Область техники
Изобретение относится к химии органических соединений, фармакологии и медицине и касается нового химического соединения, характеризующегося высокой противогрибковой активностью, которое, в частности, может использоваться для профилактики и лечения инфекционных заболеваний у субъекта.
Уровень техники
В конце 2022 года ВОЗ опубликовал список наиболее опасных грибковых патогенов, представляющих угрозу для человечества, и отнес к наиболее востребованной категории создание новых терапевтических средств для лечения инвазивных инфекций. Среди перечисленных патогенов первую строчку занимают такие штаммы грибов как С. auris, Ctropicalis, С. parapsilosis, Mucorales [WHO fungal priority pathogens list to guide research, development and public health action, https://www.who.int/publications/i/item/9789240060241, (accessed October 2022,https://www.who.intyru/news/item/25-10-2022-who-releases-first-ever-list-of-health-threatening-fungil
Одним из наиболее тревожных изменений в эпидемиологии инвазивного кандидоза является появление во всем мире особо опасного нового мультирезистентного штамма С, auris, способного к передаче инфекции через предметы обихода за пациентами.
Патоген характеризуется показателями высокой вирулентности, которые включают способность образовывать биопленки со склонностью к колонизации, что приводит к передаче инфекции в медицинских учреждениях и формированию устойчивости к существующим противогрибковым препаратам. Большинство исследованных изолятов имели высокий уровень устойчивости к флуконазолу (минимальная ингибирующая концентрация, МИК>64г/мл). Кроме того, до 30% изолятов демонстрировали сниженную чувствительность к амфотерицину В. Более 5% могут быть устойчивы к эхинокандинам, которые являются рекомендуемыми в настоящее время противогрибковыми препаратами первой линии при кандидемии. Факторами риска у пациентов отделений интенсивной терапии являются: предшествующее применение противогрибковых препаратов, сосудистые хирургические вмешательства, нахождение в больнице, основное легочное заболевание и постоянная катетеризация мочевыводящих путей. Несмотря на то, что С. auris причастен к меньшинству случаев кандидемии, смертность, связанная с С, auris, в некоторых исследованиях достигала 60% [J.Fungi, 2020, 6, 91 doi:10.3390/jof60091].
Инвазивный кандидоз - это серьезная инфекция, которая в первую очередь поражает тяжелобольных и пациентов с ослабленным иммунитетом. Candida albicans, обычно восприимчивая к флуконазолу, долгое время была наиболее распространенным видом, связанным с инвазивным кандидозом. Однако, с увеличением использования противогрибковых препаратов и новых методов диагностики, происходит изменение эпидемиологии инфекций Candida, что приводит к увеличению случаев инфекций, вызванных видами с менее предсказуемой чувствительностью к противогрибковым препаратам.
Созданная в 1997 году программа противогрибкового надзора SENTRY отслеживает глобальную эпидемиологию инвазивных инфекций Candida с учетом видового распределения и устойчивости к противогрибковым препаратам. Совсем недавно программа опубликовала данные за первые 20 лет, которые включали в себя более 20 000 клинических изолятов, собранных в ходе пассивного наблюдения в 39 странах мира.
Хотя С, albicans остается наиболее распространенным видом, вызывающим инвазивный кандидоз, общая доля инфекций, вызванных С, albicans, снизилась с 57,4 до 46,4% за 20-летний период наблюдения [Pfaller, MA; Diekema, D.J.; Turnidge, J.D.; Castanheira, M.; Jones, R.N. Twenty years of the SENTRY Antifungal Surveillance Program: Results for Candida Species from 1997-2016. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2019].
В США в настоящее время более 30% случаев кандидемии вызываются С. glabrata, что является тревожной тенденцией, учитывая растущие показатели устойчивости к противогрибковым препаратам, связанным с этим видом [Lamoth, F.; Lockhart, S.R.; Berkow, E.L.; Calandra, Т. Changes in the epidemiological landscape of invasive candidiasis. J. Antimicrob. Chemother. 2018, 1,i4-i13].
В некоторых центрах устойчивость к эхинокандинам, опосредованная мутациями в FKS1, наблюдается у 10% изолятов С, glabrata, и среди этих резистентных к эхинокандинам штаммов резистентность к применяющимся коммерческим препаратам азолового ряда встречается у 20% штаммов. И наоборот, в Великобритании изоляты С. glabrata редко были устойчивы к эхинокандинам (0,55%). Также чаще встречается С. parapsilosis. На долю этого вида приходится 15% случаев кандидемии в США и 20% в России, а в Южной Африке С. parapsilosis конкурирует с С. albicans как основная причина инвазивных заболеваний [Friedman, & Schwartz. (2019). Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. Journal of Fungi, 5(3), 67. doi:10.3390/jof5030067].
Виды Candida претерпели таксономическую ре классификацию с появлением новых классов, родов и видов за последнее десятилетие. Вместе с тем, С. albicans остается наиболее распространенным возбудителем кандидоза.
Серьезная проблема в лечении онкогематологических больных связана со слабой эффективностью применяющейся терапии и, вследствие этого, высокой летальностью. Лечение оппортунистических грибковых инфекций остается проблемой, несмотря на значительные достижения в поддерживающей терапии. Они по-прежнему являются одной из наиболее частых причин смерти у пациентов с иммуносупрессией. Помимо обнаружения грибов рода Aspergillus species, все чаще сообщается о зигомикозе, который является вторым по распространенности филаментозным грибом у пациентов с гематологическими новообразованиями [Gleissner, В., Schilling, A., Anagnostopolous, I., Siehl, I., & Thiel, E. (2004). Improved Outcome of Zygomycosis in Patients with Hematological Diseases? Leukemia & Lymphoma, 45(7), 1351-1360. doi:10.1080/10428190310001653691].
Зигомикоз - это зонтичный термин, ранее называвшийся как мукормикоз, который включает все микотические заболевания, вызываемые грибами рода Zygomycetes.
Основные известные патогены человека относятся к порядку Mucorales, семейство Мисогасеае, в которое входят роды Rhizopus, Absidia, Mucor, Rhizomucor и Apophysomyces. Большинство заболеваний человека вызывают представители рода Mucorales. Хотя чаще всего болезнь связана с Rhizopus spp. Зигомикоз, вызванный Mucorales, обычно возникает у иммунокомпрометированных больных как оппортунистическая инфекция. Факторы риска включают сахарный диабет, нейтропению, длительную иммуносупрессивную терапию, хроническое применение преднизолона, хелатную терапию железом, применение антибиотиков широкого спектра действия, первичное нарушение целостности кожного барьера, например, травмы, хирургические раны, уколы иглой или ожоги. Mucorales ассоциируются с ангиоинвазивными заболеваниями, часто приводящими к тромбозу, инфаркту вовлеченных тканей и разрушению тканей, опосредованному рядом грибковых протеаз, липаз и микотоксинов. Если диагноз не поставлен на ранней стадии, часто происходит диссеминация. Терапия, если она эффективна, должна быть начата рано и требует комбинации противогрибковых препаратов, хирургического вмешательства и устранения основных факторов риска.
Поэтому сохраняется высокая необходимость в разработке новых эффективных противогрибковых средств, для терапии широкого спектра заболеваний.
Раскрытие изобретения
Задачей настоящего изобретения является разработка и создание нового эффективного противогрибкового средства, перспективного для применения в клинической практике для терапии и/или профилактики инфекционных заболеваний, вызванных грибковой инфекцией.
Технический результат изобретения заключается в разработке нового соединения, характеризующегося высокоэффективной противогрибковой активностью широкого спектра действия, в том числе в отношении зигомицетов, и являющегося перспективным для применения в терапии инфекционных заболеваний у субъекта, вызванных грибковыми инфекциями (в том числе плесневыми грибами), например, вызванных кандидозными и филаментозными патогенами, в частности С. auris, C.albicans, C.tropicalis, С. krusei, С. parapsilosis, Cryptococcus neoformans, A.niger, A. fumigatus и других, для лечения, в частности, диссеминиров энных микозов, вагинального кандидоза, кандидозного стоматита, эндокардита, а также заболеваний, вызванных плесневидными зигомицетами (мукормикоз), и других заболеваний человека и животных. Кроме того, соединения по изобретению характеризуются высокой противомикробной активностью в отношении штаммов грибов, резистентных к применяющимся коммерческим препаратам азолового ряда.
