Область применения изобретения
Изобретение относится к реализованному на компьютере способу определения показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, способу получения по меньшей мере части последовательности и способу определения последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени.
Предпосылки создания изобретения
Возможность секвенирования молекул нуклеиновых кислот является инструментом, который очень полезен во множестве различных областей применения. Однако может быть сложно определять точные последовательности молекул нуклеиновых кислот, которые содержат проблемные структуры, такие как молекулы нуклеиновой кислоты, которые содержат повторяющиеся области. Также может быть сложно разрешать структурные особенности, такие как гаплотипная структура диплоидных и полиплоидных организмов и структурные варианты в геномах этих организмов.
Многие из более современных методик (так называемые методики секвенирования следующего поколения) способны точно секвенировать только короткие молекулы нуклеиновых кислот. Методики секвенирования следующего поколения можно использовать для секвенирования более длинных последовательностей нуклеиновых кислот, но это часто бывает сложным и дорогостоящим. Методики секвенирования следующего поколения можно использовать для генерации коротких прочтений последовательности, соответствующих последовательностям участков молекулы нуклеиновой кислоты, и полная последовательность может быть собрана из этих коротких прочтений последовательности. Если молекула нуклеиновой кислоты содержит повторяющиеся области, пользователю может быть неясно, соответствуют ли два прочтения последовательности, имеющих сходные последовательности, последовательностям двух повторов в более длинной последовательности или двух репликаций одной и той же последовательности. Аналогичным образом пользователь может желать выполнить секвенирование двух сходных молекул нуклеиновых кислот одновременно, и может быть сложно определить, соответствуют ли два прочтения последовательности, имеющих сходные последовательности, последовательностям одной и той же исходной молекулы нуклеиновой кислоты или двум разным исходным молекулам нуклеиновой кислоты.
В сборке последовательностей из коротких прочтений последовательности могут помочь методики секвенирования с использованием мутагенеза (SAM). В целом, SAM включает в себя введение мутаций в темплатные последовательности нуклеиновых кислот-мишеней. Схемы внедренных мутаций могут помочь пользователю способа при сборке последовательностей молекул нуклеиновых кислот из коротких прочтений последовательности.
Например, в тех случаях, когда темплатные молекулы нуклеиновых кислот содержат повторяющиеся области, повторы можно отличать друг от друга по различным схемам мутаций, что позволяет разрешать и собирать повторяющиеся области правильно.
В целом, SAM-методики включают в себя введение мутаций в копии молекулы темплатной нуклеиновой кислоты-мишени с получением мутированной молекулы темплатной нуклеиновой кислоты-мишени и/или одной или более содержащих мутации последовательностей, секвенирование одной или более содержащих мутации последовательностей с получением SAM-данных, включающих мутированные прочтения последовательности, и затем сборку последовательностей из мутированных прочтений последовательности на основе соответствующих схем мутаций. Поскольку различные мутированные копии будут содержать мутации в разных положениях, собранная последовательность может характеризовать исходную молекулу темплатной нуклеиновой кислоты.
Однако сохраняется потребность в более надежных и/или более вычислительно эффективных способах обработки SAM-данных.
Изложение сущности изобретения
Авторы настоящего изобретения разработали новые улучшенные способы обработки SAM-данных, включающих мутированные прочтения последовательности. Таким образом, в одном аспекте изобретения предложен реализованный на компьютере способ определения показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности. Способ включает прием множества мутированных прочтений последовательности. Каждое мутированное прочтение последовательности соответствует подпоследовательности из содержащей мутации последовательности. Содержащая мутации последовательность содержит мутации по сравнению с не содержащей мутаций последовательностью. Способ дополнительно включает применение общей минимизирующей функции для каждого мутированного прочтения последовательности с определением таким образом одного или более соответствующих минимизаторов для каждого мутированного прочтения последовательности. Способ дополнительно включает определение положений одного или более соответствующих минимизаторов в каждом мутированном прочтении последовательности. Способ дополнительно включает определение положений одной или более мутаций в каждом мутированном прочтении последовательности. Для по меньшей мере двух мутированных прочтений последовательности с общим минимизатором способ дополнительно включает подсчет количества мутаций с совпадающим положением и/или с несовпадающим положением, когда соответствующие минимизаторы выравнены.
В другом аспекте настоящего изобретения предложен способ получения по меньшей мере части последовательности молекулы темплатной нуклеиновой кислоты-мишени.
В другом аспекте настоящего изобретения предложен способ определения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени.
Дополнительные аспекты настоящего изобретения представлены в зависимых пунктах формулы изобретения и в подробном описании.
Краткое описание графических материалов
На Фиг. 1 представлен вариант осуществления способа определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в соответствии с настоящим изобретением.
На Фиг. 2 представлен вариант осуществления способа определения показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, в соответствии с настоящим изобретением.
На Фиг. 3 представлен пример стадии определения положений одной или более мутаций в мутированном прочтении последовательности.
На Фиг. 4А представлен сравнительный пример сборки из коротких чтений генома Arcobacter butzlerii размером 2,3 млн.п.н. без использования способа настоящего изобретения.
На Фиг. 4В представлен пример сборки генома Arcobacter butzlerii размером 2,3 млн.п.н. с использованием способа настоящего изобретения.
На Фиг. 5 представлены экспериментальные данные о влиянии глубины покрытия короткими чтениями длинного темплата на результаты способа настоящего изобретения.
Подробное описание изобретения
Общие определения
Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют общепринятое значение, понятное специалисту в области, к которой относится настоящее изобретение.
В целом термин «содержащий» означает «включающий, без ограничений». Например, фразу «способ, включающий [некоторые стадии]» следует интерпретировать как то, что способ включает перечисленные стадии, но могут быть выполнены дополнительные стадии.
В некоторых вариантах осуществления изобретения слово «содержащий» заменяют фразой «состоящий из». Термин «состоящий из» подразумевает ограничение. Например, фразу «способ, состоящий из [некоторых стадий] » следует понимать как то, что способ включает в себя указанные стадии, и дополнительные стадии не выполняются.
В некоторых аспектах в изобретении предложен способ определения или получения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Способ можно использовать для определения или получения полной последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. В альтернативном варианте осуществления способ может быть использован для определения или получения частичной последовательности, т.е. последовательности части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Например, если невозможно или непросто определить полную последовательность, пользователь может решить, что последовательность части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени является полезной или даже достаточной для его цели.
Для целей настоящего изобретения термин «молекула нуклеиновой кислоты» (или «немутированная молекула нуклеиновой кислоты») относится к полимерной форме нуклеотидов, имеющих любую длину. Нуклеотиды могут представлять собой дезоксирибонуклеотиды, рибонуклеотиды или их аналоги. Предпочтительно по меньшей мере одна молекула нуклеиновой кислоты состоит из дезоксирибонуклеотидов или рибонуклеотидов. Еще более предпочтительно по меньшей мере одна молекула нуклеиновой кислоты состоит из дезоксирибонуклеотидов, т.е. по меньшей мере одна молекула нуклеиновой кислоты представляет собой молекулу ДНК.
«Молекула темплатной нуклеиновой кислоты-мишени» может представлять собой любую молекулу нуклеиновой кислоты, которую пользователь желает секвенировать.
По меньшей мере одна «молекула темплатной нуклеиновой кислоты-мишени» может быть одноцепочечной или может быть частью двухцепочечного комплекса. Если по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени состоит из дезоксирибонуклеотидов, она может образовывать часть двухцепочечного комплекса ДНК. В этом случае одна цепь (например, кодирующая цепь) будет считаться по меньшей мере одной молекулой темплатной нуклеиновой кислоты-мишени, а другая цепь представляет собой молекулу нуклеиновой кислоты, которая комплементарна по меньшей мере одной молекуле темплатной нуклеиновой кислоты-мишени. По меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени может представлять собой молекулу ДНК, соответствующую гену, может содержать интроны, может представлять собой межгенную область, может представлять собой интрагенную область, может представлять собой геномную область, охватывающую множество генов, или может в действительности представлять собой весь геном организма.
Для целей настоящего изобретения термин «мутированная молекула нуклеиновой кислоты» или «мутированная молекула темплатной нуклеиновой кислоты-мишени» относится к «молекуле нуклеиновой кислоты» или «молекуле темплатной нуклеиновой кислоты-мишени», в которую были введены мутации. Мутации могут представлять собой мутации по типу замены, необязательно мутации по типу транзиции. Для целей настоящего изобретения термин «мутация по типу замены» следует интерпретировать как то, что нуклеотид заменен на другой нуклеотид. Например, превращение последовательности АТСС в последовательность AGCC вводит одну мутацию по типу замены. Для целей настоящего изобретения термин «мутация по типу транзиции» следует интерпретировать как то, что нуклеотид А заменен нуклеотидом G и наоборот (т.е. мутации A⇔G) или что нуклеотид С заменен нуклеотидом Т и наоборот (т.е. мутации С⇔Т).
Фраза «введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени» обозначает воздействие на по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором из пары образцов условий, в которых по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени мутирует. Этого можно добиться, используя любой подходящий способ. Например, мутации могут быть введены путем химического мутагенеза и/или ферментативного мутагенеза.
Для целей настоящего изобретения «содержащая мутации последовательность» соответствует по меньшей мере части нуклеотидной последовательности в «мутированной молекуле нуклеиновой кислоты» или в «мутированной молекуле темплатной нуклеиновой кислоты-мишени». «Содержащая мутации последовательность» также может называться «мутированной последовательностью». «Содержащая мутации последовательность» в настоящем документе обозначена как μi, а множество (т.е. несколько) «содержащих мутации последовательностей» обозначено как М, где μ1…μn ∈ M. «Не содержащая мутаций последовательность» соответствует по меньшей мере части последовательности нуклеотидов в «молекуле нуклеиновой кислоты» или «молекуле темплатной нуклеиновой кислоты-мишени». «Не содержащая мутаций последовательность» также может называться «немутированная последовательность». «Не содержащая мутаций последовательность» в настоящем документе обозначена как Si, а множество (т.е. несколько) «не содержащих мутаций последовательностей» обозначено как S, где S1…Sn ∈ S. «Содержащая мутации последовательность» и «не содержащая мутаций последовательность», таким образом, могут соответствовать по меньшей мере части последовательности молекулы нуклеиновой кислоты из нуклеотидов (нт) аденина (А), тимина (Т), гуанина (G) и цитозина (С). Такая хромосомная последовательность может иметь длину в диапазоне от 103 до 109 нуклеотидов (нт) и более.
Для целей настоящего изобретения «мутированное прочтение последовательности» соответствует подпоследовательности из «содержащей мутации последовательности», т.е «мутированное прочтение последовательности» может быть по существу идентичным по меньшей мере подпоследовательности «содержащей мутации последовательности», но оно содержит мутации по сравнению с содержащей мутации последовательностью и может содержать дополнительные небольшие различия из-за ошибок считывания. «Мутированное прочтение последовательности» обозначено как ρi, а множество (т.е. несколько) «мутированных прочтений последовательности» обозначены как Р, где ρ1…pn ∈ P. «Немутированное прочтение последовательности» соответствует подпоследовательности «не содержащей мутаций последовательности», т.е. «немутированное прочтение последовательности» может быть по существу идентичным подпоследовательности «не содержащей мутаций последовательности», за исключением ошибок считывания во время секвенирования. «Немутированное прочтение последовательности» обозначено как ri, а множество (т.е. несколько) «немутированных прочтений последовательности» обозначено как R, где r1…rn ∈ R. «Мутированное прочтение последовательности» может быть получено путем секвенирования области «мутированной молекулы темплатной нуклеиновой кислоты-мишени», а «немутированное прочтение последовательности» может быть получено путем секвенирования области «молекулы темплатной нуклеиновой кислоты-мишени». Прочтение последовательности может иметь длину, которая меньше, чем последовательность, например длину около 150 нт.
Способ 10 анализа последовательности
На Фиг. 1 показан способ 10 определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в соответствии с изобретением.
Способ 10 определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может включать стадию S110 подготовки образца. Стадия S110 подготовки образца может включать в себя обеспечение пары молекул темплатной нуклеиновых кислот-мишеней и введение мутаций в одну из пары молекул темплатных нуклеиновых кислот-мишеней с получением мутированной молекулы темплатной нуклеиновой кислоты-мишени. Стадия S110 подготовки образца может включать в себя любые известные методики обеспечения молекулы темплатной нуклеиновой кислоты-мишени и мутированной молекулы темплатной нуклеиновой кислоты-мишени.
Способ 10 определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может дополнительно включать стадию S120 секвенирования. Стадия S120 секвенирования включает секвенирование областей по меньшей мере одной содержащей мутации молекулы темплатной нуклеиновой кислоты-мишени с получением таким образом множества мутированных прочтений Р последовательности. Кроме того, стадия S120 секвенирования может включать секвенирование областей по меньшей мере одной (немутированной) молекулы темплатной нуклеиновой кислоты-мишени (молекулы темплатной нуклеиновой кислоты-мишени, которая соответствует мутированной молекуле темплатной нуклеиновой кислоты-мишени) с получением в результате множества немутированных прочтений R последовательности. Стадия S120 может включать любые известные методики получения множества мутированных прочтений Р последовательности.
Способ 10 определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени включает стадию 200 или способ 200 определения того, получены ли (или происходят ли) два мутированных прочтения ρi, ρj последовательности от одной и той же содержащей мутации последовательности μi. Определение того, получены ли (или происходят ли) два мутированных прочтения ρi, ρj последовательности от одной и той же содержащей мутации последовательности μi, включает определение того, получены ли (или происходят ли) два мутированных прочтения ρi, ρj последовательности от одной и той же или сходной или перекрывающейся части содержащей мутации последовательности μi, т.е. содержат ли оба мутированных прочтения ρi, ρj последовательности подпоследовательность, которая соответствует одной и той же части содержащей мутации последовательности μi. Способ 200 представляет собой реализованный на компьютере способ и может быть осуществлен процессором компьютера. Способ 200 формирует показатель, коррелированный с вероятностью того, что два мутированных прочтения ρi, ρj последовательности происходят от одной и той же содержащей мутации последовательности μi.
Способ 10 определения по меньшей мере части по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может дополнительно включать стадию S300 сборки последовательности. Стадия S300 сборки последовательности включает сборку или реконструкцию по меньшей мере части последовательности μi, Si. Содержащую мутации последовательность μi можно получать путем сборки множества мутированных прочтений Р последовательности на основе показателя, коррелированного с вероятностью того, что соответствующие два мутированных прочтения ρi, ρj последовательности происходят от одной и той же содержащей мутации последовательности μi. Это может быть достигнуто, например, путем группировки множества мутированных прочтений Р последовательности в группы, соответствующие последовательностям, содержащим мутации μi, и затем сборки каждой группы отдельно с реконструкцией части или всех отдельных содержащих мутации последовательностей μi. Не содержащая мутаций последовательность Si может быть получена путем коррекции ошибок содержащей мутации последовательности μi, например, путем выведения наиболее вероятной не содержащей мутаций последовательности Si из содержащей мутации последовательности μi с использованием множества немутированных прочтений R последовательности. Стадия S300 сборки последовательности может включать в себя любые известные способы сборки содержащей мутации последовательности μi из множества мутированных прочтений Р последовательности на основе показателя, коррелированного с вероятностью того, что соответствующие два мутированных прочтения ρi, ρj последовательности происходят от одной и той же содержащей мутации последовательности μi.
На Фиг. 2 представлен способ 200 определения того, происходят ли два мутированных прочтения ρi, ρj последовательности от одной и той же содержащей мутации последовательности μi в соответствии с настоящим изобретением.
Способ 200 включает стадию S210 приема множества мутированных прочтений ρ1…pn ∈ P последовательности. Каждое мутированное прочтение ρi последовательности соответствует подпоследовательности из содержащей мутации последовательности μ i. Содержащая мутации последовательность μi содержит мутации, например мутации по типу замены, необязательно мутации по типу транзиции, по сравнению с не содержащей мутаций последовательностью Si. Содержащая мутации последовательность μi может быть по меньшей мере частью последовательности мутированной темплатной нуклеиновой кислоты-мишени, а не содержащая мутаций последовательность может быть по меньшей мере частью (немутированной) молекулы темплатной нуклеиновой кислоты-мишени, причем мутированная молекула темплатной нуклеиновой кислоты-мишени получена путем введения мутаций, например мутаций типа замены, необязательно мутаций типа транзиции, в молекулу темплатной нуклеиновой кислоты-мишени. Каждая подпоследовательность содержащей мутации последовательности μi может быть по меньшей мере частью последовательности фрагмента мутированной молекулы темплатной нуклеиновой кислоты-мишени. Каждая подпоследовательность не содержащей мутаций последовательности Si может быть по меньшей мере частью последовательности фрагмента молекулы темплатной нуклеиновой кислоты-мишени. Стадия S210 приема множества мутированных прочтений Р последовательности может включать прием множества мутированных прочтений Р последовательности непосредственно от секвенатора, используемого для секвенирования мутированной молекулы темплатной нуклеиновой кислоты мишени, или прием множества мутированных прочтений Р последовательности из хранилища данных, в котором хранится множество мутированных прочтений Р последовательности.
Способ 200 дополнительно включает стадию S220 применения общей минимизирующей функции для каждого мутированного прочтения ρi последовательности. Применение общей минимизирующей функции определяет один или более соответствующих минимизаторов для каждого мутированного прочтения ρi последовательности. Способ 200 дополнительно включает стадию S222 определения положений одного или более соответствующих минимизаторов в каждом мутированном прочтении ρi последовательности.
В предпочтительном варианте осуществления способ 200 включает стадию S224 распределения мутированных прочтений Р последовательности по группам соответствующих минимизаторов. Мутированное прочтение ρi последовательности, для которого определено более одного минимизатора, может быть помещено в несколько соответствующих групп минимизаторов.
Способ 200 дополнительно включает стадию S230 определения положений одной или более мутаций в каждом мутированном прочтении ρi последовательности. Стадия S230 определения положений одной или более мутаций в каждом мутированном прочтении ρi последовательности может осуществляться до, после или одновременно со стадиями S220, S222 и S224, относящимися к общей минимизирующей функции.
Для по меньшей мере двух мутированных прочтений ρi, ρj последовательности с общим минимизатором способ 200 дополнительно включает подсчет количества мутаций с совпадающим положением и/или несовпадающим положением, когда соответствующие минимизаторы выравнены, т.е. когда положения нуклеотидов одного мутированного прочтения ρi последовательности смещены относительно положений нуклеотидов другого мутированного прочтения ρj последовательности таким образом, что положение минимизатора одного мутированного прочтения ρi последовательности идентично положению минимизатора другого мутированного прочтения ρj последовательности. Количество мутаций с совпадающим положением и/или несовпадающим положением может быть показателем, коррелированным с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности. В альтернативном варианте осуществления способ 200 может включать дополнительную стадию S242 определения показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от той и той же содержащей мутации последовательности, на основании количества мутаций с совпадающим положением и/или с несовпадающим положением.
