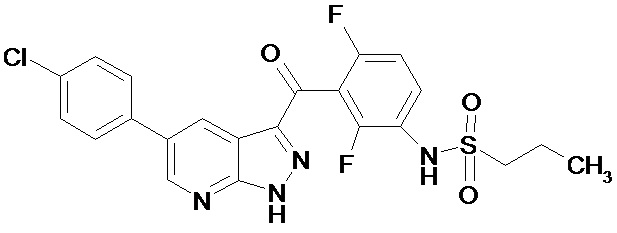
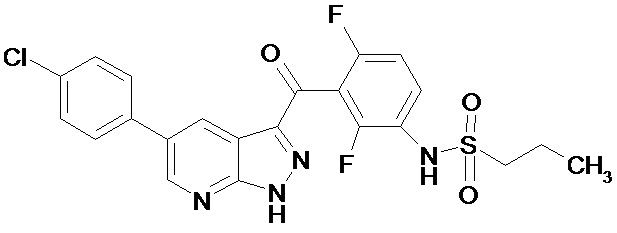
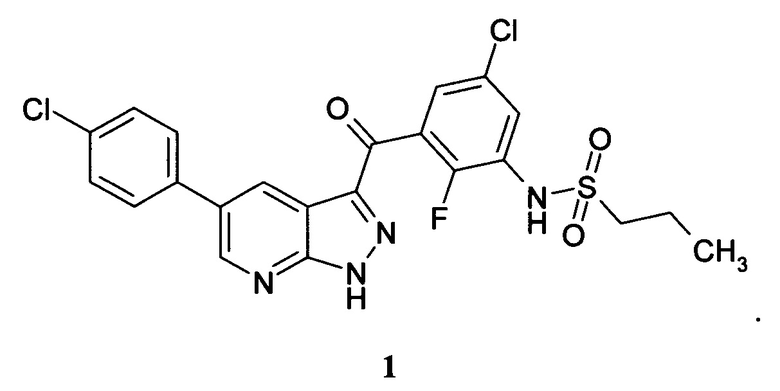
Изобретение относится к новому ингибитору BRAF киназы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамиду формулы 1, новой кристаллической форме, фармацевтической композиции и лекарственному средству для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, предпочтительно, меланомы.
Меланома - одна из наиболее злокачественных опухолей, способна быстро распространяться по организму лимфогенным и гематогенным путем. При метастазировании меланомы перспективы излечения очень безрадостны. Применение химиотерапии, хирургии и лучевой терапии может иногда несколько улучшить качество жизни больного, но это не продлевает жизнь больше, чем всего лишь на несколько месяцев. Медиана выживаемости при метастазирующей меланоме составляет 6-7 месяцев. Стандартной схемы лечения нет.
При наличии множественных метастазов меланомы (генерализация процесса) обычно предлагают провести регионарную или (и) системную химиотерапию дакарбазином (DTIC) или кармустином (BCNU), ломустином (CCNU), цисплатином, тамоксифеном или циклофосфаном. В среднем же подсчитано, что химиотерапия способна продлить жизнь пациента лишь на 2-3% в сравнении с тем сроком, который он бы прожил вовсе без какого-либо лечения.
В настоящее время меланома - один из наиболее распространенных видов рака у молодых людей в возрасте от 15 до 34 лет. Для пациентов с неоперабельной и метастатической меланомой прогноз остается неутешительным - однолетнее выживание в 25,5% случаев и пятилетнее - менее чем в 15%. Примерно 40-60% всех случаев меланомы содержат мутации BRAF в позиции 600, из них наиболее распространенные - V600E (~80%) и V600K (~20%). Они вызывают постоянную активацию сигнального пути RAF-MEK-ERK, что приводит к стимуляции клеточного роста, пролиферации и выживания в отсутствии ростовых факторов.
Идея таргетной терапии в онкологии состоит в создании лекарственного препарата, который точно поражает заданную цель - одну определенную молекулу, которая может быть ответственна либо за развитие рака, либо за формирование злокачественных признаков раковых клеток, либо поддерживающая возможность существования злокачественной опухоли в организме больного. За счет таргетного препарата в раковой клетке повреждается ключевое молекулярное звено - одна деталь (молекула) с прицелом на то, что эта поломка будет значимой и разрушит более сложную структуру - раковую клетку и, соответственно, инактивирует злокачественную опухоль. В итоге будет достигнут лечебный эффект - спасение больного раком.
Стратегия блокировки протеина - продукта протоонкогена BRAF, известного как протеин киназа (serine/threonine-protein kinase BRAF), принадлежит англичанам Ричарду Марэ и Кэролайн Спрингер. Клетка для того, чтобы активироваться и начать делиться, должна получить специальный сигнал извне. Далее сигнал передается по так называемому сигнальному пути - это каскад биохимических реакций, сопровождающих передачу внешнего сигнала с мембранного рецептора на поверхности клеточной мембраны к ядру. Передача сигналов может идти по короткой или длинной цепи (через активацию другого каскада), быть прямой или непрямой. Клеточный сигнальный путь Ras/Raf/MEK/ERK - ключевой регулятор клеточной пролиферации и выживания. Мутации вдоль всего этого пути были идентифицированы в меланоме. Активирующие мутации ведут к повышенной клеточной пролиферации и устойчивости к запрограммированной клеточной гибели. Ras-протеин присоединен к внутренней клеточной мембране, a Raf, MEK и ERK - цитозольные протеины. Ras-мутации наблюдаются у 10-20% пациентов с меланомой и могут активировать Raf и P13K (фосфоинозитид-3 киназа)-каскады.
Гены и белки BRAF были открыты около 30 лет назад. Более 13000 публикаций, связанных с изучением их молекулярной функции, роли в регуляции биологических процессов, как в норме, так и при патологиях, было опубликовано в электронной библиотеке PubMed, что говорит о важности изучения белков RAF. Тем не менее, существующие пробелы в знаниях о функции и регуляции RAF-белков белок ограничивают нашу способность ингибировать их неконтролируемую активность при лечении так смертельных заболеваний, как онкологические заболевания [Lavoie, Н. and М. Therrien, Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell. Biol, 2015. 16(5): p. 281-98.].
BRAF (B-raf или Raf киназа В) - это белок из семейства Raf киназ, главная роль которых - фосфорилирование киназ митоген активированных протеинкиназ MEK1/2 и последующей активции ERK1/2. Различные факторы роста, воспалительные стимулы, хемокины и другие факторы могут активировать BRAF.
BRAF принимает участие в различных нормальных и патологических процесах, таких как деление клеток, перестройки актинового цитоскелета, клеточная адгезия и миграция. BRAF/MEK/ERK путь в различных тканях может регулировать самые разнообразные процессы, такие как сокращение гладкомышечных клеток сосудов, в нервной ткани может принимать участие в регуляции синапсов, долговременной потенциации и депрессии, а также задействован в цитотоксичности натуральных киллеров при воспалении.
Мутантный конститутивно активный белок BRAF V600 был найден во многих опухолях с различной частотой встречаемости. Он является перспективной мишенью для таргетирования ингибиторами и, таким образом, лечения онкологических заболеваний.
Выявление мутации в гене BRAF послужило основанием для разработки нового направления таргентных препаратов - ингибиторов патологических белков, продуцируемых под воздействием мутантного гена.
В 2011 году был разрешен к применению в онкологии новейший таргетный препарата для терапии меланомы - «Вемурафениб», разработанный компанией Plexxikon (US) и выпускаемый под торговой маркой «Зелбораф» компанией Ф. Хоффманн-Ля Рош Лтд. (Швейцария) [RU 2418800, ПИРРОЛО[2,3-В]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ, 20.05.2011].
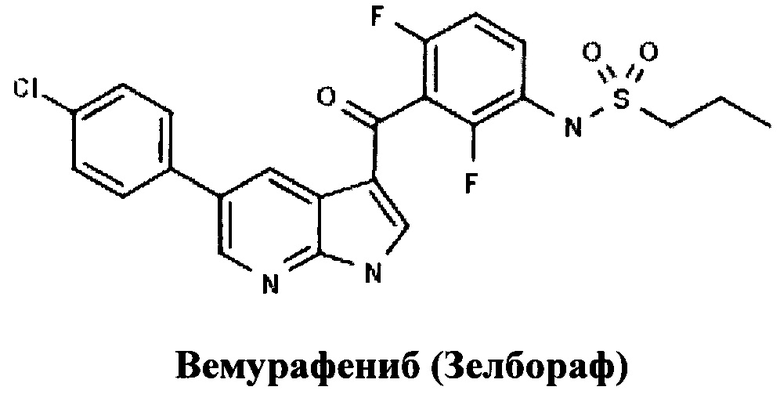
В международном многоцентровом рандомизированном открытом контролируемом исследовании III фазы BRIM3 проводилась оценка эффективности и безопасности Вемурафениба по сравнению с Дакарбазином у больных неоперабельной местно-распространенной или метастатической меланомой с мутацией гена BRAF V600, ранее не получавших лечения. В исследование было включено 675 пациентов. В данном исследовании было показано, что по сравнению с химиотерапией Вемурафениб на 63% снижает риск смерти (отношение рисков [HR]=0,37; p<0,0001) и, кроме того, на 74% (HR=0,26; p<0,0001) уменьшает риск ухудшения течения заболевания (выживаемость без прогрессирования - вторая основная цель исследования). Ответ на терапию (процент пациентов, опухоль у которых уменьшилась) в группе больных, получавших Вемурафениб (48,4%), был почти в девять раз больше, чем в группе пациентов, которые получали химиотерапию (5,5%; р<0,0001). Через шесть месяцев 84% больных из группы Вемурафениба были живы в сравнении с 64% из группы химиотерапии. Увеличение общей выживаемости, выживаемости без прогрессирования и уменьшение опухоли при применении Вемурафениба наблюдались у пациентов независимо от возраста, пола или факторов риска заболевания. Медиана общей выживаемости в группе пациентов, получавших Вемурафениб, составила 10,5 мес., в то время как общая выживаемость пациентов, получавших химиотерапию, составила 7,8 мес.
