ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает преимущества и приоритет китайской заявки на патент №201810662580.9, поданной 25 июня, 2018 г., и китайской заявки на патент №201910463156.6, поданной 30 мая, 2019 г. в Национальное управление интеллектуальной собственности, PRc, которые включены в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к ингибитору IRAK4 и его применению для получения лекарственного препарата для лечения связанных с IRAK4 заболеваний, в частности к соединению формулы (I) и его фармацевтически приемлемой соли.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Киназа 4, ассоциированная с рецептором интерлейкина-1 (IRAK4), является серин/треонин-специфической протеинкиназой, представителем семейства тирозин-подобных киназ (TLK) и ключевым элементом врожденного иммунного ответа, в котором участвуют интерлейкин-1, 18 и 33, а также toll-подобные рецепторы. После связывания внеклеточных сигнальных молекул с рецепторами интерлейкина или toll-подобными рецепторами рекрутируются белки с образованием комплекса MyD88:IRAK4:IRAK1/2, что приводит к фосфорилированию IRAK1/2, что опосредует передачу сигнала нижележащим компонентам сигнального пути. Таким образом активируются сигнальные пути р38, JNK и NF-кВ, в итоге приводя к усилению экспрессии провоспалительных цитокинов. Исследования по клинической патологии показали, что индивидуумы с мутациями IRAK4 обладают устойчивостью к хроническому заболеванию легкого и воспалительному заболеванию кишечника. Дефицит IRAK4 как таковой не является летальным, и индивидуумы могут доживать до зрелого возраста с пониженным риском инфекции в течение жизни. Следовательно, IRAK4 становится важной терапевтической мишенью, привлекающей обширный интерес к исследованиям и разработкам.
КРАТКОЕ ОПИСАНИЕ
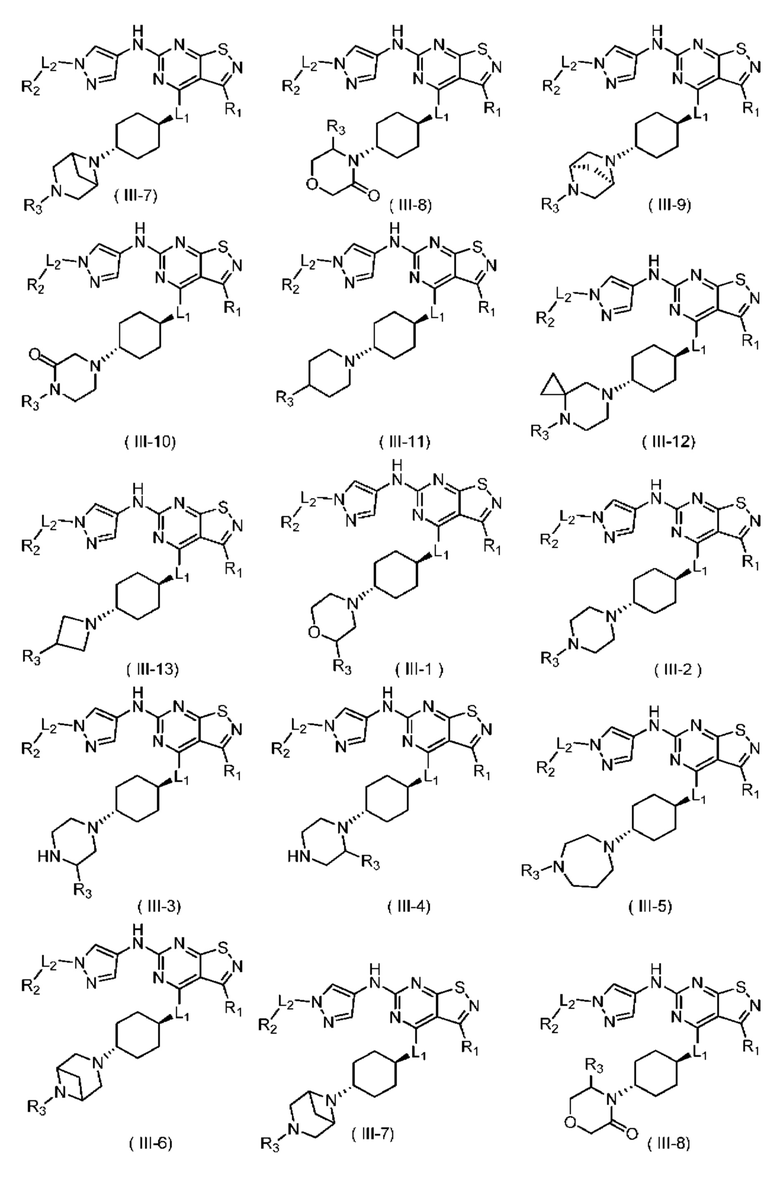
В настоящем изобретении предусмотрено соединение формулы (III), его оптический изомер или фармацевтически приемлемая соль,
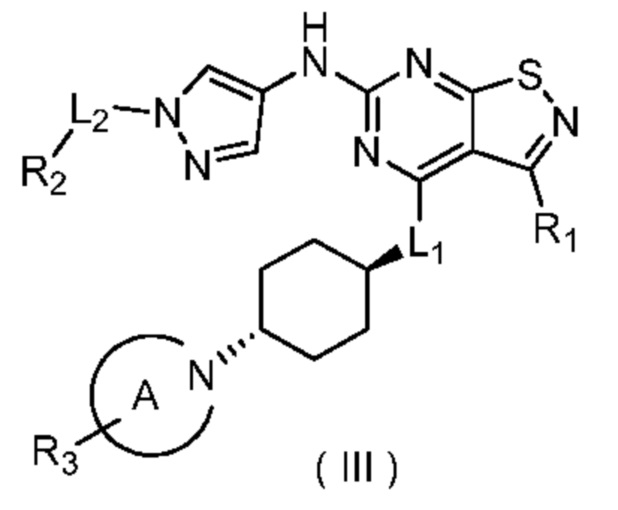
где
R1 выбран из группы, состоящей из CN, C1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra;
R2 выбран из группы, состоящей из С1-6алкила, С3-8циклоалкила и 3-6-членного гетероциклоалкила, где С1-6алкил, С3-8циклоалкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb;
R3 выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, СООН, C1-6алкила, С1-6алкиламино, -С(=O)-O-С1-6алкила, -С(=O)-С1-6алкила и С3-6циклоалкила, где C1-6алкил, C1-6алкиламино, -С(=O)-O-С1-6алкил, -С(=O)-С1-6алкил и С3-6циклоалкил необязательно замещены 1, 2 или 3 Rc;
кольцо А выбрано из группы, состоящей из 3-10-членного гетероциклоалкила, и кольцо А содержит по меньшей мере один атом азота, где 3-10-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd;
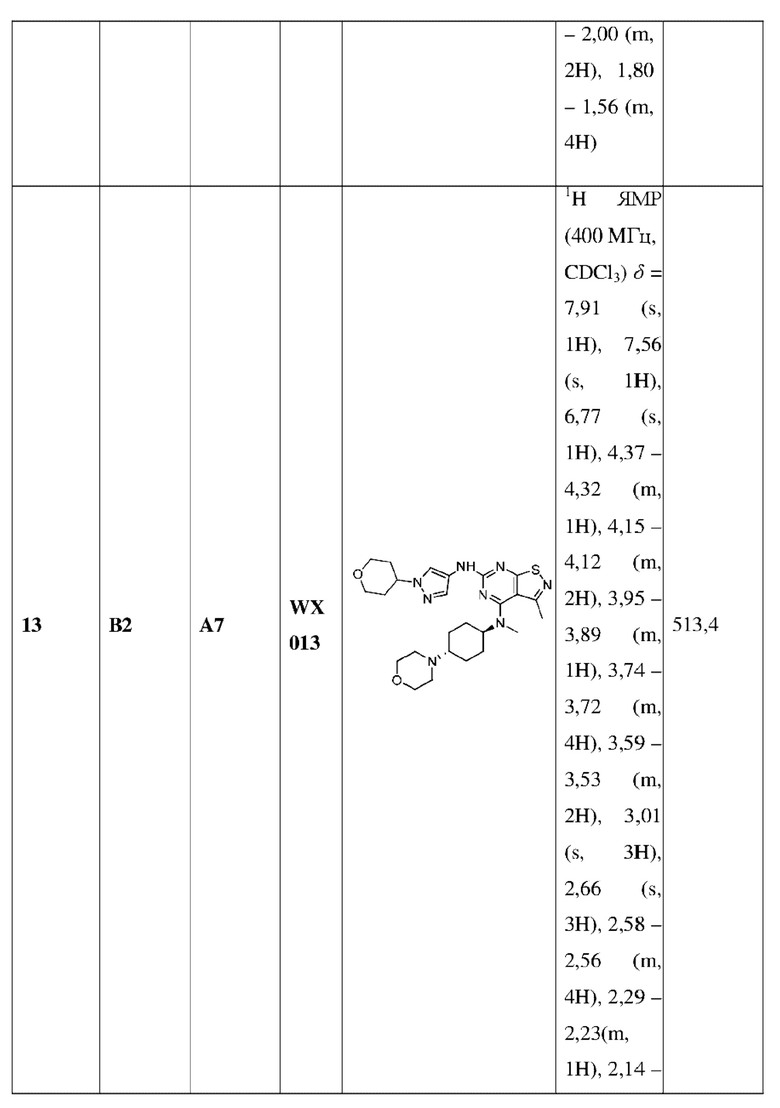
L1 выбран из группы, состоящей из О и N (R4);
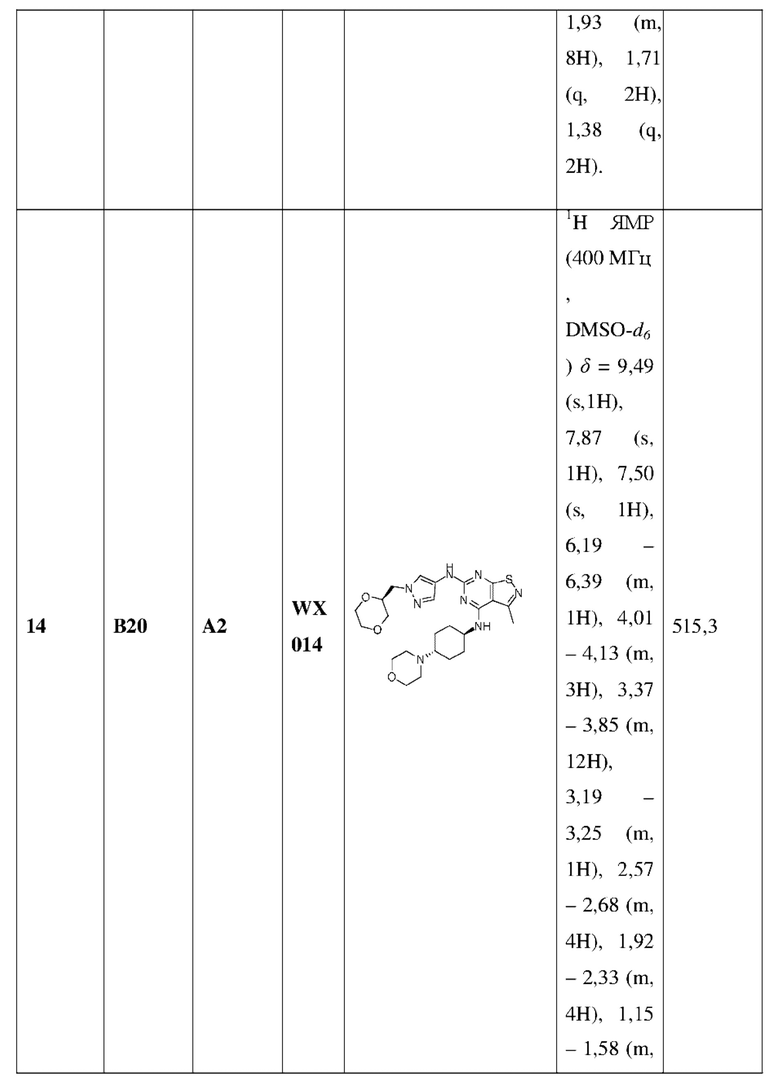
L2 выбран из группы, состоящей из одинарной связи, СН2 и СН2СН2;
R4 выбран из группы, состоящей из Н и Me;
каждый Ra независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN и СООН;
каждый Rb независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СООН и Me;
каждый Rc независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN;
каждый Rd независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN;
3-6-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -О-, -S-, -NH- и N; и
3-10-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -О-, -S-, -NH-, N и -C(=O)NH-.
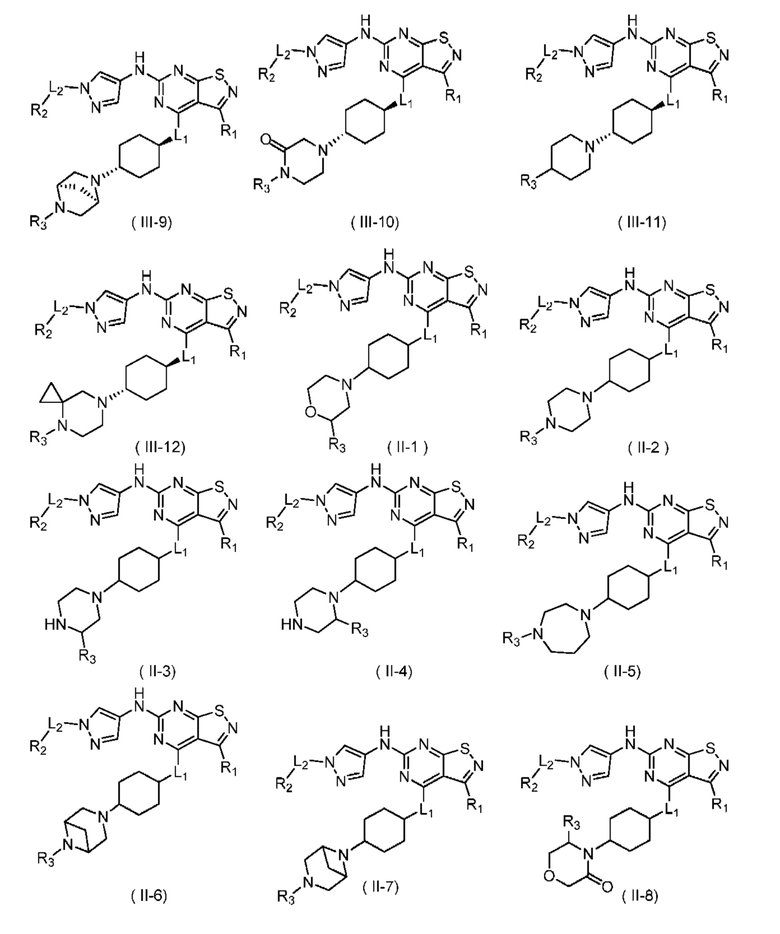
В настоящем изобретении дополнительно предусмотрено соединение формулы (II), его оптический изомер или фармацевтически приемлемая соль,
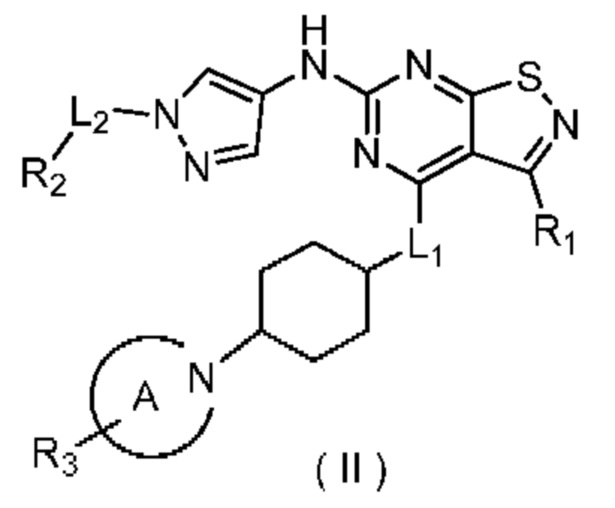
где
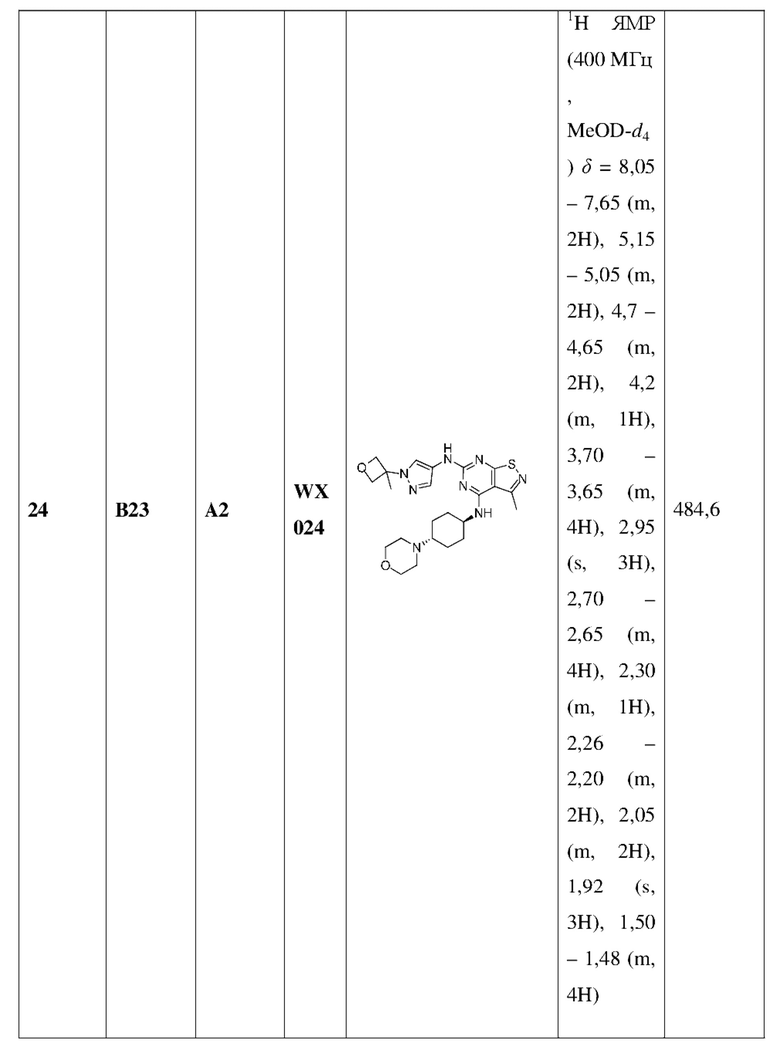
R1 выбран из группы, состоящей из C1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra;
R2 выбран из группы, состоящей из C1-6алкила и 3-6-членного гетероциклоалкила, где С1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb;
R3 выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, С1-6алкила, -С(=O)-O-С1-6алкила, -С(=O)-С1-6алкила и С3-6циклоалкила, где С1-6алкил, -С(=O)-O-С1-6алкил, -С(=O)-С1-6алкил и С3-6циклоалкил необязательно замещены 1, 2 или 3 Rc;
кольцо А выбрано из группы, состоящей из 3-10-членного гетероциклоалкила, и кольцо А содержит по меньшей мере один атом азота, где 3-10-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd;
L1 выбран из группы, состоящей из О и N (R4);
L2 выбран из группы, состоящей из одинарной связи, СН2 и СН2СН2;
R4 выбран из группы, состоящей из Н и Me;
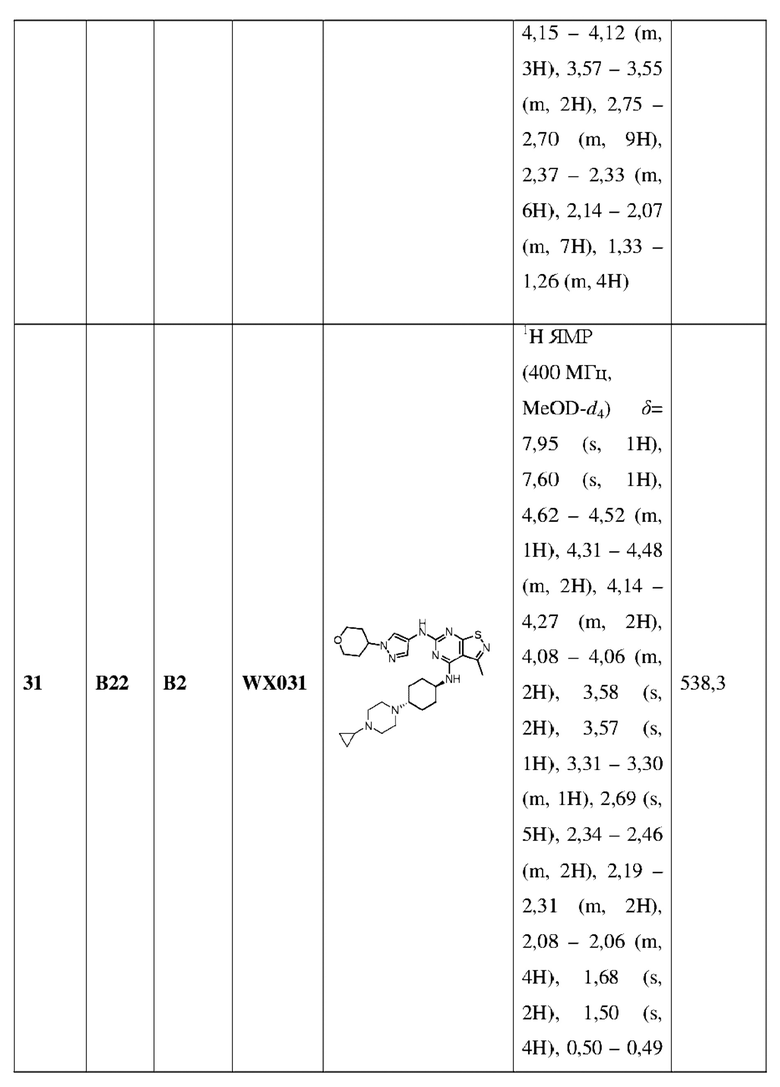
каждый Ra независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN и СООН;
каждый Rb независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СООН и Me;
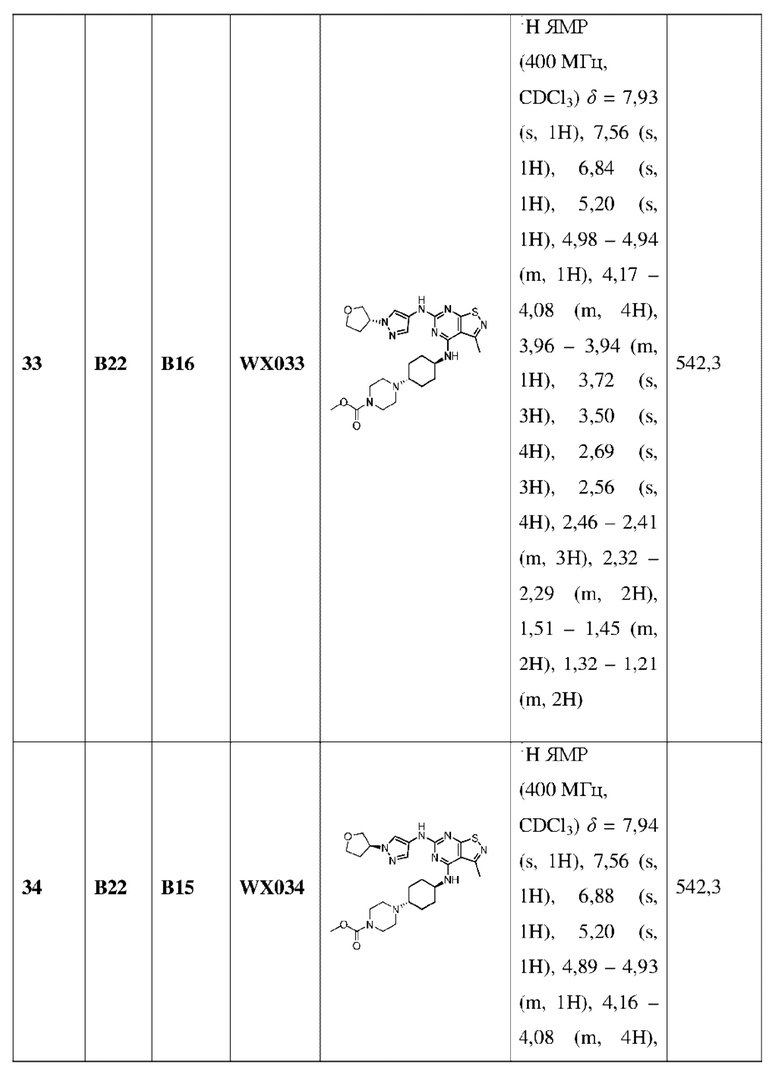
каждый Rc независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN;
каждый Rd независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и CN;
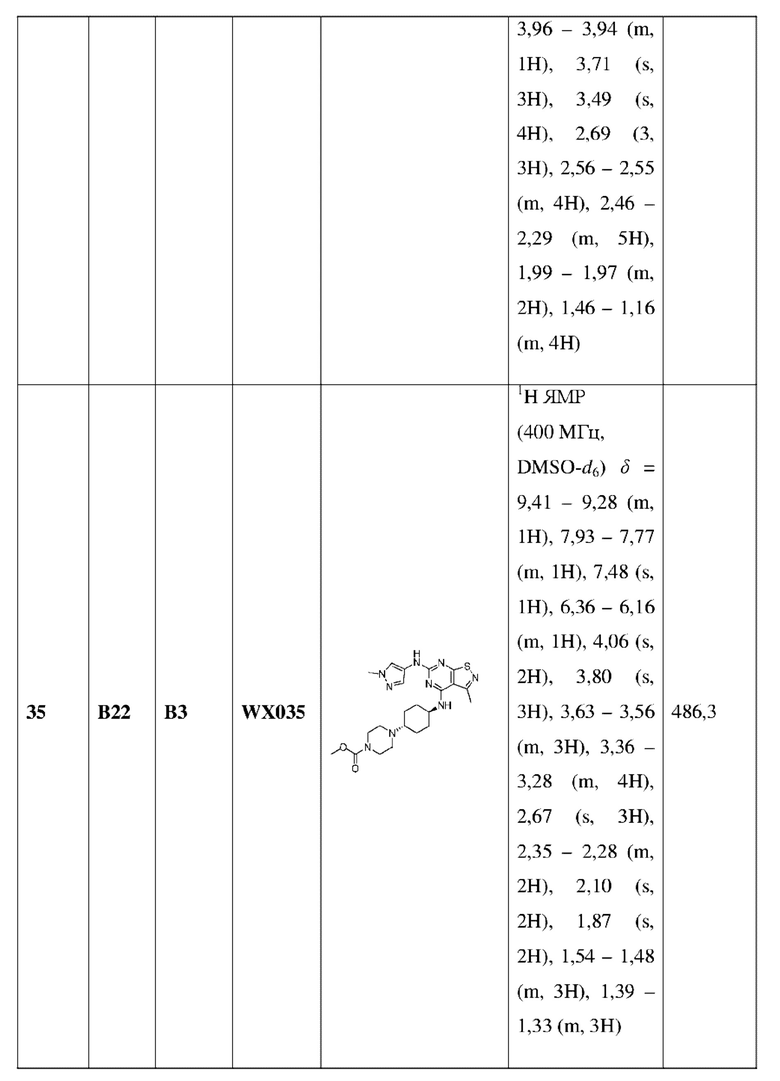
3-6-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -О-, -S-, -NH- и N; и
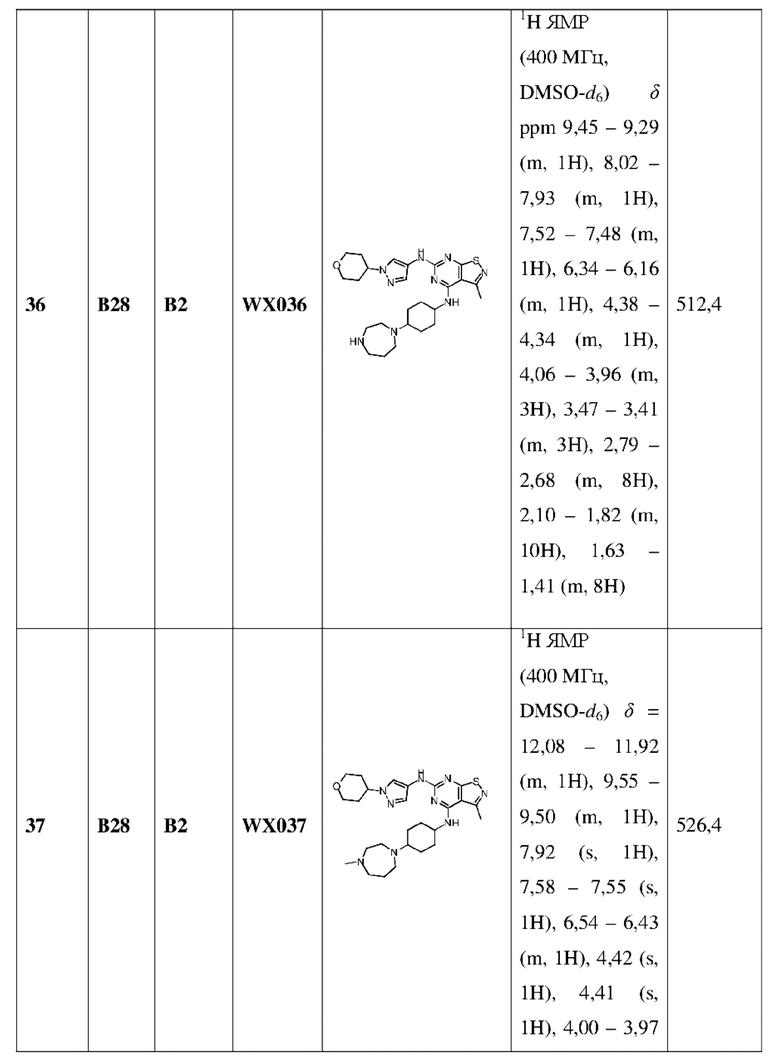
3-10-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -О-, -S-, -NH-, N и -C(=O)NH-.
В настоящем изобретении дополнительно предусмотрено соединение формулы (I), его оптический изомер или фармацевтически приемлемая соль,
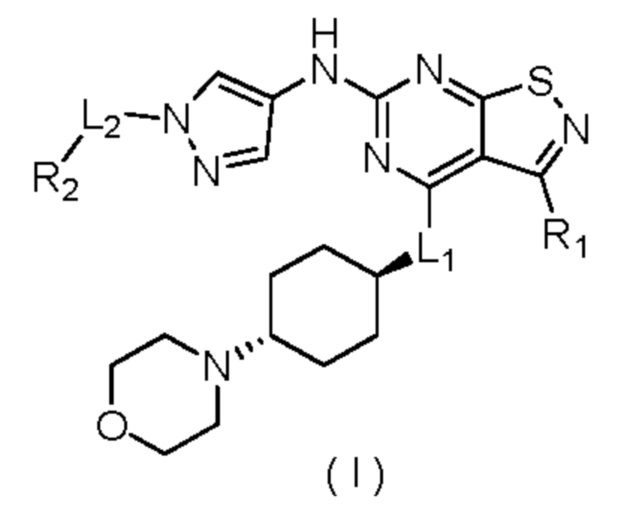
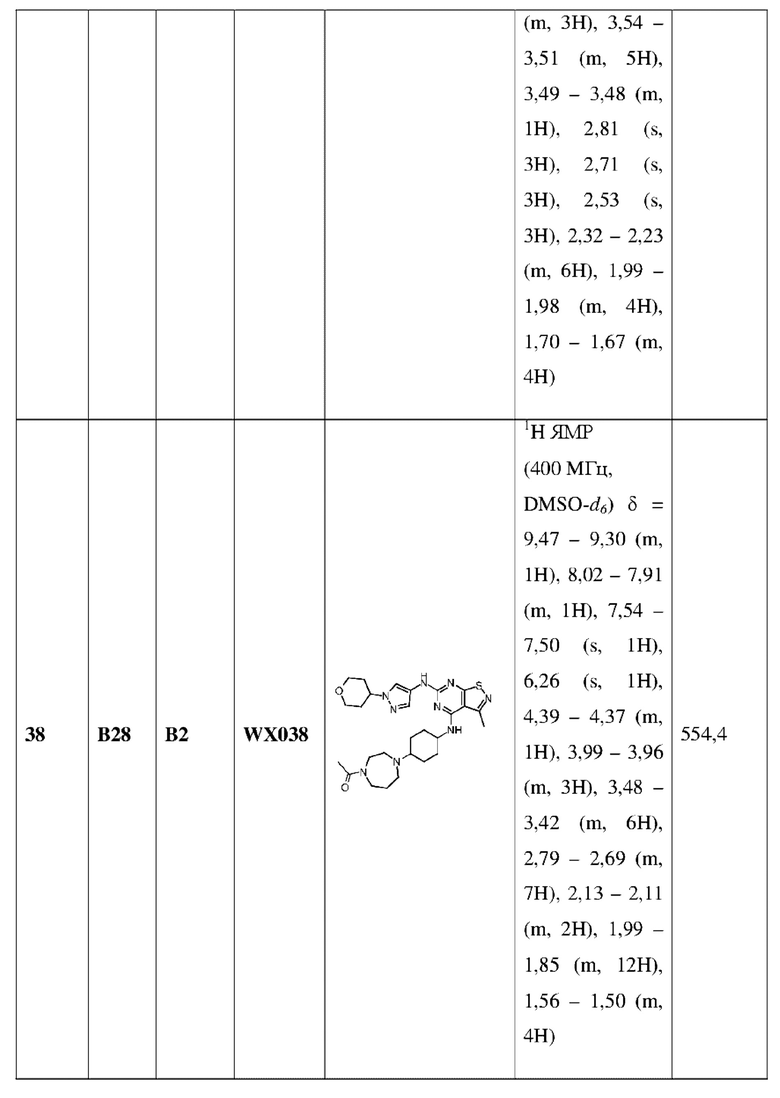
где
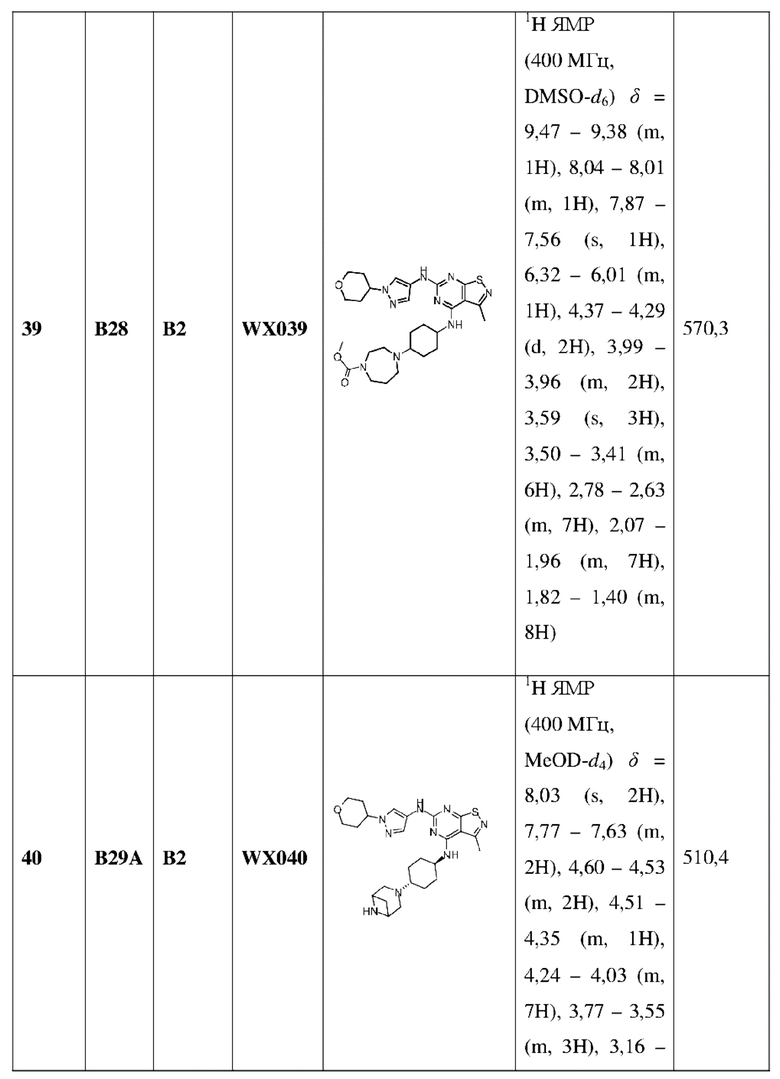
R1 выбран из группы, состоящей из C1-6алкила и 3-6-членного гетероциклоалкила, где С1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra;
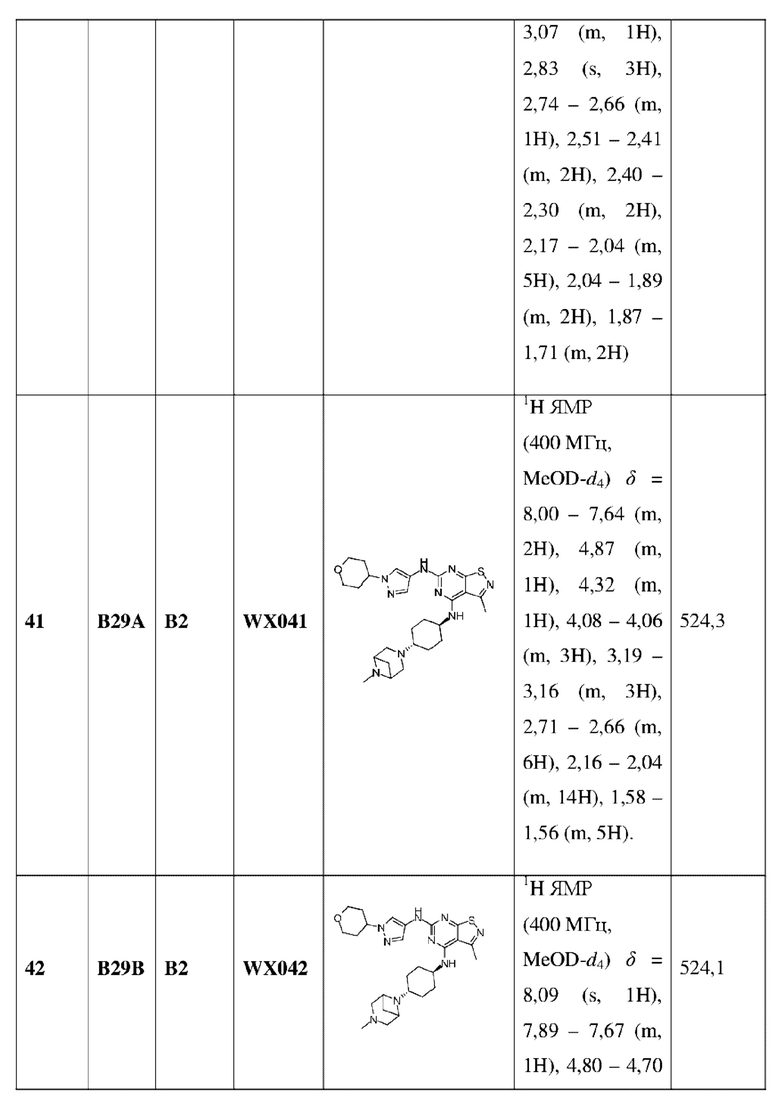
R2 выбран из группы, состоящей из С1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb;
каждый Ra независимо выбран из группы, состоящей из F, Cl, Br, I, ОН и NH2;
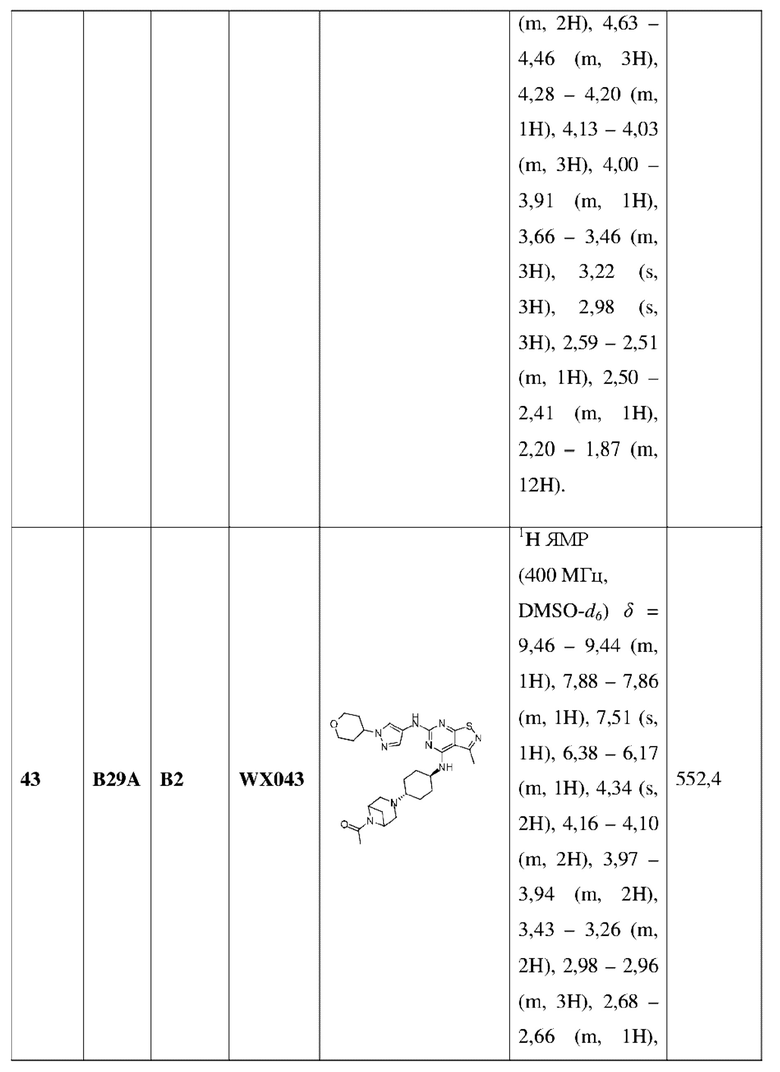
каждый Rb независимо выбран из группы, состоящей из F, Cl, Br, I, ОН и NH2;
L1 выбран из группы, состоящей из О, S и NH;
L2 выбран из группы, состоящей из одинарной связи, СН2 и СН2СН2; и
3-6-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -О-, -S-, -NH- и N.
