ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает преимущество и приоритет заявки на патент Китая №201910107947.5, поданной в Национальное управление интеллектуальной собственности КНР 2 февраля 2019 года, заявки на патент Китая №201910111576.8, поданной в Национальное управление интеллектуальной собственности КНР 12 февраля 2019 года, и заявки на патент Китая №201910684020.8, поданной в Национальное управление интеллектуальной собственности КНР 26 июля 2019 года, раскрытие каждой из которых включено в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящая заявка относится к новому типу соединений индологептамилоксима в качестве ингибитора PARP и, в частности, к соединению, представленному формулой (I), его стереоизомеру и его фармацевтически приемлемой соли.
УРОВЕНЬ ТЕХНИКИ
Поли(АДФ-рибоза)-полимераза (PARP) представляет собой семейство ферментов и может применяться для катализирования добавления остатков АДФ-рибозы к разнообразным целевым белкам. На сегодняшний день были идентифицированы и описаны в общей сложности 18 подтипов. Несмотря на широкое разнообразие ферментов в семействе PARP, PARP-1 является ответственной за более чем 90% случаев АДФ-рибозилирования в клетках, и, таким образом, ингибиторы PARP-1 являются центральным объектом при изучении ингибиторов PARP.
В живом человеческом организме постоянно происходит повреждение человеческой ДНК по причине воздействия факторов окружающей среды (таких как окислительный стресс, лучевая терапия и химиотерапия). PARP-1 тесно связана с репарацией ДНК и поддержанием функции генома. При повреждении ДНК, как правило, при однонитевом разрыве (SSB), PARP-1 сначала связывается с местом разрыва ДНК и затем активируется, и в ходе изменения структуры фермента PARP1 фермент начинает рекрутировать NAD+ (кофермент II) для синтеза поли(АДФ)рибозы, что одновременно служит в качестве сигнала для осуществления функции других репаративных ферментов, таких как ДНК-лигаза и ДНК-полимераза β. Данный процесс связывания и активации PARP-1 называется эксцизионной репарацией оснований (BER) и способствует репаративному процессу на основе амплификации ДНК. Если PARP-1 подавляется с помощью ингибитора PARP, поврежденная ДНК не может быть восстановлена посредством SSB; вместо этого активируется двухнитевой разрыв (DSB). В организме DSB восстанавливается в основном двумя путями: путем гомологичной рекомбинации (HR) и путем негомологичного соединения концов (NHEJ) ДНК, где гомологичная рекомбинация представляет собой основной путь репарации DSB и характеризуется высокой степенью надежности репарации. BRCA1 и BRCA2 играют важные роли в процессе гомологичной рекомбинации (Nature, 2005, 913-917). Результаты исследований показывают, что мутация BRCA1/2 обнаруживается при раке яичников, раке молочной железы и раке предстательной железы, и ингибитор PARP является хорошим выбором при опухолях, дефектных в отношении BRCA1/2. Ингибитор PARP может применяться по отдельности или в комбинации с химиотерапевтическими лекарственными средствами и радиотерапевтическими лекарственными средствами, вследствие чего снижается доза и повышается эффективность. На данной основе была разработана серия разных типов соединений (J. Med. Chem. 2010, 4561), и среди данных соединений олапариб, рукапариб, нирапариб (MK-4827) и талазопариб (BMN-673) были успешно выведены на рынок. Тем не менее, одновременно с продолжающимся расширением перечня показаний к применению ингибиторов PARP практическое применение ингибиторов PARP также углубляется от лечения опухоли до лечения инсульта, ишемии миокарда, воспаления и сахарного диабета. В настоящее время проводится очень большое число клинических испытаний.
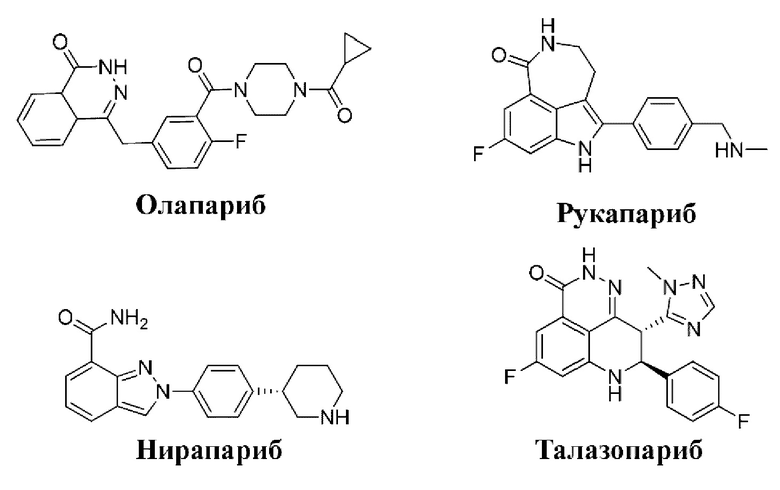
Несмотря на постоянные усилия, направленные на разработку ингибиторов PARP для лечения рака и других заболеваний, соответствующее требованиям средство для лечения не было получено и, таким образом, сохраняется острая необходимость в разработке новых ингибиторов PARP.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящей заявке предусмотрено соединение формулы (II), его стереоизомер или его фармацевтически приемлемая соль,
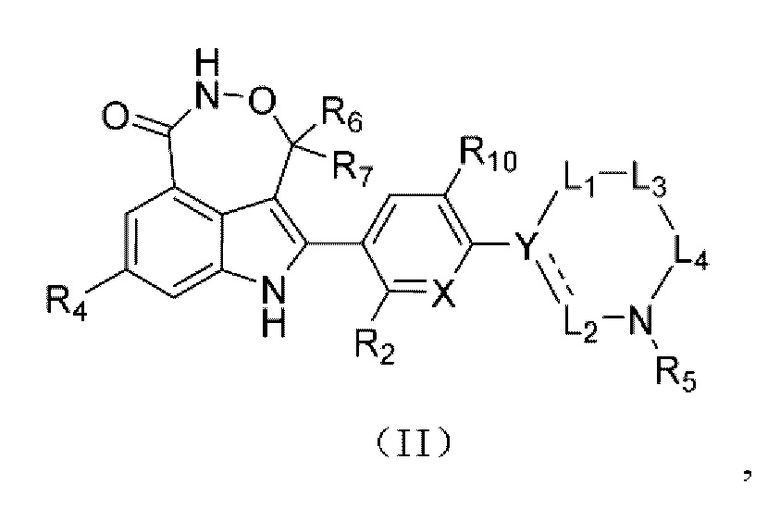
где
 выбрана из группы, состоящей из одинарной связи и двойной связи;
выбрана из группы, состоящей из одинарной связи и двойной связи;
X выбран из группы, состоящей из CR3 и N;
Y выбран из группы, состоящей из CR1 и С;
L1 выбран из группы, состоящей из одинарной связи и -(CR8R9)n-;
L2 выбран из группы, состоящей из одинарной связи, -CR8R9- и =СН-;
L1 и L2 не представляют собой одинарные связи одновременно;
если L2 выбран из одинарной связи, то  выбрана из одинарной связи;
выбрана из одинарной связи;
каждый из L3 и L4 независимо выбран из -CR8R9-;
n равняется 1 или 2;
R1 выбран из группы, состоящей из Н, D, F, Cl, Br, I и С1-3алкила, и если L2 выбран из =СН-, то R1 отсутствует;
каждый из R2 и R10 независимо выбран из группы, состоящей из Н, F, Cl, Br и I;
R3 выбран из группы, состоящей из Н, F, Cl, Br и I;
R4 выбран из группы, состоящей из Н и F;
R5 выбран из группы, состоящей из Н и С1-3алкила, где С1-3алкил необязательно замещен 1 Rd;
каждый из R6 и R7 независимо выбран из группы, состоящей из Н и D;
каждый из R8 и R9 представляет собой Н;
Rd представляет собой ОН.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, которое выбрано из формулы (II-1)
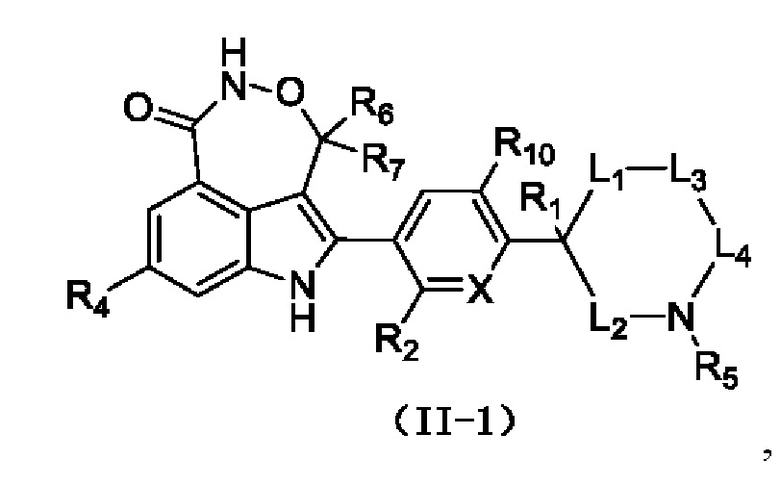 где
где
R1, R2, X, R4, R5, R6, R7, R10, L1, L2, L3 и L4 являются такими, как определено для соединения формулы (II), раскрытого в данном документе.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где R1 выбран из группы, состоящей из Н, D, F и СН3.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где каждый из R2 и R10 независимо выбран из группы, состоящей из Н и F.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где R3 выбран из группы, состоящей из Н, F и Cl.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где R3 выбран из группы, состоящей из Н и F.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где R5 выбран из группы, состоящей из Н, метила, этила, пропила и изопропила, при этом метил, этил, пропил и изопропил необязательно замещены Rd.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где R5 выбран из группы, состоящей из Н, метила,  и изопропила.
и изопропила.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где L1 выбран из группы, состоящей из одинарной связи и -CR8R9-.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где L1 выбран из -CR8R9-, L2 выбран из -CR8R9-.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где L1 выбран из -CR8R9-, L2 выбран из одинарной связи.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где оба из R6 и R7 представляют собой Н.
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где структурное звено 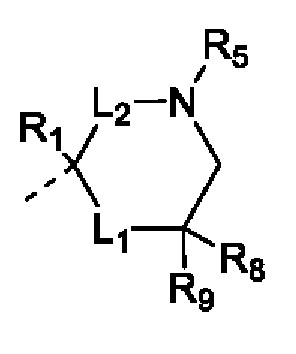 выбрано из группы, состоящей из
выбрано из группы, состоящей из  и
и 
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где структурное звено  выбрано из группы, состоящей из
выбрано из группы, состоящей из 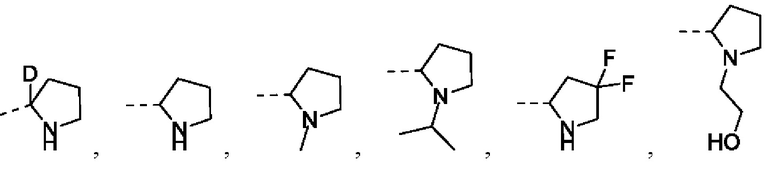 и
и 
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где структурное звено 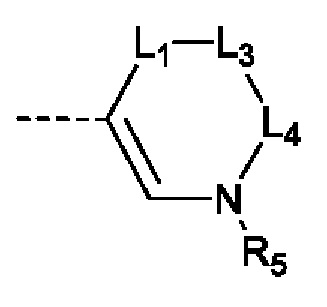 выбрано из
выбрано из 
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где структурное звено 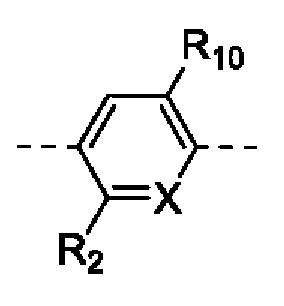 выбрано из группы, состоящей из
выбрано из группы, состоящей из  и
и 
Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, описанное выше, где структурное звено 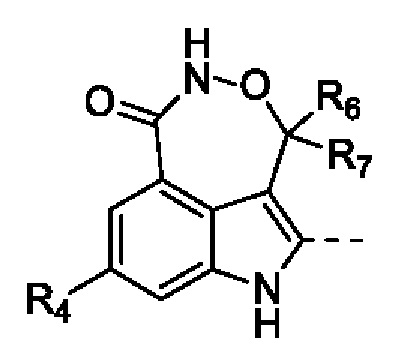 выбрано из группы, состоящей из
выбрано из группы, состоящей из 
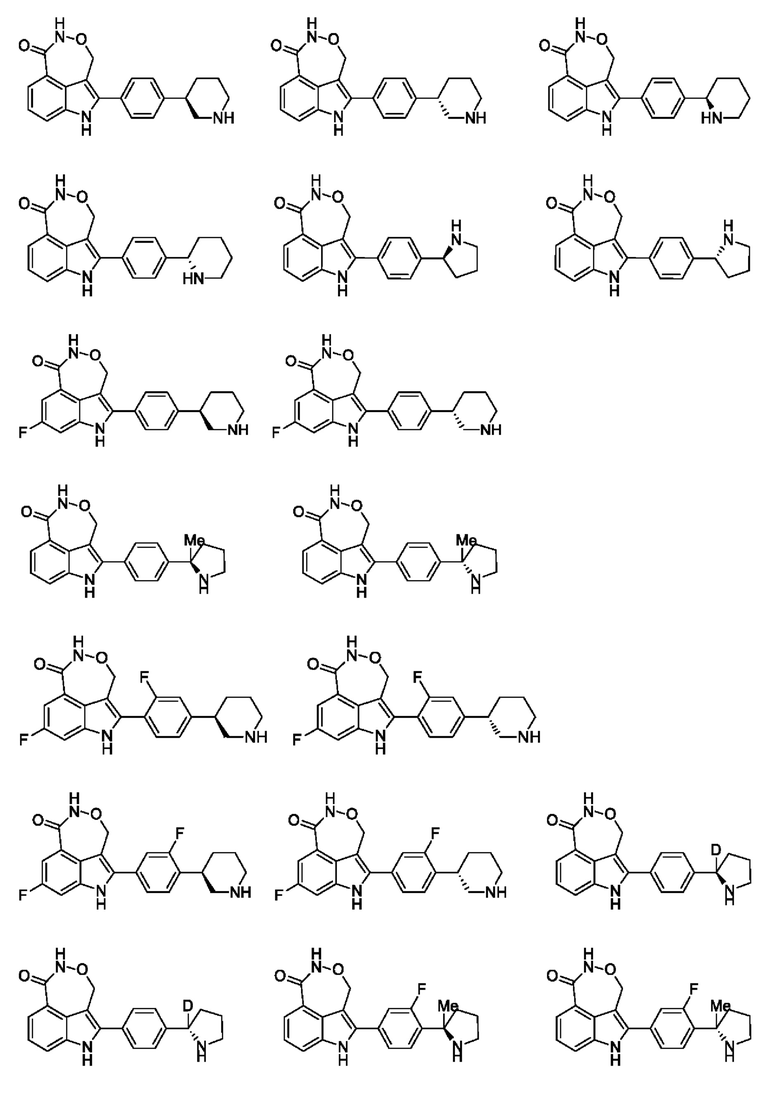
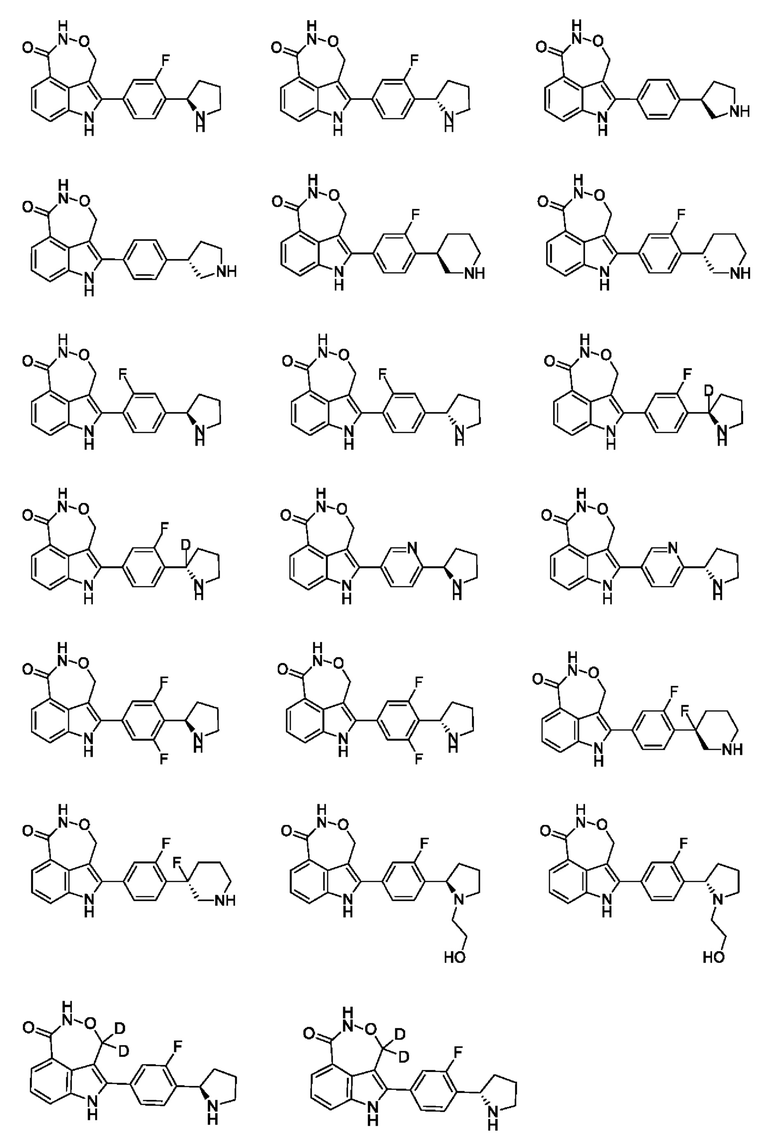
В настоящей заявке также предусмотрено соединение формулы, указанной ниже, его стереоизомер или его фармацевтически приемлемая соль, выбранное из группы, состоящей из
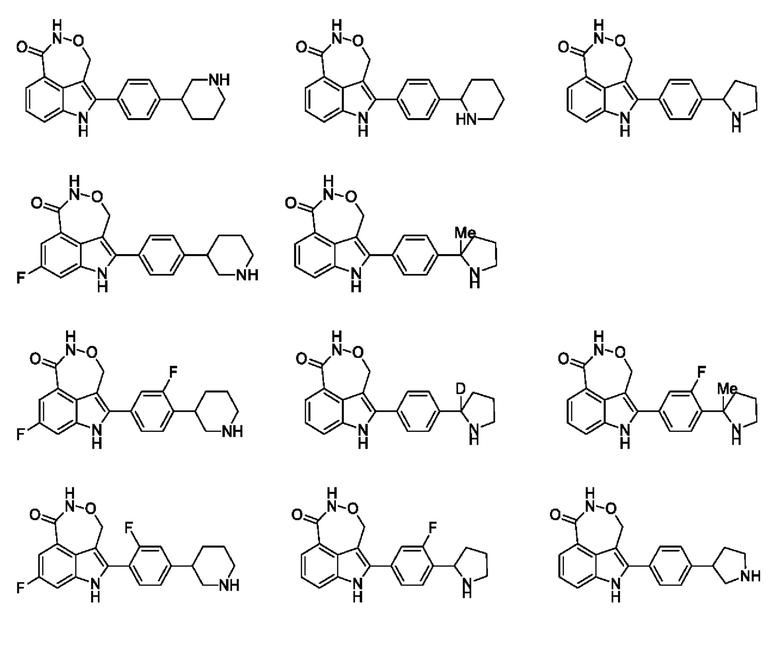

Согласно некоторым вариантам осуществления настоящей заявки предусмотрено соединение, его стереоизомер или его фармацевтически приемлемая соль, выбранное из группы, состоящей из

