Область техники, к которой относится изобретение
Настоящее изобретение относится к области медицинской радиологии и молекулярной визуализации, в частности к соединению, целенаправленно воздействующему на простатспецифический мембранный антиген, а также к его получению и применению.
Уровень техники
Рак предстательной железы является вторым по распространенности видом рака среди мужчин во всем мире и шестым по распространенности видом рака среди мужчин в Китае. За последнее десятилетие частота заболеваемости раком предстательной железы в Китае быстро возросла, среднегодовой прирост уже достиг 12,07%. Рак предстательной железы описывают как «тихий убийца», его трудно обнаружить на ранних стадиях, и около двух третей пациентов к моменту постановки диагноза уже находятся на последней стадии. Экспрессия простатспецифического мембранного антигена (PSMA) в клетках рака предстательной железы в 100-1000 раз выше, чем в здоровых клетках, при этом его экспрессия еще выше в раковых клетках на поздних стадиях рака при антиандрогенной терапии. Эти характеристики делают PSMA идеальной мишенью для таргетной диагностики и терапии рака предстательной железы.
В декабре 2020 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило меченный галлием 68 PSMA-11 (68Ga-PSMA-11), который является первым диагностическим средством для PET-визуализации, предназначенным для диагностики PSMA-положительных поражений у пациентов с раком предстательной железы. Впоследствии PSMA617, меченный с помощью 177-Lu, начали применять для терапевтического исследования PSMA-положительных поражений. Будучи низкомолекулярным препаратом 177Lu-PSMA617 быстро выводится из крови. Из-за таких метаболических характеристик доза поглощения в опухоли является низкой, и время удерживания слишком коротко. Приблизительно 30% пациентов не реагируют на терапию с использованием 177Lu-PSMA617. Для увеличения дозы, доставляемой к опухолям, были проведены исследования по соединению модифицированного малеимидом, усеченного производного синего Эванса с сульфгидрилсодержащим PSMA (177Lu-EB-PSMA617). Благодаря связыванию с альбумином в крови период полувыведения зонда, нацеленного на PSMA, из кровотока значительно продлевается. Хотя стратегия модификации увеличила поглощение дозы опухолями и продлила время удерживания в опухолях, в последующих исследованиях были обнаружены новые проблемы. Среди пациентов, получающих терапию с использованием 177Lu-EB-PSMA617 (3,52±0,58 ГБк), у 37,5% пациентов наблюдается анемия 3-4 степени, у 12,5% пациентов снижены лейкоциты, у 37,5% пациентов наблюдаются тромбоцитопения (Journal of Nuclear Medicine December 2020, 61 (12): 1772–1778). Данная ситуация указывает на то, что слишком длительный период полураспада 177Lu-EB-PSMA617 в крови приводит к гематотоксичности и миелосупрессии и особенно имеет более серьезные побочные эффекты у пациентов, страдающих раком предстательной железы с тяжелым метастазированием в кости и функцией костного мозга в критическом состоянии. Из-за таких тяжелых побочных эффектов ценность таргетного препарата в клиническом применении будет значительно снижена. Благодаря этим исследованиям было признано, что нацеленный зонд, имеющий более длительный период полувыведения из кровотока, не лучше для терапии опухолей, а наоборот, позволяет контролировать время в кровотоке в разумных пределах, обеспечивая при этом высокий уровень поглощения в опухолях.
Следовательно, необходимо дополнительно оптимизировать зонд, нацеленный на PSMA, а время нахождения в кровотоке отрегулировать в соответствующем диапазоне, при этом обеспечивается высокое поглощение в опухолях, чтобы удовлетворить потребности нуклидной терапии и достичь максимального терапевтического эффекта.
Сущность изобретения
На основании вышеизложенного основной целью настоящего изобретения является разработка соединения, целенаправленного воздействующего на простатспецифический мембранный антиген. Соединение характеризуется высокой степенью поглощения в опухолях и соответствующим временем нахождения в кровотоке. Что позволит преодолеть не только недостатки в виде слишком быстрого метаболизма и слишком короткого времени удерживания в органе-мишени существующей малой молекулы 177Lu-PSMA617, но также позволит избежать гематотоксичности и миелосупрессии, вызванных слишком длительным периодом полувыведения из крови молекулы, такой как 177Lu-EB-PSMA617. Таким образом, нуклидная диагностика и терапевтические эффекты целенаправленного воздействия на PSMA улучшаются, и представленное соединение имеет действительную ценность и потенциал для клинического применения и популяризации.
