Область применения
Изобретение относится к способу получения антимикробного пептидомиметика - трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph методом пептидного синтеза в растворе - и может быть использовано в фармацевтической промышленности в производстве лекарственных средств.
Уровень техники
Терапевтические пептиды используются людьми уже более века, с момента первого применения инсулина в 1920-х годах, и в настоящее время на рынке существует около 100 одобренных пептидных препаратов, используемых для лечения ряда заболеваний [1].
Лекарства на основе пептидов используются для нацеливания как на внутри-, так и на внеклеточные мишени, ионные каналы, GPCR и различные ферменты. В настоящее время большинство новых пептидов, поступающих на рынок, разрабатываются для лечения основных терапевтических проблем, включая сердечно-сосудистые заболевания, рак и метаболические заболевания [1, 2, 3]. Образуемые микроорганизмами пептиды, такие как грамицидин, колистин, даптомицин и липогликопептид ванкомицина, являются дополнительными примерами пептидов, которые можно использовать для борьбы с инфекционными заболеваниями [4]. Несмотря на успех этих пептидных антибиотиков микробного происхождения, продвижение многих эндогенно продуцируемых защитных антимикробных пептидов (АМП) в высших организмах еще не оказало влияния на рынок лекарств [4, 5].
АМП представляют собой ключевой компонент врожденной защитной системы, участвующей в начальной быстрой реакции на патогенные вторжения [6, 7]. Из-за острой необходимости разработки эффективных противомикробных препаратов с новыми механизмами действия в связи с появлением устойчивости к противомикробным препаратам [8], АМП были тщательно изучены и исследованы на предмет их потенциала в качестве антибиотиков следующего поколения [5]. В дополнение к прямому уничтожению микроорганизмов путем разрушения мембран или ингибирования жизненно важных внутриклеточных процессов многие АМП также способны взаимодействовать с иммунной системой хозяина, модулируя ее воспалительные реакции [9].
Поиск эффективных противомикробных препаратов, способных бороться с инфекциями, вызванными резистентными бактериями, актуален как никогда раньше. Инфекции, вызванные полирезистентными патогенами, представляют собой значительную и растущую нагрузку на здравоохранение и общество, и исследователи изучают новые классы биологически активных соединений, чтобы замедлить это развитие. Антимикробные пептиды врожденной иммунной системы представляют собой один перспективный класс, который предлагает потенциальное решение проблемы устойчивости к антибиотикам благодаря их способу действия на микробные мембраны. Однако проблемы, связанные с фармакокинетикой, биодоступностью и общей токсичностью, замедляют продвижение и использование врожденных защитных пептидов. Улучшение терапевтических свойств этих пептидов является стратегией уменьшения клинических ограничений, и синтетические имитаторы антимикробных пептидов становятся многообещающим классом молекул для различных антимикробных применений. Эти соединения могут быть значительно короче при сохранении или даже улучшении антимикробных свойств, и для ряда инфекционных заболеваний в настоящее время в клинической разработке находятся несколько уменьшенных синтетических имитаторов [10].
Ранее получение АМП основывалось на выделении из природных источников, что обычно требовало большого количества исходного биологического материала, из которого можно было извлечь небольшое количество чистого пептида [11]. В настоящее время возможно выделение АМП в больших масштабах с помощью технологии рекомбинантной ДНК или путем химического синтеза. Выбор метода производства часто диктуется размером синтезируемого АМП, при этом более крупные пептиды (> примерно 50 остатков) становятся все менее подходящими для получения химическими средствами [12]. Однако рекомбинантная экспрессия АМП обычно считается более сложной и трудоемкой, чем химический синтез, несмотря на более низкие производственные затраты и меньшее воздействие на окружающую среду. Кроме того, рекомбинантная экспрессия некоторых АМП требует включения большого слитого белка для маскировки токсичности пептида для клетки-хозяина, который позже должен быть отщеплен от АМП и удален во время очистки [13]. По этим причинам химический синтез АМП часто является более практичным подходом, особенно с появлением твердофазных методов, которые сделали этот процесс быстрым, эффективным и надежным [9]. Однако в случае трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph использование твердофазных методов химического синтеза нецелесообразно и ограничено тем фактом, что из трех аминокислот, входящих в состав рассматриваемого пептидомиметика, только одна является природной, а две другие - являются модифицированными, в связи с чем стандартные наборы реагентов для твердофазных синтезаторов не могут быть использованы, а изготовление нестандартных наборов реагентов очень трудоемко и дорого.