Указанный технический результат достигается посредством нового соединения (Z)-(5-(4-хлорбензилидeн)-3-(2-(4-((2R,3R)-3-(2,4-дифторфенил)-гидрокси-4-(1Н-1,2,4-триазол-1-ил)-2-ил) пиперззин-1-ил)-2-оксоэтил)тиазолидин-2,4-диона следующей формулы:
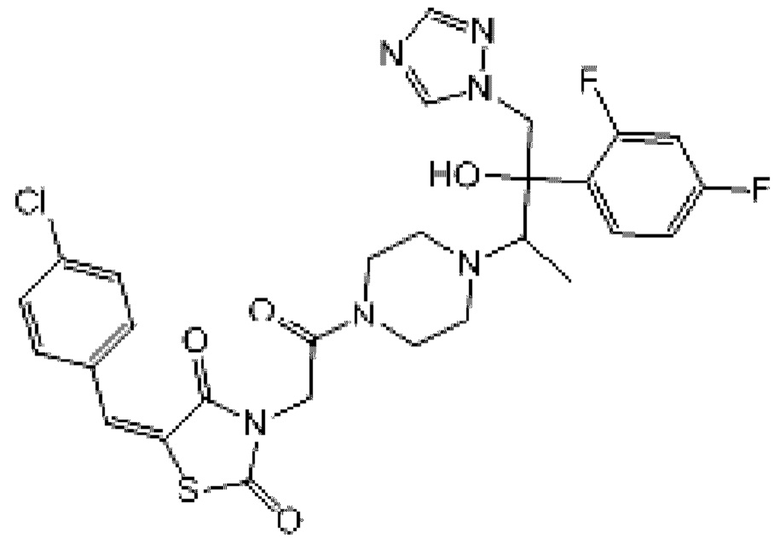
или его фармацевтически приемлемой соли.
Указанное соединение характеризуется: -
mz 617.07.
Достижение указанного технического результата обеспечивается также в результате применения соединения по изобретению или его фармацевтически приемлемой солив качестве противогрибкового средства.
Достижение указанного технического результата обеспечивается также в результате применения соединения по изобретению или его фармацевтически приемлемой соли для подавления роста и/или полной элиминации грибковых патогенов.
В некоторых вариантах воплощения изобретения патоген представляет собой патоген рода Candida spp., Rhizopus spp, Aspergillus spp., Aticrosporum spp, Trichophyton spp. и/или Epidermophyton spp.
В частных вариантах воплощения изобретения патоген представляет собой Candida auris, Candida albicans, Candida поп albicans (такие как Candida krusei, Candida glabrata, Candida parapsilosis), Aspergillus fumigatus, Aspergillus niger и/или Rhisjpus stoionifer и другие.
Достижение указанного технического результата обеспечивается также в результате применения соединения по изобретению или его фармацевтически приемлемой соли для получения фармацевтической композиции с противогрибковойактивностью для лечения и/или профилактики инфекционного заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией.
Достижение указанного технического результата обеспечивается также за счет фармацевтической композиции с противогрибковой активностью для лечения и/или предотвращения инфекционного заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией, содержащей эффективное количество соединения по изобретению и/или его фармацевтически приемлемой соли и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество.
В частных вариантах воплощения изобретения фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
В частных вариантах воплощения изобретения субъект представляет собой человека или животное.
В частных вариантах воплощения изобретения заболевание представляет собой заболевание кожи, ногтей и/или внутренних органов.
В частных вариантах воплощения изобретения заболевание представляет собой диссеминированный кандидоз, диссеминированный зигомикоз, дерматофитию, поверхностный микоз, кандидоз кожи и/или ногтей, инвазивный кандидоз, вагинальный кандидоз, кандидозный стоматит, эндокардит или мукормикоз.
Данное изобретение также относится к способу подавления роста и/или полной элиминации грибковых патогенов, в частности патогенов рода Candida spp,, Rhizopus spp, Aspergillus spp,, Microsporum spp., Trichophyton spp, и/или Epidermophyton spp, включающего введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
Изобретение также включает способ лечения и/или профилактики заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией, включающий введение терапевтически или профилактически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли, в организм субъекта, нуждающегося в лечении и/или профилактике таких заболеваний.
В частных вариантах воплощения изобретения способ подразумевает введение соединения по изобретению или его фармацевтически приемлемой соли в качестве монотерапии или в комбинации с одним или несколькими терапевтическими агентами.
Настоящее изобретение включает также получение соединений и/или композиций по изобретению.
Подробное раскрытие изобретения
Определения и термины
Для лучшего понимания настоящего изобретения ниже приведены некоторые термины, использованные в настоящем описании изобретения. Следующие определения применяются в данном документе, если иное не указано явно.
В описании данного изобретения термины «включает», «включающий» и т.п., а также «содержит», «содержащий» и т.п. интерпретируются как означающие «включает, помимо всего прочего» (или «содержит, помимо всего прочего»). Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «и/или» означает один, несколько или все перечисленные элементы.
Также здесь перечисление числовых диапазонов по конечным точкам включает все числа, входящие в этот диапазон.
Термин «необязательный» или «необязательно» или «опциональный» или «опционально», используемый в данном документе, означает, что описываемое впоследствии событие или обстоятельство может, но не обязательно, произойти, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, в которых оно не происходит.
Соединение, составляющие суть данного изобретения, может существовать в меченой радиоизотопом форме, т.е. указанное соединение может содержать один или несколько атомов, чья атомная масса или массовое число отличается от атомной массы или массового числа наиболее распространенных природных изотопов. Радиоизотопы водорода, углерода, фосфора, хлора включают 3Н, 14С, 32Р, 35S, и 36CI, соответственно. Соединение данного изобретения, которое содержит такие радиоизотопы и/или другие радиоизотопы других атомов, находятся в сфере настоящего изобретения. Тритиевые, т.е. 3Н и углеродные, т.е. 14С радиоизотопы являются особенно предпочтительными благодаря простоте приготовления и обнаружения.
Соединение настоящего изобретения, меченое радиоактивными изотопами, может быть получено с помощью методов, хорошо известных специалистам в данной области. Меченое соединение может быть получено с помощью процедур, описанных здесь, простой заменой немеченых реагентов соответствующими мечеными реагентами.
Соединение настоящего изобретения может существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты илисвободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная (хлористоводородная), бромистоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат (тозилат), ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие ал кил сульфонаты и арил сульфонаты.
Соединение настоящего изобретения может существовать в виде пролекарства. В контексте настоящей заявки термин «пролекарство» относится к предшественнику или производной форме соединения согласно изобретению, которое может обладать улучшенными свойствами, такими как лучшая растворимость, уменьшенная цитотоксичность или повышенная биодоступность, по сравнению с исходным соединением или лекарственным средством, и способно активироваться или превращаться в более активную исходную форму. «Группа пролекарства» означает группу, которая преобразуется посредством реакции с ферментом, желудочной кислотой или т.п. в физиологических условиях in vivo, с получением соединения по изобретению, т.е. группу, которая преобразуется с получением соединения по изобретению посредством гидролиза или т.п., вызванного желудочной кислотой или т.п.
Под «терапевтически эффективным количеством» подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Под «профилактически эффективным количеством» подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на профилактику инфекционных заболеваний, вызываемых грибковой инфекцией. Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п. Для профилактического лечения терапевтически или профилактически эффективное количество представляет собой то количество, которое будет эффективно в предотвращении грибковой инфекции.
