Изобретение относится к области фармацевтического анализа такого медицинского газа, как «Азота закись, газ сжатый», в частности подтверждения подлинности, количественного определения закиси азота, определения примесей углерода монооксида, углерода диоксида, кислорода и азота (неконденсирующихся газов). Способ предполагает использование портативного газового хроматографа и дает возможность проведения испытаний как в стационарных лабораторных условиях, так и с использованием передвижных лабораторий.
Закись азота (N2O; МНН - динитрогена оксид) представляет собой бесцветный негорючий газ, тяжелее воздуха, с приятным сладковатым запахом и привкусом. В малых концентрациях закись азота вызывает чувство опьянения и сонливость, в высоких - быстро развивающееся наркотическое состояние и асфиксию.
В настоящее время закись азота в смеси с кислородом используется в 60% отделений и применяется анестезиологами для наркоза в хирургической практике, оперативной гинекологии, стоматологии, в том числе и детской, для обезболивания родов, а также для купирования болевых приступов при инфаркте миокарда, острой коронарной недостаточности, остром панкреатите и других патологических состояниях, сопровождающихся болями.
Преимуществом использования закиси азота является быстрое начало анальгезирующего действия, низкая стоимость газа, отсутствие раздражения дыхательных путей и редкость кардиореспираторных осложнений. В смеси с кислородом при правильном дозировании наркоз наступает без предварительного возбуждения и побочных явлений. Кроме того, добавление закиси азота позволяет дозозависимо снизить концентрацию других анестетиков, что, в свою очередь, уменьшает частоту возникновения и тяжесть неблагоприятных эффектов этих препаратов.
В соответствии с требованиями Федерального закона №61-ФЗ от 12.04.2010 г. «Об обращении лекарственных средств» закись азота для медицинского применения относится к лекарственным средствам, качество которых оценивается по их соответствию требованиям фармакопейной статьи или нормативной документации (НД).
Закись азота медицинская поставляется в медицинские учреждения в баллонах под давлением. Согласно правилам ДОПОГ при транспортировке она относится ко 2-му классу опасности и может транспортироваться только наземным транспортом, поэтому крайне важна реализация возможности проведения анализа закиси азота непосредственно в медицинском учреждении или иной точке дистрибуции без необходимости транспортировки баллонов до специализированной лаборатории.
Существует относительно немного способов определения закиси азота. Так, например, известен газохроматографический способ определения концентрации закиси азота в газах (RU 2226688, опубл. 10.04.2004). В соответствии с этим способом анализируемую смесь разделяют на закись азота и сопутствующие газы в потоке газа-носителя на хроматографической колонке, заполненной сорбентом, в качестве которого используют цеолит СаХ с содержанием влаги 13-17 мас. %, модифицированным полиэтиленгликолем-1000, взятым в количестве 0,5-1,0% от массы цеолита. Этим способом достигается селективность и высокая чувствительность определения закиси азота - 2 мг/м3, что соответствует 0,5 ПДК. Однако данный способ требует изготовления специализированного продукта, а потому сложен, трудоемок и требует квалифицированного обслуживания.
Также известны способ газохроматографического определения закиси азота в присутствии оксида и диоксида углерода, метана, кислорода и азота с использованием цеолита СаА с программированным повышением температуры колонки от 50 до 400°С [Thorburn S. J Chromatogr., 1969, 42. №15, с. 389] и способ газохроматографического определения закиси азота в газах в присутствии других окислов азота путем разделения анализируемой смеси в потоке газа-носителя на хроматографической колонке, заполненной сорбентом, представляющим собой цеолит СаХ, прокаленный в токе гелия при температуре 350°С в течение 4 ч [Курбанбеков Э., Левчук Б.В. "Хроматографический метод анализа четырехокиси азота" в сб. Газовая хроматография в химии и нефтехимии. - М.: ИНХС АН СССР, 1985, с. 106-111]. Однако методы осложнены в аппаратурном оформлении, и предназначены для определения закиси азота в низких концентрациях в газовой среде и не предназначены для анализа медицинской закиси азота.
Кроме того, известна Фармакопейная статья ФС.2.2.0040 «Азота закись, газ сжиженный» в ГФ РФ 15, где приведены методы анализа отдельно для каждого показателя. Так подлинность азота закиси определяется по двум качественным реакциям. Углерода диоксид и углерода монооксид определяют газохроматографическим способом на насадочной колонке из нержавеющей стали 3 м х 2 мм, сорбент активированный уголь марки АС с последующим гидрированием водородом до метана в реакторе на никелевом катализаторе, при этом используют пламенно-ионизационный детектор, газ-носитель - гелий, расход газа-носителя 20 мл/мин, расход водорода 25 мл/мин, расход воздуха 250 мл/мин, скорость потока 4 мл/мин (30 см/с), температура термостата колонки 100°С, реактора (метанатора) 325°С, детектора 200°С.Количественный расчет выполняют методом абсолютной градуировки с помощью аттестованных газовых смесей. Неконденсирующиеся газы (сумма кислорода и азота) определяют также газохроматографически на насадочной колонке из нержавеющей стали 2 м × 3 мм, молекулярное сито, детектор термокондуктометрический, газ-носитель - гелий для хроматографии, скорость потока газа-носителя 30 мл/мин, температура колонки 100°С, детектора 150°С, объем пробы 1,0 мл. Количественный расчет выполняют также методом абсолютной градуировки с помощью аттестованных газовых смесей. Количественное определение закиси азота проводят с использованием инфракрасного анализатора, принцип действия которого основан на методе недисперсионного инфракрасного поглощения.
Недостатками известных способов являются длительность проведения испытаний, необходимость большого парка оборудования, реактивов и расходных материалов, и невозможность их использования в условиях передвижных лабораторий, то есть непосредственно в медицинском учреждении.
Задача настоящего изобретения заключается в создании способа, позволяющего достаточно просто и надежно в режиме экспресс-анализа и одновременно измерять содержание закиси азота и примесей в лекарственном препарате «Азота закись, газ сжатый». При этом способ должен быть воспроизводим в условиях передвижных лабораторий.
Для решения поставленной задачи предложен газохроматографический способ определения закиси азота и примесей углерода монооксида, углерода диоксида, кислорода, азота (неконденсирующихся газов), заключающийся в разделении анализируемых компонентов в лекарственном препарате в потоке газа-носителя на трехканальном портативном хроматографе с использованием планарной микронасадочной хроматографической колонки 1 м × 1 мм, заполненной сорбентом Carboxen 1000, и детектором по теплопроводности с газом-носителем гелием для разделения неразрешенного пика неконденсирующихся газов (суммы кислорода и азота), углерода диоксида и закиси азота, планарной микронасадочной хроматографической колонки 2 м × 1 мм, заполненной сорбентом Carboxen 1000, и детектором термохимическим с газом-носителем воздухом для определения углерода монооксида, планарной микронасадочной хроматографической колонки 2 м × 1 мм, заполненной сорбентом NaX, и детектором по теплопроводности с газом- носителем гелием для разделения кислорода и азота. При этом хроматографирование по всем трем каналам производится одновременно, объем пробы составляет 250 мкл, а время хроматографирования составляет не более 5 минут.
Техническим результатом является возможность проведение испытания по пяти показателям одновременно (подлинность, количественное определение, содержание примесей угарного и углекислого газов, неконденсирующихся газов) и отдельно оценить содержание примесей кислорода и азота, а также сокращение времени анализа и расхода пробы за счет применения планарных микрофлюидных компонентов хроматографической системы.
Способ проведения анализа закиси азота медицинской методом газовой хроматографии с применением портативного газового хроматографа с использованием планарных микрофлюидных хроматографических колонок и микрофлюидных детекторов предназначен для осуществления контроля качества такого лекарственного препарата, как закись азота медицинская одновременно по нескольким показателям, который можно проводить как на базе стационарных лабораторий, так и на базе передвижных лабораторий в полевых условиях.
Для осуществления данного способа используется портативный газохроматографический комплекс, включающий в себя портативные баллоны с газами-носителями, портативный насос для воздуха, портативную батарею. Хроматограф и все составные части могут быть помещены в ударопрочный кейс для удобства перемещения.
Использование для анализа газовой хроматографии позволяет повысить точность качественного и количественного определения, увеличить скорость проведения анализа и расширить спектр определяемых веществ.
Метод газо-адсорбционной хроматографии основан на разделении компонентов смесей газов на поверхности твердого пористого носителя, обусловленном различием адсорбционной способности и механизмов взаимодействия адсорбент-адсорбат.
Для отбора пробы закиси азота используют либо прямое подключение анализируемой закиси азота медицинской к хроматографу с применением редуктора и капилляра, либо отбирают пробу из баллона в пробоотборник типа ПГО-400 или газоплотный шприц, и дозируют пробу закиси азота в хроматограф из пробоотборника.
Для проведения испытаний закиси азота используется портативный газовый хроматограф, который имеет в своем составе три независимых канала:
1. Термохимический микродетектор и планарную микрофлюидную хроматографическую колонку размером 2 м × 1 мм с адсорбентом углеродные молекулярные сита (Carboxen 1000), воздух в качестве газа-носителя;
2. Микродетектор по теплопроводности и планарную микрофлюидную хроматографическую колонку размером 1 м × 1 мм с адсорбентом углеродные молекулярные сита (Carboxen 1000), гелий в качестве газа носителя;
3. Микродетектор по теплопроводности и планарную микрофлюидную хроматографическую колонку размером 2 м × 1 мм с адсорбентом молекулярные сита - цеолиты (NaX), гелий в качестве газа носителя.
В хроматографе также используется планарный микродозатор.
Суть способа заключается в следующем. Проводят подготовительные процедуры включения хроматографа и выхода хроматографа на режим. Далее проводят хроматографирование поверочной газовой смеси (стандартный образец), проверяют пригодность хроматографической системы. Затем проводят хроматографирование испытуемого образца закиси азота медицинской. Хроматографирование пробы осуществляется одновременно по трем каналам хроматографа. На первом канале происходит определение содержания примеси оксида углерода (СО). На втором канале происходит разделение пиков закиси азота и примесей углекислого газа (СО2), неконденсирующихся газов (О2 и N2). На третьем канале происходит разделение примесей кислорода (О2) и азота (N2). В случае положительных результатов пригодности хроматографической системы проводят расчет содержания разделенных компонентов по методу внешнего стандарта или по методу многоточечной градуировочной зависимости для основного вещества и всех примесей, используя площади хроматографических пиков (угарный, углекислый газ, неконденсирующиеся газы, кислород, азот и закись азота). Расчет содержания закиси азота возможен также расчетным способом:
XN20=100-ΣXi,
где XN20 - объемная доля закиси азота в испытуемом образце в %, ΣXi - сумма объемных долей примесей, определенных в испытуемом образце в %.
Подлинность закиси азота подтверждается по времени удерживания хроматографического пика закиси азота на втором канале в сравнении со стандартом (поверочной газовой смесью). Полученные значения сравнивают с требованиями, установленными в ФС (НД), и принимают решение о качестве анализируемой закиси азота медицинской.
Оптимальные условия хроматографического анализа приведены в таблице 1. 