Другой целью настоящего изобретения является создание меченного радиоактивным изотопом комплекса, целенаправленного воздействующего на простатспецифический мембранный антиген. Комплекс также характеризуется высоким поглощением в опухолях и соответствующим временем нахождения в кровотоке и может иметь такие преимущества, как высокий терапевтический эффект в отношении опухолей и низкие побочные эффекты.
Другая цель настоящего изобретения состоит в обеспечении способа получения меченного радиоактивным изотопом комплекса, целенаправленного воздействующего на простатспецифический мембранный антиген.
Другая цель настоящего изобретения состоит в обеспечении применения комплекса для нуклидной визуализации и терапии рака предстательной железы.
Технические решения для реализации вышеуказанной основной цели настоящего изобретения включают следующие два аспекта: синтез следующих лигандов и введение радиоактивных изотопов в них.
В первом аспекте в настоящем изобретении предусмотрено соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, характеризующееся высоким поглощением в опухолях и соответствующим временем нахождения в кровотоке. Соединение имеет следующую структуру, показанную формулой (I),
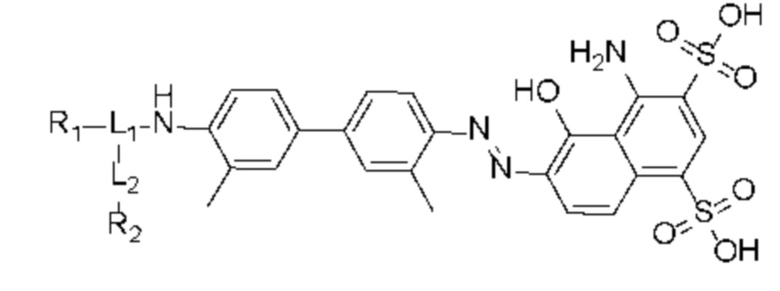 Формула (I),
Формула (I),
где
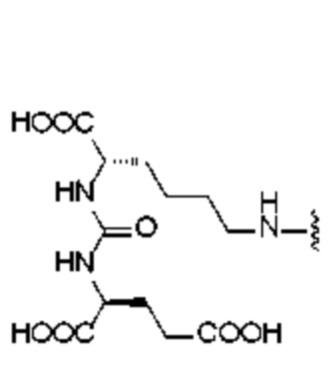
L1 представляет собой -(X)n-(CH2)m-(Y)q-, где n представляет собой целое число от 0 до 12 (предпочтительно целое число от 0 до 6), X и Y независимо выбраны из лизина, глутаминовой кислоты или производной структуры, содержащей лизин и глутаминовую кислоту, m представляет собой целое число от 0 до 60 (предпочтительно целое число от 0 до 30), q представляет собой целое число от 0 до 12 (предпочтительно целое число от 0 до 6), при этом каждый CH2 может быть независимо замещен -O-, -NH(CO)- или -(CO)-NH-;
L2 представляет собой -(CH2)p-, где p представляет собой целое число от 0 до 30 (предпочтительно целое число от 0 до 12), при этом каждый CH2 может быть независимо замещен -O-, -NH(CO)- или -(CO)-NH-, при условии, что отсутствует замещение двух смежных групп CH2;
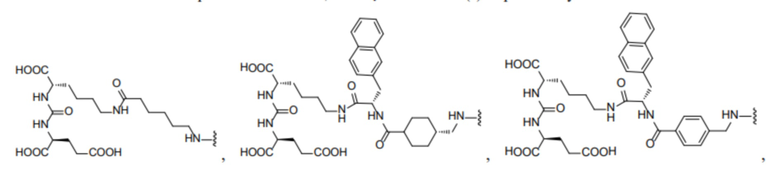
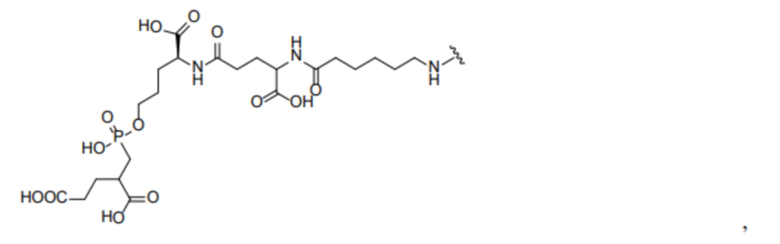
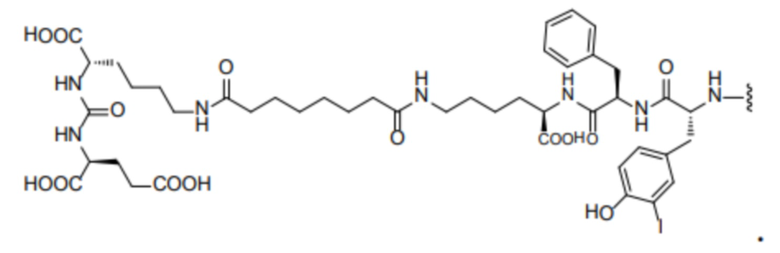
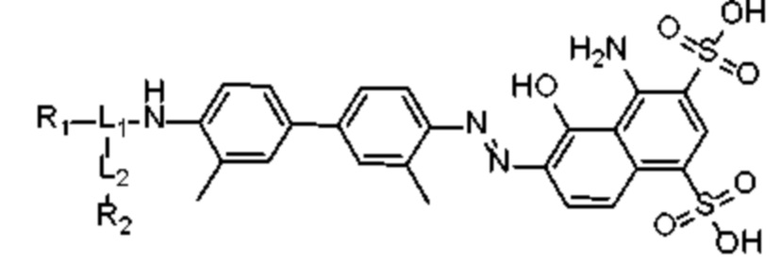
R1 происходит из соединения, целенаправленно воздействующего на простатспецифический мембранный антиген, и имеет любую из следующих структур:
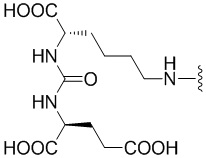 или
или 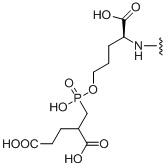 ;
;
и R2 представляет собой хелатирующую нуклиды группу и выбран из любой из следующих структур:
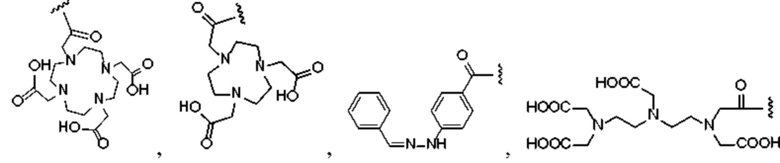 или
или
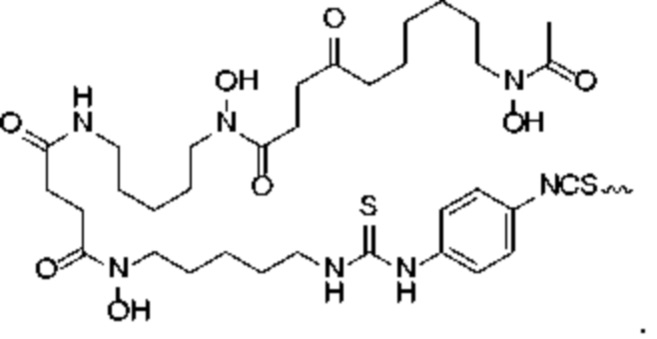
В решении настоящего изобретения R1 в формуле (I) предпочтительно выбран из:
 или
или
В предпочтительном решении настоящего изобретения R1 в формуле (I) представляет собой:
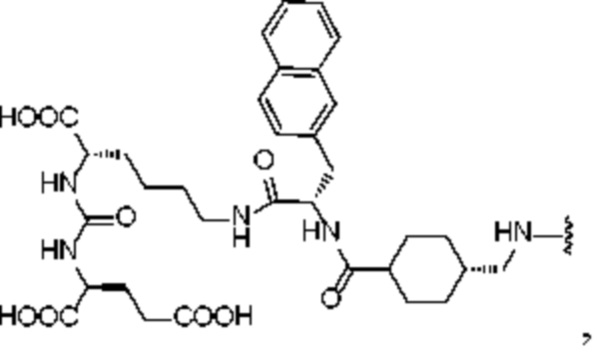 , и R2 представляет собой
, и R2 представляет собой 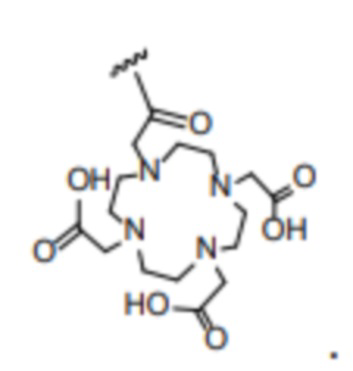 . То есть соединение имеет следующую структуру, показанную формулой (II):
. То есть соединение имеет следующую структуру, показанную формулой (II):
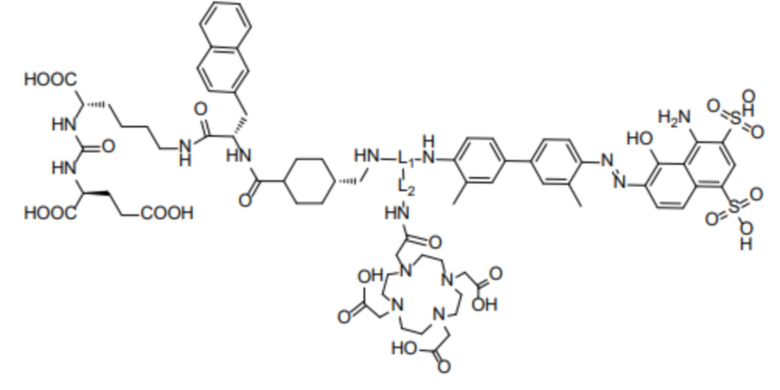 Формула (II),
Формула (II),
где L1 предпочтительно выбран из:
-Lys-(CO)-CH2CH2-(CO)-NH-CH2-(CO)-, -Lys-(CO)-CH2CH2-(OCH2CH2)-(CO)-NH-CH2-(CO)-, -Lys-(CO)-CH2CH2-(OCH2CH2)2-(CO)-NH-CH2-(CO)-, -Lys-(CO)-CH2CH2-(OCH2CH2)4-(CO)-NH-CH2-(CO)-, -(CO)-CH2CH2-(CO)-Lys-, -(CO)-CH2CH2-(OCH2CH2)-(CO)-Lys-, -(CO)-CH2CH2-(OCH2CH2)2-(CO)-Lys-, -(CO)-CH2CH2-(OCH2CH2)4-Lys-, -Lys-(CO)-CH2-(CO)-NH-CH2-(CO)-, -Lys-(CO)-CH2-(OCH2CH2)-O-CH2(CO)-NH-CH2-(CO)-, -Lys-(CO)-CH2-(OCH2CH2)3-O-CH2(CO)-NH-CH2-(CO)-, -(CO)-CH2-(CO)-Lys-, -(CO)-(OCH2CH2)-O-CH2(CO)-Lys- или -(CO)-CH2-(OCH2CH2)3-O-CH2(CO)-Lys-.
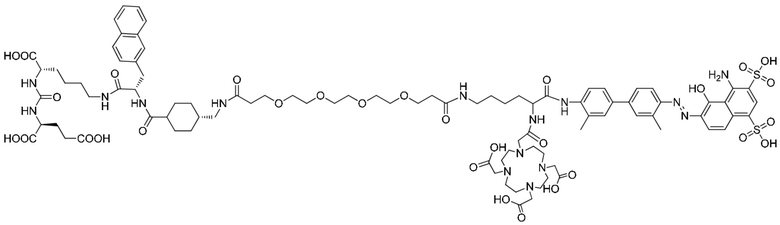
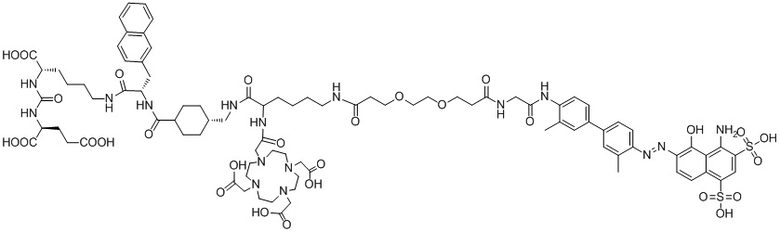
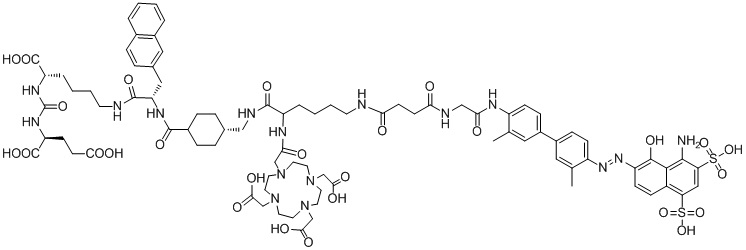
В следующем предпочтительном решении настоящего изобретения соединение имеет следующую структуру, показанную формулой (II-1):
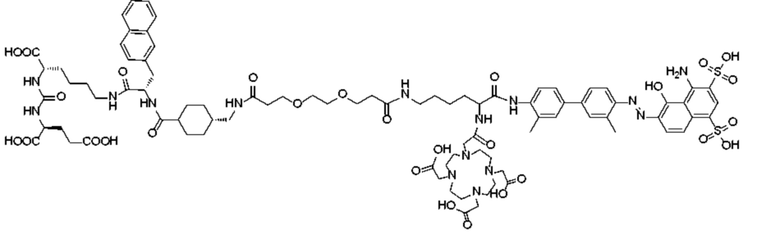
Формула (II-1).
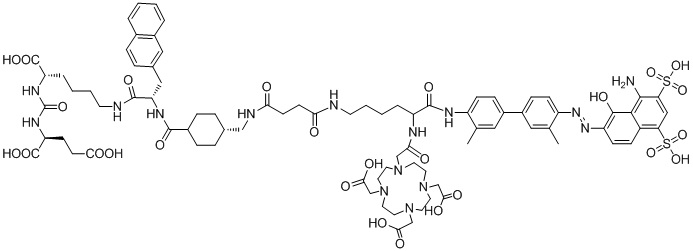
В предпочтительном решении настоящего изобретения соединение также может иметь любую из следующих структур, показанных формулами (II-2) - (II-8):
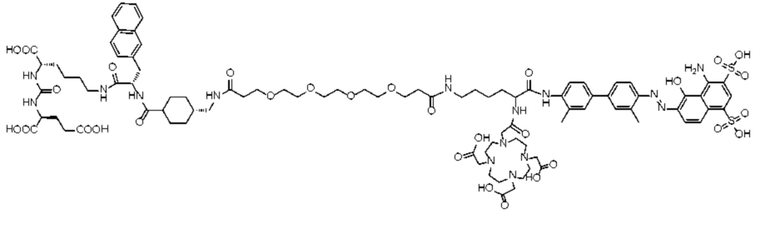
Формула (II-2),
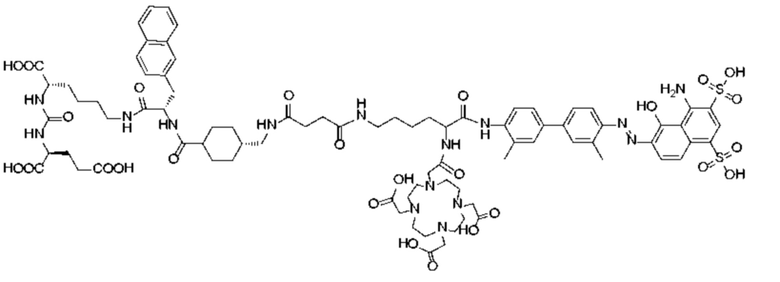
Формула (II-3),
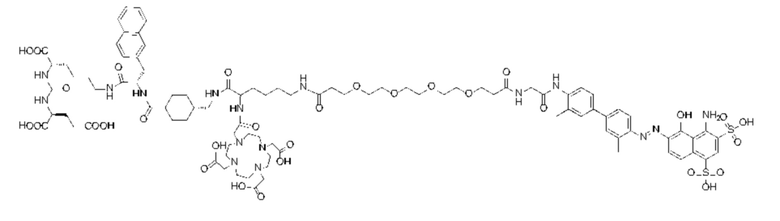
Формула (II-4),
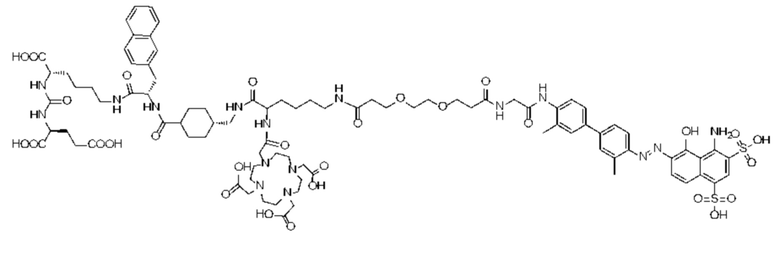
Формула (II-5)
или
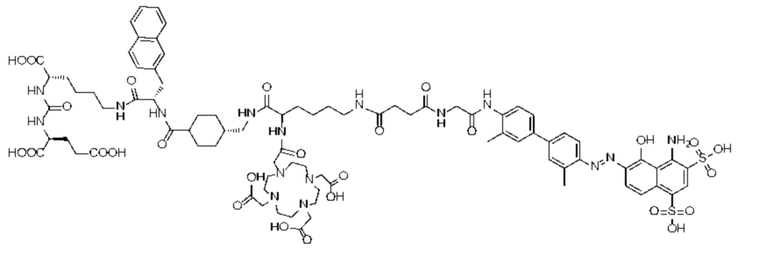
Формула (II-6).