Термин «пациент» («субъект») охватывает все виды млекопитающих, предпочтительно человека, которые используют соединения в рамках данного изобретения как путем самостоятельного введения, так и путем их введения пациенту другим лицом для лечения и/или профилактики заболевания или медицинского состояния.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) блокирование (приостановку) течения заболевания, б) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания.
Термин «профилактика», «предотвращение», «превентивная терапия» охватывает устранение факторов риска, а также профилактическое лечение суб клинических стадий заболевания у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. Пациенты для профилактической терапии отбираются на основе факторов, которые, на основании известных данных, влекут увеличение риска возникновения клинических стадий заболевания по сравнению с общим населением. К профилактической терапии относится а) первичная профилактика и б) вторичная профилактика. Первичная профилактика определяется как профилактическое лечение у пациентов, клиническая стадия заболевания у которых еще не наступила. Вторичная профилактика - это предотвращение повторного наступления того же или близкого клинического состояния заболевания.
Термин «уменьшение риска» охватывает терапию, которая снижает частоту возникновения клинической стадии заболевания. Примерами уменьшения риска заболевания является первичная и вторичная профилактика заболевания.
Термин «введение» в организм соединения настоящего изобретения включает доставку к реципиенту соединения, описанного в настоящем изобретении, пролекарства, или другого фармакологически приемлемого производного такого соединения, используя любые допустимые препараты или пути введения в организм, хорошо известные специалистам.
Термин «МИК» в настоящем документе означает минимальную ингибирующую концентрацию.
Термин «МПК» в настоящем документе означает минимальную подавляющую концентрацию.
Ссылки на методики, используемые при описании данного изобретения, относятся к хорошо известным методам, включая изменения этих методов и замену их эквивалентными методами, известными специалистам.
Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе.
Получение соединений по изобретению
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Получение соединения по изобретению возможно в результате одного из способов, представленных: на схемах ниже. 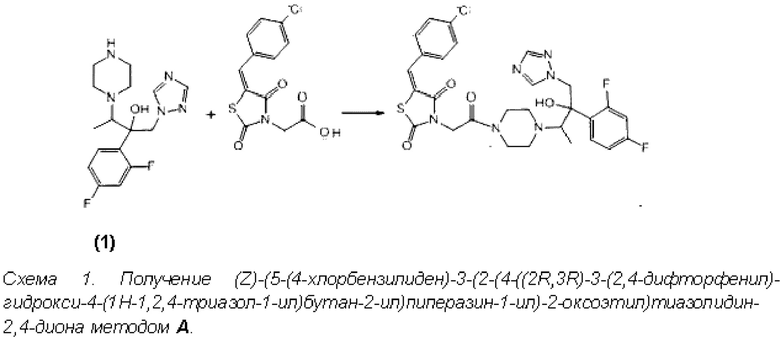
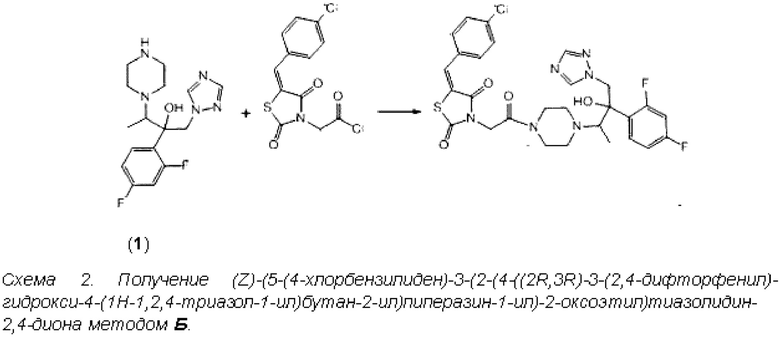
В качестве исходных продуктов в методах А и Б используется 2-(2,4-дифторфенил)-4-(пиперазин-1-ил)-3-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (1) и коммерчески доступное соединение - 3-замещенной уксусной кислоты 5-(4-хлорбензилиден)тиазолидин-2,4-дион (CAS 423153-08-6).
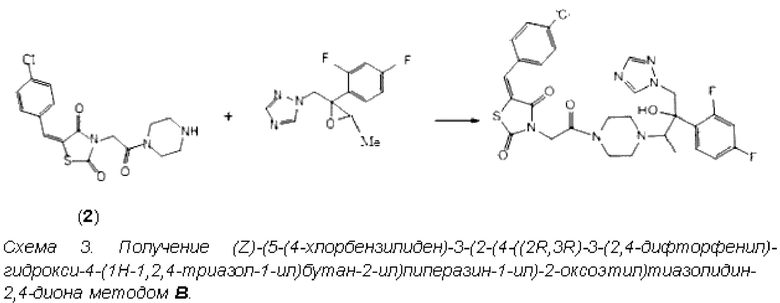
В методе В в качестве исходных компонентов используются 4-(2-(5-(4-хлорбензилиден)-2,4-диоксотиазолидин-3-ил)ацетил)пиперазин-1-ил (2) и коммерчески доступный оксиран с метальной группой в качестве заместителя (CAS 127000-90-2).
Получение соединения (1).
Получение соединения (1) описано в уровне техники [например, US 6153616; Bull. Korean Chem. Soc. 2010]. Так, например, в указанном источнике оно получено в результате длительного (48 часов) кипячения метилоксирана с пиперазином в ацетонитриле в присутствии катализатора LiCICM с выходом от 50 до 78%:
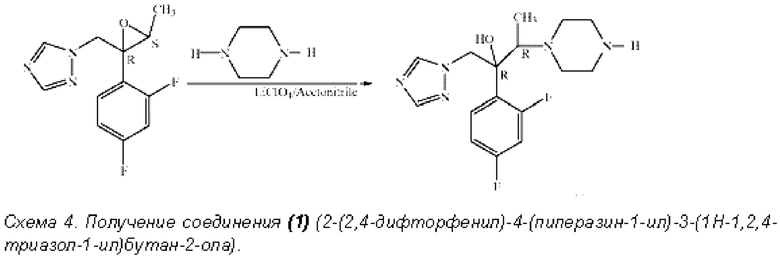
С целью оптимизации условий синтеза - сокращения времени и увеличения выхода - произведена замена растворителя ацетонитрила на диметилф ормамид или диметилсульфоксид и добавление к катализатору LiClO4 соли хлорида магния MgCl2, что позволило сократить время достижения максимального выхода целевого соединения до 12 часов и повысить выход.
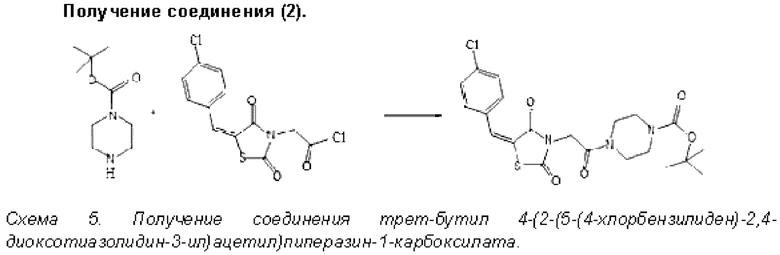
Воспиперазин (2.5 г, 13.42 ммоль, 1 экв) суспендируют в хлористом метилене (40 мл), затем добавляют триэтиламин (4.07 г, 40.27 ммоль, 3 экв). Реакционную массу охлаждают льдом, затем при перемешивании прикапывают раствор хлорангидрида 2-(5-(4-(хлорбензилиден)-2,4-диоксотиазолидин-3-ил) уксусной кислоты, полученного in situ (5.09 г, 16.11 ммоль, 1.2 экв) в хлористом метилене (20 мл). По окончании прибавления раствора хлорангидрида охлаждающую баню убирают и реакционную массу перемешивают 16 часов при комнатной температуре. Затем реакционную массу промывают насыщенным водным раствором лимонной кислоты до кислой реакции. Органический слой отделяют, сушат над сульфатом натрия и упаривают на роторном испарителе. Вещество используют далее без дополнительной очистки. Выход количественный (6.25 г), желтоватый порошок. C21H24CIN3O5S. rn/z 4655. LCMS [М+1]+=466+. Индивидуальность подтверждена хроматограф ически.