Содержание каждого из определяемых компонент в препарате в объемных процентах (Xi) может быть вычислено двумя способами:
1) согласно методу внешнего стандарта (ГФ РФ ОФС.1.2.1.2.0001.15 «Хроматография») по формуле:

где Si - измеренное значение площади пика выходного сигнала для i-го газа на хроматограмме испытуемого образца, усл.ед.;
S0 - измеренное значение площади пика выходного сигнала для i-го газа на хроматограмме стандартного образца (поверочной газовой смеси), усл.ед.;
Х0 - Объемная доля i-го газа в стандартном образце (поверочной газовой смеси), %.
2) согласно методу многоточечной градуировочной зависимости. Уравнение измерений объемных долей газов при построении многоточечной градуировочной зависимости имеет вид:
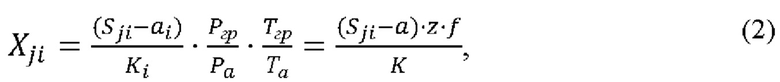
где Xij - результат измерений объемной доли i-го газа, %;
Sji - измеренное значение площади пика выходного сигнала для i-го газа, усл.ед.;
ai - свободный член градуировочной зависимости для i-го газа;
Ki - угловой член градуировочной зависимости для i-го газа;
j=1,…,3 - число параллельных результатов единичного анализа.
z - поправочный коэффициент, учитывающий различия в атмосферном давлении при градуировке и при анализе:

где Ргр и Ра - атмосферное давление при градуировке и при анализе, кПа;
ƒ - поправочный коэффициент, учитывающий различия в температуре при градуировке и при анализе:

где Тгр и Та - температура при градуировке и при анализе, К.
Построение линейной градуировочной зависимости сводят к оценке коэффициентов а и К в уравнении вида:

где Xi - результат анализа объемной доли i-го газа, %;
Si - измеренное значение площади пика выходного сигнала для i-го газа, усл.ед.;
ai - свободный член градуировочной зависимости для i-го газа;
Ki - угловой член градуировочной зависимости для i-го газа;
Коэффициенты а и К вычисляют для каждого газа по формулам:

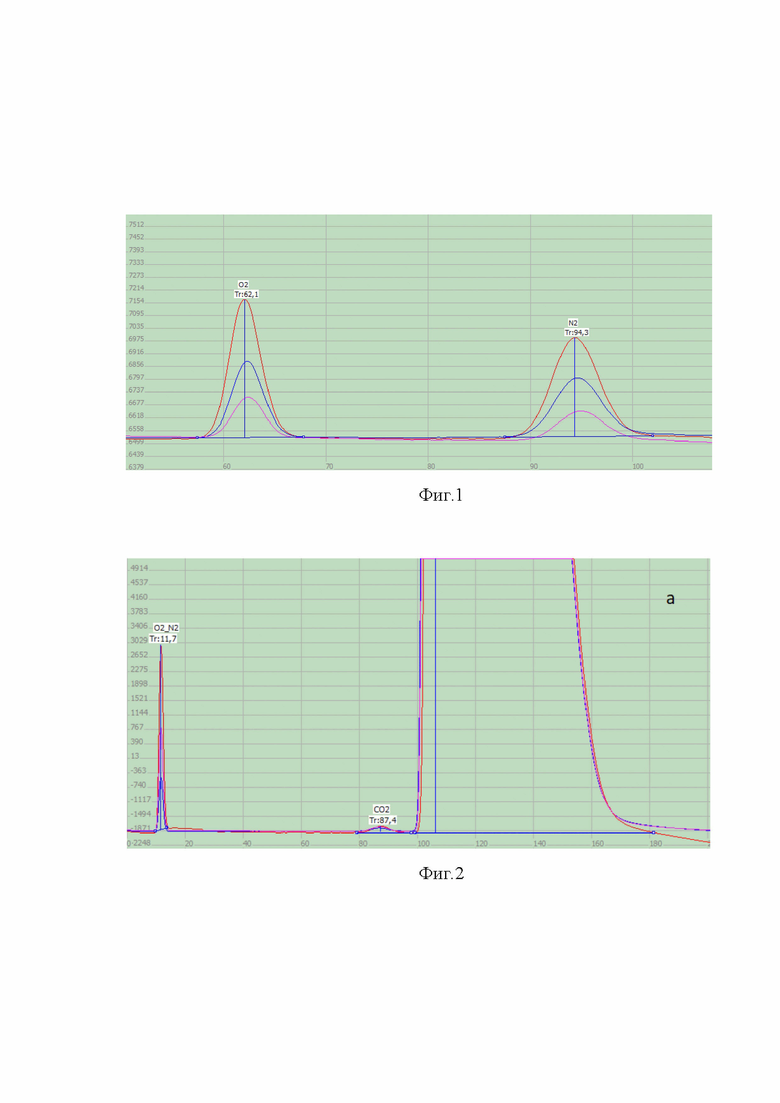
Пример хроматограмм, использованных для построения градуировочной зависимости, полученные методом наложения, приведен на фиг. 1-4, где
- на фиг. 1 представлены хроматограммы, использованные для калибровки, на примере кислорода и азота,
- на фиг. 2 представлены хроматограммы, использованные для калибровки, на примере неконденсирующихся газов (кислорода и азота), углерода диоксида и азота закиси в увеличенном масштабе,
- на фиг. 3 представлены хроматограммы, использованные для калибровки, на примере неконденсирующихся газов (кислорода и азота), углерода диоксида и азота закиси в реальном масштабе,
- на фиг. 4 представлены хроматограммы, использованные для калибровки, на примере углерода монооксида.
В качестве стандартного образца используется аттестованная поверочная газовая смесь, содержащая все определяемые компоненты.
Подлинность закиси азота медицинской определяется по времени удерживания пика закиси азота на хроматограмме испытуемого образца, которое должно совпадать со временем удерживания пика закиси азота на хроматограмме стандартного образца (поверочной газовой смеси).
Количественное содержание закиси азота может быть определено как по методу внешнего стандарта (1), так и по методу многоточечной градуировочной зависимости (2-7), и расчетным способом (8):

где XN20 - объемная доля закиси азота в испытуемом образце, %;
ΣXi - сумма объемных долей примесей, определенных в испытуемом образце, %.
Неконденсирующиеся газы могут быть определены как суммарно на первом канале в виде неразрешенного пика кислорода и азота, так и отдельно на третьем канале по пикам кислорода и азота.
Предлагаемый способ измерений объемных долей закиси азота, оксида и диоксида углерода, кислорода, азота, неконденсирующихся газов методом газовой хроматографии в закиси азота устанавливает процедуру измерений и обеспечивает получение результатов измерений в диапазонах измерений и с показателями точности измерений в относительной форме, приведенными в таблице 2, 3, 4.
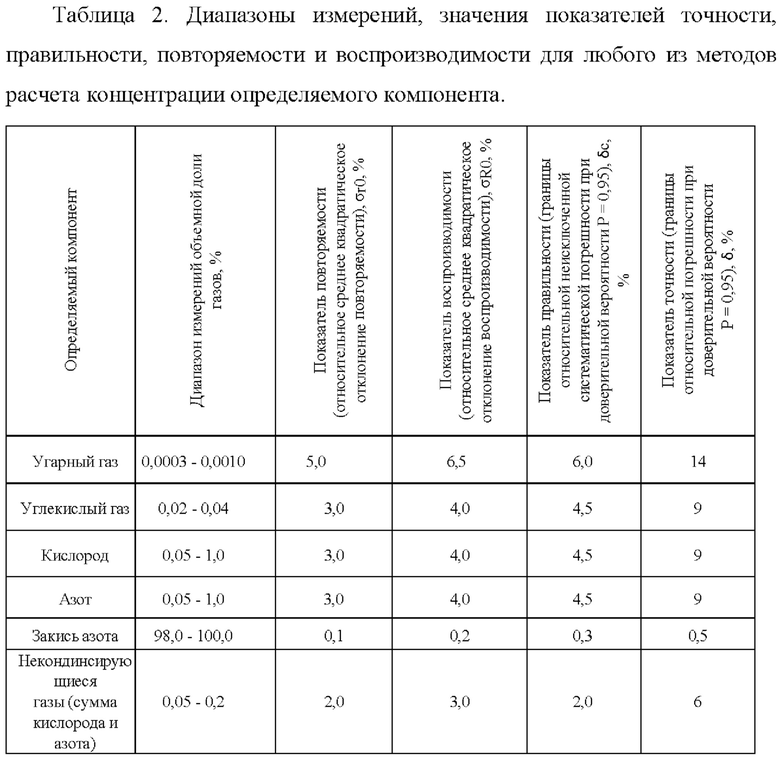
Таблица 3. Показатели точности, принятые для методов расчета концентрации определяемого компонента по методу внешнего стандарта и методу многоточечной градуировочной зависимости.
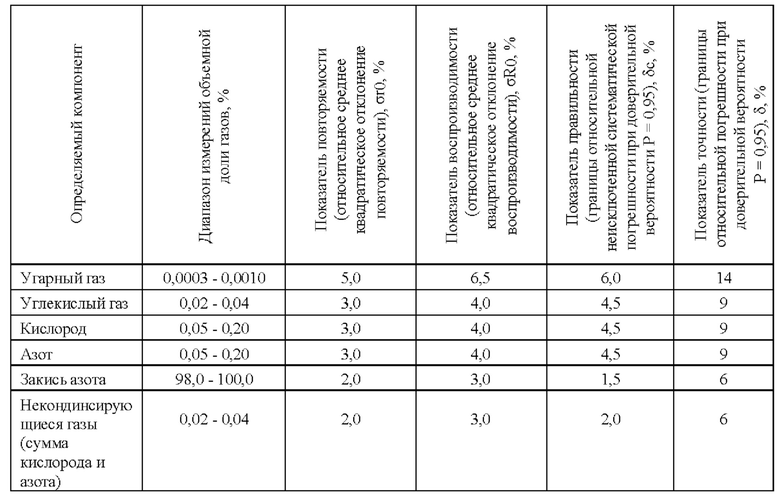

