[Область техники, к которой относится изобретение]
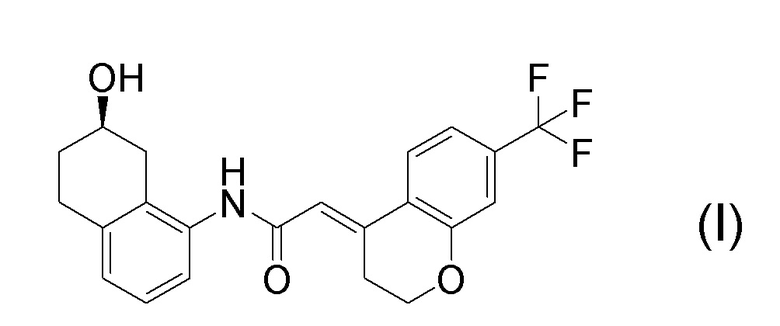
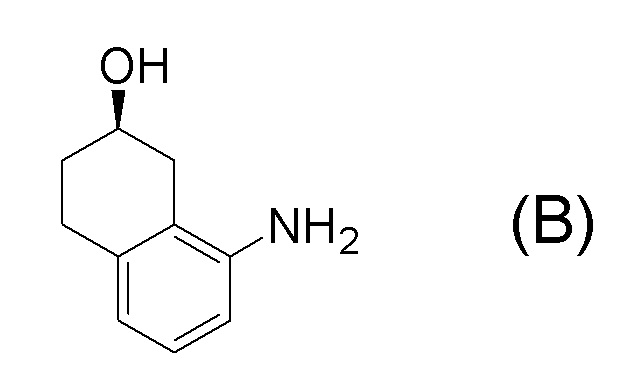
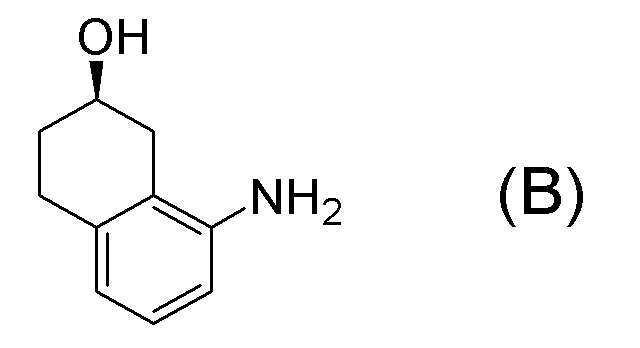
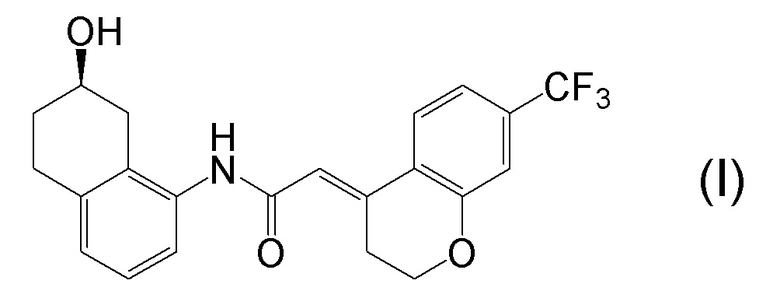
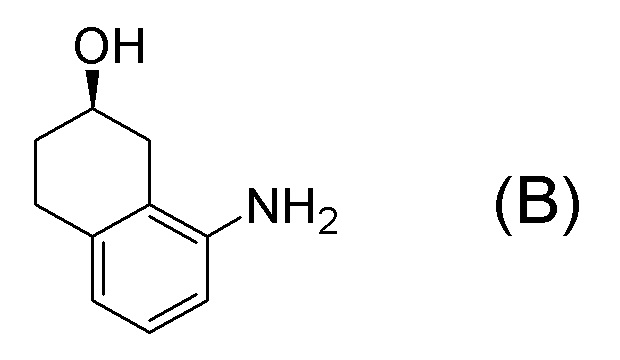
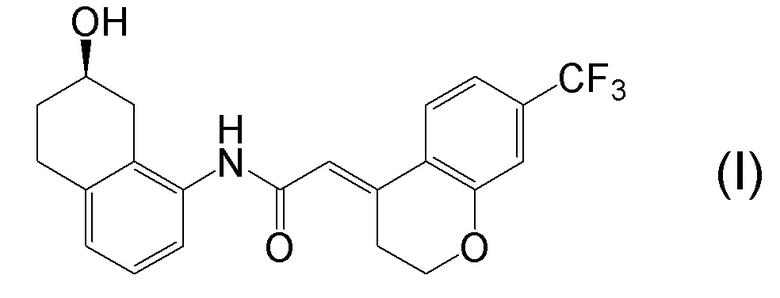
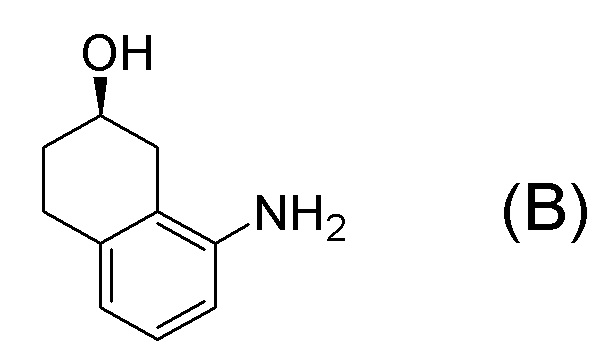
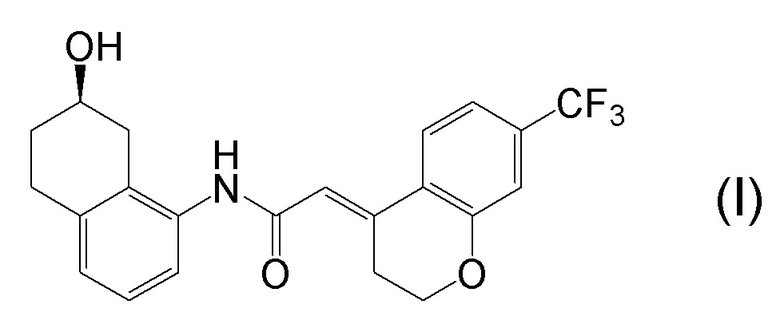
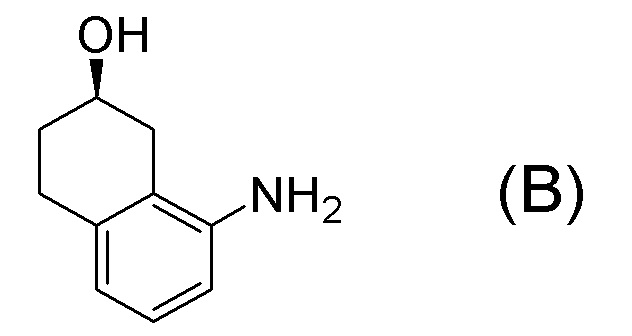
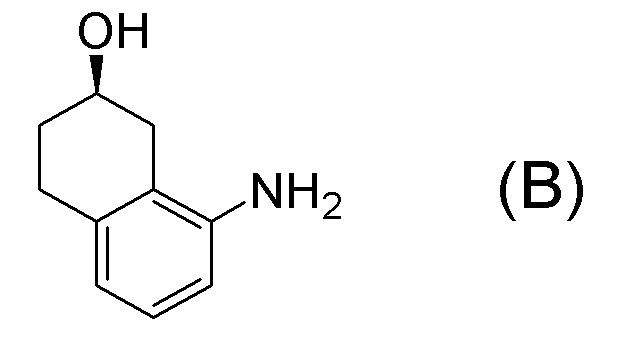
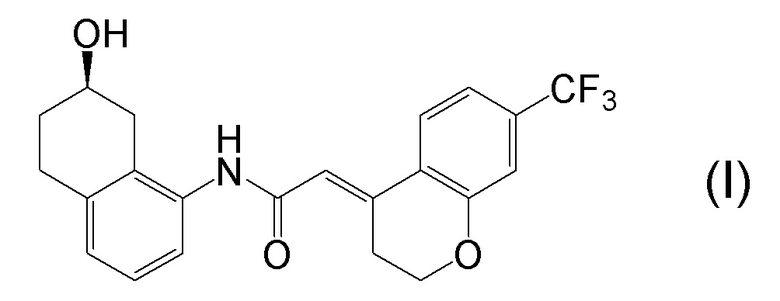
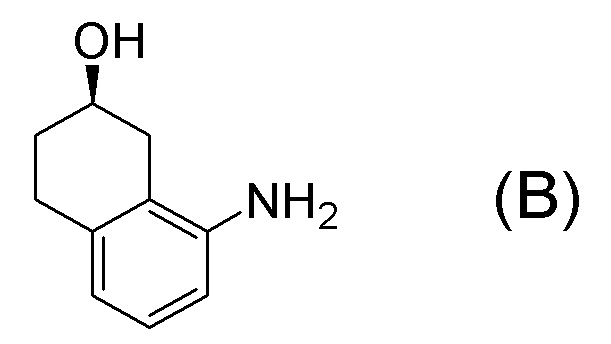
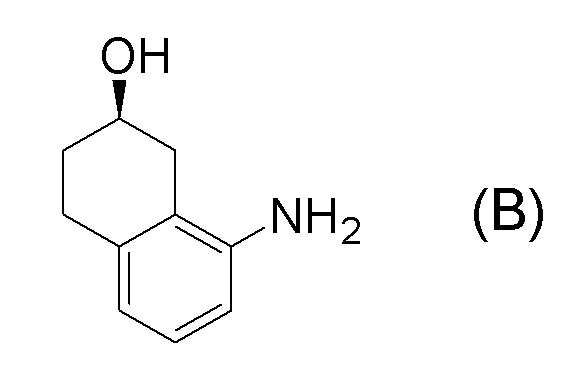
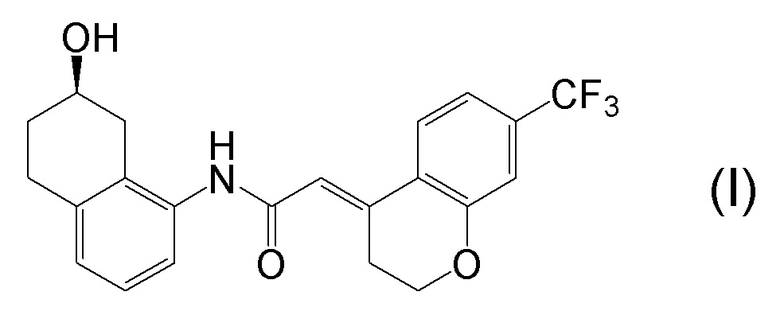
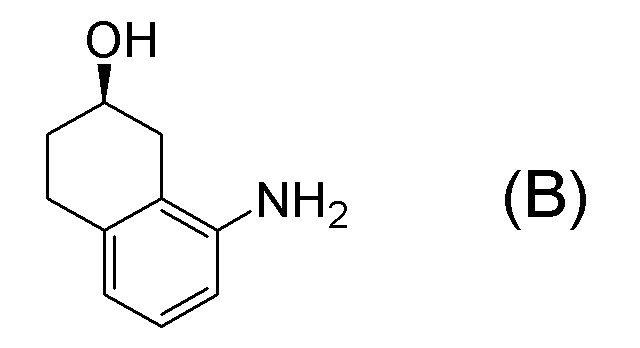
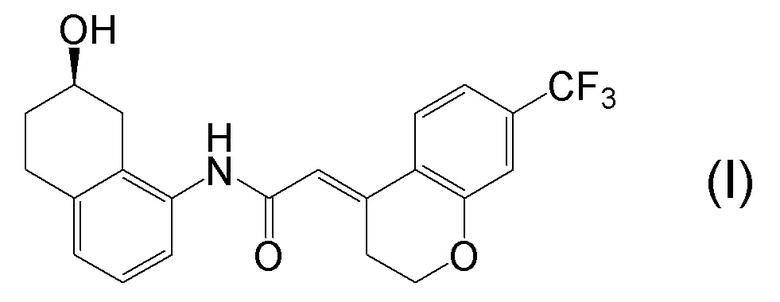
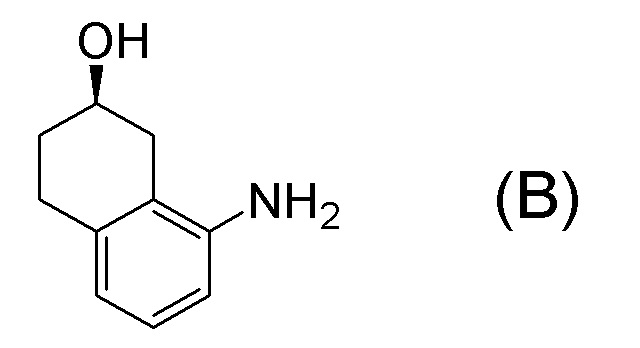
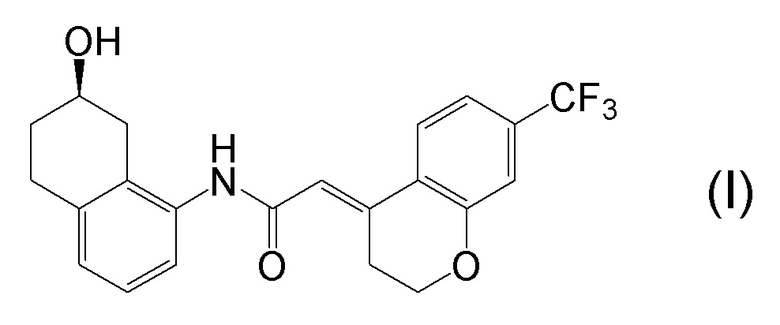
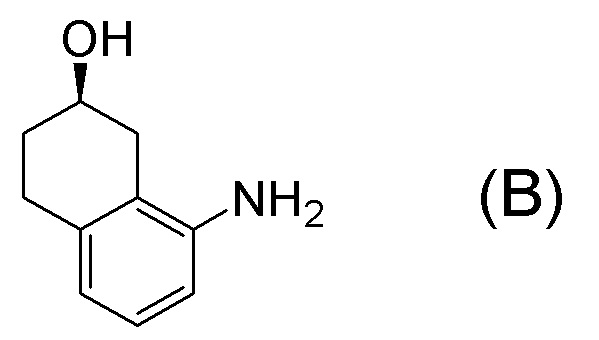
[0001] Настоящее изобретение относится к новому способу получения (E)-2-(7-трифторметилхроман-4-илиден)-N-((7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)ацетамида, представленного формулой (I), который представляет собой гетероциклиденовое ацетамидное производное. Кроме того, настоящее изобретение относится к новому способу получения (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола, представленного формулой (B), или его соли, который представляет собой промежуточное соединение пригодное в получении соединения, представленного формулой (I).
[Уровень техники]
[0002] (E)-2-(7-трифторметилхроман-4-илиден)-N-((7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)ацетамид, представленный формулой (I), представляет собой антагонист ваннилоидного канала-1 с транзиторным рецепторным потенциалом (TRPV1), и предполагается в качестве профилактического и/или терапевтического агента для заболеваний, связанных с рецептором TRPV1 (например, боли (например, нейропатической боли, диабетической невралгии, послеоперационной боли, остеоартроза, боли при ревматоидном артрите, воспалительной боли, боли при раке, мигрени и подобных), нервных расстройств, повреждения нервов, нейродегенерации, хронической обструктивной болезни легких, астмы, ринита, воспаления слизистых оболочек, например, в глазах, нервного заболевания кожи, воспалительного заболевания кожи, аллергического заболевания, недержания мочи, императивного недержания, гиперактивного мочевого пузыря, цистита, зуда и подобных) (патентная литература 1).
[C1]
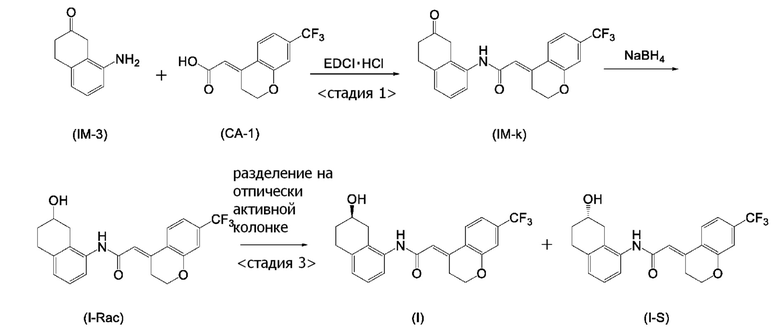
[0003] WO 2007/010383 (патентная литература 1) описывает способ получения соединения, представленного формулой (I). В данном документе, соединение, представленное формулой (I), получают стадиями <стадия 1>-<стадия 3>, показанными далее (схема A).
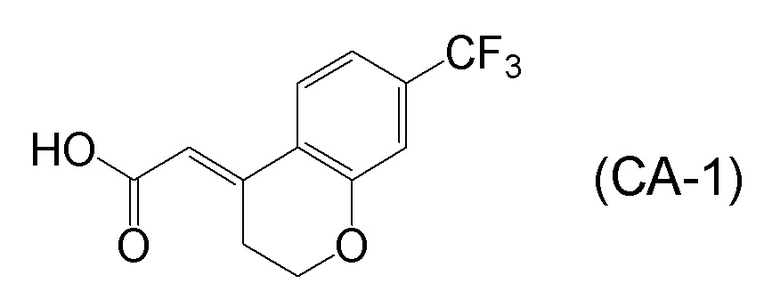
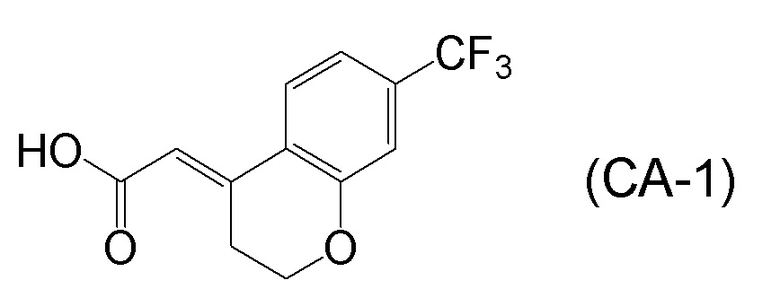
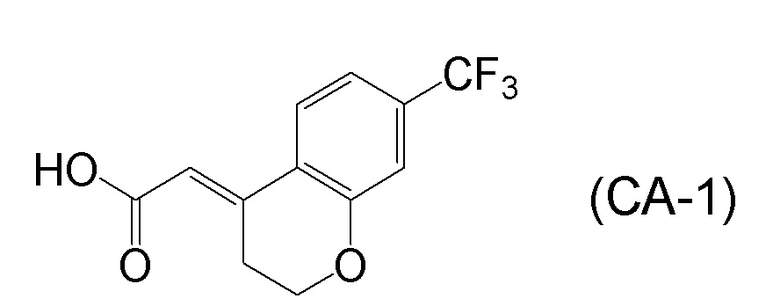
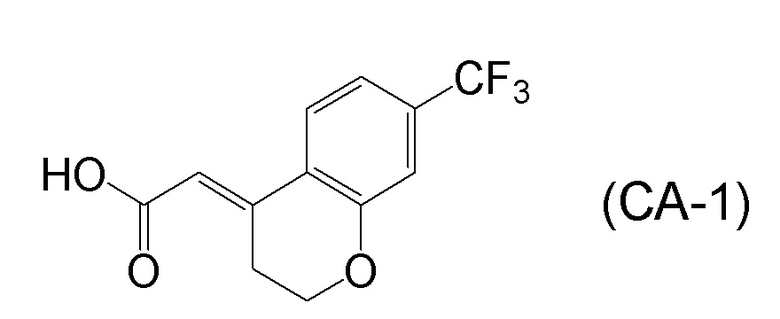
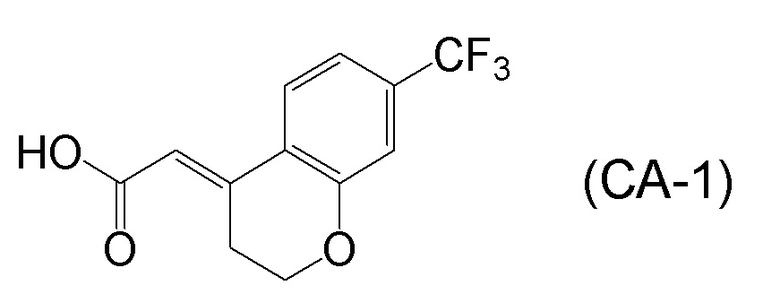
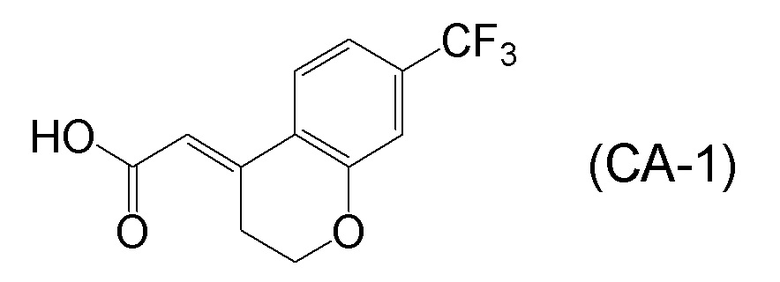
<Стадия 1> Соединение, представленное формулой (IM-k), получают проведением реакции конденсации, применяя 8-амино-3,4-дигидронафталин-2(1H)-он (формула (IM-3)), полученный согласно способу, известному из данного документа (например, WO 2005/040100 (патентная литература 2) и подобные) и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусную кислоту (формула (CA-1) в CAS No. 920334-15-2, непатентная литература 1) и конденсирующий агент гидрохлорид (1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI·HCl)).
<Стадия 2> Соединение, представленное формулой (IM-k), восстанавливают боргидридом натрия, получая соединение, представленное формулой (I-Rac).
<Стадия 3> Соединение, представленное формулой (I-Rac), оптически разделяют на оптически активной колонке, получая соединение, представленное формулой (I), и соединение, представленное формулой (I-С), которое представляет собой его изомер.
[0004] Однако, в данном способе получения соединение, представленное формулой (I), получают проведением колоночного разделения на финальной стадии, и трудно повторно применять соединение, представленное формулой (I-С), полученное после колоночного разделения.
[0005]
[C2]
(Схема А)
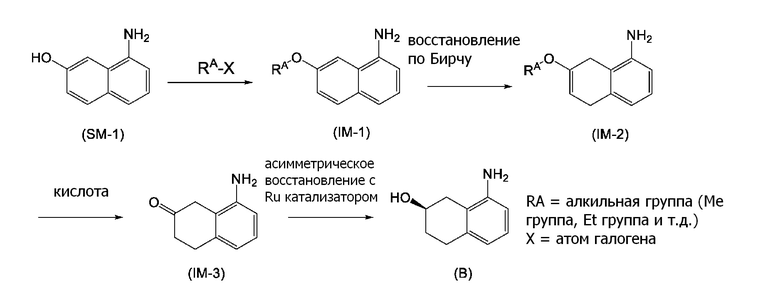
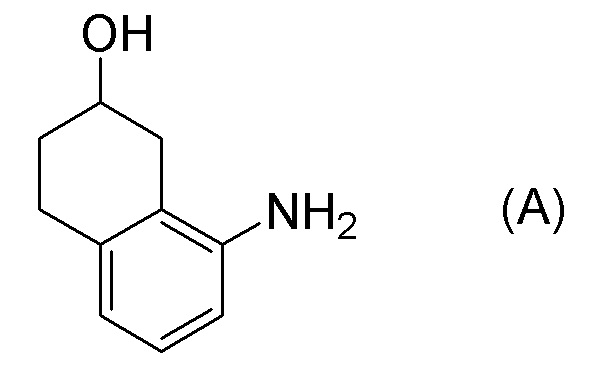
[0006] Между тем, WO 2005/040100 (патентная литература 2), WO 2003/095420 (патентная литература 3), WO 2005/040119 (патентная литература 4), и WO 2010/127855 (патентная литература 5) описывают способы получения соединения, представленного формулой (B), которое соответствует частичной структурной формуле формулы (I).
[C3]
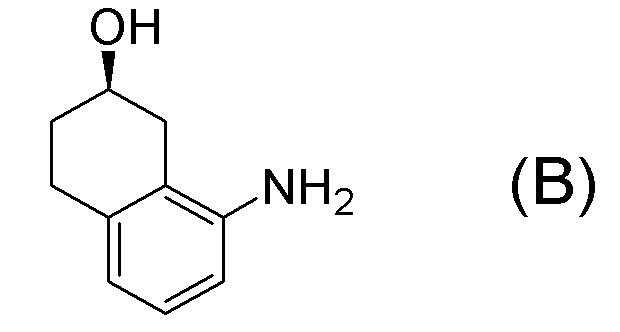
В данной литературе, 8-амино-3,4-дигидронафталин-2(1H)-он (формула (IM-3)) получают реакциями алкилирования фенольной группы, восстановления по Берчу, и деблокированием алкильной группы, применяя 8-аминонафталин-2-ол (формула (SM-1)) в качестве исходного соединения, и посредством этого, соединение, представленное формулой (B), получают его асимметрическим восстановлением в присутствии Ru катализатора (схема 1).
[0007] Однако, в данных способах получения, восстановление по Берчу применяют на одной стадии, и металлический (Ru) катализатор применяют при асимметрическом восстановлении на финальной стадии, следовательно, требуется стадия восстановления остаточного содержания металла (Ru) в полученном соединении.
[0008]
[C4]
(Схема 1)
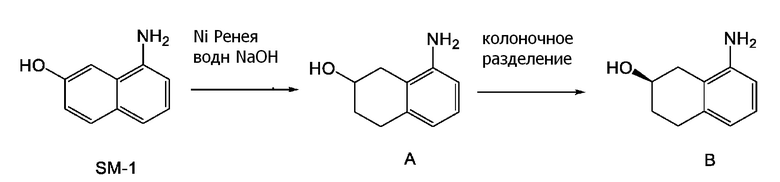
[0009] Кроме того, WO 2009/050289 (патентная литература 6), WO 2010/045401 (патентная литература 7) и WO 2010/045402 (патентная литература 8) также описывает способы получения соединения, представленного формулой (B). В данной литературе, соединение, представленное формулой (B), получают разделением, применяя оптически активную колонку после получения рацемического 8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула A) селективным восстановлением нафталинового кольца, применяя 8-аминонафталин-2-ол (формула (SM-1)) в качестве исходного соединения (схема 2).
[0010] Однако, в данном способе получения, трудно повторно применять другие изомеры (S-формы), полученные после колоночного разделения.
[0011]
[C5]
(Схема 2)
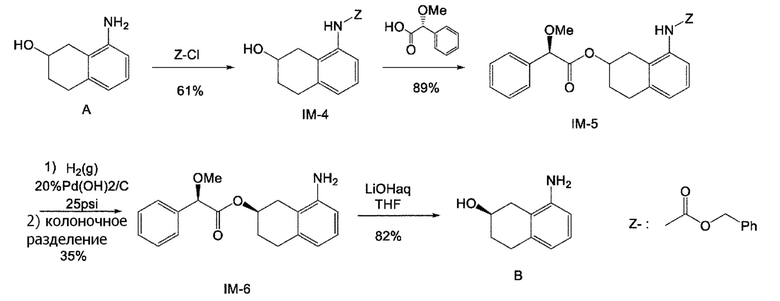
[0012] Кроме того, WO 2009/055749 (патентная литература 9) также описывает способ получения соединения, представленного формулой (B). В данной литературе, соединение, представленное формулой (B), получают колоночным отделением диастереомера, полученного после диастереомерного разделения рацемата с введенным хиральным вспомогательным веществом, представленного формулой (A) (схема 3).
[0013] Однако, также в данном способе получения трудно повторно применять другие изомеры (S-формы), полученные после колоночного разделения.
[0014]
[C6]
(Схема 3)
[0015] Способы получения соединение, представленного формулой (B), описанные в соответствующей литературе, имеют проблемы, такие как типы реакций в способе получения, реагенты, которые будут применять, трудности с повторным применением других изомеров, полученных после отделения рацемата или диастереоизомера на колонке. Следовательно, улучшенный способ получения требуется для крупномасштабного синтеза или промышленного получения соединения, представленного формулой (B). То есть при рассмотрении крупномасштабного синтеза или промышленного получения соединения, представленного формулой (B), требуется найти новый способ получения, отличный от способов получения, описанных в соответствующей литературе. Поскольку способ получения для крупномасштабного синтеза соединения, представленного формулой (B), с высоким выходом и высокой оптической чистотой еще не найден, считается, что вышеупомянутые проблемы получения соединения, представленного формулой (B), можно решить, если будет найден способ широкомасштабного синтеза соединения, представленного формулой (B), в меньшее количество стадий, с высоким химическим выходом и высокой оптической чистотой.
[0016] Патентная заявка США No. 5136103 (патентная литература 10) и подобные описывает способ окисления вторичного спирта до кетона, применяя 2,2,6,6-тетраметилпиперидин-1-оксилльный радикал (TEMPO) в качестве окислителя. Однако реакция окисления TEMPO, в которой 1,2,3,4-тетрагидронафталин (например, трет-бутил-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамат и подобные), который содержит замещенную аминогруппу и гидроксильную группу в молекуле, применяют в качестве исходного соединения, неизвестна. Кроме того, реакция окисления TEMPO химией в потоке (реакция в потоке), в которой соединение применяют в качестве исходного соединения, также неизвестна.
[0017] Патентная заявка США No. 5225339 (патентная литература 11) и патентная заявка США No. 5342767 (патентная литература 12) описывают восстановление кетонов редуктазой, полученной из кефира, полученного с помощью Lactobacillus, но ферментативная реакция, приспособленная к кето соединениям, таким как замещенные защитной группой амино-3,4-дигидронафталин-2(1H)-он или β-тетралон, не описана.