Одним из наиболее эффективных пептидомиметиков, нашедших свое применение в медицинской практике, является трипептид формулы H-Arg-Tbt-Arg-NH-C2H4-Ph (далее - трипептид). Наиболее близким к заявляемому способу получения трипептида является способ, описанный в патенте US 8598114 В2 [14], в котором пептиды получали в растворе путем поэтапного связывания аминокислот с применением стратегии использования трет-бутоксикарбонильных (далее Вос) защитных групп, в соответствии со следующей общей процедурой: С-концевую часть пептида со свободной аминогруппой (1 эквивалент), Вос-защищенную аминокислоту (1,05 эквивалента) и 1-гидроксибензотриазол (далее-HOBt) (1,8 эквивалента) растворяли в диметилформамиде (далее - DMF) (2-4 мл/ммоль аминогруппы) перед добавлением диизопропилэтиламина (далее - DIPEA) (4,8 эквивалента). Смесь охлаждали на льду и добавляли О-(бензотриазол-1-ил)-N,N,N',N'тетраметилурония гексафторфосфат (далее - HBTU) (1,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1-2 часов. Реакционную смесь разбавляли этилацетатом и промывали лимонной кислотой, бикарбонатом натрия и насыщенным раствором хлористого натрия. Растворитель удаляли в вакууме. С полученного пептида снимали Вос-защиту, используя трифторуксусную кислоту (далее - TFA) (95 масс. %) или ацетилхлорид в безводном метаноле. Далее получали амид с использованием хлортрипирролидинофосфония гексафторфосфата (далее PyCloP). Boc-Arg-N(CH2)2Ph синтезировали, растворив Boc-Arg-OH (1 эквивалент), NH(CH2)2Ph (1,1 эквивалент) и PyCloP (1 эквивалент) в сухом дихлорметане(далее - DCM), отфильтрованном через оксид алюминия, и DMF. Раствор охлаждали на льду и при перемешивании добавляли DIPEA (2 эквивалента). Раствор перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь выпаривали, повторно растворяли в этилацетате и промывали лимонной кислотой, бикарбонатом натрия и насыщенным раствором хлористого натрия. Растворитель удаляли в вакууме. С полученного пептида снимали Вос-защиту с использованием TFA (95 масс. %). Далее пептиды очищали с помощью ВЭЖХ с обращенной фазой (далее - ОФ-ВЭЖХ) на колонке Delta-Pak (Waters) C18 (100 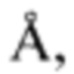 15 мкм, 25×100 мм) в системе вода-ацетонитрил (оба с добавкой 0,1 об. % TFA) в качестве элюентов. Пептиды анализировали с помощью ОФ-ВЭЖХ с использованием аналитической колонки Delta-Pak (Waters) C18 (100 А, 5 мкм, 3,9×150 мм) и масс-спектрометрии с электрораспылением положительных ионов на квадрупольном масс-спектрометре VG Quattro (VG Instruments Inc., Олтрингем, Великобритания).
15 мкм, 25×100 мм) в системе вода-ацетонитрил (оба с добавкой 0,1 об. % TFA) в качестве элюентов. Пептиды анализировали с помощью ОФ-ВЭЖХ с использованием аналитической колонки Delta-Pak (Waters) C18 (100 А, 5 мкм, 3,9×150 мм) и масс-спектрометрии с электрораспылением положительных ионов на квадрупольном масс-спектрометре VG Quattro (VG Instruments Inc., Олтрингем, Великобритания).
Недостатком данного способа получения трипептида является то, что в нем используется один протокол синтеза для всех стадий синтеза (используются С-концевой пептид/аминокислота со свободной аминогруппой и Вос-защищеннный N-компонент в соотношении 1:1,05 (избыток N-компонента 5 мольных процентов для каждой стадии конденсации), что приводит к накоплению большого количества побочных продуктов реакции, в результате чего выход целевого продукта снижается и, кроме того, требуется стадия дополнительной очистки, что увеличивает стоимость конечного продукта.
Технический результат, достигаемый данным изобретением, заключается в способе синтеза трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph, позволяющем получать целевой продукт с чистотой не менее 98 масс. % без дополнительной стадии очистки.