1Н NMR (400 MHz, drnso) δ 7.91 (d,J=23.9 Hz, 1H), 7.63 (dd,J=22.6, 7.7 Hz,5H), 4.62 (s,2H), 3.37 (s,16H), 1.40 (s,9H), 1.01 (d,J=5.9 Hz,3H).
13C NMR (101 MHz, dmso) δ 167.25, 165.60, 163.78, 154.17, 135.84, 132.66, 132.27, 132.17, 132.09, 129.91, 129.84, 122.13, 79.69, 44.27, 43.19, 41.87, {40.54, 40.33, 40.12, 39.91, 39.70, 39.49, 39.29}, 28.46, 25.92
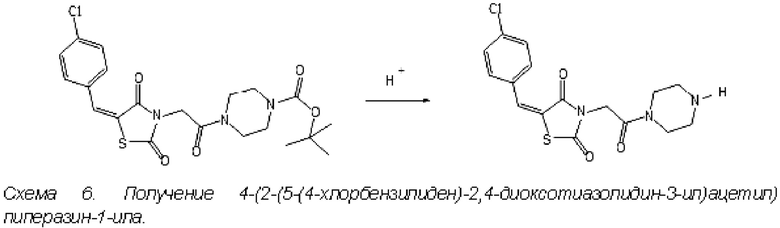
Трет- бутил 4- (2- (5-(4-хлopбензилидену 2,4-диоксотиазолидин- 3- ил )ацетил) пиперазин-1 -карбоксилат (4.65 г, 10.0 ммоль, 1 экв) растворяют в 50 мл абсолютного этилацетата, затем добавляют 50 мл насыщенного раствора хлористого водорода в этилацетате. Полученный осадок перемешивают ночь при комнатной температуре, затем отфильтровывают, промывают этил ацетатом (30 мл) и диэтиловым эфиром (50 мл). Полученное вещество переносят в колбу и высушивают на роторном испарителе до постоянной массы. Полученную соль - гидрохлорид 4-(2-(5-(4-хлорбензилиден)-2,4-диоксотиазолидин-3-ил)ацетил) пиперазин-1-ила (3,28 г) - суспензировали в воде (40 мл), добавили BuOH (60 мл) и осторожно добавили Na2CO3 (2,0 г), перемешивали при комнатной температуре 30 минут, далее органическую фазу отделили, а водную часть повторно экстрагировали BuOH (20 мл). Органические экстракты объединили и упарили в вакууме. Остаток растворили в EtOAc (50 мл), фильтровали и упарили, получили основание (2,99 г, 82 %) в виде белого порошка. C16H16CIN3O3S. rn/z 365,4. LCMS [M+1]+=366+. Индивидуальность подтверждена хроматографически.
1Н NMR (400 MHz, drnso) δ 7.94(s, 1Н),7.62 (dd,J=23.2, 8.5 Hz,4H), 4.59(s,2H), 3.43 (d,J=30.8 Hz,5H),2.79 (s, J=34.9 Hz,2H), 2.71 (s,1H).
13C NMR (101 MHz, drnso) δ 167.25, 165.62, 163.47, 135.83, 132.61, 132.25, 132.18, 129.91, 122.18, 45.59, 45.19, 43.17, 42.56.
Получение соединения по изобретению - (7)-(5-(4-хлорбензилиден)-3-(2-(4-((2R,3R-3-(2,4-д и фтор фен ил)-гид роке и-4-(1Н-1,2,4-триазол-1 -ил)бутан-2-ил)пиперазин-1 -ил)-2-оксоэтил]миазолидин-2,4-диона (L363).
Метод А
В хлористом метилене (50 мл) растворили 3-замещенной уксусной кислоты 5-(4-хлорбензилиден)тиазолидин-2,4-дион (5,94 г, 20 ммоль, 1 экв) и 2,4-д и фтор фен ил производного триазола (1) (6,74 г, 20 ммоль, 1 экв), затем добавили рассчитанное количество триэтиламина (847 мг, 5 экв). Полученный раствор перемешивали в течение30 минут, затем добавили TBTU (6,45 г, 20.1 ммоль, 1.2 экв). После рН-контроля (рН должен быть не менее 8) реакционную массу перемешивали при комнатной температуре 16 часов. Затем органический слой промыли водой, хлористый метилен отделили и высушили над сульфатом натрия. Хлористый метилен упарили на роторном испарителе. Полученное вещество почистили флеш-хроматографией на силикагеле (система хлористый метилен - 4%-ный раствор метанола в хлористом метилене). Получено 5,06 г вещества в виде сухой пены, выход 40.08%. C28H27CIF2N6O4S. m/z 617.07. LCMS [М+1]+=618+.
Метод Б
2-(2,4-дифторфенил)-4-(пиперазин-1-ил)-3-(1Н-1,2,4-триазол-1-ил)бутан-2-ол (1) (3,37 г, 10,0 ммоль, 1 экв) суспендировали в хлористом метилене (40 мл), затем добавили триэтиламин (253 мг, 3 экв). Реакционную массу охладили баней со льдом, затем по каплям добавили раствор хлорангидрида 5-(4-хлорбензилиден)тиазолидин-2,4-дион уксусной кислоты* в 2- мл хлористого метилена (4,04 г, 12 ммоль, 1.2 экв). По окончании прибавления хлорангидрида производили контроль рН смеси, он должен быть не меньше 8. Реакционную массу постепенно нагрели до комнатной температуры и перемешивали ночь при комнатной температуре. Затем промывали насыщенным раствором лимонной кислоты до кислой реакции, отделяли органический слой, сушили над сульфатом натрия и отгоняли хлористый метилен на роторном испарителе. Полученный остаток растворили в хлористом метилене и очистили вещество хроматографией на силикагеле, система хлористый метилен - 3%-ный раствор метанола в метилене. Индивидуальность подтверждена хроматографически. Выход 3,33 г 54 %, в виде желтоватой пены. LCMS [М+1]+=618+.
*Хпорангидрид 5-(4-хпорбензилиден)тиазолидин-2,4-дион уксусной кислоты получали непосредственно перед проведением реакции в результате обработки (4-хпорбензилиден)тиазолидин-2,4-дион 3-замещенной уксусной кислоты
эквимолекулярным количеством хлористого тионила при нагревании. Выход количественный. Полученный продукт использовался без очистки.
Метод В
Смесь метил-оксирана: (2R,3S)-2-(2,4-дифторфенил)-3-метил-[(1Н-1,2,4-триазол-1 -ил)метил]оксирана (5,3 г, 20 ммоль, 1 экв.), 4-(2-(5-(4-хлорбензилиден)-2,4-диоксотиазолидин-3-ил)ацетил)пиперазин-1-ила (7,3 г, 20 ммоль, 1 экв.) и диизопропилэтиламин (4,0 г, 3 экв.) кипятится в ДМСО в добавлением перхлората лития и хлористого магния в течении 16 часов. Растворитель упаривается на роторном испарителе под вакуумом и остаток растворяется в хлористом метилене и промывается водной лимонной кислотой до кислой реакции, потом водой. Органическая фаза высушивается Na2SO4 и упаривается. Продукт в виде вспененного масла при стояниизакристаллизовался. Выход 7,16 г (58%). Индивидуальность подтверждена хроматографически. LCMS [М+1 ]+=618+.