В качестве примера для оценки возможности применения метода внешнего стандарта, метода построения многоточечной градуировочной зависимости и расчетного метода в количественном определении закиси азота и примесей углерода монооксида, углерода диоксида, неконденсирующихся газов (кислорода и азота) были проведены исследования с применением метода газовой хроматографии.
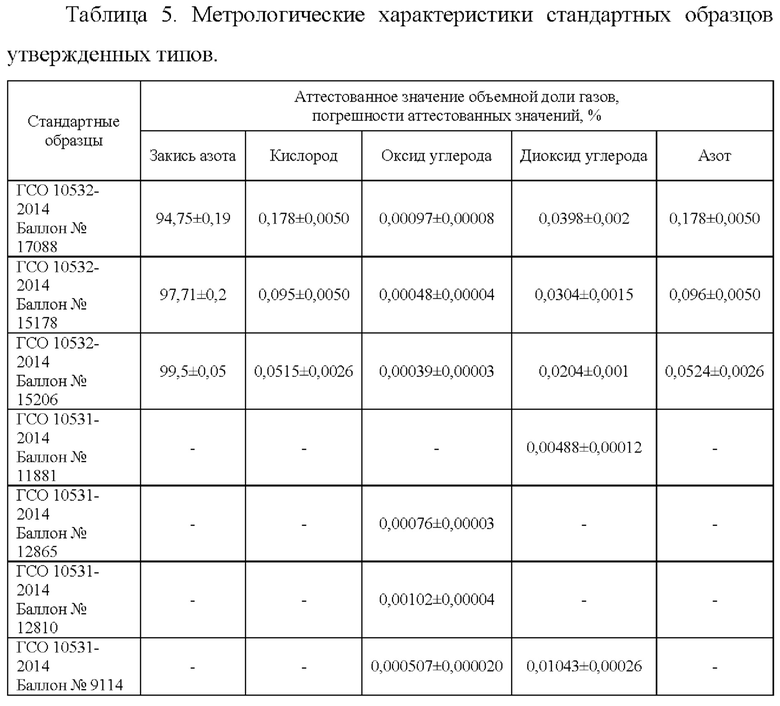
Объектами исследований являлись поверочные газовые смеси ГСО ПГС 10532-2014, состав которых моделирует возможный состав азота закиси медицинской. Стандартные образцы были подобраны таким образом, чтобы диапазон измерений методики был на 20% больше диапазона измерений, который заявлен в нормативной документации на закись азота медицинскую. Подобранные стандартные образцы приведены в табл.5.
Для осуществления способа используются портативные газохроматографические комплексы на основе портативного газового хроматографа типа «ПИА» с детектором по теплопроводности и термохимическим детектором (номер ФИФ 60785-15).
Для измерения температуры может использоваться прибор комбинированный Testo 608-Н1 («Testo Instruments Co. Ltd.», Китай) с диапазоном измерений температуры от 0 до 55°С, с абсолютной погрешностью измерений температуры ±0,5°С.
Для измерения давления может использоваться Барометр-анероид («Гидрометприбор», Россия) с диапазоном измерений от 79,5 кПа до 105 кПа и ценой деления 0,1 кПа.
Относительное среднеквадратическое отклонение выходного сигнала по времени удерживания, высоте и площади пика не превышает 2,0% для указанных детекторов газовых хроматографов.
Условия газохроматографического анализа приведены в таблице 1. В основу методики измерений положен метод газовой хроматографии без предварительной подготовки в газоадсорбционном варианте хроматографии с последующей регистрацией компонентов детектором по теплопроводности (далее - ДТП) и термохимическим детектором (далее - ДТХ).
Хроматографическое разделение компонентов пробы проводили с использованием трех модулей газового хроматографа с использованием микронасадочных хроматографических колонок планарного типа, представленных в таблице 1.
С целью определения метрологических характеристик методика оценивалась по следующим направлениям:
1. Специфичность
2. Линейность и диапазон применения (аналитическая область методики). Предел обнаружения и предел количественного определения.
3. Промежуточная (внутрилабораторная) прецизионность.
4. Правильность и точность.
Оценка показателей правильности методики измерений объемных долей закиси азота, оксида и диоксида углерода, кислорода, азота (неконденсирующихся газов) методом газовой хроматографии в закиси азота медицинской проведена с помощью образцов для оценивания в соответствии с РМГ 61. В качестве образцов для оценивания использовали стандартные образцы утвержденных типов ГСО 10532-2014, представленные в таблице 5.
Показатели прецизионности оценены в ходе проведения квазимежлабораторного эксперимента, проведение которого допускается пунктом 5.2.2 ГОСТ Р 5725-2.
Специфичность методики проводилась для определения влияния подвижной фазы (газа-носителя) на пики примесей и основного вещества (таблица 6).

