Область техники
Изобретение относится к химии органических соединений, фармакологии и медицине и касается новых химических соединений, характеризующихся высокой противомикробной активностью, которые, в частности, могут использоваться для профилактики и лечения инфекционных заболеваний у субъекта, в частности заболеваний, вызванных кандидозными и филаментозными патогенами.
Уровень техники
Недостаточная эффективность современной противомикробной терапии обусловлена в значительной степени развитием резистентности у возбудителей к имеющимся лекарственным препаратам. Именно поэтому во всем мире проводится интенсивный поиск и разработка новых эффективных лекарственных соединений. Несмотря на все усилия, современный арсенал медицинских средств недостаточен и не позволяет проводить эффективное лечение многих заболеваний, в том числе грибковых инфекций. Имеющиеся в настоящее время противогрибковые препараты - полиены и азолы, влияющие на главный компонент мембраны грибов - эргостерол, не обеспечивают должной избирательности действия. Они весьма токсичны, более того, многие штаммы возбудителей приобретают резистентность к препаратам группы азолов.
Из уровня техники известны соединения, имеющие тиазолидиндионовый фрагмент и характеризующиеся противогрибковой активностью US7105554, WO2002022612, Thiazolidine and benzylidene thiazolidinedione inhibitors of mannosyltransferease as antifungal agents // Expert Opinion Ther.pat., Patent evaluation, 2002, 12,8, 1285-7. В данных документах описаны замещенные производные 5-бензилиден-2,4-тиазолидин-3 уксусной кислоты в качестве антимикотических средств, активных в отношении многих патогенных грибов. Из уровня техники известны также производные азола, характеризующиеся противогрибковой активностью WO2005006860. Помимо этого описаны способы получения и противогрибковая активность ацильных производных азолов – широко известной группы противогрибковых средств, и, в частности, соединений, полученных путем ацилирования азольных гетероциклов по атому кислорода спиртовой части молекулы (Y.Wahbi et al. Aliphatic ethers and esters of 1-(2,4-dichlorophenyl)-2-(lH-imidazolyl)ethanol: study of antifungal activity against yeasts and hydrophobic character// Eur.J.Med.Chem, 1994, 29, 701-6.; D. De Vita et al. Synthesis and antifungal activity of a new series of 2-(1H-imidazol-1-yl)-1-phenylethanol derivatives // Eur.J.Med.Chem., 2012, 49, 334-342). Однако, как уже упоминалось выше, основном недостатком препаратов, на основе производных азолов, является растущая резистентность, что может свести на нет все прилагаемые усилия по лечению. Проблема еще усугубляется тем, что развивается перекрестная устойчивость ко всем используемым препаратам азольной группы (J.E.Parker. Resistance to antifungals that target CYP51 // J.Chem.Biol, 2014.).
Поэтому сохраняется высокая необходимость в разработке новых эффективных средств против инфекционных заболеваний, в частности противогрибковых средств, для терапии широкого спектра заболеваний, в частности, вызванных грибковыми инфекциями.
Раскрытие изобретения
Задачей настоящего изобретения является разработка и создание новых эффективных противомикробных средств, перспективных для применения в клинической практике для терапии и/или профилактики инфекционных заболеваний.
Техническим результатом изобретения является разработка и получение новых химических соединений, обладающих высокой противомикробной активностью, пониженной токсичностью и перспективных для применения в терапии инфекционных заболеваний у субъекта, в частности заболеваний, вызванных грибковыми инфекциями, например, вызванных кандидозными и филаментозными патогенами, в частности для лечения дерматофитии, поверхностного микоза, кандидоза кожи и ногтей, вагинального кандидоза, кандидозного стоматита, эндокардита и других заболеваний человека и животных. Соединения по изобретению используемые с профилактическими целями могут также снижать риск развития серьезных кандидозных инфекций у людей с ослабленным иммунитетом в результате терапии (например, химиотерапии), трансплантации органов или у людей с инфекционными заболеваниями, вызванными другими патогенами (в частности, ВИЧ).
Активность заявляемых соединений исследована на культурах микробных клеток. Соединения по изобретению в отношении Candida parapsilosis не уступают или превосходят активность Флуконазола и приближается к Кетоконазолу. Кроме того, соединения по изобретению, в отличие от флуконазола, подавляют рост филаментозных грибов M.canis B-200 и Т.rubrum 2002 и проявляют активность в отношении резистентных к Флуконазолу патогенов. Указанные виды патогенных грибов вызывают различные поражения как внутренних органов и слизистых поверхностей, так и наружных покровов кожи и ее придатков.
Выявлена высокая антикандидозная активность в отношении как к типичным грибам Candida albinans, так и устойчивым к Флуконазолу штаммов микроорганизмов Candida non-albicans.
Сравнительный эксперимент на животных в отношении Candida albicans in vivo на модели внутрибрюшинного заражения мышей (экспресс метод) с коммерческим препаратом Вориконазолом выявил явное превосходство соединений по изобретению и по применяемым дозам, и по эффекту подавления ростовых образований (ростовых трубок) дрожжевых клеток Candida albicans.
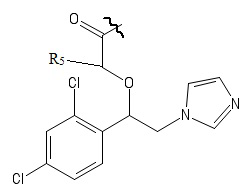
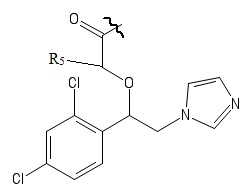
Указанный технический результат достигается посредством разработки и создания соединений общей формулы (I):
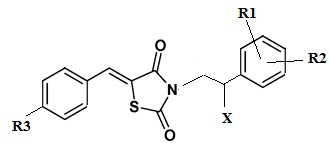 формула (I),
формула (I),
или его стереоизомер или энантиомер, фармацевтически приемлемая соль, сольват или гидрат, где:
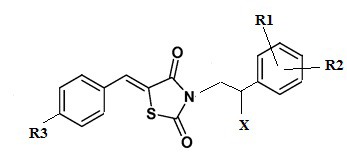
R1 выбирается независимо и представляет собой Н или галоген ;
R2 выбирается независимо и представляет собой Н или галоген;
R3 выбирается независимо и представляет собой Н или галоген;
Х выбирается независимо и представляет собой ОН, галоген, 6-членный гетeрoциклил, содержащий 2 атома N, необязательно содержащий заместитель R4;
R4 выбирается независимо и представляет собой –C1-6-алкил, - C1-6–алкил-OH, - фенил, 4-гидрокси-фенил, -COCH3-COO-tBu или представляет собой заместитель со следующей структурной формулой:
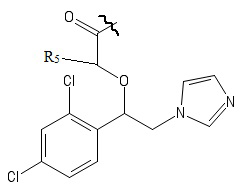 ,
,
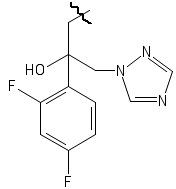 ,
,
причем волнистой линией показано место присоединения заместителя к фрагменту Х.
R5 выбирается независимо и представляет собой Н или фенил, замещенныйатомом галогена.
Отдельный подкласс соединений, представляющих интерес, включает соединения формулы (I), в которых:
R1 выбирается независимо и представляет собой Н, F или Cl;
R2 выбирается независимо и представляет собой Н, F или Cl.
R3 выбирается независимо и представляет собой F или Cl;
R4 выбирается независимо и представляет собой метил, изопропил или -(СН2)2-ОН;
X выбирается независимо и представляет собой хлор или пиперазин;
R5 выбирается независимо и представляет собой Н или фенил, замещенный атомом галогена.
В частных вариантах воплощения изобретения соединения, представляющие интерес, могут быть выбраны из следующих соединений общей формулы (I):
(E)-трет-бутил-4-[2-(5-(4-хлоробензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-хлорфенил)этил] пиперазин-1-карбоксилат;
(E)-трет-бутил-4-[2-(5-(4-фторобензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-фторфенил)этил] пиперазин-1-карбоксилат;
(E)-трет-бутил-4-[2-(5-(4-хлоробензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-фторфенил)этил] пиперазин-1-карбоксилата;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(фенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(4-хлорфенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-фторбензилиден)-тиазолидин-2,4-дион-3-ил-1-(4-фторфенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(2,4-дихлорфенил)этил]пиперазин-1-иум) гидрохлорид;
(Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-хлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-фенил-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-хлорфенил)-2-(4-метоксипиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(2-гидроксиэтил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-изо-пропил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(4-гидроксифенил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-(4-метоксипиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион;
(Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-хлорбензилиден)тиазолидин-2,4-дион;
(Z)-3-(2-(4-фторфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(2-(2-хлорфенил)-2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)ацетил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1- ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1-ил)этил)тиазолидин-2,4-дион.
Настоящее изобретение относится к применению соединений, являющихся предметом изобретения, в качестве противомикробного лекарственного средства.
Настоящее изобретение относится к применению соединений, являющихся предметом изобретения, в качестве противогрибкового лекарственного средства.
Данное изобретение также относится к применению соединений, являющихся предметом изобретения, для лечения и/или предотвращения инфекционных заболеваний, в частности вызванных грибковой инфекцией у субъекта. В частных вариантах воплощения изобретения субъект представляет собой человека или животных.
Соединения по изобретению проявляют высокую противогрибковую активность, в частности, в отношении грибов рода Candida spp, Aspergillus, Microsporum, Trichophyton и Epidermophyton. Соединения по изобретению проявляют высокую противогрибковую активность, в частности, в отношении грибов видов Aspergillus fumigatus, Aspergillus niger, Microsporum canis, Trichophyton rubrum и/или Epidermophyton floccosum.
Изобретение также относится к применению соединений по изобретению для получения фармацевтической композиции для лечения и/или профилактики инфекционных заболеваний микробной этиологии. В частных вариантах воплощения изобретения инфекционное заболевание микробной этиологии вызвано грибковой инфекцией. В частных вариантах воплощения изобретения инфекционное заболевание представляет собой заболевание кожи, ногтей или внутренних органов, в частности таких как дерматофития, поверхностный микоз, кандидоз кожи и/или ногтей, вагинальный кандидоз, кандидозный стоматит, эндокардит и других заболеваний человека или животных.