[0018] Advanced Synthesis & Catalysis, 350 (14+15), p2322-2328, 2008 (непатентная литература 2) описывает ферментативная реакция кетонов α- или β-тетралона (редуктаза: полученная из кефира, полученного с помощью Lactobacillus). Однако ясно заявлено, что восстановительная реакция кетонов β-тетралона не протекает, когда применяют редуктазу, полученную из кефира, полученного с помощью Lactobacillus.
[0019] WO 2018/205948 (патентная литература 13) описывает 8-бром-1,2,3,4-тетрагидронафталин-2-ол (CAS No. 444619-84-5, непатентная литература 3) и способ его получения, но (R)-8-бром-1,2,3,4-тетрагидронафталин-2-ол, который представляет собой его хиральную форму, и его способ получения неизвестны.
[0020] CAS Registry описывает 8-фтор-1,2,3,4-тетрагидронафталин-2-ол (CAS No. 1823867-35-1, непатентная литература 4), но (R)-8-фтор-1,2,3,4-тетрагидронафталин-2-ол, который представляет собой его хиральную форму, и способ его получения неизвестны. Кроме того, CAS Registry описывает 8-хлор-1,2,3,4-тетрагидронафталин-2-ол (CAS No. 1823929-47-0, непатентная литература 5), но (R)-8-хлор-1,2,3,4-тетрагидронафталин-2-ол, который представляет собой его хиральную форму, и способ его получения неизвестны.
[0021] Bioorganic & Medicinal Chemistry Letters, 18 (6), p1830-1834, 2008 (непатентная литература 6) описывает способ получения 8-амино-6-фтор-1,2,3,4-тетрагидронафталин-2-ола (выход 18%) восстановительным аминированием 8-бром-6-фтор-1,2,3,4-тетрагидронафталин-2-ола с Pd катализатором (Pd2(dba)3) и трет-бутилкарбаматом, и последующим удалением Boc группы.
[Список цитирований]
[Патентная литература]
[0022]
[Патентная литература 1] WO 2007/010383
[Патентная литература 2] WO 2005/040100
[Патентная литература 3] WO 2003/095420
[Патентная литература 4] WO 2005/040119
[Патентная литература 5] WO 2010/127855
[Патентная литература 6] WO 2009/050289
[Патентная литература 7] WO 2010/045401
[Патентная литература 8] WO 2010/045402
[Патентная литература 9] WO 2009/055749
[Патентная литература 10] патентная заявка США No. 5136103
[Патентная литература 11] патентная заявка США No. 5225339
[Патентная литература 12] патентная заявка США No. 5342767
[Патентная литература 13] WO 2018/205948
[0023]
[Непатентная литература]
[Непатентная литература 1] CAS No. 920334-15-2
[Непатентная литература 2] Advanced Synthesis & Catalysis, 350 (14+15), p2322-2328, 2008
[Непатентная литература 3] CAS No. 444619-84-5
[Непатентная литература 4] CAS No. 1823867-35-1
[Непатентная литература 5] CAS No. 1823929-47-0
[Непатентная литература 6] Bioorganic & Medicinal Chemistry Letters, 18(6), p1830-1834, 2008
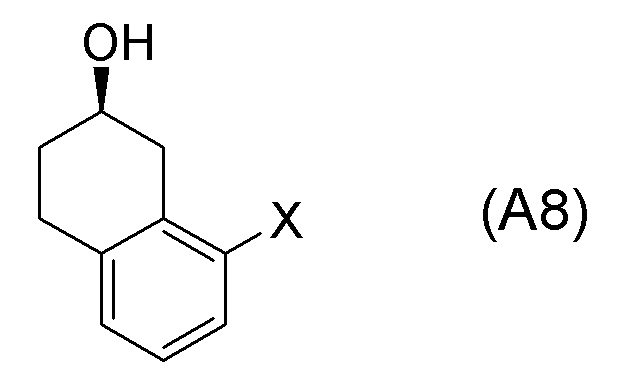
[Сущность настоящего изобретения]
[Техническая проблема]
[0024] В данных обстоятельствах, требуется новый способ получения вышеупомянутого соединения, представленного формулой (I).
[Решение проблемы]
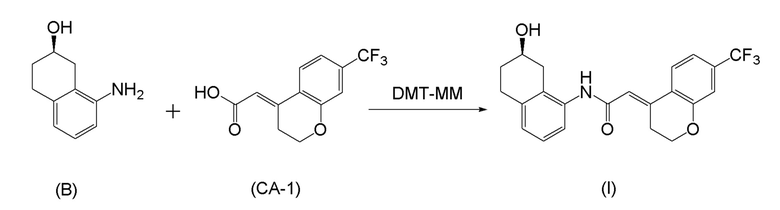
[0025] Изобретатели настоящего изобретения неоднократно проводили обширные исследования, чтобы решить вышеописанные проблемы. Как результат, изобретатели обнаружили новый способ более простого получения соединения, представленного формулой (I), с высоким выходом, и завершили настоящее изобретение на основе данной находки. То есть, изобретатели обнаружили новый способ получения соединения, представленного формулой (I), реакцией конденсации карбоновой кислоты, представленной формулой (CA-1), и аминоспирта, представленного формулой (B), или его соли, применяя DMT-MM (4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид) (CAS No: 3945-69-5) в качестве конденсирующего реагента (схема B).
[0026]
[C7]
(Схема B)
[0027] Изобретатели дополнительно обнаружили новый способ получения соединения, представленного формулой (B), или его соли, которое представляет собой промежуточное соединение, пригодное в получении соединения, представленного формулой (I), и завершили настоящее изобретение на основе данной находки (схема C-1).
[C8]
(Схема С-1)

[0028] Изобретатели еще обнаружили новый способ получения соединения, представленного формулой (B), или его соли, которое представляет собой промежуточное соединение, пригодное в получении соединения, представленного формулой (I), и завершили настоящее изобретение на основе данной находки (схема C-2).
[C9]
(Схема С-2)
[Эффект настоящего изобретения]
[0029] Настоящее изобретение обеспечивает новый способ получения соединения, представленного формулой (I), или соединения, представленного формулой (B), или его соли. Настоящее изобретение предпочтительно обеспечивает эффективный способ получения, пригодный для крупномасштабного синтеза или промышленного получения соединения, представленного формулой (I), или соединения, представленного формулой (B), или его соли. Способы получения согласно некоторым аспектам настоящего изобретения представляют собой способы, которые обеспечивают получение соединения, представленного формулой (I), или соединения, представленного формулой (B), или их соли с высоким выходом и промышленно выгодно, и, следовательно, данные способы являются очень пригодными в промышленности. Кроме того, некоторые другие аспекты обеспечивают новые соединения, представленные формулами (A-7) и (A8), которые представляют собой исходные соединения в получении соединения, представленного формулой (B), и его соли.
[Краткое описание чертежей]
[0030]
[Фигура 1] ФИГУРА 1 представляет собой пример устройства для реакции, применяемый для химии в потоке;
[Фигура 2] ФИГУРА 2 представляет собой фигуру, показывающую кристаллическую структуру HBr соли соединения, представленного формулой (B); и
[Фигура 3] ФИГУРА 3 представляет собой фигуру, показывающую кристаллическую структуру соединения, представленного формулой (I).
[Описание вариантов осуществления]
[0031] [Аспекты настоящего изобретения]
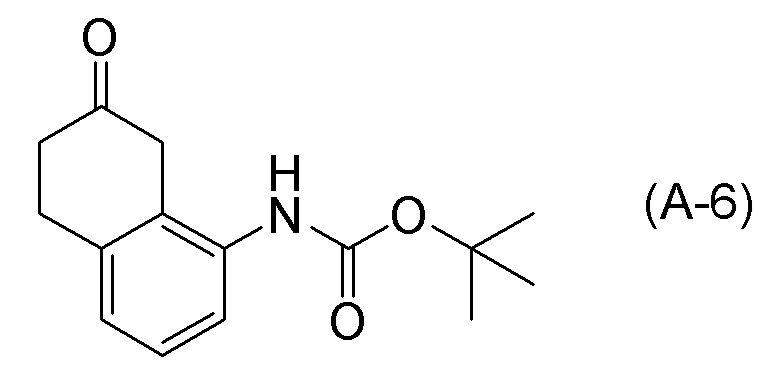
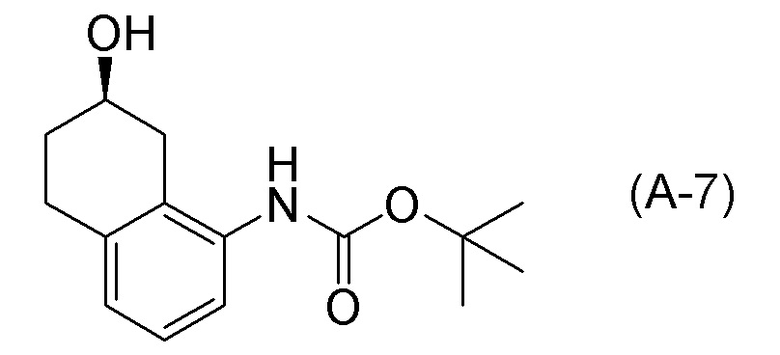
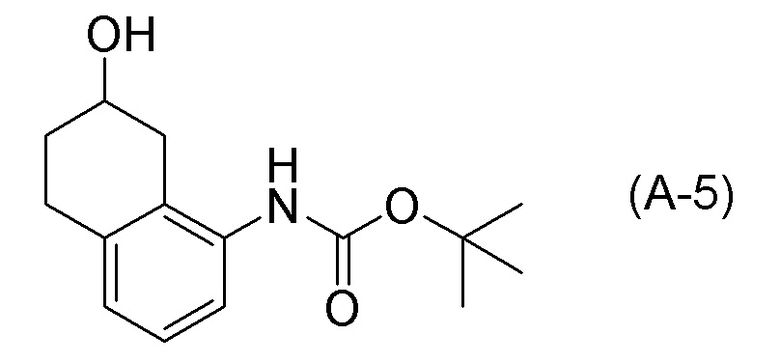
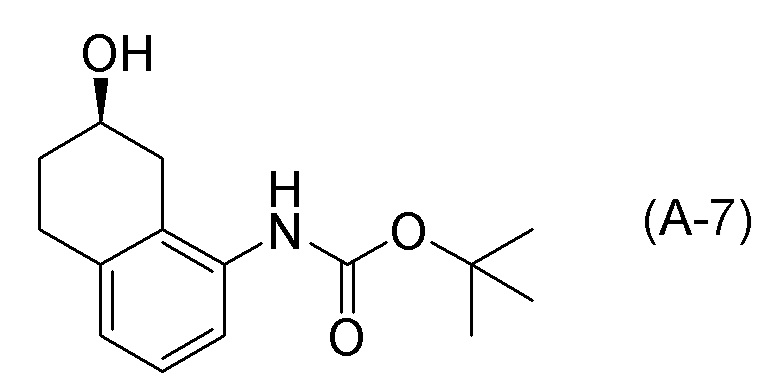
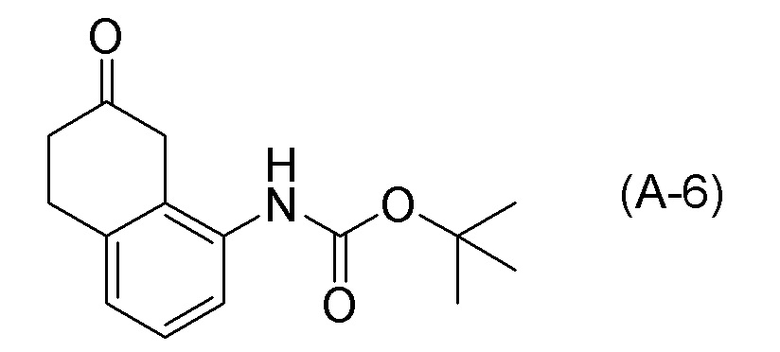
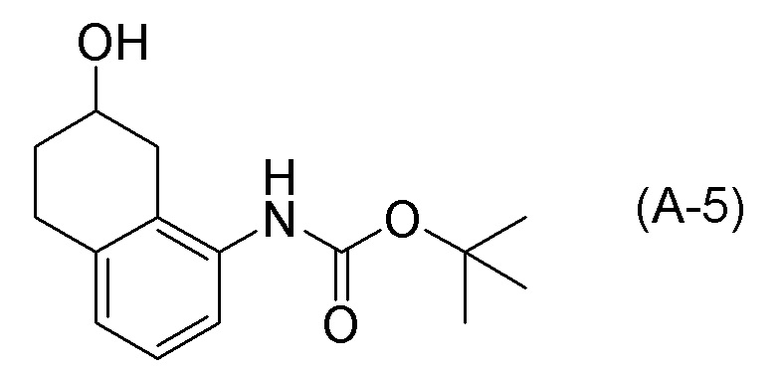
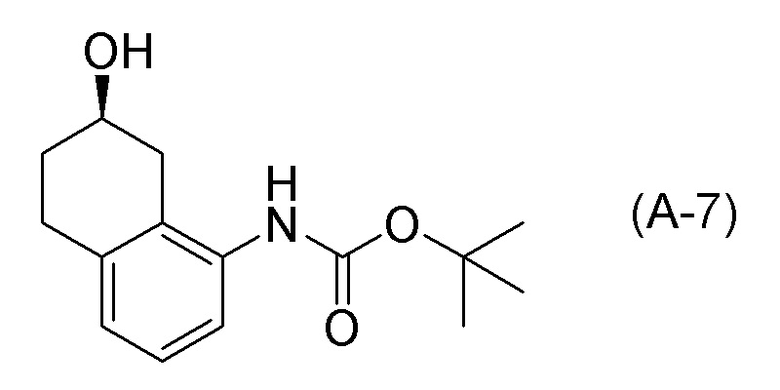
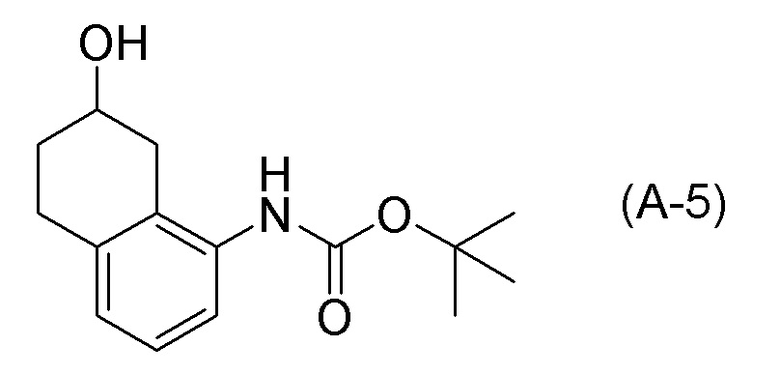
Обеспечивают способ получения соединения, представленного формулой (I). Некоторые аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A), в качестве исходного соединения. Некоторые другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A-5), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A-6), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A-7), в качестве исходного соединения.
[0032] Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (SM8), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (SM8-BR), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A8), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (A8-BR), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (I), применением соединения, представленного формулой (B), в качестве исходного соединения.
[0033] Кроме того, также обеспечивают способ получения соединения, представленного формулой (B) или его соли. Некоторые аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A), в качестве исходного соединения. Некоторые другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A-5), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A-6), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A-7), в качестве исходного соединения.
[0034] Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (SM8), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (SM8-BR), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A8), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (B), или его соли применением соединения, представленного формулой (A8-BR), в качестве исходного соединения.
[0035] Еще другие аспекты представляют собой способ получения соединения, представленного формулой (A-6), применением соединения, представленного формулой (A-5) или формулой (A-7), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (A-7), применением соединения, представленного формулой (A-6), в качестве исходного соединения. Еще другие аспекты представляют собой соединение, представленное формулой (A-7).
[0036] Еще другие аспекты представляют собой способ получения соединения, представленного формулой (A8), применением соединения, представленного формулой (SM8), в качестве исходного соединения. Еще другие аспекты представляют собой способ получения соединения, представленного формулой (A8-BR), применением соединения, представленного формулой (SM8-BR), в качестве исходного соединения. Еще другие аспекты представляют собой соединение, представленное формулой (A8), и соединение, представленное формулой (A8-BR).
Далее в настоящем изобретении каждый из аспектов будет описан конкретно.
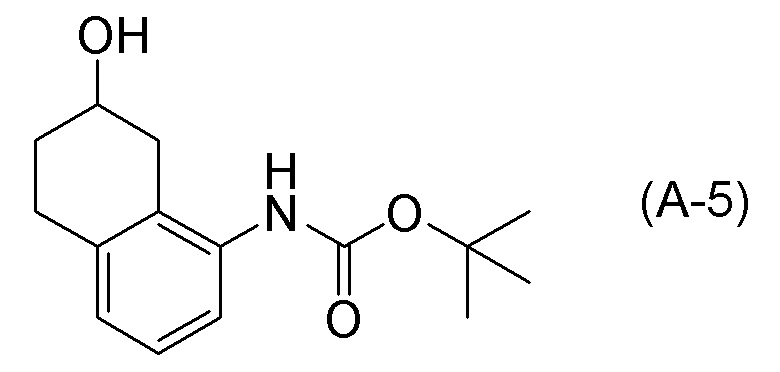
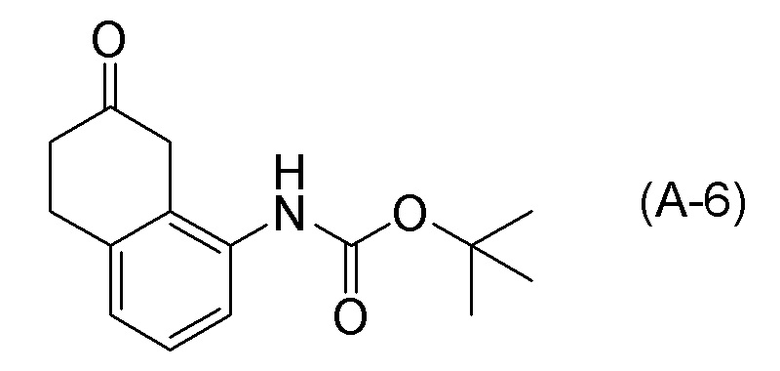
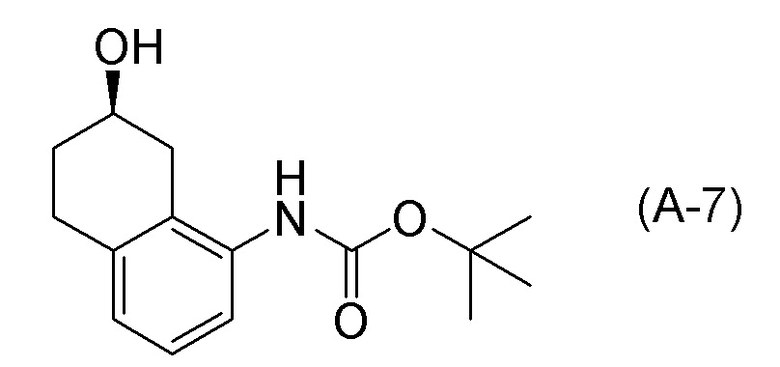
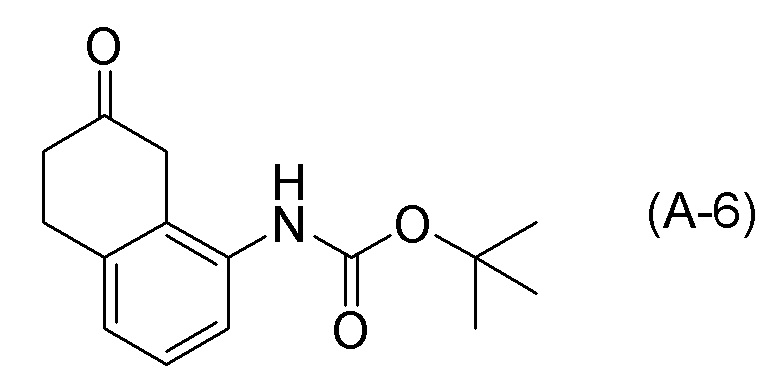
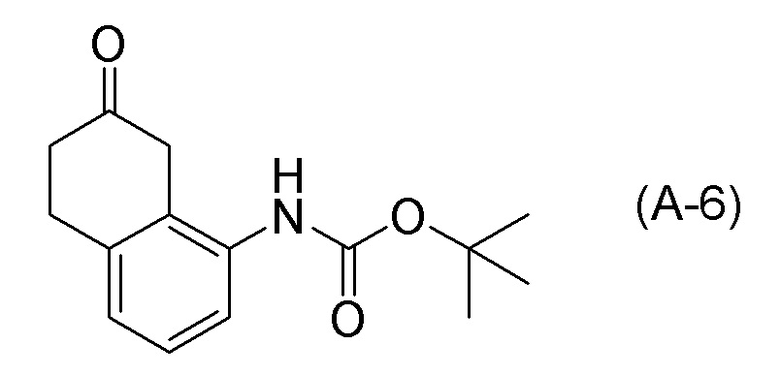
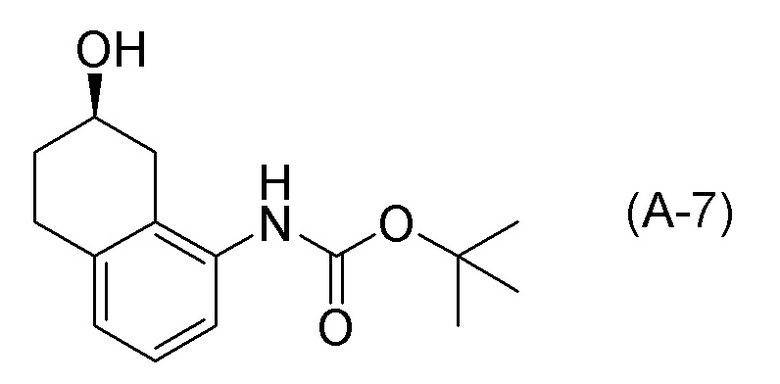
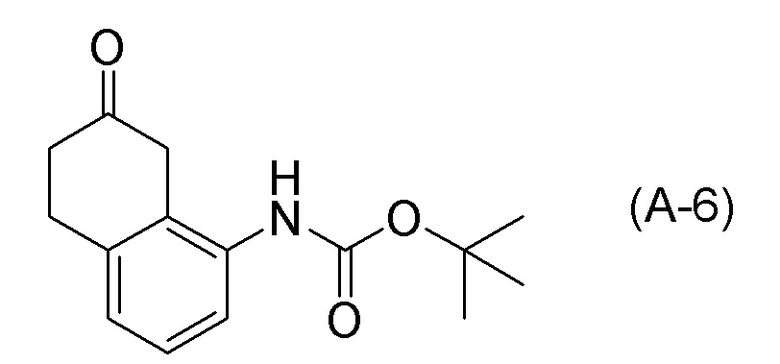
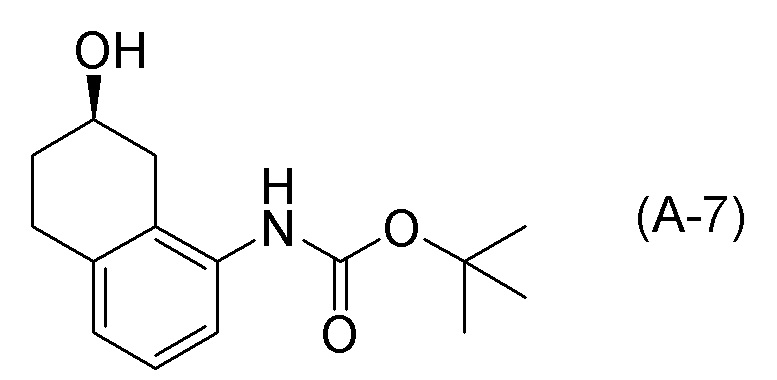
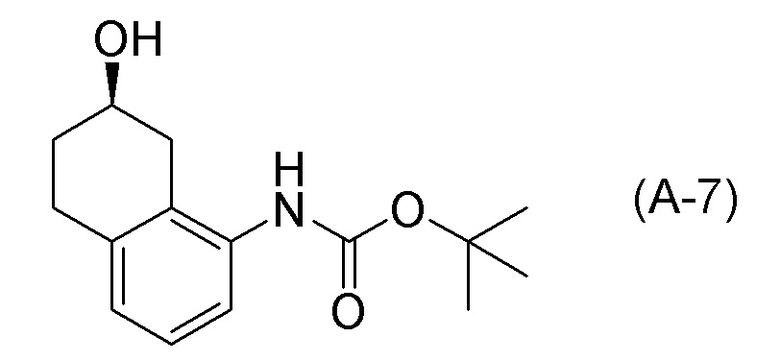
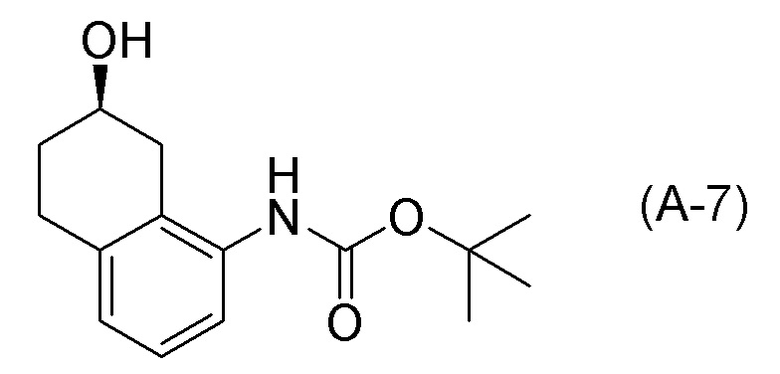
[0037] [1] Первый аспект представляет собой способ получения соединения, представленного формулой (B), или его соли, причем способ включает: трет-бутоксикарбонилирование аминогруппы соединения, представленного формулой (A), получая соединение, представленное формулой (A-5); проведение реакции окисления соединения, представленного формулой (A-5), получая соединение, представленное формулой (A-6); асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); и деблокирование a трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B) или его соль.
Формула (B):
[C10]
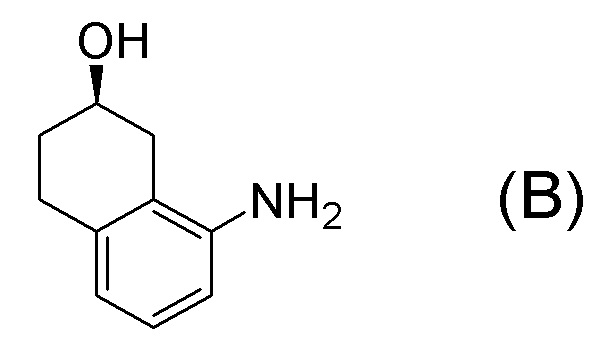
Формула (A):
[C11]
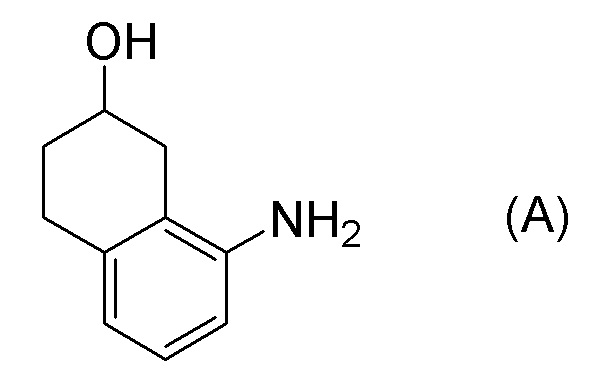
Формула (A-5):
[C12]
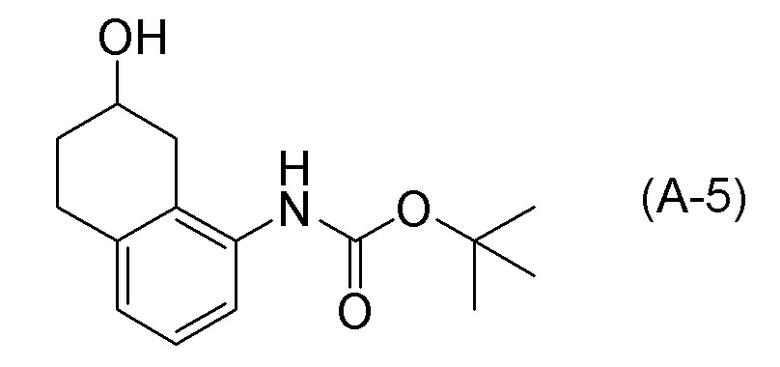
Формула (A-6):
[C13]
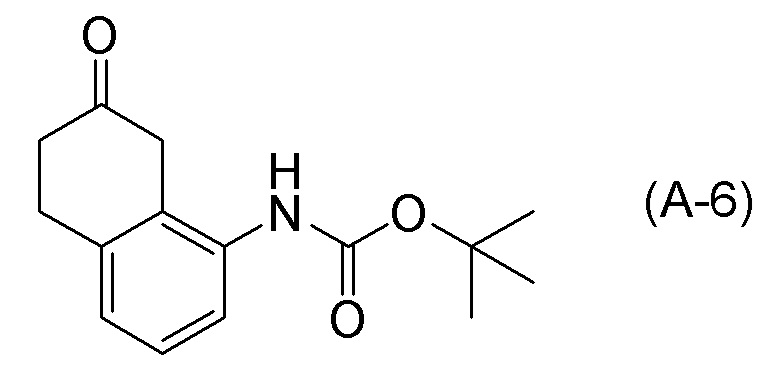
Формула (A-7):
[C14]
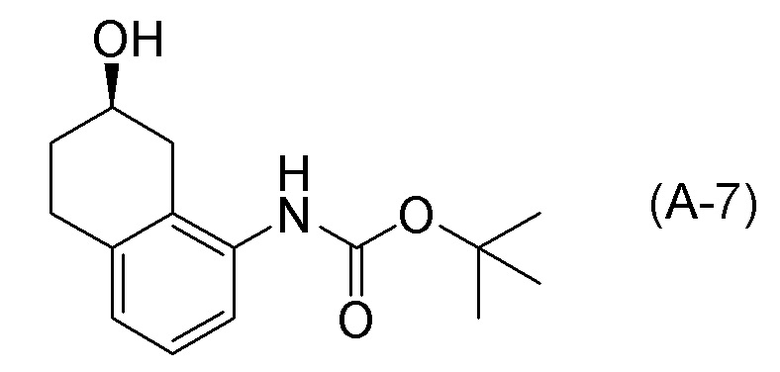
[0038] [2] Второй аспект представляет собой способ получения соединения, представленного формулой (B), или его соли, причем способ включает: проведение реакции окисления соединения, представленного формулой (A-5), получая соединение, представленное формулой (A-6); асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); и деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль.