1Н NMR (600 MHz, DMSO-d6) δ 8.29 (s,1H),7.96 (s,1H), 7.ББ (d,J=8.8 Hz, 3H), 7.63-7.59 (m,2H),7.32 (td,J=9.0,6.8 Hz,1H), 7.10 (ddd, J=11.9,9.0,2.6 Hz,1H), 6.90 (td, J=8.5, 2.6 Hz,1H),5.54 (s.1H), 4.91 (d,J=14.8 Hz,1H),4.31 (d,J=14.8 Hz,1H),4.61 (s,2H), 3.59 (d,J=16.8 Hz, 4H), 3.48 (s,2H),3.15 (q,J=7.0 Hz, 1H),2.92 (s,OH),2.81 (s,1H),2.44 (s,1H),0.75 (d,J=6.9 Hz,3H).
13C NMR (151 MHz, DMSO-d6) δ 166.92, 165.29,162.96, 161.74 (dd.J=246.0, 12.5 Hz), 158.48 (dd,J=246.6, 12.1 Hz), 150.60, 144.76, 135.49, 132.29, 131.91, 131.85, 130.29 (dd,J=9.5,6.1 Hz), 129.56, 125.69 (dd,J=13.0,3.6 Hz), 121.85, 110.84 (d,J=20.5 Hz), 103.92 (dd, J=28.7,25.6 Hz), 78.84 (d,J=6.0 Hz), 63.24 (d,J=4.4 Hz), 55.78 (d,J=5.0 Hz),50.42,44.82,42.85,42.46,39.61 (dp,J=42.1, 21.0 Hz), 7.21.
Примеры получения фармацевтически приемлемых солей соединения по изобретению
Благодаря наличию в структуре соединения атомов азота, реализуется способность под действием сильных кислот присоединять протон, что ведет к образованию соли. Подтверждением служит, например, ЯМР спектр соли тозилата, в котором имеются сигналы в области 2.28-2.44 м.д., соответствующие 2-м эквивалентам метильной группы сульфонового остатка и химические сдвиги протонов фенильного кольца при 7.50-7.47 (4Н) и 7.14-7.13 (4Н) м.д.
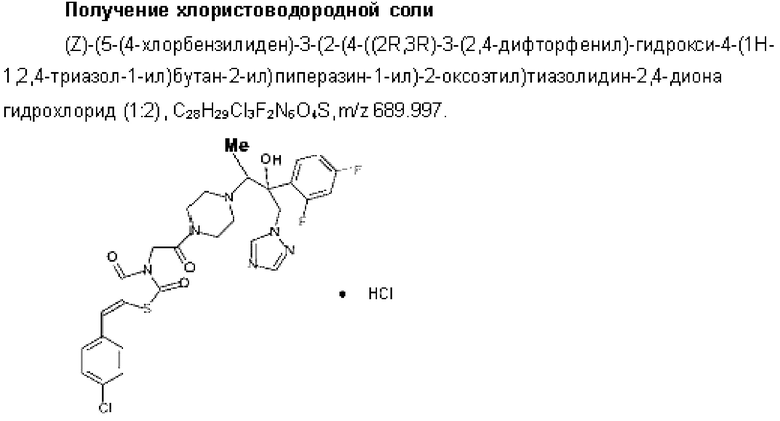
К раствору (7)-(5-(4-хлорбензилиден)-3-(2-(4-((2R3R)-3-(2,4-дифторфенил)-гидрокси-4-(1Н-1,2,4-триазол-1-ил)бутан-2-ил)пиперазин-1-ил)-2-оксоэтил)тиазолидин-2,4-диона (6,17 г, 0,01 моль) в 50 мл сухого этил ацетата прибавляют 50 мл раствора этилацетата, предварительно обработанного газообразным хлористым водородом. Приперемешивании в течение 2 часов получают суспензию, которую фильтруют и промывают последовательно сухим этилацетатом и эфиром. Выход количественный. Полученный порошок сушат при комнатной температуре в вакууме и хранят в эксикаторе с целью предотвращения сорбции атмосферной влаги.
1Н NMR (700 MHz, DMSO-d6) δ 10.39 (s,1Н), 10.30 (s,1Н), S S3 (s,1H),7.99 (s,1H), 7.97 (s, 1H), 7.69-7.66 (m,2H), 7.63-7.59 (m,2H), 7.33 (s,1H), 7.26 (t,J=10.6 Hz, 1H), 7.03 (t,J=8.6 Hz, 1H),6.85 (s,3H),5.54 (d,J=14.7 Hz, 1H),5.01 (d, J=14.5 Hz, 1H), 4.77 (s,1H), 4.63 (s,1H), 4.31 (s,1H), 4.07 (s,2H),3.86-3.68 (m,H), 3.66-3.50 (m,2H),3.49-3.39 (m,H), 3.38-3.27 (m,H), 3.22-3.07 (m,H),2.49 (p,J=1.9 Hz, 4H), 1.15-1.06 (m,3H).
13C NMR (176 MHz, DMSO-d6) 5 167.28, 165.63, 163.85,163.49, 162.12, 149.82, 145.47, 135.39, 132.74,132.32, 132.23,130.41, 129.97, 124.04,122.13,1 11.89, 111.77, 104.96,104.81,104.66,77.28,67.13,55.39,52.83,49.24,43.06,9.99.
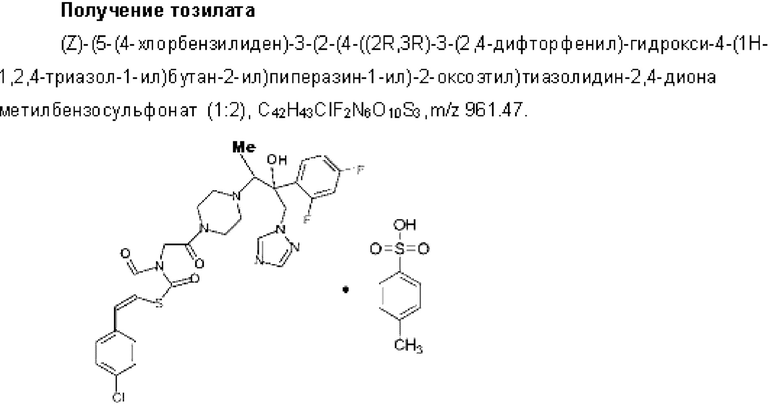
Данная соль получена аналогично описанному выше методу с использованием подходящих реагентов.
1Н NMR (700 MHz, DMSO-d6) 5 9.40 (s,1H),8.58 (s,1H),8.09 (s,1 H), 7.96 (s,1H), 7.69-7.66 (m,2H), 7.64-7.60 (m,2H), 7.50-7.47 (rn,4H)17.47-7.40 (m,1H), 7.29 (s,1H), 7.11 (d,J=7.9 Hz, 5H), 5.10 (d,J=14.6 Hz, 1H), 4.93 (d,J=14.7 Hz, 1H),4.77 (s,1H), 4.63 (s,1 H), 4.31 (s,1H), 4.07 (s,2H), 3.86-3.68 (m,OH), 3.66 - 3.50 (m,2H), 3.49-3.39 (m,1H), 3.33-3.27 (m,1H),3.22-3.07 (m,1H), 2.28 (s,6H), 1.16 (t,J=7.1 Hz, 3H).
13C NMR (176 MHz, DMSO-d6) δ 167.29, 165.61, 163.81, 150.84, 145.77, 138.37, 135.91,132.75, 132.33,132.22, 130.30, 129.98, 128.62,125.96, 124.09, 122.19,112.05,105.09,77.10,55.27,52.80,48.95,43.06,21.26,9.64.