Сущность изобретения
Заявленный технический результат достигается в способе получения трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph, включающем стадию получения фенэтиламида трет-бутоксикарбонил-L-аргинина карбодиимидным методом с последующим деблокированием трет-бутоксикарбонильной группы и получением дигидрохлорида фенэтиламида-L-аргинина, далее - стадию получения защищенного дипептида - трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида карбодиимидным методом с последующим удалением трет-бутоксикарбонильной группы и получением дигидрохлорида 2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида, и стадию получения защищенного трипептида трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида) с последующим удалением трет-бутоксикарбонильной группы, при этом на стадии получения защищенного дипептида используют избыток 20 мольных процентов аминокомпонента - дигидрохлорида фенэтиламида-L-аргинина, а на стадии получения защищенного трипептида используют избыток 20 мольных процентов карбоксильного компонента - трет-бутоксикарбонил-L-аргинина.
Таким образом, трипептид получают методом многостадийного пептидного синтеза в растворе с использованием разных тактических схем. Несмотря на то, что необходимые для этого методические приемы, реагенты и условия реакций известны в химии пептидов, их сочетание, используемое в предлагаемом изобретении, не является очевидным для специалиста в данной области.
Таким образом, предлагаемый способ получения трипептида состоит из 6 стадий синтеза, которые более детально описаны ниже.
1. Получение фенэтиламида-трет-бутоксикарбонил-L-аргинина
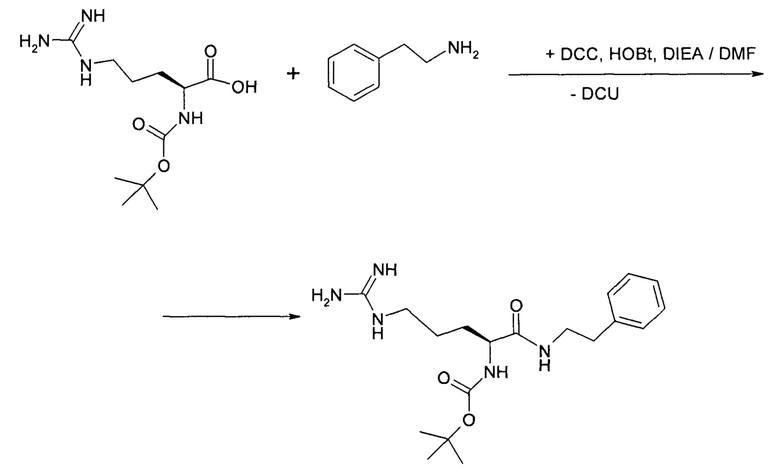
Фенэтиламид-трет-бутоксикарбонил-L-аргинина получают конденсацией трет-бутоксикарбонил-L-аргинина и декарбоксифенилаланина гидрохлорида (избыток 10 мольных процентов) в DMF в присутствии основания диизопропилэтиламина (далее - DIEA). В качестве конденсирующего агента используют избыток 10-12 мольных процентов дициклогексилкарбодиимида (далее - DCC) и HOBt в качестве катализатора (25 мольных процентов).
После удаления из реакционной массы осадка дициклогексилмочевины (далее - DCU) и DIEA х HCl, DMF отгоняют в вакууме и экстрагируют полупродукт хлороформом. После отгонки хлороформа целевой полупродукт получают в виде аморфной твердой массы.
2. Получение дигидрохлорида фенэтиламида-L-аргинина

Дигидрохлорид фенэтиламида-L-аргинина получают удалением трет-бутоксикарбонильной группы с фенэтиламида-трет-бутоксикарбонил-L-аргинина при действии ацетилхлорида (4 эквивалента) в метаноле при нагревании (40-45°С).
После отгонки метанола из реакционной массы, целевой полупродукт переупаривают повторно с метанолом и дважды с толуолом. Полупродукт выделяют в виде густого масла.
3. Получение трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида

Трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амид получают конденсацией трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофана и дигидрохлорида фенэтиламида-L-аргинина (избыток 20 мольных процентов) в DMF в присутствии основания DIEA. В качестве конденсирующего агента используют DCC (избыток 10-12 мольных процентов) и HOBt в качестве катализатора (25 мольных процентов).
После удаления из реакционной массы осадка DCU и DIEA х HCl, DMF отгоняют в вакууме и полупродукт экстрагируют этил ацетатом. После отгонки этилацетата целевой полупродукт получают в виде аморфной твердой пены.
