того, в изобретении предлагается соединение формулы II

или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, C1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления, в изобретении предлагаются твердые формы 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона.
В одном варианте осуществления, в изобретении предлагается фармацевтическая композиция, включающая соединение любой из описанных в изобретении формул или его фармацевтически приемлемая соль и фармацевтически приемлемый носитель или вспомогательное вещество. В некоторых вариантах осуществления, фармацевтическая композиция включает два или более фармацевтически приемлемых носителя и/или вспомогательных веществ.
В одном варианте осуществления, в изобретении предлагаются терапевтические способы и применения, включающие введение соединения любой из описанных в изобретении формул или его фармацевтически приемлемой соли субъекту.
В одном варианте осуществления, в изобретении предлагается способ лечения аномального роста клеток, например, опухоли, например, МЕК-ассоциированной опухоли, у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения любой из описанных в изобретении формул или его фармацевтически приемлемой соли. Соединения любой из описанных в изобретении формул могут быть введены в форме монотерапии или могут быть введены в комбинации с одним или более противораковыми препаратами.
В одном варианте осуществления, в изобретении предлагается способ лечения аномального роста клеток, например, опухоли, например МЕК-ассоциированной опухоли, у субъекта, нуждающегося в этом, включающий введение субъекту количества соединения любой из описанных в изобретении формул или его фармацевтически приемлемой соли в комбинации с количеством дополнительного противоракового средства, где вместе взятые количества являются эффективными при лечении указанного аномального роста клеток.
В одном варианте осуществления, в изобретении предлагается соединение любой из описанных в изобретении формул или его фармацевтически приемлемая соль для применения в качестве лекарственного препарата.
В одном варианте осуществления, в изобретении предлагается соединение любой из описанных в изобретении формул или его фармацевтически приемлемая соль для применения при лечении аномального роста клеток, например, опухоли, например, МЕК-ассоциированной опухоли.
В одном варианте осуществления, в изобретении предлагается применение соединения любой из описанных в изобретении формул или его фармацевтически приемлемой соли для производства лекарственного препарата для лечения аномального роста клеток, например, опухоли, например, МЕК-ассоциированной опухоли, у субъекта.
В одном варианте осуществления, в изобретении предлагается фармацевтическая композиция, включающая соединение любой из описанных в изобретении формул или его фармацевтически приемлемую соль и, по меньшей мере, один фармацевтически приемлемый носитель или вспомогательное вещество.
Каждый из вариантов осуществления соединений любой из описанных в изобретении формул может быть объединен с одним или более другими вариантами осуществления соединений любой из описанных в изобретении формул, не противоречащими варианту (вариантам) осуществления, с которым их объединяют.
Следует иметь в виду, что как предшествующее общее описание, так и следующее далее подробное описание приводятся только в качестве примера и разъяснения, и они никоим образом не ограничивают изобретение, заявленное в пунктах формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 приведена порошковая рентгенограмма кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона, форма 1.
На фигуре 2 приведена порошковая рентгенограмма кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона, форма 2.
На фигуре 3 приведена порошковая рентгенограмма кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3.
На фигуре 4 приведена порошковая рентгенограмма аморфного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона, форма 4.
На фигуре 5 приведена изотерма сорбции кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона, формы 3.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте, в изобретении предлагается соединение формулы I

или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
Используемые в изобретении в отношении заместителей формы единственного числа включают и формы множественного числа, если не указано иное. Например, "заместитель" включает один или более заместителей.
В случае используемых в изобретении сложных химических названий, замещающую группу обычно называют перед группой, к которой она присоединяется. Например, метоксиэтил включает основную цепь этила с заместителем метокси.
Термин "галоген" обозначает -F (иногда называемый в изобретении "фтором" или "атомами фтора"), -Cl, -Br и -I.
Используемый в изобретении термин "C1-С6 алкил" относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу, содержащему от одного до шести углеродных атомов. Примеры алкильных групп включают, но этим не ограничивая, метил, этил, 1-пропил, изопропил, 1-бутил, изобутил, вторбутил, третбутил, 2-метил-2-пропил, пентил, неопентил и гексил.
Используемый в изобретении термин "гидроксиС1-С6 алкил-" относится к определяемому в изобретении С1-С6 алкильному радикалу, в котором один из атомов водорода замещен гидроксильной группой.
Термин "гидрокси" относится к группе -ОН.
Термин "С3-С6 циклоалкил" обозначает полностью насыщенное карбоциклическое кольцо, имеющее 3-6 кольцевых углеродных атомов. Примеры включают циклопропил, циклобутил, циклопентил и циклогексил.
Используемый в изобретении термин "фторС1-С6 алкил" относится к определяемому в изобретении С1-С6 алкильному радикалу, в котором один, два или три атомов водорода замещены с помощью одного, двух или трех атомов фтора, соответственно. Примеры включают, но этим не ограничивая, фторметил, дифторметил, трифторметил, 2-фторэтил, 2,2-дифторэтил и 2,2,2-трифторэтил.
Используемый в изобретении термин "C1-С6 алкокси" относится к определяемому в изобретении С1-С6 алкильному радикалу, который соединен одинарной связью с атомом кислорода, где радикал находится на атоме кислорода (то есть С1-С6-O-). Примеры алкоксильных групп включают, но этим не ограничивая, метокси, этокси, пропокси и изопропокси.
Используемый в изобретении термин "фторС1-С6 алкокси" относится к определяемому в изобретении С1-С6 алкокси, в котором один, два или три атома водорода замещены с помощью одного, двух или трех атомов фтора, соответственно. Пример включает, но этим не ограничивая, трифторметокси.
Термин "(С3-С6 циклоалкил)С1-С6 алкокси-" относится к определяемому в изобретении С1-С6 алкокси-, в котором один из атомов водорода замещен с помощью определяемой в изобретении С3-С6 циклоалкильной группы.
Используемый в изобретении термин "C1-С6 алкилтио" относится к радикалу (C1-С6 алкил)S-, в котором C1-С6 алкильный фрагмент определяется в изобретении.
Используемый в изобретении термин "фторС1-С6 алкилтио" относится к определяемой в изобретении группе C1-С6 алкилтио, в которой один, два или три атома водорода замещены с помощью одного, двух или трех атомов фтора, соответственно.
В одном варианте осуществления формулы I, R1 представляет собой Н.
В одном варианте осуществления формулы I, R1 представляет собой Br.
В одном варианте осуществления формулы I, R1 представляет собой C1-С6 алкил. В одном варианте осуществления формулы I, R1 представляет собой метил.
В одном варианте осуществления формулы I, R1 представляет собой фенил.
В одном варианте осуществления формулы I, R2 представляет собой Н.
В одном варианте осуществления, R2 представляет собой галоген.
В одном варианте осуществления формулы I, R2 представляет собой F.
В одном варианте осуществления формулы I, R2 представляет собой Cl.
В одном варианте осуществления формулы I, R2 представляет собой Br.
В одном варианте осуществления формулы I, R2 представляет собой I.
В одном варианте осуществления формулы I, R2 представляет собой СН3-.
В одном варианте осуществления формулы I, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы I, R3 представляет собой Н.
В одном варианте осуществления формулы I, R3 представляет собой гидроксиС1-С6 алкил-. Неограничивающие примеры включают 2-гидроксиэтил.
В одном варианте осуществления формулы I, R3 представляет собой гидроксиС1-С6 алкокси-. Неограничивающие примеры включают 2-гидроксиэтокси и 2-гидроксипропокси, имеющие структуры:

соответственно.
В одном варианте осуществления формулы I, R3 представляет собой С1-С6 алкокси. Неограничивающие примеры включают метокси, этокси, 1-метилэтокси и 2,2-диметилэтокси.
В одном варианте осуществления формулы I, R3 представляет собой фторС1-С6 алкокси. Неограничивающий пример включает 2,2-дифторэтокси.
В одном варианте осуществления формулы I, R3 представляет собой С3-С6 циклоалкил. Неограничивающий пример представляет собой циклопропил.
В одном варианте осуществления формулы I, R3 представляет собой (С3-С6 циклоалкил)С1-С6 алкокси-. Неограничивающий пример представляет собой циклопропилметокси.
В одном варианте осуществления формулы I, R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из фтора, хлора, брома, йода, этила, пропила, изопропила, метилтио, дифторметилтио, трифторметила, метокси, дифторметокси, циклопропила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы I, R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы I, R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из фтора, хлора, брома, йода, этила, пропила, изопропила, метилтио, дифторметилтио, трифторметила, метокси, дифторметокси, циклопропила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы I, R4 представляет собой фенил, замещенный с помощью 1 заместителя, выбранного из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы I, R4 представляет собой фенил, замещенный с помощью 1 заместителя, выбранного из фтора, хлора, брома, йода, этила, пропила, изопропила, метилтио, дифторметилтио, трифторметила, метокси, дифторметокси, циклопропила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы I, R4 выбирают из структур:

В одном варианте осуществления формулы I, R4 представляет собой
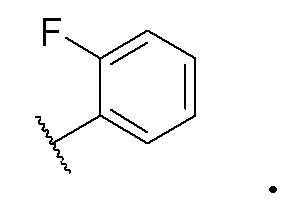
В одном варианте осуществления формулы I, R4 представляет собой
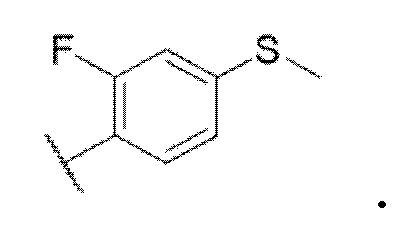
В одном варианте осуществления формулы I, R4 выбирают из структур:

В одном варианте осуществления формулы I, R4 выбирают из структур:

В одном варианте осуществления, R4 имеет структуру:

где Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-. В одном варианте осуществления, где Ra представляет собой галоген. В одном варианте осуществления, Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио или фторС1-С6 алкокси. В одном варианте осуществления, где Ra представляет собой галоген и Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио или фторС1-С6 алкокси.
В одном варианте осуществления, в изобретении предлагается соединение формулы II:

или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы II, R1 представляет собой Н.
В одном варианте осуществления формулы II, R1 представляет собой Br.
В одном варианте осуществления формулы II, R1 представляет собой С1-С6 алкил. В одном варианте осуществления формулы II, R1 представляет собой метил.
В одном варианте осуществления формулы II, R1 представляет собой фенил.
В одном варианте осуществления формулы II, R2 представляет собой Н.
В одном варианте осуществления формулы II, R2 представляет собой галоген.
В одном варианте осуществления формулы II, R2 представляет собой F.
В одном варианте осуществления формулы II, R2 представляет собой Cl.
В одном варианте осуществления формулы II, R2 представляет собой Br.
В одном варианте осуществления формулы II, R2 представляет собой I.
В одном варианте осуществления формулы II, R2 представляет собой СН3-.
В одном варианте осуществления формулы II, R2 представляет собой Н или СН3-.
В одном варианте осуществления формулы II, R1 представляет собой Н, и R2 представляет собой Н или СН3-.
В одном варианте осуществления формулы II, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, R3 представляет собой Н.
В одном варианте осуществления формулы II, R3 представляет собой гидроксиС1-С6 алкил-. Неограничивающие примеры включают 2-гидроксиэтил.
В одном варианте осуществления формулы II, R3 представляет собой гидроксиС1-С6 алкокси-. Неограничивающие примеры включают 2-гидроксиэтокси и 2-гидроксипропокси, имеющие структуры:

соответственно.
В одном варианте осуществления формулы II, R3 представляет собой C1-С6 алкокси. Неограничивающие примеры включают метокси, этокси, 1-метилэтокси и 2,2-диметилэтокси.
В одном варианте осуществления формулы II, R3 представляет собой фторС1-С6 алкокси. Неограничивающий пример включает 2,2-дифторэтокси.
В одном варианте осуществления формулы II, R3 представляет собой С3-С6 циклоалкил. Неограничивающий пример представляет собой циклопропил.
В одном варианте осуществления формулы II, R3 представляет собой (С3-С6 циклоалкил)С1-С6 алкокси-. Неограничивающий пример представляет собой циклопропилметокси.
В одном варианте осуществления формулы II, R3 представляет собой Н или гидроксиС1-С6 алкокси-.
В одном варианте осуществления формулы II, Ra представляет собой галоген. В одном варианте осуществления формулы II, Ra представляет собой фтор или хлор. В одном варианте осуществления формулы II, Ra представляет собой фтор.
В одном варианте осуществления формулы II, Rb представляет собой фтор, хлор, бром, йод, этил, пропил, изопропил, метилтио, дифторметилтио, трифторметил, метокси, дифторметокси, циклопропил или СН3С(=O)-.
В одном варианте осуществления формулы II, Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио, или фторС1-С6 алкокси.
В одном варианте осуществления формулы II, Rb представляет собой бром, йод, этил, метилтио или дифторметокси. В одном варианте осуществления формулы II, Rb представляет собой метилтио.
В одном варианте осуществления формулы II, Ra представляет собой фтор, и Rb представляет собой метилтио.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкил, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио или фторС1-С6 алкокси.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой галоген. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой галоген, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой С1-С6 алкил. В одном варианте осуществления формулы II, Ra представляет собой галоген,
Rb представляет собой C1-С6 алкил, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой С1-С6 алкилтио. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой С1-С6 алкилтио, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой фторС1-С6 алкилтио. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой фторС1-С6 алкилтио, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой фтор, Rb представляет собой метилтио, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой фторС1-С6 алкил. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой фторС1-С6 алкил, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой С1-С6 алкокси. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой С1-С6 алкокси, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой фторС1-С6 алкокси. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой фторС1-С6 алкокси, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой С3-С6 циклоалкил. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой С3-С6 циклоалкил, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, Ra представляет собой галоген, и Rb представляет собой С1-С6 алкил-С(=O)-. В одном варианте осуществления формулы II, Ra представляет собой галоген, Rb представляет собой С1-С6 алкил-С(=O)-, R1 представляет собой Н, и R2 представляет собой Н.
В одном варианте осуществления формулы II, R1 представляет собой Н, R2 представляет собой Н или СН3-, R3 представляет собой Н или гидроксиС1-С6 алкокси-, Ra представляет собой галоген, и Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкил, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или С1-С6 алкил-С(=O)-.
В одном варианте осуществления формулы II, R1 представляет собой Н, R2 представляет собой Н или СН3-, R3 представляет собой Н или гидроксиС1-С6 алкокси-, Ra представляет собой галоген, и Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио или фторС1-С6 алкокси.
В одном варианте осуществления любого из описанных выше вариантов осуществления формулы II, группу

выбирают из структур:


Подразумевается, что используемый в изобретении термин "соединение" включает все стереоизомеры, геометрические изомеры, таутомеры и изотопы изображенных структур. Предполагается, что соединения, идентифицированные в изобретении путем использования названия или структуры одной конкретной таутомерной формы, включают и другие таутомерные формы, если не указано иное.
Соединения приведенных в изобретении формул могут иметь асимметричные углеродные атомы. Связи углерод-углерод в соединениях по изобретению могут быть изображены в изобретении с использованием сплошной линии  сплошной клиновидной линии
сплошной клиновидной линии  или пунктирной клиновидной линии
или пунктирной клиновидной линии  Подразумевается, что использование сплошной линии для изображения связей с асимметричными углеродными атомами указывает, что в случае данного углеродного атома включены все возможные стереоизомеры (например, индивидуальные энантиомеры, рацемические смеси, и так далее). Подразумевается, что использование сплошной или пунктирной линии в форме клина для изображения связей с асимметричными атомами углерода означает, что должен быть включен только показанный стереоизомер. Но не исключено, что соединения по изобретению могут содержать более одного асимметрического атома углерода. Предполагается, что в этих соединениях использование сплошной линии для изображения связей с асимметричными углеродными атомами указывает, что должны быть включены все возможные стереоизомеры и присоединенный стереоцентр. Например, если не указано иное, то предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Предполагается, что использование сплошной линии для изображения связей с одним или несколькими асимметричными углеродными атомами в соединении по изобретению и использование сплошной или пунктирной линии в форме клина для изображения связей с другими асимметричными углеродными атомами в том же соединении указывает, что присутствует смесь диастереомеров.
Подразумевается, что использование сплошной линии для изображения связей с асимметричными углеродными атомами указывает, что в случае данного углеродного атома включены все возможные стереоизомеры (например, индивидуальные энантиомеры, рацемические смеси, и так далее). Подразумевается, что использование сплошной или пунктирной линии в форме клина для изображения связей с асимметричными атомами углерода означает, что должен быть включен только показанный стереоизомер. Но не исключено, что соединения по изобретению могут содержать более одного асимметрического атома углерода. Предполагается, что в этих соединениях использование сплошной линии для изображения связей с асимметричными углеродными атомами указывает, что должны быть включены все возможные стереоизомеры и присоединенный стереоцентр. Например, если не указано иное, то предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Предполагается, что использование сплошной линии для изображения связей с одним или несколькими асимметричными углеродными атомами в соединении по изобретению и использование сплошной или пунктирной линии в форме клина для изображения связей с другими асимметричными углеродными атомами в том же соединении указывает, что присутствует смесь диастереомеров.
Соединения по изобретению, которые имеют хиральные центры, могут существовать в форме стереоизомеров, таких как рацематы, энантиомеры или диастереомеры.
Стереоизомеры соединений приведенных в изобретении формул могут включать цис- и транс-изомеры, оптические изомеры, такие как (R) и (S) энантиомеры, диастереомеры, геометрические изомеры, поворотные изомеры, атропоизомеры, конформационные изомеры и таутомеры соединений по изобретению, включая соединения, проявляющие более чем один тип изомеризма, и их смеси (такие как рацематы и диастереоизомерные пары).
Кроме того, в изобретение включены соли присоединения кислоты или соли присоединения основания, в которых противоион является оптически активным, например, d-лактат или 1-лизин, или рацемическим, например, dl-тартрат или dl-аргинин.
При кристаллизации любого рацемата возможно образование кристаллов двух разных типов. Первый тип представляет собой упомянутое выше рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, в котором образуются две формы кристаллов в эквимолярных количествах, каждая из которых содержит один энантиомер.
Традиционные методы получения/выделения индивидуальных энантиомеров включают хиральный синтез из соответствующего оптически чистого прекурсора или разделение рацемата (или рацемата соли или рацемата производного) используя, например, хиральную высокоэффективную жидкостную хроматографию (HPLC) или сверхкритическую флюидную хроматографию (SFC).
В качестве варианта, рацемат (или рацемический прекурсор) может быть подвергнут реакции с соответствующим оптически активным соединением, например, со спиртом, или, в случае, когда соединение содержит фрагмент с кислотными или основными свойствами, с кислотой или основанием, такими как винная кислота или 1-фенилэтиламин. Полученная диастереоизомерная смесь может быть разделена методом хроматографии и/или фракционной кристаллизации, и один или оба диастереоизомеров превращены в соответствующий чистый энантиомер (энантиомеры) хорошо известными специалистам в данной области методами.
Хиральные соединения по изобретению (и их хиральные прекурсоры) могут быть получены в энантиомерно обогащенной форме, используя хроматографию, обычно HPLC, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно, от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюента дает обогащенную смесь.
Стереоизомерные конгломераты могут быть разделены известными специалистам в данной области методами; смотрите, например, монографию "Stereochemistry of Organic Соединения" by Е л Eliel (Wiley, New York, 1994), полное содержание которой включено в настоящее изобретение путем ссылки на нее.
Энантиомерная чистота описанных в изобретении соединений может быть охарактеризована показателем энантиомерного избытка (ее), который указывает степень, в которой образец содержит один энантиомер в больших количествах, чем другой. Рацемическая смесь имеет показатель ее, равный 0%, тогда как один практически чистый энантиомер имеет показатель ее, равный 100%. Аналогично, диастереомерная чистота может быть охарактеризована показателем диастереомерного избытка (de).
The соединения по изобретению могут проявлять таутомеризм и структурную изомерию. Например, соединения могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, и кето и енаминную форму, и в виде геометрических изомеров и их смесей. Все такие таутомерные формы включены в объем соединений по изобретению. Таутомеры существуют в форме смесей таутомерных пар в растворе. В твердой форме, обычно преобладает один таутомер. Даже в том случае, если может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений приведенных формул. Таутомер соединения формулы I может возникать, например, когда R3 представляет собой водород, то есть:

Кроме того, некоторые из соединений по изобретению могут образовывать атропоизомеры (например, замещенные биарилы). Атропоизомеры представляют собой конформационные стереоизомеры, которые возникают, когда предотвращено или в значительной степени замедленно вращение вокруг одинарной связи в молекуле в результате стерических взаимодействий с другими частями молекулы и заместители на обоих концах одинарной связи являются асимметричными. Взаимное превращение атропоизомеров происходит достаточно медленно, что позволяет их разделять и выделять при заданных условиях. Энергетический барьер для термической рацемизация может быть определен по стерическому затруднению для свободного вращения одной или более связей, образующих ось хиральности.
Настоящее изобретение также включает фармацевтически приемлемые меченые изотопами соединения, которые являются идентичными соединениям, описанным в одной из приведенных формул, за исключением того, что один или более атомов заменены на атом, имеющий атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно обнаруживаемых в природе.
Меченые изотопами соединения по изобретению могут быть получены, в большинстве случаев, традиционными методами, известными специалистам в данной области, или с помощью процессов, аналогичных процессам, описанным в изобретении, используя соответствующий меченый изотопом реагент вместо немеченого реагента.
Примеры изотопов, которые могут быть введены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как, но этим не ограничивая, 2Н, 3Н, 13С, 14С, 15N, 18О, 17О, 32Р, 35S, 18F и 36CI. Конкретные меченые изотопами соединения по изобретению, например, соединения, в которые введены радиоактивные изотопы, такие как 2Н, 3Н или 14С, могут применяться в одном или обоих исследованиях распределения в тканях лекарственного средства или субстрата. Меченые изотопами трития, то есть 3Н, и углерода 14, то есть 14С, являются особенно предпочтительными вследствие легкости их получения и возможности обнаружения. Кроме того, замещение с помощью более тяжелых изотопов, таких как дейтерий, то есть 2Н, может давать конкретные терапевтические преимущества, обусловленные более высокой метаболической стабильностью, например, увеличение периода полувыведения in vivo или снижение величины требуемой дозы, и, следовательно, может быть предпочтительным при некоторых обстоятельствах. Замещение с помощью позитронно активных изотопов, таких как 11С, 18F, 15О и 13N, может применяться при исследованиях методом позитрон-эмиссионной томографии (PET) для оценки степени занятости рецептора субстратом. Меченые изотопами соединения по изобретению могут быть получены, в большинстве случаев, с использованием методик, описанных ниже в схемах и/или в примерах и синтезах, путем замещения немеченого изотопом реагента на меченый изотопом реагент.
Фармацевтически приемлемые сольваты по изобретению включают сольваты, в которых растворитель для кристаллизации может быть изотопно замещенным, например, D2O, d6-ацетон, d6-DMSO.
Если не указано иное, то все ссылки в изобретении на патентуемые соединения включают в себя ссылки на их соли, сольваты, гидраты и комплексы, и на сольваты, гидраты и комплексы их солей, включая их полиморфы, стереоизомеры и меченые изотопами версии.
Соединения по изобретению могут существовать в форме фармацевтически приемлемых солей, таких как, например, соли присоединения кислоты и соли присоединения основания соединений одной из предлагаемых в изобретении формул. Используемый в изобретении термин "фармацевтически приемлемая соль" относится к тем солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Если не указано иное, то используемая в изобретении фраза "фармацевтически приемлемая соль (соли)" включает соли групп с кислотными или основными свойствами, которые могут присутствовать в соединениях раскрытых в изобретении формул.
Например, соединения по изобретению, которые обладают свойствами основания, способны образовывать разнообразные соли с различными неорганическими и органическими кислотами. Несмотря на то, что такие соли должны быть фармацевтически приемлемыми для введения животным, тем не менее, на практике часто является желательным сначала выделить соединение по настоящему изобретению из реакционной смеси в форме фармацевтически неприемлемой соли и затем просто превратить эту соль обратно в соединение в форме свободного основания путем обработки с помощью щелочного реагента и после этого превратить полученное соединение в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты соединений с основными свойствами по этому изобретению могут быть получены путем обработки соединения с основными свойствами по существу эквивалентным количеством выбранной неорганической или органической кислоты в среде водного растворителя или в соответствующем органическом растворителе, таком как метанол или этанол. После испарения растворителя, получают требуемую твердую соль. Требуемая соль кислоты может быть также осаждена из раствора соединения в форме свободного основания в органическом растворителе путем добавления к раствору соответствующей неорганической или органической кислоты.
Кислоты, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислоты таких соединений с основными свойствами из тех, которые образуют нетоксичные соли присоединения кислоты, представляют собой соли, содержащие фармакологически приемлемые анионы, такие как гидрохлоридные, гидробромидные, гидройодидные, нитратные, сульфатные, бисульфатные, фосфатные, кислые фосфатные, изоникотинатные, ацетатные, лактатные, салицилатные, цитратные, кислые цитратные, тартратные, пантотенатные, битартратные, аскорбатные, сукцинатные, малеатные, гентизинатные, фумаратные, глюконатные, глюкуронатные, сахаратные, формиатные, бензоатные, глутаматные, метансульфонатные, этансульфонатные, бензолсульфонатные, п-толуолсульфонатные и памоатные соли.
Примеры солей включают, но этим не ограничивая, ацетатные, акрилатные, бензолсульфонатные, бензоатные (такие как хлорбензоатные, метилбензоатные, динитробензоатные, гидроксибензоатные и метоксибензоатные), бикарбонатные, бисульфатные, бисульфитные, битартратные, боратные, бромидные, бутин-1,4-диоатные, кальцийэтилендиаминтетраацетатные, камзилатные, карбонатные, хлоридные, капроатные, каприлатные, клавуланатные, цитратные, деканоатные, дигидрохлоридные, дигидрогенфосфатные, этилендиаминтетраацетатные, эдизилатные, эстолатные, эзилатные, этилсукцинатные, формиатные, фумаратные, глюцептатные, глюконатные, глутаматные, гликолатные, гликолиларсанилатные, гептаноатные, гексин-1,6-диоатные, гексилрезорцинатные, гидрабаминные, гидробромидные, гидрохлоридные, гидроксибутиратные, йодидные, изобутиратные, изотионатные, лактатные, лактобионатные, лауратные, малатные, малеатные, малонатные, манделатные, мезилатные, метафосфатные, метансульфонатные, метилсульфатные, моногидрогенфосфатные, мукатные, напсилатные, нафталин-1-сульфонатные, нафталин-2-сульфонатные, нитратные, олеатные, оксалатные, памоатные (эмбонатные), пальмитатные, пантотенатные, фенилацетатные, фенилбутиратные, фенилпропионатные, фталатные, фосфатные/дифосфатные, полигалактуронатные, пропансульфонатные, пропионатные, пропиолатные, пирофосфатные, пиросульфатные, салицилатные, стеаратные, субацетатные, субератные, сукцинатные, сульфатные, сульфонатные, сульфитные, таннатные, тартратные, теоклатные, тозилатные и валератные соли.
Иллюстративные примеры подходящих солей включают органические соли, образованные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, образованные натрием, кальцием, калием, магнием, марганцем, железом, медью, цинком, алюминием и литием.
Соединения по изобретению, которые включают фрагмент с основными свойствами, такой как аминогруппа, могут образовывать, помимо с упомянутыми выше кислотами, фармацевтически приемлемые соли с различными аминокислотами.
В качестве варианта, применяемые соединения, которые обладают кислотными свойствами, способны образовывать соли присоединения основания с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочноземельных металлов и, в частности, соли натрия и калия. Все эти соли получают традиционными методами. Химические основания, которые используют в качестве реагентов для получения фармацевтически приемлемых солей присоединения основания по этому изобретению, являются основания, которые образуют нетоксичные соли присоединения основания с соединениями, обладающими кислотными свойствами. Эти соли могут быть получены любым подходящим методом, например, обработкой соединения в форме свободной кислоты с помощью неорганического или органического основания, такого как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла, или другие подобные основания. Эти соли могут быть также получены обработкой соответствующих соединений с кислотными свойствами водным раствором, содержащим требуемые фармакологически приемлемые катионы, и затем испарением полученного раствора досуха, предпочтительно, при пониженном давлении. В качестве варианта, они могут быть также получены путем смешения растворов соединений с кислотными свойствами в низших спиртах с требуемым алкоксидом металла и затем испарения полученного раствора досуха таким же образом, как указано выше. И в том и в другом случае, предпочтительно использовать стехиометрические количества реагентов, для того чтобы обеспечить полноту протекания реакции и максимальный выход требуемого конечного продукта.
Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых солей присоединения основания соединений по изобретению, которые обладают кислотными свойствами, являются химическими основаниями, которые образуют нетоксичные соли присоединения основания с такими соединениями. Такие нетоксичные соли присоединения основания включают, но этим не ограничивая, соли, образованные такими фармакологически приемлемыми катионами, такими как катионы щелочных металлов (например, калия и натрия) и катионы щелочноземельных металлов (например, кальция и магния), соли присоединения аммония или растворимого в воде амина, такого как N-метилглюкамин (меглумин), и низшего алканоламмония и другие соли присоединения фармацевтически приемлемых органических аминов.
Могут быть также образованы полусоли кислот и оснований, например, гемисульфатные и гемикальциевые соли.
По поводу обзора подходящих солей, смотрите справочник Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley VCH, 2002). Методы получения фармацевтически приемлемых солей соединений по изобретению и взаимопревращения солевых форм и форм свободного основания хорошо известны любому специалисту в данной области.
Соли по настоящему изобретению могут быть получены известными специалистам в данной области методами. Фармацевтически приемлемая соль патентуемых соединений может быть легко получена путем смешения растворов соединения и требуемой кислоты или основания, в зависимости от конкретного случая. Соль может быть осаждена из раствора и собрана фильтрацией или может быть извлечена путем испарения растворителя. Степень диссоциации соли может варьировать от полной диссоциации до практически неионизированного состояния.
Для специалистов в данной области является очевидным, что соединения по изобретению в форме свободного основания, имеющие функциональную группу с основными свойствами, могут быть превращены в соли присоединения кислоты путем обработки с помощью соответствующей кислоты в стехиометрическом избытке. Соли присоединения кислоты соединений по изобретению могут быть подвергнуты обратному превращению в соединение в форме свободного основания путем обработки с помощью соответствующего основания в стехиометрическом избытке, такого как карбонат калия или гидроксид натрия, обычно, в присутствии водного растворителя и при температуре в диапазоне приблизительно от 0°С до 100°С. Соединение в форме свободного основания может быть выделено традиционными способами, такими как экстракция органическим растворителем. Кроме того, соли присоединения кислоты соединений по изобретению могут быть подвергнуты реакции взаимообмена в результате использования дифференциальных растворимостей солей, летучести или кислотности кислот, или путем обработки с помощью соответственным образом загруженной ионообменной смолы. Например, взаимообмен может достигаться путем проведения реакции соли соединения по изобретению с небольшим стехиометрическим избытком кислоты с более низкой величиной рК, чем у компонента с кислотными свойствами исходной соли. Это превращение обычно проводят при температуре в интервале приблизительно от 0°С до температуры кипения растворителя, используемого в качестве среды для проведения реакции. Аналогичные обмены возможны и в случае солей присоединения основания, обычно, через промежуточное образование соединения в форме свободного основания.
Соединения по изобретению могут существовать как в несольватированной, так и в сольватированной формах. В случае, когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, независимую от влажности. Однако когда растворитель или вода связаны слабо, как в канальных сольватах и гигроскопических соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях, отклонение от стехиометрии будет нормальным явлением. Термин "сольват" используется в изобретении для описания молекулярного комплекса, включающего соединение по настоящему изобретению и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин "гидрат" используется, когда растворителем является вода. Фармацевтически приемлемые сольваты по изобретению включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например, D2O, d6-ацетон, d6-DMSO.
Изобретение также относится к пролекарствам соединений приведенных в изобретении формул. Так, например, конкретные производные соединений по изобретению, которые сами по себе могут обладать незначительной фармакологической активностью или вообще не обладать ею, могут, при введении пациенту, превращаться в соединения по изобретению, например, в результате гидролитического расщепления. Такие производные называются "пролекарствами". Дополнительную информацию по использованию пролекарств можно найти в публикациях "Prodrugs as Novel Delivery Systems", Vol.14, ACS Symposium Series (T Higuchi and W Stella); "Bioreversible Carriers in Drug Design", Pergamon Press, 1987 (ed. E В Roche, American Pharmaceutical Association); Guarino, V.R; Stella, V.J.: Biotech Pharm. Aspects 2007 5 (Pt2) 133-187; и J. Rautio et al. Nature Reviews Drug Discovery, 17, 559-587 (2018), полное содержание которых включено в настоящее изобретение путем ссылки на них.
Пролекарства по изобретению могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, определенными фрагментами, известными специалистам в данной области как "промотирующие группы", описанные, например, в монографии "Design of Prodrugs" by H Bundgaard (Elsevier, 1985), полное содержание которой включено в настоящее изобретение путем ссылки на нее.
Некоторые неограничивающие примеры пролекарств по изобретению включают:
(i) когда соединение содержит функциональную группу карбоновой кислоты (-СООН), то пролекарство образуют в форме его сложного эфира, например, заменяя водород на (С1-С6)алкил;
(ii) когда соединение содержит спиртовую функциональную группу (-ОН), то пролекарство образуют в форме его простого эфира, например, заменяя водород на (С1-С6)алканоилоксиметильную группу или на группу простого фосфатного эфира; и
(iii) когда соединение содержит первичную или вторичную аминогруппу (NH2 или NHR, где R не представляет собой Н), то пролекарство образуют в форме его амида, например, заменяя один или оба атома водорода подходящей метаболически лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и другие подобные группы.
Дополнительные примеры замещающих групп в соответствии с упомянутыми выше примерами и примеры других типов пролекарств можно найти в упомянутых выше ссылках.
И наконец, конкретные патентуемые соединения могут сами по себе действовать в качестве пролекарств других патентуемых соединений.
В объем изобретения также включены метаболиты соединений описанных в изобретении формул, то есть соединения, образующиеся in vivo после введения лекарственного средства, чаще всего в результате окисления или деалкилирования. Некоторые примеры метаболитов по изобретению включают, но этим не ограничивая,
(i) когда соединение по изобретению содержит алкильную группу, то метаболит представляет собой гидроксиалкильное производное (-СН → -СОН):
(ii) когда соединение по изобретению содержит алкоксильную группу, то метаболит представляет собой гидроксипроизводное (-OR → -ОН);
(iii) когда соединение по изобретению содержит третичную аминогруппу, то метаболит представляет собой вторичное аминопроизводное (-NRR' → -NHR или -NHR');
(iv) когда соединение по изобретению содержит вторичную аминогруппу, то метаболит представляет собой первичное аминопроизводное (-NHR → -NH2);
(v) когда соединение по изобретению содержит фенильный фрагмент, то метаболит представляет собой фенольное производное (-Ph → -PhOH);
(vi) когда соединение по изобретению содержит амидную группу, то метаболит представляет собой производное карбоновой кислоты (-CONH2 → СООН); и
(vii) когда соединение содержит гидроксильную группу или группу карбоновой кислоты, то соединение может быть метаболизировано в результате сопряжения, например, с глюкуроновой кислоты с образованием глюкуронида. Существуют и другие пути сопряженного метаболизма. Эти пути часто называют фазой 2 метаболизма и они включают, например, сульфатизацию или ацетилирование. Другие функциональные группы, такие как группы NH, могут быть также предметом сопряжения.
В одном варианте осуществления, в изобретении предлагаются твердые формы 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. В одном варианте осуществления, твердая форма представляет собой кристаллическую форму. В одном варианте осуществления, твердая форма представляет собой аморфную форму.
Используемый в изобретении термин "кристаллический" обозначает наличие регулярно повторяющееся расположение молекул или граней внешней поверхности. Одно соединение может образовывать целый ряд кристаллических форм, где каждая форма имеет разные и четко различимые физические свойства в твердом состоянии, такие как различные профили растворимости, скорости растворения, температуры плавления, сыпучесть и/или различные рентгенодифракционные пики. Различия в физических свойствах могут влиять на фармацевтические показатели, такие как стабильность при хранении, прессуемость и плотность (которые могут быть важными при разработке состава и производстве продукта), и скорость растворения (которая может быть важным фактором с точки зрения биодоступности).
Термин "аморфный" относится к состоянию, при котором материал на молекулярном уровне не имеет дальнего порядка, и в зависимости от температуры он может проявлять физические свойства твердого тела или жидкости. Обычно такие материалы не дают характерных рентгенограмм, и хотя они и проявляют свойства твердого тела, более формально их описывают как жидкость. При нагревании происходит переход от свойств твердого тела к свойствам жидкости, который характеризуется изменением состояния, обычно, второго порядка ("стеклованием").
Существует ряд аналитических методов, которые могут быть использованы обычным специалистом в области химии твердого тела для анализа твердых форм. Для количественного определения кристаллической твердой формы (или форм) в смеси может быть также использована порошковая рентгеновская дифракция. При порошковой рентгеновской дифракции, рентгеновские лучи направляют на кристаллический порошок, и измеряют интенсивность дифрагированных рентгеновских лучей в зависимости от угла между источником рентгеновских лучей и лучом, дифрагированным образцом. Интенсивность этих дифрагированных рентгеновских лучей может быть отображена графически в виде пиков, где ось X представляет собой угол (он известен как угол "2-тета") между источником рентгеновских лучей и дифрагированными рентгеновскими лучами, и ось Y представляет собой интенсивность дифрагированных рентгеновских лучей. Этот график называют порошковой рентгенограммой или порошковой дифрактограммой. Различные кристаллические твердые формы характеризуются разными порошковыми дифрактограммами, так как расположение пиков на оси х является свойством твердотельной структуры кристалла.
Для специалиста в данной области является очевидным, что обычная точность определения величины 2-тета на оси X пика на порошковой диаграмме составляет порядка плюс или минус 0,2 градуса 2-тета (±0,2 градуса 2-тета). Поэтому, например, дифракционный пик, который проявляется при "приблизительно 18,0 градусов 2-тета", означает, что пик появляется при 18,0 градусов±0,2 градуса 2-тета, то есть он может находиться между 17,8 градусами 2-тета и 18,2 градусами 2-тета, при измерении на большинстве рентгеновских дифрактометров при большинстве используемых условий. Кроме того, для специалиста в данной области является очевидным, что относительные интенсивности пиков будут характеризоваться между приборной вариабельностью, а также вариабельностью, обусловленной степенью кристалличности, предпочтительной ориентацией, подготовленной поверхностью образца и другими факторами, известными специалистам в данной области, и их следует учитывать только как качественные показатели. Соответственно, используемый в изобретении термин "по существу то же самые" применительно к положениям пиков порошковой дифракции рентгеновских лучей означает, что типичная вариабельность положения пика и интенсивности составляет порядка ±0,2 градуса 2-тета.
Порошковая дифракция рентгеновских лучей является лишь одним из нескольких аналитических методов, которые могут быть использованы для характеризации и/или идентификации твердых кристаллических форм. Спектроскопические методы, такие как рамановская спектроскопия (в том числе микроскопическая рамановская спектроскопия), инфракрасная спектроскопия и ЯМР-спектроскопия твердого тела, могут быть использованы для характеризации и/или идентификации кристаллических твердых форм. Эти методы могут быть также использованы для количественного определения одной или нескольких кристаллических твердых форм в смеси, а характеризующие пики величины могут регистрироваться с определением "приблизительно" перед величинами, характеризующими пики.
Используемый в изобретении термин "безводный" относится к кристаллической форме, не содержащей молекул какого-либо растворителя или воды в кристаллической решетке.
Термин "гидрат" относится к сольвату, включающему соединение и стехиометрическое или нестехиометрическое количество воды. Термин "моногидрат" относится к гидрату, включающему одну молекулу воды на молекулу соединения (то есть стехиометрия 1:1 воды к соединению).
В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1.
В одном варианте осуществления, кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1 характеризуется порошковой рентгенограммой (PXRD) (2-тета).
В таблице X приводится перечень пиков PXRD для кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 1 в градусах 2-тета (±0,2 градусов 2-тета).


В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1, имеющий порошковую рентгенограмму, включающую характеристические пики при 5,0, 8,7, 9,3, 10,8, 14,5, 15,3, 18,8 и 20,5 градусов 2-тета (±0,2 градусов 2-тета).
В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 1.
В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2.
В одном варианте осуществления, кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2
характеризуется порошковой рентгенограммой (PXRD) (2-тета). В одном варианте осуществления, анализ методом PXRD кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2 проводят при 25°С и при относительной влажности ниже 10%, например, как это описано в примере 77.
В таблице Y приводится перечень пиков PXRD для кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона, формы 2 в градусах 2-тета (±0,2 градусов 2-тета).

В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2, имеющий порошковую рентгенограмму, включающую характеристические пики при 7,1, 9,4, 12,4, 12,8, 14,3, 15,6, 16,4, 17,4, 18,5, 18,9, 19,5, 19,9, 21,1, 21,4, 23,2, 23,7, 24,8, 25,6, 27,6, 30,3, 33,2, 33,5 и 37,5 градусов 2-тета (±0,2 градусов 2-тета).
В одном варианте осуществления, в изобретении предлагается кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 2.
В одном варианте осуществления, в изобретении предлагается кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3.
В одном варианте осуществления, кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3
характеризуется порошковой рентгенограммой (PXRD) (2-тета). В одном варианте осуществления, анализ методом PXRD кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1, 6(2Н,7Н)-диона, формы 3 проводят при t 25°С и при относительной влажности выше 30%.
В таблице Z приводится перечень пиков PXRD для кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3 в градусах 2-тета (±0,2 градусов 2-тета).

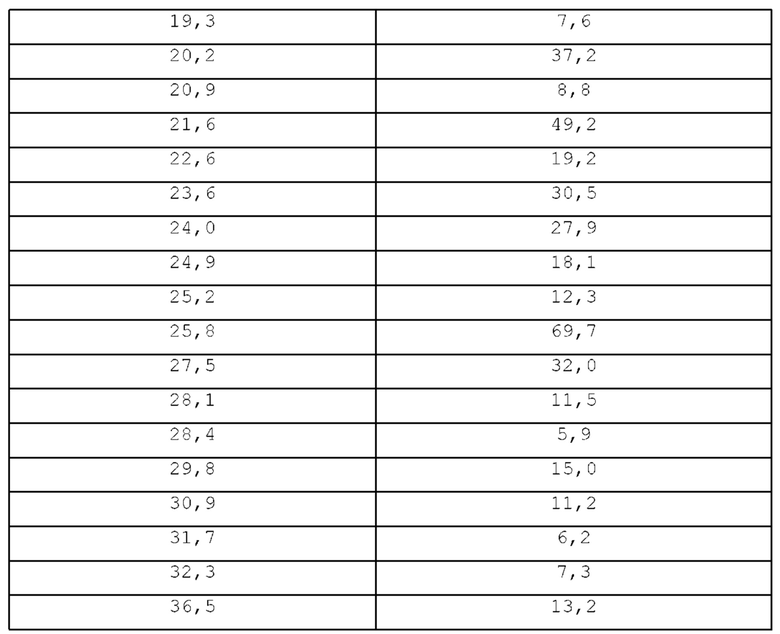
В одном варианте осуществления, в изобретении предлагается кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3, имеющий порошковую рентгенограмму, включающую характеристические пики при 13,7, 18,0 и 18,3 градусов 2-тета (±0,2 градусов 2-тета).
В одном варианте осуществления, в изобретении предлагается кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3, имеющий порошковую рентгенограмму, включающую характеристические пики при 6,9, 9,1, 13,7, 18,0 и 18,3 градусов 2-тета (±0,2 градусов 2-тета).
В одном варианте осуществления, в изобретении предлагается кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3, имеющий порошковую рентгенограмму, включающую характеристические пики при 6,9, 9,1, 11,8, 12,0, 13, 7, 14, 0, 15, 2, 15, 8, 18, 0, 18, 3, 19, 0, 19, 3, 20, 2, 20, 9, 21, 6, 22, 6, 23, 6, 24, 0, 24, 9, 25, 2, 25, 8, 27, 5, 28, 1, 28, 4, 29,8, 30,9, 31,7, 32,3 и 36,5 градусов 2-тета (±0,2 градусов 2-тета).
В одном варианте осуществления, в изобретении предлагается кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 3.
В одном варианте осуществления, в изобретении предлагается аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4.
В одном варианте осуществления, аморфный 8- ((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4 характеризуется порошковой рентгенограммой (PXRD) (2-тета).
В одном варианте осуществления, в изобретении предлагается аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 4.
В изобретении дополнительно предлагаются терапевтические методы и применения, включающие введение соединений по изобретению или их фармацевтически приемлемые соли в форме монотерапии или в комбинации с другими терапевтическими средствами или паллиативными средствами.
Соединения формулы I и формулы II, и их фармацевтически приемлемые соли могут применяться для лечения заболеваний и нарушений, которые можно лечить с помощью ингибитора киназы МЕК, таких как МЕК-ассоциированных заболеваний и нарушений, например, для лечения аномального роста клеток, такого как опухоли, например, МЕК-ассоциированных опухолей. Способность соединений формулы I и формулы II, и их фармацевтически приемлемых солей действовать в качестве ингибиторы МЕК может быть продемонстрирована путем проведения исследований, описанных в примере А. Величины IC50 приведены в таблице А.
Соответственно, в одном варианте осуществления, в изобретении предлагается способ лечения опухоли, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, опухоль представляет собой МЕК-ассоциированную опухоль.
Используемые в изобретении термины "ингибитор киназы МЕК" и "ингибитор МЕК" применяют взаимозаменяемо и они относятся к соединению, которое ингибирует ферменты киназы митоген-активированной протеинкиназы МЕК1 и/или МЕК2.
Термины "МЕК-ассоциированное" и "МЕК-опосредованное" применяют взаимозаменяемо и они относятся к заболеванию или нарушению, имеющему конститутивную активацию киназы МЕК, которые можно лечить с помощью ингибитора МЕК. Примеры включают МЕК-ассоциированный аномальный рост клеток, такой как МЕК-ассоциированные опухоли, например, МЕК-ассоциированные типы рака. В одном варианте осуществления, термин "МЕК-ассоциированное" относится к заболеванию или нарушению, имеющему дисрегуляцию экспрессии или активности киназы МЕК, или дисрегуляцию гена BRAF или BRAF киназы.
Фраза "дисрегуляция экспрессии или активности киназы МЕК" относится к амплификации гена, которая приводит к сверхэкспрессии белка МЕК, или к аутокринной деятельности, возникающей вследствие сверхэкспрессии гена МЕК в клетках, которая приводит к патогенетическому увеличению активности киназного домена белка МЕК (например, постоянно активного киназного домена белка МЕК) а клетке.
Фраза "дисрегуляция гена BRAF или BRAF киназы" относится к генетической мутации (например, к транслокации гена BRAF, которая приводит к экспрессии гибридного белка, делеции в гене BRAF, которая приводит к экспрессии белка BRAF, который включает делецию, по меньшей мере, одной аминокислоты, по сравнению с немутантным типом белка BRAF, или к мутации в гене BRAF, которая приводит к экспрессии белка BRAF с одной или более точечными мутациями по сравнению с немутантным типом белка BRAF). В качестве еще одного примера, дисрегуляция гена BRAF, белка BRAF или экспрессии или активности, или уровня любого из упомянутых процессов, может представлять собой мутацию в гене BRAF, который кодирует белок BRAF, который является постоянно активным или имеет повышенную активность по сравнению с белком, кодированным с помощью гена BRAF, который не включает мутацию. Например, дисрегуляция гена BRAF, белка BRAF, или экспрессии или активности, или уровня любого из упомянутых процессов, может быть результатом транслокации гена или хромосомы, которая приводит к экспрессии гибридного белка, который содержит первую порцию BRAF, которая включает функциональный киназный домен, и вторую порцию белка-партнера (то есть который не является BRAF).
В одном варианте осуществления, МЕК-ассоциированное заболевание или нарушение имеет активирующую мутацию BRAF. В одном варианте осуществления, МЕК-ассоциированное заболевание или нарушение представляет собой МЕК-ассоциированный рак, имеющий активирующую мутацию BRAF. Неограничивающие примеры мутаций BRAF включают мутации BRAF V600, например, V600E, V600D, V600K, V600R и V600S. В одном варианте осуществления, мутация BRAF представляет собой мутацию V600E. В одном варианте осуществления, мутация BRAF представляет собой мутацию V600K.
В одном варианте осуществления, МЕК-ассоциированное заболевание или нарушение представляет собой МЕК-ассоциированную опухоль, имеющую одно или более слияний BRAF, которые вызывают постоянную активацию и трансформацию киназы, включая но этим не ограничивая, KIAA11549-BRAF, MKRN1-BRAF, TRIM24-BRAF, AGAP3-BRAF, ZC3HAV1-BRAF, AKAP9-BRAF, CCDC6-BRAF, AGK-BRAF, EPS15-BRAF, NUP214-BRAF, ARMC10-BRAF, BTF3L4-BRAF, GHR-BRAF, ZC3HAV1-BRAF, ZNF767-BRAF, CCDC91-BRAF, DYNC112-BRAF, ZKSCAN1-BRAF, GTF2I-BRAF, MZT1-BRAF, RAD18-BRAF, CUX1-BRAF, SLC12A7-BRAF, MYRIP-BRAF, SND1-BRAF, NUB1-BRAF, KLHL7-BRAF, TANK-BRAF, RBMS3-BRAF, STRN3-BRAF, STK35-BRAF, ETFA-BRAF, SVOPL-BRAF, JHDM1D-BRAF или ВСАР29-BRAF.
В одном варианте осуществления, МЕК-ассоциированное заболевание или нарушение представляет собой МЕК-ассоциированную опухоль, имеющую BRAF-гибридный белок, где опухоль представляет собой карциному молочной железы (например, инфильтративно-протоковую карциному молочной железы), колоректальную карциному (например, аденокарциному толстой кишки), карциному пищевода (например, аденокарциному пищевода), глиому (например, десмопластическую младенческую ганглиоглиому головного мозга, пилоидную астроцитому головного мозга, плеоморфную ксантоастроцитому головного мозга, глиому спинного мозга низкой степени злокачественности (NOS), анапластическую олигодендроглиому, анапластическую ганглиоглиому), карциному головы и шеи (например, нейроэндокринную карциному головы и шеи), карциному легкого (например, аденокарциному легкого, немелкоклеточный рак легкого (NOS)), меланому (например, шпицоидную меланому кожи, нешпицоидную меланому слизистых оболочек, шпицоидную меланому кожи, меланому невыявленной первичной локализации, нешпицоидную меланому кожи), карциному поджелудочной железы (например, аденокарциному, карциному ацинарных клеток поджелудочной железы), карциному предстательной железы (например, ацинарную аденокарциному предстательной железы), саркому (злокачественную солитарную фиброзную опухоль), карциному щитовидной железы (папиллярную карциному щитовидной железы), карциному неизвестной первичной локализации (например, аденокарциному неизвестной первичной локализации), мезотелиому плевры, аденокарциному прямой кишки, карциному эндометрия матки (например, аденокарциному эндометрия матки (NOS)) или серозную карциному яичников.
В одном варианте осуществления, МЕК-ассоциированный рак выбирают из типов рака, имеющих BRAF-гибридные белки, описанных в таблице 1 (J.S. Ross, et al., Int. J. Cancer: 138, 881-890 (2016)).