Кроме того, изобретением предусматриваются фармацевтические композиции для лечения и/или предотвращения инфекционных заболеваний микробной этиологии, включающие терапевтически эффективное количество, по меньшей мере, одного соединения, являющегося предметом изобретения, и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество.
В некоторых вариантах воплощения изобретения вспомогательное вещество может представлять собой носитель, наполнитель и/или растворитель.
В некоторых вариантах воплощения изобретения инфекционное заболевание микробной этиологии вызвано грибковой инфекцией.
В частных вариантах воплощения изобретения фармацевтическая композиция предназначена для лечения инфекционных заболеваний, вызванных грибом рода Candida spp, Aspergillus, Microsporum, Trichophyton и/или Epidermophyton. В частных вариантах воплощения изобретения фармацевтическая композиция предназначена для лечения инфекционных заболеваний, вызванных грибом вида Aspergillus fumigatus, Aspergillus niger, Microsporum canis, Trichophyton rubrum или Epidermophyton floccosum.
В частных вариантах воплощения изобретения фармацевтическая композиция предназначена для лечения и/или предотвращения заболевания, которое представляет собой заболевание кожи, ногтей или внутренних органов, в частности дерматофития, поверхностный микоз, кандидоз кожи и/или ногтей, вагинальный кандидоз, кандидозный стоматит, эндокардит и других заболеваний человека или животных.
Настоящее изобретение также относится к способу лечения и/или профилактики инфекционных заболеваний, включающему введение (в качестве монотерапии или в комбинации с одним или несколькими агентами) терапевтически эффективного количества соединения, являющегося предметом изобретения, в организм человека или животного, нуждающегося в лечении и/или профилактики таких заболеваний. Термин «введение» в организм соединения настоящего изобретения включает доставку к реципиенту соединения, описанного в настоящем изобретении, пролекарства, или другого фармакологически приемлемого производного такого соединения, используя любые допустимые препараты или пути введения в организм, хорошо известные специалистам.
Изобретение также включает получение соединений общей формулы (I).
Подробное раскрытие изобретения
Определения (термины)
Для лучшего понимания настоящего изобретения ниже приведены некоторые термины, использованные в настоящем описании изобретения. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо, два вхождения могут быть как одинаковыми, так и разными.
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «алкил» сам по себе или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода (то есть, С1-6 подразумевает от одного до шести атомов углерода). Примеры алкилов включают метил, этил, н-пропил, изо-пропил.
Термин «галоген» сам по себе или в части другого термина относится к атому фтора, хлора, брома или йода.
Термин «бензилиден», если иное не оговорено, обозначает углеводородный остаток =СН-С6Н5:
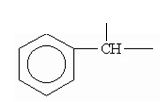 .
.
Термин «гетероцикл» или «гетероциклил» означает в настоящем документе неароматические циклические системы (насыщенные или частично ненасыщенные), имеющие шесть атомов, содержащие 2 гетероатома N. Примеры гетероциклических колец, включают, но не ограничиваются, пиперазином.
Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40° C в отсутствие химически активных условий, в течение, по крайней мере, одной недели.
Если не указано иначе, приведенные в материалах заявки структуры соединений также подразумевают и все стереоизомеры, то есть R- и S- изомеры для каждого ассиметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%; более предпочтительно, >95% мольной чистоты), так же, как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например, путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных молекул путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, дистилляцией, кристаллизаций или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
Термин «сольват» относится к ассоциации или комплексу из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин.
Термин «гидрат» относится к комплексу, где молекулами растворителя является вода.
Соединения настоящего изобретения могут существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Настоящее изобретение включает все фармацевтически приемлемые изотопно меченые соединения по настоящему изобретению, в которых один или несколько атомов замещен атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36CI, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фософора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно меченые соединения формулы (I), например, те, которые включают радиоактивный изотоп, используют в исследованиях распределения лекарственного препарата и/или субстрата в тканях. В частности, с этой целью используют радиоактивные изотопы, такие как тритий, то есть 3H, и углерод-14, то есть 14C, ввиду легкости их введения и доступности средств их обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может обеспечить определенные терапевтические эффекты, обусловленные метаболической стабильностью, например, увеличением периода полувыведения in vivo или снижением норм дозирования, и, следовательно, может быть предпочтительным в некоторых случаях.
Изотопно меченые соединения по изобретению могут быть получены обычными способами, известными специалисту в данной области или способами, аналогичными описанным в прилагаемых примерах способов синтеза, при использовании соответствующих изотопно меченых реагентов вместо немеченого ранее применяемого реагента.
Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, где растворитель кристаллизации может быть изотопно замещен, например, D2O, d6-ацетон, d6-ДМСО.
Термин "микроб", в том числе в составе сложных слов (например, антимикробный, противомикробный), в данном документе включает, например, бактерии, грибы, дрожжи и простейших.
Осуществление изобретения
Обзор методов получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Общий способ получения производных 3- (2-(R1,R2-фенил)-2-Х- пиперазин-1-ил) этил) 5-арилиден производных тиазолидин-2,4-диона:
1. Получение калиевой соли 5-(R3-бензилиден)тиазолидин-2,4-диона (1)
Калиевую соль 5-арилиден тиазолидин-2,4-диона получают известным способом путем взаимодействия 5-арилиден тиазолидин-2,4-диона с гидроокисью калия в сухом этаноле (Схема 1).
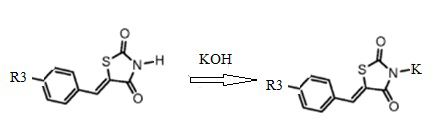
Схема 1. Общая схема синтеза калиевых солей 5-(R3-бензилиден) тиазолидин-2,4-диона.
2. Получение 3-(2-R1,R2-фенил-2-гидроксиэтил)замещенные производные 5-(R3-бензилиден) тиазолидин-2,4-диона (3).
Калиевую соль 5-арилиден тиазолидин-2,4-диона конденсируют в ДМФА при нагревании с соответствующим α-бромэтил-(R1,R2)-фенилэтанолом (2). В результате реакции получают 3-(2-фенил-2-гидроксиэтил)замещенные производные 5-(R3-бензилиден) тиазолидин-2,4-диона (Схема 2).
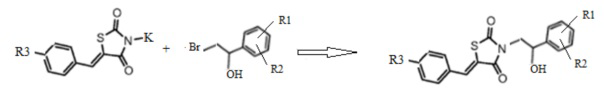
Схема 2. Общая схема синтеза 3-(2-(R1,R2-фенил)-2-гидроксиэтил)замещенных производных 5-(R3-бензилиден) тиазолидин-2,4-диона.
3. Получение 3-(2-R1,R2-фенил-2-хлорэтил)замещенных производных 5-(R3-бензилиден) тиазолидин-2,4-диона (4)
Спиртовый гидроксил 3-(2-R1,R2-фенил-2-гидроксиэтил)замещенных производных 5-(R3-бензилиден) тиазолидин-2,4-диона в результате взаимодействия с хлористым тионилом замещается на атом хлора с количественным выходом c получением неописанных ранее 3-(2-фенил-2-хлорэтил)замещенных производных 5-(R3-бензилиден) тиазолидин-2,4-диона (Схема 3).
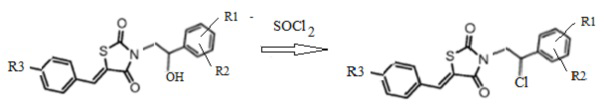
Схема 3. Общая схема синтеза 3-(2-(R1,R2-фенил)-2-хлорэтил)замещенные производные 5-(R3-бензилиден) тиазолидин-2,4-диона.
4. Получение (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона (5-8)
Хлорированные производные 3-(2-R1,R2-фенил-2-хлорэтил)замещенные производные 5-(R3-бензилиден) тиазолидин-2,4-диона аминируют Boc-защищенным пиперазином или N-замещенным пиперазином с получением соединений (6) и (8) соответственно. После снятия Вос-защиты трифторуксусной кислотой или раствором хлористого водорода в этилацетате получают целевые продукты (6) или после соответствующей обработки карбонатом натрия получают основания - соединения (7).
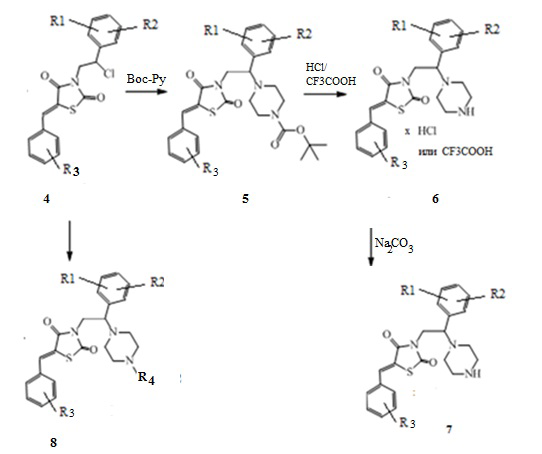
Схема 4. Общая схема синтеза (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2- (пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона.
5. Получение амидов (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона (10,11).
При взаимодействии солей (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2- (пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона с хлорангидридом имидазол уксусной кислоты (9.1), или фенилуксусной кислоты (9.2) образуются соответствующие амиды (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона (10,11).
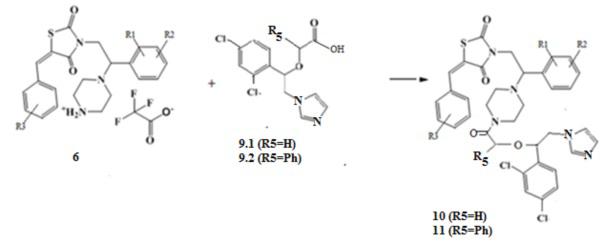
Схема 5. Общая схема синтеза амидов имидазолил уксусной и фенилуксусной кислот (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(пиперазин-1-ил)этил)тиазолидин-2,4-диона (10,11).
6. Получение (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1-ил)этил)тиазолидин-2,4-диона (13)
При взаимодействии солянокислой или трифторуксусной соли (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(пиперазин-1-ил)этил) замещенных тиазолидин-2,4-диона (6) со смесью оксирана метансульфоната и триэтиламина при кипячении в этаноле образуются (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1- ил)этил)тиазолидин-2,4-дионы.
