ОБЛАСТЬ ТЕХНИЧЕСКОГО ПРИМЕНЕНИЯ
Настоящее изобретение относится к области медицины и, в частности, имеет отношение к ингибитору JAK, а также к способу его получения.
ИСТОРИЧЕСКАЯ СПРАВКА
Сигнальный путь JAK-STAT - это путь передачи сигналов, подаваемых цитокинами для участия в процессах пролиферации, дифференциации, апоптозе, иммунорегуляции клеток, а также во множестве других важных биологических процессах. В отличие от других сигнальных путей, подобный сигнальный путь имеет относительно простой процесс передачи и главным образом состоит из трех компонентов, а именно рецептора, ассоциированного с тирозинкиназой, Янус-киназы (или JAK) и фактора транскрипции STAT.
Сигналы по сигнальному пути JAK-STAT передаются с помощью рецептора, ассоциированного с тирозинкиназой, множества цитокинов и факторов роста, включая интерлейкины 2-7 (IL-2-7), фактор, стимулирующий колонии гранулоцитов/макрофагов (GM-CSF), гормон роста (GH), эпидермальный фактор роста (EGF), фактор роста тромбоцитов (PDGF), интерферон (IFN) и т.п. Указанные цитокины и фактор роста имеют соответствующие рецепторы в цитоплазматической клеточной мембране. Общей характеристической особенностью таких рецепторов является то, что каждый такой рецептор собственной киназной активности не проявляет, но имеет в своем внутриклеточном домене центры связывания тирозинкиназы JAK. После связывания с лигандом рецептор, активируемый связанным с ним JAK, фосфорилирует тирозиновые остатки различных таргетных белков, осуществляя тем самым передачу сигнала из внеклеточного пространства внутрь клетки.
Янус-киназа (JAK), многие тирозинкиназы являются рецепторами клеточной мембраны, вместе именуемыми рецепторной тирозинкиназой (RTK); при этом JAK является разновидностью не обладающей трансмембранными свойствами тирозинкиназы. JAK - это аббревиатура от словосочетания «Janus kinase» (Янус-киназа); поскольку Янус - это божество, ведающее вопросами начала и конца. Причина, по которой ее называют киназой Януса (или Янус-киназой), заключается в том, что JAK может не только фосфорилировать связанные с ней рецепторы цитокинов, но также может фосфорилировать и многие сигнальные молекулы, содержащие специфические SH2-домены. В семейство JAK входит 4 белка: JAK1, JAK2, JAK3 и Tyk2; в их структуре имеется 7 доменов гомологии JAK (JH), где домен JH1 представляет собой домен киназы, домен JH2 - «ложный» домен киназы; JH6 и JH7 представляют собой рецептор-связывающие домены.
В понятии «фактор транскрипции STAT» аббревиатура «STAT» расшифровывается как «signal transducer and activator of transcription», или же «переносчик сигнала и активатор транскрипции». Как следует уже из самого названия, STAT играет решающую роль в передаче сигнала и активации транскрипционной деятельности. В настоящее время выявлено 6 белков семейства STAT, а именно STAT1-STAT6. По своей структуре белок STAT можно разделить на несколько функциональных участков: N-концевая консервативная последовательность, ДНК-связывающий домен, SH3-домен, SH2-домен и C-концевой домен активации транскрипции. Зоной, являющейся наиболее консервативной в последовательности, но наиболее важной по функции, выступает SH2-домен, имеющий последовательность сердцевины «GTFLLRFSS», что идентична последовательности сердцевины SH2-домен тирозинкиназы Src.
Сигнальный путь JAK-STAT выполняет обширные функции и участвует в процессах пролиферации, дифференциации, апоптозе, иммунорегуляции клеток, а также во множестве других важных биологических процессах. На сегодняшний день основной акцент в исследованиях, связанных с заболеваниями и инновационными решениями в области медикаментозного лечения, делается на воспалительных и опухолевых заболеваниях. В число таких воспалительных заболеваниям входят ревматоидный артрит, собачий дерматит, псориаз, язвенный колит или болезнь Крона; а в число таких опухолевых заболеваний главным образом - миелофиброз, истинная полицитемия и идиопатическая тромбоцитемия. Более того, мутация самой молекулы JAK также может приводить к развитию острого миелоцитарного лейкоза (AML), острошго лимфоцитарного лейкоза (ALL), протоковой карциномы молочной железы, немелкоклеточного рака легких (NSCLC) и т.п.
Ингибитор JAK может выполнять избирательное ингибирование JAK-киназы с целью блокирования сигнального пути JAK-STAT. В настоящее время в число ингибиторов JAK, которые были одобрены Федеральным управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США, входят тофацитиниб, руксолитиниб, оклацитиниб и барицитиниб. Оклацитиниб является новейшим ингибитором JAK, который, в свою очередь, способен подавлять функции JAK1-зависимых цитокинов при некоторых аллергических, воспалительных реакциях и реакциях, сопряженных с зудом. Исследования показывают, что при пероральном введении лабораторным животным (собакам) по два раза в день дозы в объеме 0,4-0,6 мг/кг оклацитиниб доказал свою безопасность и эффективность в лечении симптомов зуда, возникающих при атопическим дерматите, и продемонстрировал более высокую эффективность, чем пероральные кортикостероиды, например, преднизолон. В процессе лечения введение оклацитиниба может в течение 24 часов облегчать зудение, а на 7-й день у более чем 70% лабораторных животных более чем на 50% ослабли симптомы сопряженной с зудом реакции. Оклацитиниб в 2013 году был одобрен Федеральным управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США и по сей день используется для устранения зуда и атопического дерматита, вызываемого аллергическим дерматитом собакообразных. При этом, оклацитиниб обладает даже большей эффективностью при лечении аллергического дерматита у домашних собак. Так как оклацитиниб оказывает слабое влияние на цитокины, не участвующие в активации JAK1, оклацитиниб может лишь ингибировать высвобождение аллергической среды, провоцирующей анафилактическую реакцию, и не может непосредственно блокировать связывание такой аллергической среды с родственными рецепторами и, соответственно, не может блокировать развитие и прогрессирование аллергического дерматита, что ведет вследствие к ограничению области применения оклацитиниба. Другой ингибитор JAK, коим является барицитиниб, представляет собой избирательный ингибитор JAK1 и JAK2; причем IC50 составляет, соответственно, 5,9 и 5,7 нМ, что примерно в 70 и 10 раз превышает степень избирательности, действующей в отношении JAK3 и Tyk2, при этом он не оказывает ингибирующего действия на c-Met и Chk2. Барицитиниб - это своего рода лекарственное средство от ревматоидного артрита, разработанное компанией «Эли Лили энд Компани» совместно с партнерской компаний «Инсайт», которое имеет относительно узкие показания. Другие показания барицитиниба, в частности, псориаз и диабетическая нефропатия, находятся на этапе клинической разработки фазы II. Для устранения недостатков известного уровня технических достижений и расширения области применения подобного типа ингибиторов JAK настоящим изобретением предлагается ингибитор JAK, который можно применять, как на людях, так и на животных; причем этот ингибитор обладает такими преимуществами, как высокая эффективность, широкий диапазон действия и низкая токсичность.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ
Цель настоящего изобретения состоит в том, чтобы предложить ингибитор JAK, а другая цель настоящего изобретения состоит в том, чтобы предложить способ получения ингибитора JAK.
Указанные цели настоящего изобретения достигаются следующими техническими решениями:
Настоящим изобретением предлагается соединение, представленное общей формулой I:
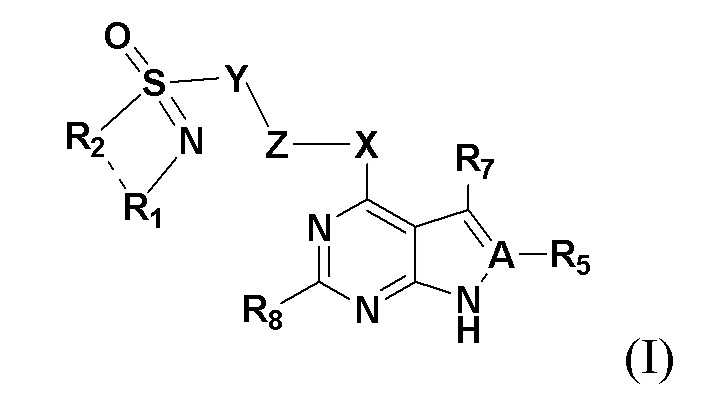
где A представляет собой на выбор C либо N; когда А представляет собой N, R5 отсутствует; когда A представляет собой C, R5 представляет собой на выбор: H, галоген, гидрокси, циано, замещенный или незамещенный алкил, замещенный или незамещенный галогеналкил, замещенный или незамещенный алкокси, замещенный или незамещенный гидроксиалкил, замещенный или незамещенный циклоалкил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный амино, замещенный или незамещенный сульфо либо замещенный или незамещенный сульфонил.
Предпочтительно R5 представляет собой на выбор: H, C1-3-алкил, а также -OC0-2-алкил.
Более предпочтительно R5 представляет собой на выбор: H, а также -CH3.
X представляет собой на выбор: -O- либо 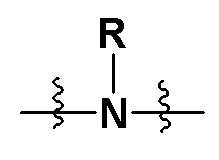 ; R представляет собой на выбор: H, C1-10 линейный или разветвленный алкил, C1-10 линейный или разветвленный алкенил, C1-10 линейный или разветвленный алкинил, C6-18-арил, C6-18 гетероциклоарил, C3-10 циклоалкил, -OC0-10-алкил либо -O-гетероциклоалкил; при этом H на атоме углерода может быть замещен такими группами, как дейтеро, гидрокси, галоген, -CN, -OCH2F, -OCHF2, -OCF3, C1-10 линейный или разветвленный алкил, -N(C0-10-алкил) (C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил или -S-гетероциклоарил; где алкильная фрагмент указанных групп может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как -SO2, -SO2N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил)SO2(C0-10-алкил), -CON (C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил), CO(C0-10-алкил), -N(C0-10-алкил), COO(C0-10-алкил), -OCON (C0-10-алкил) (C0-10-алкил), галоген, -CN, -OCH2F, -OCHF2, -OCF3, -N(C0-10-алкил) (C0-10-алкил), -OC0-10-алкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил;
; R представляет собой на выбор: H, C1-10 линейный или разветвленный алкил, C1-10 линейный или разветвленный алкенил, C1-10 линейный или разветвленный алкинил, C6-18-арил, C6-18 гетероциклоарил, C3-10 циклоалкил, -OC0-10-алкил либо -O-гетероциклоалкил; при этом H на атоме углерода может быть замещен такими группами, как дейтеро, гидрокси, галоген, -CN, -OCH2F, -OCHF2, -OCF3, C1-10 линейный или разветвленный алкил, -N(C0-10-алкил) (C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил или -S-гетероциклоарил; где алкильная фрагмент указанных групп может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как -SO2, -SO2N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил)SO2(C0-10-алкил), -CON (C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил), CO(C0-10-алкил), -N(C0-10-алкил), COO(C0-10-алкил), -OCON (C0-10-алкил) (C0-10-алкил), галоген, -CN, -OCH2F, -OCHF2, -OCF3, -N(C0-10-алкил) (C0-10-алкил), -OC0-10-алкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил;
Предпочтительно X представляет собой на выбор: ; R представляет собой на выбор: H, C1-10 линейный алкил, а также C3-10-циклоалкил; H на атоме углерода может замещаться такими группами, как дейтеро, гидрокси, галоген, -CN, -OCH2F, -OCHF2, -OCF3, C1-3 линейный алкил, -N(C0-3-алкил)(C0-3-алкил), -OC0-6-алкил либо C3-8-циклоалкил; где алкильный фрагмент указанных групп может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как -SO2, -SO2N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил) SO2(C0-10-алкил), -CON (C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил), CO(C0-10-алкил), -N(C0-10-алкил), COO(C0-10-алкил), -OCON(C0-10-алкил) (C0-10-алкил), галоген, -CN, -OCH2F, -OCHF2, -OCF3, -N(C0-10-алкил) (C0-10-алкил), -OC0-10-алкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил.
Более предпочтительно X представлял собой на выбор: ; R представляет собой на выбор C1-6 линейный алкил; H на атоме углерода может замещаться такиим группами, как дейтеро, гидрокси, -CN, -OCH2F, -OCHF2, -OCF3, C1-3 линейный алкил либо C3-6-циклоалкил;
В предпочтительных вариантах осуществления настоящего изобретения X представляет собой .
.
Y представляет собой на выбор: 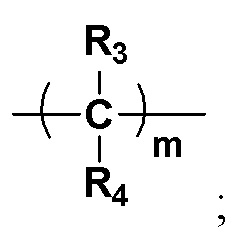 R3 и R4 представляют собой каждый на выбор: H, галоген, -CN, C1-10 линейный алкил, C3-10-циклоалкил, -CF3, -OCF3, -OCHF2, -OCH2F, замещенный или незамещенный галогеналкил, замещенный или незамещенный арил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, -OC0-10-алкил, -S(O)mC0-10-алкил, -SO2N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил) C(=O)(C0-10-алкил), -N(C0-10-алкил)C(=O)O(C0-10-алкил), -N(C0-10-алкил)C(=O)N(C0-10-алкил), -C(=O)C0-10-алкил, -C(=O)OC0-10-алкил, -C(=O)N(C0-10-алкил) (C0-10-алкил), -O-гетероциклоалкил, -N(C0-10-алкил) гетероциклоалкил, -N(C0-10-алкил) гетероциклоарил, -S-гетероциклоарил либо -O-гетероциклоарил, где гетероциклоалкил может замещаться любой одной или любыми несколькими из таких групп, как кислород, C1-10-алкил, C1-10-алкенил, C1-10-алкинил, C6-18-арил, C(=O)OC0-10-алкил, C(=O)N(C0-10-алкил) (C0-10-алкил), -SO2N(C0-10-алкил) (C0-10-алкил) либо SO2C1-10-алкил, где алкильный фрагмент может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как гидрокси, -OC1-10-алкил, -N(C0-10-алкил) (C0-10-алкил), -C(=O)N(C0-10-алкил) (C0-10-алкил), C(=O)OC0-10-алкил, C6-18-арил, гетероциклоалкил либо гетероциклоарил, m представляет собой любое целое число от 0 до 6, например, 0, 1, 2, 3, 4, 5 или 6;
R3 и R4 представляют собой каждый на выбор: H, галоген, -CN, C1-10 линейный алкил, C3-10-циклоалкил, -CF3, -OCF3, -OCHF2, -OCH2F, замещенный или незамещенный галогеналкил, замещенный или незамещенный арил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, -OC0-10-алкил, -S(O)mC0-10-алкил, -SO2N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил) (C0-10-алкил), -N(C0-10-алкил) C(=O)(C0-10-алкил), -N(C0-10-алкил)C(=O)O(C0-10-алкил), -N(C0-10-алкил)C(=O)N(C0-10-алкил), -C(=O)C0-10-алкил, -C(=O)OC0-10-алкил, -C(=O)N(C0-10-алкил) (C0-10-алкил), -O-гетероциклоалкил, -N(C0-10-алкил) гетероциклоалкил, -N(C0-10-алкил) гетероциклоарил, -S-гетероциклоарил либо -O-гетероциклоарил, где гетероциклоалкил может замещаться любой одной или любыми несколькими из таких групп, как кислород, C1-10-алкил, C1-10-алкенил, C1-10-алкинил, C6-18-арил, C(=O)OC0-10-алкил, C(=O)N(C0-10-алкил) (C0-10-алкил), -SO2N(C0-10-алкил) (C0-10-алкил) либо SO2C1-10-алкил, где алкильный фрагмент может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как гидрокси, -OC1-10-алкил, -N(C0-10-алкил) (C0-10-алкил), -C(=O)N(C0-10-алкил) (C0-10-алкил), C(=O)OC0-10-алкил, C6-18-арил, гетероциклоалкил либо гетероциклоарил, m представляет собой любое целое число от 0 до 6, например, 0, 1, 2, 3, 4, 5 или 6;
Предпочтительно R3 и R4 представляют собой каждый на выбор: H, галоген, -CN, C1-6 линейный алкил, а также C3-6-циклоалкил; при этом алкильный фрагмент может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как гидрокси, -OC1-10-алкил, -N(C0-10-алкил) (C0-10-алкил), C6-18-арил, гетероциклоалкил или гетероциклоарил; m равно на выбор 0, 1, 2, 3 либо 4;
Более предпочтительно R3 и R4 представляют собой каждый на выбор: H; m равно на выбор 0, 1 либо 2;
Z представляет собой на выбор: C1-10 линейный или разветвленный алкил, C1-10 линейный или разветвленный алкенил, C1-10 линейный или разветвленный алкинил, замещенный или незамещенный гидроксиалкил, C3-12-циклоалкил, C1-20-алкокси, C3-12-циклоалкокси, гетероциклоалкил, (-N, -O и -S), C6-18-арил, -N-гетероциклоарил, -S-гетероциклоарил либо -O-гетероциклоарил, ароматический дицикло, ароматический гетеродицикло и трицикло, где алкильный фрагмент может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как -N(C0-10-алкил) (C0-10-алкил), -CON(C0-10-алкил)(C0-10-алкил), -N(C0-10-алкил)CO(C0-10-алкил), -N(C0-10-алкил) COO(C0-10-алкил), -OCON(C0-10-алкил)(C0-10-алкил), галоген, -CN, -OCH2F, -OCHF2, -OCF3, -OC0-10-алкил, C6-18-алкил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил.
Предпочтительно Z представляет собой на выбор: C3-12-циклоалкил или C3-12-циклоалкокси; где алкильный фрагмент может в необязательном порядке замещаться любой одной или любыми несколькими из таких групп, как -N(C0-10-алкил) (C0-10-алкил), -CON(C0-10-алкил)(C0-10-алкил), -N(C0-10-алкил)CO(C0-10-алкил), -N(C0-10-алкил) COO(C0-10-алкил), -OCON(C0-10-алкил)(C0-10-алкил), галоген, -CN, -OCH2F, -OCHF2, -OCF3, -OC0-10-алкил, C6-18-алкил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил.
Более предпочтительно Z представляет собой 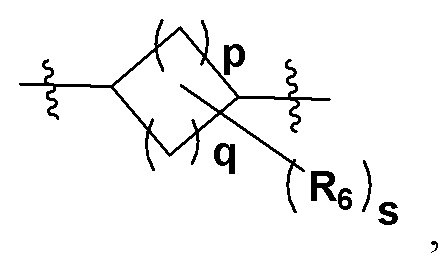 где p представляет собой любое целое число от 0 до 4; q - любое целое число от 0 до 4; p и q при этом не равны ; R6 представляет собой замещающий Н элемент на одном или нескольких атомах углерода в составе циклоалкила; R6 представляет собой на выбор C1-6-алкил либо C3-6-циклоалкил, а s представляет собой целое число от 0 до 8; например, 0, 1, 2, 3, 4 и 5.
где p представляет собой любое целое число от 0 до 4; q - любое целое число от 0 до 4; p и q при этом не равны ; R6 представляет собой замещающий Н элемент на одном или нескольких атомах углерода в составе циклоалкила; R6 представляет собой на выбор C1-6-алкил либо C3-6-циклоалкил, а s представляет собой целое число от 0 до 8; например, 0, 1, 2, 3, 4 и 5.
Предпочтительно R6 представляет собой на выбор: C1-3-алкил.
Более предпочтительно R5 представляет собой на выбор: -CH3, либо -CH2CH3.
В предпочтительных вариантах осуществления настоящего изобретения Z представляет собой на выбор: C4-10-циклоалкил, например, 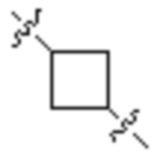 ,
, 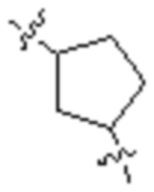 ,
, 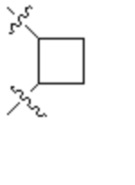 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
Более предпочтительно Z представляет собой .
R7 и R8 представляют собой каждый на выбор: H, галоген, гидрокси, циано, замещенный или незамещенный алкил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, замещенный или незамещенный галогеналкил, замещенный или незамещенный алкокси, замещенный или незамещенный гидроксиалкил, замещенный или незамещенный циклоалкил, замещенный или незамещенный негетероциклоарил, замещенный или незамещенный гетероциклоарил, замещенный или незамещенный гетероциклоалкил, замещенный или незамещенный амино, замещенный или незамещенный сульфо либо замещенный или незамещенный сульфонил.
Предпочтительно R7 и R8 представляют собой каждый на выбор: H, галоген, C1-3-алкил, а также -OC0-2-алкил.
Более предпочтительно R7 и R8 представляют собой каждый на выбор: H либо -CH3.
Соединение, описываемое в настоящим изобретении, имеет следующую структуру:
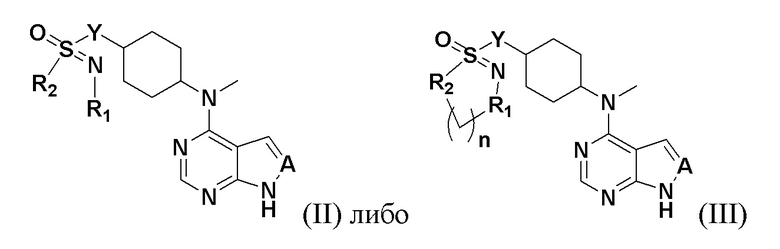
где n представляет собой положительное целое число от 1 до 4; предпочтительно n равно 1 или 2; при этом соединение, представленное цифрой III, имеет следующую структурную формулу:
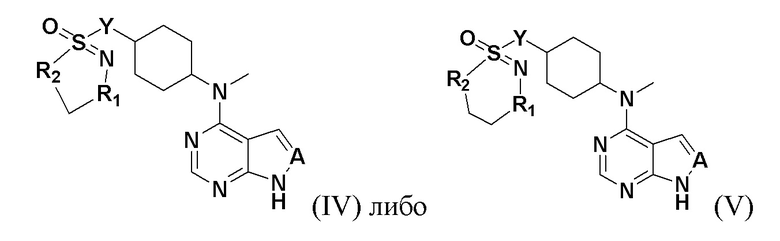
R1 и R2 представляют собой каждый на выбор: H, галоген, -CN, -OCH2F, -OCHF2, -OCF3, C1-10 линейный или разветвленный алкил, C3-10-циклоалкил, -OC0-10-алкил, -N(C0-10-алкил)(C0-10-алкил), -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил, C6-18-арил, -N-гетероциклоарил, -S-гетероциклоарил либо -O-гетероциклоарил, где H на атоме углерода может замещаться такими группами, как дейтеро, гидрокси, галоген, -CN, -OCH2F, -OCHF2, -OCF3, C1-6 линейный алкил, -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил, C6-18-арил (например, фенил), -N-гетероциклоарил, -O-гетероциклоарил или -S-гетероциклоарил; где H на C6-18-ариле (например, фениле) или гетероциклоариле может замещаться любой одной или любыми несколькими из таких групп, как галоген, C1-4 линейный алкил, -N (C0-10-алкил)SO2(C0-10-алкил), -CON(C0-10-алкил)(C0-10-алкил) , N-(C0-10-алкил)CO(C0-10-алкил), -N(C0-10-алкил)COO(C0-10-алкил), -OCON(C0-10-алкил)(C0-10-алкил), -CN, -OCH2F, -OCHF2, -OCF3, -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, -N-гетероциклоарил, -O-гетероциклоарил или -S-гетероциклоарил, или же где соседние атомы углерода на C6-18-ариле (например, фениле) и гетероциклоариле образуют C3-8-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил либо -N-гетероциклоарил, -O-гетероциклоарил, -S-гетероциклоарил; или же имеющиеся между ними атомы R1, R2, S и N образуют гетероциклическое кольцо; причем предпочтительно такое гетероциклическое кольцо представляет собой пятичленный гетероцикл либо шестичленный гетероцикл.
Предпочтительно R1 представляет собой на выбор: H, C1-6 линейный алкил либо C3-6-циклоалкил; а H на атоме углерода может замещаться такими группами, как дейтеро, гидрокси, галоген, -CN, -OCF3, -N(C0-10-алкил)(C0-10-алкил), -OC0-4-алкил, C3-10-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, а также C6-18-арил (например, фенил).
Более предпочтительно R1 представляет собой на выбор: H, -CH2-, -CH3, -CH2CH3, - CH2CH2CH3, -CH2CH2OCH3,  ;
;
R2 представляет собой на выбор: C1-6 линейный алкил, C3-6-циклоалкил, C3-8-циклоалкокси, -N(C0-10-алкил)(C0-10-алкил), C6-18-арил (например, фенил), а также -N-гетероциклоарил; H на атоме углерода или азота может замещаться любой одной или любыми несколькими из таких групп, как дейтеро, гидрокси, галоген, -CN, OCH2F, -OCHF2, -OCF3, C1-3 линейный алкил, -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, C6-18-арил (например, фенил), -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил; соседние атомы углерода на фениле либо гетероциклоариле образуют C3-8-циклоалкил, -O-гетероциклоалкил, -N-гетероциклоалкил, -S-гетероциклоалкил либо -N-гетероциклоарил и -O-гетероциклоарил.
