Ссылка на родственные заявки
Для настоящей заявки испрашивается преимущество и приоритет в соответствии с заявкой на выдачу патента Китая № 201810528259.1, поданной в Национальное управление интеллектуальной собственности КНР 25 мая 2018 года, заявкой на выдачу патента Китая № 201810843225.1, поданной в Национальное управление интеллектуальной собственности КНР 27 июля 2018 года, и заявкой на выдачу патента Китая № 201811189801.1, поданной в Национальное управление интеллектуальной собственности КНР 12 октября 2018 года, содержание каждой из которых полностью включено в настоящий документ посредством ссылки.
Область техники
Настоящая заявка относится к 2,3-дигидро-1H-пирролизин-7-формамидному производному в качестве ингибитора нуклеопротеинов и к его применению при получении лекарственного средства для лечения заболеваний, связанных с вирусом гепатита В (HBV). В частности, настоящая заявка относится к соединению, представленному формулой (II), соединению, представленному формулой (II-A), соединению, представленному формулой (II-B), соединению, представленному формулой (I), или их стереоизомерам или фармацевтически приемлемым солям, и к их применению при получении лекарственного средства для лечения заболеваний, связанных с HBV.
Предпосылки изобретения
Гепатит B является воспалительным состоянием, вызываемым инвазией вируса гепатита B. Он характеризуется тенденцией развития фиброза и цирроза печени и является прямой причиной 80% случаев первичного рака печени по всему миру.
Гепатит В является глобальной проблемой для здравоохранения. В настоящее время не существует специального лекарства для лечения гепатита B. Нуклеозиды и интерфероны занимают доминирующее положение на мировом рынке лекарственных средств против гепатита B и являются основными лекарственными средствами первой линии для лечения гепатита B. Однако лечение нуклеозидами или интерферонами характеризуется такими недостатки, как дороговизна, высокая вероятность рецидива и тому подобное. Поэтому существует потребность в разработке нового лекарственного средства против гепатита В.
Краткое описание изобретения
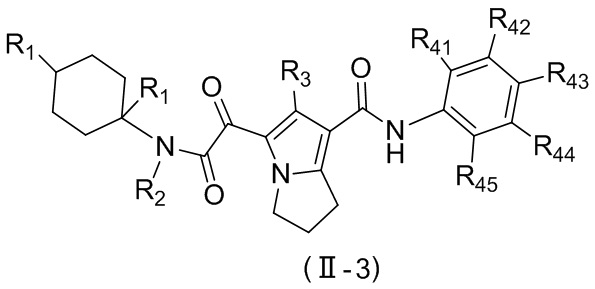
В настоящей заявке предложено соединение, представленное формулой (II), или его стереоизомер или их фармацевтически приемлемая соль:

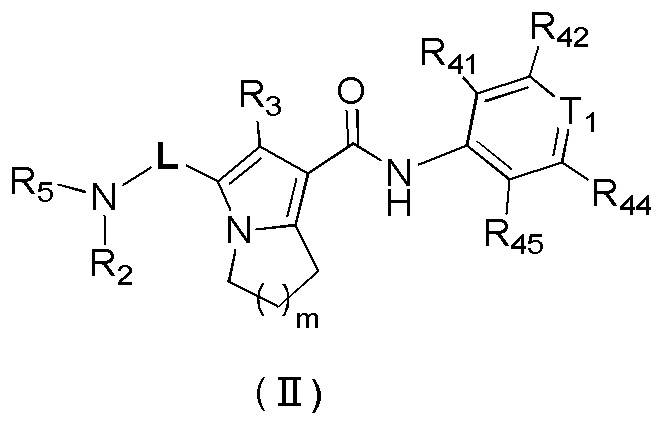 ,
,
где
m равно 1 или 2;
L выбран из 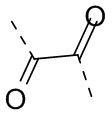 ;
;
T1 выбран из группы, состоящей из N и C (R43);
R2 выбран из группы, состоящей из H и C1-6-алкила, причем C1-6-алкил необязательно имеет 1, 2 или 3 заместителя Rb;
R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, C1-6-алкила и C1-3-алкокси, причем C1-6-алкил и C1-3-алкокси необязательно имеют 1, 2 или 3 заместителя Rc;
каждый из R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rd;
R5 выбран из группы, состоящей из R51, C3-10-циклоалкила и 3-6-членного гетероциклоалкила, причем C3-10-циклоалкил и 3-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1;
R51 выбран из группы, состоящей из C1-10-алкила и C1-6-гетероалкила, причем C1-10-алкил и C1-6-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re;
каждый R1 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-6-алкила, -COO-C1-6-алкила и -C1-3-алкил-COO-C1-6-алкила, причем C1-6-алкил, -COO-C1-6-алкил и -C1-3-алкил-COO-C1-6-алкил необязательно имеют 1, 2 или 3 заместителя Ra;
каждый Ra независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила и C1-3-алкокси, причем C1-3 необязательно имеет 1, 2 или 3 заместителя R;
каждый Rb независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Rc независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя R;
каждый Rd независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Re независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый R независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый из C1-6-гетероалкила и 3-6-членного гетероциклоалкила содержит 1, 2, 3 или 4 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Ra выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и -OCH3, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Rc выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый вышеупомянутый R1 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила, -COO-C1-3-алкила и -C1-3-алкил-COO-C1-3-алкила, причем C1-3-алкил, -COO-C1-3-алкил и -C1-3-алкил-COO-C1-3-алкил необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый вышеупомянутый R1 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3, Et,  и
и  , причем CH3, Et, и необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
, причем CH3, Et, и необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый вышеупомянутый R1 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3, CF3, Et, -CH2-COOH, -CH2-OCH3, -(CH2)2-COOH, и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H и C1-3 алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rb, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2 и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rc, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CH3, CF3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый из вышеупомянутого R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN и -COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из C1-7-алкила и C1-6-гетероалкила, причем C1-7-алкил и C1-6-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из C1-7-алкила и C1-3-гетероалкила, причем C1-7-алкил и C1-3-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из метила, этила, пропила, изопропила, 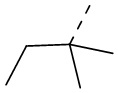 ,
, 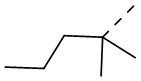 и
и 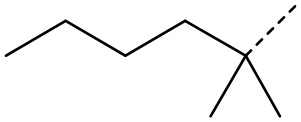 , причем метил, этил, пропил, изопропил, , и необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
, причем метил, этил, пропил, изопропил, , и необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из 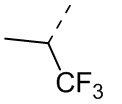 ,
,  ,
,  и
и  , а остальные переменные являются такими, как определено в настоящем документе.
, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, C3-8-циклоалкила и 5-6-членного гетероциклоалкила, причем C3-8-циклоалкил и 5-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, циклогексила, тетрагидропиранила, пиперидинила и бицикло[2.2.2]октила, причем циклогексил, тетрагидропиранил, пиперидинил и бицикло[2.2.2]октил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51,  ,
, 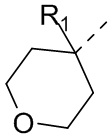 ,
, 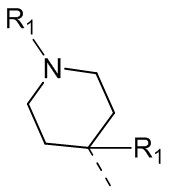 и
и 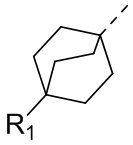 , а остальные переменные являются такими, как определено в настоящем документе.
, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из , , , , 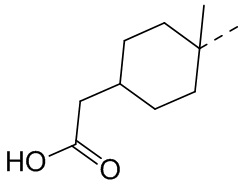 ,
,  ,
,  ,
,  ,
,  ,
, 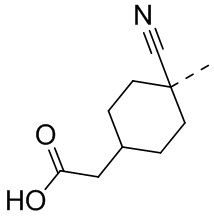 ,
, 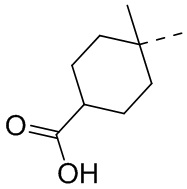 ,
, 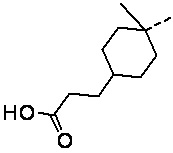 ,
, 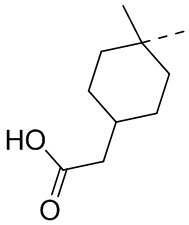 ,
,  ,
,  ,
,  и
и  , а остальные переменные являются такими, как определено в настоящем документе.
, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутая структурная единица 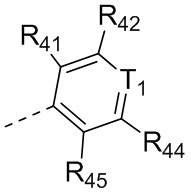 выбрана из группы, состоящей из
выбрана из группы, состоящей из  ,
,  ,
,  и
и  , а остальные переменные являются такими, как определено в настоящем документе.
, а остальные переменные являются такими, как определено в настоящем документе.
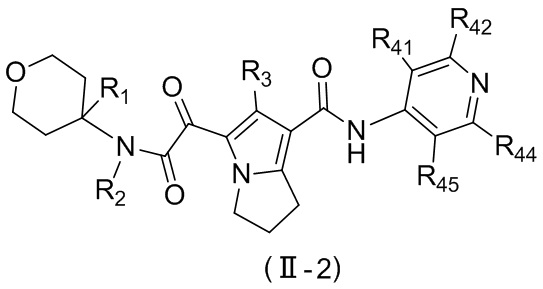
Соединение, представленное формулой (II), или его стереоизомер или их фармацевтически приемлемая соль, которые описаны выше, выбраны из соединения, представленного формулой (II-A), или его стереоизомера или их фармацевтически приемлемой соли:
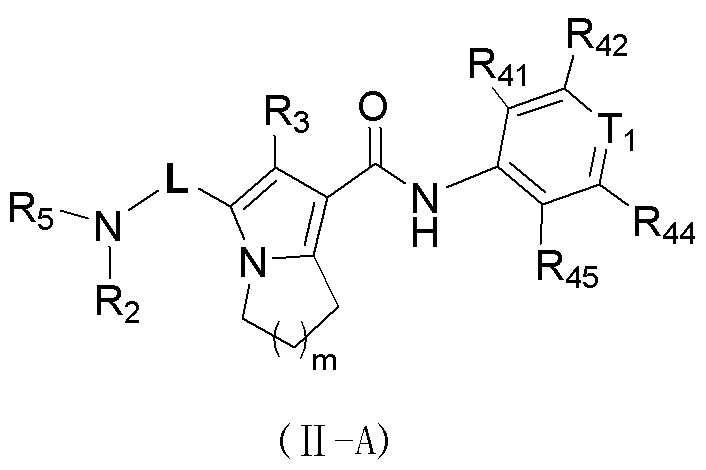 ,
,
где
m равно 1 или 2;
L выбран из ;
T1 выбран из группы, состоящей из N и C (R43);
R2 выбран из группы, состоящей из H и C1-6-алкила, причем C1-6-алкил необязательно имеет 1, 2 или 3 заместителя Rb;
R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, C1-6-алкила и C1-3-алкокси, причем C1-6-алкил и C1-3-алкокси необязательно имеют 1, 2 или 3 заместителя Rc;
каждый из R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rd;
R5 выбран из группы, состоящей из R51, C3-10-циклоалкила и 3-6-членного гетероциклоалкила, причем C3-10-циклоалкил и 3-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1;
R51 выбран из группы, состоящей из C1-7-алкила и C1-6-гетероалкила, причем C1-7-алкил и C1-6-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re;
R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-6-алкила, -COO-C1-6-алкила и -C1-3-алкил-COO-C1-6-алкила, причем C1-6-алкил, -COO-C1-6-алкил и -C1-3-алкил-COO-C1-6-алкил необязательно имеют 1, 2 или 3 заместителя Ra;
каждый Ra независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила и C1-3-алкокси, причем C1-3 необязательно имеет 1, 2 или 3 заместителя R;
каждый Rb независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Rc независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя R;
каждый Rd независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Re независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый R независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый из C1-6-гетероалкила и 3-6-членного гетероциклоалкила содержит 1, 2, 3 или 4 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Ra выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и -OCH3, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Rc выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила, -COO-C1-3-алкила и -C1-3-алкил-COO-C1-3-алкила, причем C1-3-алкил, -COO-C1-3-алкил и -C1-3-алкил-COO-C1-3-алкил необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3 и Et, причем CH3 и Et необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3, Et, -CH2-COOH, -CH2-OCH3 и -(CH2)2-COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rb, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2 и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rc, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CH3, CF3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый из вышеупомянутого R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN и -COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из C1-6-алкила и C1-3-гетероалкила, причем C1-6-алкил и C1-3-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из метила, этила, пропила, изопропила, и , причем метил, этил, пропил, изопропил, и необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из , и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, C3-8-циклоалкила и 5-6-членного гетероциклоалкила, причем C3-8-циклоалкил и 5-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, циклогексила, тетрагидропиранила, пиперидинила и бицикло[2.2.2]октила, причем циклогексил, тетрагидропиранил, пиперидинил и бицикло[2.2.2]октил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, , , и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из , , , , , , , , , , , и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-А), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутая структурная единица выбрана из группы, состоящей из , , и , а остальные переменные являются такими, как определено в настоящем документе.
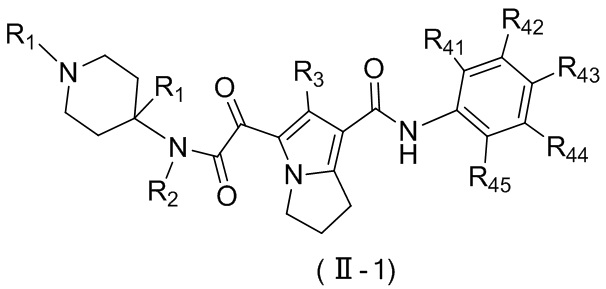
Соединение, представленное формулой (II), или его стереоизомер или их фармацевтически приемлемая соль, которые описаны выше, выбраны из соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли:
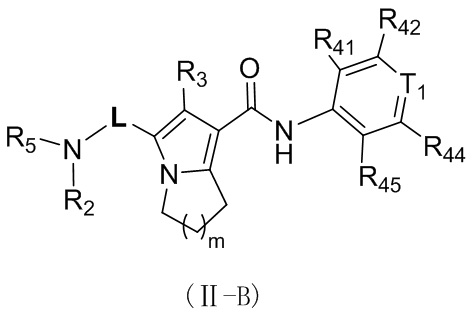 ,
,
где
m равно 1 или 2;
L выбран из ;
T1 выбран из группы, состоящей из N и C (R43);
R2 выбран из группы, состоящей из H и C1-6-алкила, причем C1-6-алкил необязательно имеет 1, 2 или 3 заместителя Rb;
R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, C1-6-алкила и C1-3-алкокси, причем C1-6-алкил и C1-3-алкокси необязательно имеют 1, 2 или 3 заместителя Rc;
каждый из R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rd;
R5 выбран из группы, состоящей из R51, C3-6-циклоалкила и 3-6-членного гетероциклоалкила, причем 5-6-членный циклоалкил и 3-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1;
R51 выбран из группы, состоящей из C1-6-алкила и C1-6-гетероалкила, причем C1-6-алкил и C1-6-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re;
R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-6-алкила, -COO-C1-6-алкила и -C1-3-алкил-COO-C1-6-алкила, причем C1-6-алкил, -COO-C1-6-алкил и -C1-3-алкил-COO-C1-6-алкил необязательно имеют 1, 2 или 3 заместителя Ra;
каждый Ra независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила и C1-3-алкокси, причем C1-3 необязательно имеет 1, 2 или 3 заместителя R;
каждый Rb независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Rc независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя R;
каждый Rd независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый Re независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый R независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
каждый из C1-6-гетероалкила и 3-6-членного гетероциклоалкила содержит 1, 2, 3 или 4 гетероатома или гетероатомные группы, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Ra выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и -OCH3, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Rc выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила, -COO-C1-3-алкила и -C1-3-алкил-COO-C1-3-алкила, причем C1-3-алкил, -COO-C1-3-алкил и -C1-3-алкил-COO-C1-3-алкил необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3 и Et, причем CH3 и Et необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3, Et, -CH2-COOH и -CH2-OCH3, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rb, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2 и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rc, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CH3, CF3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый из вышеупомянутого R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN и -COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из C1-3-алкила и C1-3-гетероалкила, причем C1-3-алкил и C1-3 -гетероалкил необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 выбран из группы, состоящей из метила, этила, пропила и изопропила, причем метил, этил, пропил и изопропил необязательно имеют 1, 2 или 3 заместителя Re, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R51 представляет собой , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, C5-6-циклоалкила и 5-6-членного гетероциклоалкила, причем 5-6 членный циклоалкил и 5-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, циклогексила, тетрагидропиранила и пиперидинила, причем циклогексил, тетрагидропиранил и пиперидинил необязательно имеют 1, 2 или 3 заместителя R1, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из R51, , и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R5 выбран из группы, состоящей из , , , , и , а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (II-B), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутая структурная единица выбрана из группы, состоящей из , , и , а остальные переменные являются такими, как определено в настоящем документе.
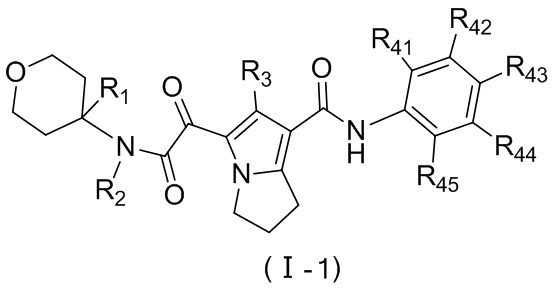
Соединение, представленное формулой (II), или его стереоизомер или их фармацевтически приемлемая соль выбраны из соединения, представленного формулой (I), или его фармацевтически приемлемой соли:
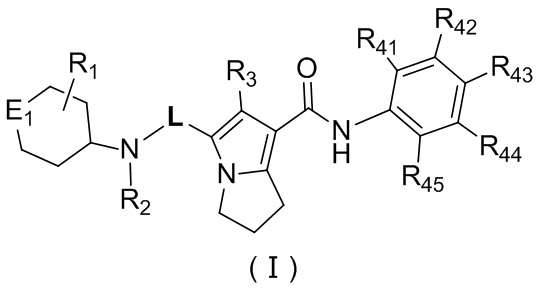 ,
,
где
L выбран из ;
E1 выбран из группы, состоящей из -O-, -S- и -NH-;
R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-6-алкила, -COO-C1-6-алкила и -C1-3-алкил-COO-C1-6-алкила, причем C1-6-алкил, -COO-C1-6-алкил и -C1-3-алкил-COO-C1-6-алкил необязательно имеют 1, 2 или 3 заместителя Ra;
R2 выбран из группы, состоящей из H и C1-6-алкила, причем C1-6-алкил необязательно имеет 1, 2 или 3 заместителя Rb;
R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, C1-6-алкила и C1-3-алкокси, причем C1-6-алкил и C1-3-алкокси необязательно имеют 1, 2 или 3 заместителя Rc;
каждый из R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rd;
Ra выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3 необязательно имеет 1, 2 или 3 заместителя R;
Rb выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
Rc выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN, COOH и C1-3-алкила, причем C1-3 необязательно имеет 1, 2 или 3 заместителя R;
каждый Rd независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH;
и каждый R независимо выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Ra выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый Rc выбран из группы, состоящей из Cl, F, Br, I, OH, NH2, CN и COOH, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, C1-3-алкила, -COO-C1-3-алкила и -C1-3-алкил-COO-C1-3-алкила, причем C1-3-алкил, -COO-C1-3-алкил и -C1-3-алкил-COO-C1-3-алкил необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3 и Et, причем CH3 и Et необязательно имеют 1, 2 или 3 заместителя Ra, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R1 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN, COOH, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rb, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R2 выбран из группы, состоящей из H, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2 и C1-3-алкила, причем C1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rc, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, вышеупомянутый R3 выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CH3 и Et, а остальные переменные являются такими, как определено в настоящем документе.
В некоторых вариантах осуществления соединения, представленного формулой (I), или его стереоизомера или их фармацевтически приемлемой соли, которые раскрыты в настоящем документе, каждый из вышеупомянутого R41, R42, R43, R44 и R45 независимо выбран из группы, состоящей из H, Cl, F, Br, I, OH, NH2, CN и -COOH, а остальные переменные являются такими, как определено в настоящем документе.
Некоторые другие варианты осуществления настоящей заявки получают в результате любого сочетания описанных выше переменных.
В некоторых вариантах осуществления настоящей заявки описанные выше соединения или их стереоизомеры или фармацевтически приемлемые соли выбраны из группы, состоящей из
 ,
, ,
,
 ,
, ,
,
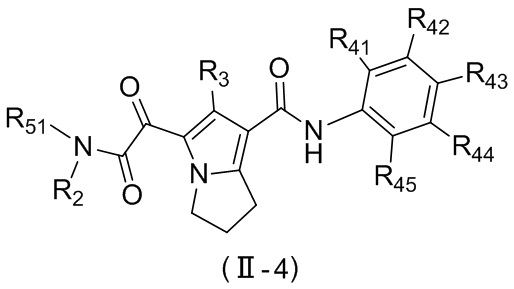 ,
,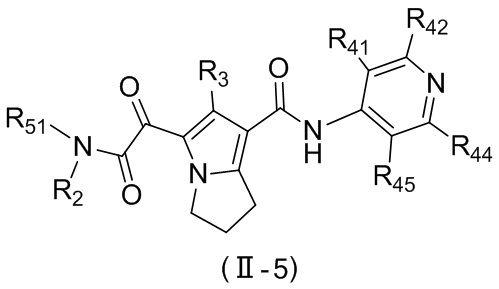 ,
,
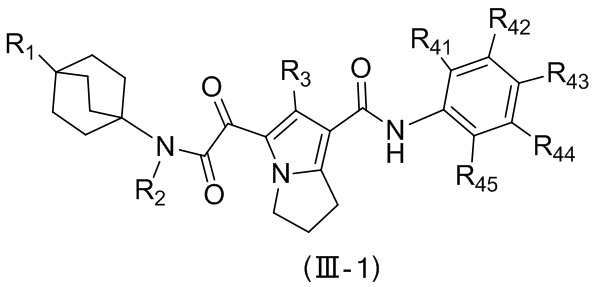 ,
,
где
R1, R2, R3, R41, R42, R43, R44, R45 и R51 являются такими, как определено в настоящем документе.
В настоящей заявке дополнительно предложены соединения, представленные приведенными далее формулами, или их стереоизомеры или фармацевтически приемлемые соли:
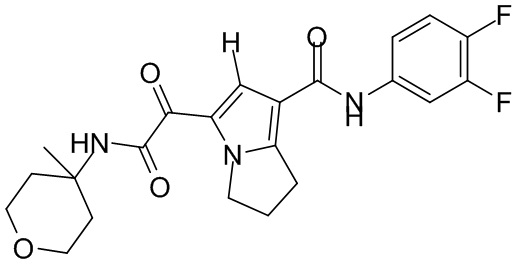 ,
, 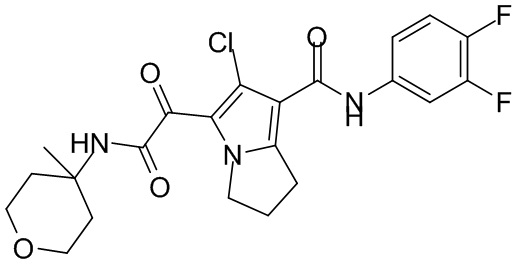 ,
, 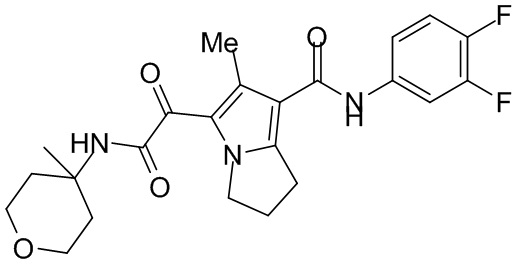 ,
, 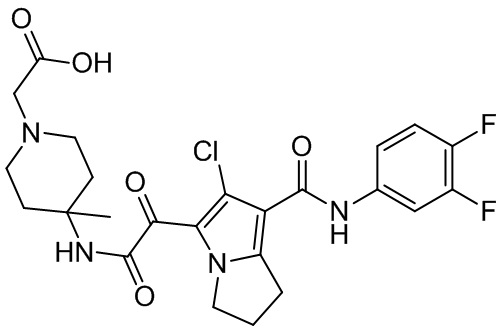 ,
, 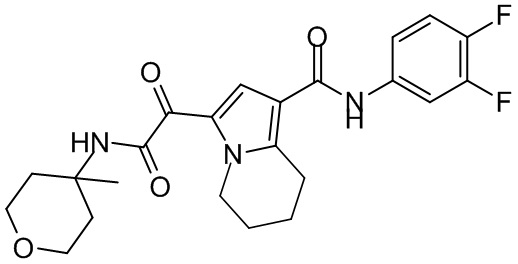 ,
, 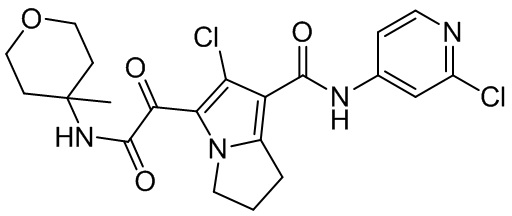 ,
, 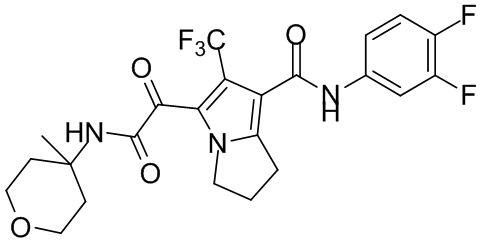 ,
, 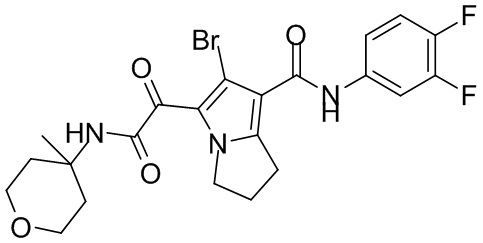 ,
, 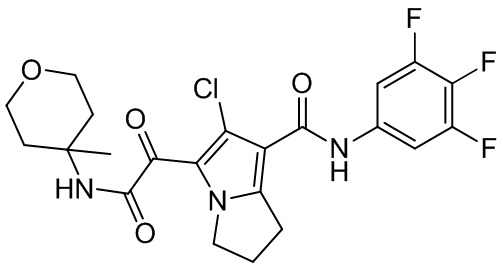 ,
, 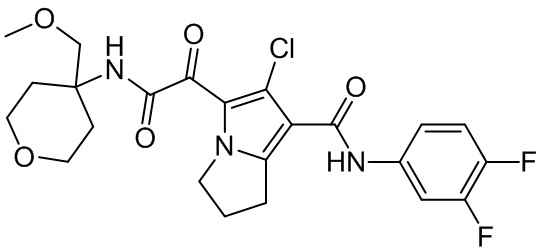 ,
, 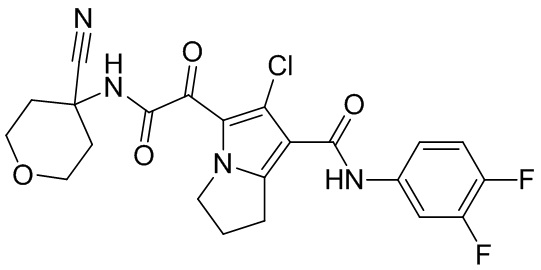 ,
, 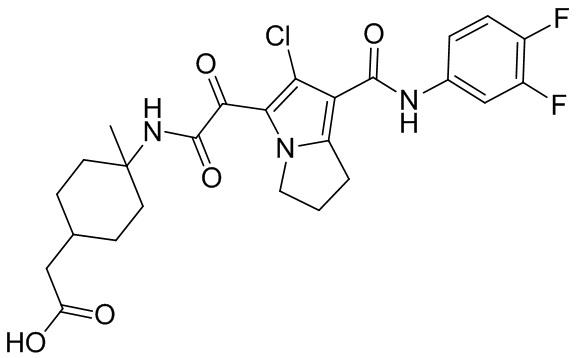 ,
, 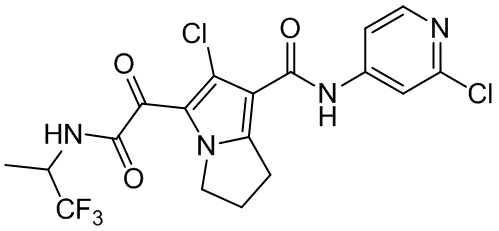 ,
, 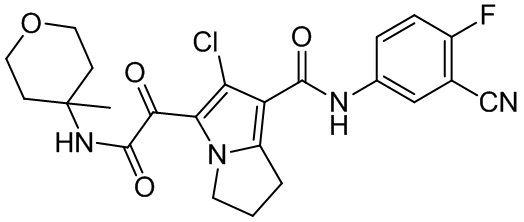 ,
, 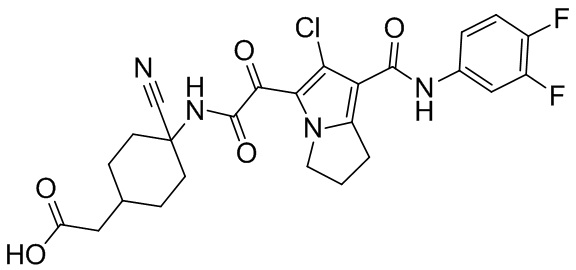 ,
, 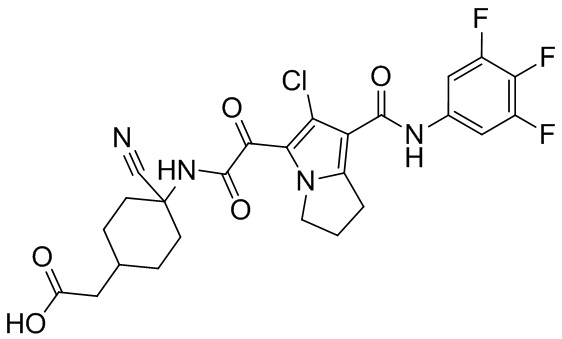 ,
,  ,
, 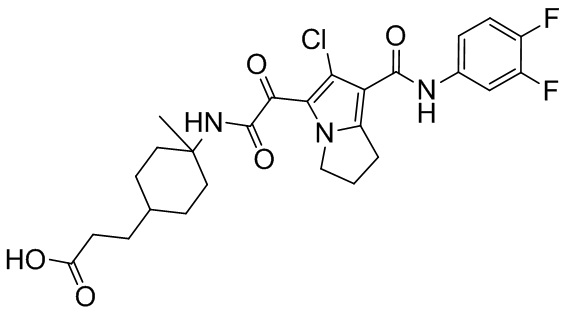 ,
, 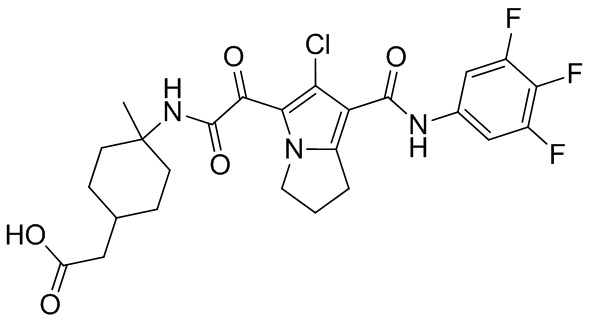 ,
, 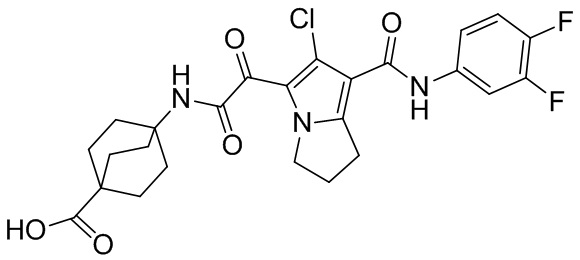 ,
, 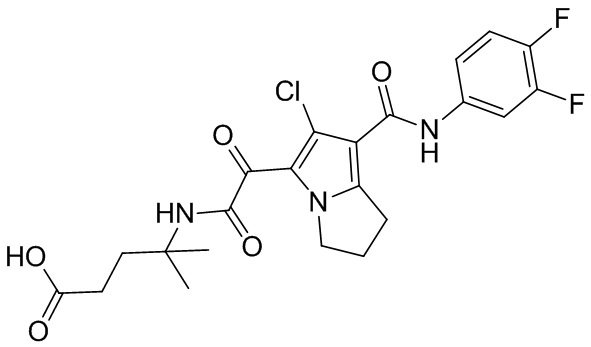 ,
, 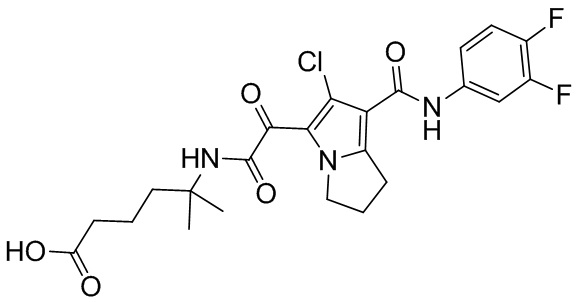 ,
,  ,
, 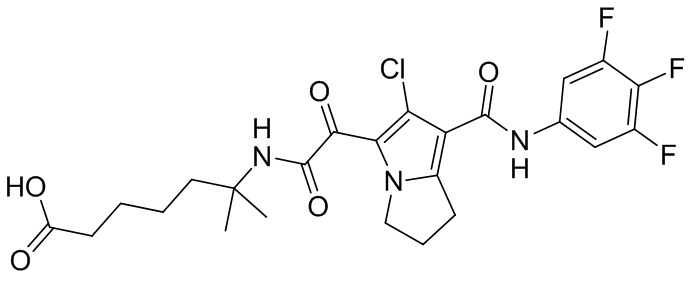 ,
, 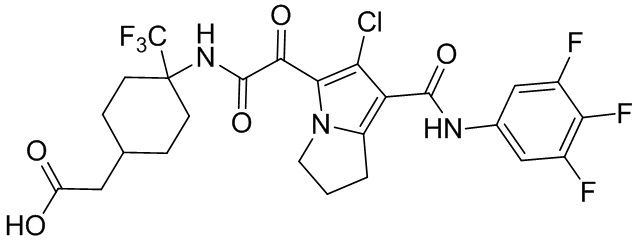 и
и 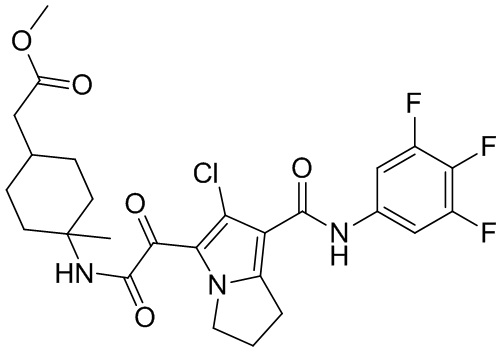 .
.
В некоторых вариантах осуществления настоящей заявки описанные выше соединения или их стереоизомеры или фармацевтически приемлемые соли выбраны из группы, состоящей из
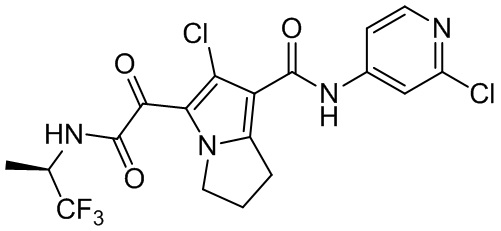 ,
, 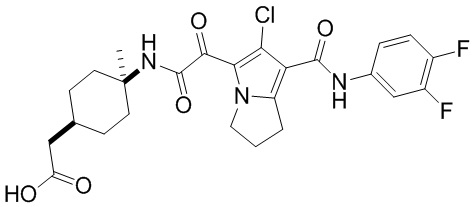 ,
, 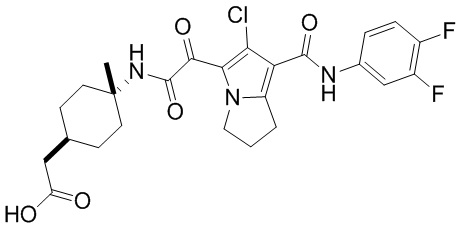 ,
,  ,
, 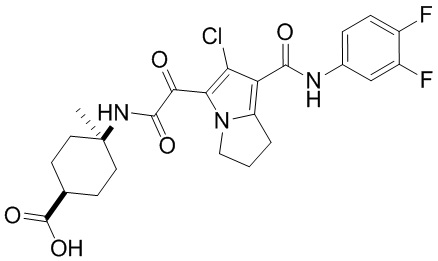 ,
, 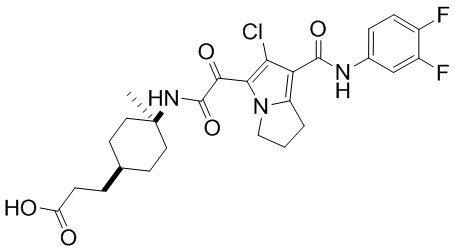 ,
, 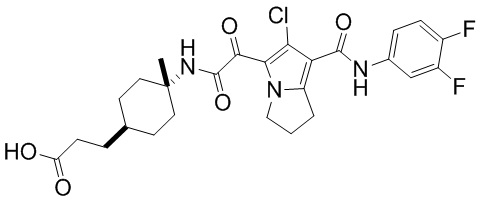 ,
, 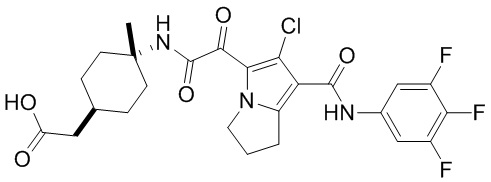 ,
, 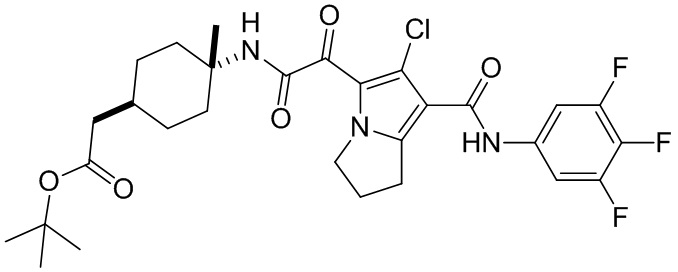 ,
, 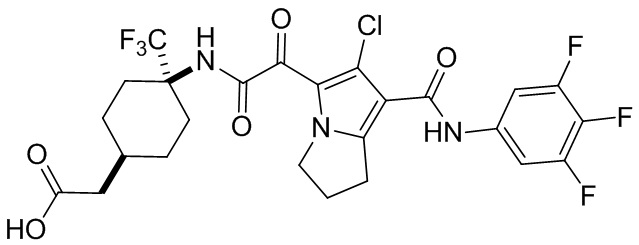 ,
, 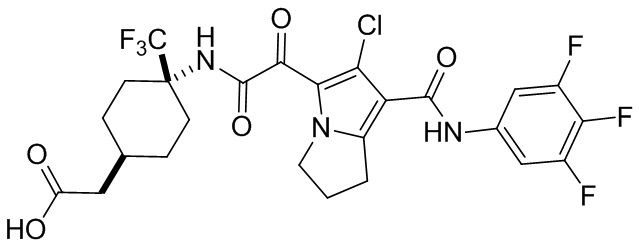 и
и 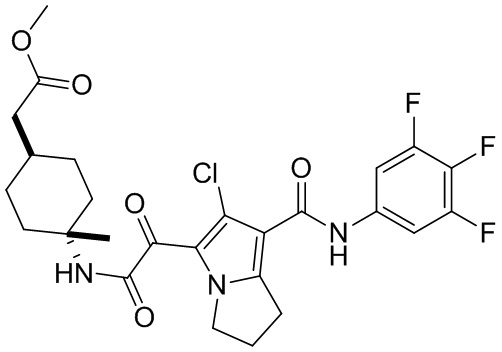 .
.
В настоящей заявке дополнительно предложена фармацевтическая композиция, содержащая терапевтически эффективное количество описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей в качестве действующего ингредиента и фармацевтически приемлемый носитель.
В настоящей заявке дополнительно предложено применение описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей при получении лекарственного средства для ингибирования нуклеопротеина.
В настоящей заявке дополнительно предложено применение описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей в качестве ингибитора нуклеопротеинов.
В настоящей заявке дополнительно предложен способ ингибирования нуклеопротеина, предусматривающий введение нуждающемуся в таком лечении или предупреждении млекопитающему, предпочтительно человеку, терапевтически эффективного количества описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей или содержащей их фармацевтической композиции.
В настоящей заявке дополнительно предложены описанные выше соединения, их стереоизомеры или фармацевтически приемлемые соли или содержащая их фармацевтическая композиция для применения в качестве ингибитора нуклеопротеинов.
В некоторых вариантах осуществления настоящей заявки вышеупомянутое применение характеризуется тем, что фармацевтический ингибитор нуклеопротеинов представляет собой лекарственное средство для применения при лечении или предупреждении заболеваний, связанных с инфекцией HBV.
В настоящей заявке дополнительно предложено применение описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей при получении лекарственного средства для лечения или предупреждения заболеваний, связанных с инфекцией HBV.
В настоящей заявке дополнительно предложено применение описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей для лечения или предупреждения заболеваний, связанных с инфекцией HBV.
В настоящей заявке дополнительно предложен способ лечения заболеваний, связанных с инфекцией HBV, предусматривающий введение нуждающемуся в таком лечении или предупреждении млекопитающему, предпочтительно человеку, терапевтически эффективного количества описанных выше соединений или их стереоизомеров или фармацевтически приемлемых солей или содержащей их фармацевтической композиции.
В настоящей заявке дополнительно предложены описанные выше соединения или их стереоизомеры или фармацевтически приемлемые соли или содержащая их фармацевтическая композиция для применения при лечении или предупреждении заболеваний, связанных с инфекцией HBV.
Технические результаты
В качестве нового лекарственного средства против гепатита B описанные в настоящем документе соединения обладают заметным ингибирующим действием, оказываемым на HBV. Раскрываемые в настоящем документе соединения проявляют хорошие фармакокинетические свойства в том, что касается всасывания, распределения in vivo, метаболизма и т.д., например, хорошее печень-ориентированное действие in vivo. Раскрываемые в настоящем документе соединения характеризуются низким уровнем токсичного побочного действия.
Определения и описание
Если не указано иное, приведенные далее термины и фразы, применяемые в настоящем документе, имеют приведенные далее значения. Конкретный термин или конкретную фразу, если явно не определено иное, не следует истолковывать как неопределенные или неясные, а следует толковать в соответствии с их общим значением. При упоминании торгового наименования подразумевают отсылку к соответствующему коммерческому продукту или его действующему ингредиенту.
В контексте настоящего документа термин «фармацевтически приемлемый» применяют в отношении тех соединений, материалов, композиций и/или лекарственных форм, которые, с медицинской точки зрения, подходят для применения в контакте с тканями людей и животных, не вызывают чрезмерную токсичность, раздражение, аллергический ответ или другие проблемы или осложнения и соответствуют разумному соотношению польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли раскрываемого в настоящем документе соединения по настоящему изобретению, которая получена из соединения, содержащего определенные заместители, раскрываемые в настоящем документе, и относительно нетоксичной кислоты или основания. Если раскрываемое в настоящем документе соединение содержит относительно кислую функциональную группу, то соль присоединения основания можно получить путем приведения нейтральной формы такого соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. К фармацевтически приемлемым солям присоединения оснований относятся соли натрия, калия, кальция, аммония, органического амина или магния или аналогичные соли. Если раскрываемое в настоящем документе соединение содержит относительно основную функциональную группу, то соль присоединения кислоты можно получить путем приведения нейтральной формы такого соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли, полученные из неорганических кислот, таких как соляная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонатный радикал, фосфорная кислота, моногидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота, фосфористая кислота и др.; а также соли, полученные из органических кислот, таких как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, винная кислота, метансульфоновая кислота и др. Также включены соли аминокислот (например, аргинина и т.д.) и соли органических кислот, таких как глюкуроновая кислота. Некоторые конкретные раскрываемые в настоящем документе соединения содержат как основные, так и кислотные функциональные группы, которые позволяют превращать соединения в соли присоединения либо основания, либо кислоты.
