Область техники
Изобретение относится к области получения кристаллических цеолитных материалов, в том числе с заданными кислотными свойствами, которые могут быть использованы в качестве компонентов катализаторов и адсорбентов.
Уровень техники
К кристаллическим элементосиликатам семейства пентасил относятся цеолиты, содержащие фрагменты пятичленных колец в структуре. Цеолиты данного семейства, такие как BEA, MEL (известный также как цеолит ZSM-12) и MFI (известный также как цеолит ZSM-5). Эти цеолиты имеют микропористую структуру, построенную из сдвоенных пятичленных колец и содержащую трехмерную систему пересекающихся каналов. Данные цеолиты характеризуются высокой гидротермальной устойчивостью и возможностью регулирования их кислотности в широком диапазоне за счет изменения отношения Si/M в кристаллическом каркасе цеолита (от 15 до ∞).
Наряду с высокой пористостью, обусловлено широкое применение цеолитов в катализе. Цеолиты являются катализаторами важных процессов нефтепереработки (каталитический крекинг, изомеризация, алкилирование алкилирование ароматических соединений, этерификация, олигомеризация олефинов). Возможность использования цеолитов в качестве носителей для различных наночастиц металлов и их соединений расширяет область их применения на процессы, включающие в себя окислительно-восстановительные реакции, такие, как гидрокрекинг прямое окисление насыщенных углеводородов в спирты, а также эпоксидирование олефинов.
Характер кристаллической решетки цеолитов определяет наличие микропор полостей и каналов, которые могут выступать как молекулярные сита, контролирующие ход реакции за счет избирательного пропускания реагентов, продуктов или даже переходных комплексов. Размер кристаллов, количества макро-, мезо- и микропор, различных включений, а также химический состав цеолита, том числе, локализация атомов алюминия и других элементов в центре кристаллов или на их поверхности, зависят от механизма и условий процесса кристаллизации.
Цеолиты способны к изоморфному замещению атомов кремния на атомы других металлов, в частности, на атомы Ti, Al, Sn, Zr, Hf и др. Это позволяет изменить некоторые физико-химические свойства цеолита и соответственно расширить область его применения в качестве катализатора.
Существует два основных подхода к синтезу элементосиликатов с гетероатомами в каркасе: прямой синтез и пост-синтетическое модифицирование. В прямом синтезе материал получают в результате гидротермальной кристаллизации гелей, содержащих источник кремния, источник металла, органический структурообразующий агент и воду во фторидной среде. Пост-синтетическое модифицирование основано на встраивании гетероатомов в структуру элементосиликатов. В подавляющем большинстве случаев на первой стадии проводят деалюминирование цеолита, после чего вводят гетероатомы.
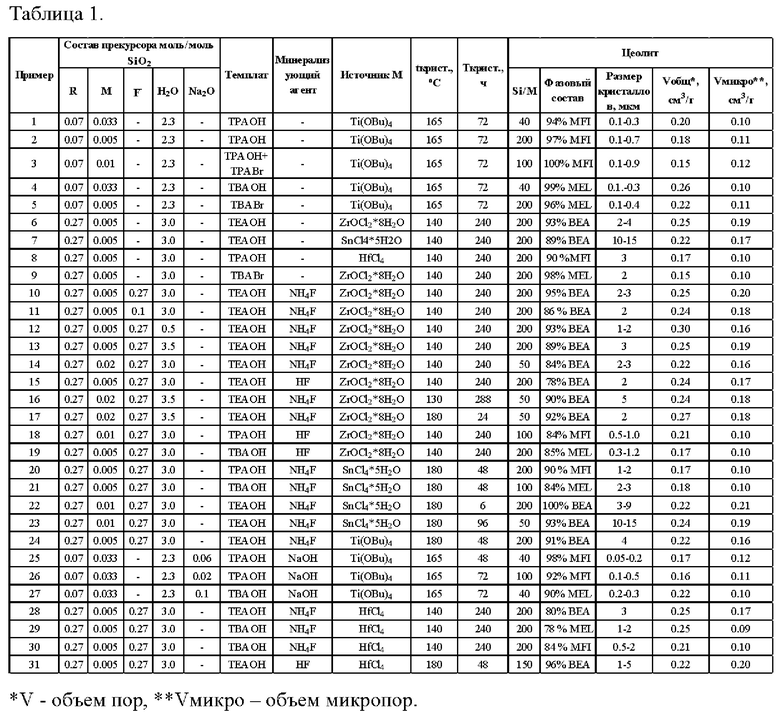
Высокую каталитическую активность кристаллических элементосиликатов с гетероатомами объясняют оптимальной льюисовской кислотностью, которая создается изоморфным замещением кремния в тетраэдрических позициях каркаса цеолита на гетероатом. Различают два типа центров: «закрытые» центры, связанные с четырьмя атомами кремния структуры цеолита через кислородные мостики, и «открытые» центры, содержащие М-ОН группу. На фиг. 1 схематически показаны «открытые» и «закрытые» центры на примере элементосиликата Zr-BEA.