Характеристика биологической активности соединения по изобретению
Сравнительное изучение противогрибковой активности соединения по изобретению на клинических изолятах Candida spp., Rhizopus spp., Aspergillus spp. микрометодом двукратных серийных разведений in vitro
Была исследована сравнительная противогрибковая активность соединения по изобретению, известного из уровня техники аналога (соединение 9 из документа RU 2703997) и микробиологических стандартов флуконазола и вориконазола по спектру противогрибкового действия в отношении клинических изолятов Candida spp. и Rhizopus spp. микрометодом двукратных серийных разведений в питательном бульоне RPMI 1640 в отношении клинических изолятов и стандартных штаммов Candida spp. Оценку активности проводили в соответствии с рекомендациями Всемирной некоммерческой организации по разработке стандартов и рекомендаций в сфере медицины - Clinical and Laboratory Standards Institute (CLSI). Использовали методы M27-A3 (Clinical and Laboratory Standards Institute. Approved standard third edition M27-A3, Wayne,PA, USA, 2008) и ISO 16256:2012 Clinical laboratory testing and in vitro diagnostic test systems — Reference method for testing the in vitro activity of antimicrobial agents against yeast fungi involved in infectious diseases (IDT) для определения значения минимальной ингибирующей концентрации (МИК), микрометодом серийных разведений в среде RPMI 1640 с добавлением глюкозы до концентрации 0,2%. Результаты представлены в таблице 1 ниже. 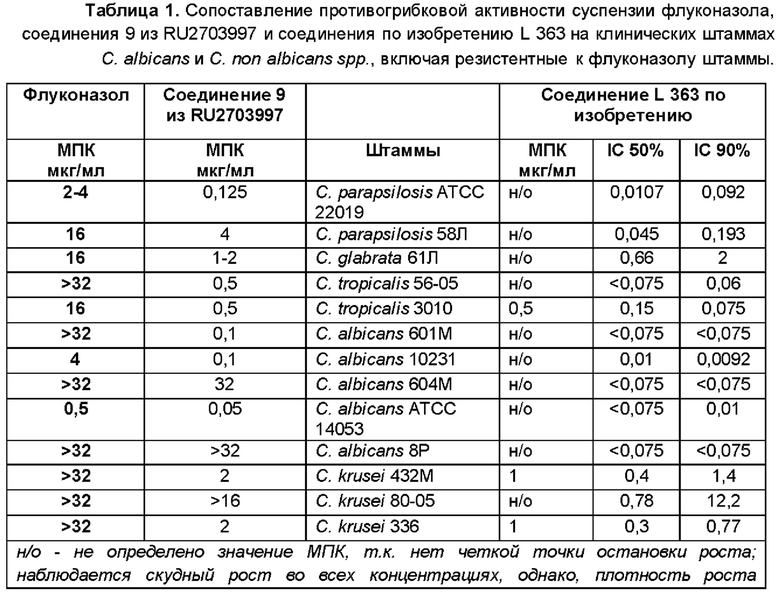

Из представленных в таблице 1 результатов видно, что соединение по изобретению (L 363) характеризуется в 2 раза более высокой активностью на клинических штаммах С. albicans и С, поп albicans spp. по сравнению с активностью аналога из RU 2703997, а также в десятки раз более высокой активностью по сравнению с микробиологическим стандартом - коммерческим препаратом флуконазол.

Из представленных в таблице 2 результатов видно, что соединение по изобретению характеризуется в 4 раза более высокой активностью по сравнению с активностью аналога из RU 2703997 и в десятки раз более высокой активностью по сравнению с микробиологическим стандартом флуконазолом.
Более того, сравнительные исследования показали, что соединение по изобретению также и гораздо более эффективно действует на особо опасный патоген вида Rhisopus spp., известный как «Черный гриб» (вспышка инфекции в 2020-21 гг. в Индии, 10 тысяч пораженных) по сравнению с соединением аналогом из документа RU 2703997 и коммерческим препаратом вориконазол. Результаты исследований приведены в таблице 3.
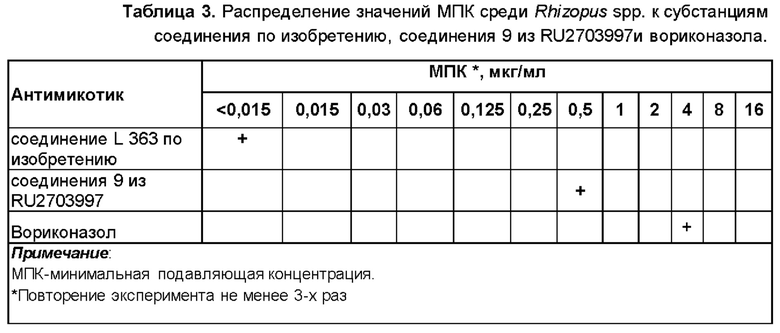
Таким образом, результаты проведенных исследований показали, что соединение по изобретению характеризуются значительно более высоким уровнем активности в отношении особо опасного патогена вида Rhisopus spp. по сравнению с известнымсоединением-аналогом из уровня техники, а также по сравнению с противогрибковым препаратом вориконазолом, а именно, уровень МПК у соединения по изобретению в, по меньше мере, 33 раза меньше, чем у соединения аналога из RU 2703997, а по сравнению вориконозолом, по меньшей мере, в 266 раз меньше.
Далее также были проведены сравнительные исследования в отношении штаммов C.auris. Результаты представлены в таблице 4. 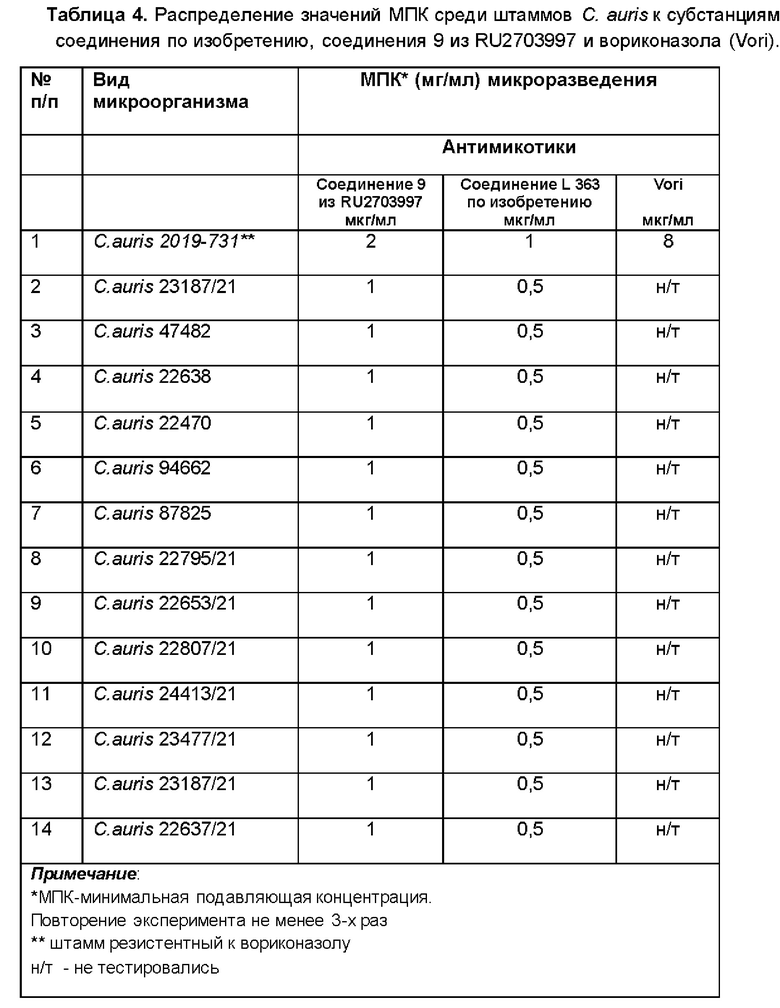
Результаты проведенных исследований показали, что соединение по изобретению L 363 характеризуются значительно более высоким уровнем активности в отношении штаммов C.auris по сравнению с известным соединением-аналогом из уровня техники (RU 2703997), а именно уровень МПК у соединения по изобретению в 2 раза меньше, чем у соединения аналога из RU 2703997 и в 8 раз превышает активность коммерческого препарата вориконазол к отношении резистентного к нему штамма C.auris 2019-731.
Проведены также сравнительные исследования на штаммах грибов-дерматофитов: Microsporum canis, Epidermophyton spp., Trichophyton spp., результаты представлены в таблице 5.
