Область техники
Изобретение относится к областям химии, фармацевтики и химико-фармацевтической промышленности, а именно к новым соединениям - производным 5-(4-метоксифенил)-3Н-1,2-дитиол-3-тиона (соединение S1), 4-метил-5-пиразинил-3Н-1,2-дитиол-3-тиона (соединение S2) и 3-трифторметил-α-этилбензгидрола (соединение S3), которые могут найти применение для лечения таких заболеваний печени, как, например, неалкогольная и алкогольная жировая болезнь печени (все стадии, включая стеатоз); стеатогепатит вирусной и невирусной этиологии; фиброз и цирроз печени; токсические поражения печени; состояния, сопровождающиеся внутри- и внепеченочным холестазом; наследственные и приобретенные состояния, сопровождающиеся гипербилирубинемией.
Уровень техники
На сегодняшний день заболевания печени являются одними из наиболее сложных и проблемных. Для них характерны неспецифические проявления, что обуславливает трудности в диагностике, особенно на ранних стадиях течения. Заболевания печени часто приводят к преждевременной смерти и наносят значительный социально-экономический ущерб обществу.
Одним из подходов к лечению и профилактике метаболических, воспалительных, инфекционных, холестатических, токсических и других заболеваний печени является применение средств, повышающих устойчивость печени к патологическим воздействиям, усиливающих ее детоксицирующую функцию, способствующих регрессу биохимических и гистологических изменений гепатобилиарной системы. Такие средства выделяют в отдельную группу и называют гепатопротекторами. Известным примером гепатопротекторов является силибилин, который представляет собой алкалоид из экстракта плодов расторопши пятнистой. Его гепатопротекторное действие объясняется антиоксидантной активностью. Другими примерами гепатопротекторов являются катерген и ацетилцистеин. Значительное распространение на территории России нашли препараты природного происхождения, например, экстракты зверобоя, солодки и семян тыквы (М.Д. Машковский, Лекарственные средства, Т.1, 2002, М.: ООО Издательство Новая Волна, ISBN:5-7864-0128-6, Глава II. Гепатопротекторные средства, с. 506-508).
В настоящей заявке раскрываются новые производные 5-(4-метоксифенил)-3H-1,2-дитиол-3-тиона (соединение S1), 4-метил-5-пиразинил-3H-1,2-дитиол-3-тиона (соединение S2) и 3-трифторметил-а-этилбензгидрола (соединение S3) в качестве гепатопротекторных средств.
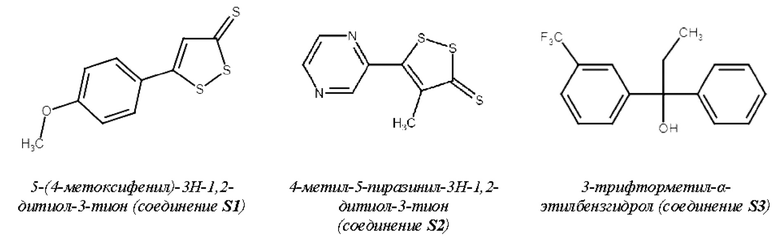
5-(4-Метоксифенил)-3Н-1,2-дитиол-3-тион (соединение S1)
Для соединения S1 известно применение в лечении ксеростомии (Т. HAMADA et al., Treatment of xerostomia with the bile secretion-stimulating drug anethole trithione: a clinical trial, THE AMERICAN JOURNAL OF THE MEDICAL SCIENCES, 1999, V. 318, N. 3, pp. 146-151, doi:10.1016/S0002-9629(l5)40606-8), некоторых видов рака (B.S.REDDY et al., Chemoprevention of colon carcinogenesis by organosulmr compounds, CANCER RESEARCH, 1993, T. 53, N. 15, pp. 3493-3498). Сообщается о применении в качестве дополнительной терапии при холецистите, желчнокаменной болезни, расстройстве желудка и хроническом гепатите (CN 1771938 А).
Механизм действия соединения S1 на организм человека достоверно неизвестен. Считается, что в процессе его метаболизма в организме человека высвобождается сероводород (H2S), который в небольших концентрациях является сигнальной молекулой, запускающей модулирующее действие на слизистые оболочки. Такое действие обеспечивает выраженный противовоспалительный эффект (L.LAZZARATO et al., New nitric oxide or hydrogen sulfide releasing aspirins, JOURNAL OF MEDICINAL CHEMISTRY, 2011, V. 54,N.15,pp. 547S-54S4, doi: 10.1021/jm2004514).
Несмотря на высокую активность и низкую токсичность, соединение S1 плохо растворимо в воде. С этим связаны ограничения в выборе лекарственных форм и в дальнейшем клиническом применении (M.BONA et al, Water/n- octanol partition coefficients of 1,2- dithiole- 3- thiones, JOURNAL OF PHARMACEUTICAL SCIENCES, 1995, V. S4, N. 9, pp. 1107-1112, doi:10.1002/jps.2600840914). Поэтому проводятся исследования, направленные на поиск производных и пролекарств соединения S1 с оптимальным балансом активности и растворимости.
В статье P.CHEN et al. Design, synthesis, and pharmacological evaluation of the aqueous prodrugs of desmethyl anethole trithione with hepatoprotective activity, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, 2010, V. 45, N. 7, p. 3005-3010 получены производные соединения S1, замещенные диметиламино-группой, пиррилидинилом, морфолинилом, пиперидинилом и другими амино-группами. Исследования in vitro и in vivo указанных соединений показали, что они способны метаболизироваться в организме до десметилированного производного (соединение S4), которое обуславливает гепатопротекгорную активность.
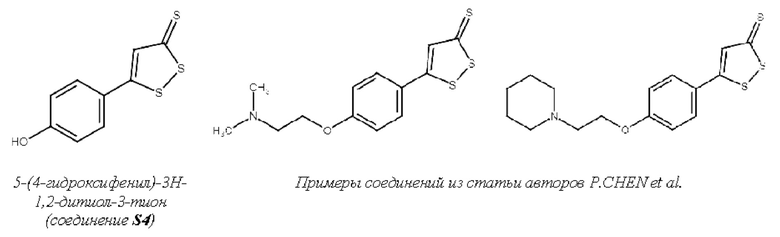
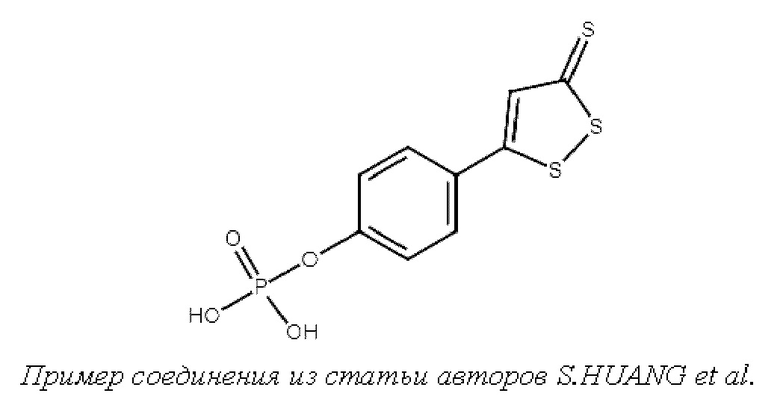
В статье S.HUANG et al. Synthesis, characterization, and in vivo evaluation of desmethyl anethole trithione phosphate prodrug for ameliorating cerebral ischemia-reperfusion injury in rats, ACS OMEGA, 2020, V. 5, N. 9, pp. 4595-4602 было получено производное S1, содержащее фосфатную группу. Соединение в условиях in vivo обладает улучшенной растворимостью и быстро претерпевает метаболическое превращение в организме до соединения S4.
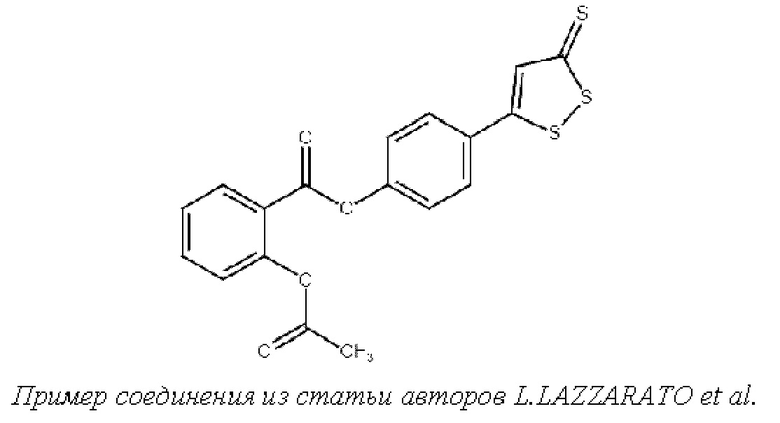
В научно-технической литературе также изучены конъюгаты соединения S1 с некоторыми другими лекарственными средствами. Так, в работе L.LAZZARATO et al., New nitric oxide or hydrogen sulfide releasing aspirins, JOURNAL OF MEDICINAL CHEMISTRY, 2011, V. 54, N. 15, pp. 5478-5484 для увеличения противовоспалительного действия был получен конъюгат с аспирином.
Известен конъюгат соединения S1 с пропаргилцистеином, для которого в заявке WO 2017202266 A1 было продемонстрировано применение для лечения и профилактики нейродегенеративных заболеваний, в частности, болезни Альцгеймера.
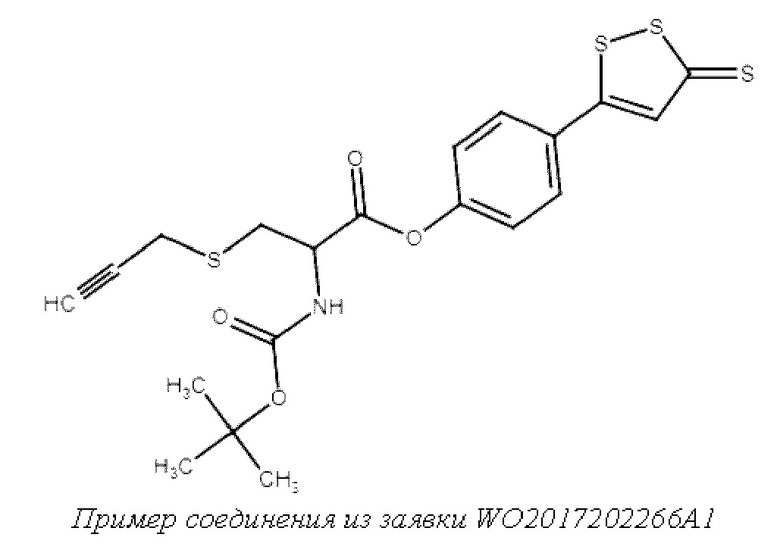
4-Метил-5-пиразинил-3Н-1,2-дитиол-3-тион (соединение S2)
Соединение S2 было впервые описано в патенте US 4110450 в качестве антишистосомозного средства. Позже были исследованы его противоопухолевые свойства (T.W.KENSLER et al, Development of cancer chemopreventive agents: oltipraz as a paradigm, CHEMICAL RESEARCH IN TOXICOLOGY, 1999, V. 12, N. 2, pp. 113-126). Также было показано, что в условиях in vivo соединение S2 метаболизируется до пирроло[1,2-а]пиразина (M.B.FLEURY, et al., Toward an understanding of the schistosomicidal effect of 4-methyl-5-(2-pyrazinyl)-1,2-dithiole-3-thione (oltipraz), BIOCHEMICAL PHARMACOLOGY, 1991, V.41, N.3, pp.361-367). Производные соединения S2 не получили широкого распространения в научно-технической литературе.
3-Трифторметил-а-этилбензгидрол (соединение S3)
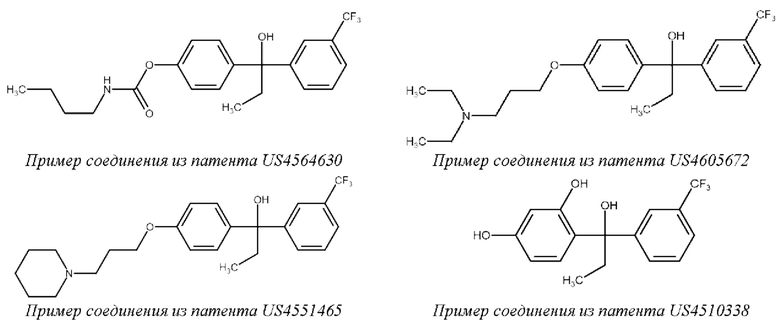
3-Трифторметил-а-этилбензгидрол (соединение S3) известен в качестве гепатопротекторного лекарственного средства зиксорин. Препарат индуцирует оксидазную ферментативную активность печени, усиливает выведение метаболитов и сам хорошо метаболизируется. Зиксорин применяют при функциональной гипербилирубинемии у больных с хроническими заболеваниями печени. Имеются данные об эффективности в терапии кожных заболеваний, например, дерматита и псориаза (М.Д. Машковский, Лекарственные средства, Т. 1, 2002, М.: ООО Издательство Новая Волна, ISBN:5-7864-0128-6, Глава П. Гепатопротекторные средства, с. 506-508).
В уровне техники известны производные соединения S3, содержащие карбаматную группу (US 4564630), диэтилалкокси-группу (US 4605672), пиперидиновую группу (US 4551465) и соединения с резорциновым фрагментом (US 4510338). Для полученных соединений раскрыто применение в лечении гиперлипидемии и алкогольной интоксикации.
Несмотря на разнообразие производных рассмотренных соединений, до сих пор ни одно из соединений не нашло широкого применения в клинической практике. Поэтому техническая задача, направленная на поиск новых гепатопротекторных средств, на настоящий момент является актуальной и своевременной.
