ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, композициям, комбинациям и лекарственным препаратам, содержащим указанные соединения, и способам их получения. Изобретение также относится к применению указанных соединений, комбинаций, композиций и лекарственных препаратов, например, для лечения заболеваний и состояний, связанных с альфа-1-антитрипсином.
УРОВЕНЬ ТЕХНИКИ
Многие генетические нарушения у человека вызваны мутациями, которые нарушают фолдинг и транспорт белков. Белки могут вырабатываться в нормальных количествах, но вследствие их нарушенного фолдинга могут возникнуть проблемы.
Альфа-1-антитрипсин или α1антитрипсин (A1AT) представляет собой ингибитор протеазы, принадлежащий к суперсемейству серпинов. Он представляет собой белок, вырабатываемый гепатоцитами и, в меньшей степени, другими клетками и секретируемый в кровь, где он действует для ограничения ферментативной активности ключевых протеаз, в частности нейтрофильной эластазы. При его отсутствии (например, при дефиците альфа-1-антитрипсина) активность ключевых протеаз, включая нейтрофильную эластазу, не контролируется, что приводит к чрезмерному разрушению эластина и соединительных тканей. Наиболее распространенная ткань, в которой это проявляется патологически, находится в легких, где повышенное разрушение соединительной ткани легких обычно приводит к респираторным осложнениям, таким как эмфизема или хроническая обструктивная болезнь легких (ХОБЛ). Реже другие ткани также могут быть затронуты дефицитом/дисфункцией A1AT, такие как кожа.
У многих пациентов дефицит альфа-1-антитрипсина вызывается мутациями, включая, например, миссенс-мутацию E342K (Glu342Lys), называемую в настоящем описании мутантом Z (Z-AT). Мутантный Z альфа-1-антитрипсин образует полимеры, которые накапливаются в клетках и могут нарушать функцию пораженной ткани. Органом, наиболее часто страдающим от накопления полимера при дефиците альфа-1-антитрипсина, является печень, вызывая повреждение печени и в тяжелых случаях приводящее к необходимости трансплантации печени. Полимеры A1AT также обнаруживаются в других тканях, включая кровь, легкие и кожу. Было показано, что полимеры обладают провоспалительным действием и могут способствовать развитию патологии в тканях, где они обнаруживаются, в частности в легких и коже.
Современная терапия состояний, связанных с дефицитом альфа-1-антитрипсина, ограничивается белково-заместительной терапией M-AT (белок альфа-1-антитрипсин дикого типа), полученным из плазмы крови человека, как правило, дозированным еженедельно. Принимая во внимание, что такая терапия является эффективной при патологии легких, включая эмфизему, она не влияет на заболевание печени, вызванное накоплением полимеризованного Z-AT в гепатоцитах ER. Для 10-15% гомозигот для заболевания печени, пораженной Z-AT, включая фиброз, цирроз и гепатоцеллюлярную карциному, трансплантация печени является единственным доступным вариантом лечения. Кроме того, накопление полимеризованного Z-AT в эпителии легких оказывает хемоаттрактантный эффект на нейтрофилы, что может вызвать дальнейшее разрушение соединительной ткани легких. Таким образом, существует потребность в терапевтических средствах и способах, направленных на лечение болезненных состояний, связанных как с дефицитом альфа-1-антитрипсина, так и с токсическим накоплением альфа-1-антитрипсина.
Авторы настоящего изобретения идентифицировали соединения, которые способны модулировать альфа-1-антитрипсин, в частности мутантную форму Z-AT, предотвращая его полимеризацию в печени, таким образом являясь потенциально пригодным для лечения заболеваний, связанных с альфа-1-антитрипсином, более определенно с мутантными формами альфа-1-антитрипсина, в частности Z-AT, включая заболевания печени.
Авторы настоящего изобретения, в частности, идентифицировали соединения, которые способны связываться с α1-антитрипсином таким образом, чтобы предотвращать полимеризацию белка и тем самым вызывать полезные терапевтические эффекты. Хотя белки α1-антитрипсина были ранее кристаллизованы и их трехмерная структура изучена, сайт связывания соединений, идентифицированных авторами изобретения, ранее не был идентифицирован. В настоящем описании обсуждается более подробно, что он является криптическим сайтом связывания, образованным посредством взаимодействия между белком и настоящими соединениями, определенное расположение и структура которого приводит к благоприятному ингибированию полимеризации белка. Кроме того, авторы настоящего изобретения идентифицировали специфические структурные мотивы, которые вносят вклад в создание релевантного криптического сайта связывания и связывание в нем соединений. Таким образом, настоящее изобретение, следовательно, направлено на давнюю потребность в предоставлении соединений, которые способны лечить заболевания или состояния, опосредованные полимеризацией α1-антитрипсина.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение предоставляет вещество для применения в способе лечения заболевания или состояния, опосредованного полимеризацией α1-антитрипсина, в котором указанное вещество представляет собой: (а) соединение, которое способно ингибировать полимеризацию α1-антитрипсина; или (b) его фармацевтически приемлемый сольват, комплекс, таутомер, меченное изотопами производное или пролекарство. Предпочтительно, заболевание или состояние опосредовано полимеризацией Z-α1-антитрипсина и соединение способно ингибировать полимеризацию Z-α1-антитрипсина. Вещество обычно представляет собой низкомолекулярное соединение, например, соединение с молекулярной массой 1000 Дальтон или менее.
Предпочтительно, соединение способно связываться с α1-антитрипсином, индуцируя образование криптического сайта связывания в структуре белка α1-антитрипсина, причем указанный α1-антитрипсин содержит последовательность SEQ ID NO: 1. Например, сайт связывания может быть расположен между β-листом-A и β-листом-B указанного α1-антитрипсина, в котором: указанный β-лист-A содержит аминокислоты, соответствующие остаткам 140-144, 111-121, 181-191, 330-340 и 292-299 SEQ ID NO: 1; и указанный β-лист-B содержит аминокислоты, соответствующие остаткам 228-231, 236-244, 248-256, 369-376, 381-389 и 49-53 SEQ ID NO: 1. Более предпочтительно, сайт связывания расположен между аминокислотными цепями, соответствующими каждому из: (а) остатков 191-194 SEQ ID NO: 1; (b) остатков 288-293 SEQ ID NO: 1; (c) остатков 371-374 SEQ ID NO: 1; (d) остатков 249-253 SEQ ID NO: 1; и (e) остатков 240-243 SEQ ID NO: 1; и необязательно также (f) остатков 338-341 SEQ ID NO: 1. Сайт связывания может включать один или более из W194, Y244, L291, P289, F252, K290, I293, L338, I340, F372 и M374 SEQ ID NO: 1.
Предпочтительно, KD соединения по отношению к M-α1-антитрипсину составляет менее приблизительно 250 нМ, причем указанный M-α1-антитрипсин содержит последовательность SEQ ID NO: 2. Предпочтительно, KD соединения по отношению к Z-α1-антитрипсину составляет менее приблизительно 25 нМ, причем указанный Z-α1-антитрипсин содержит последовательность SEQ ID NO: 3. Предпочтительно, KD соединения по отношению к Z-α1-антитрипсину, по меньшей мере, в десять раз ниже, чем KD соединения по отношению к M-α1-антитрипсину, причем указанный M-α1-антитрипсин содержит последовательность SEQ ID NO: 2 и указанный Z-α1-антитрипсин содержит последовательность SEQ ID NO: 3.
Соединение может содержать четырехвалентный фрагмент формулы (IA)
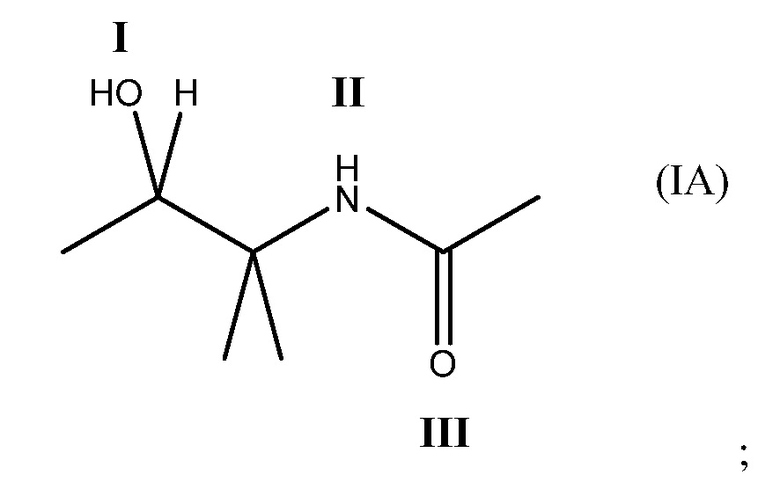
в которой соединение способно связываться с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1. Например, соединение может содержать двухвалентный фрагмент формулы (IB)
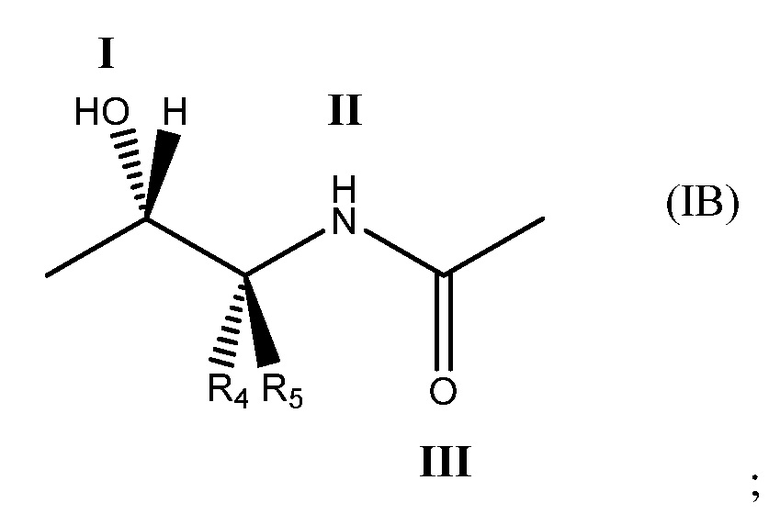
в которой: соединение способно связываться с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1; R4 выбран из водорода, C1-5алкила, C2-5алкенила, C2-5алкинила и C1-4алкокси и R5 выбран из водорода, C1-5алкила, C2-5алкенила, C2-5алкинила и C1-4алкокси. Необязательно R5 представляет собой водород. Необязательно R4 представляет собой C2-4алкил, C2-4алкенил, C2-4алкинил или C1-3алкокси. Необязательно R4 представляет собой н-пропил.
Более определенно, соединение может содержать одновалентный фрагмент формулы (IC)
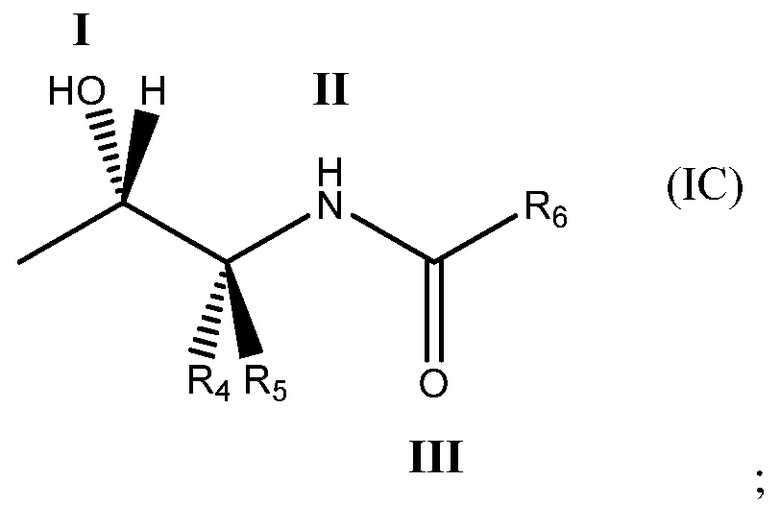
в которой: соединение способно связываться с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1; R4 и R5 представляют собой, как определено выше; и R6 представляет собой замещенную или незамещенную арильную или гетероарильную группу, способную связываться с боковой цепью W194 SEQ ID NO: 1. Необязательно, R6 представляет собой замещенную или незамещенную 4-оксиндолильную группу. Например, необязательно R6 представляет собой группу формулы R6'
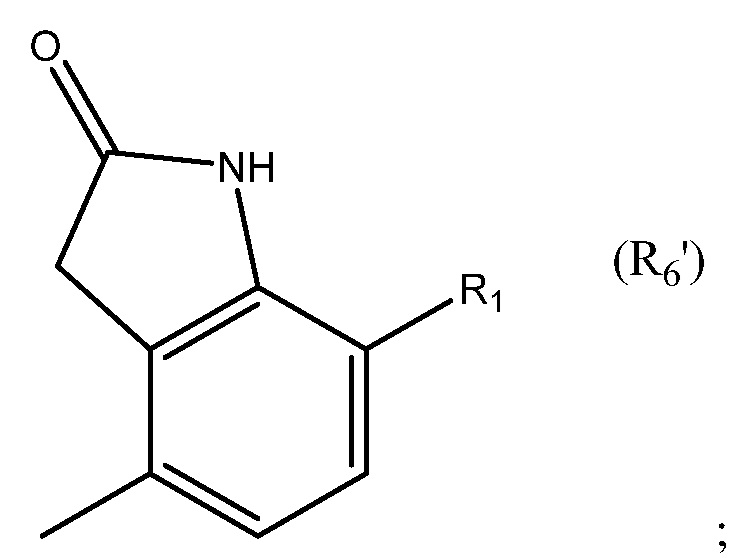
в которой R1 выбран из группы, состоящей из H, F, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Cl, Br и I. Необязательно R1 выбран из группы состоящий из H, F, CH3, NH2, OH и Cl. Например, необязательно R1 выбран из группы, состоящей из H и F.
Еще более определенно, соединение может иметь формулу (ID)
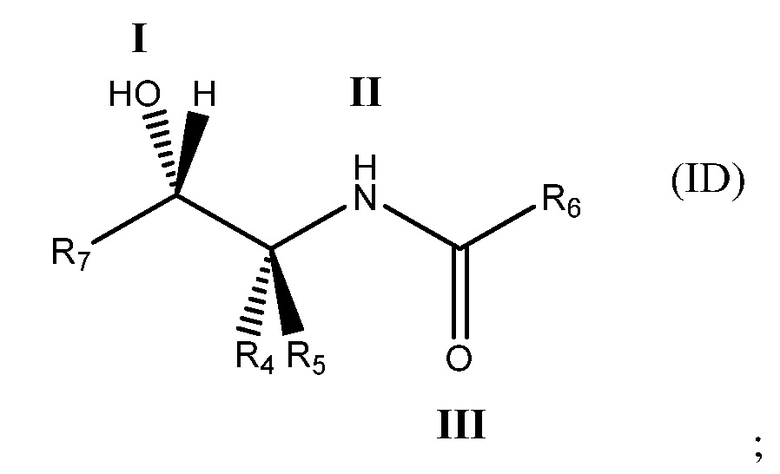
в которой: соединение способно связываться с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1; R4 и R5 представляют собой, как определено выше; R6 представляет собой, как определено выше; и R7 представляет собой замещенную или незамещенную арильную или гетероарильную группу.
Необязательно R7 представляет собой замещенную или незамещенную фенильную группу. Например, необязательно R7 представляет собой группу формулы R7'
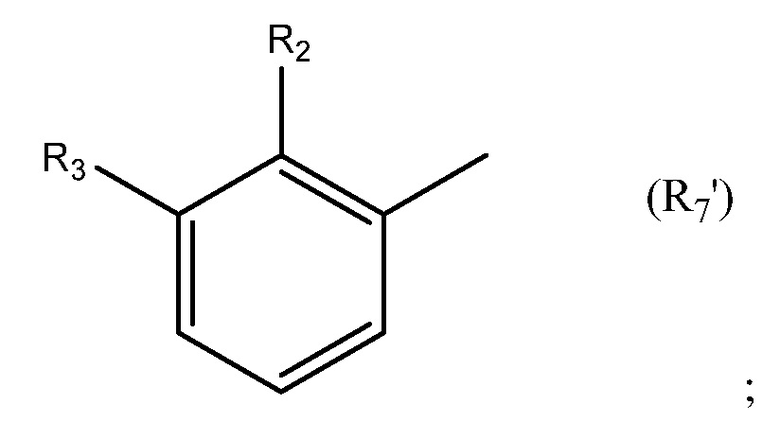
в которой: R2 выбран из группы, состоящей из CH3, Cl, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, SH, CN, F, Br и I; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Br, I и SH. Необязательно, R2 выбран из группы, состоящей из CH3, Cl, NH2, OH, SH, CN и F; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, NH2, OH и SH. Например, необязательно R2 выбран из группы, состоящей из CH3 и Cl; и R3 выбран из группы, состоящей из F, Cl и CN.
Например, соединение может иметь формулу (I)
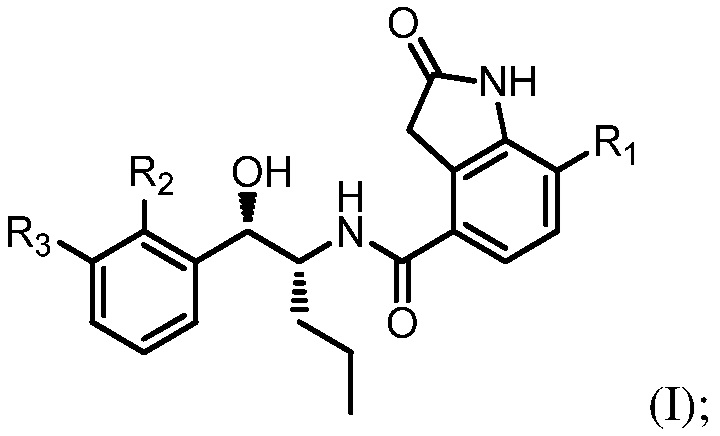
в которой: R1 выбран из группы, состоящей из H, F, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Cl, Br и I; R2 выбран из группы, состоящей из CH3, Cl, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, SH, CN, F, Br и I; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Br, I и SH.
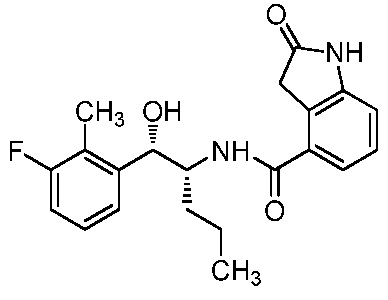
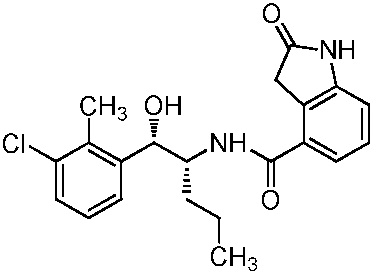
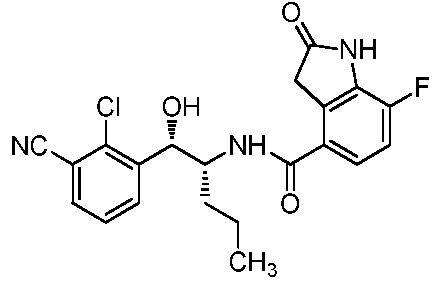
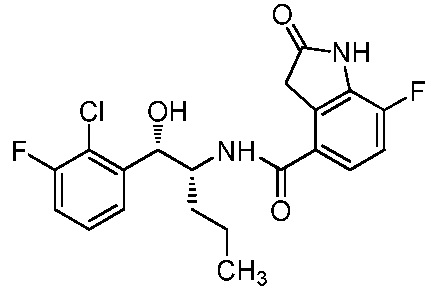
Необязательно в формуле (I): R1 выбран из группы, состоящей из H, F, CH3, NH2, OH и Cl; R2 выбран из группы, состоящей из CH3, Cl, NH2, OH, SH, CN и F; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, NH2, OH и SH. Например, необязательно: R1 выбран из группы, состоящей из H и F; R2 выбран из группы, состоящей из CH3 и Cl; и R3 выбран из группы, состоящей из F, Cl и CN. Например, необязательно: R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой F; или R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой Cl; или R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой CN; или R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой F.
Настоящее изобретение также предоставляет вещество, как определено выше. Кроме того, настоящее изобретение предоставляет способ идентификации соединения-кандидата в лекарственное средство, включающий: взаимодействие соединения-кандидата в лекарственное средство с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с образованием комплекса между соединением-кандидатом в лекарственное средство и указанным α1-антитрипсином; разрешение структуры комплекса; и определение, присутствует ли в комплексе соединение-кандидат в лекарственное средство в сайте связывания, как определено выше.
В еще дополнительных аспектах настоящее изобретение предоставляет следующие варианты осуществления [1]-[11].
[1] соединение, которое: (а) имеет формулу (I):
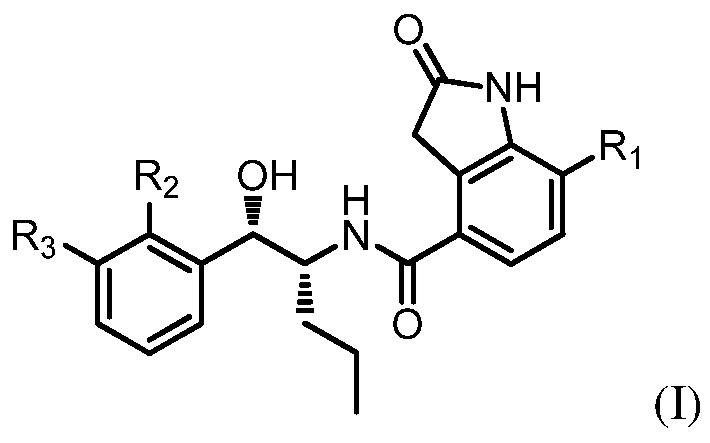
в которой: R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой F; или R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой Cl; или R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой CN; или R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой F; или
(b) представляет собой его фармацевтически приемлемый сольват, комплекс, таутомер, меченное изотопами производное или пролекарство.
[2] Соединение, как определено в [1], для применения в терапии.
[3] Соединение, как определено в [1], для применения при лечении заболевания или состояния, опосредованного альфа-1-антитрипсином.
[4] Фармацевтическая композиция, содержащая соединение, как определено в [1], и один или более фармацевтически приемлемых носителей, разбавителей и эксципиентов.
[5] Способ лечения заболевания или состояния, опосредованного альфа-1-антитрипсином, у объекта, включающий введение терапевтически эффективного количества соединения, как определено в [1].
[6] Применение соединения, как определено в [1], при изготовлении лекарственного препарата для применения при лечении заболевания или состояния, опосредованного альфа-1-антитрипсином.
[7] Комбинация, содержащая соединение, как определено в [1], и, по меньшей мере, один дополнительный терапевтический агент.
[8] Комбинация, содержащая соединение, как определено в [1], и, по меньшей мере, один дополнительный терапевтический агент для применения в терапии, в частности для лечения заболевания или состояния, опосредованного альфа-1-антитрипсином.
[9] Комбинация, содержащая соединение, как определено в [1], и, по меньшей мере, один дополнительный терапевтический агент для применения при лечении заболевания или состояния, опосредованного альфа-1-антитрипсином.
[10] Способ лечения заболевания или состояния, опосредованного альфа-1-антитрипсином, включающий введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества комбинации, содержащей соединение, как определено в [1], и, по меньшей мере, один дополнительный терапевтический агент.
[11] Применение комбинации, содержащей соединение, как определено в [1], и, по меньшей мере, один дополнительный терапевтический агент при изготовлении лекарственного препарата для лечения заболевания или состояния, опосредованного альфа-1-антитрипсином.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 представлено типичное изображение кристаллизованного комплекса, образованного между типичным соединением настоящего изобретения и белком α1-антитрипсином, как более подробно описано в Примере 2.
На Фиг. 2 представлено типичное изображение части кристаллизованного комплекса, образованного между типичным соединением настоящего изобретения и белком α1-антитрипсином, как более подробно описано в Примере 2 (в частности, часть содержит сайт связывания между белком и соединение изобретения).
На Фиг. 3 представлена схематическая двумерная визуализация типичного соединения настоящего изобретения в сайте связывания белка и выделение некоторых аминокислотных остатков, которые составляют сайт связывания, также как частичных химических структур внутри белка, которые образуют значительные межмолекулярные взаимодействия (например, водородные связи) с соединением (показано пунктирными линиями), как более подробно описано в Примере 2.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Используемая в настоящем описании алкильная группа представляет собой насыщенный углеводородный радикал с прямой или разветвленной цепью. В одном примере алкильная группа содержит от 1 до 5 атомов углерода. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, изопентил и неопентил.
Используемая в настоящем описании алкенильная группа представляет собой углеводородный радикал с прямой или разветвленной цепью, который содержит одну или более (например, одну) углерод-углеродных двойных связей. В одном примере алкенильная группа содержит от 2 до 5 атомов углерода.
Используемая в настоящем описании алкинильная группа представляет собой углеводородный радикал с прямой или разветвленной цепью, который содержит одну или более (например, одну) углерод-углеродных тройных связей. В одном примере алкинильная группа содержит от 2 до 5 атомов углерода.
Используемая в настоящем описании алкоксигруппа (например, С1-4алкоксигруппа) представляет собой группу формулы -OR, в которой R представляет собой алкильную (например, C1-4алкильную) группу.
Используемая в настоящем описании алкилтиольная группа (например, C1-4алкилтиольная группа) представляет собой группу формулы -SR, в которой R представляет собой алкильную (например, C1-4алкильную) группу.
Используемая в настоящем описании арильная группа обычно представляет собой C6-14монокарбоциклическое ароматическое кольцо или поликарбоциклическую кольцевую систему, в которой, по меньшей мере, одно из колец в ней является ароматическим. Предпочтительно такая группа представляет собой C6-10монокарбоциклическое ароматическое кольцо или бикарбоциклическую кольцевую систему, в которой, по меньшей мере, одно из колец в ней является ароматическим. Примеры включают фенил, нафтил и инданил. Если специально не указано иначе, валентность может располагаться на любом атоме любого кольца арильной группы.
Используемая в настоящем описании гетероарильная группа обычно представляет собой содержащее от 5 до 14 атомов кольца моноциклическое, ароматическое кольцо или полициклическую кольцевую систему, в которой, по меньшей мере, одно из колец в ней является ароматическим. Гетероарильная группа содержит, по меньшей мере, один (например, 1, 2, 3 или 4) кольцевой гетероатом, выбранный из O, S, N (например, образующий, по меньшей мере, одно кольцо O, S(O)x (в котором x равно 0, 1 или 2), N, NH или N+O- фрагмент), причем другие атомы кольца являются атомами углерода. Предпочтительно такая группа содержит от 5 до 10 атомов кольца. Если специально не указано иначе, валентность может располагаться на любом атоме любого кольца гетероарильной группы.
Примеры моноциклических гетероарильных групп включают тиенильную, фурильную, пирролильную, имидазолильную, тиазолильную, изотиазолильную, пиразолильную, оксазолильную, изоксазолильную, триазолильную, тиадиазолильную, оксадиазолильную, пиридинильную, пиридазинильную, пиримидинильную, пиразинильную, триазинильную и тетразолильную группы.
Примеры полициклических гетероарильных групп включают оксиндолильную, бензотиенильную, бензофуранильную, бензимидазолильную, бензотиазолильную, бензизотиазолильную, бензоксазолильную, бензизоксазолильную, бензтриазолильную, индолильную, изоиндолильную и индазолильную группы. Оксиндолил включает 2-оксиндолил (т. е. одновалентную группу, производную 2-оксиндола, также известную как 2-индолинон) и 3-оксиндолил (т. е. одновалентную группу, производную 3-оксиндола, также известную как 3-индолинон).
