ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет по следующим заявкам:
заявка на патент Китая 201810124494.2, поданная 7 февраля 2018 г.
заявка на патент Китая 201811361512.5, поданная 15 ноября 2018 г.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Предоставлены соединения в качестве ингибитора ATR и их применение для получения ингибитора ATR и, в частности, соединение формулы (I), его изомер или фармацевтически приемлемая соль.
УРОВЕНЬ ТЕХНИКИ
ATR (атаксия-телеангиэктазия мутированный и Rad3-родственный киназный белок) принадлежит к семейству (киназ, родственных по отношению к фосфатидилинозитол-3-киназе) PIKK и участвует в репарации повреждений ДНК для поддержания стабильности генов. Протеинкиназа ATR оказывает синергетический ответ на повреждение ДНК, репликативный стресс и нарушения клеточного цикла. ATR и ATM принадлежат к семейству PIKK серии/треониновых протеинкиназ, и являются общим компонентом клеточного цикла и восстановления повреждений ДНК; другими членами являются Chk1, BRCA1, р53. ATR активируется в основном в ответ на репликативный стресс ДНК (остановка вилки дупликации) и направлена на восстановление однонитевого разрыва.
Когда происходят разрыв двухцепочечной ДНК и остановка репликативной вилки, ATR активируется одноцепочечной структурой ДНК. ДНК-полимераза остается в процессе репликации ДНК, и репликативная геликаза продолжает раскрутку на ведущем конце репликативной вилки ДНК, что приводит к образованию длинной одноцепочечной ДНК (оцДНК), и затем происходит связь одноцепочечной ДНК с RPA (репликативным белком А). В месте повреждения при репликативном стрессе или повреждении ДНК RPA рекрутирует ATR/ATR действующий белковый комплекс, комплекс RPA-одноцепочечная ДНК активирует комплекс RAD17/rfc2-5, который связывается с участком повреждения, связь ДНК-оцДНК активирует гетеротример Rad9-HUS1-RAD1 (9-1-1), и 9-1-1, в свою очередь, рекрутирует TopBP1 для активации ATR. После активации ATR обеспечивает репарацию ДНК через нижестоящие мишени, стабилизацию и перезапуск остановленных репликативных вилок и временно остановленного клеточного цикла. Эти функции ATR опосредованы действием нисходящего целевого Chk1. ATR действует в качестве контрольной точки повреждения ДНК в клеточном цикле во время S-фазы. Он может опосредовать деградацию CDC25A с участием Chk1, тем самым задерживая процесс репликации ДНК и предоставляя время для восстановления репликативной вилки. ATR также является основным регулятором контрольной точки клеточного цикла G2/M, предотвращая преждевременное, до завершения репликации ДНК или повреждения ДНК, вхождение клеток в митоз. Эта ATR-зависимая остановка клеточного цикла G2/M в основном опосредуется двумя механизмами: 1. Деградацией CDC25A; 2. Chk1-опосредованным фосфорилированием Cdc25C для связывания 14-3-белка. Связывание Cdc25C с белком 14-3-3 способствует его экспорту из ядра и цитоплазматической изоляции, тем самым подавляя его способность к дефосфорилированию и активации ядерного Cdc2, что, в свою очередь, предотвращает вступление в митоз.
Мутации гена ATR очень редки, и только у нескольких пациентов с синдромом Секкеля имеются мутации гена ATR, которые характеризуются задержкой роста и микроцефалией. Нарушение путей, связанных с ATR, может привести к нестабильности генома, и белок ATR активируется большинством противораковых химиотерапевтических средств. Кроме того, дупликация гена ATR описана как фактор риска развития рабдомиосаркомы.
ATR важен для клеточной саморепликации и активируется в S-фазе для регуляции точки начала репликации и восстановления поврежденных репликативных вилок. Повреждение репликативных вилок может повысить чувствительность раковых клеток к противораковым агентам на основе платины и гидроксимочевины и снизить устойчивость раковых клеток. Следовательно, подавление ATR может стать эффективным методом лечения рака в будущем.
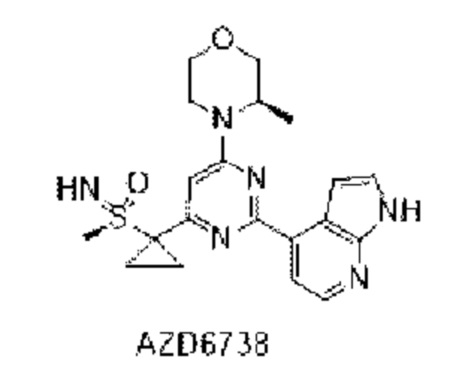
В WO 2011154737 раскрыто соединение AZD6738 в качестве ингибитора ATR, имеющего следующую структуру:
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
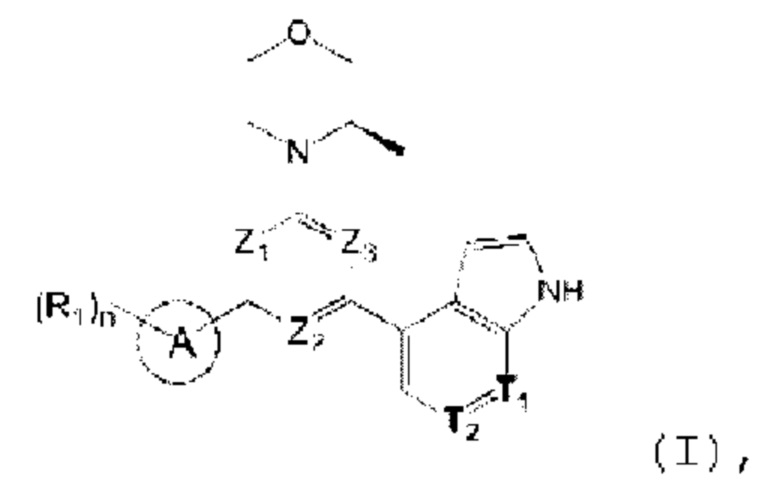
В одном из аспектов предоставлено соединение формулы (I) или его изомер или фармацевтически приемлемая соль
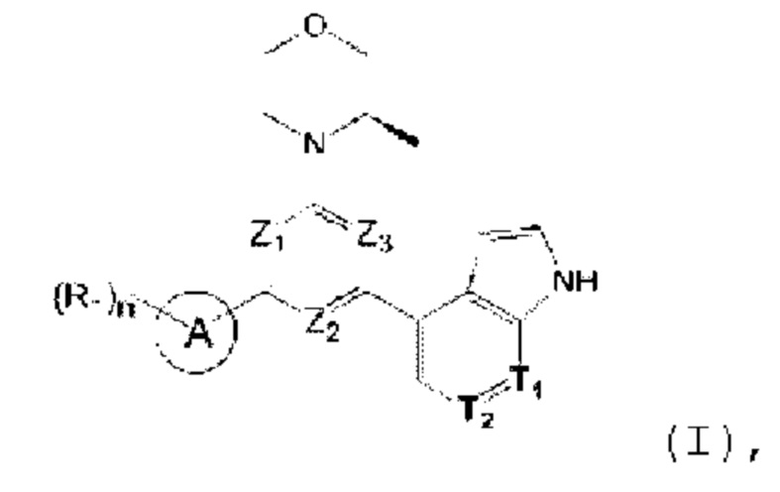
где
n равно 1, 2, 3 или 4;
каждый из Ζ1, Z2 и Z3 независимо выбирают из группы, состоящей из СН и N, и по меньшей мере один из Ζ1, Z2 и Z3 представляет собой N;
каждый из Τ1 и Τ2 независимо выбирают из группы, состоящей из С (R2) и N;
кольцо А выбирают из группы, состоящей из 5-6-членного гетероарила;
каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, С1-6 алкила, C1-6 алкокси и С3-6 циклоалкила, где С1-6 алкил, C1-6 алкокси и С3-6 циклоалкил необязательно замещены 1, 2 или 3 R;
каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СООН и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 R;
каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН, NH2, С1-3 алкила и C1-3 алкокси, где C1-3 алкил и С1-3 алкокси необязательно замещены 1, 2 или 3 R';
каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН и NH2;
5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
В некоторых вариантах осуществления изобретения каждый R независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СН3, Et и -О-СН3, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, C1-3 алкила, C1-3 алкокси и циклопропила, где C1-3 алкил, C1-3 алкокси и циклопропил необязательно замещены 1, 2 или 3 R, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СН3, CH2F, CHF2, CF3, Et, -CH2OH, -O-CH3,  а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СООН, СН3, Et и -СН2-ОН, а другие переменные определены в настоящем описании.
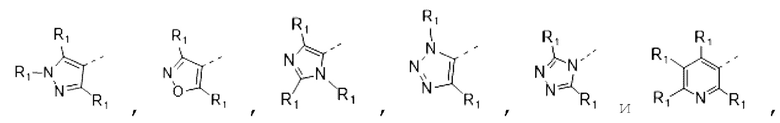
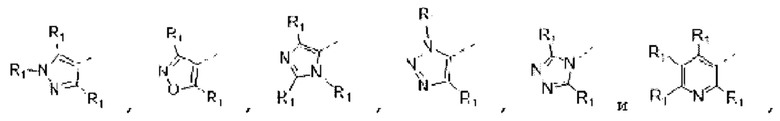
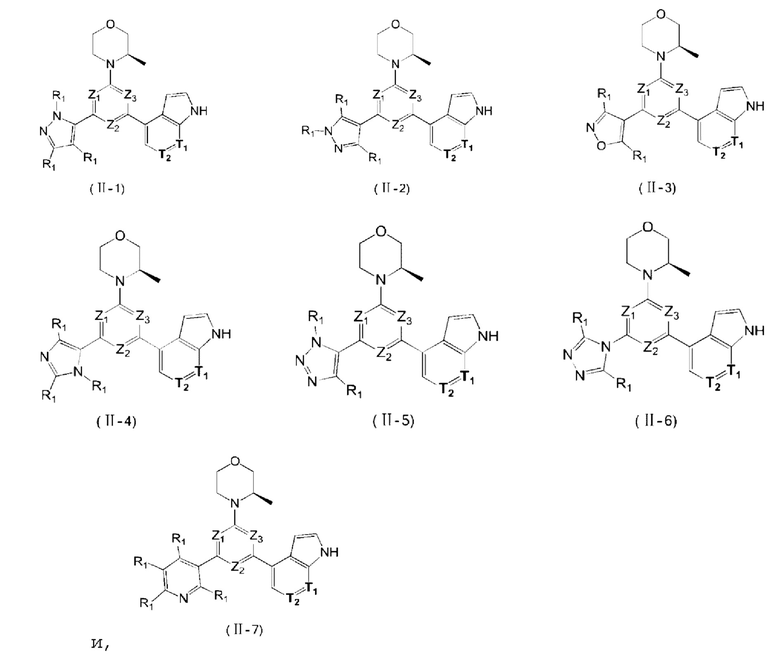
В некоторых вариантах осуществления изобретения кольцо А выбирают из группы, состоящей из пиразолила, изоксазолила, оксазолила, имидазолила, 1,2,3-триазолила, 1,2,4-триазолила и пиридила, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения кольцо А выбирают из группы, состоящей из 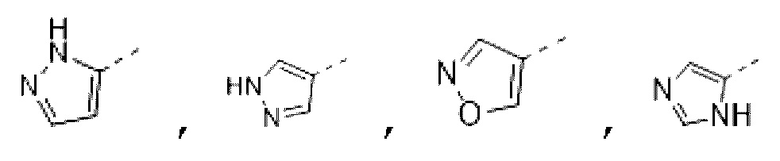
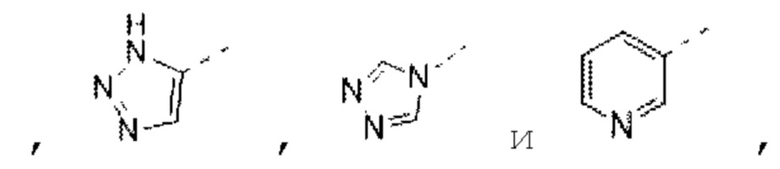 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
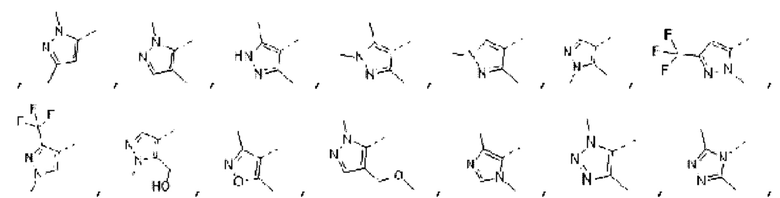
В некоторых вариантах осуществления изобретения структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 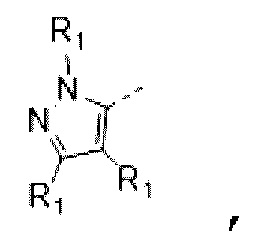
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
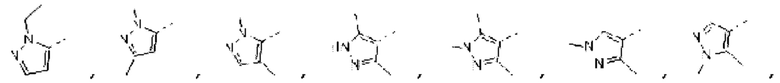
В некоторых вариантах осуществления изобретения структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
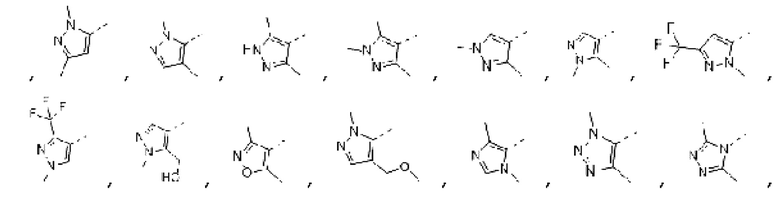
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
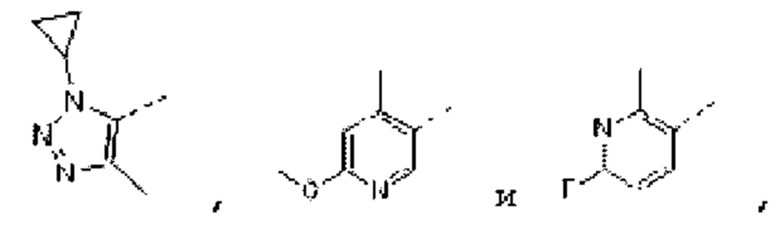
В некоторых вариантах осуществления изобретения структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
 а другие переменные определены в настоящем описании. В некоторых вариантах осуществления изобретения структурную единицу
а другие переменные определены в настоящем описании. В некоторых вариантах осуществления изобретения структурную единицу 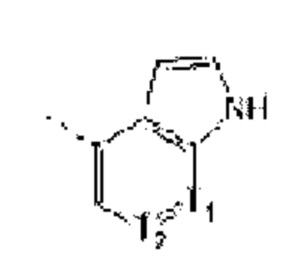 выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и 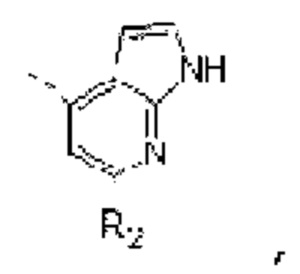 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
Также предоставлено соединение формулы (I) или его изомер или фармацевтически приемлемая соль 
где
n равно 1, 2, 3 или 4;
каждый из Ζ1, Z2, и Z3 независимо выбирают из группы, состоящей из СН и N, и по меньшей мере один из Ζ1, Z2 и Z3 представляет собой N;
каждый из Τ1 и Т2 независимо выбирают из группы, состоящей из C(R2) и N;
кольцо А выбирают из группы, состоящей из 5-6-членного гетероарила;
каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, C1-6 алкила, C1-6 алкокси и С3-6 циклоалкила, где C1-6 алкил, C1-6 алкокси и С3-6 циклоалкил необязательно замещены 1, 2 или 3 R;
каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 R;
каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН, NH2, C1-3 алкила и C1-3 алкокси, где C1-3 алкил и C1-3 алкокси необязательно замещены 1, 2 или 3 R';
каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН и NH2;
5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
В некоторых вариантах осуществления изобретения каждый R независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СН3, Et и -O-СН3, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, C1-3 алкила, C1-3 алкокси и циклопропила, где C1-3 алкил, C1-3 алкокси и циклопропил необязательно замещены 1, 2 или 3 R, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СН3, CH2F, CHF2, CF3, Et, -СН2ОН, -O-СН3, 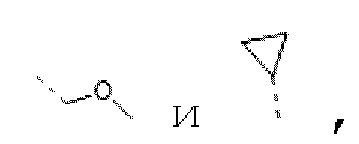 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2, СН3, Et и -СН2-ОН, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения кольцо А выбирают из группы, состоящей ив пиразолила, изоксазолила, оксазолила, имидазолила, 1,2,3-триазолила, 1,2,4-триазолила и пиридила, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения кольцо А
выбирают из группы, состоящей из 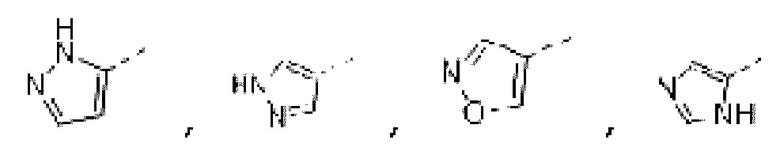
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу 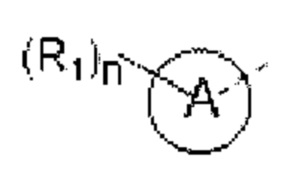 выбирают из группы, состоящей из
выбирают из группы, состоящей из 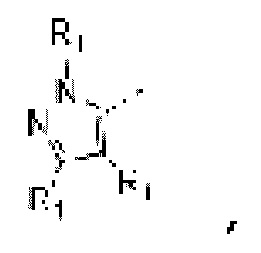
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу 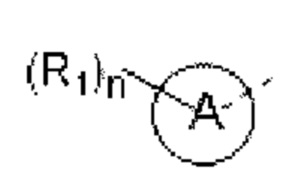 выбирают из группы, состоящей из
выбирают из группы, состоящей из 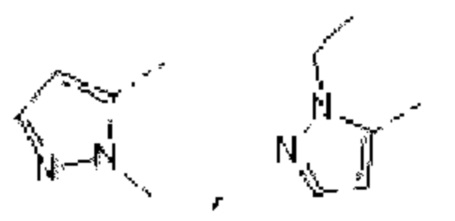
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу 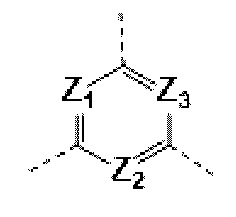 выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и  а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из  и
и  а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
Также предоставлено соединение формулы (I) или его изомер или фармацевтически приемлемая соль
где
n равно 1, 2, 3 или 4;
каждый из Ζ1, Z2 и Z3 независимо выбирают из группы, состоящей из СН и N, и по меньшей мере один из Ζ1, Z2 и Z3 представляет собой N;
каждый из Τ1 и Τ2 независимо выбирают из группы, состоящей из С(R2) и Ν;
кольцо А выбирают из группы, состоящей из 5-6 членного гетероарила;
каждый R1 независимо выбирают из группы, состоящей из Н, F, Сд, Br, I, ОН, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 R;
каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, NH2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 R;
каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, OH, ΝΗ2 и C1-3 алкила, где C1-3 алкил необязательно замещен 1, 2 или 3 R';
каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН и NH2;
5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбранные из группы, состоящей из -NH-, -O-, -S- и N.
