Настоящее изобретение относится к визуализации и эндорадиотерапии заболеваний, в которые вовлечен специфический мембранный антиген простаты (PSMA). Представлены соединения, которые связывают или ингибируют PSMA и, кроме того, имеют по меньшей мере один фрагмент, который чувствителен к введению радиоактивных меток. Также представлены медицинские применения таких соединений.
В настоящем описании процитирован ряд документов, в том числе заявки на получение патента и руководства производителей. Раскрытие этих документов, если оно не рассматривается как релевантное в отношении патентоспособности настоящего изобретения, в полном объеме включено в настоящий документ посредством ссылки. Более конкретно, все документы, на которые сделаны ссылки, включены посредством ссылки в такой же степени, как если бы каждый отдельный документ был конкретно и отдельно указан в качестве включенного посредством ссылки.
В течение последних десятилетий рак простаты (РСа) является наиболее распространенным злокачественным заболеванием у мужчин с высокой заболеваемостью и низким процентом выживаемости. Ввиду своей сверхэкспрессии при раке простаты (Silver, D.A., et al., Prostate-specific membrane antigen expression in normal and malignant human tissues. Clinical Cancer Research, 1997. 3(1): p.81-85), специфический мембранный антиген простаты (PSMA) или глутаматкарбоксипептидаза II (GCP II) доказал свою пригодность в качестве превосходной мишени для разработки высокочувствительных радиомеченых агентов для эндорадиотерапии и визуализации РСа (Afshar-Oromieh, A., et al., The diagnostic value of PET/CT imaging with the 68Ga-labelled PSMA ligand HBED-CC in the diagnosis of recurrent prostate cancer. European journal of nuclear medicine and molecular imaging, 2015. 42(2): p.197-209; 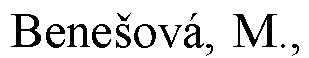 et al., Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. Journal of Nuclear Medicine, 2015. 56(6): p.914-920; Robu, S., et al., Preclinical evaluation and first patient application of 99mTc-PSMA-l&S for SPECT imaging and radioguided surgery in prostate cancer. Journal of Nuclear Medicine, 2016: p.jnumed. 116.178939; Weineisen, M., et al., Development and first in human evaluation of PSMA I&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine, 2014. 55(supplement 1): p.1083-1083; Rowe, S., et al., PET imaging of prostate-specific membrane antigen in prostate cancer: current state of the art and future challenges. Prostate cancer and prostatic diseases, 2016; Maurer, Т., et al., Current use of PSMA-PET in prostate cancer management. Nature Reviews Urology, 2016). Специфический мембранный антиген простаты представляет собой внеклеточную гидролазу, каталитический центр которой содержит два иона цинка(М) с мостиковым гидроксидным лигандом. Он сильно апрегулирован в метастатических и гормонально-рефрактерных карциномах простаты, однако было сообщено о его физиологической экспрессии также в почках, слюнных железах, тонком кишечнике, головном мозге и, в низкой степени, также в здоровой ткани простаты. В кишечнике PSMA способствует поглощению фолата за счет преобразования птероилполи-γ-глутамата в птероилглутамат (фолат). В головном мозгу он гидролизирует N-ацетил-L-аспартил-L-глутамат (NAAG) в N-ацетил-L-аспартат и глутамат. Ферментативная функция PSMA в здоровой и больной простате еще не была классифицирована.
et al., Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. Journal of Nuclear Medicine, 2015. 56(6): p.914-920; Robu, S., et al., Preclinical evaluation and first patient application of 99mTc-PSMA-l&S for SPECT imaging and radioguided surgery in prostate cancer. Journal of Nuclear Medicine, 2016: p.jnumed. 116.178939; Weineisen, M., et al., Development and first in human evaluation of PSMA I&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine, 2014. 55(supplement 1): p.1083-1083; Rowe, S., et al., PET imaging of prostate-specific membrane antigen in prostate cancer: current state of the art and future challenges. Prostate cancer and prostatic diseases, 2016; Maurer, Т., et al., Current use of PSMA-PET in prostate cancer management. Nature Reviews Urology, 2016). Специфический мембранный антиген простаты представляет собой внеклеточную гидролазу, каталитический центр которой содержит два иона цинка(М) с мостиковым гидроксидным лигандом. Он сильно апрегулирован в метастатических и гормонально-рефрактерных карциномах простаты, однако было сообщено о его физиологической экспрессии также в почках, слюнных железах, тонком кишечнике, головном мозге и, в низкой степени, также в здоровой ткани простаты. В кишечнике PSMA способствует поглощению фолата за счет преобразования птероилполи-γ-глутамата в птероилглутамат (фолат). В головном мозгу он гидролизирует N-ацетил-L-аспартил-L-глутамат (NAAG) в N-ацетил-L-аспартат и глутамат. Ферментативная функция PSMA в здоровой и больной простате еще не была классифицирована.
Нацеливающиеся на PSMA молекулы, как правило, содержат связывающую единицу, которая охватывает цинк-связывающую группу (такую как мочевина (Zhou, J., et al., NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nature Reviews Drug Discovery, 2005. 4(12): p.1015-1026), фосфинат или фосфорамидат), прикрепленную к фрагменту глутамата Р1', который гарантирует высокую аффинность и специфичность к PSMA, и, как правило, также прикреплен к функционалу эффектора (Machulkin, А.Е., et al., Small-molecule PSMA ligands. Current state, SAR and perspectives. Journal of drug targeting, 2016: p.1-15). Эффекторная часть более гибкая и в некоторой степени устойчивая к структурным модификациям. Туннель попадания PSMA охватывает два других главных структурных признака, которые важны для связывания лиганда. Первым является пэтч аргинина, положительно заряженная область на стенке туннеля попадания и структурное объяснение предпочтительных отрицательно заряженных функционалов в Р1-положении PSMA. При связывании, одновременное репозиционирование боковых цепей аргинина может привести к открытию гидрофобного вспомогательного кармана S1, второй важной структуры, которая, как было показано, включает группу йод-бензил из нескольких ингибиторов на основе мочевины, тем самым внося вклад в их высокую аффинность к PSMA (Barinka, С., et al., Interactions between Human Glutamate Carboxypeptidase // and Urea-Based Inhibitors: Structural Characterizationf. Journal of medicinal chemistry, 2008. 51(24): p.7737-7743).
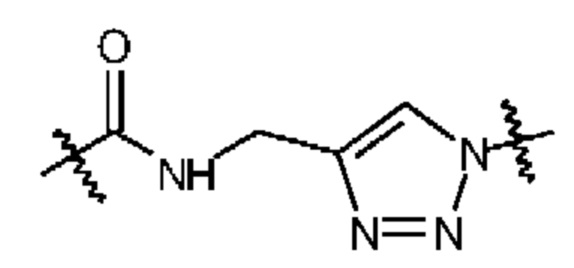
Zhang et al. был выявлен удаленный участок связывания PSMA, который может быть задействован для режима бидентатного связывания (Zhang, А.Х., et al., A remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. Journal of the American Chemical Society, 2010. 132(36): p.12711-12716). Так называемый арен-связывающий участок представляет собой простой структурный мотив, образованный боковыми цепями Arg463, Arg511 и Trp541, и он является частью крышки для входа PSMA. Зацепление арен-связывающего участка фрагмента дистального ингибитора может привести к существенному повышению аффинности ингибитора к PSMA вследствие эффектов авидности. PSMA I&T (см. Фигуру 1) был разработан с целью взаимодействия таким образом с PSMA, хотя никакого структурного анализа кристаллов режима связывания в доступности нет.Согласно Zhang et al., необходимым признаком является линкерная единица (субериновая кислота в случае PSMA I&T), которая способствует открытой конформации крышки для входа PSMA и, таким образом, обеспечивает доступность арен-связывающего участка. Кроме того, было показано, что структурная композиция линкера обладает существенным воздействием на активность нацеливания на опухоль и биологическую активность, а также на контраст визуализации и фармакокинетику (Liu, Т., et al., Spacer length effects on in vitro imaging and surface accessibility of fluorescent inhibitors of prostate specific membrane antigen. Bioorganic & medicinal chemistry letters, 2011. 21(23): p.7013-7016), свойства которых являются ключевыми как для высокого качества визуализации, так и для эффективной нацеленной эндорадиотерапии.
В настоящее время, в клинических условиях используют две категории PSMA-нацеливающих ингибиторов. С одной стороны находится метка с хелатирующими единицами для комплексирования радионуклида в качестве PSMA I&T или связанные соединения (Kiess, А.Р., et al., Prostate-specific membrane antigen asa target for cancer imaging and therapy. The quarterly journal of nuclear medicine and molecular imaging: official publication of the Italian Association of Nuclear Medicine (AIMN)[and] the International Association of Radiopharmacology (IAR),[and] Section of the Society of… 2015. 59(3): p.241). С другой стороны находятся небольшие молекулы, содержащие нацеливающую единицу и эффекторные молекулы. В зависимости от используемого радионуклида/галогена, радиомеченые ингибиторы PSMA могут быть использованы для визуализации или эндорадиотерапии. Среди низкомолекулярных ингибиторов с хелаторами для визуализации, наиболее часто используемыми агентами для селективной визуализации PSMA являются PSMA HBED-CC (Eder, М., et al., 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjugate chemistry, 2012. 23(4): p.688-697), PSMA-617 (Benešová, M., et al., Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. Journal of Nuclear Medicine, 2015. 56(6): p.914-920) и PSMA I&T (Weineisen, M., et al., Development and first in human evaluation of PSMA I&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine, 2014. 55(supplement 1): p.1083-1083). PSMA HBED-CC или PSMA-11 был одним из первых ингибиторов PSMA и он в настоящее время используется для визуализации, поскольку терапевтические применения невозможны с хелатором HBED-CC. Однако за счет уникальных физических характеристик и преимуществ 18F для ПЭТ-визуализации, таких как более длительный период полужизни, низкая позитронная энергия, которая приводит к более высокому разрешению изображения и возможности крупномасштабной продукции в циклотроне, несколько групп были сфокусированы на разработке 18F-меченых ингибиторов на основе мочевины для визуализации РСа. 18F-меченый PSMA-ингибитор на основе мочевины [18F]DCFPyl продемонстрировал обнадеживающие результаты при выявлении первичных и метастатических форм РСа (Rowe, S.P., et al., PSMA-Based [18F] DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Molecular Imaging and Biology, 2016: p.1-9) и превосходство над [68Ga]PSMA-HBED-CC в исследовании сравнения (Dietlein, М., et al., Comparison of[18F] DCFPyL and [68Ga] Ga-PSMA-HBED-CC for PSMA-PET imaging in patients with relapsed prostate cancer. Molecular Imaging and Biology, 2015.17(4): p.575-584).
PSMA DKFZ 617 ( et al., Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. Journal of Nuclear Medicine, 2015. 56(6): p.914-920, Becker, A., et al., Nephro-and hepatotoxicity after radioligand therapy of metastatic castrate-resistant prostate cancer with 177Lu-PSMA-617. Journal of Nuclear Medicine, 2016. 57(supplement 2): p.1430-1430; Rahbar, K., et al., Response and tolerability of a single dose of 177Lu-PSMA-617 in patients with metastatic castration-resistant prostate cancer: a multicenter retrospective analysis. Journal of Nuclear Medicine, 2016: p.jnumed. 116.173757) и PSMA I&T (Weineisen, M., et al., Development and first in human evaluation of PSMA I&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine, 2014. 55(supplement 1): p.1083-1083, Eiber, M., et al., Systemic radioligand therapy with 177Lu-PSMA I&T in patients with metastatic castration-resistant prostate cancer. Journal of Nuclear Medicine, 2016. 57(supplement 2): p.61-61; Schottelius, M., et al., [111 In] PSMA-I&T: expanding the spectrum of PSMA-I&T applications towards SPECT and radioguided surgery. EJNMMI research, 2015. 5(1): p.1) применяются в клинических условиях для паллиативного лечения пациентов с раком простаты. Хелатирующая единица DOTA и связанная DOTAGA обеспечивают не только визуализацию, но также терапевтические применения, поскольку объем для возможной хелатизации радиометаллами охватывает, помимо прочего, 111In, 177Lu, 90Y и 213Bi. [111In]PSMA I&T уже был реализован клинически для радиоуправляемой терапии для помощи хирургу при удалении злокачественной ткани (Schottelius, М., et al., [111 In] PSMA-I&T: expanding the spectrum of PSMA-I&Tapplications towards SPECTand radioguided surgery. EJNMMI research, 2015. 5(1): p.1). Подобным образом, недавно разработанный и клинически испытанный PSMA-ингибитор PSMA I&S (для визуализации и хирургии) продемонстрировал чрезвычайно обнадеживающие результаты (Robu, S., et al., Preclinical evaluation and first patient application of 99mTc-PSMA-I&S for SPECT imaging and radioguided surgery in prostate cancer. Journal of Nuclear Medicine, 2016: p.jnumed. 116.178939).
Эндорадиотерапевтические подходы с [177Lu]PSMA I&T продемонстрировали эффективность, устойчивость и высокий потенциал безопасности у пациентов, получавших до четырех циклов с 7,4 ГБк. Полученные дозиметрические значения для облучения органа показали, что почки и слюнные железы особенно получали наивысшую дозу после лезий опухоли. Подобные значения облучения были показаны для PSMA DKFZ 617 и [18F]DCFPyL (Rowe, S.P., et al., PSMA-Based[18F]DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Molecular Imaging and Biology, 2016: p.1-9; Delker, A., et al., Dosimetry for 177Lu-DKFZ-PSMA-617: a new radiopharmaceutical for the treatment of metastatic prostate cancer. European journal of nuclear medicine and molecular imaging, 2016. 43(1): p.42-51; Kabasakal, L, et al., Pre-therapeutic dosimetry of normal organs and tissues of 177Lu-PSMA-617 prostate-specific membrane antigen (PSMA) inhibitor in patients with castration-resistant prostate cancer. European journal of nuclear medicine and molecular imaging, 2015. 42(13): p.1976-1983; Yadav, M.P., et al., 177Lu-DKFZ-PSMA-617 therapy in metastatic castration resistant prostate cancer: safety, efficacy, and quality of life assessment. European Journal of Nuclear Medicine and Molecular Imaging, 2016: p.1-11). Эти повышенные числа можно объяснить физиологической экспрессией PSMA (Silver, D.A., et al., Prostate-specific membrane antigen expression in normal and malignant human tissues. Clinical Cancer Research, 1997. 3(1): p.81-85) и экскрецией радиомеченого соединения с мочой. Периодические случаи нефротоксичности и гематологической токсичности после введения, как правило, являются обратимыми, хотя существует обоснованный интерес в отношении хронической токсичности, особенно у пациентов с общими длительными показателями выживаемости, как у пациентов с РСа. Таким образом, необходим подходящий замысел для снижения нежелательного облучения и одновременного повышения опухолевого поглощения.
Ввиду вышесказанного, техническую задачу, лежащую в основе настоящего изобретения, можно усматривать в представлении средств и способов смягчения вызванных облучением побочных эффектов PSMA-нацеливающей радиомеченой диагностики и терапии. Еще одну техническую задачу можно усматривать в представлении средств и способов повышения опухолевого поглощения при такой диагностике и терапии. Выражаясь в более общем смысле, техническую задачу можно усматривать в представлении усовершенствованных PSMA-связывающих агентов.
Техническая задача решается объектом изобретения, резюмированным в прилагаемой формуле изобретения и более подробно разъясненным ниже.
В частности, в первом аспекте настоящего изобретения представлено соединение формулы (I) или его фармацевтически приемлемая соль,

