Область изобретения
[01] Настоящее изобретение относится к области конъюгатов антитело-лекарственное средство и, в частности, к способу получения олигопептидного линкера и его промежуточного соединения.
Предшествующий уровень техники
[02] Конъюгат антитело-лекарственное средство (ADC), как новый тип биологического "управляемого снаряда", обеспечивает выгодное сочетание нацеливающего эффекта моноклональных антител и цитотоксического эффекта низкомолекулярных лекарственных средств и в настоящее время является одной из самых быстроразвивающихся областей в терапии, направленно воздействующей на опухоли. Три компонента ADC (антитело, цитотоксин и линкер) составляют систему направленной доставки лекарственного средства, в которой конструкция и оптимизация линкеров являются решающими для разработки такой системы. Линкеры являются основой для эффективной доставки цитотоксических лекарственных средств посредством ADC и ключевым фактором в определении токсичности ADC продуктов, поскольку преждевременное высвобождение лекарственных средств в кровоток может приводить к системной токсичности и снижению терапевтического индекса. Путем оптимизации существующей технологии связывания, разработки стабильных линкеров и изыскания новых механизмов высвобождения токсинов можно решить такие проблемы, как ненаправленное действие токсинов и лекарственная устойчивость, которые обычно наблюдаются у ADC, и повысить, тем самым, эффективность и безопасность.
[03] На данном этапе в центре внимания находятся основанные на олигопептидах расщепляемые ADC, которые распадаются под действием катепсина В в лизосомах клеток-мишеней с высвобождением токсинов, которые в конечном счете уничтожают опухоли (Источник информации 1: Wang Y, Fan S, Zhong W, et al. Development and properties of valine-alanine based antibody-drug conjugates with monomethyl auristatine as the potent payload[J]. International journal of molecular sciences, 2017, 18(9): I860.). Среди них олигопептидные линкеры (такие как дипептид валин-цитруллин (Val-Cit, VC)) широко используются в различных лекарственных средствах, представляющих собой ADC. В пяти разрешенных для применения в настоящее время конъюгированных с антителом лекарственных средствах (на июль 2019 года) (Таблица 1) линкеры брентуксимаба ведотина компании Seattle & Takeda и полатузумаба ведотина-piiq компании Genentech являются олигопептидными линкерами (дипептидный линкер валин-цитруллин).
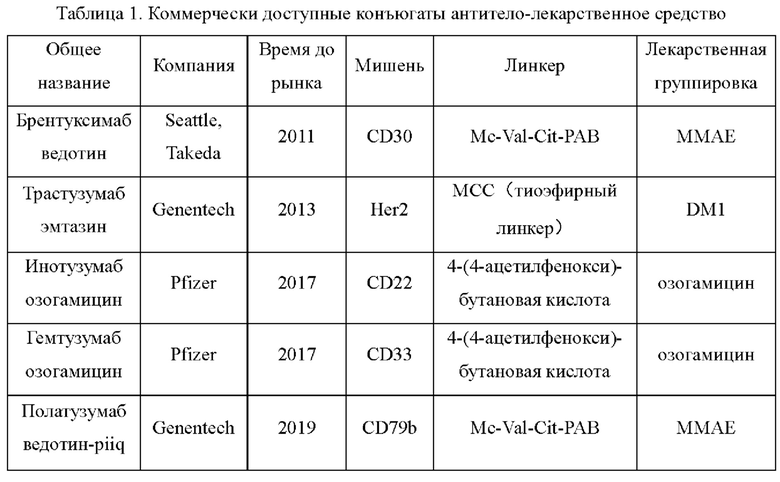
[04] В настоящее время существует три основных способа синтеза олигопептидных линкеров (Источник информации 2: Mondal D, Ford J, Pinney K G. Improved Methodology for the Synthesis of a Cathepsin В Cleavable Dipeptide Linker, Widely Used in Antibody-Drug Conjugate Research [J]. Tetrahedron letters, 2018, 59 (40): 3594-3599.), причем в первом способе используется защитная группа Fmoc, во втором используется защитная группа Cbz и в третьем используется защитная группа Вос.
[05] Вышеуказанные три способа получения описаны ниже с использованием дипептидных линкеров валин-цитруллин в качестве примеров:
[06] (1) Использование защитной группы Fmoc 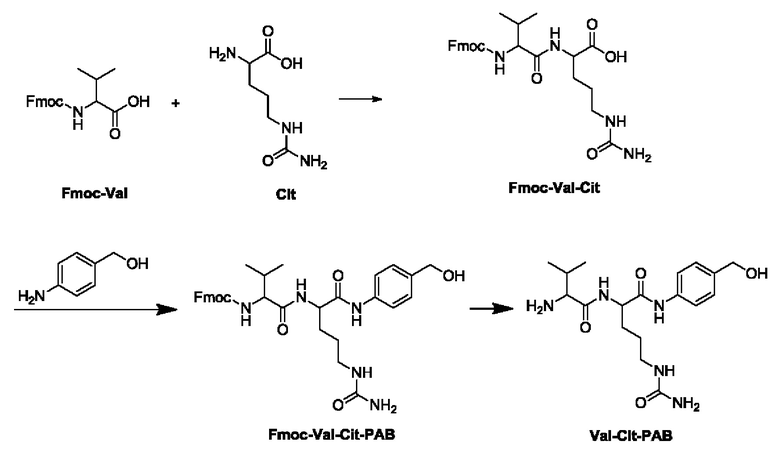
[07] В этом способе Fmoc-Val сначала подвергают реакции конденсации с Cit с получением Fmoc-Val-Cit, который затем подвергают реакции конденсации с пара-аминобензиловым спиртом с получением Fmoc-Val-Cit-PAB, и затем Fmoc удаляют с образованием Val-Cit-PAB. Однако для удаления защитной группы Fmoc в этом способе обычно используют низшие амины, поэтому легко образуются побочные продукты, которые трудно полностью удалить. Кроме того, Fmoc имеет сильное УФ поглощение, и следовые остатки оказывают большое влияние на тестирование чистоты продукта.
[08] (2) Использование защитной группы Cbz

[09] В этом способе Cbz-Val сначала подвергают реакции конденсации с Cit с получением Cbz-Val-Cit, который затем подвергают реакции конденсации с пара-аминобензиловым спиртом с получением Cbz-Val-Cit-PAB, и затем Val-Cit-PAB получают путем удаления Cbz. Однако для удаления защитной группы Cbz в этом способе в качестве катализатора требуется переходный металл, обычно Pd, который вреден для человеческого организма из-за того, что остатки тяжелого металла трудно удалять. Кроме того, требуется источник водорода, обычно газ водород, который является чрезвычайно взрывоопасным, что создает высокие риски возникновения угрозы безопасности и делает его непригодным для крупномасштабного использования. Хотя могут быть использованы другие источники водорода, газ водород также высвобождается во время использования, угрожая безопасности.
[10] (3) Использование защитной группы Вос
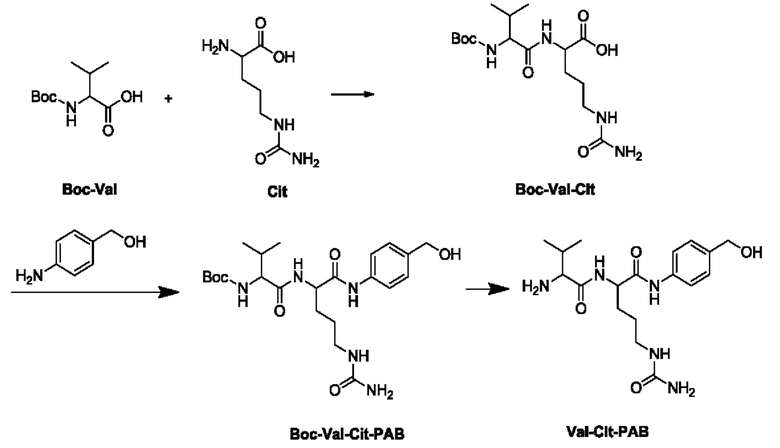
[11] В этом способе Boc-Val сначала подвергают реакции конденсации с Cit с получением Boc-Val-Cit, который затем подвергают реакции конденсации с пара-аминобензиловым спиртом с получением Вос-Val-Cit-PAB, и затем Val-Cit-PAB получают путем удаления Вос. В этом способе для удаления защитной группы Вос используется сильная кислота, обычно раствор НСl и трифторуксусная кислота. При использовании НСl гидроксил спирта в положении бензила молекулы Вос-Val-Cit-PAB будет замещаться хлором с образованием соответствующего бензилхлорида, что будет сказываться на качестве продукта. При использовании трифторуксусной кислоты спирт в положении бензила будет подвергаться реакции этерификации с образованием соответствующего трифторацетата, поэтому требуется дополнительная стадия гидролиза для получения Val-Cit-PAB. Кроме того, щелочной гидролиз создает риск рацемизации хирального центра аминокислотных единиц.
Краткое изложение сущности изобретения
[12] Для решения вышеуказанных проблем согласно настоящему изобретению предложен новый способ получения олигопептидных линкеров. Способ получения Val-Cit-PAB с использованием Теос в качестве защитной группы, предусмотренный настоящим изобретением, легко осуществляется в мягких реакционных условиях, и поскольку при реакции почти отсутствуют побочные реакции, этот способ дает высокочистый продукт с меньшим количеством примесей и легко очищается, обеспечивая достижение неожиданного технического результата.
[13] Конкретно, согласно настоящему изобретению предложено олигопептидное линкерное промежуточное соединение, имеющее структуру, представленную формулами (1)-(4):
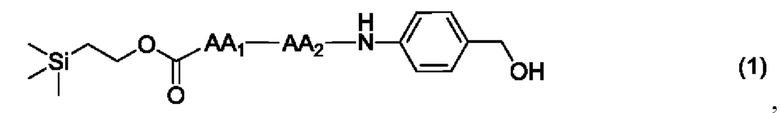
или
или
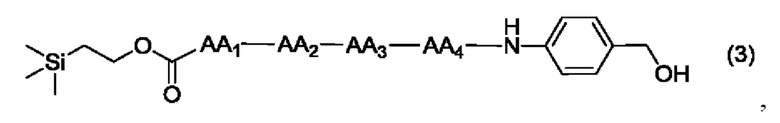
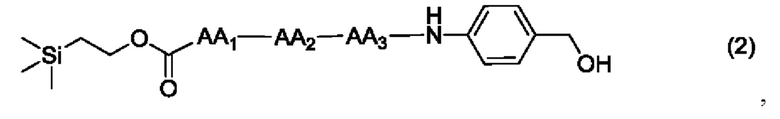
или
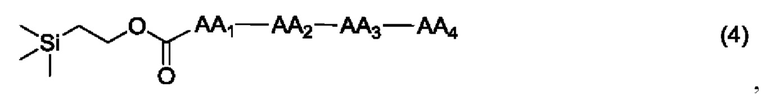
где AA1, АА2, АА3 и АА4 представляют собой любую аминокислоту.
[14] Далее, AA1, АА2, АА3 и АА4 независимо выбраны из группы, состоящей из следующих: -валин- (-Val-), -цитруллин -(-Cit-), -аланин- (-Ala-), -лизин- (-Lys-), -лизин(тритил)- (-Lys(Trt)-), -лизин(монометокситритил)- (-Lys(Mmt)-), -лизин(флуоренилметилоксикарбонил)- (-Lys(Fmoc)-), -аргинин- (-Arg-), -фенилаланин- (-Phe-), -глицин- (-Gly-), -лейцин- (-Leu-) и -изолейцин- (-Ile-).
[15] Кроме того, -АА1-АА2- выбран из группы, состоящей из следующих: -валин-цитруллин- (-Val-Cit-), -в алии-аланин- (-Val-Ala-), -валин-лизин- (-Val-Lys-), -валин-лизин(тритил)- (-Val-Lys(Trt)-), -валин-лизин(монометокситритил)- (-Val-Lys(Mmt)-), -валин-лизин(флуоренилметилоксикарбонил)- (-Val-Lys(Fmoc)-), -валин-аргинин- (-Val-Arg-), -фенилаланин-цитруллин- (-Phe-Cit-), -фенил про пил-лизин- (-Phe-Lys-), фенилаланин-лизин(тритил)- (-Phe-Lys(Trt)-), -фенилаланин-лизин(монометокситритил)- (-Phe-Lys(Mmt)-), -фенилаланин-лизин(флуоренилметилоксикарбонил)- (-Phe-Lys(Fmoc)-), лейцин-цитруллин- (-Leu-Cit-), изолейцин-цитруллин- (-Ile-Cit-) и -фенилаланин-аргинин- (-Phe-Arg-); -АА1-АА2-АА3- выбран из -фенилаланин-аргинин-аргинина- (-Ala-Arg-Arg-); -AA1-AA2-AA3-AA4- выбран из группы, состоящей из следующих: -глицин-глицин-фенилаланин-глицин- (-Gly-Gly-Phe-Gly-), -глицин-фенилаланин-лейцин-глицин- (-Gly-Phe-Leu-Gly-) и -аланин-лейцин-аланин-лейцин (-Ala-Leu-Ala-Leu-).