Раскрытие сущности изобретения
Химические соединения
Технические результаты, на достижение которых направлено настоящее изобретение, заключаются в
- повышении растворимости и биодоступности новых соединений по сравнению с известными гепатопротекторными средствами (5-(4-метоксифенил)-3Н-1,2-дитиол-3-тион - соединение S1, 4-метил-5-пиразинил-3Н-1,2-дитиол-3-тион - соединение S2 и 3-трифторметил-а-этилбензгидрол - соединение S3);
- проявлении увеличенной гепатопротекторной активности;
- снижении токсичности новых соединений по сравнению с известными гепатопротекторными средствами (5-(4-метоксифенил)-3Н-1,2-дитиол-3-тион соединение S1, 4-метил-5-пиразинил-3Н-1,2-дитиол-3-тион - соединение S2 и 3-трифторметил-а-этилбензгидрол - соединение S3).
Указанная техническая проблема решается, а заявленный технический результат достигается благодаря новым химическим соединениям. В одном из вариантов соединение выбрано из группы:
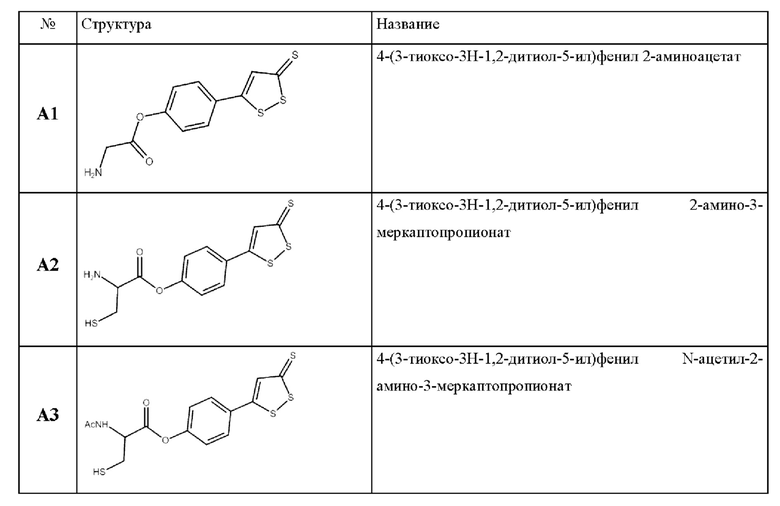
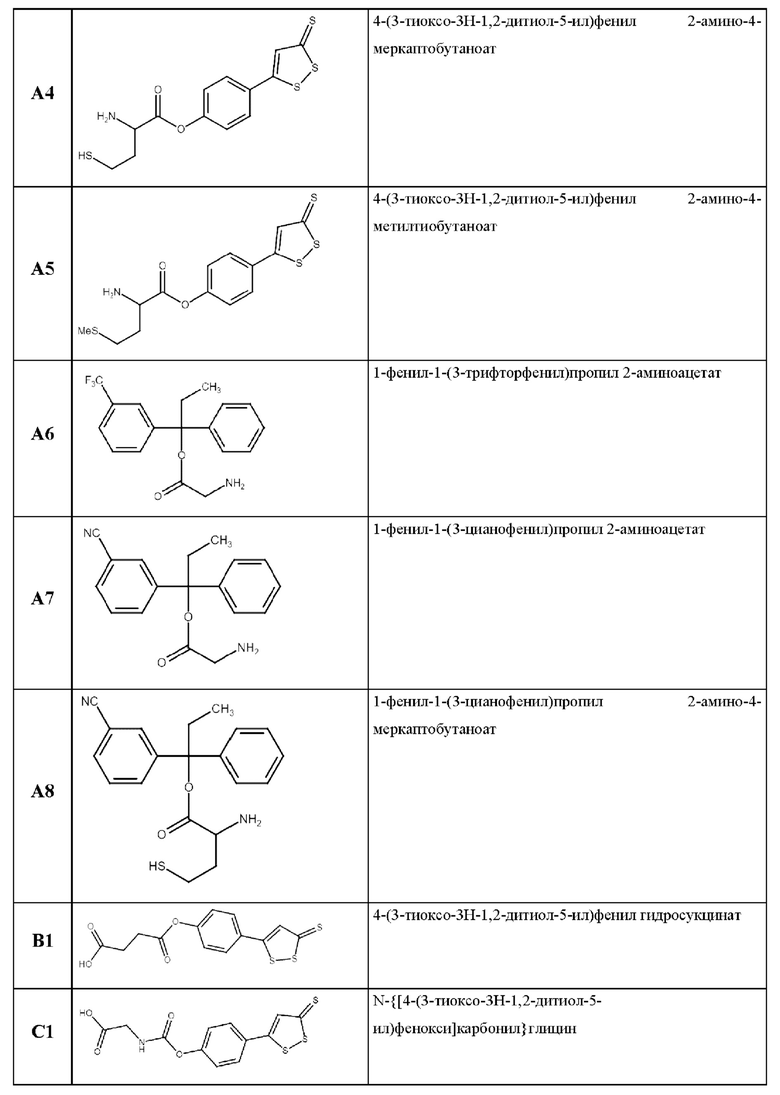
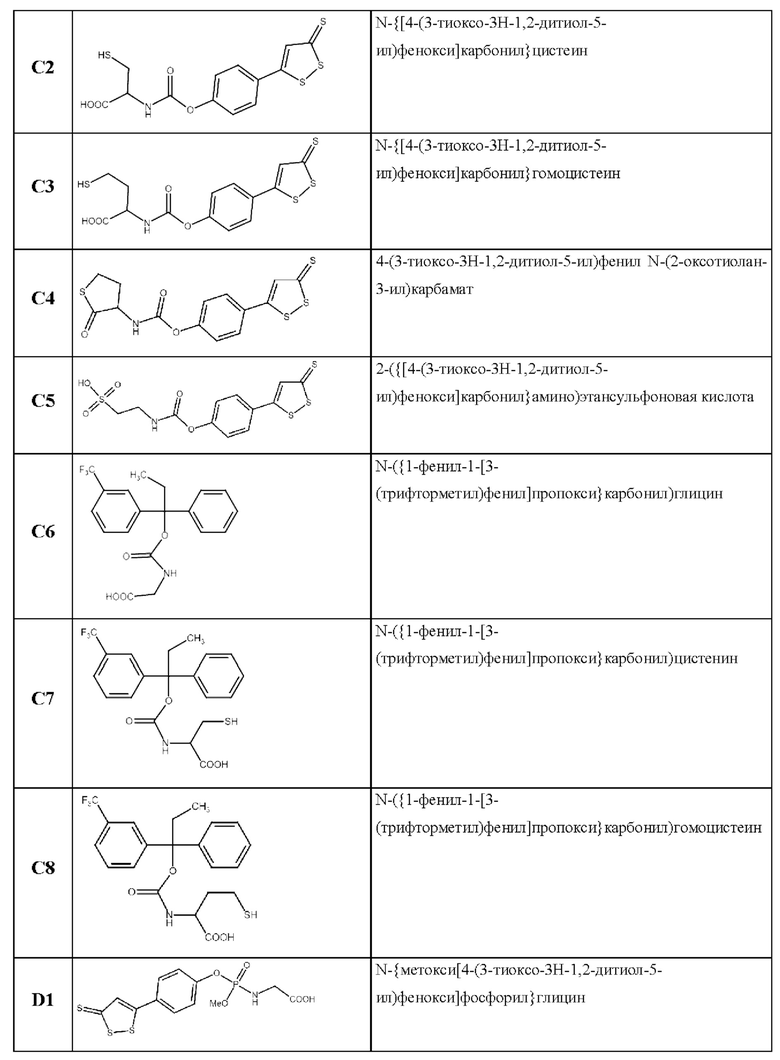
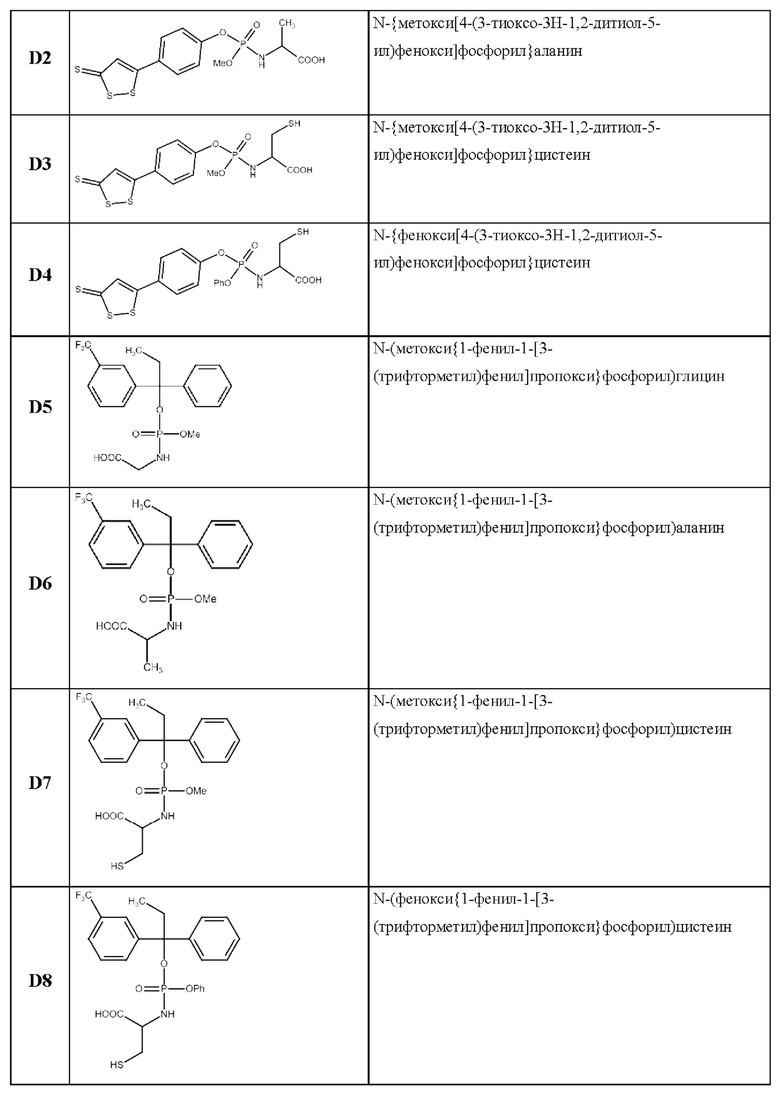
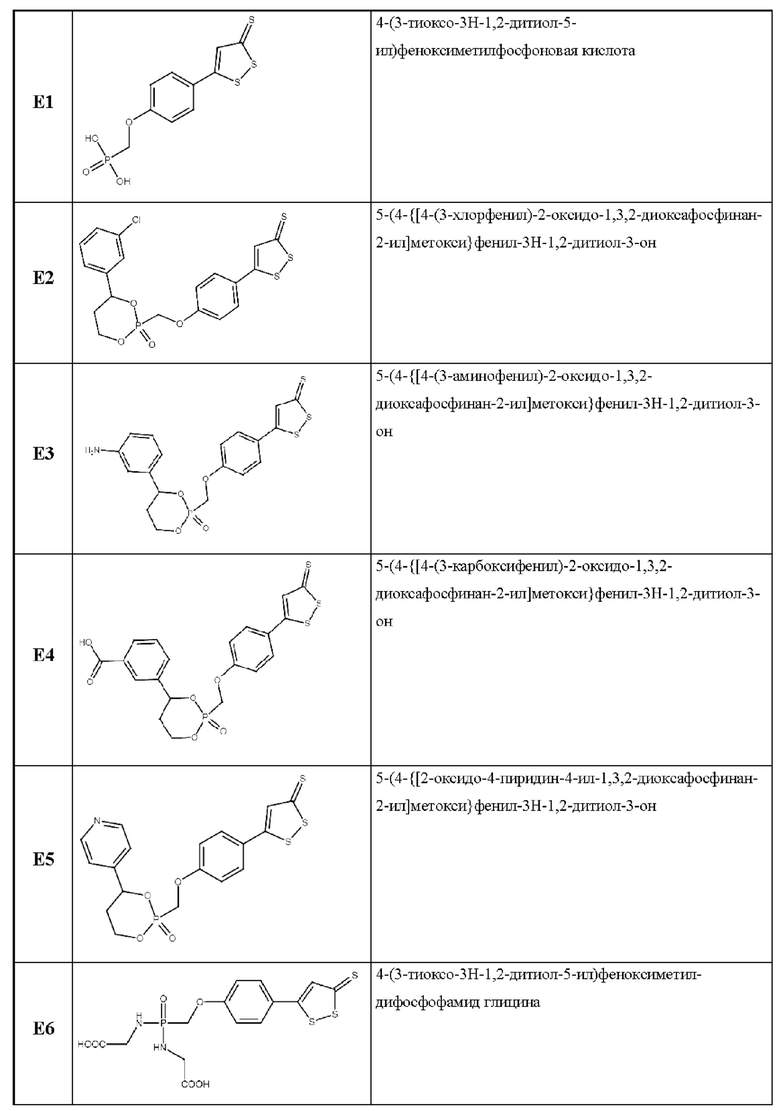
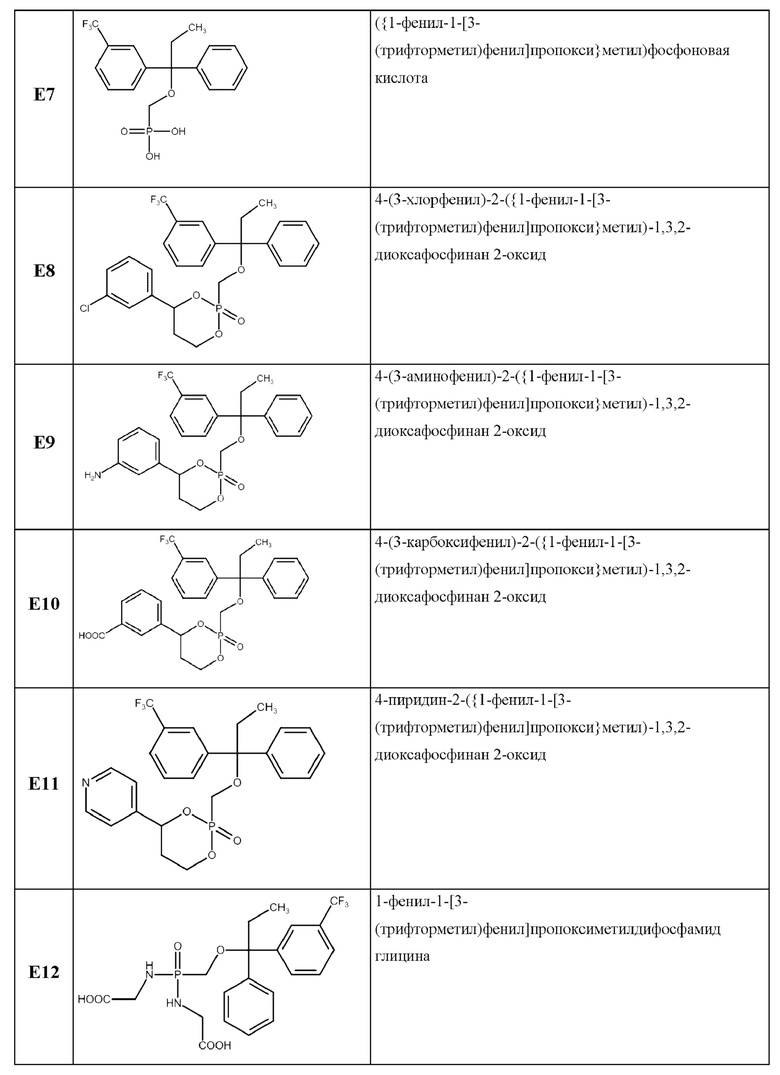
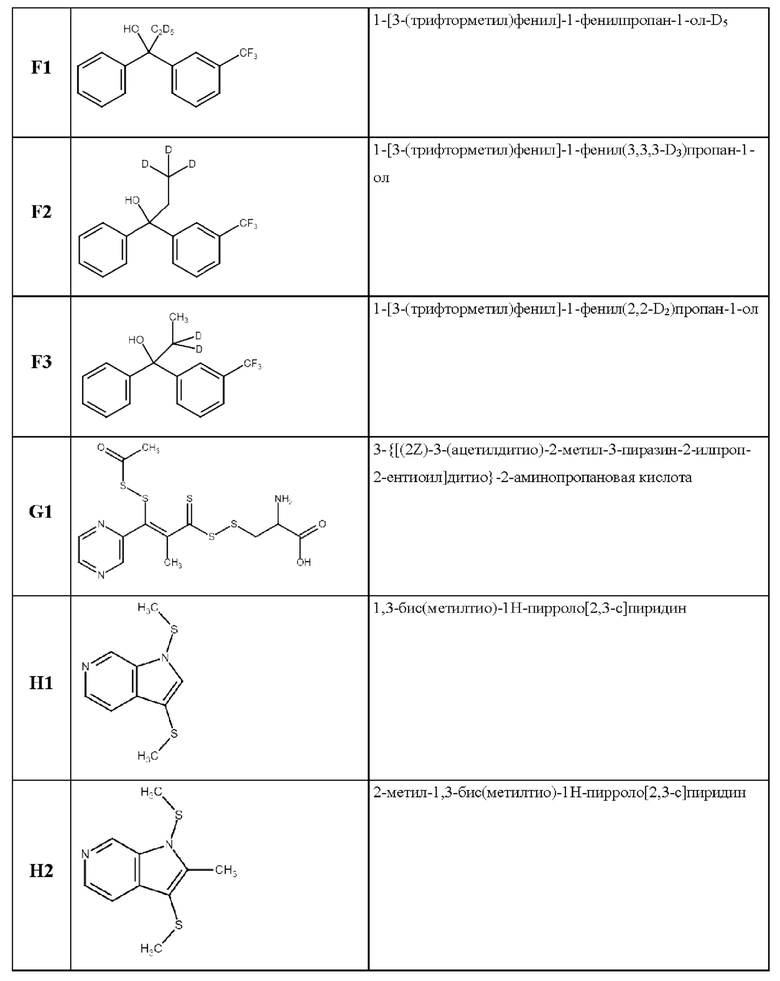
В случае возможных расхождений между названиями соединений и их структурными формулами, то приоритетными являются структурные формулы.
Настоящее изобретение также относится к стереоизомерам и фармацевтически приемлемым солям указанных соединений.
Термин стереоизомеры означает пространственные изомеры указанного соединения, то есть соединения имеющие одинаковую структуру, но отличающиеся пространственным расположением атомов. К стереоизомерам относятся энантиомеры (оптические изомеры), диастереомеры (включая цис-/транс-шомеры, Z-/E-изомеры) и конформеры. Все стереоизомеры соединений настоящего изобретения также включены в объем настоящего изобретения.
Термин конъюгат означает химическое соединение, которое содержит в своем составе две или более молекул с разными химическими свойствами.
Подразумевается, что настоящее изобретение включает все изотопы атомов, имеющихся в соединениях по настоящему изобретению. Например, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают 13С и 14С.
Настоящее изобретение относится также и к фармацевтически приемлемым солям указанных выше соединений. Под фармацевтически приемлемой солью понимают соль, состоящую из катиона (катионов) и аниона (анионов), в которой соединение является или катионом, или анионом. Кроме того, фармацевтически приемлемая соль согласно настоящему изобретению является нетоксичной для животных и/или человека.
Подходящей фармацевтически приемлемой солью соединения настоящего изобретения является, например, соль щелочного или щелочноземельного металла, например, соль натрия, калия, кальция, магния, а также соль аммония, соль с органическим основанием, например, с метиламином, диметиламином, триметиламином, пиперидином, морфолином и т.д.
Подходящей фармацевтически приемлемой солью соединения настоящего изобретения также может быть соль, полученная присоединением кислоты, например, соляной, серной, малеиновой или трифторуксусной кислоты. Могут быть выбраны и другие достаточно сильные органические и неорганические кислоты.
Фармацевтическая композиция
Настоящая техническая проблема решается, а указанные технические результаты достигаются также благодаря фармацевтической композиции, обладающей гепатопротекторной активностью, включающей соединение настоящего изобретения и по меньшей мере один фармацевтически приемлемый эксципиент.
Под фармацевтической композицией понимается пригодная для использования для людей или животных композиция (смесь, состав и т.д.), включающая активную фармацевтическую субстанцию. Активная фармацевтическая субстанция в фармацевтической композиции включает действующее вещество соединение настоящего изобретения. Специалисту в данной области будет понятно, что к фармацевтической композиции настоящего изобретения будут относиться также композиции, содержащие одно или несколько других активных фармацевтических субстанций, например, другие гепатопротекторные или противовоспалительные средства.
Понятие «включает» в контексте настоящего изобретения означает, что указанные фармацевтические композиции (лекарственные средства, группы компонентов и т.д.) включают перечисленные далее компоненты/ингредиенты, но не исключают включение других компонентов/ингредиентов.
Количественное содержание соединения настоящего изобретения в фармацевтической композиции выбирается из диапазона от 0.01 до 99.99 мас. %, в предпочтительном варианте от 1.00 до 80.00 мас. %, в более предпочтительном от 10.00 до 60.00 мас. %, например, 5.00 мас. %, 10.00 мас. %, 15.00 мас. %, 20.00 мас. %, 25.00 мас. %, 30.00 мас. %, 35.00 мас. %, 40.00 мас. %, 45.00 мас. %, 50.00 мас. %, 55.00 мас. %, 60.00 мас. %.
В другом предпочтительном варианте фармацевтическая композиция включает соединение настоящего изобретения в эффективном количестве.