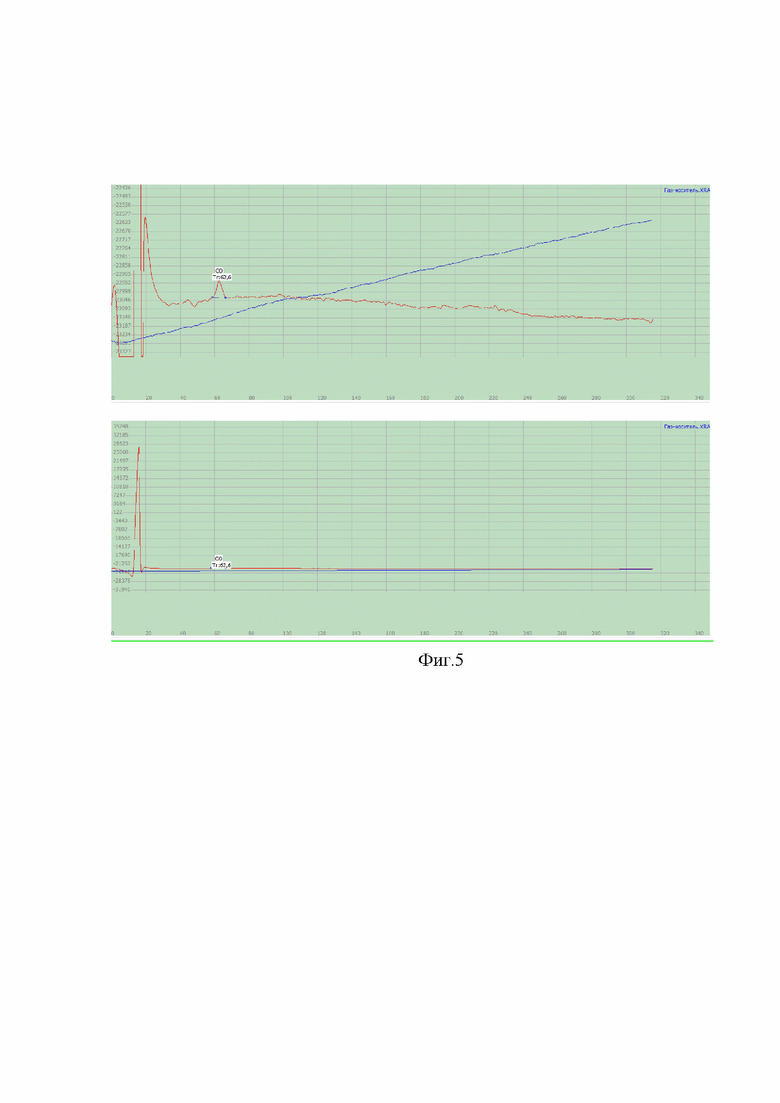
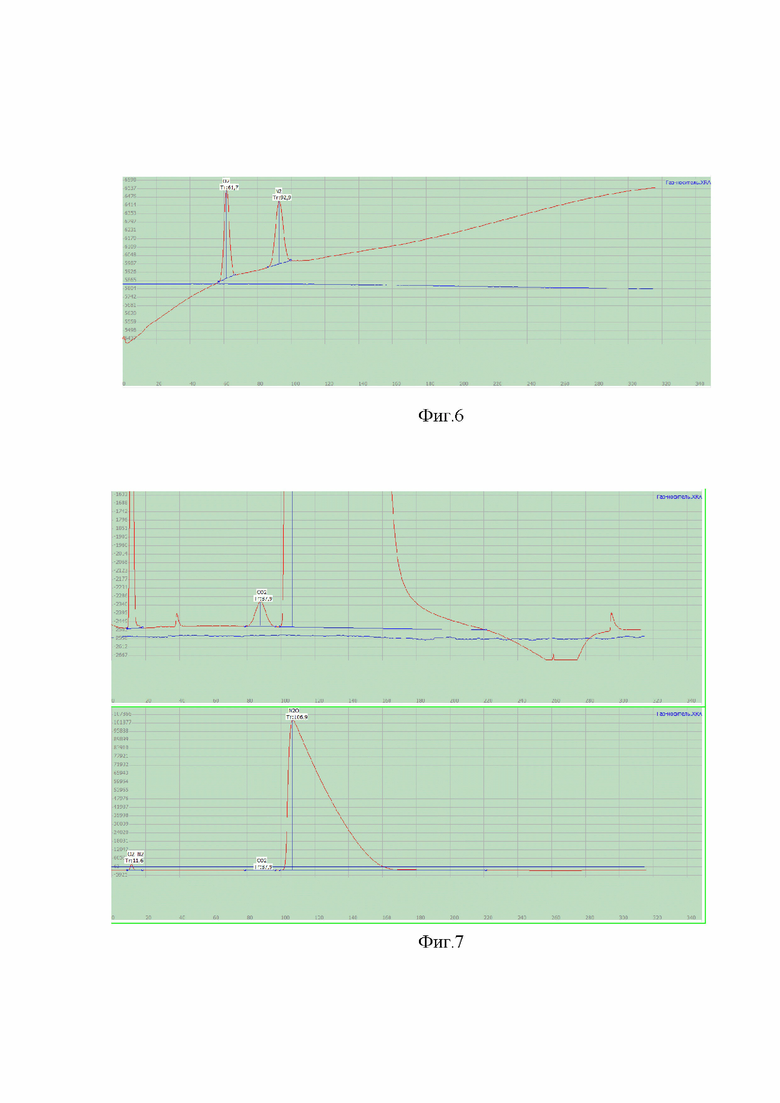
На фиг. 5-7 представлены хроматограммы, полученные на каждом из каналов хроматографа для стандартного образца азота закиси медицинской (ПГС) и газа-носителя (гелия) для проверки специфичности методики.
На фиг. 5 представлены хроматограммы газа-носителя (синяя) и ПГС (красная) на втором канале хроматографа на определение монооксида углерода. На хроматограмме газа-носителя отсутствует пик углерода монооксида, в отличие от хроматограммы ПГС, что свидетельствует о специфичности методики.
На фиг. 6 представлены хроматограммы газа-носителя (синяя) и ПГС (красная) на третьем канале хроматографа на определение неконденсирующихся газов (кислорода и азота двумя разрешенными пиками). На хроматограмме газа-носителя отсутствуют пики кислорода и азота, в отличие от хроматограммы ПГС, что свидетельствует о специфичности методики.
На фиг. 7 представлены хроматограммы газа-носителя (синяя) и ПГС (красная) на первом канале хроматографа. Определение углерода диоксида, азота закиси, неконденсирующихся газов (одним неразрешенным пиком). На хроматограмме газа-носителя отсутствуют пики углерода диоксида, азота закиси, неконденсирующихся газов (одним неразрешенным пиком), в отличие от хроматограммы ПГС, что свидетельствует о специфичности методики.
Отсутствие пиков в области времени удерживания примесей и основного действующего вещества при анализе газа-носителя и разрешение между двумя наиболее близко элюирующимися веществами не менее 0,8 свидетельствует о специфичности методики испытания.
Для оценки линейности и диапазона применения получено по три хроматограммы семи стандартных образцов поверочной газовой смеси (ПГС), а также три хроматограммы газовой смеси, полученной процедурой разбавления, содержащих определяемые компоненты, с использованием портативного газового хроматографа ПИА.
Линейность аналитической методики - это ее способность (в пределах определенного диапазона) получать результаты анализа, которые прямо пропорциональны концентрации (количеству) определяемого вещества в образце. Для ее оценки необходимо построить график зависимости аналитического сигнала как функции концентрации или количества определяемого вещества и оценить его линейность. Характеристикой линейности является показатель линейности R2, который должен быть не менее 0,99.
Диапазон аналитической методики - это интервал между наибольшей и наименьшей концентрацией (количеством) определяемого вещества в образце (включая эти концентрации), в отношении которого было продемонстрировано, что аналитическая методика имеет приемлемый уровень прецизионности, правильности и линейности.