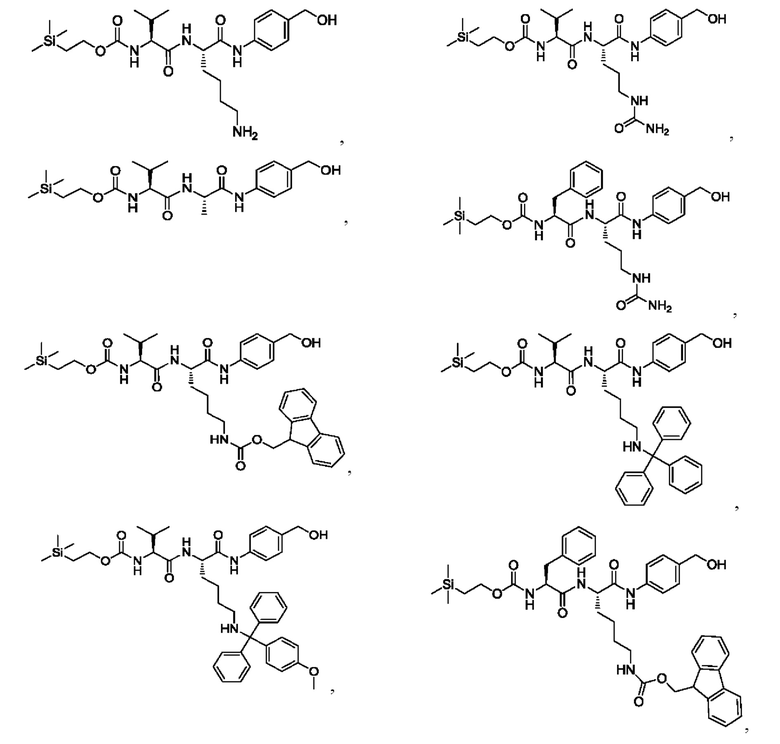
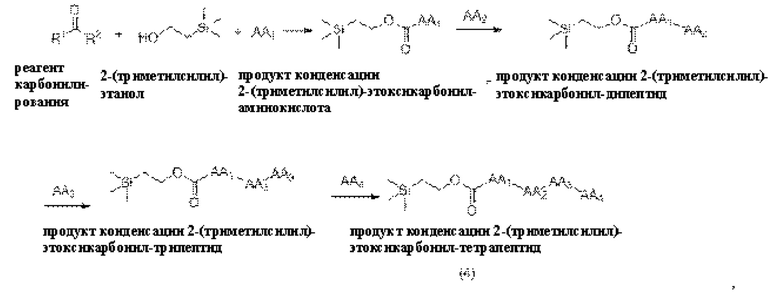
[16] Предпочтительно, олигопептидное линкерное промежуточное соединение включает в себя следующую структуру:
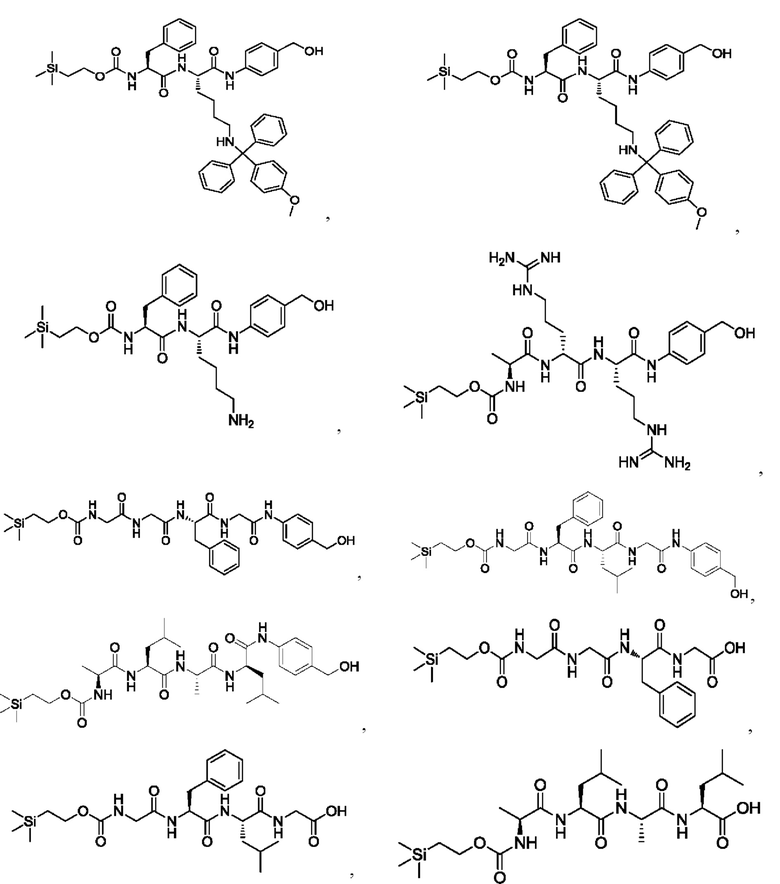
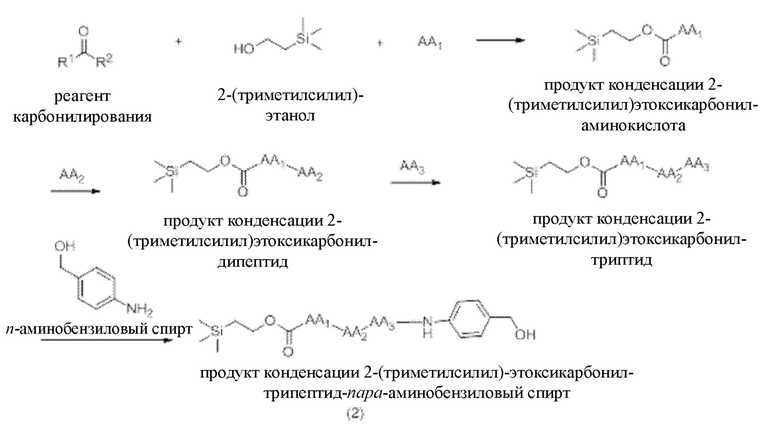
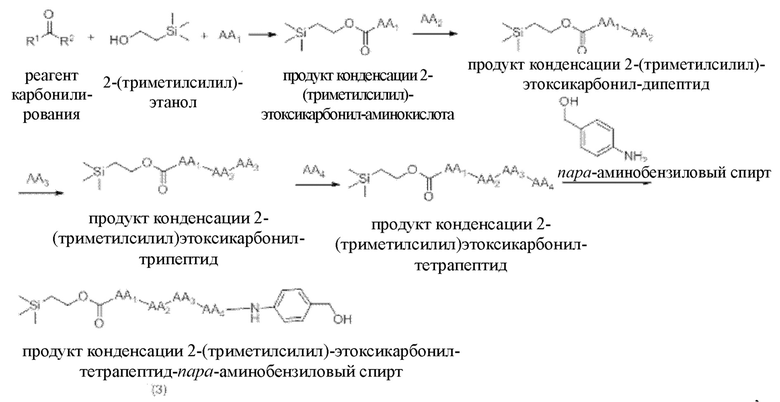
[17] Согласно настоящему изобретению предложен также способ получения олигопептидного линкерного промежуточного соединения формул (1)-(3), где реакционный путь способа следующий:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 и затем аминокислотой АА2, или аминокислотой AA1, аминокислотой АА2 и аминокислотой АА3 последовательно, или аминокислотой AA1, аминокислотой АА2, аминокислотой АА3 и аминокислотой АА4 последовательно с получением продукта конденсации, представляющего собой 2-(триметилсилил) этоксикарбонил -олигопептид;
2) взаимодействие полученного продукта конденсации 2-(триметилсилил)этоксикарбонил-олигопептида и пара-аминобензилового спирта с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил -олигопептид-пара-аминобензиловый спирт;
где реагент карбонилирования представляет собой любое соединение, содержащее карбонильную группу.
[18] Далее, олигопептидное линкерное промежуточное соединение формулы (1) получают следующим реакционным путем:
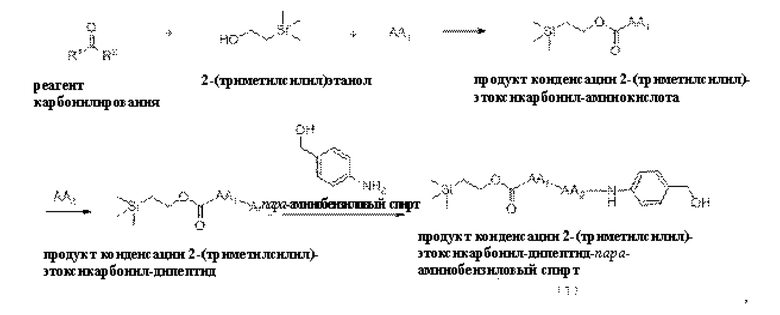
где способ получения включает в себя следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид;ии
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид-пара-аминобензиловыйспирт.
[19] Далее, олигопептидное линкерное промежуточное соединение формулы (2) получают следующим реакционным путем:
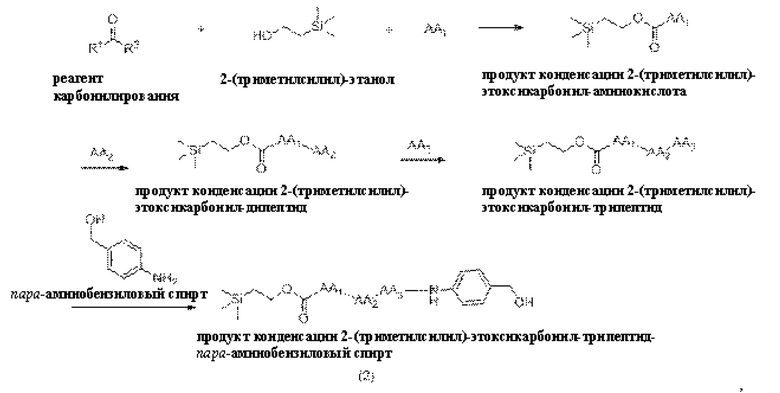
где способ получения включает в себя следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил) этанолом и аминокислотой AA1 с получением продукта реакции конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил) этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид;
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и аминокислотой АА3 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид, и
4) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил -трипептид-пара-аминобензиловый спирт.
[20] Далее, олигопептидное линкерное промежуточное соединение формулы (3) получают следующим реакционным путем:
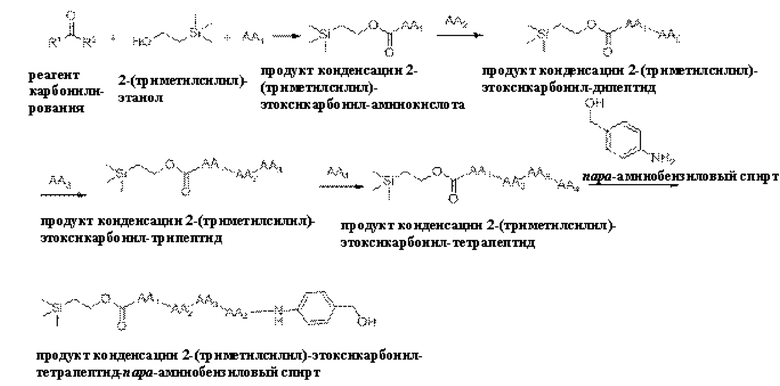
где способ получения включает в себя следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 с получением продукта реакции конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту,
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид,
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и аминокислотой АА3 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид,
4) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом и аминокислотой АА4 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид, и
5) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-тетрапептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид-пара-аминобензиловыйспирт.
[21] Согласно настоящему изобретению дополнительно предложен способ получения олиго пептидного линкерного промежуточного соединения формулы (4), где реакционный путь способа следующий: проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1, аминокислотой АА2, аминокислотой АА3 и аминокислотой АА4 последовательно с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид, где реагент карбонилирования представляет собой любое соединение, содержащее карбонильную группу.
[22] Далее, олигопептидное линкерное промежуточное соединение формулы (4) получают следующим реакционным путем:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил) этанолом и аминокислотой AA1 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил) этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид;
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и аминокислотой АА3 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид; и
4) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом и аминокислотой АА4 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид.
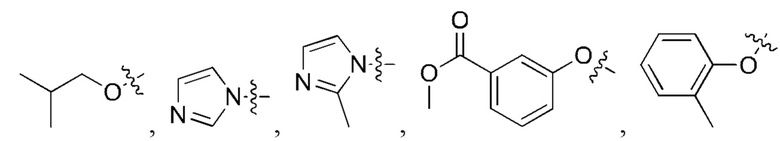
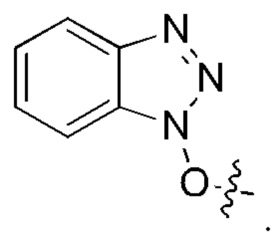
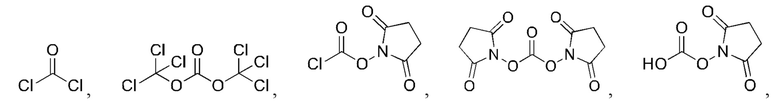
[23] Далее, реагент карбонилирования имеет структуру формулы (5):
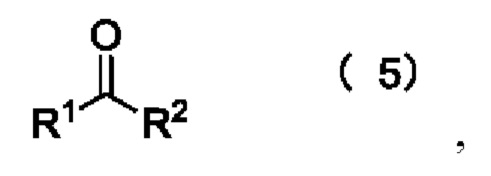
где R1 и R2 независимо выбраны из группы, состоящей из:
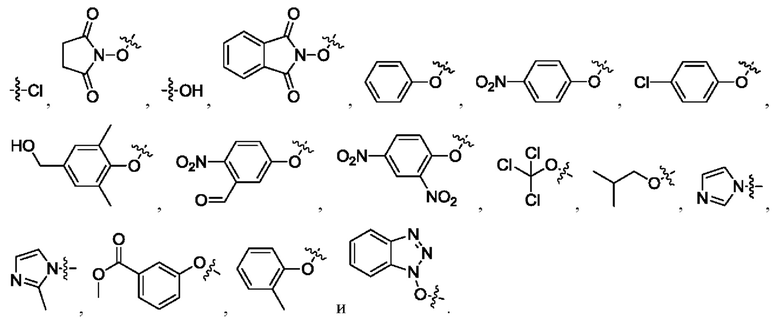
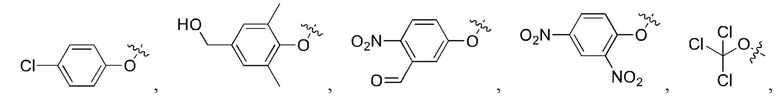
[24] Кроме того, реагент карбонилирования выбран из группы, состоящей из:
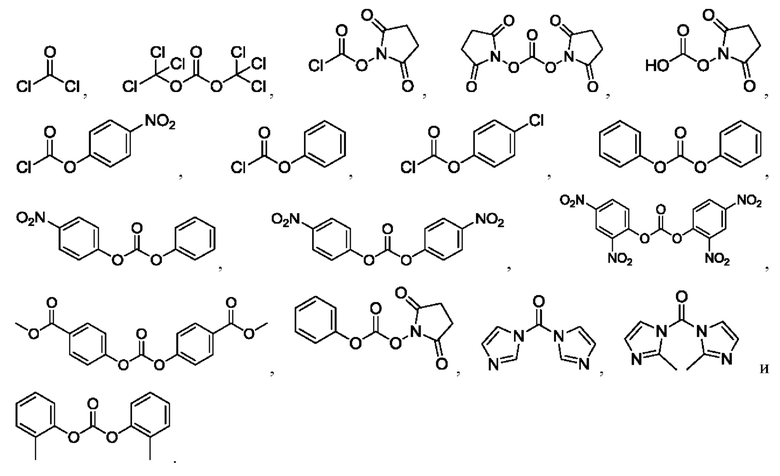
[25] Далее, АА1, АА2, АА3 и АА4 независимо выбраны из группы, состоящей из следующих: -валин- (-Val-), -цитруллин -(-Cit-), -аланин- (-Ala-), -лизин- (-Lys-), -лизин(тритил)- (-Lys(Trt)-), -лизин(монометокситритил)- (-Lys(Mmt)-), -лизин(флуоренилметилоксикарбонил)- (-Lys(Fmoc)-), -аргинин- (-Arg-), -фенилаланин- (-Phe-), -глицин- (-Gly-), -лейцин- (-Leu-) и -изолейцин- (-Ile-).
[26] Кроме того, -АА1-АА2- выбран из следующих: -валин-цитруллин- (-Val-Cit-), -валин-аланин- (-Val-Ala-), -валин-лизин- (-Val-Lys-), -валин-лизин(тритил)- (-Val-Lys(Trt)-), -валин-лизин(монометокситритил)- (-Val-Lys(Mmt)-), -валин-лизин(флуоренилметилоксикарбонил)- (-Val-Lys(Fmoc)-), -валин-аргинин- (-Val-Arg-), -фенилаланин-цитруллин- (-Phe-Cit-), -фенилпропил-лизин- (-Phe-Lys-), -фенилаланин-лизин(тритил)- (-Phe-Lys(Trt)-), -фенилаланин-лизин(монометокситритил)- (-Phe-Lys(Mmt)-), -фенилаланин-лизин(флуоренилметилоксикарбонил)- (-Phe-Lys(Fmoc)-), лейцин-цитруллин- (-Leu-Cit-), изолейцин-цитруллин- (-Ile-Cit-) и -фенилаланин-аргинин-(-Phe-Arg-); -АА1-АА2-АА3- выбран из -фенилаланин-аргинин-аргинина-(-Ala-Arg-Arg-); -АА1-АА2-АА3-АА4- выбран из группы, состоящей из следующих: -глицин-глицин-фенилаланин-глицин- (-Gly-Gly-Phe-Gly-), -глицин-фенилаланин-лейцин-глицин- (-Gly-Phe-Leu-Gly-) и -аланин-лейцин-аланин-лейцин (-Ala-Leu-Ala-Leu-).