В настоящей заявке дополнительно предусмотрена фармацевтическая композиция, обладающая ингибирующей активностью в отношении PARP-1, содержащая терапевтически эффективное количество соединения, его стереоизомера или его фармацевтически приемлемой соли, раскрытого в данном документе, и фармацевтически приемлемый носитель.
В настоящей заявке дополнительно предусмотрено применение соединения, его стереоизомера или его фармацевтически приемлемой соли, раскрытого в данном документе, в получении лекарственного препарата для применения в лечении заболевания, связанного с рецептором PARP.
В настоящей заявке дополнительно предусмотрен способ лечения заболевания, связанного с рецептором PARP, включающий введение млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения, его изомера или его фармацевтически приемлемой соли, раскрытого в данном документе.
Некоторые другие варианты осуществления настоящей заявки получены за счет любой комбинации переменных, описанных выше.
Технические эффекты
Соединение, раскрытое в данном документе, обладает выраженной ингибирующей активностью в отношении PARP1-киназы и превосходной антипролиферативной активностью в отношении клеток MDA-MB-436 с мутацией BRCA1, и в то же время оно не имеет ингибирующей активности в отношении клеток MDA-MB-231 с BRCA дикого типа, что показывает, что соединение, раскрытое в данном документе, является превосходным в отношении селективности и безопасности. Соединение, раскрытое в данном документе, также обладает некоторым ингибирующим эффектом в отношении поли-АДФ-рибозилирования. Кроме того, соединение, раскрытое в данном документе, имеет превосходные фармакокинетические свойства, так как оно является стабильным в процессе метаболизма in vivo и характеризуется высокой биологической доступностью. В целом, соединение, раскрытое в данном документе, не только является превосходным по активности и легко синтезируемым, а также является превосходным по фармакокинетическим свойствам.
Определения и описание
Если не указано иное, то предполагается, что следующие термины и фразы, применяемые в данном документе, имеют следующие значения. Определенные термин или фразу, если четко не определено иное, не следует считать неопределенными или неясными, а следует понимать в соответствии с их общепринятым значением. Предполагается, что ссылка на торговое наименование относится к соответствующему ему коммерческому продукту или его активному ингредиенту.
Термин «фармацевтически приемлемый» используется в данном документе в отношении тех соединений, материалов, композиций и/или лекарственных форм, которые в пределах объема тщательной медицинской оценки являются подходящими для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений и соизмеримо с приемлемым соотношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения, раскрытого в данном документе, которая получена из соединения, содержащего определенные заместители, раскрытые в данном документе, и относительно нетоксичных кислоты или основания. Если соединение, раскрытое в данном документе, содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения такого соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, соли органического амина или магния или подобные соли. Если соединение, раскрытое в данном документе, содержит относительно основную функциональную группу, то соль присоединения кислоты можно получать посредством приведения такого соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли, полученные из неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонатный радикал, фосфорная кислота, моногидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота и фосфористая кислота; и соли, полученные из органических кислот, таких как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, бензолсульфоновая кислота, п-толуол су ль фоновая кислота, лимонная кислота, винная кислота и метансульфоновая кислота. Также включены соли аминокислот (например, аргинина) и соли органических кислот, таких как глюкуроновая кислота. Определенные конкретные соединения, раскрытые в данном документе, содержат как основную, так и кислотную функциональную группы, которые позволяют превращать соединения либо в соли присоединения основания, либо в соли присоединения кислоты.
Фармацевтически приемлемые соли, раскрытые в данном документе, могут быть синтезированы из исходного соединения, имеющего кислотную или основную группу, с помощью традиционных химических способов. В целом такие соли получают с помощью следующего способа: соединение в форме свободной кислоты или основания приводят в реакцию со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или их смеси.
Соединения, раскрытые в данном документе, могут находиться в форме геометрического изомера или стереоизомера. Все такие соединения предусмотрены в данном документе, в том числе цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры и их рацемические смеси и другие смеси, такие как обогащенные энантиомерами или диастереоизомерами смеси, все из которых охвачены объемом настоящей заявки. Заместители, такие как алкил, могут иметь дополнительный асимметричный атом углерода. Все такие изомеры и их смеси охвачены объемом настоящей заявки.
Если не указано иное, абсолютная конфигурация стереогенного центра представлена сплошной клиновидной связью 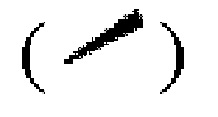 и пунктирной клиновидной связью
и пунктирной клиновидной связью  а относительная конфигурация стереогенного центра представлена прямой сплошной связью
а относительная конфигурация стереогенного центра представлена прямой сплошной связью 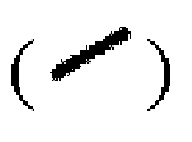 и прямой пунктирной связью
и прямой пунктирной связью 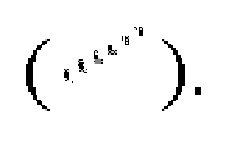 Волнистая линия
Волнистая линия 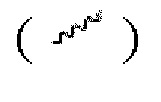 представляет собой сплошную клиновидную связь
представляет собой сплошную клиновидную связь 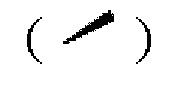 или сплошную пунктирную связь
или сплошную пунктирную связь  или волнистая линия
или волнистая линия 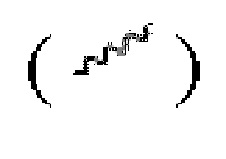 представляет собой прямую сплошную связь
представляет собой прямую сплошную связь 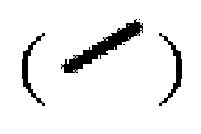 и прямую пунктирную связь
и прямую пунктирную связь 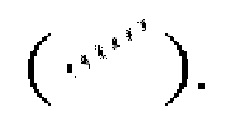
Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры могут быть получены посредством хирального синтеза, или хиральных реагентов, или других общепринятых методик. Энантиомер определенного соединения, раскрытого в данном документе, может быть получен посредством асимметричного синтеза или дериватизации с применением хирального вспомогательного средства, где полученную диастереоизомерную смесь разделяют и вспомогательную группу отщепляют таким образом, чтобы обеспечить получение требуемого чистого энантиомера. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), то соединение вступает в реакцию с подходящими оптически активными кислотой или основанием с образованием соли диастереоизомера, которую затем подвергают диастереоизомерному разделению посредством общепринятых в уровне техники способов с получением чистого энантиомера. Кроме того, энантиомер и диастереизомер, как правило, выделяют посредством хроматографии с применением хиральной неподвижной фазы, необязательно в комбинации с химической дериватизацией (например, карбамат, получаемый из аминов).
Соединение, раскрытое в данном документе, может содержать не встречающуюся в природе пропорцию атомных изотопов на одном или более атомов, которые образуют соединение. Например, соединение может быть меченным с помощью радиоактивного изотопа, такого как тритий (D, 3Н), йод-125 (125I) или С-14 (14С). В качестве другого примера, водород может быть замещен дейтерием с образованием дейтерированного лекарственного средства и связь, образованная между дейтерием и углеродом, прочнее связи, образованной между обычным водородом и углеродом. По сравнению с недейтерированным лекарственным средством дейтерированное лекарственное средство имеет преимущества в виде сниженного токсического побочного эффекта, повышенной стабильности, увеличенной эффективности, продленного биологического времени полужизни и т.п. Все изотопные варианты соединения, описанного в данном документе, являются они радиоактивными или нет, включены в объем настоящего изобретения.
«Необязательный» или «необязательно» означает, что далее описанное событие или обстоятельство может, но не обязательно, произойти, при этом описание включает случаи, когда событие или обстоятельство происходит, и случаи, когда это не происходит.
Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителями, которые могут включать варианты дейтерия и водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой кислород (т.е. =O), это означает, что замещены два атома водорода. Замещение кислородом не происходит в ароматических группах. Термин «необязательно замещенный» означает, что атом может быть замещен заместителем или не замещен заместителем. Если не указано иное, тип и количество заместителей могут быть произвольными, при условии, что это достижимо с химической точки зрения.
Если любая переменная (например, R) встречается больше одного раза в составе или структуре соединения, то в каждом случае переменная определяется независимо. Таким образом, например, если группа замещена 0-2 R, то группа может быть необязательно замещена не более чем двумя R, и определение R в каждом случае является независимым. Кроме того, комбинация заместителя и/или его варианта допустима, только если комбинация может привести к получению стабильного соединения.
Если число связывающих групп равняется 0, например -(CRR)0-, это означает, что связывающая группа представляет собой одинарную связь.
Если один из вариантов выбран из одинарной связи, то две группы, связанные с помощью такого варианта, являются связанными непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, это означает, что структура фактически представляет собой A-Z.
Если заместитель отсутствует, это означает, что заместителя не существует. Например, если X в А-Х отсутствует, то структура фактически представляет собой А.
Если не указано иное, то если группа имеет один или более соединяемых участков, то любые один или более таких участков группы могут быть присоединены к другим группам посредством химических связей. Химическая связь, которая соединяет такой участок с другой группой, может быть представлена прямой сплошной связью  прямой пунктирной линией связи
прямой пунктирной линией связи  или волнистой линией
или волнистой линией 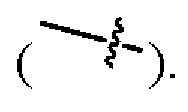 Например, сплошная прямая линия в -ОСН3 указывает на то, что группа присоединена к другой группе посредством атома кислорода; в
Например, сплошная прямая линия в -ОСН3 указывает на то, что группа присоединена к другой группе посредством атома кислорода; в 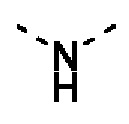 прямая пунктирная линия указывает на то, что группа присоединена к другой группе посредством атома азота с двух концов; в
прямая пунктирная линия указывает на то, что группа присоединена к другой группе посредством атома азота с двух концов; в 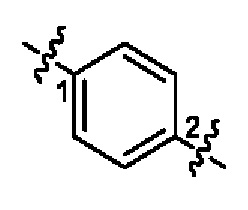 волнистая линия указывает на то, что фенильная группа присоединена к другой группе посредством атомов углерода в положениях 1 и 2.
волнистая линия указывает на то, что фенильная группа присоединена к другой группе посредством атомов углерода в положениях 1 и 2.
Если не указано иное, количество атомов в кольце, как правило, определяется как количество членов кольца. Например, «5-7-членное кольцо» относится к «кольцу», в котором 5-7 атомов расположены в виде кольца.
Если не указано иное, «С3-8циклоалкил» относится к насыщенной циклической углеводородной группе, состоящей из 3-8 атомов углерода. Он включает моноциклические и бициклические системы, где бициклическая система включает спироциклические, конденсированные и мостиковые кольца. С3-8циклоалкил включает С3-6, С3-5, С4-8, С4-6, С4-5, С5-8, С5-6циклоалкил или т.п., и может быть одновалентным, двухвалентным или поливалентным. Примеры С3-8циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил, [2.2.2]бициклооктан и т.п.
Если не указано иное, то термин «3-8-членный гетероциклоалкил», отдельно или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 3-8 атомов кольца, из которых 1, 2, 3 или 4 атома кольца являются гетероатомами, независимо выбранными из группы, состоящей из О, S и N, при этом остальные атомы представляют собой атомы углерода. Атом азота является необязательно кватернизированным, и гетероатомы углерода, азота и серы могут быть необязательно окисленными (т.е. С=O, NO и S(O)p, где р равняется 1 или 2). Включены моноциклические и бициклические системы, где бициклическая система включает спироциклические, конденсированные и соединенные мостиковой связью кольца. Кроме того, применительно к «3-8-членному гетероциклоалкилу» гетероатом может занимать положение, где гетероциклоалкил присоединен к остальной части молекулы. 3-8-членный гетероциклоалкил включает 3-6-членный, 3-5-членный, 4-6-членный, 5-6-членный, 4-членный, 5-членный и 6-членный гетероциклоалкил и т.п. Примеры 3-8-членного гетероциклоалкила включают без ограничения азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (включая тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (включая 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (включая 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил, диоксепанил или т.п.
Если не указано иное, то термин «5-6-членный гетероциклоалкил», отдельно или в комбинации с другими терминами, относится к насыщенной циклической группе, состоящей из 5 6 атомов кольца, из которых 1, 2, 3 или 4 атома кольца являются гетероатомами, независимо выбранными из группы, состоящей из О, S и N, при этом остальные атомы представляют собой атомы углерода. Атом азота является необязательно кватернизированным, и гетероатомы углерода, азота и серы могут быть необязательно окисленными (т.е. С=O, NO и S(O)p, где р равняется 1 или 2). Включены моноциклические и бициклические системы, где бициклическая система включает спироциклические, конденсированные и соединенные мостиковой связью кольца. Кроме того, применительно к «5 6-членному гетероциклоалкилу» гетероатом может занимать положение, где гетероциклоалкил присоединен к остальной части молекулы. 5 6-членный гетероциклоалкил включает 5-членный гетероциклоалкил и 6-членный гетероциклоалкил. Примеры 5-6-членного гетероциклоалкила включают без ограничения пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиен-2-ил, тетрагидротиен-3-ил и т.д.), тетрагидрофуранил (включая тетрагидрофуран-2-ил и т.д.), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и т.д.), пиперазинил (включая 1-пиперазинил, 2-пиперазинил и т.д.), морфолинил (включая 3-морфолинил, 4-морфолинил и т.д.), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил, гомопиперидинил или т.п.
Если не указано иное, то термин «С1-3алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. С1-3алкил включает без ограничения C1-2-, С2-3алкил и т.д. и может быть одновалентным (например, метил), двухвалентным (например, метилен) или многовалентным (например, метенил). Примеры С1-3алкила включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.п.
Соединения, раскрытые в данном документе, могут быть получены с помощью разнообразных способов синтеза, хорошо известных специалистам в данной области, в том числе конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, полученных путем их комбинирования с другими способами химического синтеза, и их эквивалентов, известных специалистам в данной области. Предпочтительные варианты осуществления включают без ограничения примеры, раскрытые в данном документе.
Растворители, применяемые в данном документе, могут являться коммерчески доступными.
В настоящей заявке применяют следующие сокращения: Boc обозначает трет-бутилоксикарбонил, защитную группу для аминогруппы; pht обозначает фталоил, защитную группу для первичного амина; Ms обозначает метилсульфонил.
ПОДРОБНОЕ ОПИСАНИЕ
Ниже настоящее изобретение подробно описано с помощью примеров. Однако это не ограничивает объем настоящей заявки неблагоприятным образом. Соединения, раскрытые в данном документе, могут быть получены с помощью разнообразных способов синтеза, хорошо известных специалистам в данной области, в том числе конкретных вариантов осуществления, перечисленных ниже, вариантов осуществления, полученных путем их комбинирования с другими способами химического синтеза, и их эквивалентов, известных специалистам в данной области. Предпочтительные варианты осуществления включают без ограничения примеры, раскрытые в данном документе. Специалистам в данной области будет очевидно, что могут быть сделаны различные изменения и модификации в отношении конкретных вариантов осуществления без отступления от сути и объема настоящей заявки.
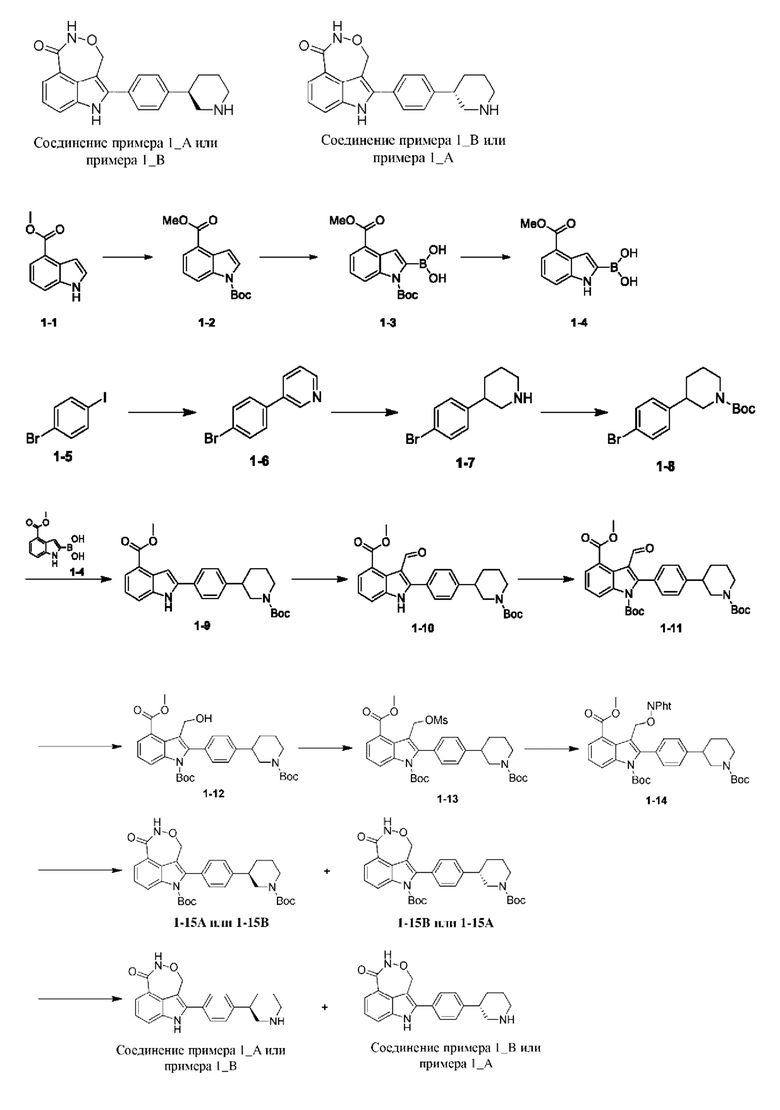
Пример 1 (1_А и 1_В)
Стадия A: 1-1 (50 г, 285,41 ммоль) растворяли в дихлорметане (500 мл) и добавляли триэтиламин (43,32 г, 428,12 ммоль), 4-диметиламинопиридин (3,49 г, 28,54 ммоль), ди-трет-бутилдикарбонат (68,52 г, 313,96 ммоль) при 0°С. Реакционную систему перемешивали при 25°С в течение 0,5 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении и затем добавляли этилацетат (500 мл). Органическую фазу промывали насыщенным водным раствором хлорида аммония (300 мл × 3) и насыщенным солевым раствором (300 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-2.
Стадия В: диизопропиламин (46,1 г, 456,23 ммоль) растворяли в тетрагидрофуране (250 мл), и к реакционной системе по каплям добавляли н-бутиллитий (2,5 М, 159,68 мл) при -78°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Реакционную систему перемешивали при 0°С в течение получаса, и затем по каплям добавляли в другую трехгорлую колбу, содержащую раствор 1-2 (78,5 г, 285,14 ммоль) и триизопропилборат (80,44 г, 427,72 ммоль) в тетрагидрофуране (750 мл), при 0°С в атмосфере азота, и добавление по каплям завершали в пределах полутора часов. Полученную реакционную систему перемешивали при 0°С в течение 1 ч. Затем к реакционной системе добавляли раствор уксусной кислоты (200 мл) для гашения реакции, разбавляли водой (600 мл) и экстрагировали с помощью этилацетата (500 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида аммония (200 мл × 3) и насыщенным солевым раствором (200 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт суспендировали с ацетонитрилом (100 мл) и водным раствором (500 мл), и осадок на фильтре высушивали с применением масляного насоса с получением 1-3.
Стадия С: 1-3 добавляли к раствору три фторуксусной кислоты (556,25 мл) при 0°С в виде трех частей и реакционную систему перемешивали при 0°С в течение одного часа в атмосфере азота. Затем реакционную систему выливали в ледяную воду (600 мл) с осаждением твердого вещества и получали осадок на фильтре и концентрировали его при пониженном давлении с применением масляного насоса с получением 1-4.
Стадия D: 1-5 (60 г, 212,09 ммоль) и 3-пиридинбороновую кислоту (26,07 г, 212,09 ммоль) растворяли в 1,4-диоксане (600 мл) и воде (120 мл), и затем добавляли дихлорид [1,1-бис(трифенилфосфино)ферроцен]палладия (573,14 мг, 879,39 мкмоль, 44,64 мл) и карбонат калия (1,48 г, 17,59 ммоль). Реакционную систему перемешивали при 90°С в течение 16 ч. в атмосфере азота. После завершения реакции реакционную систему фильтровали и фильтрат экстрагировали с помощью этилацетата (500 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали на колонке с силикагелем (элюент (V/V): петролейный эфир/этилацетат = 10/1-2/1) с получением 1-6.
Стадия Е: 1-6 (20 г, 85,44 ммоль) растворяли в метаноле (200 мл), и добавляли диоксид платины (3.88 г, 17.09 ммоль) и водный раствор хлористоводородной кислоты (1 н., 85,44 мл) при комнатной температуре, и реакционную систему перемешивали при 10°С в течение 16 ч. в атмосфере водорода при давлении 50 фунтов/кв. дюйм. После завершения реакции реакционную систему фильтровали с получением фильтрата, который концентрировали при пониженном давлении с получением 1-7.
Стадия F: 1-7 (23,63 г, 85,43 ммоль) растворяли в метаноле (300 мл) и добавляли N,N-диизопропилэтиламин (33,12 г, 256,29 ммоль, 44,64 мл) и Вос-ангидрид (37,29 г, 170,86 ммоль, 39,25 мл) при комнатной температуре. Реакционную систему перемешивали при комнатной температуре в течение 1 ч. Затем реакционную систему концентрировали при пониженном давлении с удалением растворителя и экстрагировали с помощью этилацетата (300 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 1), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-8.
Стадия G: 1-8 (4 г, 8,79 ммоль) и 4-метоксикарбонилиндол-2-бороновую кислоту (1,93 г, 8,79 ммоль) растворяли в диметиловом эфире этиленгликоля (40 мл) и воде (8 мл) и добавляли дихлорид [1,1-бис(ди-трет-бутилфосфино)ферроцен]палладия (573,14 мг, 879,39 мкмоль, 44,64 мл) и бикарбонат натрия (1,48 г, 17,59 ммоль) при комнатной температуре. Реакционную систему перемешивали при 80°С в течение 16 ч. После завершения реакции к реакционной системе добавляли воду (60 мл) и экстрагировали с помощью этилацетата (50 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 1), высушивали над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали на колонке с силикагелем (элюент (V/V): петролейный эфир/этилацетат = 10/1-3/1) с получением 1-9.
Стадия Н: оксалилхлорид (1,87 г, 14,73 ммоль, 1,29 мл) растворяли в дихлорметане (20 мл) при поддержании температуры на уровне 0°С.Добавляли N,N-диметилформамид (1,61 г, 22,09 ммоль, 1,7 мл) при 0°С в атмосфере азота. Реакционную систему перемешивали при 0°С в течение 0,25 ч. 1-9 (3,2 г, 7,36 ммоль) растворяли в дихлорметане (10 мл) и по каплям добавляли к реакционной системе при поддержании температуры на уровне 0°С. Обеспечивали протекание реакции в реакционной системе при 0-15°С в течение 0,5 ч. После завершения реакции к реакционной системе добавляли раствор ацетата аммония (10%, 150 мл) и перемешивали при 15°С в течение 1 ч. Реакционную систему концентрировали при пониженном давлении с удалением растворителя и затем экстрагировали с помощью этилацетата (60 мл × 2). Органические фазы объединяли, промывали насыщенным хлоридом аммония (30 мл × 2) и насыщенным солевым раствором (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-10.
Стадия I: 1-10 (3,5 г, 7,57 ммоль) растворяли в дихлорметане (50 мл) и добавляли Вос-ангидрид/ди-трет-бутилкарбонат (1,98 г, 9,08 ммоль, 2,09 мл), триэтиламин (1,53 г, 15,13 ммоль, 2,11 мл) и 4-диметиламинопиридин (92,44 мг, 756,70 мкмоль) при перемешивании с поддержанием температуры на уровне 20°С. Реакционную систему перемешивали при 20°С в течение 1 ч. После завершения реакции органическую фазу промывали насыщенным раствором хлорида аммония (150 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-11.
Стадия J: 1-11 (4,3 г, 7,64 ммоль) растворяли в тетрагидрофуране (40 мл) и метаноле (10 мл) и добавляли борогидрид натрия (578,22 мг, 15,28 ммоль) в атмосфере азота при поддержании температуры на уровне 0°С. Обеспечивали протекание реакции в реакционной системе при 0°С в течение 0,5 ч. После завершения реакции к реакционной системе добавляли насыщенный хлорид аммония (80 мл) для гашения реакции и экстрагировали с помощью этилацетата (100 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 1), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-12.
Стадия K: 1-12 (4,0 г, 7,08 ммоль) растворяли в дихлорметане (50 мл) и добавляли триэтиламин (2,15 г, 21,25 ммоль, 2,96 мл) и метансульфонилхлорид (1,62 г, 14,17 ммоль) при 0°С. Реакционную систему перемешивали при 15°С в течение 16 ч. После завершения реакции к реакционной системе добавляли дихлорметан (100 мл). Органическую фазу промывали насыщенным раствором хлорида аммония (100 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-13.
Стадия L: 1-13 (4,0 г, 6,22 ммоль) растворяли в N,N-диметилформамиде (50 мл) и добавляли карбонат натрия (1,32 г, 12,45 ммоль) и N-гидроксифталимид (2,03 г, 12,45 ммоль). Реакционную систему перемешивали при 50°С в течение 16 ч. После завершения реакции реакционную систему промывали этилацетатом (150 мл) и насыщенным солевым раствором (180 мл × 4); высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-14.
Стадия М: 1-14 (4,0 г, 5,64 ммоль) растворяли в метаноле (40 мл) и добавляли гидразингидрат (1,15 г, 22,54 ммоль, 1,12 мл, чистота 98%). Реакционную систему перемешивали при 70°С в течение 2 ч. в атмосфере азота. После завершения реакции реакционную систему концентрировали при пониженном давлении с удалением органического растворителя и полученное твердое вещество очищали посредством препаративной высокоэффективной жидкостной хроматографии (препаративной HPLC) (колонка: Phenomenex Synergi Max-RP (250 мм × 50 мм, 10 мкм); подвижная фаза: вода (0,225% муравьиная кислота)-ацетонитрил; градиент элюирования: 60%-90%, 29 мин.) с получением желтого твердого вещества. Затем твердое вещество разделяли на колонке для хиральной хроматографии (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1%) аммиак в изопропаноле; градиент элюирования: 30%-30%, 2,1 мин.; 300 мин.) с получением 1-15А (время удерживания = 1,704 мин., значение ее (энантиомерный избыток): 100%) и 1-15В (время удерживания = 1,782 мин., значение ее (энантиомерный избыток): 97%).
Стадия N: 1-15А (420 мг, 764,09 мкмоль) растворяли в дихлорметане (6 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную систему перемешивали при 20°С в течение 1 ч. в атмосфере азота. После завершения реакции реакционную систему концентрировали при пониженном давлении с удалением органического растворителя и полученное твердое вещество очищали посредством препаративной HPLC (колонка: Phenomenex Gemini (150 мм × 25 мм, 10 мкм); подвижная фаза: вода (10 мМ бикарбонат аммония)-ацетонитрил; градиент элюирования: 30%-51%, 7 мин.) с получением соединения примера 1А (время удерживания = 6,63 мин., значение ее (энантиомерный избыток): 94%). Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (100 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,80 (dd, J=0,86, 7,58 Гц, 1H), 7,62-7,70 (m, 1H), 7,48-7,54 (m, 2Н), 7,39-7,46 (m, 2Н), 7,31 (t, J=7,83 Гц, 1H), 5,41-5,50 (m, 1Н), 5,26-5,36 (m, 1Н), 3,16 (br t, J=12,90 Гц, 2Н), 2,68-2,86 (m, ЗН), 2,04 (br s, 1H), 1,83-1,94 (m, 1H), 1,68-1,80 (m, 2Н).
Стадия О: 1-15 В (440 мг, 803,45 мкмоль) растворяли в дихлорметане (6 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную систему перемешивали при 20°С в течение 1 ч. в атмосфере азота. После завершения реакции реакционную систему концентрировали при пониженном давлении с удалением органического растворителя и полученное твердое вещество очищали посредством препаративной HPLC (колонка: Phenomenex Gemini (150 мм × 25 мм, 10 мкм); подвижная фаза: вода (10 мМ бикарбонат аммония)-ацетонитрил; градиент элюирования: 30%-51%, 7 мин.) с получением соединения примера 1_В (время удерживания = 5,62 мин., значение ее (энантиомерный избыток): 94%). Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (100 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40%) изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,80 (dd, J=0,86, 7,58 Гц, 1H), 7,64-7,70 (m, 1Н), 7,48-7,53 (m, 2Н), 7,41-7,46 (m, 2Н), 7,31 (t, J=7,83 Гц, 1H), 5,41-5,50 (m, 1H), 5,27-5,35 (m, 1H), 3,13-3,19 (m, 2Н), 2,71-2,84 (m, 3Н), 2,04 (br s, 1H), 1,84-1,91 (m, 1H), 1,71-1,79 (m, 2Н)
Пример 2 (2_А и 2_В)
Синтез осуществляли согласно способу, представленному в примере 1.
В случае примера 2 перед удалением защитной группы Вое соединение разделяли на колонке для хиральной HPLC (разделительная колонка: WHELK-O1 (250 мм × 50 мм, 10 мкм); подвижная фаза: 0,1% аммиак в метаноле; градиент элюирования: 40%-40%, 4 мин.; 260 мин.) с получением двух изомеров с разными конфигурациями: примера 2_АА (время удерживания = 4,453 мин., значение ее (энантиомерный избыток): 100%) и примера 2_ВВ (время удерживания = 4,735 мин., значение ее (энантиомерный избыток): 98%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 2_А (время удерживания = 1,276 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 2_В (время удерживания = 1,632 мин., значение ее (энантиомерный избыток): 98%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralcel OJ-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 2_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,52 (s, 1Н), 7,82 (d, J=7,21 Гц, 1H), 7,60-7,72 (m, 5Н), 7,34 (t, J=7,76 Гц, 1H), 5,43-5,52 (m, 1H), 5,25-5,35 (m, 1Н), 4,32 (br d, J=11,86 Гц, 1H), 3,51 (br d, J=12,72 Гц, 1H), 3,16-3,29 (m, 1H), 1,95-2,21 (m, 4Н), 1,73-1,93 (m, 2Н).
Пример 2_В: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,55 (s, 1Н), 7,82 (d, J=7,34 Гц, 1H), 7,59-7,72 (m, 5Н), 7,34 (t, J=7,83 Гц, 1H), 5,43-5,56 (m, 1H), 5,24-5,36 (m, 1Н), 4,28 (br d, J=11,86 Гц, 1Н), 3,49 (br d, J=11,98 Гц, 1H), 3,11-3,26 (m, 1H), 1,92-2,21 (m, 4H), 1,71-1,90 (m, 2H).
Пример 3
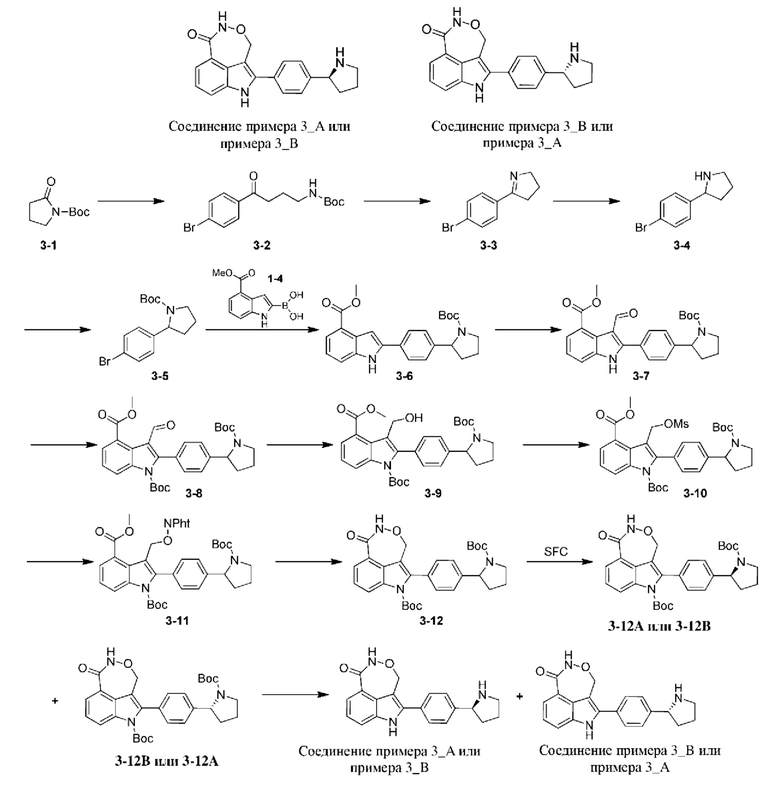
Стадия А: 1,4-дибромбензол (8,92 г, 37,79 ммоль) растворяли в тетрагидрофуране (35,00 мл), и затем по каплям добавляли в трехгорлую колбу, содержащую магниевую стружку (918,56 мг, 37,79 ммоль) и йод (137,03 мг, 539,9 мкмоль), при 70°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Реакционную систему перемешивали при 70°С в течение 1 ч. и затем охлаждали до 20°С. Реакционную систему по каплям добавляли в другую трехгорлую колбу, содержащую раствор 3-1 (5 г, 26,99 ммоль) в тетрагидрофуране (15 мл), при -70°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Полученную реакционную систему перемешивали при -70°С в течение 2 ч. и затем нагревали до 15°С и перемешивали в течение 1 ч. К реакционной системе добавляли насыщенный водный раствор хлорида аммония (60 мл) для гашения реакции и экстрагировали с помощью этилацетата (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент (V/V): петролейный эфир/этилацетат = 30/1-10/1) с получением 3-2.
Стадия В: 3-2 (5 г, 14,61 ммоль) добавляли к трифторуксусной кислоте (25 мл). Реакционную систему перемешивали при 25°С в течение 4 ч. Затем реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении и добавляли воду (40 мл). Смесь доводили до рН=14 с помощью 40% водного раствора гидроксида натрия и осаждали белое твердое вещество. Полученную смесь фильтровали и осадок на фильтре промывали небольшим количеством воды и подвергали ротационному выпариванию с получением 3-3.
Стадия С: 3-3 (2,8 г, 4,97 ммоль) растворяли в метаноле (30 мл) и воде (7 мл). Реакционную систему охлаждали до -41°С и добавляли борогидрид натрия (945,34 мг, 24,99 ммоль). Реакционную систему перемешивали при -41°С в течение 4 ч. Затем к реакционной системе добавляли хлористоводородную кислоту в концентрации 2 моль/л (50 мл) при 0°С для гашения реакции и добавляли этилацетат (100 мл). Органическую фазу промывали водой (50 мл) с последующим разделением жидкостей. Водную фазу доводили до рН=14 с помощью гидроксида натрия в концентрации 4 моль/л и экстрагировали с помощью этилацетата (150 мл × 3). Органические фазы объединяли, промывали водой (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3-4.
Стадия D: 3-4 (3 г, 13,15 ммоль) растворяли в дихлорметане (30 мл) и добавляли триэтиламин (3,99 г, 39,45 ммоль) и ди-трет-бутилдикарбонат (3,16 г, 14,47 ммоль). Реакционную систему перемешивали при 25°С в течение 1 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении и затем добавляли этилацетат (60 мл). Органическую фазу промывали насыщенным водным раствором хлорида аммония (30 мл × 3) и насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3-5.
Согласно способу в примере 1 соединение 3-5 разделяли посредством хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 30%-30%, 2,1 мин.; 85 мин.) на 3-12А (время удерживания = 1,627 мин., значение ее (энантиомерный избыток): 100%) и 3-12В (время удерживания = 1,711 мин., значение ее (энантиомерный избыток): 98%), в которых удаляли защитную группу с получением соединения примера 3А (время удерживания = 0,732 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 3_В (время удерживания = 1,402 мин., значение ее (энантиомерный избыток): 97,32%) соответственно.
Способ измерения значения ее (энантиомерного избытка): разделительная колонка: Chiralcel OJ-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 3_А: 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,76 2,00 (m, 3Н), 2,23 2,34 (m, 1H), 3,07-3,15 (m, 1H), 3,21 (dt, J=10,18, 7,26 Гц, 1H), 4,36 (br t,.7=8,01 Гц, 1H), 5,17-5,27 (m, 1H), 5,36-5,49 (m, 1H), 7,29 (t, J=7,76 Гц, 1H), 7,58 (d, J=0,86 Гц, 4H), 7,64-7,71 (m, 2H), 8,33 (s, 1H), 11,06 (br s, 1H), 11,85 (s, 1H).
Пример 3_В: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,73-1,97 (m, 3H), 2,19-2,35 (m, 1H), 3,04-3,24 (m, 2H), 4,33 (brt, J=8,07 Гц, 1H), 5,14-5,26 (m, 1H), 5,36-5,50 (m, 1H), 7,29 (t, J=7,76 Гц, 1H), 7,58 (d, J=0,98 Гц, 4H), 7,63-7,71 (m, 2H), 8,31 (s, 1H), 11,06 (br s, 1H), 11,83 (s, 1H).
Пример 4 (4_А и 4_В)
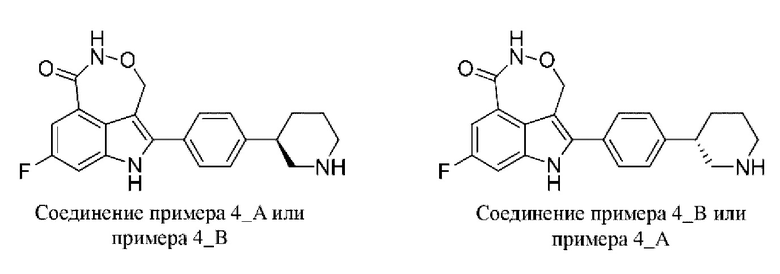
Синтез осуществляли согласно способу, представленному в примере 1.
Соединение примера 4 разделяли на колонке для хиральной HPLC (разделительная колонка: IC (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% аммиак в этаноле; градиент элюирования: 55% 55%, 3,6 мин.; 80 мин.) с получением соединения примера 4_А (время удерживания = 2,353 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 4_В (время удерживания = 3,177 мин., значение ее (энантиомерный избыток): 100%).
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak IC-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05%о диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 4_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,46-7,59 (m, 5Н), 7,38 (br d,J=7,70 Гц, 1Н), 5,38-5,50 (m, 1Н), 5,23-5,34 (m, 1Н), 3,49 (br d, J=11,00 Гц, 2Н), 3,02-3,24 (m, 3Н), 2,12 (br d,J=10,39 Гц, 2Н), 1,79-2,02 (m, 2Н).
Пример 4_В: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,30-7,55 (m, 6Н), 5,38-5,48 (m, 1Н), 5,24-5,34 (m, 1H), 3,11 (br t, J=13,82 Гц, 2Н), 2,57-2,89 (m, 3Н), 2,03 (br d,J=8,80 Гц, 1H), 1,85 (br d, J=10,27 Гц, 1Н), 1,62-1,78 (m, 2Н).
Пример 5 (5_А и 5_В)