Б некоторых вариантах осуществления изобретения каждый R независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, ΝΗ2, CH3 и Et, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, ΝΗ2, СН3, CH2F, CHF2, CF3, Et и -СН2ОН, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, ΝΗ2, CH3 и Et, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения кольцо А выбирают из группы, состоящей из пиразолила, изоксазолила, оксазолила и имидазолила, а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения кольцо А выбирают из группы, состоящей из  а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу 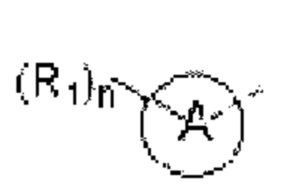 выбирают из группы, состоящей из
выбирают из группы, состоящей из 
 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу 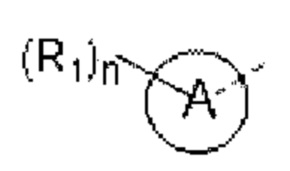 выбирают из группы, состоящей из
выбирают из группы, состоящей из 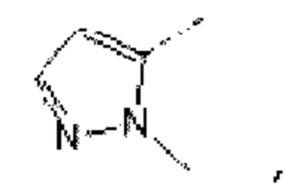
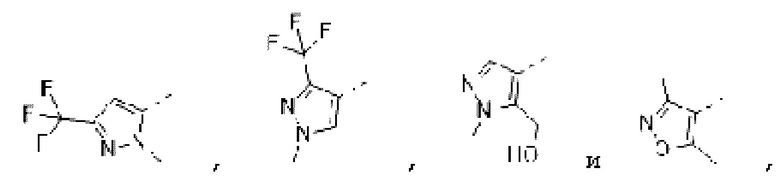 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения, структурную единицу 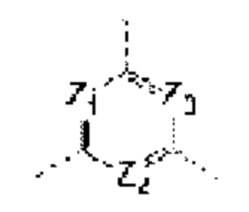 выбирают из группы, состоящей из
выбирают из группы, состоящей из 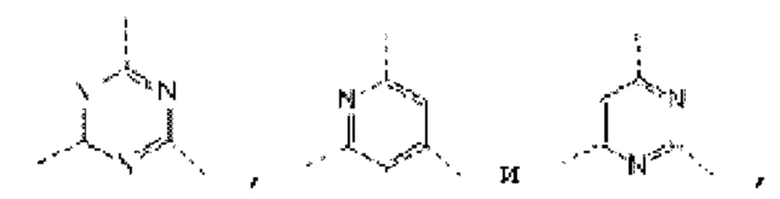 а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
В некоторых вариантах осуществления изобретения структурную единицу  выбирают из группы состоящей из
выбирают из группы состоящей из  и
и  а другие переменные определены в настоящем описании.
а другие переменные определены в настоящем описании.
Некоторые варианты осуществления настоящего раскрытия получены из комбинации вышеуказанных переменных.
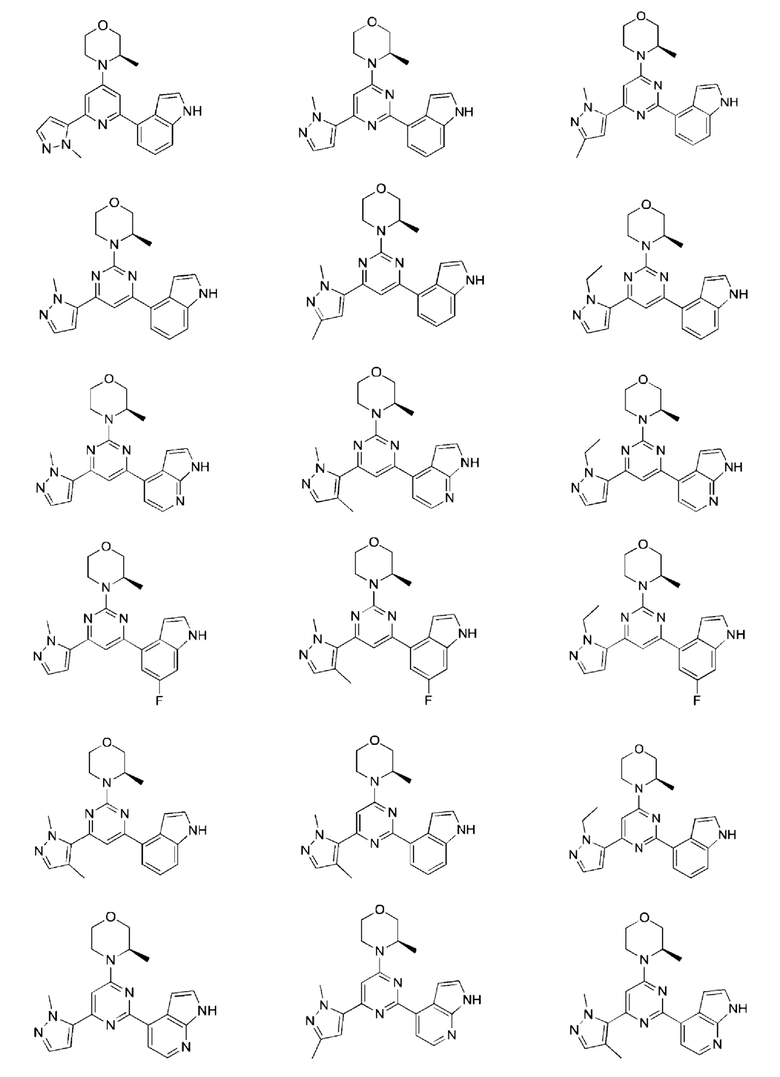
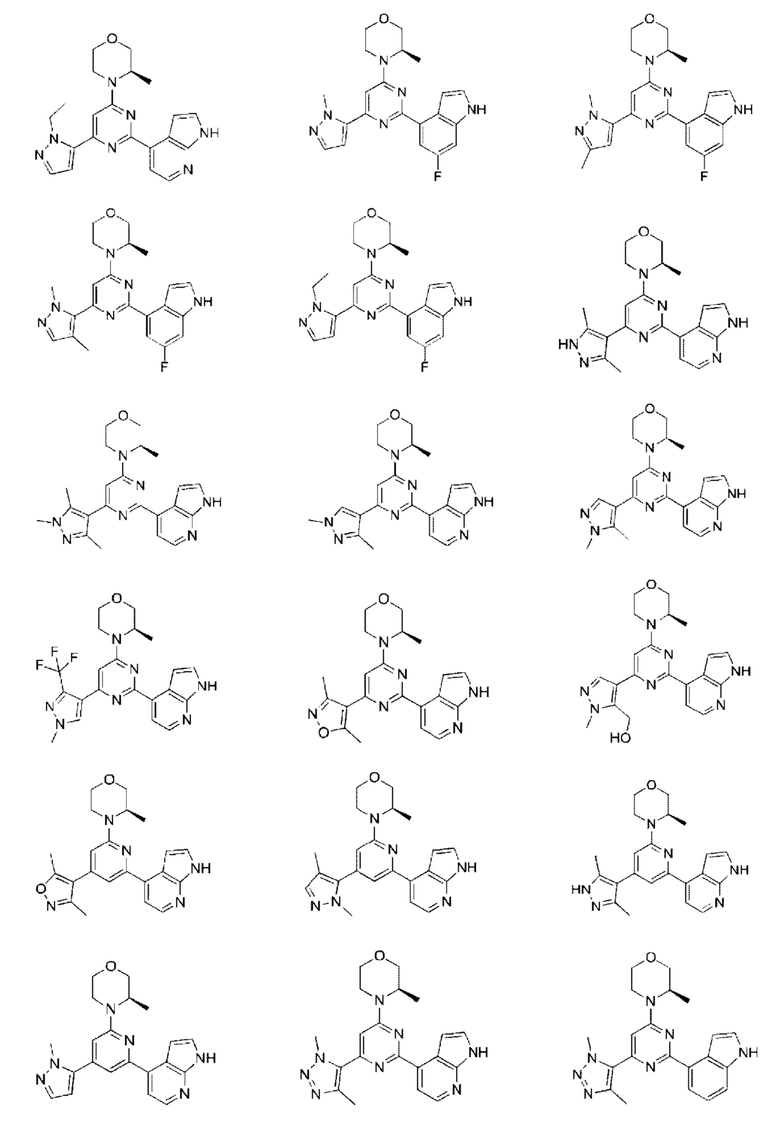
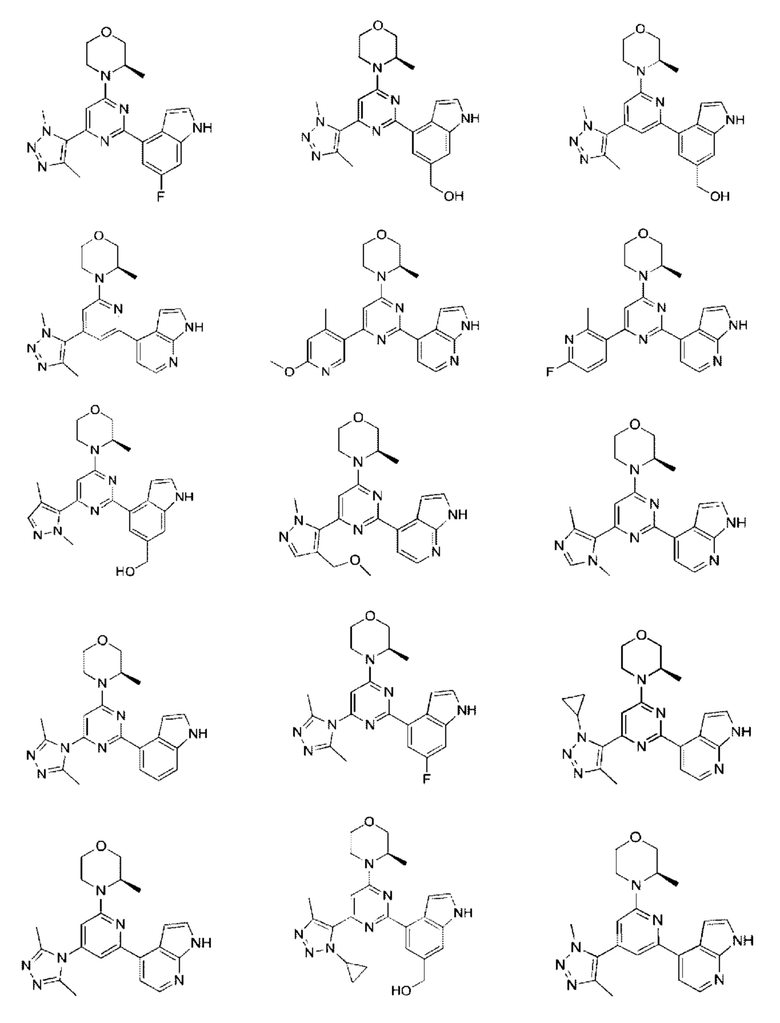
В некоторых вариантах осуществления изобретения предоставленное выше соединение или его изомер или фармацевтически приемлемую соль выбирают из:
где
каждый из Τ1 и Т2 независимо выбирают из группы, состоящей из С (R2) и N;
R1, R2, Ζ1, Z2 и Z3 являются такими, как определено в настоящем описании.
В некоторых вариантах осуществления изобретения предоставленное выше соединение или его изомер или фармацевтически приемлемую соль выбирают из: 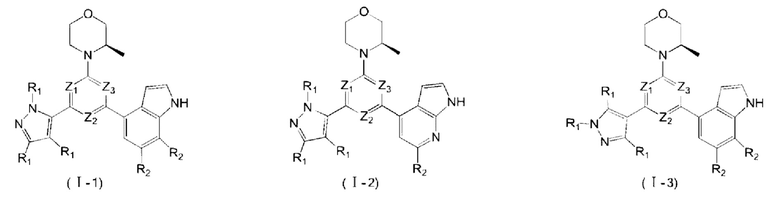
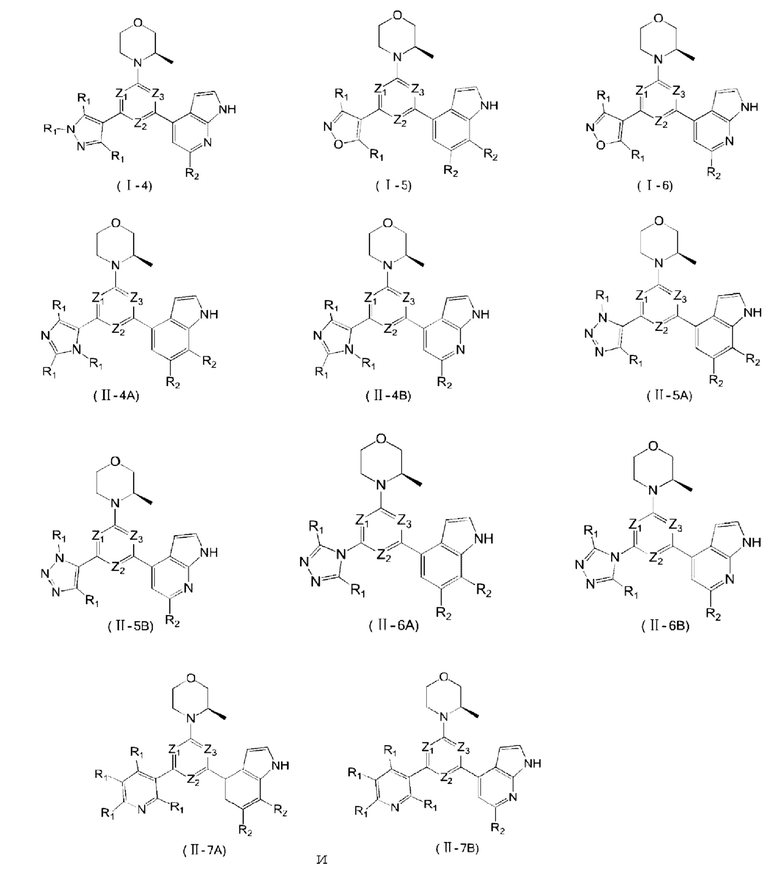
где
R1, R2, Ζ1, Z2 и Z3 являются такими, как определено в настоящем описании.