В арильной группе или гетероарильной группе, по меньшей мере, один (например, 1, 2 или 3) атом углерода в кольце может быть замещен карбонильной группой (т. е. -C(O)-). Например, бензоксазолил может необязательно содержать -C(O)- вместо углерода в его 2-положении, что приводит к образованию одновалентной гетероарильной группы, производной бензоксазолона.
Если не указано иначе, арильная или гетероарильная группа обычно является незамещенной. Однако если указано, что такая группа является незамещенной или замещенной, один или более атомов водорода необязательно замещены атомами дейтерия, атомами галогена или гидроксилом, тиолом (-SH), нитро, сульфоновой кислотой, нитрилом (-CN), амино (например, -NR2, в которой каждый R независимо выбран из H и C1-5алкила), алкилом (например, C1-5алкилом), дейтерированным алкилом (например, C1-5дейтерированным алкилом), алкенилом (например, C2-5алкенилом), алкинилом (например, C2-5алкинилом), галогеналкилом (например, C1-5галогеналкилом), галогеналкенилом (например, C2-5галогеналкенилом), алкокси (например, C1-4алкокси), алкилтио (например, C1-4алкилтио), алкилсульфонилом (например, C1-4алкилсульфонилом), галогеналкокси (например, C1-4галогеналкокси), галогеналкилтио (например, C1-4галогеналкилтио), галогеналкилсульфонилом (например, C1-4галогеналкилсульфонилом), циклоалкилом (например, C3-5циклоалкилом) или гетероциклоалкилом (например, C3-5гетероциклоалкилом).
Предпочтительными такими заместителями являются атомы дейтерия, атомы фтора, атомы хлора или гидроксил, нитро, нитрил (-CN), -NR2 (в которой каждый R независимо выбран из H и метила), C1-5алкил, -CD3, C2-3алкенил, C2-3алкинил, CF3, CHF2, CH2CF3, C2F5, CF=CF2, C1-3алкокси, C1-3алкилтио, C1-3алкилсульфонил, OCF3, SCF3, SO2CF3, циклопропил, циклобутил, оксетанил и азетидинил.
Предпочтительно замещенная арильная или гетероарильная группа имеет от 1 до 5 заместителей, более предпочтительно от 1 до 3 заместителей и наиболее предпочтительно 1 или 2 заместителя. Предпочтительно замещенная арильная или гетероарильная группа несет не более 2 нитрозаместителей и не более 2 заместителей сульфоновой кислоты.
Используемые в настоящем описании атомы галогена обычно представляют собой атомы F, Cl, Br или I, предпочтительно атомы F или Cl.
Во избежание сомнений, термины α1-антитрипсин, альфа-1-антитрипсин, альфа 1 антитрипсин и т. д. используются в настоящем описании взаимозаменяемо.
Если не указано иначе, все ссылки на органические группы, содержащие один или более атомов водорода, также включают их частично или полностью дейтерированные аналоги. Например, ссылки в настоящем описании на алкильные группы охватывают алкильные группы, состоящие из атомов углерода и водорода, алкильные группы, содержащие атомы углерода и смесь атомов водорода и дейтерия, и полностью дейтерированные алкильные группы. Однако предпочтительно, что такие органические группы не являются частично или полностью дейтерированными, за исключением случаев, когда точно указано.
Общая информация о соединениях изобретения, фармацевтических композициях, содержащих такие соединения, и комбинированных терапиях
Используемый в настоящем описании термин «соединение изобретения» включает соединения, которые способны ингибировать полимеризацию α1-антитрипсина, и все сольваты, комплексы, таутомеры, полиморфные модификации, меченные изотопами производные, стереоизомеры и оптические изомеры таких соединений.
Используемый в настоящем описании термин «эффективное количество» означает такое количество лекарственного средства или фармацевтического агента, которое будет вызывать биологический или медицинский ответ ткани, системы, животного или человека, который требуется, например, исследователю или клиницисту. Кроме того, термин «терапевтически эффективное количество» означает любое количество, которое по сравнению с соответствующим объектом, не получавшим такое количество, приводит к улучшенному лечению, исцелению, профилактике или облегчению заболевания, нарушения или побочного эффекта или снижению скорости развития заболевания или нарушения. Термин также включает в свой объем количества, эффективные для улучшения нормальной физиологической функции.
Используемый в настоящем описании термин «фармацевтически приемлемый» относится к таким соединениям, материалам, композициям и лекарственным формам, которые в рамках здравого медицинского заключения подходят для применения при взаимодействии с тканями людей и животных без чрезмерной токсичности, раздражения, или другой проблемы, или осложнения, соизмеримого с целесообразным отношением польза/риск.
Соединения настоящего изобретения могут быть в форме соли.
Обычно соли настоящего изобретения представляют собой фармацевтически приемлемые соли. Соли, охватываемые термином «фармацевтически приемлемые соли», относятся к нетоксичным солям соединений данного изобретения. Для обзора подходящих солей см. Berge et al, J. Pharm. Sci. 1977, 66, 1-19.
Соединения изобретения могут существовать в твердой или жидкой форме. В твердой форме соединение изобретения может существовать в большом разнообразии вариантов твердых состояний от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию, в котором материалу не хватает дальнего порядка на молекулярном уровне, и в зависимости от температуры он может проявлять физические свойства твердого вещества или жидкости. Обычно такие материалы не дают отличительных рентгеновских дифрактограмм и, хотя и проявляют свойства твердого вещества, более формально описываются как жидкость. При нагревании происходит изменение свойств твердого вещества в жидкость, которое характеризуется изменением состояния, обычно второго порядка («стеклование»). Термин «кристаллический» относится к твердой фазе, в которой материал имеет регулярную упорядоченную внутреннюю структуру на молекулярном уровне и дает отличительную рентгеновскую дифрактограмму с определенными пиками. Такие материалы при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния к жидкому характеризуется фазовым переходом, обычно первого порядка («температура плавления»).
Соединение настоящего изобретения может существовать в сольватированной и несольватированной формах. Используемый в настоящем описании термин «сольват» относится к комплексу переменной стехиометрии, образованному растворенным веществом (в данном изобретении соединение изобретения и растворитель). Такие растворители в целях изобретения не могут влиять на биологическую активность растворенного вещества. Специалист в данной области техники поймет, что фармацевтически приемлемые сольваты могут быть образованы для кристаллических соединений, в которых молекулы растворителя включены в кристаллическую решетку во время кристаллизации. Включенные молекулы растворителя могут представлять собой молекулы воды или неводные, такие как молекулы этанола, изопропанола, ДМСО, уксусной кислоты, этаноламина и этилацетата. Кристаллическую решетку, объединенную с молекулами воды, обычно называют «гидратами». Гидраты включают стехиометрические гидраты, также как композиции, содержащие переменные количества воды. Настоящее изобретение включает все такие сольваты.
Соединения изобретения могут иметь способность кристаллизоваться в более чем одной форме, характеристика, которая известна как полиморфизм, и понятно, что такие полиморфные формы («полиморфные модификации») находятся в пределах объема изобретения. Полиморфизм обычно может возникать в результате изменений температуры или давления или того и другого, и также может возникать в результате изменений в процессе кристаллизации. Полиморфные модификации можно отличить по различным физическим характеристикам, известным в данной области техники, таким как рентгеновские дифрактограммы, растворимость и температура плавления.
Также следует отметить, что соединения изобретения могут образовывать таутомеры. Понятно, что все таутомеры и смеси таутомеров соединений настоящего изобретения включены в объем соединений настоящего изобретения.
Изобретение также включает меченные изотопами соединения, которые идентичны соединениям изобретения, но с тем обстоятельством, что один или более атомов замещены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, наиболее часто обнаруженного в природе. Примеры изотопов, подходящих для включения в соединения изобретения, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, иода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P и серы, такие как 35S.
Определенные меченные изотопами соединения настоящего изобретения, например, соединения, включающие радиоактивный изотоп, являются пригодными в исследованиях распределения лекарственного вещества и/или субстрата в тканях. Радиоактивные изотопы трития, т. е. 3H и углерода-14, т. е. 14C являются наиболее пригодными для этой цели ввиду удобства их включения и готовых средств обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, т. е. 2H, может дать определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный период полувыведения in vivo или уменьшенные требования к дозировке, и, следовательно, может быть предпочтительным в некоторых случаях.
Меченные изотопами соединения настоящего изобретения обычно могут быть получены стандартными методами, известными специалисту в данной области техники, или способами, аналогичными тем, которые описаны в прилагаемых Примерах и Способах получения, с использованием соответствующих меченных изотопами реагентов вместо ранее применяемого немеченого реагента.
В одном варианте осуществления соединение настоящего изобретения представляет собой одно из:
Хотя возможно, что для применения в терапии соединение изобретения можно вводить в виде неочищенного химического вещества, можно представить соединение изобретения в виде активного ингредиента в фармацевтической композиции. Такие композиции могут быть приготовлены способом, хорошо известным в области фармацевтики, и содержат, по меньшей мере, одно активное соединение. Соответственно, изобретение дополнительно предоставляет фармацевтические композиции, содержащие соединение изобретения и один или более фармацевтически приемлемых эксципиентов. Эксципиент(ы) должен быть приемлемым в смысле совместимости с другими ингредиентами композиции и не быть вредным для его реципиента. В соответствии с другим аспектом изобретения также предоставлен способ получения фармацевтической композиции, включающей агент с одним или более фармацевтически приемлемыми эксципиентами. Фармацевтическая композиция может быть использована для лечения и/или профилактики любого из состояний, описанных в настоящей заявке.
Обычно соединение изобретения вводят в фармацевтически эффективном количестве. Фактически вводимое количество соединения обычно будет определяться врачом исходя из соответствующих обстоятельств, включая состояние, подлежащее лечению, выбранный путь введения, фактическое вводимое соединение, возраст, вес и ответ отдельного пациента, тяжесть симптомов пациента и подобное.
Фармацевтические композиции могут быть представлены в виде стандартных доз, содержащих предварительно определенное количество активного ингредиента на стандартную дозу. Термин «стандартные лекарственные формы» относится к физически дискретным единицам, подходящим в качестве единичных доз для людей и других млекопитающих, каждая единица содержит предварительно определенное количество активного материала, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом, наполнителем или носителем. Типичные стандартные лекарственные формы включают предварительно заполненные, предварительно отмеренные ампулы или шприцы жидких композиций или пилюли, таблетки, капсулы или подобное в случае твердых композиций.
Предпочтительными композициями стандартной дозы являются композиции, содержащие суточную дозу, или часть дозы, или ее соответствующую долю активного ингредиента. Таким образом, такие стандартные дозы можно вводить один или более одного раза в день. Такие фармацевтические композиции могут быть получены любым из способов, хорошо известных в области фармацевтики.
Фармацевтические композиции могут подходить для введения любым подходящим путем, например, пероральным (включая буккальный или сублингвальный), ректальным, ингаляционным, интраназальным, местным (включая буккальный, сублингвальный или трансдермальный), вагинальным или парентеральным (включая подкожный, внутримышечный, внутривенный или внутрикожный) путем. Такие композиции могут быть получены любым способом, известным в области фармацевтики, например, путем объединения активного ингредиента с носителем(ями) или эксципиентом(ами).
Фармацевтические композиции, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; пищевые пены или взбитые массы; или жидкие эмульсии типа «масло в воде» или жидкие эмульсии типа «вода в масле».
Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент можно комбинировать с пероральным нетоксичным фармацевтически приемлемым инертным эксципиентом, таким как этанол, глицерин, вода и подобное. Порошки получают путем измельчения соединения до подходящего маленького размера и смешивания с подобным образом приготовленным фармацевтическим эксципиентом, таким как пищевой углевод, например, крахмал или маннит. Также могут присутствовать ароматизатор, консервант, диспергирующее средство и краситель.
Капсулы получают путем приготовления порошковой смеси, как описано выше, и заполнения образованных желатиновых оболочек. Эксципиенты, включая глиданты и скользящие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль могут быть добавлены к порошковой смеси перед процессом заполнения. Дезинтегрирующее или солюбилизирующее средство, такое как агар-агар, карбонат кальция или карбонат натрия также может быть добавлено для улучшения доступности лекарственного препарата при проглатывании капсулы.
Более того, когда желательно или необходимо, в смесь также могут быть включены эксципиенты, включая подходящие связующие вещества, глиданты, скользящие вещества, подсластители, ароматизаторы, дезинтегрирующие средства и красители. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как камедь, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и подобное. Скользящие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и подобное. Разрыхлители включают, но не ограничиваются ими, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и подобное. Таблетки изготавливают, например, путем приготовления порошковой смеси, гранулирования или уплотнения, добавления скользящего вещества и разрыхлителя и прессования в таблетки. Порошковую смесь получают путем смешивания соединения, измельченного подходящим образом, с разбавителем или основанием, как описано выше, и необязательно со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем резорбции, таким как четвертичная соль и/или абсорбентом, таким как бентонит, каолин или дикальция фосфат. Порошковая смесь может быть гранулирована путем смачивания со связующим веществом, таким как сироп, крахмальная паста, мазь на основе камеди или растворы целлюлозных или полимерных материалов и продавливания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину, и в результате чего плохо сформированные куски разбиваются на гранулы. Гранулы могут быть смазаны для предотвращения прилипания к формам для таблетирования путем добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Затем смазанную смесь прессуют в таблетки. Соединения настоящего изобретения также могут быть объединены с легкосыпучим инертным носителем и напрямую спрессованы в таблетки, минуя стадии гранулирования или комкования. Может быть предоставлено прозрачное или непрозрачное защитное покрытие, состоящее из герметизирующего слоя шеллака, покрытия из сахара или полимерного материала и полирующего покрытия из воска. К этим покрытиям можно добавлять красители, чтобы различать разные однократные дозировки.
Пероральные жидкости, такие как раствор, суспензии, сиропы и эликсиры можно получить в виде единичной дозированной формы, так что заданное количество содержит предварительно определенное количество соединения. Сиропы можно получить путем растворения соединения в водном растворе с подходящим вкусом, в то время как эликсиры получают с использованием нетоксичного спиртового наполнителя. Суспензии можно приготовить путем диспергирования соединения в нетоксичном наполнителе. Также можно добавить солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и эфиры полиоксиэтиленсорбита, консерванты, вкусовые добавки, такие как масло перечной мяты, или натуральные подсластители, или сахарин, или другие искусственные подсластители и подобное.
В случае необходимости композиции единицы дозирования для перорального введения могут быть микроинкапсулированы. Композиция также может быть получена для продления или поддержания высвобождения, например, путем покрытия или внедрения материала в виде частиц в полимеры, воск или подобное. Соединения изобретения также можно вводить в форме липосомальных систем доставки, таких как малые моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть образованы из множества фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Фармацевтические композиции, подходящие для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для сохранения тесного контакта с эпидермисом реципиента в течение продолжительного периода времени.
Фармацевтические композиции, подходящие для местного введения, могут быть составлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел.
Для лечения глаз или других внешних тканей, например, рта и кожи, композиции предпочтительно наносят в виде мази или крема для местного применения. При составлении мази активный ингредиент может быть использован или с парафиновой, или с водорастворимой мазевой основой. Альтернативно, активный ингредиент может быть составлен в виде крема на основе крема типа масло в воде или основе типа вода в масле.
Фармацевтические композиции, подходящие для местного введения в глаза, включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, в частности в водном растворителе.
Фармацевтические композиции, подходящие для местного введения в ротовую полость, включают леденцы, пастилки и жидкости для полоскания рта.
Фармацевтические композиции, подходящие для ректального введения, могут быть представлены в виде суппозиториев или клизм.
Лекарственные формы для назального или ингаляционного введения могут быть удобно составлены в виде аэрозолей, растворов, суспензий, капель, гелей или сухих порошков.
Композиции для интраназального введения включают водные композиции, вводимые в нос каплями или насосом под давлением. Подходящие композиции для этой цели содержат воду в качестве разбавителя или носителя. Композиции для введения в легкие или нос могут содержать один или более эксципиентов, например, один или более суспендирующих агентов, один или более консервантов, одно или более поверхностно-активных веществ, один или более регулирующих тоничность агентов, один или более сорастворителей и могут включать компоненты для контроля pH композиции, например, буферную систему. Кроме того, композиции могут содержать другие эксципиенты, такие как антиоксиданты, например, метабисульфит натрия и маскирующие вкус агенты. Композиции также можно вводить в нос или другие области дыхательных путей путем распыления.
Интраназальные композиции могут обеспечить доставку соединения(й) изобретения во все области носовых полостей (целевую ткань) и, кроме того, могут предоставить соединению(ям) изобретения оставаться во взаимодействии с целевой тканью в течение более продолжительных периодов времени. Подходящим режимом дозирования для интраназальных композиций будет медленный вдох пациентом через нос после очищения носовой полости. Во время ингаляции композицию вводят в одну ноздрю, в то время как другую сжимают вручную. Затем эту процедуру повторяют для другой ноздри. Как правило, один или два впрыскивания в ноздрю вводят с помощью вышеописанной процедуры один, два или три раза в день, в идеале один раз в день. Особый интерес представляют интраназальные композиции, подходящие для однократного в день введения.
Композиции для введения в легкие или нос могут содержать один или более эксципиентов, которые могут защищать от микробного или грибкового загрязнения и роста путем включения одного или более консервантов. Примеры фармацевтически приемлемых антимикробных агентов или консервантов включают, но не ограничиваются ими, соединения четвертичного аммония (например, бензалкония хлорид, бензетония хлорид, цетримид, цетилпиридиния хлорид, лауралкония хлорид и миристил пиколиния хлорид), ртутьсодержащие агенты (например, фенилртути нитрат, фенилртути ацетат и тимеросал), спиртовые агенты (например, хлорбутанол, фенилэтиловый спирт и бензиловый спирт), антибактериальные эфиры (например, сложные эфиры пара-гидроксибензойной кислоты), хелатирующие агенты, такие как динатрия эдетат (ЭДТА) и другие антимикробные агенты, такие как хлоргексидин, хлоркрезол, сорбиновая кислота и ее соли (например, сорбат калия) и полимиксин. Примеры фармацевтически приемлемых противогрибковых агентов или консервантов включают, но не ограничиваются ими, бензоат натрия, сорбиновую кислоту, пропионат натрия, метилпарабен, этилпарабен, пропилпарабен и бутилпарабен. Консервант(ы), если он включен, может присутствовать в количестве от 0,001 до 1% (масс./масс.), например, от 0,015% до 0,5% (масс./масс.) на основе общей массы композиции.
Композиции (например, в которых, по меньшей мере, одно соединение находится в суспензии) могут включать одно или более поверхностно-активных веществ, которые действуют для облегчения растворения частиц лекарственного препарата в водной фазе композиции. Например, количество используемого поверхностно-активного вещества представляет собой количество, которое не вызовет образования пены во время смешивания. Примеры фармацевтически приемлемых поверхностно-активных веществ включают жирные спирты, сложные эфиры и простые эфиры, такие как полиоксиэтилен (20) сорбитан моноолеат (Полисорбат 80), макроголовые эфиры и полоксамеры. Поверхностно-активное вещество может присутствовать в количестве от приблизительно 0,01 до 10% (масс./масс.), например, от 0,01 до 0,75% (масс./масс.), например, приблизительно 0,5% (масс./масс.) на основе общей массы композиции.
Один или более регулирующих тоничность агента(ов) могут быть включены для достижения тоничности жидкостей организма, например, жидкостей из носовой полости, что снижает уровни раздражения. Примеры фармацевтически приемлемых регулирующих тоничность агентов включают, но не ограничиваются ими, хлорид натрия, декстрозу, ксилит, хлорид кальция, глюкозу, глицерин и сорбит. Регулирующий тоничность агент, если присутствует, может быть включен в количестве от 0,1 до 10% (масс./масс.), например, от 4,5 до 5,5% (масс./масс.), например, приблизительно 5,0% (масс./масс.) на основе общей массы композиции.
Композиции изобретения могут быть буферизированы путем добавления подходящих буферных агентов, таких как цитрат натрия, лимонная кислота, трометамол, фосфаты, такие как динатрия фосфат (например, додекагидрат, гептагидрат, дигидрат и безводные формы) или фосфат натрия и их смеси.
Буферный агент, если он присутствует, может быть включен в количестве от 0,1 до 5% (масс./масс.), например, от 1 до 3% (масс./масс.) на основе общей массы композиции.
Примеры маскирующих вкус агентов включают сукралозу, сахарозу, сахарин или его соль, фруктозу, декстрозу, глицерин, кукурузный сироп, аспартам, ацесульфам-K, ксилит, сорбит, эритрит, аммония глицирризинат, тауматин, неотам, маннит, ментол, эвкалиптовое масло, камфору, натуральный ароматизатор, искусственный ароматизатор и их комбинации.
Один или более сорастворителя(ей) могут быть включены для улучшения растворимости соединения(й) лекарственного препарата и/или других эксципиентов. Примеры фармацевтически приемлемых сорастворителей включают, но не ограничиваются ими, пропиленгликоль, дипропиленгликоль, этиленгликоль, глицерин, этанол, полиэтиленгликоли (например, ПЭГ300 или ПЭГ400) и метанол. В одном варианте осуществления сорастворитель представляет собой пропиленгликоль.
Сорастворитель(и), если он присутствует, может быть включен в количестве от 0,05 до 30% (масс./масс.), например, от 1 до 25% (масс./масс.), например, от 1 до 10% (масс./масс.) на основе общей массы композиции.
Композиции для ингаляционного введения включают водные, органические или водные/органические смеси, сухой порошок или кристаллические композиции, вводимые в дыхательные пути с помощью насоса под давлением или ингалятора, например, резервуарных ингаляторов сухого порошка, однодозовых ингаляторов сухого порошка, многодозовых ингаляторов предварительно отмеренного сухого порошка, назальных ингаляторов или аэрозольных ингаляторов под давлением, небулайзеров или инсуффляторов. Подходящие композиции содержат воду в качестве разбавителя или носителя для этой цели и могут быть снабжены стандартными эксципиентами, такими как буферные агенты, модифицирующие тоничность агенты и подобное. Водные композиции также можно вводить в нос и другие области дыхательных путей с помощью распыления. Такие композиции могут быть водными растворами, или суспензиями, или аэрозолями, доставляемыми из упаковок под давлением, таких как дозированные ингаляторы с использованием подходящего сжиженного пропеллента.
Композиции для местного введения в нос (например, для лечения ринита) или в легкие включают композиции аэрозолей под давлением и водные композиции, доставляемые в носовые полости с помощью насоса под давлением. Композиции, которые не находятся под давлением и подходят для местного введения в носовую полость, представляют особый интерес. Подходящие композиции для этой цели содержат воду в качестве разбавителя или носителя. Водные композиции для введения в легкие или нос могут быть снабжены стандартными эксципиентами, такими как буферные агенты, модифицирующие тоничность агенты и подобное. Водные композиции также можно вводить в нос с помощью распыления.
Дозатор жидкости обычно может использоваться для доставки жидкой композиции в носовые полости. Жидкая композиция может быть водной или неводной, но обычно водной. Такой дозатор жидкости может иметь дозирующую насадку или дозирующее отверстие, через которое отмеренная доза жидкой композиции выдается при приложении прикладываемой пользователем силы к насосному механизму дозатора жидкости. Такие дозаторы жидкости обычно снабжены резервуаром с множеством отмеренных доз жидкой композиции, причем дозы выдаются при последовательном включении насоса. Дозирующая насадка или отверстие могут иметь конфигурацию с возможностью введения в ноздри пользователя для распыления жидкой композиции в носовой полости.
Композиции сухого порошка для местной доставки в легкие с помощью ингаляции могут, например, быть представлены в капсулах и картриджах, например, из желатина или в блистерах, например, из ламинированной алюминиевой фольги для применения в ингаляторе или инсуффляторе. Композиции порошковой смеси обычно содержат порошковую смесь для ингаляции соединения изобретения и подходящую порошковую основу (носитель/разбавитель/эксципиент), такую как моно-, ди- или полисахариды (например, лактоза или крахмал). Композиции сухого порошка могут также включать, помимо лекарственного средства и носителя, дополнительный эксципиент (например, тройной агент, такой как сложный эфир сахара, например, октаацетат целлобиозы, стеарат кальция или стеарат магния.
В одном варианте осуществления композиция, подходящая для ингаляционного введения, может быть включена во множество герметичных контейнеров с дозой, снабженных упаковкой(ами) лекарственного препарата, установленных внутри подходящего ингаляционного устройства. Контейнеры могут быть разрывающимися, снимающимися или открываемыми иным способом по отдельности, и дозы композиции сухого порошка могут вводиться с помощью ингаляции через мундштук устройства для ингаляции, как известно в данной области техники. Упаковка лекарственного препарата может иметь несколько различных форм, например, форму диска или удлиненную полоску.
Дополнительный способ доставки композиции для ингаляции сухого порошка предназначен для отмеренных доз композиции, которые предоставлены в капсулах (одна доза на капсулу), которые затем загружаются в устройство для ингаляции, обычно пациентом по мере необходимости. Устройство имеет средства для разрыва, прокалывания или иного открытия капсулы, чтобы доза могла попасть в легкое пациента, когда он вдыхает через мундштук устройства.
Композиции аэрозолей под давлением, подходящие для ингаляции, могут быть или суспензией, или раствором и могут содержать соединение изобретения или его фармацевтически приемлемую соль и подходящий пропеллент, такой как фторуглерод или водородсодержащий хлорфторуглерод или их смеси, в частности гидрофторалканы, в частности 1,1,1,2-тетрафторэтан, 1,1,1,2,3,3,3-гептафтор-н-пропан или их смесь. Композиция аэрозоля может необязательно содержать дополнительные эксципиенты композиции, хорошо известные в данной области техники, такие как поверхностно-активные вещества, например, олеиновая кислота, лецитин или олигомолочная кислота или их производные, например, как описано в WO 94/21229 и WO 98/34596 (Minnesota Mining and Manufacturing Company), и сорастворители, например, этанол. Композиции под давлением обычно будут удерживаться в контейнере (например, алюминиевом контейнере), закрытом клапаном (например, клапаном-дозатором) и вставленным в дозатор, снабженный мундштуком.
Фармацевтические композиции, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или составов для распыления.
Фармацевтические композиции, подходящие для парентерального введения, включают водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, которые делают композицию изотоничной крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, в запечатанных ампулах и виалах и могут храниться при сушке сублимацией (лиофилизированном) условии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций непосредственно перед применением. Экстемпоральные инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Следует понимать, что в дополнение к ингредиентам, конкретно упомянутым выше, композиции могут включать другие агенты, стандартные в данной области техники, принимая во внимание тип рассматриваемого состава, например, агенты, которые подходят для перорального введения, могут включать ароматизаторы.
Терапевтически эффективное количество агента будет зависеть от ряда факторов, включая, например, возраст и вес объекта, точное состояние, подлежащее лечению, и его тяжесть, свойство состава и способ введения, и будет в конечном итоге на усмотрение лечащего врача или ветеринара. В частности, объект, подлежащий лечению, представляет собой млекопитающее, в частности человека.
Агент можно вводить в суточной дозе. Это количество можно вводить в виде однократной дозы в день или чаще обычно в количестве (например, двух, трех, четырех, пяти или шести) частей дозы в день, так что общая суточная доза остается такой же.
Соответственно, количество соединения изобретения, вводимое в соответствии с настоящим изобретением, будет представлять собой количество, выбранное от 0,01 мг до 5 г в день.
Кроме того, соединения изобретения можно вводить в виде пролекарств. Используемое в настоящем описании «пролекарство» соединения изобретения представляет собой функциональное производное соединения, которое при введении пациенту в конечном итоге высвобождает соединение изобретения in vivo. Введение соединения изобретения в виде пролекарства может позволить специалисту в данной области техники выполнить одно или более из следующего: (а) изменить начало активности соединения in vivo; (b) изменить продолжительность действия соединения in vivo; (c) изменить транспорт или распределение соединения in vivo; (d) изменить растворимость соединения in vivo и (e) преодолеть побочный эффект или другие трудности, с которыми сталкивается соединение. Типичные функциональные производные, используемые для получения пролекарств, включают модификации соединения, которые химически или ферментативно расщепляются in vivo. Такие модификации, которые включают получение фосфатов, амидов, сложных эфиров, тиоэфиров, карбонатов и карбаматов, хорошо известны специалисту в данной области техники.