Формула (B):
[C15]
Формула (A-5):
[C16]
Формула (A-6):
[C17]
Формула (A-7):
[C18]
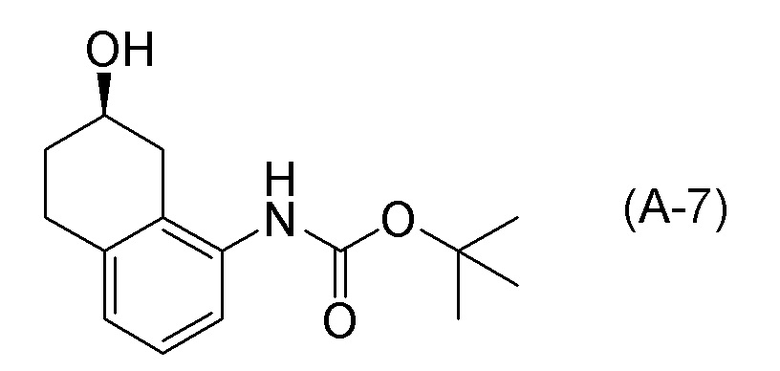
[0039] [3] Третий аспект представляет собой способ получения соединения, представленного формулой (B), или его соли, причем способ включает: асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); и деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль.
Формула (B):
[C19]
Формула (A-6):
[C20]
Формула (A-7):
[C21]
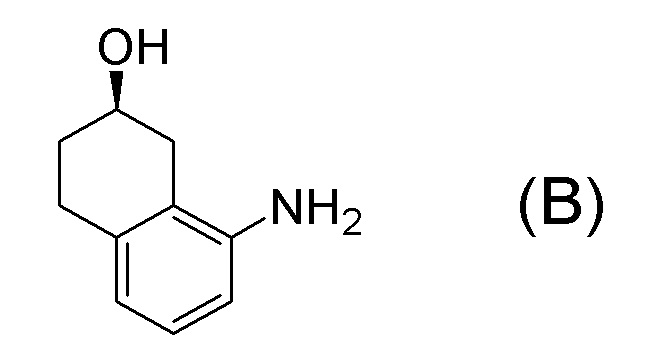
[0040] [4] Четвертый аспект представляет собой способ получения соединения, представленного формулой (B), или его соли, причем способ включает: деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль.
Формула (B):
[C22]
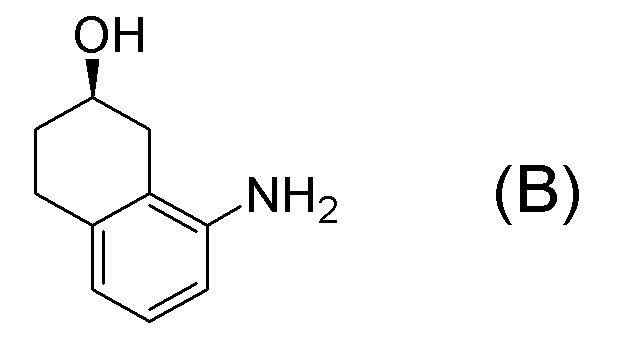
Формула (A-7):
[C23]
[0041] [4B] 4B-ый аспект представляет собой способ получения соли соединения, представленного формулой (B), причем способ включает: добавление кислоты к соединению, представленному формулой (B), получая соль соединения, представленного формулой (B).
Формула (B):
[C24]
[0042] [4B-1] В аспекте выше [4B], кислота, применяемая в получении соли соединения, представленного формулой (B), предпочтительно представляет собой неорганическую кислоту или органическую кислоту, более предпочтительно представляет собой неорганическую кислоту, и даже более предпочтительно представляет собой бромистоводородную кислоту.
[0043] [5] Пятый аспект представляет собой способ получения соединения, представленного формулой (A-6), причем способ включает: проведение реакции окисления соединения, представленного формулой (A-5), получая соединение, представленное формулой (A-6).
Формула (A-6):
[C25]
Формула (A-5):
[C26]
[0044] [6] Шестой представляет собой способ получения соединения, представленного формулой (A-7), причем способ включает: асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7).
Формула (A-7):
[C27]
Формула (A-6):
[C28]
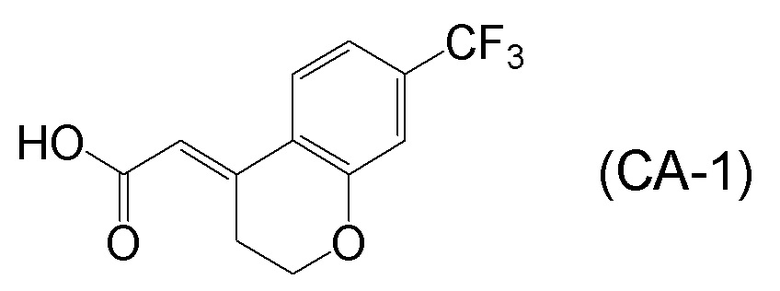
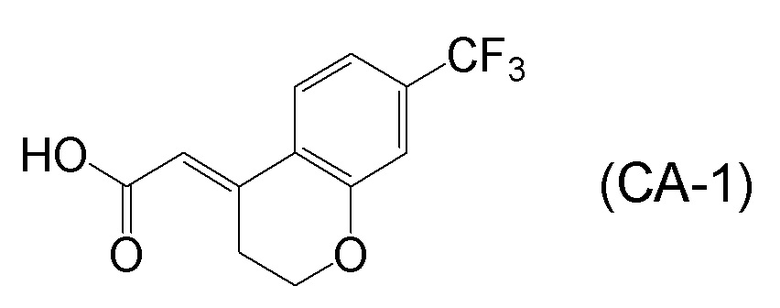
[0045] [7] Седьмой аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C29]
Формула (B):
[C30]
Формула (CA-1):
[C31]
[0046] [8] Восьмой аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: трет-бутоксикарбонилирование аминогруппы соединения, представленного формулой (A), получая соединение, представленное формулой (A-5); проведение реакции окисления соединения, представленного формулой (A-5), получая соединение, представленное формулой (A-6); асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B) или его соль; и проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C32]
Формула (A):
[C33]
Формула (A-5):
[C34]
Формула (A-6):
[C35]

Формула (A-7):
[C36]
Формула (B):
[C37]
Формула (CA-1):
[C38]
[0047] [9] Девятый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: проведение реакции окисления соединения, представленного формулой (A-5), получая соединение, представленное формулой (A-6); асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль; и проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C39]
Формула (A-5):
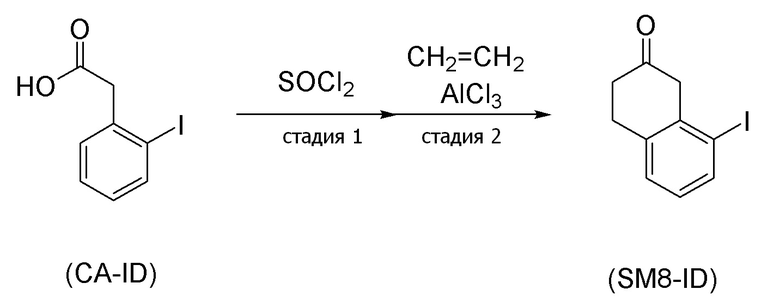
[C40]
Формула (A-6):
[C41]
Формула (A-7):
[C42]
Формула (B):
[C43]
Формула (CA-1):
[C44]
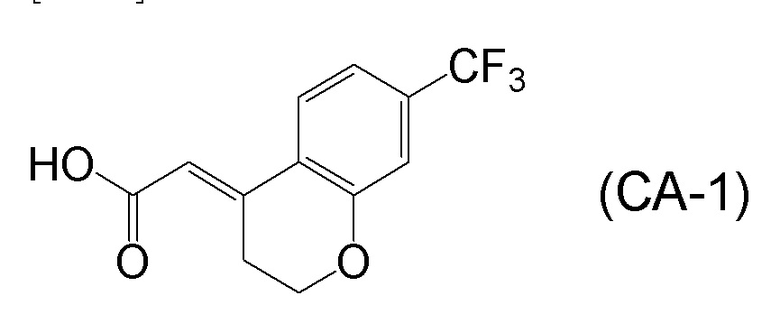
[0048] [10] Десятый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: асимметрическое восстановление соединения, представленного формулой (A-6), получая соединение, представленное формулой (A-7); деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль; и проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C45]

Формула (A-6):
[C46]
Формула (A-7):
[C47]
Формула (B):
[C48]
Формула (CA-1):
[C49]
[0049] [11] Одиннадцатый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль; и проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C50]
Формула (A-7):
[C51]
Формула (B):
[C52]
Формула (CA-1):
[C53]
[0050] [12] Двенадцатый аспект представляет собой соединение, представленное формулой (A-7).
[C54]
[0051] В каждом из аспектов выше, “деблокирование трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B), или его соль”, может дополнительно включать:
Обессоливание соли соединения, представленного формулой (B), которую получают деблокированием трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), получая соединение, представленное формулой (B); или
превращение соединения, представленного формулой (B), которое получают деблокированием трет-бутоксикарбонильной группы соединения, представленного формулой (A-7), в его соль, получая соль соединения, представленного формулой (B).
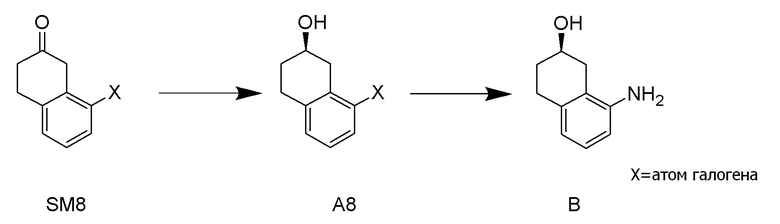
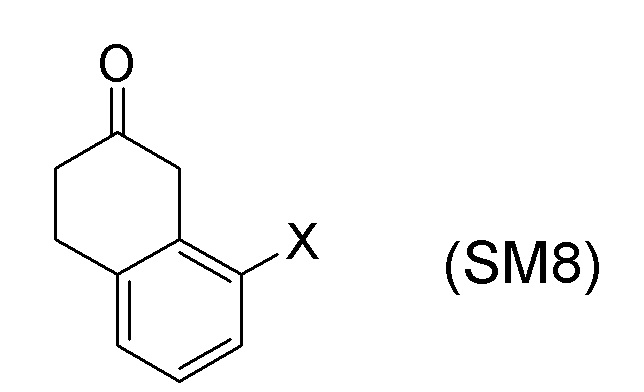
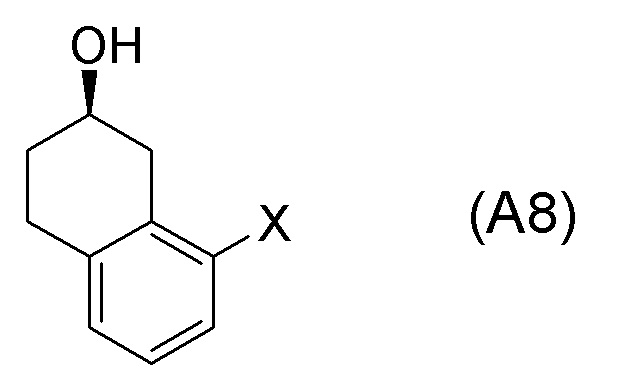
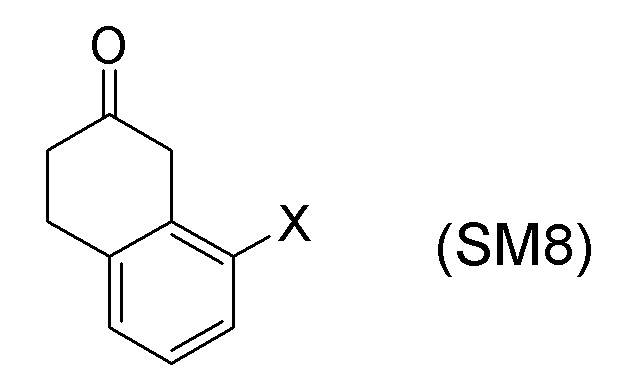
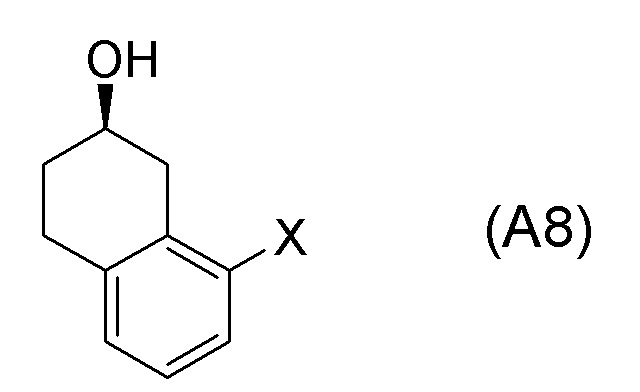
[0052] [13] Тринадцатый аспект представляет собой способ получения соединения, представленного формулой (B), причем способ включает: асимметрическое восстановление кето группы соединения, представленного формулой (SM8), получая соединение, представленное формулой (A8); и реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B).
Формула (B):
[C55]
Формула (SM8):
[C56]
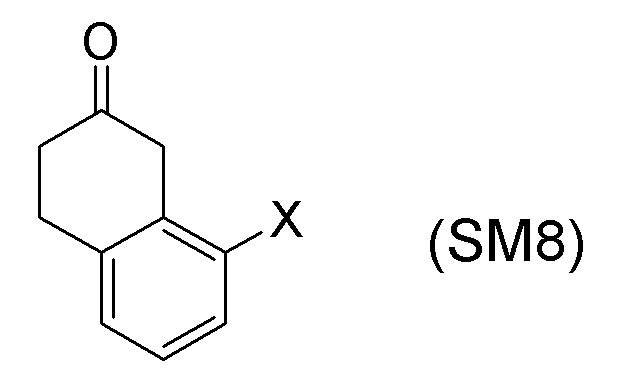
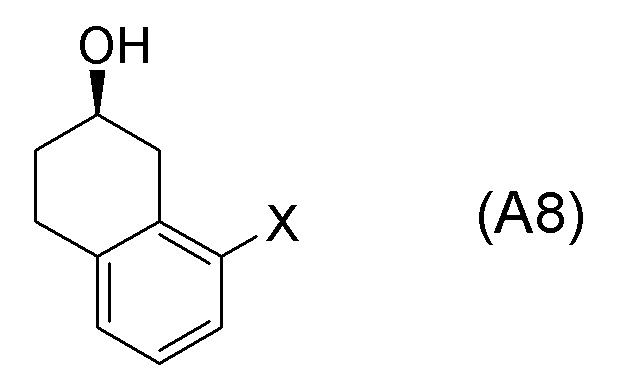
[в формуле (SM8), X представляет собой атом галогена.]
Формула (A8):
[C57]
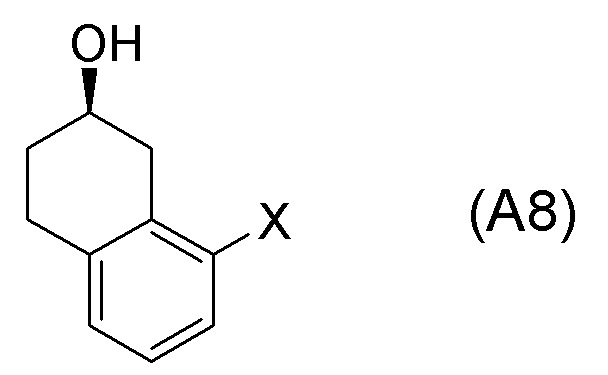
[в формуле (A8), X представляет собой атом галогена.]
[0053] [13-1] В аспекте выше [13], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора, или атом брома, и более предпочтительно представляет собой атом брома.
[0054] [13-2] В аспекте выше [13], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0055] [13-3] 13-3-ый аспект представляет собой способ получения соли соединения, представленного формулой (B), в аспекте выше [13], причем способ включает: добавление неорганической кислоты или органической кислоты к соединению, представленному формулой (B), получая соль соединения, представленного формулой (B). Неорганическая кислота или органическая кислота, применяемая для получения соли соединения, представленного формулой (B), предпочтительно представляет собой неорганическую кислоту, более предпочтительно представляет собой хлористоводородную кислоту или бромистоводородную кислоту, и даже более предпочтительно представляет собой бромистоводородную кислоту.
[0056] В настоящем изобретении, если не указано иначе, “атом галогена” относится к, например, атому фтора, атому хлора, атому брома, атому йода и подобным.
[0057] [14] Четырнадцатый аспект представляет собой способ получения соединения, представленного формулой (B), причем способ включает: реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B).
Формула (B):
[C58]
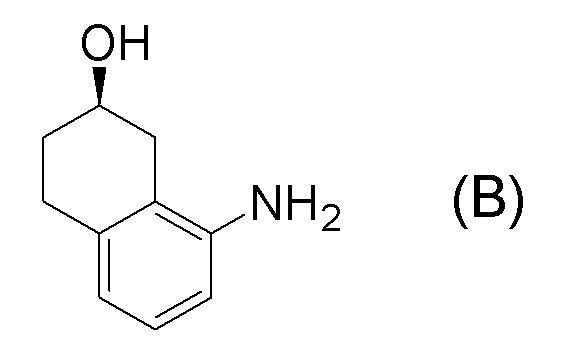
Формула (A8):
[C59]
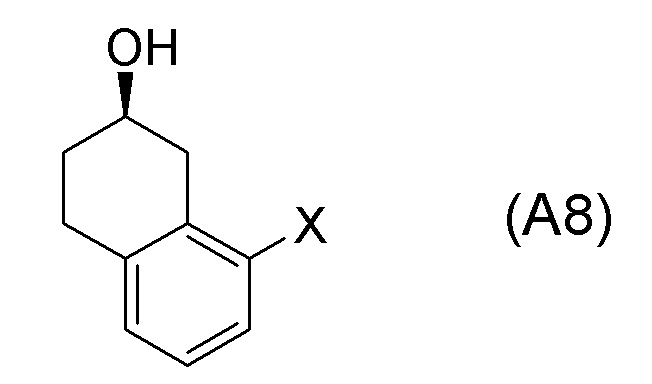
[в формуле (A8), X представляет собой атом галогена.]
[0058] [14-1] В аспекте выше [14], X в соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0059] [14-2] В аспекте выше [14], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0060] [14-3] 14-3-ый аспект представляет собой способ получения соли соединения, представленного формулой (B), в аспекте выше [14], причем способ включает: добавление неорганической кислоты или органической кислоты к соединению, представленному формулой (B), получая соль соединения, представленного формулой (B). Неорганическая кислота или органическая кислота, применяемые в получении соли соединения, представленного формулой (B), предпочтительно представляет собой неорганическую кислоту, более предпочтительно представляет собой хлористоводородную кислоту или бромистоводородную кислоту, и даже более предпочтительно представляет собой бромистоводородную кислоту.
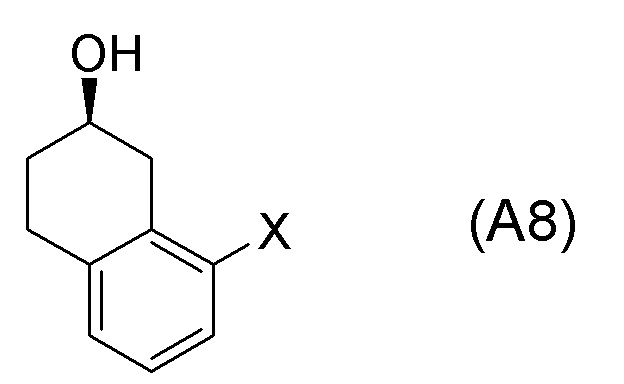
[0061] [15] Пятнадцатый аспект представляет собой способ получения соединения, представленного формулой (A8), причем способ включает: асимметрическое восстановление кето группы соединения, представленного формулой (SM8), получая соединение, представленное формулой (A8).
Формула (A8):
[C60]
[в формуле (A8), X представляет собой атом галогена.]
Формула (SM8):
[C61]
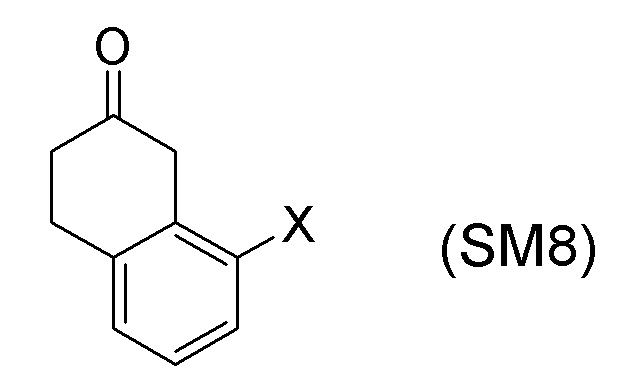
[в формуле (SM8), X представляет собой атом галогена.]
[0062] [15-1] В аспекте выше [15], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора, или атом брома, и более предпочтительно представляет собой атом брома.
[0063] [16] Шестнадцатый аспект представляет собой соединение, представленное формулой (A8).
[C62]
[в формуле (A8), X представляет собой атом галогена.]
[0064] [16-1] В аспекте выше [16], X в соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0065] [17] Семнадцатый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: асимметрическое восстановление кето группы соединения, представленного формулой (SM8), получая соединение, представленное формулой (A8); реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B); и проведение реакции конденсации соединения, представленного формулой (B), и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
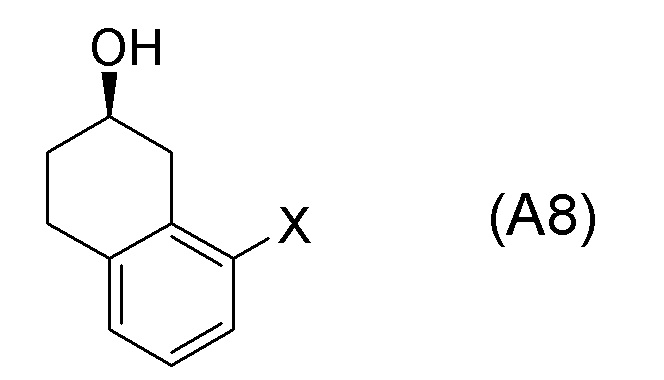
[C63]
Формула (SM8):
[C64]
[в формуле (SM8), X представляет собой атом галогена.]
Формула (A8):
[C65]
[в формуле (A8), X представляет собой атом галогена.]
Формула (B):
[C66]
Формула (CA-1):
[C67]
[0066] [17-1] В аспекте выше [17], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0067] [17-2] В аспекте выше [17], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0068] [18] Восемнадцатый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: асимметрическое восстановление кето группы соединения, представленного формулой (SM8), получая соединение, представленное формулой (A8); реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B); добавление кислоты к соединению, представленному формулой (B), получая соль соединения, представленного формулой (B); и проведение реакции конденсации соли соединения, представленного формулой (B), и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C68]

Формула (SM8):
[C69]
[в формуле (SM8), X представляет собой атом галогена.]
Формула (A8):
[C70]
[в формуле (A8), X представляет собой атом галогена.]
Формула (B):
[C71]

Формула (CA-1):
[C72]
[0069] [18-1] В аспекте выше [18], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0070] [18-2] В аспекте выше [18], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0071] [18-3] В аспекте выше [18], кислота, применяемая в получении соли соединения, представленного формулой (B), предпочтительно представляет собой неорганическую кислоту или органическую кислоту, более предпочтительно представляет собой неорганическую кислоту, даже более предпочтительно представляет собой хлористоводородную кислоту или бромистоводородную кислоту, и особенно предпочтительно представляет собой бромистоводородную кислоту.
[0072] [19] Девятнадцатый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B); и проведение реакции конденсации соединения, представленного формулой (B), и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C73]
Формула (A8):
[C74]
[в формуле (A8), X представляет собой атом галогена.]
Формула (B):
[C75]
Формула (CA-1):
[C76]
[0073] [19-1] В аспекте выше [19], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0074] [19-2] В аспекте выше [19], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0075] [20] Двадцатый аспект представляет собой способ получения соединения, представленного формулой (I), причем способ включает: реакцию соединения, представленного формулой (A8), с водным аммиаком в присутствии катализатора, получая соединение, представленное формулой (B); добавление кислоты к соединению, представленному формулой (B), получая соль соединения, представленного формулой (B); и проведение реакции конденсации соли соединения, представленного формулой (B), и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное формулой (I).
Формула (I):
[C77]
Формула (A8):
[C78]
[в формуле (A8), X представляет собой атом галогена.]
Формула (B):
[C79]
Формула (CA-1):
[C80]
[0076] [20-1] В аспекте выше [20], каждый X в соединении, представленном формулой (SM8), и соединении, представленном формулой (A8), предпочтительно представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом брома.
[0077] [20-2] В аспекте выше [20], катализатор предпочтительно представляет собой Pd катализатор или Cu катализатор, более предпочтительно представляет собой Cu катализатор, и даже более предпочтительно представляет собой Cu2O.
[0078] [20-3] В аспекте выше [20], кислота, применяемая в получении соли соединения, представленного формулой (B), предпочтительно представляет собой неорганическую кислоту или органическую кислоту, более предпочтительно представляет собой неорганическую кислоту, даже более предпочтительно представляет собой хлористоводородную кислоту или бромистоводородную кислоту, и особенно предпочтительно представляет собой бромистоводородную кислоту.
[0079] В настоящем изобретении далее, каждая из реакций в описанных выше аспектах будет описана подробно.
[0080] <Стадия получения соединения, представленного формулой (A-5)>
Соединение, представленное формулой (A-5), получают трет-бутоксикарбонилированием аминогруппы соединения, представленного формулой (A).
[0081] Примеры агентов для трет-бутоксикарбонилирования включают ди-трет-бутилдикарбонат (Boc2O), 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил (Boc-ON), N-трет-бутоксикарбонилимидазол, 2-(трет-бутоксикарбонилтио)-4,6-диметилпиримидин, 1-трет-бутоксикарбонил-1,2,4-триазол, трет-бутилфенилкарбонат, трет-бутилкарбазат, N-(трет-бутоксикарбонилокси)фталимид и подобные. Ди-трет-бутилдикарбонат (Boc2O) и 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил (Boc-ON) являются предпочтительными, и ди-трет-бутилдикарбонат (Boc2O) является более предпочтительным. Количество применяемого агента для трет-бутоксикарбонилирования обычно составляет 1,0-2,0 молярных эквивалентов, предпочтительно 1,1-1,8 молярных эквивалентов, и более предпочтительно 1,3-1,65 молярных эквивалентов по отношению к 1 молярному эквиваленту соединения, представленного формулой (A).
[0082] Реакцию можно проводить в присутствии растворителя. В качестве растворителя, можно применять, например, растворитель, не участвующий в реакции, такой как дихлорметан, ацетонитрил, диэтиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, трет-бутиловый эфир, толуол, вода и подобные, или их смешанный растворитель. Растворитель можно подходящим образом выбрать в зависимости от типа агента для трет-бутоксикарбонилирования, который будут применять. Тетрагидрофуран, 1,4-диоксан, смешанный растворитель тетрагидрофуран-вода, и смешанный растворитель 1,4-диоксан-вода являются предпочтительными, и тетрагидрофуран, смешанный растворитель тетрагидрофуран-вода и смешанный растворитель 1,4-диоксан-вода являются более предпочтительными.
[0083] Реакцию можно проводить в присутствии основания. В качестве основания, можно применять основания, такие как гидрокарбонат натрия, карбонат калия, карбонат натрия, триэтиламин, N, N-диизопропилэтиламин, пиридин и подобные. Основание можно подходящим образом выбрать в зависимости от типа агента для трет-бутоксикарбонилирования, который будут применять. Гидрокарбонат натрия, триэтиламин и пиридин являются предпочтительными, и гидрокарбонат натрия является более предпочтительным.
[0084] Применяемое количество основания составляет, например, 1,0-4,0 молярных эквивалентов, предпочтительно составляет 1,0-3,5 молярных эквивалентов, и более предпочтительно составляет 1,0-3,2 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A).
[0085] Что касается температуры реакции, реакцию можно проводить, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, или в диапазоне от 0°C до комнатной температуры. Температуру реакции можно подходящим образом выбрать в зависимости от типа агента для трет-бутоксикарбонилирования, который будут применять. Температура реакции предпочтительно находится в диапазоне 20°C-55°C.
[0086] <Стадия получения соединения, представленного формулой (A-6)>
Соединение, представленное формулой (A-6), получают проведением реакции окисления соединения, представленного формулой (A-5), или соединения, представленного формулой (A-7).
Примеры реакций окисления включают окисление по Сверну, PCC окисление (хроматное окисление), Окисление по Десс-Мартину, TPAP окисление, TEMPO окисление и подобные. TEMPO окисление является предпочтительным.
[0087] TEMPO окисление обычно представляет собой реакцию, в которой TEMPO и реоксидант смешивают в качестве окисляющего агента для окисления субстрата, такого как спирт. Кроме того, TEMPO окисление можно также проводить в присутствии основания.