В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой опухоль с BRAF немутантного типа (дикого типа).
Термин "дикий тип (немутантный тип)" описывает нуклеиновую кислоту (например, ген BRAF или мРНК BRAF), которую обычно обнаруживают у субъекта, который не имеет заболевания или нарушения, относящиеся к референсной нуклеиновой кислоте или белку.
Термин "немутантного типа (дикого типа) BRAF" описывает нуклеиновую кислоту BRAF (например, ген BRAF или мРНК BRAF) или белок BRAF, который обнаруживают у субъекта, который не имеет активирующую BRAF-мутацию.
Если не указано иное, то используемый в изобретении термин "аномальный рост клеток" относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, потеря контактного торможения), например, к опухоли. Аномальный рост клеток может быть доброкачественным (нераковым) или злокачественным (раковым).
Термины "рак" или "раковый" относятся к любому злокачественному и/или инвазивному росту или к опухоли, обусловленным аномальным ростом клеток. Рак включает первичный рак, который возникает в конкретном месте в организме, метастатический рак, который распространился из места, в котором он возник, в другие части организма, повторное проявление исходного первичного рака после ремиссии, и второй первичный рак, который представляет собой новый первичный рак у пациента с историей ранее возникшего рака другого типа, отличающегося от второго первичного рака. Рак включает солидные опухоли, называемые по типу клеток, которые их образуют, рак крови, костного мозга или лимфатической системы. Солидная опухоль представляет собой аномальный рост или массу ткани, которая обычно не содержит защитных оболочек или жидких областей. Примерами солидных опухолей являются саркомы, карциномы и лимфомы. Лейкозы (типы рака крови) обычно не образуют солидные опухоли (National Cancer Institute, Dictionary of Cancer Terms).
Используемые в изобретении термины "лечить" или "лечение" относятся к терапевтическим или паллиативным мерам. Положительные или требуемые клинические результаты включают, но этим не ограничивая, облегчение, полное или частичное, симптомов, ассоциированных с заболеванием или нарушением, или состоянием, снижение степени заболевания, стабилизацию (то есть не ухудшение) состояния при заболевании, отсрочку или замедление прогрессирования заболевания, улучшение или временное облегчение состояния при заболевании (например, одного или более симптомов заболевания) и ремиссию (либо частичную, либо полную), выявляемые или невыявляемые.
Используемый в изобретении термин "лечить" или "лечение" рака означает введение соединения по настоящему изобретению субъекту, имеющему рак или у которого диагностировали рак, с целью достижения, по меньшей мере, одного положительного терапевтического эффекта, такого как, например, уменьшение числа раковых клеток, уменьшение размера опухоли, снижение степени инфильтрации раковых клеток в периферические органы или уменьшение скорости метастазирования опухоли или роста опухоли, обратное развитие, уменьшение или торможение прогрессирования нарушения или состояния, к которым применяют такой термин, или одного или более симптомов такого нарушения или состояния. Используемый в изобретении термин "лечение", если не указано иное, относится к акту лечения, непосредственно определенному выше. Термин "лечение" также включает адъювантное и неоадъювантное лечение субъекта.
Применительно к целям настоящего изобретения, положительные или требуемые клинические результаты включают, но этим не ограничивая, один или более из следующих: уменьшение пролиферации (или сведение на нет) опухолевых или раковых клеток; ингибирование метастазирования или торможение роста опухолевых клеток; уменьшение объема или размера опухоли; увеличение периода ремиссии у субъекта (например, по сравнению с одним или более метрическим показателем (показателями) у субъекта, имеющего аналогичный тип рака, но не получающего данного лечения или получающего другое лечение, или по сравнению с одним или более метрическим показателем (показателями) у того же самого субъекта до начала лечения); облегчение симптомов, обусловленных раком; повышение качества жизни страдающих раком пациентов; уменьшение дозы других лекарственных препаратов, требующихся для лечения рака; отсрочка прогрессирования рака; излечение рака; преодоление одного или более механизмов резистентности рака к лечению противораковыми препаратами; и/или пролонгирование срока выживаемости пациентов, страдающих раком. Положительные терапевтические эффекты при лечении рака могут быть измерены несколькими способами (смотрите, например, W. А. Weber, Assessing tumor response to therapy, J. Nucl. Med. 50 Suppl. 1:1 S-10S (2009). Например, что касается ингибирования роста опухоли (Т/С), то в соответствии со стандартами Национального института рака США (NCI), показатель Т/С меньший чем или равный 42% считается минимальным уровнем противоопухолевой активности. Показатель Т/С < 10% считают высоким уровнем противоопухолевой активности, при этом Т/С (%) = медианный объем опухоли в случае проведении лечения/медианный объем опухоли в случае контрольного эксперимента × 100.
В одном варианте осуществления, результат лечения, достигаемый в результате введения соединения по изобретению, определяется в соответствии с любым из следующих показателей: частичный ответ (PR), полный ответ (CR), общий ответ (OR), выживаемость без прогрессирования заболевания (PFS), выживаемость без признаков заболевания (DFS) и общая выживаемость (OS). PFS, также называемый как "время до прогрессирования опухоли" обозначает продолжительность времени в процессе и после лечения, в течение которого опухоль не растет, и обозначает промежуток времени, в течение которого состояние пациентов было оценено показателями CR или PR, а также промежуток времени, в течение которого состояние пациентов было оценено как стабильное заболевание (SD). DFS относится к продолжительности времени в процессе и после лечения, в течение которого у пациентов не обнаруживалось заболевание. OS относится к пролонгации средней продолжительности жизни по сравнению с неподвергавшимися какому-либо воздействию или неподвергавшимися лечению субъектами или пациентами. В одном варианте осуществления, ответ на лечение с помощью соединения по изобретению является любым из PR, CR, OR, PFS, DFS или OS, который оценивает с использованием "Критериев оценки ответа солидных опухолей на терапию (RECIST), 1.1 критерии ответа".
Схема лечения с помощью соединения по изобретению, которое является эффективным для лечения рака у пациента, может изменяться в зависимости от факторов, таких как состояние при заболевании, возраст и масса тела пациента, и способность терапии достигать противоракового ответа у субъекта. Несмотря на то, что вариант осуществления любого из аспектов изобретения может быть неэффективным для достижения положительного терапевтического эффекта у каждого субъекта, но, тем не менее, он должен позволять достигать такого эффекта у статистически значимого числа субъектов, что определяется любым известным в данной области статистическим критерием, таким как критерий Стьюдента, критерий хи-квадрат, U-критерий Манна-Уитни, критерий Краскела-Уоллиса (Н-критерий), критерий Джонкхиера-Терпстра и критерий Вилкоксона.
Термины "схема лечения", "протокол дозирования" и "режим дозирования" используются взаимозаменяемо для обозначения дозы и времени введения соединения по изобретению в форме монотерапии или в комбинации с еще одним терапевтическим средством.
"Улучшение" означает ослабление или положительную динамику одного или более симптомов при лечении комбинацией лекарственных средств, описанной в изобретении, по сравнению со случаем, когда комбинацию не вводят."Улучшение" также включает сокращение или уменьшение продолжительности проявления симптома.
Используемый в изобретении термин "субъект" относится к любому животному, в том числе к млекопитающим, таким как человек. В одном варианте осуществления, субъект испытывал и/или у субъекта проявлялся, по меньшей мере, один симптом заболевания или нарушения, подлежащий лечению и/или предотвращению. В одном варианте осуществления, у субъекта была идентифицирована или диагностирована МЕК-ассоциированная опухоль (например, определенная с использованием анализа или набора, одобренных органом государственного регулирования, например, Управлением по контролю качества пищевых продуктов и лекарственных средств США
(FDA). В одном варианте осуществления, субъект имеет МЕК-ассоциированную опухоль, которая является положительной на наличие мутации BRAF (например, что определено с использованием анализа или набора, одобренных органом государственного регулирования). Субъектом может быть субъект, опухоли у которого имеют мутацию МЕК (например, если опухоль идентифицирована как таковая с использованием набора или анализа, одобренных органом государственного регулирования, например, одобренного FDA). В одном варианте осуществления, у субъекта подозревают наличие МЕК-ассоциированной опухоли. В одном варианте осуществления, субъект имеет историю болезни, в которой указано, что субъект имеет МЕК-ассоциированную опухоль с мутацией BRAF (и, необязательно, в истории болезни указано, что субъекта следует лечить любой из композиций, предлагаемых в изобретении). В одном варианте осуществления, субъектом является человек. В одном варианте осуществления, подвергаемый лечению человек является пациентом детского возраста.
Используемый в изобретении термин "пациент детского возраста" относится к субъекту в возрасте до 21 года на момент постановки диагноза или проведения лечения. Термин "детский возраст" может быть дополнительно подразделен на различные подгруппы, включающие: новорожденные (возраст с момента рождения и в течение первого месяца жизни); младенцы (возраст от 1 месяца до двух лет); дети (возраст от двух лет до 12 лет) и подростки (возраст от 12 лет до 21 года (вплоть до, но за исключением, для рождения в двадцать два года)). Berhman RE, Kliegman R, Arvin AM, Nelson WE, Nelson Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Saunders Company, 1996; Rudolph AM, et al. Rudolph's Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; и Avery MD, First LR. Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994. В одном варианте осуществления, пациент детского возраста имеет возраст от момента рождения в течение первых 28 дней жизни, возраст от 29 дней до менее двух лет, возраст от двух лет до менее 12 лет, или возраст от 12 лет до 21 года (вплоть до, но за исключением, для рождения в двадцать два года). В одном варианте осуществления, пациент детского возраста имеет возраст от момента рождения в течение первых 28 дней жизни, возраст от 29 дней до менее чем 1 года, возраст от одного месяца до менее чем четыре месяца, возраст от трех месяцев до менее чем семь месяцев, возраст от шести месяцев до менее чем 1 года, возраст от 1 года до менее чем 2 года, возраст от 2 лет до менее чем 3 года, возраст от 2 лет до менее чем семь лет, возраст от 3 лет до менее чем 5 лет, возраст от 5 лет до менее чем 10 лет, возраст от 6 лет до менее чем 13 лет, возраст от 10 лет до менее чем 15 лет, или возраст от 15 лет до менее чем 22 года.
В одном варианте осуществления, в изобретении предлагается способ лечения опухоли, где способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, опухоль представляет собой МЕК-ассоциированную опухоль. В одном варианте осуществления, МЕК-ассоциированная опухоль имеет мутацию BRAF. В одном варианте осуществления, мутация BRAF представляет собой V600E и/или V600K, и/или V600D и/или V600R, и/или V600S. В одном варианте осуществления, мутация BRAF представляет собой V600E. В одном варианте осуществления, мутация BRAF представляет собой V600K. В одном варианте осуществления, МЕК-ассоциированная опухоль имеет слияние BRAF, например, описанное в изобретении слияние BRAF. В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой опухоль с BRAF немутантного типа (дикого типа).
В одном варианте осуществления любого из описанного в изобретении способов применения, опухоль представляет собой солидную опухоль. В одном варианте осуществления любого из описанных в изобретении способов, солидная опухоль представляет собой МЕК-ассоциированную опухоль. В одном варианте осуществления, опухоль является интракраниальной. В одном варианте осуществления, опухоль является экстракраниальной. В одном варианте осуществления любого из описанного в изобретении способов применения, опухоль (например, МЕК-ассоциированная опухоль) представляет собой злокачественную опухоль (то есть рак,) например, МЕК-ассоциированный рак. В одном варианте осуществления любого из описанного в изобретении способов применения, МЕК-ассоциированный рак представляет собой меланому, рак толстой кишки, колоректальный рак, рак легкого (например, мелкоклеточную карциному легкого или немелкоклеточную карциному легкого), рак щитовидной железы (например, папиллярный рак щитовидной железы, медуллярный рак щитовидной железы, дифференцированный рак щитовидной железы, рецидивирующий рак щитовидной железы или рефрактерный дифференцированный рак щитовидной железы), рак молочной железы, рак яичников, рак центральной нервной системы (ЦНС), рак костей, рак анального отверстия, анального канала или аноректума, рак глаза, рак желчного протока, дуктальную карциному in situ, рак печени, желчного пузыря или плевры, рак ротовой полости, рак полости рта, рак губ, рак ротоглотки, рак носа, носовой полости или среднего уха, рак вульвы, рак пищевода, рак шейки матки, гастроинтестинальную карциноидную опухоль, рак гортаноглотки, рак почки, рак гортани, рак печени, рак легкого, меланому, рак носоглотки, типы рака периферической нервной системы (например, нейробластому), рак яичников, рак поджелудочной железы, рак перитонеальной полости, сальника и брыжейки тонкой кишки, рак глотки, рак предстательной железы, почечный рак (например, почечно-клеточную карциному (RCC)), рак тонкого кишечника, рак мягких тканей, рак желудка, рак яичка, рак матки, рак мочеточника или рак мочевого пузыря.
В одном варианте осуществления любого их описанных в изобретении способов применения, МЕК-ассоциированный рак представляет собой экстракраниальный рак (то есть экстракраниальную опухоль). В одном варианте осуществления, экстракраниальный рак выбирают из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников и нейробластомы. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой меланому. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой колоректальный рак. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой рак щитовидной железы. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой немелкоклеточный рак легкого. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой рак яичников. В одном варианте осуществления, МЕК-ассоциированный рак представляет собой нейробластому.
В одном варианте осуществления любого их описанных в изобретении способов применения, МЕК-ассоциированный рак представляет собой рак центральной нервной системы (ЦНС).
В одном варианте осуществления любого их описанных в изобретении способов применения, МЕК-ассоциированный рак представляет собой интракраниальный рак (рак головного мозга).
В одном варианте осуществления любого их описанных в изобретении способов применения, рак представляет собой метастатический рак.
Термин "метастазирование" является известным в данной области термином, который относится к распространению раковых клеток из места, в котором они первоначально образовались (первичного места) в одно или более других мест в организме субъекта (одно или более вторичных мест). При метастазировании, раковые клетки отделяются от исходной (первичной) опухоли, перемещаются через кровь или лимфатическую систему, и образуют новую опухоль (метастатическую опухоль) в других органах или тканях организма. Новая метастатическая опухоль включает те же самые или аналогичные раковые клетки, что и первичная опухоль. На вторичном месте, опухолевые клетки могут пролиферировать и начинать расти или образовывать колонии клеток вторичной опухоли в этом отдаленном месте.
Используемый в изобретении термин "метастатический рак" (также известный как "вторичный рак") относится к типу рака, который возникает в одном типе ткани, но затем распространяется в одну или более тканей за пределами возникновения (первичного) рака. Метастатический рак головного мозга относится к раку в головном мозге, то есть к раку, который возник в ткани, не являющейся тканью головного мозга, и метастазировал в головной мозг.
В одном варианте осуществления, соединения формулы I или их фармацевтически приемлемые соли и соединения формулы II или их фармацевтически приемлемые соли проявляют удивительную способность проникать в головной мозг и/или ЦНС. Такие соединения способны преодолевать гематоэнцефалитический барьер (ВВВ) и ингибировать киназу МЕК в головном мозге и/или других структурах ЦНС. Соответственно, в одном варианте осуществления, в изобретении предлагаются соединения, которые могут применяться для лечения опухоли ЦНС, такой как рак ЦНС.
В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой злокачественную опухоль ЦНС (то есть МЕК-ассоциированный рак ЦНС). В одном варианте осуществления, МЕК-ассоциированный рак ЦНС имеет мутацию BRAF. В одном варианте осуществления, МЕК-ассоциированный рак ЦНС имеет мутацию BRAF V600. В одном варианте осуществления, мутация BRAF является V600E и/или V600K, и/или V600D и/или V600R, и/или V600S. В одном варианте осуществления, МЕК-ассоциированный рак ЦНС имеет мутацию BRAF V600E. В одном варианте осуществления, МЕК-ассоциированный рак ЦНС имеет мутацию BRAF V60 0K. В одном варианте осуществления, МЕК-ассоциированная опухоль имеет BRAF слияние. В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой опухоль BRAF немутантного типа.
Используемые взаимозаменяемо в изобретении термин "рак ЦНС" или термин "рак центральной нервной системы (ЦНС)" относятся к раку (то есть злокачественной опухоли) ЦНС, в том числе к типам рака головного мозга (также известным как интракраниальные опухоли), типам рака спинного мозга, и типам рака мягких мозговых оболочек, окружающих головной мозг и спинной мозг. Типы рака головного мозга включают метастатические типы рака головного мозга (то есть метастатические интракраниальные типы рака) и злокачественные первичные опухоли головного мозга.
В одном варианте осуществления, МЕК-ассоциированный рак ЦНС представляет собой МЕК-ассоциированный метастатический рак головного мозга. МЕК-ассоциированный метастатический рак головного мозга может быть результатом любого описанного в изобретении рака, когда у субъекта развился, по меньшей мере, один метастаз в головном мозге. В одном варианте осуществления, метастатический рак головного мозга представляет собой меланому, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников, или нейробластома. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой метастатическую меланому, метастатический колоректальный рак или метастатический немелкоклеточный рак легкого. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой метастатическую меланому. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой метастатический колоректальный рак. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой метастатический немелкоклеточный рак легкого. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой метастатический рак яичников. В одном варианте осуществления, метастатический рак головного мозга представляет собой метастатический рак щитовидной железы. В одном варианте осуществления, МЕК-ассоциированный метастатический рак головного мозга представляет собой рак почки. В одном варианте осуществления, рак представляет собой МЕК-ассоциированный метастатический рак, по меньшей мере, с одним метастазом в головном мозге (то есть метастатический рак головного мозга). В одном варианте осуществления, рак представляет собой МЕК-ассоциированную метастатическую меланому, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления, рак представляет собой МЕК-ассоциированный метастатический колоректальный рак, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления, рак представляет собой МЕК-ассоциированный метастатический немелкоклеточный рак легкого, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления, рак представляет собой МЕК-ассоциированный метастатический рак яичников, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления, рак представляет собой МЕК-ассоциированный метастатический рак щитовидной железы, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления, рак представляет собой МЕК-ассоциированную нейробластому, по меньшей мере, с одним метастазом в головном мозге. В одном варианте осуществления одного из указанных типов МЕК-ассоциированного метастатического рака головного мозга, рак имеет мутацию BRAF. В одном варианте осуществления, рак имеет мутацию BRAF V600. В одном варианте осуществления, мутация BRAF представляет собой V600E и/или V600K, и/или V600D и/или V600R, и/или V600S. В одном варианте осуществления, рак имеет мутацию BRAF V600E. В одном варианте осуществления, рак имеет мутацию BRAF V600K. В одном варианте осуществления, МЕК-ассоциированная опухоль имеет BRAF слияние. В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой BRAF опухоль немутантного типа.
В одном варианте осуществления, МЕК-ассоциированный рак представляет собой лептоменингеальные метастазы (лептоменингеальное заболевание (LMD)). LMD представляет набор метастаз ЦНС, которые растут в оболочках головного или спинного мозга и/или в цереброспинальной жидкости (CSF), или карциноматоз мозговых оболочек. У млекопитающих, мозговые оболочки представляют собой твердую мозговую оболочку, арахноидную оболочку и мягкую оболочку мозга. Цереброспинальная жидкость (CSF) расположена в субарахноидальном пространстве между арахноидной оболочкой и мягкая оболочкой мозга. Арахноидную оболочку и мягкую оболочку мозга вместе иногда называют лептоменинксом. Когда лептоменингеальное заболевание (LMD) возникает в лептоменинксе и/или цереброспинальной жидкости (CSF), окружающей спинной мозг, его могут называть "экстракраниальным LMD". Когда LMD возникает в лептоменинксе и/или цереброспинальной жидкости головного мозга, его могут называть " интракраниальным LMD". Так как раковые клетки LMD могут быть суспендированы в CSF, они могут быстро распространяться по всей центральной нервной системе (ЦНС). В результате, лептоменингеальное заболевание (LMD) имеет неблагоприятный прогноз с периодом выживания, измеряемого обычно несколькими месяцами. В одном варианте осуществления, метастатический рак представляет собой LMD. В одном варианте осуществления, метастатический рак представляет собой МЕК-ассоциированный LMD. В одном варианте осуществления, метастатический рак представляет собой МЕК-ассоциированное интракраниальное LMD. В одном варианте осуществления, метастатический рак представляет МЕК-ассоциированное экстракраниальное LMD. В одном варианте осуществления, МЕК-ассоциированное LMD представляет собой LMD, образованное из метастазов меланомы (то есть LMD представляет собой метастатическую меланому). В одном варианте осуществления, МЕК-ассоциированное LMD представляет собой LMD, образованное из метастазов колоректального рака (то есть LMD представляет собой метастатический колоректальный рак). В одном варианте осуществления, МЕК-ассоциированное LMD представляет собой LMD, образованное из метастазов немелкоклеточного рака легкого (то есть LMD представляет собой метастатический немелкоклеточный рак легкого). В одном варианте осуществления любого из указанных МЕК-ассоциированных лептоменингеальных заболеваний (LMD), LMD имеет мутацию BRAF. В одном варианте осуществления, МЕК-ассоциированное LMD имеет мутацию BRAF V600. В одном варианте осуществления, мутация BRAF представляет собой V600E и/или V600K, и/или V600D и/или V600R, и/или V600S. В одном варианте осуществления, МЕК-ассоциированное LMD имеет мутацию BRAF V600E. В одном варианте осуществления, МЕК-ассоциированное LMD имеет мутацию BRAF V600K. В одном варианте осуществления, МЕК ассоциированное LMD имеет BRAF слияние. В одном варианте осуществления, МЕК-ассоциированное LMD представляет собой опухоль BRAF немутантного типа.
В одном варианте осуществления, МЕК-ассоциированная опухоль представляет собой рак с высокой степенью риска метастазирования. В одном варианте осуществления, опухоль с высокой степенью риска метастазирования представляет собой рак, имеющий мутацию BRAF V600E, V600D, V600K, V600R и/или V600S. В одном варианте осуществления, рак с высокой степенью риска метастазирования имеет BRAF слияние, например, любое из описанных в изобретении BRAF слияний. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой опухоль BRAF немутантного типа. В одном варианте осуществления, рак с высокой степенью риска метастазирования is меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластома. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой меланому, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластому. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой меланому. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой меланому, имеющую мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой колоректальный рак. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой колоректальный рак, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой рак щитовидной железы. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой рак щитовидной железы, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой немелкоклеточный рак легкого. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой немелкоклеточный рак легкого, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой рак яичников. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой рак яичников, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой нейробластому. В одном варианте осуществления, рак с высокой степенью риска метастазирования представляет собой нейробластому, имеющую мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, рак с высокой степенью риска метастазирования имеет KIAA11549-BRAF слияние.
В одном варианте осуществления, опухоль ЦНС представляет собой первичную опухоль головного мозга. Первичные опухоли головного мозга представляют собой опухоли, которые возникают в головном мозге и спинном мозге, или которые в совокупности называют глиомами. Термин "глиома" используют для описания опухолей, которые возникают в глиальных клетках, присутствующих в ЦНС. Согласно классификации ВОЗ опухолей головного мозга, глиомы подразделяют по активности клеток и агрессивности по шкале степени злокачественности, включающей степень I (доброкачественные опухоли ЦНС) и степени от II до IV (злокачественные опухоли ЦНС):
Глиома степени злокачественности I (пилоидная астроцитома): обычно возникает у ребенка в мозжечке или стволе головного мозга, и, изредка, в полушариях головного мозга, и является медленно растущей. Глиома степени злокачественности I может возникать у взрослых людей. Несмотря на то, что такие глиомы являются доброкачественными (степень злокачественности I по классификации ВОЗ), тем не менее, трудность излечения этого заболевания делает их рост по характеру изменения свойств злокачественным с высокими показателями заболеваемости (Rostami, Acta Neurochir (Wien). 2017; 159(11): 2217-2221).
Глиома степени злокачественности II (глиомы низкой степени злокачественности): включает астроцитому, олигодендроглиому и смешанную олигоастроцитому. Глиомы степени злокачественности II обычно возникают у молодых совершеннолетних людей (например, в возрасте 20-50 лет) и чаще всего обнаруживаются в полушариях головного мозга. Вследствие инфильтративной природы этих опухолей, может происходить их повторное проявление. Некоторые глиомы степени злокачественности II рецидивируют и развиваются в более агрессивные опухоли (степени злокачественности III или IV).
Глиома степени злокачественности III (злокачественная глиома): включает анапластическую астроцитому, анапластическую олигодендроглиому и анапластическую смешанную олигоастроцитому. Опухоли степени злокачественности III являются агрессивными типами рака высокой степени злокачественности и прорастают в близлежащие ткани головного мозга с помощью похожих на щупальцы наростов, что затрудняет их полное хирургическое удаление.
Глиомы степени злокачественности IV: включает мультиформную глиобластому (GBM) и глиосаркому; мультиформная глиобластома (GBM) является злокачественной глиомой. GBM представляет собой наиболее агрессивную и наиболее распространенную первичную опухоль головного мозга. Мультиформная глиобластома обычно быстро распространяется и прорастает в другие части головного мозга с помощью похожих на щупальцы наростов, что делает более затруднительным ее полное хирургической удаление. Глиосаркома представляет собой злокачественный тип рака и определяется как глиобластома, состоящая из глиоматозных и саркоматозных компонентов.
В одном варианте осуществления, первичная опухоль головного мозга представляет собой глиому. В одном варианте осуществления, глиома представляет собой глиому низкой степени
злокачественности. В одном варианте осуществления, глиома представляет собой глиома низкой степени злокачественности у детей.
В одном варианте осуществления, первичная опухоль головного мозга представляет собой доброкачественную первичную опухоль головного мозга. Доброкачественные первичные опухоли головного мозга могут вызывать сильную боль, постоянные повреждения головного мозга и смерть, и, в некоторых случаях, становятся злокачественными. Неограничивающие примеры доброкачественных первичных опухолей головного мозга включают глиомы степени злокачественности I, папиллярные краниофарингиомы, менингиому (в том числе рабдоидную менингиому), атипичные тератоидные/рабдоидные опухоли и дизэмбриопластическую нейроэпителиальную опухоль (DNT), пилоидную астроцитому, олигодендроглиому, смешанную олигоастроцитому, анапластическую астроцитому, анапластическую олигодендроглиому, анапластическую смешанную олигоастроцитому, диффузную астроцитому, эпендимому, плеоморфную квантоастроцитому (РХА), ганглиоглиому, глиосаркому или анапластическую ганглиоглиому.
В одном варианте осуществления, рак представляет собой рак периферической нервной системы. В одном варианте осуществления, рак периферической нервной системы представляет собой нейробластому.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли ЦНС, включающий введение (например, пероральное введение) терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли субъекту, нуждающемуся в этом. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС имеет мутацию BRAF V600. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС имеет мутацию BRAF V600E и/или V600K, и/или V600D и/или V600R, и/или V600S. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС имеет мутацию BRAF V600E. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС имеет мутацию BRAF V600K. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС имеет BRAF слияние, например, любое из описанных в изобретении BRAF слияний, например, KIAA11549-BRAF слияние. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС представляет собой опухоль BRAF немутантного типа. В одном варианте осуществления, субъект был подвергнут лечению с помощью одной или более противораковых терапий, независимо выбранных из противораковых средств, хирургического вмешательства и лучевой терапии, перед введением соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, например, как описано в изобретении ниже. В одном варианте осуществления, субъекта подвергают лечению с помощью терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в комбинации с одной или более противораковыми терапиями, независимо выбранными из одного или более противораковых средств, хирургического вмешательства и/или лучевой терапии, например, как описано в изобретении ниже. В одном варианте осуществления, субъекта подвергают лечению с помощью одной или более противораковых терапий, независимо выбранных из противоракового средства, хирургического вмешательства и лучевой терапии, после введения соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, например, как описано в изобретении ниже. В одном варианте осуществления, MEK-ассоциированная опухоль представляет собой опухоль ЦНС. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС представляет собой злокачественную опухоль ЦНС (то есть рак ЦНС). В одном варианте осуществления, злокачественная опухоль ЦНС представляет собой метастатический рак ЦНС. В одном варианте осуществления, метастатический рак ЦНС выбирают из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников. В одном варианте осуществления, метастатический рак ЦНС представляет собой метастатическую меланому. В одном варианте осуществления, метастатический рак ЦНС представляет собой колоректальный рак. В одном варианте осуществления, метастатический рак ЦНС представляет собой метастатический немелкоклеточный рак легкого. В одном варианте осуществления, метастатический рак ЦНС представляет собой метастатический рак щитовидной железы. В одном варианте осуществления, метастатический рак ЦНС представляет собой метастатический рак яичников. В одном варианте осуществления, MEK-ассоциированный рак ЦНС представляет собой лептоменингеальное заболевание (LMD). В одном варианте осуществления, LMD является интракраниальным. В одном варианте осуществления, LMD является экстракраниальным. В одном варианте осуществления, LMD представляет собой метастатическую меланому. В одном варианте осуществления, LMD выбирают из метастатической меланомы, метастатического колоректального рака и метастатического немелкоклеточного рака легкого. В одном варианте осуществления, LMD представляет собой метастатический колоректальный рак. В одном варианте осуществления, LMD представляет собой метастатический немелкоклеточный рак легкого. В одном варианте осуществления, MEK-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В одном варианте осуществления, первичная опухоль головного мозга представляет собой глиому степени злокачественности 2. В одном варианте осуществления, первичная опухоль головного мозга представляет собой глиому степени злокачественности 3. В одном варианте осуществления, первичная опухоль головного мозга представляет собой глиому степени злокачественности 4. В одном варианте осуществления, MEK-ассоциированная опухоль ЦНС представляет собой доброкачественную опухоль. В одном варианте осуществления, доброкачественная опухоль ЦНС представляет собой папиллярную краниофарингиому, менингиому (в том числе рабдоидную менингиому), атипичный тератоидную/рабдоидную опухоль или дизэмбриопластическую нейроэпителиальную опухоль (DNT). В одном варианте осуществления, соединение представляет собой соединение формулы I или его фармацевтически приемлемую соль. В одном варианте осуществления, соединение представляет собой соединение формулы II или его фармацевтически приемлемую соль. В одном варианте осуществления, соединение выбирают из соединения примеров 1-69 или его фармацевтически приемлемой соли.
Возможность применения соединение для лечения рака ЦНС может быть определена, например, путем выяснения, является ли соединение субстратом эффлюксного переносчика, и/или путем измерения клеточной проницаемости, и/или путем измерения отношения свободной концентрации соединения в крови к свободной концентрации соединения в плазме, как это описано в изобретении.
В одном варианте осуществления, соединения формулы I или их фармацевтически приемлемые соли, и соединения формулы II или их фармацевтически приемлемые соли проявляют высокую клеточную проницаемость. Методы определения проницаемости соединения по изобретению могут быть выбраны в соответствии с проводимым анализом, описанным в примере В, и коэффициенты проницаемости приведены в таблице В1.
Соединения по изобретению характеризуются низкой величиной эффлюкса. In vitro методы оценки, являются ли соединения по изобретению субстратами для эффлюксных переносчиков Р-гликопротеина (Р-gp или белка мультилекарственной резистентности 1 (MDR1)) и белка резистентности рака молочной железы (BCRP), описаны в примере В, и коэффициенты эффлюкса соединений по изобретению приведены в таблицы ВЗ.
В одном варианте осуществления, соединения по изобретению характеризуются от средних до высоких величинами отношений головной мозг (несвязанное соединение)/плазма (несвязанное соединение) (то есть от средних до высоких величинами отношений свободная концентрация соединения в головном мозге/свободная концентрация соединения в плазме). Способность соединения по изобретению проникать через гематоэнцефалитический барьер (ВВВ) субъекта (например, человека) может быть определена в соответствующей экспериментальной модели на животных (например, на грызунах, таких как мышь). Например, способность конкретных соединений проникать через гематоэнцефалитический барьер (ВВВ) у мышей определяли путем оценки отношения концентрации несвязанного соединения в головном мозге к концентрации несвязанного соединения в плазме (свободная В/Р) у мышей, например, как описано в примере С, и отношения концентрации свободного соединения в головном мозге к концентрации в плазме приведены в таблице С2. Расчет отношения концентрации свободного соединения в головном мозге к концентрации в плазме позволяет предсказывать величину эффективных концентраций, требуемых для достижения эффективного действия на периферии и в головном мозге на основе дозозависимого воздействия в экспериментальных моделях на животных. Эти данные по распределению, наряду с сопутствующими фармакокинетическими данными, могут быть использованы для моделирования и предсказания величины дозы, требуемой для достижения эффективного воздействия у больных людей.
Соответственно, в одном варианте осуществления, способы по настоящему изобретению включают способы лечения MEK-ассоциированного рака ЦНС у субъекта, нуждающегося в этом, включающие введение соединения формулы II или его фармацевтически приемлемой соли, где, по меньшей мере, часть соединения формулы II проникает через гематоэнцефалитический барьер (ВВВ), как это продемонстрировано в соответствующей экспериментальной модели на животном. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,3 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,35 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,4 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,45 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,5 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,55 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0, 6 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0, 65 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,7 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,75 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,8 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,85 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,9 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 0,95 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,0 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,1 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,2 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,3 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,4 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,5 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,6 после введения (например, перорального или внутривенного введения) субъекту. В одном варианте осуществления, отношение головной мозг/плазма для суммарного количества лекарственного средства составляет, по меньшей мере, приблизительно 1,7 после введения (например, перорального или внутривенного введения) субъекту. Следует иметь в виду, что процент соединения, которое проникает через гематоэнцефалитический барьер (ВВВ) рассчитывают на основе площади под кривой концентрация-время для данного периода времени (AUC0-t) в головном мозге в сравнении с плазмой.
Соответственно, проценты представляют отношение концентраций. То есть, если (AUC0-24h) Для соединения составляет 20 нг/мл в головном мозге и 80 нг/мл в плазме, то тогда процент соединения, которое проникает через гематоэнцефалитический барьер (ВВВ) составляет 20% (20 нг/мл в головном мозге, деленные на суммарную концентрацию (составляет 0,20). В одном варианте осуществления, проценты рассчитывают на основе площади под кривой концентрация-время для периода времени от t=0 (момент дозирования) до последнего момента времени количественного определения концентрации, то есть (AUC0-last).
Известно, что типы рака, которые часто метастазируют в головной мозг, несут в себе путь МАРК, активизирующий альтерации, такие как мутации BRAF, включая мутации BRAF, раскрытые в изобретении, или слияния BRAF, включая слияния BRAF, раскрытые в изобретении. Несмотря на то, что активирующие мутации могут возникать на различных уровнях в каноническом пути, тем не менее, они все требуют активацию сигнального пути через митоген/киназа, регулируемая внеклеточными сигналами (МЕК), для того чтобы увеличить пролиферацию и выживание (Schubbert S, Shannon К, Bollag G. Nat Rev Cancer. 2007; 7:295-308). Мутации в гене BRAF были идентифицированы при злокачественных меланомах, папиллярных карциномах щитовидной железы, колоректальных карциномах, немелкоклеточной карциноме легкого (NSCLC) и карциномах яичника и их метастатических опухолях, и при первичных опухолях головного мозга (Davies Н., et al., Nature 417(6892):949-954, 2002). Например, мутации BRAF, такие как мутации BRAF V600, были обнаружены при многочисленных метастатических опухолях ЦНС, включая метастазы меланомы в головном мозге (Flaherty КТ, et al., Nat Rev Cancer (2012) 12(5):349-61), метастазы типов колоректального рака в головном мозге и метастазы немелкоклеточного рака легкого в головном мозге (Berghoff, AS, Preusser М., Curr Opin Neurol (2014) 27(6):689-696), папиллярного рака щитовидной железы (Kim, WW et al., J Otolaryngol Head Neck Surg. 2018; 47:4, 1-6) и рака яичников (Grisham RN., et al., Cancer, 2013;119:548-554).
Мутации BRAF, например, мутации BRAF, раскрытые в изобретении, и слияния BRAF, например, слияния BRAF, раскрытые в изобретении, были также обнаружены при злокачественных первичных опухолях головного мозга, в том числе при глиомах степени злокачественности IV, например, глиобластомах и глиосаркомах, анапластических астроцитомах (опухолях высокой степени злокачественности) и анапластических ганглиоглиомах степени III по классификации ВОЗ (Berghoff, AS, Preusser М., Curr Opin Neurol (2014) 27(6):689-696); Schindler et al. (Acta Neuropathol 121 (3):397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016); K.C. Schreck, et al., Cancers, 2019, 11, 1262)) в популяциях детей и взрослых людей.
Мутации BRAF, например, мутации BRAF, раскрытые в изобретении, и слияния BRAF, например, слияния BRAF, раскрытые в изобретении, были также обнаружены в доброкачественных первичных опухолях головного мозга, например, при астроцитомах степени II по классификации ВОЗ, плеоморфных ксантоастроцитомах степени II по классификации ВОЗ (РХА), плеоморфных ксантоастроцитомах с анаплазией, пилоидной астроцитоме (РА), папиллярных
краниофарингиомах, ганглиоглиомах, астробластомах, пилоидных астроцитомах, атипичных тератоидных/рабдоидных опухолях, рабдоидных менингиомах (Berghoff, AS, Preusser М., Curr Opin Neurol (2014) 27 (6):689-696; Schindler et al. (Acta Neuropathol 121 (3):397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016); (Behling et al., Diagn Pathol 11(1):55, 2016; Brastianos et al., Nat Genet 46 (2): 161-165, 2014; Dougherty et al., Neuro Oncol 12(7):621- 630, 2010; Lehman et al., Neuro Oncol 19(1):31-42, 2017; Mordechai et al., Pediatr Hematol Oncol 32 (3):207-211, 2015; Myung et al., Transl Oncol 5 (6): 430-436, 2012; Schindler et al., Acta Neuropathol 121 (3):397-405, 2011)), в популяциях детей и взрослых людей.
Мутации BRAF были также обнаружены при рецидивирующих нейробластомах (Eleveld, TF, et al., Nat Genet 47 (8):864-871, 2015). Нейробластома представляет собой опухоль периферической нервной системы у детей. Большинство субъектов с нейробластомой имеют опухоли, которые изначально отвечают на химиотерапию, но большая часть субъектов будет сталкиваться с резистентными к терапии ответами.
Соответственно, в изобретении также предлагается способ лечения субъекта, у которого диагностировано или идентифицировано наличие MEK-ассоциированной опухоли, например, любой из приводимых в качестве примера MEK-ассоциированных опухолей, раскрытых в изобретении, включающий введение субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, где у субъекта было диагностировано или идентифицировано наличие опухоли, имеющей мутацию BRAF или BRAF слияние, например, путем использования одобренных органом государственного регулирования, например, Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) теста или анализа для идентификации мутации BRAF или слияния у субъекта или в биопсийном образце, взятым у субъекта, или путем проведения любого из неограничивающих примеров анализов, описанных в изобретении. В одном варианте осуществления, тест или анализ предлагаются в виде набора. В одном варианте осуществления, при анализе используются секвенирование нового поколения, пиросеквенирование, иммуногистохимия, флуоресцентная микроскопия, FISH анализ разъединения хромосом (метод флуоресцентной гибридизации in situ), блоттинг-метод по Саузерну, вестерн-блоттинг, анализ FACS (сортировки флуоресцентно-активированных клеток), нозерн-блоттинг или амплификация на основе ПЦР (например, ПЦР с обратной транскрипцией и количественная ПЦР с обратной транскрипцией в реальном времени). В одном варианте осуществления, анализ представляет собой одобренный органом государственного регулирования анализ, например, одобренный Управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) набор для анализа.
В одном варианте осуществления, биопсия представляет собой биопсию опухоли (например, образец опухоли, полученный при традиционном хирургическом вмешательстве или стереотаксической пункционной биопсии, например, стереотаксической пункционной биопсии под контролем компьютерной томографии (СТ) или магнитной резонансной томографии (MRI)). Методы биопсии ткани могут быть использованы для выявления общей опухолевой массы и/или мутации BRAF и/или слияния BRAF.
В одном варианте осуществления, мутация BRAF или слияние могут быть идентифицированы с использованием жидкостной биопсии (иначе называемой биопсией биологической жидкости или биопсией жидкой фазы). Смотрите, например, Karachialiou et al., "Realtime liquid biopsies become a reality in cancer treatment", Ann. Transl. Med., 3(3):36, 2016. Методы жидкостной биопсии могут быть использованы для выявления общей опухолевой массы и/или мутации BRAF. Жидкостные биопсии могут быть проведены на биологических образцах, получаемых относительно легко у субъекта (например, путем простого взятия крови), и они обычно являются менее инвазивными, чем традиционные методы, используемые для выявления опухолевой массы и/или мутации BRAF. В одном варианте осуществления, жидкостные биопсии могут быть использованы для выявления присутствия мутации BRAF на более ранней стадии, чем в случае использования традиционных методов. В одном варианте осуществления, биологический образец, который будет использоваться при жидкостной биопсии, может включать цереброспинальную жидкость (CSF), кровь, плазму, мочу, слюну, мокроту, бронхоальвеолярный лаваж, желчь, лимфатическую жидкость, жидкость в кисте, кал, перитонеальные выпоты и их комбинации. В одном варианте осуществления, жидкостная биопсия может быть использована для выявления циркулирующих опухолевых клеток (СТС). В одном варианте осуществления, жидкостная биопсия может быть использована для выявления бесклеточных ДНК. В одном варианте осуществления, бесклеточные ДНК, выявленные с использованием жидкостной биопсии, представляют собой циркулирующие опухолевые ДНК (ctDNA), которые образуются из опухолевых клеток. Анализ ctDNA (например, используя чувствительные методы детекции, такие как, но без ограничения, секвенирование нового поколения (NGS), традиционная ПНР, цифровая ПЦР или микроматричный анализ), могут быть использованы для идентификации мутации BRAF или слияния BRAF.
В одном варианте осуществления, мутация BRAF или слияние BRAF, идентифицированные с использованием жидкостной биопсии, также присутствуют в раковой клетке, которая присутствует у субъекта (например, в опухоли). В одном варианте осуществления, любой из типов мутаций или слияний BRAF может быть выявлен с использованием жидкостной биопсии. В одном варианте осуществления, генетическая мутация, идентифицированная с помощью жидкостной биопсии, может быть использована для идентификации субъекта в качестве кандидата для конкретного лечения.
"Опухолевая масса", также называемая "опухолевой нагрузкой", относится к суммарному количеству опухолевого материала, распределенного в организме. Опухолевая масса относится к суммарному количеству раковых клеток или суммарному размеру опухоли (опухолей) в организме, включая лимфатические узлы и костный мозг. Опухолевая масса может быть определена целым рядом методов, известных в данной области, таких как, например, измерение размеров опухоли (опухолей) после удаления у субъекта, например, используя штангенциркуль, или при нахождении в организме, используя методы визуализации, например, магниторезонансную визуализацию (MRI), компьютерную томографию (СТ), мультидетекторную компьютерную томографию (MDCT), позитронно-эмиссионную томографию (PET), рентгеновские снимки, ультразвуковое исследование или сканирование костей скелета.
Термин "размер опухоли" относится к суммарному размеру опухоли, который может быть измерен как длина и ширина опухоли. Размер опухоли может быть определен целым рядом методов, известных в данной области, таких как, например, измерение размеров опухоли (опухолей) после удаления у субъекта, например, используя штангенциркуль, или при нахождении в организме, используя методы визуализации, например, магниторезонансную визуализацию (MRI), сканирование костей скелета, ультразвуковое исследование или компьютерную томографию (СТ).
Жидкостные биопсии могут быть проведены множество раз в процессе постановки диагноза, в процессе мониторинга и/или в процессе лечения для определения одного или более важных с клинической точки зрения параметров, включающих, но без ограничения, прогрессирование болезни или эффективность лечения, после применения лечения к субъекту. Например, первая жидкостная биопсия может быть проведена в первый момент времени, а вторая жидкостная биопсия может быть проведена во второй момент времени в процессе постановки диагноза, в процессе мониторинга и/или в процессе лечения. В одном варианте осуществления, первым моментом времени может быть момент времени до диагностирования заболевания у субъекта (например, когда субъект является здоровым), а вторым моментом времени может быть момент времени после развития заболевания у субъекта (например, второй момент времени может быть использован для диагностирования заболевания у субъекта). В одном варианте осуществления, первым моментом времени может быть момент времени до диагностирования заболевания у субъекта (например, когда субъект является здоровым), после чего проводят мониторинг состояния здоровья пациента, а вторым моментом времени может быть момент времени после проведения мониторинга состояния здоровья пациента. В одном варианте осуществления, первым моментом времени может быть момент времени после диагностирования заболевания у субъекта, после чего применяют лечение к субъекту, а вторым моментом времени может быть момент времени после применения лечения; в таких случаях, второй момент времени может быть использован для оценки эффективности лечения (например, если генетическая мутация (мутации), выявленная в первый момент времени, уменьшается в значительной степени или не обнаруживается).
Соединение формулы I или его фармацевтически приемлемая соль, или соединение формулы II или его фармацевтически приемлемая соль могут применяться в форме монотерапии или в комбинации с одной или более различными формами терапии для лечения субъекта с аномальным ростом клеток, таким как MEK-ассоциированная опухоль, например, MEK-ассоциированный рак.
В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемая соль или соединение формулы II или его фармацевтически приемлемая соль могут применяться в комбинации с одной или более, например, одной или более дополнительными противораковыми терапиями, например, одной или более терапиями, независимо выбранными из хирургического вмешательства, лучевой терапии и применения противораковых средств, которые имеют такой же или отличающийся механизм действия. В одном варианте осуществления, лечение субъекта, имеющего MEK-ассоциированный рак, с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в комбинации с одной или более, например, одной или более дополнительными терапиями, например, независимо выбранными из хирургического вмешательства, лучевой терапии и применения противораковых средств (например, любых из противораковых средств, описанных в изобретении ниже, где противораковое средство не является соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью), может иметь повышенную терапевтическую эффективность по сравнению с лечением того же самого субъекта или аналогичного субъекта с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в форме монотерапии. Когда применяют комбинированную терапию и одну или более, например, одну, две или три противораковых терапии независимо выбирают из применения одного или более противораковых средств, таких как любое из противораковых средств, раскрытых в изобретении, противораковое средство (средства) может быть введено одновременно или раздельно с различными установленными промежутками времени и в любом порядке с соединением по изобретению и используя различные режимы дозирования. В одном варианте осуществления, противораковое средство (средства) вводят субъекту перед введением соединения по изобретению. В еще одном варианте осуществления, противораковое средство (средства) вводят субъекту после введения соединения по изобретению. В еще одном варианте осуществления, противораковое средство (средства) вводят субъекту одновременно с введением соединения по изобретению. В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемую соль, или соединение формулы II или его фармацевтически приемлемую соль используют в комбинации с одной дополнительной противораковой терапией, которая является хирургическим вмешательством, лучевой терапией или противораковым средством, которое имеет такой же или отличающийся механизм действия.
Соответственно, в одном варианте осуществления, в изобретении предлагаются способы лечения субъекта, имеющего MEK-ассоциированную опухоль (например, любую из MEK-ассоциированных опухолей, описанных в изобретении), которые включают введение субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в комбинации с одной или более дополнительными противораковыми терапиями. В одном варианте осуществления, противораковая терапия представляет собой применение одного или более противораковых средств, не являющихся соединением формулы I или его фармацевтически приемлемой солью или соединением формулы II или его фармацевтически приемлемой солью. В одном варианте осуществления, противораковая терапия представляет собой применение одного противоракового средства, не являющегося соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью. В одном варианте осуществления, дополнительной противораковой терапией является хирургическое вмешательство. В одном варианте осуществления, дополнительной противораковой терапией является лучевая терапия.
Кроме того, в изобретении предлагается соединение формулы I или его фармацевтически приемлемая соль, или соединение формулы II или его фармацевтически приемлемая соль для применения в комбинации с одной или более, например, одной или более противораковыми терапиями. В одном варианте осуществления, дополнительную противораковую терапию независимо выбирают из одной или более терапий, независимо выбранных из хирургического вмешательства, лучевой терапии и/или применения одного или более противораковых средств, которые имеют такой же или отличающийся механизм действия.
Кроме того, в изобретении предлагается одна или более, например, одна или более противораковых терапий для применения в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью. В одном варианте осуществления, дополнительную противораковую терапию независимо выбирают из одной или более терапий, независимо выбранной из хирургического вмешательства, лучевой терапии и/или применения одного или более противораковых средств, которые имеют такой же или отличающийся механизм действия.
Кроме того, в изобретении предлагается соединение формулы I или его фармацевтически приемлемая соль, или соединение формулы II или его фармацевтически приемлемая соль для применения при лечении MEK-ассоциированной опухоли в комбинации с одной или более, например, с одной или более дополнительными противораковыми терапиями.
Кроме того, в изобретении предлагается одна или более, например, одна или более дополнительных противораковых терапий, для применения при лечении MEK-ассоциированной опухоли путем совместного введения с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью.
В одном варианте осуществления, для лечения субъекта, имеющего MEK-ассоциированную опухоль, субъекта подвергают одной или более противораковым терапиям, которые не являются применением соединения формулы I или его фармацевтически приемлемой соли или соединения формулы II или его фармацевтически приемлемой соли, перед введением соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, одну или более противораковых терапий выбирают из хирургического вмешательства, лучевой терапии и применения противоракового средства, которое имеет такой же или отличающийся механизм действия. Например, в одном варианте осуществления, нуждающийся в лечении субъект может быть подвергнут, по меньшей мере, частичной резекции опухоли перед введением соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, лечение путем, по меньшей мере, частичной резекции опухоли, которое позволяет уменьшить размер опухоли (например, массу опухоли), проводят перед введением одной или более доз соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, нуждающийся в лечении субъект может быть подвергнут лучевой терапии перед введением соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, нуждающийся в лечении субъект может быть подвергнут лечению с помощью одного или более противораковых средств, не являющихся соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью, перед введением соединения формулы I или его фармацевтически приемлемой соли или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект имеет рак, который является рефрактерным или интолерантным к проводимым ранее терапии или терапиям.
Соответственно, в одном варианте осуществления, в изобретении предлагаются способы лечения субъекта, имеющего MEK-ассоциированную опухоль, включающие (i) применение одной или более, например, одной или более противораковых терапий к указанному субъекту, и (ii) после стадии (i), введение (а) соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в форме монотерапии, или (b) соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли в комбинации с одной или более, например, одной или более дополнительными противораковыми терапиями. В одном варианте осуществления, дополнительную противораковую терапию независимо выбирают из одной или более из хирургического вмешательства, лучевой терапии и/или применения одного или более противораковых средств, которые имеют такой же или отличающийся механизм действия. В одном варианте осуществления, дополнительная противораковая терапия
представляет собой применение одного или более противораковых средств, которые имеют такой же или отличающийся механизм действия. В одном варианте осуществления, дополнительная противораковая терапия представляет собой применение одного противоракового средства, которое имеет такой же или отличающийся механизм действия. В одном варианте осуществления, дополнительная противораковая терапия представляет собой хирургическое вмешательство. В одном варианте осуществления, дополнительная противораковая терапия представляет собой лучевую терапию.
Неограничивающие примеры дополнительных противораковых средств, которые могут применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, включают дополнительные киназные ингибиторы, не являющиеся соединением формулы I или его фармацевтически приемлемой солью, или соединение формулы II или его фармацевтически приемлемой солью, включающие ингибиторы МЕК, ингибиторы BRAF, ингибиторы EGFR, ингибиторы HER2 и/или HER3, ингибиторы SHP2, ингибиторы Axl, ингибиторы PI3K, ингибиторы S0S1, ингибиторы пути сигнальный трансдукции, ингибиторы контрольных точек иммунного ответа, модуляторы апоптозного пути, цитотоксические химиотерапевтические средства, лекарственные средства, таргетированные на ангиогенез, и лекарственные средства, таргетированные на иммунную систему, включая иммунотерапию.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой таргетное терапевтическое средство. Используемое в изобретении "таргетное терапевтическое средство" относится к молекуле, которая блокирует рост раковых клеток путем воздействия на определенные молекулы-мишени, необходимые для канцерогенеза и роста опухоли, а не просто путем воздействия на все быстро делящиеся клетки (например, как в случае традиционной цитотоксической химиотерапии), и включает, но этим не ограничивая, терапевтические средства, нацеленные на рецепторную
тирозинкиназу, ингибиторы пути сигнальной трансдукции (например, ингибиторы пути Ras-Raf-MEK-ERK, ингибиторы пути PI3K-Akt-mT0R-S6K ("ингибиторы PI3K")), и модуляторы апоптозного пути.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой ингибитор BRAF. Неограничивающие примеры других ингибиторов BRAF включают энкорафениб, дабрафениб, вемурафениб, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифтор-фенил]пропан-1-сульфонамид (PLX4720) и (3R)-N-(3-[[5-(2-циклопропил-пиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид (PLX8394), и их фармацевтически приемлемые соли, соединения, раскрытые в патентном документе International Application No. РСТ/IB2020/055992, опубликованном 30 декабря 2020 года в форме РСТ Publication No. WO 2020/261156 Al, включая, например, соединение, выбранное из следующих соединений:
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)фенил)-3-фторпропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)-4,5-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)-4-фторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)-4-фторфенил)-3-фторпропан-1-сульфонамид;
N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамид;
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-окси]-4-фторфенил}пропан-1-сульфонамид;
N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-окси)-5-фторпиридин-2-ил)пропан-1-сульфонамид; и
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-окси]-4-фторфенил}-3-фторпропан-1-сульфонамид; или его фармацевтически приемлемая соль;
и соединения, раскрытые в патентном документе РСТ Publication No. WO 2021/250521, опубликованном 16 декабря 2021 года, включая, например, N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)фенил)-2-азабицикло[2.1.1]гексан-2-сульфон-амид, (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид и N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид, или их фармацевтически приемлемая соль.
В одном варианте осуществления, ингибитор BRAF выбирают из энкорафениба или его фармацевтически приемлемой соли, N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида или его фармацевтически приемлемой соли и N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида или его фармацевтически приемлемой соли.
В одном варианте осуществления, ингибитор BRAF представляет собой энкорафениб или его фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор BRAF представляет собой N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фтор-фенил)-3-фторпропан-1-сульфонамид или его фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор BRAF представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид или его фармацевтически приемлемую соль.
В данной области известны и дополнительные примеры ингибиторов BRAF.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой ингибитор EGFR. Неограничивающие примеры ингибиторы EGFR включают цетуксимаб (эрбитукс®), панитумумаб (вектибикс®), осимертиниб (мерелектиниб, тагриссо®), эрлотиниб (тарцева®), гефинитиб (иресса®), нецитумумаб (портразза™), нератиниб (нерлинкс®), лапатиниб (тикерб®), вандетаниб (капрелса®), бригатиниб (алунбриг®) и ингибиторы EGFR, раскрытые в патентных документах РСТ Publication Nos. WO 2019/071351 и WO 2017/117680. В данной области известны и дополнительные примеры ингибиторов EGFR.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой ингибитор SHP2. Неограничивающие примеры ингибиторы SHP2 включают 6-(4-амино-4-метилпиперидин-1-ил)-3-(2,3-дихлорфенил)пиразин-2-амин (SHP099), [3-[(3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил]-6-(2,3-дихлорфенил)-5-метил-пиразин-2-ил]метанол (RMC-4550) RMC-4630, TN0155 и соединения, раскрытые в патентных документах WO 2020/081848, WO 2020/201991, WO 2015/107493, WO 2015/107494, WO 2015/107495 и WO 2019/075265. В одном варианте осуществления, ингибитор SHP2 представляет собой соединение, раскрытое в патентном документе WO 2020/201991. В одном варианте осуществления, ингибитор SHP2 представляет собой (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидро-спиро[инден-2,4'-пиперидин]-1-амин или его фармацевтически приемлемую соль.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой ингибитор PI3K. Неограничивающие примеры включают бупарлисиб (ВКМ120), алпелисиб (BYL719), самотолисиб (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N,N-диметил-2-(морфолин-4-ил)-4-оксо-4Н-хромен-6-карбоксамид (AZD8186), теналисиб (RP6530),воксталисиба гидрохлорид (SAR-245409), гедатолисиб (PF-05212384), панулисиб (Р-7170), таселисиб (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(б-метоксипиридин-3-ил)-4-метил-пиридо[2,3-d]пиримидин-7(8Н)-он (PF-04691502), дувелисиб (ABBV-954), N2-[4-оксо-4-[4-(4-оксо-8-фенил-4Н-1-бензопиран-2-ил)морфолин-4-ий-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серина ацетат (SF-1126), пиктилисиб (GDC-0941), 2-метил-1-[2-метил-3-(трифтор-метил)бензил]-6-(морфолин-4-ил)-1Н-бензимидазол-4-карбоновую кислоту (GSK2636771), иделалисиб (GS-1101), умбралисиб тозилат (TGR-1202), пиктилисиб (GDC-0941), копанлисиб гидрохлорид (BAY 84-1236), достолисиб (BEZ-235), 1-(4-[5-[5-амино-6-(5-третбутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1Н-1,2,4-триазол-3-ил]-пиперидин-1-ил)-3-гидрокси-пропан-1-он (AZD-8835), 5-[6,б-диметил-4-(морфолин-4-ил)-8,9-дигидро-6Н-[1,4]оксазино[4,3-е]пурин-2-ил]-пиримидин-2-амин (GDC-0084), эверолимус, рапамицин, перифосин, сиролимус и темсиролимус.
В одном варианте осуществления, противораковое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой иммунотерапевтическое средство. Термин "иммунотерапевтическое средство" относится к средству, которое оказывает модулирующее воздействие на иммунную систему. В одном варианте осуществления, иммунотерапевтическое средство может увеличивать экспрессию и/или активность регулятора иммунной системы. В одном варианте осуществления, иммунотерапевтическое средство может уменьшать экспрессию и/или активность регулятора иммунной системы. В одном варианте осуществления, иммунотерапевтическое средство может рекрутировать иммунные клетки и/или усиливать активность иммунных клеток.
В одном варианте осуществления, иммунотерапевтическое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой антитело (например, моноклональное антитело, конъюгированное антитело). В одном варианте осуществления, антитело представляет собой бевацизумаб (мвасти™, авастин®), трастузумаб (герцептин®), ритуксимаб (мабтера™, ритуксан®), эдреколомаб (панорекс), даратумуаб (дарзалекс®), оларатумаб (лартруво™), офатумумаб (арзерра®), алемтузумаб (кампат®), цетуксимаб (эрбитукс®), ореговомаб, пембролизумаб (кейтруда®), динутиксимаб (унитуксин®), обинутузумаб (газива®), тремелимумаб (СР-675,206), рамуцирумаб (цирамза®), ублитуксимаб (TG-1101), панитумумаб (вектибикс®), элотузумаб (эмплисити™), нецитумумаб (портразза™), цирмтузумаб (UC-961), ибритумомаб (зевалин®), исатуксимаб (SAR650984), нимотузумаб, фрезолимумаб (GC1008), лирилумаб (INN), могамулизумаб (потелигео®), фиклатузумаб (AV-299), деносумаб (эксджива®), ганитумаб, урелумаб, пидилизумаб, аматуксимаб, блинатумомаб (AMG103; блинцито®) или мидостаурин (ридапт). В одном варианте осуществления, иммунотерапевтическое средство, которое может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой ингибитор контрольных точек иммунного ответа. В одном варианте осуществления, иммунотерапевтическое средство включает один или более, например, один или два ингибитора контрольных точек иммунного ответа. В одном варианте осуществления, ингибитор контрольных точек иммунного ответа представляет собой ингибитор CTLA-4, ингибитор PD-1 или ингибитор PD-L1. В одном варианте осуществления, ингибитор CTLA-4 представляет собой ипилимумаб (ервой®) или тремелимумаб (СР-675,206). В одном варианте осуществления, ингибитор PD-1 представляет собой пембролизумаб (китруда®), ниволумаб (опдиво®) и сасанлимаб (RN888). В одном варианте осуществления, ингибитор PD-L1 представляет собой атезолизумаб (тецентрик®) или дурвалумаб (имфинзи™).
В одном варианте осуществления, противораковая терапия, которая может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой лучевую терапию.
Неограничивающие примеры лучевой терапии включают внешнюю лучевую терапию (например, внешнюю лучевую терапию с использованием киловольтного рентгеновского излучения или мегавольтного рентгеновского излучения) или внутреннюю лучевую терапию. Внутренняя лучевая терапия (называемая также брахитерапией) может включать использование, например, низкодозной внутренней лучевой терапии или высокодозной внутренней лучевой терапии. Низкодозная внутренняя лучевая терапия включает, например, введение небольших радиоактивных гранул в раковую ткань или в непосредственной близости от раковой ткани субъекта. Высокодозная внутренняя лучевая терапия включает, например, введение тонкой трубочки (например, катетера) или импланта в раковую ткань или в непосредственной близости от раковой ткани субъекта и доставку высокой дозы радиации в тонкую трубочку или имплант, используя радиационную установку. Методы проведения лучевой терапии на субъекте, имеющем рак, хорошо известны в данной области. В вариантах осуществления, в которых опухоль представляет собой опухоль ЦНС, лучевая терапия может включать лучевую терапию всего головного мозга (WBRT) или стереотаксическое радиохирургическое вмешательство (SRS), такое как кибернож®, икс-нож®, гамма нож®, или система ЕхасТгас®.
В одном варианте осуществления, противораковая терапия, которая может применяться в комбинации с соединением формулы I или его фармацевтически приемлемой солью, или соединением формулы II или его фармацевтически приемлемой солью в соответствии с любым из методов комбинированной терапии, описанных в изобретении, представляет собой хирургическое вмешательство. Неограничивающие примеры хирургического вмешательства включают, например, открытое хирургическое вмешательство или минимально инвазивное хирургическое вмешательство. Хирургическое вмешательство может включать, например, по меньшей мере, частичную резекцию опухоли, полное удаление опухоли, циторедуктивное удаление опухоли или удаление опухоли, которая вызывает боль или оказывает давление у субъекта. Методы проведения открытого хирургического вмешательства и минимально инвазивного хирургического вмешательства на субъекте, имеющего рак, известны в данной области.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли (например, любой из MEK-ассоциированных опухолей, описанных в изобретении), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективных количеств соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли и ингибитора BRAF (например, любого из ингибиторов BRAF, раскрытых в изобретении) в любом порядке, вместе или раздельно. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из соединений примеров 1-69, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли (например, любой из MEK-ассоциированных опухолей, описанных в изобретении), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективных количеств соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли и ингибитора EGFR (например, любого из ингибиторов EGFR, раскрытых в изобретении) в любом порядке, вместе или раздельно. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из соединений примеров 1-69, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли (например, любой из MEK-ассоциированных опухолей, описанных в изобретении), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективных количеств соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли и ингибитора SHP2 (например, любого из ингибиторов SHP2, раскрытых в изобретении) в любом порядке, вместе или раздельно. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из соединений примеров 1-69, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли (например, любой из MEK-ассоциированных опухолей, описанных в изобретении), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективных количеств соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли и ингибитора контрольных точек иммунного ответа (например, любого из ингибитором контрольных точек иммунного ответа, раскрытых в изобретении) в любом порядке, вместе или раздельно. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из соединений примеров 1-69, или его фармацевтически приемлемую соль.
Кроме того, в изобретении предлагается (i) фармацевтическая комбинация для лечения MEK-ассоциированной опухоли у субъекта, нуждающегося в этом, которая включает (а) соединение формулы I или его фармацевтически приемлемую соль, или соединение формулы II или его фармацевтически приемлемую соль, и (b) по меньшей мере, одно дополнительное противораковое средство (например, любое из типичных дополнительных противораковых средств, описанных в изобретении или известных в данной области), где соединение формулы I или его фармацевтически приемлемая соль или соединение формулы II или его фармацевтически приемлемая соль и одно или более, например, одно или более дополнительных противораковых средств приготавливают раздельно для одновременного или раздельного применения для лечения опухоли, где количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли и дополнительного противоракового средства (средств) являются вместе эффективными при лечении опухоли; (ii) применение такой комбинации для приготовления лекарственного препарата для лечения опухоли; и (iii) и продажная упаковка или продукт, включающий такую комбинацию в качестве комбинированного препарата для одновременного, раздельного или последовательного применения; и для способа лечения опухоли у субъекта, нуждающегося в этом.
Используемый в изобретении термин "фармацевтическая комбинация" относится к нефиксированной комбинации активных ингредиентов. Термин "нефиксированная комбинация" означает, что соединение формулы I или его фармацевтически приемлемая соль, или соединение формулы II или его фармацевтически приемлемая соль и одно или более, например, одно или более дополнительных противораковых средств приготавливают в форме раздельных композиций или доз, в результате чего они могут быть введены субъекту, нуждающемуся в этом, одновременно или раздельно с различными определенными интервалами времени и в любом порядке, где такое введение позволяет достигать эффективных концентраций двух или более соединений в организме субъекта. Упомянутое выше также применимо к коктейльным терапиям, например, к введению трех или более активных ингредиентов. Аналогично, термин "комбинация" применительно к соединению формулы I или его фармацевтически приемлемой соли, или соединению формулы II или его фармацевтически приемлемой соли при использовании в комбинации одного или более противораковых средств относится к нефиксированной комбинации.
Соответственно, кроме того, в изобретении предлагается способ лечения MEK-ассоциированной опухоли, включающий введение субъекту, нуждающемуся в этом, фармацевтической комбинации для лечения указанной опухоли, которая включает (а) соединение формулы I или его фармацевтически приемлемую соль, или соединение формулы II или его фармацевтически приемлемую соль, и (b) одно или более, например, одно или более дополнительных противораковых средств, для одновременного, раздельного или последовательного применения для лечения опухоли, где количества соединения формулы I или его фармацевтически приемлемой соли или соединения формулы II или его фармацевтически приемлемой соли и дополнительного противоракового средства (средств) являются вместе эффективными при лечении опухоли.
В одном варианте осуществления, в изобретении предлагается способ лечения MEK-ассоциированной опухоли (например, доброкачественной, злокачественной или метастатической опухоли), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, где субъект не подвергался лечению с помощью противораковой терапии перед введением указанного соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, где противораковую терапию выбирают из одной или более, например, одной или более противораковых терапий, независимо выбранных из хирургического вмешательства, лучевой терапии и применения противоракового средства, которое имеет такой же или отличающийся механизм действия. В одном варианте осуществления, пациента не подвергали лечению с помощью противоракового средства перед введением указанного соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, пациента не подвергали лечению путем хирургического вмешательства перед введением указанного соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, пациента не подвергали лечению путем лучевой терапии перед введением указанного соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли.
В одном варианте осуществления, в изобретении предлагается способ лечения субъекта, имеющего MEK-ассоциированную опухоль (например, доброкачественную, злокачественную или метастатическую опухоль), включающий введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли, где субъекта не подвергали предшествующей терапии или стандартной терапии (например, лечению с помощью одного или более противораковых средств, не являющихся соединением формулы I или его фармацевтически приемлемой солью, и/или путем лучевой терапии и/или хирургического вмешательства), где MEK-ассоциированная опухоль стала рефрактерной или интолерантной к указанной предшествующей терапии. В одном варианте осуществления, у субъекта возникли метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора BRAF перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]-пропан-1-сульфонамида, (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фтор-пирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF) был подвергнут лечению с помощью ингибитора BRAF и ингибитора МЕК перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида и (3R)-N-(3-[[5-(2-цикло-пропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил] карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба, и ингибитора МЕК, выбранного из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью энкорафениба и биниметиниба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью дабрафениба и траметиниба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью вемурафениба и кобиметиниба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более, например, одного или двух ингибиторов PI3K перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более, например, одного или двух ингибиторов PI3K, выбранных из бупарлисиба (ВКМ120), алпелисиба (BYL719), самотолисиба (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N, Ы-диметил-2-(морфолин-4-ил)-4-оксо-4Н-хромен-6-карбоксамида (AZD8186), теналисиба (RP6530), воксталисиба гидрохлорида (SAR-245409), гедатолисиба (PF-05212384), панулисиба (Р-7170), таселисиба (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(б-метоксипиридин-3-ил)-4-метил-пиридо[2,3-d]пиримидин-7(8Н)-она (PF-04691502), дувелисиба (ABBV-954), N2-[4-оксо-4-[4-(4-оксо-8-фенил-4Н-1-бензопиран-2-ил)-морфолин-4-ий-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серина ацетата (SF-1126), пиктилисиба (GDC-0941), 2-метил-1-[2-метил-3-(трифторметил)бензил]-6-(морфолин-4-ил)-1Н-бензимидазол-4-карбоновой кислоты (GSK2636771), иделалисиба (GS-1101), умбралисиба тозилата (TGR-1202), пиктилисиба (GDC-0941), копанлисиба гидрохлорида (BAY 84-1236), достолисиба (BEZ-235), 1-(4-[5-[5-амино-6-(5-третбутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1Н-1,2,4-триазол-3-ил]-пиперидин-1-ил)-3-гидроксипропан-1-она (AZD-8835), 5-[6,6-Диметил-4-(морфолин-4-ил)-8,9-дигидро-6Н-[1,4]-оксазино[4,3-е]-пурин-2-ил]пиримидин-2-амина (GDC-0084) эверолимуса, рапамицина, перифосина, сиролимуса и темсиролимуса. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью бупарлисиба или алпелисиба, в форме монотерапии или в комбинации. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора BRAF и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитор PD-1 и/или ингибитора PD-L1), перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил-карбонил)-2,4-дифторфенил]-пропан-1-сульфонамида и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил] карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида
(PLX8394), и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора BRAF, ингибитор МЕК и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]-карбонил]-2,4-дифторфенил)-3-фтор-пирролидин-1-сульфонамида (PLX8394), ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида
(CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733), и одного или более ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба, ингибитора МЕК, выбранного биниметиниба, траметиниба и кобиметиниба, и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатическую меланому (например, метастатическую меланому, имеющую мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более, например, одного или двух алкилирующих средств перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью алкилирующих средств, выбранных из темозоломида, фотемустина, ломустина и кармустина. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью темозоломида. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический колоректальный рак (например, метастатический колоректальный рак, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора МЕК и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфенил-амино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метил-пиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733), и одного или более ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитор CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора МЕК, выбранного из биниметиниба, траметиниба и кобиметиниба, и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора МЕК, который представляет собой биниметиниб, и ингибитора контрольных точек иммунного ответа, который представляет собой ниволумаб, ипилимумаб или пембролизумаб. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический колоректальный рак (например, метастатический колоректальный рак, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитор PD-1 и/или ингибитор PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба, пембролизумаба и сасанлимаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический колоректальный рак (например, мутантный метастатический колоректальный рак, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более цитотоксических химиотерапевтических средств перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект был подвергнут лечению с помощью оксалиплатина, иринотекана, комбинации FOLFOXIRI (оксалиплатина, иринотекана и фторурацила), комбинации FOLFIRI (фолиновой кислоты, фторурацила и иринотекана) или комбинации САРЕОХ (капецитабина и оксалиплатина) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический колоректальный рак (например, метастатический колоректальный рак, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора EGFR, ингибитора BRAF и одного или более цитотоксических химиотерапевтических средств перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект, имеющий метастатический колоректальный рак, ранее подвергался лечению с помощью ингибитора EGFR, выбранного из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефинитиба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, ингибитора BRAF, выбранного энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифтор-фенил]-пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-цикло-пропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и одного или более цитотоксических химиотерапевтических средств. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора EGFR, выбранного из цетуксимаба и панитумумаба, ингибитора BRAF, который представляет собой вемурафениб, и цитотоксического химиотерапевтического средства, которое представляет собой иринотекан. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический колоректальный рак (например, метастатический колоректальный рак, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью ингибитора EGFR и одного или более цитотоксических химиотерапевтических средств перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора EGFR, выбранного из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефинитиба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и одного или более химиотерапевтических средств. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора EGFR, выбранного из цетуксимаба и панитумумаба, и цитотоксического химиотерапевтического средства, которое представляет собой иринотекан или комбинацию FOLFIRI (фолиновая кислота, фторурацил и иринотекан). В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический немелкоклеточный рак легкого (например, метастатический немелкоклеточный рак легкого, имеющий мутацию BRAF V600 или слияние BRAF), был подвергнут лечению с помощью одного или более, например, одного или двух ингибиторов EGFR перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более, например, одного или двух ингибиторов EGFR, независимо выбранных из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефинитиба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью эрлотиниба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью гефинитиба. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью эрлотиниба и гефинитиба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический немелкоклеточный рак легкого (например, метастатический немелкоклеточный рак легкого, имеющий мутацию BRAF), ранее подвергался лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло-[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил] пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-цикло-пропилпиримидин-5-ил)-1Н-пирроло-[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфенил-амино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидрокси-пропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метил-пиридо[2,3-d]-пиримидин-4,7(3Н,8Н)-диона (ТАК-733), и ингибитора EGFR, выбранного из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефинитиба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из вемурафениба, дабрафениба и энкорафениба, и ингибитора EGFR, выбранного из цетуксимаба и панитумумаба, перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект, имеющий метастатический рак щитовидной железы (например, метастатический рак щитовидной железы, имеющий мутацию BRAF V600 или слияние BRAF), ранее подвергался лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-Ь]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фтор-пирролидин-1-сульфонамида (PLX8394), ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфенил-амино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733), и ингибитора EGFR, выбранного из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефинитиба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из вемурафениба, дабрафениба и энкорафениба, перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта образовывались метастазы в головном мозге в процессе указанного предшествующего лечения.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет лептоменингеальное заболевание (LMD) и ранее подвергался лечению с помощью ингибитора BRAF и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет лептоменингеальное заболевание (LMD) и ранее подвергался лечению с помощью ингибитор BRAF, ингибитора МЕК и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитор PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]-карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733), и ингибитора контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба, ингибитора МЕК, выбранного из биниметиниба, траметиниба и кобиметиниба, и одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет лептоменингеальное заболевание (LMD) и ранее подвергался лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа (например, любого из раскрытых в изобретении ингибиторов контрольных точек иммунного ответа, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более, например, одного или двух ингибиторов контрольных точек иммунного ответа, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имел глиому и ранее подвергался лечению путем проведения хирургического вмешательства перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имел глиому и ранее подвергался лечению путем проведения лучевой терапии (например, лучевой терапии всего головного мозга или стереотаксического радиохирургического вмешательства) перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имел глиому и ранее подвергался лечению с помощью одного или более цитотоксических химиотерапевтических средств перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью одного или более цитотоксических химиотерапевтических средств, независимо выбранных из цисплатина, пеметрекседа, винорелбина и паклитаксела. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению с помощью ингибитора орнитиндекарбоксилазы перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора орнитиндекарбоксилазы, который представляет собой эфлорнитин (в форме рацемата, или D или л энантиомера). В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению с помощью алкилирующего средства перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению с помощью алкилирующего средства, выбранного из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению с помощью алкилирующего средства и ингибитора орнитиндекарбоксилазы перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению с помощью алкилирующего средства, выбранного из темозоломида, ломустина и кармустина, и ингибитора орнитиндекарбоксилазы, который представляет собой эфлорнитин (в форме рацемата, или D или л энантиомера). В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению путем проведения лучевой терапия (например, лучевой терапии всего головного мозга или стереотаксического радиохирургического вмешательства) и с помощью алкилирующего средства перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению путем проведения лучевой терапии (например, лучевой терапии всего головного мозга или стереотаксического радиохирургического вмешательства) и с помощью алкилирующего средства, выбранного из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению путем проведения терапии антителами перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению путем проведения терапии антителами, где антитело представляет собой бевацизумаб. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению путем хирургического вмешательства и проведения лучевой терапии перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению путем хирургического вмешательства, проведения лучевой терапии с помощью алкилирующего средства перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению путем хирургического вмешательства, проведения лучевой терапии (например, лучевой терапии всего головного мозга или стереотаксического радиохирургического вмешательства) и с помощью алкилирующего средства, выбранного из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению с помощью ингибитора BRAF перед лечением с помощью соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора BRAF, выбранного из N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил-карбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]-карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную глиому и ранее подвергался лечению с помощью ингибитор BRAF и ингибитора МЕК перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, сольвата или полиморфа, или соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, субъект ранее подвергался лечению с помощью ингибитора BRAF, выбранного из N-[3-(5-хлор-1Н-пирроло[2,3-b]-пиридин-3-илкарбонил)-2,4-дифторфенил] пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]-карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамида (PD-325901), 2-(2-хлор-4-йодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфенил-амино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (ТАК-733). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба, и ингибитора МЕК, выбранного из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
В одном варианте осуществления раскрытого в изобретении способа лечения субъекта, имеющего MEK-ассоциированную опухоль, субъект имеет MEK-ассоциированную ганглиоглиому ствола головного мозга и ранее подвергался лечению с помощью ингибитора BRAF перед лечением с помощью соединения формулы I или его фармацевтически приемлемой соли, соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]-пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропил-пиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]-карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъекта ранее подвергали лечению с помощью ингибитора BRAF, выбранного из энкорафениба, дабрафениба и вемурафениба. В одном варианте осуществления, субъект становился рефрактерным к указанному предшествующему лечению.
Несмотря на то, что генетическая основа онкогенеза может различаться в зависимости от типа рака, тем не менее, клеточные и молекулярные механизмы, необходимые для метастазирования, по-видимому, являются аналогичными для всех типов солидных опухолей. В процессе метастатического каскада, раковые клетки теряют ингибирующие рост ответные реакции, подвергаются изменениям их адгезивности и продуцируют ферменты, которые могут разрушать компоненты внеклеточного матрикса. Это приводит к отделению опухолевых клеток от исходной опухоли, проникновению в кровоток через вновь сформированную сосудистую систему, миграции и экстравазации опухолевых клеток в благоприятные отдаленные участки, где они могут образовывать колонии. Ряд генов были идентифицированы в качестве промоторов или супрессоров метастазирования.
Соответственно, кроме того, в изобретении предлагаются способы лечения, ингибирования, предотвращения, способствования предотвращению или уменьшения метастазирования MEK-ассоциированной опухоли у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или соединения формулы II или его фармацевтически приемлемой соли. В одном варианте осуществления, соединение формулы I или его фармацевтически приемлемую соль применяют в комбинации с одной или более противораковыми терапиями, независимо выбранными из хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли), лучевой терапии и применения противоракового средства.
Используемый в изобретении, термин "лечение метастазирования" означает уменьшение размера, прогрессирования и/или дальнейшего распространения одного или более метастазов.
Используемый в изобретении термин "ингибирование метастазирования" означает уменьшение появления (или повторного появления) одного или более метастазов, предотвращение появления (или повторного появления) одного или более метастазов, или уменьшение распространения одного или более метастазов.
В одном варианте осуществления, может быть проведена оценка состояния субъекта, подвергаемого лечению в соответствии с любым из раскрытых в изобретении способов, в соответствии с одним или более известными в данной области стандартными критерии оценки объективного ответа на проводимое лечение, включающими RECIST
(критерии оценки ответа солидных опухолей на терапию, например, RECIST version 1.0, RECIST version 1.1, и модифицированные RECIST 1.1 (mRECIST 1.1)), RANO-BM (критерии оценки объективного ответа при лечении метастатических опухолей головного мозга), Macdonald, RANO-LMD и NANO (шкала неврологической оценки в нейроонкологии). В одном варианте осуществления любого из указанных критериев, опухоль оценивают по результатам исследования визуализации опухоли (например, магнитно-резонансной томографией (MRI), компьютерной томографией (СТ), мультидетекторной компьютерной томографией (MDCT) или позитронно-эмиссионной томографией (PET)). В одном варианте осуществления, объективный ответ на лечение оценивают в соответствии с RECIST version 1.1, где: полный ответ (CR) определяется как полное исчезновение всех опухолевых очагов; частичный ответ (PR) определяется как уменьшение суммы измерений размеров опухоли, по меньшей мере, на 30%; прогрессирование заболевания (PD) определяется как увеличение, по меньшей мере, на 20% суммы измерений размеров опухоли (где появление новых очагов или существенное прогрессирование нецелевых опухолевых очагов также определяется как PD), где увеличение, по меньшей мере, на 5 мм от исходного значения оценивается как PD; и стабильное заболевание (SD) определяется как отсутствие существенного уменьшения размеров опухоли, для того чтобы оценивать состояние как PR, так и отсутствие существенного увеличения размеров опухоли, для того чтобы оценивать состояние как PD, используя для сравнения наименьшую сумму диаметров опухоли в процессе проведения лечения. В одном варианте осуществления, оценки включают интракраниальный ответ (оцениваемый в соответствии с модифицированным RECIST, используя МРТ, усиленную путем введения контрастного вещества на основе гадолиния), экстракраниальный ответ, частоту объективного глобального ответа, частоту контроля опухолевого процесса (DCR), длительность ответа (DOR), выживаемость без прогрессирования заболевания (PFS) и общую выживаемость (OS).
Используемый в изобретении термин "эффективная доза" или "эффективное количество" лекарственного средства соединения или фармацевтической композиции относится к количеству, достаточному для того, чтобы оказывать любой одно или более воздействий, положительных или требуемых, в том числе на биохимические, гистологические и/или поведенческие симптомы заболевания, его осложнения и промежуточные патологические фенотипы, проявляющиеся в процессе развития заболевания. С точки зрения использования в терапевтических целях, "терапевтически эффективное количество" относится к количеству вводимого соединения, которое облегчает, в некоторой степени, один или более из симптомов подвергаемого лечению заболевания. Применительно к лечению рака, терапевтически эффективное количество относится к количеству, которое оказывает воздействие (1) по уменьшению размера опухоли, (2) по ингибированию (то есть, замедлению, в некоторой степени, предпочтительно, прекращению) метастазирования опухоли, (3) по ингибированию (то есть, замедлению, в некоторой степени, предпочтительно, прекращению) роста опухоли или инвазивности опухоли, (4) по облегчению, в некоторой степени (или, предпочтительно, устранению) одного или более признаков или симптомов, ассоциированных с раком, (5) по снижению дозы других лекарственных препаратов, требующихся для лечения заболевания, и/или (б) по усилению воздействия еще одного лекарственного препарата, и/или (7) по замедлению прогрессирования заболевания у пациента.
Эффективная доза может быть введена за одно или более введений. Применительно к целям этого изобретения, эффективная доза лекарственного средства, соединения или фармацевтической композиции представляет собой дозу, достаточную для осуществления профилактического или терапевтического лечения, либо непосредственно, либо опосредованно. Следует иметь в виду, что в клиническом контексте, эффективная доза лекарственного средства, соединения или фармацевтической композиции может достигаться или может не достигаться в сочетании с еще одним лекарственным средством, соединением или фармацевтической композиции.
"Фармацевтическая композиция" относится к смеси одного или более соединений по изобретению или их фармацевтически приемлемых солей, сольватов, гидратов или пролекарств в качестве активного ингредиента, и, по меньшей мере, одного фармацевтически приемлемого носителя или вспомогательного вещества. В одном варианте осуществления, фармацевтическая композиция включает два или более фармацевтически приемлемых носителей и/или вспомогательных веществ.
В одном варианте осуществления, в изобретении предлагается фармацевтическая композиция, включающая соединение по изобретению или его фармацевтически приемлемую соль и, по меньшей мере, один фармацевтически приемлемый носитель или вспомогательное вещество. В одном варианте осуществления, фармацевтическая композиция включает два или более фармацевтически приемлемых носителей и/или вспомогательных веществ.
Соответственно, в одном варианте осуществления, в изобретении предлагается фармацевтическая композиция для применения при лечении аномального роста клеток у субъекта, нуждающегося в этом, где фармацевтическая композиция включает соединение по изобретению или его фармацевтически приемлемую соль и, по меньшей мере, один фармацевтически приемлемый носитель или вспомогательное вещество.
Используемый в изобретении "фармацевтически приемлемый носитель" относится к носителю или разбавителю, который не вызывает существенного раздражающего воздействия на организм и не оказывает негативного влияния на биологическую активность и свойства вводимого соединения.
Фармацевтически приемлемый носитель может включать любой традиционный фармацевтический носитель или вспомогательное вещество. Выбор носителя и/или вспомогательного вещества будет, в значительной степени, зависеть от таких факторов, как конкретный способ введения, влияние носителя или
вспомогательного вещества на растворимость и стабильность, и свойства лекарственной формы.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как образующие гидраты и сольваты). Фармацевтические композиции могут, если это желательно, содержать дополнительные ингредиенты, такие как вещества, корригирующие вкус и запах лекарственного средства, связующие вещества, вспомогательные вещества и другие подобные ингредиенты.
Термин "вспомогательное вещество" используется в изобретении для описания любого ингредиента, не являющегося соединением (соединениями) по изобретению. Выбор вспомогательного вещества будет, в значительной степени, зависеть от таких факторов, как конкретный способ введения, влияние вспомогательного вещества на растворимость и стабильность, и свойства лекарственной формы.
Используемое в изобретении "вспомогательное вещество" включает всевозможные растворители, диспергирующие среды, покрытия, антибактериальные и противогрибковые средства, изотонические и замедляющие всасывание вещества, носители, разбавители и другие подобные вспомогательные вещества, которые являются физиологически совместимыми. Примеры вспомогательных веществ включают одно или более веществ, таких как вода, физиологический раствор, забуференный фосфатом физиологический раствор, декстроза, глицерин, этанол и другие подобные вещества, а также их комбинации, и могут включать в композиции изотонические средства, например, сахара, хлорид натрия или полиспирты, такие как маннит или сорбит. Примеры вспомогательных веществ также включают различные органические растворители (такие как образующие гидраты и сольваты). Фармацевтические композиции могут, если это требуется, содержать дополнительные вспомогательные вещества, такие как вещества, корригирующие вкус и запах лекарственного средства, связующие вещества, смазывающие вещества, разрыхлители, подсластители или ароматизаторы, окрашивающие вещества или красители, и другие подобные вспомогательные вещества. Например, для перорального введения, могут быть использованы таблетки, содержащие различные вспомогательные вещества, такие как лимонная кислота, вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и конкретные комплексные силикаты, и со связывающими средствами, такими как сахароза, желатин и аравийская камедь. Неограничивающие примеры вспомогательных веществ включают карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли. Так, например, для перорального введения могут использоваться таблетки, содержащие различные
вспомогательные вещества, такие как лимонная кислота, вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и конкретные комплексные силикаты, а также со связующими веществами, такими как сахароза, желатин и аравийская камедь. Неограничивающие примеры вспомогательных веществ включают карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли. Кроме того, для таблетирования часто используют смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа могут быть также использованы в мягких и твердых наполняемых желатиновых капсулах. Поэтому, неограничивающие примеры материалов включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. В случае, когда для перорального введения желательно использовать водные суспензии или эликсиры, содержащееся в них активное соединение можно объединять с различными подсластителями или ароматизаторами, окрашивающими веществами или красителями и, если это требуется, с эмульгаторами или суспендирующими средствами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Примеры вспомогательных веществ также включают фармацевтически приемлемые вещества, такие как смачивающие вещества, или незначительные количества вспомогательных веществ, таких как увлажняющие вещества или эмульгаторы, консерванты или буферы, которые увеличивают срок годности или эффективность соединения.
Фармацевтическая композиция может, например, находиться в форме, подходящей для перорального введения, в форме таблетки, капсулы, пилюли, порошка, составов с замедленным высвобождением, раствора суспензии, жидких растворов (например, растворов для инъекций и инфузий), подходящей для парентеральной инъекции в форме стерильного раствора, суспензии или эмульсии, подходящей для местного применения в форме мази или крема, порошков, липосом и суппозиториев (например, для ректального введения в форме суппозиториев). Типичные формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. При желании такие лекарственные формы могут быть соответствующим образом забуферены. Лекарственная форма зависит от предполагаемого способа введения и терапевтического применения.
Фармацевтическая композиция может находиться в лекарственной форме с разовой дозой, применяемой для разового введения точно отмеренных доз.
Соединения по изобретению могут быть введены перорально. Пероральное введение может включать проглатывание, вследствие чего соединение попадает в желудочно-кишечный тракт, или может быть использовано буккальное или сублингвальное введение, путем которого соединение попадает в кровоток непосредственно из ротовой полости. Лекарственные формы, применяемые для перорального введения, включают твердые формы, такие как, таблетки, капсулы, содержащие твердые частицы, жидкости или порошки, пастилки для рассасывания (включающие заполненные жидкостью), жевательные пастилки, мультичастицы и наночастицы, гели, твердый раствор, липосомы, пленки (включая мукоадгезивные), суппозитории, спреи и жидкие лекарственные формы. Такие капсулы или таблетки могут включать формы с контролируемым высвобождением. В случае капсул, таблеток и пилюль, лекарственные формы могут также содержать буферные вещества или могут быть приготовлены с энтеросолюбильными покрытиями.
Жидкие формы включают суспензии, растворы, сиропы и эликсиры. Такие формы могут быть использованы в качестве наполнителей в мягких или жестких капсулах и обычно включают носитель или вспомогательное вещество, например такое как одно или более из увлажняющих веществ, эмульгаторов, суспендирующих веществ, корригирующих вкус и запах веществ (например, подсластителей) или ароматизаторов, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгаторов и/или суспендирующих веществ. Жидкие формы могут быть также приготовлены путем растворения твердого вещества, например, из порошка для приготовления раствора.
Соединения по изобретению могут применяться в быстро растворяющихся, быстро распадающихся лекарственных формах, таких как лекарственные формы, описанные в публикации Expert Opinion in Therapeutic Patents, 11 (6), 981986 by Liang and Chen (2001), полное содержание которой включено в настоящее изобретение путем ссылки на нее.
Для лекарственных форм таблеток, в зависимости от дозы, содержание лекарственного средства может составлять от 1 масс. % до 80 масс. % от массы лекарственной формы, чаще всего, от 5 масс. % до 60 масс. % от массы лекарственной формы. Помимо лекарственного средства, таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают натрия крахмалгликолат, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. Обычно, разрыхлитель будет включать от 1 масс. % до 25 масс. %, предпочтительно, от 5 масс. % до 20 масс. % от массы лекарственной формы. Связующие вещества обычно применяют для придания когезионных свойств таблетке. Подходящие связующие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропил-целлюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводный и другие подобные формы), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двузамещенный кальция фосфата. Таблетки могут также необязательно включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. В случае присутствия, поверхностно-активные вещества обычно составляют от 0,2 масс. % до 5 масс. % от массы таблетки, а глиданты обычно составляют от 0,2 масс. % до 1 масс. % от массы таблетки. Таблетки также обычно содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие вещества обычно присутствуют в количествах от 0,25 масс. % до 10 масс. %, предпочтительно, от 0,5 масс. % до 3 масс. % от массы таблетки. Другие традиционные ингредиенты включают антиоксиданты, окрашивающие вещества, ароматизаторы, консерванты и вещества, корригирующее вкус и запах лекарственного средства. Смеси для приготовления таблеток могут быть непосредственно подвергнуты прессованию или вальцеванию с получением таблеток. Смеси для приготовления таблеток или порции смесей могут, в качестве варианта, подвергнуты увлажнению, сушке или гранулированию из расплава, отвердеванию из расплава, или экструдированию перед таблетированием. Готовая форма может включать один или более слоев и может быть покрыта оболочкой или быть без оболочки, или может быть инкапсулированной. Твердые лекарственные формы для перорального введения могут быть приготовлены для немедленного высвобождения и/или модифицированного высвобождения.
Лекарственные формы для модифицированного высвобождения включают отсроченное высвобождение, замедленное высвобождение, импульсное высвобождение, контролируемое высвобождение, таргетное высвобождение и программируемое высвобождение.
Для перорального введения, могут быть предложены композиции в форме таблеток или капсул, содержащих 0,01, 0,05, 0,1, 0,25, 0, 5, 1, 0, 2, 5, 5, 0, 10, 0, 15, 0, 25, 0, 50, 0, 75, 0 или 100 миллиграмм активного ингредиента для симптоматической корректировки дозы для пациента. Лекарственный препарат обычно содержит от приблизительно 0,01 мг до приблизительно 100 мг активного ингредиента. В еще одном варианте осуществления, лекарственный препарат содержит от приблизительно 0,01 до 0,25 мг активного ингредиента. В еще одном варианте осуществления, лекарственный препарат содержит приблизительно 0,25, 0,5, 1,0, 5,0, 15 или 25 мг активного ингредиента.
Соединения по изобретению могут быть также введены непосредственно в поток крови, в мышцу или во внутренний орган.
Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное и подкожное. Соответствующие устройства для парентерального введения включают игольчатые (в том числе микроигольчатые) шприцы, безыгольные шприцы и устройства для инфузий. Инъекционные форма препаратов (то есть стерильные инъекционные водные или масляные суспензии) могут быть приготовлены известными в данной области методами, используя одно или более из подходящих диспергирующих, смачивающих или суспендирующих веществ. Парентеральные формы обычно представляют собой водные растворы, которые могут содержать вспомогательные вещества, такие как соли, углеводы и буферные вещества (предпочтительно, с величиной рН от 3 до 9), но, в случае некоторых вариантов применения, их более удобно приготавливать в форме стерильного неводного раствора или в высушенной форме, которая будет использована в сочетании с подходящей средой, такой как стерильная апирогенная вода. Приготовление парентеральных форм в стерильных условиях, например, путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических методов, хорошо известных специалистам в данной области. Растворимость соединений по изобретению, используемых для приготовления парентеральных растворов, может быть увеличена путем использования соответствующих методов приготовления, таких как введение средств увеличения растворимости.
Лекарственные формы для парентерального введения могут быть приготовлены для немедленного высвобождения и/или модифицированного высвобождения. Лекарственные формы для модифицированного высвобождения включают отсроченное высвобождение, замедленное высвобождение, импульсное высвобождение, контролируемое высвобождение, таргетное высвобождение и программируемое высвобождение. Так, например, соединения по изобретению могут быть приготовлены в форме твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в форме имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких лекарственных форм включают стенты и микросферы из сополимера молочной и гликолевой кислот (PGLA) с нанесенным на них лекарственным средством.
Соединения по изобретению могут также применяться местно на коже или слизистой оболочке, то есть дермально или трансдермально, например, с использованием трансдермальных пластырей или ионтофоретических устройств, для интраокулярного введения или интраназального или ингаляционного введения. Лекарственная форма для местного введения может включать соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие пораженные области. Когда соединения по этому изобретению вводят с помощью трансдермального устройства, введение будет осуществляться с использованием пластыря, либо типа резервуара и типа пористой мембраны, либо типа разнообразных твердых матриц. Типичные лекарственные формы для местного введения включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, трансдермальные пластыри, капсулы, имплантаты, губки, волокна, бинты и микроэмульсии. Могут быть также использованы липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть введены вещества, способствующие проникновению. Другие методы местного применения включают доставку лекарственного средства путем электропорации, ионтофореза, фонофореза, сонофореза и микроигольчатой или безыгольной (например Powderject™, Bioject™, и так далее) инъекции.
Лекарственные формы, которые могут применяться для местного введения в глаза, включают, например, глазные капли, в которых соединение по настоящему изобретению растворено или суспендировано в подходящем вспомогательном веществе. Типичная форма, подходящая для глазного или ушного введения, может представлять собой капли микронизированной суспензии или раствор в изотоническом стерильном физиологическом растворе с корректированной величиной рН. Другие формы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (то есть рассасывающиеся гелевые губки, коллаген) и небиоразлагаемые (то есть силиконовые) имплантаты, капсулы, линзы и дисперсные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, полимер целлюлозы, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, геллановая камедь, может быть введен в лекарственную форму вместе с консервантом, таким как хлорид бензалкония. Такие составы могут быть также доставлены с использованием ионтофореза.
Лекарственные формы для местного применения могут быть приготовлены таким образом, что они могут осуществлять немедленное и/или модифицированное высвобождение. Формы с модифицированным высвобождением включает отсроченное, замедленное, импульсное, контролируемое, таргетное и программируемое высвобождение.
Соединения по изобретению могут быть также введены интраназально или ингаляционно, обычно, в форме сухого порошка (либо сами по себе, либо в форме смеси, например, в сухой смеси с лактозой, либо в виде смеси с частицами, например, смеси с фосфолипидами, такими как, фосфатидилхолин), из ингалятора сухого порошка или в форме распыляемого аэрозоля из контейнера под давлением, насоса, спрея, распылителя (предпочтительно распылителя, использующего электрогидродинамику для создания мелкодисперсного тумана) или распылителя, в котором используется или не используется подходящий пропеллент, такой как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. В случае интраназального применения, порошок может включать биоадгезивное вещество, например хитозан или циклодекстрин. Контейнер под давлением, насос, спрей, аэрозольный ингалятор или небулайзер содержит раствор или суспензию соединения (соединений) по изобретению, содержащие, например, этанол, водный раствор этанола или подходящее альтернативное вещество для диспергирования, солюбилизации или пролонгирования высвобождения активного соединения, пропеллент (пропелленты) в качестве растворителя и необязательное поверхностно-активное вещество, такое как триолеат сорбитана, олеиновая кислота или олигомолочная кислота. Перед применением в форме сухого порошка или суспензии, лекарственный препарат может быть микронизирован до размера частиц, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Это может быть достигнуто с помощью любого подходящего метода тонкого измельчения, такого как размол на спиральной струйной мельнице, размол на струйной мельнице с псевдоожиженным слоем, обработка сверхкритической жидкостью с формированием наночастиц, гомогенизация под высоким давлением или распылительная сушка.
Капсулы (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы (НРМС)), блистеры и картриджи для использования в ингаляторе или инсуффляторе могут быть приготовлены таким образом, чтобы они содержали порошковую смесь соединения по изобретению, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор эффективности, такой как лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно, в форме моногидрата. Другие подходящие вспомогательные вещества включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
В лекарственные формы по настоящему изобретению, предназначенные для ингаляционного/интраназального введения, можно добавлять подходящие ароматизаторы, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрия.
Соединения по изобретению могут быть введены ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Традиционной основой для суппозиториев является масло какао, но, в зависимости от конкретного случая, могут быть использованы и различные альтернативные основы.
Лекарственные формы для ректального/вагинального введения могут быть приготовлены в формах с мгновенным и/или модифицированным высвобождением. Формы с модифицированным высвобождением включает отсроченное, замедленное, импульсное, контролируемое, таргетное и программируемое высвобождение.
Соединения по изобретению могут быть также введены непосредственно в глаз или ухо, обычно, в форме капель микронизированной суспензии или раствор в изотоническом стерильном физиологическом растворе с корректированной величиной рН. Другие формы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (то есть рассасывающиеся гелевые губки, коллаген) и небиоразлагаемые (то есть силиконовые) имплантаты, капсулы, линзы и дисперсные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, полимер целлюлозы, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, геллановая камедь, может быть введен в лекарственную форму вместе с консервантом, таким как хлорид бензалкония. Такие составы могут быть также доставлены с использованием ионтофореза.
Лекарственные формы для введения в глаз/ухо могут быть приготовлены таким образом, что они могут обеспечивать немедленное и/или модифицированное высвобождение. Формы с модифицированным высвобождением включает отсроченное, замедленное, импульсное, контролируемое, таргетное и программируемое высвобождение.
Кроме того, могут быть использованы и другие известные в области фармацевтики вспомогательные вещества и способы введения. Фармацевтические композиции по изобретению могут быть приготовлены любым из хорошо известных в фармацевтике методов, таких как методы приготовления эффективных лекарственных форм и способы эффективного введения. Изложенные выше соображения относительно эффективных лекарственных форм и способов эффективного введения хорошо известны в данной области и описаны в общеизвестных учебниках. Приготовление лекарственных форм обсуждаются, например, в монографиях и руководствах Hoover, John Е., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Приемлемые вспомогательные вещества являются нетоксичными для субъектов в используемых дозировках и концентрациях и могут включать одно или более из следующих вспомогательных веществ: 1) буферы, такие как фосфатный, цитратный или из других органических кислот; 2) соли, такие как хлорид натрия; 3) антиоксиданты, такие как аскорбиновая кислота или метионин; 4) консерванты, такие как хлорид октадецилдиметилбензиламмония, хлорид гексаметония, хлорид бензалкония, хлорид бензетония, фенол, бутиловый или бензиловый спирт; 5) алкилпарабены, такие как метил- или пропилпарабен, катехин, резорцин, циклогексанол, 3-пентанол или м-крезол; 6) низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; 7) белки, такие как сывороточный альбумин, желатин или иммуноглобулины; 8) гидрофильные полимеры, такие как поливинилпирролидон; 9) аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; 10) моносахариды, дисахариды или другие углеводы, включая глюкозу, маннозу или декстрины; 11) хелатообразующие вещества, такие как этилендиаминтетрауксусная кислота (EDTA); 12) сахара, такие как сахароза, маннит, трегалоза или сорбит; 13) солеобразующие противоионы, такие как натрий, комплексы металлов (например, комплексы Zn-белок) или 14) неионные поверхностно-активные вещества, такие как полисорбаты (например, полисорбат 20 или полисорбат 80), полоксамеры или полиэтиленгликоль (PEG).
Липосома, содержащая соединения по изобретению, может быть получена известными в данной области способами (смотрите, например, Chang, H.I.; Yen, М.K.; Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy; Int J Nanomedicine 2012; 7; 49-60). В частности, пригодные для целей изобретения липосомы могут быть получены методом обращенно-фазового выпаривания с использованием липидной композиции, содержащей фосфатидилхолин, холестерин и PEG-дериватизированный фосфатидилэтаноламин (PEG-РЕ). Липосомы экструдируют через фильтры с определенным размером пор для получения липосом требуемого диаметра.
Соединения по изобретению могут быть также заключены в микрокапсулы, приготовленные, например, методами коацервации или межфазной полимеризации, например, в гидроксиметилцеллюлозные или желатиновые микрокапсулы и полиметилметакрилатные микрокапсулы, соответственно, в коллоидных системах доставки лекарственных средств (например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. Такие методы раскрыты в монографии Remington, The Science and Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Могут использоваться препараты с замедленным высвобождением. Подходящие примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение по изобретению, причем эти матрицы находятся в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц с замедленным высвобождением включают полиэфиры, гидрогели (например, поли(2-гидроксиэтил-метакрилат) или поливиниловый спирт), полилактиды, сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, неразлагающийся сополимер этилен-винилацетат, разлагающиеся сополимеры молочной и гликолевой кислот, такие как сополимеры, которые используют для формирования депо-суспензии лейпролида ацетата (микросферы для инъекций, состоящие из сополимера молочной и гликолевой кислот и лейпролида ацетата), изобутират ацетата сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
Лекарственные формы, используемые для внутривенного введения, должны быть стерильными. Это легко достигается, например, путем фильтрации через стерилизующие фильтрующие мембраны. Соединения по изобретению обычно помещают в контейнер, имеющий стерильное входное отверстие, например, в пакет или флакон для раствора для внутривенного введения, имеющие пробку, прокалываемую иглой для подкожных инъекций.
Подходящие эмульсии могут быть приготовлены с использованием выпускаемых промышленностью жировых эмульсий, таких как липидные эмульсии, содержащие соевое масло, жировая эмульсия для внутривенного введения (например, содержащая сафлоровое масло, соевое масло, яичные фосфатиды и глицерин в воде), эмульсии, содержащие соевое масло и триглицериды со средней длиной цепи и липидные эмульсии хлопкового масла. Активный ингредиент может быть либо растворен в предварительно смешанной эмульсионной композиции, либо, в качестве варианта, он может быть растворен в масле (например, соевом масле, сафлоровом масле, хлопковом масле, кунжутном масле, кукурузном масле или миндальном масле) с образованием эмульсии при смешивании с фосфолипидом (например, яичными фосфолипидами, соевыми фосфолипидами или соевым лецитином) и водой. Следует иметь в виду, что для корректировки тоничности эмульсии могут быть добавлены и другие ингредиенты, например глицерин или глюкоза. Подходящие эмульсии обычно содержат до 2 0% масла, например от 5 до 20%. Жировая эмульсия может содержать капли жира размером от 0,1 до 1,0 мкм, в частности от 0,1 до 0,5 мкм, и иметь величину рН в диапазоне от 5,5 до 8,0.
Например, эмульсионные композиции могут представлять собой эмульсии, приготовленные путем смешения соединения по изобретению с липидными эмульсиями, содержащими соевое масло или его компоненты (соевое масло, яичные фосфолипиды, глицерин и воду).
Промежуточный продукт формы лекарственного средства (DPI) представляет собой подвергнутый частичной обработке материал, который должен быть подвергнут последующим стадиям обработки перед тем как он станет нерасфасованной лекарственной формой препарата. Соединения по изобретению могут быть приготовлены в виде промежуточного продукта формы лекарственного средства, содержащего ингредиент в форме с более высокой свободной энергией, чем кристаллическая форма. Одной причиной для использования DPI является необходимость улучшения характеристик пероральной абсорбции, недостаточной вследствие низкой растворимости, медленного растворения, улучшения массопереноса через слизистый слой, прилегающий к эпителиальным клеткам, и, в некоторых случаях, ограничений вследствие биологических барьеров, таких как метаболизм и переносчики. Другие причины могу включать необходимость повышения стабильности в твердом состоянии и улучшения технологичности последующих этапов обработки. В одном варианте осуществления, промежуточный продукт формы лекарственного средства содержит соединение по изобретению, выделенное и стабилизированное в аморфном состоянии (например, в форме дисперсий твердого аморфного вещества (ASD)). В данной области известно много методов получения дисперсий твердого аморфного вещества, которые продуцируют материал, подходящий для интеграции в нерасфасованную лекарственную форму, например, высушенные распылением дисперсии (SDD), экструдаты из расплава (часто называемые как НМЕ), совместно осажденные осадки, аморфные наночастицы лекарственного средства и наноадсорбаты. В одном варианте осуществления, дисперсии аморфного твердого вещества включают соединение по изобретению и полимерное вспомогательное вещество. Другие вспомогательные вещества, а также концентрации указанных вспомогательных веществ, и соединение по изобретению являются хорошо известными в данной области и описаны в общеизвестных руководствах. Смотрите, например, монографию "Amorphous Solid Dispersions Theory and Practice" by Navnit Shah et al.
В другом аспекте, в изобретении предлагается соединение по изобретению или его фармацевтически приемлемая соль для применения в качестве лекарственного препарата, в частности, лекарственного препарата для лечения аномального роста клеток.
В еще одном аспекте, в изобретении предлагается применение соединения по изобретению или его фармацевтически приемлемой соли для производства лекарственного препарата для лечения аномального роста клеток у субъекта, например, опухоли, например, МЕК-ассоциированной опухоли, у субъекта.
В еще одном аспекте, в изобретении предлагается соединение любой из описанных в изобретении формул или его фармацевтически приемлемая соль для применения при лечении аномального роста клеток, например, опухоли, например МЕК-ассоциированной опухоли.
Введение соединений по изобретению может быть осуществлено любым способом, позволяющим доставить соединения к месту его действия. Эти способы включают пероральное введение, интрадуоденальные способы введения, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или инфузию), местное и ректальное введение.
Режимы дозирования могут быть скорректированы для достижения оптимального требуемого ответа. Например, может быть введена одна разовая доза, могут быть введены несколько разделенных доз в течение конкретного периода времени или доза может быть пропорционально уменьшена или увеличена в зависимости от требований терапевтической ситуации. Особенно удобно приготавливать в форме разовой дозы парентеральные композиции с точки зрения простоты введения и постоянства дозы. Используемая в изобретении форма с разовой дозой относится к физически дискретным единицам, подходящим в качестве разовых доз для подвергаемых лечению млекопитающих; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для достижения требуемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Требования к лекарственным формам с разовой дозой по изобретению обусловлены и напрямую зависят от (а) уникальных характеристик вводимого соединения и конкретного терапевтического или профилактического эффекта, который должен быть достигнут, и от (b) ограничений, которые существуют при составлении композиций такого активного соединения для лечения заболеваний у индивидуумов.
Так, например, для специалиста в данной области будет очевидным, после того как он ознакомится с описанием настоящего изобретения, что доза и режим дозирования корректируются с использованием методов, хорошо известных в области терапии. То есть может быть легко установлена максимально переносимая доза, а также может быть определено эффективное количество, позволяющее достигать обнаруживаемого положительного терапевтического эффекта у пациента, и, кроме того, могут быть определены временные требования по введения каждого лекарственного средства для достижения обнаруживаемого положительного терапевтического эффекта у пациента. Соответственно, не смотря на то, что в изобретении приведены в качестве примеров конкретные дозы и режимы введения, тем не менее, эти примеры никоим образом не ограничивают дозы и режим введения, которые могут быть предложены пациенту при применении на практике настоящего изобретения.
Следует иметь в виду, что величины доз могут изменяться в зависимости от типа и тяжести болезненного состояния, которое требуется облегчить, и они могут включать разовые или многократные дозы. Кроме того, следует иметь в виду, что для любого конкретного субъекта, конкретные режимы дозирования должны быть скорректированы в течение времени в соответствии с потребностью индивидуума и профессиональным суждением лица, осуществляющего или контролирующего введение композиций, и что диапазоны доз, представленные в изобретении, являются только примерами и они никоим образом не ограничивают разнообразие или практическое применение заявленной композиции. Например, дозы могут быть скорректированы с учетом фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или данные лабораторных анализов. Так, например, настоящее изобретение включает возможность увеличения индивидуальной дозы для пациента, определенной специалистом в данной области. Определение соответствующих доз и режимов введения химиотерапевтического средства хорошо известно в соответствующей области техники и, следует иметь в виду, что, после ознакомления с идеями настоящего изобретения, для специалиста в данной области не будет представлять особой сложности определить соответствующие дозы и режимы введения. В одном варианте осуществления, эффективная доза находится обычно в диапазоне от приблизительно 0,001 до приблизительно 100 мг на кг массы тела в сутки, и, часто, от приблизительно 0,01 до приблизительно 35 мг/кг/сутки, в разовой дозе или в разделенных дозах. Для мужчины с массой тела 70 кг, это будет составлять от приблизительно 0,07 мг/сутки до приблизительно 7000 мг/сутки, чаще всего, от приблизительно 10 мг/сутки до приблизительно 1000 мг/сутки. Иногда, доза составляет приблизительно 10, 20, 30, 40, 50, 60, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 750, 800, 900 или 1000 мг/сутки. Иногда, доза составляет от приблизительно 10 мг/сутки до приблизительно 1000 мг/сутки, от приблизительно 10 мг/сутки до приблизительно 750 мг/сутки, от приблизительно 10 мг/сутки до приблизительно 600 мг/сутки, от приблизительно 10 мг/сутки до приблизительно 300 мг/сутки, от приблизительно 10 мг/сутки до приблизительно 150 мг/сутки, от приблизительно 20 мг/сутки до приблизительно 750 мг/сутки, от приблизительно 20 мг/сутки до приблизительно 600 мг/сутки, от приблизительно 2 0 мг/сутки до приблизительно 300 мг/сутки, от приблизительно 20 мг/сутки до приблизительно 150 мг/сутки, от приблизительно 50 мг/сутки до приблизительно 750 мг/сутки, от приблизительно 50 мг/сутки до приблизительно 600 мг/сутки, от приблизительно 50 мг/сутки до приблизительно 300 мг/сутки, от приблизительно 50 мг/сутки до приблизительно 150 мг/сутки, от приблизительно 75 мг/сутки до приблизительно 750 мг/сутки, от приблизительно 75 мг/сутки до приблизительно 600 мг/сутки, от приблизительно 75 мг/сутки до приблизительно 300 мг/сутки, или от приблизительно 75 мг/сутки до приблизительно 150 мг/сутки. В некоторых случаях, уровни доз ниже нижнего предела упомянутого выше диапазона могут быть более чем достаточными, в то время как в других случаях, могут быть использованы еще более высокие дозы, не вызывая каких-либо вредных побочных эффектов, при этом такие высокие дозы обычно разделяют на несколько меньших доз для введения в течение дня. В одном варианте осуществления, субъекту вводят приблизительно 50 мг/сутки.
В случаях, когда для лечения конкретного заболевания или состояния может быть желательным введение комбинации активных соединений, в объем настоящего изобретения входит случай, когда две или более фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, удобно объединить в форме набора, применяемого для совместного введения композиций. Так, например, набор по изобретению включает две или более раздельных фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, и средства для раздельного хранения указанных композиций, такие как контейнер, разделенный на несколько отделений сосуд или разделенный на несколько отделений пакет из фольги. Примером такого набора является хорошо всем известная блистерная упаковка, используемая для упаковки таблеток, капсул и других подобных лекарственных форм.
Набор по изобретению особенно удобен в случае введения различных лекарственных форм, например, пероральных и парентеральных, для введения отдельных композиций с различными интервалами между приемами лекарственного средства или для титрования отдельных композиций при сопоставлении друг с другом. Для облегчения соблюдения предписанного режима терапии, набор обычно включает инструкции по применению и может быть снабжен памяткой. В некоторых вариантах осуществления, набор включает соединение или его фармацевтическую композицию и диагностическое средство. В других вариантах осуществления, набор включает соединение или его фармацевтическую композицию и одно или более терапевтических средств, таких как ингибитор BRAF, например, ингибитор BRAF, выбранный из N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамид; N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)-фенил)-3-фторпропан-1-сульфонамида; N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамида; N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)-4-фторфенил)-пропан-1-сульфонамида; N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фтор-фенил)-3-фторпропан-1-сульфонамида; N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-фенил)-3-фтор-пропан-1-сульфонамида; N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)окси]-4-фторфенил}пропан-1-сульфонамида; N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторпиридин-2-ил)пропан-1-сульфонамида; N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}-3-фторпропан-1-сульфонамида; N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2.1.1]гексан-2-сульфонамида, (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида и N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)-амино)-4-фторфенил)-3-фтор-азетидин-1-сульфонамида или их фармацевтически приемлемых солей;
На следующих далее схемах и описаниях представлена общая подробная информация относительно получения соединений по изобретению.
Соединения по изобретению могут быть получены любым методом, известным в данной области для получения соединений аналогичной структуры. В частности, соединения по изобретению могут быть получены с использованием методик, описанных со ссылкой на приведенные далее схемы реакций, или с использованием конкретных методов, описанных в примерах, или с использованием аналогичных им процессов.
Для специалиста в данной области является очевидным, что экспериментальные условия, указанные на приведенных далее схемах, иллюстрируют подходящие условия для осуществления показанных превращений, и что может возникнуть необходимость или желание изменить точные условия, используемые для получения соединений формулы I и соединений, подпадающие под формулу I, например, соединений формулы II и других подобных соединений.
Кроме того, для специалиста в данной области является очевидным, что на любой стадии синтеза соединений по изобретению может возникнуть необходимость или желание защитить одну или несколько реакционноспособных групп с целью предотвращения протекания нежелательных побочных реакций. В частности, может возникнуть необходимость или желание защитить аминогруппы или спиртовые группы. Защитные группы (PG), используемые при получении соединений по изобретению, могут быть использованы обычным способом. Смотрите, например, защитные группы, описанные в монографии "Greene's Protective Groups in Organic Synthesis" by Theodora W Greene and Peter G M Wuts, third edition, (John Wiley and Sons, 1999), в частности в главе 7 ("Protection for the Amino Group") и главе 2 ("Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols", полное содержание которой включено в настоящее изобретение путем ссылки на нее, в которой также описаны методы удаления таких групп.

