Изобретение относится к новым соединениям, которые являются антагонистами рецепторов аминокислот-медиаторов возбуждения, и к получению таких соединений.
Хорошо известна роль аминокислот-медиаторов возбуждения, таких как глутаминовая кислота и аспаргиновая кислота, в качестве основных медиаторов синаптической передачи возбуждения в центральной нервной системе. См. Watkins bc Evans, Ann. Rev. Pharmacol. Toxicol., 21, 165 (1981); Monaghan, Bridges, and Cotman, Ann. Rev. Pharmacol. Toxicol, 29, 365 (1989), EP заявка 0383504, Watkins, Krogsgaard - Larsen, and Honore, Trans. Pharm. Sci 11, 25 (1990). Эти аминокислоты функционируют в синаптической передаче преимущественно путем воздействия на рецепторы аминокислот-медиаторов возбуждения. Эти аминокислоты также участвуют в самых разнообразных физиологических процессах, таких как регуляция моторики, дыхания, регуляция сердечно-сосудистой системы, чувственное восприятие и распознавание.
Рецепторы аминокислот-медиаторов возбуждения подразделяются на два основных типа. Рецепторы, непосредственно соединенные с входами катионных каналов в клеточной мембране нейронов, называются "ионотропными". Рецепторы этого типа подразделяются на, по меньшей мере, три подгруппы в зависимости от деполяризующей активности селективных антагонистов: рецепторы N-метил-D-аспартата (NMDA), альфа-амино-3-гидрокси-5-метилизоксазол-4-пропионовой кислоты (АМРА) и каиновой кислоты (КА). Второй тип - это G-протеин, или связанный с вторичным посредником "метаботропный" рецептор аминокислот-медиаторов возбуждения. Эти рецепторы второго типа, будучи активированы антагонистами квизквалата, иботената или транс-1-аминоциклопентан-1,3-дикарбоновой кислоты, вызывают усиление гидролиза фосфоинозитида в постсинаптической клетке. Оба типа рецепторов не только опосредуют нормальную синаптическую передачу, но и участвуют в модификации синаптических связей в процессе развития и изменения эффективности синаптической передаче в течение жизни. См. Schoepp, BocKaert, and Sladeczek, Trends in Pharmacol. Sci, 11, 508 (1990); McDonald and Johnson, Brain Research Reviews 15, 41 (1990).
Чрезмерная или несоответствующая стимуляция рецепторов аминокислот-медиаторов возбуждения ведет к разрушению нейронов или повреждает их посредством механизма, известного как эксайтотоксичность. Предполагается, что этот процесс лежит в основе дегенерации нейронов при различных условиях. Медицинские последствия такой дегенерации нейронов заставляют признать смягчение подобных дегенеративных неврологических процессов важной терапевтической задачей.
Эксайтотоксичность аминокислот-медиаторов возбуждения является одним из патофизиологических механизмов целого ряда неврологических нарушений. Эксайтотоксичность является патофизиологическим механизмом острых и хронических нейродегенеративных нарушений, включая церебральную недостаточность вследствие операций на сердце с применением АИК, инсульта, травмы спинного мозга, травмы головы, болезни Альцгеймера, хореи Гентингтона, бокового амиотрифического склероза, слабоумия, вызванного СПИДом, перинатальной гипоксии, остановки сердца, гипогликемического поражения нейронов, поражения глаз и ретинопатии, а также болезни Паркинсона (идиопатической или вследствие приема лекарственных препаратов). При других неврологических состояниях, вызванных глутаматной дисфункцией, необходима нейромодуляция. Эти другие нейрологические состояния включают мышечные спазмы, мигрени, недержание мочи, психозы, толерантность к препаратам опия и синтром отменены, беспокойство, рвоту, отек мозга, хронические боли, конвульсии и позднюю дискинезию. Считается, что применение нейропротектора, такого как антагониста рецептора АМРА, поможет в лечении этих нарушений и в уменьшении неврологических повреждений, связанных с этими нарушениями. Антагонисты аминокислот-медиаторов возбуждения могут применяться и в качестве анальгетиков.
В последних исследованиях на моделях местной и общей ишемии показано нейропротекторное действие антагонистов рецепторов АМРА. Антагонист рецепторов АМРА - NBQX (2,3-дигидрокси-6-нитро-7-сульфамоилбензо[f]хиноксалин) конкурентного действия эффективно предотвращал повреждения тканей вследствие общей и локальной ишемии. Sheardown et al. Science, 247, 571 (1990); Buchan et. al, Neuroreport, 2, 473 (1991); Lepeillet et. al, Brain Research 571, 115 (1992). Антагонисты рецепторов АМРА - GKY1 52466 неконкурентного действия проявили себя в качестве эффективных нейропротекторов на моделях общей ишемии у крыс. LaPeillet et. al, Brain Research 571, 115 (1992). Эти исследования дают основания предположить, что поздняя дегенерация нейронов при ишемии мозга происходит в результате глутаминовой эксайтотоксичности, опосредованной, по меньшей мере, частично, активацией рецепторов АМРА. Таким образом, антагонисты рецепторов АМРА могут использоваться как нейропротекторы и улучшать неврологическое состояние при церебральной ишемии у больных.
Настоящее изобретение дает соединения, которые являются антагонистами рецепторов аминокислот-медиаторов возбуждения. Более конкретно, изобретение относится к соединениям, которые являются селективными для рецепторов АМРА. Изобретение относится к соединениям формулы I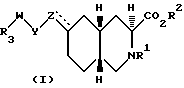
где
R1 представляет собой водород, алкил C1-C10, арилалкил, алкоксикарбонил или ацил;
R2 представляет собой водород, алкил C1-C6-замещенный алкил, циклоалкил или арилалкил;
R3 представляет собой CO2H, SO3H, CONHSO2R8 или группу формулы: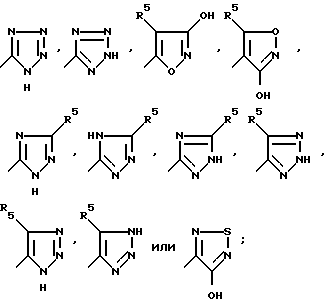
где
W - (CH2)n, S, SO, SO2;
Y - CHR7, NR4, O, S, SO, или SO2;
Z - NR6, CHR7 или CH;
W и Y вместе представляют собой HC = CH или C≡C , или Y и Z вместе представляют собой HC=CH или C≡C ;
R4 - водород, алкил C1-C4, фенил, или ацил;
R5 - водород, алкил C1-C4, CF3, фенил, гидрокси, амино, бромо, иодо или хлоро;
R6 - ацил;
R7 - независимо водород, алкил C1-C4, фенил или замещенный фенил;
R8 - алкил C1-C4 или тетразол-5-ил;
n равно 0, 1 или 2;
при условии, что когда Y представляет собой NR4, O, S, SO или SO2, W представляет собой (CH2)n и Z представляет собой CHR7 или CH; при условии, что когда W представляет собой S, SO или SO2, Y представляет собой CHR7, Z представляет собой CHR7 или CH; или Y и Z вместе являются HC=CH или C≡C ;
при условии далее, что когда W и Z являются CH2, Y не является S;
при условии далее, что когда W и Y вместе являются HC = CH или C≡C , является CHR7;
или фармацевтически приемлемая соль этих соединений.
Настоящее изобретение дает также рецептуры фармацевтических композиций, которые включают соединение формулы I и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Варианты осуществления настоящего изобретения включают способ блокирования рецептора аминокислот-медиаторов возбуждения (АМРА), а также способ лечения неврологических нарушений, связанных с рецепторами аминокислот-медиаторов возбуждения, который включает назначение соединения формулы I. В числе примеров подобных неврологических нарушений, которые подлежат лечению с применением соединения формулы I: церебральные нарушения, вызванные операцией с применением искусственного кровообращения, трансплантацией, "ударом", церебральной ишемией, травмы спинного мозга, травмы головы, болезнь Альцгеймера, хорея Гентингтона, боковой амиотрофический склероз, слабоумие, вызванное СПИДом, мышечные спазмы, мигрени, недержание мочи, психозы, конвульсии, перинатальная гипоксия, остановка сердца, поражения нейронов, вызванные гипогликемией, толерантность к препаратам опия и синдром отмены, поражение глаза и ретинопатия, идиопатическая и вызванная лекарствами болезнь Паркинсона, беспокойство, рвота, отек мозга, хронические боли или поздняя дискинезия. Соединения формулы I можно использовать также в качестве анальгетиков.
Настоящее изобретение дает также способ получения соединений формулы I, где R1 представляет собой водород, алкил C1-C10 или арилалкил, а R2 представляет собой водород, способ включает гидролиз соединения формулы I, где R1 представляет собой алкоксикарбонил или ацил, а R2 представляет собой алкил C1-C6, замещенный алкил, циклоалкил или арилалкил.
Настоящее изобретение относится также к соединениям, которые используют для получения антагонистов рецепторов АМРА. Второй аспект настоящего изобретения относится к соединению формулы II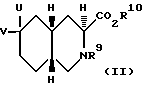
где
R9 - ацил или алкоксикарбонил;
R10 - водород, алкил C1-C6 или арил;
U - гидроксил, гидроксиметил, формил, бромметил, бромэтил или гидроксиэтил;
V - водород;
U и V вместе представляют собой метилен или метоксиметилен.
Настоящее изобретение дает также способ получения соединений формулы VIIIb: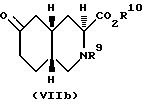
где
R9 - ацил или алкоксикарбонил;
R10 - хиральная аммониевая группа, водород, алкил C1-C6 или арил.
Четвертый аспект настоящего изобретения - это способ получения соединения формулы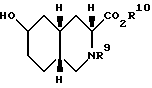
где
R9 - ацил или алкоксикарбонил;
R10 - водород, алкил C1-C6 или ацил.
Еще один аспект настоящего изобретения - способ получения соединения формулы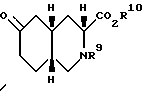
где
R9 - ацил или алкоксикарбонил;
R10 - водород, алкил C1-C6 или ацил.
Еще один аспект настоящего изобретения - способ получения соединения формулы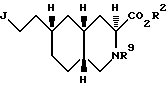
где
J - группа формулы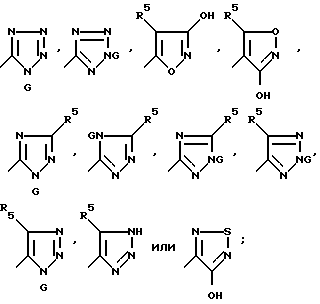
где
G - азотозащитная группа или водород;
R5 - имеет вышеуказанное значение:
R9 - ацил или алкоксикарбонил;
R10 - алкил C1-C6 или арил.
Настоящее изобретение дает также соединения, которые используют для получения ряда антагонистов рецепторов АМРА. Другой аспект настоящего изобретения связан с соединением формулы:
J v Q
где
J - группа формулы: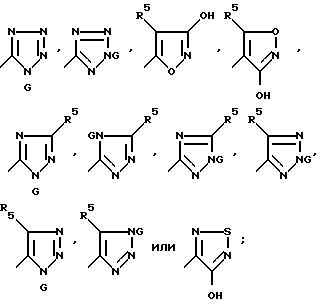
где
Q представляет собой CHR7P+(Ph)3X-, CHR7PO(Ph)2, CR7MSiP'3, CH(SiR3)PO(OR')2 или CH2SnR'3;
R' представляет собой алкил C1-C6 или фенил;
R5 и R7 имеют вышеуказанные значения;
G представляет собой азотозащитную группу или водород;
M представляет собой Li+ или M9 +2 X-;
X- - представляет собой бромид, хлорид, иодид, тетрафторборат или гексафторфосфат.
В вышеприведенной формуле термином "алкил C1-C10" обозначен алкил с прямой или разветвленной цепью, содержащий от одного до десяти атомов углерода. Типичные алкильные C1-C10 группы включают метил, этил, n-пропил, изопропил, n-бутил, изобутил, втор-бутил, трет-бутил, n-пентил, изопентил, n-гексил, 2-метилпентил, n-октил, децил и т.п. Термин "алкил C1-C10" включает в себя термины "алкил C1-C6" и "алкил C1-C4". Типичные алкильные C1-C6 группы включают метил, этил, n-пропил, изопропил, n-бутил, изобутил, втор-бутил, трет-бутил, n-пентил, изопентил и n-гексил. Типичные алкильные C1-C4 группы включают метил, этил, n-пропил, изопропил, n-бутил, изобутил, втор-бутил, трет-бутил.
Термином "ацил" обозначен водород или алкильная C1-C6 группа, присоединенная к карбонильной группе. Типичные ацильные группы включают формил, ацетил, пропионил, бутирил, валерил и капроил.
Термин "незамещенный алкил" использован здесь для обозначения алкильных C1-C6-групп, которые замещены одним или более из следующих заместителей: гидрокси, фтор, хлор-, бром - и иод-. Примеры замещенных алкильных групп включают гидроксиметил, хлорметил, бромметил, иодметил, трихлорметил, трифторметил, хлорэтил, бромэтил, перфторэтил и т.п.
Термином "алкокси C1-C4" обозначены такие группы, как метокси, этокси, n-пропокси, изопропокси, n-бутокси, трет-бутокси, и т.п. Термин "галоген" относится к фторо-, хлоро-, бромо- и иодогруппам.
Термин "замещенный фенил" здесь обозначает фенильную группу, замещенную одним или более заместителями, выбираемыми из группы, в которую входят галоген, гидрокси, циано, нитро, алкил C1-C6, алкокси C1-C4, алкоксикарбонил, защищенный карбокси, карбоксиметил, гидроксиметил, амино, аминоэтил или трифторметил. Примеры замещенных фенильных групп включают 4-хлорфенил, 2,6-дихлорфенил, 2,5-дихлорфенил, 3,4-дихлорфенил, 3-хлорфенил, 3-бромфенил, 4-бромфенил, 3,4-дибромфенил, 3-хлор-4-фторфенил, 2-фторфенил, 4-гидроксифенил, 3-гидроксифенил, 2,4-дигидроксифенил, 3-нитрофенил, 4-нитрофенил, 4-цианофенил, 4-метилфенил, 4-фенилметил, 4-этилфенил, 4-метоксифенил, 4-карбоксифенил, 4-(гидроксиметил)фенил, 4-аминфенил и т.п.
Термин "арил" обозначает группы, такие как фенильная и замещенная фенильная, которая описана выше. Термин "арилалкил" обозначает алкильную C1-C4-группу, имеющую арильную группу. Представители этой последней группы включают бензил, 1-фенилэтил, 2-фенилэтил, 3-фенилпропил, 4-фенилбутил, 2-метил-2-фенилпропил, (4-хлорфенил)метил, (2,6-дихлорфенил)метил, (4-гидроксифенил)метил, (2,4-динитрофенил)метил и т.п.
Термин "циклоалкил" обозначает циклоалкильную C3-C7-группу, например циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Термин "алкоксикарбонил" означает карбоксильную группу, имеющую алкильную C1-C6-группу, присоединенную к углероду карбонильной группы через атом кислорода. Представители этой группы включают трет-бутоксикарбонил и метоксикарбонил.
Термин "арилоксикарбонил" обозначает карбоксильную группу, несущую арильную группу, присоединенную к углероду карбонильной группы через атом кислород. Представители такое группы включают феноксикарбонил, (4-хлорфенокси)карбонил, (3-нитрофенокси)карбонил.
Термин "хиральная аммониевая группа" обозначает амин с хиральной группой, причем указанный амин образует соль с остатком карбоновой кислоты на атоме углерода, который является соседним с атомом азота декагидроизохинолинового кольца (C - 3). Примеры таких аминов, имеющих хиральную группу, способных реагировать с остатком карбоновой C-3 кислоты до образования хиральной аммониевой соли, включают R-(+)-альфа-метилбензиламин, S-(-)-альфа-метилбензиламин, (-)-альфа-(2-нафтил)этиламин, иохимбин, (+)-амфетамин, (-)-эфедрин; стрихнин, бруцин, хинин, хинидин, цинхонин, цинхонидин, и т.п.
Термин "азотозащитные группы" включают тритил, бензил, трет-бутил, трет-бутилдиметилсилил и трифенилсилил.
Несмотря на то, что все соединения формулы I по настоящему изобретению считаются антагонистами рецептор аминокислот-медиаторов возбуждения (АМРА), отдельные соединения по изобретению являются предпочтительными для этой цели. Желательно, чтобы R1 представлял собой водород или алкоксикарбонил; R2 представлял собой водород или алкил C1-C6; R3 представлял собой группу, выбираемую из группы, в которую входят CO2H, SO3H, CONHSO2R8,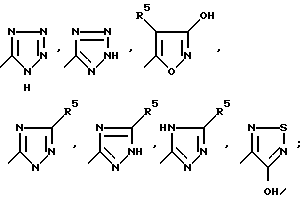
W представлял собой S или (CH2)n, где n = 0, 1 или 2; Y представлял собой CHR7, S, SO2 или O; Z представлял собой CHR7 или NR6; Y и Z вместе представляли собой HC = CH; R5 представлял собой водород, алкил C1-C4, CF3 или фенил; R6 представлял собой формил; R7 представлял собой водород, алкил C1-C4 или фенил; R8 представлял собой алкил C1-C4 или тетразол-5-ил. Представителями этой предпочитаемой группы являются следующие соединения:
6-[2-(1(2)Н-тетразол-5-ил)этил]декагидроизохинолин-3-карбоновая кислота, 6-[N-(1(2)Н-тетразол-5-ил)метилформамидо] декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-2-тиаэтил] декагидроизохинолин-3- карбоновая кислота, 6-[(1(2)Н-тетразол-5-ил)проп-1-ил] декагидроизохинолин-3-карбоновая кислота, 6-[1(2)Н-тетразол-5-ил)метоксиметил]-декагидроизохинолин-3-карбоновая кислота, [6-/3-(1(2)Н-тетразол-5-ил)]-3-тиапроп-1-ил]- декагидроизохинолин-3-карбоновая кислота, 6-[(1(2)Н-тетразол-5-ил)-бут-1-ил] декагидроизохинолин-3-карбоновая кислота, 6-(2-карбоксиэтил)декагидроизохинолин-3-карбоновая кислота, 6-(2-сульфоэтил)-декагидроизохинолин-3-карбоновая кислота, 6-[2-(3-гидроксиизоксазол-5-ил)этил]декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2-4)Н-1,2,4-триазол-5-ил)-2-тиэтил] декагидроизохинолин-3-карбоновая кислота, 6-[(1(2-4)Н-1,2,4-триазол-5-ил)сульфонилметил] -декагидроизохинолин -3-карбоновая кислота, 6-[2-(N-метансульфонил)-карбоксамидо)этил] декагидроизохинолин-3- карбоновая кислота, 6-[2-(N-(1(2)Н-тетразол-5-ил)карбоксамидо) этил] -декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)1-метилэтил/-декагидроизохинолин-3- карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-1-фенилэтил] декагидроизохинолин-3- карбоновая кислота, 6-[2-(3-гидрокси-1,2,5-тиадиазол-4-ил)этенил]декагидроизохинолин-3- карбоновая кислота и т.п.
Определенные соединения по изобретению являются более предпочитаемыми в качестве антагонистов рецепторам аминокислот-медиаторов возбуждения (АМРА). Более предпочитаемыми являются соединения, где R1 является водородом или алкоксикарбонилом; R2 представляет собой водород или C1-C6; R3 представляет собой группу, выбираемую из SO3H и группы формулы: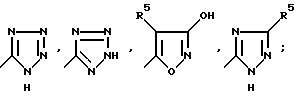
W представляет собой S, SO2 или (CH2)n; n равно 0, 1 или 2; Y представляет собой CHR7, S или SO2; Z представляет собой CHR7; R5 представляет собой водород, алкил C1-C4 или CF3; и R7 представляет собой водород, алкил C1-C4 или фенил. Представителями этой более предпочитаемой группы являются следующие соединения (в числе прочих): 6-[2-(2(2)Н-тетразол-5-ил)этил]декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-2-тиаэтил] декагидроизохинолин-3- карбоновая кислота, 6-[(1(2)Н-тетразол-5-ил)проп-1-ил] декагидроизохинолин-3-карбоновая кислота, 6-[(1(2)Н-тетразол-5-ил) метоксиметил] -декагидроизохинолин-3-карбоновая кислота, 6-[3-(1(2)Н-тетразол-5-ил)-3-тиапроп-1-ил] декагидроизохинолин-3- карбоновая кислота, 6-[(1(2)Н-тетразол-5-ил)бут-1-ил] -декагидроизохинолин-3-карбоновая кислота, 6-(2-сульфоэтил)декагидроизохинолин-3-карбоновая кислота, 6-(2-(3-гидроксиизоксазол-5-ил)этил] декагидроизохинолин-3-карбоновая кислота, 6-[(1(2-4)Н-1,2,4-триазол-5-ил)сульфонилметил] декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил) -1-метилэтил] декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н- тетразол-5-ил)-1-фенилэтил] декагидроизохинолин-3-карбоновая кислота и т.п.
Определенные соединения по изобретению являются наиболее предпочтительными в качестве антагонистов рецепторов аминокислот-медиаторов возбуждения (АМРА). Наиболее предпочтительными являются соединения, где R1 и R2 являются водородом; R3 представляет собой группу, выбираемую из группы формулы: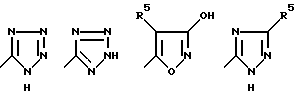
W представляет собой (CH2)n, где n равно 0; Y представляет собой CHR7, S, или SO2; Z представляет собой CHR7; R5 представляет собой водород или алкил C1-C4; R7 представляет собой водород, алкил C1-C4 или фенил. Представителями этой наиболее предпочтительной группы соединения являются следующие соединения: 6-[2-(1(2)Н-тетразол-5-ил)этил]декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-2-тиаэтил]декагидроизохинолин-3- карбоновая кислота, 6-[2-(3-гидроксиизоксазол-5-ил)этил] декагидроизохинолин-3-карбоновая кислота, 6-[(1(2-4)Н-1,2,4-триазол-5-ил)сульфонилметил] декагидроизохинолин-3- карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-1-метилэтил] декагидроизохинолин-3-карбоновая кислота, 6-[2-(1(2)Н-тетразол-5-ил)-1-фенилэтил] декагидроизохинолин-3- карбоновая кислота и т.п.
Соединениями формулы I по изобретению являются соединения, имеющие следующие стереохимическое строение: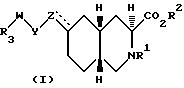
Соединения по настоящему изобретению, в которых Z не является CH, обладают по меньшей мере четырьмя асимметрическими атомами углерода. Асимметрические центры представляют собой замещенный атом углерода, смежный с кольцевой группой NR1 (3), атом углерода, когда Z присоединен к кольцу (6) и два атома углерода в голове моста (4a и 8a). В этом случае соединения могут существовать в виде диастереоизомеров, каждый из которых может существовать в виде рацемата энантиомеров. Соединения по настоящему изобретению включают не только рацематы, но и соответствующие энантиомеры. Когда Z представляет собой NR6, предпочитаемой конфигурацией диастереоизомера является следующая: 3SR, 4aSR, 6SR, 8aRS, а предпочитаемой конфигурацией энантиомера будет: 3S, 4aS, 6S, 8aR. Когда Z представляет собой CHR7, предпочитаемой конфигурацией диастереоизомера является: 3SR, 4aRS, 6SR, 8aRS, за исключением следующих случаев: когда R7 представляет собой водород, Y - CH2, W - (CH2)n, и n = 0, то предпочитаемой конфигурацией диастереоизомера является: 3SR, 4aRS, 6RS, 8aRS; когда R7 представляет собой водород и Y и Z вместе являются HC=CH, предпочитаемой конфигурацией диастереоизомера является 3SR, 4aRS, 6RS, 8aRS; когда Y представляет собой S, SO или SO2, W является (CH2)n, а n = 0, предпочитаемой конфигурацией этого диастереоизомера является 3SR, 4aSR, 6SR, 8aRS. Когда Z является CHR7, предпочитаемой конфигурацией энантиомера является 3S, 4aR, 6S, 8aR, за исключением следующих случаев: когда R7 является водородом, Y - CH2, W - (CH2)n и n = 0, то предпочитаемой конфигурацией энантиомера является 3S, 4aR, 6R, 8aR; когда R7 является водородом и Y и Z вместе являются HC = CH, то предпочитаемой конфигурацией энантиомера является 3S, 4aR, 6R, 8aR; когда Y является S, SO или SO2, W является (CH2)n, и n = 0, то предпочитаемой конфигурацией этого энантиомера является 3S, 4aS, 6S, 8aR. Когда Z является CH, то предпочитаемой конфигурацией диастереоизомера является 3SR, 4aRS, 8aRS, за исключением того случая, когда W представляет собой (CH2)n, а n = 0, тогда предпочитаемой конфигурацией диастереоизомера является 3SR, 4aSR, 8aRS. Когда Z является CH, то предпочитаемой конфигурацией энантиомера является 3S, 4aR, 8aR, за исключением случая, когда W является CH2)n, и n = 0, тогда предпочитаемой конфигурацией энантиомера будет 3S, 4aS, 8aR. Когда Z и Y вместе являются HC=CH или C≡C , то предпочитаемой конфигурацией диастереоизомера является 3SR, 4aRS, 6SR, 8aRS, за исключением случая, когда W является (CH2)n и n = 0, тогда предпочитаемой конфигурацией диастереоизомера является 3SR, 4aRS, 6SR, 8aSR. Когда Z и Y вместе являются HC = CH или C≡C, то предпочитаемой конфигурацией энантиомера является 3S, 4aR, 6S, 8aR, за исключением случая, когда W является (CH2)n и n = 0, тогда предпочитаемой конфигурацией энантиомера будет 3S, 4aR, 6S, 8aS. Более предпочитаемая относительная и абсолютная конфигурация показана следующей формулой: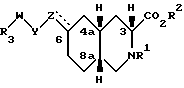
Соединения по настоящему изобретению могут содержать тетразольное кольцо, которое, как известно, может существовать в виде таутомеров. Тетразол, имеющий двойную связь на атоме азота в 1 положении и водород на атоме азота во 2 положении, называется 2Н-тетразолом и имеет следующую структуру: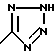
Соответствующий таутомер, имеющий водород на атоме азота в 1 положении и двойную связь на атоме азота в 4 положении, называется 1Н-тетразол. 1Н-тетразол имеет формулу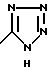
Смеси этих двух таутомеров здесь и далее обозначаются как 1(2)Н-тетразолы. Настоящее изобретение включает в себя оба данных таутомера и их комбинацию.
Соединения по настоящему изобретению могут содержать триазольное кольцо. Триазолы существуют в двух видах: 1,2,4-триазол и 1,2,3-триазол. Каждая из этих форм может существовать в виде таутомеров. Триазол, имеющий двойную связь на атоме азота в 1 положении и водород на атоме азота во 2 положении, называется 2Н-триазолом. Таутомер, в котором водород находится на атоме азота в 1 положении и двойная связь на атоме азота во 2 положении, называется 1Н-триазолом. Таутомер, в котором водород находится на атоме азота в 3 или 4 положении, называется, соответственно, 3Н-триазолом или 4Н-триазолом. Смеси этих таутомеров здесь и далее называются 1(2-4)Н-триазолами. Настоящее изобретение включает в себя оба изомера и отдельные таутомеры, а также их комбинации.
Настоящее изобретение включает фармацевтически приемлемые соли соединений формулы I. Эти соли могут иметь связь с кислотным или основным фрагментом молекулы и могут существовать в виде кислых, первичных, вторичных, третичных или четвертичных солей аммония, щелочных или щелочно-земельных металлов. Обычно кислые соли получают путем взаимодействия кислоты с соединением формулы I, где R1 представляет собой водород, алкил C1-C10 или арилалкил. Соли щелочных и щелочноземельных металлов обычно получают путем взаимодействия основной соли нужного металла с соединением формулы I, в котором R2 является водородом.
Кислоты, применяемые для получения таких солей, включают такие неорганические кислоты, как соляная, бромистоводородная, иодистоводородная, серная и фосфатная кислота, а также органические кислоты, такие как паратолуолсульфокислота, метансульфокислота, щавелевая кислота, парабромфенилсульфокислота, угольная, янтарная, лимонная, бензойная и уксусная кислота и аналогичные неорганические и органические кислоты. Такие фармацевтически приемлемые соли включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, соли аммония, кислые фосфаты, дифосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изолутираты, капронаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, гиппураты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксиленсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, альфа-гидроксибутираты, гликолаты, малеаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты, соли аммония, магния, тетраметиламмония, калия, триметиламмония, натрия, метиламмония, кальция и т.п.
Все соединения формулы II по настоящему изобретению могут использоваться для получения антагонистов рецепторов АМРА, но некоторые соединения по изобретению являются предпочтительными. Предпочтительно, чтобы R9 представлял собой алкоксикарбонил; R10 представлял собой алкил C1-C6; U представлял собой гидроксил, гидроксиметил, гидроксиэтил или формил; V представлял собой водород; или U и V вместе являлись бы метиленом или метоксиметиленом. Более предпочтительно, чтобы U представлял собой гидроксиметил или формил, или U и V вместе представляли собой метилен или метоксиметилен. Лучше всего, чтобы R9 представлял собой метоксикарбонил, а R10 -этил. В числе примеров соединений этой наиболее предпочитаемой группы этил-6-метилидин-2- метоксикарбонил-декагидроизохинолин-3-карбоксилат, этил-2-метоксикарбонил-6-(метоксиметилен)декагидроизохинолин-3- карбоксилат, этил-6-гидроксиметил-2-метоксикарбонил-декагидроизохинолин-3- карбоксилат и этил-6-формил-2-метоксикарбонилдекагидроизохинолин-3- карбоксилат.
