Область техники, к которой относится изобретение
Настоящее изобретение относится к области биофармацевтической технологии и, в частности, относится к конъюгату амантина с антителом.
Предпосылки создания изобретения
Аманитин представляет собой бициклический пептид, состоящий из 8 аминокислот, который является одним из нескольких пептидных аманитатоксинов, выделенных из высокотоксичных грибов; существует девять природных амантинов, которые были выделены и очищены, которые, соответственно, представляют собой α-амантин, β-амантин, γ-амантин, ε-амантин, аманин, аманинамид, амануллин, амануллиновую кислоту и проамануллин, при этом α-амантин и β-амантин являются основными токсинами, ответственными за смертельные случаи. Амантины представляют собой класс медленно действующих токсинов, которые ингибируют транскрипцию эукариотической РНК-полимеразы II и РНК-полимеразы III, приводя к потере белка и клеточной гибели. Этот класс токсинов обладает чрезвычайно высоким ингибиторным эффектом на РНК-полимеразу II, с KD до 3 нМ, и они периодически абсорбируются в организме в результате энтерогепатической циркуляции в желудочно-кишечном тракте, вызывая устойчивое серьезное повреждение органов в организме человека, таких как печень, почки, сердце и легкие.
Когда амантин связывают с большим биомолекулярным носителем (таким как молекула антитела), его токсичность существенно снижается, и даже амантин становится относительно инертным, и только после удаления биомолекулярного носителя в специфических физиологических условиях амантин будет проявлять цитотоксичность.
В последние годы ряд исследовательских институтов и компаний в разных странах мира структурно модифицировали молекулы амантина с получением фармацевтически ценных конъюгатов антитело-токсин, в том числе: компания Heidelberg Pharma AG (Germany), которая осуществила связывание 1-положения, 3-δ- положения или 6'-фенольной гидроксильной группы природного амантина с моноклональным антителом с использованием линкера; компания Agensys Co. (US), которая структурно модифицировала фенил на индольном кольце α-амантина для его связывания с антителом из положения 6'-фенольного гидроксила или 7'-положения с использованием линкера; компания Hangzhou Duoxi Biotechnology Co., Ltd., которая осуществила нитрование 5'- и 7'-положений амантинового производного для его связывания с антителом с использованием линкера с амидной структурой; компания Leg Chem Biosciences Co. (Korea), которая также исследовала способы для связывания с антителом с использованием линкера из положения 6'-фенольной гидроксильной группы, а также 3-δ-положения. Из вышеперечисленных способов синтеза способ, используемый для присоединения линкера в положении 6'-фенольной гидроксильной группы, требует многостадийной защиты и удаления защиты гидроксильных групп, расположенных на других участках молекулы амантина, и соответствующий путь синтеза усложняется; кроме того, неопределенное позиционирование в реакции замещения, осуществляемой по 5'- и 7'- положениям бензольного кольца, быстро приводит к трудностям при выделении продукта и низкому выходу.
Сущность изобретения
На основании описанного выше предшествующего уровня техники, целью настоящего изобретения является предоставление конъюгата бициклического октапептидного амантинового производного и биомакромолекулы, который является стабильным в кровотоке и расщепляется после эндоцитоза клетками-мишенями, высвобождая при этом амантиновое производное, которое действует как ингибитор РНК-полимеразы, оказывая сильное токсическое действие на клетки посредством специфического ингибирования синтеза мРНК у эукариот.
Для достижения указанной выше цели настоящее изобретение использует следующее техническое решение: Конъюгат токсина, представленный структурной формулой (I), или его фармацевтически приемлемая соль:
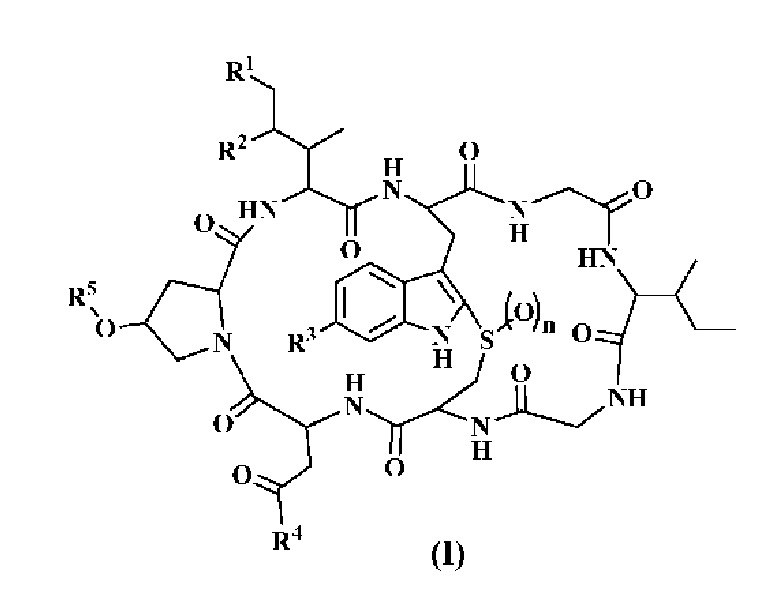
Где:
n=0, 1 или 2,
R1 представляет собой -H или -OH,
R2 представляет собой -H или -OH,
R3 представляет собой -H, -OH или C1-6 алкил,
R4 представляет собой -NH2 или -OH,
R5 представляет собой -L-A,
A представляет собой часть, являющуюся биологической макромолекулой, которая связывается с мишенью, и
L представляет собой любую химическую структуру, связывающую амантиновое производное и биологическую макромолекулу.
Предпочтительно, химическая структура L включает расщепляемую или нерасщепляемую структуру.
Предпочтительно, A включает антитело или его антиген-связывающий фрагмент или антиген-связывающий полипептид.
Предпочтительно, место конъюгации L и связывающейся с мишенью биомолекулы A включает следующую структуру:
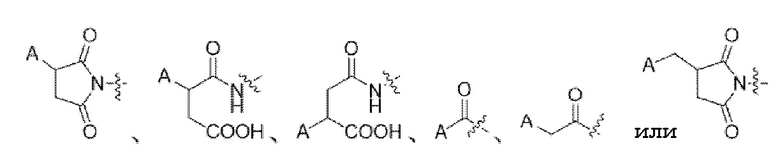 .
.
Предпочтительно, L связан с токсином через сложный эфир или простой эфир.
Настоящее изобретение также включает фармацевтическую композицию, содержащую вышеуказанный конъюгат токсина или его фармацевтически приемлемую соль в качестве активного ингредиента.
Настоящее изобретение также включает применение вышеуказанного конъюгата токсина или его фармацевтически приемлемой соли для получения противоопухолевого лекарственного средства или противоракового лекарственного средства.
Кроме того, указанное противоопухолевое лекарственное средство или противораковое лекарственное средство представляет собой лекарственное средство от рака легкого, лекарственное средство от рака почек, лекарственное средство от рака мочевыводящих путей, лекарственное средство от колоректального рака, лекарственное средство от рака предстательной железы, лекарственное средство от глиобластомы, лекарственное средство от рака яичников, лекарственное средство от рака поджелудочной железы, лекарственное средство от рака молочной железы, лекарственное средство от меланомы, лекарственное средство от рака печени, лекарственное средство от рака мочевого пузыря, лекарственное средство от злокачественной лимфомы, лекарственное средство от лейкоза, лекарственное средство от рака желудка или лекарственное средство от рака пищевода.
Определения: В соответствии со стандартной практикой в данной области техники, символ  , используемый в формулах и таблицах, относящихся к настоящему изобретению, представляет собой связь в ядре или связывание по ядру фрагмента или заместителя со структурой соединения.
, используемый в формулах и таблицах, относящихся к настоящему изобретению, представляет собой связь в ядре или связывание по ядру фрагмента или заместителя со структурой соединения.
В соответствии со стандартной практикой в данной области техники, в контексте настоящего изобретения гетеро (атом, алкил, арил, циклическая группа) относится к соответствующей химической структуре, содержащей атом, отличный от углерода.
Преимущества настоящего изобретения заключаются в следующем:
Авторы настоящего изобретения конъюгировали молекулу антитела с гидроксильной группой 2-пролина амантина или его производной молекулы, используя фармацевтически приемлемую связывающую структуру, и тем самым смогли получить конъюгат, который является стабильным и нетоксичным в плазме, но может расщепляться с высвобождением токсичных молекул в больных клетках, и были приятно удивлены, обнаружив, что указанный способ связывания прост и эффективен и в значительной степени удовлетворяет потребности в массовом производстве таких продуктов в промышленных масштабах.
Настоящее изобретение раскрывает бициклическое октапептидное производное, которое конъюгировано с соответствующей связывающейся с мишенью группой через специфическую химическую структуру, которое стабильно в плазме и расщепляется с высвобождением активного лекарственного средства в определенной биологической среде, чтобы максимизировать летальность для клеток-мишеней и минимизировать токсичные побочные эффекты на клетки, не являющиеся мишенями, и которое можно использовать при лечении различных злокачественных опухолей.
Описание чертежей
Фиг. 1 показывает экспериментальные результаты, полученные из клеточной линии BT474 в Примере 13.
Фиг. 2 показывает экспериментальные результаты, полученные из клеточной линии SKBR3 в Примере 13.
Фиг. 3 показывает экспериментальные результаты, полученные из клеточной линии N87 в Примере 13.
Конкретные варианты осуществления
Настоящее изобретение описано более подробно ниже.