На схеме 1 описан общий метод получения соединения 8, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой С3-С6 циклоалкил, и R4 определен для формулы I. Выпускаемая промышленностью 2,6-дихлор-4-метилникотиновая кислота (соединение 1) может быть превращена в ее эфирный аналог 2 при обработке с помощью (триметилсилил)диазометан. Соединение 2 может быть превращено в диметиламиновинильное промежуточное соединение 3 при обработке с помощью N,N-диметилформамида диметилацеталя. Соединение 3 может быть превращено в альдегидное промежуточное соединение 4 при обработке с помощью соответствующей кислоты, такой как хлористоводородная кислота, в подходящем растворителе, таком как эфир. Циклизация соединения 4 может быть осуществлена путем взаимодействия соединения 4 с реагентом, имеющим формулу R3NH2, где R3 представляет собой С3-С6 циклоалкил, в присутствии восстановителя (например, цианоборгидрида натрия), в подходящем растворителе, таком как метанол, с получением соединения 5. Соединение 5 может быть превращено в соединение 6 при обработке с помощью триметилсилилйодида в подходящем растворителе, таком как ацетонитрил. Соединение 6 может быть метилировано путем обработки с помощью метилйодида в присутствии соответствующего основания, такого как карбонат щелочного металла, такого как карбонат калия, в присутствии соответствующего растворителя, такого как THF, с получением соединения 7. Соединение 7 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2 где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 8.