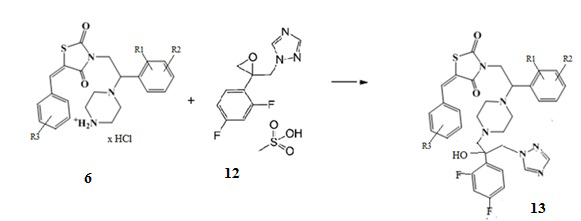
Схема 6. Общая схема синтеза (Z)-5-(R3-бензилиден)-3-(2-(R1,R2-фенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1-ил)этил)тиазолидин-2,4-диона.
Промежуточные продукты для получения некоторых соединений изобретения могут быть получены по методикам в нижеследующих примерах.
1. Пример получения калиевой соли (Z)-5-(4-фторбензилиден)тиазолидин-2,4-диона (схема 1).
В конической колбе на 100 мл, снабженной хлоркальциевой трубкой и охлаждением, в 25 мл сухого метанола суспендируют 1,12 г (Z)-5-(4-фторбензилиден)тиазолидин-2,4-диона (0,0049 моль) . При температуре 20 оС вносят 0,279 г КОН (1,00 мол. экв.) в виде раствора в 5 мл сухого метанола. Реакционную массу перемешивают при 20 оС в течение 1,5 часа, упаривают на роторном испарителе (РПИ) до 1/3 объема. Кубовый осадок отфильтровывают и промывают 5 мл холодного сухого метанола и 5 мл диэтилового эфира. Осадок сушат при пониженном давлении на роторном испарителе.
Получено 1,15 г (Z)-5-(4-фторбензилиден)тиазолидин-2,4-диона калиевая соль. Выход составил 89,29%.
Аналогичным образом получают калиевые соли других 5-замещенных бензилиден тиазолидин-2,4-диона.
2. Пример получения 2-бромо-1- (4-хлорфенил)этанола (схема 2).
В конической колбе на 100 мл, снабженной хлоркальциевой трубкой и охлаждением, в 50 мл сухого метанола растворяют 10,00 г 2- бромацетофенона (0,045моль). При температуре 0-3оС вносят 0,95 г NaBH4 (0,55 мол. экв.), наблюдают интенсивное вспенивание. Реакционную массу перемешивают при 00С в течение 0,5 часа, убирают охлаждение и перемешивают ещё 0,5 часа. Реакционную массу подкисляют 0,1 мл концентрированной соляной кислоты, упаривают на РПИ. Кубовый остаток растворяют в 100 мл хлористого метилена, промывают два раза 50 мл дистиллированной воды. Органический слой сушат Na2SO4 и упаривают на роторном испарителе.
Получено 8,16 г 2-бромо-1-(4-хлорфенил)этанола. Выход составил 74.90%
3. Пример получения 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси) уксусной кислоты (9.1), (схемы 5, 7).
К раствору вторичного спирта (2 г, 1 экв.) в 30 мл ДМФА в атмосфере аргона добавляется гидрид натрия, 60% суспензия в минеральном масле (653 мг, 2.1 экв.), после чего смесь перемешивается 20 мин. Затем добавляется порциями хлоруксусная кислота (808 мг, 1.1 экв.), после чего реакционная смесь перемешивается 24 ч при комнатной температуре. К смеси добавляется 10 мл воды, после чего она подкисляется конц. HCl до pH 7 и упаривается под вакуумом досуха. Сухой остаток экстрагируется 30 мл метанола, фильтруется, упаривается под вакуумом досуха. Кристаллический остаток растирается с эфиром. Выход 2.45 г (99%).
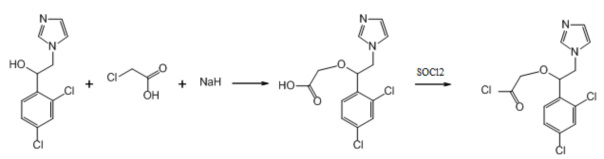
Схема 7. Схема синтеза 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси) уксусной кислоты.
4. Пример получения 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)-(2-хлорфенил) уксусной кислоты ( 9.2), (схемы 5, 8)
К раствору вторичного спирта (1 г, 1 экв.) в 30 мл ДМФА под аргоном добавляется гидрид натрия, 60% суспензия в минеральном масле (321 мг, 2 экв.), после чего смесь перемешивается 20 мин. Затем добавляется порциями бромокислота (1 г, 1 экв.), после чего реакционная смесь перемешивается 24 ч при комнатной температуре. К смеси добавляется 10 мл воды, после чего она концентрируется под вакуумом. Остаток разбавляется 50 мл воды, раствор промывается эфиром и при перемешивании подкисляется конц. HCl до pH 7. Выпавшее белое вещество при стоянии кристаллизуется, отфильтровывается, промывается водой и эфиром. Выход 1.36 г 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)-(2-хлорфенил) уксусной кислоты (80%).
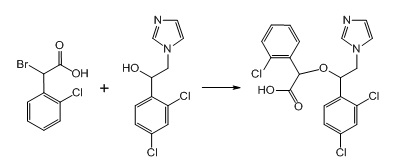
Схема 8. Схема синтеза 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)-(2-хлорфенил) уксусной кислоты
5. Пример получения 1-((2-(2,4-дифторфенил)oксиран-2-ил)метил)-1H-1,2,4-триазол метансульфоната (12) (схемы 6, 9)
К смеси 2,4-дифтор-2-(1,2,4-триазол-1-ил)ацетофенона(5 г, 1 экв.), триметилсульфоксония йодида (4.93 г, 1 экв.) и гексадецилтриметиламмония бромида (204 мг, 0.025 экв.) в 40 мл толуола добавляется 9 мл 20% NaOH (1.79 г, 2 экв.). Смесь перемешивается при 60°С ночь. Эмульсия фильтруется, толуольная фаза отделяется, упаривается, остаток разбавляется этилацетатом. К полученному раствору при перемешивании по каплям добавляется метансульфокислота (4.3 г, 2 экв.). Выпавший осадок соли отфильтровывается и промывается этилацетатом. Выход 4.9 г (66%) 1-((2-(2,4-дифторфенил)oксиран-2-ил)метил)-1H-1,2,4-триазол метансульфоната.
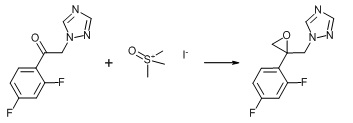
Схема 9. Схема синтеза 1-((2-(2,4-дифторфенил)oксиран-2-ил)метил)-1H-1,2,4-триазол метансульфоната.
Примеры получения соединений по изобретению
1. Пример получения (E)-3-(2-фенил-2-гидроксиэтил)-5-(4–хлорбензилиден) тиазолидин-2,4-дион (3.1)
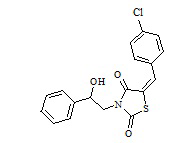
В конической колбе на 100 мл, снабженной хлоркальциевой трубкой, в 25 мл сухого ДМФА суспендируют 5,55 г калиевой соли 5-(4-хлорбензилиден)тиазолидин-2,4-диона (0,019 моль). Вносят 4,12 г 2-бромо-1-фенилэтанола (1,05 мол. экв.). Реакционную массу перемешивают при 75оС в течение 18 часов, убирают нагрев и охлаждают до 25оС. Реакционную массу выливают в 150 мл дистиллированной воды, ждут формирования устойчивого осадка. Осадок отфильтровывают, отжимают на фильтре, промывают 2 порциями по 10 мл диэтилового эфира, оставшийся осадок сушат на воздухе 24 часа. Получено 5,8 г (E)-3-(2-фенил-2-гидроксиэтил)-5-(4-хлорбензилиден)тиазолидин-2,4-диона. Высушенный осадок нагревают в 20 мл метанола до температуры кипения и выдерживают 1 час, охлаждают до комнатной температуры, осадок отфильтровывают, сушат.
Получено 5,6 г (E)-3-(2-фенил-2-гидроксиэтил)-5-(4-хлорбензилиден) тиазолидин-2,4-диона. Выход составил 74,6% .Структура соединения подтверждена совокупностью данных ЯМР и масс-спектрометрии. C18H14ClNO3S.LCMS [M+H]+=361.
1H NMR (500 MHz, DMSO-d6) δ ppm 3.23 (br s, 1 H) 3.66 (dd, J=13.42, 4.46 Hz, 1 H) 3.83 (dd, J=13.42, 9.08 Hz, 1 H) 4.88 (dt, J=8.96, 4.39 Hz, 1 H) 5.69 (d, J=4.46 Hz, 1 H) 7.25 - 7.39 (m, 5 H) 7.54 - 7.67 (m, 5 H) 7.90 (s, 1 H)
Следующие примеры соединений по изобретению, представленные в таблице 1, получены в соответствии с методикой синтеза, описанной для соединения 3.1, используя подходящие исходные вещества: калиевые соли 5-арилидентиазолидин-2,4-диона и 2-бром-1-арилэтанол.
Таблица 1. Примеры соединений по изобретению.
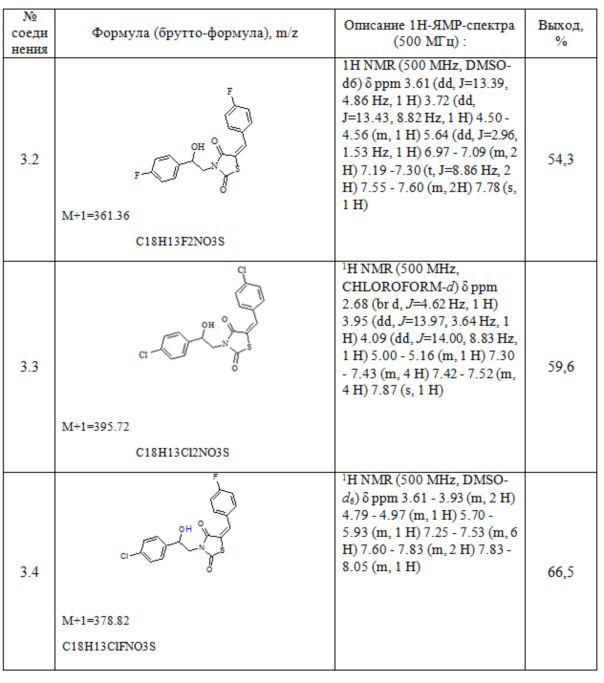
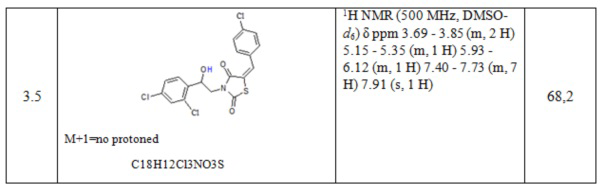
2. Пример получения (E)-3-(2-фенил-2-хлорэтил)- 5-(4-хлорбензилиден) тиазолидин-2,4-диона (4.1)
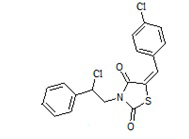
В конической колбе на 50 мл в 25 мл хлористого метилена суспендируют 1,33г (0,0037 моль) (E)-3-(2-фенил-2-гидроксиэтил)-5-(4-хлорбензилиден) тиазолидин-2,4-диона, к суспензии при перемешивании добавляют 1,6 г (4 мол. экв.) хлористого тионила, и перемешивают 18 часов при температуре 45оС.