В некоторых вариантах осуществления настоящего изобретения описанный выше L1 выбран из группы, состоящей из О и N(R4), при этом другие переменные являются такими, как определено в данном документе.
R1 выбран из группы, состоящей из CN, C1-6алкила, 3-членного гетероциклоалкила, 4-членного гетероциклоалкила, 5-членного гетероциклоалкила и 6-членного гетероциклоалкила, где C1-6алкил, 3-членный гетероциклоалкил, 4-членный гетероциклоалкил, 5-членный гетероциклоалкил и 6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из C1-3алкила и 6-членного гетероциклоалкила, где C1-3алкил и 6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из C1-3алкила и тетрагидропиранила, где C1-3алкил и тетрагидропиранил необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из метила, этила, изопропила и тетрагидропиранила, где метил, этил, изопропил и тетрагидропиранил необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из Me, Et, 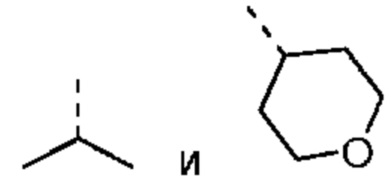 , где Me, Et,
, где Me, Et,  необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
необязательно замещены 1, 2 или 3 Ra, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения каждый Ra независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2, CN и СООН, при этом другие переменные являются такими, как определено в данном документе.
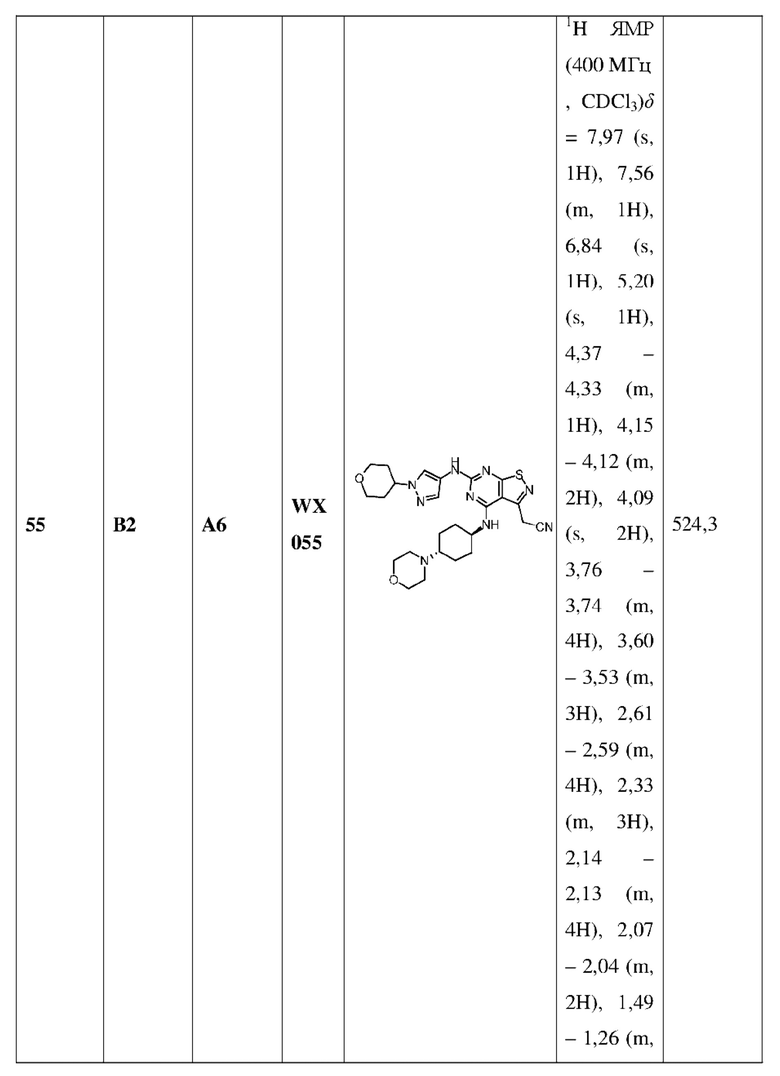
В некоторых вариантах осуществления настоящего изобретения каждый Ra независимо выбран из группы, состоящей из ОН, CN и СООН, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из Me, Et,  при этом другие переменные являются такими, как определено в данном документе. В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из Me, Et,
при этом другие переменные являются такими, как определено в данном документе. В некоторых вариантах осуществления настоящего изобретения R1 выше выбран из группы, состоящей из Me, Et,  при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из C1-3алкила, С4-6циклоалкила, тетрагидропиранила, оксетанила, тетрагидрофуранила и 1,4-диоксанила, где C1-3алкил, С4-6циклоалкил, тетрагидропиранил, оксетанил, тетрагидрофуранил и 1,4-диоксанил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из С1-6алкила, 3-членного гетероциклоалкила, 4-членного гетероциклоалкила, 5-членного гетероциклоалкила и 6-членного гетероциклоалкила, где C1-6алкил, 3-членный гетероциклоалкил, 4-членный гетероциклоалкил, 5-членный гетероциклоалкил и 6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из C1-3алкила, 4-членного гетероциклоалкила, 5-членного гетероциклоалкила и 6-членного гетероциклоалкила, где C1-3алкил, 4-членный гетероциклоалкил, 5-членный гетероциклоалкил и 6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе. В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из C1-3алкила, тетрагидропиранила, оксетанила, тетрагидрофуранила и 1,4-диоксанила, где C1-3алкил, тетрагидропиранил, оксетанил, тетрагидрофуранил и 1,4-диоксанил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из C1-3алкила и тетрагидро-2Н-пиранила, где C1-3алкил и тетрагидропиранил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе. В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из Me, Et,  и циклогексила, где Me, Et,
и циклогексила, где Me, Et, 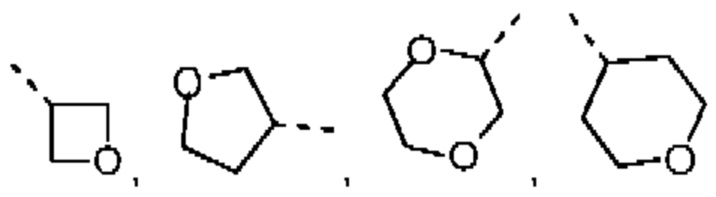 и циклогексил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
и циклогексил необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из Me, Et, 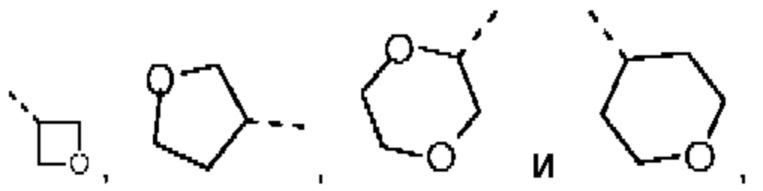 где Me, Et,
где Me, Et, 
 необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из Me, Et, 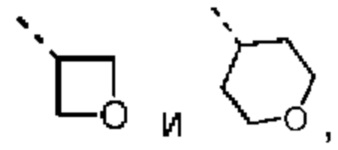 где Me, Et,
где Me, Et, 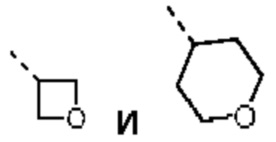 необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
необязательно замещены 1, 2 или 3 Rb, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения каждый Rb независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2, СООН и Me.
В некоторых вариантах осуществления настоящего изобретения каждый Rb независимо выбран из группы, состоящей из F, ОН, NH2, СООН и Me.
В некоторых вариантах осуществления настоящего изобретения R2 выбран из группы, состоящей из Me, -СН2ОН, Et, 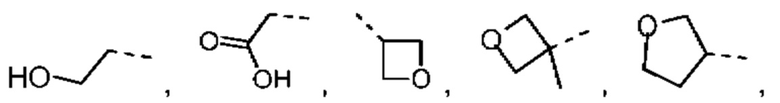
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из Me, -СН2ОН, Et, 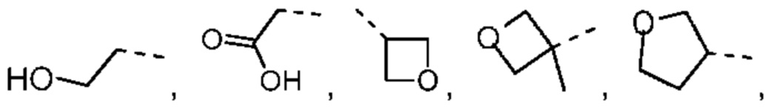
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R2 выше выбран из группы, состоящей из Me, Et,  при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения L2 выше представляет собой одинарную связь.
В некоторых вариантах осуществления настоящего изобретения структурная единица 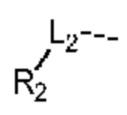 выше выбрана из группы, состоящей из Me, Et,
выше выбрана из группы, состоящей из Me, Et, 
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения структурная единица  выше выбрана из группы, состоящей из Me, Et,
выше выбрана из группы, состоящей из Me, Et, 
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения структурная единица  выше выбрана из группы, состоящей из Me, Et,
выше выбрана из группы, состоящей из Me, Et, 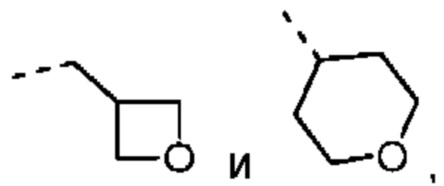 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R3 выше выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, СООН, C1-3алкила, -N(С1-3алкил)2, -С(=O)-O-С1-3алкила, -С(=O)-С1-3алкила и С3-6циклоалкила, где C1-3алкил, -N(С1-3алкил)2, -С(=O)-O-С1-3алкил, -С(=O)-С1-3алкил и С3-6циклоалкил необязательно замещены 1, 2 или 3 Rc, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R3 выше выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, C1-3алкила, -С(=O)-O-С1-3алкила, -С(=O)-С1-3алкила и С3-6циклоалкила, где C1-3алкил, -С(=O)-O-С1-3алкил, -С(=O)-С1-3алкил и С3-бциклоалкил необязательно замещены 1, 2 или 3 Rc, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения каждый Rc независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2 и CN. В некоторых вариантах осуществления настоящего изобретения R3 выше выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, СООН, Me, Et,  -C(=O)-O-Me, -C(=O)-O-Et и -С(=O)-Ме, при этом другие переменные являются такими, как определено в данном документе.
-C(=O)-O-Me, -C(=O)-O-Et и -С(=O)-Ме, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения R3 выше выбран из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, CN, Me, Et, 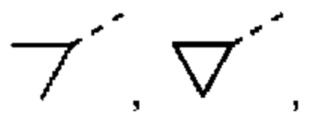 -C(=O)-O-Me, -C(=O)-O-Et и -C(=O)-Me, при этом другие переменные являются такими, как определено в данном документе.
-C(=O)-O-Me, -C(=O)-O-Et и -C(=O)-Me, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 3, 4, 5, 6, 7, 8, 9 или 10-членного гетероциклоалкила, где 3, 4, 5, 6, 7, 8, 9 или 10-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 4, 5, 6, 7 или 8-членного гетероциклоалкила, где 4, 5, 6, 7 или 8-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 4, 6, 7 или 8-членного гетероциклоалкила, где 4, 6, 7 или 8-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 6, 7 или 8-членного гетероциклоалкила, где 6, 7 или 8-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 6 или 7-членного гетероциклоалкила, где 6 или 7-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
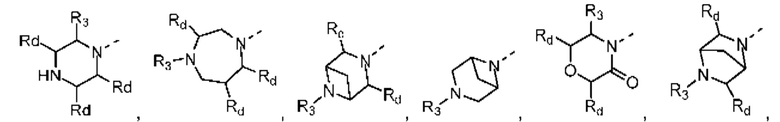
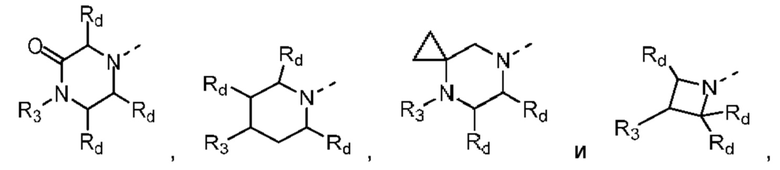
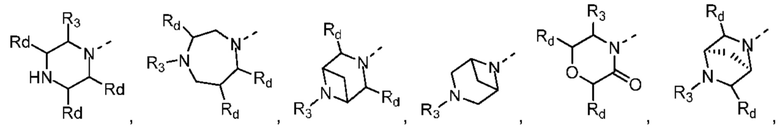
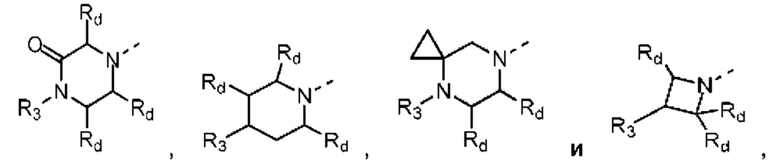
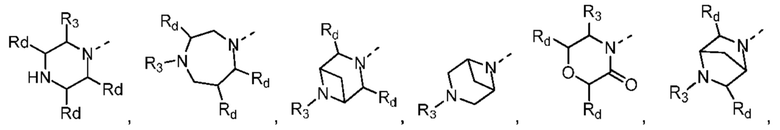
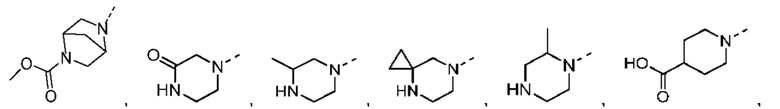
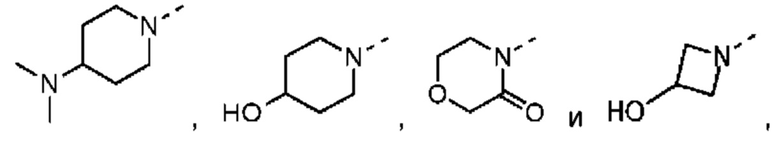
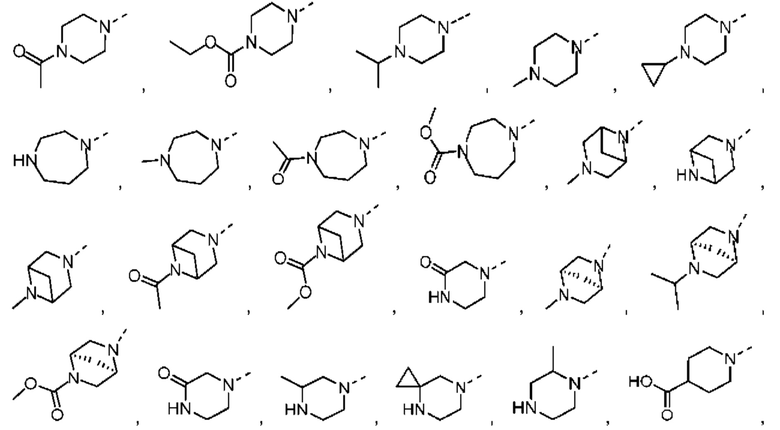
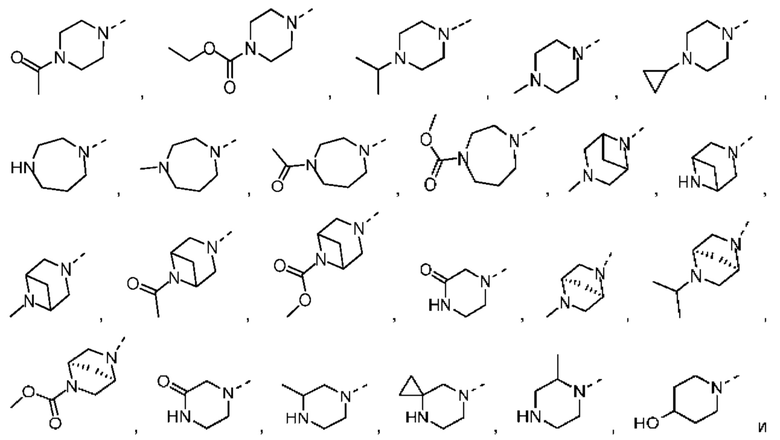
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из морфолинила, пиперазинила, 3-морфолинонила, 2-пиперазинонила, гомопиперазинила, 4,7-диазаспиро[2,5]октила, 3,6-диазабицикло[3,1,1]гептила, 2-азациклогексанонила, 2,5-диазабицикло[2.2.1]гептила и азетидинила, где морфолинил, пиперазинил, 3-морфолинонил, 2-пиперазинонил, гомопиперазинил, 4,7-диазаспиро[2,5]октил, 3,6-диазабицикло[3,1,1]гептил, 2-азациклогексанонил, 2,5-диазабицикло[2.2.1]гептил и азетидинил необязательно замещены 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из морфолинила, пиперазинила, 3-морфолинонила, 2-пиперазинонила, гомопиперазинила, 4,7-диазаспиро[2,5]октила, 3,6-диазабицикло[3,1,1]гептила, 2-азациклогексанонила и 2,5-диазабицикпо[2.2.1]гептила, где морфолинил, пиперазинил, 3-морфолинонил, 2-пиперазинонил, гомопиперазинил, 4,7-диазаспиро[2,5]октил, 3,6-диазабицикло[3,1,1]гептил, 2-азациклогексанонил и 2,5-диазабицикло[2.2.1]гептил необязательно замещены 1, 2 или 3 Rd, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения каждый Rd независимо выбран из группы, состоящей из F, Cl, Br, I, ОН, NH2 и CN.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из морфолинила, пиперазинила, 3-морфолинонила, 2-пиперазинонила, гомопиперазинила, 4,7-диазаспиро[2,5]октила, 3,6-диазабицикпо[3,1,1]гептила, 2-азациклогексанонила и 2,5-диазабицикло[2.2.1]гептила, при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 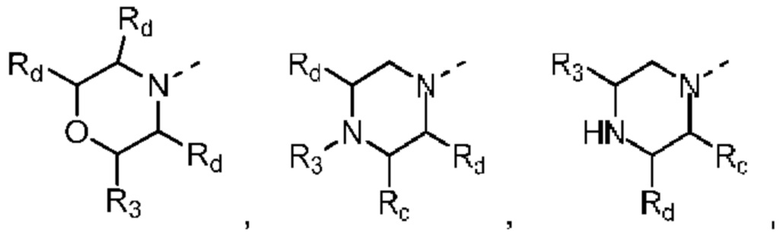
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 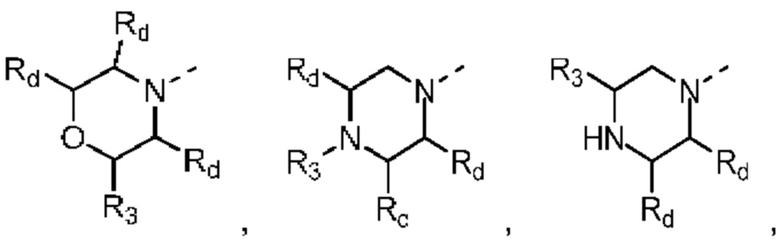
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения кольцо А выше выбрано из группы, состоящей из 
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения структурная единица 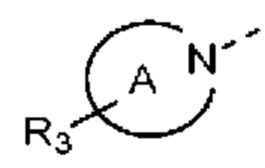 выбрана из группы, состоящей из
выбрана из группы, состоящей из 
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 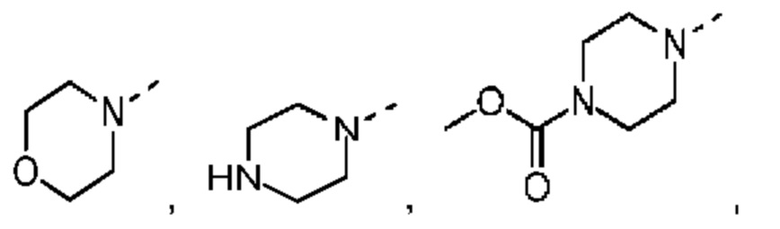
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
В некоторых вариантах осуществления настоящего изобретения структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 
 при этом другие переменные являются такими, как определено в данном документе.
при этом другие переменные являются такими, как определено в данном документе.
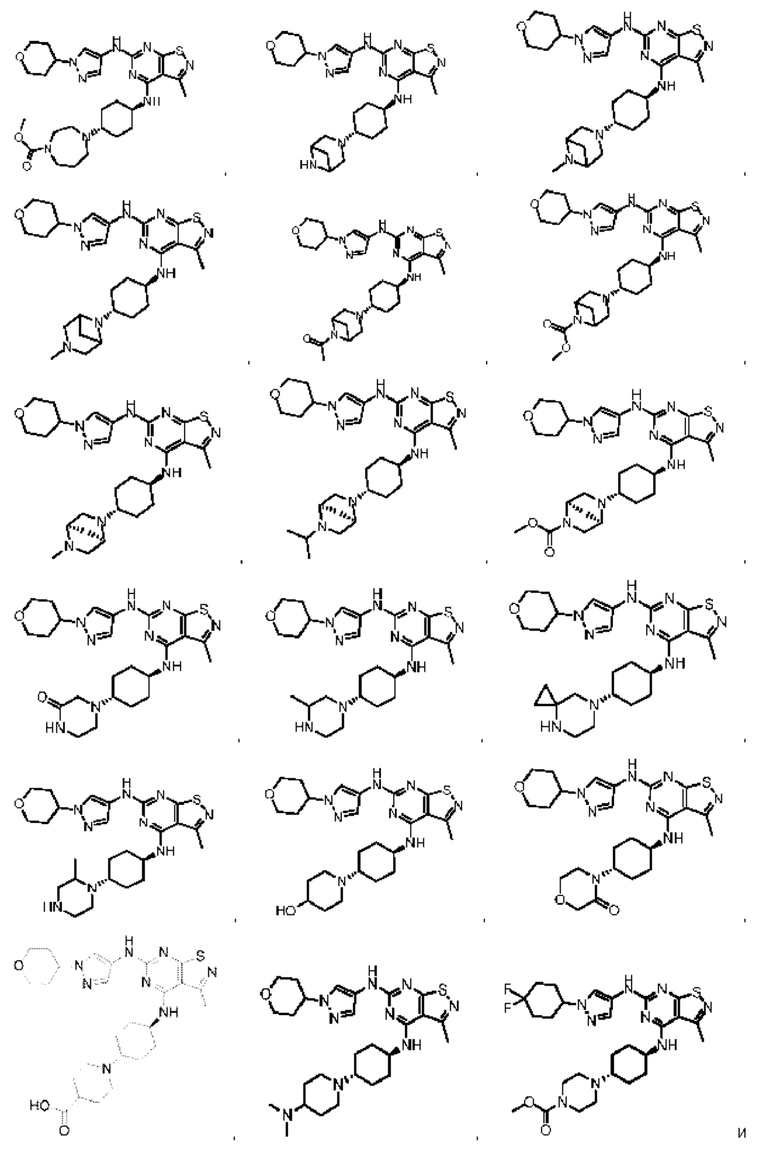
В некоторых вариантах осуществления настоящего изобретения соединение, его оптический изомер или фармацевтически приемлемая соль выбраны из

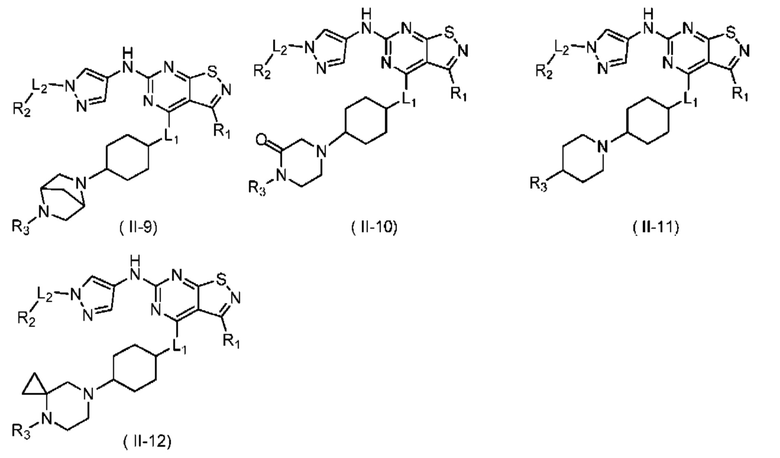
при этом R1, R2, R3, L1 и L2 являются такими, как определено в данном документе.
Еще некоторые варианты осуществления настоящего изобретения получены из любой комбинации переменных, как описано выше.
В настоящем изобретении также предусмотрено соединение следующей формулы, его оптический изомер или фармацевтически приемлемая соль, которая выбрана из:
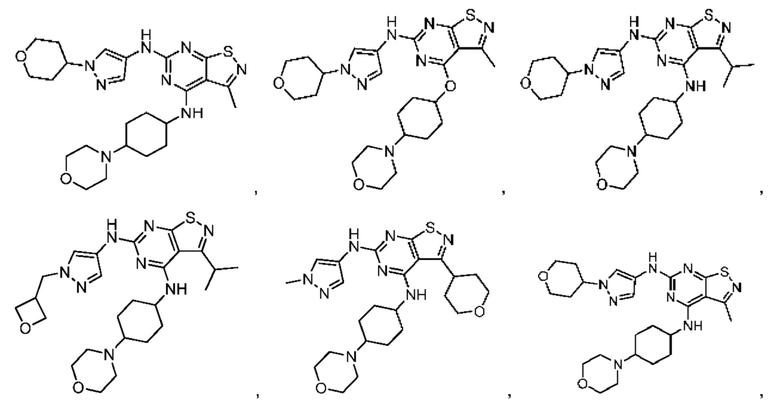
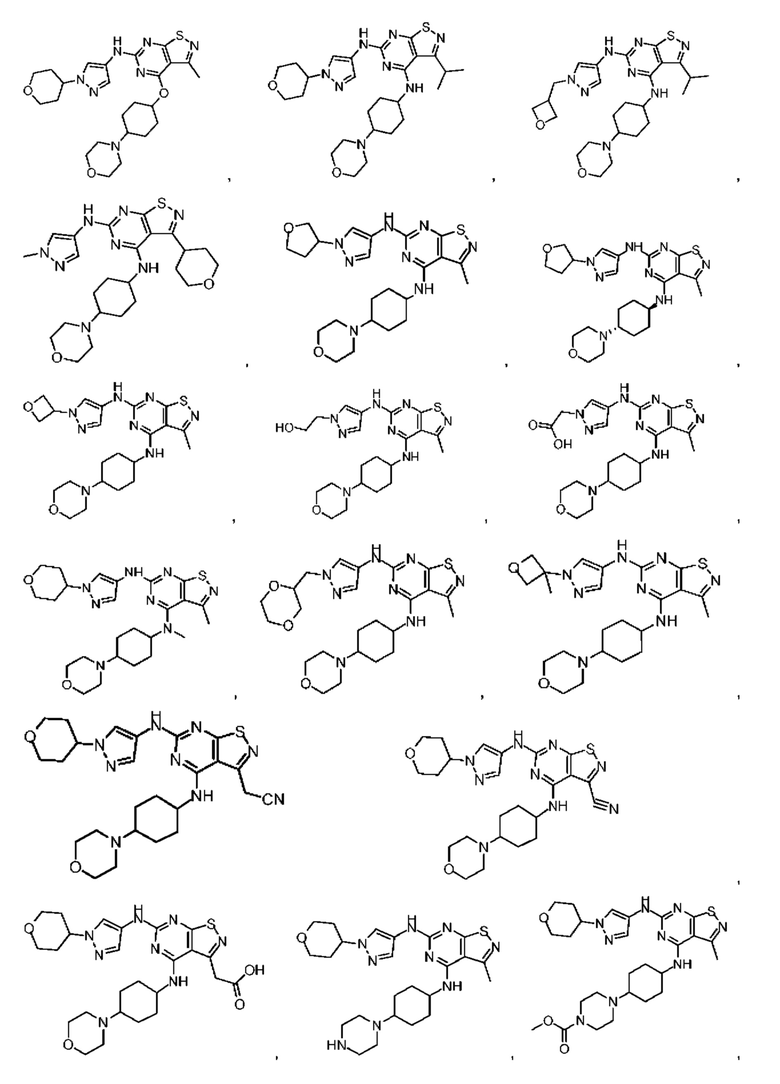
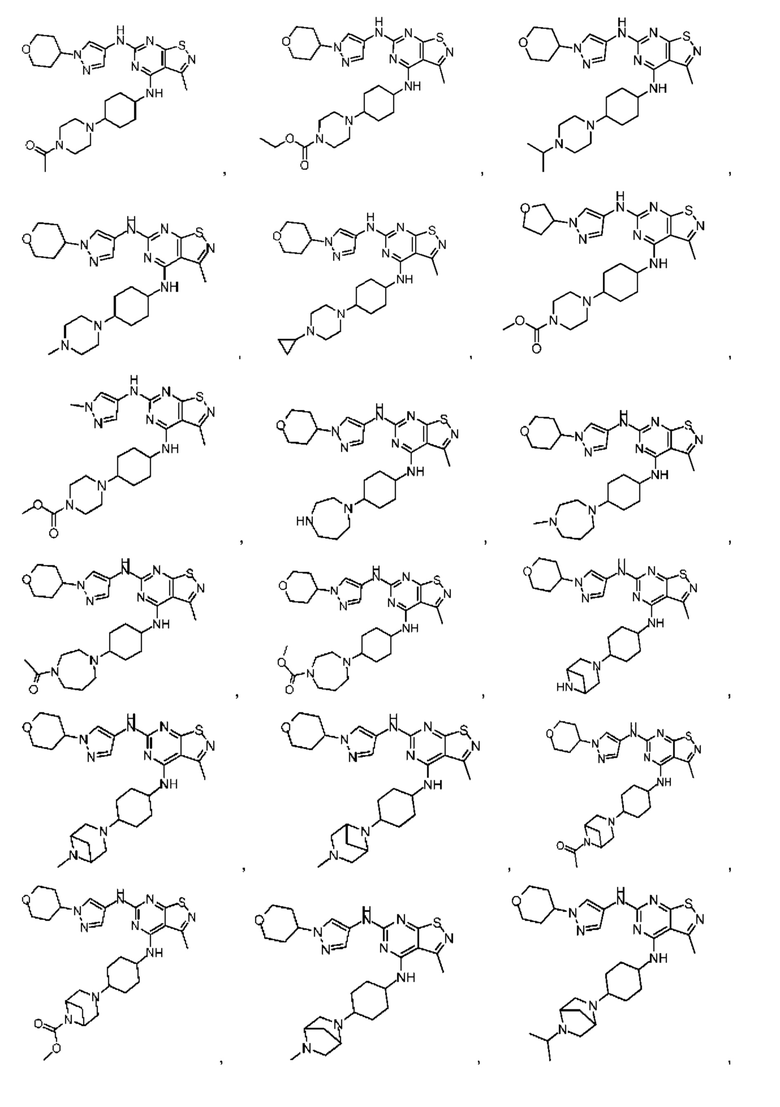
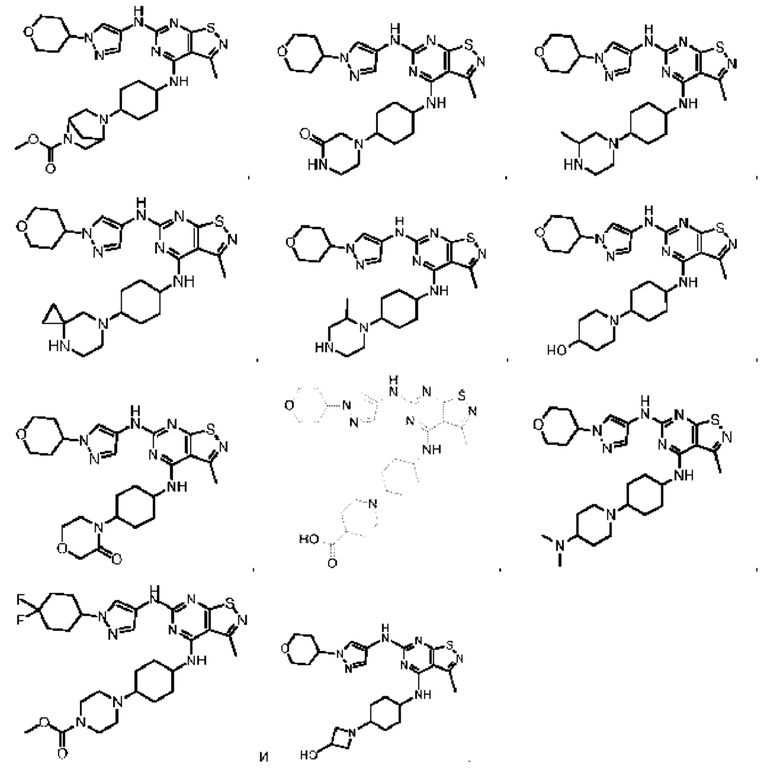
В некоторых: вариантах осуществления настоящего изобретения соединение выше, его оптический изомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из
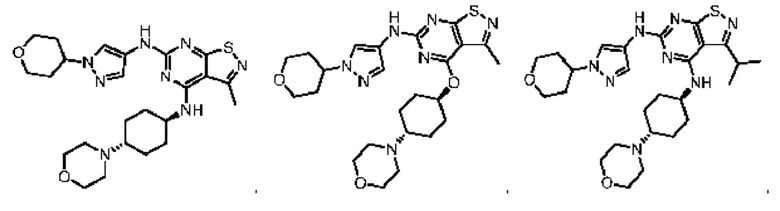
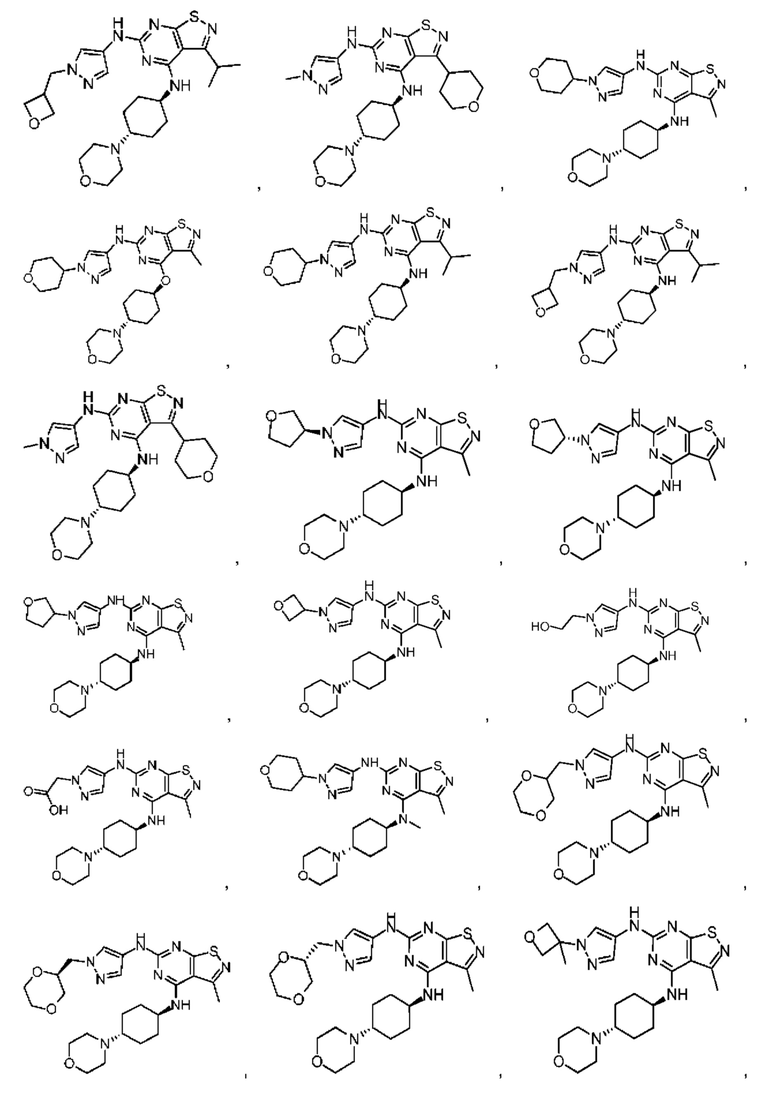
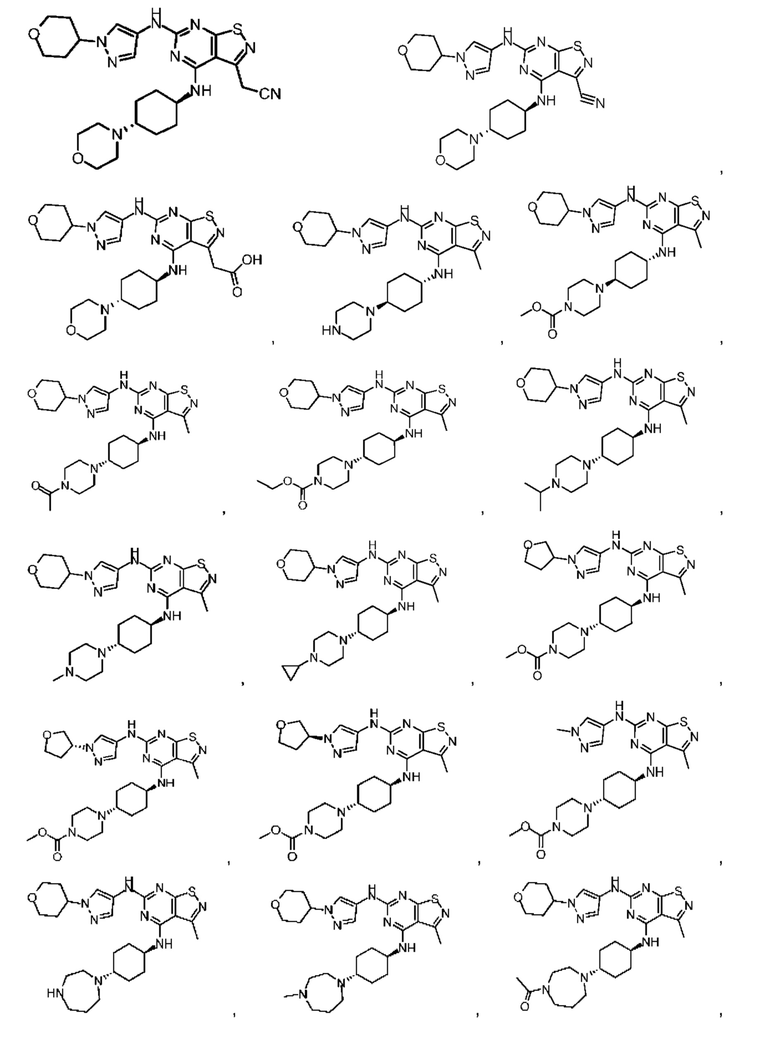
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения выше, его изомера или его фармацевтически приемлемой соли в качестве активного ингредиента, и фармацевтически приемлемые носители.
В настоящем изобретении также предусмотрен способ лечения заболеваний, связанных с IRAK4 у млекопитающего, включающий введение млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения выше, его изомера или его фармацевтически приемлемой соли, или фармацевтической композиции на их основе.
В настоящем изобретении также предусмотрено применение соединения выше, его изомера, или его фармацевтически приемлемой соли, или фармацевтической композиции на их основе, для получения лекарственного препарата для лечения заболеваний, связанных с IRAK4.
В настоящем изобретении также предусмотрено применение соединения выше, его изомера или его фармацевтически приемлемой соли, или фармацевтической композиции на его основе, для лечения заболеваний, связанных с IRAK4. В настоящем изобретении также предусмотрено соединение выше, его изомер или его фармацевтически приемлемая соль, или фармацевтическая композиция на их основе, для лечения заболеваний, связанных с IRAK4.
В настоящем изобретении также предусмотрено применение соединения выше, его изомера или его фармацевтически приемлемой соли для получения ингибитора IRAK4.
В настоящем изобретении также предусмотрено применение композиции выше для получения ингибитора IRAK4.
ОПРЕДЕЛЕНИЯ
Если не указано иное, следующие термины и фразы, применяемые в данном документе, имеют следующие значения. Конкретный термин или фраза, если другое конкретно не определено, не должен считаться неопределенным или неясным, но должен истолковываться в соответствии с его общепринятым значением. При ссылке на торговое наименование, оно относится к его соответствующему коммерческому продукту или его активному ингредиенту.
Термин «фармацевтически приемлемый» применяется в данном документе для тех соединений, материалов, композиций и/или лекарственных форм, которые, в пределах объема тщательной медицинской оценки, являются подходящими для применения в контакте с тканями человека и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, и соизмеримы с приемлемым соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения, раскрытого в данном документе, которая получена из соединения, имеющего конкретные заместители, раскрытые в данном документе, и относительно нетоксичной кислоте или основанию. Если соединение, раскрытое в данном документе, содержит относительно кислотную функциональную группу, соль присоединения основания можно получать посредством приведения в контакт нейтральной формы такого соединения с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, соли органического амина или магния, или подобные соли. Если соединение, раскрытое в данном документе, содержит относительно основную функциональную группу, соль присоединения кислоты можно получать посредством приведения в контакт нейтральной формы такого соединения с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли, полученные из неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонатный радикал, фосфорная кислота, моногидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота, фосфористая кислота; и соли, полученные из органических кислот, таких как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, винная кислота, метансульфоновая кислота. Также включены соли аминокислот (например, аргинина и т.д.) и соли органических кислот, таких как глюкуроновая кислота. Определенные конкретные соединения, раскрытые в данном документе, содержат как основную, так и кислотную функциональную группы, которые позволяют превращать соединения в соли присоединения либо основания, либо кислоты.
