Область изобретения
Настоящее изобретение касается новых промежуточных продуктов синтеза производных индолина, которые демонстрируют превосходную ACAT-ингибирующую активность, и способа их получения.
Предпосылки изобретения
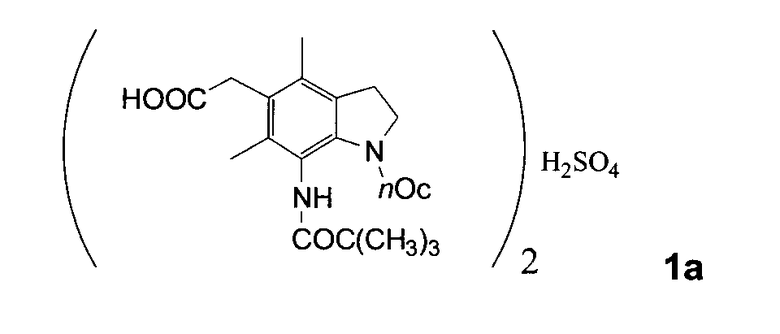
Производные индолина следующей общей формулы (1) [где R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу; nOc представляет октильную группу; предпочтительное соединение формулы (1) является соединением формулы (1a), а именно, где R2 и R3 оба представляют метильную группу] обладают превосходной ингибирующей активностью против ацил-коэнзима А: холестеринацилтрансферазы (здесь и далее обозначенный как ACAT), как раскрыто в Японском патенте № 2968050 (EP 866059 и USP 6063806).

Синтетические промежуточные продукты для получения производных индолина (1) и способ их получения раскрыты в публикации Японской патентной заявки № Hei 8-92210 (EP 782986 и USP 5990150). В частности, синтетические промежуточные продукты производных индолина формулы (1a) и способ их получения описаны в примере 3(1) и (2) публикации Японской патентной заявки № Hei 8-92210 (EP 782986 и USP 5990150), как показано на следующей схеме.

[где Ac представляет ацетильную группу]
Следующий способ получения соединения формулы (1a) раскрыт в примерах 3, 4 и 6 в Японском патенте № 2968050 (EP 866059 и USP 6063806).
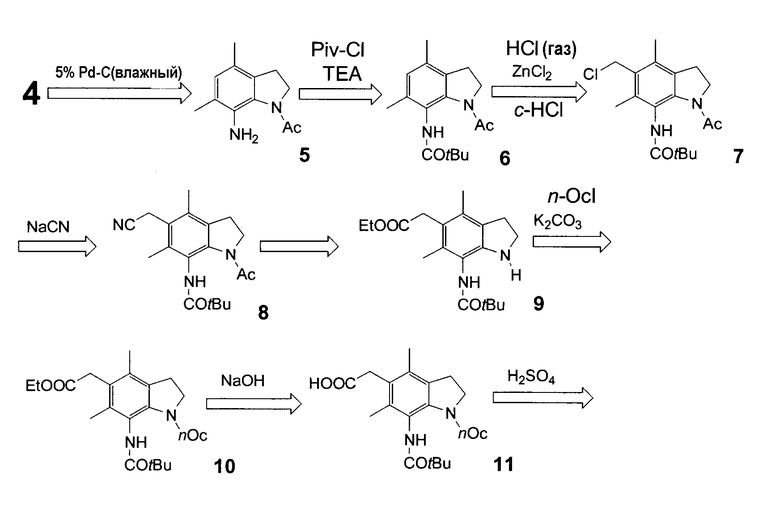
[где Et представляет этильную группу; tBu представляет трет-бутильную группу; Ac и nOc имеют такие же значения, как указано выше].
В указанном выше способе получения соединения формулы (1a) каждая из стадий реакции дает следующий выход:
стадия получения соединения (3) из соединения (2): 83,4%,
стадия получения соединения (4) из соединения (3): 63,2%,
стадия получения соединения (6) из соединения (4): 76,0%,
стадия получения соединения (7) из соединения (6): 90,0%,
стадия получения соединения (8) из соединения (7): 75,9%,
стадия получения соединения (9) из соединения (8): 59,1%,
стадия получения соединения (10) из соединения (9): 74,8%,
стадия получения соединения (11) из соединения (10): 73,2%
стадия получения соединения (1a) из соединения (10): 59,7%, и
стадия получения соединения (1a) из соединения (2): 7,2%.
Описание изобретения
В течение многих лет заявители прикладывали огромные усилия по исследованию синтетических промежуточных продуктов для получения производных индолина формулы (1), демонстрирующих превосходную ACAT-ингибирующую активность, и изучению способов их получения. Были обнаружены новые синтетические промежуточные продукты для получения производных индолина формулы (1) и новые способы их получения, что и составило данное изобретение. Новый способ получения превосходит предыдущие способы в следующем:
(1) отсутствует необходимость использовать бром или цианид натрия, при работе с которыми имеются проблемы обращения с ними и безопасности,(2) можно улучшить условия проведения реакций, в особенности, например, в процессе нитрования,
(3) можно повысить производительность, имея возможность сократить рабочее время (около 2/3),
(4) реакционные условия можно значительно смягчить, например, понижая концентрацию водного раствора гидроксида натрия на последней стадии получения производных карбоновых кислот и
(5) можно получить высокий выход соединения формулы (1) (ранее выход соединения формулы (1), получаемого из соединения формулы (2), раскрытый в указанном прототипе, составлял 7,2%, тогда как выход, получаемый в данном изобретении, составляет 27,3% или более).
Настоящее изобретение обеспечивает новые и полезные промежуточные продукты для получения производных индолина формулы (I) и способ их получения.
Новый промежуточный продукт настоящего изобретения представляет соединение общей формулы (I)
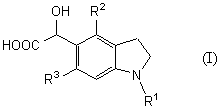
(где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу), его соль или производное амида;
или соединение общей формулы (II)
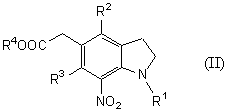
(где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу, R4 представляет атом водорода или защитную группу для карбоксильной группы), его соль или производное амида.
С другой стороны, новый способ получения соединения общей формулы (I)
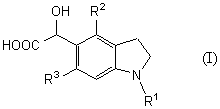
которое является новым синтетическим промежуточным продуктом в настоящем изобретении (где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу) или его соли, включает взаимодействие соединения общей формулы (IV)
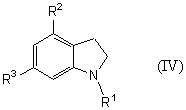
(где R1, R2 и R3 имеют такие же значения, как указано выше) с соединением общей формулы (V)
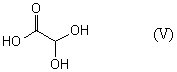
или его солью.
Новый способ получения соединения общей формулы (VI’)
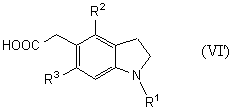
которое является новым синтетическим промежуточным продуктом в настоящем изобретении (где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу) или его соли, включает восстановление гидроксильной группы соединения общей формулы (I)
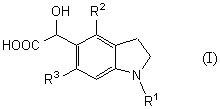
(где R1, R2 и R3 имеют такие же значения, как приведены выше) или его соли фосфорной кислотой и йодидом щелочного металла предпочтительно в органической кислоте.
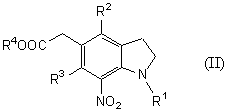
Новый способ получения соединения общей формулы (II)
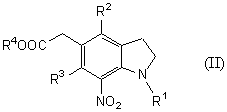
которое является новым синтетическим промежуточным продуктом в настоящем изобретении (где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу, R4 представляет атом водорода или защитную группу для карбоксильной группы) или его соли, включает нитрование соединения общей формулы (VI)
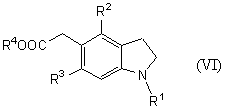
(где R1, R2, R3 и R4 имеют такие же значения, как указано выше) или его соли.
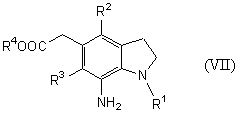
Новый способ получения соединения общей формулы (VII)
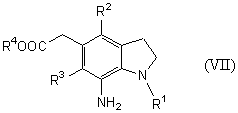
которое является синтетическим промежуточным продуктом в настоящем изобретении (где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу, R4 представляет атом водорода или защитную группу для карбоксильной группы) или его соли, включает восстановление соединения общей формулы (II)
(где R1, R2, R3 и R4 имеют такие же значения, как указано выше) или его соли.
Новый способ получения соединения общей формулы (VIII)
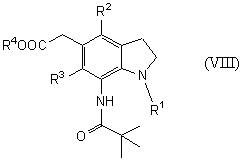
которое является синтетическим промежуточным продуктом в настоящем изобретении (где R1 представляет защитную группу для аминогруппы, R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу, R4 представляет атом водорода или защитную группу для карбоксильной группы) или его соли, включает введение пивалоила в соединение общей формулы (VII)
(где R1, R2, R3 и R4 имеют такие же значения, как указано выше) или его соль.
Новый способ получения соединения общей формулы (III)
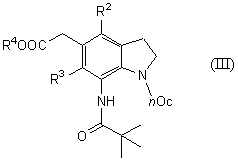
которое является синтетическим промежуточным продуктом в настоящем изобретении (где R2 и R3 являются одинаковыми или разными, и каждый представляет низшую алкильную группу, R4 представляет атом водорода или защитную группу для карбоксильной группы, nOc представляет октильную группу) или его соли, включает введение октила в соединение общей формулы (IX)
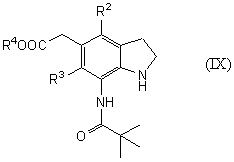
(где R2, R3 и R4 имеют такие же значения, как указано выше) или его соль предпочтительно в растворителе бутилацетате или ксилоле и более предпочтительно в присутствии диизопропилэтиламина в качестве основания.
Защитная группа для аминогруппы в определении R1 является обычной защитной группой для аминогрупп и включает:
“алифатическую ацильную группу”, например, C1-C20 алкилкарбонильную группу, такую как формил, ацетил, пропионил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, лауроил, миристоил, тридеканоил, пальмитоил или стеароил; галоген-замещенную (низший алкил)карбонильную группу, такую как хлорацетил, дихлорацетил, трихлорацетил или трифторацетил; (низший алкокси-низший алкил)карбонильную группу, такую как метоксиацетил, или (ненасыщенный алкил)карбонильную группу, такую как (E)-2-метил-2-бутеноил;
“ароматическую ацильную группу”, например, арилкарбонильную группу, такую как бензоил, α-нафтоил или β-нафтоил; галогенарилкарбонильную группу, такую как 2-бромбензоил или 4-хлорбензоил; алкилированную низшими алкилами арилкарбонильную группу, такую как 2,4,6-триметилбензоил или 4-толуоил; алкоксилированную низшими алкоксилами арилкарбонильную группу, такую как 4-анизоил; нитрованную арилкарбонильную группу, такую как 4-нитробензоил или 2-нитробензоил; (низший алкокси)карбонилированную арилкарбонильную группу, такую как 2-(метоксикарбонил)бензоил; или арилированную арилкарбонильную группу, такую как 4-фенилбензоил;
“алкоксикарбонильную группу”, например, низшую алкоксикарбонильную группу, такую как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил или изобутоксикарбонил; или низшую алкоксикарбонильную группу, замещенную галогеном(галогенами) или три(низший алкил)силилом(силилами), такую как 2,2,2-трихлорэтоксикарбонил, 2-триметилсилилэтоксикарбонил;
“алкенилоксикарбонильную группу”, такую как винилоксикарбонил или аллилоксикарбонил;
“аралкилоксикарбонильную группу”, которая может быть необязательно замещена одним или двумя заместителями, выбранными из низшей алкокси или нитро-группы, такую как бензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, 2-нитробензилоксикарбонил или 4-нитробензилоксикарбонил;
“силильную группу”, например, три(низший алкил)силильную группу, такую как триметилсилил, триэтилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, метилизопропилсилил, метил-ди-трет-бутилсилил или триизопропилсилил; или три(низший алкил)силильную группу, замещенную одной или двумя арильными группами, такую как дифенилметилсилил, дифенилбутилсилил, дифенилизопропилсилил или фенилдиизопропилсилил;
“аралкильную группу”, например, низшую алкильную группу, замещенную одной-тремя арильными группами, такую как бензил, фенетил, 3-фенилпропил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, α-нафтилдифенилметил или 9-антрилметил; или низшую алкильную группу, замещенную одной-тремя арильными группами, где указанная арильная группа имеет заместители, выбранные из низшего алкила, низшего алкокси, нитро, галогена или цианогруппы, такую как 4-метилбензил, 2,4,6-триметилбензил, 3,4,5-триметилбензил, 4-метоксибензил, 4-метоксифенилдифенилметил, 2-нитробензил, 4-нитробензил, 4-хлорбензил, 4-бромбензил, 4-цианобензил, 4-цианобензилдифенилметил, бис(2-нитрофенил)метил или пиперонил;
“ацилоксиалкильную группу”, такую как этилкарбонилоксиметил, пивалоилоксиметил, диметиламиноацетилоксиметил или 1-ацетоксиэтил;
“1-(алкоксикарбонилокси)алкильную группу, такую как 1-(метоксикарбонилокси)этил, 1-(этоксикарбонилокси)этил, этоксикарбонилоксиметил, 1-(изопропоксикарбонилокси)этил, 1-(трет-бутоксикарбонилокси)этил, 1-(этоксикарбонилокси)пропил или 1-(циклогексилоксикарбонилокси)этил;
“фталидильную группу”; или “карбонилоксиалкильную группу”, например, оксодиоксоленилметильную группу, такую как 4-метил-оксодиоксоленилметил, 4-фенил-оксодиоксоленилметил или оксодиоксоленилметил. Предпочтительной защитной группой для аминогруппы является алифатическая ацильная группа, более предпочтительна C1-C20 алкилкарбонильная группа и наиболее предпочтительной защитной группой является ацетильная группа.