4. Получение дигидрохлорида 2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида
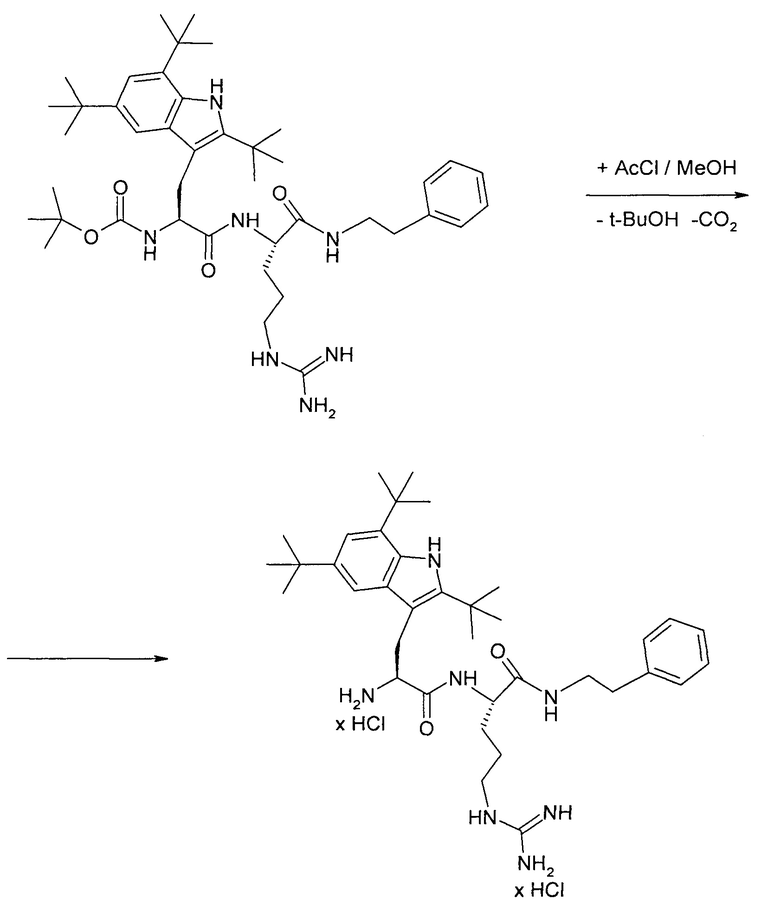
Дигидрохлорид 2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида получают удалением трет-бутоксикарбонильной группы с трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида при действии ацетилхлорида (4 эквивалента) в метаноле при нагревании (40-45°С).
После отгонки метанола из реакционной массы целевой полупродукт экстрагируют этилацетатом. Экстракт упаривают до постоянной массы (выделяют в виде густого масла).
5. Получение трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида
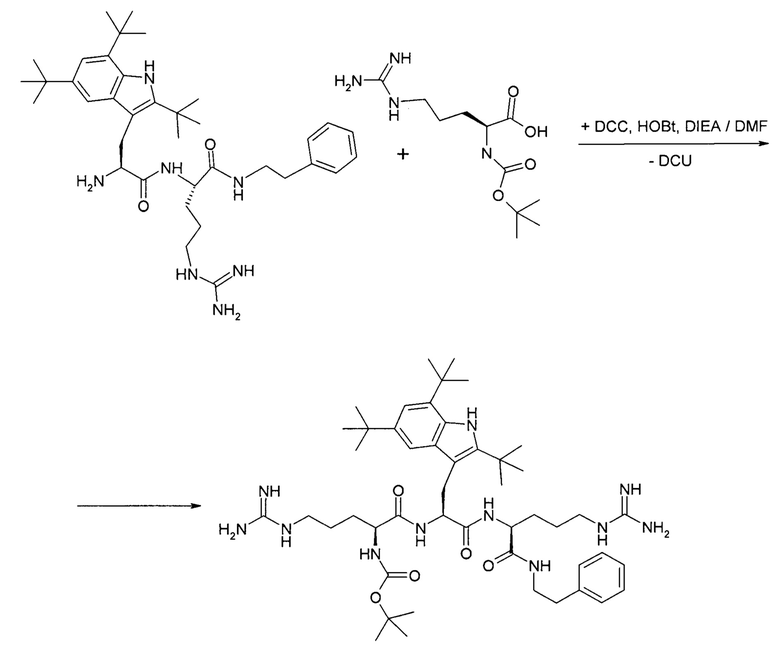
Трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амид получают конденсацией дигидрохлорида 2,5,7-три(трет-бутил)-триптофана и трет-бутоксикарбонил-L-аргинина (избыток 20 мольных процентов) в DMF в присутствии основания DIEA. В качестве конденсирующего агента используют DCC (избыток 10-12 мольных процентов) и HOBt в качестве катализатора (100 мольных процентов).
После удаления из реакционной массы осадка DCU и DIEA х HCl, DMF отгоняют в вакууме и полупродукт экстрагируют этилацетатом. После отгонки этилацетата целевой полупродукт получают в виде аморфной твердой пены.