Понятие «эффективное количество» в контексте настоящего изобретения относится к количеству фармацевтической композиции или лекарственного средства, которое при введении субъекту является достаточным для воздействия такого лечения на заболевание, нарушение или симптом. «Эффективное количество» может изменяться, например, в зависимости от того, в какой форме находится вещество, от природы заболевания, нарушения и/или симптомов заболевания или нарушения, от тяжести заболевания, нарушения и/или симптомов заболевания или нарушения, от возраста субъекта, подлежащего лечению, и/или от веса субъекта, подлежащего лечению. Надлежащее количество в каждом конкретном случае будет очевидно специалисту в данной области или может быть определено путем стандартных экспериментов.
Фармацевтическая композиция настоящего изобретения включает по меньшей мере один фармацевтически приемлемый эксципиент, являющийся носителем действующих веществ, обеспечивающий требуемый объем/массу и необходимые характеристики лекарственного средства в определенной лекарственной форме. В предпочтительном варианте фармацевтическая композиция включает фармацевтически приемлемый эксципиент, который выбирают из группы, включающей наполнитель, связывающее вещество, смазывающее вещество, разрыхляющее вещество, скользящее вещество, консервант, корригент, краситель.
Наполнители (носители, разбавители) добавляются для получения определенной массы лекарственной формы. Примерами наполнителей являются крахмал, сахара, оксид магния, целлюлоза, карбонат кальция, декстрин, амилопектин, сорбит, маннит, пектин.
Связывающие вещества добавляются для заполнения межчастичного пространства и для увеличения контактной поверхности частиц, что необходимо для таблетирования твердых лекарственных форм. Примерами связывающих веществ являются альгинат натрия, сахар, желатин, крахмал, поливиниловый спирт, производные целлюлозы, поливинилпирролидон(повидон).
Скользящие вещества добавляются для уменьшения шероховатости твердой лекарственной формы, что облегчает ее высыпание. Примерами скользящих веществ являются крахмал, тальк, полиэтиленоксид-4000, аэросил.
Смазывающие вещества облегчаются выталкивание твердой формы (например, таблетки из матрицы). Примерами смазывающих веществ являются стеариновая кислота и ее соли (стеарат магния), жиры.
Разрыхляющие вещества (дезинтегрант, диспергирующий агент) облегчают растворение фармацевтической композиции и лекарственной формы. Примерами разрыхляющих веществ являются гидрокарбонат натрия, твин-80, альгинат натрия.
Корригенты используются для улучшения вкуса (подсластитель) и запаха (ароматизатор). К ним относятся сахар, какао, ванилин.
Красители (пигменты) используются для улучшения внешнего вида фармацевтической композиции и лекарственной формы. Примерами красителей являются диоксид титана, индигокармин.
Количество, состав и форма фармацевтически приемлемого эксципиента могут быть выбраны специалистом в данной области произвольно при условии полного или частичного сохранения активности соединения настоящего изобретения.
Фармацевтическое применение
Соединения настоящего изобретения, их стереоизомеры и их фармацевтически приемлемые соли являются гепатопротекторами и пригодны для лечения или профилактики заболеваний печени. К заболеваниям печени относятся неалкогольная и алкогольная жировая болезнь печени (все стадии, включая стеатоз); стеатогепатит вирусной и невирусной этиологии; фиброз и цирроз печени; токсические поражения печени; состояния, сопровождающиеся внутри- и внепеченочным холестазом; наследственные и приобретенные состояния, сопровождающиеся гипербилирубинемией. Примерами заболеваний печени также являются ассоциированный холангит, муковисцидоз, прогрессирующий семейный внутрипеченочный холестаз, доброкачественный рецидивирующий внутрипеченочный холестаз, гемолитическая болезнь новорожденных, наследственный микросфероцитоз, дефицит глюкозо-6-фосфатдегидрогеназы, синдром Криглера-Найяра, синдром Люцея-Дрисколла, галактоземия, первичный склерозирующий холангит, первичный билиарный цирроз, физиологическая желтуха новорожденных, синдром Жильбера, синдром Кароли, синдром Алажилля.
Осуществление изобретения
Для иллюстративных целей далее представлены примеры, которые не ограничивают объем настоящего изобретения.
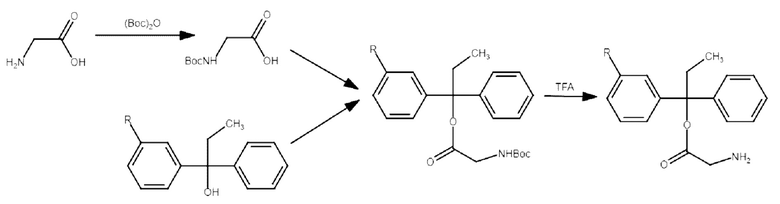
Пример 1. Получение соединений А1-А8
Соединения А1-А5 могут быть синтезированы по реакции получения сложного эфира (э тарификации) из соответствующих аминокислоты (в качестве карбоновой кислоты) и соединения S4 (в качестве спирта) согласно следующей схеме:
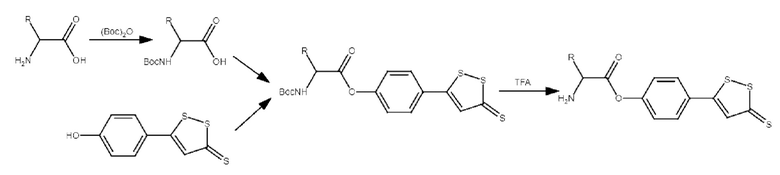
Синтез включает предварительную установку защитной группы (Вое) на аминогруппу аминокислоты; реакцию получения сложного эфира (этерификации) защищенной аминокислоты и соединения S4; удаление защитной группы. Каждая из указанных стадий известна из научно-технической литературы и может быть осуществлена специалистом в данной области. Далее представлено подробное экспериментальное описание примера получения соединения настоящего изобретения, где в качестве аминокислоты использовали глицин.
Стадия 1.
