Область техники настоящего изобретения
Настоящая заявка относится к технической области способов органического синтеза и, в частности, относится к синтезу (S)-никотина, более конкретно, к способу синтеза (S)-никотина с высокой химической чистотой и высокой оптической чистотой.
Предшествующий уровень техники настоящего изобретения
(S)-никотин (никотин) представляет собой алкалоид, присутствующий в пасленовых растениях (Solatium) и являющийся важным компонентом табака. Табачные листья содержат от 1,5% до 3,5% (S)-никотина. В настоящее время наиболее распространенный способ получения никотина представляет собой экстракцию никотина из листьев табака. Согласно имеющимся сообщениям химический синтез разработан в недостаточной степени и является значительно более дорогостоящим, чем способ экстракции. Химические способы синтеза, которые были описаны до настоящего времени, представляют собой способ химического разделения, способ асимметричного гидрирования, способ хирального вспомогательного вещества и т.д.
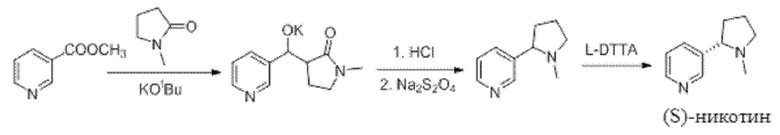
В патенте US 20160326134 описан способ, в котором метилникотинат в качестве исходного материала конденсируется с N-метилпирролидином (NMP) и подвергается внутримолекулярной перегруппировке, восстановлению и хиральному разделению с получением (S)-никотина. Путь синтеза (S)-никотина представлен ниже.
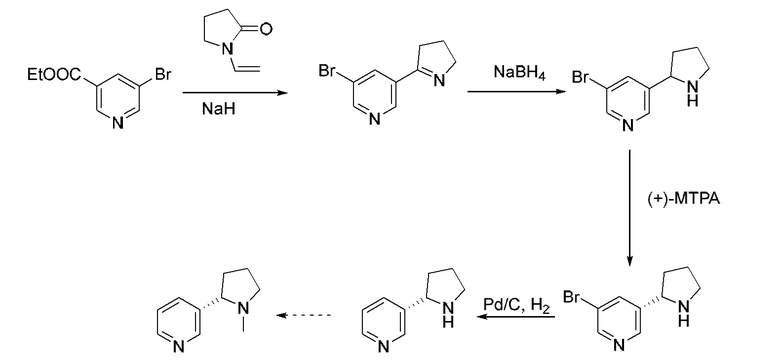
В документе (Jacob Peyton «Разделение (±)-5-бромникотина. Синтез (R)- и (S)-норникотина высокой энантиомерной чистоты». The Journal of Organic Chemistry 1982, 4165-4167) описан способ получения (S)-норникотина с хиральной кислотой в качестве разделяющего реагента с помощью процессов, представляющих собой разделение, восстановительное дебромирование и т.д. (S)-норникотин можно дополнительно подвергать метилированию для удобного получения (S)-никотина. Соответствующий путь синтеза представлен ниже. Однако способ химического разделения имеет недостатки, представляющие собой низкий выход и высокую стоимость.
В документе (Gui Guo, Dong-Wei Sun, Shuang Yang и др. «Катализируемое иридием асимметричное гидрирование 2-пиридилциклических иминов: высокоэнантиоселективный подход к производным никотина». Journal of the American Chemical Society, 2015, 90-93) описан способ синтеза в процессе асимметричного каталитического гидрирования. Путь синтеза никотина представлен ниже.
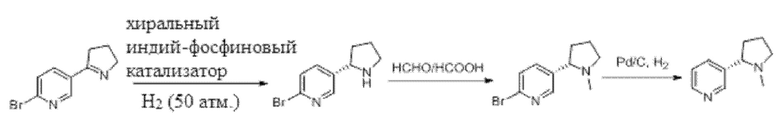
Однако в способе асимметричного каталитического гидрирования требуется применение дорогостоящего хирального металлического катализатора и, следовательно, этот способ имеет высокую стоимость, реакции необходимо проводить под высоким давлением, требуются значительные капиталовложения в оборудование, а использование водорода также представляет собой потенциальную угрозу безопасности. Кроме того, в этом документе показано, что только гексазамещенный субстрат участвует в каталитической реакции с относительно высокими эксплуатационными характеристиками, и для прямого гидрирования миосмина отсутствуют технические достижения.
В документе (Teck-Peng Loh, Jian-Rong Zhou, Xu-Ran Li, Keng-Yeow Sim «Новая восстановительная аминоциклизация для синтеза хиральных пирролидинов: стереоселективный синтез (S)-норникотина и 2-(2'-пирролидил)-пиридинов». Tetrahedron Letters, 1999, 7847-7850) описан способ синтеза (S)-норникотина с применением галактозамина в качестве хирального вспомогательного вещества. (S)-норникотин может подвергаться последующей реакции с получением (S)-никотина. Соответствующий путь синтеза представлен ниже.
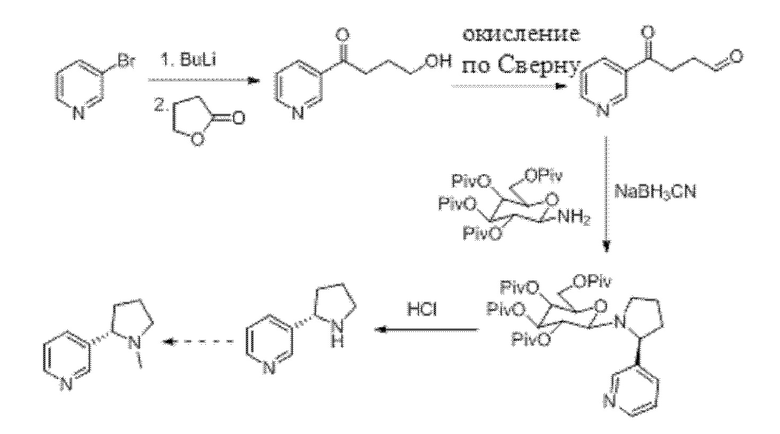
Однако в этом способе хирального вспомогательного вещества должно присутствовать в стехиометрическом количестве хиральное вспомогательное вещество, которое не может быть повторно использовано, что приводит к высокой стоимости и отсутствию практической ценности.
Таким образом, каждый способ химического синтеза (S)-никотина, описанный в литературе предшествующего уровня техники, имеет недостатки, и существует необходимость в разработке более экономичного, эффективного и безопасного способа синтеза.
Краткое раскрытие настоящего изобретения
Вследствие недостатков предшествующего уровня техники, цель настоящей заявки заключается в том, чтобы предложить способа синтеза (S)-никотина, в частности, способ синтеза (S)-никотина с высокой химической чистотой и высокой оптической чистотой.
Для достижения поставленной цели в настоящей заявке предложены технические решения, которые описаны ниже.
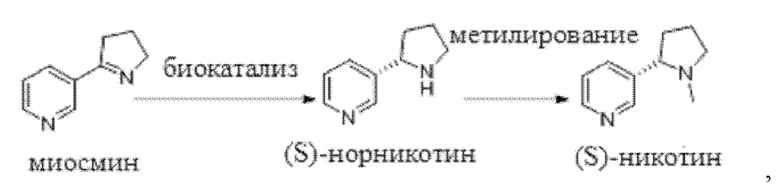
В настоящей заявке предложен способ синтеза (S)-никотина, в котором в условиях системы циркуляции кофермента с применением кофермента в качестве донора водорода и иминредуктазы в качестве катализатора осуществляют каталитическое восстановление миосмина с получением (S)-норникотина и последующее метилирование (S)-норникотина с получением (S)-никотина.