Соединения общей формулы:
J v Q
где
J и Q имеют вышеуказанные значения, используют для синтеза соединений формулы I, где Z представляет собой CH или CHR7, а Y является CH2. Отдельные соединения по изобретению предпочтительно использовать для этой цели. Предпочтительно, чтобы Q представлял собой группу формулы CH(SiR'3)PO(OR')2, CHR7PO(Rh)2 или CHR7P+(Ph)3X-; X- представлял собой тетрафторборат, гексафторфосфат, иодид, бромид или хлорид; R5 представлял собой алкил C1-C4, CF3, водород или фенил; R7 представлял собой водород, алкил C1-C4 или фенил; R' представлял собой алкил C1-C6 или фенил; G представлял собой водород или тритил; J представлял собой группу формулы: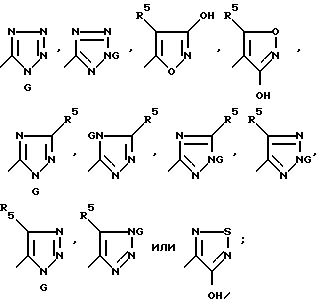
Более предпочтительно, чтобы Q представлял собой группу формулы CHR7PO(Ph)2 или CHR7P+(Ph)3X-; X- представлял собой иодид, хлорид или бромид; R5 представлял собой водород, метил или фенил; R7 представлял собой водород, метил или фенил; J представлял собой группу формулы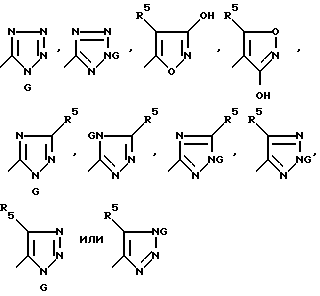
Лучше всего, чтобы Q представлял собой CHR7P+(Ph)3X-; X- представлял собой бромид или хлорид, R5 являлся водородом; R7 - водородом, J представлял собой группу формулы:
Соединения формулы I по изобретению можно синтезировать из обычного промежуточного вещества, 6-оксо-декагидроизохинолин-3-карбоксилата (VIII). Синтез данного вещества описан в Патенте США N 4 902 695, упоминаемого здесь для сведения. Усовершенствованный синтез этого промежуточного вещества из d, 1-m-тирозина показан на схеме 1.
Обычно m-тирозин (IV) конденсируют формальдегидом до образования 6-гидрокси-замещенной тетрагидроизохинолин-3-карбоновой кислоты (V). Это соединение этерифицируют на карбоксильной группе и блокируют на кольцевом азоте соответствующей защитной группой, получая дважды защищенное промежуточное соединение (VI). Это промежуточное соединение восстанавливают до получения защищенного 6-гидроксидекагидроизохинолин-3-карбоксилата (VII). 6-гидроксильную группу затем окисляют до 6-оксогруппы, получая обычное промежуточное вещество (VIII).
Настоящее изобретение включает усовершенствованный способ синтезирования промежуточного вещества VIII, в котором кольцевое присоединение образует цис-изомер. Метатирозин, предпочтительно рацемический m-тирозин, конденсируют формальдегидом до образования гидрокси-замещенного тетрагидро-изохинолин-3-карбоксилата (V). Эту реакцию предпочтительно проводить в деионизированной воде, содержащей концентрированную соляную кислоту, при температуре от около 55oC до около 70oC в течение от около 0,5 до около 2 часов. Соединение формулы V предпочтительно отделять путем охлаждения реакционной смеси до температур приблизительно 3 - 10oC и удаления продукта фильтрацией.
Желательно, чтобы соединение было защищено как на 3-карбоксильной группе, так и на кольцевом азоте. Способы защиты аминогрупп и карбоксильных групп описаны в MeOmie, Protective Groups in Organic Chemistry "Пленум Пресс", Нью-Йорк, 1973 и в Green and Wutz, Protecting Groups in Organic Synthesis, 2-е изд. , "Джон Уилей и Санз" Нью-Йорк, 1991. Карбоксильная группа может быть защищена как алкил C1-C6, замещенный алкил или арилэфир. Предпочитаемым эфиром является алкил C1-C6 эфир; наиболее предпочтительным является этилэфир. Этот эфир получают путем взаимодействия промежуточного вещества V со смесью этанола и концентрированной серной кислоты. Реакцию предпочтительно проводить при температуре перегонки растворителя в течение около 16 часов. Кольцевой азот можно защитить ацильной или алкоксикарбонильной группой. Предпочитаемыми защитными группами являются трет-бутоксикарбонил и метоксикарбонил. Наиболее предпочитаемой защитной группой является метоксикарбонил.
Добавку 2-метоксикарбонильной защитной группы производят с использованием стандартных способов синтеза органических соединений. Этилэфир промежуточного соединения V взаимодействуют с метилхлорформиатом в присутствии карбоната калия до образования промежуточного соединения VI. Эту реакцию предпочтительно проводить при температуре около 1 - 15oC в течение около 2 часов. Реакцию желательно проводить с последующей добавкой карбоната калия и метилхлорформиата в реакционную смесь. Промежуточное соединение VI, в котором R9 представляет собой метоксикарбонил, а R10 является этилом, предпочтительно отделяют экстракцией и кристаллизацией (этанол/вода).
Промежуточное соединение VII получают восстановлением промежуточного соединения VI. Предпочитаемым способом восстановления является каталитическое гидрирование. В числе подходящих катализаторов - палладий на углероде, платина на углероде, палладий на оксиде алюминия, окись платины, рутений на оксиде алюминия, родий на оксиде алюминия, или родий на углероде. Предпочитаемыми катализаторами являются рутений на оксиде алюминия, родий на оксиде алюминия, родий на углероде. Наиболее предпочтительным катализатором восстановления является родий на углероде. Подходящие для реакции растворители включают полярные органические растворители, такие как этилацетат, метанол и этанол. Этилацетат является предпочитаемым растворителем для данной реакции. Восстановление проводят под давлением водорода в около 100 - 1000 фунтов/кв. дюйм и при температуре около 80 - 150oC. Если в реакции применяют родий на оксиде алюминия, то реакция завершается через около 24 часа. Катализатор можно удалить фильтрацией и защищенный 6-гидроксидекагидроизохинолин-3-карбоксилат используют на следующем этапе без отделения.
Для получения промежуточного соединения VIII 6-гидроксильную группу промежуточного соединения VII окисляют до 6-оксогруппы. Желательно проводить эту реакцию с использованием мягкого окислителя. Подходящие мягкие окислители включают гипохлорит натрия, трихлорид рутения/периодат натрия и трихлорид рутения/иодная кислота. Для проведения этой реакции можно использовать и другие окислители, такие как хлорхромат пиридиния (РСС), реагент Джонса, диметилсульфоксид/N-хлорсукцинимид, тетрапропиламмонийперутенат (ТРАР), пиридин/SO3 и хлорноватистая кислота. Предпочтительно, профильтрованный раствор этилацетата, содержащий промежуточное соединение VII, обрабатывают трихлоридом рутения и водой, полученную смесь охлаждают до температуры от -10oC до около 25oC. Затем смесь, содержащую две фазы, обрабатывают иодной кислотой. После добавки иодной кислоты реакционная смесь разогревается до температуры приблизительно 20 - 35oC. Искомый продукт (промежуточное соединение VIII) отделяют, используя стандартные способы.
Согласно другому варианту, промежуточное соединение VI восстанавливают до получения промежуточного соединения VIII. Предпочитаемый способ восстановления - каталитическое гидрирование. Эта реакция дает смесь 6-гидрокси-промежуточное соединение VII и 6-кето-промежуточное соединение VIII. Без последующей очистки смесь этих промежуточных соединений можно использовать на следующем этапе для окисления 6-гидрокси-промежуточного соединения VII смеси до получения промежуточного соединения VIII без дальнейшей очистки. Подходящие катализаторы для этой реакции включают палладий на углероде и родий на углероде. Предпочитаемым катализатором является родий на углероде. Подходящие растворители для этой реакции включают полярные органические растворители, такие как этилацетат, метанол и этанол. Этилацетат является предпочтительным растворителем для данной реакции. Восстановление осуществляют под давлением водорода в 30 - 200 фунтов/кв.дюйм при температуре приблизительно 70 - 90oC. Предпочитаемые условия для этой реакции - давление водорода около 100 фунтов/кв.дюйм и температура около 85oC. Если в реакции используют родий на углероде, то реакция завершается приблизительно через 2 - 24 часа. Катализатор можно удалить фильтрацией, а полученные продукты используют на следующем этапе без дальнейшей очистки.
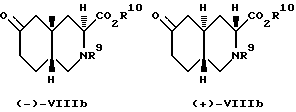
Синтез по вышеописанной схеме дает смесь диастереоизомеров, чьи относительные конфигурации представлены формулами VIIIa и VIIIb: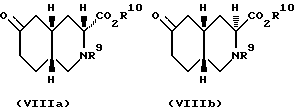
Преобладающим является диастереоизомер VIIIa. Эту смесь диастереоизомеров можно уравновесить до смеси, где VIIIb станет преобладающим диастереоизомером, путем обработки сильным основанием. Подходящими сильными основаниями для этого являются алкоксиды металлов, такие как этоксид натрия и трет-бутокси калия, диизопропиламид лития. Предпочитаемым сильным основанием является этоксид натрия. Если в качестве основания использован алкоксид металла, то в качестве растворителя может быть использован соответствующий спирт. Предпочитаемым растворителем в этом случае будет этанол. Если используют этоксид натрия и этанол, то уравновешивание можно проводить при температурах от комнатной до температуры перегонки растворителя. Предпочтительно, если проводить уравновешивание в NaOEt/EtOH, то температура должна равняться 40oC. Процесс уравновешивания займет от одного до около шести часов. Предпочитаемый диастереоизомер, промежуточное соединение VIIIb, отделяют от эфира (кристаллизацией: R9 - метоксикарбонил, а R10 - этил).
Энантиомеры каждой пары диастереоизомеров промежуточного соединения VIII пептизируют с использованием стандартных способов пептизации. См. Jacgues, Collet, and Wilen, Enantiomers, Racemates, and Resolutions. "Джон Уилей энд Санз", Нью-Йорк, 1981. Согласно предпочитаемому способу пептизации диастереоизомеров и энантиомеров для образования диастереоизомерных солей используют хиральные амины. Подходящие хиральные амины описаны в Tacquesetal Глава 5, стр. 253 - 259, упоминаемой здесь для сведения. Примеры подходящих хиральных аминов включают R-(+)-альфа-метилбензиламин, S-(-)-альфа-метилбензиламин, (-)-альфа-(2-нафтил)этиламин, иохимбин, (+)-амфетамин, (-)-эфедрин, стрихнин, бруцин, хинин, хинидин, цинхонин, цинхонидин, и т.п. Предпочитаемыми хиральными аминами являются альфа-метилбензиламин, бруцин, хинин, хинидин, цинхонин, цинхонидин. Более предпочитаемыми аминами являются альфа-метилбензиламин, бруцин и хинин. Наиболее предпочитаемым амином для пептизации соединения VIIIb является альфа-метилбензиламин.
Ниже описан предпочтительный способ пептизации энантиомера. Этил-эфир, промежуточное соединение VIIIb, где R9 представляет собой метоксикарбонил, а R10 представляет собой этил, гидрируют с использованием 5 н. гидроксида натрия при температуре от около 25 до 40oC в течение 0,5 - 2 часов. Подходящие растворители для этой реакции включают спирты, такие, как метанол и этанол. Свободную кислоту можно отделить экстракцией этилацетатом. Свободную кислоту, предпочтительно в растворе этилацетата, обрабатывают R-(+)-альфа-метилбензиламином при температуре приблизительно 25 -35oC в течение 15 - 60 минут. Промежуточное соединение (-)-VIIIb (R10-водород) выпадает в осадок из раствора в виде R-(+)-альфа-метилбензиламинной соли. Этот материал далее очищают повторным суспендированием в теплом (45 -50oC) этилацетате. Аналогичным образом получают (+)-VIIIb, используя S-(-)-альфа-метилбензиламин. Относительная и абсолютная конфигурации этих промежуточных соединений показаны ниже. Промежуточное соединение (-)-VIIIb является предпочтительным энантиомером.
Пептизированный энантиомер этерифицируют на 3-карбоксильной группе для последующих химических реакций. Предпочитаемым эфиром является этиловый эфир. Этерификацию проводят в следующих условиях: промежуточное соединение VIII (R10-водород) взаимодействуют с алкилирующим агентом в присутствии основания. Для данной реакции подходящими являются следующие алкилирующие агенты: этилиодид, этилбромид, этилхлорид, диэтилсульфат. Основание выбирают из группы, в которую входят триэтиламин, N,N-диизопропилэтиламин, пиридин, коллидин, бикарбонат натрия и карбонат натрия. Подходящими растворителями для этерификации являются полярные органические растворители, такие как диметилформамид и ацетонитрил. Этерификацию желательно осуществлять с использованием этилбромида и триэтиламина в ацетонитриле при температуре перегонки растворителя в течение от одного до двух часов.
Соединения по настоящему изобретению синтезируют из обычного промежуточного соединения VIII различными способами. Этапы синтеза, описанные в настоящем изобретении, можно скомбинировать другим образом для получения соединений формулы I. Описанные ниже способы не ограничивают рамки настоящего изобретения. Синтез соединений формулы I, где Y представляет собой CH2, Z представляет собой CHR7, а W является (CH2)n или S, n = 0, показан на схеме 2.
Промежуточное соединение VIII взаимодействует с реактивом Хенера-Эммонса до образования ненасыщенного промежуточного соединения IX, где R11 представляет собой защищенную карбоксильную группу. Это соединение восстанавливают до промежуточного соединения X. Затем карбоксильную группу восстанавливают до гидроксильной группы, которую превращают в бром-промежуточное соединение XI. Промежуточное соединение XI можно взаимодействовать с рядом нуклеофильных веществ до получения соединения формулы I, где W является (CH2)n, n = 0 и R3 представляет собой CO2H или SO3H, или же где W является S, R3 представляет собой триазол или тетразол.
Точнее, промежуточное соединение VIII взаимодействует с реактивом Хенера-Эммонса общей формулы (CH3CH2O)2POCH(R7)R11, где R7 является водородом, алкилом C1-C4, фенилом или замещенным фенилом, R11 представляет собой защищенную карбоксильную группу. Подходящие защитные группы включают этиловый и бензиловый эфиры. Эту реакцию проводят следующим образом: соответствующий диэтилфосфонат обрабатывают сильным основанием, таким как гидрид натрия, или бис(триметилсилил)амид натрия, до получения натриевой соли фосфоната, которую затем взаимодействуют в полярном органическом растворителе, таком как сухой тетрагидрофуран, с соединением VIII до получения промежуточного соединения IX. Эту реакцию проводят при температурах от 0oC до температуры перегонки реакционной смеси. Когда достигается небольшой избыток анионов фосфоната, реакция завершается приблизительно через шесть часов при комнатной температуре.
Затем промежуточное соединение IX восстанавливают до получения промежуточного соединения X. Предпочтительно проводить это восстановление путем гидрирования, желательно в присутствии катализатора. Подходящие катализаторы для такого восстановления включают палладий на углероде и платину на углероде. В числе подходящих для такого восстановления растворителей полярные органические растворители, такие как этанол и этилацетат. Восстановление обычно осуществляют при давлении водорода в приблизительно 60 - 100 фунтов/кв. дюйм. Реакция обычно завершается через четыре часа при комнатной температуре.
Затем промежуточное соединение X используют для получения соединения формулы XI. Это преобразование обычно проводят путем восстановления промежуточного соединения X, где R11 представляет собой карбоксильную или защищенную карбоксильную группу. Карбоксильная группа может быть восстановлена до спирта с помощью одного из известных способов. Один из таких способов предполагает обработку карбоксильной группы боран-метил-сульфидом. Эту реакцию обычно проводят в полярном органическом растворителе, таком как тетрагидрофуран, при температуре около 0oC. Затем гидроксильное соединение превращают в соединение формулы XI путем обработки гидроксизамещенного соединения трифенилфосфином и бромином. Эту реакцию обычно проводят в полярном органическом растворителе, таком как метиленхлорид, при температуре около 0oC.
Промежуточное соединение XI можно взаимодействовать с рядом нуклеофильных реагентов до получения соединений формулы I. В числе таких нуклеофилов тиоцианат, тиотриазол и тиотетразол. Например, реакция бром-промежуточного соединения XI с тиотетразолом в присутствии аминного основания дает соединение формулы I, где R3 представляет собой тетразол, а W является S. Подходящие аминные основания для этой реакции включают триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, пиридин и коллидин. Эту реакцию желательно проводить в полярном органическом растворителе, таком как ацетонитрил, при температуре около 50 - 100oC. Продукт этой реакции превращают в соединение формулы I, где R1 и R2 являются водородом, путем обработки 6 н. соляной кислотой, которая удаляет амин и карбоксизащитные группы.
Промежуточное соединение XI можно взаимодействовать также с сульфитионом. Реакция бром-промежуточного соединения XI с сульфитом натрия в смеси органического растворителя с водой дает соединение формулы I, где R3 является SO3H. Реакцию обычно проводят в смеси органического растворителя с водой, такой как этанол/вода, при температуре перегонки растворителя. Амино- и карбоксизащитные группы можно потом удалить обработкой 6 н. соляной кислотой.
Соединения формулы I, где W является S, используются для получения соединений формулы I, в которых W является SO, или SO2. Обычно соединение формулы I, в котором W является S, обрабатывают окислителем до получения соответствующих соединений, в которых W является SO или SO2. Подходящим окислителем для этой реакции является 3-хлорнадбензойная кислота. Окисление обычно проводят в полярном органическом растворителе, таком как метиленхлорид. Соединения формулы I, в которых W является SO, получают путем обработки соответствующего соединения формулы I, в котором W является S, окислителем при температуре приблизительно от -78 до -30oC. Обычно реакция завершается через 1 -4 часа. Соединения формулы I, в которых W является SO2, получают путем обработки соответствующего соединения формулы I, в котором W является S или SO, окислителем при температуре от приблизительно равной комнатной до 50oC. Предпочтительно проводить окисление при комнатной температуре в метиленхлориде при избыточной концентрации 3-хлорнадбензойной кислоты. Реакция обычно завершается через восемнадцать часов.
Вторую группу соединений формулы I, в которых R3 представляет собой CO2H или CONHSO2R8, W является (CH2)n, n = 0, Y и Z являются CH = CH, или Y и Z являются CH2, получают в соответствии со схемой 3.
Промежуточное соединение VIII взаимодействует с реагентом Уиттинга, а продукт этой реакции подвергают гидролизу до получения 6-формил-промежуточного соединения XII. Это соединение взаимодействуют с реагентом Хенера-Эммонса до поучения ненасыщенного соединения XIII. Промежуточное соединение XIII можно восстановить и/или подвергнуть изменениям с применением стандартных химических способов.
Конкретно, соединение VIII взаимодействуют с реагентом Уиттинга формулы Ph3PCHOCH3 до получения промежуточного соединения XI, где R7 является водородом, а R11 - метокси. Эту реакцию обычно осуществляют путем обработки метоксиметилтрифенилфосфонийхлорида сильным основанием, таким как бис(триметилсилил)амид, получая илид, который затем взаимодействуют в полярном органическом растворителе, таком как сухой тетрагидрофуран, с промежуточным соединением VIII. Эту реакцию обычно проводят при температуре от 0oC до около 25oC. Реакция завершается приблизительно через 30 мин при 0oC. Затем промежуточное соединение IX преобразуют в 6-формил-промежуточное соединение XII путем обработки кислотой, разбавленной водой. Подходящей кислотой для этой реакции является разбавленная соляная кислота, такая как 1 н. соляная кислота. Реакцию обычно проводят при комнатной температуре в течение от двух до восьми часов.
Затем промежуточное соединение XII взаимодействуют с реагентом Хенера-Эммонса до получения соединений формулы XIII. Этот реагент Хенера-Эммонса имеет общую формулу (CH3CH2O)POCH2R12, где R12 представляет собой защищенную карбоксильную группу, такую как этоксикарбонил или бензилоксикарбонил, циано, тетразол, триазол или тиодиазол. Эту реакцию осуществляют путем обработки соответствующего диэтилфосфоната сильным основанием, таким как гидрид натрия, до получения натриевой соли фосфоната, которую затем взаимодействуют с органическим растворителем, таким как сухой тетрагидрофуран, до получения соединения формулы XIII. Эту реакцию обычно осуществляют при температуре от 0 до 25oC. Реакция завершается через 0,5 - 4 часа при комнатной температуре.
Затем промежуточное соединение XIII восстанавливают до получения промежуточного соединения XIV. Предпочитаемым способом такого восстановления промежуточного соединения XIII является каталитическое гидрирование, предпочтительно, в присутствии палладия на углероде или платины на углероде в инертном растворителе. Подходящие инертные растворители: этанол и этилацетат. Если защитной карбоксильной группой является бензильная группа, то эту группу удаляют в процессе каталитического гидрирования. После этого промежуточное соединение XIV можно преобразовать в соединение формулы I, где R1 и R2 являются водородом, путем освобождения от защиты кислотного и азотного фрагментов. Обычно этого достигают обработкой промежуточного соединения XIV 6 н. соляной кислотой. Предпочитаемый способ: нагрев промежуточного соединения в 6 н. соляной кислоте с обратным холодильником в течение приблизительно 18 часов.
Промежуточное соединение XIV, где R12 представляет собой защитную карбоксильную группу, можно использовать для получения соединений формулы I, в которых R3 является CONHSO2R8. Реакция карбоксильного промежуточного соединения XIV с карбонилдиимидазолом, с последующей добавкой замещенного амина, дает соответствующий замещенный амид. Реакцию карбокси-промежуточного соединения XIV и 1,1'-карбонилдиимидазола предпочтительно проводить в безводном органическом растворителе, таком как сухой тетрагидрофуран, при температуре перегонки растворителя. Затем продукт этой реакции обрабатывают замещенным амином в присутствии основания. Одним из примеров подходящего замещенного амина является метансульфонамид. Подходящие для этой реакции основания включают N,N-диизопропилэтиламин, коллидин и 1,8-диазабицикло[5,4,0]-ундек-7-ен. Эту реакцию желательно проводить при комнатной температуре в течение около 6 - 20 часов. Затем защитную группу на 3-карбоксильной группе удаляют, обрабатывая амидпромежуточное соединение XIV 1 н. гидроксидом натрия. Эту реакцию проводят при комнатной температуре в течение около 18 часов. Защитную группу на кольцевом азоте удаляют, обрабатывая амид-промежуточное соединение иодотриметилсиланом в полярном органическом растворителе, таком как хлороформ. В данном способе освобождения от защиты желательно использовать 6 н. соляную кислоту, что позволит сохранить амидную группу.
Карбокси-промежуточное соединение XIV можно использовать также для получения дополнительных соединений по настоящему изобретению, например соединений формулы I, где R3 представляет собой CO2H, CONHSO2R8 или тетразол, W является (CH2)n, n = 2, а Y и Z являются CH2, что показано на схеме 4.
Обычно карбокси-промежуточное соединение XIV превращают в альдегид-промежуточное соединение XVI. Это соединение взаимодействует с реагентом Хенера-Эммонса до получения ненасыщенного промежуточного соединения XVII. Это соединение можно восстановить до получения соединения XVIII.
Конкретно, карбокси-промежуточное соединение восстанавливают до гидрокси-промежуточного соединения XV с помощью подходящего восстановителя, такого как боран-метилсульфид. Эту реакцию предпочтительно проводить в полярном органическом растворителе, таком как тетрагидрофуран, при температуре от 0 до 25oC.
Затем гидрокси-промежуточное соединение превращают в альдегид-промежуточное соединение XVI. Гидроксильную группу окисляют до альдегида с помощью известных реагентов. Одним из таких реагентов является комбинация оксалилхлорида и диметилсульфоксида. Обычно диметилсульфоксид и оксалилхлорид соединяют в органическом растворителе, таком как метиленхлорид, при -78oC до получения окислителя. Через 5 - 15 минут в охлажденный раствор окислителя добавляют спиртовой раствор. После этого смесь обрабатывают аминным основанием, таким как триэтиламин, и оставляют прогреваться до комнатной температуры.
После этого альдегид-промежуточное соединение XVI взаимодействуют с реагентом Хенера-Эммонса общей формулы (CH3CH2O)2POCH2R13, где R13 представляет собой защищенную карбоксильную группу или циано. Эту реакцию осуществляют путем обработки соответствующего диэтилфосфоната сильным основанием, таким как гидрид натрия, получая натриевую соль фосфоната, которую затем взаимодействуют в органическом растворителе, таком как сухой тетрагидрофуран, с промежуточным соединением XVI до получения промежуточного соединения XVII. Эту реакцию проводят при температуре от 0 до около 25oC в течение от 0,5 до 2 часов.
После этого промежуточное соединение XVII восстанавливают до получения соответствующего насыщенного аналога промежуточного соединения XVIII. Это восстановление проводят путем каталитического гидрирования, предпочтительно в присутствии палладия на углероде или платины на углероде. Это преобразование можно осуществить также методом восстановления солюционным металлом. Подходящим металлом для этой реакции является магний в полярном органическом растворителе, таком как метанол.
Циано-промежуточное соединение XVIII, где R13 представляет собой -CN, можно преобразовать в тетразол-промежуточное соединение. Цианосоединение взаимодействует с трибутиловоазидом при температуре около 80oC. Продукт можно отделить, но предпочтительно гидролизовать его прямо в соединение формулы I, в котором R1 и R2 являются водородом. Гидролиз проводят в 6 н. серной кислоте при температуре около 100oC в течение 2 - 24 часов до получения соединения формулы I, в котором R3 представляет собой тетразол. Этот способ образования тетразола из нитрилов пригоден также для превращения тиоцианата в тиотетразольное соединение.
Соединения формулы I, где Y и Z вместе являются C≡C , получают как показано на схеме 5.
Обычно 6-формил-промежуточное соединение XII преобразуют в дибромолефин-промежуточное соединение XIX, а затем в этинил-промежуточное соединение XX по способу, описанному в работе Кори и Фачс. Corey and Fuchs, Tetra. Lett. 36, 3769 - 3772 (1972). Этинил-промежуточное соединение изменяют, используя стандартные способы и способ, описанный здесь для получения соединений формулы I.
Конкретно, 6-формил-промежуточное соединение XII обрабатывают смесью трифенилфосфина с карбонтетрабромидом до получения дибромолефин-промежуточного соединения XIX. Реакцию проводят в метиленхлориде при температуре около 0oC в течение 5 - 60 мин. Согласно другому варианту, смесь цинковой пыли, трифенилфосфина и карбонтетрабромида в метиленхлориде оставляют реагировать при комнатной температуре в течение около 24 - 30 часов, а затем обрабатывают ее 6-формил-промежуточное соединение. Эту вторую реакцию проводят в метиленхлориде при температуре 20 - 30oC в течение 1 - 2 часов.
Затем дибромолефин-промежуточное соединение XIX преобразуют в этинил-промежуточное соединение XX. Обработка дибромолефинсоединения двумя эквивалентами n-бутиллития дает ацетилид лития XX, где R13 является Li. Это преобразование обычно проводят в полярном органическом растворителе, таком как тетрагидрофуран, при температуре от -78oC до 25oC. Ацетилид лития взаимодействуют с электрофилами, такими как метоксиметилхлорид, 5-бромметил-3-метоксиизоксазол, 3-дифенилметокси-4-иодометил-1,2,5-тиадиазол, двуокись углерода, тетразолдисульфид и т.п., до получения соединений формулы I. Например, ацетилид лития обрабатывают твердым CO2 при температуре от около -78oC до -60oC до получения промежуточного соединения XX в виде пропаргиловой кислоты, причем R13 представляет собой CO2H. Это соединение можно подвергнуть дальнейшим преобразованиям, как описано ниже.
В другом варианте, промежуточное соединение XX в виде пропаргиловой кислоты можно превратить в этилтетразол-промежуточное соединение XX, где R13 является тетразольной группой. Для этого преобразования карбоксильную группу превращают в карбоксамидную путем обработки хлорформиатом и аминовым основанием, после чего проводят обработку аммиаком. Типичные хлорформиаты включают метилхлорформиат, этилхлорформиат, бутилхлорформиат и т.п. Подходящие аминовые основания включают триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин и т. д. Реакцию обычно проводят при температуре от -10oC до около 25oC, предпочтительно при 0oC.
Этинилкарбоксамид-промежуточное соединение можно преобразовать в этинитрилсоединение, где R13 является CN. Стандартные способы дегидратации карбоксамидов в нитрилы описаны в работе Марча и Ларока. Marc, Advanced Organic Chemistry Reactions, Mechanism and Structure 932 - 933 (3-е изд., 1985); Compendium of Organic Synthetic Method; Larock, Comprehensive Organic Transformations (1989). Например, карбоксамид дегидратируют с помощью обработки фенилфосфоновым дихлоридом в пиридинметиленхлориде при температуре около 0oC. Нитрил-промежуточное соединение преобразуют в тетразолсоединение с помощью обработки трибутиловоазидом, как описано выше.
Соединения формулы I, где n и Y вместе являются C≡C, получают способом, аналогичным вышеописанному. Промежуточное соединение X, где R11 является гидроксильной группой, окисляют до альдегид-промежуточного соединения, где R11 представляет собой формил, используя способ, аналогичный вышеописанному. Затем этот альдегид превращают в дибромолефин и ацетилил лития, как описано выше.
Соединения формулы I, где Z представляет собой NR6, получают способом, представленным на схеме 6.
