Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к области органического химического синтеза фармацевтических промежуточных соединений, в частности к способу синтеза ингибиторов натрий-зависимого котранспортера глюкозы (SGLT) с использованием самостоятельно разработанных и инновационных промежуточных соединений.
Предшествующий уровень техники настоящего изобретения
Сахарный диабет представляет собой одно из основных заболеваний, угрожающих здоровью человека в 21 веке. Согласно оценкам, в 2019 году 116 миллионов взрослых в Китае страдают диабетом, и пациенты также могут подвергаться риску развития опасных для жизни осложнений. Из этой оценочной популяции, составляющей 116 миллионов человек, более 65 миллионов человек не диагностированы и, таким образом, подвергаются чрезвычайно высокому риску. Подсчитано, что во всем мире 463 миллиона взрослых страдают диабетом, и из них только на территориях западного побережья Тихого океана проживают 163 миллиона человек.
Ингибиторы натрий-зависимого котранспортера глюкозы (SGLT) представляют собой новый класс пер оральных препаратов для лечения диабета, которые производят гипогликемическое действие за счет увеличения экскреции глюкозы с мочой.
В публикации патента США №2015/0152075 раскрыты производные дифенилметана, которые проявляют ингибирующую активность в отношении SGLT, оказываются которые эффективными при лечении диабета и значительно снижают выделение глюкозы с мочой у животных по сравнению с основным лекарственным средством дапаглифлозином.
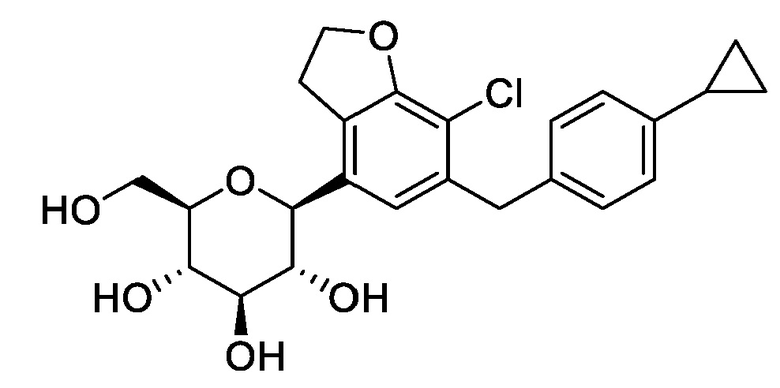
Структурная формула производных дифенилметана, раскрытых в литературе
В патенте КНР № CN 109311861 A раскрыт способ получения вышеуказанного соединения и его ключевых промежуточных соединений с применением способа, который представлен ниже.

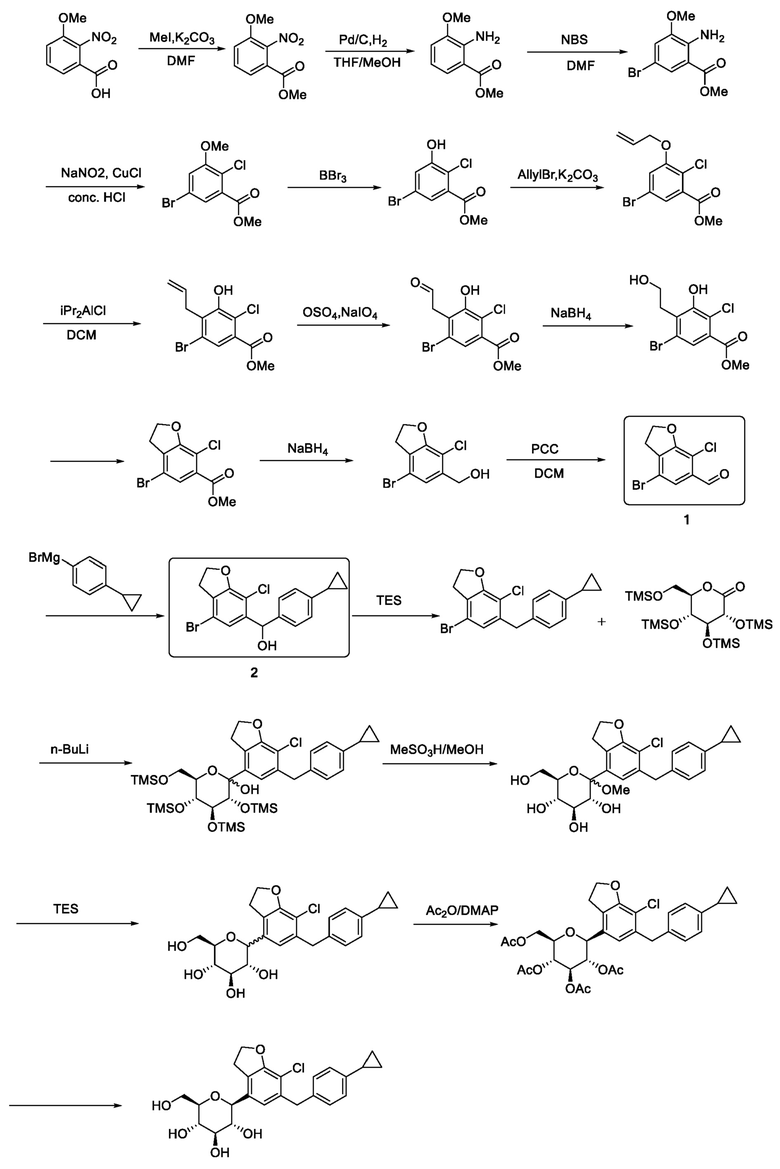