Пример 1: Синтез низкомолекулярной нагрузки ama-1
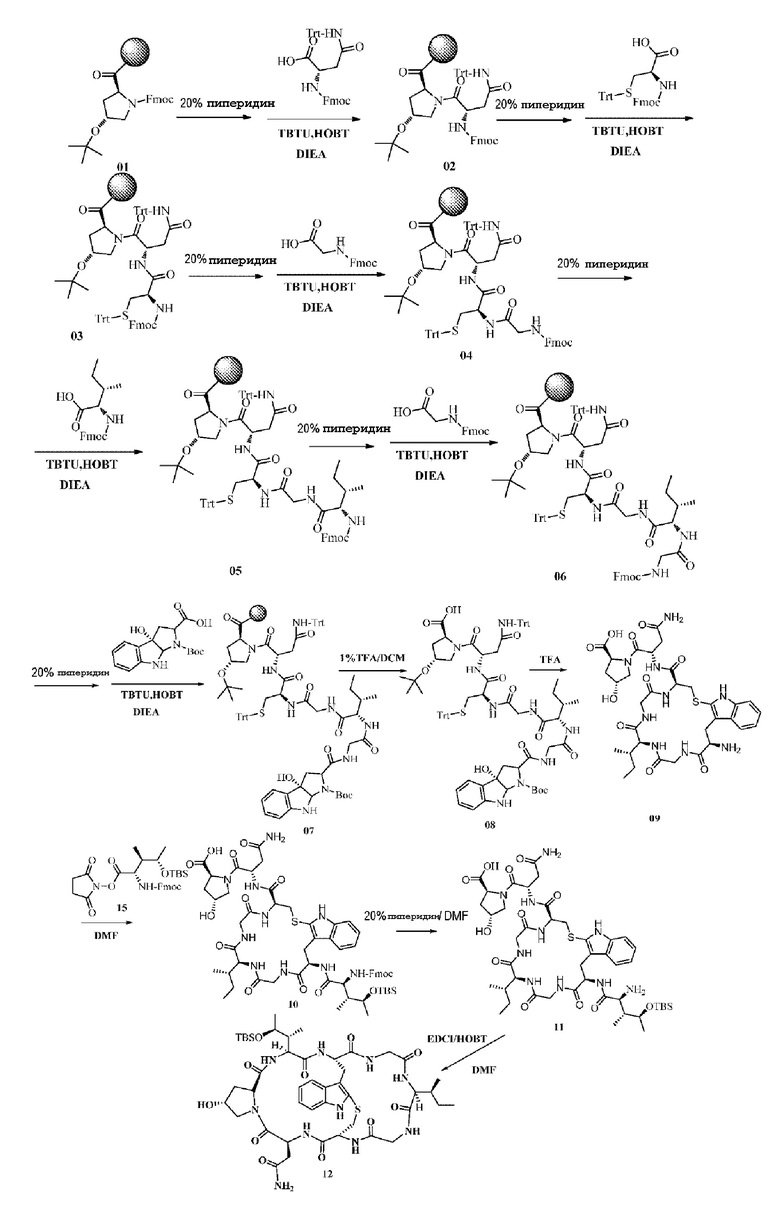
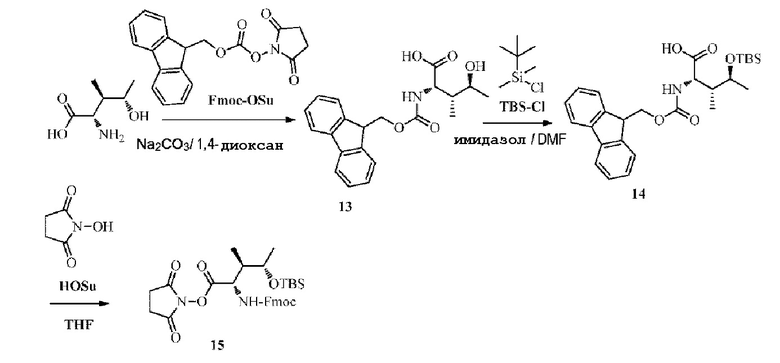
Твердофазный синтез промежуточного соединения 08:
Используя предварительно нагруженную N-флуоренилметоксикарбонил-O-трет-бутил-L-гидроксипролином смолу в качестве исходного вещества, защитную группу Fmoc удаляли с использованием 20% раствора пиперидина (20 мл 20% раствора пиперидина в 1 г смолы), затем добавляли DMF в виде раствора (20 мл/г), с последующим последовательным добавлением Fmoc-N-пропинил-L-аспарагина (Fmoc-Asn(Trt)-OH) (3 экв.), TBTU (2,5 экв.), HOBT (1,8 экв.) и DIPEA (6 экв.); реакции давали протекать в течение 2 ч при комнатной температуре (28°C), затем осуществляли промывку 3 раза при помощи DMF (20 мл DMF на 1 г смолы каждый раз), после чего осуществляли присоединение последующих аминокислот в соответствии с предыдущей процедурой; после завершения конечного связывания использовали 1% раствор TFA (по 20 мл на 1 г смолы каждый раз; 1% TFA 5 мин; повторяли три раза) для отщепления от смолы и этот раствор удаляли центрифугированием, после чего осуществляли перемешивание с метил-трет-бутиловым эфиром для индукции выделения кристаллов, с получением соединения 08 с выходом приблизительно 54% и чистотой 76%. MS: [M+H]+ 1417,6123.
Синтез соединения 09:
10 г соединения 08 растворяли с использованием TFA (10 мл/г), затем осуществляли перемешивание при комнатной температуре в течение пяти часов с последующим удалением TFA при пониженном давлении при 50°C, очищали препаративной ВЭЖХ с получением приблизительно 4,3 г чистого продукта с выходом 43% и чистотой 95,6%. MS: [M+H]+ 760,2144.
Синтез соединения 13:
2,94 г (4S)-гидроксиизолейцина, 40 мл 1,4-диоксана и 40 мл насыщенного раствора карбоната натрия добавляли в 250-мл одногорлую колбу и тщательно перемешивали, затем партиями добавляли Fmoc-OSu; через 10 минут реакцию продолжали при комнатной температуре в течение 12 часов при перемешивании до завершения взаимодействия исходного вещества; затем в реакционный раствор добавляли 50 мл воды и pH доводили до приблизительно 4 с использованием 5% раствора лимонной кислоты; осуществляли экстрагирование этилацетатом три раза (по 50 мл каждый раз) и органический слой собирали и промывали один раз с использованием 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и концентрировали с получением бледно-желтого маслянистого вещества. Никакой очистки не требовалось, и полученное вещество подавали непосредственно на следующую стадию. Выход >100%.
Синтез соединения 14:
После растворения неочищенного продукта, образованного Соединением 13, в 40 мл DMF добавляли 2,68 г имидазола (2 экв.) с последующим добавлением партиями TBS-Cl; после того, как добавление было завершено, осуществляли перемешивание при комнатной температуре в течение 12 часов до тех пор, пока исходные вещества полностью не прореагировали, затем добавляли 50 мл воды и 50 мл этилацетата и осуществляли перемешивание; органический слой затем отделяли с последующим экстрагированием водного слоя два раза этилацетатом (по 50 мл каждый раз) и органический слой собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого маслянистого вещества, которое подвергали колоночной хроматографии на силикагеле (элюент: PE:EA=5:1), с получением таким образом 4,5 г маслянистого вещества. Соответствующий выход от двух стадий составил приблизительно 46,6%.
Синтез соединения 15:
Соединение 14, HOSu (1,23 г, 1,15 экв.), DCC (2,23 г, 1,15 экв.) и 50 мл THF добавляли в 250-мл одногорлую колбу и осуществляли перемешивание при комнатной температуре в течение шести часов в атмосфере азота, затем добавляли 50 мл воды и 50 мл этилацетата и смесь перемешивали в течение 10 минут, с последующим отделением органического слоя и экстрагированием водного слоя два раза этилацетатом с использованием каждый раз по 50 мл, органический слой затем объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого маслянистого вещества, которое затем очищали препаративной ВЭЖХ, с получением приблизительно 3,24 г белого пенистого твердого вещества с выходом 60%. 1H-ЯМР(400МГц, DMSO-d6): 0,08(с, 6H), 0,86(с, 9H), 0,98(д, 3H, J=8,0Гц),1,06(д, 3H, J=5,6), 1,95(т, J=10,8), 2,83(с, 4H), 4,21(дд, 1H, J=16,8Гц, 8,0Гц), 4,34(дд, 1H, J=12Гц, 4Гц), 4,67-4,73(м, 1H), 7,31(д, 2H, J=8,0Гц), 7,34-7,46(м, 2H), 7,70-7,76(м, 2H), 7,89(т, 2H, J=12,0), 8,24(д, 1H, J=8,8Гц); MS:[M+H]+581,34。
Синтез соединения 10
После растворения 180 мг соединения 09 с использованием 1,5 мл DMF добавляли Соединение 15 (825 мг, 6 экв.) и DIPEA (0,25 мл, 6 экв.) в атмосфере азота и реакции давали осуществиться при комнатной температуре в течение пяти часов, отслеживая реакцию методом ВЭЖХ. Полученный продукт использовали непосредственно на следующей стадии без необходимости в последующей очистке.
Синтез соединения 11
0,3 мл пиперидина добавляли к указанному выше реакционному раствору и реакцию продолжали при комнатной температуре при перемешивании в течение двух часов, затем реакцию останавливали, смесь очищали с использованием препаративной ВЭЖХ(нейтральные условия, ацетонитрил/чистая водная система) и фракции, соответствующие пику целевого продукта, собирали с последующим удалением ацетонитрила при пониженном давлении, затем осуществляли лиофилизацию с получением 136 мг белого порошкообразного твердого вещества с соответствующим выходом от двух стадий приблизительно 52%; MS: [M+H]+ 1003,5654.
Синтез соединения 12
После растворения 136 мг соединения 11 безводным DMF добавляли EDCI (130 мг, 5 экв.), HOBT (367 мг, 20 экв.) и DIPEA (0,12 мл, 5 экв.) и осуществляли перемешивание при комнатной температуре в течение четырех часов, затем использовали ВЭЖХ для подтверждения завершения реакции, затем осуществляли препаративную ВЭЖХ (нейтральные условия, ацетонитрил/чистая водная система) и фракции, соответствующие пику целевого продукта, собирали с последующим удалением ацетонитрила при пониженном давлении, осуществляли лиофилизацию с получением 60 мг белого порошкообразного твердого вещества с выходом приблизительно 45%; MS: [M+H]+ 985,5421.