Стадия S210 приема множества мутированных прочтений последовательности
Стадия S210 включает прием множества мутированных прочтений ρ1…ρn ∈ Р последовательности. Стадия S210 может дополнительно включать прием множества немутированных прочтений r1… rrn ∈ R последовательности. Каждое мутированное прочтение ρi последовательности может соответствовать подпоследовательности содержащей мутации последовательности μi. Каждое немутированное прочтение ri последовательности может соответствовать подпоследовательности не содержащей мутации последовательности Si.
Содержащая мутации последовательность μi может быть получена путем введения мутаций в не содержащую мутаций последовательность Si. Каждое мутированное прочтение ρi последовательности, таким образом, может содержать мутации, т.е. соответствовать области мутированной молекулы темплатной нуклеиновой кислоты-мишени, которая включает в себя мутации, т.е. соответствовать подпоследовательности содержащей мутации последовательности. В одном варианте осуществления каждое мутированное прочтение ρi последовательности содержит мутации по типу замены, т.е. соответствует области мутированной молекулы темплатной нуклеиновой кислоты-мишени, которая включает в себя мутации по типу замены. В предпочтительном варианте осуществления мутации по типу замены представляют собой мутации по типу транзиции, так что каждое мутированное прочтение ρi последовательности содержит мутации по типу транзиции, т.е. соответствует области мутированной молекулы темплатной нуклеиновой кислоты-мишени, которая включает в себя мутации по типу транзиции.
Каждый нуклеотид каждого прочтения ρi, ri последовательности предпочтительно кодируется в двоичном формате с использованием двух битов. Это дает преимущество, в частности, когда множество мутированных прочтений Р последовательности содержат мутации типа транзиции (A⇔G и С⇔Т), так что один из двух битов (например, первый бит) определяет, является ли нуклеотид пуриновым (А или G) или пиримидиновым (Т или С). Например, нуклеотиды могут быть закодированы в двоичной форме с использованием следующего формата: А: 00, G: 01, С: 10 и Т: 11. Это кодирование будет использовано в настоящем описании. Однако будет очевидно, что настоящее изобретение не ограничивается этим кодированием, и что настоящее изобретение может быть легко осуществлено с использованием любого другого кодирования нуклеотидов.
Каждое прочтение ρi, ri последовательности может быть закодировано для учета гомополимерных ошибок в прочтении ρi, ri последовательности. Гомополимерные ошибки возникают, когда при проходе неправильно считывается длина одного и того же нуклеотида, например, последовательность TAAAAGC может быть неправильно прочитана как TAAGC, поскольку секвенатору сложно определить количество А, если при проходе встречается множество А. Для учета таких гомополимерных ошибок проходы с несколькими идентичными нуклеотидами могут быть закодированы как один экземпляр нуклеотида. В альтернативном варианте осуществления гомополимерные ошибки могут быть учтены во время последующей обработки (т.е. не при первоначальном кодировании) прочтений ρi, ri последовательности, например, путем кодирования любых k-меров, используемых в способе 200, и/или любых затравочных паттернов, используемых на стадии S230, так что проходы с несколькими идентичными нуклеотидами кодируются как один экземпляр нуклеотида.
Стадии S220 и S222: общая минимизирующая функция
Минимизатор представляет собой k-мер из набора k-меров, который удовлетворяет общей минимизирующей функции min(⋅) на наборе k-меров.
Для целей настоящей заявки k-мер представляет собой нуклеотидную подпоследовательность длины k. k-мер, начинающийся с положения i в последовательности S=[S1, S2, …, Sn-1, Sn] длиной n, обозначен как k(Si), причем k(Si)=[Si, Si+1, …, Si+k-1]. Набор k-меров в последовательности S с начальными положениями между i и j обозначен как k(Si…Sj). Минимизатор из всех k-меров с исходным положениями в диапазоне i до j последовательности S будет обозначен как min(k(Si…Sj)).
Общую минимизирующую функцию min(⋅) используют для определения одного или более минимизаторов (т.е. одного или более репрезентативных k-меров) из набора k-меров, предпочтительно из всех или по существу всех к-меров, образованных прочтением ρi, ri последовательности, т.е. k-меров предпочтительно из всех или по существу всех k-меров, которые существуют в прочтении ρi, ri последовательности. Для целей настоящего изобретения набор k-меров, которые существуют в прочтении ρi, ri последовательности, может содержать k-меры обратного комплемента прочтения ρi, ri последовательности. Предпочтительно каждый минимизатор представляет собой k-мер длиной, равной или превышающей 5 (т.е. 5-мер или более), предпочтительно равной или превышающей 10 (т.е. 10-мер или более), еще более предпочтительно равной или превышающей 15 (т.е. 15-мер или более). Каждый минимизатор может представлять собой k-мер длиной менее 50, необязательно менее 30, дополнительно необязательно менее 25. Если общую функцию минимизации min(⋅) используют для определения более длинных минимизаторов, то выше вероятность, что определенный минимизатор будет репрезентативным для конкретной части последовательности, т.е. меньше вероятность, что минимизатор появится в нескольких отдельных и несвязанных участках последовательности. Установка верхнего предела размера минимизаторов снижает риск того, что минимизаторы будут содержать ошибки секвенирования.
Стадия S220 применения общей минимизирующей функции min (⋅) может включать идентификацию в соответствующем мутированном прочтении ρi последовательности одного или более k-меров, который(-ые) указан(-ы) первым(-и) в упорядоченном списке возможных k-меров. Один или более минимизаторов, определенных для соответствующего мутированного прочтения ρi последовательности, может представлять сбой идентифицированный один или более k-меров. Упорядоченный список возможных k-меров может содержать все или некоторые возможные k-меры в заранее заданном порядке. Стадия S220 может включать генерацию упорядоченного списка возможных k-меров или может не включать генерацию упорядоченного списка возможных k-меров (например, в ситуациях, когда для определения минимизатора не требуется прямое сравнение с списком, как в некоторых примерах ниже).
Например, общая минимизирующая функция min() может определять как минимизатор k-мер с целочисленным минимальным значением из двухбитных двоичных кодирований всех k-меров в мутированном прочтении ρi последовательности. Другими словами, общая минимизирующая функция min() может идентифицировать k-мер, который указан первым в списке k-меров, которые упорядочены по целочисленному значению их двухбитных двоичных кодировок. Например, на основании двоичного кодирования А: 00, G: 01, С: 10 и Т: 11 общая минимизирующая функция может идентифицировать в мутированном прочтении последовательности 5-мер, который указан первым в примере упорядоченного списка ААААА, AAAAG, AAAAC, ААААТ, AAAGA, AAAGG,…, СТТТС, СТТТТ, ТТТТТ. Например, пример мутированного прочтения последовательности:
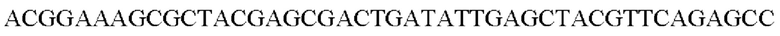
содержит 5-меры ACGGA, CGGAA, GGAAA, … AGAGC, GAGCC. 5-мер AAAGC указан первым в приведенном выше примере упорядоченного списка, и общая минимизирующая функция min() будет определять AAAGC как минимизатор для этого примера мутированного прочтения последовательности. Следует понимать, что для этой общей минимизирующей функции min(⋅) не требуется фактически генерировать упорядоченный список возможных k-меров для определения минимизатора для набора k-меров.
Определение целочисленного минимального значения двухбитных двоичных кодирований всех k-меров в мутированном прочтении ρi последовательности является лишь одним примером общей минимизирующей функции min(), которая может быть применена к мутированному прочтению ρi последовательности для определения минимизатора. Можно использовать любую другую общую минимизирующую функцию min(). Например, предпочтительно, чтобы общая минимизирующая функция min() рандомизировала упорядочивание целочисленной минимальной функции. Один из способов достижения такой рандомизации заключается в применении сначала побитового логического ИЛИ с произвольным битовым вектором к каждому k-меру, содержащемуся в мутированном прочтении ρi последовательности, после чего можно использовать целочисленную минимальную функцию.
В альтернативном варианте осуществления вместо упорядоченного списка возможных k-меров можно использовать предварительно заданный набор возможных k-меров, и применение общей минимизирующей функции min() включает идентификацию одного или более k-меров, которые существуют в предварительно заданном наборе возможных k-меров. Один или более минимизаторов, определенных для соответствующего мутированного прочтения ρi последовательности, может представлять сбой идентифицированный один или более k-меров. Предварительно заданный набор возможных k-меров может быть упорядочен или неупорядочен. Предварительно заданный набор возможных к-меров может представлять собой набор k-меров, включающий только k-меры, которые подходят или предназначены для использования в качестве минимизаторов. Стадия S220 применения общей минимизирующей функции min() может включать создание предварительно заданного набора возможных k-меров.
В предпочтительном варианте осуществления в упорядоченном списке возможных k-меров k-меры упорядочены на основании вероятности того, что k-меры встречаются в содержащей мутации последовательности μi и не встречаются в не содержащей мутаций последовательности Si, т.е. k-меры, которые относительно вероятно встречаются в содержащей мутации последовательности, но не в не содержащей мутаций последовательности, могут быть перечислены выше в упорядоченном списке, а k-меры, которые относительно маловероятно встречаются в содержащей мутации последовательности, но не в не содержащей мутаций последовательности, могут быть перечислены ниже в упорядоченном списке. В альтернативном предпочтительном варианте осуществления предварительно заданный набор возможных k-меров содержит k-меры, присутствие которых относительно вероятно в содержащей мутации последовательности, но не в не содержащей мутаций последовательности, и необязательно набор не содержит k-меров, присутствие которых относительно маловероятно в содержащей мутации последовательности, но не в не содержащей мутаций последовательности. Стадия S220 может включать определение того, какие k-меры, содержащиеся в множестве мутированных прочтений Р последовательности относительно вероятно встречаются в содержащей мутации последовательности, но не в не содержащей мутаций последовательности, например, путем сравнения количества вхождений (или наблюдений) k-мера во множестве мутированных прочтений Р последовательности с количеством вхождений k-мера во множестве немутированных прочтений R последовательности. Стадия может включать подсчет количества вхождений k-мера во множестве мутированных прочтений Р последовательности и подсчет количества вхождений k-мера во множестве немутированных прочтений R последовательности.
В обоих предпочтительных вариантах осуществления общую минимизирующую функцию min() выбирают таким образом, чтобы предпочтительно определять как один или более минимизаторов те k-меры, которые с большей вероятностью встречаются в мутированном прочтении ρi последовательности, чем в немутированном прочтении ri последовательности. Это повышает вероятность того, что каждый минимизатор содержит мутацию.
В более предпочтительном варианте осуществления упорядоченный список возможных k-меров содержит только те k-меры, т.е. состоит только из тех k-меров, которые чаще встречаются во множестве мутированных прочтений Р последовательности, чем во множестве немутированных прочтений R последовательности (или чаще - в содержащей мутации последовательности, чем в не содержащей мутаций последовательности), т.е. k-меров, для которых количество вхождений во множестве мутированных прочтений Р последовательности больше числа вхождений во множестве немутированных прочтений R последовательности. В альтернативном более предпочтительном варианте осуществления предварительно заданный набор возможных k-меров содержит только те k-меры, т.е. состоит только из тех k-меров, которые чаще встречаются во множестве мутированных прочтений Р последовательности, чем в множестве немутированных прочтений R последовательности (или чаще - в содержащей мутации последовательности, чем в не содержащей мутаций последовательности), т.е. k-меров, для которых количество вхождений во множестве мутированных прочтений Р последовательности больше числа вхождений во множестве немутированных прочтений R последовательности. Предпочтительно, упорядоченный список возможных k-меров или предварительно заданный набор возможных k-меров содержит только те k-меры, т.е. состоит из тех k-меров, которые встречаются n или более раз в множестве мутированных прочтений последовательности, и встречаются менее чем n раз во множестве немутированных прочтений последовательности, т.е. тех k-меров, для которых количество вхождений во множестве мутированных прочтений Р последовательности равно или больше n, и количество вхождений во множестве немутированных прочтений R последовательности меньше n. N может представлять собой целое число, большее или равное 1. N может представлять собой целое число, большее или равное 2. Предпочтительно «n» равно 2. Кроме того, упорядоченный список возможных k-меров или предварительно заданный набор возможных k-меров содержит только те k-меры, т.е. состоит только из тех k-меров, которые не встречаются в множестве немутированных прочтений последовательности, т.е. тех k-меров, для которых количество вхождений во множестве немутированных прочтений R последовательности равно 0.
Например, упорядоченный список возможных k-меров или предварительно заданный набор возможных k-меров может содержать только те k-меры, которые встречаются по меньшей мере два раза в наборе k-меров множества мутированных прочтений Р последовательности, но не встречаются (или встречаются редко) в наборе k-меров множества немутированных прочтений R последовательности. Это гарантирует, что с высокой вероятностью упорядоченный список возможных k-меров или предварительно заданный набор возможных k-меров будет включать в себя минимизаторы, которые содержат мутацию, присутствующую в двух или более из множества мутированных прочтений Р последовательности. Необязательно k-меры, которые чаще встречаются во множестве мутированных прочтений последовательности, чем во множестве немутированных прочтений последовательности, относительно вероятно встречаются в содержащей мутации последовательности. Причем необязательно, те k-меры, которые встречаются n или более раз во множестве мутированных прочтений последовательности и встречаются менее чем n раз во множестве немутированных прочтений последовательности, относительно вероятно встречаются в содержащей мутации последовательности.
Предварительно заданный набор возможных k-меров может быть создан путем построения набора мутационных минимизаторов UM, где UМ содержит k-меры, предпочтительно все или по существу все k-меры, для которых количество вхождений или наблюдений во множестве мутированных прочтений Р последовательности больше или равно n (предпочтительно, где n ≥2, более предпочтительно, где n равно 2), а количество вхождений или наблюдений во множестве немутированных прочтений Р последовательности меньше n (предпочтительно, где n равно 0 или 1, более предпочтительно, где n равно 0). Набор мутационных минимизаторов UM может быть создан путем подсчета частоты встречаемости каждого k-мера во множестве немутированных прочтений R последовательности и множестве мутированных прочтений Р последовательности. Набор мутационных минимизаторов UM можно эффективно рассчитать из множества немутированных прочтений R последовательности и множества мутированных прочтений Р последовательности с использованием вероятностных структур данных, таких как фильтр Блума с подсчетом или связанные с ним метод кукушки и метод фильтра с коэффициентами. Упорядоченный список возможных k-меров может быть создан из всего набора мутационных минимизаторов UM.
Набор мутационных минимизаторов UM можно использовать в качестве предварительно заданного набора возможных k-меров. В альтернативном варианте осуществления, набор мутационных минимизаторов UM можно дополнительно обрабатывать для получения предварительно заданного набора возможных k-меров. В предпочтительном варианте осуществления в качестве предварительно заданного набора возможных k-меров используют подмножество WМ набора мутационных минимизаторов UM. Подмножество WМ можно сконструировать путем разделения каждого мутированного прочтения ρi ∈ Р на две или более неперекрывающихся секции (необязательно по существу одинаковых размеров), например неперекрывающихся наборов начальных позиций k-меров размера Lw, например {1…Lw}, {Lw+1…2LW} и т.д. Типичное значение для Lw может составлять 50 при использовании мутированных прочтений последовательности длиной 150, в результате чего положения начала возможных k-меров делятся на 3 группы. Затем для каждого набора начальных положений подмножество WМ можно обозначить следующим образом:
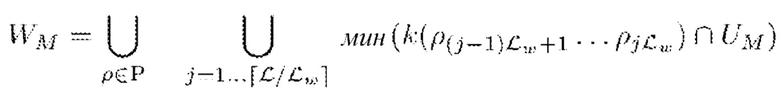
В результате каждое из множества мутированных прочтений Р последовательности может быть разделено на две или более секций (например, на 3 секции), и может быть найден минимизатор, представляющий каждую секцию. Минимизатор определяют путем выявления сначала потенциальных минимизаторов по пересечению k-меров в этой секции соответствующего мутированного прочтения последовательности с набором мутационных минимизаторов UM, и затем применяют общую минимизирующую функцию к этому набору для идентификации одного минимизатора для каждой секции.
Таким образом, в предпочтительном варианте осуществления стадия S220 применения общей минимизирующей функции min() к каждому мутированному прочтению последовательности включает:
создание набора мутационных минимизаторов UM который состоит из k-меров, предпочтительно всех или по существу всех k-меров во множестве мутированных прочтений Р последовательности, которые встречаются n или более раз во множестве мутированных прочтений Р последовательности и встречаются менее чем n раз во множестве немутированных прочтений R последовательности, где n представляет собой целое число, большее или равное 2;
необязательно создание подмножества WМ из набора мутационных минимизаторов UM путем разделения каждого из множества мутированных прочтений Р последовательности на две или более секции, идентификации k-меров, предпочтительно всех или по существу всех k-меров в каждой секции каждого из множества мутированных прочтений Р последовательности, которые встречаются в наборе мутационных минимизаторов UM, и добавления к подмножеству WМ одного из идентифицированных k-меров для каждой секции каждого из множества мутированных прочтений Р последовательности, причем необязательно один из идентифицированных k-меров для каждой секции каждого из множества мутированных прочтений Р последовательности выбирают путем применения общей минимизирующей функции min (⋅) (например, функции обнаружения целочисленного минимума или любой другой известной минимизирующей функции) к идентифицированным k-мерам каждой секции каждого из множества мутированных прочтений Р последовательности; и
использование набора мутационных минимизаторов UM или подмножества из набора мутационных минимизаторов UM (например, подмножества WМ) в качестве предварительно заданного набора возможных k-меров, и для каждого из множества мутированных прочтений Р последовательности - идентификацию k-меров, предпочтительно всех или по существу всех k-меров в соответствующем мутированном прочтении последовательности μi, которые встречаются в предварительно заданном наборе возможных k-меров, причем один или более минимизаторов, определенных для соответствующего мутированного прочтения последовательности, представляют собой идентифицированные k-меры.
Способ 200 дополнительно включает стадию S222 для определения положений] одного или более соответствующих минимизаторов в каждом мутированном прочтении ρi последовательности. Положения j каждого из минимизаторов в каждом соответствующем мутированном прочтении ρi последовательности можно хранить в виде целочисленного битового значения в ассоциации (например, в одном и том же местоположении или группе минимизатора) с соответствующим минимизатором.
Стадия S224: распределение по группам минимизаторов
В предпочтительном варианте осуществления способ 200 включает стадию S224, заключающуюся в распределении мутированных прочтений Р последовательности в одну или более групп минимизаторов. Распределение мутированных прочтений Р последовательности в одну или более групп минимизаторов включает в себя помещение указателя i, характеризующего мутированное прочтение ρi последовательности в одну или более групп минимизаторов. Каждая группа минимизатора может содержать мутированные прочтения Р последовательности, имеющие общий минимизатор, и не содержит мутированные прочтения Р последовательности, не имеющие общего минимизатора. Стадия S240 подсчета количества мутаций с совпадающим положением и/или с несовпадающим положением может быть выполнена только на мутированных прочтениях Р последовательности, находящихся в одной и той же группе минимизатора. Это улучшает вычислительную эффективность выполнения стадии S240.
