Область техники, к которой относится изобретение
Настоящее изобретение касается области органического синтеза, в частности способа получения водорастворимых производных магнолола и производных хонокиола и их интермедиатов, и родственных моногидрокси-защищенных интермедиатов.
Предшествующий уровень техники
Магнолол и хонокиол являются основными активными ингредиентами средства традиционной китайской медицины cortex magnoliae officinalis (коры магнолии лекарственной), и химические структурные формулы магнолола и хонокиола изображены ниже:
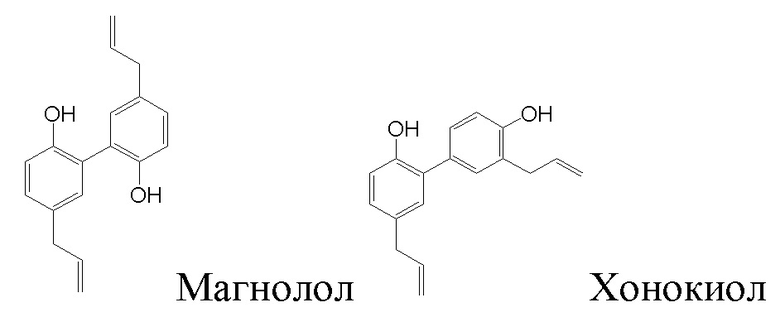
В 1930 году магнолол (Chinese herbal medicine: 2005, 36, 10, 1591-1594) впервые выделил Sugii в Японии из коры китайской magnolia officinalis. В 1989 году хонокиол (Chinese patent medicine: 1989, 11 (8): 223.) был также выделен из magnolia officinalis группой Meng Lizhen с соавторами в Китае.
Магнолол и хонокиол имеют широкий спектр фармакологической активности (Chinese herbal medicine: 2005, 36, 10, 1591-1594), например, оказывают антибактериальное, противовоспалительное, противоопухолевое действие, являются мышечными релаксантами, понижают уровень холестерина и замедляют старение. Однако растворимость магнолола и хонокиола в воде очень низкая, что серьезно ограничивает их широкое применение в медицине и требует улучшения этого параметра. Недавно растворимость магнолола и хонокиола в воде улучшали посредством химической дериватизации. Например, в патенте Китая CN103313264B описан лабораторный способ получения производного магнолола и производного хонокиола. Однако в описанном в этом патенте способе получения селективность образования целевых продуктов в процессе синтеза относительно низкая, выделение изомеров сложное, требуется многостадийная очистка методом колоночной хроматографии и т.п., поэтому сложно осуществить масштабное производство.
По указанным выше причинам необходим способ получения производного магнолола и производного хонокиола для улучшения селективности и упрощения методики, так чтобы он был пригоден для производства в промышленных масштабах.
Краткое описание изобретения
Основной целью настоящего изобретения является разработка способа получения водорастворимого производного магнолола и производного хонокиола, и его интермедиата, и родственного моногидрокси-защищенного интермедиата, для решения проблем в существующих технологиях, заключающихся в том, что при получении водорастворимого производного магнолола и производного хонокиола наблюдается низкая селективность, и методика получения сложная, поэтому сложно осуществить масштабное производство.
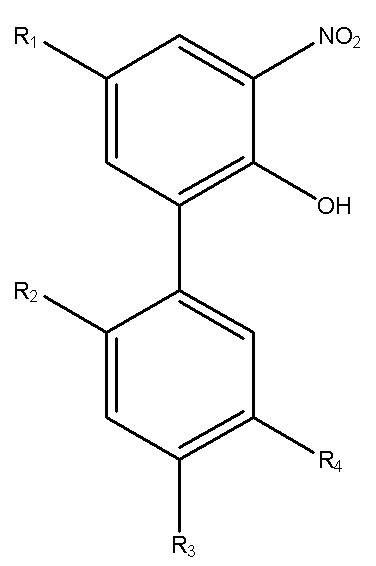
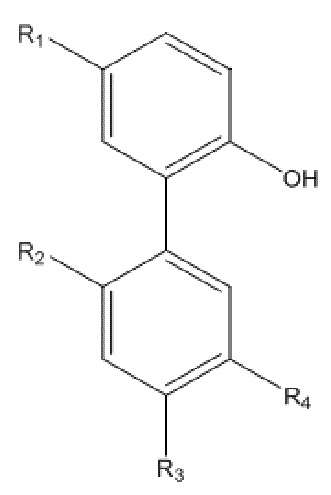
Для достижения указанной цели в одном аспекте настоящего изобретения описан способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола, где нитрованный интермедиат имеет структуру формулы I:
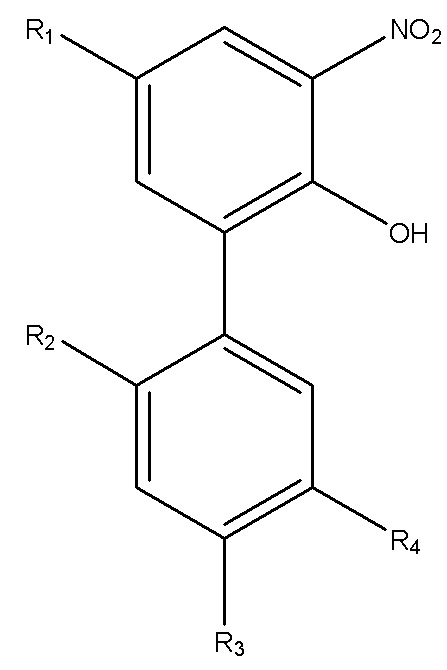
Формула I
В формуле I, R2 представляет собой гидроксил, и R3 представляет собой H; или R2 представляет собой H, и R3 представляет собой гидроксил; и R1 и R4 независимо выбраны из C1~C12 электронодонорной группы; и способ получения включает следующие стадии: проведение моногидрокси-защиты в соединении A 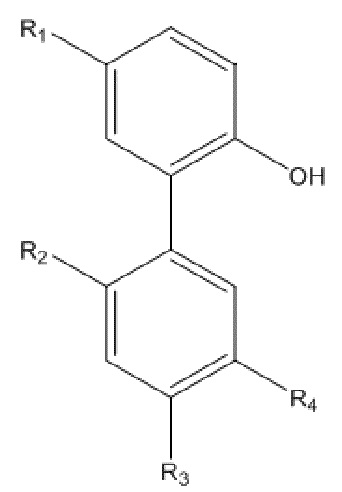 с помощью защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонил хлорид и 1-гидроксибензотриазол; и последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенным соединением с получением нитрованного интермедиата.
с помощью защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонил хлорид и 1-гидроксибензотриазол; и последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенным соединением с получением нитрованного интермедиата.
C1~C12 электронодонорная группа выбрана из C1~C12 алкила или C1~C12 алкенила; предпочтительно, C1~C12 электронодонорная группа выбрана из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, н-гексила, винила, аллила, октила, гептила, пропенила, бут-1-енила, бут-2-енила, бут-3-енила, пент-1-енила, пент-2-енила, пент-3-енила, пент-4-енила, гекс-1-енила, гекс-2-енила, гекс-3-енила, гекс-4-енила, гекс-5-енила, гепт-1-енила, гепт-2-енила, гепт-3-енила, гепт-4-енила, гепт-5-енила, гепт-6-енила, окт-1-енила, окт-2-енила, окт-3-енила, окт-4-енила, окт-5-енила, окт-6-енила или окт-7-енила; и предпочтительно, R1 и R4 одинаковые.
В реакции моногидрокси-защиты, мольное соотношение соединения A и п-толуолсульфонилхлорида составляет 1:(0.75~1), а мольное соотношение соединения A и 1-гидроксибензотриазола составляет 1:(0.75~1); предпочтительно, реакцию моногидрокси-защиты проводят в первом растворителе, и первый растворитель представляет собой нереакционноспособный гидрофобный растворитель; предпочтительно, первый растворитель выбран из одного или больше из следующих: дихлорметан, хлороформ, 1,1-дихлорэтан, метил-трет-бутиловый эфир и толуол; предпочтительно, мольное соотношение кислотосвязывающего агента и защитного реагента для гидрокси-группы составляет (2~3):1; предпочтительно, кислотосвязывающий агент представляет собой органическое основание; и более предпочтительно, органическое основание выбрано из одного или больше из следующих: пиридин, 4-диметиламино-пиридин, 1,8-диазабициклоундецено-7-ен, триэтиламин и N,N-диизопропилэтиламин; предпочтительно, температура реакции в реакции моногидрокси-защиты составляет -10°C~25°C, более предпочтительно 0~10°C, и время реакции составляет 6~10 часов; и предпочтительно, в реакции моногидрокси-защиты мольная концентрация соединения A составляет менее 0.2 моль/л относительно объема первого растворителя, и более предпочтительно, мольная концентрация соединения A составляет менее 0.1 моль/л.
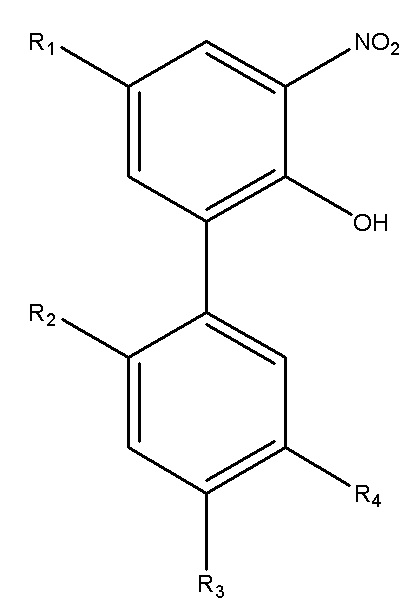
Стадия реакции нитрования включает: реакцию моногидрокси-защищенного соединения с 60~70 вес.% азотной кислотой с получением нитрованного продукта; предпочтительно, реакцию нитрования проводят во втором растворителе, и второй растворитель представляет собой нереакционноспособный растворитель; предпочтительно, второй растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, метил-трет-бутиловый эфир и уксусная кислота; предпочтительно, в процессе реакции нитрования азотную кислоту добавляют по каплям во второй растворитель, содержащий моногидрокси-защищенное соединение, и реакцию проводят при температуре 0~25°C, получая нитрованный продукт; и предпочтительно, в пересчете на 65 вес.% азотную кислоту, весовое соотношение моногидрокси-защищенного соединения и азотной кислоты составляет 3.2~4.2:1.
Стадия реакции снятия защиты включает: смешивание нитрованного продукта с третьим растворителем, с получением смешанного раствора, в настоящем изобретении третий растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, третий растворитель выбран из одного или больше из следующих: 1,4-диоксан, н-пропанол, этиленгликоль и толуол; добавление водного раствора гидроксида щелочного металла в смешанный раствор и нагревание для прохождения реакции с получением нитрованного интермедиата.