“Низшая алкильная группа” в определении R2 и R3 является линейной или разветвленной алкильной группой, имеющей от 1 до 6 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил или 2-этилбутил. Предпочтительная алкильная группа представляет линейную или разветвленную алкильную группу, имеющую от 1 до 4 атомов углерода, более предпочтительны метильная или этильная группы и наиболее предпочтительна метильная группа.
Термин “защитная группа для карбоксильной группы” в описании R4 относится к “защитной группе при химических взаимодействиях”, которую можно удалить гидрированием, гидролизом, электролизом или фотолизом. Такая “защитная группа при химических взаимодействиях” включает:
“низшую алкильную группу”, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил или 2-этилбутил;
“алкенильную группу”, такую как этенил, 1-пропенил, 2-пропенил, 1-метил-2-пропенил, 1-метил-1-пропенил, 2-метил-1-пропенил, 2-метил-2-пропенил, 2-этил-2-пропенил, 1-бутенил, 2-бутенил, 1-метил-2-бутенил, 1-метил-1-бутенил, 3-метил-2-бутенил, 1-этил-2-бутенил, 3-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 1-этил-3-бутенил, 1-пентенил, 2-пентенил, 1-метил-2-пентенил, 2-метил-2-пентенил, 3-пентенил, 1-метил-3-пентенил, 2-метил-3-пентенил, 4-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил;
“алкинильную группу”, такую как этинил, 2-пропинил, 1-метил-2-пропинил, 2-метил-2-пропинил, 2-этил-2-пропинил, 2-бутинил, 1-метил-2-бутинил, 2-метил-2-бутинил, 1-этил-2-бутинил, 3-бутинил, 1-метил-3-бутинил, 2-метил-3-бутинил, 1-этил-3-бутинил, 2-пентинил, 1-метил-2-пентинил, 2-метил-2-пентинил, 3-пентинил, 1-метил-3-пентинил, 2-метил-3-пентинил, 4-пентинил, 1-метил-4-пентинил, 2-метил-4-пентинил, 2-гексинил, 3-гексинил, 4-гексинил, 5-гексинил;
“галоген-замещенную низшую алкильную группу”, такую как трифторметил, трихлорметил, дифторметил, дихлорметил, дибромметил, фторметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 2-бромэтил, 2-хлорэтил, 2-фторэтил, 2-иодэтил, 3-хлорпропил, 4-фторбутил, 6-иодгексил или 2,2-дибромэтил;
“гидроксил-замещенную низшую алкильную группу”, такую как 2-гидроксиэтил, 2,3-дигидроксипропил, 3-гидроксипропил, 3,4-дигидроксибутил или 4-гидроксибутил;
“алифатическую ацил-замещенную низшую алкильную группу”, такую как ацетилметил;
“аралкильную группу”, например, “низшую алкильную группу”, замещенную одной-тремя арильными группами, такую как бензил, фенетил, 3-фенилпропил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, 6-фенилгексил, α-нафтилдифенилметил или 9-антрилметил, или “низшую алкильную группу”, замещенную одной-тремя арильными группами, где указанная арильная группа имеет заместители, выбранные из низшего алкила, низшего алкокси, нитро, галогена, циано или алкоксикарбонильной группы, такую как 4-метилбензил, 2,4,6-триметилбензил, 3,4,5-триметилбензил, 4-метоксибензил, 4-метоксифенилдифенилметил, 2-нитробензил, 4-нитробензил, 4-хлорбензил, 4-бромбензил, 4-цианобензил, 4-цианобензилдифенилметил, бис(2-нитрофенил)метил, пиперонил или 4-метоксикарбонилбензил; или
“силильную группу”, такую как триметилсилил, триэтилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, метилдиизопропилсилил, метил-ди-трет-бутилсилил, триизопропилсилил, метилдифенилсилил, изопропилдифенилсилил, бутилдифенилсилил или фенилдиизопропилсилил. Предпочтительной защитной группой для карбоксильной группы является низшая алкильная группа, более предпочтителен линейный или разветвленный алкил, имеющий от 1 до 4 атомов углерода, еще более предпочтителен метил, этил или н-пропил и наиболее предпочтительна в качестве защитной группы этильная группа.
Термин “амид” относится к группе, которую получают при замещении карбоксильной группы аминогруппой, где указанная аминогруппа может быть необязательно замещена одним или двумя заместителями, описанными ниже. Указанный заместитель включает описанную выше “низшую алкильную группу”;
“алкилокси-группу”, например, низшую алкокси-группу, такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси или трет-бутокси, замещенную низшим алкокси низшую алкокси-группу, такую как 2-метоксиэтокси или галогенированную низшую алкокси-группу, такую как 2,2,2-трихлорэтокси;
“аралкилокси-группу”, например, низшую алкокси-группу, замещенную одной-тремя арильными группами, такую как бензилокси, фенетилокси, 3-фенилпропокси, α-нафтилметокси, β-нафтилметокси, дифенилметокси, трифенилметокси, α-нафтилдифенилметокси или 9-антрилметокси; или низшую алкокси-группу, замещенную одной-тремя арильными группами, где указанная арильная группа имеет заместители, выбранные из низшего алкила, низшего алкокси, нитро, галогена или циано-группы, такой как 4-метилбензилокси, 2,4,6-триметилбензилокси, 3,4,5-триметилбензилокси, 4-метоксибензилокси, 4-метоксифенилдифенилметокси, 2-нитробензилокси, 4-нитробензилокси, 4-хлорбензилокси, 4-бромбензилокси, 4-цианобензилокси, 4-цианобензилдифенилметокси, бис(2-нитрофенил)метокси или пиперонилокси;
“гидрокси-замещенную низшую алкильную группу”, такую как гидроксиметил, 2-гидроксиэтил или 3-гидроксипропил;
“аминоалкильную группу”, такую как 2-аминоэтил или 3-аминопропил; или
“арильную группу, которая может быть необязательно замещенной низшим алкилом, низшим алкокси или галогеном”, такую как 4-толил, 4-метоксифенил, 4-хлорфенил или α- или β-нафтил.
Соль можно получить путем взаимодействия соединения, имеющего основную группу, такую как аминогруппа, с кислотой или путем взаимодействия соединения, имеющего кислотную группу, такую как карбоксильная группа, с основанием.
Предпочтительные соли соединения, имеющего основную группу, включают соли неорганических кислот, например, соли галогенводородных кислот, такие как соль фтористоводородной кислоты, соль соляной кислоты, соль бромистоводородной кислоты и соль йодистоводородной кислоты, соль азотной кислоты, соль перхлорной кислоты, соль серной кислоты или соль фосфорной кислоты; соли органических кислот, например, соли низших алкансульфоновых кислот, такие как соль метансульфоновой кислоты, соль трифторметансульфоновой кислоты или соль этансульфоновой кислоты, соли арилсульфоновых кислот, такие как соль бензолсульфоновой кислоты или пара-толуолсульфоновой кислоты, соль уксусной кислоты, соль яблочной кислоты, соль фумаровой кислоты, соль янтарной кислоты, соль лимонной кислоты, соль аскорбиновой кислоты, соль винной кислоты, соль щавелевой кислоты или соль малеиновой кислоты; или соли аминокислот, такие как соль глицина, соль лизина, соль альгинина, соль орнитина, соль глутаминовой кислоты или соль аспарагиновой кислоты. Более предпочтительной солью является соль галогенводородной кислоты или соль органической кислоты, еще более предпочтительной солью является соль галогенводородной кислоты или соль неорганической кислоты и наиболее предпочтительной солью является соль соляной кислоты или соль серной кислоты.
Предпочтительные соли соединения, имеющего кислотные группы, включают соли щелочных металлов, такие как соль натрия, соль калия или соль лития; соли щелочноземельных металлов, такие как соль кальция или соль магния; такие соли металлов как соль алюминия, соль железа, соль цинка, соль меди, соль никеля или соль кобальта; соли аминов, которые включают, например, неорганические соли, такие как соль аммония или органические соли, такие как соль трет-октиламина, соль дибензиламина, соль морфолина, соль глюкозамина, соль фенилглициналкилового эфира, соль этилендиамина, соль N-метилглюкамина, соль гуанидина, соль диэтиламина, соль триэтиламина, соль дициклогексиламина, соль N,N’-дибензилэтилендиамина, соль хлорпрокаина, соль прокаина, соль диэтаноламина, соль N-бензил-фенетиламина, соль пиперазина, соль тетраметиламмония или соль трис(гидроксиметил)аминометана.
Если соединение настоящего изобретения оставляют в контакте с атмосферой, оно может абсорбировать воду, или вода может присоединяться к нему, образуя гидрат. Настоящее изобретение включает такие гидраты.
Соединение настоящего изобретения может абсорбировать растворитель с образованием сольвата. Настоящее изобретение включает такие сольваты.
Если соединение настоящего изобретения имеет асимметрические углероды, каждый асимметрический углерод может иметь конфигурацию R или S. Благодаря наличию асимметрических углеродов такое соединение может существовать в виде различных стереоизомеров. Настоящее изобретение включает как индивидуальные стереоизомеры, так и смеси двух или трех из них в любом соотношении.
Способы получения синтетических промежуточных продуктов, солей и амидов настоящего изобретения включают следующую реакционную схему:

[где R1, R2, R3, R4 и nOc имеют такие же значения, как определено выше].
Стадия 1
Стадия 1 представляет способ получения соединения формулы (I), который включает взаимодействие соединения формулы (IV) (такого же как соединение формулы (2)) с глиоксиловой кислотой (V) (предпочтителен моногидрат) в присутствии кислотного катализатора.
Ограничений по кислотному катализатору, используемому в данном процессе, не существует при условии, что его можно применять в обычных реакциях как кислотный катализатор. Предпочтительные кислотные катализаторы включают кислоты Бренстеда, например, неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота или фосфорная кислота; или органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота. Предпочтительными кислотными катализаторами являются неорганические кислоты и наиболее предпочтительными соляная кислота или серная кислота.