где:
m представляет собой целое число от 2 до 6, предпочтительно, от 2 до 4, более предпочтительно, 2;
n представляет собой целое число от 2 до 6, предпочтительно, от 2 до 4, более предпочтительно, 2 или 4;
R1L представляет собой СН2, NH или О, предпочтительно, NH;
R2L представляет собой С или Р(ОН), предпочтительно, С;
R3L представляет собой СН2, NH или О, предпочтительно, NH;
X1 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика и аминной связи, и, предпочтительно, представляет собой амидную связь;
L1 представляет собой двухвалентную связывающую группу со структурой, выбранной из следующего: олигоамид, олигоэфир, олиготиоэфир, сложный олигоэфир, сложный олиготиоэфир, олигомочевина, олиго(эфир-амид), олиго(тиоэфир-амид), олиго(сложный эфир-амид), олиго(сложный тиоэфир-амид), олиго(мочевина-амид), олиго(эфир-тиоэфир), олиго(эфир-сложный эфир),олиго(эфир-сложный тиоэфир), олиго(эфир-мочевина), олиго(тиоэфир-сложный эфир), олиго(тиоэфир-сложный тиоэфир), олиго(тиоэфир-мочевина), олиго(сложный эфир-сложный тиоэфир), олиго(сложный эфир-мочевина) и олиго(сложный тиоэфир-мочевина), предпочтительно, со структурой, выбранной из олигоамида и олиго(сложный эфир-амид),
при этом связывающая группа может иметь EDS-группу;
X2 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика и аминной связи, и, предпочтительно, представляет собой амидную связь;
R2 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, при этом арильная группа или аралкильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН;
R3 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, при этом арильная группа или аралкильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН;
r равняется 0 или 1, предпочтительно, 1;
р равняется 0 или 1;
q равняется 0 или 1;
и, предпочтительно, p+q=1;
R4 выбран из арильной группы и EDS-группы;
X3 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика, аминной связи и группы, имеющей формулу
 , где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 костальной части соединения формулы (I);
, где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 костальной части соединения формулы (I);
и представляет собой, предпочтительно, амидную связь;
RM представляет собой группу метки, которая содержит хелатирующую группу, необязательно содержащую хелатированный нерадиоактивный или радиоактивный катион;
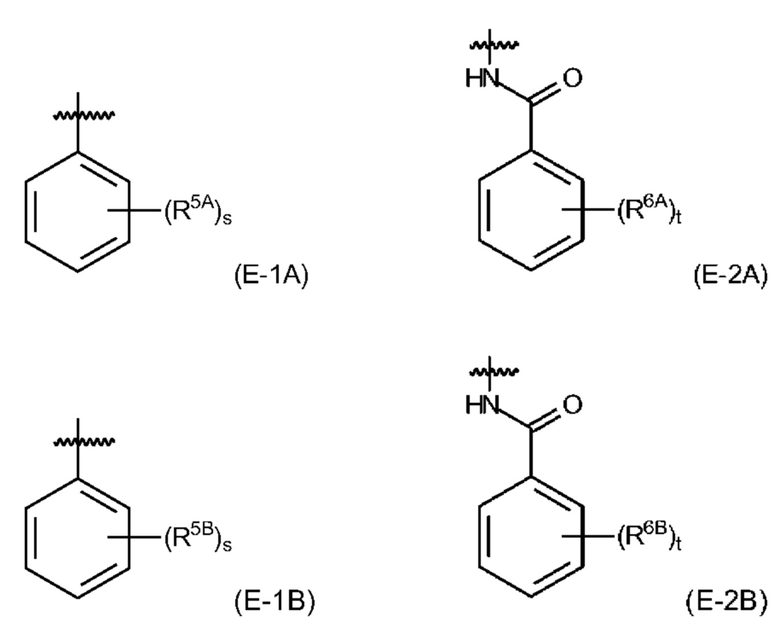
и где EDS-группа содержится по меньшей мере в одном месте в соединении формулы (I) и имеет структуру, выбранную из (Е-1А), (Е-1 В), (Е-2А) и (Е-2 В):
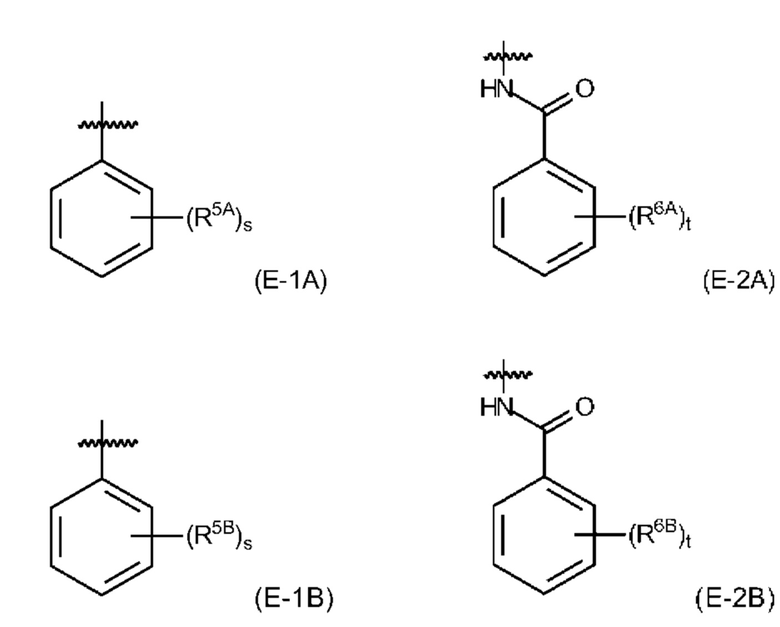
где
 помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
s равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 1;
t равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 2;
R5A представляет собой, независимо для каждого случая, когда s>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R5A и фенильным кольцом указывает на то, что s групп R5A замещают s атомов водорода в любом положении на фенильном кольце;
R5B представляет собой, независимо для каждого случая, когда s>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1 В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -NH2, и причем связь между R5B и фенильным кольцом указывает на то, что s групп R5B замещают s атомов водорода в любом положении на фенильном кольце;
R6A представляет собой, независимо для каждого случая, когда t>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R6A и фенильным кольцом указывает на то, что t групп R6A замещают t атомов водорода в любом положении на фенильном кольце;
R6B представляет собой, независимо для каждого случая, когда t>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1 В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -ОН, и причем связь между R6B и фенильным кольцом указывает на то, что t групп R6B замещают t атомов водорода в любом положении на фенильном кольце.
Включение EDS-заместителя, как показано выше, где ароматическое кольцо имеет один или более заместителей с высокой плотностью электронов, выбранных из электроноакцепторного заместителя и заместителя, имеющего неподеленную пару электронов, дает несколько неожиданных преимуществ. Эти преимущества включают повышенную аффинность, улучшенную интернализацию, повышенное удержание опухолевых клеток, пониженное неспецифическое связывание, снижение накопление в почках и повышенное опухолевое поглощение.
В особенности, пониженное неспецифическое поглощение в органах, отличных от простаты, дает менее нежелательное облучение и уменьшает вызываемые облучением побочные эффекты.
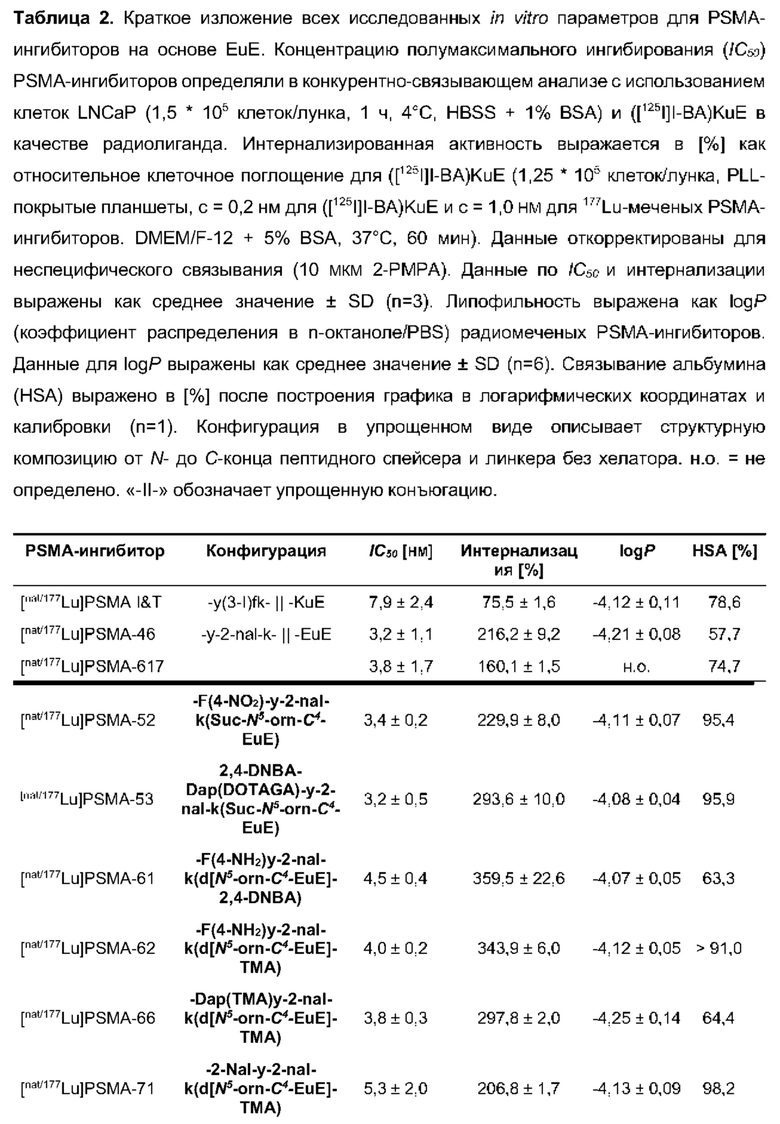
Для описания этих преимуществ на примере, рассмотрим свойства особенно предпочтительных соединений, обозначенных в настоящем документе как PSMA-71 и PSMA-66, которые более подробно описаны ниже.
В частности, наномолярная аффинность (5,3±2,0 нМ по сравнению с 7,9±2,4 нМ), значительно улучшенная интернализация (206,8±1,7% по сравнению с 75,5±1,6%) демонстрируют превосходство [177Lu]PSMA-71 при сравнении с [177Lu]PSMA I&T. Данные биораспределения показали, что [177Lu]PSMA-71 проявлял ощутимо более высокое опухолевое поглощение (14,29±0,89 по сравнению с 4,06±1,12%ВД/г, соответственно) по сравнению с [177Lu]PSMA I&T, тогда как накопление в почках было похожим (32,36±2,49 по сравнению с 34,66±17,20%ВД/г, соответственно).
Подобным образом, более низкая наномолярная аффинность (3,8±0,3 нМ по сравнению с 7,9±2,4 нМ), значительно улучшенная интернализация (297,8±2,0% по сравнению с 75,5±1,6%), повышенное удержание опухолевых клеток in vitro (90,1±3,5% по сравнению с 62,8±0,4%, инкубация в течение 60 мин) вместе с более низким неспецифическим связыванием in vivo, пониженное накопление в почках (117,5±6,9% ВД/г по сравнению с 128,9±10,7% ВД/г) и более чем двукратное повышение опухолевого поглощения (10,0±0,4% по сравнению с 4,7±1,0% ВД/г) демонстрируют превосходство [177Lu]PSMA-66 при прямом сравнении с [177Lu]PSMA I&T.
Как отмечено выше, соли соединений по изобретению, в том числе соединений формулы (I) (и в том числе предпочтительные варианты их реализации), также являются пригодными для применения в контексте изобретения. Следует понимать, что эти соли в целом являются фармацевтически приемлемыми формами соли этих соединений, которые могут быть образованы, например, путем протонирования атома, имеющего неподеленную пару электронов, которая чувствительная к протонированию, такую как аминогруппу, с органической или неорганической кислотой, или как соль карбоновокислотной группы с физиологически приемлемым катионом, поскольку они широко известны из области техники. Примеры солей присоединения оснований включают, например, соли щелочных металлов, такие как соли натрия или калия; соли щелочноземельных металлов, такие как соли кальция или магния; соли аммония; соли алифатических аминов, такие как триметиламин, триэтиламин, дициклогексиламин, этаноламин, диэтаноламин, триэтаноламин, соли прокаина, соли меглумина, соли этаноламина или соли этилендиамина; соли аралкиламина, такие как соли N,N-дибензилэтилендиамина, соли бенетамина; соли гетероциклических ароматических аминов, такие как соли пиридина, соли пиколина, соли хинолина или соли изохинолина; соли четвертичного аммония, такие как соли тетраметиламмония, соли тетраэтиламмония, соли бензилтриметиламмония, соли бензилтриэтиламмония, соли бензилтрибутиламмония, соли метилтриоктиламмония или соли тетрабутиламмония; и соли основных аминокислот, такие как соли аргинина или соли лизина. Примеры солей присоединения кислот включают, например, соли минеральных кислот, такие как гидрохлорид, гидробромид, гидройодид, сульфатные соли, нитратные соли, фосфатные соли (такие как, например, фосфатные, гидрофосфатные или дигидрофосфатные соли), карбонатные соли, гидрокарбонатные соли или перхлоратные соли; соли органических кислот, такие как ацетат, пропионат, бутират, пентаноат, гексаноат, гептаноат, октаноат, циклопентанпропионат, ундеканоат, лактат, малеат, оксалат, фумарат, тартрат, малат, цитрат, никотинат, бензоат, салицилат или аскорбат; сульфонатные соли, такие как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, бензолсульфонат, р-толуолсульфонат (тозилат), 2-нафталинсульфонат, 3-фенилсульфонат или камфорсульфонат; и кислые соли аминокислот, такие как соли аспартата или соли глутамата.
Дополнительные примеры фармацевтически приемлемых солей включают, но без ограничения, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, эдетат кальция, камфорат, камфорсульфонат, камсилат, карбонат, хлорид, цитрат, клавуланат, циклопентанпропионат, диглюконат, дигидрохлорид, додецил сульфат, эдетат, эдисилат, эстолат, эсилат, этансульфонат, формат, фумарат, глюцептат, глюкогептонат, глюконат, глутамат, глицерофосфат, гликолиларсанилат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидройодид, 2-гидрокси-этансульфонат, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метансульфонат, метилсульфат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, аммониевая соль N-метилглюкамина, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, 3-фенилпропионат, фосфат/дифосфат, пикрат, пивалат, полигалактуронат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид, ундеканоат, валерат и подобные (см., например, S. М. Berge et al., "Pharmaceutical Salts", J. Pharm. Sci., 66, pp.1-19 (1977)).
Следует понимать, что в настоящем описании термин «соединение» охватывает сольваты, полиморфы, пролекарства, комбинированные лекарственные средства, сокристаллы, таутомеры, рацематы, энантиомеры или диастереомеры, или их смеси, если не указано иное.
Когда соединения по настоящему изобретению представлены в кристаллической форме, структура может содержать молекулы растворителя. Как правило, растворители представляют собой фармацевтически приемлемые растворители и включают, помимо прочего, воду (гидраты) или органические растворители. Примеры возможных сольватов включают этанолаты и изопропанолаты.
Термин «комбинированное лекарственное средство» относится к двум или более терапевтическим соединениям, связанным через ковалентную химическую связь. Подробное определение может быть найдено, например, в N. Das et al., European Journal of Pharmaceutical Sciences, 41, 2010, 571-588.
Термин «сокристалл» относится к многокомпонентному кристаллу, в котором все компоненты являются твердыми при обычных условиях окружающей среды, когда они находятся в своей чистой форме. Эти компоненты существуют совместно как стоихиометрическое или нестоихиометрическое соотношение целевой молекулы или иона (т.е. соединения по настоящему изобретению) и одного или более нейтральных молекулярных формирователей сокристалла. Подробное описание может быть найдено, например, в Ning Shan et al., Drug Discovery Today, 13(9/10), 2008, 440-446 и в D. J. Good et al., Cryst. Growth Des., 9(5), 2009, 2252-2264.
Соединения по настоящему изобретению также могут быть представлены в форме пролекарства, а именно, соединения, которое метаболизировано in vivo до активного метаболита. Подходящими пролекарствами являются, например, сложные эфиры. Конкретные примеры подходящих групп представлены, среди прочего, в параграфах [0082] и [0118] в US 2007/0072831 под заголовками пролекарств и защитных групп.
До той степени, при которой соединения по изобретению проявляют рН-зависимое заряженное состояние, следует понимать, что охвачены все возможные заряженные состояния. В этом отношении, предпочтительный диапазон рН составляет от 0 до 14.
До той степени, при которой соединение по изобретению имеет суммарный заряд, следует понимать, что соединение представлено в электронейтральной форме. Это достигается благодаря одному или более противоионам, предпочтительные противоионы определены в настоящем документе выше в отношении термина «соль».
В формуле (I) m представляет собой целое число от 2 до 6. Предпочтительно, m составляет от 2 до 4, более предпочтительно, 2. R1L представляет собой CH2, NH или О, предпочтительно, NH. R2L представляет собой С или Р(ОН), предпочтительно, С. R3L представляет собой CH2, NH или О, предпочтительно, NH. Таким образом, также предпочтительны соединения формулы (I) или их соли, где m равняется 2, R1L представляет собой NH, R2L представляет собой С, a R3L представляет собой NH.
n представляет собой целое число от 2 до 6, предпочтительно, от 2 до 4, более предпочтительно, 2 или 4, и наиболее предпочтительно, 2.
Таким образом, особенно предпочтительны соединения формулы (I) или их соли, где m равняется 2, n равняется 2 или 4, R1L представляет собой NH, R2L представляет собой С, a R3L представляет собой NH. Более предпочтительными являются соединения формулы (I) или их соли, где m равняется 2, n равняется 2, R1L представляет собой NH, R2L представляет собой С, a R3L представляет собой NH.
X1 в формуле (I) выбран из амидной связи (т.е. -C(O)-NH-), эфирной связи (т.е. -О-), тиоэфирной связи (т.е. -S-), сложноэфирной связи (т.е. -С(O)-O-, сложной тиоэфирной связи (т.е. -C(S)-O- или -C(O)-S-), мочевинного мостика (т.е. -NH-C(O)-NH-) и амидной связи (т.е. -NH-). В качестве X1 предпочтительной является амидная связь.
Более того, в формуле (I) также предпочтительно, чтобы n равнялось 2, а X1 представлял собой амидную связь с атомом углерода в амидной связи -C(O)-NH-, прикрепленным к группе -(СН2)n-, или чтобы n равнялось 4, а X1 представлял собой амидную связь с атомом углерода в амидной связи -C(O)-NH-, прикрепленным к группе -(СН2)n-. Наиболее предпочтительной опцией среди этих является, чтобы n равнялось 2, а X1 представлял собой амидную связь с атомом углерода в амидной связи -C(O)-NH-, прикрепленным к группе -(СН2)2-.
Таким образом, особенно предпочтительными являются соединения формулы (I) и их соли, где в формуле (I) m равняется 2, n равняется 2, R1L представляет собой NH, R2L представляет собой С, R3L представляет собой NH, а X1 представляет собой амидную связь с атомом углерода в амидной связи -C(O)-NH-, прикрепленным к группе -(СН2)n-.
L1 в формуле (I) представляет собой двухвалентную связывающую группу со структурой, выбранной из следующего: олигоамид, олигоэфир, олиготиоэфир, сложный олигоэфир, сложный олиготиоэфир, олигомочевина, олиго(эфир-амид), олиго(тиоэфир-амид), олиго(сложный эфир-амид), олиго(сложный тиоэфир-амид), олиго(мочевина-амид), олиго(эфир-тиоэфир), олиго(эфир-сложный эфир),олиго(эфир-сложный тиоэфир), олиго(эфир-мочевина), олиго(тиоэфир-сложный эфир), олиго(тиоэфир-сложный тиоэфир), олиго(тиоэфир-мочевина), олиго(сложный эфир-сложный тиоэфир), олиго(сложный эфир-мочевина) и олиго(сложный тиоэфир-мочевина), предпочтительно, со структурой, выбранной из олигоамида и олиго(сложный эфир-амид), и более предпочтительно, со структурой олигоамида, связывающая группа которой может иметь EDS-группу.
Термин «олиго», используемый в определении L1 в отношении олигоамида, олигоэфира, олиготиоэфира, сложного олигоэфира, сложного олиготиоэфира, олигомочевины, олиго(эфир-амида), олиго(тиоэфир-амида), олиго(сложный эфир-амида), олиго(сложный тиоэфир-амида), олиго(мочевина-амида), олиго(эфир-тиоэфира), олиго(эфир-сложного эфира), олиго (эфир-сложно го тиоэфира), олиго(эфир-мочевины), олиго(тиоэфир-сложного эфира), олиго(тиоэфир-сложного тиоэфира), олиго(тиоэфир-мочевины), олиго(сложный эфир-сложного тиоэфира), олиго(сложный эфир-мочевины) и олиго(сложный тиоэфир-мочевины), предпочтительно, следует понимать, как относящийся к группе, где от 2 до 20, более предпочтительно, где от 2 до 10 субъединиц связаны типом связей, указанных в тех же терминах. Как поймет читатель, обладающий знаниями в данной области техники, в тех местах, где различные типы связей указаны в скобках, оба типа связей содержатся в рассматриваемой группе (например, в «олиго(сложный эфир-амиде)» содержатся сложноэфирные связи и амидные связи).
Более предпочтительно, чтобы L1 имела структуру, выбранную из олигоамида, который содержит всего от 1 до 5, более предпочтительно, всего от 1 до 3, и наиболее предпочтительно, всего 1 или 2 амидных связи в своем остове, и олиго(сложный эфир-амида), который содержит всего от 2 до 5, более предпочтительно, всего от 2 до 3, и наиболее предпочтительно, всего 2 амидных и сложноэфирных связей в своем остове. В особенно предпочтительном варианте реализации L1 представляет собой двухвалентную связывающую группу с олигоамидной структурой, которая содержит 1 или 2 амидных связи в своем остове.
Кроме того, L1 может иметь EDS-группу (т.е. группу, имеющую заместитель с высокой плотностью электронов или «электронно-плотный заместитель»), как определено в настоящем документе, т.е. EDS-группу, которая ковалентно прикреплена к L1. Предпочтительно, необязательная EDS-группа прикреплена в качестве заместителя к остову двухвалентной связывающей группы L1, при этом L1 имеет структуру, как определено выше, выбранную из следующего: олигоамид, олигоэфир, олиготиоэфир, сложный олигоэфир, сложный олиготиоэфир, олигомочевина, олиго(эфир-амид), олиго(тиоэфир-амид), олиго(сложный эфир-амид), олиго(сложный тиоэфир-амид), олиго(мочевина-амид), олиго(эфир-тиоэфир), олиго(эфир-сложный эфир),олиго(эфир-сложный тиоэфир), олиго(эфир-мочевина), олиго(тиоэфир-сложный эфир), олиго(тиоэфир-сложный тиоэфир), олиго(тиоэфир-мочевина), олиго(сложный эфир-сложный тиоэфир), олиго(сложный эфир-мочевина) и олиго(сложный тиоэфир-мочевина), предпочтительно, структуру, выбранную из олигоамида и олиго(сложный эфир-амид), и наиболее предпочтительно, структуру олигоамида, остов которой проходит между X1 и X2 в соединении формулы (I). Также, в этом отношении применимы дополнительные предпочтительные определения для L1, т.е. более предпочтительно, чтобы L1 имела структуру, выбранную из олигоамида, который содержит всего от 1 до 5, более предпочтительно, всего от 1 до 3, и наиболее предпочтительно, всего 1 или 2 амидных связи в своем остове, и олиго(сложный эфир-амида), который содержит всего от 2 до 5, более предпочтительно, всего от 2 до 3, и наиболее предпочтительно, всего 2 амидных и сложноэфирных связей в своем остове. В особенно предпочтительном варианте реализации L1 представляет собой двухвалентную связывающую группу с олигоамидной структурой, которая содержит 1 или 2 амидных связи в своем остове.
В соответствии с вышеизложенным, L1 может иметь одну или более, например, 2 или 3 EDS-групп. Однако предпочтительно, чтобы L1 не имела EDS-группу, или чтобы L1 имела одну EDS-группу, и более предпочтительно, чтобы L1 имела одну EDS-группу.
Если L1 имеет EDS-группу (в том числе более предпочтительный случай, когда EDS имеет одну EDS-группу), то предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-1А), (Е-2А) и (Е-2 В). Более предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-2А) и (Е-2 В), и наиболее предпочтительно, чтобы она имела структуру (Е-2А).
Как поймет читатель, обладающий знаниями в данной области техники, указание на то, что L1 может иметь EDS-группу, служит в качестве информации о возможном положении этой группы в соединениях согласно изобретению. Тот факт, что L1 может иметь EDS-группу, не накладывает ограничение на присутствие других групп, которые могут присутствовать, например, в виде альтернатив или дополнительных заместителей на остове L1. Например, предпочтительно, чтобы связывающая группа L1 содержала одну или более, например, две, группы, которые независимо выбраны из -ОН, -ОСН3, -СООН, -СООСН3, -NH2 и -NHC(NH)NH2, прикрепленные в качестве заместителей к остову. Более предпочтительно, связывающая группа L1 содержит одну или более, например, две, группы -СООН, прикрепленные в качестве заместителей к остову.
В формуле (I) X2 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика и аминной связи, и, предпочтительно, представляет собой амидную связь. Более предпочтительно, чтобы атом азота в амидной связи -C(O)-NH- был прикреплен к L1.
Таким образом, также предпочтительно, чтобы X1 и X2 представляли собой амидные связи, особенно, амидные связи, расположенные в предпочтительных ориентациях, дополнительно определенных выше.
В соответствии с вышеизложенным, предпочтительно, чтобы фрагмент -Х2-L1-Х1- в формуле (I) имел структуру, выбранную из:
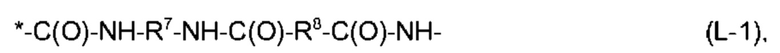
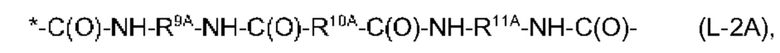 и
и
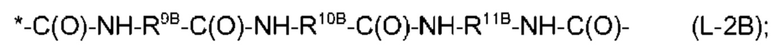
причем амидная связь, помеченная *, прикреплена к атому углерода, имеющему R2 в формуле (I), и причем
R7, R8, R9A, R9B, R11A и R11B независимо выбраны из необязательно замещенного С2-С10-алкандиила, предпочтительно, необязательно замещенного линейного С2-С10-алкандиила, причем каждая алкандиильная группа может быть замещена одним или более заместителями, независимо выбранными из -ОН, -ОСН3, -СООН, -СООСН3, -NH2, -NHC(NH)NH2 и EDS-группы, и R10A и R10B выбраны из необязательно замещенного С2-С10-алкандиила, предпочтительно, необязательного замещенного линейного С2-С10-алкандиила, и необязательно замещенного С6-С10-арендиила, предпочтительно, фенилена, причем каждая алкандиильная и арендиильная группа может быть замещена одним или более заместителей, независимо выбранными из -ОН, -ОСН3, -СООН, -СООСН3, -NH2, -NHC(NH)NH2 и EDS-группы. R10A представляет собой, предпочтительно, необязательно замещенный С2-С10-алкандиил, более предпочтительно, необязательно замещенный линейный С2-С10-алкандиил, как определено выше. R10B представляет собой, предпочтительно, необязательно замещенный С6-С10-арендиил, как определено выше, более предпочтительно, фениленовую группу, например, пара-фениленовую группу.
В группах формул (L-1), (L-2A) и (L-2B) предпочтительно, чтобы необязательный заместитель на R7 представлял собой -СООН, чтобы необязательный заместитель R8 представлял собой EDS-группу, чтобы необязательный заместитель на R9A и R9B представлял собой -СООН, чтобы необязательный заместитель на R10A представлял собой EDS-группу, и чтобы необязательный заместитель на R11A и R11B представлял собой -СООН.
Также предпочтительно, чтобы каждая из групп формул (L-1) и (L-2A) имела EDS-группу в качестве по меньшей мере одного заместителя, как разъяснено выше, предпочтительно, в качестве заместителя R8 и R10A. Также, в данном контексте предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-1А), (Е-2А) и (Е-2 В). Более предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-2А) и (Е-2 В), и наиболее предпочтительно, чтобы она имела структуру (Е-2А).
Кроме того, предпочтительно, чтобы общее количество атомов углерода в R7 и R8 формулы (L-1) составляло от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях, чтобы общее количество атомов углерода в R9A, R10A и R11A формулы (L-2A) составляло от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях, и чтобы общее количество атомов углерода в R9B, R10B и R11B формулы (L-2B) составляло от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях.
Из информации, представленной выше в отношении предпочтительных значений n и X1, будет ясно, что по-прежнему предпочтительно, чтобы -Х2-L1-Х1- в формуле (I) имел структуру (L-1), если n равняется 4, и чтобы -Х2-L1-Х1- в формуле (I) имел структуру (L-2A) или (L-2B), если n равняется 2.
В соответствии с представленными выше определениями, еще более предпочтительно, чтобы фрагмент -Х2-L1-Х1- в формуле (I) имел структуру, выбранную из:
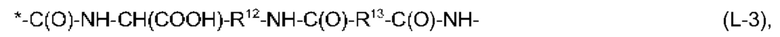
 и
и
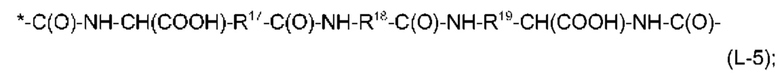
причем связь, помеченная *, прикреплена к атому углерода, имеющему R2 в формуле (I),
R12 и R14 независимо выбраны из линейного С2-С6-алкандиила, предпочтительно, из линейного С3-С6-алкандиила,
R13 представляет собой линейный С2-С10-алкандиил, предпочтительно, линейный С4-С8-алкандиил,
R15 и R16 независимо выбраны из линейного С2-С6-алкандиила, предпочтительно, из линейного С2-С4-алкандиила,
и причем каждый из R13 и R15 может иметь одну EDS-группу в качестве заместителя и, предпочтительно, каждый из R13 и R15 имеет одну EDS-группу в качестве заместителя, R17 представляет собой линейный С2-С6-алкандиил, предпочтительно, линейный С2-С4-алкандиил,
R18 представляет собой фениленовую группу, например, пара-фениленовую группу, и
R19 представляет собой линейный С2-С6-алкандиил, предпочтительно, линейный С2-С4-алкандиил.
Также, в данном контексте предпочтительно, чтобы EDS-группа, которая может быть прикреплена к R13 и R15, имела структуру, выбранную из (Е-1А), (Е-2А) и (Е-2 В). Более предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-2А) и (Е-2 В), и наиболее предпочтительно, чтобы она имела структуру (Е-2А).
Кроме того, предпочтительно, чтобы общее количество атомов углерода в R12 и R13 в формуле (L-3), без атомов углерода, содержащихся в EDS-группе в качестве заместителя, составляло от 6 до 16, более предпочтительно, от 6 до 14, а общее количество атомов углерода в R14, R15 и R16 в формуле (L-4), без атомов углерода, содержащихся в EDS-группе в качестве заместителя, составляло от 6 до 16, более предпочтительно, от 6 до 14.
Из информации, представленной выше в отношении предпочтительных значений n и X1, будет ясно, что особенно предпочтительно, чтобы -Х2-L1-Х1- в формуле (I) имел структуру (L-3), если n равняется 4, и чтобы -Х2-L1-Х1- в формуле (I) имел структуру (L-4) или (L-5), если n равняется 2.
Особенно предпочтительно, чтобы в формуле (I) n равнялось 2, а фрагмент -Х2-L1-Х1-имел одну из следующих структур:



причем связь, помеченная *, прикреплена к атому углерода, имеющему R2 в формуле (I), EDS представляет собой EDS-группу, как определено в настоящем документе, в том числе в предпочтительных вариантах ее реализации, а Ph представляет собой пара-фениленовую группу.
Также, в данном контексте предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-1А), (Е-2А) и (Е-2 В). Более предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-2А) и (Е-2 В), и наиболее предпочтительно, чтобы она имела структуру (Е-2А).
В формуле (I) R2 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, предпочтительно, необязательно замещенную аралкильную группу. Как будет понятно, термин «аралкильная группа», используемый в настоящем документе, относится к алкильной группе, в которой атом водорода замещен арильной группой в качестве заместителя. Предпочтительно, аралкильная группа представляет собой группу, в которой одна арильная группа связана с алкандиильной группой. Арильная группа или аралкильная группа, представленная R2, может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН. Арил и арильная часть аралкильной группы, предпочтительно, выбраны из фенила и нафтила, такого как 2-нафтил. Алкандиильная часть аралкильной группы, предпочтительно, представляет собой С1-С4-алкандиильную группу, более предпочтительно, -СН2-группу. Таким образом, более предпочтительно, R2 выбран из необязательно замещенного -СН2-фенила и необязательно замещенного -СН2-нафтила, в частности, необязательно замещенного -СН2-(2-нафтила). Необязательно замещенный -СН2-(2-нафтил) является особенно предпочтительной опцией для R2.
Необязательно замещенная арильная группа и арильная часть необязательно замещенной аралкильной группы, в том числе предпочтительные варианты их реализации, может быть замещена одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН. Таким образом, может присутствовать один или более одного, например, 2 или 3, заместителей, выбранных из галогена, предпочтительно, I, и -ОН. Однако предпочтительно, чтобы R2 был незамещенным.
Ввиду вышеизложенного, будет ясно, что R2, наиболее предпочтительно, представляет собой незамещенный -СН2-(нафтил), и что нафтильная группа, наиболее предпочтительно, представляет собой 2-нафтильную группу для обеспечения R2 в виде -СН2-(2-нафтила).
В формуле (I) R3 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, предпочтительно, необязательно замещенную аралкильную группу. Арильная группа или аралкильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН. Арил и арильная часть аралкильной группы, предпочтительно, выбраны из фенила и нафтила, такого как 2-нафтил. Более предпочтительно, чтобы арил и арильная часть аралкила представляли собой фенил. Алкандиильная часть аралкильной группы, предпочтительно, представляет собой С1-С4-алкандиильную группу, более предпочтительно, -СН2-группу. Таким образом, более предпочтительно, R3 представляет собой необязательно замещенный -СН2-фенил.
Необязательно замещенная арильная группа и арильная часть необязательно замещенной аралкильной группы, в том числе предпочтительные варианты их реализации, может быть замещена одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН. Таким образом, может присутствовать один или более одного, например, 2 или 3, заместителей, выбранных из галогена, предпочтительно, I, и -ОН. Предпочтительно, R3 замещен одним заместителем, который представляет собой -ОН, или комбинацией одного заместителя -ОН и одного заместителя -I.
Таким образом, особенно предпочтительно, чтобы R3 представлял собой -СН2-фенил, замещенный на фенильном кольце одним заместителем, который представляет собой -ОН, или комбинацией одного заместителя -ОН и одного заместителя -I, и наиболее предпочтительно, чтобы заместитель -ОН присутствовал в пара-положении фенильного кольца относительно -СН2- группы.
В соответствии с вышеизложенным, в формуле (I) предпочтительно, чтобы R2 представлял собой группу формулы

a R3 представлял собой группу формулы

где помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части соединения формулы (I).
Даже еще более предпочтительной является комбинация R2 и R3, в которой R2 представляет собой группу формулы
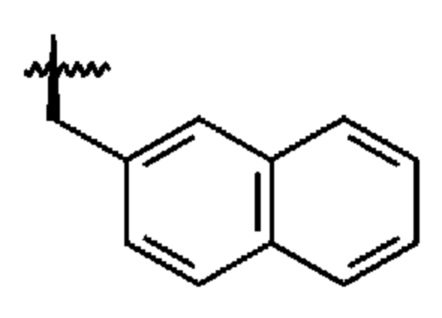
a R3 представляет собой группу формулы

где помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части соединения.
В формуле (I) r может равняться 0 или 1, и предпочтительно, чтобы r равнялось 1.
Кроме того, как разъяснено выше, р равняется 0 или 1, a q равняется 0 или 1, и предпочтительно, чтобы p+q=1. Более предпочтительно, р равняется 0, a q равняется 1.
R4 в формуле (I) выбран из арильной группы и EDS-группы. Арил, предпочтительно, выбран из фенила и нафтила, такого как 2-нафтил. Таким образом, R4, более предпочтительно, выбран из фенила, нафтила, такого как 2-нафтил, и EDS-группы. Наиболее предпочтительно, он представляет собой EDS-группу.
Если R4 представляет собой EDS-группу, то предпочтительно, чтобы EDS-группа имела структуру, выбранную из (Е-1А), (Е-2А) и (Е-1 В).
X3 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика, аминной связи и группы, имеющей формулу
 , где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части молекулы.
, где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части молекулы.
Предпочтительно, X3 выбран из амидной связи и группы формулы
 , где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части молекулы.
, где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части молекулы.
В более предпочтительном варианте реализации X3 представляет собой амидную связь -C(O)-NH- с атомом углерода, прикрепленным к RM.
RM представляет собой группу метки, которая содержит хелатирующую группу, необязательно содержащую хелатированный нерадиоактивный или радиоактивный катион.
Как поймет читатель, обладающий знаниями в данной области техники, представленное выше определение, согласно которому, RM содержит хелатирующую группу, охватывает случай, в котором RM представляет собой хелатирующую группу; в данном случае хелатирующая группа, как правило, прямо связана с X3;
и случай, в котором RM содержит, вместе с хелатирующей группой, например, дополнительный линкерный фрагмент; в этом случае хелатирующая группа может быть непрямым образом связана через этот дополнительный линкерный фрагмент с X3.
Хелатирующая группа, представленная RM, пригодна для образования хелата с радиоактивным или нерадиоактивным катионом. Подходящие хелатирующие группы для разнообразных катионов хорошо известны из области техники и могут быть использованы в контексте настоящего изобретения.
Хелатирующая группа, необязательно содержащая хелатированный нерадиоактивный или радиоактивный катион, предпочтительно, выбрана из хелатирующей группы, содержащей по меньшей мере одно из следующего:
(i) структуру макроциклического кольца с от 8 до 20 атомами кольца, из которых 2 или более, предпочтительно, 3 или более, выбраны из атомов кислорода, атомов серы и атомов азота; и
(ii) ациклическую хелатирующую структуру с открытой цепью с от 8 до 20 атомами основной цепи, из которых 2 или более, предпочтительно, 3 или более, являются гетероатомами, выбранными из атомов кислорода, атомов серы и атомов азота.
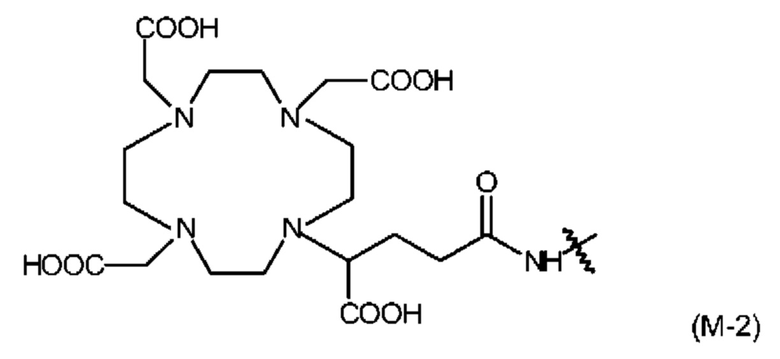
Примером хелатирующей группы и, следовательно, также примером группы RM является остаток хелатирующего агента, выбранного из следующего: бис(карбоксиметил)-1,4,8,11-тетраазабицикпо[6.6.2]гексадекан (СВТЕ2а), циклогексил-1,2-диаминтетрауксусная кислота (CDTA), 4-(1,4,8,11-тетраазациклотетрадец-1-ил)-метилбензойная кислота (СРТА), N'-[5-[ацетил(гидрокси)амино]пентил]-N-[5-[[4-[5-аминопентил-(гидрокси)амино]-4-оксобутаноил]амино]пентил]-N-гидроксибутандиамид (DFO), 4,11-бис(карбоксиметил)-1,4,8,11-тетраазабицикло[6.6.2]гексадекан (DO2A) 1,4,7,10-тетраазацикпододекан-N,N',N'',N'''-тетрауксусная кислота (DOTA), 2-[1,4,7,10-тетраазациклододекан-4,7,10-триуксусная кислота]-пентандиовая кислота (DOTAGA), N,N'-дипиридоксилэтилендиамин-N,N'-диацетат-5,5'-бис(фосфат) (DPDP), диэтилентриаминпентауксусная кислота (DTPA), этилендиамин-N,N'-тетрауксусная кислота (EDTA), этиленгликоль-O,O-бис(2-аминоэтил)-N,N,N',N'-тетрауксусная кислота (EGTA), N,N-бис(гидроксибензил)-этилендиамин-N,N'-диацетилуксусная кислота (HBED), гидроксиэтилдиаминтриуксусная кислота (HEDTA), 1-(р-нитробензил)-1,4,7,10-тетраазациклодекан-4,7,10-триацетат (HP-DOA3), 6-гидразинил-N-метилпиридин-3-карбоксамид (HYNIC), 1,4,7-триазациклононан-1-янтарная кислота-4,7-диацетилуксусная кислота (NODASA), 1-(1-карбокси-3-карбоксипропил)-4,7-(карбокси)-1,4,7-триазациклононан (NODAGA), 1,4,7-триазациклононантриуксусная кислота (NOT А), 4,11-бис(карбоксиметил)-1,4,8,11-тетраазабицикло[6.6.2]гексадекан (ТЕ2А), 1,4,8,11-тетраазациклододекан-1,4,8,11-тетрауксусная кислота (ТЕТА), терпиридин-бис(метиленаминтетрауксусная кислота (ТМТ), 1,4,7,10-тетраазациклотридекан-N,N',N'',N'''-тетрауксусная кислота (TRITA), триэтилентетраамингексауксусная кислота (ТТНА), N,N'-бис[(6-карбокси-2-пиридил)метил]-4,13-диаза-18-краун-6 (H2macropa) и 4-амино-4-{2-[(3-гидрокси-1,6-диметил-4-оксо-1,4-дигидро-пиридин-2-илметил)-карбамоил]-этил} пимелиновая кислота бис-[(3-гидрокси-1,6-диметил-4-оксо-1,4-дигидро-пиридин- 2-илметил)-амид] (ТНР);
причем остаток образуется за счет ковалентного связывания карбоксильной группы, содержащейся в хелатирующем агенте, с остальной частью соединения через сложноэфирную или амидную связь, предпочтительно, амидную связь. Читатель, обладающий знаниями в данной области техники, поймет, что в формуле (I) эта сложноэфирная или амидная связь в данном случае может быть охвачена X3 или она, предпочтительно, может быть представлена X3.
Среди этих хелатирующих агентов предпочтительными являются DOTA и DOTAGA.
Таким образом, также предпочтительно, чтобы RM-X3- в формуле (I) представлял собой группу формулы
 или
или
где группа, обозначенная  прикреплена к остальной части соединения формулы (I), и где хелатирующая группа может содержать хелатированный нерадиоактивный или радиоактивный катион.
прикреплена к остальной части соединения формулы (I), и где хелатирующая группа может содержать хелатированный нерадиоактивный или радиоактивный катион.
Примеры радиоактивных катионов, которые необязательно хелатированы хелатирующей группой, выбирают из катионов 44Sc, 47Sc, 51Cr, 52mMn, 58Co, 52Fe, 58Ni, 57Ni, 62Cu, 64Cu, 67Cu, 66Ga, 68Ga, 67Ga, 89Zr, 90Y, 89Y, 94mTc, 99mTc, 97Ru, 105Rh, 109Pd, 111Ag, 110mIn, 111In, 113mIn, 114mIn, 117mSn, 121Sn, 127Te, 142Pr, 143Pr, 149Pm, 151Pm, 149Tb, 153Sm, 157Gd, 161Tb, 166Ho, 165Dy, 169Er, 169Yb, 175Yb, 172Tm, 177Lu, 186Re, 188Re, 191Pt, 197Hg, 198Au, 199Au, 212Pb, 203Pb, 211At, 212Bi, 213Bi, 223Ra, 225Ac и 227Th, или катионной молекулы, содержащей 18F, такой как 18F-[AIF]2+.
Предпочтительные хелатированные катионы выбирают из катионов 44Sc, 47Sc, 64Cu, 67Cu, 68Ga, 90Y, 111In, 181Tb, 166Ho, 177Lu, 188Re, 212Pb, 212Bi, 213Bi, 225Ac и 227Th, или катионной молекулы, содержащей 18F.
В формуле (I) EDS-группа встречается по меньшей мере один раз, так что, например, может содержаться одна, две или три EDS-группы. Предпочтительно, соединения или соли, в соответствии с изобретением, содержат одну или две EDS-группы. Как разъяснено выше, EDS-группы(ы) может(могут) присутствовать у L1 и/или может(могут) быть представлена(ы) R4.
Наиболее предпочтительными соединениями формулы (I) или их солями являются те, что содержат одну EDS-группу, которая имеется у связывающей группы L1, в том числе в предпочтительных вариантах ее реализации, как изложено выше, и те, что содержат две EDS-группы, одна из которых представлена R4 (т.е. r равняется 1) и одна из которых имеется у L1, в том числе в предпочтительных вариантах ее реализации, как изложено выше.
Как изложено выше, EDS-группа имеет структуру, выбранную из (Е-1А), (Е-1 В), (Е-2А) и (Е-2 В):
где
 помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
s равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 1;
t равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 2;
R5A представляет собой, независимо для каждого случая, когда s>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R5A и фенильным кольцом указывает на то, что s групп R5A замещают s атомов водорода в любом положении на фенильном кольце;
R5B представляет собой, независимо для каждого случая, когда s>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -NH2, и причем связь между R5B и фенильным кольцом указывает на то, что s групп R5B замещают s атомов водорода в любом положении на фенильном кольце;
R6A представляет собой, независимо для каждого случая, когда t>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R6A и фенильным кольцом указывает на то, что t групп R6A замещают t атомов водорода в любом положении на фенильном кольце;
R6B представляет собой, независимо для каждого случая, когда t>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1 В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -ОН, и причем связь между R6B и фенильным кольцом указывает на то, что t групп R6B замещают t атомов водорода в любом положении на фенильном кольце.
В целом, предпочтительно, чтобы в EDS-группе (Е-1А) заместители R5A были одинаковыми при s>1 и были выбраны из -NO2 и -СООН, и, более предпочтительно, представляли собой -СООН, и чтобы в EDS-группе (Е-2А) заместители R6A были одинаковыми при t>1 и были выбраны из -NO2 и -СООН, и, более предпочтительно, представляли собой -СООН.
Подобным образом, в целом, предпочтительно, чтобы в EDS-группе (Е-1 В) заместители R5B были одинаковыми при s>1 и были выбраны из -ОН и -NH2, и, более предпочтительно, представляли собой -NH2, и чтобы в EDS-группе (Е-2 В) заместители R6B были одинаковыми при t>1 и были выбраны из -ОН и -NH2, и, более предпочтительно, представляли собой -ОН.
Таким образом, также предпочтительно, чтобы соединение формулы (I) содержало EDS-группу, которая имеет формулу (Е-2А):

где  помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I); и
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I); и
t равняется 1 или 2, a R6A представляет собой -NO2 или -СООН.
Наиболее предпочтительной в качестве EDS-группы в контексте настоящего изобретения является группа

В соответствии с вышеизложенным, предпочтительные соединения формулы (I) иллюстрируются следующей формулой (Ia)
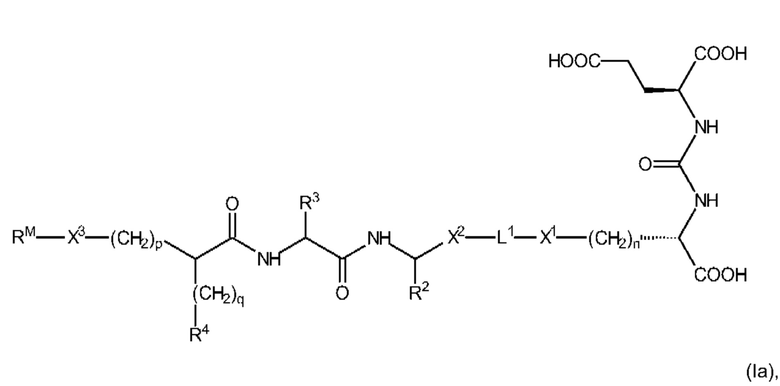
где n, X1, L1, X2, R2, R3, R4, q, р, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено выше, в том числе в предпочтительных вариантах реализации.
Более предпочтительные соединения формулы (I) иллюстрируются следующей формулой (Ib)

где n, X1, L1, X2, R2, R3, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено выше, в том числе в предпочтительных вариантах реализации.
Еще более предпочтительные соединения формулы (I) иллюстрируются следующей формулой (Ic)

где n, X1, L1, X2, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено выше, в том числе в предпочтительных вариантах ее реализации.
Еще более предпочтительные соединения формулы (I) иллюстрируются следующими формулами (Id) и (Ie):
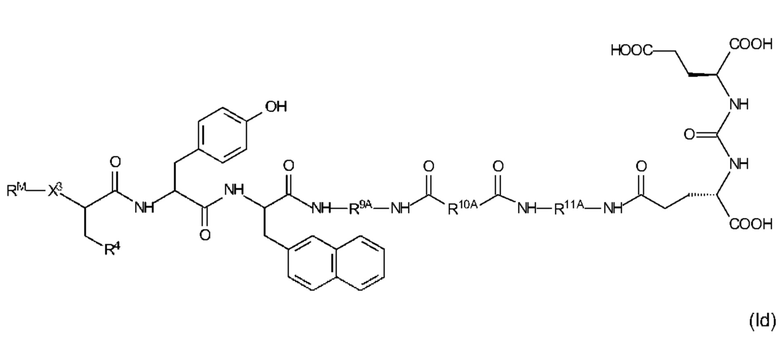
где R9A, R10A, R11A, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где (i) R4 представляет собой EDS-группу со структурой, как определено выше, в том числе в предпочтительных вариантах ее реализации, или (ii) R10A имеет одну EDS-группу со структурой, как определено выше, в том числе в предпочтительных вариантах ее реализации, или применимо как (i), так и (ii);
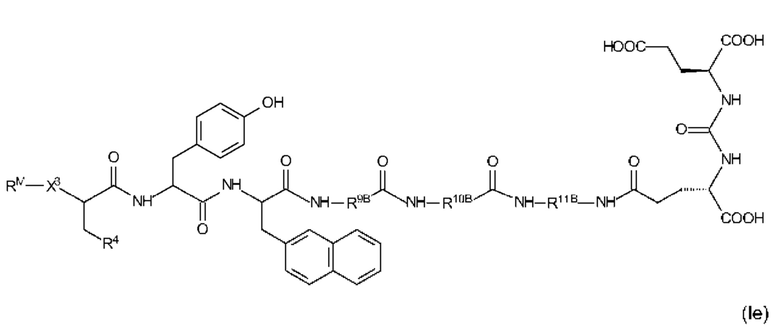
где R9B, R10B, R11B, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где R4 представляет собой EDS-группу со структурой, как определено выше, в том числе в предпочтительных вариантах ее реализации.
Особенно предпочтительные соединения формулы (I) иллюстрируются следующими формулами (If) и (Ig)

где R9A, R10A, R11A, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где (i) R4 представляет собой EDS-группу со структурой, как определено выше, в том числе в предпочтительных вариантах ее реализации, или (ii) R10A имеет одну EDS-группу со структурой, как определено выше, в том числе в предпочтительных вариантах ее реализации, или применимо как (i), так и (ii);
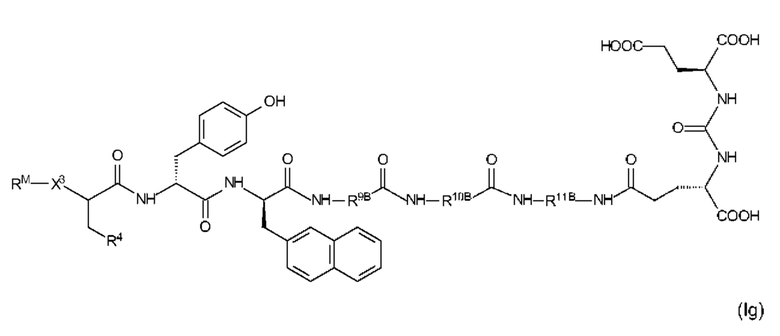
где R9B, R10B, R11B, R4, X3 и RM являются такими, как определено выше, в том числе в предпочтительных вариантах их реализации, и где R4 представляет собой EDS-группу со структурой, как определено выше, в том числе в предпочтительные варианты ее реализации.
В предпочтительном варианте реализации указанная хелатирующая группа имеет радионуклидную связь, радионуклид которой излучает а-излучение. Радионуклиды, излучающие α-излучение, включают 212Bi, 213Bi и 225Ас.
Как отмечено выше, включение электронодефицитных заместителей значительно повысило способности интернализации. Эта особенность приводит к более высокому опухолевому поглощению и особенно более длительному удержанию в опухолевой ткани, как продемонстрировано in vivo экспериментами (см. примеры). Поскольку комплекс из хелатора и радионуклида, излучающего альфа-частицу, подвержен декомплексации за счет физического эффекта отдачи, признак более длительного внутриклеточного удержания снизит вероятность свободно циркулирующих радионуклидов in vivo и, следовательно, повысит безопасность и уменьшит нежелательное облучение.
Особенно предпочтительными соединениями по изобретению являются следующие:

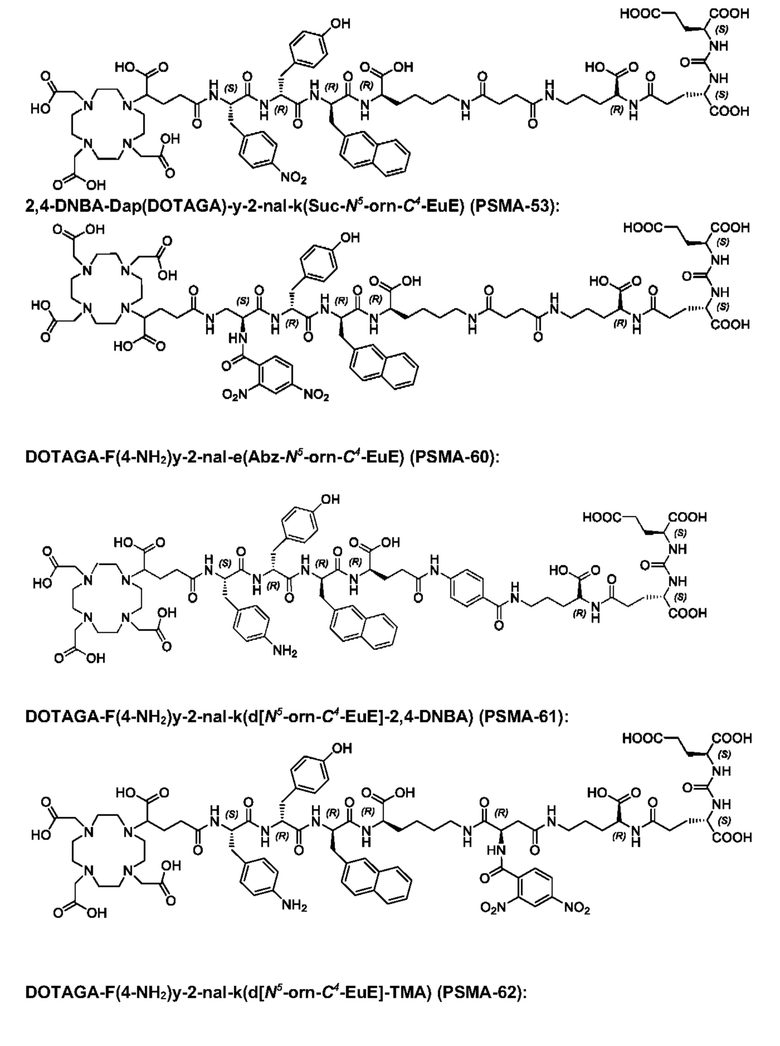
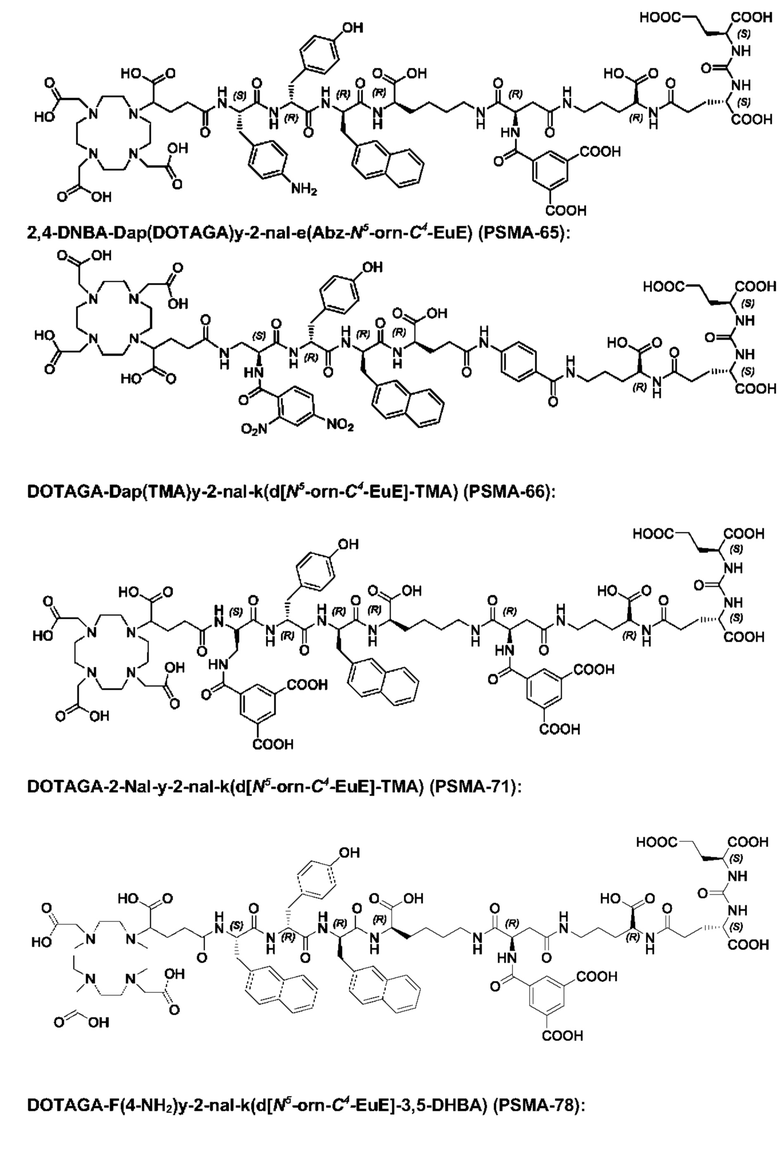

Полезные свойства этих соединений по изобретению можно увидеть из данных, представленных в таблицах 1 и 2 ниже, причем эти данные графически представлены на фигуре 1.
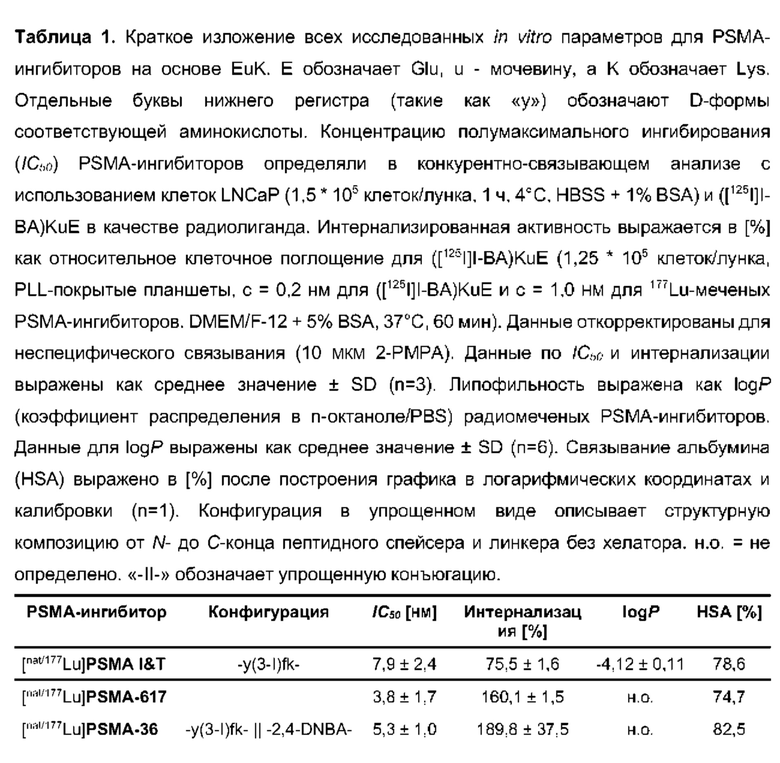