Благодаря развитой пористой структуре и наличию Льюисовской кислотности элементосиликаты используются в таких реакциях, как реакция Меервейна Понндорфа - Верлея (восстановление альдегидов и кетонов в спирты действием изопропилового спирта в присутствии изопропилата алюминия и обратная окислительная реакция); реакции Дильса-Альдера (реакция [4+2]-циклоприсоединения алкенов, алкинов или соединения, содержащие двойные связи с гетероатомом и сопряженных диенов с образованием шестичленного цикла); реакции Байера-Виллигера (окисление кетонов и альдегидов под действием перекиси водорода, мононадсерной кислоты или органических надкислот, приводящее к образованию сложных эфиров, лактонов или кислот) и др. Важную роль в таких реакциях играет льюисовская кислотность активного центра.
Кристаллические элементосиликаты применяются в качестве катализаторов в процессах переработки биомассы. Известны системы, в которых используют Zr-BEA при переработке фурфурола и левулиновой кислоты в гамма-валеролактон, фурфуроловый спирт.Элементосиликаты также применяют для получения производных молочной кислоты из С6 Сахаров, диметилфурана из гидроксиметилфурфурола.
Традиционно, синтез кристаллических элементосиликатов семейства пентасилов осуществляется в гидротермальных условиях при температуре 120-200°С и повышенном давлении. Реакционные смеси, использующиеся для синтеза, состоят из аморфных источников кремния и алюминия, минерализующего агента (фторид анионы или неорганические щелочи), органического темплата-структурообразователя и воды. В качестве органических темплатов могут быть использованы органические амины, четвертичные аммонийные основания и их соли, а также спирты и другие соединения. Наиболее предпочтительными темплатами, обеспечивающим кристаллизацию пентасилов в наиболее широком диапазоне условий, является гидроксид или бромид тетраалкилммония (далее - ТААОН или TAABr). В качестве источника оксида кремния при синтезе пентасилов используют соединения кремния различной степени дисперсности, в том числе, молекулярные (органические эфиры кремниевой кислоты), коллоидные (силиказоль и жидкое стекло) и конденсированные (силикагели). В качестве источника алюминия используют неорганические и органические соли алюминия, органические эфиры алюминия, металлический алюминий и алюминат натрия. После гидротермальной кристаллизации смесь подвергают разделению на твердую и жидкую фазы путем фильтрации или центрифугирования. Твердая фаза, состоящая из кристаллов цеолита, подвергается отмывке от темплата и минерализующего агента, просушке, прокалке для удаления темплата. Методы кристаллизации пентасилов методом гидротермальной кристаллизации обладают существенным недостатком, заключающимся в неэффективном расходовании исходных реагентов и длительном времени синтеза.
Из уровня техники известен способ получения цеолита Al-ВЕА в щелочной среде, позволяющий получать цеолит как в виде порошка, так и в виде агрегатов без связующего размером 0,1-5 мм Способ включает приготовление прекурсора путем пропитки шарикового силикагеля раствором, содержащим источник алюминия, неорганическую щелочь и темплат, выдержку прекурсора на воздухе до достижения определенной влажности и кристаллизацию в отсутствие свободной воды при температуре и давлении, соответствующих получению цеолита Al-ВЕА. Образование цеолита Al-ВЕА происходит в результате превращения прекурсора в парах воды, которая вносится только в составе прекурсора. (RU 2737895 С1, 04.12. 2020).
Однако с помощью известного способа не удается синтезировать элементосиликаты с гетероатомами Zr, Sn, Ti, Hf.
Из уровня техники известен способ получения цеолита типа Al-MFI кристаллизацией прекурсора в отсутствие свободной воды. Приготовление прекурсора проводят путем пропитки сфер силикагеля рабочим раствором, содержащим воду, темплат, источник алюминия (алюминат натрия), щелочь (гидроксид натрия). После кристаллизации прекурсора проводят отмывку и сушку полученных кристаллов. Пропитка в рамках данного метода осуществляется либо по влагоемкости, либо при поглощении из раствора в статических условиях или в условиях принудительной циркуляции. При использовании данного метода образуются частицы цеолита, форма и размер которых идентичны форме и размеру частиц исходного силикагеля (RU 2640236, 27.12.2017).
Недостатками указанного способа являются невозможность получения цеолитной структуры пентасил с гетероатомами Zr, Sn, Ti, Hf.
Из уровня техники известен способ получения титаносиликата, согласно которому реакционную смесь готовят путем перемешивания диоксида кремния, гидроксида тетрапропилармония в течение 30 минут. Далее тетрабутоксид титана растворяли в изопропиловом спирте и добавляли в специальный смеситель-миксер для перемешивания к смеси диоксида кремния и гидроксида тетрапропилармония, куда также добавляли воду. Затем смесь перемешивали до получения однородной пасты без добавления связующего вещества. Пасту высушивают до консистенции, пригодной для экструдирования с помощью пресса. Полученный пастообразный прекурсор подвергают кристаллизации в автоклаве при 150°С в течение 24 часов. Полученный продукт представляет собой экструдаты без связующего, содержащие 100% титаносилика со структурой MFI. (WO 2007033102, 22.03.2007).