Синтез осуществляли согласно способу, представленному в примере 1.
В случае примера 5 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 30%-30%, 6,0 мин.; 350 мин.) с получением двух изомеров с разными конфигурациями: 5_АА (время удерживания = 1,669 мин., значение ее (энантиомерный избыток): 98%) и 5_ВВ (время удерживания = 1,725 мин., значение ее (энантиомерный избыток): 97%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 5_А (время удерживания = 4,468 мин., значение ее (энантиомерный избыток): 97%) и соединения примера 5_В (время удерживания = 3,784 мин., значение ее (энантиомерный избыток): 94%) соответственно. Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak IC-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 5_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,54 (s, 1Н), 7,82 (d, J=7,50 Гц, 1H), 7,69 (d, J=7,63 Гц, 1Н), 7,47-7,59 (m, 1Н), 7,22-7,39 (m, 3Н), 5,26-5,38 (m, 1Н), 5,05-5,17 (m, 1Н), 3,41-3,57 (m, 2Н), 3,00-3,22 (m, 3Н), 2,10(br d, J=13,51 Гц, 2Н), 1,77-2,01 (m, 2Н).
Пример 5_В: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,54 (s, 1Н), 7,78-7,88 (m, 1H), 7,65-7,74 (m, 1Н), 7,51-7,60 (m, 1H), 7,24-7,40 (m, 3Н), 5,27-5,40 (m, 1H), 5,05-5,20 (m, 1Н), 3,42-3,55 (m, 2Н), 2,99-3,23 (m, 3Н), 2,05-2,17 (m, 2Н), 1,79-2,01 (m, 2Н).
Пример 35 (35_А и 35_В)

Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 6 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: WHELK-01 (250 мм × 50 мм, 10 мкм); подвижная фаза: 0,1% аммиак в метаноле; градиент элюирования: 40%-40%, 3,7 мин.; 450 мин.) с получением двух изомеров с разными конфигурациями: примера 6_АА (время удерживания = 2,483 мин., значение ее (энантиомерный избыток): 100%) и примера 6_ВВ (время удерживания = 2,691 мин., значение ее (энантиомерный избыток): 94%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 6_А (время удерживания = 1,955 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 6_В (время удерживания = 3,339 мин., значение ее (энантиомерный избыток): 94%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: OJ-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 30% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 6_А: 1Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,48 (s, 3Н), 1,81 (br s, 1Н), 1,93-2,17 (m, 3Н), 2,94-3,35 (m, 2Н), 5,23 (br dd, J=14,67, 3,55 Гц, 1H), 5,38-5,48 (m, 1H), 7,29 (t, J=7,76 Гц, 1Н), 7,54-7,61 (m, 2Н), 7,62-7,73 (m, 4Н), 8,18-8,33 (m, 1H), 11,07 (s, 1Н), 11,80 (br s, 1Н).
Пример 6_В: 1Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,48 (br d, J=2,69 Гц, 3Н), 1,79 (br s, 1Н) 1,95-2,18 (m, 3Н), 2,93-3,30 (m, 2Н), 5,23 (br dd, J=14,61, 3,12 Гц, 1H), 5,43 (br d, J=14,79 Гц, 1H), 7,19-7,35 (m, 1H), 7,53-7,60 (m, 2H), 7,61-7,77 (m, 4H), 8,19-8,35 (m, 1H), 11,06 (s, 1H), 11,80 (brd, J=4,65 Гц, 1H).
Пример 7 (7_A и 7_B)

Синтез осуществляли согласно способу, представленному в примере 1.
Соединение примера 7 разделяли на колонке для хиральной HPLC (разделительная колонка: IC (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% аммиак в этаноле; градиент элюирования: 50%-50%, 4,3 мин.; 120 мин.) с получением двух изомеров с разными конфигурациями: примера 7_А (время удерживания = 1,889 мин., значение ее (энантиомерный избыток): 100%) и примера 7_В (время удерживания = 2,411 мин., значение ее (энантиомерный избыток): 94%).
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: IC-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 7_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,49-7,64 (m, 2Н), 7,26-7,45 (m, 3Н), 5,40-5,48 (m, 1H), 5,25-5,34 (m, 1Н), 3,37-3,56 (m, 3Н), 2,98-3,24 (m, 2Н), 2,04-2,18 (m, 2Н), 1,81-2,01 (m, 2Н).
Пример 7_В: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 7,43 7,57 (m, 2Н), 7,38 (dd, J=2,14, 8,99 Гц, 1Н), 7,23-7,32 (m, 2Н), 5,39-5,47 (m, 1Н), 5,26-5,35 (m, 1H), 3,02-3,21 (m, 3Н), 2,60-2,82 (m, 2Н), 1,95-2,08 (m, 1Н), 1,64-1,89 (m, 3Н).
Пример 37 (37_А и 8_В)
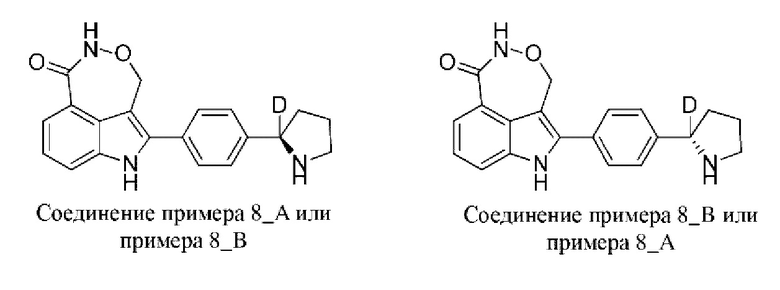
Синтез осуществляли согласно способу, представленному в примере 1.
В случае примера 8 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 30%-30%, 5,0 мин.; 110 мин.) с получением двух изомеров с разными конфигурациями: 8_АА (время удерживания = 1,570 мин., значение ее (энантиомерный избыток): 100%) и 8_ВВ (время удерживания = 1,674 мин., значение ее (энантиомерный избыток): 100%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 8_А (время удерживания = 1,262 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 8_В (время удерживания = 2,511 мин., значение ее (энантиомерный избыток): 100%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: OJ-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 30% метанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 8_А: 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,71 1,80 (m, 1Н), 1,82 1,99 (m, 2H), 2,22-2,31 (m, 1H), 3,05-3,28 (m, 2H), 5,22 (br d, J=14,55 Гц, 1H), 5,43 (br d, J=14,55 Гц, 1H), 7,29 (t, J=1,76 Гц, 1H), 7,53-7,71 (m, 6H), 8,30 (s, 1H), 11,06 (brs, 1H), 11,82 (s, 1H).
Пример 8_В: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,70-1,79 (m, 1H), 1,86-1,97 (m, 2H), 2,21-2,35 (m, 1H), 3,02-3,23 (m, 2H), 5,16-5,27 (m, 1H), 5,37-5,48 (m, 1H), 7,29 (t, J=7,76 Гц, 1H), 7,52-7,72 (m, 6H), 8,29 (s, 1H), 11,06 (br s, 1H), 11,81 (s, 1H).
Пример 9
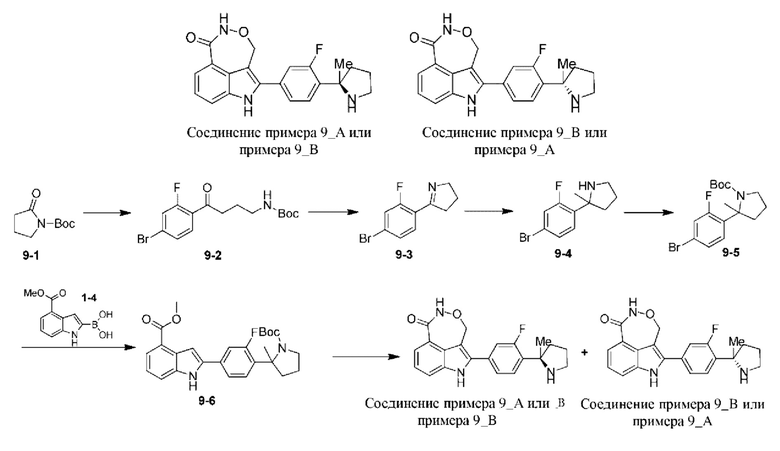
После получения соединения 9-5 согласно способу получения, представленному в примере 3, обеспечивали протекание реакции между 9-5 и 1-4 согласно примеру 1 с получением 9-6, которое разделяли на колонке для хиральной HPLC (разделительная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% аммиак в метаноле; градиент элюирования: 60%-60%, 6,6 мин.; 190 мин.) с получением двух изомеров с разными конфигурациями: 9-6_АА (время удерживания = 0,625 мин., значение ее (энантиомерный избыток): 100%) и 9-6_ВВ (время удерживания = 1,657 мин., значение ее (энантиомерный избыток): 100%), которые подвергали процедуре, схожей с процедурой, представленной в примере 1, с получением соединения примера 9_А (время удерживания = 0,716 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 9_В (время удерживания = 0,559 мин., значение ее (энантиомерный избыток): 100%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% метанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 9_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,51 (s, 1H), 7,83 (d, J=7,46 Гц, 1Н), 7,69 (d, J=8,07 Гц, 1Н), 7,61 (t, J=8,25 Гц, 1Н), 7,41-7,51 (m, 2Н), 7,37 (t, J=7,83 Гц, 1H), 5,44-5,53 (m, 1Н), 5,27-5,38 (m, 1H), 3,52-3,62 (m, 1Н), 3,40-3,50 (m, 1Н), 2,43-2,62 (m, 2Н), 2,18-2,41 (m, 2Н), 1,72 (s, 3Н).
Пример 9_В: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,49 (s, 1H), 7,83 (d, J=7,46 Гц, 1Н), 7,69 (d, J=8,07 Гц, 1Н), 7,61 (t, J=8,19 Гц, 1Н), 7,41-7,52 (m, 2Н), 7,36 (t, J=7,82 Гц, 1H), 5,42-5,58 (m, 1Н), 5,25-5,38 (m, 1H), 3,53-3,63 (m, 1Н), 3,41-3,52 (m, 1Н), 2,43-2,64 (m, 2Н), 2,18-2,42 (m, 2Н), 1,73 (s, 3Н).
Пример 10
Синтез осуществляли согласно способу, представленному в примере 1; однако рацемат в примере 10 получали непосредственно без хирального разделения.
Пример 10: 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,57-1,82 (m, ЗН), 1,93 (br s, 1H), 2,68 (br s, 1H), 2,75-2,91 (m, 2H), 2,99-3,23 (m, 2H), 5,03-5,13 (m, 1H), 5,14-5,28 (m, 1H), 7,24-7,39 (m, 2H), 7,41-7,57 (m, 3H), 8,34 (br s, 1H), 11,27 (br s, 1H), 11,87 (br s, 1H).
Пример 11 (11_А и 11_В)
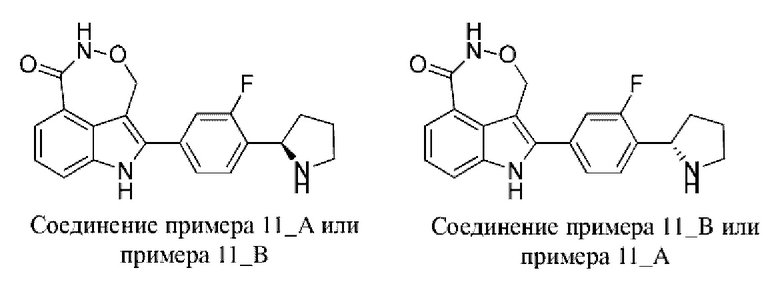
Синтез осуществляли согласно способу, представленному в примере 1.
После удаления защитной группы с помощью трифторуксусной кислоты получали соединение примера 11_А (время удерживания = 2,020 мин., значение ее (энантиомерный избыток): 96,33%) и соединение примера 11_В (время удерживания = 1,402 мин., значение ее (энантиомерный избыток): 97,32%).
Способ измерения значения ее (энантиомерного избытка): разделительная колонка: OJ-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 5%-40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 11_А: 1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,74 2,00 (m, 3Н), 2,23-2,36 (m, 1Н), 3,07-3,22 (m, 2 H), 4,57 (br t, J=7,95 Гц, 1H), 5,17-5,32 (m, 1H), 5,38-5,52 (m, 1H), 7,28-7,52 (m, 3H), 7,63-7,76 (m, 3H), 8,25 (s, 1H), 11,09 (br s, 1H), 11,90 (s, 1H).
Пример 11_B: 1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,68 2,05 (m, 3H), 2,19-2,37 (m, 1H), 3,05-3,22 (m, 2H), 4,42-4,60 (m, 1H), 5,17-5,32 (m, 1H), 5,38-5,51 (m, 1H), 7,29-7,52 (m, 3H), 7,56-7,75 (m, 3H), 8,29 (s, 1H), 11,09 (br s, 1H), 11,92 (s, 1H).
Пример 12