Способ синтеза, относящийся к настоящей заявке, может быть представлен следующей схемой реакции:
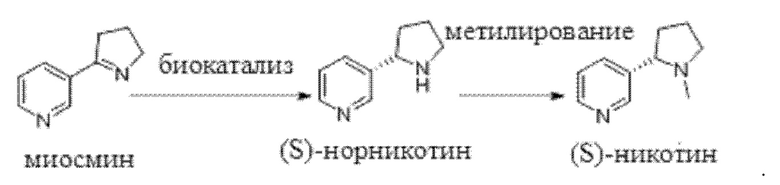
Этот способ синтеза отличается простотой осуществления, безопасностью и надежностью, обеспечивает высокий выход, имеет низкую стоимость и обеспечивает очень высокую химическую селективность с чистотой продукта, составляющей или превышающей 99,5%, а также превосходную энантиоселективность с оптической чистотой, составляющей или превышающей 99,5%.
Относящееся к настоящей заявке наименование миосмина по номенклатуре Союза теоретической и прикладной химии (IUPAC) представляет собой 3-(3,4-дигидро-2Н-пиррол-5-ил)пиридин, а наименование (S)-норникотина по номенклатуре IUPAC представляет собой (S)-3-(пирролидин-2-ил)пиридин.
Предпочтительно в системе циркуляции кофермента присутствуют кофермент, глюкоза и глюкозодегидрогеназа.
Предпочтительно кофермент содержит соль NADP (никотинамидадениндинуклеотидфосфат) и/или соль NAD (никотинамидадениндинуклеотид), предпочтительно соль NADP.
Предпочтительно глюкозодегидрогеназа представляет собой любую глюкозодегидрогеназу или комбинацию по меньшей мере двух глюкозодегидрогеназ из ES-GDH101-ES-GDH109. В частности, глюкозодегидрогеназа, представляет собой любую глюкозодегидрогеназу или комбинацию по меньшей мере двух глюкозодегидрогеназ из ES-GDH101, ES-GDH102, ES-GDH103, ES-GDH104, ES-GDH105, ES-GDH106, ES-GDH107, ES-GDH108 или ES-GDH109. Глюкозодегидрогеназа ES-GDH109 (номер партии: S20191121) является предпочтительной. Вышеуказанные глюкозодегидрогеназы могут быть приобретены у компании SyncoZymes (Shanghai) Co., Ltd.
Предпочтительно иминредуктаза представляет собой любую иминредуктазу или комбинацию по меньшей мере двух иминредуктаз из ES-IRED101-ES-IRED112, IRED1321104, IRED1321108, IRED1321110 или IRED1321114. В частности, иминредуктаза представляет собой любую иминредуктазу или комбинацию по меньшей мере двух иминредуктаз из ES-IRED101, ES-TRED102, ES-TRED103, ES-TRED104, ES-IRED105, ES-JRED106, ES-JRED107, ES-IRED108, ES-IRED109, ES-IRED110, ES-IRED111, ES-IRED112, IRED1321104, IRED1321108, IRED1321111 или I4RED13. Иминредуктаза ES-TRED103 (номер партии: S20191121) или IRED1321110 является предпочтительной. Иминредуктазы ES-IRED101-ES-IRED112 могут быть приобретены у компании SyncoZymes (Shanghai) Co., Ltd., а иминредуктазы IRED1321104, IRED1321108, IRED1321110 или IRED1321114 могут быть независимо получены из генного банка.
В частности, источник IRED1321104 представляют собой бактерии вида стрептомицет канамицетикус (Streptomyces Kanamyceticus), источник IRED 1321108 представляют собой бактерии вида L2C084 мезоризобиум (Mesorhizobium), источник IRED1321110 представляют собой бактерии вида салиниспора тихоокеанская (Salinispora pacifica), и источник IRED1321114 представляют собой бактерии вида мезоризобиум цицери (Mesorhizobium ciceri). Выбранную выше иминредуктазу отправляют в компанию по синтезу генов для синтеза, внесения в плазмиды рЕТ28(а) и введения в кишечную палочку (Escherichia coli) BL21 для конструирования рекомбинантного штамма кишечной палочки coli. Указанный выше рекомбинантный штамм инокулируют в условиях автоклава в среде Лурия-Бертани (LB) (10 г/л хлорида натрия, 10 г/л триптона и 5 г/л дрожжевого порошка) и культивируют при 37°С и 220 об/мин до тех пор, пока оптическая плотность при 600 нм (OD600) не составит от 2 до 3. Раствор 0,1 М изопропилтиогалактозида (IPTG) добавляют для индукции при 25°С в течение 16 часов. После центрифугирования клетки собирают, подвергают ультразвуковому разрушению и центрифугируют для удаления клеточного дебриса с получением раствора фермента. Раствор фермента лиофилизируют для получения порошка фермента собственного производства.
Предпочтительно каталитическое восстановление осуществляется при температуре, составляющей от 15 до 45°С, например, 15°С, 20°С, 25°С, 30°С, 35°С, 40°С или 45°С и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе. Предпочтительной является температура от 25 до 30°С.
Предпочтительно каталитическое восстановление осуществляется в течение от 8 до 72 часов, например, 8 часов, 10 часов, 12 часов, 16 часов, 24 часов, 30 часов, 48 часов, 56 часов, 60 часов или 72 часов и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе.
Предпочтительно каталитическое восстановление осуществляется в условиях перемешивания при скорости вращения, составляющей от 150 до 400 об/мин, например, 150 об/мин, 200 об/мин, 250 об/мин, 300 об/мин, 350 об/мин. мин или 400 об/мин и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе.
Предпочтительно каталитическое восстановление осуществляется в буферном растворе, причем буферный раствор представляет собой фосфатный буферный раствор, трис(гидроксиметил)метиламин-гидрохлоридный буферный раствор или триэтаноламин-гидрохлоридный буферный раствор.
Предпочтительно буферный раствор имеет значение рН, составляющее от 5,0 до 9,0, например, рН=5,0, рН=5,5, рН=6,0, рН=6,5, рН=7,0, рН=7,5, рН=8,0, рН=8,5, или рН=9,0 и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе. Предпочтительным является значение рН, составляющее от 5,8 до 6,5.
Предпочтительно для метилирования осуществляется смешивание и взаимодействие (S)-норникотина с формальдегидом и муравьиной кислотой с получением (S)-никотина.
Предпочтительно метилирование осуществляется при температуре, составляющей от 55 до 80°С, например, 55°С, 60°С, 65°С, 70°С, 75°С или 80°С и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе.
Предпочтительно метилирование осуществляется в течение от 8 до 48 ч, например, 9 ч, 12 ч, 18 ч, 26 ч, 32 ч, 38 ч, 46 ч или 48 ч и т.д. Могут быть выбраны и другие конкретные значения точек в пределах данного диапазона, и подробности не описаны в настоящем документе.
В качестве предпочтительного технического решения настоящей заявки в способе синтеза (S)-никотина предусмотрены смешивание миосмина, кофермента, глюкозы, глюкозодегидрогеназы, буфера и иминредуктазы и взаимодействие смеси при 15-45°С в течение 8-72 часов с получением (S)-норникотина, а затем смешивание (S)-норникотина с формальдегидом и муравьиной кислотой и реакция смеси при 55-80°С в течение 8-48 часов с получением (S)-никотина.
Предпочтительно массовое соотношение миосмина, кофермента и иминредуктазы составляет 1:(0,001-0,05):(0,05-0,1).