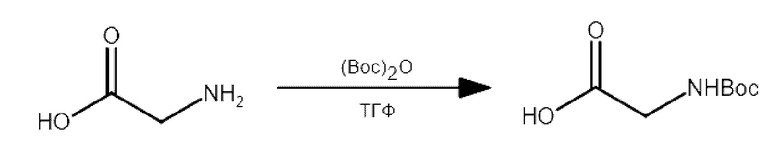
К раствору 10.0 г глицина (0.13 моль) в 100 мл тетрагидрофурана добавляли при перемешивании 28.5 г ди-трет-бутилдикарбоната (0.13 моль), затем 250 мл насыщенного раствора гидрокарбоната натрия. Полученную реакционную смесь перемешивали при 50°С в течение 12 ч. После завершения реакции смесь обрабатывали 1М HCl до рН 7. Органический слой отделяли, водный экстрагировали этилацетатом. Органические вытяжки объединяли и сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Полученный продукт хроматографировали (флеш-хроматография), в качестве элюента использовали смесь CH2Cl2:МеОН (20:1). Получено 19.2 г (82%) Вос-глицина в виде бесцветного порошка.
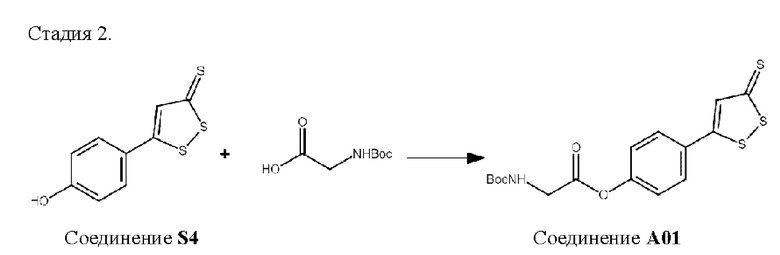
К смеси 2.3 г Вос-глицина (13.2 ммоль), 1.0 г соединения S4 (4.4 ммоль), 1.5 г 1,3-дициклокарбодиимида и 4.0 г HBTU в 50 мл тетрагидрофурана добавляли при перемешивании 2 мл триэтиламина. Полученную реакционную смесь перемешивали при комнатной температуре в течение 12 ч, после чего выливали в 100 мл воды. Органическую фазу отделяли, водный слой экстрагировали этилацетатом. Органические фракции объединяли и последовательно промывали сначала 1М HCl, далее насыщенным раствором гидрокарбоната натрия, затем насыщенным раствором хлорида натрия, после чего сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Продукт выделяли из полученной смеси методом колоночной хроматографии (силикагель, элюент - петролейный эфир: этилацетат 4: 1). Получено 1.2 г (71%) соединения А01 в виде белого порошка.
Данные ЯМР 1Н (DMSO-D6, 400.13 МГц): 1.46 (с, 9Н), 4.12 (с, 2Н), 7.23 (с, 1H), 7.45 (д, 2Н, J 8.7), 7.61 (д, 2Н, J 8.7);
Данные ESI-MS [М+Н]+: 384.04
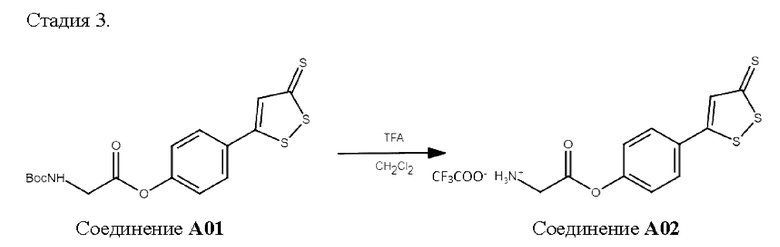
К раствору 500 мг соединения А01 в 20 мл хлористого метилена добавляли 3 мл CF3COOH. Полученную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции растворители отгоняли при пониженном давлении (20 мм.рт.ст), остаток суспендировали эфиром, взвесь центрифугировали, раствор декантировали, осадок сушили на воздухе. Получено 483 мг (93%) трифторацетата А02 в виде светло-желтого порошка.
Данные элементного анализа:
Вычислено для C13H10F3NO4S3, %: С 39.29; Н 2.54; N 3.52; S 24.21;
Найдено, %: С 39.18; Н 2.56; N 3.40; S 24.34;
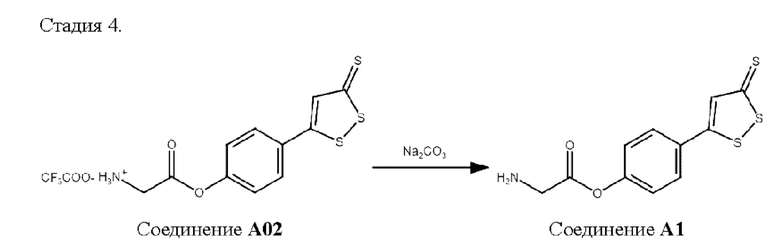
К раствору 483 мг полученного на стадии 3 соединения А02 в 10 мл воды добавляли 2 г Na2CO3. Полученную смесь перемешивали при температуре 50°С в течение 1 ч. Свободный амин экстрагировали хлористым метиленом. После удаления растворителей и высушивания продукта было получено 330 мг (95%) целевого амина (Соединение А1) в виде бесцветного порошка.
Данные ЯМР 1H (DMSO-D6, 400,13 МГц): 3.84 (с, 2Н), 7.21 (д, 2Н, J 8.7), 7.26 (с, 1Н),7.67 (д, 2Н, J 8.7);
Данные ESI-MS [М+Н]+: 283.99.
Аналогичным образом получены конъюгаты с другими аминокислотами - L-цистеином, L-гомоцистеином и L-метионином. В случае аминокислот, содержащих SH и NH-группы, использовали соответствующие защитные группы (см. WO 2017202266 A1). Для полученных соединений были зарегистрированы спектры ЯМР 1Н и/или MS.
Спектры ЯМР регистрировали на ядрах 1H с рабочей частотой 400.13 МГц и внутренним стандартом ТМС. В качестве растворителя использовали DMSO-D6. Масс-спектры регистрировали на масс-спектрометре, оснащенном источником электрораспыления с напряжением 5.5 кВ при температуре капилляра 300°С. Для ввода использовали растворы с концентрацией 0.1-10 мкг/л в ацетонитриле. Результаты измерений представлены в таблице 1 (здесь и далее сигналы нехарактеристичных подвижных протонов амино-групп и карбоксильных групп в виде уширенных синглетов и мультиплетов при описании спектров ЯМР не перечислены).
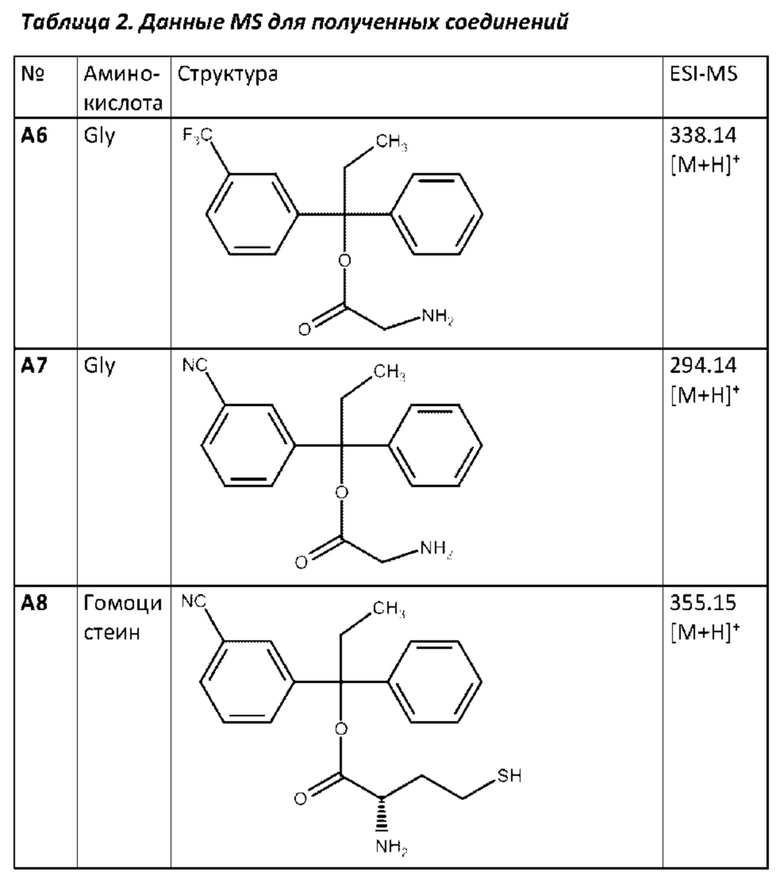
Аналогичным образом могут быть получены соединения А6-А8 (таблица 2) согласно следующей схеме:
Пример 2. Получение соединения В1
Соединение В1 получали согласно следующей схеме:
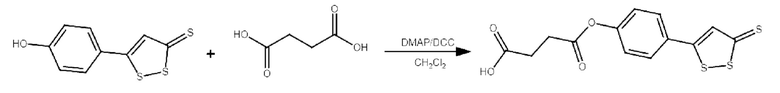
К раствору 2.26 г (10.0 ммоль) соединения S4, 0.18 г (1.5 ммоль) 4-диметиламинопиридина (DMAP) и 2.20 г (10.7 ммоль) 1,3-дициклогексилкарбодиимида (DCC) в 50 мл хлористого метилена медленно при перемешивании добавляли 1.30 г (11.0 ммоль) янтарной кислоты. Полученную реакционную смесь перемешивали при комнатной температуре в течение 12 ч, после чего выливали в 100 мл воды. Органическую фазу отделяли, водный слой экстрагировали хлористым метиленом. Органические фракции объединяли и последовательно промывали сначала 1М HCl, далее насыщенным раствором гидрокарбоната натрия, затем насыщенным раствором хлорида натрия, после чего сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Полученный продукт хроматографировали (флеш-хроматография), в качестве элюента использовали петролейный эфир. Получено 2.48 г (76%) соединения В1 в виде бесцветного порошка.
Данные ЯМР 1Н (DMSO-D6, 400.13 МГц): 2.60 (т, 2Н, J 7.4), 2.84 (т, 2Н, J 7.4), 7.16 (д, 2Н, J 8.7), 7.23 (с, 1H), 7.68 (д, 2Н, J 8.7);
Данные ESI-MS [М-Н]-: 324.97.
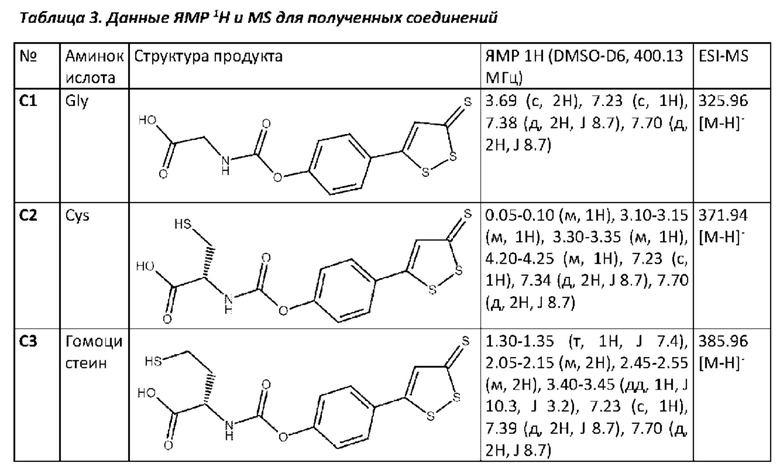
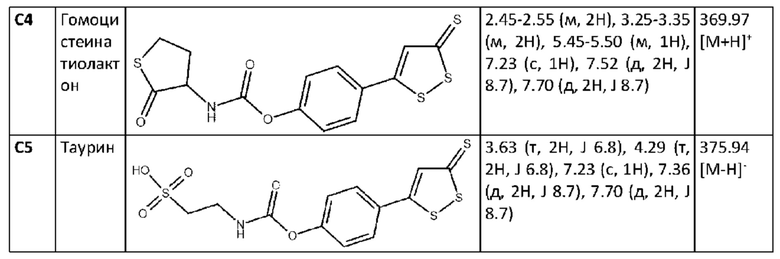
Пример 3. Получение соединений С1-С8
Соединения С1-С5 могут быть синтезированы по реакции получения карбаматов из соответствующих карбонатов и аминокислот (в качестве аминов) согласно следующей схеме:
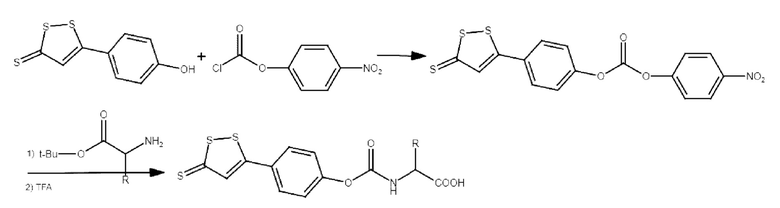
Далее представлено подробное экспериментальное описание примера получения соединения настоящего изобретения, где в качестве аминокислоты использовали глицин. Стадия 1.
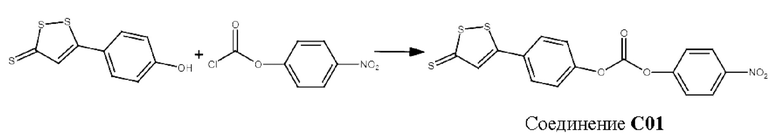
К суспензии 2.26 г (10.0 ммоль) соединения S4 в 10.0 мл CH2Cl2 добавляли при перемешивании последовательно сначала 1.34 г (11.0 ммоль) 4-диметиламинопиридин (DMAP), затем 2.21 г (11.0 ммоль) 4-нитрофенилхлорформиат.Реакционную смесь перемешивали в течение 10 ч при комнатной температуре. После этого растворитель отгоняли при пониженном давлении. Полученный остаток обрабатывали диэтиловым эфиром, выпавший осадок отфильтровывали и использовали на следующей стадии без дополнительной очистки. Получено 2.97 г (76%) соединения С01 в виде желтоватого порошка.