Более предпочтительно R2 представляет собой на выбор: -CH2-,
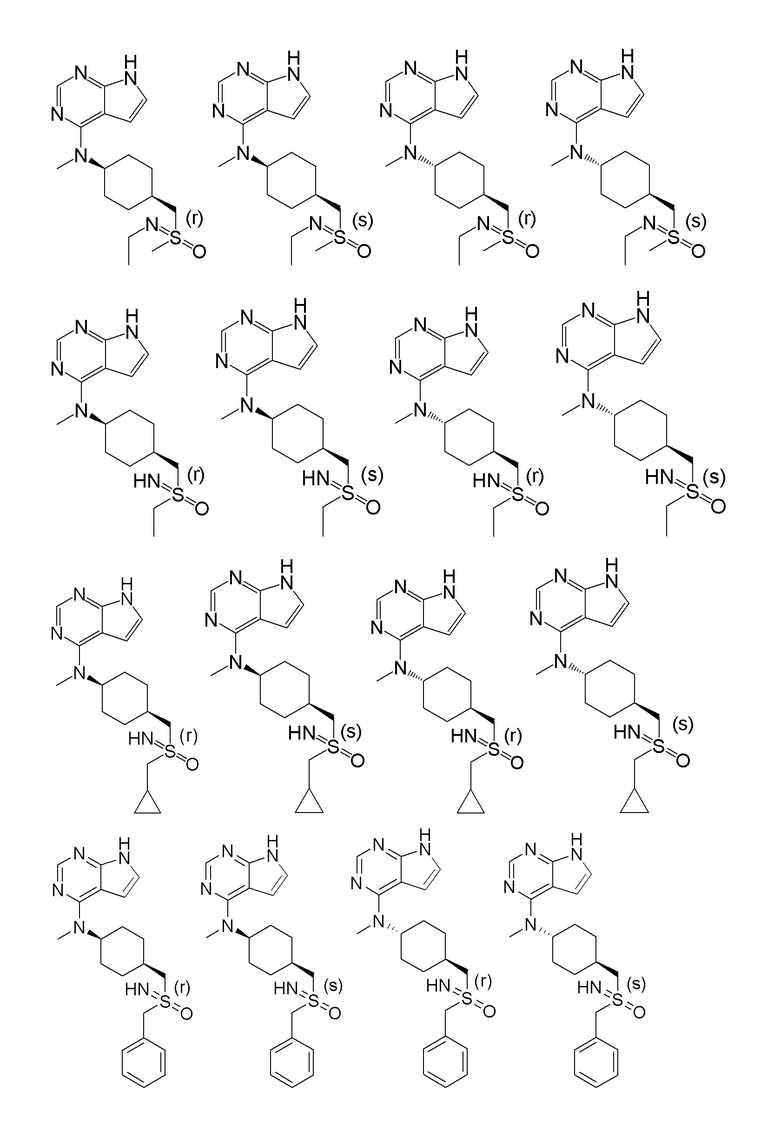
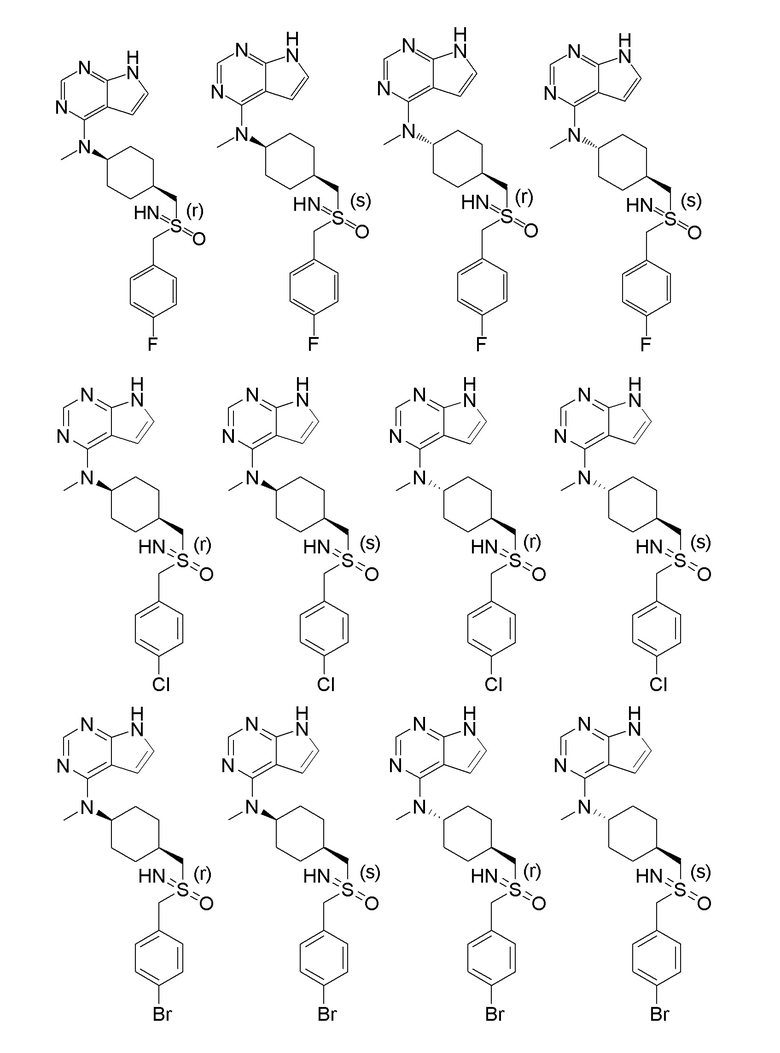
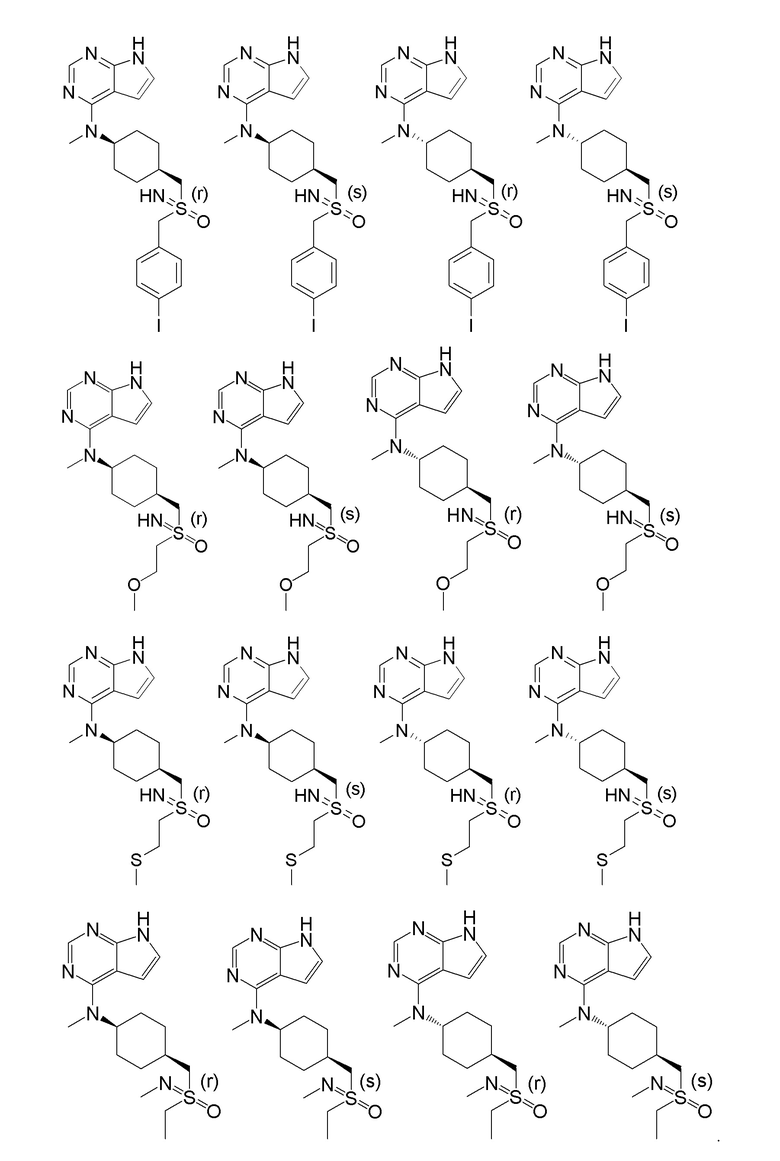
В детализированных вариантах осуществления настоящего изобретения предлагаются такие конкретные соединения:
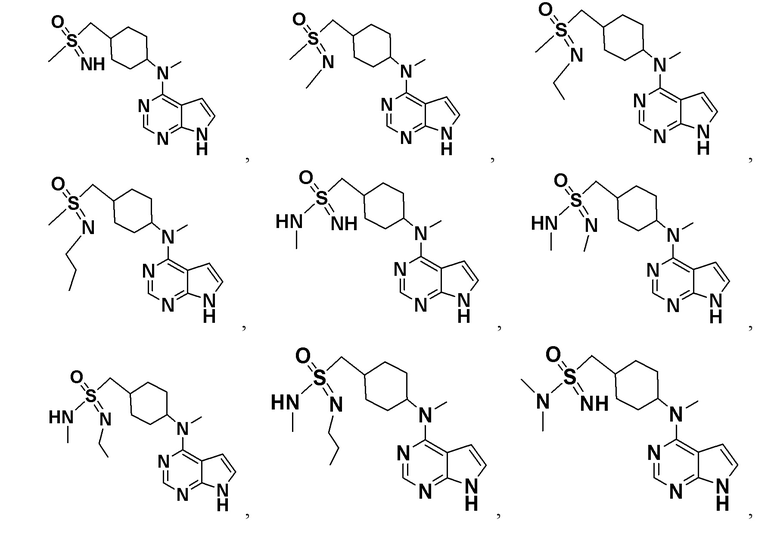
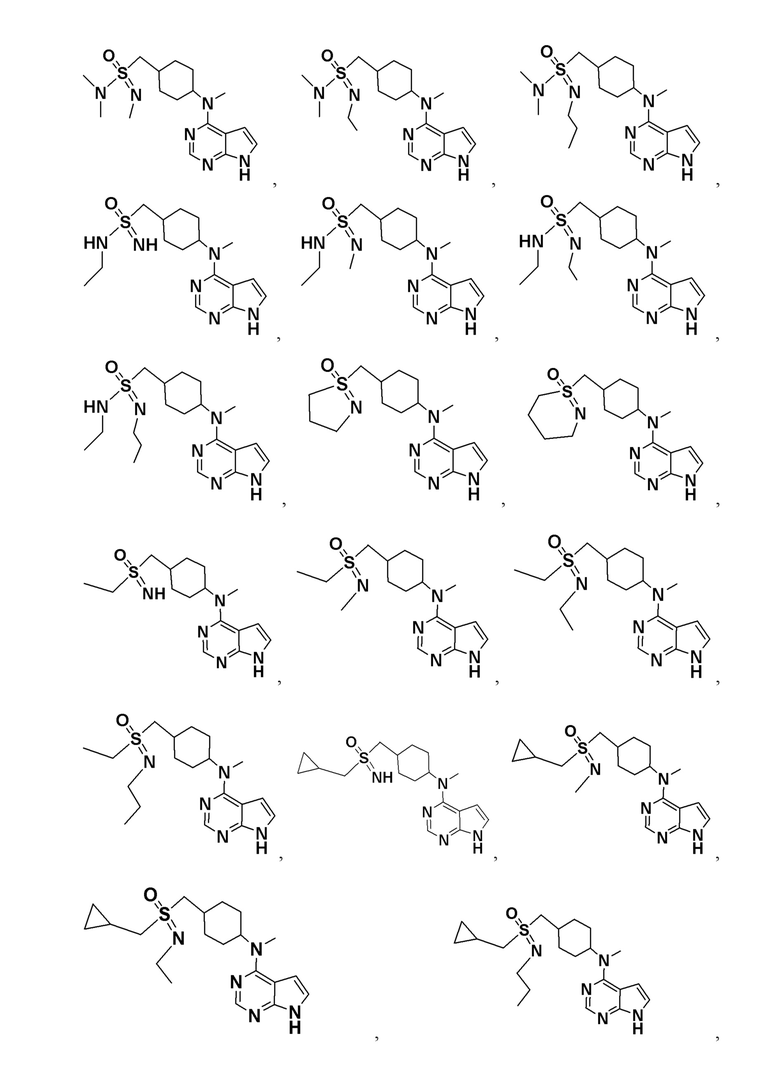
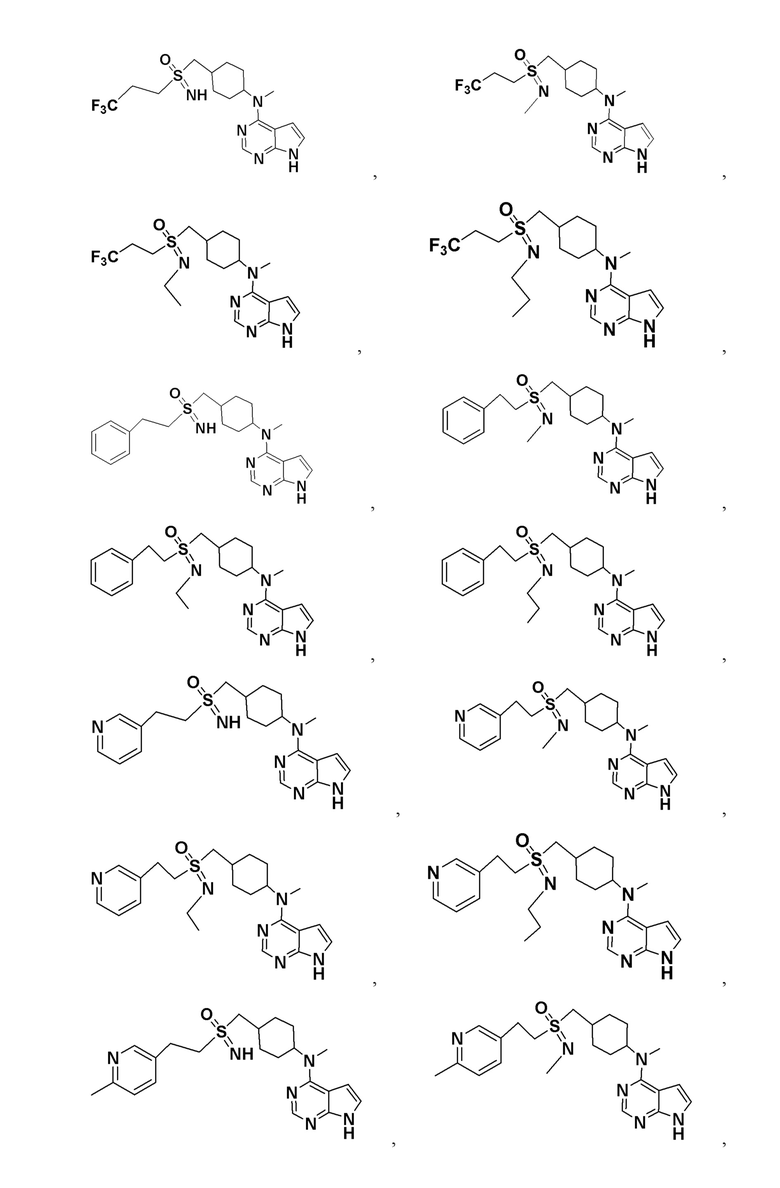
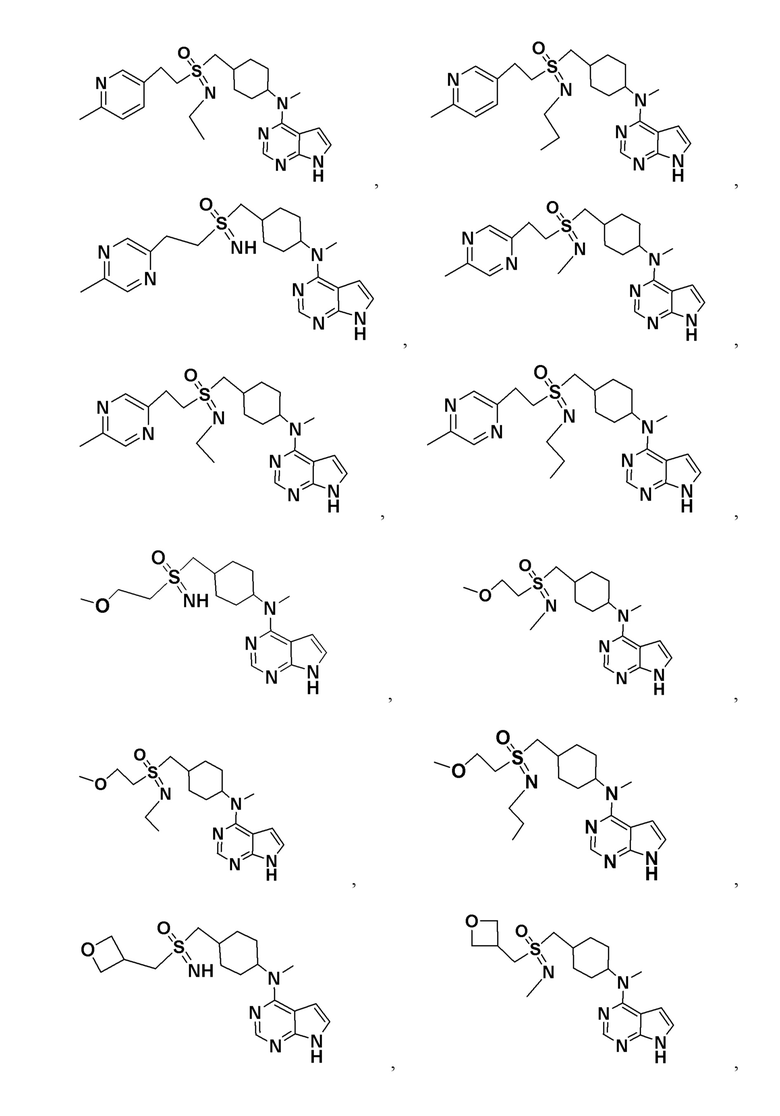
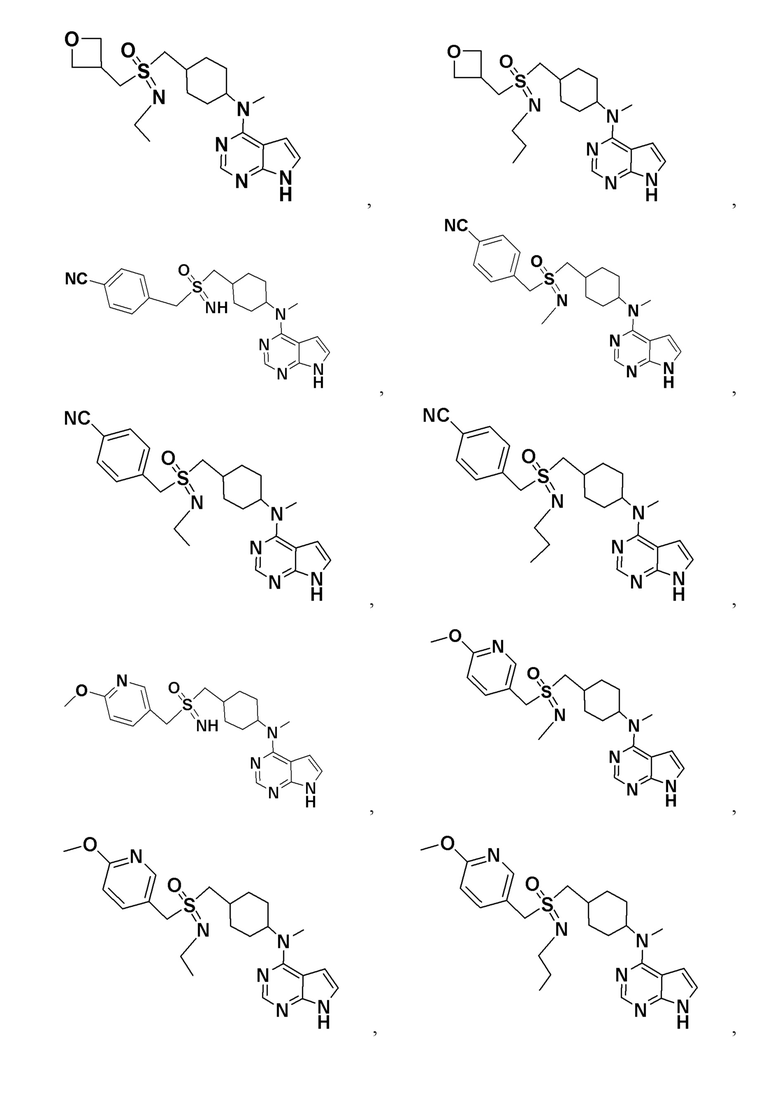
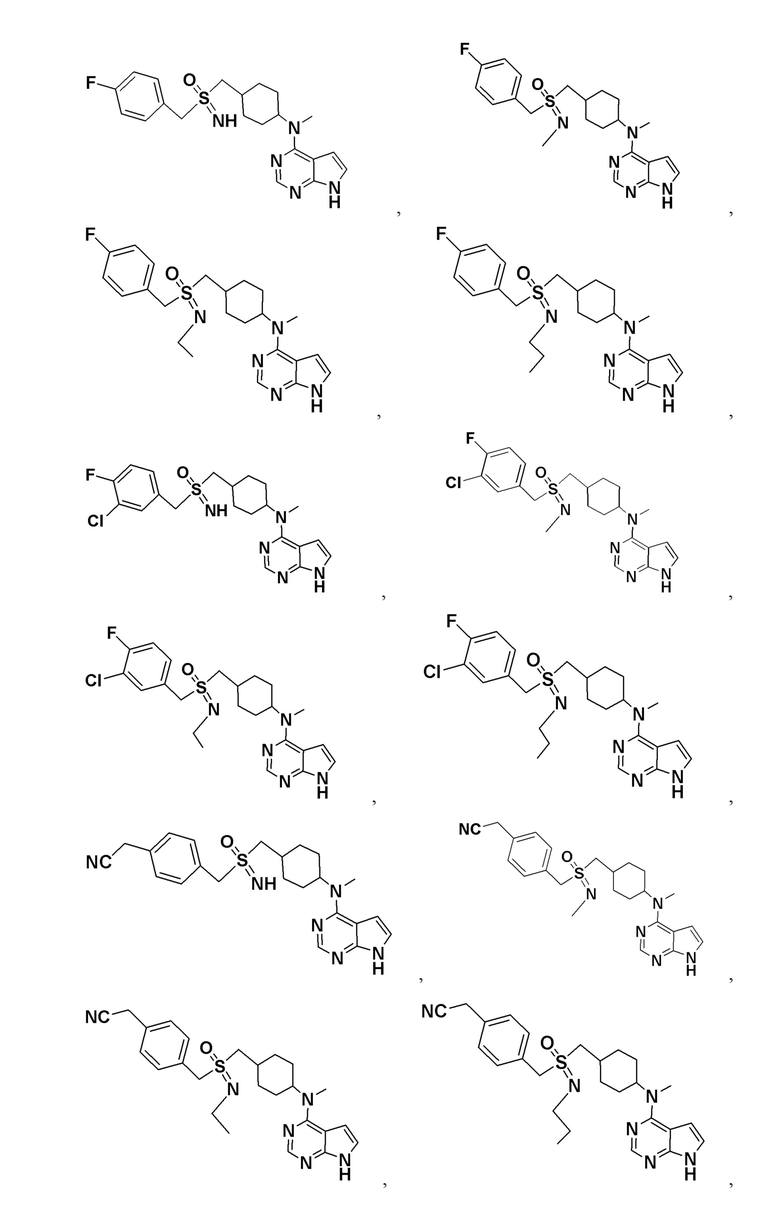
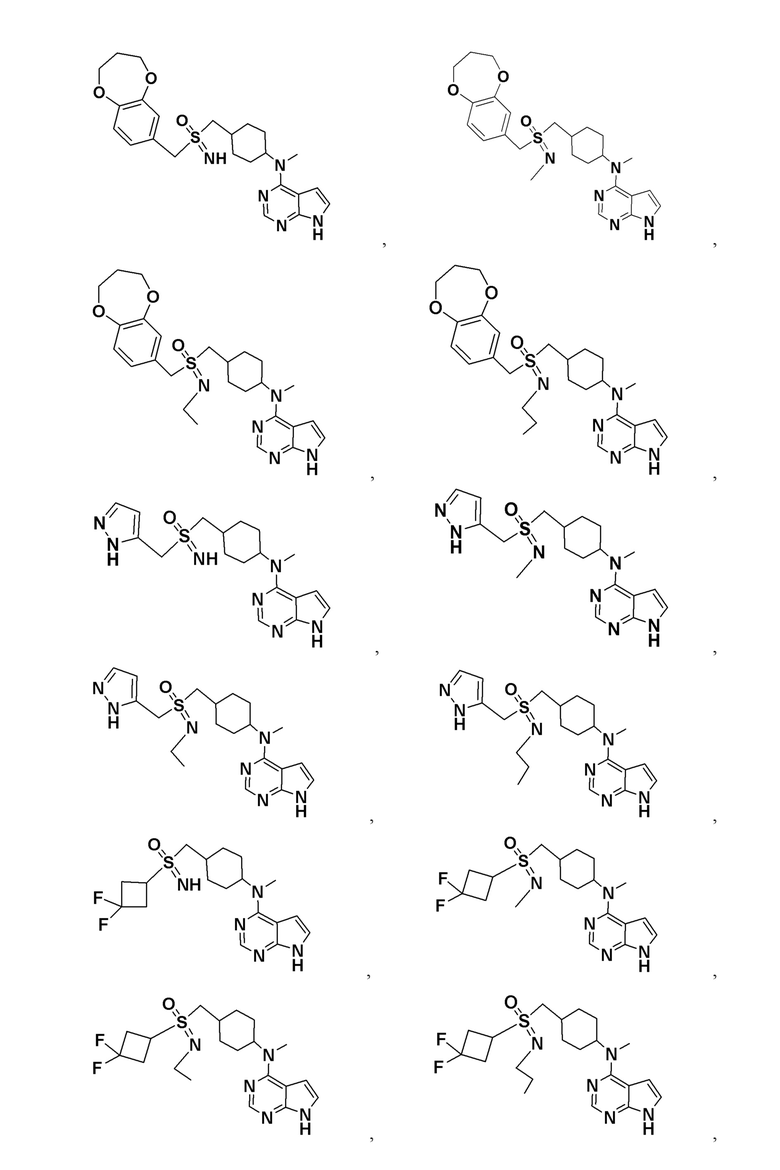
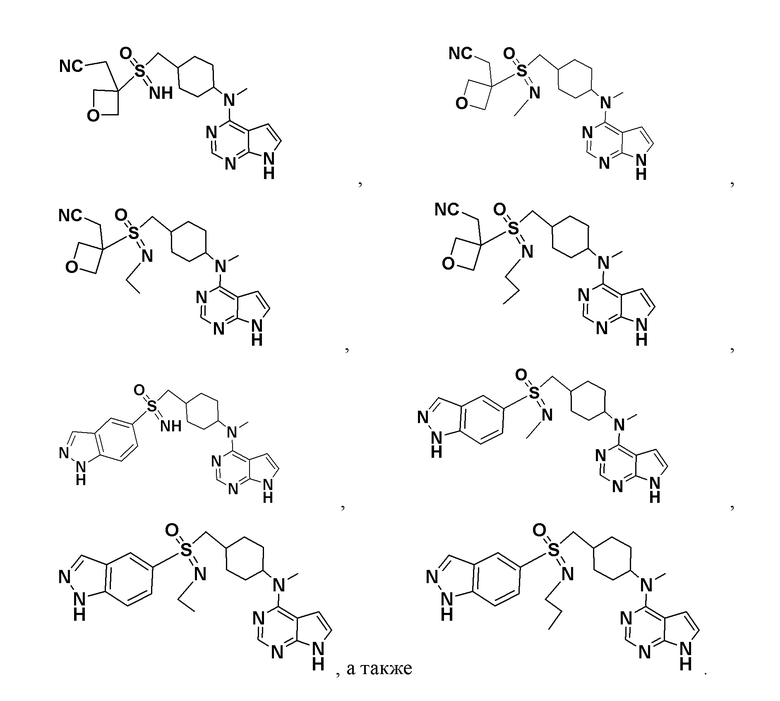
Соединение, представленное общей формулой I, также включает в себя его фармацевтически приемлемую соль, стереоизомер, сложный эфир, пролекарство, метаболит, сольват либо дейтерированное соединение.