Промежуточное соединение VIII взаимодействует с амином до образования шиффова основания. Шиффово основание восстанавливают до получения промежуточного соединения XXI. Затем азот этого соединения ацилируют до получения соединения формулы XXII. Это соединение можно подвергнуть дальнейшим преобразованиям, как описано ниже, до получения соединений формулы I, где Z представляет собой NR6.
Конкретно, промежуточное соединение VIII взаимодействует с амином общей формулы R15NH2 до образования шиффова основания, которое восстанавливают до получения соединения XXI. Радикал R15 предпочтительно представляет собой -CH(R7)WR3, где W, R7 и R3 имеют вышеуказанные значения. Радикал R15 может быть предшественником группы формулы -CH(R7)WR3, такой как цианометил. Эту реакцию обычно проводят в полярном органическом растворителе, таком как этанол, в присутствии порошковых молекулярных сит  при комнатной температуре. Промежуточное соединение VIII соединяют с амином, а затем спустя 20 мин - 2 часа добавляют восстановитель. Подходящим восстановителем для этой реакции служит цианоборогидрид натрия.
при комнатной температуре. Промежуточное соединение VIII соединяют с амином, а затем спустя 20 мин - 2 часа добавляют восстановитель. Подходящим восстановителем для этой реакции служит цианоборогидрид натрия.
После этого промежуточное соединение XXI ацилируют до получения промежуточного соединения XXII. Подходящие ацилирующие агенты включают активированные сложные эфиры и смешанные ангидриды. В числе примеров активированных эфиров эфиры, образованные с такими группами, как паранитрофенил, 2,4-динитрофенол, трихлорфенол, 1-гидрокси-1Н-бензотриазол и 1-гидрокси-6-хлор-1Н-бензотриазол. Примером смешанного ангидрида является смесь ангидридов муравьиной и уксусной кислот. Например, амино-промежуточное соединение XXI обрабатывают ацилирующим агентом в полярном органическом растворителе, таком как тетрагидрофуран, при температуре от 25oC до температуры перегонки растворителя.
Алкильную группу, выражаемую радикалом R15, можно подвергнуть химическим изменениям до получения соединений формулы I. Например, если R15 представляет собой цианометил, то обработка промежуточного соединения XII трибутилоловоазидом по вышеописанному способу позволит получить тетразолилметиловое производное. Карбоксильную защитную группу и защитную группу на кольцевом азоте селективно удаляют обработкой 1 н. гидроксида натрия и иодотриметилсилана, как описано выше, до получения соединений формулы I, в которых R1 и R2 представляют собой водород.
Соединения формулы I, где Y является NR4, получают по способу, аналогичному описанному выше. Обычно эти соединения получают путем взаимодействия 6-формил-промежуточного соединения XII с амидом до образования шиффова основания, которое потом восстанавливают. Подходящие для этого амины включают амины общей формулы R3WNH2, где R3 имеет вышеуказанное значение, W является CH2)n, n = 0, 1 или 2, или предшественник группы формулы R3W NH2.
Конкретно, 6-формил-промежуточное соединение XII взаимодействует с аминоацетонитрилом до образования соответствующего шиффова основания. Эту реакцию обычно проводят в полярном органическом растворителе, таком как этанол или метанол, в присутствии порошковых молекулярных сит  . Затем шиффово основание восстанавливают подходящим восстановителем, таким как цианоборгидрид натрия. Аминогруппу можно ацилировать, как описано выше, до получения соединений формулы I, где R4 является ацильной группой. Нитрил можно преобразовать либо в тетразол, либо в карбоксильную группу, как описано.
. Затем шиффово основание восстанавливают подходящим восстановителем, таким как цианоборгидрид натрия. Аминогруппу можно ацилировать, как описано выше, до получения соединений формулы I, где R4 является ацильной группой. Нитрил можно преобразовать либо в тетразол, либо в карбоксильную группу, как описано.
Соединения формулы I, в которых Z представляет собой CHR7, а Y - CH2, получают по способу, показанному на схеме 7.
Обычно енолтрифлат-промежуточное соединение XXIII взаимодействуют с альфа, бета-ненасыщенным карбонильным соединением или альфа, бета-ненасыщенным нитрилом в присутствии бис(трифенилфосфин)палладий (II) хлорида до получения ненасыщенного промежуточного соединения XXIV. Подходящие альфа, бета-ненасыщенные карбонильные соединения включают альфа, бета-ненасыщенные кетоны, эфиры, альдегиды и амины. Промежуточное соединение XXIV при желании можно восстановить до получения промежуточного соединения, из которого получают соединения формулы I.
Конкретно, промежуточное соединение VIII преобразуют в енолтрифлат-промежуточное соединение XXIII путем обработки сильным основанием, после чего проводят трифлиляцию. Для этой реакции подходят такие основания, как бис(триметилсили)амид лития и диизопропиламид лития. Полученный енолят-анион ацилируют либо ангидридом трифторметансульфокислоты, либо N-фенилтрифторметансульфонимидом. Это преобразование обычно проводят в полярном апротонном растворителе, таком как тетрагидрофуран.
Затем енолтрифлат-промежуточное соединение XXIII алкилируют до получения ненасыщенного промежуточного соединения XXIV. Эту реакцию обычно осуществляют следующим образом: трифлат обрабатывают замещенным альфа, бета-ненасыщенным нитрилом общей формулы HCR16CHCN, где R16 представляет собой водород, алкил C1-C4, фенил или замещенный фенил, в присутствии бис(трифенилфосфин)палладий(II)хлорида. Реакцию проводят в дегазованном полярном органическом растворителе, таком как диметилформамид, в присутствии аминного основания, такого как триэтиламин, при температуре приблизительно 70 - 80oC.
Промежуточное соединение XXIV можно восстановить до соответствующего насыщенного аналога - промежуточного соединения XXV. Промежуточное соединение XXIV можно восстановить путем восстановления солюционным металлом, таким как Mg и MeOH, или путем каталитического гидрирования. Предпочитаемый способ - каталитическое гидрирование. Подходящие катализаторы для этой реакции включают 5% палладий на углероде и 5% платину на углероде; предпочтительно, палладий на углероде. Восстановление проводят под давлением водорода в около 60 - 100 фунтов/кв.дюйм при температуре от около 25oC до температуры перегонки растворителя. Подходящие растворители для этой реакции включают полярные органические растворители, такие как этанол и этилацетат.
Затем циано-промежуточное соединение XXV превращают в соединение формулы I, как описано выше. Это промежуточное соединение можно преобразовать в соединение формулы I, в котором R3 - тетразол, с помощью реакции с трибутилоловоазидом, как описано выше. В другом варианте, циано-промежуточное соединение XXV можно превратить в соединение формулы I, в котором R3 представляет собой CO2H. Эти соединения получают с помощью реакции циано-промежуточных соединений с концентрированной соляной кислотой, которая также удаляет защитные группы.
Соединения формулы I, в которых Y представляет собой S, SO, или SO2, а Z является CH2, получают способом, показанным на схеме 8.
Обычно промежуточное соединение VIII преобразуют в 6-формилпромежуточное соединение (R7 - водород) или 6-ацил-промежуточное соединение, а затем восстанавливают до гидрокси-промежуточного соединения XXVI. Это соединение преобразуют в бром-промежуточное соединение XXVII, которое взаимодействуют с различными тиолами до получения соединений формулы I, где Y является S. Эти соединения можно окислять до получения соединений формулы I, где Y является SO или SO2. Конкретно, промежуточное соединение VIII преобразуют в промежуточное соединение IX, где R11 представляет собой метокси, как описано выше. Промежуточное соединение обрабатывают разбавленной водой кислотой в полярном органическом растворителе, таком как тетрагидрофуран, до получения промежуточного соединения XII. Это соединение восстанавливают до образования гидроксиметил-промежуточного соединения XXVI. Подходящие восстановители включают борогидрид натрия и цианоборогидрид натрия. Восстановление обычно проводят в полярном органическом растворителе, таком как этанол или изопропанол, при температуре от 2 до 25oC. Гидроксиметил-промежуточное соединение XXVI затем преобразуют в бромид, используя стандартные химические реакции. Например, гидроксиметил-промежуточное соединение обрабатывают трифенилфосфином и бромом в полярном органическом растворителе, таком как метиленхлорид, после чего добавляют аминное основание, такое как пиридин, получая бром-промежуточное соединение XXVII.
Промежуточное соединение XXVIII взаимодействует с соединением общей формулы R17SH до получения промежуточного соединения XXVIII. Радикал R17 может представлять собой группу формулы -(CH2)nR3, где n и R3 имеют вышеуказанные значения, или химический предшественник этой группы. Группа R17 предпочтительно является группой формулы -(CH2)nR3, где n и R3 имеют вышеуказанные значения. Например, промежуточное соединение XXVIII обрабатывают 1Н-1,2,4-триазол-3-тиолом и аминным основанием до получения промежуточного соединения XXVIII. Подходящие аминные основания включают триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, пиридин и коллидин. Реакцию проводят при температуре 50 - 100oC в течение 4 - 18 часов. Промежуточное соединение XXVIII можно обработать водным раствором кислоты до получения соединений формулы I, где R1 и R3 являются водородом.
В другом варианте промежуточное соединение XXVIII можно обработать окислителем до получения соединений формулы I, где Y представляет собой SO или SO2. Подходящим окислителем является 3-хлорнадбензойная кислота. Окисление проводят в полярном органическом растворителе, таком как метиленхлорид. Соединения формулы I, в которых Y является SO, получают следующим образом: соответствующее соединение формулы I, где Y является S, обрабатывают окислителем при температуре от -78oC до около -30oC. Реакция обычно завершается через 1 - 4 часа. Соединения формулы I, где Y является SO2, получают путем обработки соответствующего соединения формулы I, где Y является S, или SO, окислителем при температуре от комнатной до около 50oC. Предпочтительно проводить окисление при комнатной температуре в метиленхлориде с 3-хлорнадбензойной кислотой в избыточной концентрации. Реакция обычно завершается через 18 часов.
Соединения формулы I, в которых Y является кислородом, получают способом, указанным на схеме 9.
Обычно промежуточное соединение VIII преобразуют в 6-гидроксиметил-промежуточное соединение XXVI. Промежуточное соединение алкилируют различными алкилгалогенидами. Полученные продукты преобразуют в соединения формулы I, используя стандартные способы синтеза и описанный здесь способ.
Конкретно, гидроксиметил-промежуточное соединение XXVI ,полученное вышеописанным способом, алкилируют соединением общей формулы R18X'. Группа R18 предпочтительно является группой формулы -(CH2)nR3, где n и R3 имеют вышеуказанные значения. Группа X' представляет собой хлор, бром, иод, метилсульфонилокси или толилсульфонилокси. Предпочтительно X' представляет собой бром, хлор, или иод. Примеры таких алкилирующих агентов включают 5-бромметил-3-метоксиизоксазол, 3-дифенилметокси-4-иодометил-1,2,5-тиадиазол и т.п. В другом варианте группа R18 может представлять собой предшественник группы формулы -(CH2)nR3, такой как цианометил, метоксиэтоксиметил и метоксиэтил. Например, соединение XXVI и аминное основание в полярном органическом растворителе обрабатывают алкилирующим агентом. Подходящие аминные основания включают N,N-диизопропилэтиламин, триэтиламин, пиридин и коллидин. Примером подходящего алкилирующего агента является хлорметилметилэфир. Реакцию обычно проводят при температуре от 0 до около 10oC. Продукт этой реакции преобразуют в цианометил-промежуточное соединение XXIX путем последовательной обработки триметилсилилцианидом и бортрифторэтератом. Реакцию проводят в полярном органическом растворителе, таком как метиленхлорид, при температуре от 0 до около 10oC.
Полученное цианометил-промежуточное соединение XXIX можно преобразовать в тетразолилметил-промежуточное соединение путем обработки трибутилоловоазидом, как описано выше. Согласно другому варианту цианометил-промежуточное соединение может быть преобразовано в карбоксиметил-промежуточное соединение с помощью обработки цианометил-промежуточного соединения кислотой. Предпочтительно, в качестве кислоты используют разбавленную водой кислоту, такую как соляная кислота, и реакцию проводят при температуре перегонки раствора. Это позволяет также удалить защитные группы с карбоксильной группы и кольцевого азота, благодаря чему получают соединение формулы I, где R1 и R2 являются водородом.
Вышеприведенные примеры используют для получения соединений формулы I в виде рацематов или отдельных энантиомеров. Если синтез начинают с промежуточного соединения VIII в виде рацемата, то продуктами обычно являются рацематы. Однако, если синтез начинают с промежуточного соединения (-)-VIIIb, то продукты обычно представляют собой отдельные энантиомеры. Относительную конфигурацию атома углерода в положении C-6 кольца у соединений, где Z является CH2, можно контролировать как показано на схемах 10 и 11.
Схема 10 иллюстрирует способ получения соединения формулы XXVIa с указанной относительной стереохимической конфигурацией. Чистый энантиомер VIII преобразуют в ненасыщенное промежуточное соединение IX, используя стандартный способ Уиттинга. Продукт селективно преобразуют в промежуточное соединение XXVIa путем гидроборирования с последующим окислением.
Конкретно, промежуточное соединение VIII взаимодействуют с реагентом Уиттинга, таким как метилтрифенилфосфонийбромид, до получения промежуточного соединения IX, где R11 является водородом. Эту реакцию обычно проводят вышеописанным способом, обрабатывая бромид фосфония сильным основанием, таким как бис(триметилсилил)амид натрия, до получения илида. Этот илид затем взаимодействуют в полярном органическом растворителе, таком как сухой тетрагидрофуран, с соединением VIII до получения метиленового производного формулы IX. Эту реакцию обычно проводят при температуре от 0oC до температуры перегонки растворителя. Если реакцию проводят при слабом молярном избытке фосфониевой соли, то она обычно завершается через 6 часов.
Затем промежуточное соединение IX преобразуют (селективно) в промежуточное соединение XXVIa. Предпочтительный способ такого преобразования - гидроборирование с последующим окислением. Подходящим реагентом для гидроборирования является боран-метил-сульфид. Это гидроборирование обычно проводят в полярном органическом растворителе, таком как тетрагидрофуран, при температуре от 0oC до комнатной. Реакция обычно завершается через 2 - 4 часа. Затем продукт гидроборирования окисляют до промежуточного соединения XXVIa. Подходящим окислителем является перекись водорода. Окисление проводят следующим образом: реакционную смесь, в которой идет реакция гидроборирования, обрабатывают перекисью водорода и полученную смесь перемешивают при комнатной температуре. Реакция обычно завершается через 1 - 2 часа.
В другом варианте конфигурацию атома углерода в положении C-6 кольца изменяют до образования соединения формулы I, где атом водорода в положении C-6 связан поперечной связью с атомом водорода в положении C-4a как показано на схеме 11.
Обычно чистый энантиомер VIII взаимодействует с реагентом Уиттинга до образования промежуточного соединения IX. Затем это соединение подвергают стереоселективному гидролизу до получения 6-формил-промежуточного соединения XIIa.
Конкретно, промежуточное соединение VIII взаимодействует с реагентом Уиттинга формулы Ph3PCHOCH3 до получения промежуточного соединения IX, в котором R11 является метокси. Эту реакцию проводят вышеописанным способом. Затем промежуточное соединение IX превращают в промежуточное соединение XIIa, применяя обработку разбавленной кислотой. Подходящей кислотой для такой обработки является разбавленная соляная кислота, такая как 1 н. соляная кислота. Реакцию обычно проводят при температуре 60oC в полярном органическом растворителе, таком как ацетонитрил, в течение от двух до восьми часов.
В другом варианте промежуточное соединение XIIa можно получить из промежуточного соединения XXVIa. Чистый энантиомер спирта XXVIa окисляют до соответствующего альдегида, используя стандартную реакцию Сверна или другие реагенты на диметилсульфоксиде. Mancuso, Huang, and Swern. T. Org. Chem, 43, 2480 - 2482 (1978); Epstein and Sweat, Chem. Rev. 67, 247 - 260 (1067); и Smith Leenay, Lin, Nelson, and Ball, Tetr. Lett. 29, 49 - 52 (1988). Альдегид, в котором водород при C - 6 образует цис-изомер с атомом водорода в голове моста, обрабатывают слабым основанием до получения соединения XIIa. Подходящие мягкие основания включают третичные амины, включая триэтиламин и N, N-диизопропилэтиламин, а также бикарбонат натрия. Предпочтительно, чтобы эпимеризация водорода C-6 происходила в процессе окисления способом Сверна.
Еще одним аспектом настоящего изобретения являются соединения формулы:
J v Q
где
J представляет собой группу формулы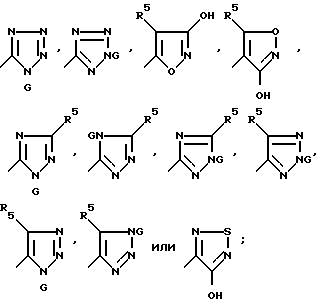
где
Q представляет собой CHP7R+(Ph)3X-, CHR7PO(Ph)2, CR7MSiR'3, CH(SiR'3)PO(OR')2 или CH2SnR'3;
R' представляет собой алкил C1-C6 или фенил;
G представляет собой азотозащитную группу или водород;
M представляет собой Li+ или Mg+X-;
X- представляет собой бромид, хлорид, иодид, тетрафторборат или гексафторфосфат;
R5 и R7 имеют вышеуказанные значения.
Обычно получение этих соединений производят в два этапа. Эти этапы включают синтез соответствующего гетероцикла и взаимопревращение функциональной группы. Гетероциклы, такие как тетразол, гидроксизамещенные изоксазолы, триазолы и гидроксизамещенные тиадиазолы, получают при помощи стандартных способов синтеза. Получение этих гетероциклов описано ниже. Взаимопревращение функциональной группы состоит в том, что группу, не принимающую участие в синтезе гетероцикла, превращают в трифенилфосфоний, триалкилстаннан, фосфонат, литий, реактив Гриньяра или дифенилфосфиноксидную группу. Примеры таких взаимопревращений описаны ниже. Триалстаннан и дифенилфосфиноксидные группы можно получить до синтеза гетероцикла.
Тетразольное кольцо получают при помощи стандартных способов синтеза. См. Butler, "Recent Advances in Tetrazole Chemistry" Advances in Heterocyclic Chemistry, 21, 354 - 361 (1977). Тетразол образуется в результате реакции нитрила с азидным реагентом в инертном растворителе. Подходящие азидные реагенты включают неорганические азиды, такие как азид натрия, азид лития или азид аммония, и такие реагенты, как 1,1,3,3-тетраметилгуанидиназид и трибутилоловоазид. Реакцию проводят с использованием азида лития или аммония в диметилформамиде, азида натрия в диглиме и N,N-диметилэтаноламингидрохлорида или трибутилоловоазида в инертном растворителе, таком как диметоксиэтан, толуол или тетрагидрофуран. Присутствие хлорида алюминия активизирует реакцию при использовании неорганических азидов. В другом варианте осуществляют взаимодействие нитрида с азидом натрия, соляной кислотой и триалкиламином. Подходящие для этой реакции триалкиламины включают триэтиламин, N,N-диизопропилэтиламин и N-метилморфолин. Проводят реакцию при температуре перегонки реакционной смеси или близкой к ней. В указанных условиях реакция завершается через 1 - 3 дня. Предпочитаемый способ преобразования нитрила в тетразол - реакция нитрила со смесью азида натрия и трибутилоловохлорида. Эту реакцию осуществляют в органическом растворителе, таком как толуол, при температуре от 75 до 100oC. Реакция обычно требует 20 - 30 часов.
Гидроксизамещенные изоксазолы получают известным способом. См. Кочетков и Соколов "Последние достижения в химии изоксазолов" Advances in Heterocyclic Chemistry, 2, 365 - 278 (1963). Обычно бета-кетоэфир или бета-кетокислоту конденсируют гидроксиламином до образования гидроксизамещенного изоксазола. Katritzky and Oksne, Proc. Chem. Soc. 387 - 388 (1961); Jacobsen, Can. I. Chem. 62, 1940, (1984). Бета-кетоэфир превращают в соответствующее оксимпроизводное путем обработки гидроксиламином и концентрированной соляной кислотой. Эту реакцию можно проводить в спиртовом сорастворителе, таком как метанол или этанол, предпочтительно использовать спирт, который соответствует эфирной группе как органический сорастворитель. Реакцию проводят при температуре от 0oC до температуры перегонки растворителя, предпочтительно при температуре от 25 до 50oC. После получения оксима, оксим-промежуточное соединение подвергают циклизации до образования изоксазольного кольца. Эту циклизацию проводят следующим образом: оксим-промежуточное соединение обрабатывают 2 н. гидроксидом натрия при pH 10. Реакцию обычно проводят при температуре от 0 до 50oC, предпочтительно при комнатной температуре. Органический сорастворитель, такой как ацетонитрил, можно использовать в тех случаях, когда оксим-промежуточное соединение нерастворимо в воде. Эта реакция занимает от 10 до 24 часов.
В другом варианте гидроксизамещенные изоксазолы получают с помощью реакции пропаргилового спирта с дибромформальдоксимом, с последующим гидролизом бромгруппы. Сначала пропаргиловый спирт взаимодействует с дибромформальдоксимом до получения циклоаддукта. Эту реакцию проводят при температуре от 15 до 50oC, предпочтительно при комнатной температуре. Подходящим для этой реакции растворителем является этилацетат. Затем циклоаддукт, 3-бром-5-гидроксиметилизоксазол, обрабатывают водным основанием до гидролиза бромгруппы. Подходящие основания включают гидроксид натрия и гидроксид калия, гидроксид калия является предпочтительным. Реакцию проводят в смеси воды и водорастворимого органического растворителя, такого как метанол. Реакцию предпочтительно проводить при температуре перегонки смеси растворителей.
Триазолы получают с использованием стандартных способов синтеза. См. Gilchrist and Gymer "1,2,3-T Advances in Heterocyclic Chemistry 16, 33 - 63 (1974); Advances in Heterocyclic Chemistry 18, 106 (1975). 1,2,3-триазолы получают с помощью реакции азида с альфа-дикетоном или замещенным ацетиленом. Подходящие азидные реагенты включают неорганические азиды, такие как азид натрия, азид лития или азид аммония. Реакцию осуществляют с использованием азида лития или аммония в диметилформамиде, и азида натрия в диглиме и N, N-диметилэтаноламингидрохлориде. В другом варианте 1,2,3-триазолы получают с помощью реакции первичного амина с N-толилсульфониламидразоном, имеющим две уходящие группы. См. Sakai, Bull. Chem. Soc. Ipn. 59, 179 (1986). Подходящие для этой реакции растворители включают спиртовые растворители, такие как метанол. Реакцию проводят при температуре от -10 до около 25oC, предпочтительно при 0oC. 1,2,4-триазолы получают путем реакции ацилгидразина либо с гидразином, либо с N-замещенным гидразином. Эту реакцию обычно проводят в смеси ацетонитрила с триэтиламином при температуре от 0oC до 50oC, предпочтительно при комнатной температуре. В другом варианте 1,2,4-триазолы получают путем реакции амидразона с ацилгидразином в сильном основании. Подходящим для этой реакции сильным основанием является алкоксид металла, такой как метилат натрия или трет-бутоксид калия. Эта реакция протекает в безводных условиях, таких как смесь сухого этанола и параксилена. Это преобразование обычно проводят при комнатной температуре. Francis, Tetr. Lett, 28, 5133 (1987).
Тетразолы и триазолы можно защитить с помощью защитных нитрогрупп, таких как тритил, бензил, трет-бутил, трет-бутилдиметилсилил и трифенилсилил. Защищенные соединения получают в результате реакции тетразола или триазола с тритил-, бензил-, трет-бутилдиметилсилил, трифенилсилилгалогенидом, таким как хлорид или бромид, в присутствии основания. Подходящие основания включают третичные амины, такие как триэтиламин, N,N-диизопропилэтиламин, пиридин, бикарбонат натрия, гидроксид натрия и гидроксид калия. Подходящими растворителями являются вода и полярные органические растворители, такие как диметилформамид, ацетонитрил и метиленхлорид. Трет-бутильную группу получают в результате реакции либо тетразола, либо триазола с изобутиленом в сильном основании. Подходящие кислоты включают серную и толуолсульфокислоту.
1,2,5-тиадиазолы получают с помощью известных способов синтеза. См. Advances in Heterocyclic Chemistry 30, 65 - 66 (1982).Обычно 1,2,5-тиадиазолы получают в результате реакции соответствующего диамина или альфа-аминоамида с двуххлористой серой или тионилхлоридом. Weinstock, Tetr. Lett. 1263 (1966). Подходящим растворителем для этой реакции является сухой диметилформамид. Реакцию проводят при температуре от -10 до около 25oC, предпочтительно при 0oC. После того как завершается добавка двухлористой серы или тионилхлорида, реакционной смеси позволяют прогреваться до комнатной температуры.
Второй этап включает преобразование функциональной группы, которая не вступала в реакцию синтеза гетероцикла, в трифенилфосфоний, триалкилстаннан, фосфонат, литий, реактив Гриньяра или дифенилфосфиноксид. Для этого преобразования подходят следующие функциональные группы: гидрокси, бром, хлор или защищенная гидрокси.
Соединения трифенилфосфония получают с помощью стандартных способов синтеза. Эти соединения получают в результате реакции трифенилфосфина с гетероциклоалкилбромидом, хлоридом или иодидом. Подходящими растворителями для этой реакции являются органические растворители, такие как ацетонитрил, толуол, ксилен и диметилформамид. Реакцию можно проводить в отсутствие растворителя. Обычно реакцию проводят при температуре от 80oC до 150oC или при температуре перегонки растворителя. В другом варианте соединения трифенилфосфония получают в результате реакции гетероциклического гидроксиалкильного соединения с трифенилфосфингидробромидом. Эту реакцию проводят в растворителе, пригодном для азиотропного удаления воды, таком как толуол или ксилол, при температуре выше температуры кипения азиотропа. Реакцию можно проводить в отсутствие растворителя. Обычно реакция длится от 1 до 5 часов.
Триалкилстаннаны получают с помощью стандартных способов с применением органометаллических соединений. Триалкилстаннаны получают в результате реакции гетероциклоалкилгалогенидов, таких как бромид или хлорида, с трибутилоловохлоридом и цинком. Knochel, Organometallics, 9, 3053 (1990). Эту реакцию проводят в полярном органическом растворителе, таком как метиленхлорид, при температуре от около -70oC до около 5oC. В другом варианте гетероциклоалкилбромид можно обработать магнием или комплексом Гриньяра, после чего проводят обработку триалкилоловохлоридом. Delmond. I. Organomet. Chem. 26, 7 (1971). Реакцию алкилбромида с магнием можно проводить в полярном органическом растворителе, таком как эфир или тетрагидрофуран, при температуре от около -10 до 50oC. Предпочтительно проводить реакцию в сухом эфире. Реакцию промежуточного соединения Гриньяра с трибутилоловохлоридом проводят при температуре перегонки растворителя.
Группу дифенилфосфиноксида получают с применением стандартных способов синтеза. Гетероциклоалкилгалогенид, тозилат или мезилат взаимодействуют с литийдифенилфосфином. Литийдифенилфосфиновый реагент получают в результате реакции дифенилфосфина с n-бутиллитием. Реакцию проводят в полярном органическом растворителе, таком как эфир или тетрагидрофуран, при температуре от -20 до 0oC. Brown, I. Chem. Soc. Perk. Trans 11, 91 (1987). Промежуточное гетероциклоалкилдифенилфосфиновое соединение окисляют до фосфиноксида, используя разбавленную известь (гидрохлорид натрия).
Альфа-силилфосфонатные соединения получают с помощью стандартных способов синтеза. Aboujaoude, Synthesis 934-937 (1986). Гетероциклоалкилгалогенид, такой как бромид или хлорид, взаимодействует с триэтилфосфатом до получения соответствующего фосфоната. Реакцию проводят в органическом растворителе, таком как толуол, при температуре перегонки растворителя. Затем промежуточный фосфонат обрабатывают сильным основанием, таким как литийдиизопропиламид, после чего добавляют триметилсилилхлорид до получения альфа-силилфосфоната. Реакцию силирования проводят в сухом органическом растворителе, таком как эфир или тетрагидрофуран, при температуре от -78oC до около -50oC.
Реактивы Петерсона, альфа-силиловый реактив Гриньяра и соединения лития получают при помощи стандартных способов синтеза. Эти реактивы получают в результате реакции гетероциклоалкил-альфа-силилгалогенида, такого как бромид или хлорид, с металлическим магнием или металлическим литием. Boaz. I. Med. Chem. 14, 1971. Для получения реактива Гриньяра предпочтительно использовать альфа-силилхлорид. Sexton. I. Org. Chem. 56, 698 (1991). Для получения гетероциклоалкиллитиевого соединения предпочтительно использовать альфа-силилбромид. Реакцию проводят в сухом органическом растворителе, таком как тетрагидрофуран или сухой эфир, при температуре от 0oC до комнатной.
Конкретно, [2-(1(2)Н-тетразол-5-ил)этил]трифенилфосфонийбромид получают из 3-гидроксипропионитрила. Гидроксинитрил сначала преобразуют в соответствующее тетразольное соединение. Предпочитаемый способ такого преобразования нитрила в тетразол - реакция нитрила со смесью азида натрия с трибутилоловохлоридом. Эту реакцию проводят в органическом растворителе, таком как толуол, при температуре от 75 до 100oC. Реакция обычно длится в течение от 20 до 30 часов. Продукт этой реакции - 5-(2-гидроксиэтил)тетразол - взаимодействуют с трифенилфосфингидромидом до получения [2-(1(2)Н-тетразол-5-ил)этил] трифенилфосфонийбромида. Эту реакцию проводят в растворителе, пригодном для азеотропного удаления воды, которая образуется в процессе реакции, таком как толуол или ксилол, при температуре выше температуры кипения азеотропа. Реакцию можно проводить и без удаления воды. Если реакцию проводят в ксилоле, то температура реакции должна быть от около 120 до 150oC. Реакция обычно протекает в течение от одного до пяти часов.