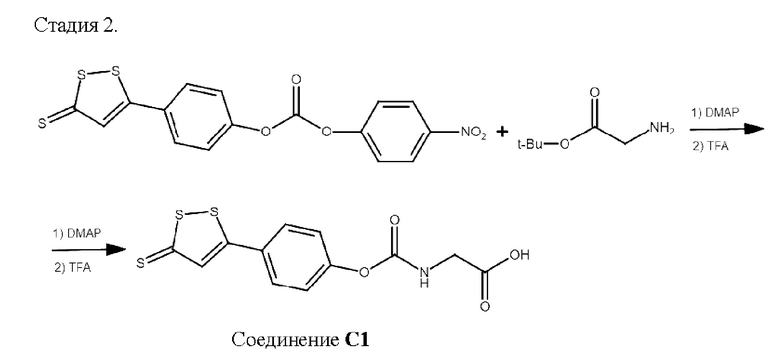
К раствору 1.95 г (5.0 ммоль) соединения С01 в 10 мл CH2Cl2 добавляли последовательно при перемешивании сначала 0.67 г (5.5 ммоль) 4-диметиламинопиридина, затем 0.72 г (5.5 ммоль) трет-бутилового эфира 2-аминоацетата (трет-бутиловый эфир глицина, получали согласно Organic Syntheses, Coll. Vol. 5, 1973, p. 586). Полученный раствор перемешивали в течение 20 ч при комнатной температуре. Затем полученную реакционную смесь промывали последовательно сначала насыщенным раствором гидрокарбоната натрия, затем насыщенным раствором хлорида натрия. После этого органический слой отделяли и сушили над безводным сульфатом натрия. Затем растворители отгоняли при пониженном давлении. Полученный промежуточный продукт без дополнительной очистки обрабатывали избытком 40%-ного раствора трифторуксусной кислоты в хлористом метилене. Процесс удаления трет-бутильной группы контролировали методом ТСХ. Через 1 ч растворители и избыток трифторуксусной кислоты удаляли при пониженном давлении (20 мм.рт.ст). Полученное соединение С1 обрабатывали 1М NaOH для окончательной нейтрализации остатков трифторуксусной кислоты. После этого соединение С1 экстрагировали хлористым метиленом. Экстракт промывали последовательно сначала насыщенным раствором NaCl, затем водой до нейтральной среды промывных вод. Растворитель снова отгоняли при пониженном давлении (20 мм.рт.ст). Остаток, представляющий собой чистое соединение С1, сушили на воздухе. Получено 1.15 г (70%) соединения С1 (таблица 3).
Данные ЯМР 1H (DMSO-D6, 400.13 МГц): 3.69 (с, 2Н), 7.23 (с, 1Н), 7.38 (д, 2Н, J 8.7), 7.70 (д, 2Н, J 8.7)
Данные ESI-MS [М-Н]-:325.96.
Аналогичным образом могут быть получены конъюгаты с другими аминокарбоновыми кислотами и аминосульфо кислотами. В качестве аминокислот использовали L-цистеин, L-гомоцистеин и (±)-гомоцистеина тиолактон. В качестве аминосульфокислот использовали таурин. В случае аминокислот, содержащих SH и NH-группы, использовали соответствующие защитные группы (см. WO 2017202266 A1). Для полученных соединений были зарегистрированы спектры ЯМР 1Н и/или MS. Результаты представлены в таблице 3.
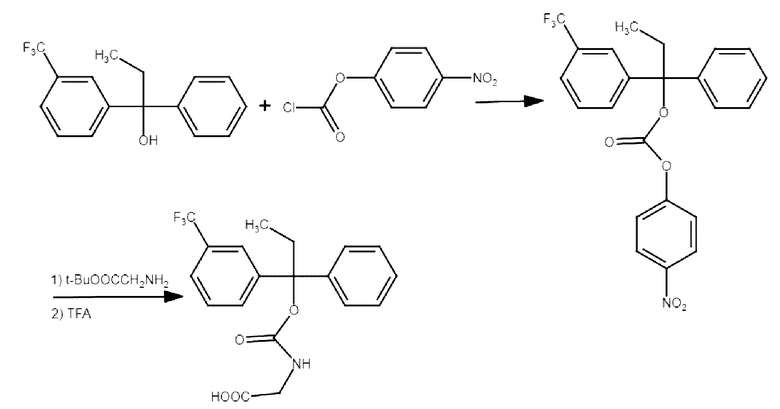
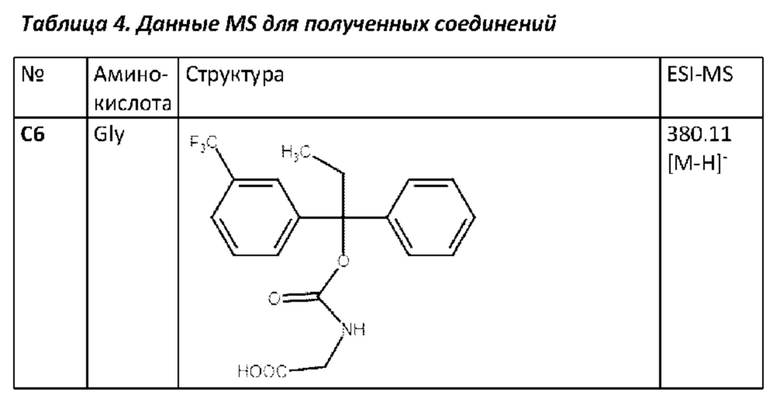
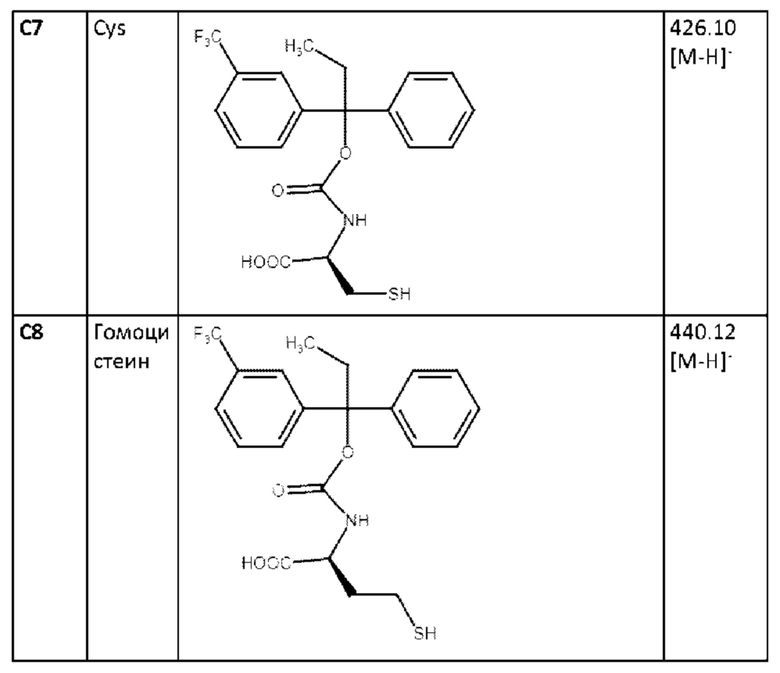
Аналогичным образом могут быть получены соединения С6-С8 (таблица 4) согласно следующей схеме:
Пример 4. Получение соединений D1-DS
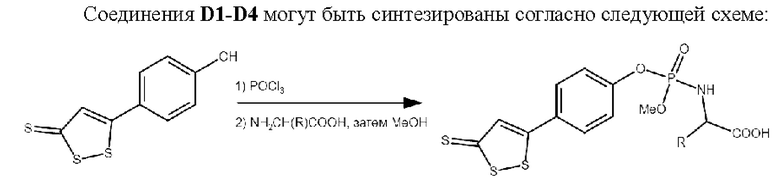
Соединения D1-D4 могут быть синтезированы согласно следующей схеме:
Далее представлено подробное экспериментальное описание примера получения соединения настоящего изобретения, где в качестве аминокислоты использовали глицин.
К охлажденному до -10°С раствору 1.00 г (4.4 ммоль) соединения S4 и 2 мл пиридина в 10.0 мл хлористого метилена медленно, по каплям, добавляли 2.70 г (17.6 ммоль) оксихлорида фосфора (POCl3). Через 30 мин к реакционной смеси добавляли 0.33 г (4.4 ммоль) глицина в 5.0 мл хлористого метилена. Смесь перемешивали еще в течение 1 ч при комнатной температуре и после завершения реакции выливали в 100 мл метанола. После этого растворители отгоняли при пониженном давлении (20 мм.рт.мт.), остаток суспендировали эфиром, взвесь центрифугировали, раствор декантировали, осадок сушили на воздухе. Получено 1.15 г (70%) соединения D1 в виде желтоватого порошка (таблица 5).
Данные ЯМР 1H (DMSO-D6, 400.13 МГц): 3.52 (д, 3Н, J 11.0), 3.95 (дд, 1H, J 5.9, J 17.4), 4.05 (дд, 1H, J 5.9, J 17.4), 7.23 (с, 1H), 7.29 (д, 2Н, J 8.7), 7.66 (д, 2Н, J 8.7);
Данные ESI-MS [М-Н]- 375.95
Аналогичным образом получены конъюгаты с другими аминокислотами. В качестве аминокислот использовали L-аланин и L-цистеин. В случае аминокислот, содержащих SH и NH-группы, использовали соответствующие защитные группы (см. WO 2017202266 А1). Для полученных соединений были зарегистрированы спектры ЯМР 1Н и/или MS. Результаты представлены в таблице 5.
Аналогичным образом могут быть получены соединения D5-D8 (таблица 6) согласно следующей схеме:
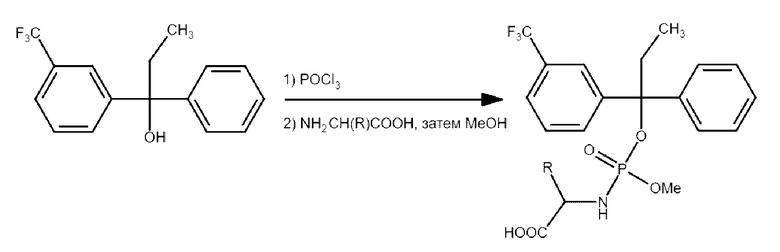
Пример 5. Получение соединений Е1-Е12
Соединения Е1-Е5 могут быть синтезированы согласно следующей схеме:
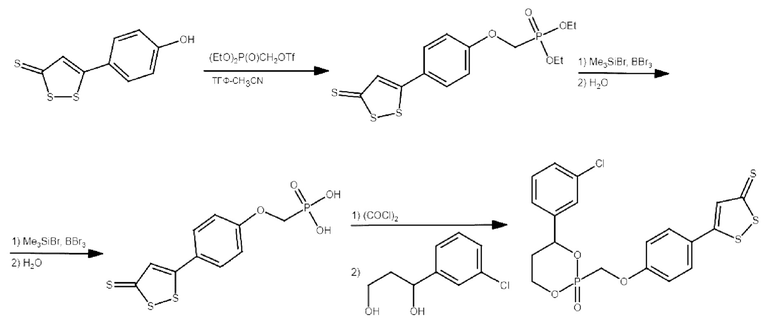
Далее представлено подробное экспериментальное описание примера получения соединения настоящего изобретения.
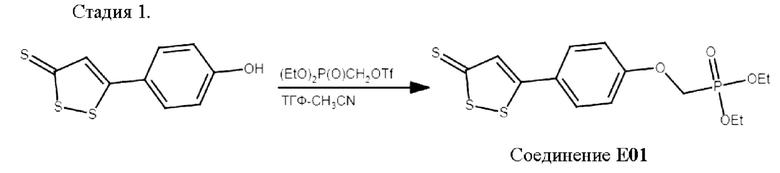
К раствору 1.00 г (4.4 ммоль) соединения S4, 2.00 г (6.6 ммоль) трифлата (EtO)2P(0)CH2OTf (получен по известной методике D.P.PHJLLION and S.S.ANDREW, Synthesis and reactivity of diethyl phosphonomethyltriflate, TETRAHEDRON LETTERS, 1986, V.27, N.13, pp.1477-1480, doi: 10.1016/S0040-4039(00)84289-6) в 50 мл смеси ТГФ-CH3CN (1:1) добавляли 2.15 г (6.6 ммоль) карбоната цезия CS2CO3. Полученную смесь перемешивали в течение 1 ч при комнатной температуре. После завершения реакции смесь выливали в 100 мл воды. Органическую фазу отделяли, водный слой экстрагировали этилацетатом. Органические фракции объединяли, после чего сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Полученный продукт хроматографировали (флеш-хроматография), в качестве элюента использовали этилацетат: петролейный эфир 1:10. Получено 1.18 г (71%) соединения Е01 в виде желтоватого масла.
Стадия 2.
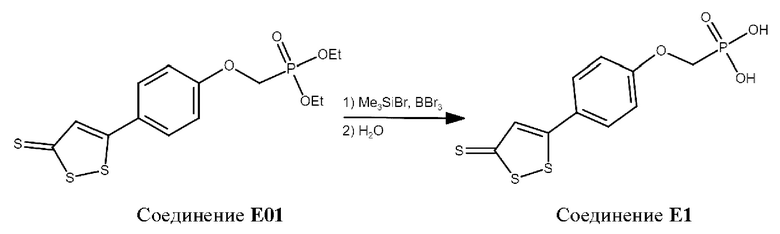
К раствору 1.00 г (2.9 ммоль) соединения Е01 в 10.0 мл CH2Cl2 добавляли при -78°С 3.80 мл (29 ммоль) бромтриметилсилана. Полученную реакционную смесь отогревали и перемешивали при комнатной температуре в течение 16 ч. После этого растворитель отгоняли при пониженном давлении (20 мм.рт.ст), а к полученному остатку добавляли 1.37 мл (14.5 ммоль) трибромида бора. Полученную реакционную смесь вновь выдерживали при комнатной температуре в течение 16 ч, после чего выливали в 25 мл воды. Органическую фазу отделяли, водный слой экстрагировали хлористым метиленом. Органические фракции объединяли, после чего сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Полученный продукт хроматографировали (флеш-хроматография), в качестве элюента использовали этилацетат: петролейный эфир 1:10. Получено 0.81 г (87%) соединения Е1 в виде желтоватого масла.