6. Получение тригидрохлорида L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида

Тригидрохлорид L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амид получают удалением трет-бутоксикарбонильной группы с трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида при действии ацетилхлорида (4 эквивалента) в метаноле при нагревании (40-45°С).
После отгонки метанола из реакционной массы целевой полупродукт экстрагируют водой из раствора в этилацетате. Водную вытяжку промывают метил-третбутиловым эфиром и упаривают. Повторно растворяют в воде. При выпадении в осадок посторонних примесей отфильтровывают их на сборном стеклянном фильтре с капроновым фильтрующим диском с диаметром пор 0,45 мкм. Упаривают водный раствор продукта до густого остатка. При необходимости проводят ионообменнную хроматографию с заменой противоиона с хлорида на ацетат. Продукт, в виде желтого порошка, получают, высушивая водный раствор на лиофильной сушилке.
Отличие заявляемого способа от прототипа заключается в том, что по сравнению с прототипом в заявляемом способе
- на стадии получения защищенного дипептида, используется избыток аминокомпонента (дигидрохлорид фенэтиламида-L-аргинина) в 20 мольных процентов, а не избыток карбоксильного компонента (трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофана) в 5 мольных процентов. За счет этого полностью расходуется карбоксильный компонент, который, в противном случае, в следующих стадиях не удастся удалить, кроме как при помощи хроматографии с соответствующими затратами. При должной последующей тщательности, этот прием дает возможность избежать хроматографической очистки и при этом достичь конечной чистоты продукта 98 масс. % и более, либо существенно повысить выход целевого продукта, что положительно сказывается на его себестоимости;
- на стадии получения защищенного трипептида используется избыток карбоксильного компонента (трет-бутоксикарбонил-L-аргинина) в 20 мольных процентов, а не 5 мольных процентов, что в пептидном синтезе используется редко ввиду нерациональности, поскольку часто дает множество побочных продуктов, однако в данном случае неожиданно приводит к увеличению скорости реакции, сопровождаемому уменьшением доли трудноудаляемых побочных продуктов.
Осуществление изобретения
Сущность изобретения иллюстрируется следующим примером:
Стадия 1. Синтез Boc-Arg-NH-C2H4-Ph (I)
2,74 г (10 ммоль) Boc-Arg-OH растворяют в 40 мл DMF, прибавляют 1,72 г (11 ммоль) декарбоксифенилаланина гидрохлорида, 0,33 г (2,5 ммоль) HOBt и 1,9 мл (11 ммоль) DIEA, охлаждают до -5°С добавляют 2,26 г (11 ммоль) DCC. Завершение реакции контролируют с помощью ТСХ в системе этилацетат:пиридин:уксусная кислота:вода (в соотношении 75:20:6:24). Реакционную смесь фильтруют, отделяя DCU, упаривают, маслообразный остаток растворяют в 80 мл хлороформа. Раствор переносят в делительную воронку, добавляют 30 мл воды и энергично встряхивают. Хлороформный слой собирают отдельно. Водную фазу отбрасывают. Промывают хлороформный слой дополнительной порцией (20 мл) воды, подкислив лимонной кислотой до рН 4, повторно встряхивают. После расслоения водную фазу отбрасывают. Хлороформный экстракт промывают водой (20 мл), насыщенным раствором хлористого натрия, пропускают через слой безводного сульфата натрия. Растворитель удаляют в вакууме. В итоге получают 3,6 г (95%) соединения I в виде аморфной пены. Rf=0,3. MW: 377,5.
Стадия 2. Синтез H-Arg-NH-C2H4-Ph х 2HCl (II)
Для удаления Вос-защитной группы растворяют соединение (I) в 40 мл метанола, затем, при помощи капельной воронки добавляют в перемешиваемый раствор по каплям 2,85 мл (40 ммоль) хлористого ацетила. Метанол упаривают. Повторно растворяют остаток в метаноле и упаривают. Продукт выделяют в виде густого масла с выходом 99% MW: 350,3.