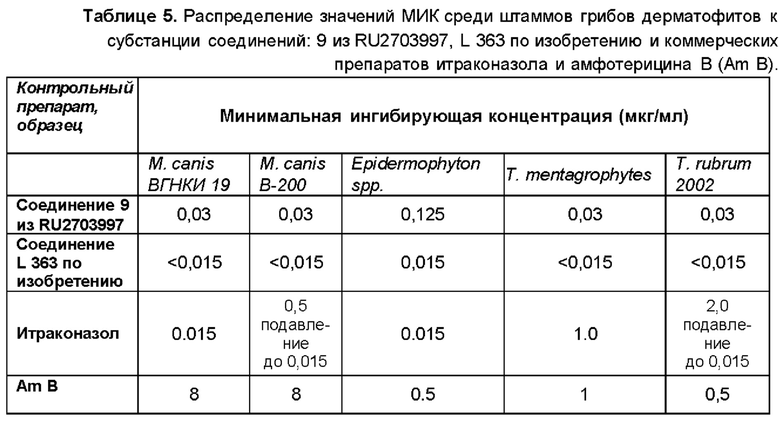
Как видно из приведенных данных, результаты проведенных исследований показали, что активность ингибирующего действия соединения по изобретению L 363 в отношении штаммов грибов дерматофитов в несколько раз выше в сравнении с препаратами сравнения (итраконазол и амфотерицин В) и соединением 9 из RU 2703997.
Таким образом, предлагаемые соединения представляют интерес для медицины и могут найти применение для лечения и профилактики инфекционных заболеваний, в частности, вызванных различными грибковыми инфекциями (в том числе особо опасными патогенами), например, таких заболеваний как мукормикоз, инвазивный кандидоз, вагинальный кандидоз, кандидозный стоматит, эндокардит, и других заболеваний человека и животных.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Изобретение также относится к фармацевтическим композициям, которые содержат соединение по изобретению (или про-лекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/илинаполнителей, которые могут быть введены в организм пациента вместе с соединением, составляющим сущность этого изобретения, не снижают фармакологическую активность этого соединения и не токсичны при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, указанные в этом изобретении, содержат соединения этого изобретения вместе с фармацевтически приемлемыми носителями, которые могут включать любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазки и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают моно- и олигосахариды, а также их производные; желатин; тальк; вспомогательные вещества, такие как масло какао и воск для суппозиториев; масла, такие как арахисовое, хлопковое, кунжутное, оливковое, кукурузное и соевое масло и другие; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; депирогенизированная вода; изотонический раствор, раствор Рингера; спиртовые и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленочные агенты, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом настоящего изобретения являются также лекарственные формы - класс фармацевтических композиций, структура которых оптимизирована для определенного способа введения в организм в терапевтической эффективной дозе, например, для введения в организм внутривенно, внутримышечно, перорально, подкожно, ингаляционно, интраназально, сублингвально, ректально, или иными общепринятыми способами в рекомендуемых дозировках.
Лекарственные формы настоящего изобретения могут содержать структуры, полученные методами использования липосом, методами микрокапсулирования, способами получения наноформ лекарственного средства или другими способами, известными в фармацевтике.
При получении композиции, например, в форме таблетки, активный компонент смешивают с одним или несколькими фармацевтическими наполнителями, такими как желатин, крахмал, лактоза, стеарат магния, тальк, диоксид кремния, арабская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки могут быть покрыты сахарозой, производными целлюлозы или другими веществами, подходящими для нанесения покрытия. Таблетки могут быть полученыразличными способами, такими как прямое прессование, сухая или влажная грануляция или горячее легирование в горячем состоянии.
Фармацевтическую композицию в виде желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным способом используются водные суспензии, изотонические физиологические растворы или стерильные растворы для инъекций и инфузий.
Примеры получения композиции по изобретению
Метод А: гранулирование (P.Bhatt. Handbook of pharmaceutical technology practical. P.Bhatt, A.Kumar Eds. Pharmatech, 2021).
Соединение по изобретению, тонко измельченные порошки лактозы, крахмала, талька тщательно перемешивают в ступке и добавляют к полученной смеси крахмальную пасту в качестве связующего ингредиента и перемешивают. Полученную смесь просеивают через сито. Полученные гранулы сушат в духовом шкафу при температуре 60°С в течение 35 мин.
Метод Б: таблетирование
Гранулы, полученные по методу А, смазывают стеаратом магния и запекают в духовом шкафу при температуре 60°С до полного высыхания (P.Bhatt. Handbook of pharmaceutical technology practical. P.Bhatt, A.Kumar Eds. Pharmatech 2021). Затем высушенные гранулы формуют в таблетки с помощью эксцентриковой (прессовальной) таблеточной машины (Меньшутина Н.В., Алвес СВ., Мишина Ю.В. Инновационные технологии и оборудование фармацевтического производства. БИНОМ, 2012).
МЕТОДЫ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ
Соединение настоящего изобретения и его фармацевтически приемлемые соли являются противогрибковым средством, то есть полезным агентом для терапии и/или профилактики заболеваний или состояний, вызванных грибковыми инфекциями, в том числе патогенами рода Candida spp., Rhizopus spp., Aspergillus spp., Microsporum spp., Trichophyton spp. Благодаря своей противогрибковой активности, соединение настоящего изобретения может быть использовано для профилактики, лечения и/или уменьшения риска развития инфекционного заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией.
Эти инфекционные заболевания хорошо охарактеризованы у человека, но также с аналогичной этиологией присутствуют у других млекопитающих, и их можно лечить фармацевтическими композициями настоящего изобретения.
Для терапевтического применения соединение по изобретению может вводиться с помощью фармацевтической композиции в любой фармацевтической лекарственной форме любым способом введения. Лекарственные формы обычно включают фармацевтически приемлемый носитель, подходящий для выбранной конкретной лекарственной формы. В частности, соединение по изобретению может вводиться пациенту ежедневно в течение периода времени, необходимого для лечения и/или профилактики заболеваний, включая курс терапии, длящийся дни, месяцы, годы. Способы введения включают, но не ограничиваются: внутривенно, внутримышечно, перорально, подкожно, ингаляционно, интраназально и сублингвально. Предпочтительными способами введения являются пероральный и внутривенный.
Изобретение также относится к фармацевтической композиции, содержащей ежедневную дозу указанного соединения в форме фиксированной единицы дозировки, и к комбинации, содержащей указанную фармацевтическую композицию или указанное соединение. В предпочтительном варианте осуществления указанную композицию для применения в соответствии с изобретением вводят один или несколько раз в день в дозировке 1 мг или более соединения по изобретению. Предпочтительная дозировка составляет 1-1500 мг. Наиболее предпочтительная дозировка составляет 10-1000 мг.