Фармацевтически приемлемые соли, раскрытые в данном документе, могут быть синтезированы из исходного соединения, имеющего кислотную или основную группу, с помощью традиционных химических способов. В целом, такие соли получают с помощью следующего способа: осуществление реакции соединения в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде, или органическом растворителе, или их смеси.
Соединения, раскрытые в данном документе, могут быть в форме геометрического изомера или стереоизомера. Все такие соединения предусмотрены в данном документе, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (5)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры и их рацемические смеси и другие смеси, такие как обогащенные энантиомерами или диастереомерами смеси, все из которых находятся в пределах объема настоящего изобретения. Заместители, такие как ал кил, могут иметь дополнительный асимметричный атом углерода. Все такие изомеры и их смеси находятся в пределах объема настоящего изобретения.
Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стерео изо мерам, которые являются зеркальными отражениями друг друга.
Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» обусловлен неспособностью двойной связи или одинарной связи атома углерода в кольце свободно вращаться.
Если не указано иное, термин «диастереомер» или «диастереоизомер» относится к стерео изо мерам, молекула которых имеет два или более хиральных центра, и которые не являются зеркальным отражением друг друга.
Если не указано иное, «(D)» или «(+)» означает вращение вправо, «(L)» или «(-)» означает вращение влево, и «(DL)» или «(±)» означает рацемизацию.
Если не указано иное, абсолютная конфигурация стереогенного центра представлена сплошной клиновидной связью  и пунктирной клиновидной связью
и пунктирной клиновидной связью  а относительная конфигурация стереогенного центра представлена прямой сплошной связью
а относительная конфигурация стереогенного центра представлена прямой сплошной связью  и прямой пунктирной связью
и прямой пунктирной связью  Волнистая линия
Волнистая линия  представляет собой сплошную клиновидную связь
представляет собой сплошную клиновидную связь  или сплошную пунктирную связь
или сплошную пунктирную связь  или волнистая линия
или волнистая линия  представляет собой прямую сплошную связь
представляет собой прямую сплошную связь  и прямую пунктирную связь
и прямую пунктирную связь 
Оптически активные (R)- и (S)-изомеры и D и L изомеры можно получать посредством хирального синтеза, или хиральных реагентов, или других традиционных методик. Энантиомер определенного соединения, раскрытого в данном документе, может быть получен посредством асимметричного синтеза или дериватизации с применением хирального вспомогательного средства, где полученную диастереомерную смесь отделяют и вспомогательную группу отщепляют таким образом, чтобы обеспечить получение требуемого чистого энантиомера. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксил), соединение реагирует с подходящей оптически активной кислотой или основанием с образованием соли диастереоизомера, которая далее подвергается диастереомерному разделению посредством общепринятых в данной области техники способов с получением чистого энантиомера. Кроме того, энантиомер и диастереомер в целом выделяют с помощью хроматографии с применением хиральной неподвижной фазы, необязательно в комбинации с химической дериватизацией (например, образованием карбамата из аминов). Соединение, раскрытое в данном документе, может содержать не встречающуюся в природе пропорцию атомных изотопов на одном или более атомов, которые образуют соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) или С-14 (14С). В качестве другого примера, водород может быть замещен дейтерием с образованием дейтерированного лекарственного средства, и связь, образованная между дейтерием и углеродом, более жесткая, чем связь, которая образована между обычным водородом и углеродом. По сравнению с недейтерированным лекарственным средством дейтерированное лекарственное средство имеет преимущества в виде пониженного токсического побочного эффекта, повышенной стабильности, повышенной эффективности, увеличенного биологического периода полужизни и т.п. Все изотопные варианты соединения, описанного в данном документе, являются они радиоактивными или нет, включены в объем настоящего изобретения. «Необязательный» или «необязательно» означает, что может, но не обязательно, возникнуть последовательно описанное событие или обстоятельство, и описание включает случаи, при которых событие или обстоятельство возникает, и случаи, при которых не возникает.
Термин «замещенный» означает, что один или несколько атомов водорода при конкретном атоме замещены заместителями, которые могут включать варианты дейтерия и водорода, при условии, что валентность конкретного атома является нормальной, и соединение после замещения является стабильным. Если заместитель представляет собой кислород (т.е. =O), это означает, что замещены два атома водорода. Замещение кислородом не происходит в ароматических группах. Термин «необязательно замещенный» означает, что атом может быть замещен заместителем или может быть не замещен заместителем. Если не указано иное, тип и количество заместителей могут быть произвольными до тех пор, пока это достижимо химически.
Если любая переменная (например, R) встречается более одного раза в составе или структуре соединения, определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, группа может быть необязательно замещена не более чем двумя R, и определение R в каждом случае является независимым. Кроме того, комбинация заместителей и/или их вариантов допускается, только если комбинация приведет к получению стабильного соединения.
Если переменная представляет собой одинарную связь, это означает, что две группы непосредственно связаны, например, в A-L-Z, если L представляет собой одинарную связь, это означает, что структура фактически представляет собой A-Z.
Для перечисленных связанных групп направление для связывания, которое не указано, является произвольным. Например, при связывании группа L, содержащаяся в 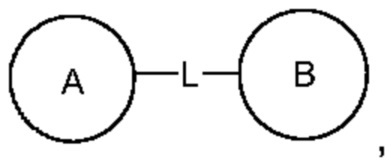 представляет собой -M-W-, -M-W- может связывать либо кольцо А, либо кольцо В в направлении, аналогичном порядку прочтения слева направо, с образованием
представляет собой -M-W-, -M-W- может связывать либо кольцо А, либо кольцо В в направлении, аналогичном порядку прочтения слева направо, с образованием 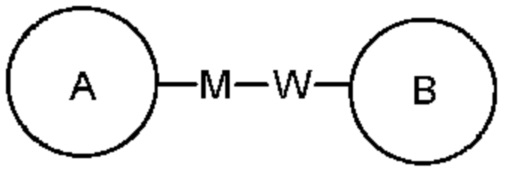 , или связывать кольцо А и кольцо В в противоположном направлении с образованием
, или связывать кольцо А и кольцо В в противоположном направлении с образованием 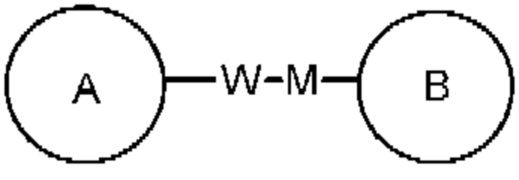 . Комбинация связывающей группы, заместителя и/или их варианта допустима только, если комбинация приведет к получению стабильного соединения.
. Комбинация связывающей группы, заместителя и/или их варианта допустима только, если комбинация приведет к получению стабильного соединения.
Если не указано иное, количество атомов в кольце в целом определено как количество членов кольца. Например, «3-6-членное кольцо» относится к «кольцу», в котором 3-6 атомов расположены в виде кольца.
Если не указано иное, термин «C1-6алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-6 атомов углерода. C1-6алкил включает С1-5, С1-4, С1-3, С1-2, С2-6, С2-4, С6 и С5алкил и т.д., и может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метенил). Примеры C1-6алкила включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил (включая н-пентил, изопентил, и неопентил), гексил и т.п.
Если не указано иное, термин «C1-3алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. С1-3алкил включает С1-2 и С2-3алкил и т.д., и может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метенил). Примеры C1-3алкила включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.п. Если не указано иное, «С2-8алкенил» применяют для обозначения линейной или разветвленной углеводородной группы, содержащей 2-8 атомов углерода и по меньшей мере одну углерод-углеродную двойную связь, которая может быть расположена в любом месте в группе. С2-8алкенил включает С2-6, С2-4, С2-3, С4, С3 и С2алкенил и т.д., и может быть одновалентным, двухвалентным или поливалентным. Примеры С2-8алкенила включают без ограничения этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, 1,3-пентадиенил, 1,3-гексадиенил и т.п.
Если не указано иное, термин «C1-6алкиламино» относится к алкильной группе, содержащей 1-6 атомов углерода, которая присоединена к остальной части молекулы посредством аминогруппы. С1-6алкиламино включает С1-4, C1-3, C1-2, С2-6, С2-4, С6, С5, С4, С3, С2алкиламино и т.п. Примеры C1-6алкиламино включают без ограничения -NHCH3, -N(CH3)2, -NHCH2CH3, -N(CH3)CH2CH3, -N(CH2CH3)(CH2CH3), -NHCH2CH2CH3, -NHCH2(CH3)2, -NHCH2CH2CH2CH3 и т.п.
Если не указано иное, термин «C1-3алкиламино» относится к алкильной группе, содержащей 1-3 атома углерода, которая присоединена к остальной части молекулы посредством аминогруппы. C1-3алкиламиногруппа включает C1-2, С3, С2алкиламино и т.п. Примеры C1-3алкиламино включают без ограничения -NHCH3, -М(СН3)2, -NHCH2CH3, -N(CH3)CH2CH3, -NHCH2CH2CH3, -NHCH2(CH3)2 и т.п.
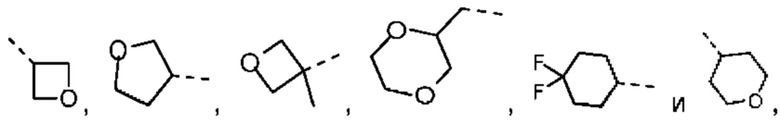
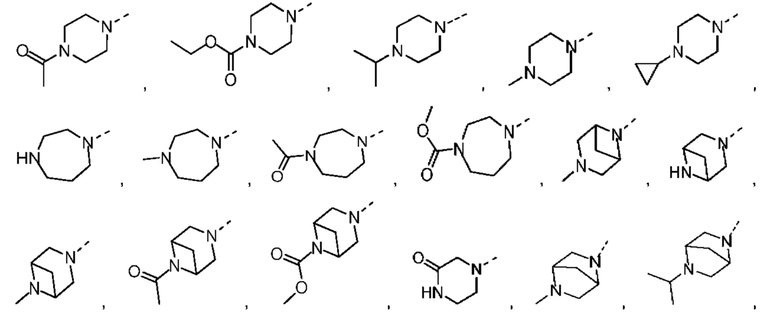
Если не указано иное, термин «3-10-членный гетероциклоалкил», применяемый отдельно или в комбинации с другими терминами, обозначает насыщенную циклическую группу, состоящую из 3-10 атомов кольца, 1, 2, 3 или 4 из которых являются гетероатомами, независимо выбранными из группы, состоящей из -О-, -S-, -NH-, N и -C(=O)NH-, при этом остальная часть представляет собой атомы углерода. Это включает моноциклические, бициклические и трициклические системы, где бициклические и трициклические системы включают спироциклические, конденсированные и мостиковые кольца. Кроме того, в случае «3-10-членного гетероциклоалкила» гетероатом может занимать положение, где гетероциклоалкил присоединен к остальной части молекулы. 3-10-членный гетероциклоалкил включает 3-9-членную, 3-8-членную, 3-7-членную, 3-6-членную, 3-5-членную, 4-6-членную, 5-6-членную, 4-членную, 5-членную, 6-членную гетероциклоалкильную группы и т.п. Примеры 3-10-членного гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (включая тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (включая 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (включая 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил или диоксепанил и т.д.
Если не указано иное, термин «3-6-членный гетероциклоалкил», применяемый отдельно или в комбинации с другими терминами, обозначает насыщенную циклическую группу, состоящую из 3-6 атомов кольца, 1,2,3 или 4 из которых являются гетероатомами, независимо выбранными из группы, состоящей из -О-, -S-, -NH- и N, при этом остальная часть представляет собой атомы углерода. Он включает моноциклические и бициклические системы, где бициклическая система включает спироциклические, конденсированные и мостиковые кольца. Кроме того, в случае «3-6-членного гетероциклоалкила» гетероатом может занимать положение, где гетероциклоалкил присоединен к остальной части молекулы. 3-6-членный гетероциклоалкил включает 4-6-членный, 5-6-членный, 4-членный, 5-членный, 6-членный гетероциклоалкил и т.п. Примеры 3-6-членного гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (включая тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (включая 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (включая 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил и т.д.
Если не указано иное, «С3-8циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-8 атомов углерода. Он включает моноциклические и бициклические системы, где бициклическая система включает спироциклические, конденсированные и мостиковые кольца. С3-8циклоалкил включает С3-6, С3-5, С4-8, C4-6, С4-5, С5-8, С5-6циклоалкил или т.п., и может быть одновалентным, двухвалентным или поливалентным. Примеры С3-8циклоалкила включают без ограничения, циклопропил, циклобутил и т.п.
Если не указано иное, «С4-6циклоалкил» обозначает насыщенную циклическую углеводородную группу, состоящую из 4-6 атомов углерода, включая моноциклические и бициклические системы. С4-6циклоалкил включает С4-5, С5-6циклоалкил и т.п., и может быть одновалентным, двухвалентным или поливалентным. Примеры С4-6циклоалкила включают без ограничения циклобутил, циклопентил, циклогексил и т.п.
Если не указано иное, Cn-n+m или Cn-Cn+m включает все из конкретных случаев атомов углерода от n до n+m, например, C1-6 включает С1, С2, С3, С4, С5 и С6, и также включает любой из диапазонов в пределах от n до n+m, например, С1-6 включает С1-2, С1-3, С1-4, С2-3, С2-4 и С3-5 и т.д.; подобным образом, от n-членного до n+m-членного означает от n до n+m атомов в кольце, например, 3-6-чле иные кольца включают 3-членные кольца, 4-членные кольца, 5-членные кольца, 6-членные кольца, и включают любые диапазоны в пределах от n до n+m, например, 3-6-членные кольца включают 3-5-членные кольца, 3-6-членные кольца, 4-6-членные кольца, 4-5-членные кольца и 5-6-членные кольца и т.д.
Соединения, раскрытые в данном документе, могут быть получены с помощью разнообразных синтетических способов, хорошо известных специалистам в данной области техники, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные их комбинациями с другими химическими синтетическими способами, и их эквиваленты, известные специалистам в данной области техники. Предпочтительные варианты осуществления включают без ограничения примеры, раскрытые в данном документе.
Растворитель, применяемый в настоящем изобретении, может быть коммерчески доступным. Следующие аббревиатуры применяют в настоящем изобретении: водн. обозначает водный; HATU обозначает гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; EDC обозначает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида; m-СРВА обозначает 3-хлорпероксибензойную кислоту; экв. обозначает эквивалент; CDI обозначает карбонилдиимидазол; DCM обозначает дихлорметан; РЕ обозначает петролейный эфир; DIAD обозначает диизопропилазодикарбоксилат; DMF обозначает N,N-диметилформамид; DMSO обозначает диметилсульфоксид; EtOAc обозначает этилацетат; EtOH обозначает этанол; МеОН обозначает метанол; CBz обозначает бензилоксикарбонил, защитную группу для аминогруппы; ВОС обозначает трет-бутокси карбонил, защитную группу для аминогруппы; НОАс обозначает уксусную кислоту; NaCNBH3 обозначает цианоборгидрид натрия; к.т. обозначает комнатную температуру; O/N обозначает в течение ночи; THF обозначает тетрагидрофуран; Вос2О обозначает ди-трет-бутилдикарбонат; TFA обозначает трифторуксусную кислоту; DIPEA обозначает диизопропилэтиламин; SOCl2 обозначает тионилхлорид; CS2 обозначает дисульфид углерода; TsOH обозначает п-толуолсульфоновую кислоту; NFSI обозначает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS обозначает 1-хлорпирролидин-2,5-дион; н-Bu4NF обозначает фторид тетрабутиламмония; iPrOH обозначает 2-пропанол; т.пл. обозначает точку плавления; и LDA обозначает диизопропиламид лития.
Соединения называются в соответствии с традиционными правилами номенклатуры из уровня техники или с применением программного обеспечения ChemDraw® и для коммерчески доступных соединений приводятся названия согласно каталогу производителя.
ТЕХНИЧЕСКИЕ ЭФФЕКТЫ
В соответствии с настоящим изобретением получают серию соединений с конденсированным кольцом с повышенной активностью, улучшенной метаболической стабильностью, улучшенными фармацевтическими свойствами и предпочтительными фармакокинетическими свойствами.
Соединения, раскрытые в данном документе, в целом демонстрируют улучшенную ингибирующую активность в отношении IRAK4. Иллюстративные соединения, раскрытые в данном документе, обладают впечатляющими преимуществами над исходным соединением (WXR1) в отношении стабильности в микросомах печени у множества видов, и в частности у некоторых видов (например, мышь) обладают превосходством в клиренсе до 20 раз. Соединения, раскрытые в данном документе, в целом демонстрируют улучшенную ингибирующую активность в отношении пролиферации клеток линии ТНР-1. Общее системное воздействие, пиковая концентрация и биологическая доступность различных соединений по данному проекту, вводимых перорально, были эквивалентными или превосходящими таковые соединения WXR2 (ND-2110) при той же дозе, демонстрируя превосходящие фармакокинетические свойства. При тех же дозах, WX001, вводимое перорально, у крыс линии SD продемонстрировало значительный ингибирующий эффект в отношении секреции TNF-α, индуцированной липополиколлагеном (LPS), который в значительной степени превосходит таковой у исходных соединений WXR2(ND-2110), WXR3(BAY-1830839) и WXR4 (BAY-1834845). В данном эксперименте эффективность WX001 была эквивалентна эффективности дексаметазона DEX. При тех же дозах, вводимые перорально соединения WX001 WX026 и WX044 у крыс SD продемонстрировали значительный ингибирующий эффект в отношении секреции TNF-α, индуцированной липополиколлагеном (LPS). В данном эксперименте эффективность WX026 была эквивалентна эффективности дексаметазона DEX.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг.1 показаны результаты фармакодинамического исследования, оценивающие секрецию TNF-α, индуцированную липополиколлагеном (LPS) у крыс SD, при этом доза соединений составляет 30 мг/кг; и
На фиг.2 показаны результаты фармакодинамического исследования, оценивающие секрецию TNF-α, индуцированную липополиколлагеном (LPS) у крыс SD, при этом доза соединений составляет 20 мг/кг.
ПОДРОБНОЕ ОПИСАНИЕ
Настоящее изобретение описано подробно ниже в качестве примеров. Однако это не означает ограничения объема настоящего изобретения, приводящего к неблагоприятным последствиям. Хотя настоящее изобретение было подробно описано в данном документе и конкретные примеры также были раскрыты, специалистам в данной области техники будет очевидно, что могут быть сделаны различные изменения и модификации в отношении конкретных примеров без отступления от сущности и объема настоящего изобретения.
Промежуточное соединение А1
Путь синтеза:
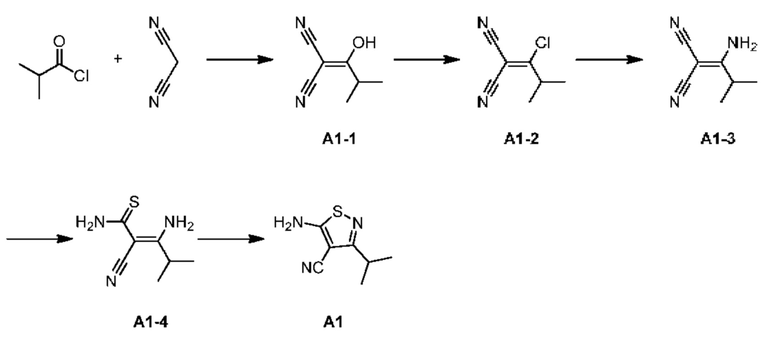
Стадия 1: Синтез соединения А1-1
Малононитрил (52,70 г) растворяли в ацетонитриле (1 л) в одногорлой колбе объемом 3 л и добавляли триэтиламин (161,45 г). Смесь хорошо перемешивали, медленно добавляли изобутирилхлорид (85 г) и инкубировали при 50°С в течение 2 часов. Затем добавляли ацилхлорид и система была экзотермической и превратилась из бесцветного прозрачного раствора в желто-зеленую суспензию. После завершения реакции растворитель удаляли с помощью роторного испарителя. Остаток растворяли в воде (100 мл) и добавляли этилацетат (30 мл) для трех экстракций. Органические фазы объединяли и высушивали с применением роторного испарителя с получением продукта А1-1. 1Н ЯМР (400 МГц, CDCl3) δ = 1,23 (d, J=10,4 Гц, 6Н), 3,16 (септет, J=10,4 Гц, 1Н).
Стадия 2: Синтез соединения А1-2
А1-1 (1,1 г) растворяли в дихлорметане (15 мл) в реакционной колбе объемом 100 мл, добавляли оксихлорид фосфора (2,26 г) по каплям при 0°С и инкубировали при 20°С в течение 16 часов. После завершения реакции смесь непосредственно добавляли в воду и органическую фазу отделяли. Водную фазу затем экстрагировали дважды дихлорметаном (10 мл) и органические фазы объединяли и высушивали с применением роторного испарителя с получением соединения А1-2. 1Н ЯМР (400 МГц, CDCl3) δ = 1,7 (d, J=10,4 Гц, 6Н), 3,09 (септет, J=10,4 Гц, 1Н).
Стадия 3: Синтез соединения А1-3
А1-2 (10 г) и водный аммиак (7,56 г) добавляли в реакционную колбу объемом 250 мл и инкубировали при 20°С в течение 1 часа. Наблюдали осаждение и в реакционную систему непосредственно добавляли воду (10 мл) с этилацетатом (10 мл) для трех экстракций. Органические фазы объединяли, высушивали с помощью роторного испарителя с получением соединения А1-3. 1Н ЯМР (400 МГц, CDCl3) δ = 1,24 (d, J=10,4 Гц, 6Н), 3,15 (септет, J=10,4 Гц, 1Н).
Стадия 4: Синтез соединения А1-4
А1-3 (1 г) и триэтиламин (2,08 г) растворяли в пиридине (10 мл) в трехгорлой колбе объемом 100 мл и затем добавляли сульфид водорода и инкубировали при 30°С в течение 0,5 часа. После завершения реакции смесь высушивали с применением роторного испарителя и в остаток добавляли воду и затем дихлорметан (10 мл) для двух экстракций. Растворитель удаляли с применением роторного испарителя с получением желтого маслянистого вещества, которое затем очищали с помощью колоночной хроматографии (элюент: дихлорметан) с получением продукта А1-4. 1Н ЯМР (400 МГц, CDCl3) δ = 1,20 (d, J=10,4 Гц, 6Н), 3,15 (s, J=10,4 Гц, 1Н), 6,04 (br, 1Н), 6,55 (br, 1Н), 6,87 (br, 1Н), 12,18 (br, 1Н).
Стадия 5: Синтез соединения А1
В одногорлую колбу объемом 250 мл с магнитной мешалкой добавляли соединение А1-4 и МеОН (80 мл), и Н2О2 (8,87 г) добавляли по каплям к смеси. Реакционную систему затем перемешивали при 10-20°С в течение 16 ч. После завершения реакции большую часть растворителя удаляли посредством концентрирования при пониженном давлении с получением твердого остатка. Этилацетат (40 мл) добавляли для растворения твердого вещества с последующим добавлением насыщенного водного раствора сульфита натрия (30 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и фазы разделяли. Водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы дегидратировали с применением безводного сульфата натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением продукта А1. 1Н ЯМР (400 МГц, CDCl3) δ = 5,38 (br s, 2Н), 3,13 (td, J=6,8, 13,8 Гц, 1H), 1,33 (d, J=6,8 Гц, 6H).
Промежуточное соединение А2
Путь синтеза:
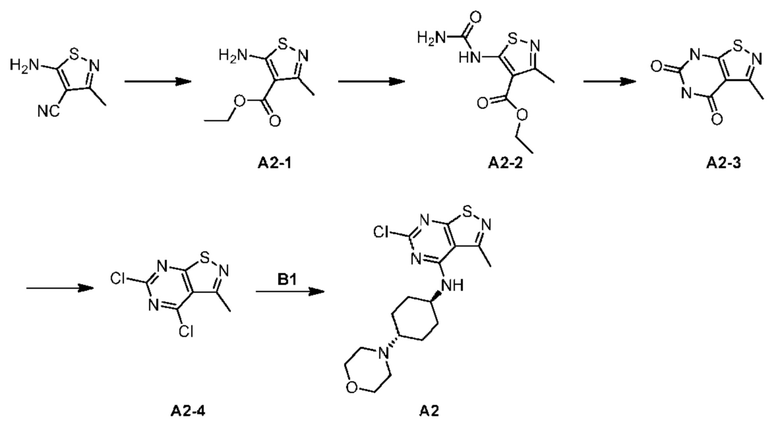
Стадия 1: Синтез соединения А2-1
В круглодонную одногорлую колбу объемом 100 мл с магнитной мешалкой добавляли 5-амино-3-метилизотиазол-4-карбонитрил (1,00 г) и абсолютный этанол (25 мл). Концентрированную серную кислоту (7,05 г) медленно добавляли в реакционную систему при 20°С. Реакционную смесь в колбе перемешивали и инкубировали на масляной бане при 100°С в течение 16 часов. После завершения реакции реакционную систему охлаждали до комнатной температуры и медленно добавляли в 100 мл раствора бикарбоната натрия. рН смеси доводили до 8, добавляли этилацетат (30 мл) для трех экстракций, дегидратировали с применением безводного сульфата натрия и фильтровали, и фильтрат концентрировали при пониженном давлении с получением желтого неочищенного продукта. Неочищенный продукт отделяли и очищали с помощью колоночной хроматографии (SiO2, петролейный эфир: этилацетат = 10:1-3:1, объем/объем) с получением соединения А2-1. 1Н ЯМР (400 МГц, CDCl3) δ = 6,49 (br s, 2Н), 4,35 (q, J=7,2 Гц, 2H), 2,53 (s, 3Н), 1,40 (t, J=7,0 Гц, 3Н).
Стадия 2: Синтез соединения А2-2
Раствор А2-1 (2,5 г) в сухом дихлорметане (25 мл) добавляли в одногорлую колбу объемом 50 мл с магнитной мешалкой в атмосфере азота, охлаждали до -60°С и добавляли изоцианат хлорсульфоновой кислоты (1,79 г) по каплям в реакцию. Затем реакционный раствор медленно нагревали до 25°С и перемешивали в течение 30 минут до того момента, пока раствор не стал прозрачным. Реакционную смесь концентрировали при пониженном давлении с получением желтого твердого вещества, которое суспендировали в воде (10 мл), и суспензию перемешивали при 75°С в течение 30 минут, фильтровали и высушивали с получением соединения А2-2. Стадия 3:
Синтез соединения А2-3
К раствору А2-2 (1,7 г) в н-бутаноле (20 мл) в одногорлой колбе объемом 50 мл с магнитной мешалкой добавляли карбонат калия (3,07 г) в атмосфере N2 и смесь инкубировали при 130°С в течение 16 часов. Растворитель удаляли с применением роторного испарителя. Остаток суспендировали с помощью 10 мл воды, фильтровали и высушивали с получением соединения А2-3. LCMS (ESI) масса/заряд: 183,8 [М+Н]+, 1Н ЯМР (400 МГц, DMSO-d6) δ = 9,43 (br s, 2Н), 2,39-2,34 (m, 3Н).
Стадия 4: Синтез соединения А2-4
Оксихлорид фосфора (2,93 г) добавляли в раствор диметиланилина (330,75 мг) и А2-3 (0,5 г) в толуоле (5 мл) в закрытой пробирке объемом 100 мл и смесь инкубировали при 120°С в течение 16 часов. Реакционную систему добавляли медленно в 30 мл воды и экстрагировали дихлорметаном (20 мл × 3). Органические фазы объединяли, дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт подвергали хроматографии с получением соединения А2-4. LCMS (ESI) масса/заряд: 219,8 [М+Н]+.
Стадия 5: Синтез соединения А2
А2-4 (0,2 г), промежуточное соединение В1 (303,85 мг) и карбонат натрия (385,28 мг) растворяли в ацетонитриле (4 мл) в одногорлой колбе объемом 50 мл с магнитной мешалкой. Реакционную систему инкубировали и перемешивали при 80°С в течение 16 часов. Реакционную систему охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением остатка. К остатку добавляли дихлорметан (10 мл) и воду (10 мл) для повторного растворения. Водную фазу экстрагировали дихлорметаном (5 мл). Объединенные органические фазы промывали насыщенным солевым раствором, дегидратировали с применением безводного сульфата натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением бледно-желтого твердого вещества. Твердое вещество суспендировали с применением РЕ/EtOAc = 10:1 (10 мл), перемешивали в течение 1 часа и фильтровали с получением соединения А2. LCMS (ESI) масса/заряд: 368,1 [М+Н]+.
Промежуточное соединение A3
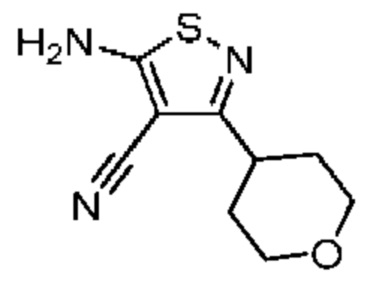
Синтез промежуточного соединения A3 подобен синтезу промежуточного соединения А1, за исключением того, что исходный материал изобутирилхлорид на стадии 1 был заменен на тетрагидропиран-4-карбонилхлорид.
1Н ЯМР (400 МГц, DMSO-d6) δ = 8,16 - 7,89 (m, 2Н), 3,99 - 3,80 (m, 2Н), 3,40 (dt, J=3.9, 10,9 Гц, 2Н), 2,99 - 2,84 (m, 1Н), 1,82 - 1,60 (m, 4Н).
Промежуточное соединение А4
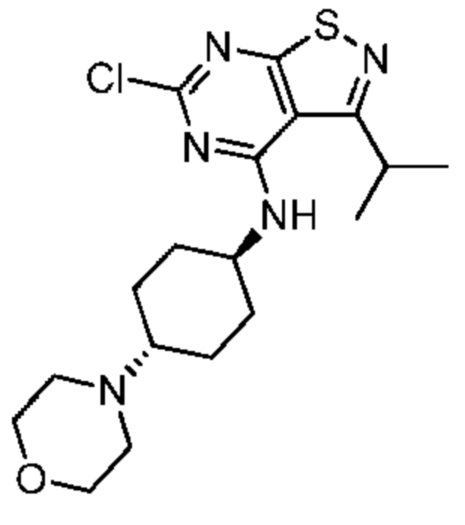
Синтез промежуточного соединения А4, начиная с А1 подобен синтезу А2, за исключением того, что исходный материал, представляющий собой 5-амино-3-метилизотиазол-4-карбонитрил, на стадии 1 был заменен на А1.
LCMS (ESI) масса/заряд: 396,2,398,2 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ = 5,52 (br d, J=7,6 Гц, 1Н), 4,31-4,19 (m, 1Н), 4,16 -4,09 (m, 1Н), 4,16-4,09 (m, 1Н), 3,79-3,70 (m, 4Н), 3,22-3,13 (m, 1Н), 2,65-2,56 (m, 4Н), 2,41 - 2,27 (m, 3Н), 2,06 - 1,96 (m, 2Н), 1,60 - 1,48 (m, 2Н), 1,46 (d, J=6,8 Гц, 4Н), 1,39- 1,29 (m, 2Н).
Промежуточное соединение А5
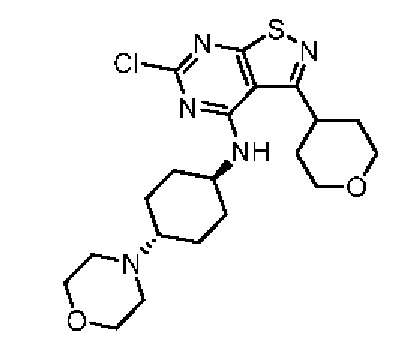
Синтез промежуточного соединения А5, начиная с A3 подобен синтезу А2, за исключением того, что исходный материал, представляющий собой 5-амино-3-метилизотиазол-4-карбонитрил, на стадии один был заменен на A3.
LCMS (ESI) масса/заряд: 438,2, 440,2 [М+Н]+
Промежуточное соединение А6
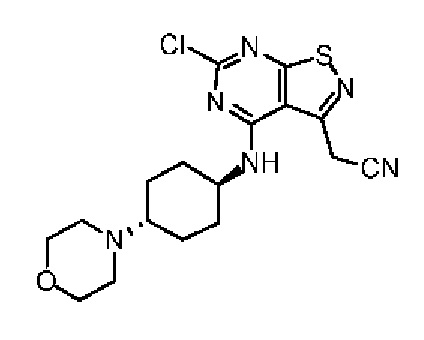
Путь синтеза:
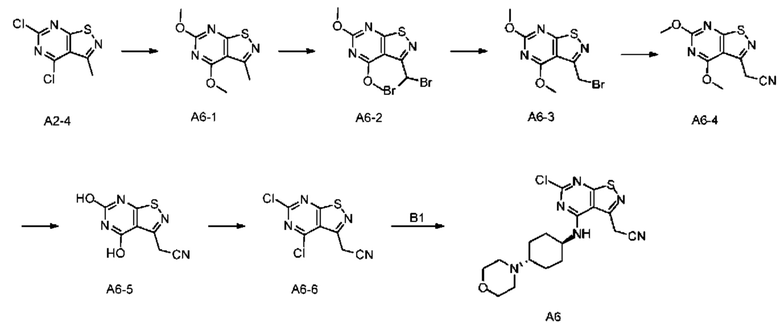
Стадия 1: Синтез соединения А6-1
А2-4 (10 г) растворяли в метаноле (200 мл) и добавляли раствор метоксида натрия в метаноле (5 М, 36,35 мл). Смесь инкубировали при 68°С в течение 12 часов, и затем добавляли в насыщенный раствор хлорида аммония (500 мл) и экстрагировали этилацетатом (150 мл). Органическую фазу дегидратировали, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонка 80 г; элюент: 0-50% этилацетат/петролейный эфир; скорость потока: 40 мл/мин) с получением соединения А6-1.
Стадия 2: Синтез соединения А6-2
Соединение А6-1 (8,79 г) смешивали с 1,2-дихлорэтаном (100 мл), Л/-бромсукцинимидом (26,66 г) и бензоилпероксидом (453,58 мг) в колбе. После трех продувок азотом смесь инкубировали при 80°С в течение 12 часов и добавляли в насыщенный водный тиосульфат натрия (200 мл), и фазы разделяли. Водную фазу экстрагировали дихлорметаном (50 мл × 2) и объединенные органические фазы высушивали, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO® для неочищенных продуктов; колонка 80 г; элюент: 0-60% этилацетат/петролейный эфир)) с получением соединения А6-2.
Стадия 3: Синтез соединения А6-3
А6-2 (4 г) растворяли в ацетонитриле (20 мл), добавляли диэтилфосфит (1,95 г) и N,N-диизопропилэтиламин (2,80 г) и смесь инкубировали при 25°С в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении и неочищенный продукт очищали с помощью колоночной хроматографии (ISCO® для неочищенных продуктов; колонка 80 г; элюент: 0-60% этилацетат/петролейный эфир)) с получением соединения А6-3.
Стадия 4: Синтез соединения А6-4
А6-3 (1,5 г) растворяли в ацетонитриле (40 мл) в колбе и последовательно добавляли триметилсил ил цианид (564,20 мг) и карбонат калия (786,00 мг). Смесь перемешивали при 10°С в течение 12 часов. Реакционную систему добавляли в 200 мл насыщенного водного хлорида аммония и экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли, дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с силикагелем (элюент: дихлорметан : этилацетат = 100:0-10:1) с получением соединения А6-4. LCMS (ESI) масса/заряд: 237,1 [М+Н]. 1Н ЯМР (400 МГц, CDCl3) δ = 4,22 (s, 3Н), 4,21 (s, 2Н), 4,13 (s, 3Н).
Стадия 5: Синтез соединения А6-5
А6-4 (0,5 г) смешивали с N,N-диметилформамидом (10 мл) и затем добавляли пиридин гидрохлорид (1,22 г) в колбу и смесь продували азотом три раза. Смесь перемешивали при 100°С в течение 12 часов. Растворитель полностью удаляли при пониженном давлении. Остаток суспендировали с помощью 10 мл дихлорметана и смесь фильтровали. Осадок на фильтре высушивали при пониженном давлении с получением соединения А6-5. LCMS (ESI) масса/заряд: 209,1 [М+Н].
Стадия 6: Синтез соединения А6-6
Синтез промежуточного соединения А6-6, начиная с А6-5 подобен стадии четыре синтеза А2, за исключением того, что исходный материал А2-3 на стадии 4 был заменен на А6-5.
Стадия 7: Синтез соединения А6
Синтез промежуточного соединения А6, начиная с А6-6 подобен стадии четыре синтеза А2, за исключением того, что исходный материал А2-4 на стадии 5 был заменен на А6-6.
Промежуточное соединение А7
Путь синтеза
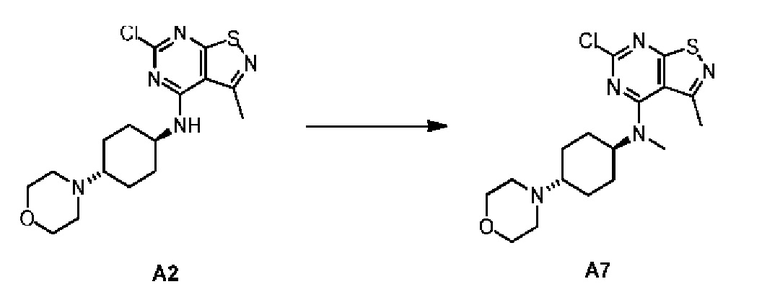
Соединение А2 (0,3 г) добавляли к N,N-диметилформамиду (3 мл) и смесь охлаждали до 0°С перед добавлением гидрида натрия (48,93 мг, чистота: 60%). Смесь перемешивали в течение 0,5 часа перед добавлением йодметана (150,47 мг). Смесь затем нагревали до 15°С с последующим перемешиванием в течение 2,5 часов. Полученный раствор добавляли в воду (80 мл) и экстрагировали этилацетатом (90 мл × 2). Органические фазы объединяли и промывали насыщенным солевым раствором (40 мл × 3). Затем органическую фазу дегидратировали с применением безводного сульфата натрия и фильтровали для удаления высушивающего средства. Фильтрат подвергали выпариванию под вакуумом с получением соединения А7. LCMS (ESI) масса/заряд: 382 [М+Н]+.
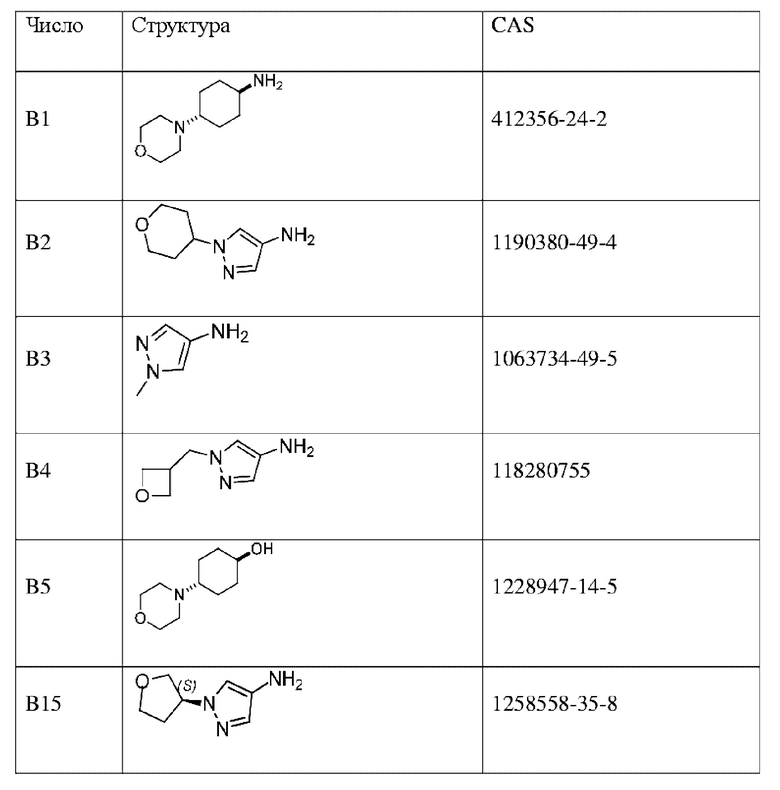
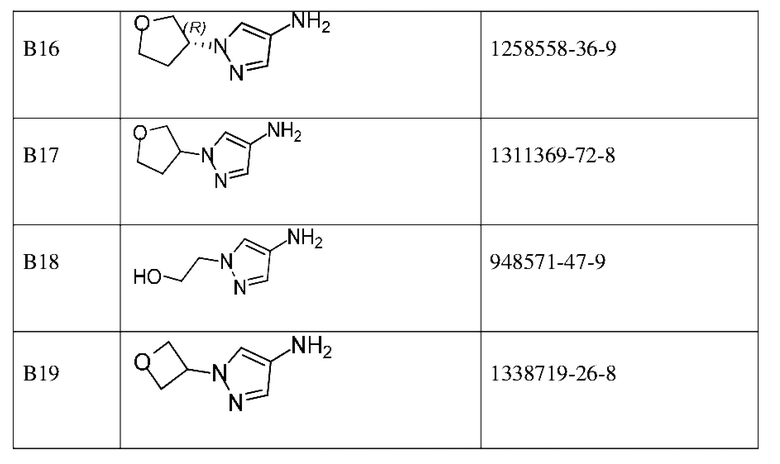
Промежуточные соединения в таблице ниже представляют собой коммерчески доступные реагенты.