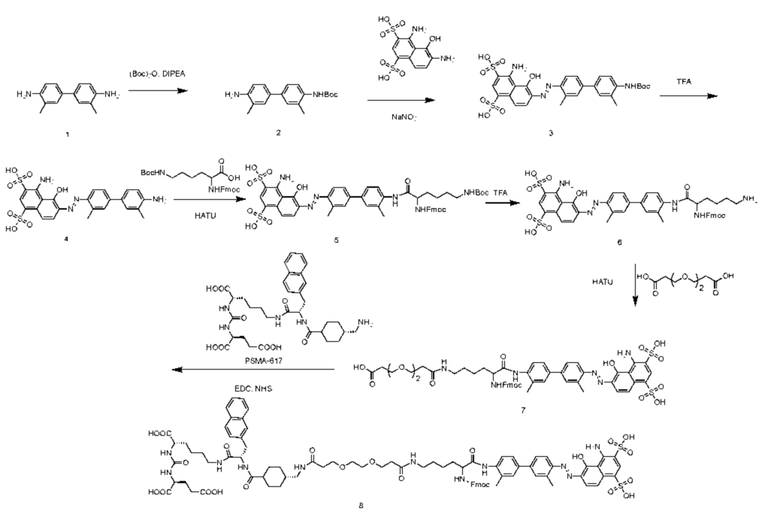
На основании вышеизложенного в настоящем изобретении дополнительно представлен способ получения соединения, представленного формулой (II-1). Представленный способ включает следующие стадии:
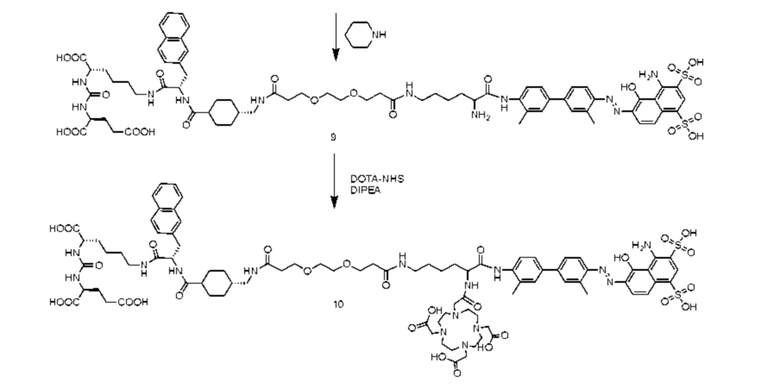
введение защитной Boc-группы на один конец 4,4'-диамино-3,3'-диметилбифенила с последующим обеспечением реакции с 4,6-диамино-5-гидрокси-1,3-нафталиндисульфоновой кислотой с получением усеченного производного синего Эванса; удаление защитной Boc-группы с обеспечением последующей реакции амидной конденсации с Nα-Fmoc-Nε-Boc-L-лизином; затем удаление защитной Boc-группы под воздействием TFA (трифторуксусной кислоты) с последующим обеспечением реакции с COOH-PEG2-COOH и реакции с PSMA-617 в присутствии EDC (1-(3-диметиламинопропил)-3-этилкарбодиимида) и NHS (N-гидроксисукцинимида); затем удаление защитной группы Fmoc с помощью пиперазина и, наконец, осуществление реакции с DOTA-NHS с получением соединения, имеющего следующую структуру, представленную формулой (II-1).
Предпочтительный способ получения соединения, представленного формулой (II-1), по настоящему изобретению, в частности, включает следующие стадии:
осуществление реакции 4,4'-диамино-3,3'-диметилбифенила (соединение 1) с ди-трет-бутилдикарбонатом с получением соединения 2; осуществление реакции соединения 2 с 4,6-диамино-5-гидрокси-1,3-нафталиндисульфоновой кислотой и нитритом натрия с получением усеченного производного синего Эванса (соединение 3); удаление защитной Вос-группы из соединения 3 с получением соединения 4; осуществление реакции соединения 4 с Nα-Fmoc-Nε-Boc-L-лизином в присутствии HATU и DIPEA посредством конденсации с получением соединения 5; растворение соединения 5 в растворе трифторуксусной кислоты для удаления защитной группы с получением соединения 6; растворение соединения 6 в N,N-диметилформамиде и осуществление реакции с COOH-PEG2-COOH в присутствии HATU с получением соединения 7; затем осуществление реакции с PSMA-617 в присутствии EDC и NHS с получением соединения 8; затем удаление Fmoc-защитной группы с помощью пиперазина с получением соединения 9 и, наконец, осуществление реакции с DOTA-NHS с получением соединения 10, имеющего следующую структуру, показанную формулой (II-1).
Путь синтеза согласно вышеуказанным конкретным стадиям выглядит следующим образом:
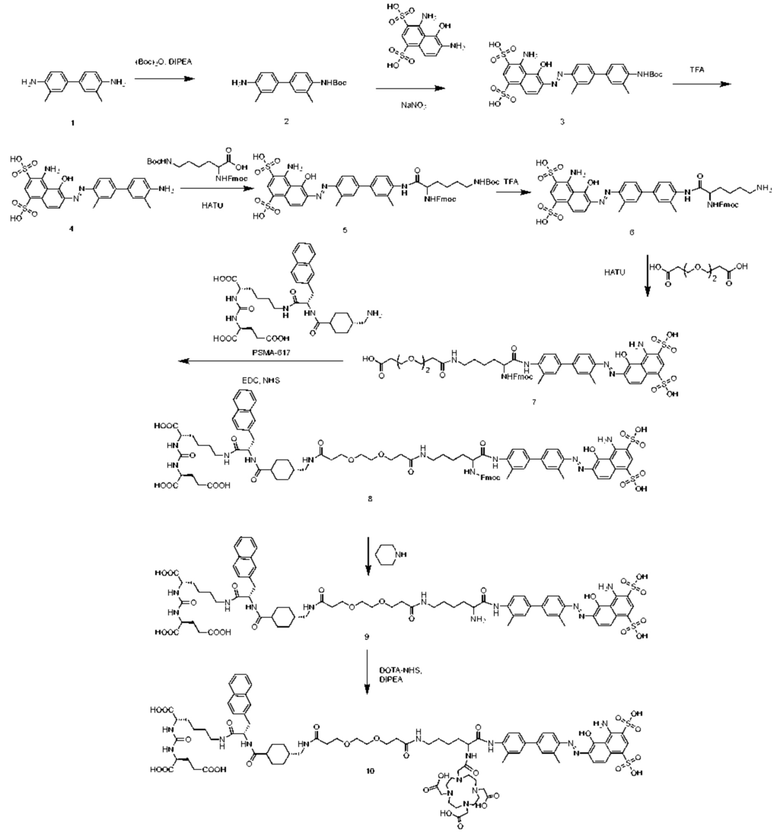
Способы получения других соединений в растворах по настоящему изобретению аналогичны способу получения соединения 10, и получение может быть осуществлено в основном исходя из существующих традиционных способов со ссылкой на путь синтеза соединения 10.
В другом аспекте в настоящем изобретении дополнительно представлен меченный радиоактивным изотопом комплекс. Комплекс получают путем использования соединения, представленного формулой (I) по настоящему изобретению, в качестве лиганда и мечения лиганда радионуклидом. Меченный радиоактивным изотопом комплекс может быть использован в качестве нового радиоактивного диагностического и терапевтического зонда для опухолей, а именно радионуклидного диагностического зонда или радионуклидного терапевтического зонда. Нуклид может быть выбран из любого из 177Lu, 90Y, 18F, 64Cu, 68Ga, 62Cu, 67Cu, 86Y, 89Zr, 99mTc, 89Sr, 153Sm, 149Tb, 161Tb, 186Re, 188Re, 212Pb, 213Bi, 223Ra, 225Ac, 226Th, 227Th, 131I, 211At или 111In и представляет собой Bi, 223Ra, 225Ac, 226Th, 227Th, 131I, 211At или 111In и предпочтительно представляет собой 68Ga, 177Lu или 90Y.
Комплекс по настоящему изобретению предпочтительно имеет следующую структуру, показанную формулой (III),
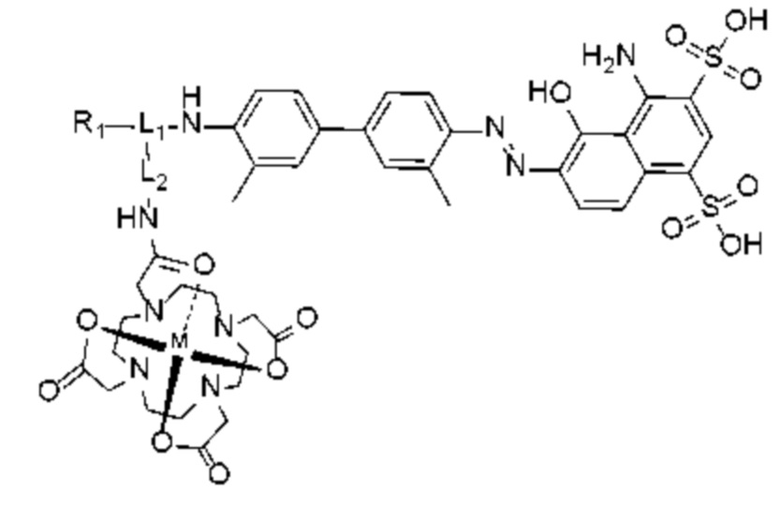 Формула (III),
Формула (III),
где
L1 представляет собой -(X)n-(CH2)m-(Y)q-, где n представляет собой целое число от 0 до 12 (предпочтительно целое число от 0 до 6), X и Y независимо выбраны из лизина, глутаминовой кислоты или производной структуры, содержащей лизин и глутаминовую кислоту, m представляет собой целое число от 0 до 60 (предпочтительно целое число от 0 до 30), q представляет собой целое число от 0 до 12 (предпочтительно целое число от 0 до 6), при этом каждый CH2 может быть независимо замещен -O-, -NH(CO)- или -(CO)-NH-;
L2 представляет собой -(CH2)p-, где p представляет собой целое число от 0 до 30 (предпочтительно целое число от 0 до 12), при этом каждый CH2 может быть независимо замещен -O-, -NH(CO)- или -(CO)-NH-, при условии, что отсутствует замещение двух смежных групп CH2;
R1 представляет собой структуру соединения, целенаправленно воздействующего на простатспецифический мембранный антиген, которая предусматривает структуру:
 или
или 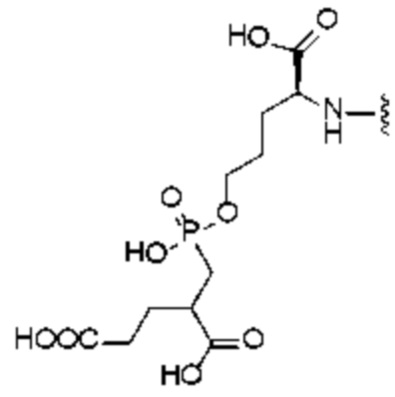 и предпочтительно выбрана из:
и предпочтительно выбрана из:
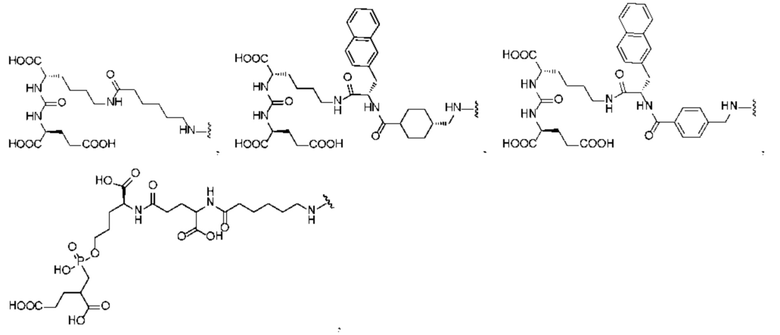
или
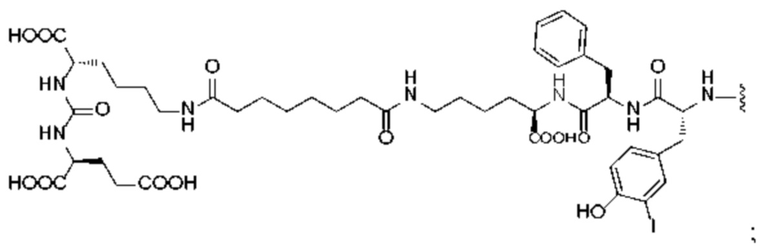
и М представляет собой радионуклид, выбранный из любого из 68Ga, 177Lu или 90Y.