Определенные и строго неограничивающие примеры пролекарств в соответствии со смыслом настоящего изобретения включают производные соединений формул (IA), (IB), (IC), (ID) и (I), в которых гидроксильный фрагмент замещен реакционноспособным фрагментом, который способен распадаться in vivo (например, путем гидролиза) с образованием гидроксильного фрагмента. Во избежание сомнений, гидроксильный фрагмент формул (IA), (IB), (IC) и (ID) обозначен меткой «I» в химических формулах, и гидроксильный фрагмент формулы (I) присоединен к атому углерода, который находится между фенильным кольцом и пропильной боковой группой соединения. Подобным образом настоящее изобретение включает производные соединений формулы (II), в которых Z представляет собой реакционноспособный фрагмент, который способен распадаться in vivo (например, путем гидролиза) с образованием гидроксильного фрагмента. Строго неограничивающие и просто типичные примеры таких реакционноспособных фрагментов включают фрагменты формулы ORz, в которой Rz имеет формулу PO3H2, CO(CH2)nNH2 и PO3H(CH2)n-NH2 (в которой n представляет собой целое число, например от 2 до 5). Специалисту в данной области техники будет очевидно, что можно также использовать множество других реакционноспособных фрагментов. В соединении, содержащем множество гидроксильных групп, конечно, возможно замещение некоторых или всех указанных гидроксильных групп такими реакционноспособными фрагментами.
Соединения изобретения можно использовать отдельно или в комбинации с другими терапевтическими агентами. Соединения изобретения и другой фармацевтически активный агент(ы) можно вводить вместе или по отдельности, и при раздельном введении введение может происходить одновременно или последовательно в любом порядке с помощью любого подходящего способа в отдельных или комбинированных фармацевтических композициях.
Количества соединения(й) изобретения и другого фармацевтически активного агента(ов) и относительные сроки введения будут выбраны для достижения желаемого комбинированного терапевтического эффекта. Соединения настоящего изобретения и дополнительный терапевтический агент(ы) можно использовать в комбинации с помощью одновременного введения в единой фармацевтической композиции, включающей оба соединения. Альтернативно, комбинацию можно вводить отдельно в отдельных фармацевтических композициях, каждая из которых включает одно из соединений последовательным образом, в которой, например, соединение изобретения вводят первым и другое вводят вторым и наоборот. Такое последовательное введение может быть близким по времени (например, одновременно) или отдаленным по времени. Кроме того, не имеет значения, вводятся ли соединения в одной и той же лекарственной форме, например, одно соединение можно вводить местно и другое соединение можно вводить перорально. Соответственно, оба соединения вводят перорально.
Комбинации могут быть представлены в виде комбинированного набора. Под термином «комбинированный набор» «или набор из частей», используемым в настоящем описании, подразумевается фармацевтическая композиция или композиции, которые используются для введения комбинации в соответствии с изобретением. Когда оба соединения вводят одновременно, комбинированный набор может содержать оба соединения в одной фармацевтической композиции, такой как таблетка или в отдельных фармацевтических композициях. Когда соединения не вводят одновременно, комбинированный набор будет содержать каждое соединение в отдельных фармацевтических композициях или в одной упаковке, или в отдельных фармацевтических композициях в отдельных упаковках.
Комбинированный набор также может быть предоставлен с инструкцией, такой как инструкции по дозировке и введению. Такие инструкции по дозировке и введению могут быть такого рода, которые предоставляются врачу, например, на этикетке лекарственного препарата, или они могут быть такого рода, которые предоставляются врачом, например, инструкции для пациента.
При введении комбинации отдельно последовательным образом, когда одну вводят первой и другую вводят второй или наоборот, такое последовательное введение может быть близким по времени или отдаленным по времени. Например, включается введение другого агента от нескольких минут до нескольких десятков минут после введения первого агента и введение другого агента от нескольких часов до нескольких дней после введения первого агента, при этом промежуток времени не ограничен. Например, один агент можно вводить один раз в день и другой агент можно вводить 2 или 3 раза в день или один агент можно вводить один раз в неделю и другой агент можно вводить один раз в день и подобное.
Специалисту в данной области техники будет очевидно, что в случае необходимости другой терапевтический ингредиент(ы) можно использовать в форме солей, например, в виде солей щелочного металла или амина, или в виде кислотно-аддитивных солей, или пролекарств, или в виде сложных эфиров, например, низших алкиловых эфиров, или в виде сольватов, например, гидратов для оптимизации активности, и/или стабильности, и/или физических характеристик, таких как растворимость терапевтического ингредиента. Также будет ясно, что в случае необходимости терапевтические ингредиенты можно использовать в оптически чистой форме.
При объединении в единую композицию следует понимать, что два соединения должны быть стабильными и совместимыми друг с другом и другими компонентами композиции и могут быть составлены для введения. При раздельном приготовлении они могут быть предоставлены в любой подходящей композиции таким образом, как известно в данной области техники для таких соединений.
Когда соединение изобретения используют в комбинации со вторым терапевтическим агентом, активным в отношении того же заболевания, состояния или нарушения, доза каждого соединения может отличаться от дозы, когда соединение используют отдельно. Соответствующие дозы легко оценит специалист в данной области техники.
В одном варианте осуществления млекопитающее в способах и применениях настоящего изобретения представляет собой человека.
Подробное описание соединений настоящего изобретения
Соединение настоящего изобретения представляет собой соединение, которое способно ингибировать полимеризацию α1-антитрипсина. Обычно соединение способно ингибировать полимеризацию Z-α1-антитрипсина.
Соединение обычно представляет собой низкомолекулярное соединение. Используемое в настоящем описании низкомолекулярное соединение обычно имеет молекулярную массу 2000 дальтон или менее, чаще 1000 дальтон или менее и предпочтительно 800 дальтон или менее. Более предпочтительно молекулярная масса составляет 600 или менее и наиболее предпочтительно 500 или менее. Например, низкомолекулярное лекарственное средство может иметь молекулярную массу от 250 до 800, например, от 300 до 600.
В одном аспекте настоящего изобретения соединение, которое способно ингибировать полимеризацию α1-антитрипсина, представляет собой соединение общей формулы (II):
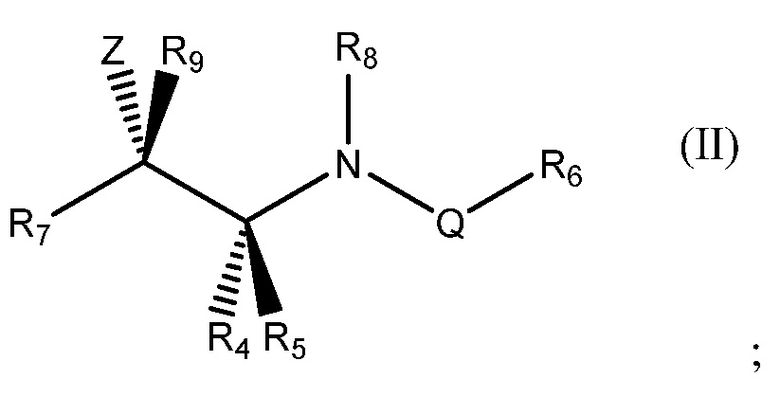
в которой:
- R4, R5, R6 и R7 представляют собой, как определено в другом месте в настоящем описании (например, в отношении общей формулы (ID));
- R8 представляет собой водород, дейтерий, алкил (например, C1-5алкил) или дейтерированный алкил (например, C1-5дейтерированный алкил);
- R9 представляет собой водород, дейтерий, алкил (например, C1-5алкил) или дейтерированный алкил (например, C1-5дейтерированный алкил);
- Z представляет собой OH, F, -NHCHO, -CH2F, -CHF2 или CF3 и
- Q представляет собой -C(=O)-, -C(=S)-, -C(=NOH)-, -C(=NNH2)- или -S(O)2-.
Примеры предпочтительных групп R4, R5, R6 и R7 представляют собой, как определено в другом месте в настоящем описании.
Предпочтительные группы R8 представляют собой водород, дейтерий и C1-2 необязательно дейтерированный алкил. Более предпочтительно R8 представляет собой водород, метил или этил, еще более предпочтительно водород или метил и наиболее предпочтительно водород.
Предпочтительные группы R9 представляют собой водород, дейтерий и C1-2 необязательно дейтерированный алкил. Более предпочтительно R9 представляет собой водород, метил или этил, еще более предпочтительно водород или метил и наиболее предпочтительно водород.
Z представляет собой предпочтительно ОН или F, наиболее предпочтительно ОН.
Q представляет собой предпочтительно -C(=O)-, -C(=S)- или -S(O)2-, более предпочтительно -C(=O)- или -C(=S)- и наиболее предпочтительно -C(=O)-.
В предпочтительных вариантах осуществления соединение общей формулы (II) представляет собой соединение общей формулы (ID) и еще более предпочтительно соединение общей формулы (I), как дополнительно определено в другом месте в настоящем описании.
Связывание соединения с α1-антитрипсином
Соединение настоящего изобретения способно связываться с α1-антитрипсином. Обычно соединение способно связываться с α1-антитрипсином через криптический сайт связывания в структуре белка α1-антитрипсина. Криптический сайт связывания, как определено в настоящем описании, относится к сайту связывания, который отсутствует, закрыт или доступен только временно в несвязанном белке, но присутствует, когда соединение связано с белком; например, криптический сайт связывания может возникать как следствие взаимодействия белка с соединением, и/или ранее только временно доступная конфигурация белка, показывающая сайт связывания, может стабилизироваться за счет присутствия белка.
Под «α1-антитрипсином» в выражении «соединение способно связываться с α1-антитрипсином» подразумевается белок, который содержит последовательность SEQ ID NO: 1, а именно
X1DPQGDAAQKTDTSHHDQDHPTFNKITPNLAEFAFSLYRQLAHQSNSTNIFFSPVSIATAFAMLSLGTKADTHDEILEGLNFNLTEIPEAQIHEGFQELLX2TLNQPDSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQX3TTVKVPMMKRLGMFNIQHX4KKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDX5KGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLMIX6QNTKSPLFMGKVVNPTQK
в которой X1 представляет собой T или E, X2 представляет собой R или H, X3 представляет собой A или V, X4 представляет собой C или S, X5 представляет собой E или K и X6 представляет собой D или E.
Во избежание сомнений, соединение представляет собой соединение изобретения, если оно способно связываться с любым таким белком, т. е. любым отдельным белком, который содержит последовательность SEQ ID NO: 1 (т. е. необязательно, что соединение должно связываться со всеми возможными вариантами, охватываемыми SEQ ID NO: 1, и такое связывание не исключает возможности, что соединение также может связываться с другими белками, такими как белки, содержащие аминокислотные последовательности, которые отличаются от SED ID NO: 1).
Кроме того, также во избежание сомнений, все номера остатков, приписываемые конкретным аминокислотам в белке α1-антитрипсина, например, в отношении определения сайта связывания соединений настоящего изобретения отсчитываются от N-конца последовательности SEQ ID NO: 1, т. е. X1 считается как остаток 1, соседний D как остаток 2, соседний P как остаток 3, соседний Q как остаток 4, соседний G как остаток 5 и т. д. Данная нумерация применяется также, когда белок содержит дополнительные аминокислоты на N-конце по отношению к SEQ ID NO: 1, т. е. любые такие дополнительные «фланкирующие» аминокислоты не изменяют систему нумерации, при этом X1 по-прежнему считается как остаток 1, D как остаток 2, P как остаток 3, Q как остаток 4, G как остаток 5 и т. д. Таким образом, нумерация всегда учитывается только со ссылкой на аминокислоты SEQ ID NO: 1 и исключает любые дополнительные аминокислотные остатки, присутствующие, например, в любом сигнальном пептиде или аффинной метке и/или любых дополнительных аминокислотах, предоставляемых вектором экспрессии, используемым в синтезе белка. Следовательно, в любом соответствующем белке X1 представляет собой аминокислоту 1, X4 представляет собой аминокислоту 232, X5 представляет собой аминокислоту 342 и т. д.
Наиболее распространенным аллелем M-AT дикого типа является аллель M1V (оценивается в 44-49% от общего числа). В аллеле M1V X1 представляет собой E, X2 представляет собой R, X3 представляет собой V, X4 представляет собой C, X5 представляет собой E и X6 представляет собой E. Полный человеческий белок M1V дополнительно содержит сигнальный пептид на N-конце, имеющий последовательность MPSSVSWGILLLAGLCCLVPVSLA. Таким образом, полная последовательность человеческого белка M1V соответствует последовательности SEQ ID NO: 2, а именно
MPSSVSWGILLLAGLCCLVPVSLAEDPQGDAAQKTDTSHHDQDHPTFNKITPNLAEFAFSLYRQLAHQSNSTNIFFSPVSIATAFAMLSLGTKADTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQPDSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHCKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLMIEQNTKSPLFMGKVVNPTQK.
Мутант Z-α1-антитрипсина отличается от аллеля M1V тем, что X5 представляет собой K, а не E (т. е. мутация определяется как E342K) и X3(213) представляет собой A. Таким образом, полная последовательность Z-A1AT соответствует последовательности SEQ ID NO: 3, а именно
MPSSVSWGILLLAGLCCLVPVSLAEDPQGDAAQKTDTSHHDQDHPTFNKITPNLAEFAFSLYRQLAHQSNSTNIFFSPVSIATAFAMLSLGTKADTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQPDSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQATTVKVPMMKRLGMFNIQHCKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDKKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLMIEQNTKSPLFMGKVVNPTQK.
Для простоты обращения (например, для получения кристаллов белка α1-антитрипсина и/или создания комплексов белка α1-антитрипсина, связанного с соединением изобретения) может быть желательно мутировать цистеин 232 белка в серин, т. е. произвести мутацию C232S. Это консервативная мутация, и она не находится в положении, непосредственно связанном с сайтом связывания соединения. SEQ ID NO: 1 охватывает оба белка, принимая во внимание его вариабельный аминокислотный остаток X4.
В предпочтительном варианте осуществления соединение способно связываться с белком, содержащим SEQ ID NO: 1, в которой X1 представляет собой E, X2 представляет собой R, X3 представляет собой A, X4 представляет собой C, X5 представляет собой K и X6 представляет собой E, т. е. последовательность имеет 100% идентичность последовательности соответствующей части Z-α1-антитрипсина.
В другом примерном варианте осуществления белок может представлять собой последовательность, которая содержит SEQ ID NO: 1 и которая дополнительно содержит аффинную метку (например, аффинную метку гексагистидина) и/или аминокислоты, предоставляемые вектором экспрессии. Например, такой белок может быть экспрессирован в E. coli. Полная последовательность одного такого примерного белка представляет собой последовательность SEQ ID NO: 4, а именно
MRGSHHHHHHTDPQGDAAQKTDTSHHDQDHPTFNKITPNLAEFAFSLYRQLAHQSNSTNIFFSPVSIATAFAMLSLGTKADTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQPDSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHSKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLMIEQNTKSPLFMGKVVNPTQK.
Этот белок, который используется в Примере 2 в настоящем описании, содержит N-концевую последовательность MRGSHHHHHHT, имеющую аффинную метку гексагистидина, и аминокислоты, предоставляемые вектором экспрессии, используемым в синтезе, и в которой X1 представляет собой T. Этот белок дополнительно содержит серин в положении 232. (т. е. X4 представляет собой S) для содействия простоте обращения. Этот белок дополнительно содержит глутамат в положении 342 (т. е. X5 представляет собой E).
Важным открытием настоящего изобретения является то, что исследуемые соединения способны к специфическому связыванию с белком α1-антитрипсином в части белка, которая ранее не была продемонстрирована как место связывания лекарственного средства (и тем более та, которая при образовании связывания соединение-белок способна ингибировать полимеризацию белка).
Более определенно, указанный сайт связывания предпочтительно расположен между β-листом-A и β-листом-B указанного α1-антитрипсина. Указанный β-лист-A содержит аминокислоты, соответствующие одному или более из: (i) остатков 140-144; (ii) остатков 111-121; (iii) остатков 181-191; (iv) остатков 330-340 и (v) остатков 292-299 SEQ ID NO: 1. Предпочтительно указанный β-лист-A содержит аминокислоты, соответствующие двум или более из, более предпочтительно трем или более, еще более предпочтительно четырем или более и наиболее предпочтительно всем пяти из (i)-(v). Указанный β-лист-B содержит аминокислоты, соответствующие одному или более из: (i') остатков 228-231; (ii') остатков 236-244; (iii') остатков 248-256; (iv') остатков 369-376; (v') остатков 381-389 и (vi') остатков 49-53 SEQ ID NO: 1. Предпочтительно указанный β-лист-B содержит аминокислоты, соответствующие двум или более из, более предпочтительно трем или более, еще более предпочтительно четырем или более, еще более предпочтительно пяти или более и наиболее предпочтительно всем шести из (i')-(vi').
Таким образом, предпочтительно сайт связывания расположен между β-листом-A и β-листом-B указанного α1-антитрипсина, в котором: указанный β-лист-A содержит аминокислоты, соответствующие остаткам 140-144, 111-121, 181-191, 330-340 и 292-299 SEQ ID NO: 1; и указанный β-лист-B содержит аминокислоты, соответствующие остаткам 228-231, 236-244, 248-256, 369-376, 381-389 и 49-53 SEQ ID NO: 1.
Под «расположенным между β-листом-A и β-листом-B» подразумевается, когда соединение связано с α1-антитрипсином, по меньшей мере, часть (например, практически все или все) соединения размещается в физическом пространстве между указанным β-листом-A и указанным β-листом-B. Специалисту в данной области техники будет очевидно, что подтверждение местоположения сайта связывания соединения на белке может быть обеспечено способами, хорошо известными в данной области техники, такими как применение стандартных кристаллографических методик (см., например, Пример 2 в настоящем описании).
Еще более определенно сайт связывания более предпочтительно расположен между аминокислотными цепями, соответствующими трем или более из: (а) остатков 191-194 SEQ ID NO: 1; (b) остатков 288-293 SEQ ID NO: 1; (c) остатков 371-374 SEQ ID NO: 1; (d) остатков 249-253 SEQ ID NO: 1 и (e) остатков 240-243 SEQ ID NO: 1. Особенно предпочтительно, что сайт связывания располагается между аминокислотными цепями, соответствующими четырем или более и наиболее предпочтительно каждому из: (a) остатков 191-194 SEQ ID NO: 1; (b) остатков 288-293 SEQ ID NO: 1; (c) остатков 371-374 SEQ ID NO: 1; (d) остатков 249-253 SEQ ID NO: 1 и (e) остатков 240-243 SEQ ID NO: 1. Кроме того, предпочтительно, что сайт связывания также располагается между аминокислотными цепями, соответствующими (f) остаткам 338-341 SEQ ID NO: 1. Под «аминокислотой цепью» подразумевается множество аминокислотных остатков в белке, как пронумеровано в настоящем описании, например, аминокислотная цепь, соответствующая остаткам 191-194 SEQ ID NO: 1, относится к аминокислотам 191, 192, 193 и 194 SEQ ID NO: 1. Под «расположенным между» указанными аминокислотными цепями подразумевается, когда соединение связано с α1-антитрипсином, по меньшей мере, часть (например, практически все или все) соединения размещается в физическом пространстве между соответствующими аминокислотными цепями.
В примерном аспекте сайт связывания содержит один или более из W194, Y244, L291, P289, F252, K290, I293, L338, I340, F372 и M374 SEQ ID NO: 1. В этом примерном аспекте сайт связывания предпочтительно содержит три или более, более предпочтительно пять или более, еще более предпочтительно семь или более и более предпочтительно девять или более из W194, Y244, L291, P289, F252, K290, I293, L338, I340, F372 и M374 SEQ ID NO: 1. Сайт связывания может, например, содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или все 11 из W194, Y244, L291, P289, F252, K290, I293, L338, I340, F372 и M374 SEQ ID NO: 1. Особенно предпочтительными аминокислотами, связанными с сайтом связывания, являются W194, Y244, L291 и P289, и, таким образом, предпочтительно сайт связывания содержит, по меньшей мере, W194, Y244, L291 и P289. Как дополнительно описано в настоящей заявке, соединение может оптимально быть способным к образованию нековалентных связей (включая, но не ограничиваясь ими, водородные связи) с функциональными группами соответствующих аминокислотных остатков белка.
Предпочтительно, KD соединения по отношению к M-α1-антитрипсину составляет менее приблизительно 250 нМ, причем указанный M-α1-антитрипсин содержит последовательность SEQ ID NO: 2 и/или KD соединения по отношению к Z-α1-антитрипсину составляет менее приблизительно 25 нМ, причем указанный Z-α1-антитрипсин содержит последовательность SEQ ID NO: 3. Предпочтительно, KD соединения по отношению к Z-α1-антитрипсину, по меньшей мере, в десять раз ниже, чем KD соединения по отношению к M-α1-антитрипсину, причем указанный M-α1-антитрипсин содержит последовательность SEQ ID NO: 2 и указанный Z-α1-антитрипсин содержит последовательность SEQ ID NO: 3. Способы измерения KD хорошо известны в данной области техники, и можно использовать любой такой способ. Один примерный такой способ описан в Примере 2. Все ссылки на KD в настоящем описании определены при комнатной температуре (например, при 20°C).
Предпочтительные структурные элементы, способствующие связыванию белка
В иллюстративном аспекте настоящего раскрытия соединение содержит β-гидроксиамидный фрагмент, т. е. фрагмент формулы -C(OH)(X)-CX2-NH-C(O)-, в которой каждый X независимо представляет собой заместитель (например, любой конкретный заместитель, как определено в другом месте в настоящем описании).
Важно отметить, что было обнаружено, что такой структурный мотив β-гидроксиамида значительно способствует способности настоящих соединений связываться с α1-антитрипсином в данном сайте связывания. В частности, такой фрагмент может обеспечивать связывание с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) β-гидроксильной группой и L291 SEQ ID NO: 1; (ii) NH карбоксамидной группы и P289 SEQ ID NO: 1 и (iii) карбонилом карбоксамидной группы и Y244 SEQ ID NO: 1.
Соответственно, предпочтительно, что соединение содержит четырехвалентный фрагмент формулы (IA)
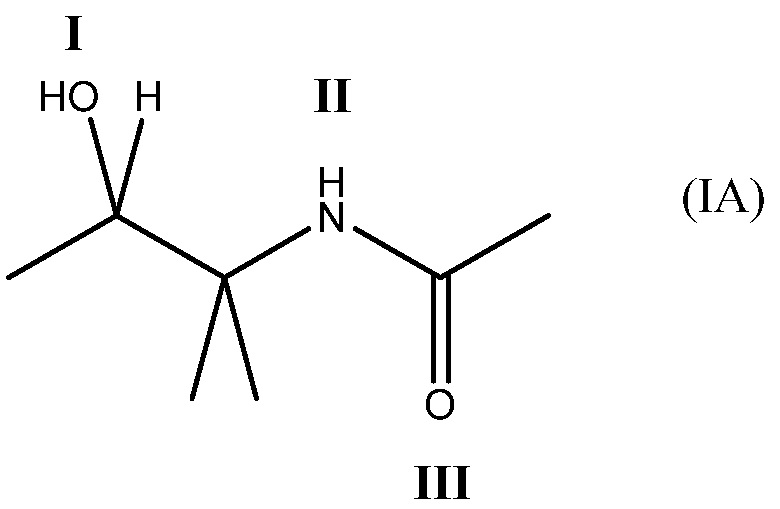
в которой соединение способно связываться с α1-антитрипсином, содержащим последовательность SEQ ID NO: 1, с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1.
Под четырехвалентным подразумевается, что фрагмент формулы (IA) содержит четыре точки связывания, т. е. в положениях 1, 2, 3 и 4, указанных ниже:
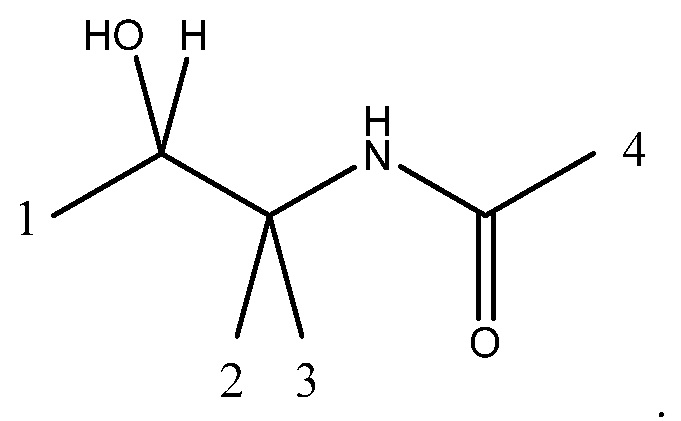
Более предпочтительно соединение содержит двухвалентный фрагмент формулы (IB)
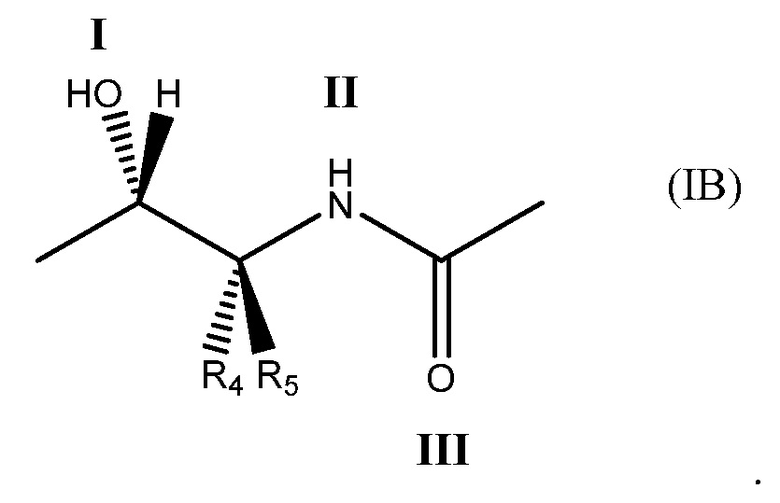
Под двухвалентным подразумевается, что фрагмент формулы (IB) содержит две точки связывания, т. е. в положениях 1 и 2 ниже.
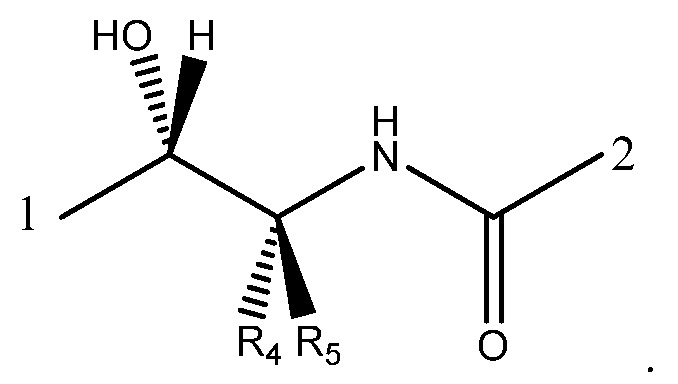
В формуле (IB), как в формуле (IA), обычно соединение способно связываться с α1-антитрипсином с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1.
R4 и R5 могут быть одинаковыми или отличаться друг от друга. Они независимо выбраны из водорода, C1-5алкила, C2-5алкенила, C2-5алкинила и C1-4алкокси.
R5 представляет собой предпочтительно водород, метил, этил, этенил, этинил или метокси. Более предпочтительно R5 представляет собой водород, метил или этил. Наиболее предпочтительно R5 представляет собой водород.