Недостатками указанного способа является неоднородность целевого продукта из-за невозможности однородного распределения тетрабутоксида титана в пасте прекурсора. Кроме того, способ не является универсальным, так как не воспроизводится в попытках синтеза элементосиликатов с другими гетероатомами в каркасе цеолита.
Наиболее близким к предложенному техническому решению является способ получения эдементосиликатов, таких, как оловосиликат, оловотитаносиликат, оловогерманийтитаносиликат методом гидротермальной золь-гель кристаллизации. Элементосиликаты готовят из реакционной смеси, содержащей тетраэтоксисилан, тетраэтиламмония гидроксид, источник гетероатомов и плавиковую кислоту. После гелирования к реакционной смеси добавляют кристаллы цеолита ВЕА, являющиеся затравкой. Смесь кристаллизуют при 140°С в течение 11-20 дней при перемешивании. Продукт промывают водой на воронке Бюхнера и сушат при 100°С. Высушенный образец прокаливают при 580°С для удаления органического темплата. (US 5968473, 19.10. 1999)
Недостатками указанного способа, принятого за прототип, являются длительное время синтеза, многостадийность из-за необходимости предварительного приготовления затравочных кристаллов Al-ВЕА с последующим деалюминированием азотной кислотой. Для данного способа характерен также высокий расход исходных реагентов.
Сущность изобретения
Задача настоящей группы изобретений состоит в разработке нового способа получения кристаллических элементосиликатов из семейства цеолитов пентасил с развитой микропористой структурой, характеризующихся высокой степенью кристалличности и фазовой чистоты,
Техническим результатом является расширение ассортимента элементосиликатов как синтезируемых типов цеолитовых структур из семейства пентасилов (MFI, MEL, ВЕА), так и замещающих гетероатомов (Ti, Sn, Zr, Hf), введенных в каркас указанных типов цеолитов, а также упрощение способа, уменьшение продолжительности процесса и снижение расхода реагентов.
Поставленная задача решается описываемым способом синтеза кристаллического элементосиликата семейства цеолитов пентасил структурного типа MFI или MEL или ВЕА, содержащего в структуре каркаса гетероатомы элементов, выбранных из Ti, Sn, Zr, Hf, согласно которому осуществляют приготовление прекурсора, характеризующегося составом, соответствующим области кристаллизации элементосиликата заданного структурного типа, путем пропитки по влагоемкости частиц силикагеля (SiC)2) раствором, содержащим воду, органический структурообразователь (R) гидроксид или бромид тетраалкиламмония, где алкил - этил, пропил или бутил, и источник гетероатома (М), с обеспечением в прекурсоре следующих мольных отношений компонентов:
M:SiO2 от 0,005 до 0,033
R:SiO2 от 0,07 до 0,27
H2O:SiO2 от 0,5 до 3,5,
после чего влажный прекурсор выдерживают в закрытой емкости при комнатной температуре в течение 1-2 часов, помещают в устройство для
кристаллизации и осуществляют парофазную кристаллизацию в отсутствие свободной воды при 130-180°С в течение 6-288 часов до образования твердых частиц, образованных сростками кристаллов элементосиликата, имеющих форму и размер частиц идентичные форме и размеру частиц исходного силикагеля, твердые частицы промывают, сушат и прокаливают.
Согласно способу, пропитке подвергают частицы силикагеля предпочтительно в виде сфер 0,2-7 мм при комнатной температуре.
Преимущественно, пропиточный раствор дополнительно содержит минерализующий агент, представленный фторидом водорода или аммония (F) при мольном отношении F:SiO2 от 0,1 до 0,27, либо гидроксидом щелочного металла (Me), где Me, предпочтительно Na2O при мольном отношении от 0,02 до 0,1.
Согласно способу, при приготовлении прекурсора в качестве источника гетероатома циркония предпочтительно используют оксид-дихлорид циркония.
Согласно способу, при приготовлении прекурсора в качестве источника гетероатома олова предпочтительно используют тетрахлорид олова.
Согласно способу, при приготовлении прекурсора в качестве источника гетероатома титана предпочтительно используют тетрабутоксид титана в присутствии спиртового растворителя.
В качестве источника гетероатома гафния предпочтительно используют тетрахлорид гафния.
Прокаливание предпочтительно проводят при 550°С в течение 10 часов.
Поставленная задача решается также описываемым кристаллическим элементосиликатом цеолитной структуры семейства пентасил типа MFI или ВЕА или MEL, который содержит в структуре гетероатомы элементов (М), выбранные из Zr, Sn, Ti, Hf, и получен способом, охарактеризованным выше в виде сростков кристаллов с размером единичных кристаллов в интервале от 0,1 до 15 мкм, характеризующийся мольным отношением Si/M от 40 до 200, объемом микропор 0,09-0,21 см3/г.
Преимущественно, кристаллический элементосиликат имеет механическую прочность 8-10 Н.