Другими словами, один или более минимизаторов можно использовать в качестве хеш-ключей для сбора прочтений последовательности, содержащих минимизатор, в общий хэш-сегмент (в настоящем документе именуемый группой минимизатора), например, при подготовке к некоторой дополнительной обработке (например, стадии S240), проводимой на этих прочтениях последовательности.
Каждый минимизатор, который определяют путем применения общей минимизирующей функции min() к мутированным прочтениям Р последовательности, можно использовать для распределения мутированных прочтений Р последовательности в одну или более групп минимизаторов. В одном варианте осуществления каждый минимизатор в упорядоченном списке возможных k-меров или каждый минимизатор в предварительно заданном наборе возможных k-меров (например, каждый минимизатор в наборе мутационных минимизаторов UM или его подмножестве, например подмножестве WМ), можно использовать для целей распределения мутированных прочтений Р последовательности в одну или более групп минимизаторов.
Стадия S224 распределения мутированных прочтений Р последовательности в одну или более групп минимизаторов может включать создание одной или более групп минимизаторов. Это может включать создание одной группы минимизатора для каждого минимизатора, определенного с помощью общей минимальной функции min(), или одной группы минимизатора для каждого минимизатора (или k-мера) в предварительно заданном наборе возможных k-меров UM или одой группы минимизатора для каждого k-мера в подмножестве WМ. Каждая группа минимизатора может быть реализована как непрерывный блок ОЗУ. Предпочтительно, чтобы коллекции минимизаторов были реализованы в виде файла на компьютерном носителе данных (таком как компьютерный диск, например вращающийся магнитный диск или твердотельный диск), позволяя каждой группе хранить большие объемы данных (что уместно в случаях анализа последовательностей).
Стадия S224 распределения мутированных прочтений Р последовательности в одну или более групп минимизаторов может включать сохранение мутированного прочтения ρi последовательности или указателя i, характеризующего мутированное прочтение ρi последовательности, в соответствующей группе минимизатора. Стадия S222 определения положений] одного или более соответствующих минимизаторов в каждом мутированном прочтении ρi последовательности может включать сохранение положения j соответствующего минимизатора в соответствующей группе минимизатора. Кроме того, положение α=morphomuts(ρi,VR) одной или более мутаций в каждом мутированном прочтении ρi последовательности, определенное на стадии S230 определения положений α одной или более мутаций в каждом мутированном прочтении последовательности, может храниться в соответствующей группе минимизатора. Необязательно в группе минимизатора можно хранить произвольные дополнительные значения, такие как последовательность мутированного прочтения ρi последовательности, информацию о качестве относительно точности последовательности или другую информацию, если эти данные полезны для последующей обработки. Эти значения, связанные с каждым мутированным прочтением ρi последовательности, можно хранить в виде кортежа в каждой группе минимизатора. Для условного обозначения элементы кортежа у-го элемента z-й группы минимизатора bz,y обозначены как bz,y.i, bz,y.j, и bz,y.α. Каждое мутированное прочтение ρi последовательности можно добавлять к нескольким группам минимизаторов.
Стадия S230: положения мутаций
Способ 200 включает стадию S230 определения положений а одной или более мутаций в каждом мутированном прочтении ρi последовательности. Стадию S230 определения положений α одной или более мутаций в каждом мутированном прочтении ρi последовательности можно выполнять с использованием способов, не включающих выравнивания.
Стадия S230 определения положений α одной или более мутаций в каждом мутированном прочтении ρi последовательности может включать получение набора маскированных затравкой немутированных k-меров VR, т.е. набора k-меров немутированного прочтения R последовательности, к которому были применены один или более затравочных паттернов ψ. Получение набора маскированных затравкой немутированных k-меров VR может включать создание или генерацию набора маскированных затравкой немутированных k-меров VR. Набор маскированных затравкой немутированных k-меров VR может быть получен или создан путем применения каждого из одного или более затравочных паттернов к каждому k-меру в не содержащей мутаций последовательности, например к каждому k-меру в немутированных прочтениях последовательности. Применение затравочного паттерна к k-меру может включать определение результата побитового логического И к затравочному паттерну и (имеющему двухбитное кодирование) k-меру. Применение затравочного паттерна к k-меру приводит к получению маскированного затравкой k-мера. Набор маскированных затравкой немутированных k-меров VR может быть обозначен как
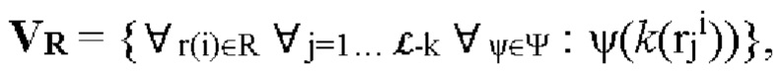
т.е. набор маскированных затравкой немутированных k-меров VR создают путем применения каждого из одного или более затравочных паттернов ψ семейства затравок ψ к каждому k-меру k(rji) для всех (или по существу всех) положений j k-мера (т.е. от 1 до 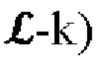 в каждом немутированном чтении ri для всех (или по существу всех) немутированных прочтений ri во множестве немутированных прочтений R последовательности.
в каждом немутированном чтении ri для всех (или по существу всех) немутированных прочтений ri во множестве немутированных прочтений R последовательности.
Затравочный паттерн можно использовать для модификации способа сравнения k-меров друг с другом. Затравочный паттерн определяется как набор положений (т.е. нуклеотидов) в пределах двух k-меров, которые должны быть идентичными в обоих k-мерах, чтобы можно было считать маскированные затравкой k-меры совпадающими. Затравочный паттерн может содержать маскирующие положения и немаскирующие положения. Применение затравочных паттернов к k-меру создает маскированный затравкой k-мер, в котором позиции маскированного затравкой k-мера, соответствующие маскирующим позициям соответствующего затравочного паттерна, игнорируются при любой дополнительной обработке (такой как сравнения), тогда как позиции маскированного затравкой k-мера, соответствующие немаскирующим позициям соответствующего затравочного паттерна, не игнорируются при любой дополнительной обработке (такой как сравнения). Например, затравочный паттерн {1, 2, 4, 6, 7} требует, чтобы первое, второе, четвертое, шестое и седьмое положения (или нуклеотиды) в двух сравниваемых k-мерах k(Si) и k(Sj) были идентичны, чтобы они считались совпадающими (для k=7). Третье и пятое положения в двух k-мерах могут представлять собой произвольные нуклеотиды. Это означает, что третье и пятое положения в двух маскированных затравкой k-мерах маскированы затравочным паттерном.
Один или более затравочных паттернов необязательно могут представлять собой один или более затравочных паттернов транзиции. Это является преимуществом, в частности, когда содержащая мутации последовательность М содержит мутации типа транзиции по сравнению с не содержащей мутаций последовательностью S, т.е. каждое из множества мутированных прочтений Р последовательности содержит одну или более мутаций типа транзиции.
Затравочный паттерн транзиции представляет собой специализированный тип затравочного паттерна, где положения делятся на три класса вместо всего двух: каждое положение должно (1) точно соответствовать, или (2) оба должны быть пуриновыми или пиримидиновыми, или (3) каждое положение должно быть любым из четырех нуклеотидов, чтобы совпадать. Затравочные паттерны транзиции являются особенно предпочтительными, когда содержащая мутации последовательность содержит мутации типа транзиции. При реализации на компьютере с использованием двухбитного кодирования нуклеотидов, предложенного выше, положение, где необходимо точное совпадение, может быть реализовано в виде битовой маски 11, в то время как положение, где разрешены только мутации типа транзиции, обозначено как 10, а положение, в котором разрешен любой нуклеотид, обозначено как 00. Затравочный паттерн {1, 2, 4, 6, 7} может быть записан как битовая маска 11110011001111. Затравочный паттерн транзиции {1, 2, 4, 6, 7} может быть записан как битовая маска 11111011101111. Два k-мера можно оценивать на совпадение путем вычисления для каждого из них результата побитового логического И для битовой маски и двухбитного кодирования k-мера и последующей проверки идентичности двух полученных маскированных затравкой k-меров. Для удобства функция, которая применяет затравочный паттерн к k-меру k(Si) путем побитового логического И, будет обозначена как функция ψ(k(Si)).
В одном варианте осуществления один или более затравочных паттернов выбраны таким образом, что вероятность получения идентичных маскированных затравкой k-меров при применении по меньшей мере одного из одного или более затравочных паттернов к любому k-меру из множества мутированных прочтений Р последовательности (или содержащих мутации последовательностей) и соответствующему k-меру из множества немутированных прочтений R последовательности (или не содержащих мутаций последовательностей) составляет более 90%, предпочтительно более 95%, дополнительно предпочтительно более 98%, наиболее предпочтительно более 99%.
Один или более затравочных паттернов могут составлять семейство затравочных паттернов ψ. Семейство затравочных паттернов ψ представляет собой набор из двух или более затравочных паттернов, которые при совместном использовании способны идентифицировать совпадения среди k-меров при конкретной процентной идентичности нуклеотидов с высокой вероятностью, например с вероятностью более 90%, предпочтительно более 95%, дополнительно предпочтительно более 98%, наиболее предпочтительно более 99%. Семейство затравочных паттернов ψ обозначается как набор из n различных функций для применения затравочных паттернов ψ1…ψn ∈ ψ. Вес затравочного паттерна w(ψ) определяется как количество позиций затравки, которое должно быть идентичным для того, чтобы два k-мера считались совпадающими, где w(ψ)≤k.
Для каждого мутированного прочтения последовательности стадия S230 определения положений α одной или более мутаций в каждом мутированном прочтении ρi последовательности может включать применение каждого из одного или более затравочных паттернов ψi к k-мерам (необязательно к каждому k-меру) в соответствующем мутированном прочтении ρi последовательности с получением множества маскированных затравкой мутированных k-меров. Положения одной или более мутаций могут быть определены путем идентификации одного или более положений в мутированном прочтении ρi последовательности, маскированном всеми затравочными паттернами, которые соответствуют маскированным затравкой мутированным k-мерам из множества маскированных затравкой мутированных к-меров, которые встречаются в наборе маскированных затравкой немутированных k-меров VR. Это означает, что положения, которые не являются мутациями в мутированном прочтении ρi последовательности могут быть идентифицированы как одно или более положений в мутированном прочтении ρi последовательности, которые не маскированы каким-либо из затравочных паттернов, которые соответствуют маскированным затравкой мутированным k-мерам из множества маскированных затравкой мутированных k-меров, которые существуют в наборе маскированных затравкой немутированных k-меров VR.
Например, положения а одной или более мутаций каждого мутированного прочтения ρi последовательности можно определять следующим образом:
• создание битового вектора а длиной 2L и установка начальных значений битового вектора α на 0;
• создание битового вектора b длиной 2k и установка всех начальных значений битового вектора b на 1;
• для каждого ψ ∈ Ψ и для каждого положения j в прочтении между 1 и 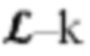 - вычисление ψ(k(ρji)). Если ψ(k(pji)) ∈ VR, то присвоение α←α | (ψ(b)>>2j), где оператор | обозначает побитовый логический оператор ИЛИ, а оператор >> обозначает оператор сдвига вправо. Запрос принадлежности набора к VR может быть реализован либо точно, с использованием чего-либо подобного хеш-таблице, либо приблизительно, с использованием высокоэффективной вероятностной структуры данных, такой как фильтр Блума, фильтр коэффициентов, или аналогичного подхода.
- вычисление ψ(k(ρji)). Если ψ(k(pji)) ∈ VR, то присвоение α←α | (ψ(b)>>2j), где оператор | обозначает побитовый логический оператор ИЛИ, а оператор >> обозначает оператор сдвига вправо. Запрос принадлежности набора к VR может быть реализован либо точно, с использованием чего-либо подобного хеш-таблице, либо приблизительно, с использованием высокоэффективной вероятностной структуры данных, такой как фильтр Блума, фильтр коэффициентов, или аналогичного подхода.
• Необязательно для простоты дальнейшей обработки преобразование битового вектора а из длины двоичного двухбитного кодирования мутированного прочтения последовательности в длину самого мутированного прочтения последовательности, путем удаления нечетных позиций, например α←{α2,α4,α6,…α2L}.
• Необязательно для простоты дальнейшей обработки применение операции логического НЕ к битам, так что значение 1 представляет собой положения, для которых не было обнаружено совпадений с затравкой.
Результатом вышеописанной процедуры будет битовый вектор α, где каждое положение, содержащее 1, с высокой вероятностью соответствует положению мутации. Для условного обозначения функция, которая вычисляет битовый вектор α для мутированного прочтения ρi последовательности, обозначена как α=morphomuts(ρi,YR).
На Фиг. 3 показан пример, иллюстрирующий, как битовый вектор α может быть получен для примера мутированного прочтения ρ=ACGCAAAGCGCTACGAGCGACTGATATT последовательности с использованием одного затравочного паттерна ψ=1110110011. 4-е, 8-е, 11-е, 12-е и 16-е положения мутированного прочтения ρ последовательности соответствуют мутациям в мутированном прочтении ρ последовательности, т.е. нуклеотиды в этих положениях в не содержащей мутаций последовательности будут отличаться. На практике мутированное прочтение ρ последовательности может быть закодировано в двухбитном двоичном формате, и каждое положение затравочного паттерна ψ может покрывать два бита (т.е. каждая 1 в затравочном паттерне ψ будет реализована в виде двух двоичных 1, а каждый 0 в затравочном паттерне будет реализован в виде двоичного 00 или двоичного 10). Набор маскированных затравкой немутированных k-меров VR был получен ранее в этом примере.
Как показано в примере, показанном на Фиг. 3, затравочные паттерны применяют к каждому k-меру в мутированном прочтении ρ последовательности, таким образом формируя один маскированный затравкой k-мер для каждого k-мера в мутированном прочтении ρ последовательности. Затем проверяют, существует ли маскированный затравкой k-мер в наборе маскированных затравкой не мутированных k-меров VR. В показанном примере все из 1-го, 5-го, 13-го, 17-го, 18-го и 19-го маскированных затравкой k-меров встречаются в наборе маскированных затравкой не мутированных k-меров VR. Эти маскированные затравкой k-меры не содержат позиций мутации, не маскированных затравочным паттерном.
Затем 1-й, 5-й, 13-й, 17-й, 18-й и 19-й маскированные затравкой k-меры используют для идентификации позиций, которые маскированы всеми затравочными паттернами, соответствующими этим маскированным затравкой k-мерам. 4-е положение мутированного прочтения ρ последовательности маскировано всеми этими затравочными паттернами, указывая на то, что 4-е положение мутированного прочтения ρ последовательности игнорируется при обработке 13-го, 17-го, 18-го и 19-го маскированных затравками k-меров, т.е. 4-е положение мутированного прочтения ρ последовательности маскируется затравочным паттерном для 13-го, 17-го, 18-го и 19-го маскированных затравкой k-меров. Ни один из этих затравочных паттернов не маскирует 4-е положение мутированного прочтения ρ последовательности. Таким образом, 4-е положение мутированного прочтения ρ последовательности идентифицируется как положение мутации. Напротив, хотя 7-е положение мутированного прочтения ρ последовательности маскируется всеми затравочными паттернами, соответствующими 1-му, 13-му, 17-му, 18-му и 19-му маскированными затравкой k-мерам, это 7-е положение мутированного прочтения ρ последовательности не маскируется затравочным паттерном, соответствующим 5-му маскированному затравкой k-меру. Таким образом, 7-е положение мутированного прочтения ρ последовательности не идентифицируется как положение мутации. Вместо этого 7-е положение мутированного прочтения ρ последовательности идентифицируется как положение, которое не является мутацией.
По существу все затравочные паттерны, соответствующие 1-му, 5-му, 13-му, 17-му, 18-му и 19-му маскированным затравкой k-мерам комбинируют с использованием логического ИЛИ. Биты полученного битового вектора могут быть зеркально отражены (например, с использованием операции логического НЕ) для получения положений мутаций в мутированном прочтении ρ последовательности в качестве битового вектора α.
Альтернативный вариант осуществления стадии 230 с использованием эталонной сборки
В описанном выше варианте осуществления стадию 230 определения положений α одной или более мутаций в каждом мутированном прочтении ρi последовательности выполняют с использованием множества мутированных прочтений Р последовательности и множества немутированных прочтений R последовательности на основании применения затравочных паттернов к каждому мутированному прочтению ρi последовательности.
В больших и сложных геномах, таких как геном человека, значительная часть генома состоит из повторяющихся последовательностей. Например, считается, что более половины генома человека является частью повторяющихся последовательностей. Эти повторяющиеся последовательности классифицируются в «семейства» сходных повторяющихся последовательностей. Наиболее распространенным в геноме человека является семейство Alu коротких диспергированных ядерных элементов (SINE), которое имеет длину около 300 нт и присутствует в приблизительно 1 миллионе копий. Другим распространенным семейством является семейство L1 длинных диспергированных ядерных элементов (LINE) с размером элементов в диапазоне от 1 до 6,5 т.п.н. и с числом копий около 10000.
Различные копии повторяющихся последовательностей в геноме могут быть неидентичными, например, они содержат различия в одиночных основаниях. Из-за биологии мутации эти различия часто являются различиями типа транзиции. В некоторых ситуациях эти различия могут выглядеть аналогично различиям, обусловленным введением мутаций между множеством мутированных прочтений Р последовательности и множеством немутированных прочтений R последовательности. Это особенно актуально для некоторых полимеразных подходов к мутагенезу, применяемых для введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени в рамках получения множества мутированных прочтений Р последовательности, поскольку при этом часто вводят мутации типа транзиции.
В результате множество немутированных прочтений R последовательности может содержать большое количество k-меров, которые отличаются друг от друга только некоторым числом различий типа транзиции. Следовательно, множество мутированных прочтений Р последовательности может включать один или более k-меров, которые идентичны k-мерам множества немутированных прочтений R последовательности, несмотря на наличие мутаций по сравнению с немутированными R прочтениями последовательности. В некоторых ситуациях возможно, что естественные различия между различными копиями повторяющейся последовательности в различных немутированных прочтениях ri последовательности будут частично «маскировать» мутации, введенные в множество мутированных прочтений Р последовательности. Это особенно выражено в отношении SINE из семейств Alu.
Таким образом, было бы предпочтительно, если бы в подобных ситуациях был предложен вариант осуществления способа, позволяющий лучше отличать намеренно введенные мутации от природных различий между копиями повторяющихся последовательностей.