R4 представляет собой предпочтительно C2-4алкил, C2-4алкенил, C2-4алкинил или C1-3алкокси. Более предпочтительно R4 представляет собой н-пропил, -CH=CH-CH3, -C-C=CH2 или этокси. Наиболее предпочтительно R4 представляет собой н-пропил.
Особенно предпочтительная комбинация R5 и R4 представляет собой, когда R5 представляет собой водород и R4 представляет собой н-пропил. Было обнаружено, что присутствие единственного н-пропильного заместителя у углерода α карбоксамида особенно предпочтительно для обеспечения возможности связывания соединения с α1-антитрипсином в соответствующем сайте связывания.
Еще более предпочтительно соединение содержит одновалентный фрагмент формулы (IC)
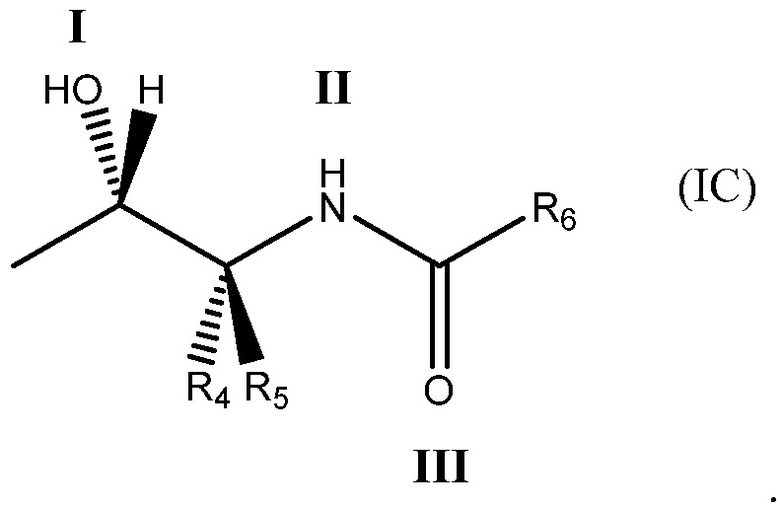
Под одновалентным подразумевается, что фрагмент формулы (IC) содержит одну точку связывания, т. е. в положении 1 ниже
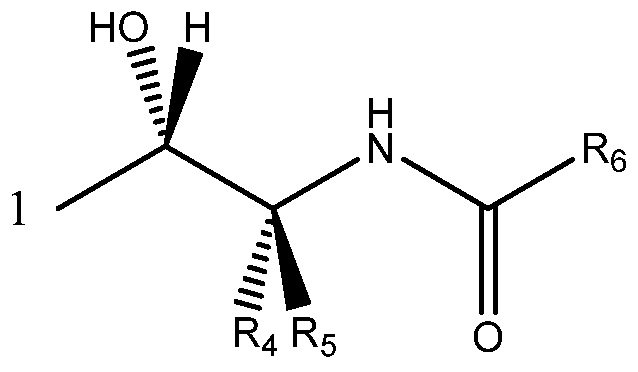
В формуле (IC), как в формулах (IA) и (IB), обычно соединение способно связываться с α1-антитрипсином с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1. R4 и R5 представляют собой, как определено со ссылкой на формулу (IB). R6 представляет собой замещенную или незамещенную арильную или гетероарильную группу. Обычно R6 способен складываться в стопку с боковой цепью W194 SEQ ID NO: 1, когда соединение связано с указанным α1-антитрипсином.
R6 является предпочтительно замещенным или незамещенным и представляет собой: (а) C6-10арильную группу или (b) гетероарильную группу, которая содержит от 5 до 10 атомов кольца. Более предпочтительно R6 является замещенным или незамещенным и представляет собой: (а) бикарбоциклическую ароматическую группу или (b) бициклическую гетероарильную группу.
Когда R6 представляет собой арильную группу, особенно предпочтительные группы включают нафтил и инданил (в обоих случаях замещенный или незамещенный и в обоих случаях, когда необязательно один атом углерода в кольце может быть замещен карбонильной группой), наиболее предпочтительно инданил (необязательно когда один атом углерода в кольце может быть замещен карбонильной группой).
Когда R6 представляет собой гетероарильную группу, предпочтительные группы включают оксиндолил (например, 2-оксиндолил или 3-оксиндолил), бензотиенил, бензофуранил, бензимидазолил, бензотиазолил, бензизотиазолил, бензоксазолил, бензизоксазолил, бензотриазолил, индолил, изоиндолил и индазолил (во всех случаях замещенный или незамещенный и во всех случаях, когда необязательно один атом углерода в кольце может быть замещен карбонильной группой) и более предпочтительные группы включают оксиндолил (например, 2-оксиндолил или 3-оксиндолил), бензотиенил, бензофуранил, бензимидазолил, бензотиазолил, бензоксазолил и индолил (во всех случаях замещенный или незамещенный и во всех случаях, когда необязательно один атом углерода в кольце может быть замещен карбонильной группой).
Для любой арильной или гетероарильной группы, в которой один атом углерода в кольце замещен карбонильной группой, полученное кольцевое звено представляет собой -C(O)-. Когда «исходный» атом углерода в кольце образует двойную связь с соседним атомом кольца, замещение этого атома углерода в кольце -C(O)- сопровождается насыщением указанного соседнего атома кольца, например, с помощью добавления к нему атома водорода или необязательного заместителя, как определено в настоящем описании.
Особенно предпочтительные R6-группы представляют собой замещенные или незамещенные бициклические арильные и гетероарильные группы, содержащие шестичленное кольцо, конденсированное с пятичленным кольцом (т. е. содержащее 9 атомов кольца). Из таких R6-групп предпочтительно атом кольца пятичленного кольца, который не является смежным ни с одним из атомов кольца, общих для пятичленного кольца и шестичленного кольца, представляет собой атом углерода, несущий карбонильную группу, т. е. представляет собой -C(O)-. Например, предпочтительные R6-группы включают следующие группы от 6-1 до 6-12:
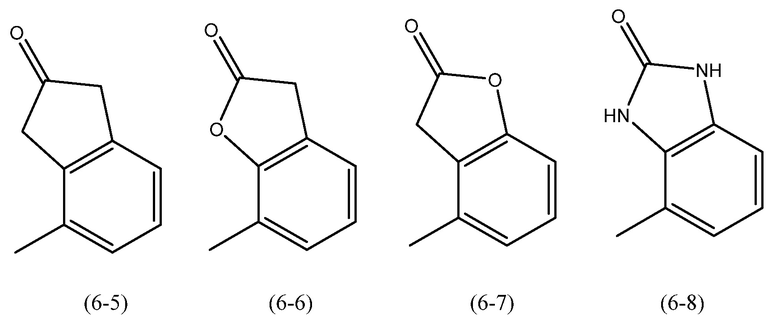
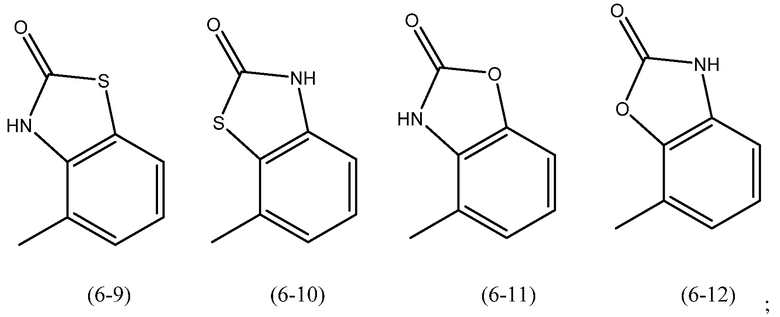
в которой одна или более H-групп, связанных с атомами кольца (предпочтительно атомами углерода кольца), могут быть замещены заместителями.
Еще более предпочтительно R6 представляет собой замещенную или незамещенную 4-оксиндолильную группу (т. е. 4-(2-оксиндолильную) группу), которая представляет собой группу 6-1, указанную выше.
Предпочтительно R6-группа является незамещенной или замещенной от 1 до 4 заместителями, например, 1 или 2 заместителями. Более предпочтительно R6-группа является незамещенной или замещенной 1 или 2 заместителями. Наиболее предпочтительно R6-группа является незамещенной или замещенной 1 заместителем.
Необязательные заместители для R6 включают заместители, определенные в разделе «Определения» в настоящем описании. Особенно предпочтительные заместители для R6 включают атомы галогена, C1-5алкил, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила) и гидроксил. Еще более особенно предпочтительные заместители включают F, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Cl, Br и I, и еще более предпочтительные заместители представляют собой F, CH3, NH2, OH и Cl. Наиболее предпочтительные заместители представляют собой F и Cl (например, F).
В примерном аспекте R6 представляет собой группу формулы R6'
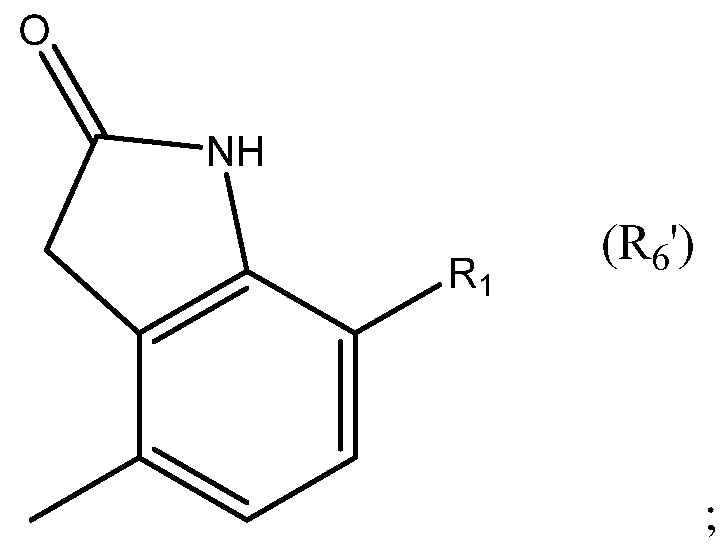
в которой R1 выбран из группы, состоящей из H, F, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Cl, Br и I. В формуле R6' R1 более предпочтительно выбран из группы, состоящей из H, F, CH3, NH2, OH и Cl, и еще более предпочтительно выбран из группы, состоящей из H и F.
Особенно предпочтительно, что соединение имеет формулу (ID)
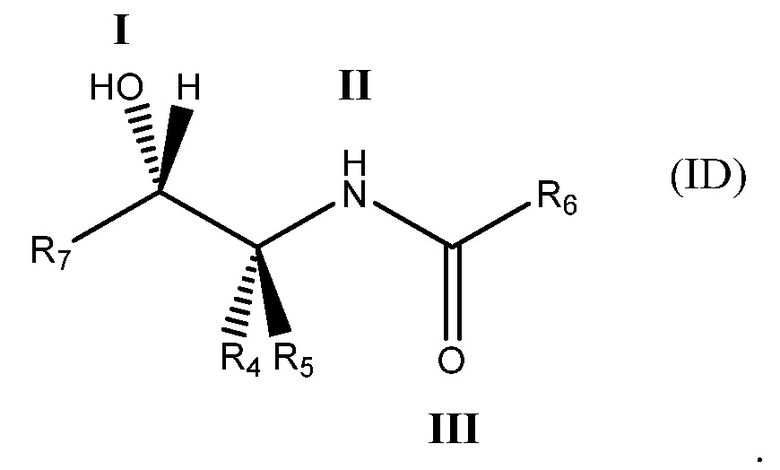
В формуле (ID), как в формулах (IA), (IB) и (IC), обычно соединение способно связываться с α1-антитрипсином с помощью образования водородной связи между: (i) гидроксильной группой I и L291 SEQ ID NO: 1; (ii) NH-группой II и P289 SEQ ID NO: 1 и (iii) карбонильной группой III и Y244 SEQ ID NO: 1. R4 и R5 представляют собой, как определено со ссылкой на формулу (IB). R6 представляет собой, как определено со ссылкой на формулу (IC).
R7 в формуле (ID) представляет собой замещенную или незамещенную арильную или гетероарильную группу. Предпочтительно R7 представляет собой замещенную или незамещенную фенильную, пиридинильную, пиридазинильную, пиримидинильную, пиразинильную или триазинильную группу. Более предпочтительно R7 представляет собой замещенную или незамещенную фенильную группу.
R7 предпочтительно замещен 1, 2, 3, 4 или 5 заместителями, более предпочтительно 1, 2 или 3 заместителями и наиболее предпочтительно 2 заместителями. Необязательные заместители для R7 включают заместители, определенные в разделе «Определения» в настоящем описании. Особенно предпочтительные заместители R7 включают атомы галогена и C1-5алкил, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксил, -SH и -CN-группы.
Особенно предпочтительный R7 представляет собой фенильную группу, замещенную 1, 2, 3, 4 или 5 (предпочтительно, по меньшей мере, 2) заместителями. Предпочтительно такая фенильная группа содержит, по меньшей мере, мета-заместитель и орто-заместитель, т. е. представляет собой формулу R7″.
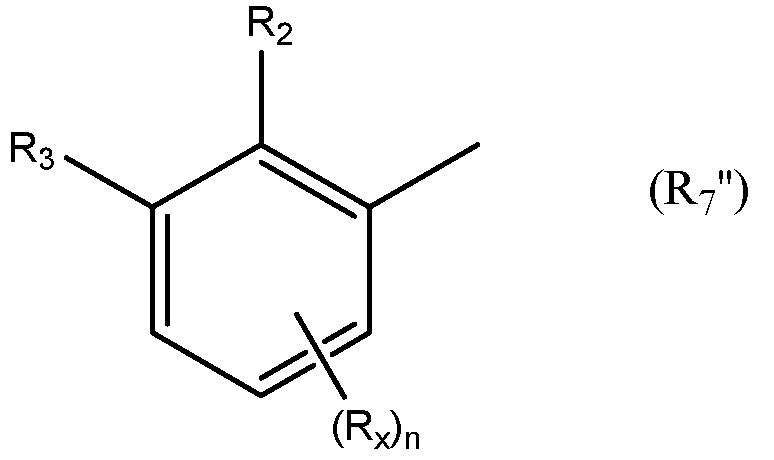
в которой R2 и R3 представляют собой заместители, каждый Rx представляет собой независимо выбранный заместитель и n представляет собой целое число от 0 до 3. Более предпочтительно такая фенильная группа имеет формулу R7'
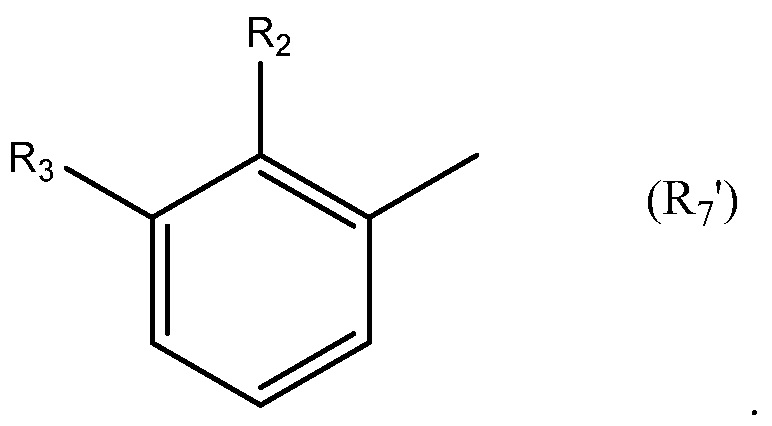
Предпочтительно в R7' и R7″ каждый из R2, R3 и любой Rx независимо выбран из атомов галогена и C1-5алкила, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, -SH и -CN-групп. Более предпочтительно R2 выбран из группы, состоящей из CH3, Cl, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, SH, CN, F, Br и I; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Br, I и SH. Еще более предпочтительно R2 выбран из группы, состоящей из CH3, Cl, NH2, OH, SH, CN и F; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, NH2, OH и SH. Наиболее предпочтительно R2 выбран из группы, состоящей из CH3 и Cl; и R3 выбран из группы, состоящей из F, Cl и CN.
Предпочтительной комбинацией R4, R5, R6 и R7 в формуле (ID) является комбинация, в которой:
- R4 и R5 независимо выбраны из водорода, C1-5алкила, C2-5алкенила, C2-5алкинила и C1-4алкоксила;
- R6 представляет собой C6-10арильную группу или гетероарильную группу, которая содержит от 5 до 10 атомов кольца, причем указанная арильная или гетероарильная группа является незамещенной или замещенной одним или более (например, от 1 до 5) заместителями, выбранными из атомов галогена и C1-5алкила, C1-4алкокси, C1-4алкилтиола, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, тиола (SH), нитрила (CN), нитро и сульфокислотных групп; и
- R7 представляет собой фенильную, пиридинильную, пиридазинильную, пиримидинильную, пиразинильную или триазинильную группу, которая является незамещенной или замещенной одним или более (например, от 1 до 4) заместителями, выбранными из атомов галогена и C1-5алкила, C1-4алкокси, C1-4алкилтиола, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, тиола (SH), нитрила (CN), нитро и сульфокислотных групп.
Более предпочтительной комбинацией R4, R5, R6 и R7 в формуле (ID) является комбинация, в которой:
- R4 представляет собой C2-4алкил, C2-4алкенил, C2-4алкинил или C1-3алкокси;
- R5 представляет собой водород, метил, этил, этенил, этинил или метокси;
- R6 выбран из следующих групп от 6-1 до 6-12:
в которой одна или более (например, от 1 до 3) H-групп, связанных с атомами кольца, необязательно замещены заместителями, выбранными из атомов галогена и C1-5алкила, C1-4алкокси, C1-4алкилтиола, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, тиола (SH), нитрила (CN), нитро и сульфокислотных групп; и
- R7 представляет собой фенильную, пиридинильную, пиридазинильную, пиримидинильную, пиразинильную или триазинильную группу, которая является незамещенной или замещенной одним или более (например, от 1 до 4) заместителями, выбранными из атомов галогена и C1-5алкила, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, -SH и -CN-групп.
Еще более предпочтительной комбинацией R4, R5, R6 и R7 в формуле (ID) является комбинация, в которой:
- R4 представляет собой н-пропил, -CH=CH-CH3, -C-C=CH2 или этокси;
- R5 представляет собой водород, метил или этил;
- R6 представляет собой 4-(2-оксиндолильную) группу, которая является незамещенной или замещенной одним или более (например, от 1 до 3) заместителями, выбранными из атомов галогена и C1-5алкила, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила) и гидроксильных групп; и
- R7 представляет собой группу формулы R7″
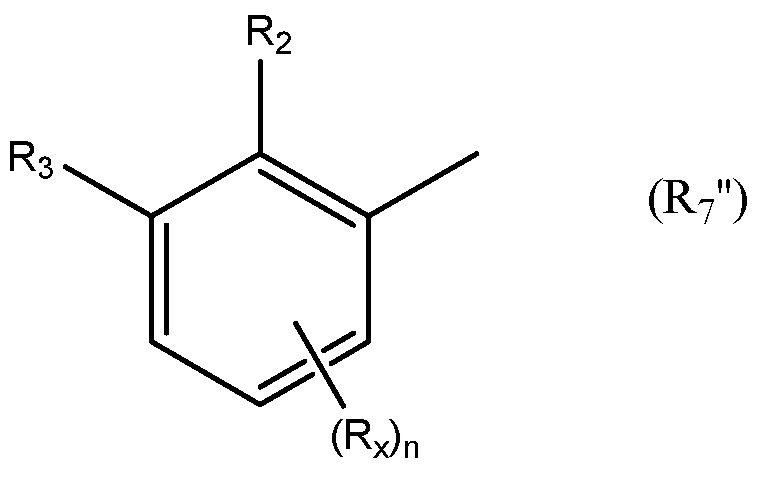
- в которой R2 и R3 представляют собой заместители, каждый Rx представляет собой независимо выбранный заместитель, n представляет собой целое число от 0 до 3 и каждый заместитель независимо выбран из атомов галогена и C1-5алкила, -NR2 (в котором каждый R независимо выбран из H и C1-5алкила), гидроксила, -SH и -CN-групп.
В одном иллюстративном аспекте соединение имеет формулу (I)
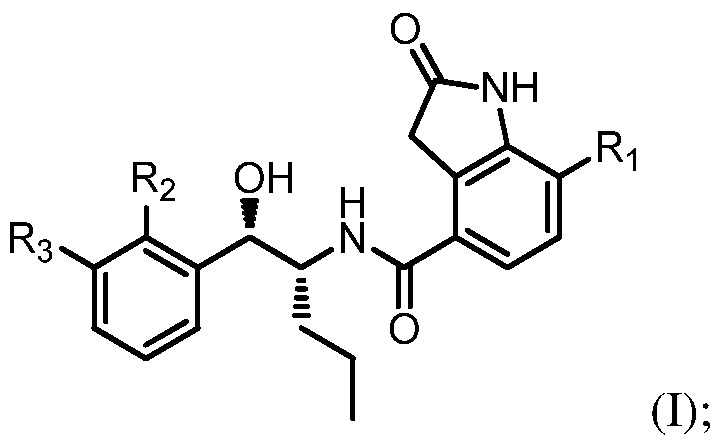
в которой
- R1 выбран из группы, состоящей из H, F, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Cl, Br и I;
- R2 выбран из группы, состоящей из CH3, Cl, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, SH, CN, F, Br и I; и
- R3 выбран из группы, состоящей из F, Cl, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, NH2, NHCH3, N(CH3)2, OH, Br, I и SH.
Предпочтительно в формуле (I) R1 выбран из группы, состоящей из H, F, CH3, NH2, OH и Cl. Предпочтительно в формуле (I) R2 выбран из группы, состоящей из CH3, Cl, NH2, OH, SH, CN и F. Предпочтительно в формуле (I) R3 выбран из группы, состоящей из F, Cl, CN, CH3, NH2, OH и SH. Предпочтительно в формуле (I) R1 выбран из группы, состоящей из H, F, CH3, NH2, OH и Cl; и R2 выбран из группы, состоящей из CH3, Cl, NH2, OH, SH, CN и F; и R3 выбран из группы, состоящей из F, Cl, CN, CH3, NH2, OH и SH.
Особенно предпочтительно в формуле (I) R1 выбран из группы, состоящей из H и F. Особенно предпочтительно в формуле (I) R2 выбран из группы, состоящей из CH3 и Cl. Особенно предпочтительно в формуле (I) R3 выбран из группы, состоящей из F, Cl и CN. Особенно предпочтительно в формуле (I) R1 выбран из группы, состоящей из H и F; и R2 выбран из группы, состоящей из CH3 и Cl; и R3 выбран из группы, состоящей из F, Cl и CN.
Неограничивающие примерные соединения формулы (I) представляют собой соединения, в которых: (a) R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой F; (b) R1 представляет собой H, R2 представляет собой CH3 и R3 представляет собой Cl; (c) R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой CN и (d) R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой F.
Терапевтические показания
Соединения изобретения могут быть пригодными при лечении заболеваний, опосредованных альфа-1-антитрипсином, включая дефицит альфа-1-антитрипсина. Заболевания, опосредованные альфа-1-антитрипсином, включают дефицит альфа-1-антитрипсина, дисфункцию печени, фиброз, цирроз, печеночную недостаточность и гепатоцеллюлярную карциному, заболевания легких, дисфункцию и воспаление, включая астму, ХОБЛ, эмфизему и рак легких, воспалительные состояния кожи, включая дерматит и кожный зуд. Обычно заболевания, опосредованные альфа-1-антитрипсином, возникают в результате мутации гена альфа-1-антитрипсина и соответствуют общему классу заболеваний, называемых «серпинопатиями».
Соединения данного изобретения могут быть особенно пригодными при лечении респираторного заболевания, включая ХОБЛ и эмфизему.
Более определенно, соединения изобретения могут быть пригодными при лечении нарушений, вызванных мутантными формами альфа-1-антитрипсина, включая мутантную Z-AT-форму альфа-1-антитрипсина, таким образом, являются потенциально пригодными для лечения заболеваний, связанных с Z-AT, в частности заболеваний печени, включая желтуху, печеночную недостаточность, цирроз печени, аутоиммунный гепатит и гепатоцеллюлярную карциному.
Дефицит альфа-1-антитрипсина затрагивает 1 из 2000 человек североевропейского происхождения, что приводит к заболеваниям печени и легких. Более 100 различных мутаций было идентифицировано в гене SERPINA1, кодирующем α1-антитрипсин, но по оценкам 95% серьезного дефицита является результатом формы Z-AT (Glu342Lys). Данная аминокислотная замена нарушает фолдинг α1-антитрипсина, что приводит к секреции только ~15% зрелого белка. Большая часть оставшегося белка расщепляется через ERAD-протеасомный путь, но приблизительно 15% образует упорядоченные полимеры, которые накапливаются в эндоплазматическом ретикулуме гепатоцитов. Последующий дефицит важного ингибитора протеазы в кровотоке приводит к недостаточной защите легких от нейтрофильной эластазы, что приводит к эмфиземе. Накопление полимеров в гепатоцитах является цитотоксичным для клеток, вызывая неонатальный гепатит, цирроз и гепатоцеллюлярную карциному. Внутрипеченочные полимеры также повышают чувствительность печени к негативным воздействиям окружающей среды, таким как алкоголь, жир или вирусный гепатит.
Ингибирование протеазы-мишени - эластазы - α1-антитрипсином включает нарушение протеолитического механизма: после нуклеофильной атаки на узнаваемую последовательность в петле реакционного центра (ПРЦ) α1-антитрипсина два белка становятся ковалентно прикрепленными через эфирную связь. В результате происходит быстрое встраивание ПРЦ в центральный β-лист A, превращая его из 5-нитевой в 6-нитевую конфигурацию. Сопутствующее перемещение протеазы от одного полюса серпина к другому и сжатие его активного сайта предотвращает гидролиз этого обычно временного связывания; оба, таким образом, необратимо захватываются ковалентным комплексом инактивированная протеаза-серпин. Мутация Z находится в начале нити 5 β-листа A. Она возмущает локальную среду, позволяя α1-антитрипсину заселять нестабильный интермедиат (который был назван M⃰-состоянием), в котором β-лист A открывается и раскручивается верхняя часть спирали F. Остается установить, является ли включение ПРЦ межмолекулярным - приводящим к димеру петли-листа, который простирается с образованием более длинных полимеров - или внутримолекулярным с заменой домена С-концевой области, обеспечивая межсубъединичную связь для патологического полимера, который откладывается в ткани печени. Другие формы полимеризации различными мутантами α1-антитрипсина могут быть возможны. Способность дестабилизированного β-листа A включать петлю реакционного центра является обязательной стадией в образовании полимеров. Это делает механизм полимеризации тесно связанным с механизмом ингибирования протеазы: в обоих случаях петля реакционного центра является центральным звеном в термодинамически управляемом переходе между умеренно стабильной и гиперстабильной конформацией.
Сходство между физиологическим механизмом ингибирования серпинов и патологическим образованием полимера также наблюдается в других членах суперсемейства серпинов и дает начало классу родственных патологий, называемых серпинопатиями. Они включают семейную энцефалопатию с включением нейросерпиновых телец (FENIB), вызванную полимерогенными мутациями в нейросерпине, и формы тромбоза, вызванные полимеризацией и дефицитом антитромбина.
Терапия и способы лечения
Данное изобретение также предоставляет соединение изобретения для применения в терапии. Данное изобретение конкретно предоставляет применение соединения изобретения в качестве активного терапевтического вещества при лечении заболеваний, опосредованных альфа-1-антитрипсином.
Изобретение также предоставляет применение соединения изобретения при изготовлении лекарственного препарата для применения при лечении заболеваний, опосредованных альфа-1-антитрипсином.
В дополнительном аспекте предоставлена комбинация, содержащая соединение изобретения и, по меньшей мере, один дополнительный терапевтический агент, пригодный для лечения заболеваний, опосредованных альфа-1-антитрипсином.