Меченный радиоактивным изотопом комплекс согласно настоящему изобретению можно получить из соединения, содержащего радионуклид, и соединения, представленного формулой (I) по настоящему изобретению, с помощью множества существующих способов мечения. Способ мечения согласно настоящему изобретению предпочтительно включает следующий влажный способ или способ лиофилизации.
Схема мечения влажным способом включает: растворение соответствующего количества соединения, представленного формулой (I) по настоящему изобретению, в буферном растворе или деионизированной воде и добавление раствора радионуклида к полученному раствору для осуществления реакции в закрытых условиях в течение 5-40 мин с получением меченного радионуклидом комплекса;
или схема мечения по способу лиофилизации включает: растворение соответствующего количества соединения, представленного формулой (I) по настоящему изобретению, в буферном растворе или деионизированной воде; обработку полученного раствора посредством асептической фильтрации с последующей загрузкой в контейнер, лиофилизацией и укупориванием пробкой с получением лиофилизированного набора; а затем добавление соответствующего количества раствора уксусной кислоты или буферного раствора в лиофилизированный набор для растворения и добавление соответствующего раствора радионуклида для осуществления реакции в закрытых условиях в течение 5-40 минут с получением меченного радионуклидом комплекса. При этом контейнер для дополнительной упаковки предпочтительно представляет собой пробирку для криоконсервации или флакон с контрольным антибиотиком. Также в набор можно добавить вспомогательное вещество, такое как маннит, аскорбиновая кислота, в соответствии с условиями формирования лиофилизированного порошка в наборе, и набор может обеспечить оптимальный эффект формования путем регулирования дозы соединения, представленного формулой (I) по настоящему изобретению, и вспомогательного вещества.
Продукты, получаемые по схеме мечения влажным способом и схеме мечения лиофилизацией, могут быть дополнительно подготовлены для инъекций посредством традиционной обработки (например, хроматографического разделения и очистки, ротационного испарения для удаления растворителя, растворения остатков PBS, воды или физиологического раствора и асептической фильтрации).
В предпочтительном варианте осуществления настоящего изобретения, где соединение 10, показанное формулой (II-1), используется в качестве лиганда, предпочтительным способом получения меченного радиоактивным изотопом соединения 10 является способ влажного мечения. Способ включает следующие стадии: растворение соединения 10 в буферном растворе или деионизированной воде; добавление свежего радиоактивного раствора для осуществления реакции в закрытых условиях при температуре 37-90°С в течение 5-40 мин с последующим охлаждением; добавление воды для разбавления реакционного раствора с последующим разделением и очисткой на хроматографической колонке Sep-Pak C18; промывка хроматографической колонки буферным раствором или водой для удаления непрореагировавших радиоактивных ионов, и осуществление промывки раствором хлористоводородной кислоты в этаноле или раствором этанола, и осуществление разведения физиологическим раствором или PBS с последующей асептической фильтрацией с получением инъекционного препарата на основе меченого радиоактивным изотопом комплекса, имеющего структуру, показанную формулой (IV), где радионуклид M представляет собой 68Ga, 177Lu или 90Y.
Другим предпочтительным способом получения меченного радиоактивным изотопом соединения 10 согласно настоящему изобретению является способ лиофилизации. Способ включает: растворение соединения 10 и других необходимых реагентов в буферном растворе и обработку полученного раствора асептической фильтрацией с последующей загрузкой в пробирку для криоконсервации, лиофилизацией и герметизацией с получением лиофилизированного набора; добавление соответствующего количества буферного раствора в лиофилизированный набор для растворения и добавление свежеприготовленного радиоактивного раствора для осуществления реакции в закрытых условиях при температуре 37-120°С в течение 5-40 мин с последующим охлаждением; добавление воды для разбавления реакционного раствора с последующим разделением и очисткой на хроматографической колонке Sep-Pak C18; промывку хроматографической колонки буферным раствором или водой для удаления непрореагировавших радиоактивных ионов, и осуществление промывки раствором хлористоводородной кислоты в этаноле или раствором этанола, и осуществление разведения физиологическим раствором или PBS с последующей асептической фильтрацией с получением инъекционного препарата на основе меченого радиоактивным изотопом комплекса, имеющего структуру, показанную формулой (IV), где радионуклид M представляет собой 68Ga, 177Lu или 90Y.
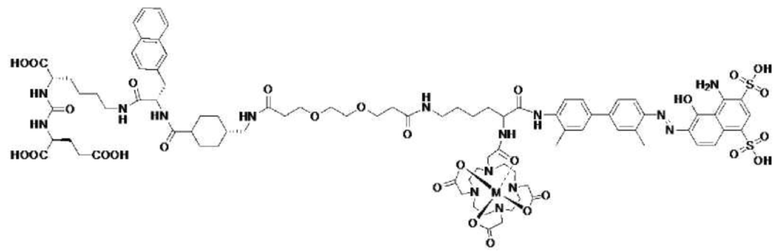
Формула (IV)
Другие химические вещества, используемые на вышеуказанных стадиях синтеза, являются коммерчески доступными продуктами.
Буферный раствор представляет собой вещество для стабилизации значения pH реакционного раствора и может представлять собой ацетат, лактат, тартрат, малат, малеат, сукцинат, аскорбат, карбонат, фосфат и их смесь.
В другом аспекте в настоящем изобретении также предусмотрено применение соединения, представленного формулой (I), или его фармацевтически приемлемой соли при получении препаратов для нуклидной терапии или средств визуализации опухолей у млекопитающих с высокой экспрессией PSMA.
В настоящем изобретении также предусмотрено применение меченного радиоактивным изотопом комплекса, представленного формулой (III), в нуклидной терапии и визуализации опухолей с высокой экспрессией PSMA у млекопитающих.
В предпочтительном применении по настоящему изобретению комплекс составляют в виде инъекционного препарата и затем вводят внутривенно пациентам-людям или млекопитающим с опухолями с высокой экспрессией PSMA.
В настоящем изобретении предусмотрены соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, характеризующееся высоким поглощением в опухолях и соответствующим временем нахождения в кровотоке, и меченный радионуклидами комплекс на его основе, а также предусмотрен способ получения и способ мечения такого соединения. Результаты биологических испытаний показывают, что соединение характеризуется соответствующим периодом полувыведения из кровотока, высоким поглощением в опухолях и долгим периодом времени удерживания. Такая превосходная эффективность в настоящее время недоступна другим средствам, целенаправленно воздействующим на PSMA, и соединение подходит для нуклидной терапии и визуализации опухолей с высокой экспрессией PSMA.
Описание графических материалов
На фиг. 1 показаны результаты поглощения в опухолях через 24 часа после инъекции различных препаратов мышам с раком предстательной железы на модели с подкожной трансплантацией опухоли.
На фиг. 2 показано сравнение поглощения в крови в разные моменты времени после инъекции здоровым мышам соединения из примера 31, 177Lu PSMA и 177Lu EB PSMA 617.
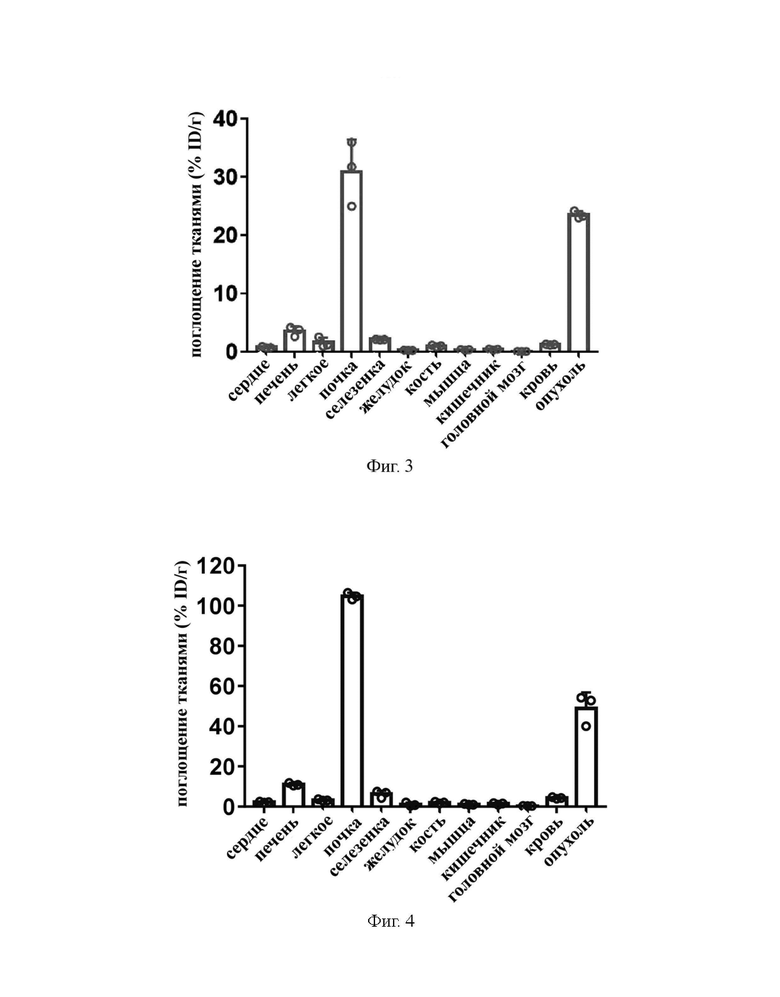
На фиг. 3 показаны результаты распределения в тканях через 24 часа после инъекции соединения из примера 31 мышам с раком предстательной железы на модели с подкожной трансплантацией опухоли.
На фиг. 4 показаны результаты распределения в тканях через 24 часа после инъекции 177Lu-EB-PSMA 617 мышам с раком предстательной железы на модели с подкожной трансплантацией опухоли.
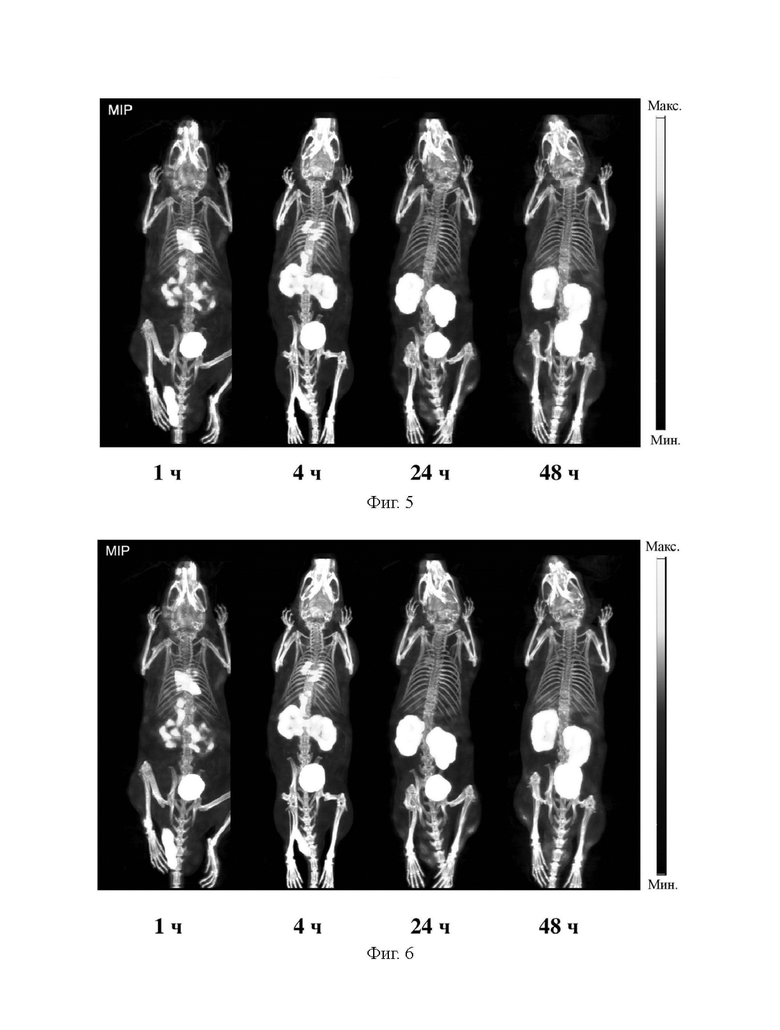
На фиг. 5 представлена диаграмма, демонстрирующая визуализацию SPECT-CT в различные моменты времени после инъекции соединения из примера 31 здоровым мышам.