Как было показано, существующие и одобренные к применению ингибиторы (вемурафениб и дебрафениб) вызывают развитие вторичной резистентности, поэтому подавляющее количество больных перестает отвечать на терапию через 6-7 месяцев. В ряде случаев назначают комбинацию ингибиторов BRAF V600 и MEK.
Также, препараты данной группы не лишены множества отмеченных в клинических исследованиях побочных эффектов и являются чрезвычайно дорогостоящими. Создание эффективного отечественного лекарственного средства с действием на основе ингибирования BRAF энзима даст возможность повысить эффективность лечения меланомы в Российской Федерации.
Возможно, ингибиторы BRAF V600 нового поколения с новым механизмом действия сумеют преодолеть проблемы вторичной резистентности и низкой эффективности ингибиторов при других нозологиях
Определение мутации BRAF V600 имеет клиническое значение, т.к. является предиктором ответа на терапию ингибитором онкогенной Raf киназы В.
Поэтому поиск новых действующих веществ, обладающих свойством ингибитора Raf киназы В, способов их получения, новых лекарственных и пролекарственных средств на их основе, для терапии метастатической меланомы является актуальной и важной задачей на сегодняшний день.
Так известно соединение PLX8394, которое является селективным ингибитором BRAF, находится в фазе I/II клинических испытаний компании Plexxikon (US) для лечения больных с твердыми опухолями или ворсистых клеток лейкемии [WO 2012109075, COMPOUNDS AND METHODS FOR KINASE MODULATION, AND INDICATIONS THEREFOR, 16.08.2012].
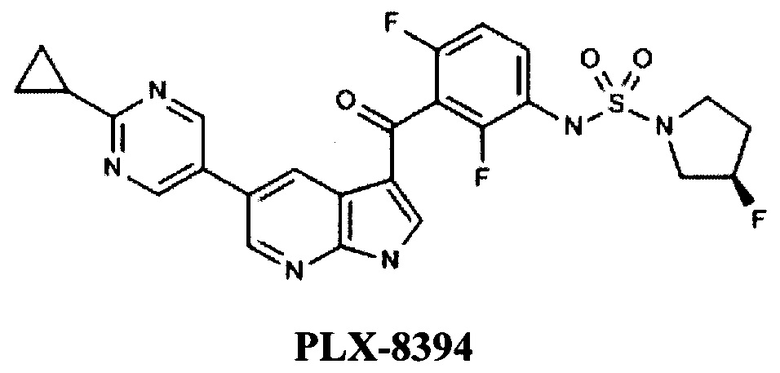
Среди наиболее известных ингибиторов Raf киназы В (включая мутацию V600E), выпущенных на рынок или находящихся в стадии активных исследований представлены различные класс химических соединений.
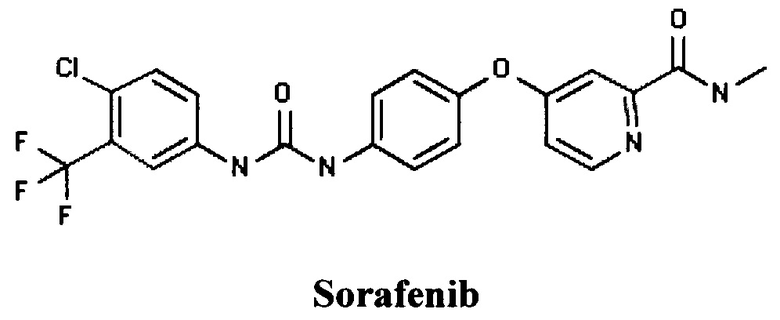
Известны бифенилы компании Bayer Pharma AG (DE), например, Сорафениб (RU 2319693, ПРОИЗВОДНЫЕ МОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, СВЯЗАННОГО С РОСТОМ РАКОВЫХ КЛЕТОК (ВАРИАНТЫ), 20.03.2008].
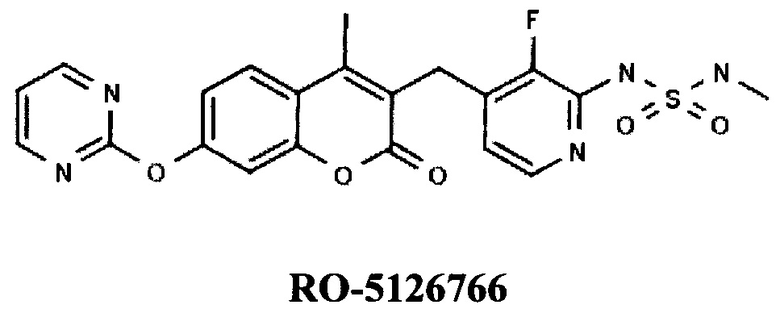
Известны пиримидиниокси-оксобензопираны компаний Roche (US) и Chugai Pharmaceutical (JP), например, RO-5126766 (RU 2428420, НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, 10.09.2011].
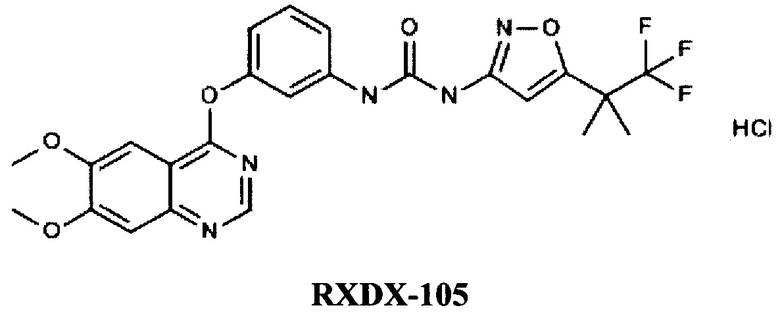
Известны хиназолиновые производные компании AMBIT BIOSCIENCES CORPORATION (US), например, RXDX-105 [EA 019350, ПРОИЗВОДНЫЕ ХИНАЗОЛИНА КАК МОДУЛЯТОРЫ RAF-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ, 31.03.2014].
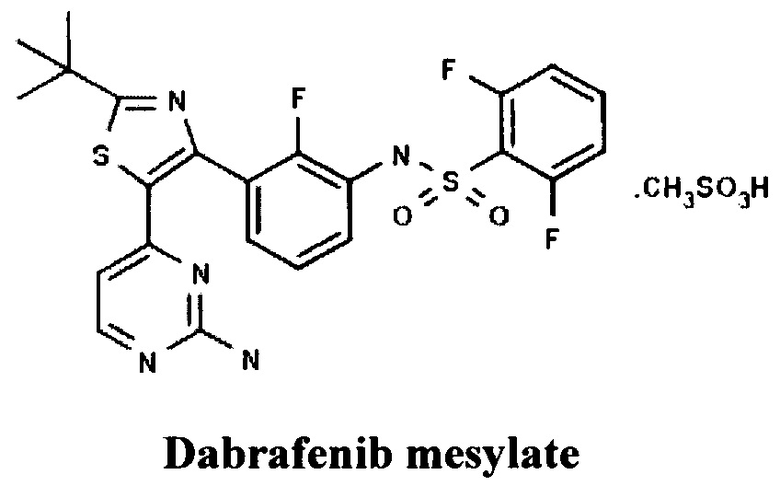
Известны фенилсульфонамидтиазольные производные компании GlaxoSmithKline (US), например, известный препарат для терапии метастатической меланомы Дабрафениб [ЕА 019349, СОЕДИНЕНИЯ БЕНЗОЛСУЛЬФОНАМИДТИАЗОЛА И ОКСАЗОЛА, 31.03.2014].
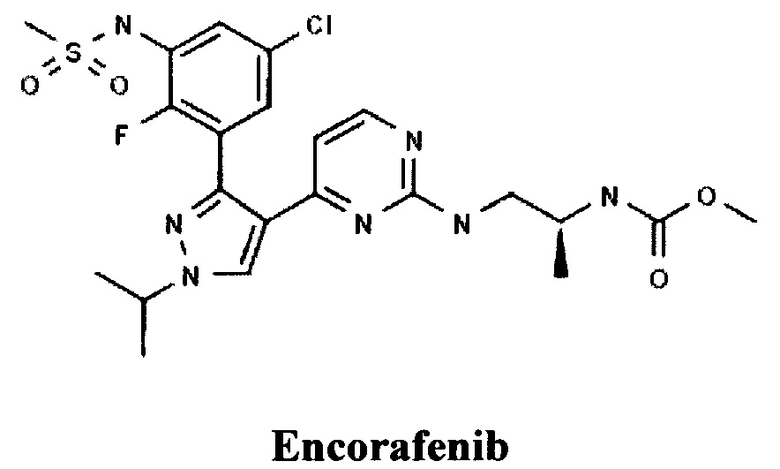
Известны пиразолилпиримидиновые производные компании NOVARTIS AG (СН), например, Энкорафениб [ЕР 2470526, COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS, 28.05.2014].