Используемый на стадии 1 растворитель является любым обычным растворителем, который не оказывает вредных воздействий на взаимодействие. В качестве растворителя можно применять на стадии 1 кислотный растворитель, который действует как кислотный катализатор. Предпочтительным растворителем является уксусная кислота, которая действует как кислотный катализатор, или вода.
Реакционная температура на стадии 1 составляет от 0°C до 110°C и предпочтительно от 60°C до 70°C.
Время взаимодействия на стадии 1 зависит, главным образом, от реакционной температуры, исходных веществ, кислотного катализатора и используемого в данной реакции растворителя и обычно составляет от 1 часа до 2 дней, предпочтительно от 3 час до 1 дня.
После взаимодействия стадии 1 можно выделить из реакционной смеси требуемое соединение формулы (I) по обычной методике. Например, можно получить требуемое соединение, добавляя к реакционной смеси подходящее количество воды с образованием осадка и отфильтровывая кристаллы. Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 2
Стадия 2 представляет способ получения соединения формулы (VI), который включает реакцию восстановительного удаления гидроксильной группы соединения формулы (I) и, если необходимо, введение защиты для карбоксильной группы соединения формулы (I). Основной способ стадии 2 можно выполнить обычным образом посредством общепринятой реакции восстановления гидроксильной группы. Предпочтительным вариантом данной реакции является:
(1) каталитическое гидрирование гидроксильной группы соединения формулы (I) в растворителе или
(2) восстановление гидроксильной группы соединения формулы (I) фосфорной кислотой и йодидом щелочного металла.
Ограничений по растворителю, используемому в реакции (1), не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол или метилцеллозольв, или органические кислоты, такие как уксусная кислота. Наиболее подходящим растворителем является этанол или уксусная кислота.
Кроме того, если в реакции применяют в качестве растворителя спирт, то получают соединение формулы (VI), имеющее группу R4, соответствующую указанному спирту (например, если применяют этанол, то получают соединение формулы (VI), имеющее в качестве группы R4 этильную группу, а если применяют метанол, то получают соединение формулы (VI), имеющее в качестве группы R4 метильную группу).
Используемый в реакции (1) катализатор восстановления включает палладий-на-угле, платину, платину-на-угле, оксид платины, гидроксид палладия или никель Ренея, предпочтительно палладий-на-угле.
Ограничений по давлению, используемому в реакции (1), не существует, и обычно оно составляет величину в диапазоне от 1 до 10 атмосфер.
Реакционная температура в реакции (1) составляет от 30°C до 90°C, предпочтительно от 60°C до 80°C.
Время взаимодействия в реакции (1) зависит, главным образом, от реакционной температуры, исходных веществ, катализатора восстановления и используемого в данной реакции растворителя и обычно составляет от 2 до 10 час, предпочтительно от 3 до 6 час.
Ограничений по растворителю, используемому в реакции (2), не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота, более подходящим растворителем является уксусная кислота.
Используемый при данном взаимодействии йодид щелочного металла представляет йодид калия или йодид натрия и более предпочтительно йодид калия.
Реакционная температура составляет от 80°C до 200°C и преимущественно от 90°C до 180°C.
Время взаимодействия зависит, главным образом, от реакционной температуры, исходных веществ и используемого при данном взаимодействии растворителя и обычно составляет от 1 до 10 час, предпочтительно от 1,5 до 6 час.Защиту карбоксильной группы на стадии 2 можно выполнить способами 1-6, что является необязательным.
Способ 1
Реакцию в способе 1 можно провести посредством взаимодействия карбоксильной группы с соединением формулы R4-X в растворителе в присутствии основания при температуре в диапазоне от -20°C до 120°C (предпочтительно от 0°C до 80°C) в течение реакционного времени от 0,5 до 10 час.
В формуле R4-X, R4 имеет такое же значение, как дано выше, и X представляет уходящую группу, например группу, нуклеофильного остатка, например, атом галогена, такой как хлор, бром или йод; низшую алкансульфонилокси-группу, такую как метансульфонилокси или этансульфонилокси; галоген-замещенную низшую алкансульфонилокси-группу, такую как трифторметансульфонилокси или пентафторэтансульфонилокси; арилсульфонилокси-группу, такую как бензолсульфонилокси, пара-толуолсульфонилокси или пара-нитробензолсульфонилокси.
R4-X включает, например, алифатический ацилоксиметилгалогенид, такой как ацетоксиметилхлорид, пивалоилоксиметилбромид или пивалоилоксиметилхлорид; низший алкоксикарбонилоксиалкилгалогенид, такой как этоксикарбонилоксиметилхлорид, изопропоксикарбонилоксиметилхлорид, 1-(этоксикарбонилокси)этилхлорид
или 1-(этоксикарбонилокси)этилйодид; фталидиилгалогенид или (5-метил-2-оксо-1,3-диоксолен-4-ил)метилгалогенид.
Ограничений по растворителю, используемому в способе 1, не существует при условии, что он не оказывает нежелательных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или ди(этиленгликоль)диметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитрилы, такие как ацетонитрил или изобутиронитрил; или амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетоамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид.
Ограничений по используемому в способе 1 основанию не существует при условии, что его можно применять обычным образом как основание. Подходящие основания включают неорганические основания, например, карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат лития; бикарбонаты щелочных металлов, такие как бикарбонат натрия, бикарбонат калия или бикарбонат лития; гидриды щелочных металлов, такие как гидрид лития, гидрид натрия или гидрид калия; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид бария или гидроксид лития; фториды щелочных металлов, такие как фторид натрия или фторид калия; алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия, метилат калия, этилат калия, трет-бутоксилат калия или метилат лития; меркаптиды щелочных металлов, такие как метилмеркаптид натрия или этилмеркаптид натрия; органические основания, такие как N-метилморфолин, триэтиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0]нон-5-ен или 1,4-диазабицикло[2.2.2]октан (DABCO) или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU); или металлоорганические основания, такие как бутиллитий, литийдиизопропиламид или литийбис(триметилсилил)амид.
Способ 2
Реакцию в способе 2 можно провести посредством взаимодействия соединения, имеющего карбоксильную группу, с соединением формулы R4-OH [где R4 имеет такие же значения, как указаны выше], применяя агент сочетания в растворителе в присутствии или в отсутствие основания.
Используемый в способе (2) агент сочетания представляет собой:
(i) комбинацию эфира фосфорной кислоты, такого как диэтилфосфорилцианид, дифенилфосфорилазид или диэтилцианфосфонат и основания, которое описано ниже;
(ii) карбодиимид, такой как 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид; комбинацию карбодиимида, приведенного выше, и основания, описанного ниже; комбинацию карбодиимида, приведенного выше, и N-гидрокси-соединения, такого как N-гидроксисукцинимид, 1-гидроксибензотриазол или N-гидрокси-5-норборнен-2,3-дикарбоксиимид;
(iii) комбинацию дисульфида, такого как 2,2’-дипиридилдисульфид или 2,2’-дибензотриазолилдисульфид, и фосфина, такого как трифенилфосфин или трибутилфосфин;
(iv) карбонат, такой как N,N’-дисукцинимидилкарбонат, ди-2-пиридилкарбонат или S,S’-бис(1-фенил-1H-тетразол-5-ил)дитиокарбонат;
(v) хлорангидрид фосфоновой кислоты, такой как N,N’-бис(2-оксо-3-оксазолидинил)фосфиновый хлорангидрид;
(vi) оксалат, такой как N,N’-дисукцинимидилоксалат, N,N’-дифталимидоксалат, N,N’-бис(5-норборнен-2,3-дикарбоксимидил)оксалат, 1,1’-бис(бензотриазолил)оксалат, 1,1’-бис(6-хлорбензотриазолил)оксалат или 1,1’-бис(6-трифторметилбензотриазолил)оксалат;
(vii) комбинацию фосфина, приведенного выше, и эфира азодикарбоновой кислоты, такого как диэтилазодикарбоксилат или 1,1’-(азодикарбонил)дипиперидин или азодикарбоксиламид; комбинацию фосфина, приведенного выше, и основания, которое описано ниже;
(viii) N-низший алкил-5-арилизоксазолий-3’-сульфонат, такой как N-этил-5-фенилизоксазолий-3’-сульфонат;
(ix) дигетероарилдиселенид, такой как ди-2-пиридилдиселенид;
(x) арилсульфонилтриазолид, такой как пара-нитробензолсульфонилтриазолид;
(xi) 2-галоген-1-низший алкилпиридинийгалогенид, такой как 2-хлор-1-метилпиридиниййодид;
(xii) имидазол, такой как 1,1’-оксалилдиимидазол или N,N’-карбонилдиимидазол;
(xiii) 3-низший алкил-2-галоген-бензотиазолийфторборат, такой как 3-этил-2-хлорбензотиазолийфторборат;
(xiv) 3-низший алкил-бензотиазол-2-селон, такой как 3-метилбензотиазол-2-селон;
(xv) фосфат, такой как фенилдихлорфосфат или сложный эфир полифосфата;
(xvi) галогенсульфонилизоцианат, такой как хлорсульфонилизоцианат;
(xvii) галогенсилан, такой как триметилсилилхлорид или триэтилсилилхлорид;
(xviii) комбинацию низшего алкалсульфонилгалогенида, такого как метансульфонилхлорид и основания, описанного ниже;
(xix) N,N,N’,N’-тетра-низший алкил-галогенформамидийхлорид, такой как N,N,N’,N’-тетра-метилхлорформамидийхлорид.
Предпочтительным агентом сочетания является карбодиимид, комбинация фосфина и сложного эфира азодикарбоновой кислоты или азодикарбоксиламида.
Ограничений по растворителю, используемому в способе 2, не существует при условии, что он не оказывает вредных воздействий на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол или
ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат, простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или ди(этиленгликоль)диметиловый эфир; нитрилы, такие как ацетонитрил или изобутиронитрил; или амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид.
Ограничений по основанию, используемому в способе 2, не существует при условии, что его можно применять обычным образом как основание. Подходящие основания включают органические основания, такие как N-метилморфолин, триэтиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин или N,N-диэтиланилин.
В способе 2 каталитическое количество 4-(N,N-диметиламино)пиридина или 4-пирролидинопиридина можно использовать в комбинации с другим основанием. Для эффективного проведения взаимодействия в реакции сочетания можно использовать дегидратирующий агент, такой как молекулярные сита; четвертичную аммониевую соль, такую как бензилтриэтиламмонийхлорид или тетрабутиламмонийхлорид; краун-эфир, такой как дибензо-18-краун-6; или улавливающий агент для кислоты, такой как 3,4-дигидро-2H-пиридо[1,2-a]пиримидин-2-он.
Реакционная температура составляет от -20°C до 80°C, предпочтительно от 0°C до комнатной температуры.
Время взаимодействия зависит, главным образом, от реакционной температуры, исходных веществ, реагентов и растворителя, используемого при данном взаимодействии, и обычно составляет от 10 мин до 3 дней, предпочтительно от 30 мин до 1 дня.
Способ 3
Способ 3 представляет процесс получения соединения, имеющего низшую алкильную группу в качестве защитной группы для карбоксильной группы. Процесс можно провести посредством взаимодействия соединения, имеющего карбоксильную группу, со спиртом в растворителе в присутствии кислотного катализатора при температуре в диапазоне от 0°C до 100°C (предпочтительно от 20°C до 60°C) в течение времени реакции от 10 мин до 24 час (предпочтительно от 15 мин до 12 час).
Ограничений по растворителю, используемому в способе 3, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают те же спирты, которые применяют в качестве реагентов данного взаимодействия, алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан
или ди(этиленгликоль)диметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитрилы, такие как ацетонитрил или изобутиронитрил; или амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид. Более подходящим растворителем является спирт, тот же, который используют в качестве реагента в данном взаимодействии.