[27] Предпочтительно, -AA1-AA2- выбран из следующих структур:
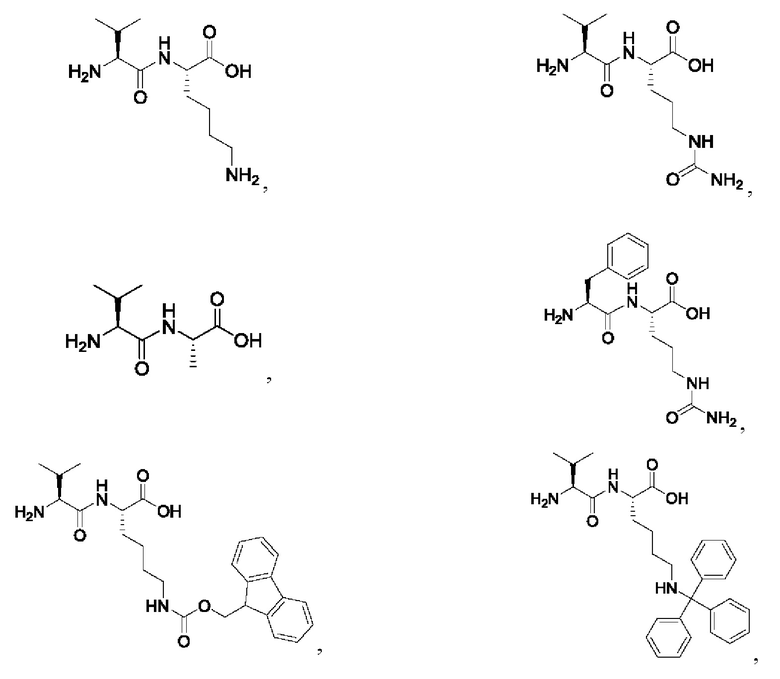

[28] Предпочтительно, -AA1-AA2-AA3- выбран из следующих структур:
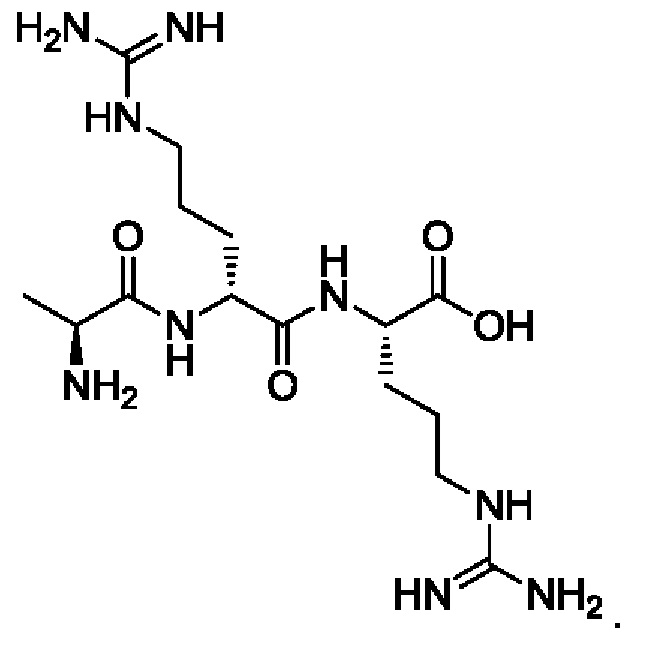
[29] Предпочтительно, -АА1-АА2-АА3-АА4- выбран из следующих структур:
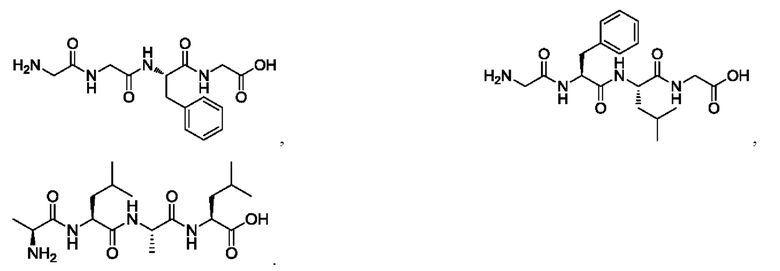
[30] Согласно настоящему изобретению дополнительно предложено применение промежуточного соединения согласно одному из вышеизложенных определений в получении конъюгата антитело-лекарственное средство.
[31] Далее, растворитель, использованный в реакции конденсации, описанной выше, представляет собой любой полярный растворитель или неполярный растворитель; предпочтительно, растворитель представляет собой один или более чем один растворитель, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, DMAc, DMPU, НМРА, диметилового эфира этиленгликоля, диэтилового эфира, трет-бутил-метилового эфира, трет-бутанола, воды, этилацетата, метанола, этанола, изопропанола, дихлорметана, хлороформа и четыреххлористого углерода; более предпочтительно, растворитель представляет собой один или более чем один растворитель, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, воды, метанола, дихлорметана, хлороформа, четыреххлористого углерода и этанола.
[32] В некоторых конкретных воплощениях при проведении реакции конденсации между реагентом карбонилирования с 2-(триметилсилил)этанолом и аминокислотой AA1 реагент, использованный в реакции конденсации, предпочтительно представляет собой один или более чем один реагент, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, DMAc, DMPU, НМРА, диметилового эфира этиленгликоля, диэтилового эфира, трет-бутил-метилового эфира, трет-бутанола, воды и этилацетата; более предпочтительно, реагент представляет собой один или более чем один реагент, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO и воды.
[33] В других конкретных воплощениях после реакции реагента карбонилирования с 2-(триметилсилил)этанолом и аминокислотой AA1 реагент, использованный в реакции конденсации продукта конденсации 2-(триметилсилил)этоксикарбонил-аминокислоты и АА2 и дополнительно с АА3 и/или АА4, в других воплощениях представляет собой один или более чем один, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, DMAc, DMPU, НМРА, диметилового эфира этиленгликоля, диэтилового эфира, трет-бутил-метилового эфира, трет-бутанола, воды и этилацетата; более предпочтительно, реагент представляет собой один или более чем один, выбранный из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, воды и метанола
[34] В других конкретных воплощениях, после вышеуказанных реакций, при проведении реакции конденсации между поли пептидным продуктом конденсации (т.е. продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом, продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом, продуктом конденсации 2-(триметилсилил)этоксикарбонил-тетрапептидом) с пара-аминобензиловым спиртом реагент, использованный в реакции конденсации, предпочтительно представляет собой один или более чем один, выбранный из группы, состоящей из дихлорметана, хлороформа, четыреххлористого углерода, метанола, этанола, изопропанола, тетрагидрофурана, диоксана, ацетонитрила, DMF, DMSO, DMAc, DMPU, НМРА, диметилового эфира этиленгликоля, диэтилового эфира, трет-бутил-метилового эфира, трет-бутанола и этилацетата; более предпочтительно, реагент представляет собой один или более чем один, выбранный из группы, состоящей из дихлорметана, хлороформа, четыреххлористого углерода, метанола, этанола и DMF.