Данные ЯМР 1Н (DMSO-D6, 400.13 МГц): 4.40 (д, 2Н, J 13.2), 7.12 (д, 2Н, J 8.7), 7.23 (с, Ш, J 8.7), 7.46 (д, 2Н, J 8.7);
Данные ESI-MS [М-Н]-: 318.93;
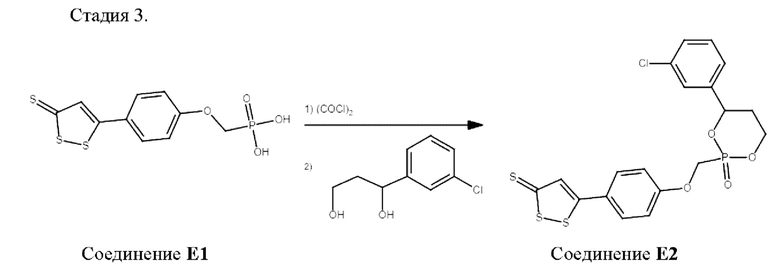
К раствору 0.81 г (2.5 ммоль) соединения Е1 и 1.0 г ДМФА в 50 мл хлористого метилена добавляли 1.0 мл (12 ммоль) оксалил хлорида (COCl)2. Полученный раствор кипятили с обратным холодильником в течение 3 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученное масло вновь растворяли в 20 мл хлористого метилена, после чего к нему добавляли при -78°С 1.0 мл (12 ммоль) пиридина и 2.24 г (12 ммоль) 1-арил-1,3-пропандиола (в виде смеси стереошомеров). Смесь отогревали до комнатной температуры и перемешивали в течение 1 ч, после чего выливали в 20 мл воды. Органическую фазу отделяли, водный слой экстрагировали хлористым метиленом. Органические фракции объединяли, после чего сушили над безводным сульфатом натрия. Осушитель отфильтровывали, растворители отгоняли при пониженном давлении (20 мм.рт.ст). Полученный продукт хроматографировали (флеш-хроматография), в качестве элюента использовали этилацетат: петролейный эфир 1:5. Получено 0.87 г (74%) пролекарства Е2 (в виде смеси стереошомеров) в виде желтоватого порошка.
Данные ESI-MS [М+Н]+: 470.97.
Аналогичным образом могут быть получены соединения Е3-Е5 (таблица 7). Для получения соединения ЕЗ использовали 1-арил-1,3-пропандиол с Вос-загдищенной аминогруппой. Соединение Е6 может быть получено путем добавления глицина (вместо 1,3-пропандиола) на стадии 3, как это описано в примере 4.
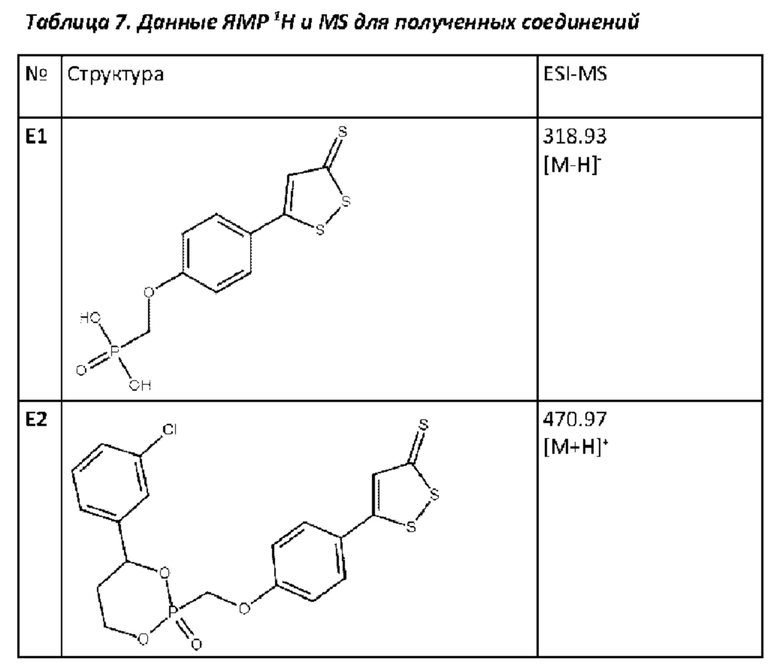
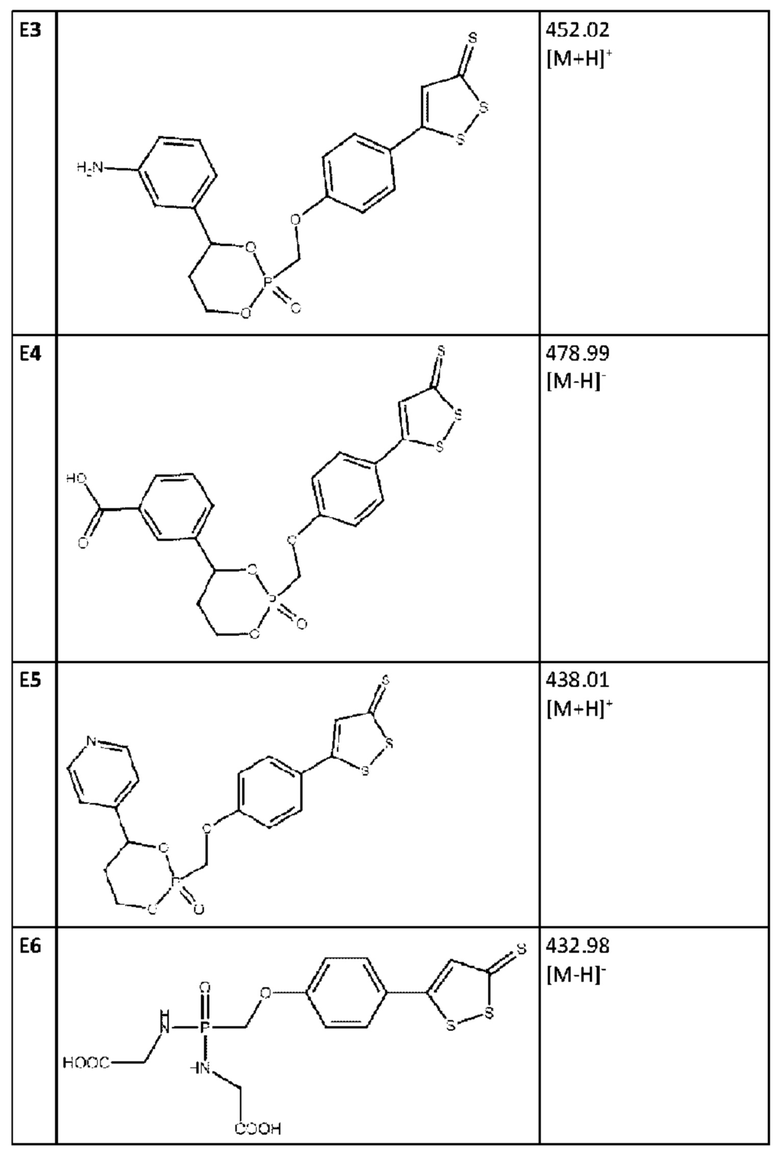
Соединения Е7-Е11 (таблица 8) могут быть получены согласно следующей схеме:
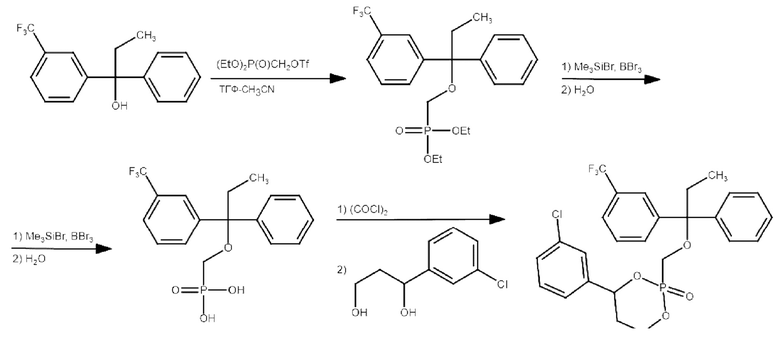
Для получения соединения Е9 использовали 1-арил-1,3-пропандиол с Вос-защищенной амино-группой. Соединение Е12 может быть получено путем добавления глицина (вместо 1,3-пропандиола) на стадии 3, как это описано в примере 4.
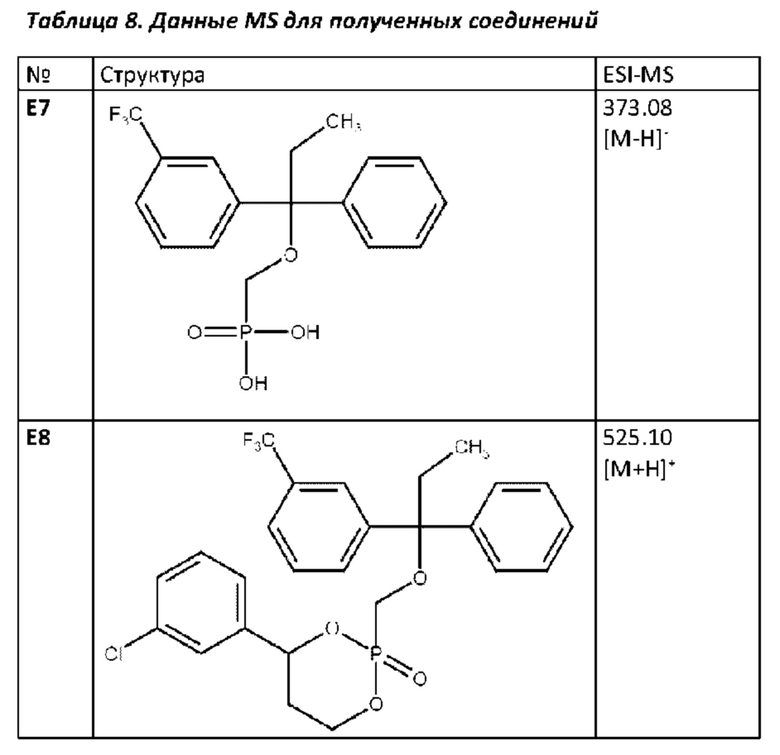
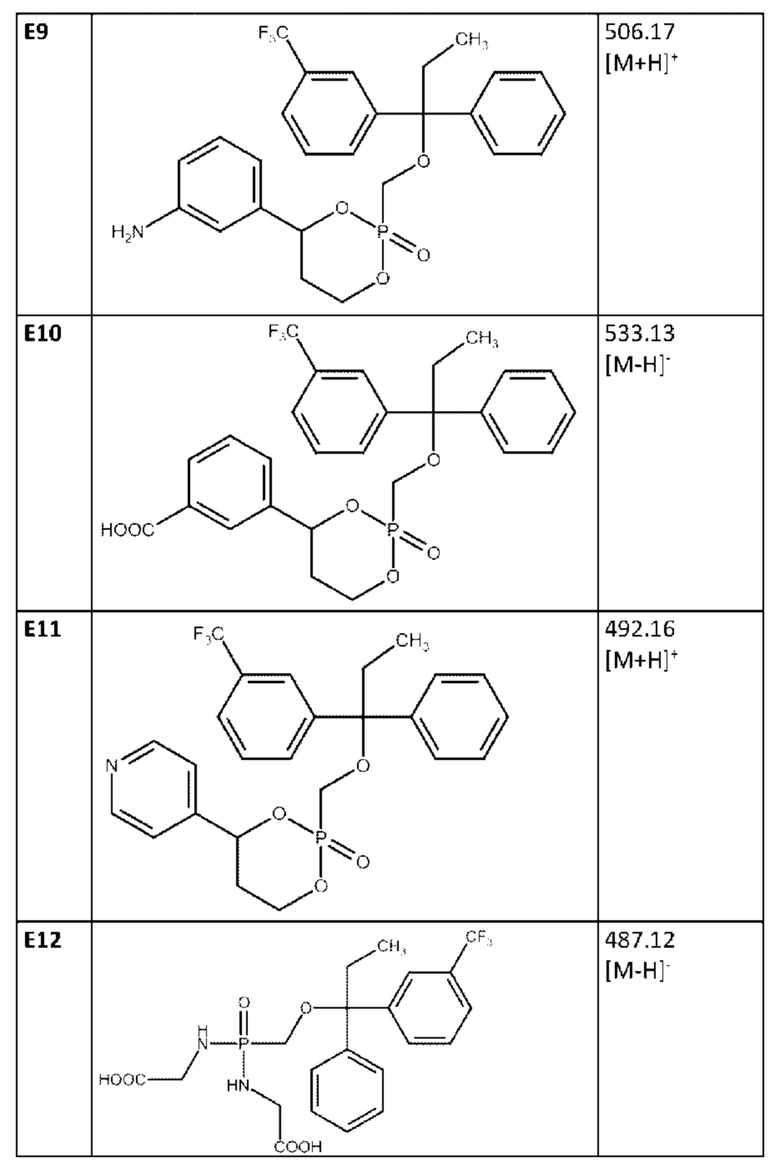
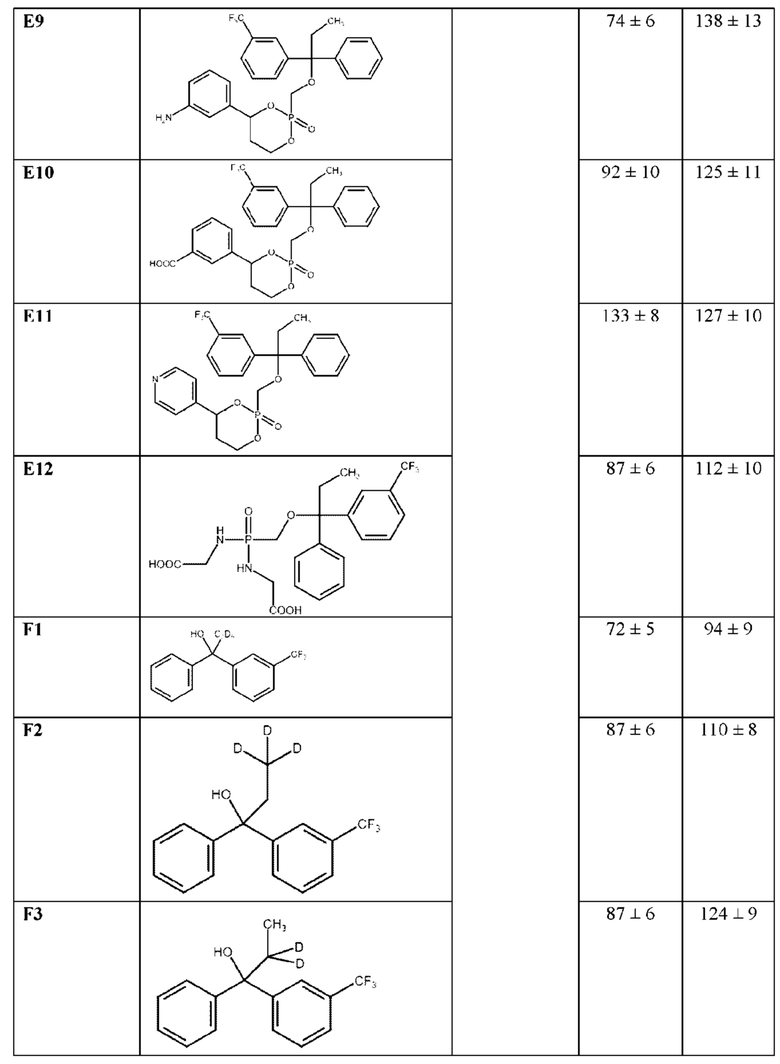
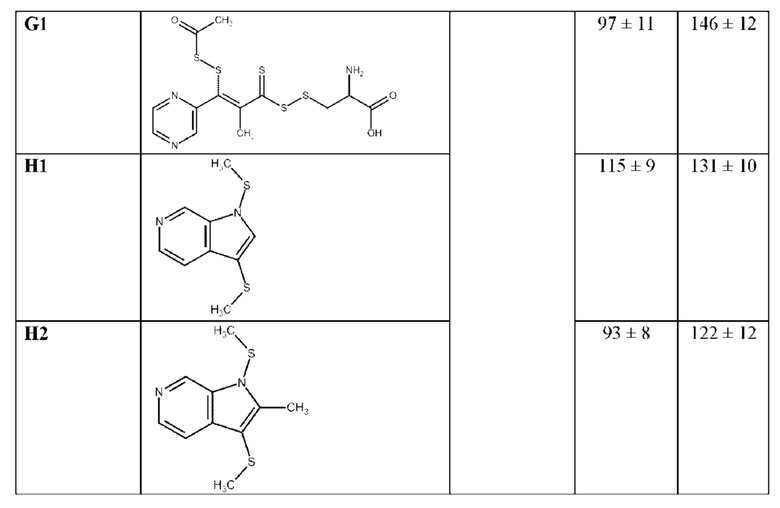
Пример 6. Получение соединений F1-F3
Соединение F1 может быть получено по реакции бензофенона с дейтероэтилмагнийбромидом (реакция Гриньяра) согласно следующей схеме: 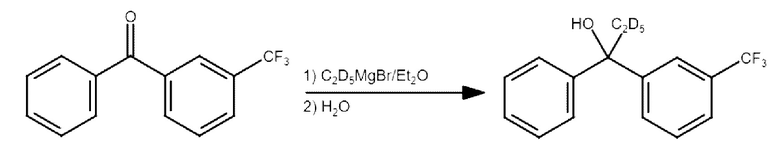
Далее представлено подробное экспериментальное описание примера получения соединения настоящего изобретения:
Реактив Гриньяра, дейтероэтилмагнийбромид, получали согласно известной методике (Н.С. ЗЕФИРОВ, Практикум по органической химии, 2012, М.Бином; с. 239-240). К раствору реактива Гриньяра, приготовленного из 22.8 г дейтероэтилбромида (0.2 моль) и 4.8 г (0.2 моль) магния в 100 мл абсолютного эфира, прибавляли по каплям раствор 37.5 г (0.15 моль) бензофенона в 50 мл абсолютного эфира. Смесь нагревали на водяной бане при перемешивании в течение 5 ч. После завершения реакции к смеси медленно по каплям добавляли сначала 30 мл ледяной воды, затем 20 мл концентрированной соляной кислоты, затем 20 мл воды. Полученную смесь перемешивали до полного растворения осадка соединений магния. Эфирный слой отделяли, водный экстрагировали эфиром. Органические вытяжки объединяли, после чего промывали 5% раствором NaHCO3. Эфирный раствор сушили над безводным сульфатом натрия. Органические растворители отгоняли при пониженном давлении (20 мм.рт.ст.), остаток перегоняли под вакуумом. Полученный остаток перекристаллизовывали из этанола. Получено 38.7 г (68%) соединения F1 в виде бесцветного порошка.