В другом аспекте настоящего изобретения, описан способ получения амино-замещенного интермедиата водорастворимого производного магнолола и производного хонокиола, в настоящем изобретении амино-замещенный интермедиат имеет структуру, изображенную на формуле II:
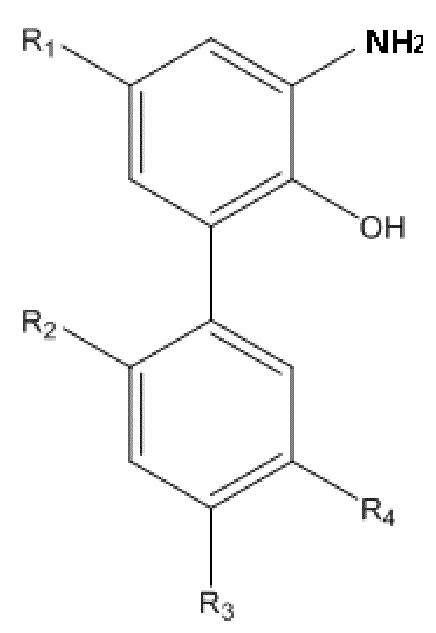
Формула II
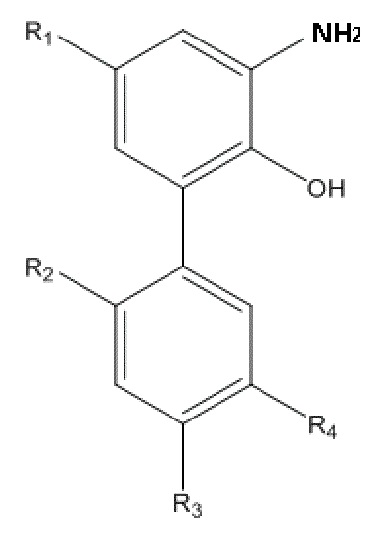
R1, R2, R3 и R4 в формуле II имеют такие же значения, как в любом из пп. 1 - 5 Формулы изобретения; и способ получения по настоящему изобретению включает следующие стадии: применение описанного выше способа получения для получения нитрованного интермедиата, изображенного на формуле I; и проведение реакции восстановления нитрованного интермедиата с получением амино-замещенного интермедиата.
Восстановитель, применяющийся на стадии реакции восстановления, включает один из следующих: хлорид олова, порошок железа, Na2S и NaHS; предпочтительно, реакцию восстановления проводят в четвертом растворителе, и четвертый растворитель представляет собой систему растворов спирт/кислота, в настоящем изобретении спирт выбран из одного или больше из следующих: метанол, этанол и этиленгликоль, и раствор кислоты выбран из соляной кислоты, уксусной кислоты и водного раствора хлорида аммония; и предпочтительно, в процессе реакции восстановления реакционную систему нагревают до кипения.
В другом аспекте настоящего изобретения, описан способ получения свободного основания интермедиата водорастворимого производного магнолола и производного хонокиола, где свободное основание интермедиата имеет структуру, изображенную на формуле III:
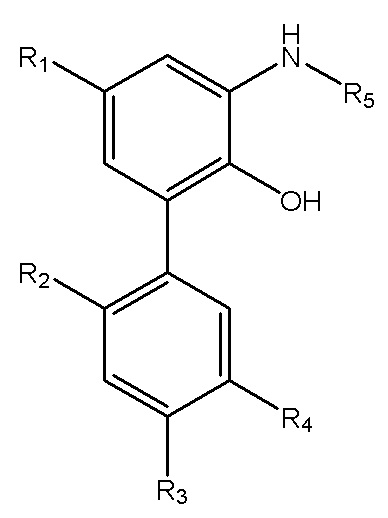
Формула III
R1, R2, R3 и R4 в формуле III имеют указанные выше значения, и R5 выбран из остатка, образующегося при удалении гидроксила из карбоксила индивидуальной аминокислоты или пептида во время реакции конденсации по карбоксилу; и способ получения включает следующие стадии: применение описанного выше способа получения амино-замещенного интермедиата, изображенного на формуле II; проведение реакции конденсации между амино-замещенным интермедиатом и индивидуальной аминокислотой, защищенной трет-бутоксикарбонилом, или пептидом, защищенным трет-бутоксикарбонилом, с получением продукта конденсации; и реакция продукта конденсации с хлороводородом, и затем подщелачивание водным раствором аммиака, экстракция и перекристаллизация с получением интермедиата в виде свободного основания.
Индивидуальная аминокислота выбрана из одной из следующих: лизин, метионин, триптофан, валин, аланин, фенилаланин, лейцин, изолейцин, глицин, гистидин, аргинин, пролин, глутамат, цистин и аспарагиновая кислота, и молекулярный вес пептида составляет ≤2500 Да; предпочтительно, мольное соотношение амино-замещенного интермедиата и индивидуальной аминокислоты, защищенной трет-бутоксикарбонилом, или пептида, защищенного трет-бутоксикарбонилом, составляет (1.2~0.8):1; предпочтительно, реакцию конденсации проводят в пятом растворителе, пятый растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, пятый растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, тетрагидрофуран и N,N-диметилформамид; предпочтительно, в процессе реакции конденсации температура реакции составляет 0~30°C; предпочтительно, реакцию между продуктом конденсации и хлороводородом проводят в шестом растворителе, шестой растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, шестой растворитель выбран из одного или больше из следующих: простой эфир, этилацетат, дихлорметан и 1,4-диоксан.
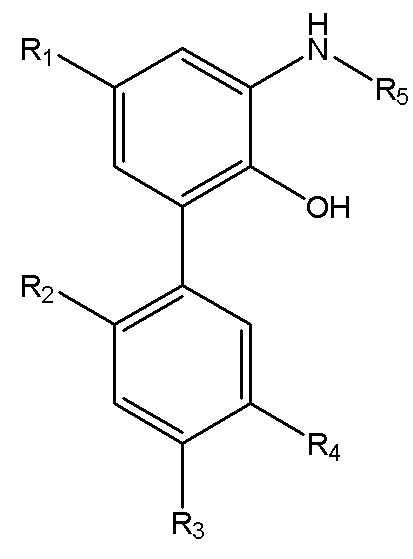
В другом аспекте настоящего изобретения, описан способ получения водорастворимого производного магнолола и производного хонокиола, где водорастворимое производное магнолола и производное хонокиола имеет структуру, изображенную на формуле IV:
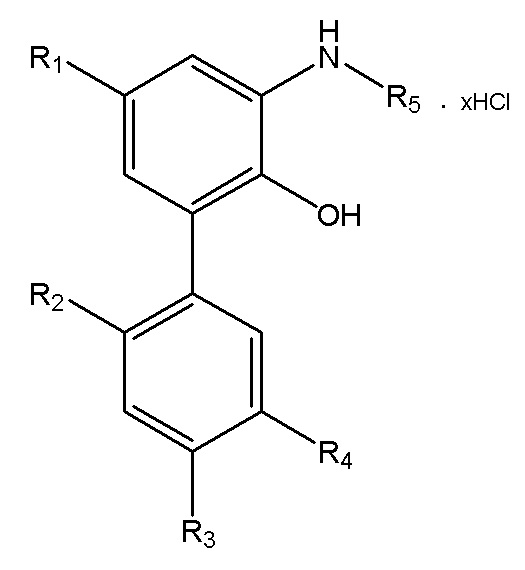
Формула IV
R1, R2, R3, R4 и R5 в формуле IV имеют указанные выше значения; x представляет собой число салифициованных аминогрупп, содержащихся в R5; и данный способ получения включает следующие стадии: применение описанного выше способа получения интермедиата в форме свободного основания, изображенного на формуле III; реакция интермедиата в форме свободного основания с хлороводородом, упаривание или лиофилизация с получением водорастворимого производного магнолола и производного хонокиола.
В другом аспекте настоящего изобретения, описан моногидрокси-защищенный интермедиат водорастворимого производного магнолола и производного хонокиола, где моногидрокси-защищенный интермедиат имеет структуру, изображенную ниже на формуле V:
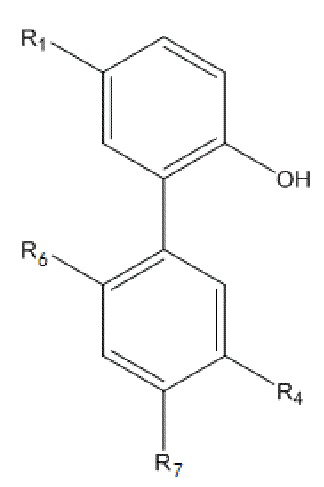
Формула V
В данном случае, в формуле V, R1 и R4 имеют указанные выше значения; R6 представляет собой 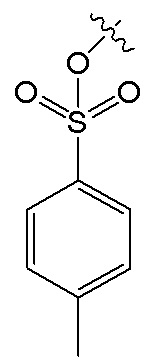 , и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой
, и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой 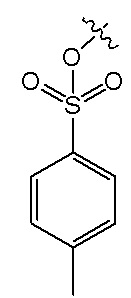 .
.
В настоящем изобретении описан способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола, в нем используется активный эфир, сформированный с применением п-толуолсульфонилхлорида и 1-гидроксибензотриазола в присутствии кислотосвязывающего агента как защитного реагента для гидрокси-группы, и после защиты моногидрокси-соединения A последовательно проводят реакцию нитрования и реакцию снятия защиты, получая нитрованный интермедиат 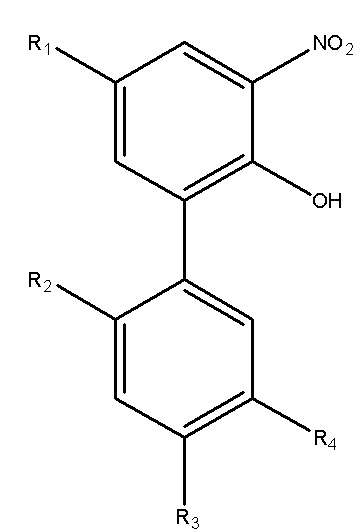 .
.
Соединение A имеет в своей структуре ядро магнолола или хонокиола, и два его бензольных кольца, соответственно, содержат гидроксил. В настоящем изобретении применяется п-толуолсульфонилхлорид и 1-гидроксибензотриазол в качестве защитного реагента для гидрокси-группы, и образуемый ими активный эфир позволяет осуществить селективную моно-защиту гидроксила, представленного R2 и R3, из двух гидроксилов в производном магнолола и производном хонокиола, тем самым существенно улучшается селективность последующей реакции нитрования, а именно реакция нитрования проходит в орто-положение к незамещенному гидроксилу. После завершения реакции нитрования можно получить нитрованный интермедиат формулы I после проведения реакции снятия защиты. Нитрованный интермедиат используется в последующем восстановлении нитрогруппы, в реакции конденсации с аминокислотой или пептидом, защищенными трет-бутоксикарбонилом, в реакции снятия трет-бутоксикарбонильной защитной группы и в других реакциях с получением водорастворимого производного магнолола и производного хонокиола.
Суммируя вышесказанное, применение способа получения по настоящему изобретению эффективно повышает селективность реакции нитрования соединения A, и соответственно улучшает эффективность синтеза водорастворимого производного магнолола и производного хонокиола; и с одной стороны это благоприятно для повышения выхода целевого продукта, а с другой стороны это может полностью исключить стадии колоночной хроматографии, существенно упрощая методику проведения синтеза, уменьшая сложность производства, что соответствует потребностям производства в промышленном масштабе.
Подробное описание вариантов осуществления
Следует отметить, что варианты осуществления настоящего изобретения и отличительные признаки разных вариантов осуществления можно комбинировать друг с другом при неконфликтности таких комбинаций. Настоящее изобретение далее описано более подробно в комбинации с вариантами осуществления.