Для приготовления прекурсора нами были использованы промышленные марки силикагелей в виде частиц сферической формы, хотя можно использовать и частицы неправильной формы, например, отходы от производства силикагелей после технологической стадии рассева. Размер частиц исходного силикагеля составляет 0,2-7 мм. Использование таких частиц силикагеля для синтеза позволяет значительно упростить технологию получения, т.к. продуктом кристаллизации являются частицы такой же формы (сферической или неправильной), образованные сростками единичных кристаллов элементосиликата. Исходная форма частиц сохраняется в процессе кристаллизации и последующих обработок. Качество полученного кристаллического элементосиликата не зависит от формы и размера частиц силикагеля, используемых в качестве источника SiO2. Поскольку после кристаллизации получают элементосиликат в виде частиц, форма которых идентична форме частиц исходного силикагеля, то, во-первых, упрощаются последующие процедуры (отмывки, прокаливания), а во-вторых можно получить продукт заданной геометрической формы.
При синтезе элементосиликатов заявленным способом большим преимуществом является отсутствие жидких продуктов после стадии кристаллизации. Продукт непосредственно после кристаллизации подают на стадию отмывки водой, что упрощает способ, исключая трудоемкую стадию разделения фаз, и, как следствие, сокращает продолжительность процесса. Кроме того, исключение обсуждаемой стадии значительно повышает выход элементосиликата относительно прототипа, так как основные потери кристаллического продукта обычно происходят на стадии разделения кристаллов и жидких продуктов, обязательной для способа-прототипа.
Количественные признаки, включенные в независимый пункт формулы (п. 1), определены нами экспериментально.
Мольные отношения компонентов M:SiO2 в интервале 0,005-0,033, и R: SiO2 в интервале 0,07-0,27 являются существенными, поскольку выход за нижнюю и верхнюю границы заявленных интервалов мольных отношений не обеспечивает решение поставленной задачи, связанное с синтезом кристаллических элементосиликатов типа MFI или ВЕА или MEL с гетероатомами Zr, Sn, Ti, Hf в каркасе, с мольным отношением Si/M от 40 до 200, обладающих высокой фазовой чистотой (на уровне 80-100%), что является принципиальным фактором, определяющим их эффективность в каталитических процессах. Заявленный интервал мольного отношения H2O:SiO2 в интервале 0,5-3,5 дает возможность проведения кристаллизации в паровой фазе в отсутствии свободной воды, что упрощает способ и обеспечивает значительное снижение расхода воды (в 3-5 раз относительно способа-прототипа).
Выдержка влажного прекурсора после пропитки в течение 1-2 часов обеспечивает необходимую влажность прекурсора, подаваемого на стадию кристаллизации, т.е. требуемые молярные отношения.
Заявленные температура и время кристаллизации обеспечивают полноту кристаллизации прекурсора без образования посторонних фаз в продукте кристаллизации.
Решение поставленной задачи подтверждено далее в описании конкретными примерами (1-31), параметры проведения которых, а также характеристики синтезированных в примерах целевых продуктов, сведены в таблицу. Представленные примеры иллюстрируют заявленную группу изобретений, но не ограничивают ее объем.
Дополнительно изобретение проиллюстрировано фигурами 13.
На фиг. 1 схематически показаны «открытые» и «закрытые» центры в структуре элементосиликата на примере Zr-BEA.
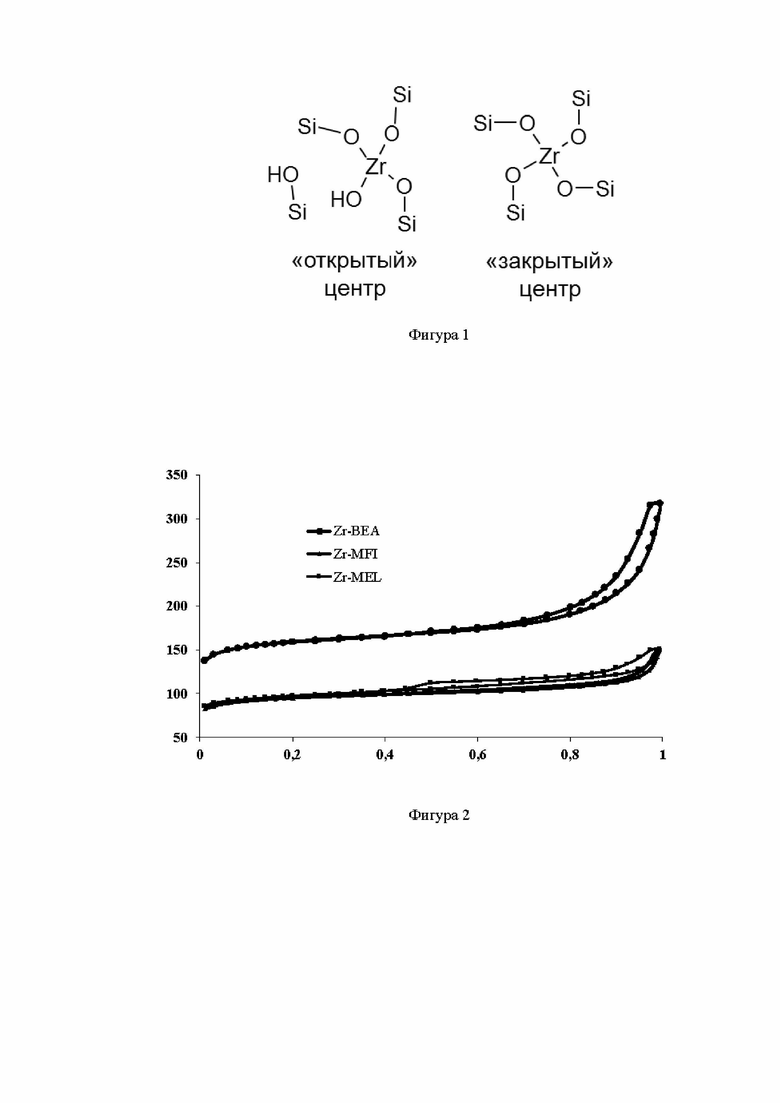
На фиг. 2 приведены изотермы низкотемпературной адсорбции-десорбции азота для синтезированных элементосиликатов Zr-BEA, Zr-MFI и Zr-MEL. Изотерма цирконийсиликатов относится к I типу по номенклатуре ИЮПАК и соответствует микропористым материалам. Гистерезис в области р/р0=0,50 для образцов Zr-BEA, Zr-MFI и Zr-MEL может быть связан с наличием незначительных количеств внутрикристалличесих полостей и дефектов, которые могут образовывать мезопоры. Изотермы однозначно показывают, что в результате превращений прекурсора в процессе парофазной кристаллизации, осуществляемой в заявленном способе, происходит образование микропористой структуры элементосиликатов.