Промежуточное соединение В20
Путь синтеза:

Стадия 1: Синтез соединения В20-1
2-Бромэтанол (40.52 г) и диэтилэфират трифторида бора (1.53 г) добавляли в толуол (80 мл). Смесь нагревали до 70°С, добавляли эпихлоргидрин (20 г) по каплям и инкубировали при 70°С в течение 1 часа. Реакционную систему охлаждали до 10°С. Водный гидроксид натрия (21,61 г, 100 мл) медленно добавляли по каплям перед инкубацией смеси при 25°С в течение 12 часов. Фазы разделяли. Водную фазу экстрагировали 2-метилтетрагидрофураном (20 мл × 3) и органические фазы объединяли. Объединенные органические фазы промывали водой (100 мл), дегидратировали, фильтровали и концентрировали при пониженном давлении с получением соединения В20-1.
Стадия 2: Синтез соединения В20-2
Водный гидроксид натрия (21,61 г, 150 мл) добавляли с соединением В20-1 (29,52 г) и инкубировали при 90°С в течение 1 часа перед охлаждением до 15°С. Затем по каплям добавляли раствор п-толуолсульфонилхлорида (41.21 г) в безводном тетрагидрофуране (150 мл) и смесь инкубировали при 25°С в течение 12 часов. Фазы разделяли. Водную фазу экстрагировали 2-метилтетрагидрофураном (100 мл × 2) и полученные органические фазы объединяли и добавляли 4 г диметиламинопиридина и 30 мл триэтиламина перед перемешиванием в течение 10 минут. Добавляли насыщенный водный хлорид аммония (200 мл) и органическую фазу отделяли и концентрировали с получением неочищенного продукта, который затем подвергали колоночной хроматографии (ISCO®; колонка 220 г; элюент: 0-80% этилацетат/петролейный эфир; скорость потока: 60 мл/мин) с получением соединения В20-2.
Стадия 3: Синтез соединения В20-3
Соединение В20-2 (2 г, 7,34 ммоль, 1 экв), 4-нитропиррол (1,25 г) и карбонат цезия (4,79 г) добавляли к N,N-диметилформамиду (20 мл) и смесь инкубировали при 70°С в течение 2 часов. Полученный раствор добавляли в воду (100 мл) и экстрагировали этилацетатом (180 мл). Полученную органическую фазу дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали на колонке с силикагелем (петролейный эфир : этилацетат = от 100:0 до 4:1) с получением соединения В20-3. LCMS (ESI) масса/заряд: 214.0 [М+Н]+
Стадия 4: Синтез соединения В20
Влажный палладий на угле (1 г, 10%) с метанолом (5 мл) и В20-3 (1,5 г) последовательно добавляли в колбу для гидрогенизации в атмосфере аргона. После трех продувок водородом (50 фунтов/кв. дюйм) смесь инкубировали при 30°С в течение 3 часов. Полученную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением соединения В20. LCMS (ESI) масса/заряд: 184,1 [М+Н]+
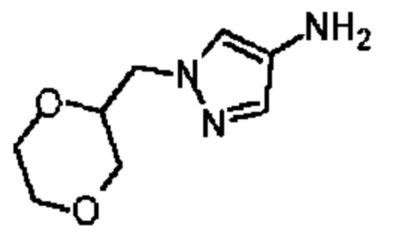
Промежуточное соединение В21
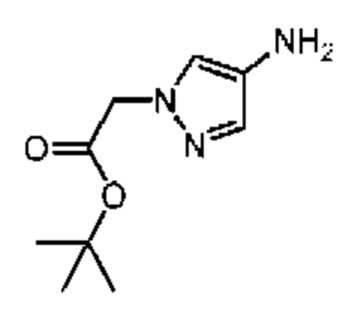
Путь синтеза
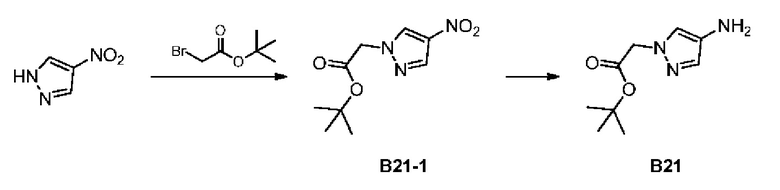
Стадия 1: Синтез соединения В21-1
4-Нитропиррол (10 г) и трет-бутилбромацетат (17,25 г) добавляли в реактор, содержащий ацетонитрил (100 мл), и затем добавляли карбонат калия (14,67 г). Смесь перемешивали при 80°С в течение 5 часов. Реакционную систему добавляли в 100 мл насыщенного водного хлорида аммония и экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли, дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонка 220 г; элюент: 0-60% этилацетат/петролейный эфир; скорость потока: 80 мл/мин) с получением соединения В21-1.
Стадия 2: Синтез соединения В21
В атмосфере аргона добавляли влажный палладий на угле (3 г, 10%) с метанолом (50 мл) в колбу для гидрогенизации и затем смешивали с В21-1 (10 г) с последующими тремя продувками водородом (50 фунтов/кв. дюйм). Смесь перемешивали при 25°С в течение 3 часов. Полученный раствор фильтровали через целит. Растворитель в фильтрате полностью удаляли при пониженном давлении и неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонка 12 г; элюент: 0-50% DCM/MeOH; скорость потока: 40 мл/мин) с получением соединения В21. LCMS (ESI) масса/заряд: 198,2[М+1]+
Промежуточное соединение В22
Путь синтеза
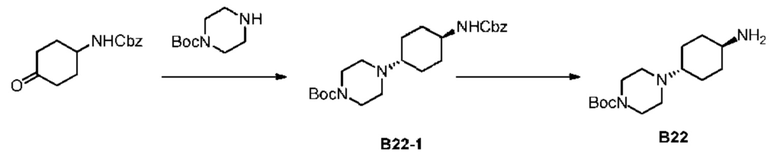
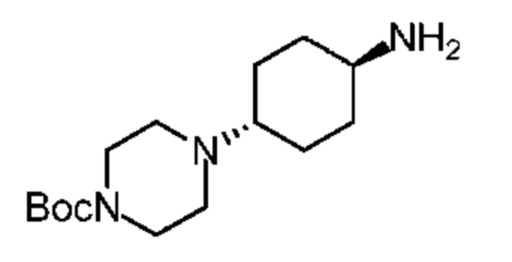
Стадия 1: Синтез соединения В22-1
Метанол (80 мл) добавляли с 4-N-бензилоксикарбониламиноциклогексаноном (5 г) и N-Вос-пиперазином (3,77 г) и затем медленно добавляли триацетоксиборгидрид натрия (6.43 г). Смесь перемешивали при 25°С в течение 12 часов. Растворитель полностью удаляли при пониженном давлении и остаток добавляли с дихлорметаном (100 мл) и водой (100 мл) для экстракции. Органическую фазу дегидратировали, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт подвергали распределительной хроматографии (колонка: Agela Durashell 10 мкм 250 × 50 мм; подвижная фаза: [вода (10 мкМ NH4HCO3)-ACN]; В%: 35% - 55%, 20 мин.) для разделения цис-транс-изомеров. (HPLC Shimadzu 20AD, X-bridge Shield RP18 2,1 × 50 мм, 5 мкм, подвижная фаза [вода + 10 ммоль/L-бикарбонат аммония-ацетонитрил]; В%: 10% - 80%, 4,5 мин., Rt = 2,615 мин) целевую фракцию концентрировали для удаления ацетонитрила и добавляли дихлорметан (500 мл) для экстракции. Органическую фазу дегидратировали, фильтровали и концентрировали при пониженном давлении с получением соединения В22-1. Продукт применяли на следующей стадии без очистки.
Стадия 2: Синтез соединения В22
В атмосфере аргона влажный палладий на угле (0,2 г, 10%) добавляли в колбу для гидрогенизации и последовательно добавляли метанол (5 мл) и соединение В22-1 (0,2 г) в реакционную систему, которую перемешивали в атмосфере водорода (50 фунтов/кв. дюйм) при 30°С в течение 2 часов. Реакционную систему фильтровали через целит, осадок на фильтре промывали метанолом (50 мл × 3) и растворитель удаляли из фильтрата при пониженном давлении с получением продукта В22. LCMS (ESI) масса/заряд: 284,1 [М+1]+
Промежуточное соединение В23
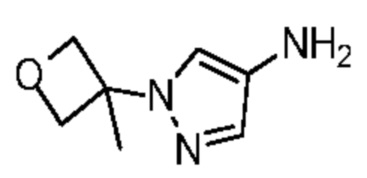
Путь синтеза:
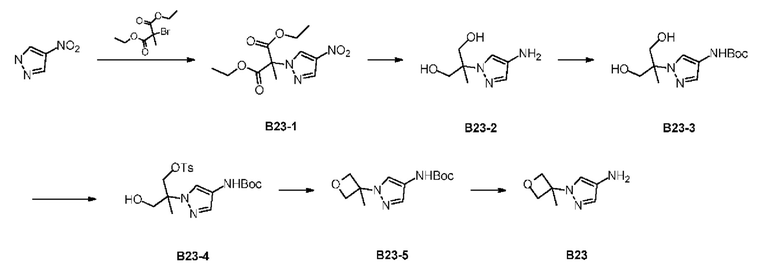
Стадия 1: Синтез соединения В23-1
4-Нитропиррол (1 г) и диэтил-2-бром-2-метилмалеат (2,69 г) растворяли в N,N-диметилформамиде (10 мл) и добавляли карбонат калия (2,44 г) для осуществления реакции. Смесь перемешивали при 100°С в течение 15 часов. Реакционную систему добавляли в этилацетат (20 мл) и промывали полунасыщенным солевым раствором (20 мл). Органическую фазу дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонка 20 г SepaFlash® Silica Flash, элюент 0-60%, градиент DCM/MeOH @ 40 мл/мин) с получением соединения В23-1. LCMS (ESI) масса/заряд: 286,1 [М+1]+
Стадия 2: Синтез соединения В23-2
Соединение В23-1 (1 г) растворяли в метаноле (10 мл) и добавляли боргидрид натрия (265,26 мг). Реакционную систему перемешивали при 25°С в течение 2 часов. В реакционную систему добавляли 2 мл насыщенного хлорида аммония, оставляли до удаления пузырьков и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонки 20 г; элюент: 0-50% DCM/MeOH, 40 мл/мин.) с получением соединения В23-2. LCMS (ESI) масса/заряд: 172,1 [М+1]+
Стадия 3: Синтез соединения В23-3
Соединение В23-2 (500 мг), ди-трет-бутилдикарбонат (956,12 мг) и триэтиламин (886.61 мг) растворяли в безводном тетрагидрофуране (10 мл) и смесь перемешивали при 40°С в течение 3 часов. Реакционную систему концентрировали при пониженном давлении и неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; 20 г; элюент: 0-50% DCM/MeOH @ 30 мл/мин.) с получением соединения В23-3. LCMS (ESI) масса/заряд: 272,1 [М-100+1]+
Стадия 4: Синтез соединения В23-4
Соединение В23-3 (340 мг) растворяли в смеси пиридина (10 мл) и дихлорметана (20 мл). Раствор охлаждали до 0°С и затем добавляли п-толуолсульфонилхлорид (238,91 мг, 1,25 ммоль, 1 экв). Смесь перемешивали при 0°С в течение 3 часов. Полученную смесь концентрировали и очищали с помощью хроматографии (колонка: Welch Xtimate С18 150×25 мм × 5 мкм; подвижная фаза: [вода (10 мкМ NH4HCO3)-ACN]; В%: 38% - 68%, 10,5 мин) с получением соединения В23-4. LCMS (ESI) масса/заряд: 426,2[М+1]+
Стадия 5: Синтез соединения В23-5
Соединение В23-4 (75 мг) растворяли в безводном тетрагидрофуране (3 мл) и добавляли гидрид натрия (21,15 мг, 60% чистота) при 0°С.Реакционную систему нагревали до 67°С и перемешивали в течение 2 часов. Реакцию гасили посредством добавления воды (0,1 мл) и непосредственно перемешивали с помощью мешалки для смешивания с силикагелем. Неочищенный продукт очищали с помощью колоночной хроматографии (ISCO®; колонка 12 г; элюент: 0-80% этилацетат/петролейный эфир; 20 мл/мин) с получением соединения В23-5. LCMS (ESI) масса/заряд: 254,2[М+1]+
Стадия 6: Синтез соединения В23
Соединение В23-5 (20 мг) растворяли в дихлорметане (1,5 мл) и добавляли трифторуксусную кислоту (1,5 мл). Смесь перемешивали при 25°С в течение 1 часа. Растворитель из полученной смеси удаляли при пониженном давлении с получением соединения В23. LCMS (ESI) масса/заряд: 154,2[М+1]+
Синтез промежуточных соединений в таблице ниже подобен таковому для промежуточного соединения В22, за исключением того, что N-Вос-пиперазин был заменен на исходный материал один.
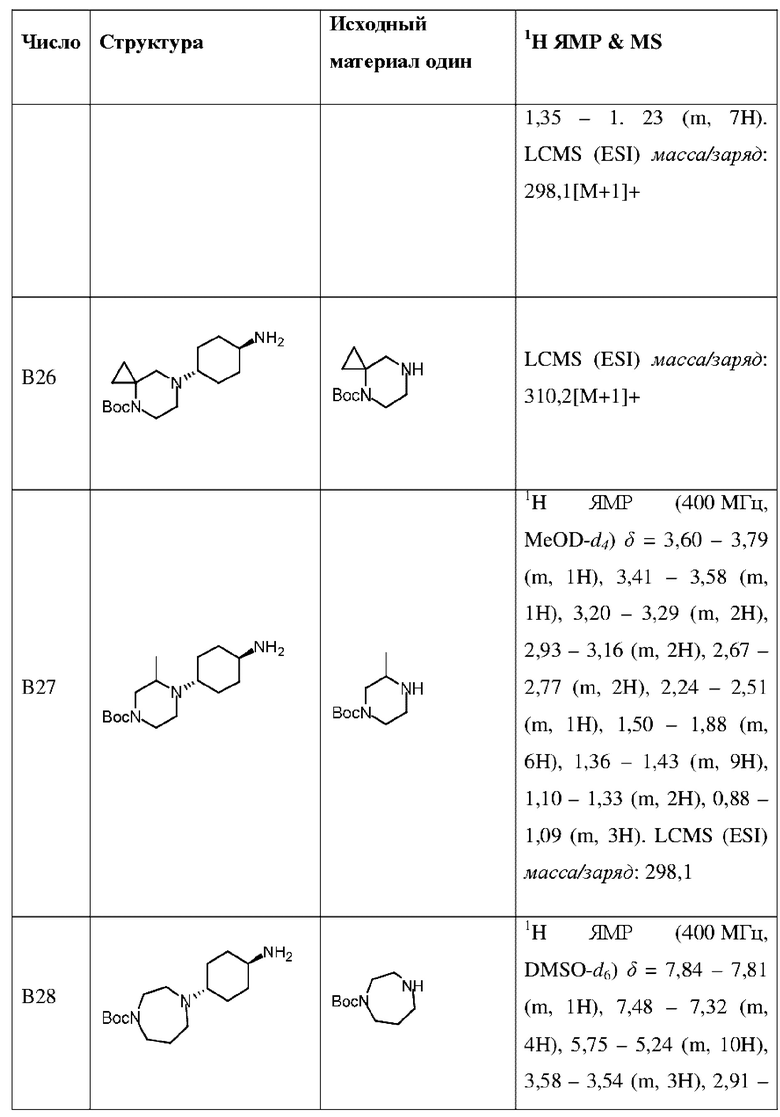
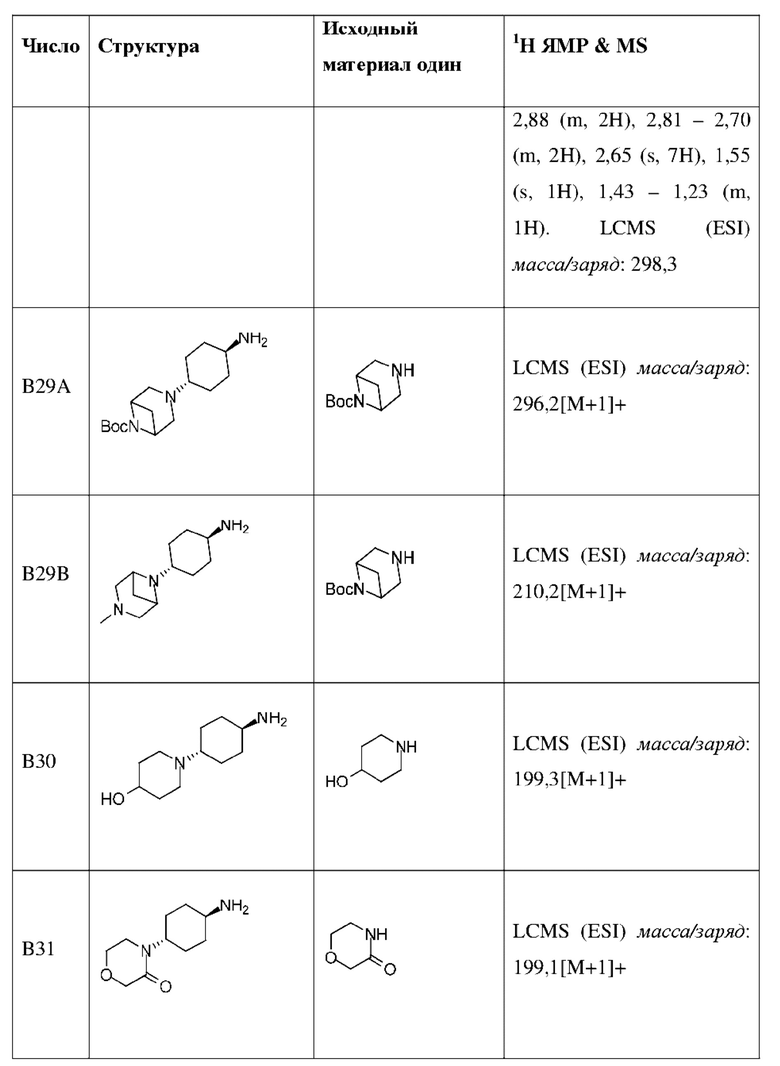

Промежуточное соединение В35
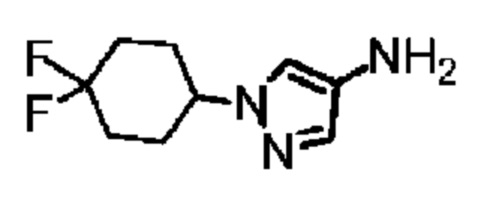
Путь синтеза:
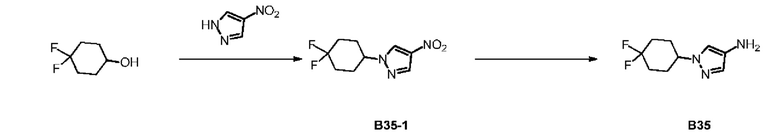
Стадия 1: Синтез соединения В35-1
Диизопропилазодикарбоксилат (854,05 мг) добавляли в раствор 4,4-дифторциклогексанола (0,5 г), 4-нитропиразола (415,29 мг) и трифенилфосфина (1,06 г) в тетрагидрофуране (20 мл) и смесь перемешивали при 20°С в течение 12 часов. Растворитель полностью удаляли при пониженном давлении и неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир: петролейный эфир : этилацетат = 3:1) с получением соединения В35-1.
Стадия 2: Синтез соединения В35
Соединение В35-1 (2 г) добавляли в метанол (70 мл) и добавляли влажный палладий на угле (1 г, чистота: 10%). После трех продувок водородом смесь перемешивали при 30°С в течение 2 часов в атмосфере водорода (30 фунтов/кв. дюйм) и фильтровали через целит. Растворитель из фильтрата полностью удаляли при пониженном давлении с получением соединения В35. LCMS (ESI) масса/заряд: 202,2[М+Н].
Эталонный пример 1: WXR1
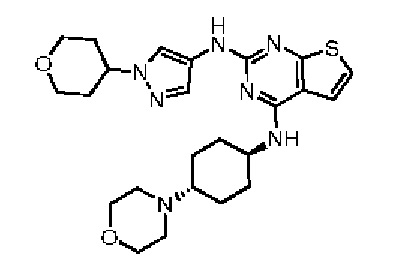
Соединение WXR1 синтезировали в соответствии с путем, указанным в патенте № WO 2017205762.
Эталонный пример 2: WXR2
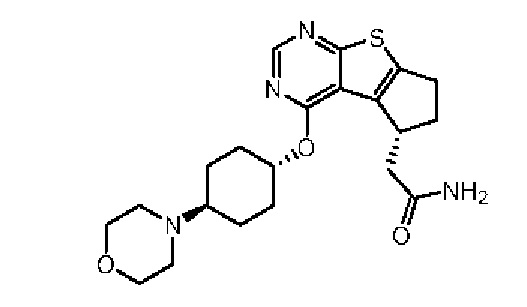
Соединение WXR2 (ND-2110) синтезировали в соответствии с путем, указанным в патенте № WO 2012097013.
Эталонный пример 3: WXR3
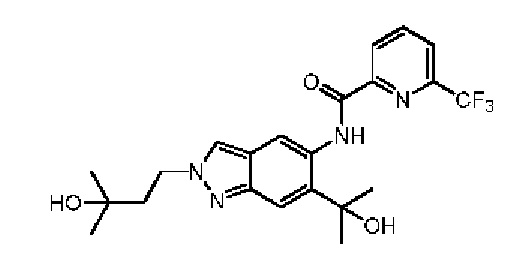
Соединение WXR33 (BAY-1830839) синтезировали в соответствии с путем, указанным в патенте № WO 2017186700.
LCMS (ESI) масса/заряд: 451,2[М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 12,35 (s, 1Н), 8,71 (s, 1Н), 8,45 (d, J=7,6 Гц, 2Н), 8,37 (t, J=6,0 Гц, 1Н), 8,16 (d, J=6,8 Гц, 1Н), 7,57 (s, 1Н), 5,93 (d, J=8,0 Гц, 1Н), 4,51 (s, 1Н), 4,49 - 4,45 (m, 2Н), 2,05 - 2,01 (m, 2Н),1,62 (s, 6Н), 1,15 (s,6H)
Эталонный пример 4: WXR4
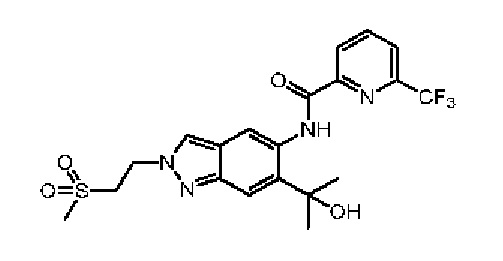
Соединение WXR4 (BAY-1834845) синтезировали в соответствии с путем, указанным в патенте № WO 2017186689.
LCMS (ESI) масса/заряд: 471,2[М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 12,37 (s, 1Н), 8,73 (s, 1Н), 8,48-8,42 (m, 2Н), 8,40 - 8,32 (m, 1Н), 8,16 (d, J=7,8 Гц, 1Н), 7,59 (s, 1H),5,99(s, 1Н), 4,86 (t, J=6,6 Гц, 2Н), 3,85 (t, J=6,6 Гц, 2Н), 2,90 (s, 3Н), 1,62 (s, 6Н).
Пример 1: Синтез соединения WX001
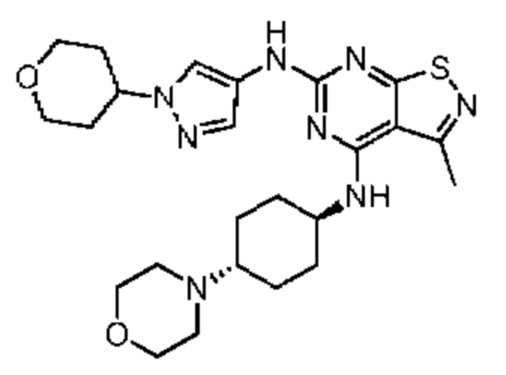
Путь синтеза:
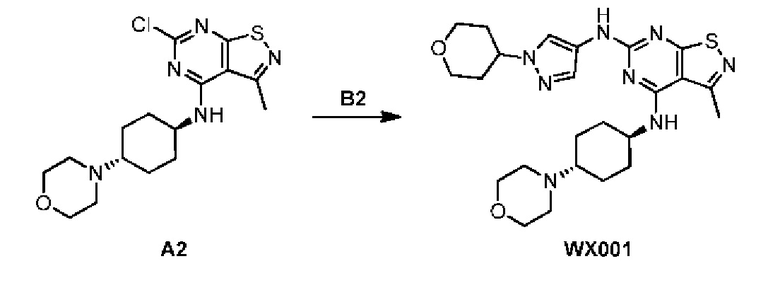
Пример 1: Синтез соединения WX001
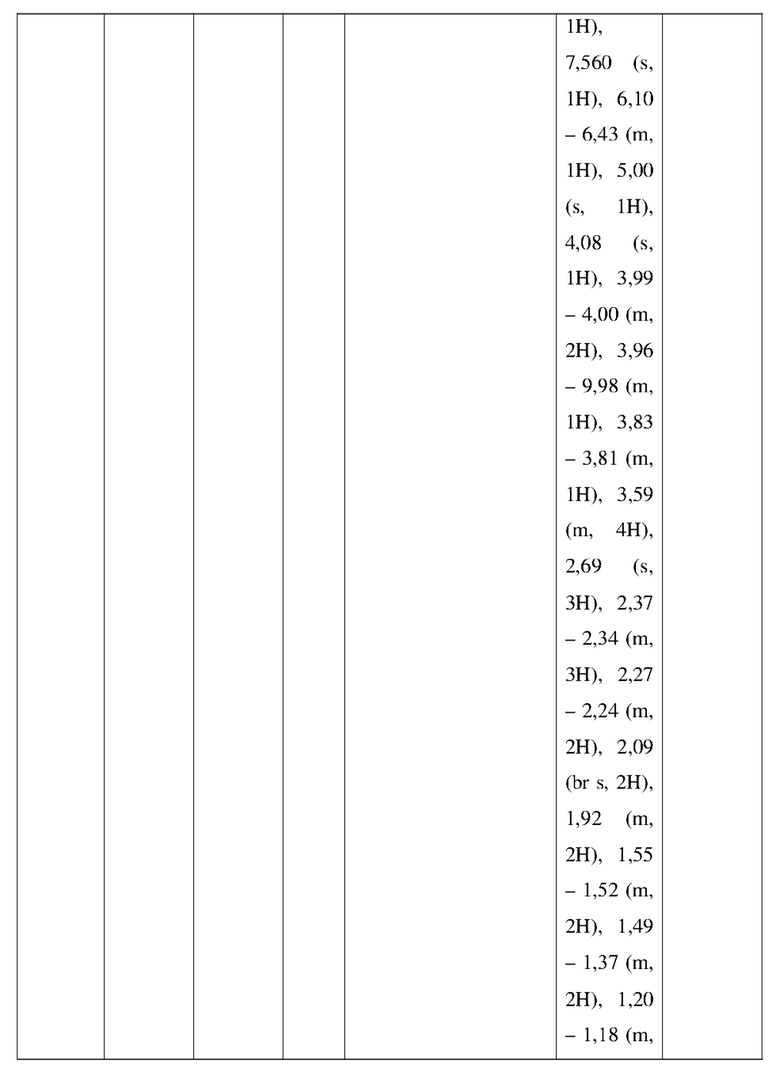
Промежуточное соединение А2 (0,15 г) растворяли в н-бутаноле (1 мл) с последующим добавлением смеси хлористоводородная кислота/этилацетат (4 М, 1,02 мл) и промежуточного соединения В2 (81,81 мг). Смесь перемешивали при 120°С в течение 16 ч. Полученную смесь фильтровали. Осадок на фильтре промывали этилацетатом (2 мл × 3) и осадок на фильтре высушивали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной хроматографии (дихлорметан : метанол = 10:1) с получением неочищенного соединения WX001, которое суспендировали с 2 мл метанола с получением соединения WX001.
LCMS (ESI) масса/заряд: 499,2 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 9,05 (s, 1Н), 7,90 (s, 1Н), 7,55 (s, 1Н), 6,10 (d, J=7,2 Гц, 1Н), 4,40-4,31 (m, 1Н), 4,16-4,05 (m, 1Н), 4,01-3,94 (m, 2Н), 3,83-3,77 (m, 1Н), 3,63-3,57 (m, 4Н), 3,48 (dt, J=3,2, 11,2 Гц, 2Н), 2,68 (s, 3Н), 2,34 - 2,24 (m, 2Н), 2,18-2,10 (m, 2Н), 2,03 - 1,91 (m, 7Н), 1,60 - 1,48 (m, 2Н), 1,44 -1,32 (m, 2Н).
Пример 2: Синтез соединения WX002
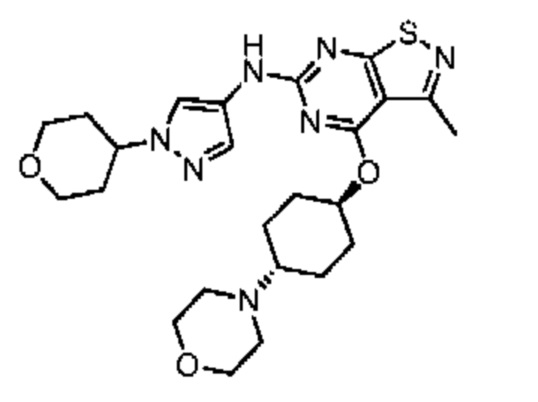
Путь синтеза:
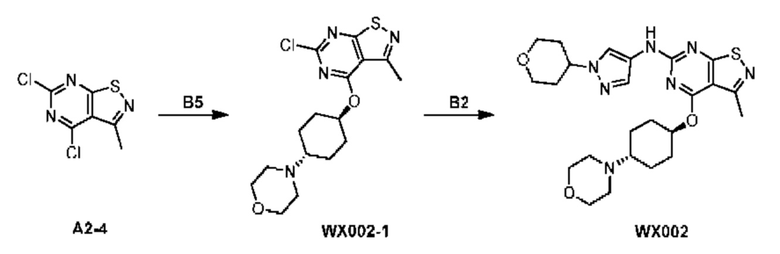
Синтез соединения WX002, начиная с промежуточных соединений А2-4, В5 и В2, подобен стадии 5 синтеза А2 и стадиям синтеза соединения WX001.
LCMS (ESI) масса/заряд: 500,2 [М+Н]+. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ = 8,03 (s, 1Н), 7,64-7,37 (m, 1Н), 7,09-6,83 (m, 1Н), 4,27 (td, J=8,0, 15,7 Гц, 1Н), 4,15-4,12 (m, 2Н), 4,06 (br d, J=11,8 Гц, 2H), 3,74 - 3,72(m, 2Н), 3,54 - 3,40 (m, 2Н), 2,60 (s, 3Н), 2,52 (br s, 3Н), 2,56-2,47 (m, 1H), 2,24 (br, J=13,1 Гц, 2H), 2,06 (br s, 4H), 1,97 (br d, J=10,8 Гц, 2H), 1,58 - 1,49 (m, 2H), 1,46 - 1,38 (m, 2H), 0,84 - 0,74 (m, 2H).
Пример 3: Синтез соединения WX003
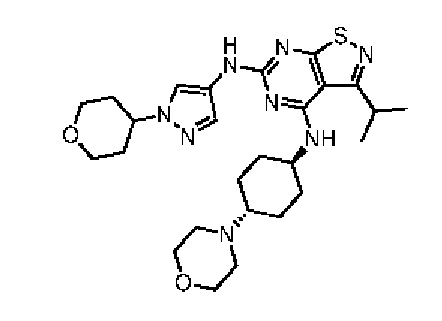
Путь синтеза:
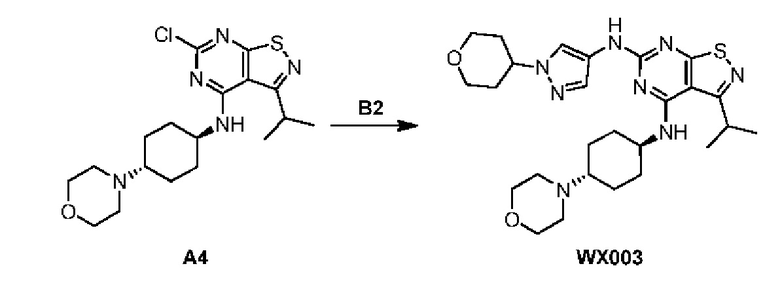
Синтез соединения WX003, начиная с промежуточных соединений А4 и В2, подобен стадиям синтеза соединения WX001.
LCMS (ESI) масса/заряд: 527,2 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 9,04 (br s, 1Н), 7.88 (s, 1Н), 7,52 (s, 1Н), 6,06 (br s, 1H), 4,38 - 4,27 (m, 1 H), 4,20 - 4,08 (m, 1 H), 4,00 - 3.89 (m, 2H), 3,60-3,40 (m, 11 H), 2,33-2,22 (m, 1H), 2,15-2,05 (m, 2H), 2,03- 1,87 (m, 6H), 1,60 - 1,46 (m, 2H), 1,42 - 1,25 (m, 8H).
Пример 4: Синтез соединения WX004
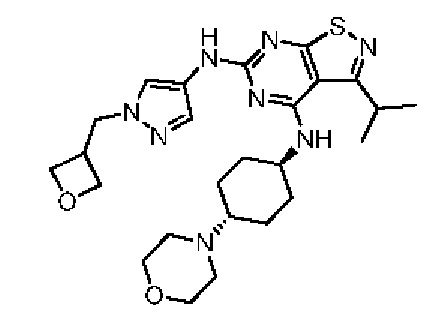
Путь синтеза:
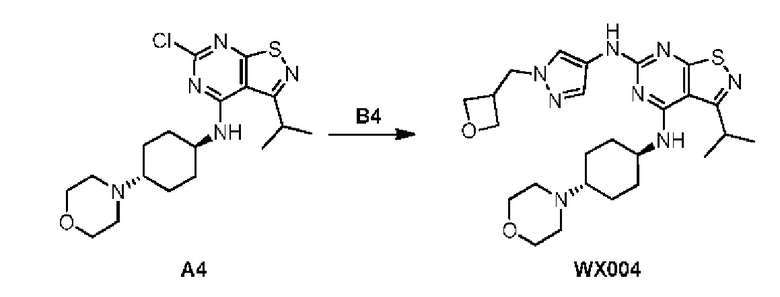
Синтез соединения WX004, начиная с промежуточных соединений А4 и В4, подобен стадиям синтеза соединения WX001.
LCMS (ESI) масса/заряд: 513,3 [М+Н]+. 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ = 8,60 (s, 2Н), 4,89 - 4,79 (m, 2Н), 4,73 - 4,60 (m, 2Н), 4,45 - 4,33 (m, 1Н), 4,14 - 4,05 (m, 2Н), 4,02 - 3,91 т, 2Н), 3,82 (d, J=5,2 Гц, 2Н), 3,72 - 3,52 (m, 5Н), 3,28 - 3,18 (m, 2Н), 2,44 - 2,25 (m, 4Н), 2,25 - 2,25 (m, 1Н), 1,94 - 1,73 (m, 4Н), 1,39 (d, J=6,8 Гц, 6Н)
Пример 5: Синтез соединения WX005
Путь синтеза:
Синтез соединения WX005, начиная с промежуточных соединений А5 и В3, подобен стадиям синтеза соединения WX001.
LCMS (ESI) масса/заряд: 499,3 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 9,54 - 9,22 (m, 1Н), 8,04 - 7,70 (m, 1Н), 7,50 (br s, 1Н), 6,51 - 6,09 (m, 1Н), 4,12 - 4,00 (br s, 1 H), 3,96 -3,86 (m, 3Н), 3,80 (s, 3Н), 3,72 - 3,49 (m, 7H), 2,67 (s, 3Н), 2,32 - 2,22 (m, 1H), 2,17 - 2,07 (m, 2H), 1,99 - 1,82 (m, 4H), 1,81 - 1,65 (m, 2H), 1,60 - 1,45 (m, 2H), 1,37 (br s, 2H)
Ссылаясь на процедуры синтеза в примере 1, соединения в следующих примерах синтезировали, начиная с фрагмента 1 и фрагмента 2 в следующей таблице.