[35] Согласно настоящему изобретению предложено также применение способа согласно любому из вышеизложенных определений в получении конъюгата антитело-лекарственное средство.
[36] Получение конъюгатов антитело-лекарственное средство дополнительно включает в себя удаление защитной группы Теос:
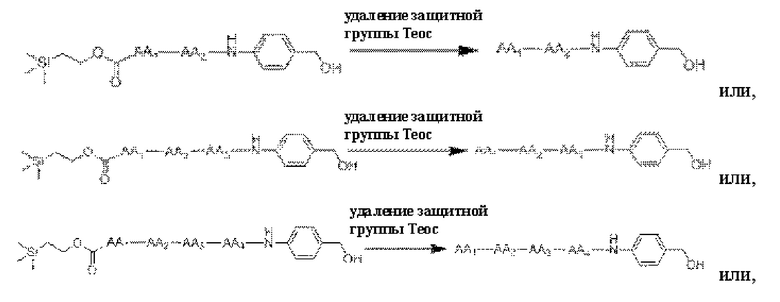
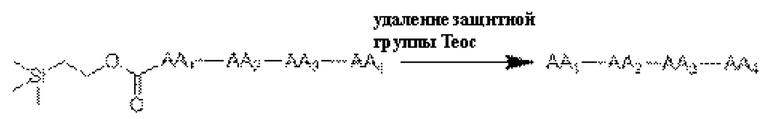
[37] Продукт, полученный путем удаления защитной группы Теос, как описано выше, часто используют в получении конъюгата антитело-лекарственное средство в качестве части линкера, ковалентно связанной с группировкой лекарственного средства. Для достижения ковалентного связывания с группировкой антитела указанный продукт часто связывают с линкером следующими конкретными реакционными путями:
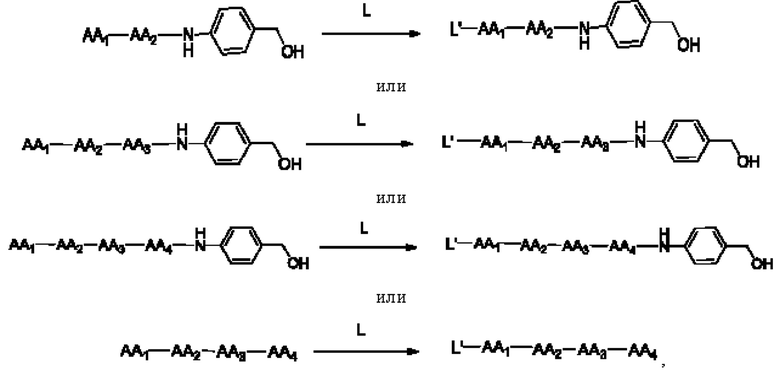
где L и L' являются линкерами и могут представлять собой любую линкерную группу.
[38] В некоторых воплощениях линкерная группа L выбрана из:
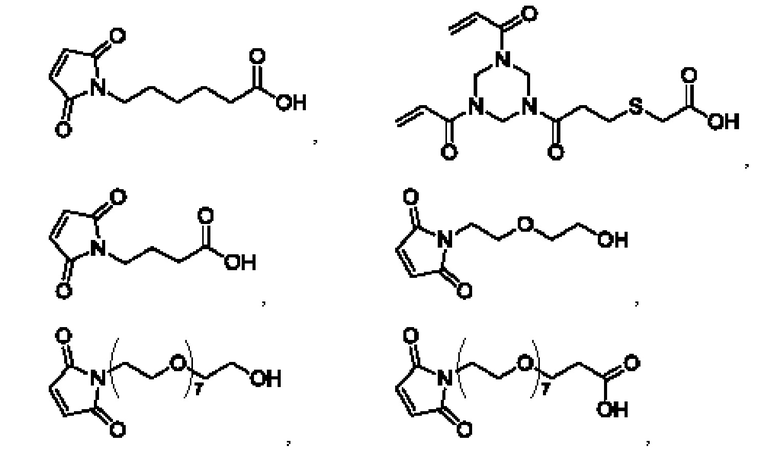
Соответственно, линкерная группа L' выбрана из:

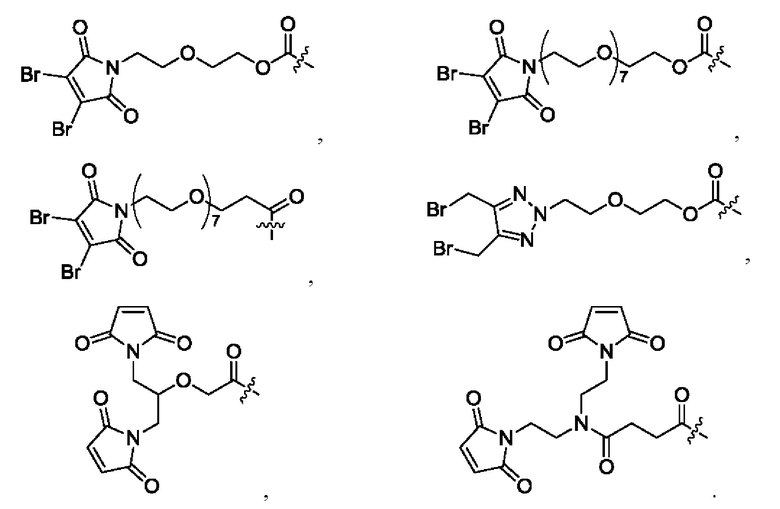
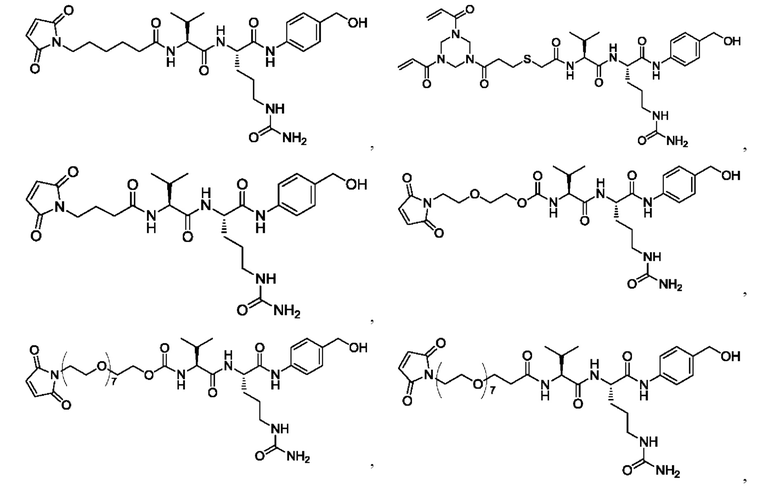
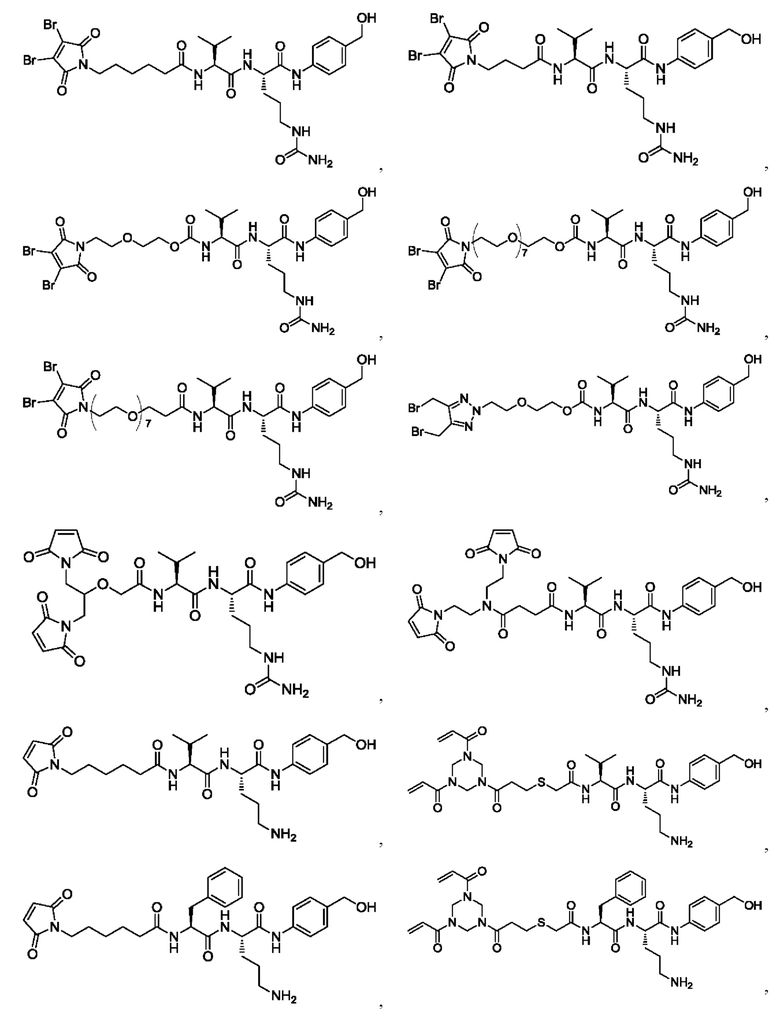
[39] В конкретном воплощении олигопептидный линкер выбран из:

Краткое описание графических материалов
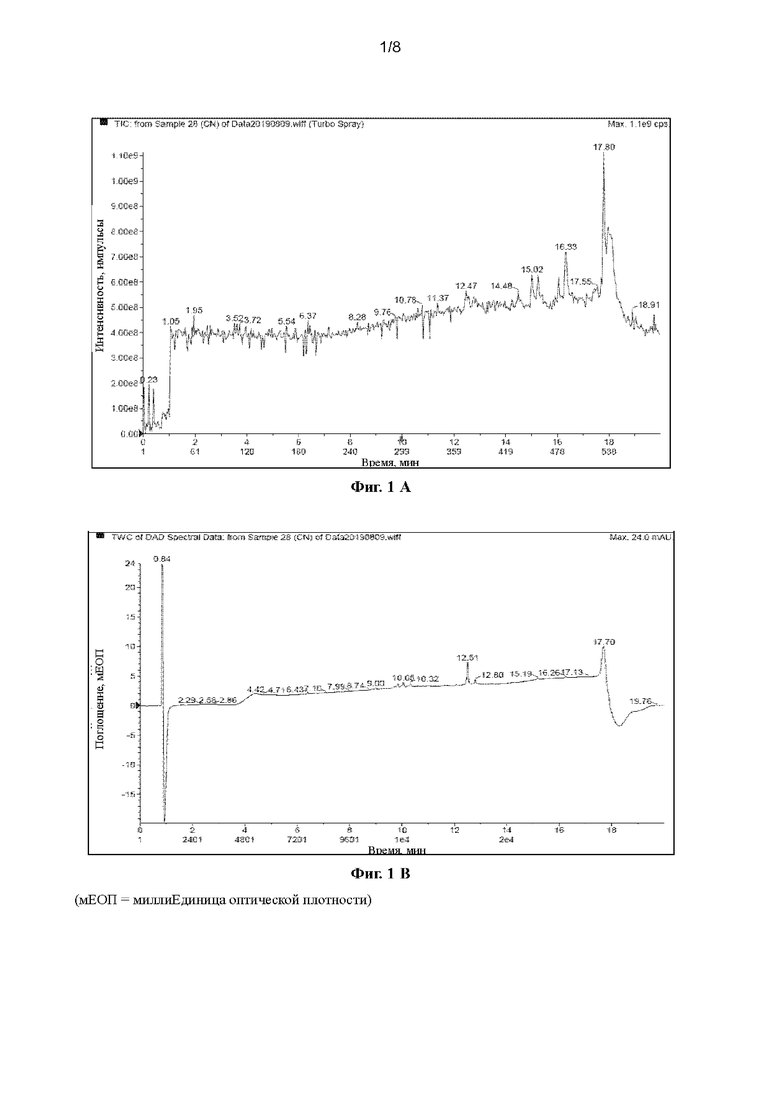
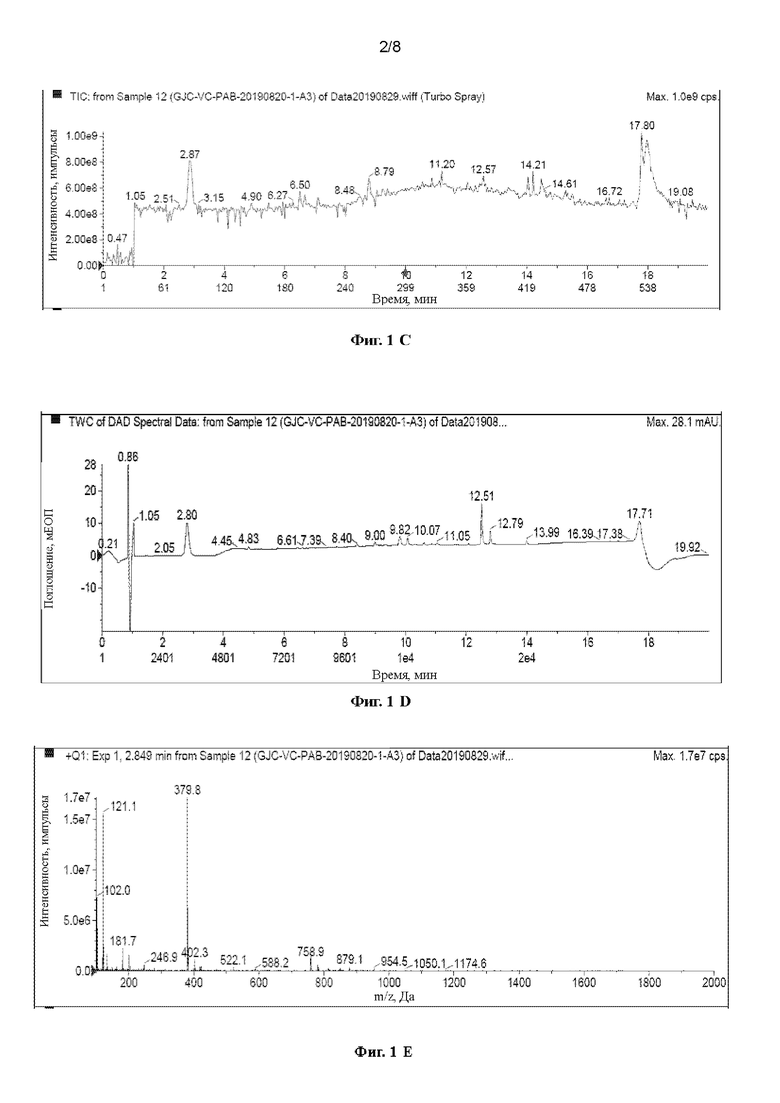
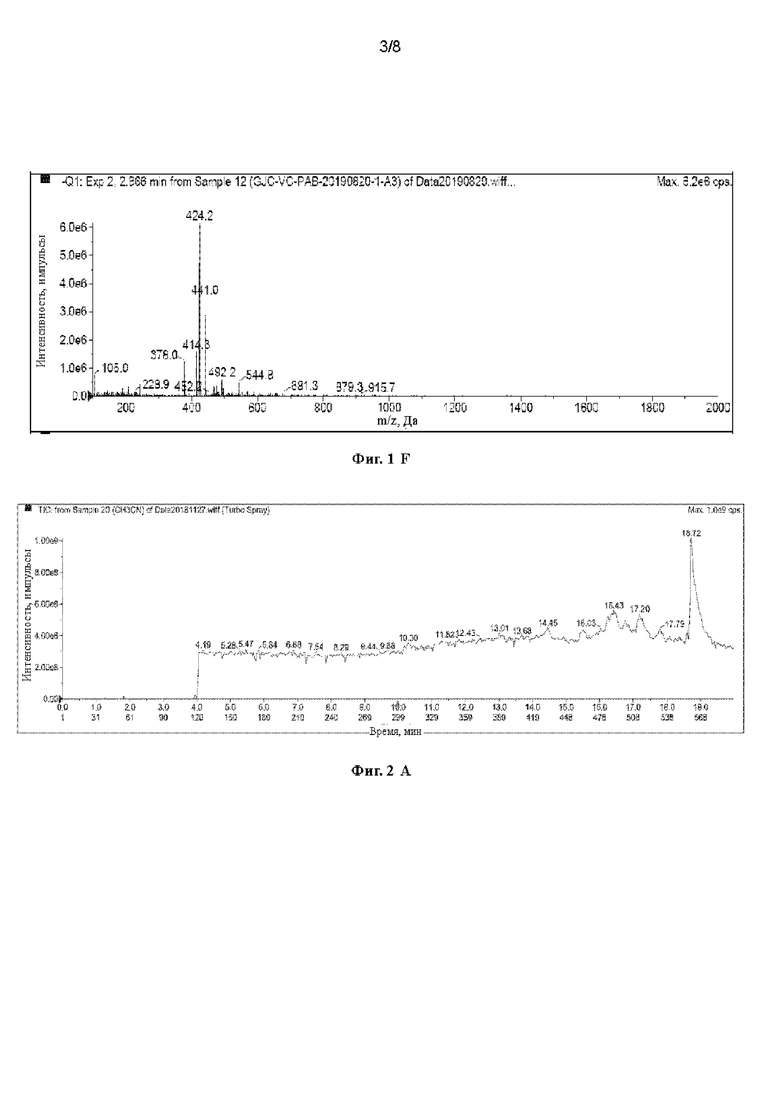
[40] На Фигурах с Фиг. 1-А по Фиг. 1-F представлены релевантные ЖХ-МС диаграммы Val-Cit-PAB, полученного способом с использованием защитной группы Теос, при этом Фиг. 1-А представляет собой масс-спектр пустого контроля, Фиг. 1-В представляет собой жидкофазный спектр пустого контроля, Фиг. 1-С представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Теос, Фиг. 1-D представляет собой жидкофазный спектр продукта, Фиг. 1-Е представляет собой масс-спектр продукта с регистрацией положительных ионов, и Фиг. 1-F представляет собой масс-спектр продукта с регистрацией отрицательных ионов.
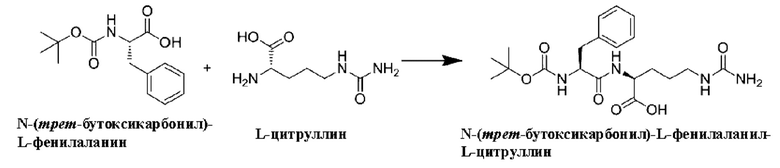
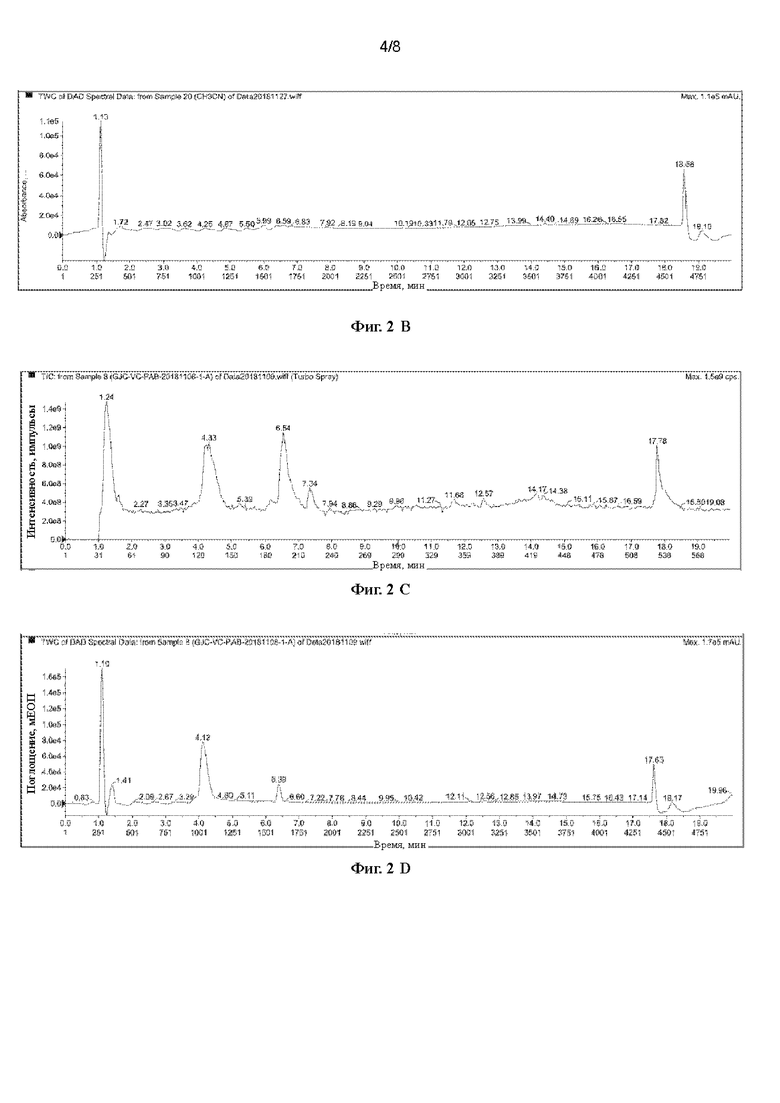
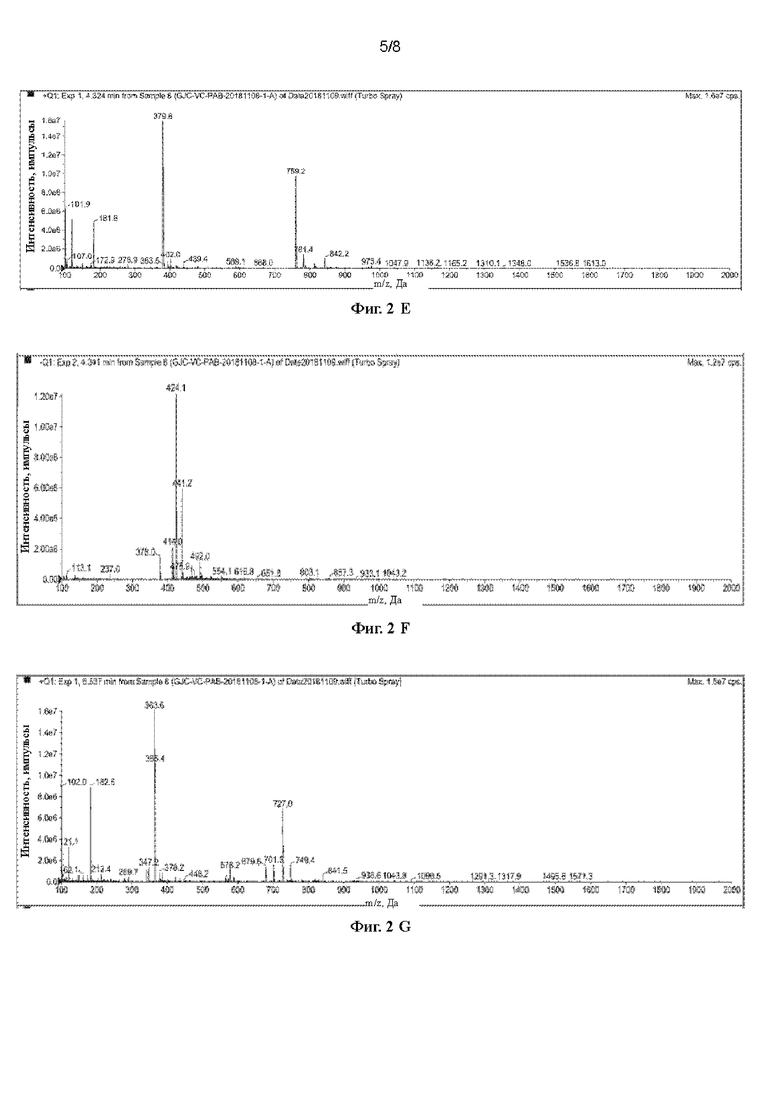
[41] На Фигурах с Фиг. 2-А по Фиг. 2-Н представлены релевантные ЖХ-МС диаграммы Val-Cit-PAB, полученного способом с использованием защитной группы Cbz, при этом Фиг. 2-А представляет собой масс-спектр пустого контроля, Фиг. 2-В представляет собой жидкофазный спектр пустого контроля, Фиг. 2-С представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Cbz, Фиг. 2-D представляет собой жидкофазный спектр продукта, Фиг. 2-Е представляет собой масс-спектр продукта с регистрацией положительных ионов, Фиг. 2-F представляет собой масс-спектр продукта с регистрацией отрицательных ионов, Фиг. 2-G представляет собой ионный масс-спектр примеси, и Фиг. 2-Н представляет собой масс-спектр примеси с регистрацией отрицательных ионов.