В одном из вариантов осуществления настоящего изобретения стереоизомер соединения, представленного общей формулой I, имеет следующую структуру:
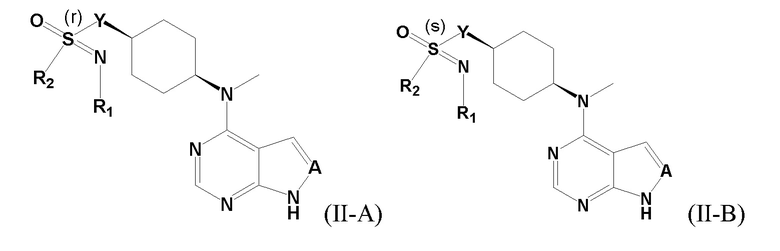
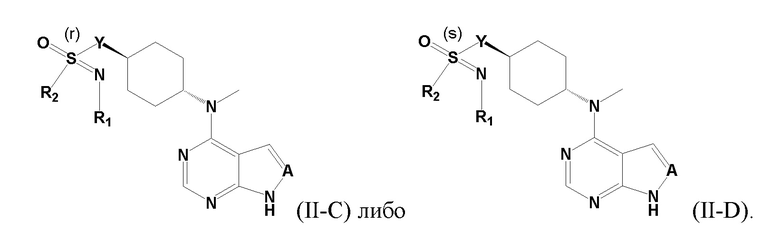
В еще одном из вариантов осуществления настоящего изобретения стереоизомер соединения, представленного приведенной выше общей формулой I, имеет следующую структуру:
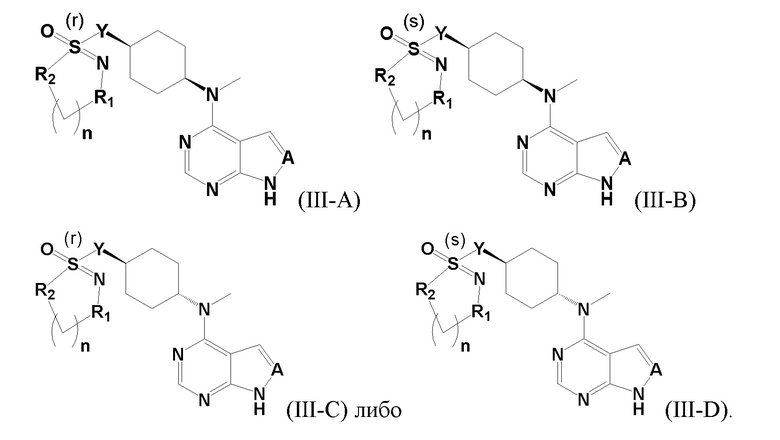
В детализированных вариантах осуществления настоящего изобретения предлагается стереоизомер такого конкретного соединения:
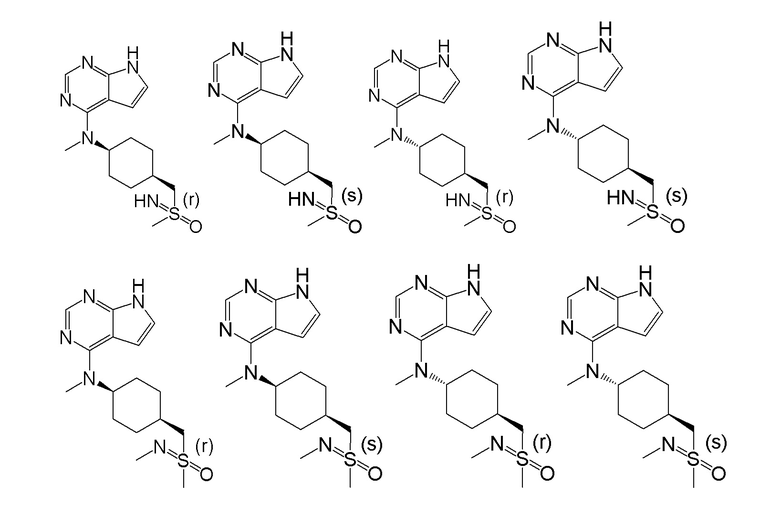
Соединение, представленное в настоящем изобретении общей формулой I, может быть получено в результате такого пути прохождения реакции:
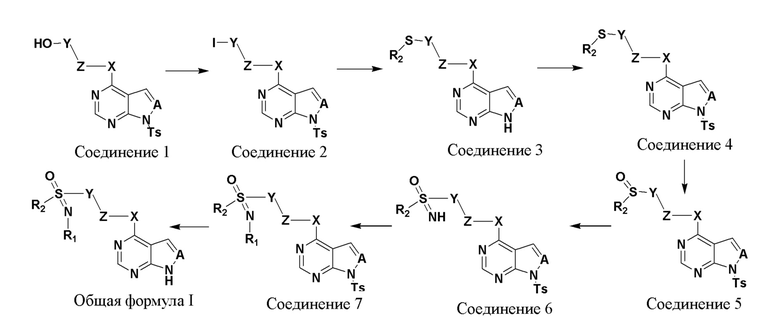
(1) растворение соединения 1 в растворителе 1, добавление триэтиламина и паратолуолсульфонилхлорида, перемешивание при комнатной температуре в течение 20-24 часов, концентрирование, добавление йодирующего реагента, нагревание до 60-70°C и перемешивание в течение 8-9 часов с получением соединения 2;
(2) растворение соединения 2 в растворителе 2, добавление алкилтиолата натрия, обеспечение прохождения реакции в течение 20-24 часов, фильтрование и концентрирование с получением соединения 3;
(3) растворение соединения 3 в растворителе 3, добавление триэтиламина и паратолуолсульфонилхлорида, перемешивание при комнатной температуре в течение 20-24 часов и сепарирование с получением соединения 4;
(4) растворение соединения 4 в растворителе 4, добавление метахлорпербензойной кислоты (m-CPBA), обеспечение прохождения реакции в течение 1-2 часов, экстракция со сбором органической фазы, промывка водой и сушка такой органической фазы, фильтрование и концентрирование с получением соединения 5;
(5) растворение соединения 5 в растворителе 5, добавление иодбензолдиацетата (PhI(OAc) 2) и карбамата аммония, обеспечение прохождения реакции в течение 30-35 мин, концентрирование при пониженном давлении с получением соединения 6;
(6) растворение соединения 6 и полиальдегида в растворителе 6, нагревание до 90-95°C, обеспечение прохождения реакции в течение 20-24 часов, концентрирование, экстракция со сбором органической фазы, промывка водой и сушка такой органической фазы, фильтрование и концентрирование с получением соединения 7; а также
(7) растворение соединения 7 и карбоната цезия (Cs2CO3) в растворителе 7, обеспечение прохождения реакции при 40-50°C в течение 3-4 часов, фильтрование и концентрирование с получением соединения, представленного общей формулой I.
Растворители 1-7 на вышеприведенном этапе получения представляют каждый одно либо сочетание двух или более из таких веществ, как дихлорметан (DCM), ацетон, тетрагидрофуран (THF), метиловый спирт и муравьиная кислота; предпочтительно растворитель 1 представляет собой DCM; растворитель 2 - THF; растворитель 3 - DCM; растворитель 4 - THF; растворитель 5 - метиловый спирт; растворитель 6 - муравьиную кислоту; а растворитель 7 - смешанный раствор THF и метилового спирта.
Предпочтительно продолжительность перемешивания при комнатной температуре на этапе (1) составляет 24 часа, а йодирующим реагентом выступает иодид натрия.
Продолжительность реакции на этапе (2) составляет 24 часа.
Сепарирование на этапе (3) выполняется путем отделения с помощью колоночной хроматографии.
После добавления m-CPBA на этапе (4) смешанный материал в течение 1 часа подвергают реакции на ледяной бане при 0°C; затем реакционную жидкость гасят насыщенным раствором NaHCO3 , экстрагируют с помощью DCM, органические слои смешивают, затем промывают насыщенным солевым раствором, сушат с помощью безводного Na2SO4 и в конечном итоге фильтруют.
Полиальдегид на этапе (6) предпочтительно представляет собой параформальдегид; реакционную жидкость концентрируют, а затем доводят насыщенным NaHCO3 до pH=7-8, экстрагируют этилацетатом; при этом органические слои смешивают, затем промывают насыщенным солевым раствором, сушат безводным Na2SO4 и в конечном итоге фильтруют.
Температура реакции на этапе (7) составляет 40°C, а продолжительность реакции составляет 3 часа.
Настоящим изобретением предлагается фармацевтический состав; причем такой фармацевтический состав содержит соединение, представленное общей формулой I, а также содержит фармацевтически приемлемое вспомогательное вещество; причем такое вспомогательное вещество представляет собой на выбор носитель, разбавитель, связывающий материал, смазывающее вещество и смачивающее вещество. Предпочтительно фармацевтический состав содержит обладающее терапевтическим эффектом количество соединения, представленного общей формулой I. В некоторых вариантах осуществления фармацевтический состав может использоваться отдельно либо в сочетании с другими ингибиторами JAK.
Предпочтительно фармацевтический состав можно вводить людям и/или животным.
Фармацевтический состав подходит для энтерального либо парентерального введения, в частности, внутривенного, внутримышечного, внутрикожного и подкожного введения. Следовательно, предпочтительно фармацевтический состав дополнительно включает в себя антиоксидант, буферное вещество, бактериостатическое вещество и растворенное вещество, делающее препараты и кровь субъекта и тому подобное изотоническими, а также водную и неводную стерильную суспензию, которая может включать в себя суспендирующее вещество, солюбилизатор, загуститель, стабилизатор и консервант.
Описываемое в настоящем изобретении соединение может быть получено в формах фармацевтических препаратов, как сироп, эликсир, суспендирующее вещество, порошок, гранулы, таблетки, капсулы, паста, водный раствор, крем, мазь, лосьон, гель, эмульсия и тому подобное.
Фармацевтический препарат предпочтительно представляет собой порционную форму. В такой форме препарат подразделяется на стандартные дозы, содержащую надлежащее количество действующих веществ. Указанная порционная форма может представлять собой капсулы, таблетки или какую-любо лекарственную форму; более того, такая порционная форма может также представлять собой упакованный препарат, например, упакованные во флакон либо ампулу таблетки, капсулы и порошок.
Количество действующих веществ в указанной порционной форме может меняться либо корректироваться от 0,1 мг до 1000 мг; причем определяется оно в соответствии с конкретной областью применения и с учетом эффективности действующих веществ. При необходимости указанный состав может дополнительно включать в себя и другие подходящие терапевтические средства.
В настоящем изобретении предлагается способ применения соединения, представленного общей формулой I, либо его фармацевтически приемлемой соли, стереоизомера, сложного эфира, пролекарства, метаболита, сольвата или кооперированного соединения при получении лекарственного средства, предназначенного для лечения заболевания, связанного с активированными Янус-киназой переносчиками сигналов и активаторами транскрипции (JAK-STAT).
В настоящем изобретении предлагается способ применения соединения, представленного общей формулой I, либо его фармацевтически приемлемой соли, стереоизомера, сложного эфира, пролекарства, метаболита, сольвата или дейтерированного соединения при получении лекарственного средства, предназначенного для профилактики и/или лечения воспалительных либо онкологических заболеваний у людей и/или животных.
Способ применения соединения, представленного общей формулой I, либо его фармацевтически приемлемой соли, стереоизомера, сложного эфира, пролекарства, сольвата или дейтерированного соединения в соответствии с п.1 формулы при получении лекарственного средства, предназначенного для профилактики и/или лечения воспалительных и онкологических заболеваний у людей и/или животных.
Предпочтительно к указанным воспалительным заболеваниям относят ревматоидный артрит, собачий дерматит, псориаз, язвенный колит либо болезнь Крона; а указанное онкологическое заболевание - миелофиброз, истинная полицитемия, эссенциальная тромбоцитемия, хронический гранулоцитарный лейкоз, рак груди, рак легких и рак поджелудочной железы.
Что касается используемого в настоящем изобретении термина «C0-10-алкил», то под C0-алкил подразумевается H. Следовательно, C0-10-алкил включает в себя H, C1-алкил, C2-алкил, C3-алкил, C4-алкил, C5-алкил, C6-алкил, C7-алкил, C8-алкил, C9-алкил и C10-алкил.
Используемый в настоящем изобретении термин «C3-10-циклоалкил» включает в себя C3-циклоалкил, C4-циклоалкил, C5-циклоалкил, C6-циклоалкил, C7-циклоалкил, C8-циклоалкил, C9-циклоалкил и C10-циклоалкил.
Используемый в настоящем изобретении термин «галоген» включает в себя фтор, хлор, бром и йод.
Описываемая в настоящем изобретении фармацевтически приемлемая соль включает в себя кислотно-аддитивную соль и щелочно-аддитивную соль.
Кислотно-аддитивную соль включает в себя, помимо прочего, соли неорганических кислот, например, хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, серной кислоты, бромистоводородной кислоты, йодистоводородной кислоты и фосфоновой кислоты; а также соли органических кислот, например, алифатической монокарбоновой кислоты и дикарбоновой кислоты, фенилзамещенной парафиновой кислоты, гидроксипарафиновой кислоты, алкандиовой кислоты, ароматической кислоты, и соли алифатических и ароматических сульфоновых кислот. Следовательно, такие соли включают в себя, помимо прочего, сульфаты, пиросульфаты, дисульфаты, сульфиты, гидросульфиты, нитраты, фосфорную кислоту, вторичные кислые фосфаты фосфат, первичные кислые фосфаты, метафосфаты, пирофосфаты, гидрохлориды, гидробромиды, йодаты, ацетаты, пропионаты, каприлаты, изобутилаты, сукцинаты, октандикарбоновую кислоту, себацаты, фумараты, малеаты, менделаты, бензоаты, хлорированные бензоаты, метилбензоаты, бинитробензоаты, фталаты, бензолсульфонаты, тозилаты, фенилацетаты, цитраты, лактаты, малеаты, тартраты и мезилаты, а также включают в себя соли аминокислот, например, соли аргинина, глюконаты, галактуронаты и тому подобное. Кислотно-аддитивная соль может быть получена традиционным способом, а именно путем приведения в соприкосновение свободной щелочной формы с достаточным количеством необходимой для образования соли кислоты. Указанную солевую форму приводят в соприкосновение с щелочью, в результате чего происходит восстановление формы свободной щелочи, а далее традиционным способом производят выделение полученной свободной щелочи.
Щелочно-аддитивная соль и металл либо амин образуют гидроксиды щелочных металлов либо щелочно-земельных металлов или же формы с содержанием органических аминов. Используемый в качестве катионного металла образец включает в себя, помимо прочего, натрий, калий, магний и кальций. Образец с содержанием подходящих аминов включает в себя, помимо прочего, N, N’-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин (этан-1,2-диамин), N-метилглюкозамин и прокаин. Щелочно-аддитивная соль может быть получена традиционным способом, а именно путем приведения в соприкосновение свободной кислотной формы с достаточным количеством необходимой для образования соли щелочи. Указанную солевую форму можно приводить в соприкосновение с кислотой, в результате чего будет происходить восстановление формы свободной кислоты, а далее традиционным способом будет производиться выделение полученной свободной кислоты.
Описываемый в настоящем изобретении стереоизомер представлен в форме энантиомеров, диастереомеров и геометрических изомеров. Некоторые из описываемых в настоящем изобретении соединения содержат циклоалкил, который может замещаться на более чем одном атоме углерода. В таком случае все их геометрические формы, включая цис-формы и транс-формы, а также их сочетания, подпадают под действие правовой охраны по настоящему изобретению.
Описываемый в настоящем изобретении сольват отсылает к физической связи описываемого настоящем изобретении соединения с одним или несколькими растворителями. Такая физическая связь включает в себя различные степени ионных и ковалентных связей, включая водородную связь. В некоторых случаях упомянутый сольват может быть отделен, например, при добавлении к решетке твердого кристаллического тела одной или нескольких молекул растворителя. Понятие «сольват» распространяется на сольваты в фазе раствора либо отделяемые сольваты. К типичным сольватам относят в себя алкоголяты, метилаты и тому подобное. Под «гидратом» понимают сольват, в котором одна или несколько молекул растворителя представляют собой H2O.
Под описываемым в настоящем изобретении пролекарство подразумевается форма соединения, представленного общей формулой I, которая подходит для введения пациенту, не обладает чрезмерной токсичностью, не вызывает раздражительных и аллергических реакций и демонстрирует эффективность при применении по назначению, включая такие формы, как ацеталь, сложный эфир и цвиттер-ион. Пролекарство преобразуется в условиях in vivo (например, гидролизуется в крови) с получением вышеуказанной формулы исходного соединения.
ПОДРОБНОЕ ОПИСАНИЕ
Техническое решение, применяемое в вариантах осуществления настоящего изобретения, будет изложено в подробном и комплексном описании; очевидным является тот факт, что описанные в настоящем документе варианты осуществления дают лишь частичную картину, не представляя собой исчерпывающий перечень вариантов осуществления настоящего изобретения. Все другие варианты осуществления, получаемые специалистами в данной области техники без изобретательских усилий, а на основе указанных вариантов осуществления настоящего изобретения, подпадают под действие правовой охраны по настоящему изобретению.
Вариант осуществления 1: Синтез Т1
Этап 1:
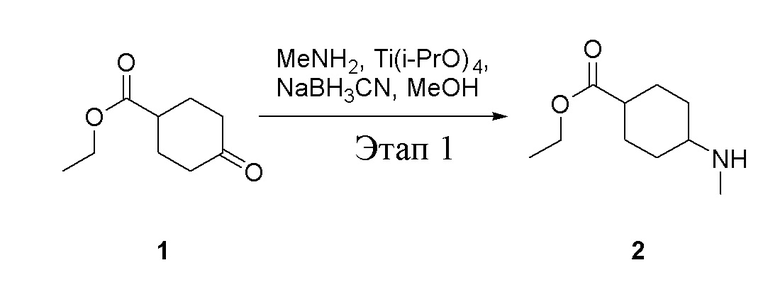
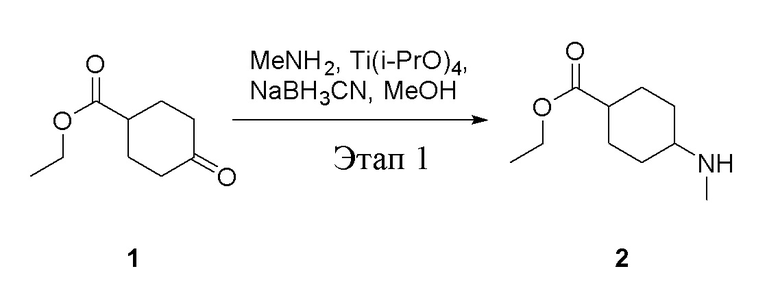
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
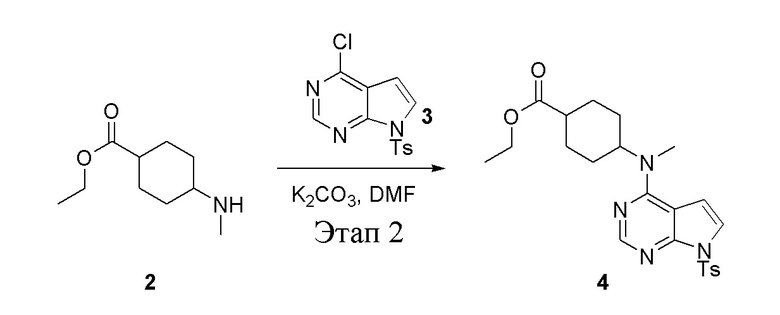
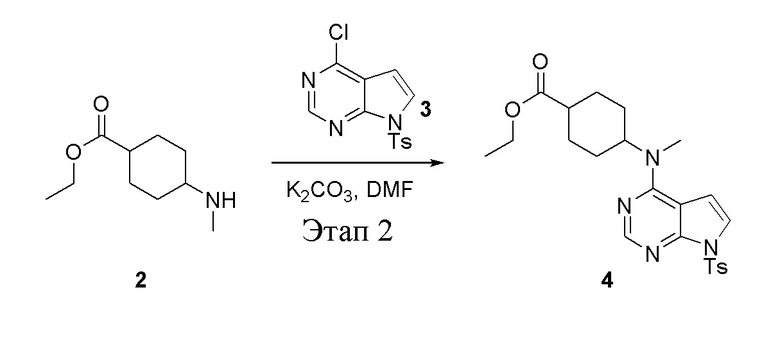
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
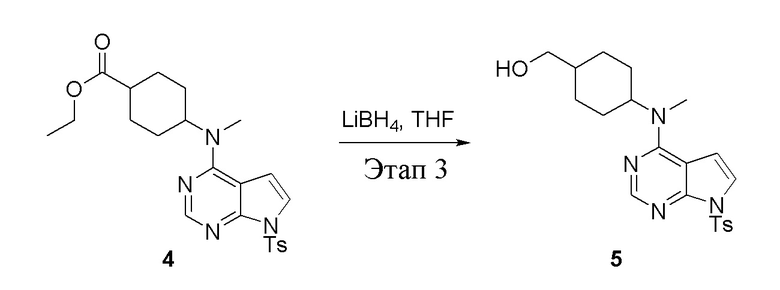
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
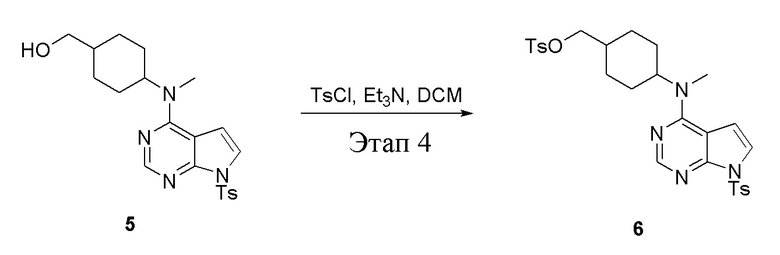
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
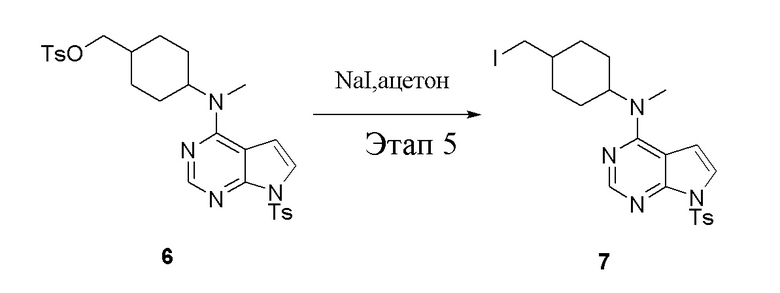
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
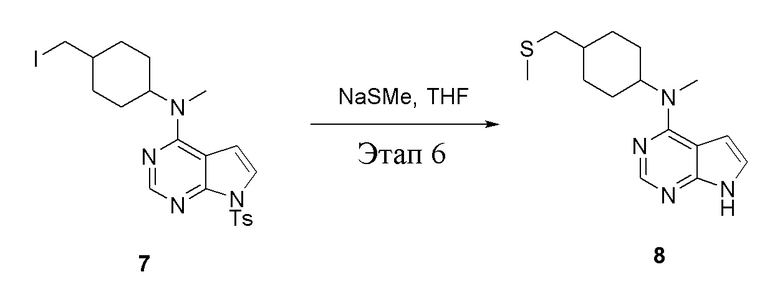
в одногорлую колбу объемом 1 л добавили 7 (25 г, 47,7 ммоль) (продукт, полученный на этапе 5), тетрагидрофуран (200 мл) и метилмеркаптид натрия (6,69 г, 95,4 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (11,5 г, выход: 83,1%). ЖХТМС: 291 [M+H]+
Этап 7:
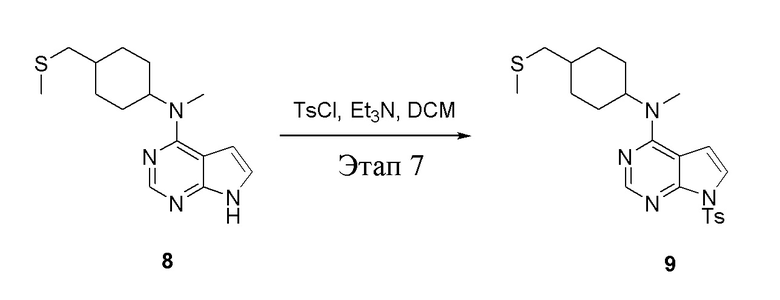
в одногорлую колбу объемом 500 л при 0°C добавили 8 (11,5 г, 39,6 ммоль) (продукт, полученный на этапе 6) и дихлорметан (110 мл), затем последовательно добавляли TsCl (11,3 г, 59,4 ммоль) и триэтиламин (8,0 г, 79,2 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (14,5 г). ЖХТМС: 445 [M+H]+
Этап 8:
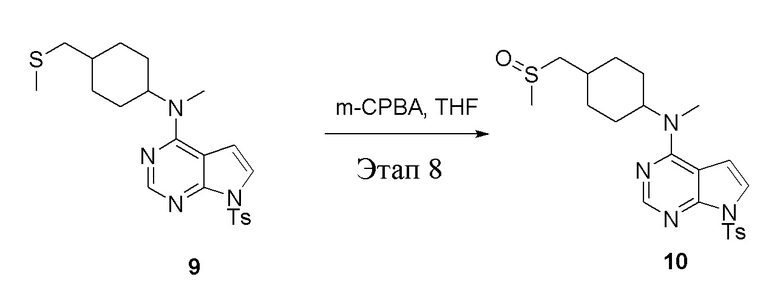
в одногорлую колбу объемом 250 мл при 0°C добавили 9 (14,5 г, 32,65 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (120 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (5,63 г, 32,65 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (100 мл), потом последовательно промыли насыщенным сульфитом натрия (50 мл × 3), бикарбонатом натрия (50 мл × 3) и физиологическим раствором (50 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (7,0 г, выход: 46,6%). ЖХТМС: 461 [M+H]+
Этап 9:
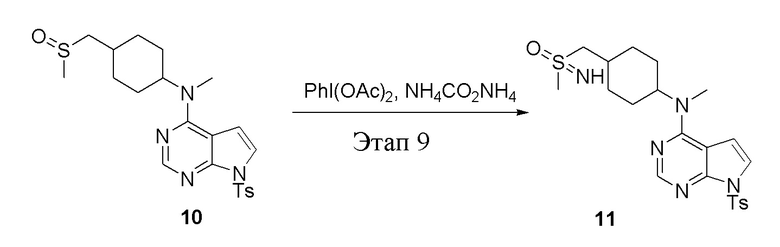
в одногорлую колбу объемом 250 л добавили 10 (7,0 г, 15,21 ммоль) (продукт, полученный на этапе 8), дихлорметан (70 мл), PhI(OAc)2 (7,35 г, 22,82 ммоль) и карбонат аммония (2,92 г, 30,42 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. Остаточные материалы после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (3,0 г, выход: 41,5%). ЖХТМС: 474 [M-H]+
Этап 10:
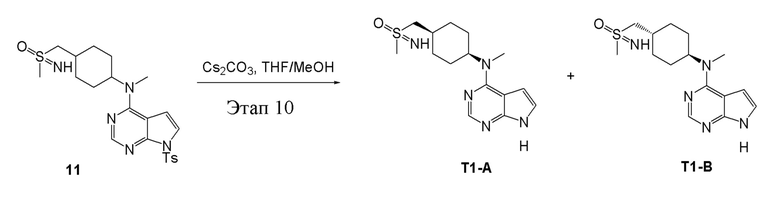
в одногорлую колбу объемом 25 мл добавили 11 (485 мг, 1,02 ммоль), тетрагидрофуран/метиловый спирт (5,0 мл) и карбонат цезия (665 мг, 2,04 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия, концентрировали, после чего с применением традиционного и хирального способов было получено белое твердое вещество, то есть продукт A (95 мг, выход: 29,0%). ЖХТМС:322 [M+H]+,H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,83 (с, 1H), 8,15 (с, 1H), 7,20 (t, J=25,5 Гц, 2H), 6,60 (d, J=2,4 Гц, 1H), 4,64 (с, 1H), 3,38-3,28 (м, 2H), 3,23 (с, 3H), 3,19 (с, 3H), 2,13-1,98 (м, 3H), 1,73 (с, 4H), 1,42-1,29 (м, 2H) и продукт B (85 мг, выход: 25,9%), ЖХТМС:322 [M+H]+, H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,79 (с, 1H), 8,14 (с, 1H), 7,28-7,00 (м, 2H), 6,59 (d, J=2,3 Гц, 1H), 4,64 (с, 1H), 3,28 (d, J=3,4 Гц, 2H), 3,19 (с, 6H), 2,14-1,96 (м, 3H), 1,72 (d, J=7,4 Гц, 4H), 1,34 (dd, J=10,7, 5,7 Гц, 2H).