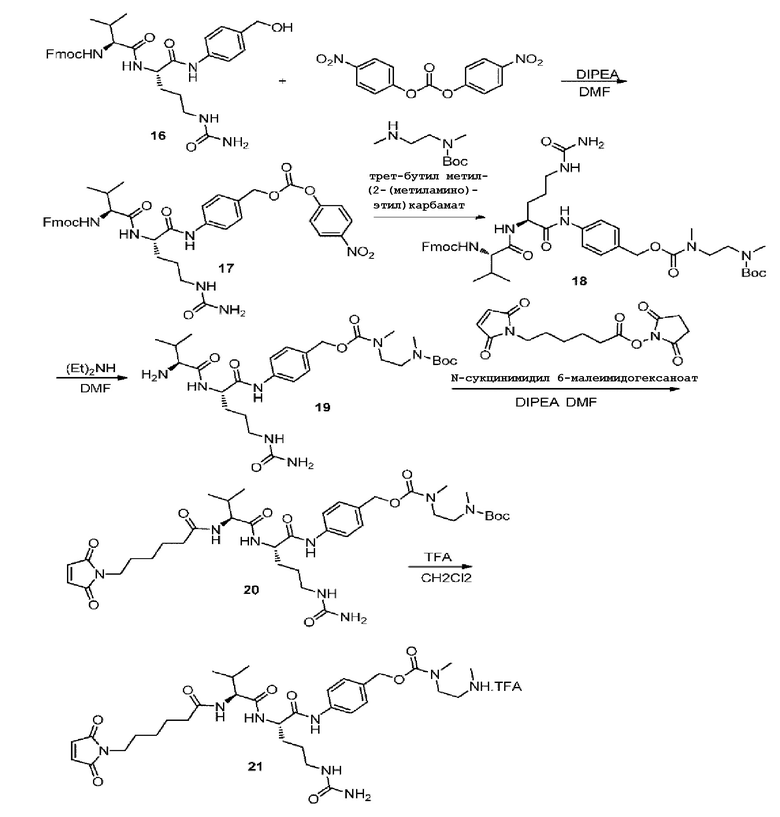
Синтез соединения 17
30 г соединения 16 (49,92 ммоль, 1,0 экв.) добавляли в 500-мл круглодонную колбу и растворяли при перемешивании с 240 мл DMF, затем добавляли 22,76 г бис(п-нитрофенил)карбонат (74,87 ммоль, 1,5 экв.) и добавляли по каплям 9,68 г DIPEA (74,87 ммоль, 1,5 экв.); затем осуществляли перемешивание при комнатной температуре и реакции давали осуществиться в течение 1,5 часов, при этом использовали ТСХ для подтверждения, что исходные вещества полностью прореагировали, и затем реакцию останавливали. Последующая обработка: 1,5 л изопропилового эфира добавляли в реакционную колбу при интенсивном перемешивании и перемешивание продолжали в течение двух часов перед удалением верхнего жидкого слоя, затем к остатку добавляли 800 мл изопропилового эфира и перемешивание продолжали в течение одного часа с последующим вакуумным фильтрованием, полученную фильтровальную лепешку затем добавляли в 600 мл изопропилового эфира и оставляли выстаиваться в течение ночи при перемешивании, затем осуществляли вакуумное фильтрование с получением 31,2 г коричневато-желтого порошкообразного твердого вещества с выходом 81,6%. MS: [M+H]+ 767,62.
Синтез соединения 18
31,2 г соединения 17 (40,73 ммоль, 1,0 экв.) добавляли в 500-мл одногорлую колбу и растворяли при перемешивании с 150 мл DMF, затем добавляли 8,1 г трет-бутилметил(2-(метиламино)этил)карбамата (36,66 ммоль, 1,1 экв.), растворенного в 50 мл DMF, и реакции давали осуществиться при комнатной температуре при перемешивании в течение 3,5 часов, использовали ТСХ для подтверждения, что Соединение 17 полностью прореагировало, и реакцию останавливали. Последующая обработка: 2 л изопропилового эфира добавляли в реакционный раствор и осуществляли перемешивание для осаждения вязкого маслянистого вещества на внутренней стенке колбы, затем супернатант сливали, добавляли 1 л изопропилового эфира при интенсивном перемешивании; супернатант затем снова сливали, добавляли 500 мл изопропилового эфира и смеси давали выстояться в течение ночи при перемешивании; в завершение, осуществляли вакуумное фильтрование с получением 27,2 г коричневато-желтого порошкообразного твердого вещества с выходом 81,9%. MS: [M+H]+ 816,73.
Синтез соединения 19
20 г соединения 18 (24,53 ммоль) отвешивали в 500-мл круглодонную колбу и в колбу добавляли 100 мл DMF для растворения соединения, затем добавляли по каплям 20 мл диэтиламина на ледяной бане в течение 10 минут, затем осуществляли перемешивание и реакции давали осуществиться в течение двух часов при комнатной температуре, при этом использовали ТСХ для подтверждения, что Соединение 18 полностью прореагировало, и затем реакцию останавливали. Последующая обработка: DMF и диэтиламин удаляли при пониженном давлении и полученный продукт непосредственно подавали на следующую стадию без осуществления очистки.
Синтез соединения 20
В 500-мл круглодонную колбу добавляли 150 мл DMF для растворения неочищенного продукта, представляющего собой Соединение 19, полученное на предыдущей стадии, с последующим добавлением 11,7 г N-сукцинимидил-6-малеимидогексаноата (37,98 ммоль, 1,55 экв.) и затем 8,4 мл DIEA (50,63 ммоль, 2,06 экв.), который добавляли по каплям, и осуществляли перемешивание при комнатной температуре, давая продолжиться реакции в течение двух часов, при этом использовали ТСХ для подтверждения, что исходное вещество, Соединение 19, полностью прореагировало, и затем реакцию останавливали. Последующая обработка: 1,5 л изопропилового эфира добавляли в реакционный раствор и осуществляли перемешивание для осаждения кристаллов, что приводило к осаждению твердого вещества, а также маслянистого вязкого вещества, которое прилипало к стенке колбы, супернатант сливали и добавляли 500 мл метил-трет-бутилового эфира и 500 мл изопропилового эфира для осуществления перемешивания-промывки один раз, с последующим вакуумным фильтрованием, полученную фильтровальную лепешку сушили при пониженном давлении при 45°C в вакуумной сушилке с получением 20 г неочищенного продукта в виде коричневато-желтого порошкообразного твердого вещества. MS: [M+H]+ 787,63.
Синтез соединения 21
0,4 г соединения 20 растворяли в 2 мл 50% раствора TFA в дихлорметане, затем осуществляли перемешивание при комнатной температуре в течение двух часов и использовали ТСХ для подтверждения завершения реакции, затем раствор удаляли при пониженном давлении для последующего использования и продукт непосредственно подавали на следующую стадию без необходимости в очистке.
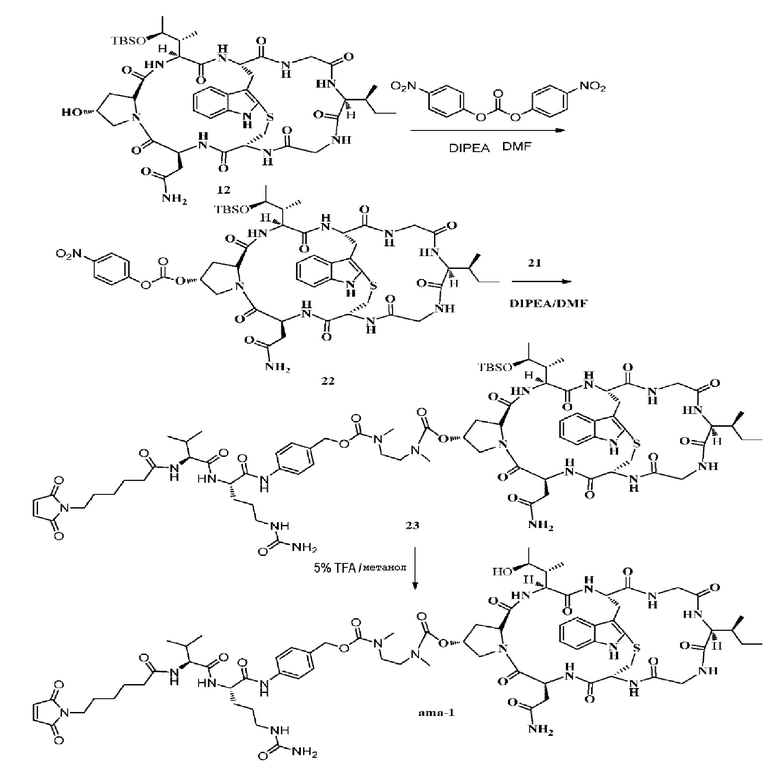
Синтез соединения 22:
100 мг (0,10 ммоль, 1,0 экв.) соединения 12, 304 мг (1,0 ммоль, 10 экв.) бис(п-нитрофенил)карбоната и 3 мл DMF добавляли в 20-мл коричневую колбу и, когда растворение было завершено, добавляли DIPEA (260 мг, 20 экв.) и температуру повышали до 28°C в течение 24 часов в атмосфере азота, при этом использовали ВЭЖХ для отслеживания реакции и подтверждения, что исходное вещество 012 и NPC полностью прореагировали, полученный неочищенный продукт не нуждался в очистке, и его непосредственно подавали в следующую реакцию.
Синтез соединения 23
Неочищенное Соединение 21 добавляли в реакционный раствор соединения 22, затем добавляли подходящее количество DIPEA, поддерживая pH реакционного раствора в пределах от 8 до 9, осуществляли перемешивание при комнатной температуре в течение трех часов в атмосфере азота, когда реакция была завершена, как показал ВЭЖХ анализ, целевой продукт очищали препаративной ВЭЖХ с получением 30 мг светло-желтого твердого вещества. MS: [M+H]+ 1697,9150.
Синтез соединения ama-1
15 мг соединения 23 добавляли в 4-мл коричневую колбу, с последующим добавлением 1 мл 5% TFA/MeOH, чтобы полностью растворить соединение, температуру реакции затем повышали до 28°C в течение одного часа в атмосфере азота и использовали ВЭЖХ (чистая вода) для отслеживания реакции, после того, как исходное вещество 23 полностью прореагировало, растворитель удаляли путем высушивания азотом и использовали ВЭЖХ для получения продукта с концентрацией ацетонитрила приблизительно 35%, который затем лиофилизировали, с получением 11 мг белого твердого вещества с выходом (Y) 42%. MS: [M+H]+ 1583,7532.