После завершения реакции реакционную смесь упаривают на роторном испарители, полученный кубовый остаток промывают 10 мл холодного гексана.
Получено 1,37 г (E)-3-(2-фенил-2-хлорэтил)-5-(4-хлорбензилиден) тиазолидин-2,4-диона. Выход составил 98,0 %. Структура соединения подтверждена совокупностью данных ЯМР и масс-спектрометрии. C18H13Cl2NO2S. М+1= 379,28
1H NMR (500 MHz, CHLOROFORM-d) δppm 3.91 (brd, J=19.75 Hz, 1 H) 3.91 (brd, J=7.76 Hz, 1 H) 4.13 (brd, J=9.17 Hz, 1 H) 4.05 - 4.24 (m, 1 H) 4.06 - 4.14 (m, 1 H) 5.15 (dd, J=8.41, 6.51 Hz, 1 H) 7.05 - 7.24 (m, 5 H) 7.25 - 7.29 (m, 2 H) 7.42 (s, 1 H) 7.43 - 7.47 (m, 1 H) 7.51 (s, 1 H) 7.58 - 7.72 (m, 1 H) 7.79 (s, 1 H) 11.95 (brs, 1 H)
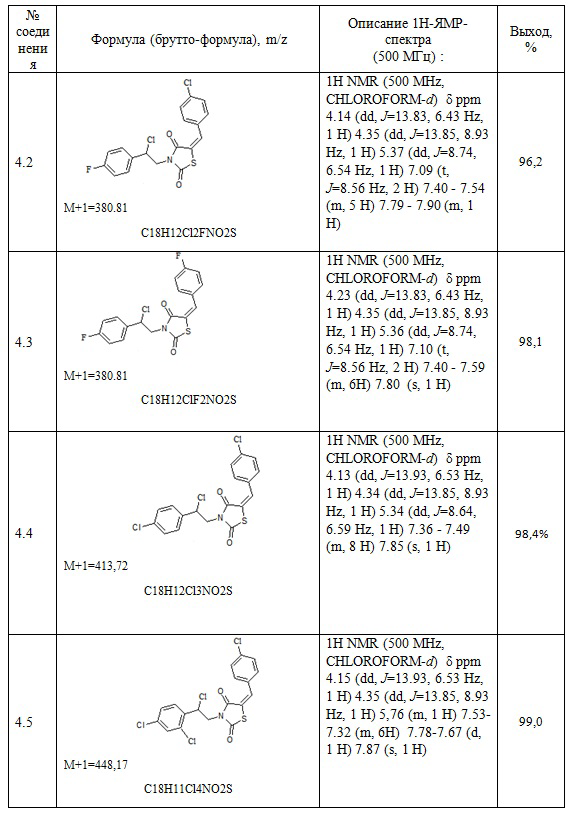
Следующие примеры соединений по изобретению, представленные в таблице 2, получены в соответствии с методикой синтеза, описанной для соединения 4.1, используя подходящие исходные вещества: (E)-3-(2-арил-2-гидроксиэтил)- 5- арилиден тиазолидин-2,4-дионы и хлористый тионил
Таблица 2. Примеры соединений по изобретению.
3. Пример получения (E)-трет-бутил 4-[2-(5-(4-хлоробензилиден) тиазолидин-2,4-дион-3-ил)-2-фенилэтил] пиперазин-1-карбоксилата (5.1)
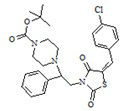
В конической колбе на 50 мл в 25 мл ДМФА растворяют 1,37г (0,0028 моль) (E)-3-(2-фенил-2-хлорэтил)-5-(4-хлорбензилиден) тиазолидин-2,4-дион, к раствору при перемешивании добавляют 0,65 г (1 мол. экв.) трет-бутил пиперазин-1-карбоксилат, и 0,693 г прокаленного карбоната калия, перемешивают 18 часов при температуре 60 оС.
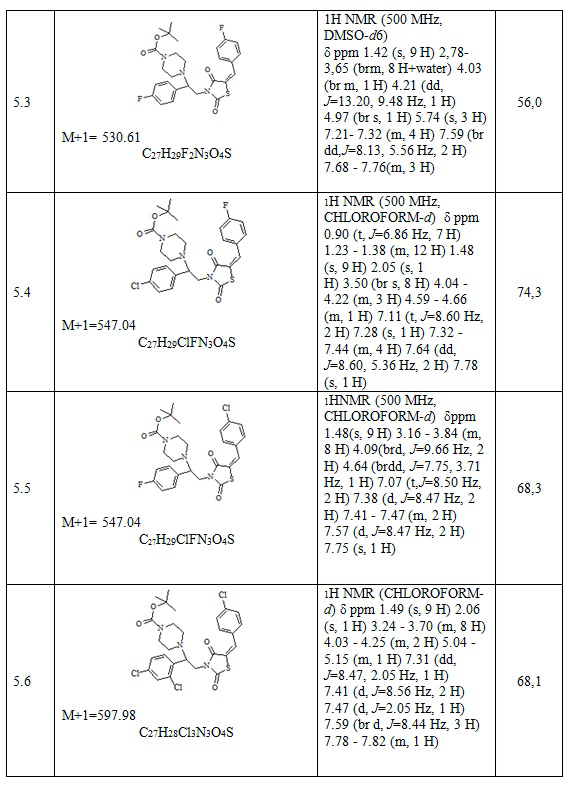
После прохождения реакции реакционную смесь отфильтровывают от неорганических солей и упаривают на роторном испарителе, полученный кубовый остаток очищают на колонке (2:1, гексан-этилацетат). Получают 0,81 г (E)-трет-бутил 4-[2-(5-(4-хлоробензилиден)-тиазолидин-2,4-дион-3-ил)2-(фенил-этил)] пиперазин-1-карбоксилат. Выход составил 55,1%. C27H30ClN3O4S. M+1=529.06.
1H NMR (500 MHz, CHLOROFORM-d) δ ppm 0.83 - 0.95 (m, 1 H) 1.23 - 1.45 (m, 3H) 1.48 (s, 9 H) 2.05 (s, 1 H) 3.50 (br s, 8 H) 4.03 - 4.24 (m, 2H) 4.59 - 4.67 (m, 1 H) 7.08-7.20 (m, 4H) 7.28-7.32 (m, 4 H) 7.41-7.52 (m, 3H) 7.86-7.98 (m, 2H).
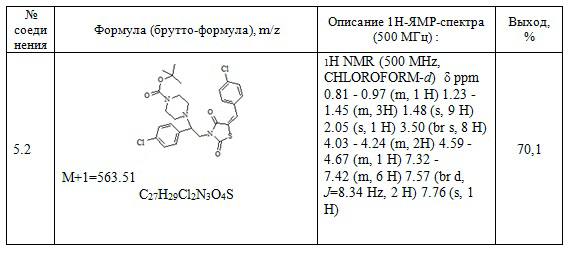
Следующие примеры соединений по изобретению, представленные в таблице 3, получены в соответствии с методикой синтеза, описанной для соединения 5.1, используя подходящие исходные вещества: (E)-3-(2-арил-2-хлорэтил)-5-арилиден тиазолидин-2,4-дионы и трет-бутил пиперазин.
Таблица 3. Примеры соединений по изобретению.
4. Пример получения (E)-4-[ (2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион -3-ил-1-(фенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат (6.1)
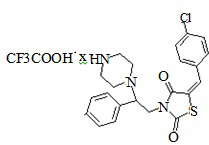
В конической колбе на 50 мл в 25 мл хлористого метилена растворяют 0,750 г (0,0013 моль) трет-бутил 4-[2-(5-(4-хлоробензилиден)-тиазолидин-2,4-дион-3-ил)-1-(фенил)этил] пиперазин-1-карбоксилата, после чего прибавляют 0,53г (3,5 мол. экв.) трифторуксусной кислоты (TFA), реакционную массу перемешивают при комнатной температуре 18 часов. После завершения реакции реакционную смесь упаривают на роторном испарителе, полученный кубовый остаток промывают 10 мл холодного сухого диэтилового эфира. Полученный осадок сушат на вакууме, продувают аргоном.
Получено 0,762 г (E)- 4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(фенил)этил] пиперазин-1-иум 2,2,2-трифторацетат.
Выход составил 54%. Структура соединения подтверждена совокупностью данных ЯМР-спектроскопии и масс-спектрометрии. M+1=428. C24H23ClF3N3O4S. 1HNMR (500 MHz, DMSO-d6) δppm 2.89 - 4.23 (m, 34 H) 4.81- 5.00 (m, 1 H) 7.21 - 7.34 (m, 4H) 7.36 - 7.46 (m, 4 H) 7.55 -7.62 (m, 3 H) 8.82 - 9.04 (m, 2H).