Вариант осуществления 2: Синтез T2
Этап 1:
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
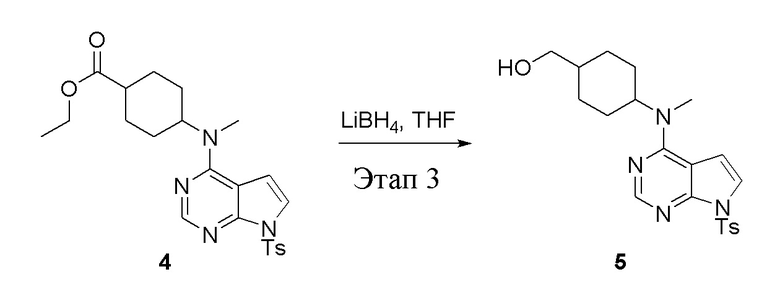
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
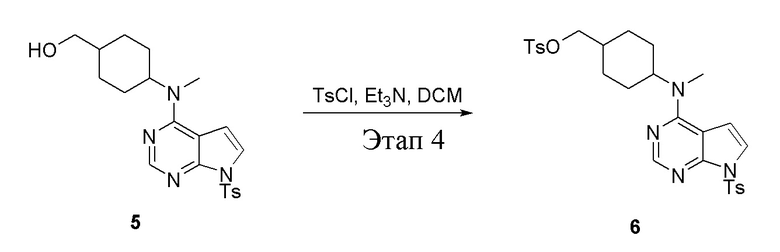
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
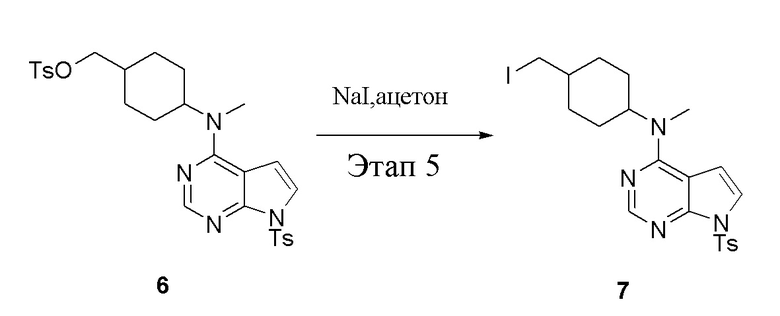
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
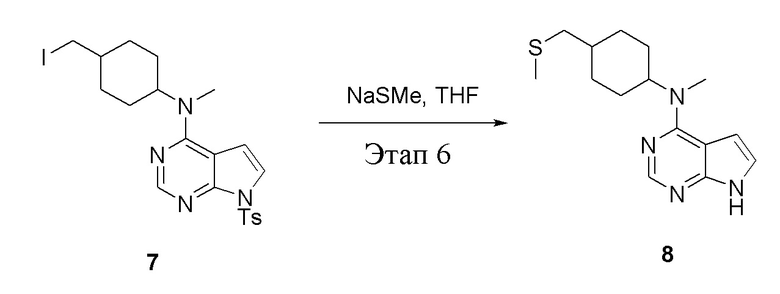
в одногорлую колбу объемом 1 л добавили 7 (25 г, 47,7 ммоль) (продукт, полученный на этапе 5), тетрагидрофуран (200 мл) и метилмеркаптид натрия (6,69 г, 95,4 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (11,5 г, выход: 83,1%). ЖХТМС: 291 [M+H]+
Этап 7:
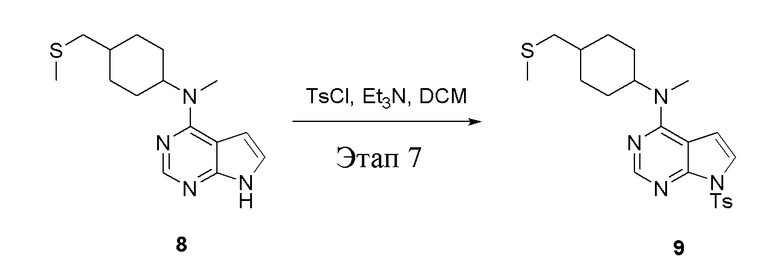
в одногорлую колбу объемом 500 л при 0°C добавили 8 (11,5 г, 39,6 ммоль) (продукт, полученный на этапе 6) и дихлорметан (110 мл), затем последовательно добавляли TsCl (11,3 г, 59,4 ммоль) и триэтиламин (8,0 г, 79,2 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (14,5 г). ЖХТМС: 445 [M+H]+
Этап 8:
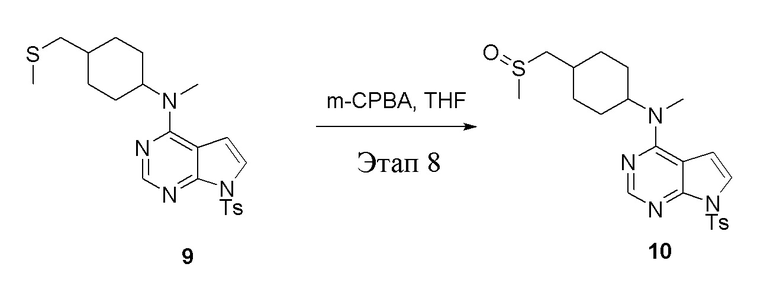
в одногорлую колбу объемом 250 мл при 0°C добавили 9 (14,5 г, 32,65 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (120 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (5,63 г, 32,65 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (100 мл), потом последовательно промыли насыщенным сульфитом натрия (50 мл × 3), бикарбонатом натрия (50 мл × 3) и физиологическим раствором (50 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (7,0 г, выход: 46,6%). ЖХТМС: 461 [M+H]+
Этап 9:
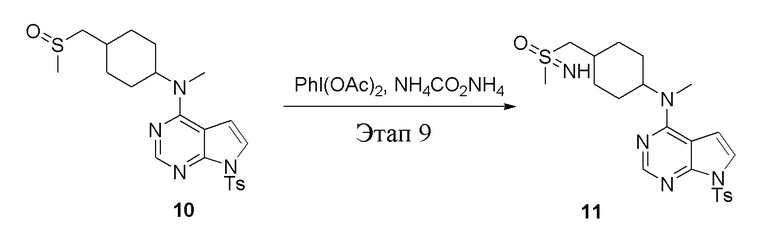
в одногорлую колбу объемом 250 л добавили 10 (7,0 г, 15,21 ммоль) (продукт, полученный на этапе 8), дихлорметан (70 мл), PhI(OAc)2 (7,35 г, 22,82 ммоль), затем последовательно добавляли TsCl (7,35 г, 22,82 ммоль) и карбонат аммония (2,92 г, 30,42 ммоль), после чего в течение 6 часов при комнатной температуре проводили реакцию. Остаточные материалы после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (3,0 г, выход: 41,5%). ЖХТМС: 474 [M-H]+
Этап 10:
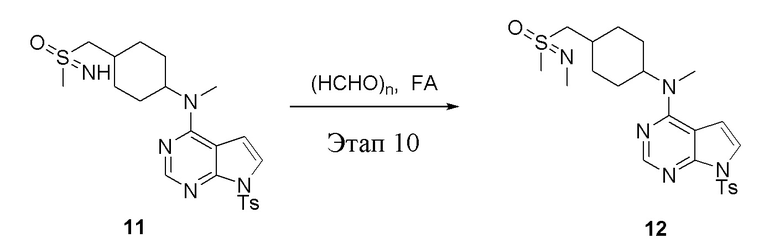
в одногорлую круглодонную колбу объемом 25 л добавили 11 (1,0 г, 2,10 ммоль) (продукт, полученный на этапе 9), параформальдегид (379 мл, 4,20 ммоль) и муравьиную кислоту (8 мл), после чего в течение 48 часов при 100°C проводили реакцию. Остаточные материалы после концентрирования влили в дихлорметан и 2N серную кислоту; водную фазу нейтрализовали бикарбонатом натрия и экстрагировали дихлорметаном (10 мл × 3), а органическую фазу высушили безводным сульфатом натрия и концентрировали с получением белого твердого вещества, то есть целевого продукта (500 мг, выход: 48,7%). ЖХТМС: 490 [M+H]+
Этап 11:
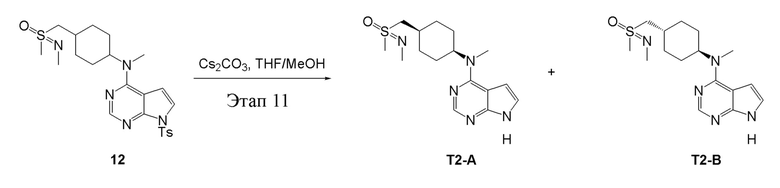
в одногорлую колбу объемом 25 мл добавили 12 (500 мг, 1,02 ммоль) (продукт, полученный на этапе 10), тетрагидрофуран/метиловый спирт (5,0 мл) и карбонат цезия (665 мг, 2,04 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия, концентрировали, после чего с применением традиционного и хирального способов было получено белое твердое вещество, то есть продукт A (100 мг, выход: 29,8%). ЖХТМС:336 [M+H]+,H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,60 (с, 1H), 8,09 (с, 1H), 7,20-7,04 (м, 1H), 6,53 (с, 1H), 4,67 (с, 1H), 3,23-2,99 (м, 5H), 2,94 (м, 3H), 2,64 (м, 3H), 2,16-1,89 (м, 3H), 1,69 (м, 4H), 1,35-1,14 (м, 2H) и продукт B (80 мг, выход: 23,4%), ЖХТМС:336 [M+H]+, H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,60 (с, 1H), 8,09 (с, 1H), 7,12 (dd, J=3,3, 2,5 Гц, 1H), 6,54 (с, 1H), 4,67 (с, 1H), 3,21-2,99 (м, 5H), 2,93 (с, 3H), 2,63 (с, 3H), 2,14-1,89 (м, 3H), 1,70 (м, 4H), 1,36-1,20 (м, 2H).
Вариант осуществления 3: Синтез T3
Этап 1:
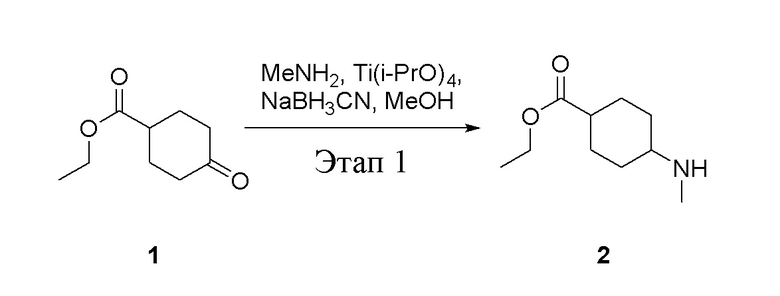
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
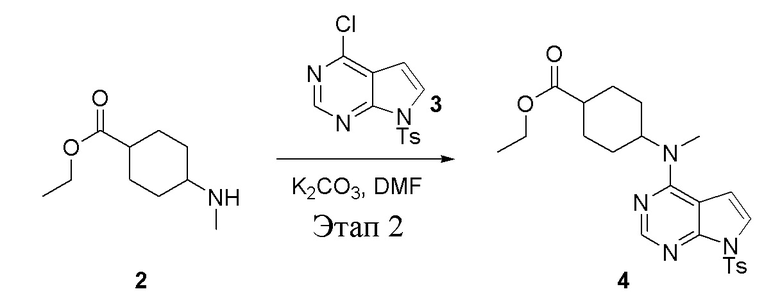
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
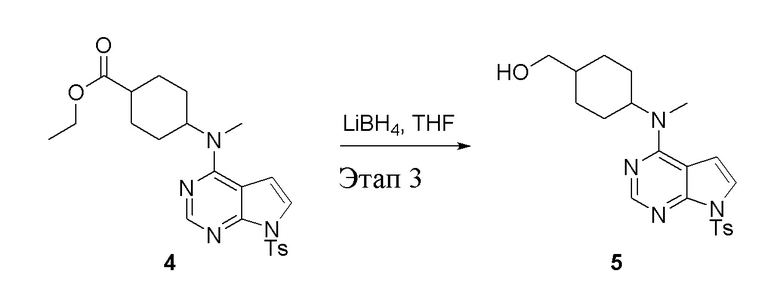
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
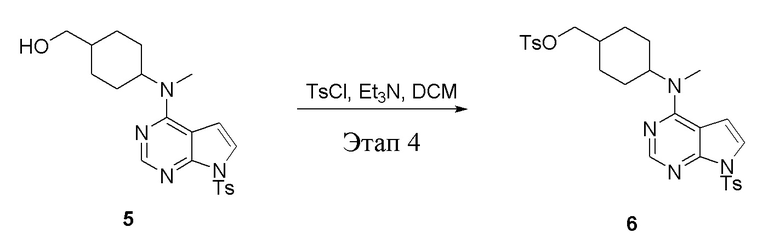
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
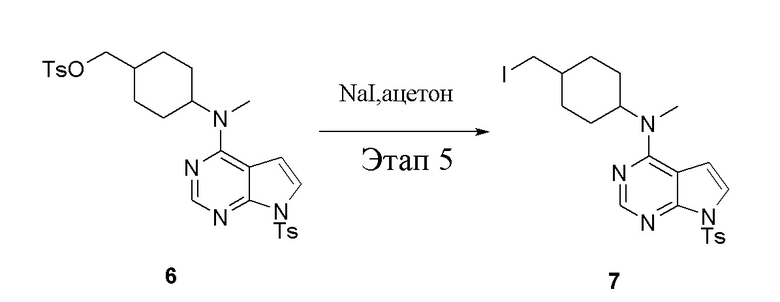
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
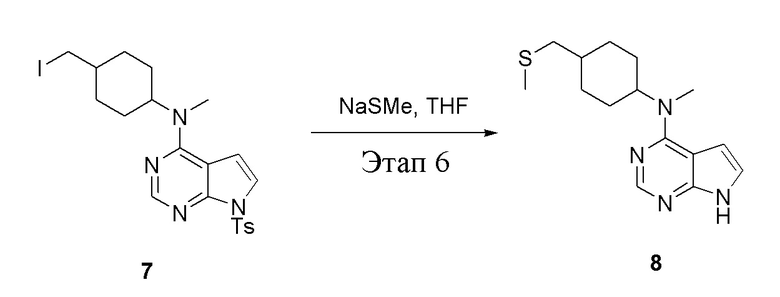
в одногорлую колбу объемом 1 л добавили 7 (25 г, 47,7 ммоль) (продукт, полученный на этапе 5), тетрагидрофуран (200 мл) и метилмеркаптид натрия (6,69 г, 95,4 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (11,5 г, выход: 83,1%). ЖХТМС: 291 [M+H]+
Этап 7:
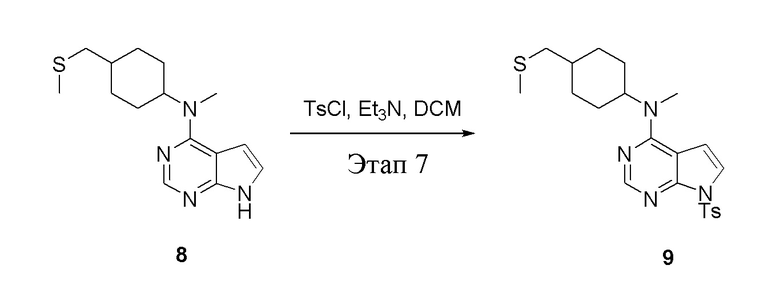
в одногорлую колбу объемом 500 л при 0°C добавили 8 (11,5 г, 39,6 ммоль) (продукт, полученный на этапе 6) и дихлорметан (110 мл), затем последовательно добавляли TsCl (11,3 г, 59,4 ммоль) и триэтиламин (8,0 г, 79,2 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (14,5 г). ЖХТМС: 445 [M+H]+
Этап 8:
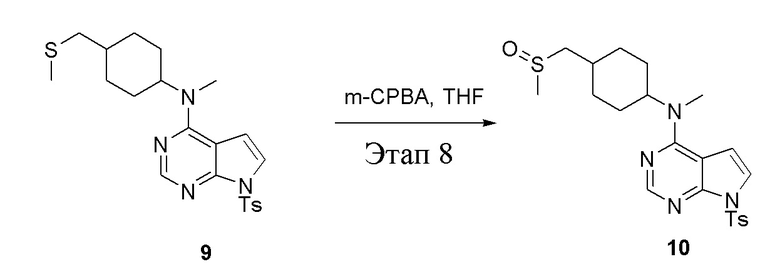
в одногорлую колбу объемом 250 мл при 0°C добавили 9 (14,5 г, 32,65 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (120 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (5,63 г, 32,65 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (100 мл), потом последовательно промыли насыщенным сульфитом натрия (50 мл × 3), бикарбонатом натрия (50 мл × 3) и физиологическим раствором (50 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (7,0 г, выход: 46,6%). ЖХТМС: 461 [M+H]+
Этап 9:
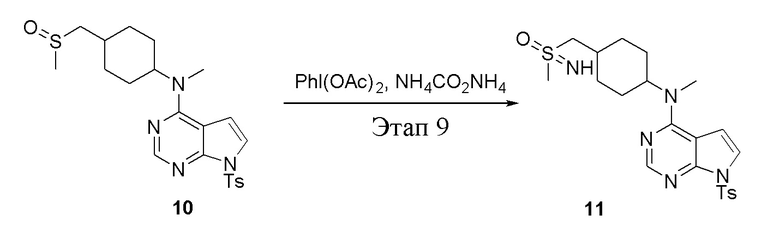
в одногорлую колбу объемом 250 л добавили 10 (7,0 г, 15,21 ммоль) (продукт, полученный на этапе 8), дихлорметан (70 мл), PhI(OAc)2 (7,35 г, 22,82 ммоль) и карбонат аммония (2,92 г, 30,42 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. Остаточную смесь после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (3,0 г, выход: 41,5%). ЖХТМС: 474 [M-H]+
Этап 10:
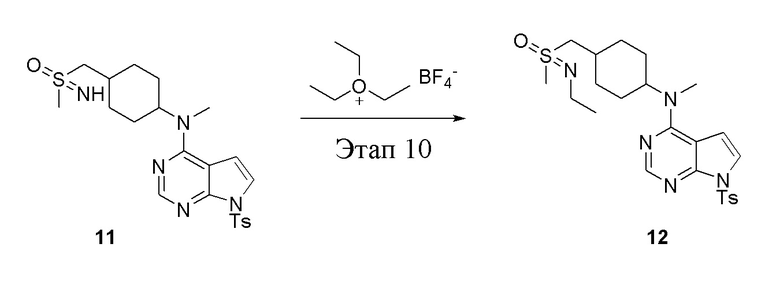
в одногорлую круглодонную колбу объемом 25 л добавили 11 (1,0 г, 2,10 ммоль) (продукт, полученный на этапе 9), тетрафторборат триэтилоксония (798 мг, 4,20 ммоль), карбонат калия (580 мг, 4,20 ммоль) и дихлорметан (10 мл), после чего при комнатной температуры в течение 18 часов проводили реакцию. После концентрирования провели обработку в хроматографической колонке (в условиях этилацетата: метилового спирта=1:1) с получением белого твердого вещества, то есть целевого соединения (500 мг, выход: 47,3%). ЖХТМС: 504 [M+H]+
Этап 11:
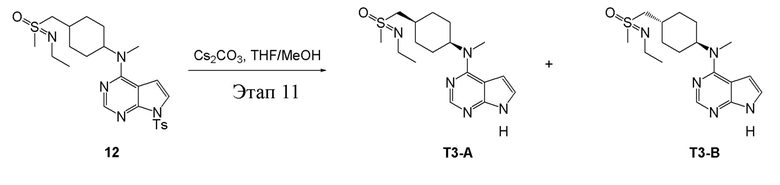
в одногорлую колбу объемом 25 мл добавили 12 (500 мг, 0,99 ммоль) (продукт, полученный на этапе 10), тетрагидрофуран/метиловый спирт (5,0 мл) и карбонат цезия (648 мг, 1,98 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия, концентрировали, после чего с применением традиционного и хирального способов было получено белое твердое вещество, то есть продукт A (75 мг, выход: 21,7%). ЖХТМС:350.1 [M+H]+, 1H-ЯМР (400 МГц, ДМСО) δ 11,60 (с, 1H), 8,09 (с, 1H), 7,15-7,10 (м, 1H), 6,53 (с, 1H), 4,66 (с, 1H), 3,23 (с, 3H), 3,12-2,96 (м, 4H), 2,93 (с, 3H), 2,07 (t, J=14,7 Гц, 2H), 1,95 (d, J=3,2 Гц, 1H), 1,76-1,68 (м, 4H), 1,32-1,23 (м, 2H), 1,08 (t, J=7,1 Гц, 3H) и продукт B (85 мг, выход: 24,6%), ЖХТМС:350.1 [M+H]+, 1H-ЯМР (400 МГц, ДМСО) δ 11,60 (с, 1H), 8,09 (с, 1H), 7,12 (d, J=1,2 Гц, 1H), 6,53 (d, J=2,6 Гц, 1H), 4,67 (с, 1H), 3,19 (d, J=19,5 Гц, 3H), 3,12-2,96 (м, 4H), 2,94 (d, J=5,2 Гц, 3H), 2,07 (t, J=14,9 Гц, 2H), 1,93 (d, J=17,4 Гц, 1H), 1,76-1,68 (м, 4H), 1,27 (dd, J=14,6, 6,9 Гц, 2H), 1,09 (dd, J=7,2, 3,9 Гц, 3H).