В соответствии с описанным выше способом требуется 22-стадийная реакция от исходного материала до производного дифенилметана, а также 14 и 15 стадий, соответственно, от исходного материала до ключевых промежуточных соединений, соединений 1 и 2, со сложными общими операциями и высокая стоимость производства, поэтому этот способ вряд ли сможет удовлетворить потребности фармацевтической промышленности.
Краткое раскрытие настоящего изобретения
Что касается характеристик длинного пути синтеза, сложных операций и высокой стоимости производства существующих производных дифенилметана, согласно настоящему изобретению предложен улучшенный способ получения соединения 1 и соединения 2.
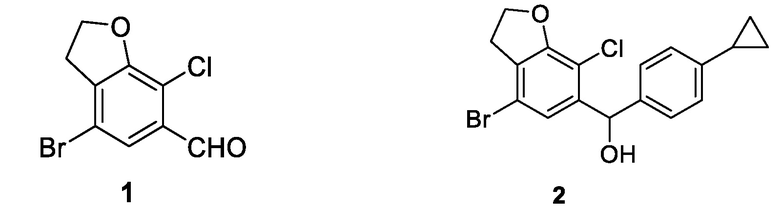
Согласно настоящему изобретению предложен способ получения соединения 1 и соединения 2, включающий следующие стадии.
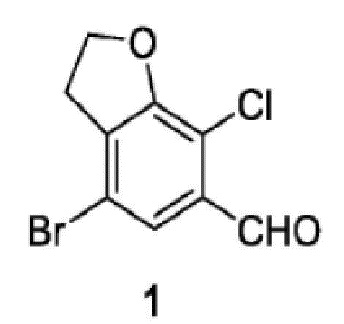
Соединение 1:
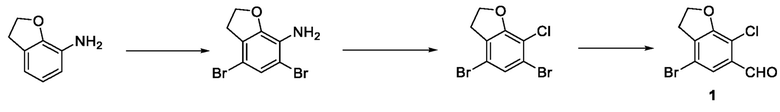
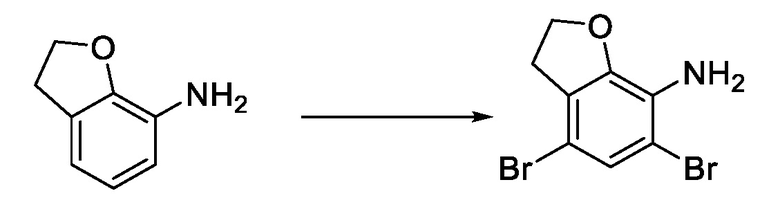
стадия 1-1: применение 2,3-дигидробензофуран-7-амина в качестве исходного материала и проведение селективного дибромирования с использованием бромирующего реагента и получением 4,6 дибром-2,3-дигидробензофуран-7-амина;
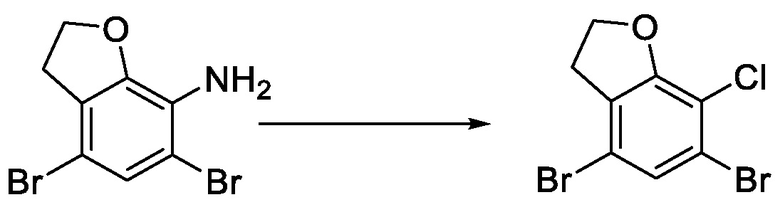
стадия 1-2: проведение реакции хлорирования 4,6-дибром-2,3-дигидробензофуран-7-амина по Зандмайеру с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана;
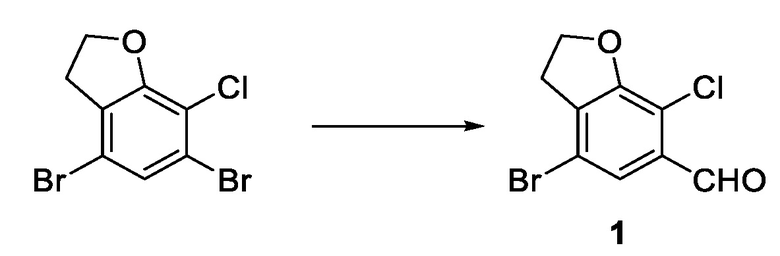
стадия 1-3: селективное удаление брома из 4,6-дибром-7-хлор-2,3-дигидробензофурана путем селективного использования сильного основания с последующим добавлением формилирующего реагента и получением соединения 1.
Способ синтеза может быть представлен следующим образом.
Соединение 2:
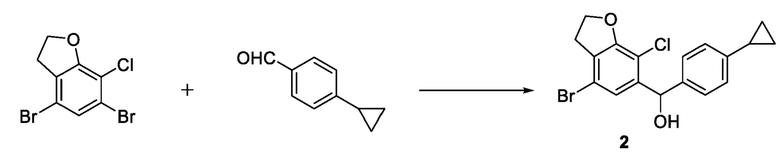
стадия 2-1: селективное удаление брома из 4,6-дибром-7-хлор-2,3-дигидробензофурана с использованием сильного основания последующим взаимодействием с 4-циклопропилбензальдегидом и получением соединения 2.
Способ синтеза может быть представлен следующим образом.
При этом на стадии 1-1 бромирующий реагент содержит одно вещество из брома, N-бромсукцинимида (NBS) и дибромгидантоина, предпочтительно N-бромсукцинимид. Селективное дибромирование проводят при температуре реакции в диапазоне от 10°С до 20°С.