Описываемые в настоящем документе соединения могут иметь форму геометрического изомера или стереоизомера. В настоящем документе предусмотрены все такие соединения, включая цис- и транс-изомеры, (-)- и (+)- энантиомеры, (R)- и (S)- энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры и рацемические смеси и другие их смеси, такие как обогащенная энантиомерами или диастереомерами смесь, все из которых входят в объем настоящей заявки. Такие заместители, как алкил, могут иметь дополнительный асимметричный атом углерода. Все такие изомеры и их смеси входят в объем настоящей заявки.
Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стереоизомерам, которые являются зеркальными отображениями друг друга.
Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» описывает следствие неспособности свободно вращаться двойной или одинарной связи кольцевого атома углерода.
Если не указанно иное, термин «диастереомер» относится к стереоизомерам, молекулы которых имеют два или более хиральных центров, и которые не являются зеркальным отображением друг друга.
Если не указанно иное, «(D)» или «(+)» обозначает правостороннее вращение, «(L)» или «(-)» обозначает левостороннее вращение, а «(DL)» или «(±)» обозначают рацемизацию.
Если не указанно иное, абсолютная конфигурация стереогенного центра представлена клиновидной сплошной связью ( ) и клиновидной пунктирной связью (
) и клиновидной пунктирной связью ( ), а относительная конфигурация стереогенного центра представлена прямой сплошной связью (
), а относительная конфигурация стереогенного центра представлена прямой сплошной связью ( ) и прямой пунктирной связью (
) и прямой пунктирной связью ( ). Волнистая линия (
). Волнистая линия ( ) обозначает клиновидную сплошную связь () или клиновидную пунктирную связь (), или волнистой линией () обозначает прямую сплошную связь () и прямую пунктирную связь ().
) обозначает клиновидную сплошную связь () или клиновидную пунктирную связь (), или волнистой линией () обозначает прямую сплошную связь () и прямую пунктирную связь ().
Если не указано иное, если в соединении присутствует структура двойной связи, такая как двойная углерод-углеродная связь, двойная углерод-азотная связь и двойная азот-азотная связь, и каждый атом двойной связи связан с двумя разными заместителями (в двойной связи, включающей атом азота, неподеленная пара электронов на атоме азота рассматривается в качестве заместителя, с которым связан атом азота), если атом на двойной связи соединения и его заместители связаны с помощью волнистой линии (), это означает, что соединение существует в форме изомера (Z)-типа, изомера (E)-типа или смеси двух изомеров. Например, представленная далее формула (A) означает, что соединение существует в форме одного изомера, представленного формулой (A-1) или формулой (A-2), или в форме смеси обоих изомеров, представленных формулой (A-1) и формулой (А-2); представленная далее формула (B) означает, что соединение существует в форме одного изомера, представленного формулой (B-1) или формулой (B-2), или в форме смеси обоих изомеров, представленных формулой (B-1) и формулой (В-2); а представленная далее формула (C) означает, что соединение существует в форме одного изомера, представленного формулой (C-1) или формулой (C-2), или в форме смеси обоих изомеров, представленных формулой (C-1) и формулой (C-2).
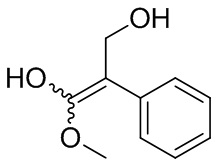 (A)
(A) 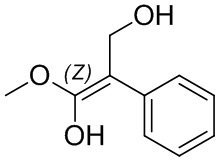 (A-1)
(A-1) 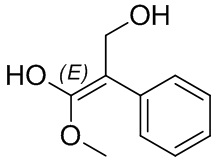 (A-2)
(A-2)
 (B)
(B) 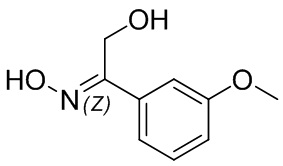 (B-1)
(B-1) 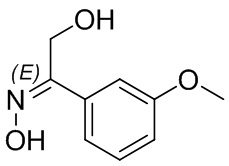 (B-2)
(B-2)
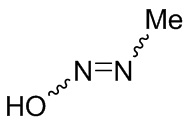 (C)
(C) 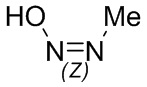 (C-1)
(C-1) 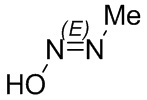 (C-2)
(C-2)
Раскрываемые в настоящем документе соединения могут присутствовать в конкретной форме. Если не указано иное, термин «таутомер» или «таутомерная форма» означает, что различные функциональные изомеры находятся в динамическом равновесии при комнатной температуре и могут легко превращаться друг в друга. Если возможны таутомеры (например, в растворе), то может быть достигнуто химическое равновесие таутомеров. Например, протонный таутомер, также известный как прототропный таутомер, включает взаимное превращение за счет переноса протона, такое как кето-енольная изомеризация и имин-енаминовая изомеризация. Валентный изомер включает взаимное превращение путем перегруппировки некоторых участвующих в формировании связи электронов. Конкретным примером кето-енольной таутомерии является взаимопревращение между двумя таутомерами: пентан-2,4-дионом и 4-гидроксипент-3-ен-2-оном.
Если не указано иное, термин «быть обогащенным по одному изомеру», «обогащенный по изомеру», «быть обогащенным по одному энантиомеру» или «обогащенный по энантиомеру» означает, что содержание одного из изомеров или энантиомеров составляет менее 100% и более или равно 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%, 99,5%, 99,6%, 99,7%, 99,8% или 99,9%.
Если не указанно иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разнице между относительным процентным содержанием двух изомеров или энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, а содержание другого изомера или энантиомера составляет 10%, изомерный или энантиомерный избыток («ee») составляет 80%.
Оптически активные (R)- и (S)-изомеры и D- и L-изомеры можно получить с помощью хирального синтеза или хиральных реагентов либо другими общепринятыми методиками. Энантиомер определенного описанного в настоящем документе соединения по настоящей заявке можно получить с помощью асимметричного синтеза или дериватизации с применением хирального вспомогательного вещества, при этом разделяют полученную смесь диастереомеров и отщепляют вспомогательную группу с получением требуемого чистого энантиомера. Альтернативно, если молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), соединение вступает в реакцию с соответствующей оптически активной кислотой или соответствующим оптически активным основанием с образованием соли диастереоизомера, которую затем подвергают диастереомерному разделению посредством традиционных в настоящей области техники способов с получением чистого энантиомера. При этом, как правило, энантиомер и диастереоизомер выделяют с помощью хроматографии с использованием хиральной неподвижной фазы, необязательно в сочетании с химической дериватизацией (например, образованием карбамата из аминов).
Раскрываемое в настоящем документе соединение может содержать неестественную долю атомного изотопа по одному или нескольким атомам, входящим в состав соединения. Например, соединение можно пометить радиоизотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). В другом примере водород можно заместить дейтерием с образованием дейтерированного лекарственного средства, и связь, образованная дейтерием и углеродом, будет более прочной, чем связь, образованная обычным водородом и углеродом. По сравнению с недейтерированным лекарственным средством дейтерированное лекарственное средство обладает рядом преимуществ, которые заключаются в снижении побочного токсического действия, повышении стабильности, повышении эффективности, увеличении биологического периода полувыведения и др. Объемом настоящей заявки охватываются все изотопные варианты описанного в настоящем документе соединения, независимо от того, радиоактивны они или нет.
«Необязательный» или «необязательно» означает, что описанное далее событие или обстоятельство может иметь место быть, но не обязательно, и описание включает случаи, когда такое событие или обстоятельство имеет место быть, и случаи, когда не имеет место быть.
Термин «замещенный» означает, что один или несколько атомов водорода на конкретном атоме замещены заместителями, куда можно отнести варианты с дейтерием и водородом, при условии, что валентность конкретного атома является нормальной, а соединение после замещения является стабильным. Если заместителем является кислород (т.е. =O), это означает, что замещены два атома водорода. В ароматических группах замещение кислородом не происходит. Термин «необязательно замещенный» означает, что атом может быть или может не быть замещен заместителем. Если не указано иное, тип и количество заместителя могут быть произвольными, если это химически достижимо.
При появлении в составе или структуре соединения любой переменной (например, R) более одного раза, определение такой переменной в каждом случае является независимым. Таким образом, например, если группа имеет 0-2 заместителя R, группа может быть необязательно замещена максимум двумя R, а определение R в каждом случае является независимым. Кроме того, сочетание заместителя и/или его варианта допустимо лишь в том случае, если такое сочетание может привести к получению стабильного соединения.
Если заместитель отсутствует, это означает, что заместителя не существует. Например, если в A-X отсутствует X, структура A-X фактически имеет форму A. Если не указано, каким именно атомом перечисленный заместитель связан с подлежащей замещению группой, заместитель может быть связан через любой атом данной группы. Например, пиридинил в качестве заместителя может быть связан с подлежащей замещению группой через любой атом углерода в пиридиновом кольце.
Если для указанной связывающей группы не указано направление для связывания, направление для связывания является произвольным. Например, если связывающая группа L, содержащаяся в 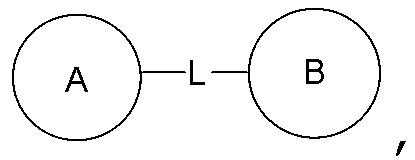 представляет собой -M-W-, -M-W- может либо связывать кольцо A и кольцо B с образованием
представляет собой -M-W-, -M-W- может либо связывать кольцо A и кольцо B с образованием 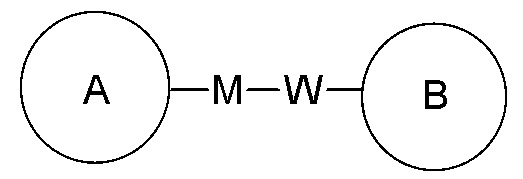 в направлении, аналогичном порядку чтения слева направо, либо связывать кольцо A и кольцо B с образованием
в направлении, аналогичном порядку чтения слева направо, либо связывать кольцо A и кольцо B с образованием 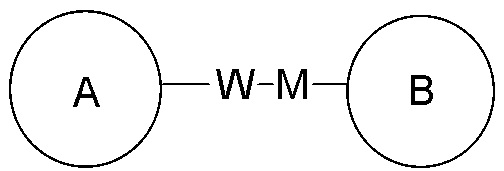 в направлении, противоположном порядку чтения слева направо. Сочетание замещающей группы, заместителя и/или его варианта допустимо лишь в том случае, если такое сочетание может привести к получению стабильного соединения.
в направлении, противоположном порядку чтения слева направо. Сочетание замещающей группы, заместителя и/или его варианта допустимо лишь в том случае, если такое сочетание может привести к получению стабильного соединения.
Если не указано иное, количество атомов в кольце, как правило, определяют как количество членов кольца. Например, «5-7-членное кольцо» относится к «кольцу», на котором в форме круга расположены от 5 до 7 атомов.
Если не указано иное, «3-10-членное кольцо» относится к циклоалкилу, гетероциклоалкилу, циклоалкенилу или гетероциклоалкенилу, состоящему из 3-10 кольцевых атомов. Кольцо может быть моноциклическим, бициклическим или полициклическим, причем бициклическая или полициклическая система включает спирокольцо, конденсированное кольцо, кольцо с внутренним мостиком и т.д. Если не указано иное, кольцо необязательно содержит 1, 2 или 3 гетероатома, независимо выбранных из группы, состоящей из O, S и N. 3-10-членное кольцо может быть 3-10-членным, 3-9-членным, 3-8-членным, 3-7-членным, 3-6-членным, 3-5-членным, 4-10-членным, 4-9-членным, 4-8-членным, 4-7-членным, 4-6-членным, 4-5-членным, 5-10-членным, 5-9-членным, 5-8-членным, 5-7-членным, 5-6-членным, 6-10-членным, 6-9-членным, 6-8-членным или 6-7-членным кольцом или тому подобным. Термин «5-7-членный гетероциклоалкил» включает пиперидинил и тому подобное, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, в которой каждое «кольцо» независимо соответствует приведенному выше определению.
Если не указано иное, «5-6-членное кольцо» относится к циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу, состоящему из 5-6 кольцевых атомов. Кольцо может быть моноциклическим или бициклическим, причем бициклическая кольцевая система включает спирокольцо, конденсированное кольцо, кольцо с внутренним мостиком и т.д. Если не указано иное, кольцо необязательно содержит 1, 2 или 3 гетероатома, независимо выбранных из группы, состоящей из O, S и N. 5-6-членное кольцо включает 5-членное кольцо, 6-членное кольцо и тому подобное. «5-6-членное кольцо» включает, например, фенил, пиридинил, пиперидинил и тому подобное. С другой стороны, термин «5-6-членный гетероциклоалкил» включает пиперидинил и тому подобное, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, в которой каждое «кольцо» независимо соответствует приведенному выше определению.
Если не указано иное, термины «5-6 членное гетероароматическое кольцо» и «5-6-членный гетероарил» в настоящем документе можно применять взаимозаменяемо. Термин «5-6-членный гетероарил» относится к моноциклической группе, которая состоит из 5-6 кольцевых атомов и имеет сопряженную по пи-электронам систему, из которых 1, 2, 3 или 4 кольцевых атома представляют собой гетероатомы, независимо выбранные из группы, состоящей из O, S и N, при этом остальные являются атомами углерода. Атом азота необязательно кватернизован, а гетероатом азота и серы необязательно окислен (т.е. NO и S(O)p, где p равно 1 или 2). 5-6-членный гетероарил может быть связан с остальной частью молекулы через гетероат или атом углерода. К 5-6-членному гетероарилу относится 5-членный гетероарил и 6-членный гетероарил. Примеры 5-6-членного гетероарила включают без ограничения пирролил (в том числе N-пирролил, 2-пирролил, 3-пирролил и т.д.), пиразолил (в том числе 2-пиразолил, 3-пиразолил и т.д.), имидазолил (в том числе N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил и т.д.), оксазолил (в том числе 2-оксазолил, 4-оксазолил, 5-оксазолил и т.д.), триазолил (в том числе 1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и т.д.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил, 5-изоксазолил и т.д.), тиазолил (в том числе 2-тиазолил, 4-тиазолил, 5-тиазолил и т.д.), фуранил (в том числе 2-фуранил, 3-фуранил и т.д.), тиенил (в том числе 2-тиенил, 3-тиенил и т.д.), пиридинил (в том числе 2-пиридинил, 3-пиридинил, 4-пиридинил и т.д.), пиразинил или пиримидинил (в том числе 2-пиримидинил, 4-пиримидинил и т.д.).
Если не указано иное, термин «алкил» относится к линейной или разветвленной насыщенной углеводородной группе. В некоторых вариантах осуществления алкил представляет собой C1-12-алкил. В других вариантах осуществления алкил представляет собой C1-6-алкил. В еще одних вариантах осуществления алкил представляет собой C1-3-алкил. Алкил может быть одновалентным (например, метилом), двухвалентным (например, метиленом) или поливалентным (например, метенилом). Примеры алкила включают метил (Me), этил (Et), пропил (в том числе н-пропил и изопропил), бутил (в том числе н-бутил, изобутил, втор-бутил и трет-бутил), пентил (в том числе н-пентил, изопентил и неопентил), гексил и тому подобное. Например, термин «C1-10-алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-10 атомов углерода. К C1-10-алкилу относится без ограничения C1-10-, C1-9-, C1-8-, C1-6-, C1-5-, C1-4-, C1-3-, C1-2-, C2-6-, C2-4-, C10-, C8-, C7-, C6- и C5-алкил и тому подобное, и он может быть одновалентным (например, метилом), двухвалентным (например, метиленом) или поливалентным (например, метенилом). Примеры C1-12-алкила включают без ограничения метил (Me), этил (Et), пропил (в том числе н-пропил и изопропил), бутил (в том числе н-бутил, изобутил, втор-бутил и трет-бутил), пентил (в том числе н-пентил, изопентил и неопентил), гексил, гептил, октил и тому подобное. Например, термин «C1-6-алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-6 атомов углерода. К C1-6-алкилу относится без ограничения C1-5-, C1-4-, C1-3-, C1-2-, C2-6-, C2-4-, C6- и C5-алкил и тому подобное, и он может быть одновалентным (например, метилом), двухвалентным (например, метиленом) или поливалентным (например, метенилом). Примеры C1-6-алкила включают без ограничения метил (Me), этил (Et), пропил (в том числе н-пропил и изопропил), бутил (в том числе н-бутил, изобутил, втор-бутил и трет-бутил), пентил (в том числе н-пентил, изопентил и неопентил), гексил и тому подобное. Например, термин «C1-3-алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. К C1-3-алкилу относится без ограничения C1-2- и C2-3-алкил и тому подобное, и он может быть одновалентным (например, метилом), двухвалентным (например, метиленом) или поливалентным (например, метенилом). Примеры C1-3-алкила включают без ограничения метил (Me), этил (Et), пропил (в том числе н-пропил и изопропил) и тому подобное.
Термин «гетероалкил», сам по себе или в сочетании с другим термином, относится к стабильному линейному или разветвленному алкильному радикалу или их комбинации, состоящему из определенного количества атомов углерода и по меньшей мере одного гетероатома или по меньшей мере одной гетероатомной группы. В некоторых вариантах осуществления гетероатом выбран из группы, состоящей из B, O, N и S, причем атомы азота и серы необязательно окислены, а гетероатом азота необязательно кватернизован. В других вариантах осуществления гетероатомная группа выбрана из группы, состоящей из -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2-, -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-. В некоторых вариантах осуществления гетероалкил представляет собой C1-6-гетероалкил. В других вариантах осуществления гетероалкил представляет собой C1-3-гетероалкил. Гетероатом или гетероатомная группа могут быть расположены в любом внутреннем положении гетероалкила, включая положение, где алкил связан с остальной частью молекулы. Тем не менее, термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) являются широко используемыми выражениями и относятся к таким алкильным группам, которые связаны с остальной частью молекулы соответственно через атом кислорода, аминогруппу или атом серы. Примеры гетероалкила включают без ограничения -OCH3, -OCH2CH3, -OCH2CH2CH3, -OCH2(CH3)2, -CH2-CH2-O-CH3, -NHCH3, -N(CH3)2, -NHCH2CH3, -N(CH3)(CH2CH3), -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -SCH3, -SCH2CH3, -SCH2CH2CH3, -SCH2(CH3)2, -CH2-S-CH2-CH3, -CH2-CH2, -S(=O)-CH3 и -CH2-CH2-S(=O)2-CH3. Последовательно могут идти максимум два гетероатома, например, -CH2-NH-OCH3.
Если не указано иное, термин «алкокси» относится к тем алкильным группам, каждая из которых связана с остальной частью молекулы через атом кислорода. Если не указано иное, C1-6-алкокси включает C1-, C2-, C3-, C4-, C5- и C6-алкокси. В некоторых вариантах осуществления алкокси представляет собой C1-3-алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентилокси. Например, термин «C1-3-алкокси» относится к тем алкильным группам, каждая из которых содержит 1-3 атома углерода и связана с остальной частью молекулы через атом кислорода. C1-3-алкокси включает без ограничения C1-2-, C2-3-, C3- и C2-алкокси и тому подобное. Примеры C1-3-алкокси включают без ограничения метокси, этокси, пропокси (в том числе н-пропокси и изопропокси) и тому подобное.
Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай, задействования от n до n+m углеводов (например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12) или любой случай задействования диапазона между n и n+m, (например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12, C9-12 и тому подобное). Аналогично, n-членный - n+m-членный предполагает, что количество атомов на кольцо составляет от n до n+m. Например, к 3-12-членному кольцу относится 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо. Также, n-членный - n+m-членный включает любой случай задействования любого диапазона между n и n+m. Например, к 3-12-членному кольцу относится 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо, 6-10-членное кольцо и тому подобное.
Если не указано иное, термин «циклоалкил» включает любой стабильный циклический алкил, который может быть моноциклической, бициклической или трициклической системой, причем бициклическая и трициклическая системы включают спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. В некоторых вариантах осуществления циклоалкил представляет собой C3-8-циклоалкил. В других вариантах осуществления циклоалкил представляет собой C3-6-циклоалкил. В еще одних вариантах осуществления циклоалкил представляет собой C5-6-циклоалкил. Циклоалкил может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил, [2.2.2]бициклооктан, [4.4.0]бициклодекан и тому подобное. Например, «C3-10-циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-10 атомов углерода, и она может быть моноциклической, бициклической или трициклической системой, причем бициклическая и трициклическая системы включают спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. C3-10-циклоалкил включает без ограничения C3-8-, C3-6-, C3-5-, C4-10-, C4-8-, C4-6-, C4-5-, C5-8- и C5-6-циклоалкил и тому подобное, и он может быть одновалентным, двухвалентным или поливалентным. Примеры C3-10-циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил, [2.2.2]бициклооктан, [4.4.0]бициклодекан и тому подобное. Например, «C3-8-циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-8 атомов углерода, и она может быть моноциклической или бициклической системой, причем бициклическая система включает спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. C3-8-циклоалкил включает без ограничения C3-6-, C3-5-, C4-8-, C4-6-, C4-5-, C5-8- или C5-6-циклоалкил или тому подобное, и он может быть одновалентным, двухвалентным или поливалентным. Примеры C3-8-циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил, [2.2.2]бициклооктан и тому подобное.
Если не указано иное, термин «гетероциклоалкил», сам по себе или в комбинации с другими терминами, относится к циклизированному «гетероалкилу», и он может быть моноциклической, бициклической или трициклической системой, причем бициклическая и трициклическая системы включают спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. Кроме того, что касается «гетероциклоалкила», гетероатом может занимать положение, где гетероциклоалкил связан с остальной частью молекулы. В некоторых вариантах осуществления гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил. В других вариантах осуществления гетероциклоалкил представляет собой 5-6-членный гетероциклоалкил. Примеры гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (в том числе тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (в том числе тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил или оксациклогептанил. Например, термин «3-6-членный гетероциклоалкил», сам себе или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 3-6 кольцевых атомов, из которых 1, 2, 3 или 4 кольцевых атома представляют собой гетероатомы, независимо выбранные из группы, состоящей из O, S, и N, при этом остальные являются атомами углерода. Атом азота необязательно кватернизован, гетероатомы азота и серы могут быть необязательно окислены (т.е. NO и S(O)p, где p равно 1 или 2). «3-6-членный гетероциклоалкил» может быть моноциклической или бициклической системой, причем бициклическая система включает спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. Более того, что касается «3-6-членного гетероциклоалкила», гетероатом может занимать положение, где гетероциклоалкил связан с остальной частью молекулы. К 3-6-членному гетероциклоалкилу относится без ограничения 4-6-членный, 5-6-членный, 4-членный, 5-членный и 6-членный гетероциклоалкил и тому подобное. Примеры 3-6-членного гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (в том числе тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (в том числе тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил и т.д. Например, термин «4-6-членный гетероциклоалкил», сам по себе или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 4-6 кольцевых атомов, из которых 1, 2, 3 или 4 кольцевых атома представляют собой гетероатомы, независимо выбранные из группы, состоящей из O, S, и N, при этом остальные являются атомами углерода. Атом азота необязательно кватернизован, гетероатомы азота и серы могут быть необязательно окислены (т.е. NO и S(O)p, где p равно 1 или 2). «4-6-членный гетероциклоалкил» может быть моноциклической или бициклической системой, причем бициклическая система включает спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. Более того, что касается «4-6-членного гетероциклоалкила», гетероатом может занимать положение, где гетероциклоалкил связан с остальной частью молекулы. К 4-6-членному гетероциклоалкилу относится без ограничения 5-6-членный, 4-членный, 5-членный и 6-членный гетероциклоалкил и тому подобное. Примеры 4-6-членного гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (в том числе тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (в том числе тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил и т.д. Например, термин «5-6-членный гетероциклоалкил», сам по себе или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 5-6 кольцевых атомов, из которых 1, 2, 3 или 4 кольцевых атома представляют собой гетероатомы, независимо выбранные из группы, состоящей из O, S, и N, при этом остальные являются атомами углерода. Атом азота необязательно кватернизован, гетероатомы азота и серы могут быть необязательно окислены (т.е. NO и S(O)p, где p равно 1 или 2). «5-6-членный гетероциклоалкил» может быть моноциклической или бициклической системой, причем бициклическая система включает спирокольцо, конденсированное кольцо и кольцо с внутренним мостиком. Более того, что касается «5-6-членного гетероциклоалкила», гетероатом может занимать положение, где гетероциклоалкил связан с остальной частью молекулы. К 5-6-членному гетероциклоалкилу относится 5-членный гетероциклоалкил и 6-членный гетероциклоалкил. Примеры 5-6-членного гетероциклоалкила включают без ограничения пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (в том числе тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (в том числе тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (в том числе 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (в том числе 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (в том числе 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил и т.д.
Термин «фармацевтически приемлемый носитель» относится к любому средству или к любой среде-носителю, которые способны доставлять эффективное количество раскрываемого в настоящем документе действующего вещества, не оказывают отрицательного влияния на биологическую активность действующего вещества и не оказывают побочного токсического действия на принимающего их индивидуума или пациента. К типичным носителям относятся вода, масло, растительное и минеральное, кремовая основа, основа лосьона, мазевая основа и тому подобное. Эти основы включают суспендирующее средство, загуститель, усилитель проникновения и др. Содержащие их составы хорошо известны специалистам в области косметики или фармацевтики препаратов для местного применения.
Термин «вспомогательное вещество» обычно относится к носителю, разбавителю и/или среде, которые необходимы для составления эффективной фармацевтической композиции.
Слово «содержать» и его варианты, такие как «содержит» или «содержащий», нужно понимать в открытом, неисключительном смысле, т.е. «включающий без ограничения».
Термин «лечение» означает введение описанного в настоящем документе соединения или состава для ослабления или устранения заболевания или одного или нескольких связанных с данным заболеванием симптомов и включает:
(i) подавление заболевания или болезненного состояния, т.е. остановку его развития;
(ii) облегчение заболевания или болезненного состояния, т.е. индукцию его регресса.
Термин «предупреждение» означает введение описанного в настоящем документе соединения или состава для предупреждения заболевания или одного или нескольких связанных с данным заболеванием симптомов и включает предупреждение возникновения заболевания или болезненного состояния у млекопитающего, особенно когда такое млекопитающее предрасположено к развитию данного заболевания, но оно все еще не было диагностировано у него.
В случае лекарственных средств и фармакологических действующих средств термин «эффективное количество» или «терапевтически эффективное количество» относится к количеству лекарственного средства или лекарственного препарата, которое является достаточным для достижения требуемого эффекта, но не является токсичным. Что касается раскрываемой в настоящем документе пероральной лекарственной формы, «эффективное количество» одного действующего вещества в композиции представляет собой количество, которое необходимо для достижения требуемого эффекта при применении действующего средства в комбинации с другим действующим средством в композиции. Результат определения эффективного количества варьирует для разных людей в зависимости от возраста и общего состояния субъекта, а также в зависимости от конкретного действующего вещества. В одном случае подходящее эффективное количество может быть определено специалистом в настоящей области техники по результатам стандартных тестов.
Термин «действующий ингредиент», «терапевтическое средство», «действующее вещество» или «действующее средство» относится к химическому веществу, которое является эффективным при лечении целевого нарушения, заболевания или патологического состояния.
Раскрываемые в настоящем документе соединения можно получить рядом способов синтеза, которые хорошо известны специалистам в настоящей области, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные их комбинациями с другими способами химического синтеза, и их эквиваленты, известные специалистам в настоящей области. Предпочтительные варианты осуществления включают без ограничения примеры, которые раскрыты в настоящем документе.
Применяемый в настоящем документе растворитель может быть коммерчески доступным. В настоящей заявке применяют следующие сокращения: водн. означает водный; HATU означает O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC означает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида; m-CPBA означает 3-хлорпероксибензойную кислоту; экв. означает эквивалент или эквивалентность; CDI означает карбонилдиимидазол; DCM означает дихлорметан; PE означает простой петролейный эфир; DIAD означает диизопропилазодиформиат; DMF означает N,N-диметилформамид; DMSO означает диметилсульфоксид; EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол; CBz означает бензилоксикарбонил, защитную группу для аминогруппы; ВОС означает трет-бутоксикарбонил, который является защитной группой для аминогруппы; HOAc означает уксусную кислоту; NaCNBH3 означает цианоборгидрид натрия; к.т. означает комнатную температуру; O/N означает в течение ночи; THF означает тетрагидрофуран; Boc2O означает ди-трет-бутилдикарбонат; TFA означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SOCl2 означает тионилхлорид; CS2 означает сероуглерод; TsOH означает п-толуолсульфоновую кислоту; NFSI означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS означает N-хлорсукцинимид; н-Bu4NF означает фторид тетрабутиламмония; iPrOH означает 2-пропанол; mp означает температуру плавления; LDA означает диизопропиламид лития; POCl3 означает оксихлорид фосфора; LiHMDS означает гексаметилдисилазид лития; DCE означает дихлорэтан; DIEA означает N,N-диизопропилэтиламин; MTBE означает простой метил-трет-бутиловый эфир; TEA означает триэтиламин; DIPEA означает N,N-диизопропилэтиламин; и LLOQ означает нижний предел количественного определения.
Краткое описание чертежей
Фиг. 1: уровни ДНК HBV в плазме крови;
Фиг. 2: уровни ДНК HBV в печени.
Подробное описание
Настоящая заявка ниже более подробно описана с помощью примеров. Тем не менее, это никоим образом не подразумевает приносящее ущерб ограничение объема настоящей заявки. Несмотря на то, что в настоящем документе была подробно описана настоящая заявка, а также были раскрыты конкретные примеры, специалистам в настоящей области техники будет очевидно, что в данные конкретные примеры можно внести различные изменения и модификации без отступления от сущности и объема настоящей заявки.
Пример 1
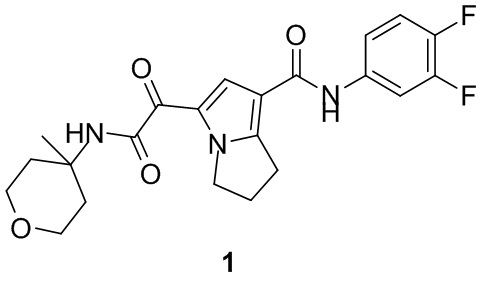
Путь синтеза:
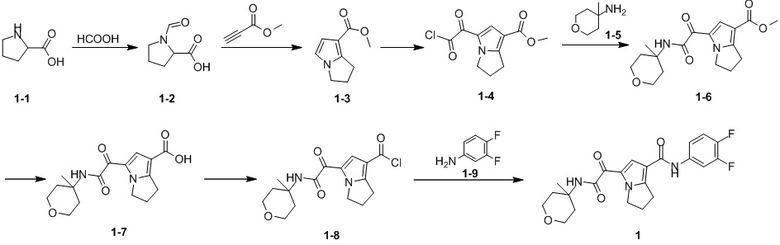
Стадия 1
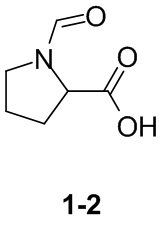
Раствор уксусного ангидрида (150 мл) в муравьиной кислоте (100 мл) добавляли к раствору соединения 1-1 (15 г, 130,29 ммоль, 1 экв.) в муравьиной кислоте (150 мл) при 0°C. Смесь перемешивали при 25°C в течение 16 ч. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 1-2, которое применяли непосредственно на следующей стадии.
1H ЯМР (400 MГц, MeOH-d4) δ = 11,51 (br s, 1H), 8,40-7,97 (m, 1H), 4,64-4,36 (m, 1H), 3,78-3,46 (m, 2H), 2,38-2,13 (m, 2H), 2,10-1,89 (m, 2H).
Стадия 2
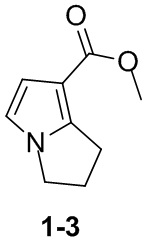
Метилпропионат (35,24 г, 419,17 ммоль, 34,89 мл, 6 экв.) в 250-мл колбе добавляли к раствору соединения 1-2 (10 г, 69,86 ммоль, 1 экв.) в уксусном ангидриде (100 мл). Смесь перемешивали при 130°C в течение 6 ч. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 1-3. MS (ESI) m/z: 165,8 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 6,61 (d, J=2,8 Гц, 1H), 6,55 (d, J=2.8 Гц, 1H), 3,97 (t, J=7,2 Гц, 2H), 3,79 (s, 3H), 3,06 (t, J=7,4 Гц, 2H), 2,54 (quin, J=7,3 Гц, 2H).
Стадия 3
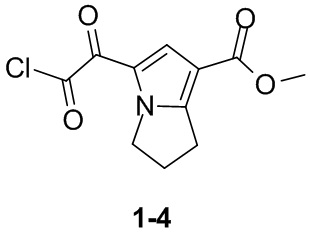
Оксалилхлорид (535,10 мг, 4,22 ммоль, 369,03 мкл, 2 экв.) добавляли к раствору соединения 1-3 (500 мг, 2,11 ммоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота, а затем добавляли N,N-диметилформамид (0,05 мл). Смесь перемешивали при 25°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 1-4, которое применяли непосредственно на следующей стадии.
Стадия 4
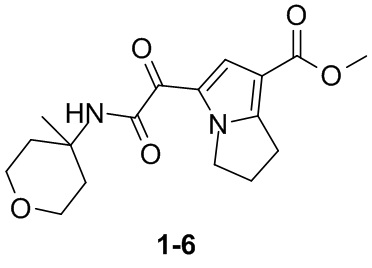
Триэтиламин (870,23 мг, 8,60 ммоль, 1,20 мл, 4 экв.) добавляли к раствору соединения 1-4 (550 мг, 2,15 ммоль, 1 экв.) в дихлорметане (10 мл), а затем добавляли соединение 1-5 (247,62 мг, 2,15 ммоль, 1 экв.). Смесь перемешивали при 20°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления. Остаток разделяли с помощью автоматической хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:0 до 80:20, об./об.) с получением соединения 1-6. MS (ESI) m/z: 357,2 [M+Na]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,23 (s, 1H), 7,10 (br s, 1H), 4,28 (t, J=7,3 Гц, 2H), 3,73 (s, 3H), 3,69-3,66 (m, 2H), 3,60-3,52 (m, 2H), 3,05 (t, J=7,7 Гц, 2H), 2,52 (q, J=7,5 Гц, 2H), 2,06 (br d, J=13,8 Гц, 2H), 1,68 (m, 2H), 1,42 (s, 3H).
Стадия 5
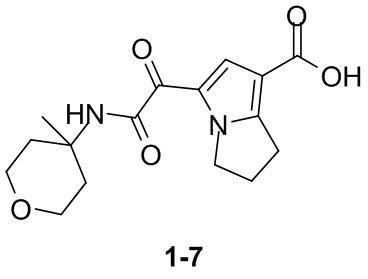
Раствор моногидрата гидроксида лития (131,78 мг, 3,14 ммоль, 3 экв.) в H2O (1 мл) добавляли к раствору соединения 1-6 (350 мг, 1,05 ммоль, 1 экв.) в тетрагидрофуране (5 мл). Смесь перемешивали при 30°C в течение 12 ч. Реакционный раствор доводили до pH = 1-2 с помощью 2 моль/л разведенной соляной кислоты и экстрагировали этилацетатом (10 мл ×2). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением соединения 1-7. MS (ESI) m/z: 343,2 [M+Na]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 7,89 (s, 1H), 4,37 (br d, J=7,0 Гц, 2H), 3,70-3,63 (m, 2H), 3,14-3,10 (m, 2H), 2,66-2,52 (m, 4H), 2,25-2,19 (m, 2H), 1,73-1,67 (m, 2H), 1,49 (s, 3H).
Стадия 6
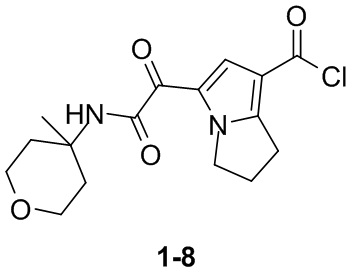
Оксалилхлорид (237,74 мг, 1,87 ммоль, 163,96 мкл, 2 экв.) добавляли к раствору соединения 1-7 (300 мг, 936,51 мкмоль, 1 экв.) в дихлорметане (3 мл) при 0°C в условиях атмосферы азота, а затем добавляли N,N-диметилформамид (0,01 мл). Смесь перемешивали при 20°C в течение 1 ч с получением соединения 1-8.
Стадия 7
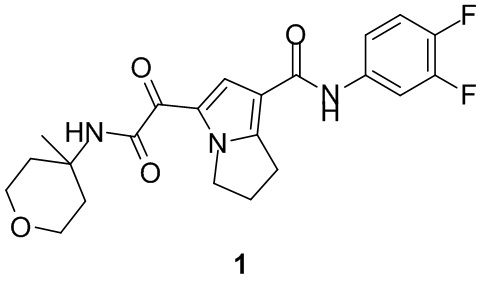
Триэтиламин (358,42 мг, 3,54 ммоль, 493,01 мкл, 4 экв.) добавляли к раствору соединения 1-8 (300 мг, 885,52 мкмоль, 1 экв.) в дихлорметане (5 мл) в условиях атмосферы азота, а затем добавляли соединение 1-9 (114,33 мг, 885,52 мкмоль, 1 экв.). Смесь перемешивали при 20°C в течение 12 ч. Реакционный раствор концентрировали в условиях пониженного давления, а остаток разделяли с помощью автоматической хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:0 до 50:50, об./об.) с получением соединения 1. MS (ESI) m/z: 432,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,18 (s, 1H), 7,73-7,64 (m, 1H), 7,59 (s, 1H), 7,23 (s, 1H), 7,09-7,02 (m, 2H), 4,31 (t, J=7,3 Гц, 2H), 3,71 (td, J=4,3, 12,0 Гц, 2H), 3,62-3,52 (m, 2H), 3,21-3,12 (m, 2H), 2,63-2,50 (m, 2H), 2,07 (br d, J=14,1 Гц, 2H), 1,70 (ddd, J=4,3, 9,9, 14,0 Гц, 2H), 1,43 (s, 3H).
Пример 2
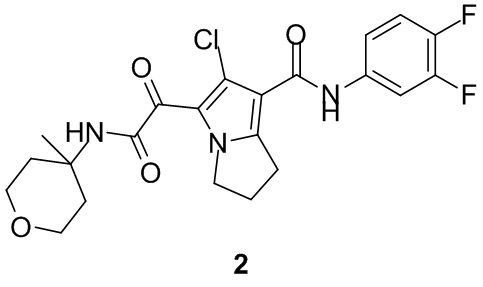
Путь синтеза:
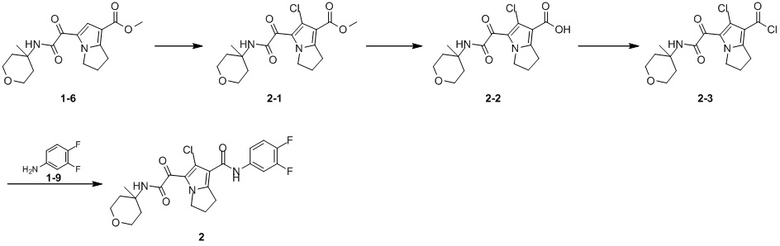
Стадия 1
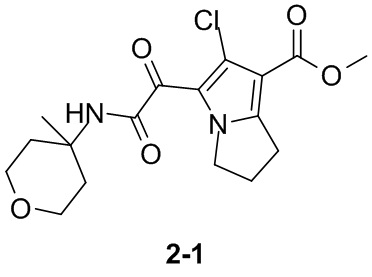
N-хлорсукцинимид (77,87 мг, 583,19 мкмоль, 1,3 экв.) добавляли к раствору соединения 1-6 (150 мг, 448,61 мкмоль, 1 экв.) в ацетонитриле (10 мл). Смесь перемешивали при 40°C в течение 6 ч. Реакционный раствор концентрировали в условиях пониженного давления. Остаток разделяли с помощью автоматической хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:0 до 80:20, об./об.) с получением соединения 2-1. MS (ESI) m/z: 369,1 [M+H]+.
Стадия 2
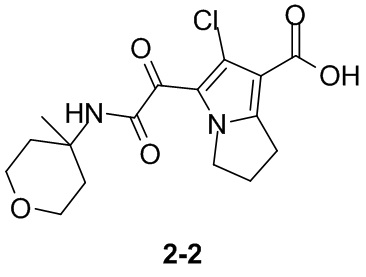
Раствор моногидрата гидроксида лития (34,13 мг, 813,42 мкмоль, 5 экв.) в H2O (0,5 мл) добавляли к раствору соединения 2-1 (60 мг, 162,68 мкмоль, 1 экв.) в этаноле (2 мл). Смесь перемешивали при 40 °C в течение 3 ч. Реакционный раствор доводили до pH = 1-2 с помощью 2 моль/л разведенной соляной кислоты и экстрагировали этилацетатом (10 мл ×2). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением неочищенного соединения 2-2. MS (ESI) m/z: 377,1 [M+Na]+.
Стадия 3
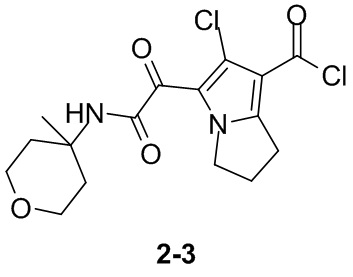
Оксалилхлорид (28,62 мг, 225,49 мкмоль, 19,74 мкл, 2 экв.) добавляли к раствору соединения 2-2 (40 мг, 112,74 мкмоль, 1 экв.) в дихлорметане (2 мл) при 0°C в условиях атмосферы азота, а затем добавляли N,N-диметилформамид (0,01 мл). Смесь перемешивали при 25°C в течение 45 мин. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 2-3.
Стадия 4
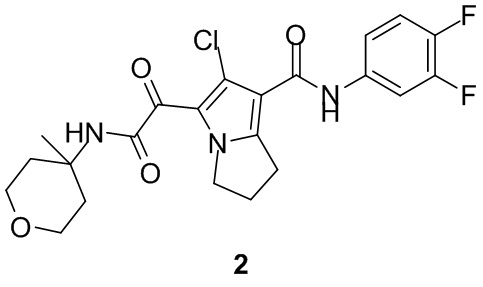
Триэтиламин (43,38 мг, 428,69 мкмоль, 59,67 мкл, 4 экв.) добавляли к раствору соединения 2-3 (40 мг, 107,17 мкмоль, 1 экв.) в дихлорметане (3 мл) в условиях атмосферы азота. Затем добавляли соединение 1-9 (13,84 мг, 107,17 мкмоль, 1 экв.). Смесь перемешивали при 25°C в течение 12 ч. Реакционный раствор концентрировали в условиях пониженного давления, а остаток разделяли с помощью препаративной HPLC (колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,225% муравьиной кислоты)-ацетонитрил; % содержания ацетонитрила: 73% - 83%, 7 мин) с получением соединения 2. MS (ESI) m/z: 466,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,40 (s, 1H), 7,72-7,61 (m, 1H), 7,11-6,99 (m, 2H), 6,23 (s, 1H), 4,25 (t, J=7,4 Гц, 2H), 3,80-3,57 (m, 4H), 3,19 (t, J=7,7 Гц, 2H), 2,46 (quin, J=7,5 Гц, 2H), 2,08 (br d, J=14,1 Гц, 2H), 1,73 (ddd, J=4,4, 9,6, 13,9 Гц, 2H), 1,48 (s, 3H).