Как описано выше в обзоре предшествующего уровня техники, в существующих методиках при получении водорастворимого производного магнолола и производного хонокиола возникают проблемы из-за низкой селективности, и методика синтеза усложняется, что препятствует производству в промышленном масштабе.
Для решения описанных выше проблем, в настоящем изобретении описан способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола, и нитрованный интермедиат имеет структуру, изображенную на формуле I:
Формула I
В формуле I, R2 представляет собой гидроксил, и R3 представляет собой H; или R2 представляет собой H, и R3 представляет собой гидроксил; и R1 и R4 независимо выбраны из C1~C12 электронодонорной группы; и способ получения включает следующие стадии: осуществление моногидрокси-защиты в соединении A 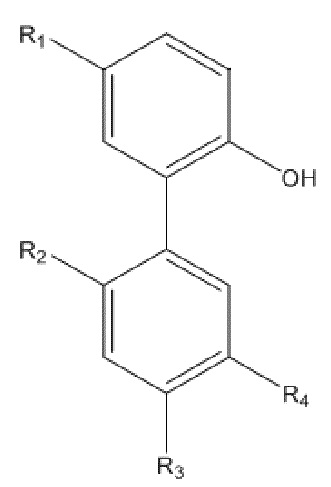 с применением защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонилхлорид и 1-гидроксибензотриазол; и последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенного соединения, с получением нитрованного интермедиата.
с применением защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонилхлорид и 1-гидроксибензотриазол; и последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенного соединения, с получением нитрованного интермедиата.
Соединение A имеет в своей структуре ядро магнолола или хонокиола, и два его бензольных кольца, соответственно, содержат гидроксил. В настоящем изобретении применяется активный эфир, образуемый п-толуолсульфонилхлоридом и 1-гидроксибензотриазолом, в присутствии кислотосвязывающего агента в качестве защитного реагента для гидрокси-группы, и этот активный эфир позволяет осуществить селективную моно-защиту гидроксила, представленного R2 и R3, из двух гидроксилов в производном магнолола и производном хонокиола, тем самым существенно улучшается селективность последующей реакции нитрования, а именно реакция нитрования проходит в орто-положение к незамещенному гидроксилу. После завершения реакции нитрования, можно получить нитрованный интермедиат формулы I после проведения реакции снятия защиты. Хонокиольное соединение A можно взять в качестве примера, и реакционная схема будет выглядеть следующим образом:
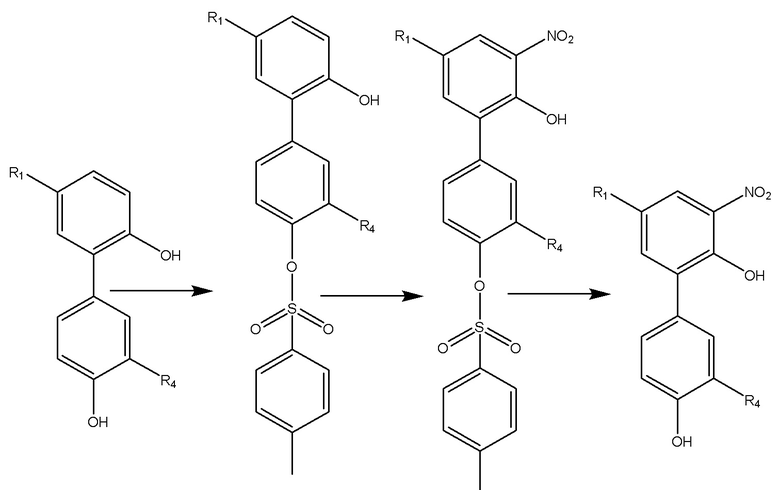
Нитрованный интермедиат используется в последующем восстановлении нитрогруппы, в реакции конденсации с аминокислотой, защищенной трет-бутоксикарбонилом, снятии защитной трет-бутоксикарбонильной группы, реакции кислоты со щелочью, и в других реакциях с получением водорастворимого производного магнолола и производного хонокиола.
Следует отметить, что активный эфир, образованный п-толуолсульфонилхлоридом и 1-гидроксибензотриазолом, менее активный, чем прямая реакция этерификации с толуолсульфонилхлоридом, и стерическая структура данного активного эфира более объемная, чем у толуолсульфонилхлорида, что дополнительно снижает вероятность реакции с более стерически затрудненным гидроксилом. Эти причины позволяют осуществить селективную моно-гидрокси-защиту соединения A, имеющего ди-гидроксильную структуру типа магнолола и хонокиола.
Кроме того, для более стерически затрудненного защитного реагента, такого как октаноилхлорид и лауроилхлорид, авторами настоящего изобретения было обнаружено, что положение гидроксила, селективно защищаемого ими, противоположно положению защиты, требуемому описанным выше соединением A. Это показывает, что активность двух гидроксилов в соединении A высокая или низкая (в особенности для соединения A, имеющего в основе материнскую структуру хонокиола). Высокая или низкая активность двух гидроксилов противоположна масштабу стерических затруднений. Хотя молекулярный размер октаноилхлорида и лауроилхлорида относительно большой, но благодаря гибкой структуре жирной цепочки разность в активности гидроксилов играет ведущую роль в данной реакции. Напротив, активный эфир, образованный гидрокси-защитным агентом по настоящему изобретению, представляет собой жесткую структуру с ароматическим кольцом, поэтому стерические затруднения играют ведущую роль в этой реакции. Основываясь на этом исследовании, в настоящем изобретении достигнут технический подход к селективной моногидрокси-защите соединения A с применением активного эфира, образованного п-толуолсульфонилхлоридом и 1-гидроксибензотриазолом, в качестве защитного реагента для гидрокси-группы, тем самым существенно улучшается селективность синтеза всего водорастворимого производного магнолола и производного хонокиола, уменьшается сложность очистки и разделения продуктов, и упрощается методика проведения синтеза, что делает ее пригодной для промышленного производства в большом масштабе.
Кроме того, в соединении A по настоящему изобретению R2 представляет собой гидроксил, и R3 представляет собой H; или R2 представляет собой H, и R3 представляет собой гидроксил; R1 и R4 независимо выбраны из C1~C12 электронодонорной группы, соответственно. Когда R2 представляет собой гидроксил, и R3 представляет собой H, образуется нитрованный интермедиат производного магнолола; и когда R2 представляет собой H, и R3 представляет собой гидроксил, образуется нитрованный интермедиат производного хонокиола. R1 и R4 независимо выбраны из C1~C12 электронодонорной группы, соответственно, и использование электронодонорного эффекта этих групп может дополнительно изменить селективность гидроксилов в положениях R2 и R3 в процессе реакции моногидрокси-защиты. Одним словом, применение способа получения по настоящему изобретению эффективно повышает селективность реакции нитрования соединения A, и соответственно улучшает селективность синтеза водорастворимого производного магнолола и производного хонокиола; и с одной стороны это благоприятно для повышения выхода целевого продукта, а с другой стороны это может упростить очистку и выделение продукта на каждой реакционной стадии, существенно упрощая методику проведения синтеза, уменьшая сложность производства, что соответствует потребностям производства в промышленном масштабе
Для улучшения селективности моногидрокси-защиты и реакции нитрования, в предпочтительном варианте осуществления C1~C12 электронодонорная группа выбрана из C1~C12 алкила или C1~C12 алкенила; предпочтительно, C1~C12 электронодонорная группа выбрана из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, н-гексила, винила, аллила, октила, гептила, пропенила, бут-1-енила, бут-2-енила, бут-3-енила, пент-1-енила, пент-2-енила, пент-3-енила, пент-4-енила, гекс-1-енила, гекс-2-енила, гекс-3-енила, гекс-4-енила, гекс-5-енила, гепт-1-енила, гепт-2-енила, гепт-3-енила, гепт-4-енила, гепт-5-енила, гепт-6-енила, окт-1-енила, окт-2-енила, окт-3-енила, окт-4-енила, окт-5-енила, окт-6-енила или окт-7-енила; и более предпочтительно, указанные выше R1 и R4 одинаковые.
Более предпочтительно, R1 и R4 независимо выбраны из C1~C8 электронодонорной группы, соответственно, такой как C1~C8 алкил или C1~C8 алкенил.
В предпочтительном варианте осуществления, в реакции моногидрокси-защиты мольное соотношение соединения A и п-толуолсульфонилхлорида составляет 1:(0.75~1), а мольное соотношение соединения A и 1-гидроксибензотриазола составляет 1:(0.75~1); и предпочтительно, кислотосвязывающий агент представляет собой органическое основание, и его мольное соотношение с п-толуолсульфонилхлоридом составляет (2~3):1. При проведении реакции моногидрокси-защиты предпочтительно соблюдать количественные соотношения между реагентами в указанных выше диапазонах, тем самым увеличивая эффективность реакции.
Для дальнейшего улучшения стабильности прохождения реакции и достижения высокой эффективности прохождения реакции, в предпочтительном варианте осуществления реакцию моногидрокси-защиты проводят в первом растворителе, и первый растворитель представляет собой нереакционноспособный гидрофобный растворитель. В реальном реакционном процессе, п-толуолсульфонилхлорид, 1-гидроксибензотриазол и кислотосвязывающий агент обычно сначала добавляют в первый растворитель для прохождения реакции с образованием активного эфира, и затем добавляют соединение A для прохождения дальнейшей реакции с получением моногидрокси-защищенного интермедиата. Более предпочтительно, температура реакции находится в диапазоне 0~25°C, и более предпочтительно, температура реакции находится в диапазоне 0~10°С, и время реакции составляет 6~10 часов. Предпочтительно, органическое основание выбрано из пиридина, 4-диметиламино-пиридина, 1,8-диазабициклоундецено-7-ена, триэтиламина и N,N-диизопропилэтиламина; и первый растворитель выбран из одного или больше из следующих: дихлорметан, хлороформ, 1,1-дихлорэтан, метил-трет-бутиловый эфир и толуол. В этом случае эффективность реакции выше, реакция проходит стабильнее, и уровень безопасности выше.
Для дальнейшего улучшения селективности и эффективности реакции нитрования, в предпочтительном варианте осуществления стадия реакции нитрования включает: реакцию моногидрокси-защищенного соединения с 60~70 вес.% азотной кислотой, с получением нитрованного продукта. Предпочтительно, реакцию нитрования проводят во втором нереакционноспособном растворителе, и предпочтительно второй растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, метил-трет-бутиловый эфир и уксусная кислота. При проведении в перечисленных выше растворителях, реакция нитрования более стабильна и дает меньше побочных продуктов. Предпочтительно, в реакции нитрования азотную кислоту добавляют по каплям во второй растворитель, содержащий моногидрокси-защищенное соединение, и реакцию проводят при температуре 0~25°C, получая нитрованный продукт; и предпочтительно, весовое соотношение моногидрокси-защищенного соединения и азотной кислоты составляет 3.2~4.2:1 (вычислено для 65 вес.% азотной кислоты). Реакция нитрования безопасна и более стабильна в указанных выше условиях реакции и при указанных соотношениях реагентов.
В предпочтительном варианте осуществления, стадия реакции снятия защиты включает: смешивание нитрованного продукта с третьим растворителем с образованием смешанного раствора, где третий растворитель представляет собой нереакционноспособный растворитель, и третий растворитель выбран из одного или больше из следующих: 1,4-диоксан, н-пропанол, этиленгликоль и толуол; добавление водного раствора гидроксида щелочного металла в полученный смешанный раствор, и проведение реакции с получением нитрованного интермедиата.