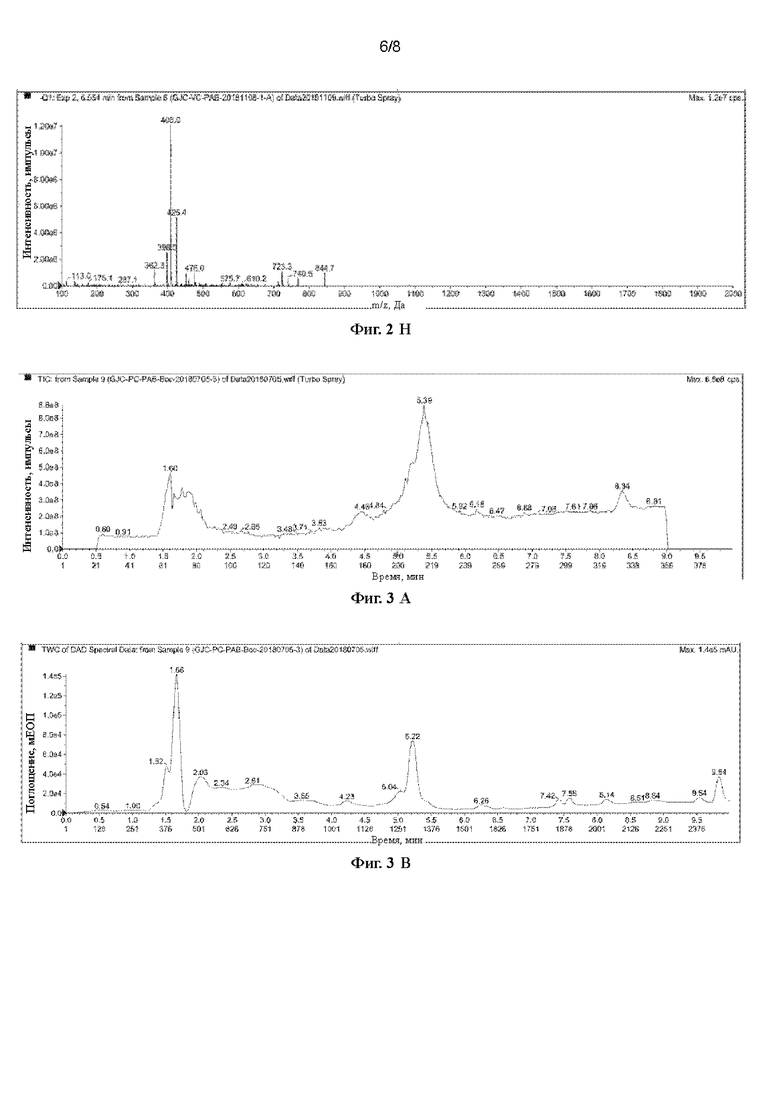
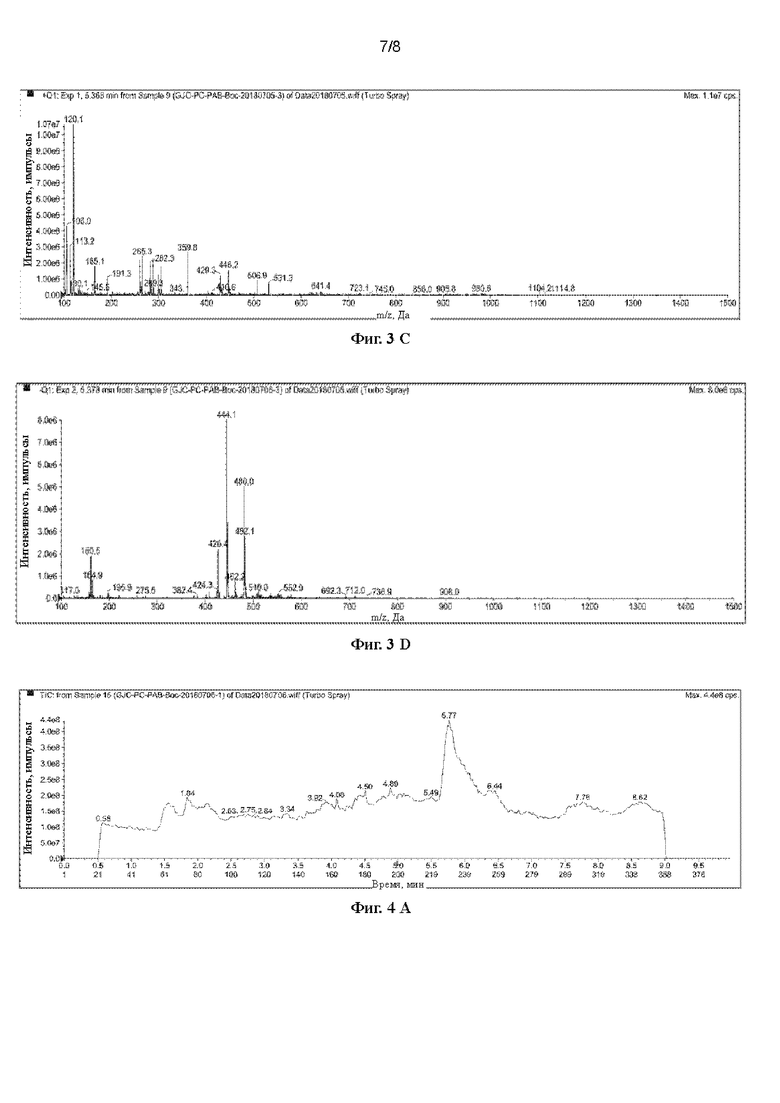
[42] На Фигурах с Фиг. 3-А по Фиг. 3-D представлены релевантные ЖХ-МС диаграммы Val-Cit-PAB, полученного способом с использованием защитной группы Вос (способ удаления защитной группы с использованием соляной кислоты), при этом Фиг. 3-А представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Вос, Фиг. 3-В представляет собой жидкофазный спектр продукта, Фиг. 3-С представляет собой спектр с регистрацией положительных ионов, и Фиг. 3-D представляет собой спектр с регистрацией отрицательных ионов.
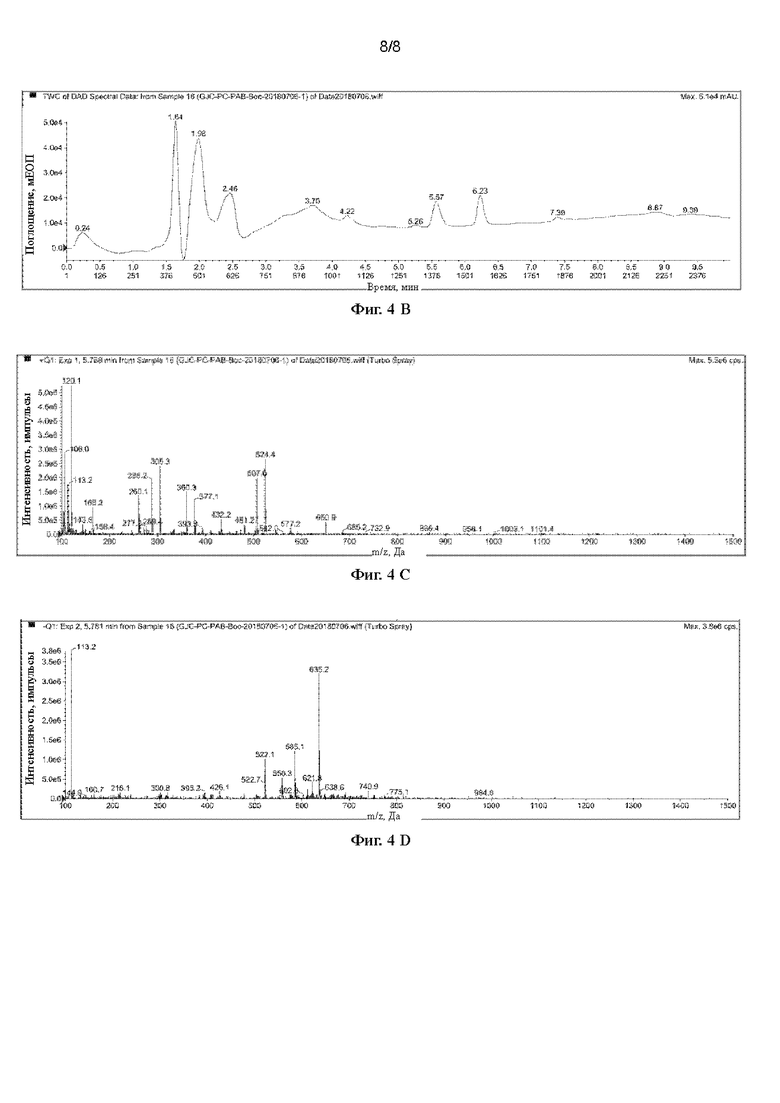
[43] На Фигурах с Фиг. 4-А по Фиг. 4-D представлены релевантные ЖХ-МС диаграммы Val-Cit-PAB, полученного способом с использованием защитной группы Вос (способ удаления защитной группы с использованием трифторуксусной кислоты), при этом Фиг. 4-А представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Вос, Фиг. 4-В представляет собой жидкофазный спектр продукта, Фиг. 4-С представляет собой спектр с регистрацией положительных ионов, и Фиг. 4-D представляет собой спектр с регистрацией отрицательных ионов.
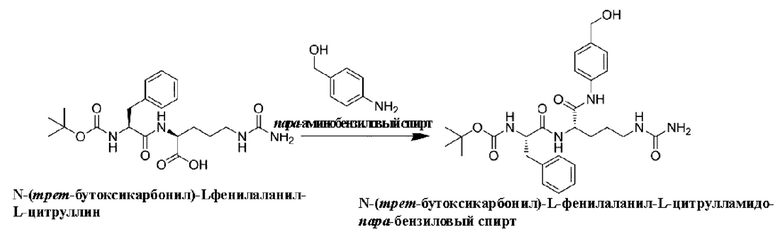
Подробное описание изобретения
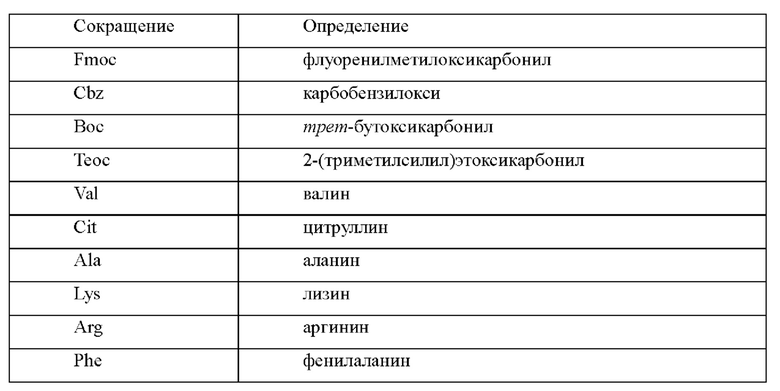
Сокращения
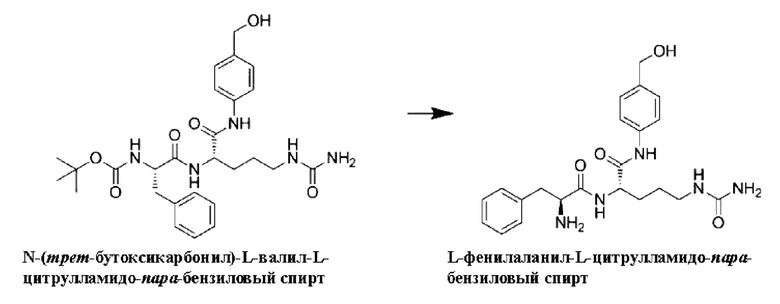
[44] Если не указано иное, все сокращения, использованные в настоящем изобретении, имеют значения, понятные специалистам в данной области. Использованные в настоящем изобретении общепринятые сокращения и их определения следующие:
Определения
[45] Различные термины, относящиеся к различным аспектам описания изобретения, использованы везде в описании и формуле изобретения. Если не указано иное, такие термины даны в их обычном значении в данной области. Другие конкретно определенные термины следует понимать в соответствии с определениями, приведенными в данном описании изобретения.
[46] В данном описании изобретения термины в единственном числе использованы в соответствии со стандартной практикой и означают один или более, если контекст не указывает иное. Так, например, ссылка на "конъюгат антитело-лекарственное средство" включает в себя комбинацию двух или более конъюгатов антитело-лекарственное средство и т.п.
[47] Следует понимать, что везде, где аспект описан в настоящем документе со словом "содержащий", он также предусматривает аналогичные аспекты, описанные выражениями "состоящий из" и "по существу состоящий из".
[48] Хотя числовые диапазоны и приблизительные параметры, представленные в широком объеме настоящего изобретения, числовые значения, указанные в конкретных примерах, описаны настолько точно, насколько это возможно. Тем не менее, любое числовое значение безусловно должно содержать некоторую погрешность, которая вызвана стандартным отклонением, которое имеет место при соответственных измерениях. Кроме того, понятно, что все диапазоны, раскрытые в данном документе, охватывают любой и все субдиапазоны, входящие в них. Например, указанный диапазон "от 1 до 10" следует рассматривать как охватывающий любой и все субдиапазоны между минимум 1 и максимум 10 (включительно); то есть все субдиапазоны, начинающиеся с минимум 1 или больше, такие как от 1 до 6,1, и субдиапазоны, заканчивающиеся максимум 10 или меньше, такие как от 5,5 до 10. Кроме того, любую ссылку, упомянутую как "включенную в данный документ", следует понимать как включенную во всей ее полноте.