Стадия А: 12-1 (5,0 г, 26,99 ммоль) растворяли в тетрагидрофуране (50 мл) и добавляли гексаметилдисилазид лития (29,69 ммоль, 1 моль/л) при перемешивании с поддержанием температуры на уровне -78°С. Реакционную систему перемешивали при -78°С в течение 0,5 ч. Затем N-фенилбис(трифторметансульфонил)имид (11,57 г, 32,39 ммоль) растворяли в тетрагидрофуране (100 мл) и медленно по каплям добавляли к реакционной системе. Полученную реакционную систему перемешивали при температуре в диапазоне от -78°С до 0°С в течение 2 ч. После завершения реакции к реакционной системе добавляли насыщенный бикарбонат натрия (500 мл) для гашения реакции и экстрагировали с помощью этилацетата (200 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (200 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 12-2.
Стадия В: 12-2 (6,0 г, 18,91 ммоль) растворяли в диоксане (60 мл) и добавляли бис(пинаколато)дибор (4,8 г, 18,91 ммоль), ацетат калия (3,71 г, 37,82 ммоль) и комплекс дихлорида [1,1-бис(трифенилфосфино)ферроцен]палладия с дихлорметаном (1,54 г, 1,89 ммоль) при комнатной температуре. Реакционную систему перемешивали при 80°С в течение 16 ч. После завершения реакции в отношении реакционной системы не проводили дополнительную обработку и получали 12-3, которое непосредственно применяли на следующей стадии.
Стадия С: 12-3 (1,2 г, 3,70 ммоль) и 1-бром-4-йодбензол (4,79 г, 16,94 ммоль) растворяли в диоксане (60 мл) и воде (12 мл) и добавляли комплекс дихлорида [1,1-бис(трифенилфосфино)ферроцен]палладия с дихлорметаном (1,38 г, 1,69 ммоль) и карбонат калия (4,68 г, 33,88 ммоль). Реакционную систему перемешивали при 80°С в течение 5 ч. После завершения реакции к реакционной системе добавляли воду (200 мл) и экстрагировали с помощью этилацетата (100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 1), высушивали над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали на колонке с силикагелем (элюент (V/V): петролейный эфир/этилацетат (объемное соотношение) = 50:1-10:1) с получением 12-4.
Стадия D: 12-4 (1,2 г, 3,70 ммоль) и 4-метоксикарбонилиндол-2-бороновую кислоту (810,59 мг, 3,70 ммоль) растворяли в диметиловом эфире этиленгликоля (15 мл) и воде (3 мл) и добавляли дихлорид [1,1-бис(ди-трет-бутилфосфино)ферроцен]палладия (241,23 мг, 370,13 мкмоль) и бикарбонат натрия (621,89 мг, 7,40 ммоль). Реакционную систему перемешивали при 80°С в течение 16 ч. После завершения реакции к реакционной системе добавляли воду (100 мл) и экстрагировали с помощью этилацетата (100 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 1), высушивали над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали на колонке с силикагелем (элюент (V/V): петролейный эфир/этилацетат (объемное соотношение) = 10:1-2:1) с получением 12-5.
Стадия Е: 12-5 (1,1 г, 32,43 ммоль) растворяли в метаноле (10 мл) и добавляли палладий на углероде (100 мг, чистота 10%) при комнатной температуре. Реакционную систему перемешивали при 25°С в течение 16 ч. в атмосфере водорода при давлении 15 фунтов/кв. дюйм. После завершения реакции реакционную систему фильтровали и концентрировали при пониженном давлении с получением 12-6.
Получение соединения 12-6 осуществляли согласно способу, представленному в примере 1, с получением соединения до удаления защитной группы Boc, которое затем разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в этаноле; градиент элюирования: 35%-35%, 2,3 мин.; 50 мин.) с получением двух изомеров с разными конфигурациями: 12_АА (время удерживания = 2,771 мин., значение ее (энантиомерный избыток): 98%) и 12_ВВ (время удерживания = 2,875 мин., значение ее (энантиомерный избыток): 98%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 12_А (время удерживания = 4,564 мин., значение ее (энантиомерный избыток): 100%о) и соединения примера 12_В (время удерживания = 4,148 мин., значение ее (энантиомерный избыток): 100%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: AS-3 (100 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 30%-45% изопропанол (0,05%) изопропиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 12_А: 1Н ЯМР (400 МГц, дейтерированный метанол) δ 8,56 (s, 1Н), 7.81 (d, J=7,09 Гц, 1H), 7,67 (d, J=7,95 Гц, 1H), 7,48-7,59 (m, 4Н), 7,32 (t, J=7.82 Гц, 1Н), 5,42-5,52 (m, 1Н), 5,25-5,35 (m, 1Н), 3,77 (br s, 1H), 3,60 (br s, 2H), 3,42 (br d,J=7,58 Гц, 1H), 3,28 (br d, J=9,66 Гц, 1H), 2,52 (br s, 1H), 2,08-2,30 (m, 1H).
Пример 12_B: 1H ЯМР (400 МГц, дейтерированный метанол) δ 8,55 (br s, 1H), 7,78-7,85 (m, 1H), 7,67 (d, J=7,58 Гц, 1H), 7,49-7,60 (m, 4H), 7,33 (t, J=7,83 Гц, 1H), 5,41-5,51 (m, 1H), 5,25-5,36 (m, 1H), 3,72-3,83 (m, 1H), 3,58-3,65 (m, 2H), 3,38-3,52 (m, 1H), 3,27 (br s, 1H), 2,53 (br d, J=4,16 Гц, 1H), 2,11-2,26 (m, 1H).
Пример 13 (13_А и 13_В)
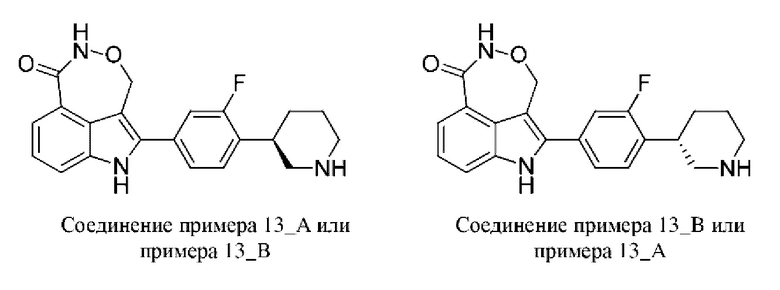
Синтез осуществляли согласно способу, представленному в примере 1.
В случае примера 13 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм х 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 25%-25%, 2,0 мин.; 500 мин.) с получением двух изомеров с разными конфигурациями: 13_АА (время удерживания = 3,090 мин., значение ее (энантиомерный избыток): 85%) и 13_ВВ (время удерживания = 3,357 мин., значение ее (энантиомерный избыток): 86%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 13_А (время удерживания = 1,477 мин., значение ее (энантиомерный избыток): 93%) и соединения примера 13_В (время удерживания = 1,162 мин., значение ее (энантиомерный избыток): 50%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 13_А: Н ЯМР (400 МГц, дейтерированный метанол) δ 7,69 (d, J=7,46 Гц, 1H), 7,55 (d, J=7,95 Гц, 1H), 7,32-7,38 (m, 1H), 7,15-7,22 (m, 3Н), 5,30-5,38 (m, 1Н), 5,16-5,25 (m, 1Н), 3,02 (br d, J=10,03 Гц, 2Н), 2,48-2,70 (m, 3Н), l,88(br d, J=12,10 Гц, 1Н), 1,66-1,78 (m, 3Н).
Пример 13_В: Н ЯМР (400 МГц, дейтерированный метанол) δ 7,69 (d, J=6,85 Гц, 1H), 7,57 (d, J=7,95 Гц, 1H), 7,36-7,43 (m, 1H), 7,18-7,27 (m, 3Н), 5,30-5,38 (m, 1H), 5,16-5,24 (m, 1H), 3,15 (br t, J=11,92 Гц, 3Н), 2,68-2,88 (m, 2H), 1,83-1,96 (m, 2H), 1,69-1,78 (m, 2H).
Пример 14 (14_A и 14_В)
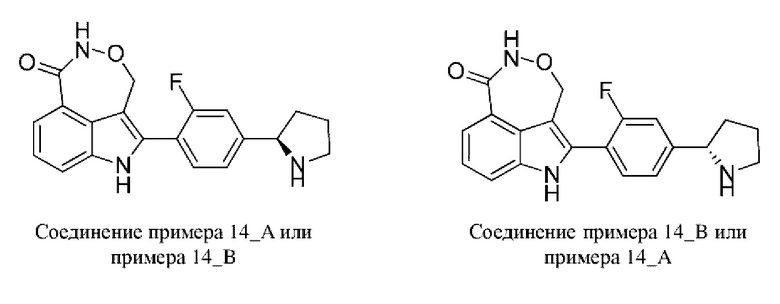
Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 14 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: Whelk-Ol (250 мм × 50 мм, 10 мкм); подвижная фаза: 0,1% аммиак в метаноле; градиент элюирования: 40%-40%, 3,6 мин.; 150 мин.) с получением двух изомеров с разными конфигурациями: 14_АА (время удерживания = 4,092 мин., значение ее (энантиомерный избыток): 100%) и 14_ВВ (время удерживания = 4,411 мин., значение ее (энантиомерный избыток): 98%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 14_А (время удерживания = 1,489 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 14_В (время удерживания = 2,700 мин., значение ее (энантиомерный избыток): 91%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 14_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,82 (s, 1H), 11,25-10,87 (m, 1Н), 8,30 (s, 1Н), 7,74-7,65 (m, 2Н), 7,60-7,54 (m, 1H), 7,52-7,45 (m, 1H), 7,44-7,39 (m, 1H), 7,36-7,28 (m, 1Н), 5,30-5,17 (m, 1H), 5,13-4,99 (m, 1H), 4,37 (br t, J=7,9 Гц, 1H), 3,21-3,07 (m, 2H), 2,34-2,25 (m, 1H), 1,97-1,72 (m, 3H).
Пример 14_B: H ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,80-11,74 (m, 1H), 11,15-11,03 (m, 1H), 8,26 (s, 1H), 7,73-7,65 (m, 2H), 7,58-7,52 (m, 1H), 7,48-7,38 (m, 2H), 7,31 (s, 1H), 5,28-5,18 (m, 1H), 5,11-5,02 (m, 1H), 4,32-4,27 (m, 1H), 3,15-3,10 (m, 1H), 3,08-3,03 (m, 1H), 2,30-2,22 (m, 1H), 1,92-1,80 (m, 2H), 1,72-1,62 (m, 1H).
Пример 15 (15_А и 15_В)
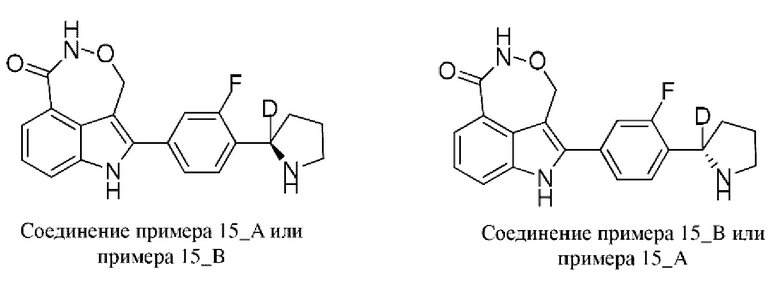
Синтез осуществляли согласно способу, представленному в примере 8.
В случае примера 15 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 20%-20%, 3,1 мин.; 250 мин.) с получением двух изомеров с разными конфигурациями: 15_АА (время удерживания = 3,191 мин., значение ее (энантиомерный избыток): 100%) и 15_ВВ (время удерживания = 3,511 мин., значение ее (энантиомерный избыток): 97%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 15_А (время удерживания = 2,28 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 15_В (время удерживания = 2,101 мин., значение ее (энантиомерный избыток): 87%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak OD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 5%-40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 15_А: Н ЯМР (400 МГц, дейтерированный диметилсульф оксид) δ ppm 1,64 (br s, 1Н), 1,75-1,89 (m, 2Н), 2,17-2,30 (m, 1H), 3,07 (br d, J=17,69 Гц, 2Н), 5,14-5,32 (m, 1H), 5,37-5,52 (m, 1H), 7,31 (t, J=7,72 Гц, 1Н), 7,39 (br s, 2Н), 7,65 (d, J=8,03 Гц, 1Н), 7,68-7,75 (m, 2Н), 8,23 (br s, 1Н), 11,08 (s, 1H), 11,85 (br s, 1H).
Пример 15_B: H ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,66 (br s, 1H), 1,86 (br s, 2H), 2,25 (br s, 1H), 3,09 (br s, 2H), 5,24 (br d, J=14,05 Гц, 1H), 5,39-5,49 (m, 1H), 7,31 (t, J=7,78 Гц, 1H), 7,39 (br s, 2H), 7,66 (d, J=8,03 Гц, 1H), 7,69 7,74 (m, 2H), 8,16 (br s, 1H), 11,08 (s, 1H), 11,83 (s, 1H).
Пример 16 (16_A и 16_В)

Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 16 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD (250 мм × 30 мм, 10 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 50%-50%, 3,7 мин.; 52 мин.) с получением двух изомеров с разными конфигурациями: 16_АА (время удерживания = 0,567 мин., значение ее (энантиомерный избыток): 100%) и 16_ВВ (время удерживания = 0,835 мин., значение ее (энантиомерный избыток): 99%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 16_А (время удерживания = 0,702 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 16_В (время удерживания = 1,301 мин., значение ее (энантиомерный избыток): 100%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Amycoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 60% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 16_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11.98 (s, 1H), 11,11 (s, 1Н), 8,79-8,73 (m, 1H), 8,04-7,98 (m, 1Н), 7,74-7,66 (m, 3Н), 7,33 (s, 1H), 5,47 (d, J=14,7 Гц, 1Н), 5,31-5,19 (m, 1Н), 4,56-4,46 (m, 1H), 3,23-3,09 (m, 3Н), 1,95-1,83 (m, 3Н).
Пример 16_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11.99 (s, 1H), 11,23-11,04 (m, 1Н), 8,77 (s, 1Н), 8,27-8,17 (m, 1H), 8,08-7,99 (m, 1H), 7,73-7,66 (m, 3Н), 7,34 (t, J=7,8 Гц, 1Н), 5,53-5,42 (m, 1H), 5,29-5,20 (m, 1H), 4,64-4,52 (m, 1H), 3,27-3,14 (m, 3Н), 1,86 (br s, 3Н).
Пример 17 (17_А и 17_В)
Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 17 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: WHELK-01 (250 мм × 50 мм, 10 мкм); подвижная фаза: 0,1%) аммиак в этаноле; градиент элюирования: 45%-45%, 3,2 мин.; 150 мин.) с получением двух изомеров с разными конфигурациями: 17_АА (время удерживания = 2,145 мин., значение ее (энантиомерный избыток): 100%) и 17_ВВ (время удерживания = 2,396 мин., значение ее (энантиомерный избыток): 93%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 17_А (время удерживания = 0,622 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 17_В (время удерживания = 1,114 мин., значение ее (энантиомерный избыток): 95%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak OD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 17_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,96 (s, 1Н), 11,16 11,06 (m, 1Н), 8,31 8,17 (m, 1Н), 7,69 (dd, J=7,6, 15,4 Гц, 2Н), 7,37-7,28 (m, 3Н), 5,51-5,40 (m, 1Н), 5,31-5,19 (m, 1Н), 4,67-4,56 (m, 1H), 3,17-3,11 (m, 1H), 3,09-3,03 (m, 1Н), 2,28-2,18 (m, 1Н), 2,03-1,82 (m, 3Н).
Пример 17_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,94-11,87 (m, 1H), 11,14-11,05 (m, 1Н), 8,24-8,21 (m, 1Н), 7,74-7,63 (m, 2Н), 7,37-7,25 (m, 3Н), 5,44 (s, 1H), 5,30-5,21 (m, 1Н), 4,54-4,47 (m, 1Н), 3,10-3,05 (m, 1H), 2,96-2,91 (m, 1H), 2,15 (br s, 1H), 1,98-1,77 (m, 3Н).
Пример 18
Синтез осуществляли согласно способу, представленному в примере 12.
Пример 18: Н ЯМР (400 МГц, дейтерированный метанол) δ 8,53 (s, 1Н), 7,79-7,89 (m, 1H), 7,57-7,73 (m, 2Н), 7,23-7,50 (m, 3Н), 6,54 (br s, 1H), 5,41-5,54 (m, 1H), 5,25-5,38 (m, 1Н), 4,51 (br s, 2Н), 4,30 (br d, J=2,08 Гц, 2H).
Пример 19 (19_А и 19_В)
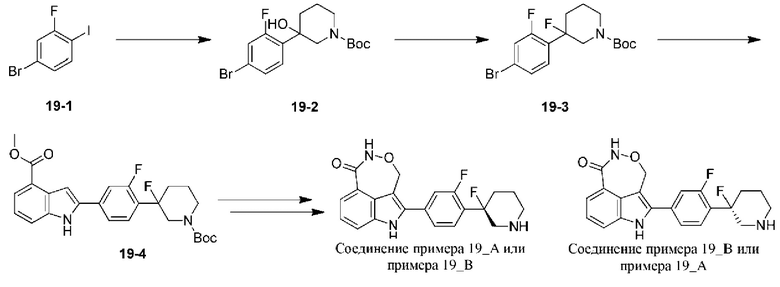
Стадия А: 19-1 (10 г, 33,23 ммоль) растворяли в тетрагидрофуране (100,00 мл) и к реакционной системе по каплям добавляли н-бутиллитий (2,5 М, 13,29 мл) при -78°С в атмосфере азота и добавление по каплям завершали в пределах получаса. Раствор N-трет-бутоксикарбонил-3-пиперидона (6,62 г, 33,23 ммоль) в тетрагидрофуране (20 мл) добавляли по каплям к реакционной системе при -78°С в атмосфере азота. Полученную реакционную систему перемешивали при -78°С в течение 1,5 ч. Затем к реакционной системе добавляли насыщенный водный раствор хлорида аммония (100 мл) для гашения реакции и экстрагировали с помощью этилацетата (100 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент: петролейный эфир / этилацетат = 15/13/1) с получением 19-2.
Стадия В: 19-2 (9,56 г, 22,83 ммоль) растворяли в дихлорметане (100 мл) и добавляли трифторид диэтиламиносеры (18,40 г, 114,14 ммоль) к реакционной системе при -78°С в атмосфере азота. Реакционную систему перемешивали при -78°С в течение 2 ч. Затем к реакционной системе добавляли водный раствор бикарбоната натрия (10 мл, рН=7-8) для гашения реакции и экстрагировали с помощью этилацетата (10 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем (элюент: петролейный эфир / этилацетат = 30/1-5/1) с получением 19-3.
Синтез 19-4 и последующих соединений осуществляли согласно примеру 1.
В случае примера 19 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в этаноле; градиент элюирования: 25%-25%, 2,7 мин.; 570 мин.) с получением двух изомеров с разными конфигурациями: 19_АА (время удерживания = 1,400 мин., значение ее (энантиомерный избыток): 100%) и 19_ВВ (время удерживания = 1,489 мин., значение ее (энантиомерный избыток): 91,2%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 19_А (время удерживания = 0,901 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 19_В (время удерживания = 1,325 мин., значение ее (энантиомерный избыток): 92%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 19_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 11,92 (s, 1H), 11,09 (s, 1Н), 8,24 (s, 1H), 7,72-7,67 (m, 1Н), 7,65-7,64 (m, 2Н), 7,474-7,465 (m, 2Н), 7,34-7,32 (m, 1Н), 5,46-5,42 (d, J=14,80 Гц, 1H), 5,28-5,25 (d, J=14,80 Гц, 1Н), 3,29-3,22 (dd, J=23,20 Гц, 4,40 Гц, 1Н), 3,08-3,04 (d, J=12,8 Гц, 1Н), 2,73 (s, 1H), 2,51-2,11 (m, 1Н), 1,68-1,64 (m, 1Н).
Пример 19_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 11,90 (s, 1H), 11,09 (s, 1Н), 8,24 (s, 1H), 7,71-7,69 (m, 1Н), 7,67-7,64 (m, 2Н), 7,463-7,457 (m, 2Н), 7,34-7,32 (m, 1Н), 5,45-5,42 (d, J=14,80 Гц, 1H), 5,28-5,25 (d, J=14,80 Гц, 1H), 3,22-3,15 (m, 2H), 3,03-3,00 (m, 2H), 2,67 (s, 1H), 2,50-2,09 (m, 2H), 1,83 1,79 (m, 1H), 1,63-1,60 (m, 1H).
Пример 20 (20_А и 20_В)