В дополнительном аспекте предоставлена комбинация, содержащая соединение изобретения и, по меньшей мере, один дополнительный терапевтический агент, пригодный для лечения заболеваний, опосредованных альфа-1-антитрипсином, для применения при лечении заболеваний, опосредованных альфа-1-антитрипсином.
В дополнительном аспекте предоставлено применение комбинации, содержащей соединение изобретения и, по меньшей мере, один дополнительный терапевтический агент, пригодный для лечения ХОБЛ, при изготовлении лекарственного препарата для лечения заболеваний, опосредованных альфа-1-антитрипсином.
В дополнительном аспекте предоставлен способ лечения ХОБЛ, включающий введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества комбинации, содержащей соединение изобретения и, по меньшей мере, один дополнительный терапевтический агент, пригодный для лечения заболеваний, опосредованных альфа-1-антитрипсином.
В дополнительном аспекте предоставлена фармацевтическая композиция, содержащая комбинацию, содержащую соединение изобретения и, по меньшей мере, один дополнительный терапевтический агент, пригодный для лечения заболеваний, опосредованных альфа-1-антитрипсином, и один или более фармацевтически приемлемых эксципиентов.
Респираторное заболевание
Для лечения респираторного заболевания, включая ХОБЛ, соединения или фармацевтические композиции изобретения можно вводить вместе с одним или более бронходилататорами или их фармацевтическими композициями. Например, соединения изобретения можно составить вместе с одним или более бронходилататорами в единой композиции, такой как сухой порошок для ингаляции. Альтернативно, фармацевтическую композицию, содержащую соединение изобретения, можно вводить в сочетании с фармацевтической композицией, содержащей один или более бронходилататоров, или одновременно, или последовательно. В дополнительном альтернативном варианте композицию, содержащую соединение изобретения и бронходилататор, можно вводить в сочетании с фармацевтической композицией, содержащей дополнительный бронходилататор. В одном варианте осуществления фармацевтическая композиция, содержащая соединение изобретения, и фармацевтическая композиция, содержащая один или более бронходилататоров, может каждая содержаться в устройстве, подходящем для одновременного введения обеих композиций посредством ингаляции. В дополнительном варианте осуществления фармацевтическая композиция, содержащая соединение изобретения вместе с бронходилататором, и фармацевтическая композиция, содержащая дополнительный бронходилататор, может каждая содержаться в устройстве, подходящем для одновременного введения обеих композиций посредством ингаляции.
Подходящие бронходилататоры для введения вместе с соединениями изобретения включают агонисты β2-адренорецепторов и антихолинергические средства. Примеры агонистов β2-адренорецепторов включают, например, вилантерол, салметерол, сальбутамол, формотерол, салмефамол, фенотерол, кармотерол, этантерол, наминтерол, кленбутерол, пирбутерол, флербутерол, репротерол, бамбутерол, индакатерол, тербуталин и их соли, например, соль ксинафоат (1-гидрокси-2-нафталинкарбоксилат) салметерола, сульфатную соль сальбутамола или фумаратную соль формотерола. Примеры антихолинергических средств включают умеклидиний (например, в виде бромида), ипратропий (например, в виде бромида), окситропий (например, в виде бромида) и тиотропий (например, в виде бромида). В одном варианте осуществления соединение изобретения можно вводить вместе с агонистом β2-адренорецепторов, таким как вилантерол и антихолинергическим средством, таким как умеклидиний.
Заболевание печени
Для лечения заболевания печени соединения или фармацевтические композиции изобретения можно вводить с другими терапевтическими агентами, пригодными для лечения этих заболеваний.
Общие методы синтеза
Соединения общей формулы (I) можно получить способами, известными в области органического синтеза. Хорошо известно, что во всех способах можно использовать защитные группы для неустойчивых или реакционноспособных групп в случае необходимости в соответствии с общими принципами химии. С защитными группами работают в соответствии со стандартными методами органического синтеза (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons). Эти группы удаляют на удобной стадии синтеза соединения с использованием способов, которые очевидны специалисту в данной области техники. Выбор методов, также как условий реакции и порядка их выполнения должны соответствовать получению соединений Формулы (I). Другие соединения изобретения также можно получить с использованием способов, обычно известных в данной области техники, и, где это применимо, со ссылкой на принципы и конкретные реакции, описанные в настоящей заявке.
Способ идентификации соединения-кандидата в лекарственное средство
Открытие авторов настоящего изобретения, что соединения изобретения ингибируют полимеризацию α1-антитрипсина путем связывания с белком α1-антитрипсином в конкретном сайте связывания, также дает начало способу анализа, основанному на определении способности произвольного соединения-кандидата в лекарственное средство связываться с тем же сайтом на белке.
Таким образом, настоящее изобретение дополнительно предоставляет способ идентификации соединения-кандидата в лекарственное средство. В данном способе соединение-кандидат в лекарственное средство взаимодействует с α1-антитрипсином с образованием комплекса между соединением-кандидатом в лекарственное средство и α1-антитрипсином. α1-антитрипсин представляет собой белок, содержащий SEQ ID NO: 1, как определено в другом месте в настоящем описании (или производное этой последовательности с одной или более заменами аминокислот). Предпочтительно стадия взаимодействия соединения-кандидата в лекарственное средство с α1-антитрипсином включает образование кристаллического вещества α1-антитрипсина и взаимодействие указанного кристаллического вещества с указанным кандидатом в лекарственное средство.
В дальнейшем способ включает разрешение структуры комплекса, которое может быть выполнено стандартно с использованием методов разрешения кристаллической структуры, хорошо известных в данной области техники (см. также, например, Пример 2 данной заявки).
Наконец, способ включает определение, присутствует ли в комплексе соединение-кандидат в лекарственное средство в сайте связывания, как определено в настоящем описании. Если соединение-кандидат в лекарственное средство присутствует в сайте связывания, то это указывает на соединение, которое может быть способно ингибировать полимеризацию α1-антитрипсина и, таким образом, иметь предпочтительные терапевтические свойства по аналогии с соединениями настоящего изобретения, как обсуждается в другом месте в настоящем описании.
ПРИМЕРЫ
Пример 1
Общие методы
Если не указано иначе, исходные материалы представляли собой коммерчески доступные. Все растворители и коммерческие реагенты были лабораторного качества и использовались в том виде, в котором они были получены.
Соединения формулы (I) могут быть синтезированы по существу в соответствии со Схемой реакции 1 путем амидного сочетания соответствующего аминоспирта и соответствующей бензойной кислоты.
Схема реакции 1
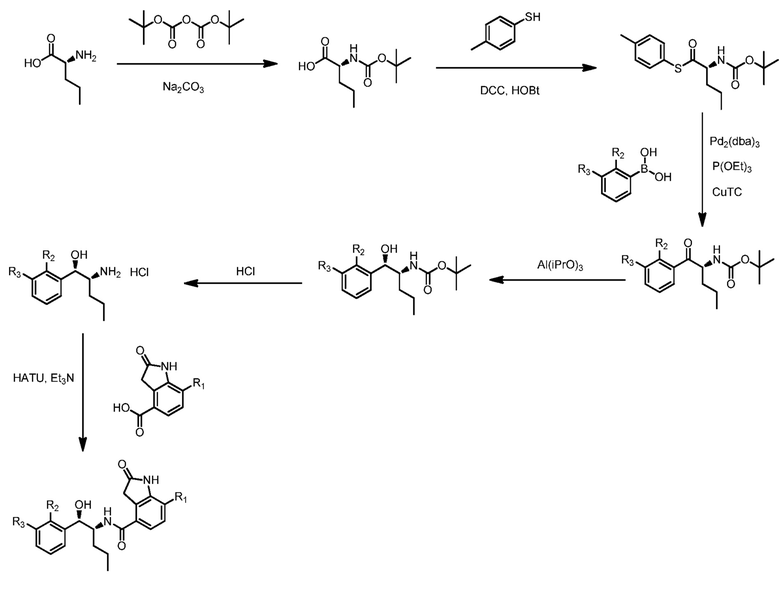
Сокращения:
ДМСО - Диметилсульфоксид
HATU - N-[(Диметиламино)-1H-1,2,3-триазоло-[4,5-b]пиридин-1-илметилен]-N-метилметанаминий гексафторфосфат N-оксид
к.т. - Комнатная температура
Rt - Время удерживания
Способы ЖХМС
Подвижная фаза: A: 0,1% муравьиная кислота в воде; B: 0,1% муравьиная кислота в ацетонитриле
Время (мин)/%B: 0/3%, 1,5/99,9%, 1,9/99,9%, 2,0/3,0%
Темп. колонки: 40°C
Скорость потока: 1,0 мл/мин
Подвижная фаза: A: 0,1% об./об. водный раствор аммиака pH 10; B: ацетонитрил
Время (мин)/%B: 0/3%, 1,5/99,9%, 1,9/99,9%, 2,0/3,0%
Темп. колонки: 40°C
Скорость потока: 1,0 мл/мин
Подвижная фаза: A: 0,1% муравьиная кислота в воде; B: 0,1% муравьиная кислота в ацетонитриле
Время (мин)/%B: 0/3,0%, 1,5/100,0%, 1,9/100,0%, 2,0/3%
Темп. колонки: 40°C
Скорость потока: 1,0 мл/мин
Способ хиральной ВЭЖХ
Подвижная фаза: н-гексан/(этанол+0,1% изопропиламин) 65/35% об./об.
Скорость потока: 1,0 мл/мин
Соединение 1:
N-[(1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил]-2-оксо-2,3-дигидро-1H-индол-4-карбоксамид
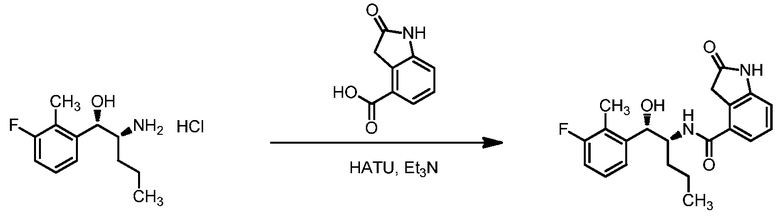
В круглодонной колбе к раствору (1S,2R)-2-амино-1-(3-фтор-2-метилфенил)пентан-1-ола гидрохлорида (интермедиат 1, 12,9 г, 51,9 ммоль), 2-оксоиндолин-4-карбоновой кислоты (9,2 г, 51,9 ммоль) и триэтиламина (10,5 г, 103,9 ммоль) в диметилформамиде (100 мл) при 0°C добавляли HATU (19,8 г, 51,9 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 ч и затем разбавляли этилацетатом (200 мл). Раствор промывали насыщенным раствором NaHCO3 (100 мл) и солевым раствором (2×100 мл). Органическую часть высушивали над сульфатом натрия, отфильтровывали и испаряли при пониженном давлении с получением неочищенного материала, который очищали с помощью флэш-хроматографии (Biotage Isolera, картридж 340 г, дихлорметан/ацетонитрил от 80/20 до 40/60 в качестве элюента) с получением N-[(1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил]-2-оксо-2,3-дигидро-1H-индол-4-карбоксамида в виде белого твердого вещества (12,5 г, Y=65,0%).
В круглодонной колбе (12,5 г, 33,74 ммоль) суспендировали в этилацетате (100 мл) при к.т. Суспензию нагревали до 60°C и добавляли этилацетат до тех пор, пока смесь не стала гомогенной (≈200 мл растворителя всего). Раствор оставляли охлаждаться при к.т. в течение ночи и образовавшийся белый осадок собирали фильтрованием с отсасыванием. Твердое вещество хранили в сушильном шкафу при 40°C под вакуумом с получением кристаллического N-[(1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил]-2-оксо-2,3-дигидро-1H-индол-4-карбоксамида в виде белого твердого вещества (10,8 г, ˃98% э.и., выход 87%).
ЖХМС: Rt 0,90 мин, MH+ 371 (Способ A).
1H ЯМР (500 МГц, ДМСО-d6) δ м.д. 0,80 (т, J=7,34 Гц, 3H), 1,07-1,21 (м, 1H), 1,30-1,43 (м, 1H), 1,45-1,54 (м, 1H), 1,56-1,67 (м, 1H), 2,31 (д, J=1,51 Гц, 3H), 3,46 (с, 2H), 4,06-4,17 (м, 1H), 4,85 (т, J=5,21 Гц, 1H), 5,44 (д, J=4,80 Гц, 1H), 6,89 (д, J=7,68 Гц, 1H), 6,99 (т, J=8,92 Гц, 1H), 7,11-7,25 (м, 3H), 7,28 (д, J=7,68 Гц, 1H), 7,97 (д, J=8,92 Гц, 1H), 10,45 (с, 1H).
Хиральная ВЭЖХ: Rt 6,3 мин
Интермедиат 1:
(1S,2R)-2-амино-1-(3-фтор-2-метилфенил)пентан-1-ола гидрохлорид
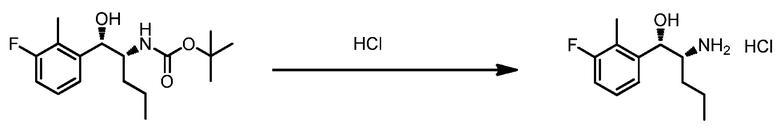
В круглодонной колбе растворяли трет-бутил-((1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил)карбамат (интермедиат 2, 15,8 г, 50,74 ммоль) в HCl в диоксане 4 M (100 мл) при 0°C. Смесь перемешивали в течение 2 ч при той же температуре и растворитель испаряли. Остаток суспендировали в 300 мл диэтилового эфира и перемешивали в течение 1 ч. Суспензию отфильтровывали с получением (1S,2R)-2-амино-1-(3-фтор-2-метилфенил)пентан-1-ола гидрохлорида в виде белого твердого вещества (11,9 г, Y=95,0%).
ЖХМС: Rt 0,85 мин, MH+ 212 (Способ B).
Интермедиат 2:
трет-бутил-((1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил)карбамат
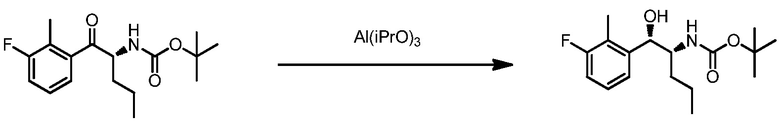
(R)-трет-бутил-(1-(3-фтор-2-метилфенил)-1-оксопентан-2-ил)карбамат (интермедиат 3, 18,7 г, 60,4 ммоль) растворяли в толуоле (150 мл) в круглодонной колбе и добавляли изопропанол (100 мл) при к.т. и изопропоксид алюминия (49,4 г, 241,8 ммоль). Раствор перемешивали при 50°C в течение 16 ч, и затем выливали в нас. раствор NH4Cl, и экстрагировали этилацетатом (4×300 мл). Органические части собирали, высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении с получением неочищенного вещества, названного бледно-желтым маслом. Этот неочищенный материал очищали с помощью флэш-хроматографии (Biotage Isolera, картридж 340 г, циклогексан/этилацетат от 80/20 до 50/50 в качестве элюента) с получением трет-бутил-((1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил)карбамата в виде бесцветного масла (15,8 г, Y=84,0%)
ЖХМС: Rt 1,16 мин, MH+ 312 (Способ A)
Интермедиат 3:
(R)-трет-бутил-(1-(3-фтор-2-метилфенил)-1-оксопентан-2-ил)карбамат
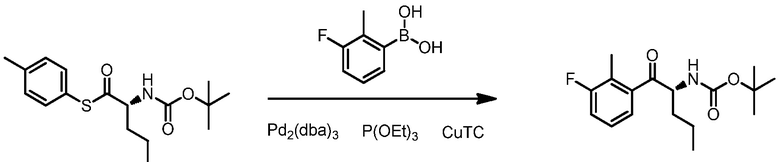
К смеси (3-фтор-2-метилфенил)бороновой кислоты (интермедиат 4, 50 г, 0,32 моль) в круглодонной колбе добавляли (R)-S-п-толил-2-((трет-бутоксикарбонил)амино)пентантиоат (53,2 г, 0,16 моль), трис(дибензилиденацетон)дипалладий(0) (7,3 г, 8,0 ммоль) и тиофен-2-карбоксилат меди (I) (61,0 г, 0,32 моль) в 1,4-диоксане (500 мл) при к.т., триэтилфосфит (10,6 г, 64,0 ммоль). Реакционную смесь перемешивали при к.т. в течение 4 ч и затем отфильтровывали через слой целита. Растворитель испаряли при пониженном давлении и неочищенный продукт растворяли в этилацетате (500 мл); раствор промывали водой (500 мл) и солевым раствором (2×500 мл). Органическую часть высушивали над сульфатом натрия, отфильтровывали и испаряли при пониженном давлении с получением неочищенного материала (100 г) в виде черного масла. Партию неочищенного материала 65 г очищали с помощью флэш-хроматографии (Biotage Isolera, картридж 3×340 г, циклогексан/дихлорметан от 60/40 до 0/100 в качестве элюента) с получением (R)-трет-бутил-(1-(3-фтор-2-метилфенил)-1-оксопентан-2-ил)карбамата в виде бесцветного масла (18,7 г, 60,4 ммоль).
ЖХМС: Rt 1,26 мин, MH+ 310 (Способ A)
Интермедиат 4:
(R)-S-п-толил-2-((трет-бутоксикарбонил)амино)пентантиоат
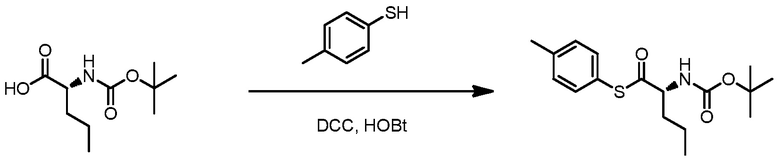
К раствору (R)-2-((трет-бутоксикарбонил)амино)пентановой кислоты (интермедиат 5, 85,0 г, 0,39 моль) в этилацетате (800 мл) в круглодонной колбе добавляли 4-метилбензол-1-тиол (53,5 г, 0,43 моль) и 1-гидроксибензотриазол (50,0 г, 0,59 моль). Смесь охлаждали до 0°C и добавляли N, N'-дициклогексилкарбодиимид (80,5 г, 0,39 моль). Реакционную смесь перемешивали при к.т. в течение 16 ч; образовавшееся белое твердое вещество затем удаляли фильтрованием с отсасыванием. Фильтрат промывали насыщенным раствором NaHCO3 (700 мл), водой (700 мл) и солевым раствором (700 мл). Органическую часть высушивали над сульфатом натрия, отфильтровывали и растворитель испаряли при пониженном давлении с получением неочищенного вещества в виде белого твердого вещества. Неочищенный материал очищали с помощью флэш-хроматографии (Biotage Isolera, картридж 340 г, циклогексан/дихлорметан от 60/40 до 0/100 в качестве элюента) с получением (R)-S-п-толил-2-((трет-бутоксикарбонил)амино)пентантиоата в виде белого твердого вещества (111,0 г, Y=88,0%).
ЖХМС: Rt 1,30 мин, MH+ 324 (Способ A)
Интермедиат 5:
(R)-2-((трет-бутоксикарбонил)амино)пентановая кислота
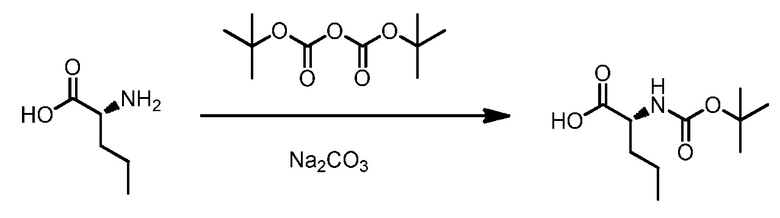
В круглодонной колбе к суспензии (2R)-2-аминопентановой кислоты (75,0 г, 0,64 моль) в воде (250 мл) при к.т. добавляли карбонат натрия (68,0 г, 0,64 моль). Раствор ди-трет-бутилдикарбоната (139,9 г, 0,64 моль) в тетрагидрофуране (400 мл) добавляли по каплям более одного часа. Реакционную смесь перемешивали при к.т. в течение 24 ч и затем концентрировали при пониженном давлении (приблиз. 400 мл). Добавляли насыщенный раствор лимонной кислоты (300 мл) до тех пор, пока pH не достигал значения ≈3. Водный слой промывали этилацетатом (4×500 мл). Органические части собирали, высушивали над сульфатом магния и испаряли при пониженном давлении с получением (R)-2-((трет-бутоксикарбонил)амино)пентановой кислоты в виде бледно-желтого масла (137,0 г, Y=98,6%).
ЖХМС: Rt 0,82 мин, MH+ 218 (Способ A).
Аналогичным образом были получены:
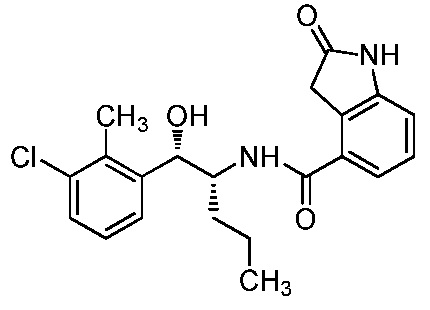
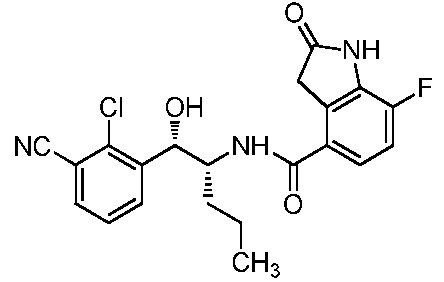
Эффективность соединений изобретения в отношении ингибирования полимеризации Z-альфа-1-антитрипсина (Z-A1AT) может быть определена с помощью анализа флуоресцентного резонансного переноса энергии с временным разрешением (TR-FRET), выполняемого на очищенном Z-A1AT, как описано в настоящей заявке. Все соединения формулы (I) продемонстрировали ингибирующую активность в отношении полимеризации Z-A1AT in vitro с концентрацией полумаксимального ингибирования (IC50) менее одного микромоля (1 мкМ). Средние данные активности для анализа, описанного ниже для соединений 1-4:
Протокол анализа ингибирования полимеризации Z-альфа-1-антитрипсина
Z-альфа-1-антитрипсин очищали из плазмы крови доноров, гомозиготных по форме Z-A1AT. Очистку проводили в соответствии с опубликованным протоколом (Lomas et al. Biochemistry 1993; 32:500-508), включающим осаждение сульфатом аммония, фракционирование с обменом тиола и фракционирование с обменом аниона.
Очищенный Z-A1AT разбавляли до 5 нМ в буфере ФСБ с 0,01% Твин-20 и смешивали с различными концентрациями соединений-ингибиторов, растворенных в ДМСО, в 384-луночных планшетах для анализа. После инкубации в течение 72 часов при 37°C количество полимера Z-A1AT определяли с использованием формата анализа флуоресцентного резонансного переноса энергии с временным разрешением (TR-FRET) путем добавления смеси для обнаружения антител в планшеты для анализа. Смесь для обнаружения содержала моноклональное антитело, специфичное к полимеру A1AT (Miranda et al. Hepatology 2010; 52: 1078-1088), меченное тербием антимышиное антитело, поликлональное кроличье анти-A1AT антитело и меченое Alexa488 антикроличье антитело. Данные TR-FRET были нормализованы к контролю (контроль наполнитель ДМСО, обеспечивающий 0% ингибирования, насыщающие концентрации активного соединения, обеспечивающие 100% ингибирование), и значения IC50 были получены путем подгонки 4-параметрических логистических кривых к нормализованным данным.
Пример 2
Введение
Целью данной работы была разработка низкомолекулярного корректора фолдинга Z-α1-антитрипсина, способного блокировать образование полимера в эндоплазматическом ретикулуме гепатоцитов, и который был пригоден для перорального дозирования в качестве потенциального лечения дефицита α1-антитрипсина. Для достижения этого необходимо было преодолеть ряд проблем: (i) специфическая форма белка, представляющего лекарственную мишень, представляет собой высокоподвижный интермедиат фолдинга, расположенный в эндоплазматическом ретикулуме; (ii) целевая фармакология включает предотвращение сильного взаимодействия белок-белок, и требование, чтобы молекула обладала лекарственными свойствами для обеспечения возможности перорального дозирования, значительно ограничивает подходящее химическое пространство; (iii) в качестве неклассической лекарственной мишени низкомолекулярные связывающие вещества могут быть плохо представлены в библиотеках скрининга соединений; (iv) относительно высокая концентрация циркулирующего мономерного Z-α1-антитрипсина (~5 мкМ) даже у лиц с тяжелым дефицитом плазмы представляет собой поглотитель высокого сродства с соединением, ограничивая его доступ к мишени в гепатоците и требуя высоких общих концентраций лекарственного средства в крови для достижения достаточной концентрации свободного лекарственного средства и целевого воздействия на печень.
В настоящем описании сообщается о первых низкомолекулярных подобных лекарственным средствам корректоров фолдинга Z-α1-антитрипсина, полученным путем оптимизации попаданий из анализа технологии кодированных библиотек, которые подходят для пероральной доставки, правильного фолдинга в гепатоцитах, полученных из ИПСК пациента, и увеличения циркулирующих уровней Z-α1-антитрипсина в модели дефицита α1-антитрипсина у трансгенных мышей.
Материалы и методы
Альфа1-антитрипсин очищали из плазмы гомозигот ZZ и MM-α1-антитрипсина, как описано в Lomas et al., Biochemistry. 1993;32:500-8. Рекомбинантный Cys232Ser α1-антитрипсин экспрессировали и очищали, как подробно описано в Irving et al. Methods Enzymol. 2011;501:421-66 and Haq et al. Biosci Rep. 2013;33(3):e00046.
«Соединение 1» представляет собой N-[(1S,2R)-1-(3-фтор-2-метилфенил)-1-гидроксипентан-2-ил]-2-оксо-2,3-дигидро-1H-индол-4-карбоксамид:
Анализ технологии ДНК-кодированных библиотек (ELT)
Анализ технологии кодированных библиотек (ELT) приблизительно 10 миллионов соединений использовали для идентификации малых молекул, которые будут стабилизировать мономерный Z-α1-антитрипсин при 37 и 41°C. Самые сильные связывающие вещества элюировали, идентифицировали по их ДНК-метке и синтезировали.
Оценка полимеризации Z-α1-антитрипсина на основе антител
Анализ на основе антител флуоресцентного резонансного переноса энергии с временным разрешением (TR-FRET) использовали для контроля полимеризации 5 нМ Z-α1-антитрипсина после инкубации с различными концентрациями соединений при 37°C в течение 72 часов. В этом анализе использовали моноклональное антитело 2C1, которое специфично к патологическим полимерам α1-антитрипсина (Miranda et al. Hepatology 2010; 52: 1078-1088), поликлональное антитело, которое связывается со всеми формами α1-антитрипсина, анти-мышиный IgG флуоресцентный донор (Eu-W1024) и анти-кроличий IgG акцептор (APC).
Биохимический анализ конкурентного связывания для Z-α1-антитрипсина и M-α1-антитрипсина
Флуоресцентное производное соединения 1 получали путем дериватизации соединения 1 по его кольцевому атому азота с помощью метки Alexa Fluor 488 через короткую органическую линкерную группу.
Последовательное разведение исследуемых соединений в ДМСО разбавляли в 25 раз буфером для анализа и 2,5 мкл этого промежуточного разведения переносили в черный 384-луночный микротитровальный планшет с малым объемом (Greiner BioOne; продукт № 784076). 5 мкл предварительно смешанного раствора буфера для анализа, содержащего 2,3 нМ моноклонального антитела 3Cl 1, связывающегося с антитрипсином, 3 нМ меченого тербием анти-мышиного IgG LanthaScreen, который будет связываться с антителом 3C11 (Invitrogen), также как или 2 нМ Z-alAT или 10 нМ M-alAT, добавляли с использованием диспенсера Thermo Multidrop (ThermoFisher). Планшеты инкубировали при комнатной температуре в течение 30 мин, затем добавляли 2,5 мкл раствора лиганда, содержащего или 40 нМ (для Z-α1-антитрипсина), или 2 мкМ (для M-α1-антитрипсина) флуоресцентного производного соединения 1, с использованием диспенсера Thermo Multidrop.