[49] В настоящем изобретении 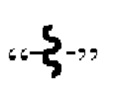 означает, что группа, содержащая
означает, что группа, содержащая 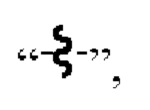 связана с другой группой через химическую связь в этом месте.
связана с другой группой через химическую связь в этом месте.
[50] Термин "линкерная группа", использованный в настоящем изобретении, относится к бифункциональной или многофункциональной молекуле, которая может взаимодействовать соответственно с молекулой белка/антитела и дипептидным линкером, и поэтому функционирует в качестве "мостика" для связывания белка/антитела с дипептидным линкером.
[51] Термин "олигопептидный линкер", использованный в настоящем изобретении относится, как правило, к связывающей структуре, содержащей два или более аминокислотных остатков, которая дополнительно содержит остаток пара-бензилового спирта. Две аминокислоты связываются вместе в результате реакции конденсации с дегидратацией. Аминокислоты, упомянутые в данном документе, обычно относятся к тем органическим соединениям, которые содержат как амино, так и карбоксильные группы, включая все незаменимые аминокислоты и не являющиеся незаменимыми аминокислоты. Предпочтительно, аминокислота включает, без ограничения, -валин- (-Val-), -цитруллин- (-Cit-), -аланин- (-Ala-), -лизин- (-Lys-), -лизин(тритил)- (-Lys(Trt)-), -лизин(монометокситритил)- (-Lys(Mmt)-), -лизин(флуоренилметилоксикарбонил)- (-Lys(Fmoc)-), -аргинин- (-Arg-), -фенилаланин- (-Phe-), -глицин- (-Gly-), -лейцин- (-Leu-) и -изолейцин- (-Ile-).
Конкретные примеры
[52] Ниже настоящее изобретение дополнительно описано в сочетании с конкретными примерами. Следует иметь в виду, что эти примеры использованы только для иллюстрации настоящего изобретения и не ограничивают объем настоящего изобретения. Экспериментальные методы без конкретных условий в нижеследующих примерах осуществляют, как правило, в стандартных условиях или в условиях, рекомендованных производителем. Реагенты без конкретных источников являются стандартными реагентами, продаваемыми на рынке. Если не указано иное, все проценты, соотношения, пропорции или части являются массовыми.
[53] Единицы в масс-объемных процентах в настоящем изобретении известны специалистам в данной области и, например, относятся к массе растворенного вещества в 100 мл раствора.
[54] Если не дано иного определения, все специальности и науки в данном документе использованы в том смысле, в котором они знакомы специалистам в данной области. Кроме того, любой способ или любое вещество, подобный(ое) или равный(ое) описанному, может быть использован в способе по настоящему изобретению. Предпочтительные воплощения и вещества, описанные в данном документе, приведены только в целях иллюстрации.
[55] Пример 1. Получение Val-Cit-PAB способом с использованием защитной группы Теос
[56] (1) Получение Teoc-Val-Cit
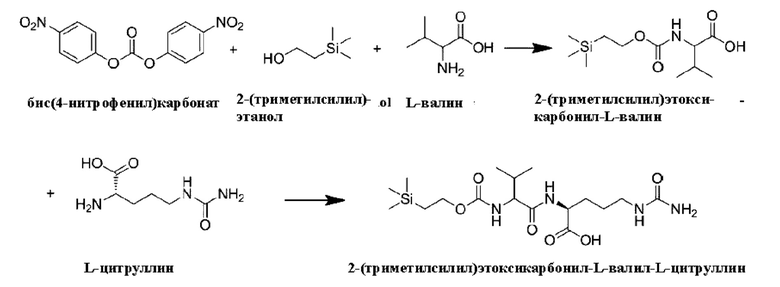
[57] 14,16 г 2-(триметилсилил)этанола (120 ммоль), 38,7 г N,N-диизопропилэтиламина (300 ммоль), 30,4 г бис(4-нитрофенил)карбоната (100 ммоль) и 400 мл ацетонитрила последовательно добавляли в одногорлую колбу вместимостью 500 мл. После добавления смесь перемешивали при комнатной температуре в течение 16 ч с образованием реакционного раствора 1. 14,08 г L-валина (120 ммоль) и 25,8 г N,N-диизопропилэтиламина (200 ммоль) добавляли в одногорлую колбу вместимостью 1 л и растворяли в 400 мл воды с образованием реакционного раствора 2. Реакционный раствор 1 добавляли к реакционному раствору 2 при перемешивании, и после перемешивания при комнатной температуре в течение 16 h, завершение реакции определяли методом ЖХ-МС. После подтверждения завершения реакции реакционный раствор выпаривали на роторном испарителе для удаления растворителя. После выпаривания добавляли 500 мл воды и добавляли по каплям 1 моль/л соляной кислоты при перемешивании до установления рН1. Затем водную фазу экстрагировали дважды по 300 мл этилацетата каждый раз.
Экстракты объединяли и промывали дважды по 300 мл воды каждый раз. После промывки водой экстракт сушили путем добавления безводного сульфата магния, и растворитель выпаривали на роторном испарителе с получением 54 г бледно-желтого вязкого твердого продукта (т.е. 2-(триметилсилил)этоксикарбонил-L-валильной смеси, содержащей 4-нитрофенол (просто упоминаемый как 2-(триметилсилил)этоксикарбонил-L-валильная смесь)). ЖХ-МС (М-Н)-: 260,1. 2-(Триметилсилил)этоксикарбонил-L-валильную смесь использовали на следующей стадии без очистки.
[58] 10,5 г 2-(триметилсилил)этоксикарбонил-L-валильной смеси (40 ммоль), полученной выше, 24,3 г N,N,N',N'-тетраметил-O-(N-сукцинимид)мочевины тетрафторбората (80 ммоль), 25,8 г N,N-диизопропилэтиламина (200 ммоль) и 200 мл N,N-диметилформамида последовательно добавляли в одногорлую колбу вместимостью 500 мл. После добавления смесь перемешивали при комнатной температуре в течение 6 ч с образованием реакционного раствора 3. 21 г L-цитруллина (120 ммоль) добавляли в одногорлую колбу вместимостью 1 л и полностью растворяли при перемешивании с добавлением 400 мл воды с образованием реакционного раствора 4. 15,5 г N,N-диизопропилэтиламина (120 ммоль) и 200 мл N,N-диметилформамида добавляли в мерный стакан на 500 мл с образованием реакционного раствора 5. Реакционный раствор 5 добавляли к реакционному раствору 4, затем переносили в низкотемпературную баню и перемешивали при -10°С в течение 0,5 ч. Затем реакционный раствор 3 медленно добавляли по каплям в вышеуказанную реакционную систему при перемешивании при -10°С, и контролировали, чтобы добавление закончилось примерно через 2 ч. После завершения добавления смесь выдерживали при -10°С в течение 16 ч при перемешивании. Затем завершение реакции определяли методом ЖХ-МС. После подтверждения завершения реакции систему выпаривали на роторном испарителе для удаления растворителя, и после выпаривания на роторном испарителе добавляли 500 мл 0,1 моль/л соляной кислоты и хорошо перемешивали. Водную фазу экстрагировали дважды по 300 мл этилацетата каждый раз. Экстракты объединяли и промывали дважды по 300 мл 0,1 моль/л соляной кислоты каждый раз и затем промывали дважды 300 мл воды. После промывки водой экстракт сушили путем добавления безводного сульфата магния, и растворитель выпаривали на роторном испарителе, после чего добавляли 100 мл этилацетата и 1000 мл толуола при перемешивании в течение 16 ч и затем фильтровали. Осадок на фильтре растворяли в 50 мл THF и добавляли 1 л смеси метил-трет-бутиловый эфир:н-гексан = 1:1 с получением раствора. Смесь перемешивали при комнатной температуре в течение 16 часов и затем фильтровали. Осадок на фильтре растворяли в 50 мл THF, после чего добавляли 1 л толуола. После перемешивания при комнатной температуре в течение 6 ч смесь фильтровали, и осадок на фильтре сушили с получением 11,1 г не совсем белого порошкообразного твердого продукта с выходом 68%. ЖХ-МС(М+Н)+: 418,7, ЖХ-МС(М-Н)-: 416,7.
[59] (2) Получение Теос-Val-Cit-PAB
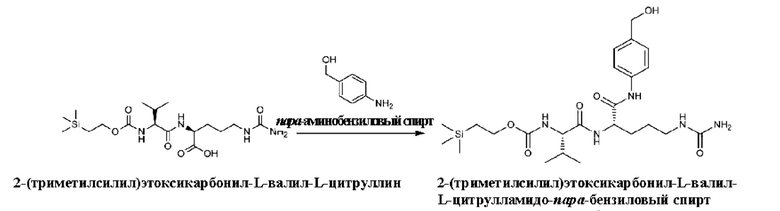
[60] 7,5 г 2-(триметилсилил)этоксикарбонил-L-валил-L-цитруллина (18 ммоль), 9 г N-этоксиацил-2-этокси-1,2-дигидрохинолина (36 ммоль), 4,5 г пара-аминобензилового спирта (36 ммоль), 150 мл дихлорметана и 75 мл метанола последовательно добавляли в одногорлую колбу вместимостью 500 мл. После добавления смесь перемешивали при комнатной температуре в течение 16 ч. Затем завершение реакции определяли методом ЖХ-МС. После подтверждения завершения реакционный раствор сушили на роторном испарителе и после выпаривания добавляли 200 мл дихлорметана и 200 мл этилацетата и перемешивали в течение 16 ч и затем фильтровали. К осадку на фильтре добавляли 100 мл дихлорметана, 100 мл этилацетата и 200 мл н-гексана при перемешивании в течение 16 ч и затем фильтровали. Осадок на фильтре сушили с получением 7,5 г не совсем белого порошкообразного твердого продукта (этим продуктом был 2-(триметилсилил)этоксикарбонил-L-валил-L-цитрулламидо-пара-бензиловый спирт, а именно Teoc-Val-Cit-PAB) с выходом 80%. ЖХ-МС(М+Н)+: 523,5, ЖХ-МС(М-Н)-: 522.
[61] (3) Получение Val-Cit-PAB (относительная молекулярная масса: 379,46)
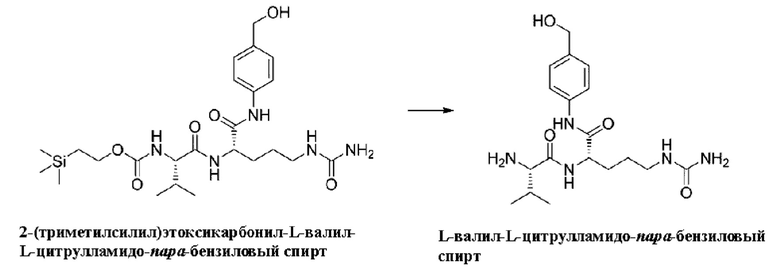
[62] 3,15 г 2-(триметилсилил)этоксикарбонил-L-валил-L-цитрулламидо-пара-бензилового спирта (6,2 ммоль), 2,16 г фторида калия (37,2 ммоль), 12,4 мл 1,0 моль/л раствора тетрабутиламмонийфторида в тетрагидрофуране и 100 мл N,N-диметилформамида последовательно добавляли в одногорлую колбу вместимостью 500 мл. После добавления смесь перемешивали при комнатной температуре в течение 24 ч и затем завершение реакции определяли методом ЖХ-МС. После завершения реакции реакционный раствор фильтровали. После выпаривания растворителя на ротоном испарителе из фильтрата 100 мл безводного этанола добавляли до полного растворения остатка. Добавляли 18 мл 1 моль/л разбавленного раствора соляной кислоты и хорошо перемешивали, после чего смесь сушили путем добавления безводного сульфата магния и затем фильтровали. Фильтрат сушили выпариванием на роторном испарителе, и затем добавляли 30 мл этанола до полного растворения остатка. При перемешивании при комнатной температуре добавляли смешанный раствор 200 мл метил-трет-бутилового эфира и 200 мл дихлорметана и перемешивали в течение 1 ч, после чего фильтровали. Осадок на фильтре сушили с получением 1,83 г не совсем белого порошкообразного твердого продукта (этим продуктом был L-валил-L-цитрулламидо-пара-бензиловый спирт, т.е. Val-Cit-PAB) с выходом 80%.
[63] При регистрации ЖХ-МС Фиг. 1-А представляет собой масс-спектр пустого контроля, Фиг. 1-В представляет собой жидкофазный спектр пустого контроля, Фиг. 1-С представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Теос (продукт в этом примере), Фиг. 1-D представляет собой жидкофазный спектр продукта, Фиг. 1-Е представляет собой масс-спектр продукта с регистрацией положительных ионов, и Фиг. 1-F представляет собой масс-спектр продукта с регистрацией отрицательных ионов. Сравнивая Фиг. 1-А с Фиг. 1-С, можно видеть, что пик в точке времени 2,87 мин является ионным пиком продукта. Сравнивая Фиг. 1-В с Фиг. 1-D, можно видеть, что продукт имеет сильный ВЭЖХ сигнал, и в сочетании с Фиг. 1-А и Фиг. 1-С можно видеть, что Val-Cit-PAB, полученный способом с использованием защитной группы Теос, имеет меньше примесей. Анализируя спектр с регистрацией положительных ионов (2,849 мин) и спектр с регистрацией отрицательных ионов (2,866 мин) при 2,87 мин, можно узнать, что: ЖХ-МС(М+Н)+: 379,6, ЖХ-МС(М-Н)-: 378,0.
[64] Пример 2. Получение Val-Cit-PAB способом с использованием защитной группы Cbz
[65] (1) Получение Cbz-Val-Cit
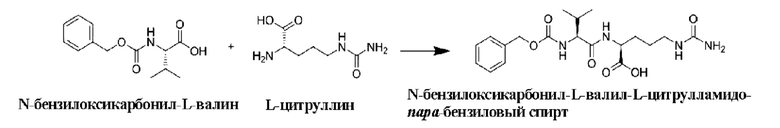
[66] 10 г N-бензилоксикарбонил-L-валина (т.е. Cbz-Val) (40 ммоль), 18,2 г N,N,N',N'-тетраметил-O-(N-сукцинимид)мочевины тетрафторбората (60 ммоль), 15,5 г N,N-диизопропилэтиламина (120 ммоль) и 300 мл ацетонитрила последовательно добавляли в одногорлую колбу вместимостью 1 л. После добавления смесь перемешивали при комнатной температуре в течение 4 ч. После растворения 7,7 г L-цитруллина (44 ммоль) в 300 мл воды, смесь добавляли в вышеуказанную реакционную систему и перемешивали при комнатной температуре в течение 16 ч. Отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали. При перемешивании добавляли по каплям 0,1 моль/л соляную кислоту до достижения рН 1. После сушки выпариванием растворителя на роторном испарителе добавляли 800 мл воды, перемешивали при комнатной температуре в течение 16 ч и фильтровали. После сушки осадка на фильтре в вакууме добавляли 400 мл этилацетата и 400 мл дихлорметана и перемешивали в течение 16 ч, после чего фильтровали. После сушки осадка на фильтре в вакууме добавляли 200 мл этилацетата, 200 мл дихлорметана и 400 мл н-гексана, и смесь перемешивали в течение 16 часов и затем фильтровали. Осадок на фильтре сушили с получением 12,65 г не совсем белого порошкообразного твердого продукта (т.е. N-бензилоксикарбонил-L-валил-L-цитруллина) с выходом 77%. ЖХ-МС(М+Н)+: 408,7, ЖХ-МС(М-Н)-: 406,9.