Соединение примера 11_А (50 мг, 142,3 мкмоль) и 2-бромэтанол (21,34 мг, 170,76 мкмоль) растворяли в N,N-диметилформамиде и затем добавляли карбонат калия (23,60 мг, 0,17 ммоль). Полученную реакционную систему перемешивали при 60°С в течение 16 ч. После выявления завершения реакции реакционную систему фильтровали с удалением нерастворимого материала и оставшуюся жидкость высушивали посредством ротационного выпаривания и подвергали препаративному разделению (колонка: Phenomenex Synergi С18 (150 мкм × 25 мкм, 10 мкм); подвижная фаза: вода (0,225% муравьиная кислота)-ацетонитрил; В%: 15%-45%, 9 мин.) с получением соединения примера 20_А (время удерживания = 2,523 мин., значение ее (энантиомерный избыток): 99%). Соединение примера 20_В (время удерживания = 2,130 мин., значение ее (энантиомерный избыток): 99%) может быть получено таким же путем.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Cellucoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 5%-40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 20_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,91-11,77 (m, 1Н), 11,08 (br s, 1Н), 7,75-7,63 (m, 3Н), 7,43-7,28 (m, 3Н), 5,50-5,37 (m, 1H), 5,30-5,19 (m, 1Н), 4,50-4,38 (m, 1H), 3,74-3,65 (m, 1Н), 3,51-3,43 (m, 1Н), 3,40-3,23 (m, 1Н), 3,16-2,94 (m, 2Н), 2,31-2,13 (m, 2Н), 2,04-1,71 (m, 3Н), 1,71-1,58 (m, 1Н).
Пример 20_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=11,84 (s, 1H), 11,42-10,86 (m, 1Н), 8,22 (s, 1Н), 7,77-7,62 (m, 3Н), 7,46-7,24 (m, 3Н), 5,51-5,37 (m, 1H), 5,34-5,18 (m, 1H), 3,67 (s, 1H), 3,47 (br d, J=3,2 Гц, 2Н), 3,33 (br s, 1H), 2,37-2,14 (m, 5H), 1,88-1,79 (m, 2H), 1,58-1,49 (m, 1H).
Пример 21 (21_А и 21_В)
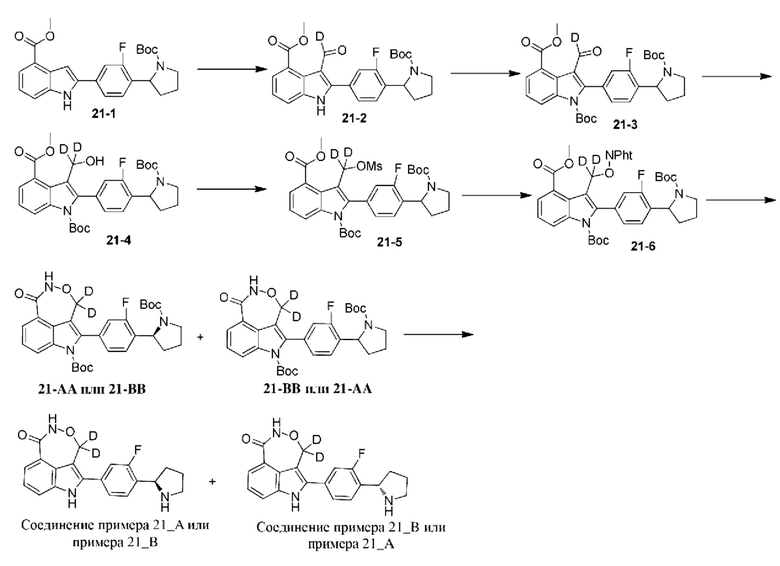
Стадия А: оксалилхлорид (0,579 г, 4,56 ммоль) добавляли к дихлорметану (15 мл) и медленно по каплям добавляли дейтерированный N,N-диметилформамид (0,5 г, 6,84 ммоль) при 0°С в атмосфере азота. Реакционную систему перемешивали при 0°С в течение 15 мин. 21-1 (1 г, 2,28 ммоль) затем растворяли в дихлорметане (5 мл) и добавляли к реакционной системе при 0°С. Реакционную систему перемешивали при 15°С в течение 0,5 ч. После выявления завершения реакции к реакционной системе добавляли 10% водный раствор ацетата аммония (30 мл) и тетрагидрофуран (20 мл) для гашения реакции. Органический растворитель удаляли посредством ротационного выпаривания, и остальную часть экстрагировали с помощью этилацетата (30 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида аммония (30 мл × 3) и насыщенным солевым раствором (30 мл × 3); высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 21-2.
Стадия В: 21-2 (2,15 г, 5,08 ммоль) растворяли в дихлорметане (10 мл) и добавляли триэтиламин (0,65 г, 6,42 ммоль), ди-трет-бутилдикарбонат (0,7 г, 3,21 ммоль) и 4-диметиламинопиридин (26 мг, 0,214 ммоль). Реакционную систему перемешивали при 15°С в течение 1 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении и затем добавляли этилацетат (60 мл). Органическую фазу промывали насыщенным водным раствором хлорида аммония (30 мл × 3) и насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 21-3.
Стадия С: 21-3 (1,2 г, 2,11 ммоль) растворяли в тетрагидрофуране (8 мл) и метаноле (2 мл). Реакционную систему охлаждали до 0°С и добавляли дейтерированный борогидрид натрия (120 мг, 3,17 ммоль). Реакционную систему перемешивали при 0°С в течение 0,5 ч. К реакционной системе добавляли насыщенный водный раствор хлорида аммония (30 мл) для гашения реакции и экстрагировали с помощью этилацетата (30 мл × 2). Органические фазы объединяли, промывали водой (30 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 21-4.
Можно ссылаться на способ, представленный в примере 3, для получения из соединения 21-4 двух изомеров с разными конфигурациями: 21_АА (время удерживания = 1,489 мин., значение ее (энантиомерный избыток): 100%) и 21_ВВ (время удерживания = 1,558 мин., значение ее (энантиомерный избыток): 97%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 21_А (время удерживания = 1,857 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 21_В (время удерживания = 2,663 мин., значение ее (энантиомерный избыток): 97%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak IG-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% метанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 21_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,58-1,74 (m, 1Н), 1,85 (br dd, J=13,63, 7,27 Гц, 2Н), 2,25 (td, J=12,17, 7,09 Гц, 1H), 2,99-3,16 (m, 2Н), 4,38^,53 (m, 1Н), 7,24-7,48 (m, 3Н), 7,60-7,80 (m, 3Н), 8,22 (br s, 1H), 11,07 (br s, 1H), 11,85 (br s, 1H).
Пример 21_B: H ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,61-2,01 (m, 3Н), 2,21-2,35 (m, 1H), 3,04-3,22 (m, 2H), 4,54 (br s, 1H), 7,27-7,47 (m, 3H), 7,60-7,81 (m, 3H), 8,26 (br s, 1H), 11,08 (br s, 1H), 11,89 (br s, 1H).
Пример 22 (22_А и 22_В)
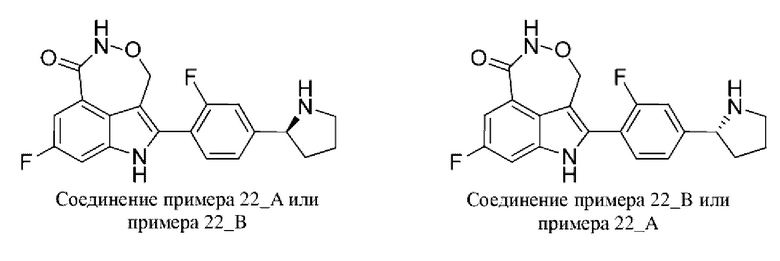
Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 22 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: WHELK-O1 (250 мм × 50 мм, 10 мкм); подвижная фаза: 0,1% аммиак в этаноле; градиент элюирования: 40%-40%, 2,8 мин.; 120 мин.) с получением двух изомеров с разными конфигурациями: 22_АА (время удерживания = 1,411 мин., значение ее (энантиомерный избыток): 100%) и 22_ВВ (время удерживания = 1,613 мин., значение ее (энантиомерный избыток): 99%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 22_А (время удерживания = 1,523 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 22_В (время удерживания = 3,001 мин., значение ее (энантиомерный избыток): 100%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% метанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 22_А: Н ЯМР (400 МГц, дейтерированный метанол) δ ppm 8,49 (s, 1H), 7,63-7,61 (m, 1H), 7,55-7,53 (m, 1H), 7,473-7,468 (m, 2Н), 7,45-7,38 (m, 1H), 5,32-5,28 (d, J=14,4 Гц, 1Н), 5,11-5,08 (d, J=14,4 Гц, 1H), 4,70-4,68 (m, 1H), 3,50-3,44 (m, 2Н), 2,57-2,51 (m, 1H), 2,26-2,18 (m, 3Н).
Пример 22_В: Н ЯМР (400 МГц, дейтерированный метанол) δ ppm 8,53 (s, 1H), 7,63-7,57 (m, 1H), 7,553-7,547 (m, 1Н), 7,49-7,47 (m, 2Н), 7,43-7,40 (m, 1Н), 5,34 5,31 (d, J=14,8 Гц, 1Н), 5,14-5,10 (d, J=14,8 Гц, 1H), 4,70-4,66 (m, 1H), 3,49-3,44 (m, 2Н), 2,55 (s, 1H), 2,30-2,22 (m, 3Н).
Пример 23 (23_А и 23_В)

Синтез осуществляли согласно способу, представленному в примере 21.
В случае примера 23 после удаления защитной группы с помощью трифторуксусной кислоты получали соединение примера 23_А (время удерживания = 3,712 мин., значение ее (энантиомерный избыток): 99%) и соединение примера 23_В (время удерживания = 3,397 мин., значение ее (энантиомерный избыток): 97%).
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Cellucoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 10%-40% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 23_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,50 (s, 3Н), 1,68-1,79 (m, 1Н), 1,87-1,98 (m, 1Н), 2,07-2,18 (m, 2Н), 2,91-3,01 (m, 1Н), 3,13-3,28 (m, 1Н), 7,22-7,49 (m, 3Н), 7,61-7,82 (m, 3Н), 8,20 (s, 1H), 11,07 (s, 1H), 11,85 (s, 1Н).
Пример 23_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,50 (s, 3Н), 1,69-1,81 (m, 1Н), 1,88-1,98 (m, 1Н), 2,08-2,18 (m, 2Н), 2,89-3,06 (m, 1Н), 3,14-3,31 (m, 1Н), 7,25-7,48 (m, 3Н), 7,62-7,82 (m, 3Н), 8,23 (s, 1H), 11,08 (s, 1H), 11,87 (s, 1Н).
Пример 24 (24_А и 24_В)

Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 24 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 30%-30%, 1,5 мин.; 45 мин.) с получением двух изомеров с разными конфигурациями: 24_АА (время удерживания = 1,474 мин., значение ее (энантиомерный избыток): 99%) и 24_ВВ (время удерживания = 1,560 мин., значение ее (энантиомерный избыток): 94%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 24_А (время удерживания = 2,298 мин., значение ее (энантиомерный избыток): 99%) и соединения примера 24_В (время удерживания = 2,115 мин., значение ее (энантиомерный избыток): 88%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Cellucoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 5%-40% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 24_А: Н ЯМР (400 МГц, дейтерированный метанол) δ 8,54 (br s, 1Н), 7,60-7,70 (m, 4H), 7,54 (dd, J=2,26, 10,54 Гц, 1H), 7,40 (dd, J=2,26, 9,03 Гц, 1H), 5,40-5,55 (m, 1H), 5,23-5,37 (m, 1H), 4,70 (br dd, J=6,90, 9,54 Гц, 1H), 3,40-3,61 (m, 2H), 2,48-2,64 (m, 1H), 2,14-2,43 (m, 3H).
Пример 24_B: H ЯМР (400 МГц, дейтерированный метанол) δ 8,52 (br s, 1H), 7,63 (q, J=8,41 Гц, 4H), 7,53 (dd, J=2,20, 10,48 Гц, 1H), 7,38 (dd, J=2,26, 9,03 Гц, 1Н), 5,38-5,53 (m, 1H), 5,24-5,35 (m, 1H), 4,71 (br dd, J=6,90, 9,41 Гц, 1H), 3,43-3,61 (m, 2H), 2,48-2,62 (m, 1H), 2,18-2,39 (m, 3H).
Пример 25 (25_А и 25_В)