Ограничений по кислотному катализатору, используемому в способе 3, не существует при условии, что его можно применять обычным образом в качестве кислотного катализатора. Подходящий кислотный катализатор включает кислоты Бренстеда, например, неорганические кислоты, такие как хлористый водород, бромистоводородная кислота, серная кислота, перхлорная кислота или фосфорная кислота; или органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота; кислоты Льюиса, такие как трихлорид бора, трифторид бора или трибромид бора; или кислотные ионообменные смолы. Более подходящим кислотным катализатором является неорганическая кислота и наиболее подходящим хлористый водород.
Спирт, используемый в способе 3 в качестве реагента, представляет метанол, этанол, пропанол или бутанол.
Способ 4
Реакцию в способе 4 можно провести посредством:
(i) взаимодействия соединения, имеющего карбоксильную
группу, с агентом галогенирования (например, пентахлоридом фосфора, тионилхлоридом или оксалилхлоридом) при комнатной температуре в течение времени реакции в диапазоне от 30 мин до 5 час, получая галогенангидрид кислоты, или посредством
(ii) взаимодействия соединения, имеющего карбоксильную группу, с эфиром хлормуравьиной кислоты, таким как метилхлорформиат или этилхлорформиат в присутствии органического основания, такого как триэтиламин, получая ангидрид кислоты, с последующим взаимодействием указанного галогенангидрида кислоты или ангидрида кислоты со спиртом (если получают трет-бутиловый эфир, то используют трет-бутилат калия) в инертном растворителе в присутствии основания (например, триэтиламина) при температуре от -10°C до 150°C (предпочтительна комнатная температура) в течение времени от 10 мин до 15 час (предпочтительно от 30 мин до 10 час).
Ограничений по растворителю, используемому в способе 4, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Подходящие растворители включают ароматические углеводороды, такие как бензол, толуол или ксилол; галогенированные углеводороды, такие как метиленхлорид или хлороформ, сложные эфиры, такие как этилацетат или пропилацетат; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; или нитрилы, такие как ацетонитрил.
Способ 5
Реакцию в способе 5 можно провести путем взаимодействия соединения, имеющего карбоксильную группу, с диазоалканом, таким как диазометан или диазоэтан (раствор диазоалкана в эфире) при комнатной температуре (в некоторых случаях, если необходимо, взаимодействие можно проводить при нагревании).
Способ 6
Способ 6 представляет процесс получения соединения, имеющего низшую алкильную группу в качестве защитной группы для карбоксильной группы. Данный способ можно осуществить путем взаимодействия соединения, имеющего карбоксильную группу, с диалкилсульфатом, таким как диметилсульфат или диэтилсульфат, по обычной методике.
После взаимодействия стадии 2 требуемое соединение формулы (VI) можно выделить из реакционной смеси по обычной методике. Например, реакционную смесь нейтрализуют подходящим образом или отделяют осадок, если таковой присутствует, фильтрованием и нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав
которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 3
Стадия 3 представляет способ получения соединения формулы (II), указанный способ включает нитрование 7-положения соединения формулы VI в растворителе.
Ограничений по реакции нитрования не существует при условии, что ее можно применять обычным образом для нитрования. Предпочтительной реакцией нитрования является способ, использующий азотную кислоту, нитрат натрия или дымящую азотную кислоту в качестве агента нитрования, наиболее предпочтительно применять дымящую азотную кислоту.
Ограничений по растворителю, используемому на стадии 3, не существует при условии, что он не оказывает на данную реакцию вредного воздействия. Подходящим растворителем является кислота, такая как уксусная кислота, серная кислота или смесь уксусной и серной кислот, более подходящей является смесь уксусной и серной кислот.
Реакционная температура стадии 3 составляет от -20°C до 30°C и предпочтительно от -10°C до 20°C.
Время реакции стадии 3 зависит, главным образом, от реакционной температуры, исходных веществ и используемого в данной реакции растворителя и обычно составляет от 0,5 час до 5 час, предпочтительно от 1 до 3 час.
После взаимодействия стадии 3 требуемое соединение формулы (II) можно выделить из реакционной смеси по обычной методике. Например, реакционную смесь нейтрализуют подходящим образом или отделяют осадок, если таковой присутствует, фильтрованием и нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы,
такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 4
На стадии 4 соединение (VII) получают путем восстановления нитрогруппы соединения (II) в растворителе.
Ограничений по растворителю, используемому в данной реакции, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Примеры предпочтительных растворителей включают сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; спирты, такие как метанол, этанол, н-пропанол,
изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол или метилцеллозольв; или смеси данных растворителей. Наиболее предпочтительными являются этилацетат, этанол или смеси указанных выше растворителей.
Ограничений по методике восстановления, применяемой в данной реакции, обычно не существует, но предпочтительным является каталитическое гидрирование.
Примеры катализаторов восстановления, применяемых при каталитическом гидрировании, включают палладий-на-угле, платину, платину-на-угле, оксид платины, гидроксид палладия и никель Ренея, более предпочтителен никель Ренея.
Ограничений по давлению при каталитическом гидрировании не существует, но обычно каталитическое гидрирование проводят при давлении от 1 до 10 атмосфер.
Реакционная температура обычно составляет от 20°C до 80°C и предпочтительно от 40°C до 60°C.
Время реакции зависит, главным образом, от реакционной температуры, исходных веществ и природы растворителя, используемого при данном взаимодействии. Время реакции обычно составляет от 0,5 до 10 час и предпочтительно от 1 до 5 час.
По завершении реакции соединение (VII), которое является требуемым продуктом данного взаимодействия, выделяют из реакционной смеси, применяя обычные методики.
Например, реакционную смесь нейтрализуют подходящим образом или удаляют фильтрованием осадок, если таковой присутствует, и нейтрализованную реакционную смесь или фильтрат реакционной
смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 5
На стадии 5 соединение (VIII) получают посредством введения пивалоила в аминогруппу соединения (VII) в присутствии основания в растворителе.
Ограничений по реагенту для введения пивалоила, используемому в данной реакции, обычно не существует. Примеры предпочтительных реагентов, используемых в данной реакции, включают пивалоилгалогенид, такой как пивалоилхлорид, и ангидрид пивалиновой кислоты. Среди данных реагентов более предпочтителен пивалоилгалогенид и наиболее предпочтителен пивалоилхлорид.
Ограничений по основанию, используемому при данном взаимодействии, не существует при условии, что его можно обычным образом применять в органической химии как основание. Примеры предпочтительных оснований включают гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия, гидроксид бария или гидроксид лития; и органические амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO) или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Органические основания являются более предпочтительными, и наиболее предпочтительны диизопропилэтиламин или триэтиламин.
Ограничений по растворителю, используемому в данной реакции, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и растворяет исходные вещества, по меньшей мере, в некоторой степени. Примеры предпочтительных растворителей включают галогенированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, метилацетат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; нитрилы, такие как ацетонитрил или изобутиронитрил; и спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол или метилцеллозольв, или смеси данных растворителей. Более предпочтительными среди данных растворителей являются галогенированные углеводороды или спирты и наиболее предпочтительны дихлорметан или этанол.
Реакционная температура обычно составляет от -10°C до 20°C и предпочтительно от 0°C до 10°C.
Время реакции зависит, главным образом, от реакционной температуры, исходных веществ и основания или растворителя, используемого при данном взаимодействии. Время реакции обычно составляет от 0,5 до 4 час и предпочтительно от 0,5 до 2 час.
По завершении реакции соединение (VIII), которое является требуемым продуктом данного взаимодействия, выделяют из реакционной смеси, применяя обычные методики.
Например, реакционную смесь нейтрализуют подходящим образом или удаляют фильтрованием осадок, если таковой присутствует, и
нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя
подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 6
На стадии 6 соединение (IX) получают при удалении амино-защитной группы R1 соединения (VIII) в растворителе.
Методики удаления группы R1 зависят от природы используемой защитной группы, но обычно удаление защитной группы проводят в соответствии с методиками, широко известными в данной области, которые описаны ниже.
Если группа R1 представляет силильную группу, то реакцию удаления защиты обычно проводят при обработке соединением, которое генерирует анион фтора, таким как тетрабутиламмонийфторид.
Ограничений по растворителю, используемому в указанной выше реакции, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие. Примеры предпочтительных растворителей включают простые эфиры, такие как тетрагидрофуран или диоксан.
Не существует ограничений по реакционной температуре, и время реакции конкретно не ограничено. Однако обычно удаление защиты проводят при комнатной температуре в течение периода от 10 до 18 час.
Если группа R1 представляет алифатический ацил или алкоксикарбонильную группу, то их можно удалить обработкой кислотой или основанием в присутствии растворителя.
Ограничений по кислоте, используемой в указанной выше реакции, не существует при условии, что она не оказывает вредных эффектов на данное взаимодействие. Примеры предпочтительных кислот включают неорганические кислоты, такие как соляная кислота, серная кислота, фосфорная кислота, бромистоводородная кислота, хлористый водород или бромистый водород. Наиболее предпочтителен хлористый водород.
Ограничений по основанию, используемому в указанной выше реакции, не существует при условии, что оно не оказывает вредного воздействия на другие структурные фрагменты, кроме защитной группы. Примеры предпочтительных оснований включают алкоголяты щелочных металлов, такие как метилат натрия, этилат натрия, метилат калия, этилат калия, трет-бутоксилат калия, метилат лития или этилат лития; карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат лития; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия или гидроксид лития; и аммиачные растворы, такие как водный аммиачный раствор или концентрированный аммиачный раствор в метаноле. Наиболее предпочтительны алкоголяты щелочных металлов.
Ограничений по растворителю, используемому в указанной выше реакции, не существует при условии, что его обычно применяют в реакциях гидролиза или сольволиза. Примеры предпочтительных растворителей включают воду; органические растворители, включая ароматические углеводороды, такие как бензол, толуол или ксилол; спирты, такие как метанол, этанол или н-пропанол; простые эфиры, такие как тетрагидрофуран или диоксан; сложные эфиры, такие как этилформиат, метилацетат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; или смешанные растворители, такие как смеси
органических растворителей или смеси воды и одного или большего количества из этих органических растворителей. В частности, примеры предпочтительных растворителей, используемых при обработке неорганической кислотой, включают смеси органических растворителей, выбранных из ароматических углеводородов, спиртов и сложных эфиров, перечисленных выше. Более предпочтительны смеси органических растворителей, выбранных из спиртов и сложных эфиров, и наиболее предпочтительна смесь этанола и бутилацетата.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, растворителя и кислоты или основания, используемого в данной реакции, но особо не ограничены. Удаление защиты обычно проводят при температуре от 0°C до 150°C в течение периода от 1 до 20 час для минимизации побочных реакций.
Если группа R1 представляет аралкильную или аралкилоксикарбонильную группу, то обычно и предпочтительно удаляют защитную группу посредством обработки восстановительным агентом в растворителе (предпочтительно путем каталитического гидрирования с катализатором при комнатной температуре) или окислительным агентом.
Ограничений по растворителю, используемому при удалении защиты, не существует при условии, что он не оказывает вредного воздействия на данную реакцию. Примеры предпочтительных растворителей включают спирты, такие как метанол, этанол или изопропанол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан; ароматические углеводороды, такие как толуол, бензол, или ксилол; алифатические углеводороды,
такие как гексан или циклогексан; сложные эфиры, такие как этилацетат или пропилацетат; алифатические кислоты, такие как уксусная кислота; или смеси воды и одного или большего количества из этих органических растворителей.
Ограничений по катализатору, применяемому при удалении защиты, не существует при условии, что он обычно применяется в реакциях каталитического гидрирования. Примеры предпочтительных катализаторов, применяемых при каталитическом гидрировании, включают палладий-на-угле, никель Ренея, оксид платины, платиновую чернь, родий-алюминий оксид, трифенилфосфин-родий хлорид и палладий-барий сульфат.