Представлено также следующее соединение или его изомер или фармацевтически приемлемая соль:
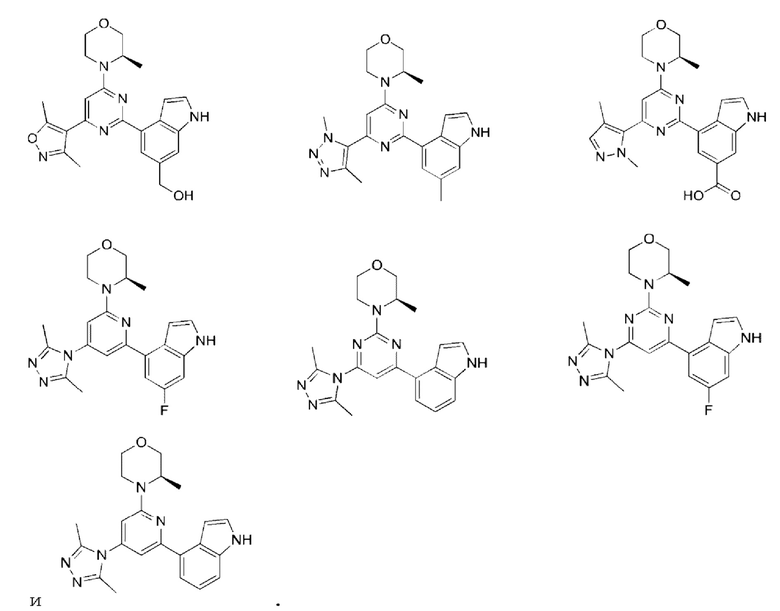
В другом аспекте предоставлено применение указанного выше соединения или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения ATR-ассоциированного заболевания.
В некоторых вариантах осуществления изобретения указанное лекарственное средство применяют для лечения солидной опухоли или гематологической опухоли.
ТЕХНИЧЕСКИЙ ЭФФЕКТ
В качестве нового ингибитора ATR, заявленные соединения проявляют хорошую ингибирующую активность в отношении ATR киназы. Более того, они демонстрируют хорошие эффекты подавления опухолей на животных моделях и могут применяться в качестве новых противоопухолевых агентов.
ОПРЕДЕЛЕНИЕ И ОПИСАНИЕ
Если не указано иное, приведенные ниже термины и фразы имеют следующие определения. Конкретный термин или фразу не следует рассматривать как неопределенную или неясную, если не приведено конкретное определение, и следует понимать в соответствии с обычным значением. Используемое в настоящем описании торговое наименование относится к соответствующему изделию или активному ингредиенту. Термин «фармацевтически приемлемый» означает, что с точки зрения надежной медицинской оценки соединения, материалы, композиции и/или лекарственная форма подходят для применения в контакте с тканями людей и животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, и имеют соразмерное разумное отношение польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают в результате взаимодействия соединения по настоящему изобретению, которое имеет конкретный заместитель, с относительно нетоксичной кислотой или основанием. Когда соединение по настоящему изобретению содержит относительно кислотную функциональную группу, аддитивная соль основания может быть получена путем контактирования нейтральной формы такого соединения с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые аддитивные соли оснований включают соли натрия, калия, кальция, аммония, органического амина или магния и т.п. Когда соединение по настоящему изобретению содержит относительно основную функциональную группу, аддитивная соль кислоты может быть получена путем контактирования нейтральной формы такого соединения с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых аддитивных солей кислот включают соли неорганических кислот, включая, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту, и т.д.; и соли органических кислот, включая, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту, метансульфоновую кислоту и т.д.; и также включают соли аминокислот (таких как аргинин и т.д.) и соли органических кислот, таких как глюкуроновая кислота. Некоторые конкретные соединения по настоящему изобретению содержат основные и кислотные функциональные группы, которые могут быть преобразованы в любую аддитивную соль основания или кислоты.
Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, содержащего кислотные радикалы или основные группы, в результате обычных химических процессов. В общем, процесс получения таких солей выполняют в воде или органическом растворителе или их смеси путем взаимодействия этих соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты.
Соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение охватывает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомер, (L)-изомер и их рацемические смеси и другие смеси, такие как смеси, обогащенные энантиомером или диастереомером. Все эти смеси включены в объем настоящего раскрытия. Могут присутствовать дополнительные асимметричные атомы углерода в алкиле и других заместителях. Все эти изомеры и их смеси включены в объем настоящего изобретения.
Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стереоизомерам, которые являются зеркальным отображением друг друга.
Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» обусловлен двойной или одинарной связью образующего кольцо атома углерода, который не может свободно вращаться.
Если не указано иное, термин «диастереомер» относится к стереоизомеру, в котором молекула имеет два или более хиральных центра, при этом молекулы не находятся в зеркальном отношении.
Если не указано иное, «(D)» или «(+)» означает правосторонний, «(L)» или «(-)» означает левосторонний, а «(DL)» или «(+)» означает рацемический.
Если не указано иное, связь, обозначенная сплошной линией в форме клина  и связь, обозначенная пунктирной линией в форме клина
и связь, обозначенная пунктирной линией в форме клина  указывают на абсолютную конфигурацию стереоцентра; связь, обозначенная прямой сплошной линией
указывают на абсолютную конфигурацию стереоцентра; связь, обозначенная прямой сплошной линией  и связь, обозначенная прямой пунктирной линией
и связь, обозначенная прямой пунктирной линией  указывают на относительную конфигурацию стереоцентра; волнистая линия
указывают на относительную конфигурацию стереоцентра; волнистая линия  указывает на связь, обозначенную сплошной линией в форме клина
указывает на связь, обозначенную сплошной линией в форме клина  или связь, обозначенную пунктирной линией в форме клина
или связь, обозначенную пунктирной линией в форме клина  или волнистая линия в форме клина указывают на связь, обозначенную прямой сплошной линией
или волнистая линия в форме клина указывают на связь, обозначенную прямой сплошной линией  и связь, обозначенную прямой пунктирной линией
и связь, обозначенную прямой пунктирной линией
Соединения по настоящему изобретению могут присутствовать в конкретных таутомерных формах. Если не указано иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре различные функциональные группы изомера находятся в динамическом равновесии и могут быстро превращаться друг в друга. При наличии таутомера (например, в растворе) возможно достижение химического равновесия таутомеров. Например, протонный таутомер (также известный как прототропный таутомер) включает взаимопревращение в ходе протеолиза, такого как кетон-енольная изомеризация и имин-енаминная изомеризация. Валентный таутомер включает некоторую рекомбинацию связывающих электронов, участвующих во взаимопревращениях. Конкретным примером кето-енольной таутомеризации является взаимопревращение двух таутомеров, пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
Если не указано иное, термин «обогащенный изомером», «изомерно обогащенный», «обогащенный энантиомером» или «энантиомерно обогащенный» означает, что содержание изомера или энантиомера составляет менее 100%, и это содержание изомера или энантиомера составляет 60% или более, или 70% или более, или 80% или более, или 90% или более, или 95% или более, или 96% или более, или 97% или более, или 98% или более, или 99% или более, или 99,5% или более, или 99,6% или более, или 99,7% или более, или 99,8% или более, или 99,9% или более.
Если не указано иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разнице между относительным процентным содержанием двух изомеров или двух энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, а содержание другого изомера или энантиомера составляет 10%, избыток изомера или энантиомера (значение ее) составляет 80%.
Оптически активные (R)- и (S)-изомеры и D- и L-изомеры могут быть получены синтезом хиральных соединений или путем использования хиральных реагентов, или другими традиционными способами. Если необходим энантиомер соединения по настоящему изобретению, он может быть получен асимметричным синтезом или дериватизацией с помощью хирального вспомогательного вещества, при этом полученную смесь диастереомеров разделяют, и вспомогательную группу отщепляют с получением чистого и необходимого энантиомера. Альтернативно, когда молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная группа), диастереомерную соль получают, используя подходящую оптически активную кислоту или основание, и разделение диастереомеров выполняют обычными способами, известными в данной области, а затем извлекают чистый энантиомер. Кроме того, разделение энантиомеров и диастереомеров обычно осуществляют с помощью хроматографии, в которой используется хиральная неподвижная фаза необязательно с процессами химической дериватизации (например, образование карбамата из амина). Соединения по настоящему изобретению могут содержать не встречающиеся в природе пропорции атомных изотопов одного или более атомов, составляющих соединение. Например, соединения могут быть помечены радиоактивными изотопами, такими как тритий (3Н), йод-125 (125I) или С-14 (14С). В качестве другого примера водород можно заменить тяжелым водородом с образованием дейтерированного лекарственного вещества. Связь между дейтерием и углеродом прочнее, чем между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными веществами дейтерированные лекарственные вещества имеют такие преимущества, как меньшее количество побочных эффектов, повышенная стабильность, улучшенная эффективность, увеличенный биологический период полувыведения и т.п. В объем изобретения входит возможность применения в соединении любого из радиоактивных изотопов, как радиоактивных, так и не радиоактивных.
«Необязательный» или «необязательно» означает, что описанное далее событие или условие может, но не обязательно, иметь место, и описание включает ситуацию, в которой это событие или условие возникает, и ситуацию, в которой это событие или условие не возникает.
Термин «замещенный» означает, что любой один или более атомов водорода в конкретном атоме замещены заместителем, который может включать тяжелый водород и варианты водорода при условии, что валентное состояние конкретного атома является нормальным и соединение после замещения является стабильным. Заместитель в виде кислорода (т.е.=О) означает, что замещены два атома водорода. Замещение кислорода в ароматической группе не происходит. Термин «необязательное замещение» или «необязательно замещенный» охватывает случаи, которые могут быть незамещенными или замещенными. Если не указано иное, тип и количество заместителей могут быть произвольными, при условии, что они могут быть получены химическим путем.
Когда любая переменная (например, R) появляется более одного раза в составе или структуре соединения, в каждом случае она определяется независимо. Таким образом, например, если группа замещена 0-2 R, то группа необязательно может быть замещена не более чем двумя R, и R в каждом случае имеет независимые варианты. Кроме того, допустимы комбинации заместителей и/или их вариантов при условии, что такие комбинации дают стабильные соединения.
Когда количество связывающих групп равно 0, -(CRR)0-, это означает, что связывающая группа представляет собой одинарную связь.
Когда одну из переменных выбирают из группы, состоящей из одинарных связей, это означает, что две связанные ею группы связаны напрямую. Например, когда L представляет собой одинарную связь в A-L-Z, фактическая структура представляет собой -A-Z.
Когда заместитель отсутствует, это означает, что заместитель не присутствует. Например, когда X в А-Х отсутствует, это означает, что фактическая структура представляет собой А. Когда не указано с каким атомом связаны перечисленные заместители, такие заместители могут быть связаны через любой из атомов. Например, пиридил в качестве заместителя может быть присоединен к замещенной группе через любой атом углерода в пиридиновом кольце.
Если для указанной связывающей группы не указано направление соединения, она может быть присоединена в любом направлении. Например, связывающая группа L в 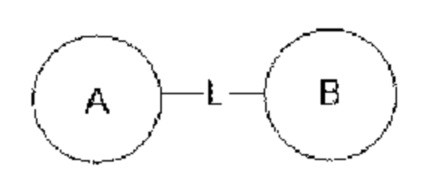 представляет собой -MW-, причем -MW- может соединять кольцо А с кольцом В в том же направлении, как она показана, слева направо, с образованием
представляет собой -MW-, причем -MW- может соединять кольцо А с кольцом В в том же направлении, как она показана, слева направо, с образованием 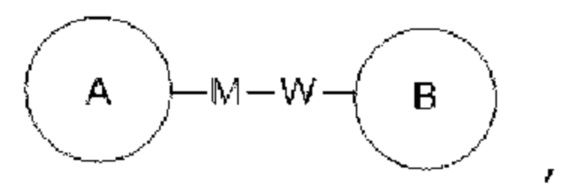 или может соединять кольцо А с кольцом В в противоположном показанному слева направо направлении с образованием
или может соединять кольцо А с кольцом В в противоположном показанному слева направо направлении с образованием 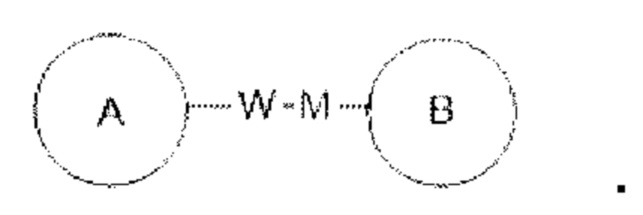 Комбинация связывающей группы, заместителей и/или их вариантов допускается при условии, что такая комбинация дает стабильное соединение.
Комбинация связывающей группы, заместителей и/или их вариантов допускается при условии, что такая комбинация дает стабильное соединение.
Если не указано иное, термин «гетеро» относится к гетероатому или гетерорадикалу (т.е. радикалу, содержащему гетероатом), включая атомы, отличные от углерода (С) и водорода (Η), и радикалам, содержащим такие гетероатомы, включая, например, кислород (О), азот (Ν), серу (S), кремний (Si), германий (Ge), алюминий (Αl), бор (В), -O-, -S-, -С (=O)O-, С(=O)-, -C(=S)-, -S(=O), -S(=O)2-, and -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- или -S(=O)N(H)-, которые являются необязательно замещенными.
Если не указано иное, «цикло» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Кольцо включает одно кольцо, а также включает бициклические или полициклические кольцевые системы, такие как спирокольцо, конденсированное кольцо, мостиковое кольцо и т.п.Количество атомов в кольце обычно определяют как количество членов кольца. Например, «5-7-членное кольцо» относится к 5-7 атомам, образующим кольцевую структуру. Если не указано иное, кольцо необязательно содержит 1-3 гетероатома. Соответственно, «5-7-членное кольцо» включает, например, фенил, пиридил и пиперидинил. В другом аспекте термин «5-7-членный гетероциклоалкил» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую, по меньшей мере одно кольцо, где каждое «кольцо» независимо соответствует приведенному выше определению.
Если не указано иное, термин «алкил» относится к линейной или разветвленной насыщенной углеводородной группе. В некоторых вариантах осуществления изобретения алкил представляет собой C1-6 алкил; в других вариантах осуществления алкил представляет собой С1-3 алкил; в других вариантах осуществления алкил представляет собой C1-3 алкил. Алкил может быть моно замещенным (например, CH2F) или полизамещенным (например, -CF3), может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры алкила включают, без ограничения, метил (Me), этил (Et), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил (включая н-пентил, изопентил и неопентил), гексил или т.п.
Если не указано иное, термин «C1-6 алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-6 атомов углерода. C1-6 алкил включает С1-5, С1-4, C1-3, С1-2, С2-6, С2-4, С6 и С5 алкил или т.п. Алкил может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры C1-6 алкила включают, без ограничения, метил (Me), этил (Et), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил (включая н-пентил, изопентил и неопентил), гексил или т.п.
Если не указано иное, термин «C1-3 алкил» относится к линейной или разветвленной насыщенной углеводородной группе, состоящей из 1-3 атомов углерода. C1-3 алкил включает С1-2 и С2-3 алкил или т.п. Алкил может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры C1-3 алкила включают, без ограничения, метил (Me), этил (Et), пропил (включая н-пропил и изопропил) или т.п.
Если не указано иное, «алкенил» относится к линейной или разветвленной углеводородной группе, содержащей одну или более двойных углерод-углеродных связей. Двойная углерод-углеродная связь может находиться в любом положении группы. В некоторых вариантах осуществления алкенил представляет собой С2-8 алкенил; в других вариантах осуществления алкенил представляет собой С2-6 алкенил; в других вариантах осуществления алкенил представляет собой С2-4 алкенил. Алкенил может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкенила включают, без ограничения, этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т.п.