Пример 2: Синтез низкомолекулярной нагрузки ama-2
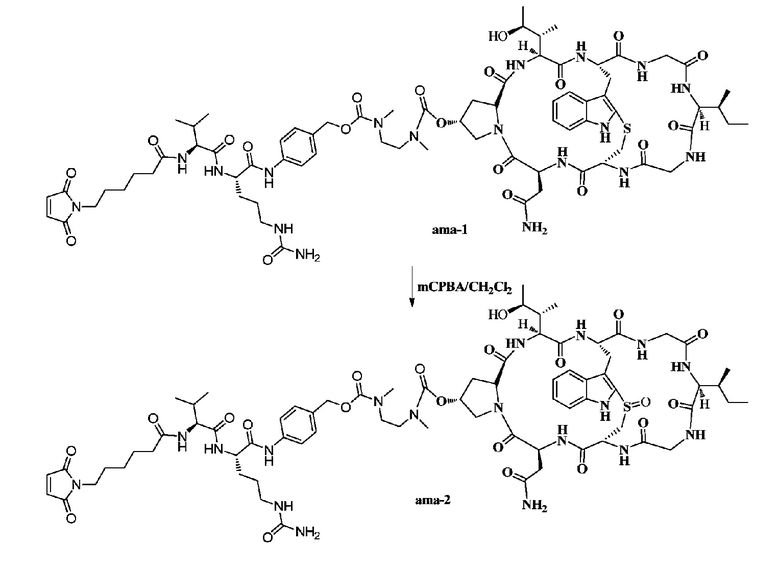
Приблизительно 6 мг ama-1 растворяли с использованием дихлорметана, с последующим добавлением 13 микролитров дихлорметанового раствора 1,0 экв. mCPBA (0,05 г/мл), затем осуществляли перемешивание при комнатной температуре в течение двух часов и использовали ВЭЖХ для подтверждения, что исходные вещества полностью прореагировали, затем осуществляли очистку методом препаративной ВЭЖХ с последующей лиофилизацией с получением приблизительно 3 мг белого порошкообразного твердого вещества. Выход составил 49,5%. MS: [M+H]+ 1599,8450.
Пример 3: Синтез низкомолекулярной нагрузки ama-3
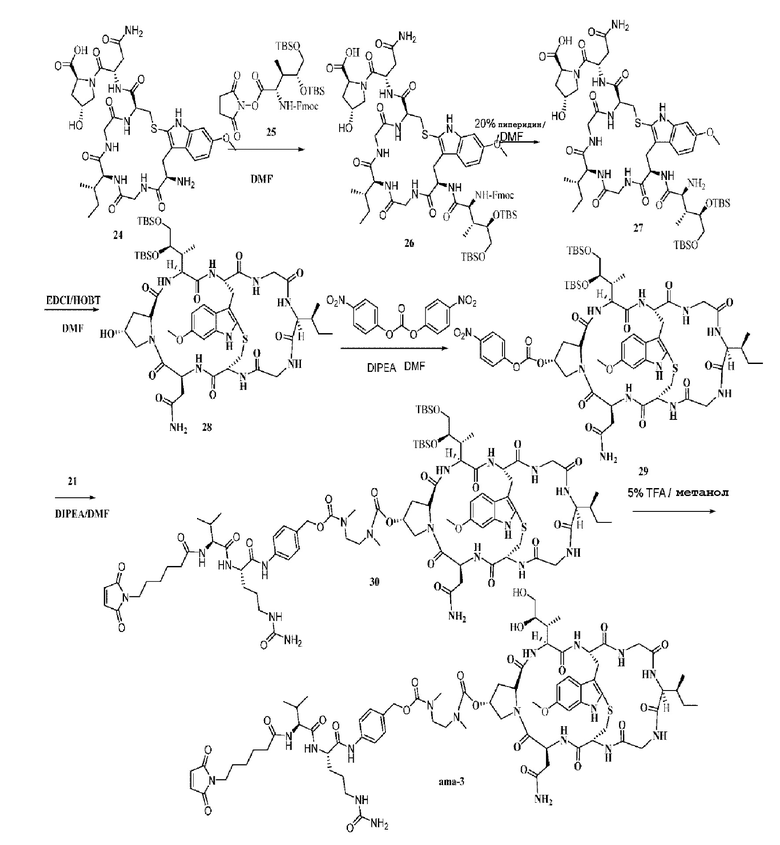
Синтез соединения 24: Ссылаясь на синтез соединения 09, получали приблизительно 1,2 г белого твердого вещества. Выход составил 25,7%. MS: [M+H]+ 790,4254.
Синтез соединения 25: Синтез осуществляли со ссылкой на Liang Zhao, et al. Synthesis of a Cytotoxic Amanitin for Biorthogonal Conjugation. ChemBionchem. 2015, 16, 1420-1425.
Синтез соединения 26: Ссылаясь на способ синтеза соединения 10, добавляли 300 мг соединения 24 и продукт непосредственно использовали на следующей стадии без очистки.
Синтез соединения 27: Ссылаясь на способ синтеза соединения 11, очищали препаративной ВЭЖХ с последующей лиофилизацией с получением 195 мг белого твердого вещества с общим выходом для двух стадий 44,1%. MS: [M+H]+ 1163,6341.
Синтез соединения 28: Ссылаясь на способ синтеза соединения 12, добавляли 150 мг соединения 27 с получением 96 мг целевого соединения в виде белого твердого вещества с выходом 65%. MS: [M+H]+ 1145,6124.
Синтез соединения 29: Ссылаясь на синтез соединения 22, добавляли 80 мг соединения 28 и продукт непосредственно использовали на следующей стадии без очистки.
Синтез соединения 30: Ссылаясь на синтез соединения 23, после получения и очистки было получено приблизительно 59 мг светло-желтого твердого вещества с выходом 45,4%. MS: [M+H]+ 1857,9813.
Синтез соединения ama-3: Ссылаясь на синтез соединения ama-1, добавляли 40 мг соединения 30, и после получения и очистки было получено приблизительно 8,4 мг светло-желтого твердого вещества. Выход составил 24%. MS: [M+H]+ 1629,8021.
Пример 4: Синтез низкомолекулярной нагрузки ama-4
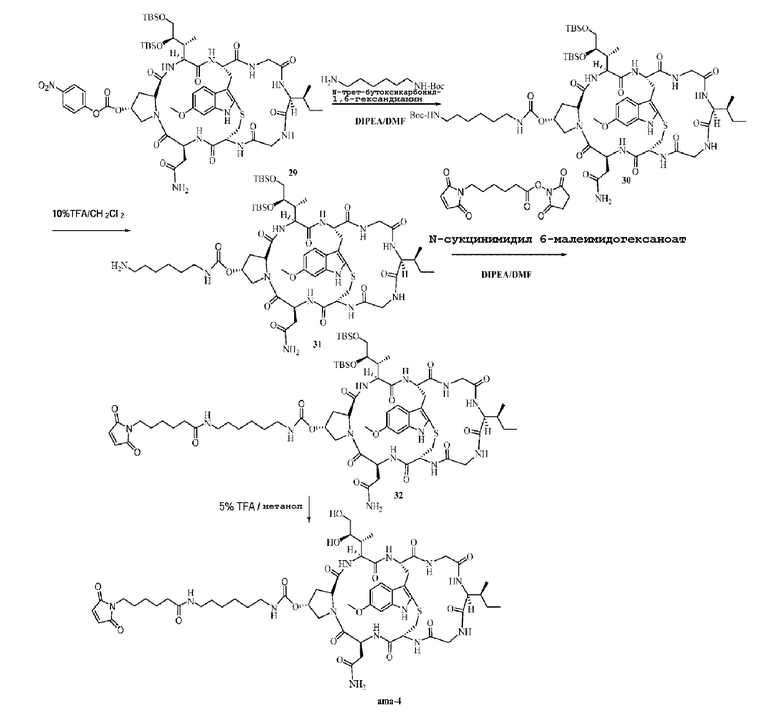
Синтез соединения 30: 49 мг N-трет-бутоксикарбонил-1,6-гександиамина (2 экв.) добавляли к реакционному раствору соединения 29 (при этом Соединение 28 пополняли до 500 мг, что соответствовало добавлению в этом случае 130 мг, без очистки, и рассчитано в соответствии с 100% выходом) и добавляли DIPEA по каплям, при этом pH раствора поддерживали в пределах 8-9, и осуществляли перемешивание при комнатной температуре в атмосфере азота в течение четырех часов, после подтверждения при помощи ВЭЖХ, что Соединение 29 полностью прореагировало, реакционную смесь очищали препаративной ВЭЖХ, фракции, соответствующие целевым пикам, собирали и органический растворитель удаляли центрифугированием, с последующей лиофилизацией с получением приблизительно 83,4 мг белого твердого вещества с выходом 53%. [M+H]+ 1387,7541.
Синтез соединения 31: 80 мг соединения 30 растворяли в 1 мл 20% раствора TFA в дихлорметане и осуществляли перемешивание при комнатной температуре в течение двух часов в атмосфере азота, использовали ВЭЖХ для подтверждения, что Соединение 30 полностью прореагировало, затем органический растворитель удаляли центрифугированием при пониженном давлении и полученный продукт откладывали для последующего использования.
Синтез соединения 32: После растворения неочищенного продукта, соответствующего Соединению 31, при помощи 2 мл DMF pH доводили до 8-9 при помощи DIPEA, затем добавляли 38 мг (2 экв.) N-сукцинимидил-6-малеимидогексаноата и осуществляли перемешивание при комнатной температуре в течение шести часов в атмосфере азота, после подтверждения методом ВЭЖХ, что Соединение 31 полностью прореагировало, реакционную смесь очищали препаративной ВЭЖХ, фракции, соответствующие целевым пикам, собирали и органический растворитель удаляли центрифугированием, с последующей лиофилизацией с получением приблизительно 46 мг белого твердого вещества с выходом 54%; [M+H]+ 1480,7841.
Синтез соединения ama-4: Ссылаясь на синтез соединения ama-3, добавляли 44 мг соединения 32 и осуществляли лиофилизацию с получением 21 мг целевого соединения с выходом 56,4%; [M+H]+ 1152,5431.
Пример 5: Синтез низкомолекулярной нагрузки ama-5
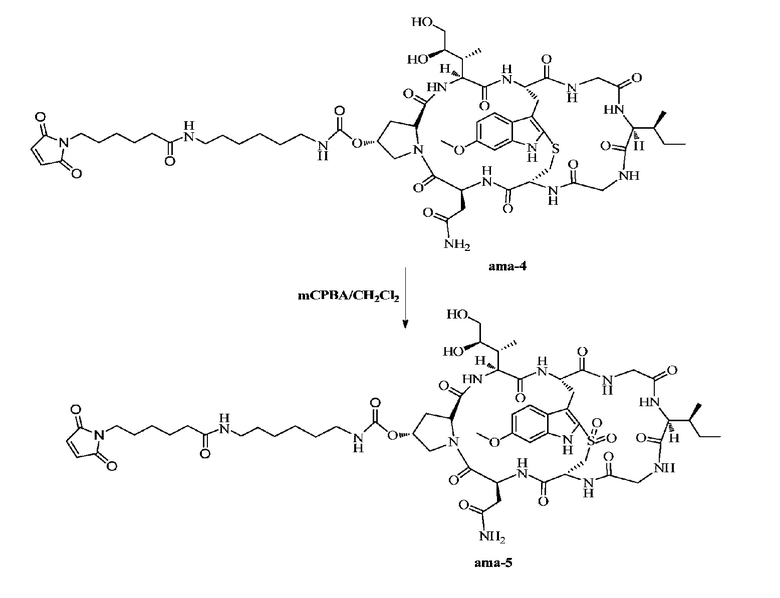
12 мг ama-4 растворяли с использованием дихлорметана с последующим добавлением 165 микролитров дихлорметанового раствора 5,0 экв. mCPBA (0,05 г/мл), затем осуществляли перемешивание при комнатной температуре в течение двух часов и использовали ВЭЖХ для подтверждения, что исходные вещества полностью прореагировали, затем осуществляли очистку при помощи препаративной ВЭЖХ с последующей лиофилизацией с получением приблизительно 4 мг желтого порошкообразного твердого вещества. Выход составил 32,9%. MS: [M+H]+ 1268,5821.