Пример 3
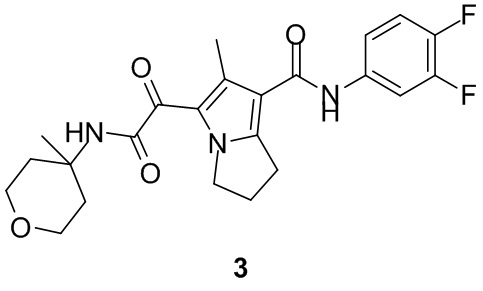
Путь синтеза:
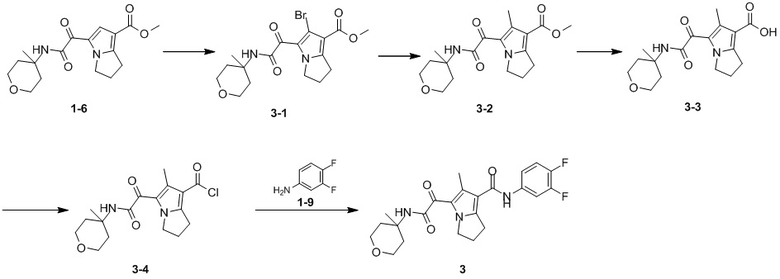
Стадия 1
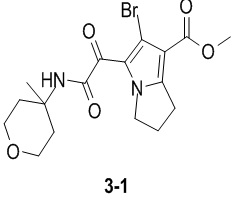
В бутылку с рифленым горлышком добавляли соединение 1-6 (300 мг, 897,22 мкмоль, 1 экв.) и ацетонитрил (3 мл), а затем добавляли туда N-бромсукцинимид (191,62 мг, 1,08 ммоль, 1,2 экв.). Смесь перемешивали при 25°C в течение 12 ч. Реакционный раствор концентрировали в условиях пониженного давления. Остаток разделяли с помощью автоматической хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:0 до 80:20, об./об.) с получением соединения 3-1. MS (ESI) m/z: 435,0 [M+Na]+.
1H ЯМР (400 MГц, CDCl3) δ = 4,30 (t, J=7,4 Гц, 2H), 3,83 (s, 3H), 3,80-3,67 (m, 4H), 3,13 (t, J=7,7 Гц, 2H), 2,50 (q, J=7,5 Гц, 2H), 2,15 (d, J=14,1 Гц, 2H), 1,78 (m, 2H), 1,55 (s, 3H).
Стадия 2
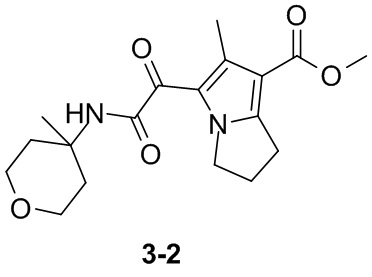
В бутылку с рифленым горлышком в условиях атмосферы азота добавляли соединение 3-1 (100 мг, 241,98 мкмоль, 1 экв.), метилбориновую кислоту (28,97 мг, 483,95 мкмоль, 2 экв.), карбонат калия (100,33 мг, 725,93 мкмоль, 3 экв.), 1,4-диоксан (2,5 мл) и воду (0,5 мл), а затем добавляли Pd(dppf)Cl2 (17,71 мг, 24,20 мкмоль, 0,1 экв.). Смесь перемешивали при 80°C в течение 2 ч. В реакционный раствор добавляли воду (5 мл) и экстрагировали этилацетатом (10 мл ×3). Объединенные органические фазы промывали разбавленной соляной кислотой (10 мл) и насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением соединения 3-2. MS (ESI) m/z: 348,9 [M+H]+.
Стадия 3
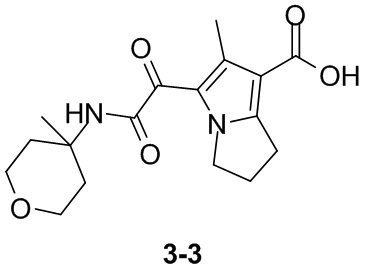
В бутылку с рифленым горлышком добавляли соединение 3-2 (80 мг, 229,63 мкмоль, 1 экв.) и этанол (2 мл), а затем добавляли раствор гидрохлорида натрия (27,56 мг, 688,88 мкмоль, 3 экв.) в воде (0,5 мл). Смесь перемешивали при 25°C в течение 12 ч. В реакционный раствор добавляли воду (5 мл) и промывали этилацетатом (5 мл). Водную фазу доводили до pH = 1-2 с помощью 2 моль/л разбавленной соляной кислоты и экстрагировали этилацетатом (10 мл × 2). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением соединения 3-3.
Стадия 4
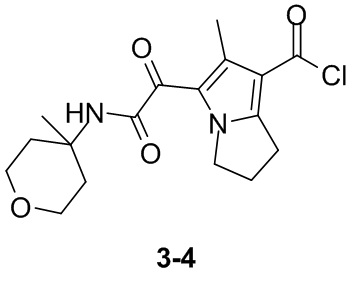
Оксалилхлорид (26,57 мг, 209,35 мкмоль, 18,33 мкл, 1 экв.) добавляли к раствору соединения 3-3 (70 мг, 209,35 мкмоль, 1 экв.) в дихлорметане (3 мл) при 0°C, а затем добавляли N,N-диметилформамид (15,30 мг, 209,35 мкмоль, 16,11 мкл, 1 экв.). Смесь перемешивали при 25°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 3-4.
Стадия 5
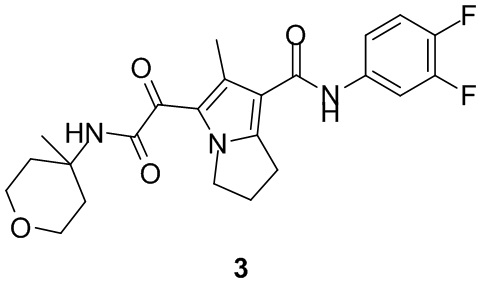
Триэтиламин (86,04 мг, 850,31 мкмоль, 118,35 мкл, 4 экв.) добавляли к раствору соединения 3-4 (75 мг, 212,58 мкмоль, 1 экв.) в дихлорметане (3 мл), а затем добавляли соединение 1-9 (27,45 мг, 212,58 мкмоль, 1 экв.). Смесь перемешивали при 25°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления. Остаток разделяли с помощью препаративной HPLC-хроматографии (колонка: Boston Green ODS 150мм × 30 мм × 5 мкм; подвижная фаза: вода (содержащая 0,075% трифторуксусную кислоту)-ацетонитрил; % содержания ацетонитрила: 47% - 67%, 7 мин) с получением соединения 3. MS (ESI) m/z: 446,1 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,76-7,66 (m, 1H), 7,37 (s, 1H), 7,19-7,05 (m, 1H), 6,64-6,46 (m, 1H), 6,56 (s, 1H), 4,30 (t, J=7,3 Гц, 2H), 3,87-3,76 (m, 2H), 3,75-3,66 (m, 2H), 3,15 (t, J=7,5 Гц, 2H), 2,62-2,52 (m, 5H), 2,15 (d, J=13,8 Гц, 2H), 1,81 (m, 2H), 1,56 (s, 3H).
Пример 4
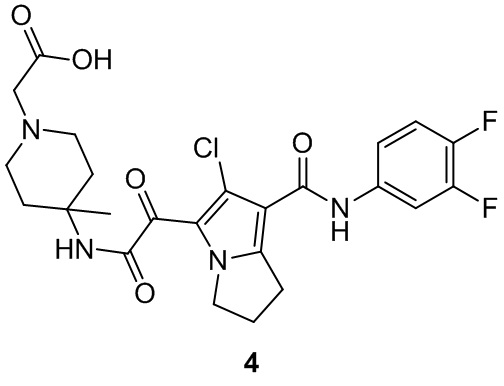
Путь синтеза:
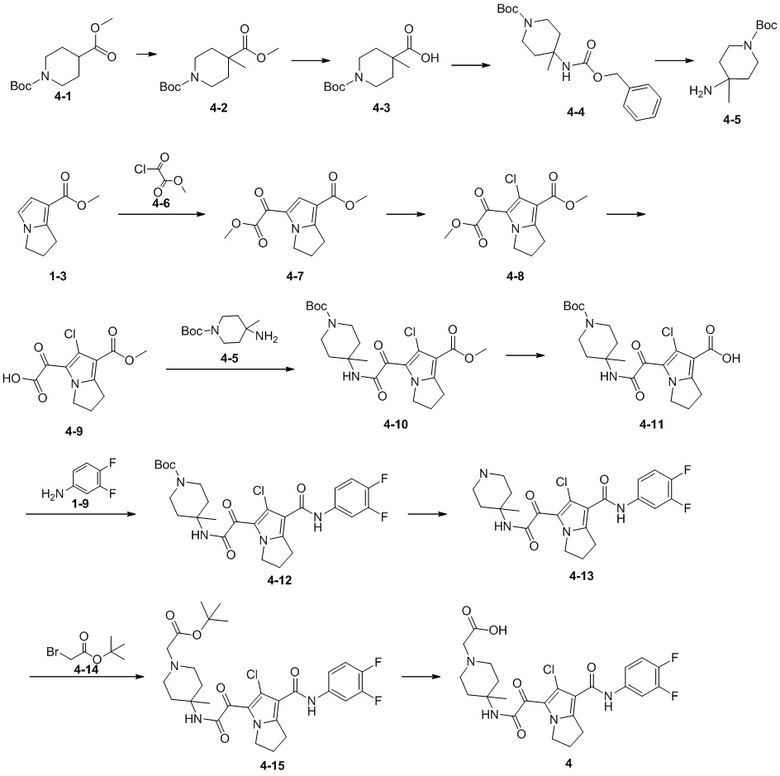
Стадия 1

Диизопропиламин (49,91 г, 493,22 ммоль, 69,71 мл, 1,2 экв.) растворяли в тетрагидрофуране (1000 мл), а затем добавляли н-бутиллитий (2,5 M, 197,29 мл, 1,2 экв.) при -78°C. После перемешивания в течение 0,5 ч добавляли соединение 4-1 (100 г, 411,02 ммоль, 1 экв.) при -78°C. После перемешивания в течение еще 0,5 ч добавляли йодметан (58,34 г, 411,02 ммоль, 25,59 мл, 1 экв.) при -78°C. Реакционную систему перемешивали при 25°C в течение 16 ч. Реакционный раствор выливали в 1 л насыщенного раствор хлорида аммония для остановки реакции и экстрагировали этилацетатом (500 мл ×4). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 4-2.
1H ЯМР (400 MГц, CDCl3) δ = 3,79-3,73 (m, 2H), 3,70 (s, 3H), 2,99 (t, J=11,2 Гц, 2H), 2,07-2,04 (m, 2H), 1,45 (s, 9H), 1,40-1,33 (m, 2H), 1,20 (s, 3H).
Стадия 2
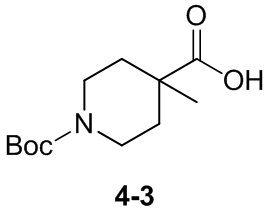
Соединение 4-2 (107 г, 415,81 ммоль, 1 экв.) растворяли в воде (500 мл) и этаноле (1000 мл), а затем добавляли гидроксид калия (116,65 г, 2,08 моль, 5 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам TLC (дихлорметан : метанол = 20:1) было видно, что соединение 4-2 полностью израсходовано и были получены новые пятна. Реакционный раствор концентрировали в условиях пониженного давления при 40°C для удаления растворителя, а остаток экстрагировали с помощью 500 мл этилацетата. Водную фазу доводили до pH = 3-5 с помощью 1 M соляной кислоты и экстрагировали этилацетатом (1 л ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 4-3.
1H ЯМР (400 MГц, MeOH-d4) δ = 3,77 (td, J=4,0, 13,8 Гц, 2H), 3,02 (s, 2H), 2,05-2,00 (m, 2H), 1,45 (s, 9H), 1,38-1,31 (m, 2H), 1,22 (s, 3H).
Стадия 3
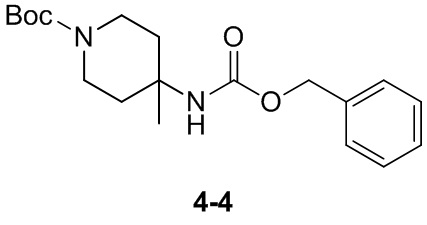
Соединение 4-3 (10 г, 41,10 ммоль, 1 экв.) растворяли в толуоле (100 мл), а затем добавляли триэтиламин (4,99 г, 49,32 ммоль, 6,87 мл, 1,2 экв.) и бензиловый спирт (6,67 г, 61,65 ммоль, 6,41 мл, 1,5 экв.). Затем добавляли дифенилфосфорилазид (16,97 г, 61,65 ммоль, 13,36 мл, 1,5 экв.) при 25°C в условиях атмосферы азота. Реакционную систему перемешивали при 110°C в течение 3 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что соединение 4-3 полностью израсходовано и были получены новые пятна. Реакционный раствор выливали в 100 мл насыщенного раствора хлорида аммония для остановки реакции, а затем экстрагировали этилацетатом (50 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 5:1, об./об.). Очищенный продукт концентрировали в условиях пониженного давления с получением соединения 4-4.
1H ЯМР (400 MГц, CDCl3) δ = 7,34-7,30 (m, 3H), 7,20-7,17 (m, 2H), 4,70-4,64 (m, 2H), 3,64 (s, 2H), 3,37-3,21 (m, 2H), 1,95 (s, 2H), 1,62-1,51 (m, 2H), 1,46 (s, 9H), 1,38 (s, 3H).
Стадия 4
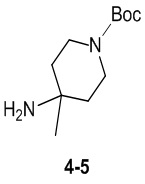
Соединение 4-4 (14 г, 40,18 ммоль, 1 экв.) растворяли в метаноле (150 мл), а затем добавляли влажный палладиевый катализатор на углеродном носителе (5 г, содержание 10%). Реакционную систему перемешивали при 25°C в течение 6 ч в условиях атмосферы водорода (50 psi). По результатам TLC (дихлорметан : метанол = 10:1) было видно, что соединение 4-4 полностью израсходовано и были получены новые пятна. Реакционный раствор концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, дихлорметан : метанол = 10:1 до 5:1, об./об.) и концентрировали в условиях пониженного давления с получением соединения 4-5.
1H ЯМР (400 MГц, CDCl3) δ = 7,02 (br s, 2H), 3,59-3,49 (m, 4H), 1,98-1,51 (m, 4H), 1,46-1,44 (m, 12H).
Стадия 5
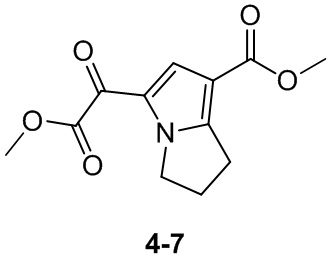
Соединение 1-3 (20 г, 121,07 ммоль, 1 экв.) в сухой колбе с одним горлышком растворяли в DCM (400 мл). По каплям добавляли соединение 4-6 (29,66 г, 242,15 ммоль, 22,30 мл, 2 экв.) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 20°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в 100 мл воды. Затем водную фазу экстрагировали посредством DCM (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 1:1, об./об.) с получением соединения 4-7. MS(ESI) m/s: 252,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,76 (s, 1H), 4,38 (t, J=7,6 Гц, 2H), 3,93 (s, 3H), 3,82 (s, 3H), 3,14-3,10 (m, 2H), 2,62-2,55 (m, 2H).
Стадия 6
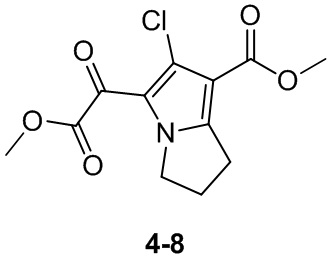
Соединение 4-7 (5 г, 19,90 ммоль, 1 экв.) в сухой 50-мл колбе с одним горлышком растворяли в ацетонитриле (100 мл), а затем добавляли N-хлорсукцинимид (3,59 г, 26,87 ммоль, 1,35 экв.). Реакционную систему перемешивали при 40°C в течение 16 ч. По результатам LCMS и TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор выливали в насыщенной раствор бикарбоната натрия (200 мл), а затем концентрировали в условиях пониженного давления для удаления ацетонитрила. Водную фазу экстрагировали дихлорметаном (50 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 3:1, об./об.) с получением соединения 4-8. MS(ESI) m/s: 286,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 4,40 (t, J=7,6 Гц, 2H), 3,95 (s, 3H), 3,85 (s, 3H), 3,15 (t, J=7,6 Гц, 2H), 2,59-2,49 (m, 2H).
Стадия 7
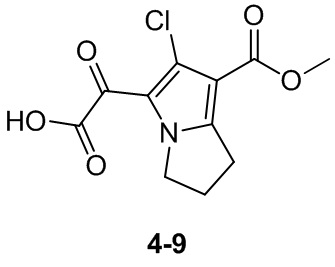
Соединение 4-8 (2,5 г, 8,75 ммоль, 1 экв.) добавляли к смеси тетрагидрофурана (40 мл), этанола (10 мл) и воды (10 мл) при 20°C, а затем к реакционной системе добавляли карбонат калия (3,63 г, 26,25 ммоль, 3 экв.). Реакционную систему перемешивали при 45°C в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 2:1) было видно, что реакция завершилась. К реакционному раствору добавляли 100 мл воды, доводили до pH = 3 с помощью 1 моль/л соляной кислоты и экстрагировали этилацетатом (50 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 4-9.
1H ЯМР (400 MГц, DMSO-d6) δ = 4,29 (t, J=7,2 Гц, 2H), 3,75 (s, 3H), 3,05 (t, J=7,6 Гц, 2H), 2,48-2,40 (m, 2H).
Стадия 8
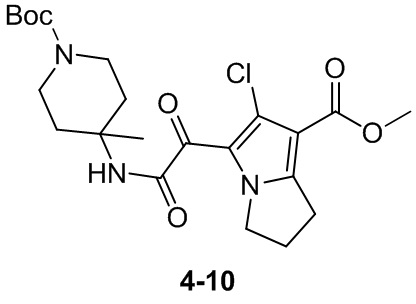
Соединение 4-9 (0,4 г, 1,47 ммоль, 1 экв.), N,N-диизопропилэтиламин (380,61 мг, 2,94 ммоль, 512,95 мкл, 2 экв.) и гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (1,68 г, 4,42 ммоль, 3 экв.) растворяли в тетрагидрофуране (10 мл). После перемешивания в течение 0,5 ч добавляли соединение 4-5 (473,34 мг, 2,21 ммоль, 1,5 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно, что соединение 4-9 полностью израсходовано и были получены новые пятна. Результаты LCMS подтверждали получение соединения 4-10. Реакционный раствор выливали в насыщенный раствор хлорида аммония (20 мл) для остановки реакции, а затем экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 1:1, об./об.) и концентрировали в условиях пониженного давления с получением соединения 4-10. MS(ESI) m/s: 412,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, CDCl3) δ = 6,03 (s, 1H), 4,32 (t, J=7,2 Гц, 2H), 3,85 (s, 3H), 3,70 (s, 2H), 3,25-3,12 (m, 4H), 2,54-2,47 (m, 2H), 1,69-1,62 (m, 4H), 1,52 (s, 3H), 1,47 (s, 9H).
Стадия 9
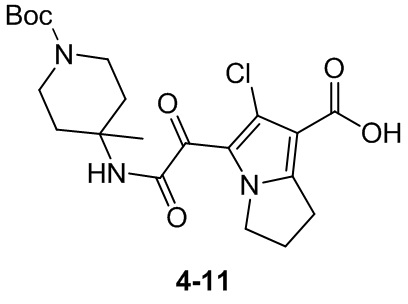
Соединение 4-10 (0,5 г, 1,07 ммоль, 1 экв.) растворяли в смеси метанола (5 мл), тетрагидрофурана (5 мл) и воды (5 мл), а затем добавляли моногидрат гидроксида лития (134,52 мг, 3,21 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 3 ч. По результатам TLC (дихлорметан : метанол = 10:1) было видно, что соединение 4-10 полностью израсходовано и были получены новые пятна. Реакционный раствор доводили до pH = 1-3 с помощью 1 моль/л соляной кислоты и экстрагировали этилацетатом (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 4-11.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,36 (t, J=7,2 Гц, 2H), 3,72-3,67 (m, 2H), 3,24 (s, 2H), 3,13 (t, J=4,4 Гц, 2H), 2,56-2,48 (m, 2H), 2,20 (d, J=13,6 Гц, 2H), 1,60-1,53 (m, 2H), 1,49 (s, 3H), 1,46 (s, 9H).
Стадия 10
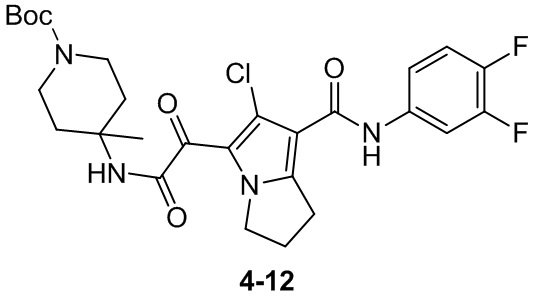
Соединение 4-11 (0,38 г, 837,16 мкмоль, 1 экв.), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (954,94 мг, 2,51 ммоль, 3 экв.) и триэтиламин (254,14 мг, 2,51 ммоль, 349,57 мкл, 3 экв.) растворяли в N,N-диметилформамиде (10 мл). После перемешивания при 25°C в течение 0,5 ч добавляли соединение 1-9 (216,17 мг, 1,67 ммоль, 2 экв.). Реакционную систему перемешивали при 55°C в течение 5 ч, затем нагревали до 70°C и перемешивали в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно, что соединение 4-11 полностью израсходовано и были получены новые пятна. Результаты LCMS подтверждали, что соединение 4-11 полностью израсходовано и было получено соединение 4-12. Реакцию останавливали с помощью 20 мл воды, а затем реакционный раствор экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 1:1, об./об.) и концентрировали в условиях пониженного давления с получением соединения 4-12. MS(ESI) m/s: 465,1 [M-Boc+H]+.
Стадия 11
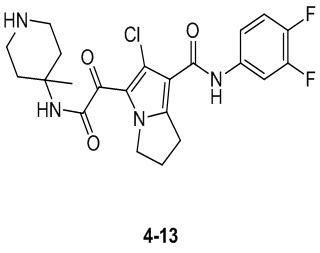
Соединение 4-12 (0,3 г, 530,97 мкмоль, 1 экв.) растворяли в этилацетате (15 мл), а затем добавляли соляную кислоту/этилацетат (4 моль/л, 5 мл, 37,67 экв.). Реакционную систему перемешивали при 25°C в течение 0,5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно, что соединение 4-12 полностью израсходовано и были получены новые пятна. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 4-13.
Стадия 12
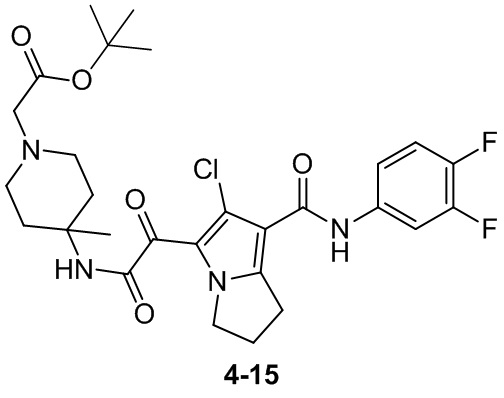
Соединение 4-13 (0,2 г, 430,21 мкмоль, 1 экв.) растворяли в тетрагидрофуране (10 мл), а затем добавляли триэтиламин (174,13 мг, 1,72 ммоль, 239,52 мкл, 4 экв.) и соединение 4-14 (83,91 мг, 430,21 мкмоль, 63,57 мкл, 1 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS было видно, что соединение 4-13 полностью израсходовано и было получено соединение 4-15. В реакционный раствор добавляли 20 мл воды для остановки реакции и экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 4-15. MS(ESI) m/s: 579,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,49 (s, 1H), 7,77-7,72 (m, 1H), 7,15-7,09 (m, 2H), 6,20 (s, 1H), 4,33 (t, J=7,6 Гц, 2H), 3,27 (t, J=7,2 Гц, 2H), 3,14 (s, 2H), 2,78-2,75 (m, 2H), 2,57-2,44 (m, 4H), 2,20-2,16 (m, 2H), 1,89-1,84 (m, 2H), 1,52 (s, 3H), 1,47 (s, 9H).
Стадия 13
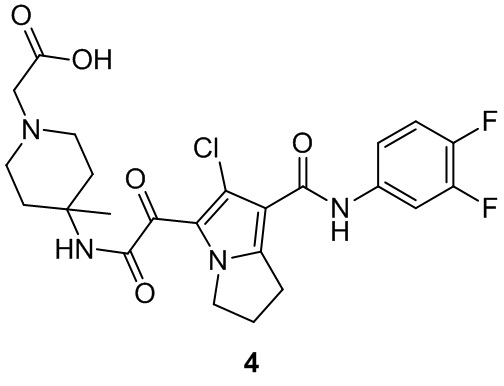
Соединение 4-15 (0,2 г, 345,40 мкмоль, 1 экв.) растворяли в дихлорметане (10 мл), а затем добавляли трифторуксусную кислоту (7,70 г, 67,53 ммоль, 5 мл, 195,51 экв.). Реакционную систему перемешивали при 25°C в течение 0,5 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 22% - 42%, 10,5 мин) с получением соединения 4. MS(ESI) m/s: 523,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,95 (s, 1H), 8,40 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,39 (m, 2H), 4,29 (t, J=7,2 Гц, 2H), 3,18 (s, 2H), 3,09 (t, J=7,6 Гц, 2H), 2,88-2,71 (m, 4H), 2,46-2,43 (m, 2H), 2,18 (d, J=14,0 Гц, 2H), 1,73 (t, J=10,0 Гц, 2H), 1,39 (s, 3H).
Пример 5
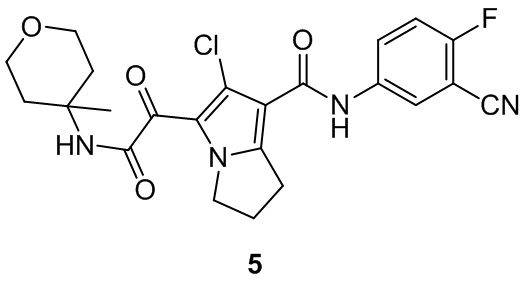
Путь синтеза:
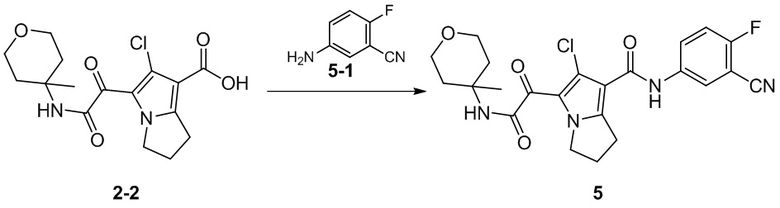
Стадия 1
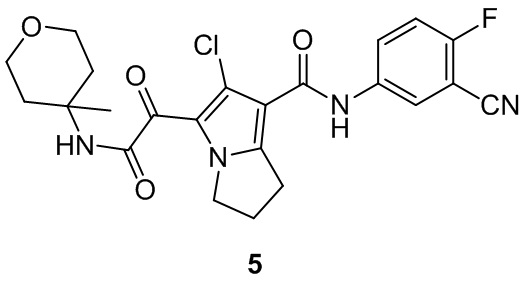
Соединение 2-2 (0,13 г, 366,42 мкмоль, 1 экв.), триэтиламин (55,62 мг, 549,63 мкмоль, 76,50 мкл, 1,5 экв.) и гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (167,19 мг, 439,70 мкмоль, 1,2 экв.) растворяли в N,N-диметилформамиде (3 мл). После перемешивания при 25°C в течение 0,5 ч добавляли соединение 5-1 (99,76 мг, 732,84 мкмоль, 2 экв.). Реакционную систему нагревали до 70°C и перемешивали в течение 3 ч. Результаты LCMS и HPLC подтверждали, что соединение 2-2 полностью израсходовано и было получено соединение 5. В реакционный раствор добавляли 2 капли воды для остановки реакции, а затем фильтровали. Фильтрат очищали с помощью препаративной HPLC (нейтральная система; колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 30% - 50%, 5 мин) с получением соединения 12. MS(ESI) m/s: 473,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,05 (s, 1H), 8,42 (s, 1H), 8,18 (dd, J=2,8, 5,8 Гц, 1H), 7,97 (ddd, J=2,8, 5,0, 9,2 Гц, 1H), 7,52 (t, J=9,2 Гц, 1H), 4,30 (t, J=7,2 Гц, 2H), 3,65-3,53 (m, 4H), 3,10 (t, J=7,6 Гц, 2H), 2,48-2,41 (m, 2H), 2,07 (d, J=13,6 Гц, 2H), 1,56 (ddd, J=4,8, 8,8, 13,6 Гц, 2H), 1,40 (s, 3H).
Пример 6
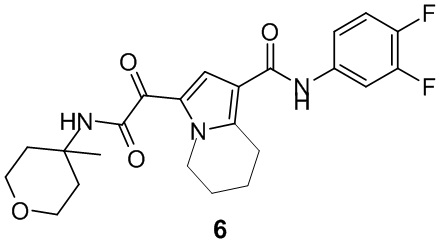
Путь синтеза:
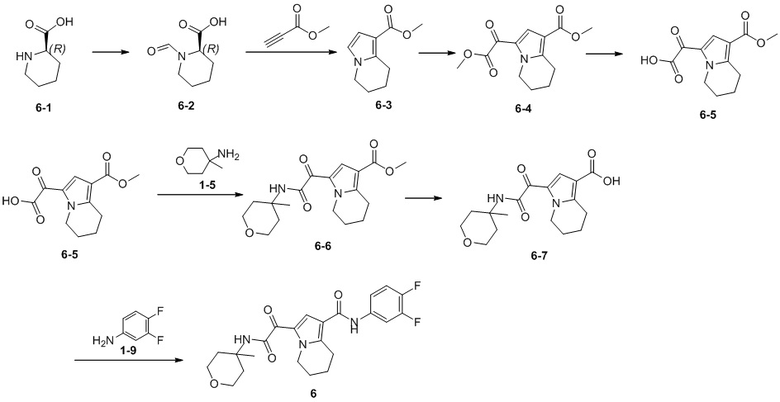
Стадия 1

Соединение 6-1 (10 г, 77,43 ммоль, 1 экв.) растворяли в смеси уксусного ангидрида (40 мл) и муравьиной кислоты (60 мл). Проводили реакцию реакционной смеси при 100°C в течение 16 ч в условиях атмосферы азота. По результатам LCMS наблюдали получение соединения 6-2. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 6-2.
Стадия 2
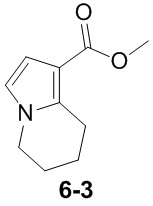
Соединение 6-2 (12,10 г, 77 ммоль, 1 экв.) растворяли в уксусном ангидриде (120 мл), а затем к реакционной смеси добавляли метилпропиолат (32,37 г, 385,00 ммоль, 32,05 мл, 5 экв.). Проводили реакцию полностью всей реакционной системы при 120°C в течение 16 ч в условиях атмосферы азота. По результатам LCMS наблюдали получение соединения 6-3. Реакционный раствор немедленно концентрировали в условиях пониженного давления и неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 50:1 до 5:1, об./об.) с получением соединения 6-3.
1H ЯМР (400 MГц, CDCl3) δ = 6,36 (d, J=3,2 Гц, 1H), 6,30 (d, J=3,2 Гц, 1H), 3,95-3,90 (m, 2H), 3,78 (s, 3H), 3,10-3,05 (m, 2H), 1,96-1,93 (m, 2H), 1,85-1,83 (m, 2H).
Стадия 3
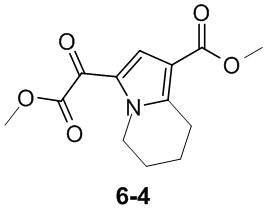
В бутылку с рифленым горлышком с дихлорэтаном (5 мл) добавляли соединение 6-3 (0,5 г, 2,79 ммоль, 1 экв.). Реакционную систему охлаждали до 0°C, а затем в реакционную систему по каплям добавляли метилоксалилхлорид (512,69 мг, 4,18 ммоль, 385,48 мкл, 1,5 экв.). Реакцию в реакционной системе проводили при 0°C 1 ч, и по результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 6-3 практически полностью израсходовано. Реакционный раствор выливали в ледяной насыщенной раствор бикарбоната натрия (20 мл) и экстрагировали этилацетатом (20 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:0 до 10:1, об./об.) с получением соединения 6-4. MS(ESI) m/s: 266,1 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,68 (s, 1H), 4,40 (t, J=6,0 Гц, 2H), 3,96 (s, 3H), 3,84 (s, 3H), 3,17 (t, J=6,0 Гц, 2H), 2,05-1,94 (m, 2H), 1,91-1,81 (m, 2H).
Стадия 4
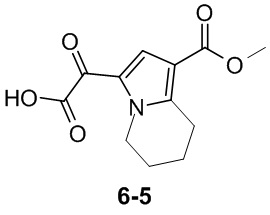
В бутылку с рифленым горлышком с этанолом (5 мл) и водой (2 мл) при 25°C добавляли соединение 6-4 (0,3 г, 1,13 ммоль, 1 экв.), а затем к реакционной системе добавляли карбонат калия (468,93 мг, 3,39 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 2:1) было видно, что соединение 6-4 полностью израсходовано. Реакционный раствор экстрагировали простым метил-трет-бутиловым эфиром (20 мл ×2), доводили до pH = 3 с помощью 1 M соляной кислоты, а затем экстрагировали этилацетатом (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 6-5.
Стадия 5
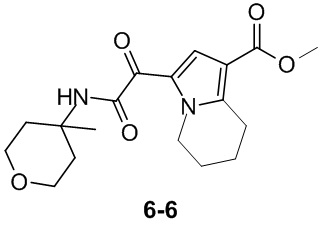
Соединение 1-5 (55,01 мг, 477,64 мкмоль, 1,2 экв.) добавляли в колбу с дихлорэтаном (3 мл) при 20°C, а затем в реакционную систему добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (227,02 мг, 597,05 мкмоль, 1,5 экв.) и N,N-диизопропилэтиламин (154,33 мг, 1,19 ммоль, 207,99 мкл, 3 экв.). После это проводили реакцию в реакционной системе при 0°C в течение 10 мин, соединение 6-5 (0,1 г, 398,03 мкмоль, 1 экв.) растворяли в дихлорэтане (3 мл) и по каплям добавляли в реакционную систему. В полученной в результате реакционной системе проводили реакцию при 20°C в течение 20 мин, и по результатам LCMS было видно получение соединения 6-6. Реакционный раствор промывали с помощью насыщенного раствора хлорида аммония (10 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной TLC (простой петролейный эфир : этилацетат = 2:1, об./об.) с получением соединения 6-6.
MS(ESI) m/s: 349,1 [M+H]+.
Стадия 6
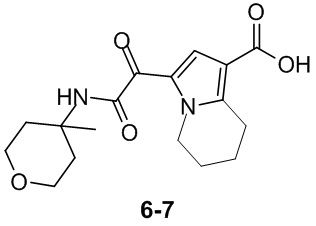
В бутылку с рифленым горлышком с тетрагидрофураном (2 мл), этанолом (2 мл) и водой (2 мл) добавляли соединение 6-6 (0,1 г, 287,03 мкмоль, 1 экв.), а затем к реакционной системе добавляли моногидрат гидроксида лития (48,18 мг, 1,15 ммоль, 4 экв.). Реакционную систему перемешивали при 20°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 2:1) было видно, что соединение 6-6 полностью израсходовано. Реакционный раствор разводили посредством 10 мл воды, доводили до pH = 3 с помощью 1 M соляной кислоты и экстрагировали этилацетатом (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 6-7.
Стадия 7
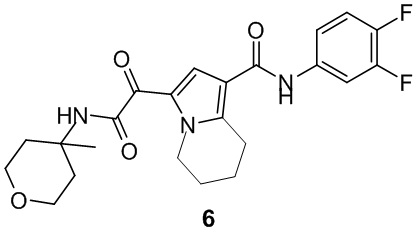
В бутылку с рифленым горлышком с дихлорметаном (2 мл) при 20°C добавляли соединение 1-9 (42,28 мг, 327,49 мкмоль, 1,5 экв.), а затем реакционную систему охлаждали до 0°C. Затем к реакционной системе добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (107,92 мг, 283,82 мкмоль, 1,3 экв.) и N,N-диизопропилэтиламин (84,65 мг, 654,97 мкмоль, 114,08 мкл, 3 экв.). После этого реакционную систему перемешивали при 0°C в течение 10 мин, соединение 6-7 (73 мг, 218,32 мкмоль, 1 экв.) растворяли в дихлорметане (2 мл) и по каплям добавляли в реакционную систему. В полученной в результате реакционной системе проводили реакцию при 0°C в течение 20 мин, затем нагревали до 70°C и перемешивали в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно получение основных пятен, а по результатам LCMS было видно получение соединения 6. Реакционный раствор промывали с помощью насыщенного хлорида аммония (10 мл), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 45% - 65%, 10,5 мин) с получением соединения 6.
MS(ESI) m/s: 446,2 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 8,04 (s, 1H), 7,79-7,69 (m, 1H), 7,38-7,30 (m, 1H), 7,26-7,16 (m, 1H), 4,42 (t, J=6,8 Гц, 2H), 3,79-3,62 (m, 4H), 3,20 (t, J=6,8 Гц, 2H), 2,23 (m, 2H), 2,00 (m, 2H), 1,88 (m, 2H), 1,71 (m, 2H), 1,50 (s, 3H).
Пример 7
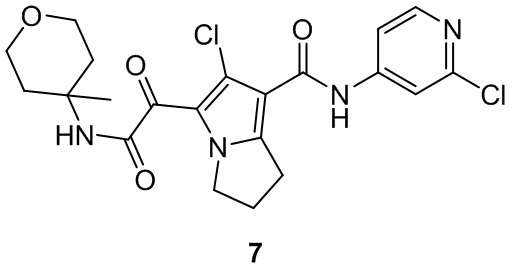
Путь синтеза:

Стадия 1
Соединение 7-1 (36,17 мг, 281,33 мкмоль, 1,5 экв.) и триэтиламин (56,94 мг, 562,65 мкмоль, 78,32 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем при 0°C добавляли соединение 2-3 (70 мг, 187,55 мкмоль, 1 экв.), растворенное в дихлорметане (5 мл). Реакционную систему перемешивали при 25°C в течение 2 ч. По результатам LCMS и HPLC было видно, что реакция завершена. Реакцию останавливали с помощью 15 мл воды, а затем реакционный раствор экстрагировали дихлорметаном (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 28% - 58%, 10,5 мин) с получением соединения 7. MS(ESI) m/s: 465,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,28 (s, 1H), 8,42 (s, 1H), 8,29 (d, J=5,6 Гц, 1H), 7,81 (d, J=1,6 Гц, 1H), 7,62-7,60 (m, 1H), 4,30 (t, J=7,2 Гц, 2H), 3,63-3,57 (m, 4H), 3,10 (t, J=7,2 Гц, 2H), 2,47-2,45 (m, 2H), 2,07 (d, J=13,2 Гц, 2H), 1,59-1,53 (m, 2H), 1,41 (s, 3H).
Пример 8
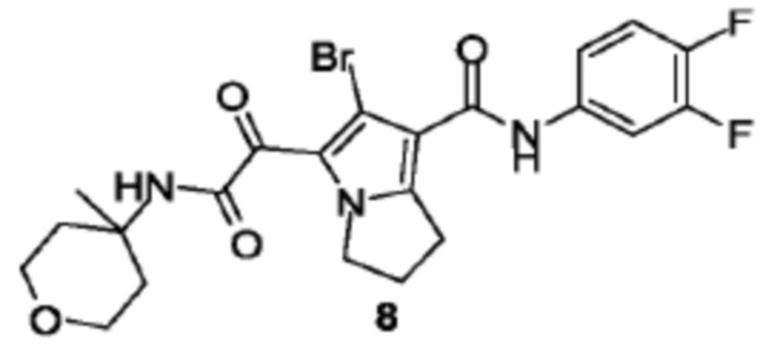
Путь синтеза:
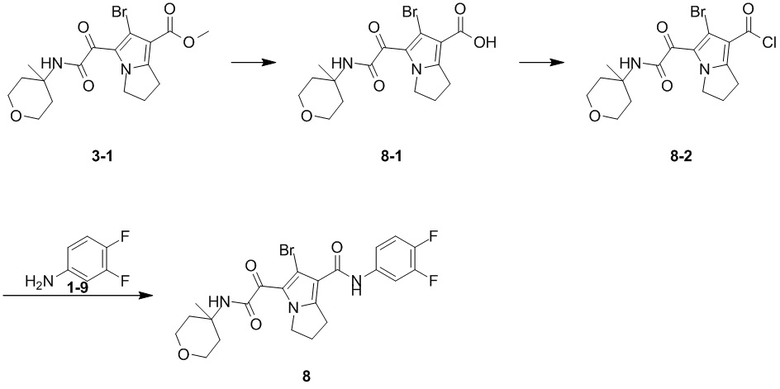
Стадия 1
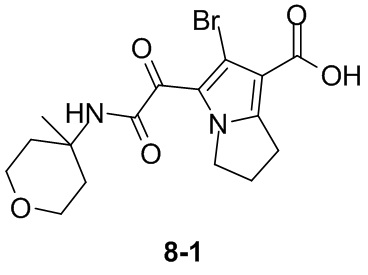
Раствор гидрохлорида натрия (87,11 мг, 2,18 ммоль, 3 экв.) в воде (2 мл) добавляли к раствору соединения 3-1 (300 мг, 725,93 мкмоль, 1 экв.) в тетрагидрофуране (5 мл) и смесь перемешивали при 40°C в течение 12 ч. Реакционный раствор доводили до pH = 1-2 с помощью 2 моль/л разбавленной соляной кислоты и экстрагировали этилацетатом (10 мл ×3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления с получением соединения 8-1. MS m/s (ESI): 398,9 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,34 (t, J=7,2 Гц, 2H), 3,82-3,70 (m, 4H), 3,15 (t, J=7,8 Гц, 2H), 2,50-2,48 (m, 2H), 2,22 (d, J=13,8 Гц, 2H), 1,77-1,65 (m, 2H), 1,55 (s, 3H).
Стадия 2
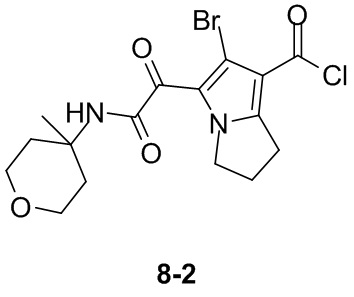
Тионил хлорид (166,88 мг, 1,40 ммоль, 101,75 мкл, 2 экв.) по каплям добавляли к раствору соединения 8-1 (280 мг, 701,34 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота, а затем добавляли N,N-диметилформамид (25,63 мг, 350,67 мкмоль, 26,98 мкл, 0,5 экв.). Смесь перемешивали при 25°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 8-2, которое применяли непосредственно на следующей стадии.