[0088] В реакции окисления, например, применяют периодический способ и химию в потоке (реакция в режиме потока, применяя химический проточный реактор с мешалкой (CSTR)).
[0089] Применяемое количество окислителя в реакции окисления обычно составляет 1,0-2,2 молярных эквивалентов, предпочтительно составляет 1,2-2,1 молярных эквивалентов, и более предпочтительно составляет 1,4-2,0 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-5), или соединения, представленного формулой (A-7).
[0090] Применяемое количество TEMPO в TEMPO окислении обычно составляет 0,01-1,0 молярных эквивалентов, предпочтительно составляет 0,05-0,7 молярных эквивалентов, и более предпочтительно составляет 0,5 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-5) или Соединение, представленное формулой (A-7).
[0091] Примеры реоксидантов в TEMPO окислении включают e гипохлорит натрия (NaClO), йодбензолдиацетат и подобные. Применяемое количество гипохлорита натрия в TEMPO окислении обычно составляет 1,0-2,5 молярных эквивалентов, предпочтительно составляет 1,1-2,2 молярных эквивалентов, и более предпочтительно составляет 1,2-2,0 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-5), или соединения, представленного формулой (A-7).
[0092] TEMPO окисление можно проводить в присутствии основания, и, например, применяемое количество NaHCO3 в качестве основания обычно составляет 1,0-5,0 молярных эквивалентов, предпочтительно составляет 2,0-4,5 молярных эквивалентов, и более предпочтительно составляет 4,0 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-5) или Соединение, представленное формулой (A-7).
[0093] Применяемое количество KBr в TEMPO окислении обычно составляет 0,01-0,30 молярных эквивалентов, предпочтительно составляет 0,02-0,25 молярных эквивалентов, и более предпочтительно составляет 0,05-0,2 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-5), или соединения, представленного формулой (A-7).
[0094] Реакцию окисления (например, TEMPO окисление) можно проводить в присутствии растворителя. В качестве растворителя, можно применять, например, растворитель, не участвующий в реакции, такой как дихлорметан, 1,2-дихлорэтан, хлороформ, ацетонитрил, ацетон, вода и подобные, или их смешанный растворитель. Растворитель можно подходящим образом выбрать в зависимости от типа реакции окисления, которую будут применять. В TEMPO окислении, дихлорметан, ацетонитрил, ацетон, вода или их смешанный растворитель является предпочтительным; дихлорметан, ацетонитрил, ацетон, вода, дихлорметан-вода, ацетонитрил-вода или ацетон-вода является более предпочтительным; и дихлорметан, вода или дихлорметан-вода является даже более предпочтительным.
[0095] Что касается реакции окисления (например, температуры реакции в TEMPO окислении), реакцию можно проводить, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, или в диапазоне от 0°C до комнатной температуры. Температуру реакции можно подходящим образом выбрать в зависимости от типа реакции окисления, которую будут применять. В TEMPO окислении, температура реакции предпочтительно находится в диапазоне от -2°C до 5°C.
[0096] Также можно применять восстанавливающий агент, такой как Na2S2O4 (водный раствор), удаляя TEMPO после реакции окисления TEMPO.
[0097] <Стадия получения соединения, представленного формулой (A-7)>
Соединение, представленное формулой (A-7), получают асимметрическим восстановлением кетонового соединения, представленного формулой (A-6).
[0098] Примеры асимметрического восстановления включают асимметрическое восстановление, применяя химический катализатор или подобные, асимметрическое восстановление, применяя биокатализатор (дрожжи, грибы, плесень, фермент и подобные) и подобные. Асимметрическое восстановление, применяя фермент, является предпочтительным, асимметрическое восстановление, применяя кетонредуктазу (кеторедуктаза: KRED) в качестве фермента, является более предпочтительным, и асимметрическое восстановление, применяя кетонредуктаза, полученную из Lactobacillus sp., в качестве фермента, является особенно предпочтительным. Асимметрическую реакцию, применяя кетонредуктаза проводят, применяя кетонредуктазу, кофермент и систему регенерации кофермента. Типичные примеры коферментов для кетонредуктаз включают NADP. Кроме того, в качестве типичного примера системы регенерации кофермента, которая регенерирует NADP, который представляет собой кофермент, известно окисление глюкозы глюкозадегидрогеназой (GDH). Кроме того, асимметрическую реакцию, применяя кетонредуктаза, предпочтительно проводят в растворителе в присутствии буферного раствора.
[0099] Применяемое количество восстанавливающего агента в асимметрической реакции, например, в асимметрической реакции, применяя химический катализатор или подобные, обычно составляет 1,0-2,2 молярных эквивалентов, и предпочтительно составляет 1,2-2,0 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A-6).
[0100] В асимметрической реакции, применяя фермент, применяемое количество фермента обычно составляет 1,0-25 кратное, предпочтительно составляет 5-20 кратное, и более предпочтительно составляет 10 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0101] В асимметрической реакции, применяя кетонредуктазу, полученную из Lactobacillus sp., применяемое количество фермента обычно составляет 1,0-25 кратное, предпочтительно составляет 5-20 кратное, и более предпочтительно составляет 10 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0102] D-глюкозу можно применять в асимметрической реакции, применяя фермент. Когда применяют D-глюкозу, применяемое количество D-глюкозы обычно составляет 1,0-5,0 кратное, предпочтительно составляет 1,5-3,5 кратное, и более предпочтительно составляет 2,0 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0103] В асимметрической реакции, применяя фермент, можно применять глюкозадегидрогеназу (GDH). Когда применяют глюкозадегидрогеназу (GDH), применяемое количество глюкозадегидрогеназы (GDH) обычно составляет 0,01-0,5 кратное, предпочтительно составляет 0,05-0,2 кратное, и более предпочтительно составляет 0,05 кратное или 0,2 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0104] В асимметрической реакции, применяя фермент, можно применять кофермент, и, например, можно применять никотинамидадениндинуклеотидфосфат (NADP). Когда применяют никотинамидадениндинуклеотидфосфат (NADP), применяемое количество никотинамидадениндинуклеотидфосфата (NADP) обычно составляет 0,01-0,5 кратное, предпочтительно составляет 0,025-0,1 кратное, и более предпочтительно составляет 0,025 кратное или 0,1 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0105] Асимметрическую реакцию можно проводить в присутствии растворителя. В качестве растворителя, можно применять, например, растворитель, не участвующий в реакции, такой как спиртовые растворители, такие как метанол, этанол, пропанол, бутанол и подобные; углеводородные растворители, такие как гептан, гексан, октан, толуол и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; полярные растворители, такие как ацетонитрил, диметилсульфоксид, диметилформамид и подобные; и воду, или их смешанный растворитель. Растворитель можно подходящим образом выбрать в зависимости от типа фермента, который будут применять.
В асимметрической реакции, применяя фермент, в качестве буферного раствора, можно применять, например, буферные растворы, такие как фосфатный буферный раствор, калийфосфатный буферный раствор (который можно получить, например, из реагентов, таких как K2HPO4·3H2O, KH2PO4 и подобных), Tris/HCl буферный раствор, буферный раствор на основе тетрабората натрия -хлористоводородной кислоты, триэтаноламиновый буферный раствор и подобные. Буферный раствор можно подходящим образом выбрать в зависимости от типа фермента, который будут применять.
[0106] В асимметрической реакции, применяя кетонредуктазу, полученную из Lactobacillus sp., растворитель предпочтительно представляет собой диметилсульфоксид, толуол, воду или их смешанный растворитель, и более предпочтительно представляет собой толуол, воду или смешанный растворитель толуол-вода.
[0107] В асимметрической реакции, применяя фермент, применяемое количество органического растворитель обычно составляет 1,0-15 кратное, предпочтительно составляет 2-13 кратное, и более предпочтительно составляет 5,0 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0108] В асимметрической реакции, применяя фермент, применяемое количество буферного раствора обычно составляет 10-40 кратное, предпочтительно составляет 15-30 кратное, и более предпочтительно составляет 30 кратное относительно количества 1 г соединения, представленного формулой (A-6).
[0109] В асимметрической реакции, применяя фермент, pH реакционного раствора обычно составляет 6,0-7,5, предпочтительно составляет 6,0-6,5, 6,5-7,0 или 6,0-7,0, и более предпочтительно составляет 6,0-7,0.
[0110] Температура реакции при проведении асимметрической реакции можно подходящим образом выбрать из температуры реакции, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, в диапазоне от 0°C до комнатной температуры, и подобные. Температура реакции предпочтительно находится в диапазоне от 0°C до комнатной температуры.
[0111] Температура реакции при проведении асимметрической реакции, применяя фермент, обычно находится в диапазоне температур, при которых фермент не дезактивирован, и она предпочтительно находится в диапазоне 20°C-60°C, более предпочтительно находится в диапазоне 20°C-25°C или в диапазоне 50°C-60°C, и даже более предпочтительно находится в диапазоне 20°C-25°C.
[0112] <Стадия получения соединения, представленного формулой (B), или его соли из формулы (A-7)>
Соединение, представленное формулой (B), или его соль получают деблокированием трет-бутоксикарбонильной группы хирального спиртового соединения, представленного формулой (A-7), обессоливанием соли соединения, представленного формулой (B), полученной деблокированием трет-бутоксикарбонильной группы, или превращением соединения, представленного формулой (B), полученного деблокированием трет-бутоксикарбонильной группы, в его соль.
[0113] Примеры реагентов, применяемых для деблокирования трет-бутоксикарбонильной группы, обычно включают кислотные реагенты, и реагент предпочтительно представляет собой хлороводород (который представляет собой хлористоводородную кислоту, или который генерируют в системе растворителей, применяя ацетилхлорид и спиртовой растворитель, такой как метанол, этанол, пропанол и подобные), бромоводород, и трифторуксусную кислоту; более предпочтительно представляет собой хлороводород (который представляет собой хлористоводородную кислоту, или который генерируют в системе растворителей, применяя ацетилхлорид и спиртовой растворитель, такой как метанол, этанол, пропанол и подобные) и трифторуксусную кислоту; и особенно предпочтительно представляет собой хлороводород (который представляет собой хлористоводородную кислоту, или который генерируют в системе растворителей, применяя ацетилхлорид и спиртовой растворитель, такой как метанол, этанол, пропанол и подобные).
[0114] Деблокирование трет-бутоксикарбонильной группы можно проводить в присутствии растворителя. Примеры растворителей для деблокирования трет-бутоксикарбонильной группы включают растворители, не участвующие в реакции, такие как галогеновые растворители, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные; спиртовые растворители, такие как метанол, этанол, пропанол, бутанол и подобные; углеводородные растворители, такие как гептан, гексан, октан, толуол и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; полярные растворители, такие как ацетон, ацетонитрил, диметилсульфоксид, диметилформамид и подобные; и воду, или их смешанный растворитель, и галогеновые растворители, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные, и спиртовые растворители, такие как метанол, этанол, пропанол, бутанол и подобные, являются предпочтительными, и пропанол (н-пропанол) является более предпочтительным.
[0115] Температура реакции при деблокировании трет-бутоксикарбонильной группы можно подходящим образом выбрать из температуры реакции, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, в диапазоне от 0°C до комнатной температуры, и подобные. Температура реакции предпочтительно находится в диапазоне 0°C-55°C.
[0116] Можно обессоливать соль соединения, представленного формулой (B), применением основания. В качестве основания для обессоливания соли соединения, представленного формулой (B), можно применять основания, такие как гидрокарбонат натрия, карбонат калия, карбонат натрия, триэтиламин, N, N-диизопропилэтиламин, пиридин и подобные, и гидрокарбонат натрия, калия карбонат и карбонат натрия являются предпочтительными, и гидрокарбонат натрия является более предпочтительным.
[0117] Обессоливание соли соединения, представленного формулой (B), можно проводить в присутствии растворителя. Примеры растворителей для обессоливания соли соединения, представленного формулой (B), включают растворители, не участвующие в реакции, такие как галогеновые растворители, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; полярные растворители, такие как этилацетат, изопропилацетат, ацетонитрил, диметилсульфоксид, диметилформамид и подобные; и воду, или их смешанный растворитель, и этилацетат, изопропилацетат, вода и смешанный растворитель этилацетат-вода или изопропилацетат-вода являются предпочтительными, и смешанный растворитель этилацетат-вода является более предпочтительным.
[0118] <Стадия получения соли из соединения, представленного формулой (B)>
Соединение, представленное формулой (B), можно превратить в соль, применяя органическую кислоту или неорганическую кислоту. В качестве кислоты для превращения соединения, представленного формулой (B), в его соль, можно применять, например, кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, масляная кислота, валериановая кислота, энантовая кислота, каприновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, молочная кислота, сорбиновая кислота, миндальная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, яблочная кислота, винная кислота, лимонная кислота, бензойная кислота, салициловая кислота, фталевая кислота, коричная кислота, гликолевая кислота, пировиноградная кислота, оксиловая кислота, салициловая кислота, N -ацетилцистеин, метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, аспарагиновая кислота, глутаминовая кислота и подобные, и хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, уксусная кислота, фталевая кислота, фумаровая кислота, щавелевая кислота винная кислота, малеиновая кислота, лимонная кислота, янтарная кислота, метансульфоновая кислота и п-толуолсульфоновая кислота являются предпочтительными, хлористоводородная кислота и бромистоводородная кислота являются более предпочтительными, и бромистоводородная кислота является даже более предпочтительной.
[0119] Превращение соединения, представленного формулой (B), в его соль можно проводить в присутствии растворителя. Примеры растворителей для превращения соединения, представленного формулой (B), в его соль включают растворители, не участвующие в реакции, такие как галогеновые растворители, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; спиртовые растворители, такие как метанол, этанол и подобные; полярные растворители, такие как этилацетат, изопропилацетат, ацетонитрил, диметилсульфоксид, диметилформамид и подобные; и воду, или их смешанный растворитель, и растворитель, не участвующий в реакции, такой как дихлорметан, хлороформ, 1,2-дихлорэтан, тетрагидрофуран, 1,4-диоксан, метанол, этанол, Этилацетат и вода, или их смешанный растворитель является предпочтительным, и этилацетат, вода, или этилацетат-вода является более предпочтительным.
[0120] <Стадия получения соединения, представленного формулой (A8)>
Соединение, представленное формулой (A8), получают асимметрическим восстановлением кетонового соединения, представленного формулой (SM8).
[0121] Примеры асимметрического восстановления включают асимметрическое восстановление, применяя химический катализатор или подобные, асимметрическое восстановление, применяя биокатализатор (дрожжи, грибы, плесень, фермент и подобные), и подобные. Асимметрическое восстановление, применяя фермент, является предпочтительным, асимметрическое восстановление, применяя кетонредуктазу (KRED: кеторедуктаза) в качестве фермента, является более предпочтительным, и асимметрическое восстановление, применяя кетонредуктазу, полученную из Escherichia coli sp. в качестве фермента, является особенно предпочтительным. Асимметрическую реакцию, применяя кетонредуктазу, проводят, применяя кетонредуктазу, кофермент и систему регенерации кофермента. Типичные примеры коферментов для кетонредуктаз включают NADP. Кроме того, в качестве типичного примера системы регенерации кофермента, который регенерирует NADP, который представляет собой кофермент, известно окисление глюкоза глюкозадегидрогеназой (GDH). Кроме того, асимметрическую реакцию, применяя кетонредуктазу, предпочтительно проводят в растворителе в присутствии буферного раствора.
[0122] Применяемое количество восстанавливающего агента асимметрической реакции, например, в асимметрической реакции, применяя химический катализатор или подобные, обычно составляет 1,0-2,2 молярных эквивалентов, и предпочтительно составляет 1,2-2,0 молярных эквивалентов относительно 1 молярного эквивалента соединения, представленного формулой (SM8).
[0123] В асимметрической реакции, применяя фермент, применяемое количество фермента обычно составляет 0,01-0,1 кратное, предпочтительно составляет 0,02-0,07 кратное, и более предпочтительно составляет 0,047-0,05 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0124] В асимметрической реакции, применяя кетонредуктазу (KRED), полученную из Escherichia coli sp., применяемое количество фермента составляет 0,01-0,1 кратное, предпочтительно составляет 0,02-0,07 кратное, и более предпочтительно составляет 0,047-0,05 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0125] D-глюкозу можно применять в асимметрической реакции, применяя фермент. Когда применяют D-глюкозу, применяемое количество D-глюкозы обычно составляет 1,0-5,0 кратное, предпочтительно составляет 1,5-3,5 кратное, и более предпочтительно составляет 1,9-2,0 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0126] В асимметрической реакции, применяя фермент, можно применять глюкозадегидрогеназу (GDH). Когда применяют глюкозадегидрогеназу (GDH), применяемое количество глюкозадегидрогеназы (GDH) обычно составляет 0,01-0,1 кратное, предпочтительно составляет 0,01-0,05 кратное, и более предпочтительно составляет 0,019-0,02 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0127] В асимметрической реакции, применяя фермент, можно применять никотинамидадениндинуклеотидфосфат (NADP). Когда применяют никотинамидадениндинуклеотидфосфат (NADP), применяемое количество никотинамид дениндинуклеотидфосфата (NADP) обычно составляет 0,00-0,1 кратное, предпочтительно составляет 0,005-0,05 кратное, и более предпочтительно составляет 0,009-0,01 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0128] Асимметрическую реакцию можно проводить в присутствии растворителя. В качестве растворителя, можно применять, например, растворитель, не участвующий в реакции, такой как спиртовые растворители, такие как метанол, этанол, пропанол, бутанол и подобные; углеводородные растворители, такие как гептан, гексан, октан, толуол и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; полярные растворители, такие как ацетон, ацетонитрил, диметилсульфоксид, диметилформамид и подобные; и воду, или их смешанный растворитель, и растворитель можно подходящим образом выбрать в зависимости от типа фермента, который будут применять.
В асимметрической реакции, применяя фермент, в качестве буферного раствора можно применять, например, буферные растворы, такие как фосфатный буферный раствор, калийфосфатный буферный раствор (который можно получить, например, из реагентов, таких как K2HPO4·3H2O и KH2PO4), Tris/HCl буферный раствор, буферный раствор на основе тетрабората натрия-хлористоводородной кислоты, триэтаноламиновый буферный раствор и подобные, и буферный раствор можно подходящим образом выбрать в зависимости от типа фермента, который будут применять.
[0129] В асимметрической реакции, применяя кетонредуктазу (KRED), полученную из Escherichia coli sp., растворитель предпочтительно представляет собой диметилсульфоксид, воду или смешанный растворитель диметилсульфоксид-вода.
[0130] В асимметрической реакции, применяя кетонредуктазу (KRED), полученную из Escherichia coli sp., применяемое количество органического растворителя обычно составляет 1,0-10 кратное, предпочтительно составляет 2-5 кратное, и более предпочтительно составляет 2,5-3,0 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0131] В асимметрической реакции, применяя кетонредуктазу (KRED), полученную из Escherichia coli sp., применяемое количество буферного раствора обычно составляет 10-40 кратное, предпочтительно составляет 15-30 кратное, и более предпочтительно составляет 28-30 кратное относительно количества 1 г соединения, представленного формулой (SM8).
[0132] В асимметрической реакции, применяя фермент, pH реакционного раствора обычно составляет 6,0-7,5, и предпочтительно составляет 6,5-7,0.
[0133] Температуру реакции при проведении асимметрической реакции можно подходящим образом выбрать из температуры реакции, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, в диапазоне от 0°C до комнатной температуры, и подобные. Температура реакции предпочтительно находится в диапазоне от 0°C до комнатной температуры.
[0134] Температура реакции при проведении асимметрической реакции, применяя фермент, обычно находится в диапазоне температур, при которых фермент не дезактивирован, и она предпочтительно находится в диапазоне 20°C-60°C, более предпочтительно находится в диапазоне 20°C-35°C, и даже более предпочтительно находится в диапазоне 20°C-30°C.
[0135] Если не указано иначе в настоящем изобретении, когда ссылаются на формулу (SM8), она включает ее формулы младшего порядка (например, формулу (SM8-FL), формулу (SM8-CL), формулу (SM8-BR), формулу (SM8-ID) и подобные). Аналогично, если не указано иначе в настоящем изобретении, когда ссылаются на формулу (A8), она включает ее формулы младшего порядка (например, формулу (A8-FL), формулу (A8-CL), формулу (A8-BR), формулу (A8-ID) и подобные).
Кроме того, формула (SM8-FL) представляет собой соединение, в котором X=атом фтора в соединении, представленном формулой (SM8). Формула (SM8-CL) представляет собой соединение, в котором X=атом хлора в соединении, представленном формулой (SM8). Формула (SM8-BR) представляет собой соединение, в котором X=атом брома в соединении, представленном формулой (SM8). Формула (SM8-ID) представляет собой соединение, в котором X=атом йода в соединении, представленном формулой (SM8).
Кроме того, Формула (A8-FL) представляет собой соединение, в котором X=атом фтора в соединении, представленном формулой (A8). Формула (A8-CL) представляет собой соединение, в котором X=атом хлора в соединении, представленном формулой (A8). Формула (A8-BR) представляет собой соединение, в котором X=атом брома в соединении, представленном формулой (A8). Формула (A8-ID) представляет собой соединение, в котором X=атом йода в соединении, представленном формулой (A8).
[0136] <Стадия получения соединения, представленного формулой (B), из формулы (A8)>
Соединение, представленное формулой (B), получают проведением реакции аминирования соединения, представленного формулой (A8), в присутствии металлического катализатора, применяя аммиак (водный аммиак (например, 25%, 28%, 30%, и подобные)). Концентрация (%)водного аммиака представляет собой вес/вес% или вес/об%.
[0137] Примеры катализаторов для реакции аминирования соединения, представленного формулой (A8), применяя аммиак в качесвте источника азота, включают Pd катализатор, Cu катализатор и подобные. Примеры Pd катализаторов включают Pd2(dba)3 PdCl2-Josiphos комплекс и подобные, и примеры Cu катализаторов включают CuI, Cu(OAc)2, Cu2O, CuO, CuBr, CuCl, CuSO4, CuFe2O4 и подобные, и Cu катализатор является предпочтительным, и Cu2O является более предпочтительным.
[0138] Примеры растворителей для реакции аминирования включают растворители, такие как диметилсульфоксид, N, N-диметилформамид, N-метилпирролидон (NMP), 1,4-диоксан, ацетонитрил, толуол, их смешанный растворитель и подобные, где N-метилпирролидон (NMP) является предпочтительным.
[0139] Основание может присутствовать в реакции аминирования, и примеры оснований включают основания, такие как карбонат калия, фосфат калия, карбонат цезия, N, N-диизопропилэтиламин, триэтиламин и подобные.
[0140] Реакцию аминирования проводят нагреванием герметично закрытой пробирки, применяя, реактор для герметичных пробирок (изготовленный, например, нержавеющей стали, стекла или подобных). Когда проводят реакцию с нагреванием, обычно не проводят нагревание выше температуры кипения применяемого растворителя или реагента, и нагревание проводят в закрытой системе, применяя реактор для герметичных пробирок или подобные, когда реакцию проводят при температуре выше температуры кипения применяемого растворителя или реагента.
[0141] Примеры растворителей, который можно применять при проведении реакции аминирования и их температуры кипения являются следующими: диметилсульфоксид (температура кипения 189°C), N, N-диметилформамид (температура кипения 153°C), N-метилпирролидон (NMP) (температура кипения 202°C), 1,4-диоксан (температура кипения 101°C), ацетонитрил (температура кипения 82°C) и толуол (температура кипения 110,6°C). Кроме того, температура кипения водного аммиака составляет 37,7°C для 25% водного аммиака и 24,7°C для 32% водного аммиака.
[0142] Температура реакции при проведении реакции аминирования можно подходящим образом выбрать, например, из температуры реакции, находящейся в диапазоне 100°C-250°C, в диапазоне 100°C-200°C, в диапазоне 100°C-150°C и подобных. Температура реакции предпочтительно находится в диапазоне 100°C-120°C.
[0143] В реакции аминирования, когда применяют Cu2O, применяемое количество металлического катализатора обычно составляет 0,1-1,0 молярных эквивалентов, предпочтительно составляет 0,2-0,8 молярных эквивалентов, и более предпочтительно составляет 0,5-0,7 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (A8).
[0144] В реакции аминирования, применяемое количество органического растворителя обычно составляет 0,1-30 кратное, и предпочтительно составляет 0,5-20 кратное относительно количества 1 г соединения, представленного формулой (A8).
[0145] В реакции аминирования, применяемое количество водного аммиака обычно составляет 1,0-50 кратное, предпочтительно составляет 2,5-30 кратное, и более предпочтительно составляет 2,5-3,5 кратное относительно количества 1 г соединения, представленного формулой (A8).
[0146] Если не указано иначе, диапазон числовых значений, описанный в настоящем изобретении, также включает значения±10% от данных значений. Например, когда упоминается фраза «0,1-1,0 молярных эквивалентов», это означает от 0,1 ± 0,01 до 1,0 ± 0,1 молярных эквивалентов, и когда применяют фразу «0,1-30-кратное количество ...», это означает от 0,1±0,01 до 30±3-кратное.
[0147] <Стадия получения соединения, представленного формулой (I)>
Соединение, представленное формулой (I), получают реакцией конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента.
[0148] Реакции конденсации можно проводить в присутствии растворителя. Примеры растворителей включают растворители, не участвующие в реакции, такие как спиртовые растворители, такие как метанол, этанол, пропанол, изопропанол, бутанол и подобные; эфирные растворители, такие как тетрагидрофуран, 1,4-диоксан, бутиловый эфир и подобные; и воду, или их смешанный растворитель, и спиртовые растворители, вода или их смешанный растворитель является предпочтительным; метанол, этанол, изопропанол, вода, или их смешанный растворитель является более предпочтительным; метанол, этанол или изопропанол является даже более предпочтительным; и метанол или изопропанол являются особенно предпочтительными.
[0149] В реакции конденсации, применяемое количество соединения, содержащего карбоновую кислоту, представленного формулой (CA-1), обычно составляет 0,5-2,0 молярных эквивалентов, предпочтительно составляет 0,5-1,5 молярных эквивалентов и более предпочтительно составляет 0,7-1,25 молярных эквивалентов, относительно 1 молярного эквивалента соединения, представленного формулой (B) или его соль. Как будет описано ниже, изобретатели настоящего изобретения обнаружили, что применение DMT-MM в качестве конденсирующего агента обеспечивает селективную реакцию конденсации карбоксильной группы соединения, представленного формулой (CA-1), и аминогруппы соединения, представленного формулой (B), и что, следовательно, не требуется защищать гидроксильные группы соединения, представленного формулой (B), в реакции конденсации.
[0150] В реакции конденсации, соль соединения, представленного формулой (B), предпочтительно представляет собой HCl соль или HBr соль.
[0151] В реакции конденсации, применяемое количество DMT-MM в качестве конденсирующего реагента обычно составляет 1,0-2,0 молярных эквивалентов, предпочтительно составляет 1,1-1,8 молярных эквивалентов и более предпочтительно составляет 1,2-1,5 молярных эквивалентов относительно 1 молярного эквивалента соединения, представленного формулой (B), или его соли.
[0152] Когда соль соединения, представленного формулой (B), применяют в реакции конденсации, можно добавлять основание. Примеры оснований включают органические основания, такие как триэтиламин, N, N-диизопропилэтиламин, пиридин и подобные; и неорганические основания, такие как гидроксид лития (моногидрат гидроксида лития), гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия и подобные. Триэтиламин, N, N-диизопропилэтиламин, пиридин, карбонат натрия или карбонат калия является предпочтительным, и триэтиламин является более предпочтительным.
[0153] Количество основания, которое можно добавлять, когда соль соединения, представленного формулой (B), применяют в реакции конденсации, обычно составляет 1,0-2,5 молярных эквивалентов, предпочтительно составляет 1,05-2,3 молярных эквивалентов и более предпочтительно составляет 1,05-2,1 молярных эквивалентов, относительно 1 молярного эквивалента соли соединения, представленного формулой (B).
[0154] В реакции конденсации, применяемое количество растворителя обычно составляет 5,0-100 кратное, предпочтительно составляет 5-40 кратное и более предпочтительно составляет 5-30 кратное относительно количества 1 г соединения, представленного формулой (B), или его соли.
[0155] Температура реакции при проведении реакции конденсации можно подходящим образом выбрать из температуры реакции, например, в диапазоне от -78°C до температуры, при которой кипит растворитель, в диапазоне от -78°C до комнатной температуры, в диапазоне от 0°C до температуры, при которой кипит растворитель, в диапазоне от 0°C до комнатной температуры, и подобные. Температура реакции предпочтительно находится в диапазоне от 0°C до комнатной температуры.
[0156] 8-амино-1,2,3,4-тетрагидронафталин-2-ол [CAS No. 624729-66-4], представленный формулой (A) в описанном выше аспекте [1], можно получить селективным восстановлением нафталинового кольца, применяя 8-аминонафталин-2-ол (формула (SM-1)) в качестве исходного соединения согласно способу получения, известному из литературы, например, способу получения ниже, который описан в WO 2009/050289 (патентная литература 6).
[0157]
[C81]
(Схема 4-1)
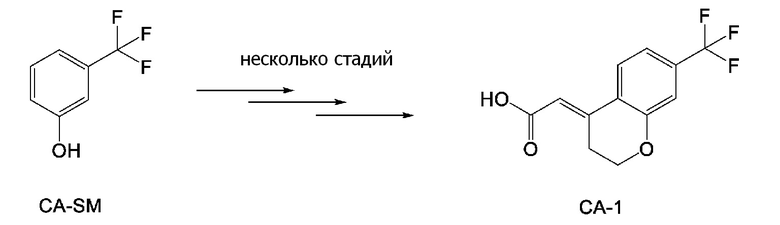
[0158] (E)-2-(7-(трифторметил)хроман-4-илиден)уксусная кислота [CAS No. 920334-15-2], представленная формулой (CA-1), в описанных выше аспектах [7]-[11] и описанных выше аспектах [17]-[20], можно получить проведением нескольких стадий, применяя 3-гидроксибензотрифторид (формула (CA-SM)) в качестве исходного соединения согласно способу получения, известному из литературы, например, способу получения ниже, который описан в WO 2007/010383 (патентная литература 1).