На фиг. 6 представлена диаграмма, демонстрирующая визуализацию SPECT-CT в различные моменты времени после инъекции здоровым мышам соединения (II-2), меченного с помощью 177Lu, из примера.
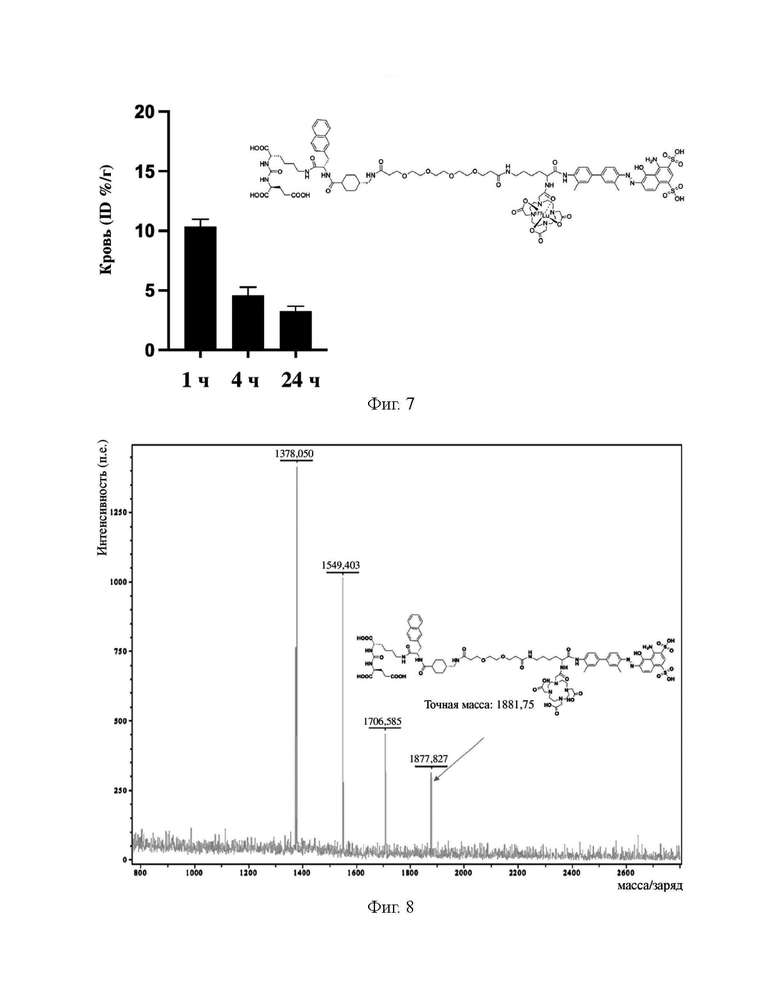
На фиг. 7 показаны результаты поглощения в крови в различные моменты времени после инъекции здоровым мышам соединения (II-2), меченного с помощью177Lu.
На фиг. 8 представлена диаграмма, демонстрирующая масс-спектр соединения 10, полученного в примере 1.
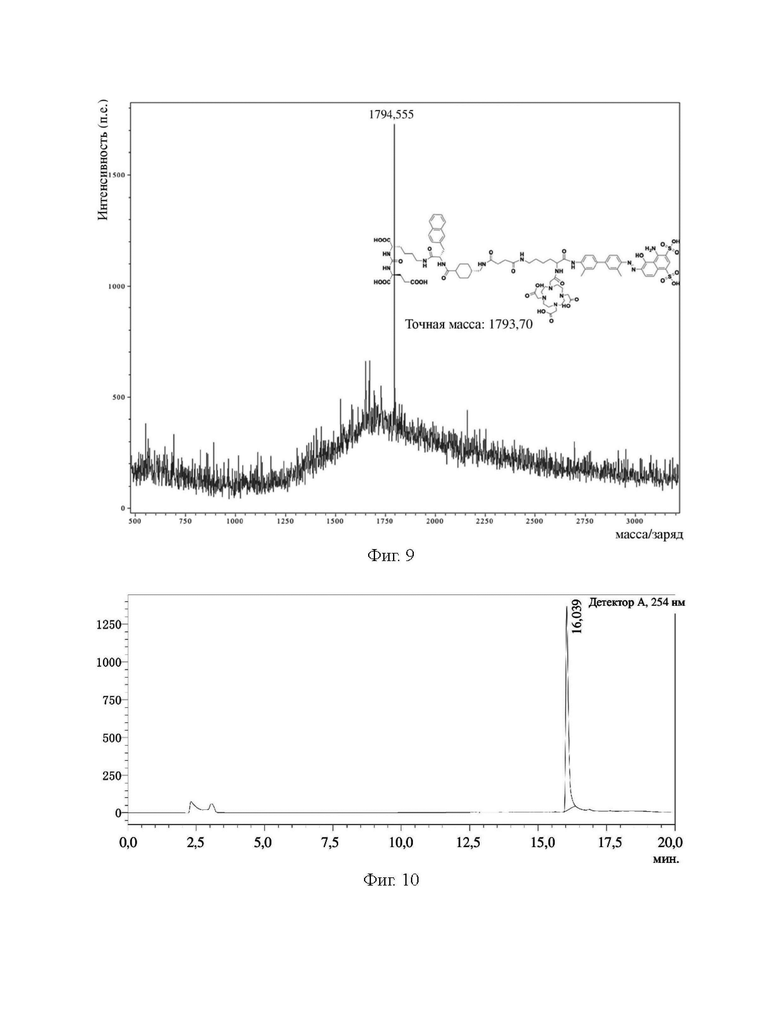
На фиг. 9 представлена диаграмма, демонстрирующая масс-спектр соединения (II-3), полученного в примере 3.
На фиг. 10 представлена HPLC-хроматограмма соединения 5, полученного в примере 1.
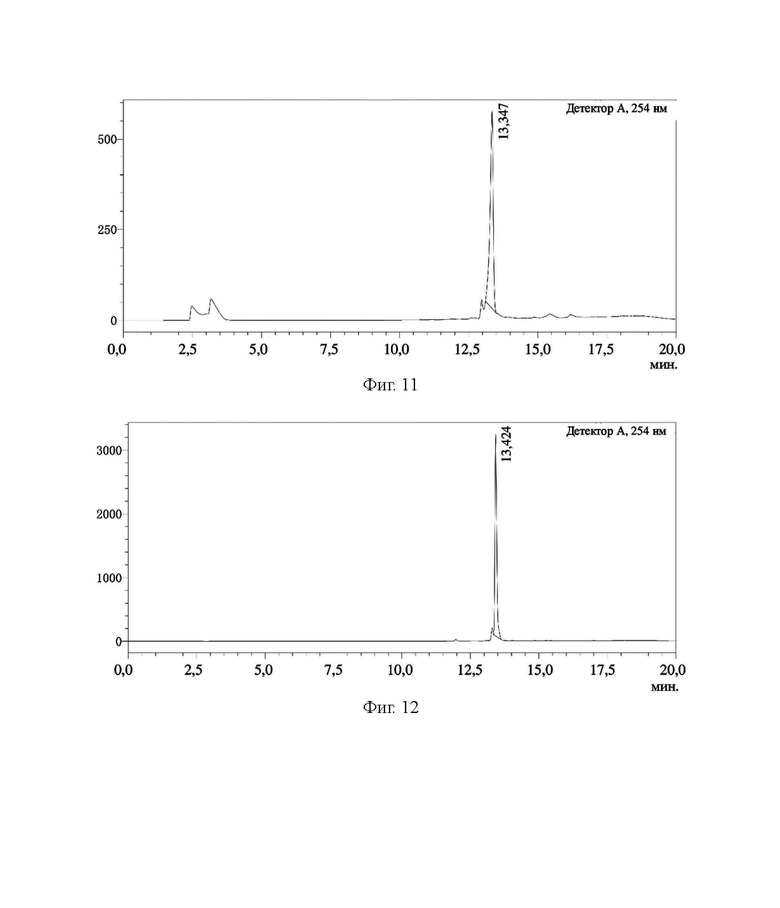
На фиг. 11 представлена HPLC-хроматограмма соединения 6, полученного в примере 1.
На фиг. 12 представлена HPLC-хроматограмма соединения 7, полученного в примере 1.
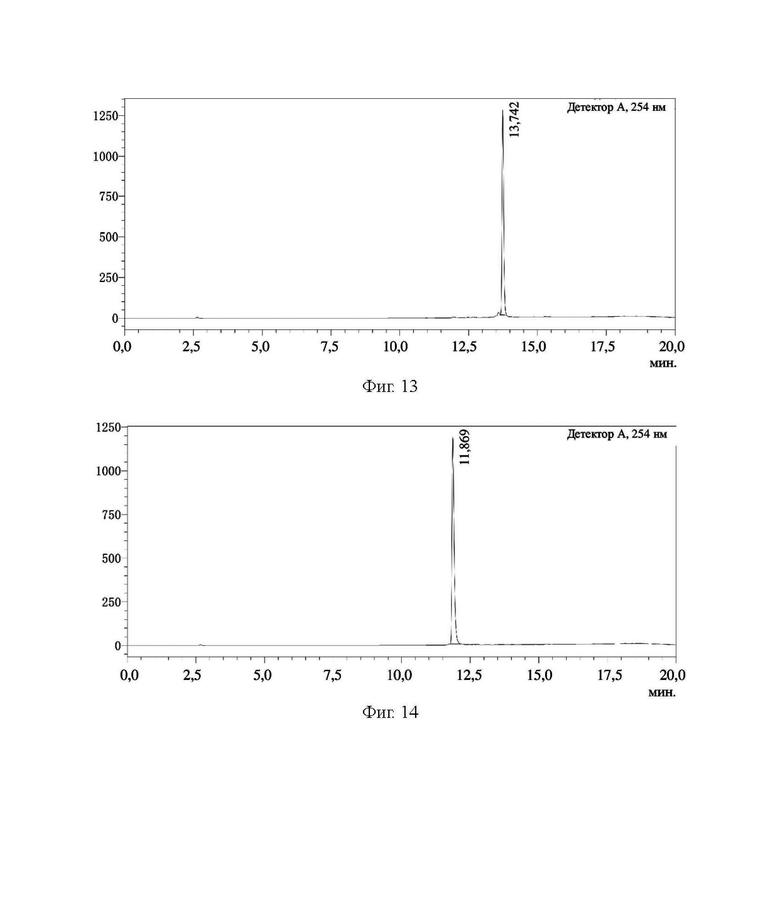
На фиг. 13 представлена HPLC-хроматограмма соединения 8, полученного в примере 1.
На фиг. 14 представлена HPLC-хроматограмма соединения 9, полученного в примере 1.