В другом аспекте настоящего изобретения, описан способ получения амино-замещенного интермедиата водорастворимого производного магнолола и производного хонокиола, где амино-замещенный интермедиат имеет структуру, изображенную на формуле II:
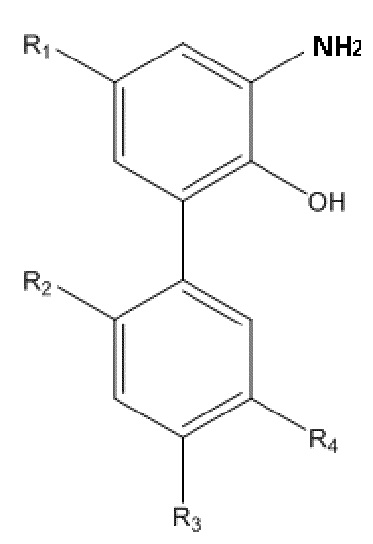
Формула II
R1, R2, R3 и R4 в формуле II имеют указанные выше значения; и способ получения включает следующие стадии: применение описанного выше способа получения нитрованного интермедиата, изображенного на формуле I; и проведение реакции восстановления нитрованного интермедиата, с получением амино-замещенного интермедиата.
В реальном практическом процессе, для улучшения эффективности реакции восстановления, в предпочтительном варианте осуществления, применяющийся на стадии восстановления восстановитель включает один из следующих: хлорид олова, порошок железа, Na2S и NaHS; предпочтительно, реакцию восстановления проводят в четвертом растворителе, и четвертый растворитель представляет собой смешанный растворитель спирт/ кислота, где спирт выбран из одного или больше из следующих: метанол, этанол и этиленгликоль, и раствор кислоты выбран из соляной кислоты, уксусной кислоты и водного раствора хлорида аммония; и предпочтительно, в реакции восстановления реакционную систему нагревают до состояния кипения.
В другом аспекте настоящего изобретения, описан способ получения интермедиата в форме свободного основания водорастворимого производного магнолола и производного хонокиола, где интермедиат в форме свободного основания имеет структуру, изображенную на формуле III:
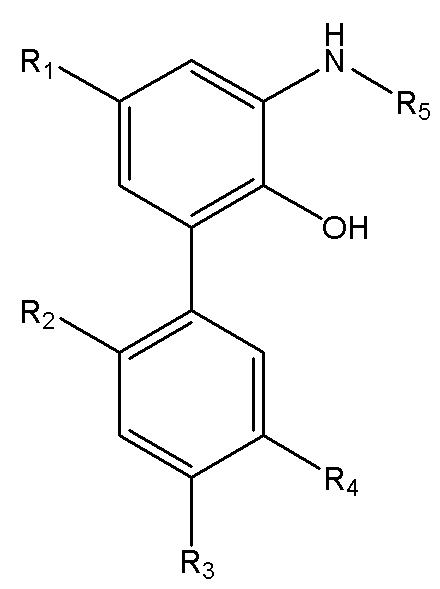
Формула III
R1, R2, R3 и R4 в формуле III имеют указанные выше значения, и R5 выбран из остатка, образующегося при удалении гидроксила из карбоксила отдельной аминокислоты или пептида во время реакции конденсации по карбоксилу; и способ получения включает следующие стадии: применение описанного выше способа получения амино-замещенного интермедиата, изображенного на формуле II; проведение реакции конденсации между амино-замещенным интермедиатом и индивидуальной аминокислотой, защищенной трет-бутоксикарбонилом, или пептидом, защищенным трет-бутоксикарбонилом, с получением продукта конденсации; и реакция продукта конденсации с хлороводородом, и затем подщелачивание водным раствором аммиака, экстракция и перекристаллизация с получением интермедиата в виде свободного основания.
В предпочтительном варианте осуществления, индивидуальная аминокислота выбрана из одной из следующих: лизин, метионин, триптофан, валин, аланин, фенилаланин, лейцин, изолейцин, глицин, гистидин, аргинин, пролин, глутамат, цистин и аспарагиновая кислота, и молекулярный вес пептида составляет ≤2500 Да (предпочтительно образован из перечисленных выше аминокислот). Для дальнейшего улучшения эффективности реакции и повышения выхода целевого соединения, в предпочтительном варианте осуществления, мольное соотношение амино-замещенного интермедиата и индивидуальной аминокислоты, защищенной трет-бутоксикарбонилом, или пептида, защищенного трет-бутоксикарбонилом, составляет (1.2~0.8):1; предпочтительно, реакцию конденсации проводят в пятом растворителе, пятый растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, пятый растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, тетрагидрофуран и N,N-диметилформамид; предпочтительно, в процессе реакции конденсации температура реакции составляет 0~30°C; предпочтительно, реакцию между продуктом конденсации и хлороводородом проводят в шестом растворителе, и шестой растворитель выбран из одного или больше из следующих: простой эфир, этилацетат, дихлорметан и 1,4-диоксан.
В другом аспекте настоящего изобретения, описан способ получения водорастворимого производного магнолола и производного хонокиола, где водорастворимое производное магнолола и производное хонокиола имеет структуру, показанную на формуле IV:
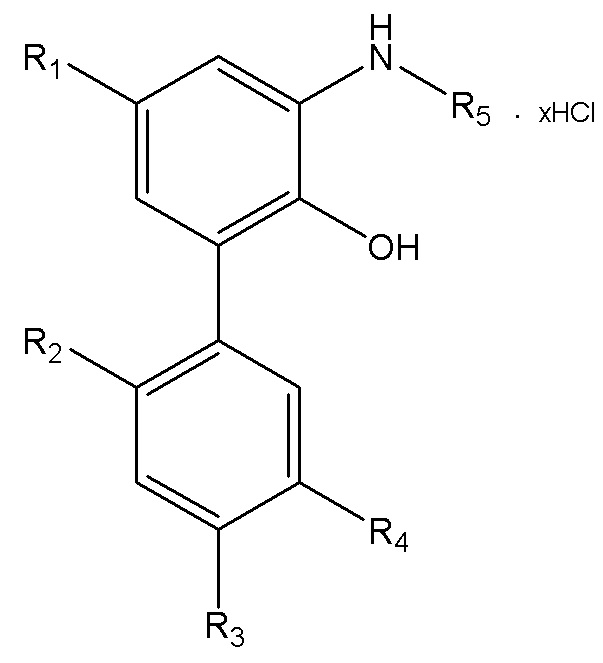
Формула IV
R1, R2, R3, R4 и R5 в формуле IV имеют указанные выше значения; x представляет собой число салифициованных аминогрупп, содержащихся в R5; и данный способ получения включает следующие стадии: получение интермедиата в форме свободного основания, изображенного на формуле III; и реакция интермедиата в форме свободного основания с хлороводородом, с получением водорастворимого производного магнолола и производного хонокиола.
В реальном процессе получения, интермедиат в форме свободного основания может быть подкислен соляной кислотой и лиофилизован с получением водорастворимого производного магнолола и производного хонокиола, изображенного на формуле IV. Концентрация соляной кислоты может быть любой, например, можно применять коммерчески доступную соляную кислоту с концентрацией 37 вес.%, и более предпочтительно можно использовать разбавленную соляную кислоту с концентрацией 1~10 вес.%.
С учетом более высокой селективности ранее описанной в настоящем тексте реакции моногидрокси-защиты и реакции нитрования, являются простыми и удобными в реализации следующие реакции: реакция снятия защиты с использованием гидроксида щелочного металла, реакция конденсации, реакция снятия защиты с использованием хлороводорода, операция подщелачивания с использованием водного раствора аммиака, экстракция, кристаллизация, подкисление и лиофилизация, так что получение водорастворимого производного магнолола и производного хонокиола, показанного на формуле IV, протекает с более высокой конверсией и выходом, и их легко реализовать в промышленном масштабе.
В другом аспекте настоящего изобретения, описан моногидрокси-защищенный интермедиат водорастворимого производного магнолола и производного хонокиола, и он имеет структуру, изображенную ниже на формуле V:
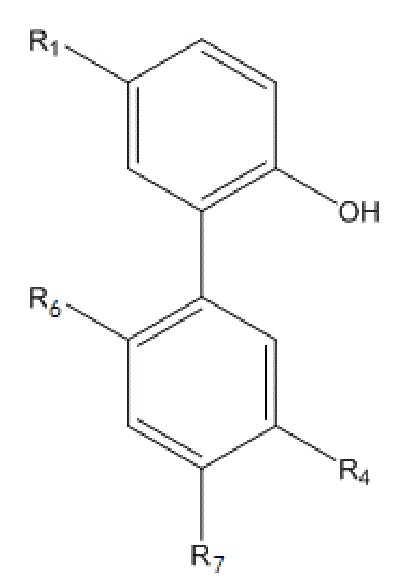
Формула V
В формуле V, R1 и R4 имеют указанные выше значения; R6 представляет собой 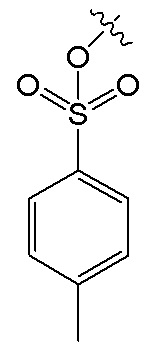 , и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой
, и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой 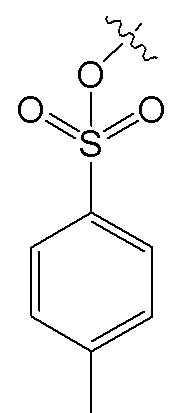 .
.
Описанное выше моногидрокси-защищенное соединение получают, используя следующий способ получения: проводят моногидрокси-защиту описанного выше соединения A 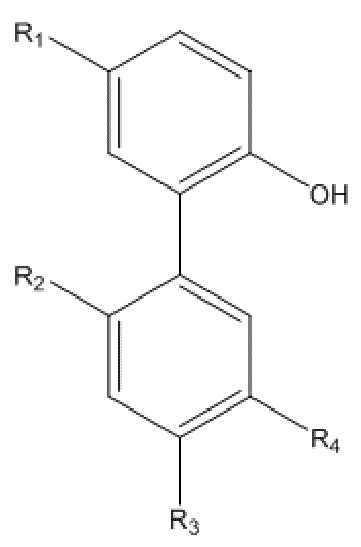 с использованием защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с образованием моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-группы представляет собой п-толуолсульфонилхлорид и 1-гидроксибензотриазол.
с использованием защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с образованием моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-группы представляет собой п-толуолсульфонилхлорид и 1-гидроксибензотриазол.
Настоящее изобретение дополнительно описано подробнее ниже в комбинации с частными вариантами осуществления, и эти варианты осуществления не следует понимать как ограничивающие объем притязаний, заявленный в Формуле изобретения.
В описании вариантов осуществления применяются следующие аббревиатуры: DCC: 1,3-дициклогексилкарбодиимид; TsCl: п-толуолсульфонилхлорид; HOBT: 1-гидроксибензотриазол; DIEA: N,N-диизопропилэтиламин; SnCl2: хлорид олова; и 1,4-диоксан.
Вариант осуществления 1
Нитрованный интермедиат (соединение 6) производного хонокиола получали согласно следующей схеме реакции.