[0159]
[C82]
(Схема 4-2)
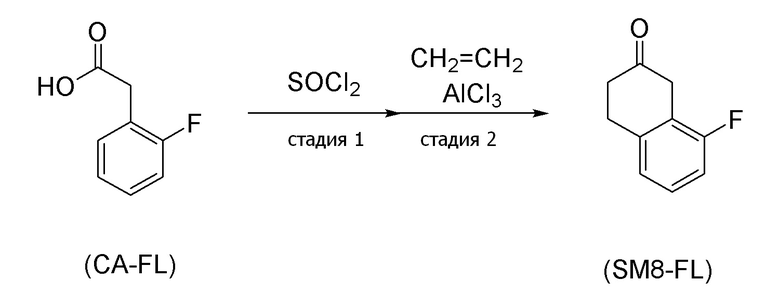
[0160] Для соединения, представленного формулой (SM8), в описанных выше аспектах [13], [15], [17] и [18], можно применять имеющееся в продаже соединение. Альтернативно, его можно получить согласно способу получения, известному из литературы, применяя имеющееся в продаже соединение.
[0161] В соединении, представленном формулой (SM8), соединение, в котором X=атом фтора (формула (SM8-FL)), можно получить согласно, например, способу получения (схемы 4-3) ниже, который описан в публикации европейской патентной заявки No. 343830.
[C83]
(Схема 4-3)
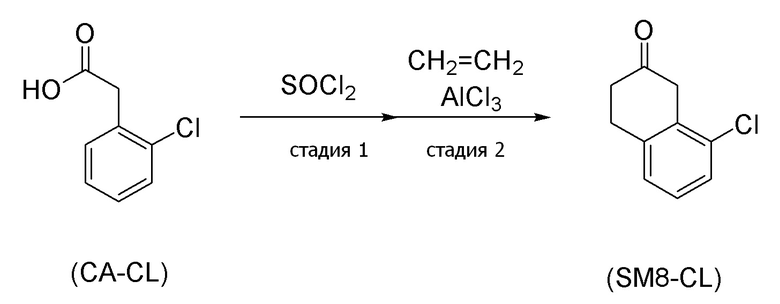
[0162] В соединении, представленном формулой (SM8), соединение, в котором X=атом хлора (формула (SM8-CL)), можно получить согласно, например, способу получения (схемы 4-4) ниже, который описан в публикации европейской патентной заявки No. 343830.
[C84]
(Схема 4-4)
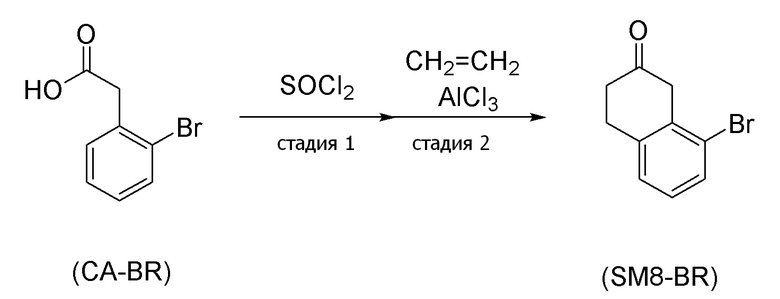
[0163] В соединении, представленном формулой (SM8), соединение, в котором X=атом брома (формула (SM8-BR)), можно получить согласно, например, способу получения (схемы 4-5) ниже, который описан в Journal of Medicinal Chemistry, 36(17), стр. 2485-93, 1993 и European Journal of Medicinal Chemistry (1993), 28(9), стр. 693-701.
[C85]
(Схема 4-5)
[0164] В формуле (SM8), соединение, в котором X=атом йода (формула (SM8-ID)), можно получить согласно способам получения для формулы (SM8-FL), формулы (SM8-CL) и формулы (SM8-BR) (схема 4-6).
[C86]
(Схема 4-6)
[0165] Исходное соединение каждой из стадий способа получения можно применять на следующей стадии в виде самого реакционного раствора или в виде неочищенного продукта. Кроме того, исходное соединение также можно выделить из реакционной смеси согласно общепринятому способу, и его можно легко очистить известным способом, например, способами разделения, такими как экстракция, концентрирование, нейтрализация, фильтрация, дистилляция, перекристаллизация, хроматография и подобные.
[0166] Когда смешанный растворитель применяют в описанных выше реакциях, его можно применять смешением двух или более видов растворителей в подходящем соотношении, например, в соотношении 1:1-1:10 в виде объемного соотношения или массового соотношения.
[0167] Если не указано иначе, продолжительность реакции для каждой из стадий в способе получения не ограничена при условии, что она представляет собой продолжительность времени, которое обеспечивает протекание реакции в достаточной степени реакцию, например, продолжительность реакции может составлять любую из 0,1 часа, 0,5 часов, 1 часа, 1,5 часов, 2 часов, 3 часов, 4 часов, 5 часов, 10 часов, 12 часов, 18 часов, 24 часов, 36 часов, 48 часов, 60 часов, 72 часов или 115 часов, и она может представлять собой продолжительность, находящуюся в пределах нижнего предельного значения и верхнего предельного значения данных продолжительностей.
[0168] Что касается температуры реакции, температура, когда ссылаются на фразу “в диапазоне от -78°C до температуры, при которой кипит растворитель” обозначает температуру, находящуюся в диапазоне от -78°C до температуры, при которой кипит растворитель (или смешанный растворитель), применяемый в реакции, например, когда метанол применяют в качестве растворителя, фраза “от -78°C до температуры, при которой кипит растворитель” обозначает то, что температура находится в диапазоне от -78°C до температуры, при которой кипит метанол.
[0169] То же самое применимо, когда ссылаются на фразу “от 0°C до температуры, при которой кипит растворитель”, и она обозначает температуру в диапазоне от 0°C до температуры, при которой кипит растворитель (или смешанный растворитель), применяемый для реакции. Нижнее предельное значение температуры составляет, например, -78°C или 0°C, как описано выше, но она может также представлять собой другие температуры, такие как 20°C, 23°C, 25°C, 40°C, 50°C, 70°C, 80°C, 90°C, 100°C, 150°C и подобные.
[0170] Что касается температуры реакции, нижнее предельное значение и верхнее предельное значение температуры реакции может составлять, например, ±1°C, ±2°C, ±3°C, ±4°C и ±5°C соответствующих температур.
[0171] Если не указано иначе, в способе получения настоящего изобретения, “комнатная температура” обозначает температуру лаборатории, экспериментальной лаборатории или подобную, и “комнатная температура” в примерах настоящего изобретения обычно обозначает температуру от приблизительно 1°C до приблизительно 30°C (определенную фармакопеей Японии). Она предпочтительно указывает на температуру обычно от приблизительно 5°C до приблизительно 30°C, более предпочтительно указывает на температуру обычно от приблизительно 15°C до приблизительно 25°C, и даже более предпочтительно указывает на температуру 20°C±3°C.
[0172] Соединения в настоящем изобретении могут образовывать соль присоединения кислоты в зависимости от типа заместителя. Данная соль конкретно не ограничена при условии, что она представляет собой фармацевтически приемлемую соль, и ее примеры включают соль неорганической кислоты, соль органической кислоты и подобные. Предпочтительные примеры солей неорганических кислот включают соли хлористоводородной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, азотной кислоты, серной кислоты, фосфорной кислоты и подобных. Предпочтительные примеры солей органических кислот включают соли алифатических монокарбоновых кислот, таких как муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, масляная кислота, валериановая кислота, энантовая кислота, каприновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, молочная кислота, сорбиновая кислота, миндальная кислота и подобные; соли алифатических дикарбоновых кислот, таких как щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, яблочная кислота, винная кислота и подобные; соли алифатических трикарбоновых кислот, таких как лимонная кислота и подобные; соли ароматических монокарбоновых кислот, таких как бензойная кислота, салициловая кислота и подобные; соли ароматических дикарбоновых кислот, таких как фталевая кислота и подобные; соли органических карбоновых кислот, таких как коричная кислота, гликолевая кислота, пировиноградная кислота, оксиловая кислота, салициловая кислота, N-ацетилцистеин и подобные; соли органических сульфоновых кислот, такие как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и подобные; и соль присоединения кислот кислых аминокислот, таких как аспарагиновая кислота, глутаминовая кислота и подобные.
[0173] Соль, описанную выше, модно получить согласно общепринятому способу, например, смешиванием раствора, содержащего подходящее количество кислоты, с соединением, описанным в настоящем изобретении, получая требуемые соли, и затем фракционированием солей и сбором их фильтрацией; или путем отгонки смешанного растворителя. Кроме того, соединение в настоящем изобретении или его соль может образовывать сольват с растворителем, таким как вода, этанол, глицерин и подобные. В качестве обзорной статьи по солям опубликован «Handbook of Pharmaceutical Salts: Properties, Selection, и Use. Stahl & Wermuth (Wiley-VCH, 2002), и данная книга включает подробное описание.
[0174] Как показано на (схеме 5) ниже, применяя соединение, представленное формулой (A), в качестве исходного соединения, соединение, представленное формулой (B), или его соль можно получить через соединения формул (A-5), (A-6) и (A-7).
[0175]
[C87]
(Схема 5)
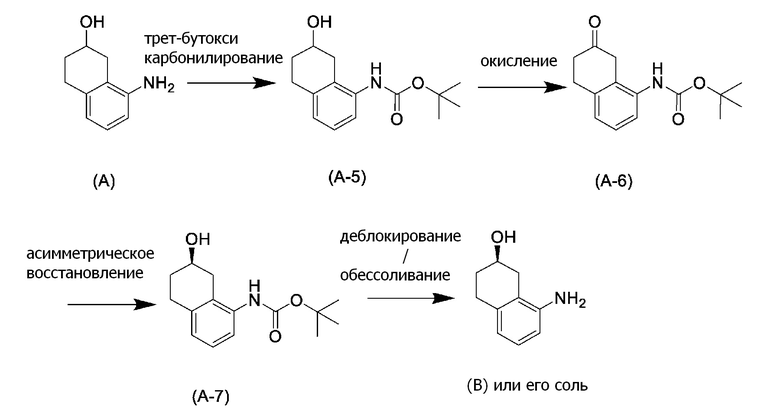
[0176] Кроме того, как показано на (схеме 6) ниже, соединение, представленное формулой (B), или его соль можно получить способом согласно (схеме 5), описанной выше, заменой защитной группы аминогруппы соединения, представленного формулой (A), на защитную группу, которая является отличной от трет-бутоксикарбонильной группы, например, защитную группу P1, такую как карбаматные защитные группы, такие как диметоксикарбонильная группа, этоксикарбонильная группа, бензилоксикарбонильная группа, 2,2,2-трихлорэтоксикарбонильная группа, 9-флуоренилметилоксикарбонильная группа, аллилоксикарбонильная группа и подобные; сульфонильные защитные группы, такие как метансульфонильная группа, этансульфонильная группа, бензолсульфонильная группа, тозильная группа, нитробензолсульфонильная группа и подобные; и алкилкарбонильные или арилкарбонильные защитные группы, такие как ацетильная группа, этилкарбонильная группа, трифторацетильная группа, бензоильная группа и подобные.
[0177]
[C88]
(Схема 6)
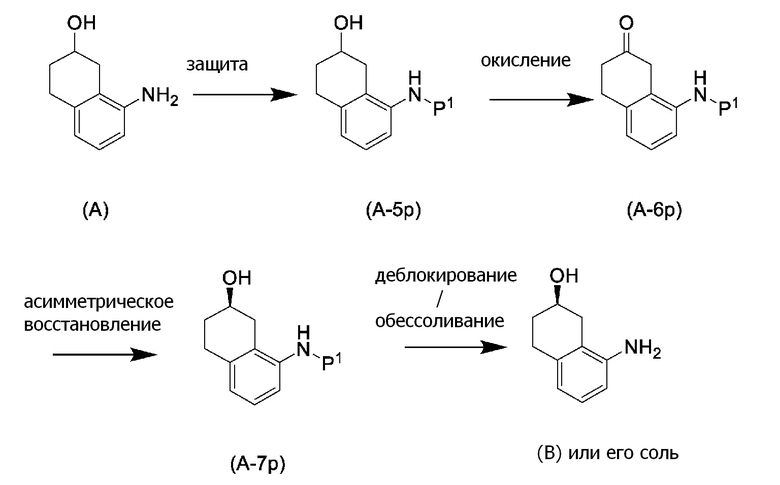
P1=метоксикарбонильная группа, этоксикарбонильная группа, бензилоксикарбонильная группа, 2,2,2-трихлорэтоксикарбонильная группа, 9-флуоренилметилоксикарбонильная группа, аллилоксикарбонильная группа, метансульфонильная группа, этансульфонильная группа, бензолсульфонильная группа, тозильная группа, нитробензолсульфонильная группа, ацетильная группа, этилкарбонильная группа, трифторацетильная группа, бензоильная группа и т.д.
[0178] Условия защиты соединения, представленного формулой (A), защитной группой P1 или удаления защитной группы P1 соединения, представленного формулой (A-7p), можно выбрать в зависимости от типа защитной группы P1 способом, известным из литературы, например, способ защиты и удаления, описанный в книге, “Protective Groups in Organic Synthesis, 4th Edition, 2007, John Wiley & Sons, Greene et al.”
[0179] Как показано на (схеме 7) ниже, применяя соединение, представленное формулой (SM8), в качестве исходного соединения, соединение, представленное формулой (B), можно получить через соединение, представленное формулой (A8).
[0180]
[C89]
(Схема 7)
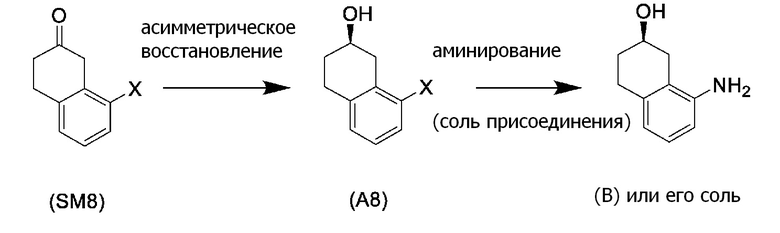
[0181] Кроме того, как показано на (схеме 8) ниже, применяя соединение, представленное формулой (SM8-BR), в качестве исходного соединения, соединение, представленное формулой (B), можно получить через соединение, представленное формулой (A8-BR).
[C90]
(Схема 8)

[0182] Как показано на (схеме 9) ниже, применяя соединение, представленное формулой (A), в качестве исходного соединения, соединение, представленное формулой (I), можно получить через соединение, представленное формулой (B). На (схеме 9), соединение, представленное формулой (B-HA), представляет собой соль кислоты HA соединения, представленного формулой (B), где HA представляет собой кислоту.
[C91]
(Схема 9)
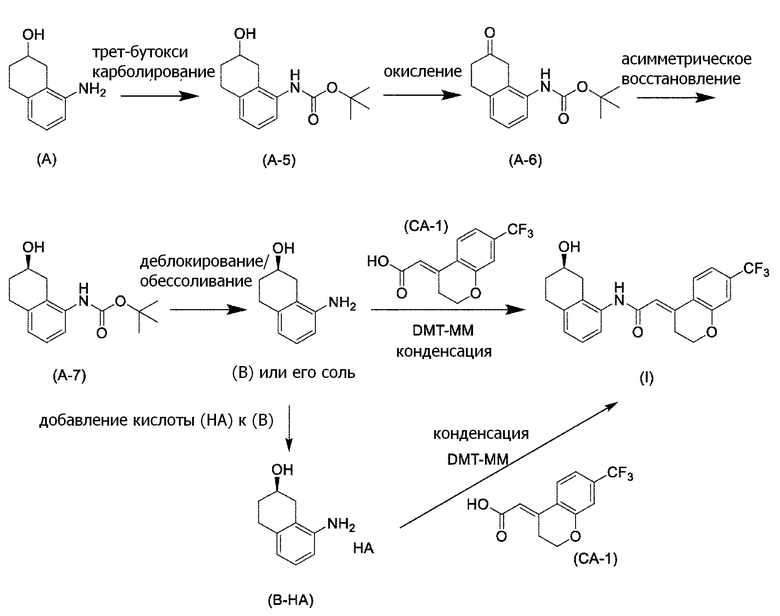
[0183] Кроме того, как показано на (схеме 10) ниже, применяя соединение, представленное формулой (A), в качестве исходного соединения, соединение, представленное формулой (I), можно получить через соединение, представленное формулой (B). Определение заместителя P1 на (схеме 10) является таким же, как определение на (схеме 6), описанной выше. На (схеме 10), соединение, представленное формулой (B-HA), представляет собой соль кислоты HA соединения, представленного формулой (B), где HA представляет собой кислоту.
[C92]
(Схема 10)
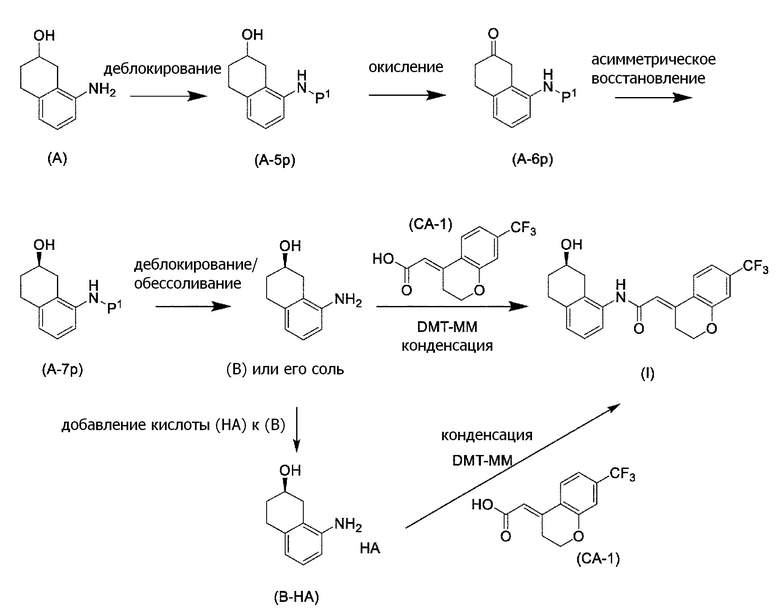
[0184] Кроме того, как показано на (схеме 11) ниже, применяя соединение, представленное формулой (SM8), в качестве исходного соединения, соединение, представленное формулой (I), можно получить через соединение, представленное формулой (B). На (схеме 11), соединение, представленное формулой (B-HA), представляет собой соль кислоты HA соединения, представленного формулой (B), где HA представляет собой кислоту.
[C93]
(Схема 11)
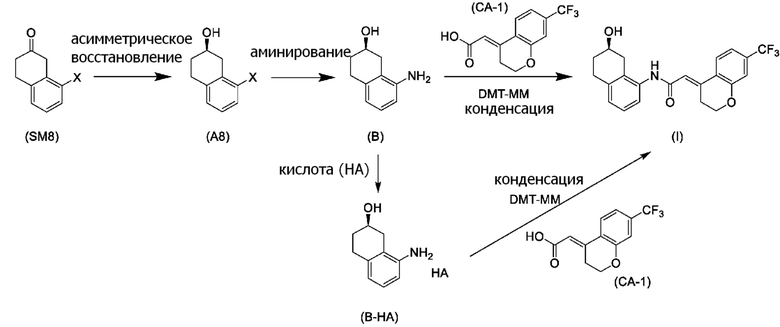
[0185] Кроме того, как показано на (схеме 12) ниже, применяя соединение, представленное формулой (SM8-BR), в качестве исходного соединения, соединение, представленное формулой (I), можно получить через соединение, представленное формулой (B). На (схеме 12), соединение, представленное формулой (B-HA), представляет собой соль кислоты HA соединения, представленного формулой (B), где HA представляет собой кислоту.
[C94]
(Схема 12)
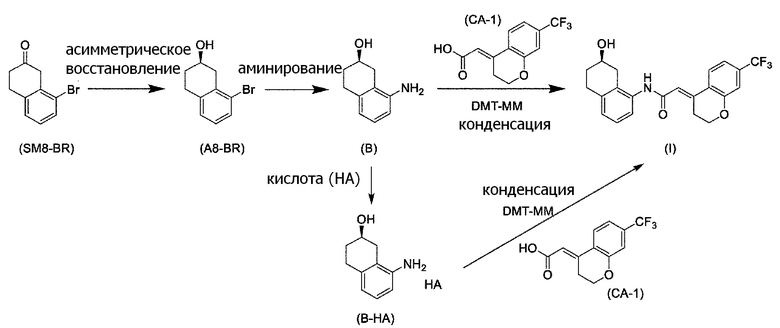
[0186] В настоящем изобретении, соединения формулы (A), формулы (A-5) и формулы (A-5p), которые представляют собой рацематы, включают (R) форму и (S) форму. Это обозначает, что, например, формула (A-5) включает формулу (A-5S) и формулу (A-5R) (=формула (A-7)).
[C95]
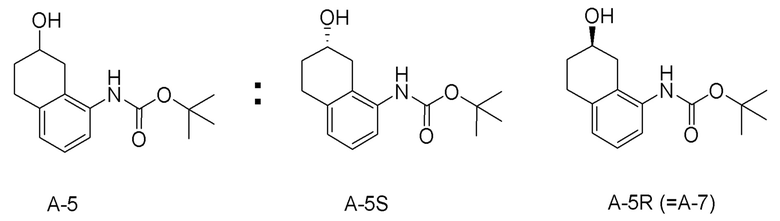
[0187] [Асимметрическое восстановление кетонов]
Различные реакции являются известными как способ превращения кето группы в молекуле в хиральную спиртовую группу. Например, существует способ, в котором кето группу превращают в рацемическую спиртовую группу, применяя восстанавливающий агент (боргидрид натрия, алюмогидрид лития (LAH), боран-тетрагидрофуран (BH3-THF) и подобные), и после этого получают хиральную спиртовую группу способом, таким как способ фракционной перекристаллизации (способ, в котором кристаллический диастереомер получают ионным связыванием оптического агента для разделения с рацематом, и данный кристаллический диастереомер фракционируют перекристаллизацией и нейтрализуют, при необходимости, получая свободное хиральное соединение), диастереомерный способ (смотри WO 2009/055749) и способ с хиральной колонкой (смотри WO 2009/050289).
[0188] Кроме того, следующие реакции являются известными: асимметрическая восстановительная реакция, применяя катализатор на основе переходного металла (например, Ru, Rh и подобные) (WO 2009/050289; Organometallics 10, p500-, 1991; и подобные), асимметрическая восстановительная реакция, в которой смешивают Al(CH3)3 и BINOL в качестве лиганда (Angew. Chem. Int. Ed., 41, p1020-, 2002), асимметрическая восстановительная реакция, применяя хиральный Ru (BINAP) катализатор (J. Am. Chem. Soc. 110, p629-, 1988), асимметрическая восстановительная реакция, применяя оксазаборолидин (J. Am. Chem. Soc. 109, p5551-1987), асимметрическая восстановительная реакция, применяя биокатализатор (дрожжи, грибы, плесень, фермент и подобные) (смотри таблицу 1) и подобные.
[0189] В некоторых аспектах, асимметрическую реакцию предпочтительно проводят, применяя биокатализатор. Асимметрическая реакция, применяя биокатализатор, имеет преимущества не только в том, что она имеет высокую стереоселективность, органический растворитель и/или воду можно применять в качестве растворителя для реакции, реакция протекает в мягких условиях (нормальная температура, нормальное давление), и он дешевле, чем химический катализатор и подобные, но она также представляет собой реакцию, которая в последние годы привлекает внимание как экологически безопасная реакция, поскольку можно снизить количество отходов после реакции, и также является подходящей реакцией для легкого получения хирального соединения.
[0190] Обычно, в асимметрической восстановительной реакции, применяя фермент, химический выход (%) и оптически активный выход (ee%) хирального соединения, которое получают, изменяется в зависимости от специфичности реакции (селективности для типа фермент-специфической реакции), субстратной специфичности (селективности для типа субстрата) и условий реакции (температура реакции, pH, растворитель, продолжительность реакции и подобные). Многие ферменты имеют очень высокую специфичность реакции, и реакции, катализируемые одним ферментом, ограничены, но существуют различные ферменты, то есть ферменты с более высокой субстратной специфичностью или ферменты с более низкой субстратной специфичностью. Соответственно, например, когда кето группу асимметрически восстанавливают до хиральной спиртовой группы, даже если выбирают фермент, для которого получают благоприятный химический выход и оптически активный выход из соединений, имеющих структуру, аналогичную структуре субстрата (кетоновое соединение), который будут применять, и ферментативную реакцию проводят в тех же условиях, требуемое хиральное спиртовое соединение нельзя получить с таким же химическим выходом и оптически активным выходом.
[0191] Например, биокатализаторы, которые могут селективно восстанавливать кето группы β-тетралона [CAS номер: 530-93-8] до хирального спирта, показанные в таблице 1, являются известными.
[0192]
[Таблица 1]
[0193] [Химия в потоке]
Для синтетических реакций, обычно существуют способ в потоке (химия в потоке) и периодический способ. Химия в потоке представляет собой непрерывный способ синтеза, применяя реакционное устройство, которое направляет жидкость из сосуда, содержащего два или более отличных видов растворов (например, сырье+растворитель, реагент+растворитель и подобные) через трубу в реактор, и затем в бак для извлечения при постоянной скорости потока, применяя насос.
[0194] Химию в потоке можно применять при превращении соединения, представленного формулой (A-5), в соединение, представленное формулой (A-6), реакцией окисления. Фигура 1 показывает пример реакционного устройства, применяемого в химии в потоке. Реакционное устройство, показанное на фигуре 1, has впускные отверстия для азота (L1, L2, L3, L4); сосуд (M1), содержащий сырье, TEMPO и дихлорметан; сосуд (M2), содержащий KBr, NaHCO3 и воду; сосуд (M3), содержащий 5,0% по весу NaClO; насосы (P1, P2, P3); трубы предварительного охлаждения (T1, T2, T3); мешалки (S1, S2, S3); и реактор (R1, R2, R3).
[0195] Реакционное устройство фигуры 1 применяют, например, следующим образом. Сначала, сырье (соединение, представленное формулой (A-5)), TEMPO и дихлорметан подают в сосуд M1, KBr, гидрокарбонат натрия и воду подают в сосуд M2, и 5,0% по весу NaClO подают в сосуд M3. При ток газообразного азота из соответствующих впускных отверстий для азота L1, L2, L3, L4, реагенты перекачивают из соответствующих сосудов M1, M2, M3 при предварительно определенной скорости потока, применяя соответствующие насосы P1, P2, P3, затем пропускают через соответствующие трубы предварительного охлаждения T1, T2, T3, затем последовательно пропускают через реактор R1, реактор R2 и реактор R3, и, таким образом, выливают в бак для извлечения CD. Затем, целевой продукт (соединение, представленное формулой (A-6)) получают из бака для извлечения CD.
Химию в потоке можно также применять к реакциям, для которых трудно сохранять безопасность нормальным периодическим способом (смотри обзорную статью по химии в потоке ChemSusChem, 5(2), Special Issue; Flow Chemistry, стр. 213-439, February 13, 2012).
[0196] Периодический способ представляет собой обычную синтетическую реакцию и представляет собой способ очистки продукта, полученного после проведения реакции, применяя реактор. Периодический способ обладает преимуществом, заключающимся в том, что соединение можно получать в несколько стадий.
В способе с потоком (химии в потоке), реакцию проводят в поточном режиме, применяя, например, химический проточный реактор с мешалкой (CSTR) в качестве реакционного устройства. В способе с потоком, поскольку реакцию можно проводить в небольшом реакторе, эффективность реакции является высокой, и реакционные условия можно точно контролировать, так что целевой продукт может стабильно поставлять.
[0197] [Реакция аминирования]
Способ (реакция аминирования) превращения атома галогена галогенированного арила в аминогруппу можно проводить в присутствии металлического катализатора и в присутствии или отсутствии лиганда, применяя в качестве источника азота, соединение, представленное NHRARB (где каждый RA и RB независимо представляет собой атом водорода или заместитель, такой как метильная группа, этильная группа, бензильная группа и подобные), RCCONH2 (где RC независимо представляет собой заместитель, такой как метильная группа, этильная группа, бензильная группа, диметокси группа, этокси группа, a трет-бутокси группа, бензилокси группа и подобные), или подобные.
[0198] Что касается реакции аминирования галогенированного арила, применяя аммиак в качестве источника азота, например, следующий способ, применяя металлический катализатор, является известным как способ, известный из литературы, но способ не ограничивается им. Pd2(dba)3 (J. Am. Chem. Soc., 129(34), p10354-10355, 2007), PdCl2-Josiphos complex (J. Am. Chem. Soc., 128(31), p10028-10029, 2006), CuI (Chem. Commun., 26, p3052-3054, 2008; J. Org. Chem., 74(12), p4542-4546, 2009), Cu(OAc)2 (Angew. Chem. Int. Ed., 48(2), p337-339, 2009), Cu2O (Ukrainskii Khimiche skii Zhurnal (Russian Edition), 53(12), P1299-302, 1987).
[0199] Например, для реакции аминирования 8-галоген-1,2,3,4-тетрагидронафталин-2-ола, в котором вторичный спирт присутствует в молекуле, является известной реакция аминирования, применяя Pd2(dba)3 в качестве металлического катализатора и трет-бутилкарбамат в качестве источника азота, но примеры, применяя другие металлические катализаторы, неизвестны.