На фиг. 3 приведены микрофотографии элементосиликатов с гетероатомами Ti, Sn, Zr, Hf, которые иллюстрируют морфологию и размер кристаллов синтезированных элементосиликатов.
Конкретные примеры.
Примеры 1-9 показывают возможность осуществление заявленного способа в отсутствие минерализующего агента.
Пример 1.
Пропитывающий раствор готовят смешением 9,2 г 25% раствора ТРАОН и 1,87 г тетрабутоксида титана. Пропитывающий раствор наносят пропиткой по влагоемкости на 10 г шарикового силикагеля. Полученный влажный прекурсор подсушивают при 80°С для достижения остаточного содержания воды 20% масс. в течение 3 часов. Полученный прекурсор засыпают в автоклав, автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в статических условиях при температуре 165°С в течение 3 суток. По окончании кристаллизации продукт в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Ti-MFI со степенью кристалличности 94% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 0.1-0.3 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 2.
Пропитывающий раствор и прекурсор готовят аналогично примеру 1. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 1. Анализ показал, что синтезирован элементосиликат Ti-MFI со степенью кристалличности 97% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 3.
Пропитывающий раствор и прекурсор готовят аналогично примеру 1. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 1. Анализ показал, что синтезирован элементосиликат Ti-MFI со степенью кристалличности 100% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 4.
Пропитывающий раствор и прекурсор готовят аналогично примеру 1. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 1. Анализ показал, что синтезирован элементосиликат Ti-MEL со степенью кристалличности 99% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 5.
Пропитывающий раствор и прекурсор готовят аналогично примеру 1. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 1. Анализ показал, что синтезирован элементосиликат Ti-MEL со степенью кристалличности 96% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 6.
Пропитывающий раствор готовят из 16.52 г водного 35% раствора гидроксида тетраэтиламмония, 0.23 г октогидрата цирконил хлорида. Полученным прозрачным раствором пропитывают по влагоемкости 8.65 г аморфного макропористого силикагеля. Мольные соотношения компонентов в прекурсоре приведены в таблице. Полученный влажный прекурсор выдерживают в закрытом виде 1.5 ч, по истечении которых влажные сферы прекурсора засыпают в автоклав. Автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в автоклаве при температуре 140°С в течение 10 суток. По окончании кристаллизации продукт цеолит ВЕА в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Zr-BEA со степенью кристалличности 93% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 2-4 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 7.
Пропитывающий раствор и прекурсор готовят аналогично примеру 6. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 6. Анализ показал, что синтезирован элементосиликат Sn-BEA со степенью кристалличности 89% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 8.
Пропитывающий раствор и прекурсор готовят аналогично примеру 6. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 6. Анализ показал, что синтезирован элементосиликат Hf-MFI со степенью кристалличности 90% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 9.
Пропитывающий раствор и прекурсор готовят аналогично примеру 6. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 6. Анализ показал, что синтезирован элементосиликат Zr-MEL со степенью кристалличности 98% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 10, 11 показывают возможность осуществления заявляемого способа при различных мольных отношениях SiO2/F- в прекурсоре.
Пример 10.
Пропитывающий раствор готовят из 16.52 г водного 35% раствора гидроксида тетраэтиламмония, 0.23 г октогидрата цирконил хлорида, 1.46 г фторида аммония. Полученным прозрачным раствором пропитывают по влагоемкости 8.65 г аморфного макропористого силикагеля. Мольные соотношения компонентов в прекурсоре приведены в таблице. Полученный влажный прекурсор выдерживают в закрытом виде 1.5 ч, по истечении которых влажные сферы прекурсора засыпают в автоклав. Автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в автоклаве при температуре 140°С в течение 10 суток. По окончании кристаллизации продукт цеолит ВЕА в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Zr-BEA со степенью кристалличности 95% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 2-3 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 11.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 86% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 10, 12, 13 показывают возможность осуществления заявляемого способа при различных мольных отношениях Н2О/SiO2 в прекурсоре.