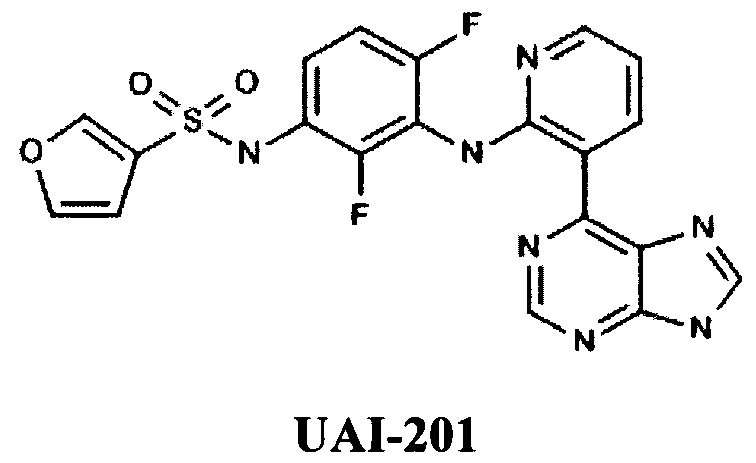
Известны новые пуринилпиридиниламино-2,4-дифторфенилсульфонамид производные компании Youai (KR), например, UAI-201 [RU 2550038, НОВОЕ ПРОИЗВОДНОЕ ПУРИНИЛПИРИДИНИЛАМИНО-2,4-ДИФТОРФЕНИЛСУЛЬФОНАМИДА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ RAF-КИНАЗЫ, СОДЕРЖАЩАЯ ДАННОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА, 10.05.2015].
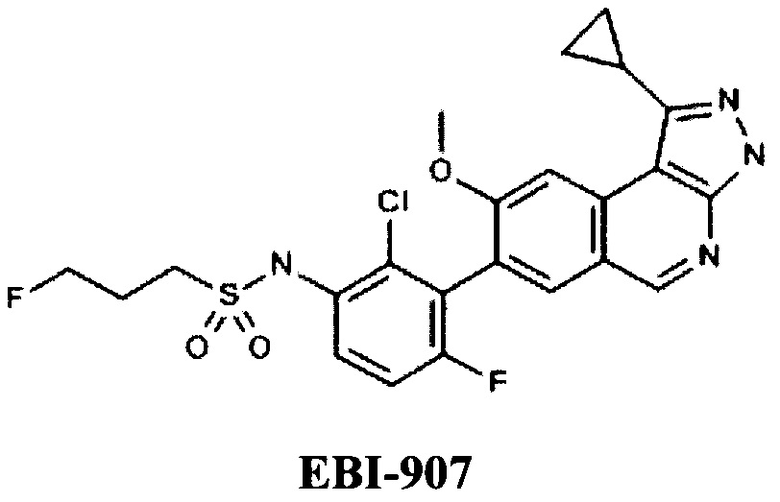
Из пиразолопиримидинов известны производные компании Eternity Bioscience (US), например, EBI-907 [WO 2014043296, AMINOISOQUINOLINE DERIVATIVES AS PROTEIN KINASE INHIBITORS, 20.03.2014].
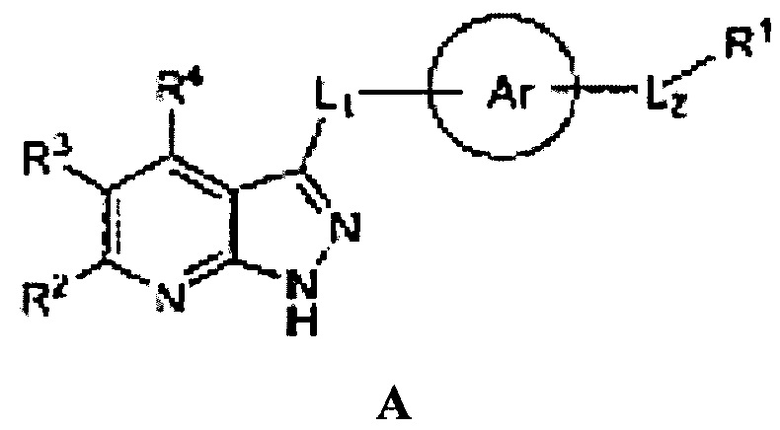
Наиболее близкими к настоящему изобретению являются пиразолопиримидины общей формулы А,
где Ar выбран из группы
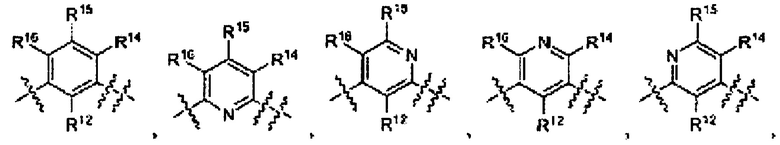
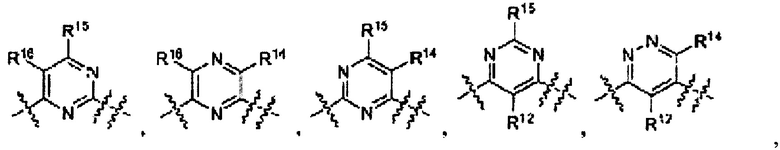
L1 выбран из группы -C(R5R6)-, -С(O)-, -C(S)-, -N(R7)-, -О-, -S-, -S(O)-, и -S(O)2-;
L2 выбран из группы -N(RS)-C(O)-, -N(R8)-C(S)-, -N(RS)-S(O)-, -N(R8)-S(O)2-, -N(R8)-C(O)-K(RS)-, -N(RS)-C(S)-N(R8)-, и -N(R8)-S(O)2-N(R8)- и т.д. [WO 2010111527, PYRAZOLO[3,4-B] PYRIDINES AS KINASE INHIBITORS AND THEIR MEDICAL USE, 30.09.2010], которые также известны как ингибиторы хотя бы одной Raf киназы.
Однако при этом среди соединений общей формулы А, не описан N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамид. Ни его свойства, ни кристаллическая форма его неизвестны. В отношении киназной активности в вышеуказанном источнике эксперимент проводился всего с тремя соединениями.
Поэтому N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамид является новым соединением. Необходимо выяснить, способно ли данное соединение кристаллизоваться и иметь кристаллические формы. Затем, если кристаллические формы существуют, необходимо найти воспроизводимые способы получения химически стабильных кристаллических форм и выявить новые кристаллические формы данного соединения.
Стабильность в твердом состоянии и срок хранения активных компонентов являются очень важными факторами. Лекарственное соединение и композиции, включающие его, должны обладать способностью эффективно храниться в течение значительных периодов времени, не проявляя значительного изменения физико-химических свойств активного компонента (например, его химического состава, плотности, гигроскопичности и растворимости). Кроме того, также является важным представить лекарственный препарат в форме, которая является как можно более чистой. В этом отношении аморфные соединения могут представлять значительные проблемы. Например, с такими соединениями труднее обращаться и включать их в лекарственные формы по сравнению с кристаллическим соединением, за счет сомнительной растворимости, и часто оказывается, что они нестабильны и химически загрязнены. Специалисту в данной области будет понятно, что, если лекарственный препарат можно легко получать в стабильной кристаллической форме, то можно решить вышеуказанные проблемы. Обычно невозможно предсказать только на основе молекулярной структуры, каким будет поведение соединения при кристаллизации, и обычно это можно установить только эмпирически.
Таким образом, задача настоящего изобретения заключается в получении нового ингибитора BRAF киназы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамида и нахождении его новых стабильных кристаллических форм.
Технический результат настоящего изобретения заключается в улучшенных свойствах данного соединения при его использовании, в частности более высокая скорость растворения и повышенная стабильность при хранении.
В результате работы было обнаружено, что N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамид формулы 1 обладает выраженной активностью по отношению к клеточным линиям, экспрессирующим BRAF.
Данный ингибирующий эффект был подтвержден в ходе реакции фосфорилирования внеклеточной сигналрегулирующей киназы и клеточной антипролиферации в доступных клеточных линиях меланомы, экспрессирующих ген BRAF с мутациями V600.