Стадия 3. Синтез Boc-Tbt-Arg-NH-C2H4-Ph (III)
3,78 г (8 ммоль) Boc-Tbt-OH растворяют в 40 мл DMF, прибавляют 3,36 г (9,6 ммоль) дигидрохлорида аргинин(2-фенилэтил)амида (И), 0,33 г (2,5 ммоль) HOBt и 1,9 мл (11 ммоль) DIEA. Реакционную смесь охлаждают до -5°С и добавляют 1,85 г (9 ммоль) DCC. Завершение реакции контролируют с помощью ВЭЖХ, до исчезновения пика Boc-Tbt-OH. Реакционную смесь фильтруют, отделяя DCU, упаривают, маслообразный остаток растворяют в 80 мл этилацетата. Раствор переносят в делительную воронку, добавляют 30 мл воды и энергично встряхивают. Водную фазу сливают, добавляют еще порцию (20 мл) воды, подкисляют лимонной кислотой до рН 4, повторно встряхивают. После расслоения водную фазу отбрасывают. Этилацетатный экстракт промывают водой (20 мл), насыщенным раствором хлористого натрия, пропускают через слой безводного сульфата натрия. Растворитель удаляют в вакууме. В итоге получают 5,84 г (95%) соединения III в виде аморфной пены. MW:768,5, ВЭЖХ-МС анализ: m/z (М+Н+) (молекулярный ион) 733,0.
Стадия 4. Синтез H-Tbt-Arg-NH-C2H4-Ph х 2HCl (IV)
Дигидрохлорид IV получают полностью аналогично, следуя протоколу стадии 2. Выход 99%, MW:704,81, ВЭЖХ-МС анализ: m/z (М+Н)+(молекулярный ион) 632,89.
Стадия 5. Синтез Boc-Arg-Tbt-Arg-NH-C2H4-Ph х 2HCl (V)
5,28 г (7,5 ммоль) H-Tbt-Arg-NH-C2H4-Ph х 2HCl растворяют в 40 мл DMF, прибавляют 2,47 г (9,0 ммоль) Boc-Arg-OH, 0,33 г (2,5 ммоль) HOBt и 2,6 мл (15 ммоль) DIEA. Реакционную смесь охлаждают до -5°С и добавляют 2,06 г (10 ммоль) DCC. Завершение реакции контролируют с помощью реактива Сарина, до исчезновения свободных аминогрупп. Реакционную смесь фильтруют, отделяя DCU, упаривают, маслообразный остаток растворяют в 80 мл этилацетата. Раствор переносят в делительную воронку, добавляют 30 мл воды и энергично встряхивают. Водную фазу сливают, добавляют еще порцию (20 мл) воды, подкисляют лимонной кислотой до рН 4, повторно встряхивают. После расслоения водную фазу отбрасывают. Этилацетатный экстракт промывают водой (20 мл), насыщенным раствором хлористого натрия, пропускают через слой безводного сульфата натрия. Растворитель удаляют в вакууме. В итоге получают 6,85 г соединения V (выход 95%). MW 961,11, ВЭЖХ-МС анализ: m/z (М+Н)+ (молекулярный ион) 889,19.
Стадия 6. Синтез H-Arg-Tbt-Arg-NH-C2H4-Ph х 3HCl (VI)
Тригидрохлорид VI получают полностью аналогично, следуя протоколу стадии 2. Выход 99%, MW: 897,46.
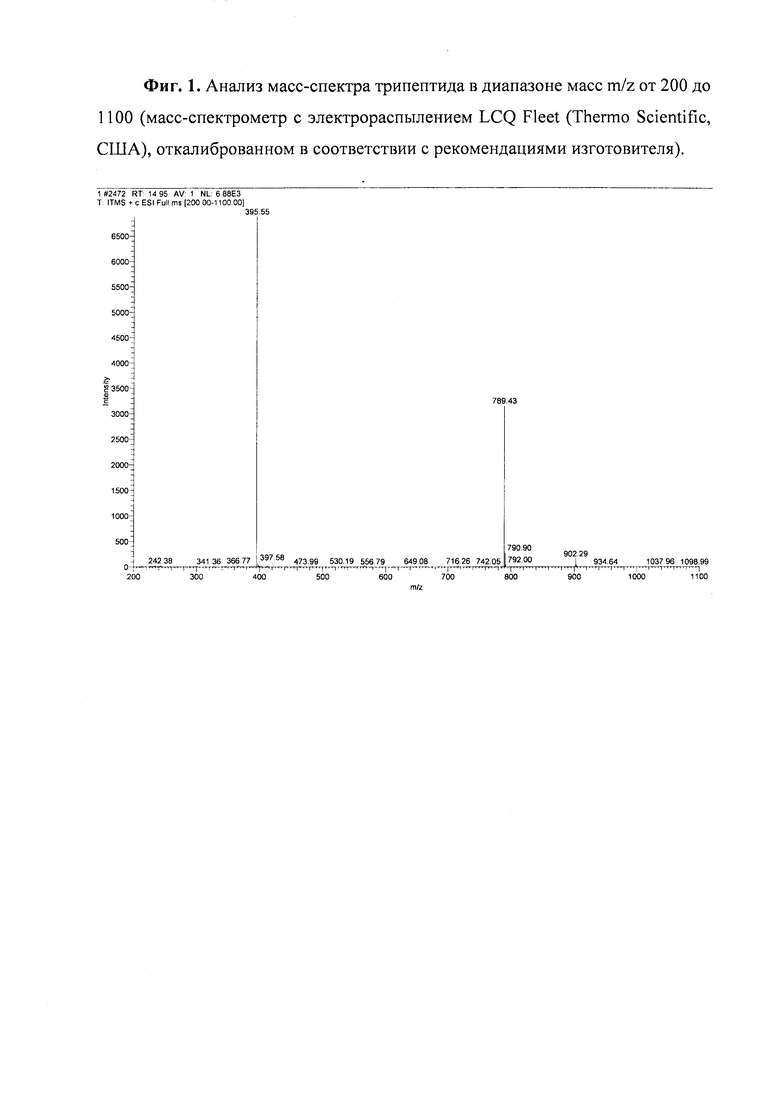
Анализ полученного трипептида. Анализ производится на масс-спектрометре с электрораспылением LCQ Fleet (Thermo Scientific, США), или на аналогичном. Прибор должен быть откалиброван в соответствии с рекомендациями изготовителя. Для указанной модели срок с момента последней калибровки не должен превышать 3 месяца.
Анализируют диапазон масс m/z от 200 до 1100 Да при следующих параметрах:

Результаты МС анализа представлены на Фиг. 1: m/z (М+Н+)+ (молекулярный ион): 789,08 (рассчитанный) / 789,43 (получено в спектре), (М+2Н+)2+: 395,04/395,5.
Источники
1. Muttenthaler, М, King, G. F., Adams, D. J., & Alewood, P. F. (2021). Trends in peptide drug discovery. Nature reviews. Drug discovery, 20(4), 309-325. https://doi.org/10.1038/s41573-020-00135-8