Пример 12.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 93% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 13.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 89% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 10, 14 показывают возможность осуществления заявляемого способа при различных мольных отношениях SiO2/ZrO2 в прекурсоре.
Пример 14.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 84% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 15-17 показывают возможность осуществления заявляемого способа при различных температурах и временах кристаллизации прекурсора.
Примеры 10, 16 показывают возможность осуществления заявляемого способа при использовании различных источников фтора.
Пример 15.
Пропитывающий раствор готовят из 16.52 г водного 35% раствора гидроксида тетраэтиламмония, 0.23 г октогидрата цирконил хлорида, 1.62 г плавиковой кислоты (47.5% масс, HF). Полученным прозрачным раствором пропитывают по влагоемкости 8.65 г аморфного макропористого силикагеля. Мольные соотношения компонентов в прекурсоре приведены в таблице. Полученный влажный прекурсор выдерживают в закрытом виде 1.5 ч, по истечении которых влажные сферы прекурсора засыпают в автоклав. Автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в автоклаве при температуре 140°С в течение 10 суток. По окончании кристаллизации продукт цеолит ВЕА в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Zr-BEA со степенью кристалличности 78% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 2 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 16.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 130°С в течение 12 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 90% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 17.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 1 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Zr-BEA со степенью кристалличности 92% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 10, 18, 19 показывают возможность осуществления заявляемого способа при использовании различных темплатов в прекурсоре.
Пример 18.
Пропитывающий раствор готовят из 31.61 г водного 25% раствора гидроксида тетрапропиламмония, 0.23 г октогидрата цирконил хлорида, 1.6 г плавиковой кислоты (47.5% масс. HF). Полученным прозрачным раствором пропитывают по влагоемкости 8.65 г шарикового силикагеля. Мольные соотношения компонентов в прекурсоре приведены в таблице. Полученный влажный прекурсор выдерживают в закрытом виде 1.5 ч, по истечении которых влажные сферы прекурсора засыпают в автоклав. Автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в автоклаве при температуре 140°С в течение 10 суток. По окончании кристаллизации продукт цеолит ВЕА в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Zr-MFI со степенью кристалличности 84% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 0.5-1.0 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 19.
Пропитывающий раствор готовят из 50.42 г водного 25% раствора гидроксида тетрабутиламмония, 0.23 г октогидрата цирконил хлорида, 1.6 г плавиковой кислоты (47.5% масс. HF). Полученным прозрачным раствором пропитывают по влагоемкости 8.65 г шарикового силикагеля. Мольные соотношения компонентов в прекурсоре приведены в таблице. Полученный влажный прекурсор выдерживают в закрытом виде 1.5 ч, по истечении которых влажные сферы прекурсора засыпают в автоклав. Автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в автоклаве при температуре 140°С в течение 10 суток. По окончании кристаллизации продукт цеолит ВЕА в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Zr-MEL со степенью кристалличности 85% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 0.3-1.2 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Примеры 20-31 показывают возможность осуществления заявляемого способа при включении гетероатомов Sn, Ti, Hf в каркас элементосиликатов структурных типов MFI, ВЕА, MEL.
Пример 20.
Пропитывающий раствор и прекурсор готовят аналогично примеру 18. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 2 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 18. Анализ показал, что синтезирован элементосиликат Sn-MFI со степенью кристалличности 90% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 21.
Пропитывающий раствор и прекурсор готовят аналогично примеру 19. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 2 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 19. Анализ показал, что синтезирован элементосиликат Sn-MEL со степенью кристалличности 84% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 22.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 0.25 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Sn-BEA со степенью кристалличности 100% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 23.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 4 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Sn-BEA со степенью кристалличности 93% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 24.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 2 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Ti-BEA со степенью кристалличности 91% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 25.
Пропитывающий раствор готовят смешением 9,2 г 25% раствора ТРАОН и 0,8 г NaOH с последуюим прикапыванием 1,87 г тетрабутоксида титана. Полученный влажный прекурсор подсушивают при 80°С для достижения остаточного содержания воды 20% масс. в течение 3 часов. Полученный прекурсор засыпают в автоклав, автоклав герметизируют и помещают в нагревательное устройство. Кристаллизацию проводят в статических условиях при температуре 165°С в течение 2 суток. По окончании кристаллизации продукт в виде сфер выгружают из автоклава, отмывают дистиллированной водой до нейтрального рН. Продукт высушивают при температуре 60°С в течение 12 ч и прокаливают при 550°С в течение 10 ч. Анализ показал, что получен цеолит Ti-MFI со степенью кристалличности 98% (фазовая чистота) в виде изолированных кристаллов с размером кристаллов 0.05-0.2 мкм. Состав прекурсора, условия синтеза, а также характеристики пористой структуры элементосиликата представлены в таблице.
Пример 26.
Пропитывающий раствор и прекурсор готовят аналогично примеру 25. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 25. Анализ показал, что синтезирован элементосиликат Ti-MFI со степенью кристалличности 92% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 27.
Пропитывающий раствор и прекурсор готовят аналогично примеру 25. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 165°С в течение 3 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 25. Анализ показал, что синтезирован элементосиликат Ti-MEL со степенью кристалличности 90% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 28.