На схеме 2 описан общий метод получения соединения 17, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I. Выпускаемый промышленностью 4-бром-2,6-дихлорпиридин может быть подвергнут литированию с помощью реагента, такого как диизопропиламид лития, и обработан с помощью диоксида углерода с получением карбоновой кислоты 10. Соединение 10 может быть превращено в соединение 11 путем кипячения с обратным холодильником в водном растворе основания, такого как 4М гидроксид натрия. Соединение 11 может быть метилировано путем обработки с помощью метилйодида в присутствии соответствующего основания, такого как карбонат щелочного металла, такого как карбонат калия, в присутствии соответствующего растворителя, такого как DMF, с получением соединения 12. Соединение 12 может быть превращено в промежуточное соединение винилового эфира 13 путем проведения реакции Сузуки с (Е)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксабороланом (соединение (1)), используя катализатор, такой как метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II) и основание щелочного металла (например, карбонат щелочного металла, например, водный раствор карбоната калия), в подходящем растворителе, таком как 1,4-диоксан. Соединение 13 может быть подвергнуто реакции образования оксимина при обработке с помощью реагента формулы R3a-NH2HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, или (С3-С6 циклоалкил)С1-С6 алкокси-, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя триэтиламин и HCl и нагревание в подходящем растворителе, таком как 1,4-диоксан, с получением соединения 14. Соединение 14 может быть восстановлено до промежуточного соединения алкоксиамина 15, используя соответствующий восстановитель, такой как цианоборгидрид натрия, в подходящем растворителе, таком как изопропанол. Соединение 15 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, и соответствующим образом циклизируют, затем проводят необязательное удаление защитной группы, если соединение 16 содержит защитную группу Р1 (используя стандартные условия проведения реакции удаления защитной группы для спиртов, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмония фторид), с получением соединения 17, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-.

На схеме 3 описан общий метод получения соединения 20, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой СН3-, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I. Соединение 15, полученное как описано на схеме 2, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, или (С3-С6 циклоалкил)С1-С6 алкокси-, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, и где R4 определен для формулы I, может быть циклизировано при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 16. Соединение 16 может быть йодировано, используя н-йодсукцинимид и п-толуолсульфоновую кислоту, в подходящем растворителе, таком как 1:1 MeOH:THF, с получением соединения 18. Соединение 18 может быть подвергнуто реакции сочетания Негиши с метилцинка(II) хлоридом, используя катализатор, например, палладиевый катализатор, такой как бис (тритретбутилфосфин), затем необязательному удалению защитной группы, если соединение 19 содержит защитную группу (используя стандартные условия проведения реакции удаления защитной группы для спиртов, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 20, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси.

На схеме 4 описан общий метод получения соединения 24, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой Н, и R4 определен для формулы I. Соединение 13, полученное как описано на схеме 2, может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 21. Соединение 21 может быть подвергнуто гидролизу с образованием промежуточного соединения альдегида 22 при кислотных условиях, используя кислоту, такую как трифторуксусная кислота, в подходящем растворителе, таком как дихлорметан. Соединение 22 может быть циклизировано при обработке с помощью реагента, имеющего формулу P2-NH2, where Р2 представляет собой защитную группу для аминогруппы, такую как бензил, п-метоксибензил или 2,4-диметоксибензил, используя восстановитель, такой как триацетоксиборгидрид натрия, в подходящем растворителе, таком как дихлорэтан, с получением соединения 23. Удаление защитной группы с соединения 2 3 может быть достигнуто путем использования стандартных условий для удаления защитной группы, известных специалистам в данной области, таких как нагревание с трифторуксусной кислотой или хлористоводородной кислотой, с получением соединения 24.

На схеме 5 описан общий метод получения соединения 27, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидроксиС1-С6 алкил, и R4 определен для формулы I. Соединение 13, полученное как описано на схеме 2, может быть подвергнуто гидролизу в промежуточное соединение альдегида 25, используя соответствующую кислоту, такую как трифторуксусная кислота. Соединение 25 может быть подвергнуто реакции с реагентом формулы P1O-(C1-С6 алкил)-ONH2HCl, где Р1 представляет собой защитную группу для спиртовой группы, такую как третбутилдиметилсили, используя соответствующий восстановитель, такой как триацетоксиборгидрид натрия и уксусную кислоту в подходящем растворителе, таком как дихлорэтан, при 60°С с получением циклизированного с удаленной защитной группой соединения 26. Соединение 26 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 27.
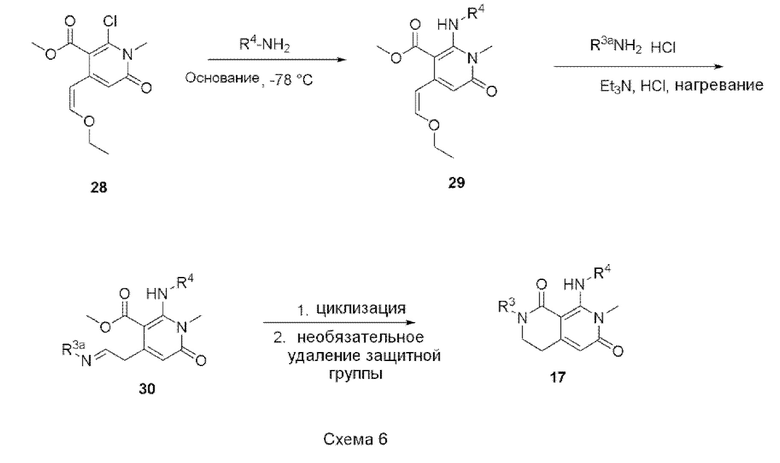
На схеме 6 описан альтернативный общий метод получения соединения 17, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I. Соединение 28, полученное методом, аналогичным методу, описанному для соединения 13, может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 29. Соединение 29 может быть подвергнуто реакции образования оксимина при обработке с помощью реагента формулы R3aNH2 HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя триэтиламин и HCl и нагревание, в подходящем растворителе, таком как 1,4-диоксан, с получением соединения 30. Соединение 30 может быть циклизировано с помощью соответствующего восстановителя, такого как цианоборгидрид натрия, и уксусной кислоты в подходящем растворителе, таком как изопропанол, и затем подвергнуто необязательному удалению защитной группы, если соединение 30 содержит защитную группу Р1 (используя стандартные условия удаления защитной группы со спиртовой группы, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 17, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси.

На схеме 7 описан альтернативный общий метод получения соединение 17, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидроксиС1-С6 алкокси, и R4 определен для формулы I. Соединение 2 8 может быть подвергнуто реакции образования оксимина при обработке с помощью реагента формулы TBSO-(С1-С6 алкил)-ONH2 HCl, используя триэтиламин и HCl и нагревание, в подходящем растворителе, таком как 1,4-диоксан, с получением соединения 32. Соединение 32 может быть защищено с помощью соответствующей защитной группы для спиртовой группы Р1, такой как третбутилдиметилсилил, используя третбутилдиметилсилилхлорид и соответствующее основание, такое как имидазол, в подходящем растворителе, таком как DMF, с получением соединения 14. Соединение 14 может быть восстановлено до промежуточного соединения алкоксиамина 15, используя соответствующий восстановитель, такой как цианоборгидрид натрия, в подходящем растворителе, таком как изопропанол. Соединение 15 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 16. С соединения 16 может быть удалена защитная группа, используя стандартные условия для удаления защитной группы для спиртовой группы, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид, с получением соединения 17.

На схеме 8 описан альтернативный общий метод получения соединение 20, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой СН3-, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I. Соединение 28 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 29. Соединение 29 может быть подвергнуто гидролизу в промежуточное соединение альдегида 22, используя соответствующую кислоту, такую как трифторуксусная кислота. Соединение 22 может быть подвергнуто реакции с реагентом формулы R3aNH2 HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя соответствующий восстановитель, такой как триацетоксиборгидрид натрия, и уксусную кислоту в растворителе, таком как дихлорэтан, при 60°С с получением циклизированного соединения 16. Соединение 16 может быть подвергнуто йодированию, используя п-йодсукцинимид и п-толуолсульфоновую кислоту в подходящем растворителе, таком как 1:1 MeOH:THF, с получением соединения 18. Соединение 18 может быть подвергнуто реакции сочетания Негиши с метилцинка(II) хлоридом, используя катализатор, такой как бис(тритретбутилфосфин)палладий (0), в подходящем растворителе, таком как THF, затем необязательному удалению защитной группы, если соединение 18 содержит защитную группу Р1 (используя стандартные условия для удаления защитной группы для спиртовой группы, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 20, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси.

На схеме 9 описан общий метод синтеза соединения 35, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой галоген, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I. Соединение 12 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 33. Соединение 33 может быть превращено в промежуточное соединения винилого эфира 21 путем проведения реакции Сузуки с (Е)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диокса-бороланом используя катализатор, например, палладиевый катализатор, такой как метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II), и основанием щелочного металла, например, карбоната щелочного металла, такого как водный раствор карбоната калия, в подходящем растворителе, таком как 1,4-диоксан. Соединение 21 может быть подвергнуто реакции образования оксимина при обработке с помощью реагента формулы R3aNH2 HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя триэтиламин и HCl, и нагревание, в подходящем растворителе, таком как 1,4-диоксан, с получением соединения 30. Соединение 30 может быть обработано с помощью соответствующего восстановителя, такого как цианоборгидрид натрия, и уксусной кислоты в подходящем растворителе, таком как изопропанол, с получением циклизированного соединения 31. Соединение 31 может быть подвергнуто галогенированию, используя условия, такие как обработка с помощью N-йодсукцинимида и п-толуолсульфоновой кислоты в 1:1 THF/MeOH, или с помощью N-бромсукцинимида в подходящем растворителе, таком как DMF, или с помощью N-хлорсукцинимида в подходящем растворителе, таком как DMF, или с помощью электрофильного фторирующего реагента Selectfluor в подходящем растворителе, таком как ацетонитрил, затем необязательному удалению защитной группы, если соединение 31 имеет защитную группу Р1 (такую как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 35, где R2 представляет собой йод, бром, хлор или фтор, соответственно, и R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси

На схеме 10 описан альтернативный общий метод получения соединение 17, которое представляет собой соединение формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I. Соединение 12 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии соответствующего основания, такого как третбутоксид калия, в подходящем растворителе, таком как THF, с получением соединения 36. Соединение 36 может быть превращено в промежуточное соединение винилового эфира 29 путем проведения реакции Сузуки с (Z)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксабороланом, используя катализатор, например, палладиевый катализатор, такой как метансульфонато(2-дицикло-гексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II), и соответствующее основание, такое как основание щелочного металла, например, карбонат щелочного металла, например, водный раствор карбонат калия, в растворителе, таком как 2-метилтетрагидрофуран. Соединение 29 может быть подвергнуто реакции с реагентом формулы R3aNH2 HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя триэтиламин и хлористоводородную кислоту в подходящем растворителе, таком как 1,4-диоксан, затем подвергнуто обработке с помощью соответствующего восстановителя, такого как пиридинборан, и хлористоводородной кислоты и нагревания при 60°С, затем необязательному удалению защитной группы, если соединение 29 имеет защитную группу Р1 (такую как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 17, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси.

На схеме 11 описан метод получения соединения 43, которое представляет собой соединение формулы I, где R1 представляет собой фенил, R2 представляет водород, R3 представляет водород, и R4 описан для формулы I. Выпускаемый промышленностью 2,6-дихлор-4-йодпиридин может быть подвергнут литированию с помощью реагента, такого как диизопропиламид лития, и обработан диоксидом углерода с получением соединение 37. Соединение 37 может быть превращено в соединение 3 8 путем кипячения с обратным холодильником в водном растворе основания, такого как 4М гидроксида натрия. Соединение 3 8 может быть метилировано путем обработки с помощью метилйодида в присутствии соответствующего основания, такого как карбонат щелочного металла, такого как карбонат калия, в присутствии соответствующего растворителя, такого как DMF, с получением соединения 39. Соединение 3 9 может быть превращено в промежуточное соединение винилового эфира 4 0 путем проведения реакции Сузуки с 4,4,5,5-тетраметил-2-(1-фенилвинил)-1,3,2-диоксабороланом, используя соответствующий катализатор, например палладиевый катализатор (например Pd(dppf)Cl2), и основание, такое как карбонат щелочного металла (например водный раствор карбоната калия), в подходящем растворителе, таком как 1,4-диоксан. Соединение 40 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, с получением соединения 41. Соединение 41 может быть циклизировано с помощью (2,4-диметоксифенил)метанамина в присутствии кислоты Льюиса, такой как триметилалюминий, путем нагревания в подходящем растворителе, таком как толуол, с получением соединение 42. Соединение 42 может быть подвергнуто реакции удаления защитной группы путем нагревания с подходящей кислотой, такой как TFA, с получением соединение 43.
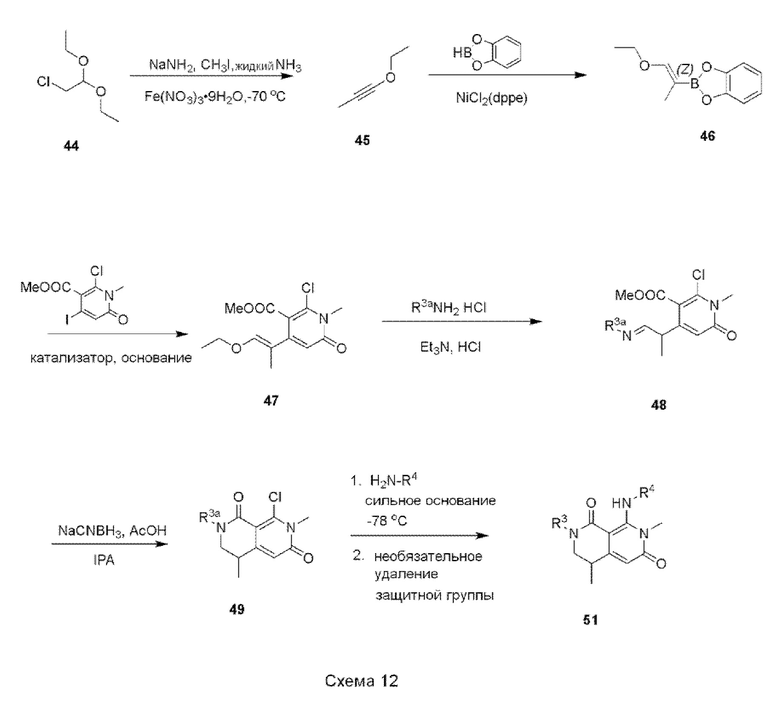
На схеме 12 описан метод получения соединения 51, которое представляет собой соединение формулы I, где R1 представляет собой метил, R2 представляет собой водород, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I. Соединение 44 может быть подвергнуто обработке с помощью NH3, Fe(NO3)3 ⋅ 9H2O и NaNH2 при низкой температуре, затем обработке с помощью метилйодида с получением соединения 45. Соединение 45 может быть нагрето вместе с бензо[d] [1, 3, 2] диоксабороланом в подходящем растворителе, таком как толуол, с катализатором, таким как NiCl2 (dppe), с получением соединения 46. Соединение 46 может быть превращено в соединение 47 путем проведения реакции с соединением 39, используя условия проведения реакции Сузуки, например, в присутствии катализатора, например, палладиевого катализатора, такого как Pd(dppf)Cl2, и основания, такого как K3PO4, K2CO3, KOtBu, Cs2CO3, NaOH или триэтиламин, в подходящем растворителе, например, в смеси растворителей, такой как толуол/THF. Соединение 47 может быть подвергнуто реакции образования оксимина при обработке с помощью реагента формулы R3aNH2 HCl, где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и Р1 представляет собой защитную группу для спиртовой группы, такую как третбутил, бензил или третбутилдиметилсилил, используя триэтиламин и HCl, и нагревание, в подходящем растворителе, таком как 1,4-диоксан, с получением соединения 48. Соединение 48 может быть обработано с помощью соответствующего восстановителя, такого как цианоборгидрид натрия, и уксусной кислоты в подходящем растворителе, таком как изопропанол, с получением циклизированного продукта 49. Соединение 49 может быть подвергнуто нуклеофильному ароматическому замещению при обработке с помощью реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, такого как гексаметилдисилазид лития, в подходящем растворителе, таком как THF, затем необязательному удалению защитной группы, если соединение 49 содержит защитную группу Р1 (используя стандартные условия для удаления защитной группы для спиртовой группы, известные специалистам в данной области, такие как фосфорная кислота, трифторуксусная кислота или тетрабутиламмонияфторид) с получением соединения 51, где R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси.
Используемый в изобретении термин "защитная группа для аминогруппы" относится к производному групп, которые обычно используются для блокирования или защиты аминогруппы, в тот момент времени, когда проводят реакции с участием других функциональных группах на соединении. Примеры подходящих защитных групп для использования в любом из методов синтеза, описанных в изобретении, включают карбаматы, амиды, алкильные и арильные группы, имины, а также многие N-гетероатомные производные, которые могут быть удалены для регенерации требуемой аминогруппы. Неограничивающие примеры защитных групп для аминогруппы представляют собой третбутилоксикарбонил ("Boc"), 2-триметилсилилэтоксиметил (SEM) и п-метоксибензил (РМВ). Дополнительные примеры этих групп и других защитных групп можно найти в монографии Т.W. Greene, et al., Greene's Protective Groups in Organic Synthesis. New York: Wiley Interscience, 2006.
Используемый в изобретении термин "защитная группа для спиртовой группы" относится к производному групп, обычно применяемых для блокирования гидроксильной группы, в тот момент времени, когда проводят реакции с участием других функциональных группах на соединении. Примеры подходящих защитных групп для использования в любом из методов, описанных в изобретении, включают бензиловые, тритиловые, силиловые эфиры и другие подобные защитные группы.
Промежуточные соединения 7, 15, 18, 22, 26, 29, 30 и 31, представленные на приведенных выше схемах, являются также новыми промежуточными соединения, применяемыми для получения соединений формулы I, и предлагаются в качестве дополнительных вариантов осуществления изобретения.
В одном варианте осуществления, в изобретении предлагается способ получения соединений формулы I, включающий:
(a) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой С3-С6 циклоалкил, и R4 определен для формулы I, проведение реакции соединения формулы 7

с реагентом, имеющим формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания; или
(b) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I, циклизацию соединения формулы 15

где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и Р1 представляет собой защитную группу для спиртовой группы, в присутствии реагента, имеющего формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, затем необязательное удаление защитной группы для спиртовой группы, если она присутствует; или
(c) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой СН3-, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I, обработку соединения формулы 18

где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, Р1 представляет собой защитную группу для спиртовой группы, и R4 определен для формулы I, с помощью метилцинка(II) хлорида в присутствии палладиевого катализатора, затем удаление защитной группы для спиртовой группы, если она присутствует; или
(d) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой Н, и R4 определен для формулы I, циклизацию соединения формулы 22

где R4 определен для формулы I, в присутствии реагента, имеющего формулу P2-NH2, где Р2 представляет собой защитную группу для аминогруппы, затем удаление защитной группы для аминогруппы; или
(e) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидроксиС1-С6 алкил, и R4 определен для формулы I, реакцию соединения формулы 26
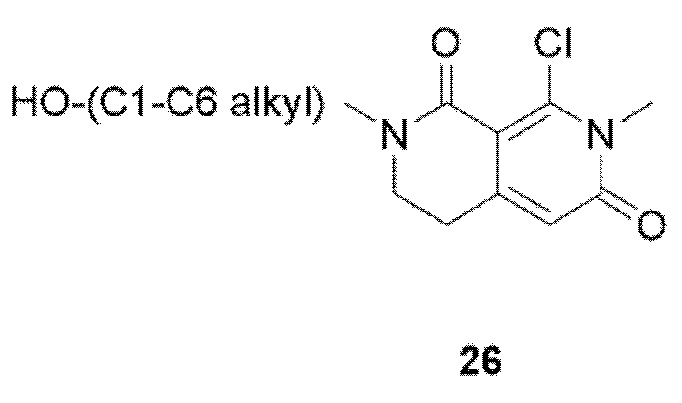
В присутствии реагента, имеющего формулу R4NH2, в присутствии сильного основания, где R4 определен для формулы I; или
(f) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидроксиС1-С6 алкил, и R4 определен для формулы I, циклизацию соединения, имеющего формулу 30

где Р1 представляет собой защитную группу для спиртовой группы, и R4 определен для формулы I, в присутствии восстановителя, затем удаление защитной группы для спиртовой группы; или
(г) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой галоген, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I, галогенирование соединения формулы 31

где R4 определен для формулы I, R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и Р1 представляет собой защитную группу для спиртовой группы, затем удаление защитной группы для спиртовой группы, если она присутствует; или
(h) для соединения формулы I, где R1 представляет собой Н, R2 представляет собой Н, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и R4 определен для формулы I, реакцию соединение формулы 29

где R4 определен для формулы I, с реагентом формулы P1O-(С1-С6 алкил)-ONH2HCl, где Р1 представляет собой защитную группу для спиртовой группы, в присутствии триэтиламина и хлористоводородной кислоты, затем удаление защитной группы для спиртовой группы; или
(i) для соединения формулы I, где R1 представляет собой фенил, R2 представляет собой водород, R3 представляет собой водород, и R4 описан для формулы I, циклизацию соединения, имеющего формулу 41

где R4 определен для формулы I, с (2,4-диметоксифенил)метанамином в присутствии кислоты Льюиса при повышенной температуре с получением соединения, имеющего формулу 42:

затем обработку соединения 42 с помощью кислоты; или
(j) для соединения формулы I, где R1 представляет собой метил, R2 представляет собой водород, R3 представляет собой гидрокси-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси, и R4 определен для формулы I, реакцию соединения, имеющего формулу 49

где R3a представляет собой P1O-С1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси или (С3-С6 циклоалкил)С1-С6 алкокси-, и Р1 представляет собой защитную группу для спиртовой группы, с реагентом, имеющим формулу R4NH2, где R4 определен для формулы I, в присутствии сильного основания, затем удаление защитной группы Р1, если она присутствует; и, необязательно, превращение соединения формулы I в его фармацевтически приемлемую соль.
Кроме того, предполагается, что синтетические промежуточные соединения 3, 4, 5, 6, 7, 13, 14, 15, 16, 18, 21, 22, 23, 25, 26, 28, 29, 30, 32, 33, 36, 37, 38, 39, 40, 41, 42, 47, 48 и 49 являются новыми и представляют собой дополнительные варианты осуществления этого изобретения.
Для более глубокого понимания настоящего изобретения, далее приводится подробное описание вариантов осуществления изобретения и примеры, включенные в настоящее изобретение. Следует иметь в виду, что данное изобретение не ограничивается конкретными примерами синтезов соединений, которые, несомненно, могут быть изменены. Следует также иметь в виду, что используемая в изобретении терминология служит только для описания конкретных вариантов осуществления и никоим образом не ограничивает изобретение.
Е1. Соединение формулы I:

или его фармацевтически приемлемая соль, где:
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, C1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
Е2. Соединение по варианту осуществления Е1 или его фармацевтически приемлемая соль, где R1 представляет собой Н.
Е3. Соединение по варианту осуществления Е1 или Е2 или его фармацевтически приемлемая соль, где R2 представляет собой Н.
Е4. Соединение по варианту осуществления Е1 или Е2 или его фармацевтически приемлемая соль, где R2 представляет собой СН3-.
Е5. Соединение по любому одному из вариантов осуществления E1 - Е4 или его фармацевтически приемлемая соль, где R3 представляет собой гидроксиС1-С6 алкил-.
Е6. Соединение по любому одному из вариантов осуществления E1 - Е5 или его фармацевтически приемлемая соль, где R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
Е7. Соединение по любому одному из вариантов осуществления Е1 - Е6 или его фармацевтически приемлемая соль, где R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из галогена и С1-С6 алкилтио.
Е8. Соединение формулы II:
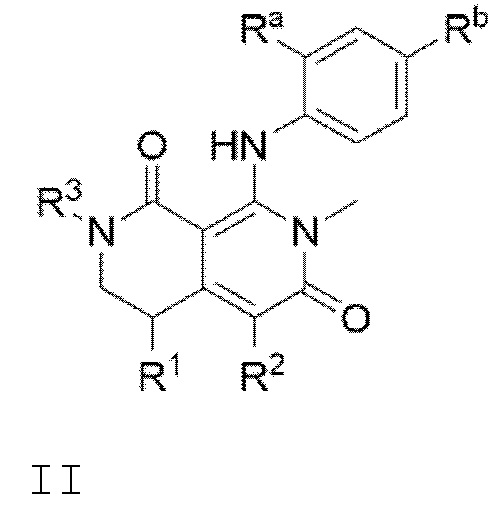
или его фармацевтически приемлемая соль, где
R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, C1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, C1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и C1-С6 алкил-С(=O)-.
Е9. Соединение по варианту осуществления Е8 или его фармацевтически приемлемая соль, где R1 представляет собой Н.
Е10. Соединение по варианту осуществления Е8 или Е9 или его фармацевтически приемлемая соль, где R2 представляет собой Н.
Е11. Соединение по варианту осуществления Е8 или Е9 или его фармацевтически приемлемая соль, где R2 представляет собой СН3-.
Е12. Соединение по любому одному из вариантов осуществления Е8 - Е11 или его фармацевтически приемлемая соль, где R3 представляет собой гидроксиС1-С6 алкил-.
Е13. Соединение по любому одному из вариантов осуществления Е8 - Е11 или его фармацевтически приемлемая соль, где R3 представляет собой Н.
Е14 Соединение по любому одному из вариантов осуществления Е8 - Е13 или его фармацевтически приемлемая соль, где Ra представляет собой галоген.
Е15. Соединение по любому одному из вариантов осуществления Е8 - Е14 или его фармацевтически приемлемая соль, где Rb представляет собой галоген, C1-С6 алкил, C1-С6 алкилтио или фторС1-С6 алкокси.
Е16. Соединение по варианту осуществления Е15 или его фармацевтически приемлемая соль, где Rb представляет собой C1-С6 алкилтио.
Е17. Соединение по варианту осуществления Е1, выбранное из следующих соединений:
8-((4-бром-2-фторфенил)амино)-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(циклопропилметокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-этокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-циклопропил-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-циклопропил-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-хлорфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2,3-дифторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-3-хлор-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(трифторметил)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-метоксифенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-((трифторметил)тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-изопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-циклопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-ацетил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-этилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(трифторметокси)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-((дифторметил)тио)-2-фторфенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-этокси-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-метокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-(третбутокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(S)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(S)-8- ((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5-хлор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
5-хлор-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5-фтор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
5-бром-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-йод-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-пропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-хлорфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-(2,2-дифторэтокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
или его фармацевтически приемлемая соль.
Е18. Соединение, которое представляет собой 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, имеющий структуру:

или его фармацевтически приемлемая соль.
Е19. Соединение, которое представляет собой 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, имеющий структуру:

Е20. Кристалл, включающий 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион.
Е21. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1.
Е22. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1 по варианту осуществления Е21, имеющий порошковую рентгенограмму, включающую характеристические пики при 5,0, 8,7, 9,3, 10,8, 14,5, 15,3, 18,8 и 20,5 градусов 2-тета (±0,2 градусов 2-тета).
Е23. Кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 1 по варианту осуществления Е21, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 1.
Е24. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2.
Е25. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 по варианту осуществления Е24, имеющий порошковую рентгенограмму, включающую характеристические пики при 7,1, 9,4, 12,4, 12,8, 14,3, 15,6, 16,4, 17,4, 18,5, 18,9, 19,5, 19,9, 21,1, 21,4, 23,2, 23,7, 24,8, 25,6, 27,6, 30,3, 33,2, 33,5 и 37,5 градусов 2-тета (±0,2 градусов 2-тета).
Е26. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 по варианту осуществления Е24, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 2.
Е27. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 по варианту осуществления Е25 или Е26, где порошковую рентгенограмму получают путем проведения анализа методом порошковой рентгеновской дифракции при 25°С и при относительной влажности ниже 10%.
Е28. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3.
Е29. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 по варианту осуществления Е28, имеющий пики на порошковой рентгенограмме при 13,7, 18,0 и 18,3 градусов 2-тета (±0,2 градусов 2-тета).
Е30. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)-фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 по варианту осуществления Е2 8, имеющий пики на порошковой рентгенограмме при 6,9, 9,1, 13,7, 18,0 и 18,3 градусов 2-тета (±0,2 градусов 2-тета).
Е31. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 по варианту осуществления Е2 8, имеющий пики на порошковой рентгенограмме в величинах угла 2-тета 6,9, 9,1, 11,8, 12,0, 13,7, 14,0, 15,2, 15,8, 18,0, 18,3, 19,0, 19,3, 20,2, 20,9, 21,6, 22,6, 23,6, 24,0, 24,9, 25,2, 25,8, 27,5, 28,1, 28,4, 29,8, 30,9, 31,7, 32,3 и 36,5 градусов 2-тета (±0,2 градусов 2-тета).
Е32. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 по варианту осуществления Е28, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 3.
Е33. Кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 по любому одному из вариантов осуществления Е29 - Е32, где порошковую рентгенограмму получают путем проведения анализа методом порошковой рентгеновской дифракции при 25°С и при относительной влажности выше 35%.
Е34. Аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4.
Е35. Аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4 по варианту осуществления Е34, имеющий порошковую рентгенограмму, включающую характеристические пики при величинах угла 2-тета, по существу таких же, как приведенные на фигуре 4.
Е36. Фармацевтическая композиция, включающая соединение по любому одному из вариантов осуществления E1 - Е35 или его фармацевтически приемлемую соль и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество.
Е37. Способ лечения МЕК-ассоциированной опухоли, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому одному из вариантов осуществления E1 - Е35 или его фармацевтически приемлемой соли.
Е38. Способ по варианту осуществления Е37, где опухоль имеет мутацию BRAF V600, выбранную из V600E, V600K, V600D, V600R и V600S.
Е39. Способ по варианту осуществления Е37 или Е38, где опухоль имеет мутацию BRAF V600.
Е40. Способ по любому из вариантов осуществления Е37 - Е39, где опухоль представляет собой экстракраниальную опухоль.
Е41. Способ по варианту осуществления Е40, где экстракраниальную опухоль выбирают из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников и нейробластомы.
Е42. Способ по любому из вариантов осуществления Е37 - Е39, где опухоль представляет собой опухоль ЦНС.
Е43. Способ по варианту осуществления Е42, где опухоль ЦНС представляет собой интракраниальную опухоль.
Е44. Способ по варианту осуществления Е43, где интракраниальная опухоль представляет собой рак головного мозга.
Е45. Способ по варианту осуществления Е44, где рак головного мозга представляет собой метастатический рак головного мозга.
Е46. Способ по варианту осуществления Е45, где метастатический рак головного мозга выбирают из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников.
Е47. Способ по варианту осуществления Е42, где опухоль ЦНС представляет собой интракраниальное лептоменингеальное заболевание (LMD) или экстракраниальное лептоменингеальное заболевание (LMD).
Е48. Способ по варианту осуществления Е47, где LMD выбирают из метастатической меланомы, метастатического колоректального рака и метастатического немелкоклеточного рака легкого.
Е49. Способ по варианту осуществления Е43, где интракраниальная опухоль представляет собой первичную опухоль.
Е50. Способ по варианту осуществления Е49, где первичная опухоль головного мозга представляет собой злокачественную опухоль.
Е51. Способ по варианту осуществления Е50, где первичная опухоль головного мозга представляет собой глиому степени злокачественности 2, глиому степени злокачественности 3 или глиому степени злокачественности 4.
Е52. Способ по варианту осуществления Е49, где первичная опухоль головного мозга представляет собой доброкачественную опухоль.
Е53. Способ по варианту осуществления Е37, где опухоль имеет слияние BRAF.
Е54. Способ по варианту осуществления Е53, где опухоль имеет слияние BRAF, выбранное из KIAA11549-BRAF, MKRN1-BRAF, TRIM24-BRAF, AGAP3-BRAF, ZC3H7AV1-BRAF, AKAP9-BRAF, CCDC6-BRAF, AGK-BRAF, EPS15-BRAF, NUP214-BRAF, ARMC10-BRAF, BTF3L4-BRAF, GHR-BRAF, ZC3HAV1-BRAF, ZNF767-BRAF, CCDC91-BRAF, DYNC112-BRAF, ZKSCAN1-BRAF, GTF2I-BRAF, MZT1-BRAF, RAD18-BRAF, CUX1-BRAF, SLC12A7-BRAF, MYRIP-BRAF, SND1-BRAF, NUB1-BRAF, KLHL7-BRAF, TANK-BRAF, RBMS3-BRAF, STRN3-BRAF, STK35-BRAF, ETFA-BRAF, SVOPL-BRAF и JHDM1D-BRAF.
E55. Способ по варианту осуществления E54, где опухоль представляет собой карциному молочной железы (например, инвазивную дуктальную карциному молочной железы), колоректальную карциному (например, аденокарциному толстой кишки), карциному пищевода (например, аденокарциному пищевода), глиому (например, десмопластическую младенческую ганглиоглиому головного мозга, пилоидную астроцитому головного мозга, плеоморфную ксантоастроцитому головного мозга, глиому спинного мозга низкой степени злокачественности (NOS), анапластическую олигодендроглиому, анапластическую ганглиоглиому), карциному головы и шеи (например, нейроэндокринную карциному головы и шеи), карциному легкого (например, аденокарциному легкого, немелкоклеточный рак легкого (NOS)), меланому (например, шпицоидную меланому кожи, нешпицоидную меланому слизистых оболочек, шпицоидную меланому кожи, меланому невыявленной первичной локализации, нешпицоидную меланому кожи), карциному поджелудочной железы (например, аденокарциному, карциному ацинарных клеток поджелудочной железы), карциному предстательной железы (например, ацинарную аденокарциному предстательной железы), саркому (злокачественную солитарную фиброзную опухоль), карциному щитовидной железы (папиллярную карциному щитовидной железы), карциному неизвестной первичной локализации (например, аденокарциному неизвестной первичной локализации), мезотелиому плевры, аденокарциному прямой кишки, карциному эндометрия матки (например, аденокарциному эндометрия матки (NOS)) или серозную карциному яичников.
Е56. Способ по варианту осуществления Е37, где опухоль представляет собой опухоль с BRAF немутантного типа (дикого типа).
Е57. Способ по любому из вариантов осуществления Е37 - Е56, где способ дополнительно включает применение одной или более противораковых терапий.
Е58. Способ по варианту осуществления Е57, где одну или более дополнительных противораковых терапий независимо выбирают из хирургического вмешательства, лучевой терапии и применения противораковых средств.
Е59. Способ по варианту осуществления Е58, где дополнительную противораковую терапию выбирают из применения одного или более противораковых средств.
Е60. Способ по варианту осуществления Е59, где противораковое средство выбирают из ингибиторов МЕК, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов SHP2, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути сигнальной трансдукции, ингибиторов контрольных точек иммунного ответа, модуляторов апоптозного пути, цитотоксических химиотерапевтических средств, нацеленных на ангиогенез терапевтических средств и нацеленных на иммунную систему лекарственных средств, включая иммунотерапию.
Е61. Способ по варианту осуществления Е60, где противораковое средство представляет собой ингибитор BRAF.
Е62. Способ по варианту осуществления Е61, где ингибитор BRAF представляет собой энкорафениб или его фармацевтически приемлемую соль.
Е63. Способ по варианту осуществления Е61, где ингибитор BRAF выбирают из следующих соединений:
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид;
N- (2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-фенил)-3-фторпропан-1-сульфонамид;
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-окси]-4-фторфенил}пропан-1-сульфонамид;
N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторпиридин-2-ил)пропан-1-сульфонамид; и
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)-окси]-4-фторфенил}-3-фторпропан-1-сульфонамид;
или их фармацевтически приемлемой соли.
Е64. Способ по варианту осуществления Е63, где ингибитор BRAF представляет собой N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид или его фармацевтически приемлемую соль.
Е65. Способ по варианту осуществления Е61, где ингибитор BRAF выбирают из следующих соединений:
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2.1.1]гексан-2-сульфон-амид,
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид и
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид,
или их фармацевтически приемлемой соли.
Е66. Способ по варианту осуществления Е65, где ингибитор BRAF представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид или его фармацевтически приемлемую соль.
Е67. Способ по варианту осуществления Е60, где противораковое средство представляет собой ингибитор SHP2.
Е68. Способ по варианту осуществления Е67, где ингибитор SHP2 представляет собой (S)-1'-(6-((2-амино-3-хлорпиридин-4-ил)тио)-1,2,4-триазин-3-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин или его фармацевтически приемлемую соль
Е69. Способ по любому из вариантов осуществления Е37 - Е68, где соединение представляет собой 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион и где субъекту вводят 50 мг 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона один раз в сутки.
Е70. Соединение по любому одному из вариантов осуществления E1 - Е35 или его фармацевтически приемлемая соль для применения в качестве лекарственного препарата.
Е71. Соединение по одному из вариантов осуществления Е1 - Е35 или его фармацевтически приемлемая соль для применения при лечении МЕК-ассоциированной опухоли.
Е72. Применение соединения по любому одному из вариантов осуществления E1 - Е35 или его фармацевтически приемлемой соли для производства лекарственного препарата для лечения МЕК-ассоциированной опухоли у субъекта.
Для более глубокого понимания этого изобретения, далее приводятся примеры. Эти примеры приведены в изобретении только с целью иллюстрации, и они никоим образом не ограничивают объем изобретения.
Описанные ниже соединения и промежуточные соединения были названы, используя правило о названиях, применяемое в компьютерной программе ChemDraw, Version 20.1.1.125 (Perkin Elmer Informatics). Правило о названиях, применяемое в компьютерной программе ChemDraw, Version 20.1.1.125, хорошо известно специалистом в данной области и, полагают, что правило о названиях, применяемое в ChemDraw, Version 20.1.1.125, в целом соответствует рекомендациям по номенклатуре органических соединений IUPAC (Международного союза теоретической и прикладной химии) и по названиям и индексации CAS (Chemical Abstracts Service). Если не указано иное, то все химические реагенты приобретались у фирм-производителей и не подвергались дополнительной очистке, или их синтезировали, используя методы, известные в литературе.
ПРИМЕРЫ
Примеры исследований биологической активности
Пример А
Клеточный Phospho-p44/42 МАРК (Erk1/2) (Thr202/Tyr204) анализ
Ингибирование фосфорилирования ERK1/2(Thr202/Tyr204) определяли с использованием следующего клеточного анализа, который включает инкубацию клеток с соединением в течение 1 часа и количественную оценку сигнала pERK с помощью In-Cell Western на фиксированных клетках и нормализацию к сигналу GAPDH. Клетки A375 получали из Американской коллекции типовых культур (АТСС) и выращивали в среде DMEM с добавлением 10% фетальной бычьей сыворотки, пенициллина/стрептомицина, глутамакса©, неосновных аминокислот и пирувата натрия. Клетки высевали в 96-луночные планшеты по 30000 клеток на лунку и давали им возможность прикрепиться в течение ночи при 37°С/5% CO2. Клетки обрабатывали соединениями, приготовленными в виде последовательных разбавлений 1:3 с получением 10 концентраций (диапазон: 20 мкМ - 0,05 нМ; максимальная концентрация варьировалась от 20 мкМ до 1 мкМ) с конечной концентрацией DMSO 0,5%. Контрольные лунки содержали либо только 0,5% DMSO (без контроля ингибирования), либо 1 мкМ сильнодействующего контрольного соединения (контроль полного ингибирования). После инкубации в течение 1 часа, клетки фиксировали в 3,7% формальдегиде в dPBS (забуференном фосфатом физиологическом растворе Дульбекко) при комнатной температуре в течение 20 минут. Затем клетки промывали dPBS и пермеабилизировали в 100% МеОН при комнатной температуре в течение 10 минут. После пермеабилизации, клетки промывали dPBS и инкубировали в блокирующем буфере LI-COR (LI-COR Biosciences, Cat # 927-40000) в течение 1 часа или более. Затем планшеты инкубировали с антителом, специфичным к МЕК-зависимым сайтам фосфорилирования ERK1/2, треонину 202 и тирозину 204 (Cell Signaling Technologies; Cat # 9101), расположенным ниже МЕК в пути передачи сигнала МАР-киназы, а также с GAPDH (Millipore; Cat # МАВ374). Антитело pErk1/2 (Thr202/Tyr204) разводили в блокирующем буфере LI-COR, содержащем 0,05% Tween-20, в соотношении 1:250; GAPDH разводили в соотношении 1:2500. Планшеты инкубировали в течение ночи при 4°С. После промывки с помощью PBS/0,05% Tween-20, клетки инкубировали с флуоресцентно меченными вторичными антителами (Anti-rabbit-Alexa Flour680, Invitrogen Cat # A21109; Anti-mouse-IRDye800CW, LI-COR Biosciences Cat # 926-32210, оба в разведении 1:1000) в течение 1 часа. Затем клетки промывали, как указано выше, и анализировали флуоресценцию при длинах волн 680 и 800 нм с использованием системы инфракрасной визуализации Odyssey CLx (LI-COR Biosciences). Сигнал фосфорилированного Erk1/2 (Thr202/Tyr204) нормализовали к сигналу GAPDH для каждой лунки. Величины IC50 рассчитывали на основе нормализованных значений с использованием 4-параметрической аппроксимации в программном обеспечении BioAssay, и эти величины представлены в таблице А.


Пример В
Анализ проницаемости MDR1 LLC-PK1 и BCRP MDCKII
Клетки LLC-PK1, как LLC-PK1, так и трансфицированные с помощью MDR1, культивировали и высевали в соответствии с рекомендациями фирмы-производителя, за исключением того, что среда для пассажа содержала только 2% фетальной бычьей сыворотки, чтобы продлить время пассажа до семи дней.
Линию клеток собак с нокаутом Р-gp, трансфицированных с помощью BCRP, культивировали и высевали в соответствии с рекомендациями фирмы-производителя.
Для оценки функциональности Р-gp или BCRP эффлюкса, в анализах использовали как положительный, так и отрицательный контроль. Исходные растворы для контрольных анализов и тестируемого препарата готовили в DMSO до конечных испытуемых концентраций 10 и 1 мкМ, соответственно. Конечная концентрация органических веществ в анализе составляла 1%. Все дозируемые растворы содержали 10 мкМ люциферового желтого для контроля целостности монослоя клеток LLC PK1 или MDCKII.
Для апикально-базолатерального определения (от А до В), 75 мкл испытуемого образца в буфере для переноса добавляли к апикальной стороне отдельных трансвеллов, и в каждую лунку добавляли 250 мкл базолатеральной среды без соединения или люциферового желтого. Для определения от базолатерального до апикального (от В до А), в каждую лунку добавляли 250 мкл испытуемого образца в буфере для переноса, и в каждую транслунку добавляли 75 мкл буфера для переноса без соединения или люциферового желтого. Все испытания проводили в трех параллельных опытах, и каждое соединение испытывали как на апикально-базолатеральный, так и на базолатерально-апикальный перенос.Планшеты инкубировали в течение 2 часов на орбитальном шейкере Lab-Line Instruments Titer Orbital Shaker (VWR, West Chester, PA) при 50 об/мин и 37°C с 5% CO2. Все культуральные планшеты извлекали из инкубатора, из апикальной и базолатеральной части каждой лунки удаляли по 50 мкл среды и добавляли к 150 мкл 1 мкМ лабеталола в смеси 2:1 по объему ацетонитрил : H2O.
Планшеты считывали с использованием флуоресцентного считывающего устройства Molecular Devices (Sunnyvale, СА) Gemini для оценки концентраций люциферового желтого при длинах волн возбуждения/эмиссии 425/535 нм. Эти значения признавали приемлемыми, когда обнаруживали, что они составляют ниже 2% для апикально-базолатерального потока и 5% базолатерально-апикального потока через монослои клеток LLC-PK1, трансфицированных с помощью MDR1, или клеток MDCKII, трансфицированных с помощью BCRP. Планшеты герметично закрывали, и содержимое каждой лунки анализировали методом жидкостной хроматография с тандемной масс-спектрометрией (LC MS/MS). Концентрации соединения определяли по отношению площадей пиков соединения к внутреннему стандарту (лабеталолу) по сравнению с дозируемым раствором.
Анализ методом жидкостной хроматографии с масс-спектрометрией (LC-MS)
Система для жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS/MS) состояла из автоматического дозатора HTS-PAL (Leap Technologies, Carrboro, NC), хроматографа для высокоэффективной жидкостной хроматографии НР1200 HPLC (Agilent, Palo Alto, СА) и системы ионной ловушки MDS Sciex 4000 Q Trap (Applied Biosystems, Foster City, СА). Хроматографическое разделение определяемого вещества и внутреннего стандарта достигалось при комнатной температуре с использованием колонки С18 (Kinetics®, 50×300 мм, размер частиц 2,6 мкм, Phenomenex, Torrance, СА) в сочетании с градиентными условиями с использованием подвижных фаз А (вода, содержащая 1% изопропилового спирта и 0,1% муравьиной кислоты) и В (0,1% муравьиной кислоты в ацетонитриле). Суммарное время записи хроматограммы, включая повторное приведение к равновесному состоянию, для одной вводимой в хроматограф пробы составляло 1,2 минуты. Масс-спектрометрическое обнаружение определяемых веществ осуществляли с использованием ионизации электрораспылением в режиме регистрации положительных ионов. Ответные реакции определяемых веществ измеряли путем мониторинга множественных реакций (MRM) переходов, уникальных для каждого соединения (протонированный ион-предшественник и выбранные ионы-продукты для каждого испытуемого образца и m/z от 329 до m/z 162 для лабеталола, внутреннего стандарта).
Коэффициент проницаемости (Рарр) вычисляют по следующему уравнению:
Papp=[((Cd × V × (1×106))/(t × 0,12 cm2 × С)]
где Cd, V, t и С0 представляют собой обнаруживаемую концентрацию (мкМ), объем на стороне дозирования (мл), время инкубации (сек) и исходную дозируемую концентрацию (мкМ), соответственно. Расчеты для Рарр выполняли для каждого из повторяющихся экспериментов и затем усредняли. Коэффициенты проницаемости соединений формулы I приведены в таблице В1. В этом анализе, соединение определяют как имеющее высокую проницаемость, если его проницаемость составляет больше чем 8×10-6 см/сек, соединение определяют как имеющее среднюю проницаемость, если его проницаемость составляет от 2×10-6 см/сек до 8×10-6 см/сек, и соединение определяют как имеющее низкую проницаемость, если его проницаемость составляет менее чем 2×10-6 см/сек.
Коэффициент эффлюкса рассчитывают из средних значений данных Рарр по проницаемости от апикальной мембраны к базолатеральной мембране (А-В) и данных Рарр от базолатеральной мембраны к апикальной (В-А):
Коэффициент эффлюкса = Рарр (В-А) /Рарр (А-В)



Коэффициент эффлюкса рассчитывают из средних значений данных Рарр по проницаемости от апикальной мембраны к базолатеральной мембране (А-В) и данных Рарр от базолатеральной мембраны к апикальной (В-А):
Коэффициент эффлюкса = Рарр (В-А) /Рарр (А-В)
В таблице В2 приведены коэффициенты эффлюкса для соединений формулы I при их испытании в этом анализе.


N/A: данные отсутствуют
Пример С
Фармакокинетика (отношение концентрации соединения в свободном состоянии в головном мозге к концентрации соединения в свободном состоянии в плазме) (у мышей)
Способность типичных соединений проникать через гематоэнцефалитический барьер (ВВВ) у мышей определяли путем оценки отношения концентрации несвязанного соединения в головном мозге к концентрации несвязанного соединения в плазме (также называемого отношением концентрации свободного соединения в головном мозге к концентрации свободного соединения в плазме) у самцов мышей линии CD-I.
Концентрации соединений в головном мозге определяли путем перорального введения соединений мышам для исследования фармакокинетики (РК) с типичными моментами времени отбора проб 2, 4, 8, 12 и 24 часа после перорального введения дозы 10 мг/кг через зонд. До проведения анализа, образцы мозга хранились при температуре -20±5°С. Концентрации испытуемого соединения в гомогенате мозга мши определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS/MS) после осаждения белка ацетонитрилом. Строили калибровочную кривую по 12 точкам в диапазоне от 0,5 до 10000 нг/мл в двух экземплярах. Раствор 4 00 мкг/мл испытуемого соединения в диметилсульфоксиде (DMSO) последовательно разводили (трехкратно) в 100% DMSO, и затем по 2,5 мкл каждого стандартного раствора добавляли к 100 мкл гомогената мозга наивных самцов мышей линии CD-I. Для имитации экстракции в стандартной кривой, ко всем испытуемым образцам добавляли 2,5 мкл DMSO. В калибровочные и испытуемые образцы гомогената головного мозга добавляли по 10 мкл IS (1 мкг/мл структурного аналога). Гомогенат головного мозга получали путем добавления 0,7 5 мл смеси вода : МеОН 4:1 к каждому образцу головного мозга с последующей гомогенизацией в течение 1 минуты с помощью трубчатых шариковых дробилок со скоростью 6 м/сек с использованием MP Fast Prep-24®. Белки осаждали из 100 мкл образца гомогената головного мозга путем добавления 300 мкл ацетонитрила. Образцы перемешивали на вортексе в течение 5 минут и центрифугировали в центрифуге Allegra X-12R (Beckman Coulter, Fullerton, CA; SX4750A rotor) в течение 15 минут при приблизительно 1500 г при 4°С. Аликвоту каждой надосадочной жидкости объемом 100 мкл переносили с помощью персональной пипетки объемом 550 мкл (Apricot Designs, Monrovia, СА) в 96-луночные планшеты и разбавляли 1:1 водой, квалификации "для ВЭЖХ". Полученные планшеты герметизировали алюминиевой фольгой для анализа методом LC-MS/MS.
Соотношения "головной мозг: плазма" рассчитывали, используя концентрацию соединения, измеренную в головном мозге, деленную на концентрацию соединения, измеренную в плазме. Соотношения "головной мозг: плазма" всегда рассчитывали для одного животного и в определенный момент времени. Отношение концентрации свободного соединения в головном мозге к концентрации свободного соединения в плазме рассчитывали путем умножения отношения концентрации свободного соединения в головном мозге к концентрации свободного соединения в плазме на свободную фракцию гомогената головного мозга in vitro, деленную на свободную фракцию плазмы in vitro, используя следующее уравнение: (В/Р)*(Bfu/Pfu).
В Таблице С представлены отношения концентрации свободного соединения в головном мозге к концентрации свободного соединения в плазме для типичных соединений примеров 6, 7, 8, 14, 22, 36 и 46, раскрытых в изобретении..

Примеры синтеза
Промежуточное соединение 1

Метил (Е)-2,6-дихлор-4-(2-(диметиламино)винил)никотинат
Стадия 1. Получение метил 2,6-дихлор-4-метилникотината. К раствору 2,6-дихлор-4-метилникотиновой кислоты (1,0 г, 4,9 ммоль) в смеси 1:1 МеОН : диоксан (10 мл) при 0°С добавляли (триметилсилил)диазометан (3,3 мл, 2М в гексанах, 6,6 ммоль). Смесь удаляли из ледяной бани и перемешивали в течение 10 минут, затем концентрировали до половина объема и распределяли между водой (20 мл) и EtOAc (20 мл). Водный слой экстрагировали с помощью EtOAc (2×20 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и осторожно концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% EtOAc/петролейный эфир с получением метил 2,6-дихлор-4-метилникотината (0,81 г, 76%). 1Н ЯМР (400 МГц, CDCl3) δ 7,2 (с, 1Н), 4,0 (с, 3Н), 2,3 (с, 3Н) ppm.
Стадия 2. Получение метил (Е)-2,6-дихлор-4-(2-(диметил-амино)-винил)никотината. К раствору метил 2,6-дихлор-4-метилникотината (810 мг, 3,68 ммоль) в DMF (5 мл) добавляли N, N-диметилформамида диметилацеталь (978 мл, 7,36 ммоль), и смесь перемешивали при 100°С в течение 16 часов. Охлажденную смесь обрабатывали водой (40 мл), перемешивали в течение 10 минут и затем твердые вещества собирали фильтрацией, промывали водой и сушили под вакуумом с получением метил (Е)-2,6-дихлор-4-(2-(диметиламино) винил) никотината (764 мг, 75%). 1H ЯМР (400 МГц, CDCl3) δ 7,3 (с, 1Н), 7,1 (д, 1Н), 4,8 (д, 1Н), 3,1 (с, 3Н), 2,9 (с, 6Н) ppm.
Промежуточное соединение 2

Метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Стадия 1. Получение 4-бром-2,6-дихлорникотиновой кислоты. Раствор 4-бром-2,6-дихлорпиридина (100 г, 440,7 ммоль) в THF (1000 мл) охлаждали до -78°С. Добавляли по каплям LDA (2 М в THF, 242,4 мл, 484,8 ммоль) при -78°С и продолжали перемешивание в течение 1 часа при -78°С. К реакционной смеси добавляли порциями твердый CO2 (155,1 г, 3,53 моль) и продолжали перемешивание в течение 2 часов при -7 8°С. Реакцию прерывали путем добавления 1М Na2CO3 (1600 мл), затем воды (500 мл) и перемешивали в течение 10 минут. Водный слой экстрагировали с помощью EtOAc (300 мл). Величину рН водного раствора корректировали с помощью 2N HCl с получением раствора с рН 2. Водный раствор затем экстрагировали с помощью EtOAc (3×300 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 4-бром-2,6-дихлорникотиновой кислоты (540 г, 75%). 1Н ЯМР (400 МГц, DMSO-d6) δ 15,51-12,87 (м, 1Н), 8,13 (с, 1Н) ppm.
Стадия 2. Получение 4-бром-2-хлор-6-оксо-1,6-дигидро-пиридин-3-карбоновой кислоты. Раствор NaOH (4 М, 1,62 л, 6,46 моль) нагревали до 110°С, и добавляли одной порцией 4-бром-2,6-дихлорникотиновую кислоту (70 г, 2 58,4 ммоль). Смесь перемешивали в течение 8 часов, затем охлаждали до 0°С. Корректировали величину рН реакционной смеси до 1 с помощью HCl (6 М) и перемешивали в течение 30 минут. Твердые вещества собирали фильтрацией и сушили под вакуумом с получением 4-бром-2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (по расчету 100%).
Стадия 3. Получение метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К смеси 4-бром-2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (100 г, 396 ммоль) в DMF (800 мл) одной порцией добавляли метилйодид (168,6 г, 1,19 моль, 73, 98 мл) и K2CO3 (164, 2 г, 1,19 моль). Смесь перемешивали при 25°С в течение 3 часов, затем выливали в насыщенный водный раствор NH4Cl (1800 мл), и водную фазу экстрагировали с помощью EtOAc (2×500 мл). Объединенные органические слои промывали солевым раствором (400 мл), сушили над Na2SO4, фильтровали и концентрировали. Объединенные остатки (5 партий) очищали колоночной хроматографией, элюируя с использованием 2-100% EtOAc/петролейный эфир, с получением метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (из 5 партий, 141,83 г, 24,7%). 1Н ЯМР (400 МГц, CDCl3) δ 6, 86 (с, 1Н), 3,94 (с, 3Н), 3,69-3,66 (м, 3Н); MS (химическая ионизация при атмосферном давлении (apci), m/z)=280,0, 282,0 (М+Н).
Промежуточное соединение 3

Метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (20,0 г, 71,3 ммоль, метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II) (5,96 г, 7,13 ммоль) и (Z)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (14,8 г, 74,9 ммоль) суспендировали в 1,4-диоксане (700 мл) и добавляли K2CO3 (53, 5 мл, 107 ммоль) (2 N водный раствор). Смесь перемешивали при 60°С в течение 6 часов, затем при температуре окружающей среды в течение 12 часов в атмосфере аргона. Реакционную смесь распределяли между водой (1500 мл) и EtOAc (500 мл). Водный слой экстрагировали с помощью EtOAc (2×400 мл), и объединенные органические слои промывали солевым раствором (500 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-4 0-60% EtOAc/гептаны, с получением метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоксилата (13,3 г, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 7, 23 (с, 1Н), 6,45 (д, 1Н), 4,86 (д, 1Н), 4,40 (кварт, 2Н), 3,89 (с, 3Н), 3,67 (с, 3Н), 1,35 (т, 3Н); MS (химическая ионизация при атмосферном давлении (химическая ионизация при атмосферном давлении (apci)), m/z)=272,0 (М+Н).
Промежуточное соединение 4

Метил 4-(2-((2-(третбутокси)этокси)амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Стадия 1. Получение метил (E/Z)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,95 г, 3,50 ммоль) и О-(2-(третбутокси)этил)гидроксиламина гидрохлорид (593 мг, 3,50 ммоль) объединяли в 1,4-диоксане (10 мл). Добавляли Et3N (487 мл, 3,50 ммоль) и HCl (1,75 мл, 6,99 ммоль) (4N/диоксан). Суспензию нагревали до 60°С в течение 1 часа, затем охлаждали и фильтровали. Фильтрат концентрировали с получением метил (E/Z)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (по расчетам 100%) в форме 1:1 смеси изомеров. MS (химическая ионизация при атмосферном давлении (apci), m/z)=359,1 (М+Н).
Стадия 2. Получение метил 4-(2-((2-(третбутокси)этокси)-амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору метил (E/Z)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (1,25 г, 3,48 ммоль) в IPA (20 мл) добавляли цианоборгидрид натрия (1,09 г, 17,4 ммоль), затем уксусную кислоту (1,0 мл, 17,4 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 часов, затем распределяли между насыщенным раствором NaHCO3 (50 мл) и EtOAc (50 мл). Водный слой экстрагировали с помощью EtOAc (2×30 мл), и объединенные органические слои промывали солевым раствором (30 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/DCM с получением метил 4-(2-((2-(третбутокси)этокси)амино)-этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,66 г, 53%). 1H ЯМР (400 МГц, CDCl3) δ 6, 43 (с, 1Н), 3,90 (с, 3Н), 3,78 (т, 2Н), 3,68 (с, 3Н), 3,50 (т, 2Н), 3,10 (т, 2Н), 2,71 (т, 2Н), 1,20 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=361,1, 363, 1 (М+Н).
Промежуточное соединение 5

2-Хлор-4-этиланилин
Стадия 1. Получение N-(4-этилфенил)ацетамида. 4-Этил-анилин (513 мкл, 4,13 ммоль) растворяли в DCM (10,3 мл). После добавления триэтиламина (690 мкл, 4,95 ммоль) и охлаждение до 0°С, добавляли по каплям уксусный ангидрид (467 мкл, 4,95 ммоль). Через 3 0 минут, реакцию прерывали путем добавления насыщенного водного раствора NaHCO3 (50 мл). Органические слои отделяли, и водный слой экстрагировали с помощью DCM (2×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток растирали с гексанами, и твердые вещества собирали фильтрацией с получением N-(4-этилфенил) ацетамида (600 мг, 89%). 1Н ЯМР (400 МГц, CDCl3) δ 7, 40-7, 38 (д, 2Н), 7,16-7,14 (д, 2Н), 7,07 (уш. с, 1Н), 2,64-2,58 (кварт, 2Н), 2,16 (с, 3Н), 1,23-1,20 (т, 3Н) ppm.
Стадия 2. Получение N-(2-хлор-4-этилфенил)ацетамида. N-(4-этилфенил)ацетамид (50 мг, 0,31 ммоль) растворяли в DMF (613 мкл). После добавления N-хлорсукцинимида (65 мг, 0,49 ммоль), раствор нагревали до 70°С в течение 6 часов, затем охлаждали до температуры окружающей среды в течение 16 часов. Реакционную смесь выливали в 2N HCl (4 мл) и перемешивали в течение 15 минут. Смесь экстрагировали с помощью EtOAc (10 мл). Органические слои промывали водой (3×10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-25% EtOAc/гексаны, с получением N-(2-хлор-4-этилфенил)ацетамида (36 мг, 59%). 1H ЯМР (400 МГц, CDCl3) δ 8,23-8,21 (д, 1Н), 7,51 (уш. с, 1Н), 7,20-7,19 (д, 1Н), 7,11-7,08 (дд, 1Н), 2, 63-2, 57 (кварт, 2Н), 2,23 (с, 3Н), 1, 23-1, 20 (т, 3Н) ppm.
Стадия 3. Получение 2-хлор-4-этиланилина. N-(2-хлор-4-этилфенил)ацетамид (36 мг, 0,18 ммоль) растворяли в EtOH (0,5 мл) и добавляли 12N HCl (0,5 мл, 6,0 ммоль). Смесь перемешивали в течение 2 часов при 120°С. Реакционную смесь охлаждали до температуры окружающей среды, доводили до рН 10 путем добавления 6N NaOH, затем экстрагировали с помощью МТВЕ (3×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 2-хлор-4-этиланилина (26 мг, 92%). 1Н ЯМР (400 МГц, CDCl3) δ 7,09-7,08 (д, 1Н), 6,91-6,88 (дд, 1Н), 6,71-6,69 (д, 1Н), 2,55-2,49 (кварт, 2Н), 1,20-1,16 (т, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=156,1 (М+Н).
Промежуточное соединение 6

4-((Дифторметил)тио)-2-фторанилин
LiBF4 (196 мг, 2,10 ммоль) и LiH (17,5 мг, 2,10 ммоль) объединяли в DMF (8,8 мл, 1,75 ммоль). Добавляли 4-амино-3-фторбензолтиол (250 мг, 1,75 ммоль), и смесь перемешивали в течение 5 минут при температуре окружающей среды. Быстро добавляли (трифторметил)-триметилсилан (0,644 мл, 4,37 ммоль), затем раствор перемешивали в течение 10 минут при этой же температуре. Быстро добавляли TBAF (6 мл, 1N/THF, 6,00 ммоль), затем раствор перемешивали в течение 10 минут при этой же температуре. Реакционную смесь гасили водой (50 мл). Смесь экстрагировали с помощью EtOAc (2×25 мл), и объединенные органические слои промывали водой (3×50 мл) и солевым раствором (50 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% EtOAc/гексаны, с получением 4-((дифторметил)тио)-2-фторанилина (56 мг, 17%). 1H ЯМР (400 МГц, CDCl3) δ 7,25-7,22 (дд, 1Н), 7,19-7,16 (ддд, 1Н), 6,86-6,58 (т, 1Н), 6,77-6,73 (дд, 1Н), 3,95 (уш. с, 1Н) ppm.
Промежуточное соединение 7

2-Хлор-4-(метилтио)анилин
2-Хлор-4-йоданилин (250 мг, 0,986 ммоль), NiBr2 (22 мг, 0,099 ммоль), порошок Zn (129 мг, 1,97 ммоль) и 2,2'-бипиридин (15 мг, 0,099 ммоль) растворяли в THF (1,6 мл) в атмосфере Ar. Добавляли 1,2-диметилдисульфан (44 мкл, 0,493 ммоль), и смесь герметизировали и нагревали до 65°С в течение 16 часов. Реакционную смесь разбавляли с помощью EtOAc (50 мл) и промывали концентрированным раствором NH4OH (50 мл), затем 10% раствором лимонной кислоты (50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% EtOAc/гексаны, с получением 2-хлор-4-(метилтио)анилина (116 мг, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 7,27-7,26 (м, 1Н), 7,09-7,06 (дд, 1Н), 6,71-6,69 (д, 1Н), 4,02 (уш. с, 2Н), 2,41 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=174,0 (М+Н).
Промежуточное соединение 8

Метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат
Метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (1,0 г, 3,565 ммоль), метансульфонато(2-дициклогексил-фосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)палладий(II) (0,2982 г, 0,3565 ммоль) и (Е)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,7414 г, 3,743 ммоль) суспендировали в 1,4-диоксане (35,65 мл) и добавляли карбонат калия (2,674 мл, 2N водный раствор, 5,35 ммоль). После дегазирования с помощью аргона, смесь перемешивали при 60°С в течение 4 часов. Охлажденную реакционную смесь распределяли между водой (150 мл) и EtOAc (50 мл). Водный слой промывали с помощью EtOAc (2×40 мл). Объединенные органические слои промывали солевым раствором (150 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/гептан, с получением метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (379 мг, 39%). 1H ЯМР (400 МГц, CDCl3) δ 7,27 (с, 1Н), 7,03 (д, 1Н), 6,43 (с, 1Н), 5,53 (д, 1Н), 3,95-3,83 (м, 5Н), 3,67 (с, 3Н), 1,34 (т, 2Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=272,1 (М+Н).
Пример 1

8-((4-бром-2-фторфенил)амино)-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил 2,6-дихлор-4-(2-оксоэтил)-никотината. Суспензию метил (Е)-2,6-дихлор-4-(2-(диметиламино)-винил) никотината (0,797 г, 2,90 ммоль) в Et2O (30 мл) и IN HCl (30 мл) интенсивно перемешивали при температуре окружающей среды в течение 1 часа. Полученный раствор обрабатывали солевым раствором (20 мл) и экстрагировали с помощью Et2O (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над MgSO4, фильтровали и осторожно концентрировали с получением метил 2,6-дихлор-4-(2-оксоэтил)никотината (по расчетам 100%), который тут же использовали.
Стадия 2. Получение 8-хлор-2-циклопропил-6-метокси-3,4-дигидро-2,7-нафтиридин-1(2Н)-она. К раствору метил 2,6-дихлор-4-(2-оксоэтил)никотината (0,719 г, 2,90 ммоль) в 1:1 IPA:МеОН (20 мл) при 0°С добавляли циклопропиламин (201 мл, 2,90 ммоль). Смесь перемешивали при температуре окружающей среды в течение 10 минут, затем добавляли цианоборгидрид натрия (546 мг, 8,70 ммоль) и уксусную кислоту (498 мл, 8,70 ммоль). После перемешивания при температуре окружающей среды в течение 16 часов, смесь распределяли между насыщенным раствором NaHCO3 (50 мл) и EtOAc (20 мл), и водный слой экстрагировали с помощью EtOAc (2×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в метаноле (10 мл), обрабатывали водным 1N раствором NaOH (3,33 мл, 3,33 ммоль) и перемешивали при температуре окружающей среды в течение 1 часа. Смесь концентрировали до половины объема, затем распределяли между водой (20 мл) и DCM (20 мл). Водный слой экстрагировали с помощью DCM (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-10% (20% MeOH/DCM)/DCM с получением 8-хлор-2-циклопропил-6-метокси-3,4-дигидро-2,7-нафтиридин-1(2Н)-она (310 мг, 42%). 1Н ЯМР (400 МГц, CDCl3) δ 6,5 (с, 1Н), 4,0 (с, 3Н), 3,5 (т, 2Н), 2,9 (м, 3Н), 0,9 (м, 2Н), 0,7 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=253,1, 255,1 (М+Н).
Стадия 3. Получение 8-хлор-2-циклопропил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-хлор-2-циклопропил-6-метокси-3,4-дигидро-2,7-нафтиридин-1(2Н)-она (99 мг, 0,39 ммоль) в ацетонитриле (2 мл) добавляли триметилсилилйодид (2,94 мл, 1,9 6 ммоль). °Смесь перемешивали при температуре окружающей среды в течение 48 часов, затем концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15-20% (20% MeOH/DCM)/DCM, с получением 8-хлор-2-циклопропил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (по расчетам 100%), который тут же использовали. MS (химическая ионизация при атмосферном давлении (apci), m/z)=239,0 (М+Н).
Стадия 4. Получение 8-хлор-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-хлор-2-циклопропил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (93 мг, 0,39 ммоль) в THF (2 мл) добавляли K2CO3 (110 мг, 0,78 ммоль), затем метилйодид (29 мл, 0,47 ммоль). Смесь перемешивали при температуре окружающей среды в течение 2 часов, затем обрабатывали дополнительным количеством метилйодида (50 мл, 0,84 ммоль) и перемешивали в течение еще 16 часов. Смесь фильтровали, промывали с помощью (4:1) DCM/MeOH (10 мл), и фильтрат концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM, с получением 8-хлор-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (31 мг, 31%). 1Н ЯМР (400 МГц, CDCl3) δ 6,29 (с, 1Н), 3,73 (с, 3Н), 3,45 (т, 2Н), 2,87-2,81 (м, 1Н), 2,77 (т, 2Н), 0,94-0,89 (м, 2Н), 0,72-0,68 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=253,1 (М+Н).
Стадия 5. Получение 8-((4-бром-2-фторфенил) амино)-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 4-бром-2-фторанилина (24 мг, 0,12 ммоль) в THF (1 мл) при -78°С в атмосфере N2 добавляли LiHMDS (1,84 мл, 1,0 M/THF, 0,184 ммоль). Смесь перемешивали в течение 4 5 минут, затем добавляли суспензию 8-хлор-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (31 мг, 0,13 ммоль) в THF (2 мл). Смесь перемешивали при -78°С в течение 20 минут, затем гасили насыщенным водным раствором NH4Cl (10 мл), перемешивали при температуре окружающей среды в течение 10 минут, затем экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гексаны, с получением 8-((4-бром-2-фторфенил)амино)-2-цикло-пропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (24,9 мг, 50%). 1Н ЯМР (400 МГц, CDCl3) δ 11,6 (с, 1Н), 7,29 (дд, 1Н), 7,19 (ддд, 1Н), 6,71 (т, 1Н), 5,99 (с, 1Н), 3,50 (т, 2Н), 3,20 (с, 3Н), 2,77 (т, 2Н), 2,74-2,70 (м, 1Н), 0,93-0,87 (м, 2Н), 0,72-0,67 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=406,0, 408,0 (М+Н).
Пример 2

8-((4-бром-2-фторфенил)амино)-2-(циклопропилметокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 1, за исключением того, что применяли О-(циклопропилметил)гидроксиламина гидрохлорид вместо циклопропиламина на стадии 2, с получением 8-((4-бром-2-фторфенил)амино)-2-(циклопропилметокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (20 мг, 57%). 1Н ЯМР (400 МГц, CDCl3) δ 11,32 (с, 1Н), 7,30 (дд, 1Н), 7,21 (м, 1Н), 6,72 (т, 1Н), 6,00 (с, 1Н), 3,85 (д, 2Н), 3,77 (т, 2Н), 3,18 (с, 3Н), 2,98 (т, 2Н), 1,22-1,14 (м, 1Н), 0,64-0,59 (м, 2Н), 0,36-0,31 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=436,0, 438,0 (М+Н).
Пример 3

8-((4-бром-2-фторфенил)амино)-2-этокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 1, за исключением того, что применяли О-этилгидроксиламина гидрохлорид вместо циклопропиламина на стадии 2, с получением 8-((4-бром-2-фторфенил)амино)-2-этокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н, 7Н)-диона (22 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 11,34 (с, 1Н), 7,30 (дд, 1Н), 7,21 (м, 1Н), 6,72 (т, 1Н), 5,99 (с, 1Н), 4,08 (кварт, 2Н), 3,73 (т, 2Н), 3,19 (с, 3Н), 2,99 (т, 2Н), 1,32 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=410,0, 412,0 (М+Н).
Пример 4

2-циклопропил-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 1, за исключением того, что применяли 2-фтор-4-(метилтио)анилин вместо 4-бром-2-фторанилина на стадии 5, с получением 2-циклопропил-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (50,4 мг, 66%). 1Н ЯМР (400 МГц, CDCl3) δ 11,62 (с, 1Н), 7,01 (дд, 1Н), 6,95 (дд, 1Н), 6,79 (т, 1Н), 5,95 (с, 1Н), 3,49 (т, 2Н), 3,18 (с, 3Н), 2, 80-2, 68 (м, 3Н), 2,47 (с, 3Н), 0,93-0,87 (м, 2Н), 0,72-0,67 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=374,1 (М+Н).
Пример 5

2-циклопропил-8-((2-фтор-4-йодфенил)амино)-7-метил-З,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 1, за исключением того, что применяли 2-фтор-4-йоданилин вместо 4-бром-2-фторанилина на стадии 5, с получением 2-циклопропил-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (46,7 мг, 52%). 1Н ЯМР (400 МГц, CDCl3) δ 11,60 (с, 1Н), 7,46 (дд, 1Н), 7,37 (дт, 1Н), 6,55 (т, 1Н), 6,00 (с, 1Н), 3,50 (т, 2Н), 3,21 (с, 3Н), 2,81-2,69 (м, 3Н), 0,93-0,87 (м, 2Н), 0,72-0,67 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=454,0 (М+Н).
Пример 6