Вариант осуществления 4: Синтез T4
Этап 1:
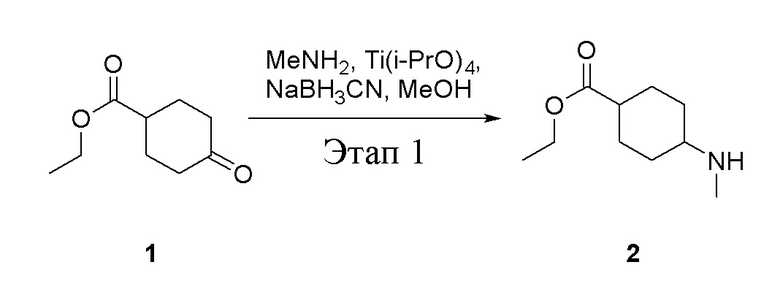
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
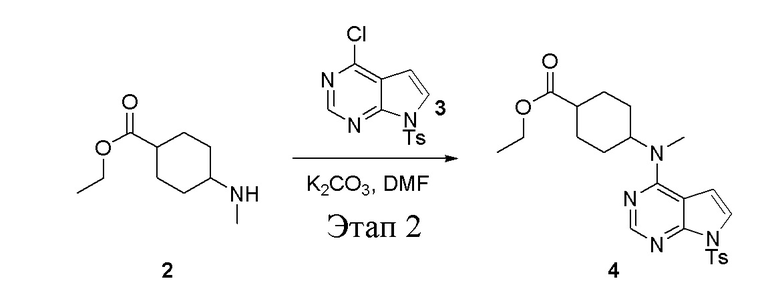
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
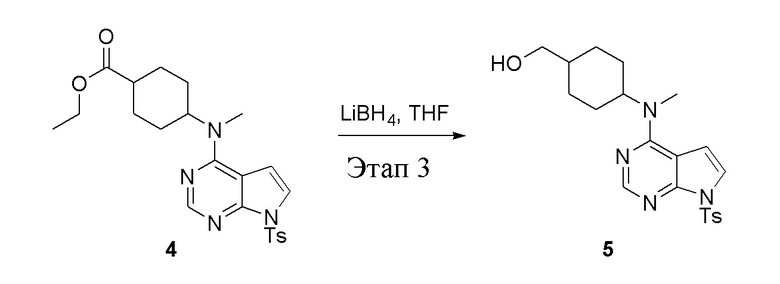
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
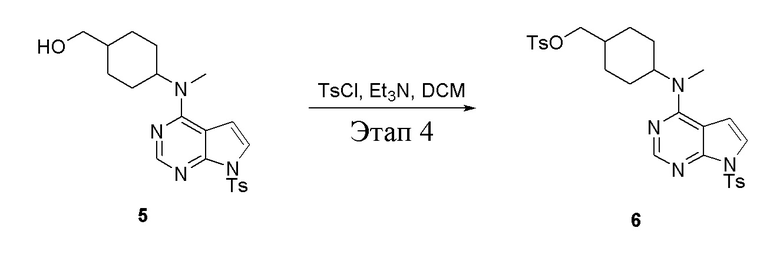
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
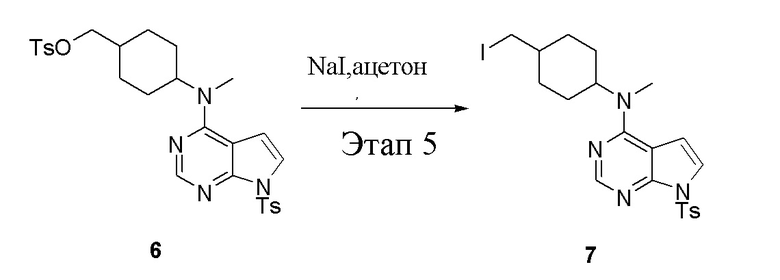
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
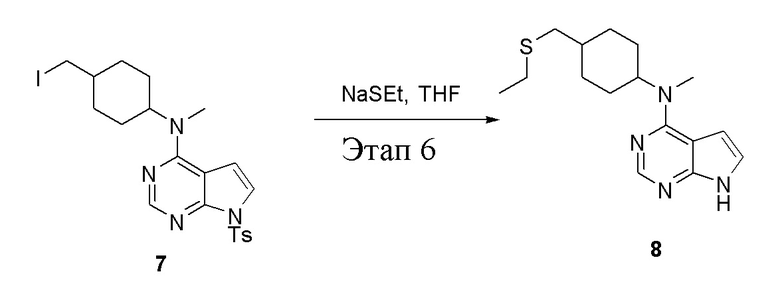
в одногорлую колбу объемом 1 л добавили 7 (15 г, 28,62 ммоль) (продукт, полученный на этапе 5), тетрагидрофуран (120 мл) и этантиолат натрия (4,82 г, 57,24 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (7,5 г, выход: 86,2%). ЖХТМС: 305 [M+H]+
Этап 7:
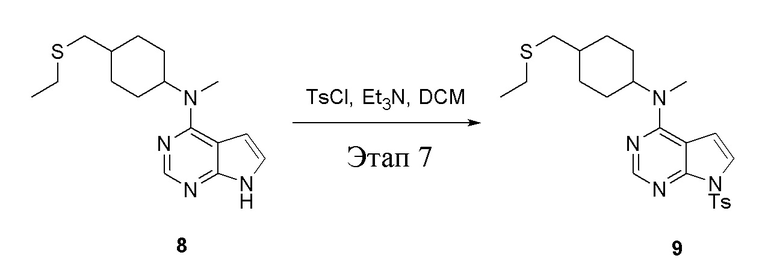
в одногорлую колбу объемом 500 л при 0°C добавили 8 (7,5 г, 39,6 ммоль) (продукт, полученный на этапе 6) и дихлорметан (80 мл), затем последовательно добавляли TsCl (7,06 г, 37,01 ммоль) и триэтиламин (5,0 г, 49,34 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором (50 мл × 3), высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (5,5 г). ЖХТМС: 459 [M+H]+
Этап 8:
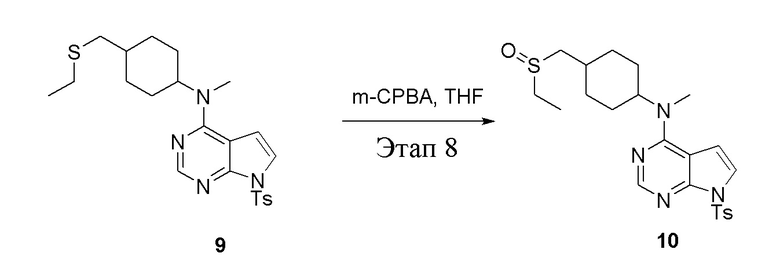
в одногорлую колбу объемом 250 мл при 0°C добавили 9 (2,6 г, 5,68 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (30 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (980 мг, 5,68 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (30 мл), потом последовательно промыли насыщенным сульфитом натрия (20 мл × 3), бикарбонатом натрия (20 мл × 3) и физиологическим раствором (20 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (2,0 г, выход: 74,3%). ЖХТМС: 475 [M+H]+
Этап 9:
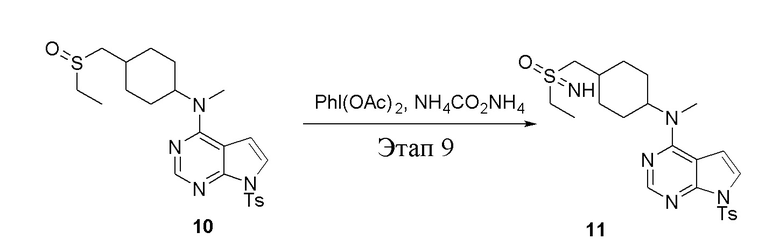
в одногорлую колбу объемом 250 л добавили 10 (2,0 г, 4,22 ммоль) (продукт, полученный на этапе 8), дихлорметан (20 мл), PhI(OAc)2 (2,04 г, 6,33 ммоль), PhI(OAc)2(2,04 г, 6,33 ммоль) и карбонат аммония (811 мг, 8,44 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. Остаточные материалы после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (1,2 г, выход: 58,1%). ЖХТМС: 488 [M-H]+
Этап 10:
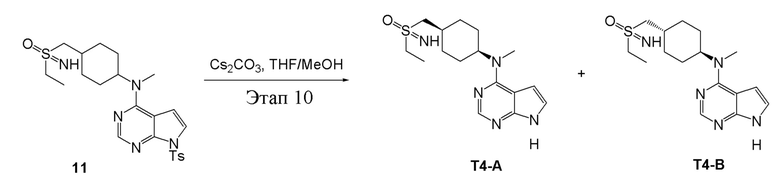
в одногорлую колбу объемом 25 мл добавили 11 (1,0 г, 2,04 ммоль) (продукт, полученный на этапе 9), тетрагидрофуран/метиловый спирт (10 мл) и карбонат цезия (1,33 г, 4,08 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия, концентрировали, после чего с применением традиционного и хирального способов было получено белое твердое вещество, то есть продукт A (20 мг, выход: 2,9%). ЖХТМС:336 [M+H]+,H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,61 (с, 1H), 8,09 (с, 1H), 7,13 (с, 1H), 6,54 (с, 1H), 4,67 (с, 1H), 3,90-3,83 (м, 1H), 3,17 (с, 3H), 3,06-2,93 (м, 4H), 2,12-2,01 (м, 3H), 1,73-1,70 (м, 4H), 1,31-1,22 (м, 5H) и продукт B (25 мг, выход: 3,7%), ЖХТМС:336 [M+H]+, H1-ЯМР: 1H-ЯМР (400 МГц, ДМСО) δ 11,59 (с, 1H), 8,09 (с, 1H), 7,12 (dd, J=3,3, 2,6 Гц, 1H), 6,54 (с, 1H), 4,67 (с, 1H), 3,58 (с, 1H), 3,17 (с, 3H), 3,06-2,89 (м, 4H), 2,16-1,93 (м, 3H), 1,74-1,69 (м, 4H), 1,25-1,23 (м, 5H).
Вариант осуществления 5: Синтез T5
Этап 1:
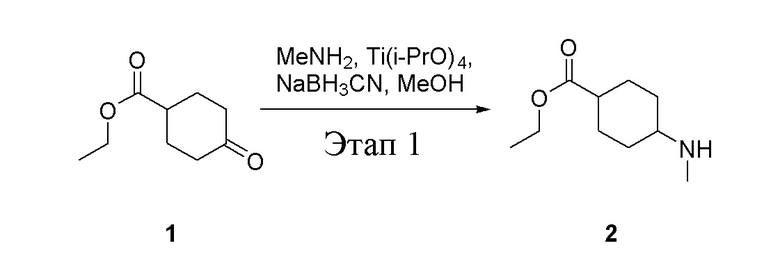
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
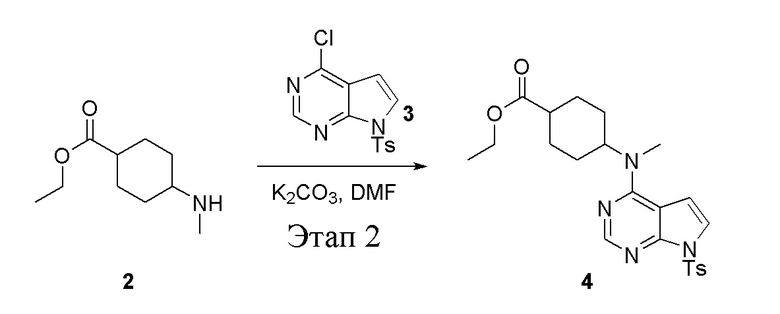
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
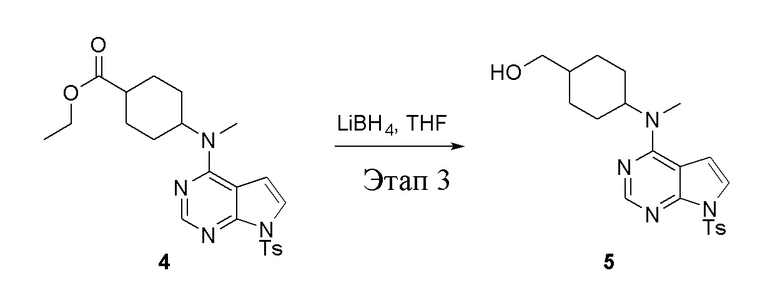
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
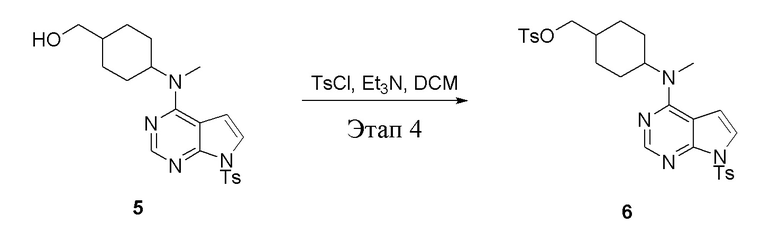
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
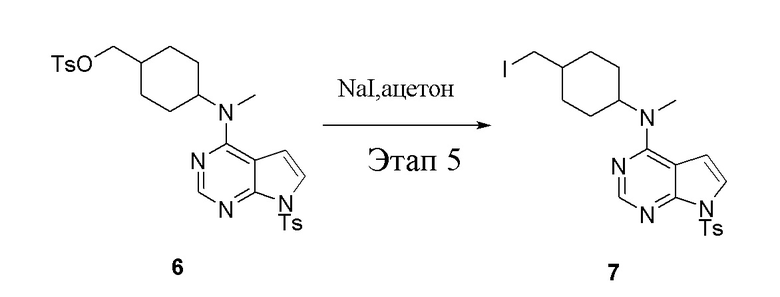
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
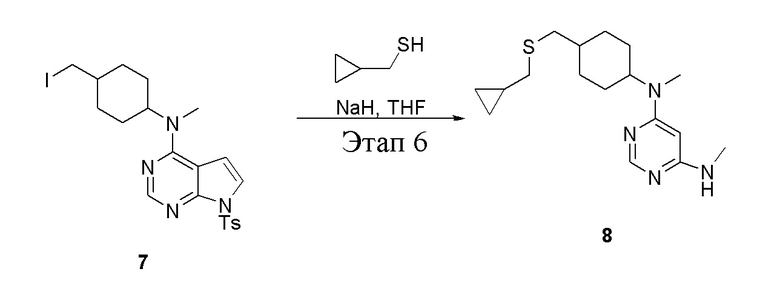
в одногорлую колбу объемом 50 мл добавили 7 (1,1 г, 2,10 ммоль) (продукт, полученный на этапе 5), циклопропилметантиол (154 мг, 1,75 ммоль), тетрагидрофуран (12 мл) и гидрид натрия (140 мг, 3,50 ммоль), после чего при комнатной температуре в течение 30 минут проводили реакцию, а затем смесь в течение 12 часов подвергали нагреву с обратным холодильником, далее просушили с помощью центробежной сушилки с получением желтого твердого вещества (1,2 г, неочищенный продукт). ЖХТМС: 331 [M+H]+
Этап 7:
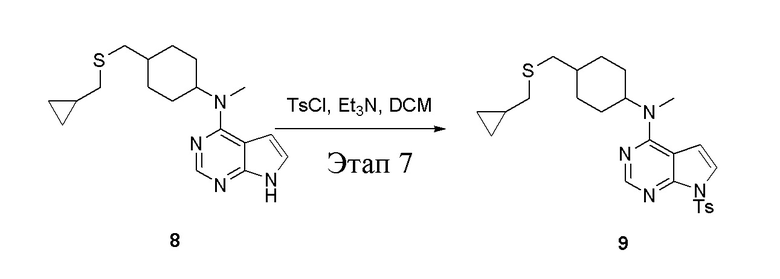
в одногорлую колбу объемом 50 л при 0°C добавили 8 (1,2 г, 3,63 ммоль) (продукт, полученный на этапе 6) и дихлорметан (12 мл), затем последовательно добавляли TsCl (831 мг, 4,36 ммоль) и триэтиламин (735 мг, 7,26 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (1,0 г). ЖХТМС: 485 [M+H]+
Этап 8:
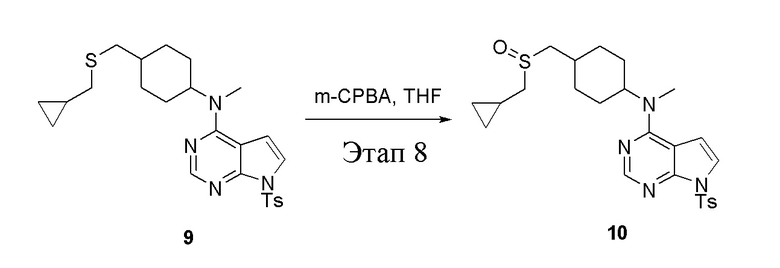
в одногорлую колбу объемом 50 мл при 0°C добавили 9 (1,0 г, 2,07 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (10 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (713 мг, 4,13 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (20 мл), потом последовательно промыли насыщенным сульфитом натрия (10 мл × 3), бикарбонатом натрия (10 мл × 3) и физиологическим раствором (10 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (320 мг, выход: 30,9%). ЖХТМС: 501 [M+H]+
Этап 9:
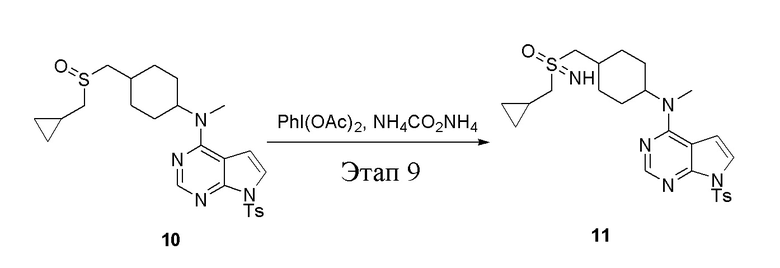
в одногорлую колбу объемом 250 л добавили 10 (320 мг, 0,54 ммоль) (продукт, полученный на этапе 8), дихлорметан (5 мл), PhI(OAc)2 (309 мг, 0,96 ммоль) и карбонат аммония (123 мг, 1,28 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. Остаточные материалы после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (300 мг, выход: 91,0%). ЖХТМС: 514 [M-H]+
Этап 10:
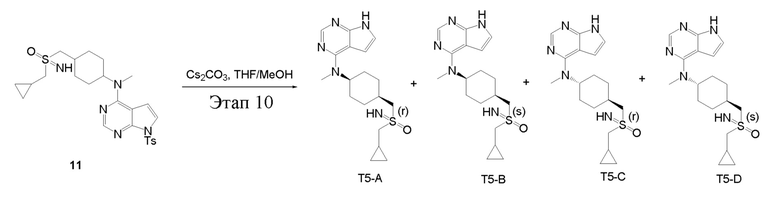
в одногорлую колбу объемом 25 мл добавили 11 (300 мг, 0,58 ммоль), тетрагидрофуран/метиловый спирт (5,0 мл) и карбонат цезия (380 мг, 1,16 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия, концентрировали, после чего с применением традиционного и хирального способов было получено белое твердое вещество, то есть продукт A (4 мг, выход: 1,9%), ЖХТМС: 362,2 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,12 (с, 1H), 7,14 (d, J=3,6 Гц, 1H), 6,68 (d, J=3,6 Гц, 1H), 4,72 (с, 1H), 3,34 (с, 3H), 3,16 (d, J=7,1 Гц, 2H), 2,08 (dd, J=28,6, 9,0 Гц, 3H), 1,90 (т, J=8,4 Гц, 4H), 1,72 (d, J=8,7 Гц, 2H), 1,35-1,21 (м, 5H), 0,80-0,74 (м, 2H), 0,52-0,45 (м, 2H); причем белое твердое вещество представляло собой продукт B (5 мг, выход: 2,4%), ЖХТМС 362,2 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,11 (с, 1H), 7,11 (d, J=3,6 Гц, 1H), 6,65 (d, J=3,6 Гц, 1H), 4,73 (d, 1H), 3,30 (d, J=6,8 Гц, 3H), 3,15 (d, J=7,1 Гц, 2H), 2,08 (dd, J=28,8, 9,0 Гц, 3H) , 1,90 (t, J=8,6 Гц, 4H), 1,72 (с, 2H), 1,41-1,15 (м, 3H), 0,81-0,71 (м, 2H), 0,49 (dd, J=7,6, 4,6 Гц, 2H); причем белое твердое вещество представляло собой продукт C (12 мг, выход: 5,7%), ЖХТМС:362,2 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,10 (с, 1H), 7,11 (d, J=3,6 Гц, 1H), 6,65 (d, J=3,6 Гц, 1H), 4,73 (с, 1H), 3,28 (с, 3H), 3,22-3,09 (м, 4H), 2,30-2,12 (м, 3H), 1,84 (ddd, J=15,7, 9,8, 3,2 Гц , 4H), 1,46 (qd, J=13,0, 6,3 Гц, 2H), 1,21 (tdd, J=7,2, 6,4, 2,3 Гц, 1H), 0,79-0,72 (м, 2H), 0,50-0,42 (м, 2H); причем белое твердое вещество представляло собой продукт D (15 мг, выход: 7,2%), ЖХТМС:362,2 [M+H] +, 1H-ЯМР (400 МГц, ЭУОО) δ 8,10 (с, 1H), 7,11 (d, J=3,6 Гц, 1H), 6,65 (d, J=3,6 Гц, 1H), 4,73 (с, 1H), 3,28 (с, 3H), 3,22-3,09 (м, 4H), 2,30-2,23 (м, 1H), 2,16 (dd, J=13,1, 3,1 Гц, 2H), 1,92-1,78 (м, 4H), 1,46 (qd, J=13,0, 6,3 Гц, 2H), 1,25-1,17 (м, 1H), 0,79-0,71 (м, 2H), 0,47 (td, J=4,7, 2,1 Гц, 2H).