Поставленная задача и технический результат достигаются новым ингибитором BRAF киназы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамид формулы 1
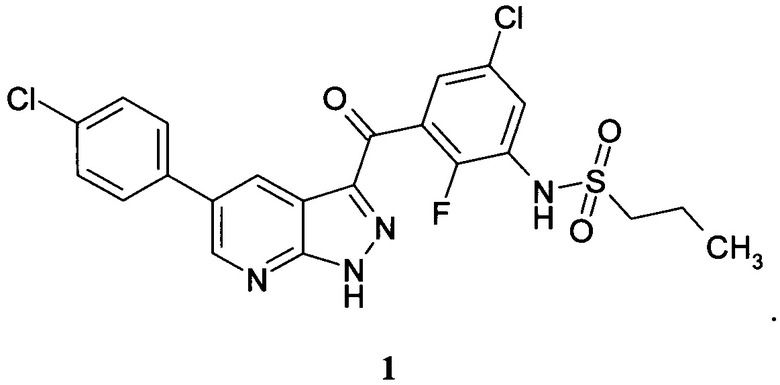
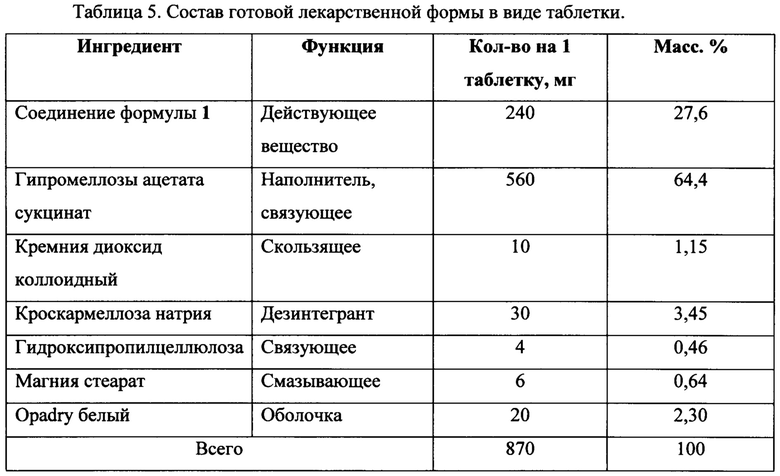
Предметом настоящего изобретения является новая кристаллическая форма соединения N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, имеющая на порошковой рентгеновской дифрактограмме характеристические пики при следующих углах 2θ (±0,2°): 8,9; 13,0; 14,5; 16,4; 18,5; 19,6; 21,4; 22,3; 24,3; 25,4; 28,0.
Предметом данного изобретения также является активный компонент, обладающий свойством ингибитора BRAF киназы, представляющий собой соединение формулы 1 или его новую кристаллическую форму, или его фармацевтически приемлемую соль.
Предметом данного изобретения также является фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, содержащая в эффективном количестве активный компонент, представляющий собой соединение формулы 1 или его новую кристаллическую форму или его фармацевтически приемлемую соль.
Более предпочтительной является фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, в которой мутацией BRAF является мутация V600E.
Более предпочтительной является фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, где пролиферативное заболевание представляет собой меланому.
Более предпочтительной является фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, где пролиферативное заболевание представляет собой колоректальную аденокарциному.
Фармацевтическая композиция может включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с активным компонентом по настоящему изобретению может включать и другие активные ингредиенты, при условии, что они не вызывают нежелательных эффектов, например, аллергических реакций. При необходимости использования фармацевтических композиций по настоящему изобретению в клинической практике они могут смешиваться для изготовления различных форм, при этом они могут включать в свой состав традиционные фармацевтические носители; например, пероральные формы (такие как, таблетки, желатиновые капсулы, пилюли, растворы или суспензии); формы для инъекций (такие как, растворы или суспензии для инъекций, или сухой порошок для инъекций, который требует лишь добавления воды для инъекций перед использованием); местные формы (такие как, мази или растворы).
Носители, используемые в фармацевтических композициях по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Фармацевтическая композиции по настоящему изобретению может быть получена смешением активного компонента, Н-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамида, с инертным наполнителем, растворителем и/или другими фармацевтически приемлемыми эксципиентами.
Предметом настоящего изобретения также является лекарственное средство для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, в виде таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, содержащее в эффективном количестве соединение формулы 1 или его новую кристаллическую форму, или его фармацевтически приемлемую соль, или активный компонент, или фармацевтическую композицию по настоящему изобретению.
Более предпочтительным является лекарственное средство в виде таблетки, содержащей, масс. %:
Предметом настоящего изобретения является способ профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, согласно которому нуждающемуся субъекту вводят эффективное количество активного компонента, обладающего свойством ингибитора BRAF киназы, представляющий собой соединение формулы 1 или его новую кристаллическую форму, или его фармацевтически приемлемую соль, или фармацевтической композиции, или лекарственного средства по настоящему изобретению.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно, местно или ректально). Клиническая дозировка активного компонента (субстанции), фармацевтической композиции или лекарственного комбинированного средства, включающих фармацевтически эффективное количество активного компонента, у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 10~1500 мг, предпочтительно - 50~1200 мг. Поэтому во время приготовления фармацевтических композиций по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 10~1500 мг, предпочтительно 50~1200 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
BRAF (Raf киназа В) - белок, состоящий из 766 аминокислот, - ключевой элемент сигнального пути RAS-RAF, обеспечивающий рост и существование клеток.
«Азагетероцикл» означает ароматическую или неароматическую моноциклическую или полициклическую систему, содержащую в цикле, по крайней мере, один атом азота.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Ароматический цикл» (ароматическая система) означает планарную циклическую систему, в которой все атомы цикла участвуют в образовании единой системы сопряжения, включающей, согласно правилу Хюккеля, (4n+2) π-электронов (n - целое неотрицательное число). Примерами ароматических циклов являются бензол, нафталин, антрацен и т.п. В случае «гетероароматических циклов» в системе сопряжения участвуют π-электроны и р-электроны гетероатомов, их суммарное число также равняется (4n+2). Примерами таких циклов являются пиридин, тиофен, пиррол, фуран, тиазол и т.п. Ароматический цикл может иметь один или более «заместителей циклической системы» и может быть анелирован с неароматическим циклом, гетероароматической или гетероциклической системой.
«Гетероциклил» означает ароматическую или неароматическую насыщенную моноциклическую или полициклическую систему, включающую от 3 до 10 атомов углерода, преимущественно от 5 до 6 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера. Приставка «аза», «окса» или «тиа» перед гетероциклилом означает наличие в циклической системе, атома азота, атома кислорода или атома серы, соответственно. Гетероциклил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероциклиле могут быть окислены до N-оксида, S-оксида или S-диоксида. Представителями гетероциклилов являются пиперидинил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксан-2-ил, тетрагидрофуранил, тетрагидротиофенил и др.
«Гидрат» означает стехиометрическую или нестехиометрическую композицию соединения или его соли с водой.
«Заместитель» означает химический радикал, который присоединяется к скэффолду (фрагменту), например, «заместитель алкильный», «заместитель аминогруппы», «заместитель карбамоильный», «заместитель циклической системы».
«Ингибиторы» (лат. inhibere - задерживать) - общее название веществ, подавляющих или задерживающих течение физиологических и физико-химических (главным образом ферментативных) процессов, изучение ингибирования ферментов играет важную роль в создании лекарств, в изучении механизма действия и структуры ферментов.
«Лекарственное средство (препарат)» - комбинация нескольких лекарственных веществ для одновременного использования в виде таблеток, капсул, инъекций, мазей, ректальных суспензий и гелей и др. готовых форм, предназначенный для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего. Лекарственные вещества в одном комплекте могут быть представлены в виде различных готовых форм, предназначенных для введения в организм животного или человека различными способами, например перорально и ректально.
«Лиганды» (от латинского ligo - связывать) представляют собой химические вещества (малая молекула, неорганический ион, пептид, белок и прочее), способные взаимодействовать с рецепторами, которые трансформируют это взаимодействие в специфический сигнал.
«Рецепторы» (от латинского recipere - получать, узнавать) представляют собой биологические макромолекулы, расположенные на цитоплазматической мембране клетки или внутриклеточно, способные специфически взаимодействовать с ограниченным набором физиологически активных веществ (лигандов) и трансформировать сигнал об этом взаимодействии в определенный клеточный ответ.
«Сигнальный каскад» (сигнальная система, каскад передачи сигнала) означает совокупность взаимосвязанных последовательных и параллельных молекулярных процессов регуляции клеточного метаболизма внешними (первичными) сигналами, несущими в клетку информацию, что принципиально отличает их от других поступающих в клетку химических соединений, служащих для нее источником материи и энергии. Молекулярные механизмы передачи (трансдукции) внешних сигналов в клетку подразумевают не только передачу сигналов как таковую, но и весь комплекс событий, с ней сопряженных, в том числе усиление, ослабление и подавление (или выключение) сигналов.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как, консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Фармацевтические композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонко измельченным твердым носителем. Для изготовления суппозиториев помимо активных компонентов используют также масло какао, сплавы его с парафином и гидрогенизированными жирами, растительные и животные гидрогенизированные жиры, твердый жир, ланоль, сплавы гидрогенизированных жиров с воском, твердым парафином и другие основы, разрешенные для медицинского применения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные. (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Фармацевтически приемлемые эксципиенты» под фармацевтически приемлимыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители.
Также изобретение поясняется чертежами.
Фиг. 1. Порошковая рентгеновская дифрактограмма новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамид формулы 1, полученной согласно настоящему изобретению. Зависимость интенсивность от угла 2θ, °.