Пример 6: Синтез низкомолекулярной нагрузки ama-6
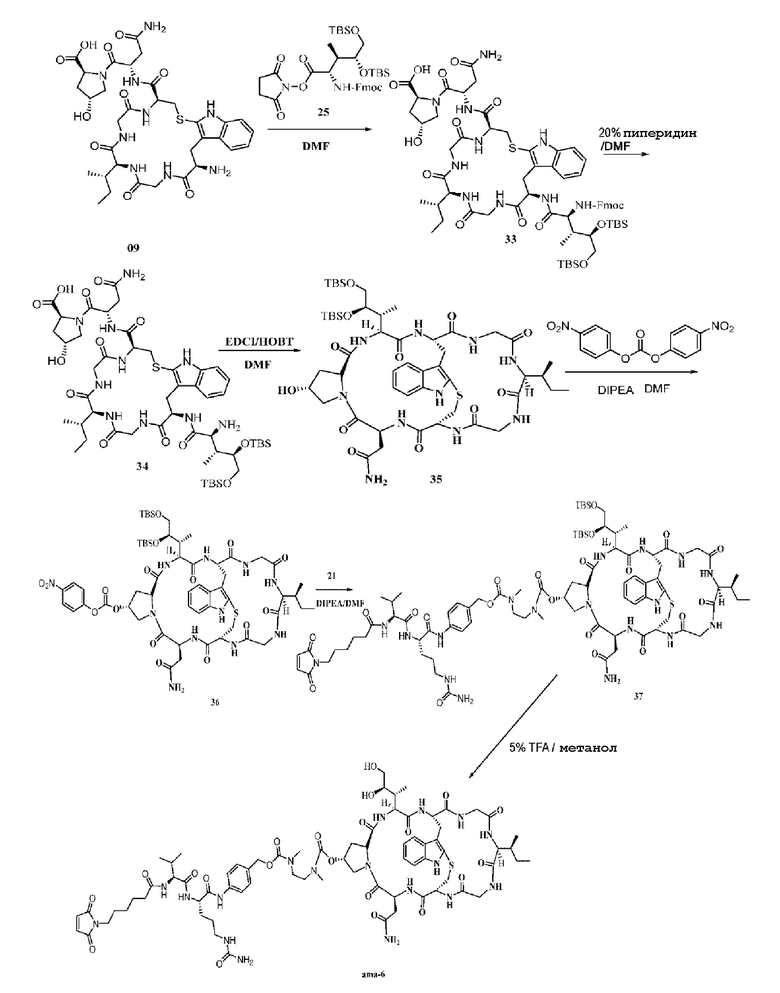
Синтез соединения 33: Ссылаясь на синтез соединения 10, добавляли 300 мг соединения 09 и продукт непосредственно использовали на следующей стадии без очистки.
Синтез соединения 34: Ссылаясь на синтез соединения 11, после очистки при помощи препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 248,9 мг белого твердого вещества с выходом 55,6%; MS: [M+H]+ 1133,6348.
Синтез соединения 35: Ссылаясь на синтез соединения 12, после добавления 220 мг исходного вещества 34 и очистки при помощи препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 137,3 мг белого твердого вещества с выходом 63,4%; MS: [M+H]+ 1115,6147.
Синтез соединения 36: Ссылаясь на синтез соединения 22, добавляли 120 мг исходного вещества 35 и продукт непосредственно использовали на следующей стадии без очистки.
Синтез соединения 37: Ссылаясь на синтез соединения 23, после очистки при помощи препаративной ВЭЖХ органическую фазу удаляли центрифугированием и осуществляли лиофилизацию с получением приблизительно 112,4 мг белого твердого вещества с выходом 57,1%; MS: [M+H]+ 1827,9857.
Синтез соединения ama-6
Синтез осуществляли со ссылкой на синтез соединения ama-1, после добавления 20 мг соединения 37 и очистки при помощи препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 7 мг светло-желтого твердого вещества с выходом 40%. MS: [M+H]+ 1599,8324.
Пример 7: Синтез низкомолекулярной нагрузки ama-7
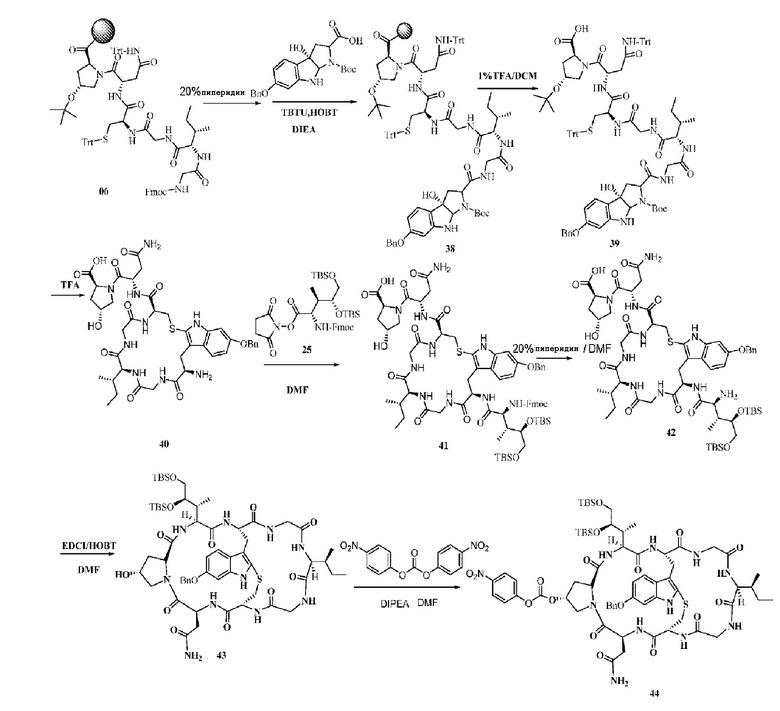
Синтез соединения 38: Ссылаясь на синтез соединения 07, нагрузку рассчитывали до приблизительно 0,32 ммоль/г.
Синтез соединения 39: Ссылаясь на синтез соединения 08, осуществляли сушку центрифугированием с получением коричневого маслянистого вещества, которое непосредственно использовали на следующей стадии без очистки.
Синтез соединения 40: Ссылаясь на синтез соединения 09, после очистки препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 526,4 мг светло-желтого твердого вещества с выходом 34%, рассчитанным исходя из используемого количества соединения 38. MS: [M+H]+ 866,3512.
Синтез соединения 41: Ссылаясь на синтез соединения 10, добавляли 500 мг соединения 40 и фракции, соответствующие целевым пикам, собирали с последующей лиофилизацией с получением 531,6 мг не совсем белого твердого вещества с выходом 63%. MS: [M+H]+ 1461,6624.
Синтез соединения 42: Ссылаясь на синтез соединения 11, добавляли 525 мг соединения 41, осуществляли очистку и фракции, соответствующие целевым пикам, собирали с последующей лиофилизацией с получением 368,5 мг светло-желтого твердого вещества с выходом 82,8%. MS: [M+H]+ 1239,5764.
Синтез соединения 43: Ссылаясь на синтез соединения 12, добавляли 350 мг соединения 42, фракции, соответствующие целевым пикам, собирали и осуществляли лиофилизацию с получением 223,4 мг светло-желтого твердого вещества с выходом 64,7%. MS: [M+H]+ 1221,5748.
Синтез соединения 44: Ссылаясь на синтез соединения 22, добавляли 220 мг соединения 43 и продукт непосредственно использовали на следующей стадии без очистки.
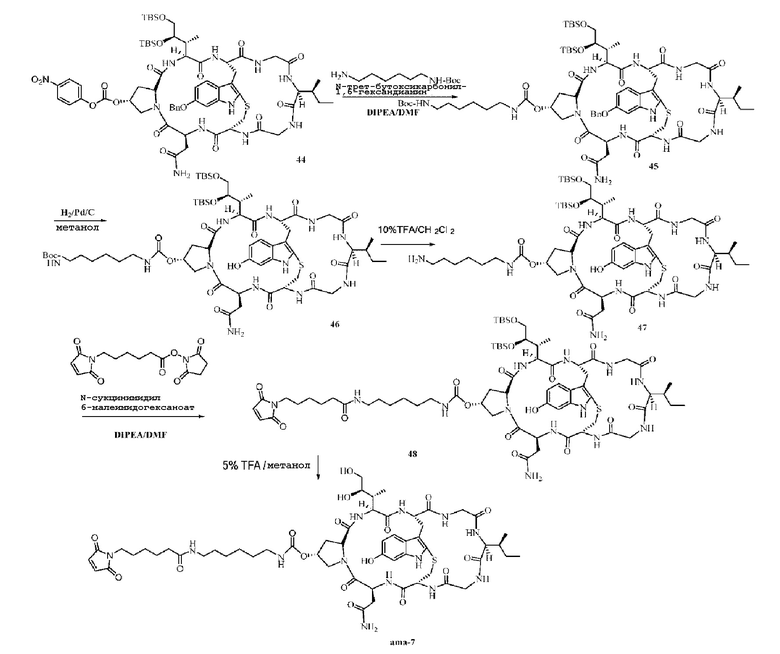
Синтез соединения 45: Ссылаясь на синтез соединения 30, после очистки препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 130,2 мг светло-желтого твердого вещества с выходом 49,4%. MS: [M+H]+ 1463,7524.
Синтез соединения 46: Приблизительно 125 мг соединения 45 растворяли в 1 мл метанола, затем добавляли 12,5 мг 10% Pd/C и осуществляли реакцию восстановительного гидрирования при 40°C и 0,5 мПа в течение 12 часов, используя ВЭЖХ для подтверждения завершения реакции, затем Pd/C удаляли фильтрованием и фильтрат собирали и концентрировали с получением коричневого маслянистого вещества, которое подавали непосредственно на следующую стадию без очистки.