Если не указано иное, «алкинил» относится к линейной или разветвленной углеводородной группе, содержащей одну или более тройных углерод-углеродных связей. Тройная углерод-углеродная связь может находиться в любом положении группы. В некоторых вариантах осуществления алкинил представляет собой С2-8 алкинил; в других вариантах осуществления алкинил представляет собой С2-6 алкинил; в других вариантах осуществления алкинил представляет собой С2-4 алкинил. Алкинил может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкинила включают, без ограничения, этинил, пропинил, бутинил, пентинил и т.п.
Если не указано иное, термин «гетероалкил», сам по себе или в комбинации с другим термином, относится к стабильному линейному или разветвленному алкильному радикалу или его композиции, которая состоит из определенного количества атомов углерода и по меньшей мере одного гетероатома или гетерорадикала. В некоторых вариантах осуществления гетероатом выбирают из группы, состоящей из В, О, N и S, где атомы N и S необязательно окислены, гетероатом N необязательно является кватернизованным. В некоторых других вариантах осуществления гетерорадикал выбирают из группы, состоящей из -С(=О)О-, -С(=О)-, -C(=S)-, -S(=О), -S(=О)2-, -C(=О)N(H)-, -Ν(Η)-, -C(=NH)-, -S(=О)2N(H)- и -S(=О)N(H)-. В некоторых вариантах осуществления гетероалкил представляет собой C1-6 гетероалкил; в некоторых других вариантах осуществления гетероалкил представляет собой C1-3 гетероалкил. Гетероатом или гетерорадикал может находиться в любом внутреннем положении гетероалкила, включая положение соединения алкила с остальной частью молекулы, но термины «алкокси», «алкилаамино» и «алкилатио» (или тиоалкокси) являются общепринятыми выражениями и относятся к алкильным группам, которые связаны с остальной частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Примеры гетероалкила включают, без ограничения, -ОСН3, -ОСН2СН3, ОСН2СН2СН3, -ОСН2(СН3)2, -СН2-СН2-О-СН3, -NHCH3, -N(CH3)2, -NHCH2CH3, -N(CH3)(СН2СН3), -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-СН3, -SCH3, SCH2CH3, -SCH2CH2CH3, -SCH2(CH3)2, -CH2-S-CH2-CH3, -CH2-CH2, -S(=О)-CH3, -CH2-CH2-S(=О)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(СН3)-СН3. Не более двух гетероатомов могут быть присоединены непосредственно друг к другу, например -CH2-NH-OCH3.
Если не указано иное, термин «гетероалкенил» сам по себе или в комбинации с другим термином относится к стабильному линейному или разветвленному алкенильному радикалу или его композиции, которая состоит из определенного количества атомов углерода и по меньшей мере одного гетероатома или гетерорадикала. В некоторых вариантах осуществления гетероатом выбирают из группы, состоящей из В, О, N и S, где атомы N и S необязательно окислены, гетероатом N необязательно является кватернизованным. В некоторых других вариантах осуществления гетерорадикал выбирают из группы, состоящей из -С(=О)O-, -С(=О)-, -C(=S)-, -S(=О), -S(=О)2-, -C(=О)N(H)-, -Ν(Η)-, -C(=NH)-, -S(=О)2N(H)- и -S(=О)N(H)-. В некоторых вариантах осуществления гетероалкенил представляет собой С2-6 гетероалкенил; в некоторых других вариантах осуществления гетероалкил представляет собой С2-4 гетероалкенил. Гетероатом или гетерорадикал может находиться в любом внутреннем положении гетер «алкенилокси», «алкениламино» и «алкенилтио» являются общепринятыми выражениями и относятся к алкенильным группам, которые связаны с остальной частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Примеры гетероалкенила включают, без ограничения, -O-СН=СН2, -O-СН=СНСН3, -O-СН=С(СН3)2, -СН=СН-O-СН3, -O-СН=СНСН2СН3, -СН2-СН=СН-ОСН3, -NH-СН=СН2, -N(СН=СН2)-СН3, -CH=CH-NH-CH3, -CH=CH-N(СН3)2, -S-CH=CH2, -S-CH=CHCH3, -S-CH=C(СН3)2, -CH2-S-CH=CH2, -S(=О)-CH=CH2 и -СН=СН-S(=О)2-СН3. Не более двух гетероатомов могут быть присоединены непосредственно друг к другу, например -СН=СН-NH-ОСН3.
Если не указано иное, термин «гетероалкинил», сам по себе или в комбинации с другим термином, относится к стабильному линейному или разветвленному алкинильному радикалу или его композиции, которая состоит из определенного количества атомов углерода и по меньшей мере одного гетероатома или гетерорадикала. В некоторых вариантах осуществления гетероатом выбирают из группы, состоящей из В, О, N и S, где атомы N и S необязательно окислены, гетероатом N необязательно является кватернизованным. В некоторых других вариантах осуществления гетерорадикал выбирают из группы, состоящей из -С(=О)O-, -С(=О)-, -C(=S)-, -S(=О), -S(=О)2-, -C(=О)N(H)-, -Ν(Η)-, -C(=NH)-, -S(=О)2N(H)- и -S(=О)N(H)-. В некоторых вариантах осуществления гетероалкинил представляет собой С2-6 гетероалкинил; в некоторых других вариантах осуществления гетероалкил представляет собой С2-4 гетероалкинил. Гетероатом или гетерорадикал может находиться в любом внутреннем положении гетероалкинила, включая положение соединения алкинила с остальной частью молекулы, но термины «алкинилокси», «алкиниланино» и «алкинилтио» являются общепринятыми выражениями и относятся к алкинильным группам, которые связаны с остальной частью молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Примеры гетероалкинила включают, без ограничения, 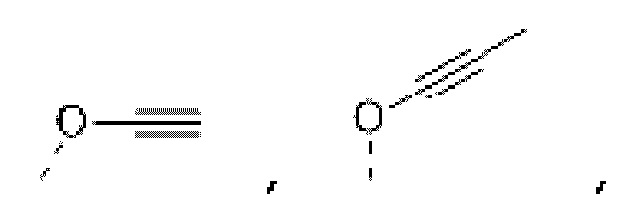
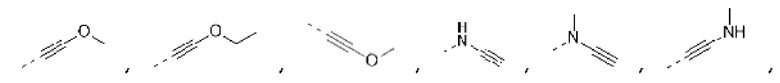
 Не более двух гетероатомов могут быть присоединены непосредственно друг к другу, например,
Не более двух гетероатомов могут быть присоединены непосредственно друг к другу, например, 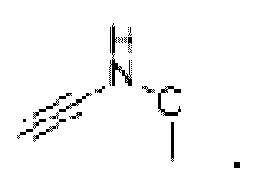
Если не указано иное, «циклоалкил» содержит любой стабильный циклический алкил, включая моноциклические,
бициклические или трициклические системы, причем бициклические и трициклические системы включают спирокольцо, конденсированное кольцо и мостиковое кольцо. В некоторых вариантах осуществления циклоалкил представляет собой С3-8 циклоалкил. В некоторых вариантах осуществления циклоалкил представляет собой С3-6 циклоалкил. В некоторых вариантах осуществления циклоалкил представляет собой C5-6 циклоалкил. Циклоалкил может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил алкил, [2. 2. 2]бициклооктан, [4. 4. 0] бицикл одекан или т.п.
Если не указано иное, «С3-6 циклоалкил» представляет собой насыщенную циклическую углеводородную группу, состоящую из 3-6 атомов углерода, которая представляет собой моноциклическую и бициклическую систему. С3-6 циклоалкил включает С3-5, C4-5 и C5-6 циклоалкил или т.п. Циклоалкил может быть одновалентным, двухвалентным или поливалентным. Примеры С3-6 циклоалкила включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил или т.п.
Если не указано иное, «циклоалкенил» содержит любой стабильный циклический алкенил, содержащий одну или более ненасыщенных двойных углерод-углеродных связей в любом положении, который включает моноциклические, бициклические или трициклические системы, причем бициклические и трициклические системы включают спирокольцо, конденсированное кольцо и мостиковое кольцо, но все кольца в этой системе являются неароматическими. В некоторых вариантах осуществления циклоалкенил представляет собой С3-8 циклоалкенил. В некоторых других вариантах осуществления циклоалкенил представляет собой С3-6 циклоалкенил. В некоторых других вариантах осуществления циклоалкенил представляет собой С5-6 циклоалкенил. Циклоалкенил может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкенила включают, без ограничения, циклопентенил, циклогексенил и т.п.
Если не указано иное, «циклоалкинил» содержит любой стабильный циклический алкинил, содержащий одну или более тройных углерод-углеродных связей в любом положении, который включает моноциклическую, бициклическую или трициклическую систему, причем бициклические и трициклические системы включают спироциклическое, конденсированное кольцо и мостиковое кольцо. Циклоалкинил может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным.
Если не указано иное, термин «гетероциклоалкил», сам по себе или в комбинации с другим термином, относится к циклическому «гетероалкилу», включая моноциклические,
бициклические и трициклические системы, причем бициклические и трициклические системы включают спирокольцо, конденсированное кольцо и мостиковое кольцо. Кроме того, что касается «гетероциклоалкила», гетероатом может занимать положение, соединяющее гетероциклоалкил с остальной частью молекулы. В некоторых вариантах осуществления гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил. В некоторых других вариантах осуществления гетероциклоалкил представляет собой 5-6-членный гетероциклоалкил. Примеры гетероциклоалкила включают, без ограничения, азетидинил, оксетанил, тиетанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиен-2-ил и тетрагидротиен-3-ил или т.п.), тетрагидрофуранил (включая тетрагидрофуран-2-ил или т.п.), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил и 3-пиперидинил или т.п.), пиперазинил (включая 1-пиперазинил и 2-пиперазинил или т.п.), морфолинил (включая 3-морфолинил и 4-морфолинил или т.п.), диоксанил, дитианил, изоксазолалкил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, пексапидропиридазинил, гомопиперазинил, помопиперидинил или оксепанил.
Если не указано иное, термин «гетероциклоалкенил», сам по себе или в комбинации с другим термином, относится к циклическому «петероалкенилу», включая моноциклические, бициклические и трициклические системы, причем бициклические и трициклические системы включают спироколвцо, конденсированное кольцо и мостиковое кольцо, но все кольца в этой системе являются неароматическими. Кроме того, что касается «гетероциклоалкенила», гетероатом может занимать положение, соединяющее гетероциклоалкенил с остальной частью молекулы. В некоторых вариантах осуществления гетероциклоалкенил представляет собой 4-6-членный гетероциклоалкенил. В некоторых других вариантах осуществления гетероциклоалкенил представляет собой 5-6-членный гетероциклоалкенил. Примеры гетероциклоалкенила включают, без ограничения, 
Если не указано иное, термин «гетероциклоалкинил», сам по себе или в комбинации с другим термином, относится к циклическому «гетероалкинилу», включая моноциклические, бициклические и трициклические системы, причем бициклические и трициклические системы включают спирокольцо, конденсированное кольцо и мостиковое кольцо. Кроме того, что касается «гетероциклоалкинила», гетероатом может занимать положение, соединяющее гетероциклоалкинил с остальной частью молекулы. В некоторых вариантах осуществления гетероциклоалкинил представляет собой 4-6-членный гетероциклоалкинил. В некоторых других вариантах осуществления гетероциклоалкинил представляет собой 5-6-членный гетероциклоалкинил.
Если не указано иное, термин «галоген» или «гало», сам по себе или как часть другого заместителя, относится к атому F, Cl, Br или I. Кроме того, термин «галоалкил» включает моногалоалкил и полигалоалкил. Например, термин «гало(С1-С4)алкил» включает, без ограничения, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил и 3-бромпропил или т.п. Если не указано иное, примеры галогеналкила включают, без ограничения, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
«Алкокси» относится к вышеуказанному алкилу, имеющему определенное количество атомов углерода, связанных через кислородный мостик. Если не указано иное, C1-6 алкокси включает C1, С2, С3, С4, С5 и С6 алкокси. В некоторых вариантах осуществления алкокси представляет собой C1-3 алкокси. Примеры алкокси включают, без ограничения, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентокси и втор-пентокси.
Если не указано иное, термин «C1-6 алкокси» относится к алкильной группе, содержащей 1-6 атомов углерода, связанной с остальной частью молекулы через атом кислорода. C1-6 алкокси включает С1-4, С1-3, С1-2, С2-6, С2-4, С6, С5, С4 и С3 алкокси или т.п. Примеры C1-6 алкокси включают, без ограничения, метокси, этокси, пропокси (включая н-пропокси и изопропокси), бутокси (включая н-бутокси, изобутокси, втор-бутокси и трет-бутокси), пентокси (включая н-пентокси, изопентокси и неопентокси), гексилокси или т.п.
Если не указано иное, термин "C1-3 алкокси" относится к алкильной группе, содержащей 1-3 атомов углерода, соединенных с остальной частью молекулы через атом кислорода. C1-3 алкокси включает С1-2, С2-3, С3 и С2 алкокси или т.п. Примеры C1-3 алкокси включают, без ограничения, метокси, этокси, пропокси (включая н-пропокси и изопропокси) или т.п.
Если не указано иное, термины «ароматическое кольцо» и «арил» могут использоваться в настоящем описании взаимозаменяемо. Термин «ароматическое кольцо» или «арил» относится к полиненасыщенной карбоциклической системе, которая может быть моноциклической, бициклической или полициклической системой, причем по меньшей мере одно кольцо является ароматическим. Каждое кольцо в бициклической и полициклической системе слиты вместе. Оно может быть моно- или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. В некоторых вариантах осуществления арил представляет собой С6-12 арил. В некоторых вариантах осуществления арил представляет собой С6-10 арил. Примеры арила включают, без ограничения, фенил, нафтил (включая 1-нафтил и 2-нафтил или т.п.). Заместитель любой из указанной выше системы, содержащей арильное кольцо, может быть выбран из группы, состоящей из приемлемых заместителей, раскрытых в настоящем описании.
Если не указано иное, термины «гетероароматическое кольцо» и «гетероарил» могут использоваться в настоящем описании взаимозаменяемо. Термин «гетероарил» относится к арилу (или ароматическому кольцу), содержащему 1, 2, 3 или 4 гетероатома, независимо выбираемых из группы, состоящей из В, N, О и S, и может быть моноциклической, бициклической или трициклической системой, причем атом азота может быть замещенным или незамещенным (например, N или NR, где R представляет собой Η или другие заместители, определенные в настоящем описании) и необязательно кватернизованным; и гетероатомы, азот и сера, могут быть необязательно окисленными (т.е. NO и S(O)p, где ρ равно 1 или 2). Гетероарил может быть связан с остальной частью молекулы через гетероатом. В некоторых вариантах осуществления гетероарил представляет собой 5-10-членный гетероарил. В некоторых других вариантах осуществления гетероарил представляет собой 5-6-членный гетероарил. Примеры гетероарила включают, без ограничения, пирролил (включая N-пирролил, 2-пирролил и 3-пирролил или т.п.), пиразолил (включая 2-пиразолил и 3-пиразолил или т.п.), имидазолил (включая N-имидазолил, 2-имидазолил, 4-имидазолил и 5-имидазолил или т.п.), оксазолил (включая 2-оксазолил, 4-оксазолил и 5-оксазолил или т.п.), триазолил (1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил, 1Н-1,2,4-триазолила и 4Н-1,2,4-триазолила или т.п.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил и 5-изоксазолил или т.п.), тиазолил (включая 2-тиазолил, 4-тиазолил и 5-тиазолил или т.п.), фурил (включая 2-фурил и 3-фурил или т.п.), тиенил (включая 2-тиенил и 3-тиенил или т.п.), пиридил (включая 2-пиридил, 3-пиридил и 4-пиридил или т.п.), пиразинил, пиримидинил (включая 2-пиримидинил и 4-пиримидинил или т.п.), бензотиазолил (включая 5-бензотиазолил или т.п.), пуринил, бензимидазолил (включая 2-бензимидазолил или т.п.), индолил (включая 5-индолил или т.п.), изохинолинил (включая 1-изохинолинил и 5-изохинолинил или т.п.), хиноксалинил (включая 2-хиноксалинил и 5-хиноксалинил или т.п.), хинолинил (включая 3-хинолинил и 6-хинолинил или т.п.), пиразинил, пуринил, бензоксазолил. Заместитель любой из вышеуказанных гетероарильных кольцевых систем может быть выбран из группы, состоящей из приемлемых заместителей, раскрытых в настоящем описании.