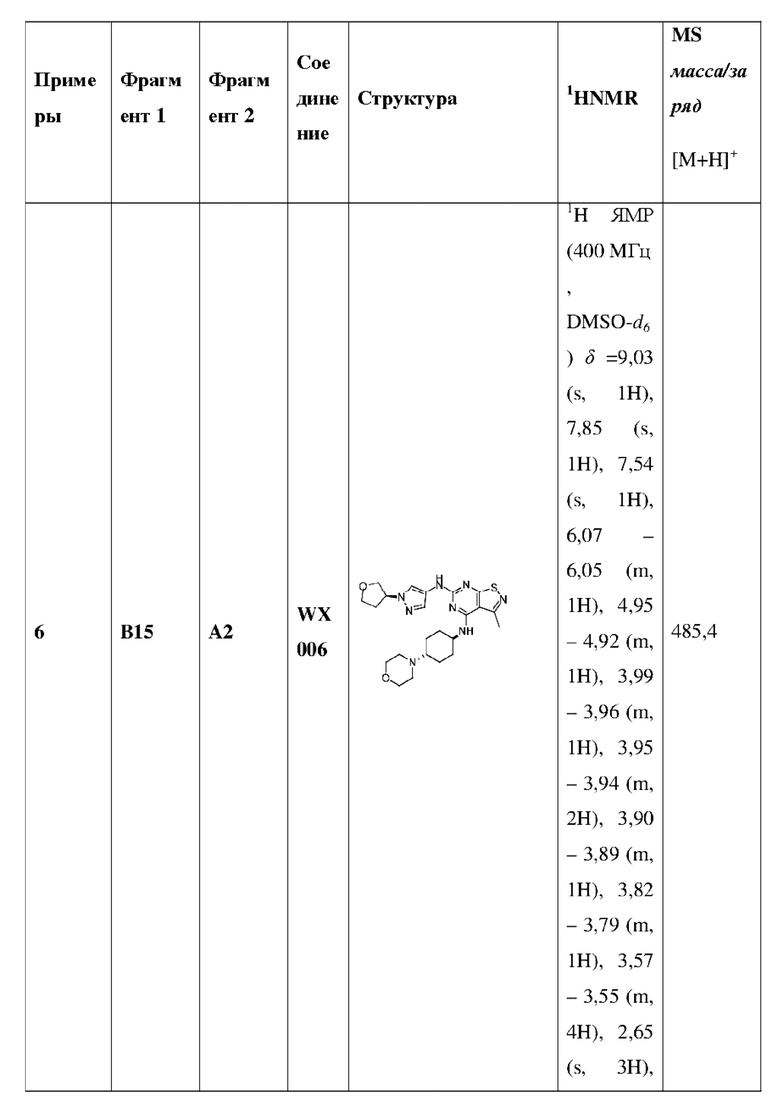

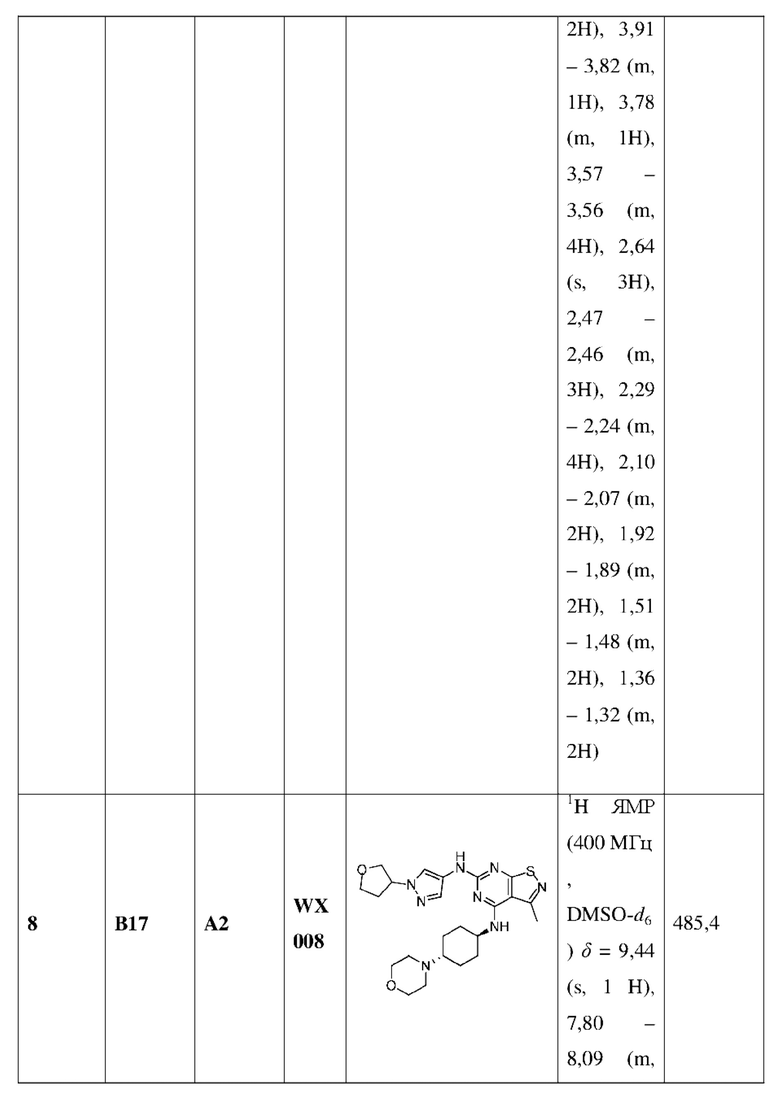
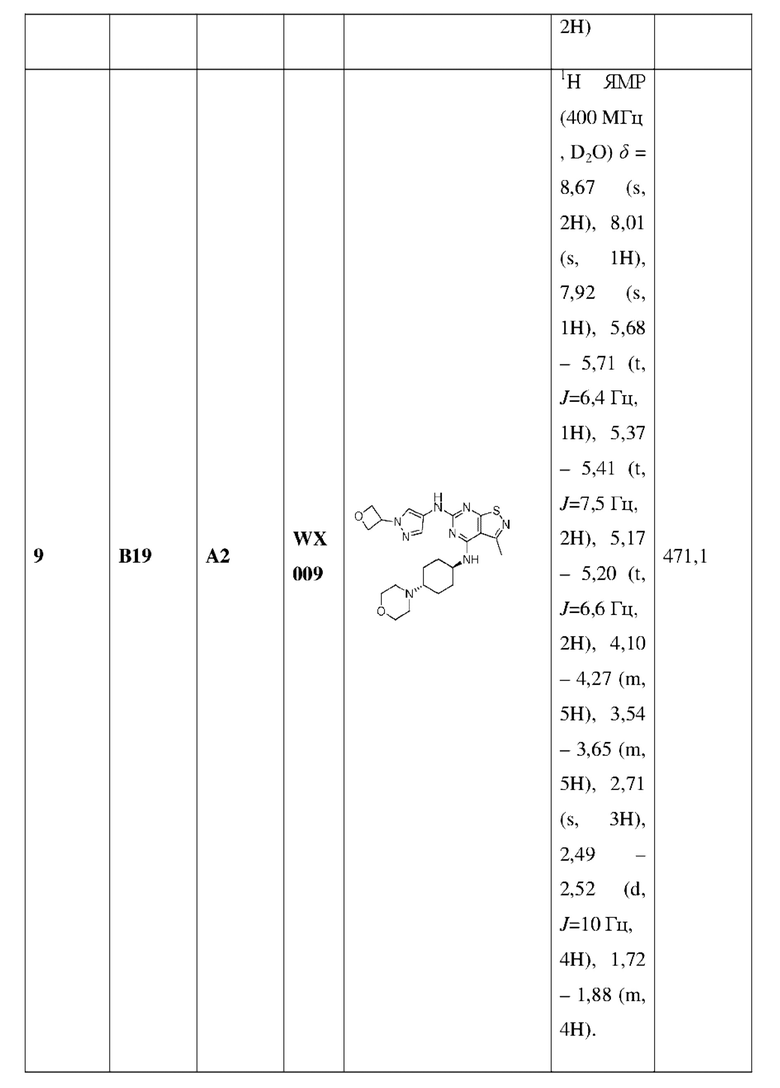
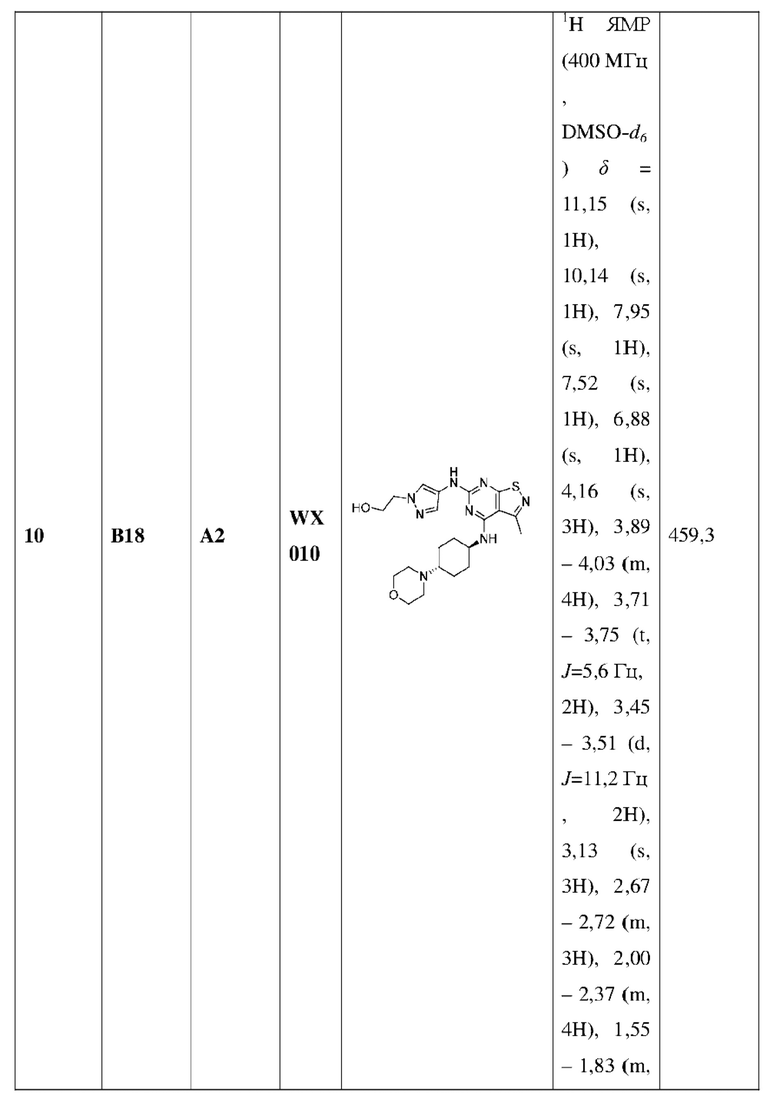
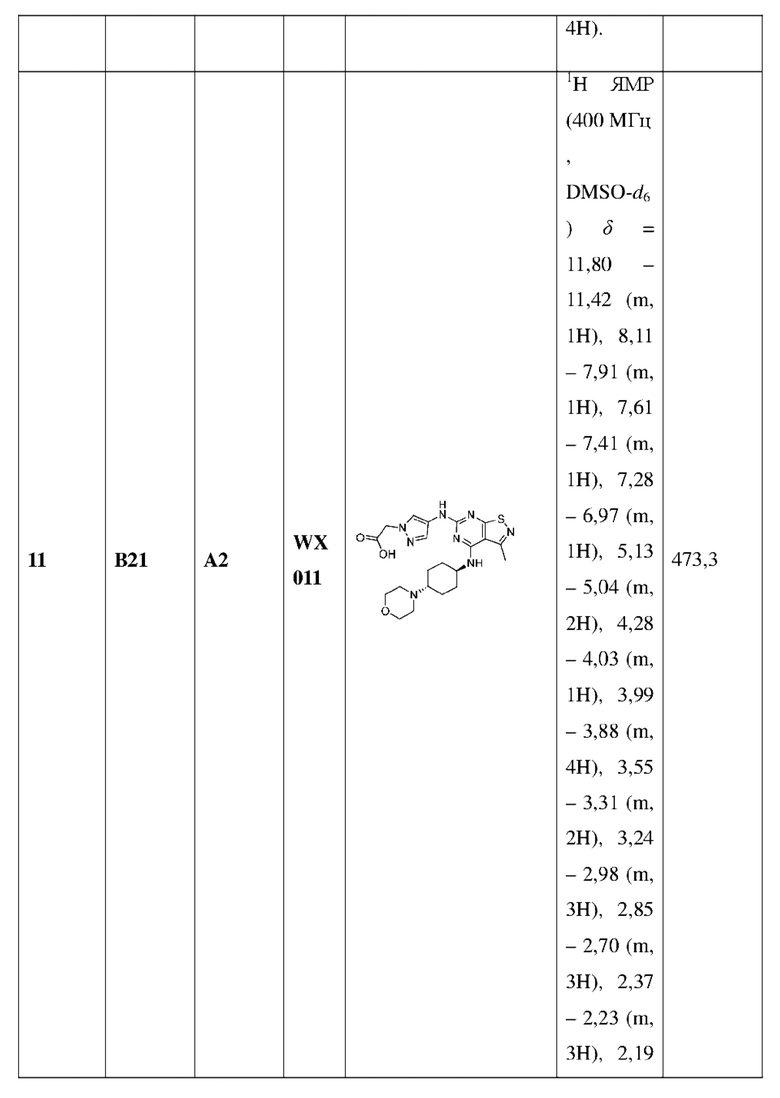

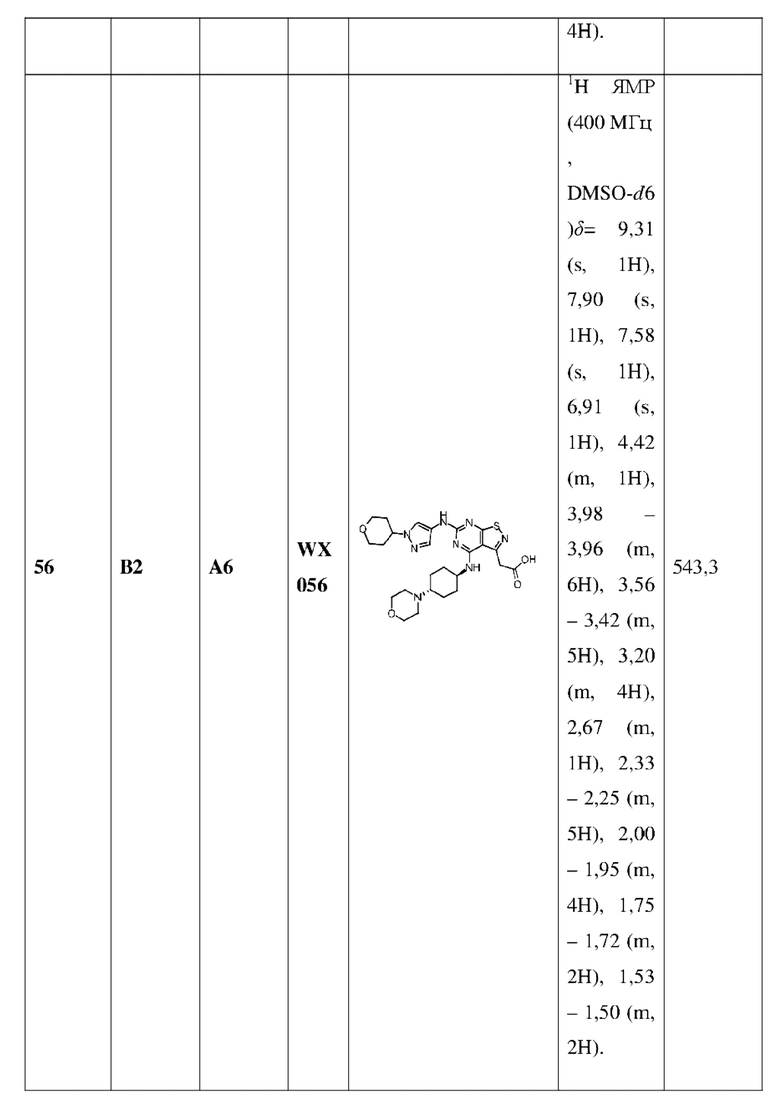
Пример 25: Синтез соединения WX025
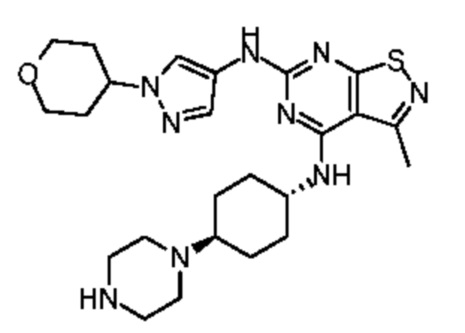
Путь синтеза:
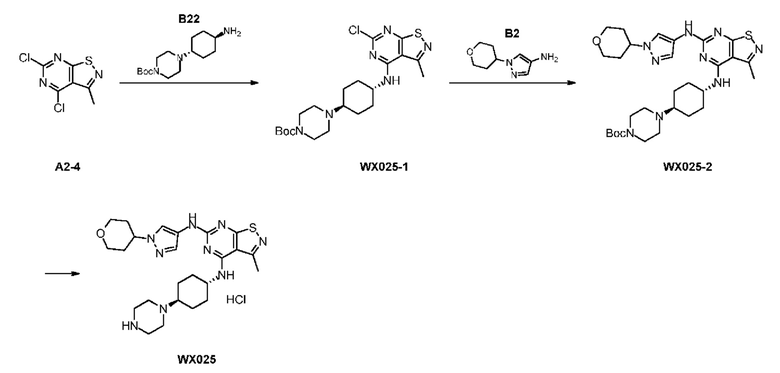
Стадия 1:
Ссылаясь на синтез промежуточного соединения А2, соединение WX025-1 синтезировали, начиная с промежуточных соединений А2-4 и В22.
Стадия 2:
Ссылаясь на синтез WX001, соединение WX025-2 синтезировали, начиная с промежуточных соединений WX025-1 и В2.
Стадия 3: Синтез соединения WX025
Соединение WX025-2 (0,18 г) смешивали с раствором хлористоводородной кислоты в метаноле (15 мл, 4 М) в колбе. Смесь перемешивали при 15°С в течение 12 часов и реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии (колонка: Boston PR1me С18 150×30 мм × 5 мкм; подвижная фаза: [вода (0,05% HCl)-ACN]; В%: 1%-22%, 10 мин) с получением соединения WX025. LCMS (ESI) масса/заряд: 498,1 [М+Н]. 1Н ЯМР (400 МГц, MeOD-d4) δ = 7.99 (s, 1H), 7,80-7,65 (m, 1H), 4,49 - 4,23 (m, 2H), 4,10-4,08 (d, J=8,0 Гц, 2H), 3,72-3,50 (m, 11H), 2,80 (s, 3H), 2,40-2,36 (m, 4H), 2,10 (s, 4H), 1,83-1,75 (m, 4H).
Пример 26: Синтез соединения WX026
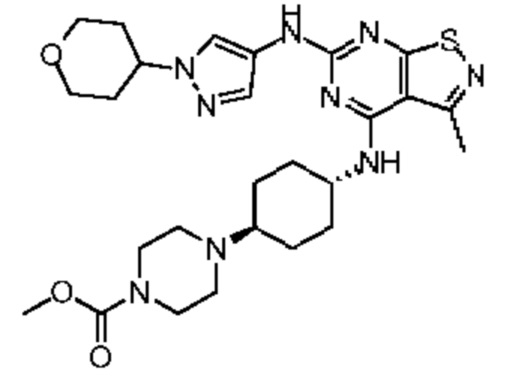
Путь синтеза:
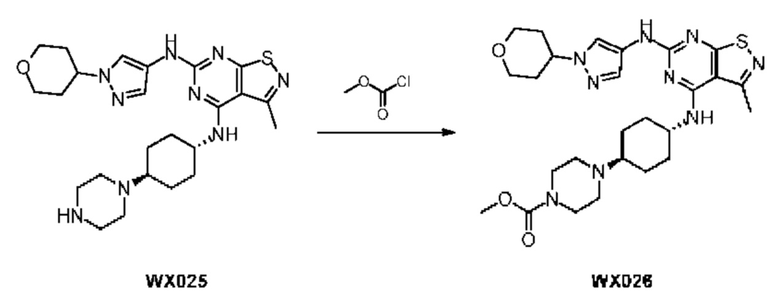
Стадия 1: Синтез соединения WX026
В реакционную колбу добавляли соединение WX025 (0,03 г, соль HCl) и дихлорметан (2 мл) с последующим добавлением триэтиламина (14,21 мг). Смесь перемешивали и добавляли по каплям метилхлорформиат (6,37 мг, 5,22 мкл). Полученную смесь перемешивали при 15°С в течение 2 часов. Реакцию гасили 30 мл воды и добавляли этилацетат (30 мл × 2) для экстракции. Объединенные органические фазы дегидратировали с применением безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью хроматографа (колонка: Xtimate С18 150x25 ммх5 мкм; подвижная фаза: [вода (0,04% NH3H2O + 10 мМ NH4HCO3)-ACN]; В%: 30-55%, 10,5 мин) с получением соединения WX026. LCMS (ESI) масса/заряд: 556,4 [М+Н], 1Н ЯМР (400 МГц, CDCl3) δ = 8,00 (s, 1Н), 7,55 (s, 1Н), 6,81 (s, 1Н), 5,19 (s, 1Н), 4,34 (m, 1Н), 4,14 - 4,11 (m, 3Н), 3,72 (s, 3Н), 3,55 - 3,50 (m, 6Н), 2,70 (s, 3Н), 2,56 (s, 4Н), 2,41 - 2,30 (m, 3Н), 2,13 (s, 4Н), 2,01 - 1,98 (m, 2Н), 1,50- 1,44(m, 2Н), 1,33-1,30 (m, 2Н).
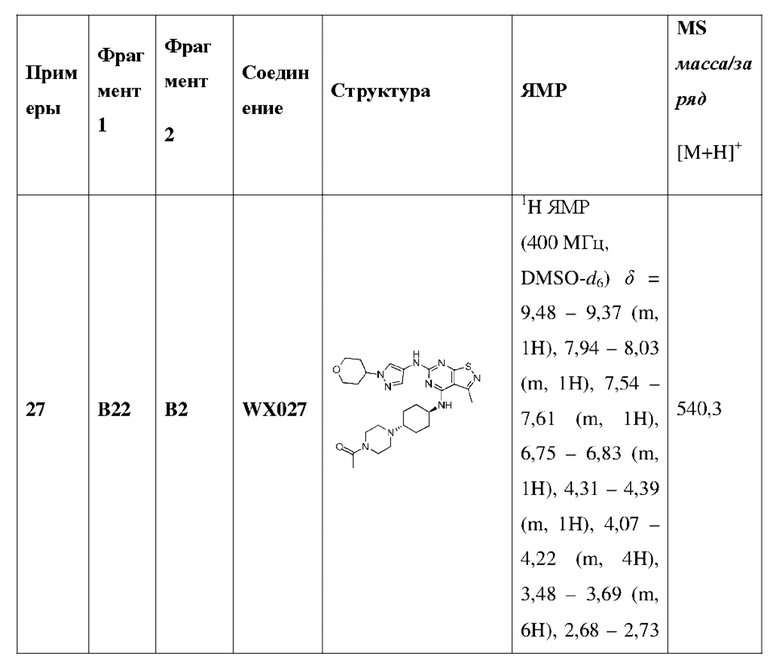
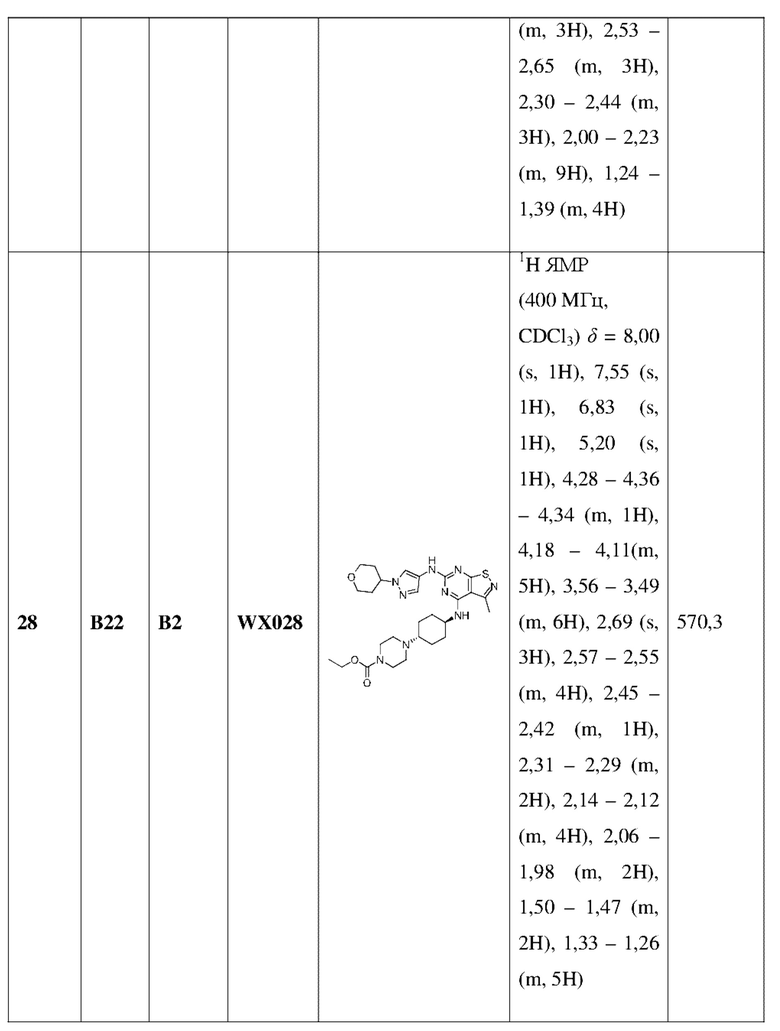
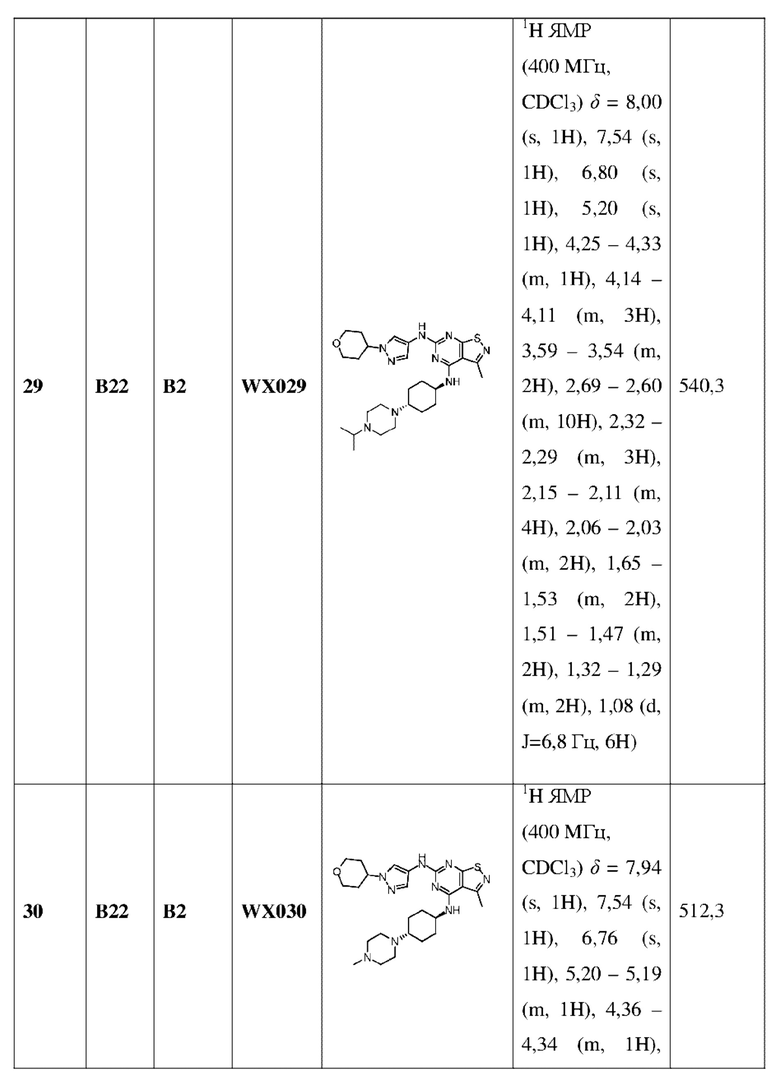
Ссылаясь на процедуры синтеза в примере 25 и примере 26, соединения в следующих примерах синтезировали, начиная с промежуточного соединения А2-4, фрагмента 1 и фрагмента 2 в следующей таблице.

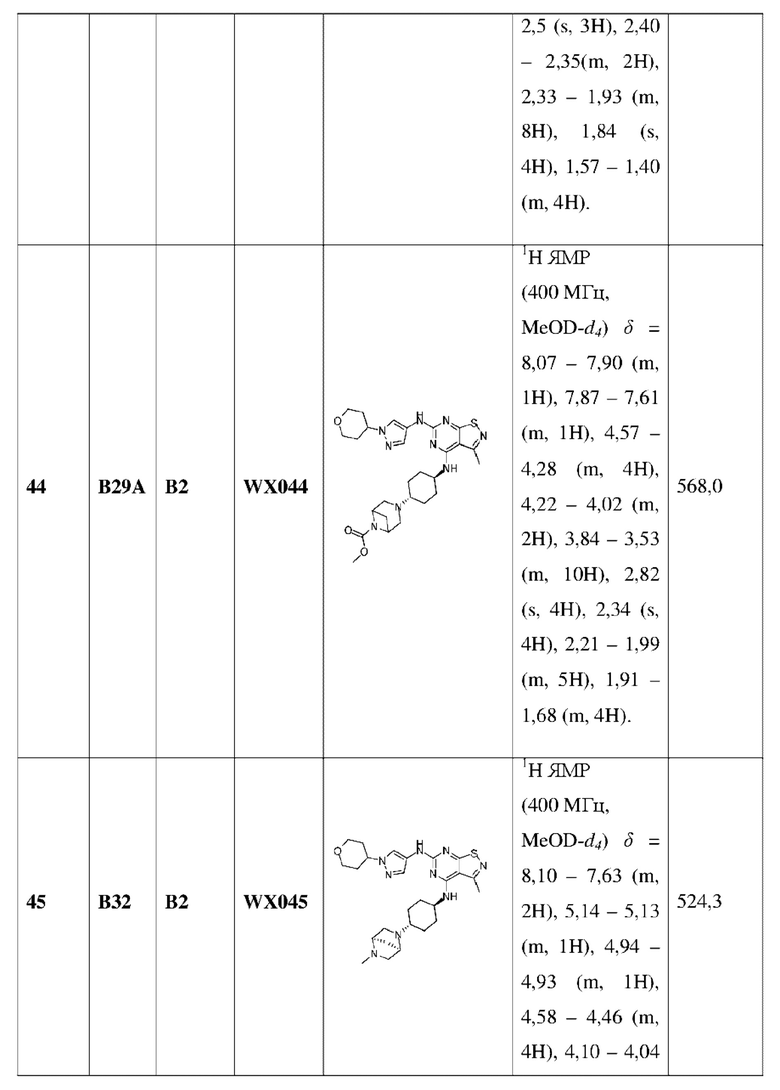
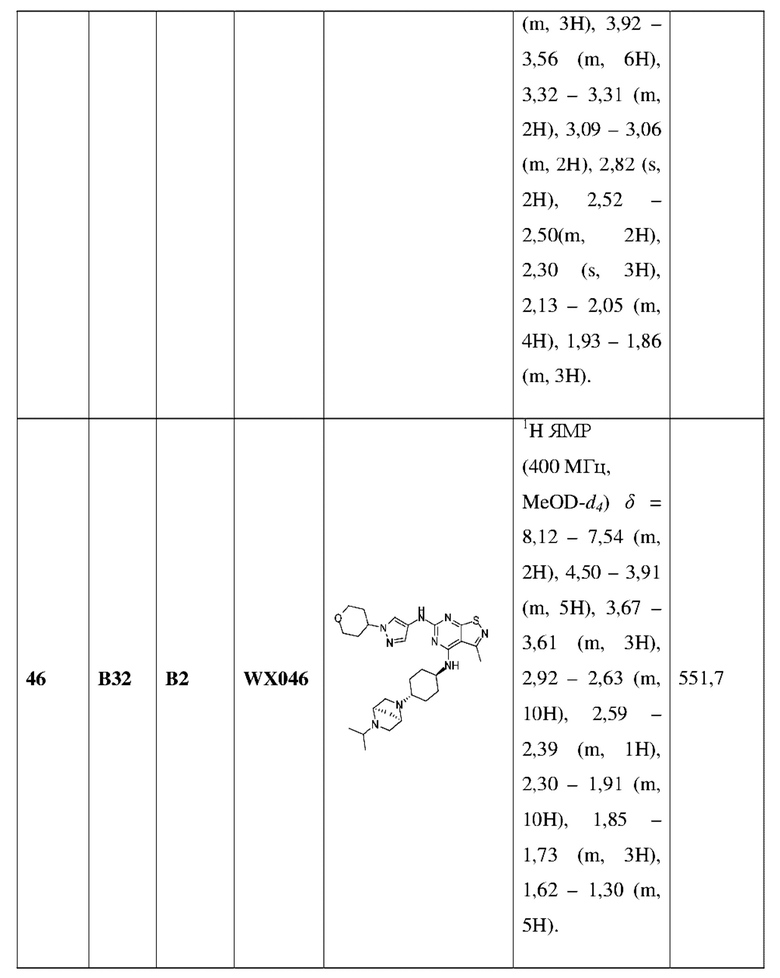
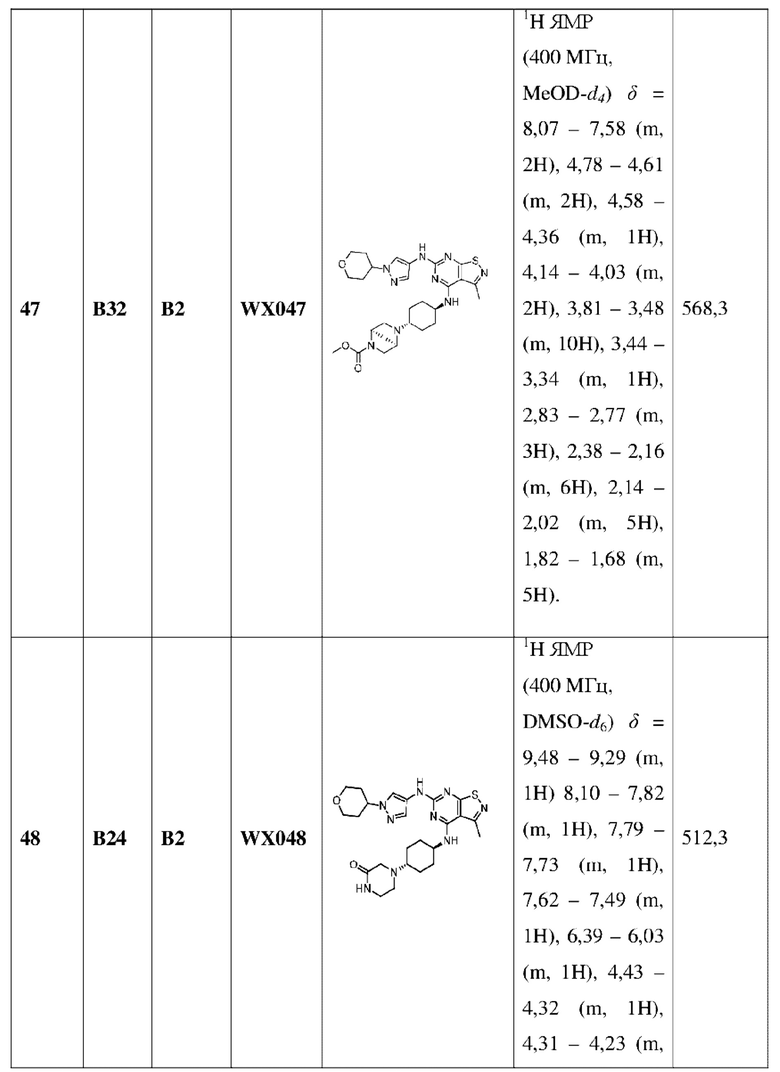
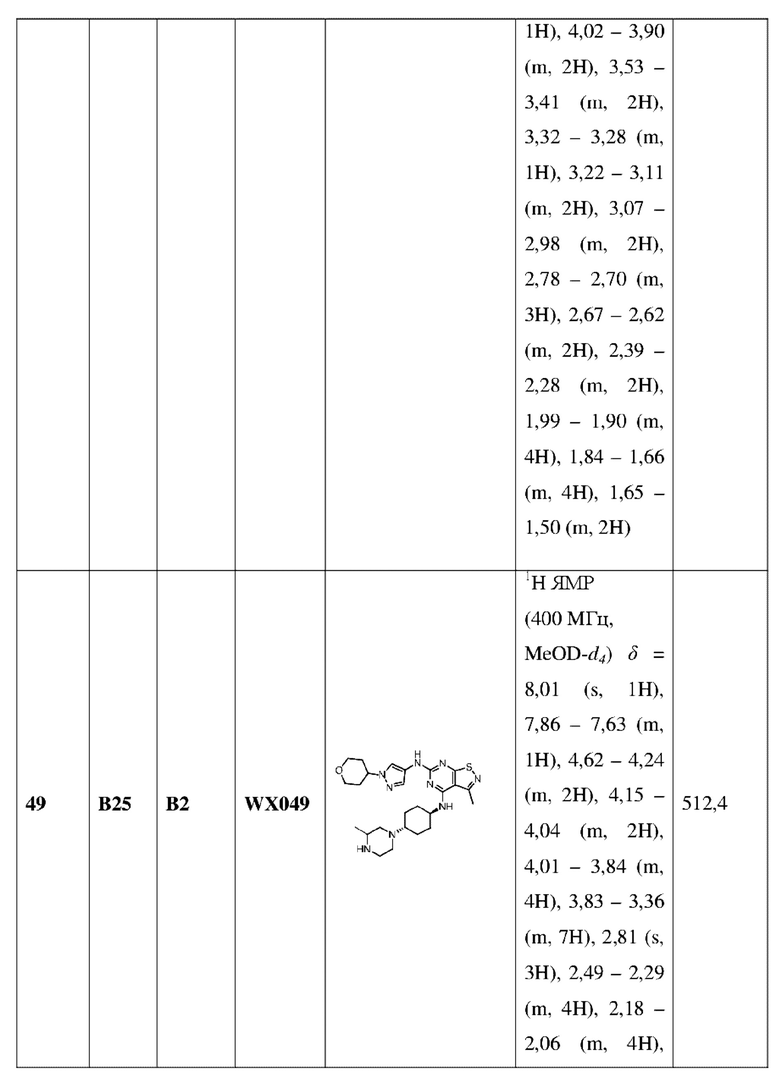

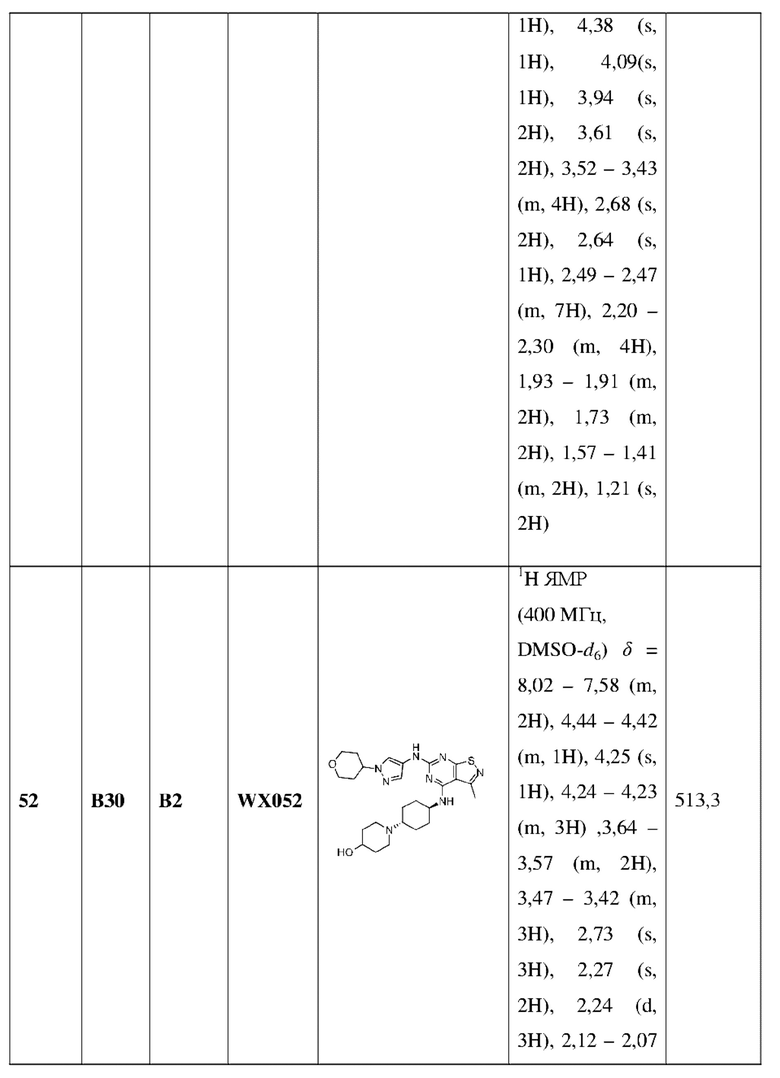
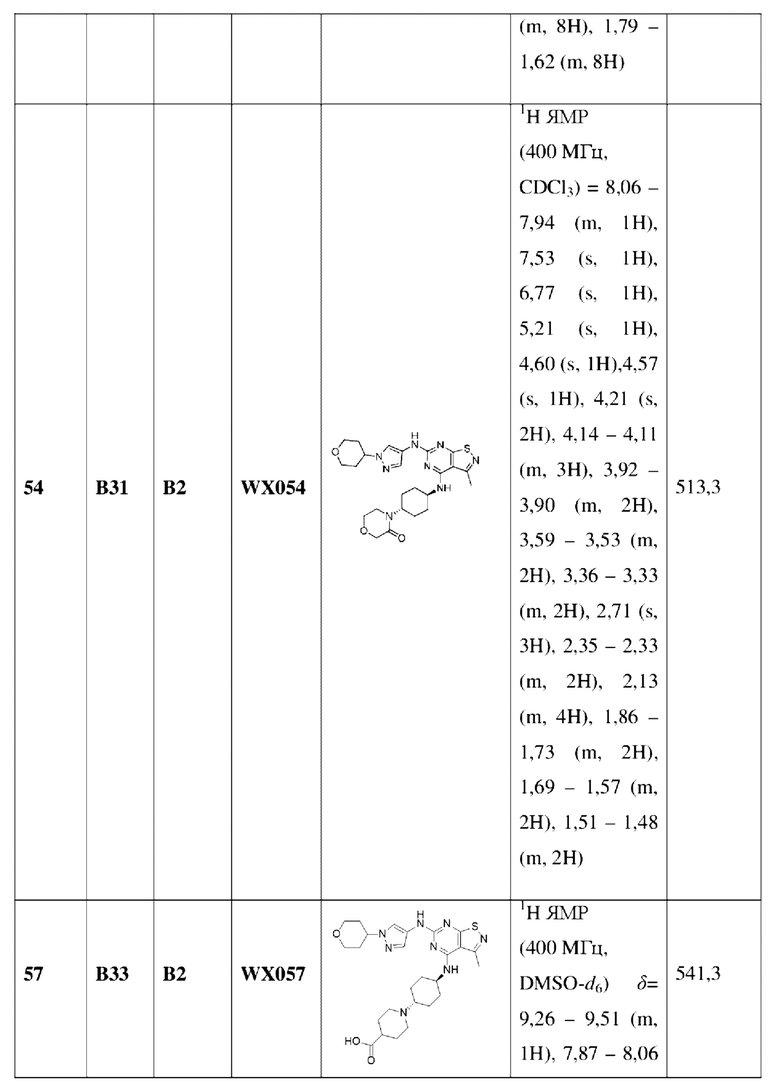
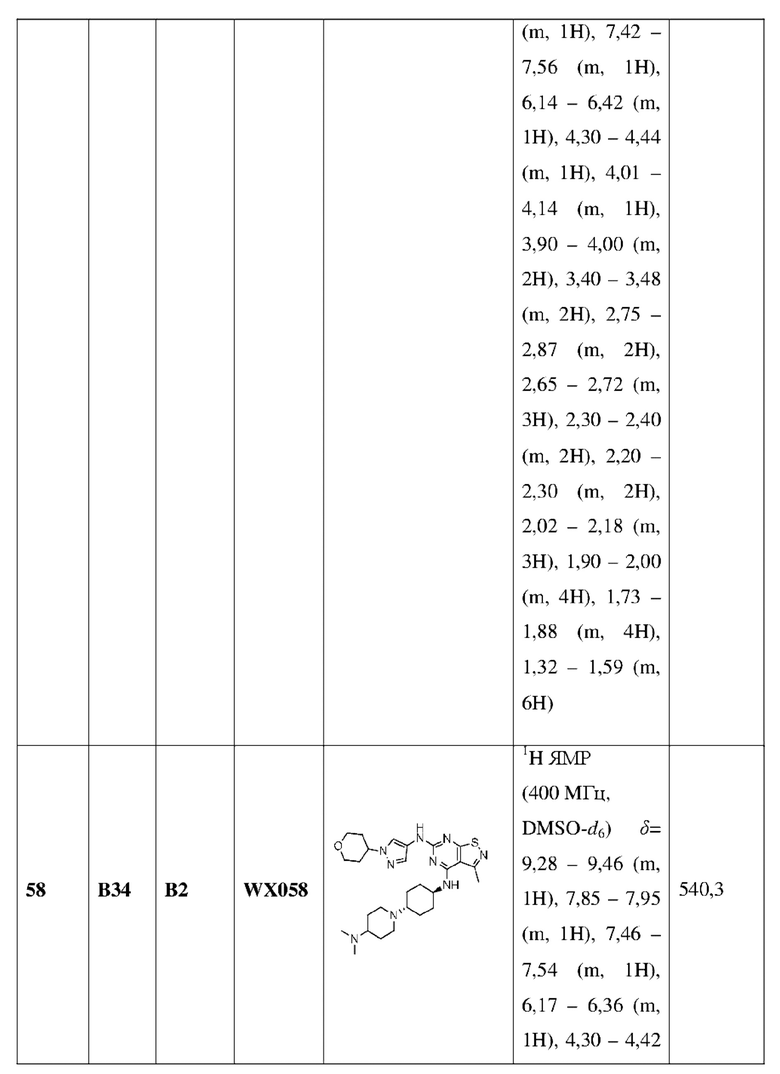
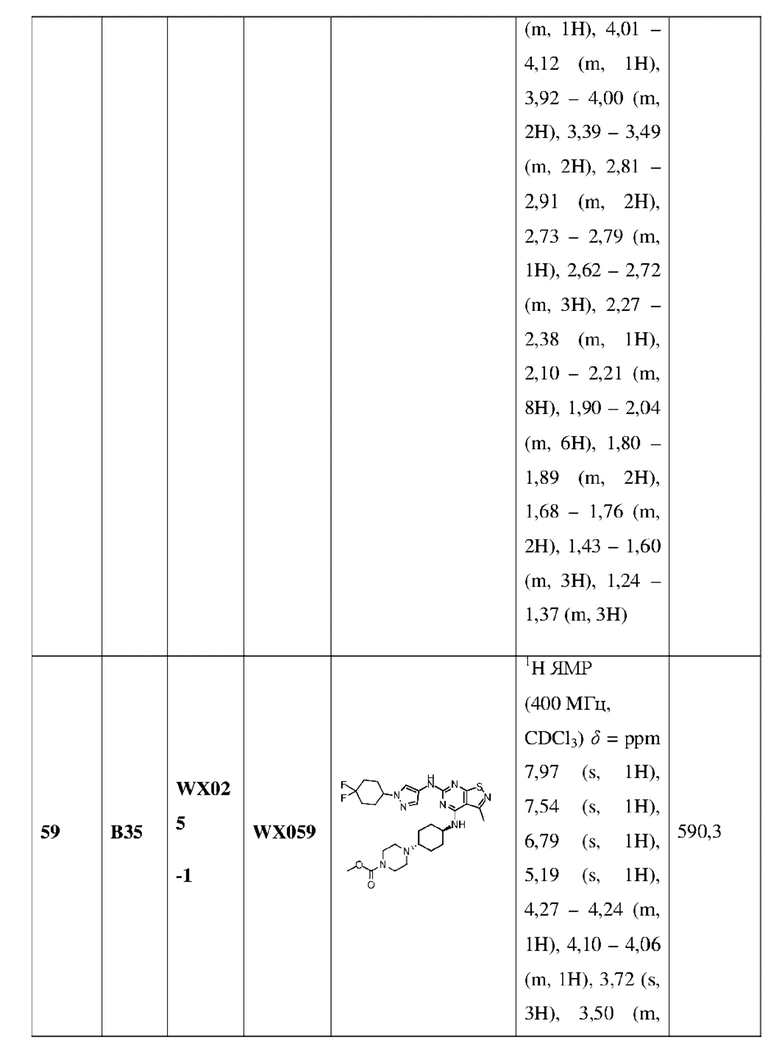

Пример 60: Синтез соединения WX060
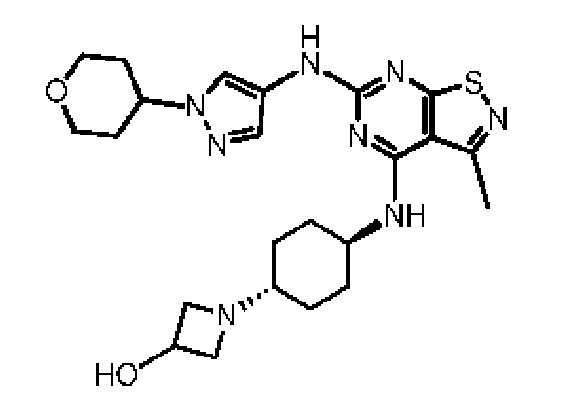
Путь синтеза:
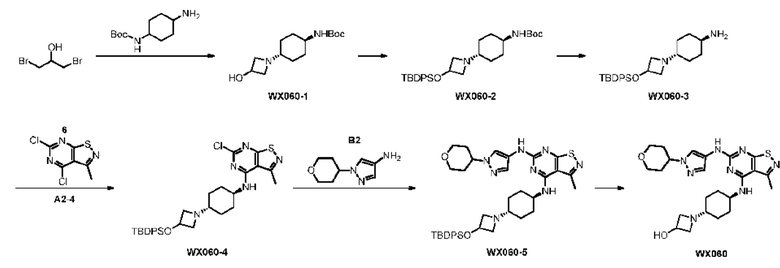
Стадия 1: Синтез соединения WX060-1
1,3-дибром-2-пропанол (5 г), N-Boc-1,4-циклогександиамин (4,47 г) и карбонат натрия (19,90 г) смешивали в колбе, содержащей этанол (300 мл), и смесь перемешивали при 80°С в течение 12 часов. Растворитель в смеси полностью удаляли при пониженном давлении. Неочищенный продукт суспендировали с применением этилацетата (100 мл) и фильтровали. Растворитель в фильтрате полностью удаляли при пониженном давлении с получением соединения WX060-1.