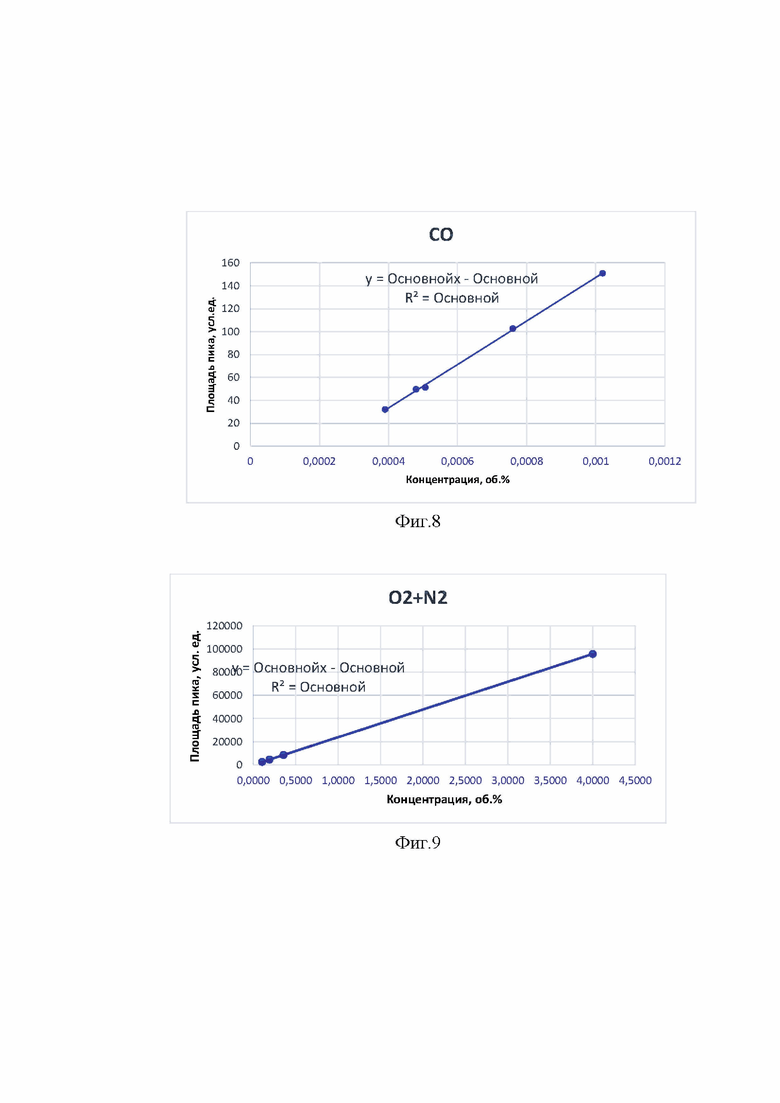
Примеры калибровочных графиков, полученных для определяемых компонентов приведены на фиг. 8-15.
На фиг. 8 представлен калибровочный график для пика углерода монооксида.
На фиг. 9 - калибровочный график для неразрешенного пика неконденсирующихся газов.
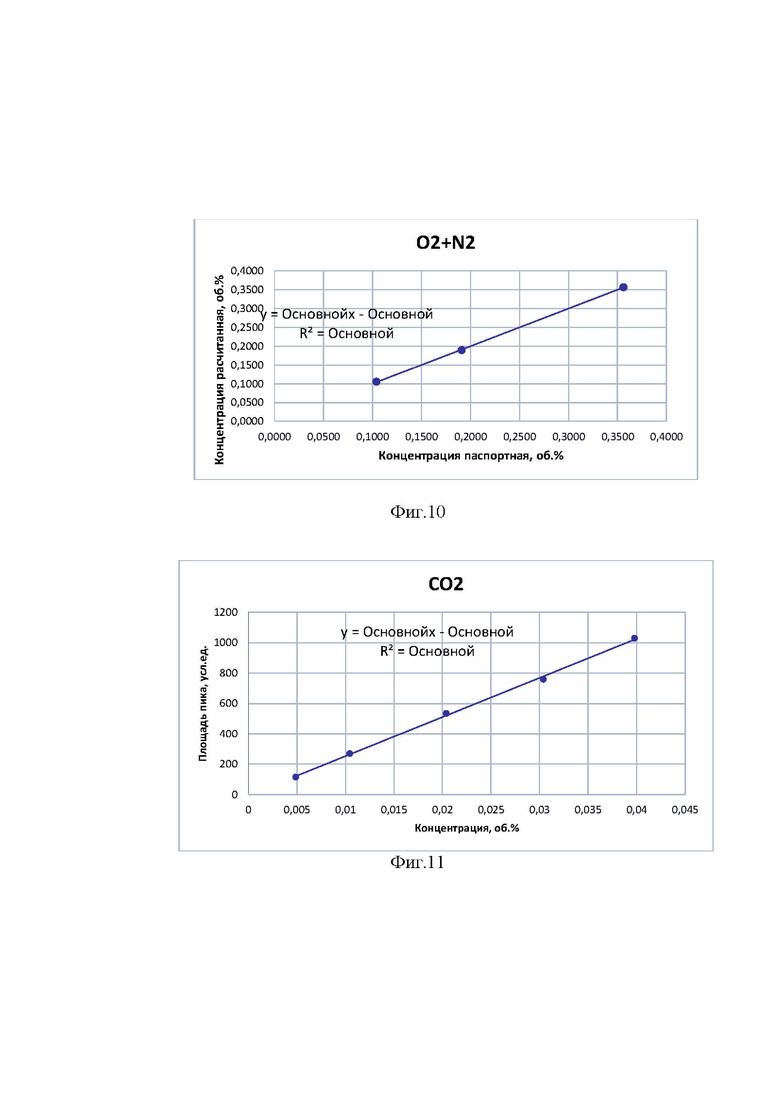
На фиг. 10 - калибровочный график для неконденсирующихся газов (двумя разрешенными пиками кислорода и азота). Поскольку количественное содержание неконденсирующихся газов определяется расчетным способом график линейной зависимости строился по данным паспортной и рассчитанной концентрации.
На фиг. 11 - калибровочный график для пика углерода диоксида.
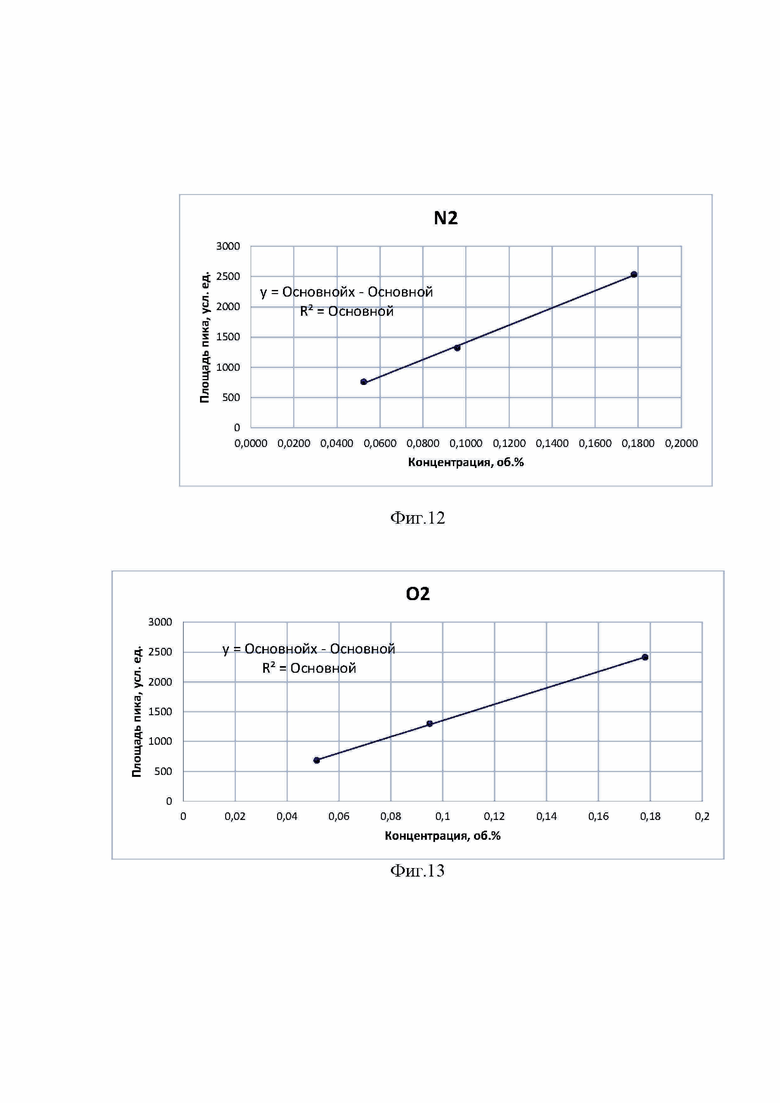
На фиг. 12 - калибровочный график для пика азота.
На фиг. 13 - калибровочный график для пика кислорода.
На фиг. 14 - калибровочный график для пика азота закиси.
На фиг. 15 - калибровочный график для пика азота закиси (расчетный метод).
В таблице 8 приведены результаты определения линейности.
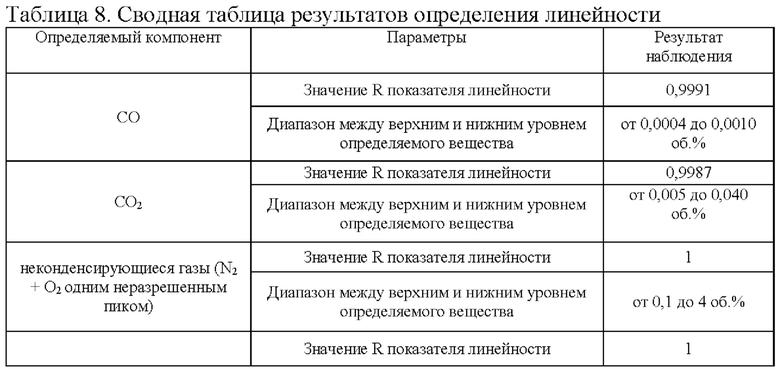

Исследование подтверждает, что площади пиков основного определяемого вещества азота закиси линейны в диапазоне от 97% стандартной концентрации до 102% стандартной концентрации. А также, что площади пиков определяемых примесей линейны в диапазоне от LOQ до 120% от предельно допустимого содержания.
Предел обнаружения (LOD) и предел количественного определения (LOQ) были определены по калибровочной функции, полученной в ходе оценки линейности, по следующим выражениям:
LOD=3,3⋅S/b,
LOQ=10⋅S/b,
где S - стандартное отклонение аналитического сигнала;
b - коэффициент чувствительности, представляющий собой отношение аналитического сигнала к определяемой величине (тангенс угла наклона калибровочной кривой).
Результаты определения значений LOD и LOQ представлены в таблице 9. 

Прецизионность способа исследовалась в двух вариантах:
- как повторяемость (сходимость);
- как внутрилабораторная (промежуточная) прецизионность.
Сходимость методики характеризуется разбросом результатов определенного числа измерений, полученных в одинаковых условиях в одной лаборатории в пределах короткого промежутка времени.
Промежуточная прецизионность отражает разброс результатов в рамках лаборатории: при варьировании некоторых факторов (разные операторы, различные дни, разное оборудование и т.д.).
Для оценки прецизионности проводили измерения стандартного образца ПГС, характеристики и состав которой приведены в таблице 10, по 10 параллельных измерений на двух приборах (портативные газовые хроматографы «ПИА» зав. номера 9004, 3003) два разных дня. Рассчитанные значения показателей прецизионности методики, полученные при обработке результатов данных измерений, приведены в таблице 11, где
Sr - стандартное отклонение повторяемости;
SR - стандартное отклонение воспроизводимости;
r - предел повторяемости;
R - предел воспроизводимости;
RSD - относительное стандартное отклонение.