Если не указано иное, термины «5-6-членное гетероароматическое кольцо» и «5-6-членный гетероарил» могут использоваться в настоящем описании взаимозаменяемо. Термин «5-6-членный гетероарил» относится к моноциклической группе, состоящей из 5-6 кольцевых атомов с сопряженной п-электронной системой, где 1, 2, 3 или 4 кольцевых атома являются гетероатомами, независимо выбираемыми из группы, состоящей из О, S и N, а остальные представляют собой атомы углерода, причем атом азота необязательно является кватернизованным, и гетероатомы азота и серы необязательно могут быть окисленными (т.е. NO и S(O)p, где ρ равно 1 или 2). 5-6-членный гетероарил может быть связан с остальной частью молекулы через гетероатом или атом углерода. 5-6-членный гетероарил включает 5-членный и 6-членный гетероарил. Примеры 5-6 членного гетероарила включают, без ограничения, пирролил (включая N-пирролил, 2-пирролил и 3-пирролил или т.п.), пиразолил (включая 2-пиразолил и 3-пиразолил или т.п.), имидазолил (включая N-имидазолил, 2-имидазолил, 4-имидазолил и 5-имидазолил или т.п.), оксазолил (включая 2-оксазолил, 4-оксазолил и 5-оксазолил или т.п.), триазолил (1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил и 4H-1,2,4-триазолил или т.п.), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил и 5-изоксазолил или т.п.), тиазолил (включая 2-тиазолил, 4-тиазолил и 5-тиазолил или т.п.), фурил (включая 2-фурил и 3-фурил или т.п.), тиенил (включая 2-тиенил и 3-тиенил или т.п.), пиридил (включая 2-пиридил, 3-пиридил и 4-пиридил или т.п.), пиразинил или пиримидинил (включая 2-пиримидинил и 4-пиримидинил или т.п.).
Если не указано иное, термин «аралкил» включает группы, в которых арил присоединен к алкилу. В некоторых вариантах осуществления аралкил представляет собой C6-10 арил-С1-4 алкил. В некоторых других вариантах осуществления аралкил представляет собой С6-10 арил-С1-2 алкил. Примеры аралкила включают, без ограничения, бензил, фенетил, нафтилметил или т.п."Арилокси" и "арилтио" относится к группам, в которых атом углерода (такой как метил) в аралкиле замещен атомом О или S. В некоторых вариантах осуществления арилокси представляет собой С6-10 арил-О-С1-2 алкил. В некоторых других вариантах осуществления арилокси представляет собой С6-10 арил-С1-2 алкил-О-. В некоторых вариантах осуществления арилтио представляет собой C6-10 арил-S-C1-2 алкил. В некоторых других вариантах осуществления арилтио представляет собой С6-10 арил-С1-2 алкил-S-. Примеры арилокси и арилтио включают, без ограничения, феноксиметил, 3-(1-нафтилокси)пропил, фенилтиометил или т.п.
Если не указано иное, термин «гетероаралкил» включает группы, в которых гетероарил присоединен к алкильной группе. В некоторых вариантах осуществления гетероаралкил представляет собой 5-8-членный гетероарил-С1-4 алкил. В некоторых других вариантах осуществления гетероаралкил представляет собой 5-6-членный гетероарил-С1-2 алкил. Примеры гетероаралкила включают, без ограничения, пирролилметил, пиразолилметил, пиридилметил, пиримидинилметил или т.п.«Гетероарилокси» и «гетероарилтио» относятся к группам, в которых атом углерода (такой как метил) в группе гетероаралкила замещен атомом О или S. В некоторых вариантах осуществления гетероарилокси представляет собой 5-8-членный гетероарил-O-С1-2 алкил. В некоторых вариантах осуществления гетероарилокси представляет собой 5-6-членный гетероарил-С1-2 алкил-О-. В некоторых вариантах осуществления гетероарилтио представляет собой 5-8-членный гетероарил-S-С1-2 алкил. В некоторых других вариантах осуществления гетероарилтио представляет собой 5-6-членный гетероарил-С1-2 алкил-S-. Примеры гетероарилокси и гетероарилтио включают, без ограничения, пирролилоксиметил, пиразолилоксиметил, 2-пиридилоксиметил, пирролилтиометил, пиразолилтиометил, 2-пиридилтиометил или т.п.
Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай, в котором количество атомов углерода составляет от n до n+m. Например, С1-12 охватывает C1, С2, С3, С4, С5, С6, С7, C8, C9, С10, С11 и С12, а также любое значение в пределах от n до n+m, например, С1-12 охватывает С1-3, C1-6, С1-9, С3-6, С3-9, С3-12, С6-9, С6-12 и С9-12 и т.п. Аналогично, значение от n-членного до n+m-членного означает, что количество атомов в кольце составляет от n до n+m, например, 3-12-членное кольцо включает 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7 членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо, а также включает любое количество в пределах от n до n+m, например, 3-12-членное кольцо содержит 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо и 6-10-членное кольцо или т.п.
Термин «уходящая группа» относится к функциональной группе или атому, который может быть заменен другой функциональной группой или атомом в результате реакции замещения (например, реакции аффинного замещения). Например, репрезентативные уходящие группы включают трифлат; Cl, Br, I; сульфонат, такой как мезилат, тозилат, п-бромбезилат, п-толуолсульфонат или т.п.; ацилокси, например ацетокси, трифторацетокси или т.п.
Термин «защитная группа» включает, без ограничения, «аминозащитную группу», «гидроксильную защитную группу» или «меркаптозащитную группу». Термин «аминозащитная группа» относится к защитной группе, подходящей для предотвращения побочных реакций в положении азота аминогруппы. Типичные аминозащитные группы включают, без ограничения, формил; ацил, такой как алканоил (такой как ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Вос); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-фторметоксикарбонил (Fmoc); арилметил, такой как бензил (Βn), трифенилметил (Tr), 1,1-бис-(4'-метоксилфенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) или т.п.
Соединения по настоящему изобретению могут быть получены различными способами синтеза, хорошо известными специалисту в данной области, включая конкретные варианты осуществления, перечисленные ниже. Варианты осуществления, образованные комбинацией с другими процессами химического синтеза и эквивалентностью, хорошо известными специалисту в данной области, и предпочтительные варианты осуществления включают, без ограничения, примеры, приведенные в настоящем описании.
Соединения по настоящему изобретению могут иметь множество применений или показаний, включая, без ограничения, конкретные перечисленные в настоящем описании соединения.
Используемые в настоящем описании растворители являются коммерчески доступными. В настоящем описании используются следующие сокращения: aq.: вода; HATU: О-(7-азабензотриазол-1-ил)-N, Ν,Ν',N'-гексафторфосфат тетраметилмочевины; EDC: N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; m-СРВА: 3-хлорпероксибензойная кислота; eq.: эквивалент, эквивалентность; CDI: карбонилдиимидазол; DCM: дихлорметан; РЕ: петролейный эфир; DIAD: диизопропилазодикарбоксилат; DMF: N, N-диметилформамид; DMSO: диметилсульфоксид; EtOAc: этилацетат; EtOH: этанол; МеОН: метанол; CBz: бензилоксикарбонил, аминозащитная группа; ВОС: трет-бутоксикарбонил, аминозащитная группа; НОАс: уксусная кислота; NaCNBH3: цианоборгидрид натрия; г.t.: комнатная температура; O/N: в течение ночи; THF: тетрагидрофуран; Вос2О: ди-трет-бутилдикарбонат; TFA: трифторацетат; DIPEA: диизопропилэтиламин; SOCl2: тионилхлорид; CS2: дисульфид углерода; TsOH: р-толуолсульфоновая кислота; NFSI: Ν-фтор-Ν-(бензолсульфонил)бензолсульфонамид; NCS: N-хлорсукцинимид; n-Bu4NF: тетрабутиламмония фторид; iPrOH: 2-пропанол; mp: температура плавления; LDA: диизопропиламид лития.
Названия соединений заданы вручную или сгенерированы с помощью программного обеспечения ChemDraw®. Сложные названия использованы из каталога поставщиков.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1: Кривая роста опухоли у мышей, несущих опухоль, на модели подкожного ксенотрансплантата из клеток колоректального рака человека LoVo после введения соединения по настоящему изобретению.
Примеры
Настоящее изобретение описано подробно в приведенных ниже примерах, которые не следует рассматривать как ограничение. Настоящее изобретение подробно описано в настоящем описании, в котором также раскрыты его конкретные варианты осуществления. Для специалиста в данной области техники будет очевидно, что в конкретные варианты осуществления настоящего изобретения могут быть внесены различные изменения и модификации без отклонения от сущности и объема изобретения.
Промежуточное соединение 1
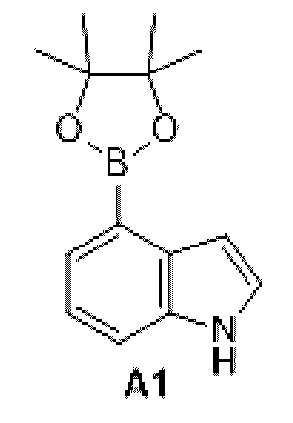
Схема синтеза:
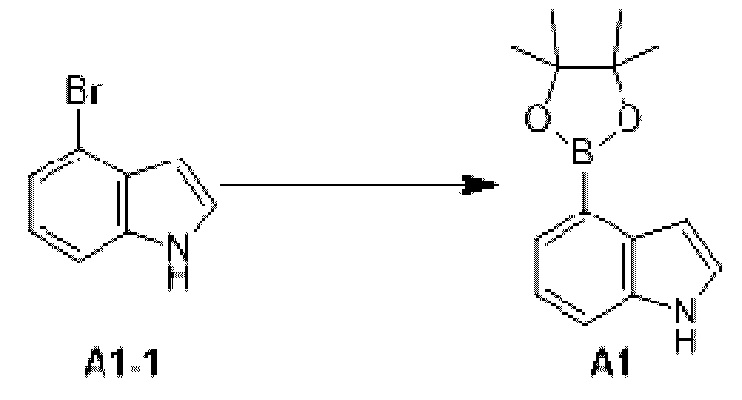
Этап 1: Синтез соединения А1
К раствору соединения А1-1 (65 г, 331,56 ммоль) в диметилсульфоксиде (1 л) добавляли биспинакол борат (126,29 г, 497,34 ммоль), 1,1-бис(дифенилфосфино)ферроцен палладия хлорид (12,13 г, 16,58 ммоль) и ацетат калия (113,89 г, 1,16 моль). Реакционную смесь перемешивали под защитой азота при 90°С в течение 16 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 1 л этилацетата (500 мл ×2), и органическую фазу промывали 3 л воды (1 л ×3) и сушили над безводным сульфатом натрия. Затем десиккант (обезвоживающее вещество) отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 4:1) с получением соединения А1.
МС-ЭСИ м/з: 243,9 [М+Н]+. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (с, 13 Н) 7,09 (т, J=2,13 Гц, 1 Н) 7,21-7,25 (м, 1 Н) 7,52 (д, J=8,03 Гц, 1 Н) 7,67 (д, J=7, 03 Гц, 1 Н) 8,23 (уш.с, 1 Н).
Промежуточное соединение 2
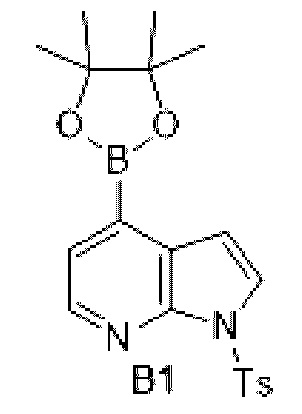
Схема синтеза:

Этап 1: Синтез соединения В1-2
К раствору соединения В1-1 (90 г, 456,78 ммоль) в дихлорметане (1 л) добавляли раствор гидроксида натрия (2М, 685,17 мл) и гидросульфат тетрабутиламмония (7,75 г, 22,84 ммоль), и затем медленно добавляли р-толуолсульфонилхлорид (174,17 г, 913,56 ммоль). Реакционную смесь перемешивали при 25°С в течение 15 ч и экстрагировали 500 мл дихлорметана (250 мл ×2), и органическую фазу промывали 3л воды (1 л ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 1:0) с получением соединения В1-2.
МС-ЭСИ м/з: 352,9 [М+Н]+.
Этап 2: Синтез соединения В1
К раствору соединения В1-2 (25 г, 71,18 ммоль) в N,N-диметилформамиде (500 мл) добавляли биспинакол борат (36,15 г, 142,36 ммоль), 1, 1-бис (дифенилфосфино) ферроцен палладия хлорид (5,21 г, 7,12 ммоль) и ацетат калия (20,96 г, 213,54 ммоль). Реакционную смесь перемешивали под защитой азота при 90°С в течение 16 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 1 л этилацетата (500 мл ×2), и органическую фазу промывали 3 л воды (1 л*3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 4:1) с получением соединения В1.
МС-ЭСИ м/з: 399,1 [М+Н]+. 1Н ЯМР (400 МГц, ХПОРОФОРМ-d) δ ppm 1,27 (д, J=2,76 Гц, 3 Н) 1,32-1,39 (м, 1 Н) 1,33-1,38 (м, 1 Н) 1,36 (с, 10 Н) 6,95-7,05 (м, 1 Н) 7,02 (д, J=4, 02 Гц, 1 Н) 7,20-7,26 (м, 1 Н) 7,24 (д, J=8,03 Гц, 1 Н) 7,52 (д, J=4,77 Гц, 1 Н) 7,72-7,78 (м, 1 Н) 7,75 (д, J=3,76 Гц, 1 Н) 8,02-8,04 (м, 2 Н) 8,43 (д, J=4,77 Гц, 1 Н).
Промежуточное соединение 3

Схема синтеза:
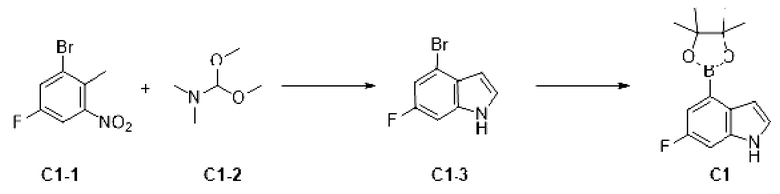
Этап 1: Синтез соединения С1-3
При комнатной температуре к раствору соединения С1-1 (3,00 г, 12,82 ммоль) в N,N-диметилформамиде (30, 00 мл) добавляли С1-2 (7,65 г, 64,23 ммоль, 8,50 мл), который перемешивали в атмосфере азота при 160°С в течение 8 ч. Реакционную систему охлаждали, разбавляли дихлорметаном (50 мл), промывали водой (20 мл ×5) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта. Неочищенный продукт растворяли уксусной кислотой (5 мл) и добавляли по каплям в кипящий раствор железного порошка (7,16 г, 128,19 ммоль) в уксусной кислоте (5 мл). Реакционную смесь нагревали с обратным холодильником в течение 40 мин. Реакционную смесь охлаждали до комнатной температуры, доводили рН до основного значения с помощью насыщенного раствора карбоната натрия и экстрагировали дихлорметаном (30 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/дихлорметан=3/1) с получением соединения С1-3.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 6,50 (т, J=2,26 Гц, 1 Н) 6,96-7,00 (м, 1 Н) 7,05 (дд, J=9,04, 2,01 Гц, 1 Н) 7,16 (т, J=2,76 Гц, 1 Н) 7,47 (дд, J=8,52, 5, 52 Гц, 1 Н) 8,20 (уш.с, 1 Н).
Этап 2: Синтез соединения С1
При комнатной температуре к раствору соединения C1-3 (1,00 г, 4,67 ммоль) в 1,4-диоксане (15,00 мл) добавляли биспинакол борат (1,78 г, 7,00 ммоль), 1,1-бис(дифенилфосфино)ферроцен палладия хлорид (341,71 мг, 467,00 мкмоль), ацетат калия (1,37 г, 14,01 ммоль), который перемешивали в атмосфере азота в течение 12 ч. После охлаждения реакционную систему разбавляли этилацетатом (40 мл) и фильтровали. Органическую фазу промывали водой (20 мл ×2) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/дихлорметан=3/1) с получением соединения С1.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,34 (с, 12 Н) 6,74 (уш.с, 1 Н) 7,12 (дд, 47=10,04, 2,51 Гц, 1 Н) 7,30 (дд, J=10,04, 2,01 Гц, 1 Н) 7,38 (т, J=2,76 Гц, 1 Н) 11,18 (уш.с, 1 Н).