Предпочтительно массовое соотношение кофермента, глюкозы и глюкозодегидрогеназы составляет (0,001-0,05):(1,2-2,4):(0,01-0,05). При вышеуказанном массовом соотношении способ получения обеспечивает улучшенную химическую селективность.
По сравнению с предшествующим уровнем техники настоящая заявка обеспечивает положительные эффекты, которые описаны ниже.
Способ синтеза (S)-никотина согласно настоящей заявке является простым в осуществлении, безопасным и надежным, обеспечивает высокий выход и низкую стоимость, а также проявляет очень высокую химическую селективность с чистотой продукта, составляющей более или равной 99,5%, а также превосходную энантиоселективность с оптической чистотой, составляющей более или равной 99,5%.
Краткое описание фигур
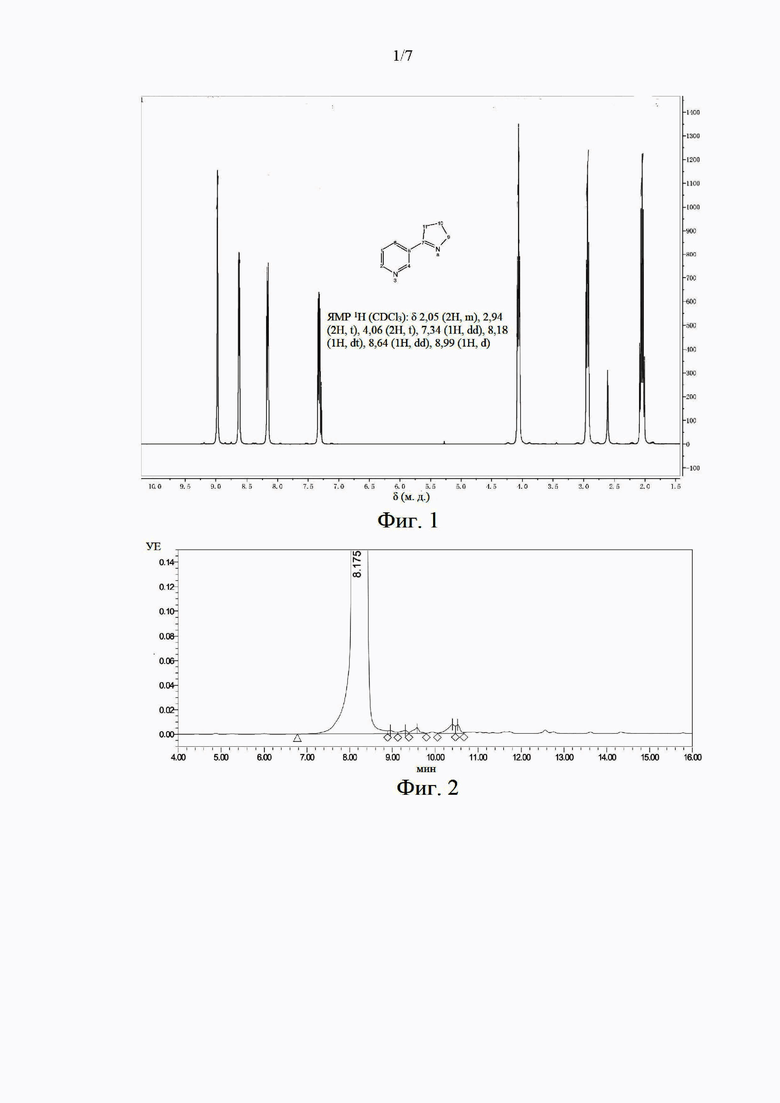
На фиг. 1 представлен характеристический спектр протонного ядерного магнитного резонанса (ЯМР 1H) образца миосмина в примере 1;
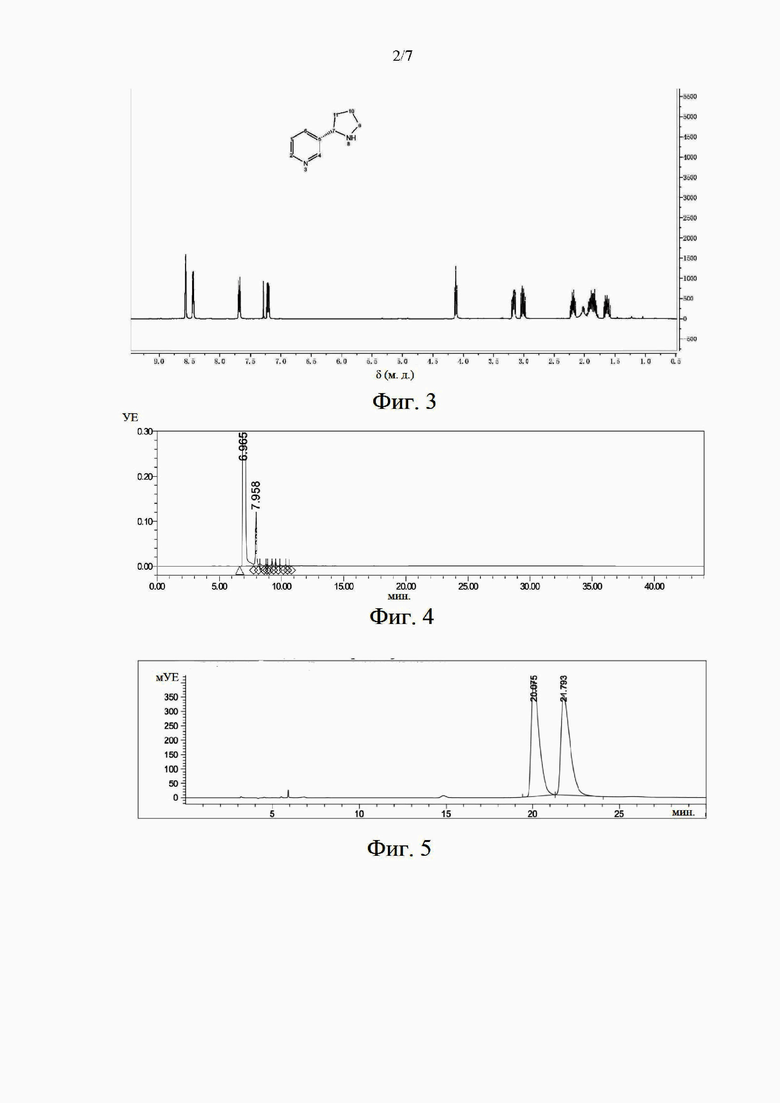
на фиг. 2 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца миосмина в примере 1;
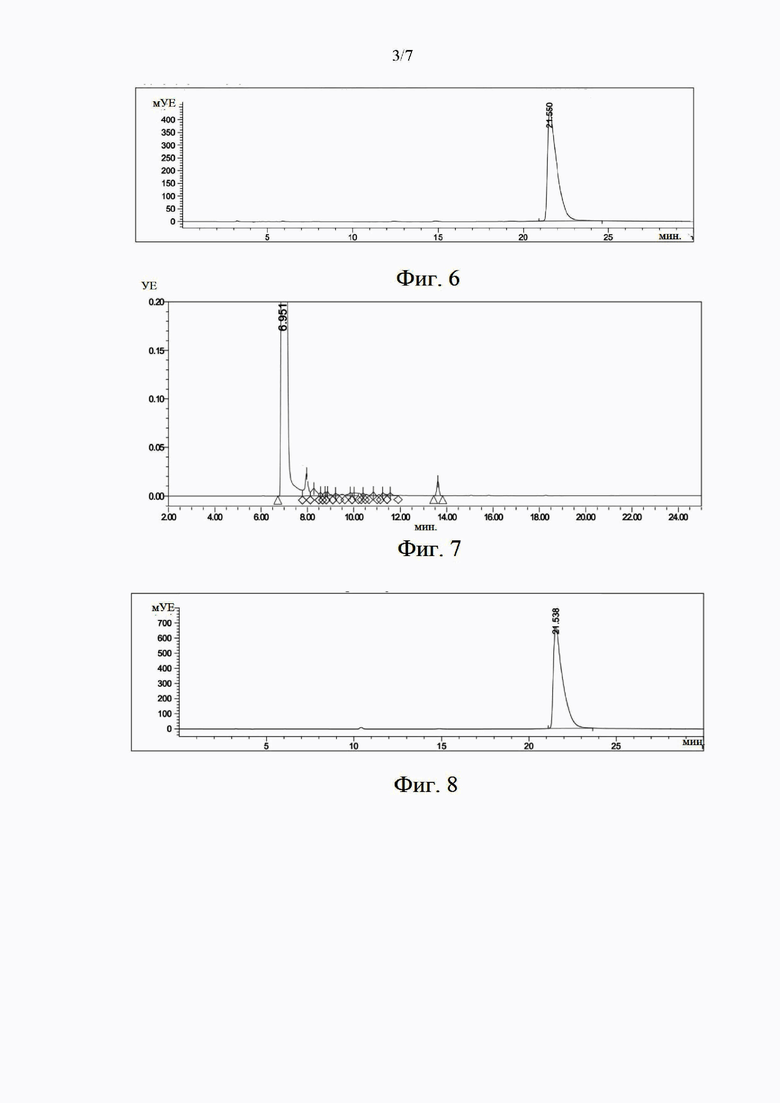
на фиг. 3 представлен собой характеристический спектр ЯМР 1Н образца (S)-норникотина в примере 2;