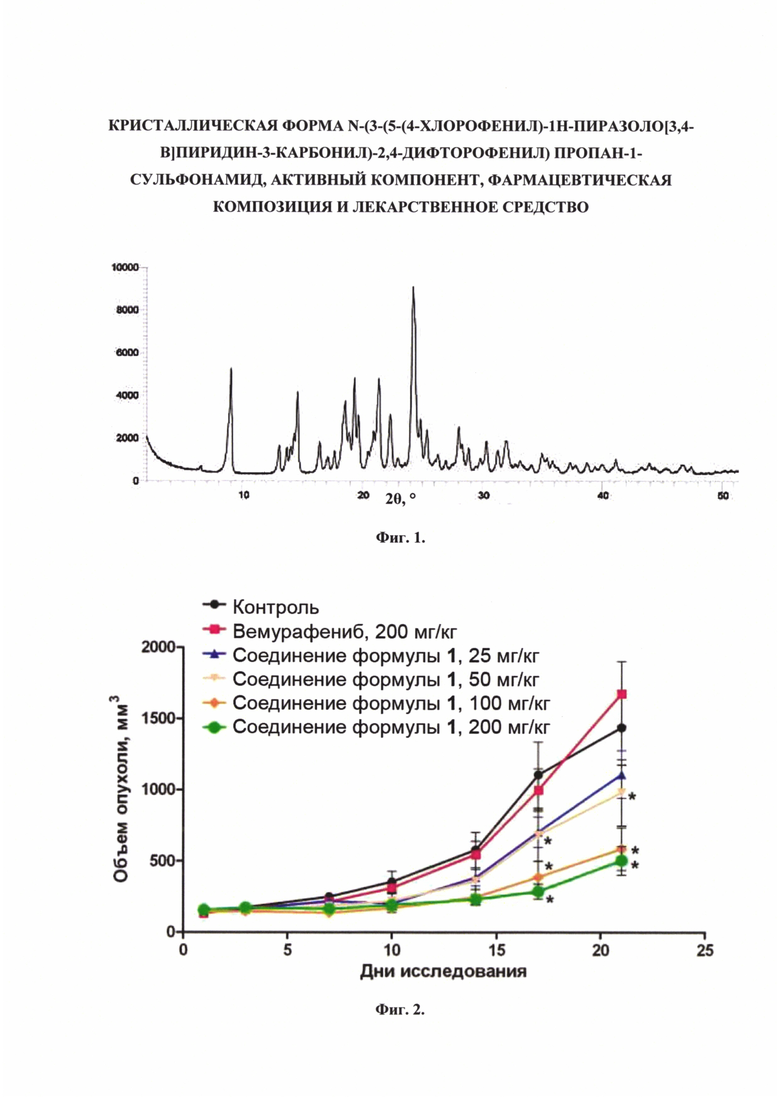
Фиг. 2. Влияние исследуемых веществ на рост опухоли клеточной линии А375 в модели ксенографтов * - р<0,05 по сравнению с группой «Контроль»).
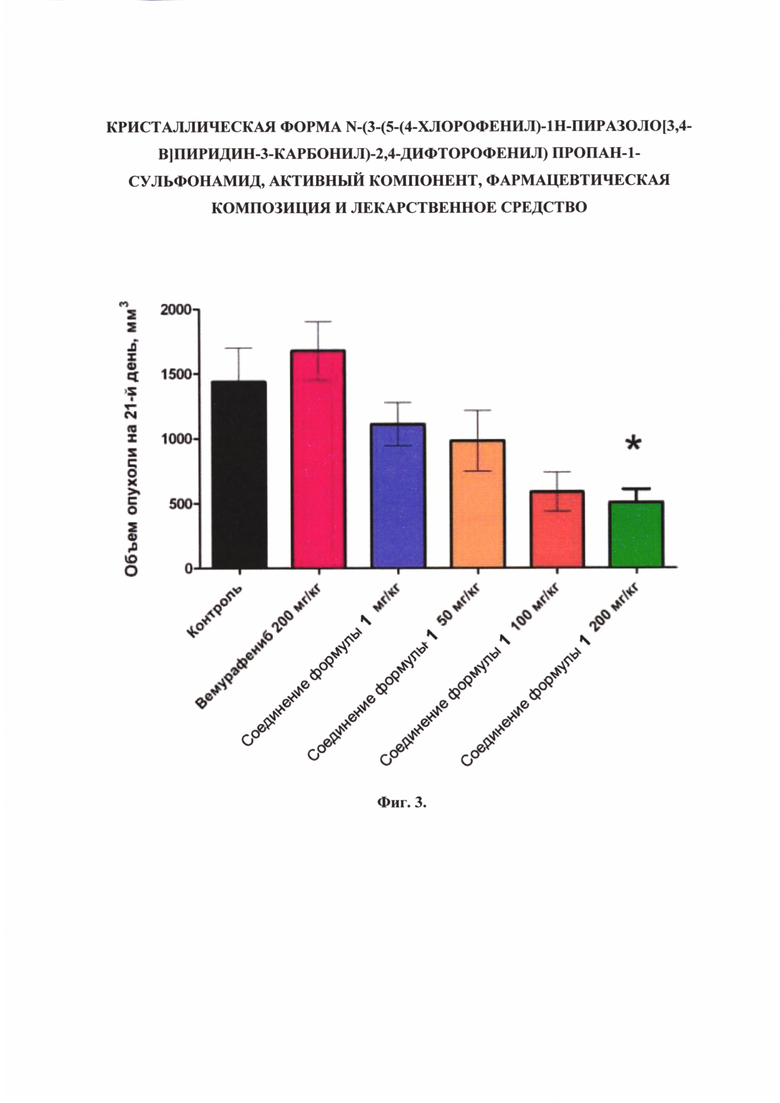
Фиг. 3. Объем опухоли на 21-й день исследований в модели ксенографтов A375.
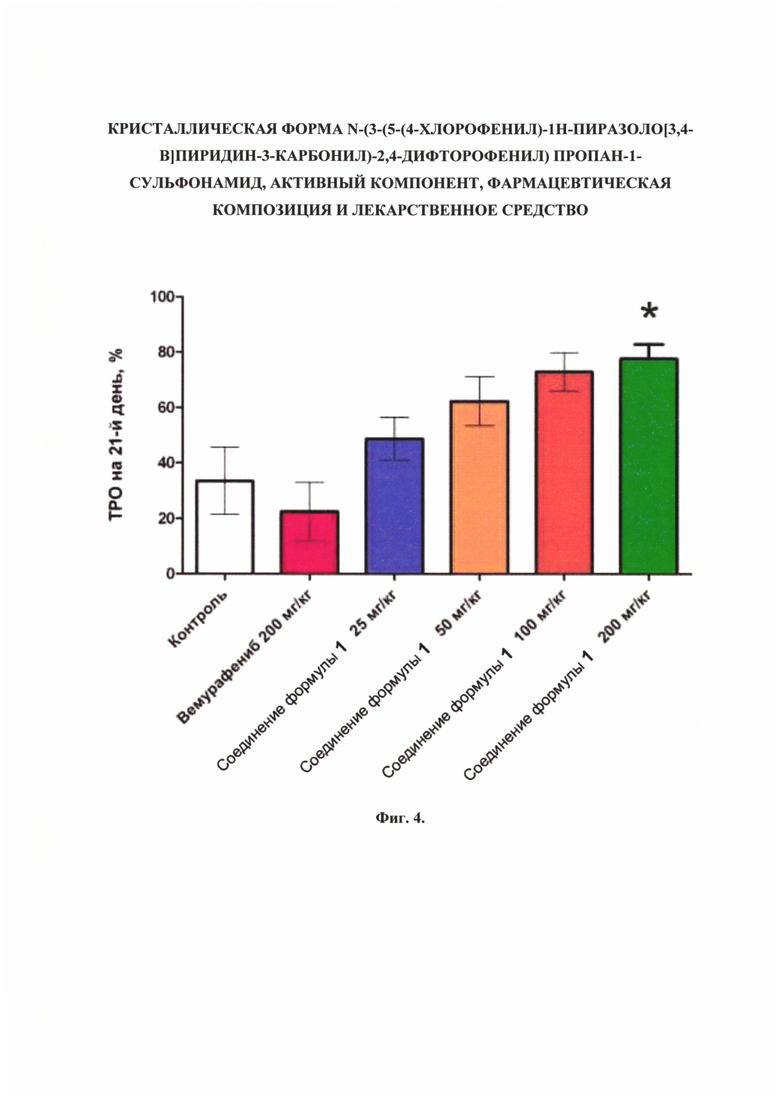
Фиг. 4. Торможение роста опухоли (ТРО) на 21-й день исследований роста опухолевых клеток линии А375 в модели ксенографтов (* - p<0,05 по сравнению с группой «Контроль»).
Представленные ниже примеры иллюстрируют, но не ограничивают изобретение.
Пример 1. Метод синтеза соединения формулы 1.
В круглодонной четырехгорлой колбе, продутой азотом, в 981 мл безводного тетрагидрофурана в токе азота (0,5-0,6 л/мин) растворяют 98,14 г 5-бромо-3-йодо-1-(4-метокси-бензил)-1Н-пиразоло[3,4-b]пиридина, полученный раствор охлаждают до -40°С, добавляют 118 мл 2М раствора изопропилмагний хлорида в тетрагидрофуране. Через 20 минут в круглодонной четырехгорлой колбе в 736 мл безводного тетрагидрофурана растворяют 58 г N-(2,4-дифторо-3-формилфенил) пропан-1-сульфонамида. Полученный раствор охлаждают до -78°С, при перемешивании добавляют 240 мл 1М раствора о-толилмагний хлорида в тетрагидрофуране. Полученные растворы сливают, поддерживая температуру реакционной смеси -40°С. К реакционной массе при перемешивании добавляют при комнатной температуре 556 мл 1N соляной кислоты и 491 мл этилацетата. Прохождение реакции контролируют методом тонкослойной хроматографии, элюент этилацетат : гексан 1:2. Полученную эмульсию переносят в делительную воронку и отделяют органический слой. Органическую фазу сушат над сульфатом натрия и упаривают растворитель на ротационном испарителе.