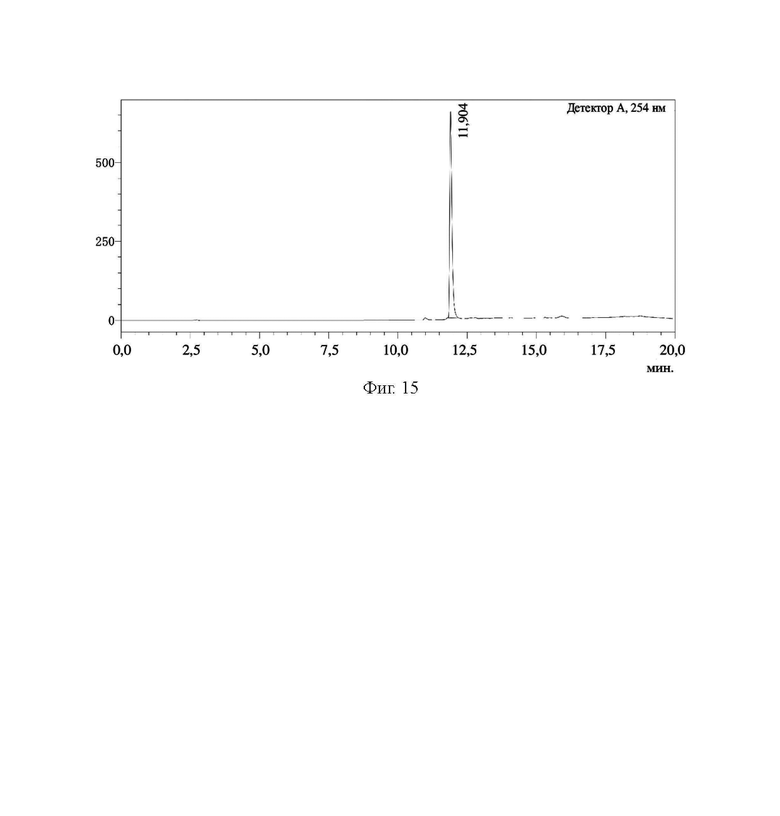
На фиг. 15 представлена HPLC-хроматограмма соединения 10, полученного в примере 1.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Технические решения по настоящему изобретению будут дополнительно проиллюстрированы и описаны ниже посредством конкретных вариантов осуществления вместе с прилагаемыми графическими материалами.
Пример 1. Получение соединения 10, представленного формулой (II-1)
Синтез соединения 2
В колбу объемом 100 мл по отдельности помещали 4,4'-диамино-3,3'-диметилбифенил (соединение 11) (2,12 г, 10,0 ммоль), ди-трет-бутилдикарбонат (2,2 г, 10,0 ммоль), N,N-диизопропилэтиламин (1,3 г, 10,0 ммоль) и 20 мл дихлорметана и перемешивали в течение ночи при комнатной температуре. После контроля завершения реакции посредством HPLC (время удерживания составляло 10,13 мин) осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем осуществляли очистку на колонке с силикагелем (соотношение петролейного эфира и этилацетата составляло 5:1) с получением соединения 2 в виде белого твердого вещества с выходом 59%.
Синтез соединения 3
Соединение 2 (0,31 г, 1,0 ммоль) и 4 мл ацетонитрила по отдельности помещали в колбу объемом 50 мл на ледяной бане, в реакционную колбу по каплям добавляли 1,5 мл 2 М раствора хлористоводородной кислоты для осуществления реакции в течение 15 мин, к 2 мл воды добавляли нитрит натрия (0,068 г, 1,0 ммоль) для растворения и затем добавляли по каплям в реакционную колбу для осуществления реакции в течение получаса с получением раствора А для последующего использования. В другую реакционную колбу емкостью 50 мл на ледяной бане добавляли 4,6-диамино-5-гидрокси-1,3-нафталиндисульфоновую кислоту (0,33 г, 1,0 ммоль), карбонат натрия (0,105 г, 1,0 ммоль) и 5 мл воды с получением раствора B, раствор A медленно по каплям добавляли к раствору B и перемешивали для осуществления реакции в течение 2 ч на ледяной бане. Затем проводили очистку на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 3 с выходом 47%.
Синтез соединения 4
В условиях ледяной бани соединение 3 (0,52 г, 1,0 ммоль) растворяли в трифторуксусной кислоте. Систему нагревали до комнатной температуры для осуществления реакции в течение 2 ч, а после завершения реакции осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 4 с выходом 73%.
Синтез соединения 5
В колбу объемом 100 мл по отдельности помещали соединение 4 (0,54 г, 1,0 ммоль), Nα-Fmoc-Nε-Boc-L-лизин (0,46 г, 1,0 ммоль), HATU (0,38 г, 1,0 ммоль), N,N-диизопропилэтиламин (0,26 г, 2,0 ммоль) и 10 мл N,N-диметилформамида. Реакционную смесь перемешивали до завершения реакции и осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 5 с выходом 57%.
Синтез соединения 6
трет-Бутильную и Boc-защитные группы из соединения 5 удаляли с помощью смеси тиоанизола, 1,2-этандитиола, анизола и TFA (в соотношении 5:3:2:90) при комнатной температуре с получением соединения 6. После завершения реакции TFA удаляли потоком аргона и полученный продукт растворяли в 10 мл N,N-диметилформамида для дальнейшего использования.
Синтез соединения 7
К N,N-диметилформамидному раствору соединения 6 по отдельности добавляли COOH-PEG2-COOH (0,23 г, 1,10 ммоль), HATU (0,38 г, 1,0 ммоль) и N,N-диизопропилэтиламин (0,39 г, 3,0 ммоль). Систему перемешивали в течение ночи при комнатной температуре и завершение реакции контролировали по данным HPLC (время удерживания составляло 10,84 мин). Осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 7 с выходом 50% за две стадии.
Синтез соединения 8
В колбу объемом 50 мл по отдельности помещали соединение 7 (0,21 г, 0,2 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,04 г, 0,2 ммоль), NHS (0,02 г, 0,2 ммоль) и 10 мл N,N-диметилформамида. После осуществления реакции в течение 4 ч добавляли N,N-диизопропилэтиламин (0,06 г, 0,5 ммоль) и PSMA-617 (0,13 г, 0,2 ммоль). Реакционную смесь перемешивали и завершение реакции контролировали по данным HPLC (время удерживания составляло 12,16 мин). Осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 8 с выходом 59%.
Синтез соединения 9
В колбу объемом 25 мл с 5 мл DMF по отдельности помещали соединение 8 (0,16 г, 0,1 ммоль) и пиперидин (0,08 г, 10,0 ммоль). Процесс удаления защитных групп контролировали посредством HPLC до завершения реакции (время удерживания составляло 10,47 мин). Осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 9 с выходом 63%.
Синтез соединения 10
В колбу объемом 25 мл с 5 мл N,N-диметилформамида по отдельности помещали соединение 9 (0,13 г, 0,1 ммоль), DOTA-NHS (0,05 г, 0,1 ммоль) и N,N-диизопропилэтиламин (0,04 г, 0,3 ммоль). Реакционную систему перемешивали для осуществления реакции при комнатной температуре и процесс удаления защитных групп контролировали посредством HPLC до завершения реакции (время удерживания составляло 11,35 мин). Осуществляли перегонку при пониженном давлении для удаления растворителя с получением неочищенного продукта. Затем проводили очистку неочищенного продукта на колонке с обращенной фазой с последующей лиофилизацией с получением чистого соединения 10 с выходом 61%. Описание характеристик структуры полученного соединения представлено на фиг. 8.
Путь синтеза согласно вышеуказанным стадиям выглядит следующим образом:
Примеры 2-6
Соединения в примерах 2-6 имеют структуры, показанные формулами (II-2)-(II-6) соответственно, и способы получения соединений могут ссылаться на способ получения в примере 1. Например, соединение COOH-PEG2-COOH, реагирующее с соединением 6 в примере 1, было заменено на COOH-PEG4-COOH, малоновую кислоту или другие подходящие соединения для получения соединений, представленных формулой (II-2) и формулой (II-3). Nα-Fmoc-Nε-Boc-L-лизин, реагирующий с соединением 4 в примере 1, заменяли Вос-глицином для получения соединений, представленных формулами (II-4)-(II-6), или PSMA-617, реагирующий с соединением 7 в примере 1, заменяли на PSMA-617-(Fmoc)Lys- с получением соответствующих структур следующим образом:
Формула (II-2),
Формула (II-3),
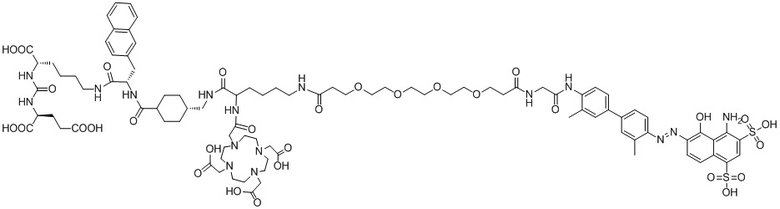
Формула (II-4),
Формула (II-5)
или
Формула (II-6).
Описание характеристик структуры соединения (II-3) представлено на фиг. 9.
Примеры 7-30
Обращаясь к способам получения примеров 1-6, получали соединение, представленное следующей формулой (I),
 Формула (I).
Формула (I).