[0200] В некоторых аспектах, реакция аминирования предпочтительно представляет собой реакцию, которое обеспечивает непосредственное введение аминогруппы аммиаком. Альтернативно, можно вводить аминогруппу деблокированием защитной группы после замещения, например, защищенным амино соединением, таким как NHRA1RB1 (где RA1 представляет собой атом водорода, и RB1 представляет собой защитную группу аминогруппы, такую как бензильная группа, 4-диметоксибензил и подобные), и RCCONH2 (где RC независимо представляет собой заместитель, такой как метильная группа, этильная группа, бензильная группа, диметокси группа, этокси группа, трет-бутокси группа, бензилокси группа и подобные). Однако, поскольку реакция аминирования, применяя защищенное амино соединение, требует стадии деблокирования защитной группы, реакция, которая обеспечивает непосредственное введение аминогруппы аммиаком, является предпочтительной при рассмотрении крупномасштабного синтеза или промышленного получения.
[0201] [Реакции конденсации]
В общем, что касается реакции конденсации соединения, содержащего карбоксильную группу, и соединения, содержащего аминогруппу, амидную связь можно получить проведением реакции конденсации, применяя, например, конденсирующий агент, такой как 1,3-дициклогексилкарбодиимид (DCC), гидрохлорид 1-этил-3-(3'-диметиламинопропил)карбодиимида (WSC·HCl), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (BOP реагент), бис(2-оксо-3-оксазолидинил)фосфинхлорид (BOP-Cl), гексафторфосфат 2-хлор-1,3-диметилимидазолиния (CIP) и подобные (смотри, например, Experimental Chemistry Course 22, 4th Edition, Organic Synthesis IV: Acids, Amino Acids, Peptides, pp.193-309, 1992, Maruzen; и подобные).
[0202] Как результат примера конденсирующего агента, изобретатели настоящего изобретения обнаружили, что соединение, представленное формулой (I), можно легко получить с высоким выходом применением, а частности DMT-MM в качестве конденсирующего реагента в реакции конденсации соединения, представленного формулой (B), которое содержит и гидроксильную группу и анилиновую аминогруппу, и гетероциклиденуксусной кислоты, представленной формулой (CA-1), которая содержит has a карбоксильную группу.
[0203] Все публикации, цитируемые в настоящем изобретении, такие как документы предшествующего уровня техники, нерассмотренные патентные публикации, патентные публикации и другие патентные документы, включены в настоящее изобретение посредством ссылки во всей своей полноте. Настоящее изобретение включает описание объема формулы изобретения, спецификации и чертежи китайской патентной заявки № 201910783254.8 (поданной 23 августа 2019 г), международной патентной заявки № PCT/JP2019/036451 (поданной 18 сентября 2019 г) и китайской патентной заявки № 202010355546.4 (подана 29 апреля 2020 г), которые являются основанием для спрашивания приоритета настоящей заявки.
Примеры
[0204] Далее настоящее изобретение будет конкретно описано со ссылкой на примеры, но настоящее изобретение не ограничивается ими.
[0205] Bruker AVANCE III 400 МГц ЯМР спектрометр (оборудованный 5 мм Bruker PABBO Z-градиентным датчиком и TOPSPIN 3.5 программным обеспечением) применяли для получения спектров ядерного магнитного резонанса (ЯМР) соединений, представленных формулой (A-5), формулой (A-6), формулой (A-7) и формулой (B). Кроме того, JEOL JNM-LA300 FT-ЯМР (JEOL Ltd.) применяли для получения спектров ядерного магнитного резонанса (ЯМР) броматной соли соединения, представленного формулой (B), и соединения, представленного формулой (I).
Соединения, представленные формулой (A-5), формулой (A-6), формулой (A-7) и формулой (B), проверяли высокоэффективной жидкостной хроматографией(ВЭЖХ) в следующих условиях.
[0206]
[Таблица 2]
[условия ВЭЖХ анализа для соединения, представленного формулой (A-5), формулой (A-6) и формулой (A-7)]
Соединение, представленное формулой (A-6): 0,25 мг/мл
(способ получения: 770 мг NH4Ac точно взвешивают и тщательно смешивают с 2000 мл чистой воды, и смесь дегазируют ультразвуковыми волнами)
[Таблица 3]
[Время удерживания (RT) каждого соединения]
[Таблица 4]
[Условия ВЭЖХ анализа для соединения, представленного формулой (B)]
Гидрохлоридная соль соединения, представленного формулой B: 0,12 мг/мл
[Таблица 5]
[Время удерживания (RT) для каждого соединения]
[Таблица 6]
[Условия хирального анализа ВЭЖХ для соединения, представленного формулой (A-7)]
(Способ получения: 900 мл н-гексана и 100 мл этанола тщательно смешивали, и затем 1 мл диэтиламина добавляли и смешивали со смешанным раствором н-гексана и этанола)
[Таблица 7]
[Время удерживания (RT) для каждого соединения]
[0207] Кроме того, спектр жидкостной хроматографии-масс-спектрометрии (LC-Mass) броматной соли соединения, представленного формулой (B), и соединения, представленного формулой (I), получали следующим способом.
[UPLC] Применяли Waters AQUITY UPLC систему и BEH C18 колонку (2,1 мм×50 мм, 1,7 мкм) (Waters), и применяли подвижную фазу и градиентные условия ацетонитрил:водный раствор 0,05% трифторуксусной кислоты=5:95 (0 минут)-95:5 (1,0 минута)-95:5 (1,6 минут)-5:95 (2,0 минут).
[0208] В 1H-ЯМР данных, с обозначает синглет, д обозначает дуплет, т обозначает триплет, кв обозначает квартет, м обозначает мультиплет, ушс обозначает уширенный в качестве паттерна ЯМР сигналов, J обозначает константу связывания, Гц обозначает Герц, DMSO-d6 представляет собой дейтерированный диметилсульфоксид, и CDCl3 обозначает дейтерированный хлороформ. В 1H-ЯМР данных, сигналы, которые нельзя подтвердить из-за широкополосных сигналов, такие как протоны гидроксильной группы (OH), аминогруппы (NH2) и амидной группы (CONH), не описывают в данных.
[0209] В LC-Mass данных, M обозначает молекулярный вес, RT обозначает время удерживания, и [M+H]+ обозначает пик молекулярного иона.
[0210] (примеры 1a-1d) получение трет-бутил(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамата (A-5)
[0211]
[C96]
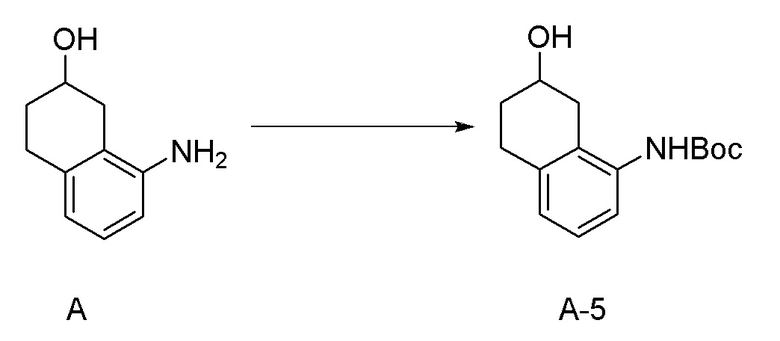
[0212] (пример 1a)
Ди-трет-бутилдикарбонат (Boc2O) (0,74 г) добавляли к раствору соединения, представленного формулой A (полученного согласно способу получения, описанному в WO 2009/050289) (0,5 г) и гидрокарбоната натрия (0,154 г) в 1,4-диоксан (5 мл)-вода (5 мл), и смесь перемешивали при температуре реакции 20°C-30°C в течение 22 часов. Гидрокарбонат натрия (0,104 г) дополнительно добавляли к смеси, и смесь перемешивали при температуре реакции 20°C-30°C в течение 18 часов. Ди-трет-бутилдикарбонат (0,15 г) дополнительно добавляли к смеси, и смесь перемешивали при температуре реакции 20°C-30°C в течение 5 часов. Ди-трет-бутилдикарбонат (0,15 г) дополнительно добавляли к смеси, и смесь перемешивали при температуре реакции 20°C-30°C в течение 16 часов. Ди-трет-бутилдикарбонат (0,1 г) дополнительно добавляли к смеси, и смесь дополнительно перемешивали при температуре реакции 20°C-30°C в течение 1 часа. Этилацетат добавляли к реакционному раствору, и органический слой фракционировали. Водный слой промывали этилацетатом, и органический слой комбинировали с ранее полученным органическим слоем, и затем промывали соляным раствором. Органический слой концентрировали при пониженном давлении, и полученный остаток отверждали дихлорметаном и н-гептаном, получая заявленное в заголовке соединение (0,46 г).
[0213] (пример 1b)
Ди-трет-бутилдикарбонат (Boc2O) (1,61 г) добавляли к раствору соединения, представленного формулой A (WO 2009/050289) (1,0 г), и гидрокарбоната натрия (1,55 г) в тетрагидрофуран (10 мл)-вода (10 мл), и смесь перемешивали при температуре реакции 45°C-55°C в течение 17 часов. Ди-трет-бутилдикарбонат (0,13 г) дополнительно добавляли к смеси, и смесь перемешивали при температуре реакции 45°C-55°C в течение 2 часов. После охлаждения реакционного раствора до комнатной температуры, метил трет-бутиловый эфир (MTBE) добавляли к реакционному раствору, и pH доводили до 5-6 10 вес/об% раствором лимонной кислоты, затем органический слой фракционировали. Водный слой экстрагировали метил трет-бутиловым эфиром, и органический слой комбинировали с ранее полученным органическим слоем, и затем промывали водой и соляным раствором. Органический слой концентрировали при пониженном давлении, и полученный остаток отверждали дихлорметаном и н-гептаном, получая заявленное в заголовке соединение (1,34 г).
[0214] (пример 1c)
Ди-трет-бутилдикарбонат (Boc2O) (17,4 г) добавляли к раствору соединения, представленного формулой A (полученного согласно способу получения, описанному в WO 2009/050289) (10 г), и гидрокарбоната натрия (15,5 г) в тетрагидрофуран (100 мл)-вода (100 мл), и смесь перемешивали при температуре реакции 45°C-55°C в течение 17 часов. Ди-трет-бутилдикарбонат (1,3 г) дополнительно добавляли к смеси, и смесь перемешивали при температуре реакции 45°C-55°C в течение 2 часов. Ди-трет-бутилдикарбонат (1,3 г) дополнительно добавляли к смеси, и смесь дополнительно перемешивали при температуре реакции 45°C-55°C в течение 1 часа. После охлаждения реакционного раствора до комнатной температуры, метил трет-бутиловый эфир (MTBE) добавляли к реакционному раствору, и pH доводили до 5-6 10% раствором лимонной кислоты, затем органический слой фракционировали. Водный слой экстрагировали метил трет-бутиловым эфиром, и органический слой комбинировали с ранее полученным органическим слоем, и затем промывали водой и соляным раствором. Органический слой концентрировали при пониженном давлении, и полученный остаток отверждали дихлорметаном и н-гептаном, получая заявленное в заголовке соединение (15,1 г).
[0215] (пример 1d)
Раствор соединения, представленного формулой A (полученного согласно способу получения, описанному в WO 2009/050289) (218,5 г), в тетрагидрофуране (1,9 л) доводили до температуры 20°C-30°C, и водный раствор гидрокарбоната натрия (319 г, (3,2 экв. карбоната натрия и 1,9 л воды) добавляли к раствору в течение 10 минут при температуре 20°C-30°C. Температура смешанного раствора выше устанавливали на 0°C-10°C, и ди-трет-бутилдикарбонат (413 г) добавляли к смешанному раствору в течение 15 минут, поддерживая ту же температуру. Температуру реакции устанавливали на 45°C-55°C, и смешанный раствор перемешивали при той же температуре в течение 18 часов. После охлаждения температуры реакции до 20°C-30°C, метил трет-бутиловый эфир (1,9 л) добавляли к реакционному раствору, и смешанный раствор перемешивали при 20°C-30°C в течение 10 минут. 10% раствор лимонной кислоты (2,5 л) добавляли к смешанному раствору, и органический слой фракционировали. Водный слой экстрагировали метил трет-бутиловым эфиром (1 л×2 раза), и органический слой комбинировали с ранее полученным органическим слоем, и затем промывали водой (1 л×2 раза). После концентрирования органического слоя при пониженном давлении до того, как он составлял 500 мл, операцию добавления дихлорметана (1 л) и концентрирования органического слоя при пониженном давлении до того, как органический слой составлял приблизительно 500 мл проводили дважды, затем добавляли н-гептан (1 л), и органический слой концентрировали при пониженном давлении до того, как органический слой не составлял приблизительно 500 мл, добавляли н-гептан (800 л), и органический слой концентрировали при пониженном давлении до того, как органический слой составлял приблизительно 600 мл, и дихлорметан (300 мл) и н-гептан (600 мл) добавляли, и органический слой концентрировали при пониженном давлении до того, как органический слой составлял приблизительно 600 мл, затем добавляли дихлорметан (1 л), и активированный уголь (21 г) добавляли к ней, и смешанный раствор перемешивали при 20°C-30°C в течение 2 часов. Затем, смешанный раствор фильтровали, и фильтрат концентрировали при пониженном давлении до того, как он составлял приблизительно 500 мл, затем добавляли к нему дихлорметан (800 мл), и раствор концентрировали при пониженном давлении до того, как он составлял приблизительно 500 мл. Добавляли дихлорметан (800 мл), и раствор фильтровали, получая твердый остаток. Полученный твердый остаток сушили при 35°C в течение 15 часов, получая заявленное в заголовке соединение (303,3 г) в виде серовато-черного твердого остатка.
[0216] [Данные физических свойств соединения, представленного формулой (A-5)]
(1H ЯМР, 400 МГц, изготовитель: Bruker, DMSO-d6, δ ppm)
8,36 (с, 1H), 7,09 (д, 1H, J=7 Гц), 7,03 (т, 1H, J=7 Гц), 6,87 (д, 1H, J=7 Гц), 4,78 (д, 1H, J=4 Гц), 3,90-3,84 (м, 1H), 2,89-2,81 (м, 2H), 2,75-2,65 (м, 1H), 2,42 (дд, 1H, J=7 Гц, 17 Гц), 1,90-1,80 (м, 1H), 1,62-1,53 (м, 1H), 1,46 (с, 9H)
[0217] (примеры 2a-2f) получение трет-бутил(7-оксо-5,6,7,8-тетрагидронафталин-1-ил)карбамата (A-6)
[0218]
[C97]
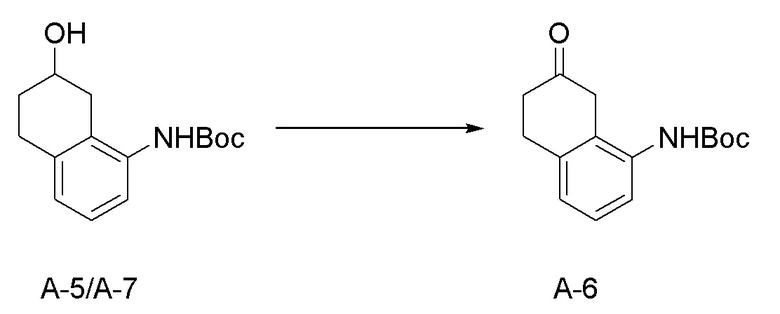
[0219] (пример 2a)
Применяя трет-бутил (R)-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамат (формула (A-7)) (0,5 г), полученный из (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола тем же способом, как в способах (примеров 1a-1d), реакцию окисления проводили в условиях реагентов, показанных в следующей таблице, в растворителе (дихлорметан (12,5 мл)-вода (7,5 мл)) при температуре реакции (0°C-5°C), и подтверждали, что заявленное в заголовке соединение получали (IPC чистота, применяя ВЭЖХ (IPC=контроль при получении)).
[0220]
[Таблица 8]
(экв.)
(экв.)
(экв.)
(экв.)
(мин)
(мин)
(мин)
(ч)
[0221] (пример 2b)
Применяя трет-бутил (R)-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамат (формула (A-7)) (0,5 г), полученный из(R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола тем же способом, как в способах (примеров 1a-1d), реакцию окисления проводили в условиях реагентов, показанных в следующей таблице, при температуре реакции (0°C-5°C), получая заявленное в заголовке соединение (IPC чистота, применяя ВЭЖХ).
[0222]
[Таблица 9]
вода: 15 кратный ее объем
Растворитель: 10 кратный его объем
(экв.)
(экв.)
(экв.)
(экв.)
7,5 мл
5 мл
7,5 мл
5 мл
[0223] (пример 2c)
Раствор трет-бутил (R)-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамата (формула (A-7)) (10 г), который получали тем же способом, как в способах (примеров 1a-1d) в дихлорметане (250 мл, 25 кратный его объем)-вода (150 мл, 15 кратный его объем) охлаждали до -2°C-2°C, и TEMPO (0,5 экв.), KBr (0,2 экв.), NaHCO3 (4,0 экв.) и NaClO ((8,1%), 1,4 экв.) добавляли к раствору при той же температуре. Когда после этого подтверждали IPC чистоту, подтверждали, что реакция завершилась с чистотой 95,5%. Реакционный раствор обрабатывали, и посредством этого, получали заявленное в заголовке соединение (1H-ЯМР выход 77,1%).
[0224] (пример 2d)
TEMPO окисление проводили в поточном режиме, применяя химический проточный реактор с мешалкой (CSTR), показанный на фигуре 1. Раствор (500 мл) трет-бутил-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамата (формула (A-5)) (25 г), полученный тем же способом, как в способах (примеров 1a-1d) и TEMPO (0,5 экв.) в дихлорметане добавляли в сосуд M1, и KBr (0,05 экв.), гидрокарбонат натрия (4 экв.) и воду (375 мл) добавляли в сосуд M2, и 5,0 вес.% NaClO (1,3 экв.) добавляли в сосуд M3. При токе газообразного азота из соответствующих впускных отверстий для азота L1, L2, L3, и L4, реагенты подавали из соответствующих сосудов M1, M2, и M3 при каждой скорости потока 13,67 мл/мин, 9,83 мл/мин и 4,27 мл/мин, применяя соответствующие насосы P1, P2, и P3, затем пропускали через соответствующие трубы для предварительного охлаждения T1, T2 и T3 (температура 0°C-5°C, длина трубы: 2 м, диаметр трубы: 1/16 SС), затем последовательно пропускали через реактор R1, реактор R2 и реактор R3 (где каждый из реакторов 1, 2, и 3 имел емкость 25 мл, и соответствующие реакторы охлаждали до 0°C-5°C), и посредством этого выливали в бак для извлечения CD (где продолжительность реакции в каждом из реакторов составляла 0,9 минут). Целевой продукт получали с IPC чистотой 96,5% реакционного раствора, полученного из бака для извлечения CD.
[0225] (пример 2e)
TEMPO окисление проводили в поточном режиме, применяя химический проточный реактор с мешалкой (CSTR), показанный на фигуре 1. Трет-бутил (R)-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамат (формула (A-5)) (35 г), полученный тем же способом, как в способах (примеров 1a-1d), TEMPO (0,5 экв.) и дихлорметан (700 мл) добавляли в сосуд M1, и KBr (0,05 экв.), гидрокарбонат натрия (4 экв.) и воду (525 мл) добавляли в сосуд M2, и 5,0 вес.% NaClO (1,3 экв.) добавляли в сосуд M3. При токе газообразного азота из соответствующих впускных отверстий для азота L1, L2, L3, и L4, реагенты подавали из соответствующих сосудов M1, M2, и M3 при каждой скорости потока 13,67 мл/мин, 9,83 мл/мин и 4,27 мл/мин, применяя соответствующие насосы P1, P2, и P3, затем пропускали через соответствующие трубы для предварительного охлаждения T1, T2, и T3 (температура 0°C-5°C, длина трубы: 2 м, диаметр трубы: 1/16 SС), затем последовательно пропускали через реактор R1, реактор R2 и реактор R3 (где каждый из реакторов 1, 2 и 3 имел вместимость 25 мл, и соответствующие реакторы охлаждали до 0°C-5°C), и затем выливали в бак для извлечения CD (где продолжительность реакции в каждом из реакторов составляла 0,9 минут, и она требовала 47 минут для завершения потока). IPC чистота реакционного раствора (850 г), полученного из бака для извлечения CD, составляла 95,0%. Водный раствор Na2S2O4 (Na2S2O4: 10 г, вода: 250 мл) добавляли к реакционному раствору, и смешанный раствор перемешивали в течение 30 минут. После разделения органического слоя и водного слоя, органический слой промывали водой (300 мл×2 раза). После концентрирования органического слоя при пониженном давлении до объема 1,5-2,5 объема, добавляли к нему н-гептан (30-50 мл), и смесь перемешивали при комнатной температуре в течение 1 часа, и добавляли к нему н-гептан (200 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Полученный в результате твердый остаток собирали фильтрованием и промывали н-гептаном (70 мл), и посредством этого получали заявленное в заголовке соединение (25,2 г) в виде грязно-белого твердого остатка.
[0226] (пример 2f)
TEMPO окисление проводили в поточном режиме, применяя химический проточный реактор с мешалкой (CSTR), показанный на фигуре 1. Трет-бутил-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамат (формула (A-5)) (276,6 г), полученный тем же способом, как в способах (примеров 1a-1d), TEMPO (82,588 г, 0,5 экв.) и дихлорметан (5,535 мл, 20 об) добавляли в сосуд M1, и KBr (6,251 г), гидрокарбонат натрия (352,943 г) и воду (4,149 мл) добавляли в сосуд M2, и 5,0 вес% NaClO (1,849 л) добавляли в сосуд M3. При токе газообразного азота из соответствующих впускных отверстий для азота L1, L2, L3, и L4, реагенты подавали из M1, M2 и M3 при каждой скорости потока 13,67 мл/мин, 9,83 мл/мин и 4,27 мл/мин, применяя соответствующие насосы P1, P2, и P3, затем пропускали через соответствующие трубы для предварительного охлаждения T1, T2, и T3 (температура 0°C-5°C, длина трубы: 2 м, диаметр трубы: 1/16 SС), затем последовательно пропускали через реактор R1, реактор R2 и реактор R3 (где каждый из реакторов 1, 2, и 3 имел вместимость 25 мл, и соответствующие реакторы охлаждали до 0°C-5°C), и затем выливали в бак для извлечения CD (где продолжительность реакции в каждом из реакторов в поточном режиме составляла 0,9 минут, и она требовала 410 минут для завершения потока). 3,7% раствор Na2S2O4 (2,597 г) добавляли к 6,845 г полученного реакционного смешанного раствора, и смешанный раствор перемешивали в течение 30 минут. После разделения органического слоя и водного слоя, органический слой промывали водой (3 л×2 раза). После концентрирования органического слоя при пониженном давлении до объема 2,5 об, н-гептан (205 мл) и один фрагмент уже полученного соединения, представленного формулой A-6, добавляли к нему, и смесь перемешивали при комнатной температуре в течение 1 часа. Кроме того, добавляли к нему н-гептан (2,1 л), и полученный в результате твердый остаток собирали фильтрованием, промывали н-гептаном (800 мл) и сушили при пониженном давлении в течение 13 часов, получая заявленное в заголовке соединение (190 г) в виде коричневого твердого остатка.
[0227] Данные физических свойств соединения, представленного формулой (A-6)]
(1H ЯМР, 400 МГц, изготовитель: Bruker, CDCl3, δ ppm)
7,42 (д, 1H, J=7 Гц), 7,14 (т, 1H, J=7 Гц), 6,97 (д, 1H, J=7 Гц), 6,12 (с, 1H), 3,41 (с, 2H), 3,01 (т, 2H, J=6 Гц), 2,51 (дд, 2H, J=6 Гц, 7 Гц), 1,44 (с, 9H)
[0228] (примеры 3a-3g) получение трет-бутил (R)-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)карбамата (A-7)
[0229]
[C98]
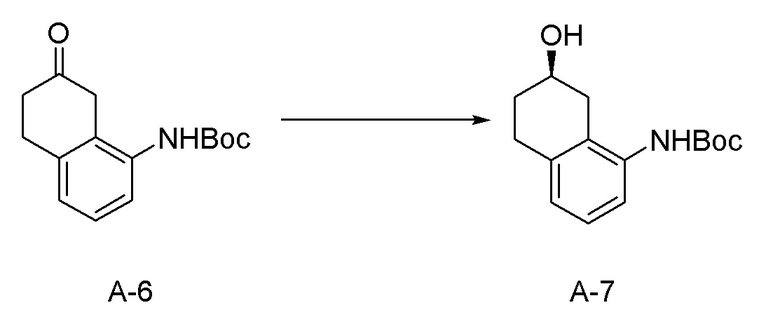
[0230] (пример 3a)
KRED (кетонредуктазу, полученную из Lactobalius sp., 2,0 г), D-глюкозу (0,2 г), глюкозадегидрогеназу (GDH) (0,02 г), никотинамидадениндинуклеотидфосфат (NADP) (0,01 г) и фосфатный буферный раствор (3,0 мл, полученный добавлением 21,25 г K2HPO4 и 10,62 г KH2PO4 к 1000 мл воды) смешивали в стеклянной пробирке (емкость 8 мл) и перемешивали для получения смешанного раствора A. Смешанный раствор, полученный растворением соединения (0,1 г) формулы (A-6), полученного тем же способом, как в способах, (примеров 2a-2f) в диметилсульфоксиде (DMSO) (0,2 мл) добавляли в ранее полученный смешанный раствор A. Смесь перемешивали при температуре реакции 23°C (20°C-25°C) в течение 43 часов (при 250 об/мин в круговой качалке). Некоторую часть реакционного раствора отбирал и подвергали ВЭЖХ анализу, и подтверждали, что получали заявленное в заголовке соединение.
KRED, применяемая в примерах 3a-3g, представляет собой кетонредуктазу, полученную из Lactobacillus sp. (EnzymeWorks, Inc., номер продукта: EW-KRED-172).
[0231] (пример 3b)
KRED (кетонредуктазу, полученную из Lactobalius sp., 20 г), D-глюкозу (2 г), глюкозадегидрогеназу (GDH) (0,2 г), никотинамидадениндинуклеотидфосфат (NADP) (0,1 г) и буферный раствор (полученный добавлением 0,86 г K2HPO4·3H2O и 0,3 г KH2PO4 к 30 мл воды) смешивали в реакторе и перемешивали для получения смешанного раствора B. Смешанный раствор, полученный растворением соединения (1 г) формулы (A-6), полученного тем же способом, как в способах (примеров 2a-2f), в толуоле (13 мл) добавляли к предварительно полученному смешанному раствору B. Смесь перемешивали при температуре реакции 23°C (20°C-25°C) в течение 15 часов. Реакционного раствора фильтровали, применяя целит, и водный слой и органический слой разделяли, затем водный слой экстрагировали толуолом (30 мл), и органический слой комбинировали с ранее полученным органическим слоем, и затем промывали водой (30 мл×2 раза), затем концентрировали, получая заявленное в заголовке соединение (0,5 г, оптическая чистота 99,9% ee) в виде коричневого масла.
[0232] (пример 3c)
Исследование количества KRED и pH проводили в условиях реакции, показанных в таблице ниже. Соединение, представленное формулой (A-6), получали тем же способом, как в способах (примеров 2a-2f). Буферный раствор получали из K2HPO4·3H2O и KH2PO4 тем же способом, как в примере 3b.
[0233]
[Таблица 10]
(g)
(кратный=кратный его вес)
(кратный=кратный его объем)
(°C)
(ч)
(%)
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
[0234] (пример 3d)
Исследование количества растворителя проводили в условиях реакции, показанных в таблице ниже. Соединение, представленное формулой (A-6), получали тем же способом, как в способах (примеров 2a-2f). Буферный раствор получали из K2HPO4·3H2O и KH2PO4 тем же способом, как в примере 3b.
[0235]
[Таблица 11]
(g)
(кратный=кратный его вес)
(кратный=кратный его объем)
(°C)
(ч)
(%)
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
[0236] (пример 3e)
Исследование количества буферного раствора, количества исходного материала и количества KRDE проводили в условиях реакции, показанных в таблице ниже. Соединение, представленное формулой (A-6), получали тем же способом, как в способах (примеров 2a-2f). Буферный раствор получали из K2HPO4·3H2O и KH2PO4 тем же способом, как в примере 3b.
[0237]
[Таблица 12]
(g)
(кратный=кратный его вес)
(кратный=кратный его объем)
(°C)
(ч)
(%)
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 30 кратный
GDH 0,2 кратный
NADP 0,1 кратный
Буферный раствор 15 кратный
[0238] (пример 3f)
Ферментативную реакцию проводили в течение 23 часов с pH реакционного раствора=6,0-7,0 и при температуре реакции 23°C (20°C-25°C), и затем в течение 16 часов при 50°C-60°C, применяя соединение (10 г) формулы (A-6), полученное тем же способом, как в способах (примеров 2a-2f), толуол (50 мл), буферный раствор (300 мл, состав K2HPO4·3H2O и KH2PO4 был тем же как в описанных выше примерах), KRED (100 г), D-глюкозу (20 г), NADP (0,25 г) и GDH (0,5 г), затем обработку проводили согласно упомянутому выше способу обработки, и посредством этого получали заявленное в заголовке соединение (11,75 г) в виде темно-красного масла.