Полученный N-(3-((5-бромо-1-(4-метоксибензил)-1Н-пиразоло[3,4-b]пиридин-3-ил)(гидрокси)метил)-2,4-дифторофенил)пропан-1-сульфонамид растворяют в 1164 мл дихлорметана. При перемешивании на ледяной охлаждающей бане порциями добавляют 56,06 г 1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензиододиоксол-3-(1Н)-она, поддерживая температуру 20°С. Ледяную баню убирают и перемешивают реакционную массу при 20°С в течение 2 часов. Полученный продукт смешивают с диоксаном (1052 мл), водой (349 мл), 4-хлорофенил бороновой кислотой (14,96 г) и калия карбонатом (52,88 г). Реакционную массу выдерживают под вакуумом в течение 30 минут, затем продувают азотом (0,5-0,6 л/мин), доводя давление до атмосферного. Потом вносят тетракис(трифенилфосфин)палладий (5,53 г). Полученную смесь снова вакуумируют (при перемешивании в течение 5 минут, затем продувают азотом (0,5-0,6 л/мин), доводя давление до атмосферного, потом нагревают до 80°С Реакционную массу выдерживают при температуре 80°С в течение 16 часов, затем фильтруют через цеолит.
Полученный N-(3-(5-(4-хлорофенил)-1-(4-метокси-бензил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамид промывают гексаном и растворяют в 388 мл концентрированной серной кислоты, поддерживая температуру 20°С. Реакционную массу выливают при перемешивании в 2717 г льда и нейтрализуют, внося небольшими порциями 1085 г карбоната калия. Затем добавляют 388 мл этилацетата. Выпавший осадок неорганических солей отфильтровывают и промывают 388 мл этилацетата. Эмульсию переносят в делительную воронку отделяют органическую фазу и сушат над сульфатом натрия и упаривают растворитель на ротационном испарителе. Получают 37,69 г N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1. Выход 94,0%. LCMS m/z 492 (М+1)+.
Для получения фармацевтически приемлемой соли к N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамиду формулы 1 в метаноле (30 мл) добавляют 1,25 М раствор HCl в метаноле или растворы других соответствующих кислот согласно известным методикам.
Пример 2. Получение кристаллической формы соединения формулы 1.
10 мг N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1 растворяют в 10 мл метанола, нагревая смесь до 42°С. К раствору, полученному в результате этого, по каплям добавляют 10 мл воды при той же температуре в течение 20 минут и полученную смесь охлаждают до 5°С в течение суток, после чего отфильтровывают и собирают кристаллическое вещество. Кристаллы сушат при 50°С при пониженном давлении до тех пор, пока масса кристаллов не перестает уменьшаться при дальнейшем высушивании, в результате чего получают 9,52 мг белого твердого вещества.
Порошковый рентгеноструктурный анализ полученной кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1 проводят при помощи дифрактометра Siemens D500 (медное излучение CuKα, графитовый монохроматор на вторичном пучке). Полнопрофильная дифрактограмма измерена в интервале углов 2<2θ<60° с шагом 0,01°. В качестве внешнего стандарта использован гексаборид лантана.
На спектре порошковой рентгеновской дифрактограммы кристаллического вещества (Фиг. 1) наблюдают характеристические пики при углах дифракции 2θ (±0,2°): 8,9; 13,0; 14,5; 16,4; 18,5; 19,6; 21,4; 22,3; 24,3; 25,4; 28,0.
Данная кристаллическая форма не образует сольватов с органическими растворителямим, Тпл=222°С, практически не растворима в воде.
Пример 3. Определение специфической противоопухолевой активности соединения формулы 1 in vitro.
В качестве модельного объекта на котором должно быть проведено исследование специфической противоопухолевой активности соединения формулы 1 были выбраны клеточные линии, экспрессирующие BRAF:
А375 - клетки меланомы человека;
НСТ116 - клетки колоректальной карциномы человека;
НТ29 - клетки колоректальной аденокарциномы человека.
Протоколы культивирования, субкультивирования и посадки клеток выполняют в соответствии с инструкцией к клеточным линиям. Клетки считают с помощью автоматического клеточного счетчика, и концентрацию клеток А375 доводят до 1.6*104 клеток/мл (3000 клеток на лунку), концентрацию клеток НСТ116 доводят до 1.1*104 клеток/мл (2000 клеток на лунку), концентрацию клеток НТ29 доводят до 2.7*104 клеток/мл (5000 клеток на лунку)
В качестве контрольного соединения в исследовании специфической противоопухолевой активности соединения формулы 1 in vitro был выбран препарат туберцидин. Это связано с тем, что для проверки качества эссея in vitro используются соединения, вызывающие 100% гибель клеток в независимости от наличия или отсутствия у них определенных мутаций. Это позволяет более точно построить дозозависимые кривые и рассчитать значение IC50.
Цитотоксичность этих вещества оценивали с помощью стандартного МТТ-теста с (3-(4,5-диметилтиазол-2-ил)-2,5-дифенил) тетразолий бромидом.
Тестируемое соединение и контрольное вещество туберцидин растворяют в ДМСО в концентрации 10 мМ. Готовят разведение тестируемого соединения в ДМСО с шагом 3 (трехкратное последовательное разведение) с помощью Biomek 2000, начиная с 10 мМ, контрольное вещество туберцидин, начиная с 1 мМ. Минимальный объем в каждой лунке составляет 100 мкл. С помощью многоканального дозатора к 180 мкл клеток добавляют тестируемое соединение и контрольное вещество туберцидин из промежуточного разведения по 20 мкл в каждую лунку в трех повторах В контрольные лунки добавляют по 20 мкл среды, содержащей ДМСО вместо тестируемого соединения.
Центрифугируют при 500 rpm, инкубируют при 37°С, 5% СО2, на 5 суток, после чего добавляют 10 мкл раствора МТТ в PBS до концентрации 5 мг/мкл. Планшеты с реагентом МТТ обрабатывают на планшетном ридере CLARIOstar, измеряя поглощение на длине волны 570 нм для каждой лунки. Реагент МТТ позволяет оценить общую жизнеспособность клеток относительно контрольных условий.
В качестве количественного параметра для оценки цитотоксичности использовалась величина IC50, которая соответствует концентрации вещества, при которой жизнеспособность клеток составляет 50%. Значения IC50 рассчитываются при помощи программы GraphPadPrism 6 (GraphPadSoftware, Inc.) на основании первичных данных.
По результатам теста исследуемое соединение формулы 1 активно подавляет пролиферацию клеток линии А375 (меланома человека), IC50 для двух партий препарата представлены в таблице 1.
Исследуемое соединение формулы 1 активно подавляет пролиферацию клеток линии НТ29 (колоректальная аденокарцинома человека), IC50 для двух партий препарата представлены в таблице 1.
Исследуемое соединение формулы 1 неактивно в отношении клеток линии НСТ116 (колоректальная карцинома человека), установить IC50 в ходе эксперимента не удалось.
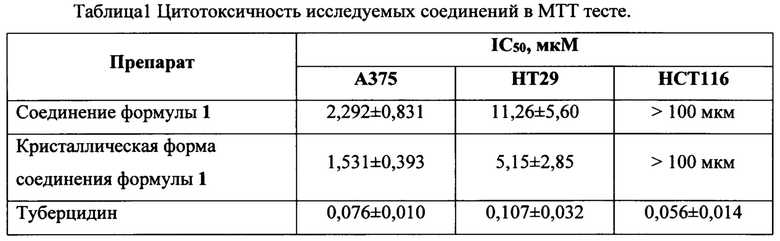
По результатам проведенных исследований установлено, что соединение формулы 1 проявляет сильный цитотоксический эффект в отношении клеточной линии А375 и менее выраженный в отношении линии НТ29. Клеточная линия НСТ116 обладает устойчивостью к воздействию соединения формулы 1.
Пример 4. Определение специфической противоопухолевой активности соединения формулы 1 in vivo.
Изучение противоопухолевой активности соединение формулы 1 проводят на модели ксенографтов меланомы человека линии А375 в сравнении с препаратом Вемурафениб.
Исследование включает в себя:
1) Подсаживание иммунодефицитным самцам мышей линии SCID в возрасте 13 недель клеток меланомы человека линии A375,
2) изучение влияния соединения формулы 1 на массу животных,
3) изучение влияния соединения формулы 1 на рост опухоли у животных,
4) Расчет ТРО (торможения роста опухоли).
Иммунодефицитные мыши являются удобной моделью для изучения противоопухолевой активности лекарственных средств. Отсутствие иммунитета позволяет перевивать им раковые клетки человека, что увеличивает прогностическую достоверность активности исследуемых веществ.
Животных содержат в контролируемых условиях окружающей среды (22-26°С и 30-70% относительная влажность) в индивидуально вентилируемых клетках. В комнатах содержания животных поддерживается 12 часовой цикл освещения и 10-15-ти кратная смена объема воздуха в час. Животные имеют свободный доступ к воде и корму.
Клетки меланомы человека линии A375 наращивали в флаконах в среде ДМЕМ с добавлением 10% FBS во влажной атмосфере при 95% воздуха и 5% СО2 при 37°С. По мере достижения 100% конфлюентности, клетки пересеивали: в среднем раз в 3-4 дня. При откреплении удаляли культуральную жидкость, промывали в фосфатно-солевом буфере и добавляли раствор трипсина-ЭДТА 0,25% с солями Хенкса. Инкубировали 5 минут при 37°С.