Первый подход к улучшению способности способа отличать намеренно введенные мутации от природных различий между копиями повторяющихся последовательностей заключается в использовании затравочных паттернов с гораздо более высокой массой, так чтобы мутированные маскированные затравкой k-меры с большей вероятностью включали одну или более позиций, содержащих различие, отличающее копии повторяющейся последовательности. В одном варианте осуществления, который задействует первый подход, масса w(ψ) каждого затравочного паттерна ψ находится в диапазоне от 50 до 100, предпочтительно в диапазоне от 70 до 90. Для генома человека для первого подхода будет достаточной масса приблизительно 80.
Однако первый подход не может быть идеальным во всех случаях. Затравочный паттерн с массой 80 будет очень длинным, вероятно, более длинным, чем типичная длина мутированного прочтения ρi последовательности. Кроме того, размер семейства Ψ затравочных паттернов, необходимый для обеспечения высокой чувствительности, может стать очень большим, что требует значительных дополнительных вычислительных ресурсов для обработки всех затравочных паттернов. Наконец, будет расти вероятность покрытия затравочным паттерном ошибки вставки-делеции, и для адаптации к возможности ошибок вставки-делеции потребуется дополнительная алгоритмическая сложность. Таким образом, в некоторых обстоятельствах этот первый подход может не быть предпочтительным.
Второй подход к улучшению способности способа отличать намеренно введенные мутации от природных различий между копиями повторяющихся последовательностей заключается в использовании подхода на основе выравнивания (или сопоставления) множества мутированных прочтений Р последовательности с эталонной сборкой (или эталонным геномом). Эталонная сборка может быть либо сгенерирована независимо, как, например, геном человека hg38, полученный Консорциумом референсного генома (GRC), или может представлять собой сборку de-novo на основании множества немутированных прочтений R последовательности. Для одного или более мутированных прочтении последовательности во втором подходе стадия определения положений одной или более мутаций в каждом мутированном прочтении последовательности включает выравнивание соответствующих мутированных прочтений последовательности с эталонной сборкой.
Этот подход может быть особенно подходящим, когда мутированные прочтения ρi последовательности представляют собой прочтения последовательности со спаренными концами. Преимущество выравнивания мутированных прочтений последовательности со спаренными концами с эталонной сборкой, в частности, применительно к повторам SINE, состоит в том, что размер фрагмента в библиотеке коротких прочтений метода дробовика, как правило, больше длины повторяющихся последовательностей. Типичный размер фрагмента при секвенировании спаренных концов составляет 400 600 п.н., при этом около 150 п.н. секвенированы с каждого конца фрагмента. Таким образом, если одно прочтение последовательности со спаренными концами из пары прочтений последовательности со спаренными концами приходится на повторяющуюся последовательность, то другое из прочтений последовательности со спаренными концами в паре прочтений последовательности со спаренными концами, вероятно, придется на уникальную последовательность за пределами повторяющейся последовательности. Таким образом, стандартная программа выравнивания спаренных концов (например, выравниватель Барроуза-Уилера, такой как BWA-MEM) способна надежно совместить пару прочтений последовательности со спаренными концами с правильным местом в эталонной сборке, включая правильную копию повторяющейся последовательности. Затем можно регистрировать положения любых различий между выравненными мутированными прочтениями ρi последовательности и эталонной сборкой и сохранять их в битовой матрице α, аналогичной той, которая получена с использованием подхода, основанного на применении затравочных паттернов к каждому мутированному прочтению ρi последовательности. Таким образом, определение положений одной или более мутаций в соответствующем мутированном прочтении последовательности обеспечивают путем идентификации в соответствующем мутированном прочтении последовательности положений различий между соответствующим мутированным прочтением последовательности и эталонной сборкой.
Однако выравнивание множества мутированных прочтений Р последовательности с эталонной сборкой в некоторых ситуациях может быть не идеальным, поскольку любая указанная молекула темплатной нуклеиновой кислоты-мишени, как правило, будет иметь области, которые не представлены в эталонной сборке. Следовательно, невозможно выравнять мутированные прочтения ρi последовательности с теми областями, которые не представлены в эталонной сборке и получить битовый вектор α по различиям между выравненными мутированными прочтениям ρi последовательности и эталонной сборкой. Кроме того, области, которые не представлены в эталонной сборке, часто представляют клинический интерес, поскольку они представляют собой структурные варианты-вставки относительно эталонной сборки. В дополнение к крупным вставочным областям, любые возникающие в небольших вставках мутации относительно эталонной сборки, также будут пропущены при подходе, основанном на выравнивании множества мутированных прочтений Р последовательности с эталонной сборкой.
Таким образом, третий гибридный подход к улучшению способности способа отличать намеренно введенные мутации от природных различий между копиями повторяющихся последовательностей заключается в объединении подхода на основе выравнивания множества мутированных прочтений Р последовательности с эталонной сборкой и подхода, основанного на применении затравочных паттернов к каждому мутированному прочтению ρi последовательности. Этот третий подход может быть использован в качестве альтернативного варианта осуществления стадии 230 настоящего способа.
В третьем подходе положение одной или более мутаций в каждом мутированном прочтении последовательности определяют с использованием обоих подходов: на основе выравнивания множества мутированных прочтений Р последовательности с эталонной сборкой и на основе применения затравочных паттернов к каждому мутированному прочтению ρi последовательности. Если положение в соответствующем мутированном прочтении последовательности выравнено с эталонной сборкой, то положение в соответствующем мутированном прочтении последовательности определяется как положение мутации в соответствующем мутированном прочтении последовательности, если положение в соответствующем мутированном прочтении последовательности представляет собой положение, в котором соответствующее мутированное прочтение последовательности отличается от эталонной сборки. Если положение в соответствующем мутированном прочтении последовательности не выравнено с эталонной сборкой, то положение в соответствующем мутированном прочтении последовательности определяется как положение мутации в соответствующем мутированном прочтении последовательности, если положение в соответствующем мутированном прочтении последовательности представляет собой положение, которое маскируется всеми затравочными паттернами, которые соответствуют маскированным затравками мутированным k-мерам из множества маскированных затравками мутированных k-меров, которые встречаются в наборе маскированных затравками немутированных k-меров.
Для достижения этого битовый вектор а описанного выше типа независимо получают посредством обоих подходов: основанного на выравнивании и основанного на применении затравочных паттернов. Битовый вектор из подхода, основанного на применении затравочных паттернов к каждому мутированному прочтению ρi последовательности обозначен αmmd, а битовый вектор, основанный из подхода, основанного на применении выравнивания множества мутированных прочтений Р последовательности с эталонной сборкой обозначен αmap.Также сконструирован дополнительный битовый вектор выравнивающей маски, обозначенный αamask, который регистрирует те положения каждого мутированного прочтения последовательности, которые успешно выравниваются с эталонной сборкой. Битовый вектор выравнивающей маски αamask будет иметь 1 в каждом положении, которое выравнено успешно, и 0 - в положениях, которые не были успешно выравнены с эталонной сборкой.
Затем конструируют итоговый гибридный битовый вектор αhybrid, который объединяет битовый вектор из подхода на основе применения затравочных паттернов к каждому мутированному прочтению ρi последовательности, αmmd и битовый вектор из подхода, основанного на выравнивании множества мутированных прочтений Р последовательности с эталонной сборкой, αmap, следующим образом:
αhybrid=αmap | (α.mmd & ~ αamask)
Где | обозначает побитовый логический оператор ИЛИ, & обозначает побитовый логический оператор И, и ~ обозначает побитовый оператор НЕ.
Таким образом, в третьем подходе используют положения мутаций, определенные по выравниванию с эталонной сборкой, в тех положениях мутированного прочтения последовательности, где выравнивание было успешным, и положения мутаций, определенные путем применения затравочных паттернов, во всех других положениях. Это обеспечивает преимущество, заключающееся в возможности включения в анализ высококачественной эталонной сборки при одновременной обработке всех типов вставок относительно эталонной сборки. Выравнивание по независимой высококачественной эталонной сборке, такой как эталонный геном человека, может быть гораздо более точным, чем выравнивание по сборке из коротких чтений de novo. Использование положений мутаций, определенных по выравниванию с эталонной сборкой, может обеспечивать более точные оценки положений мутаций, особенно в областях повторяющихся последовательностей, тогда как способ без выравнивания, основанный на затравочных паттернах, может идентифицировать положения мутаций в областях, которые не представлены в эталонной сборке. Последнее может происходить без необходимости вычислять сборку, что представляет собой требовательную к вычислительным ресурсам задачу. Таким образом, гибридный подход обеспечивает улучшение точности идентификации положений мутаций и вычислительной эффективности относительно применения любого из подходов по отдельности.
Также возможно «увеличивать» эталонную сборку вариантами и локально собранными областями из конкретной молекулы темплатной нуклеиновой кислоты-мишени с получением графа сборки, специфичного для данной молекулы темплатной нуклеиновой кислоты-мишени. Битовый вектор из подхода, основанного на выравнивании множества мутированных прочтений Р последовательности с эталонной сборкой (обозначенный αmap) может быть получен по выравниванию мутированных прочтений последовательности с увеличенным графом сборки, а затем комбинирования с подходом, основанным на применении затравочных паттернов к каждому мутированному прочтению ρi последовательности для любых областей молекулы темплатной нуклеиновой кислоты-мишени, которая остается сложной для выравнивания по техническим или другим причинам.
Стадии S240 и S240: определение показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности
Способ 200 включает стадию S240, на которой для по меньшей мере двух мутированных прочтений последовательности с общим минимизатором выполняют подсчет количества мутаций с совпадающим положением и/или с несовпадающим положением, когда соответствующие минимизаторы выравнены.
Это может быть достигнуто путем первоначального определения разницы в положении j минимизатора, определенного на стадии S222 для каждого из двух мутированных прочтений последовательности. Например, разница в положении j минимизатора для каждого из двух мутированных прочтений ρа и ρс последовательности сохраненных в группе минимизатора в виде а=bz,y.i и с=bz,x.i может быть определена как d=bz,y.j-bz,x.j.
Подсчет количества мутаций с совпадающими положениями может включать определение размера пересечения множеств для положений мутаций, определенных на стадии S230, когда положения мутаций, определенные для одного из двух мутированных прочтений ρх и ρу последовательности, сохраненные как bz,y и bz,x, имеют правый сдвиг d. Например, для двух мутированных прочтений ρх и ρу последовательности, сохраненных как bzy и bz,x, число мутаций с совпадающими положениями может быть определено следующим образом:
λх,у=|Ω(bz,x.α) ∩ (Ω(bz,y.α) - d)|, где Ω(α) определяется как набор указателей положения в α, которые являются ненулевыми (т.е. набор положений мутаций в соответствующем мутированном прочтении ρi последовательности) и где Ω(bz,y.α) - d понимается как поэлементное вычитание d из Ω(bz,y.α). Пересечение множеств может быть эффективно реализовано на компьютере с использованием таких команд ЦП, как побитовый сдвиг и popcount.
Подсчет количества мутаций с несовпадающими положениями может включать определение размера симметричной разницы множеств для положений мутаций, определенных на стадии S230, когда положения мутаций, определенные для одного из двух мутированных прочтений ρх и ρу последовательности, сохраненные как bz,y и bz,x, имеют правый сдвиг d. Например, для двух мутированных прочтений ρх и ρу последовательности, сохраненных как bz,y и bz,x, число мутаций с несовпадающими положениями может быть определено следующим образом:
δх,у=|(Ω(bz,х.α) \ (Ω(bz,y.α) - d))∪ ((Ω(bz,y.α) - d) \ Ω(bz,x.α))|.
Стадия S242 определения показателя, коррелированного с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, может быть основана на количестве мутаций с совпадающим положением λх,у и/или с несовпадающим положением δx,У. В одном варианте осуществления показатель, коррелированный с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, соответствует количеству мутаций с совпадающим положением λx,y. Чем выше количество мутаций с совпадающими положениями λx,y, тем выше вероятность того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности. В альтернативном варианте осуществления показатель, коррелированный с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, соответствует количеству мутаций с несовпадающим положением δx,y. Чем ниже количество мутаций с несовпадающими положениями δx,y, тем выше вероятность того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности.
В предпочтительном варианте осуществления указанный показатель, коррелированный с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, представляет собой один из: i) плотности вероятности, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, и ii) оценочной функции, которая коррелирована с плотностью вероятности, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности.
Например, количество мутаций с совпадающими положениями λx,y и количество мутаций с несовпадающими положениями δx,y можно использовать для вычисления в модели плотности вероятности, что два прочтения получены от одной и той же содержащей мутации последовательности М, или оценочной функции, которая коррелирована с такой плотностью вероятности. Одна такая оценочная функция оценки представляет собой ωа,с=Δ(λx,y) - Δ(δx,y), где Δ(n)=(0,5n)(n+1) для а=bz,x.i и с=bz,y.i. Таким образом, ωа,c представляет собой балльную оценку или вес линии связи между двумя мутированными прочтениями последовательности ρа и ρс. Коллекция таких линий связи может быть получена для всех пар мутированных прочтений ρi последовательности в соответствующей группе минимизатора bz, или, если в группе минимизатора bz имеется большое количество записей, вычисление или создание отчета по линиям связи может быть ограничено случайно выбранными парами записей в группе минимизатора bz.
Стадия S300: сборка последовательности или реконструкция последовательности
Способ 10 может дополнительно включать стадию S300 сборки или реконструкции последовательности или по меньшей мере части последовательности, например содержащей мутации последовательности или не содержащей мутаций последовательности. Собранная или реконструированная последовательность может представлять собой содержащую мутации последовательность или не содержащую мутаций последовательность.
Способ 200, например стадия S300 реконструкции или сборки последовательности, может включать создание ненаправленного взвешенного графа из множества мутированных прочтений последовательности. Ненаправленный взвешенный граф содержит узлы, соответствующие множеству мутированных прочтений последовательности. Например, каждый узел может соответствовать соответствующему мутированному прочтению последовательности в том смысле, что он представлен указателем i прочтения соответствующего мутированного прочтения последовательности или последовательностью соответствующего мутированного прочтения последовательности. Ребра между узлами ассоциированы с соответствующими весовыми значениями ребер, причем вес каждого ребра может быть определен на основании количества мутаций с совпадающим положением и/или с несовпадающим положением, определенным для двух мутированных прочтений последовательности, соответствующих двум узлам, связанным с соответствующим ребром. Вес каждого ребра может соответствовать показателю, коррелированному с вероятностью того, что по меньшей мере два мутированных прочтения последовательности (т.е. два мутированных прочтения последовательности, соответствующие узлам, связанным с ребром, ассоциированным с весом ребра) происходят от одной и той же содержащей мутации последовательности. Таким образом, вес ребра, соединяющего два мутированных прочтения последовательности (узлы), представляет вероятность того, что эти два мутированных прочтения последовательности были получены от одной и той же содержащей мутации последовательности, или какую-либо другую произвольную функцию, которая коррелирована с этой вероятностью.
Ненаправленный взвешенный граф может быть построен путем обработки каждой из групп минимизаторов последовательно или параллельно с вычислением таким образом ребер между мутированными прочтениями последовательности в каждой группе минимизатора. Вес ребра может представлять собой оценочную функцию ωа,с.
Ненаправленный взвешенный граф, включающий в себя веса ребер ωа,с, затем можно использовать для обработки SAM-данных (например, мутированных прочтений последовательности), например, с использованием любых известных или неизвестных методик применения такого ненаправленного взвешенного графа для сборки последовательности. Сборка последовательности из ненаправленного взвешенного графа может включать, например, создание кластеров мутированных прочтений последовательности и сборку мутированных прочтений последовательности в каждом кластере для реконструкции темплата, соответствующего по меньшей мере части содержащей мутации последовательности.
Например, способ 200 или стадия S300 реконструкции или сборки по меньшей мере части последовательности могут включать в себя выполнение операции кластеризации графа на ненаправленном взвешенном графе, с получением таким образом кластеров мутированных прочтений последовательности, которые, как ожидается, происходят от одной и той же содержащей мутации последовательности. Кластеризация графа может быть выполнена с использованием любого стандартного поточного алгоритма кластеризации графов, такого как кластеризация Маркова (MCL) или Infomap.В альтернативном варианте осуществления ребра ненаправленного взвешенного графа могут быть отфильтрованы по некоторому минимальному пороговому весу, а затем для представления мутированных прочтений последовательности могут быть взяты соединенные компоненты графа, которые происходят от одной и той же содержащей мутации последовательности.
Стадия S300 реконструкции или сборки по меньшей мере части последовательности может дополнительно включать реконструкцию по меньшей мере части содержащей мутации последовательности путем сборки мутированных прочтений последовательности в кластеры. Например, мутированные прочтения последовательности в кластерах могут быть подвергнуты обработке методами стандартной сборки de novo для реконструкции содержащей мутации последовательности. Такие методы сборки de novo включают, например, алгоритм IDBA-UD из публикации «IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth», Peng Y et al., Bioinformatics. 2012 Jun l;28(11):1420-8. doi: 10.1093/bioinformatics/bts174. Epub 2012 Apr 11, или метод SPAdes из публикации SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing", Benkevich A et al., J Comput Biol. 2012 May; 19(5): 455-477, или метод A5-miseq из публикации «A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data», Coil D et al., Bioinformatics. 2015 Feb 15;31(4):587-9. doi: 10.1093/bioinformatics/btu661. Epub 2014 Oct 22.
Стадия S300 реконструкции или сборки последовательности может дополнительно включать реконструкцию по меньшей мере части не содержащей мутаций последовательности с использованием коррекции ошибок на реконструированной части содержащей мутации последовательности, т.е. путем выведения наиболее вероятной не содержащей мутаций последовательности из реконструированной части содержащей мутации последовательности, с использованием множества немутированных прочтений последовательности. Способы такой коррекции ошибок включают, например, метод FMLRC из публикации «FMLRC: Hybrid long read error correction using an FM-index», Jeremy R. Wang et al., BMC Bioinformatics volume 19, Article number: 50 (2018). Например, содержащая мутации последовательность может быть подвергнута коррекции ошибок с использованием немутированных прочтений последовательности для удаления внедренных мутаций с реконструкцией таким образом участков не содержащей мутаций последовательности. Коррекция ошибок может включать, например, определение возможных наборов редактирований содержащей мутации последовательности, которые потребуются для преобразования содержащей мутации последовательности, в не содержащую мутаций последовательность, совместимую с немутированными прочтениями последовательности, определение набора редактирований, имеющих наименьший размер (т.е. содержащего наименьшие редактирования) из возможных наборов редактирований, и применение определенного набора редактирований, имеющих наименьший размер, к содержащей мутации последовательности, с получением вероятной оценки не содержащей мутаций последовательности. Части не содержащей мутаций последовательности затем могут быть собраны с использованием стандартных инструментов для сборки из длинных чтений de novo, таких как Canu, Flye или PEREGRINE, или в комбинации с короткими чтениями в R с использованием такого инструмента, как Unicycler или MaSuRCA, со сборкой таким образом не содержащей мутаций последовательности.