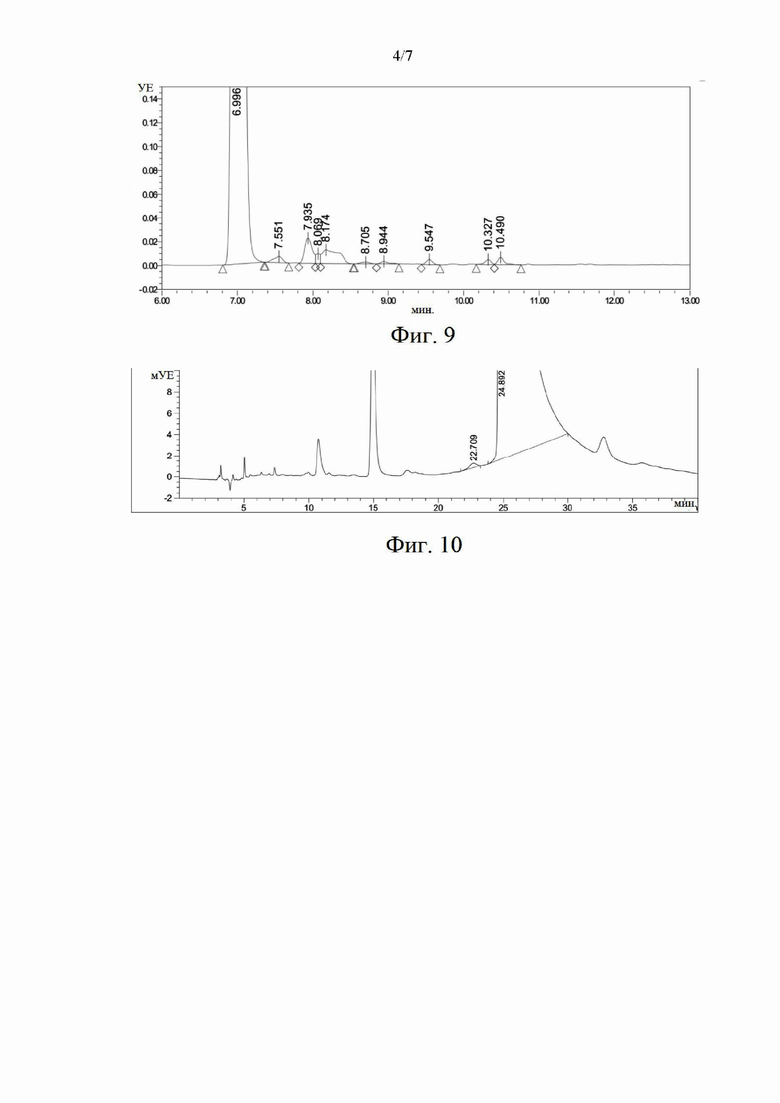
на фиг. 4 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца (S)-норникотина в примере 2;
на фиг. 5 представлена хиральная хроматограмма образца рацемата норникотина;
на фиг. 6 представлена хиральная хроматограмма образца (S)-норникотина в примере 2;
на фиг. 7 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца (S)-норникотина в примере 3;
на фиг. 8 представлена хиральная хроматограмма образца (S)-норникотина в примере 3;
на фиг. 9 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца (S)-норникотина в примере 6;
на фиг. 10 представлена хиральная хроматограмма образца (S)-норникотина в примере 6;
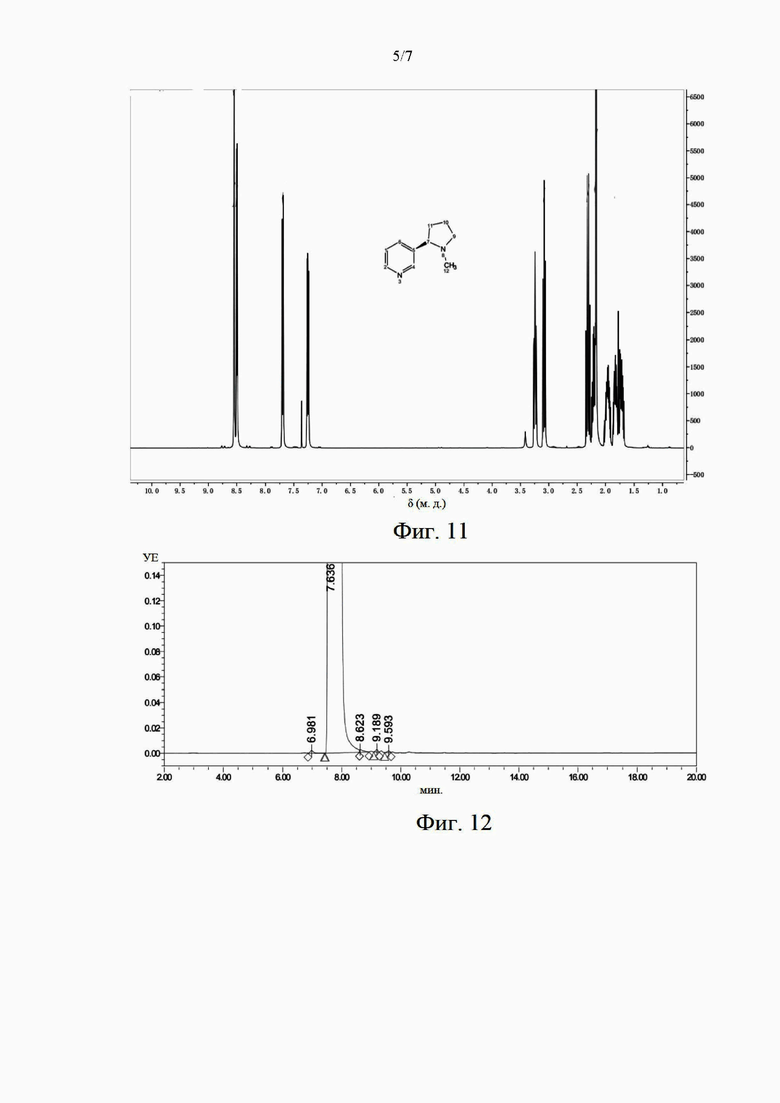
на фиг. 11 представлен собой характеристический спектр ЯМР 1Н образца (S)-никотина в примере 7;
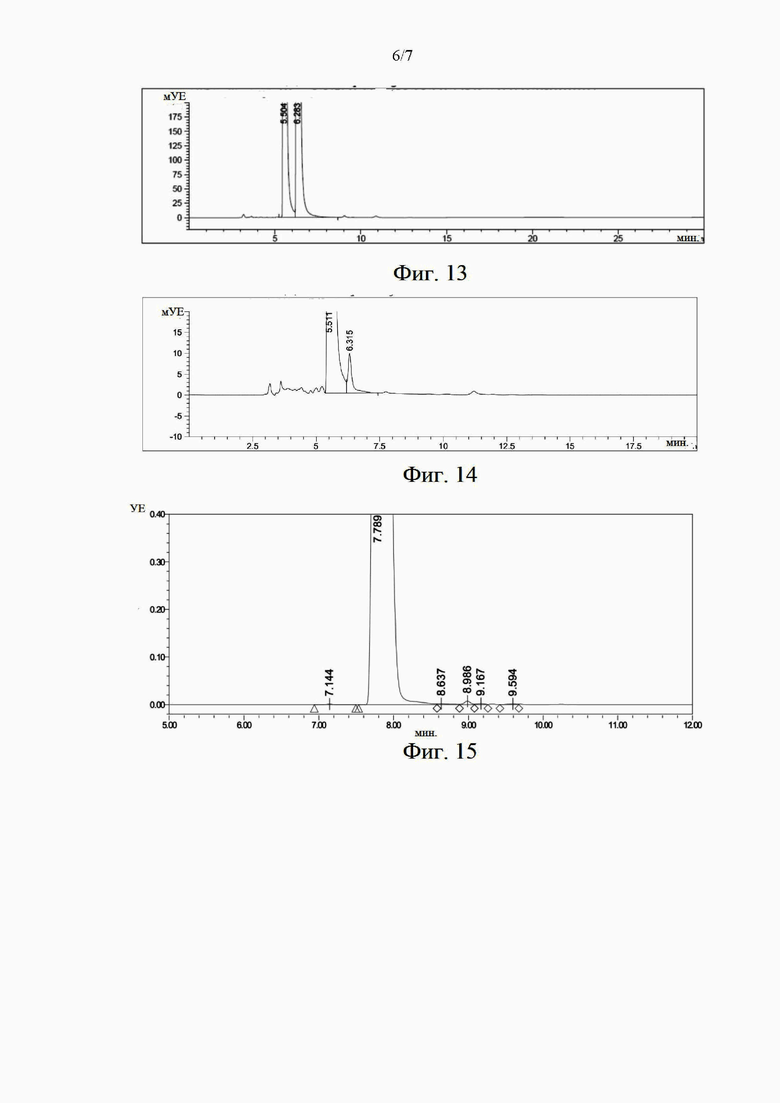
на фиг. 12 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца (S)-норникотина в примере 7;
на фиг. 13 представлена хиральная хроматограмма образца рацемата никотина;
на фиг. 14 представлена хиральная хроматограмма образца (S)-никотина в примере 7;
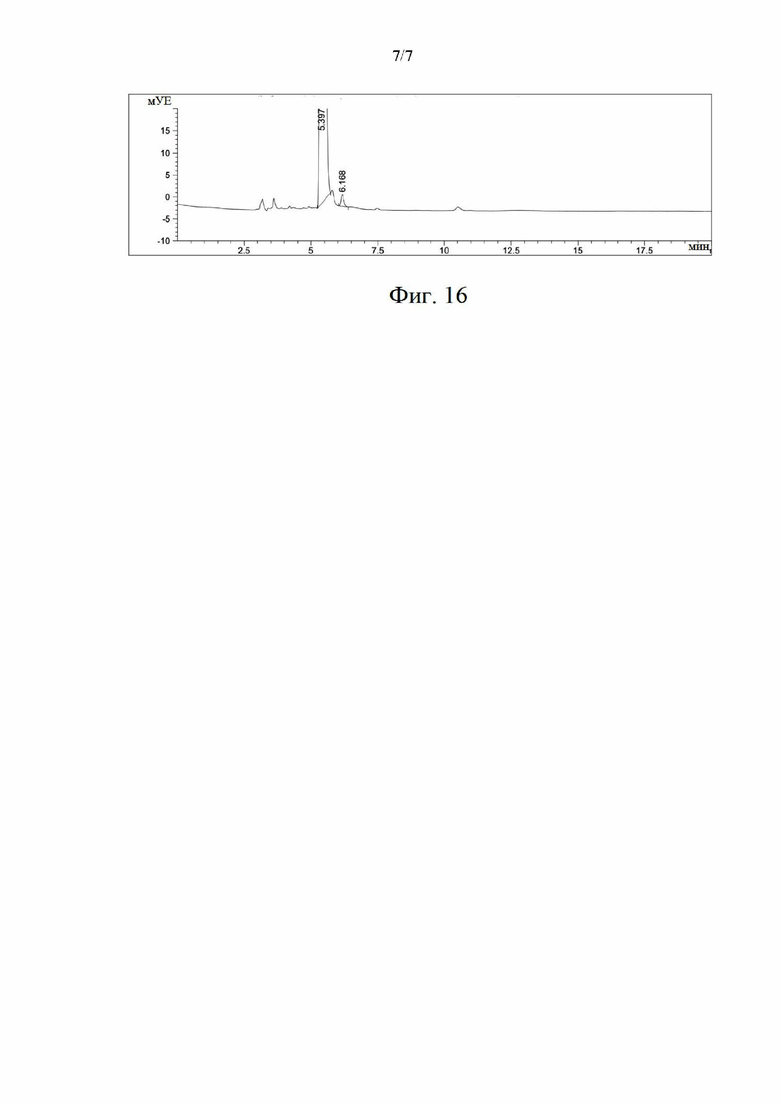