Давление при каталитическом гидрировании не имеет особых ограничений, но обычно при удалении защиты каталитическое гидрирование проводят при давлении от 1 до 10 атмосфер.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, природы катализатора и растворителя, используемого в данной реакции. Удаление защиты обычно проводят при температуре от 0°C до 100°C в течение периода от 5 мин до 24 час.
Ограничений по растворителю, используемому при удалении защиты реакцией окисления, не существует при условии, что он не оказывает вредного воздействия на данную реакцию. Предпочтительно проводить данную реакцию в органическом растворителе, содержащем воду. Примеры предпочтительных органических растворителей, используемых в данной реакции, включают кетоны, такие как ацетон; галогенированные углеводороды, такие как дихлорметан, хлороформ или
четыреххлористый углерод; нитрилы, такие как ацетонитрил; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; или сульфоксиды, такие как диметилсульфоксид.
Применяемый в данной реакции окислитель не имеет особых ограничений при условии, что его обычно используют для реакций окисления. Примеры предпочтительных окислителей, применяемых в реакциях окисления, включают персульфат калия, персульфат натрия, аммонийцерийнитрат (CAN) и 2,3-дихлор-5,6-дициано-пара-бензохинон (DDQ).
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, природы окислителя или растворителя, используемого в данной реакции. Удаление защиты обычно проводят при температуре от 0°C до 150°C в течение периода от 10 мин до 24 час.
Если группа R1 представляет алкенилоксикарбонил, то удаление защиты обычно проводят посредством обработки основанием в таких же реакционных условиях, как описанные для удаления защиты аминогруппы, защищенной алифатическим ацилом, ароматическим ацилом или алкоксикарбонильной группой.
Однако если группа R1 представляет аллилоксикарбонил, то удаление защиты обычно проводят, используя палладий/трифенилфосфин или никель тетракарбонил, так как данная методика удаления защиты проста, и можно предотвратить побочные реакции.
По завершении реакции соединение (IX), которое является
требуемым продуктом данной реакции, выделяют из реакционной смеси, применяя обычные методики.
Например, реакционную смесь нейтрализуют подходящим образом или удаляют фильтрованием осадок, если таковой присутствует, и нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из
используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 7
На стадии 7 соединение (III) получают, вводя октил по аминогруппе соединения (IX) в присутствии основания в растворителе.
Ограничений по агенту октилирования не существует при условии, что его обычно применяют в реакциях введения октила. Примеры предпочтительных реагентов в данной реакции включают октилгалогениды, такие как октилхлорид, октилбромид или октилйодид. Более предпочтителен октилбромид.
Ограничений по основанию, используемому при данном взаимодействии, не существует при условии, что его можно обычным образом применять в органической химии как основание. Примеры предпочтительных оснований включают карбонат щелочного металла, такой как карбонат натрия, карбонат калия или карбонат лития; бикарбонат щелочного металла, такой как бикарбонат натрия, бикарбонат калия или бикарбонат лития; гидрид щелочного металла, такой как гидрид лития, гидрид натрия или гидрид калия; алкоголят металла, такой как метилат натрия, этилат натрия, метилат калия, этилат калия, трет-бутилат калия, метилат лития или этилат лития; или органические основания, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин,
диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO)
или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Органические основания являются более предпочтительными, и наиболее предпочтителен диизопропилэтиламин.
Ограничений по растворителю, используемому в указанном выше взаимодействии, не существует при условии, что он не оказывает вредных эффектов на данное взаимодействие и в некоторой степени растворяет исходные вещества. Примеры предпочтительных растворителей включают ароматические углеводороды, такие как бензол, толуол или ксилол; сложные эфиры, такие как этилформиат, метилацетат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид; или сульфоксиды, такие как диметилсульфоксид или сульфолан. Более предпочтительными среди данных растворителей являются ароматические углеводороды или сложные эфиры, еще более предпочтительны бутилацетат, толуол и ксилол и наиболее предпочтительны бутилацетат или ксилол.
Реакционная температура обычно составляет от 0°C до 160°C и предпочтительно от 100°C до 150°C.
Время реакции зависит, главным образом, от реакционной температуры, исходных веществ и природы основания или
растворителя, используемого при данном взаимодействии. Время реакции обычно составляет от 2 час до одного дня и предпочтительно от 4 до 10 час.
По завершении реакции соединение (III), которое является требуемым продуктом данного взаимодействия, выделяют из реакционной смеси, применяя обычные методики.
Например, реакционную смесь нейтрализуют подходящим образом или удаляют фильтрованием осадок, если таковой присутствует, и нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с
использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография); или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
В случае, когда необходимо разделить изомеры, каждый из изомеров можно выделить на подходящей стадии в конце каждой указанной выше стадии или в конце необходимой стадии, используя подходящую методику разделения/очистки, указанную выше, или их комбинацию.
Стадия 8
На стадии 8 соединение (1), которое является соединением, полезным в качестве ACAT-ингибитора, получают путем удаления группы R4 соединения (III) и последующего превращения в сульфатное производное.
Методика удаления группы R4 зависит от природы используемой защитной группы, но, как правило, удаление защитной группы проводят в соответствии с методиками, широко известными в данной области, которые описаны ниже.
Если группа R4 представляет низшую алкильную группу или арильную группу, то удаление защиты обычно проводят обработкой кислотой или основанием.
Примеры предпочтительных кислот, используемых в указанных выше реакциях, включают соляную кислоту, серную кислоту, фосфорную кислоту или бромистоводородную кислоту.
Ограничений по основанию, используемому в указанной выше реакции, не существует при условии, что оно не оказывает вредных
воздействий на структурные фрагменты других групп, кроме защитной группы. Примеры предпочтительных оснований включают карбонаты щелочных металлов, такие как карбонат натрия или карбонат калия; органические основания, такие как диизопропилэтиламин; гидроксиды щелочных металлов, такие как гидроксид натрия или гидроксид калия; или концентрированный аммиачный раствор в метаноле.
Ограничений по растворителю, используемому в указанной выше реакции, не существует при условии, что его обычно применяют в реакциях гидролиза, и он не оказывает вредных эффектов на данное взаимодействие. Примеры предпочтительных растворителей включают воду и смесь воды и одного или большего количества органических растворителей, включая спирты, такие как метанол, этанол или н-пропанол; или простые эфиры, такие как тетрагидрофуран или диоксан.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, растворителя и реагента, используемого в данной реакции, но не имеют особых ограничений. Удаление защиты обычно проводят при температуре от 0°C до 150°C в течение периода от 1 до 10 час для минимизации побочных реакций.
Если группа R4 представляет диарил-замещенную метильную группу, такую как дифенилметил, то удаление защиты обычно проводят посредством обработки кислотой в растворителе.
Примеры предпочтительных растворителей, используемых в указанной выше реакции, включают ароматические углеводороды, такие как анизол. С другой стороны, примеры предпочтительных кислот, используемых в указанной выше реакции, включают
фторированные органические кислоты, такие как трифторуксусная кислота.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, растворителя и кислоты, используемой в данном взаимодействии. Данное взаимодействие обычно проводят при комнатной температуре в течение периода от 30 мин до 10 час.
Если группа R4 представляет аралкильную группу или галоген-замещенную низшую алкильную группу, то удаление защиты обычно проводят посредством реакции восстановления в растворителе.
Как правило, если защитная группа карбоксильной группы представляет галоген-замещенный низший алкил, то предпочтительным удалением защиты является химическое восстановление, такое как реакция с использованием цинка-уксусной кислоты. С другой стороны, если защитная группа карбоксильной группы является аралкильной группой, то удаление защиты предпочтительно проводить либо путем каталитического гидрирования с применением катализатора, такого как палладий-на-угле или платина, либо химическим восстановлением с использованием сульфида щелочного металла, такого как сульфид калия или сульфид натрия.
Ограничений по растворителю, используемому в указанной выше реакции, не существует при условии, что он не оказывает вредного воздействия на данную реакцию. Примеры предпочтительных растворителей включают спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; алифатические кислоты, такие как уксусная кислота; или смесь
воды и одного или большего количества данных органических растворителей.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, растворителя и методики восстановления, используемой в данной реакции. Удаление защиты обычно проводят при температуре от 0°C до комнатной температуры в течение периода от 5 мин до 12 час.
Если группа R4 представляет алкоксиметильную группу, то удаление защитной группы обычно проводят посредством обработки кислотой в растворителе.
Ограничений по кислоте, используемой в данной реакции, не существует при условии, что она обычно применяется как кислота Бренстеда. Примеры предпочтительных кислот Бренстеда включают неорганические кислоты, такие как соляная кислота или серная кислота; и органические кислоты, такие как уксусная кислота или пара-толуолсульфоновая кислота.
Ограничений по растворителю, используемому в указанной выше реакции, не существует при условии, что он не оказывает вредного воздействия на данную реакцию. Примеры предпочтительных растворителей включают спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; или смесь воды и одного или большего количества из этих органических растворителей.
Реакционная температура и время реакции зависят, главным образом, от исходных веществ, растворителя и природы кислоты, используемой в данной реакции. Удаление защиты обычно проводят при температуре от 0°C до 50°C в течение периода от 10 мин до 18 час.
По завершении реакции соединение (1), которое является требуемым продуктом данного взаимодействия, выделяют из реакционной смеси, применяя обычные методики.
Например, реакционную смесь нейтрализуют подходящим образом или удаляют фильтрованием осадок, если таковой присутствует, и нейтрализованную реакционную смесь или фильтрат реакционной смеси распределяют между органическим растворителем, таким как этилацетат, который не смешивается с водой, и водой или подобным. Отделенный органический слой, содержащий требуемое соединение, промывают водой, сушат над безводным сульфатом магния или подобным и концентрируют, получая требуемый продукт.
Полученный таким образом продукт можно, если необходимо, выделить и очистить обычными способами, такими как перекристаллизация, переосаждение, или способами, которые обычно применяют для выделения и очистки органических соединений. Примеры указанных выше способов включают адсорбционную колоночную хроматографию с использованием стационарной фазы, такой как силикагель, оксид алюминия или флорисил, в состав которого входит магний-силикагель; распределительную колоночную хроматографию с использованием синтетического адсорбента, такого как Sephadex LH-20 (продукт Pharmacia Co., Ltd.), Amberlite XAD-11 (продукт Rohm & Haas Co., Ltd) или Diaion HP-20 (продукт Mitsubishi Chemical Corporation); ионообменную хроматографию; жидкостную хроматографию с нормальной и обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна высокоэффективная жидкостная хроматография);
или подходящую комбинацию данных способов, и требуемое соединение можно выделить и очистить, проводя элюирование из используемой колонки подходящим растворителем.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Новые промежуточные продукты данного изобретения полезны для производства указанных выше производных индолина (1), обладающих превосходной ACAT-ингибирующей активностью, и новые способы получения данного изобретения превосходят обычные общеизвестные способы по нескольким пунктам, описанным ниже:
(1) отсутствует необходимость использовать бром или цианид натрия, при работе с которыми имеются проблемы обращения с ними и безопасности,
(2) можно улучшить условия проведения реакций, в особенности, например, в процессе нитрования,
(3) можно повысить производительность, имея возможность сократить рабочее время (примерно 2/3),
(4) реакционные условия можно значительно смягчить, например, понижая концентрацию водного раствора гидроксида натрия на последней стадии получения производных карбоновых кислот, и
(5) можно получить высокий выход соединения формулы (1) (ранее выход соединения формулы (1), получаемого из соединения формулы (2), раскрытый в указанном прототипе, составлял 7,2%, тогда как выход, получаемый в данном изобретении, составляет 27,3% или более).