Обработка пулов образцов
При обработке партий образцов, содержащих множество образцов, можно вводить штрихкоды образцов в виде заданных маркерных последовательностей для каждого образца. Если пользователь способа 200 желает использовать способ на множестве образцов, при этом каждый образец содержит одну или более мутированных молекул темплатных нуклеиновых кислот-мишеней, один из возможных вариантов заключается в обработке каждого образца (например, мутанта и/или фрагмента) в лаборатории по отдельности, а затем введении штрихкодов образцов только на конечной стадии перед секвенированием. Другой альтернативный вариант заключается во введении штрихкодов образцов только на концах молекул темплатных нуклеиновых кислот-мишеней, в этом случае появляется возможность объединить все меченные штрихкодом молекулы темплатных нуклеиновых кислот-мишеней в начале процесса подготовки образца, таким образом значительно уменьшая затраты на реагенты и трудозатраты (так называемый подход с объединением образцов в пулы на ранней стадии). Таким образом, подготовка образцов может включать введение соответствующих штрихкодов образцов в концы молекул темплатных нуклеиновых кислот-мишеней в каждом образце таким образом, чтобы каждый образец содержал молекулы темплатных нуклеиновых кислот-мишеней, имеющие отличающийся штрихкод образца относительно молекул темплатных нуклеиновых кислот-мишеней в других образцах. Подготовка образцов может дополнительно включать объединение образцов в пулы с получением пула образцов, введение мутаций и необязательно фрагментацию молекул темплатных нуклеиновых кислот-мишеней в пуле образцов, а также секвенирование частей мутированных молекул темплатных нуклеиновых кислот-мишеней в пуле образцов.
Однако подход с ранним объединением образцов в пулы создает дополнительную проблему при обработке данных, поскольку полученное множество мутированных прочтений Р последовательности содержит немеченную смесь мутированных Р прочтений последовательности из множества различных образцов. Образцы могут быть обработаны по отдельности для конструирования немутированных прочтений R последовательности, в этом случае немутированные прочтения R последовательности содержат множество наборов немутированных прочтений R1…Rζ последовательности, где ζ представляет собой количество образцов, обработанных в партии. Каждый набор немутированных прочтений последовательности может быть ассоциирован с соответствующим образцом. Способ 200 может включать прием немутированного прочтения R последовательности во множестве наборов немутированных прочтений R1…Rζ последовательности, причем каждый набор немутированных прочтений R1…Rζ последовательности ассоциирован с соответствующим одним или несколькими образцами.
Таким образом, каждое из множества мутированных прочтений последовательности может представлять собой подпоследовательность содержащей мутации последовательности, ассоциированной с одним из множества образцов. Каждое из множества немутированных прочтений последовательности может соответствовать подпоследовательности не содержащей мутации последовательности, ассоциированной с одним из множества образцов. Каждая содержащая мутации последовательность может содержать мутации по сравнению с соответствующей не содержащей мутаций последовательностью. Получение набора немутированных маскированных затравкой к-меров может включать получение соответствующего набора немутированных маскированных затравкой k-меров для каждого образца.
Простой подход к обработке данных из образцов ζ будет заключаться в применении способа 200 по одному разу для каждого из образцов ζ. Альтернативным подходом является расширение способа 200 таким образом, чтобы все образцы ζ можно было обрабатывать одновременно. Этого можно достигнуть в соответствии с представленным ниже описанием.
Способ 200 (например, стадия S230) может включать создание набора битовых векторов немутированных образцов, причем для соответствующего k-мера в наборе немутированных маскированных затравкой k-меров VR каждый битовый вектор немутированного образца определяет, в каком из множества образцов соответствующий k-мер встречается (или встречается по меньшей мере х раз, где х представляет собой целое число, большее или равное 1) и в каком из множества образцов соответствующий k-мер не встречается (или встречается менее чем х раз). Набор немутированных маркированных затравкой k-меров VR может быть создан из множества немутированных k-меров способом, уже описанным выше. Множество немутированных k-меров может быть определено как объединение всех k-меров в каждом из множества образцов R1…Rζ, т.е. множество немутированных k-меров R может быть определено как R=∪R1…Rζ⋅
Например, способ 200 может включать определение сюръекции VR на коллекцию битовых векторов, содержащих двоичные индикаторы присутствия маскированных затравкой k-меров в каждом образце. Каждый битовый вектор может иметь 1 в положении i, если i-й образец во множестве образцов содержит k-мер (или содержит его по меньшей мере X раз), в противном случае он имеет 0 в положении i. В программном варианте реализации сюръекцию можно хранить с использованием неупорядоченной структуры данных карты, такой как хеш-карта, или структуры приблизительного запроса членства, такой как счетный фильтр с коэффициентами. Сюръекция может быть обозначена как Z: VR→ν где ν - это пространство битовых векторов длиной ζ⋅
Стадия S230 определения положений одной или более мутаций в каждом мутированном прочтении последовательности может быть расширена для конструирования битового вектора а одновременно для множества образцов. Для каждого мутированного прочтения последовательности и для каждого набора и/или каждой комбинации наборов немутированных маскированных затравкой k-меров определение положений одной или более мутаций может включать идентификацию одного или более положений в мутированном прочтении последовательности, которые маскированы всеми затравочными паттернами, соответствующими мутированным маскированным затравкой k-мерам из множества мутированных маскированных затравкой k-меров, которые встречаются в соответствующем наборе или комбинации наборов немутированных маскированных затравкой k-меров, и связывание идентифицированного одного или более положений с одним или более образцами, ассоциированными с соответствующим набором или комбинацией наборов немутированных маскированных затравкой k-меров. Это может быть достигнуто, например, с помощью ориентированного на множество образцов варианта morphomutsMS(ρi,VR) функции morphomuts(ρi,VR), что включает следующие стадии:
1. Инициализация набора А битовых векторов а одним исходным битовым вектором а0 длиной 2, содержащим только биты 0; инициализация битового вектора b длиной 2k, содержащего только биты 1; инициализация набора С битовых векторов одним исходным элементом с0 длиной ζ, содержащим только биты 1; инициализация сопоставления Г: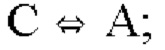
2. Для каждого положения j в прочтении между 
a. Для каждого затравочного паттерна  определить ψ(k(ρji))
определить ψ(k(ρji))
b. Если 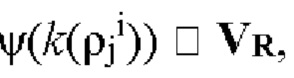 выполнить следующие стадии:
выполнить следующие стадии:
i. Для каждого элемента с (т.е. для каждого  ) вычислить d←c∧Z(ψ(k(ρji));
) вычислить d←c∧Z(ψ(k(ρji));
ii. Если d содержит только биты 0, то вернуться к 2b.i для обработки следующего элемента С (или, если их более нет, для обработки следующего затравочного паттерна, или следующего положения j), в противном случае продолжать с 2b.iii;
iii. присвоить α←Г(с) | (ψ(b)>>2j), где | означает побитовое логическое ИЛИ и >> означает оператор битового сдвига вправо, и удалить с из С;
iv. Прибавить d к С и α к А и определить сопоставление d→а в Г;
v. Если 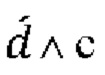 не равно нулю, то прибавить
не равно нулю, то прибавить 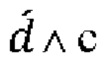 к С и Г(с) к А и определить сопоставление
к С и Г(с) к А и определить сопоставление 
vi. вернуться к 2b.i для обработки следующего элемента с из С. Если в С больше нет с, возврат к 2а для обработки следующего затравочного паттерна ψ⋅ Если в Ψ больше нет ψ, возврат к 2 для обработки следующего положения j. В противном случае продолжить с 3, иначе:
3. Преобразовать битовые векторы в А с использованием трансформации, применяемой для создания α в функции morphomuts(⋅); и
4. Вернуть С, А и сопоставление Г как результат функции.
Необязательно, если в битовом векторе А слишком мало совпадающих положений (например, меньше заданного числа у, где у представляет собой целое число, большее или равное 1, предпочтительно большее или равное 2, 3, 4 или 5), соответствующие записи в С и А могут быть отброшены. Это является преимуществом, поскольку такие записи могут появляться из-за случайного сходства между входными образцами, и, таким образом, полученные битовые векторы являются результатом ошибочных совпадений с неверным образцом. Путем отбрасывания этих положений перед дальнейшей обработкой можно избежать ненужных вычислений. Способ 200 может включать сравнение количества идентифицированных положений с предварительно заданным числом у, где у представляет собой целое число, которое больше или равно 1, предпочтительно, больше или равно 2, и если количество идентифицированных положений меньше предварительно заданного числа у, отбрасывание (или игнорирование при дальнейшей обработке) идентифицированного одного или более положений и ассоциации идентифицированного одного или более положений с одним или более образцами.
Кортежи, которые хранятся в группах минимизаторов, могут быть расширены путем включения информации о битовом векторе образца в С. В частности, хранящийся кортеж может представлять собой показатель i чтения, положение j минимизатора в мутированном прочтении последовательности, а также с и α, где с представляет собой битовый вектор образцов, а α представляет собой битовый вектор мутаций, вычисленный функцией morphomutsMS(pi,VR).
Впоследствии при обработке групп минимизаторов для вычисления весов ребер к каждому значению веса ребра может быть добавлена аннотация с соответствующим набором образцов. Если побитовое логическое И для битовых векторов образцов, связанных с парой мутированных прочтений последовательности, дает нуль, то соответствующее ребро может быть отброшено. Если балльная оценка ребра меньше предварительно заданного порогового значения балльной оценки, ребро может быть отброшено. Когда между парой мутированных прочтений последовательности имеется несколько возможных ребер, становится возможным сохранить только самый высокий вес ребра, и ассоциированный набор битовых векторов для этого ребра можно вычислять побитовым логическим И для битовых векторов образцов. Этот подход имеет преимущество, заключающееся в том, что естественную вариацию последовательностей в различных образцах можно отличить от мутаций, введенных во время обработки образца. Стадию S240 подсчета числа мутаций с совпадающим положением и/или несовпадающим положением при выравнивании минимизаторов двух мутированных прочтений последовательности можно выполнять для любой пары из одного или более положений мутаций, идентифицированных для двух мутированных прочтений последовательности, только если существует перекрытие в образцах, ассоциированных с соответствующей парой из одного или более положений мутаций, идентифицированных для двух мутированных прочтений последовательности, т.е. только если пара из одного или более положений мутаций, идентифицированных для двух мутированных прочтений последовательности, ассоциирована с по меньшей мере одним общим образцом.
Если метку образца содержат только концы мутированных молекул темплатных нуклеиновых кислот-мишеней, то некоторые из множества мутированных прочтений Р последовательности будут нести эту метку образца. В частности, мутированные прочтения последовательности, полученные в результате секвенирования концов мутированных молекул темплатной нуклеиновой кислоты-мишени, будут нести метку образца. После кластеризации мутированных прочтений последовательности становится возможным связать образцы с кластерами прочтений просто путем оценки наличия прочтений с метками образца в каждом кластере. Когда в кластере встречается только одна метка образца, связывание с образцом является простым и однозначным. Выполнение кластеризации графа ненаправленного взвешенного графа может включать ассоциирование с каждым кластером из мутированных прочтений последовательности метки образца, содержащейся в по меньшей мере одном из мутированных прочтений последовательности в соответствующем кластере.
Иногда в одном кластере может встретиться несколько меток образца либо из-за шума, либо из-за ошибки в процедурах секвенирования или анализа данных. В этом случае может сохраняться возможность достоверного связывания с образцом, если существует большой избыток одной метки образца по сравнению с другими метками. В тех случаях, когда однозначное связывание невозможно, может сохраняться возможность устранения неопределенности для образца путем применения процедуры полуконтролируемого разложения графа, которая разлагает кластер из нескольких образцов на ряд меньших кластеров, по одному кластеру на метку образца. Даже если кластер не содержит прочтений, несущих метку образца, все еще может быть возможно связать кластер с образцом, если большинство масок образцов, ассоциированных с соединениями между прочтениями, указывают на один образец. Выполнение кластеризации графа ненаправленного взвешенного графа может включать идентификацию в каждом кластере из мутированных прочтений последовательности одной или более меток образцов, содержащихся в мутированных прочтениях последовательности в соответствующем кластере мутированных прочтений последовательности. Каждый кластер мутированных прочтений последовательности можно ассоциировать с меткой образца, которая встречается чаще всего в мутированных прочтениях последовательности в соответствующем кластере. Необязательно, если в кластере мутированных прочтений последовательности идентифицированы две или более разных меток образцов, кластер мутированных прочтений последовательности может быть разделен на два или более кластеров, причем каждый из двух или более кластеров ассоциируется с соответствующей одной из двух или более разных меток образцов и содержит различные последовательности мутированных прочтений последовательности.
Подготовка образцов и секвенирование
Способ 10 определения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может включать секвенирование 100 областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени, содержащей мутации, с получением множества мутированных прочтений последовательности. Способ 10 определения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может дополнительно включать выполнение способа 200 определения показателя, коррелированного с вероятностью того, что два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, на основе множества мутированных прочтений последовательности, полученных посредством секвенирования 100.
Стадия секвенирования может включать:
а) обеспечение пары образцов, причем каждый образец содержит по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени;
(b) секвенирование областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением множества немутированных прочтений последовательности;
(c) введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором образце из пары образцов с получением по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени;
(d) секвенирование областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением множества мутированных прочтений последовательности.
В предпочтительном варианте осуществления стадия введения мутаций включает введение мутаций типа транзиции в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором из пары образцов.
Стадия секвенирования может включать:
(a) обеспечение множества пар образцов, причем каждая пара образцов содержит первый образец и второй образец, при этом каждый образец содержит по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени;
(b) введение штрихкода образца в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени из каждой пары образцов таким образом, что каждая пара образцов ассоциируется с соответствующим штрихкодом;
(c) секвенирование областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в каждом первом образце с получением множества немутированных прочтений последовательности, причем секвенирование выполняют отдельно для каждого первого образца, таким образом обеспечивая соответствующий набор немутированных прочтений последовательности для каждого первого образца;
(d) объединение вторых образцов в пулы с получением пула вторых образцов;
(e) введение мутаций в молекулы темплатных нуклеиновых кислот-мишеней в пуле образцов с получением мутированных молекул темплатной нуклеиновой кислоты-мишени;
(d) секвенирование областей мутированных молекул темплатной нуклеиновой кислоты-мишени с получением множества мутированных прочтений последовательности.
Стадия секвенирования необязательно может дополнительно включать фрагментирование по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в каждом первом образце после введения штрихкода образца и перед секвенированием областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Стадия секвенирования необязательно может дополнительно включать фрагментирование по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени или мутированных молекул темплатной нуклеиновой кислоты-мишени в пуле образцов перед секвенированием областей мутированных молекул темплатной нуклеиновой кислоты-мишени.
В способах изобретения одновременно можно секвенировать любое количество по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Таким образом, в одном варианте осуществления изобретения по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени содержит множество молекул темплатной нуклеиновой кислоты-мишени. По меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени необязательно содержит по меньшей мере 10, по меньшей мере 20, по меньшей мере 50, по меньшей мере 100 или по меньшей мере 250 молекул темплатной нуклеиновой кислоты-мишени. По меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени необязательно содержит от 10 до 1000, от 20 до 500 или от 50 до 100 молекул темплатной нуклеиновой кислоты-мишени.
Стадия S110: подготовка образца
Поскольку первый образец из пары образцов и второй образец из пары образцов оба содержат по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени, пара образцов может быть получена из одного и того же целевого организма или взята из одного и того же исходного образца.
Например, если пользователь планирует секвенировать по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени в образце, пользователь может взять пару образцов из одного и того же исходного образца. Пользователь необязательно может реплицировать эту по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени в исходном образце перед извлечением из нее пары образцов. Пользователь может планировать секвенировать различные молекулы нуклеиновых кислот конкретного организма, например E.coli. В этом случае первый образец из пары образцов может представлять собой образец E.coli из одного источника, а второй образец из пары образцов может представлять собой образец E.coli из второго источника.
Пара образцов может происходить из любого источника, который содержит или предположительно содержит по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени. Пара образцов может содержать образец молекул нуклеиновой кислоты, полученный от человека, например образец, взятый мазком с кожи пациента-человека. В альтернативном варианте осуществления пара образцов может быть получена из других источников, таких как источник воды. Такие образцы могут содержать миллиарды молекул темплатной нуклеиновых кислот. Может быть возможно секвенировать каждую из этих миллиардов молекул нуклеиновых кислот одновременно с использованием способов изобретения, и поэтому верхнего предела по количеству молекул темплатной нуклеиновой кислоты-мишени, которые можно использовать в способах изобретения, не существует.
В одном варианте осуществления может быть предусмотрено множество пар образцов. Например, может быть предусмотрено по меньшей мере 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 15, 20, 25, 50, 75, 100, 500, 1000 или 5000 пар образцов. Необязательно обеспечивают менее 10000, менее 5000, менее 1000, менее 100, менее 75, менее 50, менее 25, менее 20, менее 15, менее 11, менее 10, менее 9, менее 8, менее 7, менее 6, менее 5 или менее 4 образцов. Необязательно обеспечивают от 2 до 100, от 2 до 75, от 2 до 50, от 2 до 25, от 5 до 15 или от 7 до 15 пар образцов.
При обеспечении множества пар образцов, по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени в разных парах образцов может быть помечена различными метками образца (также называемыми в настоящем документе штрихкодами). Например, если пользователь планирует обеспечить 2 пары образцов, то все или по существу все из по меньшей мере одной молекулы темплатных нуклеиновых кислот-мишеней в первой паре образцов могут быть помечены меткой А образца, а все или по существу все из по меньшей мере одной молекулы темплатных нуклеиновых кислот-мишеней во второй паре образцов могут быть помечены меткой В образца.
В данной области известны подходящие способы амплификации по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Например, обычно используют ПЦР. ПЦР более подробно описана ниже под заголовком «введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени».
Введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени
Способ может включать стадию введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором образце из пары образцов с получением по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени.
Мутации могут представлять собой мутации по типу замены, мутации по типу вставки или мутации по типу делеции. Для целей настоящего изобретения термин «мутация по типу замены» следует интерпретировать как то, что нуклеотид заменен на другой нуклеотид. Например, превращение последовательности АТСС в последовательность AGCC вводит одну мутацию по типу замены. Для целей настоящего изобретения термин «мутация по типу вставки» следует интерпретировать как то, что в последовательность добавляют по меньшей мере один нуклеотид. Например, превращение последовательности АТСС в последовательность АТТСС представляет собой пример мутации по типу вставки (с вставкой дополнительного нуклеотида Т). Для целей настоящего изобретения термин «мутация по типу делеции» следует интерпретировать как то, что по меньшей мере один нуклеотид удаляют из последовательности. Например, превращение последовательности АТТСС в АТСС является примером мутации по типу делеции (с удалением нуклеотида Т). Предпочтительно мутации представляют собой мутации по типу замены.