В варианте осуществления настоящего изобретения стадия 1-1 может включать: фильтрование после дибромирования, добавление отфильтрованного осадка к смешанному растворителю, состоящему из воды и этил ацетата, последующее добавление в капельном режиме раствора гидроксида натрия для доведения значения рН до диапазона от 8 до 10, отделение и высушивание органического слоя над безводным сульфатом натрия и концентрирование остатка при пониженном давлении с получением 4,6-дибром-2,3-дигидробензофуран-7-амина.
Кроме того, на стадии 1-2 реагент, используемый в реакции Зандмайера, содержит одну систему из системы хлористоводородной кислоты, нитрита натрия и хлорида меди(1), системы изоамилнитрита и хлорида меди(11) и системы трет-бутилнитрита и хлорида меди (II).
В варианте осуществления настоящего изобретения стадия 1-2 может включать: добавление 4,6-дибром-2,3-дигидробензофуран-7-амина и концентрированной хлористоводородной кислоты в реакционную колбу и охлаждение до температуры 0-5°С; добавление в капельном режиме раствора нитрита натрия при поддержании постоянной температуры; после добавления в капельном режиме проведение реакции в течение 20-60 минут при поддержании постоянной температуры; затем добавление порциями хлорида меди(1) и проведение реакции в течение 1-3 часов при комнатной температуре; экстрагирование этилацетатом, промывание органического слоя раствором гидроксида натрия и концентрирование с получением неочищенного продукта; и разделение и очистку неочищенного продукта с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана.
Кроме того, на стадии 1 -3 сильное основание содержит одно или два соединения из н-бутиллития, трет-бутиллития и изопропилмагнийхлорида, предпочтительно изопропилмагнийхлорид. Изопропилмагнийхлорид является предпочтительным вследствие его высокой безопасности.
Кроме того, на стадии 1-3 формилирующий реагент содержит одно соединение из N,N-диметилформамида, N-формилморфолина и этилформиата, предпочтительно N,N-диметилформамид.