Планшеты закрывали и инкубировали в течение ночи при комнатной температуре или в течение 3 дней при 4°C, затем 6 часов при комнатной температуре. В отсутствие конкурирующего соединения перенос энергии может происходить между флуорофорами Тербий- и Alexa488. Соединения, конкурирующие с флуоресцентным производным соединения 1, ингибируют этот сигнал. Сигнал TR-FRET считывали на планшет-ридере Envision путем возбуждения Тербия при 337 нм и определения эмиссии при 520 нм и 495 нм.
Константы диссоциации (KD) для аффинности соединений к белку Z-a1AT или M-a1AT получали из значений pIC50 в соответствии с уравнением Ченга-Прусоффа с учетом концентрации используемого флуоресцентного лиганда по отношению к его аффинности, как определено в независимых экспериментах флуоресцентной поляризации и TR-FRET:
KD (соединение) = 10-pIC50(соединение)/(1-([Лиганд]/KD(Лиганд)).
Эксперименты на выявление ассоциации соединений
Собственная флуоресценция триптофана α1-антитрипсина доминирует Trp194 в области нарушения (Tew et al. J Mol Biol. 2001 Nov 9;313(5):1161-9) и может быть использована для контроля прогресса связывания соединения. Плазму M и Z-α1-антитрипсина разбавляли до 0,2 мг/мл в 100 мкМ ФСБ и добавляли Соединение 1 (или эквивалентный объем ДМСО) до концентрации 10 мкМ. Флуоресценцию (λвозб=280 нм, λэм=330 нм) контролировали в течение, по меньшей мере, 2 часов и рассчитывали полупериод изменения.
Кинетические данные в экспериментах с остановленным потоком подгоняли к кривым экспоненциального затухания со смещением и линейным наклоном, чтобы учесть постоянный дрейф фона, приводя к наблюдаемым константам скорости псевдопервого порядка k (набл):
y=A0 ⋅ e-k(набл)t+наклон ⋅ t+смещение
в которой t=время и A0=амплитуда при t=0. Затем была получена скорость связывания второго порядка как наклон при нанесении на график значений k (набл) в зависимости от концентрации соединения. Скорости диссоциации были рассчитаны на основе скоростей связывания и значений KD, определенных из анализов связывания.
Клеточная биология
Клетки CHO Tet-On, которые условно экспрессируют Z-α1-антитрипсин (Ordóñez A et al. Hepatology 2013;57(5):2049-60), одновременно обрабатывали 0,5 мкг/мл оксициклина и различными концентрациями соединений в течение 48 часов в 6-луночных планшетах. Клетки без доксициклина или обработанные 1% об./об. ДМСО действовали в качестве контроля. Клетки лизировали и внутриклеточный полимер Z-α1-антитрипсина оценивали с помощью сэндвич-варианта ELISA с моноклональным антителом 2C1 (Miranda et al. Hepatology. 2010;52:1078-88). Свежесинтезированный α1-антитрипсин оценивали путем выращивания клеток, индуцированных экспрессией Z-α1-антитрипсина в DMEM без метионина (Met) и цистеина (Cys) в течение 1 ч, и затем мечения 1,3 МБк 35S-Met/Cys в течение 15 мин. Затем клетки промывали и инкубировали в среде чейза, содержащей избыток немеченого Met и Cys, в течение 0, 2 и 6 часов. После периода чейза среду собирали и получали клетки. α1-Антитрипсин из среды и клеточных лизатов иммунопреципитировали с поликлональным антителом α1-антитрипсина или моноклональным антителом 2C1, которое обнаруживает полимеры путем разделения каждого образца на две равные части. Белки с радиоактивной меткой ресуспендировали в загрузочном буфере SDS, кипятили в течение 5 мин, разделяли с помощью 10% масс./об. акриламида SDS-PAGE и количественно определяли авторадиографией.
Иммунофлуоресцентная микроскопия
Клетки CHO-K1 инкубировали с/без доксициклина для экспрессии α1-антитрипсина и с или без низкомолекулярного соединения, представляющего интерес, фиксировали 3,7% об./об. формальдегида в ФСБ в течение 15 мин, пермеабилизировали 0,1% об./об. Тритона X-100 в ФСБ и затем блокировали 10% об./об. ФБС в ФСБ в течение 30 мин. Клетки метили кроличьим поликлональным антителом к общему α1-антитрипсину или моноклональным антителом 2С1, которое выявляет полимеры с последующим введением конъюгированных с техасским красным анти-кроличьих или меченных FITC анти-мышиных вторичных антител соответственно. Клетки визуализировали с использованием широкопольного флуоресцентного микроскопа Axiovert 200 (Carl Zeiss Microimaging Inc., Thornwood, NY).
Эксперименты по термостабильности
Стабильность нативного состояния серпинов при добавлении соединений исследовали термической денатурацией в присутствии 5-кратной концентрации раствора красителя SYPRO Orange (Life Technologies). Использовали конечную концентрацию белка 0,1 мг/мл и 50 мкМ соединения, суспендированного в ФСБ (Nettleship et al. Methods Mol Biol. 2008;426:299-318). Образцы нагревали от 25°C до 95°C со скоростью 1°C/мин в трех отдельных экспериментах на приборе для количественной ПЦР в реальном времени Eppendorph Mastercycler Realplex4 и регистрировали флуоресценцию в интервале 605±15 нм. Средняя точка денатурации (Tm) представляет собой температуру, при которой первая производная интенсивности флуоресценции в зависимости от температуры достигает максимума.
Устойчивость к полимеризации, вызванной нагреванием, также определяли с использованием анализа конечной точки при постоянной температуре. Очищенный из плазмы M и Z-α1-антитрипсин разбавляли до 0,2 мг/мл в ФСБ 5% об./об. глицерина, инкубировали с 10 мкМ Соединения 1 (или только с ДМСО) в течение 2 часов и затем нагревали в 20 мкл аликвотах в течение дополнительных 4 часов в термоциклере с градиентом 48-65°C, применяемым к планшету. Образцы анализировали на 3-12% масс./об. акриламидном Bis-Tris Native-PAGE геле (Life Technologies).
Равновесное разворачивание
Bis-ANS представляет собой краситель, который сообщает о появлении разворачивающегося интермедиата α1-антитрипсина (Dafforn et al. J Biol Chem. 1999;274:9548-55). В микропланшете 10 мкл аликвоты M и Z-α1-антитрипсина при 0,5 мг/мл с 100 мкМ bis-ANS и 100 мкМ Соединения 1 (или эквивалентный объем ДМСО) быстро смешивали с различными концентрациями гуанидина гидрохлорида, обеспечивая конечную концентрацию 0-6 М денатурата в ФСБ. Измерения интенсивности флуоресценции bis-ANS (λвозб=385 нм, λэм=490 нм) регистрировали после достижения стабильного сигнала, и значения масштабировали в диапазоне от 0 до 1,0.
Быстрый рефолдинг
Рефолдинг α1-антитрипсина in vitro не является полностью обратимым вследствие потери материала из-за аморфной агрегации, полимеризации и неправильного сворачивания. Эти вклады не могут быть легко деконволюированы методами объемной спектроскопии, и поэтому рефолдинг оценивали неденатурирующим PAGE. M и Z-α1-антитрипсин при 9,6 мг/мл денатурировали в 6 М мочевине и 15 мМ фосфате натрия pH 8,0 в течение 4 часов при комнатной температуре, и 1,25 мкл мгновенно повторно сворачивался в 250 мкл ФСБ, содержащего 0-50 мкМ Соединения 1 и нормализованную концентрацию 5% об./об. ДМСО. После инкубации в течение 1 часа при комнатной температуре образцы анализировали с помощью окрашенного кумасси 3-12% масс./об. акриламидного Bis-Tris Native PAGE геля (Life Technologies).
Кристаллография
Кристаллы рекомбинантного Cys232Ser α1-антитрипсина выращивали с использованием метода диффузии паров в варианте висячей капли путем объединения 1 мкл 12,1 мг/мл белка с 1 мкл резервуарного буфера и уравновешивания с резервуарным буфером, который содержал 0,1 М буфер MES pH 6,0 с 20-22,25% масс./об. PEG 1500. Кристалл, который образовался в 0,1 М MES pH 6,0 с 22,25% масс./об. PEG 1500, переносили в буфер, содержащий 0,09 M MES pH 6,0, 18% PEG1500, 13,5% глицерина, 5% d6 ДМСО и 25 мМ соединения для инкубации при 20°C в течение 24 часов. Кристаллы мгновенно замороживали в жидком азоте и данные собирали в пучке синхротронного излучения I03 синхротрона Diamond.
В частности, белок α1-антитрипсин, используемый для экспериментов по кристаллизации, соответствовал варианту M1V M-AT со следующими модификациями: (a) Cys232 мутирован в Ser (для простоты обращения, устраняя необходимость в восстанавливающих агентах во время различных анализов; замена является консервативной); (b) компонент сигнального пептида (MPSSVSWGILLLAGLCCLVPVSLA) и Glu1 на N-конце M-AT млекопитающих не присутствуют на N-конце используемого белка; (c) N-конец используемого белка имеет аффинную метку гексагистидина для облегчения очистки и несколько аминокислот, предоставляемых вектором экспрессии, последовательность которого представляет собой MRGSHHHHHHT.
Таким образом, полная последовательность используемого белка представляла собой:
MRGSHHHHHHTDPQGDAAQKTDTSHHDQDHPTFNKITPNLAEFAFSLYRQLAHQSNSTNIFFSPVSIATAFAMLSLGTKADTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQPDSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHSKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLMIEQNTKSPLFMGKVVNPTQK;
в котором подчеркнутая часть соответствует SEQ ID NO: 1.
Результаты
Идентификация Соединения 1 посредством скрининга с использованием технологии кодированной библиотеки, структурно-ориентированного дизайна лекарственных средств и клеточного профилирования.
Z-α1-антитрипсин представляет собой конформационно динамическую молекулу, которая полимеризуется из почти нативной конформации на поздних этапах пути фолдинга и, следовательно, представляет собой неклассическую мишень для открытия лекарственных средств. Таким образом, было рассмотрено, что обнаружение попадания может иметь положительный результат от исследования широкого и минимального смещенного химического пространства. Анализ технологии кодированных библиотек (ELT) использовали для поиска связывающих веществ с Z-α1-антитрипсином. Использовали гликозилированный Z-α1-антитрипсин, очищенный из плазмы гомозигот Z-α1-антитрипсина, поскольку он представляет собой патофизиологическую мишень лекарственного средства для человека, имеющую отношение к заболеванию. Отбор ELT осуществляли путем иммобилизации Z-α1-антитрипсина на аффинной смоле на основе антител для инкубации с библиотеками ДНК-кодированных соединений с вторичным скринингом с помощью экспериментов по тепловому сдвигу.
Один основной ряд хиральных гидроксикарбоксамидов был идентифицирован с помощью ELT-скрининга, который продемонстрировал функциональную активность при блокировании полимеризации в иммуноанализе TR-FRET. Оптимизация начального попадания сопровождалась подходом к дизайну на основе структуры с использованием данных о кристаллических структурах низкомолекулярных лигандов, образующих комплекс с α1-антитрипсином. Было обнаружено, что центральный гидроксикарбоксамид облегчает связывание с Z-α1-антитрипсином. Было также обнаружено, что алкильная боковая цепь (у атома углерода, смежного с атомом N карбоксамида) является предпочтительной, в частности пропильная боковая цепь. Развитие медицинской химии было сосредоточено на модификациях, таких как ароматические группы на концевых областях соединения. Это привело к открытию Соединения 1.
Соединение 1 является сильным ингибитором полимеризации in vitro и на клеточных моделях заболевания
Соединение 1 связывается с Z-α1-антитрипсином с высокой аффинностью (KD) 1,5 нМ, как определено с помощью анализа конкурентного связывания с флуоресцентно меченым производным; аналогичное значение было получено с помощью термофореза на микромасштабном уровне. Связывание демонстрирует селективность, по меньшей мере, в 50 раз с меньшей аффинностью в отношении очищенного от плазмы M-α1-антитрипсина дикого типа.
Форма кривых и данные нативной масс-спектрометрии соответствовали одному сайту связывания соединения. Связывание флуоресцентного производного с полимерами Z-варианта не наблюдалось, что указывает на потерю связывающего кармана при полимеризации и полученную в результате заметную конформационную селективность. Скорость взаимодействия соединения с мишенью можно было отслеживать по изменениям собственной флуоресценции триптофана; это свойство использовали для определения скоростей ассоциации второго порядка для связывания Соединения 1 с Z (4,1 × 104 М-1 с-1) и M-α1-антитрипсином (2,1 × 102 М-1 с-1). По этим значениям были рассчитаны скорости диссоциации первого порядка для Z (6,1 × 10-5 с-1) и M-α1-антитрипсина (1,6 × 10-5 с-1) и был обнаружен одинаковый порядок величин. Следовательно, в селективности соединения в отношении Z по сравнению с M-α1-антитрипсином преобладает разница в скорости ассоциации, а не диссоциации.
Было обнаружено, что Соединение 1 подавляет полимеризацию in vitro Z-α1-антитрипсина, полученного из плазмы, дозозависимым образом с измеренным pIC50, равным 8,3, что близко к пределу прочного связывания анализа TR-FRET. Его способность блокировать полимеризацию Z-α1-антитрипсина в ЭР во время фолдинга оценивали путем добавления Соединения 1 к клеткам CHO-TET-ON-Z-A1AT (Ordóñez et al. Hepatology. 2013;57(5):2049-60) с одновременной индукцией экспрессии Z-α1-антитрипсина с применением доксициклина. По сравнению с контролем Соединение 1 полностью блокировало внутриклеточное образование полимеров Z-α1-антитрипсина, как измерялось окрашиванием 2C1 анти-α1-антитрипсин полимер моноклонального антитела (pIC50=6,3). Для исследования было ли способно Соединение 1 также увеличивать секрецию Z-α1-антитрипсина кроме блокирования полимеризации, экспрессию Z-α1-антитрипсина индуцировали в клетках CHO-TET-ON-Z-A1AT, добавляли Соединение 1 и общее количество Z-α1-антитрипсина в супернатанте измеряли с помощью иммуноанализа через 6 часов. Соединение 1 увеличивало секретируемые уровни приблизительно в 3 раза по сравнению с контрольным наполнителем с pEC50 6,3. Подобную активность между эффектами на секрецию и полимеризацию наблюдали для других соединений изобретения, подтверждая гипотезу о том, что эти эффекты вызваны одним и тем же фармакологическим механизмом действия.
Чтобы подтвердить, что активность и эффективность Соединения 1 в CHO-TET-ON-Z-A1AT была разумной оценкой того, что можно было ожидать в гепатоцитах с эндогенными уровнями экспрессии, влияние Соединения 1 на секрецию и полимеризацию Z-α1-антитрипсина измеряли в гепатоцитах, полученных из ИПСК с генотипом ZZ-α1-антитрипсина (Yusa et al. Nature. 2011;478:391-4). Соединение 1 ингибировало полимеризацию и повышенную секрецию с подобной активностью в ИПСК-гепатоцитах, как в клетках CHO-TET-ON-Z-A1AT с pIC50 6,4 и pEC50 6,5 соответственно и с аналогичной эффективностью, вызывая приблизительно 3-кратное увеличение уровней секретируемого Z-α1-антитрипсина.
Поскольку все люди с Z-аллелем α1-антитрипсина обнаруживают полимер в своей печени, но только у относительно небольшой части развивается клиническое заболевание печени, вероятно, α1-антитрипсиновое заболевание печени у взрослых представляет собой двухударный процесс, в результате которого образование Z-α1-антитрипсинового полимера в печени повышает чувствительность печени к вторичным воздействиям, таким как алкоголь, лекарственные средства или жир печени. Для исследования способности Соединения 1 защитить клетки от сенсибилизации к вторичному токсину, экспрессию Z-α1-антитрипсина индуцировали в клетках CHO-TET-ON-Z-A1AT в присутствии или при отсутствии 10 мкМ Соединения 1 перед воздействием различных концентраций ЭР стрессора туникамицина. В анализе жизнеспособности клеток клетки, экспрессирующие M-α1-антитрипсин дикого типа, были менее чувствительны к туникамицину, чем клетки, экспрессирующие Z-α1-антитрипсин. Соединение 1 полностью отменяет образование полимера в клетках, экспрессирующих Z-α1-антитрипсин, и восстанавливает чувствительность клеток, экспрессирующих α1-антитрипсин, до чувствительности контрольных клеток дикого типа.
Соединение 1 связывается с новым криптическим сайтом связывания, который чаще выбирается Z-вариантом
Кристаллические структуры высокого разрешения α1-антитрипсина в комплексе с Соединением 1 были получены путем просачивания соединения в кристаллы апо α1-антитрипсина (Таблица 1).
Таблица 1 Статистика сбора и уточнения данных
⃰ Значения в скобках относятся к оболочке с самым высоким разрешением.
Структуры показывают, что взаимодействие с соединениями индуцирует образование криптического сайта связывания, не очевидного в апо структурах, наверху β-листа-A за нитью 5. Эта область называется «разрывом», поскольку является точкой, в которой петля реакционного центра сначала вставляется во время ингибирования протеазы (Whisstock et al. J Mol Biol. 2000;295(3):651-65), и включает сайт Z-мутации E342K.
Типовые изображения идентифицированного сайта связывания показаны на Фиг. 1-3. На Фиг. 1 представлен обзор белка, связанного с Соединением 1 (остатки 342-356 неупорядочены и, следовательно, отсутствуют на изображении). На Фиг. 2 представлен более подробный вид сайта связывания, включая некоторые из ближайших остатков, с водородными связями, показанными пунктирными линиями. На Фиг. 3 представлена схематическая двумерная визуализация Соединения 1 в сайте связывания белка и выделение некоторых аминокислотных остатков, которые составляют сайт связывания, также как частичные химические структуры внутри белка, которые образуют значительные межмолекулярные взаимодействия (например, водородные связи) с Соединением 1 (показано пунктирными линиями).
Более подробно структура α1-антитрипсин в комплексе с Соединением 1 показывает, что оксиндолильное кольцо складывается в стопку с боковой цепью Trp194, в то время как карбонильная группа образует водородную связь с основной цепью Trp194 (см. Фиг. 2 и Фиг. 3), что согласуется с изменением собственной флуоресценции триптофана, вызванной связыванием. Фенильное кольцо и пропильная цепь занимают два высокогидрофобных кармана. Водородные связи образуются между гидроксильной группой Соединения 1 и основной цепью Leu291, амидным азотным водородом и карбонильным кислородом основной цепи Pro289, и между амидным карбонилом и гидроксильной группой Tyr244 (см. Фиг. 2 и Фиг. 3). Как следствие, происходит смещение остатков 189-195, 290-295 и 336-342 нити 5A с соответствующей потерей электронной плотности для проксимального шарнира петли реакционного центра, поворот на ~170° боковой цепи Trp194, и сдвиг на 3,8 Å в гидроксильной группе Tyr244, и смещение остатков 196-203. Сравнение структуры, связанной с Соединением 1, с апо-формой показывает общее среднеквадратичное отклонение 0,64 Å для 335 пар остатков.
В частности, следовательно, было обнаружено, что Соединение 1 способно связываться с α1-антитрипсином в криптическом сайте связывания, как определено в другом месте в настоящем описании. Для полноты следует отметить, что аффинная метка гексагистидина и аминокислоты, предоставляемые вектором экспрессии в используемом белке, не были очевидны в профиле электронной плотности сайта связывания и, следовательно, не были ассоциированы со связыванием Соединения 1. Подобным образом, мутировавший Ser232 (заменяющий Cys232 в белке дикого типа) не находился в положении, непосредственно связанном с сайтом связывания соединения.
Соединение 1 препятствует переходу к склонному к полимеризации интермедиату M⃰ с помощью стабилизации β-листа A
Полимеризация α1-антитрипсина происходит из переходного состояния интермедиата, известного как M⃰, которое легко заполняется Z-вариантом. Одним из отличительных признаков M⃰ является его распознавание чувствительными к окружающей среде флуоресцентными репортерными красителями. Анализы теплового сдвига, в которых используется краситель SYPRO Orange, показывают стабильность нативного состояния белка по сравнению с вызванным нагреванием разворачиванием. Серия экспериментов, проведенных с использованием различных температурных градиентов в присутствии и при отсутствии 50 мкМ Соединения 1, продемонстрировала заметное увеличение температуры средней точки перехода; в α1-антитрипсине это указывает на то, что нативное состояние стабилизировано по сравнению с переходом в M⃰. Соответственно, в эксперименте с постоянной температурой олигомеры генерировались при более высоких температурах в присутствии соединения, чем при его отсутствии при визуализации с помощью неденатурирующего PAGE. Стабильность нативного состояния также может быть исследована путем равновесного разворачивания с использованием химических денатуратов, где пик флуоресценции bis-ANS соответствует максимально заселенному интермедиату разворачивания. Измеренные профили показали, что для разворачивания, индуцированного гуанидиния гидрохлоридом, эта точка возникает при 1,8 М денатурата в присутствии 50 мкМ Соединения 1, что отражает повышение стабильности в нативном состоянии по сравнению с его отсутствием (1,2 М).
Мутации, которые мешают открытию β-листа A или нарушают его взаимодействие с N-концевой частью петли реакционного центра, изменяют способность серпинов ингибировать протеазы-мишени. Стехиометрию ингибирования (SI) определяли для M и Z-α1-антитрипсина в периодических экспериментах по сравнению с моделью протеазы-мишени химотрипсина. Предварительная инкубация обоих вариантов с Соединением 1 привела к потере ˃98% активности ингибирования протеазы. Разрешение продуктов взаимодействия с помощью SDS-PAGE показало полное расщепление петли реакционного центра; следовательно, это не является следствием того, что сайт распознавания протеазы в петле реакционного центра становится недоступным для фермента. Эти данные согласуются с механизмом, в котором соединение стабилизирует β-лист A по сравнению с конформационным изменением, которое опосредует как ингибирующую активность, так и патологическое неправильное сворачивание.
Для исследования согласуется ли эта активность с действием в качестве химического шаперона, M и Z-α1-антитрипсин были развернуты in vitro в 6 M гуанидина гидрохлориде и быстро повторно свернуты путем мгновенного разбавления в буфере без денатурата в присутствии или при отсутствии Соединения 1. Электрофорез продуктов с помощью неденатурирующего PAGE показал анодно-сдвинутую миграцию для Z-варианта при отсутствии соединения, что согласуется с неправильно свернутым побочным продуктом M⃰ (Ekeowa et al. Proc Natl Acad Sci USA. 2010;107(40):17146-51), который был исправлен при стехиометрических концентрациях и выше.
Характеристика лекарствоподобных свойств Соединения 1
Чтобы исследовать пригодность Соединения 1 для продвижения к исследованиям in vivo и потенциал для использования в качестве клинического кандидата для исследования на людях, Соединение 1 было отпрофилировано исходя из панели анализов in vitro в отношении отклонений от целей, считающихся предсказывающими потенциальную ответственность за безопасность. Соединение 1 было неактивным в отношении большинства этих мишеней с низким уровнем активности, близким к нижнему пределу чувствительности в 16 анализах. Измеренная эффективность этих предельных активностей была, по меньшей мере, в 10 раз ниже клеточной активности Соединения 1 при блокировании полимеризации Z-α1-антитрипсина (pEC50 6.3).
Потеря ингибирующей активности представляет собой средство, с помощью которого можно оценить возможное нецелевое взаимодействие с другими серпинами. Однако в отличие от α1-антитрипсина при инкубации с 50 мкМ Соединения 1 ингибирующая активность гомологов антитромбина, нейросерпина и антихимотрипсина не подвергалась влиянию.
Поскольку Соединение 1 показало хороший уровень селективности по отношению к нецелевым панелям и другим серпинам in vitro и in vivo, ФК свойства молекулы были исследованы с целью изучения взаимодействия с мишенью in vivo. Соединение 1 имеет измеренный ChromLogD (pH 7,4) 3,8, низкое связывание с человеческим сывороточным альбумином 84,2% и хорошую растворимость аморфного лекарственного вещества в FaSSIF.
Соединение 1 имело свойства ADME in vitro, подходящие для оценки in vivo доклинической и клинической фармакологии молекулы. Проницаемость была высокой, и клиренс in vitro в криоконсервированных гепатоцитах был низким у человека и собаки (0,3±0,05 и ˂0,65 мл/мин/г соответственно) и от среднего до высокого у мышей и крыс (4,6 и 7,4±0,7 мл/мин/кг соответственно). Среднее воздействие Соединения 1 в крови самцов мышей CD-1 увеличивалось с дозой после однократного п/о введения 10, 30 или 100 мг/кг (нормализованная по средней дозе Cmax 58±112, 113±27 и 113±27; DNAUCinf 202±101, 294±47 и 403±246 соответственно).
Соединение 1 увеличивает секрецию Z-α1-антитрипсина на модели дефицита 1-антитрипсина у трансгенных мышей
Для исследования терапевтического потенциала Соединения 1 при дефиците Z-α1-антитрипсина, соединение было протестировано на модели трансгенных мышей, сконструированной со случайной вставкой гена Z-α1-антитрипсина человека (Teckman et al. Am J Physiol Gastrointest Liver Physiol. 2004;286:G851-G62). Для определения подходящего режима дозирования Соединения 1 в этих исследованиях общее и несвязанное воздействие на трансгенных мышей оценивали на модели in silico. Это было основано на наблюдаемых ФК-параметрах у мышей дикого типа и включении циркулирующего стока с высоким сродством к лекарственному средству, представляющего среднее значение 5 мкМ общего количества Z-α1-антитрипсина в крови мышей на исходном уровне со сродством к лекарственному средству 1,5 нМ. На основании этого моделирования было предсказано, что режим перорального дозирования 100 мг/кг три раза в день будет поддерживать концентрацию свободного лекарственного средства выше EC50, измеренную для секреции Z-α1-антитрипсина (~300 нМ) в течение периода дозирования. Напротив, можно ожидать, что несвязанные концентрации Соединения 1 после 30 и 10 мг/кг три раза в день будут значительно ниже ЕС50 для секреции в течение большей части периода дозирования.
Для исследования взаимосвязи ФК-ФД Соединения 1 Z-α1-антитрипсиновым трансгенным животным вводили дозу 100, 30 или 10 мг/кг Соединения 1 три раза в день, и на 6-й день кровь и печень собирали через 3 часа (~Cmax) и через 8 часов (Cmin) после введения дозы для измерения общего и свободного лекарственного средства в обеих тканях. Также брали кровь для измерения мономерного Z-α1-антитрипсина в плазме. Общие концентрации Соединения 1 определяли с помощью специфического анализа ЖХ-МС/МС, и свободное несвязанное лекарственное средство в обеих тканях определяли с помощью равновесного диализа и использовали для получения несвязанных концентраций. Свободные и общие концентрации в крови соответствовали прогнозам и подтвердили, что уровни Cmin концентрации свободного лекарственного средства были на уровне или выше 300 нМ в течение большей части периода дозирования после дозирования 100 мг/кг, тогда как дозы 30 мг/кг и 10 мг/кг приводили к уровням свободного лекарственного средства в крови значительно ниже клеточных концентраций ЕС50 в течение большей части периода дозирования. Концентрации как свободного, так и общего лекарственного средства Соединения 1 в целевом участке действия в печени были эквивалентны концентрациям в крови.