Лучший способ осуществления изобретения
Приведенные далее примеры предназначены для дополнительной
иллюстрации настоящего изобретения и никоим образом не ограничивают области данного изобретения.
Пример 1
Получение гидрокси(1-ацетил-4,6-диметилиндолин-5-ил)уксусной кислоты
К суспензии 1-ацетил-4,6-диметилиндолина (50 г) в концентрированной соляной кислоте (400 мл) добавляют моногидрат глиоксиловой кислоты (48,6 г, 2,0 эквивалента) и полученную смесь перемешивают в течение 4,5 час при 60-65°C. После охлаждения до 0-5°C реакционную смесь перемешивают далее в течение более 0,5 час при данной температуре и осадившиеся кристаллы отделяют фильтрованием и промывают водой (500 мл).
Полученные кристаллы (около 126,4 г, вес во влажном состоянии) растворяют в 1M водном растворе гидроксида натрия (800 мл) при перемешивании, и полученную смесь промывают дихлорметаном (700 мл). К полученному водному слою добавляют метанол (700 мл) и доводят рН полученной смеси до 2, добавляя по каплям 1M соляную кислоту (около 500 мл), при перемешивании и поддерживая температуру при 0-5°C. После дополнительного перемешивания в течение более 0,5 час при охлаждении выпаривают в вакууме только метанол. К полученной таким образом суспензии добавляют метанол (130 мл, метанол добавляют, доводя соотношение вода/метанол до 10/1) и полученную смесь перемешивают более 0,5 час при 0-5°C.
Осадившиеся кристаллы отделяют фильтрованием, промывают водой (800 мл) и сушат при пониженном давлении (в течение более 10 час при 40°C). В конце данного периода высушенные кристаллы
извлекают из сушилки и хорошо размалывают, а затем сушат при пониженном давлении (в течение более 10 час при 40°C) получая указанное в заголовке соединение (59,8 г, выход: 86%) в виде бесцветных кристаллов.
1H-ЯМР (ДМСО-d6) δ м.д.
7,70 (1H, синглет);
5,23 (1H, синглет);
4,04 (2H, триплет, J=8,28Гц);
2,99 (2H, триплет, J=8,28Гц);
2,27 (3H, синглет);
2,16 (3H, синглет);
2,12 (3H, синглет).
Пример 2
Получение 1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолина
К гидрокси(1-ацетил-4,6-диметилиндолин-5-ил)уксусной кислоте (3,0 г), полученной в примере 1, и 7,5% палладию-на-угле (1,3 г, влажный (содержание воды: 53,1%)), помещенным в 100-мл автоклав, добавляют насыщенный раствор хлористого водорода в этаноле (30 мл). После 3-кратной дегазации азотом (5 кг/см2 для каждой) и 3-кратного последовательного замещения азота водородом (5 кг/см2 для каждого) полученную смесь перемешивают в течение 5 час при 70°C в атмосфере сжатого водорода (5 кг/см2). Реакционную смесь фильтруют, удаляя палладий-на-угле, который промывают этанолом (45 мл). Фильтрат и смыв объединяют и концентрируют в вакууме, а после добавления к остатку ацетона (30 мл) полученную смесь концентрируют в вакууме еще раз. Затем к полученному остатку добавляют последовательно ацетон (30 мл),
диизопропилэтиламин (4,4 г, 3,0 эквивалента) и ацетилхлорид (0,81 мл, 1,0 эквивалент) и полученную смесь кипятят с обратным холодильником в течение 10 мин. Реакционную смесь концентрируют в вакууме. К полученному остатку добавляют последовательно воду (30 мл) и этилацетат (60 мл) и после перемешивания полученную смесь фильтруют, удаляя нерастворимые осадки, которые последовательно промывают водой (30 мл) и этилацетатом (30 мл). Фильтрат и смыв объединяют и оба слоя, органический и водный, разделяют распределением. Отделенные водные слои снова экстрагируют этилацетатом (30 мл) и экстракт объединяют с отделенным ранее органическим слоем. Органический слой промывают водой (30 мл), сушат над безводным сульфатом магния и концентрируют при пониженном давлении, получая указанное в заголовке соединение (2,86 г, выход: 91%) в виде бесцветных кристаллов.
1H-ЯМР (CDCl3) δ м.д.
7,93 (1H, синглет);
4,11 (2H, квартет, J=7,08 Гц);
4,01 (2H, триплет, J=8,28 Гц);
3,62 (2H, синглет);
3,07 (2H, триплет, J=8,28 Гц);
2,29 (3H, синглет);
2,18 (3H, синглет);
2,17 (3H, синглет);
1,21 (3H, триплет, J=7,08 Гц).
Пример 3
Получение 1-ацетил-5-метоксикарбонилметил-4,6-диметилиндолина
Указанное в заголовке соединение получают в виде бесцветных кристаллов (выход: 82%) способом, аналогичным указанному в примере 2, применяя в качестве реакционного растворителя насыщенный раствор хлористого водорода в метаноле вместо насыщенного раствора хлористого водорода в этаноле.
1H-ЯМР (CDCl3) δ м.д.
7,93 (1H, синглет);
4,02 (2H, триплет, J=8,28 Гц);
3,65 (3H, синглет);
3,64 (2H, синглет);
3,07 (2H, триплет, J=8,28 Гц);
2,29 (3H, синглет);
2,18 (3H, синглет);
2,17 (3H, синглет).
Пример 4
Получение 1-ацетил-5-н-пропилоксикарбонилметил-
4,6-диметилиндолина
Указанное в заголовке соединение получают в виде бесцветных кристаллов (выход: 93%) способом, аналогичным указанному в примере 2, применяя в качестве реакционного растворителя насыщенный раствор хлористого водорода в н-пропиловом спирте вместо насыщенного раствора хлористого водорода в этаноле.
1H-ЯМР (CDCl3) δ м.д.
7,92 (1H, синглет);
4,0 (4H, мультиплет);
3,63 (2H, синглет);
3,06 (2H, триплет, J=8,28 Гц);
2,30 (3H, синглет);
2,17 (6H, синглет);
1,60 (2H, мультиплет);
0,87 (3H, триплет, J=7,32 Гц).
Пример 5
Получение 1-ацетил-7-нитро-5-этоксикарбонилметил-
4,6-диметилиндолина
К раствору 1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолина, полученного в примере 2 (3,0 г), в уксусной кислоте (30 мл) добавляют по каплям концентрированную серную кислоту (10,8 мл) при перемешивании и охлаждении на бане со льдом, поддерживая реакционную температуру ниже 20°C.
Далее к полученной смеси добавляют по каплям дымящую азотную кислоту (0,81 мл, 1,8 эквивалента) при –5°C и перемешивают реакционную смесь в течение 1 час при данной температуре. Реакционную смесь выливают в холодную воду (90 мл) и дважды экстрагируют дихлорметаном (60 мл каждый раз). Объединенный органический слой последовательно промывают насыщенным водным раствором бикарбоната натрия (60 мл), 5% водным раствором бикарбоната натрия (60 мл) и водой (60 мл), сушат над безводным сульфатом магния и концентрируют при пониженном давлении, получая указанное в заголовке соединение (3,17 г, выход: 91%) в виде бесцветных кристаллов.
1H-ЯМР (CDCl3) δ м.д.
4,13 (2H, квартет, J=7,08 Гц);
4,13 (2H, триплет, J=8,04 Гц);
3,68 (2H, синглет);
2,28 (3H, синглет);
2,23 (3H, синглет);
2,21 (3H, синглет);
1,23 (3H, триплет, J=7,08 Гц).
Пример 6
Получение 1-ацетил-7-нитро-5-метоксикарбонилметил-
4,6-диметилиндолина
Указанное в заголовке соединение получают в виде бесцветных кристаллов (выход: 93%) способом, аналогичным указанному в примере 5, используя в качестве исходного вещества 1-ацетил-5-метоксикарбонилметил-4,6-диметилиндолин, полученный в примере 3.
1H-ЯМР (CDCl3) δ м.д.
4,14 (2H, триплет, J=8,04 Гц);
3,70 (2H, синглет);
3,67 (3H, синглет);
3,07 (2H, триплет, J=7,84 Гц);
2,28 (3H, синглет);
2,23 (3H, синглет);
2,21 (3H, синглет).
Пример 7
Получение 1-ацетил-7-амино-5-этоксикарбонилметил-
4,6-диметилиндолина
В 100-мл автоклаве суспендируют 1-ацетил-7-нитро-5-этоксикарбонилметил-4,6-диметилиндолин (5,0 г), полученный в примере 5, и 7,5% палладий-на-угле (влажный, 3,76 г) в этаноле (50 мл). После 3-кратной дегазации азотом (5 кг/см2 для каждой) и 3-кратного последовательного замещения азота водородом (5 кг/см2
для каждого) полученную смесь перемешивают в течение 2 час при 55°C в атмосфере сжатого водорода (5 кг/см2). Реакционную смесь фильтруют, удаляя палладий-на-угле, который промывают этанолом (50 мл). Фильтрат и смыв объединяют и концентрируют при пониженном давлении, получая фракцию указанного в заголовке соединения. Далее палладий-на-угле, отделенный ранее, суспендируют в дихлорметане (50 мл) и после перемешивания при комнатной температуре катализатор удаляют фильтрованием и промывают дихлорметаном (25 мл). Фильтрат и смыв объединяют и концентрируют при пониженном давлении, получая другую фракцию указанного в заголовке соединения. Указанное в заголовке соединение (4,1 г, выход: 90%) получают, объединяя две полученные ранее фракции указанного в заголовке соединения.
1H-ЯМР (CDCl3) δ м.д.
4,10 (2H, квартет, J=7,08 Гц);
4,02 (2H, триплет, J=7,46 Гц);
3,64 (2H, синглет);
2,96 (2H, триплет, J=7,56 Гц);
2,29 (3H, синглет);
2,14 (3H, синглет);
2,14 (3H, синглет);
1,21 (3H, триплет, J=7,08 Гц).
Пример 8
Получение 1-ацетил-7-амино-5-метоксикарбонилметил-4,6-диметилиндолина
Указанное в заголовке соединение с выходом 93% получают способом, аналогичным указанному в примере 7, используя
1-ацетил-7-нитро-5-метоксикарбонилметил-4,6-диметилиндолин, полученный в примере 6, в качестве исходного вещества и метанол в качестве растворителя.
1H-ЯМР (CDCl3) δ м.д.
4,02 (2H, триплет, J=7,8 Гц);
3,68 (2H, синглет);
3,64 (3H, синглет);
2,96 (2H, триплет, J=7,8 Гц);
2,29 (3H, синглет);
2,13 (6H, синглет).
Пример 9
Получение N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида
К раствору 1-ацетил-7-амино-5-этоксикарбонилметил-4,6-диметилиндолина, полученного в примере 7 (4,0 г), в дихлорметане (40 мл) добавляют диизопропилэтиламин (2,14 г, 1,2 эквивалента) при перемешивании и полученную смесь охлаждают до 0-5°C. Затем к реакционной смеси добавляют по каплям пивалоилхлорид (1,74 г, 1,05 эквивалента) при 0-5°C и полученную смесь перемешивают в течение 1,0 час при данной температуре. Реакционную смесь промывают водой (40 мл) и полученную смесь разделяют. Отделенный органический слой промывают дважды насыщенным раствором бикарбоната натрия (20 мл каждый раз) и концентрируют при пониженном давлении. К полученному остатку добавляют этилацетат (20 мл) и полученную смесь концентрируют при пониженном давлении примерно до 12 мл. Такое добавление и выпаривание этилацетата повторяют еще раз и полученную смесь перемешивают при комнатной
температуре. После перемешивания добавляют этилциклогексан (28 мл) и полученную смесь перемешивают более 10 мин при комнатной температуре, а затем более 30 мин при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, промывают смешанным растворителем: этилацетат/этилциклогексан (1:4)(40 мл) и сушат при пониженном давлении (50°C), получая указанное в заголовке соединение (3,92 г, выход: 76%) в виде бесцветных кристаллов.