В варианте осуществления настоящего изобретения стадия 1-3) может включать: помещение в реакционную колбу 4,6-дибром-7-хлор-2,3-дигидробензофурана и тетрагидрофурана, затем добавление в капельном режиме раствора изопропилмагнийхлорида в тетрагидрофуране в атмосфере азота; после добавления в капельном режиме перемешивание в течение 0,5-1,5 часов при поддержании постоянной температуры; затем добавление в капельном режиме N,N-диметилформамида при поддержании постоянной температуры и продолжение реакции в течение 20-60 минут при поддержании постоянной температуры; выливание реакционного раствора в хлористоводородную кислоту, экстрагирование этилацетатом, высушивание органического слоя над безводным сульфатом натрия; и концентрирование и разделение с получением соединения 1.
Кроме того, на стадии 2-1 сильное основание содержит одно или два соединения из н-бутиллития, трет-бутиллития и изопропилмагнийхлорида, предпочтительно изопропилмагнийхлорид.
В варианте осуществления настоящего изобретения стадия 2-1) может включать: помещение 4,6-дибром-7-хлор-2,3-дигидробензофурана и тетрагидрофурана в реакционную колбу, охлаждение до температуры -30°С в атмосфере азота; добавление в капельном режиме раствора изопропилмагнийхлорида в тетрагидрофуране с последующим добавлением в капельном режиме раствора н-бутиллития в н-гексане; после добавления в капельном режиме перемешивание в течение 20-60 минут при поддержании постоянной температуры; затем добавление в капельном режиме раствора 4-циклопропилбензальдегида в тетрагидрофуране при поддержании постоянной температуры; после добавления в капельном режиме продолжение реакции в течение 20-60 минут при поддержании постоянной температуры; выливание реакционного раствора в хлористоводородную кислоту, экстрагирование этилацетатом и высушивание органического слоя над безводным сульфатом натрия; а затем концентрирование, разделение и очистку с получением соединения 2.
По сравнению с существующими способами синтеза настоящее изобретение имеет следующие преимущества:
1. Способ получения соединения 1 и соединения 2 согласно настоящему изобретению обеспечивает общий выход более 50% и имеет характеристики короткого пути, относительно мягких условий реакции и т.п.
2. Способ синтеза, предложенный согласно настоящему изобретению, характеризуют применение легкодоступных исходных материалов, низкая стоимость, отсутствие специальных операций для процесса и низкие требования к оборудованию, и, таким образом, он является подходящим для крупномасштабного промышленного производства.
Подробное раскрытие настоящего изобретения
Ниже подробно описаны предпочтительные варианты осуществления настоящего изобретения, чтобы специалистам в данной области техники было легче понять преимущества и признаки настоящего изобретения и, таким образом, более четко определить объем патентной охраны настоящего изобретения.
Пример 1: 4,6-дибром-2,3-дигидробензофуран-7-амина
В реакционную колбу объемом 1000 мл помещали 8,7 г 2,3-дигидробензофуран-7-амина и 200 мл этилацетата, а затем в капельном режиме добавляли 41,4 г брома при контролируемой температуре от 10°С до 20°С, при этом количество добавляемого брома было оптимизировано экспериментально. Если добавить чрезмерно малое количество брома, реакция будет неполной, а если добавить чрезмерно большое количество брома, система станет более сложной, и будет производиться больше побочных продуктов. Температуру реакции контролировали, главным образом, для предотвращения увеличения количества побочных продуктов. После завершения добавления в капельном режиме систему фильтровали. Отфильтрованный осадок добавляли к смеси растворителей, состоящей из 100 мл воды и 100 мл этилацетата, с последующим добавлением в капельном режиме раствора 10% гидроксида натрия для доведения значения рН до уровня от 8 до 10, при котором система является кислой после реакции, а некоторые продукты образуют соли. Значение рН доводили до уровня от 8 до 10 для высвобождения солей. Если значение рН является чрезмерно низким, высвобождение солей происходит не полностью; а если значение рН является чрезмерно высоким, будет израсходовано большее количество щелочи. После этого органический слой отделяли, сушили безводным сульфатом натрия и концентрировали при пониженном давлении с получением 4,6-дибром-2,3-дигидробензофуран-7-амина (15,73 г, выход 83,4%) в виде белого твердого вещества.