[0239] (пример 3g)
KRED (1,279 г), D-глюкозу (253 г), NADP (12,61 г) и GDH (25,26 г) добавляли в буферный раствор (3,780 мл), полученный растворением K2HPO4·3H2O (108,4 г) и KH2PO4 (37,82 г) в воде (3,780 мл), получая смешанный раствор (MS-6-1), и затем смешанный раствор перемешивали в течение 1 часа. Смешанный раствор, полученный растворением соединения (126,05 г) формулы (A-6), полученного тем же способом, как в способах (примеров 2a-2f), в толуоле (630 мл) добавляли к предварительно полученному смешанному раствору (MS-6-1). Реакционного раствора перемешивали при температуре реакции 23°C (20°C-25°C) в течение 26 часов, поддерживая pH реакционного раствора равным pH=6,0-7,0. Трет-амиловый спирт (500 мл) и изоамиловый спирт (130 мл) добавляли к реакционному раствору, и смешанный раствор перемешивали при температуре реакции 23°C (20°C-25°C) в течение 16 часов. Добавляли к нему этилацетат (1300 мл) и целит (126 г), температура повышали до 60°C, и смесь перемешивали при той же температуре в течение 1 часа. После охлаждения до 20°C, проводили фильтрование, и водный слой и органический слой разделяли, и водный слой экстрагировали этилацетатом (1300 мл), затем органический слой комбинировали с ранее полученным органическим слоем, и затем промывали водой (1300 мл), получая органический слой (OP-6-1). Кроме того, этилацетат (1300 мл) добавляли к отфильтрованному целиту, и смесь перемешивали при 20°C-30°C в течение 10 часов и фильтровали, получая органический слой (OP-6-2), и еще раз этилацетат (1,300 мл) добавляли к отфильтрованному целиту, и смесь перемешивали при 20°C-30°C в течение 2 часов и фильтровали, получая органический слой (OP-6-3). Органический слой (OP-6-1), органический слой (OP-6-2) и органический слой (OP-6-3) комбинировали, получая органический слой (OP-6A). Кроме того, реакцию проводили тем же способом как в способе, описанном выше, применяя соединение (113 г) формулы (A-6), полученное тем же способом, как в способах (примеров 2a-2f), и посредством этого получали органический слой (OP-6B). Органический слой (OP-6A) и органический слой (OP-6B) комбинировали и затем концентрировали, и посредством этого получали заявленное в заголовке соединение (331 г) в виде красновато-коричневого масла.
[0240] [Данные физических свойств соединения, представленного формулой (A-7)]
(1H ЯМР, 400 МГц, изготовитель: Bruker, CDCl3, δ ppm)
7,51 (д, 1H, J=7 Гц), 7,05 (т, 1H, J=7 Гц), 6,80 (д, 1H, J=7 Гц), 6,19 (уш с, 1H), 4,10-4,05 (1H, м), 2,92-2,81 (2H, м), 2,80-2,69 (1H, м), 2,43 (дд, 1H, J=7 Гц, 16 Гц), 2,03-1,88 (1H, м), 1,78-1,63 (1H, м), 1,45 (9H, с)
[0241] Абсолютную конфигурацию соединения, представленного формулой (A-7) определяли превращением соединения, представленного формулой (A-7) в соединение, представленное формулой (B), затем, сравнением его аналитических данных с данными соединения, представленного формулой (B), полученного отдельно способом, описанным в WO 2003/095420 и подобных, и подтверждением того, совпадают ли аналитические данные. Кроме того, имеют ли гидроксильные группы в соединении, представленном формулой (B), (R) конфигурацию, определяли превращением соединения, представленного формулой (B), в его гидробромид, и анализом гидробромида рентгеновской кристаллической структурой (смотри (пример 5) и фигуру 2).
[0242] (пример 4a) получение гидрохлорида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула B-HCl)
[0243]
[C99]
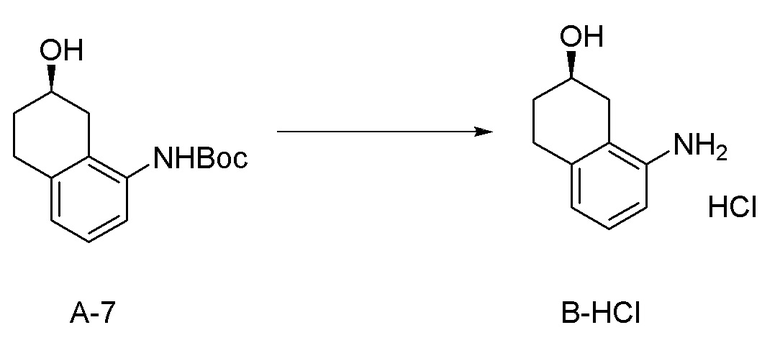
[0244] н-PrOH (2 г) добавляли в реакторе и перемешивали при -5°C-5°C. Ацетилхлорид (0,76 г) добавляли по каплям при той же температуре в течение 10 минут. После повышения температуры реакции до 50°C-55°C, раствор соединения (0,5 г) формулы (A-7), полученного тем же способом, как в способах (примера 3a-g), в н-PrOH (6 г) добавляли к смеси в течение 45 минут. После перемешивания при температуре реакции 50°C-55°C в течение 45 минут, смесь охлаждали так, что температура реакции достигала 20°C-30°C, и смесь перемешивали при 20°C-30°C в течение 16 часов. Полученный твердый остаток фильтровали, промывали н-PrOH (5 мл×2 кратный) и сушили при 40°C-50°C в течение 6,5 часов, получая заявленное в заголовке соединение (0,23 г).
[0245] (пример 4b) получение (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула B)
[0246]
[C100]
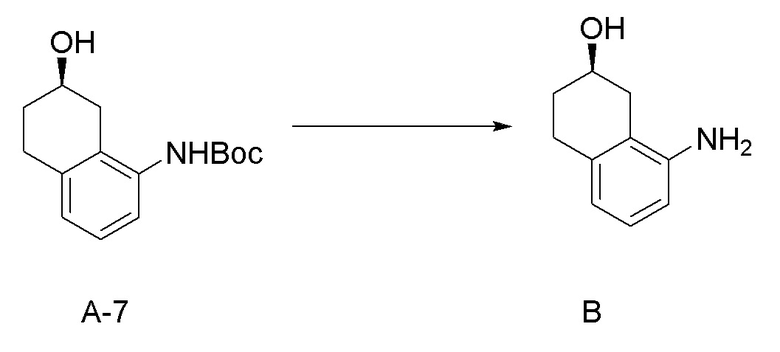
[0247] н-PrOH (1,81 г) добавляли в реакторе и перемешивали при -5°C-5°C. Ацетилхлорид (2,35 г) добавляли по каплям при той же температуре в течение 10 минут. После повышения температура реакции до 33°C-37°C, раствор соединения (4,25 г) формулы (A-7), полученного тем же способом, как в способах (примера 3a-g), в n-PrOH (12,75 г) добавляли к смеси в течение 45 минут. После перемешивания при температуре реакции 33°C-37°C в течение 49,5 часов, смесь перемешивали при температуре реакции 20°C-25°C в течение19 часов. Сгенерированный твердый остаток фильтровали, промывали i-PrOAc (10 мл×2 кратный) и сушили при 30°C-40°C в течение 4 часов, получая гидрохлорид (2,04 г). После смешения отдельно полученного гидрохлорида (0,07 г), получая 2,11 г, объединенный продукт суспендировали в этилацетата (12 мл), и pH водного слоя доводили до 7-8, применяя водный раствор гидрокарбоната натрия. После разделения водного слоя и органического слоя, водный слой экстрагировали этилацетатом (12 мл×2 кратный), и органические слои комбинировали, промывали водой (10 мл×2 кратный), затем концентрировали при пониженном давлении, получая заявленное в заголовке соединение (1,34 г).
[0248] (пример 4c) получение (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула B)
[0249] н-PrOH (138 г) добавляли в реакторе и перемешивали при -5°C-5°C. Ацетилхлорид (180,1 г) добавляли по каплям при той же температуре в течение 1 часа. После повышения температура реакции до 33°C-37°C, раствор неочищенного соединения (331 г) формулы (A-7), полученного тем же способом, как в способах (примера 3a-g), в н-PrOH (750 мл) добавляли к смеси в течение 45 минут. После перемешивания при температуре реакции 33°C-37°C в течение 15 часов, смесь перемешивали при температуре реакции 50°C-55°C в течение 2 часов и 10 минут. После того как температуру реакции устанавливали на 33°C-37°C, смешанный раствор, полученный добавлением HCl (газ) (26 г) к изо-PrOAc (192 г), добавляли к смеси при 33°C-37°C в течение 30 минут. Температуру реакции повышали до 50°C-55°C, и смесь перемешивали в течение 1,5 часов. Полученный твердый остаток фильтровали, промывали изо-PrOAc и сушили при пониженном давлении при 20°C-30°C в течение36 часов, получая гидрохлорид (158 г). Гидрохлорид (158 г) суспендировали в этилацетате (1,000 мл), и водный раствор гидрокарбоната натрия (76 г гидрокарбоната натрия, 1000 мл воды) добавляли к нему в течение 30 минут. После разделения водного слоя и органического слоя, водный слой экстрагировали этилацетатом (1,000 мл×2 кратный), и органические слои комбинировали, промывали водой (1000 мл) и концентрировали при пониженном давлении, получая заявленное в заголовке соединение (136 г).
[0250] [Данные физических свойств соединения, представленного формулой (B)]
(1H-ЯМР, 400 МГц, изготовитель: Bruker, CDCl3, δ ppm)
6,91 (1H, т, J=7 Гц), 6,52-6,46 (2H, м), 4,19-4,04 (2H, м), 3,51 (1H, уш с), 2,93-2,65 (3H, м), 2,31 (1H, дд, J=7 Гц, 16 Гц), 2,02-1,89 (1H, м), 1,85-1,65 (1H, m)
[0251] (пример 5) получение гидробромида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула (B-HBr)):
[0252]
[C101]
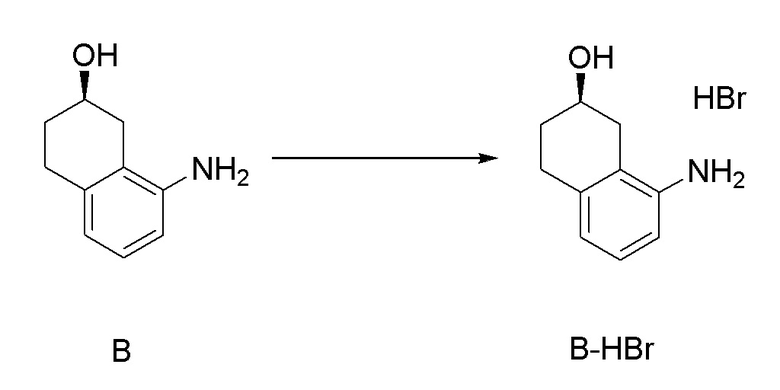
[0253] Неочищенное соединение (99,9 г) (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ол, полученный тем же способом, как в (примере 4b) или (примере 4c), растворяли в этилацетате (1 л), и водный раствор 48% бромистоводородной кислоты (80 мл) добавляли к смеси при охлаждении ледяной водой. Выпавший твердый остаток собирали фильтрованием и промывали последовательно изопропанолом (300-400 мл) и этилацетатом (500 мл). Полученный очищенный гидробромид (123,9 г, 98,3% ee) растворяли в горячей воде (250 мл) и добавляли к нему активированный уголь (20 г). Активированный уголь отфильтровали, применяя целит, когда он был горячим, и промывали водой. Фильтрат концентрировали при пониженном давлении, и полученный остаток перекристаллизовывали из воды. Полученные кристаллы собирали фильтрованием, и промывали изопропанолом и этилацетатом, получая заявленное в заголовке соединение (34,2 г, 98,4% ee). Фильтрат собирали, концентрировали при пониженном давлении, и перекристаллизовывали дважды из воды, получая заявленное в заголовке соединение (29,1 г, 98,9% ee).
[0254] Оптическую чистоту гидробромида соединения, представленного формулой (B), измеряли, применяя ВЭЖХ LC-10 систему Shimadzu Corporation в следующих условиях.
[0255]
[Таблица 13]
[0256] Кроме того, анализировали рентгеноструктурную кристаллическую структуру одного кристалла полученного гидробромида соединения, представленного формулой (B), ((R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола), применяя AFC-7 Rigaku, и получали следующие результаты (смотри фигуру 2).
[0257]
[Таблица 14]
b=8,681 (3) Å, β=103,47°,
c=8,017 (2) Å, γ=90,00 (2)°
[0258] (пример 6) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида (формула (I)):
[0259]
[C102]
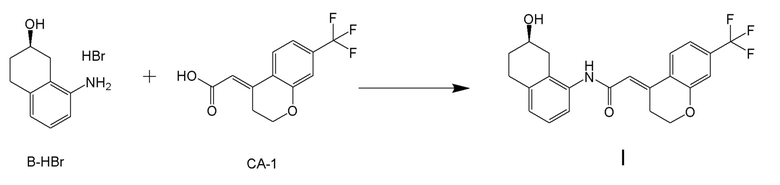
[0260] Триэтиламин (0,27 мл, 1,0 экв.) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (0,80 г, 1,5 экв.) добавляли к суспензии гидробромида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула (B-HBr)) (0,47 г), полученного в (примере 5) и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (формула (CA-1)) (0,50 г, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383, в метаноле (5,00 мл: приблизительно 10 кратный его объем относительно 1 г соединения, представленного формулой (B-HBr)), и смесь перемешивали при комнатной температуре в течение 3 часов. После охлаждения на льду реакционного раствора, выпавшие кристаллы собирали фильтрованием и промывали холодным метанолом. Полученный твердый остаток растворяли в этаноле (6 мл) нагреванием, и затем воду (6 мл) добавляли при нагревании. После охлаждения смеси, выпавший твердый остаток фильтровали, промывали последовательно 50% вода-этанол и водой, и затем сушили при пониженном давлении, получая заявленное в заголовке соединение (0,56 г) в виде твердого остатка.
[0261] [Данные физических свойств соединения, представленного формулой (I)]
(1H-ЯМР данные (CDCl3) (δ: ppm)):
7,80-7,58 (м, 1H), 7,24-6,92 (м, 5H), 6,45 (с, 1H), 4,29 (т, 2H, J=6 Гц), 4,28-4,15 (м, 1H), 3,51 (т, 2H, J=5 Гц), 3,10-2,78 (м, 3H), 2,69-2,53 (м, 1H), 2,14-2,00 (м, 1H), 1,90-1,67 (м, 2H)
(LC-MS):
RT=4,73 (минут), [M+H]+=404
Оптическая чистота: 97,9% ee
[0262] Оптическую чистоту соединения, представленного формулой (I), определяли, применяя ВЭЖХ LC-VP систему Shimadzu Corporation в следующих условиях.
[Таблица 15]
Энантиомер соединения, представленного формулой (I) - 18,6 минут
[0263] Кристаллическую структуру соединения, представленного формулой (I), анализировали при SPring-8 beamline BL32B2, применяя R-AXIS V детектор Rigaku (смотри фигуру 3).
[0264]
[Таблица 16]
b=7,272 Å, β=113,80°,
c=19,088 Å, γ=90°
[0265] (пример 7) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида: (Пример (1) условий для реакции конденсации)
[0266]
[C103]
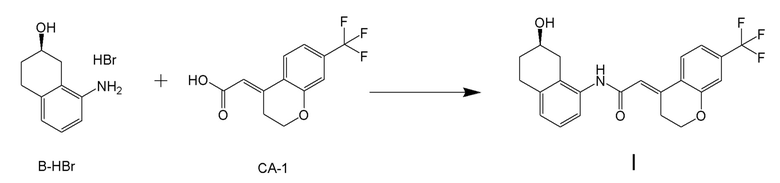
[0267] <Пример 1> Триэтиламин (120 мкл, 1,05 экв.) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (339 мг, 1,5 экв.) добавляли к суспензии гидробромида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (200 мг), полученного в (примере 5), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (212 мг, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383, в метаноле (3,15 мл: 15,75 кратный его объем относительно 1 г соединения, представленного формулой (B-HBr)), и смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли к ней воду, и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (291 мг) в виде твердого остатка.
[0268] <Пример 2> Триэтиламин (240 мкл, 2,1 экв.) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (339 мг, 1,5 экв.) добавляли к суспензии гидробромида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (200 мг), полученного в (примере 5), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (212 мг, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383 в метаноле (3,15 мл: 15,75 кратный его объем относительно 1 г соединения, представленного формулой (B-HBr)), и смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли к ней воду, и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (273 мг) в виде твердого остатка.
[0269] <Пример 3> Триэтиламин (120 мкл, 1,05 экв.) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (339 мг, 1,5 экв.) добавляли к суспензии гидробромид (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (200 мг), полученного в (примере 5), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (212 мг, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383 в метаноле (1,60 мл:8 кратный его объем относительно 1 г соединения, представленного формулой (B-HBr)), и смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли к ней воду, и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (298 мг) в виде твердого остатка.
[0270] <Пример 4> Триэтиламин (120 мкл, 1,05 экв.) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (339 мг, 1,5 экв.) добавляли к суспензии гидробромида (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (200 мг), полученного в (примере 5), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (212 мг, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383, в метаноле (3,15 мл: 15,75 кратный его объем относительно 1 г соединения, представленного формулой (B-HBr)), и смесь перемешивали при кипячении в течение 3 часов. После охлаждения смесь, добавляли к ней воду, и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (291 мг) в виде твердого остатка.
[0271] В <примере 1>-<примере 4> (примера 7), описанного выше, подавляли генерирование эндо продукта соединения, представленного формулой (I) ((R)-N-(7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)-2-(7-(трифторметил)-2H-хромен-4-ил)ацетамида).
[0272] Химическую чистоту соединения, представленного формулой (I), измеряли, применяя ВЭЖХ LC-VP систему Shimadzu Corporation в следующих условиях.
[Таблица 17]
Соединение, представленное формулой (I) - 9,1 минут
[0273] (пример 8) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида: (Пример (2) условий для реакции конденсации)
[0274]
[C104]
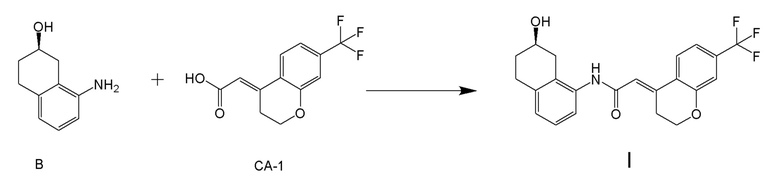
[0275] <Пример 1> хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (257 мг, 1,5 экв.) добавляли к суспензии (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (101 мг), полученного тем же способом, как в (примере 4b) или (пример 4c), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоте (200 мг, 1,25 экв.), полученной способом получения, описанным в WO 2007/010383, в изопропаноле (3,0 мл: приблизительно 30 кратный его объем относительно 1 г соединения, представленного формулой (B)), и смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли к ней (3 мл), и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (209 мг) в виде твердого остатка.
[0276] <Пример 2> хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (257 мг, 1,5 экв.) добавляли к суспензии (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (126 мг), полученного тем же способом, как в (примере 4b) или (пример 4c), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (200 мг, 1,0 экв.), полученной способом получения, описанным в WO 2007/010383, в изопропаноле (3,00 мл: приблизительно 24 кратный его объем относительно 1 г формулы (B)), и смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли к ней воду (3 мл), и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (238 мг) в виде твердого остатка.
[0277] <Пример 3> Хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (257 мг, 1,5 экв.) добавляли к суспензии (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (152 мг), полученного тем же способом, как в (примере 4b) или (примере 4c), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (200 мг, 0,83 экв.), полученной способом получения, описанным в WO 2007/010383, в изопропаноле (3,00 мл: приблизительно 20 кратный его объем относительно 1 г соединения, представленной формулой (B)), и смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли к ней воду (3 мл), и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (243 мг) в виде твердого остатка.
[0278] <Пример 4> хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (257 мг, 1,5 экв.) добавляли к суспензии (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (177 мг), полученного тем же способом, как в (примере 4b) или (пример 4c), и (E)-2-(7-(трифторметил)хроман-4-илиден)уксусной кислоты (200 мг, 0,71 экв.), полученной способом получения, описанным в WO 2007/010383, в изопропаноле (3,00 мл: приблизительно 20 кратный его объем относительно 1 г соединения, представленного формулой (B)), и смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли к ней воду (3 мл), и выпавший твердый остаток фильтровали, промывали водой и сушили, получая заявленное в заголовке соединение (238 мг) в виде твердого остатка.
[0279] (пример 9a) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида:
После проведения реакции конденсации (условия реакции: 20°C-25°C, перемешивание в течение 22 часов), применяя (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ол (1,04 г, чистота 99,35%), полученный тем же способом, как в (примере 4b) или (пример 4c), (E)-2-(7-(трифторметил)хроман-4-илиден)уксусную кислоту (1,05 экв.), полученную способом получения, описанным в WO 2007/010383, изопропанол (25 кратный его объем относительно соединения, представленного формулой (B)), и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (1,3 экв.), обработку проводили согласно (примеру 8), и посредством этого получали заявленное в заголовке соединение (1,87 г) в виде твердого остатка.
[0280] (пример 9b) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида (масштабирование):
После проведения реакции конденсации (условия реакции: 20°C-25°C, перемешивание в течение 22 часов), применяя (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ол (110 г), полученный тем же способом, как в (примере 4b) или (пример 4c), (E)-2-(7-(трифторметил)хроман-4-илиден)уксусную кислоту (173,9 г, 1,05 экв.), полученную способом получения, описанным в WO 2007/010383, изопропанол (2,600 мл: 25 кратный его объем относительно соединения, представленного формулой (B)), и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (243,7 г, 1,3 экв.), обработку проводили согласно (примеру 8), и посредством этого получали заявленное в заголовке соединение (183,3 г) в виде серовато-белого твердого остатка.
[0281] Оптическую чистоту соединения, представленного формулой (I), измеряли, применяя ВЭЖХ LC-VP систему Shimadzu Corporation в следующих условиях.
[Таблица 18]
Энантиомер соединения, представленного формулой (I) - 18,6 минут
[0282] Bruker AV 400 применяли для получения спектров ядерного магнитного резонанса (ЯМР) соединений, представленных формулой (A8-BR) и формулой (B).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) соединений, представленных формулой (A8-BR) и формулой (B), проводили следующим способом.
[Таблица 19]
Условия измерения для соединения, представленного формулой (A8-BR)
[Таблица 20]
Время удерживания (RT)
[Таблица 21]
Условия измерения для соединения, представленного формулой (B)
[Таблица 22]
Время удерживания (RT)
[Таблица 23]
Способ хирального анализа для соединения, представленного формулой (A8-BR)
[Таблица 24]
Время удерживания (RT)
[Таблица 25]
Способ хирального анализа для соединения, представленного формулой (B)
[Таблица 26]
Время удерживания (RT)
[0283] (пример 10A)-(пример 10D) получение (R)-8-бром-1,2,3,4-тетрагидронафталин-2-ола (A8-BR)
[0284]
[C105]
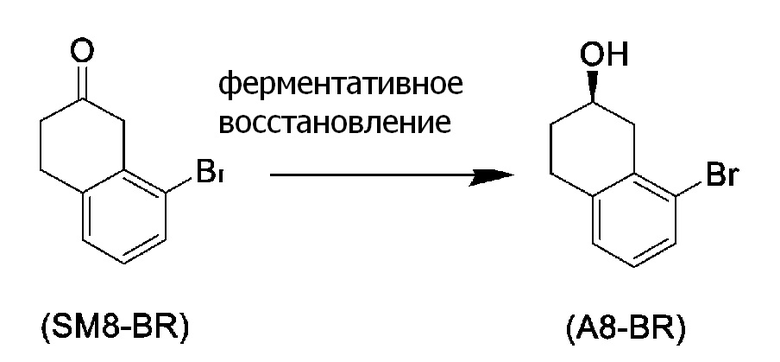
[0285] (пример 10A)
KRED (кетонредуктазу, полученную из Escherichia coli sp., 5 мг), D-глюкозу (200 мг), глюкозадегидрогеназу (GDH) (2 мг), никотинамидадениндинуклеотидфосфат (NADP) (1 мг) и фосфатный буферный раствор (3 мл, полученный добавлением 10,62 г KH2PO4 и 21,25 г K2HPO4 к 1000 мл воды) смешивали в колбе, оснащенной орбитальным встряхивателем (изготовленным Shanghai Nanrong Laboratory Equipment Co., Ltd., номер модели: NRY-200) и перемешивали, получая смешанный раствор. Затем, смешанный раствор, полученный растворением 8-бром-3,4-дигидронафталин-2(1H)-она (формула (SM8-BR)) (100 мг) в диметилсульфоксиде (DMSO) (0,3 мл), добавляли к предварительно полученному смешанному раствору, и смесь перемешивали при температуре реакции 30°C в течение20 часов (где a скорость вращения орбитального встряхивателя составляла 250 об/мин). Некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализу, и подтверждали, что заявленное в заголовке соединение получали с IPC выходом (IPC=контроль при получении) 97,8% и оптической чистотой 99,7%.
[0286] (пример 10B)
KRED (кетонредуктазу, полученную из Escherichia coli sp., 0,25 г), D-глюкозу (10 г), глюкозадегидрогеназу (GDH) (0,1 г), никотинамидадениндинуклеотидфосфат (NADP) (0,05 г) и буферный раствор (1,55 г KH2PO4 и 4,06 г K2HPO4·3H2O добавляли к 145 мл воды) смешивали в реакторе, получая смешанный раствор, и его перемешивали при 20°C-25°C. Затем, смешанный раствор, полученный растворением 8-бром-3,4-дигидронафталин-2(1H)-она (формула (SM8-BR)) (5 г) в диметилсульфоксиде (DMSO) (15 мл), добавляли по каплям к предварительно полученному смешанному раствору. После перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, после перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Кроме того, после перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, реакционный раствор перемешивали при температуре реакции 20°C-25°C в течение16 часов (некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализ, и подтверждали, что IPC выход заявленного в заголовке соединения составлял 99,6%).
Метил трет-бутиловый эфир (MTBE) (50 мл) добавляли к реакционному раствору, и дополнительно добавляли к нему диатомит (диатомовую землю) (5 г), содержащий воду (5 г), затем смешанный раствор перемешивали при температуре 50°C-60°C в течение30 минут. Температуру смешанного раствора понижали до 20°C-25°C, и смешанный раствор дополнительно перемешивали при той же температуре в течение 1 часа. Смешанный раствор, описанный выше, фильтровали, и отфильтрованный материал (влажный остаток на фильтре) промывали MTBE (5 мл), получая фильтрат A. Влажный остаток на фильтре, описанный выше, помещали в реактор, и добавляли в него MTBE (40 мл), затем смесь перемешивали при 20°C-25°C в течение 2 часов. Суспензию, содержащую влажный остаток на фильтре, фильтровали, и влажный остаток на фильтре промывали MTBE (5 мл), получая фильтрат B. После смешения фильтрата A и фильтрата B и перемешивали смесь при 20°C-30°C в течение 5 минут, водный слой и органический слой разделяли, и водный слой экстрагировали MTBE (45 мл), затем органический слой комбинировали с ранее полученным органическим слоем, промывали водой (30 мл) и концентрировали, получая неочищенное заявленное в заголовке соединение (4,61 г). Полученное неочищенное заявленное в заголовке соединение подвергали колоночной хроматографии на силикагеле (н-гептан:этилацетат=1:1), получая заявленное в заголовке соединение (4,15 г, оптическая чистота 99,9%).
[0287] (пример 10C)
KRED (кетонредуктазу, полученную из Escherichia coli sp., 0,83 г), D-глюкозу (33,28 г), глюкозадегидрогеназу (GDH) (0,33 г), никотинамидадениндинуклеотидфосфат (NADP) (0,17 г) и буферный раствор (5,31 г KH2PO4 и 13,89 г K2HPO4·3H2O добавляли к 499 мл воды) смешивали в реакторе, получая смешанный раствор, и его перемешивали при 20°C-25°C. Затем, смешанный раствор, полученный растворением 8-бром-3,4-дигидронафталин-2(1H)-она (формула (SM8-BR)) (17,3 г) в диметилсульфоксиде (DMSO) (50 мл), добавляли по каплям к предварительно полученному смешанному раствору. После перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, после перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Кроме того, после перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, смесь перемешивали при температуре реакции 20°C-25°C в течение16 часов. Некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализу, и подтверждали, что IPC выход заявленного в заголовке соединения составлял 97,4%. Проводили такую же обработку, как в (примере 1B), получая заявленное в заголовке соединение (17,12 г, оптическая чистота 99,9%).
[0288] (пример 10D)
KRED (кетонредуктазу, полученную из Escherichia coli sp., 5,55 г), D-глюкозу (220 г), глюкозадегидрогеназу (GDH) (2,20 г), никотинамидадениндинуклеотидфосфат (NADP) (1,12 г) и буферный раствор (35,12 г KH2PO4 и 91,80 г K2HPO4·3H2O добавляли к 3300 мл воды) смешивали в реакторе, получая смешанный раствор, и его перемешивали при 20°C-25°C. Затем, смешанный раствор, полученный растворением 8-бром-3,4-дигидронафталин-2(1H)-она (формула (SM8-BR)) (110,31 г) в диметилсульфоксиде (DMSO) (330 мл), добавляли по каплям к предварительно полученному смешанному раствору. После перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, после перемешивания при температуре реакции 20°C-25°C в течение 1 часа, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Кроме того, после перемешивания при температуре реакции 20°C-25°C в течение 2 часов, pH реакционного раствора доводили до диапазона pH=6,5-7,0, применяя водный раствор 2M карбоната натрия. Затем, смесь перемешивали при температуре реакции 20°C-25°C в течение 16 часов. Некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализ, и подтверждали, что IPC выход заявленного в заголовке соединения составлял 97,4%.