Стадия 3
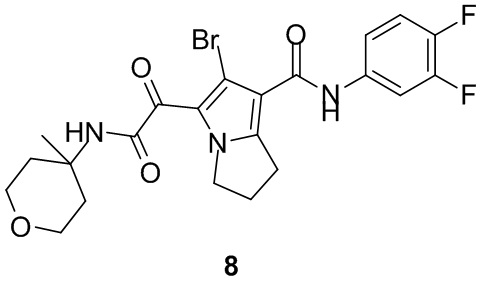
Триэтиламин (290,72 мг, 2,87 ммоль, 399,89 мкл, 4 экв.) добавляли к раствору соединения 8-2 (300 мг, 718,25 мкмоль, 1 экв.) в дихлорметане (5 мл), а затем добавляли соединение 1-9 (92,73 мг, 718,25 мкмоль, 1 экв.). Смесь перемешивали при 25°C в течение 1 ч. Реакционный раствор концентрировали в условиях пониженного давления. Остаток разделяли с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = 100:0 до 40:60, об./об.) и затем препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,225% муравьиной кислоты)-ацетонитрил; % содержания ацетонитрила: 48% - 68%, 7 мин) с получением соединения 8. MS m/s (ESI): 510,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,54 (s, 1H), 7,73-7,62 (m, 1H), 7,11-7,01 (m, 2H), 6,30 (s, 1H), 4,25 (t, J=7,4 Гц, 2H), 3,76-3,57 (m, 4H), 3,21 (t, J=7,8 Гц, 2H), 2,46-2,43(m, 2H), 2,08 (d, J=13,8 Гц, 2H), 1,74 (m, 2H), 1,49 (s, 3H).
Пример 9
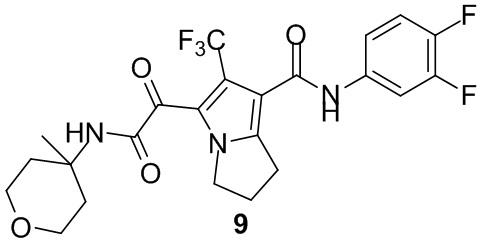
Путь синтеза:
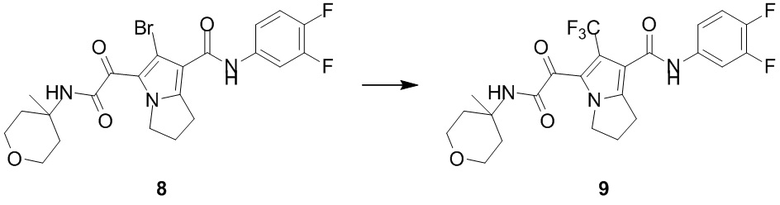
Стадия 1
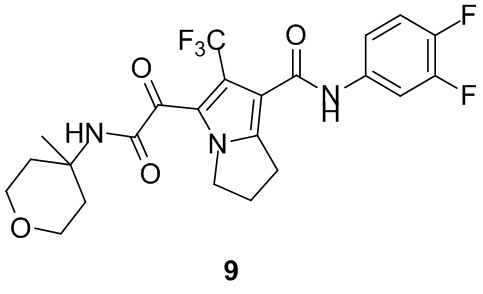
Метилфторсульфонилдифторацетат (180,70 мг, 940,57 мкмоль, 119,67 мкл, 6 экв.) и триамид гексаметилфосфорной кислоты (28,09 мг, 156,76 мкмоль, 27,54 мкл, 1 экв.) добавляли к раствору соединения 8 (80 мг, 156,76 мкмоль, 1 экв.) в DMF (3 мл). После трех продувок азотом добавляли йодид меди(I) (29,86 мг, 156,76 мкмоль, 1 экв.). Смесь перемешивали при 80°C в течение 12 ч. Реакционный раствор разводили водой (10 мл) и затем экстрагировали этилацетатом (10 мл ×3). Объединенные органические фазы промывали с помощью насыщенного солевого раствора (10 мл), сушили над безводным сульфатом натрия и концентрировали в условиях пониженного давления. Остаток разделяли с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,225% муравьиной кислоты)-ацетонитрил; % содержания ацетонитрила: 50% - 70%, 7 мин) с получением соединения 9. MS m/s (ESI): 500,2 [M+H1]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,68-7,59 (m, 1H), 7,55 (s, 1H), 7,12-6,95 (m, 2H), 6,51 (s, 1H), 4,16 (t, J=7,2 Гц, 2H), 3,77-3,66 (m, 2H), 3,64-3,51 (m, 2H), 3,09 (t, J=7,4 Гц, 2H), 2,50-2,48 (m, 2H), 2,06 (d, J=13,6 Гц, 2H), 1,71 (t, J=9,6 Гц, 2H), 1,44 (s, 3H).
Пример 10
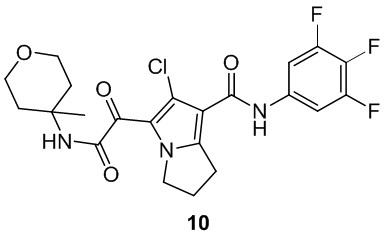
Путь синтеза:
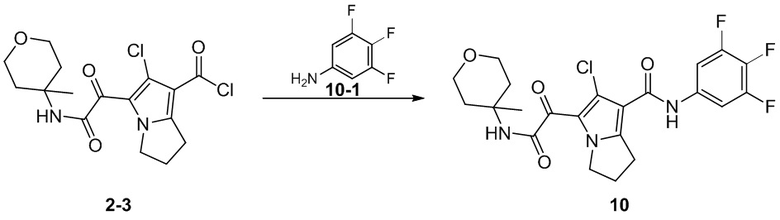
Стадия 1
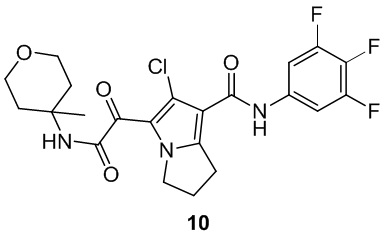
Соединение 10-1 (59,12 мг, 401,90 мкмоль, 1,5 экв.) и триэтиламин (81,34 мг, 803,79 мкмоль, 111,88 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем при 0°C добавляли соединение 2-3 (0,1 г, 267,93 мкмоль, 1 экв.), растворенное в дихлорметане (5 мл). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что соединение 2-3 полностью израсходовано и было получено соединение 10. Реакцию останавливали с помощью воды (10 мл), а затем реакционный раствор экстрагировали дихлорметаном (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 42% - 62%, 10,5 мин) с получением соединения 10. MS(ESI) m/s: 484,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,03 (s, 1H), 8,42 (s, 1H), 7,64-7,60 (m, 2H), 4,30 (t, J=7,2 Гц, 2H), 3,63-3,55 (m, 4H), 3,08 (t, J=7,6 Гц, 2H), 2,47-2,43 (m, 2H), 2,07 (d, J=13,2 Гц, 2H), 1,59-1,52 (m, 2H), 1,40 (s, 3H).
Пример 11
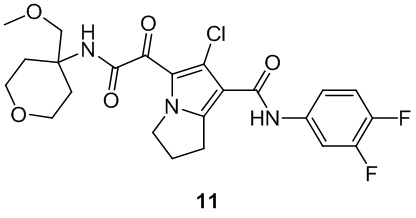
Путь синтеза:
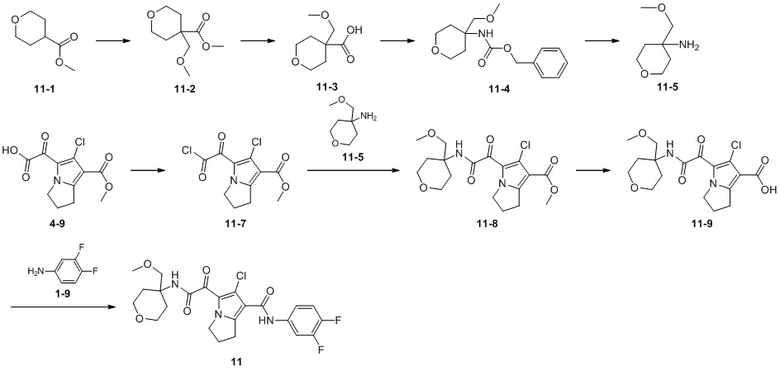
Стадия 1
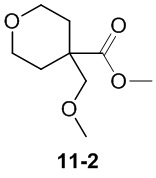
Соединение 11-1 (10 г, 69,36 ммоль, 9,26 мл, 1 экв.) растворяли в тетрагидрофуране (100 мл), а затем добавляли диизопропиламид лития (2 M, 52,02 мл, 1,5 экв.) при -78°C. После перемешивания в течение 0,5 ч добавляли простой хлорметиловый эфир (8,38 г, 104,05 ммоль, 7,90 мл, 1,5 экв.) при -78°C. Реакционную систему перемешивали при 25°C в течение 2 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что соединение 11-1 полностью израсходовано и были получены новые пятна. Реакционный раствор выливали в 50 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом (50 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 11-2.
1H ЯМР (400 MГц, CDCl3) δ = 3,82 (td, J=4,0, 11,8 Гц, 2H), 3,76 (s, 3H), 3,52-3,46 (m, 2H), 3,40 (s, 2H), 3,30 (s, 3H), 2,09-2,05 (m, 2H), 1,62-1,54 (m, 2H).
Стадия 2
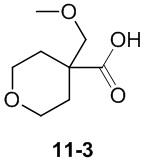
Соединение 11-2 (12 г, 63,76 ммоль, 1 экв.) растворяли в смеси метанола (100 мл) и воды (50 мл), а затем добавляли гидроксид натрия (5,10 г, 127,51 ммоль, 2 экв.). Реакционную систему перемешивали при 40°C в течение 16 ч. Реакционный раствор экстрагировали этилацетатом (50 мл ×2). Водную фазу доводили до pH = 1-3 с помощью 1 моль/л соляной кислоты и экстрагировали этилацетатом (50 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 11-3.
1H ЯМР (400 MГц, MeOH-d4) δ =3,80 (td, J=4,0, 11,6 Гц, 2H), 3,58-3,51 (m, 2H), 3,43 (s, 2H), 3,31 (s, 3H), 2,04-1,99 (m, 2H), 1,62-1,54 (m, 2H).
Стадия 3
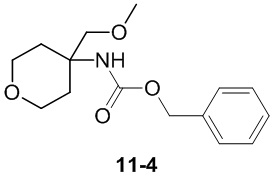
Соединение 11-3 (8,5 г, 48,80 ммоль, 1 экв.), триэтиламин (7,41 г, 73,19 ммоль, 10,19 мл, 1,5 экв.) и бензиловый спирт (10,55 г, 97,59 ммоль, 10,15 мл, 2 экв.) растворяли в толуоле (100 мл), а затем добавляли дифенилфосфорилазид (16,11 г, 58,56 ммоль, 12,69 мл, 1,2 экв.) при 25°C в условиях атмосферы азота. Реакционную систему перемешивали при 110°C в течение 5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что были получены новые пятна. Реакционный раствор выливали в 100 мл насыщенного раствора хлорида аммония, а затем экстрагировали этилацетатом (80 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:0 до 10:1, об./об.). Очищенный продукт концентрировали в условиях пониженного давления с получением соединения 11-4.
1H ЯМР (400 MГц, CDCl3) δ = 7,34-7,29 (m, 5H), 3,74-3,49 (m, 4H), 3,36 (s, 2H), 3,33 (s, 3H), 2,18 (s, 2H), 2,04-1,96 (m, 2H), 1,74-1,66 (m, 2H).
Стадия 4
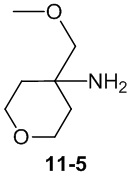
Соединение 11-4 (14 г, 50,12 ммоль, 1 экв.) в сухой 250-мл гидрогенизационной колбе растворяли в метаноле (200 мл), а затем добавляли влажный палладиевый катализатор на углеродном носителе (3 г, степень чистоты: 10%). Реакционную систему перемешивали при 25°C в течение 16 ч в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что реакция завершилась. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт гомогенизировали с простым метил-трет-бутиловым эфиром (30 мл) и фильтровали. Остаток на фильтре концентрировали в условиях пониженного давления с получением соединения 11-5.
1H ЯМР (400 MГц, MeOH-d4) δ =3,84-3,81 (m, 2H), 3,68-3,62 (m, 4H), 3,45 (s, 3H), 1,93-1,88 (m, 2H), 1,84-1,80 (m, 2H).
Стадия 5
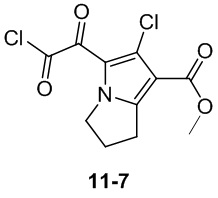
Соединение 4-9 (0,2 г, 736,23 мкмоль, 1 экв.) растворяли в дихлорметане (10 мл), а затем добавляли оксалилхлорид (186,89 мг, 1,47 ммоль, 128,89 мкл, 2 экв.) и N,N-диметилформамид (5,38 мг, 73,62 мкмоль, 5,66 мкл, 0,1 экв.) при 0°C. Реакционную систему перемешивали в течение 0,5 ч при 25°C в условиях атмосферы азота. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно, что соединение 4-9 полностью израсходовано и были получены новые пятна. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 11-7.
Стадия 6
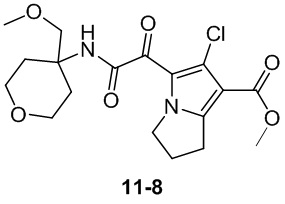
Соединение 11-5 (150,16 мг, 826,58 мкмоль, 1,20 экв., HCl) и триэтиламин (209,29 мг, 2,07 ммоль, 287,88 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем добавляли раствор соединения 11-7 (0,2 г, 689,42 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 1 ч. Результаты LCMS подтверждали, что соединение 11-7 полностью израсходовано и было получено соединение 11-8. Реакцию останавливали с помощью 15 мл воды, а затем реакционный раствор экстрагировали дихлорметаном (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 11-8.
MS(ESI) m/s: 399,1 [M+H]+.
Стадия 7
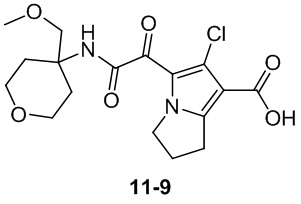
Соединение 11-8 (0,4 г, 1,00 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (8 мл) и воды (4 мл), а затем добавляли моногидрат гидроксида лития (126,26 мг, 3,01 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 12 ч. Результаты LCMS подтверждали, что соединение 11-8 полностью израсходовано и было получено соединение 11-9. Реакционный раствор доводили до pH = 1-3 с помощью 1 M разбавленной соляной кислоты и экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 11-9. MS(ESI) m/s: 385,0 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,36 (t, J=7,2 Гц, 2H), 3,77-3,72 (m, 4H), 3,64 (s, 2H), 3,37 (s, 3H), 3,15 (t, J=7,6 Гц, 2H), 2,54-2,50 (m, 2H), 2,19 (d, J=12,8 Гц, 2H), 1,84-1,77 (m, 2H).
Стадия 8
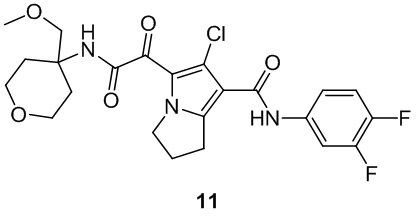
Соединение 11-9 (0,2 г, 519,74 мкмоль, 1 экв.), триэтиламин (210,37 мг, 2,08 ммоль, 289,37 мкл, 4 экв.) и гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (592,86 мг, 1,56 ммоль, 3 экв.) растворяли в N,N-диметилформамиде (3 мл). После перемешивания при 25°C в течение 0,5 ч добавляли соединение 1-9 (134,20 мг, 1,04 ммоль, 2 экв.). Реакционную систему перемешивали при 55°C в течение 3 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. В реакционный раствор добавляли 2 капли воды для остановки реакции, а затем фильтровали с получением фильтрата. Фильтрат очищали с помощью препаративной HPLC (нейтральная система; колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 30% - 60%, 11 мин) с получением соединения 12. MS(ESI) m/s: 496,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,95 (s, 1H), 8,42 (s, 1H), 7,87-7,81 (m, 1H), 7,43-7,39 (m, 2H), 4,29 (t, J=7,2 Гц, 2H), 3,67-3,55 (m, 6H), 3,28 (s, 3H), 3,08 (t, J=7,6 Гц, 2H), 2,43 (s, 2H), 2,07 (d, J=13,4 Гц, 2H), 1,68-1,60 (m, 2H).
Пример 12
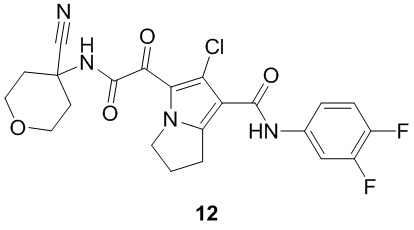
Путь синтеза:
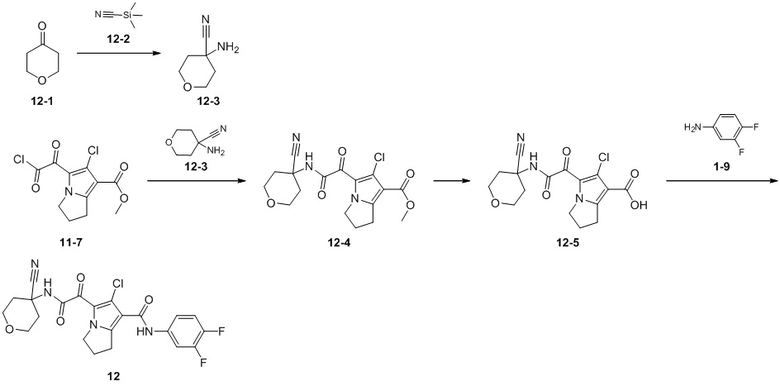
Стадия 1
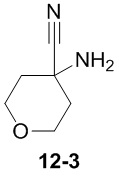
Тетраизопропанолат титана (3,41 г, 11,99 ммоль, 3,54 мл, 1,2 экв.) в сухой 40-мл колбе растворяли в растворе aммoния/метаноле (7 M, 5,71 мл, 4 экв.), а затем по каплям добавляли соединение 12-1 (1 г, 9,99 ммоль, 917,43 мкл, 1 экв.) в условиях атмосферы азота при 30°C. После перемешивания в течение 2 ч, смесь охлаждали до -5°C, а затем по каплям при такой температуре добавляли соединение 12-2 (1,04 г, 10,49 ммоль, 1,31 мл, 1,05 экв.). После перемешивания в течение 2 ч полученную смесь нагревали до 30°C и перемешивали в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) было видно, что реакция завершилась. Реакцию останавливали с помощью воды (1 мл), а затем в реакционный раствор добавляли этанол (20 мл), перемешивали в течение 30 мин и фильтровали. Исходный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 1:1, об./об.) с получением соединения 12-3.
1H ЯМР (400 MГц, CDCl3) δ = 4,02-3,95 (m, 2H), 3,68-3,62 (m, 2H), 2,01-1,96 (m, 2H), 1,87 (s, 2H), 1,78-1,74 (m, 2H).
Стадия 2
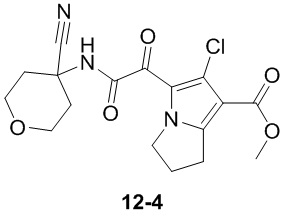
Соединение 12-3 (97,85 мг, 775,60 мкмоль, 1,5 экв.) в сухой 40-мл колбе растворяли в дихлорметане (10 мл) и по каплям добавляли раствор триэтиламина (156,97 мг, 1,55 ммоль, 215,91 мкл, 3 экв.) и соединения 11-7 (150 мг, 517,06 мкмоль, 1 экв.) в дихлорметане (5 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 30°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор разводили дихлорметаном (50 мл) и промывали разбавленной соляной кислотой (0,5 моль/л) (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 12-4. MS(ESI) m/s: 380,1 [M+H]+.
Стадия 3
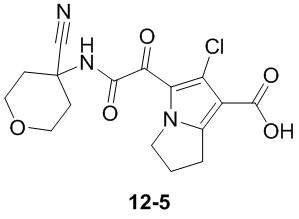
Соединение 12-4 (200 мг, 526,60 мкмоль, 1 экв.) в сухой 50-мл колбе с одним горлышком растворяли в смеси тетрагидрофурана (3 мл), метанола (3 мл) и воды (3 мл), а затем добавляли моногидрат гидроксида лития (44,19 мг, 1,05 ммоль, 2 экв.). Реакционный раствор перемешивали при 30°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Тетрагидрофуран и метанол концентрировали в условиях пониженного давления и разводили посредством 20 мл воды. Водную фазу промывали этилацетатом (5 мл ×2), доводили до pH = 1 с помощью 0,5 M разбавленной соляной кислоты и экстрагировали этилацетатом (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 12-5. MS(ESI) m/s: 366,1 [M+H]+.
Стадия 4
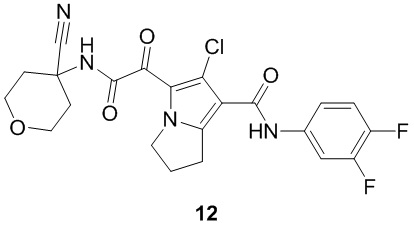
Соединение 12-5 (130 мг, 355,42 мкмоль, 1 экв.) в сухой 50-мл колбе с одним горлышком растворяли в N,N-диметилформамиде (5 мл). В условиях атмосферы азота при 30°C добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (270,28 мг, 710,83 мкмоль, 2 экв.) и триэтиламин (107,89 мг, 1,07 ммоль, 148,41 мкл, 3 экв.). После перемешивания в течение 0,5 ч добавляли соединение 1-9 (68,83 мг, 533,13 мкмоль, 1,5 экв.). После реакции смесь нагревали до 60°C и перемешивали в течение 15,5 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. В реакционный раствор добавляли воду (1 мл) и затем концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 30% - 50%, 12 мин) с получением соединения 12. MS(ESI) m/s: 477,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,96 (s, 1H), 9,58 (s, 1H), 7,85-7,80 (m, 1H), 7,42-7,38 (m, 2H), 4,29 (t, J=7,2 Гц, 2H), 3,86-3,82 (m, 2H), 3,57 (t, J=10,0 Гц, 2H), 3,09 (t, J=7,6 Гц, 2H), 2,46-2,44 (m, 2H), 2,30-2,27 (m, 2H), 1,95-1,90 (m, 2H).
Пример 13
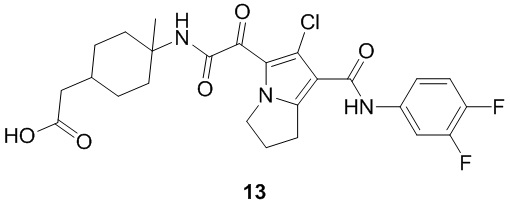
Путь синтеза:
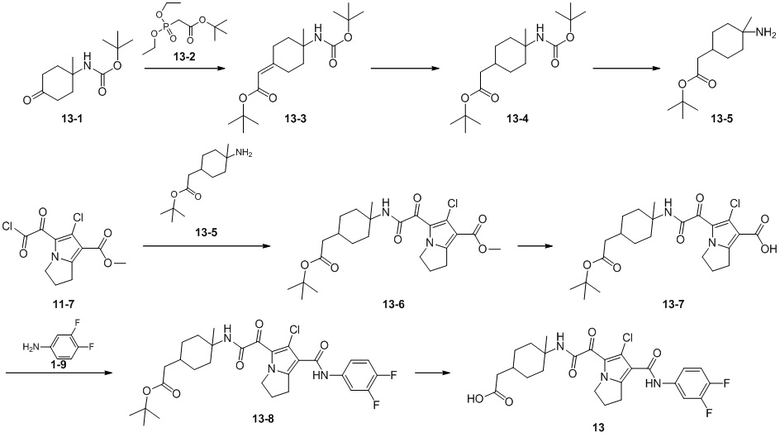
Стадия 1
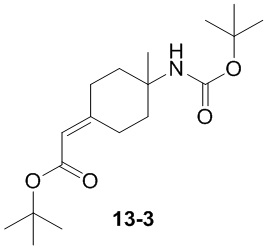
Соединение 13-1 (1 г, 4,40 ммоль, 1 экв.) в колбе с одним горлышком растворяли в тетрагидрофуране (20 мл) и добавляли гидрид натрия (615,93 мг, степень чистоты: 60%, 3,5 экв.) при 0°C в условиях атмосферы азота. После перемешивания в течение 0,5 ч добавляли соединение 13-2 (2,33 г, 9,24 ммоль, 2,1 экв.) и реакционную систему перемешивали в течение 30 мин при 30°C. По результатам TLC (простой петролейный эфир : этилацетат = 10:1) было видно, что реакция завершилась. Реакционную систему выливали в разбавленную соляную кислоту с концентрацией 0,5 моль/л (100 мл). Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 50:1 до 20:1) с получением соединения 13-3.
1H ЯМР (400 MГц, CDCl3) δ = 5,55 (s, 1H), 4,43 (s, 1H), 3,32-3,26 (m, 1H), 2,41-2,38 (m, 1H), 2,30-2,28 (m, 1H), 2,15-2,14 (m, 1H), 2,13-2,04 (m, 1H), 2,03-2,01 (m, 1H), 1,54-1,49 (m, 2H), 1,46 (s, 9H), 1,43 (s, 9H), 1,33 (s, 3H).
Стадия 2
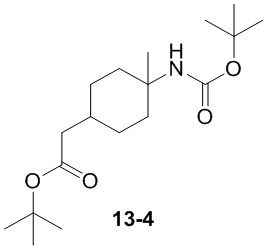
Влажный палладиевый катализатор на углеродном носителе (200 мг, содержание: 10%) в гидрогенизационной колбе растворяли в тетрагидрофуране (40 мл) и добавляли соединение 13-3 (1,35 г, 4,15 ммоль, 1 экв.). Реакционную систему перемешивали при 25°C в течение 2 ч в условиях атмосферы водорода (15 psi). По результатам TLC (простой петролейный эфир : этилацетат = 10:1) было видно, что реакция завершилась. Реакционный раствор фильтровали и концентрировали с получением соединения 13-4.
Стадия 3
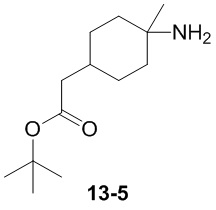
Соединение 13-4 (1,3 г, 3,97 ммоль, 1 экв.) в сухой колбе с одним горлышком растворяли в этилацетате (30 мл) и добавляли соляную кислоту/этилацетат (4 моль/л, 10 мл, 10,08 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч. Реакционный раствор немедленно концентрировали с получением соединения 13-5.
1H ЯМР (400 MГц, MeOH-d4) δ = 1,85-1,83 (m, 4H), 1,77-1,76 (m, 2H), 1,63-1,60 (m, 3H), 1,45 (s, 9H), 1,35-1,31 (m, 5H).
Стадия 4
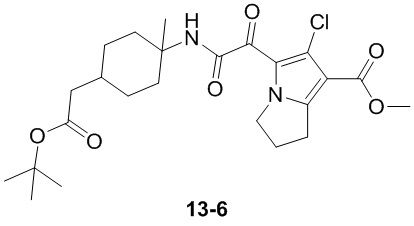
Соединение 13-5 (250 мг, 861,77 мкмоль, 1 экв.) в сухой колбе с одним горлышком растворяли в дихлорметане (10 мл) и добавляли триэтиламин (261,61 мг, 2,59 ммоль, 359,85 мкл, 3 экв.). В полученную смесь по каплям добавляли раствор соединения 11-7 (293,88 мг, 1,29 ммоль, 1,5 экв.) в дихлорметане (10 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 30°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в воду (50 мл). Затем водную фазу экстрагировали дихлорметаном (20 мл ×3), а органическую фазу промывали 0,5 моль/л разбавленной соляной кислотой (20 мл ×3), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 13-6. MS(ESI) m/s: 425,2 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, CDCl3) δ = 5,99 (s, 1H), 4,32 (t, J=7,6 Гц, 2H), 3,84 (s, 3H), 3,14 (t, J=7,6 Гц, 2H), 2,52-2,48 (m, 2H), 2,31-2,28 (m, 2H), 2,14-2,12 (m, 2H), 1,68-1,65 (m, 2H), 1,45-1,42 (m, 2H), 1,39-1,36 (m, 12H), 1,24-1,22 (m, 2H), 1,18-1,14 (m, 2H).
Стадия 5
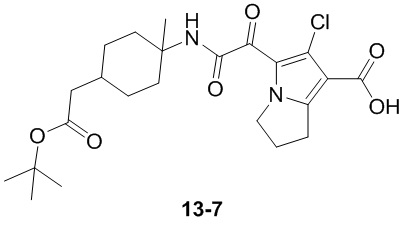
Соединение 13-6 (90 мг, 187,12 мкмоль, 1 экв.) в колбе растворяли в смеси тетрагидрофурана (3 мл), метанола (3 мл) и воды (3 мл) и добавляли моногидрат гидроксида лития (23,55 мг, 561,35 мкмоль, 3 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления для удаления тетрагидрофурана и метанола и разводили посредством 20 мл воды. Затем водную фазу промывали этилацетатом (5 мл ×2), доводили до pH = 1 с помощью 0,5 моль/л разбавленной соляной кислоты и экстрагировали этилацетатом (5 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной TLC (дихлорметан : метанол = 10:1, об./об.) с получением соединения 13-7. MS(ESI) m/s: 411,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,38-4,31 (m, 2H), 3,16-3,12 (m, 2H), 2,55-2,48 (m, 2H), 2,32-2,30 (m, 2H), 2,14-2,11 (m, 2H), 1,75-1,70 (m, 1H), 1,65-1,55 (m, 2H), 1,44 (s, 9H), 1,35-1,31 (m, 4H), 1,22 (s, 3H).
Стадия 6
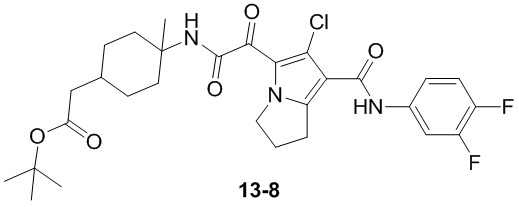
Соединение 13-7 (70 мг, 149,91 мкмоль, 1 экв.) в сухой колбе растворяли в N,N-диметилформамиде (4 мл) и добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (114,00 мг, 299,82 мкмоль, 2 экв.) и триэтиламин (45,51 мг, 449,72 мкмоль, 62,60 мкл, 3 экв.) в условиях атмосферы азота при 30°C. После перемешивания в течение 30 мин добавляли соединение 1-9 (29,03 мг, 224,86 мкмоль, 1,5 экв.) и реакционную систему нагревали до 60°C и перемешивали в течение 15,5 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в 0,2 моль/л разбавленной соляной кислоты (50 мл), а затем экстрагировали этилацетатом (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 13-8. MS(ESI) m/s: 522,1 [M+H-(трет-Bu)]+.
Стадия 7
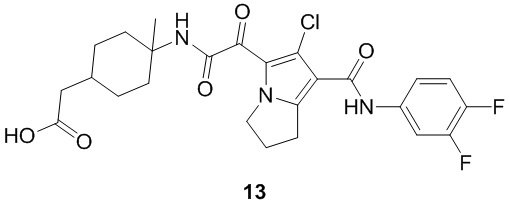
Соединение 13-8 (50 мг, 86,50 мкмоль, 1 экв.) в сухой колбе с одним горлышком растворяли в смеси трифторуксусной кислоты (1 мл) и дихлорметана (2 мл) и реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 20% - 40%, 13 мин) с получением соединения 12. MS(ESI) m/s: 522,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,93 (s, 1H), 8,10 (s, 1H), 7,88-7,83 (m, 1H), 7,44-7,42 (m, 2H), 4,29 (t, J=6,8 Гц, 2H), 3,09 (t, J=8,0 Гц, 2H), 2,47-2,45 (m, 2H), 2,24-2,22 (m, 2H), 2,09-2,07 (m, 2H), 1,65-1,60 (m, 1H), 1,54-1,51 (m, 2H), 1,35 (s,3H), 1,24-1,23 (m, 4H).
Пример 14
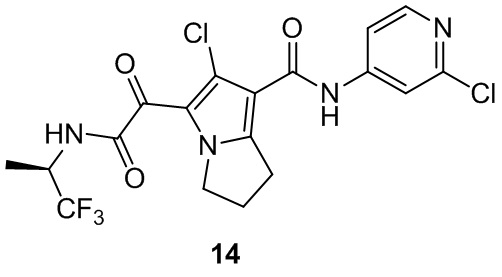
Путь синтеза:
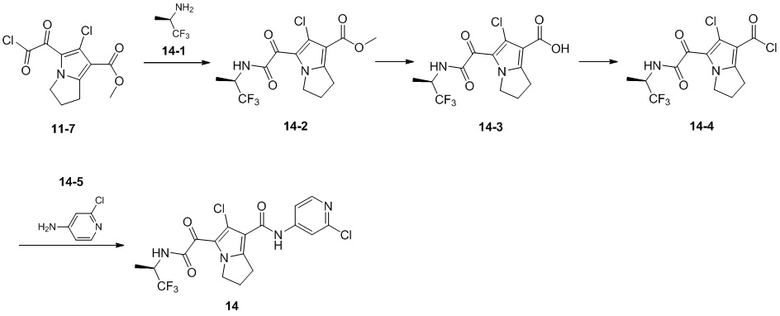
Стадия 1
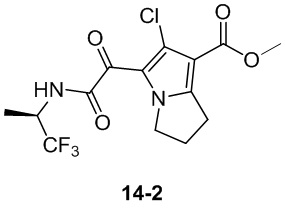
Соединение 14-1 (124,74 мг, 1,10 ммоль, 2 экв.) и триэтиламин (167,43 мг, 1,65 ммоль, 230,30 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем добавляли раствор соединения 11-7 (0,16 г, 551,54 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам LCMS было видно, что соединение 11-7 полностью израсходовано и было получено соединение 14-2. Реакцию останавливали с помощью 20 мл воды, а затем реакционный раствор экстрагировали дихлорметаном (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 14-2. MS(ESI) m/s: 367,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 6,53 (d, J=9,6 Гц, 1H), 4,38-4,25 (m, 2H), 3,85 (s, 3H), 3,15-3,10 (m, 2H), 2,55-2,48 (m, 2H), 1,45-1,42 (m, 4H).
Стадия 2
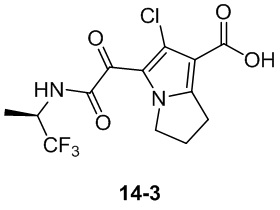
Соединение 14-2 (0,19 г, 518,11 мкмоль, 1 экв.) растворяли в смеси метанола (5 мл), тетрагидрофурана (5 мл) и воды (5 мл), а затем добавляли моногидрат гидроксида лития (65,22 мг, 1,55 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 2 ч. По результатам TLC (дихлорметан : метанол = 20:1) было видно, что соединение 14-2 полностью израсходовано и были получены новые пятна. Реакционный раствор доводили до pH = 1-3 с помощью 1 M соляной кислоты и экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 14-3.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,76-4,69 (m, 1H), 4,36 (t, J=7,2 Гц, 2H), 3,15 (t, J=7,6 Гц, 2H), 2,56-2,49 (m, 2H), 1,40 (d, J=7,0 Гц, 3H).
Стадия 3
Соединение 14-3 (0,1 г, 283,53 мкмоль, 1 экв.) растворяли в дихлорметане (5 мл), а затем добавляли оксалилхлорид (71,98 мг, 567,07 мкмоль, 49,64 мкл, 2 экв.) и N,N-диметилформамид (2,07 мг, 28,35 мкмоль, 2,18 мкл, 0,1 экв.) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 0,5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 14-3 полностью израсходовано и были получены новые пятна. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 14-4.
Стадия 4
Соединение 14-5 (69,28 мг, 538,88 мкмоль, 2 экв.) и триэтиламин (81,79 мг, 808,32 мкмоль, 112,51 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем добавляли раствор соединения 14-4 (0,1 г, 269,44 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему нагревали до 25°C и перемешивали в течение 1 ч. Результаты LCMS и HPLC подтверждали, что соединение 14-4 полностью израсходовано и было получено соединение 14. Реакционный раствор выливали в 20 мл воды для остановки реакции и экстрагировали дихлорметаном (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью препаративной HPLC (нейтральная; колонка: Waters Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 37% - 57%, 10,5 мин) с получением соединения 14. MS(ESI) m/s: 463,0 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,37 (s, 1H), 9,45 (s, 1H), 8,25 (d, J=6,0 Гц, 1H), 7,79 (d, J=1,6 Гц, 1H), 7,57 (dd, J=2,0, 6,0 Гц, 1H), 4,72-4,67 (m, 1H), 4,30 (t, J=7,2 Гц, 2H), 3,13-3,10 (m, 2H), 2,47-2,43 (m, 2H), 1,31 (d, J=6,8 Гц, 3H).
Пример 15
Путь синтеза:
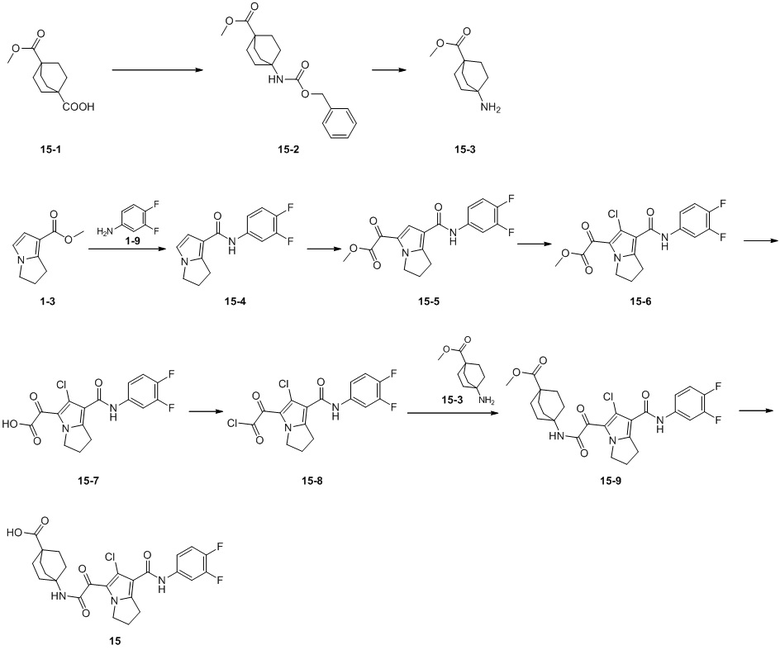
Стадия 1
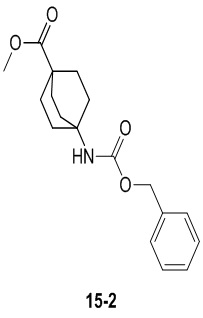
В толуол (15 мл) добавляли соединение 15-1 (1,5 г, 7,07 ммоль, 1 экв.), триэтиламин (1,07 г, 10,60 ммоль, 1,48 мл, 1,5 экв.) и бензиловый спирт (1,53 г, 14,13 ммоль, 1,47 мл, 2 экв.), а затем добавляли дифенилфосфорилазид (2,33 г, 8,48 ммоль, 1,84 мл, 1,2 экв.). Реакционную систему перемешивали при 110°C в течение 16 ч. Результаты LCMS подтверждали, что соединение 15-1 полностью израсходовано и было получено соединение 15-2; по результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что были получены новые пятна. Реакционный раствор выливали в насыщенной раствор хлорида аммония (30 мл) для остановки реакции и экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:1 до 10:1, об./об.) с получением соединения 15-2. MS(ESI) m/s: 318,1 [M+H]+.
Стадия 2
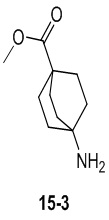
Влажный палладиевый катализатор на углеродном носителе (1 г, степень чистоты: 10%) растворяли в метаноле (20 мл), а затем добавляли соединение 15-2 (2 г, 6,30 ммоль, 1 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что соединение 15-2 полностью израсходовано и были получены новые пятна. После этого реакционный раствор фильтровали, добавляли соляную кислоту в метаноле (4 моль/л, 50 мл). Полученную в результате смесь перемешивали в течение 0,5 ч и концентрировали в условиях пониженного давления с получением соединения 15-3.
1H ЯМР (400 MГц, MeOH-d4) δ = 3,65 (s, 3H), 1,99-1,81 (m, 12H).
Стадия 3
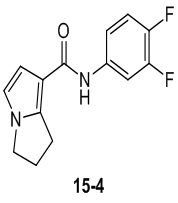
Соединение 1-3 (15 г, 90,81 ммоль, 1 экв.) и соединение 1-9 (17,59 г, 136,21 ммоль, 1,5 экв.) в сухой колбе растворяли в толуоле (450 мл) и медленно добавляли гексаметилдисилазид лития (1 моль/л, 181,61 мл, 2 экв.) при 0°C. Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) наблюдали, что соединение 1-3 полностью израсходовано. Реакционный раствор выливали в насыщенный раствор хлорида aммoния (200 мл) и экстрагировали этилацетатом (200 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 5:1 до 1:1, об./об.) с получением соединения 15-4.
1H ЯМР (400 MГц, CDCl3) δ = 7,70-7,66 (m, 2H), 7,13-7,11 (m, 1H), 7,02-7,00 (m, 1H), 6,54 (d, J=2,8 Гц, 1H), 6,48 (d, J=2,8 Гц, 1H), 3,93 (t, J=7,6 Гц, 2H), 3,09 (t, J=7,2 Гц, 2H), 2,56-2,48 (m, 2H).
Стадия 4
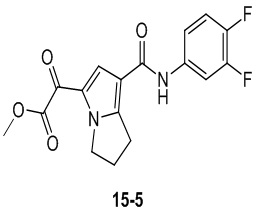
Соединение 15-4 (12 г, 45,76 ммоль, 1 экв.) растворяли в дихлорметане (20 мл), а затем добавляли метилоксалилхлорид (11,21 г, 91,51 ммоль, 8,43 мл, 2 экв.). Реакцию в реакционной системе проводили при 25°C в течение 16 ч. По результатам LCMS наблюдали, что соединение 15-4 полностью израсходовано и было получено соединение 15-5. Реакционный раствор выливали в насыщенной раствор бикарбоната натрия (200 мл) и экстрагировали этилацетатом (200 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 5:1 до 1:1, об./об.) с получением соединения 15-5. MS(ESI) m/s: 349,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,74-7,69 (m, 3H), 7,16-7,09 (m, 2H), 4,39 (t, J=7,6 Гц, 2H), 3,93 (s, 3H), 3,21 (t, J=7,6 Гц, 2H), 2,65-2,55 (m, 2H).
Стадия 5
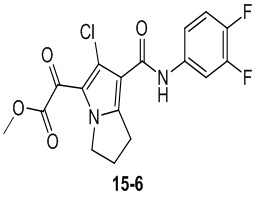
Соединение 15-5 (5,37 г, 15,42 ммоль, 1 экв.) растворяли в уксусной кислоте (110 мл), а затем добавляли N-хлорсукцинимид (3,29 г, 24,67 ммоль, 1,6 экв.). Реакцию в реакционной системе проводили при 40°C в течение 16 ч. По результатам LCMS наблюдали, что было получено соединение 15-6. Реакционный раствор выливали в воду (200 мл) и экстрагировали этилацетатом (200 мл ×3). Органическую фазу промывали насыщенным бикарбонатом натрия (1000 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 3:1 до 1:5, об./об.) с получением соединения 15-6. MS(ESI) m/s: 383,0 [M+H]+.