Пример 2: синтез 4,6-дибром-2,3-дигидробензофуран-7-амина
В реакционную колбу объемом 100 мл помещали 5 г 2,3-дигидробензофуран-7-аминаи 100 мл N,N-диметилформамида, охлаждали до температуры от -5°С до 0°С, после чего в капельном режиме порциями добавляли 13,17 г N-бромсукцинимида и осуществляли реакцию в процессе перемешивания в течение 30 минут, при этом N-бромсукцинимид, представляющий собой твердое вещество, имеет преимущества легкого расчета и контроля, в то время как бром представляет собой коррозионную жидкость, раздражающую и неудобную для взвешивания и использования. После этого реакционный раствор добавляли к 200 мл воды и дважды экстрагировали этилацетатом. Органические фазы объединяли, промывали раствором 10% гидроксида натрия, сушили безводным сульфатом натрия и концентрировали при пониженном давлении с получением 4,6-дибром-2,3-дигидробензофуран-7-амина (8,47 г, выход 78,2%).
Пример 3: синтез 4,6-дибром-7-хлор-2,3-дигидробензофурана
В реакционную колбу объемом 250 мл помещали 10 г 4,6-дибром-2,3-дигидробензофуран-7-амина и 50 мл концентрированной хлористоводородной кислоты, охлаждали до температуры 0-5°С и в капельном режиме добавляли раствор 2,47 г нитрита натрия в 8 мл воды при поддерживаемой температуре. После завершения добавления в капельном режиме система реагировала в течение 30 минут при поддерживаемой температуре, а затем порциями добавляли 6,76 г хлорида меди(I), нагревали до комнатной температуры и осуществляли реакцию в течение 2 часов. После этого добавляли этилацетат для экстракции, органический слой промывали раствором 5% гидроксида натрия и концентрировали с получением неочищенного продукта, который подвергали колоночной хроматографии на силикагеле с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана (9,01 г, выход 84,5%) в виде белого твердого вещества.
Пример 4: синтез 4,6-дибром-7-хлор-2,3-дигидробешофурана
В реакционную колбу объемом 250 мл помещали 10 г 4,6-дибром-2,3-дигидробензофуран-7-амина, 100 мл ацетонитрила и 9,18 г хлорида меди(II), нагревали до температуры 50°С и в капельном режиме добавляли 8,0 г изоамилнитрита. После завершения добавления в капельном режиме система реагировала в течение 30 минут при поддерживаемой температуре. После этого систему охлаждали до комнатной температуры и концентрировали для удаления растворителя с последующим добавлением этилацетата и фильтрованием. Раствор фильтрата промывали раствором 5% гидроксида натрия и концентрировали с получением неочищенного продукта, который подвергали колоночной хроматографии на силикагеле с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана (8,78 г, выход 82,3%) в виде белого твердого вещества.
Пример 5: синтез соединения 1
В реакционную колбу объемом 100 мл помещали 3,5 г 4,6-дибром-7-хлор-2,3-дигидробензофурана и 35 мл тетрагидрофурана. В капельном режиме добавляли 18 мл раствора 2,5 М изопропилмагнийхлорида в тетрагидрофуране в атмосфере азота. После завершения добавления в капельном режиме систему перемешивали в течение 1 часа при поддерживаемой температуре. Добавляли по каплям 5 мл N,N-диметилформамида при поддерживаемой температуре и затем выдерживали при поддерживаемой температуре для осуществления реакции в течение 30 минут. После этого реакционный раствор добавляли в раствор 3 М хлористоводородной кислоты и экстрагировали этилацетатом. Органический слой сушили безводным сульфатом натрия, концентрировали и подвергали колоночной хроматографии с получением соединения 1 (2,73 г, выход 93,2%) в виде белого твердого вещества.