Предпочтительные схемы мечения для этих наиболее предпочтительных соединений являются такими, как определено в настоящем документе выше.
Еще в одном аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая или состоящая из одного или более соединений или солей по изобретению, как раскрыто в настоящем документе выше.
Еще в одном аспекте настоящего изобретения представлена диагностическая композиция, содержащая или состоящая из одного или более соединений или солей по изобретению, как раскрыто в настоящем документе выше.
Еще в одном аспекте настоящего изобретения представлена терапевтическая композиция, содержащая или состоящая из одного или более соединений или солей по изобретению, как раскрыто в настоящем документе выше.
Фармацевтическая композиция может дополнительно содержать фармацевтически приемлемые носители, вспомогательные вещества и/или разбавители. Примеры подходящих фармацевтически приемлемых носителей, вспомогательных веществ и/или разбавителей широко известны из уровня техники и включают фосфатно-солевые буферные растворы, воду, эмульсии, такие как эмульсии масло/вода, различные типы смачивающих агентов, стерильных растворов и т.д. Композиции, содержащие такие носители, могут быть составлены широко известными традиционными способами. Эти фармацевтические композиции могут вводиться субъекту в подходящей дозировке. Введение подходящих композиций может осуществляться различными способами, например, путем внутривенного, интраперитонеального, подкожного, внутримышечного, местного, интрадермального, интраназального или интрабронхиального введения. Особенно предпочтительно, чтобы указанное введение осуществлялось путем инъекции и/или доставки, например, в участок в поджелудочной железе или в артерию головного мозга, или непосредственно в ткань головного мозга. Композиции также могут вводиться непосредственно в участок-мишень, например, путем биолистической доставки во внешний или внутренний участок-мишень, такой как поджелудочная железа или головной мозг. Режим дозировки будет определен лечащим врачом и клиническими факторами. Как широко известно из области медицины, дозировки для любого одного пациента зависят от множества факторов, в том числе габаритов пациента, области на поверхности тела, возраста, конкретного соединения, подлежащего доставке, пола, времени и пути введения, общего состояния здоровья и других одновременно вводимых лекарственных средств. Фармацевтически активное вещество может присутствовать в количествах от 0,1 нг до 10 нг/кг массы тела на дозу; однако предусмотрены дозы, которые ниже или выше этого примера диапазона, особенно при учете вышеуказанных факторов.
В части того, что раскрытая выше фармацевтическая композиция, диагностическая композиция и терапевтическая композиция содержит одно или более соединений по изобретению, предпочтительно, чтобы какие-либо дополнительные фармацевтически активные соединения, диагностически активные соединения или терапевтически активные соединения отсутствовали. В качестве альтернативы, могут присутствовать дополнительные фармацевтически активные соединения, диагностически активные соединения или терапевтически активные соединения, например, противоопухолевые агенты.
Комбинация терапевтического воздействия с соединениями по настоящему изобретению может иметь синергетическое или кумулятивное лечебное действие наподобие лечения нейроэндокринных опухолей с помощью радиотерапии [177Lu]DOTATATE в комбинации с химиотерапией или иммунотерапией. Первое исследование 3-й фазы, сравнивающее комбинацию 177Lu PRRT и капецитабина (Xeloda; Genentech), перорального химиотерапевтического агента, с взятым отдельно [177Lu]DOTATATE было инициировано в Erasmus МС, Роттердам в 2017 году (van Essen М, Krenning ЕР, Kam BL, de Herder WW, van Aken MO, Kwekkeboom DJ. Report on short-term side effects of treatments with 177Lu-octreotate in combination with capecitabine in seven patients with gastroenteropancreatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2008; 35:743-748).
Недавно были опубликованы дополнительные исследования комбинированной терапии под названием пептидная рецепторная химиорадионуклидная терапия (PRCRT) (Kong G, Callahan J, Hofman MS, et al. High clinical and morphologic response using 90Y-DOTA-octreotate sequenced with 177Lu-DOTA-octreotate induction peptide receptor chemoradionuclide therapy (PRCRT) for bulky neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2017; 44:476-489). Подобные «подходы комбинированного лечения» будут осуществляться в ближайщем будущем для улучшения эффективности PSMA-нацеленных радиолигандных видов терапии.
Еще в одном аспекте настоящего изобретения представлено одно или более соединений или солей по изобретению, как раскрыто в настоящем документе выше, для применения в медицине.
Предпочтительные варианты применения в медицине включают применение в ядерной медицине, такой как ядерная диагностическая визуализация, которая также называется ядерной молекулярной визуализацией, и/или нацеленной радиотерапии заболеваний, связанных со сверхэкспрессией, предпочтительно, PSMA на пораженной заболеванием ткани.
Еще в одном аспекте настоящего изобретения представлено соединение или соль по изобретению, как определено в настоящем документе выше, для применения в способе диагностики и/или определения стадии рака, предпочтительно, рака простаты.
Предпочтительными показаниями являются обнаружение или определение стадии рака, такого как, но без ограничения, глиомы высокой степени злокачественности, рак легких и, в особенности, рак простаты и метастазирующий рак простаты, обнаружение метастатического заболевания у пациентов с первичным раком простаты с риском от среднего до высокого, а также обнаружение метастатических участков даже при низких значениях PSA в сыворотке у пациентов с биохимически рецидивирующим раком простаты. Другим предпочтительным показанием является получение изображений и визуализация неоангиогенеза.
В части медицинских показаний, подлежащих воздействию терапией, особенно радиотерапией, предпочтительным показанием является рак. Особенно предпочтительным показанием является рак простаты.
Еще в одном аспекте настоящего изобретения представлено соединение или соль по изобретению, как определено в настоящем документе выше, для применения в способе диагностики и/или определения стадии рака, предпочтительно, рака простаты.
Что касается вариантов реализации, охарактеризованных в настоящем описании, в частности, в формуле изобретения, предполагается, что каждый вариант реализации, указанный в зависимом пункте формулы изобретения, взят в комбинации с каждым вариантом реализации каждого пункта формулы изобретения (независимого или зависимого), от которого зависит указанный зависимый пункт формулы изобретения. Например, в случае, если в независимом пункте 1 формулы изобретения указано 3 альтернативы А, В и С, в зависимом пункте 2 формулы изобретения указано 3 альтернативы D, Е и F, а в зависимом пункте 3 формулы изобретения, который зависит от пунктов 1 и 2, и в котором указано 3 альтернативы G, Н и I, следует понимать, что в описании однозначным образом раскрыты варианты реализации, соответствующие комбинациям A, D, G; A, D, Н; A, D, I; А, Е, G; А, Е, Н; А, Е, I; A, F, G; A, F, Н; A, F, I; В, D, G; В, D, Н; В, D, I; В, Е, G; В, Е, Н; В, Е, I; В, F, G; В, F, Н; В, F, I; С, D, G; С, D, Н; С, D, I; С, Е, G; С, Е, Н; С, Е, I; С, F, G; С, F, Н; С, F, I, если иное конкретно не указано.
Подобным образом, а также в случаях, когда в независимых и/или зависимых пунктах формулы изобретения не указаны альтернативы, следует понимать, что если зависимые пункты формулы изобретения ссылаются на несколько предыдущих пунктов, любая комбинация объекта изобретения, охваченная им, будет рассматриваться как раскрытая явным образом. Например, в случае, когда имеется независимый пункт 1 формулы изобретения, зависимый пункт 2 формулы изобретения, ссылающийся на пункт 1, и зависимый пункт 3 формулы изобретения, ссылающийся на оба пункта 2 и 1, из этого следует, что комбинация объекта изобретения по пунктам 3 и 1 явным и однозначным образом раскрыта как комбинация объекта изобретения по пунктам 3, 2 и 1. В случае, когда имеется еще один зависимый пункт 4 формулы изобретения, который ссылается на любой из пунктов 1-3 формулы изобретения, из этого следует, что комбинация объекта изобретения по пунктам 4 и 1, по пунктам 4, 2 и 1, по пунктам 4, 3 и 1, а также по пунктам 4, 3, 2 и 1 раскрыта явным и однозначным образом.
В частности, в изобретении представлен объект, резюмированный в следующих пунктах.
1. Соединение формулы (I) или его фармацевтически приемлемая соль,

где:
m представляет собой целое число от 2 до 6, предпочтительно, от 2 до 4, более предпочтительно, 2;
n представляет собой целое число от 2 до 6, предпочтительно, от 2 до 4, более предпочтительно, 2 или 4;
R1L представляет собой СН2, NH или О, предпочтительно, NH;
R2L представляет собой С или Р(ОН), предпочтительно, С;
R3L представляет собой СН2, NH или О, предпочтительно, NH;
X1 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика и аминной связи, и, предпочтительно, представляет собой амидную связь;
L1 представляет собой двухвалентную связывающую группу со структурой, выбранной из следующего: олигоамид, олигоэфир, олиготиоэфир, сложный олигоэфир, сложный олиготиоэфир, олигомочевина, олиго(эфир-амид), олиго(тиоэфир-амид), олиго(сложный эфир-амид), олиго(сложный тиоэфир-амид), олиго(мочевина-амид), олиго(эфир-тиоэфир), олиго(эфир-сложный эфир),олиго(эфир-сложный тиоэфир), олиго(эфир-мочевина), олиго(тиоэфир-сложный эфир), олиго(тиоэфир-сложный тиоэфир), олиго(тиоэфир-мочевина), олиго(сложный эфир-сложный тиоэфир), олиго(сложный эфир-мочевина) и олиго(сложный тиоэфир-мочевина), предпочтительно, со структурой, выбранной из олигоамида и олиго(сложный эфир-амид),
при этом связывающая группа может иметь EDS-группу;
X2 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика и аминной связи, и, предпочтительно, представляет собой амидную связь;
R2 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, при этом арильная группа или аралкильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН;
R3 представляет собой необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, при этом арильная группа или аралкильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, и -ОН;
r равняется 0 или 1, предпочтительно,1;
р равняется 0 или 1;
q равняется 0 или 1;
и, предпочтительно, p+q=1;
R4 выбран из необязательно замещенной арильной группы и EDS-группы, при этом арильная группа может быть замещена на своем ароматическом кольце одним или более заместителями, выбранными из галогена, предпочтительно, I, -ОН и -NH2;
X3 выбран из амидной связи, эфирной связи, тиоэфирной связи, сложноэфирной связи, тиосложноэфирной связи, мочевинного мостика, аминной связи и группы, имеющей формулу
 , где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части соединения формулы (I);
, где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 к остальной части соединения формулы (I);
и представляет собой, предпочтительно, амидную связь;
RM представляет собой группу метки, которая содержит хелатирующую группу, необязательно содержащую хелатированный нерадиоактивный или радиоактивный катион;
и где EDS-группа содержится по меньшей мере в одном месте в соединении формулы (I) и имеет структуру, выбранную из (Е-1А), (Е-1 В), (Е-2А) и (Е-2 В):

где
 помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I);
s равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 1;
t равняется 1, 2 или 3, предпочтительно, 1 или 2, и более предпочтительно, 2;
R5A представляет собой, независимо для каждого случая, когда s>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R5A и фенильным кольцом указывает на то, что s групп R5A замещают s атомов водорода в любом положении на фенильном кольце;
R5B представляет собой, независимо для каждого случая, когда s>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -NH2, и причем связь между R5B и фенильным кольцом указывает на то, что s групп R5B замещают s атомов водорода в любом положении на фенильном кольце;
R6A представляет собой, независимо для каждого случая, когда t>1, электроноакцепторный заместитель, который, предпочтительно, выбран из -NO2 и -СООН, и который, более предпочтительно, представляет собой -СООН, и причем связь между R6A и фенильным кольцом указывает на то, что t групп R6A замещают t атомов водорода в любом положении на фенильном кольце;
R6B представляет собой, независимо для каждого случая, когда t>1, заместитель, имеющий неподеленную пару электронов на атоме, непосредственно прикрепленном к фенильному кольцу, показанному в формуле (Е-1 В), при этом заместитель, предпочтительно, выбран из -ОН и -NH2, и который, более предпочтительно, представляет собой -ОН, и причем связь между R6B и фенильным кольцом указывает на то, что t групп R6B замещают t атомов водорода в любом положении на фенильном кольце.
2. Соединение или соль по пункту 1, где m равняется 2, n равняется 2 или 4, R1L представляет собой NH, R2L представляет собой С, a R3L представляет собой NH.
3. Соединение или соль по пункту 1 или 2, где n равняется 2.
4. Соединение или соль по любому из пунктов 1-3, где X1 представляет собой амидную связь.
5. Соединение или соль по пункту 4, где n равняется 2, а X1 представляет собой амидную связь с атомом углерода в амидной связи -C(O)-NH-, прикрепленным к группе -(СН2)n-.
6. Соединение или соль по любому из пунктов 1-5, где L1 представляет собой двухвалентную связывающую группу со структурой, выбранной из олигоамида, который содержит всего от 1 до 5, более предпочтительно, всего от 1 до 3, и наиболее предпочтительно, всего 1 или 2 амидных связи в своем остове, и олиго(сложный эфир-амида), который содержит всего от 2 до 5, более предпочтительно, всего от 2 до 3, и наиболее предпочтительно, всего 2 амидных и сложноэфирных связей в своем остове, при этом связывающая группа может иметь EDS-группу.
7. Соединение или соль по пункту 6, где L1 представляет собой двухвалентную связывающую группу с олигоамидной структурой, которая содержит 1 или 2 амидных связи в своем остове, при этом связывающая группа может иметь EDS-группу.
8. Соединение или соль по любому из пунктов 1-7, где связывающая группа L1 имеет одну EDS-группу.
9. Соединение или соль по любому из пунктов 1-8, где X2 представляет собой амидную связь.
10. Соединение или соль по пункту 9, где X2 представляет собой амидную связь с атомом азота в амидной связи -C(O)-NH-, прикрепленным к L1.
11. Соединение или соль по любому из пунктов 1-10, где фрагмент -Х2-L1-Х1- в формуле (I) имеет структуру, выбранную из:
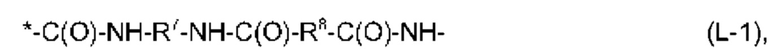
 и
и

причем амидная связь, помеченная *, прикреплена к атому углерода, имеющему R2 в формуле (I), и причем
R7, R8, R9A, R9B, R11A и R11B независимо выбраны из необязательно замещенного С2-С10-алкандиила, предпочтительно, необязательно замещенного линейного С2-С10-алкандиила, причем каждая алкандиильная группа может быть замещена одним или более заместителями, независимо выбранными из -ОН, -ОСН3, -СООН, -СООСН3, -NH2, -NHC(NH)NH2 и EDS-группы, и
R10A и R10B выбраны из необязательно замещенного С2-С10-алкандиила, предпочтительно, необязательного замещенного линейного С2-С10-алкандиила, и необязательно замещенного С6-С10-арендиила, предпочтительно, фенилена, причем каждая алкандиильная и арендиильная группа может быть замещена одним или более заместителей, независимо выбранными из -ОН, -ОСН3, -СООН, -СООСН3, -NH2, -NHC(NH)NH2 и EDS-группы. R10A представляет собой, предпочтительно, необязательно замещенный С2-С10-алкандиил, более предпочтительно, необязательно замещенный линейный С2-С10-алкандиил, как определено выше. R10B представляет собой, предпочтительно, необязательно замещенный С6-С10-арендиил, как определено выше, более предпочтительно, фениленовую группу, например, пара-фениленовую группу.
12. Соединение или соль по пункту 11, где общее количество атомов углерода в R7 и R8 формулы (L-1) составляет от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях, общее количество атомов углерода в R9A, R10A и R11A формулы (L-2A) составляет от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях, и общее количество атомов углерода в R9B, R10B и R11B формулы (L-2B) составляет от 6 до 20, более предпочтительно, от 6 до 16, без атомов углерода, содержащихся в необязательных заместителях.
13. Соединение или соль по пункту 11 или 12, где
фрагмент -Х2-L1-Х1- имеет структуру (L-1), a R8 имеет EDS-группу в качестве по меньшей мере одного заместителя, или
фрагмент -Х2-L1-Х1- имеет структуру (L-2A), a R10A имеет EDS-группу в качестве по меньшей мере одного заместителя.
14. Соединение или соль по пункту 7, где фрагмент -Х2-L1-Х1- имеет структуру, выбранную из:
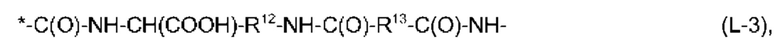
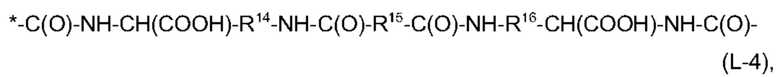 и
и

причем связь, помеченная *, прикреплена к атому углерода, имеющему R2 в формуле (I),
R12 и R14 независимо выбраны из линейного С2-С6-алкандиила, предпочтительно, из линейного С3-С6-алкандиила,
R13 представляет собой линейный С2-С10-алкандиил, предпочтительно, линейный С4-С8-алкандиил,
R15 и R16 независимо выбраны из линейного С2-С6-алкандиила, предпочтительно, из линейного С2-С4-алкандиила,
и причем каждый из R13 и R15 может иметь одну EDS-группу в качестве заместителя и, более предпочтительно, каждый из R13 и R15 имеет одну EDS-группу в качестве заместителя,
R17 представляет собой линейный С2-С6-алкандиил, предпочтительно, линейный С2-С4-алкандиил,
R18 представляет собой фениленовую группу, например, пара-фениленовую группу, и
R19 представляет собой линейный С2-С6-алкандиил, предпочтительно, линейный С2-С4-алкандиил.
15. Соединение или соль по пункту 14, где общее количество атомов углерода в R12 и R13 в формуле (L-3), без атомов углерода, содержащихся в EDS-группе в качестве заместителя, составляет от 6 до 16, более предпочтительно, от 6 до 14, а общее количество атомов углерода в R14, R15 и R18 в формуле (L-4), без атомов углерода, содержащихся в EDS-группе в качестве заместителя, составляет от 6 до 16, более предпочтительно, от 6 до 14.
16. Соединение или соль по любому из пунктов 1-15, где R2 представляет собой необязательно замещенную аралкильную группу, выбранную из необязательно замещенного -СН2-фенила и необязательно замещенного -СН2-нафтила, более предпочтительно, необязательно замещенного -СН2-(2-нафтила), причем фенильная и нафтильная группа необязательно замещены заместителем, выбранным из галогена, предпочтительно, I, и -ОН.
17. Соединение или соль по пункту 16, где R2 представляет собой аралкильную группу формулы -СН2-нафтил, более предпочтительно, -СН2-(2-нафтил).
18. Соединение или соль по любому из пунктов 1-17, где R3 представляет собой необязательно замещенную аралкильную группу, выбранную из необязательно замещенного -СН2-фенила и необязательно замещенного -СН2-нафтила, более предпочтительно, необязательно замещенного -СН2-фенила, причем фенильная и нафтильная группа необязательно замещены заместителем, выбранным из галогена, предпочтительно, I, и -ОН.
19. Соединение или соль по пункту 18, где R3 представляет собой аралкильную группу формулы -СН2-фенил, причем фенильное кольцо замещено одним заместителем, который представляет собой -ОН, или комбинацией одного заместителя -ОН и одного заместителя -I.
20. Соединение или соль по любому из пунктов 1-15, где R2 представляет собой группу формулы
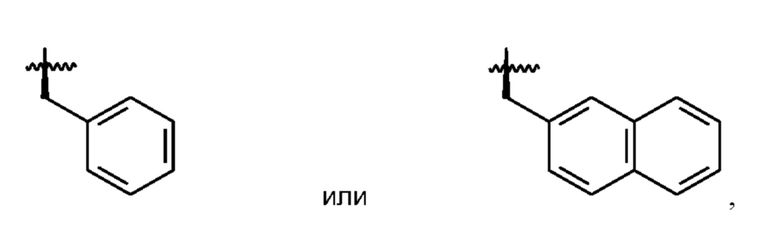
a R3 представляет собой группу формулы

где  помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части соединения формулы (I).
помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части соединения формулы (I).
21. Соединение или соль по пункту 20,
где R2 представляет собой группу формулы
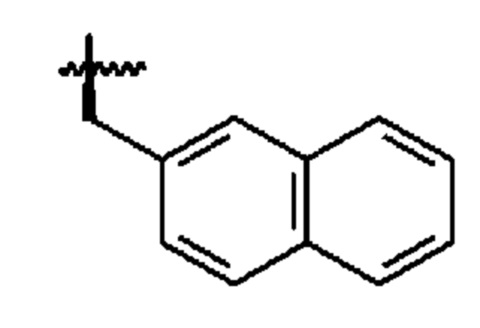
a R3 представляет собой группу формулы

где  помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части молекулы.
помечает связь, которая прикрепляет R2 и R3, соответственно, к остальной части молекулы.
22. Соединение или соль по любому из пунктов 1-21, где r равняется 1.
23. Соединение или соль по любому из пунктов 1-22, где р равняется 0, a q равняется 1.
24. Соединение или соль по любому из пунктов 1-23, где R4 выбран из фенила, необязательно нафтила, и EDS-группы.
25. Соединение или соль по пункту 24, где R4 выбран из нафтила, более предпочтительно, 2-нафтила, и EDS-группы.
26. Соединение или соль по любому из пунктов 1-25, где X3 представляет собой амидную связь или группу формулы
 , где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 костальной части молекулы.
, где помеченная связь в карбонильной группе прикрепляет X3 к RM, а другая помеченная связь прикрепляет X3 костальной части молекулы.
27. Соединение или соль по пункту 26, где X3 представляет собой амидную связь -C(O)-NH- с атомом углерода, прикрепленным к RM.
28. Соединение или соль по любому из пунктов 1-27, где RM представляет собой хелатирующую группу, необязательно содержащую хелатирующий нерадиоактивный или радиоактивный катион.
29. Соединение или соль по любому из пунктов 1-28, где хелатирующая группа содержит по меньшей мере одно из
(i) структуры макроциклического кольца с от 8 до 20 атомами кольца, из которых 2 или более, предпочтительно, 3 или более, выбраны из атомов кислорода, атомов серы и атомов азота; и
(i) ациклической хелатирующей структуры с открытой цепью с от 8 до 20 атомами основной цепи, из которых 2 или более, предпочтительно, 3 или более, являются гетероатомами, выбранными из атомов кислорода, атомов серы и атомов азота.
Соединение или соль по любому из пунктов 1-29, где хелатирующая группа представляет собой остаток хелатирующего агента, выбранного из следующего: бис(карбоксиметил)-1,4,8,11-тетраазабицикпо[6.6.2]гексадекан (СВТЕ2а), цикпогексил-1,2-диаминтетрауксусная кислота (CDTA), 4-(1,4,8,11-тетраазациклотетрадец-1-ил)-метилбензойная кислота (СРТА), N'-[5-[ацетил(гидрокси)амино]пентил]-N-[5-[[4-[5-аминопентил-(гидрокси)амино]-4-оксобутаноил]амино]пентил]-N-гидроксибутандиамид (DFO), 4,11-бис(карбоксиметил)-1,4,8,11 -тетраазабицикпо[6.6.2]гексадекан (DO2A) 1,4,7,10-тетраазациклододекан-N,N',N'',N'''-тетрауксусная кислота (DOTA), 2-[1,4,7,10-тетраазациклододекан-4,7,10-триуксусная кислота]-пентандиовая кислота (DOTAGA), N,N'-дипиридоксилэтилендиамин-N,N'-диацетат-5,5'-бис(фосфат) (DPDP), диэтилентриаминпентауксусная кислота (DTPA), этилендиамин-N,N'-тетрауксусная кислота (EDTA), этиленгликоль-O,O-бис(2-аминоэтил)-N,N,N',N'-тетрауксусная кислота (EGTA), N,N-бис(гидроксибензил)-этилендиамин-N,N'-диацетилуксусная кислота (HBED), гидроксиэтилдиаминтриуксусная кислота (HEDTA), 1-(р-нитробензил)-1,4,7,10-тетраазацикподекан-4,7,10-триацетат (НР-DOA3), 6-гидразинил-N-метилпиридин-3-карбоксамид (HYNIC), 1,4,7-триазацикпононан-1-янтарная кислота-4,7-диацетилуксусная кислота (NODASA), 1-(1-карбокси-3-карбоксипропил)-4,7-(карбокси)-1,4,7-триазациклононан (NODAGA), 1,4,7-триазациклононантриуксусная кислота (NOTA), 4,11-бис(карбоксиметил)-1,4,8,11-тетраазабицикпо[6.6.2]гексадекан (ТЕ2А), 1,4,8,11-тетраазациклододекан-1,4,8,11-тетрауксусная кислота (ТЕТА), терпиридин-бис(метиленаминтетрауксусная кислота (ТМТ), 1,4,7,10-тетраазацикпотридекан-N,N',N'',N'''-тетрауксусная кислота (TRITA) и триэтилентетраамингексауксусная кислота (ТТНА), N,N'-бис[(6-карбокси-2-пиридил)метил]-4,13-диаза-18-краун-6 (H2macropa) и 4-амино-4-{2-[(3-гидрокси-1,6-диметил-4-оксо-1,4-дигидро-пиридин-2-илметил)-карбамоил]-этил}пимелиновая кислота бис-[(3-гидрокси-1,6-диметил-4-оксо-1,4-дигидро-пиридин- 2-илметил)-амид] (ТНР); причем остаток образуется за счет ковалентного связывания карбоксильной группы, содержащейся в хелатирующем агенте, с остальной частью соединения через сложноэфирную или амидную связь, более предпочтительно, амидную связь.
31. Соединение или соль по пункту 30, где хелатирующий агент выбран из DOTA и DOTAGA.
32. Соединение или соль по пункту 30 или 31, где X3 представляет собой амидную связь, прикрепляющую хелатирующую группу костальной части молекулы.
33. Соединение или соль по пункту 32, где RM-X3- представляет собой группу формулы
 или
или

где группа, обозначенная  , прикреплена к остальной части соединения формулы (I), и где хелатирующая группа может содержать хелатированный нерадиоактивный или радиоактивный катион.
, прикреплена к остальной части соединения формулы (I), и где хелатирующая группа может содержать хелатированный нерадиоактивный или радиоактивный катион.
34. Соединение или соль по любому из пунктов 1-33, где хелатирующая группа содержит хелатированный катион, предпочтительно, хелатированный радиоактивный катион, выбранный из катионов 44Sc, 47Sc, 51Cr, 52mMn, 58Co, 52Fe, 56Ni, 57Ni, 62Cu, 64Cu, 67Cu, 66Ga, 68Ga, 67Ga, 89Zr, 90Y, 89Y, 94mTc, 99mTc, 97Ru, 105Rh, 109Pd, 111Ag, 110mIn, 111In, 113mIn, 114mIn, 117mSn, 121Sn, 127Te, 142Pr, 143Pr, 149Pm, 151Pm, 149Tb, 153Sm, 157Gd, 161Tb, 166Ho, 165Dy, 169Er, 169Yb, 175Yb, 172Tm, 177Lu, 186Re, 188Re, 191Pt 197Hg, 198Au, 199Au, 212Pb, 203Pb, 211At, 212Bi, 213Bi, 223Ra, 225Ac и 227Th, или катионной молекулы, содержащей 18F, такой как 18F-[AIF]2+.
35. Соединение или соль по пункту 34, где хелатирующая группа содержит хелатированный катион, выбранный из катионов 44Sc, 47Sc, 64Cu, 67Cu, 68Ga, 90Y, 111In, 161Tb, 166Ho, 177Lu, 188Re, 212Pb, 212Bi, 213Bi, 225Ac и 227Th, или катионной молекулы, содержащей 18F.
36. Соединение или соль по любому из пунктов 1-35, где соединение формулы (I) содержит одну или две EDS-группы.
37. Соединение или соль по пункту 36, где соединение формулы (I) содержит одну EDS-группу, которая имеется у связывающей группы L1, или содержит две EDS-группы, одна из которых представлена R4, а другая имеется у L1.
38. Соединение или соль по любому из пунктов 1-37, где в EDS-группе (Е-1А) заместители R5A являются одинаковыми при s>1 и выбраны из -NO2 и -СООН; и где в EDS-группе (Е-2А) заместители R6A являются одинаковыми при t>1 и выбраны из -NO2 и -СООН.
39. Соединение или соль по любому из пунктов 1-38, где в EDS-группе (Е-1В) заместители R5B являются одинаковыми при s>1 и выбраны из -ОН и -NO2; в EDS-группе (Е-2В) заместители R6B являются одинаковыми при t>1 и выбраны из -ОН и -NO2.
40. Соединение или соль по любому из пунктов 1-39, которое содержит EDS-группу, которая имеет формулу (Е-2А):
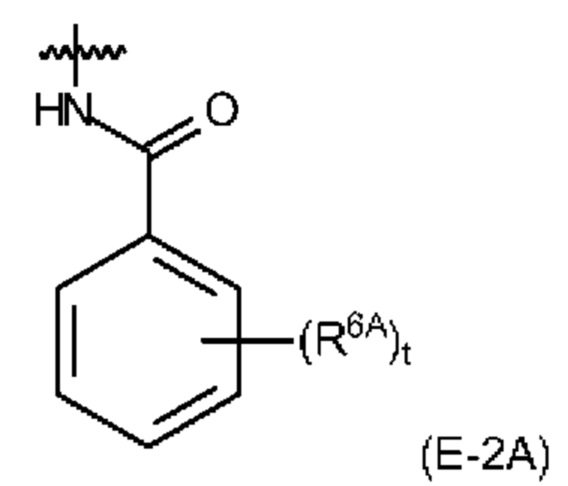
где  помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I); и
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I); и
t равняется 1 или 2, a R6A выбран из -NO2 и -СООН.
41. Соединение по любому из пунктов 1-38, где EDS-группа имеет формулу (Е-3)