Промежуточное соединение 4
Схема синтеза:
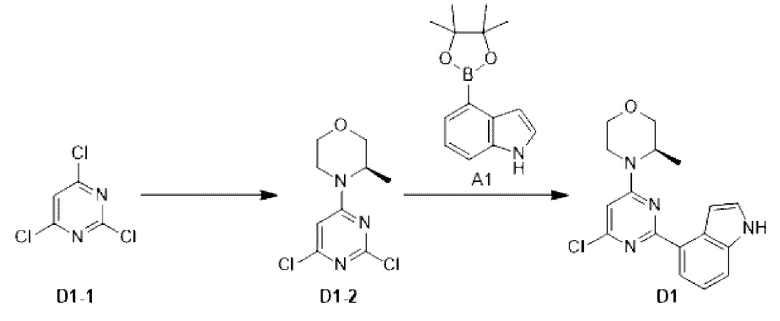
Этап 1: Синтез соединения D1-2
К раствору соединения D1-1 (2,00 г, 10,90 ммоль, 1,25 мл) в дихлорметане (20 мл) добавляли триэтиламин (3,15 г, 31,14 ммоль, 4,32 мл), и медленно по каплям добавляли [R)-3-метилморфолин при -5°С. Реакционную смесь медленно нагревали до 15°С и перемешивали в течение 15 ч. Соединение концентрировали до сухого состояния, и неочищенный продукт очищали на колонке с силикагелем (петролейный эфир/этилацетат=10:1, 5:1) с получением соединения D1-2.
МС-ЭСИ м/з: 247, 9 [М+Н]+.
Этап 2: Синтез соединения D1
К раствору соединения D1-2 (1,5 г, 6,05 ммоль) в 1,4-диоксане (40 мл) добавляли А1 (1,62 г, 6,65 ммоль), бистрифенилфосфин палладия дихлорид (424,35 мг, 604,57 мкмоль) и карбонат натрия (2 М, 9,07 мл). Реакционную смесь перемешивали под защитой азота при 110°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 50 мл этилацетата (25 мл ×2). Органическую фазу промывали 60 мл воды (20 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 1:1) с получением соединения D1.
МС-ЭСИ м/з: 32 8,9 [М+Н]+.
Промежуточное соединение 5
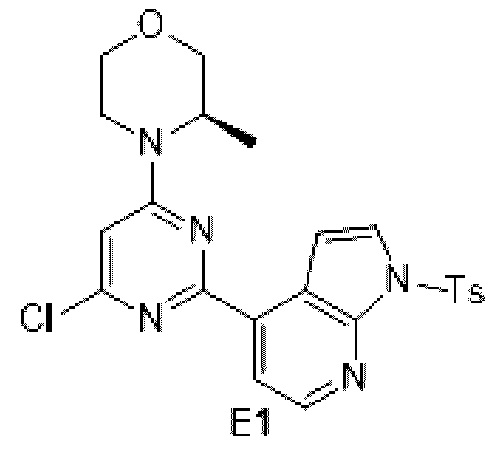
Схема синтеза:
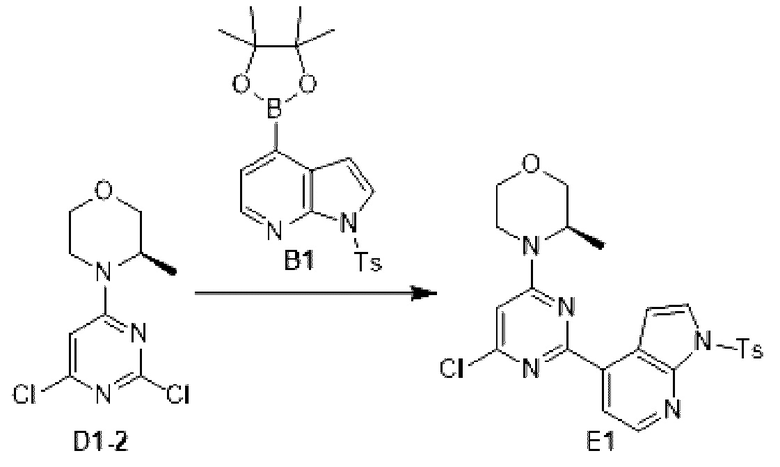
Этап 1: Синтез соединения Е1
К раствору соединения D1-2 (9,03 г, 36,41 ммоль) в 1,4-диоксане (100 мл) добавляли В1 (14,5 г, 36,41 ммоль), бистрифенилфосфин палладия дихлорид (2,555 г, 3,641 ммоль) и карбонат натрия (2 М, 54,61 мл). Реакционную смесь перемешивали под защитой азота при 110°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 600 мл этилацетата (200 мл ×3). Органическую фазу промывали 600 мл воды (200 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=4:1, 4:3) с получением соединения Е1.
МС-ЭСИ м/з: 484,2 [М+Н]+.
Промежуточное соединение 6
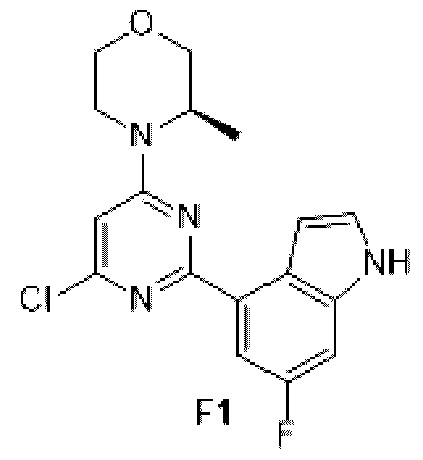
Схема синтеза:
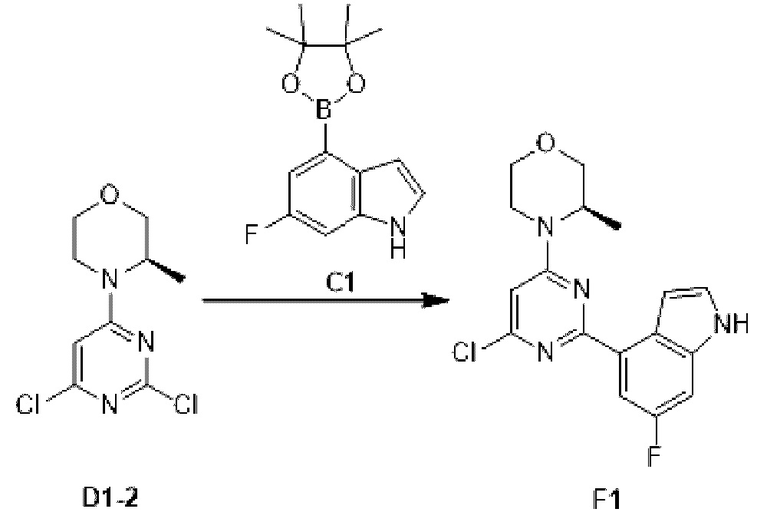
Этап 1: Синтез соединения F1
К раствору соединения D1-2 (0,5 г, 2,02 ммоль) в 1,4-диоксане (10 мл) добавляли С1 (578, 80 мг, 2,22 ммоль), бистрифенилфосфин палладия дихлорид (70,73 мг, 100,76 мкмоль) и карбонат натрия (2 М, 3,02 мл). Реакционную смесь перемешивали под защитой азота при 110°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 60 мл этилацетата (2 0 мл ×3). Органическую фазу промывали 60 мл воды (20 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=4:1, 1:1) с получением соединения F1.
МС-ЭСИ м/з: 347, 1 [М+Н]+.
Промежуточное соединение 7
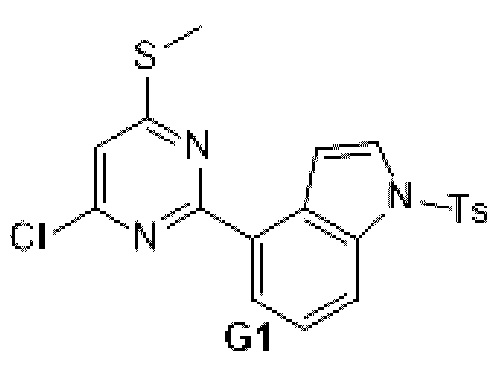
Схема синтеза:
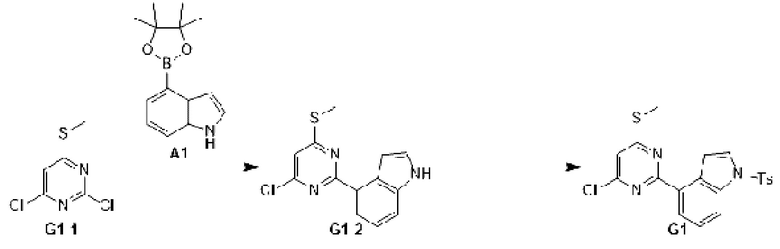
Этап 1: Синтез соединения G1-2
К раствору соединения G1-1 (1 г, 5,13 ммоль) в 1,4-диоксане (25 мл) добавляли А1 (1,37 г, 5,64 ммоль), бистрифенилфосфин палладия дихлорид (359,82 мг, 512,64 мкмоль) и карбонат натрия (2 М, 7,69 мл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 90 мл этилацетата (30 мл ×3). Органическую фазу промывали 90 мл воды (30 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 4:1) с получением соединения G1-2.
МС-ЭСИ м/з: 275,9 [М+Н]+
Этап 2: Синтез соединения G1
К раствору соединения G1-2 (1,09 г, 3,95 ммоль) в дихлорметане (20 мл) добавляли раствор гидроксида натрия (2 М, 5,93 мл) и гидросульфат тетрабутиламмония (671,36 мг, 1,98 ммоль) и затем медленно добавляли p-толуолсульфонилхлорид (1, 13 г, 5,93 ммоль). Реакционную смесь перемешивали при 25°С в течение 15 ч. Реакционную смесь экстрагировали 90 мл дихлорметана (30 мл ×3). Органическую фазу промывали 90 мл воды (30 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 4:1) с получением соединения G1.
МС-ЭСИ м/з: 429,8 [М+Н]+.
Промежуточное соединение 8
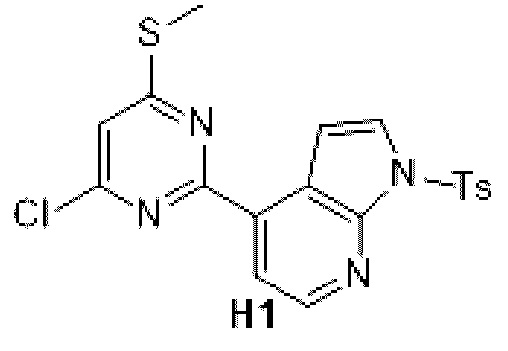
Схема синтеза:
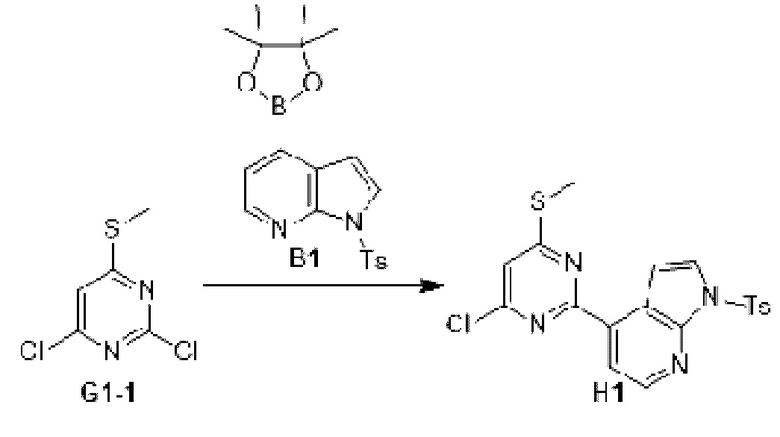
Этап 1: Синтез соединения H1
К раствору соединения G1-1 (48 7, 08 мг, 2,50 ммоль) в 1,4-диоксане (20 мл) добавляли Б1 (1 г, 2,50 ммоль), бистрифенилфосфин палладия дихлорид (87,63 мг, 124,85 мкмоль) и карбонат натрия (2 М, 3,75 мл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 90 мл этилацетата (30 мл ×3). Органическую фазу промывали 90 мл воды (30 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 5:1) с получением соединения H1.
МС-ЭСИ м/з: 431,0 [М+Н]+.
Промежуточное соединение 9
Схема синтеза:
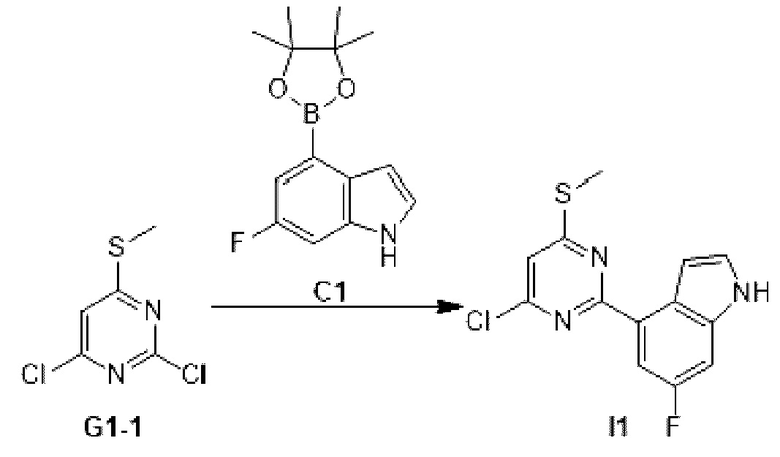
Этап 1: Синтез соединения I1
К раствору соединения G1-1 (493,09 мг, 2,53 ммоль) в 1,4-диоксане (20 мл) добавляли С1 (0,66 г, 2,53 ммоль), бистрифенилфосфин палладия дихлорид (88,71 мг, 126,39 мкмоль) и карбонат натрия (2 М, 3,79 мл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 90 мл этилацетата (30 мл ×3). Органическую фазу промывали 90 мл воды (30 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 5:1) с получением соединения I1.
МС-ЭСИ м/з: 2 9 3,9 [М+Н]+.
Промежуточное соединение 10
Схема синтеза:

Этап 1: Синтез соединения ХХ1-2
При комнатной температуре к раствору соединения ХХ1-1 (500,00 мг, 2,7 4 ммоль) в N,N-диметилформамиде (10,00 мл) добавляли (R)-3-метилморфолин (304,87 мг, 3,01 ммоль), карбонат калия (946,74 мг, 6,85 ммоль), который перемешивали в атмосфере азота при 100°С в течение 12 ч. Реакционную систему разбавляли этилацетатом (30 мл). Органическую фазу промывали водой (20 мл ×3) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=10/1, 5/1) с получением соединения ХХ1-2.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,14 (д, J=6,53 Гц, 3 Н) 3,09 (тд, J=12,80, 3,51 Гц, 1 Н) 3,44 (тд, J=11,80, 3,01 Гц, 1 Н) 3,56-3,62 (м, 1 Н) 3,67-3,73 (м, 1 Н) 3,82-3,96 (м, 2 Н) 4,28 (уш.дд, J=6,52, 2,51 Гц, 1 Н) 6,83 (д, J=1,00 Гц, 1 Н) 6,87 (д, J=1,5 0 Гц, 1 Н).
Этап 2: Синтез соединения XX1
При комнатной температуре, к раствору соединения ХХ1-2 (1,05 г, 4,25 ммоль) в 1,4-диоксане (10,00 мл) добавляли соединение В1 (1,69 г, 4,25 ммоль), дихлорбис(трифенилфосфин) палладия (298,23 мг, 424,89 мкмоль), раствор карбоната натрия (2 М, 6,37 мл), который перемешивали в атмосфере азота при 100°С в течение 9 ч. Реакционную систему разбавляли 20 мл воды и экстрагировали этилацетатом (30 мл). Органическую фазу промывали водой (20 мл) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=3/1, 1/1) с получением соединения XX1.
МС м/з: 483,1 [М+Н]+
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,19 (д, J=6,78 Гц, 3 Н) 2,34 (с, 3 Н) 3,17 (тд, J=12, 74, 3, 89 Гц, 1 Н) 3, 45-3, 54 (м, 1 Н) 3,62-3,67 (м, 1 Н) 3,71-3,78 (м, 1 Н) 3,92-3,99 (м, 2 Н) 4,42 (уш.д, J=6,27 Гц, 1 Н) 6,99 (д, J=1,00 Гц, 1 Н) 7,23 (д, J=4,02 Гц, 1 Н) 7,33 (д, J=1,2 5 Гц, 1 Н) 7,43 (д, J=8,03 Гц, 2 Н) 7,74 (д, J=5,27 Гц, 1 Н) 7,99 (д, J=4,02 Гц, 1 Н) 8,02 (д, J=8,53 Гц, 2 Н) 8,44 (д, J=5,02 Гц, 1 Н).