Пропитывающий раствор и прекурсор готовят аналогично примеру 10. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 10. Анализ показал, что синтезирован элементосиликат Hf-BEA со степенью кристалличности 80% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 29.
Пропитывающий раствор и прекурсор готовят аналогично примеру 19. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 19. Анализ показал, что синтезирован элементосиликат Hf-MEL со степенью кристалличности 78% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 30.
Пропитывающий раствор и прекурсор готовят аналогично примеру 18. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 140°С в течение 10 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 18. Анализ показал, что синтезирован элементосиликат Hf-MFI со степенью кристалличности 84% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Пример 31.
Пропитывающий раствор и прекурсор готовят аналогично примеру 15. Мольные соотношения компонентов, используемые исходные источники для приготовления прекурсора приведены в таблице. Парофазную кристаллизацию прекурсора проводят при 180°С в течение 2 суток. Порошкообразный продукт кристаллизации промывают, высушивают и прокаливают аналогично примеру 15. Анализ показал, что синтезирован элементосиликат Hf-BEA со степенью кристалличности 96% (фазовая чистота). Размер кристаллов и характеристики пористой структуры элементосиликата представлены в таблице.
Проведенное нами измерение механической прочности методом раздавливания показало, что все элементосилиты, полученные по примерам 1-31, имеют высокую механическую прочность, находящуюся в интервале от 8 до 10Н.
Как следует из описания, поставленная заявителем задача решена в объеме совокупности признаков независимых пунктов формулы. Как следует из примеров, технический результат относительно прототипа достигается, поскольку снижено расхода исходных реагентов, включая воду. Отсутствие трудоемких и продолжительных операций, таких, как разделение твердой и жидкой фаз после кристаллизации продукта и предварительный синтез затравочных кристаллов, сокращает количество стадий при синтезе и, соответственно, продолжительность процесса и его трудоемкость. Полнота использования исходных реагентов в заявляемом способе составила 92-97%. Относительно прототипа полнота использования кремнийсодержащего сырья повышена в 1,5-3 раза, а полнота использования органического темплата в 5-6 раз.
Дополнительным преимуществом группы изобретений является, то, что элементосиликаты структурного типа MFI или ВЕА или MEL, содержащие в структуре каркаса гетероатомы элементов (М), выбранные из Zr, Sn, Ti, Hf, полученные предложенным нами способом, непосредственно после синтеза имеют высокую механическую прочность (8-10 Н), что позволяет использовать их в различных каталитических процессах без введения связующего. Кроме того, изобретение обеспечивает синтез элементосиликатов в виде сростков кристаллов, форма и размер которых идентичны форме и размеру частиц исходного силикагеля, что позволяет получить целевой продукт с заранее заданными геометрическими параметрами.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО ЭЛЕМЕНТОСИЛИКАТА СЕМЕЙСТВА ЦЕОЛИТОВ ПЕНТАСИЛ И КРИСТАЛЛИЧЕСКИЙ ЭЛЕМЕНТОСИЛИКАТ | 2023 |
|
RU2814249C1 |
| Способ получения кристаллического цеолита семейства пентасил путем межцеолитных превращений. | 2021 |
|
RU2778923C1 |
| Способ получения кристаллического цеолита MEL и цеолит MEL | 2023 |
|
RU2805757C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО ЦЕОЛИТА ВЕА (варианты) И ПОЛУЧЕННЫЙ ЦЕОЛИТ ВЕА (варианты) | 2020 |
|
RU2737895C1 |
| МИКРОМЕЗОПОРИСТЫЙ КРИСТАЛЛИЧЕСКИЙ МАТЕРИАЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2393992C1 |
| ГРАНУЛИРОВАННЫЙ БЕЗ СВЯЗУЮЩЕГО КРИСТАЛЛИЧЕСКИЙ ЦЕОЛИТ MFI И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2675018C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦЕОЛИТА MFI | 2017 |
|
RU2640236C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦЕОЛИТА ТИПА MEL | 2018 |
|
RU2712549C1 |
| СПОСОБ КОНВЕРСИИ УГЛЕВОДОРОДОВ, КАТАЛИЗАТОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ С МИКРО-МЕЗОПОРИСТОЙ СТРУКТУРОЙ И СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 2005 |
|
RU2288034C1 |
| ГРАНУЛИРОВАННЫЙ ЦЕОЛИТ BEA БЕЗ СВЯЗУЮЩЕГО С ИЕРАРХИЧЕСКОЙ ПОРИСТОЙ СТРУКТУРОЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2023 |
|
RU2830125C1 |

Изобретение относится к синтезу цеолитных структур, которые могут быть использованы в области катализа и сорбционных технологий. Предложен способ синтеза кристаллического элементосиликата семейства цеолитов пентасил структурного типа MFI, или ВЕА, или MEL, содержащего в структуре гетероатомы элементов (М), выбранные из Ti, Sn, Zr, Hf, включает приготовление прекурсора, характеризующегося составом, соответствующим области кристаллизации соответствующего элементосиликата выбранного структурного типа. Проводят пропитку частиц силикагеля с размером частиц 0,2-7,0 мм раствором, содержащим структурообразователь (R) - гидроксид или бромид тетраалкиламмония, где алкил этил, пропил, или бутил и источник гетероатома (М), выбранный из Ti, Sn, Zr, Hf. Получают прекурсор в виде сфер со следующими мольными отношениями компонентов: М:SiO2 = 0,005 до 0,033, R:SiO2 = 0,07 до 0,27, H2O:SiO2 = 0,5-3,5. Приготовленный прекурсор подвергают парофазной кристаллизации в отсутствии свободной воды при 130-180°С в течение 12-288 часов до образования элементосиликата в виде сфер без связующего, имеющих исходную форму силикагеля 0,2-7,0 мм, состоящих из сростков кристаллов с размером единичных кристаллов в интервале от 0,1 до 15 мкм. Непосредственно после кристаллизации проводят стадии промывки, сушки и прокаливания. Технический результат - упрощение синтеза при снижении расхода реагентов, повышение степени использования исходных реагентов до 97%, расширение ассортимента способов получения кристаллических элементосиликатов структуры пентасил, обладающих высокой фазовой чистотой, развитой микропористостью и достаточной прочностью, а также расширение ассортимента металлов-гетероатомов, введенных в состав каркаса пентасила в результате прямого синтеза. 2 н. и 8 з.п. ф-лы, 3 ил., 1 табл., 31 пр.