Соединения формулы I, где Z представляет собой CH, получают способом, представленным на схеме 12.
Обычно промежуточное соединение VIII взаимодействует с реактивом Уиттинга, реактивом Хенера-Эммонса или вариантами этих реактивов, до получения соединения формулы XXX. Конкретно, промежуточное соединение VIII взаимодействует с реактивом общей формулы QCH2R19, где Q имеет вышеуказанное значение, а R19 представляет собой -YWR3. Соединение формулы QCH2R19 предпочтительно представляет собой соединение формулы JCH2Q, где J и Q имеют вышеуказанные значения. Например, Q представляет собой CH2P(Ph)
Соединение формулы XXX можно восстановить до получения соединения формулы I, где Z представляет собой CH2. Если R9 представляет собой алкоксикарбонил, а R10 - алкил C1-C6, то это восстановление можно провести стереоселективно до получения соединения формулы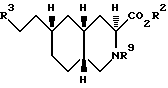
Восстановление для селективного изменения стереохимической структуры соединения можно осуществить путем каталитического гидрирования. Предпочтительным катализатором является оксид платины в количестве 5 мас.%. Подходящими растворителями для этого восстановления являются этанол и этилацетат. Восстановление предпочтительно проводить под давлением водорода в одну атмосферу и при температуре 20 - 30oC. Предпочтительно проводить восстановление при комнатной температуре. Реакция обычно завершается через 24 часа. Когда R3 представляет собой тетразольную группу, стереоселективность составляет около 6 : 1 (6-H бета : 6-H альфа).
Соединения формулы I, где R1 представляет собой алкил C1-C10, арилалкил или ацил, получают из соответствующих соединений, где R1 является водородом. Соединения, где R1 представляет собой алкил C1-C10 или арилалкил, получают путем восстановительного алкилирования. Обычно альдегид или кетон соответствующей алкильной C1-C10 группы или арилалкильной группы взаимодействуют с соединением формулы I, где R1 является водородом, до образования промежуточного шиффова основания. Реакцию проводят в полярном органическом растворителе, таком как метанол, или в смеси полярных органических растворителей, такой как смесь диметилформамида с метанолом, при температуре от 25 до 100oC. Реакцию образования шиффова основания предпочтительно проводить при температуре от около 25 до 30oC в течение от 30 минут до 2 часов в метаноле.
Затем промежуточное шиффово основание восстанавливают, предпочтительно без выделения, до получения алкильных C1-C10 или арилалкильных производных. Восстановление шиффова основания можно осуществить с помощью химического восстановителя, такого как цианоборгидрид натрия. Реакцию проводят в полярном органическом растворителе, таком как метанол, или в смеси полярных органических растворителей, такой как смесь диметилформамида с метанолом. Восстановление проводят при температуре от 25 до 100oC в течение 1 - 5 часов. Восстановление предпочтительно проводить с использованием цианоборгидрида натрия в избыточной концентрации в метаноле при температуре 25 - 40oC в течение 1 - 2 часов.
Соединения формулы I, где R1 представляет собой ацил, получают в результате реакции соединения формулы I, где R1 - водород, с активированным сложным эфиром нужной ацильной группы. Термин "активированный эфир" означает сложный эфир, который выполняет карбоксильную функцию ацилирующей группы, вступающей в реакцию до соединения с аминогруппой декагидроизохинолинового кольца. Предпочитаемым активированным эфиром является 2,4,5-трихлорфенилэфир. Реакцию проводят в полярном органическом растворителе, таком как диметилформамид или тетрагидрофуран, при температуре от 25 до 110oC в течение 1 - 5 часов. Реакцию образования ацильных производных соединений формулы I предпочтительно проводить при температуре от около 30 до 70oC в течение от 2 до 4 часов.
Соединения формулы I, где R2 представляет собой замещенный алкил, циклоалкил или арилалкил, получают из соответствующих соединений, где R2 является водородом. Эти соединения обычно получают с помощью стандартных способов синтеза. Например, соединение формулы I, где R1 является водородом, взаимодействуют с арилалкильным галогенидом, таким как бензилбромид, в присутствии основания до получения производных арилалкилэфира. Подходящие основания для этого преобразования включают третичные алкиламины, такие как триэтиламин, N-диизопропилэтиламин, N-метилморфолин, пиридин и коллидин, а также карбонат натрия. Реакцию осуществляют в органическом растворителе, таком как тетрагидрофуран, ацетонитрил и диметилформамид. В другом варианте соединение формулы I, где R1 является водородом, взаимодействуют с замещенным алкильным, циклоалкильным или арилалкильным спиртом в присутствии кислоты до получения соответствующего эфира. Обычно эту реакцию проводят при избыточной концентрации спирта в присутствии концентрированной серной кислоты.
Соединения формулы I по настоящему изобретению являются антагонистами аминокислот-медиаторов возбуждения. В частности, эти соединения являются селективными для подкласса АМРА рецепторов аминокислот-медиаторов возбуждения. Таким образом, еще одним аспектом настоящего изобретения является способ блокирования рецепторов аминокислот АМРА у млекопитающих, который включает в себя назначение млекопитающему фармацевтически эффективной дозы соединения формулы I, необходимой для уменьшения возбуждающей аминокислотной нейротрансмиссии.
Используемый термин "фармацевтически эффективная доза" обозначает дозу соединения по изобретению, необходимую для блокирования рецепторов аминокислот-медиаторов возбуждения АМРА. Конкретная доза соединения по изобретению, естественно, будет определяться конкретными условиями: какое соединение вводится, способ введения, особенности состояния, при котором вводится соединение, и т.п.
Соединение может вводиться различными способами, включая оральный, ректальный, чрескожный, подкожный, внутривенный, внутримышечный и интраназальный способы. Кроме того, соединения могут вводиться путем длительной инфузии. Типичная дневная доза будет содержать примерно от 0,01 мг/кг до 20 мг/кг активного компонента. Предпочтительной будет суточная доза 0,05 - 10 мг/кг, более предпочтительно - 0,1 - 5 мг/кг.
Чрезмерная или неадекватная стимуляция нейропередачи, осуществляемой аминокислотами-медиаторами возбуждения, влияет на самые различные физиологические функции. Мы считаем, что с помощью соединения формулы I по изобретению можно лечить различные неврологические расстройства у млекопитающих, возникающие вследствие церебральной недостаточности, которая является результатом операций на сердце с применением АИК, "ударов", церебральной ишемии, травмы спинного мозга и головы, перинатальной гипоксии, остановки сердца и гипогликемических повреждений нейронов. С помощью соединения по изобретению возможно лечить различные хронические неврологические заболевания, такие как болезнь Альцгеймера, хорея Гентингтона, боковой амиотрофический склероз, СПИД-индуцированную дементацию, повреждения глаз и ретинопатию, идиопатическую и вызванную применением лекарственных препаратов болезнь Паркинсона.
Настоящее изобретение включает в себя способ лечения этих болезней с помощью введения пациентам эффективной дозы соединения формулы I.
Соединение формулы I по настоящему изобретению также может использоваться для лечения целого ряда других неврологических расстройств у млекопитающих, связанных с глутаминовой дисфункцией: мышечных спазмов, судорог, головных болей при мигрени, недержания мочи, психозов, толерантности к препаратам опия и синдрома отмены, беспокойства, рвоты, отека мозга, хронических болезней и поздней дискинезии. Соединение также может быть использовано в качестве анальгетика.
Для демонстрации селективной ингибирующей активности соединения формулы I по настоящему изобретению в отношении рецепторов аминокислот-медиаторов возбуждения субтипа АМРА были проведены следующие эксперименты. С помощью метода связывания с радиолигандом с использованием [3H]CGS19755, [3H]АМРА и [3H] КА соединения формулы I были протестированы на предмет исследования их способности ингибировать связывание NMDA, АМРА и каиновой кислоты в рецепторах. Во всех тестах на связывание с радиолигандом использовали мужские особи крыс Spraque-Dawley. Для определения аффинности NMDA-рецепторов использовали метод вытеснения специфической связи [3H]CGS 19755 (10 нМ) в обработанных Тритоном-X синаптосомальных мембранах переднего мозга у крыс. Неспецифическое связывание определяли, используя 10 мкМ 1-глутамат. Образцы инкулировали в ледяной бане в течение 30 минут. Связанные лиганды отделяли от свободных с помощью быстрой фильтрации через WHATMAN GF/B (стекловолоконные фильтры). Murphy et al. British I. Pharmacol. 95, 932 - 938 (1988).
Исследование связывания с каинатом было проведено с использованием отмытых синаптосомальных мембран из переднего мозга крыс, как описано в Simon et al. Simon et al, I. Neurochem. 26, 141 - 147 (1976). Меченый тритием каинат (5 нМ) добавляли к 50 мМ трис-HCl буфера (pH 7,4 при 4oC), содержащего 200 - 300 мкг/мл тканевых белков. Образцы инкулировали в течение 30 мин в ледяной ванне, а затем быстро фильтровали, используя клеточный харвестер Брандела и фильтры WHATMAN GF/C. Фильтры промывали дважды 3 мл холодного буфера. Неспецифическое связывание определяли, используя 100 мкМ немеченого каината. Связывание [3H] АМРА (5 нМ) изучали, используя необработанные мембраны переднего мозга крыс в присутствии 100 нМ KSCN, как описано в Nielson et al. Nielson et al, Eur. I. Med. Chem. Chim. Ther, 21, 433 - 437 (1986). Неспецифическое связывание определяли с помощью 10 мкМ немеченого АМРА. Концентрация соединения формулы I, которая ингибировала 50% связывания (IC50, средняя ± средняя ошибка средней, n = 3) была рассчитана с помощью линейной регрессии данных по замещению с помощью формулы Хилла, как описано в работе Bennett, Neurotransmitter Receptor Binding, 57 - 90 (1978). Результаты, полученные с помощью теста связывания радиолигандов, показаны в таблицах 1 и 2.
Для изучения селективности и активности соединений формулы I как антагонистов АМРА исследовали деполяризацию участков коры головного мозга крыс с помощью техники, подобной описанной в работе Harrison and Simmonds, Bri. I. Pharmacol, 84, 381 - 391 (1984). Суть методики заключается в следующем: 4 мл аликвоты NMDA (40 мкМ), АМРА (40 мкМ) и каината (10 мкМ) наносили (2 мл/мин) на серое вещество коры головного мозга с интегралом 15 - 20 минут до получения стабильного ответа. Ткань подвергали воздействию (в течение 15 мин) различных концентраций соединения формулы I, а затем проводили повторное тестирование агонистами. Значение IC50 высчитывали из линейной регрессии логарифма кривой доза-эффект. Значение каждой точки высчитывалось из, по крайней мере, трех наблюдений на различных средах от нескольких животных. Результаты этих исследований показаны в таблицах 3 и 4.
Результаты исследований показывают, что соединение формулы I обладает селективной аффинностью к АМРА ионотропных глутаматным рецепторам. Метод связывания радиолигандов является предпочтительным для различения селективности АМРА и КА. Соединения формулы I, в частности соединения 1, 4, 5, 13, 15, 18 и 19, селективно замещали 3H - АМРА со значением IC50 меньше, чем 15 мкМ (табл. 1). Метод изучения участков коры головного мозга является предпочтительным для различения селективности АМРА и NMDA. Этот метод также позволяет различить активность вещества как агониста или антагониста. Показано, что соединения формулы I, в частности 1, 4, 5, 13 и 19, являются селективными антагонистами АМРА рецепторов (табл. 3). Результаты также показывают, что предпочтительны стереоизомеры соединений формулы I с S у атома C-3 (табл. 3 и 4).
До назначения соединений по изобретению из них предпочтительно приготовить лекарственные средства. Таким образом, еще один аспект настоящего изобретения - приготовление фармацевтических препаратов, включающих соединение формулы I и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Фармацевтические препараты по изобретению приготавливают известными способами, с использованием хорошо известных ингредиентов, выпускаемых промышленностью. При изготовлении композиции по настоящему изобретению активный ингредиент обычно смешивают с носителем, разбавляют носителем или заключают внутрь носителя, который может иметь форму капсулы, саше, бумажной или иной упаковки. Если носитель служит разбавителем, то он может представлять собой твердый, полужидкий или жидкий материал, который служит связующим, наполнителем или средом для активного ингредиента. Композиции могут быть в виде таблеток, пилюль, порошков, лепешек, саше, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей, мазей, содержащих, например, до 10 мас.% активного соединения, мягких и твердых желатиновых капсул, свечей, стерильных растворов для инъекций, стерильных расфасованных порошков.
Примеры подходящих носителей, наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, смолу, акацию, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, металцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Фармацевтические препараты могут включать дополнительно смазывающие вещества, смачивающие вещества, эмульгаторы и суспендирующие агенты или ароматизаторы. Композиции по изобретению могут входить в состав таких фармацевтических препаратов, которые обеспечат быстрое продолжительное или замедленное высвобождение активного ингредиента после назначения этих препаратов пациентам, для чего используются известные способы.
Предпочтительно, чтобы композиции по изобретению входили в состав фармацевтических препаратов в виде отдельных доз, причем каждая доза должна содержать от 5 до 500 мг, обычно от 25 до 300 мг активного ингредиента. Термин "в виде отдельных доз" предполагает некую физическую единицу, пригодную в качестве отдельной дозы для человека или других млекопитающих, причем каждая единица содержит определенное количество активного материала, которое, согласно расчетам, должно оказывать требуемое терапевтическое действие, в сочетании с подходящим фармацевтическим носителем. Нижеследующие примеры рецептур являются чисто иллюстративными и не ограничивают рамки настоящего изобретения.
Рецептура 1. Твердые желатиновые капсулы изготавливают с использованием следующих ингредиентов, мг/капсула:
6-[2-(1(2)Н-тетразол-5-ил)-этил] декагидроизохинолин-3- карбоновая кислота - 250
Крахмал, высушенный - 200
Стеарат магния - 10
Всего - 460
Вышеперечисленные ингредиенты смешивают и наполняют ими твердые желатиновые капсулы по 460 мг.
Рецептура 2. Таблетки изготавливают с использованием следующих ингредиентов, мг/таблетка:
6-[2-(1(2)Н-тетразол-5-ил)-2-тиаэтил] декагидроизохинолин-3- карбоновая кислота - 250
Целлюлоза, микрокристаллическая - 400
Двуокись кремния, пыль - 10
Стеариновая кислота - 5
Всего - 665
Компоненты смешивают и прессуют до получения таблеток массой 665 мг каждая.
Рецептура 3. Изготавливают аэрозольный раствор, содержащий следующие компоненты, мас.%:
6-[2(3-гидроксиизоксазол-5-ил)-этил] декагидроизохинолин-3- карбоновая кислота - 0,25
Этанол - 29,75
Диспергатор "Propellant 22" (хлордифторметан) - 70,00
Всего - 100,00
Активное соединение смешивают с этанолом и смесь добавляют в "Propellant 22", охлажденный до -30oC, и затем помещают в наполняющее устройство. После этого нужное количество подают в контейнер из нержавеющей стали и разбавляют оставшимся диспергатором. После этого к контейнеру прикрепляют клапан.
Рецептура 4. Таблетки, каждая из которых содержит 60 мг активного ингредиента, изготавливают следующим способом, мг:
6-[(1(2-4)-Н-1,2,4-триазол-5-ил)сульфонилметил] - декагидроизохинолин-3-карбоновая кислота - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон - 4
Карбоксиметилнатриевый порошок - 4,5
Стеарат магния - 0,5
Тальк - 1
Всего - 150
Активный ингредиент, крахмал и целлюлозу пропускают через сито N 45 U.S. и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученным порошком, который затем пропускают через сито N 14 U.S. Полученные гранулы высушивают при 50oC и пропускают через сито N 18 U.S. Затем карбоксиметилнатриевый порошок, стеарат магния и тальк, предварительно пропущенные через сито N 60 U. S, добавляют в гранулы, которые после перемешивания прессуют в машине для изготовления таблеток до получения таблеток массой 150 мг каждая.
Рецептура 5. Капсулы, содержащие по 80 мг медикамента каждая, изготавливают следующим способом, мг:
6-[2-(1(2)Н-тетразо-5-ил)-1-метилэтил] декагидроизохинолин-3-карбоновая кислота - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито N 45 и полученной смесью заполняют твердые желатиновые капсулы по 200 мг каждая.
Рецептура 6. Свечи, содержащие по 225 мг активного ингредиента каждая, изготавливают следующим способом, мг:
6-[2-(1(2)Н-тетразол-5-ил)-1-фенилэтил] декагидроизохинолин-3-карбоновая кислота - 225
Насыщенные глицериды жирной кислоты - 2000
Всего - 2225
Активный ингредиент пропускают через сито N 60 U.S. и суспендируют в насыщенных глицеридах жирной кислоты, расплавленных при минимальном нагреве. Затем смесь разливают в формы для свечей на 2 г и оставляют охлаждаться.
Рецептура 7. Суспензии, каждая из которых содержит 50 мг медикамента на дозу в 5 мл, изготавливают следующим способом, мг:
6-[2-(1(2)Н-тетразол-5-ил)-2-тиаэтил] - декагидроизохинолин-3-карбоновая кислота - 50
Карбоксиметилнатриевая целлюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты - 0,10
Ароматизатор - по желанию
Краситель - по желанию
Очищенная вода до объема - 5 мл
Медикамент пропускают через сито N 45 U.S. и смешивают с карбоксиметилнатриевой целлюлозой и сиропом до образования однородной пасты. Раствор бензойной кислоты, ароматизатор и краситель разбавляют некоторым количеством воды и добавляют в смесь при перемешивании. Затем доливают воду до нужного объема.
Рецептура 8. Раствор для внутривенных вливаний изготавливают следующим способом:
6-[2-(3-гидроксиизоксазол-5-ил)этил] -декагидроизохинолин-3- карбоновая кислота - 100 мг
Маннит - 100 мг
5 н. гидроксид натрия - 200 мкл
Очищенная вода до объема - 5 мл
Нижеследующие примеры иллюстрируют соединения по настоящему изобретению и способы их синтеза. Примеры ни в коей мере не ограничивают рамки настоящего изобретения. Все эксперименты проводили под избыточным давлением сухого азота. Тетрагидрофуран перед использованием отгоняли от натрия. Все прочие растворители и реактивы использовали в том виде, в каком их получали. Спектры протонного ядерного магнитного резонанса (1H ЯМР) получали на спектрометре "GE QE-300" при 300,15 МГц или спектрометре "Bruker AM-500" при 500 МГц. Там, где указано, добавляли небольшие количества KOD, чтобы улучшить обработку образцов ЯМР в D2O. Хроматографическое разделение на жидкостном хроматографе "Waters Prep 500" проводили с применением линейного градиента гексана к растворителю, указанному в тексте. Наблюдение за реакциями до их завершения проводили с использованием тонкослойной хроматографии. Тонкослойную хроматографию проводили с использованием пластин "E.Merck Kieselgel 60 F254", 5 • 10 см, толщиной 0,25 мм. Определение пятен производили с применением ультрафиолетового и химического способов обнаружения (пластины погружали в раствор церийаммониймолибдата (75 г аммониймолибдата и 4 г церий (IV) сульфата в 500 мл 10% водного раствора серной кислоты), а затем нагревали на горячей пластине). Экспресс-хроматографию производили согласно Still et al. Still, Kahn, and Mitra, I. Org. Chem. 43, 2923 (1978). Элементарные анализы на углерод, водород и азот проводились на Control Egnipment Corporation 440 Elemantal Analyzer". Температуры плавления определяли в открытых стеклянных капиллярах на приборе Галленкампа для определения температуры плавления с использованием горячей воздушной бани и корректировке не подвергали.
Пример 1. 6-Гидрокситетрагидроизохинолин-3-карбоновая кислота (V).
Суспензию d,1-m-тирозина (1,91 кг) в разбавленной соляной кислоте (76 мл конц. HCl, 11,5 л воды) нагревали до 55 - 60oC и обрабатывали формальдегидом (1,18 л). Нагрев до 55 - 70oC продолжали в течение 2 часов, затем реакционную смесь охлаждали до 3 - 10oC в течение 2 часов. Полученную смесь фильтровали, фильтрат промывали деионизированной водой и ацетоном. Фильтровальный осадок высушивали в вакуумной печи при 55 - 60oC до получения 1,88 кг искомого соединения.
1H ЯМР (D2O/KOD): δ 6,75 (d, 1H), 6,35 (d, 1H), 6,30 (s, 1H), 3,77 (d, 1H), 3,69 (d, 1H), 3,26 (dd, 1H), 2,79 (dd, 1H), 2,60 (dd, 1H).
Расчетные данные по C10H11NO3 • 0,85 H2O, %: C 57,60; H 6,13; N 6,71. Обнаружено, %: C 57,70; H 6,43; N 6,69.
Пример 2. Этил-6-гидрокси-2-метоксикарбонилтетрагидроизохинолин-3-карбоксилат (VI).
В смесь соединения из примера 1 (91,2 г) в этаноле (455 мл) добавляли концентрированную серную кислоту (27,5 мл) в течение двух минут. После начальной экзотермической реакции раствор нагревали с обратным холодильником в течение 16 часов. Полученный раствор охлаждали на ледяной водяной бане и добавляли раствор карбоната калия (130,5 г) в воде (130,5 мл). В этот раствор добавляли метилхлорформиат (36,5 мл) с такой скоростью, чтобы pH было выше 6,9 и температура была ниже 14oC. Спустя еще два часа реакционную смесь разделяли между этилацетатом (250 мл) и водой (500 мл). Слои отделяли и водный слой экстрагировали двумя порциями этилацетата (100 мл каждая). Органические слои соединяли и концентрировали под вакуумом до получения твердого осадка. Осадок кристаллизовали следующим образом: растворяли в дефлегмирующем этаноле (180 мл), разбавляли раствор этанола водой (360 мл) и перемешивали полученную смесь при 4oC в течение 24 часов. Кристаллическое твердое вещество собирали фильтрацией и высушивали в вакуумной печи (40oC, 23 часа) до получения 93,9 г искомого соединения.
1H ЯМР (CDCl3): δ 6,95 (m, 1H, 6,67 (d, 1H), 6,61 (s, 1H), 5,76 (s, 1H), 5,06 и 4,85 (m, 1H), 4,65 (dd, 1H), 4,48 (d, 1H), 4,05 (m, 2H, 3,78 и 3,73 (s, 3H), 3,11 (m, 2H), 1,11 (t, 3H) (дублирование из-за амидных ротамеров).
Расчетные данные по C14H17NO5, %: C 60,21; H 6,14; N 5,02. Обнаружено, %: C 60,49; H 6,24; N 4,98.
Пример 3. Этил-2-метоксикарбонил-6-оксодекагидроизохинолин-3- карбоксилат (VIII).
Вариант 1.
A. Получение этил-6-гидрокси-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата (VII).
В смесь 3% родия на окиси алюминия (6,9 г) в этилацетате (350 мл) добавляли соединение из примера 2 (69,03 г). Сосуд закрывали герметически и атмосферу азота заменяли водородом. Реакционную смесь нагревали до 85oC под давлением в 100 фунтов/кв.дюйм в течение 23 часов. Добавляли дополнительную порцию родия на окиси алюминия (1,4 г) и возобновляли нагрев при пониженном давлении еще в течение двух часов. Катализатор удаляли фильтрацией, а фильтрат, содержащий искомое соединение, использовали на следующем этапе.
B. Получение этил-2-метоксикарбонил-6-оксодекагидроизохинолин-3 -карбоксилата (VIII).
В раствор хлорида рутения (III) (69 мг) в воде (9,8 мл) добавляли фильтрат из примера 3A. Полученную двухфазную смесь охлаждали на ледяной бане и обрабатывали раствором периодной кислоты (69 г) в воде (26,9 мл). Раствор периодной кислоты добавляли с такой скоростью, чтобы температура реакционной смеси была ниже 7,8oC. После добавки периодной кислоты водяную баню удаляли и реакционной смеси позволяли прогреться до комнатной температуры. Спустя 1 1/4 часа водную фазу удаляли и органическую фазу промывали двумя порциями воды (50 мл каждая). Органическую фазу концентрировали досуха под вакуумом до получения 67,7 г искомого соединения в виде масла.
Вариант 2.
Получение этил-6-гидрокси-2-метоксикрабонилдекагидроизохинолин-3-карбоксилата (VII) и этил-2-метоксикарбонил-6-оксодекагидроизохинолин-3-карбоксилата (VIII).
Смесь 3% родия на углероде+ (1,0 кг) и соединения из примера 2 (13,2 кг) в этилацетате (67 л) гидрировали под давлением водорода в 100 фунтов/кв.дюйм при около 85oC. Спустя 23 часа реакционную смесь охлаждали до комнатной температуры и удаляли катализатор фильтрацией. Осадок катализатора промывали дополнительным количеством этилацетата (10 л), затем соединяли этилацетатные фильтраты.
Часть раствора этилацетата из предыдущего абзаца концентрировали под вакуумом до получения 3,295 г бесцветного масла, которое представляло собой смесь кетона C-6 и спиртов C-6. Эту смесь разделяли экспресс-хроматографией на силикагеле, элюировали линейным градиентом метиленхлорида к метиленхлоридэтилацетату (9:1), затем этилацетатом, до получения двух продуктов. Фракции, содержащие первый продукт, соединяли и концентрировали под вакуумом до получения 1,18 г соединения VIII. Фракции, содержащие второй продукт, соединяли и концентрировали под вакуумом до получения 1,36 г соединения VII.
Пример 4. (3S,4a,S8aR)-(-)-этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилат ((-)-VIIIb).
A. Получение 2-метоксикарбонил-6-оксодекагидроизохинолин-3- карбоновой кислоты.
Соединение из примера 3B (1,913 кг) добавляли в 21% раствор этилата натрия (509 г) в этаноле (8 л). Полученный раствор разогревали с обратным холодильником в течение шести часов, затем оставляли остывать до комнатной температуры в течение 24 часов. Этот раствор обрабатывали 5 н. раствором едкого натра (2,4 л) и оставляли при температуре от 25 до 40oC в течение двух часов. Реакционную смесь концентрировали под вакуумом до удаления этанола. Остаток экстрагировали двумя порциями трет-бутилметилового эфира (5 л каждая) и с помощью добавки концентрированной соляной кислоты (1,7 л) доводили pH до приблизительно 1,5 -2,5. Искомое соединение экстрагировали из водного раствора этилацетатом (4 • 3 л). Соединенные экстракты этилацетата обрабатывали "FLORISI L" (960 г) и сульфатом натрия (960 г). Фильтрат этилацетата, содержащий искомое соединение, использовали на следующем этапе без дальнейшей очистки.
B. Получение (3S, 4aS, 8aR)-(-)-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилат-альфа-метилбензиламиновой соли.
В фильтрат этилацетата из примера 4A добавляли R-(+)-альфа-метилбензиламин при температуре от около 25oC до около 30oC в течение одного часа. Полученную суспензию оставляли при комнатной температуре на 24 часа, после чего собирали осадок фильтрацией. Твердый материал промыли несколькими порциями этилацетата, пока промывочный раствор не стал бесцветным. Твердый остаток от фильтрования высушивали в вакуумной печи при температуре около 45 - 50oC. Этот материал повторно суспендировали в 10 объемах этилацетата при температуре около 45 - 50oC в течение четырех часов, после чего раствор оставляли охлаждаться до температуры окружающей среды, и удалили твердый материал фильтрацией. Твердые материалы высушивали под вакуумом при температуре около 45 - 50oC до получения 1,092 г искомого соединения.
[α]D = -57,0oC (c = 1, H2O)
Расчетные данные по C20H28N2O5, %: C 63,81; H 7,50; N 7,44. Обнаружено, %: C 63,87; H 7,33; N 7,33.
C. Получение (3S, 4aS, 8aR)-(-)-этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилата ((-(-VIIIb).
Смесь соединения из примера 4B (50 г) и ацетонитрила (250 мл) обрабатывали триэтиламином (26,8 г) и бромистым этилом (73 г). Полученную смесь нагревали до температуры перегонки, вызывая растворение реактивов. Через 1 - 2 часа реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток обрабатывали этилацетатом (250 мл). Полученную смесь фильтровали и твердые продукты промывали дополнительным этилацетатом. Фильтрат экстрагировали 3 н. соляной кислотой, высушивали MgSO4, фильтровали и концентрировали под вакуумом до получения 34,9 г искомого соединения.
[α]D = -51,3oC (c = 1, CH2Cl2)
Расчетные данные по C14H21NO5, %: C 59,35; H 7,47; N 4,94. Обнаружено, %: C 59,11; H 7,20; N 4,90.
D. Получение (3R4aR8aS)-(+)-этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилата ((+)-VIIIb).
Искомое соединение получали из рацемической смеси из примера 4A, используя способ, описанный в примерах 4B и 4C, с использованием S-альфа-метилбензиламина.
Пример 5. (3SR, 4aS R, 8aRS)-этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилата ((+)-VIIIb).
A. Получение этил-6-гидрокси-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 2 (158,9 г) и 5% рутения на окиси алюминия (80 г) в этаноле (1760 мл) гидрировали под давлением 2000 фунтов/кв.дюйм. Через 16 часов при температуре около 180oC охлажденную реакционную смесь фильтровали через CELITE и фильтрат концентрировали под вакуумом. Остаток разбавляли этилацетатом. Смесь фильтровали через CELITE и концентрировали под вакуумом до получения 156,7 г искомого соединения.