Стадия 6
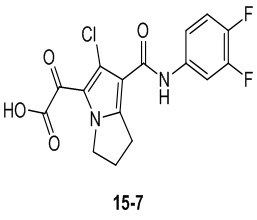
Соединение 15-6 (6,6 г, 17,24 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (40 мл), этанола (40 мл) и воды (40 мл) и добавляли моногидрат гидроксида лития (2,17 г, 51,73 ммоль, 3 экв.). Реакционную систему перемешивали при 40°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления для удаления растворителя, затем разводили водой (100 мл) и промывали этилацетатом (30 мл ×2). Водную фазу доводили до pH = 1 с помощью 0,5 M разбавленной соляной кислоты, а затем немедленно фильтровали. Остаток на фильтре суспендировали и промывали дихлорметаном (50 мл) и фильтровали. Полученный в результате остаток на фильтре концентрировали в условиях пониженного давления с получением соединения 15-7. MS(ESI) m/s: 369,1 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 7,78-7,73 (m, 1H), 7,31-7,25 (m, 1H), 7,24-7,19 (m, 1H), 4,40-4,39 (m, 2H), 3,19-3,13 (m, 2H), 2,63-2,54 (m, 2H).
Стадия 7
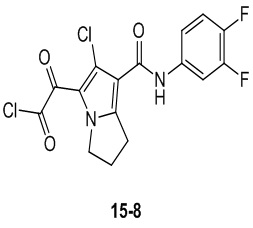
Соединение 15-7 (1,1 г, 2,98 ммоль, 1 экв.) растворяли в дихлорметане (20 мл) и по каплям добавляли оксалилхлорид (757,34 мг, 5,97 ммоль, 522,30 мкл, 2 экв.) и N,N-диметилформамид (21,80 мг, 298,33 мкмоль, 22,95 мкл, 0,1 экв.) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 0,5 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 15-8.
Стадия 8
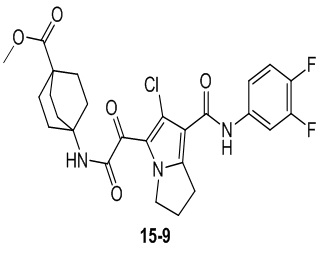
Соединение 15-3 (170,24 мг, 774,86 мкмоль, 1,5 экв., HCl) и триэтиламин (156,82 мг, 1,55 ммоль, 215,70 мкл, 3 экв.) растворяли в дихлорметане (10 мл), а затем добавляли раствор соединения 15-8 (0,2 г, 516,58 мкмоль, 1 экв.) в дихлорметане (10 мл) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 30°C в течение 16 ч. Результаты LCMS подтверждали, что соединение 15-8 полностью израсходовано и было получено соединение 15-9. Реакционный раствор разводили 1 M разбавленной соляной кислотой (20 мл) и экстрагировали дихлорметаном (20 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 15-9. MS(ESI) m/s: 534,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,48 (s, 1H), 7,76-7,71 (m, 1H), 7,47-7,40 (m, 1H), 7,15-7,11 (m, 2H), 4,32 (t,J=7,6 Гц, 2H), 3,67 (s, 3H), 3,26 (t,J=8,0 Гц, 2H), 2,56-2,48 (m, 2H), 2,07-1,97 (m, 12H).
Стадия 9
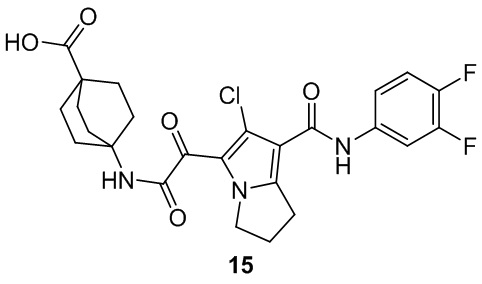
Соединение 15-9 (0,3 г, 561,85 мкмоль, 1 экв.) растворяли в смеси воды (3 мл) и диметилсульфоксида (6 мл), а затем добавляли моногидрат гидроксида лития (70,73 мг, 1,69 ммоль, 3 экв.). Реакционную систему перемешивали при 30°C в течение 2 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. Реакционный раствор фильтровали и собирали фильтрат. Фильтрат очищали с помощью препаративной HPLC (нейтральная, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 5% - 50%, 20 мин) с получением соединения 15. MS(ESI) m/s: 518,2 [M-H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,92 (s, 1H), 8,35 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,39 (m, 2H), 4,26 (t, J=7,0 Гц, 2H), 3,07 (t, J=7,4 Гц, 2H), 2,45-2,42 (m, 2H), 1,92-1,75 (m, 12H).
Пример 16

Путь синтеза:
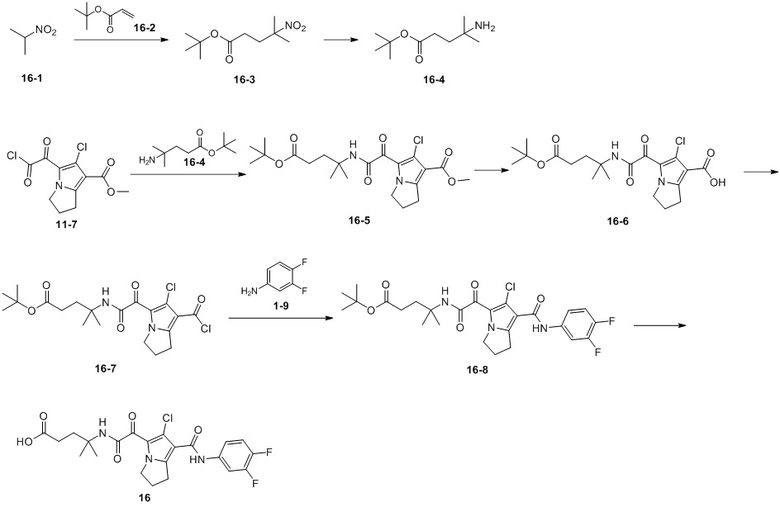
Стадия 1
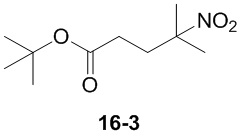
Соединение 16-1 (1 г, 11,22 ммоль, 1,01 мл, 1 экв.) и 1,8-диазабицикло[5.4.0]ундец-7-ен (3,42 г, 22,45 ммоль, 3,38 мл, 2 экв.) добавляли к безводному дихлорметану (10 мл), а затем в реакционную систему добавляли соединение 16-2 (1,73 г, 13,47 ммоль, 1,2 экв.) и перемешивали при 45°C в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) наблюдали, что соединение 16-1 полностью израсходовано. Реакционный раствор выливали в насыщенной раствор хлорида аммония (20 мл) и экстрагировали дихлорметаном (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:1 до 5:1, об./об.) с получением соединения 16-3.
Стадия 2
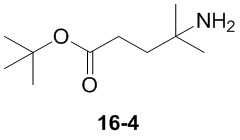
В гидрогенизационную колбу вносили скелетный никелевый катализатор гидрирования (266,67 мг, 3,11 ммоль, 1,69 экв.) и ополаскивали этанолом (10 мл ×2). Добавляли этанол (5 мл) и соединение 16-3 (0,4 г, 1,84 ммоль, 1 экв.). Реакционную систему перемешивали при 25°C в течение 4 ч в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что соединение 16-3 полностью израсходовано и были получены новые пятна. Реакционный раствор немедленно фильтровали, а фильтрат концентрировали в условиях пониженного давления с получением соединения 16-4.
1H ЯМР (400 MГц, DMSO-d6) δ = 2,21(t, J=8,0 Гц, 2H), 1,49 (t, J=8,0 Гц, 2H), 1,39 (s, 9H), 0,96 (s, 6H).
Стадия 3
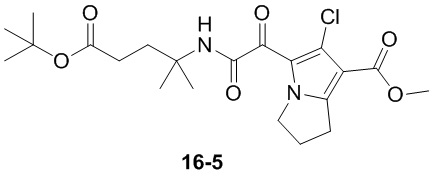
Соединение 16-4 (0,32 г, 1,71 ммоль, 1,24 экв.) добавляли к дихлорметану (10 мл), а затем к реакционной системе добавляли триэтиламин (697,62 мг, 6,89 ммоль, 959,59 мкл, 5 экв.). Затем реакционную систему охлаждали до 0°C. Соединение 11-7 (0,4 г, 1,38 ммоль, 1 экв.) растворяли в безводном дихлорметане (10 мл) и полученный в результате раствор по каплям добавляли в реакционную систему. Реакционную систему перемешивали при 20°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор промывали насыщенным хлоридом аммония (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 16-5. MS(ESI) m/s: 385,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 8,29 (s, 1H), 4,26 (t, J=7,6 Гц, 2H), 3,74 (s, 3H), 3,05 (t, J=7,6 Гц, 2H), 2,46-2,40 (m, 2H), 2,25-2,21(m, 2H), 1,93-1,88(m, 2H), 1,39 (s, 9H), 1,28 (s, 6H).
Стадия 4
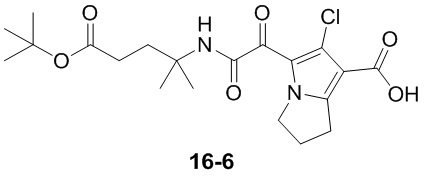
Соединение 16-5 (0,1 г, 226,80 мкмоль, 1 экв.) добавляли в колбу с тетрагидрофураном (3 мл), метанолом (3 мл) и водой (3 мл), а затем в реакционную систему добавляли моногидрат гидроксида лития (47,58 мг, 1,13 ммоль, 5 экв.) и перемешивали при 25°C в течение 12 ч. По результатам LCMS наблюдали, что соединение 16-5 полностью израсходовано. В реакционный раствор добавляли воду (20 мл), доводили до pH = 3 с помощью 1 M соляной кислоты и экстрагировали этилацетатом (10 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 15% - 45%, 10,5 мин) с получением соединения 16-6. MS(ESI) m/s: 371,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 12,50 (s, 1H), 8,26 (s, 1H), 4,25 (t, J=6,8 Гц, 2H), 3,09 (t, J=6,8 Гц, 2H), 2,43-2,41 (m, 2H), 2,26-2,22 (m, 2H), 1,93-1,89 (m, 2H), 1,40 (s, 9H), 1,29 (s, 6H).
Стадия 5
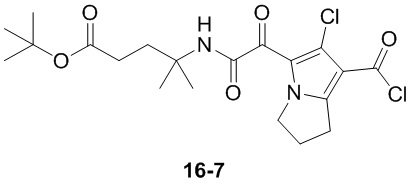
Соединение 16-6 (20 мг, 46,85 мкмоль, 1 экв.) добавляли к безводному дихлорметану (2 мл). Реакционную систему охлаждали до 0°C, последовательно добавляли оксалилхлорид (11,89 мг, 93,70 мкмоль, 8,20 мкл, 2 экв.) и N,N-диметилметанамид (3,42 мг, 46,85 мкмоль, 3,60 мкл, 1 экв.) и перемешивали при 0°C в течение 10 мин. Брали каплю реакционного раствора и добавляли к ней 1 мл метанола для гашения реакции. По результатам LCMS наблюдали, что соединение 16-6 полностью израсходовано и были получены новые пятна. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 16-7.
Стадия 6
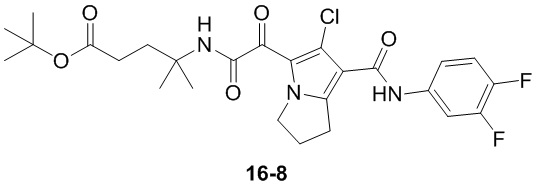
Соединение 1-9 (18,14 мг, 140,52 мкмоль, 3 экв.) и триэтиламин (23,70 мг, 234,21 мкмоль, 32,60 мкл, 5 экв.) добавляли к безводному дихлорметану (5 мл) и охлаждали реакционную систему до 0°C. Соединение 16-7 (20,86 мг, 46,84 мкмоль, 1 экв.) растворяли в безводном дихлорметане (5 мл), а полученный в результате раствор по каплям добавляли в реакционную систему. Реакцию в реакционной системе проводили при 0°C в течение 1 ч. По результатам LCMS наблюдали, что соединение 16-7 полностью израсходовано. Реакционный раствор выливали в насыщенной раствор хлорида аммония (10 мл) и экстрагировали дихлорметаном (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 16-8, которое применяли непосредственно на следующей стадии.
MS(ESI) m/s: 482,1 [M+H-(трет-Bu)]+.
Стадия 7
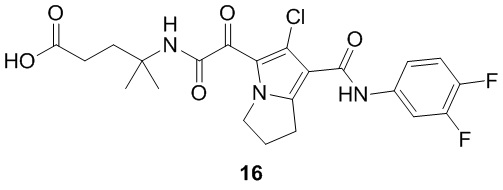
Соединение 16-8 (25,2 мг, 46,84 мкмоль, 1 экв.) добавляли к безводному дихлорметану (2 мл). Затем в реакционную систему добавляли триэтиламин (267,04 мг, 2,34 ммоль, 173,41 мкл, 50 экв.) и перемешивали при 25°C в течение 0,5 ч. По результатам LCMS наблюдали, что соединение 16-8 полностью израсходовано и было получено соединение 16. Реакционный раствор промывали водой (20 мл ×2), а полученную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 15% - 35%, 20 мин) с получением соединения 16.
MS(ESI) m/s: 482,0 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6)δ = 9,97 (s, 1H), 8,49 (s, 1H),7,57 (d, J=11,6 Гц, 1H), 7,43 (s, 2H),4,28-4,26 (m, 2H), 3,07 (m, 2H), 2,25 (m, 4H), 1,94-1,92 (m, 2H),1,38 (s, 6H).
Пример 17
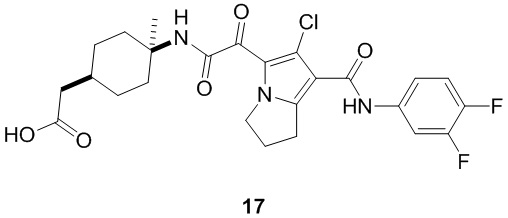
Путь синтеза:
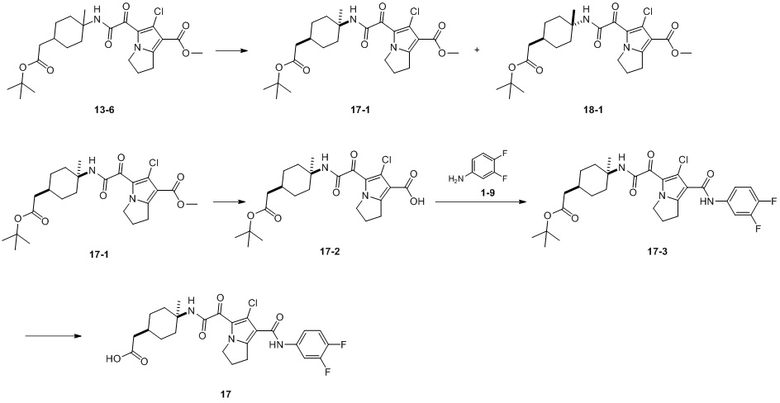
Стадия 1
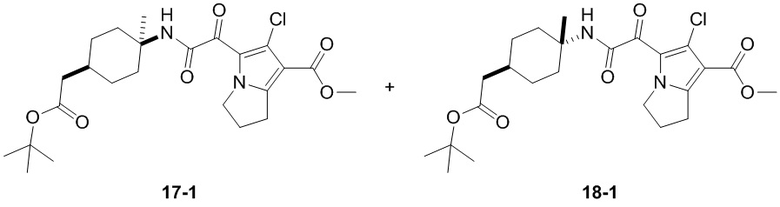
Соединение 13-6 отделяли с помощью SFC (колонка: DAICELCHIRALCELOJ (250 мм × 30 мм, 10 мкм); подвижная фаза: Neu-ETOH; % B: 30% - 30%, 9 мин). Получали соединение 17-1 (время удержания в колонке для SFC: 1,16 мин) и соединение 18-1 (время удержания в колонке для SFC: 1,7 мин). Условия SFC-анализа: колонка: хиральная колонка Daicel OJ-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 5 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 4 мин; противодавление системы: 100 бар.
Соединение 17-1: 1H ЯМР (400 MГц, CDCl3) δ = 6,00 (s, 1H), 4,32 (t, J=7,2 Гц, 2H), 3,84 (s, 3H), 3,14 (t, J=7,8 Гц, 2H), 2,52-2,48 (m, 2H), 2,31-2,28 (d, J=12,8 Гц, 2H), 2,14-2,12 (d, J=7,2 Гц, 2H), 1,76-1,74 (m, 1H), 1,74-1,67 (m, 2H), 1,48 (s, 3H), 1,44 (s, 9H), 1,42-1,38 (m, 2H), 1,35-1,18 (m, 2H).
Соединение 18-1: 1H ЯМР (400 MГц, CDCl3) δ = 6,08 (s, 1H), 4,31 (t, J=7,4 Гц, 2H), 3,83 (s, 3H), 3,12 (t, J=7,6 Гц, 2H), 2,51-2,47 (m, 2H), 2,17-2,15 (m, 2H), 2,14-2,00 (m, 2H), 1,83-1,70 (m, 3H), 1,48 (s, 3H), 1,47-1,45 (m, 10H), 1,45-1,26 (m, 3H).
Стадия 2
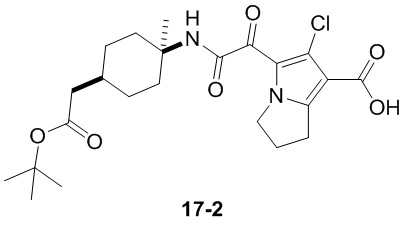
Соединение 17-1 (360,00 мг, 748,47 мкмоль, 1 экв.) в колбе с одним горлышком растворяли в смеси тетрагидрофурана (10 мл), метанола (10 мл) и воды (10 мл) и добавляли моногидрат гидроксида лития (157,03 мг, 3,74 ммоль, 5 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор доводили до pH = 1 с помощью 0,5 M разбавленной соляной кислоты. Водную фазу экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 17-2.
MS(ESI) m/s: 411,1 [M+H-(трет-Bu)]+.
Стадия 3
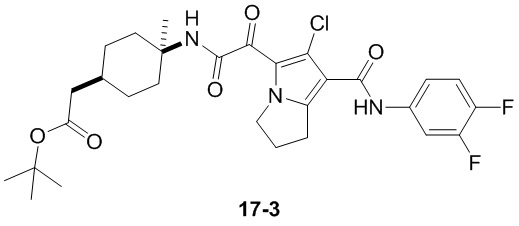
Соединение 17-2 (330,00 мг, 706,71 мкмоль, 1 экв.) в сухой колбе растворяли в N,N-диметилформамиде (15 мл) и добавляли триэтиламин (214,54 мг, 2,12 ммоль, 295,10 мкл, 3 экв.), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (537,42 мг, 1,41 ммоль, 2 экв.) и соединение 1-9 (182,48 мг, 1,41 ммоль, 2 экв.). Реакционную систему перемешивали при 60°C в течение 16 ч в условиях атмосферы азота. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор разводили этилацетатом (50 мл), а затем промывали 0,5 M разбавленной соляной кислотой (10 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 1:1, об./об.) с получением соединения 17-3. MS(ESI) m/s: 578,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,48 (d, 7,6 Гц, 1H), 7,77-7,72 (m, 1H), 7,16-7,09 (m, 2H), 6,20 (s, 1H), 4,33 (t, J=7,6 Гц, 2H), 3,26 (t, J=7,6 Гц, 2H), 2,56-2,49 (m, 2H), 2,32-2,30 (m, 2H), 1,78-1,67 (m, 5H), 1,48 (s, 3H), 1,45 (s, 9H), 1,41-1,38 (m, 2H), 1,37-1,17 (m, 2H).
Стадия 4
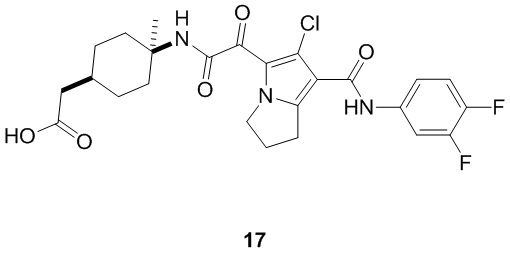
Соединение 17-3 (280 мг, 484,39 мкмоль, 1 экв.) в сухой колбе с одним горлышком растворяли в дихлорметане (6 мл) и добавляли трифторуксусную кислоту (4,62 г, 40,52 ммоль, 3 мл, 83,65 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 16% - 46%, 10,5 мин) с получением соединения 17 (колонка: хиральная колонка Daicel AS-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 10 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 5 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 2,18 мин. MS (ESI) m/s: 522,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,91 (s, 1H), 8,08 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,39 (m, 2H), 4,29 (t, J=7,2 Гц, 2H), 3,08 (t, J=7,4 Гц, 2H), 2,46-2,45 (m, 2H), 2,23 (d, J=10,4 Гц, 2H), 2,07 (d, J=6,8 Гц, 2H), 1,63 (s, 1H), 1,50 (d, J=7,6 Гц, 2H), 1,34 (s, 3H), 1,29-1,20 (m, 4H).
Пример 18
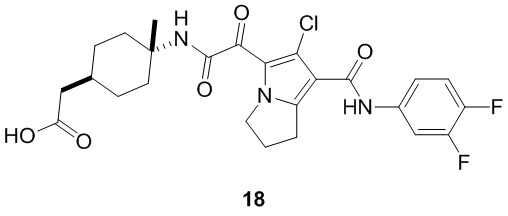
Путь синтеза:
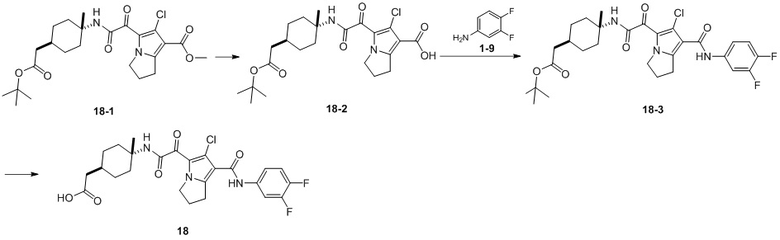
Стадия 1
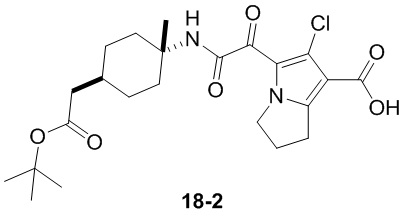
Соединение 18-1 (550,00 мг, 1,14 ммоль, 1 экв.) в колбе с одним горлышком растворяли в смеси тетрагидрофурана (10 мл), воды (10 мл) и метанола (10 мл) и добавляли моногидрат гидроксида лития (239,91 мг, 5,72 ммоль, 5 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор доводили до pH = 1 с помощью разбавленной соляной кислоты с концентрацией 0,5 моль/л. Водную фазу экстрагировали этилацетатом (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 18-2.
MS(ESI) m/s: 467,1 [M+H]+.
Стадия 2
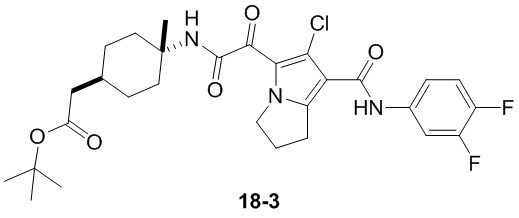
Соединение 18-2 растворяли в N,N-диметилформамиде (15 мл) и добавляли гексафторфосфат O- (7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (879,42 мг, 2,31 ммоль, 2 экв.) и триэтиламин (351,06 мг, 3,47 ммоль, 482,89 мкл, 3 экв.) при 30°C в условиях атмосферы азота. После перемешивания в течение 30 мин добавляли соединение 1-9 (298,61 мг, 2,31 ммоль, 2 экв.) и реакционную систему нагревали до 60°C и перемешивали в течение 15,5 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в 0,2 моль/л разбавленной соляной кислоты (50 мл), а затем экстрагировали этилацетатом (10 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 1:1, об./об.) с получением соединения 18-3. MS(ESI) m/s: 578,2 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,48 (s, 1H), 7,76-7,71 (m, 1H), 7,27-7,11 (m, 2H), 6,25 (s, 1H), 4,32-4,30 (m, 2H), 3,25 (t, J=7,6 Гц, 2H), 2,55-2,50 (m, 2H), 2,18-2,15 (m, 3H), 2,01-1,92 (m, 2H), 1,84-1,81 (m, 2H), 1,72-1,71 (m, 2H), 1,50 (s, 3H), 1,46 (s, 9H), 1,28-1,25 (m, 2H).
Стадия 3
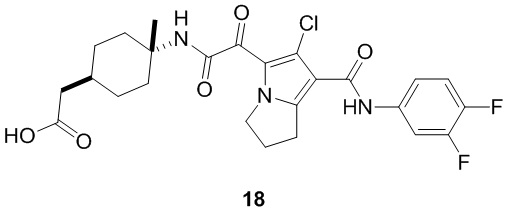
Соединение 18-3 растворяли в дихлорметане (6 мл) и добавляли трифторуксусную кислоту (4,62 г, 40,52 ммоль, 3,00 мл, 78,07 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 16% - 46%, 10,5 мин) с получением соединения 18 (колонка: хиральная колонка Daicel AS-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 10 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 5 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 2,40 мин. MS (ESI) m/s: 522,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,90 (s, 1H), 8,29 (s, 1H), 7,87-7,81 (m, 1H), 7,45-7,38 (m, 2H), 4,28 (t, J=7,2 Гц, 2H), 3,08 (t, J=7,6 Гц, 2H), 2,48-2,44 (m, 2H), 2,15 (d, J=6,8 Гц, 2H), 1,85 (d, J=12,8 Гц, 2H), 1,76-1,61 (m, 5H), 1,37 (s, 3H), 1,18-1,15 (m, 2H).
Пример 19
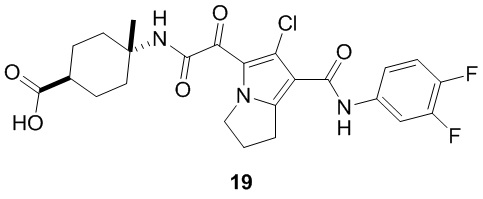
Путь синтеза:
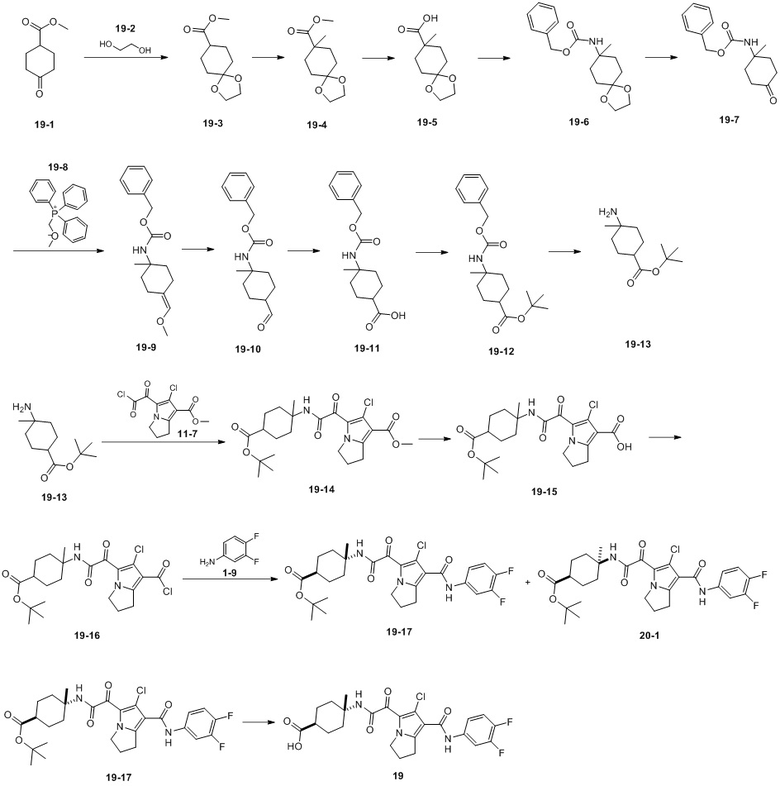
Стадия 1
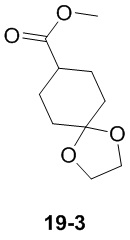
Соединение 19-1 (65 г, 416,19 ммоль, 1 экв.) и соединение 19-2 (32,29 г, 520,24 ммоль, 29,09 мл, 1,25 экв.) растворяли в толуоле (1 л) и добавляли соединение (7,17 г, 41,62 ммоль, 0,1 экв.). Реакционную систему нагревали до 130°C и перемешивали в течение 48 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали и неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 5:1, об./об.) с получением соединения 19-3.
1H ЯМР (400 MГц, CDCl3) δ = 3,95 (s, 4H), 3,68 (s, 3H), 2,37-2,35 (m, 1H), 1,93-1,91 (m, 2H), 1,82-1,76 (m, 4H), 1,58-1,54 (m, 2H).
Стадия 2
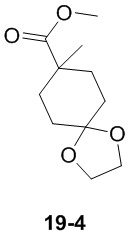
Диизопропилэтиламин (47,00 г, 464,46 ммоль, 65,64 мл, 1,5 экв.) растворяли в тетрагидрофуране (800 мл) и по каплям добавляли н-бутиллитий (2,5 M, 185,79 мл, 1,5 экв.) при -78°C в условиях атмосферы азота. После перемешивания в течение 30 мин по каплям добавляли раствор соединения 19-3 (62 г, 309,64 ммоль, 1 экв.) в тетрагидрофуране (200 мл). После перемешивания в течение еще 30 мин по каплям добавляли йодметан (65,93 г, 464,46 ммоль, 28,91 мл, 1,5 экв.). Наконец, реакционную систему нагревали до 25°C и перемешивали в течение 16 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор выливали в 0,5 M разбавленную соляную кислоту (500 мл). Водную фазу экстрагировали этилацетатом (100 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 10:1, об./об.) с получением соединения 19-4.
1H ЯМР (400 MГц, CDCl3) δ = 3,93 (s, 4H), 3,66 (s, 3H), 2,14-2,10 (m, 2H), 1,64-1,60 (m, 4H), 1,59-1,50 (m, 2H), 1,18 (s, 3H).
Стадия 3
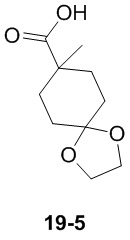
Соединение 19-4 (70 г, 326,71 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (150 мл), метанола (150 мл) и воды (150 мл) и добавляли гидроксид калия (91,66 г, 1,63 моль, 5 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления для удаления растворителя и разводили водой (100 мл). Водную фазу промывали этилацетатом (20 мл ×2), доводили до pH = 4 с помощью 0,5 М разбавленной соляной кислоты и экстрагировали этилацетатом (50 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 19-5.
1H ЯМР (400 MГц, CDCl3) δ = 3,92 (s, 4H), 2,13-2,08 (m, 2H), 1,67-1,64 (m, 4H), 1,55-1,50 (m, 2H), 1,23 (s, 3H).
Стадия 4
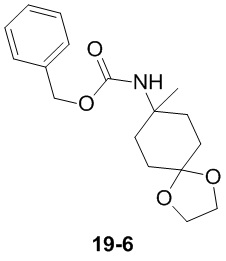
Соединение 19-5 (54 г, 269,69 ммоль, 1 экв.) растворяли в толуоле (500 мл) и добавляли триэтиламин (81,87 г, 809,07 ммоль, 112,61 мл, 3 экв.), дифенилфосфорилазид (89,06 г, 323,63 ммоль, 70,13 мл, 1,2 экв.) и бензиловый спирт (35,00 г, 323,63 ммоль, 33,65 мл, 1,2 экв.). Реакционную систему перемешивали при 100°C в течение 16 ч в условиях атмосферы азота. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что реакция завершилась. Реакционный раствор выливали в воду (500 мл). Водную фазу экстрагировали этилацетатом (100 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 5:1, об./об.) с получением соединения 19-6.
1H ЯМР (400 MГц, CDCl3) δ = 7,37-7,27 (m, 5H), 5,05 (s, 1H), 4,69 (s, 2H), 3,95 (s, 4H), 2,04 (s, 2H), 1,68-1,59 (m, 6H), 1,40-1,37 (m, 3H).
Стадия 5
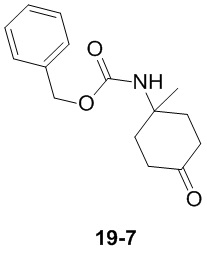
Соединение 19-6 (80 г, 261,98 ммоль, 1 экв.) растворяли в тетрагидрофуране (1 л) и добавляли соляную кислоту (2 M, 1 л, 7,63 экв.). Реакционную систему перемешивали при 50°C в течение 16 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 25:1 до 5:1, об./об.) с получением соединения 19-7.
1H ЯМР (400 MГц, CDCl3) δ = 7,40-7,29 (m, 5H), 5,09 (s, 2H), 4,87 (s, 1H), 2,45-2,38 (m, 4H), 2,31-2,27 (m, 2H), 1,85-1,82 (m, 2H), 146 (s, 3H).
Стадия 6
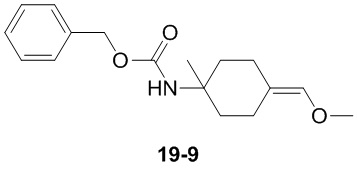
Соединение 19-8 (13,16 г, 38,27 ммоль, 2 экв., HCl) растворяли в тетрагидрофуране (100 мл) и добавляли трет-бутоксид калия (4,29 г, 38,27 ммоль, 2 экв.) при -10°C в условиях атмосферы азота. После перемешивания реакционной системы при -10°C в течение 1 ч в указанный выше реакционный раствор по каплям добавляли раствор соединения 19-7 (5 г, 19,13 ммоль, 1 экв.) в тетрагидрофуране (100 мл) и реакционный раствор перемешивали при 25°C в течение 2 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор разводили этилацетатом (500 мл), а затем фильтровали. Исходный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 30:1 до 10:1, об./об.) с получением соединения 19-9.
1H ЯМР (400 MГц, CDCl3) δ = 7,37-7,27 (m, 5H), 5,78 (s, 1H), 5,07 (s, 2H), 4,67 (s, 1H), 4,55 (s, 3H), 2,45-2,40 (m, 1H), 2,04-1,93 (m, 5H), 1,47-1,44 (m, 2H), 1,36 (s, 3H).
Стадия 7
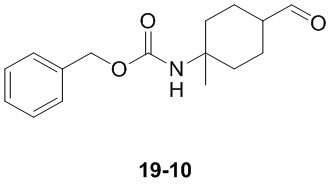
Соединение 19-9 (4 г, 13,82 ммоль, 1 экв.) растворяли в тетрагидрофуране (6,5 мл) и по каплям добавляли соляную кислоту (6 M, 3,23 мл, 1,4 экв.) при 0°C. Реакционную систему перемешивали при 0°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор разводили водой (10 мл). Водную фазу экстрагировали этилацетатом (5 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 19-10.
1H ЯМР (400 MГц, CDCl3) δ = 9,65 (d, J=17,2 Гц, 2H), 7,37-7,27 (m, 5H), 5,06 (s, 2H), 4,61 (d, J= 56,4 Гц, 1H), 2,33-2,21 (m, 2H), 1,87-1,81 (m, 3H), 1,80-1,68 (m, 2H), 1,37 (s, 3H).
Стадия 8
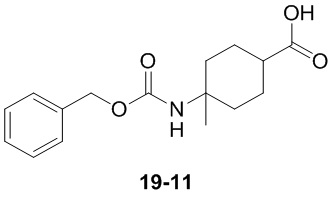
Соединение 19-10 (1,8 г, 6,54 ммоль, 1 экв.) растворяли в смеси трет-бутанола (27 мл) и изоамилена (2,58 г, 36,74 ммоль, 3,89 мл, 5,62 экв.) и медленно добавляли раствор дигидрофосфата натрия (831,41 мг, 6,93 ммоль, 1,06 экв.) и раствор хлорита натрия (709,48 мг, 7,84 ммоль, 1,2 экв.) в воде (36 мл) при 0°C. Реакционную систему перемешивали при 0°C в течение 10 мин. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор разводили водой (20 мл). Водную фазу экстрагировали этилацетатом (5 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 3:1, об./об.) с получением соединения 19-11.
1H ЯМР (400 MГц, CDCl3) δ = 7,37-7,27 (m, 5H), 5,06 (s, 2H), 4,65 (d, J= 4,0 Гц, 1H), 2,44-2,30 (m, 1H), 2,20-2,17 (m, 1H), 1,88-1,66 (m, 6H), 1,36-1,31 (m, 3H).
Стадия 9
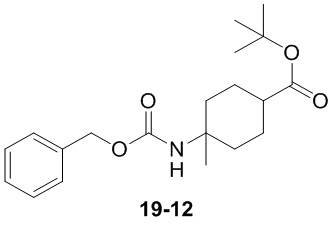
Соединение 19-11 (1,7 г, 5,84 ммоль, 1 экв.) растворяли в дихлорметане (15 мл) и по каплям добавляли оксалилхлорид (1,48 г, 11,67 ммоль, 1,02 мл, 2 экв.) и N,N-диметилформамид (42,65 мг, 583,51 мкмоль, 44,89 мкл, 0,1 экв.) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 1 ч, а затем концентрировали в условиях пониженного давления. Неочищенный продукт растворяли в дихлорметане (5 мл) и полученный в результате раствор медленно по каплям добавляли к раствору трет-бутанола ( 4,32 г, 58,35 ммоль, 5,58 мл, 10 экв.) в дихлорметане (10 мл). Реакционную систему перемешивали при 50°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 30:1 до 10:1, об./об.) с получением соединения 19-12.
1H ЯМР (400 MГц, CDCl3) δ = 7,37-7,27 (m, 5H), 5,05 (s, 2H), 4,64 (d, J= 14,8 Гц,1H), 2,27-2,14 (m, 2H), 1,82-1,64 (m, 6H), 1,44 (d, J= 3,2 Гц, 9H), 1,36-1,31 (m, 3H).
Стадия 10
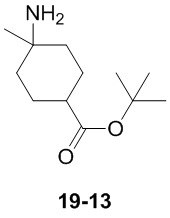
Влажный палладиевый катализатор на углеродном носителе (200 мг, содержание: 10%), метанол (20 мл) и соединение 19-12 (1 г, 2,88 ммоль, 1 экв.) добавляли в гидрогенизационную колбу. После 3 продувок водородом реакционную систему перемешивали в течение 16 ч при 25°C в условиях атмосферы водорода (50 psi). Реакционный раствор фильтровали и концентрировали в условиях пониженного давления с получением соединения 19-13.
1H ЯМР (400 MГц, MeOH-d4) δ = 2,18-2,15 (m, 2H), 1,79-1,55 (m, 4H), 1,45 (d, J=2,4, Гц, 9H), 1,43-1,37 (m, 2H), 1,12 (s, 3H).
Стадия 11
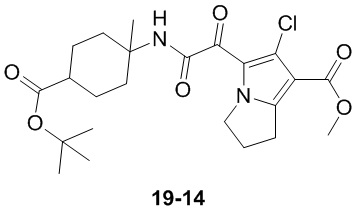
Соединение 19-13 (294,13 мг, 1,38 ммоль, 1 экв.) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (418,57 мг, 4,14 ммоль, 575,76 мкл, 3 экв.). По каплям добавляли раствор соединения 11-7 (400 мг, 1,38 ммоль, 1 экв.) в дихлорметане (10 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту (0,5 моль/л, 50 мл) и экстрагировали дихлорметаном (20 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 19-14.
MS (ESI) m/z: 467,2 [M+H]+
Стадия 12
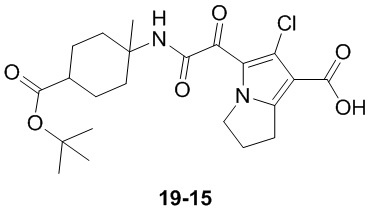
Соединение 19-14 (700 мг, 1,50 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (10 мл), метанола (10 мл) и воды (10 мл) и добавляли моногидрат гидроксида лития (188,70 мг, 4,50 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления для удаления растворителя. Водную фазу доводили до pH = 1 с помощью разбавленной соляной кислоты с концентрацией 0,5 моль/л и экстрагировали этилацетатом (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 19-15. MS (ESI) m/z: 453,1 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 4,39-4,33 (m, 2H), 3,31-3,11 (m, 2H), 2,53-2,49 (m, 2H), 2,38-2,35 (m, 2H), 1,88-1,70 (m, 6H), 1,46-1,29 (m, 13H).
Стадия 13
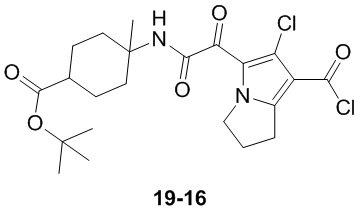
Соединение 19-15 (470 мг, 1,04 ммоль, 1 экв.) растворяли в дихлорметане (20 мл) и по каплям добавляли оксалилхлорид (263,43 мг, 2,08 ммоль, 181,68 мкл, 2 экв.) и N,N-диметилформамид (7,58 мг, 103,77 мкмоль, 7,98 мкл, 0,1 экв.) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 30 мин. Брали 1 мл реакционного раствора и добавляли метанол для остановки реакции, и по результатам LCMS-детекции было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 19-16.
Стадия 14
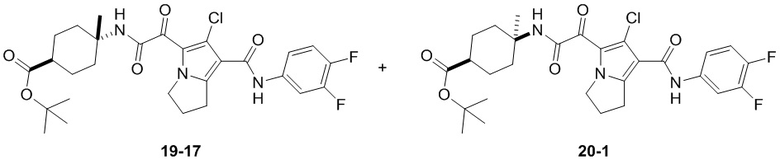
Соединение 1-9 (273,90 мг, 2,12 ммоль, 2 экв.) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (322,01 мг, 3,18 ммоль, 442,92 мкл, 3 экв.). По каплям добавляли раствор соединения 19-16 (500 мг, 1,06 ммоль, 1 экв.) в дихлорметане (10 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту (0,5 моль/л, 50 мл). Водную фазу экстрагировали этилацетатом (20 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной TLC (простой петролейный эфир : этилацетат = 1:1, об./об.) с получением соединения 19-17 (Rf = 0,35) и соединения 20-1 (Rf = 0,32).
Соединение 19-17: MS (ESI) m/z: 508,1 [M+H-(трет-Bu)]+.
Соединение 20-1:MS (ESI) m/z: 508,1 [M+H-(трет-Bu)]+.