Один или несколько дополнительных фармакологически активных агентов могут вводиться в комбинации с соединением по изобретению. Как правило, любые дополнительные одиночные или множественные активные агенты, отличные от соединения по изобретению, могут использоваться в любой комбинации с соединением по изобретению в одной или отдельной лекарственной форме, позволяющей обеспечить одновременное или последовательное терапевтическое действие активных агентов. Такие дополнительные один или несколько агентов, можно вводить одновременно, или как часть фармацевтической композиции, или в отдельные моменты времени.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,5-Замещенные производные тиазолидин-2,4-диона, обладающие противомикробной активностью | 2018 |
|
RU2690161C1 |
| Композиция на основе противогрибкового средства и модифицированного циклодекстрина | 2023 |
|
RU2831569C1 |
| ГИБРИДНЫЕ ЭФИРЫ НА ОСНОВЕ ПРОИЗВОДНЫХ ТИАЗОЛИДИН-2,4-ДИОНА И АЗОЛОВ (1Н-1,3-ИМИДАЗОЛА И 1Н-1,3,4-ТРИАЗОЛА) И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2662153C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2021 |
|
RU2824066C1 |
| Гибридные амиды на основе триазола и тиазолидина, обладающие антимикробной активностью | 2018 |
|
RU2703997C1 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| ИСПОЛЬЗОВАНИЕ СОЕДИНЕНИЙ ПРИ ЛЕЧЕНИИ ГРИБКОВЫХ ИНФЕКЦИЙ | 2021 |
|
RU2805930C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ 5-НИТРОПИРИДИН-2-ИЛ-ТИОАЛКЕНИЛ-4-ДИТИОКАРБАМАТОВ, ОБЛАДАЮЩИЕ ПРОТИВОГРИБКОВОЙ АКТИВНОСТЬЮ, И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2487132C1 |
| Химерный рекомбинантный интерферон альфа-2б, обладающий противогрибковой активностью | 2024 |
|
RU2823607C1 |
| АРАЛКИЛБЕНЗИЛОВЫЕ ПРОСТЫЕ ЭФИРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЕНИЕ ТАКИХ СОЕДИНЕНИЙ, СПОСОБЫ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 2010 |
|
RU2548009C2 |
Изобретение относится к химии органических соединений, фармакологии и медицине. Предложено новое химическое соединение, представляющее собой (Z)-(5-(4-хлорбензилиден)-3-(2-(4-((2R,3R)-3-(2,4-дифторфенил)-гидрокси-4-(1Н-1,2,4-триазол-1-ил)бутан-2-ил)пиперазин-1-ил)-2-оксоэтил)тиазолидин-2,4-дион и его фармацевтически приемлемые соли, характеризующееся высокой противогрибковой активностью. Также предложена его фармацевтическая композиция и применение в качестве противогрибкового средства. Проведенные эксперименты показали, что предлагаемое соединение активно в отношении многих штаммов грибов, в том числе резистентных к применяющимся коммерческим препаратам азолового ряда. 4 н. и 10 з.п. ф-лы, 5 табл., 3 пр.
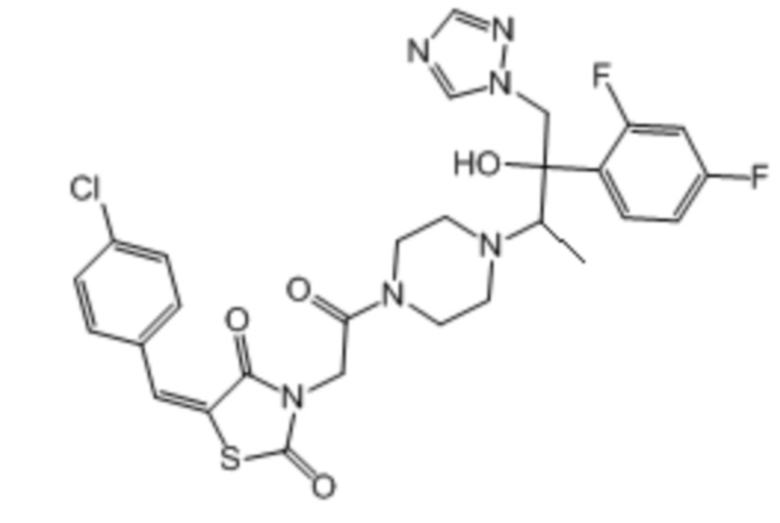
1. Соединение (Z)-(5-(4-хлорбензилиден)-3-(2-(4-((2R,3R)-3-(2,4-дифторфенил)-гидрокси-4-(1Н-1,2,4-триазол-1-ил)бутан-2-ил)пиперазин-1-ил)-2-оксоэтил)тиазолидин-2,4-дион, имеющее следующую структурную формулу:
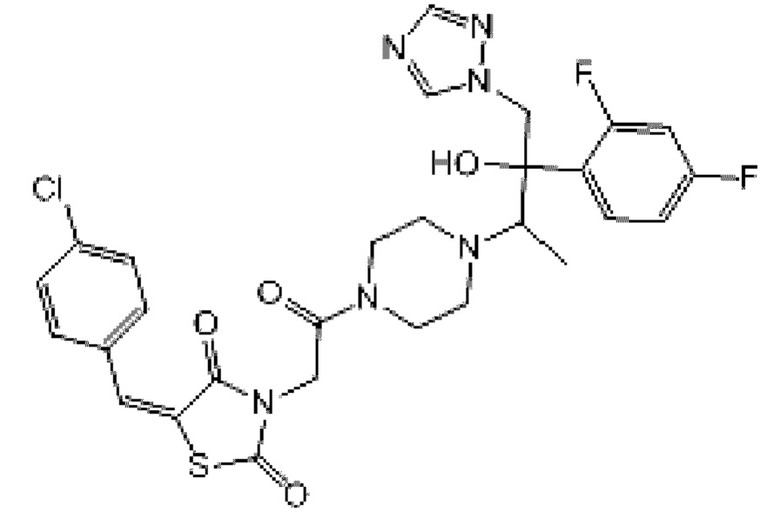
или его фармацевтически приемлемая соль.
2. Применение соединения по п. 1 в качестве противогрибкового средства.
3. Применение соединения по п. 1 для получения фармацевтической композиции с противогрибковой активностью для лечения и/или предотвращения инфекционного заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией.
4. Применение по п. 3, в котором заболевание вызвано патогеном рода Candida spp., Rhizopus spp., Aspergillus spp., Microsporum spp., Trichophyton spp. и/или Epidermophyton spp.
5. Применение по п. 3, в котором заболевание вызвано патогеном, представляющим собой Candida auris, Candida albicans, Candida non albicans Aspergillus fumigatus, Aspergillus niger и/или Rhizopus stolonifer.
6. Применение по п. 5, в котором Candida non albicans представляют собой С. krusei, С. glabrata и/или С. parapsillosis.
7. Фармацевтическая композиция с противогрибковой активностью для лечения и/или предотвращения инфекционного заболевания микробной этиологии у субъекта, вызванного грибковой инфекцией, содержащая эффективное количество соединения по п. 1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
8. Фармацевтическая композиция по п. 7, в которой заболевание вызвано патогеном рода Candida spp., Rhizopus spp., Aspergillus spp., Microsporum spp., Trichophyton spp. и/или Epidermophyton spp.
9. Фармацевтическая композиция по п. 7, в которой заболевание вызвано патогеном, представляющим собой Candida auris, Candida albicans, Candida non albicans, Aspergillus fumigatus, Aspergillus niger и/или Rhizopus stolonifer.
10. Фармацевтическая композиция по п. 9, в которой Candida non albicans представляют собой C. krusei, С. glabrata и/или C. parapsillosis.
11. Фармацевтическая композиция по п. 7, в которой фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
12. Фармацевтическая композиция по п. 7, в которой субъект представляет собой человека или животное.
13. Фармацевтическая композиция по п. 7, в которой заболевание представляет собой заболевание кожи, ногтей и/или внутренних органов.
14. Фармацевтическая композиция по п. 7, характеризующаяся тем, что заболевание представляет собой диссеминированный кандидоз, диссеминированный зигомикоз, дерматофитию, поверхностный микоз, кандидоз кожи и/или ногтей, инвазивный кандидоз, вагинальный кандидоз, кандидозный стоматит, эндокардит или мукормикоз.
| Гибридные амиды на основе триазола и тиазолидина, обладающие антимикробной активностью | 2018 |
|
RU2703997C1 |
| 3,5-Замещенные производные тиазолидин-2,4-диона, обладающие противомикробной активностью | 2018 |
|
RU2690161C1 |
| ГИБРИДНЫЕ ЭФИРЫ НА ОСНОВЕ ПРОИЗВОДНЫХ ТИАЗОЛИДИН-2,4-ДИОНА И АЗОЛОВ (1Н-1,3-ИМИДАЗОЛА И 1Н-1,3,4-ТРИАЗОЛА) И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2662153C1 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| US 7105554 B2, 12.09.2006. | |||