8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
8-((2-Фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион получали либо методом А, либо методом В.
Метод А.
Стадия 1. Получение 4-бром-2,6-дихлорникотиновой кислоты. Раствор 4-бром-2,6-дихлорпиридина (100 г, 440,7 ммоль) в THF (1000 мл) охлаждали до -78°С. Добавляли по каплям LDA (242,4 мл, 2M/THF, 484,8 ммоль) при -78°С и продолжали перемешивание в течение 1 часа при -78°С. К реакционной смеси добавляли порциями твердый CO2 (155,1 г, 3,53 моль) и продолжали перемешивание в течение 2 часов при -78°С. Реакцию прерывали путем добавления 1М Na2CO3 (1600 мл), затем воды (500 мл), и перемешивали в течение 10 минут. Водный слой экстрагировали с помощью EtOAc (300 мл). Величину рН водного раствора корректировали с помощью 2N HCl с получением раствора с рН 2. Водный раствор затем экстрагировали с помощью EtOAc (3×300 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 4-бром-2,6-дихлорникотиновой кислоты (540 г, 75%). 1H ЯМР (400 МГц, DMSO-d6) δ 15,51-12,87 (м, 1Н), 8,13 (с, 1Н) ppm.
Стадия 2. Получение 4-бром-2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты. Раствор NaOH (1,62 л, 4М, 6,46 моль) нагревали до 110°С и добавляли одной порцией 4-бром-2, 6-дихлорникотиновую кислоту (70 г, 258, 4 ммоль), полученную на стадии 1. Смесь перемешивали в течение 8 часов, затем охлаждали до 0°С. Корректировали величину рН реакционной смеси до 1 с помощью HCl (6 М) и перемешивали в течение 30 минут. Твердые вещества собирали фильтрацией и сушили под вакуумом с получением 4-бром-2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (по расчетам 100%).
Стадия 3. Получение метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К смеси 4-бром-2-хлор-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (100 г, 396 ммоль) в DMF (800 мл) добавляли одной порцией метилйодид (168,6 г, 1,19 моль, 73, 98 мл) и K2CO3 (164, 2 г, 1,19 моль). Смесь перемешивали при 25°С в течение 3 часов, затем выливали в насыщенный водный раствор NH4Cl (1800 мл), и водную фазу экстрагировали с помощью EtOAc (2×500 мл). Объединенные органические слои промывали солевым раствором (400 мл), сушили над Na2SO4, фильтровали и концентрировали. Объединенные остатки (5 партий) очищали колоночной хроматографией, элюируя с использованием 2-100% EtOAc/петролейный эфир, с получением метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (из 5 партий, 141,83 г, 24,7%). 1Н ЯМР (400 МГц, CDCl3) δ 6,86 (с, 1Н), 3,94 (с, 3Н), 3,69-3,66 (м, 3Н); MS (химическая ионизация при атмосферном давлении (apci), m/z)=280,0, 282,0 (М+Н).
Стадия 4. Получение метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (20,0 г, 71,3 ммоль), полученный на стадии 3, метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)(2-амино-1,1'-бифенил-2-ил)-палладий(II) (5,96 г, 7,13 ммоль) и (Z)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (14,8 г, 74,9 ммоль) суспендировали в 1,4-диоксане (700 мл) и добавляли K2CO3 (53,5 мл, 2N водный раствор, 107 ммоль). Смесь перемешивали при 60°С в течение 6 часов, затем при температуре окружающей среды в течение 12 часов в атмосфере аргона. Реакционную смесь распределяли между водой (1500 мл) и EtOAc (500 мл). Водный слой экстрагировали с помощью EtOAc (2×400 мл), и объединенные органические слои промывали солевым раствором (500 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40-60% EtOAc/гептаны, с получением метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (13,3 г, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 7, 23 (с, 1Н), 6,45 (д, 1Н), 4,86 (д, 1Н), 4,40 (кварт, 2Н), 3,89 (с, 3Н), 3,67 (с, 3Н), 1,35 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=272,0 (М+Н).
Стадия 5. Получение метил (E/Z)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (0,95 г, 3,50 ммоль), полученный на стадии 4, и О-(2-(третбутокси)этил)гидроксиламина гидрохлорид (593 мг, 3,50 ммоль) объединяли в 1,4-диоксане (10 мл). Добавляли Et3N (487 мл, 3,50 ммоль) и HCl (1,75 мл, 4N/диоксан, 6,99 ммоль). Суспензию нагревали до 60°С в течение 1 часа, затем охлаждали и фильтровали. Фильтрат концентрировали с получением метил (E/Z)-4-(2-((2-(третбутокси)этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (по расчетам 100%) в форме 1:1 смеси изомеров. MS (химическая ионизация при атмосферном давлении (apci), m/z)=359,1 (М+Н).
Стадия 6. Получение метил 4-(2-((2-(третбутокси)-этокси)амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору метил (E/Z)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (1,25 г, 3,48 ммоль) в IPA (20 мл), полученного на стадии 5, добавляли цианоборгидрид натрия (1,09 г, 17,4 ммоль), затем уксусную кислоту (1,0 мл, 17,4 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 часов, затем распределяли между насыщенным раствором NaHCO3 (50 мл) и EtOAc (50 мл). Водный слой экстрагировали с помощью EtOAc (2×30 мл), и объединенные органические слои промывали солевым раствором (30 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/DCM, с получением метил 4-(2-((2-(третбутокси)этокси)амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,66 г, 53%). 1H ЯМР (400 МГц, CDCl3) δ 6,43 (с, 1Н), 3,90 (с, 3Н), 3,78 (т, 2Н), 3,68 (с, 3Н), 3,50 (т, 2Н), 3,10 (т, 2Н), 2,71 (т, 2Н), 1,20 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=361,1, 363,1 (М+Н).
Стадия 7. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-Фтор-4-(метилтио)анилин (114 мг, 0,73 ммоль) растворяли в THF (5,0 мл, 0,69 ммоль) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (1,39 мл, 1,0 m/THF, 1,3 9 ммоль), затем смесь перемешивали в течение 45 минут. Затем добавляли по каплям раствор метил 4- (2-((2-(третбутокси)этокси)амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (250 мг, 0,69 ммоль), полученный на стадии 6, в THF (5,0 мл). После перемешивания в течение 10 минут, добавляли дополнительное количество LiHMDS (1,0 мл, 1,0 ммоль) и продолжали перемешивание при -78°С в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (20 мл), затем экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM), с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (111 мг, 36%). 1Н ЯМР (400 МГц, CDCl3) δ 11,35 (с, 1Н), 7,02-6,99 (дд, 1Н), 6,97-6,94 (м, 1Н), 6,82-6,77 (т, 1Н), 5,95 (с, 1Н), 4,16-4,14 (м, 2Н), 3,80-3,77 (т, 2Н), 3,62-3,60 (м, 2Н), 3,17 (с, 3Н), 2,99-2,95 (т, 2Н), 2,47 (с, 3Н), 1,21 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=450,2 (М+Н).
Стадия 8. Получение 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метил-тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (111 мг, 0,247 ммоль), полученного на стадии 7, в ацетонитриле (2,0 мл) добавляли H3PO4 (2,0 мл, 38,5 ммоль). Раствор нагревали до 60°С в течение 15 минут, затем охлаждали и гасили насыщенным водным раствором NaHCO3 (20 мл). Смесь экстрагировали с помощью EtOAc (3×10 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-20% (20% MeOH/DCM)/DCM, затем методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 30 минут). Объединяли фракции, содержащие очищенный требуемый продукт, и продукт превращали в форму свободного основания с помощью DCM/NaHCO3 с получением 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (62 мг, 64%). 1Н ЯМР (400 МГц, CDCl3) δ 11,17 (с, 1Н), 7,03-6,97 (м, 2Н), 6,85 (т, 1Н), 5,95 (с, 1Н), 4,03 (т, 2Н), 3,74-3,70 (м, 4Н), 3,16 (с, 3Н), 2,98 (т, 2Н), 2,48 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=394,1 (М+Н). Анализ методом порошковой рентгеновской дифракции (PXRD) проводили, как описано в примере 77. Порошковая рентгенограмма, которая приведена на фигуре 4, подтверждала, что полученный материал является аморфным. На этом основании, материал называли как аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 4.
Метод В.
Стадия 1. Получение метил 4-бром-2-((2-фтор-4-(метилтио)-фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоксилата (полученного в соответствии с любым из методов, описанных в изобретении; 50,0 г, 87,2%, 155 ммоль) и 2-фтор-4-(метилтио) анилина (24,4 г, 55 ммоль) в THF (50 мл) при 0°С добавляли третбутоксид калия (34,9 г, 311 мл, 1,0 молярный раствор, 311 ммоль) в течение 40 минут. Реакционную массу выдерживали при 0°С в течение 50 минут, затем разбавляли насыщенным водным раствором NH4Cl (600 мл) и EtOAc (500 мл). Добавляли воду для растворения твердых веществ, и слои разделяли. Водный слой экстрагировали с помощью EtOAc (2×300 мл), и объединенные органические слои промывали водой (500 мл) и солевым раствором (500 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток суспендировали в МеОН (57 мл) и ненадолго подвергали ультразвуковой обработке, затем нагревали до 60°С в течение 10 минут. Смесь медленно охлаждали до температуры окружающей среды, затем охлаждали до 0°С в течение 30 минут. Твердые вещества фильтровали и собирали с получением метил 4-бром-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (40,14 г, 64,4%). 1H ЯМР CDCl3 δ 9, 03 (с, 1Н), 7,04 (дд, 1Н), 6, 96 (м, 1Н), 6,71-6,65 (м, 2Н), 3,85 (с, 3Н), 3,24 (с, 3Н), 2,47 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=401,0, 403,0 (М+Н).
Стадия 2. Получение метил (Z)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. (Z)-2-(2-Этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (80,25 г, 82,3 мл, 405,17 ммоль) добавляли к перемешиваемому раствору метил 4-бром-2-((2-фтор-4-(метилтио)-фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (130,06 г, 324,14 ммоль), полученного в соответствии с методикой, используемой на стадии 1, K2CO3 (67,2 г, 243,10 мл, 2М, 486,21 ммоль) и метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)'2-амино-1,1'-бифенил-2-ил)палладия(II) (6,78 г, 8,10 ммоль) в 2-метилтетрагидрофуране (860 мл) при температуре окружающей среды в атмосфере аргона. Через реакционную смесь барботировали аргон в течение 30 минут. Реакционную смесь нагревали до 60°С и перемешивали в течение 13 часов, затем перемешивали при температуре окружающей среды в течение 6 часов. Реакционную смесь разбавляли водой (1800 мл) и EtOAc (1100 мл), затем фильтровали через фильтровальную бумагу GF/F. Слои фильтрата разделяли, и водный слой экстрагировали с помощью EtOAc (2×650 мл). Объединенные органические слои промывали солевым раствором (1500 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток обрабатывали с помощью МТВЕ (30 мл), концентрировали и сушили под вакуумом. Остаток суспендировали в МТВЕ (330 мл) и перемешивали при 60°С в течение 1 часа. Смесь медленно охлаждали до температуры окружающей среды, затем перемешивали при 0°С в течение 40 минут. Твердые вещества фильтровали, промывали холодным МТВЕ (110 мл), затем холодной смесью МТВЕ : гептан (1:1 смесь, 150 мл) и сушили под вакуумом с получением метил (Z)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (98, 54 г, 77, 46%). 1Н ЯМР CDCl3 δ 8, 65 (с, 1Н), 7,04 (дд, 1Н), 6,97 (с, 1Н), 6,92 (м, 1Н), 6,56 (т, 1Н), 6,37 (д, 1Н), 5,36 (д, 1Н), 4,01 (кварт, 2Н), 3,79 (с, 3Н), 3,31 (с, 3Н), 2,46 (с, 3Н), 1,35 (т, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=393,1 (М+Н).
Стадия 3. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Добавляли триэтиламин (2,84 г, 3,91 мл, 28,0 ммоль) к перемешиваемому раствору метил (Z)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (10,0 г, 25,5 ммоль), полученного в соответствии с методикой, используемой на стадии 2, и О-(2-(третбутокси)этил)гидроксиламина гидрохлорида (4,95 г, 96%, 28,0 ммоль) в 1,4-диоксане (255 мл). Реакционную смесь помещали в баню с холодной водой, продували Аг, и добавляли по каплям через капельную воронку HCl (2,04 г, 14,0 мл, 4М/диоксаны, 56,1 ммоль). Смесь нагревали при 60°С в течение 1 часа, затем охлаждали до температуры окружающей среды, используя баню с холодной водой. Медленно добавляли пиридинборан (4,74 г, 5,42 мл, 51,0 ммоль), и смесь перемешивали в течение 10 минут при температуре окружающей среды. Добавляли HCl (1,11 г, 7,64 мл, 4М/диоксан, 30,6 ммоль) в течение 5 минут, и продолжали перемешивание в течение 20 минут. Реакционную смесь нагревали при 60°С в течение 21 часа, затем охлаждали до температуры окружающей среды. Охлажденную смесь нейтрализовывали до приблизительно рН 7 путем медленного добавления насыщенного водного раствора NaHCO3 (140 мл), затем разбавляли с помощью EtOAc (400 мл) и H2O (300 мл). Слои разделяли, и водный слой экстрагировали с помощью EtOAc (2×150 мл). Объединенные органические слои промывали солевым раствором (600 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в минимальном количестве DCM и фильтровали через слой силикагеля высотой 12,7 см, элюируя с использованием 3% MeOH : DCM (1 л). Фильтрат концентрировали с получением 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (по расчетам 100%), который использовали без дополнительной очистки. MS (химическая ионизация при атмосферном давлении (apci), m/z)=450,2 (М+Н).
Стадия 4. Получение 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (14,18 г, 31,54 ммоль), полученного в соответствии с методикой, используемой на стадии 3, в ацетонитриле (63,09 мл, 31,54 ммоль) при температуре окружающей среды добавляли по каплям фосфорную кислоту (36,36 г, 21,60 мл, 8 5%, 315,4 ммоль) в течение 20 минут. Реакционную смесь нагревали до 60°С в течение 4 часов, затем медленно охлаждали до температуры окружающей среды. Реакционную смесь медленно выливали в холодный раствор фосфата калия (66,95 г, 157,7 мл, 2М, 315,4 ммоль) на ледяной бане, при интенсивном перемешивании. После перемешивания в течение 10 минут, добавляли EtOAc (300 мл), и слои разделяли. Органический слой промывали солевым раствором (300 мл), затем водный слой экстрагировали с помощью EtOAc (100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% (20% MeOH/DCM)/МТВЕ, с получением 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9,48 г, 76,4%). Анализ методом порошковой рентгеновской дифракции (PXRD) подтверждал, что полученный материал является аморфным 1H ЯМР CDCl3 δ 11,19 (с, 1Н), 7,06-6,93 (м, 2Н), 6,91-6,76 (м, 1Н), 5,95 (т, 1Н), 4,44 (с, 1Н), 4,21-3,87 (м, 2Н), 3,84-3,54 (м, 4Н), 3,16 (с, 3Н), 2,98 (td, 2Н), 2,48 (с, 3Н). MS (химическая ионизация при атмосферном давлении (apci), m/z)=394,1 (М+Н).
Пример 7

8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 4-бром-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (15 мг, 63%). 1Н ЯМР (400 МГц, CDCl3) δ 11,14 (с, 1Н), 7,33 (дд, 1Н), 7,24 (м, 1Н), 6,78 (т, 1Н), 5,99 (с, 1Н), 4,38 (т, 1Н), 4,05-4,01 (м, 2Н), 3,75-3,70 (м, 4Н), 3,17 (с, 3Н), 3,00 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=426,0, 428,0 (М+Н).
Пример 8

8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 4-йод-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (12 мг, 74%). 1Н ЯМР (400 МГц, CDCl3) δ 11,11 (с, 1Н), 7,49 (дд, 1Н), 7,42 (м, 1Н), 6,63 (т, 1Н), 6,00 (с, 1Н), 4,42-4,33 (м, 1Н), 4,05-4,00 (м, 2Н), 3,76-3,69 (м, 4Н), 3,18 (с, 3Н), 2,99 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=474,0 (М+Н).
Пример 9
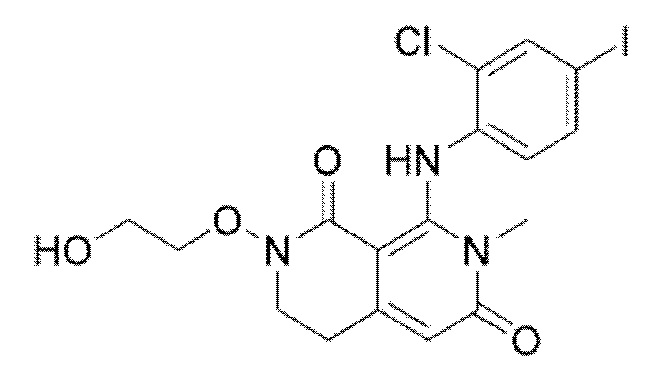
8-((2-хлор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 2-хлор-4-йоданилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((2-хлор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (6,4 мг, 50%). 1Н ЯМР (400 МГц, CDCl3) δ 11,09 (с, 1Н), 7,79 (с, 1Н), 7,48 (д, 1Н), 6,46 (д, 1Н), 6,02 (с, 1Н), 4,03 (т, 2Н), 3,78-3,70 (м, 4Н), 3,14 (с, 3 Н), 3,00 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=490,0 (М+Н).
Пример 10

8-((4-бром-2-хлорфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 4-бром-2-хлоранилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-2-хлорфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (7,9 мг, 52%). 1Н ЯМР 11,10 (с, 1Н), 7,62 (д, 1Н), 7,31 (дд, 1Н), 6,61 (д, 1Н), 6,02 (с, 1Н), 4,03 (т, 2Н), 3,77-3,68 (м, 4Н), 3,14 (с, 3Н), 3,00 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=442,0, 444,0 (М+Н).
Пример 11

8-((4-бром-2,3-дифторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 4-бром-2,3-дифторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-2,3-дифторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (12,2 мг, 75%). 1Н ЯМР 11,17 (с, 1Н), 7,30-7,22 (м, 1Н), 6,65 (дт, 1Н), 6,04 (с, 1Н), 4,32 (уш. с, 1Н), 4,04 (т, 2Н), 3,78-3,69 (м, 4Н), 3,21 (с, 3Н), 3,01 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=444,0 (М+Н).
Пример 12

8-((4-бром-3-хлор-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, методе А, за исключением того, что применяли 4-бром-3-хлор-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-3-хлор-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (10,6 мг, 68%). 1H ЯМР 11,17 (с, 1Н), 7,37 (д, 1Н), 6,68 (т, 1Н), 6,04 (с, 1Н), 4,06-4,00 (м, 2Н), 3,77-3,69 (м, 4Н), 3,20 (с, 3Н), 3,01 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=460,0, 462,0 (М+Н).
Пример 13

8-((2-фтор-4-(трифторметил)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(трифторметил)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-фтор-4-(трифторметил)анилин вместо 2-фтор-4-(метилтио)анилина, с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(трифторметил)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (11 мг, 22%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=472,2 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-(трифторметил)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(трифторметил)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((2-фтор-4-(трифторметил)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (6 мг, 62%) 1Н ЯМР (400 МГц, CDCl3) δ 11,18 (с, 1Н), 7,44-7,41 (дд, 1Н), 7,38-7,36 (д, 1Н), 6,92-6,88 (т, 1Н), 6,08 (с, 1Н), 4,05-4,03 (м, 2Н), 3,76-3,72 (м, 4Н), 3,23 (с, 3Н), 3, 03-3, 00 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=416,1 (М+Н).
Пример 14

8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 4-этил-2-фторанилин вместо 2-фтор-4-(метилтио)анилина, с получением 2-(2-(третбутокси)-этокси)-8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (19 мг, 42%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=432,2 (М+Н).
Стадия 2. Получение 8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 61%). 1Н ЯМР (400 МГц, CDCl3) δ 11,17 (с, 1Н), 7,00-6,91 (м, 2Н), 6,87-6,83 (т, 1Н), 5,93 (с, 1Н), 4,03-4,01 (м, 2Н), 3,74-3,70 (м, 4Н), 3,15 (с, 3Н), 3,00-2,97 (т, 2Н), 2,66-2,61 (кварт, 2Н), 1,25-1,21 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=376,2 (М+Н).
Пример 15

8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((4-цикло-пропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадия 7 в методе А, за исключением того, что применяли 4-циклопропил-2-фторанилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)этокси)-8-((4-циклопропил-2-фтор-фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (32 мг, 52%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=444,2 (М+Н).
Стадия 2. Получение 8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((4-циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона с получением 8-((4-циклопропил-2-фтор-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (19 мг, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 11,18 (с, 1Н), 6,83-6,78 (м, 3Н), 5,92 (с, 1Н), 4,49 (уш. с, 1Н), 4,03-4,01 (м, 2Н), 3,74-3,69 (м, 4Н), 3,13 (с, 3Н), 3,0-2,96 (т, 2Н), 1,90-1,84 (м, 1Н), 1,03-0,98 (м, 2Н), 0,69-0,65 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=388,1 (М+Н).
Пример 16

8-((2-фтор-4-метоксифенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-метоксифенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-фтор-4-метоксианилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-метоксифенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (8 мг, 17%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=434,2 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-метоксифенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-метоксифенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((2-фтор-4-метоксифенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (3 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 11,18 (с, 1Н), 6,94-6,90 (т, 1Н), 6,07-6,64 (м, 2Н), 5,91 (с, 1Н), 4,04-4,01 (м, 2Н), 3,80 (с, 3Н), 3,75-3,69 (м, 4Н), 3,12 (с, 3Н), 3,00-2,96 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=378,1 (М+Н).
Пример 17

8-((2-фтор-4-((трифторметил)тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-((трифторметил)тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадия 7 в методе А, за исключением того, что применяли 2-фтор-4-((трифторметил)тио)анилин вместо 2-фтор-4-(метилтио)анилина, с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-((трифторметил)-тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (17 мг, 31%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=504,1 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-((трифторметил)тио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)этокси)-8-((2-фтор-4-((трифторметил)тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((2-фтор-4-((трифторметил)тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 66%). 1H ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,49-7,46 (дд, 1Н), 7,40-7,38 (д, 1Н), 6,86-6,82 (т, 1Н), 6,06 (с, 1Н), 4,31 (уш. с, 1Н), 4,05-4,02 (т, 2Н), 3,76-3,72 (м, 4Н), 3,23 (с, 3Н), 3,03-3,00 (т, 2Н); MS (химическая ионизация при атмосферном давлении (apci), m/z)=448,1 (М+Н).
Пример 18

8-((2-фтор-4-изопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-изопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-фтор-4-изопропиланилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-изопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (17 мг, 34%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=446,2 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-изопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-изопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона с получением 8-((2-фтор-4-изопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 67%). 1H ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,00-6,93 (м, 2Н), 6,97-6,83 (т, 1Н), 5,93 (с, 1Н), 4,51-4,48 (т, 1Н), 4,03-4,01 (т, 2Н), 3,75-3,69 (м, 4Н), 3,14 (с, 3Н), 3,00-2,96 (т, 2Н), 2,92-2,85 (м, 1Н), 1,24-1,22 (д, 6Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=390,2 (М+Н).
Пример 19
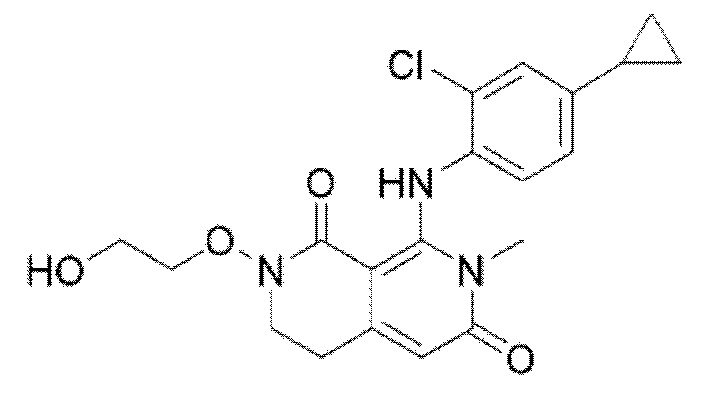
8-((2-хлор-4-циклопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-хлор-4-циклопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-хлор-4-циклопропиланилин вместо 2-фтор-4-(метилтио)анилина, с получением 2-(2-(третбутокси)этокси)-8-((2-хлор-4-циклопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (21 мг, 41%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=460,2 (М+Н).
Стадия 2. Получение 8-((2-хлор-4-циклопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-хлор-4-циклопропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((2-хлор-4-циклопропил-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (12 мг, 65%). 1Н ЯМР (400 МГц, CDCl3) δ 11,13 (с, 1Н), 7,14 (д, 1Н), 6,91-6,88 (дд, 1Н), 6,69-6,67 (д, 1Н), 5,95 (с, 1Н), 4,47-4,44 (т, 1Н), 4,04-4,01 (т, 2Н), 3,75-3,69 (м, 4Н), 3,09 (с, 3Н), 3,01-2,98 (т, 2Н), 1,89-1,82 (м, 1Н), 1,02-0,97 (м, 2Н), 0,69-0,65 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=404,1 (М+Н).
Пример 20

8-((4-ацетил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((4-этинил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-Фтор-4-((триметилсилил)этинил)анилин (30,2 мг, 0,145 ммоль) растворяли в THF (1,0 мл, 0,139 ммоль) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (0,277 мл, 1N/THF, 0,277 ммоль), и смесь перемешивали в течение 45 минут при этой же температуре. Метил 4-(2-((2-(третбутокси)этокси)амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (50 мг, 0,139 ммоль) растворяли в THF (1,0 мл, 0,139 ммоль) и добавляли по каплям к реакционной смеси. После перемешивания в течение 10 минут, добавляли LiHMDS (0,160 мл, 1N/THF, 0,160 ммоль), и реакционную смесь перемешивали в течение 1 часа при этой же температуре. Реакционную смесь гасили насыщенным водным раствором NH4Cl (25 мл), затем экстрагировали с помощью EtOAc (3×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Материал растворяли в THF (1,0 мл, 0,139 ммоль) и обрабатывали с помощью TBAF (0,1 мл, 1N/THF, 0,100 ммоль) при температуре окружающей среды в течение 30 минут. Смесь распределяли между EtOAc (25 мл) и насыщенным водным раствором NH4Cl (25 мл). Органический слой удаляли, и водный слой промывали с помощью EtOAc (2×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM, с получением 2-(2-(третбутокси)этокси)-8-((4-этинил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (19 мг, 32%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=428,2 (М+Н).
Стадия 2. Получение 8-((4-ацетил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((4-этинил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((4-ацетил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (11 мг, 64%). 1Н ЯМР (400 МГц, CDCl3) δ 11,19 (с, 1Н), 7,76-7,73 (дд, 1Н), 7,71-7,69 (дд, 1Н), 6,87-6,83 (т, 1Н), 6,07 (с, 1Н), 4,31 (уш. с, 1Н), 4,05-4,03 (м, 2Н), 3,76-3,73 (м, 4Н), 3,24 (с, 3Н), 3,03-3,00 (т, 2Н), 2,58 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=390,1 (М+Н).
Пример 21

8-((2-хлор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-хлор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-хлор-4-(метилтио)анилин вместо 2-фтор-4-(метилтио)анилина, с получением 2-(2-(третбутокси)этокси)-8-((2-хлор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (15 мг, 29%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=466,1 (М+Н).
Стадия 2. Получение 8-((2-хлор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-хлор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона с получением 8-((2-хлор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9 мг, 68%). 1H ЯМР (400 МГц, CDCl3) δ 11,15 (с, 1Н), 7,33 (д, 1Н), 7,09-7,06 (дд, 1Н), 6,71-6,69 (д, 1Н), 5,97 (с, 1Н), 4,42 (уш. с, 1Н), 4,04-4,02 (т, 2Н), 3,75-3,71 (м, 4Н), 3,12 (с, 3Н), 3,01-2,98 (т, 2Н), 2,48 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=410,1 (М+Н).
Пример 22

8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси) этокси)-8-((4-(дифтор-метокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 4-(дифторметокси)-2-фторанилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)этокси)-8-((4-(дифторметокси)-2-фтор-фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (17 мг, 33%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=470,2 (М+Н).
Стадия 2. 8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона с получением 8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 67%). 1Н ЯМР (400 МГц, CDCl3) δ 11,18 (с, 1Н), 7,00-6,91 (м, 3Н), 6,68-6,31 (т, 1Н), 5,98 (с, 1Н), 4, 43-4, 40 (т, 1Н), 4,04-4,02 (т, 2Н), 3,75-3,71 (м, 4Н), 3,16 (с, 3Н), 3,01-2,98 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=414,1 (М+Н).
Пример 23
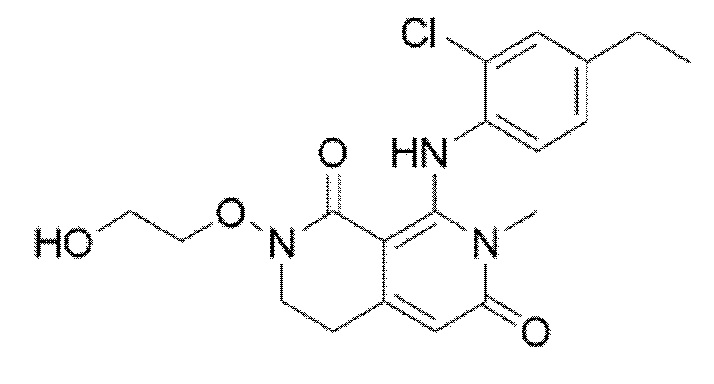
8-((2-хлор-4-этилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-хлор-4-этилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-хлор-4-этиланилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)-этокси)-8-((2-хлор-4-этилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9 мг, 14%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=448,2 (М+Н).
Стадия 2. Получение 8-((2-хлор-4-этилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-хлор-4-этилфенил)амино)-7-метил-3,4-дигидро-2, 7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((2-хлор-4-этилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона (6 мг, 80%). 1Н ЯМР (400 МГц, CDCl3) δ 11,13 (с, 1Н), 7,29 (д, 1Н), 7,02-7,00 (дд, 1Н), 6,72-6,70 (д, 1Н), 5,95 (с, 1Н), 4,46 (уш. с, 1Н), 4,04-4,01 (м, 2Н), 3,75-3,70 (м, 4Н), 3,10 (с, 3Н), 3,01-2,97 (т, 2Н), 2,65-2,59 (кварт, 2Н), 1,24-1,21 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=392,1 (М+Н).
Пример 24

8-((2-фтор-4-(трифторметокси)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(трифторметокси)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 7 в методе А, за исключением того, что применяли 2-фтор-4-(трифторметокси)анилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(трифторметокси)-фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (22 мг, 41%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=488,2 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-(трифторметокси)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-(трифторметокси)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)-этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона с получением 8-((2-фтор-4-(трифторметокси)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (13 мг, 67%). 1Н ЯМР (400 МГц, CDCl3) δ 11,17 (с, 1Н), 7,08-7,06 (д, 1Н), 7,02-6,99 (д, 1Н), 6, 94-6, 90 (т, 1Н), 6,01 (с, 1Н), 4,40-4,36 (т, 1Н), 4,05-4,02 (т, 2Н), 3,75-3,72 (т, 4Н), 3,18 (с, 3Н), 3,02-2,99 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=432,1 (М+Н).
Пример 25

8-((4-((дифторметил)тио)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((4-((дифтор-метил)тио)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадия 7 в методе А, за исключением того, что применяли 4-((дифтор-метил)тио)-2-фторанилин вместо 2-фтор-4-(метилтио)анилина с получением 2-(2-(третбутокси)этокси)-8-((4-((дифторметил)тио)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (19 мг, 35%). δ 11,31 (с, 1Н), 7,40-7,37 (дд, 1Н), 7,30-7,28 (м, 1Н), 6,94-6,66 (т, 1Н), 6,80-6,76 (т, 1Н), 6,04 (с, 1Н), 4,17-4,14 (м, 2Н), 3,82-3,79 (т, 2Н), 3,62-3,59 (м, 2Н), 3,24 (с, 3Н), 3,01-2,97 (т, 2Н), 1,21 (с, 9Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=486,2 (М+Н).
Стадия 2. Получение 8-((4-((дифторметил)тио)-2-фторфенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадии 8 в методе А, за исключением того, что применяли 2-(2-(третбутокси)этокси)-8-((4-((дифторметил)тио)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион вместо 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, с получением 8-((4-((дифторметил)тио)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (11 мг, 57%). 1Н ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,42-7,39 (дд, 1Н), 7,33-7,32 (д, 1Н), 6,96-6,68 (т, 1Н), 6,87-6,82 (т, 1Н), 6,04 (с, 1Н), 4,37-4,33 (т, 1Н), 4,05-4,02 (т, 2Н), 3,75-3,72 (т, 4Н), 3,22 (с, 3Н), 3,03-2,99 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=430,1 (М+Н).
Пример 26

8-((4-бром-2-фторфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, на стадиях 1-7 в методе А, за исключением того, что применяли О-изопропилгидроксил-амина гидрохлорид вместо О-(2-(третбутокси)этил)гидроксиламина гидрохлорида на стадии 5, и 4-бром-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-2-фтор-фенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10,3 мг, 14%). 1Н ЯМР (400 МГц, CDCl3) δ 11,37 (с, 1Н), 7,30 (дд, 1Н), 7,21 (dq, 1Н), 6,73 (т, 1Н), 6,00 (с, 1Н), 4,33 (м, 1Н), 3,68 (т, 2Н), 3,18 (с, 3Н), 2,99 (т, 2Н), 1,30 (д, 6Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=424,1, 426,1 (М+Н).
Пример 27

8-((2-фтор-4-йодфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, на стадиях 1-7 в методе А, за исключением того, что применяли 0-изопропилгидроксил-амина гидрохлорид вместо О-(2-(третбутокси)этил)гидроксиламина гидрохлорида на стадии 5, и 2-фтор-4-йоданилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((2-фтор-4-йодфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (15,2 мг, 23%). 1H ЯМР (400 МГц, CDCl3) δ 11,36 (с, 1Н), 7,46 (дд, 1Н), 7,38 (дт, 1Н), 6,58 (т, 1Н), 6,00 (с, 1Н), 4,33 (м, 1Н), 3,68 (т, 2Н), 3,19 (с, 3Н), 2,99 (т, 2Н), 1,30 (д, 6Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=472,1 (М+Н).
Пример 28

2-этокси-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, на стадиях 1-7 в методе А, за исключением того, что применяли О-этилгидроксиламина гидрохлорид вместо О-(2-(третбутокси)этил)гидроксиламина гидро-хлорида на стадии 5, и 2-фтор-4-йоданилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 2-этокси-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (24,3 мг, 25%). 1H ЯМР 11,33 (с, 1Н), 7,46 (д, 1Н), 7,38 (д, 1Н), 6,57 (т, 1Н), 6,00 (с, 1Н), 4,07 (кварт, 2Н), 3,72 (т, 2Н), 3,19 (с, 3Н), 2,99 (т, 2Н), 1,31 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=458,0 (М+Н).
Пример 2 9

8-((2-фтор-4-йодфенил)амино)-2-метокси-7-метил-3,4-дигидро-2, 7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 6, на стадиях 1-7 в методе А, за исключением того, что применяли O-метилгидроксиламина гидрохлорид вместо О-(2-(третбутокси)этил)гидроксиламина гидро-хлорида на стадии 5, и 2-фтор-4-йоданилин вместо 2-фтор-4-(метилтио)-анилина на стадии 7, с получением 8-((2-фтор-4-йодфенил)амино)-2-метокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (16,7 мг, 33%). 1H ЯМР (400 МГц, CDCl3) δ 11,32 (с, 1Н), 7,47 (д, 1Н), 7,39 (д, 1Н), 6,58 (т, 1Н), 6,00 (с, 1Н), 3,84 (с, 3Н), 3,73 (т, 2Н), 3,19 (с, 3Н), 3,00 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=444,0 (М+Н).
Пример 30

2-(третбутокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадии 1-7. Получение метил 4-(2-(третбутоксиамино)этил)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Получали таким же методом, как в примере 6, на стадиях 1-7, за исключением того, что применяли O-(третбутил)гидроксиламина гидрохлорид вместо О-(2-(третбутокси)этил)гидроксиламина гидро-хлорида на стадии 5, и 2-фтор-4-йоданилин вместо 2-фтор-4-(метилтио)-анилина на стадии 7, с получением метил 4-(2-(третбутоксиамино)этил)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (40,3 мг, 62%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=518,1 (М+Н).
Стадия 8. Получение 2-(третбутокси)-8-((2-фтор-4-йодфенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Раствор метил 4-(2-(третбутоксиамино)этил)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (40,3 мг, 0,078 ммоль) в THF (1 мл) обрабатывали с помощью Et3N (10,9 мкл, 0,078 ммоль). Реакционную смесь нагревали до 50°С в течение 1 часа. К реакционной смеси добавляли еще Et3N (10,9 мкл, 0,078 ммоль) и продолжали перемешивать при 50°С в течение еще одного часа. Добавляли дополнительную аликвоту Et3N (10,9 мкл, 0,078 ммоль), и реакционную смесь перемешивали при 50°С в течение 5 дней. Реакционную смесь распределяли между водой (40 мл) и EtOAc (20 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции объединяли, промывали насыщенным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (3×15 мл). Объединенные органические слои промывали насыщенным раствором бикарбоната натрия (20 мл) и солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом с получением 2-(третбутокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (16,5 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 11,49 (с, 1Н), 7,46 (дд, 1Н), 7,39 (td, 1Н), 6,59 (т, 1Н), 5,99 (с, 1Н), 3,70-3,60 (м, 2Н), 3,18 (с, 3Н), 3,10-2,87 (м, 2Н), 1,34 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=486,1 (М+Н).
Пример 31

(S)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил (S, Е)-2-хлор-4-(2-((2-гидроксипропокси) имино)этил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (200 мг, 0,736 ммоль) и (S)-O-(2-((третбутилдиметилсилил)окси)пропил)гидроксиламин (151 мг, 0,736) помещали в 1,4-диоксан (5 мл). TEA (103 мкл, 0,736 ммоль) и добавляли HCl (368 мкл, 4N/диоксан, 1,47 ммоль). Суспензию нагревали до 60°С в течение 3 часов. Реакционную смесь фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гексаны, с получением метил (S, Е)-2-хлор-4-(2-((2-гидроксипропокси)имино)-этил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (66,6 мг, 29%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=317,1 (М+Н).
Стадия 2. Получение метил (S, Е)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадец-2-ен-1-ил)-1,6-дигидропиридин-3-карбоксилата. Метил (S, Е)-2-хлор-4-(2-((2-гидроксипропокси)имино)этил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (66,6 мг, 0,210 ммоль) и имидазол (0,043 г, 0,631 ммоль) растворяли в DMF (2 мл) и охлаждали до 0°С. Затем добавляли TBS-C1 (63,4 мг, 0,421 ммоль), и реакционную смесь подогревали до комнатной температуры в течение 2 часов. Реакционную смесь разбавляли водой (40 мл) и EtOAc (20 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали водой (5×20 мл) и солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/гексаны, с получением метил (S, Е)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадец-2-ен-1-ил)-1,6-дигидропиридин-3-карбоксилата (63,4 мг, 70%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=431,2 (М+Н).
Стадия 3. Получение метил (S)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадецил)-1,6-дигидро-пиридин-3-карбоксилата. Метил (S, Е)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадец-2-ен-1-ил)-1,6-дигидропиридин-3-карбоксилат (63,4 мг, 0,147 ммоль) растворяли в 2-пропаноле (1 мл). Добавляли цианоборгидрид натрия (46,2 мг, 0,735 ммоль) и уксусную кислоту (42,1 мкл, 0,735 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (30 мл) и EtOAc (20 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/гексаны, с получением метил (S)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадецил)-1,6-дигидро-пиридин-3-карбоксилата (30,6 мг, 48%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=433,2 (М+Н).
Стадия 4. Получение (S)-2-(2-((третбутилдиметилсилил)окси)-пропокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-фтор-4-йоданилина (16,9 мг, 0,071 ммоль) в безводном THF (1 мл) при -78°С в атмосфере N2 добавляли LiHMDS (0,141 мл, 1M/THF, 0,141 ммоль). Смесь перемешивали в течение 45 минут, затем добавляли раствор метил (S)-2-хлор-1-метил-6-оксо-4-(6,8,8,9,9-пентаметил-4,7-диокса-3-аза-8-силадецил)-1,6-дигидропиридин-3-карбоксилата (30,6 мг, 0,071 ммоль) в THF (0,5 мл). Смесь перемешивали при -78°С в течение 10 минут. Смесь гасили насыщенным водным раствором NH4Cl (30 мл), перемешивали при температуре окружающей среды в течение 10 минут, затем экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гексаны, с получением (S)-2-(2-((третбутилдиметилсилил)окси)пропокси)-8-((2-фтор-4-йодфенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (15,2 мг, 36%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=602,2 (М+Н).
Стадия 5. Получение (S)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. (S)-2-(2-((третбутилдиметилсилил)окси)пропокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (15,2 мг, 0,025 ммоль) растворяли в МеОН (0,5 мл). Реакционную смесь обрабатывали с помощью HCl (12,6 мкл, 4N/диоксан, 0,051 ммоль) и перемешивали в течение 1 часа. Реакционную смесь концентрировали, и остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции объединяли, промывали насыщенным водным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (3×20 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (15 мл) и солевым раствором (15 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением (S)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (6,8 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,48 (дд, 1Н), 7,42 (дт, 1Н), 6,62 (т, 1Н), 6,00 (с, 1Н), 4,05-3,91 (м, 2Н), 3,78-3,60 (м, 3Н), 3,17 (с, 3Н), 3,10-2,90 (м, 2Н), 1,15 (д, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=488,0 (М+Н).
Пример 32

(R)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 31, за исключением того, что применяли (R)-O-(2-((третбутилдиметилсилил)окси)пропил)-гидроксиламин вместо (S)-O-(2-((третбутилдиметилсилил)окси)-пропил)гидроксиламина на стадии 1, с получением (R)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (8,6 мг, 49%). 1Н ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,48 (дд, 1Н), 7,42 (дт, 1Н), 6,62 (т, 1Н), 5,99 (с, 1Н), 4,05-3,91 (м, 2Н), 3, 78-3, 60 (м, 3Н), 3,17 (с, 3Н), 3,09-2, 90 (м, 2Н), 1,15 (д, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=488,0 (М+Н).
Пример 33

(S)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 31, за исключением того, что применяли 4-бром-2-фторанилин вместо 2-фтор-4-йоданилина на стадии 4, с получением (S)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9,2 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 11,16 (с, 1Н), 7,32 (дд, 1Н), 7,26-7,22 (м, 1Н), 6,78 (т, 1Н), 5,99 (с, 1Н), 4,05-3,92 (м, 2Н), 3,78-3,60 (м, 3Н), 3,17 (с, 3Н), 3,10-2,90 (м, 2Н), 1,15 (д, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=440,1, 442,1 (М+Н).
Пример 34

(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 31, за исключением того, что применяли (R)-O-(2-((третбутилдиметилсилил)окси)пропил)-гидроксиламин вместо (S)-O-(2-((третбутилдиметилсилил)окси)пропил)-гидроксиламина на стадии 1, и 4-бром-2-фторанилин вместо 2-фтор-4-йоданилина на стадии 4 с получением (R)-8-((4-бром-2-фторфенил)-амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9,2 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 11,17 (с, 1Н), 7,32 (дд, 1Н), 7,24 (дт, 1Н), 6,78 (т, 1Н), 5,99 (с, 1Н), 4,06-3,91 (м, 2Н), 3,78-3,60 (м, 3Н), 3,17 (с, 3Н), 3,10-2,90 (м, 2Н), 1,15 (д, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=440,0 (М+Н).
Пример 35

(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадии 1-4: Получение (R)-8-((4-бром-2-фторфенил)амино)-2-(2-((третбутилдиметилсилил)окси)пропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 31, на стадиях 1-4 за исключением того, что применяли (R)-О-(2-((третбутилдиметилсилил)окси)пропил)гидроксиламин вместо (S)-О-(2-((третбутилдиметилсилил)окси)пропил)гидроксил-амина на стадии 1, с получением (R)-8-((4-бром-2-фторфенил)-амино)-2-(2-((третбутилдиметилсилил)окси)пропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (63,5 мг, 62%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=554,1 (М+Н).
Стадия 5. Получение (R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. (R)-8-((4-бром-2-фторфенил)амино)-2-(2-((третбутилдиметилсилил) окси)пропокси)-7-метил-3,4-дигидро-2, 7-нафтиридин-1,6(2Н,7Н)-дион (42,3 мг, 0,076 ммоль) растворяли в THF (1 мл) и МеОН (1 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (21,8 мг, 0,114 ммоль). Через 5 минут, добавляли N-йодсукцинимид (17,2 мг, 0,076 ммоль) и перемешивали в течение 3 0 минут. Реакционную смесь распределяли между EtOAc (15 мл) и насыщенным водным раствором бикарбоната натрия (30 мл), и водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гептан, с получением (R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидрокси-пропокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (23,8 мг, 54%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=566,0 (М+Н).
Стадия 6. Получение (R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору (R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (21,7 мг, 0,038 ммоль) в THF (400 мкл) при 0°С добавляли Pd(Pt-Bu3)2 (1,96 мг, 0,004 ммоль), затем метилцинка(II) хлорид (19,4 мкл, 0,039 ммоль). Смесь удаляли из ледяной бани и перемешивали в течение 5 минут. Смесь распределяли между насыщенным водным раствором NH4Cl (20 мл) и EtOAc (15 мл). Водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции объединяли, промывали насыщенным водным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (25 мл) и солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением (R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (7,5 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 10,97 (с, 1Н), 7,30 (дд, 1Н), 7,19 (дт, 1Н), 6,68 (т, 1Н), 4,05-3,91 (м, 2Н), 3,71 (м, 2Н), 3,64 (м, 1Н), 3,22 (с, 3Н), 3,13-3,04 (м, 1Н), 3,03-2,92 (м, 1Н), 2,08 (с, 3Н), 1,14 (д, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=454,1 (М+Н).
Пример 36

8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 4-бром-2-фторанилина (88 мг, 0,47 ммоль) в безводном THF (5 мл) при -78°С в атмосфере N2 добавляли LiHMDS (694 мл, 1,0 M/THF, 0,694 ммоль). Смесь перемешивали в течение 45 минут, затем добавляли раствор метил 4-(2-((2-(третбутокси)этокси)-амино)этил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (167 мг, 0,46 ммоль) в THF (2 мл). Продолжали перемешивание при -78°С в течение 10 минут, затем добавляли LiHMDS (300 мл, 1,0 M/THF, 0,300 ммоль). После перемешивания в течение еще 10 минут при -78°С, добавляли LiHMDS (100 мл, 1,0 M/THF, 0,100 ммоль) и продолжали перемешивание в течение 10 минут. Смесь гасили насыщенным водным раствором NH4Cl (10 мл), перемешивали при температуре окружающей среды в течение 10 минут, затем экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (78 мг, 35%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=482,1 (М+Н).
Стадия 2. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н, 7Н)-диона (78 мг, 0,16 ммоль) в THF (2 мл) и МеОН (2 мл) добавляли моногидрат п-толуолсульфоновой кислоты (46 мг, 0,24 ммоль). Через 5 минут, добавляли N-йодсукцинимид (36 мг, 0,16 ммоль). После перемешивания при температуре окружающей среды в течение 30 минут, реакционную смесь распределяли между EtOAc (10 мл) и насыщенным водным раствором NaHCO3 (10 мл). Водный раствор экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-30% EtOAc/гексаны, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (62 мг, 63%). 1Н ЯМР (400 МГц, CDCl3) δ 11,54 (с, 1Н), 7,31 (дд, 1Н), 7,22 (м, 1Н), 6,73 (т, 1Н), 4,17-4,13 (м, 2Н), 3,80 (т, 2Н), 3,63-3,59 (м, 2Н), 3,27 (с, 3Н), 3,17 (т, 2Н), 1,22 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=608,0 (М+Н).
Стадия 3. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(трет-бутокси)этокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)-этокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (62 мг, 0,10 ммоль) в безводном THF (2 мл) при 0°С добавляли Pd(Pt-Bu3)2 (5 мг, 0,01 ммоль), затем метилцинка(II) хлорид (61 мл, 2M/THF, 0,12 ммоль). Смесь удаляли из ледяной бани и перемешивали в течение 5 минут, затем распределяли между насыщенным водным раствором NH4Cl (10 мл) и EtOAc (10 мл). Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-60% EtOAc/гексаны, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (39 мг, 77%). 1Н ЯМР (400 МГц, CDCl3) δ 11,11 (с, 1Н), 7,28 (дд, 1Н), 7,16 (дт, 1Н), 6,61 (т, 1Н), 4,17-4,13 (м, 2Н), 3,78 (т, 2Н), 3, 64-3, 59 (м, 2Н), 3,25 (с, 3Н), 3,02 (т, 2Н), 2,09 (с, 3Н), 1,22 (с, 9Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=496,1 (М+Н).
Стадия 4. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)-этокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (39 мг, 0,079 ммоль) в ацетонитриле (0,5 мл) добавляли фосфорную кислоту (0,5 мл). Смесь подогревали до 60°С в течение 1 часа, затем охлаждали до температуры окружающей среды. Смесь перемешивали с насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл) в течение 10 минут, и слои разделяли. Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (0-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции обрабатывали насыщенным водным раствором NaHCO3/EtOAc с получением 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (18 мг, 52%). 1Н ЯМР (400 МГц, CDCl3) δ 10,96 (с, 1Н), 7,31 (дд, 1Н), 7,20 (дт, 1Н), 6,68 (т, 1Н), 4,05-4,01 (м, 2Н), 3,75-3,69 (м, 4Н), 3,23 (с, 3Н), 3,03 (т, 2Н), 2,08 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=440,0 (М+Н).
Пример 37