1H-ЯМР (CDCl3) δ м.д.
9,18 (1H, синглет);
4,16 (1H, широкий синглет);
4,11 (2H, квартет, J=7,08 Гц);
4,00 (1H, широкий синглет);
3,68 (2H, похожий на дублет);
3,15 (1H, широкий синглет);
2,84 (1H, широкий синглет);
2,28 (3H, синглет);
2,17 (3H, синглет);
2,11 (3H, синглет);
1,25 (9H, синглет);
1,21 (3H, триплет, J=7,08 Гц).
Пример 10
Получение N-(1-ацетил-5-метоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида
Указанное в заголовке соединение получают в виде кристаллов с выходом 98% способом, аналогичным указанному в примере 9, используя 1-ацетил-7-амино-5-метоксикарбонилметил-4,6-
диметилиндолин, полученный в примере 8, в качестве исходного вещества.
1H-ЯМР (CDCl3) δ м.д.
9,18 (1H, синглет);
4,16 (1H, широкий синглет);
4,01 (1H, широкий синглет);
3,70 (2H, дублет, J=12,2 Гц);
3,15 (1H, широкий синглет);
2,85 (1H, широкий синглет);
2,28 (3H, синглет);
2,16 (3H, синглет);
2,11 (3H, синглет);
1,25 (9H, синглет).
Пример 11
Получение N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида
К смеси N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (3,5 г), и этанола (35 мл) добавляют по каплям этилат натрия (20% масс этанольный раствор), поддерживая реакционную температуру ниже 30°C, и полученную смесь кипятят с обратным холодильником в течение 1 час. После охлаждения до 0-5°C добавляют по каплям концентрированную серную кислоту (6,88 г, 7,5 эквивалента) при перемешивании и охлаждении, поддерживая реакционную температуру ниже 30°C, и полученную смесь кипятят с обратным холодильником в течение 2 час. К реакционной смеси добавляют воду (17,5 мл) и выпаривают этанол в вакууме. К полученному остатку добавляют этилацетат (35 мл) и pH полученной смеси регулируют 25% водным раствором гидроксида натрия (рН водного слоя доводят до 12-13), полученную смесь распределяют между этилацетатом и водой. Отделенный органический слой дополнительно промывают водой (35 мл). Смыв и отделенный ранее водный слой объединяют и повторно экстрагируют этилацетатом (35 мл). Данный экстракт и отделенный ранее органический слой объединяют и концентрируют при пониженном давлении, получая указанное в заголовке соединение (3,26 г) в виде сырого продукта.
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного на стадии (4) примера 3, описанного в Японском патенте № 2968050.
Пример 12
Получение N-(5-метоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида
Указанное в заголовке соединение с выходом 83% получают способом, аналогичным указанному в примере 11, используя N-(1-ацетил-5-метоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамид, полученный в примере 10, и метилат натрия в качестве исходных веществ и метанол в качестве растворителя.
1H-ЯМР (CDCl3) δ м.д.
7,05 (1H, синглет);
3,64 (2H, синглет);
3,61 (2H, синглет);
3,53 (2H, триплет, J=8,56 Гц);
2,99 (2H, триплет, J=8,56 Гц);
2,16 (3H, синглет);
2,13 (3H, синглет);
1,33 (9H, синглет).
Пример 13
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
К N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамиду, полученному в примере 11 (2,8 г), добавляют ксилол (28 мл), диизопропилэтиламин (1,72 мл, 1,2 эквивалента) и 1-октилбромид (1,80 мл, 1,3 эквивалента) и полученную смесь кипятят с обратным холодильником более 8 час. После охлаждения до температуры ниже 70°C добавляют воду (28 мл) и полученную смесь делят. Затем отделенный органический слой дважды промывают водой (28 мл каждый раз) и концентрируют в вакууме до снижения объема ксилола примерно до 6 мл, получая раствор указанного в заголовке соединения в ксилоле. К смеси раствора указанного в заголовке соединения в ксилоле и 90% водного раствора этанола (28 мл) добавляют гидроксид натрия (1,66 г, 5,0 эквивалентов) и полученную смесь перемешивают в течение 1 час при реакционной температуре 80°C. После охлаждения ниже 30°C добавляют воду (15,5 мл) и полученную смесь концентрируют в вакууме, пока объем раствора не уменьшится до 18 мл. После этого добавляют ацетон (15,5 мл) к оставшемуся раствору и регулируют рН полученной смеси до 1,4-1,6 при помощи 4н. серной кислоты при комнатной температуре. Затем добавляют воду до общего объема воды 46 мл и перемешивают полученную смесь более 30 мин при 25-30°C (температура окружающей среды). После осаждения кристаллов выпаривают только ацетон в вакууме. К остатку добавляют ацетон (5,0 мл) и полученную смесь перемешивают более 30 мин при 25-30°C (температура окружающей среды), затем отделяют кристаллы фильтрованием. Полученные таким образом кристаллы промывают 10% водным раствором ацетона и сушат при пониженном давлении, получая указанное в заголовке соединение (2,88 г, выход: 86%) в виде кристаллов.
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 14
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
К N-(5-метоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамиду, полученному в примере 12 (2,3 г) добавляют ксилол (23 мл), диизопропилэтиламин (1,12 мл, 1,2 эквивалента) и 1-октилбромид (1,81 мл, 1,3 эквивалента) и полученную смесь кипятят с обратным холодильником более 8,5 час. После охлаждения ниже 70°C, добавляют воду (23 мл) и полученную смесь делят. Затем отделенный органический слой промывают дважды водой (10 мл каждый раз) и концентрируют при пониженном давлении, получая кристаллы. К полученным кристаллам добавляют этилацетат (4 мл) и гексан (8 мл), полученную смесь перемешивают некоторое время и отделяют кристаллы фильтрованием.
Далее к раствору полученных ранее кристаллов (1,7 г) в 75% водном метаноле (37,4 мл) добавляют гидроксид натрия (0,79 г, 5,0 эквивалентов) и полученный раствор перемешивают в течение 1,5 час на бане с регулируемой температурой 60°C. После охлаждения ниже 30°C метанол выпаривают в вакууме и к остатку добавляют ацетон (9,3 мл), затем доводят рН полученной смеси до 1,4-1,6 4н. серной кислотой при комнатной температуре. После этого добавляют воду до общего объема воды 38 мл и полученную смесь перемешивают более 30 мин при 25-30°C (температура окружающей среды). После осаждения кристаллов выпаривают только ацетон в вакууме. К остатку добавляют ацетон (5,0 мл), полученную смесь перемешивают более 30 мин при 25-30°C (температура окружающей среды) и кристаллы отделяют фильтрованием. Полученные кристаллы промывают 10% водным раствором ацетона и сушат при пониженном давлении, получая указанное в заголовке соединение (1,72 г, выход: 56%) в виде кристаллов.
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 15
Получение (1-ацетил-4,6-диметилиндолин-5-ил)уксусной кислоты
К раствору гидрокси(1-ацетил-4,6-диметилиндолин-5-ил)уксусной кислоты, полученной в примере 1 (280 г), в уксусной кислоте (1120 мл) добавляют фосфорную кислоту (130 г) и йодид калия (17,6 г) и полученную смесь перемешивают в течение 2 час при 100-107°C. После охлаждения до 50°C добавляют воду (1120 мл) и осадившиеся кристаллы отделяют фильтрованием и сушат, получая (1-ацетил-4,6-диметилиндолин-5-ил)уксусную кислоту (223 г, выход: 85%).
Температура плавления: при 250°C внешних изменений не происходит.
1H-ЯМР (ДМСО-d6) δ м.д.
7,75 (1H, синглет);
4,02 (2H, триплет, J=8,0);
3,53 (2H, синглет);
2,98 (2H, триплет, J=8,0);
2,22 (3H, синглет);
2,12 (6H, синглет);
ИК-спектр (KBr)см-1: 1715, 1620.
Пример 16
Получение 1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолина
К раствору хлористого водорода (665 г) в этаноле (2000 мл) добавляют (1-ацетил-4,6-диметилиндолин-5-ил)уксусную кислоту, полученную в примере 15 (150 г), и полученную смесь перемешивают в течение 30 мин при 45-50°C и затем этанольную фракцию, содержащую соляную кислоту (1000 мл), выпаривают в вакууме. Примерно при 20°C добавляют воду (2000 мл) и выпавшие кристаллы отделяют фильтрованием и сушат, получая 1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин (146 г, выход: 87%), физико-химические свойства которого аналогичны свойствам продукта, полученного в примере 2.
Пример 17
Получение N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-
2,2-диметилпропанамидгидрохлорида
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-
диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (20 г), в смешанном растворителе: ксилол (200 мл) и этанол (40 мл) перемешивают в течение 5,5 час при 80-85°C при барботаже реакционного раствора газообразным хлористым водородом (23,1 г). После охлаждения добавляют ксилол (200 мл) и полученную смесь концентрируют в вакууме до тех пор, пока общий объем раствора не снизится примерно до 200 мл. После этого оставшийся раствор перемешивают в течение 0,5 час при 0-5°C и осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (19,25 г, выход: 98%).
1H-ЯМР (ДМСО-d6) δ м.д.
9,39 (1H, синглет)
4,07 (2H, квартет, J=7,1 Гц)
3,77 (2H, синглет)
3,68 (2H, триплет, J=7,4 Гц)
3,17 (2H, триплет, J=7,4 Гц)
2,19 (3H, синглет)
2,07 (3H, синглет)
1,28 (9H, синглет)
1,17 (3H, триплет, J=7,1 Гц).
Пример 18
Получение N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-
2,2-диметилпропанамидгидрохлорида
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (20 г), в смешанном растворителе: толуол (200 мл) и этанол (40 мл) барботируют газообразным хлористым водородом
(19,8 г) в герметичных условиях реакции и полученную смесь перемешивают в течение 10,5 час при 80-85°C. После охлаждения реакционную смесь концентрируют в вакууме до тех пор, пока общий объем не снизится примерно до 140 мл и осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (18,1 г, выход: 92%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 17.
Пример 19
Получение N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-
2,2-диметилпропанамидгидрохлорида
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (50 г), в смешанном растворителе: бутилацетат (400 мл) и этанол (8,6 мл) барботируют газообразным хлористым водородом (7,3 г) в герметичных условиях и полученную смесь перемешивают в течение 9,5 час при 90-95°C. После охлаждения реакционную смесь концентрируют в вакууме до тех пор, пока общий объем раствора не уменьшится примерно до 300 мл и осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (46,8 г, выход: 95%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 17.