Фраза «введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени» обозначает воздействие на по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором из пары образцов условий, в которых по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени мутирует. Этого можно добиться, используя любой подходящий способ. Например, мутации могут быть введены путем химического мутагенеза и/или ферментативного мутагенеза.
Стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно приводит к мутации от 1% до 50%, от 3% до 25%, от 5% до 20% или около 8% нуклеотидов по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. По меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени необязательно содержит от 1% до 50%, от 3% до 25%, от 5% до 20% или около 8% мутаций.
Пользователь может определить, сколько мутаций введено в по меньшей мере одну мутированную молекулу темплатной нуклеиновой кислоты-мишени и/или уровень, до которого на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени вводят мутации в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени путем выполнения стадии введения мутаций в молекулу нуклеиновой кислоты с известной последовательностью, секвенируя полученную молекулу нуклеиновой кислоты и определяя процент от общего количества нуклеотидов, подвергшихся изменению по сравнению с исходной последовательностью.
Стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно приводит к мутированию по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени по существу случайным образом. По меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени необязательно содержит по существу случайный рисунок мутаций.
По меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени содержит по существу случайный рисунок мутаций, если она содержит мутации по всей своей длине в по существу аналогичных количествах. Например, пользователь может определить, содержит ли по меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени по существу случайный рисунок мутаций, путем введения мутаций в тестовую молекулу нуклеиновой кислоты с известной последовательностью с получением мутированной тестовой молекулы нуклеиновой кислоты. Последовательность мутированной тестовой молекулы нуклеиновой кислоты можно сравнивать с молекулой тестовой нуклеиновой кислоты и определять положения каждой из мутаций. Затем пользователь может определить, встречаются ли мутации по всей длине мутированной тестовой нуклеиновой кислоты в по существу аналогичных количествах следующим образом:
(i) вычислить расстояния между каждой из мутаций;
(ii) вычислить среднее значение для расстояний;
(iii) сформировать подвыборку расстояний без замены на меньшее число, например из 500 или 1000 расстояний;
(iv) построить модельный набор из 500 или 1000 расстояний по геометрическому распределению, с получением среднего методом моментов, чтобы сопоставить с ранее полученным на наблюдаемых расстояниях; и
(v) провести тест Колмогорова-Смирнова на этих двух распределениях.
По меньшей мере одну мутированную молекулу темплатной нуклеиновой кислоты-мишени можно считать содержащей по существу случайный рисунок мутаций при D<0,15, D<0,2, D<0,25 или D<0,3 в зависимости от длины немутированных прочтений.
Аналогичным образом на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени вводят мутацию в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени по существу случайным образом, если полученная по меньшей мере одна мутированная темплатная нуклеиновая кислота-мишень содержит по существу случайный рисунок мутаций. Определить, действительно ли на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени мутации вводят в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени по существу случайным образом, можно путем выполнения стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени на тестовой нуклеиновой кислоте с известной последовательностью с получением мутированной тестовой молекулы нуклеиновой кислоты. Затем пользователь может секвенировать мутированную тестовую молекулу нуклеиновой кислоты, чтобы идентифицировать, какие мутации были введены, и определить, содержит ли мутированная молекула нуклеиновой кислоты по существу случайный рисунок мутаций.
По меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени необязательно содержит несмещенный рисунок мутаций. На стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно вводят мутации несмещенным образом. По меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени содержит несмещенный рисунок мутаций, если типы вводимых мутаций являются случайными. Если вводимые мутации представляют собой мутации по типу замены, то указанные мутации являются случайными, если вводят сходные доли А (аденозина), Т (тимина), С (цитозина) и G (гуанина). Фраза «вводят сходные доли А (аденозина), Т (тимина), С (цитозина) и G (гуанина)» означает, что количества введенных аденозиновых, тиминовых, цитозиновых и гуаниновых нуклеотидов находятся в пределах 20% друг от друга (например, 20 А-нуклеотидов, 18 Т-нуклеотидов, 24 С-нуклеотида и 22 G-нуклеотида).
Определить, действительно ли на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени мутации вводят в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени несмещенным образом, можно путем выполнения стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени на тестовой нуклеиновой кислоте с известной последовательностью с получением мутированной тестовой молекулы нуклеиновой кислоты. Затем пользователь может секвенировать мутированную тестовую молекулу нуклеиновой кислоты, идентифицировать, какие мутации были введены, и определить, содержит ли мутированная молекула нуклеиновой кислоты несмещенный рисунок мутаций.
В целом способы получения последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени могут быть использованы даже при условии, что стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени вводит неравномерно распределенные мутации. Таким образом, в одном варианте осуществления по меньшей мере одна мутированная молекула темплатной нуклеиновой кислоты-мишени содержит неравномерно распределенные мутации. На стадии введения мутаций в по меньшей мере одну мутированную молекулу темплатной нуклеиновой кислоты-мишени необязательно вводят мутации, которые распределены неравномерно. Мутации считаются «неравномерно распределенными» если мутации вводятся смещенным образом, т.е. количества введенных аденозиновых, тиминовых, цитозиновых и гуаниновых нуклеотидов не находятся в пределах 20% друг от друга. Определить, действительно ли по меньшей мере одна мутированная темплатная молекула нуклеиновой кислоты-мишени содержит неравномерно распределенные мутации или на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени вводят мутации, которые распределены неравномерно, можно аналогично тому, как описано выше в отношении определения того, вводят ли мутации на стадии введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени несмещенным образом.
Аналогичным образом способы получения последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени могут быть использованы, даже если мутированные прочтения последовательности и/или немутированные прочтения последовательности содержат неравномерно распределенные ошибки секвенирования. Таким образом, в одном варианте осуществления мутированные прочтения последовательности и/или немутированные прочтения последовательности содержат ошибки секвенирования, которые распределены неравномерно. Аналогичным образом в одном варианте осуществления стадия секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени и/или секвенирования областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени вводит ошибки секвенирования, которые распределены неравномерно.
Действительно ли конкретная стадия секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени и/или секвенирования областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени вводит ошибки секвенирования, которые распределены неравномерно, вероятно, будет зависеть от точности секвенирующего инструмента и, вероятно, будет известно пользователю. Однако пользователь может выяснить, действительно ли стадия секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени и/или секвенирования областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени вводит ошибки секвенирования, которые распределены неравномерно, путем выполнения способа секвенирования на молекуле нуклеиновой кислоты с известной последовательностью и сравнения полученных прочтений последовательности с прочтениями последовательности, полученными с использованием исходной молекулы нуклеиновой кислоты с известной последовательностью. Затем пользователь может применить вероятностную функцию, описанную в примере 6, и определить значения для М и Е. Если значения Е и матричной модели неравны или по существу неравны (в пределах 10% друг от друга), то стадия секвенирования по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени вводит ошибки секвенирования, которые распределены неравномерно.
Введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени посредством химического мутагенеза может быть достигнуто путем воздействия на по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени химическим мутагеном. Подходящие химические мутагены включают в себя митомицин С (ММС), н-метил-N-нитрозомочевину (MNU), азотистую кислоту (NA), диэпоксибутан (DEB), 1,2,7,8,-диэпоксиоктан (DEO), этилметансульфонат (EMS), метилметансульфонат (MMS), N-метил-N'-нитро-N-нитрозогуанидин (MNNG), 4-нитрохинолин-1-оксид (4-NQO), 2-метилокси-6-хлор-9(3-[этил-2-хлорэтил]-аминопропиламино)-акридиндигидрохлорид (ICR-170), 2-аминопурин (2А), бисульфит и гидроксиламин (НА). Например, когда молекулы нуклеиновых кислот подвергаются воздействию бисульфита, бисульфит дезаминирует цитозин с образованием урацила, эффективно вводя мутацию замены С-Т.
Как отмечено выше, стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени может быть осуществлена ферментативным мутагенезом. Ферментативный мутагенез необязательно осуществляют с использованием ДНК-полимеразы. Например, некоторые ДНК-полимеразы являются подверженными ошибкам (представляют собой низкоточные полимеразы), и репликация по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени с использованием подверженной ошибкам ДНК-полимеразы будет вводить мутации. Taq-полимераза является примером низкоточной полимеразы, а стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени может быть осуществлена путем репликации по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени с использованием Taq-полимер азы, например, с помощью ПЦР.
ДНК-полимераза может представлять собой ДНК-полимер азу с низким смещением.
Если стадию введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени осуществляют с использованием ДНК-полимеразы, по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени может быть инкубирована с ДНК-полимеразой и подходящими праймерами в условиях, подходящих для того, чтобы ДНК-полимераза катализировала образование по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени.
Подходящие праймеры содержат короткие молекулы нуклеиновой кислоты, комплементарные областям, фланкирующим по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени или областям, фланкирующим молекулы нуклеиновой кислоты, которые комплементарны по меньшей мере одной молекуле темплатной нуклеиновой кислоты-мишени. Например, если по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени является частью хромосомы, праймеры будут комплементарны областям хромосомы непосредственно от 3' и до 3'-конца по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени и непосредственно от 5' и до 5'-конца по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени, или праймеры будут комплементарны областям хромосомы непосредственно от 3' и до 3'-конца молекулы нуклеиновой кислоты, комплементарной по меньшей мере одной молекуле темплатной нуклеиновой кислоты-мишени, и непосредственно от 5' и до 5'-конца молекулы нуклеиновой кислоты, комплементарной по меньшей мере одной молекуле темплатной нуклеиновой кислоты-мишени.
Подходящие условия включают в себя температуру, при которой ДНК-полимераза может реплицировать по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени. Например, температуру от 40°С до 90°С, от 50°С до 80°С, от 60°С до 70°С или около 68°С.
Стадия введения мутаций в по меньшей мере одну молекулу нуклеиновой кислоты может включать множество циклов репликации. Например, стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени предпочтительно включает:
i) цикл репликации по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени с получением по меньшей мере одной молекулы нуклеиновой кислоты, которая комплементарна по меньшей мере одной молекуле темплатной нуклеиновой кислоты-мишени; и
ii) цикл репликации по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени с получением реплик по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени.
Стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно включает по меньшей мере 2, по меньшей мере 4, по меньшей мере 6, по меньшей мере 8, по меньшей мере 10, менее 10, менее 8, около 6, от 2 до 8 или от 1 до 7 циклов репликации по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени. Пользователь может выбрать использование небольшого количества циклов репликации для снижения вероятности введения систематической ошибки амплификации.
Стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно включает по меньшей мере 2, по меньшей мере 4, по меньшей мере 6, по меньшей мере 8, по меньшей мере 10, менее 10, менее 8, около 6, от 2 до 8 или от 1 до 7 циклов репликации при температуре от 60°С до 80°С.
Стадию введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно осуществляют с использованием полимеразной цепной реакции (ПЦР). ПЦР представляет собой процесс, который включает множество циклов из следующих стадий репликации молекулы нуклеиновой кислоты:
a) плавление;
b) отжиг; и
c) достройка и удлинение.
Молекулу нуклеиновой кислоты (например, по меньшей мере одна молекула темплатной нуклеиновой кислоты-мишени) смешивают с подходящими праймерами и полимеразой. На стадии плавления молекулу нуклеиновой кислоты нагревают до температуры выше 90°С, так чтобы молекула двухцепочечной нуклеиновой кислоты денатурировалась (разделилась на две цепи). На стадии отжига молекулу нуклеиновой кислоты охлаждают до температуры ниже 75°С, например от 55°С до 70°С, около 55°С или около 68°С, чтобы праймеры прикрепились к молекуле нуклеиновой кислоты. На стадиях достройки и удлинения молекулу нуклеиновой кислоты нагревают до температуры более 60°С, чтобы ДНК-полимераза катализировала достройку праймера - добавление нуклеотидов, комплементарных матричной цепи.
Стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени необязательно включает репликацию по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени с использованием Taq-полимеразы в условиях реакции, делающих ее подверженной ошибкам. Например, стадия введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени может включать в себя ПЦР с использованием Taq-полимеразы в присутствии Mn2+, Mg2+ или неравных концентраций дНТФ (например, избытка цитозина, гуанина, аденина или тимина).
Стадия S120: секвенирование
Получение данных, содержащих немутированные прочтения последовательности и мутированные прочтения последовательности
Способы изобретения могут включать стадию приема мутированных прочтений последовательности и необязательно приема немутированных прочтений последовательности. Немутированные прочтения последовательности и мутированные прочтения последовательности могут быть получены из любого источника.
Немутированные прочтения последовательности необязательно получают путем секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов. Мутированные прочтения последовательности необязательно получают путем введения мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором образце из пары образцов с получением по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени и секвенирования областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени.
Немутированные прочтения последовательности необязательно содержат последовательности областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов, а мутированные прочтения последовательности содержат последовательности областей по меньшей мере одной мутированной темплатной нуклеиновой кислоты-мишени второго образца из пары образцов, и пара образцов была взята из одного и того же исходного образца или была получена из одного и того же организма.
Секвенирование областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени или по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени
Способ определения последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени может включать стадию секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением немутированных прочтений последовательности и/или стадию секвенирования по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением мутированных прочтений последовательности.
Стадии секвенирования могут быть осуществлены с использованием любого способа секвенирования. Примеры возможных способов секвенирования включают в себя секвенирование по Максаму - Гилберту, секвенирование по Сэнгеру, секвенирование, включающее мостиковую амплификацию (например, мостиковую ПЦР) или любой метод высокопроизводительного секвенирования (HTS), как описано в публикациях Maxam AM, Gilbert W (February 1977), «A new method for sequencing DNA», Proc. Natl. Acad. Sci. U.S.A. 74 (2): 560-4, Sanger F, Coulson AR (May 1975), «A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase)}, J. Mol. Biol. 94 (3): 441-8; и Bentley DR, Balasubramanian S, et al. (2008), ((Accurate whole human genome sequencing using reversible terminator chemistry», Nature, 456 (7218): 53-59.
В типичном варианте осуществления по меньшей мере один или предпочтительно обе стадии секвенирования включают мостиковую амплификацию. Стадию мостиковой амплификации необязательно осуществляют с использованием времени достройки более 5, более 10, более 15 или более 20 секунд. Примером использования мостиковой амплификации являются секвенаторы для анализа генома Illumina. Предпочтительно применяют секвенирование спаренных концов.
Стадии (i) секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением немутированных прочтений последовательности и (ii) секвенирования по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением мутированных прочтений последовательности необязательно осуществляют с использованием одного и того же способа секвенирования.
Стадии (i) секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением немутированных прочтений последовательности и (ii) секвенирования по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением мутированных прочтений последовательности необязательно осуществляют с использованием разных способов секвенирования.
Стадии (ii) секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением немутированных прочтений последовательности и (ii) секвенирования по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением мутированных прочтений последовательности необязательно могут быть осуществлены с использованием более одного способа секвенирования. Например, фракция по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов может быть секвенирована с использованием первого способа секвенирования, и фракция по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов может быть секвенирована с использованием второго способа секвенирования. Аналогичным образом фракция по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени может быть секвенирована с использованием первого способа секвенирования, и фракция по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты мишени может быть секвенирована с использованием второго способа секвенирования.
Стадии (i) секвенирования областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением немутированных прочтений последовательности и (ii) секвенирования по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением мутированных прочтений последовательности необязательно осуществляют в разное время. В альтернативном варианте осуществления стадии (i) и (ii) можно проводить относительно одновременно, например в течение 1 года друг от друга. Первый образец из пары образцов и второй образец из пары образцов не обязательно должны быть взяты одновременно друг с другом. Если два образца получают из одного и того же организма, они могут быть обеспечены в по существу разные моменты времени, даже с многолетним интервалом, и поэтому две стадии секвенирования также могут разделяться периодом в несколько лет. Кроме того, даже если первый образец из пары образцов и второй образец из пары образцов были получены из одного и того же исходного образца, биологические образцы можно хранить в течение некоторого времени, и поэтому нет необходимости выполнять стадии секвенирования в одно и то же время.
Мутированные прочтения последовательности и/или немутированные прочтения последовательности могут представлять собой прочтения последовательности с одним концом или со спаренными концами.
Мутированные прочтения последовательности и/или немутированные прочтения последовательности необязательно имеют длину более 50 нт, более 100 нт, более 500 нт, менее 200000 нт, менее 15000 нт, менее 1000 нт, от 50 до 200000 нт, от 50 до 15000 нт или от 50 до 1000 нт.
Стадии секвенирования необязательно осуществляют с использованием глубины секвенирования от 0,1 до 500 прочтений, от 0,2 до 300 прочтений или от 0,5 до 150 прочтений на нуклеотид на по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени. Чем больше глубина секвенирования, тем выше будет точность определенной/сгенерированной последовательности, но сборка может быть более сложной.
Выбор параметров
Предпочтительно параметры, используемые в способе 200, выбраны так, как указано ниже.
В предпочтительном варианте осуществления вес w(ψ) каждого затравочного паттерна находится в диапазоне от 5 до 50, предпочтительно от 10 до 30, дополнительно предпочтительно от 13 до 23. Это обеспечивает, что каждый затравочный паттерн будет достаточно большим, чтобы гарантировать, что каждый k-мер, маскированный каждым затравочным паттерном ψ, с высокой вероятностью является уникальным. Например, для бактериальных геномов с типичной длиной 5 миллионов нуклеотидов вес w(ψ) каждого затравочного паттерна ψ предпочтительно находится в диапазоне 13-19, с учетом того, что 413>5 миллионов. Для геномов, по размеру сходных с человеческим, с типичной длиной около 3 миллиардов нуклеотидов вес w(ψ) каждого затравочного паттерна предпочтительно находится в диапазоне 19-23, с учетом того, что 419>3×109.
В предпочтительном варианте осуществления размер k каждого k-мера, используемого на стадии S230 определения положений одной или более мутаций в каждом мутированном прочтении последовательности, превышает вес w(ψ) каждого затравочного паттерна. Размер к каждого k-мера может быть менее чем в 5 раз, менее чем в 4 раза, менее чем в 3 раза или менее чем 2 раза меньше веса w(ψ) каждого затравочного паттерна ψ. Размер k каждого k-мера, используемого на стадии S230 определения положений одной или более мутаций в каждом мутированном прочтении последовательности, может находиться в диапазоне от 10 до 250, предпочтительно от 13 до 100, дополнительно предпочтительно от 15 до 50, наиболее предпочтительно от 20 до 40. Это гарантирует, что размер k будет достаточно малым, чтобы обеспечивать низкую вероятность того, что любой k-мер будет включать инсерционную или делеционную ошибку секвенирования, что является недостатком в контексте способа 200.