8-((4-бром-2-фторфенил)амино)-5-хлор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 6, на стадиях 1-7 в методе А, за исключением того, что применяли 4-бром-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 7, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (28 мг, 35%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=482,1 (М+Н).
Стадия 2. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-хлор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (28 мг, 0, 058 ммоль) в DMF (0,5 мл) добавляли NCS (8 мг, 0,058 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 часа, затем при 50°С в течение 2 часов. Охлажденную смесь распределяли между насыщенным раствором NaHCO3 (10 мл) и EtOAc (10 мл), и водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали водой (5×10 мл) и солевым раствором (10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гексаны, с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-хлор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (14 мг, 47%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=516,0 (М+Н).
Стадия 3. Получение 8-((4-бром-2-фторфенил)амино)-5-хлор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)-этокси)-5-хлор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (14 мг, 0,027 ммоль) в ацетонитриле (0,5 мл) добавляли фосфорную кислоту (0,5 мл). Смесь подогревали до 60°С в течение 30 минут, затем охлаждали до температуры окружающей среды. Смесь перемешивали с насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл) в течение 10 минут, и слои разделяли. Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM, с получением 8-((4-бром-2-фторфенил)амино)-5-хлор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (11 мг, 88%). 1Н ЯМР (400 МГц, CDCl3) δ 11,22 (с, 1Н), 7,33 (дд, 1Н), 7,26 (дт, 1Н), 6,78 (т, 1Н), 4,07-4,02 (м, 2Н), 3,78-3,70 (м, 4Н), 3,25 (с, 3Н), 3,22 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=460,0 (М+Н).
Пример 38

5-хлор-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 37, за исключением того, что применяли 4-йод-2-фторанилин вместо 4-бром-2-фторанилина на стадии 1, с получением 5-хлор-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 93%). 1Н ЯМР (400 МГц, CDCl3) δ 11,21 (с, 1Н), 7,49 (дд, 1Н), 7,43 (дт, 1Н), 6,63 (т, 1Н), 4,25-4,17 (м, 1Н), 4,07-4,02 (м, 2Н), 3,79-3,70 (м, 4Н), 3,25 (с, 3Н), 3,22 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=508,0 (М+Н).
Пример 39

8-((4-бром-2-фторфенил)амино)-5-фтор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-фтор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Раствор 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (77 мг, 0,16 ммоль) и 1-(хлорметил)-4-фтор-1,4-диазабицикло-[2.2.2]октан-1,4-дия тетрафторбората (57 мг, 0,16 ммоль) в ацетонитриле (9,0 мл) перемешивали при 0°С в течение 1 часа. Реакционную смесь разбавляли с помощью EtOAc (50 мл) и промывали водой (25 мл), затем солевым раствором (25 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут). Содержащие требуемый продукт фракции объединяли и разбавляли насыщенным водным раствором NaHCO3 (15 мл), затем экстрагировали с помощью EtOAc (2×25 мл). Органические фазы объединяли и сушили над Na2SO4, фильтровали и концентрировали с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-5-фтор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 13%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=500,1 (М+Н).
Стадия 2. Получение 8-((4-бром-2-фторфенил)амино)-5-фтор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Раствор 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)-этокси)-5-фтор-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (13 мг, 0,026 ммоль) и концентрированной H3PO4 (0,20 мл, 3,0 ммоль) в ацетонитриле (1,0 мл) перемешивали при 60°С в течение 1 часа. Реакционную смесь концентрировали, затем разбавляли раствором K3PO4 (1М в воде, 3,0 мл, 3,0 ммоль). Водную фазу экстрагировали с помощью EtOAc (2×15 мл), и органические фазы объединяли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом HPLC (10-95% ацетонитрил/вода/0,1% TFA). Содержащие требуемый продукт фракции объединяли и разбавляли насыщенным водным раствором NaHCO3 (15 мл). Водный раствор экстрагировали с помощью EtOAc (2×15 мл), и органические фазы объединяли, сушили над Na2SO4, фильтровали и концентрировали с получением 8-((4-бром-2-фторфенил)амино)-5-фтор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (5,0 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 10,79 (с, 1Н), 7,34-7,30 (м, 1Н), 7,25-7,21 (м, 1Н), 6,72 (т, 1Н), 4,23 (т, 1Н), 4,06-4,02 (м, 2Н), 3, 80-3, 70 (м, 4Н), 3,25 (с, 3Н), 3,17-3,12 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=444,0 (М+Н).
Пример 40

8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-4-йодфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((4-Бром-2-фторфенил)амино)-2-(2-(трет-бутокси) этокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 36, на стадиях 1-3; 65 мг, 0,13 ммоль), йодид меди (I) (6,2 мг, 0,033 ммоль), йодид натрия (39 мг, 0,2 6 ммоль) и (1S,2S)-N1,N2-диметилциклогексан-1,2-диамин (9,3 мг, 0,065 ммоль) объединяли в 1,4-диоксане (1 мл) и подвергали барботированию аргоном. Смесь нагревали при 110°С в течение 18 часов, затем при температуре окружающей среды в течение 48 часов. Смесь распределяли между EtOAc (20 мл) и раствором гидроксида аммония (10 мл/вода 10 мл). Органическую фазу отделяли, и водную фазу экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические фазы промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гептан, с получением 2-(2-(третбутокси)этокси)-8-((2-фтор-4-йодфенил)-амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (49 мг, 69%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=544,1 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион. К раствору 2-(2-(третбутокси)этокси)-8-((2-фтор-4-йод-фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (49 мг, 0,09 ммоль) в ацетонитриле (1 мл) добавляли фосфорную кислоту (0,5 мл). Смесь подогревали до 60°С в течение 1 часа, затем охлаждали до температуры окружающей среды. Смесь перемешивали с насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл) в течение 10 минут, и слои разделяли. Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0, 1% TFA в течение 20 минут), и очищенные фракции обрабатывали с помощью EtOAc/насыщенный водный раствор NaHCO3 с получением 8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (23 мг, 52%). 1Н ЯМР (400 МГц, CDCl3) δ 10,95 (с, 1Н), 7,47 (дд, 1Н), 7,37 (дт, 1Н), 6,52 (т, 1Н), 4,05-4,02 (м, 2Н), 3,75-3,65 (м, 4Н), 3,24 (с, 3Н), 3,03 (т, 2Н), 3,02 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=488,0 (М+Н).
Пример 41

8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил 4-бром-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору 2-фтор-анилина (100 мг, 0,90 ммоль) в безводном THF (5 мл) при -78°С в атмосфере N2 добавляли LiHMDS (1,34 мл, 1,0 M/THF, 1,34 ммоль). Смесь перемешивали в течение 45 минут, затем добавляли раствор метил 4-бром-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (250 мг, 0,89 ммоль) в THF (5 мл). Смесь перемешивали при -78°С в течение 50 минут. Добавляли дополнительные количества LiHMDS (0,5 мл, 1,0 M/THF, 0,5 ммоль) и продолжали перемешивание в течение еще 10 минут. Смесь гасили насыщенным водным раствором NH4Cl (20 мл), перемешивали при температуре окружающей среды в течение 10 минут, затем экстрагировали с помощью EtOAc (3×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением метил 4-бром-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (291 мг, 92%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=355,0 (М+Н).
Стадия 2. Получение метил (Е)-4-(2-этоксивинил)-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил 4-бром-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоксилат (188 мг, 0,529 ммоль), метансульфонато(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)'2-амино-1,1'-бифенил-2-ил)палладий(II) (44,3 г, 0,053 ммоль) и (Е)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диокса-боролан (123 мкл, 0,582 ммоль) суспендировали в 1,4-диоксане (5 мл) и добавляли карбонат калия (0,397 мл, 2N водный раствор, 0,794 ммоль). После дегазирования с помощью аргона, смесь перемешивали при температуре окружающей среды в течение 2 3 часов. Реакционную смесь распределяли между водой (40 мл) и EtOAc (30 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гексаны, с получением метил (Е)-4-(2-этоксивинил)-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (91,5 мг, 50%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=347,1 (М+Н).
Стадия 3. Получение метил (Е)-4-(2-((2-(третбутокси)-этокси)имино)этил)-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Е)-4-(2-этоксивинил)-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (91,5 мг, 0,264 ммоль) и О-(2-(третбутокси)этил)-гидроксиламина гидрохлорид (44,8 мг, 0,264 ммоль) помещали в 1,4-диоксан (2 мл). Добавляли TEA (36,8 мкл, 0,264 ммоль) и HCl (66 мкл, 4N/диоксан, 0,264 ммоль). Суспензию нагревали до 60°С. Через 2 часа, добавляли дополнительное количество HCl (66 мкл, 4N/диоксан, 0,264 ммоль) и продолжали перемешивание при 60°С в течение еще 2 часов. Реакционную смесь фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/гексаны, с получением метил (Е)-4-(2-((2-(третбутокси)этокси)имино)этил)-2-((2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (67,6 мг, 59%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=434,2 (М+Н).
Стадия 4. Получение 2-(2-(третбутокси)этокси)-8-((2-фтор-фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил (Е)-4-(2-((2-(третбутокси)этокси)имино)этил)-2-((2-фтор-фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (67,6 мг, 0,156 ммоль) растворяли в МеОН (1,0 мл) и 2-пропаноле (1,0 мл). Добавляли цианоборгидрид натрия (49 мг, 0,780 ммоль) и уксусную кислоту (44,6 мкл, 0,780 ммоль), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционную смесь нагревали при 40°С в течение 16 часов. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (20 мл) и EtOAc (10 мл). Водный слой промывали EtOAc (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гексаны, с получением 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (37,8 мг, 60%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=404,2 (М+Н).
Стадия 5. Получение 2-(2-(третбутокси) этокси)-8-((2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-(2-(третбутокси)этокси)-8-((2-фторфенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (19 мг, 0,047 ммоль) растворяли в THF (0,5 мл) и МеОН (0,5 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (13 мг, 0,071 ммоль). Через 5 минут, добавляли N-йодсукцинимид (11 мг, 0,047 ммоль) и перемешивали в течение 1 часа. Реакционную смесь распределяли между EtOAc (20 мл) и насыщенным водным раствором бикарбоната натрия (40 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-30% EtOAc/гексаны, с получением 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9, 9 мг, 40%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=530,1 (М+Н).
Стадия 6. Получение 8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н, 7Н)-диона. 2-(2-(Третбутокси)этокси)-8-((2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (9,9 мг, 0,019 ммоль) растворяли в ацетонитриле (0,5 мл) и добавляли фосфорную кислоту (0,5 мл). Раствор нагревали до 60°С в течение 1 часа. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (15 мл) и EtOAc (10 мл). Водный слой промывали с помощью EtOAc (2×8 мл). Объединенные органические слои промывали солевым раствором (15 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции объединяли, промывали насыщенным водным раствором бикарбоната натрия и экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (20 мл) и солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением (8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (4,5 мг, 51%). 1Н ЯМР (400 МГц, CDCl3) δ 11,41 (с, 1Н), 7,21-7,08 (м, 3Н), 6,94 (т, 1Н), 4,04 (т, 2Н), 3,76-3,69 (м, 4Н), 3,25 (с, 3Н), 3,19 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=474,0 (М+Н).
Пример 42

5-бром-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 41, за исключением того, что применяли 4-бром-2-фторанилин вместо 2-фторанилина на стадии 1, и N-бромсукцинимид на стадии 5, с получением 5-бром-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (6,5 мг, 29%). 1H ЯМР (400 МГц, CDCl3) δ 11,28 (с, 1Н), 7,33 (дд, 1Н), 7,25 (м, 1Н), 6,80 (т, 1Н), 4,05 (т, 2Н), 3,78-3,70 (м, 4Н), 3,25 (с, 3Н), 3,22 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=504,0 (М+Н).
Пример 4 3

4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фторфенил) амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (75 мг, 0,14 ммоль) в безводном THF (5 мл) при температуре окружающей среды добавляли метилцинка(II) хлорид (140 мл, 2М, 0,14 ммоль). После перемешивания в течение 5 минут, смесь концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гексаны, с получением 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (38 мг, 64%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=418,2 (М+Н).
Стадия 2. Получение 4-бром-2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (38 мг, 0,09 ммоль) в безводном DMF (1 мл) добавляли NBS (17 мг, 0,09 ммоль). Смесь перемешивали при температуре окружающей среды в течение 2 часов, затем распределяли между насыщенным раствором NaHCO3 (10 мл) и EtOAc (10 мл). Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали водой (5×10 мл) и солевым раствором (10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-60% EtOAc/гексаны, с получением 4-бром-2-(2-(третбутокси)этокси)-8-((2-фторфенил)-амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (30 мг, 66%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=496,2 (М+Н).
Стадия 3. Получение 4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-
4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (75 мг, 0,14 ммоль) в безводном THF (5 мл) при температуре окружающей среды добавляли метилцинка(II) хлорид (140 мл, 2 М, 0,14 ммоль). После перемешивания в течение 5 минут, смесь концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-80% EtOAc/гексаны, с получением 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (38 мг, 64%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=418,2 (М+Н).
Стадия 2. Получение 4-бром-2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (38 мг, 0,09 ммоль) в безводном DMF (1 мл) добавляли NBS (17 мг, 0,09 ммоль). Смесь перемешивали при температуре окружающей среды в течение 2 часов, затем распределяли между насыщенным раствором NaHCO3 (10 мл) и EtOAc (10 мл). Водный слой экстрагировали с помощью EtOAc (2×10 мл), и объединенные органические слои промывали водой (5×10 мл) и солевым раствором (10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-60% EtOAc/гексаны, с получением 4-бром-2-(2-(третбутокси)этокси)-8-((2-фторфенил)-амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (30 мг, 66%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=496,2 (М+Н).
Стадия 3. Получение 4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 4-бром-2-(2-(третбутокси)этокси)-8-((2-фтор-фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (30 мг, 0,060 ммоль) в ацетонитриле (0,5 мл) добавляли фосфорную кислоту (0,5 мл). Смесь подогревали до 60°С в течение 1 часа, затем охлаждали и перемешивали с насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл) в течение 10 минут. Слои разделяли, и водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-15% (20% MeOH/DCM)/DCM, с получением 4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (10 мг, 38%). 1H ЯМР (400 МГц, CDCl3) δ 11,08 (с, 1Н), 7,15-7,05 (м, 2Н), 6,86 (т, 1Н), 5,36 (т, 1Н), 4,17-4,04 (м, 3Н), 3,99-3,92 (м, 1Н), 3,88 (дд, 1Н), 3,68 (дт, 1Н), 3,21 (с, 3Н), 2,17 (с, 3Н), 1,26 (т, 1Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=440,l (М+Н).
Пример 44

8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил (Z)-4-(2-этоксивинил)-2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору 4-этил-2-фторанилина (155 мг, 1,12 ммоль) в безводном THF (2 мл) при -78°С в атмосфере N2 добавляли LiHMDS (2,21 мл, 1M/THF, 2,21 ммоль). Смесь перемешивали в течение 40 минут, затем добавляли через шприц раствор метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (300 мг, 1,10 ммоль) в THF (1 мл). Смесь перемешивали при -78°С в течение 45 минут. Смесь гасили насыщенным водным раствором NH4Cl (35 мл), перемешивали при температуре окружающей среды в течение 5 минут, затем экстрагировали с помощью EtOAc (3×15 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гептан, с получением метил (Z)-4-(2-этоксивинил)-2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (298 мг, 72%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=375,2 (М+Н).
Стадия 2. Получение метил 2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. Метил (Z)-4-(2-этоксивинил)-2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (298 мг, 0,797 ммоль) растворяли в DCM (800 мкл) и добавляли TFA (921 мкл, 11,9 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов. Смесь распределяли между DCM (20 мл) и насыщенным водным раствором бикарбоната натрия (35 мл). Водный слой промывали с помощью DCM (2×15 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением метил 2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата (276 мг, 100%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=347,1 (М+Н).
Стадия 3. Получение 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил 2-((4-этил-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилат (276 мг, 0,797 ммоль), О-(2-(третбутокси)этил)гидроксиламина гидрохлорид (117 мг, 0,876 ммоль), триацетоксигидроборат натрия (203 мг, 0,956 ммоль) и уксусную кислоту (45,6 мкл, 0,797 ммоль) помещали в DCE (8 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Реакционную смесь нагревали до 55°С в течение 20 минут. К реакционной смеси добавляли еще триацетоксигидроборат натрия (400 мг) и уксусную кислоту (90 мкл), и перемешивали при температуре окружающей среды в течение 16 часов. К реакционной смеси добавляли цианоборгидрид натрия (250 мг, 3,98 ммоль) и уксусную кислоту (250 мкл) и перемешивали при температуре окружающей среды в течение 3 часов. Раствор распределяли между DCM (40 мл) и насыщенным водным раствором бикарбоната натрия (70 мл). Водный слой промывали с помощью DCM (2×20 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (137 мг, 40%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=432,2 (М+Н).
Стадия 4. Получение 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-(2-(Третбутокси)этокси)-8-((4-этил-2-фтор-фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (137 мг, 0,317 ммоль) растворяли в THF (1 мл)/МеОН (1 мл). Добавляли моногидрат п-толуолсульфоновой кислоты (90,4 мг, 0,475 ммоль). Через 5 минут, добавляли N-йодсукцинимид (71,3 мг, 0,317 ммоль) и перемешивали в течение 30 минут. Реакционную смесь распределяли между EtOAc (20 мл) и насыщенным водным раствором бикарбоната натрия (30 мл), и водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (143 мг, 81%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=558,1 (М+Н).
Стадия 5. Получение 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)-амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (143 мг, 0,256 ммоль) в THF (2,5 мл, 0,256 ммоль) при 0°С добавляли Pd(Pt-Bu3)2 (13,1 мг, 0,026 ммоль), затем метилцинка(II) хлорид (130 мкл, 0,259 ммоль). Смесь удаляли из ледяной бани и перемешивали в течение 45 минут. Смесь распределяли между насыщенным водным раствором NH4Cl (50 мл) и EtOAc (20 мл), и водный слой экстрагировали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гептан, с получением 2-(2-(третбутокси)этокси)-8-((4-этил-2-фторфенил)-амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (53,7 мг, 47%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=446,2 (М+Н).
Стадия 6. Получение 8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-(2-(Третбутокси)этокси)-8-((4-этил-2-фтор-фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (53,7 мг, 0,121 ммоль) растворяли в ацетонитриле (0,75 мл) и добавляли фосфорную кислоту (0,75 мл). Реакционную смесь нагревали до 60°С в течение 60 минут. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (40 мл) и EtOAc (20 мл). Водный слой промывали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (30 мл) и солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (40,6 мг, 87%). 1H ЯМР (400 МГц, CDCl3) δ 11,01 (с, 1Н), 6,95 (дд, 1Н), 6,89 (дд, 1Н), 6,77 (т, 1Н), 4,53 (т, 1Н), 4,02 (т, 2Н), 3,75-3,67 (м, 4Н), 3,20 (с, 3Н), 3,02 (т, 2Н), 2,62 (кварт, 2Н), 2,08 (с, 3Н), 1,22 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=390,2 (М+Н).
Пример 45

8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 44, за исключением того, что применяли 4-циклопропил-2-фторанилин вместо 4-этил-2-фторанилина на стадии 1, с получением 8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (30,7 мг, 75%). 1Н ЯМР (400 МГц, CDCl3) δ 11,00 (с, 1Н), 6,81-6,71 (м, 3Н), 4,52 (т, 1Н), 4,02 (м, 2Н), 3,75-3,66 (м, 4Н), 3,19 (с, 3Н), 3,02 (т, 2Н), 2,07 (с, 3Н), 1,86 (м, 1Н), 1,02-0,95 (м, 2Н), 0,69-0,63 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=402,2 (М+Н).
Пример 46

8-((4-бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетат
Стадия 1. Получение метил (Е)-2-((4-бром-2-фторфенил)-амино)-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. 4-Бром-2-фторанилин (492 мг, 2,59 ммоль) и метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (670 мг, 2,47 ммоль) растворяли в THF (12,3 мл, 0,2 М) и охлаждали до -78°С в атмосфере азота. Добавляли по каплям LiHMDS (4,9 мл, 1,0 M/THF, 4,9 ммоль), затем смесь подогревали от -78°С до температуры окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (15 мл), затем экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением метил (Е)-2-((4-бром-2-фторфенил)амино)-4-(2-этокси-винил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (810 мг, 77%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=425,0 (М+Н).
Стадия 2. Получение метил 2-((4-бром-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-2-((4-бром-2-фторфенил)амино)-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (810 мг, 1,90 ммоль) в DCM (5,0 мл) добавляли TFA (5,0 мл). Раствор перемешивали при температуре окружающей среды в течение 16 часов, затем концентрировали. Неочищенную смесь затем разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением метил 2-((4-бром-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата, который использовали без дополнительной очистки (751 мг, 99%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=397,0, 399,0 (М+Н).
Стадия 3. Получение 8-((4-бром-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил 2-((4-бром-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилат (751 мг, 1,89 ммоль), (4-метоксифенил)метанамин (272 мг, 1,99 ммоль), триацетоксиборгидрид натрия (441 мг, 2,08 ммоль) и уксусную кислоту (11,4 мг, 0,189 ммоль) растворяли в DCE (19 мл) и перемешивали при температуре окружающей среды в течение 1 часа. Реакционную смесь подогревали до 60°С и перемешивали в течение 1 часа. Реакционную смесь затем охлаждали до температуры окружающей среды, разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (15 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((4-бром-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (630 мг, 69%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=486,0 (М+Н).
Стадия 4. Получение 8-((4-бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетата. 8-((4-бром-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (630 мг, 1,30 ммоль) растворяли в TFA (8,0 мл) и нагревали до 80°С в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, лиофилизировали с получением 8-((4-бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона трифторацетата (226 мг, 48%). 1H ЯМР (400 МГц, CDCl3) δ 11,28 (с, 1Н), 7,32-7,29 (м, 1Н), 7,23-7,20 (м, 1Н), 6,74 (т, 1Н), 6,05 (с, 1Н), 5,62 (с, 1Н), 3,51-3,47 (м, 2Н), 3,20 (с, 3Н), 2,84 (т, 2Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=366,0, 368,0 (М+Н).
Пример 47

8-((4-Бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 8-((4-бром-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((4-Бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 46; 41 мг, 0,11 ммоль) и п-толуолсульфоновую кислоту (29 мг, 0,17 ммоль) растворяли в 1 мл раствора 1:1 THF/MeOH. Через 5 минут, добавляли NIS (25 мг, 0,11 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Неочищенную реакционную смесь затем распределяли между EtOAc (10 мл) и насыщенным водным раствором NaHCO3 (15 мл), и экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением 8-((4-бром-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона который использовали без дополнительной очистки (55 мг, 100%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=493,9 (М+Н).
Стадия 2. Получение 8-((4-бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((4-Бром-2-фтор-фенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (55 мг, 0,112 ммоль) и бис(тритретбутил-фосфин)палладий(0) (5,71 мг, 0,0112 ммоль) растворяли в THF (1,1 мл) и добавляли по каплям метилцинка(II) хлорид (55,9 мкл, 2M/THF, 0,112 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NaHCO3 (2 мл), затем экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом жидкостной хроматографии с обращенной фазой, элюируя с использованием 5-95% ацетонитрил/Н20, с получением 8-((4-бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (24 мг, 56%). 1Н ЯМР (400 МГц, CDCl3) δ 11,08 (с, 1Н), 7,30-7,27 (м, 1Н), 7,18-7,15 (м, 1Н), 6,63 (т, 1Н), 5,72 (с, 1Н), 3,50-3,46 (м, 2Н), 3,26 (с, 3Н), 2,88 (т, 2Н), 2,11 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=380,0 (М+Н).
Пример 48

8-((2-Фтор-4-йодфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
8-((4-бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 46; 20 мг, 0,053 ммоль), йодид меди(I) (2,5 мг, 0,013 ммоль), йодид натрия (16 мг, 0,11 ммоль), и (1S,2S)-N1,N2-диметилциклогексан-1,2-диамин (3,7 мг, 0,026 ммоль) объединяли в 1,4-диоксане (0,5 мл). Реакционную смесь нагревали до 120°С в течение 16 часов. Реакционную смесь затем охлаждали до температуры окружающей среды, гасили насыщенным водным раствором NH4Cl (2 мл), затем экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, объединяли, и продукт превращали в форму свободного основания с помощью DCM/NaHCO3 с получением 8-((2-фтор-4-йодфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (5,0 мг, 22%). 1Н ЯМР (400 МГц, CDCl3) δ 11,07 (с, 1Н), 7,46-7,43 (м, 1Н), 7,36-7,33 (м, 1Н), 6,48 (т, 1Н), 5,82 (с, 1Н), 3,50-3,46 (м, 2Н), 3,27 (с, 3Н), 2,88 (т, 2Н), 2,11 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=428,10 (М+Н).
Пример 49

8-((4-Йод-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетат
Стадия 1. Получение метил (Е)-4-(2-этоксивинил)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. 4-Йод-2-фторанилин (266 мг, 1,12 ммоль) и метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (290 мг, 0,920 ммоль) растворяли в THF (9,2 мл) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (2,1 мл, 1,0 M/THF, 2,1 ммоль), затем смесь подогревали от -78°С до температуры окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (10 мл), затем экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением метил (Е)-4-(2-этоксивинил)-2-((2-фтор-4-йодфенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (389 мг, 77%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=473,1 (М+Н).
Стадия 2. Получение метил 2-((4-йод-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-2-((4-йод-2-фторфенил)амино)-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (389 мг, 0,824 ммоль) в DCM (4,0 мл) добавляли TFA (4,0 мл). Раствор перемешивали при температуре окружающей среды в течение 16 часов, затем концентрировали. Неочищенную смесь затем разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением метил 2-((4-йод-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидро-пиридин-3-карбоксилата, который непосредственно использовали на следующей стадии без дополнительной очистки (351 мг, 96%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=445,0 (М+Н).
Стадия 3. Получение 8-((4-йод-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил 2-((4-йод-2-фторфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилат (351 мг, 0,790 ммоль), (4-метоксифенил)метанамин (114 мг, 0,830 ммоль), триацетоксиборгидрид натрия (184 мг, 0,869 ммоль) и уксусную кислоту (4,8 мг, 0,079 ммоль) растворяли в DCE (10 мл) и перемешивали при температуре окружающей среды в течение 1 часа. После этого, реакционную смесь подогревали до 60°С и перемешивали в течение 1 часа. Реакционную смесь затем охлаждали до температуры окружающей среды, разбавляли с помощью DCM (5 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((4-йод-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (321 мг, 76%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=534,1 (М+Н).
Стадия 4. Получение 8-((4-йод-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетата. 8-((4-йод-2-фторфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (321 мг, 0,602 ммоль) растворяли в TFA (6,0 мл) и нагревали до 60°С в течение 16 часов. Реакционную смесь затем охлаждали до температуры окружающей среды и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, лиофилизировали с получением 8-((4-йод-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона трифторацетата (102 мг, 41%). 1H ЯМР (400 МГц, CDCl3) δ 11,27 (с, 1Н), 7,48-7,45 (м, 1Н), 7,40-7,38 (м, 1Н), 6,58 (т, 1Н), 6,05 (с, 1Н), 5,84 (с, 1Н), 3,51-3,47 (м, 2Н), 3,20 (с, 3Н), 2,84 (т, 2Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=414,0 (М+Н).
Пример 50

8-((2-Фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетат
Стадия 1. Получение метил (Е)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (400 мг, 1,47 ммоль) и 2-фтор-4-(метилтио)анилин (243 мг, 1,55 ммоль) растворяли в THF (9,8 мл) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (2,9 мл, 1,0 M/THF, 2,9 ммоль), затем смесь подогревали от -78°С до температуры окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (10 мл), затем экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением метил (Е)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (495 мг, 86%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=393,1 (М+Н).
Стадия 2. Получение метил 2-((2-фтор-4-(метилтио)фенил)-амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)-амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (495 мг, 1,26 ммоль) в DCM (4,0 мл) добавляли TFA (4,0 мл). Раствор перемешивали при температуре окружающей среды в течение ночи, затем концентрировали. Неочищенную смесь затем разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением метил 2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата, который использовали без дополнительной очистки (391 мг, 85%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=365,1 (М+Н).
Стадия 3. Получение 8-((2-фтор-4-(метилтио)фенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил 2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-4-(2-оксо-этил)-1,6-дигидропиридин-3-карбоксилат (391 мг, 1,07 ммоль), (4-метоксифенил)метанамин (155 мг, 1,13 ммоль), триацетоксиборгидрид натрия (250 мг, 1,18 ммоль) и уксусную кислоту (6,4 мг, 0,079 ммоль) растворяли в DCE (11 мл) и перемешивали при температуре окружающей среды в течение 1 часа. После это, реакционную смесь подогревали до 60°С и перемешивали в течение 1 часа. Реакционную смесь затем охлаждали до температуры окружающей среды, разбавляли с помощью DCM (5 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((2-фтор-4-(метил-тио)фенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (401 мг, 82%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=454,1 (М+Н).
Стадия 4. Получение 8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетата. 8-((2-Фтор-4-(метилтио)фенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (401 мг, 0,884 ммоль) растворяли в TFA (8,0 мл) и нагревали до 60°С в течение 16 часов. Реакционную смесь затем охлаждали до температуры окружающей среды и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, объединяли и лиофилизировали с получением 8-((2-фтор-4-(метилтио)фенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона трифторацетата (131 мг, 44%). 1Н ЯМР (400 МГц, CDCl3) δ 11,10 (с, 1Н), 7,04-6,96 (м, 2Н), 6,84 (т, 1Н), 6,49 (с, 1Н), 6,11 (с, 1Н), 3,53-3,49 (м, 2Н), 3,20 (с, 3Н), 2,85 (т, 2Н), 2,48 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=334,1 (М+Н).
Пример 51

8-((4-Этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 50, за исключением того, что применяли 4-этил-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 1, с получением 8-((4-этил-2-фторфенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (14,9 мг, 47%). 1Н ЯМР (400 МГц, CDCl3) δ 11,25 (с, 1Н), 6,95 (м, 1Н), 6,90 (м, 1Н), 6,81 (т, 1Н), 6,06 (с, 1Н), 5,99 (с, 1Н), 3,48 (м, 2Н), 3,17 (с, 3Н), 2,82 (т, 2Н), 2,62 (кварт, 2Н), 1,22 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=316,1 (М+Н).
Пример 52

8-((4-Циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 50, за исключением того, что применяли 4-циклопропил-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 1, с получением 8-((4-циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (5,8 мг, 17%). 1Н ЯМР (400 МГц, CDCl3) δ 11,24 (с, 1Н), 6,83-6,75 (м, 3Н), 5,99 (с, 1Н), 5,94 (с, 1Н), 3,48 (дт, 2Н), 3,16 (с, 3Н), 2,82 (т, 2Н), 1,86 (м, 1Н), 1,02-0,95 (м, 2Н), 0,69-0,62 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=328,1 (М+Н).
Пример 53

8-((4-(Дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 50, за исключением того, что применяли 4-(дифторметокси)-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 1, с получением 8-((4-(дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (30,6 мг, 47%). 1Н ЯМР (400 МГц, CDCl3) δ 11,29 (с, 1Н), 6,97 (м, 1Н), 6,91-6,84 (м, 2Н), 6,49 (т, 1Н), 6,03 (с, 1Н), 5,79 (с, 1Н), 3,49 (дт, 2Н), 3,19 (с, 3Н), 2,84 (т, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=354,1 (М+Н).
Пример 54

8-((2-Фтор-4-пропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадии 1-4. Получение 8-((4-аллил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 50, за исключением того, что применяли 4-аллил-2-фторанилин вместо 2-фтор-4-(метилтио)анилина на стадии 1, с получением 8-((4-аллил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (3,8 мг, 10%). 1Н ЯМР (400 МГц, CDCl3) δ 11,27 (с, 1Н), 7,09 (дд, 1Н), 7,01 (дд, 1Н), 6,80 (т, 1Н), 6,35-6,28 (м, 1Н), 6,25-6,15 (м, 1Н), 6,01 (с, 1Н), 5,85 (с, 1Н), 3,49 (дт, 2Н), 3,19 (с, 3Н), 2,83 (т, 2Н), 1,88 (дд, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=328,1 (М+Н).
Стадия 5. Получение 8-((2-фтор-4-пропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Перемешиваемую смесь 8-((4-аллил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (3,8 мг, 0,012 ммоль) и Pd/C (5% типа Degussa, 1 мг, 0,009 ммоль) в МеОН (0,5 мл) дегазировали и перемешивали в атмосфере Н2 в течение 24 часов. Реакционную смесь разбавляли с помощью DCM, фильтровали, и фильтрат концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/вода/0,1% TFA в течение 20 минут), и очищенные фракции объединяли, промывали насыщенным водным раствором бикарбоната натрия, и экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (30 мл) и солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением 8-((2-фтор-4-пропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (2,5 мг, 65%). 1Н ЯМР (400 МГц, CDCl3) δ 11,22 (с, 1Н), 6,93 (дд, 1Н), 6,88 (м, 1Н), 6,81 (т, 1Н), 5,99 (с, 1Н), 5,81 (с, 1Н), 3,48 (дт, 2Н), 3,17 (с, 3Н), 2,83 (т, 2Н), 2,55 (т, 2Н), 1,62 (м, 2Н), 0,92 (т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=330,1 (М+Н).
Пример 55
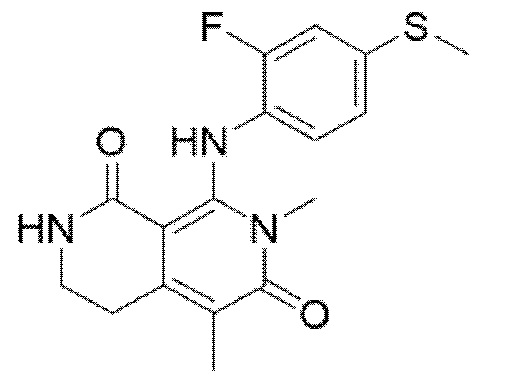
8-((2-Фтор-4-(метилтио)фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. 8-((2-фтор-4-(метилтио)фенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((2-Фтор-4-(метил-тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (41,2 мг, 0,124 ммоль) и п-толуолсульфоновую кислоту (31,9 мг, 0,185 ммоль) растворяли in 1 мл раствора 1:1 THF/MeOH. Через 5 минут, добавляли NIS (27,8 мг, 0,124 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Неочищенную реакционную смесь затем распределяли между EtOAc (10 мл) и насыщенным водным раствором NaHCO3 (15 мл), и экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 10-80% EtOAc/гептан, с получением 8-((4-бром-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (37 мг, 65%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=460,0 (М+Н).
Стадия 2. Получение 8-((2-фтор-4-(метилтио)фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((2-фтор-4-(метилтио)фенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (37,0 мг, 0,0806 ммоль) и бис(тритретбутилфосфин)-палладий(0) (4,12 мг, 0,00806 ммоль) растворяли в THF (0,8 мл) и добавляли по каплям метилцинка(II) хлорид (80,6 мкл, 2M/THF, 0,161 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (2 мл), затем экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, объединяли, и продукт превращали в форму свободного основания с помощью DCM/NaHCO3 с получением 8-((2-фтор-4-(метилтио)фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (15,5 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 11,10 (с, 1Н), 7,04-7,01 (м, 1Н), 6,95-6,93 (м, 1Н), 6,71 (т, 1Н), 5,96 (с, 1Н), 3,49-3,46 (м, 2Н), 3,25 (с, 3Н), 2,87 (т, 2Н), 2,46 (с, 3Н), 2,11 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=348,1 (М+Н).
Пример 56

8-((4-этил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 55, за исключением того, что применяли 8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 51) вместо 8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона на стадии 1, с получением 8-((4-этил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (6,7 мг, 62%). 1Н ЯМР (400 МГц, CDCl3) δ 11,06 (с, 1Н), 6,94 (дд, 1Н), 6,86 (дд, 1Н), 6,72 (т, 1Н), 5,87 (с, 1Н), 3,47 (дт, 2Н), 3,23 (с, 3Н), 2,87 (т, 2Н), 2,61 (кварт, 2Н), 2,11 (с, 3Н), 1,21(т, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=330,2 (М+Н).
Пример 57

8-((4-циклопропил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 55, за исключением того, что применяли 8-((4-циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 52) вместо 8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона на стадии 1, с получением 8-((4-циклопропил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (2,3 мг, 47%). 1Н ЯМР (400 МГц, CDCl3) δ 11,05 (с, 1Н), 6,80-6,75 (м, 2Н), 6,69 (т, 1Н), 5,80 (с, 1Н), 3,47 (дт, 2Н), 3,22 (с, 3Н), 2,87 (т, 2Н), 2,10 (с, 3Н), 1,85 (м, 1Н), 1,00-0,94 (м, 2Н), 0,67-0,61 (м, 2Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=342,2 (М+Н).
Пример 58
8-((4-(Дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 55, за исключением того, что применяли 8-((4-(дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (получали таким же методом, как в примере 53) вместо 8-((2-фтор-4-(метилтио)фенил)-амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона на стадии 1, с получением 8-((4-(дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (9,5 мг, 41%). 1Н ЯМР (400 МГц, CDCl3) δ 11,09 (с, 1Н), 6,96 (дд, 1Н), 6,87-6,82 (м, 1Н), 6,76 (т, 1Н), 6,47 (т, 1Н), 6,01 (с, 1Н), 3,48 (дт, 2Н), 3,25 (с, 3Н), 2,88 (т, 2Н), 2,11 (с, 3Н) ppm; MS (химическая ионизация при атмосферном давлении (apci), m/z)=368,1 (М+Н).
Пример 59

8-((4-Бром-2-хлорфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил (Е)-2-((4-бром-2-хлорфенил)амино)-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоксилат (140 мг, 0,515 ммоль) и 4-бром-2-хлоранилин (108 мг, 0,520 ммоль) растворяли в THF (5,2 мл) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (1,0 мл, 1,0 M/THF, 1,0 ммоль), затем смесь подогревали от -78°С до температуры окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (5 мл), затем экстрагировали с помощью EtOAc (2×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением метил (Е)-2-((4-бром-2-хлорфенил)амино)-4-(2-этокси-винил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (145 мг, 64%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=441,0, 443,0 (М+Н).
Стадия 2. Получение метил 2-((4-бром-2-хлорфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-2-((4-бром-2-хлорфенил)амино)-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (145 мг, 0,328 ммоль) в DCM (2,0 мл) добавляли TFA (2,0 мл). Раствор перемешивали при температуре окружающей среды в течение ночи, затем концентрировали. Неочищенную смесь затем разбавляли с помощью DCM (5 мл) и промывали насыщенным водным раствором NaHCO3 (5 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением метил 2-((4-бром-2-хлорфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидро-пиридин-3-карбоксилата, который использовали без дополнительной очистки (136 мг, 100%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=415,2 (М+Н).
Стадия 3. Получение 8-((4-бром-2-хлорфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Метил 2-((4-бром-2-хлорфенил)амино)-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилат (136 мг, 0,329 ммоль), (4-метокси-фенил)метанамин (47,4 мг, 0,345 ммоль), триацетоксиборгидрид натрия (76,6 мг, 0,362 ммоль) и уксусную кислоту (2,0 мг, 0,033 ммоль) растворяли в DCE (3,3 мл) и перемешивали при температуре окружающей среды в течение 1 часа. После этого, реакционную смесь подогревали до 60°С и перемешивали в течение 1 часа. Реакционную смесь затем охлаждали до температуры окружающей среды, разбавляли с помощью DCM (5 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((4-бром-2-хлорфенил)амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (104 мг, 63%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=502,l, 504,1 (М+Н).
Стадия 4. Получение 8-((4-бром-2-хлорфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 8-((4-Бром-2-хлорфенил)-амино)-2-(4-метоксибензил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (79,1 мг, 0,207 ммоль) растворяли в TFA (3,0 мл) и нагревали до 60°С в течение 16 часов. Реакционную смесь затем охлаждали до температуры окружающей среды и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, объединяли, и продукт превращали в форму свободного основания с помощью DCM/NaHCO3 с получением 8-((4-бром-2-хлорфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (28 мг, 35%). 1Н ЯМР (400 МГц, CDCl3) δ 11,27 (с, 1Н), 7,61 (д, 1Н), 7,31-7,28 (м, 1Н), 6,60-6,57 (м, 1Н), 6,07 (т, 1Н), 5,63 (с, 1Н), 3,52-3,48 (м, 2Н), 3,17 (с, 3Н), 2,85 (т, 2Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=382,0, 384,0 (М+Н).
Пример 60

8-((4-Бром-2-фторфенил)амино)-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион трифторацетат
Стадия 1. Получение метил 2-хлор-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (331 мг, 1,22 ммоль) в DCM (5,0 мл) добавляли TFA (5,0 мл). Раствор перемешивали при температуре окружающей среды в течение 16 часов, затем концентрировали. Неочищенную смесь затем разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (10 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением метил 2-хлор-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидро-пиридин-3-карбоксилата, который использовали без дополнительной очистки (236 мг, 80%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=244,1 (М+Н).
Стадия 2. Получение 8-хлор-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион. Метил 2-хлор-1-метил-6-оксо-4-(2-оксоэтил)-1,6-дигидропиридин-3-карбоксилат (122 мг, 0,501 ммоль), 2-((третбутилдиметилсилил)окси)этан-1-амин (105 мг, 0,601 ммоль), триацетоксиборгидрид натрия (127 мг, 0,601 ммоль) и уксусную кислоту (6,0 мг, 0,10 ммоль) растворяли в DCE (5 мл) и перемешивали при температуре окружающей среды в течение 2 часов. Реакционную смесь подогревали до 60°С и перемешивали в течение 16 часов. Реакционную смесь затем охлаждали до температуры окружающей среды, разбавляли с помощью DCM (10 мл) и промывали насыщенным водным раствором NaHCO3 (15 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Фракции, содержащие очищенный требуемый продукт, объединяли, и продукт превращали в форму свободного основания с помощью DCM/NaHCO3 с получением 8-хлор-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (20 мг, 16%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=257,0 (М+Н).
Стадия 3. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона трифторацетата. 4-Бром-2-фторанилин (17,7 мг, 0,093 ммоль) и 8-хлор-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-дион (20 мг, 0,077 ммоль) растворяли в THF (0,7 мл) и охлаждали до -78°С в атмосфере N2. Добавляли по каплям LiHMDS (233 мкл, 1,0 M/THF, 0,233 ммоль), затем смесь подогревали от -78°С до температуры окружающей среды в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (5 мл), затем экстрагировали с помощью EtOAc (3×5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-95% ацетонитрил/H2O/0,1% TFA). Очищенные фракции лиофилизировали с получением 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона трифторацетата (10,1 мг, 32%). 1Н ЯМР (400 МГц, CDCl3) δ 11,36 (с, 1Н), 7,32-7,29 (м, 1Н), 7,22-7,19 (м, 1Н), 6,70 (т, 1Н), 6,06 (с, 1Н), 3,85 (т, 2Н), 3,67 (т, 2Н), 3,61 (т, 2Н), 3,22 (с, 3Н), 2,86 (т, 2Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=410,1, 412,1 (М+Н).
Пример 61

8-((2-Фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 44, за исключением того, что применяли 2-фтор-4-(метилтио)анилин вместо 4-этил-2-фторанилина на стадии 1, с получением 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (2,1 мг, 21%). 1Н ЯМР (400 МГц, CDCl3) δ 11,01 (с, 1Н), 7,02 (дд, 1Н), 6,95 (м, 1Н), 6,77 (м, 1), 4,48 (т, 1Н), 4,03 (т, 2Н), 3,76-3,67 (м, 4Н), 3,21 (с, 3Н), 3,02 (т, 2Н), 2,47 (с, 3Н), 2,08 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=408,1 (М+Н).
Пример 62

8-((4-(Дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадии 1-5. Получение 2-(2-(третбутокси)этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Получали таким же методом, как в примере 41, стадии 1-5, за исключением того, что применяли 4-(дифторметокси)-2-фторанилин вместо 2-фторанилина на стадии 1, с получением 2-(2-(третбутокси)этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (156 мг, 94%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=596,1 (М+Н).
Стадия 6. Получение 2-(2-(третбутокси)этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 2-(2-(третбутокси)-этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (156 мг, 0,262 ммоль) в THF (2,5 мл) при 0°С добавляли Pd(Pt-Bu3)2 (13,4 мг, 0,026 ммоль), затем метилцинка(II) хлорид (132 мкл, 0,265 ммоль). Смесь удаляли из ледяной бани и перемешивали в течение 10 минут. Реакционную смесь распределяли между насыщенным водным раствором NH4Cl (40 мл) и EtOAc (20 мл), и водный слой экстрагировали с помощью EtOAc (2×10 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/гептан, с получением 2-(2-(третбутокси)этокси)-8-((4-(дифтор-метокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (89,9 мг, 71%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=484,2 (М+Н).
Стадия 7. Получение 8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. 2-(2-(третбутокси)этокси)-8-((4-(дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион (89,9 мг, 0,186 ммоль) растворяли в ACN (750 мкл) и добавляли фосфорную кислоту (750 мкл). Реакционную смесь нагревали до 60°С в течение 30 минут. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (50 мл) и EtOAc (20 мл). Водный слой промывали с помощью EtOAc (2×15 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (25 мл), солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-100% EtOAc/гептан, с получением 8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидрокси-этокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6 (2Н,7Н)-диона (65,9 мг, 83%). 1Н ЯМР (400 МГц, CDCl3) δ 11,00 (с, 1Н), 6,97 (дд, 1Н), 6,90-6,79 (м, 2Н), 6,48 (т, 1Н), 4,44 (т, 1Н), 4,03 (м, 2Н), 3,76-3,68 (м, 4Н), 3,22 (с, 3Н), 3,03 (т, 2Н), 2,09 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=428,1 (М+Н).
Пример 63

2-(2,2-Дифторэтокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение метил (Z)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Получали таким же методом, как в примере 44, на стадии 1, за исключением того, что применяли 2-фтор-4-(метилтио)анилин вместо 4-этил-2-фторанилина с получением метил (Z)-4-(2-этокси-винил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,373 г, 69%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=393,1 (М+Н).
Стадия 2. Получение метил (Е)-4-(2-((2,2-дифторэтокси)-имино)этил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. Получали таким же методом, как в примере 6, на стадии 5, за исключением того, что применяли метил (Z)-4-(2-этоксивинил)-2-((2-фтор-4-(метилтио)-фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат вместо метил (Z)-2-хлор-4-(2-этоксивинил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата и О-(2,2-дифторэтил)гидроксиламина гидрохлорид вместо О-(2-(трет-бутокси)этил)гидроксиламина гидрохлорида с получением метил (Е)-4-(2-((2,2-дифторэтокси)-имино)этил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (по расчетам 100%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=444,1 (М+Н).
Стадия 3. Получение 2-(2,2-дифторэтокси)-8-((2-фтор-4-(метил-тио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору метил (Е)-4-(2-((2,2-дифтор-этоксиимино)этил)-2-((2-фтор-4-(метилтио)фенил)амино)-1-метил-6-оксо-1,6-дигидро-пиридин-3-карбоксилата (36 мг, 0,081 ммоль) в МеОН (0,8 мл) добавляли цианоборгидрид натрия (26 мг, 0,406 ммоль) и уксусную кислоту (23 мкл, 0,406 ммоль). Смесь перемешивали при 45°С в течение 2 часов, затем при температуре окружающей среды в течение 72 часов. Смесь разбавляли насыщенным водным раствором NaHCO3 (25 мл) и EtOAc (25 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 5-100% EtOAc/гептан, с получением 2-(2,2-дифторэтокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (11,8 мг, 35,2%). 1H ЯМР (400 МГц, CDCl3) δ 11,26 (с, 1Н), 7,08-6,94 (м, 2Н), 6,91-6,76 (м, 1Н), 6,51-5,64 (м, 2Н), 4,22 (td, 2Н), 3,73 (т, 2Н), 3,16 (с, 3Н), 3,04-2,97 (м, 2Н), 2,48 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=414,1 (М+Н).
Пример 64