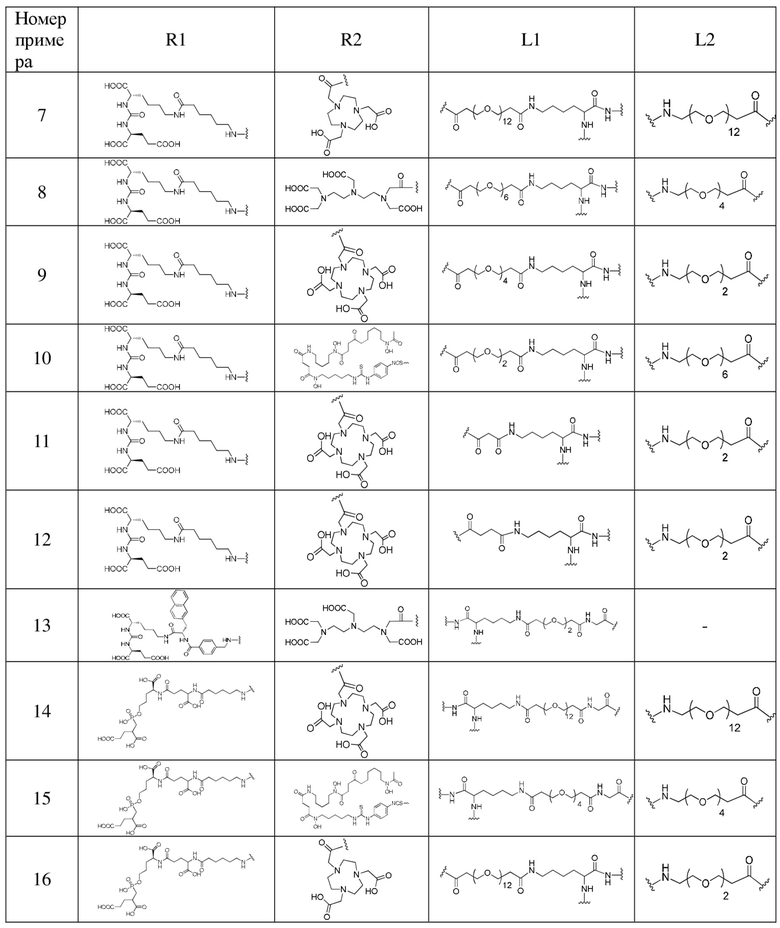
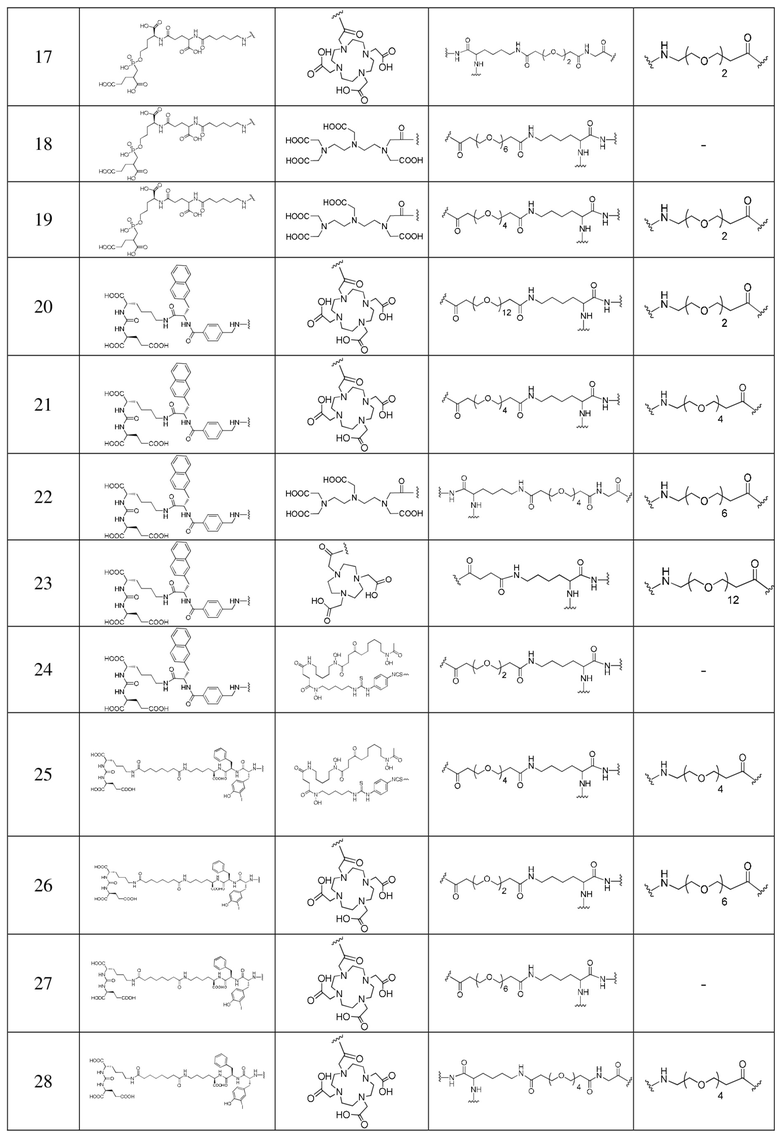
Пример 31. Получение комплекса, меченного с помощью 177Lu
Влажный способ. Раствор ацетата натрия, приблизительно 18,5-1,850 МБк 177LuCl3, добавляли к раствору уксусной кислоты-ацетата (1,0 г/л), содержащему 0,5 мл соединения 10 из примера 1, в центрифужной пробирке и реакцию осуществляли при 90°C в течение 20 мин. Небольшую колонку для разделения C18 сначала медленно промывали с помощью 10 мл безводного этанола, а затем промывали с помощью 10 мл воды. Полученный меченый раствор разбавляли с помощью 10 мл воды и затем отбирали образец в разделительную колонку. Немеченые ионы 177Lu удаляли с помощью 10 мл воды и промывали с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением комплекса, меченного с помощью 177Lu. Промытый раствор разбавляли физиологическим раствором с последующей асептической фильтрацией с получением комплекса, меченного с помощью 177Lu.
Способ лиофилизации. Раствор ацетата натрия, приблизительно 18,5-1,850 МБк 177LuCl3, добавляли в лиофилизированный набор, содержащий соединение 10 из примера 1, и равномерно перемешивали для осуществления реакции при 90°C в течение 20 мин. Небольшую колонку для разделения C18 сначала медленно промывали с помощью 10 мл безводного этанола, а затем промывали с помощью 10 мл воды. Полученный меченый раствор разбавляли с помощью 10 мл воды и затем отбирали образец в разделительную колонку. Немеченые ионы 177Lu удаляли с помощью 10 мл воды и промывали с помощью 0,3 мл 10 мМ этанольного раствора HCl с получением промытого раствора комплекса, меченного с помощью 177Lu. Промытый раствор разбавляли физиологическим раствором с последующей асептической фильтрацией с получением комплекса, меченного с помощью 177Lu.
Экспериментальный пример. Анализ и эффект применения
1. HPLC-анализ и идентификация
HPLC-система была следующей. Для анализа использовали SHIMADZULC-20A и хроматографическую колонку C18 (YMC, 3 мкм, 4,6 × 150 мм). Детекцию выполняли при длине волны 254 нм и скорости потока 1 мл/мин в соответствии со следующим градиентом промывки: в промежутке 0-3 мин 10% ацетонитрила и 90% воды (50 мМ ацетат аммония) оставались неизменными; в промежутке 3-16 мин систему доводили до 90% ацетонитрила и 10% воды (50 мМ ацетат аммония); в промежутке 16-18 мин оставалось 90% ацетонитрила и 10% воды (50 мМ ацетат аммония); в промежутке 18-20 мин систему восстанавливали до 10% ацетонитрила и 90% воды (50 мМ ацетат аммония); в промежутке 20-22 мин удерживалось 10% ацетонитрила и 90% воды (50 мМ ацетат аммония).
В соответствии с вышеуказанной системой соединение 5, соединение 6, соединение 7, соединение 8, соединение 9 и соединение 10 из примера 1 были идентифицированы и проанализированы. Результаты идентификации и анализа показаны на фиг. 10, фиг. 11, фиг. 12, фиг. 13, фиг. 14 и фиг. 15 соответственно.
Меченный радиоактивным изотопом зонд, приготовленный в примере 31, использовали в качестве экспериментального средства ниже, а эксперименты по определению свойств зонда описаны следующим образом.
2. Эксперимент по поглощению комплекса, меченного с помощью 177Lu, в модели с подкожной трансплантацией опухоли у мышей c раком предстательной железы.
Соединение из примера 31 или другой предшествующий радиоактивный зонд, целенаправленно воздействующий на PSMA, вводили мышам с раком предстательной железы на модели с подкожной трансплантацией опухоли и сравнивали результаты поглощения в опухолях и результаты распределения в тканях. Конкретный план заключается в следующем.
Мышей с раком предстательной железы на модели с подкожной трансплантацией опухоли (22RV1) случайным образом разделяли на 3 группы, а именно экспериментальную группу, контрольную группу А и контрольную группу В, по 3 мыши в каждой группе.
В соответствии со способом из примера 31 получали комплекс 177Lu с чистотой более 95%. Комплекс получали путем мечения соединения 10 из примера 1 с помощью 177Lu, который использовался в качестве препарата B для экспериментальной группы в эксперименте.
177Lu-PSMA 617 с чистотой более 95% получали согласно известному способу, который использовался в качестве препарата А для контрольной группы А в эксперименте.
177Lu-EB-PSMA 617 с чистотой более 95% получали согласно способу по примеру 8 из WO 2019/165200, который использовался в качестве препарата C для контрольной группы B в эксперименте.
5 МБк препарата B, препарата A и препарата C вводили внутривенно в хвосты мышей экспериментальной группы, контрольной группы A и контрольной группы B соответственно. После завершения инъекции в течение 24 часов мышей в каждой группе умерщвляли и препарировали для получения опухолевых тканей, крови или других тканей. Полученные ткани взвешивали и измеряли с помощью γ-счетчика для получения результатов радиоактивности образцов в экспериментальной группе, контрольной группе A и контрольной группе B. Измеренные данные вычитались из фона, время распада корректировалось, а затем были получены средние значения. Данные выражали как процент введенной дозы на грамм ткани (% ID/г) от поглощения дозы в тканях на грамм введенной дозы. Результаты показаны на фиг. 1, фиг. 3 и фиг. 4.
Из фиг. 1 можно видеть, что поглощение в опухолях комплекса 177Lu (B) из примера 31 по настоящему изобретению составляет 23,46±0,63% ID/г через 24 часа после инъекции, что намного выше, чем поглощение в опухолях 177Lu-PSMA 617 (A), введенного в контрольной группе A (7,60±1,22% ID/г), и в то же время ниже, чем поглощение в опухолях 177Lu-EB-PSMA 617 (C), введенного в контрольной группе B (48,97±7,77% ID/г).
На фиг. 3 и 4 соответственно показано распределение в основных тканях через 24 ч после инъекции комплекса 177Lu (В) из примера 31 по настоящему изобретению в экспериментальной группе и инъекции 177Lu-EB-PSMA 617 (С) в контрольной группе B. Можно заметить, что поглощение комплекса 177Lu из примера 31 по настоящему изобретению (фиг. 3) в почках через 24 ч после инъекции намного ниже, чем в группе 177Lu-EB-PSMA-617 (фиг. 4).
3. Эксперимент с использованием комплекса, меченного с помощью 177Lu, в случае здоровых мышей
Здоровых мышей случайным образом разделяли на экспериментальную группу I, экспериментальную группу II, контрольную группу А и контрольную группу В по 3 мыши в каждой группе.
В соответствии со способом из примера 31 получали комплекс 177Lu с чистотой более 95%. Комплекс получали путем мечения соединения 10 из примера 1 с помощью 177Lu, который использовался в качестве препарата B для экспериментальной группы I в эксперименте.
Что касается способа из примера 31, меченное с помощью 177Lu соединение (II-2) получали путем замены соединения 10 на соединение (II-2) из примера 2, которое использовали в качестве препарата D для экспериментальной группы II в эксперименте.
177Lu-PSMA 617 с чистотой более 95% получали согласно известному способу, который использовался в качестве препарата А для контрольной группы А в эксперименте.
177Lu-EB-PSMA 617 с чистотой более 95% получали согласно способу по примеру 8 из WO 2019/165200, который использовался в качестве препарата C для контрольной группы B в эксперименте.
5 МБк препарата B, препарата D, препарата A и препарата C вводили внутривенно в хвосты мышей первой экспериментальной группы, второй экспериментальной группы, контрольной группы A и контрольной группы B соответственно. Поглощение в крови измеряли через 1 ч, 4 ч и 24 ч после завершения инъекции. Результаты показаны на фиг. 2 и фиг. 7. Визуализацию SPECT-CT выполняли через 1 ч, 4 ч, 24 ч и 48 ч часов после завершения инъекции. Результаты показаны на фиг. 5 и фиг. 6.
Из фиг. 2 видно, что во всех тестируемых временных точках (1 ч, 4 ч и 24 ч) поглощение в крови комплекса 177Lu (В) из примера 31 по настоящему изобретению было выше, чем таковое в группе 177Lu-PSMA 617 (А), но намного ниже, чем таковое в группе 177Lu-EB-PSMA 617 (С). Из сравнения фиг. 7 и фиг. 2 видно, что во всех тестируемых временных точках (1 ч, 4 ч и 24 ч) поглощение в крови меченного с помощью 177Lu соединения (II-2) намного ниже, чем таковое в группе 177Lu-EB-PSMA 617 (C).
На фиг. 5 и фиг. 6 представлены диаграммы, демонстрирующие визуализацию SPECT-CT для здоровых мышей, которым вводили комплекс 177Lu и меченное с помощью 177Lu соединение (II-2) из примера 31 соответственно.