B. Получение этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилата (VIII).
Раствор соединения из примера 5A (156,7 г) в метиленхлориде (300 мл) добавляли в смесь хлорхромата пиридиния (260,5 г) и порошковых молекулярных сит  в метиленхлориде (1400 мл), каковую перемешивали в течение часа перед добавкой спирта. Через два часа реакционную смесь разбавляли эфиром и фильтровали через слои CELITE и силикагеля. Твердые продукты промывали эфиром и соединенные эфирные растворы концентрировали под вакуумом. Остаток растворяли в эфире, фильтровали через CELITE и силикагель, затем фильтрат концентрировали под вакуумом до получения 128,8 г смеси VIIIa и VIIIb (VIIIa:VIIIb = 78:22).
в метиленхлориде (1400 мл), каковую перемешивали в течение часа перед добавкой спирта. Через два часа реакционную смесь разбавляли эфиром и фильтровали через слои CELITE и силикагеля. Твердые продукты промывали эфиром и соединенные эфирные растворы концентрировали под вакуумом. Остаток растворяли в эфире, фильтровали через CELITE и силикагель, затем фильтрат концентрировали под вакуумом до получения 128,8 г смеси VIIIa и VIIIb (VIIIa:VIIIb = 78:22).
C. Получение (3SR, 4aSR, 8aRS)-(+)-этил-2-метоксикарбонил-6- оксодекагидроизохинолин-3-карбоксилата (VIIIb).
Раствор смеси из примера 5B (128,8 г) в этаноле (1000 мл) обрабатывали раствором гидрида натрия (1,82 г) в этаноле (100 мл), а полученную смесь нагревали до температуры перегонки. Через 1 1/2 часа смесь оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток растворяли в метиленхлориде/ эфире и промывали 10% водным раствором бисульфата натрия. Водную фазу экстрагировали эфиром, органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюировали линейным градиентом гексана к 25% этилацетат/гексана до получения 106,9 г смеси соединений VIIIa и VIIIb (VIIIa:VIIIb = 13:87). Рекристаллизация этой смеси из эфира дала 67,0 г искомого соединения. Температура плавления: 78 - 79oC.
1Н ЯМР (DMSO) δ : 4,76 (d, 1H), 4,124 (g, 2H), 3,80 (d, 1H), 3,61 (s, 3H), 3,21 (bd, 1H), 2,65 (dd, 1H), 2,43 (dt, 1H), 2,19 (m, 1H), 2,14 (m, 2H), 1,98 (ddd, 1H), 1,85 (m, 1H), 1,75 (m, 1H), 1,65 (dt, 1H), 1,20 (t, 3H).
Расчетные данные по C14H21NO5, %: C 59,35; H 7,47; N 4,94. Обнаружено, %: C 59,62; H 7,61; N 4,97.
Пример 6. (3SR, 4aRS, 6RS, 8aRS)-6-(2-карбоксиэтил)- декагидроизохинолин-3-карбоновая кислота (II).
A. Получение этил-2-метоксикарбонил-6-(метоксиметилен) декагидроизохинолин-3-карбоксилата.
В суспензию (метоксиметил)трифенилфосфонийхлорида (37,8 г), предварительно промытую тетрагидрофураном и пентаном, затем высушенную под вакуумом при комнатной температуре, в тетрагидрофуране (150 мл) при температуре 0oC, добавляли 1 М раствор бис(триметилсилил)амида натрия в тетрагидрофуране (100 мл). Через 30 минут этот раствор добавляли в раствор соединения из примера 5C (20,2 г) в тетрагидрофуране (100 мл) при температуре около 0oC. Реакцию останавливали добавлением воды (150 мл). Полученный раствор разбавляли эфиром (150 мл) и органическую фазу отделяли и промывали водой (150 мл). Соединенные водные фазы экстрагировали эфиром (2 • 150 мл). Соединенные органические растворы промывали рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом досуха. Остаток суспендировали в этилацетате/гексане (1:1), и полученную суспензию перемешивали при комнатной температуре в течение 10 мин, фильтрат концентрировали под вакуумом. Твердый материал суспендировали в дополнительном количестве этилацетата/гексана (1: 1), перемешивали при комнатной температуре и фильтровали. Фильтрат этилацетат/гексана соединяли и концентрировали под вакуумом. Продукт очищали силикагель-хроматографией на "WATERS PREP 500LC", используя градиент 8-L гексана к 25% этилацетат/гексану до получения 20,9 г искомого соединения.
B. Получение этил-6-(этил-2-карбоксиэтенил)-2-метоксикарбонил декагидроизохинолин-3-карбоксилата.
Соединение из примера 6A (3,33 г) добавляли в смесь тетрагидрофурана (58 мл) и 1н. соляной кислоты (85 мл). Через четыре часа раствор разделяли на порции между метиленом (125 мл) и водой (100 мл). Органическую фазу удаляли и водный раствор экстрагировали дополнительным количеством метиленхлорида (2 • 65 мл). Соединенные органические растворы промывали насыщенным бикарбонатом натрия (50 мл), высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток растворяли в тетрагидрофуране (10 мл) и использовали в реакции, описанной в следующем абзаце, без дальнейшей очистки.
В суспензию гидрида натрия (0,6 г), предварительно промытого гексаном, в тетрагидрофуране (20 мл) добавляли триэтилфосфонацетат (3,36 г). Через 30 мин при комнатной температуре эту смесь обрабатывали раствором тетрагидрофурана из предыдущего абзаца. Еще через полчаса при комнатной температуре реакционную смесь обрабатывали поочередно водой (25 мл) и эфиром. Органическую фазу удаляли и водную фазу экстрагировали эфиром (2 раза). Соединенные органические экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток использовали на последующем этапе без дальнейшей очистки.
C. Получение этил-6-(этил-2-карбоксиэтил)-2-метоксикарбонил- декагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 6B (3,9 г) и 5% палладия на углероде (1,0 г) в этаноле (96 мл) гидрировали под давлением водорода в 60 фунтов/кв.дюйм при комнатной температуре. Через три часа смесь фильтровали через "CELITE", твердый остаток на фильтре "CELITE" промывали эфиром и соединяли фильтраты, которые затем концентрировали под вакуумом. Остаток очищали хроматографией на колонке "LOBARC", элюировали 25% этилацетатом/ гексаном до получения фракций, содержащих диастереоизомеры 3SR, 4aRS, 6RS, 8aRS (третья фракция) и 3SR, 4aRS, 6SR, 8aRS (первая фракция), а также смесь этих диастереоизомеров (вторая фракция). Общий выход искомого соединения составил 3,22 г.
D. Получение (3SR, 4aRS, 6RS, 8aRS)-6-(2-карбоксиэтил) декагидроизохинолин-3-карбоновой кислоты.
Изомер 3SR, 4aRS, 6RS, 8aRS из примера 6C добавляли в 6 н. соляную кислоту (50 мл) и полученную смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь оставляли остывать до комнатной температуры, затем концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50x8" (100 - 200 меш) элюировали 10% пиридин/вода до получения 0,63 г искомого соединения. Температура плавления: 190 - 191oC.
Расчетные данные по C13H21O4 • H2O, %: C 57,12; H 8,48; N 5,12. Обнаружено, %: C 57,14; H 8,30; N 5,07.
Пример 7. (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2)-Н-тетразол-5-ил)-2- тиэтил] декагидроизохинолин-3-карбоновая кислота (4) и (3SR, 4aRS, 6RS, 8aRS)-6-[2(1(2)Н-тетразол-5-ил)-2-тиаэтил] декагидроизохинолин-3- карбоновая кислота (5).
A. Получение этил-6-формил-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 6A (11,1 г) в тетрагидрофуране (125 мл) обрабатывали 1 н. соляной кислотой (160 мл). Через 4 3/4 часа реакционную смесь разбавляли водой (100 мл) и экстрагировали эфиром (3 раза). Органические экстракты соединяли, промывали насыщенным бикарбонатом натрия, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Этот материал использовали непосредственно на следующем этапе без дальнейшей очистки.
B. Получение этил-6-гидркосиметил-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 7A (10,62 г) в этаноле (108 мл) охлаждали до 0oC и обрабатывали натрийбороводородом (1,34 г). Через десять минут раствор концентрировали под вакуумом. Остаток разделяли на части между 10% бисульфатом натрия и метиленхлоридом. Органическую фазу удалили, а водную фазу экстрагировали дополнительным количеством метиленхлорида (3 раза). Органические экстракты соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток использовали на следующем этапе без дальнейшей очистки.
C. Получение этил-6-бромметил-2-метоксикарбонил-декагидроизохинолин-3-карбоксилата.
Раствор трифенилфосфина (14,05 г) в метиленхлориде (225 мл) обрабатывали бромом до устойчивого желтого цвета (приблизительно 8,56 г). Дополнительное количество трифенилфосфина добавляли до тех пор, пока раствор не становился бесцветным. Этот раствор обрабатывали раствором соединения из примера 7B (10,69 г) и пиридина (5,65 г) в метиленхлориде (185 мл). Спустя два часа при комнатной температуре реакционную смесь экстрагировали 10% бисульфатом натрия. Водный экстракт экстрагировали эфиром (3 раза). Эфирные экстракты соединяли с органической фазой, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток обрабатывали эфиром и осаждали трифенилфосфиноксидом, который удаляли фильтрацией. Фильтрат концентрировали под вакуумом и остаток растворяли в дополнительном количестве эфира, до выпадения в осадок оставшихся следовых количеств трифенилфосфиноксида. Смесь фильтровали, а фильтрат концентрировали под вакуумом. Остаток очищали силикагельной хроматографией на "WATERS PREP 500 LC", элюируя градиентом 8-L 10% этилацетата/гексана к 30% этилацетат/гексан, до получения фракций, содержащих диастереоизомеры 3SR, 4aRS, 6SR, 8aRS (третья фракция) и 3SR, 4aRS, 6RS, 8aRS (первая фракция) и смеси этих диастереоизомеров (вторая фракция). Общий выход искомого соединения составил 10,73 г.
D. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6-[2-(1(2)-Н-тетразол- 5-ил)-2-тиаэтил]-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор 3SR, 4aRS, 6SR, 8aRS-изомера из примера 7C (8,33 г), тиотетразола (2,58 г) и триэтиламина (4,65 г) в безводном ацетонитриле (70 мл) нагревали до 80oC. Спустя 20 часов, реакционную смесь разделяли на части между этилацетатом и 10% бисульфатом натрия. Фазы разделяли и водную фазу экстрагировали дополнительным количеством этилацетата (3 раза). Этилацетатные экстракты соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя уксусной кислотой/этилацетатом/толуолом (4:36:60). Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли метанолом и концентрировали под вакуумом, затем разбавляли хлороформом и концентрировали под вакуумом до получения 8,91 г искомого соединения.
E. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2)-Н-теразол-5-ил)- 2-тиаэтил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 7D (8,91 г) в 6 н. соляной кислоты (100 мл) нагревали до 90oC в течение трех часов. Затем раствор оставляли охлаждаться до комнатной температуры, фильтровали, а остаток с фильтра промывали водой и ацетоном. Твердые продукты высушивали под вакуумом при комнатной температуре в течение 18 часов до получения 5,36 г искомого соединения в виде хлористоводородной соли. Температура плавления 285oC.
Расчетные данные по C12H19N5O2S • HCl, %: C 43,71; H 6,09; N 20,98. Обнаружено, %: C 43,43; H 6,17; N 20,77.
F. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[1(2)Н-тетразол-5- илтиаметил]декагидроизохинолин-3-карбоновой кислоты.
Изомер 3SR, 4aRS, 6RS, 8aRS из примера 7C превращали в искомое соединение по способу, описанному в примерах 7D и 7E. Искомое соединение очищали ионообменной хроматографией на "DOWEX 50X8", элюировали 10% пиридин/вода. Температура плавления 257oC.
Расчетные данные по C12H19N5O2S, %: C 48,46; H 6,44; N 23,55; S 10,78. Обнаружено, %: C 48,21; H 6,55; N 23,25; S 11,08.
Пример 8. (3SR, 4aRS, 6RS, 8aRS)-6-[3-(1(20-Н-тетразол-5-ил)-3- типроп-1-ил] -декагидроизохинолин-3-карбоновая кислота (8) и (3SR, 4aRS, 6SR, 8aRS)-6-[3-(1(2)Н-тетразол-5-ил)-3-тиапроп-1-ил] - декагидроизохинолин-3-карбоновая кислота (9).
A. Получение этил-6-(бензилоксикарбонилметилен)-2- метоксикарбонил-декагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (3,11 г), предварительно промытого в гексане, в тетрагидрофуране (200 мл), обрабатывали раствором бензилдиэтилфосфонацетата (22,2 г) в тетрагидрофуране (50 мл). Через 30 минут полученный прозрачный раствор обрабатывали раствором соединения из примера 5C (20 г) в тетрагидрофуране (80 мл). Спустя 5 часов при комнатной температуре реакционный раствор обрабатывали водой (100 мл) и рассолом (100 мл). Органическую фазу удаляли, а водную фазу экстрагировали эфиром (2 • 100 мл). Органические фазы соединяли, промывали рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной хроматографией на "WATERS PREP 500 LC", элюируя градиентом 8-1 гексана к 50% этил/гексан до получения 26,3 г искомого соединения.
B. Получение этил-6-карбоксиметил-2- метоксикарбонилдекагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 8A (26,15 г) и 5% палладия на углероде (5 г) в этилацетате (270 мл) гидрировали при давлении водорода в 60 фунтов/кв.дюйм при комнатной температуре. Через четыре часа катализатор удаляли фильтрацией, а фильтрат концентрировали под вакуумом. Остаток (20,5 г) использовали на последующем этапе без дальнейшей очистки.
C. Получение этил-6-(2-гидроксиэтил)-2-метоксикарбонилдекагидроизохинолин-3- карбоксилата.
Раствор соединения из примера 8B (20,5 г) в тетрагидрофуране (200 мл) при температуре 0oC обрабатывали 2 М раствором боранметилсульфида в тетрагидрофуране (61 мл). Через четыре часа этот раствор тщательно обрабатывали насыщенным раствором бикарбоната натрия. Полученную смесь экстрагировали эфиром (300 мл), а эфирный экстракт промывали насыщенным раствором хлорида натрия. Раствор бикарбоната натрия экстрагировали эфиром (3 раза). Эфирные экстракты соединения высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 19,34 г искомого соединения в виде масла. Это соединение использовали на последующем этапе без дополнительной очистки.
D. Получение этил-6-(2-бромэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор трифенилфосфина (24,3 г) в метиленхлориде (300 мл) охлаждали до 0oC и обрабатывали бромом (14,9 г). Дополнительное количество трифенилфосфина добавляли до тех пор, пока раствор не стал бесцветным. Этот раствор обрабатывали раствором соединения из примера 8C (19,34 г) и пиридином (9,76 г) в метиленхлориде (225 мл). Через 15 минут при 0oC реакционный раствор экстрагировали 10% бисульфатом натрия (2 раза). Для того, чтобы растворить осадок, добавляли дополнительное количество воды, затем водный раствор промывали эфиром (2 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток разбавляли эфиром и удаляли фильтрацией осадок трифенилфосфиноксида. Эту процедуру повторяли дважды. Остаток очищали силикагельной хроматографией на "NATERS PREP 500 LC", элюируя линейным градиентом гексана к 30% этилацетат/гексан до получения фракций, содержащих диастереоизомеры: 3SR, 4aRS, 6RS, 8aRS (первая фракция) и 3SR, 4aRS, 6SR, 8aRS (третья фракция), а также смеси этих диастереоизомеров (вторая фракция). Фракции, содержащие изомер 3SR, 4aRS, 6RS, 8aRS, соединяли и концентрировали под вакуумом до получения 3,71 г. Фракции, содержащие изомер 3SR, 4aRS, 6SR, 8aRS, соединяли и концентрировали под вакуумом до получения 5,27 г.
E. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6-[3-(1(2)Н-тетразол-5- ил)-3-тиапроп-1-ил]-2-метоксикарбонилдекагидроизохинолин-3- карбоксилата.
Раствор изомера 3SR, 4aRS, 6SR, 8aRS из примера 8D (2,0 г), тиотетразола (0,6 г) и триэтиламина (1,5 мл) в ацетонитриле (18 мл) нагревали до 85oC в течение 18 часов. Дополнительное количество тиотетразола (0,12 г) и триэтиламин (0,2 мл) добавляли в реакционную смесь при продолжающемся нагреве. Через четыре часа реакционный раствор оставляли охлаждаться и обрабатывали как описано в примере 7D. Остаток очищали силикагельной экспресс-хроматографией, элюируя уксусной кислотой/этилацетатом/гексаном (4:36:60) до получения 2,05 г искомого соединения.
F. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[3-(1(2)Н-тетразол-5-ил)- 3-тиапроп-1-ил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 8E (1,9 г) в 6 н. соляной кислоты (25 мл) нагревали с обратным холодильником в течение около 18 часов. Раствор оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридином/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли водой, что вызывало кристаллизацию искомого соединения. Кристаллический материал удаляли фильтрацией, фильтрат промывали водой, ацетоном и эфиром. Маточный раствор концентрировали под вакуумом, а остаток подвергали аналогичной процедуре до образования кристаллов во второй раз. Кристаллический материал соединяли и высушивали при 60oC под вакуумом до получения 0,94 г искомого соединения. Температура плавления: 185oC.
Расчетные данные по C13H21N5O2S • 0,5H2O • 0,25C3H6O, %: C 49,30; H 7,07; N 20,90. Обнаружено, %: C 49,48; H 6,98; N 21,25.
G. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[3-(1(2)-Н-тетразол-5-ил)- 3-тиапроп-1-ил]декагидроизохинолин-3-карбоновой кислоты.
Изомер 3SR, 4aRS, 6RS, 8aRS из примера 8D превращали в искомое соединение, используя способ, аналогичный способу, описанному в примерах 8E и F. Температура плавления: 201oC.
Расчетные данные по C13H21N5O2S • 0,8H2O, %: C 47,92; H 6,99; N 21,49. Обнаружено, %: C 47,95; H 6,91; N 21,47.
Пример 9. (3SR, 4aRS, 6SR, 8aRS)-6-[N-1(2)Н-тетразол-5-ил) метилформамидо]-декагидроизохинолин-3-карбоновая кислота (3).
A. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6-(цианометил)амино-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 5C (8,0 г) и аминоацетонитрилгидрохлорида (25,15 г) в этаноле (100 мл) обрабатывали порошковым молекулярным ситом  (38,0 г) при комнатной температуре. Спустя 20 минут смесь обрабатывали цианоборгидрилом натрия (1,7 г). Спустя еще 18 часов при комнатной температуре реакционную смесь фильтровали через "CELITE", твердый материал промывали этанолом. Фильтрат концентрировали под вакуумом. Остаток растворяли в метиленхлориде, а раствор метиленхлорида промывали 15% гидроксидом натрия. Фазы разделяли и водную фазу экстрагировали метиленхлоридом и эфиром (2 раза). Органические экстракты соединяли, промывали водой и рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя смесью этилацетат/гексан/метанол (50:49:1) до получения 7,0 г искомого соединения.
(38,0 г) при комнатной температуре. Спустя 20 минут смесь обрабатывали цианоборгидрилом натрия (1,7 г). Спустя еще 18 часов при комнатной температуре реакционную смесь фильтровали через "CELITE", твердый материал промывали этанолом. Фильтрат концентрировали под вакуумом. Остаток растворяли в метиленхлориде, а раствор метиленхлорида промывали 15% гидроксидом натрия. Фазы разделяли и водную фазу экстрагировали метиленхлоридом и эфиром (2 раза). Органические экстракты соединяли, промывали водой и рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя смесью этилацетат/гексан/метанол (50:49:1) до получения 7,0 г искомого соединения.
B. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6[N-(цианометил) формамидо]-2-метоксикарбонил-декагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 9A (1,5 г) в сухом тетрагидрофуране (100 мл) обрабатывали муравьиным ацетангидридом (1,3 г). Через час при комнатной температуре реакционный раствор концентрировали под вакуумом. Остаток разделяли на части между водой и этилацетатом. Фазы отделяли, органическую фазу высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток растворяли в этилацетате и концентрировали под вакуумом. Эту процедуру повторяли четыре раза. Остаток очищали силикагельной экспресс-хроматографией, элюируя смесью метанол/гексан/этилацетат (2:23:75) до получения 1,0 г искомого соединения.
C. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-[N-(1(2)Н-тетразол- 5-ил)метилформамидо]-2-метоксикарбонилдекагидроизохинолин-3- карбоксилата.
Смесь соединения из примера 9B и трибутилоловоазида (10 мл) нагревали до 80oC. Через четыре дня в реакционную смесь добавляли дополнительное количество трибутилоловоазида (3 мл). Еще через три дня реакционную смесь оставляли охлаждаться до комнатной температуры и разбавляли эфиром (100 мл). Этот раствор обрабатывали газообразным хлористым водородом до получения белого твердого вещества. Смесь разбавляли ацетонитрилом (100 мл) и экстрагировали гексаном (пять раз). Ацетонитриловую фазу концентрировали под вакуумом до получения искомого соединения в виде твердого вещества белого цвета.
D. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[N-(1(2)Н-тетразол-5-ил)- метилформамидо]-2-карбоксиметоксикарбонилдекагидроизохинолин-3- карбоновой кислоты.
Раствор соединения из примера 9C (1 г) в этаноле (50 мл) обрабатывали 1 н. гидроксидом натрия (2,75 мл). Через 18 часов реакционную смесь концентрировали под вакуумом. Остаток растворяли в этилацетате с добавлением этанола (1 мл), затем добавляли гексан до тех пор, пока раствор не становился мутным. Полученный раствор помещали в морозильную камеру на четыре часа. Жидкость удаляли, а кристаллическое вещество промывали гексаном (три раза). Кристаллическое вещество растворяли ацетоном и концентрировали под вакуумом. Остаток растворяли в этаноле (20 мл) и обрабатывали 1 н. гидроксидом натрия (5 мл). Через 18 часов pH раствора доводили до 4,0, и полученную смесь разделяли на части между этилацетатом и водой. Фазы отделяли и водную фазу экстрагировали этилацетатом (три раза). Водный слой концентрировали под вакуумом до получения искомого соединения.
E. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[N-(1(2)Н-тетразол-5-ил)- метилформамидо]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 9D в хлороформе (10 мл) обрабатывали триметилсилилиодидом (1,1 мл), а полученный раствор нагревали с обратным холодильником. Через два часа реакционную смесь оставляли охлаждаться до комнатной температуры. Эту смесь делили на части между водой и эфиром. Фазы отделяли и водную фазу экстрагировали эфиром (три раза), затем концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8", элюируя 10% раствором пиридин/вода до получения 110 мг искомого соединения. Температура плавления: 117 - 122oC.
Расчетные данные по C13H20N6O3 • 1,3H2O, %: C 47,06; H 6,86; N 23,33. Обнаружено, %: C 46,63; H 6,71; N 25,98.
Пример 10. (3SR, 4aRS, 6SR, 8aRS)-6-[(1-(2)Н-тетразол-5-ил) проп-1-ил] декагидро-изохинолин-3-карбоновая кислота (6).
A. Получение этил-6-метилидин-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Метилтрифенилфосфонийбромид (76,3 г) добавляли в тетрагидрофуран (800 мл). Эту смесь перемешивали при комнатной температуре в течение 15 минут, затем фильтровали. Фильтрат концентрировали досуха под вакуумом при около 50oC в течение 30 минут. Остаток суспендировали в тетрагидрофуране (220 мл) и полученную смесь охлаждали до 0oC. Холодную смесь обрабатывали 1 М раствором натрийбис(триметилсилил)амида в тетрагидрофуране (213,6 мл). Через 15 минут полученный раствор добавляли в охлажденный (0oC) раствор рацемического соединения из примера 5C (43,23 г) в тетрагидрофуране (320 мл) до тех пор, пока раствор не приобретал устойчивый желтый цвет. Реакционную смесь обрабатывали водой (250 мл) и эфиром (500 мл), фазы разделяли. Органическую фазу экстрагировали водой (10 мл), водную фазу экстрагировали эфиром (2 раза). Органические фазы соединяли, высушивали и концентрировали под вакуумом. Остаток суспендировали в 25% этилацетате/гексане и полученную смесь перемешивали при комнатной температуре. Через один час смесь фильтровали и твердые продукты промывали 25% этилацетатом/гексаном. Фильтрат концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 25% этилацетатом/гексаном до получения 40,67 г искомого соединения.
B. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-гидроксиметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Охлажденный (0oC) раствор соединения из примера 10A (40,67 г) в тетрагидрофуране (285 мл) обрабатывали 10 М раствором боранметилсульфида (9,7 мл). Через два часа при 0oC реакционной смеси позволяли прогреться до комнатной температуры. Еще через 2 1/2 часа реакционную смесь охлаждали до 0oC и обрабатывали этинолом (25 мл), 3 н. гидроксидом натрия (200 мл) и 30% перекисью водорода (200 мл). Через 30 минут при 0oC реакционной смеси позволяли прогреться до комнатной температуры. Еще через два часа при комнатной температуре эту смесь экстрагировали эфиром (3 раза). Соединенные органические экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной хроматографией на "WATERS PREP 500 LC", элюируя градиентом гексана до 60% этилацетат/гексана до получения 40,36 г искомого соединения.
C. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-формил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор диметилсульфоксида (22,1 мл) в метиленхлориде (250 мл) охлаждали до -78oC и обрабатывали оксалилхлоридом (13,05 мл). Через пять минут этот охлажденный раствор обрабатывали раствором соединения из примера 10B (37,34 г) в метиленхлориде (150 мл). Еще через 15 минут эту смесь обрабатывали триэтиламином (86,9 мл). Смесь выдерживали 45 минут при -78oC, после чего позволяли прогреться до комнатной температуры, а затем обрабатывали 10% раствором бисульфата натрия (500 мл) и эфиром (500 мл). Фазы разделяли и водную фазу экстрагировали эфиром (2 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток использовали на следующем этапе без дальнейшей очистки.
D. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-(бензил-2-карбоксиэтилен)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
В суспензию гидрида натрия (1,13 г), предварительно промытого гексаном, в тетрагидрофуране (50 мл) добавляли бензилдиэтилфосфонацетат (1,8 г). Через 15 минут выдерживания при комнатной температуре эту смесь охлаждали до около 0oC. Охлажденную смесь обрабатывали раствором соединения из примера 10C (5,60 г) в тетрагидрофуране (25 мл). Через еще 15 минут реакционной смеси позволяли прогреться до комнатной температуры. Эту смесь обрабатывали водой и эфиром. Органическую фазу удаляли, а водную экстрагировали эфиром (два раза). Комбинированные органические экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/гексаном до получения 7,35 г искомого соединения.
E. Получение (3SR, 4aRS, 6RS, 8aRS) этил-6-(2-карбоксиэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 10D (7,21 г) и 5% палладия на углероде (2,5 г) в этилацетате гидрировали при давлении водорода в 60 фунтов/кв.дюйм при комнатной температуре. Через четыре часа смесь фильтровали через CELITE", а фильтрат концентрировали под вакуумом до получения 6,18 г смеси искомого соединения и исходного материала. Эту смесь подвергали повторному гидрированию до получения искомого соединения. Этот материал использовали на следующем этапе без дальнейшей очистки.
F. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-(3-гидроксипроп-1- ил)-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 10E (5,7 г) в тетрагидрофуране (40 мл) обрабатывали раствором 2 М боранметилсульфида в тетрагидрофуране (17 мл). Через три часа выдерживания при температуре 0oC этот раствор обрабатывали водой. Обработка проводилась так же, как в примере 8C. Остаток очищали силикагельной экспресс-хроматографией, элюируя 50% смесью этилацетата/гексана до получения 3,71 г искомого соединения.
C. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-(3-цианопроп-1-ил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор трифенилфосфина (3,15 г) в метиленхлориде (10 мл) охлаждали до 0oC и обрабатывали бромом (1,92 г). Добавляли дополнительное количество трифенилфосфина до тех пор, пока раствор не обеспечивался. Этот раствор обрабатывали раствором соединения из примера 10F (1,96 г) и пиридином (1,5 мл) в метиленхлориде (10 мл). Реакционной смеси позволяли прогреться до комнатной температуры. Через 2 часа выдерживания при комнатной температуре раствор экстрагировали 10% раствором бисульфата нитрия (дважды). Для того, чтобы растворить образовавшийся осадок, добавили дополнительное количество воды, затем соединенные водные фазы промывали эфиром. Органические фазы соединяли, высушивали сульфатом магния, профильтровывали и концентрировали под вакуумом. Остаток разбавляли эфиром, а трифенилфосфиноксид, выпавший в осадок, удаляли фильтрацией. Эту процедуру повторили дважды.
Раствор продукта предыдущей реакции и цианида натрия (0,59 г) в диметилсульфоксиде (10 мл) нагревали до 60oC. Через два часа раствору позволили охладиться до комнатной температуры и обработали его 50% раствором рассола (50 мл). Полученную смесь экстрагировали метиленхлоридом (пять раз) и эфиром. Соединенные органические экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 50% раствором этилацетат/гексана до получения 1,72 г искомого соединения.
H. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[(1(2)Н-тетразол-5-ил) проп-1-ил] декагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 10G (1,62 г) и трибутилоловоазида (4,1 г) нагревали до 90oC. Через три дня эту смесь обрабатывали 6 н. соляной кислотой (50 мл), а полученную смесь нагревали до 100oC. Через 18 часов реакционной смеси позволили охладиться до комнатной температуры и экстрагировали ее метиленхлоридом и эфиром. Водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридин/ вода до получения 390 г искомого соединения. Температура плавления: 207oC.