Синтез соединения 47: Неочищенный продукт, представляющий собой Соединение 46, растворяли в 10% растворе TFA в дихлорметане, затем реакции давали осуществиться при перемешивании и в атмосфере азота в течение одного часа при комнатной температуре, ВЭЖХ использовали для подтверждения завершения реакции, раствор удаляли центрифугированием и полученный продукт непосредственно подавали на следующую стадию без очистки.
Синтез соединения 48: Ссылаясь на синтез соединения 32 и рассчитывая исходя из 100% выхода для предыдущей стадии, после очистки препаративной ВЭЖХ фракции, соответствующие целевым пикам, собирали и осуществляли лиофилизацию с получением приблизительно 20,6 мг светло-желтого твердого вещества с общим выходом от трех стадий 16%. MS: [M+H]+ 1466,7436.
Синтез соединения ama-7
Ссылаясь на синтез соединения ama-1, после добавления 10 мг исходного вещества 48 и очистки при помощи препаративной ВЭЖХ фракции, соответствующие целевым пикам, собирали и осуществляли лиофилизацию с получением приблизительно 3,2 мг не совсем белого твердого вещества с выходом 37,9%. MS: [M+H]+ 1238,6512.
Пример 8: Синтез низкомолекулярной нагрузки ama-8
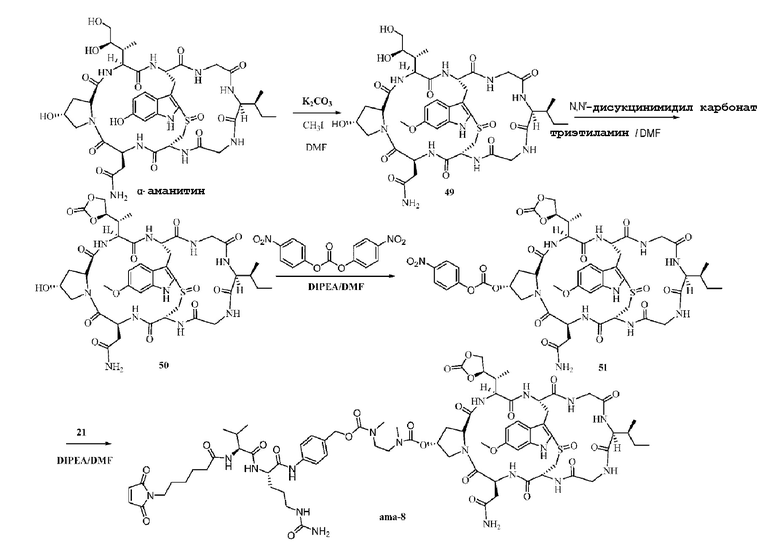
Синтез соединения 49: 30 мг α-аманитина растворяли с использованием 1 мл DMF, затем добавляли 9 мг карбоната калия (3 экв.), и смесь перемешивали в течение одного часа при комнатной температуре с последующим добавлением 13 мкл иодметана (10 экв.) и перемешиванием в течение 12 часов при комнатной температуре, а затем очисткой при помощи препаративной ВЭЖХ и лиофилизацией с получением приблизительно 9,1 мг целевого соединения с выходом 30%. MS: [M+H]+ 933,4031.
Синтез соединения 50: Соединение 49, полученное на предыдущей стадии, растворяли в 1 мл DMF с последующим добавлением 25 мг (10 экв.) N, N'-дисукцинимидилкарбоната и добавлением 0,14 мл (20 экв.) триэтиламина, затем осуществляли перемешивание при комнатной температуре в течение 12 часов, затем, после того как исходное вещество полностью прореагировало, осуществляли очистку при помощи препаративной ВЭЖХ, фракции, соответствующие целевым пикам, собирали и осуществляли лиофилизацию с получением 5,6 мг белого твердого вещества с выходом 59,9%; MS:[M+H]+ 959,4501.
Синтез соединения 51: Ссылаясь на синтез соединения 22, все 5,6 мг соединения 50, полученного на предыдущей стадии, добавляли к реакционной смеси и продукт непосредственно подавали на следующую стадию без очистки.
Синтез соединения ama-8:
Ссылаясь на синтез соединения 23, после очистки препаративной ВЭЖХ осуществляли лиофилизацию с получением приблизительно 3,2 мг белого твердого вещества с выходом 32,8%; MS: [M+H]+ 1671,7211.
Пример 9: Синтез низкомолекулярной нагрузки ama-9
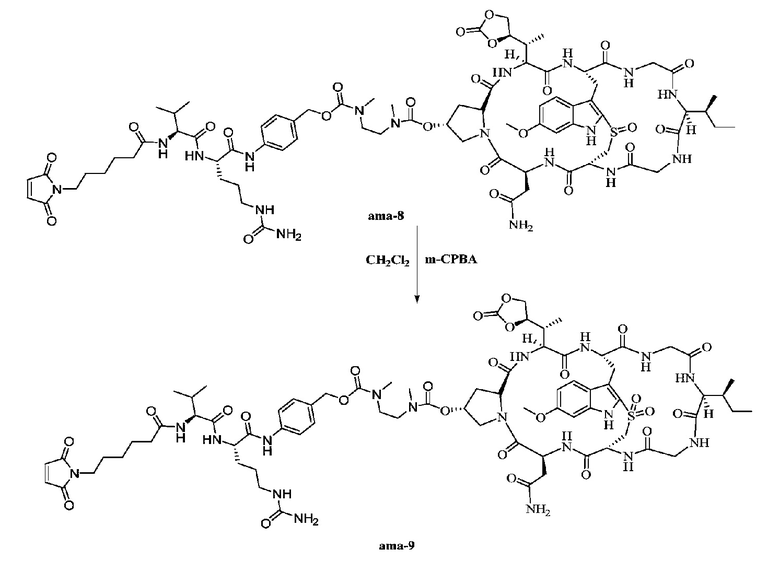
Приблизительно 2 мг соединения ama-8 растворяли в 0,5 мл дихлорметана с последующим добавлением 0,6 мг (3 экв.) м-хлорпероксибензойной кислоты (m-CPBA), затем реакции давали осуществиться при перемешивании в течение трех часов при комнатной температуре, затем осуществляли очистку при помощи препаративной ВЭЖХ и собирали фракции, соответствующие целевым пикам, с последующей лиофилизацией с получением приблизительно 0,8 мг белого твердого вещества с выходом 39,6%; MS: [M+H]+ 1687,7142.
Пример 10: Синтез низкомолекулярной нагрузки ama-10
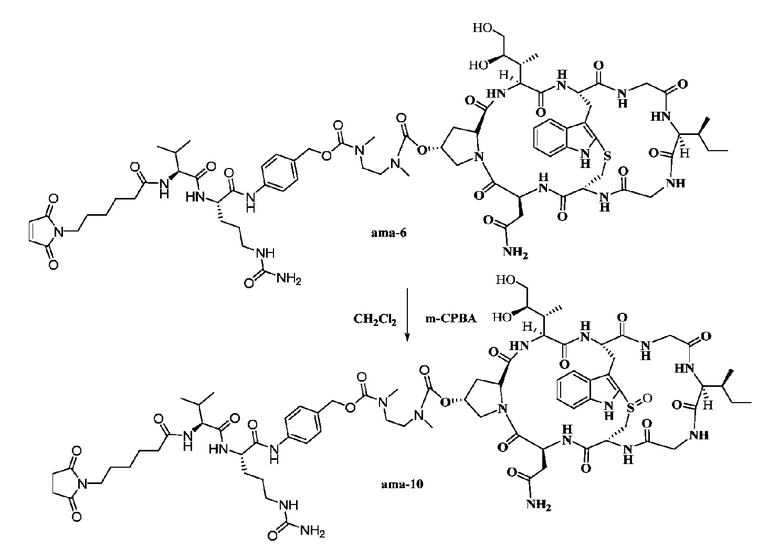
Приблизительно 4 мг соединения ama-6 растворяли в 0,5 мл дихлорметана с последующим добавлением 0,5 мг (1,2 экв.) м-хлорпероксибензойной кислоты (m-CPBA), затем реакции давали осуществиться при перемешивании в течение двух часов при комнатной температуре, затем осуществляли очистку при помощи препаративной ВЭЖХ и собирали фракции, соответствующие пику целевого продукта, с последующей лиофилизацией с получением приблизительно 1,2 мг белого твердого вещества с выходом 29,7%; MS: [M+H]+ 1615,7320.