По окончании эксперимента все животные были подвержены эвтаназии ингаляцией СО2.
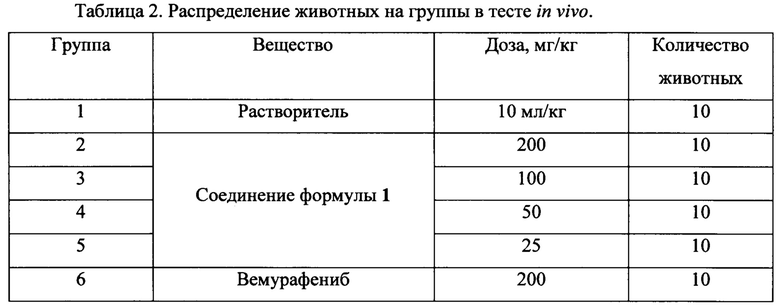
Перед имплантацией клеток соответствующий участок кожи мышей выбривали и обрабатывали дезинфицирующим раствором АХД-2000. После имплантации клеток раз в 2 дня проводили измерение опухолей. Когда средний размер опухолей достиг 100-200 мм3, животных разделили на экспериментальные группы и начали лечение исследуемыми препаратами. Препараты вводили внутрижелудочно, ежедневно 2 раза в сутки, в течение 21-го дня. Контрольная группа получала растворитель. Распределение животных на группы приведено в Таблице 2.
Объем опухоли определяли по формуле:
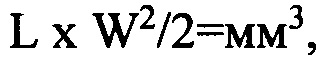
где L - соответствует наибольшему диаметру опухоли, W - наименьшему. Измерения проводили с помощью штангенциркуля.
Противоопухолевую активность исследуемых препаратов определяли по замедлению роста опухоли относительно опухолей в контрольной группе, получающей растворитель. Для этого рассчитывали значения торможения роста опухоли:
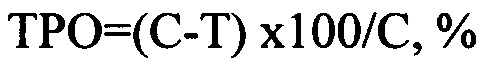
где С - средний размер опухоли в контрольной группе, получавшей растворитель,
Т - размер опухоли в экспериментальной группе.
Для статистического сравнения повторяющихся измерений - графиков массы тела животных и изменения объема опухоли в течение эксперимента использовали Repeated measures ANOVA, в случае выявления отличий между группами, применяли постанализ Ньюмана-Кейлса. Различия определяют при уровне статистической значимости p<0,05.
По результатам клинического осмотра на протяжении всего эксперимента введение исследуемых веществ не влияет на изменение массы тела животных
На Фиг. 2 видно, что у животных в группе «Контроль» размер опухоли равномерно и постоянно увеличивается. Ежедневное введение соединения формулы 1 в дозах 50, 100 и 200 мг/кг 2 раза в сутки дозозависимо снижало темпы роста опухолей, начиная с 17-го дня. При этом Вемурафениб в дозе 200 мг/кг и соединение формулы 1 в дозе 25 мг/кг также не оказали существенного влияния на рост опухолей.
При сравнении размеров опухолей и торможения роста опухоли (ТРО) на 21-й день исследования (Фиг. 3 и 4) показано, что ежедневное введение соединения формулы 1 в дозе 200 мг/кг приводит к достоверному снижению размеров опухоли, значение ТРО составило 77,5%.
Результаты исследования показывают, что ежедневное внутрижелудочное введение соединения формулы 1 в дозе 200 мг/кг 2 раза в сутки (400 мг/кг/сутки) приводит к статистически достоверному торможению роста опухоли клеточной линии A375 в модели ксенографтов.
Применение соединения формулы 1 в суммарной дозе 400 мг/кг/сутки привело к статистически достоверному снижению объема опухоли на 21-й день исследования по сравнению с контрольной группой. Параметр ТРО (торможение роста опухоли) составил около 80%.
На основании приведенных результатов, можно сделать заключение, что эффективная разовая доза соединения формулы 1 составляет 200 мг/кг при пересчете на человека.
Пример 5. Определение острой токсичности соединения формулы 1.
Целью исследования острой токсичности является определение переносимых токсических и летальных доз соединения формулы 1 при однократном внутрижелудочном и внутрибрюшинном введении самцам и самкам мышей.
Доклиническое исследование острой токсичности соединения формулы 1 проведено по стандартной методике в соответствии с Методическими указаниями, изложенными в «Руководстве по проведению доклинических исследований лекарственных средств», М., Гриф и К, 2012 на аутбредных мышах. В общей сложности в эксперименте участвовало 96 животных (48 самцов и 48 самок). Масса животных варьировалась в зависимости от пола (20-22 г для самцов и 19-21 г для самок).
Использовались пути введения:
- Внутрижелудочный (путь введения, аналогичный планируемому клиническому);
- Внутрибрюшинный (как метод, аналогичный внутривенному введению).
При изучении острой токсичности дозы исследуемого вещества подбирались таким образом, чтобы определить дозу, не вызывающую гибель животных; дозу, вызывающую 100% смертность, и дозу вызывающую смертность, не достигающую 100%. Для исследования были выбраны следующие дозы соединения формулы 1:
Для внутрижелудочного введения - 2000 мг/кг, 1000 мг/кг и 500 мг/кг
Для внутрибрюшинного введения - 750 мг/кг, 500 мг/кг и 250 мг/кг
Растворы соединения формулы 1 готовили на 0,1%-ом водном растворе твина-80. Исследуемое соединения формулы 1 вводят внутрижелудочно с помощью зонда в объеме 10 мл/кг, внутривенное введение осуществляют через шприц в брюшную полость.
На основании полученных данных были рассчитаны ЛД50, ЛД10, ЛД84 и ЛД100 при помощи пробит-анализа (метод Finney, программное обеспечение Biostat 2009).
Согласно данным пробит-анализа ЛД50 соединения формулы 1 для самцов мышей при внутрижелудочном введении составляет 975,5±280,5 мг/кг; для самок - 1095,1±267,2 мг/кг; для самцов мышей при внутрибрюшинном введении 462,3±83,3 мг/кг; для самок - 425,1±75,1 мг/кг.
Пример 6. Определение кинетики растворения кристаллической формы соединения формулы 1.
Кинетику растворения новой кристаллической формы N-(3-(5 -(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, полученной в Примере 2, сравнивают с кинетикой растворения аморфного соединения формулы 1, полученного в Примере 1. Прибор для определения скорости растворения представляет собой трехгорлый сосуд емкостью 1 л. В один из тубусов вводят термометр, в другой - стеклянную трубку для взятия проб и их комплексирования, а в третий - основную деталь прибора - цилиндрическую корзинку высотой 3,6 см и диаметром 2,5 см, сделанную из нержавеющей стали в виде сетки с отверстиями диаметром 40 меш (около 0,351 мм). Корзинка насажена на ось мотора.
В сосуд наливают растворяющую среду (1000 мл), в данном эксперименте ДМСО поскольку он является важным биполярным апротонным растворителем и, благодаря своей сильной растворяющей способности, ДМСО часто используется как растворитель в химических реакциях и система биологического скрининга. Исследуемый образец помещают в цилиндрическую корзинку, которую устанавливают на расстоянии 2 см от дна сосуда.
Для сравнительного образца 1 используют 200 мг новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, полученной в Примере 2, а для сравнительного образца 2-200 мг аморфного соединения формулы 1, полученного в Примере 1, с получением после полного растворения раствора, содержащего 200 м.д. соединения.
Температуру растворяющей среды во время опыта поддерживают постоянной (37±0,5°С). Скорость вращения корзинки в среде регулируют с точностью ±5%, она составляет 200 об/мин. Через установленные интервалы времени отбирают для анализа пробы по 1-2 мл для определения содержания лекарственного вещества. Взятый объем растворителя тотчас же восполняют новым.
Полученные результаты приведены в таблице 3 (где величины представляют собой количество (м.д.) образцов 1 или 2 в растворе).
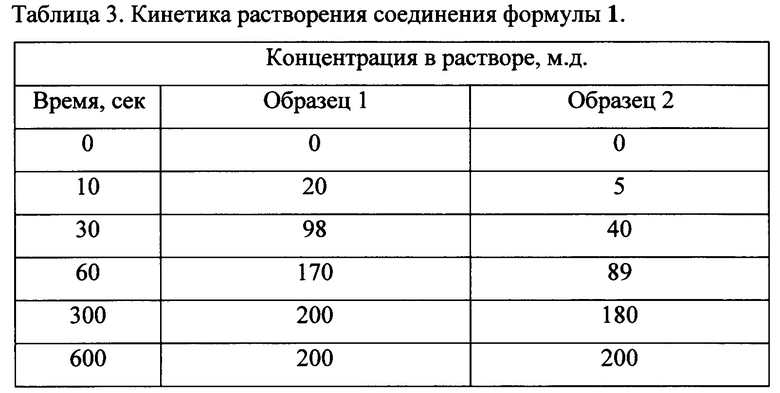
Результаты демонстрируют, что скорость растворения новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, полученной в Примере 2 в ДМСО выше, чем скорость растворения аморфного соединения формулы 1, полученного в Примере 1. В частности, время, в течение которого происходит 50% растворение новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, в два раза меньше, чем для сравнительного образца аморфного соединения формулы 1.
Пример 7. Исследование хранения кристаллической формы.