Расчетные данные по C14H23N5O2 • 0,75 H2O, %: C 54,79; H 8,05; N 22,82. Обнаружено, %: C 55,08; H 7,85; N 22,86.
Пример 11. (3SR, 4aRS, 6SR, 8aRS)-6-[(1(2)Н-тетразол-5-ил) метоксиметил] декагидроизохинолин-3-карбоновая кислота (7).
A. Получение этил-6-гидроксиметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 6A (20 г) в 1 н. соляной кислоте (85 мл) и ацетонитриле (335 мл) оставили при комнатной температуре. Спустя 45 часов реакционную смесь разделили между эфиром (1 л) и насыщенным бикарбонатом натрия (200 мл). Фазы отделили, водную фазу экстрагировали эфиром (3 • 80 мл). Органические фазы соединили, высушили, профильтровали и концентрировали под вакуумом.
Раствор остатка от предыдущей реакции в этаноле (170 мл) охладили до 0oC. Охлажденный раствор обработали борогидридом натрия (2,4 г). Через 10 минут реакционный раствор концентрировали под вакуумом. Остаток разделили между насыщенным бикарбонатом натрия и метиленхлоридом. Органическую фазу высушили, профильтровали и концентрировали под вакуумом до получения искомого соединения в виде масла. Этот материал использовали на следующем этапе без дальнейшей очистки.
B. Получение (3SR, 4aRS, 6SR, 8aRS) и (3SR, 4aRS, 6RS, 8aRS) этил-6-(цианометил)метил-2-метоксикарбонилдекагидроизохинолин-3- карбоксилата.
Раствор соединения из примера 11A (4,79 г) и N,N-диизопропилэтиламина (5,2 г) в метиленхлориде (50 мл) охлаждали до 0oC и обрабатывали хлорметилметиловым эфиром (1,54 г). Реакционную смесь выдерживали при температуре около 0oC в течение 30 минут, затем ей позволили прогреться до комнатной температуры. Спустя три часа добавили дополнительное количество хлорметилметилового эфира (0,5 мл). Через 18 часов реакционную смесь обработали 10% бисульфатом натрия. Фазы разделили и водную фазу экстрагировали эфиром (2 раза). Органические фазы соединили, высушили, профильтровали и концентрировали под вакуумом. Остаток в виде масла растворяли в метиленхлориде (50 мл), а полученный раствор обработали триметилсилилцианидом (9,63 мл). Этот раствор охладили до 0oC и обработали этератом треххлористого бора (5,92 мл). Полученный раствор оставляли прогреваться до комнатной температуры. Через час выдерживания при комнатной температуре реакционную смесь обрабатывали 10% карбонатом калия (100 мл). Фазы разделяли и водную фазу экстрагировали метиленхлоридом (2 раза) и эфиром. Органические фазы соединяли, промывали насыщенным раствором бикарбоната натрия, высушивали, профильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/гексаном до получения диастереоизомеров: 3SR, 4aRS, 6SR, 8aRS (вторая фракция) и 3SR, 4aRS, 6RS, 8aRS (первая фракция). Суммарный выход искомого соединения составил 2,85 г.
C. Получение (3SR, 4aRS, 6SR, 8ARS)-6-[(1(2)Н-тетразол-5-ил)- метоксиметил]декагидроизохинолин-3-карбоновой кислоты.
Смесь изомера 3SR, 4aRS, 6SR, 8aRS из примера 11B (1,95 г) и трибутилоловоазида (3,83 г) нагревали до 80oC. Через три дня реакционную смесь обрабатывали 6 н. соляной кислотой (20 мл) и нагревали до 90oC. Через 18 часов реакционной смеси позволили охладиться до комнатной температуры. Остывшую смесь, содержащую белый осадок, разбавляли эфиром и фильтровали. Твердые продукты промыли эфиром (3 раза) и ацетоном, затем высушили под вакуумом при 60oC до получения 1,16 г искомого соединения. Температура плавления 263oC.
Расчетные данные по C13H21N5O3 • HCl, %: C 47,06; H 6,68; N 21,11. Обнаружено, %: C 46,80; H 6,85; N 21,07.
Пример 12. (3SR, 4aRS, 6SR, 8aRS)-6-[(1(2)Н-тетразол-5-ил)бут-1- ил]декагидроизохинолин-3-карбоновая кислота (10).
A. Получение этил-6-(3-оксопроп-1-ил)-2-метоксикарбонил- декагидроизохинолин-3-карбоксилата.
Раствор диметилсульфоксида (0,98 г) в метиленхлориде (10,2 мл) охлаждали до -78oC и обрабатывали оксалилхлоридом (0,53 мл). Спустя две минуты раствор соединения из примера 10F (1,65 г) в метиленхлориде (6 мл) добавляли в холодный раствор. Еще через 15 минут раствор обрабатывали триэтиламином (3,5 мл) и полученную смесь оставляли прогреваться до комнатной температуры в течение около 45 минут. Затем реакционную смесь обрабатывали 10% раствором бисульфата натрия и эфиром. Фазы разделяли и водную фазу экстрагировали эфиром (2 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток использовали на следующем этапе без дальнейшей очистки.
B. Получение этил-6(4-циано-3-ен-1-ил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (0,28 г, 60%), предварительно промытую гексаном, в тетрагидрофуране (7,5 мл) охлаждали до 0oC. Охлажденную смесь обрабатывали диэтилцианометилфосфонатом (1,25 г) при 0oC. Через 30 минут смесь обрабатывали раствором соединения из примера 12A (1,72 г) в безводном тетрагидрофуране (5 мл), а полученную смесь оставляли прогреваться до комнатной температуры. Спустя 30 минут реакционную смесь обрабатывали водой (30 мл) и экстрагировали эфиром (3 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток использовали на следующем этапе без дальнейшей очистки.
C. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-(4-цианобут-1-ил)- 2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 12B (1,76 г) в метаноле (50 мл) обрабатывали магнием (2, 45 г). Спустя четыре часа реакционную смесь обрабатывали 1 н. соляной кислотой (250 мл) и полученную смесь экстрагировали эфиром (3 раза). Органические экстракты соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат-гексаном до получения двух продуктов. Фракции, содержащие первый продукт, который представлял собой искомое соединение, концентрировали под вакуумом до получения 0,56 г. Фракции, содержащие второй продукт, который представлял собой метиловый эфир искомого соединения, соединяли до получения 0,41 г. Эти продукты соединяли и использовали на следующем этапе.
D. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[(1(2)Н-тетразол-5-ил)- бут-1-ил] декагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 12C (0,97 г) и трибутилоловоазида (1,78 г) нагревали до 60oC. Спустя три дня смесь обрабатывали 6 н. соляной кислотой (60 мл) и нагревали до 100oC. После нагревания в течение около 18 часов смесь оставляли остывать до комнатной температуры. Эту смесь экстрагировали эфиром (6 раз) и водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли водой и концентрировали под вакуумом. Эту процедуру повторяли. Остаток разбавляли смесью воды и ацетона (1:1) и фильтрацией собирали твердый материал. Этот материал высушивали под вакуумом при 60oC до получения 0,70 г искомого соединения.
Расчетные данные по C15H25N5O2 • 1,3 H2O, %: C 54,46; H 8,41; N 21,17. Обнаружено, %: C 54,48; H 8,30; N 20,99.
Пример 13. (3SR, 4aRS, 6RS, 8aRS)-6-(2-сульфоэтил)декагидроизохинолин-3- карбоновая кислота (22).
A. Получение (3SR, 4aRS, 6RS, 8aRS) этил-6-(2-сульфоэтил)-2- метоксикарбонил-декагидроизохинолин-3-карбоксилата.
Раствор изомера 3SR, 4aRS, 6RS, 8aRS из примера 8D (1,8 г) в этаноле (11 мл) и воде (18 мл) обрабатывали сульфитом натрия (0,64 г), а полученную смесь нагревали до температуры перегонки. Через 18 часов нагрева в реакционную смесь добавляли дополнительную порцию сульфита натрия (0,59 г). Спустя еще 18 часов реакционную смесь концентрировали под вакуумом. Остаток разделяли на части между эфиром и водой. Фазы отделяли, эфирную фазу экстрагировали водой. Водные фазы соединяли и концентрировали под вакуумом. Этот материал использовали на следующем этапе без дальнейшей очистки.
B. Получение (3SR, 4aRS, 6RS, 8aRS)-6-(2-сульфоэтил) декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 13A в 6 н. соляной кислоте (80 мл) нагревали до температуры перегонки. Спустя 18 часов раствор оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "BIO RAD AG IX8" (в виде гидроксида), элюируя 6 н. уксусной кислотой до получения 0,88 г искомого соединения. Температура плавления 265oC.
Расчетные данные по C12H21NO5S • 0,25H2O, %: C 48,71; H 7,32; N 4,73. Обнаружено, %: C 48,53; H 7,39; N 4,50.
Пример 14. (3SR, 4aRS, 6RS, 8aRS)-6-[2-(3-гидроксиизоксазол-5-ил)этил] декагидроизохинолин-3- карбоновая кислота (13).
A. Получение 3-бром-5-гидроксиметилизоксазола.
В смесь бикарбоната калия (32,5 г), воды (4,4 мл) и этилацетата (395 мл) добавляли пропоргиловый спирт (12,1 г). Полученную смесь обрабатывали раствором дибромформальдоксима (21,97 г) в этилацетате (44 мл) в течение семи часов. Спустя еще 18 часов реакционную смесь обрабатывали водой (150 мл). Фазы отделяли, органическую фазу экстрагировали водой с рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Этот материал использовали на следующем этапе без дальнейшей очистки.
B. Получение 3-бром-5-карбоксиизоксазола.
Раствор соединения из примера 14A (35,2 г) в ацетоне обработали реактивом Джоунса (950 мл). Спустя шесть часов реакционную смесь обработали изопропанолом (1 л). Полученную смесь профильтровали через CELITE, фильтрат концентрировали под вакуумом. Остаток растворили в эфире и экстрагировали водой. Соединенные водные экстракты экстрагировали эфиром. Соединенные органические фазы высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 34,1 г искомого соединения.
C. Получение 5-карбокси-3-метоксиизоксазола.
Раствор соединения из примера 14B (34,1 г), гидроксида калия (169 г), метанола (580 мл) и воды (103 мл) нагревали до температуры перегонки. Спустя шесть часов реакционной смеси позволили охладиться до комнатной температуры. Эту смесь обрабатывали концентрированной соляной кислотой (450 мл) и разрабатывали водой (350 мл). Полученный раствор экстрагировали эфиром (6 раз). Органические экстракты соединили, высушили сульфатом магния, профильтровали и концентрировали под вакуумом. Остаток разбавили толуолом и метанолом и концентрировали под вакуумом до получения 20,2 г искомого соединения.
D. Получение 5-гидроксиметил-3-метоксиизоксазола.
Раствор соединения из примера 14C (20,2 г) в тетрагидрофуране обрабатывали триэтиламином (14,3 г), охладили до 0oC. Раствор изобутилхлорформиата (19,3 г) в тетрагидрофуране (35 мл) добавили в охлажденный раствор. Спустя 1 1/4 часа осадок удалили фильтрацией (210 мл) и промыли тетрагидрофураном. Фильтрат аккуратно добавили в раствор борогидрида натрия (13,4 г) в воде (140 мл). Спустя 4 1/2 часа реакционную смесь обработали 1 н. соляной кислотой, полученную смесь экстрагировали эфиром. Соединенные органические экстракты высушили сульфатом магния, профильтровали и концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюируя 35% этилацетат/гексаном до получения 9,10 г искомого соединения и 1,38 г 4-гидроксиметил-3-метоксиизоксазола.
E. Получение 5-бромметил-3-метоксиизоксазола.
Раствор трифенилфосфина (27,7 г) в метиленхлориде (425 мл) охладили до 0oC и обработали бромом (16,9 г) до устойчивого желтого цвета. Добавили дополнительное количество трифенилфосфина до исчезновения желтого цвета. Бесцветный раствор обработали раствором соединения из примера 14D (9,10 г) и пиридином (11,2 г) в метиленхлориде (11,4 мл). Через 10 минут реакционную смесь экстрагировали 10% раствором бисульфата натрия (2 раза). Органические фазы соединили и экстрагировали метиленхлоридом (2 раза). Органические фазы соединили, высушили сульфатом натрия, профильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя градиентом 10% этилацетат/гексаном, а затем 20% этилацетат/гексаном. Фракцию, содержащие искомое соединение, соединили и концентрировали под вакуумом до получения 10,8 г.
F. Получение диэтил-[(3-метоксиизоксазол-5-ил)метил]фосфоната.
Раствор соединения из примера 14E (10,8 г) в толуоле (150 мл) обрабатывали триэтилфосфитом (18,7 г). Этот раствор нагревали до температуры в около 120oC. Спустя 18 часов реакционную смесь концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюируя линейным градиентом этилацетата к 5% этанол/этилацетату до получения 11,77 г искомого соединения.
G. Получение этил-6-/2-(3-метоксиизоксазола-5-ил)этенил/-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 14F (3,71 г) в тетрагидрофуране (10 мл) охлаждали до -17oC и обрабатывали 1 М раствором бис(триметилсилил)амида натрия в тетрагидрофуране (14,9 г). Спустя 30 минут этот раствор обрабатывали раствором соединения из примера 7A (3,15 г) в тетрагидрофуране (10 мл). Полученный раствор оставляли нагреваться до комнатной температуры. Спустя 1 1/2 часа реакционную смесь обрабатывали водой и экстрагировали эфиром. Соединенные органические экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 15% этиленацетат/толуолом до получения 2,95 г искомого соединения.
H. Получение (3SR, 4aRS, 6RS, 8aRS) этил-6-/2-(3-метоксиизоксазол-5-ил)этил/-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 14G (2,33 г) и 5% палладия на углероде (2,33 г) в этилацетате (50 мл) гидрировали при давлении водорода в 60 фунтов/кв. дюйм. Через шесть часов выдерживания при комнатной температуре реакционную смесь фильтровали через CELITE, фильтрат концентрировали под вакуумом. Остаток разбавляли хлороформом и концентрировали под вакуумом. Этот остаток очищали силикагельной экспресс-хроматографией, элюируя линейным градиентом 5% этилацетат/толуола к 15% этилацетат/толуолу до получения 1,0 г искомого соединения.
I. Получение (3SR, 4aRS, 6RS, 8aRS)-6-/2-(3-гидроксиизоксазол-5-ил)этил/декагидроизохинолин-3- карбоновой кислоты.
Смесь соединения из примера 14H (0,92 г) и 48% бромистоводородной кислоты нагревали с обратным холодильником. Через три часа реакционной смеси позволили охладиться до комнатной температуры и концентрировали под вакуумом. Остаток разбавляли водой и концентрировали под вакуумом. Этот остаток разбавили водой и профильтровали, чтобы удалить твердые продукты. Твердые продукты промыли ацетоном и высушили под вакуумом при комнатной температуре до получения 0,26 г. Полученный материал очистили ионообменной хроматографией на "DOWEX 50X8", элюируя 10% пиридин/водой до получения 0,115 г искомого соединения. Температура плавления 256oC.
Расчетные данные по C15H22N2O4 • 1,0 H2O, %: C 57,67; H 7,74; N 8,96. Обнаружено, %: C 57,78; H 7,75; N 9,07.
Пример 15. (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2)Н-1,2,4-триазол-5- ил)-2-тиаэтил]-декагидроизохинолин-3-карбоновая кислота (14).
A. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-[2-(1(2-4)Н-1,2,4- триазол-5-ил)-2-тиаэтил]-2-метоксикарбонилдекагидроизохинолин-3- карбоксилата.
Раствор соединения из примера 7C (9,22 г) в безводном диметилформамиде (92 мл) обрабатывали 1Н-1,2,4-триазол-3-тиолом (3,09 г) и триэтиламином (6,19 г). Полученный раствор нагревали до 100oC в течение около 18 часов. Охлажденный реакционный раствор обрабатывали 10% раствором бисульфата натрия (200 мл), экстрагировали хлороформ/этилацетатом (1:1) и эфиром. Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток нагревали до около 50oC под вакуумом до удаления остаточного диметилформамида. Этот остаток очищали силикагель-хроматографией на "WATERS PREP LC 2000", элюируя линейным градиентом 35% этилацетат/гексана к этилацетату. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли этилацетатом и экстрагировали 1 н. соляной кислотой и рассолом. Органический слой высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 9,37 г искомого соединения.
B. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2-4)Н-1,2,4- триазол-5-ил)-2-тиаэтил]декагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 15A (1,77 г) в 6 н. соляной кислоте (10 мл) нагревали до 100oC. Спустя 18 часов реакционную смесь концентрировали под вакуумом. Остаток очищали ионообменной хроматографией, элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли водой и концентрировали под вакуумом. Эту процедуру повторяли четыре раза, затем остаток высушивали под вакуумом в течение ночи. Полученный материал растворяли в ацетоне и нагревали до температуры перегонки в течение часа. Полученную смесь оставляли остывать до комнатной температуры, затем искомое соединение удаляли фильтрацией. Твердые продукты промывали ацетоном и эфиром, затем высушивали под вакуумом при 40oC до получения 0,37 г искомого соединения.
Расчетные данные по C13H20N4O2S • 2,0H2O, %: C 46,97; H 7,27; N 16,85. Обнаружено, %: C 46,88; H 7,33; N 16,74.
Пример 16. (3SR, 4aRS, 6SR, 8aRS)-6-[(1(2-4)Н-1,2,4-триазол-5- ил)сульфонилметил]декагидроизохинолин-3-карбоновая кислота (15).
Раствор соединения из примера 15A (9,37 г) в метиленхлориде (105 мл) обрабатывали 3-хлорнадбензойной кислотой (13,25 г) (тремя порциями) в течение 30 минут. Спустя 18 часов выдерживания при комнатной температуре реакционную смесь концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя ступенчатым градиентом 50% этилацетат/гексана (500 мл), затем - этилацетатом (2 л) до получения 8,98 г прозрачного масла.
Прозрачное масло обрабатывали 6 н. соляной кислотой (250 мл) и нагревали до 110oC. Спустя 18 часов реакционную смесь оставляли остывать до комнатной температуры и концентрировали под вакуумом. Остаток растворяли в воде (100 мл), полученный раствор экстрагировали эфиром. Фазы отделяли и водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли водой и концентрировали под вакуумом. Этот процесс повторяли дважды, затем остаток концентрировали под вакуумом в течение около 18 часов. Полученный остаток разбавляли ацетоном (250 мл) и перегоняли в течение одного часа. Искомое соединение удаляли фильтрацией, затем промывали ацетоном и эфиром. Твердые продукты высушивали под вакуумом при 60oC в течение около 18 часов до получения 3,56 г искомого соединения.
Расчетные данные по C13H20N4O4S • H2O, %: C 45,08; H 6,40; N 16,17. Обнаружено, %: C 45,40; H 6,31; N 16,39.
Пример 17. (3SR, 4aRS, 6RS, 8aRS)-6-[2-((N-метансульфонил)карбоксамидо)этил]декагидроизохинолин-3- карбоновая кислота (16).
A. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-[2-((N-метансульфонил)-карбоксамидо)этил]-2-метоксикарбонил- декагидроизохинолин-3-карбоксилата.
Раствор 1,1'-карбонилдиимидазола (1,9 г) в безводном тетрагидрофуране (25 мл) обрабатывали раствором соединения из примера 10E (4 г) в тетрагидрофуране (25 мл). Полученный раствор нагревали с обратным холодильником в течение часа, затем раствор оставили остывать до комнатной температуры. Остывший раствор обработали метансульфонамидом (1,11 г). Через 10 минут этот раствор обработали раствором 1,8-диазабицикло[5,4,0]ундек-7-ена (1,8 г) в тетрагидрофуране (10 мл). Спустя 18 часов этот раствор обработали 1 н. соляной кислотой (150 мл). Полученную смесь экстрагировали эфиром. Эфирные экстракты соединяли и экстрагировали насыщенным раствором бикарбоната натрия. Фазы отделяли, и водную фазу подкисляли 5 н. соляной кислотой. Подкисленный водный слой экстрагировали эфиром. Эфирные экстракты соединяли, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом до получения 4,35 г искомого соединения.
B. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-((N-метансульфонил) карбоксамидо)этил]-2-метоксикарбонил-декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 17A в этаноле (45 мл) обрабатывали 1 н. гидроксидом натрия (22,6 мл). Через около 18 часов выдерживания при комнатной температуре раствор частично концентрировали под вакуумом до удаления этанола. Остаток экстрагировали этилацетатом. Водный слой подкислили 5 н. HCl, затем экстрагировали этилацетатом. Этилацетатные экстракты соединяли, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом до получения 3,7 г искомого соединения. Этот материал использовали на следующем этапе без дальнейшей очистки.
C. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-((N-метансульфонил)- карбоксамидо)этил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 17B (3,7 г) в хлороформе (40 мл) обрабатывали иодотриметилсиланом (11,4 г). Этот раствор нагревали с обратным холодильником в течение двух часов, затем концентрировали под вакуумом. Остаток обрабатывали водой (25 мл), экстрагировали эфиром и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридин/ водой до получения 2,75 г искомого соединения. Температура плавления 208- 215oC.
Расчетные данные по C14H24N2O5S, %: C 50,59; H 7,28; N 8,43. Обнаружено, %: C 50,36; H 7,47; N 8,55.
Пример 18. (3SR, 4aRS, 6RS, 8aRS)-6-[2-(N-(1(2)Н-тетразол-5-ил) карбоксамидо)этил]-декагидроизохинолин-3-карбоновая кислота (17).
A. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-[2-(N-(1(2)Н-тетразол- 5-ил)карбоксамидо)этил]декагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 10E (4 г) в сухом тетрагидрофуране (25 мл) обрабатывали раствором 1,1'-карбонилдимидазола (1,9 г) в сухом тетрагидрофуране (25 мл). Полученный раствор нагревали с обратным холодильником в течение одного часа и обрабатывали 5-аминотетразолом (1 г). После того как раствор нагревали с обратным холодильником в течение 18 часов, его оставляли остывать до комнатной температуры. Остывший раствор обрабатывали 1 н. соляной кислотой (150 мл) и экстрагировали эфиром. Органические экстракты соединяли и экстрагировали раствором бикарбоната натрия. Водные экстракты бикарбоната соединяли, подкисляли 5 н. соляной кислотой и экстрагировали эфиром. Эфирные экстракты соединяли, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом до получения 4,6 г искомого соединения.
B. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-(N-(1(2)Н-тетразол-5-ил)карбоксамидо)этил] -2- метоксикарбонил-декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 18A (4,4 г) в этаноле (45 мл) обрабатывали 1 н. гидроксидом натрия (23,7 мл). После 18 часов выдерживания при комнатной температуре раствор частично концентрировали под вакуумом до удаления этанола. Остаток подкисляли 5 н. соляной кислотой и экстрагировали этилацетатом. Органические фазы соединяли, экстрагировали насыщенным раствором хлорида натрия, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом до получения 3,9 г искомого соединения.
C. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-(N-(1(2)Н-тетразол-5- ил)-карбоксамидо)этил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 18B (3,9 г) в хлороформе (45 мл) обрабатывали иодотриметилсиланом (12,3 г). Полученный раствор нагревали с обратным холодильником в течение двух часов. Раствор затем концентрировали под вакуумом, остаток обрабатывали водой (30 мл). Эту смесь экстрагировали эфиром, затем водный слой концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридин/водой до получения 3,1 г искомого соединения. Температура плавления > 220oC.
Расчетные данные по C14H22N6O3 • 2,3 H2O, %: C 46,22; H 7,37; N 23,10. Обнаружено, %: C 46,13; H 7,65; N 23,14.
Пример 19. (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1)2)Н-тетразол-5-ил)-1- метилэтил]декагидроизохинолин-3-карбоновая кислота (18).
A. Получение этил-2-метоксикарбонил-6-трифторметансульфонилоктагидроизохинолин-3- карбоксилата.
Раствор бис(триметилсилил)амида лития (100 мл 1 М раствора в тетрагидрофуране) в безводном тетрагидрофуране (180 мл) охлаждали до -78oC и обрабатывали раствором рацемического соединения из примера 5C (25,8 г) в безводном тетрагидрофуране (60 мл). Через час выдерживания при -78oC охлажденный раствор обрабатывали раствором N-фенилтрифторметансульфониламида (32,5 г) в тетрагидрофуране (100 мл). Этому раствору позволили прогреться до комнатной температуры. Спустя три часа раствор разбавляли эфиром (100 мл) и экстрагировали 10% раствором бисульфата натрия. Водные экстракты соединяли и экстрагировали эфиром (3 раза). Органические фазы соединяли, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюируя градиентом 8-L гексана к 25% этилацетат/гексан до получения 24,4 г искомого соединения.
B. Получение этил-6-(2-циано-1-метилэтенил)-2- метоксикарбонилоктагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 19A (2,5 г) в диметилформамиде (21 мл), который был дегазован азотом перед его использованием, обрабатывали кротононитрилом (1 г), триэтиламином (2,1 г) и бис(трифенилфосфин)палладий (II) хлоридом (97 мг). Эту смесь нагревали до температуры 70 - 80oC в азотной атмосфере. Через 18 часов выдерживали при температуре около 75oC, реакционную смесь обрабатывали водой (100 мл). Полученную смесь экстрагировали эфир/гексаном (1:1). Органические экстракты соединяли, промывали насыщенным раствором хлорида натрия, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 25% этилацетатом/гексаном до получения 1,17 г искомого соединения.
C. Получение этил-6-(2-циано-1-метилэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 19B (1,1 г) и 5% палладия на углероде в этаноле (80 мл) гидрировали под давлением водорода в 60 фунтов/кв.дюйм при комнатной температуре. Спустя шесть часов катализатор удаляли фильтрацией, а фильтрат концентрировали под вакуумом. Остаток растворяли в этилацетате, а полученный раствор фильтровали через "ACCOD ISC" 0,2 мкм. Остаток очищали силикагельной экспресс-хроматографией, элюируя линейным градиентом этилацетат/гексана (1: 4) к этилацетат/гексану (3:7) до получения 0,66 г искомого соединения.
D. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил) -1-метилэтил]декагидроизохинолин-3-карбоновой кислоты.
Соединение из примера 19C (600 мг) добавляли в трибутилоловоазид (1,18 г) и нагревали смесь до около 80oC. Через четыре дня смесь обрабатывали 6 н. соляной кислотой (5 мл) и нагревали с обратным холодильником. Через 18 часов смеси позволили остыть до комнатной температуры. Смесь экстрагировали эфиром, водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридин/водой до получения 0,37 г искомого соединения.
Расчетные данные по C14H23N5O2 • 1,1 H2O, %: C 53,69; H 8,11; N 22,36. Обнаружено, %: C 53,63; H 8,01; N 22,16.
Пример 20. (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил)- 1-фенилэтил]декагидроизохинолин-3-карбоновая кислота (19).
A. Получение этил-6-(2-циано-1-фенилэтенил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 19A (2,5 г) в диметилформамиде (21 мл), который был дегазован азотом перед употреблением, обрабатывали циннамонитрилом (1,94 г), триэтиламином (2,1 г) и бис(трифенилфосфин)палладий(II)хлоридом (97 г). Полученную смесь нагревали до температуры около 70 - 80oC в азотной атмосфере. Через 18 часов нагрева до около 75oC реакционную смесь обрабатывали водой (100 мл). Полученную смесь экстрагировали эфир/ гексаном (1:1). Органические экстракты соединяли, промывали насыщенным раствором хлорида натрия, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя линейным градиентом этилацетат/гексана (1: 4) к этилацетат/гексану (3:7) до получения 1,45 г искомого соединения.
B. Получение этил-6-(2-циано-1-фенилэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 20A (1,35 г) и 5% палладия на углероде в этаноле (85 мл) гидрировали под давлением водорода в 60 фунтов/кв.дюйм при комнатной температуре. Спустя шесть часов катализатор удаляли фильтрацией. Фильтрат концентрировали под вакуумом, растворяли в этилацетате, фильтровали через "ACCODISC" 0,2 мкм и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя линейным градиентом этилацетат/гексана (1:3) к этилацетат/гексану (2:3) до получения 0,61 г искомого соединения.
C. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил)- 1-фенилэтил]декагидроизохинолин-3-карбоновой кислоты.
Соединение из примера 20B (0,59 г) добавляли в трибутилоловоазид (6 г) и нагревали смесь до около 80oC. Через четыре дня смесь обрабатывали 6 н. соляной кислотой (5 мл) и нагревали с обратным холодильником. Полученную смесь экстрагировали эфиром и водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридином до получения 0,38 г искомого соединения.
Расчетные данные по C19H25N5O2 • 1,8H2O • 0,25C3H6O, %: C 58,95; H 7,54; N 17,40. Обнаружено, %: C 59,29; H 6,71; N 17,01.
Пример 21. (3SR, 4aRS, 6SR, 8aRS)-6-[2-(3-гидрокси-1,2,5- тиадиазол-4-ил)этенил]декагидроизохинолин-3-карбоновая кислота (20).