8-((4-Бром-2-фторфенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 2,6-дихлор-4-йодникотиновой кислоты. Раствор 2,6-дихлор-4-йодпиридина (4,0 г, 14,6 ммоль) в THF (30 мл) охлаждали до -78°С. К раствору добавляли по каплям LDA (10,95 мл, 2 М, 21,9 ммоль) при -78°С и перемешивали 2 часа при -78°С. Через раствор реакционной смеси барботировали избыток CO2 в течение 20 минут при -78°С. Реакционную смесь затем выливали на сухой лед (10 г) и гасили водой (30 мл). Корректировали величину рН реакционной смеси до рН 3-4, используя 2N HCl, и экстрагировали с помощью EtOAc (3×100 мл). Органические слои объединяли и сушили над сульфатом натрия, фильтровали и концентрировали с получением 2,6-дихлор-4-йодникотиновой кислоты (3,0 г, 64,6%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=317,9, 319,8 (М+Н).
Стадия 2. Получение 2-хлор-4-йод-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты. Раствор NaOH (100 мл, 4 М, 236 ммоль) нагревали до 110°С и затем добавляли одной порцией 2,6-дихлор-4-йодникотиновую кислоту (3,0 г, 9,44 ммоль) и перемешивали в течение 8 часов. Корректировали величину рН реакционной смеси до рН 1 с помощью HCl (б М) при 0°С и перемешивали в течение 30 минут. Полученные твердые вещества собирали фильтрацией и сушили под вакуумом с получением 2-хлор-4-йод-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (3,0 г, assumed 100%), которую использовали без дополнительной очистки. MS (химическая ионизация при атмосферном давлении (apci), m/z)=299,9, 301,9 (М+Н).
Стадия 3. Получение метил 2-хлор-4-йод-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К смеси 2-хлор-4-йод-6-оксо-1,6-дигидропиридин-3-карбоновой кислоты (3,0 г, 10,02 ммоль) в DMF (30 мл) добавляли одной порцией MeI (4,27 г, 30,06 ммоль) и K2CO3 (4,15 г, 30,06 ммоль) при 25°С. После перемешивания в течение 3 часов, реакционную смесь выливали в насыщенный водный раствор NH4Cl (10 мл), и водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 2-100% EtOAc/петролейный эфир, с получением метил 2-хлор-4-йод-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (850 мг, 25,9%). 1Н ЯМР (400 МГц, CDCl3) δ 7,16 (с, 1Н), 3,93 (с, 3Н), 3,65 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=327,9 (М+Н).
Стадия 4. Получение метил 2-хлор-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата. К раствору метил 2-хлор-4-йод-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (0,2 г, 0,611 ммоль) в 1,4-диоксане (10 мл) и воде (1 мл) добавляли 4,4,5,5-тетраметил-2-(1-фенилвинил)-1,3,2-диокса-боролан (0,148 г, 0,641 ммоль), Pd(dppf)Cl2 (50 мг, 0,061 ммоль), Na2CO3 (0,194 г, 1,83 ммоль) и H2O (1 мл). Затем смесь дегазировали 3 раза и перемешивали при 80°С в течение 3 часов. Добавляли 4,4,5,5-тетраметил-2-(1-фенилвинил)-1,3,2-диокса-боролан (37 мг, 0,153 ммоль) и Pd(dppf)Cl2 (50 мг, 0,061 ммоль). Смесь дегазировали 3 раза и перемешивали при 80°С в течение еще 1,5 часов. Охлажденную смесь фильтровали, и фильтрат концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-20% EtOAc/петролейный эфир, с получением метил 2-хлор-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата (95 мг, 51%). 1H ЯМР (400 МГц, CDCl3) δ 7,33-7,32 (м, 3Н), 7,32-7,25 (м, 2Н), 6,58 (с, 1Н), 5,62 (с, 1Н), 5,42 (с, 1Н), 3,75 (с, 3Н), 3,36 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=303,7 (М+Н).
Стадия 5. Получение метил 2-((4-бром-2-фторфенил)амино)-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата. К раствору 4-бром-2-фторанилина (56 мг, 0,29 ммоль) в THF (5 мл) добавляли LiHMDS (0,74 мл, 1,0 М, 0,74 ммоль) при -78°С и перемешивали в течение 0,5 часа при -78°С. Затем добавляли раствор метил 2-хлор-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата (90 мг, 0,296 ммоль) в THF (0,5 мл), и смесь перемешивали при -78°С в течение 30 минут. Смесь гасили насыщенным водным раствором NH4Cl (5 мл) и экстрагировали с помощью EtOAc (3×50 мл). Объединенные органические слои промывали солевым раствором (5 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-20% EtOAc/петролейный эфир, с получением метил 2-((4-бром-2-фторфенил)амино)-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата (120 мг, 89%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=459,2 (М+Н).
Стадия 6. Получение 8-((4-бром-2-фторфенил)амино)-2-(2,4-диметоксибензил)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору метил 2-((4-бром-2-фторфенил)-амино)-1-метил-6-оксо-4-(1-фенилвинил)-1,6-дигидропиридин-3-карбоксилата (120 мг, 0,29 ммоль) в толуоле (10 мл) добавляли DMB-NH2 (43,8 мг, 0,26 ммоль) и А1Ме3 (0,4 мл, 2,0 М, 0,79 ммоль) при 25°С в атмосфере N2, и смесь перемешивали при 80°С в течение 48 часов. Смесь гасили насыщенным водным раствором NH4Cl (5 мл) и фильтровали. Фильтрат экстрагировали с помощью EtOAc (3×10 мл), и органический слой промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-50% EtOAc/петролейный эфир, с получением 8-((4-бром-2-фтор-фенил)амино)-2-(2,4-диметоксибензил)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (80,0 мг, 52%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=594,2 (М+Н).
Стадия 7. 8-((4-Бром-2-фторфенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион. Смесь 8-((4-бром-2-фтор-фенил)амино)-2-(2,4-диметоксибензил)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (80 мг, 0,14 ммоль) в TFA (5 мл) перемешивали в течение 1 часа при 80°С. Охлажденную смесь концентрировали, и остаток растворяли в МеОН (5 мл). Добавляли твердый NaHCO3 (~1 г), и смесь перемешивали в течение 15 минут, затем обрабатывали с помощью DCM (20 мл) и фильтровали. Фильтрат концентрировали и очищали методом препаративной HPLC (условия проведения препаративной HPLC: колонка: Boston Prime С18 150×25 мм × 5 мкм; подвижная фаза: вода (0,225% гидроксида аммония по объему)-ACN; В%: 40%-70%, расход (мл/мин): 25). Фракции лиофилизировали с получением 8-((4-бром-2-фторфенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (5,49 мг, 9%). 1Н ЯМР (400 МГц, MeOD) δ 7,49 (д, 1Н), 7,40-7,32 (м, 6Н), 6,93 (т, 1Н), 5,76 (с, 1Н), 4,19 (т, 1Н), 3,66 (д, 2Н), 3,24 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=442,2, 444,1 (М+Н).
Пример 65
8-((2-Фтор-4-(метилтио)фенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 64, за исключением того, что применяли 2-фтор-4-(метилтио)анилин вместо 4-бром-2-фторанилина на стадии 5, с получением 8-((2-фтор-4-(метилтио)-фенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (21 мг, 29%). 1Н ЯМР (400 МГц, d6-DMSO) δ 11,71 (с, 1Н), 8,15 (с, 1Н), 7,40-7,25 (м, 6Н), 7,08 (д, 1Н), 6,97 (т, 1Н), 5,59 (с, 1Н), 4,13 (т, 1Н), 3,54 (уш. с, 2Н), 3,30 (с, 3Н), 3,05 (с, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=410,1 (М+Н).
Пример 66

8-((4-Бром-2-фторфенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 64, за исключением того, что применяли 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолан вместо 4,4,5,5-тетраметил-2-(1-фенилвинил)-1,3,2-диоксаборолана на стадии 4, с получением 8-((4-бром-2-фторфенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (3 мг, 13%). 1Н ЯМР (400 МГц, MeOD) δ 7,48 (д, 1Н), 7,35 (д, 1Н), 6,88 (т, 1Н), 6,15 (с, 1Н), 3,51 (дд, 1Н), 3,21 (с, 3Н), 3,20-3,15 (м, 1Н), 2,99-2,86 (м, 1Н), 1,33 (д, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=380,0, 382,2 (М+Н).
Пример 67

8-((2-Фтор-4-(метилтио)фенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 64, за исключением того, что применяли 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксаборолан вместо 4,4,5,5-тетраметил-2-(1-фенилвинил)-1,3,2-диоксаборолана на стадии 4, и 2-фтор-4-(метилтио)анилин вместо 4-бром-2-фторанилина на стадии 5, с получением 8-((2-фтор-4-(метилтио)фенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (3,8 мг, 22%). 1Н ЯМР (400 МГц, MeOD) δ 7,16 (д, 1Н), 7,09 (д, 1Н), 6,92 (т, 1Н), 6,09 (с, 1Н), 3,51 (дд, 1Н), 3,19 (с, 3Н), 3,17-3,15 (м, 1Н), 2,99-2,94 (м, 1Н), 2,51 (с, 3Н), 1,33 (д, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=348,1 (М+Н).
Пример 68

8-((4-Бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Стадия 1. Получение 1-этоксипроп-1-ина. Безводный NH3 (300 мл) конденсировали при -70°С в реакционной колбе и добавляли при медленном перемешивании измельченный Fe(NO3)3⋅9H2O (252 мг, 0,625 ммоль). Добавляли твердый NaNH2 (34,14 г, 0,875 моль) при температуре от -60 до -40°С в течение 10 минут, затем добавляли по каплям 2-хлор-1,1-диэтоксиэтан (38,15 г, 0,25 моль) при этой температуре в течение 30 минут. Реакционную смесь кипятили с обратным холодильником при -30°С в течение 1 часа, затем добавляли по каплям MeI (177,42 г, 1,25 моль) в течение 30 минут. Реакционную смесь интенсивно перемешивали в течение 90 минут при этой температуре, затем осторожно гасили путем добавления по каплям охлажденного водного насыщенного раствора NH4Cl (40 мл), затем Et2O (100 мл) и дополнительного количества водного насыщенного раствора NH4Cl (50 мл). Охлаждающую баню удаляли, и реакционную смесь подогревали до 15°С. Добавляли воду(50 мл), и органический слой отделяли и промывали водой (50 мл) и солевым раствором (50 мл). Объединенные водные растворы экстрагировали с помощью Et2O (50 мл), и объединенные органические слои сушили над Na2SO4 и фильтровали с получением ~1,3 М эфирного раствора 1-этоксипроп-1-ина (100 мл, 52%). 1H ЯМР (400 МГц, CDCl3) δ 3,99 (кварт, 2Н), 1,73 (с, 3Н), 1,32 (т, 3Н) ppm.
Стадия 2. Получение (Z)-2-(1-этоксипроп-1-ен-2-ил)бензо[d]-[1,3,2]диоксаборолана. К раствору бензо[d][1,3,2]-диоксаборолана (1,43 г, 11,89 ммоль) в толуоле (50 мл) добавляли 1-этоксипроп-1-ин (7,7 мл, 1,3 M/Et2O, 11,89 ммоль) и NiCl2(dppe) (314 мг, 0,59 ммоль) в атмосфере N2. Реакционную смесь дегазировали три раза и перемешивали при 50°С в течение 3 часов. Смесь фильтровали с получением а толуольного раствора (Z)-2-(1-этоксипроп-1-ен-2-ил)бензо[d][1,3,2]диоксаборолана (1,5 г, 62%), который использовали без дополнительной очистки.
Стадия 3. Получение метил (Е)-2-хлор-4-(1-этоксипроп-1-ен-2-ил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору (Z)-2-(1-этоксипроп-1-ен-2-ил)бензо[d][1,3,2]диокса-боролана (1,5 г, 7,33 ммоль) в толуоле (50 мл) и THF (30 мл) добавляли метил 2-хлор-4-йод-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (1,2 г, 3,66 ммоль), Pd(dppf)Cl2 (299 мг, 0,37 ммоль), K3PO4 (2,33 г, 10,99 ммоль) и H2O (3 мл). Смесь дегазировали 3 раза и перемешивали при 75°С в течение 3 часов. Охлажденную смесь фильтровали. Фильтрат разбавляли с помощью EtOAc (50 мл), затем промывали водой (3×10 мл) и солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/петролейный эфир, с получением метил (Е)-2-хлор-4-(1-этоксипроп-1-ен-2-ил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (650 мг, 62%). MS (химическая ионизация при атмосферном давлении (apci), m/z)=286,О (М+Н).
Стадия 4. Получение метил (E/Z)-4-(1-((2-(третбутокси)-этокси)имино)пропан-2-ил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата. К раствору метил (Е)-2-хлор-4-(1-этоксипроп-1-ен-2-ил)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (600 мг, 2,1 ммоль) и О-(2-(третбутокси)этил)гидроксиламина гидрохлорида (356 мг, 2,1 ммоль) в диоксане (10 мл) добавляли TEA (212 мг, 2,1 ммоль) и HCl (1 мл, 4М/диоксан, 4,2 ммоль) Смесь нагревали до 60°С и перемешивали в течение 2 часов. Добавляли 1,4-диоксан (20 мл), и смесь фильтровали и концентрировали с получением метил (E/Z)-4-(1-((2-(третбутокси)этокси)имино)пропан-2-ил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (780 мг, 99,6%), который использовали без дополнительной очистки. MS (химическая ионизация при атмосферном давлении (apci), m/z)=317,1 (М+Н - t-Bu).
Стадия 5. Получение 2-(2-(третбутокси)этокси)-8-хлор-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору метил (E/Z)-4-(1-((2-(третбутокси)этокси)имино)пропан-2-ил)-2-хлор-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата (780 мг, 2,09 ммоль) в IPA (10 мл) добавляли NaCNBH3 (657 мг, 10,46 ммоль) и АсОН (628 мг, 10,46 ммоль). Смесь перемешивали при 20°С в течение 48 часов. Добавляли EtOAc (100 мл), и смесь промывали насыщенным водным раствором NaHCO3 (10 мл) и солевым раствором (10 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией, элюируя с использованием 0-40% EtOAc/петролейный эфир, с получением 2-(2-(третбутокси)-этокси)-8-хлор-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (200 мг, 28%). 1Н ЯМР (400 МГц, CDCl3) δ 6,31 (с, 1Н), 4,10-4,06 (м, 2Н), 3,84 (дд, 1Н), 3,70 (с, 3Н), 3,59-3,53 (м, 3Н), 3,06-3,05 (м, 1Н), 1,31 (д, 3Н), 1,15 (с, 9Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=343,1 (М+Н).
Стадия 6. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. К раствору 4-бром-2-фторанилина (55 мг, 0,29 ммоль) в THF (8 мл) добавляли по каплям LiHMDS (0,73 мл, 1 М, 0,73 ммоль) при -78°С в атмосфере N2. Смесь перемешивали при -78°С в течение 30 минут, затем добавляли по каплям раствор 2-(2-(третбутокси)этокси)-8-хлор-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (100 мг, 0,29 ммоль) в THF (2 мл) и продолжали перемешивание при -78°С в течение 30 минут. Смесь обрабатывали насыщенным водным раствором NH4Cl (5 мл) и экстрагировали с помощью EtOAc (2×30 мл). Объединенные органические слои промывали солевым раствором (5 мл), сушили над MgSO4, фильтровали и концентрировали с получением 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)этокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (144,8 мг, 100%), который использовали без дополнительной очистки. MS (химическая ионизация при атмосферном давлении (apci), m/z)=496,2 (М+Н).
Стадия 7. Получение 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Смесь 8-((4-бром-2-фторфенил)амино)-2-(2-(третбутокси)-этокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (144,8 мг, 0,292 ммоль) в TFA (5 мл) перемешивали в течение 2 часов при 25°С. Смесь концентрировали, растворяли в МеОН (10 мл) и обрабатывали твердым NaHCO3 (73 мг, 0,876 ммоль). После перемешивания в течение 30 минут, добавляли DCM (50 мл). Смесь фильтровали, концентрировали и очищали методом препаративной HPLC (условия проведения препаративной HPLC: колонка: Waters XBridge 150×25 мм 10 мкм; подвижная фаза: вода (0,05% гидроксида аммония по объему)-ACN; B%: 24%-64%, расход (мл/мин): 25). Очищенные фракции лиофилизировали с получением 8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (34,8 мг, 27%). 1Н ЯМР (400 МГц, MeOD) δ 7,38 (д, 1Н), 7,26 (д, 1Н), 6,82 (т, 1Н), 6,02 (с, 1Н), 3,98 (т, 2Н), 3,82 (дд, 1Н), 3,66 (т, 2Н), 3,64-3,50 (м, 1Н), 3,12 (с, 3Н), 3,09-3,08 (м, 1Н), 1,29 (д, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=440,1 (М+Н).
Пример 69

8-((2-Фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион
Получали таким же методом, как в примере 68, за исключением того, что применяли 2-фтор-4-(метилтио)анилин вместо 4-бром-2-фторанилина на стадии 6, с получением 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона (27,5 мг, 23%). 1Н ЯМР (400 МГц, MeOD) δ 7,16 (д, 1Н), 7,10 (д, 1Н), 6,99 (т, 1Н), 6,07 (с, 1Н), 4,05 (т, 2Н), 3,91 (дд, 1Н), 3,76 (т, 2Н), 3,60 (дд, 1Н), 3,21 (с, 3Н), 3,19-3,15 (м, 1Н), 2,50 (с, 3Н), 1,40 (д, 3Н) ppm. MS (химическая ионизация при атмосферном давлении (apci), m/z)=408,2 (М+Н).
Пример 70
Получение безводного кристаллического 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 1
Фосфорную кислоту (37,4 г, 25,8 мл, 14,8 молярный раствор, 50 экв, 382 ммоль) добавляли к перемешиваемому раствору 2-(2-(третбутокси)этокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, который может быть получен любым из описанных в изобретении методов (3,43 г, 1 экв, 7,63 ммоль), в ацетонитриле (ACN) на воздухе. Смесь подогревали до 60°С и перемешивали в течение 15 минут, после чего анализ методом LCMS указывал на завершение реакции. Реакционную смесь охлаждали до 0°С и обрабатывали с помощью K3PO4 (382 мл, 1 М, 382 ммоль). Смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом с получением ярко-желтой пены. Неочищенный остаток очищали на картридже, содержащем 80 г силикагеля, элюируя с использованием градиента от 0% до 15% метанола в дихлорметане (DCM) на первом этапе, затем повышали концентрацию метанола в DCM до 20% с получением продукта в виде ярко-желтой пены (2,17 г). Этот продукт растворяли в DCM (50 мл), обрабатывали активированным углем Norit СА1 (600 мг) и перемешивали в течение 15 минут. Смесь фильтровали через фильтровальную бумагу GF, и остаток концентрировали с получением ярко-желтой пены. Эту процедуру с использованием активированного угля Darco G-60 повторяли и получали продукт в виде бледно-розовой пены. Этот материал обрабатывали 2-пропанолом (20 мл) и добавляли DCM (5 мл) для полной солюбилизации. Смесь концентрировали, во время чего начинали образовываться твердые вещества, но материал становился маслом после полного концентрирования. Остаток обрабатывали 2-пропаном (10 мл) и подогревали до 40°С, после чего образовывался густой осадок. Смесь охлаждали до комнатной температуры и разбавляли колодным 2-пропанолом (5 мл) для облегчения перемешивания. Смесь затем фильтровали, и использовали 15 мл холодного 2-пропанола для ее споласкивания и промывки с получением кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 1 (1,61 г, 4,09 ммоль, 53,6%) в виде желтовато-белого твердого вещества после сушки под вакуумом. На фигуре 1 приведена порошковая рентгенограмма кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 1, полученного этим методом, где анализ методом порошковой рентгеновской дифракции (PXRD) проводили с использованием инструментальных методов, описанных в примере 77.
Пример 71
Получение затравочных кристаллов кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2
Стадия 1. Метил 4-бром-2-((2-фтор-4-(метилтио)фенил)-амино)-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (полученный любым из описанных в изобретении методов) (1,0 экв, 7,5 кг), XPhos (0,075 экв) и MeThF (10 объемов) добавляли в реактор, и реакционную смесь дегазировали. Загружали в реактор Pd (II) ацетат (0,015 экв), и реакционную смесь дегазировали. Загружали в реактор (Z)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,8 экв) и заливали 45 масс. % раствор KOH (4 экв). Реакционную смесь нагревали при 50°С в течение 1 часа, затем добавляли (Z)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,3 экв), и продолжали нагревание в течение еще одного часа. Реакционную смесь промывали водой (2 объема), затем 1М раствором уксусной кислоты (2 экв), затем перемешивали с тиолом SiliaMetS® (SiliCycle Inc., Quebec City, Quebec, Canada) (0,3 г/г) при 50°C в течение 18 часов перед фильтрованием. Осадок на фильтре промывали с помощью MeTHF (5 объемов). Органический слой концентрировали до 3 объемов и небольшую часть удаляли и охлаждали для образования затравочных кристаллов. Затравочные кристаллы загружали обратно в реактор и перемешивали в течение 1 часа при 50°С, затем медленно охлаждали до 30°С. Затем медленно загружали в реактор MeTHF/гептан 1:4 (5 объемов), и смесь перемешивали в течение 1 часа. Смесь затем медленно охлаждали до 10°С и перемешивали в течение 18 часов, после чего фильтровали. Осадок на фильтре промывали с помощью MeTHF/гептан 1:1 (5 объемов). Твердые вещества затем сушили в сушильном шкафу с получением метил 4-[(Z)-2-этоксиэтенил]-2-[2-фтор-4-(метилсульфанил)анилино]-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилата
Стадия 2. Метил 4-[(Z)-2-этоксиэтенил]-2-[2-фтор-4-(метилсульфанил)анилино]-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат (5,0 г, 12,5 ммоль) объединяли с 2-метилтетрагидро-фураном (20 объемов) и О-(2-(третбутокси)этил)гидроксиламина гидрохлоридом (1,3 экв, 16,3 ммоль) и нагревали до приблизительно 75°С. Полученную смесь охлаждали до 20°С. Добавляли комплекс боран-пиридин (2,0 экв, 25,0 ммоль),а также HCl в циклопентилметиловом эфире (СРМЕ) (3 М, 1,3 экв, 16,3 ммоль). Полученную смесь перемешивали при 20°С в течение 45 минут, затем нагревали до 60°С. После перемешивания в течение ночи, смесь охлаждали до 20°С и гасили с помощью HCl (1 М, 30 мл). За это время, наблюдалось некоторое выделение пузырьков и выделение тепла. После того, как выделение пузырьков газа заканчивалось (~5 минут), органический слой отделяли, промывали водой (8 объемов) и концентрировали до приблизительно 6 объемов путем проведения дистилляции при пониженном давлении. После чего, добавляли фосфорную кислоту (14,6 М, 15 экв, 187 ммоль), и смесь перемешивали при 20°С и разбавляли с помощью 2-метилтетрагидрофурана (7 объемов). Медленно добавляли раствор гидроксида калия (11,5 М, 20 экв). После перемешивания в течение приблизительно 10 минут, слои разделяли. Органическую фазу промывали водой (10 объемов), затем концентрировали до приблизительно 5 объемов путем дистилляции при пониженном давлении с получением раствора 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона в 2-метилтетрагидрофуране. Циклопентилметиловый эфир (2 объема) подогревали до приблизительно 40°С и затем добавляли к раствору 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. Смесь перемешивали в течение 1 часа. Во время перемешивания выпадал твердый осадок. Растворитель заменяли на СРМЕ (приблизительно 8 объемов) путем дистилляции при пониженном давлении, и полученную суспензию перемешивали при 15°С в течение 2 часов. Полученное твердое вещество собирали и сушили при 45°С под вакуумом с получением кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2. Кристаллическую форму подтверждали путем проведения анализа методом порошковой рентгеновской дифракции (PXRD) с использованием инструментальных методов, описанных в примере 77.
Пример 72
Получение кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 2
Метил 4-[(Z)-2-этоксиэтенил]-2-[2-фтор-4-(метилсульфанил)-анилино]-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат, полученный, как описано в примере 71, стадия 1 (2,0 г, 5,0 ммоль), объединяли с 2-метилтетрагидрофураном (20 объемов) и нагревали до приблизительно 75°С. К смеси добавляли О-(2-(третбутокси)этил)-гидроксиламина гидрохлорид (1,3 экв, 6,50 ммоль), и полученную смесь охлаждали до 20°С. Добавляли комплекс боран-пиридин (2,0 экв, 10,0 ммоль), а также HCl в циклопентилметиловом эфире (СРМЕ) (3 М, 1,3 экв, 6,5 ммоль). Полученную смесь перемешивали при 60°С. После перемешивания в течение ночи, смесь охлаждали до 20°С, гасили с помощью HCl (1 М, 12 мл). За это время, наблюдалось некоторое выделение пузырьков и выделение тепла. После окончания реакции, (~5 минут), органический слой отделяли, промывали водой (8 объемов) и концентрировали до приблизительно 6 объемов путем дистилляции при пониженном давлении. После чего, добавляли фосфорную кислоту (14,6 М, 15 экв, 75,0 ммоль), и смесь перемешивали при 65°С. После перемешивания в течение ночи, смесь охлаждали до 20°С и разбавляли 2-метилтетрагидрофураном (7 объемов). Медленно добавляли раствор гидроксида калия (11,5 М, 20 экв). После перемешивания в течение приблизительно 10 минут, слои разделяли. Органическую фазу промывали водой (10 мл) и затем концентрировали до приблизительно 5 объемов путем дистилляции при пониженном давлении. Смесь затем нагревали до 50°С и добавляли циклопентилметиловый эфир (2 объема). После чего добавляли затравки кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2 (полученного как описано в примере 71), и полученную суспензию охлаждали до 30°С. Растворитель заменяли на циклопентилметиловый эфир (приблизительно 9 объемов) путем дистилляции при пониженном давлении, и полученную суспензию перемешивали при 40°С в течение 1 часа. Смесь охлаждали до 20°С и перемешивали в течение ночи. Полученное твердое вещество собирали и промывали циклопентилметиловым эфиром (4 объемов) и сушили при 45°С под вакуумом с получением кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2. На фигуре 2 приведена порошковая рентгенограмма кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2, полученного этим методом, где анализ методом порошковой рентгеновской дифракции (PXRD) проводили с использованием инструментальных методов, описанных в примере 77.
Пример 73
Получение затравочных кристаллов кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3
В реактор загружали 2-метилтетрагидрофуран (20 объемов), содержащий метил 4-[(Z)-2-этоксиэтенил]-2-[2-фтор-4-(метил-сульфанил)анилино]-1-метил-6-оксо-1,6-дигидропиридин-3-карбоксилат, полученный, как описано в примере 71, стадия 1 (5,0 кг; лимитирующий реагент - все последующие загрузки основаны на этом количестве) и O-(2-(третбутокси)этил)гидроксиламина гидрохлорид (2,89 кг). Затем смесь нагревали до 70°С. После завершения реакции, смесь охлаждали до 20°С и добавляли комплекс боран-пиридин (8 М, 3,3 л) и HCl в СРМЕ (3 М, 5,95 л). Смесь перемешивали при 20°С. После завершения реакции восстановления, смесь нагревали до 65°С до завершения реакции циклизации. Реакционную смесь охлаждали до 25°С, гасили с помощью 1 М HCl (б объемов) и промывали водой (8 объемов). Органический слой концентрировали до приблизительно 7 объемов. Загружали фосфорную кислоту (13,1 л), и полученную смесь нагревали до 60°С до завершения реакции удаления защитной группы. Реакционную смесь гасили с помощью водного 45 масс. % раствора гидроксида калия (22,8 л), (7 объемов), и затем промывали водой (10 объемов). Органический слой затем фильтровали через фильтр. При температуре реакции 45°С, органический слой концентрировали до приблизительно 5 объемов, и определяли содержание воды методом Карла Фишера KF и проводили коррекцию до 5% содержания воды. Приблизительно 5% реакционной смеси удаляли и охлаждали до 20°С, получая суспензию, которую загружали обратно в реактор для затравки всего содержимого в реакторе. После перемешивания при 45°С в течение трех часов, растворитель заменяли на СРМЕ (8 объемов), при этом остаточное содержание 2-MeTHF составляло приблизительно 5%-10%, а остаточное содержание воды, определенной по Карлу Фишеру, составляло, по меньшей мере, 0,5%. Реакционную смесь затем охлаждали до 15°С в течение 3 часов и гранулировали в течение 8 часов. Твердые вещества собирали и промывали с помощью СРМЕ (4 объемов). Твердые вещества сушили под вакуумом для удаления остаточного растворителя. Процесс сушки позволял дегидратировать твердую форму до кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2. Полученный кристаллический безводный 8-((2-фтор-4-(метил-тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 затем повторно гидратировали в шкафу (без вакуума) при 25-40°С с использованием следов воды. Конечные твердые вещества становились регидратированными с получением кристаллического моногидрата 8-((2-фтор-4-(метил-тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3, на основе результатов анализа воды по Карлу Фишеру и анализа методом порошковой рентгеновской дифракции (PXRD), где анализ методом PXRD проводили с использованием инструментальных методов, описанных в примере 77.
Пример 74
Получение кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3 путем кристаллизации с затравкой
В реактор загружали 2-MeTHF (б объемов, 19 л) и воду (0,27 объемов, 0,87 л). Добавляли аморфный 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, полученный таким же методом, как в примере 6, метод А или В (3,22 кг), и смесь нагревали до 65°С. В течение этого времени, твердое вещество растворялось и получали раствор. Смесь охлаждали до 45°С и добавляли затравку кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3, полученную в примере 73. Смесь перемешивали в течение приблизительно 2 часов, затем медленно загружали гептан (4 объемов) в течение 3 часов. Смесь охлаждали до 15°С на протяжении 4 часов и перемешивали в течение, по меньшей мере, 2 часов. Твердые вещества затем собирали и промывали смесью 50:50 гептан/2-MeTHF (4 объемов, 13 л). Твердые вещества сушили под вакуумом для удаления остаточного растворителя. Процесс сушки приводил к дегидратированию твердой формы с образованием кристаллического безводного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 2. Полученный кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 затем подвергали регидратации в шкафу (без вакуума) при 25-40°С с использованием следов воды. После выдерживания в шкафу в течение 8-24 часов, твердое вещество регидратировалось с образованием кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3, что подтверждалось результатами анализа воды по Карлу Фишеру и анализа методом порошковой рентгеновской дифракции (PXRD).
Пример 75
Альтернативное получение кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3
Аморфный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, полученный таким же методом, как в примере 6, метод А или В (404,88 мг), объединяли с 2-пропанолом (1,50 мл, 3,7 объемов) при перемешивании с магнитной мешалкой. Смесь нагревали до 60°С. В течение этого времени, смесь становилась гомогенной, темно-красного цвета, и все твердые вещества растворились. Добавляли воду (2,5 мл, 6,2 объемов), и смесь охлаждали до комнатной температуры и перемешивали в течение 2 часов. В течение этого времени, происходило осаждение твердого вещества, смесь становилась достаточно густой. После чего, применяли более крупную магнитную мешалку. Отбирали аликвоту для проведения анализа методом порошковой рентгеновской дифракции (PXRD), и смесь перемешивали при комнатной температуре. После перемешивания в течение ночи, твердое вещество собирали и промывали смесью 9:12-пропанол:вода (2×1,00 мл). Сушили в предварительно нагретом сушильном шкафу при 50°С с кюветой с водой в течение 3 часов, затем выдерживали в течение ночи без крышки на этом месте. 371 мг, 91% извлечение. Путем анализа методом PXRD определяли, что выделенный материал представляет собой кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3. На фигуре 3 приведена порошковая рентгенограмма кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3, полученного этим методом, где анализ методом PXRD проводили с использованием инструментальных методов, описанных в примере 77.
Пример 76
Анализ на поглощение влаги кристаллическим моногидратом 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3
Исследования по сорбции и десорбции воды проводили на автоматизированном анализаторе сорбции паров (ТА instruments Q5000 SA). Микровесы калибровали с использованием эталонной гири массой 100 мг. Сенсор относительной влажности калибровали при значениях относительной влажности (RH) 5,0, 11,3, 32,8, 52,8, 75,3 и 84,3% (25°С), используя насыщенные солевые растворы. Приблизительно 10-20 мг кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3 помещали в кварцевой держатель для проб и сушили при относительной влажности (RH)≤3% при 25°С. Величину RH затем постепенно повышали от 0% до 40% RH с шагом приращения 5%, затем понижали до конечной величины RH 0% с шагом уменьшения 5%. На всех стадиях использовали максимальное время установления равновесия 120 минут. Приращение массы на каждой из стадий % RH определяется относительно массы после стадия изначальной сушки при 0% RH. Для оценки достижения равновесия не использовали изменение массы образца, и вместо этого программировали, что продолжительность всех стадий должна составлять 120 минут. После завершения сбора данных, проводили анализ данных, используя выпускаемое фирмой ТА универсальную компьютерную программу для анализа данных и программу Microsoft Excel.
На фигуре 5 приведена изотерма сорбции для кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 3. Как показано на фигуре 5, кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 дегидратируется в кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 при 15% RH и 25°С, что подтверждается с помощью анализа методом PXRD. Кристаллический безводный 8-((2-фтор-4-(метилтио)-фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 регидратируется в кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форму 3 при 25% RH при 25°С, что подтверждается с помощью анализа методом PXRD. На основе этого анализа, кристаллический безводный 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 2 является стабильным при RH ниже 10% при 25°С, а кристаллический моногидрат 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, форма 3 является стабильным при RH выше 30% при 25°С.
Пример 77
Общие методики анализа твердых форм методом порошковой рентгеновской дифракции (PXRD)
Инструментальные методы
Анализ методом порошковой рентгеновской дифракции безводного кристаллического 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидрокси-этокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 1, безводного кристаллического 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, форма 2 и аморфного 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона форма 4 проводили на дифрактометре Rigaku MiniFlex 6G, оснащенным источником Cu излучения. Образец приготавливали, используя кремниевый держатель для образца с нулевым фоном (2 мм × 0,5 мм лунка). Дифрагированное излучение регистрировали с помощью детектора D/teX Ultra2. Напряжение и силу тока на рентгеновской трубке устанавливали 40 кВ и 15 ма, соответственно. Данные собирали в гониометр Miniflex при длине волны Cu от 3,0 до 45,0° 2-тета, используя шаг с 0,02° и скорость шага 2,00°/минута. Первичную щель устанавливали на 1,25° и длину огранивающей щели устанавливали на 10 мм. Образец вращали при скорости 10 оборотов в минуту в процессе сбора данных. Данные экспортировали в файл в формате *txt, используя программное обеспечение SmartLab Studio II фирмы Rigaku, и анализ данных проводили с использованием программного обеспечения EVA diffract plus. Как продемонстрировано в примере 76, безводная форма 2 регидратируется в моногидрат формы 3 при 25% RH при 25°С. Поэтому, анализ безводной формы 2 методом PXRD проводили при приблизительно 25°С и относительной влажности ниже 10%.
Анализ методом порошковой рентгеновской дифракции кристаллического моногидрата 8-((2-фтор-4-(метилтио)фенил)-амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, формы 3 проводили на дифрактометре Bruker AXS D8 Endeavor, снабженном источником Cu излучения. Щель расходимости устанавливали на 10 мм с постоянной подсветкой. Дифрагированное излучение регистрировали детектором PSD-Lynx Eye, при этом открывание детектора PSD устанавливали на 4,11 градусов. Напряжение и силу тока на рентгеновской трубке устанавливали 40 кВ и 40 ма, соответственно. Данные собирали при длине волны Cu  в гониометре Theta-Theta от 3,0 до 40,0 градусов 2-тета. Использовали размер шага 0,02 градуса и временной шаг 0,3 секунды. Антирассеивающий экран устанавливали на фиксированном расстоянии 1,5 мм. В процессе сбора данных образцы вращали. Образцы приготавливали путем помещения их в кремниевый держатель для образцов с нулевым фоном и вращали их в процессе сбора данных. Данные собирали, используя программное обеспечение Bruker DIFFRAC Plus, и анализ проводили, используя программу EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программе EVA, пики, выбранные с пороговой величиной 1, использовали для предварительных отнесений пиков. Для обеспечения достоверности, поправки вносились вручную; выходные данные автоматических отнесений контролировались визуально, и расположения пиков корректировали по максимуму пика. Обычно выбирали пики с относительной интенсивностью ≥3%. Обычно, пики, которые не были разрешены или были сопоставимы с шумом, не выбирали. Типичная ошибка, связанная с положением пика на порошковой рентгенограмме, официально принятая в фармакопеи США (USP), должна составлять +/- 0,2° 2-тета (USP-941). Как показано в примере 76, моногидрат формы 3 дегидратируется в безводную форму 2 при 15% RH и 25°С. Соответственно, анализ методом PXRD моногидрата Form 3 проводили при приблизительно 25°С и относительной влажности выше 30%.
в гониометре Theta-Theta от 3,0 до 40,0 градусов 2-тета. Использовали размер шага 0,02 градуса и временной шаг 0,3 секунды. Антирассеивающий экран устанавливали на фиксированном расстоянии 1,5 мм. В процессе сбора данных образцы вращали. Образцы приготавливали путем помещения их в кремниевый держатель для образцов с нулевым фоном и вращали их в процессе сбора данных. Данные собирали, используя программное обеспечение Bruker DIFFRAC Plus, и анализ проводили, используя программу EVA diffract plus. Файл данных PXRD не обрабатывали перед поиском пиков. Используя алгоритм поиска пиков в программе EVA, пики, выбранные с пороговой величиной 1, использовали для предварительных отнесений пиков. Для обеспечения достоверности, поправки вносились вручную; выходные данные автоматических отнесений контролировались визуально, и расположения пиков корректировали по максимуму пика. Обычно выбирали пики с относительной интенсивностью ≥3%. Обычно, пики, которые не были разрешены или были сопоставимы с шумом, не выбирали. Типичная ошибка, связанная с положением пика на порошковой рентгенограмме, официально принятая в фармакопеи США (USP), должна составлять +/- 0,2° 2-тета (USP-941). Как показано в примере 76, моногидрат формы 3 дегидратируется в безводную форму 2 при 15% RH и 25°С. Соответственно, анализ методом PXRD моногидрата Form 3 проводили при приблизительно 25°С и относительной влажности выше 30%.
Параметра отбора пиков PXRD
Используя алгоритм поиска пиков в программе EVA, пики, выбранные с пороговой величиной 1, использовали для предварительных отнесений пиков. Для обеспечения достоверности, поправки вносились вручную; выходные данные автоматических отнесений контролировались визуально, и расположения пиков корректировали по максимуму пика. Обычно выбирали пики с относительной интенсивностью ≥3%. Обычно, пики, которые не были разрешены или были сопоставимы с шумом, не выбирали. Типичная ошибка, связанная с положением пика на порошковой рентгенограмме, официально принятая в фармакопеи США (USP), должна составлять +/- 0,2° 2-тета (USP-941).
Для специалистов в данной области является очевидным, что в настоящее изобретение могут быть внесены различные модификации и вариации без отклонения от объема или сущности изобретения. После ознакомления с описанием и практическим осуществлением изобретения, для специалистов в данной области является очевидным, что существуют и другие варианты осуществления изобретения. Предполагается, что описание и примеры служат только для иллюстрации, при этом истинный объем и сущность изобретения определяются приведенной далее формулой изобретения.
Полное содержание всех цитируемых в изобретении литературных источников, включая патенты, патентные заявки, статьи, учебники и другие подобные публикации, а также содержание цитированных в этих источниках публикаций, включено в настоящее изобретение путем ссылок на них. В случае, если определяемые термины, применение терминов, описываемые методы и другие подобные определения в цитируемых в изобретении литературных источниках отличаются от изложенных в настоящем изобретении или противоречат им, то преимущество имеет настоящее изобретение.
Полное содержание предварительной заявки на патент США No. 63/168456, зарегистрированной 31 марта 2021 года, и предварительной заявки на патент США No. 63/309346, зарегистрированной 11 февраля 2022 года, включено в настоящее изобретение путем ссылки на них.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| КОНДЕНСИРОВАННЫЕ ТРИАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ Р2Х7 | 2010 |
|
RU2533122C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| СОЕДИНЕНИЕ КАК ИНГИБИТОР ПЕРЕДАЧИ СИГНАЛОВ WNT, ЕГО КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2012 |
|
RU2627712C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |





Группа изобретений относится к области органической химии и включает соединение формулы I, соединение формулы II, 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион и их фармацевтически приемлемые соли, фармацевтические композиции, их содержащие, способ лечения и применение на основе 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона. В формуле I R1 представляет собой Н, Br, С1-С6 алкил или фенил; R2 представляет собой Н, галоген или СН3-; R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-. В формуле II R1 представляет собой Н, Br, С1-С6 алкил или фенил; R2 представляет собой Н, галоген или СН3-; R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-. Технический результат – производные 3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-диона, обладающие МЕК ингибирующей активностью. 7 н. и 29 з.п. ф-лы, 5 ил., 8 табл., 77 пр.


1. Соединение формулы I

или его фармацевтически приемлемая соль,
где R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
R4 представляет собой фенил, замещенный с помощью 1, 2 или 3 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой Н.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где R2 представляет собой Н.
4. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где R2 представляет собой СН3-.
5. Соединение по любому одному из пп. 1-4 или его фармацевтически приемлемая соль, где R3 представляет собой гидроксиС1-С6 алкил-.
6. Соединение по любому одному из пп. 1-5 или его фармацевтически приемлемая соль, где R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
7. Соединение по любому одному из пп. 1-5 или его фармацевтически приемлемая соль, где R4 представляет собой фенил, замещенный с помощью 1 или 2 заместителей, независимо выбранных из галогена и С1-С6 алкилтио.
8. Соединение формулы II

или его фармацевтически приемлемая соль,
где R1 представляет собой Н, Br, С1-С6 алкил или фенил;
R2 представляет собой Н, галоген или СН3-;
R3 представляет собой Н, гидроксиС1-С6 алкил-, гидроксиС1-С6 алкокси-, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкил или (С3-С6 циклоалкил)С1-С6 алкокси-; и
Ra и Rb независимо выбирают из галогена, С1-С6 алкила, С1-С6 алкилтио, фторС1-С6 алкилтио, фторС1-С6 алкила, С1-С6 алкокси, фторС1-С6 алкокси, С3-С6 циклоалкила и С1-С6 алкил-С(=O)-.
9. Соединение по п. 8 или его фармацевтически приемлемая соль, где Ra представляет собой галоген.
10. Соединение по п. 8 или 9 или его фармацевтически приемлемая соль, где Rb представляет собой галоген, С1-С6 алкил, С1-С6 алкилтио или фторС1-С6 алкокси.
11. Соединение по п. 1, выбранное из следующих соединений:
8-((4-бром-2-фторфенил)амино)-2-циклопропил-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(циклопропилметокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-этокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-циклопропил-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-циклопропил-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-хлорфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2,3-дифторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-3-хлор-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(трифторметил)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-метоксифенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-((трифторметил)тио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-изопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-циклопропилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-ацетил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-хлор-4-этилфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(трифторметокси)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-((дифторметил)тио)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-изопропокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-этокси-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-метокси-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-(третбутокси)-8-((2-фтор-4-йодфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(S)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(S)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
(R)-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксипропокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5-хлор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
5-хлор-8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5-фтор-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5-йод-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
5-бром-8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
4-бром-8-((2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-йодфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-йод-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-пропилфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-этил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-циклопропил-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-хлорфенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтил)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-(дифторметокси)-2-фторфенил)амино)-2-(2-гидроксиэтокси)-5,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
2-(2,2-дифторэтокси)-8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-7-метил-4-фенил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((4-бром-2-фторфенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-4,7-диметил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион;
или его фармацевтически приемлемая соль.
12. Соединение, которое представляет собой 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, имеющий структуру

или его фармацевтически приемлемая соль.
13. Соединение, которое представляет собой 8-((2-фтор-4-(метилтио)фенил)амино)-2-(2-гидроксиэтокси)-7-метил-3,4-дигидро-2,7-нафтиридин-1,6(2Н,7Н)-дион, имеющий структуру
14. Фармацевтическая композиция, обладающая МЕК ингибирующей активностью, включающая эффективное количество соединения по любому одному из пп. 1-13 или его фармацевтически приемлемую соль и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
15. Способ лечения МЕК-ассоциированной опухоли, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по п. 12.
16. Способ по п. 15, где опухоль имеет мутацию BRAF V600, выбранную из V600E, V600K, V600D, V600R и V600S.
17. Способ по п. 15 или 16, где опухоль имеет мутацию BRAF V600.
18. Способ по любому одному из пп. 15-17, где опухоль представляет собой опухоль ЦНС.
19. Способ по п. 18, где опухоль ЦНС представляет собой интракраниальную опухоль.
20. Способ по п. 19, где интракраниальная опухоль представляет собой рак головного мозга.
21. Способ по п. 20, где рак головного мозга представляет собой метастатический рак головного мозга.
22. Способ по п. 21, где метастатический рак головного мозга выбирают из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников.
23. Способ по п. 18, где опухоль ЦНС представляет собой интракраниальное лептоменингеальное заболевание (LMD) или экстракраниальное лептоменингеальное заболевание (LMD).
24. Способ по п. 23, где лептоменингеальное заболевание (LMD) выбирают из метастатической меланомы, метастатического колоректального рака и метастатического немелкоклеточного рака легкого.
25. Способ по п. 19, где интракраниальная опухоль представляет собой первичную опухоль.
26. Способ по п. 25, где первичная опухоль головного мозга представляет собой злокачественную опухоль.
27. Способ по п. 26, где первичная опухоль головного мозга представляет собой глиому степени злокачественности 2, степени злокачественности 3 или степени злокачественности 4.
28. Способ по п. 27, где первичная опухоль головного мозга представляет собой доброкачественную опухоль.
29. Способ по п. 15, где опухоль имеет BRAF слияние.
30. Способ по п. 29, где опухоль имеет BRAF слияние, выбранное из KIAA1154 9-BRAF, MKRN1-BRAF, TRIM24-BRAF, AGAP3-BRAF, ZC3HAV1-BRAF, AKAP9-BRAF, CCDC6-BRAF, AGK-BRAF, EPS15-BRAF, NUP214-BRAF, ARMC10-BRAF, BTF3L4-BRAF, GHR-BRAF, ZC3HAV1-BRAF, ZNF767-BRAF, CCDC91-BRAF, DYNC112-BRAF, ZKSCAN1-BRAF, GTF2I-BRAF, MZT1-BRAF, RAD18-BRAF, CUX1-BRAF, SLC12A7-BRAF, MYRIP-BRAF, SND1-BRAF, NUB1-BRAF, KLHL7-BRAF, TANK-BRAF, RBMS3-BRAF, STRN3-BRAF, STK35-BRAF, ETFA-BRAF, SVOPL-BRAF и JHDM1D-BRAF.
31. Способ по п. 30, где опухоль представляет собой карциному молочной железы, колоректальную карциному, карциному пищевода, глиому, карциному головы и шеи, карциному легкого, меланому, карциному поджелудочной железы, карциному предстательной железы, саркому, карциному щитовидной железы, карциному неизвестной первичной локализации, мезотелиому плевры, аденокарциному прямой кишки, карциному эндометрия матки или серозную карциному яичников.
32. Способ по п. 15, где опухоль представляет собой опухоль с BRAF немутантного типа (дикого типа).
33. Способ по любому одному из пп. 15-32, где субъектом является человек.
34. Соединение по любому одному из пп. 1-13 или его фармацевтически приемлемая соль для применения в качестве лекарственного препарата для лечения МЕК-ассоциированной опухоли.
35. Соединение по любому одному из пп. 1-13 или его фармацевтически приемлемая соль для применения при лечении МЕК-ассоциированной опухоли.
36. Применение соединения по п. 12 или его фармацевтически приемлемой соли для производства лекарственного препарата для лечения МЕК-ассоциированной опухоли у субъекта.
| WO 2008079814 A2, 03.07.2008 | |||
| WO 2008115890 A2, 25.09.2008 | |||
| WO 2013136249 A1, 19.09.2013 | |||
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ МЕК И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2414455C2 |