Пример 11:
Следующие соединения были получены как часть данного примера:
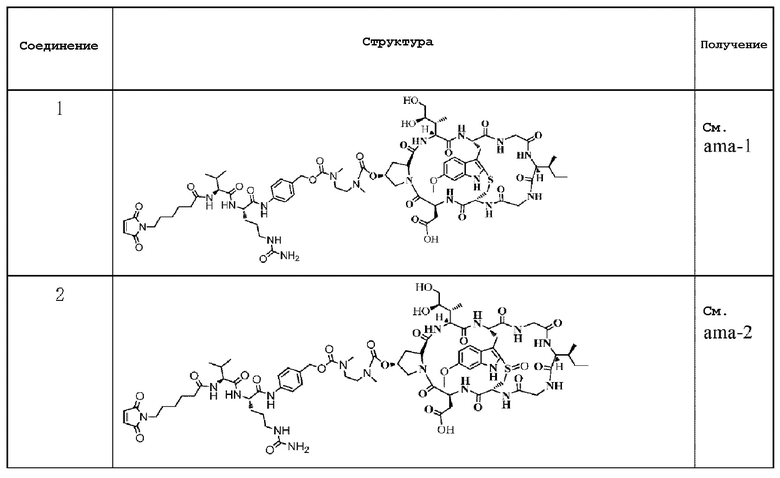
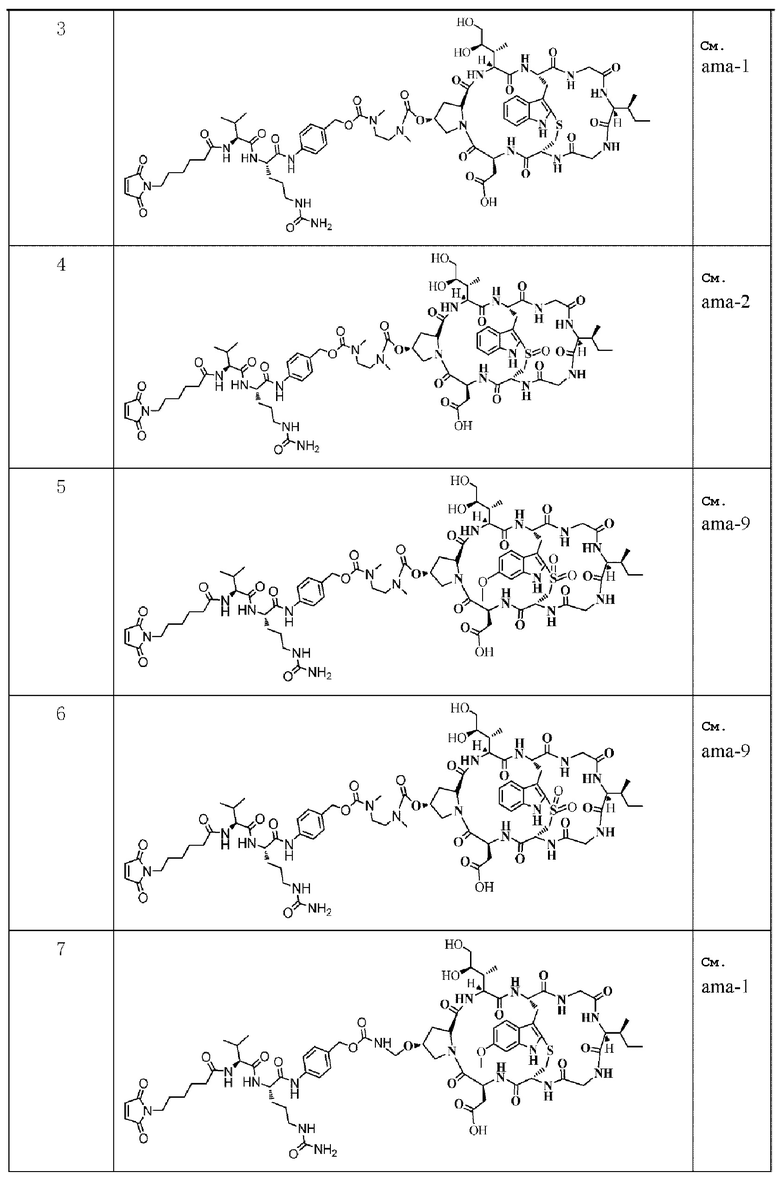
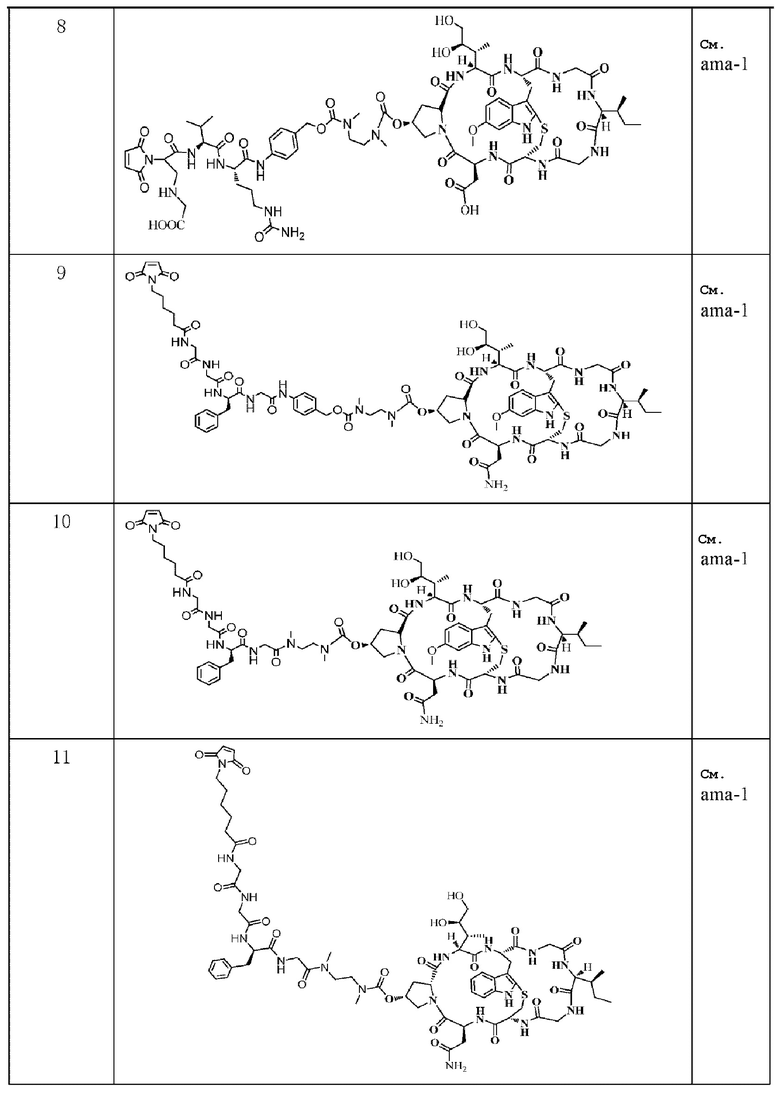
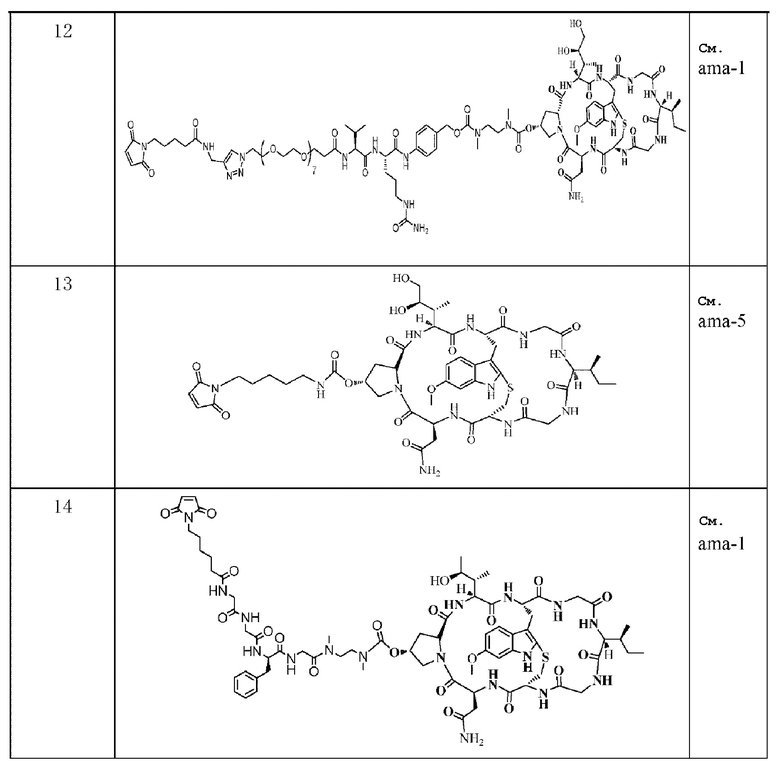
Пример 12: Получение конъюгата антитело-лекарственное средство
1) Общий способ конъюгации: После предварительной очистки молекулы антител с содержанием мономеров больше чем 95% подвергали процедуре обмена среды на фосфатный буфер, содержащий EDTA при концентрации 10 мг/мл, с использованием ультрафильтрационной центрифужной пробирки. Добавляли TCEP в 10-кратном молярном количестве относительно антитела и осуществляли взаимодействие в течение двух часов. Затем, используя ультрафильтрационную центрифужную пробирку для обмена раствора на pH 6,5 фосфатный буфер, добавляли DHAA в 10-кратном молярном количестве относительно антитела и реакции давали осуществиться в течение двух часов. Затем добавляли полезную нагрузку в 3-кратном молярном количестве относительно антитела и реакции давали осуществиться в течение четырех часов. Когда реакция завершилась, использовали ультрафильтрационную центрифужную пробирку с отсечкой по молекулярной массе 30 кДа для обмена раствора на PBS и несвязанную нагрузку удаляли.
2) Анализ DAR конъюгата антитело-лекарственное средство
Условия анализа для определения содержания мономера:
Образцы центрифугировали при 14000 об/мин в течение 5 минут и супернатант брали для анализа.
Устройство: Waters e2695 (2489 УФ/видимая область спектра)
Хроматографическая колонка: TSKgel G3000SWXL (7,8 × 300 мм, 5 мкм)
Подвижная фаза: A: 50 мМ PB, 300 мМ NaCl, 200 мМ Arg, 5% IPA, pH 6,5
Осуществляли элюирование с подвижной фазой A в изократическом режиме в течение 30 мин; скорость потока: 0,714 мл/мин; температура колонки: 25°C; и длина волны детекции: 280 нм.
Условия анализа DAR:
Образцы центрифугировали при 14000 об/мин в течение 5 минут и супернатант брали для анализа.
Устройство: Waters H-class (TUV)
Хроматографическая колонка: Proteomix HIC Butyl-NP5 (4,6 × 35 мм, 5 мкм)
Подвижная фаза: A: 1,5 M сульфата аммония, 0,025 M безводного фосфата натрия, pH 7,0
B: 0,025 M безводного фосфата натрия, 25% IPA, pH 7,0
Подвижную фазу A использовали для уравновешивания хроматографической колонки; после этого осуществляли градиентное элюирование с подвижной фазой A и B; скорость потока: 0,8 мл/мин; температура колонки: 25°C; и длина волны детекции: 214 нм.