Вариант осуществления 6: Синтез T6
Этап 1:
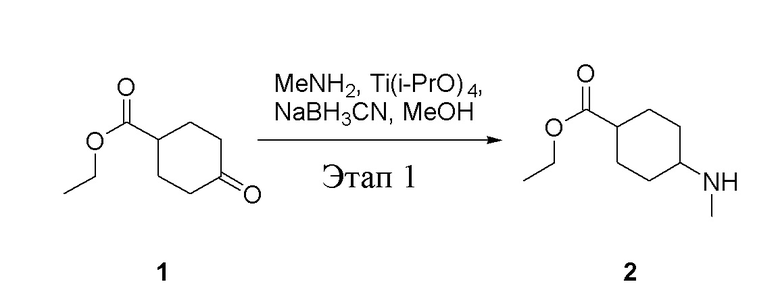
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
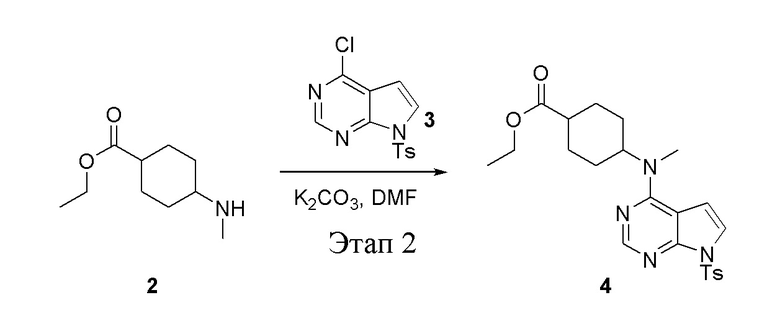
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
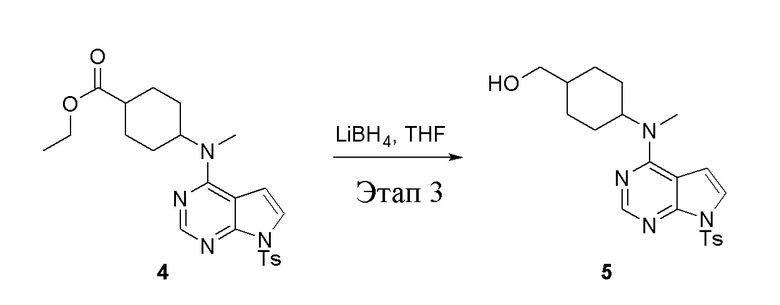
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
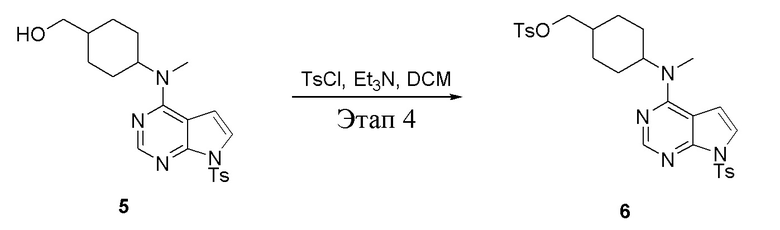
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
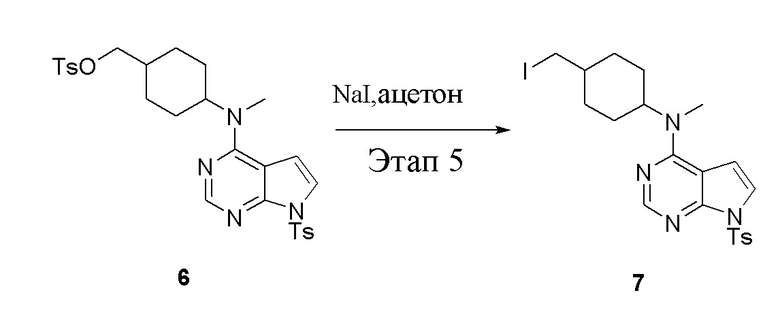
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
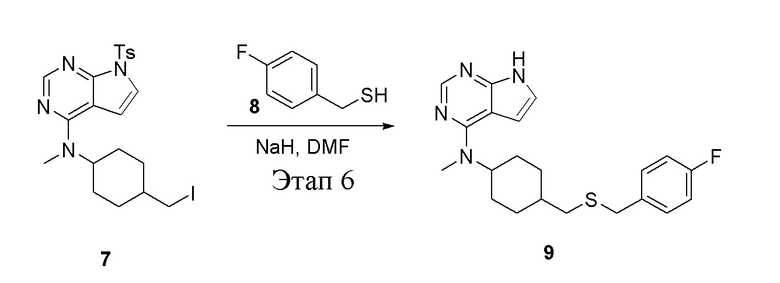
в одногорлую колбу объемом 50 мл добавили 8 (473 мг, 3,33 ммоль) и DMF (5 мл), далее, добавив при 0°C гидрид натрия (266 мг, 6,66 ммоль), перемешивали при комнатной температуре в течение 30 мин, добавили 7 (2,1 г, 4,0 ммоль) (продукт, полученный на этапе 5), после чего при комнатной температуре в течение 12 часов проводили реакцию. Для целей гашения добавили воду, после чего смесь экстрагировали этилацетатом (1,0 л × 3), промыли насыщенным солевым раствором (30 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки, а затем подвергли обработке в хроматографической колонке (в условиях дихлорметана: метилового спирта=30:1) с получением белого твердого вещества, то есть целевого продукта (1,2 г, выход: 93,8%). ЖХТМС: 385 [M+H]+
Этап 7:
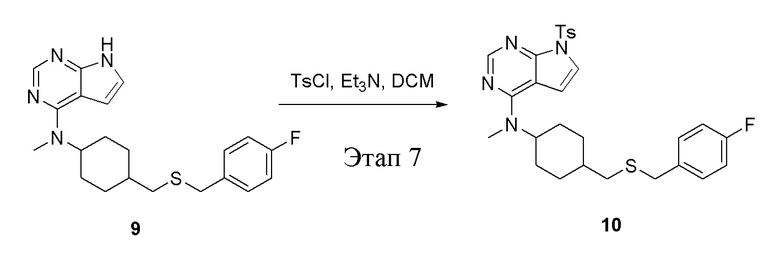
в одногорлую колбу объемом 50 л при 0°C добавили 9 (1,2 г, 3,12 ммоль) (продукт, полученный на этапе 6) и дихлорметан (15 мл), затем последовательно добавляли TsCl (893 мг, 4,69 ммоль) и триэтиламин (948 мг, 9,36 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=2:1) с получением белого твердого вещества, то есть целевого продукта (400 мг, выход: 23,8%). ЖХТМС: 539 [M+H]+
Этап 8:
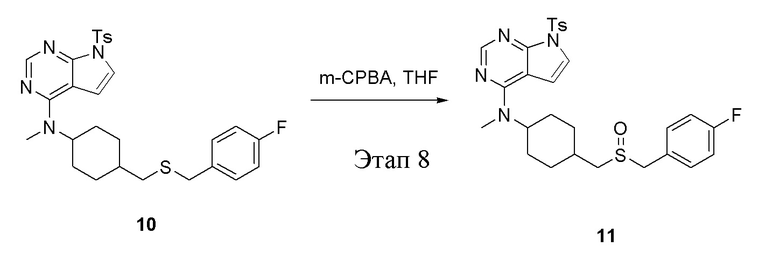
в одногорлую колбу объемом 25 мл при 0°C добавили 10 (400 мг, 0,74 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (4 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (257 мг, 1,49 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (10 мл), потом последовательно промыли насыщенным сульфитом натрия (10 мл × 3), бикарбонатом натрия (10 мл × 3) и физиологическим раствором (10 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (300 мг, выход: 73,2%). ЖХТМС: 555 [M+H]+
Этап 9:
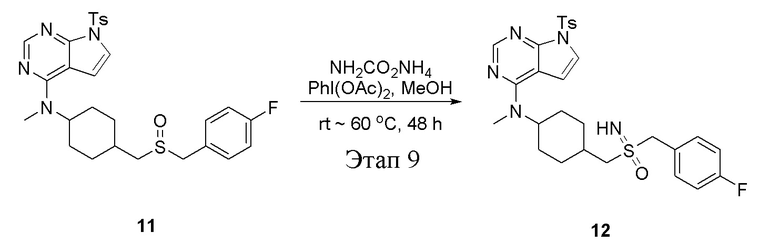
в одногорлую колбу объемом 25 л добавили 11 (300 мг, 0,54 ммоль) (продукт, полученный на этапе 8), метиловый спирт (5 мл), PhI(OAc)2 (261 мг, 0,81 ммоль) и карбонат аммония (104 мг, 1,08 ммоль), после чего при температуре 60°C в течение 48 часов проводили реакцию. Остаточные материалы после фильтрации и концентрирования влили в метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (276 мг, выход: 89,8%). ЖХТМС: 570 [M+H]+
Этап 10:
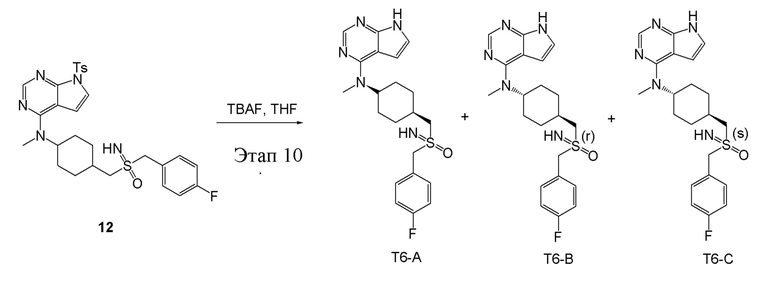
в одногорлую колбу объемом 25 мл добавили 12 (276 мг, 0,48 ммоль) (продукт, полученный на этапе 9), тетрагидрофуран (3,0 мл), карбонат цезия (251 мг, 0,96 ммоль) и фторид тетрабутиламмония (251 мг, 0,96 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия и концентрировали, после чего обработали традиционным способом и подвергли обработке хиральным раствором (в колонке: DAICEL CHRAL OD (250 мм × 30 мм, 10 мкм); градиентное время: 20 мин; в условиях: 0,2% ДЭА, этанол и гексан; скорость потока: 18 мл/мин; 75% этанол) с получением белого твердого вещества, то есть продукта-T6-A(8 мг, выход: 4,0%). ЖХТМС:416,2 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,10 (с, 1H), 7,59-7,50 (м, 2H), 7,24-7,15 (м, 2H), 7,10 (d, J=3,6 Гц, 1H), 6,64 (d, J=3,6 Гц, 1H), 4,71 (с, 1H), 4,48 (dd, J=29,1, 13,5 Гц, 2H), 3,41-3,34 (м, 1H), 3,27 (d, J=10,9 Гц, 3H), 3,22-3,17 (м, 1H), 2,60 (с, 1H), 2,09-1,98 (м, 2H), 1,91-1,83 (м, 4H), 1,68 (d, J=8,4 Гц, 2H), 1,31 (с, 1H); продукт T6-B (27 мг, выход: 13,5%), ЖХТМС: 416,1 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,09 (с, 1H), 7,52 (dd, J=8,7, 5,3 Гц, 2H), 7,21-7,15 (м, 2H), 7,10 (d, J=3,6 Гц, 1H), 6,63 (d, J=3,6 Гц, 1H), 4,71 (с, 1H), 4,45 (q, J=13,6 Гц, 2H), 3,26 (с, 3H), 3,06-2,98 (м, 2H), 2,23-2,19 (м, 1H), 2,16-2,08 (м, 2H), 1,88-1,77 (м, 4H), 1,42-1,31 (м, 3H); продукт Т6-С (28 мг, выход: 14.1%), ЖХТМС: 416,1 [M+H]+, 1H-ЯМР (400 МГц, ЭУОО) δ 8,09 (с, 1H), 7,55-7,47 (м, 2H), 7,22-7,15 (м, 2H), 7,10 (d, J=3,6 Гц, 1H), 6,63 (d, J=3,6 Гц, 1H), 4,71 (с, 1H), 4,45 (q, J=13,6 Гц, 2H), 3,26 (с, 3H), 3,07-2,97 (м, 2H), 2,21 (d, J=13,0 Гц, 1H), 2,16-2,07 (м, 2H), 1,89-1,76 (м, 4H), 1,49-1,26 (м, 3H).
Вариант осуществления 7: Синтез T7
Этап 1:
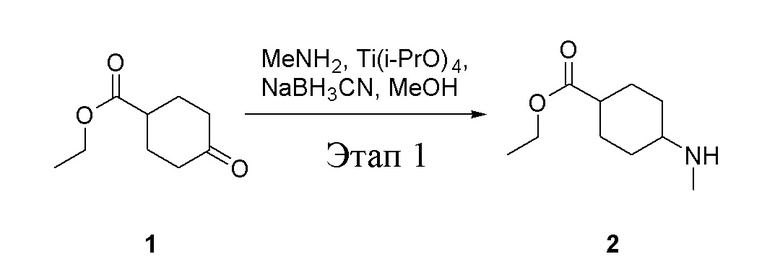
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
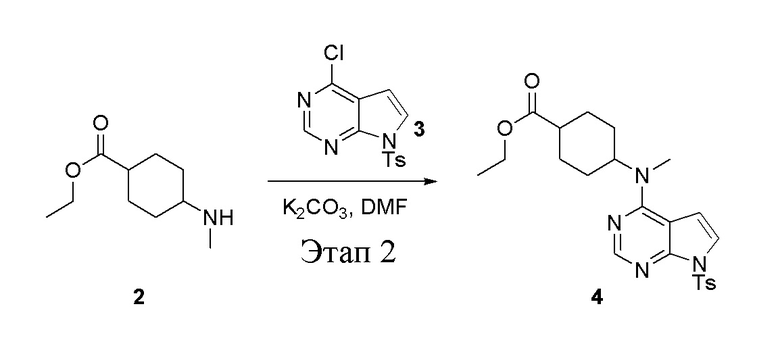
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
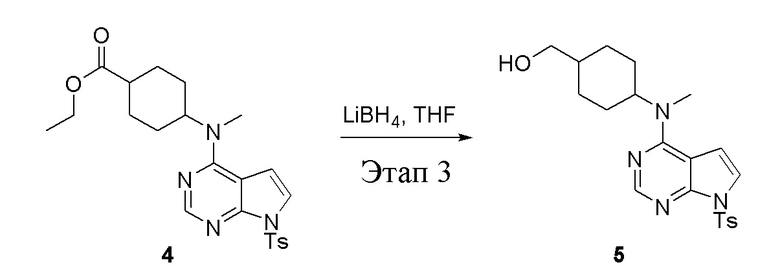
в одногорлую колбу объемом 2 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
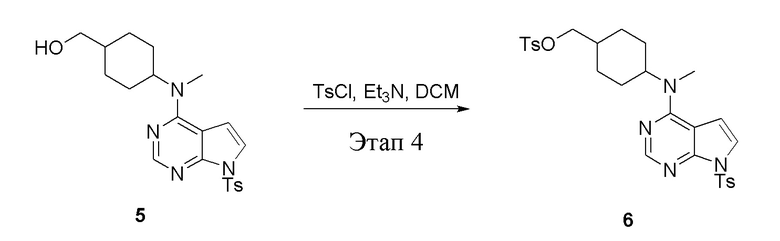
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
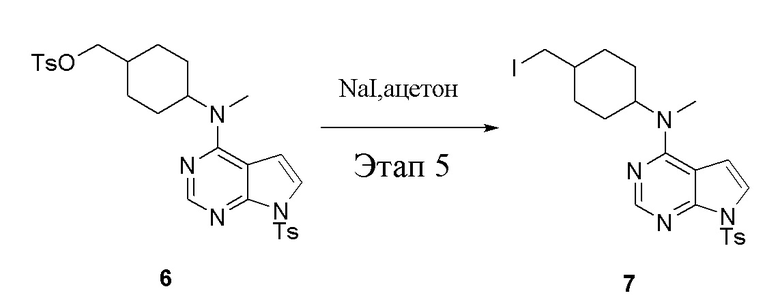
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
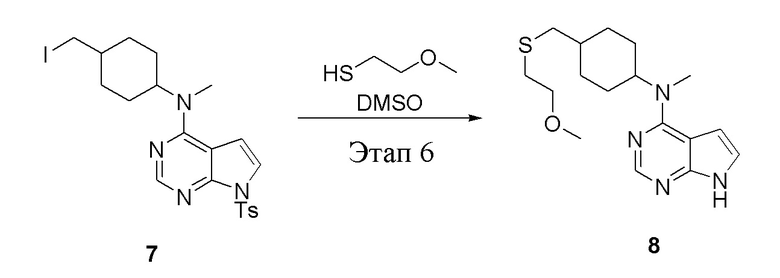
в одногорлую колбу объемом 100 л добавили 7 (1 г, 1,908 ммоль) (продукт, полученный на этапе 5), 2-метилоксиэтантиол (263 мг, 2,862 ммоль), диметилсульфоксид (15 мл) и карбонат калия (395 мг, 2,862 ммоль), после чего при температуре 130°C в течение всей ночи проводили реакцию, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (438 мг, выход: 47%). ЖХТМС: 335 [M+H]+
Этап 7:
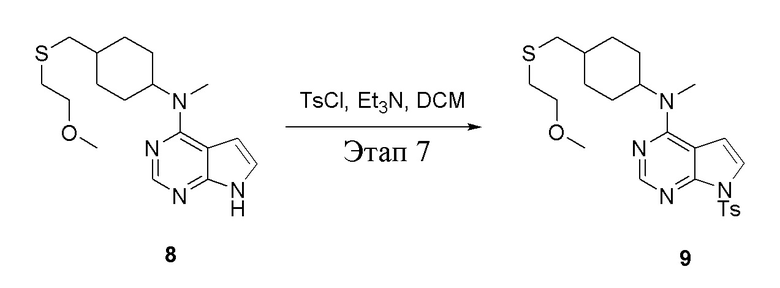
в одногорлую колбу объемом 100 л при 0°C добавили 8 (410 мг, 1,2289 ммоль) (продукт, полученный на этапе 6) и дихлорметан (10 мл), затем последовательно добавляли TsCl (468 мг, 2,4478 ммоль) и триэтиламин (371 мг, 79,2 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем сепарировали методом тонкослойной хроматографии (в условиях петролейного эфира: этилацетата=1:1) с получением белого твердого вещества, то есть целевого продукта (300 мг, выход: 50%). ЖХТМС: 489 [M+H]+
Этап 8:
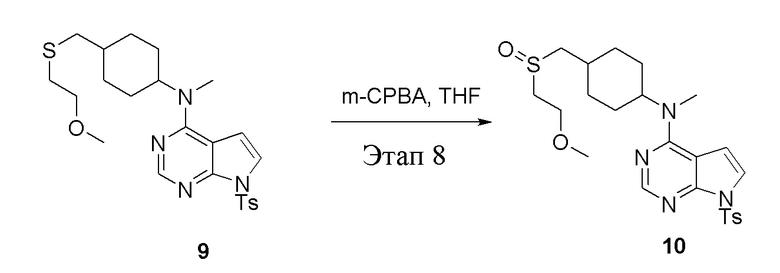
в одногорлую колбу объемом 100 мл при 0°C добавили 9 (300 мг, 0,6135 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (10 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (106 мг, 0,6135 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (20 мл), потом последовательно промыли насыщенным сульфитом натрия (50 мл × 3), бикарбонатом натрия (50 мл × 3) и физиологическим раствором (50 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке методом тонкослойной хроматографии (в условиях дихлорметана: метилового спирта=30:1) с получением желтого твердого вещества, то есть целевого продукта (200 мг, выход: 64,5%). ЖХТМС: 505 [M+H]+
Этап 9:
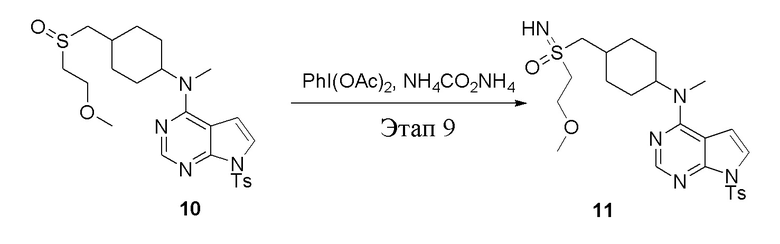
в одногорлую колбу объемом 100 мл добавили 10 (200 мг, 0,3963 ммоль) (продукт, полученный на этапе 8), метиловый спирт (10 мл), PhI(OAc)2 (382,6 мг, 0,7926 ммоль) и карбонат аммония (192 мг, 1,1889 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. После концентрирования провели обработку методом тонкослойной хроматографии (в условиях дихлорметана: метилового спирта=15:1) с получением белого твердого вещества, то есть целевого соединения (145 мг, выход: 70,4%). ЖХТМС: 520 [M-H]+
Этап 10:
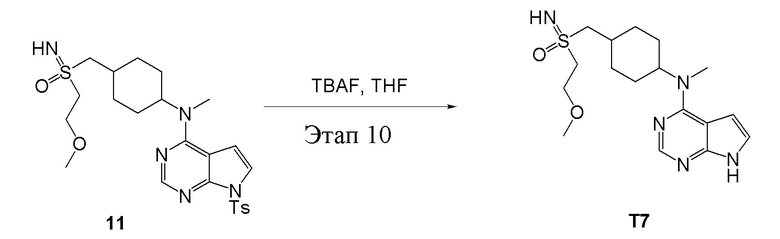
в одногорлую колбу объемом 25 мл добавили 11 (145 мг, 0,2788 ммоль) (продукт, полученный на этапе 9), тетрагидрофуран (5,0 мл), тетрагидрофурановый раствор TBAF (2,23 мл, 2,23 ммоль, 1 ммоль/мл), после чего при 60°C в течение 12 часов проводили реакцию, а затем смесь концентрировали и с применением традиционного способа получили белое твердое вещество (25 мг, 31%). 1H-ЯМР (400 МГц, ДМСО) δ 12,67 (с, 1H), 8,32 (с, 1H), 7,44 (с, 1H), 6,85 (с, 1H), 4,02 (с, 2H), 3,90-3,82 (м, 2H), 3,65 (d, J=5,8 Гц, 2H), 3,34 (с, 3H), 3,30 (с, 3H), 2,20 (с, 1H), 2,03 (dd, J=18,1, 10,7 Гц, 2H), 1,82 (с, 4H), 1,46 (с, 2H), 1,33-1,23 (м, 2H).