5. Пример получения (Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2- (пиперазин-1-ил)этил) тиазолидин-2,4-дион гидрохлорида ( 6.6)
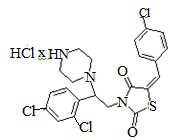
В конической колбе на 50 мл в 25 мл этилового эфира уксусной кислоты и 25 мл этилового спирта растворяют 0,68 г, (E)-трет-бутил-4-[2-(5-(4-хлоробензилиден)- тиазолидин-2,4-дион-3-ил)-1-(2,4-дихлорфенил)этил] пиперазин-1-карбоксилата (0,0011 моль) после чего прибавляют 2 мл 6Н раствора HCl в этиловом эфире уксусной кислоты, реакционную массу перемешивают при комнатной температуре 18 часов.
После прохождения реакции реакционную смесь упаривают на роторном испарителе, полученный кубовый остаток промывают 10 мл холодного сухого диэтилового эфира. Полученный осадок сушат на вакууме, продувают аргоном.
Получено 0.54 г (Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-(пиперазин-1-ил)этил)тиазолидин-2,4-дион гидрохлорид. Выход составил 88,9%. Данные, подтверждающие структуру соединения, представлены в таблице 4.
6. Пример получения (Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-диона (7.1).
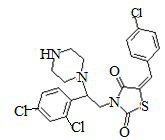
В конической колбе на 100 мл суспедируют 1,7 г (0.0032 моль) (Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-(пиперазин-1-ил)этил)тиазолидин-2,4-дион гидрохлорида в 25 мл дистиллированной воды и 25 мл хлористого метилена, к полученной суспензии при перемешивании добавляют 0,84 г (2,5 мольных эквивалента) карбоната натрия растворенного в 10 мл дистиллированной воды и перемешивают 1 час. Органическую фазу отделяют, водную промывают 25 мл хлористого метилена и объединенную органическую фазу дополнительно промывают 20 мл дистиллированной воды, после чего сушат над 5г сульфата натрия. Хлористый метилен отгоняют на РПИ, полученный осадок сушат под вакуумом.
Получают 1,45 (Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-(пиперазин-1-ил)этил) тиазолидин-2,4-дион. Выход 86%. Данные, подтверждающие структуру соединения, представлены в таблице 4.
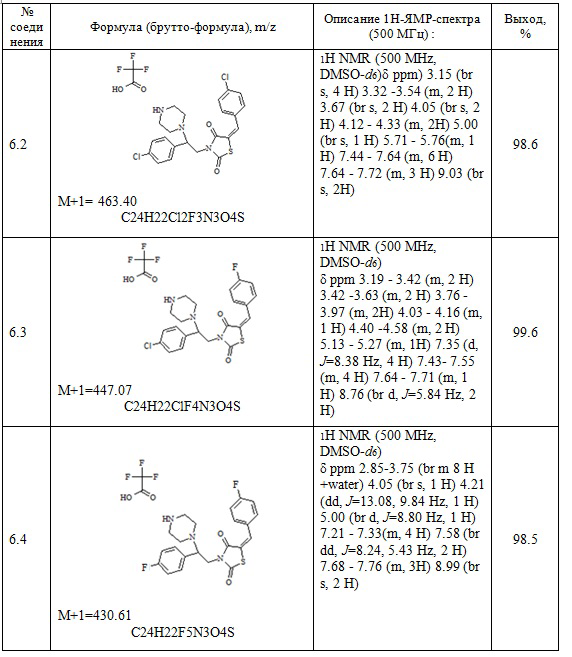
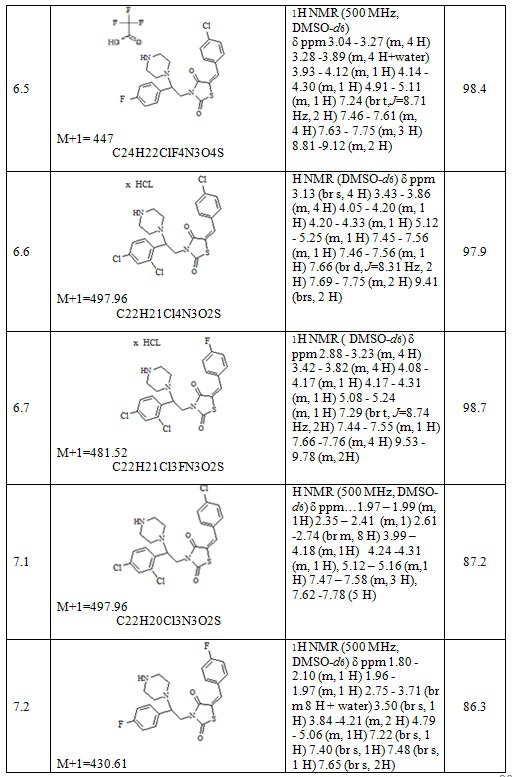
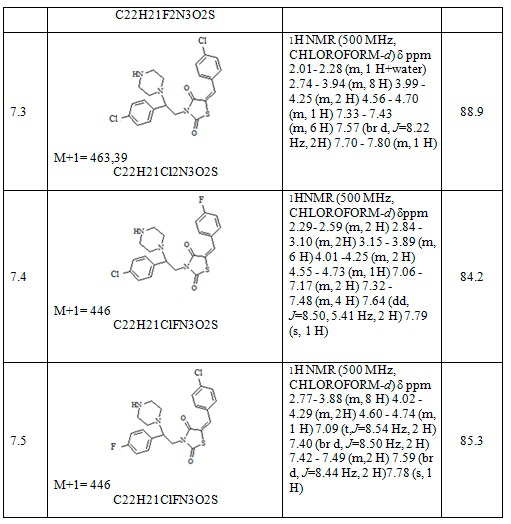
Следующие примеры соединений по изобретению, представленные в таблице 4, получены в соответствии с методиками синтеза, описанными для соединений 6.1, 6.6 и 7.1 используя подходящие исходные вещества: (E)-трет-бутил 4-[2-(5-арилидентиазолидин-2,4-дион-3-ил)-2-фенилэтил]пиперазин-1-карбоксилаты, трифторуксусная кислота, соляная кислота, карбонат натрия.
Таблица 4. Примеры соединений по изобретению.
7. Пример получения (Z)-5-(4-хлорбензилиден)-3-(2-фенил-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-дион (8.1)
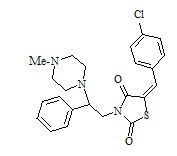
В конической колбе на 50 мл в 25 мл ДМФА растворяют 0.5 г (0,0013 моль) (E)-3-(2-фенил-2-хлорэтил)-5-(4-хлорбензилиден)тиазолидин-2,4-дион, к раствору при перемешивании добавляют 0,134 г (1 мол. экв.) 4-метилпиперазина и 0,274 г прокаленного карбоната калия, перемешивают 18 часов при температуре 60 оС.
После прохождения реакции реакционную смесь отфильтровывают от неорганических солей и упаривают на роторном испарителе, полученный кубовый остаток очищают на колонке (2:1, гексан-этилацетат). Получают 0,15 г (Z)-5-(4-хлорбензилиден)-3-(2-фенил-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-диона.
Выход составил 25,69%. Структура соединения подтверждена совокупностью данных ЯМР-спектроскопии и масс-спектрометрии.
1H NMR (500 MHz, CHLOROFORM-d) δ ppm 2.32 (s, 3 H) 2.35-2.65 (m, 4 H) 3.25-3.85 (m, 4 H) 4.10-4.15 (m, 1 H) 4.67 (m, 1 H) 7.33-739 (m, 5H) 7.45 (d, J=10Hz, 2H) 7.59 (d, J=5Hz, 2H) 7.76 (s, 1H). M+1=443. C23H24ClN3O2S.
Следующие примеры соединений по изобретению, представленные в таблице 5, получены в соответствии с методикой синтеза, описанной для соединения 5.1, используя подходящие исходные вещества: (E)-3-(2-арил-2-хлорэтил)-5-арилиден тиазолидин-2,4-дионы и соответствующие N-замещенные пиперазины.
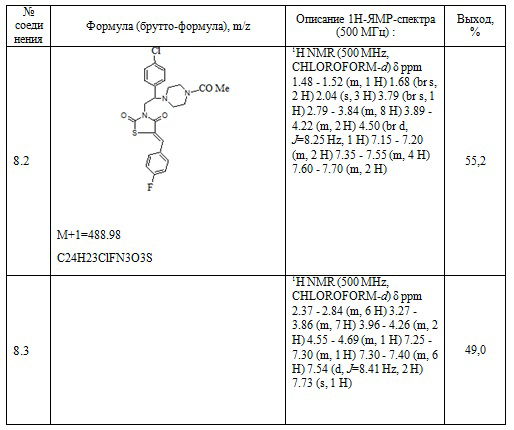
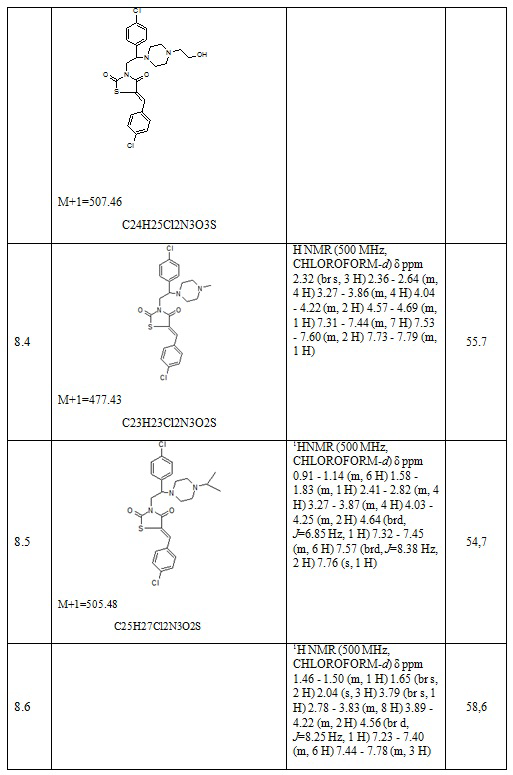
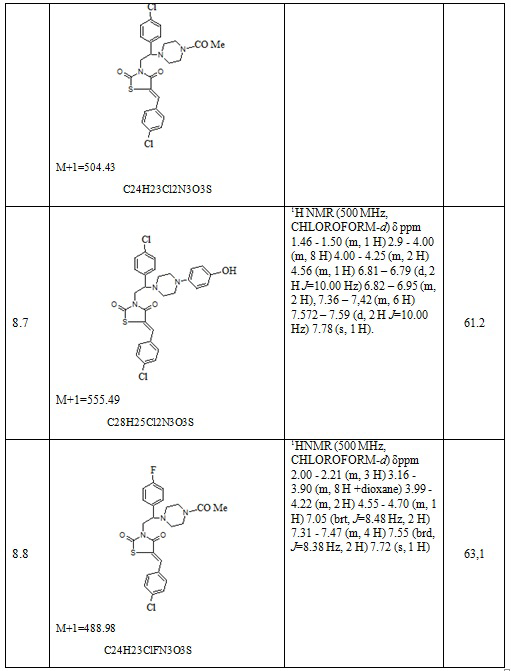
Таблица 5. Примеры соединений по изобретению.