на фиг. 15 представлена жидкостная хроматограмма, иллюстрирующая химическую чистоту образца (S)-норникотина в примере 8; и
на фиг. 16 представлена хиральная хроматограмма образца (S)-никотина в примере 8.
Подробное раскрытие настоящего изобретения
Технические решения настоящей заявки дополнительно описаны ниже с представлением конкретных примеров. Для специалистов в данной области техники должно быть понятно, что эти примеры использованы для лучшего понимания настоящей заявки и не должны рассматриваться в качестве конкретных ограничений настоящей заявки.
Пример 1
В этом примере было получено соединение миосмин. В отношении способа синтеза миосмина сделана ссылка на способ в патентной публикации № ЕР 2487172 А1. Полученный продукт охарактеризован с помощью ЯМР 1Н. На фиг. 1 представлен характеристический спектр ЯМР, содержащий следующие данные: ЯМР 1Н (400 МГц, CDCl3), δ (м. д.): 8,97 (1H, d), 8,64 (1H, dd), 8,17 (1H, dt), 7,29-7,34 (m, 1H), 4,06 (2Н), 2,91-2,96 (m, 2Н), 2,01-2,09 (m, 2Н). Показано, что миосмин был успешно синтезирован. Образец был проанализирован и обнаружен с применением высокоэффективной жидкостной хроматографии. Чистота образца составила 99,3%. Хроматограмма представлена на фиг. 2.