A. Получение 3-гидроксиметил-4-гидрокси-1,2,5-тиадиазола.
Раствор 2-амино-3-гидроксипропанамидгидрохлорида (20,25 г) в ацетонитриле (270 мл) обрабатывали N-метил-N-(триметилсилил)- трифторацетамидом (172 г). Спустя 1 час полученный раствор охлаждали до 0oC и обрабатывали триэтиламином (20 мл). Через пять минут полученный раствор обрабатывали раствором конденсированной двуокиси серы (30,75 мл) в ацетонитриле (113,25 мл). После завершения добавки двуокиси серы ледяную баню удаляли и реакционной смеси позволили прогреться до комнатной температуры. Спустя три часа реакционную смесь помещали в холодильник на ночь. Еще через два часа выдерживания при комнатной температуре реакционную смесь обрабатывали водой (8,55 мл). Через 15 минут полученную смесь концентрировали под вакуумом. Остаток обрабатывали водой (400 мл) и экстрагировали метиленхлоридом. Органические экстракты убирали, а водную фазу концентрировали под вакуумом. Остаток обрабатывали тетрагидрофураном (250 мл), помещали на ультразвуковую баню, затем фильтровали. Фильтрат тетрагидрофурана концентрировали под вакуумом. Остаток растворяли в горячем ацетоне (36 мл) и полученный раствор обрабатывали хлороформом (165 мл). Полученную смесь концентрировали на паровой бане до объема в около 165 мл, затем фильтровали. Фильтрат концентрировали до около 150 мл, охлаждали, помещали в ультразвуковую баню, замораживали до получения 5,9 г искомого соединения.
B. Получение 3-гидрокси-4-иодометил-1,2,5-тиадиазола.
Раствор соединения из примера 21A (5,90 г) в безводном ацетонитриле (90 мл) обрабатывали N-метил-N-(триметилсилил)трифторацетамидом (18,59 г) и иодотриметилсиланом (26,80 г). Полученный раствор нагревали до 55oC в течение 15 часов, затем оставляли при комнатной температуре на 3 1/2 часа. Реакционную смесь концентрировали под вакуумом, растворяли в хлороформе (300 мл). Органический раствор промывали водой, 1 н. бисульфитом натрия и еще водой. Органический слой концентрировали под вакуумом, остаток обрабатывали ацетонитрилом (62 мл) и водой (6,4 мл). Через 40 мин выдерживания при комнатной температуре полученную смесь концентрировали под вакуумом. Остаток обрабатывали ацетонитрилом (8,3 мл), фильтровали и твердый материал промывали ацетонитрилом. Фильтрат концентрировали под вакуумом, а затем обрабатывали водой (20,8 мл). Через 15 мин выдерживания при комнатной температуре полученную смесь фильтровали и твердый материал промывали водой. Полученный материал высушивали под вакуумом при 60oC до получения 5,99 г искомого соединения.
C. Получение 3-дифенилметокси-4-иодометил-1,2,5-тиадиазола.
Раствор соединения из примера 21B (5,99 г) в метиленхлориде (100 мл) обрабатывали дифенилдиазометаном (4,80 г). Через 10 мин добавляли дополнительное количество дифенилдиазометана. Спустя еще 10 минут раствор обработали уксусной кислотой, замет концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюируя градиентом 8 - 1 гексана к 50% этилацетат/гексану до получения 6,35 г искомого соединения.
D. Получение 3-(диэтилфосфонметил)-4-дифенилметокси-1,2,5-тиадиазола.
Раствор соединения из примера 21C (6,05 г) и триэтилфосфита (4,92 г) в толуоле (120 мл) нагревали с обратным холодильником. Спустя 18 часов добавляли дополнительное количество триэтилфосфита (0,25 эквивалентов). Спустя еще три часа реакционной смеси позволили остыть до комнатной температуры и концентрировали смесь под вакуумом. Остаток очищали силикагель-хроматографией, элюируя этилацетат/гексаном (1:1) до получения 5,71 г искомого соединения.
E. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6-[2-(3- дифенилметокси-1,2,5-тиадиазол-4-ил)этенил] -2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (0,49 г), предварительно промытого гексаном, в свежедистиллированном тетрагидрофуране (25 мл) охлаждали до 0oC и обрабатывали раствором соединения из примера 21D (5,10 г) в тетрагидрофуране (5 мл). Спустя десять минут этот раствор обрабатывали раствором соединения из примера 7A (3,45 г) в тетрагидрофуране (15 мл). Полученной смеси позволили прогреться до комнатной температуры. Через 30 минут реакционную смесь обработали как описано в примере 14G до получения 5,43 г искомого соединения.
F. Получение (3SR, 4aRS, 6SR, 8aRS) этил-6-[2-(3-гидрокси-1,2,5- тиадиазол-4-ил)этенил]декагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 21E (5,43 г) в метиленхлориде (119 мл) охлаждали до 0oC и обрабатывали триэтилсиланом (11,22 г) и трифторуксусной кислотой (22,0). Через час выдерживания при 0oC реакционную смесь концентрировали под вакуумом. Остаток очищали силикагель-хроматографией, элюируя этилацетат/гексаном (1:1). Фракции, содержащие искомое соединение, концентрировали под вакуумом. Остаток растворяли в хлороформе и концентрировали под вакуумом до получения 3,55 г искомого соединения.
C. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[2-(3-гидрокси-1,2,5- тиадиазол-4-ил)этенил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 21F (1,10 г) в 6 н. соляной кислоте (около 25 мл) нагревали до около 90oC. Спустя 18 часов раствору позволили остыть до комнатной температуры. Полученную смесь обработали водой (15 мл). pH этого раствора довели до 10 добавлением 5 н. гидроксида натрия. Затем раствор подкислили (pH 5)добавлением 5 н. соляной кислоты. Осадок удаляли фильтрацией, затем промывали водой, ацетоном и эфиром. Полученный материал высушивали под вакуумом при комнатной температуре до получения 0,33 г искомого соединения. Температура плавления 239 - 242oC.
Расчетные данные по C17H24N2O4, %: C 63,73; H 7,55; N 8,74. Обнаружено, %: C 63,68; H 7,65; N 8,85.
Пример 22. (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил) этил]декагидроизохинолин-3-карбоновая кислота (1).
A. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-(2-циано-1-этенил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (6,98 г, 60%), предварительно промытую гексаном, в тетрагидрофуране (185 мл) обрабатывали диэтилцианометилфосфонатом (30,93 г). Полученную смесь охлаждали до 0oC и обрабатывали раствором соединения из примера 10C (37,1 г) в тетрагидрофуране (185 мл). Спустя 45 минут реакционную смесь обрабатывали 10% бисульфатом натрия (200 мл) и эфиром (400 мл). Фазы отделяли и водную фазу экстрагировали эфиром (2 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/гексаном до получения 37,84 г искомого соединения.
B. Получение (3SR, 4aRS, 6RS, 8aRS)-этил-6-(2-цианоэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 22A (37,8 г) в метаноле (1150 мл) обрабатывали магнием (57,4 г). Через 15 минут при комнатной температуре реакционную смесь охлаждали на ледяной бане. Через 1 1/2 часа реакционную смесь обрабатывали метиленхлоридом (1,5 л) и фильтровали через CELITE. Фильтрат разделяли на две порции, и каждую порцию экстрагировали 10% сульфатом натрия ( 2 л). Фазы отделяли и водную фазу экстрагировали метиленхлоридом (3 раза) и эфиром (1 раз). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500LC", элюируя линейным градиентом гексана к 35% этилацетат/гексану до получения фракций, содержащих искомое соединение, и соответствующий метиловый эфир, а также смеси этих соединений. Эти фракции соединяли и концентрировали под вакуумом до получения 28 г смеси искомого соединения и соответствующего метилового эфира.
C. Получение (3SR, 4aRS, 6RS, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил)- этил]декагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 22B (27,96 г) и трибутилоловоазида (57,96 г) нагревали до 80oC. Через 48 часов смесь обрабатывали 6 н. соляной кислотой (200 мл) и нагревали до 90oC. После 18 часов нагрева смеси позволили остыть до комнатной температуры. Полученную смесь обработали водой и довели pH приблизительно до 5. Смесь концентрировали под вакуумом до получения раствора, содержащего белый осадок. Этот твердый материал удаляли фильтрацией и промывали водой и ацетоном до получения 13,42 г искомого соединения. Температура плавления 220oC.
Расчетные данные по C13H21N5O2 • 0,5H2O, %: C 54,15; H 7,69; N 24,29. Обнаружено, %: C 53,81; H 7,25; N 24,26.
Пример 23. (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил) этил]декагидроизохинолин-3-карбоновой кислоты (2).
A. Получение этил-6-формил-2-метоксикарбонилдекагидроизохинолин -3-карбоксилата.
Раствор соединения из примера 6A (28,85 г) и 1 н. соляной кислотой (52 мл) в ацетонитриле (155 мл) оставили стоять при комнатной температуре. Спустя 4 1/2 часа реакционную смесь разбавили эфиром (1,5 л) и насыщенным раствором бикарбоната натрия (500 мл). Фазы разделили и водную фазу экстрагировали эфиром (2 раза). Органические фазы соединили, высушили сульфатом магния, фильтровали, концентрировали под вакуумом. Полученный материал использовали на следующем этапе без дальнейшей очистки.
B. Получение этил-6-(2-цианоэтенил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (5,91 г, 60%), предварительно промытого гексаном, в тетрагидрофуране (114 мл) обрабатывали диэтилцианометилфосфонатом (22,98 г). Через 20 минут полученную смесь охлаждали до 0oC. Охлажденный раствор обрабатывали раствором соединения из примера 23A (27,56 г) в тетрагидрофуране (143 мл), полученной смеси позволили прогреться до комнатной температуры. Через 1 час реакционную смесь обрабатывали водой (200 мл) и экстрагировали эфиром. Эфирные экстракты соединяли, промывали насыщенным раствором хлорида натрия, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 33,7 г искомого соединения. Этот материал использовали на следующем этапе без дальнейшей очистки.
C. Получение (3SR, 4aRS, 6SR, 8aRS)-этил-6-[2-(1(2)Н-тетразол- 5-ил)этил]-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 22B (9,17 г) и 5% палладия на углероде (3,0 г) в этаноле (285 мл) гидрировали под давлением водорода в 60 фунтов/кв.дюйм при комнатной температуре. Через шесть часов смесь концентрировали под вакуумом. Остаток растворяли в этилацетате, фильтровали через CELITE, а фильтрат концентрировали под вакуумом. Полученный материал разбавляли хлороформом, смесь фильтровали, а фильтрат концентрировали под вакуумом. Остаток очищали силикагель-хроматографией на "WATERS PREP 500 LC", элюируя линейным градиентом гексана к 50% этилацетат/гексану до получения фракций, содержащих диастереоизомеры: 3SR, 4aRS, 6SR, 8aRS и 3SR, 4aRS, 6RS, 8aRS, и их смесь. Выход искомого продукта составил 0,74 г.
D. Получение (3SR, 4aRS, 6SR, 8aRS)-6-[2-(1(2)Н-тетразол-5-ил) этил] декагидроизохинолин-3-карбоновой кислоты.
Смесь рацемической смеси 3SR, 4aRS, 6SR, 8aRS из примера 23C (0,74 г) и трибутилоловоазида (1,52 г) нагревали до 80oC. Через 66 часов смесь обрабатывали 6 н. соляной кислотой (11 мл) и нагревали до 100oC. Через 18 часов такого нагрева смеси позволили остыть до комнатной температуры и затем концентрировали ее под вакуумом. Остаток обрабатывали водой (20 мл) и нагревали до около 80oC до растворения. Полученную смесь оставляли остывать до комнатной температуры, фильтровали через CELITE, твердые продукты промывали водой. Фильтрат и промывную воду соединяли и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток суспендировали в ацетоне, нагревали с обратным холодильником в течение часа, затем позволили смеси остыть до комнатной температуры. Полученную смесь фильтровали, осадок промывали ацетоном и эфиром. Полученный материал высушивали под вакуумом при 80oC до получения 0,36 г искомого соединения. Температура плавления 254- 255oC.
Расчетные данные по C13H21N5O2 • 0,5H2O • 0,2C3H6O, %: C 54,45; H 7,79; N 23,34. Обнаружено, %: C 54,36; H 7,41; N 23,33.
Пример 24. (3SR, 4aR, 6R, 8aR)-(-)-6-[2-(1(2)Н-тетразол-5-ил) этил]декагидроизохинолин-3-карбоновая кислота (21).
A. Получение 5-(2-гидроксиэтил)-1(2)Н-тетразола.
Смесь азида натрия (34,4 г) и толуола (150 мл) обрабатывали трибутилоловохлоридом (153 мл). Через 15 минут выдерживания при комнатной температуре полученную смесь обрабатывали 3-гидроксипропионитрилом (48 мл). Полученную смесь нагревали до около 90oC. Через 20 часов добавляли 2 молярных эквивалента 6 М HCl, полученную смесь нагревали с обратным холодильником в течение 12 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и переливали ее в делительную воронку. Водный слой удаляли и промывали 1,2-дихлорэтаном (4 • 50 мл) и этилацетатом (100 мл). Водный слой концентрировали под вакуумом до образования густой суспензии. Полученный материал обрабатывали этанолом (180 мл), твердый материал удаляли фильтрацией (NaCl). Фильтрат концентрировали под вакуумом до получения 29,4 г искомого соединения.
B. Получение [2-(1(2)Н-тетразол-5-ил)этил]трифенилфосфонийбромида.
Смесь соединения из примера 24A (12,80 г) в ксилоле (76 мл) обрабатывали трифенилфосфонгидробромидом (38,48 г). Полученную густую суспензию нагревали до около 150oC и удаляли воду азеотропной перегонкой. Спустя два часа реакционную смесь охлаждали до около 100oC и обрабатывали 1,2-дихлорэтаном (100 мл). Полученную смесь нагревали до около 100oC в течение тридцати минут, а затем позволили остывать до комнатной температуры. Смесь фильтровали до получения 18,1 г искомого соединения. Фильтрат концентрировали под вакуумом досуха. Дополнительное количество искомого соединения (22,3 г) получали из этого остатка рекристаллизацией из этилацетата. Температура плавления 222,2oC.
1Н ЯМР (d6-DMSO): δ 7,82 (m, 15H), 4,26 (m, 2H), 3,30(m, 2H).
13C ЯМР (d6-DMSO): δ 135,08, 135,05, 133,73, 133,59, 130,34, 130,17, 118,11, 116,97.
Масс-спектр высокого разрешения (FAB), расчетные данные по C21H20N4P+ : 359,14256. Обнаружено: 359,14320.
C. Получение (3S, 4aR, 8aR)-6-[2-(1(2)Н-тетразол-5-ил)этенил]-2- метоксикарбонилдекагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 4C (0,65 г) и соединения из примера 24B (1,19 г) и диметилформамида (10 мл) охлаждали до 0oC. Охлажденную суспензию обрабатывали 1 М раствором бис(триметилсилил)-амидом натрия в тетрагидрофуране (6,0 мл). Спустя 2 часа при 0oC полученную суспензию обрабатывали водой (30 мл). Полученную смесь экстрагировали этилацетатом (4 • 25 мл). Водный слой подкисляли 1 М HCl до pH 2, затем экстрагировали дополнительным количеством этилацетата (4 • 25 мл). Этилацетаты соединяли, высушивали сульфатом натрия, фильтровали и концентрировали под вакуумом до получения искомого соединения.
D. Получение (3S, 4aR, 6R, 8aR)-(-)-6-[2-(1(2)-тетразол-5-ил)- этил]декагидроизохинолин-3-карбоновой кислоты.
Смесь соединения из примера 24C (0,220 г) и 10% палладия на углероде (0,20 г) и этанола (5 мл) гидрировали под давлением водорода в 50 фунтов/кв. дюйм при комнатной температуре. Спустя три дня реакционную смесь фильтровали через CELITE, фильтрат концентрировали под вакуумом. Остаток обрабатывали 6 М соляной кислотой (6 мл) и нагревали с обратным холодильником. Спустя три часа реакционной смеси позволили остыть до комнатной температуры. Полученную смесь экстрагировали этилацетатом и водную фазу концентрировали под вакуумом до получения 0,179 г искомого соединения. Температура плавления 250 - 257oC.
[α]D = -30,0o (c = 1, 1 N HCl)
Расчетные данные по C13H21N5O2 • 0,6H2O • 0,1C3H6O, %: C 53,98; H 7,77; N 23,66. Обнаружено, %: C 53,62; H 7,38; N 23,32.
Пример 25. (3S, 4aR, 6S, 8aR)-6-[2-(1(2)Н-тетразол-5-ил)этил] декагидроизохинолин-3-карбоновая кислота (22).
A. Получение (3S, 4aS, 8aR)-этил-2-метоксикарбонил-6- (метоксиметилен)декагидроизохинолин-3-карбоксилата.
Суспензию (метоксиметил)трифенилфосфонийхлорида (11,9 г), предварительно промытую тетрагидрофураном, в пентане и высушенную под вакуумом при комнатной температуре в тетрагидрофуране (35 мл) при температуре 0oC добавляли в 1 М раствор бис(триметилсилил)амида натрия в тетрагидрофуране (34,6 мл). Через 30 минут реакционный раствор добавляли в раствор соединения из примера 4C (7,00 г) в тетрагидрофуране (50 мл) при температуре около 0oC. Реакцию прекращали добавлением воды. Полученный раствор разбавляли эфиром, органическую фазу отделяли и промывали водой. Соединенный органический раствор промывали рассолом, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток суспендировали в этилацетат/гексане (1:1) и полученную суспензию хранили при комнатной температуре в течение 10 минут, фильтровали, а фильтрат концентрировали под вакуумом. Твердый материал суспендировали в дополнительном количестве этилацетат/гексане (1:1), перемешивали при комнатной температуре и фильтровали. Соединенные фильтраты этилацетат/гексана концентрировали под вакуумом. Продукт очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/гексаном до получения 6,72 г искомого соединения.
B. Получение (3S, 4aR, 6R, 8aR)-этил-6-формил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 25A (6,72 г) в ацетонитриле (200 мл) обрабатывали 1 н. соляной кислотой (50,5 мл) и нагревали до 60oC. Спустя 18 часов реакционный раствор оставляли остывать до комнатной температуры. Полученную смесь обрабатывали насыщенным раствором бикарбоната натрия, а полученную смесь экстрагировали эфиром (3 раза). Соединенные эфирные экстракты высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 5,66 г искомого соединения.
C. Получение (3S, 4aR, 6S, 8aR)-этил-6-(2-цианоэтенил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Суспензию гидрида натрия (0,38 г 60%), предварительно промытого гексаном, в тетрагидрофуране (10 мл) охлаждали до 0oC. Охлажденную суспензию обрабатывали диэтилцианометилфосфонатом (1,67 г) при 0oC. Через 30 минут полученную смесь оставляли прогреваться до комнатной температуры, а затем обрабатывали раствором соединения из примера 25B (2,0 г) в тетрагидрофуране (5 мл). Спустя 16 минут реакционную смесь обрабатывали водой (30 мл) и экстрагировали эфиром (3 раза). Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 2,11 г искомого соединения.
D. Получение (3S, 4aR, 6S, 8aR)-этил-6-(2-цианоэтил)-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Смесь соединения из примера 25C (62,6 г) и 10% палладия на углероде (15,5 г) в этилацетате (125 мл) гидрировали при атмосферном давлении. Спустя шесть часов при комнатной температуре катализатор удаляли фильтрацией через HYFLOW, остаток на фильтре HYFLOW промывали этилацетатом, а соединенные фильтраты концентрировали под вакуумом до получения 58,13 г искомого соединения. Это соединение использовали на следующем этапе без дальнейшей очистки.
E. Получение (3S, 4aR, 6S, 8aR)-6-/2-(1(2)Н-тетразол-5-ил) этил/-декагидроизохинолин-3-карбоновой кислоты.
Смесь соединений из примера 25D (1,48 г) и трибутилоловоазида (2,96 г) нагревали до 80oC. Спустя три дня реакционную смесь обрабатывали толуолом (15 мл) и дополнительным количеством трибутилоловоазида (2 г) и возобновляли нагрев. Спустя шесть дней реакционную смесь обрабатывали 6 н. соляной кислотой (50 мл) и нагревали с обратным холодильником. Через 18 часов нагрева смесь оставляли охлаждаться до комнатной температуры. Полученную смесь экстрагировали эфиром (6 раз) и водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX50x8", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавляли водой и концентрировали под вакуумом. Эту процедуру повторяли. Остаток разбавляли ацетоном и нагревали с обратным холодильником. Через час такого нагрева смесь фильтровали, твердые материалы промывали эфиром. Твердый материал высушивали под вакуумом при 60oC в течение около 18 часов до получения 1,13 г искомого соединения. Температура плавления 201 - 209oC.
[α]D = +20,4o (c = 1, 1 н. HCl).
Расчетные данные по C13H21N5O2 • 0,75 H2O, %: C 53,32; H 7,74; N 23,91. Обнаружено, %: C 53,29; H 7,80; N 24,09.
Пример 26. (3S, 4aR, 6S, 8aR)-6-[2-(1(2)Н-тетразол-5-ил)-2- тиаэтил]декагидроизохинолин-3-карбоновая кислота (23).
A. Получение (3S, 4aR, 8aR)-этил-6-метиленил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Реакция метилтрифенилфосфонийбромида (6,66 г), 1 М раствора бис(триметилсилил)амида натрия в тетрагидрофуране (18 мл) и соединения из примера 4C (3,1 г), описанная в примере 10A, дает сырое искомое соединение. Этот материал очищают силикагельной экспресс-хроматографией, элюируя 30% этилацетат/гексаном до получения 2,88 г искомого соединения.
B. Получение (3S, 4aR, 6R, 8aR)этил-6-гидроксиметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Охлажденный (0oC) раствор соединения из примера 26A (2,82 г) в тетрагидрофуране (40 мл) обрабатывали 2,0 молярным раствором боранметилсульфида в тетрагидрофуране (7,5 мл). Спустя три часа реакционный раствор обрабатывали эфиром и смесью 3 н. гидроксида натрия (15 мл) с 30% перекисью водорода (15 мл). Через 15 мин выдерживания при 0oC эту смесь обрабатывали 10% бисульфатом натрия (40 мл). Органическую фазу отделяли, а водную фазу экстрагировали эфиром. Органические фазы соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 65% этилацетат/ гексаном до получения 1,34 г искомого соединения.
C. Получение (3S, 4aR, 6S, 8aR)-этил-6-бромметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор трифенилфосфина (2,23 г) в метиленхлориде (18 мл) обрабатывали бромом до устойчивого желтого цвета (1,14 г). Затем добавляли дополнительное количество трифенилфосфина до тех пор, пока раствор не обесцвечивался. Этот раствор обрабатывали раствором соединения из примера 26B (1,33 г) и пиридина (0,88 г) в метиленхлориде (5 мл). Реакционную смесь выдерживали при 0oC в течение 25 минут, затем позволяли ей прогреться до комнатной температуры. Спустя еще 25 минут при комнатной температуре реакционную смесь обрабатывали 10% бисульфатом натрия (50 мл). Органическую фазу удаляли, а водную экстрагировали эфиром. Органические фазы соединяли, высушивали, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/ гексаном до получения 1,40 г искомого соединения.
D. Получение (3S, 4aR, 6S, 8aR)-этил-6-[2-(1(2)-Н-тетразол-5- ил)-2-тиаэтил]-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 26C (1,31 г), тиотетразола (0,41 г) и триэтиламина (0,73 г) в безводном ацетонитриле (12 мл) нагревали до 80oC. Спустя 18 часов реакционный раствор разбавляли этилацетатом, полученную смесь экстрагировали 10% бисульфатом натрия. Органическую фазу удаляли, а водную экстрагировали этилацетатом (два раза). Органические фазы соединяли, высушивали, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя смесью уксусная кислота/ этилацетат/гексан (4: 36:60). Фракции, содержащие требуемое соединение, соединяли и концентрировали под вакуумом. Остаток обрабатывали толуолом и концентрировали под вакуумом. Остаток обрабатывали метанолом и концентрировали под вакуумом, после чего обрабатывали этилацетатом и еще раз концентрировали под вакуумом. Полученный остаток обрабатывали хлороформом, а полученную смесь фильтровали и фильтрат концентрировали под вакуумом до получения 1,37 г искомого соединения.
E. Получение (3S, 4aR, 6S, 8aR)-6-[2(1(2)Н-тетразол-5-ил)-2- тиаэтил]декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 26D (1,30 г) в 6 н. соляной кислоте (20 мл) нагревали с обратным холодильником в течение 4 часов. Раствор оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50x8-100", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, концентрировали под вакуумом. Остаток обрабатывали ацетоном, затем нагревали с обратным холодильником в течение получаса. Охлажденную смесь фильтровали и твердый материал промывали ацетоном и эфиром, затем высушивали под вакуумом в течение приблизительно 18 часов при 60oC до получения 0,73 г искомого соединения. Температура плавления 199 - 207oC.
[α]D = -53,2o (C = 1, 1 н. HCl).
Расчетные данные по C12H19N5O2S, %: C 48,47; H 6,44; N 23,55. Обнаружено, %: C 48,37; H 6,74; N 23,80.
Пример 27. (3R, 4aS, 6R, 8aS)-6-[2-(1(2)Н-тетразол-5-ил)-2- тиаэтил]декагидроизохинолин-3-карбоновая кислота (24).
Искомое соединение (0,81 г) получали из соединения из примера 4A, используя способ, описанный в примере 26. Температура плавления 165 -172oC.
[α]D = +49,3o (c = 1, 1н. HCl).
Расчетные данные по C12H19N5O2S • 0,5H2O • 0,1C5H5, %: C 47,77; H 6,57; N 22,72. Обнаружено, %: C 47,97; H 6,75; N 22,75.
Пример 28. (3S, 4aR, 6R, 8aR)-6-[2(1(2)Н-тетразол-5-ил)-2- тиаэтил]декагидроизохинолин-3-карбоновая кислота (25).
A. Получение (3S, 4aR, 6R, 8aR)-этил-6-гидроксиметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 25B (1,92 г) в этаноле (19,5 мл) охлаждали до 0oC и обрабатывали борогидридом натрия (0,24 г). Спустя 20 минут раствор осторожно обрабатывали 10% бисульфатом натрия (15 мл). Полученную смесь экстрагировали метиленхлоридом (дважды) и эфиром (дважды). Органические экстракты соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом до получения 1,94 г искомого соединения. Этот материал использовали на следующем этапе без дальнейшей очистки.
B. Получение (3S, 4aR, 6R, 8aR)-этил-6-бромметил-2- метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор трифенилфосфина (4,72 г) в метиленхлориде (10,5 мл) обрабатывали бромом до устойчивого желтого цвета (2,58 г). Добавляли дополнительное количество трифенилфосфина до тех пор, пока раствор не обеспечивался. Полученный раствор обрабатывали раствором соединения из примера 28A (1,93 г) и пиридина (1,53 г) в метиленхлориде (10,5 мл). Через два часа при комнатной температуре реакционную смесь экстрагировали 10% бисульфатом натрия. Водный экстракт экстрагировали эфиром (трижды). Эфирные экстракты соединяли с органической фазой, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток обрабатывали эфиром и выпавший в осадок трифенилфосфиноксид удаляли фильтрацией. Фильтрат концентрировали под вакуумом, остаток обрабатывали дополнительным количеством эфира до выпадения в осадок оставшихся следовых количеств трифенилфосфиноксида. Остаток очищали силикагельной экспресс-хроматографией, элюируя 35% этилацетат/гексаном до получения 1,56 г искомого соединения.
C. Получение (3S, 4aR, 6R, 8aR)-этил-6-[2-(1(2)Н-тетразол-5-ил)- 2-тиаэтил]-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 28B (1,56 г), тиотетразола (0,53 г) и триэтиламина (0,96 г) в безводном ацетонитриле (13 мл) нагревали до 80oC. Через четыре часа реакционный раствор разделяли на части между этилацетатом и 10% бисульфатом натрия. Фазы отделяли, водную фазу экстрагировали дополнительным количеством этилацетата (трижды). Этилацетатные экстракты соединяли, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток делили на две порции, каждую очищали силикагельной радиальной хроматографией на "CHROMATOTRON" (пластины 4 мм), уравновешивая пластину 35 этилацетат/гексаном и элюируя смесью уксусная кислота/этилацетат/гексан (4: 36:60) до получения 1,14 г искомого соединения.
D. Получение (3S, 4aR, 6R, 8аR)-6-[2-(1(2)Н-тетразол-5-ил)- 2-тиаэтил] декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 28C (1,14 г) в 6 н. соляной кислоте (50 мл) нагревали до 100oC. После нагрева в течение около 18 часов реакционный раствор оставляли охлаждаться до комнатной температуры и концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, концентрировали под вакуумом. Остаток обрабатывали водой и концентрировали под вакуумом. Эту процедуру повторяли. Остаток обрабатывали ацетоном, а полученную смесь нагревали с обратным холодильником в течение часа. После охлаждения до комнатной температуры смесь фильтровали. Твердый материал промывали ацетоном и эфиром, затем высушивали под вакуумом при 60oC до получения 0,197 искомого соединения.
[α]D = +34,6o (c = 1, 1 н. HCl).
Расчетные данные по C12H19N5O2S • 0,3H2O, %: C 47,6; H 6,52; N 23,13. Обнаружено, %: C 47,35; H 6,23; N 23,10.
Пример 29. (3S, 4aR, 6R, 8aR)-6-/(1(2-4)Н-1,2,4-триазол-5-ил) сульфонилметил/декагидроизохинолин-3-карбоновая кислота (26).
A. Получение (3S, 4aR, 6S, 8aR)-этил-6-[2-(1(2)Н-триазол-5-ил) -2-тиаэтил]-2-метоксикарбонилдекагидроизохинолин-3-карбоксилата.
Раствор соединения из примера 26C (2,80 г) в безводном диметилформамиде (23 мл) обрабатывали 1Н-1,2,4-триазол-3-тиолом (0,94 г) и триэтиламином (1,88 г). Полученный раствор нагревали до 100oC в азотной атмосфере. Спустя четыре часа реакционный раствор оставили остывать до комнатной температуры и обработали 10% бисульфатом натрия. Полученную смесь экстрагировали смесью хлороформ/этилацетат (1: 1). Органические фазы соединили, высушивали сульфатом магния, фильтровали и концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя ступенчатым градиентом 35% этилацетат/гексаном (500 мл), а затем 75% этилацетат/гексаном (2 л) до получения 2,72 г искомого соединения.