Промежуточное соединение 11
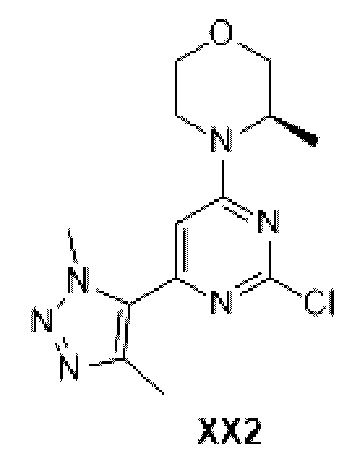
Схема синтеза:
Этап 1: Синтез соединения XX2-1
При 0°С к раствору 4-метоксибензилового спирта (1,11 г, 8,06 ммоль) в тетрагидрофуране (30 мл) добавляли гидрид натрия (386,89 мг, 9,67 ммоль, 60%) при перемешивании в течение 0,5 ч. К реакционной смеси добавляли D1-2 (2 г, 8,09 ммоль) и продували азотом три раза. Реакционную смесь перемешивали при 20°С при нагревании в течение 12 ч, реакцию останавливали добавлением воды (30 мл) и экстрагировали этилацетатом (50 мл). Органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ2-1.
МС-ЭСИ м/з: 350,2 [М+Н]+.
Этап 2: Синтез соединения ХХ2-2
К раствору соединения ХХ2-1 (4,5 г, 12,86 ммоль) в N,N-диметилформамиде (50 мл) добавляли 1,4-диметилтриазол (1,87 г, 19,30 ммоль) бис(трифенилфосфин) палладия дихлорид (451,46 мг, 643,2 мкмоль) и ацетат тетраметиламмония (2,06 г, 15,44 ммоль). Реакционную смесь перемешивали в герметично закрытой пробирке при 130°С при нагревании в течение 12 ч и затем разбавляли этилацетатом (200 мл), промывали водой (ВО мл ×2) и насыщенным солевым раствором (80 мл ×2), сушили над безводным сульфатом натрия и фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ2-2.
МС-ЭСИ м/з: 411,3 [М+Н]+.
Этап 3: Синтез соединения ХХ2-3
К раствору соединения ХХ2-2 (0,85 г, 2,07 ммоль) в этаноле (2 0 мл) добавляли влажный Pd/С (0,2 г, 2,07 ммоль, 10%) и продували водородом три раза. Реакционную смесь перемешивали при 30°С при нагревании в течение 12 ч и затем фильтровали. Фильтрат концентрировали с получением неочищенного соединения ХХ2-3.
МС-ЭСИ м/з: 291,2 [М+Н]+.
Этап 4: Синтез соединения ХХ2
К оксихлориду фосфора (20,35 г, 132,72 ммоль) добавляли соединение ХХ2-3 (0,6 г, 2,07 ммоль), и реакционную смесь перемешивали при 100°С в течение 1 ч. Реакционную смесь гасили добавлением насыщенного раствора бикарбоната натрия при 0°С, доводили рН до 9, экстрагировали дихлорметаном (100 мл), промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением неочищенного соединения ХХ2.
МС-ЭСИ м/з: 309,1 [М+Н]+.
Промежуточное соединение 12
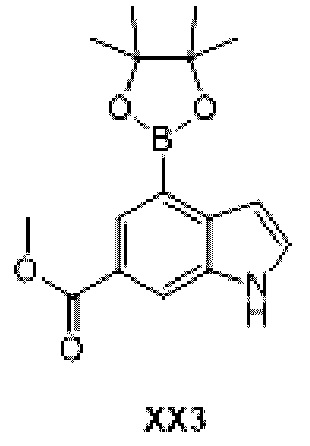
Схема синтеза:
Этап 1: Синтез соединения ХХ3
К раствору соединения ХХ3-1 (2 г, 7,87 ммоль), биспинакол бората (4,00 г, 15,74 ммоль) и 1,1-бис(дифенилфосфино)ферроцен палладия хлорида (0,3 г, 410,00 мкмоль) в 1,4-диоксане (25 мл) добавляли ацетат калия (2,32 г, 23,61 ммоль) и продували азотом три раза. Реакционную смесь перемешивали при 100°С при нагревании в течение 8 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ3.
МС-ЭСИ м/з: 302,1 [М+Н]+.
Промежуточное соединение 13
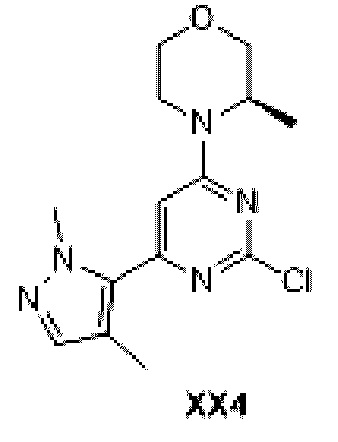
Схема синтеза:

Этап 1: Синтез соединения 1
К раствору соединения D1-2 (3,70 г, 14,91 ммоль), 1,4-диметилпиразол-5-пинакол бората (3,31 г, 14,91 ммоль) и бис (трифенилфосфин) палладия дихлорида (523,3 6 мг, 745,64 мкмоль) в 1,4-диоксане (90 мл) добавляли 2М водный раствор карбоната натрия (22,37 мл), и раствор продували азотом три раза. Реакционную смесь перемешивали при 110°С при нагревании в течение 15 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ4.
МС-ЭСИ м/з: 3 08,2 [М+Н]+.
Промежуточное соединение 14
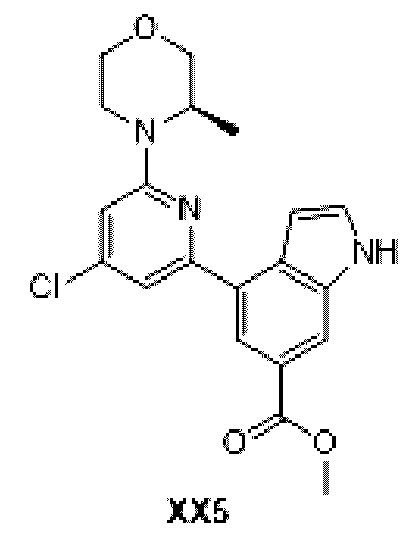
Схема синтеза:
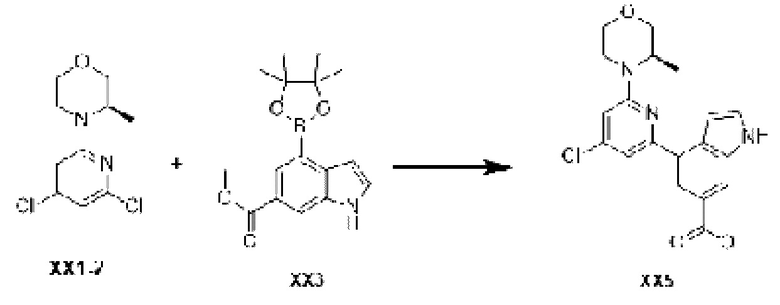
Этап 1: Синтез соединения ХХ5
Кроме использования соответствующих исходных материалов, процедура получения соединения ХХ5 идентична процедуре, использованной для соединения D1 в примере синтеза промежуточного соединения D1.
МС-ЭСИ м/з: 386,2 [М+Н]+.
Промежуточное соединение 15
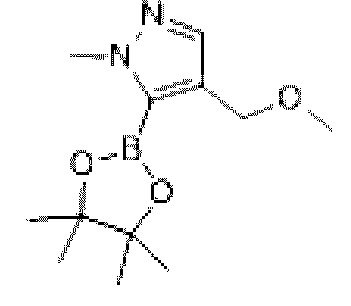
Схема синтеза:
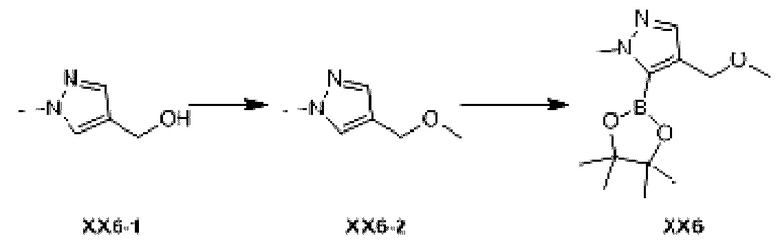
Этап 1: Синтез соединения ХХ6-2
При 0°С к раствору соединения ХХ6-1 (2 г, 17,84 ммоль) в тетрагидрофуране (20 мл) добавляли гидрид натрия (856, 07 мг, 21,40 ммоль, чистота: 60%). Реакционную смесь перемешивали при 25°С в течение 1 ч и затем охлаждали до 0°С, и добавляли метилиодид (11,4 г, 80,32 ммоль, 5,00 мл). Реакционную смесь перемешивали при 25°С в течение 10 ч. В реакционную смесь добавляли насыщенный солевой раствор (30 мл) и экстрагировали этилацетатом (50 мл ×3). Органические фазы объединяли и последовательно промывали (70 мл) и солевым раствором (70 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного соединения ХХ6-2.
МС-ЭСИ м/з: 253,1 [М+Н]+.
Этап 2: Синтез соединения ХХ6
При 0°С к раствору соединения ХХ6-2 (0,5 г, 3,96 ммоль) в те трап идрофу ране (15 мл) добавляли n-бутиллитий (2,5 М, 4,76 мл). Реакционную смесь перемешивали при 25°С в течение 1 ч и затем охлаждали до -78°С, и добавляли изопропанол пинакол борат (818,52 мг, 4,40 ммоль). Реакционную смесь перемешивали при -78°С в течение 0,5 ч и нагревали до 0°С при перемешивании в течение 1 ч. Реакцию останавливали добавлением насыщенного солевого раствора при 0-5°С, доводили значение рН до 6-7 с помощью 1М хлористоводородной кислоты и экстрагировали этилацетатом (40 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ6.
МС-ЭСИ м/з: 127,0 [М+Н]+.
Промежуточное соединение 16
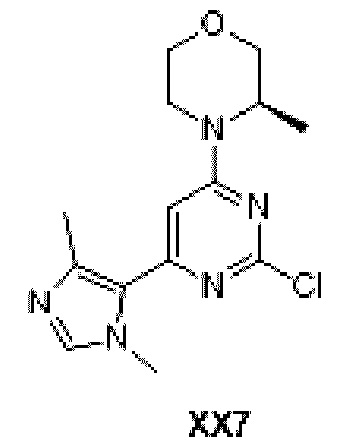
Схема синтеза:
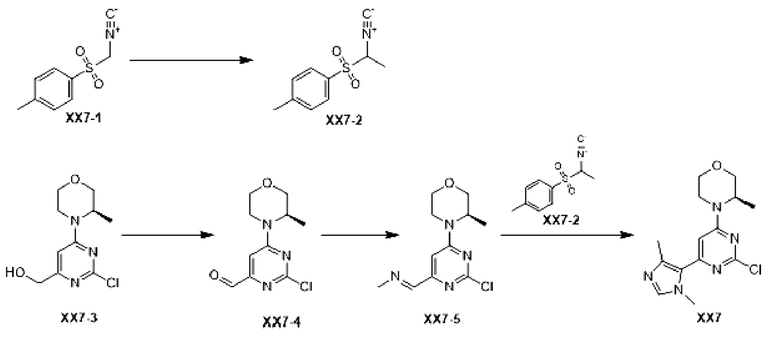
Этап 1: Синтез соединения ХХ7-2
При 0°С к раствору соединения ХХ7-1 (1 г, 5,12 ммоль) в дихлорметане (10 мл) последовательно добавляли бензилтриэтиламмония хлорид (233,33 мг, 1,02 ммоль), метилиодид (2,06 г, 14,51 ммоль, 903,51 мкл) и водный раствор гидроксида натрия (10 мл) с концентрацией 30%. Реакционную смесь перемешивали при 0°С в течение 3 ч и при 25°С в течение 2 ч. Реакционную смесь разбавляли водой (130 мл) и экстрагировали дихлорметаном (75 мл ×2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ7-2.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=7,90 (д, J=8, 3 Гц, 2Н), 7,46 (д, J=8,3 Гц, 2Н), 4,61 (к, J=6,9 Гц, 1Н), 2,52 (с, 3Н), 1,77 ppm (д, J=6, 8 Гц, 3Н)
Этап 2: Синтез соединения ХХ7-4
К раствору соединения ХХ7-3 (1 г, 4, 10 ммоль) в дихлорметане (20 мл) добавляли периодинан Десса-Мартина (2,61 г, 6,16 ммоль). Реакционную смесь перемешивали при 30°С в течение 8 ч, разбавляли водой (20 мл), экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ7-4.
МС-ЭСИ м/з: 242,0 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=9,86 (с, 1Н), 6,95 (с, 1Н), 4,38 (уш.с, 1Н), 4,06 (дд, J=11,8, 3,8 Гц, 1Н), 4,14 (уш.д, J=7,5 Гц, 1Н), 3,80-3,87 (м, 1Н), 3,69-3,76 (м, 1Н), 3,58 (тд, J=12,0, 2,9 Гц, 1Н), 3,37 (уш.т, J=11,8 Гц, 1Н), 1,38 ppm (д, J=6, 8 Гц, 3Н).
Этап 3: Синтез соединения ХХ7-5
К раствору соединения ХХ7-4 (0,51 г, 2,11 ммоль) и метиламин гидрохлорида (712,41 мг, 10,55 ммоль) в толуоле (20 мл) последовательно добавляли триэтиламин (2,14 г, 21,10 ммоль) и безводный сульфат натрия (4,50 г, 31,65 ммоль). Реакционную смесь перемешивали при 50°С в течение 13 ч, и органический растворитель отфильтровывали и концентрировали с получением неочищенного соединения ХХ7-5.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=8,13 (д, J=1,8 Гц, 1Н), 6,97 (с, 1Н), 4,37 (уш.с, 1Н), 4,08 (уш.с, 1Н), 4,00 (дд, J=11,4, 3,6 Гц, 1Н), 3,75-3,81 (м, 1Н), 3,64-3,71 (м, 1Н), 3,49-3,57 (м, 4Н), 3,25-3,35 (м, 1Н), 1,33 ppm (д, J=6, 8 Гц, 3Н)
Этап 4: Синтез соединения ХХ7
К раствору соединения ХХ7-5 (0,535 г, 2,10 ммоль) и ХХ7-2 (439,54 мг, 2,10 ммоль) в этаноле (25 мл) добавляли карбонат калия (725,71 мг, 5,25 ммоль). Реакционную смесь перемешивали при 25°С в течение 48 ч и нагревали до 70°С при перемешивании в течение 12 ч. Реакционную смесь фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ7.
МС-ЭСИ м/з: 308,1 [М+Н]+.
Промежуточное соединение 17

Схема синтеза:

Этап 1: Синтез соединения XX8-2
При 0°C к раствору соединения XX8-1 (10 г, 60,98 ммоль) и 4-диметиламинопиридина (744,96 мг, 6,10 ммоль) в дихлорметане (100 мл) медленно добавляли ди-трет-бутил декарбонат (29,28 г, 134,15 ммоль). Реакционную смесь перемешивали при 30°С в течение 36 ч и добавляли ледяную воду (120 мл) и экстрагировали дихлорметаном (150 мл ×2). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ8-2.
МС-ЭСИ м/з: 364, 1 [М+Н]+.
Этап 2: Синтез соединения ХХ8
К раствору соединения ХХ8-2 (6 г, 16,47 ммоль) и (R)-3-метилморфолина (1,83 г, 18,12 ммоль) в 1, 4-диоксане (50 мл) добавляли N,N-диизопропилэтиламин (2,13 г, 16,47 ммоль). Реакционную смесь перемешивали при 50°С в течение 10 ч и затем концентрировали при пониженном давлении с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ8.
МС-ЭСИ м/з: 429,3 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=6,76 (с, 1Н), 4,30 (уш.с, 1Н), 3,99 (уш.дд, J=11,5, 3,5 Гц, 2Н), 3,74-3,81 (м, 1Н), 3,64-3,72 (м, 1Н), 3,54 (тд, J=11,9, 3,0 Гц, 1Н), 3,28 (тд, J=12,9, 3,9 Гц, 1Н), 1,54 (с, 18Н), 1,31 ppm (д, J=6, 8 Гц, 3Н).