Пример 2
В этом примере предложен способ синтеза (S)-норникотина. Ниже описано его осуществление.
В трехгорлую круглодонную колбу объемом 50 мл помещали 3 г миосмина, добавляли 10 мл 0,1 М фосфатного буферного раствора и доводили уровень рН до 6,0. Затем в реакционную колбу добавляли 5,5 г глюкозы и перемешивали до полного растворения. В другую колбу объемом 50 мл добавляли 0,2 г иминредуктазы ES-IRED103, 0,04 г глюкозодегидрогеназы ES-GDH109 и 0,008 г соли NADP и перемешивали до полного растворения. Затем раствор из второй колбы медленно добавляли в первую колбу, нагревали до 25°С, перемешивали со скоростью 300 об/мин и осуществляли реакцию в течение 24 часов. Раствор фильтровали, фильтрат экстрагировали хлороформом, сушили над безводным сульфатом натрия и концентрировали с получением 2,2 г (S)-норникотина.
Полученный (S)-hophhkothh охарактеризован с помощью ЯМР 1Н. На фиг. 3 представлен характеристический спектр ЯМР, содержащий следующие данные: ЯМР 1Н (400 МГц, CDCl3), 5 (м. д.): 8,56 (1H, d), 8,44 (1H, dd), 7,68 (1H, dt), 7,19-7,22 (m, 1Н), 4,12 (1H, t), 3,13-3,19 (1H, m), 2,98-3,00 (1H, m), 2,15-2,23 (1H, m), 2,02 (1H, s), 1,80-1,93 (2H, m), 1,58-1,67 (1H, m). Показано, что (S)-норникотин был успешно синтезирован.
Химическую чистоту полученного (S)-норникотина определяли с помощью высокоэффективной жидкостной хроматографии. Как представлено на фиг. 4, чистота составляет 93,6%. Оптическую чистоту образца определяли с помощью высокоэффективного жидкостного хроматографа и хиральной хроматографической колонки. На фиг. 5 представлен спектр хирального разделения рацемата норникотина, где время появления пика (К)-норникотина составляет 20,08 мин, время появления пика (S)-норникотина составляет 21,79 мин, а площадь этих пиков составляет 49,9%. и 50,1% соответственно; на фиг. 6 представлена хиральная хроматограмма продукта, на которой (R)-норникотин не обнаружен, а обнаружен только пик (S)-норникотина со временем появления 21,6 мин, то есть оптическая чистота образца приближается к 100%.
Пример 3
В этом примере предложен способ синтеза (S)-норникотина. Ниже описано его осуществление.
В трехгорлую круглодонную колбу объемом 50 мл помещали 3 г миосмина, добавляли 15 мл 0,1 М фосфатного буферного раствора и доводили уровень рН до 5,8. В реакционную колбу добавляли 5,5 г глюкозы и перемешивали до полного растворения. В другую колбу объемом 50 мл добавляли 0,3 г иминредуктазы ES-IRED103, 0,04 г глюкозодегидрогеназы ES-GDH102 и 0,01 г соли NADP и перемешивали до полного растворения. Затем раствор из второй колбы медленно добавляли в первую колбу, нагревали до 37°С и перемешивали в течение 24 часов. Раствор фильтровали, фильтрат экстрагировали хлороформом, сушили над безводным сульфатом натрия и концентрировали с получением 2,4 г (S)-норникотина.
Химическую чистоту и оптическую чистоту полученного (S)-норникотина определяли отдельно с помощью высокоэффективного жидкостного хроматографа. Конкретный метод работы является таким же, как и в примере 2. Хроматограмма представлена на фиг. 7, где расчетная химическая чистота составляет 97,2%; и хиральная хроматограмма представлена на фиг. 8, где оптическая чистота приближается к 100%.
Пример 4
В этом примере предложен способ синтеза (S)-норникотина. Ниже описано его осуществление.
В трехгорлую кругл одонную колбу объемом 50 мл помещали 3 г миосмина, добавляли 15 мл 0,1 М фосфатного буферного раствора и доводили уровень рН до 5,8. В реакционную колбу добавляли 5,5 г глюкозы и перемешивали до полного растворения. В другую колбу объемом 50 мл добавляли 0,3 г иминредуктазы ES-IRED103, 0,03 г глюкозодегидрогеназы ES-GDH109 и 0,01 г соли NADP и перемешивали до полного растворения. Затем раствор из второй колбы медленно добавляли в первую колбу, нагревали до 37°С и перемешивали в течение 24 часов. Раствор фильтровали, фильтрат экстрагировали хлороформом, сушили над безводным сульфатом натрия и концентрировали с получением 2,2 г (S)-норникотина. Оптическая чистота составляет 99,5%.
Пример 5
В этом примере предложен способ синтеза (S)-норникотина. Ниже описано его осуществление.
В трехгорлую кругл одонную колбу объемом 50 мл помещали 3 г миосмина, добавляли 15 мл 0,1 М фосфатного буферного раствора и доводили уровень рН до 5,8. В реакционную колбу добавляли 5,5 г глюкозы и перемешивали до полного растворения. В другую колбу объемом 50 мл добавляли 0,3 г иминредуктазы IRED1321110, 0,03 г глюкозодегидрогеназы ES-GDH102 и 0,01 г соли NADP и перемешивали до полного растворения. Затем раствор из второй колбы медленно добавляли в первую колбу, нагревали до 37°С и перемешивали в течение 24 часов. Раствор фильтровали, фильтрат экстрагировали хлороформом, сушили над безводным сульфатом натрия и концентрировали с получением 2,3 г (S)-норникотина. Оптическая чистота составляет 99,5%.
Пример 6
В этом примере предложен способ синтеза (S)-норникотина. Ниже описано его осуществление.
В трехгорлую круглодонную колбу объемом 1000 мл помещали 100 г миосмина, добавляли 650 мл 0,1 М фосфатного буферного раствора и доводили уровень рН до 6,2. В реакционную колбу добавляли 180 г глюкозы и перемешивали до тех пор, пока раствор не становился прозрачным. В колбу объемом 500 мл добавляли 100 г раствора иминредуктазы ES-TRED103 с концентрацией 10%, 50 г раствора глюкозодегидрогеназы ES-GDH102 с концентрацией 10% и 5 г соли NADP в колбе объемом 500 мл и перемешивали до получения прозрачного раствора. Раствор из колбы медленно добавляли в трехгорлую круглодонную колбу на 1000 мл, нагревали до 37°С и перемешивали в течение 36 часов. Раствор фильтровали, фильтрат экстрагировали хлороформом, сушили над безводным сульфатом натрия и концентрировали, получая 75 г (S)-норникотина.
Химическую чистоту и оптическую чистоту полученного (S)-норникотина определяли отдельно с помощью высокоэффективного жидкостного хроматографа.
Конкретный метод работы является таким же, как и в примере 2. Хроматограмма представлена на фиг. 9, где расчетная химическая чистота составляет 95,9%; и хиральная хроматограмма представлена на фиг. 10, где оптическая чистота составляет 99,9%. Пример 7
В этом примере предложен способ синтеза (S)-никотина. Ниже описано его осуществление.