2. Lau JL, Dunn MK. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg Med Chem (2018) 26:2700-7. doi: 10.1016/j.bmc.2017.06.052
3. King GF. Venoms as a Platform for Human Drugs: Translating Toxins Into Therapeutics. Expert Opin Biol Ther(2011) 11:1469-84. doi: 10.1517/14712598.2011.621940
4. Chen CH, Lu TK. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics (2020) 9:24. doi: 10.3390/antibiotics9010024
5. Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, loannidis A, et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect Dis (2020) 20:e216-30. doi: 10.1016/S1473-3099(20)30327-3
6. Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. Antimicrobial Peptides: General Overview and Clinical Implications in Human Health and Disease. Clin Immunol (2010) 135:1-11. doi: 10.1016/j.clim.2009.12.004
7. Zasloff M. Antimicrobial Peptides of Multicellular Organisms. Nature (2002) 415:389-95. doi: 10.1038/415389a; Mookherjee N, Hancock R. Cationic Host Defence Peptides: Innate Immune Regulatory Peptides as a Novel Approach for Treating Infections. Cell Mol Life Sci (2007) 64:922-33. doi: 10.1007/s00018-007-6475-6
8. Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect Dis (2018) 18:318-27. doi: 10.1016/S1473-3099(17)30753-3
9. Gan, В.H., Gaynord, J., Rowe, S.M., Deingruber, Т., & Spring, D.R. (2021). The multifaceted nature of antimicrobial peptides: current synthetic chemistry approaches and future directions. Chemical Society reviews, 50(13), 7820-7880. https://doi.org/10.1039/d0cs00729c
10. Svenson, J., Molchanova, N., & Schroeder, С.I. (2022). Antimicrobial Peptide Mimics for Clinical Use: Does Size Matter?. Frontiers in immunology, 13, 915368. https://doi.org/10.3389/fimmu.2022.915368
11. Brand, G.D., Santos, R.C, Arake, L.M., Silva, V.G., Veras, L.M., Costa, V., Costa, С.H., Kuckelhaus, S.S., Alexandre, J.G., Feio, M.J., & Leite, J.R. (2013). The skin secretion of the amphibian Phyllomedusa nordestina: a source of antimicrobial and antiprotozoal peptides. Molecules (Basel, Switzerland), 18(6), 7058-7070. https://doi.org/10.3390/moleculesl8067058
12. Guzman F., Barberis S., Illanes A. Peptide synthesis: chemical or enzymatic //Electronic Journal of Biotechnology. - 2007. - Т. 10. - №. 2. - C. 279-314
13. Wibowo, D., & Zhao, С.X. (2019). Recent achievements and perspectives for large-scale recombinant production of antimicrobial peptides. Applied microbiology and biotechnology, 103(2), 659-671. https://doi.org/10.1007/s00253-018-9524-1
14. Патент US 8598114 B2, МПК A01N 037/18 (20060101); A61K 38/00 (20060101); A61P 031/10 (20060101), опубл. 03.12.2013, https://www.lens.org/lens/patent/075-368-438-403-442/fulltext
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕМИНА, ОБЛАДАЮЩИЕ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2009 |
|
RU2415868C1 |
| Способ получения полипептидов или их солей | 1977 |
|
SU910116A3 |
| БИОДЕГРАДИРУЕМЫЕ АРГИНИНСОДЕРЖАЩИЕ ПОЛИМЕРЫ | 2014 |
|
RU2549908C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДОВ, ПЕПТИДЫ, ИММУНОМОДУЛИРУЮЩАЯ КОМПОЗИЦИЯ И СПОСОБ РЕГУЛЯЦИИ НЕДОСТАТОЧНОЙ ИЛИ ИЗБЫТОЧНОЙ ФУНКЦИИ Т-КЛЕТОК У ПАЦИЕНТА | 1989 |