Структурная формула A0301 является следующей:
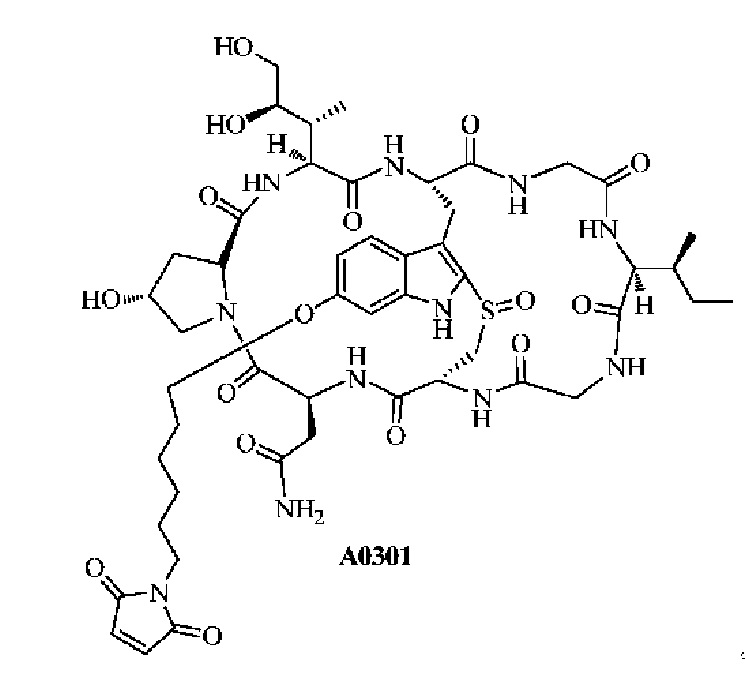
3) Результаты99
Таблица 1 показывает, что конъюгат антитела, связанный из сайта гидроксипролина, демонстрирует относительно благоприятный процент мономера и относительно высокое значение DAR. Из-за схожести их структур Соединения 1-14 также демонстрируют относительно благоприятный процент мономера и относительно высокое значение DAR.
Пример 13: Испытание на активность in vitro
1) Материалы, используемые в эксперименте
Клетки: получены от Cell Bank of Chinese Academy of Sciences
Среда для культивирования опухолевых клеток: Gibco
FBS: BIOWEST
2) Получение культуральной среды
Питательная среда (с 10% FBS, Пенициллин/стрептомицин (100 Ед/мл)
Среда для детекции (с 1% FBS, Пенициллин/стрептомицин (100 Ед/мл)
3) Процедура
Ультрафиолетовую лампу бокса биологической безопасности включали заранее за 30 минут для облучение, а затем в течение 3 минут осуществляли воздухообмен. Питательную среду, среду для детекции, D-PBS и трипсин предварительно нагревали на водяной бане с термостатом 37°C и после стерилизации поверхности их помещали в бокс биологической безопасности. Клетки с конфлюэнтностью около 80% помещали в бокс биологической безопасности, старую среду удаляли путем аспирации, клетки промывали D-PBS с последующей аспирацией и расщеплением трипсином в течение 2-3 минут, после этого добавляли питательную среду для нейтрализации реакции и осуществляли центрифугирование при 1200 об/мин в течение трех минут. Полученный после центрифугирования супернатант удаляли путем аспирации, и добавляли 4 мл среды для количественного определения и гомогенно смешивали, после этого 100 мкл раствора использовали для подсчета (при этом 50 мкл содержащего клетки раствора тщательно смешивали с 50 мкл красителя трипанового синего и затем осуществляли подсчет). Клетки высевали, в соответствии с предварительно установленным количеством клеток, при плотности 80 мкл на лунку в 96-луночные планшеты; в лунки E11, F11 и G11 добавляли только 80 мкл среды для детекции, а в крайние лунки добавляли 150 мкл DPBS. Разбавление раствора антител: 300 мкл раствора испытываемого продукта с начальной концентрацией 5 мкМ получали в первой колонке 96-луночного планшета V-типа, при этом в каждую из колонок 2-10 добавляли 210 мкл среды для детекции; затем брали 30 мкл предварительной смеси из первой колонки и добавляли во вторую колонку, смешивали пипеткой 10 раз вверх и вниз и наконечник пипетки затем выбрасывали; и затем последовательно получали семь последующих концентраций с использованием такой же процедуры. Через 24 часа после посева добавляли разбавленное антитело при 20 мкл на лунку и устанавливали дополнительные контроли; только 20 мкл среды для детекции добавляли в колонку 11, для каждой концентрации устанавливали две дублирующие лунки и после добавления клетки подвергали вихревому перемешиванию при 550 об/мин в течение трех минут.
4) Анализ
Через 4 дня реагент MTS удаляли и образцы размораживали при комнатной температуре, защищая от света, и осуществляли интенсивное перемешивание вихревым способом; 20 мкл CellTiter 96® One Solution Reagen MTS реагента добавляли вдоль боковой стенки лунок на каждые 100 мкл объема клеточной культуры и осторожно похлопывали по поверхности планшета для гомогенного смешивания раствора MTS, после чего образцы помещали в инкубатор для клеточных культур для статической инкубации в течение двух часов, защищая при этом от воздействия света. После завершения реакции 96-луночный планшет извлекали, определяли значение оптической плотности при OD 490 нм с использованием считывающего устройства для микропланшетов и соответствующие данные регистрировали, сортировали и хранили.
5) Результаты
Как показано в Таблице 2 и на Фиг. 1-3, хорошая противоопухолевая активность была продемонстрирована в клеточных линиях BT474/SKBR3/N87. Из-за схожести их структур Соединения 1-14 также демонстрируют относительно благоприятную противоопухолевую активность.
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
В предыдущем разделе варианты осуществления настоящего изобретения были подробно описаны вместе с примерами, чтобы полностью пояснить, как настоящее изобретение использует определенные технические средства для решения соответствующих технических проблем и достижения технической эффективности, и они могут быть использованы в качестве основы для его осуществления. Следует отметить, что отдельные варианты осуществления настоящего изобретения и индивидуальные характерные признаки каждого варианта осуществления могут быть объединены друг с другом, и полученные технические решения должны оставаться в пределах объема настоящего изобретения при условии отсутствия каких-либо противоречий.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2840926C2 |
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2798981C2 |
| КОНЪЮГАТЫ АМАТОКСИН - АНТИТЕЛО | 2016 |
|
RU2724328C2 |
| КОНЪЮГАТЫ АМАТОКСИНОВ С УЛУЧШЕННЫМИ ЛИНКЕРАМИ | 2011 |
|
RU2601411C2 |
| КОНЪЮГАТЫ, СОДЕРЖАЩИЕ ГИДРОФИЛЬНЫЕ СПЕЙСЕРЫ ЛИНКЕРОВ | 2008 |
|
RU2523909C2 |
| ПРОИЗВОДНЫЕ АМАТОКСИНА | 2014 |
|
RU2695370C2 |
| КОНЪЮГАТЫ ДИСОРАЗОЛОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, НАЗВАННЫЕ КОНЪЮГАТЫ ДЛЯ ИЗГОТОВЛЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА, НАБОР И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2473562C2 |
| НАЦЕЛЕННЫЕ КОНЪЮГАТЫ И ЧАСТИЦЫ И ИХ СОСТАВЫ | 2015 |
|
RU2695220C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА АМАНИТИНОВ | 2018 |
|
RU2792210C2 |
| НЕЛИНЕЙНЫЕ САМОРАСЩЕПЛЯЮЩИЕСЯ ЛИНКЕРЫ И ИХ КОНЪЮГАТЫ | 2017 |
|
RU2755899C2 |

Изобретение относится к области биотехнологии. Описана группа изобретений, включающая конъюгат токсина, обладающий ингибирующей активностью в отношении РНК-полимеразы, или его фармацевтически приемлемая соль, противоопухолевое лекарственное соединение, обладающее ингибирующей активностью в отношении РНК-полимеразы, включающее конъюгат токсина или его фармацевтически приемлемую соль, и способ применения конъюгата токсина или его фармацевтически приемлемой соли для получения противоопухолевого лекарственного средства, обладающего ингибирующей активностью в отношении РНК-полимеразы. Изобретение расширяет арсенал средств, обладающих ингибирующей активностью в отношении РНК-полимеразы. 3 н. и 4 з.п. ф-лы, 3 ил., 2 табл., 13 пр.
1. Конъюгат токсина, обладающий ингибирующей активностью в отношении РНК-полимеразы, содержащий фрагмент токсина и биомакромолекулу A, заявленный в структурной формуле (I), или его фармацевтически приемлемая соль

где:
n=0, 1 или 2;
R1 представляет собой -H или -OH;
R2 представляет собой -H или -OH;
R3 представляет собой -H, -OH или -OC1-6 алкил;
R4 представляет собой -NH2 или -OH;
R5 представляет собой -L-A, где A представляет собой антитело или его антиген-связывающий фрагмент, или его антиген-связывающий полипептид, которые сконфигурированы для связывания с мишенью, и L представляет собой химическую структуру, выбранную из группы из сукцинимида и его формы с раскрытием цикла, -C(=O)-, -C(=O)NH- и C1-6 алкила, связывающую аманитиновое производное и биологическую макромолекулярную часть А.
2. Конъюгат токсина по п. 1 или его фармацевтически приемлемая соль, где L включает расщепляемую или нерасщепляемую химическую структуру.
3. Конъюгат токсина по п. 1 или его фармацевтически приемлемая соль, где место конъюгации L и связывающейся с мишенью биомолекулы A включает следующую структуру:
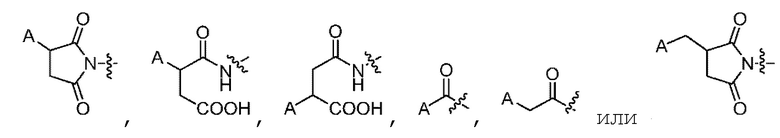 .
.
4. Конъюгат токсина по п. 1 или его фармацевтически приемлемая соль, где L связан с аманитиновым производным через сложноэфирную или простую эфирную связь.
5. Противоопухолевое лекарственное соединение, обладающее ингибирующей активностью в отношении РНК-полимеразы, включающее конъюгат токсина по п. 1 или его фармацевтически приемлемую соль.
6. Способ применения конъюгата токсина по п. 1 или его фармацевтически приемлемой соли для получения противоопухолевого лекарственного средства, обладающего ингибирующей активностью в отношении РНК-полимеразы, путем включения конъюгата токсина или его фармацевтически приемлемой соли.
7. Способ по п. 6, где указанное противоопухолевое лекарственное средство содержит лекарственное средство от рака легкого, лекарственное средство от рака почек, лекарственное средство от рака мочевыводящих путей, лекарственное средство от колоректального рака, лекарственное средство от рака предстательной железы, лекарственное средство от глиобластомы, лекарственное средство от рака яичников, лекарственное средство от рака поджелудочной железы, лекарственное средство от рака молочной железы, лекарственное средство от меланомы, лекарственное средство от рака печени, лекарственное средство от рака мочевого пузыря, лекарственное средство от злокачественной лимфомы, лекарственное средство от лейкоза, лекарственное средство от рака желудка или лекарственное средство от рака пищевода.
| WO 2011130580 A1, 20.10.2011 | |||
| КОНЪЮГАТЫ ЛИГАНДА С НЕСКОЛЬКИМИ ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2006 |
|
RU2470668C2 |
| US 10245326 B2, 02.04.2019. | |||