8. Пример получения (Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион (10.1).
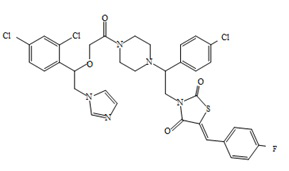
В пробирке с винтовой крышкой и магнитной мешалкой на 15 мл, в 7 мл хлористого метилена суспендируют 0,200 г ((Z)-3-(2-(4-хлорфенил)-2-(пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион 2,2,2-трифторацетат (0,34 ммоль). Вносят 0,120 г (1,1 ммол. экв.) 2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси) уксусной кислоты, 0,155 мл (3,2 ммол. экв,) триэтиамина (ТЭА), и 0,128 г (1,1 ммол. экв.) TBTU.
Реакционную массу выдерживают при перемешивании и комнатной температуре 18 часов, метилен промывают 5 мл раствора бикарбоната натрия, 5 мл раствора лимонной кислоты, 5 мл воды и упаривают на РПИ. К кубовому остатку добавляют 10 мл сухого диэтилового эфира, сформировавшийся осадок отфильтровывают и сушат под вакуумом.
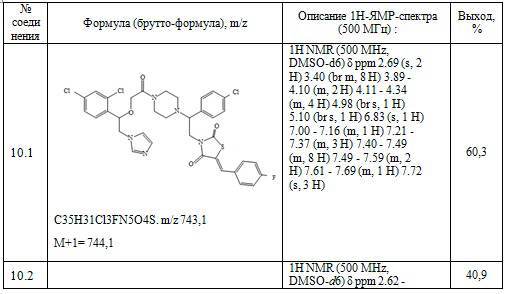
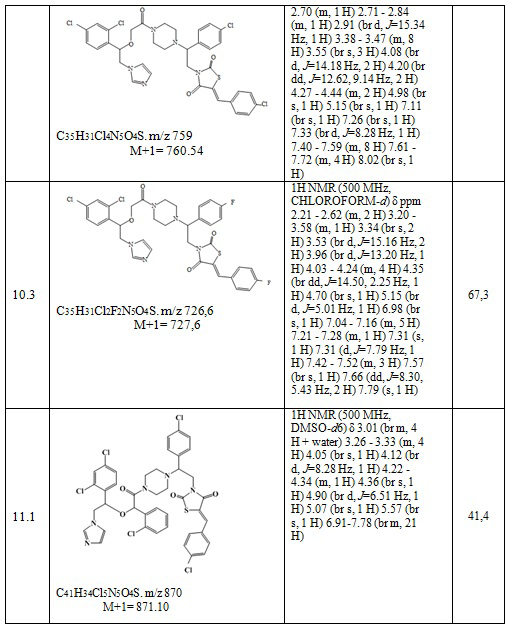
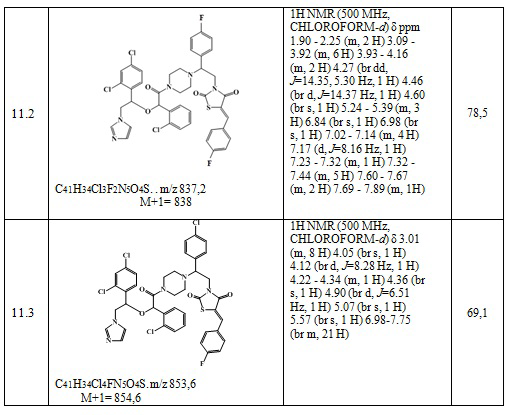
Получают 0,108 г (Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-диона. Выход составил 41,25%. Данные, подтверждающие структуру полученного соединения, представлены в таблице 6. Следующие примеры соединений по изобретению, представленные в таблице 6, получены аналогичным образом.
Таблица 6. Примеры соединений по изобретению.
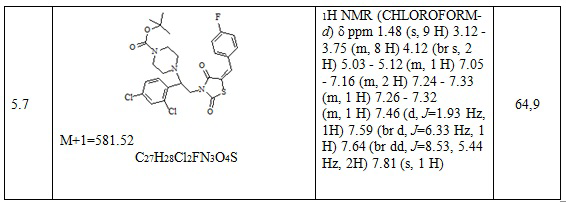
9. Пример получение (Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1- ил)этил)тиазолидин-2,4-дион, (13.1).
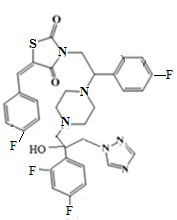
Смесь оксирана метансульфоната (123 мг, 1 экв.), (Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-диона трифторацетата (200 мг, 1 экв.) и триэтиламина (128 мкл, 2.5 экв.) кипятят в этаноле в течении 8 часов. Этанол упаривается, остаток растворяется в хлористом метилене и промывается водной и лимонной кислотой до кислой реакции, потом снова водой. Органическая фаза высушивается Na2SO4 и упаривается. Продукт получен в виде масла, постепенно закристаллирующегося при стоянии. Выход 207 мг (83,9%).
Следующие примеры соединений по изобретению, представленные в таблице 7, получены аналогичным образом.
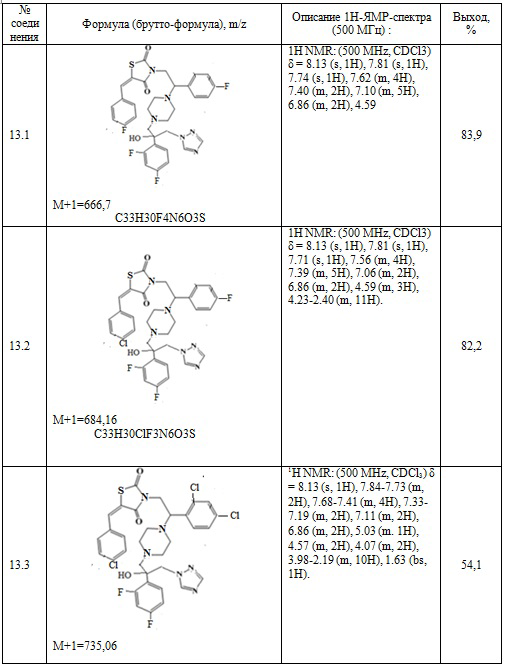
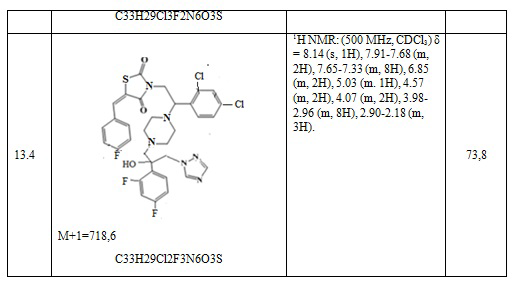
Таблица 7. Примеры соединений по изобретению
Применение химических соединений по изобретению
Применение соединений по медицинским показаниям
Соединения, описанные в данном изобретении, могут применяться для лечения и/или профилактики инфекционных заболеваний.
Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения общей формулы (I).
Под терапевтически эффективным количеством подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Соединение по изобретению или фармацевтическая композиция, содержащая соединение, может быть введено в организм пациента в любом количестве и любым путем введения, эффективным для лечения или профилактики заболевания.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, местно и т.п.
В том случае, когда соединение по изобретению используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции
Изобретение также относится с фармацевтическим композициям, которые содержат соединение общей формулы (I) (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не разрушают фармакологической активности этого соединения, и являются нетоксичными при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахариды, а также их производные; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определённого пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, местно, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственные формы данного изобретения могут содержать составы, полученные методами использования липосом, методами микрокапсулирования, методами приготовления наноформ препарата, или другими методами, известными в фармацевтике.
При получении композиции, например, в форме таблетки, активное начало смешивают с одним или несколькими фармацевтическими эксципиентами, такими как желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки можно покрыть сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например, пропиленгликоль или бутиленгликоль.
Характеристика биологической активности соединений
Сравнительное исследование спектра противогрибкового действия соединений по изобретению в опыте in vitro методом двукратных микроразведений в бульоне.
Эксперименты проводили с использованием стандартных штаммов и клинических изолятов. Чувствительность к референсным штаммам и стандартным препаратам, используемым в исследовании, нормирована по ГОСТ Р ИСО 16256-2015 и служит критерием точности проведенного эксперимента.
Для контроля проводимых исследований и сравнительной активности синтезированных соединений использовали стандартные препараты флуконазол (Fluconazole, Sigma-Аldrich), противогрибковый антибиотик Нистатин и контрольный штамм Candida parapsilosis (ATCC 22019).
Анализ осуществляли методом серийных микроразведений в соответствии с ГОСТ Р ИСО 16256-2015 «Клинические лабораторные исследования и диагностические тест-системы in vitro. Референтный метод для тестирования активности in vitro антимикробных препаратов в отношении дрожжевых грибов, вызывающих инфекционные заболевания» и рекомендациями Всемирной некоммерческой организации по разработке стандартов и рекомендаций в сфере медицины Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, 2nd ed. CLSI document M38-A, Antifungal Susceptibility Testing of Yeasts:M27-A2. 2008. Clinical and Laboratory Standards Institute, Wayne, PA.).
Все изучаемые образцы соединений растворяли в диметилсульфоксиде (ДМСО), концентрация основных растворов составляла 10000 мкг/мл. Для получения рабочих растворов основные растворы разводили в питательной среде до концентрации 64 мкг/мл. Диапазон значений изучаемых концентраций составлял от 32,0 до 0,015 мкг/мл.