Ниже показан пример семейства затравочных паттернов содержащих затравочные паттерны с весом w(ψ)=16 и k=27:
ψ1={0, 1, 2, 3, 5, 6, 9, 12, 13, 14, 16, 18, 20, 21, 22, 23},
ψ2={0, 1, 2, 4, 5, 9, 10, 11, 13, 18, 19, 21, 23, 24, 25, 26},
ψ3={0, 1, 2, 3, 4, 5, 7, 8, 9, 10, 13, 15, 16, 18, 19, 20},
ψ4={0, 1, 2, 4, 6, 7, 12, 14, 16, 17, 20, 21, 23, 24, 25, 26},
В одном варианте осуществления k-меры, используемые на стадии S220 применения общей минимизирующей функции, т.е. одного или более минимизаторов, определенных для каждого мутированного прочтения последовательности, имеют размер k, отличный от k-меров, используемых на стадии S230 определения положений одной или более мутаций в каждом мутированном прочтении последовательности. Размер k каждого минимизатора может находиться в диапазоне от 5 до 50, предпочтительно от 10 до 30, дополнительно предпочтительно от 13 до 23. Размер k каждого минимизатора может быть выбран на основе тех же соображений, что и выбор веса w(ψ) затравочных паттернов. Размер k каждого минимизатора может находиться в диапазоне от 13 до 19 для бактерий и от 19 до 23 для геномов, по размеру сходных с человеческим.
Реализация способа 200
Способ 200 может быть иметь разные варианты реализации. Предпочтительный подход заключается в том, чтобы сначала вычислить набор UM в первоначальный проход через некоторые или все мутированные прочтения Р последовательности и немутированные прочтения R последовательности, затем вычислить WМ во второй проход через мутированные прочтения Р последовательности и немутированные прочтения R последовательности. Имея WМ, в третьем проходе через мутированные Р прочтения последовательности можно вычислить положения минимизаторов вместе с положениями одной или более мутаций, и эти положения можно сохранить в группах минимизаторов либо в ОЗУ, либо в устройстве постоянного хранения (например, на диске). Множество групп минимизаторов необязательно могут храниться в одном файле либо отсортированными, либо неотсортированными. Затем каждую группу минимизатора (или каждый файл) можно считывать последовательно или параллельно, обрабатывая группы минимизаторов для вычисления веса ребер. Поскольку каждое мутированное прочтение последовательности может встречаться в нескольких группах минимизаторов, существует возможность, что пара мутированных прочтений последовательности может иметь несколько вычисленных оценок веса. В этом случае необходимо использовать некоторый показатель для выбора предпочтительного веса, как правило, максимум. Наконец, если химия секвенирования выдала прочтения спаренных концов, и каждое прочтение в паре прочтений спаренных концов имеет общие минимизаторы, тогда балльные оценки для двух концов можно суммировать и получить одну балльную оценку для пары прочтений спаренных концов.
Экспериментальные данные
Способ 200 использовали для обработки нескольких реальных наборов SAM-данных, причем каждый набор SAM-данных содержал немутированные прочтения последовательности и мутированные прочтения последовательности.
Обрабатывали набор SAM-данных Arobacter butzleri JV22. Этот организм имеет геном размером 2,3 млн.п.н., который существует в виде одной кольцевой хромосомы. Реализацию способа 200 на С++ выполняли на экземпляре службы Amazon AWS. Набор SAM-данных состоит из 956133 пар эталонных (немутированных) прочтений и 2154909 пар мутированных прочтений, полученных из приблизительно 8000 мутированных длинных темплатов. 2087506 мутированных прочтений (96,9%) происходят из внутренних частей мутированных длинных темплатов, в то время как 67403 (3,1%) происходят из концов длинных темплатов и содержат штрихкоды образцов. Каждое отдельное прочтение имеет длину 150 нт или менее. Пары прочтений предварительно прошли адаптерную обрезку и обрезку для повышения качества. Способ 200 потребовал 12 минут времени ЦП и 1,2 ГБ ОЗУ для обработки набора данных с получением 30033939 потенциальных связей между прочтениями. Затем эти связи подвергали кластеризации графа с использованием кластеризации Маркова (mcl), и полученные 6779 групп прочтений были собраны de novo собрано с использованием MEGAHIT (см. Dinghua Li, Chi-Man Liu, Ruibang Luo, Kunihiko Sadakane, and Tak-Wah Lam. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, Oxford, England, 31(10): 1674{1676, May 2015) с получением реконструкций длинных мутированных темплатов. Наконец, длинные мутированные темплаты использовали вместе с немутированными прочтениями при гибридной сборке генома, вычисленной программным обеспечением Unicycler. Полученная сборка показана на Фиг. 4В в сравнении со сборкой, полученной только по коротким прочтениям (показана на Фиг. 4А). На Фиг. 4А показана сборка по коротким прочтениям генома Arcobacter butzlerii размером 2,3 млн.п.н. с использованием сборочного конвейера Shovill перед выполнением способа 200. Это дало сборку в 78 каркасов, причем наибольший каркас покрывал 342 т.п.н., и каркас N50 в приблизительно 127 т.п.н. На Фиг. 4В показана сборка генома Arcobacter butzlerii размером 2,3 млн.п.н. с использованием способа 200. Кольцевая хромосома в значительной степени разрешена в один контиг, причем неразрешенным осталось количество копий небольшого участка 200 нт.
Масштабируемость и разрешающую мощность подхода, реализованного в способе 200, измеряли с помощью смоделированных данных. Последовательность 50 т.п.н. из гена CFTR использовали для моделирования повышающихся величин покрытия мутированными длинными темплатами и соответствующими мутированными короткими прочтениями из этих темплатов. Моделирование осуществляли с использованием только что разработанных скриптов, которые сначала генерируют длинные мутированные темплаты, затем активируют хорошо известный имитатор прочтений Illumina под названием artsim, чтобы моделировать секвенирование из мутированных темплатов по коротким прочтениям. В дополнение к мутированным данным в artsim было смоделировано 30-кратное покрытие немутированной последовательности. Мы моделировали покрытие длинными мутированными темплатами в диапазоне от 101 до 106 с инкрементом в порядок величины. Долю мутаций фиксировали на уровне 6%. Для каждого длинного темплата было смоделировано 10-кратное покрытие короткими прочтениями. Результаты для смоделированных данных оценивали путем измерения доли ложноположительных связей в способе 200.
На Фиг. 5 показано влияние глубины покрытия короткими прочтениями длинного темплата. Количество данных с короткими прочтениями на длинный темплат показано на оси х, а на оси у показаны различные показатели эффективности для результатов способа 200. Видно, что при низком покрытии темплата короткими прочтениями, например <4х, получены плохие и неполные реконструкции исходных длинных темплатов. Однако, когда покрытие мутированного темплата находится в диапазоне 5-10х, могут быть получены хорошие реконструкции.
• links num: количество связей между мутированными прочтениями по данным способа 200. links fp: количество отмеченных ложноположительных связей.
• links fp rate: доля ложноположительных связей от всех полученных связей.
• mcl num: количество кластеров, созданных кластеризацией Маркова для графа, по данным mmdreaming.
• idba scaf num: количество каркасных последовательностей, реконструированных путем сборки кластеров мутированных коротких прочтений.
• idba scaf bp: сумма длин всех собранных каркасов.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОНЕНТЫ СИСТЕМЫ CRISPR-CAS, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ | 2013 |
|
RU2701662C2 |
| СПОСОБ ДЕТЕКЦИИ ВАРИАНТА ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВОЙ КИСЛОТЫ С ПОМОЩЬЮ АНАЛИЗА ТЕРМИНАЦИИ СО СДВИГОМ | 2000 |
|
RU2200762C2 |
| ОБНАРУЖЕНИЕ МУТАЦИЙ И ПЛОИДНОСТИ В ХРОМОСОМНЫХ СЕГМЕНТАХ | 2015 |
|
RU2717641C2 |
| ГЕНОМНЫЙ ОТБОР И СЕКВЕНИРОВАНИЕ С ПОМОЩЬЮ КОДИРОВАННЫХ МИКРОНОСИТЕЛЕЙ | 2010 |
|
RU2609630C2 |
| БИБЛИОТЕКИ ДЛЯ СЕКВЕНИРОВАНИЯ НОВОГО ПОКОЛЕНИЯ | 2014 |
|
RU2698125C2 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В СЛОЖНЫХ СМЕСЯХ ДНК | 2014 |
|
RU2613489C2 |
| НУКЛЕОТИДНЫЕ ПОСЛЕДОВАТЕЛЬНОСТИ, УЧАСТВУЮЩИЕ В УВЕЛИЧЕНИИ ИЛИ УМЕНЬШЕНИИ СКОРОСТИ ОВУЛЯЦИИ У МЛЕКОПИТАЮЩИХ | 2001 |
|
RU2283866C2 |
| СПОСОБЫ СЕКВЕНИРОВАНИЯ ТРЕХМЕРНОЙ СТРУКТУРЫ ИССЛЕДУЕМОЙ ОБЛАСТИ ГЕНОМА | 2011 |
|
RU2603082C2 |
| ФРЕЙМВОРК НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ ДЛЯ ИДЕНТИФИКАЦИИ ПАТТЕРНОВ ПОСЛЕДОВАТЕЛЬНОСТИ, КОТОРЫЕ ВЫЗЫВАЮТ ПОСЛЕДОВАТЕЛЬНОСТЬ-СПЕЦИФИЧНЫЕ ОШИБКИ (SSE) | 2019 |
|
RU2745733C1 |
| ИНСТРУМЕНТ НА ОСНОВЕ ГРАФОВ ПОСЛЕДОВАТЕЛЬНОСТЕЙ ДЛЯ ОПРЕДЕЛЕНИЯ ВАРИАЦИЙ В ОБЛАСТЯХ КОРОТКИХ ТАНДЕМНЫХ ПОВТОРОВ | 2020 |
|
RU2825664C2 |
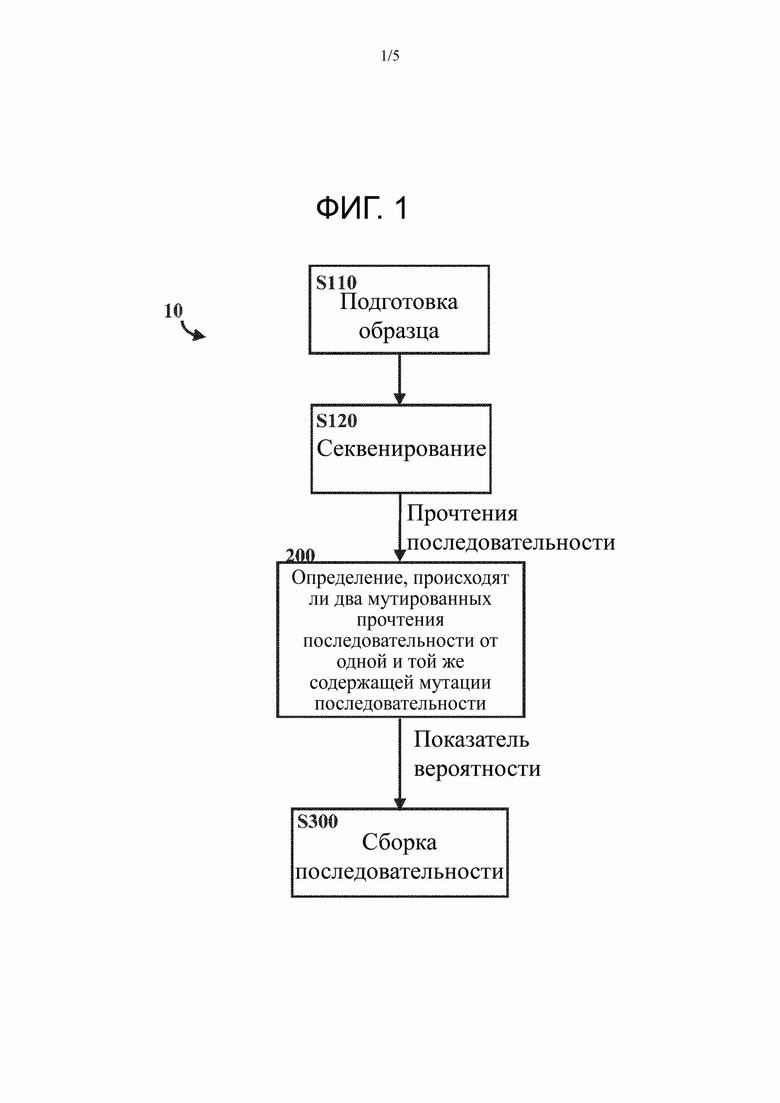
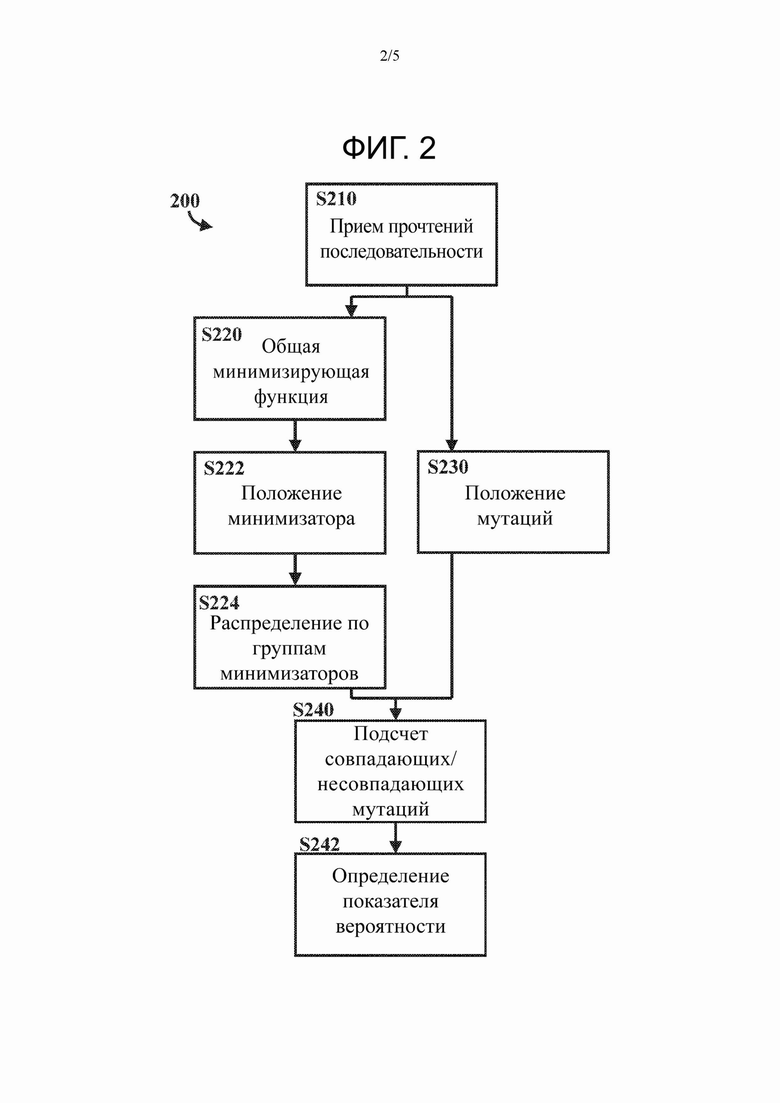
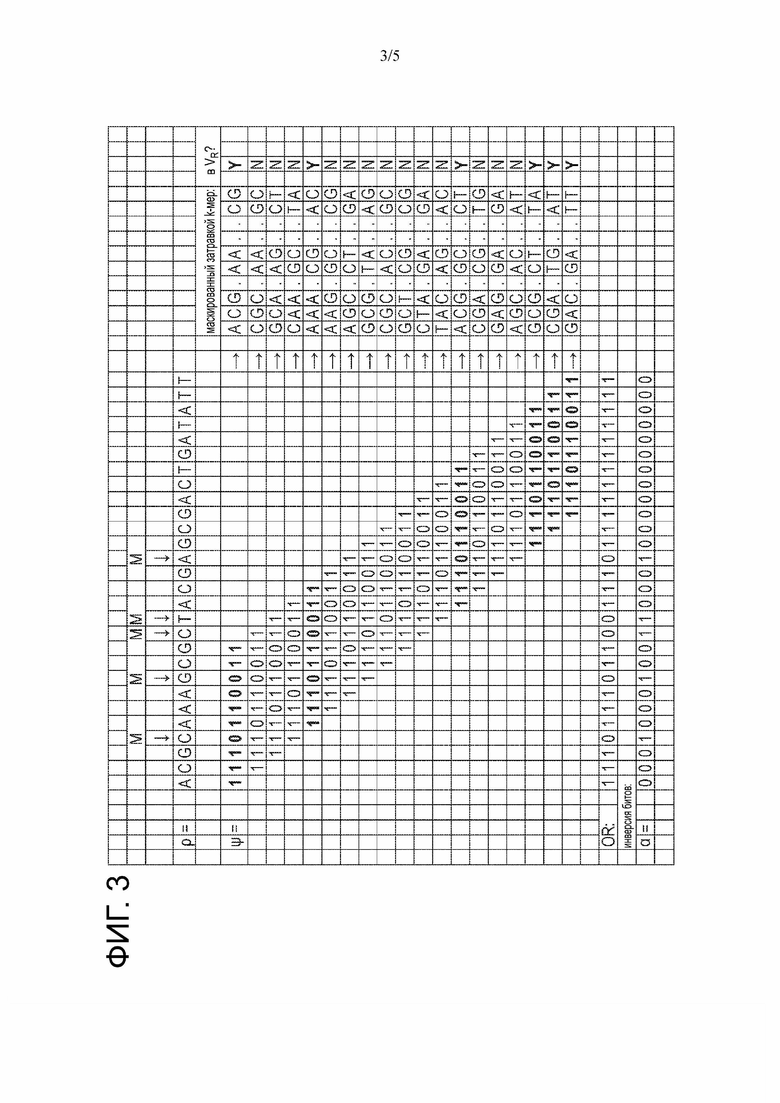
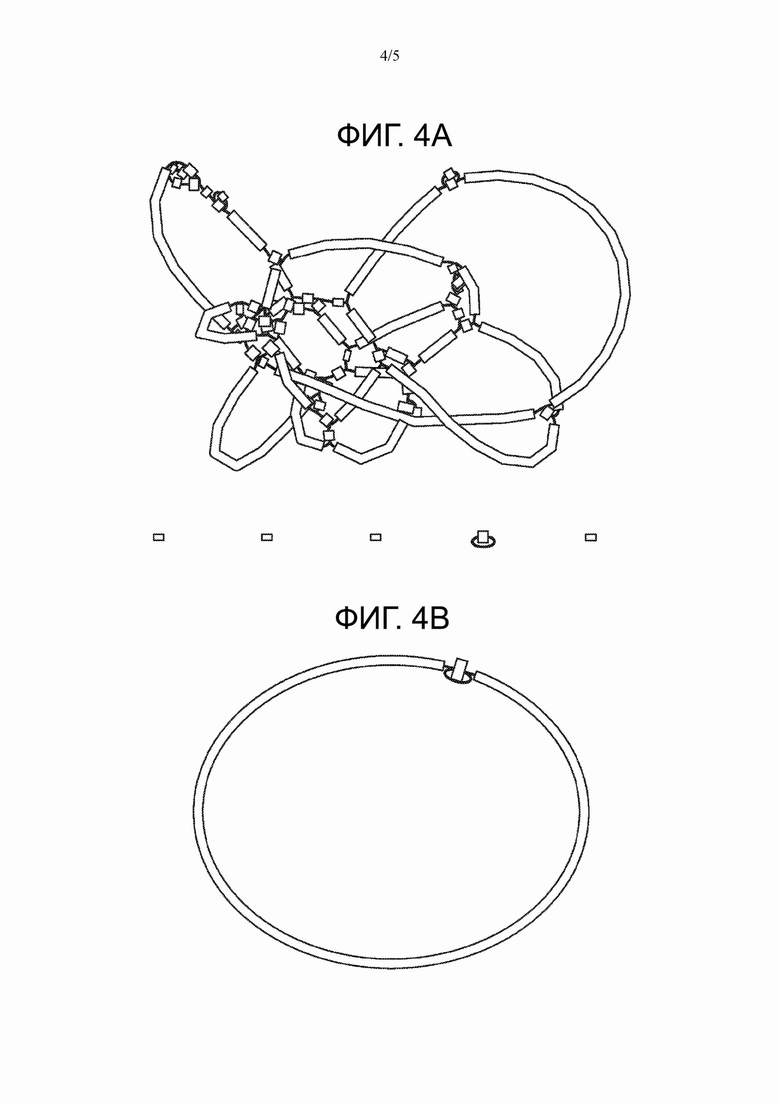
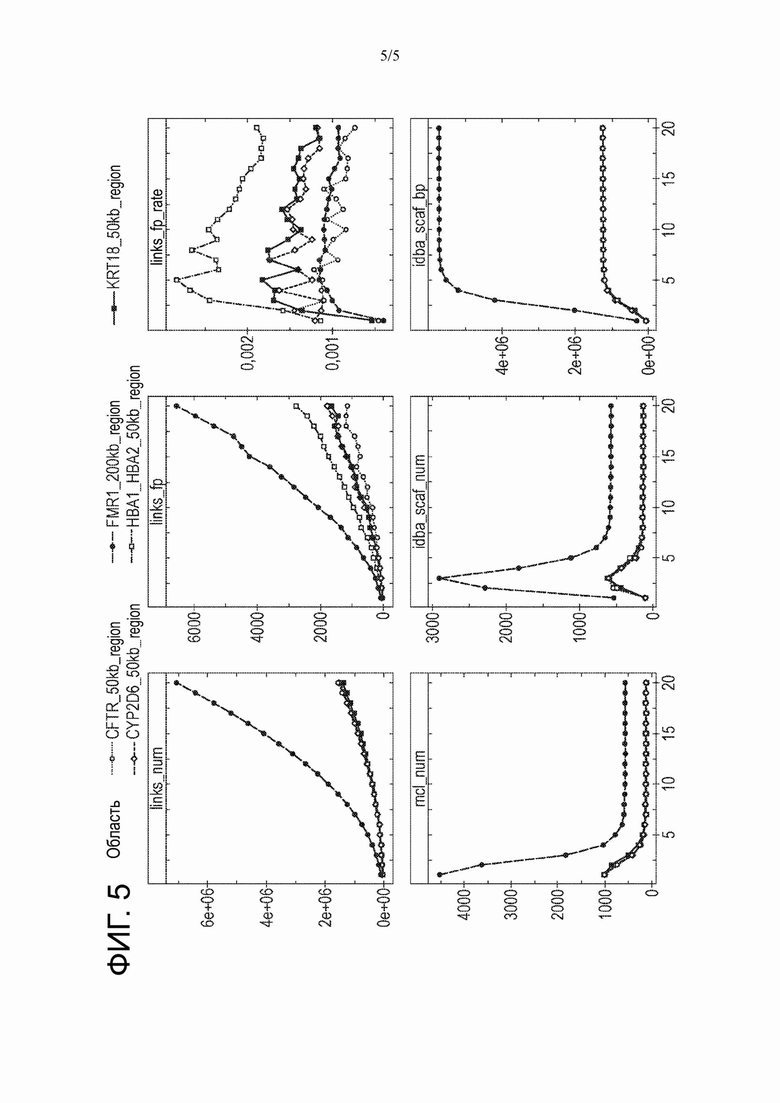
Изобретение относится к биоинформатике и биотехнологии. Описан реализованный на компьютере способ определения последовательности по меньшей мере части по меньшей мере одной темплатной нуклеиновой кислоты-мишени посредством определения, происходят ли два мутированных прочтения последовательности от одной и той же содержащей мутации последовательности. Осуществляют множество мутированных прочтений последовательности. Причем каждое мутированное прочтение последовательности соответствует подпоследовательности содержащей мутации последовательности. Применяют общую минимизирующую функцию для каждого мутированного прочтения последовательности с определением таким образом одного или более соответствующих минимизаторов для каждого мутированного прочтения последовательности. Определяют положения одного или более соответствующих минимизаторов в каждом мутированном прочтении последовательности. Определяют положения одной или более мутаций в каждом мутированном прочтении последовательности. При этом осуществляют для по меньшей мере двух мутированных прочтений последовательности с общим минимизатором подсчет количества мутаций с совпадающим положением и/или с несовпадающим положением, когда соответствующие минимизаторы выравнены для того, чтобы определить показатель, коррелированный с вероятностью того, что указанные по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности. Осуществляют сборку указанных по меньшей мере двух мутированных прочтений последовательности на основе указанного показателя. Определяют последовательности по меньшей мере части по меньшей мере одной темплатной нуклеиновой кислоты-мишени на основании указанной сборки. Также описан способ определения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени, включающий использование указанного выше способа после стадии секвенирования областей по меньшей мере одной содержащей мутации молекулы темплатной нуклеиновой кислоты-мишени с получением множества мутированных прочтений последовательности. Изобретение позволяет точно секвенировать нуклеиновые кислоты, а также получать быструю и точную сборку последовательности из коротких прочтений последовательности. 2 н. и 27 з.п. ф-лы, 5 ил.
1. Реализованный на компьютере способ определения последовательности по меньшей мере части по меньшей мере одной темплатной нуклеиновой кислоты-мишени посредством определения, происходят ли два мутированных прочтения последовательности от одной и той же содержащей мутации последовательности, включающий:
прием множества мутированных прочтений последовательности, причем каждое мутированное прочтение последовательности соответствует подпоследовательности содержащей мутации последовательности, при этом содержащая мутации последовательность содержит мутации по сравнению с не содержащей мутаций последовательностью;
применение общей минимизирующей функции для каждого мутированного прочтения последовательности с определением таким образом одного или более соответствующих минимизаторов для каждого мутированного прочтения последовательности;
определение положений одного или более соответствующих минимизаторов в каждом мутированном прочтении последовательности;
определение положений одной или более мутаций в каждом мутированном прочтении последовательности; и
для по меньшей мере двух мутированных прочтений последовательности с общим минимизатором подсчет количества мутаций с совпадающим положением и/или с несовпадающим положением, когда соответствующие минимизаторы выравнены для того, чтобы определить показатель, коррелированный с вероятностью того, что указанные по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности,
осуществление сборки указанных по меньшей мере двух мутированных прочтений последовательности на основе указанного показателя; и
определение последовательности по меньшей мере части по меньшей мере одной темплатной нуклеиновой кислоты-мишени на основании указанной сборки.
2. Способ по п. 1, дополнительно включающий прием множества немутированных прочтений последовательности, причем каждое немутированное прочтение последовательности соответствует подпоследовательности не содержащей мутаций последовательности.
3. Способ по п. 1 или 2, в котором стадия применения общей минимизирующей функции к каждому мутированному прочтению последовательности включает идентификацию i) одного или более k-меров в соответствующем мутированном прочтении последовательности, который(-ые) указан(-ы) первым(-и) в упорядоченном списке возможных k-меров, или ii) одного или более k-меров, которые встречаются в предварительно заданном наборе возможных k-меров, причем один или более минимизаторов, определенных для соответствующего мутированного прочтения последовательности, представляют собой идентифицированные один или более k-меров.