где  помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I).
помечает связь, которая прикрепляет EDS-группу к остальной части соединения формулы (I).
42. Соединение по любому из пунктов 1-41, которое имеет следующую формулу (Ia), или его фармацевтически приемлемая соль:

где n, X1, L1, X2, R2, R3, R4, q, р, X3 и RM являются такими, как определено в предыдущих пунктах, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено в предыдущих пунктах.
43. Соединение по пункту 42, которое имеет следующую формулу (Ib), или его фармацевтически приемлемая соль:
где n, X1, L1, X2, R2, R3, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено в предыдущих пунктах.
44. Соединение по пункту 43, которое имеет следующую формулу (Ic), или его фармацевтически приемлемая соль:

где n, X1, L1, X2, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где EDS-группа встречается по меньшей мере один раз и имеет структуру, как определено в предыдущих пунктах.
45. Соединение по пункту 44, которое имеет следующую формулу (Id) или (Ie), или его фармацевтически приемлемая соль:

где R9A, R10A, R11A, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где (i) R4 представляет собой EDS-группу со структурой, как определено в предыдущих пунктах, или (ii) R10A имеет одну EDS-группу со структурой, как определено в предыдущих пунктах, или применимо как (i), так и (ii);

где R9B, R10B, R11B, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где R4 представляет собой EDS-группу со структурой, как определено в предыдущих пунктах.
46. Соединение по пункту 45, которое имеет следующую формулу (If) или (Ig), или его фармацевтически приемлемая соль:

где R9A, R10A, R11A, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где (i) R4 представляет собой EDS-группу со структурой, как определено в предыдущих пунктах, или (ii) R10A имеет одну EDS-группу со структурой, как определено в предыдущих пунктах, или применимо как (i), так и (ii);
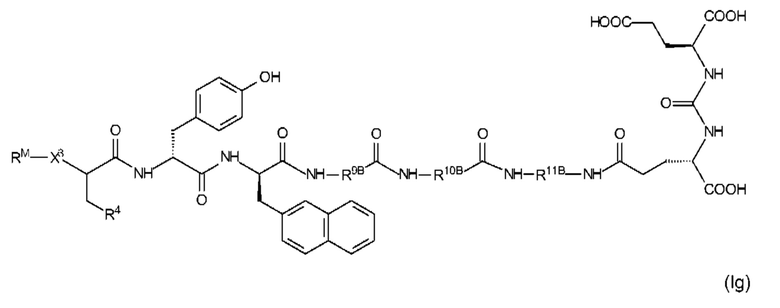
где R9B, R10B, R11B, R4, X3 и RM являются такими, как определено в предыдущих пунктах, и где R4 представляет собой EDS-группу со структурой, как определено в предыдущих пунктах.
47. Соединение или его соль по пункту 1, где указанное соединение или соль имеет одну из следующих формул:
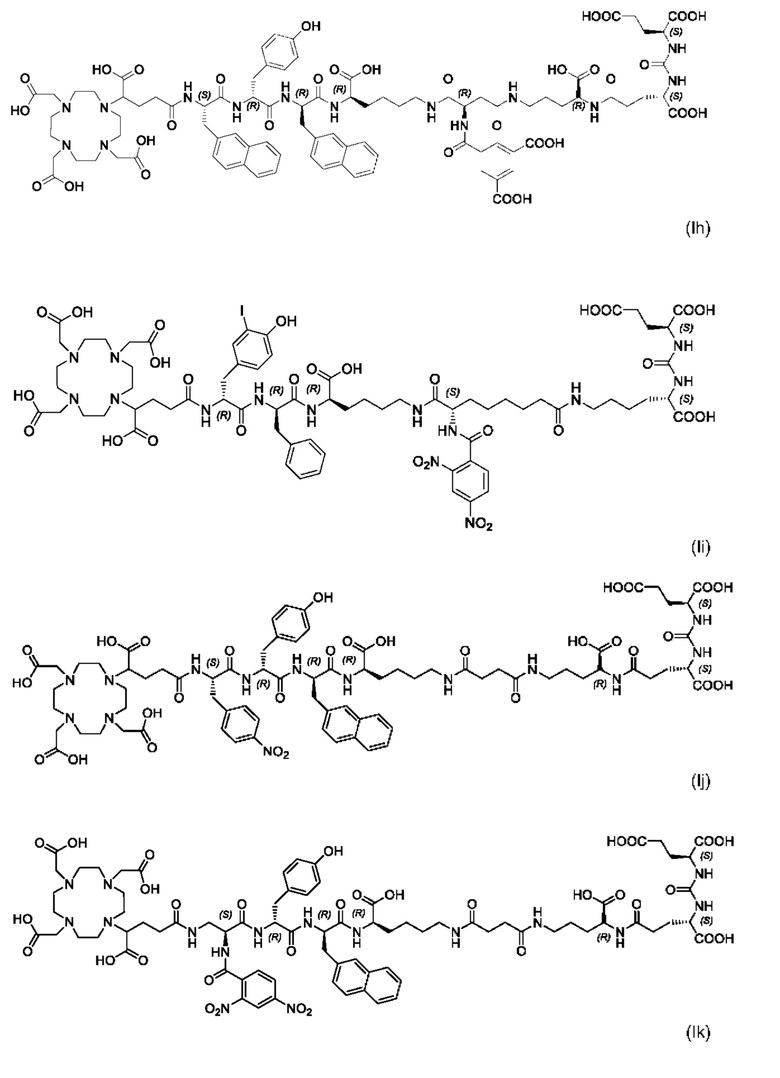

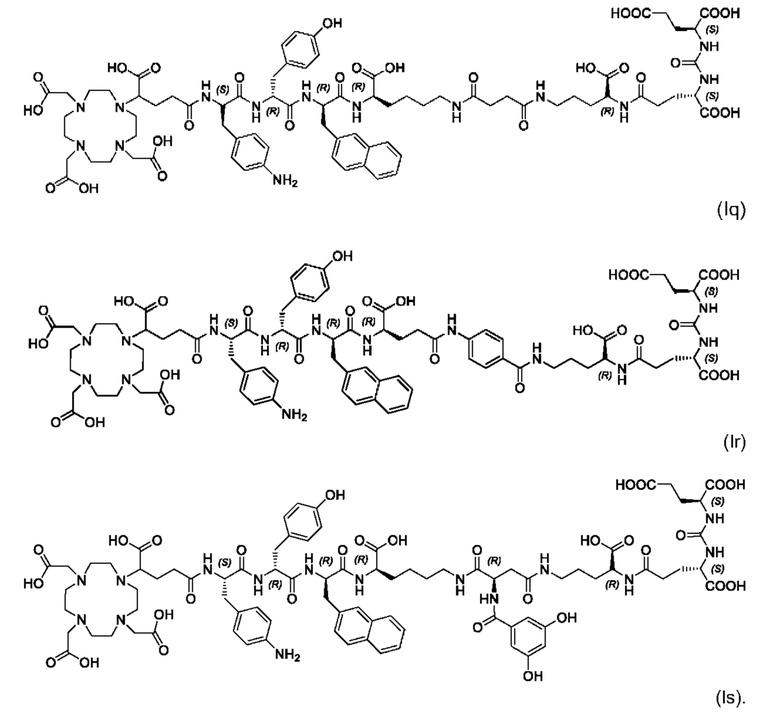
48. Фармацевтическая или диагностическая композиция, содержащая или состоящая из одного или более соединений или солей в соответствии с любым из пунктов 1-47.
49. Соединение или соль по любому из пунктов 1-47 для применения в способе диагностики и/или лечения
(a) рака, в том числе рака простаты; или
(b) нейроангиогенеза/ангиогенеза.
На фигурах показано:
Фигура 1: Совокупная диаграмма характеристик для [nat/177Lu]PSMA I&T и
[nat/177Lu]PSMA-62 и [nat/177Lu]PSMA-66.
Фигура 2: Кинетика экстернализации выбранных 177Lu-меченых PSMA-ингибиторов из клеток LNCaP. 1,25 * 105 клеток/лунку инкубировали в течение 1 ч с соответствующим радиолигандом (с=1,0 нм) при 37°С в DMEM-растворе (5% BSA). Затем удаляли супернатант и промывали один раз с помощью DMEM-раствора (5% BSA, 37°С). После этого, для замещения добавляли или А) только DM ЕМ-раствор (5% BSA), или В) блокадный DMEM-раствор (5% BSA, 10 мкм 2-РМРА). Общую клеточную интернализированную активность при t=0 мин корректировали по неспецифическому связыванию (10 мкм 2-РМРА) и нормализовали до 100%. Все данные выражены как среднее значение ±SD (n=3).
Фигура 3: Биораспределение (в % ВД/г) от 2,5 до 3,0 МБк (от 0,15 до 0,25 нмоль) [177Lu]PSMA-66 и [177Lu]PSMA I&T у мышей СВ-17 SCID с опухолью LNCaP (n=4, соответственно).
Фигура 4: Проекция максимальной интенсивности (MIP) сканированного изображения мкРЕТ у мышей СВ-17 SCID с опухолью LNCaP после инъекции приблизительно 10,3 МБк (0,19 нмоль меченого вещества) [68Ga]PSMA-36 (динамическое сканирование, суммирование кадров через от 1 до 1,5 ч после инъекции) (вверху слева). Несколько ТАС (логарифмический график) в % ВД/мл [68Ga]PSMA-36, полученные из данных динамического PET (время обнаружения 90 мин, 3D-реконструирование OSEM) у мыши СВ-17 SCID с опухолью LNCaP, по пулу крови (сердце), почкам, опухоли, мышце, слезной и слюнной железе.
Фигура 5: Проекция максимальной интенсивности (MIP) сканированных изображений мкРЕТ у мышей СВ-17 SCID с опухолью LNCaP после инъекции приблизительно 11 и 13 МБк (0,15-0,25 нмоль меченого вещества) 68Ga-меченого PSMA-ингибитора PSMA-62 и PSMA-66, соответственно (динамическое сканирование, суммирование кадров через от 1 до 1,5 ч после инъекции) (вверху слева). Несколько ТАС (логарифмический график) в % ВД/мл соответствующего 686а-меченого PSMA-ингибитора, полученные из данных динамического PET (время обнаружения 90 мин, 3D-реконструирование OSEM) у мышей СВ-17 SCID с опухолью LNCaP, по пулу крови (сердце), почкам, опухоли и мышце для обоих 68Ga-меченых веществ.
Фигура 6: Биораспределение (в % ВД/г) от 2,5 до 6,0 МБк (от 0,15 до 0,25 нмоль) [177Lu]PSMA-62, [177Lu]PSMA-66, [177Lu]PSMA-71 и [177Lu]PSMA I&T у мышей СВ-17 SCID с опухолью LNCaP (n=4, соответственно).
Изобретение проиллюстрировано на примерах.
Пример 1: Материалы и способы
1. Общая информация
Fmoc-(9-флуоренилметоксикарбонил-) и все другие аналоги защищенной аминокислоты были приобретены у «Bachem» (Бубендорф, Швейцария) или «Iris Biotech» (Марктредвиц, Германия). Смола 2-хлортритил хлорид (2-СТС) была получена у «PepChem» (Тюбинген, Германия). «Chematech» (Дижон, Франция) доставила хелатор DOTAGA-ангидрид. PSMA-DKFZ-617 был приобретен у «АВХ advanced chemical compounds)) (Радеберг, Германия). Все необходимые растворители и другие органические реагенты были приобретены у «Alfa Aesar» (Карлсруэ, Германия), «Sigma-Aldrich» (Мюнхен, Германия) или «VWR» (Дармштадт, Германия). Твердофазный синтез пептидов осуществляли вручную с использованием шприцевого шуттель-аппарата Intelli-Mixer («Neolab», Гейдельберг, Германия). Аналитическую высокоэффективную жидкостную хроматографию с обращенными фазами (RP-HPLC) проводили на колонне Nucleosil 100 С18 (5 мкм, 125 × 4,0 мм, «CS GmbH», Лангервеэ, Германия) с использованием системы Shimadzu для градиентной RP-HPLC («Shimadzu Deutschland GmbH», Нойфарн, Германия). Анализ пептидов выполняли путем применения различных градиентов 0,1% (об./об.) трифторуксусной кислоты (TFA) в Н2О (растворитель А) и 0,1% TFA вместе с (об./об.) в ацетонитриле (MeCN) (растворитель В) с постоянным потоком 1 мл/мин (конкретные градиенты указаны в тексте). УФ/ВИД-детектор выдающегося значения Shimadzu SPD 20 A («Shimadzu Deutschland GmbH») использовали при λ=220 нм и 254 нм. HSA-связывание определяли с использованием аналитической колонны Chiralpak HSA (5 мкм, 50 × 3 мм), соединенной с защитным картриджем Chiralpak HSA (5 мкм, 10 × 3 мм) («Daicel Chemical Industries))), приобретенным у «Chiral Technologies Еигоре» (Илькирш, Франция). Нелинейную регрессию для HSA-связывания выполняли с использованием OriginPro 2016G (Нортгемптон, США). Время удерживания tR, а также коэффициенты использования K', указаны в тексте. Препаративную RP-HPLC пептидов достигали на системе Shimadzu RP-HPLC с использованием колонны Multospher 100 RP 18-5 (250 × 20 мм, CS GmbH) с постоянным потоком 5 мл/мин. Аналитическую и препаративную радио RP-HPLC эталонного лиганда, меченого радиоактивным йодом, проводили с использованием колонны Nucleosil 100 С18 (5 мкм, 125 × 4,0 мм). Радиоактивность определяли посредством соединения выпускного отверстия УФ-фотометра со сцинцилляционным счетчиком луночного типа NaI(TI) от «EG&G Ortec» (Мюнхен, Германия). 68Ga- и 177Lu-меченые соединения анализировали так, как опубликовано ранее [1, 2]. Спектры масс-спектрометрии с электроионизацией распылением (ESI-MS) получали на масс-спектрометре expressionL- CMS («Advion Ltd.», Харлоу, Соединенное Королевство) и на масс-спектрометре Varian 500-MS IT («Agilent Technologies», Санта-Клара, США). Для анализа методом Брэдфорда использовали спектрофотометр V-630 UV-Vis от «JASCO Germany GmbH» (Грос-Умштадт, Германия), а центрифугирование S9-фракций проводили в центрифуге Avanti JXN-26 от «Beckman Coulter GmbH» (Крефельд, Германия). Центрифугирование радиоактивных образцов 39-метаболита проводили с использованием центрифуги Heraeus PICO 17 от «Thermo Fisher Scientific Messtechnik GmbH» (Мюнхен, Германия). Данные ЯМР получали с применением 300 K с использованием AV 300 (300 МГц) или AV400 (400 МГц) от «Bruker» (Биллерика, США). Инкубацию S9-фракций для ex vivo анализа метаболитов проводили в Biometra UNO Thermoblock («Biometra», Геттинген, Германия).
2. Протоколы синтеза (SP)
SP-1: Загрузка 2-СТС-смолы: 2-СТС-смолу (1,6 ммоль/г) нагружали Fmoc-AA-OH (1,5 экв.) в безводном дихлорметане (DCM) с N,N-диизопропилэтиламином (DIPEA) (4,5 экв.) при комнатной температуре (к.т.) на 2 ч. Оставшийся тритилхлорид блокировали путем добавления 2 мл/г метанола (МеОН) в течение 15 мин. После этого, смолу фильтровали и тщательно промывали с помощью DCM (2х), с помощью диметилформамида (DMF) (2х) и МеОН (2х), соответственно, и хранили в условиях вакуума в течение ночи. Нагрузку определяли с использованием различий в весе:

Формула 1. Определение нагрузки смолы: гпобщая: масса нагруженной смолы (Fmoc-AA-OH и HCl); MAs: молярная масса аминокислоты; гпнетто вес: масса использованной смолы; MHCI: молярная масса хлористоводородной кислоты
SP-2: Пептидный синтез посредством связывания TBTU/HOBt: Раствор Fmoc-AA-OH (2,0 экв.), N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)уроний тетрафторборат (TBTU) (2,0 экв.), W-гидроксибензотриазол (HOBt) (2,0 экв.), DIPEA (4,5 экв.) в DMF (8 мл/г смолы) добавляли в связанный смолой пептид со свободным амином и взбалтывали в течение 2 ч при к.т. и промывали с помощью DMF (6х). Связывание с вторичными или ароматическими аминами проводили с использованием другого протокола. Fmoc-AA-OH (3,0 экв.) растворяли в DMF (8 мл/г смолы) вместе с 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафтор-фосфатом (HATU) (3,0 экв.), 1-гидрокси-7-азабензотриазол (HOAt) (3,0 экв.) и DIPEA (6,0 экв.) и перемешивали в течение 15 мин. Предварительно активированный раствор добавляли к связанному смолой пептиду и взбалтывали в течение 2 ч при к.т. После завершения реакции смолу промывали с помощью DMF (6х). В целом, все пептидные каркасы синтезировали так, как было описано ранее (Weineisen, М.; Schottelius, М.; Simecek, J.; Eiber, М.; Schwaiger, М.; Wester, Н. Development and first in human evaluation of PSMA l&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine 2014, 55, 1083-1083; Weineisen, M.; Simecek, J.; Schottelius, M.; Schwaiger, M.; Wester, H.-J. Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. EJNMMI research 2014, 4, 1.).
SP-3: Fmoc-депротекция на смоле: Связанный смолой Fmoc-защищенный пептид обрабатывали с помощью 20% пиперидина в DMF (об./об.) в течение 5 мин и второй раз в течение 15 мин. После этого смолу тщательно промывали с помощью DMF (8х).
SP-4: Dde-депротекция на смоле: Защищенный N-(1-(4,4-диметил-2,6-диоксоциклогексилиден)этилом) (Dde) пептид (1,0 экв.) растворяли в растворе 2,0% моногидрата гидразина (N2H4⋅H2O) в DMF (об./об.). По прошествии 15 мин защищенный пептид, если он связан со смолой, промывали с помощью DMF (6х) или осаждали в диэтиловом эфире (Et2O) для получения неочищенного продукта. Если присутствовали Fmoc- и Dde-защитные группы и была необходима только Dde-депротекция, то нагруженный смолой пептид обрабатывали раствором, содержащим NH2OH⋅HCl (630 мг), имидазол (460 мг), DCM (0,5 мл), DMF (0,5 мл) и N-метил-2-пирролидон (NMP) (2,5 мл) в течение 3 ч при к.т. После этого, нагруженный смолой пептид промывали с помощью DMF (6х).
SP-5: Alloc/Allyl-депротекция на смоле: Alloc/Allyl-защитную группу удаляли из связанного смолой пептида с использованием раствора DCM (6,0 мл), содержащего триизопропилсилан (TIPS) (50,0 экв.) и (трифенил)палладий(О) (Pd(PPh3)4) (0,3 экв.).
Смолу обрабатывали этим раствором в течение 1,5 ч при к.т. Наконец, смолу промывали с помощью DCM (3х) для удаления Pd(PPh3)4.
SP-6: fBu/Boc-депротекция: Удаление трет-бутил (tBu)/трет-бутилоксикарбонил (Вос)-защитных групп осуществляли путем растворения неочищенного продукта в TFA (прибл. 500 мкл) и перемешивания в течение 40 мин при к.т. После этого, TFA почти полностью удаляли с использованием потока азота. После осаждения в Et2O, неочищенный продукт центрифугировали и удаляли супернатант. Высушенный осадок использовали в дальнейшем для следующих этапов синтеза.
SP-7.1: А) Отщепление пептида от смолы с сохранением защитных групп боковой цепи: Полностью защищенный, связанный смолой пептид растворяли в смеси DCM/трифторэтанол (ТРЕ)/уксусная кислота (АсОН) (6/3/1; об/об/об) и взбалтывали в течение 30 мин. Раствор отфильтровывали, а смолу растворяли в другом растворе для отщепления в течение дополнительных 30 мин. Фракции объединяли, и растворитель концентрировали при пониженном давлении. Фильтрат повторно растворяли в толуоле и концентрировали при пониженном давлении для удаления АсОН. Осаждение в воде Et2O в результате давало неочищенный пептид с защитой на боковой цепи.
SP-7.2: В) Отщепление пептида от смолы с одновременной депротекцией всех кислото-неустойчивых защитных групп: Полностью защищенный, связанный смолой пептид растворяли в смеси TFA/TIPS)/вода (95/2,5/2,5; об./об./об.) и взбалтывали в течение 30 мин. Раствор отфильтровывали, а смолу обрабатывали таким же образом в течение дополнительных 30 мин. После этого фракции объединяли, и растворитель концентрировали при постоянном потоке азота. Неочищенный пептид осаждали в Et2O и оставляли сушиться в течение ночи.
SP-8: Деацетилирование углеводных фрагментов: Деацетилирование проводили путем растворения PSMA-ингибитора в МеОН, содержащем KCN (0,5 экв.) (Herzig, J.; Nudelman, A.; Gottlieb, Н. Е.; Fischer, В. Studies in sugar chemistry. 2. A simple method for O-deacylation of polyacylated sugars. The Journal of Organic Chemistry 1986, 51, 727-730.) с сопутствующим перемешиванием в течение ночи при к.т. Конечный продукт очищали путем RP-HPLC.
SP-9: Получение нерадиоактивных комплексированных металлом PSMA-ингибиторов: SP-9.1: natGa-соединения: Для получения natGa111-комплексов, 2,0 мм водный (водн.) раствор PSMA-ингибитора (50 мкл) и 2,0 мм водн. раствор Ga(NO3)3 (50 мкл) смешивали и нагревали при 40°С в течение 30 мин. Образование хелата оценивали с использованием RP-HPLC и ESI-MS. Полученный в результате 1,0 мм раствор растворяли и использовали для in vitro определения IC50 и HSA-связывания.
SP-9.2: natLu-соединения: Соответствующие natLuIII комплексы получали из 2,0 мм водного раствора PSMA-ингибитора с 2,5 молярным избытком LuCl3 (20 мм водн. раствор) и нагревали до 95°С в течение 30 мин. После охлаждения подтверждали образование natLuIII-хелата с использованием RP-HPLC и ESI-MS. Полученные в результате 1,0 мм водные растворы соответствующих natLu-комплексов затем растворяли и использовали в in vitro исследованиях IC50 без дальнейшей обработки.
3. Построение блоков для ингибиторов PSMA-36 и PSMA-ингибиторов на основе EuE
Ди-трет-бутил(((s)-6-амино-1-(трет-бутокси)-1-оксогексан-2-ил)карбамоил)-L-глутамат
((OtBu)KuE(OtBu)2) (1): Синтез трет-бутил-защищенного Lys-мочевина-Glu связывающего мотива (EuK) проводили так, как было описано ранее, путем синтеза в жидкой фазе [3]. Вкратце, раствор DCM, содержащий L-ди-трет-бутил-глутамат-HCl (2,0 г, 7,71 ммоль, 1,0 экв.), охлаждали на льду в течение 30 мин и после этого обрабатывали триметиламином (TEA) (2,69 мл, 19,28 ммоль, 2,5 экв.) и 4-(диметиламино)пиридином (DMAP) (3,3 мг, 0,3 ммоль, 0,04 экв.). После дополнительного перемешивания в течение 5,0 мин, 1,1'-карбонилдиимидазол (CDI) (1,38 г, 8,84 ммоль, 1,1 экв.) растворяли в DCM и медленно добавляли в течение периода 30 мин. Реакционную смесь дополнительно перемешивали в течение ночи и позволяли нагреться до к.т. Реакцию останавливали с использованием насыщенного (нас.) раствора NaHCO3 (8 мл) с сопутствующими этапами промывания водой (2х) и солевым раствором (2х), и сушили над нас.раствором Na2SO4. Оставшийся растворитель удаляли в вакууме и неочищенный продукт (s)-ди-трет-бутил 2-(1Н-имидазол-1-карбоксамидо)пентандиоат использовали без дальнейшей очистки. RP-HPLC (от 10 до 90% В в течение 15 мин): tR=12,2 мин; K'=5,8. Вычисленная моноизотопная масса (C17H27N3O5): 353,4; найденная: м/з=376,1 [M+Na]+. Неочищенный продукт (5)-ди-трет-бутил 2-(1Н-имидазол-1-карбоксамидо)пентандиоат (2,72 г, 7,71 ммоль, 1,0 экв.) растворяли в 1,2-дихлорэтане (DCE) и охлаждали на льду в течение 30 мин. К этому раствору добавляли TEA (2,15 мл, 15,42 ммоль, 2,0 экв.) и H-Lys(Cbz)-OtBu⋅HCl (2,87 г, 7,71 ммоль, 1,0 экв.) и раствор перемешивали в течение ночи при 40°С. Оставшийся растворитель выпаривали, а неочищенный продукт очищали с использованием флэш-хроматографии на силикагеле с помощью смеси элюента, содержащей этилацетат (EtOAc)/гексанТЕА (500/500/0,8; об./об./об.). После удаления растворителя получали (9R,13S)-Три-трет-бутил-3,11-диоксо-1-фенил-2-окса-4,10,12-триазапентандекан-9,13,15-трикарбоксилат в виде бесцветного масла. RP-HPLC (от 40 до 100% В в течение 15 мин): tR=14,5 мин; K'=6,25. Вычисленная моноизотопная масса (C32H51N3O9)=621,8; найдено: м/з=622,3 [М+Н]+. Для синтеза (OtBu)KuE(OtBu)2 (1), (9R,13S)-Три-трет-бутил-3,11-диоксо-1-фенил-2-окса-4,10,12-триазапентадекан-9,13,15-трикарбоксилат (3,4 г, 5,47 ммоль, 1,0 экв.) растворяли в этаноле (EtOH) (75 мл), и в этот раствор добавляли палладий на активированном угле (0,34 г, 0,57 ммоль, 0,1 экв.) (10%). Сосуд, содержащий реакционную смесь, сначала очищали потоком водорода, и раствору позволяли перемешиваться в течение ночи при к.т. под давлением легкого водорода (в баллоне). Неочищенный продукт очищали через целит и растворитель выпаривали в вакууме. Желаемый продукт 1 получали в виде воскообразного твердого вещества (1,9 г, 3,89 ммоль, выход 71,6%). RP-HPLC (от 10 до 90% В в течение 15 мин): tR=12,6 мин; K'=6,4. Вычисленная моноизотопная масса (C24H45N3O7)=487,6; найдено: м/з=488,3 [М+Н]+, 510,3 [M+Na]+.
(s)-5-(трет-бутокси)-4-(3-((s)-1,5-ди-трет-бутокси-1,5-диоксопентан-2-ил)уреидо)-5-оксопентановая кислота ((OtBu)EuE(OtBu)2) (2):