Пример 6: синтез соединения 2
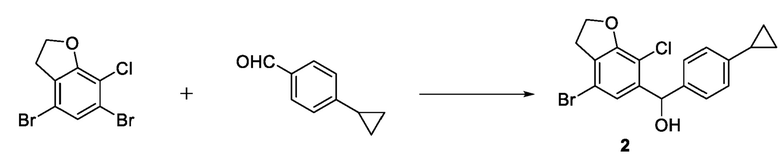
В реакционную колбу объемом 100 мл помещали 3,5 г 4,6-дибром-7-хлор-2,3-дигидробензофурана и 35 мл тетрагидрофурана и охлаждали до температуры -30°С в атмосфере азота. В капельном режиме добавляли 2,25 мл раствора 2,5 М изопропилмагнийхлорида в тетрагидрофуране, затем в капельном режиме добавляли 4,6 мл раствора 2,5 М н-бутиллития в н-гексане. После завершения добавления в капельном режиме систему перемешивали в течение 30 минут при поддерживаемой температуре, а затем добавляли 10 мл раствора 1,96 г 4-циклопропилбензальдегида в тетрагидрофуране при поддерживаемой температуре. После завершения добавления по каплям система реагировала в течение 30 минут при поддерживаемой температуре. Затем реакционный раствор добавляли в раствор 3 М хлористоводородной кислоты и экстрагировали этилацетатом. Органический слой сушили безводным сульфатом натрия, концентрировали и подвергали колоночной хроматографии с получением соединения 2 (3,70 г, выход 86,9%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ПРОИЗВОДНЫХ АМИНА | 2005 |
|
RU2385318C2 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ПРИМЕНЯЕМОЕ ДЛЯ СИНТЕЗА ИНГИБИТОРА SGLT, И СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА SGLT С ПРИМЕНЕНИЕМ УКАЗАННОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2021 |
|
RU2802443C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2014326C1 |
| ПРОИЗВОДНЫЕ АМИНОКУМАРАНА ИЛИ ИХ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ОБРАЗОВАНИЯ ЛИПОПЕРОКСИДА | 1992 |
|
RU2087473C1 |
| НОВЫЕ АНТАГОНИСТЫ РЕЦЕПТОРОВ ДВОЙНОГО ДЕЙСТВИЯ (DARA) В ОТНОШЕНИИ РЕЦЕПТОРОВ AT1 И ETA | 2007 |
|
RU2425833C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АЛКОКСИ-3-ГИДРОКСИПИКОЛИНОВЫХ КИСЛОТ | 2017 |
|
RU2744834C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОПТЕРИДИНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2010 |
|
RU2559881C2 |
| Фунгицидная композиция | 1988 |
|
SU1836016A3 |
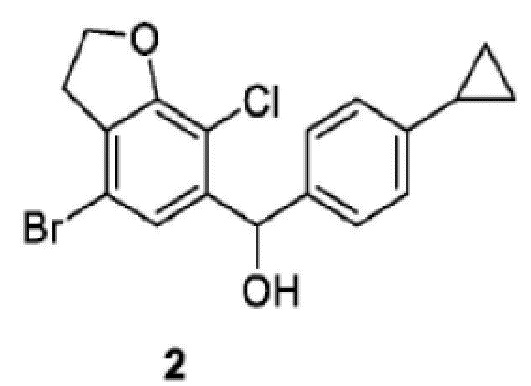
Изобретение относится к способу получения соединения 2, имеющего структуру, которая представлена ниже, который включает следующие стадии: стадия 1-1) селективное дибромирование 2,3-дигидробензофуран-7-амина в качестве исходного материала с применением бромирующего реагента и получением 4,6-дибром-2,3-дигидробензофуран-7-амина; стадия 1-2) введение 4,6-дибром-2,3-дигидробензофуран-7-амина, полученного на стадии 1-1), в реакцию Зандмайера для хлорирования с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана и стадия 2-1) селективное дебромирование 4,6-дибром-7-хлор-2,3-дигидробензофурана, полученного на стадии 1-2), с применением сильного основания, а затем взаимодействие с 4-циклопропилбензальдегидом с получением соединения 2. Изобретение также относится к способу получения соединения 1. Технический результат: разработан способ получения промежуточных соединений 1 и 2 с высоким выходом для получения ингибитора натрий-зависимого котранспортера глюкозы, характеризующийся мягкими условиями синтеза, меньшим количеством стадий и доступностью реактивов. 2 н. и 15 з.п. ф-лы, 6 пр.