[67] (2) Получение Cbz-Val-Cit-PAB
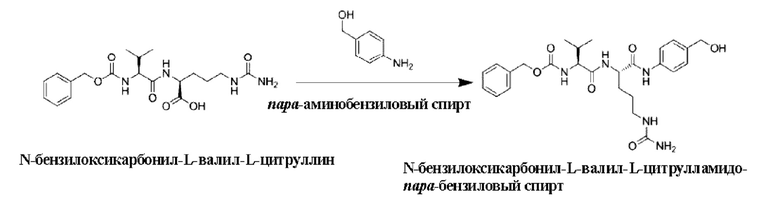
[68] 12,65 г N-бензилоксикарбонил-L-валил-L-цитруллина (31 ммоль), 15,74 г N-этоксиацил-2-этокси-1,2-дигидрохинолина (62 ммоль), 7,53 г пара-аминобензилового спирта (62 ммоль), 200 мл дихлорметана и 100 мл метанола последовательно добавляли в одногорлую колбу вместимостью 500 мл. После перемешивания при комнатной температуре в течение 16 ч отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали. После сушки реакционного раствора выпариванием на роторном испарителе добавляли 200 мл дихлорметана и 200 мл этилацетата, перемешивали в течение 16 ч и фильтровали. К осадку на фильтре добавляли 100 мл дихлорметана, 100 мл этилацетата и 200 мл н-гексана, перемешивали в течение 16 ч и фильтровали. Осадок на фильтре сушили с получением 9,1 г не совсем белого порошкообразного твердого продукта с выходом 60%. ЖХ-МС(М+Н)+: 513,6.
[69] (3) Получение Val-Cit-PAB (относительная молекулярная масса: 379,46)
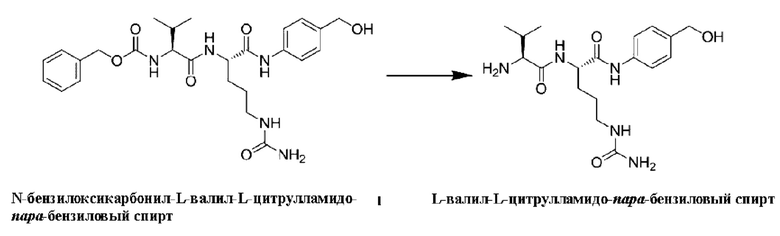
[70] 1,12 г N-бензилоксикарбонил-L-валил-L-цитрулламидо-пара-бензилового спирта (2 ммоль), 0,3 г палладия на углероде и 100 мл метанол последовательно добавляли в одногорлую колбу вместимостью 250 мл и перемешивали при 10°С. Реакционную систему подключали к баллону, заполненному газом водородом, и смесь перемешивали при 10°С в течение 16 ч. Отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали. Реакционный раствор фильтровали, и фильтрат сушили выпариванием на роторном испарителе. Добавляли 400 мл н-гексана, перемешивали в течение 16 ч и затем фильтровали. Осадок на фильтре сушили с получением 0,69 г не совсем белого порошкообразного твердого продукта с выходом 91%.
[71] При регистрации ЖХ-МС Фиг 2-А представляет собой масс-спектр пустого контроля, Фиг. 2-В представляет собой жидкофазный спектр пустого контроля, Фиг 2-С представляет собой масс-спектр Val-Cit-PAB, полученного способом с использованием защитной группы Cbz (продукт в этом примере), Фиг 2-D представляет собой жидкофазный спектр продукта, Фиг. 2-Е представляет собой масс-спектр продукта с регистрацией положительных ионов, Фиг 2-F представляет собой масс-спектр продукта с регистрацией отрицательных ионов, Фиг 2-G представляет собой ионный масс-спектр примеси, и Фиг. 2-Н представляет собой масс-спектр примеси с регистрацией отрицательных ионов. Сравнивая Фиг 2-А с Фиг 2-С, можно видеть, что имеется два более сильных ионных пика: при 4,33 мин и при 6,54 мин. Сравнивая Фиг 2-В и 2-D, можно видеть, что имеется два пика с сильными ВЭЖХ сигналами при 4,12 мин и при 5,39 мин, и в сочетании с Фиг 2-А и Фиг 2-С, можно сделать вывод, что один из двух пиков является пиком продукта, и один из двух пиков является пиком примесей, и содержание примесей выше. Анализируя масс-спектр с регистрацией положительных ионов и масс-спектр с регистрацией отрицательных ионов при 4,33 мин и при 5,54 мин, можно обнаружить, что пик при 4,33 мин является пиком продукта (ЖХ-МС(М+Н)+: 379,6, ЖХ-МС(М-Н)-: 378,0), а пик при 5,54 мин является пиком примеси (ЖХ-МС(М+Н)+: 363,6), которая предположительно является побочным продуктом дегидроксилирования Val-Cit-PAB (с разницей в молекулярной массе лишь 16), и его содержание выше. Поскольку химические свойства побочного продукта очень схожи со свойствами продукта, этот побочный продукт очень трудно удалить, и это сильно сказывается на последующих реакциях и качестве продукта. Кроме того, в процессе удаления защитной группы используется газ водород, что создает риск для возникновения угрозы безопасности.
[72] Пример 3. Получение Phe-Cit-PAB способом с использованием защитной группы Вос
[73] (1) Получение Boc-Phe-Cit
[74] 2,65 г N-(трет-бутоксикарбонил)-L-фенилаланина (10 ммоль) (т.е. Boc-Phe), 3,61 г N,N,N',N'-тетраметил-O-(N-сукцинимид)мочевины тетрафторбората (12 ммоль), 3,87 г N,N-диизопропилэтиламина (30 ммоль) и 50 мл N,N-диметилформамида последовательно добавляли в одногорлую колбу вместимостью 250 мл и перемешивали при комнатной температуре в течение 4 ч.
[75] 1,75 г L-цитруллина (10 ммоль) растворяли в 50 мл воды, и этот раствор добавляли в вышеуказанную реакционную систему и перемешивали при комнатной температуре в течение 16 ч. Отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали. При перемешивании раствор 0,1 моль/л соляной кислоты добавляли по каплям до достижения рН 1. После сушки системы выпариванием на роторном испарителе добавляли 200 мл воды, перемешивали при комнатной температуре в течение 16 ч и фильтровали. После сушки осадка на фильтре добавляли 100 мл этилацетата и 100 мл дихлорметана, перемешивали в течение 16 ч и фильтровали. Осадок на фильтре сушили с получением 3 г желтого порошкообразного твердого продукта (то есть N-(трет-бутоксикарбонил)-L-фенилаланил-L-цитруллина, Boc-Phe-Cit) с выходом 71%. ЖХ-МС(М-Н)-: 421,4.
[76] (2) Получение Boc-Phe-Cit-PAB
[77] 3 г N-(трет-бутоксикарбонил)-L-фенилаланил-L-цитруллина (7,1 ммоль) (т.е. Boc-Phe-Cit), 3,5 г N-этоксиацил-2-этокси-1,2-дигидрохинолина (14,2 ммоль), 1,75 г пара-аминобензилового спирта (14,2 ммоль), 60 мл дихлорметана и 30 мл метанола последовательно добавляли в одногорлую колбу вместимостью 250 мл и перемешивали при комнатной температуре в течение 16 ч. Затем отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали. После сушки реакционного раствора выпариванием на роторном испарителе добавляли 100 мл дихлорметана и 100 мл этилацетата, перемешивали в течение 16 ч и центрифугировали. 100 мл н-гексана добавляли к белому твердому веществе в нижнем слое, перемешивали в течение 2 ч и центрифугировали. Белое твердое вещество в нижнем слое сушили с получением 2,3 г не совсем белого порошкообразного твердого продукта (то есть N-(трет-бутоксикарбонил)-L-фенилаланил-L-цитрулламидо-пара-бензилового спирта, Boc-Phe-Cit-PAB) с выходом 62%. ЖХ-МС(М-Н)-: 526,2.
[78] (3) Получение Phe-Cit-PAB (относительная молекулярная масса: 427,51)
[79] Защитную группу Вос удаляли способом с использованием соляной кислоты и способом с использованием трифторуксусной кислоты, соответственно, как показано далее.
[80] (а) Способ удаления защитной группы с использованием соляной кислоты
[81] 0,5 г N-(трет-бутоксикарбонил)-L-фенилаланил-L-цитрулламидо-пара-бензилового спирта (1 ммоль) (т.е. Boc-Phe-Cit-PAB), 3 мл 4 моль/л раствора хлористого водорода в диоксане и 3 мл диоксана последовательно добавляли в одногорлую колбу на 10 мл. После добавления смесь перемешивали при комнатной температуре в течение 3 ч. Отбирали образцы для регистрации ЖХ-МС, и затем реакцию завершали.
[82] При регистрации ЖХ-МС Фиг. 3-А представляет собой масс-спектр Phe-Cit-РАВ, полученного способом с использованием защитной группы Вос (продукт в этом примере), Фиг. 3-В представляет собой жидкофазный спектр продукта, Фиг. 3-С представляет собой спектр с регистрацией положительных ионов, и Фиг. 3-D представляет собой спектр с регистрацией отрицательных ионов. Из Фиг. 3-А можно видеть, что имеется сильный ионный пик при 5,39 мин. Анализируя масс-спектр с регистрацией положительных ионов (5,390 мин) и масс-спектр с регистрацией отрицательных ионов (5,403 мин) в этом положении было получено значение ЖХ-МС (М+Н)+: 446,2, ЖХ-МС (М-Н)-: 444,1. Можно сделать предположение, что большинство продуктов, полученных этим способом, являются побочными продуктами, в которых гидроксильная группа в положении бензила замещена хлором.
[83] (б) Способ удаления защитной группы с использованием трифторуксусной кислоты
[84] 0,5 г N-(трет-бутоксикарбонил)-L-фенилаланил-L-цитрулламидо-пара-бензилового спирта (1 ммоль) и 3 мл трифторуксусной кислоты последовательно добавляли в одногорлую колбу на 10 мл. После добавления смесь перемешивали при комнатной температуре в течение 2 ч, и завершение реакции определяли методом ЖХ-МС.
[85] При регистрации ЖХ-МС Фиг. 4-А представляет собой масс-спектр Phe-Cit-РАВ, полученного способом с использованием защитной группы Вос (продукт в этом примере), Фиг. 4-В представляет собой жидкофазный спектр продукта, Фиг. 4-С представляет собой спектр с регистрацией положительных ионов, и Фиг. 4-D представляет собой спектр с регистрацией отрицательных ионов. Можно видеть из Фиг. 4-А, что имеется сильный ионный пик при 5,77 мин. Анализируя масс-спектр с регистрацией положительных ионов (5,768 мин) и масс-спектр с регистрацией отрицательных ионов (5,781 мин) в этой позиции, было получено значение ЖХ-МС (М+Н)+: 524,4, ЖХ-МС (М-Н)-: 522,1. Предполагается, что большинство продуктов, полученных этим способом, являются побочными продуктами, в которых гидроксильная группа в положении бензила образует трифторацетат.
[86] В итоге, способ получения Val-Cit-PAB с использованием Теос в качестве защитной группы, предложенный согласно настоящему изобретению, легко осуществляется в мягких реакционных условиях, и поскольку почти не происходит побочных реакций в этой реакции, этот способ дает высокочистый продукт, который имеет меньше примесей и легко очищается. Способ получения Val-Cit-PAB с использованием Cbz в качестве защитной группы дает большое количество побочных продуктов дегидроксилирования. Поскольку химические свойства этих побочных продуктов очень схожи со свойствами продуктов, их трудно удалять, что сильно сказывается на последующих реакциях и качестве продукта. Кроме того, в процессе удаления защитной группы используют газ водород в качестве реагента, что создает риск возникновения угрозы безопасности. Способ получения Val-Cit-PAB с использованием Вос в качестве защитной группы имеет проблемы в том, что гидроксильная группа в положении бензила замещена хлором и образует трифторацетат с высокой степенью конверсии. Следовательно, по сравнению с другими двумя способами способ с использованием Теос, предложенный согласно настоящему изобретению, легче осуществляется и дает немного примесей, что обеспечивает достижение неожиданного технического результата.
[87] Изобретение было проиллюстрировано различными конкретными воплощениями. Однако специалисты в данной области могут понять, что настоящее изобретение не ограничено этими конкретными воплощениями. Специалисты в данной области могут делать различные изменения или модификации в рамках объема настоящего изобретения, и различные технические признаки, упомянутые в разных местах в данном описании изобретения, можно сочетать друг с другом, не отклоняясь от замысла и объема настоящего изобретения. Такие модификации и варианты также входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, НАГРУЖЕННЫЙ БИНАРНЫМИ ТОКСИНАМИ, И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2795148C2 |
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| ПЕПТИДНЫЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ С ПОВЫШЕННОЙ ЭФФЕКТИВНОСТЬЮ ПРОТИВ ИНСУЛИНОРЕЗИСТЕНТНОСТИ | 2012 |
|
RU2602801C2 |
| НОВОЕ СОЕДИНЕНИЕ С ЭФФЕКТАМИ ТРОМБОЛИЗИСА, АКЦЕПТИРОВАНИЯ СВОБОДНЫХ РАДИКАЛОВ И НАПРАВЛЕННОГО ДЕЙСТВИЯ НА ТРОМБ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2013 |
|
RU2604193C2 |
| КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ОСНОВЕ ЭРИБУЛИНА И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2754369C2 |
| АНТИ-МЕЗОТЕЛИН АНТИТЕЛО И ЕГО КОНЪЮГАТ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2019 |
|
RU2747995C1 |
| АГОНИСТЫ И АНТАГОНИСТЫ УРОТЕНЗИНА-II | 2001 |
|
RU2263679C2 |
| ЛИНКЕР ДЛЯ КОНЪЮГАТОВ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2792201C2 |
| АНТИТЕЛО ПРОТИВ КЛАУДИНА 18.2 И ЕГО КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2022 |
|
RU2814164C2 |
| ТРИ-, ТЕТРА-, ПЕНТА- И ПОЛИПЕПТИДЫ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИДЕПРЕССИВНОГО АГЕНТА | 1995 |
|
RU2182910C2 |
Изобретение относится к способу получения олигопептидного линкерного промежуточного соединения формул (1)-(3), где каждая из AA1, АА2, АА3 и АА4 независимо выбрана из группы, состоящей из следующих: -валин- (-Val-), -цитруллин- (-Cit-), -аланин- (-Ala-), -лизин- (-Lys-), -фенилаланин- (-Phe-), -глицин- (-Gly-), -лейцин- (-Leu-) и -изолейцин- (-Ile-). Способ включает: 1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 и затем аминокислотой АА2, или аминокислотой AA1, аминокислотой АА2 и аминокислотой АА3 последовательно, или аминокислотой AA1, аминокислотой АА2, аминокислотой АА3 и аминокислотой АА4 последовательно с получением 2-(триметилсилил)этоксикарбонил-олигопептидного продукта конденсации; 2) взаимодействие полученного 2-(триметилсилил)этоксикарбонил-олигопептидного продукта конденсации и пара-аминобензилового спирта с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-олигопептид-пара-аминобензиловый спирт. Реагент карбонилирования имеет структуру формулы (5), где каждый из R1 и R2 независимо выбран из группы указанных под формулой (5) значений. Предлагаемый способ осуществляется в мягких реакционных условиях и обеспечивает получение продукта высокой чистоты. Изобретение относится также к применению указанного способа для получения конъюгата антитело-лекарственное средство. 2 н. и 8 з.п. ф-лы, 4 ил., 1 табл., 3 пр.