Инокулят дрожжевых культур содержал 103 КОЕ/мл, филаментозных грибов – 104 КОЕ/мл.
Серию двукратных разведений испытуемых образцов готовили в 96-луночных планшетах для иммунологических исследований в объеме 100 мкл среды RPMI-1640 с L-глютамином (ФГУП ПИПВЭ им. Чумакова, Россия), содержащей 0,2% глюкозы.
После посева дрожжевых культур планшеты инкубировали 24-48 часов при 35°C, филаментозные – 48-96 часов.
За минимальную ингибирующую концентрацию (МИК) принимали наименьшую концентрацию растворов образцов, при которой наблюдали не менее, чем 80%-ное ингибирование роста. Все эксперименты повторяли трехкратно.
Результаты исследований представлены в таблице 8.
Таблица 8. Результаты исследования in vitro спектра противогрибкового действия соединений по изобретению методом двухкратных микроразведений в бульоне.
Сравнительная оценка активности заявляемых соединений по изобретению в условиях in vitro в отношении грибных штаммов дерматофитов и препаратов сравнения (флуконазола и нистатина, а также кетоконазола) показала, что соединения по изобретению не уступают широко используемым коммерческим препаратам. Соединения по изобретению обладают широким спектром противогрибковой активности, что подтверждает их перспективность для использования в клинической практике для лечения инфекционных заболеваний, вызванных грибковыми инфекциями, в частности в дерматологии.
Активность большинства заявляемых соединений по изобретению в отношении Candida parapsilosis не уступает или превосходит активность Флуконазола и приближается к Кетоконазолу. Кроме того, соединения по изобретению, в отличие от Флуконазола, подавляют рост филаментозных грибов M.canis B-200 и Т.rubrum 2002. Высокую активность показывают, в частности, 3-(2,4-дихлорфенил)этил) производные- (соединения 6.6 и 7.1), которые в отличие от Флуконазола воздействуют и на дрожжевые и на филаментозные грибы. Аналогичное действие оказывали и гибридные соединения с триазолом (13.1 и 13.2). Соединения, полученные комбинацией с имидазольным фрагментом, в частности соединения 10.1 - 10.3., проявляют высокую активность только в отношении кандидозных патогенов. Причем, лучшую активность проявляют гибриды с триазолым производным (соединения 13.1 и 13.2)
Сравнительное изучение противогрибковой активности соединений по изобретению на клинических изолятах Candida spp. микрометодом двухкратных серийных разведений in vitro
Была исследована противогрибковая активность соединений по изобретению и микробиологических стандартов кетоконазола и итраконазола по спектру противогрибкового действия в отношении клинических изолятов Candida spр. микрометодом двукратных серийных разведений в питательном бульоне RPMI 1640 в отношении 10 клинических изолятов и стандартных штаммов Candida spр.
Оценку активности проводили в соответствии с рекомендациями Всемирной некоммерческой организации по разработке стандартов и рекомендаций в сфере медицины – Clinical and Laboratory Standards Institute (CLSI). Использовали методы M27-A3 (Clinical and Laboratory Standards Institute. Approved standard third edition M27-A3, Wayne, PA, USA, 2008) иISO 16256:2012 Clinical laboratory testing and in vitro diagnostic test systems — Reference method for testing the in vitro activity of antimicrobial agents against yeast fungi involved in infectious diseases (IDT) для определения значения минимальной ингибирующей концентрации (МИК), микрометодом серийных разведений в среде RPMI 1640 с добавлением глюкозы до концентрации 0,2%.
Для получения посевного материала, штаммы выращивали на агаре ГРМ 2 (ВФС 42-3068-98, Биохолд, Россия) при 35 °С в течение 48 часов. Посевную суспензию приготавливали в среде RPMIc 0,2% глюкозой по стандарту мутности 0,5 McFarland (~5х 106 КОЕ/мл для дрожжевых культур), титр микробных клеток приготовленной суспензии оценивали денситометрически (Densimat, Biomerieux). Полученную суспензию разводили от 1:1000 до ~2,5 х 103 колоний образующих единиц (КОЕ/мл) в среде RPMIc 0,2% глюкозой.
Для получения основных растворов образцов с концентрацией 10000 мкг/мл навески растворяли в диметилсульфоксиде (ДМСО). Субстанции соединений в количестве 18 мг растворяли в 1,8 мл ДМСО, кетоконазол в количестве 1,8 мг растворяли в 0,18 мл ДМСО. Для получения рабочих растворов с концентрацией 64 мкг/мл основные растворы в количестве 0,64 мл доводили до 10 мл в питательном бульоне RPMI 1640 с глюкозой 0,2%.
В работе использовали 96 луночные планшеты для иммунологических исследований (Медполимер, Санкт-Петербург). В лунки планшет вносили 100 мкл суспензии дрожжевых культур в питательной среде и образцы в диапазоне концентраций 32 - 0,125 мкг/мл, стандартный образец кетоконазола в диапазоне 64-0,015 мкг/мл. После внесения исследуемых образцов плотность суспензии тест-культур составила ~2,5 х 103 КОЕ/мл. Для контроля роста культуры в питательный бульон засевали тест-микроорганизмы без образцов.
Для соблюдения точности проводимой процедуры определения значений МИК исследуемых штаммов, в опыт включен эталонный штамм Candida parapsilosis ATCC 22019, для которого ингибирующая рост концентрация кетоконазола (МИК) не должна выходить за доверительные пределы, предусмотренные документом (Clinical and Laboratory Standards Institute: References; informational supplement (M27-S3). Wayne, PA: Clinical and Laboratory Standards Institute; 2007; ISO 16256:2012 Clinical laboratory testing and in vitro diagnostic test systems — Reference method for testing the in vitro activity of antimicrobial agents against yeast fungi involved in infectious diseases (IDT):
Оценку чувствительности проводили визуально после инкубации при 35°С в течение 24 и 48 часов.
Рост культуры в бульоне в присутствии препаратов оценивали в сравнении с интенсивностью роста без препаратов (контролем роста).
Таблица 9. Сравнительные результаты соединений по изобретению в отношении клинических штаммов Candida spp.
соединения
Таблица 10. Сравнительные результаты некоторых соединений по изобретению в отношении клинических штаммов Сandida spp., включая резистентные * - С.albicans 80R и C.albicans 604M.
соединения
Сравнительный анализ соединений по изобретению и препаратов сравнения (кетоконазола, итраконазола и флуконазола) выявил преимущества заявляемых соединений в отношении различных штаммов грибов Candida spp., в том числе и резистентных (C.albicans 604M (R), C.albicans 80 (R) или не чувствительных к флуконазолу (C. tropicalis 3019, C.glabrata 61Л, C. krusei 432M). По оценке активности, в отношении C. tropicalis 3019, соединения, в частности соединения 6.6, 7.1 и 7.3 показали лучшие значения, чем кетоконазол и итраконазол. По активности в отношении C. albicans ATCC 24433 соединения были сопоставимы с кетоконазолом, а с учетом того, что кетоконазол обладает значительной токсичностью, заявляемые соединения могут составить серьезную конкуренцию, т.к. соединения по изобретению, характеризуются пониженной токсичностью.
С другой стороны, заявляемые соединения составляют серьезную конкуренцию флуконазолу, к которому зафиксирована постоянного растущая резистентность. А так как соединения по изобретению имеют совершенно другой механизм действия на патогенную клетку, возможность развития резистентности к ним существенно снижена.
Сравнительное исследование in vivo соединений по изобретению (на примере соединения № 6.6) в отношении Candida albicans на модели внутрибрюшинного заражения мышей (экспресс метод) с коммерческим препаратом Вориконазолом.
Препарат сравнения
В качестве препарата сравнения использовали субстанцию вориконазола (SIGMA-ALDRICH Co., USA.).
Растворитель
В связи с нерастворимостью в воде соединения № 6.6 и вориконазола использовали в качестве растворителя диметилсульфоксид (ДМСО) (ЛенРеактив).
Тест-культура
Штамм Candida albicans РКПГ Y 1274 был получен из "Российской коллекции патогенных грибов" НИИ медицинской микологии им. П.Н. Кашкина СЗГМУ им. И.И. Мечникова. Штамм характеризуется резистентностью к флуконазолу и чувствительностью к вориконазолу.
Питательная среда
Среда Сабуро. Состав: вода дистиллированная – 1л, пептон ферментативный – 10 г, глюкоза – 40 г, агар-агар – 18 г.
Лабораторные животные
Для проведения исследования использовали самцов белых беспородных мышей, полученных в питомнике Рапполово. Масса тела к началу эксперимента – 20 г.
Животные были адаптированы в течение 7 дней после поступления в лабораторию. Во время этого периода осуществляли ежедневный осмотр состояния животных. Перед включением в исследование животные были осмотрены ветеринарным врачом. Животных с обнаруженными в ходе осмотра отклонениями не включали в исследование.
Распределение животных по группам
Для проведения эксперимента были сформированы пять групп животных (по 6 мышей в каждой группе), включая 3 экспериментальных (№ 1, 2, 3), одна группа для введения вориконозола (В), и контрольная группа (Д - внутрибрюшинное введение 0,5 мл 20% ДМСО). Животных распределяли по группам методом случайного отбора.
Для сравнительной оценки эффективности подавления прорастания дрожжевых клеток C. albicans в сальниках мышей различными дозами соединения № 6.6 и вориконазолом рассчитали процент прорастания дрожжевых клеток в каждой из этих групп животных по отношению к контрольной группе Д (20% ДМСО). Затем рассчитали процент подавления прорастания клеток C. albicans: 100% - % прорастания (Табл. 11).
Таблица 11. Подавление процесса формирования ростовых трубок C. albicans (штамм РКПГ Y 1274) в сальниках мышей различными дозами соединения № 6.6 и вориконазолом.
** p – сравнение с вориконазолом.
Как следует из результатов, представленных в таблице 11, соединение № 6.6 при введении в дозе в 2,4 раза меньшей (5 мг/кг), чем вориконазол (12 мг/кг), подавлял процесс образования ростовых трубок дрожжевыми клетками C. albicans в 2 раза эффективнее (34,86 против 17,00 %). При введении в равных дозах (12 мг/кг) соединение № 6.6. также было эффективнее, чем вориконазол (23,16 против 17,00 %). При увеличении дозы соединение № 6.6 до 25 мг/кг, что в 2,1 раза превышало дозу вориконазола (12 мкг/кг), был получен сопоставимый ингибирующий эффект на процесс прорастания дрожжевых клеток C. albicans (15,61 против 17,00).