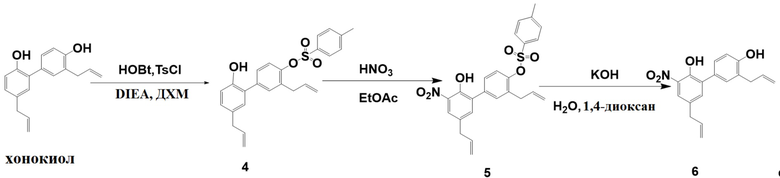
Стадия I:
Получение 3,5'-диаллил-2'-гидрокси-[1,1'-бифенил]-4-ил 4-метилбензолсульфоната (соединение 4)
Дихлорметан (160 кг) добавляли в 200-литровый реактор, перемешивали и понижали внутреннюю температуру до 10°C. Добавляли последовательно HOBT (0.744 кг), DIEA (1.61 кг) и TsCl (0.954 кг). Через 30 минут ТСХ показала, что TsCl полностью израсходовался. Добавляли хонокиол (1.6 кг), внутреннюю температуру поддерживали равной 5±5°C, и непрерывно перемешивали 6 часов. Медленно добавляли 1н. водн. HCl (20 л), и полученную смесь перемешивали в течение 30 минут, затем оставляли для разделения слоев. Органический слой снова промывали 1н. водной HCl (20 л). Затем органический слой помещали в реактор, добавляли 1н. водн. NaOH (20 л), полученную смесь перемешивали и оставляли для разделения слоев. Органический слой снова промывали 1н. водным раствором NaOH (20 л). После этого органический слой помещали в реактор, добавляли насыщенный водный раствор хлорида натрия (20 л), смесь перемешивали и оставляли для разделения слоев. Органический слой сушили над безводным Na2SO4, фильтровали и упаривали. Добавляли в остаток изопропанол (1.91 л), нагревали и перемешивали при растворении. После охлаждения выпадал твердый осадок, его отфильтровывали. Материнский раствор упаривали при пониженном давлении, получая соединение 4 (1.28 кг, 87% чистота) в виде желтого масла с выходом 61%.
Соединение 4: C25H24O4S = 420.14, MS: 438 [M + NH4]+. 1H-ЯМР (300 МГц, CDCl3) δ 9.46 (1H, с), 7.83-7.81 (2H, д), 7.52-7.49 (2H, д), 7.39-7.37 (2H, д), 7.00-6.97 (3H, м), 6.85-6.82 (1H, м), 5,98-5,69 (2H, м), 5.06-4.96 (4H, м), 3.34-3.17 (4H, м), 2.48-2.35 (3H, м).
Стадия II:
Получение 3,5'-диаллил-2'-гидрокси-3'-нитро-[1,1'-бифенил]-4-ил 4-метилбензолсульфоната (соединение 5)
Дихлорметан (5 л) помещали в 10-литровый реактор при перемешивании, добавляли соединение 4 (1.28 кг), и полученную смесь охлаждали до 0°C. По каплям добавляли 65 вес.% водн. HNO3 (0.304 кг) в течение 30 минут, затем реакционную смесь перемешивали еще 2 часа. ТСХ показала, что реакция прошла полностью. Добавляли чистую воду (2.6 л), и полученную смесь перемешивали в течение 15 минут, оставляли для разделения слоев. Органический слой последовательно промывали насыщенным водным раствором NaHCO3 (2.5 л) и насыщенным водным раствором хлорида натрия (2.5 л), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли в остаток метанол (8 л), нагревали, перемешивали до растворения, и затем охлаждали до 0°C. Смесь фильтровали, и осадок на фильтре сушили, получая соединение 5 (863.5 г, 97% чистота) в виде желтого твердого вещества с выходом 61%.
Соединение 5: C25H23NO6S2 = 465.52, MS: 483 [M + NH4]+. 1H-ЯМР (400 МГц, CDCl3) δ 11.00 (1H, с), 7.93 (1H, д), 7.82 (2H, д), 7.43-7.32 (5H, м), 7.12 (2H, д), 5.98-5.75 (2H, м), 5.16-5.02 (4H, м), 3.40-3.29 (4H, дд), 2.47 (3H, с).
Стадия III:
Получение 3',5-диаллил-3-нитро-[1,1'-бифенил]-2,4'-диола (соединение 6)
1,4-диоксан (5 л) и соединение 5 (0.862 кг) помещали в 10-литровый реактор, и полученную смесь перемешивали до растворения. KOH (0.521 кг) растворяли в воде (1.7 л) и добавляли по каплям в течение 20 минут, и реакционную смесь перемешивали при 85°C в течение 4 часов. ТСХ показала, что реакция прошла полностью. Смесь охлаждали, и упаривали при пониженном давлении. Дихлорметан (5 л) и воду (2.5 л) добавляли в остаток, и при перемешивании добавляли по каплям конц.водн. HCl до рH = 5, затем полученную смесь оставляли для разделения слоев. Органическую фазу последовательно промывали насыщенным водным раствором NaHCO3 (2.5 л) и насыщенным водным раствором хлорида натрия (2.5 л), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли дихлорметан (1 л), и остаток растворяли при нагревании. Добавляли н-гексан (8 л), и полученную смесь 1 час перемешивали при 0°C, затем фильтровали. Осадок на фильтре сушили, получая соединение 3 (0.425 кг, 95% чистота) в виде темно-красного твердого вещества с выходом 74%.
Соединение 6: C18H17NO4 = 311.33, MS: 329 [M + NH4]+. 1H-ЯМР (400 МГц, CDCl3) δ 10.03 (1H, с), 7.90 (1H, д), 7.35-7.31 (2H, м), 6.90 (1H, д), 6.11-5.90 (2H, м), 5.24-5.11 (5H, м), 3.48 (2H, д), 3.40 (2H, д).
Вариант осуществления 2
Амино-замещенный интермедиат (соединение 7) производного хонокиола получали согласно следующей схеме реакции.
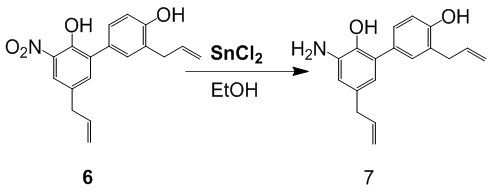
Получение 3',5-диаллил-3-амино-[1,1'-бифенил]-2,4'-диола (соединение 7)
В 10-литровый реактор добавляли этанол (2 л) и соединение 6 (0.248 кг) при перемешивании, и затем добавляли хлорид олова (0.629 кг). Результирующую смесь кипятили 2 часа. ТСХ показала, что реакция прошла полностью. Смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Добавляли этилацетат (5 л) и перемешивали смесь. Медленно добавляли по каплям насыщенный водный раствор NaHCO3 до pH = 8, при этом выпадало большое количество твердого осадка. Смесь фильтровали, и фильтрат оставляли для разделения слоев. Органическую фазу отделяли, осадок на фильтре промывали этилацетатом (5 л x 3), и объединенные органические фазы упаривали при пониженном давлении, получая соединение 7 (0.179 кг, 96% чистота) в виде твердого вещества землисто-желтого цвета с выходом 80%.
Соединение 7: C18H19NO2 = 281.14, MS: 282 [M + H]+, 1H-ЯМР (400 МГц, ДМСО) δ 9.925 (1H, с), 9.59 (1H, с), 9.22 (1H, с), 9.22, 7.12 (1H, т), 7.06 (1H, д), 7.03 (1H, д), 5.99-5.86 (2H, м), 5.10-4.98 (4H, м), 3.32 (4H, дд).
Вариант осуществления 3
Гидрохлорид (соединение 10) производного хонокиола получали согласно следующей схеме реакции
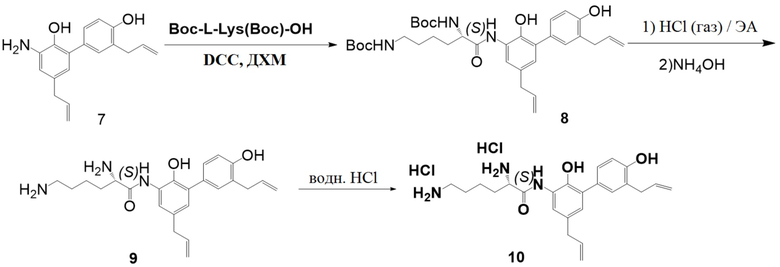
Стадия I:
Получение ди-трет-бутил (6-((3',5-диаллил-2,4'-дигидрокси-[1,1'-бифенил]-3- ил)амино)-6-оксогексан-1,5-диил)(S)-дикарбамата (соединение 8)
Дихлорметан (2.5 л), DCC (103.3 г) и Boc-L-lys (Boc)-OH (172.3 г) последовательно добавляли в 5-литровую реакционную колбу, и полученную смесь перемешивали при 0°C. Через 30 минут добавляли соединение 7 (139.5 г), и реакционную смесь перемешивали в течение 2 часов. Смесь фильтровали, и фильтрат упаривали, получая соединение 8 (324 г, 87% чистота) в виде серой твердой пены.
Соединение 8: C34H47N3O7 = 609.75, MS: 610 [M + H]+. 1H ЯМР (300 МГц, CDCl3) δ 8.637 (1H, с), 7.316 (5H, м), 6.892 (2H, м), 6.099 (3H, м), 5.357 (1H, с), 5.206 (4H, м), 4.670 (1H, с), 4.288 (1H, т), 3.445 (2H, д), 3.314 (2H, д), 3.133 (2H, т), 2.056 (2H, м), 1.460 (22H, м).
Стадия II:
Получение (S)-2,6-диамино-N-(3',5-диаллил-2,4'-дигидрокси-[1,1'-бифенил]-3- ил)гексанамида (соединение 9)
В 5-литровом реакторе заменяли атмосферу на азот три раза и добавляли 4.0 М раствор хлороводорода в этилацетате (1.5 л), перемешивали и охлаждали до 0°C. Медленно добавляли соединение 8 (321.1 г), реакционную смесь перемешивали при 0°C 5 часов, при этом выпадал осадок. ТСХ показала, что соединение 8 полностью израсходовалось. Добавляли воду (1 л), органическую фазу отделяли, и водную фазу доводили до pH = 9 водным раствором аммиака. Выпадал твердый осадок, его экстрагировали этилацетатом (1 л x 2). Объединенные органические фазы упаривали, и остаток дважды перекристаллизовывали из смеси этилацетата и н-гексана, получая соединение 10 (96.8 г, 98% чистота, и 48% выход за две стадии) в виде желтого твердого вещества.
Соединение 9: C24H31N3O3 = 409.53, MS: 410. 410 [M + H]+, 1H -ЯМР (400 МГц, MeOH) δ 7.52 (1H, д), δ 7.24 (2H, м), δ 6.84 (2H, т), δ 6.08 (2H, м), 5.11-4.94 (4H, м), 3.41 (1H, т), 3.39 (2H, д), 3.32 (2H, д), 2.66 (2H, т), 1.83 (1H, м), 1.66 (1H, м), 1.53 (4H, м).
Стадия III:
Получение (S)-2,6-диамино-N-(3',5-диаллил-2,4'-дигидрокси-[1,1'-бифенил]-3- ил)гексанамид дигидрохлорида (соединение 10)
Соединение 9 (41 г) растворяли в 1М разбавленном водном растворе HCl (210 мл) при перемешивании при 0°C, и полученный раствор лиофилизировали, получая соединение 10 (48.2 г, 97.8% чистота, и 100% выход) в виде желтоватого твердого вещества.