Недавняя работа показала, что значительная часть общего количества Z-α1-антитрипсина в циркуляции находится в полимерной конформации (Tan et al. Eur Respir J. 2014;43(5):1501-4). Поскольку не существует антител, специфичных к мономерному Z-α1-антитрипсину для непосредственного определения его концентрации, был разработан метод деконволюции, основанный на иммуноанализе с антителами или к общему, или к полимерному α1-антитрипсину и калибровочных кривых с очищенным мономерным и полимерным Z-α1-антитрипсином. Мономерный Z-α1-антитрипсин измеряли в образцах плазмы через 6 дней после введения дозы, и уровни нормализовали по отношению к контрольным уровням каждого животного до введения дозы, чтобы учесть естественные изменения Z-α1-антитрипсина между животными. 100 мг/кг Соединения 1 приводили к 6-7-кратному увеличению уровней циркулирующего мономерного Z-α1-антитрипсина, демонстрируя надежное целевое воздействие в печени. Группы 30 мг/кг и 10 мг/кг также давали значительное дозозависимое увеличение циркулирующего Z-α1-антитрипсина, несмотря на то, что свободные концентрации были ниже клеточной ЕС50 для секреции в течение большей части или всего периода введения дозы. Общие уровни лекарственного средства и изменения Z-α1-антитрипсина после 3 дней введения дозы были неотличимы от уровней после 6 дней введения дозы.
Взятые вместе, эти результаты подтверждают связывание с мишенью в печени после перорального введения дозы Соединения 1 и демонстрируют, что циркулирующие уровни Z-α1-антитрипсина могут быть увеличены на терапевтически релевантную величину на модели дефицита α1-антитрипсина. Увеличение циркулирующего Z-α1-антитрипсина при более низких дозах может указывать на то, что достижение устойчивых уровней свободного лекарственного средства выше клеточной EC50 для секреции может не потребоваться in vivo для значительного повышения уровней Z-α1-антитрипсина.
Поскольку Соединение 1 блокирует образование полимера в клетках, исследовали влияние дозирования Соединения 1 на уровни полимера в печени. Недавно было показано, что Z-α1-антитрипсиновые полимеры, образованные в CHO-TET-ON-Z-A1AT и ZZ-ИПСК-гепатоцитах, выводятся из клеток с t1/2 от 8 до 48 часов в зависимости от того, разделяются ли они на растворимые или нерастворимые фракции. Поскольку соединение не связывает полимер Z-α1-антитрипсин, влияние на общие уровни полимера в печени будет зависеть от скорости, с которой печень может очистить уже присутствующий полимер, и скорости, с которой полимер продолжает накапливаться у животных, не получавших лекарственное средство. Поэтому Соединение 1 дозировали 100 мг/кг три раза в день, как указано выше, в течение 20 дней. На 15-й и 21-й дни введения дозы определяли изменения в образцах плазмы мономерного Z-α1-антитрипсина, как указано выше, и наблюдали повышение в 7-8 раз по сравнению с исходными уровнями перед введением дозы, аналогично эффекту у животных, которым вводили дозу 3 или 6 дней, что соответствует устойчивому связыванию с мишенью в течение периода дозирования. Уровни полимера в печени были исследованы путем окрашивания 2С1 антиполимерным моноклональным антителом и оценены вслепую или cпециалистом по лабораторной диагностике, или количественной оценкой с использованием алгоритма для измерения всех областей положительного окрашивания. Не было обнаружено различий в общей полимерной нагрузке печени с помощью ручной или количественной оценки. У старых Z-α1-антитрипсиновых мышей обнаруживаются плотные полимерные включения, которые не наблюдаются в печени объектов с дефицитом α1-антитрипсина.
Для исследования есть ли в печени субпопуляция полимера, которая реагировала бы на соединение в течение периода времени эксперимента, области окрашивания были разделены на области низкой, средней и высокой интенсивности и отдельно оценивались в отношении ответа на соединение. Никакого влияния соединения не было заметно ни в одной из этих подразделенных областей. Учитывая, что образование полимера полностью блокируется Соединением 1 in vitro, был сделан вывод, что потребуется более длительный период дозирования, чтобы повлиять на общую полимерную нагрузку у Z-α1-антитрипсиновых трансгенных мышей. Однако еще предстоит определить, обладает ли печень мыши способностью очищать Z-α1-антитрипсиновый полимер после продолжительного поражения образования Z-α1-антитрипсина в печени на протяжении всей жизни или действительно ли это требуется для получения положительного действия в отношении чувствительности к вторичным триггерам дисфункции печени.
Обсуждение
Сокристаллические структуры с α1-антитрипсином дают представление о механизме действия, посредством которого Соединение 1 ингибирует полимеризацию Z-α1-антитрипсина. Хотя механизм, который приводит к патологическому образованию полимера в печени, не установлен, облигатная и центральная роль петли реационного центра в этом процессе является хорошо известной (см. (a) Lomas et al. Nature. 1992;357:605-7; (b) Ekeowa et al. Proc Natl Acad Sci USA. 2010;107(40):17146-5; и (c) Yamasaki et al. EMBO Rep. 2011;12(10):1011-7). Все существующие модели основаны на вставке дополнительной β-нити, полученной из петли реационного центра, между нитями 3 и 5 A-листа, чему способствует дестабилизирующее влияние Z-мутации на этот структурный элемент. Сокристаллическая структура α1-антитрипсина и Соединения 1 предполагает, что соединение может улучшать эффект Z-мутации E342K вследствие: (i) оптимизации гидрофобной упаковки в области разрыва; (ii) образования водородных связей со скрытыми полярными атомами и также (iii) смещения основной цепи в верхней части нити 5A в конфигурацию, менее совместимую с частичной вставкой петли, что, как предполагается, является ранней стадией в полимеризации петля-лист (Gooptu et al. Proc Natl Acad Sci (USA). 2000;97(1):67-72) и облигатной стадией в С-концевой полимеризации (Yamasaki et al. EMBO Rep. 2011;12(10):1011-7). Смещение, очевидное в верхней части нити 5A, согласуется с обусловленным скоростью ассоциации предпочтением Z-варианта, который дестабилизирует эту область по отношению к белку дикого типа. В кристаллических структурах боковая цепь при 342 ориентирована в направлении растворителя и не взаимодействует непосредственно с Соединением 1; таким образом, ключевым параметром, определяющим предпочтение соединения, вероятно, будет повышенная доступность криптического кармана. После образования карман оказывается структурно эквивалентным в обоих вариантах, что отражается сходной скоростью диссоциации. Этот способ действия также совместим с отсутствием связывания с полимерами, в котором частичная или полная вставка петли реакционного центра завершает поворот бета-шпильки, которая перекрывает сайт связывания соединения.
При связывании Соединения 1 в области разрыва наблюдается заметная стабилизация нативного состояния α1-антитрипсина по сравнению с конформационным изменением, связанным с образованием интермедиата M⃰. Это, в свою очередь, предотвращает образование полимеров. Прецедент этого общего механизма может быть обнаружен в инструментальном моноклональном антителе, которое оказывало аналогичное действие на α1-антитрипсин (Ordóñez et al. FASEB J. 2015;29:2667-78; Motamedi-Shad et al. Biochem J. 2016;473(19):3269-90).
Недостаток активности серпина в нижних дыхательных путях, приводящий к расщеплению паренхимы легких локально высвобождающейся нейтрофильной эластазой, способствует возникновению эмфиземы, наблюдаемой у объектов с дефицитом α1-антитрипсина, поскольку у редких пациентов, у которых полностью отсутствует α1-антитрипсин, развивается панлобулярная эмфизема (Fregonese et al. Respir Med. 2008;102(6):876-84), и заместительная терапия α1-антитрипсином продемонстрировала некоторое положительное действие в отношении прогрессирования заболевания (Chapman et al. Lancet. 2015;386(9991):360-8). Соединение 1 увеличивает секрецию Z-α1-антитрипсина, корректируя дефект фолдинга, и увеличивает концентрации серпина в плазме до 8 раз на модели дефицита α1-антитрипсина у трансгенных мышей.
Поскольку Соединение 1 ингибирует активность серпина α1-антитрипсина, будучи связанным с белком, не следует ожидать значительного увеличения активности серпина в течение периода введения дозы, действительно, Соединение 1 будет ингибировать активность серпина остаточного α1-антитрипсина у пациентов и можно ожидать, что он будет блокировать активность заместительной терапии, если ее вводить совместно с Соединением 1. Однако период полувыведения мономерного Z-α1-антитрипсина у человека, как полагают, составляет ~3-5 дней, тогда как лекарственное средство, как ожидается, будет очищенным с t1/2 через несколько часов после введения дозы, повышая вероятность пульсирующего режима дозирования, который приведет к увеличению активности серпина. Определение того, какое время воздействия потребуется лекарственному средству для значительного повышения уровней, сокращения воспалительного полимера в легких и периода вымывания между дозами для сохранения активности серпина, требует дальнейшего изучения. Подобным образом необходимо будет оценить соотношение риск-польза от блокирования остаточной активности серпина во время периода введения дозы; однако медленное развитие заболевания легких у объектов с дефицитом α1-антитрипсина в течение многих десятилетий позволяет предположить, что острые эффекты, связанные с ингибированием активности серпина, маловероятны. α1-антитрипсиновые полимеры были идентифицированы в легких пациентов с ХОБЛ с двумя нормальными аллелями α1-антитрипсина, что позволяет предположить, что α1-антитрипсиновые полимеры могут быть преобладающим признаком воспаления при ХОБЛ независимо от генотипа α1-антитрипсина (Bazzan et al. Chest,. 2018;154:607-16).
Соединение 1 блокирует полимеризацию Z-α1-антитрипсина в бесклеточной среде и в ЭР клеток, и свежеобразованный полимер выводится клетками посредством секреции, протеасомального разрушения или аутофагии с периодом полувыведения 8-48 часов. Однако не наблюдалось снижения Z-α1-антитрипсиновых полимеров в печени после 20 дней введения дозы Соединения 1 у Z-α1-антитрипсиновых трансгенных мышей, несмотря на 7-кратное увеличение циркулирующих уровней в течение 3 дней после введения дозы, которое сохранялось на протяжении всего периода эксперимента, указывая на устойчивое связывание с мишенью. Вероятное объяснение этого заключается в том, что полимеры, которые накапливаются в печени трансгенных мышей, не выводятся так быстро, как те, которые могут быть созданы в модельных системах, таких как клетки CHO и ИПСК-гепатоциты в течение нескольких дней. Хотя это, вероятно, является результатом хронического поражения, молекулярный или клеточный механизм этого неясен, но клеточное старение может способствовать данному эффекту. Предыдущие исследования активатора аутофагии карбамазапина продемонстрировали выраженные эффекты на полимер печени у Z-α1-антитрипсиновых трансгенных мышей после 2 недель введения дозы, что позволяет предположить, что печень действительно сохраняет способность очищать полимер после хронического поражения накопления полимера Z-α1-антитрипсина (Hidvegi et al. Science.2010; 329: 229-32). Напротив, подходы РНКи, которые ингибируют экспрессию Z-α1-антитрипсина и образование полимера, сообщили о снижении уровня Z-α1-антитрипсина в печени трансгенных мышей после 12-33 недель лечения, хотя и без сообщений о данных в более ранние сроки (Guo et al. J Clin Invest 2014;124(1):251-61). Вместе эти данные предполагают, что печень сохраняет способность очищать полимер после хронического поражения и что вполне вероятно, что Соединение 1 необходимо будет вводить трансгенным Z-α1-антитрипсиновым мышам в течение значительно более длительного периода, чем 20 дней, чтобы продемонстрировать влияние на общие уровни полимера в печени. Еще неизвестно, действительно ли образовавшийся полимер токсичен для клеток и его необходимо вывести из печени, чтобы иметь некоторое функциональное положительное действие, или же накопленные полимерные включения действительно инертны, и прекращение выработки полимера является достаточным для восстановления функционирования ЭР и жизнеспособности клеток (Ordóñez et al. Hepatology. 2013;57(5):2049-60; Dickens et al. FASEB J. 2016;30(12):4083-97).
Наблюдаемые стабильные уровни общего и свободного соединения Соединения 1 у трансгенных Z-α1-антитрипсиновых мышей были хорошо предсказаны с помощью ФК-модели in silico, построенной на данных метаболического клиренса in vitro и данных связывания с белками плазмы, данных ФК in vivo мышей дикого типа и величины, включающей 5 мкМ циркулирующий сток для лекарственного средства с аффинностью 1,5 нМ, представляющей Z-α1-антитрипсин в крови. Целевая концентрация свободного лекарственного средства была выбрана на основе наблюдаемой активности в анализах секреции in vitro, в которых общее лекарственное средство приближается к свободному лекарственному средству в анализе. Однако увеличение Z-α1-антитрипсина в кровотоке при более низких дозах Соединения 1 является неожиданным, учитывая, что системные уровни свободного лекарственного средства находятся между EC10-EC20 и EC20-EC30 в течение периода введения дозы для доз 10 и 30 мг/кг соответственно. Причина этого неясна, однако возможно, что связывание с мишенью in vivo является больше, чем прогнозировалось на основе эффективности в клеточных анализах in vitro. Альтернативно, возможно, что эффект первого прохождения лекарственного средства, достигающего печени сразу после абсорбции, обеспечивает некоторую эффективность по сравнению с предсказанной на основе моделирования концентрации соединения на уровнях устойчивого состояния. Вместе эти данные предполагают потенциальные преимущества для необходимого воздействия соединения для обеспечения эффективности в клинике.
Новый механизм лечения конформационного заболевания
Полагают, что соединения изобретения стабилизируют мономерный Z-α1-антитрипсин путем связывания в начале нити 5 β-листа A, таким образом подавляя локальный эффект Z-мутации (Lomas et al. Biochemistry. 1993;32:500-8). В частности, они смещают нить 5 в направлении нити 3 и таким образом «корректируют» локальное возмущение, вызванное остатком лизина в положении 342. Сайт связывания Соединения 1 не был идентифицирован в предыдущих кристаллических структурах α1-антитрипсина (Elliott et al. Protein Science. 2000;9:1274-81). Однако их влияние на блокирование перехода полностью свернутого Z-α1-антитрипсина в полимерогенный интермедиат и их эффективность в клеточных и животных моделях заболевания предполагает, что они действуют на Z-α1-антитрипсин в почти нативной или нативной конформации. Это означает, что полимеры образуются из почти нативной или нативной конформации, а не из более протяженного интермедиата, и что небольшие молекулы препятствуют связыванию мономера с образованием полимера. Эта низкомолекулярная коррекция Z-мутации предоставляет новую стратегию лечения заболеваний печени у лиц с дефицитом α1-антитрипсина.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ЮСиЭл БИЗНЕС ЛТД
<120> НОВЫЕ СОЕДИНЕНИЯ
<130> N414701WO
<140> PCT/GB2019/051761
<141> 2019-06-21
<150> GB1810290.5
<151> 2018-06-22
<150> GB1906708.1
<151> 2019-05-13
<160> 6
<170> PatentIn version 3.5
<210> 1
<211> 394
<212> Белок
<213> Искусственная последовательность
<220>
<223> альфа-1-антитрипсин
<220>
<221> MISC_FEATURE
<222> (1)..(1)
<223> Xaa означает T или E
<220>
<221> MISC_FEATURE
<222> (101)..(101)
<223> Xaa означает R или H
<220>
<221> MISC_FEATURE
<222> (213)..(213)
<223> Xaa означает A или V
<220>
<221> MISC_FEATURE
<222> (232)..(232)
<223> Xaa означает C или S
<220>
<221> MISC_FEATURE
<222> (342)..(342)
<223> Xaa означает E или K
<220>
<221> MISC_FEATURE
<222> (376)..(376)
<223> Xaa означает D или E
<400> 1
Xaa Asp Pro Gln Gly Asp Ala Ala Gln Lys Thr Asp Thr Ser His His
1 5 10 15
Asp Gln Asp His Pro Thr Phe Asn Lys Ile Thr Pro Asn Leu Ala Glu
20 25 30
Phe Ala Phe Ser Leu Tyr Arg Gln Leu Ala His Gln Ser Asn Ser Thr
35 40 45
Asn Ile Phe Phe Ser Pro Val Ser Ile Ala Thr Ala Phe Ala Met Leu
50 55 60
Ser Leu Gly Thr Lys Ala Asp Thr His Asp Glu Ile Leu Glu Gly Leu
65 70 75 80
Asn Phe Asn Leu Thr Glu Ile Pro Glu Ala Gln Ile His Glu Gly Phe
85 90 95
Gln Glu Leu Leu Xaa Thr Leu Asn Gln Pro Asp Ser Gln Leu Gln Leu
100 105 110
Thr Thr Gly Asn Gly Leu Phe Leu Ser Glu Gly Leu Lys Leu Val Asp
115 120 125
Lys Phe Leu Glu Asp Val Lys Lys Leu Tyr His Ser Glu Ala Phe Thr
130 135 140
Val Asn Phe Gly Asp Thr Glu Glu Ala Lys Lys Gln Ile Asn Asp Tyr
145 150 155 160
Val Glu Lys Gly Thr Gln Gly Lys Ile Val Asp Leu Val Lys Glu Leu
165 170 175
Asp Arg Asp Thr Val Phe Ala Leu Val Asn Tyr Ile Phe Phe Lys Gly
180 185 190
Lys Trp Glu Arg Pro Phe Glu Val Lys Asp Thr Glu Glu Glu Asp Phe
195 200 205
His Val Asp Gln Xaa Thr Thr Val Lys Val Pro Met Met Lys Arg Leu
210 215 220
Gly Met Phe Asn Ile Gln His Xaa Lys Lys Leu Ser Ser Trp Val Leu
225 230 235 240
Leu Met Lys Tyr Leu Gly Asn Ala Thr Ala Ile Phe Phe Leu Pro Asp
245 250 255
Glu Gly Lys Leu Gln His Leu Glu Asn Glu Leu Thr His Asp Ile Ile
260 265 270
Thr Lys Phe Leu Glu Asn Glu Asp Arg Arg Ser Ala Ser Leu His Leu
275 280 285
Pro Lys Leu Ser Ile Thr Gly Thr Tyr Asp Leu Lys Ser Val Leu Gly
290 295 300
Gln Leu Gly Ile Thr Lys Val Phe Ser Asn Gly Ala Asp Leu Ser Gly
305 310 315 320
Val Thr Glu Glu Ala Pro Leu Lys Leu Ser Lys Ala Val His Lys Ala
325 330 335
Val Leu Thr Ile Asp Xaa Lys Gly Thr Glu Ala Ala Gly Ala Met Phe
340 345 350
Leu Glu Ala Ile Pro Met Ser Ile Pro Pro Glu Val Lys Phe Asn Lys
355 360 365
Pro Phe Val Phe Leu Met Ile Xaa Gln Asn Thr Lys Ser Pro Leu Phe
370 375 380
Met Gly Lys Val Val Asn Pro Thr Gln Lys
385 390
<210> 2
<211> 418
<212> Белок
<213> Homo sapiens
<400> 2
Met Pro Ser Ser Val Ser Trp Gly Ile Leu Leu Leu Ala Gly Leu Cys
1 5 10 15
Cys Leu Val Pro Val Ser Leu Ala Glu Asp Pro Gln Gly Asp Ala Ala
20 25 30
Gln Lys Thr Asp Thr Ser His His Asp Gln Asp His Pro Thr Phe Asn
35 40 45
Lys Ile Thr Pro Asn Leu Ala Glu Phe Ala Phe Ser Leu Tyr Arg Gln
50 55 60
Leu Ala His Gln Ser Asn Ser Thr Asn Ile Phe Phe Ser Pro Val Ser
65 70 75 80
Ile Ala Thr Ala Phe Ala Met Leu Ser Leu Gly Thr Lys Ala Asp Thr
85 90 95
His Asp Glu Ile Leu Glu Gly Leu Asn Phe Asn Leu Thr Glu Ile Pro
100 105 110
Glu Ala Gln Ile His Glu Gly Phe Gln Glu Leu Leu Arg Thr Leu Asn
115 120 125
Gln Pro Asp Ser Gln Leu Gln Leu Thr Thr Gly Asn Gly Leu Phe Leu
130 135 140
Ser Glu Gly Leu Lys Leu Val Asp Lys Phe Leu Glu Asp Val Lys Lys
145 150 155 160
Leu Tyr His Ser Glu Ala Phe Thr Val Asn Phe Gly Asp Thr Glu Glu
165 170 175
Ala Lys Lys Gln Ile Asn Asp Tyr Val Glu Lys Gly Thr Gln Gly Lys
180 185 190
Ile Val Asp Leu Val Lys Glu Leu Asp Arg Asp Thr Val Phe Ala Leu
195 200 205
Val Asn Tyr Ile Phe Phe Lys Gly Lys Trp Glu Arg Pro Phe Glu Val
210 215 220
Lys Asp Thr Glu Glu Glu Asp Phe His Val Asp Gln Val Thr Thr Val
225 230 235 240
Lys Val Pro Met Met Lys Arg Leu Gly Met Phe Asn Ile Gln His Cys
245 250 255
Lys Lys Leu Ser Ser Trp Val Leu Leu Met Lys Tyr Leu Gly Asn Ala
260 265 270
Thr Ala Ile Phe Phe Leu Pro Asp Glu Gly Lys Leu Gln His Leu Glu
275 280 285
Asn Glu Leu Thr His Asp Ile Ile Thr Lys Phe Leu Glu Asn Glu Asp
290 295 300
Arg Arg Ser Ala Ser Leu His Leu Pro Lys Leu Ser Ile Thr Gly Thr
305 310 315 320
Tyr Asp Leu Lys Ser Val Leu Gly Gln Leu Gly Ile Thr Lys Val Phe
325 330 335
Ser Asn Gly Ala Asp Leu Ser Gly Val Thr Glu Glu Ala Pro Leu Lys
340 345 350
Leu Ser Lys Ala Val His Lys Ala Val Leu Thr Ile Asp Glu Lys Gly
355 360 365
Thr Glu Ala Ala Gly Ala Met Phe Leu Glu Ala Ile Pro Met Ser Ile
370 375 380
Pro Pro Glu Val Lys Phe Asn Lys Pro Phe Val Phe Leu Met Ile Glu
385 390 395 400
Gln Asn Thr Lys Ser Pro Leu Phe Met Gly Lys Val Val Asn Pro Thr
405 410 415
Gln Lys
<210> 3
<211> 418
<212> Белок
<213> Искусственная последовательность
<220>
<223> полная последовательность Z-A1AT
<400> 3
Met Pro Ser Ser Val Ser Trp Gly Ile Leu Leu Leu Ala Gly Leu Cys
1 5 10 15
Cys Leu Val Pro Val Ser Leu Ala Glu Asp Pro Gln Gly Asp Ala Ala
20 25 30
Gln Lys Thr Asp Thr Ser His His Asp Gln Asp His Pro Thr Phe Asn
35 40 45
Lys Ile Thr Pro Asn Leu Ala Glu Phe Ala Phe Ser Leu Tyr Arg Gln
50 55 60
Leu Ala His Gln Ser Asn Ser Thr Asn Ile Phe Phe Ser Pro Val Ser
65 70 75 80
Ile Ala Thr Ala Phe Ala Met Leu Ser Leu Gly Thr Lys Ala Asp Thr
85 90 95
His Asp Glu Ile Leu Glu Gly Leu Asn Phe Asn Leu Thr Glu Ile Pro
100 105 110
Glu Ala Gln Ile His Glu Gly Phe Gln Glu Leu Leu Arg Thr Leu Asn
115 120 125
Gln Pro Asp Ser Gln Leu Gln Leu Thr Thr Gly Asn Gly Leu Phe Leu
130 135 140
Ser Glu Gly Leu Lys Leu Val Asp Lys Phe Leu Glu Asp Val Lys Lys
145 150 155 160
Leu Tyr His Ser Glu Ala Phe Thr Val Asn Phe Gly Asp Thr Glu Glu
165 170 175
Ala Lys Lys Gln Ile Asn Asp Tyr Val Glu Lys Gly Thr Gln Gly Lys
180 185 190
Ile Val Asp Leu Val Lys Glu Leu Asp Arg Asp Thr Val Phe Ala Leu
195 200 205
Val Asn Tyr Ile Phe Phe Lys Gly Lys Trp Glu Arg Pro Phe Glu Val
210 215 220
Lys Asp Thr Glu Glu Glu Asp Phe His Val Asp Gln Ala Thr Thr Val
225 230 235 240
Lys Val Pro Met Met Lys Arg Leu Gly Met Phe Asn Ile Gln His Cys
245 250 255
Lys Lys Leu Ser Ser Trp Val Leu Leu Met Lys Tyr Leu Gly Asn Ala
260 265 270
Thr Ala Ile Phe Phe Leu Pro Asp Glu Gly Lys Leu Gln His Leu Glu
275 280 285
Asn Glu Leu Thr His Asp Ile Ile Thr Lys Phe Leu Glu Asn Glu Asp
290 295 300
Arg Arg Ser Ala Ser Leu His Leu Pro Lys Leu Ser Ile Thr Gly Thr
305 310 315 320
Tyr Asp Leu Lys Ser Val Leu Gly Gln Leu Gly Ile Thr Lys Val Phe
325 330 335
Ser Asn Gly Ala Asp Leu Ser Gly Val Thr Glu Glu Ala Pro Leu Lys
340 345 350
Leu Ser Lys Ala Val His Lys Ala Val Leu Thr Ile Asp Lys Lys Gly
355 360 365
Thr Glu Ala Ala Gly Ala Met Phe Leu Glu Ala Ile Pro Met Ser Ile
370 375 380
Pro Pro Glu Val Lys Phe Asn Lys Pro Phe Val Phe Leu Met Ile Glu
385 390 395 400
Gln Asn Thr Lys Ser Pro Leu Phe Met Gly Lys Val Val Asn Pro Thr
405 410 415
Gln Lys
<210> 4
<211> 404
<212> Белок
<213> Искусственная последовательность
<220>
<223> SEQ ID NO: 1, содержащая аффинную метку
<400> 4
Met Arg Gly Ser His His His His His His Thr Asp Pro Gln Gly Asp
1 5 10 15
Ala Ala Gln Lys Thr Asp Thr Ser His His Asp Gln Asp His Pro Thr
20 25 30
Phe Asn Lys Ile Thr Pro Asn Leu Ala Glu Phe Ala Phe Ser Leu Tyr
35 40 45
Arg Gln Leu Ala His Gln Ser Asn Ser Thr Asn Ile Phe Phe Ser Pro
50 55 60
Val Ser Ile Ala Thr Ala Phe Ala Met Leu Ser Leu Gly Thr Lys Ala
65 70 75 80
Asp Thr His Asp Glu Ile Leu Glu Gly Leu Asn Phe Asn Leu Thr Glu
85 90 95
Ile Pro Glu Ala Gln Ile His Glu Gly Phe Gln Glu Leu Leu Arg Thr
100 105 110
Leu Asn Gln Pro Asp Ser Gln Leu Gln Leu Thr Thr Gly Asn Gly Leu
115 120 125
Phe Leu Ser Glu Gly Leu Lys Leu Val Asp Lys Phe Leu Glu Asp Val
130 135 140
Lys Lys Leu Tyr His Ser Glu Ala Phe Thr Val Asn Phe Gly Asp Thr
145 150 155 160
Glu Glu Ala Lys Lys Gln Ile Asn Asp Tyr Val Glu Lys Gly Thr Gln
165 170 175
Gly Lys Ile Val Asp Leu Val Lys Glu Leu Asp Arg Asp Thr Val Phe
180 185 190
Ala Leu Val Asn Tyr Ile Phe Phe Lys Gly Lys Trp Glu Arg Pro Phe
195 200 205
Glu Val Lys Asp Thr Glu Glu Glu Asp Phe His Val Asp Gln Val Thr
210 215 220
Thr Val Lys Val Pro Met Met Lys Arg Leu Gly Met Phe Asn Ile Gln
225 230 235 240
His Ser Lys Lys Leu Ser Ser Trp Val Leu Leu Met Lys Tyr Leu Gly
245 250 255
Asn Ala Thr Ala Ile Phe Phe Leu Pro Asp Glu Gly Lys Leu Gln His
260 265 270
Leu Glu Asn Glu Leu Thr His Asp Ile Ile Thr Lys Phe Leu Glu Asn
275 280 285
Glu Asp Arg Arg Ser Ala Ser Leu His Leu Pro Lys Leu Ser Ile Thr
290 295 300
Gly Thr Tyr Asp Leu Lys Ser Val Leu Gly Gln Leu Gly Ile Thr Lys
305 310 315 320
Val Phe Ser Asn Gly Ala Asp Leu Ser Gly Val Thr Glu Glu Ala Pro
325 330 335
Leu Lys Leu Ser Lys Ala Val His Lys Ala Val Leu Thr Ile Asp Glu
340 345 350
Lys Gly Thr Glu Ala Ala Gly Ala Met Phe Leu Glu Ala Ile Pro Met
355 360 365
Ser Ile Pro Pro Glu Val Lys Phe Asn Lys Pro Phe Val Phe Leu Met
370 375 380
Ile Glu Gln Asn Thr Lys Ser Pro Leu Phe Met Gly Lys Val Val Asn
385 390 395 400
Pro Thr Gln Lys
<210> 5
<211> 24
<212> Белок
<213> Искусственная последовательность
<220>
<223> Сигнальный пептид
<400> 5
Met Pro Ser Ser Val Ser Trp Gly Ile Leu Leu Leu Ala Gly Leu Cys
1 5 10 15
Cys Leu Val Pro Val Ser Leu Ala
20
<210> 6
<211> 11
<212> Белок
<213> Искусственная последовательность
<220>
<223> аффинная метка гексагистидина и аминокислоты, предоставляемые вектором экспрессии
<400> 6
Met Arg Gly Ser His His His His His His Thr
1 5 10
<---
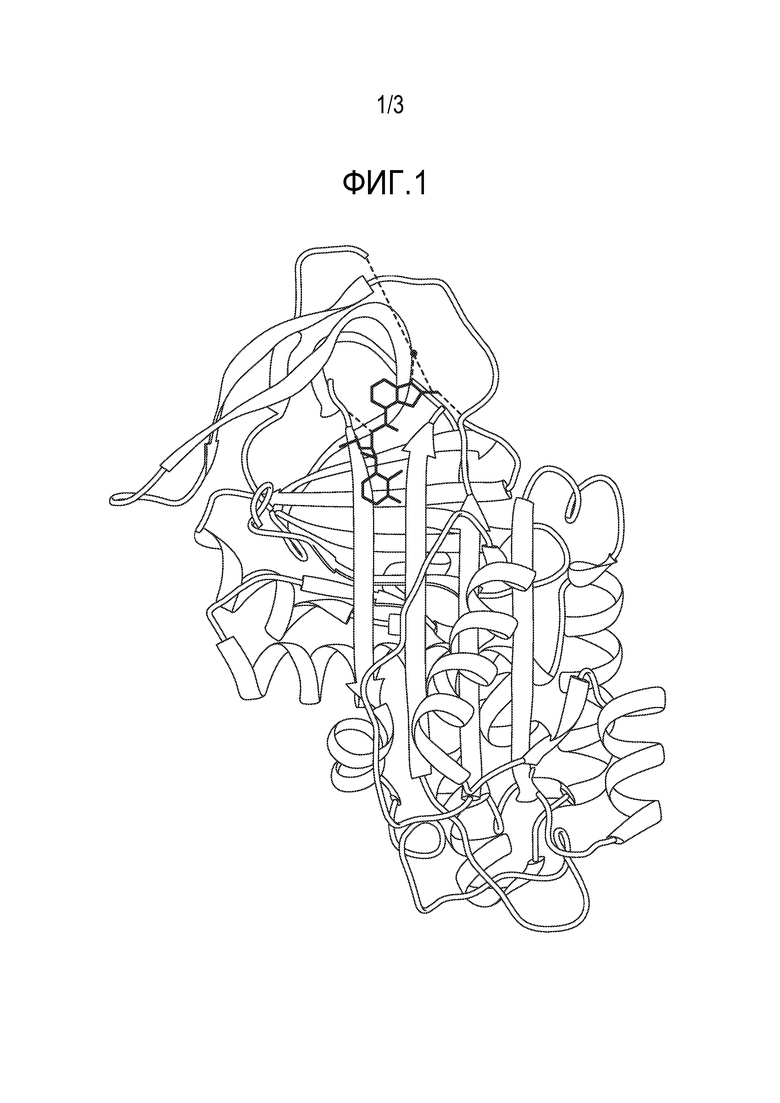
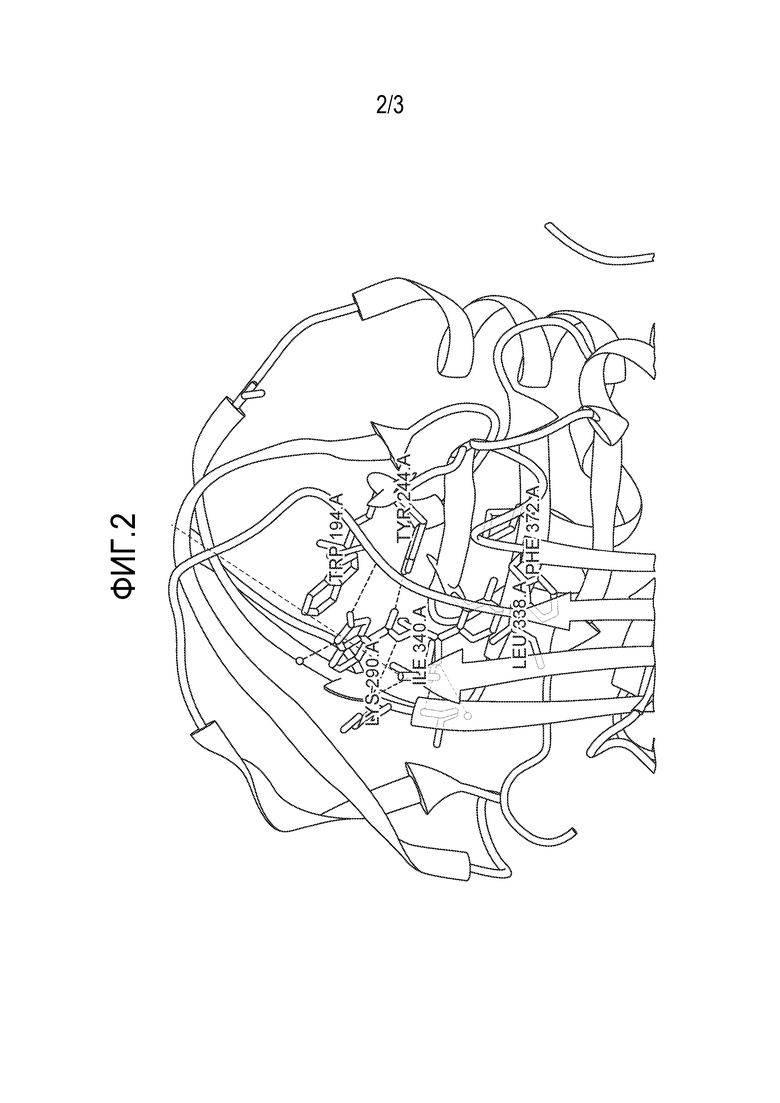

Изобретение относится к области биотехнологии. Описана группа изобретений, включающая соединение, фармацевтическую композицию для лечения заболевания или состояния, опосредованного α1-антитрипсином, включающую вышеуказанное соединение, способ лечения заболевания или состояния, опосредованного α1-антитрипсином, применение соединения или фармацевтической композиции для лечения заболевания или состояния, опосредованного α1-антитрипсином, и применение соединения или фармацевтической композиции при изготовлении лекарственного средства для применения при лечении заболевания или состояния, опосредованного альфа-1-антитрипсином. Изобретение расширяет арсенал средств для лечения заболевания или состояния, опосредованного α1-антитрипсином. 5 н. и 26 з.п. ф-лы, 3 ил., 6 табл., 2 пр.