B. Получение (3S, 4aR, 6S, 8aR)-6-/(1(2-4)Н-1,2,4-триазол-5-ил)- сульфонилметил/декагидроизохинолин-3-карбоновой кислоты.
Раствор соединения из примера 29A (2,72 г) в метиленхлориде обрабатывали 3-хлорнадбензойной кислотой (3,84 г) в трех порциях в течение 30 минут. Через 18 часов выдерживания при комнатной температуре реакционную смесь концентрировали под вакуумом. Остаток очищали силикагельной экспресс-хроматографией, элюируя ступенчатым градиентом 50% этилацетат/гексана (200 мл), а затем этилацетатом (200 мл) и 2,5% смесью уксусная кислота/этилацетат (500 мл). Фракции, содержащие искомый продукт, соединяли и концентрировали под вакуумом. Остаток обрабатывали смесью толуол/метанол и концентрировали под вакуумом. Полученный остаток обрабатывали метанолом и концентрировали под вакуумом, затем обрабатывали хлороформом и концентрировали под вакуумом до получения 2,50 г.
Продукт реакций из предыдущего абзаца обрабатывали 6 н. соляной кислотой (50 мл) и нагревали до 110oC. Через три часа реакционную смесь оставляли остывать до комнатной температуры, затем экстрагировали эфиром. Водную фазу концентрировали под вакуумом. Остаток очищали ионообменной хроматографией на "DOWEX 50X8-100", элюируя 10% пиридин/водой. Фракции, содержащие искомое соединение, соединяли и концентрировали под вакуумом. Остаток разбавили водой и концентрировали под вакуумом. Этот процесс повторяли дважды, затем остаток концентрировали под вакуумом в течение около 18 часов. Полученный остаток разбавляли ацетоном и перегоняли в течение часа. Искомое соединение удаляли фильтрацией, затем промывали ацетоном и эфиром. Твердые материалы высушивали под вакуумом при 60oC в течение около 18 часов до получения 1,60 г искомого соединения. Температура плавления 286 - 287oC.
[α]D = -39,4o (c = 1, 1 н. HCl).
Расчетные данные по C13H20N4O4S • 0,6H2O, %: C 46,03; H 6,30; N 16,52. Обнаружено, %: C 46,13; H 6,37; N 16,17.
Пример 30. (3R, 4aS, 6R, 8aS)-6-/(1(2-4)Н-1,2,4-триазол-5-ил) сульфонилметил/-декагидроизохинолин-3-карбоновая кислота (27).
Искомое соединение (1,16 г) получали из соединения из примера 4A, используя способ, описанный в примерах 26A-26C и 29. Температура плавления 267 - 270oC.
[α]D = + 33,8oC (c = 1, 1 н. HCl).
Расчетные данные по C13H20N4O4S • H2O, %: C 45,08; H 6,40; N 16,17. Обнаружено, %: C 45,47; H 6,57; N 16,16.
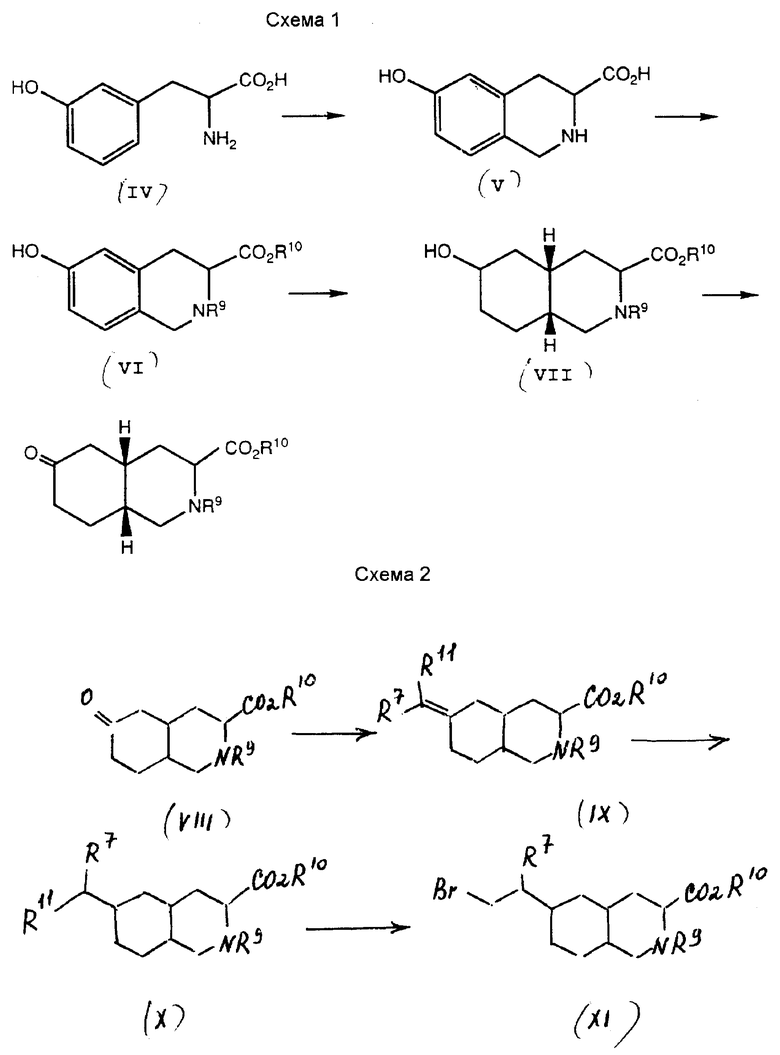
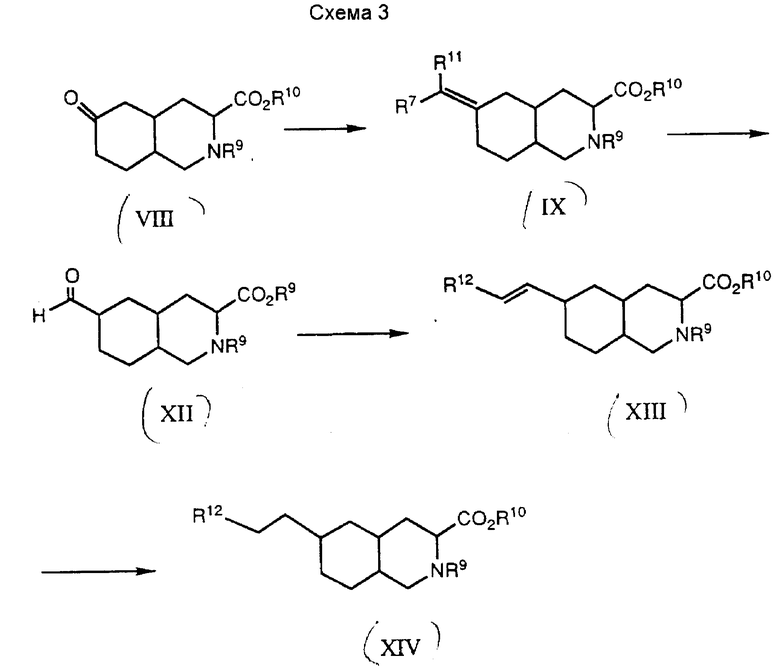
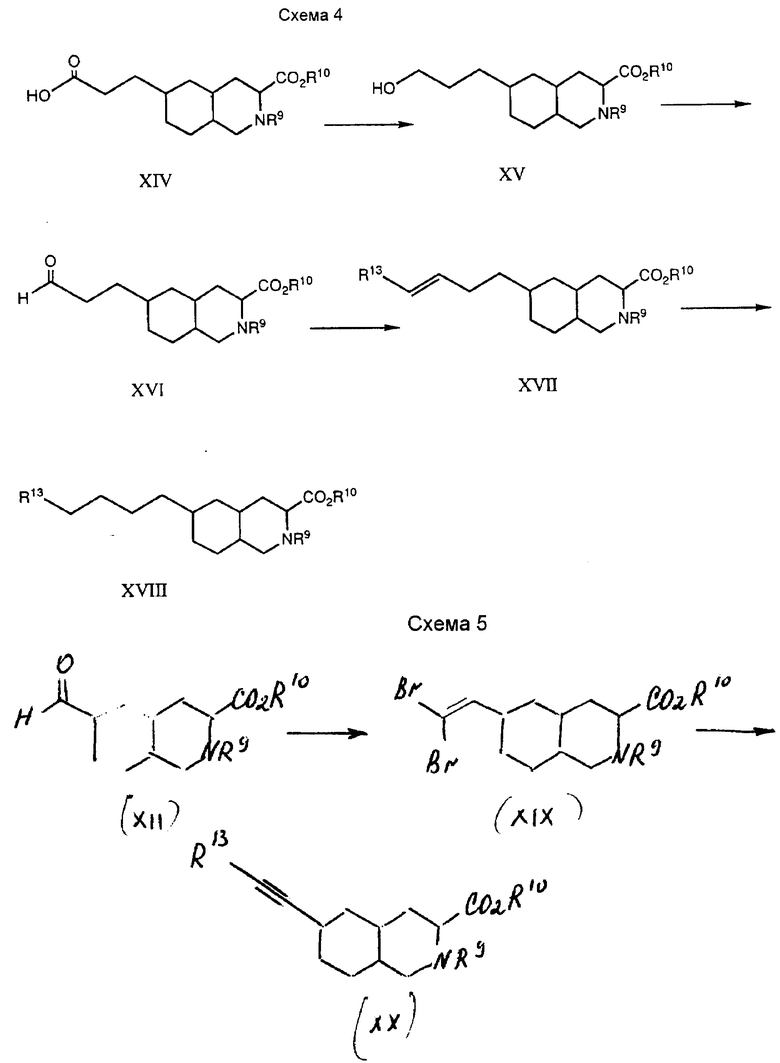
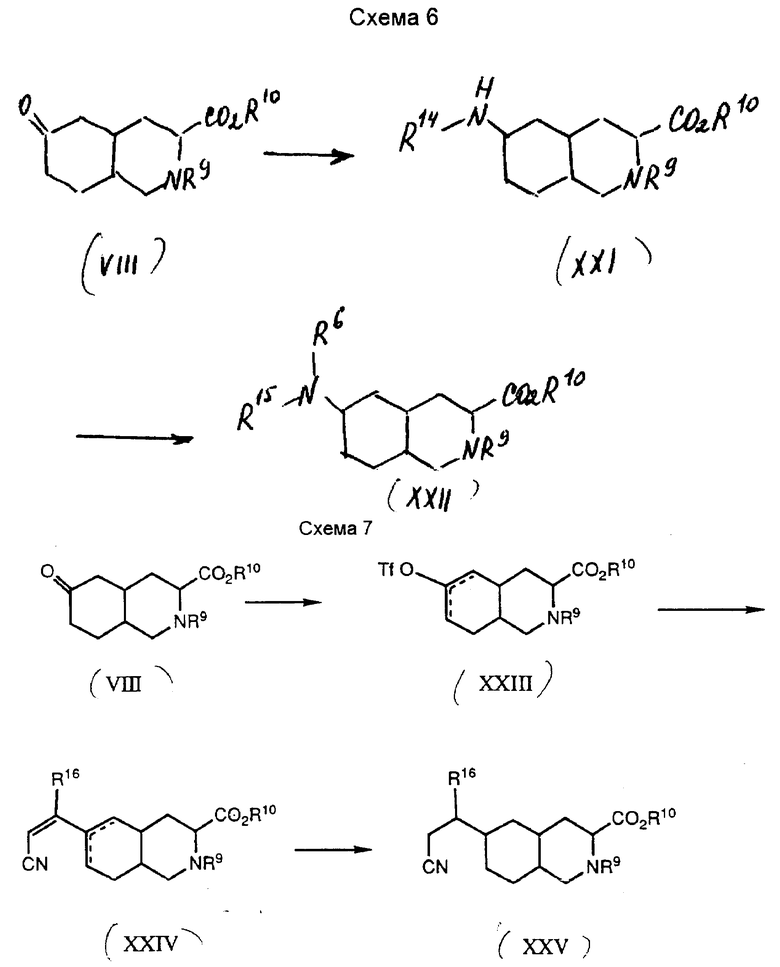
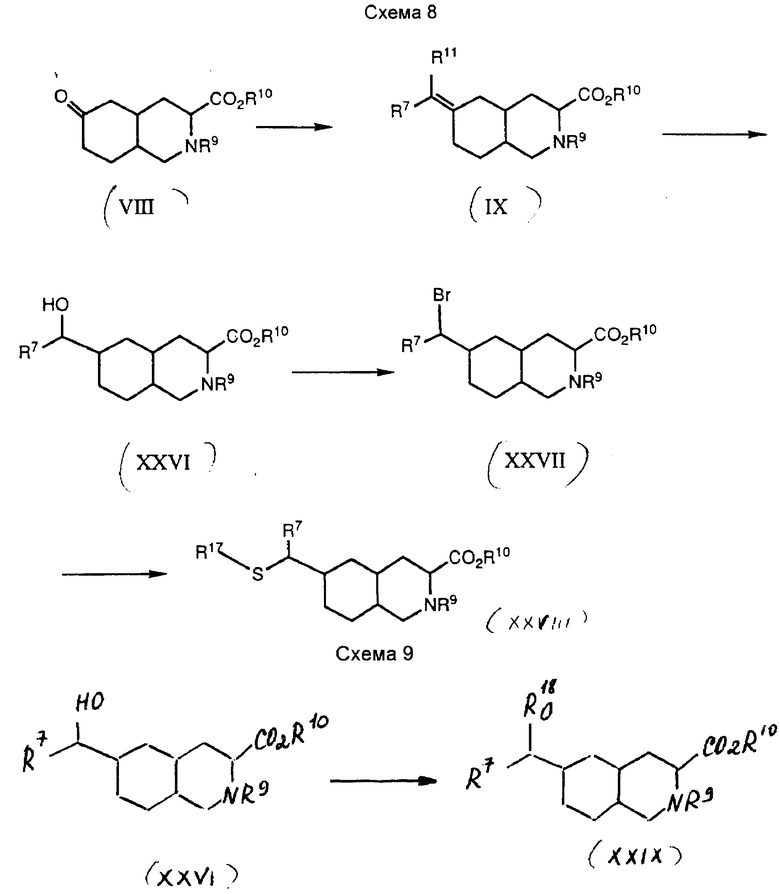
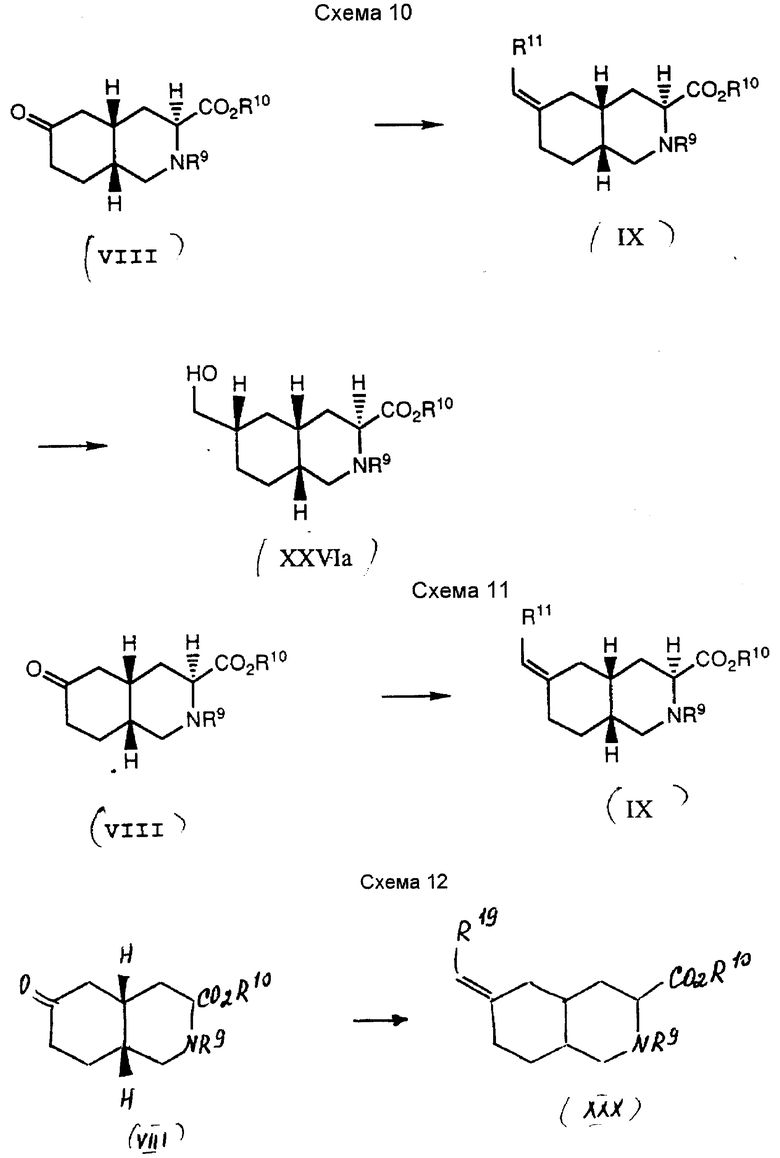
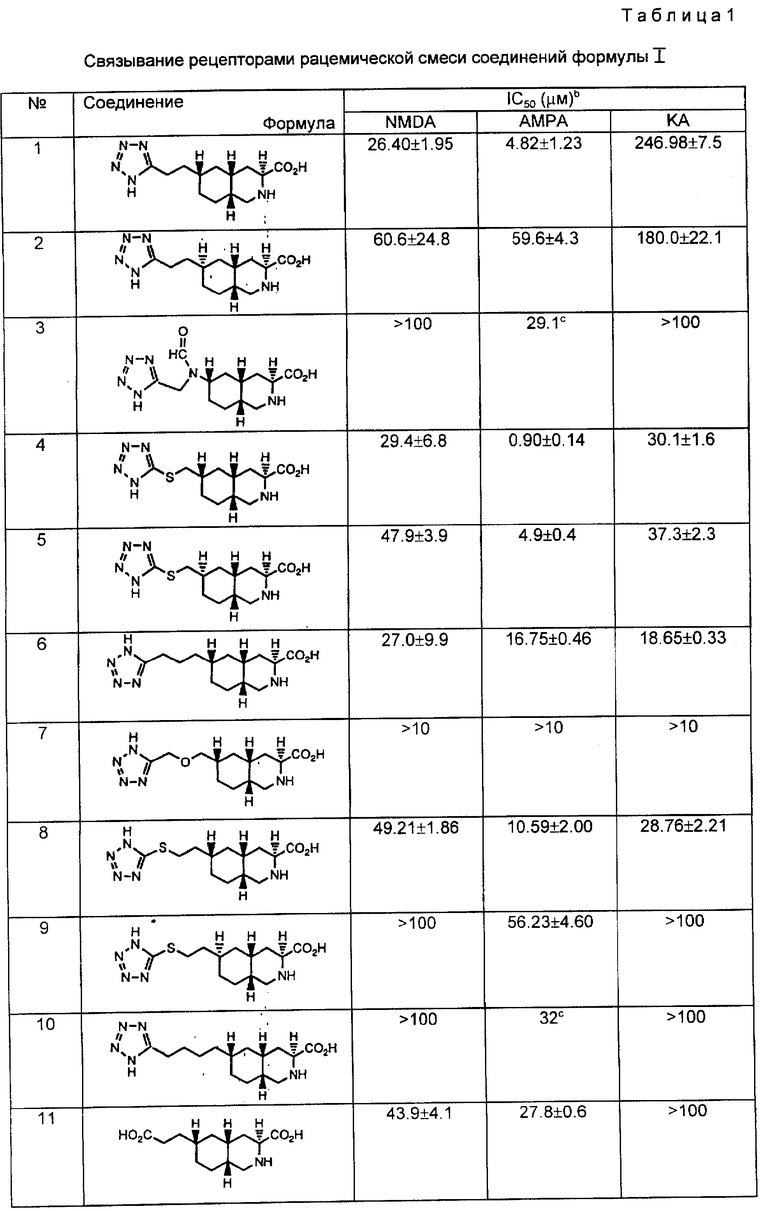

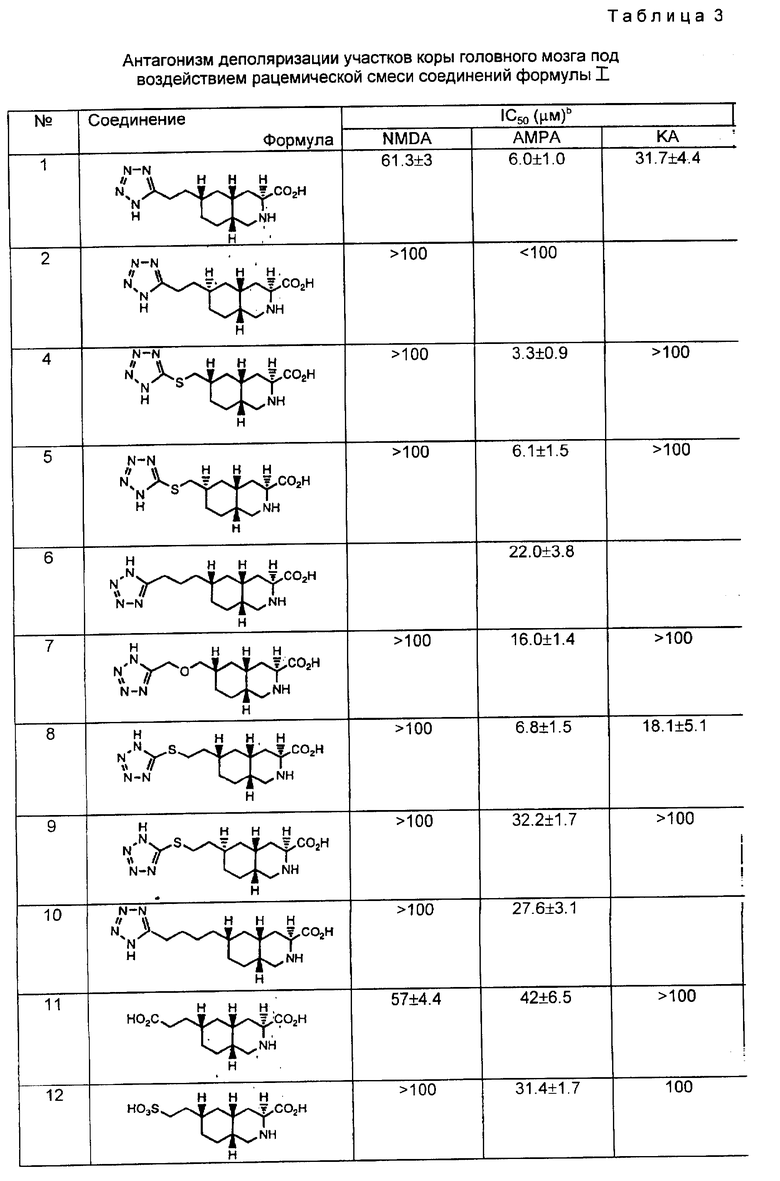
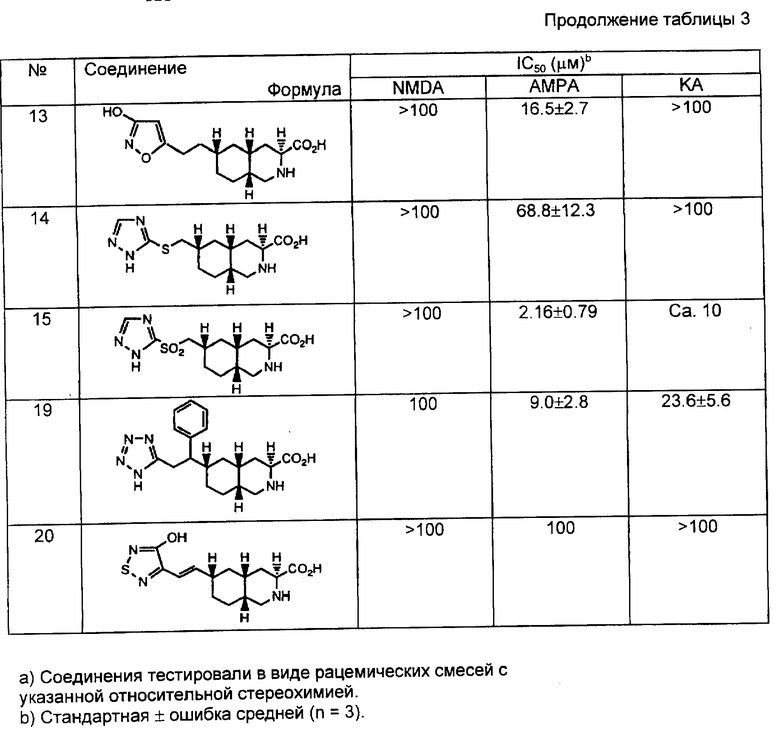
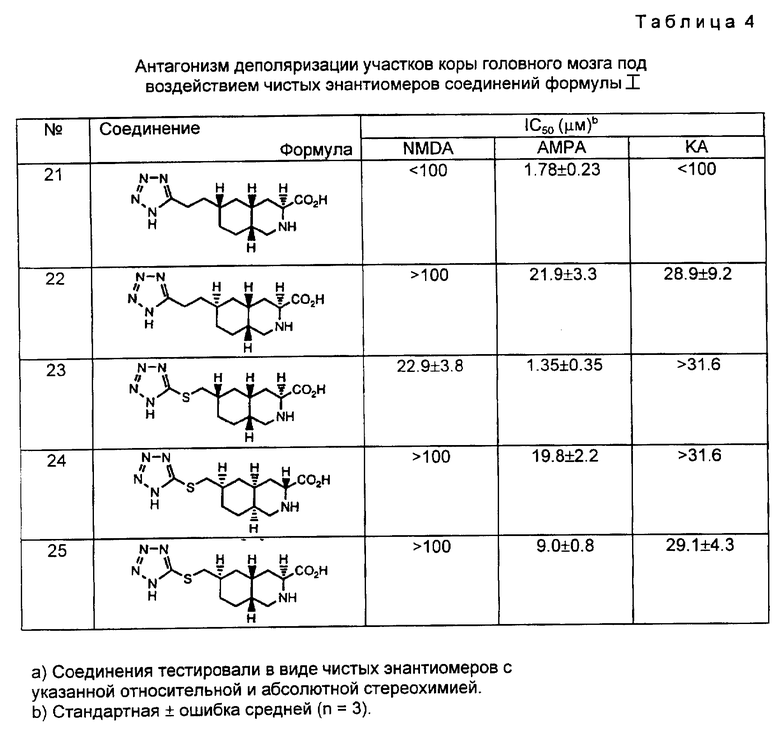
Изобретение относится к производным декагидроизохинолинов формулы , проявляющим активность антагониста рецептора аминокислот-медиаторов возбуждения, которые могут найти применение в медицине, и к фармацевтической композиции на их основе. 3 с. и 5 з.п. ф-лы, 4 табл.
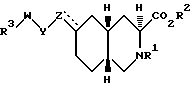
R1 - водород, алкил C1 - C10, арилалкил, алкоксикарбонил или ацил;
R2 - водород, алкил C1 - C6, замещенный алкил, циклоалкил или арилалкил;
R3 - CO2H, SO3H, CONHSO2R8 или группу формулы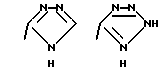
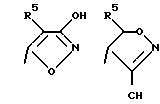
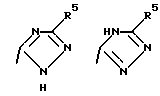
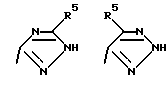
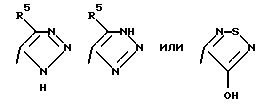
W - (CH2)n, S, SO, SO2;
Y - CHR7, NR4, O, S, SO или SO2;
Z - NR6, CHR7 или CH;
или W и Y вместе представляют собой HC = HC или C ≡ C, или Y и Z вместе представляют собой HC = HC или C ≡ C;
R4 - водород, алкил C1 - C4, фенил или ацил;
R5 - водород, алкил C1 - C4, CF3, фенил, гидрокси, амино, бром, йод или хлор;
R6 - ацил;
R7 независимо - водород, алкил C1 - C4, фенил или замещенный фенил;
R8 - алкил C1 - C4 или тетразол-5-ил;
n = 0, 1 или 2,
при условии, что, когда Y - NR4, O, S, SO или SO2, то W - (CH2)n и Z - CHR7 или CH; при условии, что, когда W - S, SO или SO2, то Y - CHR7, Z - CHR7 или CH, или Y и Z вместе представляют собой HC = CH или C ≡ C; при условии, что, когда W и Z - CH2, то Y не является S; при условии, что, когда W и Y вместе представляют собой HC = CH или C ≡ C, Z - CHR7,
или их фармацевтически приемлемые соли.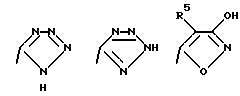
W - S, SO2 или (CH2)n, n = 0, 1 или 2; Y - CHR7, S или SO2, Z - CHR7, R5 - водород, алкил C1 - C4 или CF3, R7 - водород, алкил C1 - C4 или фенил, или фармацевтически приемлемая соль этого соединения.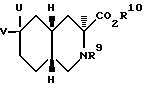
где R9 - алкоксикарбонил, ацил;
R10 - водород алкил C1 - C6 или арил;
U - гидроксил, гидроксиметил, формил, бромэтил, бромметил или гидроксиэтил;
V - водород,
или U и V вместе представляют собой метилен или метоксиметилен.
| EP, заявка, 0383504A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