Стадия А: 25-1 (25 г, 129,42 ммоль) растворяли в дихлорметане (250 мл) и добавляли триэтиламин (26,19 г, 258,83 ммоль), 4-диметиламинопиридин (3,16 г, 25,88 ммоль), ди-шреш-бутилдикарбонат (31,07 г, 142,36 ммоль) при 0°С. Реакционную систему перемешивали при 25°С в течение 2 ч. Реакционную систему промывали насыщенным водным раствором хлорида аммония (80 мл × 3) и насыщенным солевым раствором (50 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-2.
Стадия В: диизопропиламин (13,80 г, 136,38 ммоль) растворяли в тетрагидрофуране (60 мл), и к реакционной системе по каплям добавляли н-бутиллитий (2,5 М, 47,73 мл) при -78°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Реакционную систему перемешивали при 0°С в течение получаса и затем по каплям добавляли в другую трехгорлую колбу, содержащую раствор 25-2 (25 г, 285,14 ммоль) и триизопропилбората (24,05 г, 127,86 ммоль) в тетрагидрофуране (200 мл), при 0°С в атмосфере азота, и добавление по каплям быстро завершали при 0°С. Полученную реакционную систему перемешивали при 0°С в течение 1 ч. Затем к реакционной системе добавляли раствор уксусной кислоты (50 мл) для гашения реакции, разбавляли водой (60 мл) и экстрагировали с помощью этилацетата (60 мл × 3). Органические фазы объединяли, промывали насыщенным водным раствором хлорида аммония (50 мл × 3) и насыщенным солевым раствором (40 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт суспендировали с ацетонитрилом (100 мл) и водным раствором (300 мл) и осадок на фильтре высушивали с применением масляного насоса с получением 25-3.
Стадия С: 25-3 (45 г, 133,49 ммоль) добавляли к раствору трифторуксусной кислоты (200 мл) при 0°С в виде трех частей и реакционную систему перемешивали при 0°С в течение 1 ч. в атмосфере азота. Затем реакционную систему выливали в ледяную воду (300 мл) с осаждением твердого вещества и получали осадок на фильтре и концентрировали его при пониженном давлении с применением масляного насоса с получением 25-4.
Стадия D: 25-5 (25 г, 105,98 ммоль) растворяли в тетрагидрофуране (100,00 мл) и затем по каплям добавляли в трехгорлую колбу, содержащую магниевую стружку (2,58 г, 105,98 ммоль) и йод (489,05 мг, 1,93 мкмоль) при 70°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Реакционную систему перемешивали при 70°С в течение 1 ч. и затем охлаждали до 20°С. Реакционную систему по каплям добавляли в другую трехгорлую колбу, содержащую раствор трет-бутил-2-оксопирролидин-1-карбоксилата (17,84 г, 96,34 ммоль) в тетрагидрофуране (150 мл) при -70°С в атмосфере азота, и добавление по каплям завершали в пределах получаса. Полученную реакционную систему перемешивали при -70°С в течение 2 ч. и затем нагревали до 10°С и перемешивали в течение 1 ч. К реакционной системе добавляли насыщенный водный раствор хлорида аммония (60 мл) для гашения реакции и экстрагировали с помощью этилацетата (60 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке с силикагелем с получением 25-6.
Стадия Е: 25-6 (50 г, 146,10 ммоль) добавляли к трифторуксусной кислоте (250 мл) при 0°С. Реакционную систему перемешивали при 15°С в течение 12 ч. Реакционную систему доводили до значения рН=14 с помощью 40% водного раствора гидроксида натрия и осаждали желтое твердое вещество. Полученную смесь фильтровали и осадок на фильтре промывали небольшим количеством воды и подвергали ротационному выпариванию с получением 25-7.
Стадия F: 25-7 (15 г, 66,94 ммоль) растворяли в тетрагидрофуране (150 мл). Реакционную систему охлаждали до -78°С в атмосфере азота, и к реакционной системе по каплям добавляли диэтилэфират три фторида бора (19 г, 133,87 ммоль), и добавление по каплям завершали в пределах получаса. Реакционную систему перемешивали при -78°С в течение получаса. Затем к реакционной системе по каплям добавляли раствор метиллития (1,6 М, 83,67 мл). Реакционную систему медленно нагревали до 78°С и перемешивали в течение 19,5 ч. Реакционную систему охлаждали до комнатной температуры, добавляли насыщенный водный раствор бикарбоната натрия (100 мл) для гашения реакции, добавляли воду (30 мл) и экстрагировали с помощью этилацетата (80 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-8.
Стадия G: 25-8 (16 г, 66,63 ммоль) растворяли в дихлорметане (150 мл) и добавляли триэтиламин (20,23 г, 199,88 ммоль) при 0°С с последующим добавлением ди-трет-бутилдикарбоната (29,08 г, 133,26 ммоль). Реакционную систему перемешивали при 15°С в течение 1 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении, добавляли насыщенный водный раствор хлорида аммония (60 мл) и экстрагировали с помощью этилацетата (80 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-9.
Стадия Н: 25-9 (9 г, 26,45 ммоль) и 25-4 (7,52 г, 31,74 ммоль) растворяли в диметиловом эфире этиленгликоля (100 мл) и воде (20 мл) и добавляли бикарбонат натрия (6,67 г, 79,35 ммоль) и дихлорид [1,1-бис(ди-трет-бутилфосфино)ферроцен]палладия (1,72 г, 2,65 ммоль). После трехкратной продувки азотом реакционную систему перемешивали при 80°С в течение 12 ч. в атмосфере азота. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении, добавляли насыщенный солевой раствор (60 мл) и экстрагировали с помощью этилацетата (60 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали на колонке с силикагелем с получением 25-10.
Стадия I: оксалилхлорид (5,61 г, 44,20 ммоль) добавляли к дихлорметану (150 мл) и медленно по каплям добавляли N,N-диметилформамид (4,85 г, 66,30 ммоль) при 0°С в атмосфере азота. Реакционную систему перемешивали при 0°С в течение 15 мин. Затем 25-10 (10 г, 22,10 ммоль) растворяли в дихлорметане (50 мл) и добавляли к реакционной системе при 0°С. Реакционную систему перемешивали при 15°С в течение 0,5 ч. К реакционной системе добавляли 10% водный раствор ацетата аммония (100 мл) и тетрагидрофуран (100 мл) для гашения реакции и экстрагировали с помощью этилацетата (45 мл × 2). Органические фазы объединяли, промывали насыщенным водным раствором хлорида аммония (50 мл × 3) и насыщенным солевым раствором (50 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-11.
Стадия J: 25-11 (10,62 г, 22,10 ммоль) растворяли в дихлорметане (80 мл) и добавляли триэтиламин (6,71 г, 66,30 ммоль), ди-трет-бутилдикарбонат (9,65 г, 44,20 ммоль) и 4-диметиламинопиридин (810,01 мг, 6,63 ммоль) при 0°С. Реакционную систему перемешивали при 15°С в течение 1 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении, добавляли насыщенный водный раствор хлорида аммония (60 мл) и экстрагировали с помощью этилацетата (60 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-12.
Стадия К: 25-12 (12,75 г, 21,96 ммоль) растворяли в тетрагидрофуране (100 мл) и метаноле (25 мл). Реакционную систему охлаждали до 0°С и добавляли борогидрид натрия (1,25 г, 32,94 ммоль). Реакционную систему перемешивали при 0°С в течение 40 мин. К реакционной системе добавляли насыщенный водный раствор хлорида аммония (80 мл) для гашения реакции и экстрагировали с помощью этилацетата (80 мл × 2). Органические фазы объединяли, промывали водой (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-13.
Стадия L: 25-13 (12,79 г, 21,95 ммоль) растворяли в дихлорметане (150 мл) и добавляли триэтиламин (4,44 г, 43,90 ммоль) с последующим добавлением метансульфонилхлорида (3,02 г, 26,34 ммоль) при 0°С в атмосфере азота. Реакционную систему перемешивали при 0°С в течение 1 ч. Реакционную систему концентрировали посредством ротационного выпаривания при пониженном давлении и затем добавляли этилацетат (80 мл). Органическую фазу промывали насыщенным водным раствором хлорида аммония (30 мл × 2) и насыщенным солевым раствором (20 мл × 2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 25-14.
Стадия М: 25-14 (14,45 г, 21,87 ммоль) растворяли в N,N-диметилформамиде (150 мл) и добавляли карбонат натрия (4,64 г, 43,74 ммоль) и N-гидроксифталимид (5,35 г, 32,80 ммоль). Реакционную систему перемешивали при 65°С в течение 12 ч. К реакционной системе добавляли водный раствор (60 мл) и экстрагировали с помощью этилацетата (80 мл × 2). Органическую фазу промывали насыщенным водным раствором хлорида аммония (50 мл × 3) и насыщенным солевым раствором (50 мл × 3), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали на колонке с силикагелем с получением 25-15.
Стадия N: 25-15 (15 г, 20,61 ммоль) растворяли в метаноле (200 мл) и добавляли 98% гидразингидрат (3,10 г, 61,83 ммоль). Реакционную систему перемешивали при 65°С в течение 2 ч. в атмосфере азота. Реакционную систему фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали на колонке с силикагелем с получением 25-16.
Стадия О: соединение 25-16 разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 25%-25%, 2,7 мин.; 400 мин.) с получением двух изомеров с разными конфигурациями: 25_АА (время удерживания = 2,161 мин., значение ее (энантиомерный избыток): 100%) и 25_ВВ (время удерживания = 2,353 мин., значение ее (энантиомерный избыток): 97%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 25_А (время=3,461 мин., значение ее (энантиомерный избыток): 98%) и соединения примера 25_В (время=3,128 мин., значение ее (энантиомерный избыток): 90%) соответственно.
Способ измерения значения ее (энантиомерного избытка): разделительная колонка: Chiralcel Cellucoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 10%-40% изопропанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 25_А: 1Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=12.00 (br s, 1H), 11,30 (br s, 1Н), 8,23 (br s, 1Н), 7,62 (br d, J=7,75 Гц, 4H), 7,45 (br d, J=9,13 Гц, 2H), 5,44 (br d, J=14,51 Гц, 1H), 5,14-5,31 (m, 1H), 2,99-3,42 (m, 2H), 1,89-2,23 (m, 4H), 1,53 (br s, 3H).
Пример 25_B: 1H ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ=12.01 (br s, 1H), 11,30 (s, 1H), 8,17 (s, 1H), 7,63 (s, 4H), 7,40-7,49 (m, 2H), 5,45 (br d,J=14,55 Гц, 1H), 5,22 (dd, J=7,40, 14,73 Гц, 1H), 3,28 (br d, J=8,44 Гц, 2H), 1,98-2,31 (m, 4H), 1,56 (s, 3H).
Пример 26 (26_А и 26_В)
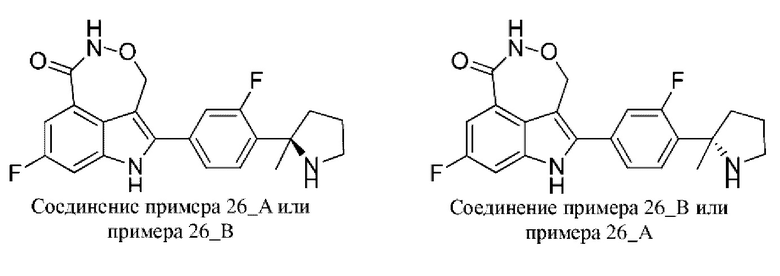
Синтез осуществляли согласно способу, представленному в примере 6.
Наконец, получали соединение примера 26_А (время удерживания = 11,430 мин., значение ее (энантиомерный избыток): 100%) и соединение примера 26_В (время удерживания = 8,159 мин., значение ее (энантиомерный избыток): 100%) посредством удаления защитной группы с помощью трифторуксусной кислоты.
Способ хиральной HPLC: разделительная колонка: Chiralpak IA-3 (50 мм × 4;6 мм, 3 мкм); подвижная фаза: фаза А, н-гептан (0,05%) диэтиламин), фаза В, 8% изопропанол + ацетонитрил (4:1) (0,05%) диэтиламин); скорость потока: 1 мл/мин.; длина волны: 220 нм.
Пример 26_А: Н ЯМР (400 МГц, дейтерированный метанол) δ 8,54 (s, 1Н), 7,63 (t, J=8,19 Гц, 1Н), 7,55 (dd, J=2,25, 10,51 Гц, 1Н), 7,37-7,48 (m, 3Н), 5,41-5,52 (m, 1H), 5,27-5,37 (m, 1Н), 3,47-3,56 (m, 1H), 3,37-3,45 (m, 1Н), 2,39-2,58 (m, 2Н), 2,11-2,38 (m, 2Н), 1,70 (s, 3Н).
Пример 26_В: Н ЯМР (400 МГц, дейтерированный метанол) δ 8,52 (s, 1Н), 7,63 (t, J=8,25 Гц, 1Н), 7,56 (dd, J=2,31, 10,44 Гц, 1Н), 7,37-7,50 (m, 3Н), 5,43-5,51 (m, 1H), 5,28-5,36 (m, 1H), 3,55 (ddd, J=4,82, 9,35, 11,85 Гц, 1H), 3,38-3,48 (m, 1H), 2,41-2,62 (m, 2Н), 2,16-2,39 (m, 2Н), 1,72 (s, 3Н).
Пример 27 (27_А и 27_В)
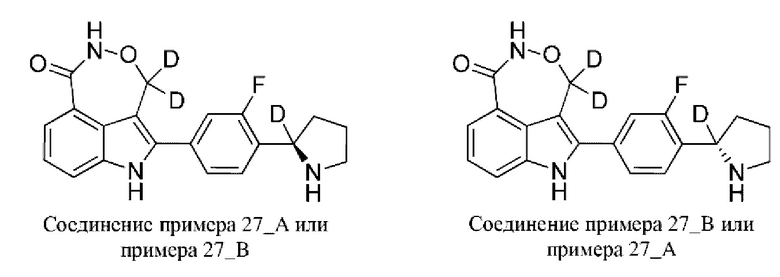
Синтез осуществляли согласно способу, представленному в примере 21 и примере 8.
В случае примера 27 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 25%-25%, 4,1 мин.; 104 мин.) с получением двух изомеров с разными конфигурациями: 27_АА (время удерживания = 1,391 мин., значение ее (энантиомерный избыток): 98%) и 27_ВВ (время удерживания = 1,482 мин., значение ее (энантиомерный избыток): 94%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 27_А (время удерживания = 2,294 мин., значение ее (энантиомерный избыток): 99%) и соединения примера 27_В (время удерживания = 2,116 мин., значение ее (энантиомерный избыток): 93%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Cellucoat (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 5%-40% этанол (0,05% диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 27_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,76-2,06 (m, 3Н), 2,30 (ddd, J=11,57, 7,13, 4,19 Гц, 1Н), 3,11-3,23 (m, 2Н), 7,32 (t, J=7,75 Гц, 1H), 7,40-7,53 (m, 2Н), 7,63-7,79 (m, 3Н), 8,26 (s, 1Н), 11,11 (brs, 1H), 11,93 (s, 1Н).
Пример 27_В: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,77-2,04 (m, 3Н), 2,24-2,40 (m, 1H), 3,09-3,33 (m, 2Н), 7,33 (X, J=1,15 Гц, 1H), 7,40-7,51 (m, 2Н), 7,64-7,78 (m, 3Н), 8,21 (s, 1H), 11,11 (s, 1Н), 11,92 (s, 1Н).
Пример 28 (28_А и 28_В)
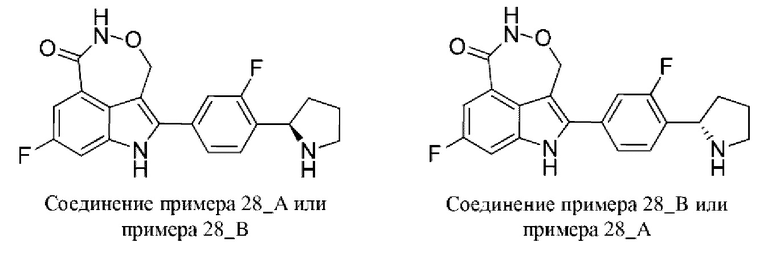
Синтез осуществляли согласно способу, представленному в примере 3.
В случае примера 28 перед удалением защитной группы Boc соединение разделяли на колонке для хиральной HPLC (разделительная колонка: AD-H (250 мм × 30 мм, 5 мкм); подвижная фаза: 0,1% аммиак в изопропаноле; градиент элюирования: 20%-20%, 2,3 мин.; 960 мин.) с получением двух изомеров с разными конфигурациями: 28_АА (время удерживания = 1,474 мин., значение ее (энантиомерный избыток): 99%) и 28_ВВ (время удерживания = 1,560 мин., значение ее (энантиомерный избыток): 94%), в которых удаляли защитную группу с помощью трифторуксусной кислоты с получением соединения примера 28_А (время удерживания = 2,619 мин., значение ее (энантиомерный избыток): 100%) и соединения примера 28_В (время удерживания = 2,350 мин., значение ее (энантиомерный избыток): 96%) соответственно.
Способ SFC (сверхкритическая флюидная хроматография): разделительная колонка: Chiralpak AD-3 (50 мм × 4,6 мм, I.D. 3 мкм); подвижная фаза: 40% изопропанол (0,05%) диэтиламин) в CO2; скорость потока: 3 мл/мин.; длина волны: 220 нм.
Пример 28_А: Н ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,56-1,70 (m, 1Н), 1,77-1,93 (m, 2Н), 2,18-2,32 (m, 1Н), 2,93 3,18 (m, 2Н), 4,45 (br t, J=7,47 Гц, 1 Н), 5,18-5,50 (m, 2Н), 7,31-7,52 (m, 4Н), 7,72 (t, J=7,97 Гц, 1Н), 8,25 (s, 1H), 11,29 (br s, 1Н), 12,01 (s, 1H).
Пример 28_B: H ЯМР (400 МГц, дейтерированный диметилсульфоксид) δ ppm 1,67 (dq, J=12,25, 8,17 Гц, 1Н), 1,78-1,98 (m, 2Н), 2,18-2,33 (m, 1Н), 2,99-3,15 (m, 2Н), 4,48 (br t, J=7,65 Гц, 1Н), 5,15-5,30 (m, 1H), 5,35-5,50 (m, 1H), 7,31-7,52 (m, 4Н), 7,64-7,79 (m, 1H), 8,27 (s, 1Н), 11,30 (br s, 1Н), 12,04 (s, 1H).
Экспериментальный пример 1. Эксперимент с применением фермента PARP-1
Материалы для осуществления эксперимента: тестируемые соединения; набор для анализа НТ Universal Chemiluminescent PARP (приобретен в TREVIGEN); PBS (приобретен в Wisent); Triton X-100 (приобретен в Macklin); многомаркерный анализатор Envision (PerkinElmer).
Методики проведения эксперимента
(I) Получение реагентов.
1. Промывочный раствор: Triton Х-100 добавляли к 1-кратному PBS, и конечная концентрация Triton Х-100 составляла 0,1%.
2. 1-кратный буфер PARP: 20-кратный буфер PARP из набора подвергали 20-кратному разбавлению водой с получением 1-кратного буфера PARP. Данный буфер применяли для получения соединения, раствора фермента и раствора субстрата.
3. 1-кратный раствор Strep-Diluent: 10-кратный Strep-Diluent из набора подвергали 10-кратному разбавлению водой с получением 1-кратного раствора Strep-Diluent.
(II) Получение тестируемых соединений.
Тестируемые соединения последовательно 5-кратно разбавляли с помощью многоканальной пипетки до получения 8-й концентрации, т.е. от 200 мкМ до 2,56 нМ, при этом концентрация DMSO составляла 100%. 2 мкл каждого из ингибиторов при различных градиентах концентрации добавляли в промежуточный планшет, содержащий соединение, и затем добавляли 38 мкл 1-кратного буфера PARP; их двоих хорошо перемешивали для применения, и концентрация DMSO составляла 5%.
(III) Способ проведения эксперимента.
a) 1-кратный буфер PARP добавляли в тестовый планшет в расчете 50 мкл на лунку и инкубировали при 25°С в течение 30 мин.
b) После завершения инкубации жидкость в тестовом планшете отбрасывали и каждое из соединений с различными градиентами концентрации пипетировали из промежуточного планшета, содержащего соединение, и добавляли в тестовый планшет в расчете 10 мкл на лунку. Эксперимент проводили в двух повторностях для каждой лунки.
c) В тестовый планшет добавляли раствор фермента (0,5 ME) в расчете 15 мкл на лунку. Соединение и фермент инкубировали совместно при 25°С в течение 10 мин.
d) После завершения инкубации 25 мкл 1-кратной смеси PARP (содержащей 2,5 мкл 10-кратной смеси PARP, 2,5 мкл 10-кратной активированной ДНК и 20 мкл 1-кратного буфера PARP) добавляли в каждую лунку тестового планшета. Тестовый планшет инкубировали при 25°С в течение 1 ч. Конечная концентрация соединения составляла от 2 мкМ до 0,0256 нМ, и концентрация DMSO составляла 1%.
e) После завершения инкубации тестовый планшет дважды промывали промывочным раствором в расчете 200 мкл на лунку и затем дважды промывали PBS в расчете 200 мкл на лунку.
f) Strep-HRP из набора подвергали 500-кратному разбавлению с помощью 1-кратного раствора Strep-Diluent, добавляли в тестовый планшет в расчете 50 мкл на лунку и затем инкубировали при 25°С в течение 1 ч.
g) После завершения инкубации тестовый планшет дважды промывали промывочным раствором в расчете 200 мкл на лунку и затем дважды промывали PBS в расчете 200 мкл на лунку.
PeroxyGlow А и В из набора смешивали в соотношении 1:1 и смешанный раствор добавляли в тестовый планшет в расчете 100 мкл на лунку. Результаты хемилюминесценции непосредственно считывали с применением многомаркерного анализатора PerkinElmer Envision со временем интеграции, составляющим 0,5 с.
Анализ данных. Исходные данные преобразовывали в степень ингибирования с применением уравнения (образец мин.) / (макс. мин.) × 100% и аппроксимацию кривой значения IC50 осуществляли с применением четырех параметров (полученных из модели «вариабельный наклон кривой log(ингибитор) в сравнении с вариабельным наклоном кривой отклика» в GraphPad Prism). В таблице 1 представлена ингибирующая активность соединений, раскрытых в данном документе, в отношении фермента PARP1.
Результаты эксперимента
Показатели ингибирующей активности соединений, раскрытых в данном документе, в отношении PARP-1-киназы определяли посредством вышеописанного способа проведения эксперимента и показатели ингибирующей активности (IC50) соединений в отношении фермента in vitro показаны в таблице 1.
Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, демонстрируют превосходную ингибирующую активность в отношении PARP1.
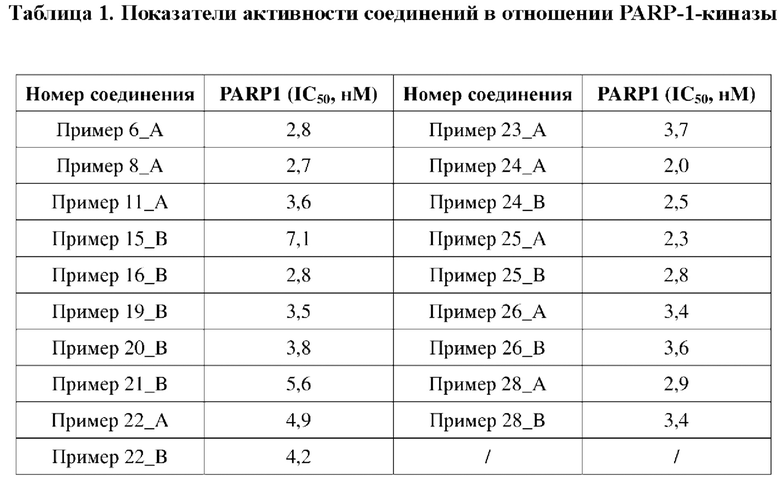
Экспериментальный пример 2. Эксперимент по изучению антипролиферативной активности с использованием клеток MDA-MB-436 CTG
Материалы для осуществления эксперимента: тестируемые соединения; среда RPMI-1640; фетальная бычья сыворотка; антибиотик пенициллин/стрептомицин; линия клеток MDA-MB-436; многомаркерный анализатор Envision (PerkinElmer).
Методики проведения эксперимента
1. Способ проведения эксперимента.
Клетки MDA-MB-436 высевали в белый 96-луночный планшет посредством добавления 80 мкл суспензии клеток (содержащих 3000 клеток MDA-MB-436) в каждую лунку. Планшет с клетками инкубировали в инкубаторе с CO2 в течение ночи.
Тестируемые соединения последовательно 5-кратно разбавляли с помощью многоканальной пипетки до получения 8-й концентрации, т.е. от 2 мМ до 26 нМ, и эксперимент проводили в двух повторностях для каждой лунки. 78 мкл среды добавляли в промежуточный планшет, 2 мкл каждого из последовательно разбавленных соединений переносили в соответствующие лунки промежуточного планшета и после перемешивания смесь переносили в планшет с клетками в расчете 20 мкл на лунку. Планшет с клетками инкубировали в инкубаторе с CO2 в течение 7 дней. Другой планшет с клетками предоставляли для считывания значений сигнала в день добавления соединения и такие значения сигнала применяли в качестве максимальных значений при проведении анализа данных. Promega CellTiter-Glo добавляли в данный планшет с клетками в расчете 25 мкл на лунку и сигналы люминесценции стабилизировали посредством инкубации в течение 10 мин. при комнатной температуре. Считывание осуществляли с применением многомаркерного анализатора PerkinElmer Envision.
Promega CellTiter-Glo добавляли в планшет с клетками в расчете 25 мкл на лунку и сигналы люминесценции стабилизировали посредством инкубации в течение 10 мин. при комнатной температуре. Считывание осуществляли с применением многомаркерного анализатора PerkinElmer Envision.
2. Анализ данных. Исходные данные преобразовывали в степень ингибирования с применением уравнения (образец - мин.) / (макс.- мин.) × 100% и аппроксимацию кривой значения IC50 осуществляли с применением четырех параметров (полученных из модели «вариабельный наклон кривой log(ингибитор) в сравнении с вариабельным наклоном кривой отклика» в GraphPad Prism). В таблице 2 представлена ингибирующая активность соединений, раскрытых в данном документе, в отношении пролиферации клеток MDA-MB-436.
Результаты эксперимента: показатели антипролиферативной активности соединений, раскрытых в данном документе, в отношении клеток MDA-MB-436 с мутацией BRCA1 определяли посредством способа проведения эксперимента, описанного выше, и полумаксимальные ингибирующие концентрации (IC50) соединений для антипролиферативной активности in vitro показаны в таблице 2.
Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, обладают превосходной антипролиферативной активностью в отношении клеток MDA-MB-436 с мутацией BRCA1.

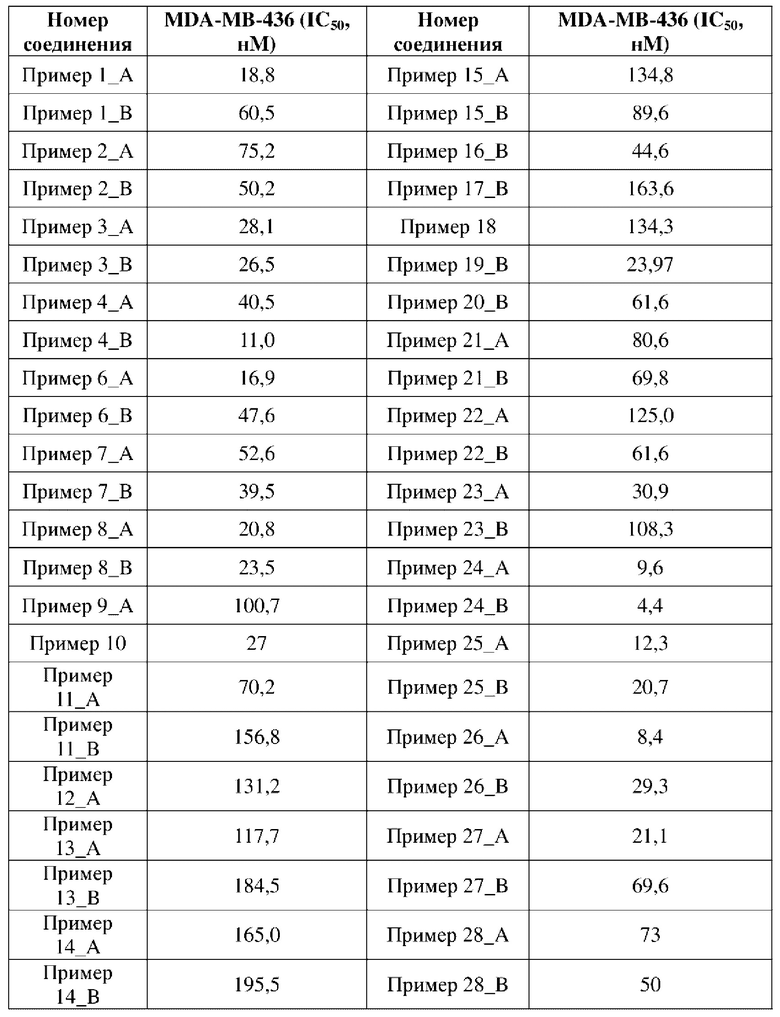
Экспериментальный пример 3. Эксперимент по изучению антипролиферативной активности с использованием клеток MDA-MB-231 CTG
Материалы для осуществления эксперимента: тестируемые соединения; среда R DMEM; фетальная бычья сыворотка; антибиотик пенициллин/стрептомицин; линия клеток MDA-MB-231; многоканальный анализатор Envision.
Способ проведения эксперимента.
Клетки MDA-MB-231 высевали в белый 96-луночный планшет посредством добавления 80 мкл суспензии клеток (содержащих 5000 клеток MDA-MB-231) в каждую лунку. Планшет с клетками инкубировали в инкубаторе с CO2 в течение ночи.
Восемь точек концентрации подготавливали для каждого соединения, тестируемые соединения последовательно 3-кратно разбавляли с помощью многоканальной пипетки до получения 8-й концентрации, т.е. от 2 мМ до 920 нМ, и эксперимент проводили в двух повторностях для каждой лунки. 78 мкл среды добавляли в промежуточный планшет, 2 мкл каждого из последовательно разбавленных соединений переносили в соответствующие лунки промежуточного планшета и после перемешивания смесь переносили в планшет с клетками в расчете 20 мкл на лунку. Планшет с клетками инкубировали в инкубаторе с CO2 в течение 3 дней. Другой планшет с клетками предоставляли для считывания значений сигнала в день добавления соединения и такие значения сигнала применяли в качестве максимальных значений при проведении анализа данных. Promega CellTiter-Glo добавляли в данный планшет с клетками в расчете 25 мкл на лунку и сигналы люминесценции стабилизировали посредством инкубации в течение 10 мин. при комнатной температуре. Считывание осуществляли с применением многомаркерного анализатора PerkinElmer Envision.
Promega CellTiter-Glo добавляли в планшет с клетками в расчете 25 мкл на лунку и сигналы люминесценции стабилизировали посредством инкубации в течение 10 мин. при комнатной температуре. Считывание осуществляли с применением многомаркерного анализатора PerkinElmer Envision.
Анализ данных. Исходные данные преобразовывали в степень ингибирования с применением уравнения (образец - мин.) / (макс.- мин.) × 100% и аппроксимацию кривой значения IC50 осуществляли с применением четырех параметров (полученных из модели «вариабельный наклон кривой log(ингибитор) в сравнении с вариабельным наклоном кривой отклика» в GraphPad Prism). В таблице 1 представлена ингибирующая активность соединений, раскрытых в данном документе, в отношении пролиферации клеток MDA-MB-231.
Результаты эксперимента: показатели антипролиферативной активности соединений, раскрытых в данном документе, в отношении клеток MDA-MB-231 с BRCA дикого типа определяли посредством способа проведения эксперимента, описанного выше, и полумаксимальные ингибирующие концентрации (IC50) соединений для антипролиферативной активности in vitro показаны в таблице 3.
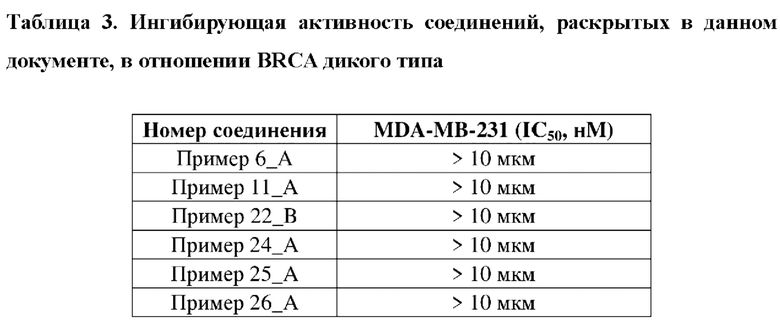
Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, обладают низкой ингибирующей активностью в отношении клеток MDA-MB-231 с BRCA дикого типа, что показывает, что соединения обладают превосходной селективностью.
Экспериментальный пример 4. Эксперимент по изучению антипролиферативной активности в отношении рибозилирования, опосредованного PAR
Материалы для осуществления эксперимента: тестируемые соединения; среда F12K; клетки LoVo; мышиные моноклональные антитела к поли(АДФ-рибоза); козьи антитела к IgG мыши, меченные FITC; пероксид водорода; DAPI; PBS; метанол; ацетон; Tween-20; сухое обезжиренное молоко; многомаркерный анализатор Envision.
Получение реагентов
День первый: клетки LoVo высевали в планшет в расчете 60000 клеток/лунка и затем инкубировали в течение ночи при 37°С/5% CO2.
День второй: получали реагенты.
1. Промывочный раствор: Tween-20 добавляли к 1-кратному PBS, и конечная концентрация Tween-20 составляла 0,05%.
2. Блокирующий раствор: сухое обезжиренное молоко добавляли в промывочный раствор, и конечная концентрация сухого обезжиренного молока составляла 5%.
3. Раствор для фиксации клеток: метанол и ацетон смешивали в соотношении 7:3.
Получение тестируемых соединений: промежуточный планшет 1, содержащий соединение: соединения разбавляли с помощью DMSO и PBS до получения конечных концентраций, составляющих от 10 мкМ до 0,13 нМ, и концентрация DMSO составляла 1%. Промежуточный планшет 2, содержащий соединение: соединения разбавляли с помощью DMSO и PBS, содержащего 50 мМ пероксида водорода, до получения конечных концентраций, составляющих от 10 мкМ до 0,13 нМ, и концентрация DMSO составляла 1%.
Методики проведения эксперимента
1. Клеточный супернатант удаляли и соединение переносили из промежуточного планшета 1, содержащего соединение, в планшет с клетками в расчете 40 мкл на лунку, и смесь инкубировали при 37°С в течение 30 мин.
Лунки, содержащие соединение: соединение, 1% DMSO.
Отрицательный и положительный контроль: добавляли 1% DMSO.
Холостой контроль: лунки без клеток, добавляли PBS.
2. После завершения инкубации соединение переносили из промежуточного планшета 2, содержащего соединение, в планшет с клетками в расчете 40 мкл на лунку, и конечная концентрация H2O2 составляла 25 мМ.
Лунки, содержащие соединение: соединение + 25 мМ H2O2.
Положительный и отрицательный контроль: 1% DMSO + 25 мМ H2O2.
Холостой контроль: лунки без клеток, добавляли PBS.
4. После завершения инкубации планшет с клетками промывали один раз с помощью PBS, предварительно охлажденного на льду и добавляли предварительно охлажденный раствор для фиксации клеток в расчете 100 мкл на лунку. После того, как планшет с клетками оставляли в покое при -20°С в течение 10 мин., раствор для фиксации клеток стряхивали.
5. После сушки на воздухе планшет с клетками промывали с помощью PBS в расчете 200 мкл на лунку и затем PBS отбрасывали.
6. В планшет с клетками добавляли блокирующий раствор в расчете 100 мкл на лунку и инкубировали при 25°С в течение 30 мин., после чего блокирующий раствор стряхивали.
7. Антитела к PAR, которые разбавляли в блокирующем растворе в соотношении 1:50, добавляли в планшет с клетками в расчете 25 мкл на лунку, и затем смесь инкубировали при 25°С в течение 60 мин.
Лунки с отрицательным контролем: блокирующий раствор добавляли в расчете 25 мкл/лунка.
Лунки с холостым контролем: блокирующий раствор добавляли в расчете 25 мкл/лунка.
8. После завершения инкубации планшет с клетками 4 раза промывали промывочным раствором в расчете 200 мкл на лунку, каждый раз составлял 3 мин., и затем промывочный раствор стряхивали.
9. Блокирующий раствор, содержащий разбавленные в соотношении 1:50 козьи антитела к IgG мыши, меченные FITC, и 0,5 мкг/мл DAPI, добавляли в планшет с клетками в расчете 25 мкл на лунку, и смесь инкубировали при 25°С в течение 60 мин.
10. После завершения инкубации планшет с клетками 4 раза промывали промывочным раствором в расчете 200 мкл на лунку, каждый раз составлял 3 мин.
11. После удаления жидкости соответствующие показатели флуоресценции считывали с применением Envision: FITC: 480 нм и 530 нм; DAPI: 360 нм и 460 нм.
Анализ данных: исходные данные нормализовали с применением уравнения (FITC - отрицательный контроль) / (DAPI - холостой контроль), и нормализованные данные преобразовывали в показатели ингибирования с применением уравнения (образец положительный контроль) / (отрицательный контроль - положительный контроль) × 100%, и затем осуществляли аппроксимацию кривой значения IC50 с применением четырех параметров (полученных из модели «вариабельный наклон кривой log(ингибитор) в сравнении с вариабельным наклоном кривой отклика» в GraphPad Prism). В таблице 1 представлена ингибирующая активность соединений, раскрытых в данном документе.
Результаты эксперимента: полумаксимальные ингибирующие концентрации (IC50) соединений, раскрытых в данном документе, в отношении рибозилирования, опосредованного PAR, показаны в таблице 4.

Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, обладают значительной ингибирующей активностью в отношении рибозилирования, опосредованного PAR.
Экспериментальный пример 5. Исследование степени связывания с белками плазмы крови
Определяли показатели степени связывания с белками для соединений, раскрытых в данном документе, в плазме крови человека, мышей CD-1 и крыс SD. 796 мкл необработанной плазмы крови получали от человека, мышей CD-1 и крыс SD и добавляли 4 мкл рабочего раствора тестируемого соединения (400 мкМ) или рабочего раствора варфарина (400 мкМ) с получением конечной концентрации, составляющей 2 мкМ, как тестируемого соединения, так и варфарина в образце плазмы крови. Образцы хорошо перемешивали. Конечная концентрация органической фазы DMSO составляла 0,5%; 50 мкл образца плазмы крови, содержащего тестируемое соединение и варфарин, пипетировали в планшет для загрузки образцов (в трех параллелях) и соответствующий объем необработанной плазмы крови или буфера непосредственно добавляли таким образом, чтобы конечный объем в каждой лунке, содержащей образец, составлял 100 мкл. Объемное соотношение плазмы крови и буфера для диализа составляло 1:1, и к данным образцам добавляли 400 мкл останавливающего раствора, их применяли в качестве образцов Т0 для определения объема извлечения и стабильности. Образцы Т0 хранили при 2-8°С для последовательной обработки совместно с другими образцами, подвергнутыми диализу; 150 мкл образца плазмы крови, содержащего тестируемое соединение и варфарин, добавляли в конец для загрузки лекарственного средства каждой лунки для диализа и 150 мкл необработанного буфера для диализа добавляли в соответствующий принимающий конец лунки для диализа. Затем планшет для диализа закрывали с помощью газопроницаемой мембраны и помещали в инкубатор при влажных условиях, 5%CO2 и инкубировали при 37°С в течение 4 ч. со встряхиванием при приблизительно 100 об./мин. После завершения диализа 50 мкл образца буфера, подвергнутого диализу, и образца плазмы крови, подвергнутого диализу, пипетировали в новый планшет для загрузки образцов. Соответствующий объем соответствующего образца необработанной плазмы крови или буфера добавляли к образцам таким образом, чтобы конечный объем в каждой лунке, содержащей образец, составлял 100 мкл, и объемное соотношение плазмы крови к буферу для диализа составляло 1:1. Все образцы подвергали анализу посредством LC/MS после осаждения белка и показатели степени связывания с белками и объема извлечения рассчитывали с применением следующих формул: степень отсутствия связывания с белками (%) = 100 × концентрация лекарственного средства, пропускаемая через мембрану для диализа/концентрация лекарственного средства, не пропускаемая через диализат; степень связывания с белками (%)=100 - степень отсутствия связывания с белками (%); объем извлечения (%) = 100 × (концентрация лекарственного средства, пропускаемая через мембрану для диализа + концентрация лекарственного средства, не пропускаемая через диализат) / общая концентрация лекарственного средства перед проведением диализа.
Результаты эксперимента: результаты показаны в таблице 5.
Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, характеризуются высокой степенью связывания с белками плазмы крови.
Вывод, сделанный в ходе эксперимента: соединения, раскрытые в данном документе, характеризуются подходящей степенью связывания с белками плазмы крови.
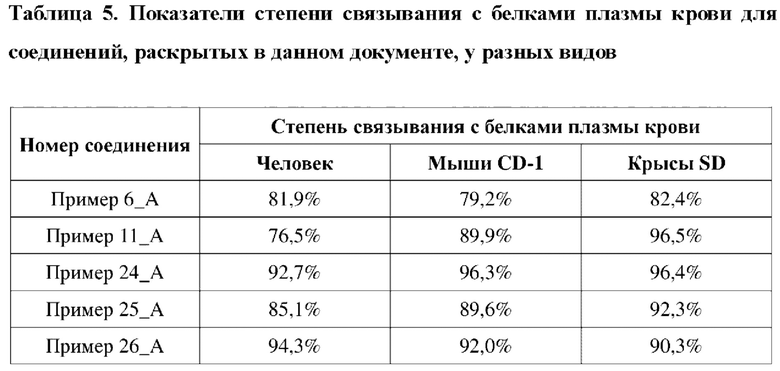
Экспериментальный пример 6. Исследование ингибирующей активности в отношении изофермента цитохрома Р450
Определяли ингибирующую активность тестируемых соединений в отношении разных подтипов человеческого изофермента цитохрома Р450. Получали тестируемые соединения, стандартный ингибитор (100 × конечная концентрация) и рабочий раствор смешанного субстрата; замороженные в холодильнике при -80°С микросомы доставали и размораживали. 2 мкл раствора тестируемого соединения и стандартного ингибитора добавляли в соответствующие лунки, и 2 мкл соответствующего растворителя добавляли в лунку с контролем без ингибитора (NIC) и в лунку с холостым контролем (холостой); затем 20 мкл раствора смешанного субстрата добавляли в соответствующие лунки за исключением лунки с холостым контролем (в лунку с холостым контролем добавляли 20 мкл РВ); получали раствор микросом печени человека (помечали дату после применения и непосредственно возвращали в холодильник) и затем добавляли во все лунки в расчете 158 мкл на лунку; планшет для образцов помещали на водяную баню при 37°С для предварительной инкубации и затем получали раствор коферментного фактора (NADPH); через 10 мин. раствор NADPH добавляли во все лунки в расчете 20 мкл на лунку и встряхивали планшет для образцов для хорошего перемешивания и затем инкубировали на водяной бане при 37°С в течение 10 мин.; в соответствующие моменты времени добавляли 400 мкл холодного раствора ацетонитрила (внутренний стандарт: 200 нг/мл толбутамида и лабеталола) для остановки реакции; после хорошего перемешивания смесь в планшете для образцов центрифугировали при 4000 об./мин. в течение 20 мин. с осаждением белков; собирали 200 мкл надосадочной жидкости и добавляли к 100 мкл воды, и смесь хорошо перемешивали и затем анализировали посредством LC/MS/MS.
Результаты эксперимента: результаты показаны в таблице 6.
Вывод: соединения, раскрытые в данном документе, демонстрируют отсутствие или наличие слабого ингибирующего эффекта в отношении 5 ферментов CYP.
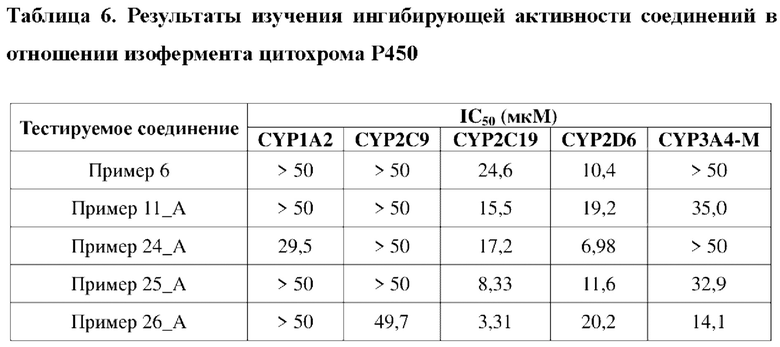
Экспериментальный пример 7. Метаболическая стабильность в микросомах печени
Цель эксперимента: испытание метаболической стабильности тестируемых соединений в микросомах печени, полученных от трех видов.
Способ проведения эксперимента. 1 мкМ тестируемого соединения и микросому (0,5 мг/мл) инкубировали при 37°С в присутствии системы для регенерации NADPH; положительные контроли представляли собой тестостерон (субстрат 3А4), пропиламин-пропиофенон (субстрат 2D6) и диклофенак (субстрат 2С9), и также при 37°С инкубировали положительный контроль с микросомой (0,5 мг/мл) в присутствии системы для регенерации NADPH; реакцию останавливали посредством прямого смешивания образца с холодным ацетонитрилом, содержащим внутренний стандарт, в различные моменты времени (0, 5, 10, 20, 30 и 60 мин.); соединение и микросому инкубировали в течение 60 мин. при отсутствии системы для регенерации NADPH; подготавливали одну параллель (n=1) в каждый момент времени; образцы анализировали посредством LC/MS/MS; концентрация соединения характеризовалась отношением площади пика к площади пика внутреннего стандарта.
Результаты эксперимента: результаты показаны в таблице 7.

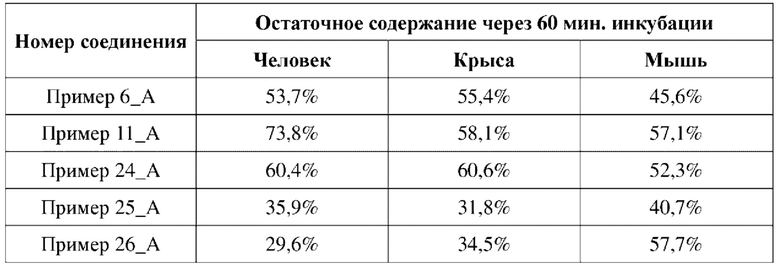
Экспериментальный пример 8. Исследование фармакокинетики однократной дозы у мышей
Цель эксперимента: оценка фармакокинетических свойств с использованием самцов мышей C57BL/6 в качестве подопытных животных и определение концентраций соединений в плазме крови, печени и спинномозговой жидкости после однократного введения дозы.
Способ проведения эксперимента: выбирали здоровых взрослых самцов мышей C57BL/6 для осуществления внутрижелудочного введения. Кандидатное соединение смешивали с подходящим количеством 10% DMSO/90% (20% гидроксипропил-β-циклодекстрин), перемешивали вихревым способом и подвергали воздействию ультразвука с получением 0,5 мг/мл прозрачного раствора для дальнейшего применения. После введения мышам дозы 1 мг/кг внутривенно и дозы 5 мг/кг перорально собирали цельную кровь в определенные моменты времени и отделяли плазму крови, а также собирали печень и спинномозговую жидкость. После предварительной обработки образцов измеряли концентрацию лекарственного средства посредством LC-MS/MS, и рассчитывали фармакокинетические параметры с применением программного обеспечения Phoenix WinNonlin.
Результаты эксперимента: результаты показаны в таблице 8.
Вывод, сделанный в ходе эксперимента: тестируемые соединения характеризуются хорошей AUC0-послед. и биологической доступностью у мышей.
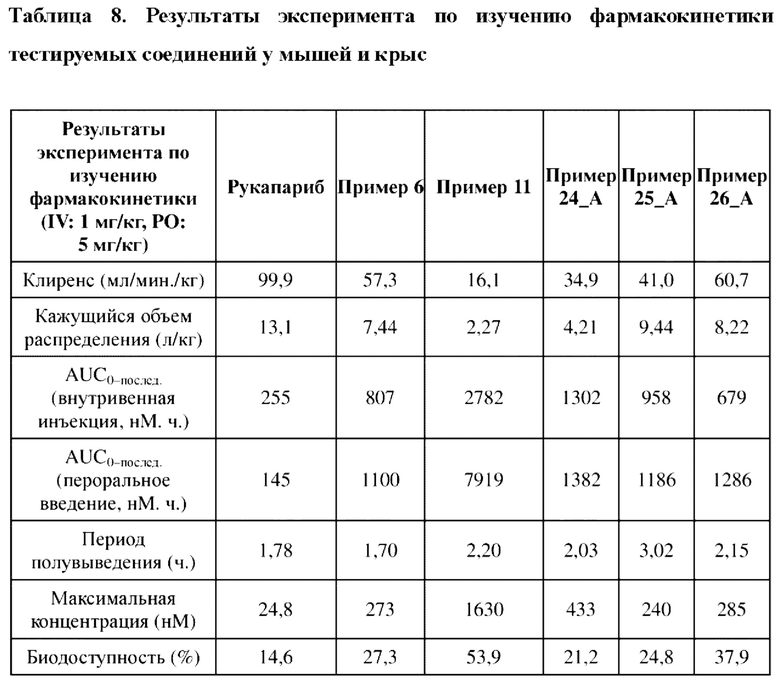
Экспериментальный пример 9. Исследование фармакодинамики соединений in vivo с применением модели, представляющей собой «голых» мышей BALB/c с подкожным ксенотрансплантатом клеток рака молочной железы человека MDA-MB-436
Цель эксперимента: исследование эффективности in vivo тестируемого соединения с применением модели, представляющей собой «голых» мышей BALB/c с подкожным ксенотрансплантатом клеток рака молочной железы человека MDA-MB-436.
План эксперимента
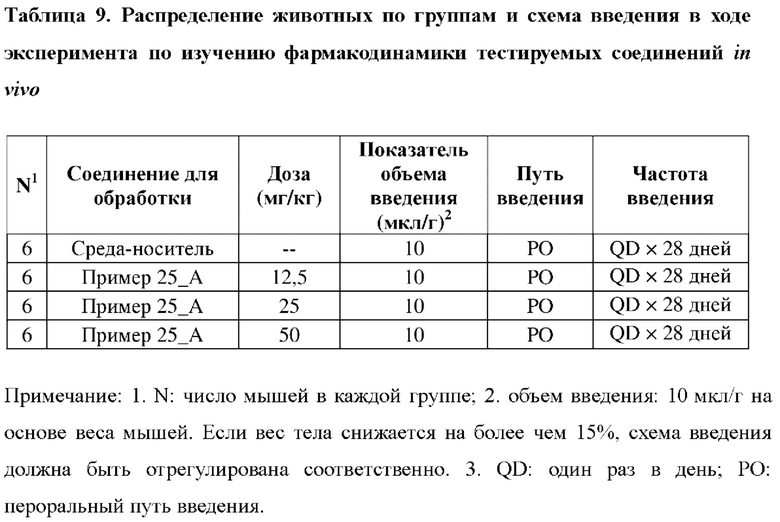
Материалы для осуществления эксперимента: возраст в неделях и вес тела: самки «голых» мышей BALB/c в возрасте 6-8 недель с весом тела 18-22 г. Эксперимент начинали после 3-7-дневного периода адаптивного кормления. В информационной карте животных каждой клетки представлена следующая информация о животных: число особей, пол, линия, дата прибытия, схема введения, номер эксперимента, группа и дата начала эксперимента. Все клетки, подстилку и питьевую воду стерилизовали перед применением. Клетки, корм и питьевую воду меняли два раза в неделю. Подопытных животных идентифицировали с помощью ушных бирок. Тестируемые образцы: пример 24_А и пример 25_А. Все тестируемые образцы получали с 10% DMSO + 90% (20%) HP-β-CD) в качестве среды-носителя и группе холостого контроля вводили только среду-носитель.
Способ проведения эксперимента.
1. Культивирование клеток. Клетки рака молочной железы человека MDA-MB-436 (АТСС, Манассас, штат Вирджиния, США, номер по каталогу: НТВ-130) культивировали в среде для культивирования RPMI-1640, содержащей 10% фетальной бычьей сыворотки и 1% антибиотика-антимикотика, из монослойной культуры in vitro в инкубаторе при 37°С/5% CO2. Клетки расщепляли трипсином-EDTA два раза в неделю для пересевания согласно традиционной практике. При достижении насыщенности клеток, составлявшей 80%-90%, и требуемого количества, клетки собирали, подсчитывали и инокулировали.
2. Инокуляция опухолевых клеток (инокуляция опухоли). 0,2 мл (1 × 10 клеток) клеток MDA-MB-436 (вместе с матригелем в объемном соотношении 1:1) инокулировали подкожно в правый бок каждой мыши, и мышей произвольным образом распределяли по группам при достижении среднего объема опухоли, составлявшего 318 мм.
3. Ежедневное наблюдение за подопытными животными: ежедневно велось наблюдение за состоянием здоровья и случаями гибели животных, и регулярные обследования включали наблюдение за влиянием роста опухоли и обработки лекарственным средством на ежедневные показатели животных, такие как поведение, потребление корма и воды, изменение веса, внешний вид или наличие других отклоняющихся от нормы состояний.
4. Измерение опухоли и экспериментальные индексы: экспериментальные индексы требовались для определения подавления роста опухоли, замедления роста опухоли или излечения опухоли. Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Объем опухоли рассчитывали с применением следующей формулы: V=0,5а×b2, где а и b обозначают диаметр по длинной оси и диаметр по короткой оси опухоли соответственно. Противоопухолевый терапевтический эффект соединения оценивали с помощью TGI (%) или относительной скорости пролиферации опухоли Т/С (%). TGI (%) относится к степени подавления роста опухоли. Расчет TGI (%) : TGI (%) = [(1 - (средний объем опухоли в конце введения в группе с обработкой средний объем опухоли в начале введения в группе с обработкой)) / (средний объем опухоли в конце обработки в контрольной группе, получавшей растворитель - средний объем опухоли в начале обработки в контрольной группе, получавшей растворитель)] × 100%. Формула для расчета относительной скорости пролиферации опухоли Т/С (%) являлась следующей: Т/С(%)=TRTV/CRTV×100%) (TRTV: RTV для группы с обработкой; CRTV: RTV для группы отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали на основе результатов измерения опухоли. Формула для расчета являлась следующей: RTV=Vt/V0, где V0 представлял собой средний объем опухоли, измеренный при разделении на группы и при введении (т.е. d0), Vt представлял собой средний объем опухоли при конкретном измерении, и используемые данные TRTV и CRTV получали в тот же день.
5. Статистический анализ включал среднее значение и стандартное отклонение среднего значения (SEM) объема опухоли в каждый момент времени для каждой группы. Группа с обработкой демонстрировала самый лучший эффект от обработки в день 27 после введения в конце эксперимента и, следовательно, статистический анализ осуществляли на основе данных для оценки различий между группами. Экспериментальные данные анализировали с помощью способа однофакторного ANOVA и способа Геймса-Ховелла. Анализ всех данных осуществляли с помощью SPSS 17.0. р<0,05 определяли как значительное отличие.
Результаты эксперимента: результаты показаны в таблице 10.
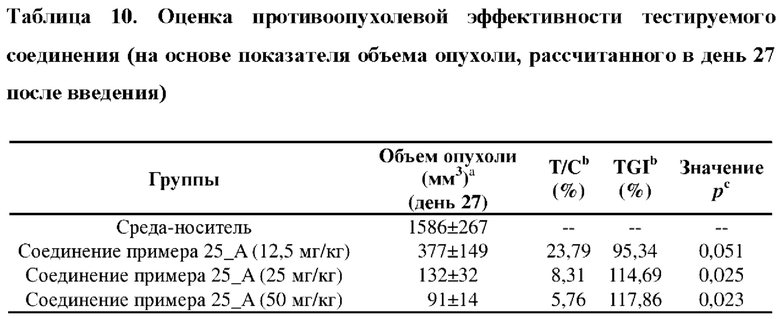