MTBE (1,100 мл) добавляли к реакционному раствору, и дополнительно добавляли к нему диатомит (диатомовую землю) (110 г), содержащий воду (110 г), и смешанный раствор перемешивали при температуре 50°C-60°C в течение30 минут. Температуру смешанного раствора понижали до 20°C-25°C, и смешанный раствор дополнительно перемешивали при той же температуре в течение 2 часов. Смешанный раствор, описанный выше, фильтровали, и отфильтрованный материал (влажный остаток на фильтре) промывали MTBE (110 мл), получая фильтрат C. Влажный остаток на фильтре, описанный выше, помещали в реактор, и добавляли к нему MTBE (900 мл), затем смесь перемешивали при 20°C-25°C в течение 12 часов. Суспензию, содержащую влажный остаток на фильтре, фильтровали, и влажный остаток на фильтре промывали MTBE (110 мл), получая фильтрат D. Фильтрат C и фильтрат D смешивали, и водный слой и органический слой разделяли, и водный слой экстрагировали MTBE (1000 мл), затем органический слой комбинировали с ранее полученным органическим слоем, промывали водой (675 мл) и концентрировали, получая неочищенное заявленное в заголовке соединение (108,03 г, оптическая чистота 99,8%).
[0289] [Данные физических свойств формулы (A8)]
(1H ЯМР, 400 МГц, изготовитель: Bruker, DMSO-d6, δ ppm)
7,40 (д, 1H, J=8 Гц), 7,10 (д, 1H, J=8 Гц), 7,04 (т, 1H, J=8 Гц), 4,89 (д, 1H, J=4 Гц), 3,99-3,95 (м, 1H), 2,92-2,86 (м, 2H), 2,70-2,60 (м, 1H), 1,83-1,75 (м, 1H), 1,65-1,55 (м, 1H))
[0290] KRED (кетонредуктаза, полученная из Escherichia coli sp.), применяемая в (примере 10A)-(примере 10D), представляет собой фермент, изготовленный EnzymeWorks, Inc. (номер продукта: HQ-K-105).
[0291] Абсолютную конфигурацию соединения, представленного формулой (A8-BR), полученного в (примере 10A)-(примере 10D) определяли превращение формулы (A8-BR) в формулу (B), и затем подтверждали, соответствуют ли его аналитические данные аналитическим данным соединения, представленного формулой (B), полученного отдельно способом, описанным в WO 2003/095420, и подобных.
[0292] (Справочный пример 1) получение 8-бром-1,2,3,4-тетрагидронафталин-2-ола (формула (A8-BR-Rac))
[C106]
8-Бром-3,4-дигидронафталин-2(1H)-он (формула (SM8-BR)) (20,0 г) и метанол (200 мл) добавляли в реактор, и NaBH4 (8,28 г) добавляли при температуре реактора 0°C-5°C, и смесь перемешивали при той же температуре в течение 1 часа (некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализу, и подтверждали, что IPC выход составлял 98,5%). Водный раствор 10% гидрокарбоната натрия (1,5 л) добавляли по каплям, когда температура реакционного раствора составляла 5°C или ниже, и смешанный раствор перемешивали в течение 0,2 часов при температуре смешанного раствора 0°C-5°C. Добавляли этилацетат (1,5 л), и водный слой и органический слой разделяли, затем водный слой экстрагировали этилацетатом (1,5 л), и органический слой комбинировали с ранее полученным органическим слоем, промывали водным раствором 25 вес % хлорида натрия (1,5 л) и концентрировали, получая неочищенное заявленное в заголовке соединение (21,53 г). Полученное неочищенное заявленное в заголовке соединение подвергали колоночной хроматографии на силикагеле (н-гептан:этилацетат=1:1), получая заявленное в заголовке соединение (21,29 г). Подтверждали, что полученное соединение, представленное формулой (A8-BR-Rac), соответствовало данным о физических свойствах, известным из литературы.
[0293] (пример 11A)-(пример 11G) получение (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола (формула (B))
[0294]
[C107]
(пример 11A)
(R)-8-бром-1,2,3,4-тетрагидронафталин-2-ол (формула (A8-BR)) (100 мг), полученный ферментативной реакцией тем же способом, как в способах примера 10A-примера 10D, Cu2O (40 мг), N-метилпирролидон (NMP) (2 мл) и водный аммиак (3 мл) смешивали в герметично закрытом реакторе, и реакцию в герметично закрытом реакторе проводили при температуре 105°C-115°C в течение20 часов. После разбавления смеси водой, смесь экстрагировали этилацетатом, и органический слой промывали водным раствором 25 вес % хлорида натрия, сушили Na2SO4, фильтровали и концентрировали, получая неочищенное заявленное в заголовке соединение (106 мг). Тонкослойную хроматографию (н-гептан:этилацетат=1:1) проводили для разделения, и посредством этого получали заявленное в заголовке соединение (10 мг) (оптическая чистота 96,8%).
[0295] (пример 11B)
(R)-8-бром-1,2,3,4-тетрагидронафталин-2-ол (формула (A8-BR)) (2,2 г), полученный ферментативной реакцией тем же способом, как в способах примера 10A-примера 10D, Cu2O (700 мг), NMP (3,5 мл, 1,6 об) и водный аммиак (5,5 мл) смешивали в герметично закрытом реакторе, и реакцию в герметично закрытом реакторе проводили при температуре 105°C-115°C в течение37 часов (подтверждали, что IPC выход составлял 83,75% после 16 часов, 88,91% после 21 часа и 93,12% после 37 часов). После разбавления смеси водой (17 мл) и этилацетатом (11 мл), смесь фильтровали, и отфильтрованный материал промывали этилацетатом (4 мл, 3 кратный), затем водный слой и органический слой разделяли. Затем, водный слой экстрагировали этилацетатом (11 мл, 5 кратный), и органический слой комбинировали с ранее полученным органическим слоем, промывали водой (20 мл, 2 кратный), водным раствором 10% Na2SO4 и концентрировали, получая неочищенное заявленное в заголовке соединение (1,25 г, 61,27%, оптическая чистота 95,6%).
[0296] (пример 11C)
Реакцию в герметично закрытом реакторе проводили в условиях, показанных в таблице ниже, и проверяли растворитель реакции.
[Таблица 27]
@6,91 мин
(1,0 экв.)
(0,50 экв.)
(2,5 об)
(0,5 об)
(1,0 экв.)
(0,51 экв.)
(2,5 об)
(1 об)
(1,0 экв.)
(0,50 экв.)
(2,5 об)
(1,5 об)
[0297] В примере 11C-2 получали 0,74 г (выход 50%) (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола, и в примере 11C-3 получали 0,5 г (36,6%) (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ола.
[0298] (пример 11D)
Реакцию в герметично закрытом реакторе проводили в условиях, показанных в таблице ниже, и проверяли количество водного аммиака.
[Таблица 28]
@6,83 мин
(0,51 экв.)
(2,5 об)
(1 об)
(0,51 экв.)
(3,5 об)
(1 об)
В таблице (B)-Rac обозначает рацемическое соединение, представленное формулой (B).
[0299] (пример 11E)
(R)-8-бром-1,2,3,4-тетрагидронафталин-2-ол (формула (A8-BR)) (3,00 г), полученный ферментативной реакцией тем же способом, как в способах примера 10A-примера 10D, Cu2O (0,96 г, 0,51 экв.), NMP (3 мл, 1 об) и водный аммиак (10,5 мл, 3,5 об) смешивали в герметично закрытом реакторе, и реакцию в герметично закрытом реакторе проводили при температуре 105°C-115°C в течение 21 часов (некоторую часть реакционного раствора отбирали и подвергали ВЭЖХ анализу, и подтверждали, что IPC выход составлял 92,93%). После охлаждения реакционного раствора, водный раствор 25 вес% хлорида натрия (23 мл) и 2-метилтетрагидрофуран (2-MeTHF) (15 мл) добавляли к реакционному раствору, и смешанный раствор фильтровали, затем отфильтрованный материал промывали 2-MeTHF (15 мл). Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (15 мл, 4 кратный), затем органический слой комбинировали с ранее полученным органическим слоем, промывали водным раствором 10 вес% Na2SO4 (15 мл), затем обесцвечивали CUNO (торговое название) (фильтер) в течение 1 часа. CUNO промывали 2-MeTHF (15 мл), и растворитель концентрировали, затем добавляли изопропилацетат (6 мл), и добавляли по каплям н-гептан (1,5 мл) при температуре 30°C-40°C, и смесь перемешивали при той же температуре в течение 0,5 часов. Кроме того, добавляли по каплям н-гептан (10,5 мл), и смесь перемешивали при температуре 30°C-40°C в течение 0,5 часа. Кроме того, добавляли по каплям н-гептан (3,0 мл), и смесь перемешивали при температуре 30°C-40°C в течение0,5 часа. Кроме того, добавляли по каплям н-гептан (3,0 мл), и смесь перемешивали при температуре 30°C-40°C в течение 0,5 часа. Смешанный раствор, описанный выше, охлаждали при 20°C в течение 30 минут и перемешивали при температуре 15°C-25°C в течение 1 часа. Смешанный раствор фильтровали, и отфильтрованный материал промывали н-гептаном (3 мл) и сушили, получая заявленное в заголовке соединение (1,375 г, 60,1%).
[0300] (пример 11F)
(R)-8-бром-1,2,3,4-тетрагидронафталин-2-ол (формула (A8-BR)) (16,32 г, NMP раствор, содержание 61,1%), полученный ферментативной реакцией тем же способом, как в способах примера 10A-примера 10D, Cu2O (3,18 г, 0,51 экв.), NMP (5 мл, 0,5 об) и водный аммиак (35 мл, 3,5 об) смешивали в герметично закрытом реакторе, и реакцию в герметично закрытом реакторе проводили при температуре 105°C-115°C в течение40 часов (подтверждали, что IPC выход за 40 часов составлял 90,16%). После охлаждения реакционного раствора, добавляли к реакционному раствору водный раствор 25 вес% хлорида натрия (75 мл) и 2-MeTHF (50 мл), и смешанный раствор фильтровали, применяя диатомит (диатомовую землю) (20,00 г), затем отфильтрованный материал (остаток на фильтре) промывали 2-MeTHF (50 мл). Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (50 мл, 2 кратный), затем органический слой комбинировали с ранее полученным органическим слоем, промывали водным раствором 8 вес% Na2SO4 (50 мл, 2 кратный), и отбирали органический слой (92,5% суммарного количества), затем водный раствор 0,5 M хлористоводородной кислоты (111 мл) добавляли по каплям при 5°C-15°C (в данном случае, pH=1,2). Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (30 мл). Водный раствор 10% гидроксида натрия (22 мл) добавляли к водному слою, и водный слой экстрагировали 2-MeTHF (100 мл, 50 мл, 50 мл). После смешения с ранее полученным органическим слоем, органический слой концентрировали при 40°C или ниже, и добавляли по каплям н-гептан (80 мл) при 35°C-45°C, затем смесь охлаждали до 5°C. Затем, смесь перемешивали при 0°C-10°C в течение 24 часов и собирали фильтрованием, и отфильтрованный материал промывали н-гептаном (10 мл) и сушили, получая заявленное в заголовке соединение (4,50 г, 67,8%, оптическая чистота 99,9%).
[0301] (пример 11G)
Реакцию в герметично закрытом реакторе проводили в условиях, показанных в таблице ниже.
[Таблица 29]
@7,34 мин
(NMP раствор, содержание 63,8%)
(0,50 экв.)
(3,5 об)
(0,5 об)
(NMP раствор, содержание 63,8%)
(0,50 экв.)
(3,5 об)
(0,5 об)
[0302] (Обработка 11G-1)
После завершения реакции примера 11G-1 и охлаждения реакционного раствора, добавляли к реакционному раствору водный раствор 25 вес% хлорида натрия (345 мл) и 2-MeTHF (250 мл), и смешанный раствор фильтровали, применяя диатомит, затем отфильтрованный материал (остаток на фильтре) промывали 2-MeTHF (230 мл). Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (230 мл, 2 кратный), затем органический слой комбинировали с ранее полученным органическим слоем, получая органическую фазу (11G-1).
[0303] (Обработка 11G-2)
После завершения реакции примера 11G-2 и охлаждения реакционного раствора, добавляли к реакционному раствору водный раствор 25 вес % хлорида натрия (375 мл) и 2-MeTHF (250 мл), и смешанный раствор фильтровали, применяя диатомит, затем отфильтрованный материал (остаток на фильтре) промывали 2-MeTHF (250 мл). Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (250 мл, 2 кратный), затем органический слой комбинировали с ранее полученным органическим слоем, получая органическую фазу (11G-2).
[0304] (Work up 11G-3)
После смешения ранее полученной органической фазы (11G-1) и органической фазы (11G-2), смесь промывали водным раствором 8 вес% Na2SO4 (480 мл, 2 кратный), затем добавляли по каплям водный раствор 0,5 M хлористоводородной кислоты (1,156 мл), и pH доводили до 0,88. Водный слой и органический слой разделяли, и водный слой экстрагировали 2-MeTHF (290 мл). Добавляли к водному слою водный раствор 10% гидроксида натрия (230 мл), и водный слой экстрагировали 2-MeTHF (1000 мо, 500 мо, 500 мо, 3 кратный). После объединения с ранее полученным органическим слоем, органический слой концентрировали при 40°C или ниже, и добавляли по каплям н-гептан (576 мл) при 35°C-45°C, и затем смесь охлаждали до 0°C-10°C, перемешивали при той же температуре, собирали фильтрованием, и отфильтрованный материал промывали н-гептаном (96 мл) и сушили, получая заявленное в заголовке соединение (53,55 г, 70,8%, оптическая чистота 99,9%).
[0305] [Данные физических свойств формулы (B)]
(1H-ЯМР, 400 МГц, изготовитель: Bruker, CDCl3, δ ppm)
6,91 (1H, т, J=7 Гц), 6,52-6,46 (2H, м), 4,19-4,04 (2H, м), 3,51 (1H, ушс), 2,93-2,65 (3H, м), 2,31 (1H, дд, J=7,16 Гц), 2,02-1,89 (1H, м), 1,85-1,65 (1H, м)
[0306] Водный аммиак, применяемый в (примере 11A)-(примере 11G) представляет собой 25%-28% водный аммиак.
[0307] (пример 12A)-(пример 12B) получение (E)-2-(7-трифторметилхроман-4-илиден)-N-[(7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил]ацетамида
[C108]
[0308] (пример 12A)
Реакцию конденсации (условия реакции: 20°C-25°C, перемешивание в течение 17 часов) проводили, применяя (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ол (3 г), полученный тем же способом, как в (примере 11G), (E)-2-(7-(трифторметил)хроман-4-илиден)уксусную кислоту (4,83 г, 1,0 экв.), полученную способом получения, описанным в WO 2007/010383, изопропанол (60,01 г: 20 кратный его вес относительно соединения, представленного формулой (B)), и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (6,67 г, 1,3 экв.). После добавления воды (75 мл) к реакционному раствору, смешанный раствор охлаждали до 10°C-15°C, перемешивали при той же температуре в течение 1 часа, фильтровали, и отфильтрованный материал промывали водой и сушили при 36°C в течение 16 часов. 2-MeTHF (90 г) добавляли к полученному отфильтрованному материалу, и смесь перемешивали при 70°C-80°C в течение 0,5 часов, затем раствор концентрировали при пониженном давлении при 40°C или ниже до того, как объем раствора составлял 6 мл. Затем, после перемешивания концентрированного раствора при 70°C-80°C в течение 1 часа, его охлаждали до 0°C-5°C, и добавляли по каплям 60 г н-гептана при той же температуре, затем смесь дополнительно перемешивали при той же температуре в течение 30 минут. Смесь собирали фильтрованием, и отфильтрованный материал промывали н-гептаном (9 г) и сушили, получая заявленное в заголовке соединение (5,96 г, оптическая чистота 99,9%).
[0309] (пример 12B) [масштабирование]:
Реакцию конденсации (условия реакции: 20°C-25°C, перемешивание в течение 13 часов) проводили, применяя (R)-8-амино-1,2,3,4-тетрагидронафталин-2-ол (44,5 г), полученный тем же способом, как в (примере 11G), (E)-2-(7-(трифторметил)хроман-4-илиден)уксусную кислоту (72,5 г, 1,0 экв.), полученную способом получения, описанным в WO 2007/010383, изопропанол (900 г: 18 кратный его вес относительно соединения, представленного формулой (B)) и хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMT-MM) (100,05 г, 1,3 экв.). Обработку проводили согласно (примеру 12a), и посредством этого получали заявленное в заголовке соединение (95,7 г, оптическая чистота 100%).
[0310] [Данные физических свойств соединения, представленного формулой (I)]
(1H-ЯМР данные (CDCl3) (δ: ppm)):
7,80-7,58 (м, 1H), 7,24-6,92 (м, 5H), 6,45 (с, 1H), 4,29 (т, 2H, J=6 Гц), 4,28-4,15 (м, 1H), 3,51 (т, 2H, J=5 Гц), 3,10-2,78 (м, 3H), 2,69-2,53 (м, 1H), 2,14-2,00 (м, 1H), 1,90-1,67 (м, 2H)
(LC-MS):
RT=4,73 (минут), [M+H]+=404
[0311] Оптическую чистоту соединения, представленного формулой (I), измеряли, применяя ВЭЖХ LC-VP систему Shimadzu Corporation в следующих условиях.
[Таблица 30]
Энантиомер соединения, представленного (I), - 18,6 минут
[Пояснение к сноскам]
[0312] L1, L2, L3, L4: впускное отверстие для азота
M1: Емкость, содержащая сырье, TEMPO и дихлорметан
M2: Емкость, содержащая KBr, NaHCO3 и воду
M3: Емкость, содержащая NaClO
P1, P2, P3: насос
T1, T2, T3: труба предварительного охлаждения
S1, S2, S3: мешалка
R1, R2, R3: реактор
CD: Бак для извлечения
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединение ингибитора FGFR в твердой форме и способ его получения | 2020 |
|
RU2810067C2 |
| ОКСАСПИРОПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2016 |
|
RU2733373C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ПИРИМИДОИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДНК-PK | 2020 |
|
RU2796163C1 |
| Фторвинилбензамидное соединение в качестве иммуномодулятора PD-L1 | 2020 |
|
RU2789450C1 |
| АЛЬФА- И БЕТА-НЕНАСЫЩЕННОЕ АМИДНОЕ СОЕДИНЕНИЕ - ПРОИЗВОДНОЕ БЕНЗОТРИАЗОЛА, ПРИМЕНЯЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА TGF-βRI | 2017 |
|
RU2737737C2 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| АГОНИСТ TLR8 | 2019 |
|
RU2800877C2 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ АРОМАТИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2729999C1 |
| ПИРИДОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, ВЫПОЛНЯЮЩИЕ ФУНКЦИЮ ДВОЙНЫХ ИНГИБИТОРОВ mTORC 1/2 | 2018 |
|
RU2771201C2 |
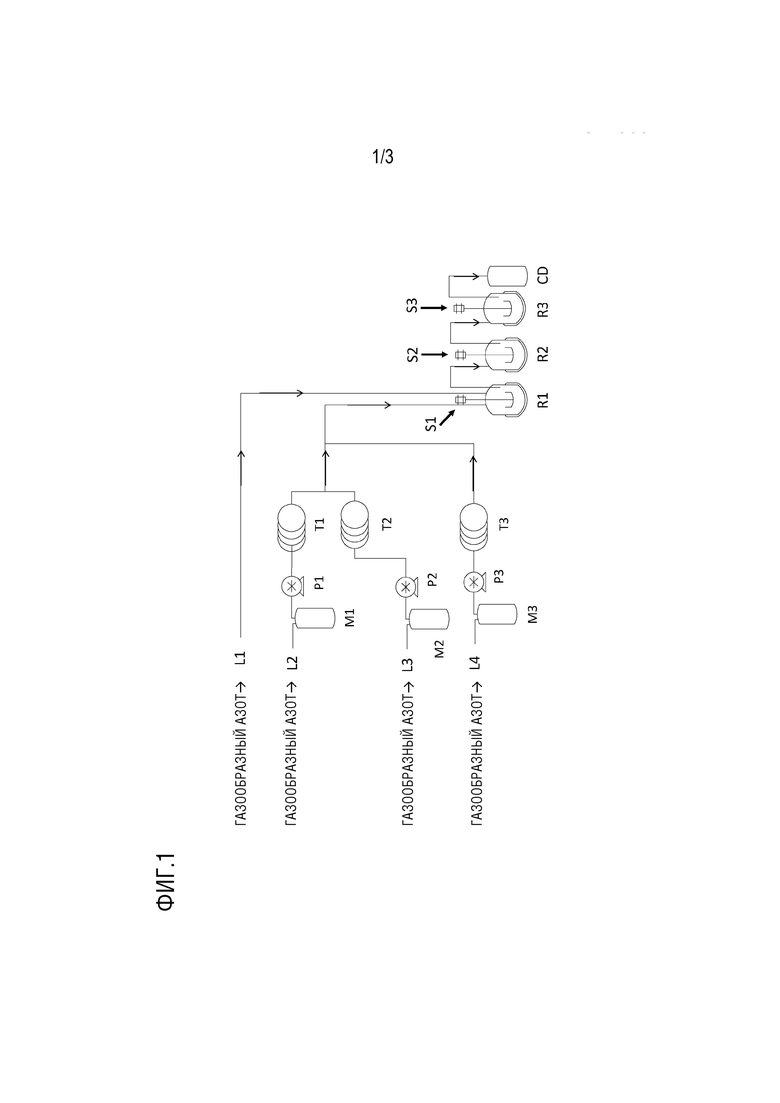

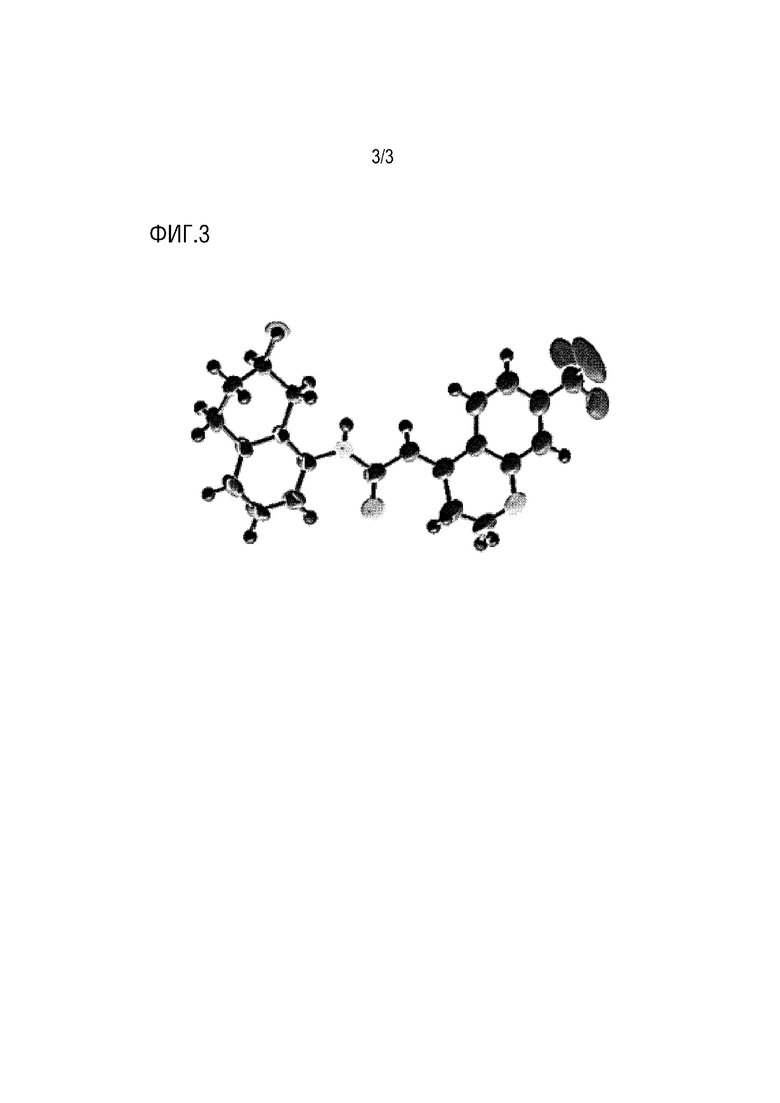
Изобретение относится к новому способу получения (E)-2-(7-трифторметилхроман-4-илиден)-N-((7R)-7-гидрокси-5,6,7,8-тетрагидронафталин-1-ил)ацетамида, представленного формулой (I), который представляет собой гетероциклиденовое ацетамидное производное. Способ включает проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное Формулой (I)
Формула (I):
[C118]
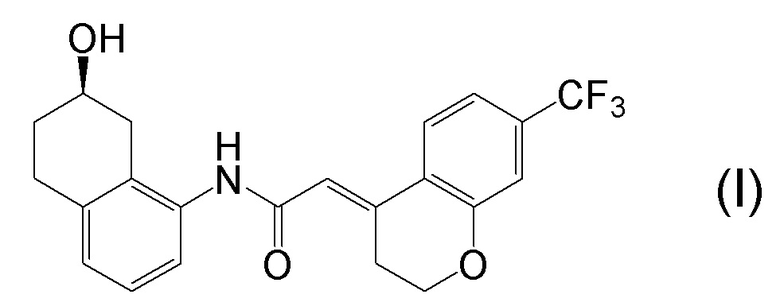
Формула (B):
[C119]
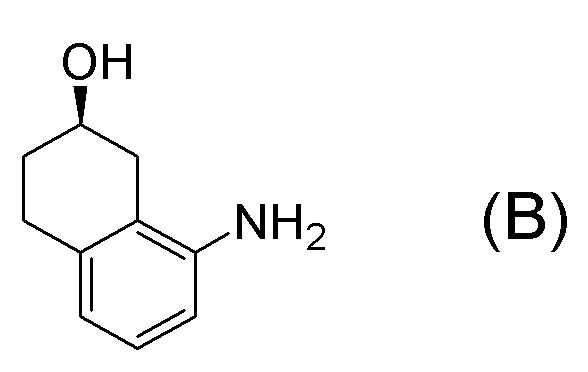
Формула (CA-1):
[C120]
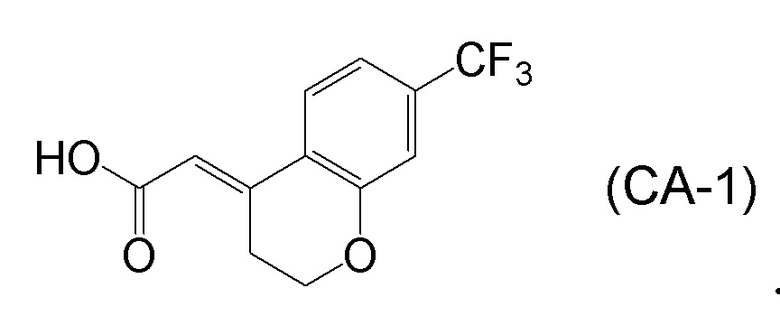 Техническим результатом заявленного изобретения является новый способ более простого получения соединения, представленного формулой (I), с высоким выходом. 3 ил., 30 табл., 12 пр.
Техническим результатом заявленного изобретения является новый способ более простого получения соединения, представленного формулой (I), с высоким выходом. 3 ил., 30 табл., 12 пр.
Способ получения соединения, представленного формулой (I), причем способ включает:
проведение реакции конденсации соединения, представленного формулой (B), или его соли и соединения, представленного формулой (CA-1), применяя DMT-MM в качестве конденсирующего реагента, получая соединение, представленное Формулой (I)
Формула (I):
[C118]
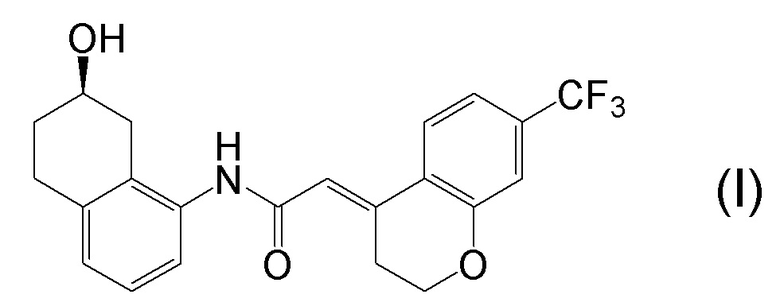
Формула (B):
[C119]
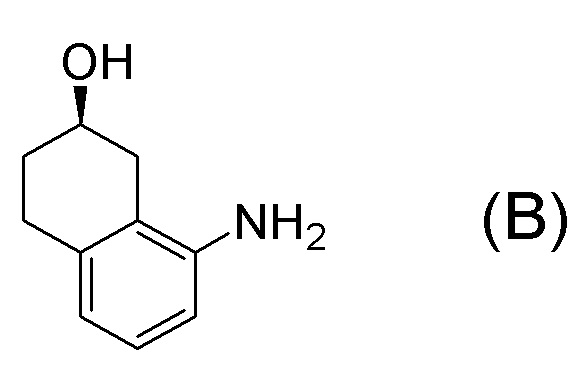
Формула (CA-1):
[C120]
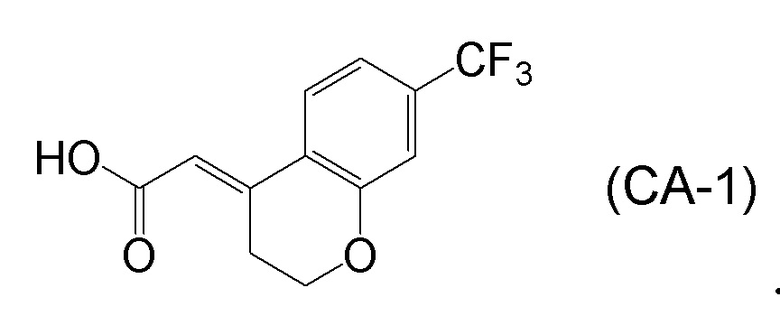
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| WO 2010127855 A1, 11.11.2010 | |||
| CN 109956872 A, 02.07.2019 | |||
| CN 108276380 A, 13.07.2018 | |||
| 1,4,5-ТРИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2196139C2 |