Стадия 2: Синтез соединения WX060-2
Соединение WX060-1 (8 г) растворяли в N,N-диметилформамиде (100 мл) при 0°С и затем добавляли трет-бутилдифенилхлорсилан (6,31 г) и имидазол (3,44 г). Смесь инкубировали при 25°С в течение 4 часов. Полученную смесь фильтровали и добавляли 200 мл воды в фильтрат с последующей экстракцией этилацетатом (150 мл × 4). Органические фазы объединяли и дегидратировали с применением соответствующего количества безводного сульфата натрия. Дегидратированную органическую фазу фильтровали для удаления высушивающего средства и растворитель в фильтрате полностью удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разделяли с применением автоматической хроматографической системы COMBI-FLASH (колонка SepaFlash® Silica Flash, подвижная фаза: 0-80% петролейный эфир/этил ацетат, скорость потока: 80 мл/мин) и очищали с получением соединения WX060-2.
Стадия 3: Синтез соединения WX060-3
Соединение WX060-2 (1 г) растворяли в дихлорметане (10 мл) при 0°С и добавляли трифторуксусную кислоту (2 мл). После 1 часа инкубации дополнительно добавляли трифторуксусную кислоту (2 мл) и реакционную систему инкубировали в течение еще 2 часов. После этого добавляли 30 мл воды в реакционную систему и в смесь добавляли этилацетат (4 × 20 мл) для экстракции. Органические фазы объединяли и дегидратировали с применением соответствующего количества безводного сульфата натрия. Дегидратированную органическую фазу фильтровали для удаления высушивающего средства и растворитель в фильтрате полностью удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт разделяли с применением автоматической хроматографической системы COMBI-FLASH (колонка SepaFlash® silicon Flash, подвижная фаза: 0-7% дихлорметан/метанол-петролейный эфир/этилацетат, скорость потока: 80 мл/мин) с получением соединения WX060-3. LCMS (ESI) масса/заряд: 409,1 [М+Н]+
Стадия 4: Синтез соединения WX060-4
Ссылаясь на синтез промежуточного соединения А2, соединение WX060-4 синтезировали, начиная с промежуточного соединения А2-4 и соединения WX060-3.
Стадия 5: Синтез соединения WX060-5
Ссылаясь на синтез WX001, соединение WX060-5 синтезировали, начиная с соединения WX060-4 и промежуточного соединения В2.
Стадия 6: Синтез соединения WX060
Соединение WX060-5 (0,03 г) растворяли в безводном тетрагидрофуране (2 мл) и добавляли раствор фторида тетрабутиламмония в тетрагидрофуран (1 М, 82,99 мкл). Реакционную систему инкубировали при 15°С в течение 12 часов в атмосфере азота. Растворитель полностью удаляли при пониженном давлении и неочищенный продукт отделяли с помощью HPLC (колонка: Welch XTimate С18 150×25 мм × 5 мкм; подвижная фаза: А: 10 мМ водного NH4HCO3, В: ацетонитрил; градиент: В%: 20%-50%, 10,5 мин) с получением соединения WX060. LCMS (ESI) масса/заряд: 485,3 [М+1]+. 1Н ЯМР (400 МГц, DMSO-d6) δ = 9,48-9,36 (m, 1Н), 8,10 - 7,81 (m, 1Н), 7,63 (s, 1Н), 6,48 - 6,43 (m, 1Н), 5,29 (d, J=6,8 Гц, 1Н), 4,41 - 4,40 (m, 1Н), 4,20 - 4,13 (m, 2Н), 4,04 - 4,01 (m, 2Н), 3,56 -3,47(m, 4Н), 3,45 - 3,42 (m, 3Н), 2,74 (s, 5Н), 2,08 - 1,97 (m, 6Н), 1,88- 1,86 (m, 2Н), 1,58-1,55(m, 1Н), 1,11 - 1,10 (m, 1Н).
Экспериментальный пример 1: Оценка ферментативной активности in vitro
Ингибирующую активность тестируемых соединений в отношении IRAK4 человека оценивали посредством измерения значений IC50 в анализе активности киназы, меченой 33Р (Reaction Biology Corp).
Состав буфера: 20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO.
Процедуры: Тестируемое соединение растворяли в DMSO при комнатной температуре с получением 10 мМ раствора для последующего применения. Субстрат растворяли в свежем буфере, к которому добавляли киназу и хорошо перемешивали. Раствор DMSO, содержащий тестируемое соединение, добавляли к вышеописанной перемешанной реакционной системе с помощью акустической методики (Echo 550). Концентрации соединений в реакционной системе были следующими: 10 мкМ, 3,33 мкМ, 1,11 мкМ, 0,370 мкМ, 0,123 мкМ, 41,2 нМ, 13,7 нМ, 4,57 нМ, 1,52 нМ и 0,508 нМ. После 15 минут инкубации реакцию начинали посредством добавления 33Р-АТР (активность: 0,01 мкКи/мкл; соответствующая концентрация указана в таблице 1). Информация относительно номера по каталогу поставщика, номера партии и концентрации в реакционной системе для IRAK4 и ее субстрата указана в таблице 1. После 120 минут осуществления реакции при комнатной температуре полученный раствор загружали на бумажный лист для ионообменной хроматографии Р81 (Whatman №3698-915). После повторного промывания 0,75% раствором фосфорной кислоты измеряли радиоактивность фосфорилированного остатка субстрата на бумажном листе. Данные по киназной активности показаны в виде сравнения киназной активности тестируемого соединения и киназной активности холостого раствора (только DM SO), и аппроксимацию кривой осуществляли с применением программного обеспечения Prism4 (GraphPad) с получением значений IC50, при этом результаты эксперимента показаны в таблице 2.
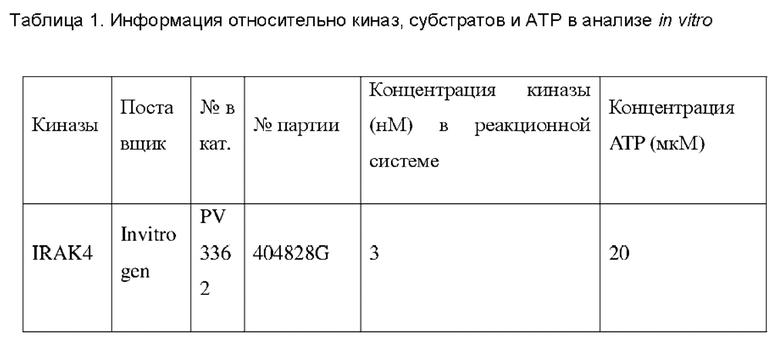
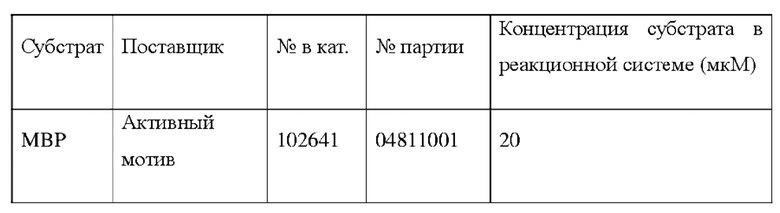

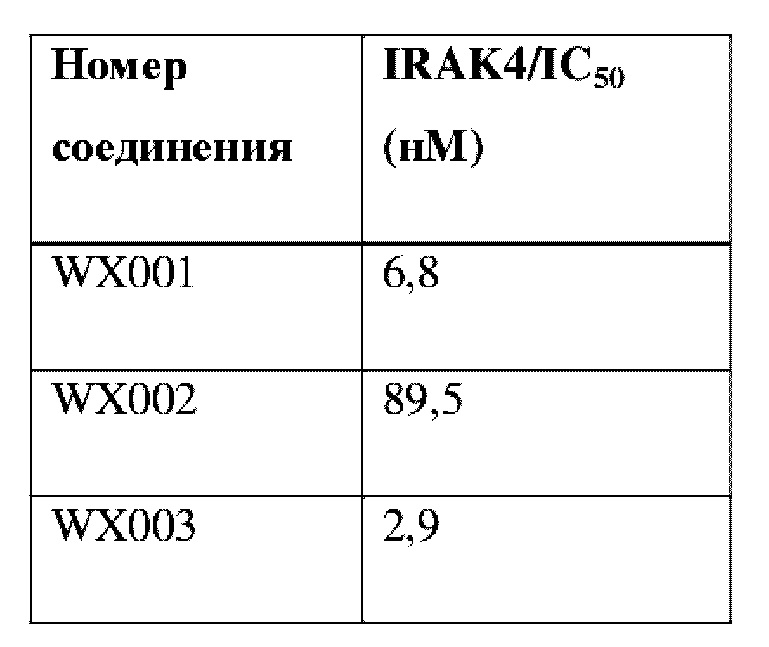
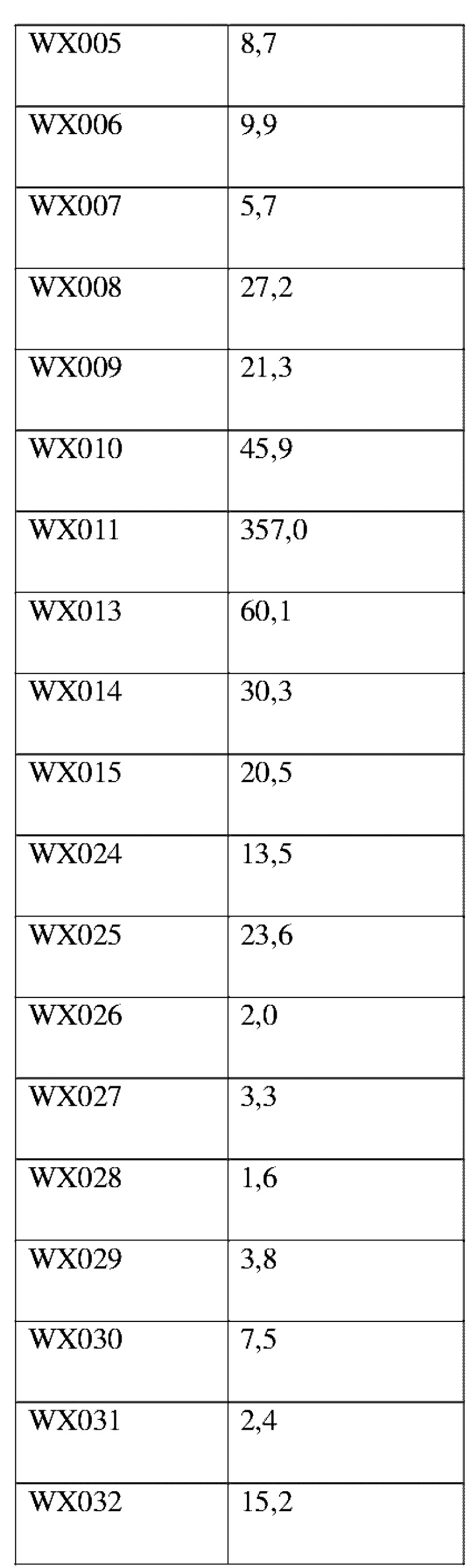
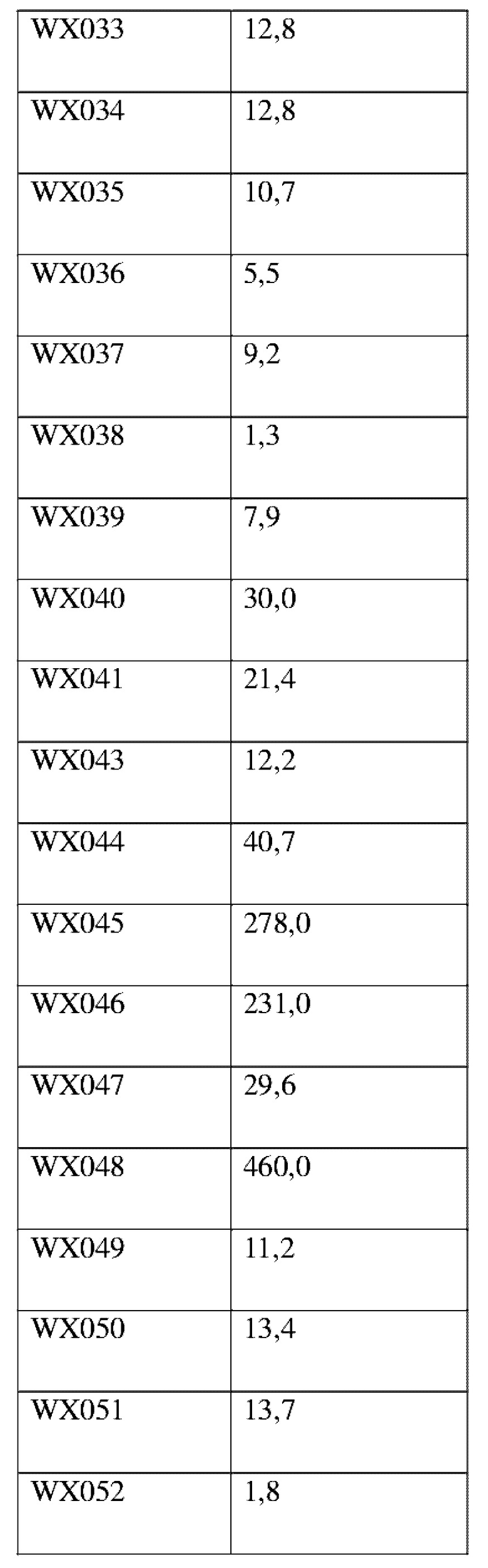
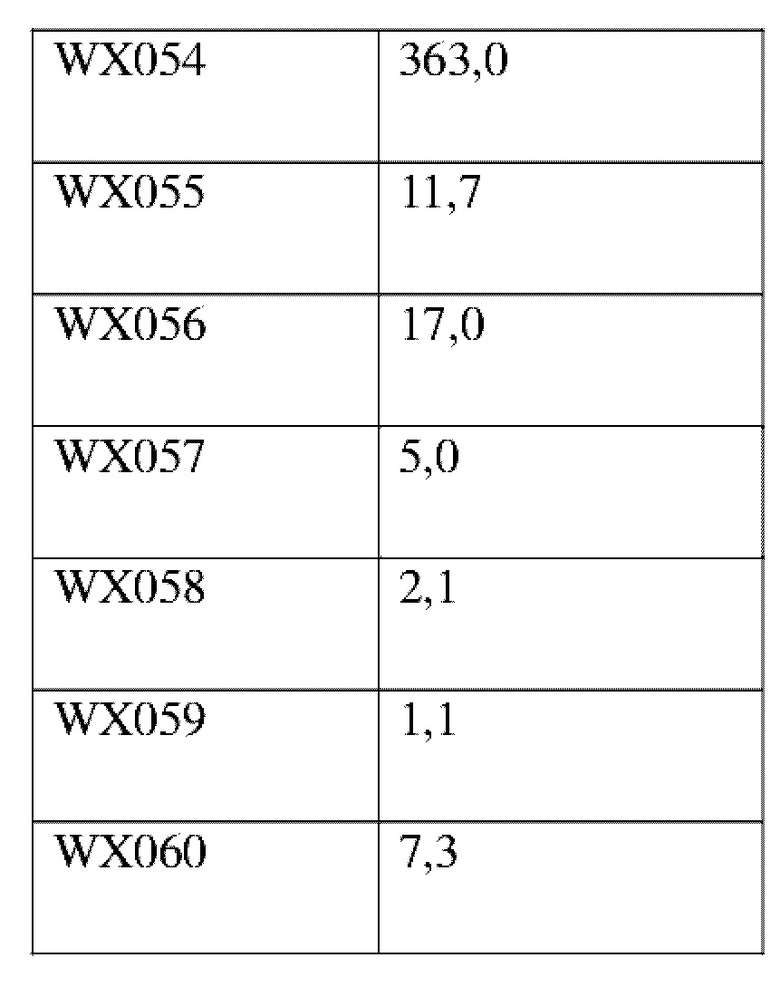
Вывод: Раскрытые в данном документе соединения в целом демонстрируют приемлемый уровень ингибирующей активности в отношении IRAK4.
Экспериментальный пример 2: Анализ стабильности в микросомах печени in vitro
Метаболическая стабильность в микросомах печени
Тестируемые соединения при 1 мкМ инкубировали совместно с микросомами печени, содержащими белок в концентрации 0,5 мг/мл под действием системы регенерации восстановительного кофермента II на водяной бане при 37°С.
1) Положительные контроли включали: тестостерон (субстрат 3А4), пропиламин-пропиофенон (субстрат 2D6) и диклофенак (субстрат 2С9). Положительные контроли инкубировали при тех же условиях, что и тестируемые соединения.
2) Реакцию останавливали с применением стоп-реагента, содержащего внутренний стандарт, в моменты времени 0, 5, 10, 20, 30 и 60 минут. Соединения инкубировали вместе с микросомами в течение 60 минут в отсутствие системы регенерации восстановительного кофермента II и они служили в качестве отрицательных контролей.
3) Для каждого из моментов времени повторность отсутствовала (n = 1).
4) Образцы анализировали с помощью LC/MS/MS и концентрации показаны в виде отношения площади пика соединения к площади пика внутреннего стандарта (нестандартная кривая).
5) В кратком описании были рассчитаны период полужизни и клиренс.
6) Для расчета клиренса применяли следующую формулу:
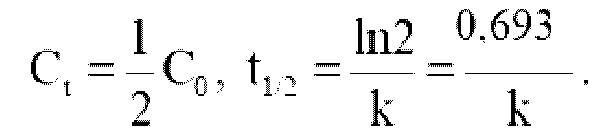

Отношение веса печени к весу тела: 88 г/кг для мышей
Клиренс в цельной печени рассчитывали с применением 

 внутренний микросомальный клиренс;
внутренний микросомальный клиренс;
Микросомальный белок при инкубации: микросомальный белок при инкубации;
Микросомы: микросомы;
Печень: печень;
Вес тела: вес тела
Результаты обобщенно представлены в таблице 3.

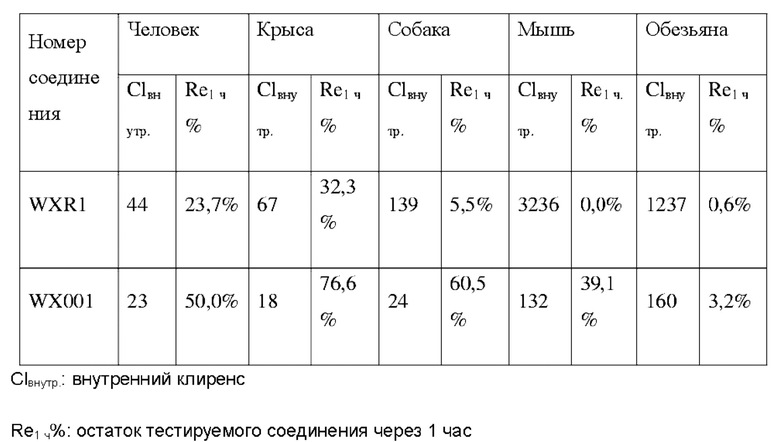
Вывод: Иллюстративные соединения, раскрытые в данном документе, обладают впечатляющими преимуществами над исходным соединением (WXR1) в отношении стабильности в микросомах печени у множества видов, и в частности у некоторых видов (например, мыши) обладают превосходством в клиренсе до 20 раз.
Экспериментальный пример 3: Анализ активности в клетках in vitro
ELISA-анализ TNF-α в клеткахТНР-1
1. Материалы:
Клетки ТНР-1, полученные от человека с острым моноцитарным лейкозом, приобретали у АТСС (№ в кат. TIB-202) и инкубировали в инкубаторе при 37°С, 5% СО2.
Среда представляла собой RPMI1640 (Gibco, № в кат. 22400-105), дополненную 10% FBS (Gibco, № в кат. 10091148); 1% PenStrep (Gibco, № в кат. 15140); 0,05 мМ 2-меркаптоэтанол (Sigma, № в кат. М6250).
2. Способ:
Содержание TNF-a в супернатанте культуры клеток измеряли с помощью набора для Elisa-определения TNF-α. TNF-α получали посредством стимуляции клеток ТНР-1 с помощью 150 нг/мл LPS (Sigma, №в кат.L6529).
Нормальные клетки ТНР-1 в логарифмической фазе роста вносили в 96-луночные планшеты (Corning №3599) при определенной концентрации (1 × 105/100 мкл) и затем инкубировали в инкубаторе. Через два часа добавляли по 16,7 мкл тестируемого соединения в разных концентрациях (8 × конечных концентраций) и смесь инкубировали. Через один час добавляли 16,7 мкл LPS в концентрации 1200 нг/мл и смесь инкубировали. Через 18 часов культуру клеток центрифугировали и собирали супернатант. Содержание TNF-α измеряли с помощью набора для Elisa-определения TNF-α. Наконец, OD-сигналы (OD450-OD570) считывали на планшет-ридере Envision.
3. Анализ данных:
OD450-CD570-сигналы преобразовывали в процент ингибирования.
% Ингибирования = (ZPE - образец)/(ZPE - НРЕ) × 100.
"НРЕ" представляет собой значение CD450-OD570-сигнала для контроля без LPS-стимулированных клеток, и "ZPE" представляет собой значение OD450-CD570-сигнала для контроля с LPS-стимулированными клетками. Значения IC50 для соединений рассчитывали с помощью XLFit в Excel.
Уравнение: Y = минимум + (максимум - минимум)/(1 + (IC50/X) ∧ наклон).
Результаты обобщенно представлены в таблице 4.

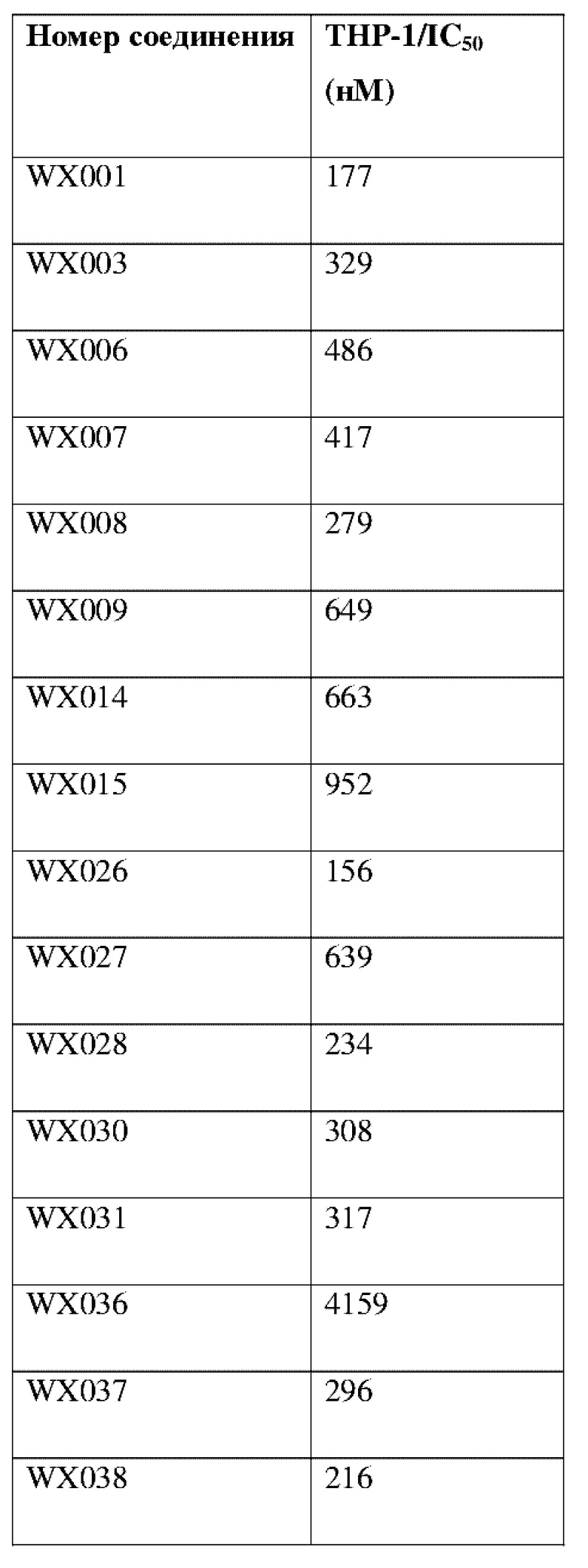
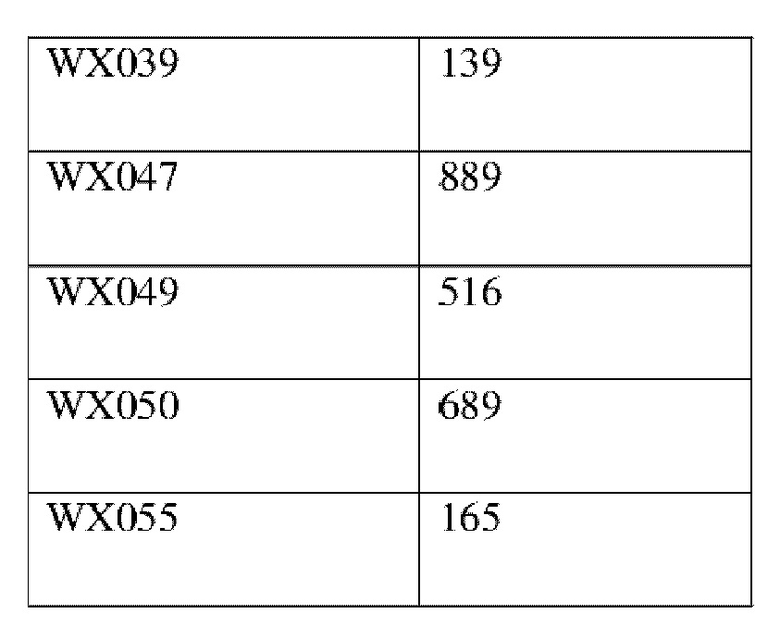
Вывод: Раскрытые в данном документе соединения в целом демонстрируют приемлемый уровень ингибирующей активности в отношении пролиферации клеток ТНР-1.
Экспериментальный пример 4: Фармакокинетическое исследование
Фармакокинетическое исследование тестируемых соединений, вводимых перорально и внутривенно, на крысах SD
Тестируемое соединение смешивали с раствором, содержащим 10% метилпирролидона, 10% стеарата полиэтиленгликоля и 80% воды. Смесь перемешали вихревым способом и подвергали воздействию ультразвука с получением 1,5 мг/мл осветленного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Самцов крыс SD в возрасте 7-10 недель отбирали и им внутривенно вводили дозу кандидатного соединения, составляющую 3 мг/кг Тестируемое соединение смешивали с 0,5% водным раствором метилцеллюлозы. Смесь перемешивали вихревым способом и подвергали воздействию ультразвука с получением 2 мг/мл суспензии для последующего применения. Самцов крыс SD в возрасте 7-10 недель отбирали и им перорально вводили дозу кандидатного соединения, составляющую 10 мг/кг. Цельную кровь собирали в определенные моменты времени и отделяли плазму крови. Концентрацию лекарственного средства измеряли с помощью LC-MS/MS и фармакокинетические параметры рассчитывали с применением программного обеспечения Phoenix WinNonlin (Pharsight, США). Результаты экспериментов показаны в таблице 7.
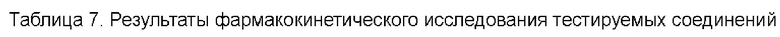
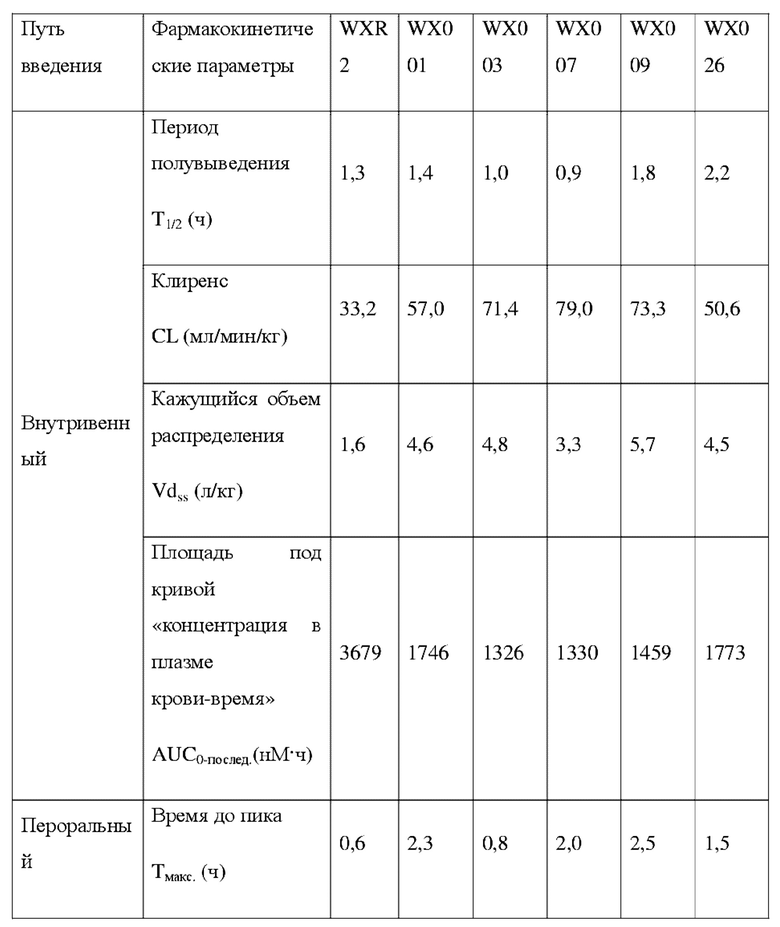
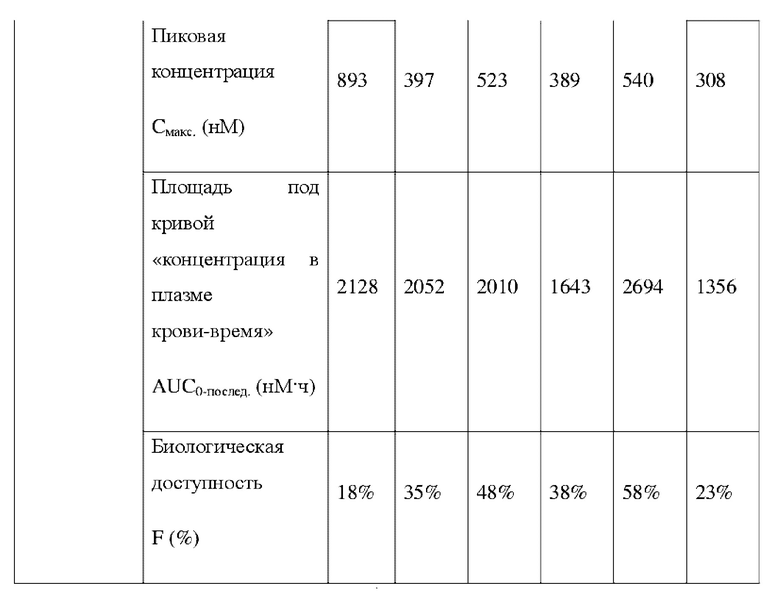
Результаты демонстрируют, что общее системное воздействие, пиковая концентрация и биологическая доступность различных соединений по данному проекту, вводимых перорально, были эквивалентными или превосходящими по сравнению с исходным соединением WXR2 (ND-2110) при той же дозе, демонстрируя превосходящие фармакокинетические свойства.
Экспериментальный пример 5: Фармакодинамическое исследование
Фармакодинамическое исследование для оценки секреции TNF-α, индуцированной липополиколлагеном (LPS), у KpbicSD
1. Моделирование и введение
Крысам SD перорально вводили растворитель, дексаметазон (DEX, 0,5 мг/кг) в качестве положительного контроля, и тестируемое соединение, и через 0,5 часа после введения внутрибрюшинно вводили LPS путем инъекции (1 мг/кг). Животных умерщвляли с помощью СО2 через 2 часа после введения LPS путем инъекции. Кровь из сердца собирали в вакуумные контейнеры с EDTA-K2, и часть антикоагулированной крови центрифугировали, и плазму крови замораживали при -80°С.
2. Анализ TNF-α и II-1b
Замороженную плазму крови размораживали при комнатной температуре и концентрацию TNF-α в плазме крови измеряли с применением набора для ELISA.
3. Статистические данные
Экспериментальные данные представляли с применением среднего ± SEM и уровни TNF-α анализировали с помощью однофакторного ANOVA. Отличия считали значимыми при р < 0,05. Результаты фармакодинамического исследования для оценки секреции TNF-α, индуцированной LPS, у крыс SD, показаны на фиг.1 и 2.
На фиг.1 показано, что при тех же дозах, WX001, вводимое перорально, продемонстрировало значительный ингибирующий эффект в отношении секреции TNF-α, индуцированной липополиколлагеном (LPS), который в значительной степени превосходил таковой в случае исходных соединений WXR2(ND-2110), WXR3(BAY-1830839) и WXR4 (BAY-1834845). В данном эксперименте эффективность WX001 была эквивалентна эффективности дексаметазона DEX.
На фиг.2 показано, что при тех же дозах, WX001, WX026 и WX044, вводимые перорально, продемонстрировали значительный ингибирующий эффект в отношении секреции TNF-α, индуцированной липополиколлагеном (LPS). В данном эксперименте эффективность WX026 была эквивалентной таковой у дексаметазона DEX.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| АГОНИСТ S1P1 И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2754845C2 |
| СОДЕРЖАЩЕЕ ЗАМЕСТИТЕЛЬ, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ БУТАН, В ГЕТЕРОЦИКЛИЧЕСКОМ КОЛЬЦЕ ПРОИЗВОДНОЕ ПИРИДОНА ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2738844C2 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2014 |
|
RU2663999C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ВЫПОЛНЯЮЩЕЕ ФУНКЦИИ ИНГИБИТОРА WEE1, И ВАРИАНТЫ ЕГО ПРИМЕНЕНИЯ | 2018 |
|
RU2783243C2 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
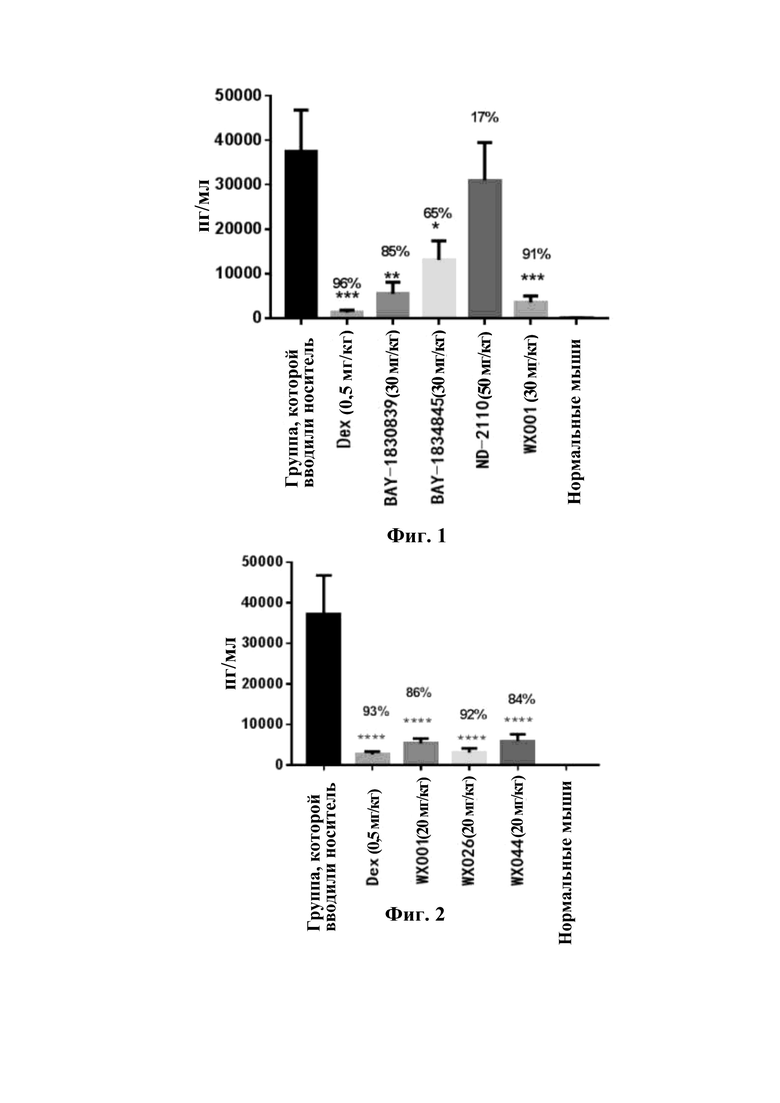
Изобретение относится к соединению, представленному формулой (III), где R1-R3, L1, L2, кольцо A определены в формуле изобретения, и его фармацевтически приемлемой соли, содержащей его фармацевтической композиции и применению соединения. Соединение может быть использовано для получения лекарственного средства для лечения заболеваний, связанных с IRAK4. 6 н. и 14 з.п. ф-лы, 2 ил., 7 табл., 60 пр.