1. Способ синтеза кристаллического элементосиликата семейства цеолитов пентасил структурного типа MFI, или BEA, или MEL, содержащего в структуре гетероатомы элементов (M), выбранных из Zr, Sn, Ti, Hf, включающий приготовление прекурсора, характеризующегося составом, соответствующим области кристаллизации элементосиликата выбранного структурного типа, при этом прекурсор готовят путём пропитки по влагоёмкости частиц силикагеля (SiO2) раствором, содержащим воду, органический структурообразователь (R) - гидроксид или бромид тетраалкиламмония, где алкил - этил, пропил или бутил, и источник гетероатома (M), с обеспечением в прекурсоре следующих мольных отношений компонентов:
затем влажный прекурсор выдерживают в закрытой ёмкости при комнатной температуре в течение 1-2 часов, помещают в устройство для кристаллизации и осуществляют парофазную кристаллизацию в отсутствие свободной воды при 130-180°С в течение 6-288 часов до образования твёрдых частиц, образованных сростками кристаллов элементосиликата, имеющих форму и размер частиц идентичные форме и размеру частиц исходного силикагеля, твёрдые частицы промывают, сушат и прокаливают.
2. Способ по п. 1, отличающийся тем, что приготовление прекурсора осуществляют путем пропитки аморфных частиц силикагеля в виде сфер 0,2-7 мм при комнатной температуре.
3. Способ по п. 1, отличающийся тем, что пропиточный раствор дополнительно содержит минерализующий агент, представленный фторидом водорода или аммония (F) при мольном отношении F:SiO2 от 0,1 до 0,27, либо гидроксидом щелочного металла (Ме), где Ме, предпочтительно, Na2O при мольном отношении от 0,02 до 0,1.
4. Способ по п. 1, отличающийся тем, что в качестве источника гетероатома Zr при приготовлении прекурсора предпочтительно используют оксид-дихлорид циркония.
5. Способ по п. 1, отличающийся тем, что в качестве источника гетероатома Sn при приготовлении прекурсора используют тетрахлорид олова.
6. Способ по п. 1, отличающийся тем, что в качестве источника гетероатома Ti при приготовлении прекурсора предпочтительно используют тетрабутоксид титана в присутствии спиртового растворителя.
7. Способ по п. 1, отличающийся тем, что в качестве источника гетероатома Hf при приготовлении прекурсора предпочтительно используют тетрахлорид гафния.
8. Способ по п. 1, отличающийся тем, что прокаливание высушенного элементосиликата осуществляют при 550°С в течение 10 часов.
9. Кристаллический элементосиликат семейства цеолитов пентасил структурного типа MFI, или BEA, или MEL, содержащего в структуре гетероатомы элементов (M), выбранные из Zr, Sn, Ti, Hf, полученный способом, охарактеризованным в пп. 1-8, в виде сфер размером 0,2-7,0 мм, состоящих из сростков кристаллов с размером единичных кристаллов в интервале от 0,1 до 15 мкм, характеризующийся мольным отношением Si/M от 40 до 200 и объёмом микропор 0,09-0,21 см3/г.
10. Кристаллический элементосиликат по п. 9, отличающийся тем, что характеризуется механической прочностью 8-10 Н.
| US 5698473 A1, 19.10.1999 | |||
| Способ получения кристаллического цеолита семейства пентасил путем межцеолитных превращений. | 2021 |
|
RU2778923C1 |
| WO 2007033102 A2, 22.03.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЦЕОЛИТА MFI | 2017 |
|
RU2640236C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОКРИСТАЛЛИЧЕСКОГО ЦЕОЛИТА ВЕА (варианты) И ПОЛУЧЕННЫЙ ЦЕОЛИТ ВЕА (варианты) | 2020 |
|
RU2737895C1 |