Синтез трет-бутил-защищенного Glu-мочевина-Glu связывающего мотива (EuE) проводили подобным образом, как описано для 1 [3], с использованием H-L-Glu(OBzl)-OtBu⋅HCl вместо H-L-Lys(Cbz)-OtBu⋅HCl. Желаемый продукт получали в виде воскообразного и сильно гигроскопичного твердого вещества (4,10 г, 8,39 ммоль, выход 84%). RP-HPLC (от 10 до 90% В в течение 15 мин): tR=11,3 мин; K'=7,69. Вычисленная моноизотопная масса (C23H49N2O9)=488,3; найдено: м/з=489,4 [М+Н]+, 516,4 [M+Na]+.
(s)-NHFmoc-Asu(OtBu)-OBzl (5): В раствор (s)-Fmoc-Asu(OtBu)-OH (50 мг, 107,0 мкмоль, 1,0 экв.) в DMF добавляли HOAt (21,8 мг, 0,16 ммоль, 1,5 экв.), HATU (61,0 мг, 161,0 мкмоль, 1,5 экв.) и DIPEA (73,2 мкл, 0,48 ммоль, 4,5 экв.). После 15 мин перемешивания при к.т.дополнительно добавляли бензиловый спирт (22,2 мкл, 0,32 ммоль, 3,0 экв.) и раствор перемешивали в течение ночи. В конце растворитель удаляли в вакууме. Завершение реакции 5 анализировали путем RP-HPLC (от 10 до 90% В в течение 15 мин): tR=17,1 мин; K'=7,55. Вычисленная моноизотопная масса для 5 (C34H39NO6)=557,28; найдено: м/з=580,7 [M+Na]+.
(s)-NHFmoc-Asu-OBzl (6): tBu-депротекцию неочищенного продукта 5 проводили при перемешивании смеси (об/об) TFA (95%) и DCM (5%) при к.т.в течение 45 мин. После выпаривания растворителя неочищенный продукт 6 очищали с использованием препаративной RP-HPLC (от 60 до 80% В в течение 15 мин): tR=9,3 мин; K'=8,9. Вычисленная моноизотопная масса для 6 (C30H31NO6)=501,22; найдено м/з=524,5 [M+Na]+.
OBzl-(s)-Fmoc-Asu[(OtBu)KuE(OtBu)2] (7): В раствор 6 (51,8 мг, 10,3 мкмоль, 1,0 экв.) в DMF добавляли HOBt (20,9 мг, 0,15 ммоль, 1,5 экв.), TBTU (36,3 мг, 15,5 мкмоль, 1,5 экв.) и DIPEA (79,4 мкл, 59,7 мг, 0,46 ммоль, 4,5 экв.). После перемешивания в течение 15 мин добавляли 1 (75,6 мг, 15,5 мкмоль, 1,5 экв.) и дополнительно перемешивали в течение 20 ч при к.т. Неочищенный продукт 7 очищали с использованием препаративной RP-HPLC (от 70 до 80% В в течение 15 мин): tR=8,9 мин; K'=1,97. Вычисленная моноизотопная масса для 7 (C54H74N4O12)=970,53; найдено: м/з=971,8 [М+Н]+.
(s)-Fmoc-Asu[(OtBu)KuE(OtBu)2] (8): Для депротекции бензилового спирта (Bzl) 7 (57,2 мг, 65,0 мкмоль, 1,0 экв.) растворяли в EtOH (2,0 мл) и добавляли палладий на активированном угле (10%) (5,72 мг, 9,0 мкмоль, 0,1 экв.). Колбу заранее очищали потоком водорода, и раствор перемешивали под давлением легкого водорода (в баллоне). После перемешивания в течение 70 мин неочищенный продукт фильтровали через целит, EtOH выпаривали в вакууме, а продукт очищали с использованием препаративной RP-HPLC (от 70 до 70,5% В в течение 15 мин): tR=6,5 мин; K'=0,54. Вычисленная моноизотопная масса для 5 (C47H68N4O12)=880,48; найдено: м/з=881,8 [М+Н]+.
OPfp-(s)-Fmoc-Asu[(OtBu)KuE(OtBu)2] (9):

8 раствор 8 (13,6 мг, 15,4 мкмоль, 1,0 экв.) в сухом DMF добавляли DIC (4,77 мкл, 1,94 мг, 30,8 мкмоль, 2,0 экв.) и PfpOH (5,67 мг, 30,8 мкмоль, 2,0 экв.). После перемешивания в течение 5 мин добавляли пиридин (2,49 мкл, 31,0 мкмоль, 2,0 экв.) и раствор оставляли перемешиваться в течение ночи при к.т.
Завершение реакции
9 анализировали путем RP-HPLC (от 10 до 90% В в течение 15 мин): tR=17,2 мин; K'=7,6. Вычисленная моноизотопная масса для 9 (C53H67F5N4O12)=1046,47; найдено: м/з=1069,8 [M+Na]+.
MHS-2,4-динитробензоат (NHS-DNBA) (27):

В раствор 2,4-динитробензойной кислоты (DNBA) (10,0 мг, 47,1 мкмоль, 1,0 экв.) в сухом THF добавляли N,N'-дициклогексилкарбодиимид (DCC) (9,7 мг, 47,1 мкмоль, 1,0 экв.) и N-гидроксисукцинимид (NHS) (10,8 мг, 94,3 мкмоль, 2,0 экв.), и реакционную смесь оставляли перемешиваться в течение ночи. Неочищенный продукт очищали с использованием RP-HPLC. RP-HPLC (от 10 до 90% В в течение 15 мин): tR=10,21 мин K'=4,1. Вычисленная моноизотопная масса (C11H7N3O8)=309,02; найдено: не обнаруживается при ESI-MS.
DOTAGA-3-йод-Tyr-D-Phe-D-Lys-OH (DOTAGA-y(3-I)fk) (30):
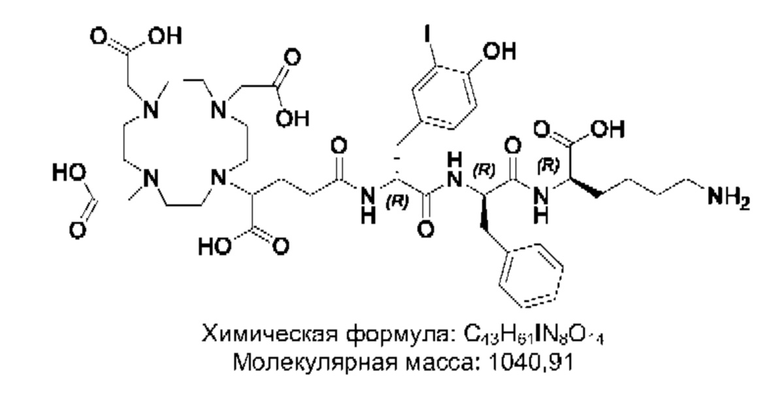
Синтез 30 проводили по твердофазной стратегии, как было описано ранее [2, 3]. Вкратце: Изначальной стартовой точкой была нагрузка 2-СТС-смолы Fmoc-D-Lys(Boc)-OH в соответствии с SP-1. После конъюгации лизина Fmoc подвергали депротекции в соответствии с SP-3 и связывали Fmoc-D-фенилаланин, применяя SP-2. Эту же процедуру использовали для связывания Fmoc-D-Tyr(3-1)-OH. После завершения реакции, Fmoc-защитную группу отщепляли в соответствии с SP-3, а связанный смолой пептид конденсировали с хелатором с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.) в DMF. Реакции позволяли перемешиваться в течение 48 ч при к.т. В конце неочищенный продукт отщепляли от смолы в соответствии cSP-7.2 и осаждали в Et2O, а также центрифугировали. Супернатант удаляли, и 30 очищали с использованием RP-HPLC. RP-HPLC (от 10 до 90% В в течение 15 мин): tR=6,2 мин K'=2,1. Вычисленная моноизотопная масса (C43H61IN8O14)=1040,34; найдено: м/з=1040,5 [М+Н]+, м/з=521,3 [М+2Н]2+, м/з=1063,4=[M+Na]+.
DOTAGA-y(3-l)fk(L-Asu[KuE]) (PSMA-8):

В раствор DMF, содержащий 30 (5,0 мг, 4,8 мкмоль, 1,0 экв.) добавляли 9 (7,5 мг, 7,2 мкмоль, 1,5 экв.) и DIPEA (3,3 мкл, 21,6 мкмоль, 4,0 экв.). Реакционному раствору позволяли перемешиваться в течение ночи при к.т. После завершения реакции растворитель удаляли в вакууме и неочищенный продукт обрабатывали смесью пиперидина в DMF (20/80; об./об.) в течение 15 мин для достижения Fmoc-депротекции. Растворитель восстанавливали до прибл. 300 мкл посредством выпаривания в вакууме, осаждали в Et2O и центрифугировали. Полученным продуктом обрабатывали пеллету в соответствии с SP-6 для удаления tBu. Конечный продукт очищали посредством RP-HPLC (от 10 до 90% В в течение 15 мин): tR=6,09 мин K'=2,05. Вычисленная моноизотопная масса (C63H93IN12O23)=1512,55; найдено: м/з=1513,9 [М+Н]+, 757,8 [М+2Н]2+.
DOTAGA-y(3-l)fk(L-Asu[KuE]-2,4-DNBA) (PSMA-36):
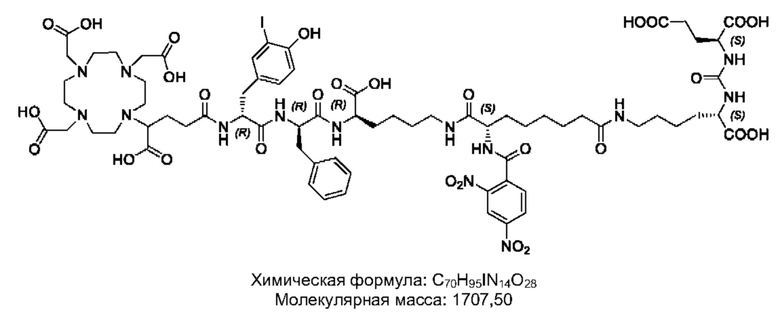
Синтез PSMA-36 достигали путем растворения PSMA-8 (3,0 мг, 3,3 мкмоль, 1,0 экв.) в DMF и добавления 27 (4,1 мг, 13,2 мкмоль, 4,0 экв.) и DIPEA (2,3 мкл, 13,2 мкмоль, 4,0 экв.). Раствор перемешивали в течение 10 ч при к.т. и конечный продукт очищали путем RP-HPLC (от 10 до 50% В в течение 15 мин): tR=12,12 мин K'=5,06. Вычисленная моноизотопная масса (C70H95IN14O28)=1706,55; найдено: м/з=1707,8 [М+Н]+, 854,7 [М+2Н]2+.
[natLu]DOTAGA-y(3-l)fk(L-Asu[KuE]-2,4-DNBA) ([natLu]PSMA-36): RP-HPLC (от 10 до 60% В в течение 15 мин): tR=9,81 мин K'=3,91. Вычисленная моноизотопная масса (C70H92IN14O28Lu)=1878,47; найдено: м/з=1879,9 [М+Н]+.
Схематическая иллюстрация синтеза PSMA-36. (a) HOAt, HATU, DIPEA, бензиловый спирт, [DMF]; (b) 95% TFA, 5% DCM; (с) 1, HOBt, TBTU, DIPEA, [DMF]; (d) Pd/C (10%), H2, [EtOH]; (e) DIC, PFP, пиридин, [DMF]; (f) 30, DIPEA, [DMF]; (g) 20% пиперидин в DMF, [DMF]; (h) TFA; (i) 27, DIPEA [DMF]
4. Синтез PSMA-ингибиторов на основе EuE PSMA-52 и PSMA-53
DOTAGA-F(4-NO2)-y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-52):

Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу, обработанную янтарным ангидридом (4,0 экв.) и DIPEA (1,5 экв.), растворяли в DMF. Реакционной смеси позволяли реагировать в течение ночи при к.т. Затем Fmoc-D-Lys-OtBu⋅HCl (1,5 экв.) связывали в соответствии с SP-2 и подвергали Fmoc-депротекции, как описано в SP-3. Следующие конъюгации с Fmoc-защищенными аминокислотами Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-Phe(4-NO2)-OH проводили так, как описано в SP-2. N-концевую подвергнутую Fmoc-депротекции аминокислоту конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант.Конечный продукт очищали посредством RP-HPLC. RP-HPLC (от 10 до 60% В в течение 15 мин): fR=9,71 мин K'=3,86. Вычисленная моноизотопная масса (C76H100N14O29)=1672,68; найдено: м/з=1673,0 [М+Н]+.
[natLu]DOTAGA-F(4-NO2)-y-2-nal-k(Suc-N5-orn-C4-EuE) ([natLu]PSMA-52): RP-HPLC (от 10 до 60% В в течение 15 мин): fR=9,4 мин K'=3,7. Вычисленная моноизотопная масса (C76H97N14O29Lu)=1844,6; найдено: м/з=1846,0 [М+Н]+.
2,4-DNBA-Dap(DOTAGA)-y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-53):
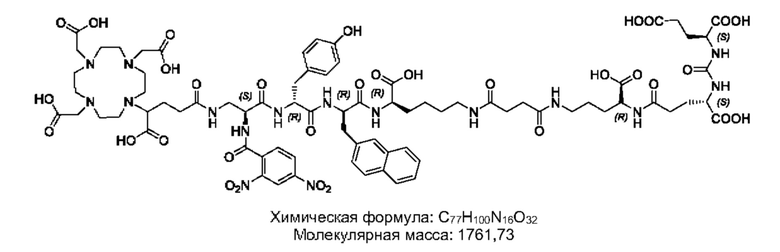
Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу, обработанную янтарным ангидридом (4,0 экв.) и DIPEA (1,5 экв.), растворяли в DMF. Реакционной смеси позволяли реагировать в течение ночи при к.т. Затем, Fmoc-D-Lys-OtBu⋅HCl (1,5 экв.) связывали в соответствии с SP-2 и подвергали Fmoc-депротекции, как описано в SP-3. Следующие конъюгации с Fmoc-защищенными аминокислотами Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(Ort3u)-OH и Fmoc-L-Dap(NHDde)-OH проводили так, как описано в SP-2. После связывания Fmoc-L-Dap(NHDde)-OH обеспечивали Fmoc-депротекцию так, как описано в SP-3. Затем свободную аминогруппу конъюгировали с 2,4-динитробензойной кислотой (2,4-DNBA) с использованием 2,4-DNBA (2,0 экв.), HOBt (2,0 экв.), TBTU (2,0 экв.) и DIPEA (4,0 экв.) в DMF. После завершения реакции обеспечивали Dde-депротекцию с использованием SP-5. N-концевую свободную аминокислоту L-Dap конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант. Конечный продукт очищали посредством RP-HPLC.
RP-HPLC (от 10 до 60% В в течение 15 мин): tR=11,71 мин K'=4,86. Вычисленная моноизотопная масса (C77H100N16O32)=1760,67; найдено: м/з=1762,1 [М+Н]+.
[natLu]2,4-DNBA-Dap(DOTAGA)-y-2-nal-k(Suc-N5-orn-C4-EuE) ([natLu]PSMA-53): RP-HPLC (от 10 до 60% В в течение 15 мин): tR=8,3 мин K'=3,15. Вычисленная моноизотопная масса (C77H97N16O32Lu)=1932,59; найдено: м/з=1933,7 [М+Н]+.
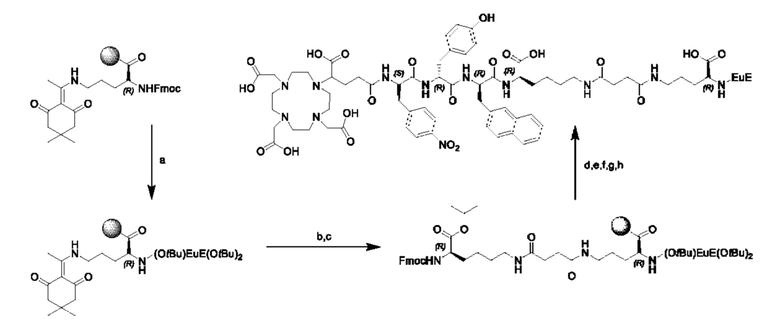
Схематическая иллюстрация общей процедуры синтеза PSMA-ингибиторов на основе EuE PSMA-52 и PSMA-53, представленных в качестве примера как PSMA-52. (а) 20% пиперидина в DMF, 2, HOBt, TBTU, DIPEA [DMF]; (b) янтарный ангидрид, DIPEA [DMF]; (с) Fmoc-D/L-Lys-OAII-HCI, HOBt, TBTU, DIPEA [DMF]; (d) 20% пиперидина в DMF, Fmoc-D-2-Nal-OH, HOBt, TBTU, DIPEA [DMF]; (e) 20% пиперидина в DMF, Fmoc-D-Tyr(OtBu)-OH, HOBt, TBTU, DIPEA [DMF]; (f) 20% пиперидина в DMF, Fmoc-D-Phe(4-NH2)-OH, HOBt, TBTU, DIPEA [DMF]; (g) DOTAGA-ангидрид, DIPEA [DMF]; (h)TFA;
5. Синтез PSMA-61 и PSMA-62
DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-С4-EuE]-2,4-DNBA) (PSMA-61):

Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу обрабатывали Fmoc-D-Asp-OAII⋅HCl (1,5 экв.) в соответствии с SP-2. Аминогруппу Fmoc-D-Asp-OAIIHCI подвергали депротекции в соответствии с SP-3 и конъюгировали с 2,4-DNBA с использованием 2,4-DNBA (1,5 экв.), HOBt (2,0 экв.), TBTU (2,0 экв.) и DIPEA (4,0 экв.) в DMF. После завершения реакции обеспечивали Allyl-депротекцию в соответствии с SP-5. Следующие этапы включали повторную конъюгацию с Fmoc-D-Lys-OtBu⋅HCl (1,5 экв.), Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-Phe(4-NHBoc)-OH в соответствии с SP-2. N-концевую подвергнутую Fmoc-депротекции аминокислоту L-Phe(4-NHBoc)-OH конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант.Конечный продукт очищали посредством RP-HPLC.
RP-HPLC (от 10 до 90% В в течение 15 мин): tR=6,40 мин K'=2,2. Вычисленная моноизотопная масса (C83H105N17O32)=1851,71; найдено: м/з=1852,5 [М+Н]+, 926,7 [М+2Н]2+.
[natLu]DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-2,4-DNBA) ([natLu]PSMA-61):
RP-HPLC (от 10 до 90% В в течение 15 мин): tR=8,22 мин K'=3,11. Вычисленная моноизотопная масса (C83H102N17O32Lu)=2023,63; найдено: м/з=1013,1 [М+2Н]2+.
DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-62):

Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу обрабатывали Fmoc-D-Asp-OAII⋅HCl (1,5 экв.) в соответствии с SP-2. Аминогруппу Fmoc-D-Asp-OAII⋅HCl подвергали Fmoc-депротекции в соответствии cSP-3 и защищали с помощью Dde-ОН (2,0 экв.) и DIPEA (4,0 экв.) в DMF при к.т. Реакции позволяли перемешиваться в течение ночи. После этого обеспечивали Allyl-депротекцию d-Asp, применяя SP-5. Следующие этапы включали повторную конъюгацию с Fmoc-D-Lys-OtBu⋅HCl (1,5 экв.), Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-Phe(4-NHBoc)-OH в соответствии с SP-2. Для конъюгации ТМА с d-Asp обеспечивали селективную Dde-депротекцию, применяя SP-4, для получения свободной аминогруппы. ТМА связывали с использованием ТМА (2,0 экв.), HOBt (1,5 экв.), TBTU (1,5 экв.) и DIPEA (10 экв.) в DMF. Реакции позволяли перемешиваться в течение 8 ч при к.т. После конъюгации ТМА обеспечивали Fmoc-депротекцию Fmoc-L-Phe(4-NHBoc)-OH с использованием SP-3. N-концевую подвергнутую Fmoc-депротекции аминокислоту L-Phe(4-NHBoc)-OH конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант. Конечный продукт очищали посредством RP-HPLC. RP-HPLC (от 10 до 70% В в течение 15 мин): tR=7,48 мин K'=2,74. Вычисленная моноизотопная масса (C85H107Ni5O32)=1849,72; найдено: м/з=1850,5 [М+Н]+, 925,7 [М+2Н]2+.
[natLu]DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) ([natLu]PSMA-62): RP-HPLC (от 10 до 70% В в течение 15 мин): tR=7,27 мин K'=2,64. Вычисленная моноизотопная масса (C85H104N15O32Lu)=2021,64; найдено: м/з=1012,3 [М+2Н]2+.
6. Синтез PSMA-65. PSMA-66 и PSMA-71
2,4-DNBA-Dap(DOTAGA)y-2-nal-e(Abz-N5-orn-C4-EuE) (PSMA-65):
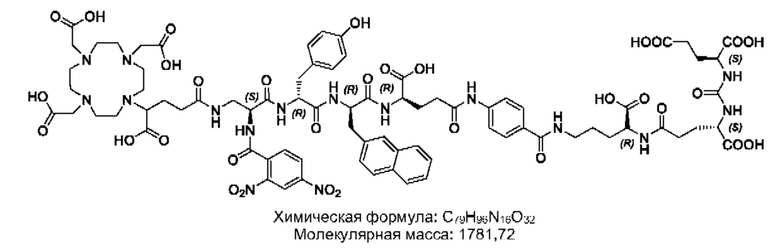
Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу обрабатывали Fmoc-4-Abz-OH (1,5 экв.), HOAt (1,5 экв.), HATU (1,5 экв.) и DIPEA (4,0 экв.) в DMF. Реакции позволяли перемешиваться в течение ночи при к.т.На следующем этапе Abz-остаток подвергали Fmoc-депротекции в соответствии с SP-3. Следующие этапы включали повторную конъюгацию с Fmoc-D-Glu-Offiu, Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-Dap(Dde)-OH в соответствии с SP-2. После Fmoc-депротекции Fmoc-L-Dap(Dde)-OH в соответствии с SP-3, 2,4-DNBA связывали с использованием 2,4-DNBA (1,5 экв.), HOBt (2,0 экв.), TBTU (2,0 экв.) и DIPEA (4,0 экв.) в DMF. После завершения реакции L-Dap(Dde)-остаток подвергали Dde-депротекции в соответствии с SP-4 и конъюгировали с хелатором с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант. Конечный продукт очищали посредством RP-HPLC. RP-HPLC (от 10 до 60% В в течение 15 мин): tR=10,2 мин K'=4,1. Вычисленная моноизотопная масса (C79H96N16O32)=1780,64; найдено: м/з=1781,3 [М+Н]+.
[natLu]2,4-DNBA-Dap(DOTAGA)y-2-nal-e(Abz-N5-orn-C4-EuE) ([natLu]PSMA-65): RP-HPLC (от 10 до 60% В в течение 15 мин): tR=9,8 мин K'=3,9. Вычисленная моноизотопная масса (C79H93N16O32Lu)=1952,56; найдено: м/з=1954,0 [М+Н]+.
DOTAGA-Dap(TMA)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-66):

Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу обрабатывали Fmoc-D-Asp-OAII⋅HCl (1,5 экв.) в соответствии с SP-2. Аминогруппу Fmoc-D-Asp-OAII⋅HCl подвергали Fmoc-депротекции в соответствии с SP-3 и защищали с помощью 2,0 экв. Dde-OH и 4,0 экв. DIPEA в DMF. Реакции позволяли перемешиваться в течение ночи. После этого обеспечивали Allyl-депротекцию d-Asp, применяя SP-5. Следующие этапы включали повторную конъюгацию с Fmoc-D-Lys-OtBu⋅HCl (1,5 экв.), Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-Dap(Dde)-OH в соответствии с SP-2. Для конъюгации ТМА с d-Asp и L-Dap обеспечивали селективную Dde-депротекцию, применяя SP-4, для получения свободных аминогрупп.ТМА связывали с использованием ТМА (4,0 экв.), HOBt (3,0 экв.), TBTU (3,0 экв.) и DIPEA (20 экв.) в DMF. Реакции позволяли перемешиваться в течение 8 ч при к.т. После конъюгации ТМА обеспечивали Fmoc-депротекцию Fmoc-L-Dap(TMA)-OH с использованием SP-3. N-концевую подвергнутую Fmoc-депротекции аминокислоту L-Dap конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант. Конечный продукт очищали посредством RP-HPLC. RP-HPLC (от 10 до 70% В в течение 15 мин): tR=7,48 мин K'=2,74. Вычисленная моноизотопная масса (C88H107N15O37)=1965,70; найдено: м/з=1966,4 [М+Н]+, 984,1 [М+2Н]2+.
[natLu]DOTAGA-Dap(TMA)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-66) RP-HPLC (от 10 до 70% В в течение 15 мин): tR=7,46 мин K'=2,73. Вычисленная моноизотопная масса (C88H108N15O37Lu)=2137,62; найдено: м/з=1070,4 [М+2Н]2+.
DOTAGA-2-Nal-y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-71):
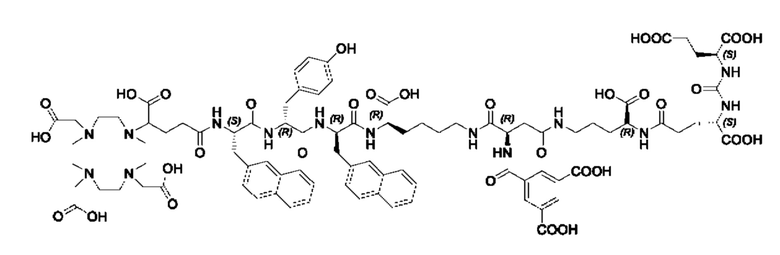
Исходную нагрузку смолы Fmoc-D-Orn(NHDde)-OH проводили так, как описано в SP-1. После Fmoc-депротекции в соответствии с SP-3, 2 (1,5 экв.) связывали с D-Orn(NHDde) в соответствии с SP-2. На следующем этапе Dde-защитную группу отщепляли в соответствии с SP-4, а свободную аминогруппу обрабатывали Fmoc-D-Asp-OAII⋅HCl (1,5 экв.) в соответствии с SP-2. Аминогруппу Fmoc-D-Asp-OAII⋅HCl подвергали Fmoc-депротекции в соответствии cSP-3 и защищали с помощью Dde-ОН (2,0 экв.) и DIPEA (4,0 экв.) в DMF при к.т. Реакции позволяли перемешиваться в течение ночи. После этого обеспечивали Allyl-депротекцию d-Asp, применяя SP-5. Следующие этапы включали повторную конъюгацию с Fmoc-D-Lys-Ofl3u-HCI (1,5 экв.), Fmoc-D-2-Nal-OH, Fmoc-D-Tyr(OtBu)-OH и Fmoc-L-2-Nal-OH в соответствии с SP-2. Для конъюгации ТМА с d-Asp обеспечивали селективную Dde-депротекцию, применяя SP-4, для получения свободной аминогруппы. ТМА связывали с использованием ТМА (2,0 экв.), HOBt (1,5 экв.), TBTU (1,5 экв.) и DIPEA (10 экв.) в DMF. Реакции позволяли перемешиваться в течение 8 ч при к.т. После конъюгации ТМА обеспечивали Fmoc-депротекцию Fmoc-L-2-Nal-OH с использованием SP-3. N-концевую подвергнутую Fmoc-депротекции аминокислоту L-2-Nal-OH конъюгировали с хелатором на конечном этапе с использованием DOTAGA-ангидрида (2,0 экв.) и DIPEA (2,0 экв.). Реакции позволяли перемешиваться в течение 48 ч при к.т. После завершения реакции с DOTAGA-ангидридом пептид отщепляли от смолы в соответствии с SP-7.2, неочищенный продукт осаждали в Et2O, центрифугировали, и удаляли супернатант. Конечный продукт очищали посредством RP-HPLC. RP-HPLC (от 10 до 80% В в течение 15 мин): tR=7,57 мин К' =2,79. Вычисленная моноизотопная масса (C89H108N14O32)=1884,73; найдено: м/з=1886,1 [М+Н]+, 943,5 [М+2Н]2+.
[natLu]DOTAGA-2-Nal-y-2-nal-k[d[N5-orn-C4-EuE]-TMA) ([natLu]PSMA-67): RP-HPLC (от 10 до 90% В в течение 15 мин): fR=7,81 мин K'=2,91. Вычисленная моноизотопная масса (C89H105N14O32Lu)=2056,64; найдено: м/з=1029,7 [М+2Н]2+.
7. Введение радиоактивной метки
68Ga-мечение: Генератор 68Ge/68Ga элюировали с водн. HCl (1,0 м), из которого фракцию 1,25 мл, содержащую приблизительно 80% активности (от 600 до 800 МБк), переносили в реакционную пробирку (ALLTECH, 5 мл). В пробирку заранее загружали соответствующее соединение (5,0 нмоль) и водн. раствор 2-(4-(2-Гидроксиэтил)-1-пиперазинил)-этансульфоновой кислоты (HEPES) (950 мкл, 2,7 м). Реакционную пробирку нагревали в течение 5 мин при 95°С с последующей фиксацией радиомеченого соединения на прекондиционном SPE-картридже (С8 light, SepPak). После предварительного промывания картриджа водой (10 мл) обеспечивали элюирование радиомеченого PSMA-ингибитора из картриджа со смесью EtOH и воды (1/1; об/об), фосфатно-солевым буферным раствором (PBS) (1,0 мл) и снова водой (1,0 мл). В конце введения радиоактивной метки, EtOH выпаривали в вакууме и метку использовали без какой-либо дальнейшей очистки. Радиохимическую чистоту контролировали с использованием радио-TLC (1,0 м буфера цитрата натрия и 0,06 м буфера NH4OAc/MeOH (1/1; об/об)).
177Lu-мечение: 177Lu-меченые соединения получали так, как описано ранее [5], с незначительными модификациями и использовали без дальнейшей очистки. Вкратце, в NH4OAc-буфер (10 мкл, 1,0 м, рН=5,9) добавляли соответствующую метку (от 0,75 до 1,0 нмоль, от 7,5 до 10 мкл), 177LuCl3 (от 10 до 40 МБк; AS>3000 ГБк/мг, 740 МБк/мл, 0,04 м HCl, «ITG», Гархинг, Германия) и в конце заполняли очищенной от метки водой (вплоть до 100 мкл) (Merck, Дармштадт, Германия). Реакционную смесь нагревали в течение 40 мин при 95°С, и радиохимическую чистоту определяли с использованием радио-TLC.
125I-мечение: Вкратце, станнилированный предшественник (SnBu3-BA)(OtBu)KuE(OtBu)2 (PSMA-45) (прибл. 0,1 мг) растворяли в растворе, содержащем перуксусную кислоту (20 мкл), [125I]Nal (5,0 мкл, прибл. 21,0 МБк) (74 ТБк/ммоль, 3,1 ГБк/мл, 40 мм NaOH, «Hartmann Analytic)), Брауншвейг, Германия), MeCN (20 мкл) и АсОН (10 мкл). Реакционный раствор инкубировали в течение 10 мин при к.т., загружали на картридж (С18 Sep Рак Plus, прекондиционированный с помощью 10 мл МеОН и 10 мл воды) и промывали водой (10 мл). После элюирования со смесью 1/1 (об/об) EtOH и MeCN (2,0 мл), раствор выпаривали насухо под слабым потоком азота и обрабатывали TFA (200 мкл) в течение 30 мин с последующим выпариванием TFA. Неочищенный продукт ([125I]I-BA)KuE очищали посредством радио-RP-HPLC (от 20 до 40% В в течение 20 мин): tR=13,0 мин; K'=6,2.
8. Определение HSA-связывания
Эксперименты HSA-связывания выполняли так, как было описано ранее [6]. Подвижная фаза состояла из системы бинарного градиента с постоянной общей скоростью потока 0,5 мл/мин. Подвижная фаза А представляла собой 50 мм рН 6,9 NH4⋅ОАс-раствор, подвижная фаза В представляла собой 2-пропанол (шкала RP-HPLC, «VWR», Германия). Градиент подвижной фазы А составлял 100% от 0 до 3 мин, а от 3 мин до конца каждого прогона подвижную фазу устанавливали на 20%. В каждый день эксперимента колонну калибровали с помощью девяти эталонных веществ для подтверждения производительности и для установления нелинейной регрессии. PSMA-ингибиторы растворяли при концентрации 0,5 мг/мл в смеси 2-пропанола и NH4OAc-буфера (50 мм рН 6,9) (1/1; об/об). Для каждого прогона, 10 мкл раствора, содержащего ингибитор, вводили в систему RP-HPLC и измеряли время удерживания. Литературное HSA-связывание [%] получали от Valko et. al. или Yamazaki et al. [6, 7]. Нелинейную регрессию устанавливали с помощью OriginPro 2016G.
9. Определение липофильности
Липофильность: Радиомеченый PSMA-ингибитор (от 0,5 до 1,0 МБк), растворенный в PBS (500 мкл, рН=7,4), добавляли в н-октанол (500 мкл) в реакционной пробирке (1,5 мл), которую тщательно встряхивали в течение 3 мин (n=6). Для отделения количественной фазы, смесь центрифугировали при 6000 г в течение 5 мин (Biofuge 15, «Heraus Sepatech», Остероде, Германия). Активность из образцов каждой фазы (100 мкл) измеряли в у-счетчике для получения значения logP(o/w).
10. Эксперименты на клетках
Клеточная культура: PSMA-положительные LNCAP-клетки (300265; «Cell Lines Service GmbH») культивировали в среде Игла/питательной смеси, модифицированной по Дульбекко, F-12 (1/1) (DMEM-F12, Biochrom) с добавлением фетальной телячьей сыворотки (FCS) (10%, Biochrom) и хранили при 37°С в атмосфере влажного СО2 (5%). За одни сутки (24 ч±2 ч) перед всеми экспериментами с LNCaP-клетками, культивированные клетки собирали с использованием смеси трипсина/этилендиаминтетраацетата (0,05%/ 0,02%) и PBS, и центрифугировали. После центрифугирования, супернатант размещали и клеточный осадок повторно суспендировали в культуральной среде. После этого, проводили подсчет клеток с помощью гемоцитометра («Neubauer») и высевали в 24-луночные планшеты. Значения IC50 определяли, перенося 150000 клеток/мл на лунку в 24-луночные планшеты, тогда как скорости интернализации получали путем переноса 125000 клеток/мл на лунку в 24-луночные PLL-покрытые планшеты.
11. Аффинность (IС50)
После удаления культуральной среды, клетки обрабатывали один раз HBSS (500 мкл, сбалансированный солевой раствор Хенкса, «Biochrom», Берлин, Германия, с добавлением 1% BSA) и оставляли на 15 мин на льду для уравновешивания в HBSS (200 мкл, 1% BSA). Затем, растворы (25 мкл на лунку), содержащие HBSS (1% BSA, контроль) или соответствующий лиганд в увеличивающейся концентрации (от 10-10 до 10-4 м в HBSS (1% BSA)) добавляли с последующим добавлением ([125I]l-BA)KuE (25 мкл, 2,0 нм) в HBSS (1% BSA). Все эксперименты проводили по меньшей мере три раза для каждой концентрации. После 60 мин инкубации на льду, эксперимент прерывали путем удаления среды и последующего промывания HBSS (200 мкл). Среды с обоих этапов объединяли в одну фракцию, и они представляли количество свободного лиганда. После этого, клетки лизировали с помощью NaOH (250 мкл, 1,0 м) и объединяли с HBSS (200 мкл) со следующего этапа промывания. Количественную оценку связанного и свободного радиолиганда проводили в v-счетчике.
12. Интернализация
После удаления культуральной среды, клетки промывали один раз раствором DMEM-F12 (500 мкл, 5% BSA) и оставляли на уравновешивание по меньшей мере на 15 мин при 37°С в растворе DMEM-F12 (200 мкл, 5% BSA). После этого, каждую лунку обрабатывали раствором DMEM-F12 (25 мкл, 5% BSA) или раствором 2-РМРА (25 мкл, 100 мкм) для блокады. Затем добавляли соответствующий 68Ga- или 177Lu-меченый PSMA-ингибитор (25 мкл; 2,0 нм и 10 нм, соответственно) и клетки инкубировали при 37°С в течение 5, 15, 30 и 60 мин, соответственно. Эксперимент прекращали путем помещения 24-луночного планшета на лед на 3 мин и последующего удаления среды. Каждую лунку промывали HBSS (250 мкл), а фракции с этих первых двух этапов объединяли, представляя количество свободного радиолиганда. Удаление активности связывания поверхности проводили путем инкубации клеток с ледяным раствором 2-РМРА (250 мкл, 10 мкм в PBS) на 5 мин и последующего промывания ледяным PBS (250 мкл).
Интернализованную активность определяли посредством инкубации клеток в NaOH (250 мкл, 1,0 м) и объединения с фракцией последующего этапа промывания вновь с NaOH (250 мкл, 1,0 м). Каждый эксперимент (контроль и блокаду) проводили в трех повторениях для каждой точки во времени. Количественную оценку свободной, поверхностно-связанной и интернализованной активности выполняли в у-счетчике.
13. Экстернализация
Кинетику экстернализации радиомеченых PSMA-ингибиторов определяли с использованием LNCaP-клеток, которые получали подобным образом, как было описано для анализа интернализации. После первичного этапа промывания клеток раствором DMEM-F12 (5% BSA), клетки оставляли восстанавливаться по меньшей мере на 15 мин при 37°С. Затем, LNCaP-клетки инкубировали с соответствующим радиомеченым пептидом (25 мкл, 10,0 нм) при 37°С в течение 60 мин при общем объеме 250 мкл в каждой лунке. По прошествии 60 мин, супернатант с несвязанной свободной фракцией удаляли и измеряли в у-счетчике. Для подсчета общей добавленной радиоактивности. Этапа промывания кислотой избегали для того, чтобы гарантировать целостность фермента в ходе последующей экстернализации и исследования рециркуляции. Для определения скорости рециркуляции, в клетки добавляли свежий раствор DMEM-F12 (250 мкл, 5% BSA) для обеспечения возможности повторной интернализации. В отличие от этого, повторную интернализацию ингибировали путем добавления раствора DMEM-F12, содержащего 2-РМРА (225 мкл DMEM-F12 (5% BSA) и 25 мкл 100 мкм раствора 2-РМРА (PBS)). Затем, клетки инкубировали в течение 0, 20, 40 и 60 мин при 37°С. После этого, супернатант удаляли, а клетки промывали ледяным HBSS (250 мкл). Объединение супернатанта и объема сопутствующего этапа промывания HBSS (200 мкл) учитывали для экстернализованного радиолиганда в исследуемой точке времени. Далее, клетки затем дважды промывали ледяным раствором 2-РМРА HBSS (250 мкл, 10 мкм), объединяли и, таким образом, представляли фракцию связанного с мембраной радиолиганда. Интернализованную фракцию определяли посредством лизиса, как было описано для анализа интернализации с помощью NaOH (250 мкл, 1,0 м). Количественную оценку значений активности свободного, экстернализированного, связанного с мембраной и интернализированного радиолиганда проводили в у-счетчике.
14. Эксперименты на животных
Все эксперименты на животных проводили в соответствии с общим протоколом обращения с животными в Германии (Deutsches Tierschutzgesetz, разрешение #55.2-1-54-2532-71-13). Для опухолевой модели, LNCaP-кпетки (прибл. 107 клеток) суспендировали в свободной от сыворотки среде DMEM-F12 и матригеле (1/1; об/об) («BD Biosciences)), Германия) и инокул провал и на правое плечо мышей-самцов СВ-17 SCID возрастом от 6 до 8 недель («Charles River Laboratories)), Зульцфельд, Германия). Животных использовали для экспериментов после того, как размер опухоли достиг от 4 до 8 мм в диаметре.
15. ПЭТ
Эксперименты с визуализацией проводили с использованием ПЭТ «Siemens Inveon small animal РЕТ», и данные анализировали с помощью соответствующего программного обеспечения «lnveon Research Workplace)). Мышам вводили анестезию изофлураном и инъецировали прибл. от 4,0 до 17 МБк 68Ga-меченых соединений через хвостовую вену (прибл. от 150 до 300 мкл). Динамическую визуализацию проводили после инъекции на подушке в течение 90 мин. Статическое изображение блокады получали через 1 ч после инъекции при времени обнаружения 15 мин. PSMA-блокаду обеспечивали путем одновременной инъекции 8 мг/кг раствора 2-РМРА (PBS). Все изображения реконструировали с использованием алгоритма OSEM3D без сканера и коррекции ослабления.
16. Биораспределение
Приблизительно от 4,0 до 12,0 МБк (прибл. от 150 до 300 мкл) соответствующих 68Ga- или 177Lu-меченых PSMA-ингибиторов инъецировали в хвостовую вену мышей-самцов СВ-17 SCID с LNCaP-опухолью, которых умерщвляли по прошествии конкретного периода времени (n=4, соответственно). Выбранные органы удаляли, взвешивали и измеряли в у-счетчике.
17. Ссылки в Примере 1
1. Simecek, J., et al., A Monoreactive Bifunctional Triazacyclononane Phosphinate Chelator with High Selectivity for Gallium-68. ChemMedChem, 2012. 7(8): p.1375-1378.
2. Weineisen, M., et al., Development and first in human evaluation ofPSMA l&T-A ligand for diagnostic imaging and endoradiotherapy of prostate cancer. Journal of Nuclear Medicine, 2014. 55(supplement 1): p.1083-1083.
3. Weineisen, M., et al., Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. EJNMMI research, 2014. 4(1): p.1.
4. Weineisen, M., et al., 68Ga- and 177Lu-Labeled PSMA l&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. Journal of Nuclear Medicine, 2015. 56(8): p. 1169-1176.
5. Sosabowski, J.K. and S.J. Mather, Conjugation of DOTA-like chelating agents to peptides and radiolabeling with trivalent metallic isotopes. Nat. Protocols, 2006. 1(2): p. 972-976.
6. Valko, K., et al., Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. Journal of pharmaceutical sciences, 2003. 92(11): p. 2236-2248.
7. Yamazaki, K. and M. Kanaoka, Computational prediction of the plasma protein-binding percent of diverse pharmaceutical compounds. Journal of pharmaceutical sciences, 2004. 93(6): p. 1480-1494.
Пример 2: Результаты
1. Эффект от введения 2,4-динитробензойной кислоты в линкерную область PSMA I&T
DOTAGA-y(3-l)fk(Sub-KuE) (PSMA I&T):


2. Связывающий мотив изменился с EuK на ЕиЕ, а пептидный спейсер - с -y(3-l)fk- на -y-2-nal-k-
DOTAGA-y(3-l)fk(Sub-KuE) (PSMA I&T):
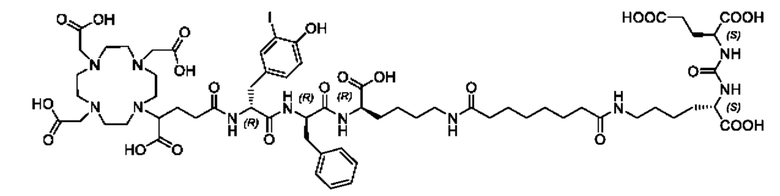
DOTAGA-y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-46):

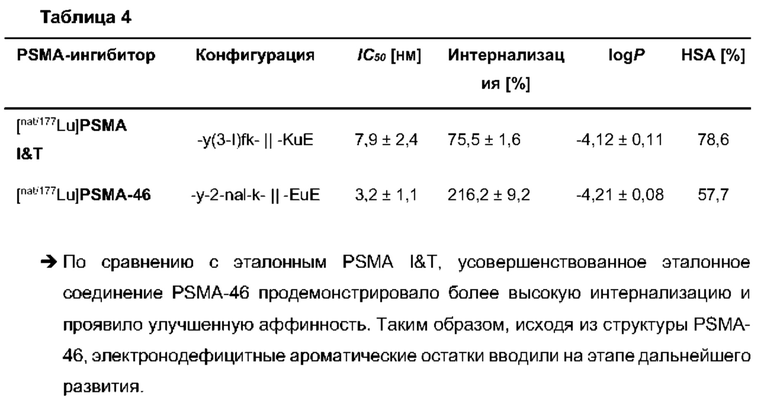
3. 4-нитрофенилаланин и 2,4-DNBA вводили в пептидный спейсер PSMA-46 DOTAGA-y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-46):
DOTAGA-F(4-N02)-y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-52):
2,4-DNBA-Dap(DOTAGA)-y-2-nal-k(Suc-N5-orn-C4-EuE)(PSMA-53):
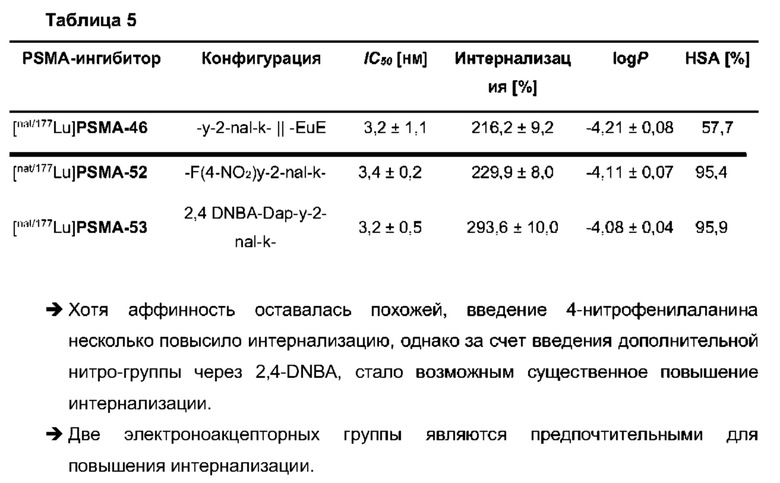
4. Введение 4-амино-фенилаланина
DOTAGA-F(4-NH2)y-2-nal-k(Suc-N5-orn-C4-EuE) (PSMA-49):
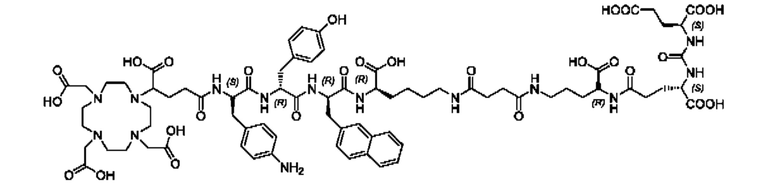
DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-2,4-DNBA) (PSMA-61):

DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-62):


5. Введение электронодефицитной группы в пептидный спейсер DOTAGA-F(4-NH2)y-2-nal-e(Abz-N5-orn-C4-EuE)(PSMA-60):

2,4-DNBA-Dap(DOTAGA)y-2-nal-e(Abz-N5-orn-C4-EuE) (PSMA-65):


6. Тримезиновую кислоту вводили в линкер и пептидный спейсер PSMA-ингибиторов DOTAGA-F(4-NH2)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-62):
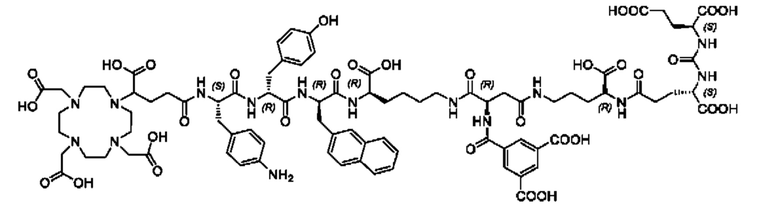
DOTAGA-Dap(TMA)y-2-nal-k(d[N5-orn-C4-EuE]-TMA) (PSMA-66):
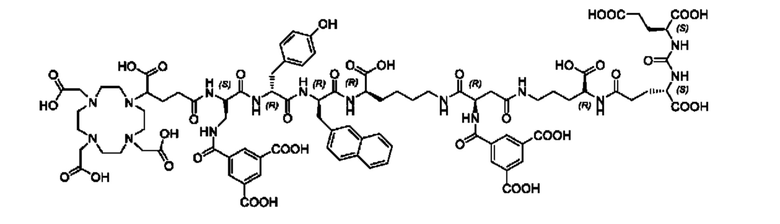

7. Влияние интернализации на удержание клеток in vitro оценивали для соединений [177Lu]PSMA-62 и [177Lu]PSMA-66 в сравнении с [177Lu]PSMA I&T и [177Lu]PSMA-617
[177Lu]PSMA-66 продемонстрировал наивысшую внутриклеточную активность в опухолевых клетках через 1 ч после [177Lu]PSMA-62, хотя было обнаружено, что интернализация [177Lu]PSMA-62 была выше чем для [177Lu]PSMA-66 (343,9% по сравнению с 297,8%; соответственно). Интересным является то, что даже когда повторная интернализация была заблокирована 100 мкм раствора 2-РМРА, внутриклеточный клиренс [177Lu]PSMA-66 был ниже чем для всех других исследуемых соединений. Разница по сравнению с эталонным [177Lu]PSMA I&T была более чем двухкратная, если повторная интернализация была заблокирована.
[177Lu]PSMA-66 имеет девять свободных карбоциклических групп, которые равняются девяти отрицательным зарядам in vivo (рН=7,4). Обширно заряженный характер этого соединения может быть возможным объяснением затянувшегося внутриклеточного удержания ввиду электростатических отталкивающих эффектов от отрицательно заряженных клеточных мембран.
8. In vivo эксперименты: Биораспределение
Таблица 9: Данные биораспределения [177Lu]PSMA-49, [177Lu]PSMA-62 и [177Lu]PSMA-66 (в % ВД/г) в ксенотрансплантатной LNCaP-опухоли у мышей СВ-17 SCID через 1 ч после инъекции (n=4, соответственно). Инъецировали от 3,5 МБк до 5,5 МБк соответствующего 177Lu-меченого радиолиганда (от 0,15 до 0,25 нмоль метки).
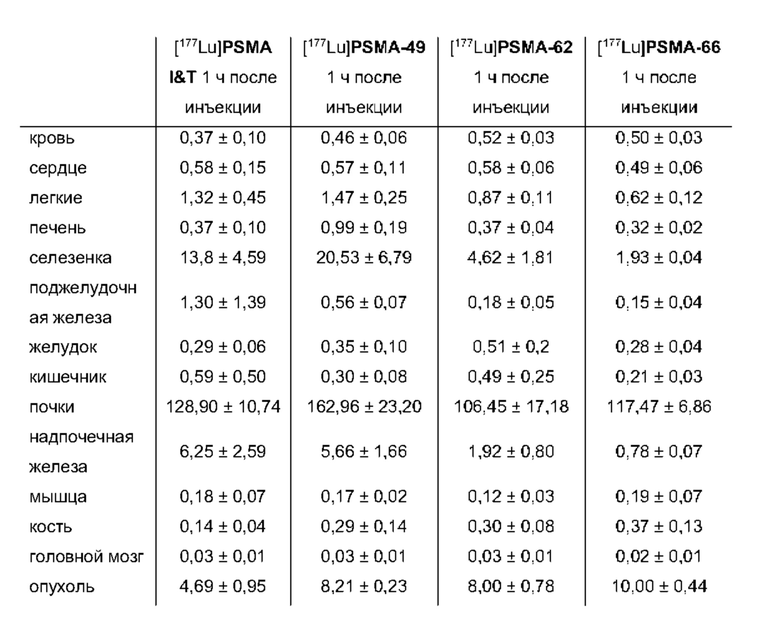