4. Способ по п. 3, в котором i) в упорядоченном списке возможных k-меров, k-меры упорядочены на основании вероятности того, что k-меры встречаются в содержащей мутации последовательности и не встречаются в не содержащей мутации последовательности, или ii) предварительно заданный набор возможных k-меров содержит k-меры, которые относительно вероятно встречаются в содержащей мутации последовательности, но не в не содержащей мутаций последовательности, причем необязательно предварительно заданный набор возможных k-меров не содержит k-меров, которые относительно маловероятно встречаются в содержащей мутации последовательности.
5. Способ по пп. 2 и 3 или 4, в котором упорядоченный список возможных k-меров или предварительно заданный набор возможных k-меров состоит из k-меров, которые чаще встречаются во множестве мутированных прочтений последовательности, чем во множестве немутированных прочтений последовательности, причем необязательно k-меры, которые чаще встречаются во множестве мутированных прочтений последовательности, чем во множестве немутированных прочтений последовательности, относительно вероятно встречаются в содержащей мутации последовательности.
6. Способ по п. 2 и любому из пп. 3–5, в котором предварительно заданный набор возможных k-меров состоит из k-меров, которые встречаются n или более раз во множестве мутированных прочтений последовательности, и встречаются менее чем n раз во множестве немутированных прочтений последовательности, где n представляет собой целое число, которое больше или равно 1, причем необязательно k-меры, которые встречаются n или более раз во множестве мутированных прочтений последовательности и встречаются менее чем n раз во множестве немутированных прочтений последовательности, относительно вероятно встречаются в содержащей мутации последовательности.
7. Способ по п. 6, в котором предварительно заданный набор возможных k-меров состоит из k-меров, которые не встречаются во множестве немутированных прочтений последовательности.
8. Способ по п. 6 или 7, в котором n равно 2.
9. Способ по п. 2 и любому из пп. 3–8, дополнительно включающий генерацию упорядоченного списка возможных k-меров или предварительно заданного набора возможных k-меров на основе сравнения k-меров во множестве мутированных прочтений последовательности и k-меров во множестве немутированных прочтений последовательности.
10. Способ по любому из предшествующих пунктов, в котором каждый минимизатор представляет собой k-мер длиной более 5, предпочтительно более 10.
11. Способ по любому из предшествующих пунктов, дополнительно включающий распределение мутированных прочтений последовательности в одну или более групп минимизаторов, так что каждая группа минимизатора содержит мутированные прочтения последовательности, имеющие общий минимизатор, и не содержит мутированные прочтения последовательности, не имеющие общего минимизатора, и
при этом стадия подсчета количества мутаций с совпадающим положением и/или с несовпадающим положением может быть выполнена только на мутированных прочтениях последовательности, находящихся в одной и той же группе минимизатора.
12. Способ по любому из пп. 1–11, в котором стадия определения положений одной или более мутаций в каждом мутированном прочтении последовательности включает:
получение набора немутированных маскированных затравкой k-меров путем применения каждого из одного или более затравочных паттернов к k-мерам во множестве немутированных прочтений последовательности;
для каждого мутированного прочтения последовательности применение одного или более затравочных паттернов к k-мерам в соответствующем мутированном прочтении последовательности с получением множества мутированных маскированных затравкой k-меров и определение положений одной или более мутаций путем идентификации одного или более положений в мутированном прочтении последовательности, которые маскированы всеми затравочными паттернами, соответствующими мутированным маскированным затравкой k-мерам из множества мутированных маскированных затравкой k-меров, которые встречаются в наборе немутированных маскированных затравкой k-меров.
13. Способ по п. 12, в котором один или более затравочных паттернов выбраны таким образом, что вероятность получения идентичных маскированных затравкой k-меров при применении по меньшей мере одного из одного или более затравочных паттернов к любому k-меру из множества мутированных прочтений последовательности и соответствующему k-меру из множества немутированных прочтений последовательности составляет более 90%, предпочтительно более 99%.
14. Способ по п. 12 или 13, в котором содержащая мутации последовательность содержит мутации типа транзиции по сравнению с не содержащей мутаций последовательностью; и
при этом один или более затравочных паттернов могут представлять собой один или более затравочных паттернов транзиции.
15. Способ по любому из пп. 12–14, в котором каждое из множества мутированных прочтений последовательности соответствует подпоследовательности содержащей мутации последовательности, ассоциированной с одним из множества образцов, и каждое из множества немутированных прочтений последовательности соответствует подпоследовательности не содержащей мутаций последовательности, ассоциированной с одним из множества образцов, причем каждая содержащая мутации последовательность содержит мутации по сравнению с соответствующей не содержащей мутаций последовательностью;
при этом получение набора немутированных маскированных затравкой k-меров включает получение соответствующего набора немутированных маскированных затравкой k-меров для каждого образца;
причем способ дополнительно включает создание набора немутированных битовых векторов образцов, при этом каждый немутированный битовый вектор определяет для соответствующего k-мера в наборе немутированных маскированных затравкой k-меров в каком из множества образцов соответствующий k-мер встречается; и
при этом для каждого мутированного прочтения последовательности и для каждого набора и/или каждой комбинации наборов немутированных маскированных затравкой k-меров определение положений одной или более мутаций включает идентификацию одного или более положений в мутированном прочтении последовательности, которые маскированы всеми затравочными паттернами, соответствующими мутированным маскированным затравкой k-мерам из множества мутированных маскированных затравкой k-меров, которые встречаются в соответствующем наборе или комбинации наборов немутированных маскированных затравкой k-меров, и связывание идентифицированного одного или более положений с одним или более образцами, ассоциированными с соответствующим набором или комбинацией наборов немутированных маскированных затравкой k-меров.
16. Способ по любому из пп. 2–15, в котором стадия определения положений одной или более мутаций в каждом мутированном прочтении последовательности включает:
для одного или более из мутированных прочтений последовательности выравнивание соответствующего мутированного прочтения последовательности с эталонной сборкой; и
определение положений одной или более мутаций в соответствующем мутированном прочтении последовательности путем идентификации в соответствующем мутированном прочтении последовательности положений различий между соответствующим мутированным прочтением последовательности и эталонной сборкой.
17. Способ по п. 16 и в зависимости от любого из пп. 12–15, в котором для каждого мутированного прочтения последовательности стадия определения положений одной или более мутаций в каждом мутированном прочтении последовательности включает:
если положение в соответствующем мутированном прочтении последовательности выравнено с эталонной сборкой, определение положения в соответствующем мутированном прочтении последовательности как положения мутации в соответствующем мутированном прочтении последовательности, если положение в соответствующем мутированном прочтении последовательности представляет собой положение, в котором соответствующее мутированное прочтение последовательности отличается от эталонной сборки; и
если положение в соответствующем мутированном прочтении последовательности не выравнено с эталонной сборкой, определение положения в соответствующем мутированном прочтении последовательности как положения мутации в соответствующем мутированном прочтении последовательности, если положение в соответствующем мутированном прочтении последовательности представляет собой положение, которое маскируется всеми затравочными паттернами, которые соответствуют маскированным затравками мутированным k-мерам из множества маскированных затравками мутированных k-меров, которые встречаются в наборе маскированных затравками немутированных k-меров.
18. Способ по любому из предшествующих пунктов, включающий определение показателя, коррелированного с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, на основании количества мутаций с совпадающим положением и/или с несовпадающим положением.
19. Способ по п. 18, в котором показатель, коррелированный с вероятностью того, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, представляет собой один из: i) плотности вероятности, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности, и ii) оценочной функции, которая коррелирована с плотностью вероятности, что по меньшей мере два мутированных прочтения последовательности происходят от одной и той же содержащей мутации последовательности.
20. Способ по любому из предшествующих пунктов, дополнительно включающий создание ненаправленного взвешенного графа из множества мутированных прочтений последовательности,
причем ненаправленный взвешенный граф содержит узлы, соответствующие множеству мутированных прочтений последовательности, и при этом ребра между узлами ассоциированы с соответствующими весовыми значениями ребер, причем вес каждого ребра определен на основании количества мутаций с совпадающим положением и/или с несовпадающим положением, определенным для двух мутированных прочтений последовательности, соответствующих двум узлам, связанным с соответствующим ребром.
21. Способ по п. 20, в котором весовые коэффициенты ребер соответствуют показателю, коррелированному с вероятностью того, что по меньшей мере два мутированных прочтения последовательности, соответствующие двум узлам, связанным с соответствующим ребром, происходят от одной и той же содержащей мутации последовательности.
22. Способ по п. 20 или 21, дополнительно включающий выполнение операции кластеризации графа на ненаправленном взвешенном графе, с получением таким образом кластеров мутированных прочтений последовательности, которые, как ожидается, происходят от одной и той же содержащей мутации последовательности.
23. Способ по п. 22, в котором кластеризация графа включает кластеризацию Маркова или Infomap.
24. Способ по п. 22 или 23, дополнительно включающий реконструкцию по меньшей мере части содержащей мутации последовательности путем сборки мутированных прочтений последовательности в кластеры.
25. Способ по пп. 2 и 24, дополнительно включающий реконструкцию по меньшей мере части не содержащей мутаций последовательности путем выведения по меньшей мере части вероятной не содержащей мутации последовательности из реконструированной части содержащей мутации последовательности, необязательно с использованием множества немутированных прочтений последовательности.
26. Способ получения по меньшей мере части последовательности молекулы темплатной нуклеиновой кислоты-мишени, включающий способ по любому из пп. 20–25.
27. Способ определения по меньшей мере части последовательности по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени, включающий
секвенирование областей по меньшей мере одной содержащей мутации молекулы темплатной нуклеиновой кислоты-мишени с получением множества мутированных прочтений последовательности,
выполнение способа по любому предшествующему пункту на полученном множестве мутированных прочтений последовательности.
28. Способ по п. 27, в котором стадия секвенирования включает
(a) обеспечение пары образцов, причем каждый образец содержит по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени;
(b) секвенирование областей по меньшей мере одной молекулы темплатной нуклеиновой кислоты-мишени в первом образце из пары образцов с получением множества немутированных прочтений последовательности;
(c) введение мутаций в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором образце из пары образцов с получением по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени;
(d) секвенирование областей по меньшей мере одной мутированной молекулы темплатной нуклеиновой кислоты-мишени с получением множества мутированных прочтений последовательности.
29. Способ по п. 28, в котором стадия введения мутаций включает введение мутаций типа транзиции в по меньшей мере одну молекулу темплатной нуклеиновой кислоты-мишени во втором из пары образцов.
| US 20180365375 A1, 20.12.2018 | |||
| US 20190164627 A1, 30.05.2019 | |||
| RU 2015153453 A, 20.06.2017. |