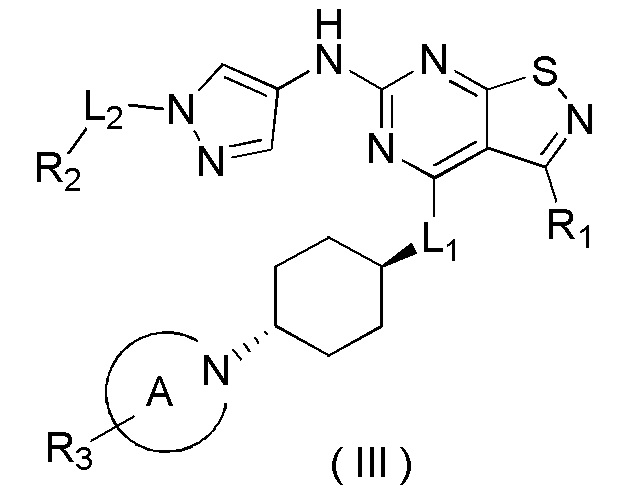
1. Соединение формулы (II), его оптический изомер или фармацевтически приемлемая соль,
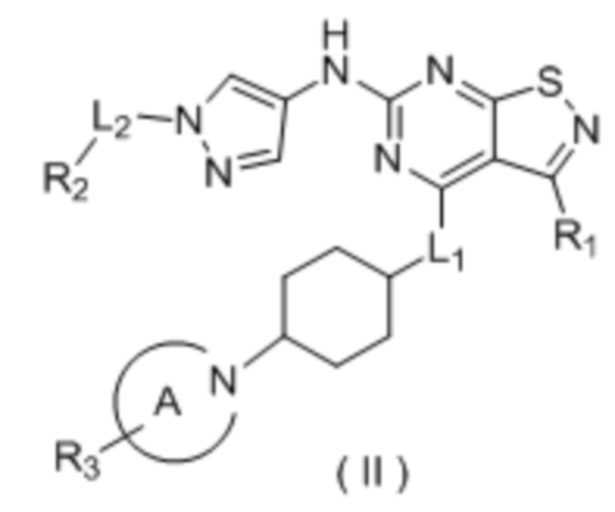
где
R1 выбран из группы, состоящей из CN, C1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra;
R2 выбран из группы, состоящей из C1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb;
R3 выбран из группы, состоящей из H, OH, C1-6алкила, -C(=O)-O-C1-6алкила, -C(=O)-C1-6алкила и C3-6циклоалкила;
кольцо A выбрано из группы, состоящей из 3-10-членного гетероциклоалкила, и 3-10-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd;
L1 выбран из группы, состоящей из O и N(R4);
L2 выбран из группы, состоящей из одинарной связи, CH2 и CH2CH2;
R4 выбран из группы, состоящей из H и Me;
каждый Ra независимо выбран из группы, состоящей из CN и COOH;
каждый Rb независимо выбран из группы, состоящей из F, OH, COOH и Me;
каждый Rd независимо представляет собой OH;
3-6-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -O-, -NH- и N; и
3-10-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -O-, -NH-, N и -C(=O)NH-.
2. Соединение формулы (III), его оптический изомер или фармацевтически приемлемая соль,
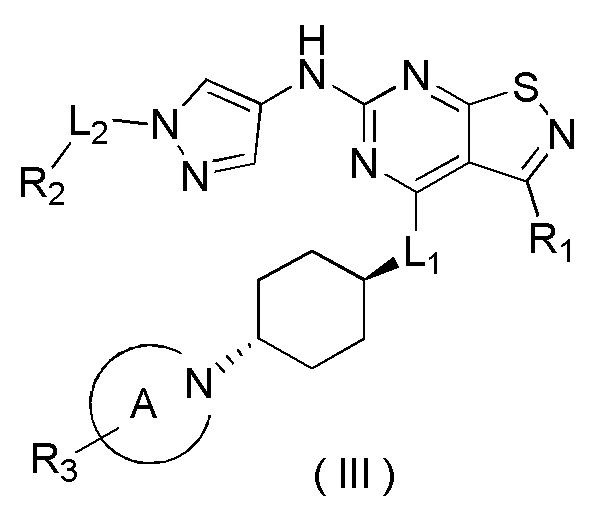
где
R1 выбран из группы, состоящей из CN, C1-6алкила и 3-6-членного гетероциклоалкила, где C1-6алкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Ra;
R2 выбран из группы, состоящей из C1-6алкила, C3-8циклоалкила и 3-6-членного гетероциклоалкила, где C1-6алкил, C3-8циклоалкил и 3-6-членный гетероциклоалкил необязательно замещены 1, 2 или 3 Rb;
R3 выбран из группы, состоящей из H, OH, COOH, C1-6алкила, C1-6алкиламино, -C(=O)-O-C1-6алкила, -C(=O)-C1-6алкила и C3-6циклоалкила;
кольцо A выбрано из группы, состоящей из 3-10-членного гетероциклоалкила, и 3-10-членный гетероциклоалкил необязательно замещен 1, 2 или 3 Rd;
L1 выбран из группы, состоящей из O и N(R4);
L2 выбран из группы, состоящей из одинарной связи, CH2 и CH2CH2;
R4 выбран из группы, состоящей из H и Me;
каждый Ra независимо выбран из группы, состоящей из CN и COOH;
каждый Rb независимо выбран из группы, состоящей из F, OH, COOH и Me;
каждый Rd независимо представляет собой OH;
3-6-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -O-, -NH- и N; и
3-10-членный гетероциклоалкил содержит 1, 2 или 3 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -O-, -NH-, N и -C(=O)NH-.
3. Соединение, его оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-2, где R1 выбран из группы, состоящей из C1-3алкила и тетрагидропиранила, где C1-3алкил и тетрагидропиранил необязательно замещены 1, 2 или 3 Ra.
4. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 3, где R1 выбран из группы, состоящей из Me, Et, 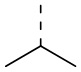 и
и  , где Me, Et, и необязательно замещены 1, 2 или 3 Ra.
, где Me, Et, и необязательно замещены 1, 2 или 3 Ra.
5. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 4, где R1 выбран из группы, состоящей из Me, Et, , , 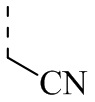 и
и  .
.
6. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 2, где R2 выбран из группы, состоящей из C1-3алкила, C4-6циклоалкила, тетрагидропиранила, оксетанила, тетрагидрофуранила и 1,4-диоксанила, где C1-3алкил, C4-6циклоалкил, тетрагидропиранил, оксетанил, тетрагидрофуранил и 1,4-диоксанил необязательно замещены 1, 2 или 3 Rb.
7. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 6, где R2 выбран из группы, состоящей из Me, Et,  и циклогексила, где Me, Et,
и циклогексила, где Me, Et,  и циклогексил необязательно замещены 1, 2 или 3 Rb.
и циклогексил необязательно замещены 1, 2 или 3 Rb.
8. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 7, где R2 выбран из группы, состоящей из Me, -CH2OH, Et, 
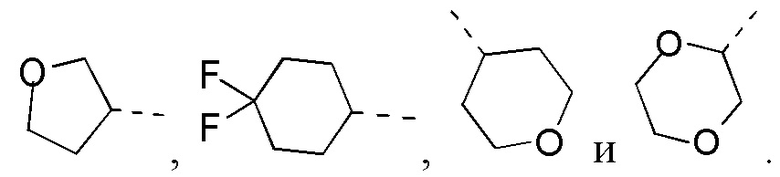
9. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 2, где структурная единица  выбрана из группы, состоящей из Me, Et,
выбрана из группы, состоящей из Me, Et, 
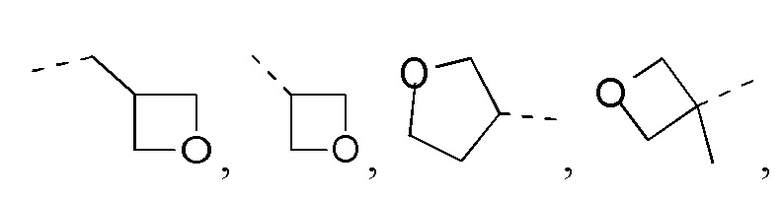
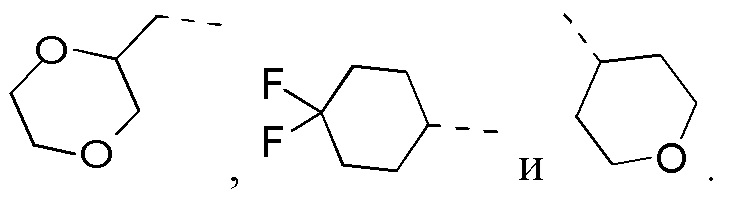
10. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 2, где R3 выбран из группы, состоящей из H, OH, COOH, C1-3алкила, -N(C1-3алкил)2, -C(=O)-O-C1-3алкила, -C(=O)-C1-3алкила и C3-6циклоалкила.
11. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 10, где R3 выбран из группы, состоящей из H, OH, COOH, Me, Et,  -C(=O)-O-Me, -C(=O)-O-Et и -C(=O)-Me.
-C(=O)-O-Me, -C(=O)-O-Et и -C(=O)-Me.
12. Соединение, его оптический изомер или фармацевтически приемлемая соль по любому из пп. 1-2, где кольцо A выбрано из группы, состоящей из морфолинила, пиперазинила, 3-морфолинонила, 2-пиперазинонила, гомопиперазинила, 4,7-диазаспиро[2,5]октила, 3,6-диазабицикло[3,1,1]гептила, 2-азациклогексанонила, 2,5-диазабицикло[2.2.1]гептила и азетидинила, где морфолинил, пиперазинил, 3-морфолинонил, 2-пиперазинонил, гомопиперазинил, 4,7-диазаспиро[2,5]октил, 3,6-диазабицикло[3,1,1]гептил, 2-азациклогексанонил, 2,5-диазабицикло[2.2.1]гептил и азетидинил необязательно замещены 1, 2 или 3 Rd.
13. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 12, где кольцо A выбрано из группы, состоящей из
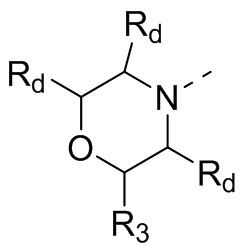 ,
, 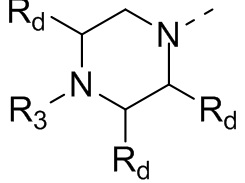 ,
, 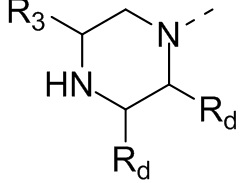 ,
, 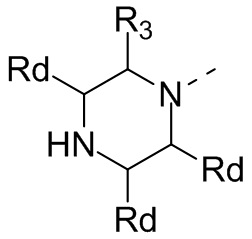 ,
, 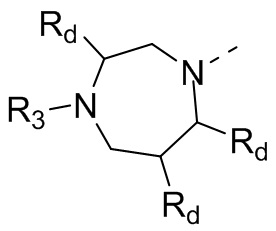 ,
, 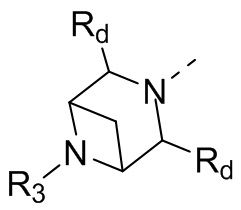 ,
, 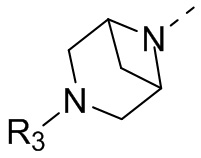 ,
, 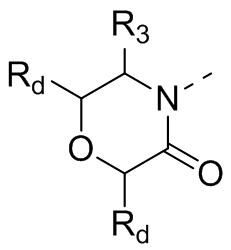 ,
, 
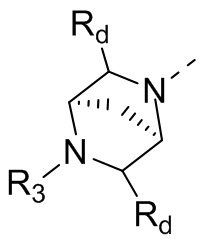 ,
, 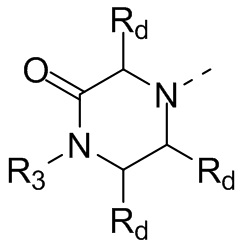 ,
, 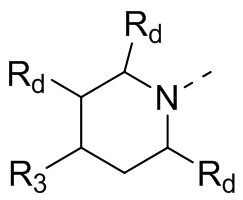 ,
, 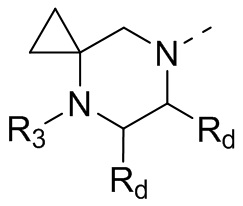 и
и 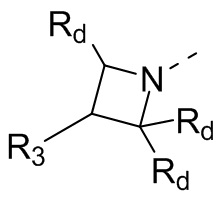 .
.
14. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 13, где структурная единица 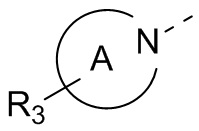 выбрана из группы, состоящей из