Данные ESI-MS [М+Н]+: 286.15
Аналогичным образом могут быть получены соединения:
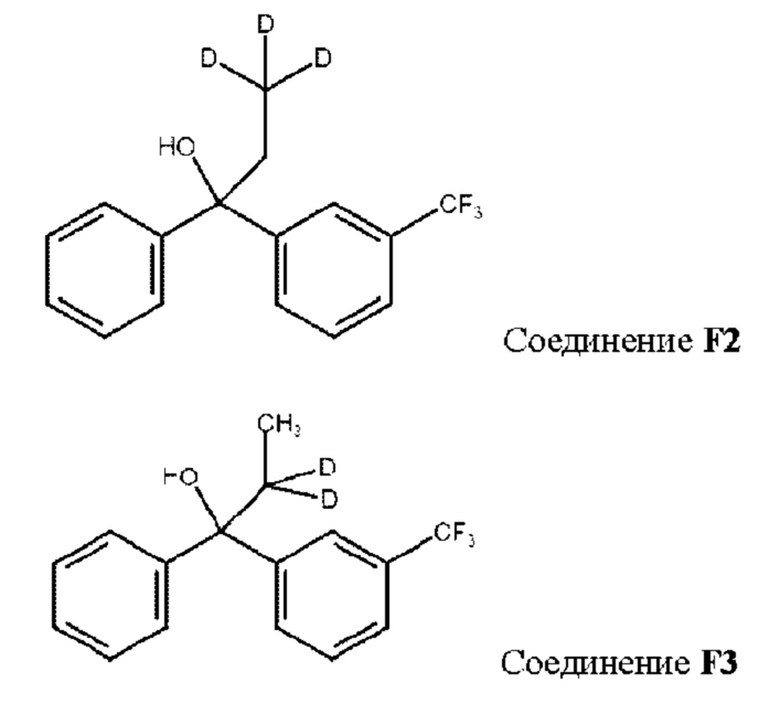
Пример 7. Получение соединения G1
Соединение G1 может быть получено согласно следующей схеме:
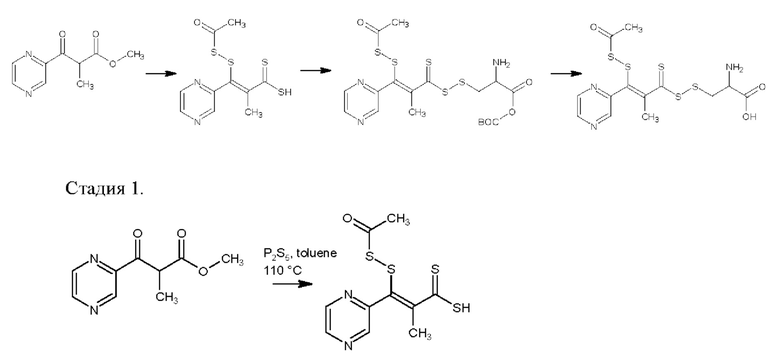
Эфир 2-метил-3-(пиразин-2-ил)-3-оксопропионата получали, руководствуясь WO 2001009118 A2 (пример 7).
К 50 мл толуола в атмосфере азота добавляли 16.9 г сульфида фосфора P2S5 при перемешивании при температуре 20°С. Далее добавляли метиловый эфир 2-метил-3-(пиразин-2-ил)-3-оксопропионата (7.67 г, 39.5 ммоль) в 20 мл толуола и перемешивали смесь 30 мин. Далее постепенно добавляли тиоацетат калия (4.6 г, 40.0 ммоль) и при перемешивании медленно нагревали до 110°С, в таком режиме оставляли реакционную смесь на 18 ч. После охлаждения реакционной смеси к ней добавляли приблизительно 15 мл гидроксида аммония до рН 7-7.5 Органическую фазу отделяли, концентрировали в вакууме.
Продукт хроматографировали (силикагель, элюент - петролейный эфир: этилацетат 4: 1) и далее без дополнительной очистки использовали на следующей стадии. Получено 4.90 г (41%) продукта стадии 1 в виде белого порошка.
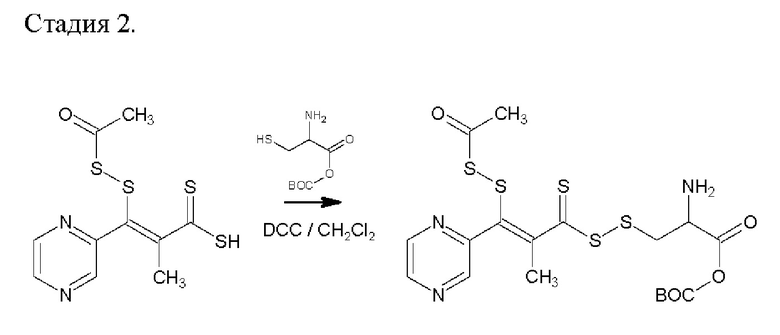
Продукт стадии 1 (3.03 г, 10 ммоль) растворяли в безводном дихлорметане (25 мл), добавляли цистеин-ВОС (2.21 г, 10 ммоль) и 1,3-дициклогексилкарбодиимид (0.21 г, 1.03 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Продукт отфильтровывали и сушили при пониженном давлении, после чего использовали на следующей стадии без дополнительной очистки. Получено 3.97 г (76%) Вос-продукта в виде белого порошка.
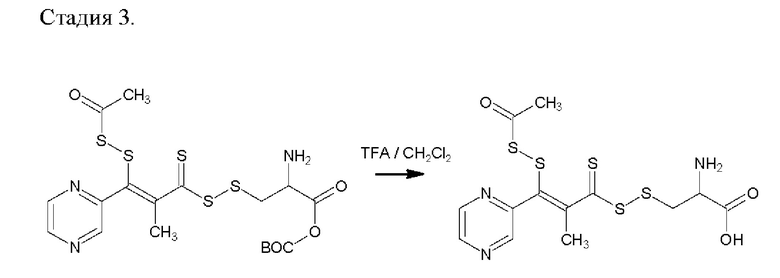
Продукт стадии 2 (5.2 г, 10 ммоль) растворяли в безводном дихлорметане (10 мл) и добавляли трифторуксусную кислоту (2.5 мл), реакцию проводили при комнатной температуре 3 ч. Растворитель отгоняли при пониженном давлении, продукт реакции промывали охлажденным диэтиловым эфиром (5°С) и сушили при пониженном давлении (20 мм.рт.ст.). Получено 3.58 г (85%) целевого продукта в виде белого порошка.
Данные ESI-MS [М+Н]+: 421.97
Пример 8. Получение соединений H1 и Н2
Соединения H1 и Н2 могут быть получены согласно следующей схеме:
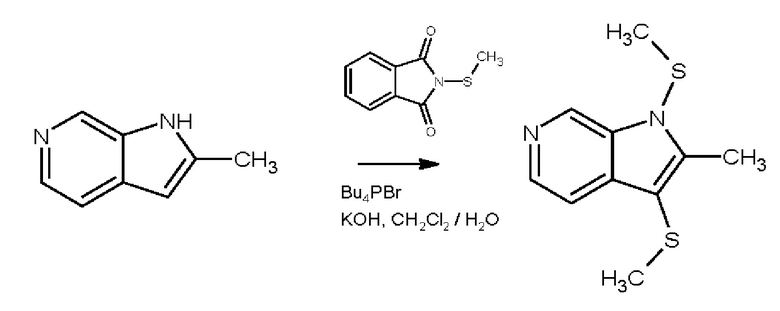
К N-метилтиофталимиду (3.86 г, 20 ммоль), растворенному в 150 мл хлористого метилена, при интенсивном перемешивании при комнатной температуре в течение 10-15 мин добавляли смесь 2-метилпирролопиридина (1.32 г, 10 ммоль), тетрабутилфосфоний бромида (0.34 г, 1 ммоль), 10 мл 50%-ного водного раствора гидроксида калия. Далее к реакционной смеси добавляли дополнительно тетрабутилфосфоний бромид (0.34 г, 1 ммоль). Смесь перемешивали в течение 12 ч при комнатной температуре, после чего выливали в 200 мл воды. Органический слой отделяли, водный - экстрагировали хлористым метиленом (3 × 100 мл). Органические вытяжки объединяли и промывали водой (3 × 100 мл), после чего сушили над безводным сульфатом натрия. Растворители отгоняли, полученный продукт представлял собой соединение HI.