|
RU2060998C1 |
| Олигопептидное линкерное промежуточное соединение и способ его получения | 2019 |
|
RU2775973C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕПТАПЕПТИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2303603C2 |
| ПЕПТИД, ОБЛАДАЮЩИЙ НЕЙРОТРОПНОЙ АКТИВНОСТЬЮ | 2008 |
|
RU2394836C2 |
| СПОСОБ ПОЛУЧЕНИЯ НОНАПЕПТИДЭТИЛАМИДА | 1995 |
|
RU2086561C1 |
| АМИД ОКТАПЕПТИДА, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ ПОВЫШАТЬ АРТЕРИАЛЬНОЕ ДАВЛЕНИЕ И ЧАСТОТУ СЕРДЕЧНЫХ СОКРАЩЕНИЙ | 2007 |
|
RU2346001C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
Изобретение относится к способу получения антимикробного пептидомиметика-трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph методом пептидного синтеза в растворе - и может быть использовано в фармацевтической промышленности в производстве лекарственных средств. Заявляемый способ характеризуется тем, что включает стадию получения фенэтиламида трет-бутоксикарбонил-L-аргинина карбодиимидным методом с последующим деблокированием трет-бутоксикарбонильной группы и получением дигидрохлорида фенэтиламида-L-аргинина, далее проводят стадию получения защищенного дипептида - трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида карбодиимидным методом с последующим удалением трет-бутоксикарбонильной группы и получением дигидрохлорида 2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида, и стадию получения защищенного трипептида - трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида, с последующим удалением трет-бутоксикарбонильной группы, при этом на стадии получения защищенного дипептида используют избыток 20 мольных процентов дигидрохлорида фенэтиламида-L-аргинина, а на стадии получения защищенного трипептида используют избыток 20 мольных процентов трет-бутоксикарбонил-L-аргинина. 1 ил., 1 пр.
Способ получения трипептида формулы H-Arg-Tbt-Arg-NH-C2H4-Ph, включающий стадию получения фенэтиламида трет-бутоксикарбонил-L-аргинина карбодиимидным методом с последующим деблокированием трет-бутоксикарбонильной группы и получением дигидрохлорида фенэтиламида-L-аргинина, далее проводят стадию получения защищенного дипептида - трет-бутоксикарбонил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида карбодиимидным методом с последующим удалением трет-бутоксикарбонильной группы и получением дигидрохлорида 2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида, и стадию получения защищенного трипептида - трет-бутоксикарбонил-L-аргинил-2,5,7-три(трет-бутил)-триптофанил-аргинин(2-фенилэтил)амида, с последующим удалением трет-бутоксикарбонильной группы, при этом на стадии получения защищенного дипептида используют избыток 20 мольных процентов дигидрохлорида фенэтиламида-L-аргинина, а на стадии получения защищенного трипептида используют избыток 20 мольных процентов трет-бутоксикарбонил-L-аргинина.
| WO 2009081152 A2, 02.07.2009 | |||
| WO 2001066147 A2, 13.09.2001 | |||
| Isaksson, J | |||
| Шкив для канатной передачи | 1920 |
|
SU109A1 |
| Haug, B | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