Вывод, сделанный в ходе эксперимента: соединение, раскрытое в данном документе, обладает хорошим эффектом в отношении подавления роста опухоли.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2013 |
|
RU2650107C2 |
| ОКСОХИНАЗОЛИНИЛБУТАНАМИДНЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2669393C2 |
| ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2663623C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, СОЕДИНЕНИЯ, КОМПЛЕКСНЫЙ МЕТАЛЛООРГАНИЧЕСКИЙ КАТАЛИЗАТОР | 2012 |
|
RU2652807C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
Группа изобретений относится к новому типу соединений индологептамилоксима в качестве ингибитора PARP. Раскрыто соединение формулы (II), его стереоизомер или его фармацевтически приемлемая соль,
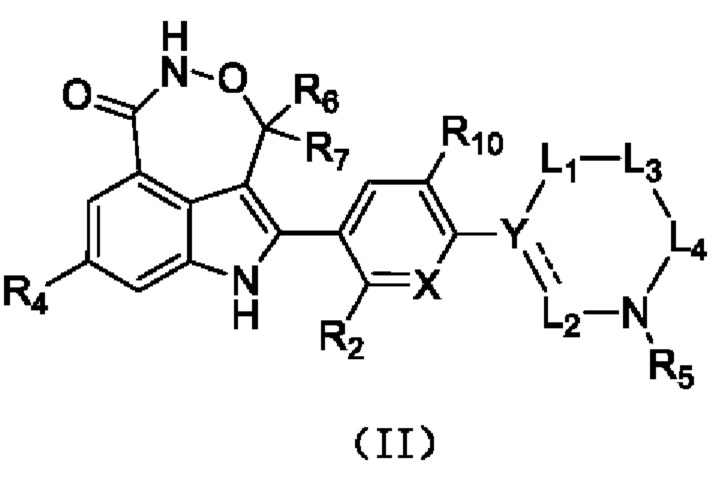 ,
,
где  выбрана из группы, состоящей из одинарной связи и двойной связи; X выбран из группы, состоящей из CR3 и N; Y выбран из группы, состоящей из CR1 и С; L1 выбран из группы, состоящей из одинарной связи и -(CR8R9)n; L2 выбран из группы, состоящей из одинарной связи, -CR8R9- и =СН-; L1 и L2 не представляют собой одинарные связи одновременно; если L2 выбран из одинарной связи, то
выбрана из группы, состоящей из одинарной связи и двойной связи; X выбран из группы, состоящей из CR3 и N; Y выбран из группы, состоящей из CR1 и С; L1 выбран из группы, состоящей из одинарной связи и -(CR8R9)n; L2 выбран из группы, состоящей из одинарной связи, -CR8R9- и =СН-; L1 и L2 не представляют собой одинарные связи одновременно; если L2 выбран из одинарной связи, то  выбрана из одинарной связи; каждый из L3 и L4 независимо выбран из -CR8R9-; n равняется 1 или 2; R1 выбран из группы, состоящей из Н, D, F, Cl, Br, I и С1-3алкила, и если L2 выбран из =СН-, то R1 отсутствует; каждый из R2 и R10 независимо выбран из группы, состоящей из Н, F, Cl, Br и I; R3 выбран из группы, состоящей из Н, F, Cl, Br и I; R4 выбран из группы, состоящей из Н и F; R5 выбран из группы, состоящей из Н и С1-3алкила; каждый из R6 и R7 независимо выбран из группы, состоящей из Н и D; каждый из R8 и R9 представляет собой Н. Также раскрыты соединения, попадающие под формулу (II), фармацевтическая композиция, содержащая соединение формулы (II), способ лечения заболевания, связанного с рецептором PARP, с использованием соединения формулы (II) и применение соединения формулы (II) для лечения заболевания, связанного с рецептором PARP. Группа изобретений обеспечивает предоставление соединений индолгептамилоксима в качестве ингибитора PARP. 5 н. и 19 з.п. ф-лы, 10 табл., 89 пр.
выбрана из одинарной связи; каждый из L3 и L4 независимо выбран из -CR8R9-; n равняется 1 или 2; R1 выбран из группы, состоящей из Н, D, F, Cl, Br, I и С1-3алкила, и если L2 выбран из =СН-, то R1 отсутствует; каждый из R2 и R10 независимо выбран из группы, состоящей из Н, F, Cl, Br и I; R3 выбран из группы, состоящей из Н, F, Cl, Br и I; R4 выбран из группы, состоящей из Н и F; R5 выбран из группы, состоящей из Н и С1-3алкила; каждый из R6 и R7 независимо выбран из группы, состоящей из Н и D; каждый из R8 и R9 представляет собой Н. Также раскрыты соединения, попадающие под формулу (II), фармацевтическая композиция, содержащая соединение формулы (II), способ лечения заболевания, связанного с рецептором PARP, с использованием соединения формулы (II) и применение соединения формулы (II) для лечения заболевания, связанного с рецептором PARP. Группа изобретений обеспечивает предоставление соединений индолгептамилоксима в качестве ингибитора PARP. 5 н. и 19 з.п. ф-лы, 10 табл., 89 пр.
1. Соединение формулы (II), его стереоизомер или его фармацевтически приемлемая соль,
 ,
,
где  выбрана из группы, состоящей из одинарной связи и двойной связи;
выбрана из группы, состоящей из одинарной связи и двойной связи;
X выбран из группы, состоящей из CR3 и N;
Y выбран из группы, состоящей из CR1 и С;
L1 выбран из группы, состоящей из одинарной связи и -(CR8R9)n;
L2 выбран из группы, состоящей из одинарной связи, -CR8R9- и =СН-;
L1 и L2 не представляют собой одинарные связи одновременно;
если L2 выбран из одинарной связи, то  выбрана из одинарной связи;
выбрана из одинарной связи;
каждый из L3 и L4 независимо выбран из -CR8R9-;
n равняется 1 или 2;
R1 выбран из группы, состоящей из Н, D, F, Cl, Br, I и С1-3алкила, и если L2 выбран из =СН-, то R1 отсутствует;
каждый из R2 и R10 независимо выбран из группы, состоящей из Н, F, Cl, Br и I;
R3 выбран из группы, состоящей из Н, F, Cl, Br и I;
R4 выбран из группы, состоящей из Н и F;
R5 выбран из группы, состоящей из Н и С1-3 алкила;
каждый из R6 и R7 независимо выбран из группы, состоящей из Н и D;
каждый из R8 и R9 представляет собой Н.
2. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где С1-3алкил замещен одним Rd, где Rd представляет собой ОН.
3. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1 или 2, выбранные из формулы (II-1)
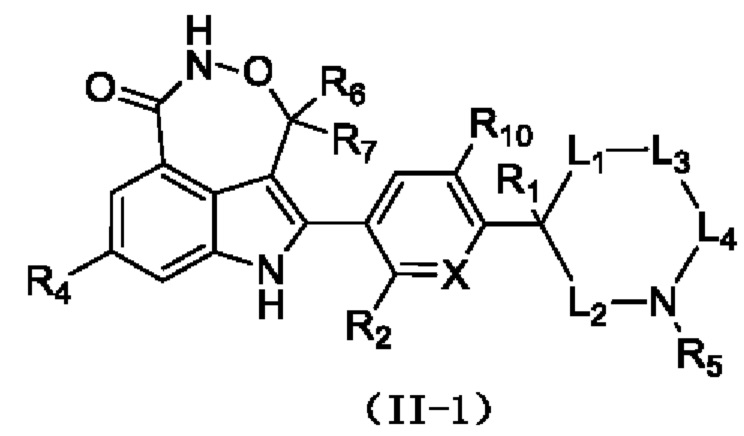 ,
,
где R1, R2, X, R4, R5, R6, R7, R10, L1, L2, L3 и L4 являются такими, как определено в п. 1 или 2.
4. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-3, где R1 выбран из группы, состоящей из Н, D, F и СН3.
5. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп.1-4, где каждый из R2 и R10 независимо выбран из группы, состоящей из Н и F.
6. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-5, где R3 выбран из группы, состоящей из Н, F и Cl.
7. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.6, где R3 выбран из группы, состоящей из Н и F.
8. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-7, где R5 выбран из группы, состоящей из Н, метила, этила, пропила и изопропила, при этом метил, этил, пропил и изопропил необязательно замещены Rd.
9. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 8, где R5 выбран из группы, состоящей из Н, метила,  и изопропила.
и изопропила.
10. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-9, где L1 выбран из группы, состоящей из одинарной связи и -CR8R9-.
11. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-9, где L1 выбран из -CR8R9- и L2 выбран из -CR8R9-.
12. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-9, где L1 выбран из -CR8R9- и L2 выбран из одинарной связи.
13. Соединение, его стереоизомер или его фармацевтически приемлемая соль по любому из пп. 1-12, где оба из R6 и R7 представляют собой Н.
14. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где структурное звено  выбрано из группы, состоящей из
выбрано из группы, состоящей из
 .
.
15. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.1, где структурное звено  выбрано из группы, состоящей из
выбрано из группы, состоящей из
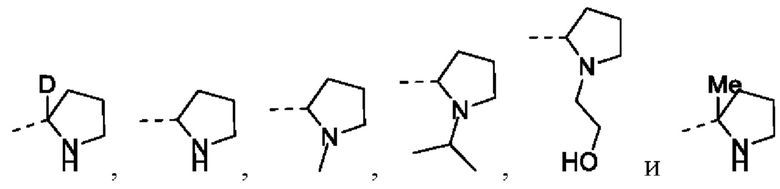 .
.
16. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где структурное звено 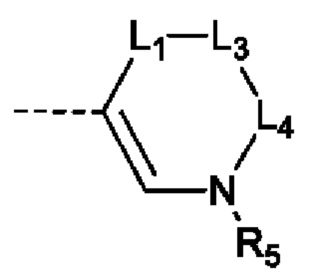 выбрано из
выбрано из  .
.
17. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где структурное звено 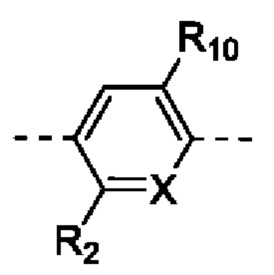 выбрано из группы, состоящей из
выбрано из группы, состоящей из
 .
.
18. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 1, где структурное звено 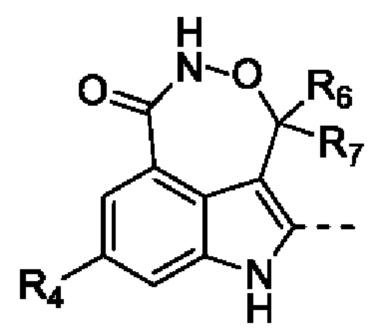 выбрано из группы, состоящей из
выбрано из группы, состоящей из
 ,
,  и
и  .
.
19. Соединение формулы, указанной ниже, его стереоизомер или его фармацевтически приемлемая соль, выбранные из группы, состоящей из
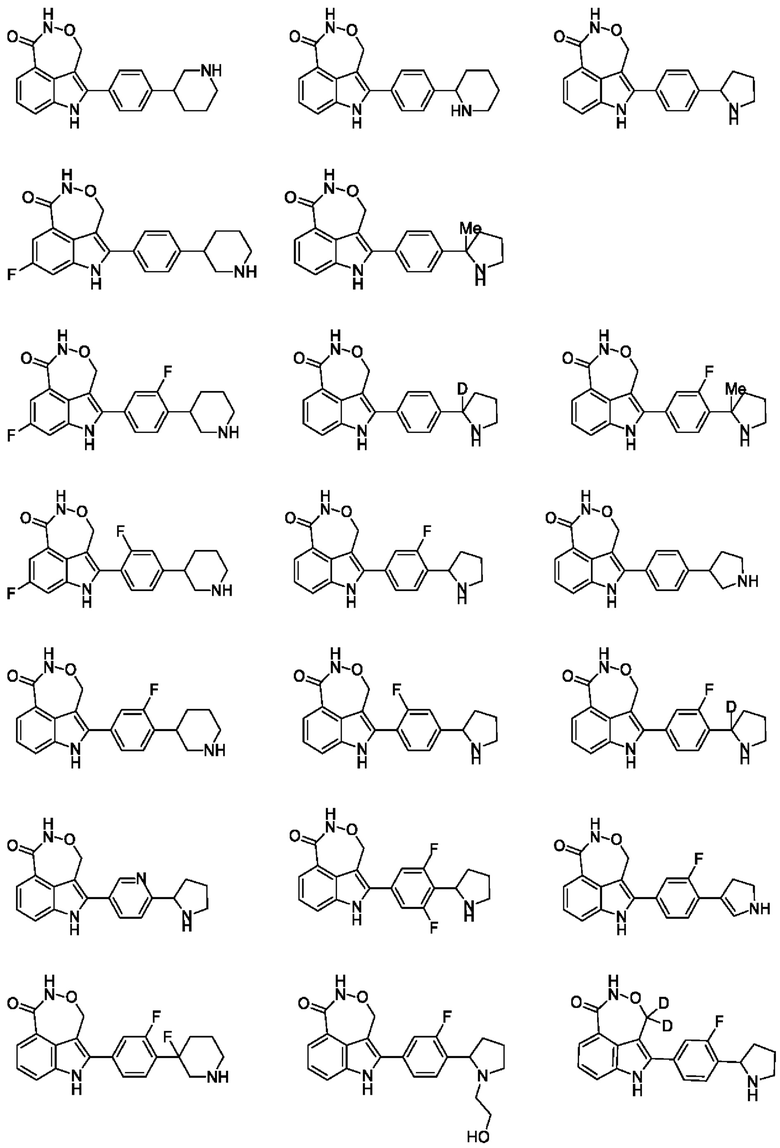
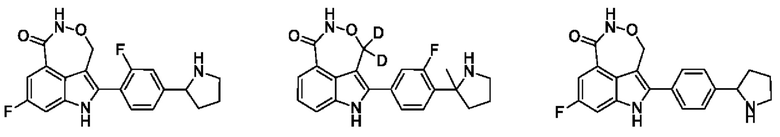
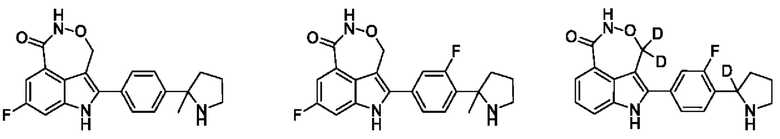
 .
.
20. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п. 19, выбранные из группы, состоящей из
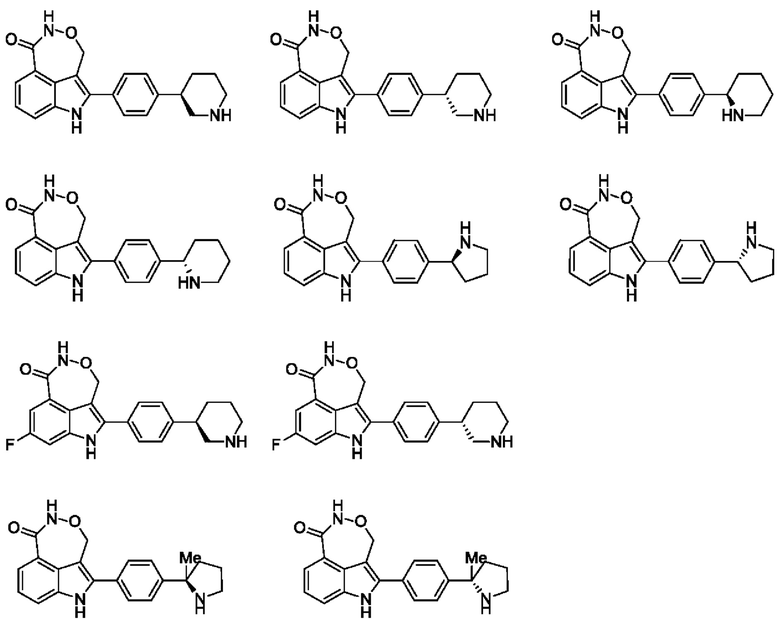
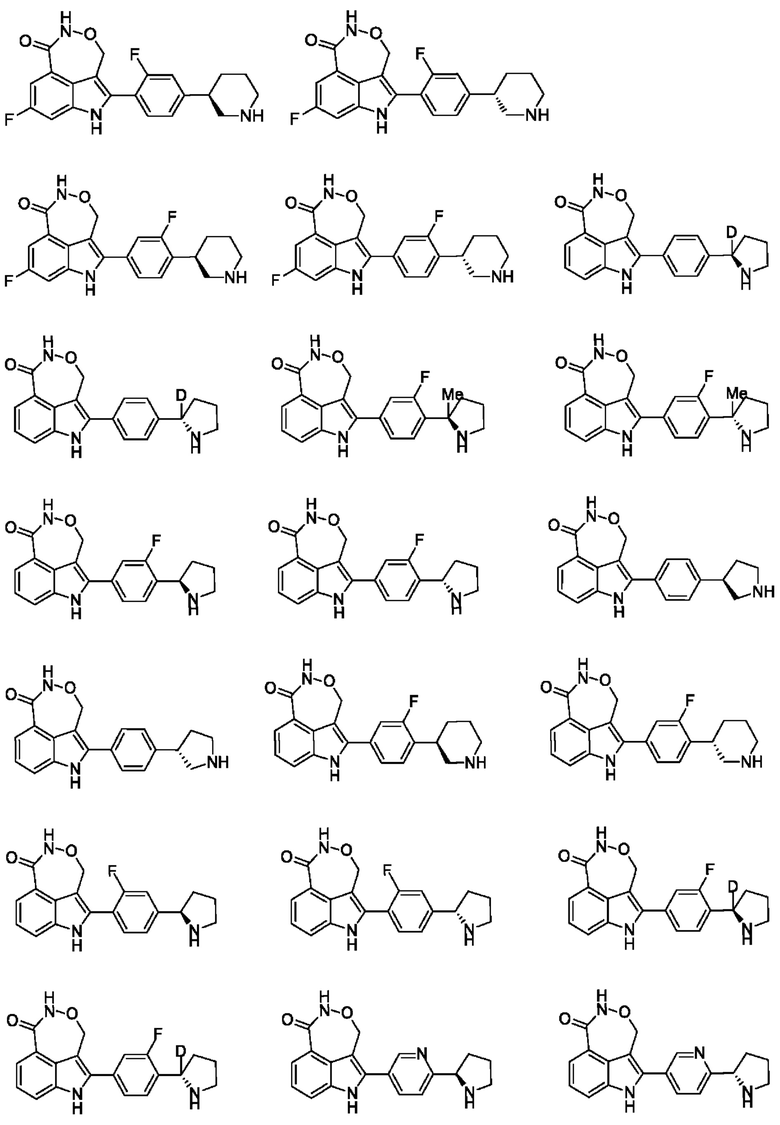
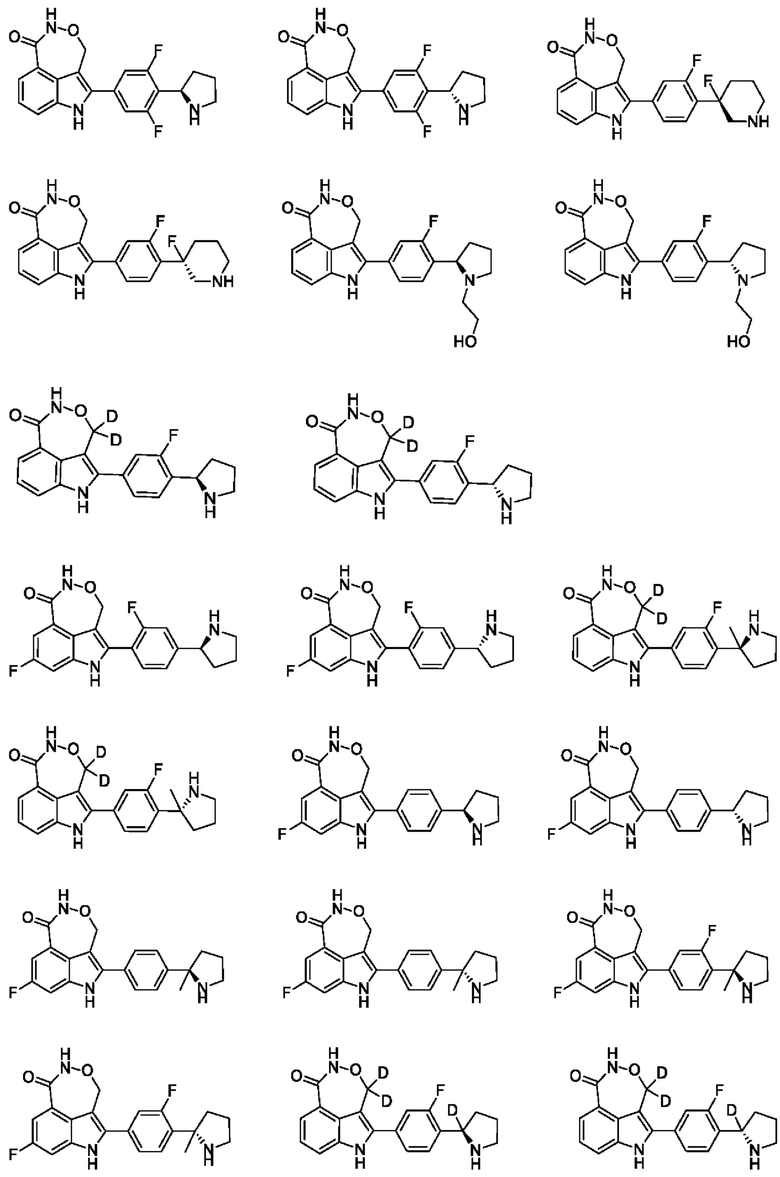
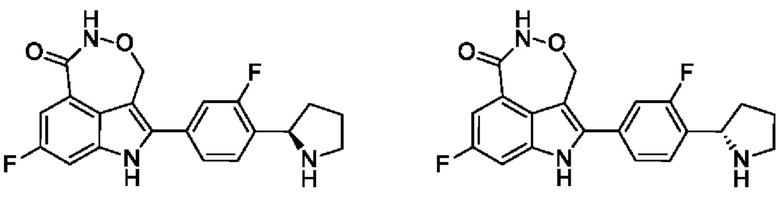 .
.
21. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении PARP-1, содержащая терапевтически эффективное количество соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-20 и фармацевтически приемлемый носитель.
22. Применение соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-20 для получения лекарственного препарата для лечения заболевания, связанного с рецептором PARR.
23. Способ лечения заболевания, связанного с рецептором PARP, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп. 1-20.
24. Способ по п. 23, где млекопитающее представляет собой человека.
| WO 200123390 A2, 05.04.2001 | |||
| WO 2000042040 A1, 20.07.2000 | |||
| WO 2015166398 A1, 05.11.2015 | |||
| FERRARIS D.V | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