Данные ЯМР 1Н (DMSO-D6, 400.13 МГц): 2.38 (с, 3H), 2.54 (с, 3H), 2.74 (с, 3H), 7.09 (д, Ш, J 6.1), 8.04 (с, 1Н), 8.16(д, 1Н, J 6.1);
ESI-MS [М+Н]+: 225.05.
Аналогичным образом может быть получено соединение Н2:
Данные ЯМР 1Н (DMSO-D6, 400.13 МГц): 2.59 (с, 3H), 2.78 (с, 3H), 6.62 (с, 1Н), 7.20 (д, 1H, J 6.1), 8.15 (с, 1H), 8.20 (д, 1H, J 6.1); ESI-MS [М+Н]+: 211.03.
Пример 9. Исследование гепатопротекторной активности на модели неалкогольного стеатогепатита (НАСГ), индуцированного метионин-холин дефицитной (МХД) диетой у мышей.
Метионин-холин дефицитная (МХД) диета наиболее часто используется для воспроизведения экспериментальной модели неалкогольного стеатогепатита (НАСГ). Применение данной диеты вызывает повышение уровня трансаминаз и гистологические изменения печени, характеризующие стеатоз, фокусное воспаление и некроз гепатоцитов (M.E.RTNELLA and R.M.GREEN, The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance, JOURNAL OF HEPATOLOGY, 2004, V. 40, N. 1, pp. 47-51; E.IP et al., Administration of the potent PPARa agonist, Wy- 14,643, reverses nutritional fibrosis and steatohepatitis in mice, HEPATOLOGY, 2004, V. 39, N. 5, pp. 1286-1296; K.YAMAGUCHI et al., Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis, HEPATOLOGY, 2007, V. 45, N. 6, pp. 1366-1374).
В данном исследовании в качестве тест-системы были использованы половозрелые самцы мышей линии С57Ы, так как данная линия имеет выраженную генетическую предрасположенность к развитию НАСГ. Так, установлено, что самцы более чувствительны к нарушениям питания, и у них легче и быстрее развивается экспериментальная патология, чем у самок (М.Н. МАКАРОВА и В.Г. МАКАРОВ, Диет индуцированные модели метаболических нарушений. Сообщение 5: экспериментальная артериальная гипертензия, Лабораторные животные для научных исследований, 2019, N. 1, с. 1-9). Возраст животных к началу эксперимента составлял 12 недель.
Сообщается, что применение МХД диеты приводит к патологическим структурным и функциональным нарушениям печени. В том числе к статистически значимому увеличению активности аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (ACT) уже через 2 недели содержания на МХД диете (более чем в 2 раза) и спустя 3 недели (более чем в 3 раза) (H.ITAGAKI et al. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice, INTERNATIONAL JOURNAL OF CLINICAL AND EXPERIMENTAL PATHOLOGY, 2013, V.6, N.12, pp.2683-2696).
Количество животных, используемое в исследовании: 1 интактная группа 10 животных и 23 группы по 16 мышей, получающих модифицированную метионин-холин дефицитную диету.
Предварительно был проведен этап постепенной адаптации животных к МХД диете. Длительность полного перевода животных на МХД диету составляла 15 дней. Приучение животных к диете осуществлялось следующим образом: в первый-третий день адаптации животные получат 75% стандартной диеты и 25% МХД, на 4-7-й день 50% МХД и 50% стандартной диеты, на 8-й - 75% МХД и 25% стандартной диеты и т.д. На протяжении всего предварительного этапа животные из интактной группы получали стандартную диету.
После завершения периода адаптации к МХД исследуемые препараты вводили экспериментальным животным ежедневно, внутрижелудочно, поскольку этот способ является аналогом перорального, который планируется использовать в клинической практике. Введение препаратов начали на следующий день после распределения по группам и проводили в течение 28 дней, на 29-ый день определяли уровень аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (ACT) в крови экспериментальных животных.
Для исследуемых препаратов была выбрана доза 10 мг/кг.
Исследуемые препараты вводили животным многократно (при необходимости дробно), ежедневно в виде предварительно приготовленной суспензии в носителе, с помощью специальных зондов и шприцев. В качестве носителя использовали 1% раствор крахмала в воде.
Контрольные животные группы №1 и животные, получающие МХД диеты группы №2, получали носитель тестируемых соединений в объеме, соответствующем объему введения дозы тестируемого препарата.
На 29-ый день исследования проводили биохимический анализ крови и определяли уровень аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (ACT). Для проведения биохимического анализа образцы крови центрифугировали для получения сыворотки.
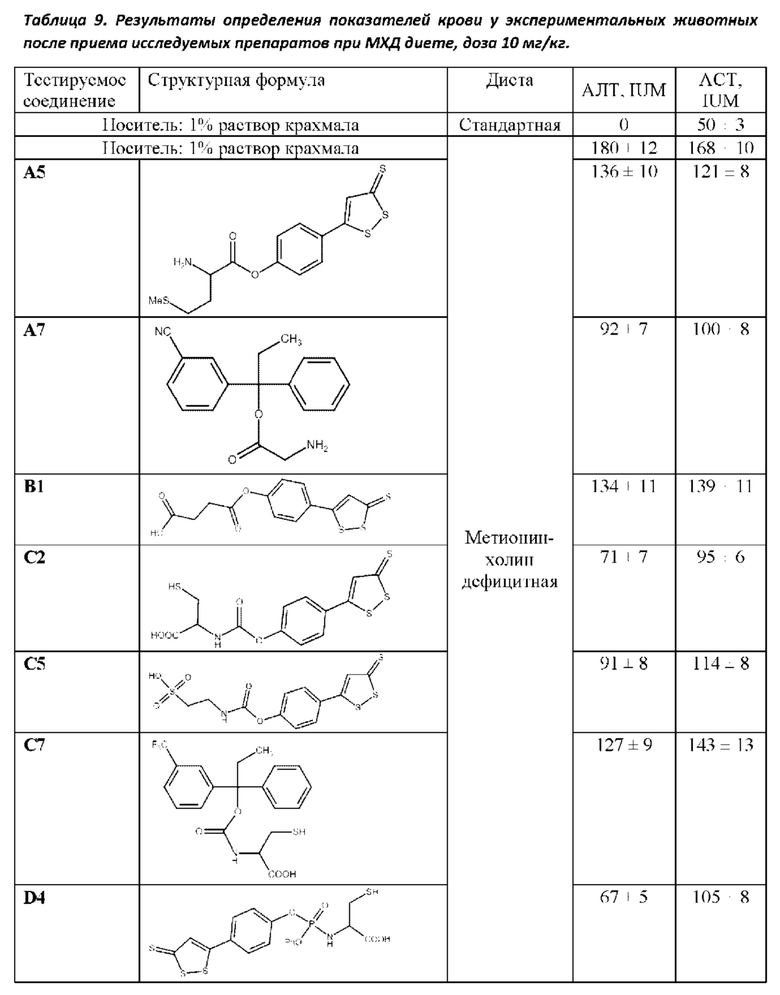
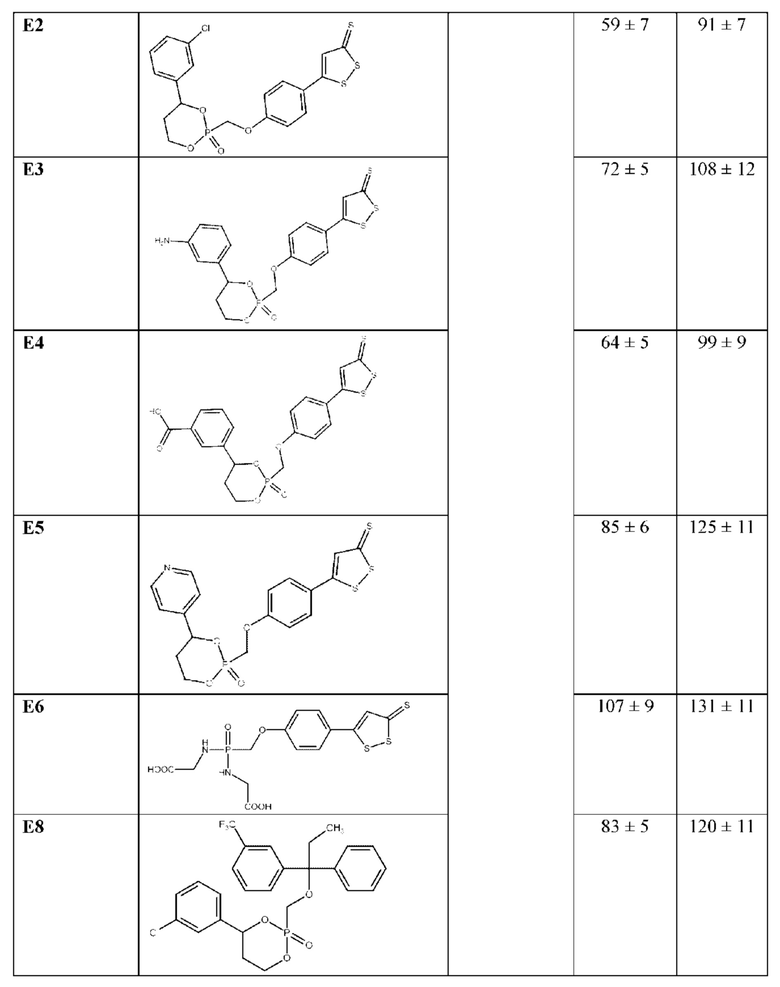
Эти данные были подтверждены в ходе настоящего эксперимента, а также показано, что исследуемые соединения способствуют снижению последствий МХД диеты и обладают гепатопротекторными свойствами (Таблица 9).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ГЕПАТОПРОТЕКТОРНЫЕ СРЕДСТВА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2815370C1 |
| ПРИМЕНЕНИЕ ТРИЦИКЛИЧЕСКОГО СЕРУСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-CoV-2 | 2021 |
|
RU2780247C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ТРИЦИКЛИЧЕСКОЕ СЕРУСОДЕРЖАЩЕЕ ПРОИЗВОДНОЕ 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2, И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2814434C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИЦИКЛИЧЕСКОГО СЕРУСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2 | 2021 |
|
RU2819783C1 |
| СЕРОВОДОРОДНЫЕ ПРОИЗВОДНЫЕ НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2007 |
|
RU2468019C2 |
| ПРОИЗВОДНЫЕ 4- ИЛИ 5-АМИНОСАЛИЦИЛОВОЙ КИСЛОТЫ | 2006 |
|
RU2414476C2 |
| СТРУКТУРНО УСИЛЕННЫЕ ЖИРНЫЕ КИСЛОТЫ, СОДЕРЖАЩИЕ КИСЛОРОД, ДЛЯ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОГО СТЕАТОГЕПАТИТА | 2018 |
|
RU2820995C2 |
| Азопроизводные 5-аминосалициловой кислоты, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2837878C1 |
| Соединение (6-{ [(1S)-1(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)пропил]амино} -9Н-пурин-9-ил)метилацетат как ингибитор p110δ - дельта-изоформы фосфоинозитид-3-киназы (PI3K), способы его получения (варианты) и применения | 2018 |
|
RU2675237C1 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
Изобретение относится к соединению
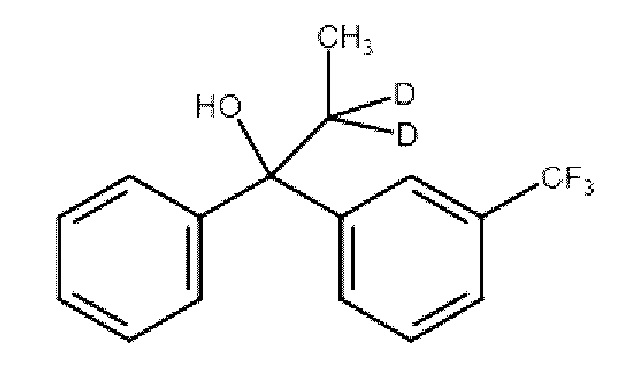
или к его стереоизомеру, или к его фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции, обладающей гепатопротекторной активностью, на основе указанного соединения. Технический результат – получено новое соединение, которое может найти применение в медицине в качестве эффективного гепатопротекторного средства. 3 н.п. ф-лы, 9 табл., 9 пр.
1. Соединение
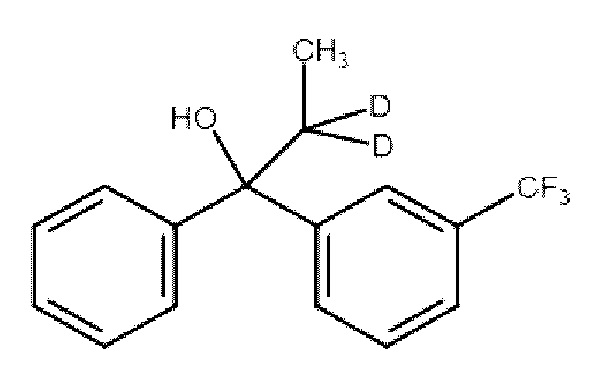
или его стереоизомер, или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция, обладающая гепатопротекторной активностью, включающая соединение по п. 1 в эффективном количестве и по меньшей мере один фармацевтически приемлемый эксципиент.
3. Применение соединения по п. 1 в качестве гепатопротекторного средства.
| Gachalyi, B | |||
| et al | |||
| European Journal of Clinical Pharmacology, 1978, vol | |||
| Насос | 1917 |
|
SU13A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| СЫРОЕШКИН А | |||
| В., и др | |||
| Влияние дейтерия на свойства фармацевтических субстанций (обзор), Разработка и регистрация лекарственных средств | |||
| Способ восстановления спиралей из вольфрамовой проволоки для электрических ламп накаливания, наполненных газом | 1924 |
|
SU2020A1 |
| P.CHEN | |||
| et al | |||
| Design, synthesis, and pharmacological evaluation of the aqueous prodrugs of | |||