Стадия 15
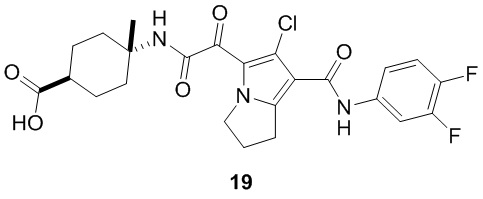
Соединение 19-17 (90 мг, 159,57 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (363,88 мг, 3,19 ммоль, 236,29 мкл, 20 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления, а неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания В (ацетонитрила): 23% - 43%, 10,5 мин) с получением соединения 19 (колонка: хиральная колонка Daicel AS-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 10 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 5 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 2,44 мин. MS (ESI) m/z: 508,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,96 (s, 1H), 8,15 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,36 (m, 2H), 4,29 (t, J=7,0 Гц, 2H), 3,08(t, J=7,6 Гц, 2H), 2,50-2,46 (m, 2H), 2,22 (d, J=12,8 Гц, 2H), 2,11 (s, 1H), 1,63-1,58 (m, 4H), 1,34 (s, 3H), 1,29-1,23 (m, 2H).
Пример 20
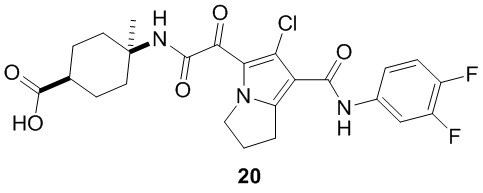
Путь синтеза:
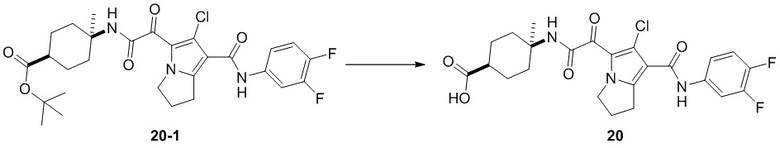
Соединение 20-1 (140 мг, 248,22 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (566,04 мг, 4,96 ммоль, 367,56 мкл, 20 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления, а неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Waters Xbridge 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания В (ацетонитрила): 18% - 38%, 10,5 мин) с получением соединения 20 (колонка: хиральная колонка Daicel AS-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 10 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 5 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 2,20 мин. MS (ESI) m/z: 508,2 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,95 (s, 1H), 8,30 (s, 1H), 7,87-7,82 (m, 1H), 7,45-7,39 (m, 2H), 4,28 (t, J=7,2 Гц, 2H), 3,08 (t, J=7,6 Гц, 2H), 2,50-2,46 (m, 2H), 2,24 (s,1H), 1,83-1,74 (m, 6H), 1,59-1,55 (m, 2H), 1,36 (s, 3H).
Пример 21
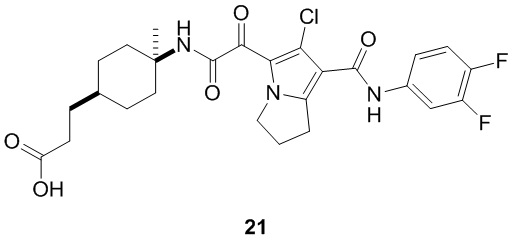
Путь синтеза:
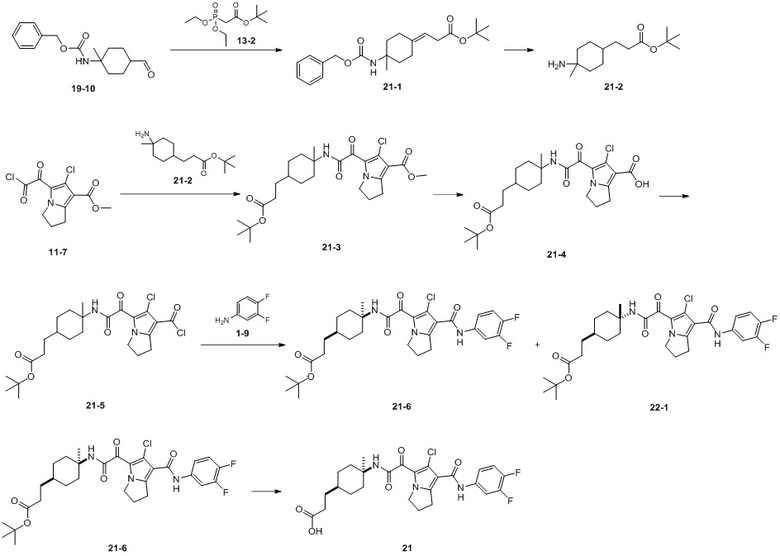
Стадия 1
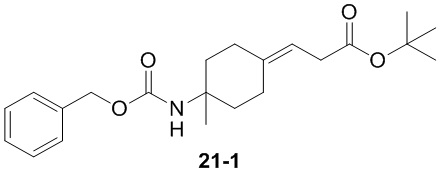
Соединение 13-2 (3,63 г, 14,38 ммоль, 2,2 экв.) растворяли в тетрагидрофуране (20 мл) и медленно добавляли гидрид натрия (784,48 мг, 19,61 ммоль, степень чистоты: 60%, 3 экв.) при 0°C в условиях атмосферы азота. Реакционный раствор перемешивали в течение 30 мин, а затем медленно по каплям добавляли раствор соединения 19-10 (1,8 г, 6,54 ммоль, 1 экв.) в THF (20 мл). Реакционную систему нагревали до 25°C и перемешивали в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту с концентрацией 0,5 моль/л (10 мл). Водную фазу экстрагировали этилацетатом (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 10:1, об./об.) с получением соединения 21-1.
1H ЯМР (400 MГц, CDCl3) δ = 7,37-7,33 (m, 5H), 6,83-6,77 (m, 1H), 5,75-5,69 (m, 1H), 5,06 (s, 2H), 4,71-4,53 (m, 2H), 2,21-2,16 (m, 2H), 1,87-1,82 (m, 1H), 1,76-1,71 (m, 2H), 1,63 (s, 2H), 1,49 (s, 9H), 1,38-1,32 (m, 4H).
Стадия 2
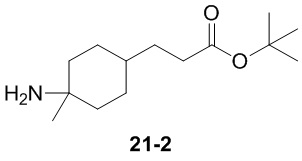
Палладиевый катализатор на углеродном носителе (200 мг, содержание: 10%) в гидрогенизационной колбе растворяли в метаноле (30 мл) и добавляли соединение 21-1 (2 г, 5,35 ммоль, 1 экв.). После 3 продувок водородом реакционную систему перемешивали в течение 16 ч при 25°C в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор фильтровали и концентрировали в условиях пониженного давления с получением соединения 21-2.
1H ЯМР (400 MГц, MeOH-d4) δ = 2,27-2,22 (m, 2H), 1,70-1,50 (m, 6H), 1,44 (s, 9H), 1,39-1,36 (m, 2H), 1,24-1,18 (m, 2H), 1,13-1,11 (m, 4H).
Стадия 3
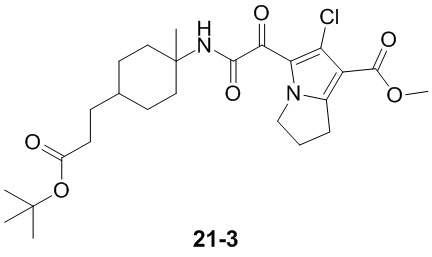
Соединение 21-2 (332,81 мг, 1,38 ммоль, 1 экв.) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (418,57 мг, 4,14 ммоль, 575,76 мкл, 3 экв.). По каплям добавляли раствор соединения 11-7 (400 мг, 1,38 ммоль, 1 экв.) в дихлорметане (10 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту с концентрацией 0,5 моль/л (50 мл). Водную фазу экстрагировали дихлорметаном (20 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 21-3.
MS(ESI) m/s: 439,2 [M+H-(трет-Bu)]+.
Стадия 4
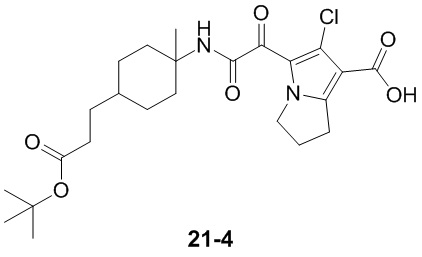
Соединение 21-3 (700 мг, 1,41 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (10 мл), метанола (10 мл) и воды (10 мл) и добавляли моногидрат гидроксида лития (178,01 мг, 4,24 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления для удаления растворителя. Водную фазу доводили до pH = 1 с помощью разбавленной соляной кислоты с концентрацией 0,5 моль/л и экстрагировали этилацетатом (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 21-4.
MS(ESI) m/s: 425,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, MeOH-d4) δ 4,39-4,33 (m, 2H), 3,17-3,11 (m, 2H), 2,53-2,51 (m, 2H), 2,32-2,23 (m, 3H), 2,03-2,00 (m, 1H), 1,81-1,55 (m, 2H), 1,58-1,52 (m, 6H), 1,44 (s, 9H), 1,30-1,27 (m, 4H).
Стадия 5
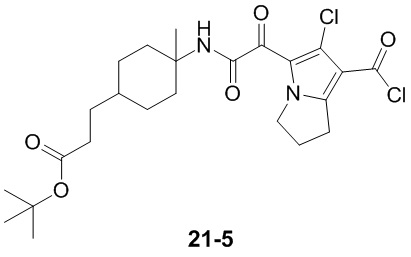
Соединение 21-4 (570 мг, 1,19 ммоль, 1 экв.) растворяли в дихлорметане (30 мл) и по каплям добавляли оксалилхлорид (300,84 мг, 2,37 ммоль, 207,48 мкл, 2 экв.) и N,N-диметилформамид (8,66 мг, 118,51 мкмоль, 9,12 мкл, 0,1 экв.) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 30 мин. Брали две капли реакционного раствора и добавляли в них метанол для остановки реакции, и по результатам LCMS было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 21-5.
Стадия 6
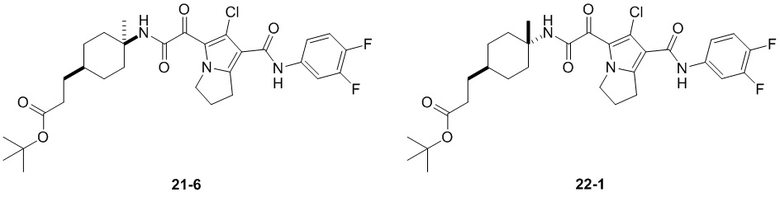
Соединение 1-9 (310,21 мг, 2,40 ммоль, 2 экв.) растворяли в дихлорметане (20 мл) и добавляли триэтиламин (364,70 мг, 3,60 ммоль, 501,65 мкл, 3 экв.). По каплям добавляли раствор соединения 21-5 (600 мг, 1,20 ммоль, 1 экв.) в дихлорметане (10 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 1:1) и LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту (0,5 моль/л, 50 мл). Водную фазу экстрагировали этилацетатом (20 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной TLC (простой петролейный эфир : этилацетат = 1:1, об./об.) с получением соединения 21-6 (Rf = 0,35) и соединения 22-1 (Rf = 0,32).
Соединение 21-6: MS(ESI) m/s: 536,2 [M+H-(трет-Bu)]+.
Соединение 21-6: MS(ESI) m/s: 536,2 [M+H-(трет-Bu)]+.
Стадия 7
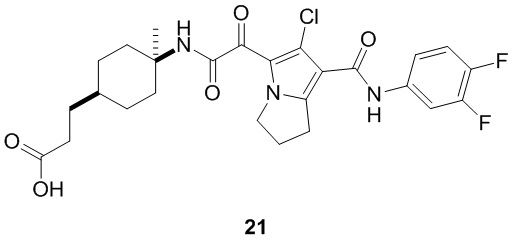
Соединение 21-6 (70 мг, 118,23 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (269,61 мг, 2,36 ммоль, 175,07 мкл, 20 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 10% - 50%, 20 мин) с получением соединения 21 (колонка: хиральная колонка Daicel OJ-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 5 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 3 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 1,27 мин. MS (ESI) m/s: 536,2 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,93 (s, 1H), 8,09 (s, 1H), 7,88-7,83 (m, 1H), 7,46-7,40 (m, 2H), 4,31-4,28 (m, 2H), 3,11-3,07 (m, 2H), 2,47-2,43 (m, 2H), 2,23-2,20 (m, 4H), 1,50-1,41 (m, 4H), 1,34 (s, 3H), 1,24-1,19 (m, 5H).
Пример 22
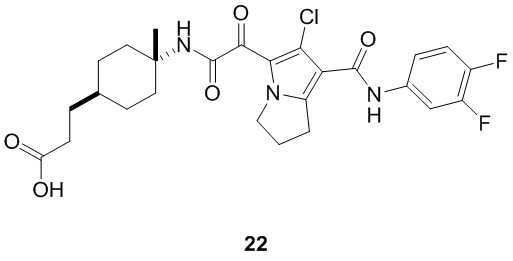
Путь синтеза:
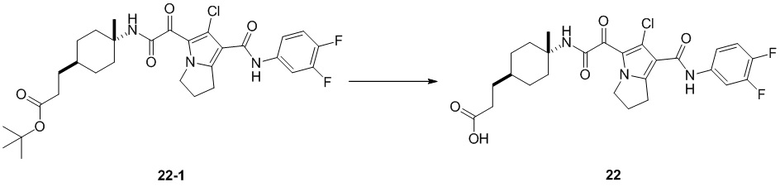
Соединение 22-1 (80 мг, 135,12 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (308,12 мг, 2,70 ммоль, 200,08 мкл, 20 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 10% - 50%, 20 мин) с получением соединения 22 (колонка: хиральная колонка Daicel OJ-3 со следующей спецификацией: 0,46 см внутреннего диаметра × 5 см; подвижная фаза: A: диоксид углерода, B: этанол для хроматографии (содержащий 0,05% изопропиламина); % B: 5% - 40%; скорость потока: 4 мл/мин; 3 мин; противодавление системы: 100 бар), а время удержания в колонке для SFC составляло 1,39 мин. MS (ESI) m/s: 536,2 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,92 (s, 1H), 8,29 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,39 (m, 2H), 4,29-4,25 (m, 2H), 3,09-3,04 (m, 2H), 2,47-2,44 (m, 2H), 2,19-2,15 (m, 2H), 1,86-1,83 (m, 2H), 1,69-1,59 (m, 4H), 1,45-1,43 (m, 2H), 1,36 (s, 3H), 1,17 (s, 1H), 1,09-1,06 (m, 2H).
Пример 23
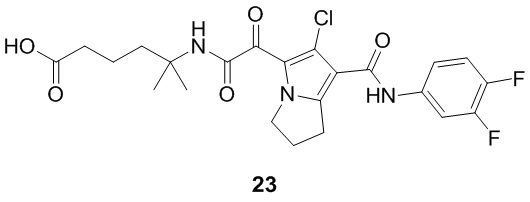
Путь синтеза:
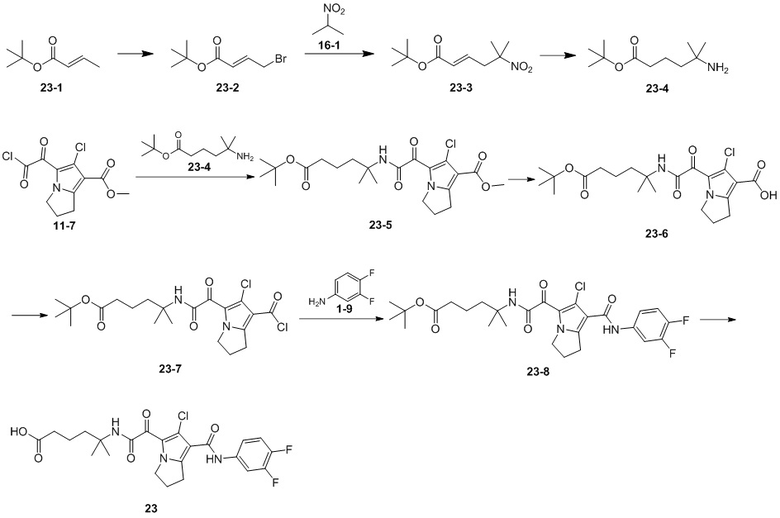
Стадия 1
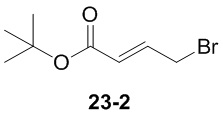
Соединение 23-1 (5 г, 35,16 ммоль, 1 экв.) растворяли в тетрахлорметане (50 мл), а затем добавляли N-бромсукцинимид (7,51 г, 42,20 ммоль, 1,2 экв.) и азобисизобутиронитрил (1,73 г, 10,55 ммоль, 0,3 экв.). Реакционный раствор нагревали до 90°C и перемешивали в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что оставалось незначительно количество соединения 23-1 и были получены новые пятна. Реакционный раствор охлаждали до комнатной температуры, добавляли воду (50 мл) и перемешивали, а затем оставляли отстаиваться до отделения органической фазы. Отделенную органическую фазу промывали насыщенным водным раствором хлорида натрия (50 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:1 до 15:1, об./об.) с получением соединения 23-2.
1H ЯМР (400 MГц, CDCl3) δ = 6,95-6,88 (m, 1H), 5,99 (dd, J=1,2, 1,2 Гц, 1H), 4,02 (t, J=3,8 Гц, 2H), 1,52 (s, 9H).
Стадия 2
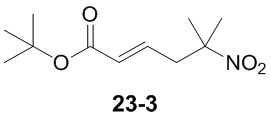
Соединение 23-2 (1,60 г, 17,91 ммоль, 1,61 мл, 1,1 экв.) растворяли в метаноле (50 мл) и добавляли тетракис(трифенилфосфин)палладий (940,79 мг, 814,14 мкмоль, 0,05 экв.), метилат натрия (967,63 мг, 17,91 ммоль, 1,1 экв.) и соединение 16-1 (3,6 г, 16,28 ммоль, 1 экв.) при 26°C в условиях атмосферы азота. Реакционную смесь нагревали до 60°C и перемешивали в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что соединение 23-2 полностью израсходовано и были получены новые пятна. Реакционный раствор выливали в воду (100 мл), а затем экстрагировали этилацетатом (50 мл ×2). Органическую фазу промывали насыщенным водным раствором хлорида натрия (100 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 100:1 до 10:1, об./об.) с получением соединения 23-3.
1H ЯМР (400 MГц, CDCl3) δ = 6,71-6,63 (m, 1H), 5,86-5,82 (m, 1H), 2,78 (dd, J=1,2, 1,2 Гц, 2H), 1,61 (s, 6H), 1,48 (s, 9H).
Стадия 3
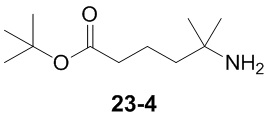
Соединение 23-3 (200 мг, 872,33 мкмоль, 1 экв.) и этанол (10 мл) вносили в сухую гидрогенизационную колбу, добавляли скелетный никелевый катализатор гидрирования (200 мг, 2,33 ммоль, 2,68 экв.) в условиях атмосферы азота и продували водородом. Реакционную систему перемешивали в течение 12 ч при 50°C в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что превращение исходных материалов завершилось и были получены новые пятна. Реакционный раствор фильтровали через целит, а затем промывали этанолом (20 мл). Фильтрат сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 23-4.
1H ЯМР (400 MГц, CDCl3) δ = 2,22 (t, J=7,4 Гц, 2H), 1,55-1,52 (m, 2H), 1,31 (s, 9H), 1,30-1,28 (m, 2H), 1,03 (s, 6H).
Стадия 4
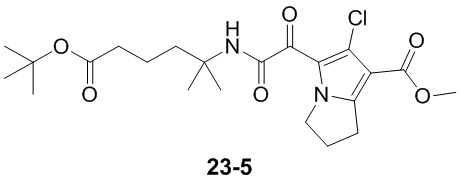
Соединение 23-4 (150 мг, 745,61 мкмоль, 7,21e-1 экв.) растворяли в дихлорметане (5 мл) при 26°C и добавляли бикарбонат натрия (434,37 мг, 5,17 ммоль, 201,10 мкл, 5 экв.). Медленно при помешивании по каплям добавляли смесь соединения 11-7 (300 мг, 1,03 ммоль, 1 экв.) и дихлорметана (5 мл), а затем реакционную систему перемешивали при 26°C в течение 0,5 ч. По результатам LCMS было видно, что было получено соединение 23-5. В реакционный раствор при помешивании добавляли воду (10 мл) и оставляли отстаиваться для разделения жидкостей с получением органических фаз. Отделенную водную фазу экстрагировали дихлорметаном (10 мл ×1), а органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 50:1 до 2:1, об./об.) с получением соединения 23-5. MS (ESI) m/z: 477,1 [M+Na]+.
1H ЯМР (400 MГц, CDCl3) δ = 6,11 (s, 1H), 4,24 (t, J=7,4 Гц, 2H), 3,76 (s, 3H), 3,05 (t, J=7,6 Гц, 2H), 2,43-2,40 (m, 2H), 2,17 (t, J=7,0 Гц, 2H), 1,70-1,69 (m, 2H), 1,57-1,55 (m, 2H), 1,37 (s, 9H), 1,36 (s, 6H).
Стадия 5
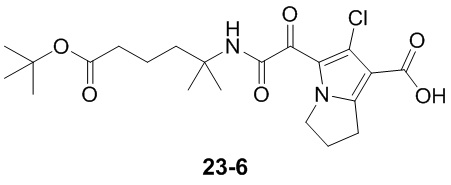
Соединение 23-5 (480 мг, 1,06 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (5 мл), метанола (5 мл) и воды (5 мл) и добавляли моногидрат гидроксида лития (221,37 мг, 5,28 ммоль, 5 экв.). Реакционную смесь перемешивали при 50°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. К реакционному раствору добавляли водный раствор гидросульфата калия с концентрацией 2,0 моль/л до pH = 5 и смесь экстрагировали этилацетатом (10 мл ×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 23-6. MS (ESI) m/z: 463,3 [M+Na]+.
1H ЯМР (400 MГц, CDCl3) δ = 4,19 (t, J=7,0 Гц, 2H), 3,22 (s, 1H), 3,01 (t, J=7,6 Гц, 2H), 2,38-2,36 (m, 2H), 2,18-2,09 (m, 2H), 1,63-1,60 (m, 2H), 1,51-1,49 (m, 2H), 1,30 (d, J=6,4 Гц, 15H).
Стадия 6
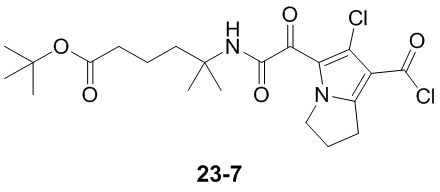
Соединение 23-6 (100 мг, 226,80 мкмоль, 1 экв.) растворяли в дихлорметане (5 мл) и добавляли оксалилхлорид (57,57 мг, 453,60 мкмоль, 39,71 мкл, 2 экв.) и N,N-диметилформамид (1,66 мг, 22,68 мкмоль, 1,75 мкл, 0,1 экв.) при 0°C. После завершения добавления реакционную систему нагревали до 26°C и перемешивали в течение 0,5 ч. Для остановки реакции в 1 мл метанола добавляли небольшое количество реакционного раствора. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что исходный материал полностью израсходован и образовались новые пятна. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 23-7.
Стадия 7
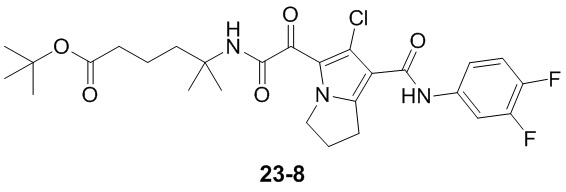
Соединение 1-9 (56,21 мг, 435,39 мкмоль, 2 экв.) растворяли в дихлорметане (5 мл) и добавляли бикарбонат натрия (91,44 мг, 1,09 ммоль, 42,33 мкл, 5 экв.). По каплям добавляли смесь соединения 23-7 (100 мг, 217,69 мкмоль, 1 экв.) и дихлорметана (5 мл) при 26°C. После завершения добавления реакционную систему перемешивали при 26°C в течение 0,5 ч. По результатам LCMS было видно, что реакция завершилась. В реакционный раствор при помешивании добавляли воду (10 мл) и оставляли отстаиваться для разделения жидкостей с получением дихлорметановой фазы. Водную фазу экстрагировали дихлорметаном (10 мл ×1), а органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 23-8. MS (ESI) m/z: 574,4 [M+Na]+.
Стадия 8
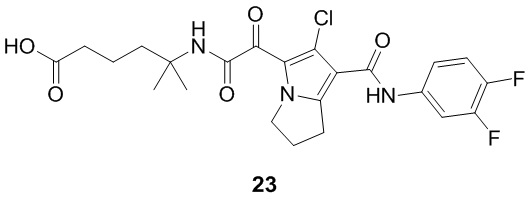
Соединение 23-8 (150 мг, 271,73 мкмоль, 1 экв.) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (309,84 мг, 2,72 ммоль, 201,19 мкл, 10 экв.) при 26°C. После завершения добавления реакционную систему перемешивали при 26°C в течение 0,5 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали в условиях пониженного давления, а неочищенный продукт очищали с помощью препаративной HPLC (колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,225% муравьиной кислоты)-ацетонитрил; % содержания ацетонитрила: 47% - 77%, 7 мин) с получением соединения 23. MS (ESI) m/z: 518,3 [M+Na]+.
1H ЯМР (DMSO-d6) δ = 12,04 (br s, 1H), 9,93 (s, 1H), 8,30 (s, 1H), 7,87-7,82 (m, 1H), 7,43-7,40 (m, 2H), 4,28 (t, J=7,2 Гц, 2H), 3,08 (t, J=7,6 Гц, 2H), 2,47-2,45 (m, 2H), 2,19 (t, J=7,2 Гц, 2H), 1,68 (t, J=8,2 Гц, 2H), 1,56-1,52 (m, 2H), 1,32 (s, 6H).
Пример 24
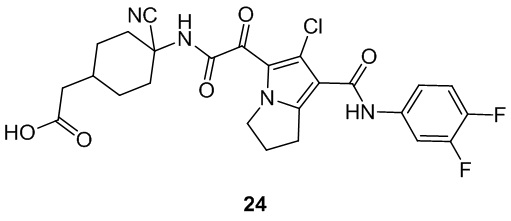
Путь синтеза:
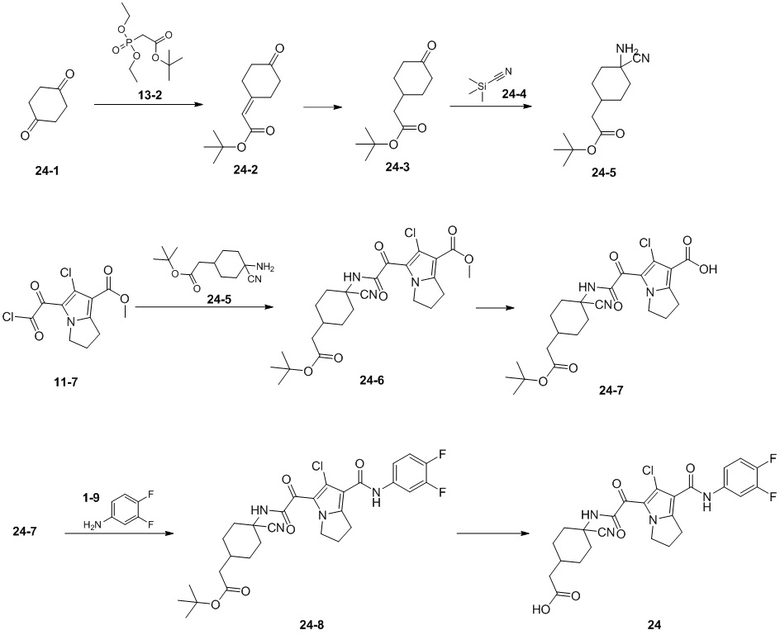
Стадия 1
Соединение 13-2 (25 г, 99,11 ммоль, 1 экв.) растворяли в тетрагидрофуране (100 мл), а затем добавляли гидрид натрия (5,95 г, 148,67 ммоль, степень чистоты: 60%, 1,5 экв.). Реакцию в реакционной системе проводили при 0°C в течение 1 ч, а затем добавляли в нее соединение 24-1 (22,23 г, 198,22 ммоль, 2 экв.) и проводили реакцию при 25°C в течение 1 ч. По результатам LCMS было видно, что соединение 24-1 было полностью израсходовано и было получено соединение 24-2. Реакционный раствор выливали в 500 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом (500 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 20:1 до 5:1, об./об.) с получением соединения 24-2.
1H ЯМР (400 MГц, CDCl3) δ = 5,77 (s, 1H), 3,17 (t, J=7,2 Гц, 2H), 2,63 (t, J=6,4 Гц, 2H), 2,52-2,47 (m, 4H), 1,49 (s, 9H).
Стадия 2
Соединение 24-2 (6 г, 28,53 ммоль, 1 экв.) растворяли в этаноле (20 мл), а затем добавляли влажный палладиевый катализатор на углеродном носителе (0,2 г, 28,53 ммоль, содержание: 10%, 1 экв.). Реакцию в реакционной системе проводили при 25°C в течение 1 ч в условиях атмосферы водорода (15 psi). По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 24-2 полностью израсходовано. Реакционный раствор немедленно фильтровали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 3:1, об./об.) с получением соединения 24-3.
1H ЯМР (400 MГц, CDCl3) δ = 3,45 (s, 1H), 2,40-2,37 (m, 4H), 2,23 (s, 3H), 2,07-2,06 (m, 3H), 1,47 (s, 9H).
Стадия 3
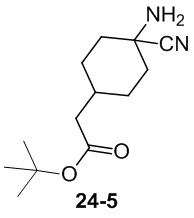
Тетраизопропоксид титана (4,50 г, 15,83 ммоль, 4,67 мл, 1,2 экв.) растворяли в растворе аммония/метанола (7 M, 7,54 мл, 4 экв.), а затем добавляли соединение 24-3 (2,8 г, 13,19 ммоль, 1 экв.). Реакцию в реакционной системе проводили при 25°C в течение 2 ч, а затем охлаждали ее до -5°C, добавляли в нее соединение 24-4 (1,37 г, 13,85 ммоль, 1,73 мл, 1,05 экв.) и затем проводили реакцию при 25°C в течение 14 ч. По результатам HPLC было видно, что соединение 24-3 было полностью израсходовано. В реакционный раствор вносили воду (5 мл), перемешивали в течение 0,5 ч и фильтровали. Фильтрат концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 3:1 до 1:1, об./об.) с получением соединения 24-5.
1H ЯМР (400 MГц, CDCl3) δ = 2,19-2,06 (m, 2H), 2,06-2,03 (m, 2H), 1,86-1,82 (m, 5H), 1,60-1,53 (m, 2H), 1,52 (s, 9H), 1,45-1,38 (m, 2H).
Стадия 4
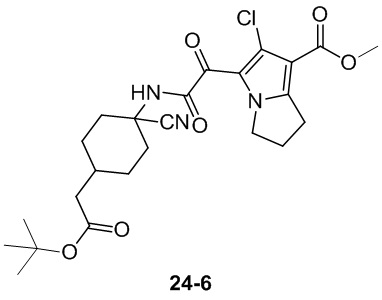
Соединение 24-5 (1,23 г, 5,17 ммоль, 1,5 экв.) и триэтиламин (1,05 г, 10,34 ммоль, 1,44 мл, 3 экв.) растворяли в дихлорметане (5 мл), а затем добавляли раствор соединения 11-7 (1 г, 3,45 ммоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему нагревали до 30°C и перемешивали в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 11-7 полностью израсходовано и были получены новые пятна. Результаты LCMS подтверждали получение соединения 24-6. Реакцию останавливали с помощью воды (30 мл), а затем реакционный раствор экстрагировали дихлорметаном (30 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:1 до 2:1, об./об.) с получением соединения 24-6. MS(ESI) m/s: 436,0 [M+H-(трет-Bu)]+.
Стадия 5
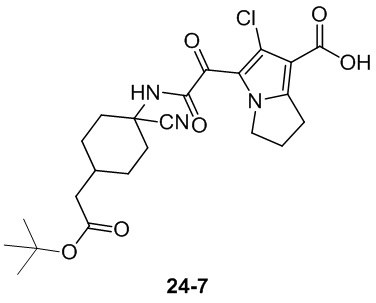
Соединение 24-6 (1 г, 2,03 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (10 мл) и воды (5 мл), а затем добавляли моногидрат гидроксида лития (255,87 мг, 6,10 ммоль, 3 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч. По результатам LCMS было видно, что соединение 24-6 полностью израсходовано и было получено соединение 24-7. Реакционный раствор доводили до pH = 3-5 с помощью 1,0 моль/л разбавленной соляной кислоты и экстрагировали этилацетатом (30 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 24-7. MS(ESI) m/s: 476,1 [M-H]-.
1H ЯМР (400 MГц, DMSO-d6) δ = 12,43 (br s, 1H), 9,40 (s, 1H), 4,27 (t, J=7,2 Гц, 2H), 3,06 (t, J=7,5 Гц, 2H), 2,45-2,40 (m, 4H), 2,15 (d, J=6,4 Гц, 2H), 1,91 (s, 1H), 1,79 (d, J=12,8 Гц, 2H), 1,64-1,58 (m, 2H), 1,41 (s, 9H), 1,31-1,23 (m, 2H).
Стадия 6
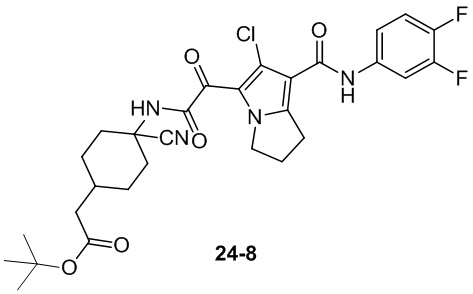
Соединение 24-7 (0,4 г, 836,93 мкмоль, 1 экв.), триэтиламин (127,03 мг, 1,26 ммоль, 174,74 мкл, 1,5 экв.) и гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (381,87 мг, 1,00 ммоль, 1,2 экв.) растворяли в N,N-диметилформамиде (5 мл). Реакционную систему перемешивали при 25°C в течение 0,5 ч, а затем в нее добавляли соединение 1-9 (324,16 мг, 2,51 ммоль, 3 экв.). Реакционную систему нагревали до 60°C и перемешивали в течение 5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 24-7 было полностью израсходовано и были получены новые пятна; по результатам LCMS было видно, что было получено соединение 24-8. Реакцию останавливали с помощью воды (30 мл), а затем реакционный раствор экстрагировали этилацетатом (20 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 100:1 до 2:1, об./об.) с получением соединения 24-8. MS(ESI) m/s: 587,1 [M-H]-.
Стадия 7
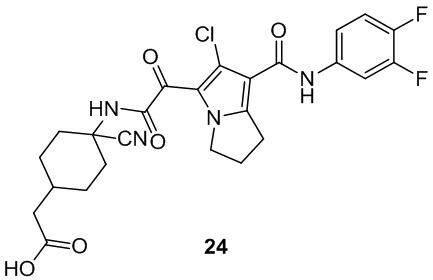
Соединение 24-8 (0,3 г, 509,31 мкмоль, 1 экв.) растворяли в дихлорметане (5 мл), а затем добавляли трифторуксусную кислоту (5 мл). Реакционную систему перемешивали при 30°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. В реакционный раствор добавляли воду (0,1 мл) для остановки реакции, а затем фильтровали. Фильтрат очищали с помощью препаративной HPLC (нейтральная, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 10% - 50%, 20 мин) с получением соединения 24. MS(ESI) m/s: 531,1 [M-H]-.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,96 (s, 1H), 9,45 (br s, 1H), 7,86-7,81 (m, 1H), 7,45-7,39 (m, 2H), 4,30 (t, J=7,2 Гц, 2H), 3,09 (t, J=7,6 Гц, 2H), 2,53-2,52 (m, 2H), 2,46-2,41 (m, 2H), 2,15 (d, J=6,8 Гц, 2H), 1,83 (d, J=12,8 Гц, 2H), 1,75 (br s, 1H), 1,65-1,59 (m, 2H), 1,29-1,20 (m, 2H).
Пример 25
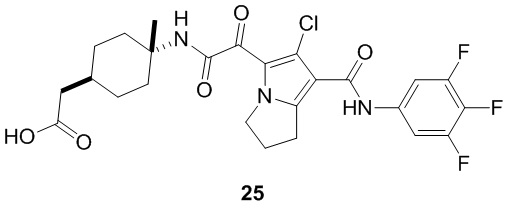
Путь синтеза:
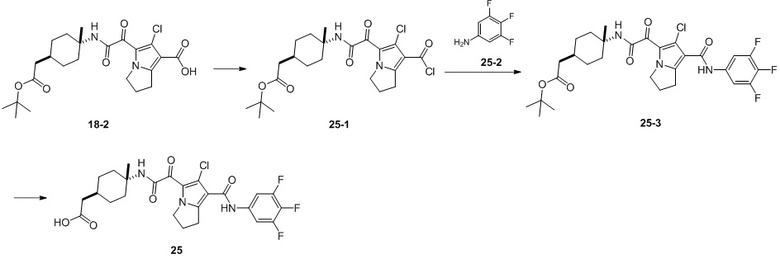
Стадия 1
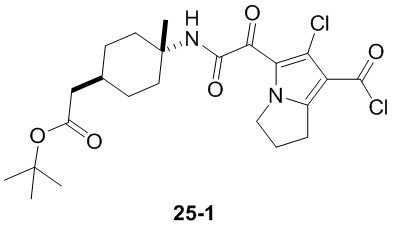
Соединение 18-2 (500 мг, 1,07 ммоль, 1 экв.) растворяли в дихлорметане (20 мл) и по каплям добавляли к нему оксалилхлорид (271,83 мг, 2,14 ммоль, 187,47 мкл, 2 экв.) и N,N-диметилформамид (7,83 мг, 107,08 мкмоль, 8,24 мкл, 0,1 экв.) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. Брали две капли реакционного раствора и добавляли в них метанол для остановки реакции, и по результатам LCMS было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 25-1.
Стадия 2
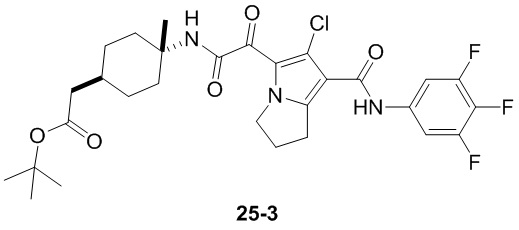
Соединение 25-1 растворяли в дихлорметане (15 мл) и добавляли TEA (312,70 мг, 3,09 ммоль, 430,12 мкл, 3 экв.). По каплям добавляли раствор соединения 25-2 (500,00 мг, 1,03 ммоль, 1 экв.) в дихлорметане (5 мл) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту с концентрацией 0,5 моль/л (50 мл). Водную фазу экстрагировали дихлорметаном (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = от 10:1 до 4:1, об./об.) с получением соединения 25-3. MS(ESI) m/s: 540,1 [M+H-(трет-Bu)]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,47 (s, 1H), 7,38-7,34 (m, 2H), 6,23 (s, 1H), 4,35-4,31 (m, 2H), 3,28-3,23 (m, 2H), 2,52-2,49 (m, 2H), 2,18-2,14 (m, 2H), 2,05-2,00 (m, 2H), 1,93-1,81(m, 3H), 1,80-1,71 (m, 2H), 1,55 (s, 3H), 1,50-1,45 (m, 9H), 1,32-1,26 (m, 2H).
Стадия 3
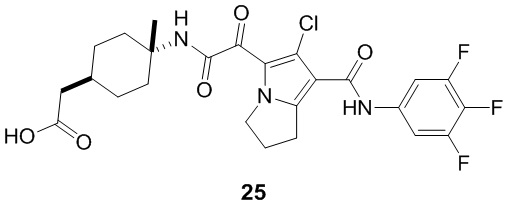
Соединение 25-3 (100,00 мг, 167,78 мкмоль, 1 экв.) растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (382,59 мг, 3,36 ммоль, 248,44 мкл, 20 экв.). Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 10% - 50%, 20 мин) с получением соединения 25. MS(ESI) m/s: 540,2 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 9,99 (s, 1H), 8,29 (s, 1H), 7,65-7,57 (m, 2H), 4,30-4,26 (m, 2H), 3,09-3,05 (m, 2H), 2,47-2,43 (m, 2H), 2,16-2,12 (m, 2H), 1,85-1,80 (m, 2H), 1,75-1,61 (m, 5H), 1,35 (s, 3H), 1,21-1,16(m, 2H).
Пример 26
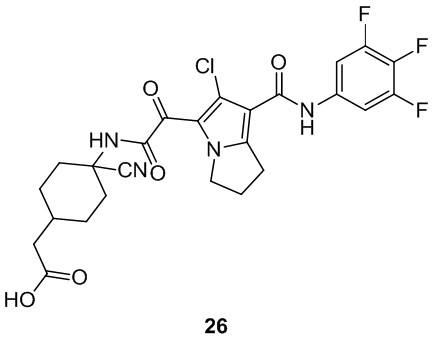
Путь синтеза:
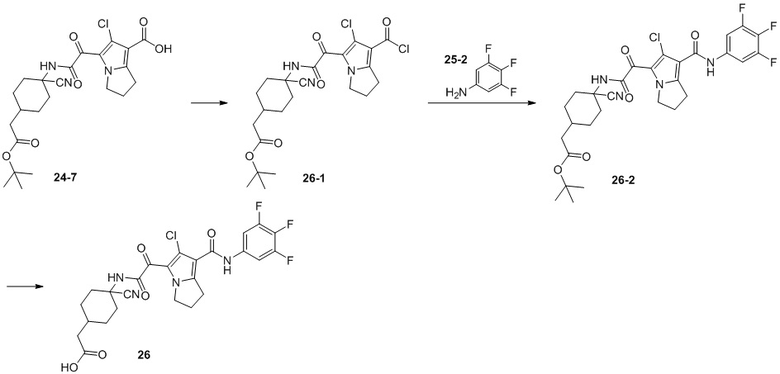
Стадия 1
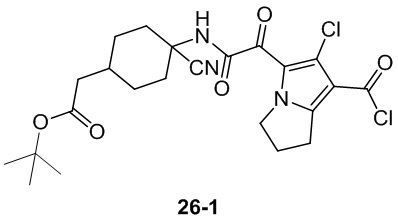
Соединение 24-7 (0,3 г, 627,70 мкмоль, 1 экв.) растворяли в дихлорметане (5 мл), а затем добавляли оксалилхлорид (159,34 мг, 1,26 ммоль, 109,89 мкл, 2 экв.) и N,N-диметилформамид (4,59 мг, 62,77 мкмоль, 4,83 мкл, 0,1 экв.) при 0°C в условиях атмосферы азота. Реакционную систему нагревали до 30°C и перемешивали в течение 0,5 ч. Результаты LCMS подтверждали, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления с получением соединения 26-1, которое применяли непосредственно на следующей стадии.
Стадия 2
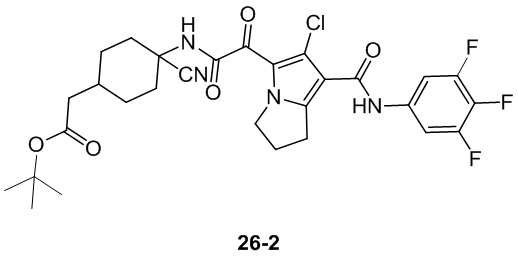
Соединение 25-2 (133,35 мг, 906,56 мкмоль, 1,5 экв.) и триэтиламин (183,47 мг, 1,81 ммоль, 252,36 мкл, 3 экв.) растворяли в дихлорметане (5 мл), а затем добавляли раствор соединения 26-1 (0,3 г, 604,37 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему нагревали до 30°C и перемешивали в течение 1 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. Реакционный раствор выливали в воду (20 мл) и экстрагировали дихлорметаном (15 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью препаративной HPLC (система HCl, колонка: Xtimate C18 100 мм × 30 мм × 5 мкм; подвижная фаза: вода (содержащая 0,05% соляной кислоты)-ацетонитрил; % содержания ацетонитрила: 50% - 75%, 10 мин) с получением соединения 26-2. MS(ESI) m/s: 605,1 [M-H]-.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,07 (s, 1H), 9,46 (s, 1H), 7,64-7,59 (m, 2H), 4,30 (t, J=7,2 Гц, 2H), 3,09 (t, J=7,4 Гц, 2H), 2,53-2,51 (m, 2H), 2,47-2,44 (m, 1H), 2,42-2,41 (m, 2H), 2,15 (d, J=6,8 Гц, 2H), 1,80 (d, J=12,8 Гц, 2H), 1,65-1,56 (m, 2H), 1,41 (s, 9H), 1,32-1,23 (m, 2H).