1. Соединение с формулой (I)
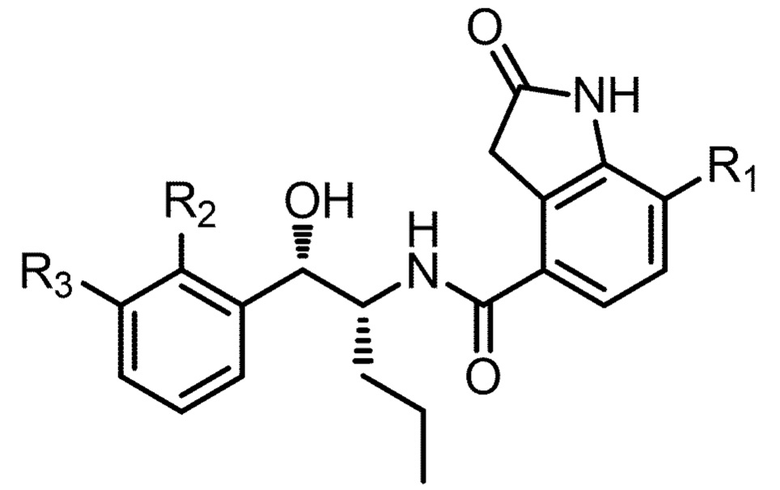
или его фармацевтически приемлемая соль, где:
R1 выбран из группы, состоящей из Н, F, СН3, СН2СН3, СН2СН2СНз, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, Cl, Br и I;
R2 выбран из группы, состоящей из СН3, Cl, СН2СН3, СН2СН2СН3, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, SH, CN, F, Br и I; и
R3 выбран из группы, состоящей из F, Cl, CN, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, Br, I и SH.
2. Соединение по п. 1, где:
R1 выбран из группы, состоящей из Н, F, СН3, NH2, ОН и Cl; R2 выбран из группы, состоящей из СН3, Cl, NH2, ОН, SH, CN и F; и
R3 выбран из группы, состоящей из F, Cl, CN, СН3, NH2, ОН и SH.
3. Соединение по п. 2, где:
R1 выбран из группы, состоящей из Н и F;
R2 выбран из группы, состоящей из СН3 и Cl; и
R3 выбран из группы, состоящей из F, Cl и CN.
4. Соединение по п. 3, где:
R1 представляет собой Н, R2 представляет собой СН3 и R3 представляет собой F, или
R1 представляет собой Н, R2 представляет собой СН3 и R3 представляет собой Cl, или
R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой CN, или
R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой F.
5. Соединение или фармацевтически приемлемая соль по п. 1, где соединение представляет собой
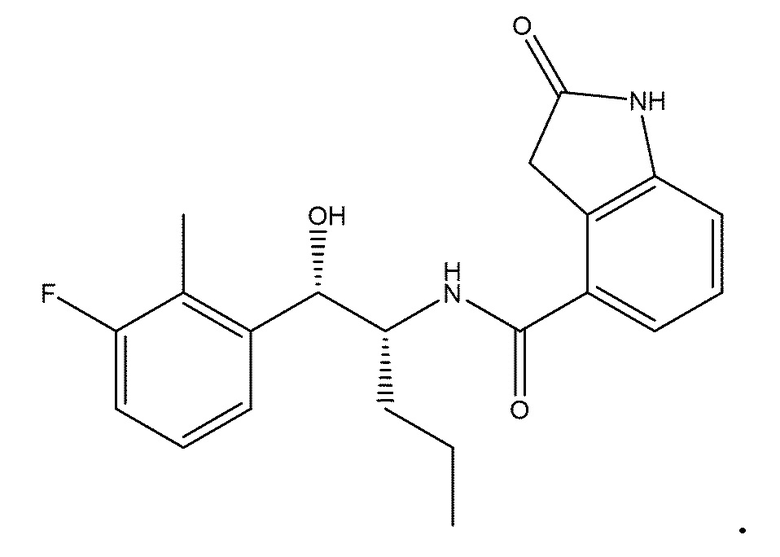
6. Соединение или фармацевтически приемлемая соль по п. 1, где соединение представляет собой
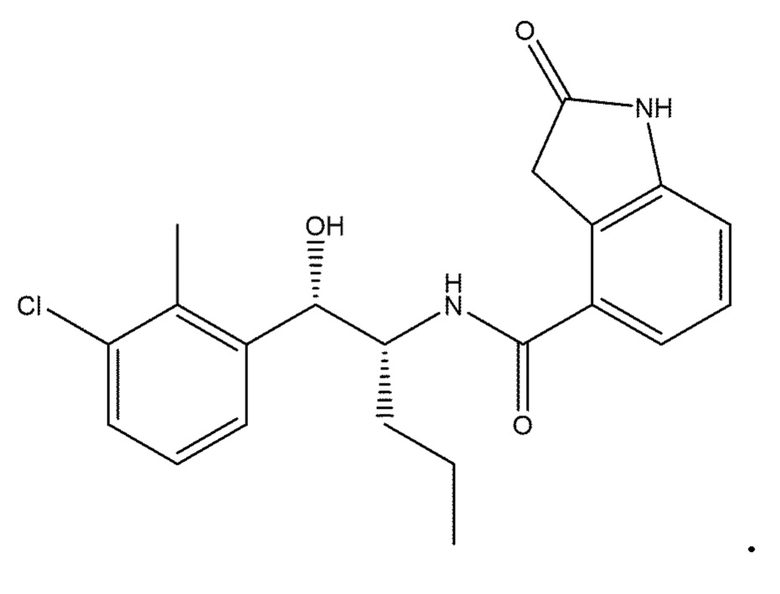
7. Соединение или фармацевтически приемлемая соль по п. 1, где соединение представляет собой
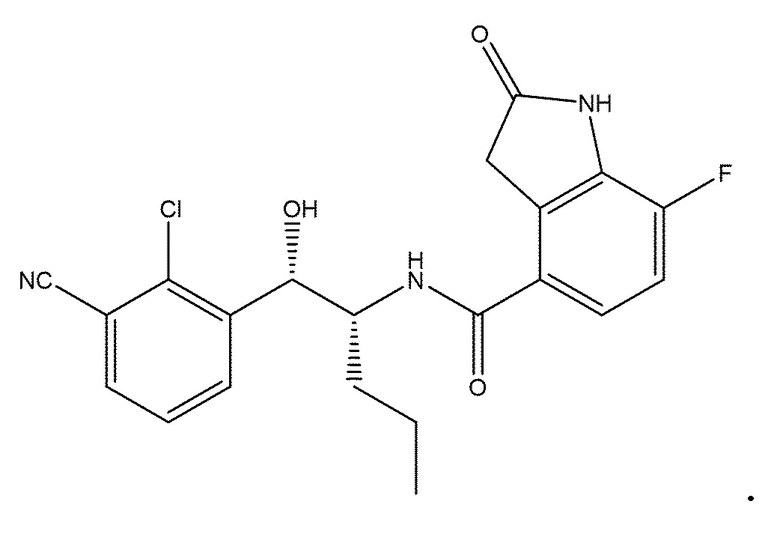
8. Соединение или фармацевтически приемлемая соль по п. 1, где соединение представляет собой
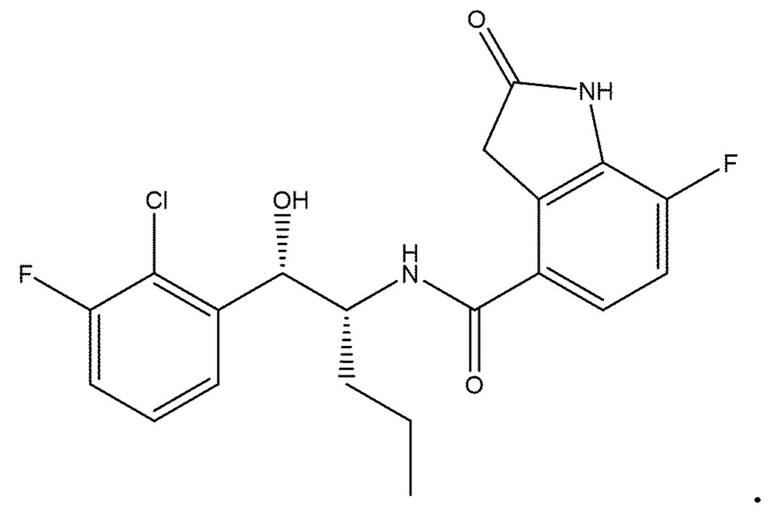
9. Фармацевтическая композиция для лечения заболевания или состояния, опосредованного α1-антитрипсином, содержащая соединение или фармацевтически приемлемую соль по любому из пп. 1-8 и фармацевтически приемлемый эксципиент.
10. Способ лечения заболевания или состояния, опосредованного α1-антитрипсином, включающий введение субъекту, нуждающемуся в этом, соединения с Формулой (I):
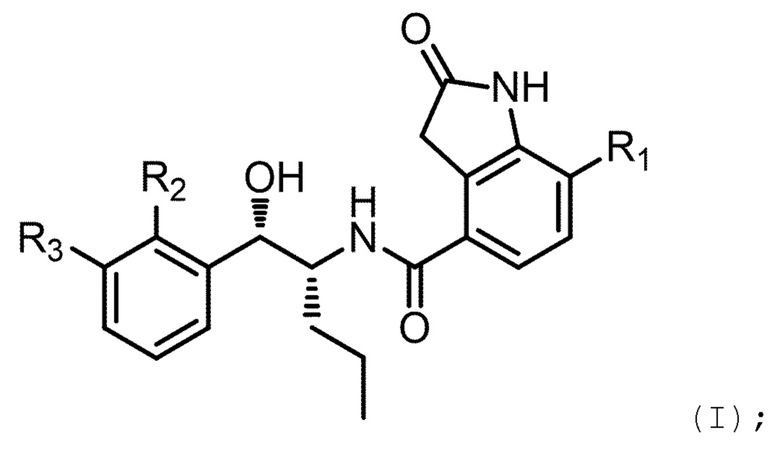
или его фармацевтически приемлемой соли, где:
R1 выбран из группы, состоящей из Н, F, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, Cl, Br и I;
R2 выбран из группы, состоящей из СН3, Cl, СН2СН3, СН2СН2СН3, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, SH, CN, F, Br и I; и
R3 выбран из группы, состоящей из F, Cl, CN, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, NH2, NHCH3, N(CH3)2, ОН, Br, I и SH.
11. Способ по п. 10, где:
R1 выбран из группы, состоящей из Н, F, СН3, NH2, ОН и Cl; R2 выбран из группы, состоящей из СН3, CI, NH2, ОН, SH, CN и F; и
R3 выбран из группы, состоящей из F, Cl, CN, СН3, NH2, ОН и SH.
12. Способ по п. 11, где:
R1 выбран из группы, состоящей из Н и F;
R2 выбран из группы, состоящей из СН3 и Cl; и R3 выбран из группы, состоящей из F, Cl и CN.
13. Способ по п. 12, где:
R1 представляет собой Н, R2 представляет собой СН3 и R3 представляет собой F, или
R1 представляет собой Н, R2 представляет собой СН3 и R3 представляет собой Cl, или
R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой CN, или
R1 представляет собой F, R2 представляет собой Cl и R3 представляет собой F.
14. Способ по п. 10, где соединение представляет собой
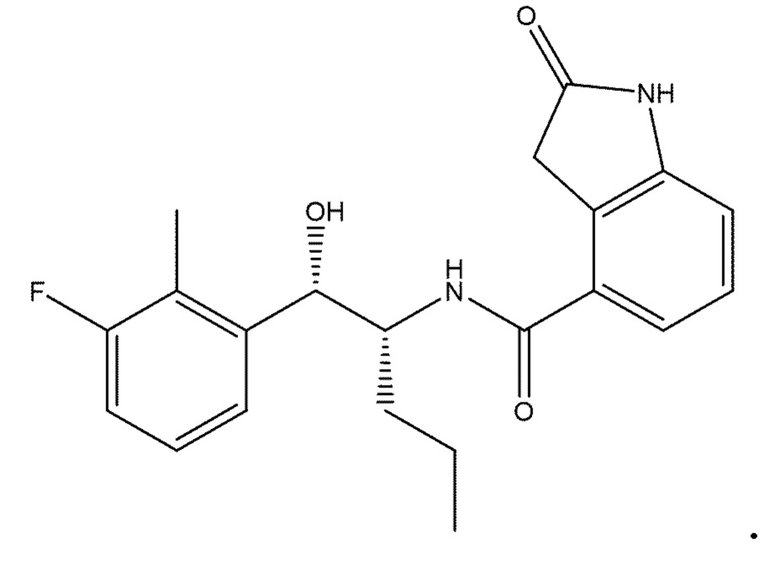
15. Способ по п. 10, где соединение представляет собой
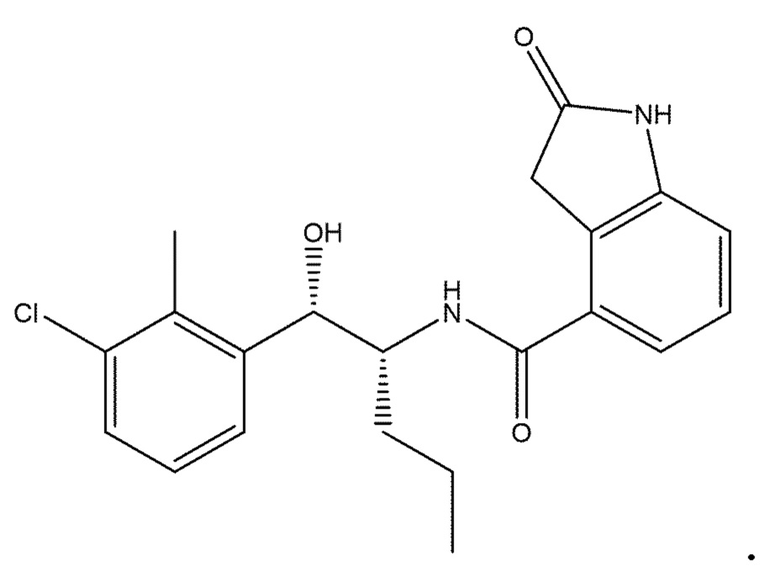
16. Способ по п. 10, где соединение представляет собой
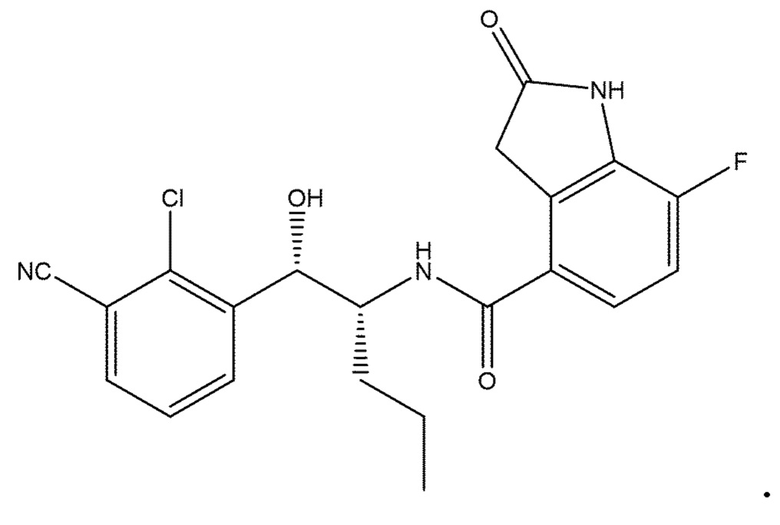
17. Способ по п. 10, где соединение представляет собой
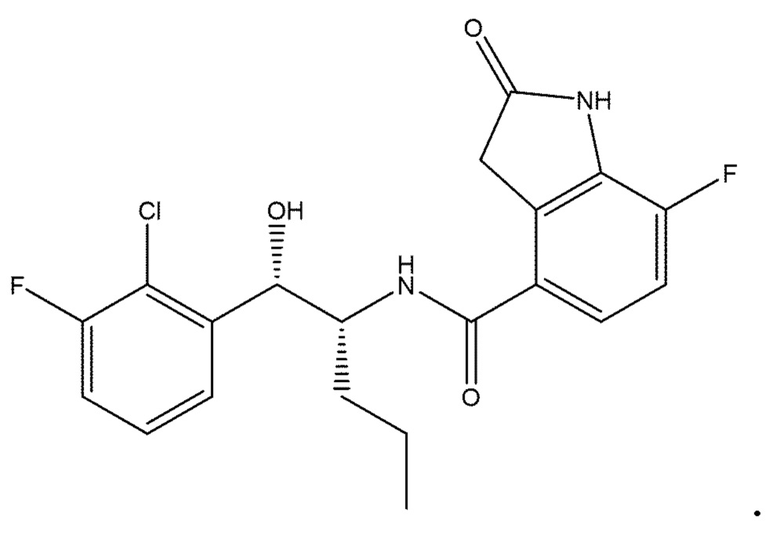
18. Способ по любому из пп. 10-17, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой серпинопатию.
19. Способ по любому из пп. 10-17, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой дефицит альфа-1-антитрипсина, дисфункцию печени, фиброз, цирроз, печеночную недостаточность, гепатоцеллюлярную карциному, хроническую обструктивную болезнь легких (ХОБЛ), астму, эмфизему, рак легких, дерматит или кожный зуд.
20. Способ по любому из пп. 10-17, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой желтуху, печеночную недостаточность, цирроз печени, аутоиммунный гепатит и гепатоцеллюлярную карциному.
21. Способ по любому из пп. 10-17, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой респираторное заболевание, и способ дополнительно включает введение субъекту, нуждающемуся в этом, бронходилататора.
22. Способ по любому из пп. 10-21, где субъект представляет собой человека.
23. Применение соединения по любому из пп. 1-8 или фармацевтической композиции по п. 9 для лечения заболевания или состояния, опосредованного α1-антитрипсином.
24. Применение по п. 23, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой дефицит альфа-1-антитрипсина, дисфункцию печени, фиброз, цирроз, печеночную недостаточность, гепатоцеллюлярную карциному, хроническую обструктивную болезнь легких (ХОБЛ), астму, эмфизему, рак легких, дерматит или кожный зуд.
25. Применение по п. 23, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой желтуху, печеночную недостаточность, цирроз печени, аутоиммунный гепатит и гепатоцеллюлярную карциному.
26. Применение по п. 23, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой респираторное заболевание, и соединение или фармацевтическая композиция предназначены для применения с в комбинации с бронходилататором.
27. Применение соединения по любому из пп. 1-8 или фармацевтической композиции по п. 9 при изготовлении лекарственного средства для применения при лечении заболевания или состояния, опосредованного альфа-1-антитрипсином.
28. Применение по п. 27, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой дефицит альфа-1-антитрипсина, дисфункцию печени, фиброз, цирроз, печеночную недостаточность, гепатоцеллюлярную карциному, хроническую обструктивную болезнь легких (ХОБЛ), астму, эмфизему, рак легких, дерматит или кожный зуд.
29. Применение по п. 27, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой желтуху, печеночную недостаточность, цирроз печени, аутоиммунный гепатит и гепатоцеллюлярную карциному.
30. Применение по п. 27, где заболевание или состояние, опосредованное α1-антитрипсином, представляет собой респираторное заболевание, и лекарственное средство предназначено для применения в комбинации с бронходилататором.
31. Применение по любому из пп. 23-30, где субъект представляет собой человека.
| Mitchell E | |||
| L., Khan Z | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Вагонетка для кабельной висячей дороги, переносной радиально вокруг центральной опоры | 1920 |
|
SU243A1 |
| Berthelier V | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление для разборки шпал | 1959 |
|
SU126256A1 |
| US 9801637 B2, 31.10.2017 | |||
| НОВЫЙ ВАРИАНТ АЛЬФА-1-АНТИТРИПСИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2012 |
|
RU2567007C1 |