Все образцы хранились в стеклянных виалах, укупоренных резиновыми пробками с алюминиевыми колпачками, в холодильной камере при температуре ~4,7°С и в морозильной камере при температура около -23°С. Количество примесей определялось методом внутренней нормализации, детектирование проводилось УФ детектором при длине волны 220 нм.
В таблице 4 представлены результаты испытаний новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, полученной в Примере 2 (Образец 1), свидетельствующие об улучшении ее стабильности при пониженной температуре по сравнению с аморфным соединением формулы 1, полученным в Примере 1 (Образец 2).
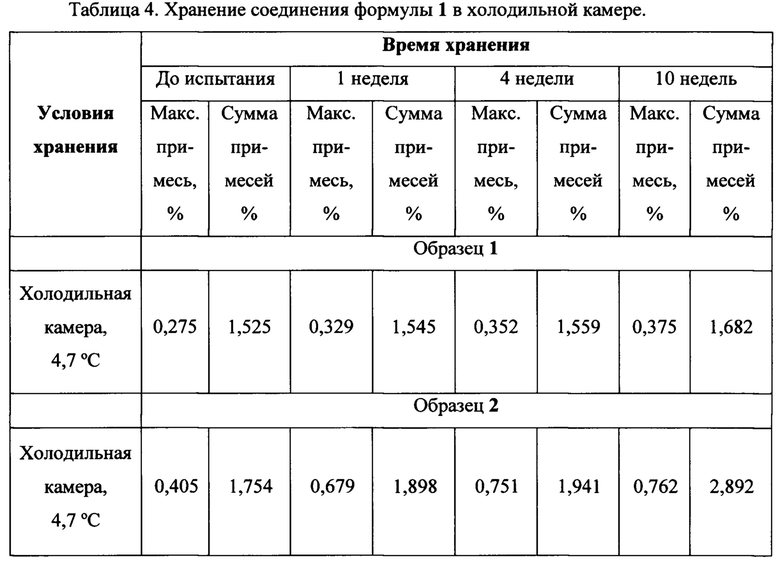
Результаты однозначно свидетельствуют о перспективности заявляемой новой кристаллической формы N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1.
Пример 8. Получение готовой лекарственной формы соединения формулы 1.
480,0 г соединения формулы 1, 1120,0 г гипромеллозы ацетата сукцината, 20,0 г кремния диоксида коллоидного, 60,0 г кроскармеллозы натрия, 8,0 г гипролозы (гидроксипропил целлюлозы), 12,0 г магния стеарата просеивают на лабораторном сите с номинальным диаметром отверстий 0,5 мм, загружают подготовленное сырье в рабочую камеру миксера-гранулятора и перемешивают.
Через форсунку подают в рабочую камеру увлажнитель в количестве 500 мл (вода очищенная - 313,0 г, спирт 96% - 187,0 г). Полученную влажную массу выгружают с помощью лопатки и сушат в сушилке-грануляторе.
В смесительную установку СТД-12 осторожно, избегая пылеобразования, загружают полученный гранулят, и перемешивают 20 мин при скорости перемешивания 10±) об/мин.
Полученную массу для таблетирования выгружают в технологическую тару. Содержание соединения формулы 1 в пересчете на среднюю массу одной таблетки должно быть в пределах от 228,0 мг до 252,0 мг.
Таблетирование полученной массы производят на таблетпрессе ТП-13 с обеспылевателем и устройством для отбраковки таблеток. По окончании таблетирования таблетки - ядра покрывают пленочной оболочкой.
Для получения пленочной оболочки при включенной мешалке со скоростью 400±5 об/мин в емкость загружают сырье:
- воду очищенную - 300,0 г;
- Opadry белый - 40,0 г.
Полученную массу перемешивают в течение 45±5 минут, полученную суспензию фильтруют через сито с номинальным диаметром отверстий 0,25 мм. Профильтрованная суспензия должна быть однородной, не иметь комков.
В машину для нанесения пленочного покрытия загружают таблетки-ядра в количестве 1640,0 г, нагревают 5 минут при температуре входящего воздуха 60±2°С и скорости вращения барабана 6±1 об/мин.
С помощью перистальтического насоса начинают подачу пленкообразующей суспензии в количестве 354,0 г. Процесс покрытия таблеток - ядер ведут до получения таблеток массой 870,0±42,0 мг, после чего сушат 10 минут и охлаждают.
Фасовку таблеток осуществляют по 8 таблеток в контурную ячейковую упаковку из пленки поливинилхлоридной и фольги алюминиевой печатной лакированной на блистерной машине БМ-17 и упаковывают по 7 блистеров вместе с инструкцией по применению помещают в пачку из картона.
Пример 9. Получение лекарственного средства в форме капсул. Тщательно смешивают соединение формулы 1 с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 300 мг в желатиновые капсулы подходящего размера.
Пример 10. Получение лекарственного средства в форме инъекционных композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивают 500 мг соединения формулы 1 с 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл инъекционной воды. Полученный раствор фильтруют и помещают по 1 мл в ампулы, которые запаивают.
Настоящее изобретение может быть использовано в медицине, ветеринарии, биохимии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР BRAF КИНАЗЫ N-(3-(5-(4-ХЛОРОФЕНИЛ)-1H-ПИРАЗОЛО[3,4-B]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИД | 2018 |
|
RU2687107C1 |
| КРИСТАЛЛИЧЕСКИЕ СОЛИ ИНГИБИТОРА B-RAF-КИНАЗЫ | 2018 |
|
RU2798091C2 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ, ВКЛЮЧАЮЩАЯ ВЕМУРАФЕНИБ И ИНТЕРФЕРОН, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РАКА | 2011 |
|
RU2592983C2 |
| ПРОТИВООПУХОЛЕВОЕ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО | 2012 |
|
RU2580609C2 |
| АГЕНТ, ИНДУЦИРУЮЩИЙ КЛЕТОЧНУЮ ГИБЕЛЬ, ДЛЯ КЛЕТОК, ИМЕЮЩИХ МУТАЦИИ ГЕНА BRAF, АГЕНТ, ПОДАВЛЯЮЩИЙ РОСТ ТАКИХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ТЕРАПИИ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ ДЕФЕКТОМ РОСТА ТАКИХ КЛЕТОК | 2016 |
|
RU2760835C2 |
| 3,4-ДИГИДРО-2,7-НАФТИРИДИН-1,6(2H,7H)-ДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ MEK | 2022 |
|
RU2826000C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПИРАЗОЛО-ПИРИДИНОВЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ СТИМУЛИРОВАНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2020 |
|
RU2837173C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
Изобретение относится к области органической химии, а именно к N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил)пропан-1-сульфонамиду формулы 1 или его фармацевтически приемлемой соли. Также изобретение относится к новой кристаллической форме, активному компоненту, фармацевтической композиции и лекарственному средству на основе соединения 1. Технический результат: получено новое соединение, используемое для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы. 6 н. и 4 з.п. ф-лы, 4 ил., 5 табл., 10 пр.
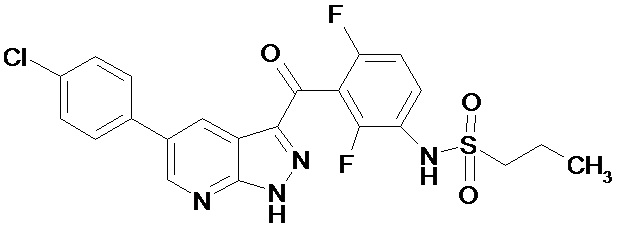
1
1. Соединение N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамид формулы 1, или его фармацевтически приемлемая соль,
2. Кристаллическая форма соединения N-(3-(5-(4-хлорофенил)-1Н-пиразоло[3,4-b]пиридин-3-карбонил)-2,4-дифторофенил) пропан-1-сульфонамида формулы 1, имеющая на порошковой рентгеновской дифрактограмме характеристические пики при следующих углах 2θ (±0,2°): 8,9; 13,0; 14,5; 16,4; 18,5; 19,6; 21,4; 22,3; 24,3; 25,4; 28,0.
3. Активный компонент, обладающий свойством ингибитора BRAF киназы, представляющий собой соединение формулы 1 по п. 1 или кристаллическую форму по п. 2.
4. Фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, содержащая в эффективном количестве активный компонент по п.3.
5. Фармацевтическая композиция по п. 4, отличающаяся тем, что мутацией BRAF является мутация V600E.
6. Фармацевтическая композиция по п. 4, отличающаяся тем, что пролиферативное заболевание представляет собой меланому.
7. Фармацевтическая композиция по п. 4, отличающаяся тем, что пролиферативное заболевание представляет собой колоректальную аденокарциному.
8. Лекарственное средство для профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, в виде таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, содержащее в эффективном количестве соединение формулы 1, или его фармацевтически приемлемую соль, по п. 1 или кристаллическую форму по п. 2, или активный компонент по п. 3, или фармацевтическую композицию по любому из пп. 4-7.
9. Лекарственное средство по п. 8 в виде таблетки, содержащей, мас.%:
10. Способ профилактики или лечения пролиферативного заболевания, характеризующегося мутацией BRAF киназы, согласно которому нуждающемуся субъекту вводят соединение формулы 1, или его фармацевтически приемлемую соль, по п. 1 или кристаллическую форму по п. 2, или активный компонент по п. 3, или фармацевтическую композицию по любому из пп. 4-7 или лекарственное средство по любому из пп. 8 и 9.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ FGFR | 2007 |
|
RU2466130C2 |