В каждую из трех трехгорлых колб объемом 500 мл помещали 40 г (S)-норникотина, 32 г 37% раствора формальдегида и 25 г 85% раствора муравьиной кислоты, нагревали до 60°С и осуществляли реакцию в течение 24 ч. После завершения реакции добавляли гидроксид натрия, чтобы довести уровень рН до 12. Водные фазы экстрагировали метил-трет-бутиловым эфиром, экстракты объединяли, концентрировали и дистиллировали при пониженном давлении с получением 29 г (S)-никотина в виде бесцветной жидкости.
Полученный продукт охарактеризован с помощью ЯМР 1Н. На фиг. 1 представлен характеристический спектр ЯМР, содержащий следующие данные: ЯМР 1Н (400 МГц, CDCl3), 5 (м. д.): 8,54 (1H, d), 8,50 (1H, dd), 7,70 (1H, dt), 7,24-7,27 (1H, m), 3,22-3,27 (1H, m), 3,08 (1H, t), 2,27-2,34 (1H, m), 2,17-2,24 (1H, m), 2,16 (3Н, m), 1,91-2,02 (1H, m), 1,79-1,87 (1H, m), 1,68-1,76 (1H, m). Показано, что (S)-никотина был успешно синтезирован.
Химическую чистоту приготовленного образца (S)-никотина определяли с помощью высокоэффективного жидкостного хроматографа. Как представлено на фиг. 12, обнаруженная чистота составляет 99,9%. Оптическую чистоту образца определяли с помощью высокоэффективного жидкостного хроматографа и хиральной хроматографической колонки. На фиг. 13 представлена хиральная хроматограмма рацемата никотина, где пик при 5,43 мин соответствует (S)-никотину, пик при 6,17 мин соответствует (Я)-никотину, и площади этих пиков составляют 50,1% и 49,9% соответственно. На фиг. 14 представлена хиральная хроматограмма образца, и из детектирующей хроматограммы видно, что площадь пика при 5,43 мин составляет 99,65%, то есть чистота (S)-никотина составляет 99,65%.
Пример 8
В этом примере предложен способ синтеза (S)-никотина. Ниже описано его осуществление.
В каждую из трех трехгорлых колб объемом 500 мл помещали 107 г (S)-норникотина и 80 г 37% раствора формальдегида и нагревали до 75°С.Добавляли в капельном режиме 60 г 85% раствора муравьиной кислоты. После добавления раствор реагировал в течение 24 ч при поддержании температуры. После завершения реакции добавляли гидроксид натрия для доведения уровня рН до 12. Водные фазы экстрагировали метил-трет-бутиловым эфиром, экстракты объединяли, концентрировали и дистиллировали при пониженном давлении с получением 80 г (S)-никотина в виде бесцветной жидкости.
Химическую чистоту и оптическую чистоту полученного (S)-никотина определяли отдельно с помощью высокоэффективного жидкостного хроматографа. Конкретные операции такие же, как и в примере 5. Хроматограмма представлена на фиг. 15, где расчетная химическая чистота составляет 99,7%; и хиральная хроматограмма представлена на фиг. 16, где оптическая чистота составляет 99,87%.
Пример 9
В этом примере обсуждается предпочтительный тип иминредуктазы. Ниже описано осуществление соответствующего способа.
Порошок фермента IRED взвешивали и помещали по 10 мг в каждый из реакционных сосудов объемом 2 мл. В каждый сосуд добавляли по 9,2 мг глюкозы, 1 мг натриевой соли NAD, 1 мг натриевой соли NADP, 2 мг глюкозодегидрогеназы ES-GDH105 и 900 мкл 0,1 М фосфатного буферного раствора (рН 7,0) и встряхивали до тех пор, пока раствор не становился прозрачным. Затем в каждый сосуд добавляли 5 мг миосмина и 100 мкл DMSO. Сосуды плотно закрывали, помещали во встряхивающее устройство с постоянной температурой 30°С и встряхивали в течение ночи. Образцы анализировали с помощью ВЭЖХ в отношении степени превращения и хиральности. Результаты представлены в таблице 1.
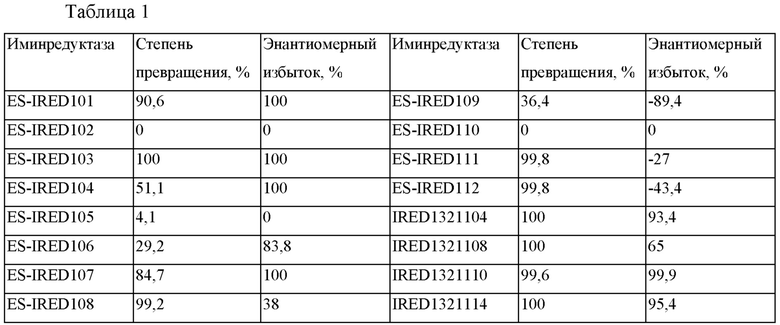
Как видно из данных таблицы 1, ES-TRED103 и IRED1321110 представляют собой более предпочтительные типы иминредуктазы.
Пример 10
В этом примере обсуждается предпочтительный тип глюкозодегидрогеназы. Ниже описано осуществление соответствующего способа.
Порошок фермента GDH взвешивали и помещали по 2 мг в каждый из реакционных сосудов объемом 2 мл. В каждый сосуд добавляли 9,2 мг глюкозы, 1 мг натриевой соли NAD, 1 мг натриевой соли NADP, 10 мг ES-IRED103 и 900 мкл 0,1 М фосфатного буферного раствора (рН 7,0) и встряхивали до тех пор, пока раствор не становился прозрачным. Затем в каждый сосуд добавляли 150 мг миосмина и 100 мкл DMSO. Сосуды плотно закрывали, помещали во встряхивающее устройство с постоянной температурой 30°С и встряхивали в течение ночи. Образцы анализировали с помощью ВЭЖХ для определения степени превращения. Результаты представлены в таблице 2.
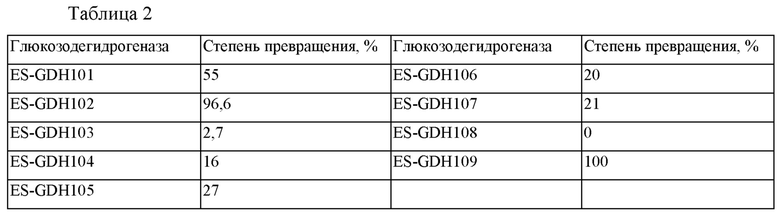
Как можно видеть из данных таблицы 2, ES-GDH109 представляет собой более предпочтительный тип глюкозодегидрогеназы.
Пример 11
В этом примере обсуждается предпочтительный тип кофермента. Ниже описано осуществление соответствующего способа.
Навески 1 мг натриевой соли NAD и 1 мг натриевой соли NADP помещали в каждый из реакционных сосудов объемом 2 мл, соответственно. В каждый сосуд добавляли 9,2 мг глюкозы, 2 мг порошка фермента ES-GDH109, 10 мг ES-IRED103 и 900 мкл 0,1 М фосфатного буферного раствора (рН 7,0) и встряхивали до тех пор, пока раствор не становился прозрачным. Затем в каждый сосуд добавляли 150 мг миосмина и 100 мкл DMSO. Сосуды плотно закрывали, помещали во встряхивающее устройство с постоянной температурой 30°С и встряхивали в течение ночи. Образцы анализировали с помощью ВЭЖХ для определения степени превращения. Результаты представлены в таблице 3.
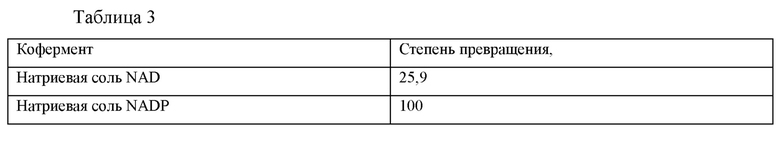
Как можно видеть из данных таблицы 3, натриевая соль NADP представляет собой более предпочтительный тип кофермента.