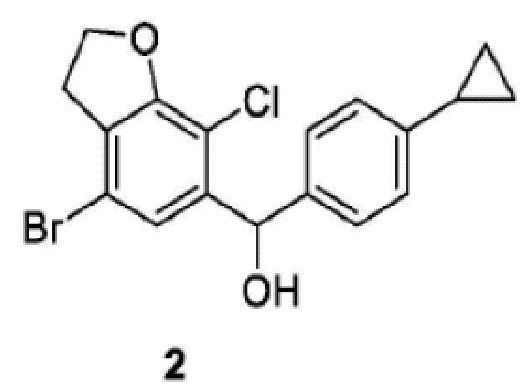
1. Способ получения соединения 2, имеющего структуру, которая представлена ниже, причем включает следующие стадии:
Стадия 1-1) селективное дибромирование 2,3-дигидробензофуран-7-амина в качестве исходного материала с применением бромирующего реагента и получением 4,6-дибром-2,3-дигидробензофуран-7-амина;
Стадия 1-2) введение 4,6-дибром-2,3-дигидробензофуран-7-амина, полученного на стадии 1-1), в реакцию Зандмайера для хлорирования с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана и
Стадия 2-1) селективное дебромирование 4,6-дибром-7-хлор-2,3-дигидробензофурана, полученного на стадии 1-2), с применением сильного основания, а затем взаимодействие с 4-циклопропилбензальдегидом с получением соединения 2.
2. Способ по п. 1, отличающийся тем, что на стадии 1-1) бромирующий реагент представляет собой по меньшей мере одно вещество из брома, N-бромсукцинимида (NBS) и дибромгидантоина.
3. Способ по п. 1, в котором селективное дибромирование проводят при температуре реакции в диапазоне от 10°С до 20°С.
4. Способ по п. 1, отличающийся тем, что на стадии 1-2) реагент, используемый в реакции Зандмайера, представляет собой по меньшей мере одну систему из системы хлористоводородной кислоты, нитрита натрия и хлорида меди(I), системы изоамилнитрита и хлорида меди(II) и системы трет-бутилнитрита и хлорида меди(II).
5. Способ по п. 1, отличающийся тем, что на стадии 2-1) сильное основание представляет собой по меньшей мере одно соединение из н-бутиллития, трет-бутиллития и изопропилмагнийхлорида.
6. Способ по п. 1, отличающийся тем, что стадия 1-1) включает: фильтрование после дибромирования, добавление отфильтрованного осадка к смешанному растворителю, состоящему из воды и этилацетата, последующее добавление в капельном режиме раствора гидроксида натрия для доведения значения рН до диапазона от 8 до 10, отделение и высушивание органического слоя над безводным сульфатом натрия и концентрирование остатка при пониженном давлении с получением 4,6-дибром-2,3-дигидробензофуран-7-амина.
7. Способ по п. 1, отличающийся тем, что стадия 1-2) включает: добавление 4,6-дибром-2,3-дигидробензофуран-7-амина и концентрированной хлористоводородной кислоты в реакционную колбу и охлаждение до температуры 0-5°С; добавление в капельном режиме раствора нитрита натрия при поддержании постоянной температуры; после добавления в капельном режиме проведение реакции в течение 20-60 минут при поддержании постоянной температуры; затем добавление порциями хлорида меди(I) и проведение реакции в течение 1-3 часов при комнатной температуре; экстрагирование этилацетатом, промывание органического слоя раствором гидроксида натрия и концентрирование с получением неочищенного продукта; и разделение и очистку неочищенного продукта с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана.
8. Способ по п. 1, отличающийся тем, что, стадия 2-1) включает: помещение 4,6-дибром-7-хлор-2,3-дигидробензофурана и тетрагидрофурана в реакционную колбу, охлаждение до температуры -30°С в атмосфере азота, добавление в капельном режиме раствора изопропилмагнийхлорида в тетрагидрофуране с последующим добавлением в капельном режиме раствора н-бутиллития в н-гексане; после добавления в капельном режиме перемешивание в течение 20-60 минут при поддержании постоянной температуры; затем добавление в капельном режиме раствора 4-циклопропилбензальдегида в тетрагидрофуране при поддержании постоянной температуры; после добавления в капельном режиме продолжение реакции в течение 20-60 минут при поддержании постоянной температуры; выливание реакционного раствора в хлористоводородную кислоту, экстрагирование этилацетатом и высушивание органического слоя над безводным сульфатом натрия; а затем концентрирование, разделение и очистку с получением соединения 2.