Таким образом, по сравнению с существующими зондами, целенаправленно воздействующими на PSMA, соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, представленное в настоящем изобретении, характеризуется высокой степенью поглощения в опухолях и, что более важно, характеризуется соответствующим временем нахождения в кровотоке, таким образом, когда меченное радионуклидом соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, по настоящему изобретению используется в терапии рака предстательной железы не только могут быть удовлетворены терапевтические потребности в поглощении в крови и в опухолях, но также значительно снижаются риски гематотоксичности и миелосупрессии. Соединение имеет большую ценность для клинического применения и популяризации и, как ожидается, будет применяться для нуклидной терапии и визуализации рака предстательной железы.
Хотя настоящее изобретение было подробно описано с помощью общих описаний, конкретных вариантов осуществления и испытаний, приведенных выше, специалистам в данной области очевидно, что на основании настоящего изобретения могут быть сделаны некоторые модификации или улучшения. Следовательно, все модификации или улучшения, внесенные без отклонения от сущности настоящего изобретения, попадают в объем защиты настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СВЯЗЫВАЮЩИЕ ОПУХОЛЕВЫЙ АНТИГЕН АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2831681C2 |
| КОМПЛЕКС, СОДЕРЖАЩИЙ НАЦЕЛИВАЮЩЕЕСЯ НА PSMA СОЕДИНЕНИЕ, СВЯЗАННОЕ С РАДИОНУКЛИДОМ СВИНЦА ИЛИ ТОРИЯ | 2018 |
|
RU2795398C2 |
| НОВЫЕ ПСА-СВЯЗЫВАЮЩИЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2787105C2 |
| ПСМА-ТАРГЕТНОЕ СОЕДИНЕНИЕ И ЕГО КОМПЛЕКС С РАДИОНУКЛИДАМИ ДЛЯ ТЕРАНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА | 2022 |
|
RU2803734C1 |
| Лиофилизат на основе лигандов к простат-специфическому мембранному антигену (ПСМА) для приготовления радиофармацевтической композиции в форме раствора для инъекций для лечения рака предстательной железы, радиофармацевтическая композиция на ее основе для лечения рака предстательной железы и способ приготовления радиофармацевтической композиции | 2023 |
|
RU2817970C1 |
| КОМПЛЕКС ПРОИЗВОДНОГО МОЧЕВИНЫ С РАДИОНУКЛИДНОЙ МЕТКОЙ Tс ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПРОСТАТСПЕЦИФИЧЕСКИЙ МЕМБРАННЫЙ АНТИГЕН, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2023 |
|
RU2825402C1 |
| ИНГИБИТОР БЕЛКА АКТИВАЦИИ ФИБРОБЛАСТОВ, МОДИФИЦИРОВАННЫЙ УСЕЧЕННЫМ СИНИМ ЭВАНСА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2021 |
|
RU2841304C2 |
| СОЕДИНЕНИЕ ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА, И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2019 |
|
RU2730507C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСА ПСМА-ТАРГЕТНОГО СОЕДИНЕНИЯ НА ОСНОВЕ МОЧЕВИНЫ Lu-PS-161 И КОМПЛЕКС | 2023 |
|
RU2808636C1 |
| СОЕДИНЕНИЕ ДВОЙНОГО НАЦЕЛИВАНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2022 |
|
RU2838179C2 |
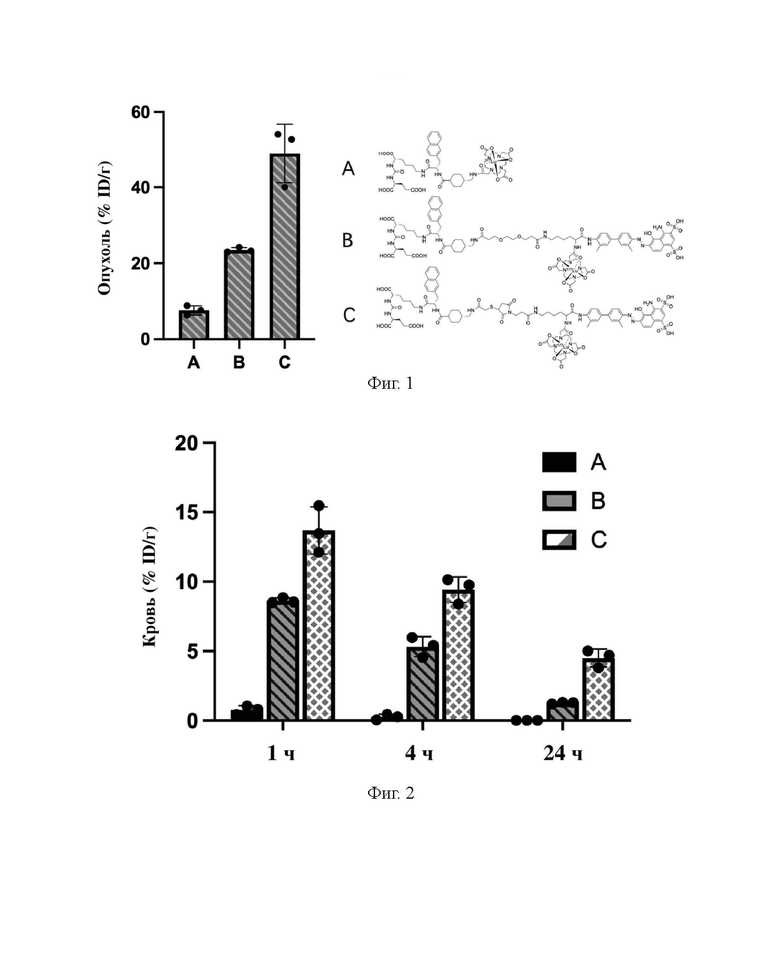
В настоящем изобретении предложено соединение, целенаправленно воздействующее на простатспецифический мембранный антиген (PSMA), или его фармацевтическая соль. Указанное соединение имеет структуру, представленную формулой (II-1). В настоящем изобретении дополнительно предложены способ получения указанного соединения, меченный радиоактивным изотопом комплекс на основе соединения формулы (II-1) и способ его получения, а также фармацевтическая композиция. Указанное соединение и меченный радиоактивным изотопом комплекс характеризуются соответствующим временем нахождения в кровотоке, сравнительно высоким уровнем поглощения и удержания опухолью и подходят для использования в нуклидной терапии и визуализации опухолей с высокой экспрессией PSMA. 5 н. и 2 з.п. ф-лы, 15 ил., 32 пр. 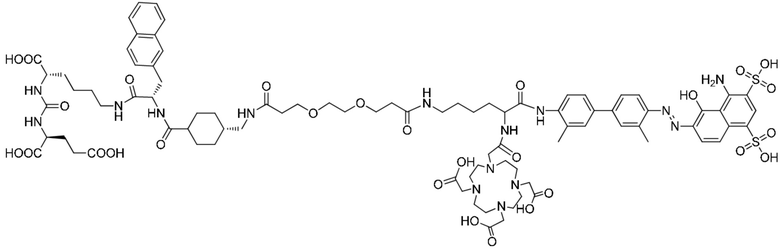 Формула (II-1)
Формула (II-1)
1. Соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, или его фармацевтически приемлемая соль, где молекулярная структура соединения, целенаправленно воздействующего на простатспецифический мембранный антиген, представлена формулой (II-1)
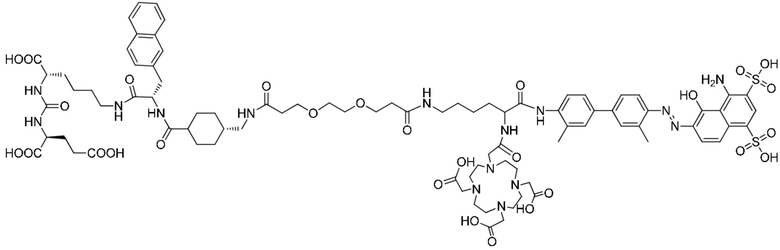
Формула (II-1).
2. Способ получения соединения, целенаправленно воздействующего на простатспецифический мембранный антиген, включающий следующие стадии:
введение защитной Boc-группы на один конец 4,4'-диамино-3,3'-диметилбифенила с последующим обеспечением реакции с 4,6-диамино-5-гидрокси-1,3-нафталиндисульфоновой кислотой с получением усеченного производного синего Эванса, представленного формулой:
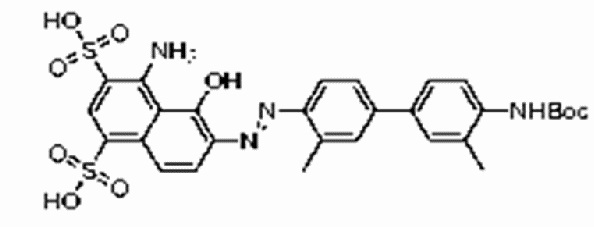 ;
;
удаление защитной Boc-группы с обеспечением последующей реакции амидной конденсации с Nα-Fmoc-Nε-Boc-L-лизином; затем удаление защитной Boc-группы под воздействием TFA с обеспечением последующей реакции амидной конденсации с COOH-PEG2-COOH и реакции с PSMA-617 в присутствии EDC и NHS; затем удаление защитной группы Fmoc с помощью пиперидина и, наконец, осуществление реакции с DOTA-NHS с получением соединения, имеющего следующую структуру, представленную формулой (II-1):
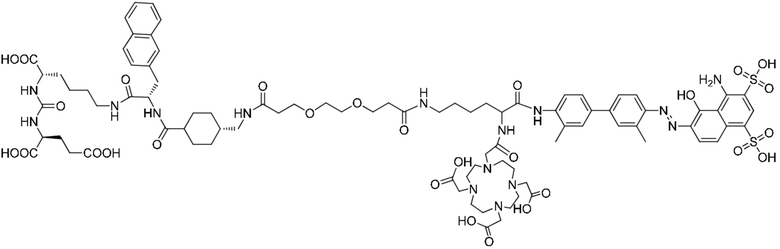
Формула (II-1).
3. Меченное радиоактивным изотопом соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, где соединение представляет собой комплекс, полученный посредством использования соединения, представленного формулой (I), по п. 1 в качестве лиганда и мечения лиганда радионуклидом,
при этом радионуклид представляет собой любой из 177Lu, 90Y, 18F, 64Cu, 68Ga, 62Cu, 67Cu, 86Y, 89Zr, 89Sr, 153Sm, 149Tb, 161Tb, 186Re, 188Re, 212Pb, 213Bi, 223Ra, 225Ac, 131I, 211At или 111In.
4. Меченное радиоактивным изотопом соединение по п. 3, где радионуклид представляет собой 68Ga, 177Lu или 90Y.
5. Способ получения меченного радиоактивным изотопом соединения, целенаправленно воздействующего на простатспецифический мембранный антиген, включающий следующие стадии: растворение соединения, представленного формулой (II-1), по п. 1 в буферном растворе или деионизированной воде и добавление раствора радионуклида к полученному раствору для осуществления реакции в закрытых условиях в течение 5-40 мин с получением меченного радионуклидом комплекса.
6. Фармацевтическая композиция для терапии рака предстательной железы, содержащая меченное радиоактивным изотопом соединение, целенаправленно воздействующее на простатспецифический мембранный антиген, по п. 3 или п. 4 и фармацевтически приемлемый носитель.
7. Композиция по п. 6, где фармацевтически приемлемый носитель выбран из адгезива, буфера, красителя, разбавителя, разрыхлителя, эмульгатора, ароматизатора, вещества, способствующего скольжению, смазывающего вещества, консерванта, стабилизатора, поверхностно-активного вещества, вспомогательного вещества для таблетирования, смачивающего средства или их комбинации.
| WO 2019165200 A1, 29.08.2019 | |||
| TW 202005669 A, 01.02.2020 | |||
| WO 2017196806 A1, 16.11.2017 | |||
| WO 2017192874 A1, 09.11.2017 | |||
| EA 201991812 A1, 23.01.2020. |