Заявитель утверждает, что, хотя способ синтеза (S)-никотина в настоящей заявке описан посредством примеров, которые представлены выше, настоящая заявка не ограничивается описанными выше примерами, и это означает, что реализация настоящей заявки не должна обязательно зависеть от примеров, описанных выше. Для специалистов в данной области техники должно быть очевидно, что любые улучшения, внесенные в настоящую заявку, эквивалентные замены исходных материалов продукта, добавление вспомогательных ингредиентов, выбор конкретных методов и другие условия в настоящей заявке во всех случаях находятся в пределах объема правовой защиты и объема раскрытия настоящей заявки.
Хотя предпочтительные варианты осуществления настоящей заявки подробно описаны выше, настоящая заявка не ограничивается подробностями вариантов осуществления, которые описаны выше, и в технические решения настоящей заявки могут быть внесены различные простые модификации без отклонения я от технической концепции настоящей заявки. Все эти простые модификации входят в объем правовой защиты настоящей заявки.
Кроме того, следует отметить, что если отсутствует противоречие, конкретные технические признаки, описанные в вариантах осуществления, которые представлены выше, могут быть объединены любым подходящим образом. Во избежание ненужного повторения различные возможные варианты объединения дополнительно не описаны в настоящей заявке.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРИМЕНЕНИЯ ВОССТАНОВЛЕНИЯ ДЛЯ ПОЛУЧЕНИЯ (S)-НИКОТИНА | 2022 |
|
RU2833281C2 |
| СПОСОБ ПОЛУЧЕНИЯ L-НИКОТИНА | 2022 |
|
RU2836890C2 |
| СПОСОБ ПРОИЗВОДСТВА НИКОТИНА | 2018 |
|
RU2753548C1 |
| РЕАГЕНТНЫЕ МАТЕРИАЛЫ И СООТВЕТСТВУЮЩИЕ ТЕСТОВЫЕ ЭЛЕМЕНТЫ | 2013 |
|
RU2652888C2 |
| ХИРАЛЬНОЕ РАЗДЕЛЕНИЕ СМЕСИ ЭНАНТИОМЕРОВ НИКОТИНА | 2018 |
|
RU2753492C1 |
| СПОСОБ ПОЛУЧЕНИЯ L-BPA | 2016 |
|
RU2688676C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИДЕНОВОГО АЦЕТАМИДНОГО ПРОИЗВОДНОГО | 2020 |
|
RU2813203C2 |
| СТАБИЛИЗАЦИЯ ДЕГИДРОГЕНАЗ СТАБИЛЬНЫМИ КОФЕРМЕНТАМИ | 2009 |
|
RU2499834C2 |
| ПРОИЗВОДНЫЕ КАРБОКСИЗАМЕЩЕННЫХ (ГЕТЕРО)АРОМАТИЧЕСКИХ КОЛЕЦ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2733750C2 |
| СИСТЕМА ВОССТАНОВЛЕНИЯ КОНФЕРМЕНТА, НАБОР ДЛЯ ФЕРМЕНТАТИВНОГО ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ АНАЛИЗИРУЕМОГО ВЕЩЕСТВА И ФЕРМЕНТАТИВНЫЙ СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ АНАЛИЗИРУЕМОГО ВЕЩЕСТВА | 1996 |
|
RU2184778C2 |
Предложен способ синтеза (S)-никотина, в котором в условиях системы циркуляции кофермента, включая кофермент, глюкозу и глюкозодегидрогеназу, с применением кофермента в качестве донора водорода и иминредуктазы в качестве катализатора миосмин подвергают каталитическому восстановлению с получением (S)-норникотина, а затем (S)-норникотин подвергают метилированию с получением (S)-никотина, где кофермент представляет собой соль NADP и/или соль NAD. Технический результат изобретения заключается в осуществлении способа синтеза (S)-никотина с высокой химической чистотой и высокой оптической чистотой. 8 з.п. ф-лы, 3 табл., 16 ил., 11 пр.
1. Способ синтеза (S)-никотина, в котором в условиях системы циркуляции кофермента, включая кофермент, глюкозу и глюкозодегидрогеназу, с применением кофермента в качестве донора водорода и иминредуктазы в качестве катализатора миосмин подвергают каталитическому восстановлению с осуществлением следующих реакций с получением (S)-никотина:
где кофермент представляет собой соль NADP и/или соль NAD.
2. Способ синтеза (S)-никотина по п. 1, в котором кофермент включает соль NADP.
3. Способ синтеза (S)-никотина по п. 1 или 2, в котором глюкозодегидрогеназа представляет собой любую глюкозодегидрогеназу или комбинацию по меньшей мере двух глюкозодегидрогеназ из ES-GDH101, ES-GDH102 или ES-GDH109, предпочтительно ES-GDH109.
4. Способ синтеза (S)-никотина по любому из пп. 1-3, в котором иминредуктаза представляет собой любую иминредуктазу или комбинацию по меньшей мере двух иминредуктаз из ES-TRED101, ES-IRED103, ES-IRED104, ES-IRED107, IRED1321104, IRED1321108, IRED1321110 или IRED1321114, предпочтительно ES-TRED103 или IRED1321110.
5. Способ синтеза (S)-никотина по любому из пп. 1-4, в котором каталитическое восстановление осуществляют при температуре, составляющей от 15 до 45°С и предпочтительно от 25 до 30°С; предпочтительно каталитическое восстановление осуществляют в течение от 8 до 72 ч; предпочтительно каталитическое восстановление проводят в условиях перемешивания при скорости вращения от 150 до 400 об/мин.
6. Способ синтеза (S)-никотина по любому из пп. 1-5, в котором каталитическое восстановление осуществляют в буферном растворе, причем буферный раствор представляет собой фосфатный буферный раствор, трис(гидроксиметил)метиламингидрохлоридный буферный раствор или триэтаноламингидрохлоридный буферный раствор; предпочтительно буферный раствор имеет значение рН, составляющее от 5,0 до 9,0 и предпочтительно от 5,8 до 6,5.
7. Способ синтеза (S)-никотина по любому из пп. 1-6, в котором метилирование включает смешивание и взаимодействие (S)-норникотина с формальдегидом и муравьиной кислотой с получением (S)-никотина.
8. Способ синтеза (S)-никотина по любому из пп. 1-7, в котором метилирование осуществляют при температуре от 55 до 80°С; предпочтительно метилирование осуществляют в течение от 8 до 48 часов.
9. Способ синтеза (S)-никотина по любому из пп. 1-8, включающий смешивание миосмина, кофермента, глюкозы, глюкозодегидрогеназы, буферного раствора и иминредуктазы и взаимодействие смеси при температуре от 15 до 45°С в течение от 8 до 72 часов с получением (S)-норникотина, причем массовое соотношение миосмина, кофермента и иминредуктазы составляет 1:(0,001-0,05):(0,05-0,1); а также последующее смешивание (S)-норникотина с формальдегидом и муравьиной кислотой и взаимодействие смеси при температуре от 55 до 80°С в течение от 8 до 48 часов с получением (S)-никотина.
| US 20160326134 A1, 10.11.2016 | |||
| WO 2014174505 A2, 30.10.2014 | |||
| PEYTON J | |||
| et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Synthesis of (R) and (S)-Nornicotine of High Enantiomeric Purity, J | |||
| Org | |||
| Chem., 1982, vol.47, p.4165-4167 | |||
| GUO C | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