Таким образом, в результате проведенного исследования на модели внутрибрюшинного заражения мышей (экспресс-метод) установлено, что соединения по изобретению, в частности соединение № 6.6 при введении в дозах 5 мг/кг и 12 мг/кг активнее подавляет образование ростовых трубок дрожжевыми клетками Candida albicans, чем вориконазол в дозе 12 мкг/кг. При введении в дозе 25 мг/кг соединение № 6.6 оказывал ингибирующий эффект, не отличающийся от воздействия вориконазола в дозе 12 мг/кг.
Таким образом, заявляемые соединения представляют интерес для медицины и могут найти применение для лечения и профилактики инфекционных заболеваний, в частности, вызванных различными грибковыми инфекциями, например таких заболеваний как дерматофития, поверхностный микоз, кандидоз кожи и ногтей, вагинальный кандидоз, кандидозный стоматит, эндокардит и других заболеваний человека и животных.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Гибридные амиды на основе триазола и тиазолидина, обладающие антимикробной активностью | 2018 |
|
RU2703997C1 |
| ГИБРИДНЫЕ ЭФИРЫ НА ОСНОВЕ ПРОИЗВОДНЫХ ТИАЗОЛИДИН-2,4-ДИОНА И АЗОЛОВ (1Н-1,3-ИМИДАЗОЛА И 1Н-1,3,4-ТРИАЗОЛА) И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2662153C1 |
| Гибридные производные (1Н-1,2,4) триазола и серосодержащих гетероциклов: производных тиазолидин-2,4-диона, тиоморфолин-3-она и 1,4-тиазепан-3-она, обладающих антимикробной активностью | 2020 |
|
RU2771027C1 |
| Новые потенциальные противогрибковые средства на основе тиазолидин-2,4-диона и триазола | 2023 |
|
RU2814730C1 |
| 5-[(ПИПЕРАЗИН-1-ИЛ)-3-ОКСО-ПРОПИЛ]-ИМИДАЗОЛИДИН-2,4-ДИОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ADAMTS ДЛЯ ЛЕЧЕНИЯ ОСТЕОАРТРИТА | 2015 |
|
RU2693459C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 5-АМИНО-1-ПЕНТЕН-3-ОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2001 |
|
RU2289571C2 |
| ДИСПИРОИНДОЛИНОНЫ НА ОСНОВЕ РОДАНИНОВ КАК ИНГИБИТОРЫ Р53-MDM2 БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ | 2019 |
|
RU2730286C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ | 2018 |
|
RU2682678C1 |
| Замещенные 4-(азол-1-илметил)-1-фенил-5,5-диалкилспиро-[2.5]октан-4-олы, способ их получения (варианты), фунгицидная и рострегуляторная композиции на их основе | 2016 |
|
RU2648240C1 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ПИРИМИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2293731C2 |
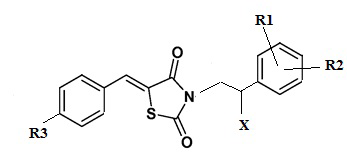
Изобретение относится к соединению общей формулы (I) или его фармацевтически приемлемой соли, где R1 выбирается независимо и представляет собой Н, галоген; R2 выбирается независимо и представляет собой Н, галоген; R3 выбирается независимо и представляет собой Н, галоген; Х выбирается независимо и представляет собой ОН, галоген, 6-членный гетeрoциклил, содержащий 2 атома N, необязательно содержащий заместитель R4; R4 выбирается независимо и представляет собой –C1-6-алкил, -C1-6–алкил-OH, 4-гидрокси-фенил, -COCH3-, -COO-tBu или представляет собой заместитель со следующей структурной формулой (а) или (б), причем волнистой линией показано место присоединения заместителя к фрагменту Х, R5 выбирается независимо и представляет собой Н или фенил, замещенный атомом галогена. Соединения по изобретению предназначены для лечения и/или предотвращения инфекционного заболевания микробной этиологии, вызванного грибковой инфекцией. 4 н. и 9 з.п. ф-лы, 11 табл., 9 пр.
формула (I),
 (а),
(а),  (б)
(б)
1.Соединение общей формулы (I):
 формула (I):
формула (I):
или его фармацевтически приемлемая соль,
где R1 выбирается независимо и представляет собой Н, галоген;
R2 выбирается независимо и представляет собой Н, галоген;
R3 выбирается независимо и представляет собой Н, галоген;
Х выбирается независимо и представляет собой ОН, галоген, 6-членный гетeрoциклил, содержащий 2 атома N, необязательно содержащий заместитель R4;
R4 выбирается независимо и представляет собой –C1-6-алкил, -C1-6–алкил-OH, 4-гидрокси-фенил, -COCH3-, -COO-tBu или представляет собой заместитель со следующей структурной формулой:
 ,
,
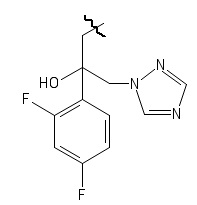 ,
,
причем волнистой линией показано место присоединения заместителя к фрагменту Х,
R5 выбирается независимо и представляет собой Н или фенил, замещенный атомом галогена.
2. Соединение по п.1, в котором:
R1 выбирается независимо и представляет собой Н, F или Cl;
R2 выбирается независимо и представляет собой Н, F или Cl;
R3 выбирается независимо и представляет собой F или Cl;
R4 выбирается независимо и представляет собой метил, изопропил или -(СН2)2-ОН;
X выбирается независимо и представляет собой хлор или пиперазин;
R5 выбирается независимо и представляет собой Н или фенил, замещенный атомом галогена.
3. Соединение по п.1, выбранное из группы:
(E)-трет-бутил-4-[2-(5-(4-хлоробензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-хлорфенил)этил] пиперазин-1-карбоксилат;
(E)-трет-бутил-4-[2-(5-(4-фторобензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-фторфенил)этил] пиперазин-1-карбоксилат;
(E)-трет-бутил-4-[2-(5-(4-хлоробензилиден)тиазолидин-2,4-дион-3-ил)-2-(4-фторфенил)этил] пиперазин-1-карбоксилат;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(фенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(4-хлорфенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-фторбензилиден)-тиазолидин-2,4-дион-3-ил-1-(4-фторфенил)этил]пиперазин-1-иум) 2,2,2-трифторацетат;
(E)-4-[(2-(5-(4-хлорбензилиден)-тиазолидин-2,4-дион-3-ил-1-(2,4-дихлорфенил)этил]пиперазин-1-иум) гидрохлорид;
(Z)-5-(4-хлорбензилиден)-3-(2-(2,4-дихлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-хлорфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-пиперазин-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-фенил-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-хлорфенил)-2-(4-метилкарбонилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(2-гидроксиэтил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-метилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-изо-пропил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(4-гидроксифенил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-(4-метилкарбонилпиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион;
(Z)-3-(2-(4-хлорфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-хлорбензилиден)тиазолидин-2,4-дион;
(Z)-3-(2-(4-фторфенил)-2-(4-(2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил) этокси)ацетил)пиперазин-1-ил)этил)-5-(4-фторбензилиден)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-хлорфенил)-2-(4-(2-(2-хлорфенил)-2-(1-(2,4-дихлорфенил)-2-(1H-имидазол-1-ил)этокси)ацетил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-фторбензилиден)-3-(2-(4-фторфенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1-ил)этил)тиазолидин-2,4-дион;
(Z)-5-(4-хлорбензилиден)-3-(2-(4-фторфенил)-2-(4-(2-(2,4-дифторфенил)-2-гидрокси-3-(1H-1,2,4-триазол-1-ил)пропил)пиперазин-1-ил)этил)тиазолидин-2,4-дион.
4. Применение соединения по любому из пп.1-3 в качестве противогрибкового средства.
5. Применение соединения по любому из пп.1-3 для получения фармацевтической композиции для лечения и/или предотвращения инфекционного заболевания микробной этиологии, вызванного грибковой инфекцией, у субъекта .
6. Применение соединения по п.5, характеризующееся тем, что заболевание микробной этиологии вызвано грибом рода Candida spp, Aspergillus spp., Microsporum spp. Trichophyton spp. и/или Epidermophyton spp.
7. Применение по п.5, характеризующееся тем, что заболевание микробной этиологии вызвано грибом вида Aspergillus fumigatus, Aspergillus niger, Microsporum canis, Trichophyton rubrum или Epidermophyton floccosum.
8. Применение по п.5, характеризующееся тем, что заболевание представляет собой заболевание кожи, ногтей и/или внутренних органов.
9. Применение по п.8, в котором заболевание представляет собой дерматофитию, поверхностный микоз, кандидоз кожи и/или ногтей, вагинальный кандидоз, кандидозный стоматит или эндокардит.
10. Фармацевтическая композиция для лечения и/или предотвращения инфекционного заболевания микробной этиологии, вызванного грибковой инфекцией, у субъекта, содержащая эффективное количество соединения по п.1-3 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
11. Фармацевтическая композиция по п.10, характеризующаяся тем, что фармацевтически приемлемое вспомогательное вещество представляет собой носитель, наполнитель и/или растворитель.
12. Фармацевтическая композиция по п.10, характеризующаяся тем, что заболевание представляет собой заболевание кожи, ногтей и/или внутренних органов.
13. Фармацевтическая композиция по п.12, характеризующаяся тем, что заболевание представляет собой дерматофитию, поверхностный микоз, кандидоз кожи и/или ногтей, вагинальный кандидоз, кандидозный стоматит или эндокардит.
| US 7105554 B2, 12.09.2006 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОГРИБКОВОЙ АКТИВНОСТЬЮ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2012 |
|
RU2497521C1 |
| 3-АЛКОКСИКАРБОНИЛ-5-ЗАМЕЩЕННЫЙ БЕНЗИЛИДЕНТИАЗОЛИДИН-2,4-ДИОНЫ, ОБЛАДАЮЩИЕ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1986 |
|
SU1417436A1 |