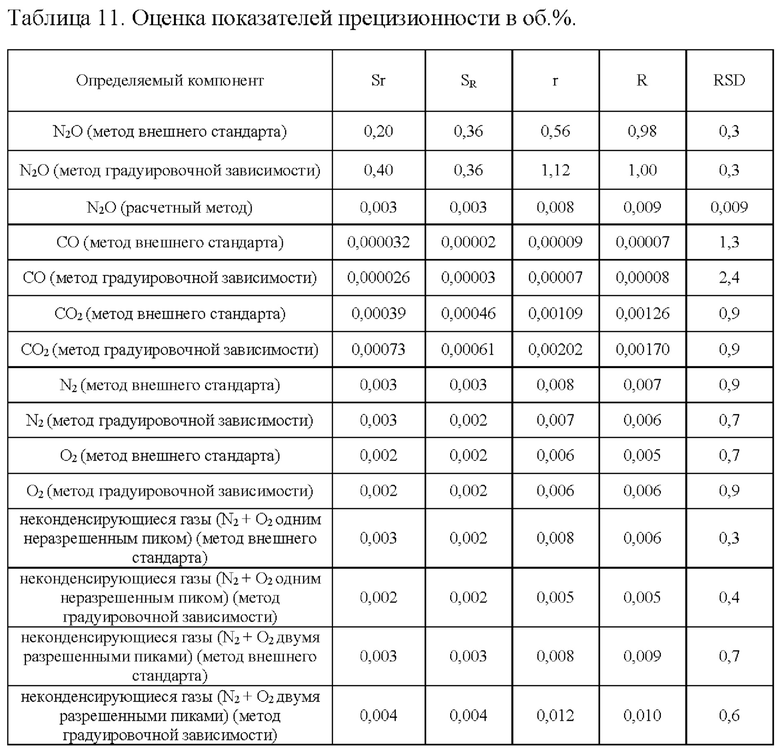
Относительное стандартное отклонение (RSD) рассчитанных концентраций определяемых веществ в стандартном образце ПГС находится в допустимых пределах и не превышает для количественного определения 2%, для примесей 10%. Таким образом, методика для определения количественного содержания азота закиси, примесей монооксида углерода, диоксида углерода, неконденсирующихся газов методом газовой хроматографии отвечает критериям приемлемости по показателям прецизионности.
Точность аналитической методики отражает степень близости результата измерений к принятому опорному значению.
Правильность аналитической методики выражается в уровне приближенности между номинальным значением и рассчитанным значением концентрации. В таблице 12 представлены результаты оценки показателей правильности и точности, где
Θ1 - оценка смещения результатов анализа;
ΔС - показатель правильности методики;
Δ - систематическая погрешность лаборатории при реализации методики;
BIAS% - оценка смещения результатов анализа в процентах (систематическая ошибка);
Sr0, % - относительное среднее квадратическое отклонение повторяемости;
SR0, % - относительное среднее квадратическое отклонение воспроизводимости;
δ, % - показатель точности (границы, в которых находится относительная погрешность измерения с вероятностью Р=0,95).
Аналитическое смещение (систематическая ошибка) определяемых веществ находится в допустимых пределах:
- Для количественного определения азота закиси: BIAS %<5.0%
- Для родственных примесей: BIAS %<20.0%
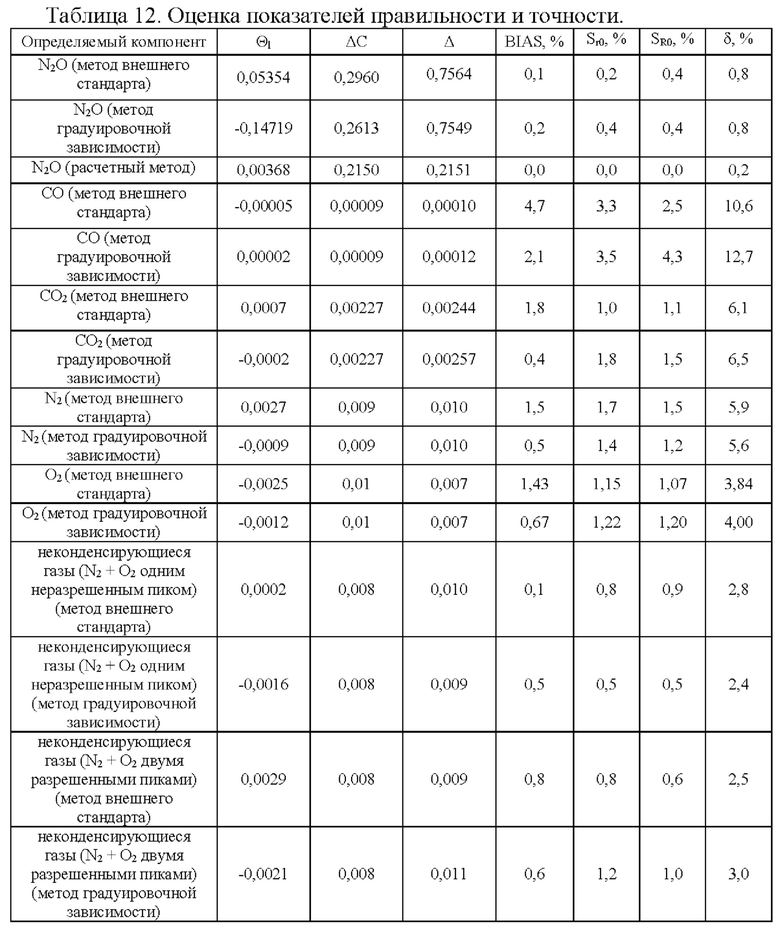
Таким образом, методика для определения количественного содержания азота закиси, примесей монооксида углерода, диоксида углерода, неконденсирующихся газов методом газовой хроматографии отвечает критериям приемлемости по показателю точность и правильность.
Так же был поведен квазимежлабораторный многофакторный эксперимент. В ходе которого проверялось влияние на величину неопределенности результатов измерений факторов, не входящих в уравнение измерений, но оказывающих влияние на результат измерений: расход газа носителя, объем аликвоты, температура термостата колонок.
Расчет неопределенности результатов измерений проведен для метода построения градуировочной зависимости с учетом положений ЕВРАХИМ/СИТАК.
Уравнение измерений для объемной доли (Φni) i-го газа имеет следующий вид
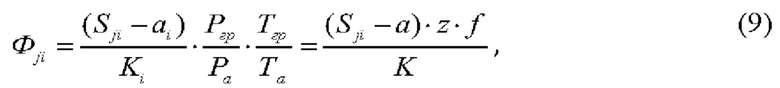
Стандартную неопределенность типа А, uA, результатов измерений объемной доли газов (Φij) i-го газа вычисляют по формуле (на основе экспериментальных данных):

Стандартную неопределенность типа В, uв, измерений объемной доли (Φni) i-го газа, исходя из анализа уравнения измерения определяют композицией составляющих неопределенности, обусловленных неопределенностью определения - площади пика Sij; неопределенностью определения коэффициентов градуировочной зависимости K, а; неопределенностью измерений давления при градуировке и при анализе Ргр, Ра; неопределенностью измерений температуры Тгр и Та.
Поскольку в ходе эксперимента одновременно измеряемыми величинами являются независимые друг от друга величины, то их оценки приняты некоррелированными.
Для оценки влияющих факторов, не входящих в уравнение измерений, методом множественной линейной регрессии с помощью «Пакета анализа» в Microsoft Excel строится модель вида
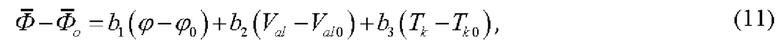
где b1, b2, b3 - коэффициенты чувствительности для следующих факторов влияния соответственно: расход газа носителя, объем аликвоты, температура термостата колонок;
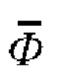 - среднеарифметическое результатов измерений объемной доли газа в закиси азота медицинской при значениях факторов ϕ, Val, Tk;
- среднеарифметическое результатов измерений объемной доли газа в закиси азота медицинской при значениях факторов ϕ, Val, Tk;
 - среднеарифметическое результатов измерений объемной доли газа в закиси азота медицинской при оптимальных значениях факторов ϕ0, Val0, Tk0.
- среднеарифметическое результатов измерений объемной доли газа в закиси азота медицинской при оптимальных значениях факторов ϕ0, Val0, Tk0.
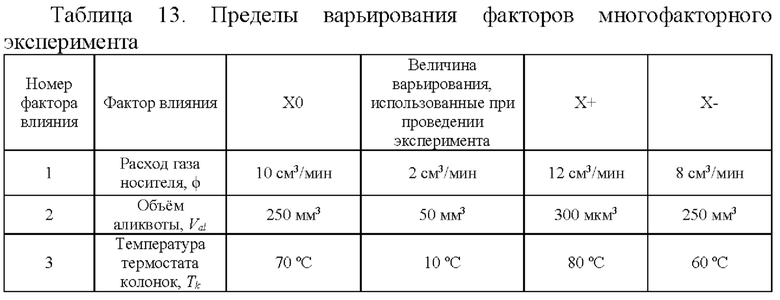
Значения коэффициентов чувствительности и величины их неопределенностей установлены в ходе проведения многофакторного эксперимента. Пределы варьирования факторов и план многофакторного эксперимента приведены в таблицах 13 и 14 соответственно.


Стандартная неопределенность измерений объемной доли i-го газа, оцениваемая по типу В, uВ, рассчитывается по формуле

где b1, b2, b3 - коэффициенты чувствительности модели, оцененные с помощью «Пакета анализа» Microsoft Excel;
u(b2), u(b2). u(b3) - стандартные неопределенности коэффициентов модели по типу А, оцененные с помощью «Пакета анализа» Microsoft Excel;
(ϕ-ϕ0), (Val-Val0), (Tk-Tk0) - допускаемые отклонения расхода газа носителя, объема аликвоты, температуры термостата колонок (оптимальные значения параметров ϕ0=10 мг/см3; Val0=250 мкл; =70°С и допускаемые отклонения приведены в методике измерений);
u(ϕ) - суммарная стандартная неопределенность отклонения расхода газа носителя от оптимального расхода газа носителя;
u(Val) - суммарная стандартная неопределенность отклонения объема аликвоты от оптимального объема аликвоты;
u(Tk) - суммарная стандартная неопределенность отклонения температуры термостата колонок от оптимальной температуры термостата колонок;
u(Si) - суммарная стандартная неопределенность измерений площади пика,
u(Ki) - суммарная стандартная неопределенность углового коэффициента градуировочной зависимости,
u(Ргр) - суммарная стандартная неопределенность измерения давления при градуировке,
u(Ра) - суммарная стандартная неопределенность измерения давления при анализе,
u(Tгр) - суммарная стандартная неопределенность измерений температуры при градуировке,
u(Та) - суммарная стандартная неопределенность измерений температуры при анализе,
u(a) - суммарная стандартная неопределенность свободного члена градуировочной зависимости,
c1, c2, c3, c4, c5, c6, c7, - коэффициенты чувствительности, определяемые из уравнения измерения объемной доли (Φi) i-го газа и определяются как частные производные формулы (19)
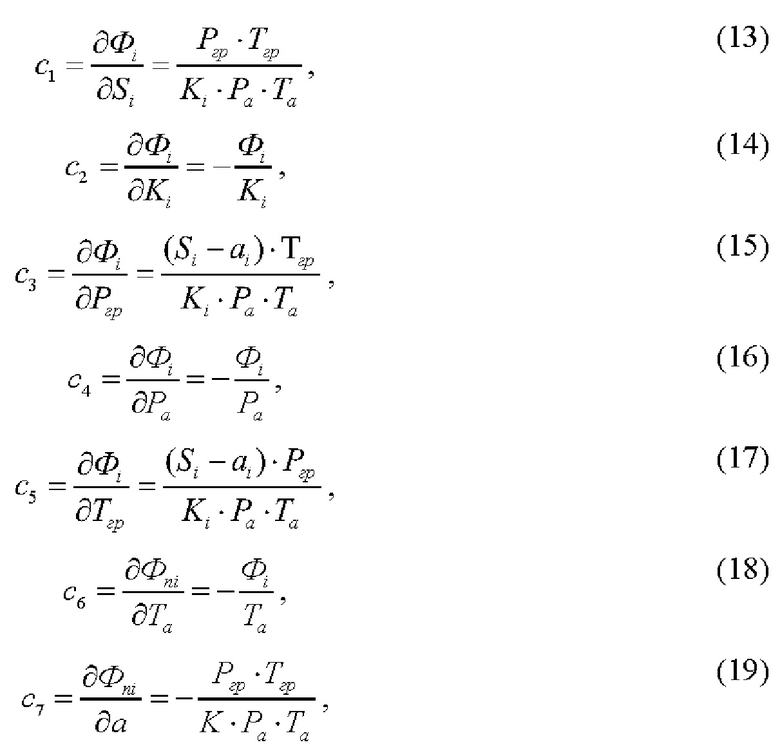
Неопределенность определения площади пика оценивают, исходя из разброса результатов измерений, полученных при анализе в виде среднего квадратического отклонения.
Неопределенность коэффициентов градуировочной зависимости определяют методом регрессионного анализа по формулам (14) и (15).
Неопределенность определения давления оценивают, исходя из свидетельства о поверке и описания типа на используемое средство измерений по формуле, предполагая равномерное распределение