Пример 20
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-
2,2-диметилпропанамидсульфата
К суспензии N-(5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамидгидрохлорида, полученного в примере 17 (49,3 г), в ксилоле (300 мл) добавляют последовательно диизопропилэтиламин (34,5 г) и октилбромид (51,6 г) и полученную смесь перемешивают в течение 5 час при 140-145°C. После охлаждения реакционную смесь промывают последовательно дважды 3% водным раствором серной кислоты (150 мл каждый раз) и 5% водным раствором гидроксида натрия (150 мл). Затем к отделенному органическому слою добавляют этанол (75 мл) и 25% водный раствор гидроксида натрия (36,5 г) и полученную смесь перемешивают в течение 3 час при 50°C. После перемешивания добавляют воду (300 мл) и полученную смесь перемешивают в течение 0,15 час при комнатной температуре. После распределения реакционной смеси отделенный водный слой концентрируют в вакууме, удаляя этанол, и к остатку добавляют ацетон (75 мл), а затем рН полученной смеси доводят до 1,4-1,6 при помощи серной кислоты и вносят затравку кристаллов аутентичного образца. После осаждения кристаллов добавляют по каплям воду (300 мл) и далее полученную смесь перемешивают в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая сырой продукт - указанное в заголовке соединение (58,9 г). Затем указанный в заголовке сырой продукт, полученный выше (20 г), суспендируют в смешанном растворителе: этилацетат (95 мл) и вода (5 мл) и перемешивают в течение 3 час при 40-50°C, а далее в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (19,2 г, общий выход: 91%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 21
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (50 г), в смешанном растворителе: бутилацетат (400 мл) и этанол (8,6 мл) барботируют газообразным хлористым водородом (15,2 г) в герметичных условиях реакции и полученную смесь перемешивают в течение 9 час при 70-75°C. После охлаждения реакционную смесь концентрируют в вакууме до тех пор, пока общий объем раствора не уменьшится примерно до 300 мл. К полученной смеси последовательно добавляют диизопропилэтиламин (47 мл), октилбромид (47 мл) и бутилацетат (50 мл) и полученную смесь перемешивают в течение 9 час при 130-135°C. После охлаждения реакционную смесь последовательно промывают дважды 3% водным раствором серной кислоты (150 мл каждый раз) и 5% водным раствором бикарбоната натрия (300 мл). Отделенный органический слой концентрируют в вакууме, удаляя растворитель, и к остатку добавляют этанол (100 мл) и 25% водный раствор гидроксида натрия (36,3 г), полученную смесь перемешивают в течение 3 час при 50-70°C. После перемешивания добавляют воду (300 мл) и полученную смесь концентрируют в вакууме, удаляя этанол. К остатку добавляют воду (200 мл) и ацетон (200 мл), доводят рН полученной
смеси до 1,4-1,6 при помощи серной кислоты и вносят затравку кристаллов аутентичного продукта. После осаждения кристаллов добавляют по каплям воду (375 мл) и затем полученную смесь перемешивают в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая сырой продукт - указанное в заголовке соединение (57,1 г).
После этого указанное в заголовке сырое соединение, полученное выше (50 г), суспендируют в смешанном растворителе: этилацетат (235 мл) и вода (12,5 мл) и перемешивают в течение 3 час при 40-50°C, а затем в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (48,6 г, общий выход: 88%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 22
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (50 г), в смешанном растворителе: ксилол (460 мл) и этанол (8,4 мл) барботируют газообразным хлористым водородом (12,2 г) в герметичных реакционных условиях и полученную смесь перемешивают в течение 8,5 час при 70-75°C. После охлаждения реакционную смесь концентрируют в вакууме до тех пор, пока общий объем раствора не уменьшится примерно до 350 мл. К полученной смеси добавляют последовательно диизопропилэтиламин (34,9 мл),
октилбромид (46 мл) и ксилол (100 мл) и полученную смесь перемешивают в течение 9 час при 135-140°C. После охлаждения реакционную смесь последовательно промывают дважды 3% водным раствором серной кислоты (150 мл каждый раз) и 5% водным раствором гидроксида натрия (150 мл). Затем к отделенному органическому слою добавляют этанол (125 мл) и 25% водный раствор гидроксида натрия (42,7 г) и полученную смесь перемешивают в течение 1 час при 70-80°C. После перемешивания добавляют воду (300 мл) и полученную смесь перемешивают в течение 0,15 час при комнатной температуре. После распределения реакционной смеси отделенный водный слой концентрируют в вакууме, удаляя этанол, добавляют к остатку воду (200 мл) и ацетон (200 мл), доводят рН полученной смеси до 1,4-1,6 при помощи серной кислоты и вносят затравку кристаллов аутентичного продукта. После выпадения кристаллов добавляют по каплям воду (375 мл) и полученную смесь перемешивают 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая сырой продукт - указанное в заголовке соединение (53,1 г).
Затем указанное в заголовке сырое соединение, полученное выше (50 г), суспендируют в смешанном растворителе: этилацетат (237,5 мл) и вода (12,5 мл) и перемешивают в течение 3 час при 40-50°C, а затем в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (47,5 г, общий выход: 81%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта,
полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 23
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
Раствор N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамида, полученного в примере 9 (50 г), в смешанном растворителе: этилацетат (400 мл) и этанол (8,6 мл) барботируют газообразным хлористым водородом (19,5 г) в герметичных реакционных условиях и полученную смесь перемешивают в течение 9 час при 60-65°C. Реакционную смесь концентрируют в вакууме, удаляя этилацетат, и последовательно добавляют к остатку ксилол (400 мл), диизопропилэтиламин (58 мл) и октилбромид (46 мл), полученную смесь перемешивают в течение 18 час при 120-130°C. После охлаждения реакционную смесь последовательно промывают дважды 3% водным раствором серной кислоты (150 мл каждый раз) и 5% водным раствором гидроксида натрия (150 мл). Затем отделенный органический слой выпаривают в вакууме, удаляя растворители, к остатку добавляют этанол (450 мл) и 25% водный раствор гидроксида натрия (36,3 г) и полученную смесь перемешивают в течение 3,5 час при 50°C. Добавляют воду (275 мл) и полученную смесь концентрируют в вакууме, удаляя этанол. К остатку добавляют воду (200 мл) и ацетон (200 мл), доводят рН полученной смеси до 1,4-1,6 при помощи серной кислоты и вносят затравку кристаллов аутентичного продукта. После осаждения кристаллов добавляют по каплям воду (375 мл) и далее перемешивают полученную смесь в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая
сырой продукт - указанное в заголовке соединение (60,4 г). После этого полученное выше сырое соединение, указанное в заголовке (50 г), суспендируют в смешанном растворителе: этилацетат (238 мл) и вода (12,5 мл) и перемешивают в течение 3 час при 40-50°C, а затем в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (46,4 г, общий выход: 90,2%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
Пример 24
Получение N-(5-карбоксиметил-4,6-диметил-1-октилиндолин-7-ил)-2,2-диметилпропанамидсульфата
К N-(1-ацетил-5-этоксикарбонилметил-4,6-диметилиндолин-7-ил)-2,2-диметилпропанамиду, полученному в примере 9 (3 г), добавляют 1M раствор этилата лития в этаноле (16,0 мл) и этанол (2 мл) и полученную смесь перемешивают в течение 2,5 час при 75-80°C. К реакционной смеси добавляют октилбромид (4,64 г) и полученную смесь перемешивают в течение 21 час при 85-90°C. К реакционной смеси добавляют 25% водный раствор гидроксида натрия (6,41 г) и полученную смесь перемешивают в течение 1 час при 80-85°C. После охлаждения до комнатной температуры добавляют последовательно толуол (18 мл), воду (18 мл) и этанол (3 мл) и полученную смесь перемешивают. После распределения полученной смеси отделенный водный слой концентрируют в вакууме, удаляя этанол. К остатку добавляют воду (7,5 мл) и ацетон (12 мл), доводят рН полученной смеси до 1,4-1,6 при помощи серной кислоты
и вносят затравку кристаллов аутентичного продукта. К полученной смеси добавляют по каплям воду (15 мл), и полученную смесь перемешивают в течение 1 час при комнатной температуре, а затем в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая сырой продукт - указанное в заголовке соединение (3,45 г). Затем полученное сырое соединение, указанное в заголовке (3 г), суспендируют в смешанном растворителе: этилацетат (14,3 мл) и вода (0,75 мл) и перемешивают в течение 3 час при 40-50°C, а затем в течение 1 час при 0-5°C. Осадившиеся кристаллы отделяют фильтрованием, получая указанное в заголовке соединение (2,77 г, общий выход: 85,3%).
Физико-химические свойства полученного соединения, указанного в заголовке, аналогичны свойствам продукта, полученного в примере 6, описанном в Японском патенте № 2968050.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СИНТЕТИЧЕСКИХ ПРОМЕЖУТОЧНЫХ ИНДОЛИНОВЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2283304C1 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ | 1996 |
|
RU2173316C2 |
| НОВОЕ СОЕДИНЕНИЕ ИНДОЛИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2003 |
|
RU2318808C2 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ДЕЙСТВИЯ ТАХИКИНИНА | 1993 |
|
RU2119922C1 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| ПРОИЗВОДНЫЕ ТИОФОРМАМИДА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2125055C1 |
| ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПРЕРЫВИСТОГО ВВЕДЕНИЯ ИНГИБИТОРА FGFR | 2014 |
|
RU2664118C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2128170C1 |
| ПРОИЗВОДНЫЕ ОКТАГИДРОНАФТАЛИНОКСИМА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2125043C1 |
Изобретение касается новых промежуточных производных индолина формул I и II
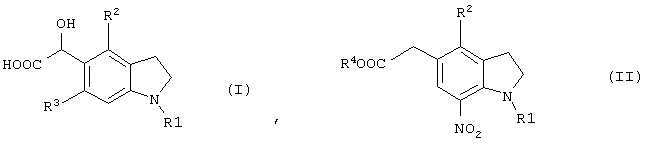
где R1 означает C1-C20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый означает низшую алкильную группу; R4 означает Н, низшую алкильную группу, его соль или производное амида и способов их получения. 6 с. и 1 з.п. ф-лы.
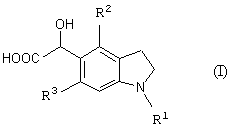
где R1 представляет C1-C20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу,
его соль или производное амида.
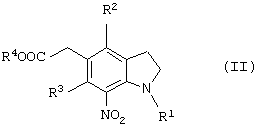
где R1 представляет С1-С20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу; R4 представляет атом водорода или низшую алкильную группу,
его соль или производное амида.
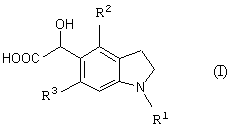
где R1 представляет C1-С20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу
или его соли, который включает взаимодействие соединения общей формулы (IV)
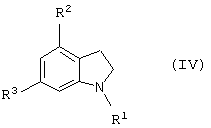
где R1, R2 и R3 имеют такие же значения, как указано выше,
с соединением общей формулы (V)
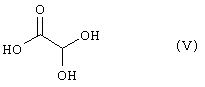
или его солью.

где R1 представляет C1-C20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу
или его соли, который включает восстановление гидроксильной группы соединения общей формулы (I)
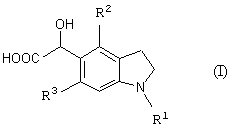
где R1, R2 и R3 имеют такие же значения, как приведены выше,
или его соли фосфорной кислотой и йодидом щелочного металла.
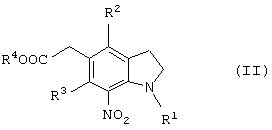
где R1 представляет C1-С20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу; R4 представляет атом водорода или низшую алкильную группу,
или его соли, который включает нитрование соединения общей формулы (VI)
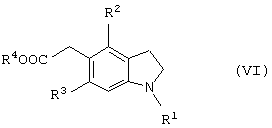
где R1, R2, R3 и R4 имеют такие же значения, как указано выше,
или его соли.
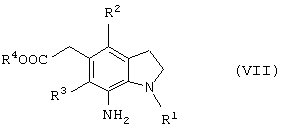
где R1 представляет С1-С20 алкилкарбонильную группу; R2 и R3 являются одинаковыми или разными и каждый представляет низшую алкильную группу; R4 представляет атом водорода или низшую алкильную группу
или его соли, который включает восстановление соединения общей формулы (II)
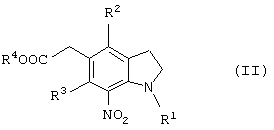
где R1, R2, R3 и R4 имеют такие же значения, как указано выше,
или его соли.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Автоматическая линия обработки труб | 1979 |
|
SU782986A2 |
| Направляющий механизм арматурно-навивочной машины | 1981 |
|
SU1004579A1 |
| RU 2052457 С1, 20.01.1996 | |||
| GB 991937, 21.08.1965 | |||
| Способ получения алифатических трихлорметилкарбинолов | 1976 |
|
SU726078A1 |