Соединение 10: C24H33Cl2N3O3 = 482.44, MS: 410 [M + H]+ (свободная форма). 1H-ЯМР (400 МГц, ДМСО) δ 10.43 (1H, с), δ 9.46 (1H, с), δ 8.61 (1H, с), δ 8.43 (3H, с), 7.99 (3H, с), 7.33 (1H, с), 7.16 (2H, т), 6.86 (2H, д), 5.99 (2H, м), 5.09 (4H, м), 4.15 (1H, т), 3.29 (4H, м), 2.77 (2H, т), 1.89 (2H, кв), 1.63 (2H, м), 1.47 (2H, м).
Вариант осуществления 4
Нитрованный интермедиат (соединение 13) производного магнолола получали согласно следующей схеме реакции:
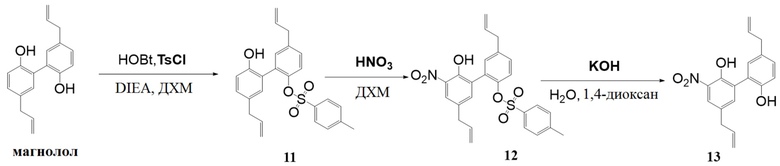
Стадия I:
Получение 5,5'-диаллил-2'-гидрокси-[1,1'-бифенил]-2-ил 4-метилбензолсульфоната (соединение 11)
Дихлорметан (3 л) помещали в 5-литровую реакционную колбу, перемешивали и понижали внутреннюю температуру до 10°C. Последовательно добавляли HOBT (18.5 г), DIEA (40.1 г) и TsCl (23.7 г). Через 30 минут ТСХ показала, что TsCl полностью израсходовался. Добавляли магнолол (40.0 г) и реакционную смесь далее перемешивали при 5±5°C еще 6 часов. Медленно добавляли 1н. водн. HCl (500 мл). После 30 минут перемешивания смесь оставляли для разделения слоев. Органический слой промывали 1н. водной HCl (500 мл) еще раз. Добавляли 1н. водный раствор NaOH (500 мл), смесь перемешивали и оставляли для разделения слоев. Органический слой промывали 1н. водным раствором NaOH (500 мл) еще раз. Добавляли насыщенный водный раствор хлорида натрия (500 мл). После перемешивания в течение 30 минут, смесь оставляли для разделения слоев. Органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали. Добавляли изопропанол (50 мл) в остаток, смесь нагревали и перемешивали до растворения. После охлаждения выпадал твердый осадок, который отфильтровывали. Материнский раствор упаривали при пониженном давлении, получая соединение 11 (33 кг, 87% чистота) в виде желтого масла с выходом 57.3%.
Соединение 11: C25H24O4S = 420.14, MS: 438 [M + NH4]+.
Стадия II:
Получение 5,5'-диаллил-2'-гидрокси-3'-нитро-[1,1'-бифенил]-2-ил 4-метилбензолсульфоната (соединение 12)
В 250-миллилитровую реакционную колбу при перемешивании добавляли дихлорметан (120 мл) и соединение 11 (30.1 г), и полученную смесь охлаждали до 0°C. По каплям добавляли 65 вес.% водную HNO3 (7.15 г) в течение 30 минут, и реакционную смесь далее перемешивали 2 часа. ТСХ показала, что реакция прошла полностью. Добавляли воду (60 мл). После перемешивания в течение 15 минут, смесь оставляли для разделения слоев. Органическую фазу последовательно промывали насыщенным водным раствором NaHCO3 (60 мл) и насыщенным водным раствором хлорида натрия (60 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли в остаток метанол (8 л), смесь нагревали, перемешивали до растворения и затем охлаждали до 0°C. Смесь фильтровали, и осадок на фильтре сушили, получая соединение 12 (20.1 г, 98% чистота) в виде желтого твердого вещества с выходом 60%.
Стадия III:
Получение 5,5'-диаллил-3-нитро-[1,1'-бифенил]-2,2'-диола (соединение 13)
В 250-миллилитровую реакционную колбу добавляли 1,4-диоксан (125 мл) и соединение 12 (20 г), и полученную смесь перемешивали до растворения. Гидроксид калия (2.8 г) растворяли в воде (50 мл) и по каплям добавляли в течение 20 минут, затем смесь перемешивали при 85°C в течение 4 часов. ТСХ показала, что реакция прошла полностью. После охлаждения смесь упаривали при пониженном давлении. Добавляли в остаток дихлорметан (250 мл) и воду (120 мл), по каплям добавляли конц. водн. HCl при перемешивании до рH=5 и оставляли смесь для разделения слоев. Органическую фазу последовательно промывали насыщенным водным раствором NaHCO3 (120 мл) и насыщенным водным раствором хлорида натрия (120 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение 13 (13 г, 97% чистота) в виде темно-красного твердого вещества с выходом 97%.
Соединение 13: C18H17NO4 = 311.12, MS: 329 [M + NH4]+, 1H-ЯМР (300 МГц, CDCl3) δ 8.01 (1H, д), 7.50 (1H, д), 7.18 (1H, дд), 7.01 (1H, дд), 6.91 (1H, дд), 6.04-5.88 (2H, м), 5.18-5.06 (4H, м), 3.42 (4H, дд).
Вариант осуществления 5
Влияние температуры реакции на соотношение (селективность) 3,5'-диаллил-2'-гидрокси-[1,1'-бифенил]-4-ил 4-метилбензолсульфоната (соединение 4) и 3,5'-диаллил-4'-гидрокси-[1,1'-бифенил]-2-ил 4-метилбензолсульфоната (соединение 4a)
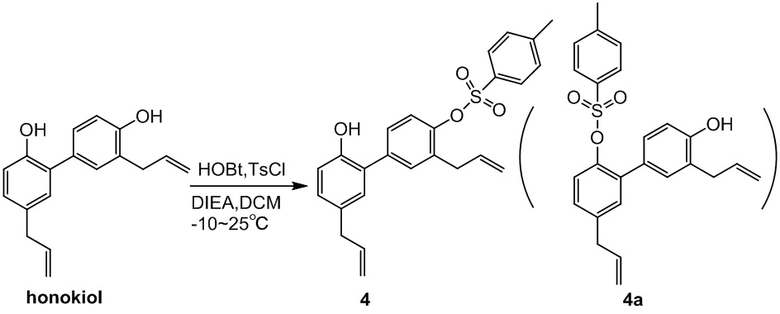
Методика: Дихлорметан (1 л) помещали при перемешивании в 2-литровую трехгорлую колбу и охлаждали до -10°C (другие три партии охлаждали до 0°C, 10°C и 25°C, соответственно), через 30 минут добавляли последовательно HOBT (12.4 г), DIEA (26.9 г) и TsCl (15.9 г). Добавляли хонокиол (26.6 г), и реакционную смесь непрерывно перемешивали 6 часов. Методом LC-MS отслеживали соотношение соединения 4 и соединения 4a.
Результаты эксперимента показаны ниже в таблице:
Вывод: в диапазоне от -10°C до 25°C соотношение соединений 4 и 4a всегда больше чем 10:1, и селективность хорошая. В диапазоне от -10°C до 10°C, селективность реакции составляет 14:1. Селективность при 25°C немного ниже. Учитывая селективность реакции, безопасность для оборудования и экономию энергии, предпочтительным диапазоном температур является диапазон от 0°C до 10°C.
Вариант осуществления 6
Влияние концентрации реагентов на соотношение (селективность) 3,5'-диаллил-2'-гидрокси-[1,1'-бифенил]-4-ил 4-метилбензолсульфоната (соединение 4) и 3,5'-диаллил-4'-гидрокси-[1,1'-бифенил]-2-ил 4-метилбензолсульфоната (соединение 4a)
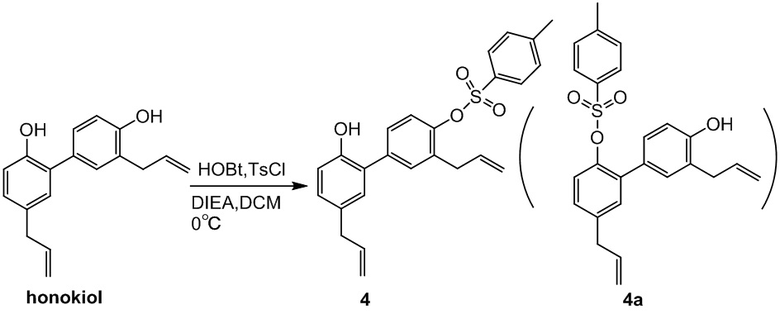
Методика: 1) 500 мл дихлорметана (в двух других партиях 1 л и 2 л, соответственно) помещали при перемешивании в 2-литровую трехгорлую колбу и охлаждали до 0°C, затем последовательно добавляли HOBT (12.4 г) DIEA (26.9 г) и TsCl (15.9 г). Через 30 минут добавляли хонокиол (26.6 г), и полученную смесь непрерывно перемешивали 6 часов. Методом LC-MS отслеживали соотношение соединения 4 и соединения 4a.
Результаты эксперимента показаны ниже в таблице:
Заключение: до тех пор, пока концентрация реагентов ниже 0.2 моль/л, соотношение соединения 4 к 4a выше 10:1, и селективность хорошая; и более предпочтительно, чтобы концентрация была ниже 0.1 моль/л.
Вариант осуществления 7:
Соединение 7 использовалось в качестве исходного, и приведенные ниже соединения 14-17 также были получены способом по настоящему изобретению.
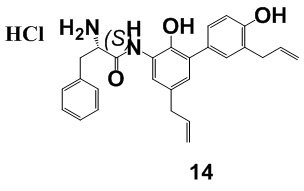
MS: 429.1 [M + H]+ (свободное основание).
1H-ЯМР (400 МГц, CDCl3) δ 10.29 (1H, с), 9.46 (1H, с), 8.51 (1H, с), 8.42 (3H, т), 7.31-7.29 (5H, м), 7.18-7.14 (2H, м), 7.09-7.08 (1H, д), 6.87-6.85 (2H, т), 6.00-5.88 (2H, м), 5.11-4.99 (4H, м), 4.40-4.37 (2H, т), 3.33-3.10 (6H, м).