Вариант осуществления 8: Синтез T8
Этап 1:
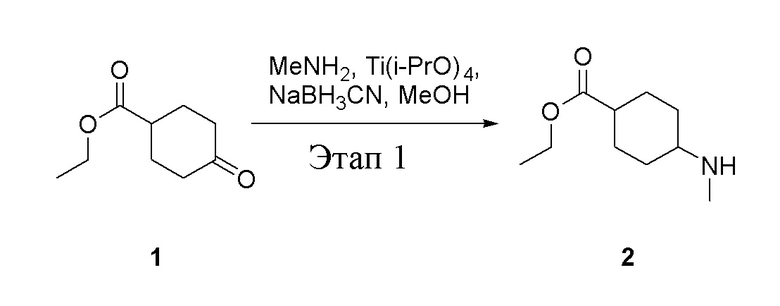
в одногорлую колбу объемом 5 л добавили 1 (220 г, 1,29 моль) и МеОН (40 мл), после чего в нее последовательно добавили тетрагидрофурановый раствор метиламина (0,78 л, 1,55 моль и 2 М в THF), тетраизопропилтитанат (733 г, 2,58 моль) и NaBH3CN (162 г, 2,58 моль), перемешивали при комнатной температуре в течение 18 часов, высушили с помощью центробежной сушилки и затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением белого маслянистого вещества, то есть целевого продукта (135 г, выход: 56,5%). ЖХТМС: 186[M+H]+
Этап 2:
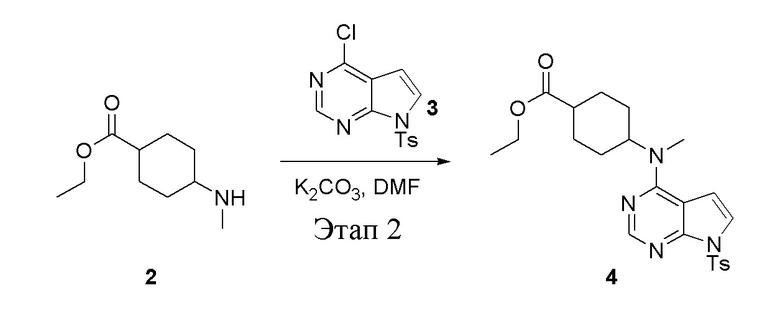
в одногорлую круглодонную колбу объемом 1 л добавили 2 (70 г, 378 ммоль) (продукт, полученный на этапе 1), 3 (96,7 г, 315 ммоль), карбонат калия (86,9 г, 630 ммоль) и DMF (300 мл), после чего в течение 18 часов при 100°C проводили реакцию. После добавления ледяной воды остаточные материалы подвергнули вакуумной фильтрации, а фильтровальный осадок высушили с получением желтого твердого вещества, то есть целевого продукта (136 г, выход: 94,6%). ЖХТМС: 457[M+H]+
Этап 3:
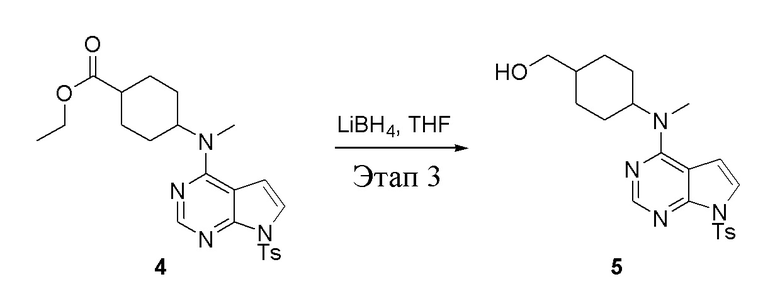
в одногорлую колбу объемом 5 л добавили 4 (136 г, 298 ммоль) (продукт, полученный на этапе 2) и тетрагидрофуран (1,0 л), затем порциями добавили боргидрид лития (13,0 г, 596 ммоль), после чего в течение 5 часов при 50°C проводили реакцию. Для целей гашения добавили метиловый спирт (20 мл), после чего смесь перелили в воду и экстрагировали дихлорметаном (1,0 л × 3), промыли насыщенным солевым раствором (500 мл × 3) и высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением белого твердого вещества, то есть целевого продукта (75 г, выход: 60,8%). ЖХТМС: 415 [M+H]+
Этап 4:
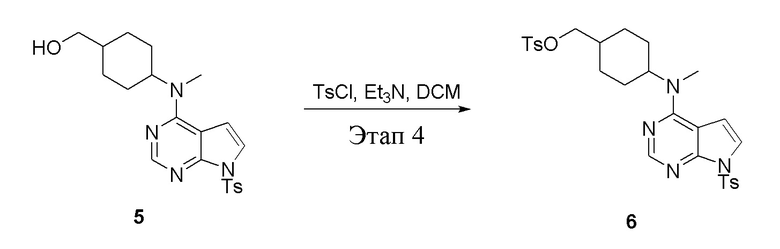
в одногорлую колбу объемом 2 л при 0°C добавили 5 (75 г, 181 ммоль) (продукт, полученный на этапе 3) и дихлорметан (750 мл), затем последовательно добавляли TsCl (51,8 г, 272 ммоль) и триэтиламин (36,7 г, 362 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (80 г, выход: 77,8%). ЖХТМС: 569 [M+H]+
Этап 5:
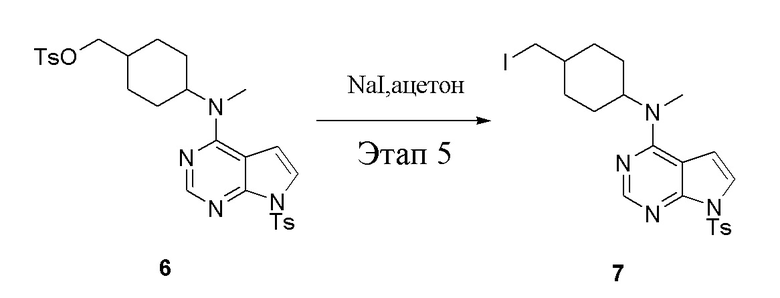
в одногорлую колбу объемом 1 л добавили 6 (80 г, 141 ммоль) (продукт, полученный на этапе 4), ацетон (500 мл) и йодид натрия (42,2 г, 282 ммоль), которые в течение 16 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (70 г, выход: 94,7%). ЖХТМС: 525 [M+H]+
Этап 6:
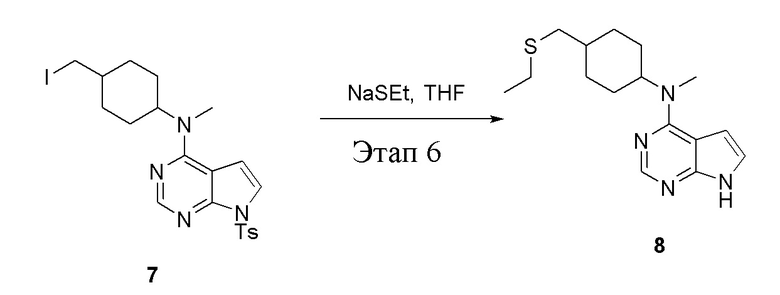
в одногорлую колбу объемом 500 мл добавили 7 (15 г, 28,62 ммоль) (продукт, полученный на этапе 5), тетрагидрофуран (200 мл) и этантиолат натрия (4,82 г, 57,24 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, а затем просушили с помощью центробежной сушилки и подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:1) с получением желтого твердого вещества, то есть целевого продукта (7,5 г, выход: 86,2%). ЖХТМС: 305 [M+H]+
Этап 7:
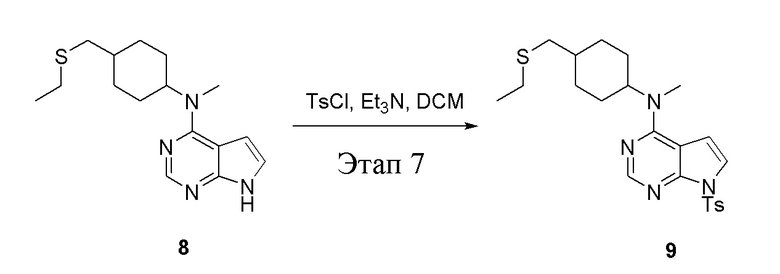
в одногорлую колбу объемом 250 л при 0°C добавили 8 (7,5 г, 24,67 ммоль) (продукт, полученный на этапе 6) и дихлорметан (80 мл), затем последовательно добавляли TsCl (7,06 г, 37,01 ммоль) и триэтиламин (5,0 г, 49,34 ммоль), после чего в течение 3 часов при комнатной температуре проводили реакцию. Реакционную жидкость промыли насыщенным солевым раствором, высушили безводным сульфатом натрия, а также просушили с помощью центробежной сушилки с получением неочищенного продукта (5,5 г). ЖХТМС: 459 [M+H]+
Этап 8:
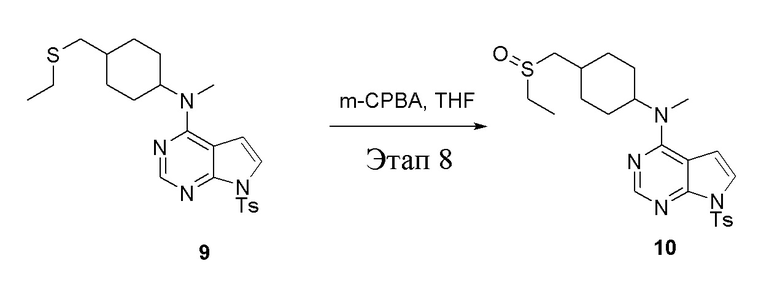
в одногорлую колбу объемом 250 мл при 0°C добавили 9 (14,5 г, 32,65 ммоль) (продукт, полученный на этапе 7) и тетрагидрофуран (120 мл); по каплям добавили тетрагидрофурановый раствор метахлорпербензойной кислоты (5,63 г, 32,65 ммоль), после чего в течение 30 мин проводили реакцию и концентрировали смесь, далее влили ее в этилацетат (100 мл), потом последовательно промыли насыщенным сульфитом натрия (50 мл × 3), бикарбонатом натрия (50 мл × 3) и физиологическим раствором (50 мл × 3), высушили безводным сульфатом натрия и просушили с помощью центробежной сушилки, затем подвергли обработке в хроматографической колонке (в условиях петролейного эфира: этилацетата=1:2) с получением желтого твердого вещества, то есть целевого продукта (7,0 г, выход: 46,6%). ЖХТМС: 461 [M+H]+
Этап 9:
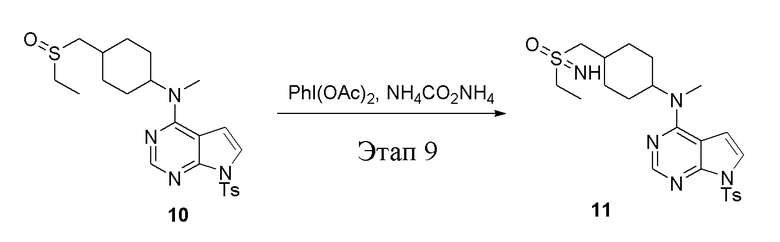
в одногорлую колбу объемом 25 л добавили 10 (400 мг, 0,84 ммоль) (продукт, полученный на этапе 8), дихлорметан (5 мл), PhI(OAc)2 (408 мг, 1,27 ммоль) и карбонат аммония (161 мг, 1,68 ммоль), после чего при комнатной температуре в течение 6 часов проводили реакцию. После фильтрации и концентрирования в смесь влили метиловый спирт и добавили карбонат калия, перемешивали в течение 30 минут для достижения нужной концентрации, а затем подвергли обработке в хроматографической колонке (в условиях этилацетата: метилового спирта=4:1) с получением белого твердого вещества, то есть целевого соединения (400 мг, выход: 97,4%). ЖХТМС: 488 [M-H]+
Этап 10:
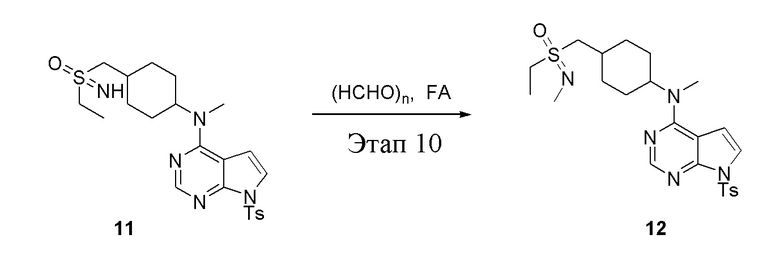
в одногорлую круглодонную колбу объемом 25 л добавили 11 (400 мг, 0,82 ммоль) (продукт, полученный на этапе 9), параформальдегид (147 мл, 1,64 ммоль) и муравьиную кислоту (4 мл), после чего в течение 48 часов при 100°C проводили реакцию. Остаточные материалы после концентрирования влили в дихлорметан и 2N серную кислоту; водную фазу нейтрализовали бикарбонатом натрия и экстрагировали дихлорметаном (10 мл × 3), а органическую фазу высушили безводным сульфатом натрия и концентрировали с получением белого твердого вещества, то есть целевого продукта (170 мг, выход: 41,1%). ЖХТМС: 504 [M+H]+
Этап 11:
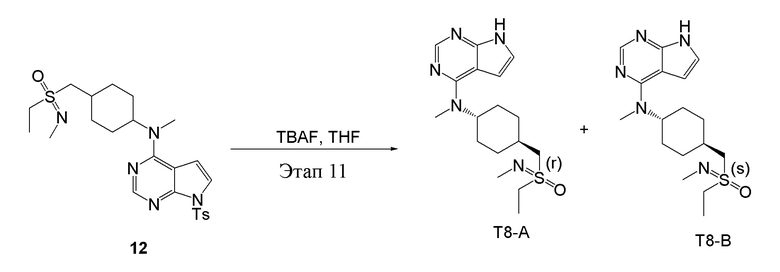
в одногорлую колбу объемом 25 мл добавили 12 (170 мг, 0,34 ммоль) (продукт, полученный на этапе 10), тетрагидрофуран (2,0 мл) и фторид тетрабутиламмония (177 мг, 0,68 ммоль), которые в течение 12 часов подвергали нагреву с обратным холодильником, концентрировали и влили в дихлорметан и насыщенный солевой раствор, при этом органическую фазу высушили безводным сульфатом натрия и концентрировали, после чего обработали традиционным и хиральным способами (в колонке: DAICEL CHRAL IC (250 мм × 30 мм, 10 мкм); градиентное время: 20 мин; в условиях: 0,2% ДЭА, этанол и гексан; скорость потока: 18 мл/мин; 80% этанол) с получением белого твердого вещества, то есть продукта-T8-A(20 мг, выход: 16,8%). ЖХТМС:350,2 [M+H]+,H1-ЯМР: 1H-ЯМР (400 МГц, ЭУОО) δ 8,13 (с, 1H), 7,16 (d, J=3,6 Гц, 1H), 6,70 (d, J=3,6 Гц, 1H), 4,69 (с, 1H), 3,32-3,25 (м, 5H), 3,18 (dd, J=6,2, 1,9 Гц, 2H), 2,79 (с, 3H), 2,27-2,01 (м, 3H), 1,93-1,82 (м, 4H), 1,45 (d, J=12,0 Гц, 2H), 1,37 (t, J=7,4 Гц, 3H), а также продукт T8-B (30 мг, выход: 25,3%), ЖХТМС: 350,2 [M+H]+, H1-ЯМР: 1H-ЯМР (400 МГц, ЭУОО) δ 8,13 (с, 1H), 7,16 (d, J=3,6 Гц, 1H), 6,70 (d, J=3,6 Гц, 1H), 4,69 (с, 1H), 3,27-3,21 (м, 3H), 3,11-3,10 (м, 2H), 3,18 (dd, J=6,2, 1,9 Гц, 2H), 2,79 (с, 3H), 2,19 (dd, J=24,0, 12,8 Гц, 2H), 2,07 (с, 1H), 1,93-1,82 (м, 4H), 1,51-1,42 (м, 2H), 1,37 (t, J=7,4 Гц, 3H).
Вариант осуществления 9: эксперимент по энзимологии JAK1
Экспериментальные материалы
JAK1 представляет собой разновидность протеинтирозинкиназы, которую можно приобрести у компании «Thermo» (№ по каталогу: PV4774). АТФ был приобретен у компании «Thermo» (№ по каталогу: PV4774), тофацитиниб был приобретен у компании «Selleck» (№ по каталогу: S5001), набор HTRF KinEASE-TK был приобретен у компании «Cisbio» (№ по каталогу: 62TKOPEC).
Экспериментальный способ
HTRF®KinEASE™-TK был универсальным способом измерения активности тирозинкиназы с применением субстрата и универсальной системы обнаружения. Реакцию проводили в 384-луночном планшете (марки Greiner, № по каталогу: 784075), а общая реакционная система составляла 20 мкл/лунку. Реакционная система главным образом состояла из 1 × киназного буферного раствора, 5 мМ MgCl2, 0,625 мМ EGTA, 0,06 мкМ SEB, 1 мМ DTT, 0,01% Brij-35, 1 мкМ ТК-суббиотина и 10 мкМ АТФ. Соединения, полученные в вариантах осуществления 1-8, и контрольное соединение оклацитиниба непрерывно разбавляли с помощью ДМСО до уровня 10 точек концентрации, после чего по 100 нл каждоого раствора переносили в измерительный планшет для проведения эксперимента. После добавления 19,76 нМ JAk1 началась реакция, через 30 минут прохождения которой при 25°C для прекращения реакции добавляли детекторный реагент (0,25 X TK-антитела, 62,5 нМ Стрептавидина-XL665). После выдерживания при комнатной температуре в течение 60 минут с помощью аппарата для прочтения планшетов марки Spark 10M либо Envision было выполнено считывание сигнала передачи энергии резонанса флуоресценции (FRET). (Технология HTRF (однородной флуоресценции с временным разрешением) 665/615=значение сигнала с частотой 665 нм/значение сигнала с частотой 615 нм).
Анализ данных
Соотношение сигналов 665/615 преобразовывали в коэффициент ингибирования в процентах.
Коэффициент ингибирования, % = (max-образец)/(max-min)*100, где «min» обозначает соотношение значений сигналов с частотой 665/615 для контрольной лунки без ферментов, «max» обозначает соотношение 665/615 значений сигналов с частотой для контрольной лунки с ДМСО.
Результат
Исходя из описанного выше экспериментального способа, в таблице ниже были приведены измерения по IC50 соединения
Таблица 1. Результаты испытаний по ферментной активности JAK1
Вариант осуществления 10. Анализ индуцированной IL-2 и ConA пролиферации мононуклеаров периферической крови у собак.
Экспериментальные материалы
Рекомбинантный собачий белок IL-2 (Cys147Ser) был приобретен у компании «R&D» (№ по каталогу: 1815-CL-020/CF). Собачьи конъюгированные CD4 Alexa Fluor 647 антитела были приобретены у компании «R&D» (№ по каталогу: 1815-CL-020/CF), конканавалин А, полученный из растения «Canavalia ensiformis» (канавалия мечевидная), был приобретен у компании «Sigma» (№ по каталогу: C5275-5MG). Малеат оклацитиниба был приобретен у компании «AbMole Bioscience» (№ по каталогу: M5827). Мононуклеары периферической крови были приобретены у компании «AllCells» (№ по каталогу: DPB-002).
Экспериментальный способ
Проточная цитометрия представляет собой определенную технологию, применяемую для обнаружения и измерения физических и химических характеристик той или иной популяции клеток либо того или иного роя частиц. Собачьи мононуклеары периферической крови собирали и окрашивали CFSE, а затем мононуклеары (1×10*5 клеток/лунку) инокулировали в 96-луночный планшет для инкубации при 37°C в течение 1 часа в инкубаторе с использованием 5%CO2. Соединение в соответствующей концентрации (соединение имело начальную концентрацию на уровне 5 мкМ и было троекратно разбавлено до 9%, причем ДМСО имел конечную концентрацию на уровне 0,1%) было введено в соответствующие лунки, через 4 часа в них было добавлено по 50 нг/мкл IL-2 и 1 мкг/мл конканавалина А, для активации клетки в течение 120 часов инкубировали при 37°C в инкубаторе с использованием 5%CO2. После этого клетки собирали и окрашивали конъюгированными CD4 Alexa Fluor 647 собачьими антителами.
Образцы с содержанием клеток ресуспендировали в буферном растворе для окрашивания и вводили в устройство для проведения проточной цитометрии. Когда клеточная суспензия пропускается через устройство для проведения проточной цитометрии, вытекающая из небольшого раструба клеточная суспензия под действием жидкостной механики внешней части жидкости агрегируется по направлению к центру. Вследствие образования подобной струйки клетки последовательно проходят сквозь лазерные лучи, причем исключительно по одной за раз. При прохождении клеток сквозь лазерные лучи детектором выполняется обнаружение рассеянного света клеток либо частиц. Передний детектор обнаруживал направленное вперед рассеяние (НВР), несколько размещенных сбоку детекторов обнаружили боковое рассеяние (БР); а детектор флуоресценции обнаруживал испускаемое окрашенными клетками либо частицами флуоресцентное свечение. Следовательно, такие клетки удалось разделить на разные популяции клеток, исходя из разных значений НВР и БР.
Анализ данных
Была классифицирована единичная клетка, у которой SSC-A и SSC-H демонстрировали линейную связь. CD4+находилась в круге, сформировавшемся из субпопуляций лимфоцитов. Были выполнен расчет среднекратного прироста (СКП) популяции CD4 +.
Коэффициент ингибирования = (СКП соединения - СКП отрицательных лунок)/(СКП положительных лунок - СКП отрицательных лунок). На графике показателей ингибирования ось X указывает на уровень концентрации, а ось Y указывает на процент ингибирования.
График показателей ингибирования был составлен для получения соединения IC50. Результаты были представлены в Таблице 2.
Таблица 2. Результаты испытаний:
В заключение следует указать на то, что приведенные выше варианты осуществления используются только для описания представленного настоящим изобретением технического решения, но не служат для обозначения его пределов. Даже несмотря на то, что настоящее изобретение подробно описано со ссылкой на приведенные выше варианты осуществления, любой специалист в данной области техники должен понимать, что в техническое решение, представленное в приведенных выше вариантах осуществления, все еще могут вноситься поправки, или же его технические характеристики все еще могут в определенной части либо в своей полноте становиться предметом эквивалентных замен; более того, такие поправки или замены не лишают характер соответствующего технического решения сущности и предмета технических решений, реализованных в вариантах осуществления настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ КАРБОКСИЗАМЕЩЕННЫХ (ГЕТЕРО)АРОМАТИЧЕСКИХ КОЛЕЦ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2733750C2 |
| ПРОИЗВОДНОЕ 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2804127C2 |
| ИНГИБИТОРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2019 |
|
RU2806033C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2799340C2 |
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2683325C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ МЕТАЛЛОФЕРМЕНТЫ | 2017 |
|
RU2764666C2 |
Изобретение относится к соединению, представленному общей формулой I, или его фармацевтически приемлемой соли или стереоизомеру, которые обладают ингибирующим действием в отношении киназы JAK. В формуле I A представляет собой C; R5 представляет собой H; X представляет собой 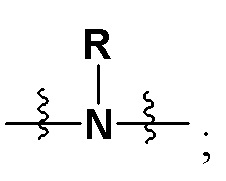 R представляет собой C1-10 линейный или разветвленный алкил; Y представляет собой
R представляет собой C1-10 линейный или разветвленный алкил; Y представляет собой 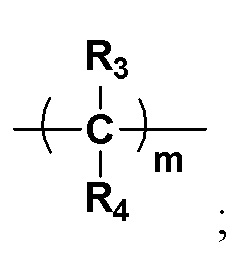 R3 и R4 представляют собой H; m представляет собой любое целое число от 1 до 3, в частности 1, 2 или 3; Z представляет собой C3-12-циклоалкил; R1 и R2 представляют собой каждый на выбор H, C1-10 линейный или разветвленный алкил, где H на атоме углерода может замещаться такими группами, как -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил; где H на C6-18-ариле либо гетероциклоариле может замещаться группой галогена или C1-4 линейным алкилом; каждый R7 и R8 представляет собой H. Изобретение относится также к способу получения соединения, представленного общей формулой I, фармацевтическому составу, включающему указанное соединение, и его применению при получении лекарственного средства, предназначенного для лечения заболевания, связанного с сигнальным путем JAK-STAT, в том числе для профилактики и/или лечения воспалительных либо онкологических заболеваний. 5 н. и 7 з.п. ф-лы, 2 табл., 9 пр.
R3 и R4 представляют собой H; m представляет собой любое целое число от 1 до 3, в частности 1, 2 или 3; Z представляет собой C3-12-циклоалкил; R1 и R2 представляют собой каждый на выбор H, C1-10 линейный или разветвленный алкил, где H на атоме углерода может замещаться такими группами, как -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил; где H на C6-18-ариле либо гетероциклоариле может замещаться группой галогена или C1-4 линейным алкилом; каждый R7 и R8 представляет собой H. Изобретение относится также к способу получения соединения, представленного общей формулой I, фармацевтическому составу, включающему указанное соединение, и его применению при получении лекарственного средства, предназначенного для лечения заболевания, связанного с сигнальным путем JAK-STAT, в том числе для профилактики и/или лечения воспалительных либо онкологических заболеваний. 5 н. и 7 з.п. ф-лы, 2 табл., 9 пр.
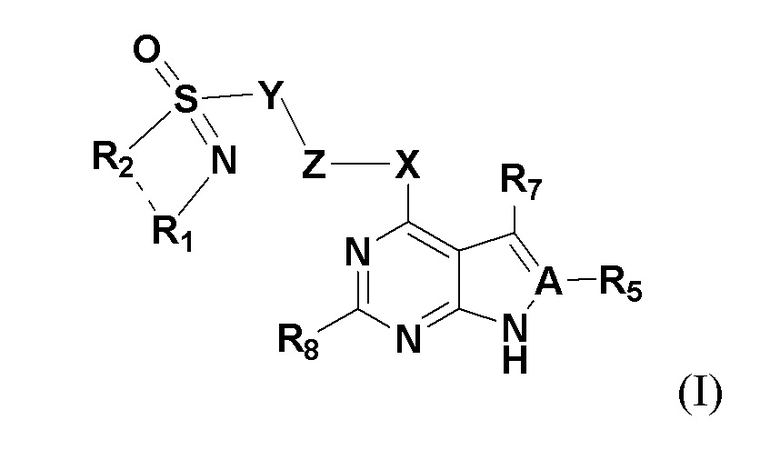
1. Соединение, представленное общей формулой I, или его фармацевтически приемлемая соль или стереоизомер
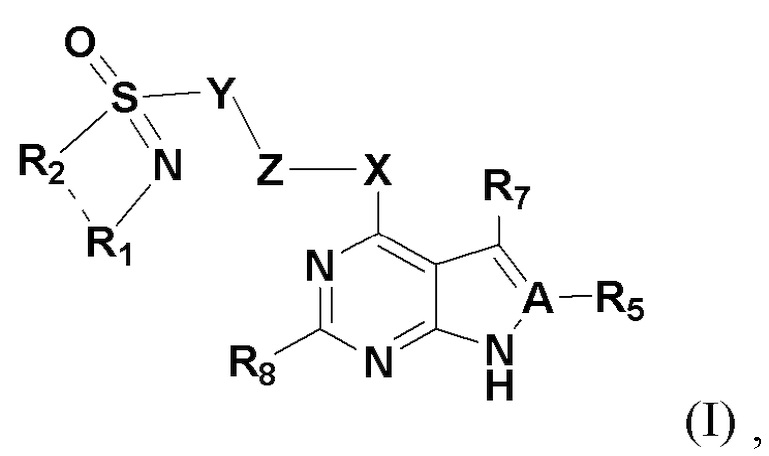
где A представляет собой C;
R5 представляет собой H;
X представляет собой  R представляет собой C1-10 линейный или разветвленный алкил;
R представляет собой C1-10 линейный или разветвленный алкил;
Y представляет собой 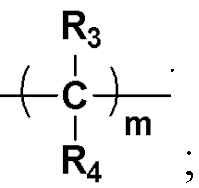 R3 и R4 представляют собой H;
R3 и R4 представляют собой H;
m представляет собой любое целое число от 1 до 3, в частности 1, 2 или 3;
Z представляет собой C3-12-циклоалкил;
R1 и R2 представляют собой каждый на выбор H, C1-10 линейный или разветвленный алкил, где H на атоме углерода может замещаться такими группами, как -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил, C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил; где H на C6-18-ариле либо гетероциклоариле может замещаться группой галогена или C1-4 линейным алкилом;
каждый R7 и R8 представляет собой H.
2. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 1, отличающиеся тем, что R представляет собой C1-10 линейный алкил;
R1 представляет собой на выбор H, C1-6 линейный алкил; где H на атоме углерода может замещаться такими группами, как -N(C0-10-алкил)(C0-10-алкил), -OC0-4-алкил, C3-10-циклоалкил и C6-18-арил;
R2 представляет собой C1-6 линейный алкил; H на атоме углерода может замещаться следующими группами, такими как -N(C0-10-алкил)(C0-10-алкил), -OC0-10-алкил, C3-10-циклоалкил или C6-18-арил, -N-гетероциклоарил, -O-гетероциклоарил либо -S-гетероциклоарил.
3. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 2, отличающиеся тем, что R представляет собой C1-6 линейный алкил.
4. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 3, отличающиеся тем, что Z представляет собой на выбор C4-10-циклоалкил, например
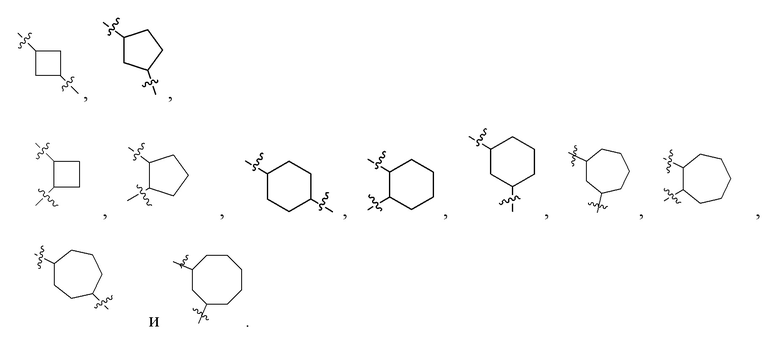
5. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 4, отличающиеся тем, что указанное соединение имеет следующую структурную формулу:
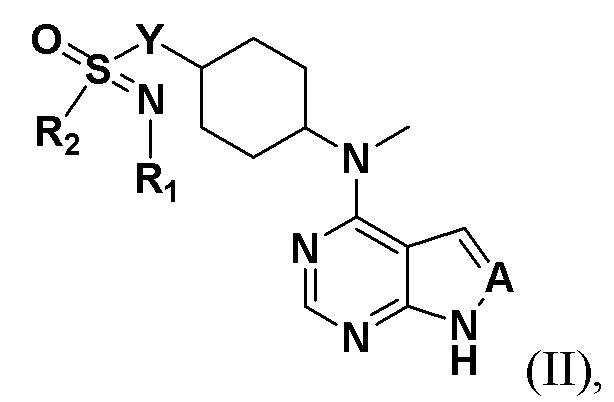
где R1 представляет собой на выбор H, -CH3, -CH2CH3, -CH2CH2CH3 и -CH2CH2OCH3; и
R2 представляет собой на выбор -CH3-,
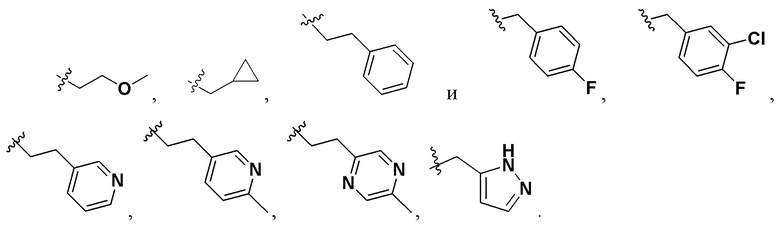
6. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 5, отличающиеся тем, что указанное соединение имеет следующую характерную структурную формулу:
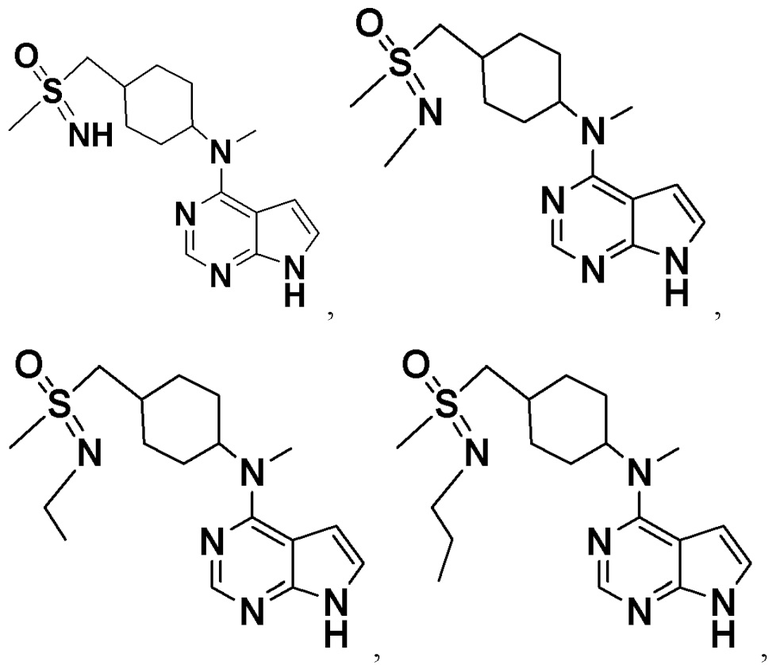
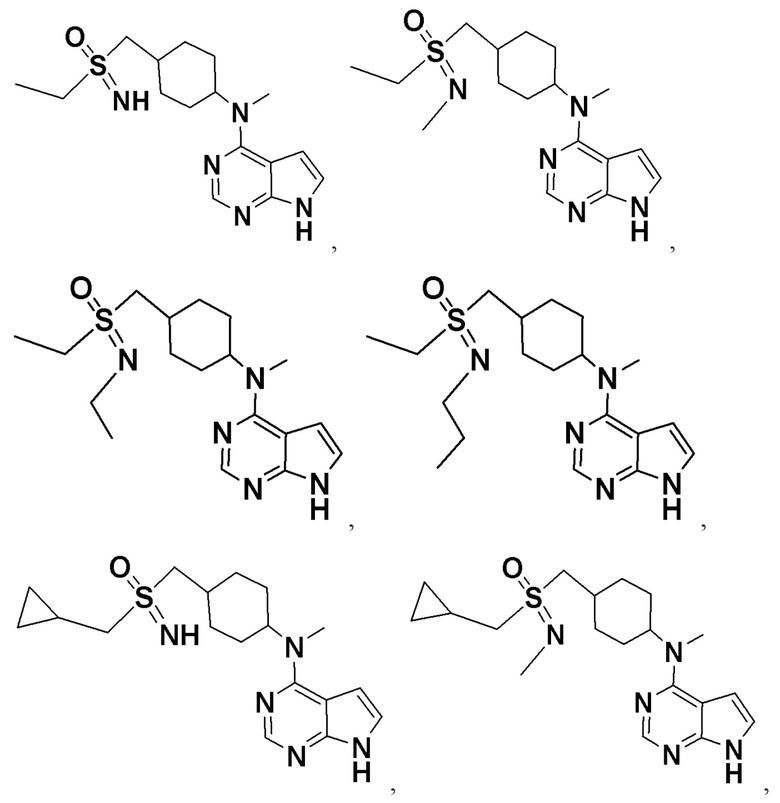
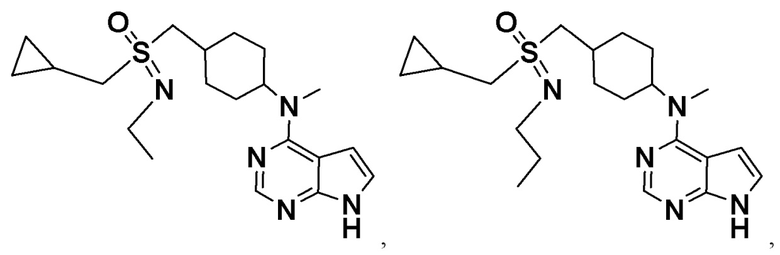
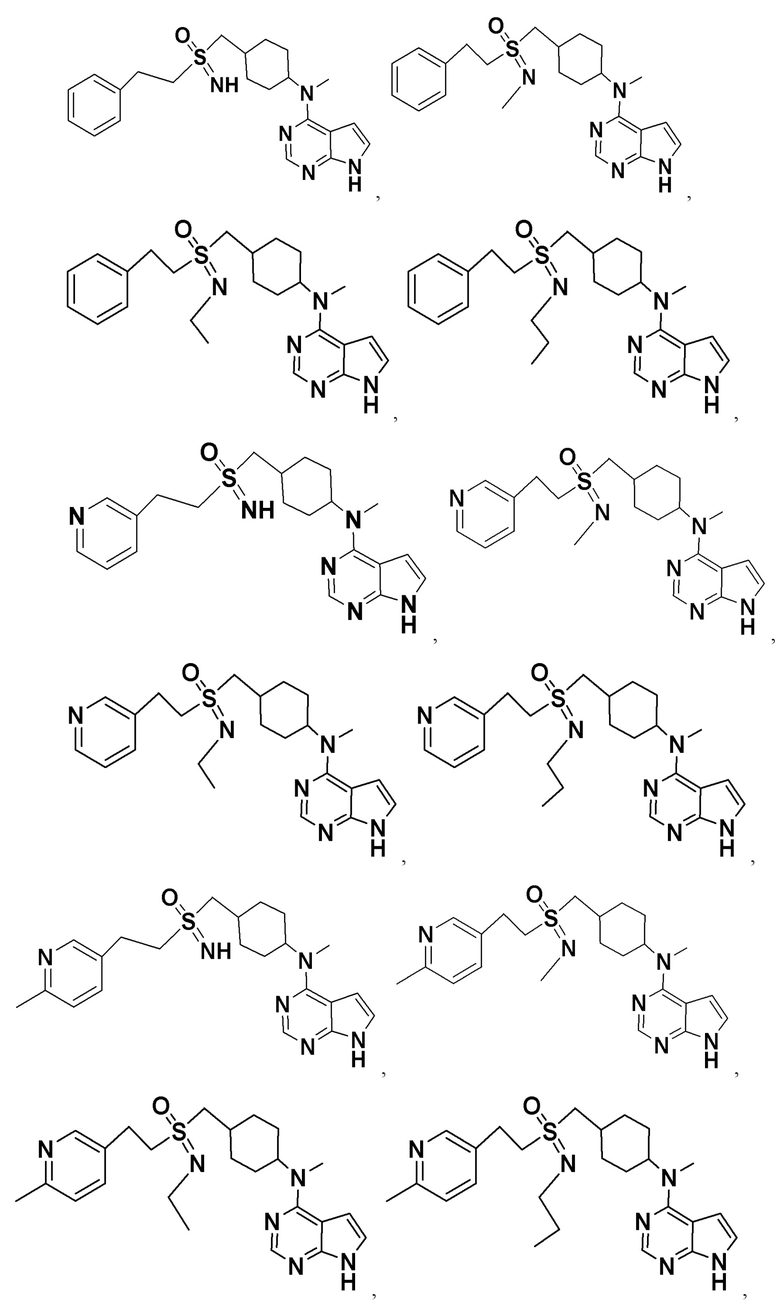
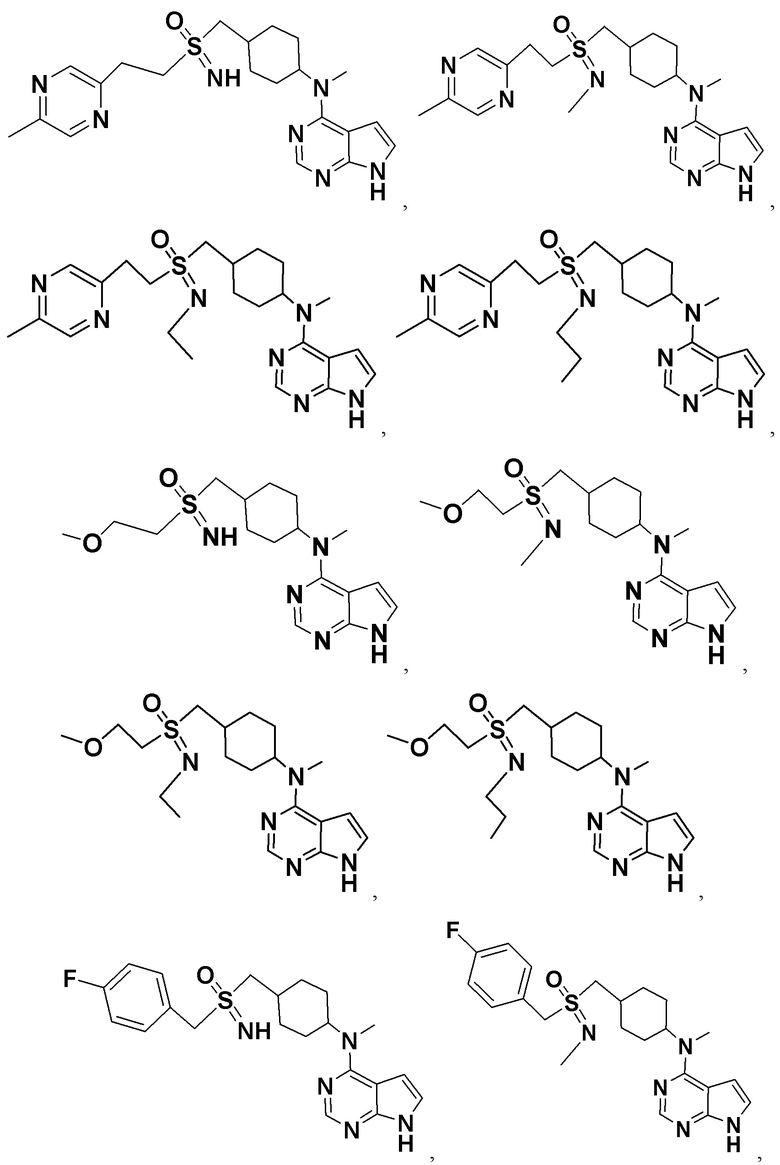
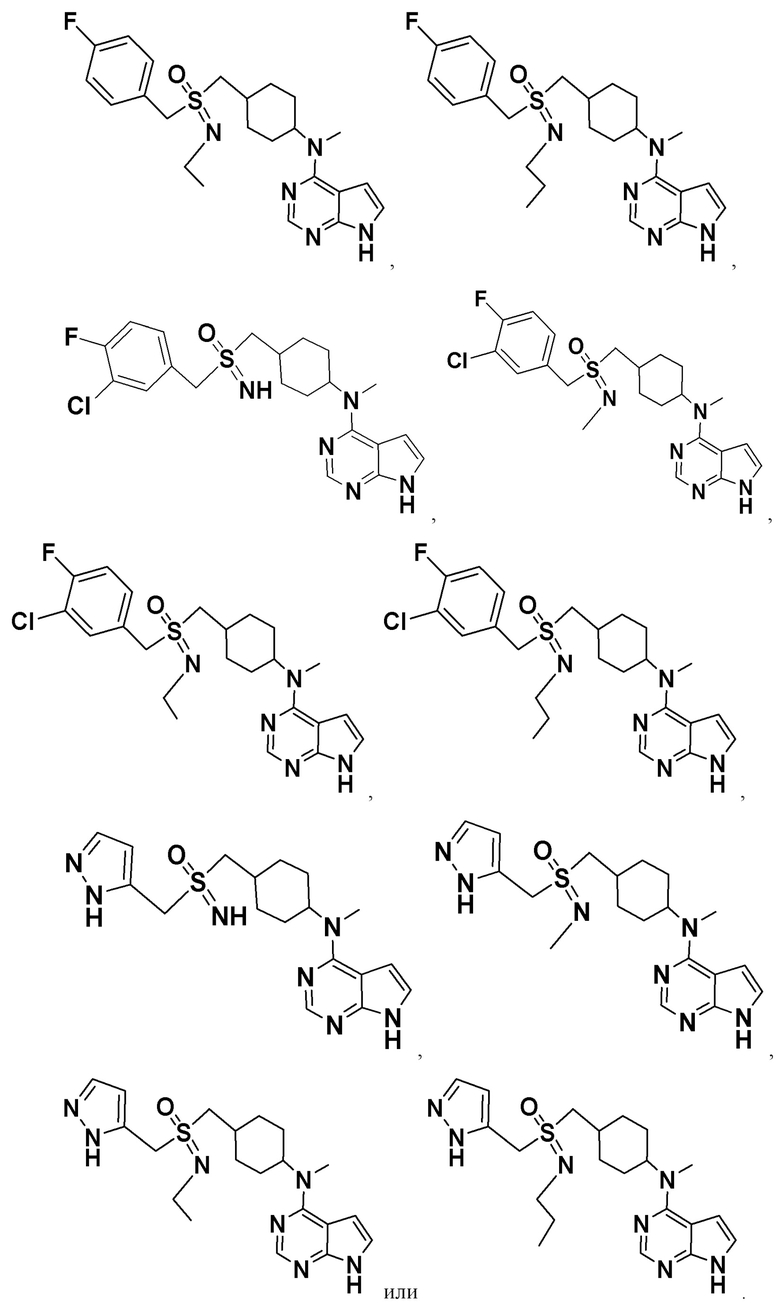
7. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 1, отличающиеся тем, что указанный стереоизомер имеет следующую структуру:
8. Соединение либо его фармацевтически приемлемая соль или стереоизомер по п. 7, отличающиеся тем, что указанный стереоизомер имеет следующую структуру:
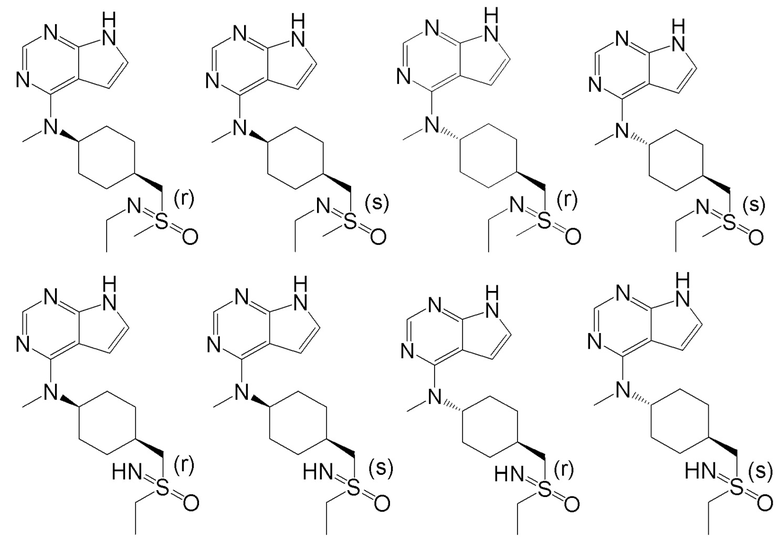
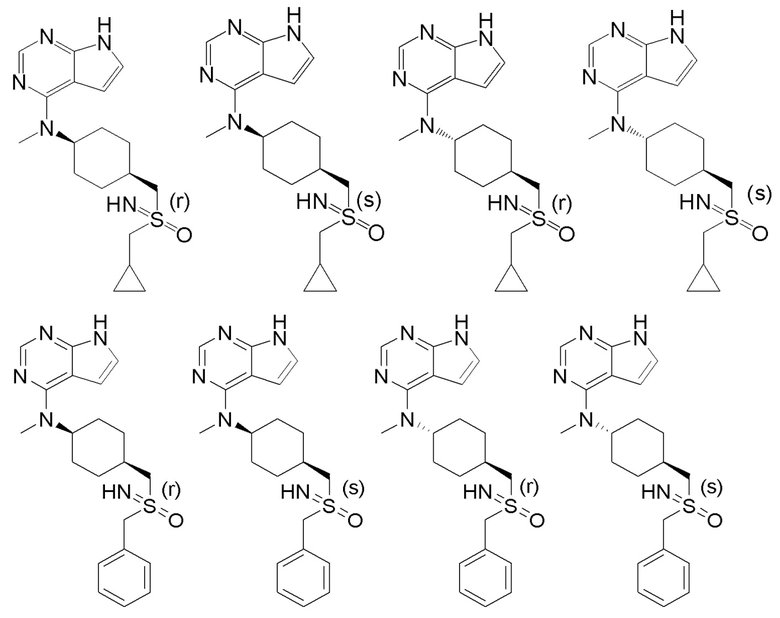
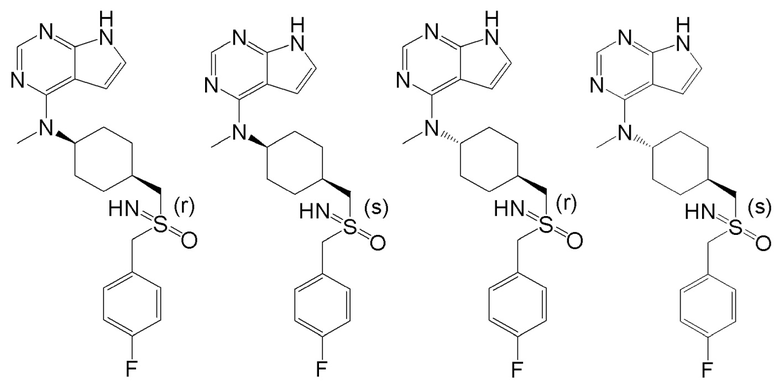
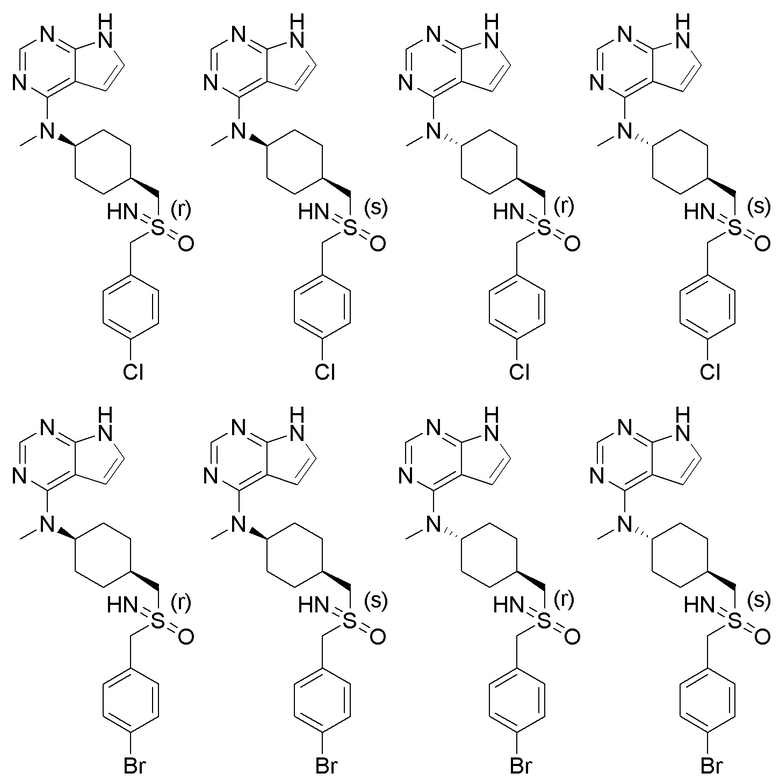
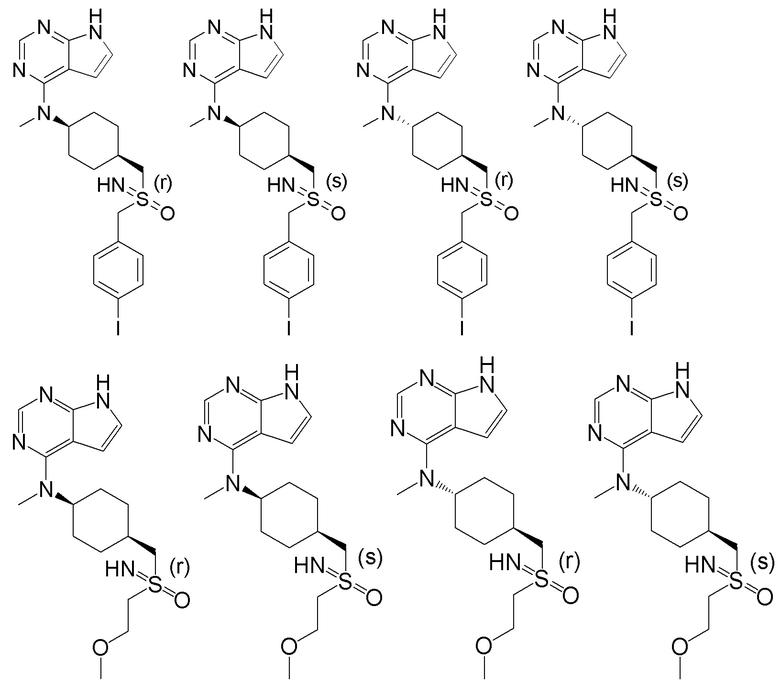
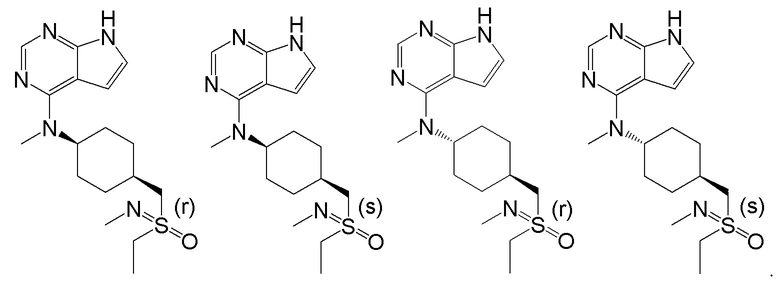
9. Способ получения соединения, представленного общей формулой I по п. 1, включающий в себя следующий путь прохождения реакции:
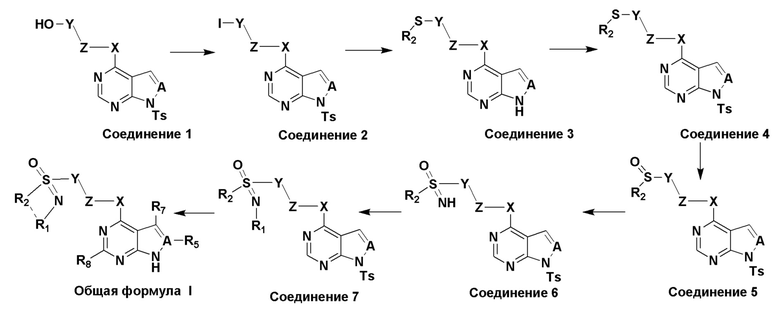
где значения A, X, Y, Z, R1, R2, R3, R4, R5, R7 и R8 определены в п. 1;
(1) растворение соединения 1 в растворителе 1, добавление триэтиламина и пара-толуолсульфонилхлорида, перемешивание при комнатной температуре в течение 20-24 ч, концентрирование, добавление йодирующего реагента, нагревание до 60-70°C и перемешивание в течение 8-9 ч с получением соединения 2;
(2) растворение соединения 2 в растворителе 2, добавление алкилтиолата натрия, обеспечение прохождения реакции в течение 20-24 ч, фильтрование и концентрирование с получением соединения 3;
(3) растворение соединения 3 в растворителе 3, добавление триэтиламина и пара-толуолсульфонилхлорида, перемешивание при комнатной температуре в течение 20-24 ч и сепарирование с получением соединения 4;
(4) растворение соединения 4 в растворителе 4, добавление мета-хлорпербензойной кислоты (m-CPBA), обеспечение прохождения реакции в течение 1-2 ч, экстракция со сбором органической фазы, промывка водой и сушка такой органической фазы, фильтрование и концентрирование с получением соединения 5;
(5) растворение соединения 5 в растворителе 5, добавление иодбензолдиацетата (PhI(OAc)2) и карбамата аммония, обеспечение прохождения реакции в течение 30-35 мин, концентрирование при пониженном давлении с получением соединения 6;
(6) растворение соединения 6 и полиальдегида в растворителе 6, нагревание до 90-95°C, обеспечение прохождения реакции в течение 20-24 ч, концентрирование, экстракция со сбором органической фазы, промывка водой и сушка такой органической фазы, фильтрование и концентрирование с получением соединения 7; а также
(7) растворение соединения 7 и карбоната цезия (Cs2CO3) в растворителе 7, обеспечение прохождения реакции при 40-50°C в течение 3-4 ч, фильтрование и концентрирование с получением соединения, представленного общей формулой I;
где растворители 1-7 представляют собой на выбор одно или сочетание двух или более из таких веществ, как хлористый метилен, ацетон, тетрагидрофуран, метиловый спирт и муравьиная кислота.
10. Фармацевтический состав, обладающий ингибирующим действием в отношении JAK, где такой фармацевтический состав включает эффективное количество соединения, представленного общей формулой I, либо его фармацевтически приемлемую соль или стереоизомер по п. 1, а также включает фармацевтически приемлемое вспомогательное вещество.
11. Применение соединения, представленного общей формулой I, либо его фармацевтически приемлемой соли или стереоизомера по п. 1 при получении лекарственного средства, предназначенного для лечения заболевания, связанного с сигнальным путем JAK-STAT.
12. Применение соединения, представленного общей формулой I, либо его фармацевтически приемлемой соли или стереоизомера по п. 1 при получении лекарственного средства, предназначенного для профилактики и/или лечения воспалительных либо онкологических заболеваний у людей и/или животных, где указанное воспалительное заболевание представляет собой ревматоидный артрит, собачий дерматит, псориаз, язвенный колит либо болезнь Крона; а указанное онкологическое заболевание - миелофиброз, истинную полицитемию, эссенциальную тромбоцитемию, хронический гранулоцитарный лейкоз, рак груди, рак легких и рак поджелудочной железы.
| US 2014243312 A1, 28.08.2014 | |||
| WO 2011075334 A1, 23.06.2011 | |||
| WO 2014015107 A1, 23.01.2014 | |||
| IRVENT J | |||
| ET AL, Novel Pieces for the Emerging Picture of Sulfoximines in Drug Discovery: Synthesis and Evaluation of Sulfoximine Analogues of Marketed Drugs and Advanced Clinical Candidates, CHEMMEDCHEM, 2017, 12(7), pp | |||
| Кренометр | 1923 |
|
SU487A1 |