Неопределенность определения температуры оценивают, исходя из свидетельства о поверке и описания типа на используемое средство измерений по формуле, предполагая равномерное распределение

Суммарную стандартную неопределенность результатов измерений объемной доли (Φi) i-го оценивают по формуле

Расширенную неопределенность результатов измерений объемной доли (Φni) i-го оценивают по формуле

где k - коэффициент охвата, равный 2 при вероятности Р=0,95.
Для оценивания неопределенности необходимы данные для построения многоточечной градуировочной зависимости, расчета углового коэффициента, свободного члена градуировочной зависимости и их стандартных неопределенностей.
Результаты измерений получены при реализации квазимежлаборатоного эксперимента. Результаты измерений, полученные лабораториями, в соответствии с РМГ 61-2010 обрабатывались совместно. Пример преставления результатов измерения при измерении объемной доли оксида углерода тремя «лабораториями» ГСО 10532-2014 с аттестованным значением оксида углерода 0,00039% приведен в табл.15.
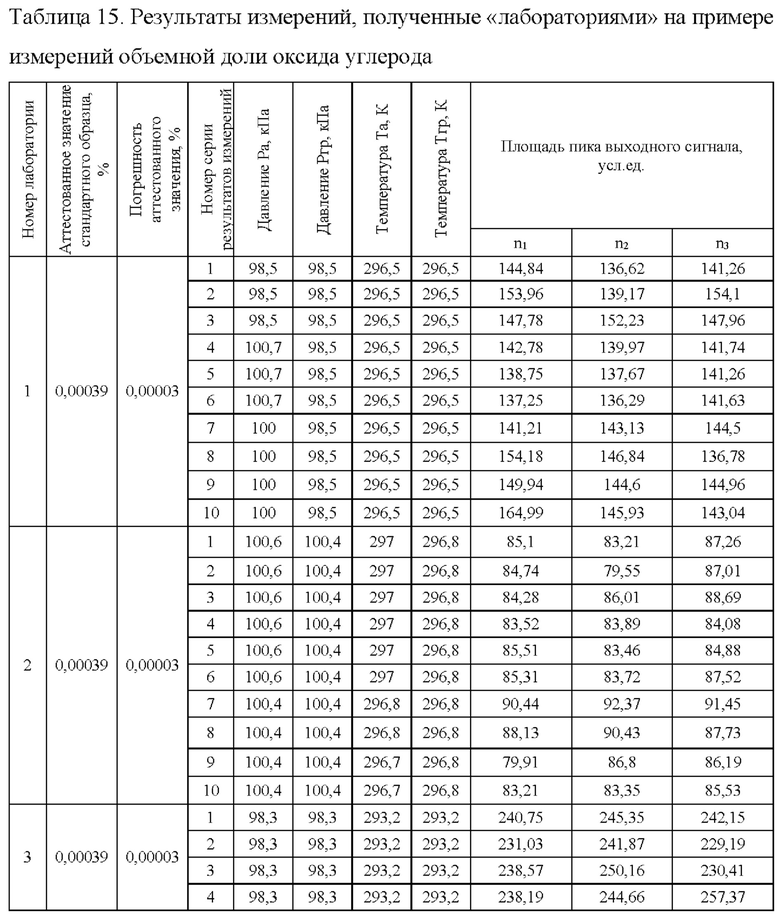