выбрана из группы, состоящей из 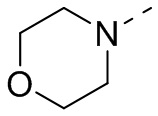 ,
, 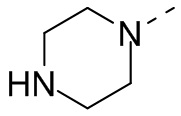 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 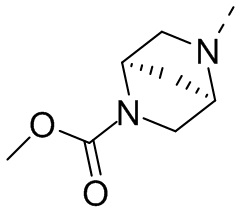 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
15. Соединение, его оптический изомер или фармацевтически приемлемая соль по любому из пп. 2-11, которые выбраны из:
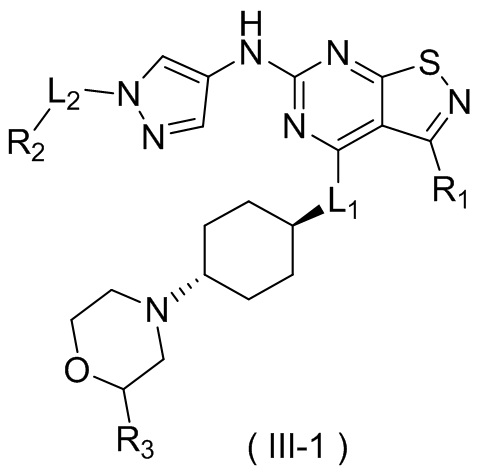
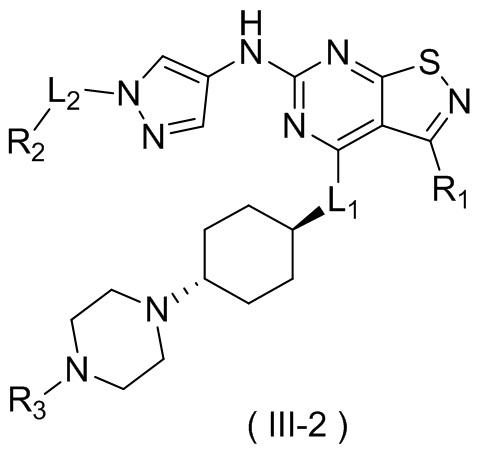
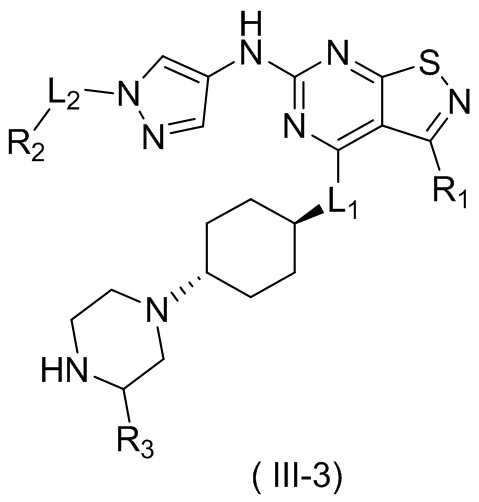
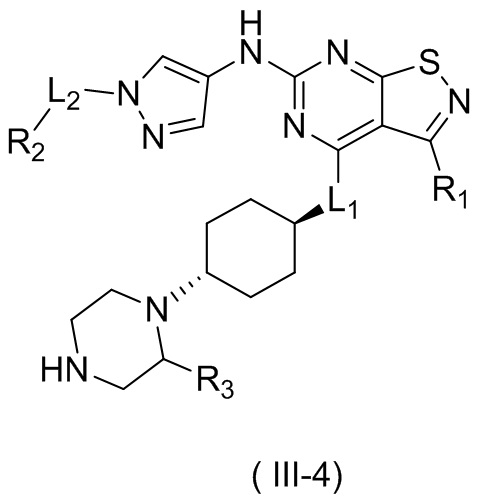
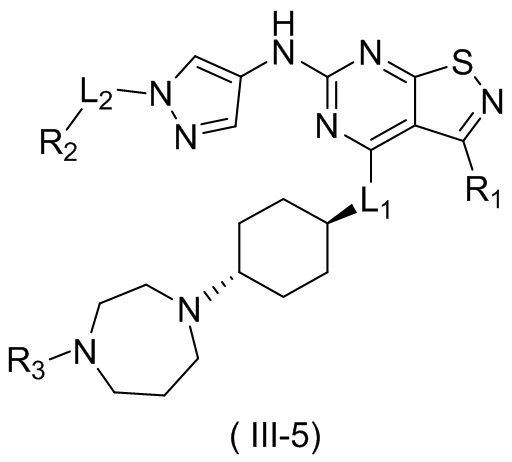
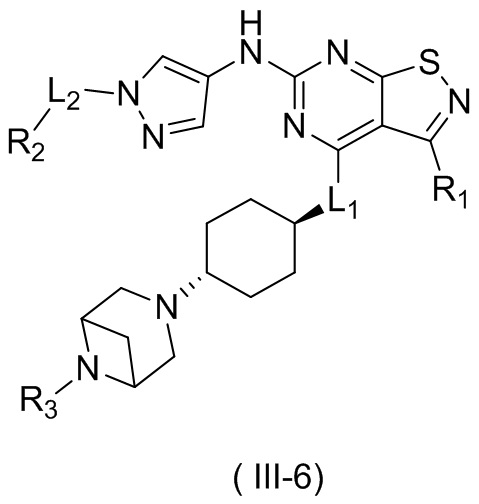
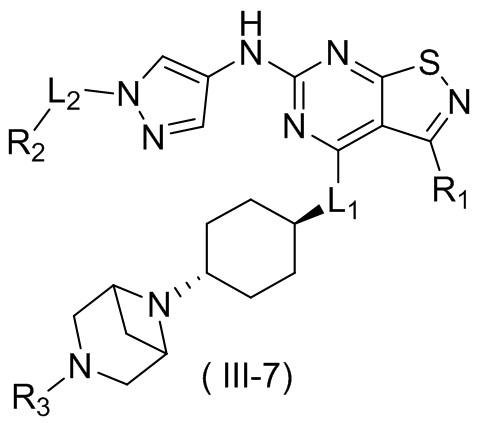
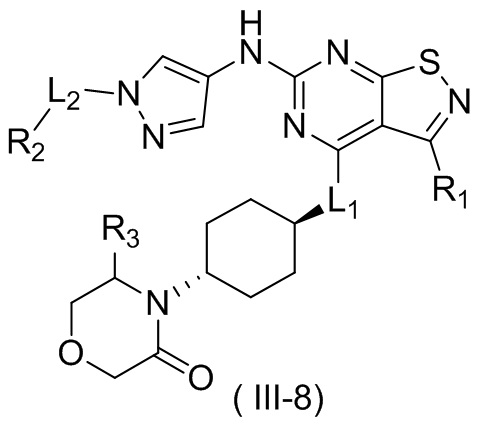
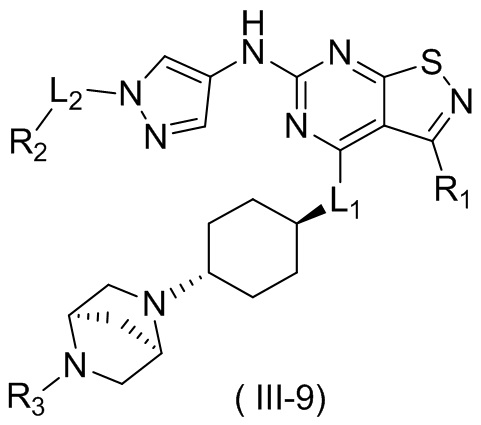
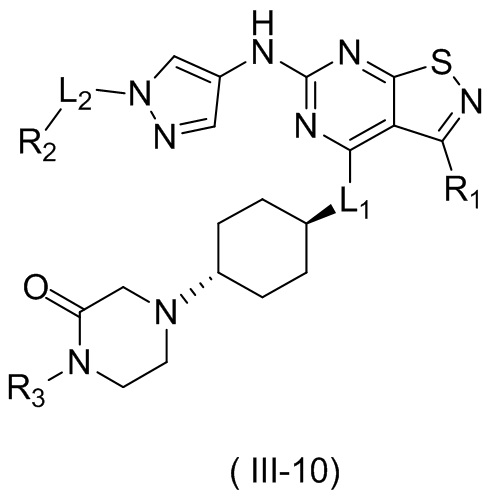
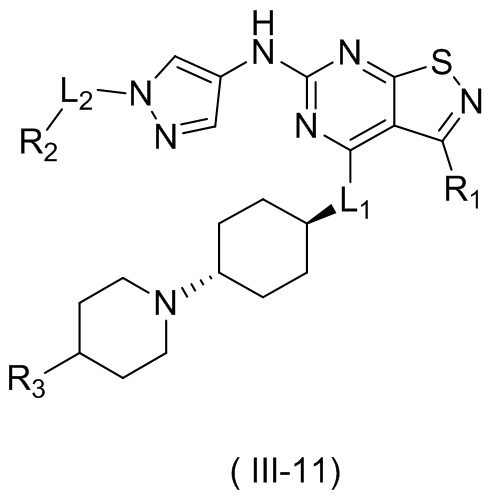
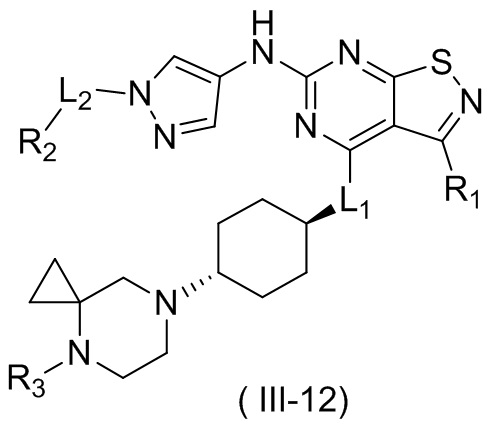
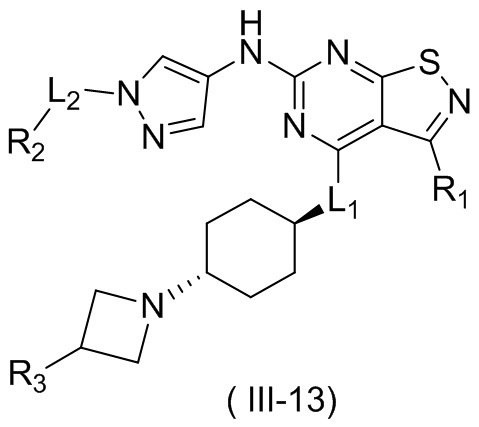
R1, R2, R3, L1 и L2 определены в любом из пп. 2-11.
16. Соединение следующей формулы, его оптический изомер или фармацевтически приемлемая соль, которые выбраны из:
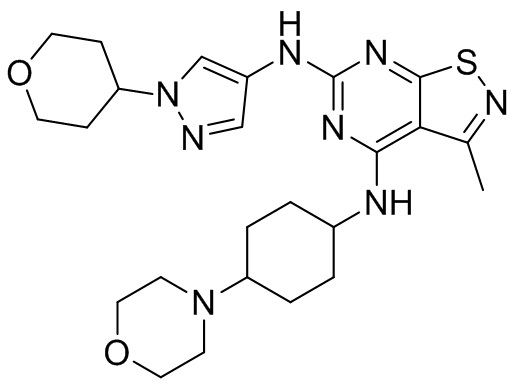 ,
, 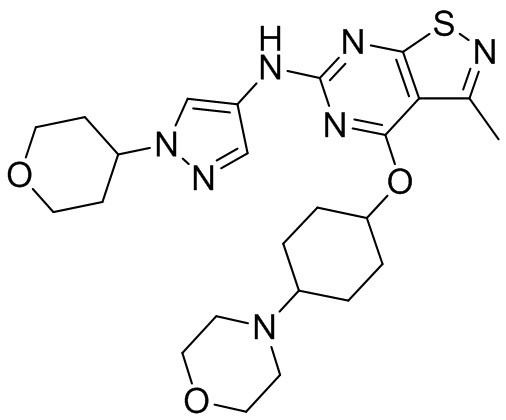 ,
, 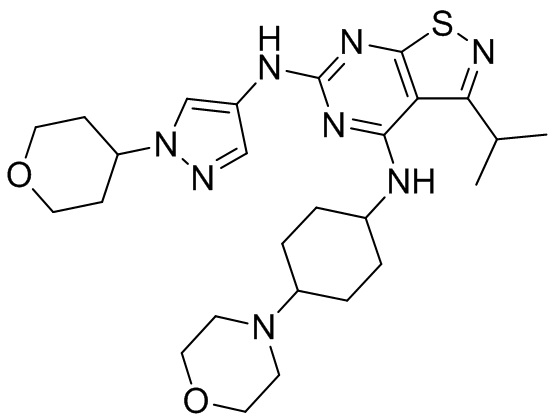 ,
, 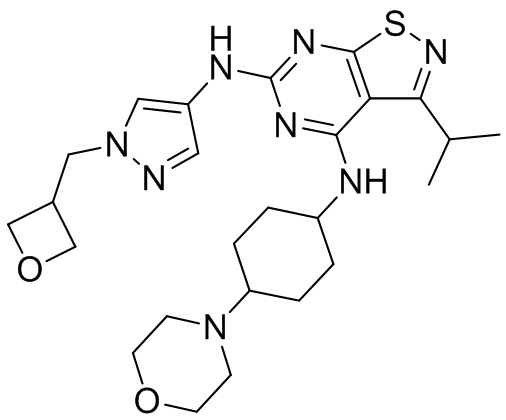 ,
, 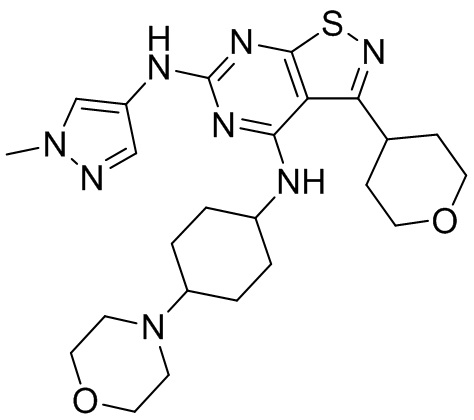 ,
,  ,
, 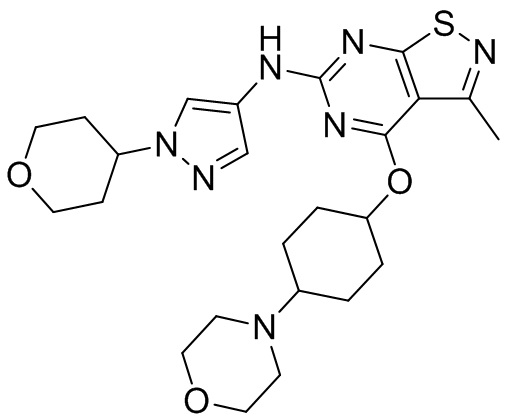 ,
, 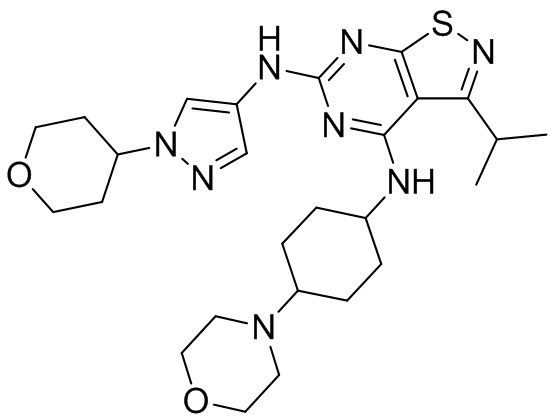 ,
, 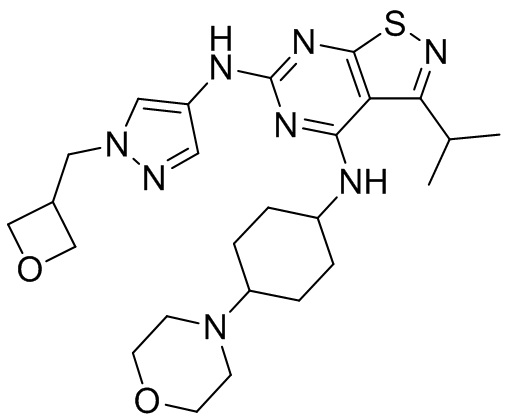 ,
, 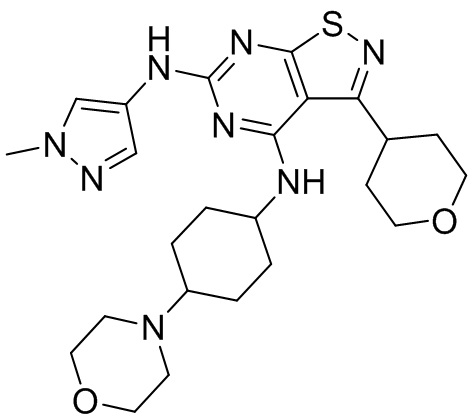 ,
, 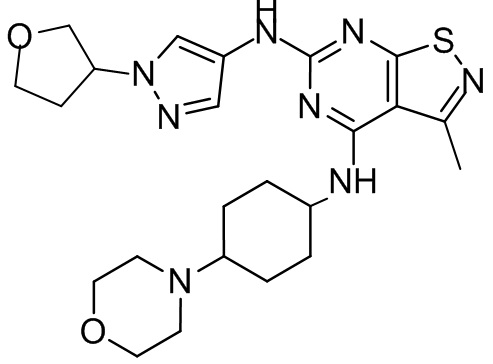 ,
, 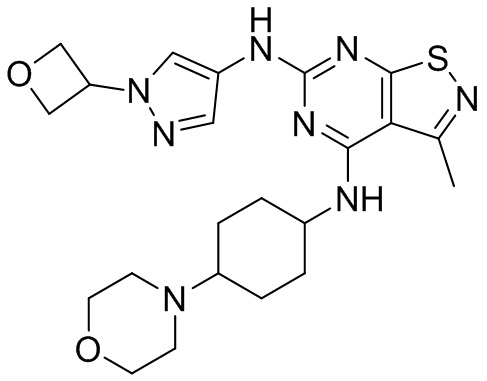 ,
, 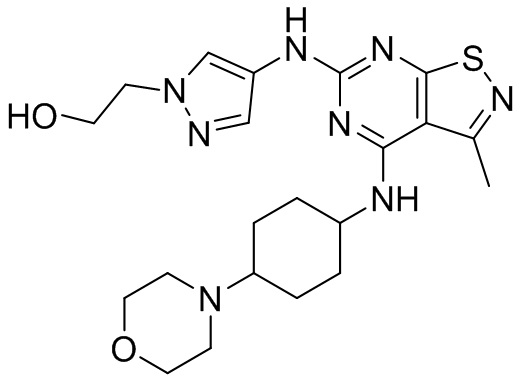 ,
,  ,
, 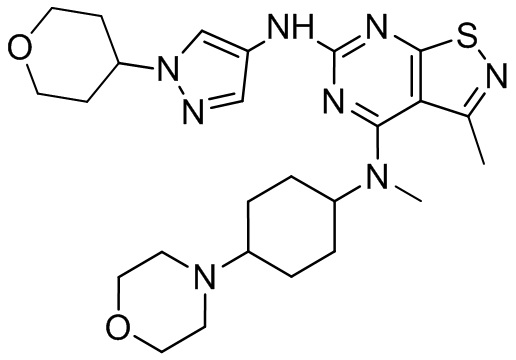 ,
, 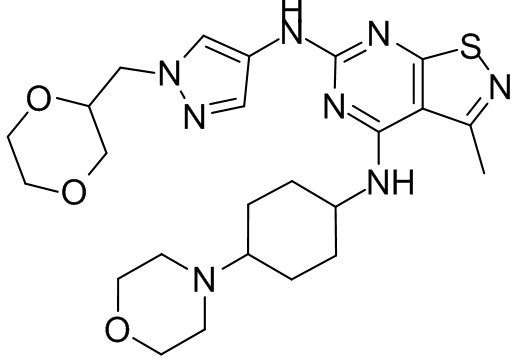 ,
, 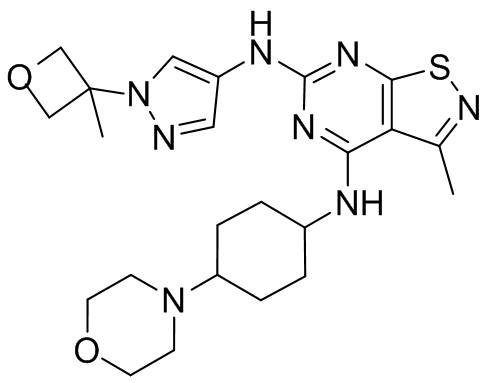 ,
,  ,
,  ,
, 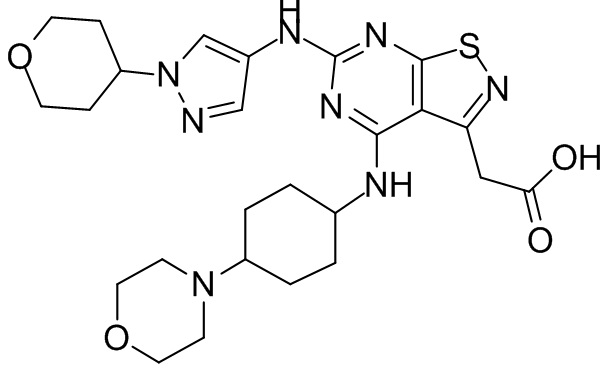 ,
, 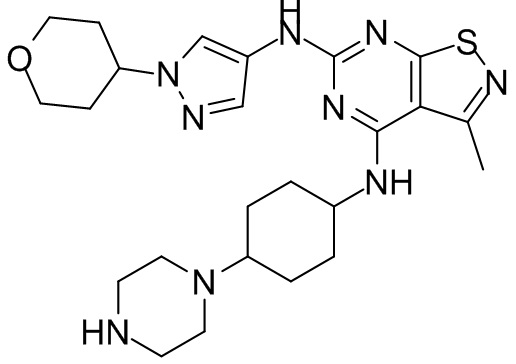 ,
, 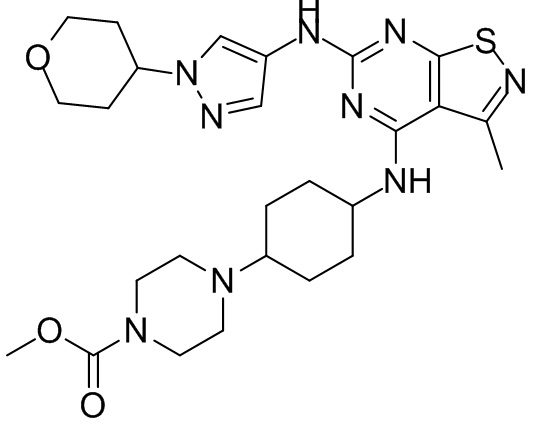 ,
, 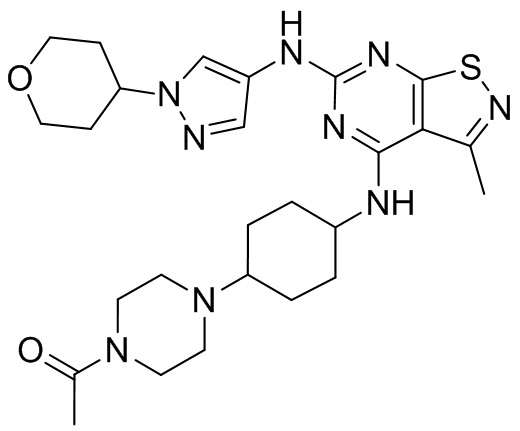 ,
, 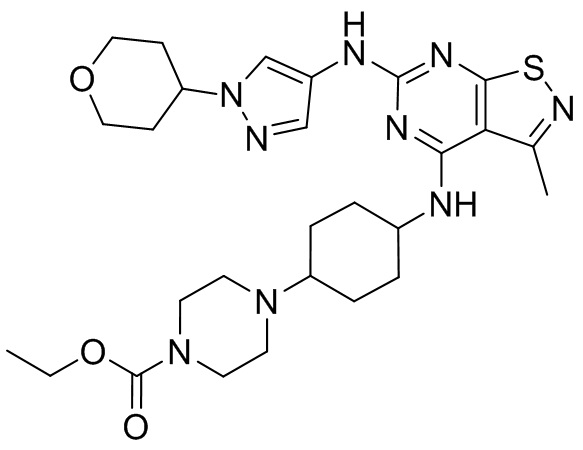 ,
,  ,
, 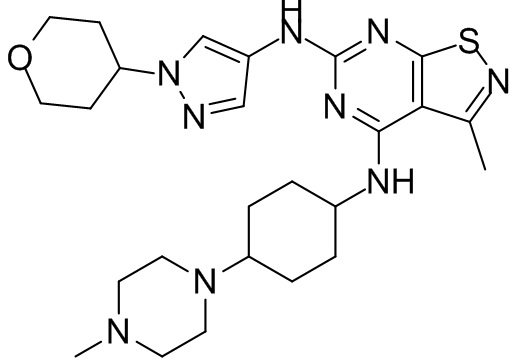 ,
, 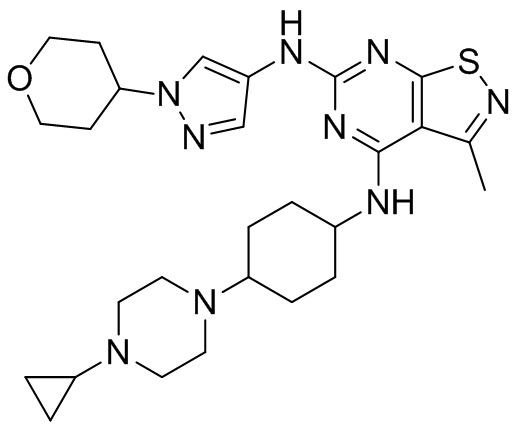 ,
, 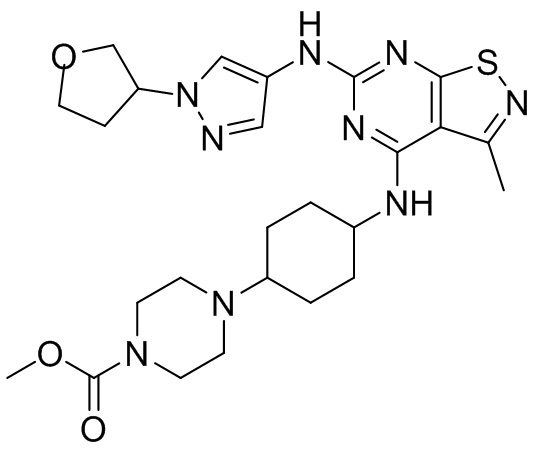 ,
, 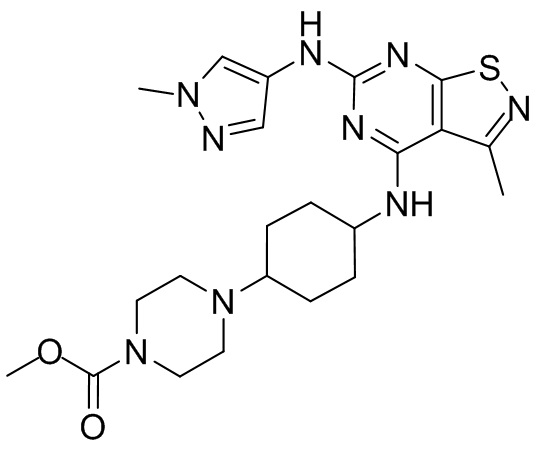 ,
, 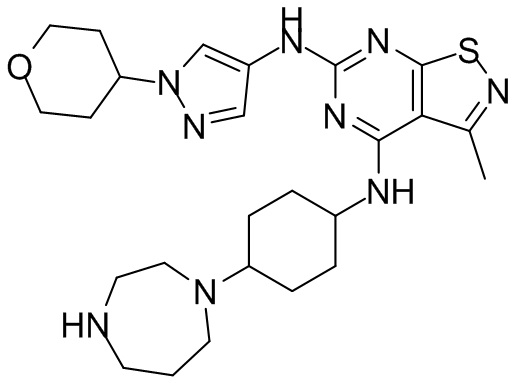 ,
, 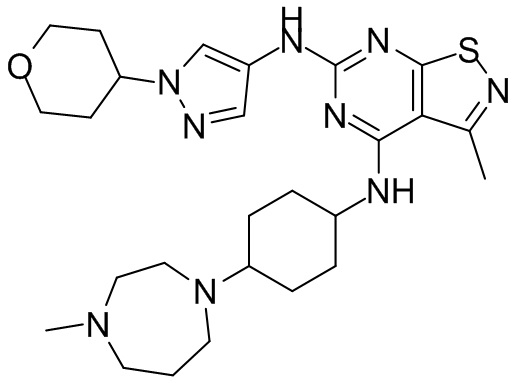 ,
, 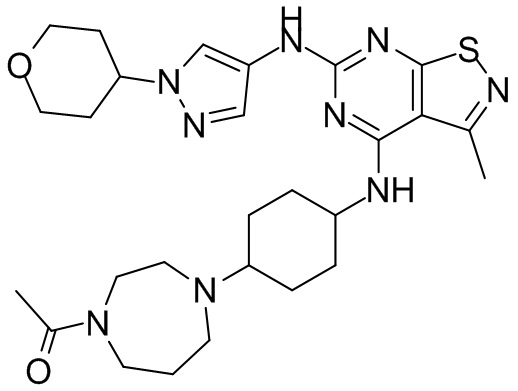 ,
, 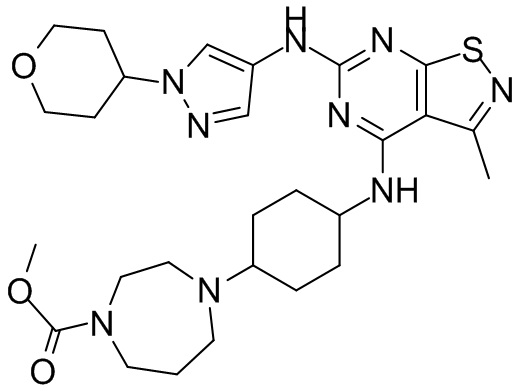 ,
, 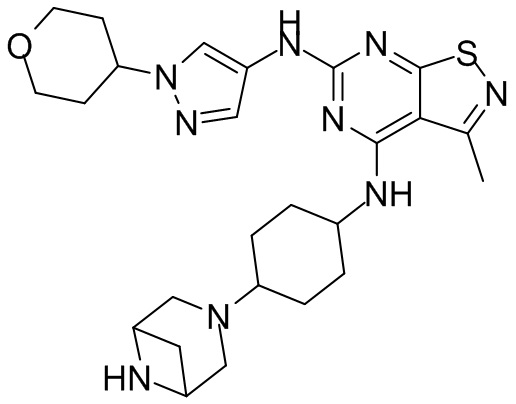 ,
,  ,
, 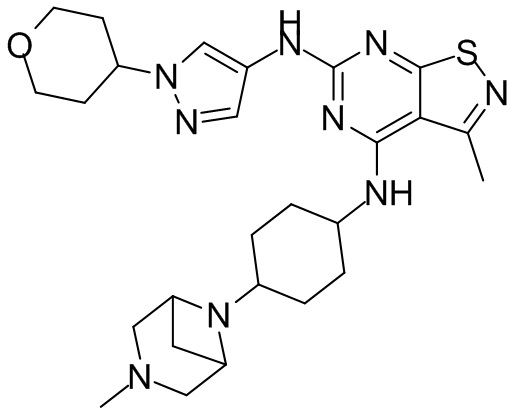 ,
, 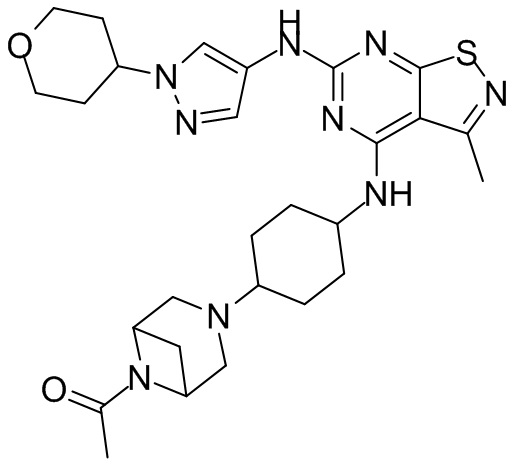 ,
, 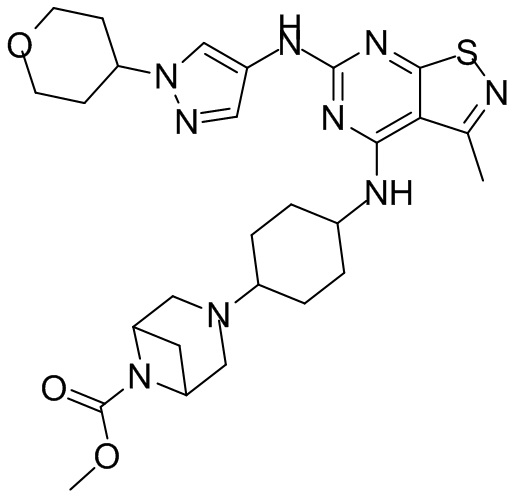 ,
, 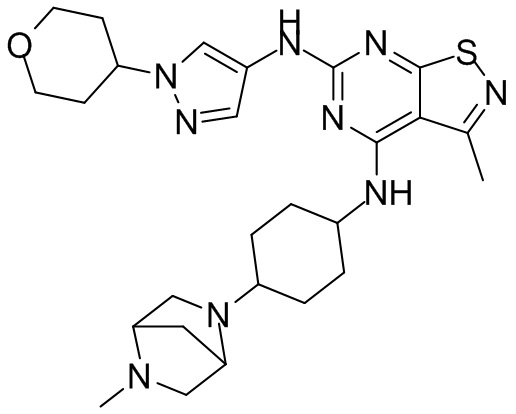 ,
,  ,
, 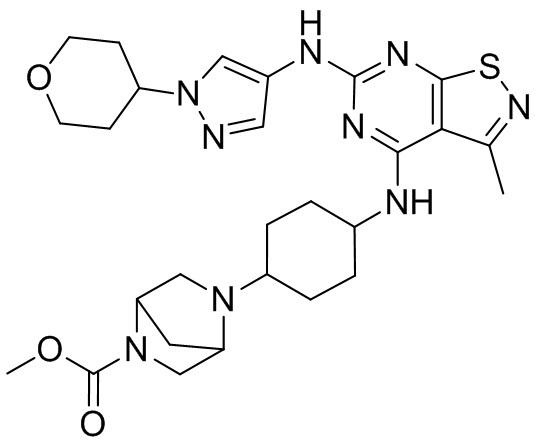 ,
, 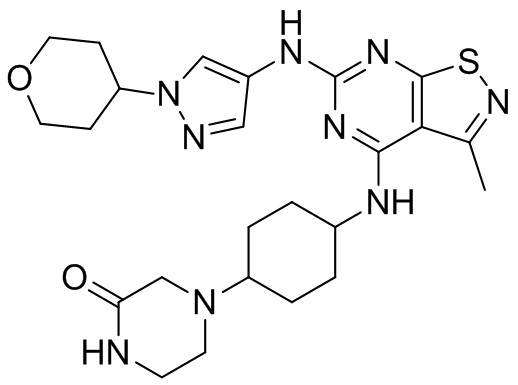 ,
, 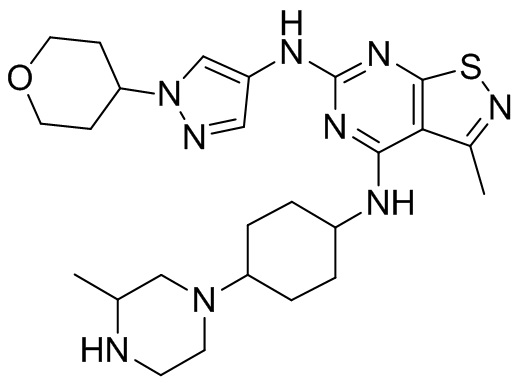 ,
, 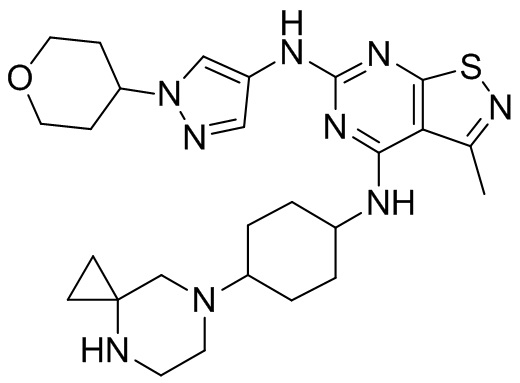 ,
, 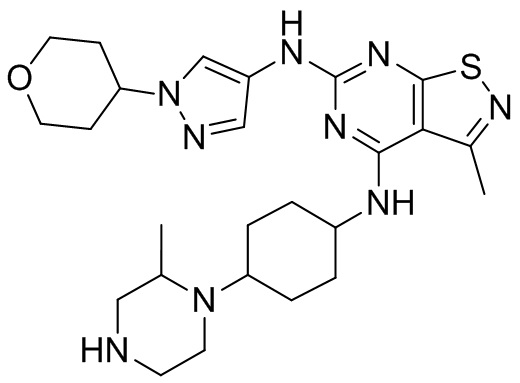 ,
, 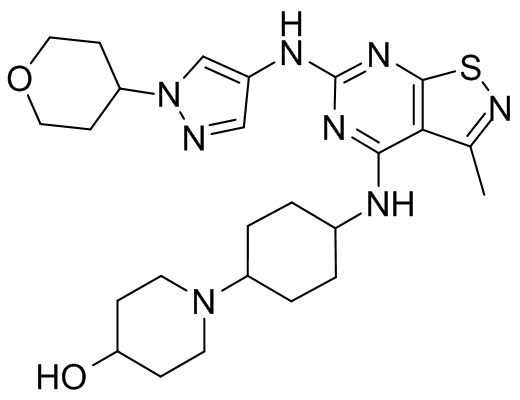 ,
, 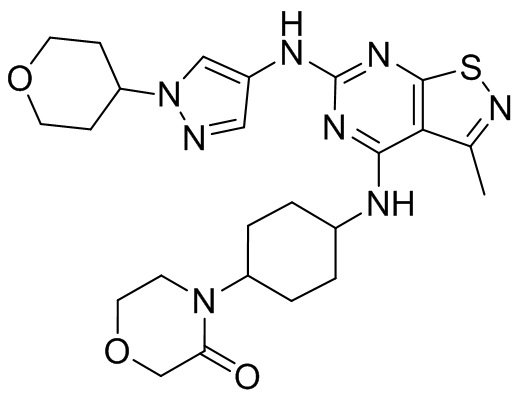 ,
, 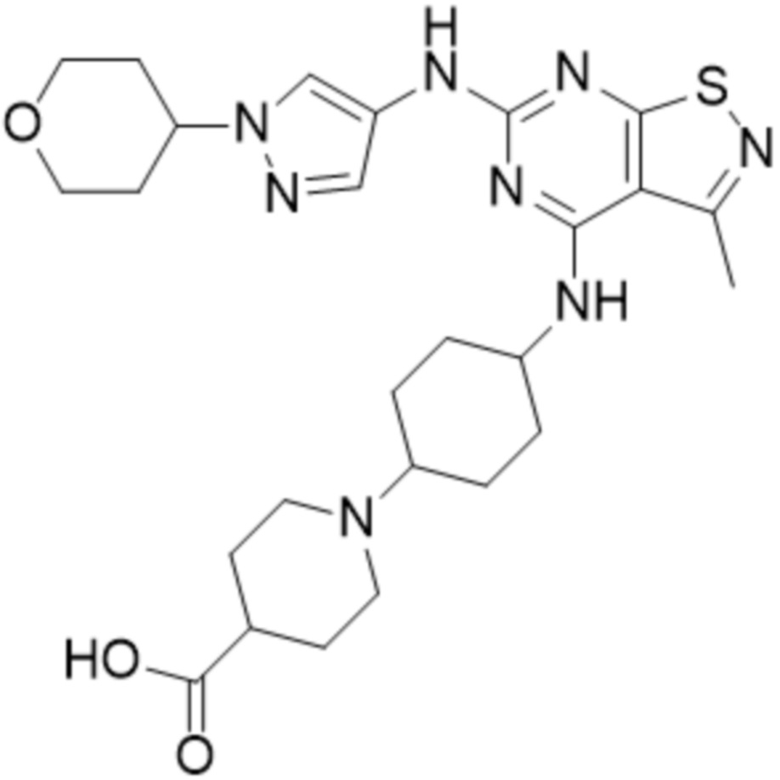 ,
, 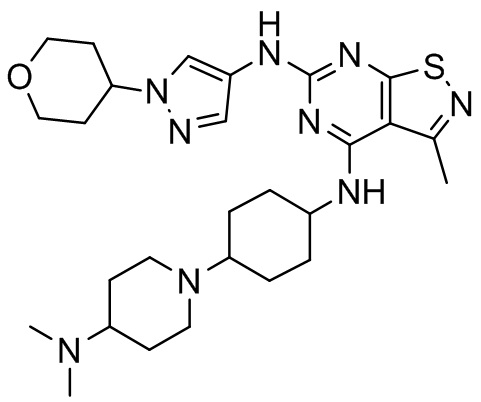 ,
, 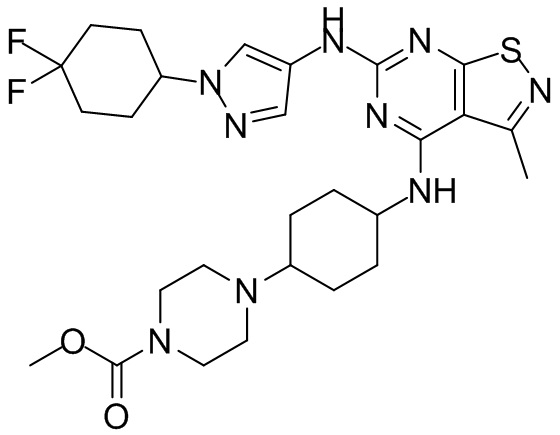 и
и 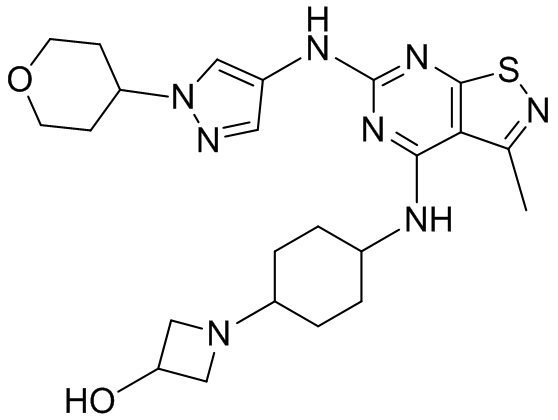 .
.
17. Соединение, его оптический изомер или фармацевтически приемлемая соль по п. 16, которые выбраны из:
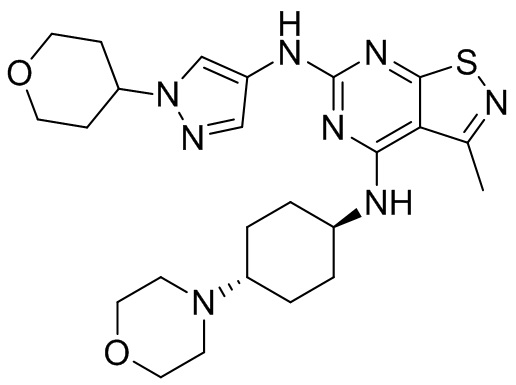 ,
, 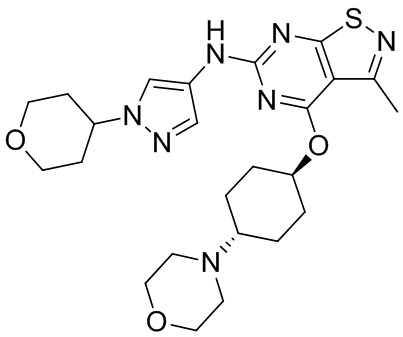 ,
, 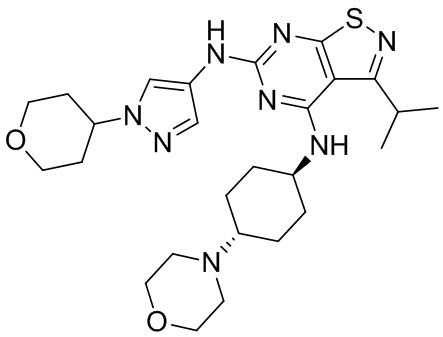 ,
, 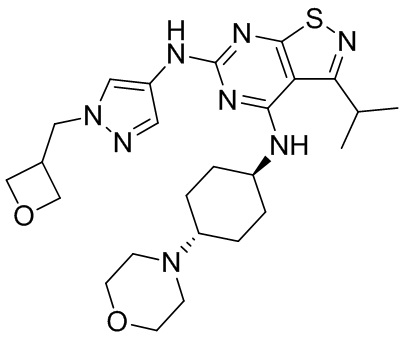 ,
, 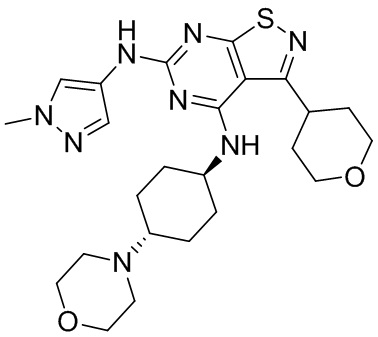 , ,
, , 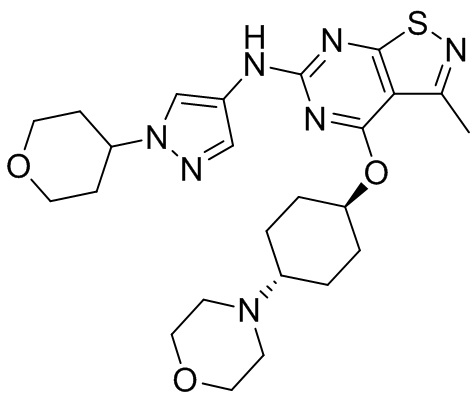 ,
, 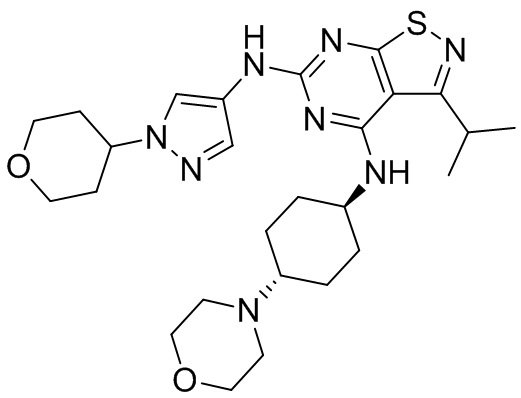 ,
, 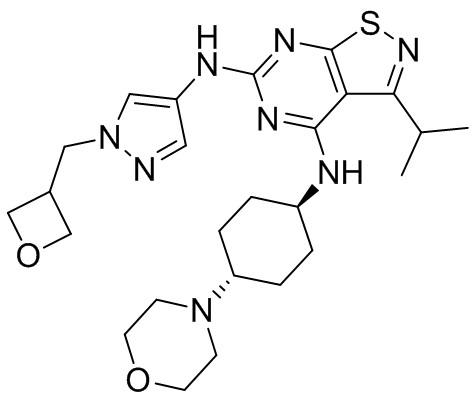 ,
, 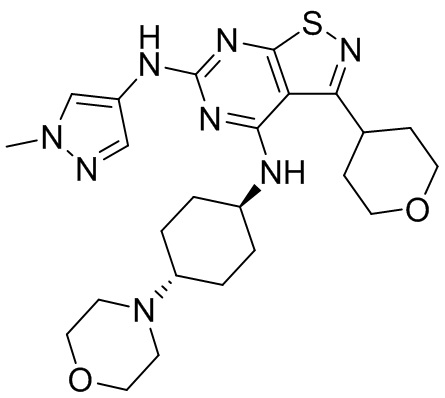 ,
, 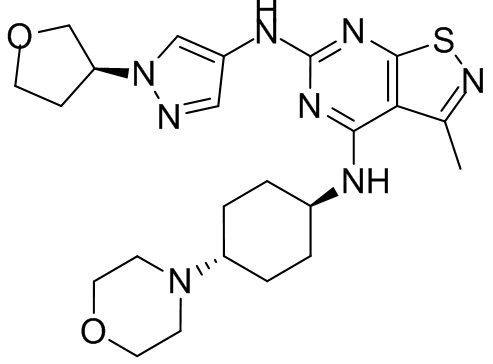 ,
, 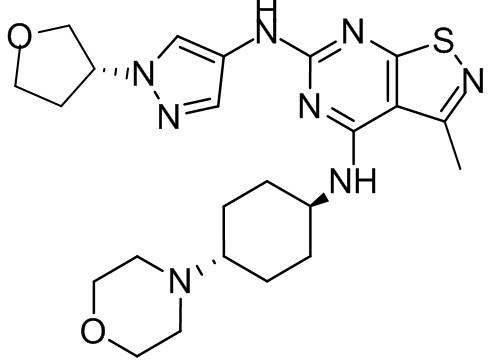 ,
, 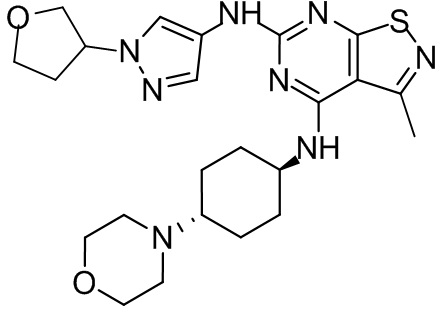 ,
,  ,
,  ,
, 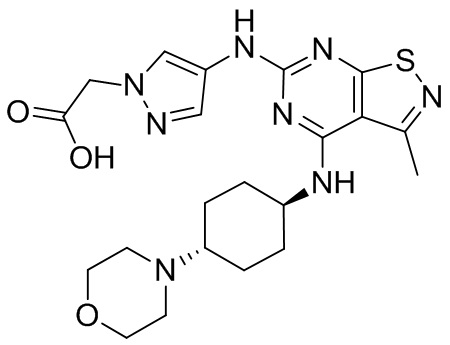 ,
, 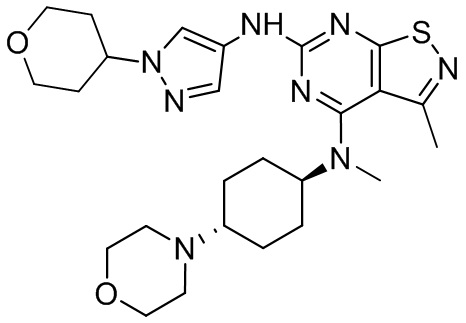 ,
, 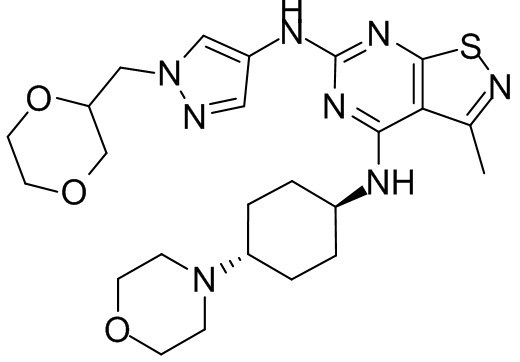 ,
, 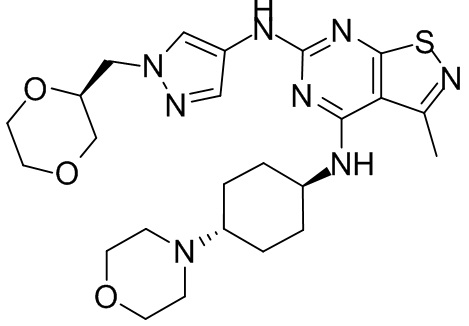 ,
,  ,
, 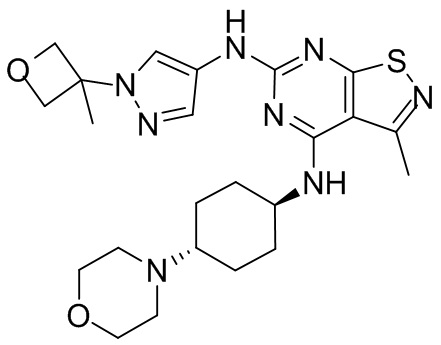 ,
, 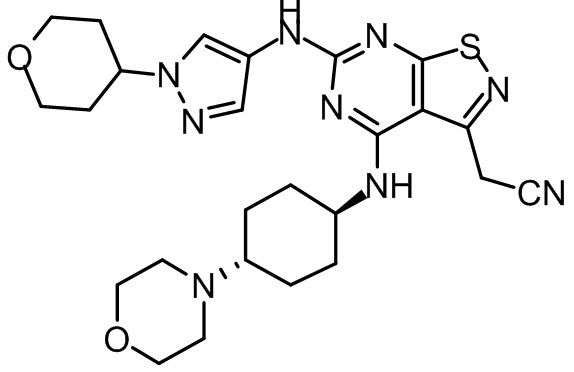
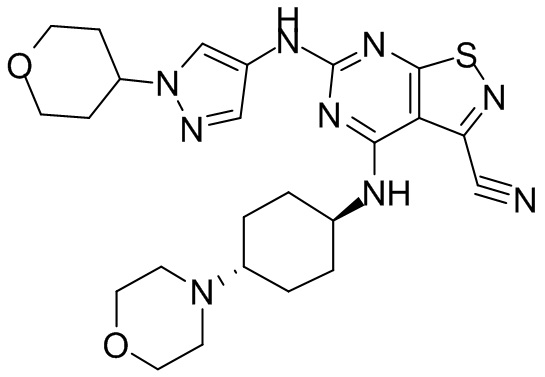 ,
, 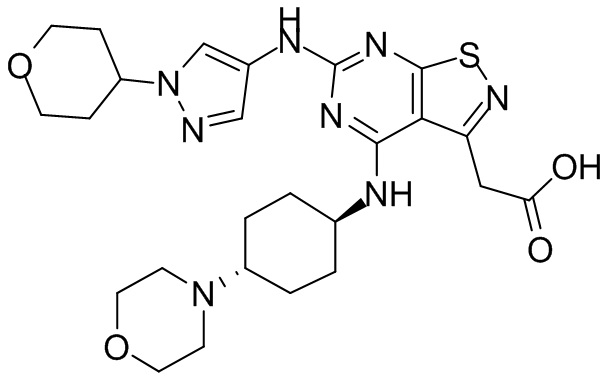 ,
, 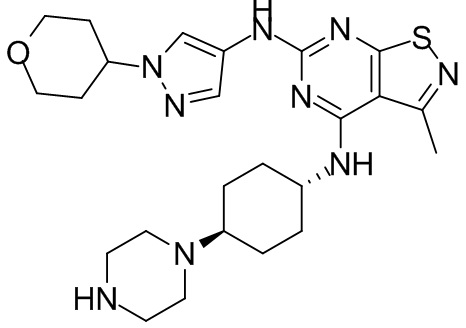 ,
, 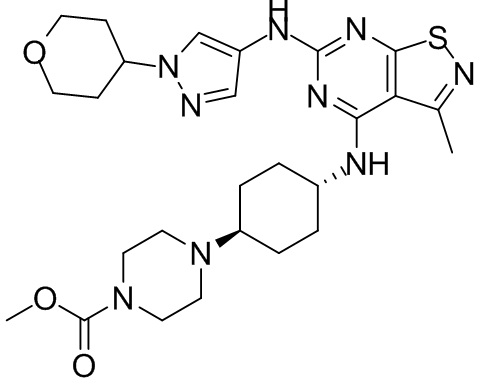 ,
, 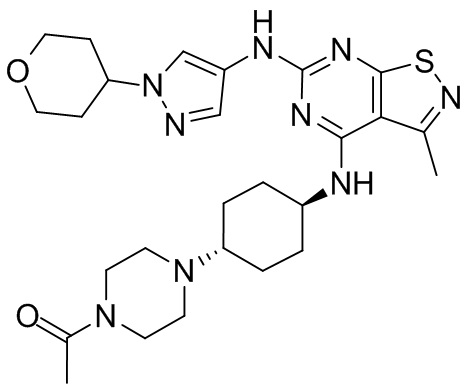 ,
, 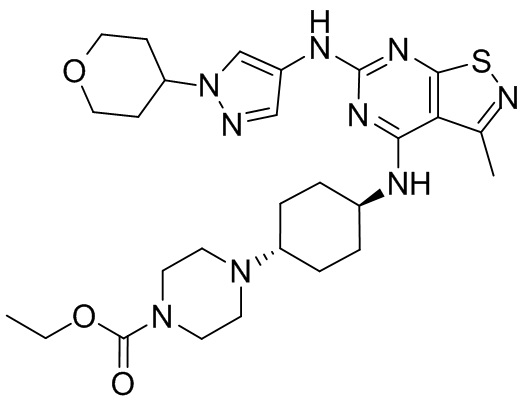 ,
, 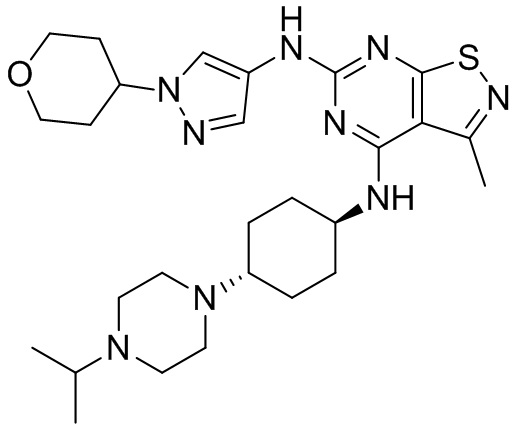 ,
, 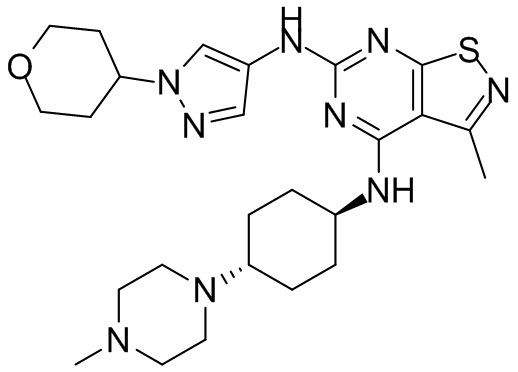 ,
, 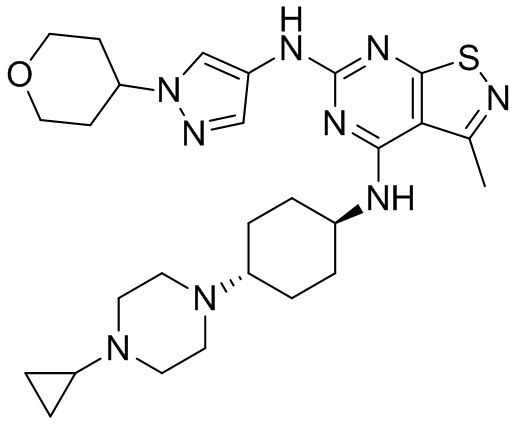 ,
, 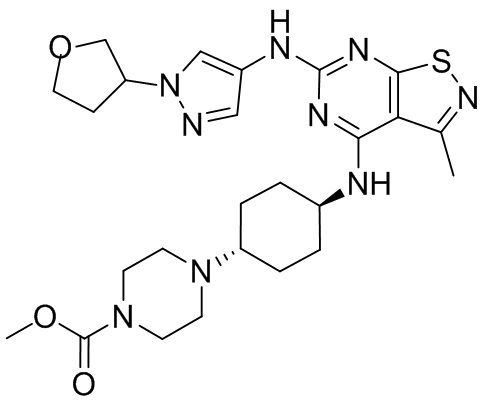 ,
, 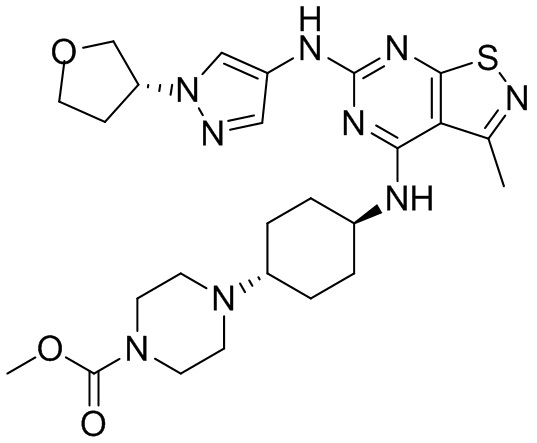 ,
, 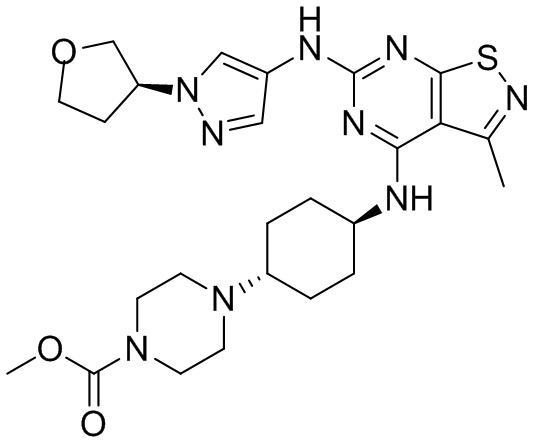 ,
, 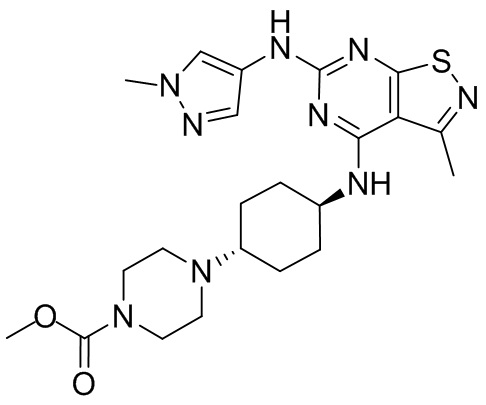 ,
,  ,
,  ,
, 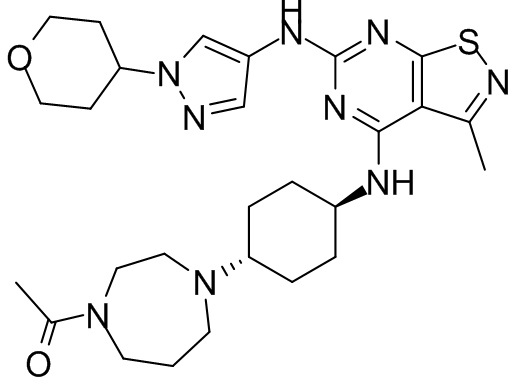 ,
,  ,
,  ,
,  ,
,  ,
, 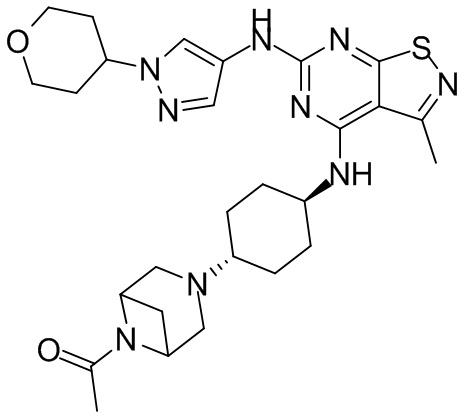 ,
, 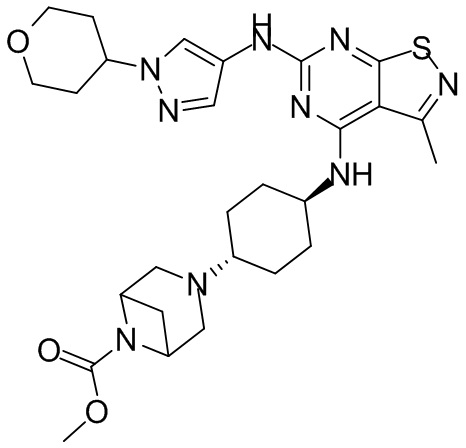 ,
, 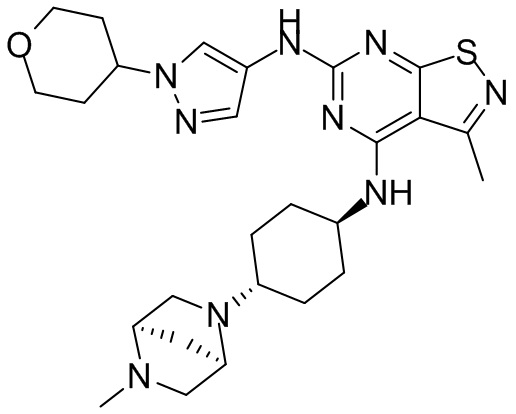 ,
,  ,
, 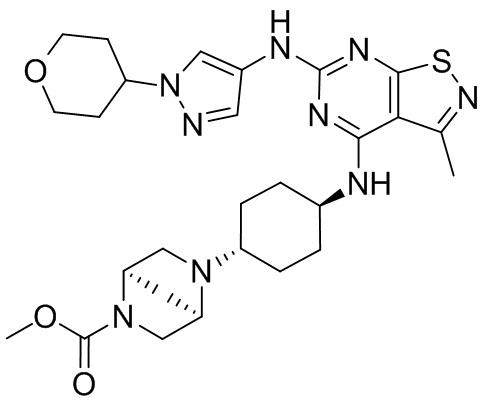 ,
, 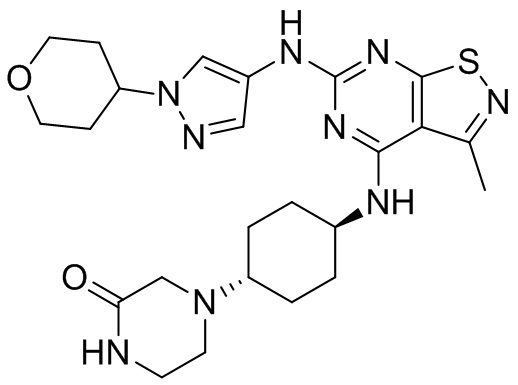 ,
,  ,
,  ,
, 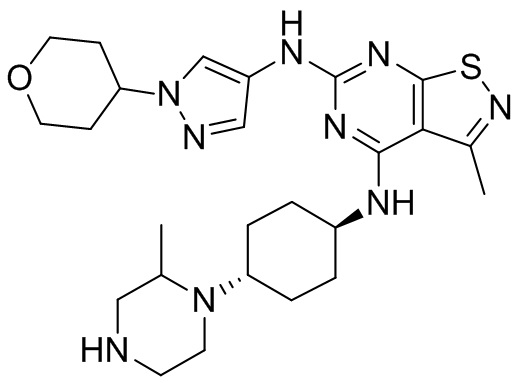 ,
, 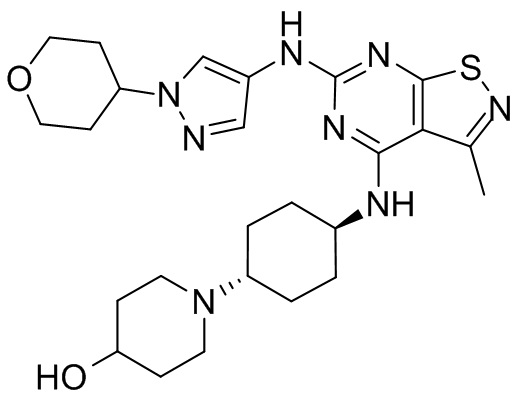 ,
, 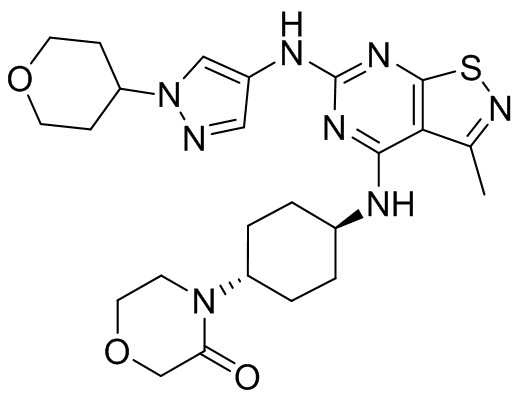 ,
, 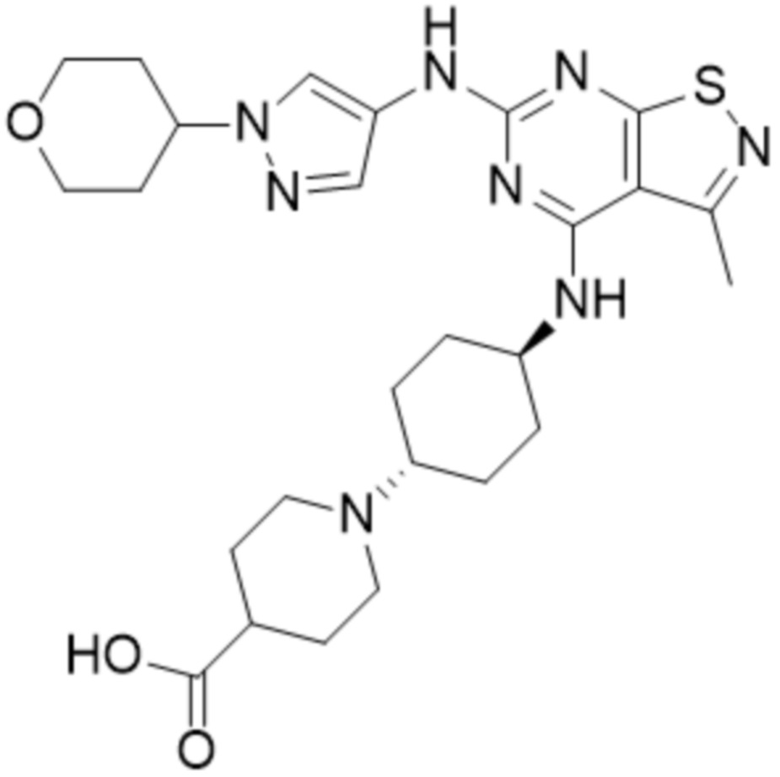 ,
, 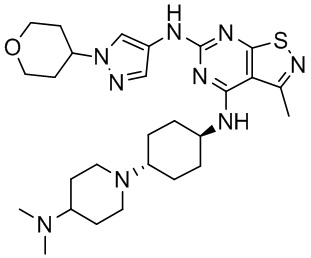 ,
, 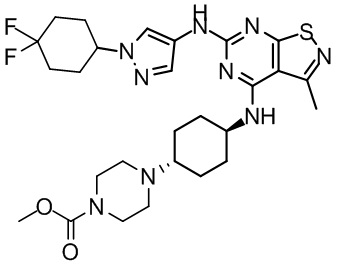 и
и 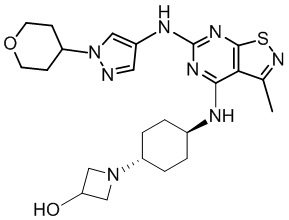 .
.
18. Фармацевтическая композиция для получения ингибитора IRAK4, содержащая терапевтически эффективное количество соединения, его изомера или фармацевтически приемлемой соли по любому из пп. 1-17 в качестве активного ингредиента и фармацевтически приемлемый носитель.
19. Применение соединения, его изомера или его фармацевтически приемлемой соли по любому из пп. 1-17 для получения ингибитора IRAK4.
20. Применение композиции по п. 18 для получения ингибитора IRAK4.
| RU 2016110852 A, 30.10.2017 | |||
| WO 2017205766 A1, 30.11.2017 | |||
| WO 2017004133 A1, 05.01.2017. |