 (1),
(1),
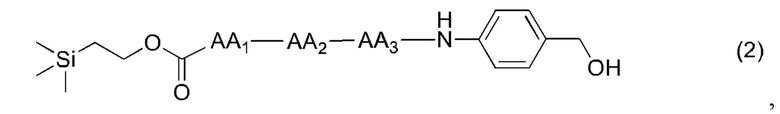 (2),
(2),
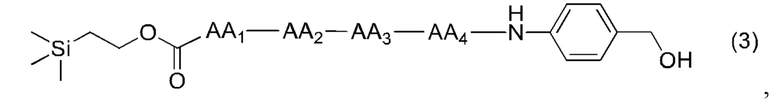 (3),
(3),
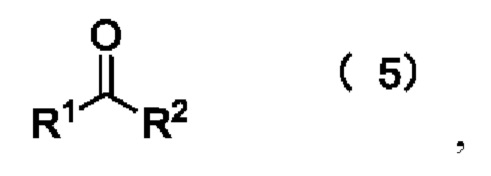
где R1 и R2 независимо выбраны из группы, состоящей из:

 ,
, .
.
1. Способ получения олигопептидного линкерного промежуточного соединения формул (1)-(3)
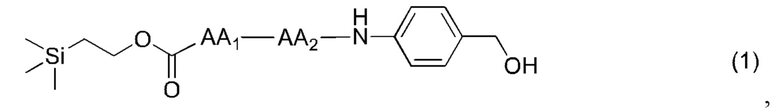
или

или
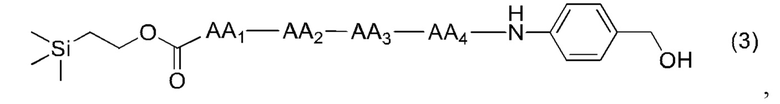
где каждая из AA1, АА2, АА3 и АА4 независимо выбрана из группы, состоящей из следующих: -валин- (-Val-), -цитруллин- (-Cit-), -аланин- (-Ala-), -лизин- (-Lys-), -фенилаланин- (-Phe-), -глицин- (-Gly-), -лейцин- (-Leu-) и -изолейцин- (-Ile-); включающий:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 и затем аминокислотой АА2, или аминокислотой AA1, аминокислотой АА2 и аминокислотой АА3 последовательно, или аминокислотой AA1, аминокислотой АА2, аминокислотой АА3 и аминокислотой АА4 последовательно с получением 2-(триметилсилил)этоксикарбонил-олигопептидного продукта конденсации;
2) взаимодействие полученного 2-(триметилсилил)этоксикарбонил-олигопептидного продукта конденсации и пара-аминобензилового спирта с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-олигопептид-пара-аминобензиловый спирт,
где реагент карбонилирования имеет структуру формулы (5)
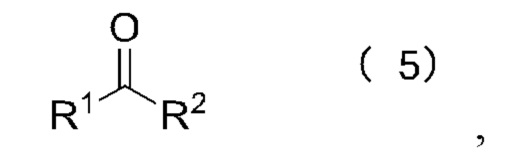
где каждый из R1 и R2 независимо выбран из группы, состоящей из
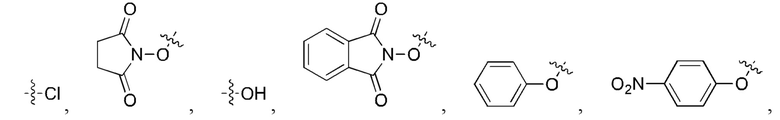
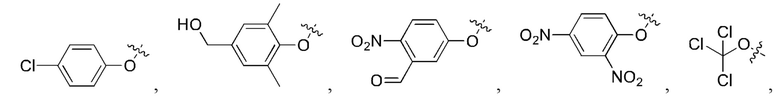
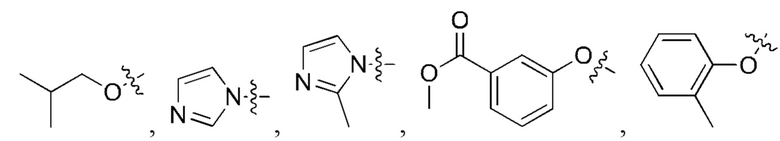 и
и 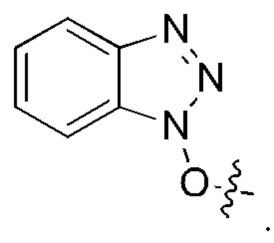
2. Способ по п. 1, где олигопептидное линкерное промежуточное соединение формулы (1) получают следующим реакционным путем:

включающий следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид; и
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид-пара-аминобензиловый спирт.
3. Способ по п. 1, где олигопептидное линкерное промежуточное соединение формулы (2) получают следующим реакционным путем:
 ,
,
включающий следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид;
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и аминокислотой АА3 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид; и
4) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид-пара-аминобензиловый спирт.
4. Способ по п. 1, где олигопептидное линкерное промежуточное соединение формулы (3) получают следующим реакционным путем:
включающий следующие стадии:
1) проведение реакции конденсации между реагентом карбонилирования, 2-(триметилсилил)этанолом и аминокислотой AA1 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-аминокислоту;
2) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-аминокислотой и аминокислотой АА2 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-дипептид;
3) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-дипептидом и аминокислотой АА3 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-трипептид;
4) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-трипептидом и аминокислотой АА4 с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид; и
5) проведение реакции конденсации между продуктом конденсации 2-(триметилсилил)этоксикарбонил-тетрапептидом и пара-аминобензиловым спиртом с получением продукта конденсации, представляющего собой 2-(триметилсилил)этоксикарбонил-тетрапептид-пара-аминобензиловый спирт.
5. Способ по п. 1, где реагент карбонилирования выбран из группы, состоящей из:
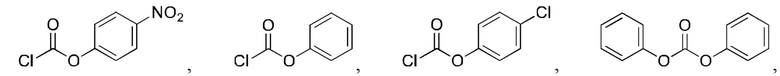
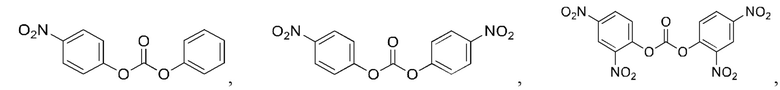
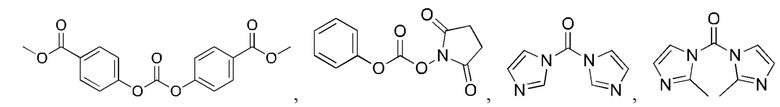 и
и
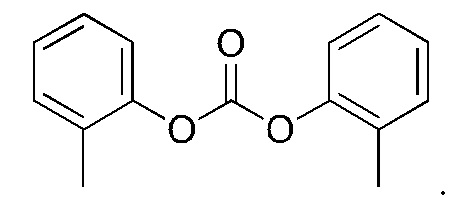
6. Способ по п. 1, где -АА1-АА2- выбран из следующих структур:
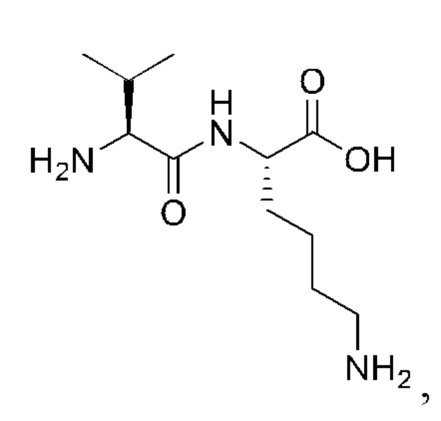
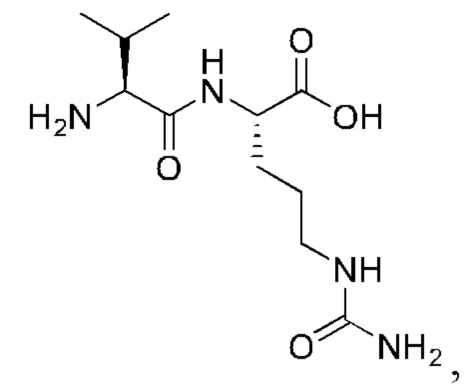
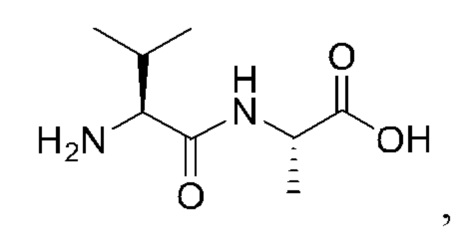

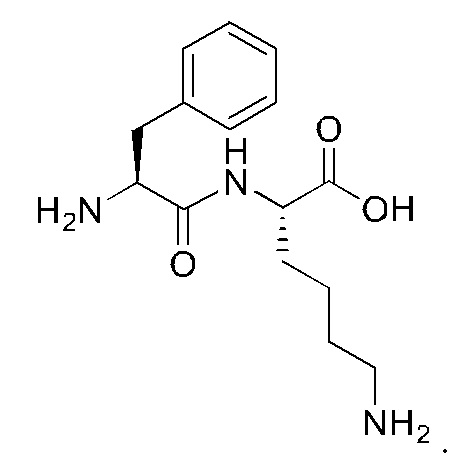
7. Способ по п. 1, где -АА1-АА2-АА3- представляет собой
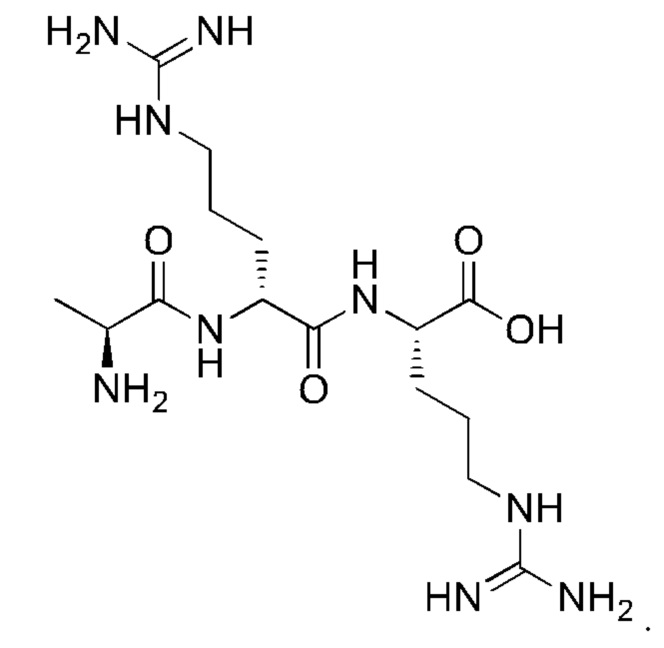
8. Способ по п. 1, где -АА1-АА2-АА3-АА4- выбран из следующих структур:
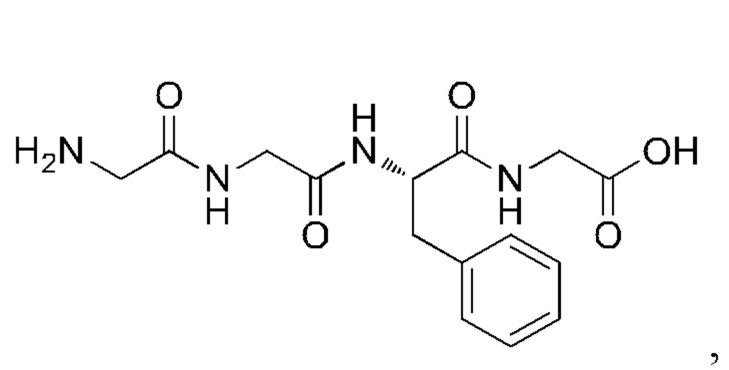
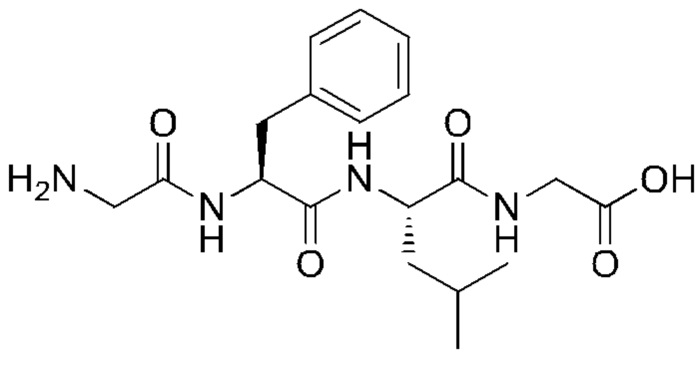 и
и
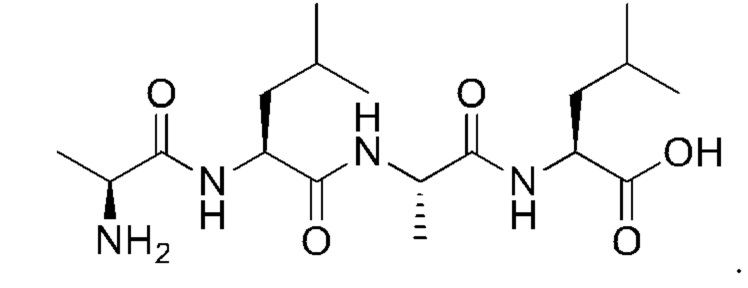
9. Способ по п. 1, где растворитель, использованный в реакции конденсации, выбран из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, DMF (диметилформамид), DMSO (диметилсульфоксид), воды, метанола, дихлорметана, хлороформа, четыреххлористого углерода и этанола.
10. Применение способа по любому из пп. 1-9 в получении конъюгата антитело-лекарственное средство.
| D | |||
| MONDAL ET AL | |||
| Improved Methodology for the Synthesis of a Cathepsin B Cleavable Dipeptide Linker, Widely Used in Antibody-Drug Conjugate Research, TETRAHEDRON LETTERS, 2018, Vol | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| Прибор для определения наивыгоднейших условий работы токарных и других станков | 1923 |
|
SU3594A1 |
| A | |||
| DAL CORSO ET AL | |||
| Protease-Cleavable Linkers Modulate the Anticancer Activity of Non-Internalizing Antibody-Drug Conjugates, BIOCONJUGATE | |||