Стадия 3
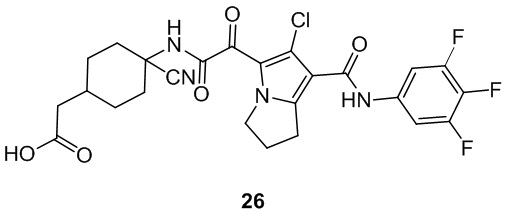
Соединение 26-2 (15 мг, 24,71 мкмоль, 1 экв.) растворяли в дихлорметане (2 мл), а затем добавляли трифторуксусную кислоту (2 мл). Реакционную систему перемешивали при 30°C в течение 0,5 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. Реакционный раствор концентрировали в условиях пониженного давления с получением остатка. Остаток очищали с помощью препаративной HPLC (нейтральная, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 5% - 50%, 20 мин) с получением соединения 26. MS(ESI) m/s: 549,1 [M-H]-.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,06 (s, 1H), 9,46 (s, 1H), 7,64-7,60 (m, 2H), 4,30 (t, J=7,2 Гц, 2H), 3,09 (t, J=7,8 Гц, 2H), 2,52-2,51 (m, 2H), 2,45-2,41 (m, 2H), 2,17 (d, J=6,8 Гц, 2H), 1,83 (d, J=13,6 Гц, 2H), 1,75 (br s, 1H), 1,65-1,59 (m, 2H), 1,30-1,24 (m, 2H).
Пример 27
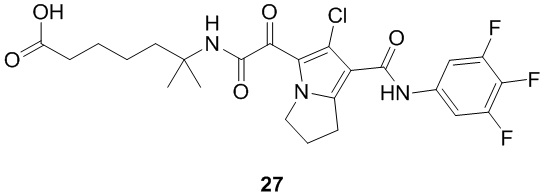
Путь синтеза:
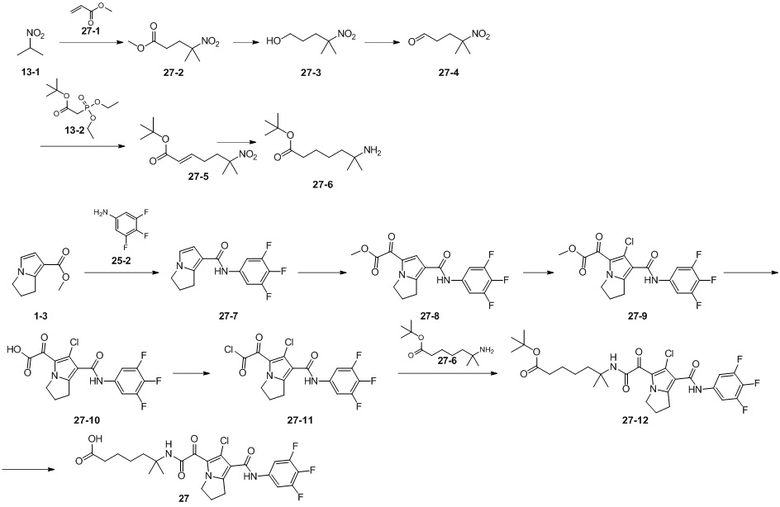
Стадия 1
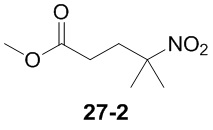
Соединение 13-1 (20 г, 224,48 ммоль, 20,16 мл, 1 экв.) растворяли в дихлорметане (100 мл), а затем добавляли соединение 27-1 (23,19 г, 269,38 ммоль, 24,26 мл, 1,2 экв.) и 1,8-диазабицикло[5.4.0]ундец-7-ен (68,35 г, 448,97 ммоль, 67,67 мл, 2 экв.). Реакционную систему перемешивали при 45°C в течение 12 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что было получено соединение 27-2. Реакционный раствор выливали в насыщенной раствор хлорида аммония (100 мл) и экстрагировали дихлорметаном (100 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением неочищенного соединения. Неочищенное соединение очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 20:1 до 5:1, об./об.) с получением соединения 27-2.
1H ЯМР (400 MГц, CDCl3) δ = 3,69 (s, 3H), 2,35-2,32 (m, 2H), 2,29-2,26 (m, 2H), 1,60 (s, 6H).
Стадия 2
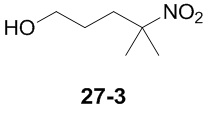
В условиях атмосферы азота соединение 27-2 (18 г, 102,75 ммоль, 1 экв.) в сухой колбе растворяли в тетрагидрофуране (1 л) и медленно добавляли алюмогидрид лития (2,14 г, 56,51 ммоль, 0,55 экв.) при 0°C. Реакционную систему медленно нагревали до 25°C и перемешивали в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, реакция завершилась. В реакционный раствор медленно при помешивании по каплям добавляли насыщенный раствор сегнетовой соли (9 мл), а затем фильтровали. Фильтрат немедленно концентрировали в условиях пониженного давления с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = 4:1, об./об.) с получением соединения 27-3.
1H ЯМР (400 MГц, CDCl3) δ = 3,68-3,63 (m, 2H),2,04-1,97 (m, 2H), 1,59 (s, 6H),1,55-1,51 (m, 2H).
Стадия 3
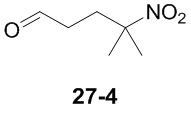
Соединение 27-3 (2 г, 13,59 ммоль, 1 экв.) в сухой колбе растворяли в дихлорметане (50 мл) и добавляли хлорохромат пиридиния (4,39 г, 20,38 ммоль, 1,5 экв.) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 1 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакцию останавливали с помощью воды (20 мл), а затем реакционный раствор экстрагировали дихлорметаном (10 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 30:1 до 10:1) с получением соединения 27-4.
1H ЯМР (400 MГц, CDCl3) δ = 9,78 (s, 1H), 2,52-2,49 (m, 2H), 2,25-2,23 (m, 2H), 1,60 (s, 6H).
Стадия 4
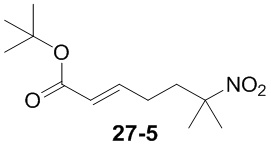
Соединение 13-2 (2,68 г, 10,61 ммоль, 2,2 экв.) в сухой колбе растворяли в тетрагидрофуране (15 мл) и добавляли гидрид натрия (578,69 мг, 14,47 ммоль, степень чистоты: 60%, 3 экв.) при 0°C в условиях атмосферы азота. После перемешивания в течение 30 мин добавляли раствор соединения 27-4 (700 мг, 4,82 ммоль, 1 экв.) в тетрагидрофуране (5 мл) и реакционную систему перемешивали при 25°C в течение 16 ч. По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту (0,5 моль/л, 20 мл). Водную фазу экстрагировали этилацетатом (20 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 30:1 до 10:1, об./об.) с получением соединения 27-5.
1H ЯМР (400 MГц, CDCl3) δ = 6,83-6,76 (m, 1H), 5,77 (d, J=15,6 Гц, 1H), 2,16-2,06 (m, 2H), 2,05-2,04 (m, 2H), 1,60 (s, 6H), 1,48 (s, 9H).
Стадия 5
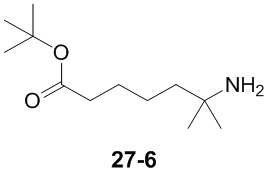
В условиях слабого потока аргона палладиевый катализатор на углеродном носителе (150 мг, степень чистоты: 10%) вносили в гидрогенизационную колбу, медленно вносили метанол (20 мл) по стенке колбы, а затем добавляли соединение 27-5 (800 мг, 3,29 ммоль, 1 экв.). После 3 продувок водородом реакционную систему перемешивали при 20°C в течение 2 ч в условиях атмосферы водорода (50 psi). По результатам TLC (простой петролейный эфир : этилацетат = 5:1) было видно, что реакция завершилась. Реакционный раствор немедленно фильтровали и концентрировали в условиях пониженного давления с получением соединения 27-6.
1H ЯМР (400 MГц, CDCl3) δ = 2,22 (t, J=7,6 Гц, 2H), 1,60-1,57 (m, 2H), 1,44 (s, 9H), 1,37-1,28 (m, 4H), 1,10 (s, 6H).
Стадия 6
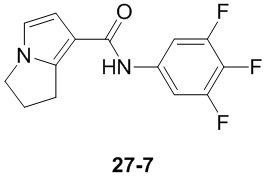
Соединение 1-3 (20 г, 121,07 ммоль, 1 экв.) и соединение 25-2 (26,71 г, 181,61 ммоль, 1,5 экв.) растворяли в толуоле (300 мл), а затем добавляли к ним гексаметилдисилазид лития (1 моль/л, 242,15 мл, 2 экв.) при 0°C в условиях атмосферы азота. Реакционную систему нагревали до 25°C и перемешивали в течение 16 ч. Результаты TLC подтверждали, что соединение 1-3 было полностью израсходовано и были получены новые пятна. Результаты LCMS подтверждали, что соединение 1-3 полностью израсходовано и было получено соединение 27-7. Реакционный раствор выливали в насыщенной раствор хлорида аммония (500 мл) для остановки реакции и экстрагировали этилацетатом (200 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 100:1 до 1:1,5, об./об.) с получением соединения 27-7. MS(ESI) m/s: 281,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 7,44 (s, 1H), 7,37-7,28 (m, 2H), 6,61 (d, J=2,8 Гц, 1H), 6,45 (d, J=3,2 Гц, 1H), 3,99 (t, J=2,8 Гц, 2H), 3,13 (t, J=3,2 Гц, 2H), 2,62-2,55 (m, 2H).
Стадия 7
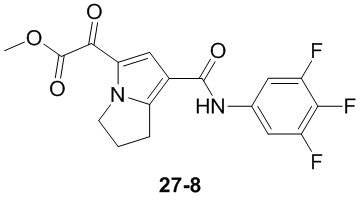
Соединение 27-7 (31 г, 110,62 ммоль, 1 экв.) растворяли в дихлорметане (300 мл), а затем добавляли метилоксалилхлорид (27,10 г, 221,23 ммоль, 20,38 мл, 2 экв.) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 25°C в течение 1,5 ч. Результаты TLC (простой петролейный эфир : этилацетат = 3:1) подтверждали, что реакция завершилась. Реакционный раствор выливали в ледяную воду (300 мл) для остановки реакции, а затем экстрагировали этилацетатом (100 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 100:1 до 2,5:1, об./об.) с получением соединения 27-8. MS(ESI) m/s: 367,1 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 9,92 (s, 1H), 7,80 (s, 1H), 7,43-7,32 (m, 2H), 4,37 (t, J=7,4 Гц, 2H), 3,91 (s, 3H), 3,19 (t, J=7,6 Гц, 2H), 2,65-2,57 (m, 2H).
Стадия 8
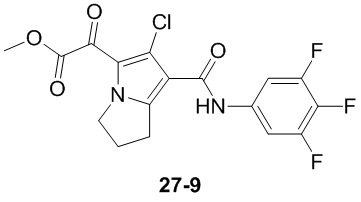
Соединение 27-8 (11 г, 21,02 ммоль, 1 экв.) растворяли в уксусной кислоте (110 мл), а затем добавляли N-хлорсукцинимид (4,21 г, 31,53 ммоль, 1,5 экв.). Реакционную систему перемешивали при 40°C в течение 16 ч. По результатам TLC было видно, что соединение было полностью израсходовано. Реакционный раствор выливали в воду (100 мл) для остановки реакции и экстрагировали этилацетатом (50 мл ×3). Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (50 мл ×3), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 100:1 до 4:1, об./об.) с получением соединения 27-9. MS(ESI) m/s: 401,0 [M+H]+.
1H ЯМР (400 MГц, CDCl3) δ = 8,29 (s, 1H), 7,36-7,30 (m, 2H), 4,40 (t, J=7,4 Гц, 2H), 3,98 (s, 3H), 3,26 (t, J=7,6 Гц, 2H), 2,61-2,51 (m, 2H).
Стадия 9
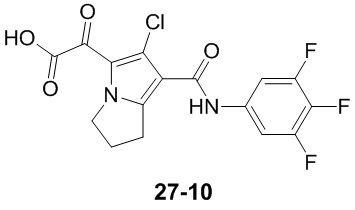
Соединение 27-9 (8 г, 19,96 ммоль, 1 экв.) растворяли в смеси тетрагидрофурана (100 мл) и воды (100 мл), а затем добавляли моногидрат гидроксида лития (2,51 г, 59,89 ммоль, 3 экв.). Реакционную систему перемешивали при 25°C в течение 16 ч. Результаты LCMS подтверждали, что соединение было полностью израсходовано. Реакционный раствор выливали в воду (50 мл), экстрагировали этилацетатом (50 мл ×2), доводили до pH = 2 с помощью соляной кислоты с концентрацией 1,0 моль/л, а затем экстрагировали этилацетатом (50 мл ×3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 27-10. MS(ESI) m/s: 387,1 [M+H]+.
1H ЯМР (400 MГц, MeOH-d4) δ = 7,63-7,41 (m, 2H), 4,38 (t, J=7,6 Гц, 2H), 3,15 (t, J=7,6 Гц, 2H), 2,62-2,50 (m, 2H).
Стадия 10
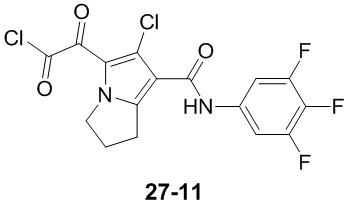
Соединение 27-10 (150 мг, 387,89 мкмоль, 1 экв.) в сухой колбе растворяли в дихлорметане (10 мл) и по каплям добавляли оксалилхлорид (98,47 мг, 775,78 мкмоль, 67,91 мкл, 2 экв.) и N,N-диметилформамид (2,84 мг, 38,79 мкмоль, 2,98 мкл, 0,1 экв.) в условиях атмосферы азота при 0°C. Реакционную систему перемешивали при 20°C в течение 1 ч. Брали две капли реакционного раствора и добавляли в них метанол для остановки реакции, а затем производили детекцию. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что превращение исходных материалов завершилось. Реакционный раствор немедленно концентрировали в условиях пониженного давления с получением соединения 27-11.
Стадия 11
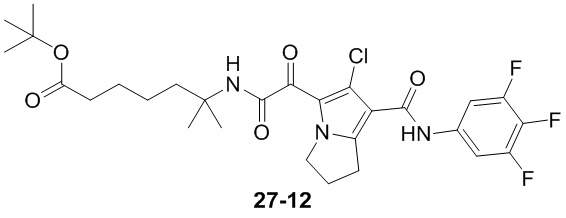
Соединение 27-6 (87,69 мг, 407,25 мкмоль, 1,1 экв.) в сухой колбе растворяли в дихлорметане (5 мл) и добавляли триэтиламин (112,39 мг, 1,11 ммоль, 154,59 мкл, 3 экв.). Добавляли раствор соединения 27-11 (150 мг, 370,23 мкмоль, 1 экв.) в дихлорметане (5 мл) при 0°C в условиях атмосферы азота. Реакционную систему перемешивали при 20°C в течение 2 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту (0,5 моль/л, 20 мл). Водную фазу экстрагировали дихлорметаном (10 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 27-12. MS(ESI) m/s: 528,2 [M+H-(трет-Bu)]+.
Стадия 12
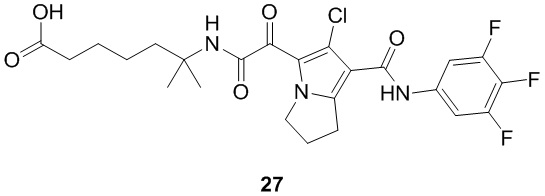
Соединение 27-12 (150 мг, 256,84 мкмоль, 1 экв.) в сухой колбе растворяли в дихлорметане (1 мл) и добавляли трифторуксусную кислоту (585,69 мг, 5,14 ммоль, 380,32 мкл, 20 экв.). Реакционную систему перемешивали при 20°C в течение 1 ч. По результатам LCMS и HPLC было видно, что реакция завершилась. Реакционный раствор немедленно концентрировали. Неочищенный продукт очищали с помощью препаративной HPLC (нейтральная система, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 0,04% гидрохлорида аммония + 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 28% - 58%, 10,5 мин) с получением соединения 27. MS(ESI) m/s: 528,1 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,02 (s, 1H), 8,25 (s, 1H), 7,64-7,60 (m, 2H), 4,28 (t, J=6,8 Гц, 2H), 3,08 (t, J=7,2 Гц, 2H), 2,50-2,46 (m, 2H), 2,20 (t, J=7,2 Гц, 2H), 1,68-1,64 (m, 2H), 1,50-1,46 (m, 2H), 1,35-1,28 (m, 8H).
Пример 28
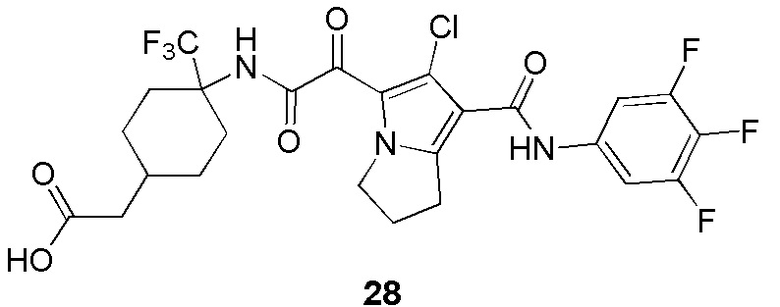
Путь синтеза:
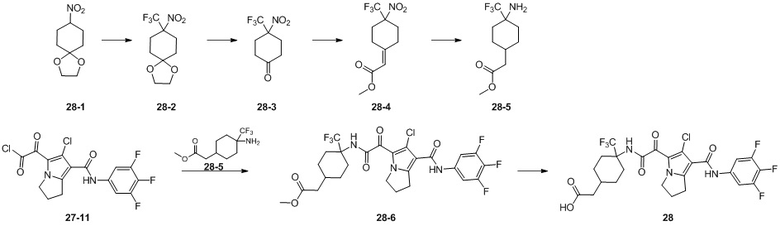
Стадия 1
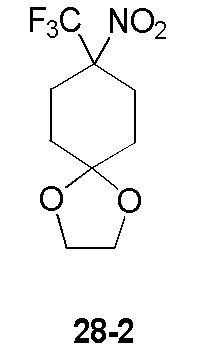
Соединение 28-1 (500 мг, 2,67 ммоль, 1 экв.) и трифторметансульфонат S-(трифторметил)дибензотиофения (1,40 г, 3,47 ммоль, 1,3 экв.) добавляли к дихлорметану (10 мл). Полученную смесь охлаждали до -25°C и к ней добавляли 1,8-диазабицикло[5,4,0]ундец-7-ен (813,28 мг, 5,34 ммоль, 805,23 мкл, 2,0 экв.). Реакционную систему перемешивали при -25°C в течение 1,5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 28-1 полностью израсходовано и были получены новые пятна. В реакционный раствор при помешивании добавляли воду (10 мл) и оставляли отстаиваться для разделения жидкостей с получением дихлорметановой фазы. В дихлорметановую фазу добавляли насыщенной водный раствор хлорида натрия (10 мл ×1), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью хроматографической системы COMBI-FLASH (градиентное элюирование: простой петролейный эфир : этилацетат = от 50:1 до 5:1, об./об.) с получением соединения 28-2.
1H ЯМР (400 MГц, CHCl3) δ = 3,99-3,92 (m, 4H), 2,82-2,77 (m, 2H), 2,22- 2,17 (m, 2H), 1,83-1,79 (m, 2H), 1,61-1,55 (m, 2H).
Стадия 2
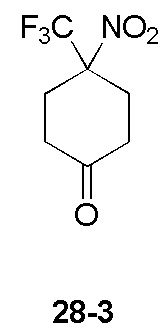
Соединение 28-2 (500 мг, 1,96 ммоль, 1 экв.) растворяли в дихлорметане (30 мл) и добавляли серную кислоту (3 мл) при 0°C. Реакционную систему нагревали до 26°C и перемешивали в течение 5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 28-2 было полностью израсходовано и были получены новые пятна. Реакционный раствор выливали в ледяную воду (30 мл) и медленно добавляли насыщенный водный раствор карбоната натрия до pH = 7. Затем полученную смесь экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (50 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 50:1 до 3:1, об./об.) с получением соединения 28-3.
1H ЯМР (400 MГц, CHCl3) δ = 3,12-3,09 (m, 2H), 2,56-2,48 (m, 2H), 2,45-2,39 (m, 4H).
Стадия 3
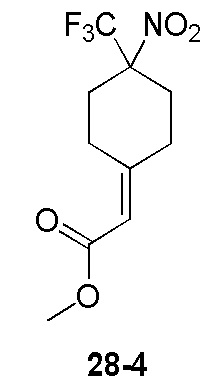
Соединение 28-3 (270 мг, 1,28 ммоль, 1 экв.) растворяли в дихлорметане (5 мл) и добавляли метил(трифенилфосфоранилиден)ацетат (427,56 мг, 1,28 ммоль, 1 экв.) при 26°C в условиях атмосферы азота. Реакционную систему нагревали до 50°C и перемешивали в течение 6 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что соединение 28-3 полностью израсходовано и были получены новые пятна. Реакционный раствор охлаждали до комнатной температуры, в него добавляли воду (10 мл), а затем экстрагировали дихлорметаном (10 мл ×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью хроматографической системы COMBI-FLASH (простой петролейный эфир : этилацетат = от 50:1 до 5:1) с получением соединения 28-4.
1H ЯМР (400 MГц, CHCl3) δ = 5,76 (s, 1H), 4,01-3,98 (m, 1H), 3,71 (s, 3H), 2,97-2,92 (m, 2H), 2,43-2,37 (m, 1H), 2,32-2,31 (m, 1 H), 2,02-1,96 (m, 3H).
Стадия 4
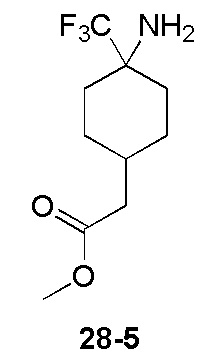
Соединение 28-4 (250 мг, 935,63 мкмоль, 1 экв.) и метанол (10 мл) вносили в сухую гидрогенизационную колбу и добавляли палладиевый катализатор на углеродном носителе (20 мг, 935,63 мкмоль, степень чистоты: 5%, 1 экв.) в условиях атмосферы водорода. Реакционную систему перемешивали при 60°C в течение 16 ч в условиях атмосферы водорода (50 psi). По результатам LCMS было видно, что соединение 28-4 было полностью израсходовано. Реакционный раствор фильтровали через целит для удаления палладиевого катализатора на углеродном носителе, а фильтрат концентрировали в условиях пониженного давления с получением соединения 28-5. MS (ESI) m/z: 239,7 [M+H]+.
Стадия 5
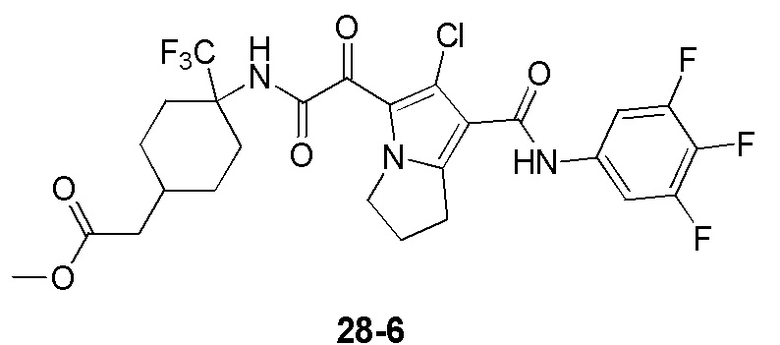
Соединение 28-5 (67,90 мг, 283,84 мкмоль, 1 экв.) и бикарбонат натрия (143,07 мг, 1,70 ммоль, 66,24 мкл, 6 экв.) добавляли к дихлорметану (2 мл) и к ним медленно добавляли смесь соединения 27-11 (115 мг, 283,84 мкмоль, 1 экв.) и дихлорметана (2 мл) при 0°C. Реакционную систему нагревали до 26°C и перемешивали в течение 0,5 ч. По результатам TLC (простой петролейный эфир : этилацетат = 2:1) было видно, что соединение 27-11 было полностью израсходовано. В реакционный раствор добавляли воду (10 мл), а затем экстрагировали дихлорметаном (5 мл ×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл ×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт отделяли с помощью хроматографической системы COMBI-FLASH (градиентное элюирование: простой петролейный эфир : этилацетат = от 50:1 до 3:1, об./об.) с получением соединения 28-6. MS (ESI) m/z: 608,3 [M+H]+.
1H ЯМР (400 MГц, CHCl3) δ = 8,47 (s, 1H), 7,34 (dd, J=9,0, 6,4 Гц, 2H), 6,22-6,16 (m, 1H), 4,33 (t, J=7,2 Гц, 2H), 3,70-3,69 (m, 3H), 3,27 (t, J=7,6 Гц, 2H), 2,62-2,52 (m, 3H), 2,41 (d, J=7,2 Гц, 1H), 2,40-2,26 (m, 2H), 2,04-1,96 (m, 1H), 1,86-1,75 (m, 3H), 1,28-1,24 (m, 3H).
Стадия 6
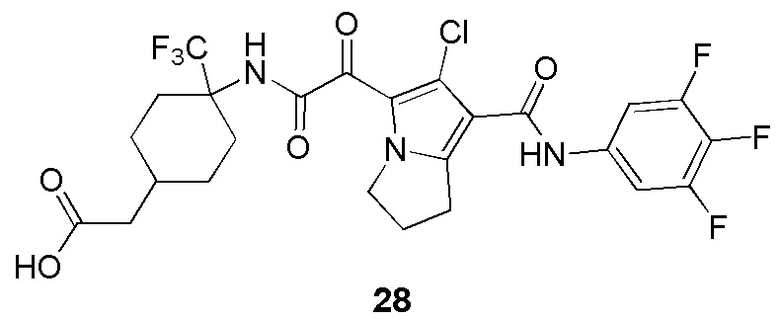
Соединение 28-6 (60,00 мг, 98,70 мкмоль, 1 экв.) растворяли в смеси тетрагидрофурана (1 мл), метанола (1 мл) и воды (1 мл) и добавляли моногидрат гидроксида лития (24,85 мг, 592,18 мкмоль, 6 экв.) при 40°C. Реакционную систему перемешивали при 40°C в течение 0,5 ч. По результатам LCMS было видно, что соединение 28-6 было полностью израсходовано. В реакционный раствор добавляли 2 моль/л водный раствор гидросульфата калия до pH = 4, а затем экстрагировали дихлорметаном (10 мл ×2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью препаративной HPLC (система на основе муравьиной кислоты, колонка: Waters Xbridge 150 мм × 18 мм × 5 мкм; подвижная фаза: вода (содержащая 0,225% муравьиной кислоты)-ацетонитрил; % содержания ацетонитрила: 55% - 85%, 7 мин) с получением соединения 28. MS (ESI) m/z: 594,3 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 12,12 (s, 1H), 10,07 (d, J=8,2Гц, 1 H), 8,70 (d, J=16,2Гц, 1 H), 7,63 (dd, J=10,2, 6,2Гц, 2 H), 4,31 (t, J=7,2Гц, 2 H), 3,10 (t, J=7,6Гц, 2H), 2,49-2,43 (m, 3H), 2,35-2,25 (m, 2H), 2,16-2,07 (m, 2H), 1,78-1,65 (m, 3H), 1,59-1,42 (m, 2H), 1,28-1,17 (m, 1H).
Примеры 29 и 30
Путь синтеза:
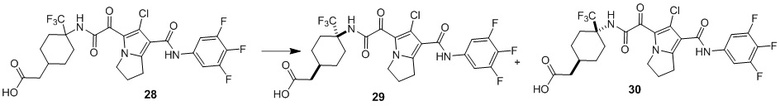
Соединение 28 (15 мг, 25,26 мкмоль, 1 экв.) разделяли с помощью SFC (колонка: Phenomenex-Amylose-1 (250 мм × 30 м, 5 мкм); подвижная фаза: 0,1% NH3H2O EtOH; % B: 35% - 35%, мин) с получением соединения 29 (время удержания в колонке для SFC: 1,763 мин) и соединение 30 (время удержания в колонке для SFC: 2,086 мин). Условия SFC-анализа: колонка: хиральная колонка Agilent 1260 с DAD-детектором со следующей спецификацией: AD-3 50 ×3 мм внутреннего диаметра, 3 мкм; подвижная фаза: A: диоксид углерода, B: этанол (содержащий 0,05% диэтаноламина); градиент: B, изменяющийся с 5% до 40% за 2,5 мин, удерживаемый на уровне 40% в течение 0,35 мин, а затем оставляли плавно изменяться на 1,65 мин; скорость потока: 2,5 мл/мин; температура колонки: 40°C.
Соединение 29: 1H ЯМР (DMSO-d6) δ = 10,14 (br s, 1 H),8,65 (br s, 1 H), 7,68-7,62 (m, 2 H), 4,35-4,33 (m, 2 H), 3,13-3,09 (m, 2 H), 2,37-2,31 (m, 2 H), 2,09-2,03 (m, 2 H), 1,70-1,62 (m, 3 H), 1,59-1,48 (m, 2 H), 1,27-1,21 (m, 4 H).
Соединение 30: 1H ЯМР (DMSO-d6) δ = 10,09 (s, 1 H), 8,70 (s, 1 H), 7,64 (dd, J=10,2, 6,27 Гц, 2 H), 4,31 (t, J=7,2 Гц, 2 H), 3,10 (t, J=7,2 Гц, 2 H), 2,34-2,25 (m, 4 H), 2,14-2,09 (m, 1 H), 1,75-1,32 (m, 4 H), 1,46 (br s, 2 H), 1,24 (s, 2 H).
Пример 31
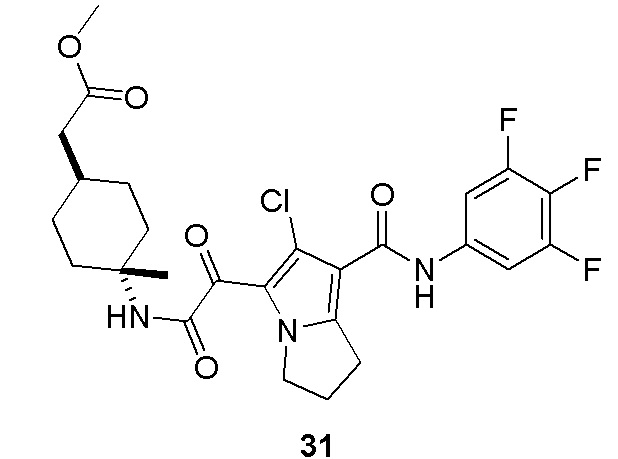
Путь синтеза:
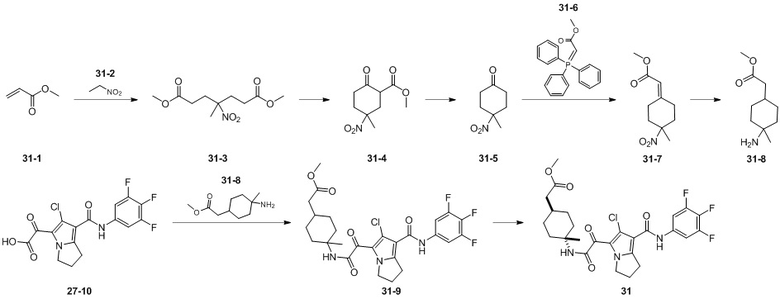
Стадия 1
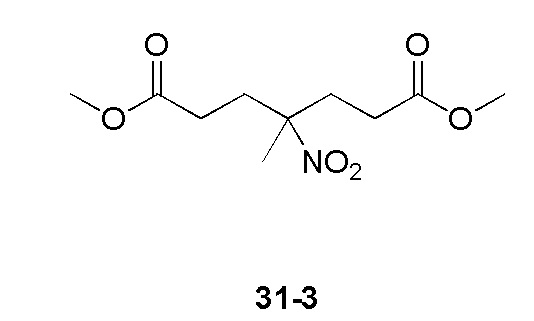
Соединение 31-1 (22,94 г, 266,43 ммоль, 23,99 мл, 2 экв.), соединение 31-2 (10 г, 133,22 ммоль, 9,52 мл, 1 экв.) и 1,8-диазабицикло[5.4.0]ундец-7-ен (42,59 г, 279,75 ммоль, 42,17 мл, 2,1 экв.) в сухой колбе растворяли в дихлорметане (200 мл) и реакционную систему перемешивали при 40°C в течение 3 ч. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в воду (500 мл) для разделения жидкостей. Водную фазу экстрагировали дихлорметаном (100 мл ×2), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, простой петролейный эфир : этилацетат = 3:1, об./об.) с получением соединения 31-3.
1H ЯМР (400 MГц, CDCl3) δ = 3,62 (s, 6H), 2,32-2,27 (m, 6H), 2,22- 2,20 (m, 2H), 1,51 (s, 3H).
Стадия 2
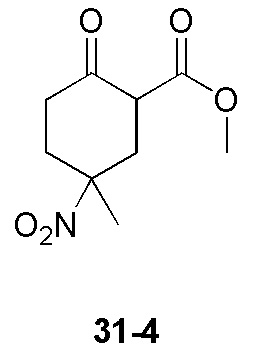
Соединение 31-3 (10 г, 40,45 ммоль, 1 экв.) в сухой колбе растворяли в тетрагидрофуране (100 мл) и добавляли метилат натрия (5 M, 9,71 мл, 1,2 экв.). Реакционную систему перемешивали при 15°C в течение 16 ч. По результатам LCMS было видно, что было получено соединение 31-4. Реакционный раствор медленно выливали в 1 моль/л разбавленную соляную кислоту (100 мл) для разделения жидкостей. Водную фазу промывали полунасыщенным солевым раствором (50 мл ×3), а органическую фазу собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением соединения 31-4. MS (ESI) m/z: 216,1 [M+H]+
1H ЯМР (400 MГц, CDCl3) δ = 12,12 (s, 1H), 4,11 (s, 3H), 3,36-3,24 (m, 1H), 2,54-2,45 (m, 2H), 2,40-2,35 (m, 2H), 2,19-1,54 (m, 1H), 1,26 (s, 3H).
Стадия 3
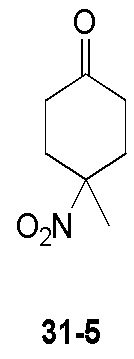
Соединение 31-4 (4 г, 18,59 ммоль, 1 экв.) в колбе растворяли в смеси 2-метилтетрагидрофурана (20 мл) и воды (20 мл) и добавляли гидроксид калия (3,13 г, 55,76 ммоль, 3 экв.). Реакционную систему перемешивали при 85°C в течение 16 ч. По результатам TLC (простой петролейный эфир : этилацетат = 3:1) было видно, что реакция завершилась. Реакционный раствор оставляли расслаиваться. Водную фазу экстрагировали этилацетатом (10 мл ×6), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 20:1 до 3:1, об./об.) с получением соединения 31-5.
1H ЯМР (400 MГц, CDCl3) δ = 2,89-2,78 (m, 2H), 2,50-2,41 (m, 2H), 2,39-2,27 (m,2 H), 2,06-1,82 (m, 2H), 1,56 (s, 3H).
Стадия 4
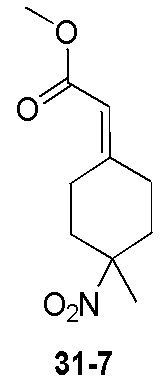
Соединение 31-5 (11,4 г, 72,53 ммоль, 1 экв.) в сухой колбе растворяли в толуоле (100 мл) и добавляли соединение 31-6 (26,68 г, 79,79 ммоль, 1,1 экв.). Реакционную систему перемешивали при 110°C в течение 16 ч в условиях атмосферы азота. По результатам LCMS было видно, что реакция завершилась. Реакционный раствор выливали в разбавленную соляную кислоту с концентрацией 0,5 моль/л (50 мл). Водную фазу экстрагировали этилацетатом (10 мл ×3), а органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель с размером пор 100-200 меш, градиентное элюирование: простой петролейный эфир : этилацетат = от 100:0 до 10:1, об./об.) с получением соединения 31-7.
1H ЯМР (400 MГц, CDCl3) δ = 5,70 (s, 1H), 3,68 (s, 3H), 3,51-3,48 (s, 1H), 2,68-2,63 (m, 2H), 2,35-2,04 (m, 3H), 1,77-1,60 (m, 2H), 1,59 (s, 3H).
Стадия 5
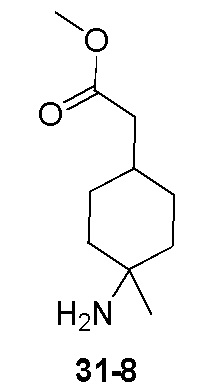
Скелетный никелевый катализатор гидрирования (7,00 г, 119,26 ммоль, 3,63 экв.) растворяли в метаноле (60 мл), а затем добавляли соединение 31-7 (7 г, 32,83 ммоль, 1 экв.). Реакционную систему перемешивали при 30°C в течение 16 ч в условиях атмосферы водорода (30 psi). По результатам TLC (дихлорметан : метанол =10:1) было видно, что соединение 31-7 было полностью израсходовано. Реакционный раствор фильтровали и концентрировали в условиях пониженного давления с получением соединения 31-8.
1H ЯМР (400 MГц, MeOD-d4) δ = 3,65 (s, 3H), 2,29-2,22 (m, 2H), 1,73-1,67 (m, 2H), 1,56-1,51 (m, 4H), 1,39-1,38 (m, 1H), 1,35 (s, 3H), 1,19-1,14 (m, 2H).
Стадия 6
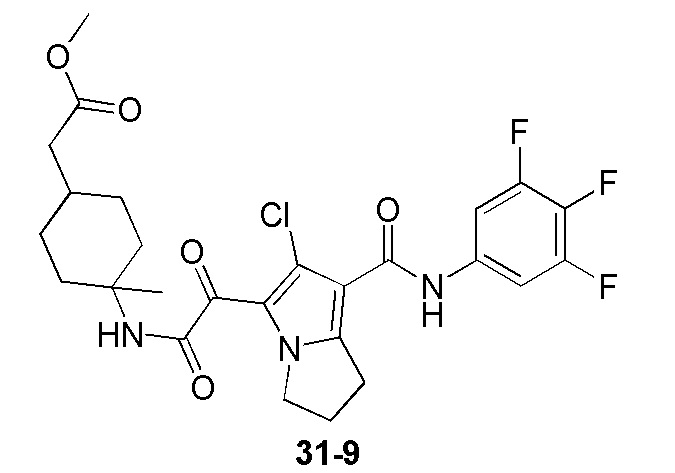
Соединение 27-10 (0,5 г, 1,29 ммоль, 1 экв.) растворяли в N,N-диметилформамиде (10 мл), а затем добавляли гексафторфосфат бензотриазол-1-илокситрипирролидинфосфония (1,08 г, 2,07 ммоль, 1,6 экв.), 1-гидроксибензотриазол (297,00 мг, 2,20 ммоль, 1,7 экв.) и N,N-диизопропилэтиламин (668,41 мг, 5,17 ммоль, 900,82 мкл, 4 экв.). Полученную в результате смесь перемешивали при 20°C в течение 0,5 ч, а затем в нее добавляли соединение 31-8 (287,44 мг, 1,55 ммоль, 1,2 экв.). Реакционную систему перемешивали при 40°C в течение 15,5 ч. Результаты LCMS и HPLC подтверждали, что реакция завершилась. Реакционный раствор выливали в воду (50 мл) для остановки реакции, а затем фильтровали. Фильтрат экстрагировали этилацетатом (20 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Неочищенный продукт очищали с помощью колоночной хроматографии (градиентное элюирование: простой петролейный эфир : этилацетат = от 100:1 до 3:1, об./об.). Затем неочищенный продукт отделяли с помощью SFC (время удержания = 2,35 мин) (колонка: DAICEL CHIRALPAKIC (250 мм × 30 мм, 5 мкм); подвижная фаза: Neu-MeOH; % B: 40% - 40%, 10 мин) с получением соединения 31-9. MS (ESI) m/z: 554,2 [M+H]+.
Стадия 7
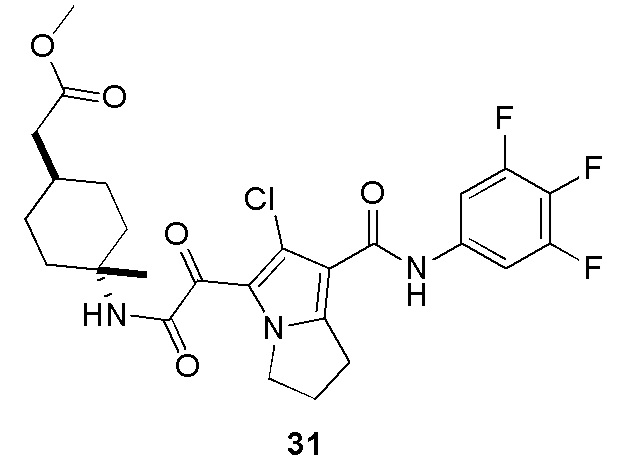
Соединение 31-9 (0,1 г, 180,52 мкмоль, 1 экв.) очищали с помощью препаративной HPLC (нейтральная система, колонка: Xtimate C18 150 мм × 25 мм × 5 мкм; подвижная фаза: вода (содержащая 10 мМ бикарбоната аммония)-ацетонитрил; % содержания ацетонитрила: 50% - 80%, 10,5 мин) с получением соединения 31. MS (ESI) m/z: 554,2 [M+H]+.
1H ЯМР (400 MГц, DMSO-d6) δ = 10,01 (s, 1H), 8,30 (s, 1H), 7,66-7,58 (m, 2H), 4,28 (t, J=7,6 Гц, 2H), 3,58 (s, 3H), 3,08 (t, J=7,6 Гц, 2H), 2,49-2,43 (m, 2H), 2,26 (d, J=7,0 Гц, 2H), 1,88-1,56 (m, 7H), 1,36 (s, 3H), 1,24-1,11 (m, 2H).
Экспериментальный пример 1: кПЦР-тест в in vitro тесте на HBV
1. Цель эксперимента:
Оценить ингибирующее действие соединения на HBV путем детектирования содержания ДНК HBV в клетках HepG2.2.15 в кПЦР-тесте в режиме реального времени с ЕС50 соединения в качестве индикатора.