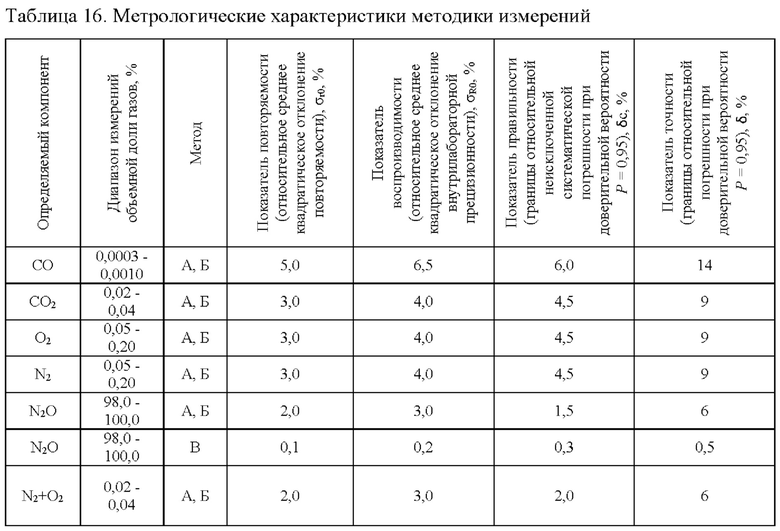
После обработки результатов измерений всеми лабораториями всех газов во всем диапазоне измерений получены следующие обобщенные метрологические характеристики методики измерений (таблица 16), где
А - с использованием метода многоточечной градуировочной зависимости;
Б - с использованием метода внешнего стандарта;
В - с использованием расчетного метода.
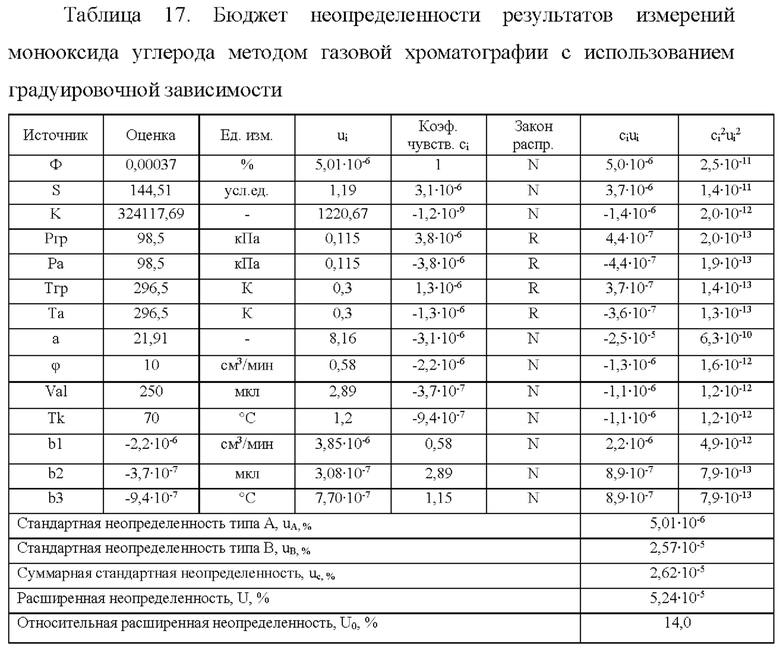
Пример расчета неопределенности результатов измерений на примере измерений объемной доли монооксида углерода приведен в таблице 17.
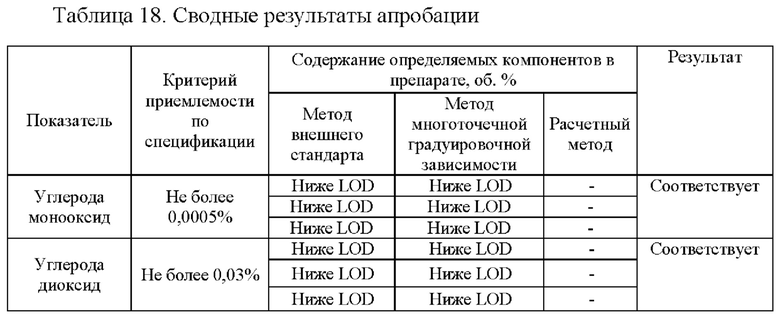
Для апробации предлагаемого способа осуществили анализ образца для контроля «Азота закись ОСЧ» в баллоне вместимостью 10 дм3, № партии 19403, произведенного ООО «Фессен Эм Ай И». Полученные результаты по трем параллельным определениям представлены в таблице 18.
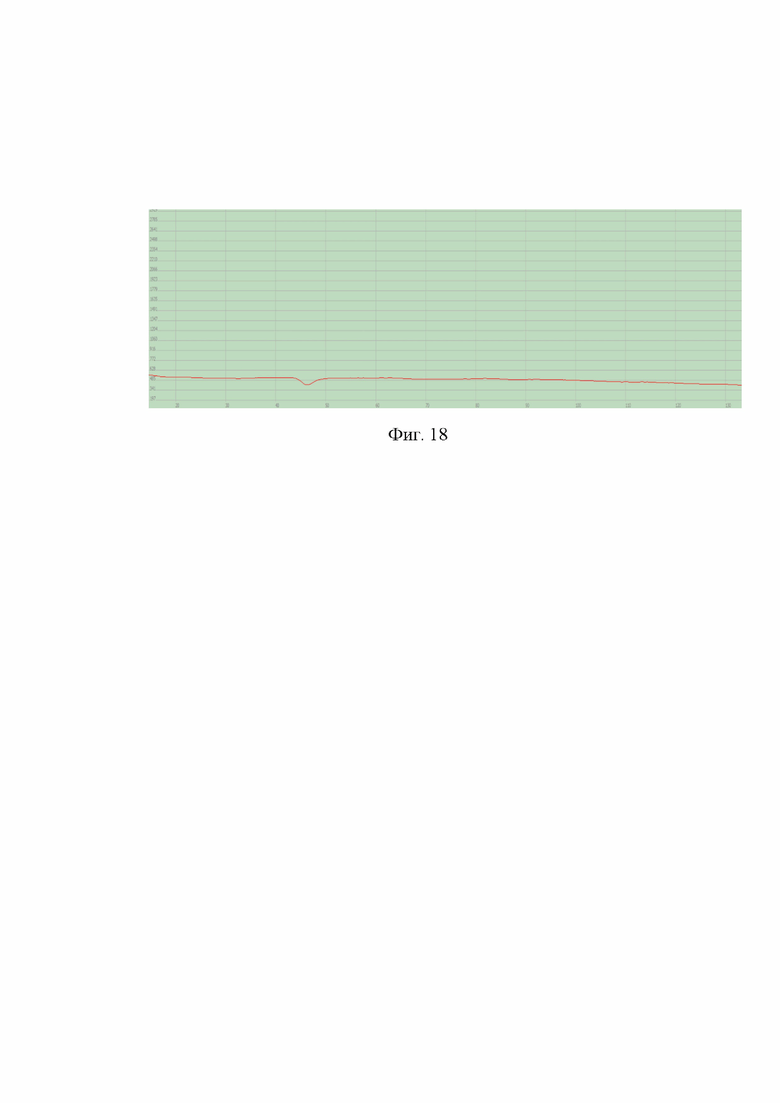
Хроматограммы апробации представлены на фиг. 16-18.
На фиг. 16 представлена хроматограмма апробации образца для контроля «Азота закись ОСЧ» на первом канале, на котором происходит разделение пиков закиси азота (N2O) и примесей углекислого газа (СО2), неконденсирующихся газов одним неразрешенным пиком.
На фиг. 17 - хроматограмма апробации образца для контроля «Азота закись ОСЧ» на третьем канале, на котором происходит разделение примесей кислорода (О2) и азота (N2).
На фиг. 18 - хроматограмма апробации образца для контроля «Азота закись ОСЧ» на втором канале, на котором происходит определение содержания примеси монооксида углерода (СО).
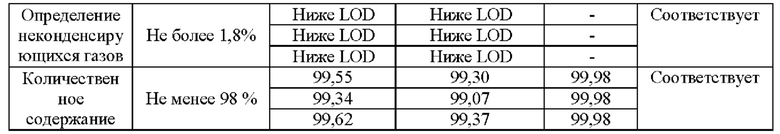
Как видно из сводной таблицы и хроматограмм анализируемый образец «Азота закись ОСЧ» в баллоне вместимостью 10 дм3, № партии 19403, произведенного ООО «Фессен Эм Ай И» соответствует требованиям. Количественное содержание азота закиси не менее 98%, содержание углерода монооксида не превышает 0,0005%), углерода диоксида не превышает 0,03%), неконденсирующихся газов не превышает 1,8%, водяных паров - 0,0041%.
Таким образом, преимущество предложенного способа заключается в том, что с его применением возможно провести испытания по пяти показателям одновременно (подлинность, количественное определение, содержание примесей угарного и углекислого газов, неконденсирующихся газов). Более того применение данного способа позволяет существенно сократить время анализа и расход пробы. Также благодаря использованию портативного газового хроматографа возможно проведение испытаний в полевых условиях на базе передвижной лаборатории или непосредственно в медицинском учреждении (больнице).
| название | год | авторы | номер документа |
|---|---|---|---|
| СИСТЕМА И СПОСОБ ДЛЯ ИЗМЕРЕНИЯ И КОЛИЧЕСТВЕННОГО АНАЛИЗА КИСЛОРОДА И ПРИМЕСЕЙ, СОДЕРЖАЩИХСЯ В КИСЛОРОДЕ МЕДИЦИНСКОМ ГАЗООБРАЗНОМ | 2022 |
|
RU2797786C1 |
| СПОСОБ ГАЗОХРОМАТОГРАФИЧЕСКОГО ОПРЕДЕЛЕНИЯ ЗАКИСИ АЗОТА В ГАЗАХ | 2003 |
|
RU2226688C1 |
| Способ определения содержания кислорода в газах | 1990 |
|
SU1772707A1 |
| Газохроматографический способ раздельного определения окислов азота в газовой смеси | 1981 |
|
SU965998A1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ ЗАКИСИ АЗОТА | 2002 |
|
RU2214863C1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ ЗАКИСИ АЗОТА | 2002 |
|
RU2214862C1 |
| КАТАЛИЗАТОР ПОЛУЧЕНИЯ ЗАКИСИ АЗОТА И СПОСОБ | 2002 |
|
RU2212934C1 |
| НЕПОДВИЖНАЯ ФАЗА ДЛЯ ГАЗОВОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2570705C1 |
| КАТАЛИЗАТОР ПОЛУЧЕНИЯ ЗАКИСИ АЗОТА И СПОСОБ | 2002 |
|
RU2214306C1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ ЗАКИСИ АЗОТА | 2002 |
|
RU2213615C1 |


Изобретение относится к аналитической химии, а именно к анализу медицинского газа «Азота закись, газ сжатый». Способ определения закиси азота, углерода монооксида, углерода диоксида, кислорода, азота в лекарственном препарате «азота закись, газ сжатый» включает отбор пробы для анализа, проведение анализа содержащихся закиси азота, углерода монооксида, углерода диоксида, неконденсирующихся газов: кислорода и азота при помощи портативного хроматографа, проведение расчёта содержания закиси азота по методу внешнего стандарта или по методу многоточечной градуировочной зависимости для основного вещества и всех примесей, используя площади хроматографических пиков, или расчетным способом. В качестве портативного газового хроматографа используется газовый хроматограф, имеющий три независимых канала. Первый канал включает планарную микрофлюидную хроматографическую колонку 1 м × 1 мм, заполненную сорбентом Carboxen 1000, и детектор по теплопроводности с газом-носителем гелием для разделения неразрешенного пика неконденсирующихся газов, углерода диоксида и закиси азота. Второй канал включает планарную микрофлюидную хроматографическую колонку 2 м × 1 мм, заполненную сорбентом Carboxen 1000, и детектор термохимический с газом-носителем воздухом для определения углерода монооксида. Третий канал включает планарную микрофлюидную хроматографическую колонку 2 м × 1 мм, заполненную сорбентом NaX, и детектор по теплопроводности с газом-носителем гелием для разделения кислорода и азота, при этом хроматографирование по всем трем каналам производится одновременно. Техническим результатом является возможность проведения испытания по пяти показателям одновременно стационарно и в условиях передвижной лаборатории, а также сокращение времени анализа и расхода пробы. 18 ил., 18 табл.
Способ определения закиси азота, углерода монооксида, углерода диоксида, кислорода, азота в лекарственном препарате «азота закись, газ сжатый», включающий отбор пробы для анализа, проведение анализа содержащихся закиси азота, углерода монооксида, углерода диоксида, неконденсирующихся газов: кислорода и азота при помощи портативного хроматографа, проведение расчёта содержания закиси азота по методу внешнего стандарта или по методу многоточечной градуировочной зависимости для основного вещества и всех примесей, используя площади хроматографических пиков, или расчетным способом, при этом в качестве портативного газового хроматографа используется газовый хроматограф, имеющий три независимых канала, где первый канал включает планарную микрофлюидную хроматографическую колонку 1 м × 1 мм, заполненную сорбентом Carboxen 1000, и детектор по теплопроводности с газом-носителем гелием для разделения неразрешенного пика неконденсирующихся газов, углерода диоксида и закиси азота, второй канал включает планарную микрофлюидную хроматографическую колонку 2 м × 1 мм, заполненную сорбентом Carboxen 1000, и детектор термохимический с газом-носителем воздухом для определения углерода монооксида, третий канал включает планарную микрофлюидную хроматографическую колонку 2 м × 1 мм, заполненную сорбентом NaX, и детектор по теплопроводности с газом-носителем гелием для разделения кислорода и азота, при этом хроматографирование по всем трем каналам производится одновременно.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СПОСОБ ГАЗОХРОМАТОГРАФИЧЕСКОГО ОПРЕДЕЛЕНИЯ ЗАКИСИ АЗОТА В ГАЗАХ | 2003 |
|
RU2226688C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОБЪЕМНОЙ ДОЛИ ОКСИДА АЗОТА (I) В ГАЗОВЫХ СМЕСЯХ | 2003 |
|
RU2255333C1 |
| СПОСОБ ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ОКСИДОВ АЗОТА | 1992 |
|
RU2035040C1 |