По сравнению с опухолевым поглощением [177Lu]PSMA I&T через 1 ч после инъекции (4,69±0,95%), было достигнуто существенное повышение активности опухоли за счет улучшения интернализации и аффинности. Как уже наблюдалось для [177Lu]PSMA-16, [177Lu]PSMA-40 и [177Lu]PSMA-41, расширение пептидного спейсера посредством 4-амино-о-фенилаланина привело к высокому почечному поглощению и подтвердило то, что эта модификация увеличивает накопление в почках.
Введение тримезиновой кислоты в линкер [177Lu]PSMA-62 привело к снижению почечного поглощения (106,45±17,18% по сравнению с 162,96±23,20%, соответственно) и несколько пониженному опухолевому поглощению по сравнению с эталоном. Поскольку интернализация для [177Lu]PSMA-62 была более высокой при прямом сравнении с [177Lu]PSMA-49, более низкое опухолевое поглощение было неожиданным. Неясно, до какой степени интернализация вносит вклад в опухолевое поглощение, и является ли она менее важной чем аффинность. Прямое сравнение [177Lu]PSMA-49 и [177Lu]PSMA-62 указывает на то, что аффинность является более значимой, поскольку [177Lu]PSMA-49 был более аффинным к PSMA (2,5±0,6 нм по сравнению с 4,0±0,2 нм, соответственно).
Таблица 10: Данные биораспределения [177Lu]PSMA I&T, [177Lu]PSMA-62 и [177Lu]PSMA-66 и [177Lu]PSMA-71 (в % ВД/г) в ксенотрансплантатной LNCaP-опухоли у мышей СВ-17 SCID через 24 ч после инъекции (n=4, соответственно). Инъецировали от 3,5 МБк до 5,5 МБк соответствующего 177Lu-меченого радиолиганда (от 0,15 до 0,25 нмоль метки).
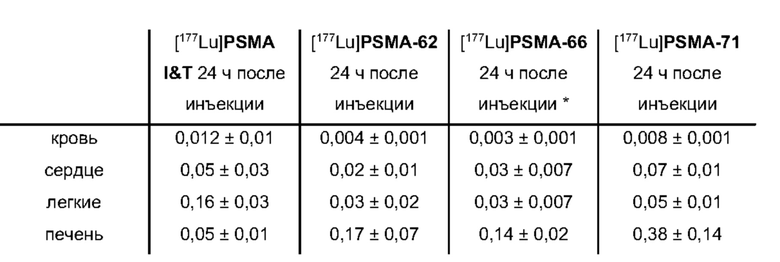
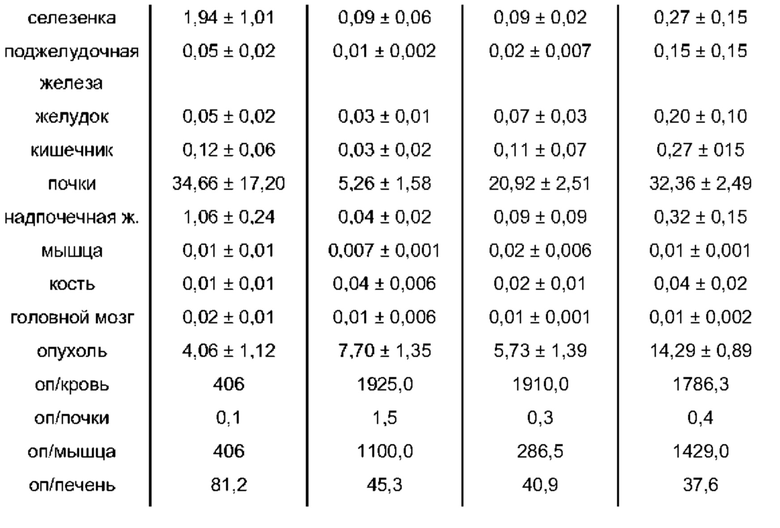
Результаты в Таблице 10 показывают явные отличия между оцененной меткой [177Lu]PSMA-62, [177Lu]PSMA-66 и [177Lu]PSMA-71. Что касается почечного клиренса, было видно, что для всех лигандов наблюдалось снижение почечного поглощения по сравнению с 1 ч после инъекции (Таблица 9). Несмотря на то, что [177Lu]PSMA I&T через 24 ч после инъекции продемонстрировал наивысшее почечное поглощение, [177Lu]PSMA-62 показал наинизшее, что соответствует наблюдаемому почечному клиренсу в ПЭТ-исследовании. Опухолевое поглощение [177Lu]PSMA-61 через 24 ч после инъекции оставалось почти устойчивым в течение 23 ч (8,00±0,75 по сравнению с 7,70±1,35% ВД/г, через 1 ч после инъекции и 24 ч после инъекции, соответственно). Несмотря на то, что [177Lu]PSMA-62 и [177Lu]PSMA-66 продемонстрировали похожий in vitro параметр в отношении интернализации и аффинности, опухолевое поглощение [177Lu]PSMA-66 снизилось в большей степени с 1 ч после инъекции до 24 ч после инъекции. (10,00±0,44 по сравнению с 5,73±1,39% ВД/г, через 1 ч после инъекции и 24 ч после инъекции, соответственно) по сравнению с [177Lu]PSMA-62. Более сильное опухолевое удержание вместе с более выгодными соотношениями опухоль-печень и опухоль-мышца делают [177Lu]PSMA-62 превосходным по сравнению с [177Lu]PSMA-66. Наивысшее опухолевое поглощение наблюдали для PSMA-71, который также проявлял наивысшее значение HSA-связывания. Хотя почечное поглощение через 24 ч после инъекции было подобно [177Lu]PSMA I&T, опухолевое поглощение было более чем в три раза выше для [177Lu]PSMA-71 (4,06±1,12 по сравнению с 14,29±0,89% ВД/г, [177Lu]PSMA I&T и [177Lu]PSMA-71, соответственно).
В этом отношении, [177Lu]PSMA-71 можно рассматривать как особенно ценную метку для применения в эндорадиотерапии и как кандидата для применения в медицине.
9. In vivo эксперименты: ПЭТ-визуализация
Эффект от замены 2,4-динитробензойного линкера на ингибиторы на основе EuK
Ингибитор на основе EuK PSMA-36 оценивали на сканированном ПЭТ-изображении небольшого животного для исследования влияния 2,4-динитробензойной кислоты в линкере на распределение in vivo.
График логарифмических ТАС показывает конкретное почечное и опухолевое поглощение [68Ga]PSMA-36. Линейное снижение активности пула крови и области мышцы предполагает низкое неспецифическое связывание и быструю экскрецию. Накопление в опухоли оставалось стойким в течение всего периода наблюдения. Несмотря на то, что [177Lu]PSMA-36 проявлял скорость интернализации, которая была выше более чем в три раза чем [177Lu]PSMA l&T, опухолевое поглощение было умеренным только при 3,5% ВД/мл через 85 мин после инъекции. Наиболее существенное отличие по сравнению с [88Ga]PSMA l&T заключалось в высоком и стойком поглощении в слезной и слюнной железе, отображая прибл. 2% ВД/мл в обеих областях. Поскольку единственным структурным отличием от эталонного [68Ga]PSMA l&T является введение 2,4-динитробензойной кислоты, модификация линкера должна быть причиной этого усиленного поглощения. Однако для подтверждения этого эффекта необходимы дополнительные исследования.
Также интересно то, что клиренс в этих областях был медленнее, чем в пуле крови и мышце, что предполагает участие другого механизма удержания. Было сообщено, что PSMA принимает участие в ангиогенезе при глазной неоваскуляризации у мышей и, следовательно, может объяснять поглощение [88Ga]PSMA-36 [1]. Накопление метки в слюнных железах является общей проблемой в ходе клинических терапевтических подходах с 177Lu-мечеными PSMA-ингибиторами [2]. Поглощение лекарственного средства в слюнных железах зависит от внутри- и внешнеклеточных путей и, в наиболее общем случае, от простой диффузии среди фосфолипидного двойного слоя ациноцитов. Концентрации лекарственного средства в слюне преимущественно отражаются свободной, неионизированной фракцией в плазме крови относительно пассивной диффузии [3-5]. В этом отношении, по всей видимости, очень маловероятно, что пассивная диффузия ответственна за поглощение в слюнной железе. Должны быть вовлечены другие механизмы, поскольку PSMA-ингибиторы на основе EuK являются сильно заряженными in vivo и, следовательно, проявляют высокую полярность. Кроме того, пассивная диффузия была бы визуализирована в ходе получения сканированных ПЭТ-изображений в каждой области в виде высокой фоновой активности, что не происходит для большинства лигандов PSMA, поскольку быстрый клиренс удаляет метку из пула крови.
Эффект тримезиновой кислоты на ингибиторы на основе ЕuЕ
Замена лигандов PSMA на электронодефицитные ароматические системы в результате дала повышенные скорости интернализации [177Lu]PSMA-62 и [177Lu]PSMA-66 (343,9% и 297,8%, соответственно). Таким образом, оба лиганда были оценены и сравнены между собой в ПЭТ-исследованиях.
Обе метки проявляли превосходную кинетику метки в отношении поглощения почками, мышцей и пулом крови. Конкретное поглощение в почках было несколько более высоким для [68Ga]PSMA-66 по сравнению с [68Ga]PSMA-62 (45,3% ВД/мл по сравнению с 34,8% ВД/мл, соответственно). Более высокое почечное поглощение на сканированном ПЭТ-изображении [68Ga]PSMA-66 по сравнению с [68Ga]PSMA-62 четко коррелировало с экспериментами биораспределения. ТАС для активности в мышце и пуле крови показали линейное поглощение и непрерывный клиренс из этих отделов.
10. Ссылки в Примере 2
1. Grant, C.L., et al., Prostate specific membrane antigen (PSMA) regulates angiogenesis independently of VEGF during ocular neovascularization. PloS one, 2012. 7(7): p.e41285.
2. Kulkarni, H.R., et al., PSMA-Based Radioligand Therapy for Metastatic Castration-Resistant Prostate Cancer: The Bad Berka Experience Since 2013. Journal of Nuclear Medicine, 2016. 57(Supplement 3): p.97S-104S.
3. Haeckel, R., Factors influencing the saliva/plasma ratio of drugs. Annals of the New York Academy of Sciences, 1993. 694(1): p.128-142.
4. Jusko, W.J. and R.L. Milsap, Pharmacokinetic Principles of Drug Distribution in Salivaa. Annals of the New York Academy of Sciences, 1993. 694(1): p.36-47.
5. Aps, J.K. and L.C. Martens, Review: the physiology of saliva and transfer of drugs into saliva. Forensic science international, 2005.150(2): p.119-131.
6. Young, J.D., et al., 68Ga-THP-PSMA: a PET imaging agent for prostate cancer offering rapid, room temperature, one-step kit-based radioiabeiing. Journal of Nuclear Medicine, 2017: p.jnumed. 117.191882.
7. Wustemann, Т., et al., Design of Internalizing PSMA-specific Glu-ureido-based Radiotherapeuticals. Theranostics, 2016. 6(8): p.1085.
8. Hao, G., et al., A multivalent approach of imaging probe design to overcome an endogenous anion binding competition for noninvasive assessment of prostate specific membrane antigen. Molecular pharmaceutics, 2013. 10(8): p.2975-2985.
9. Soret, M., S.L. Bacharach, and I. Buvat, Partial-volume effect in PET tumor imaging. Journal of Nuclear Medicine, 2007. 48(6): p. 932-945.
10. Bao, Q., et al., Performance evaluation of the inveon dedicated PET preclinical tomograph based on the NEMA NU-4 standards. Journal of Nuclear Medicine, 2009. 50(3): p. 401-408.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПСА-СВЯЗЫВАЮЩИЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2787105C2 |
| НОВЫЕ СВЯЗЫВАЮЩИЕ ОПУХОЛЕВЫЙ АНТИГЕН АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2831681C2 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
| РАДИОФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА, РАДИОВИЗУАЛИЗИРУЮЩИЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2804297C2 |
| ПСМА-ТАРГЕТНОЕ СОЕДИНЕНИЕ И ЕГО КОМПЛЕКС С РАДИОНУКЛИДАМИ ДЛЯ ТЕРАНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА | 2022 |
|
RU2803734C1 |
| ЛИГАНДЫ МЕЛАНОКОРТИНОВЫХ РЕЦЕПТОРОВ | 2001 |
|
RU2246501C2 |
| СОЕДИНЕНИЕ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩЕЕ НА ПРОСТАТСПЕЦИФИЧЕСКИЙ МЕМБРАННЫЙ АНТИГЕН, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2832300C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДЫ, УМЕНЬШАЮЩИЕ ИЛИ УСТРАНЯЮЩИЕ МЕШКИ, ОБРАЗУЮЩИЕСЯ ПОД НИЖНИМ КРАЕМ ГЛАЗ, И ИХ ПРИМЕНЕНИЕ В КОСМЕТИЧЕСКИХ ИЛИ ДЕРМОФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЯХ | 2005 |
|
RU2387666C2 |
| АНТАГОНИСТЫ СОМАТОСТАТИНА | 2002 |
|
RU2263678C2 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
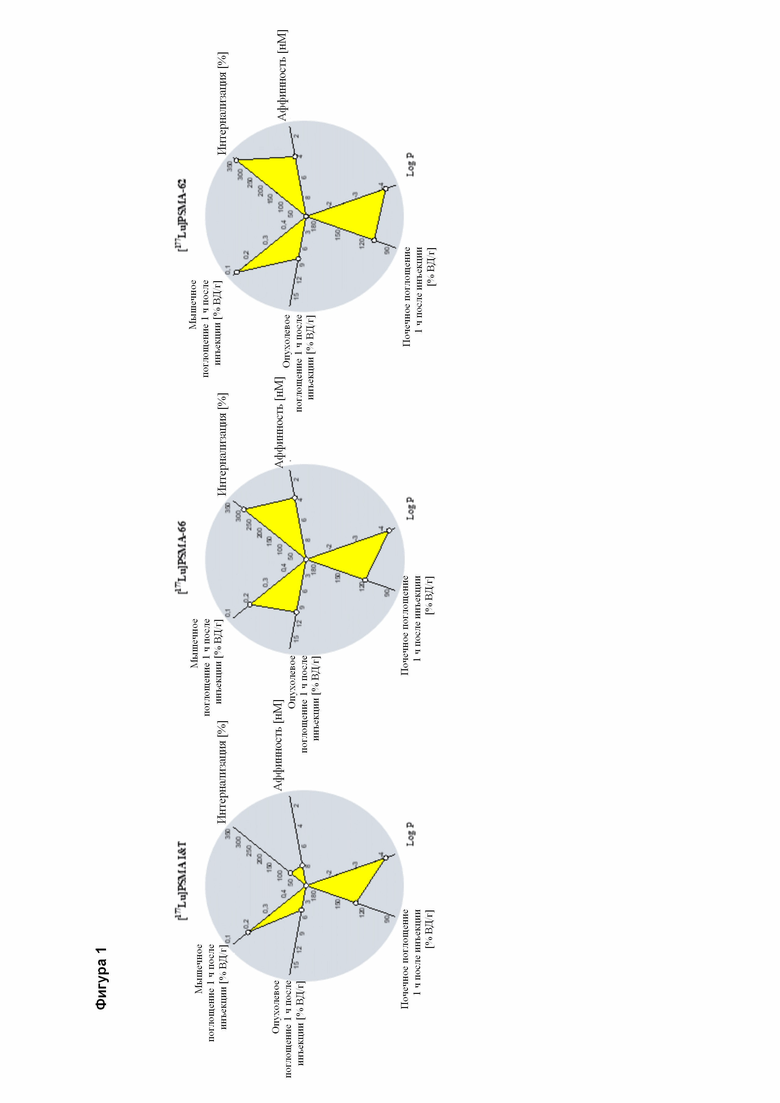


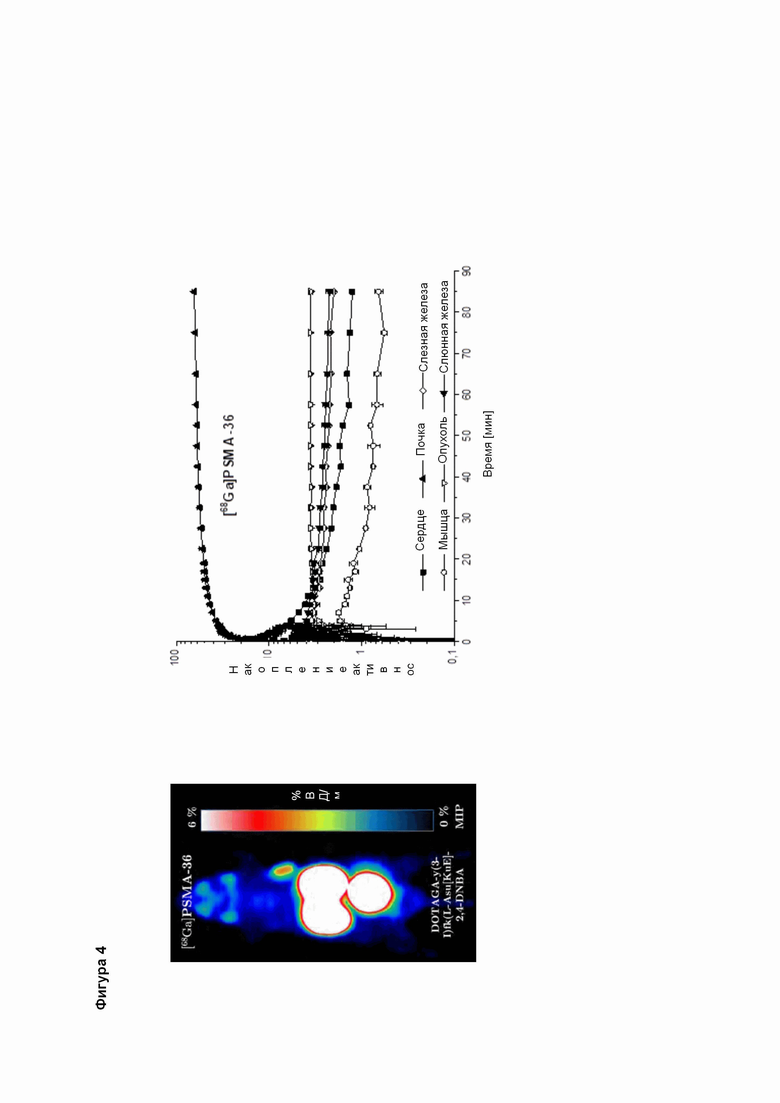

Изобретение относится к визуализации и эндорадиотерапии заболеваний, в которые вовлечен специфический мембранный антиген простаты (PSMA). Представлены соединения, которые связывают или ингибируют PSMA и, кроме того, имеют по меньшей мере один фрагмент, способный к мечению радиоактивными метками. Также представлены медицинские применения таких соединений. 5 н. и 8 з.п. ф-лы, 6 ил., 10 табл., 2 пр.
1. Соединение или его фармацевтически приемлемая соль, где соединение выбрано из соединения формулы (Ih)
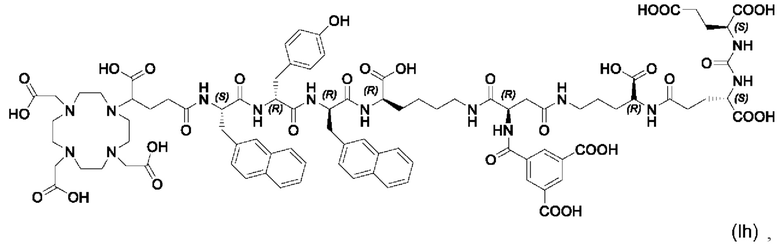
соединения формулы (Ii)

соединения формула (Ij)

соединения формулы (Ik)

соединения формулы (Im)

соединения формулы (In)
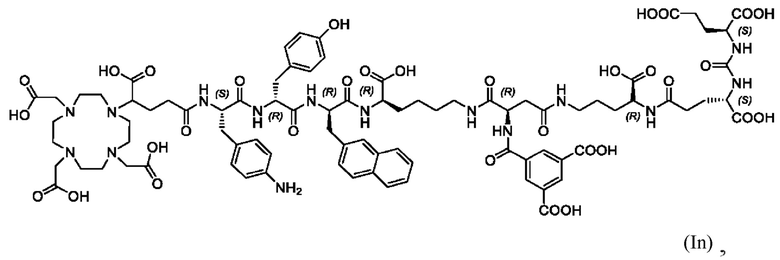
соединения формулы (Iо)

соединения формулы (Iр)
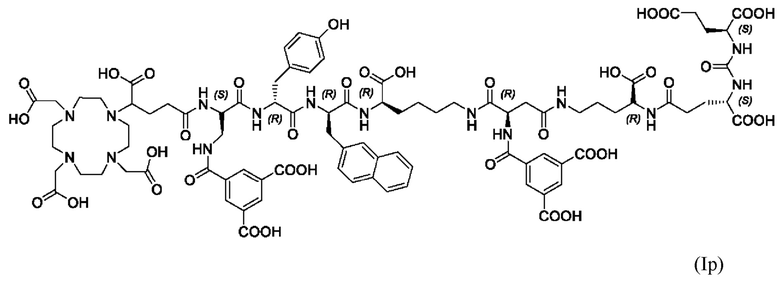
и соединения формулы (Iq)
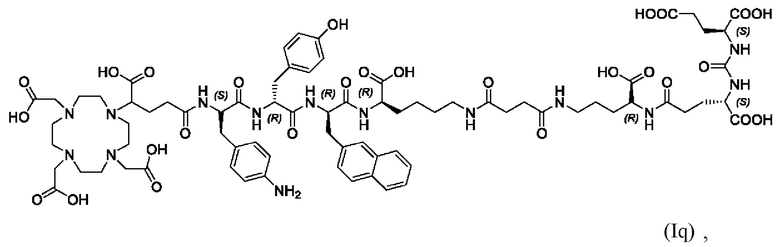
причем остаток 2-[1,4,7,10-тетраазациклододекан-4,7,10-триуксусная кислота]-пентандиовая кислота содержит хелатирующую группу, необязательно содержащую хелатированный нерадиоактивный или радиоактивный катион.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где хелатирующая группа содержит хелатированный радиоактивный катион выбран из катионов 44Sc, 47Sc, 51Cr, 52mMn, 58Co, 52Fe, 56Ni, 57Ni, 62Cu, 64Cu, 67Cu, 66Ga, 68Ga, 67Ga, 89Zr, 90Y, 89Y, 94mTc, 99mTc, 97Ru, 105Rh, 109Pd, 111Ag, 110mIn, 111In, 113mIn, 114mIn, 117mSn, 121Sn, 127Te, 142Pr, 143Pr, 149Pm, 151Pm, 149Tb, 153Sm, 157Gd, 161Tb, 166Ho, 165Dy, 169Er, 169Yb, 175Yb, 172Tm, 177Lu, 186Re, 188Re, 191Pt, 197Hg, 198Au, 199Au, 212Pb, 203Pb, 211At, 212Bi, 213Bi, 223Ra, 225Ac и 227Th, или катионной молекулы, содержащей 18F.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где хелатированный радиоактивный катион выбран из катионов 44Sc, 47Sc, 64Cu, 67Cu, 68Ga, 90Y, 111In, 161Tb, 166Ho, 177Lu, 188Re, 212Pb, 212Bi, 225Ac и 227Th или катионной молекулы, содержащей 18F.
4. Соединение или его фармацевтически приемлемая соль по п. 2, где хелатированный радиоактивный катион представляет собой 177Lu.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где соединение выбрано из
соединения формулы (Ih)
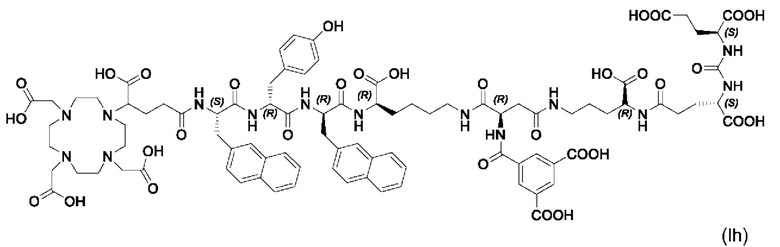
и соединение формулы (Ih) помечено 177Lu.
6. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где соединение выбрано из
соединения формулы (In)
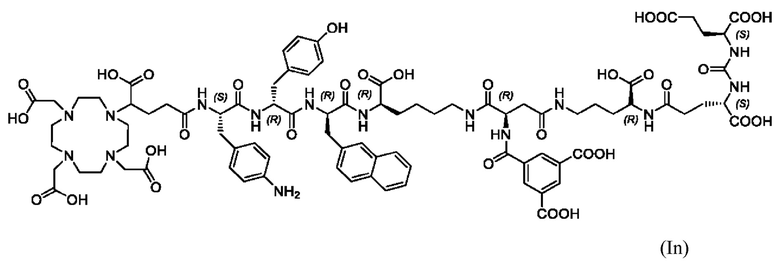
и соединение формулы (In) помечено 177Lu.
7. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где соединение выбрано из
соединения формулы (Ip)
и соединение формулы (Ip) помечено 177Lu.
8. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где соединение выбрано из:
соединения формулы (Iq):

и соединение формулы (Iq) помечено 177Lu.
9. Соединение или его фармацевтически приемлемая соль по п. 8, где соединение представляет собой соединение формулы (Iq), помеченное 177Lu:

10. Фармацевтическая композиция для лечения рака простаты, содержащая или
состоящая из одного или более соединений по любому из пп. 1-9 или одной или более фармацевтически приемлемых солей указанных соединений, где соединение или соль присутствуют в эффективном количестве.
11. Диагностическая композиция для диагностики рака простаты, содержащая или
состоящая из одного или более соединений по любому из пп. 1-9 или одной или более фармацевтически приемлемых солей указанных соединений, где соединение или соль присутствуют в эффективном количестве.
12. Применение соединения по любому из пп. 1-9 или фармацевтически приемлемой соли указанного соединения для диагностики рака простаты.
13. Применение соединения по любому из пп. 1-9 или фармацевтически приемлемой соли указанного соединения для лечения рака простаты.
| WO 2017116994 A1 (FIVE ELEVEN PHARMA INC.), 06.07.2017 | |||
| WO 2017165473 A1 (THE JOHNS HOPKINS UNIVERSITY ), 28.09.2017 | |||
| Eleni Gourni et al., "Metal-Based PSMA Radioligands", Molecules, 2017 Apr; 22(4): 523 (doi: 10.3390/molecules22040523) | |||
| Martina Weineisen et al., "Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for |