9. Способ получения соединения 1, имеющего структуру, которая представлена ниже, причем включает следующие стадии:
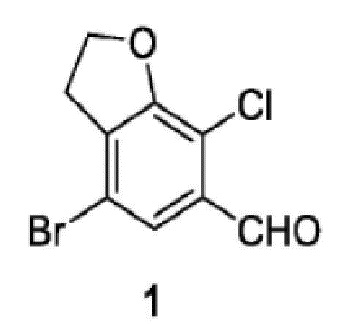
Стадия 1-1) селективное дибромирование 2,3-дигидробензофуран-7-амина в качестве исходного материала с применением бромирующего реагента и получением 4,6-дибром-2,3-дигидробензофуран-7-амина;
Стадия 1-2) введение 4,6-дибром-2,3-дигидробензофуран-7-амина, полученного на стадии 1-1), в реакцию Зандмайера для хлорирования с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана;
Стадия 1-3) селективное дебромирование 4,6-дибром-7-хлор-2,3-дигидробензофурана, полученного на стадии 1-2), с применением сильного основания, последующим добавлением формилирующего реагента и получением соединения 1.
10. Способ по п. 9, отличающийся тем, что на стадии 1-1) бромирующий реагент представляет собой по меньшей мере одно вещество из брома, N-бромсукцинимида (NBS) и дибромгидантоина.
11. Способ по п. 9, в котором селективное дибромирование проводят при температуре реакции в диапазоне от 10°С до 20°С.
12. Способ по п. 9, отличающийся тем, что на стадии 1-2) реагент, используемый в реакции Зандмайера, представляет собой по меньшей мере одну систему из системы хлористоводородной кислоты, нитрита натрия и хлорида меди(I), системы изоамилнитрита и хлорида меди(II) и системы трет-бутилнитрита и хлорида меди(II).
13. Способ по п. 9, отличающийся тем, что на стадии 1-3) сильное основание представляет собой по меньшей мере одно соединение из н-бутиллития, трет-бутиллития и изопропилмагнийхлорида.
14. Способ по п. 9, отличающийся тем, что, на стадии 1-3) формилирующий реагент представляет собой одно соединение из N,N-диметилформамида, N-формилморфолина и этилформиата.
15. Способ по п. 9, отличающийся тем, что стадия 1-1) включает: фильтрование после дибромирования, добавление отфильтрованного осадка к смешанному растворителю, состоящему из воды и этилацетата, последующее добавление в капельном режиме раствора гидроксида натрия для доведения значения рН до диапазона от 8 до 10, отделение и высушивание органического слоя над безводным сульфатом натрия и концентрирование остатка при пониженном давлении с получением 4,6-дибром-2,3-дигидробензофуран-7-амина.
16. Способ по п. 9, отличающийся тем, что стадия 1-2) включает: добавление 4,6-дибром-2,3-дигидробензофуран-7-амина и концентрированной хлористоводородной кислоты в реакционную колбу и охлаждение до температуры 0-5°С; добавление в капельном режиме раствора нитрита натрия при поддержании постоянной температуры; после добавления в капельном режиме проведение реакции в течение 20-60 минут при поддержании постоянной температуры; затем добавление порциями хлорида меди(I) и проведение реакции в течение 1-3 часов при комнатной температуре; экстрагирование этилацетатом, промывание органического слоя раствором гидроксида натрия и концентрирование с получением неочищенного продукта; и разделение и очистку неочищенного продукта с получением 4,6-дибром-7-хлор-2,3-дигидробензофурана.
17. Способ по п. 9, отличающийся тем, что стадия 1-3) включает: помещение в реакционную колбу 4,6-дибром-7-хлор-2,3-дигидробензофурана и тетрагидрофурана, затем добавление в капельном режиме раствора изопропилмагнийхлорида в тетрагидрофуране в атмосфере азота; после добавления в капельном режиме перемешивание в течение 0,5-1,5 часов при поддержании постоянной температуры; затем добавление в капельном режиме N,N-диметилформамида при поддержании постоянной температуры и продолжение реакции в течение 20-60 минут при поддержании постоянной температуры; выливание реакционного раствора в хлористоводородную кислоту, экстрагирование этилацетатом, высушивание органического слоя над безводным сульфатом натрия; и концентрирование и разделение с получением соединения 1.
| RU 2019101056 А3, 17.07.2020 | |||
| Якорь механический | 2019 |
|
RU2714032C1 |
| US 20140274918 A1, 18.09.2014 | |||
| US 8039441 B2, 18.10.2011. | |||