2. Материалы, использованные в ходе эксперимента:
2.1. Линия клеток: клетки HepG2.2.15
Среда для культивирования клеток HepG2.2.15 (DMEM/F12, Invitrogen-11330057; 10% сыворотка, Invitrogen-10099141; 100 единиц/мл пенициллина и 10 мкг/мл стрептомицина, Invitrogen-15140122; 1% незаменимых аминокислот, Invitrogen-11140076; 2 мМ L-глутамин, Invitrogen-25030081; 300 мкг/мл генетицина, Invitrogen-10131027)
2.2. Реактивы:
Панкреатин (Invitrogen-25300062)
DPBS (Hyclone-SH30028.01B)
DMSO (Sigma-D2650-100ML)
Набор для высокоэффективной очистки ДНК (QIAamp 96 DNA Blood Kit, Qiagen-51162)
Реактив с универсальным зондом для количественного определения faststart (FastStart Universal Probe Master, Roche-04914058001)
2.3. Расходные материалы и приборы:
96-луночный планшет для культивирования клеток (Corning-3599)
CO2-термостат (HERA-CELL-240)
Оптическая герметизирующая пленка (ABI-4311971)
96-луночный планшет для количественной ПЦР (Applied Biosystems-4306737)
Прибор для флуоресцентной количественной ПЦР (система для ПЦР в режиме реального времени Applied Biosystems-7500)
3. Стадии эксперимента и способы:
3.1. Клетки HepG2.2.15 (4×104 клеток/лунка) инокулировали в 96-луночный планшет и культивировали в течение ночи при температуре 37°C в атмосфере 5% CO2.
3.2. На 2-е сутки соединение разводили в 3-кратном градиенте с получением итого 8 градиентов разведения. В лунки планшета для культивирования в двух повторностях добавляли соединение в различных концентрациях. Конечная концентрация DMSO в среде для культивирования составляла 1%. В качестве контроля 100% ингибирования использовали 1 мкM GLS4 (структура GLS4, раскрытая в публикации WO2008154817A1, имеет следующий вид:  в качестве контроля 0% ингибирования использовали 1% DMSO.
в качестве контроля 0% ингибирования использовали 1% DMSO.
3.3. На 5-е сутки среду для культивирования заменяли свежей средой для культивирования, содержащей соединение.
3.4. На 8-е сутки собирали среду для культивирования в лунках с культурой и использовали набор для высокоэффективной очистки ДНК (Qiagen-51162) для экстракции ДНК. Конкретные стадии см. в руководстве по применению продукта.
Приготовление реакционного раствора для ПЦР показано в таблице 1.
Таблица 1. Приготовление реакционного раствора для ПЦР
Последовательность прямого праймера: GTGTCTGCGGCGTTTTATCA
Последовательность обратного праймера: GACAAACGGGCAACATACCTT
Последовательность зонда: 5' + FAM + CCTCTKCATCCTGCTGCTATGCCTCATC + TAMRA - 3'
3.6. В каждую лунку 96-луночного планшета для ПЦР вносили 15 мкл реакционной смеси, а затем 10 мкл образца ДНК или стандарта ДНК HBV.
3.7. Реакционные условия при ПЦР: нагревание до 95°C в течение 10 минут, разрушение при 95°C в течение 15 секунд и элонгация при 60°C в течение 1 минуты, итого 40 циклов.
3.8. Анализ данных:
3.8.1. Расчет процента ингибирования: % инг. = [1 - (количество копий ДНК в образце - количество копий ДНК в 1 мкM GLS4)/(количество копий ДНК в контроле с DMSO - количество копий ДНК в 1 мкM GLS4)]×100.
3.8.2. Расчет ЕС50: полумаксимальную ингибирующую концентрацию (ЕС50) соединения для HBV рассчитывали с применением программного обеспечения GraphPad Prism.
3.8.3. Результаты экспериментов показаны в таблице 2.
Таблица 2. Результаты по EC50 согласно кПЦР-тесту
(титульное соединение)
(EC50) HBV
Экспериментальный пример 2: тест на ингибирование калиевого канала hERG
1. Цель эксперимента:
Оценить влияние раскрываемого в настоящем документе соединения на калиевый канал hERG с использованием способа автоматической фиксации потенциала.
2. Способ проведения эксперимента
2.1. Культура клеток
Используемыми в эксперименте клетками, стабильно экспрессирующими калиевый канал hERG, были CHO-hERE от компании Aviva Biosciences. CHO-hERG культивировали при температуре 37°C в атмосфере 5% CO2. Среда для культивирования CHO hERG представлена в таблице 3.
Таблица 3. Среда для культивирования CHO hERG
2.2. Предварительная подготовка клеток
Клетки CHO-hERG, использованные в данном эксперименте, культивировали в течение по меньшей мере двух суток, и плотность клеток достигала 75% или более. Перед проведением эксперимента клетки отщепляли посредством TrypLE, а затем собранные клетки ресуспендировали во внеклеточной жидкости.
2.3. Приготовление внутриклеточной и внеклеточной жидкости
Внеклеточную жидкость нужно было готовить один раз в месяц. Внутриклеточную жидкость необходимо было замораживать аликвотами при -20°C. Составы внутриклеточной и внеклеточной жидкости показаны в таблице 4.
Таблица 4. Составы внутриклеточной и внеклеточной жидкости
2.4. Получение соединения
Тестируемое соединение и его положительный контроль под названием амитриптилин растворяли в DMSO с получением исходного раствора с определенной концентрацией. Затем исходный раствор разводили до различных градиентов и, наконец, добавляли во внеклеточную жидкость в определенном соотношении, необходимом для разведения до концентрации для теста. Перед началом эксперимента невооруженным глазом проверяли наличие осадка. Наконец, концентрация DMSO в подлежащем тестировании растворе и положительном контроле под названием амитриптилин не должна была превышать 0,3%.
2.5. Схема стимуляции напряжением
При исходном потенциале -80 мВ стимуляцию напряжением -50 мВ подавали в течение 80 мс для регистрации значения остаточного тока клетки; затем канал hERG открывали деполяризацией до +20 мВ в течение 4800 мс, возбуждали следовой ток hERG реполяризацией до -50 мВ в течение 5000 мс и регистрировали данные; и, наконец, напряжение восстанавливали до исходного потенциала -80 мВ и поддерживали в течение 3100 мс. Вышеописанную стимуляцию напряжением повторяли каждые 15000 мс.
2.6. Регистрация данных в ходе фиксации потенциала у цельных клеток с помощью технологии QPatchHTX
Эксперимент с hERG с помощью QPatchHTX проводили при комнатной температуре. Схема применения цельных клеток, схема стимуляции напряжением и схема детектирования соединений были созданы с помощью программного обеспечения QPatch Assay Software 5.2 (Sophion Bioscience).
Сначала проводили 30 повторяющихся заданных стимуляций напряжением, в результате чего данный участок на спектре тока принимали за базовый участок для последующего анализа. Затем вносили 5 мкл внеклеточной жидкости и стимуляцию напряжением повторяли три раза. Последовательно в объеме 5 мкл вносили каждое соединение в действующей концентрации и три раза повторяли стимуляцию напряжением. Клетки в течение по меньшей мере 5 минут инкубировали с соединением в каждой тестируемой концентрации. В течение всего процесса регистрации результатов все индикаторы должны были соответствовать стандарту приемлемости для анализа данных. Если отсутствовало соответствие требованиям стандарта, клетку не включали в диапазон анализа, а соединение необходимо было протестировать снова. Вышеупомянутый процесс регистрации результатов выполнялся автоматически аналитическим программным обеспечением Qpatch. Тестируемыми концентрациями соединений были 0,24 мкМ, 1,20 мкМ, 6,00 мкМ и 30,00 мкМ, и каждую концентрацию дублировали по меньшей мере для двух клеток.
2.7. Анализ данных
В каждой полной регистрации данных тока процент ингибирования у каждого соединения в действующей концентрации можно рассчитать, исходя из процента пикового тока в отрицательном контроле. Кривую зависимости доза-эффект получали путем аппроксимации с помощью стандартного уравнения Хилла, а само уравнение выглядит следующим образом:
I(C) = Ib + (Ifr - Ib) × cn/(IC50n + cn)
C означает тестируемую концентрацию соединения, n означает угловой коэффициент, I означает ток
Аппроксимацию кривой и расчет степени ингибирования полностью производили с помощью аналитического программного обеспечения Qpatch. Если степень ингибирования превышала 50% при наиболее низкой концентрации или степень ингибирования не достигала 50% при наиболее высокой концентрации, соответствующая IC50 соединения была ниже наиболее низкой концентрации или больше наиболее высокой концентрации.
2.8. Результаты теста
В таблице 5 представлены значения IC50 hERG у соединений из примеров.
Таблица 5. Значения IC50 hERG у соединений из примеров
Экспериментальный пример 3: эксперимент по ингибированию изозимов цитохрома Р450
Цель эксперимента: определить ингибирующее действие тестируемого соединения на активность микросомальных изозимов цитохрома P450 печени человека (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4).
Порядок проведения эксперимента: сначала тестируемое соединение (10 мМ) градиентно разводили для приготовления рабочих растворов (100× конечная концентрация) со следующими концентрациями: 5, 1,5, 0,5, 0,15, 0,05, 0,015 и 0,005 мМ, и одновременно готовили рабочие растворы положительных ингибиторов изозимов P450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4) и смеси их специфических субстратов (5 в 1); микросомы печени человека, замороженные в холодильнике при температуре -80°C, размораживали на льду, а после размораживания микросомы печени человека разводили в PB для приготовления рабочего раствора с конкретной концентрацией (0,253 мг/мл); 20 мкл смеси субстратов (в контрольную лунку вносили 20 мкл PB) и 158 мкл рабочего раствора микросом печени человека вносили в реакционный планшет, который затем помещали на лед для использования; затем в соответствующую лунку вносили 2 мкл тестируемого соединения в каждой концентрации (N=1) и специфический ингибитор (N=2) и вносили группу без ингибитора (тестируемое соединение или положительный ингибитор) с соответствующим органическим растворителем в качестве контрольного образца (контрольный образец тестируемого соединения представлял собой 1:1 DMSO : МеОН; положительный контрольный образец представлял собой 1:9 DMSO : МеОН); после предварительной инкубации на водяной бане при 37°C в течение 10 минут в реакционный планшет вносили 20 мкл раствора дигидролипоамиддегидрогеназы (NADPH) и инкубировали на водяной бане при 37°C в течение 10 мин, для прекращения реакции добавляли 400 мкл холодного раствора ацетонитрила (внутренний стандарт представлял собой 200 нг/мл толбутамида и лабеталола) и реакционный планшет помещали на шейкер и встряхивали в течение 10 минут; после центрифугирования на скорости 4000 об/мин в течение 20 минут собирали 200 мкл надосадочной жидкости, и добавляли к 100 мкл воды для разведения образца, и, наконец, планшет герметично закрывали, перемешивали покачиванием, равномерно встряхивали и подвергали детекции с помощью LC/MS/MS. Результаты экспериментов показаны в таблице 6.
Таблица 6. Результаты ингибирующего действия тестируемого соединения на активность изозимов цитохрома P450 микросом печени человека
Экспериментальный пример 4: эксперимент на цитотоксичность
1. Соединение разводили посредством DMSO (диметилсульфоксида) в 3-кратном градиенте для получения 9 концентраций и вносили в 96-луночный планшет в двух повторностях. Концентрация соединения в 200 раз превышала конечную тестируемую концентрацию.
2. Клетки промывали один раз посредством PBS (фосфатно-солевого буферного раствора), вносили 0,25% трипсина и расщепляли в течение примерно 2-5 минут в термостате с атмосферой 5% CO2 при температуре 37°C. Затем расщепление останавливали посредством среды для культивирования клеток, а клетки разделяли на отдельные клетки путем пипетирования пипеткой.
3. Плотность клеток подсчитывали с помощью счетчика клеток и доводили до требуемой плотности с помощью среды.
4. Клетки вносили в 96-луночный планшет, в который было внесено соединение. Конечная концентрация DMSO в каждой лунке составляла 0,5%. Лунки, содержащие 0,5% DMSO, использовали в качестве нетоксичных отрицательных контролей, а лунки, содержащие среду для культивирования клеток, использовали в качестве контролей со 100% цитотоксичностью. Затем планшет для клеток течение 3 суток инкубировали в термостате для инкубирования клеток в атмосфере 5% CO2 при температуре 37°C.
5. Сигнал хемилюминесценции (RLU — относительная единица хемилюминесценции) каждой лунки в планшете для культивирования клеток детектировали с помощью набора для детекции жизнеспособности клеток CellTiter-Glo с применением многофункционального микропланшетного ридера Envision согласно инструкциям, прилагаемым к набору.
6. Для расчета жизнеспособности клеток в каждой лунке (% жизнеспособности клеток) необработанные данные (RLU) подставляли в следующую формулу:
Жизнеспособность клеток, % =
RLUобразца: значение сигнала из лунки с образцом; Среднее RLUклеточного контроля: среднее значение сигнала из лунки с клеточным контролем; Среднее RLUсредового контроля: среднее значение сигнала из лунки со средовым контролем.
7. С помощью программного обеспечения GraphPad Prism данные по жизнеспособности клеток нелинейно экстраполировали для построения кривой зависимости дозы от ответа и получали значение 50% цитотоксической концентрации (CC50) для соединения. Результаты показаны в таблице 7.
Таблица 7. Результаты теста 50% цитотоксической концентрации (CC50)
Экспериментальный пример 5: эксперимент по изучению микросомальной стабильности in vitro
Метаболическая стабильность соединения в микросомах печени человека и CD-1 мышей
Цель эксперимента: оценить метаболическую стабильность соединения в микросомах печени человека и CD-1 мышей
Процедуры, проводимые в ходе эксперимента: сначала тестируемое соединение (10 мМ) подвергали двухэтапному разведению, при этом соединение разводили до промежуточной концентрации 100 мкМ посредством 100% метанола, а рабочий раствор разводили до 10 мкМ калий-фосфатным буфером; подготавливали восемь 96-луночных инкубационных планшетов, которым соответственно присваивали названия «Т0», «Т5», «Т10», «Т20», «Т30», «Т60», «холостой» и «NCF60»; моментами времени прохождения реакции, соответствующими первым 6 инкубационным планшетам, были соответственно 0, 5, 10, 20, 30 и 60 минут; в случае холостого планшета не добавляли ни тестируемое соединение, ни контрольное соединение, а в случае планшета NCF60 использовали калий-фосфатный буфер при инкубации в течение 60 минут вместо раствора системы регенерации NADPH; в планшеты T0, T5, T10, T20, T30, T60 и NCF60 вносили 10 мкл рабочего раствора с тестируемым соединением и 80 мкл рабочего раствора с микросомами (концентрация белка микросом печени составляла 0,625 мг/мл), тогда как в холостой планшет вносили только рабочий раствор с микросомами, а затем все инкубационные планшеты помещали в водяную баню с температурой 37°C для предварительной инкубации в течение приблизительно 10 минут; после предварительной инкубации для запуска реакции вносили 10 мкл рабочего раствора системы регенерации NADPH в каждую лунку с образцом всех планшетов, за исключением планшета NCF60 и планшета T0, а в каждую лунку планшета NCF60 вносили 10 мкл калий-фосфатного буфера; таким образом, в образцах с тестируемым соединением или контрольным соединением конечные концентрации соединения, тестостерона, диклофенака и пропафенона в реакционной смеси составляли 1 мкМ, концентрация микросом печени составляла 0,5 мг/мл, а конечные концентрации DMSO и ацетонитрила в реакционной системе соответственно составляли 0,01% (об./об.) и 0,99% (об./об.); после инкубации в течение соответствующего времени (например, 5, 10, 20, 30 и 60 минут) для остановки реакции в каждую лунку с образцом вносили 300 мкл стоп-раствора (раствора ацетонитрила, содержащего 100 нг/мл толбутамида и 100 нг/мл лабеталола); в планшет T0 вносили 300 мкл стоп-раствора, а затем 10 мкл рабочего раствора NADPH, все планшеты с образцами перемешивали встряхиванием и центрифугировали на центрифуге (3220×g) в течение 20 минут, а затем из каждой лунки отбирали 100 мкл надосадочной жидкости и разводили посредством 300 мкл чистой воды для анализа в системе жидкостная хроматография - тандемная масс-спектрометрия.
Результаты экспериментов показаны в таблице 8.
Таблица 8. Результаты по метаболической стабильности теста соединения в микросомах печени человека и CD-1 мышей
.
Метаболическая стабильность соединения в микросомах печени SD крыс, собак породы бигль и яванских макаков
Цель эксперимента: оценить метаболическую стабильность тестируемых соединений 18 и 25 в микросомах печени крыс, собак породы бигль и яванских макаков.
Процедуры, проводимые в ходе эксперимента: сначала тестируемое соединение (10 мМ) подвергали двухэтапному разведению, при этом соединение разводили до промежуточной концентрации 100 мкМ посредством 100% метанола, а рабочий раствор разводили до 10 мкМ калий-фосфатным буфером; подготавливали восемь 96-луночных инкубационных планшетов, которым соответственно присваивали названия «Т0», «Т5», «Т10», «Т20», «Т30», «Т60», «холостой» и «NCF60»; моментами времени прохождения реакции, соответствующими первым 6 инкубационным планшетам, были соответственно 0, 5, 10, 20, 30 и 60 минут; в случае холостого планшета не добавляли ни тестируемое соединение, ни контрольное соединение, а в случае планшета NCF60 использовали калий-фосфатный буфер при инкубации в течение 60 минут вместо раствора системы регенерации NADPH; в планшеты T0, T5, T10, T20, T30, T60 и NCF60 вносили 10 мкл рабочего раствора с тестируемым соединением и 80 мкл рабочего раствора с микросомами (концентрация белка микросом печени составляла 0,625 мг/мл), тогда как в холостой планшет вносили только рабочий раствор с микросомами, а затем все инкубационные планшеты помещали в водяную баню с температурой 37°C для предварительной инкубации в течение приблизительно 10 минут; после предварительной инкубации для запуска реакции вносили 10 мкл рабочего раствора системы регенерации NADPH в каждую лунку с образцом всех планшетов, за исключением планшета NCF60 и планшета T0, а в каждую лунку планшета NCF60 вносили 10 мкл калий-фосфатного буфера; таким образом, в образцах с тестируемым соединением или контрольным соединением конечные концентрации соединения, тестостерона, диклофенака и пропафенона в реакционной смеси составляли 1 мкМ, концентрация микросом печени составляла 0,5 мг/мл, а конечные концентрации DMSO и ацетонитрила в реакционной системе соответственно составляли 0,01% (об./об.) и 0,99% (об./об.); после инкубации в течение соответствующего времени (например, 5, 10, 20, 30 и 60 минут) для остановки реакции в каждую лунку с образцом вносили 300 мкл стоп-раствора (раствора ацетонитрила, содержащего 100 нг/мл толбутамида и 100 нг/мл лабеталола); в планшет T0 вносили 300 мкл стоп-раствора, а затем 10 мкл рабочего раствора NADPH, все планшеты с образцами перемешивали встряхиванием и центрифугировали на центрифуге (3220×g) в течение 20 минут, а затем из каждой лунки отбирали 100 мкл надосадочной жидкости и разводили посредством 300 мкл чистой воды для анализа в системе жидкостная хроматография - тандемная масс-спектрометрия.
Результаты экспериментов показаны в таблице 9.
Таблица 9. Результаты по метаболической стабильности тестируемого соединения в микросомах печени SD крыс, собак породы бигль и яванских макаков
Экспериментальный пример 6: фармакокинетическое исследование
Фармакокинетическое исследование тестируемого соединения на Balb/c мышах путем перорального введения и внутривенной инъекции:
Тестируемое соединение смешивали с раствором, содержащим диметилсульфоксид (10%), полиэтиленгликоль 400 (60%) и воду (30%), и данную смесь встряхивали на вортексе и обрабатывали ультразвуком до получения 0,2 мг/мл прозрачного раствора, который фильтровали через фильтр Millipore для более позднего использования. Самкам Balb/c мышей возрастом 7-10 недель внутривенно вводили раствор соединения-кандидата дозой 1 мг/кг.
Тестируемое соединение смешивали с водным раствором, содержащим 10% полиоксиэтиленстеарата, и данную смесь встряхивали на вортексе и обрабатывали ультразвуком до получения 0,3 мг/мл прозрачного раствора для более позднего использования. Самкам Balb/c мышей возрастом 7-10 недель перорально вводили раствор соединения-кандидата дозой 3 мг/кг.
Собирали цельную кровь и получали плазму крови. Концентрацию лекарственного средства анализировали с помощью LC-MS/MS, а фармакокинетические параметры рассчитывали с помощью программного обеспечения Phoenix WinNonlin. Результаты показаны в таблице 10.
Таблица 10. Результаты фармакокинетического исследования тестируемого соединения
Экспериментальный пример 7: эксперимент по изучению соотношения содержания в печени и крови у мышей
Эксперимент по изучению соотношения содержания в печени и крови у Balb/c мышей, которым перорально вводили тестируемое соединение.
Соединение 18 смешивали с водным раствором, содержащим 10% гидроксистеарата полиэтиленгликоля-15, и данную смесь встряхивали на вортексе и обрабатывали ультразвуком до получения 0,3 мг/мл однородной суспензии для более позднего использования. Самкам Balb/c мышей возрастом 7-10 недель перорально вводили раствор соединения-кандидата дозой 3 мг/кг.
В определенный момент времени собирали цельную кровь и получали плазму крови. В соответствующий момент времени собирали ткани печени для приготовления гомогената тканей. Концентрацию лекарственного средства анализировали с помощью LC-MS/MS, а фармакокинетические параметры рассчитывали с помощью программного обеспечения Phoenix WinNonlin.
Соединение 25 смешивали с водным раствором, содержащим 10% гидроксистеарата полиэтиленгликоля-15, и данную смесь встряхивали на вортексе и обрабатывали ультразвуком до получения 0,3 мг/мл прозрачного раствора для более позднего использования. Самкам Balb/c мышей возрастом 7-10 недель перорально вводили раствор соединения-кандидата дозой 3 мг/кг.
В определенный момент времени собирали цельную кровь и получали плазму крови. В соответствующий момент времени собирали ткани печени для приготовления гомогената тканей. Концентрацию лекарственного средства анализировали с помощью LC-MS/MS, а фармакокинетические параметры рассчитывали с помощью программного обеспечения Phoenix WinNonlin. Результаты показаны в таблице 11.
Таблица 11. Соотношение содержания тестируемого соединения в печени и крови
Соотношение концентрации в печени/плазме крови
Экспериментальный пример 8: эксперимент по изучению эффективности in vivo
Модель HDI/HBV
Цель эксперимента: оценить эффективность соединения в отношении вируса гепатита В у мышей с помощью мышиной модели HDI/HBV.
Приготовление соединения: растворитель представлял собой водный раствор, содержащий 10% гидроксистеарата полиэтиленгликоля-15; определенное количество тестируемых соединений 18 и 25 раздельно растворяли в водном растворе, содержащем 10% гидроксистеарата полиэтиленгликоля-15, и полученные смеси встряхивали на вортексе и обрабатывали ультразвуком до получения однородных суспензий. Суспензии хранили при температуре 4°C для более позднего использования.
Инъекция под высоким давлением раствора ДНК HBV в плазмиде через хвостовую вену мышей: день инъекции плазмиды приходился на 0-е сутки, спустя сутки после инъекции шли 1-е сутки и так далее. На 0-е сутки всем мышам вводили физиологический раствор, содержащий 10 мкг плазмидной ДНК, через их хвостовые вены дозой 8% от массы тела, данную инъекцию совершали за 5 секунд.
Способ введения: всем мышам внутрижелудочное введение производили дважды в сутки (интервал 8/16 ч) на 1-6-е сутки и один раз на 7-е сутки. Всех мышей умерщвляли во второй половине дня на 7-е сутки. Масса тела мышей, которую отслеживали каждые сутки, оставалась неизменной на протяжении всего эксперимента.
Сбор образцов: кровь собирали из поднижнечелюстной вены через 4 часа после первого введения утром на 1-е, 3-е и 5-е сутки. Все образцы крови собирали в пробирки с антикоагулянтом K2-ЭДТА и центрифугировали в течение 10 минут при температуре 4°C, 7000g, с получением приблизительно 40 мкл плазмы крови. Спустя четыре часа после введения утром на 7-е сутки всех мышей умерщвляли при помощи CO2. Забирали кровь из сердца, а способ получения плазмы был таким же, как указано выше. Забирали две ткани печени, по 70-100 мг каждой, и подвергали быстрой заморозке жидким азотом. После взятия всех образцов их хранили в холодильнике с температурой -80°C для определения содержания ДНК HBV.
Анализ образцов: все образцы плазмы крови и печени подвергали детекции в отношении ДНК HBV с помощью количественной ПЦР.
Результаты эксперимента: результаты эксперимента показаны в таблице 12, на фигуре 1 и на фигуре 2.
Таблица 12
Δ Log10 копий/мкл
Δ Log10 копий/100 нг
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
| АНТАГОНИСТ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2019 |
|
RU2832735C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ПИРИМИДИНЫ И ИХ ВАРИАНТЫ, И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2760733C2 |
| ГЛУТАРИМИДНОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ КОЛЬЦОМ, КОНДЕНСИРОВАННЫМ С ФУРАНОМ | 2022 |
|
RU2836921C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
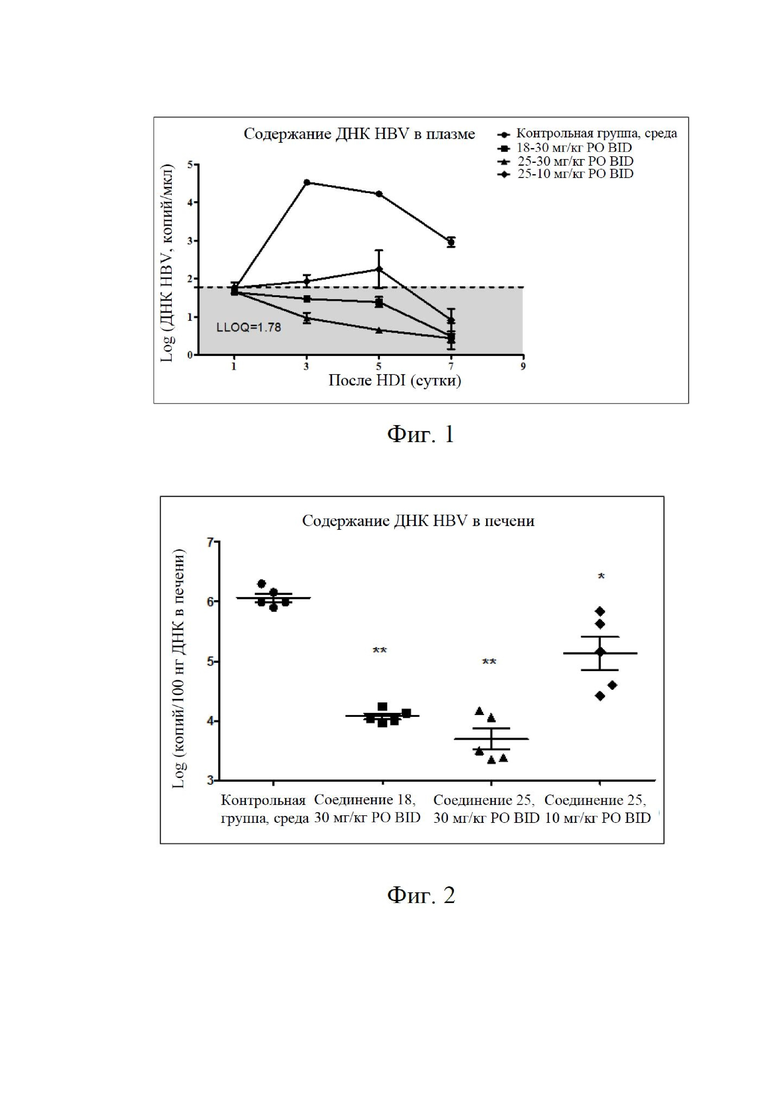
Изобретение относится к 2,3-дигидро-1H-пирролизин-7-формамидному производному в качестве ингибитора нуклеопротеинов. Технический результат: получены новые соединения формулы (II), которые могут найти применение при получении лекарственного средства для лечения заболеваний, связанных с HBV. 4 н. и 16 з.п. ф-лы, 12 табл., 39 пр., 2 ил.
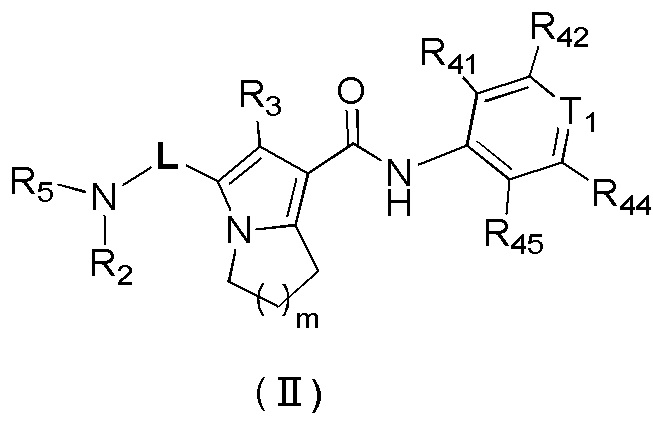
1. Соединение, представленное формулой (II), или его фармацевтически приемлемая соль:
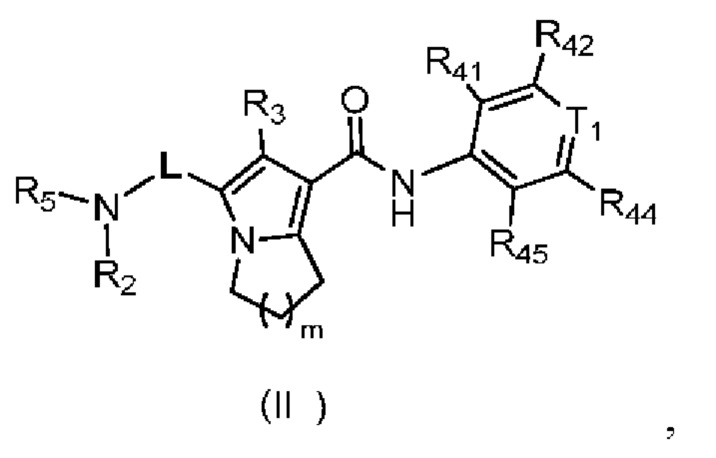
где
m равно 1;
L выбран из 
Т1 выбран из группы, состоящей из N и С (R43);
R2 представляет собой Н;
R3 выбран из группы, состоящей из Cl, F, Br, I и С1-6-алкила, причем С1-6-алкил имеет 1, 2 или 3 заместителя Rc;
каждый из R41 и R45 независимо выбран из группы, состоящей из Н;
каждый из R42, R43 и R44 независимо выбран из группы, состоящей из Н, Cl, F, Br, I, CN и С1-3-алкила, причем С1-3-алкил необязательно имеет 1, 2 или 3 заместителя Rd;
R5 выбран из группы, состоящей из R51, С3-8-циклоалкила и 5-6-членного гетероциклоалкила, причем 5-6-членный гетероциклоалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из группы, состоящей из -NH-, -О- и N, причем С3-8-циклоалкил и 5-6-членный гетероциклоалкил необязательно имеют 1, 2 или 3 заместителя R1;
R51 выбран из группы, состоящей из С1-7-алкила и С1-3-гетероалкила, причем С1-3-гетероалкил содержит 1, 2, 3 или 4 гетероатома, независимо выбранные из группы, состоящей из -NH-, -О- и N, причем С1-7-алкил и С1-3-гетероалкил необязательно имеют 1, 2 или 3 заместителя Re;
каждый R1 независимо выбран из группы, состоящей из Н, Cl, F, Br, I, ОН, CN, СООН, C1-3-алкила, -СОО-С1-4-алкила и -С1-3-алкил-СОО-С1-4-алкила, причем C1-3-алкил, -СОО-С1-4-алкил и -С1-3-алкил-СОО-С1-4-алкил необязательно имеют 1, 2 или 3 заместителя Ra;
каждый Ra независимо выбран из группы, состоящей из Cl, F, Br, I, CN, СООН, С1-3-алкила и С1-3-алкокси, причем С1-3 необязательно имеет 1, 2 или 3 заместителя R;
каждый Rc независимо выбран из группы, состоящей из Cl, F, Br и I;
каждый Rd независимо выбран из группы, состоящей из Cl, F, Br и I;
каждый Re независимо выбран из группы, состоящей из Cl, F, Br, I, ОН, NH2, CN и СООН;
каждый R независимо выбран из группы, состоящей из Cl, F, Br, I, ОН, NH2, CN и СООН.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Ra выбран из группы, состоящей из Cl, F, Br, I, CN, СООН и -ОСН3.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где Rc выбран из группы, состоящей из Cl, F и Br.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый R1 независимо выбран из группы, состоящей из Н, Cl, F, ОН, CN, СООН, С1-3-алкила, -СОО-С1-3-алкила и -С1-3-алкил-СОО-С1-3-алкила, причем С1-3-алкил, -СОО-С1-3-алкил и -С1-3-алкил-СОО-С1-3-алкил необязательно имеют 1, 2 или 3 заместителя Ra.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый R1 независимо выбран из группы, состоящей из Н, Cl, F, ОН, CN, СООН, СН3, Et,  причем СН3, Et,
причем СН3, Et,  необязательно имеют 1, 2 или 3 заместителя Ra.
необязательно имеют 1, 2 или 3 заместителя Ra.
6. Соединение или его фармацевтически приемлемая соль по п. 5, где каждый R1 независимо выбран из группы, состоящей из Н, Cl, F, ОН, CN, СООН, СН3, CF3, Et, -CH2-COOH, -СН2-ОСН3, -(СН2)2-СООН, 
7. Соединение или его фармацевтически приемлемая соль по п. 1, где R3 выбран из группы, состоящей из Cl, F и С1-3алкила, причем С1-3алкил необязательно имеет 1, 2 или 3 заместителя Rc.
8. Соединение или его фармацевтически приемлемая соль по п. 7, где R3 выбран из группы, состоящей из Cl, F, СН3, CF3 и Et.
9. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый из R42, R43 и R44 независимо выбран из группы, состоящей из Н, Cl, F и CN.
10. Соединение или его фармацевтически приемлемая соль по п. 1, где R51 выбран из группы, состоящей из метила, этила, пропила, изопропила, 
 причем метил, этил, пропил, изопропил,
причем метил, этил, пропил, изопропил,  и
и  необязательно имеют 1, 2 или 3 заместителя Re.
необязательно имеют 1, 2 или 3 заместителя Re.
11. Соединение или его фармацевтически приемлемая соль по п. 10, где R51 выбран из группы, состоящей из  и
и 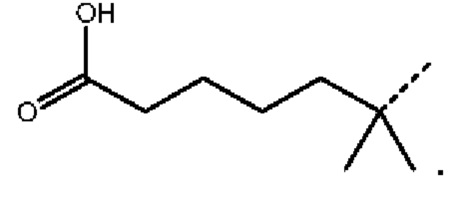
12. Соединение или его фармацевтически приемлемая соль по п. 1, где R5 выбран из группы, состоящей из R51, циклогексила, тетрагидропиранила, пиперидинила и бицикло[2.2.2]октила, причем циклогексил, тетрагидропиранил, пиперидинил и бицикло[2.2.2]октил необязательно имеют 1, 2 или 3 заместителя R1.
13. Соединение или его фармацевтически приемлемая соль по п. 12, где R5 выбран из группы, состоящей из R51, 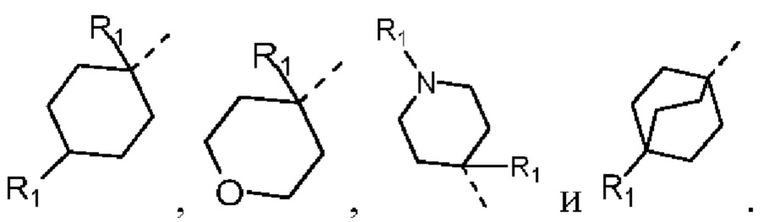
14. Соединение или его фармацевтически приемлемая соль по п. 13, где R5 выбран из группы, состоящей из 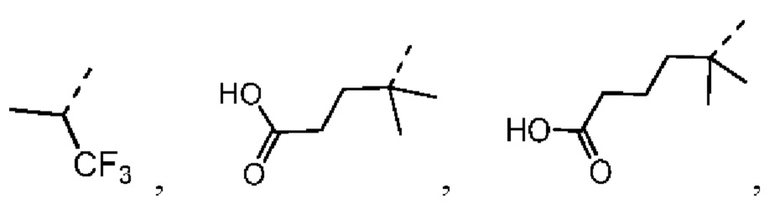
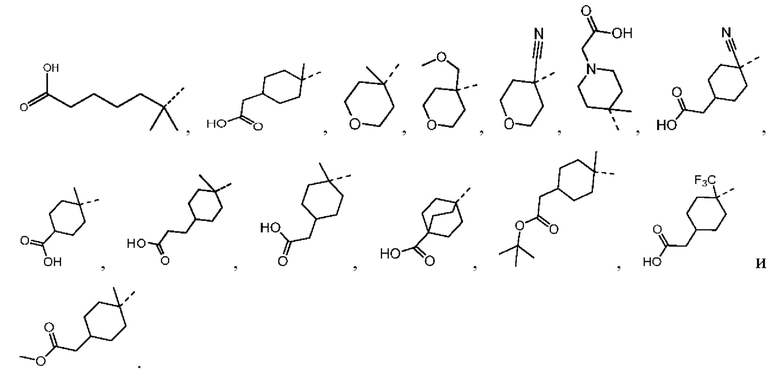
15. Соединение или его фармацевтически приемлемая соль по п. 1, где структурная единица 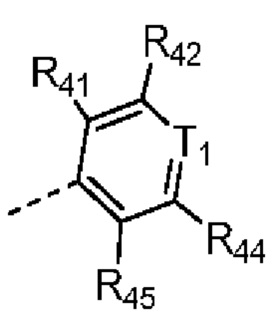 выбрана из группы, состоящей из
выбрана из группы, состоящей из 
16. Соединение, представленное следующими формулами, его стереоизомер или их фармацевтически приемлемая соль:
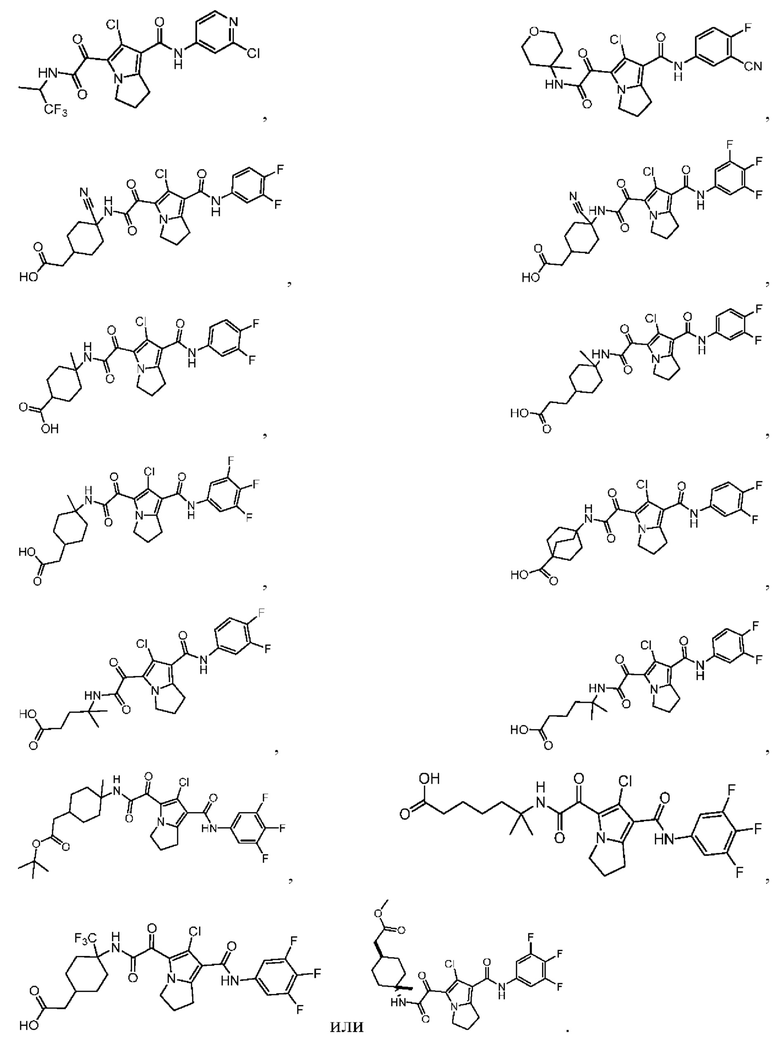
17. Соединение, его стереоизомер или их фармацевтически приемлемая соль по п. 16, выбранные из группы, состоящей из
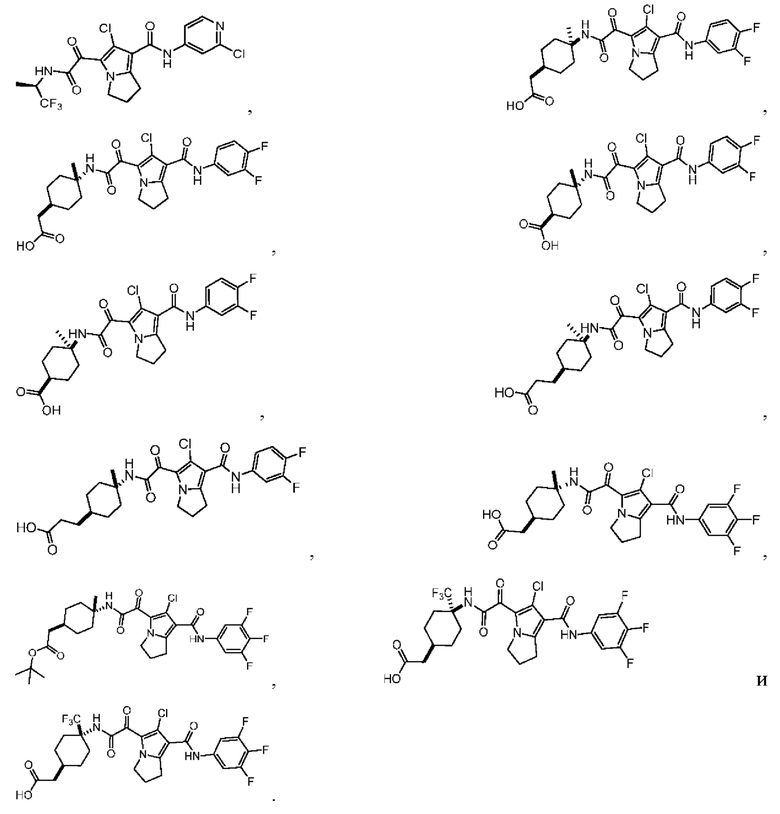
18. Фармацевтическая композиция для лечения или предупреждения заболеваний, связанных с инфекцией HBV, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-17.
19. Фармацевтическая композиция для лечения или предупреждения заболеваний, связанных с инфекцией HBV, по п. 18, дополнительно содержащая фармацевтически приемлемый носитель.
20. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-17 в качестве ингибитора нуклеопротеинов для лечения или предупреждения заболеваний, связанных с инфекцией HBV.
| WO2018039531A1, 01.03.2018 | |||
| CN101743246A, 16.06.2010 | |||
| АННЕЛИРОВАННЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ПРОТОНОВОГО НАСОСА ДЛЯ ЛЕЧЕНИЯ ЯЗВЫ | 2003 |
|
RU2336872C2 |