дигидрохлорид (соединение 15)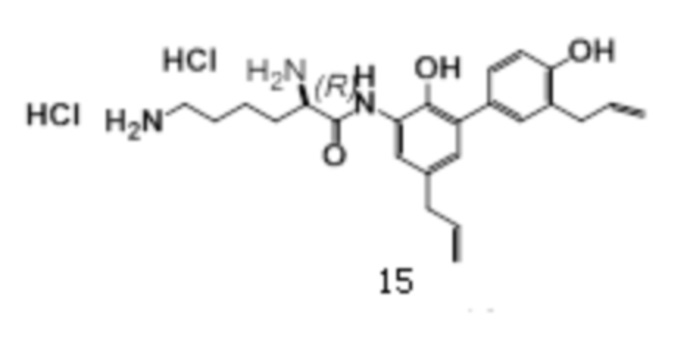
1H-ЯМР (400 МГц, ДМСО) δ 10.43 (1H, с), δ 9.46 (1H, с), δ 8.61 (1H, с), δ 8.43 (3H, с), 7.99 (3H, с), 7.36 (1H, с), 7.18-7.14 (2H, т), 6.87-6.85 (2H, д), 6.00-5.90 (2H, м), 5.11-4.99 (4H, м), 4.16 (1H, д), 3.29 (4H, м), 2.78 (2H, т), 1.89 (2H, кв), 1.64 (2H, м), 1.48 (2H, м).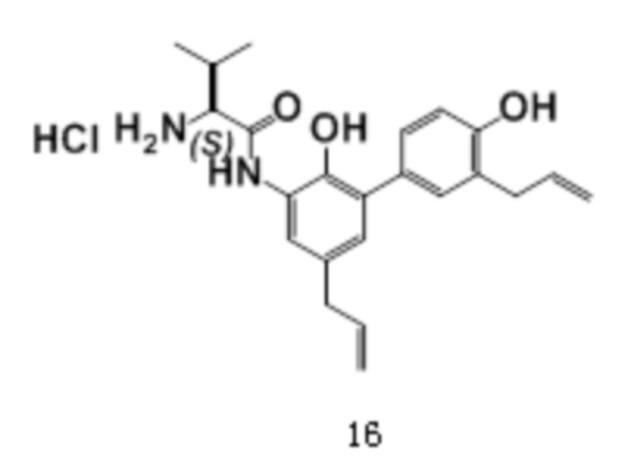
MS: 381.2 [M + H]+ (свободное основание).
1H -ЯМР (400 МГц, ДМСО) δ 10.32 (1H, с), δ 9.45 (1H, с), δ 8.56 (1H, с), δ 8.34 (3H, с), 7.34 (1H, с), 7.19-7.14 (2H, м), 6.87-6.85 (2H, кв), 6.01-5.90 (2H, м), 5.11-5.00 (4H, м), 4.00 (1H, т), 3.33-3.29 (4H, м), 2.23 (1H, м), 1.17 (6H, д).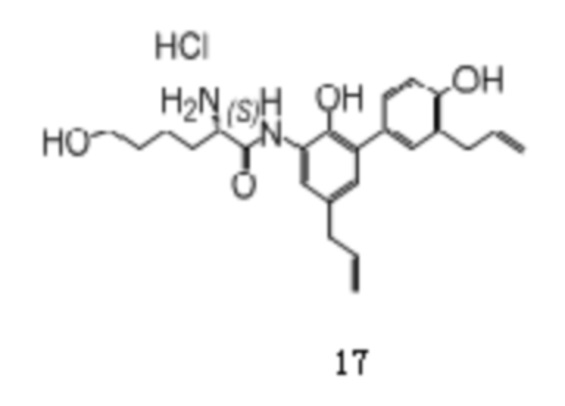
MS: 413.3 [M + H]+ (свободное основание).
1H -ЯМР (400 МГц, ДМСО) δ 10.19 (1H, с), δ 9.45 (1H, с), δ 8.55 (1H, с), δ 8.36 (3H, с), 7.34 (1H, с), 7.19-7.14 (2H, м), 6.86-6.84 (2H, т), 6.00-5.90 (2H, м), 5.11-5.00 (4H, м), 4.19 (1H, т), 3.39-3.29 (4H, м), 2.61-2.57 (2H, м), 1.84 (2H, кв), 2.14-2.09 (5H, м).
Сравнительный пример 1
Нитрованный интермедиат (соединение 3) производного хонокиола получали согласно следующей схеме реакции.
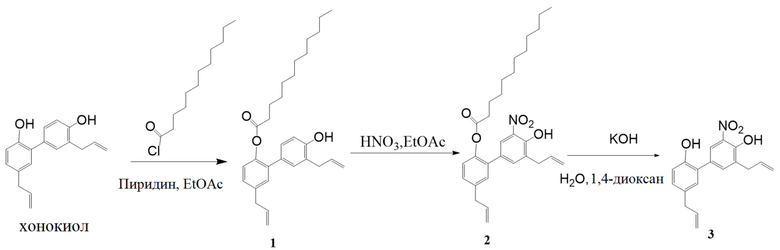
Стадия I:
Получение 3',5-диаллил-4'-гидрокси-[1,1'-бифенил]-2-ил додеканоата (соединение 1)
Хонокиол (5 г, 1 экв.) растворяли в этилацетате (50 мл), и раствор перемешивали при 0°C. Добавляли пиридин (3.9 г, 2 экв.), затем по каплям добавляли лауроил хлорид (4.5 г, 1.1 экв.). После этого реакционную смесь перемешивали в течение ночи. Разбавляли смесь этилацетатом (50 мл) и промывали последовательно 1н. водной HCl, насыщенным водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия. Органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали. Полученный сырой продукт очищали методом колоночной хроматографии (этилацетат: петролейный эфир = 1:20), получая соединение 1 (3.6 г, 95% чистота) с выходом 43%.
Соединение 1: C30H40O3 = 448.30, MS: 446 [M + NH4]+. 1H-ЯМР (300 МГц, CDCl3) δ 7.19 (4H, м), 7.03 (1H, д), 6.81 (1H, д), 6.01 (2H, м), 5.18 (4H, м), 3.42 (4H, м), 2.36 (2H, т), 1.66 (2H, м), 1.25 (16H, м), 0.89 (3H, т).
Стадия II:
Получение 3',5-диаллил-4'-гидрокси-5'-нитро-[1,1'-бифенил]-2-ил додеканоата (соединение 2)
Соединение 1 (2 г, 4.5 ммоль) растворяли в уксусном ангидриде (5 мл), и раствор перемешивали при 0°C. 65 вес.% водную HNO3 (0.8 мл) растворяли в уксусной кислоте (6 мл) и добавляли по каплям. По окончании добавления реакционную смесь непрерывно перемешивали при 0°C в течение 1 часа. Выливали смесь в ледяную воду и экстрагировали этилацетатом (50 мл x 2). Органическую фазу сушили над безводным Na2SO4, фильтровали и упаривали, получая сырой продукт, который очищали методом колоночной хроматографии (петролейный эфир: этилацетат = 30:1), получая соединение 2 (410 мг) с выходом 18%.
Соединение 2: C30H39NO5 = 493.28, MS: 511 [M + NH4]+.
Стадия III:
Получение 3', 5-диаллил-5'-нитро-[1,1'-бифенил]-2,4'-диола (соединение 3)
Соединение 2 (300 мг, 0.6 ммоль) растворяли в 1,4-диоксане (5 мл) и воде (5 мл), добавляли NaOH (100 мг, 2.5 ммоль), и полученную смесь перемешивали при комнатной температуре 2 часа. Смесь доводили до кислой реакции среды добавлением 1н. водной HCl и экстрагировали этилацетатом. Органическую фазу промывали последовательно насыщенным водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над безводным Na2SO4, фильтровали и упаривали. Полученный сырой продукт очищали методом колоночной хроматографии (петролейный эфир: этилацетат = 15:1), получая соединение 3 (140 мг, 95% чистота) с выходом 70%.
Соединение 3: C18H17NO4 = 311.12, MS: 329 [M + NH4]+. 1H ЯМР (300 МГц, ДМСО) δ 10.385 (1H, с), 9.579 (3H, с), 7.707 (1H, с), 7.422 (1H, с), 7.200 (2H, м), 6.869 (1H, с), 5.974 (2H, м), 5.062 (4H, м), 3.398 (4H, м).
Из приведенных выше результатов видно, что в описанных выше вариантах осуществления настоящего изобретения достигнуты следующие технические эффекты.
При использовании способа по настоящему изобретению существенно улучшена селективность реакции, и методика проведения значительно проще. В пилотном процессе получения по данному варианту осуществления, селективность и выход по-прежнему выше, и эффективность синтеза лучше.
Выше описаны только предпочтительные варианты осуществления настоящего изобретения, которые не ограничивают объем притязаний настоящей заявки. Квалифицированному специалисту в данной области будет понятно, что настоящее изобретение допускает различные изменения и вариации. Любые модификации, эквивалентные замены, улучшения и т.п., сделанные в рамках духа и сути настоящего изобретения, входят в объем защиты настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БИФЕНИЛАМИДИНА | 1998 |
|
RU2197478C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БАРБИТУРОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВНОСТЬЮ ИНГИБИРОВАНИЯ МЕТАЛЛОПРОТЕАЗ | 1996 |
|
RU2177475C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109736C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 2011 |
|
RU2575129C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ | 1996 |
|
RU2173316C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2252213C2 |
| БИФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ И КОСМЕТИЧЕСКАЯ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 1998 |
|
RU2193552C2 |
Настоящее изобретение относится к способу получения водорастворимого производного магнолола и производных хонокиола, к способам получения интермедиатов водорастворимых производных магнолола и производных хонокиола, а также к моногидрокси-защищенному интермедиату водорастворимого производного магнолола и производных хонокиола. Один из способов получения интермедиата относится к способу получения нитрованного интермедиата
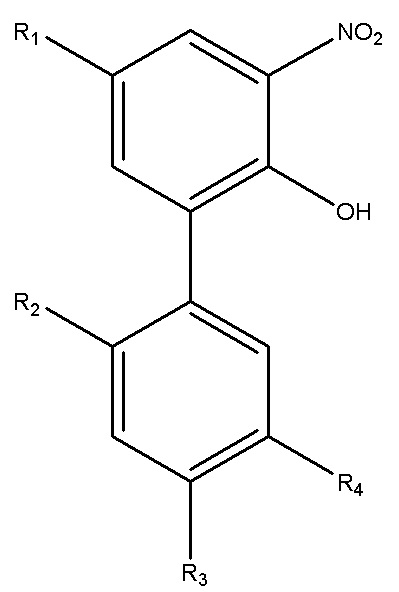 Формула I,
Формула I,
где R2 представляет собой гидроксил и R3 представляет собой H; или R2 представляет собой H и R3 представляет собой гидроксил; и R1 и R4 независимо выбраны из C1~C12 электронодонорных групп. Данный способ включает следующие стадии: проведение моногидрокси-защиты в соединении A
с помощью защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, при этом защитный реагент для гидрокси-групп представляет собой п-толуолсульфонил хлорид и 1-гидроксибензотриазол, и последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенным соединением с получением нитрованного интермедиата. Технический результат – разработка способа получения производного магнолола и производного хонокиола с улучшенной селективностью, при использовании упрощенной методики, пригодного для производства в промышленных масштабах. 5 н. и 5 з.п. ф-лы.
1. Способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола, в котором нитрованный интермедиат имеет структуру, изображенную на формуле I
Формула I
в формуле I, R2 представляет собой гидроксил и R3 представляет собой H; или R2 представляет собой H и R3 представляет собой гидроксил; и R1 и R4 независимо выбраны из C1~C12 электронодонорных групп; и
способ получения включает следующие стадии:
проведение моногидрокси-защиты в соединении A 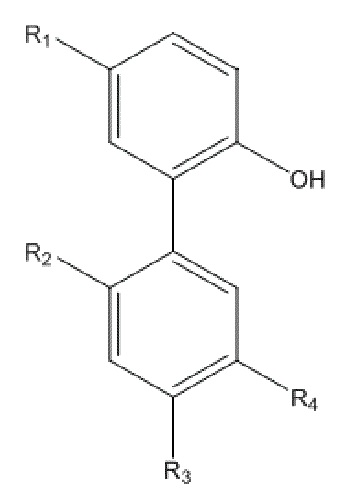 с помощью защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонил хлорид и 1-гидроксибензотриазол; и
с помощью защитного реагента для гидрокси-группы в присутствии кислотосвязывающего агента, с получением моногидрокси-защищенного соединения, где R1, R2, R3 и R4 в соединении A имеют указанные выше значения, и защитный реагент для гидрокси-групп представляет собой п-толуолсульфонил хлорид и 1-гидроксибензотриазол; и
последовательное проведение реакции нитрования и реакции снятия защиты с моногидрокси-защищенным соединением с получением нитрованного интермедиата.
2. Способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола по п. 1, в котором C1~C12 электронодонорная группа выбрана из C1~C12 алкила или C1~C12 алкенила;
предпочтительно, C1~C12 электронодонорная группа выбрана из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, н-гексила, винила, аллила, октила, гептила, пропенила, бут-1-енила, бут-2-енила, бут-3-енила, пент-1-енила, пент-2-енила, пент-3-енила, пент-4-енила, гекс-1-енила, гекс-2-енила, гекс-3-енила, гекс-4-енила, гекс-5-енила, гепт-1-енила, гепт-2-енила, гепт-3-енила, гепт-4-енила, гепт-5-енила, гепт-6-енила, окт-1-енила, окт-2-енила, окт-3-енила, окт-4-енила, окт-5-енила, окт-6-енила или окт-7-енила; и
предпочтительно, R1 и R4 одинаковые.
3. Способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола по п. 1, в котором в реакции моногидрокси-защиты мольное соотношение соединения A и п-толуолсульфонилхлорида составляет 1:(0.75~1), а мольное соотношение соединения A и 1-гидроксибензотриазола составляет 1:(0.75~1);
предпочтительно, реакцию моногидрокси-защиты проводят в первом растворителе, и первый растворитель представляет собой нереакционноспособный гидрофобный растворитель;
предпочтительно, первый растворитель выбран из одного или больше из следующих: дихлорметан, хлороформ, 1,1-дихлорэтан, метил-трет-бутиловый эфир и толуол;
предпочтительно, мольное соотношение кислотосвязывающего агента и защитного реагента для гидрокси-группы составляет (2~3):1;
предпочтительно, кислотосвязывающий агент представляет собой органическое основание; и более предпочтительно, органическое основание выбрано из одного или больше из следующих: пиридин, 4-диметиламино-пиридин, 1,8-диазабициклоундецено-7-ен, триэтиламин и N,N-диизопропилэтиламин;
предпочтительно, температура реакции в реакции моногидрокси-защиты составляет -10~25°C, более предпочтительно 0~10°C, и время реакции составляет 6~10 часов; и
предпочтительно, в реакции моногидрокси-защиты, мольная концентрация соединения A составляет меньше 0.2 моль/л относительно объема первого растворителя, и более предпочтительно, мольная концентрация соединения A составляет меньше 0.1 моль/л.
4. Способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола по любому из пп. 1-3, в котором стадия реакции нитрования включает: реакцию моногидрокси-защищенного соединения с 60~70 вес.% азотной кислотой с получением нитрованного продукта;
предпочтительно, реакцию нитрования проводят во втором растворителе, и второй растворитель представляет собой нереакционноспособный растворитель;
предпочтительно, второй растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, метил-трет-бутиловый эфир и уксусная кислота;
предпочтительно, в реакции нитрования азотную кислоту добавляют во второй растворитель, содержащий моногидрокси-защищенное соединение, по каплям, и проводят реакцию в диапазоне температур 0~25°C, получая нитрованный продукт; и
предпочтительно, в пересчете на 65 вес.% азотную кислоту, весовое соотношение моногидрокси-защищенного соединения и азотной кислоты составляет 3.2~4.2:1.
5. Способ получения нитрованного интермедиата водорастворимого производного магнолола и производного хонокиола по п. 1, в котором стадия реакции снятия защиты включает:
смешивание нитрованного продукта с третьим растворителем, с получением смешанного раствора, где третий растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, третий растворитель выбран из одного или больше из следующих: 1,4-диоксан, н-пропанол, этиленгликоль и толуол; и
добавление водного раствора гидроксида щелочного металла в смешанный раствор и нагревание для прохождения реакции с получением нитрованного интермедиата.
6. Способ получения амино-замещенного интермедиата водорастворимого производного магнолола и производного хонокиола, в котором амино-замещенный интермедиат имеет структуру, изображенную на формуле II
Формула II
R1, R2, R3 и R4 в формуле II имеют такие же значения, как в любом из пп. 1-5; и
способ получения включает следующие стадии:
применение способа получения по любому из пп. 1-5 для получения нитрованного интермедиата, изображенного на формуле I; и
проведение реакции восстановления нитрованного интермедиата с получением амино-замещенного интермедиата.
7. Способ получения амино-замещенного интермедиата водорастворимого производного магнолола и производного хонокиола по п. 6, в котором восстановитель, используемый на стадии реакции восстановления, включает один из следующих: хлорид олова, порошок железа, Na2S и NaHS;
предпочтительно, реакцию восстановления проводят в четвертом растворителе, и четвертый растворитель представляет собой смешанный растворитель спирт/кислота, где спирт выбран из одного или больше из следующих: метанол, этанол и этиленгликоль, и кислота выбрана из соляной кислоты, уксусной кислоты и водного раствора хлорида аммония; и
предпочтительно, в процессе реакции восстановления реакционную систему нагревают до кипения.
8. Способ получения свободного основания интермедиата водорастворимого производного магнолола и производного хонокиола, где свободное основание интермедиата имеет структуру, изображенную на формуле III
Формула III
R1, R2, R3 и R4 в формуле III имеют значения, указанные в любом из пп. 1-5, и R5 выбран из ацильного остатка, образующегося при удалении гидроксила из карбоксила индивидуальной аминокислоты или пептида во время реакции конденсации по карбоксилу; и
способ получения включает следующие стадии:
применение способа получения по п. 6 или 7 для получения амино-замещенного интермедиата, изображенного на формуле II;
проведение реакции конденсации между амино-замещенным интермедиатом и индивидуальной аминокислотой, защищенной трет-бутоксикарбонилом, или пептидом, защищенным трет-бутоксикарбонилом, с получением продукта конденсации; и
реакция продукта конденсации с хлороводородом, и затем подщелачивание водным раствором аммиака, экстракция и перекристаллизация с получением интермедиата в виде свободного основания,
предпочтительно
индивидуальная аминокислота выбрана из одной из следующих: лизин, метионин, триптофан, валин, аланин, фенилаланин, лейцин, изолейцин, глицин, гистидин, аргинин, пролин, глутамат, цистин и аспарагиновая кислота, и молекулярный вес пептида составляет ≤2500 Да;
предпочтительно, мольное соотношение амино-замещенного интермедиата и индивидуальной аминокислоты, защищенной трет-бутоксикарбонилом, или пептида, защищенного трет-бутоксикарбонилом, составляет (1.2~0.8):1;
предпочтительно, реакцию конденсации проводят в пятом растворителе, пятый растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, пятый растворитель выбран из одного или больше из следующих: дихлорметан, 1,2-дихлорэтан, этилацетат, тетрагидрофуран и N,N-диметилформамид;
предпочтительно, в процессе реакции конденсации температура реакции составляет 0~30°С;
предпочтительно, реакцию между продуктом конденсации и хлороводородом проводят в шестом растворителе, шестой растворитель представляет собой нереакционноспособный растворитель, и предпочтительно, шестой растворитель выбран из одного или больше из следующих: простой эфир, этилацетат, дихлорметан и 1,4-диоксан.
9. Способ получения водорастворимого производного магнолола и производного хонокиола, в котором водорастворимое производное магнолола и производное хонокиола имеет структуру, изображенную на формуле IV
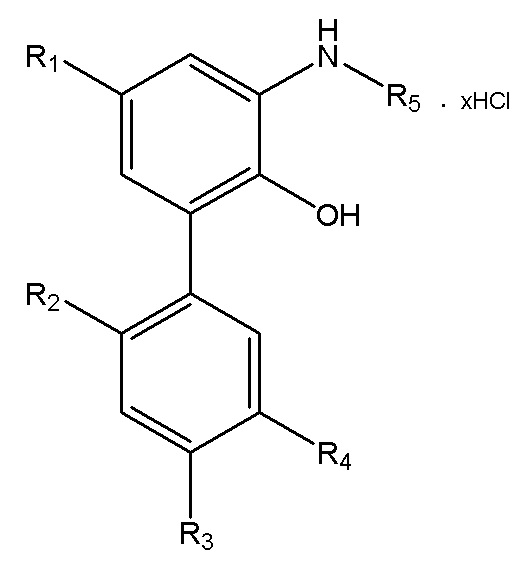
Формула IV
R1, R2, R3, R4 и R5 в формуле IV имеют значения, указанные в п. 8; x представляет собой число салифицированных аминогрупп, содержащихся в R5; и данный способ получения включает следующие стадии:
применение способа получения по п. 8 для получения интермедиата в форме свободного основания, изображенного на формуле III;
реакция интермедиата в форме свободного основания с хлороводородом, упаривание или лиофилизация с получением водорастворимого производного магнолола и производного хонокиола.
10. Моногидрокси-защищенный интермедиат водорастворимого производного магнолола и производного хонокиола, где моногидрокси-защищенный интермедиат имеет структуру, изображенную ниже на формуле V
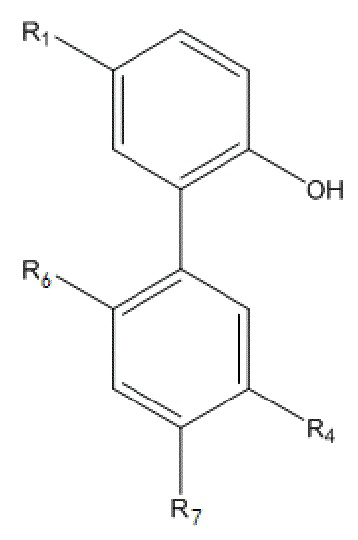
Формула V
где, в формуле V, R1 и R4 имеют указанные выше значения; R6 представляет собой 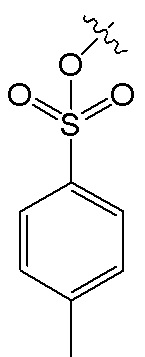 , и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой
, и R7 представляет собой H; или R6 представляет собой H, и R7 представляет собой 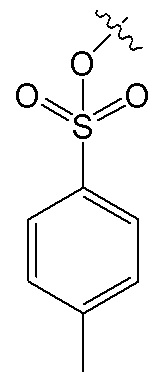 .
.
| CN 103113264 B, 14.01.2015 | |||
| РАЗГРУЗОЧНАЯ РЕШЕТКА МЕЛЬНИЦЫ | 2009 |
|
RU2423181C1 |
| WO 2008099994 A1, 21.08.2008 | |||
| Zhang, Wu et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| The Journal of Organic Chemistry, 2020, 85, 13, 8702-8713 | |||
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-ДИАЛКИЛАМИНОАЛКИЛБИФЕНИЛА | 2000 |
|
RU2259349C2 |
| EA 201070497 A1, 30.12.2010. | |||