Промежуточное соединение 18
Схема синтеза:
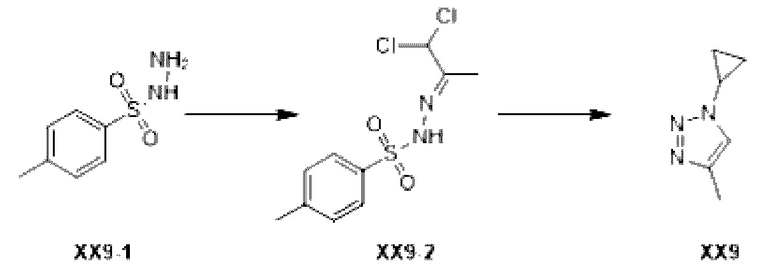
Этап 1: Синтез соединения ХХ9-2
К раствору соединения ХХ9-1 (6,2 г, 33,29 ммоль) в пропионовой кислоте (10 мл) добавляли 1, 1-дихлорацетон (4,57 г, 35,96 ммоль). Реакционную смесь перемешивали при 30°С в течение 14 ч и фильтровали. Осадок промывали циклогексаном (100 мл), и твердое вещество центрифугировали для удаления растворителя с получением неочищенного соединения ХХ9-2.
1H ЯМР (DMSO-d6, 400 МГц): δ=11,88 (уш.с, 1Н), 9,19 (с, 1Н), 7,80 (д, J=8,3 Гц, 2Н), 7,43 (д, J=8,0 Гц, 2Н), 2,39 (с, 3Н), 1,84 ppm (с, 3Н)
Этап 2: Синтез соединения ХХ9
При 0°С к раствору циклопропиламина (1,25 г, 21,89 ммоль, 1,52 мл) в этаноле (50 мл) добавляли триэтиламин (11,08 г, 109,45 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 ч, затем добавляли раствор ХХ9-2 (7,11 г, 24,08 ммоль) в ацетонитриле (50 mL) и нагревали до 30°С при перемешивании в течение 16 ч. Реакционную смесь концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ9.
МС-ЭСИ м/з: 124,0 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=7,32 (с, 1Н), 3,73 (м, 1Н), 2,34 (с, 3Н), 1,20-1,27 (м, 2Н), 1,10-1,17 ppm (м, 2Н).
Промежуточное соединение 19
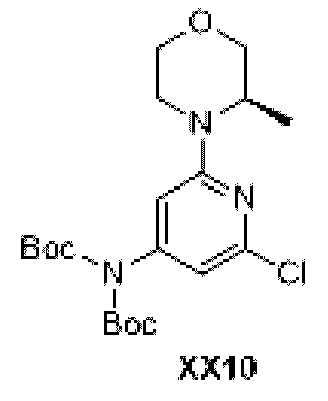
Схема синтеза:
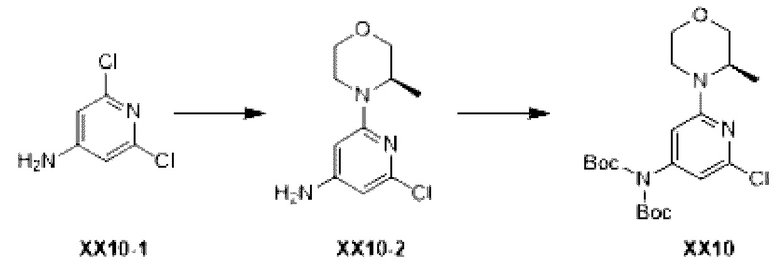
Этап 1: Синтез соединения ХХ10-2
К раствору соединения ХХ10-1 (2 г, 12,27 ммоль) и (R)-3-метилморфолин (1,61 г, 15,95 ммоль) в 1-метил-2-пирролидон (5 мл) добавляли N,N-диизопропилэтиламин (1,74 г, 13,50 ммоль). Реакционную смесь нагревали при воздействии микроволнового излучения при 180°С в течение 1 ч, и реакционную смесь концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения ХХ10-2.
МС-ЭСИ м/з: 228,0 М+Н]+.
Этап 2: Синтез соединения ХХ10
При 0-5°С к раствору соединения ХХ10-2 (1,4 г, 6,15 ммоль) и 4-диметиламинопиридина (1 г, 8,19 ммоль) в дихлорметане (30 мл) медленно добавляли ди-трет-бутил декарбонат (4,03 г, 18,45 ммоль). Реакционную смесь перемешивали при 30°С в течение 5 ч, затем добавляли воду (30 мл) и экстрагировали дихлорметаном (50 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (70 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения XX10.
МС-ЭСИ м/з: 428,6 М+Н]+.
Пример 1: Соединение WX01
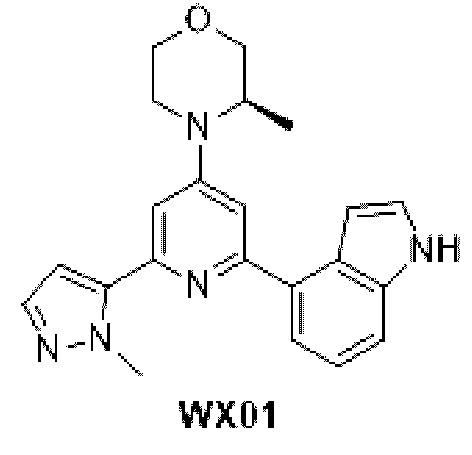
Схема синтеза:
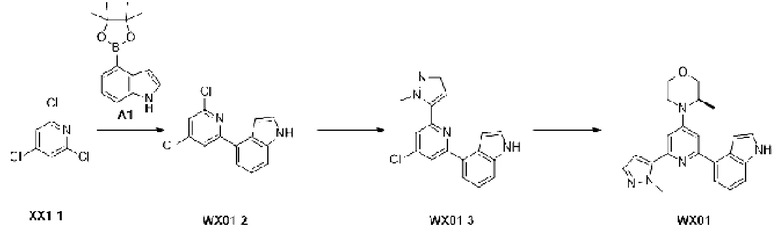
Этап 1: Синтез соединения WX01-2
При комнатной температуре к раствору соединения А1 (300,00 мг, 1,64 ммоль) в 1,4-диоксане (10,00 мл) добавляли соединение ХХ1-1 (398,70 мг, 1,64 ммоль), дихлорбис(трифенилфосфин) палладия (115,11 мг, 164,00 мкмоль), раствор карбоната натрия (2 М, 2,46 мл) и перемешивали при 90°С в течение 12 ч. Реакционную систему разбавляли 20 мл воды и экстрагировали этилацетатом (40 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл ×2) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=10/1, 6/1) с получением соединения WX01-2.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 7,03 (уш.с, 1 Н) 7,23 (т, J=7,78 Гц, 1 Н) 7,51 (т, J=2,76 Гц, 1 Н) 7,57 (д, J=8,03 Гц, 1 Н) 7,63 (д, J=7,53 Гц, 1 Н) 7,71 (д, J=1,51 Гц, 1 Н) 8,07 (д, J=1,51 Гц, 1 Н) 11,41 (уш.с, 1 Н).
Этап 2: Синтез соединения WX01-3
При комнатной температуре к раствору соединения WX01-2 (100,00 мг, 380,05 мкмоль) в 1,4-диоксане (3,00 мл) добавляли 1-метил-1Н-пиразол-5-борную кислоту (47,86 мг, 380,05 мкмоль), дихлорбис(трифенилфосфин) палладия (26,68 мг, 38,00 мкмоль) и раствор карбоната натрия (2 М, 570, 08 мкл) и перемешивали при 90°С в течение 12 ч. Реакционную систему разбавляли 20 мл воды и экстрагировали этилацетатом (30 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл ×2) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который отделяли и очищали на пластинке со слоем силикагеля (петролейный эфир/этилацетат=2/1) с получением соединения WX01-3.
МС м/з: 308,9 [М+Н]+
Этап 3: Синтез соединения WX01
При комнатной температуре к раствору соединения WX01-3 (70,00 мг, 226,71 мкмоль) в 1,4-диоксане (3,00 мл) добавляли (R)-3-метилморфолин (45,86 мг, 453,43 мкмоль), ацетат палладия (26,68 мг, 38,00 мкмоль), 2-дициклогексилфосфоно-2,4,6-триизопропилбифенил (21,62 мг, 45,34 мкмоль), карбонат цезия (221,60 мг, 680,14 мкмоль) и перемешивали при 100°С в атмосфере азота в течение 12 ч. Реакционную систему разбавляли 20 мл воды и экстрагировали этилацетатом (25 мл). Органическую фазу промывали насыщенным солевым раствором (15 мл ×2) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который отделяли с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX01.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,34 (д, J=6,52 Гц, 3 Н) 3,31-3,38 (м, 1 Н) 3,49 (уш.с, 1 Н) 3,70-3,77 (м, 1 Н) 3,88 (с, 2 Н) 4,04-4,13 (м, 2 Н) 4,32 (с, 3 Н) 6,60 (д, J=2,00 Гц, 1 Н) 6,93 (д, J=2,52 Гц, 1 Н) 7,00 (уш.с, 1 Н) 7,19 (д, J=2,00 Гц, 1 Н) 7,31-7,36 (м, 2 Н) 7,50 (д, J=8,52 Гц, 1 Н) 7,53 (д, J=2,00 Гц, 1 Н) 7,61 (д, J=7,52 Гц, 1 Н) 8,33 (уш.с, 1 Н). МС м/з: 374,0 [М+Н]+.
Пример 2: Соединение WX02
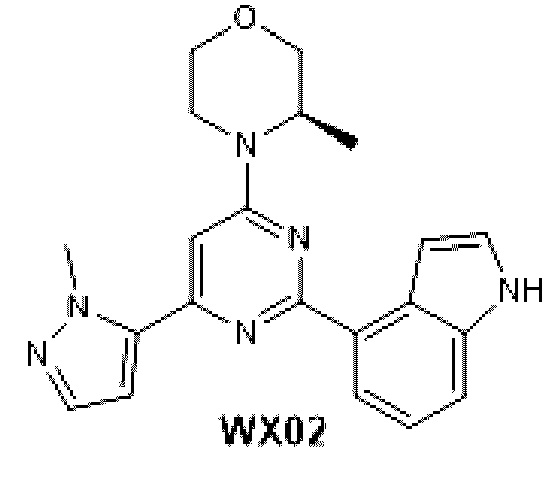
Схема синтеза:
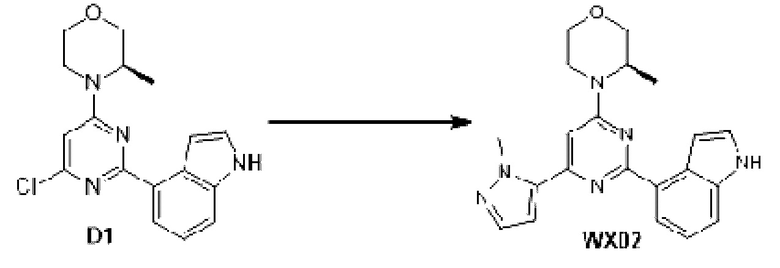
Этап 1: Синтез соединения WX02
К раствору соединения D1 (0,08 г, 2 43,31 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (75,94 мг, 364,97 мкмоль), бистрифенилфосфин палладия дихлорид (17,08 мг, 24,33 мкмоль) и карбонат натрия (2 М, 364,97 мкл). Реакционную смесь перемешивали при 90°С под защитой азота в течение 15 ч. Реакционную смесь фильтровали через целит, и фильтрат экстрагировали 30 мл этилацетата (10 мл ×3). Органическую фазу промывали 3 0 мл воды (10 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который отделяли с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX02.
МС-ЭСИ м/з: 375,0 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,30 (д, J=6,78 Гц, 3 Н) 3,28-3,32 (м, 1 Н) 3,31 (с, 1 Н) 3,55 (тд, J=11,80, 2, 76 Гц, 1 Н) 3,70 (дд, J=11,29, 2,76 Гц, 1 Н) 3,79-3,86 (м, 1 Н) 4,03 (дд, J=11,17, 3,14 Гц, 1 Н) 4,29 (с, 4 Н) 4,68 (уш.с, 1 Н) 6,98-7,04 (м, 1 Н) 7,01 (с, 1 Н) 7,00 (д, J=2,01 Гц, 1 Н) 7,22 (т, J=7,78 Гц, 1 Н) 7,30 (уш.с, 1 Н) 7,47 (т, J=2,64 Гц, 1 Н) 7,52-7,57 (м, 2 Н) 8,13 (д, J=7,28 Гц, 1 Н) 11,22-11,34 (м, 1 Н) 11,29 (уш.с, 1 Н).
Пример 3: Соединение WX03
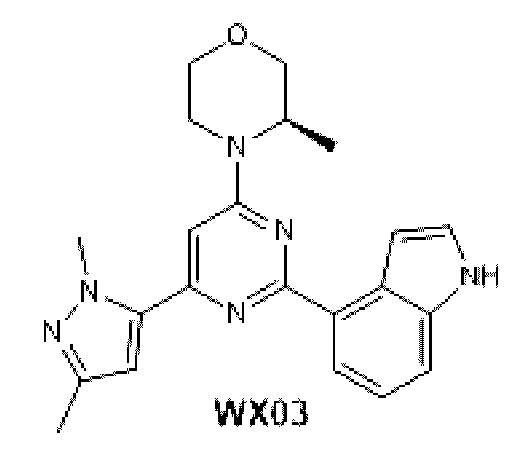
Схема синтеза:

Этап 1: Синтез соединения WX03
К раствору соединения D1 (0,08 г, 243,31 мкмоль) в 1,4-диоксане (5 мл) добавляли 1,3-диметилпиразол-5-пинакол борат (54,04 мг, 243,31 мкмоль), бистрифенилфосфин палладия дихлорид (17,08 мг, 24, 33 мкмоль) и карбонат натрия (2 М, 364, 97 мкл). Реакционную смесь перемешивали при 90°С под защитой азота в течение 15 ч. Реакционную смесь фильтровали через целит, и фильтрат экстрагировали 30 мл этилацетата (10 мл ×3). Органическую фазу промывали 3 0 мл воды (10 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который отделяли с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX03.
МС-ЭСИ м/з: 389,1 [М+Н]+.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,29 (д, J=6,53 Гц, 3 Н) 2,18-2,26 (м, 1 Н) 2,21 (с, 1 Н) 2,26-2,27 (м, 1 Н) 3,23-3,30 (м, 1 Н) 3,24-3,31 (м, 1 Н) 3, 48-3, 59 (м, 1 Н) 3,69 (дд, J=11,42, 2,64 Гц, 1 Н) 3,77-3,88 (м, 1 Н) 3,77-3,84 (м, 1 Н) 4,02 (уш.дд, J=11,29, 3,01 Гц, 1 Н) 4,20 (с, 2 Н) 4,17-4,22 (м, 1 Н) 4,26 (уш.д, J=13,55 Гц, 1 Н) 4,54-4,80 (м, 1 Н) 4,65 (уш.с, 1 Н) 6, 64-6, 85 (м, 1 Н) 6,69-6,83 (м, 1 Н) 6,71-6,82 (м, 1 Н) 6,78 (с, 1 Н) 6,96 (с, 1 Н) 6, 92-7, 03 (м, 1 Н) 7,21 (т, J=7,78 Гц, 1 Н) 7,27-7,33 (м, 1 Н) 7,29 (уш.с, 1 Н) 7,46 (т, J=2,64 Гц, 1 Н) 7,55 (д, J=8,03 Гц, 1 Н) 8,11 (д, J=7,28 Гц, 1 Н) 11,27 (уш.с, 1 Н).
Пример 4: Соединение WX04
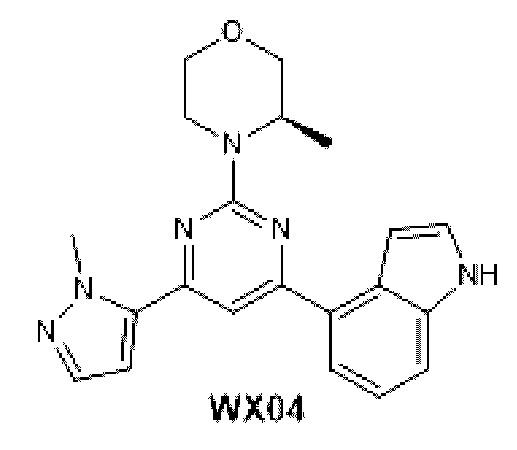
Схема синтеза:

Этап 1: Синтез соединения WX04-1
К раствору соединения G1 (0,3 г, 69 7,77 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (188,74 мг, 907,10 мкмоль), бистрифенилфосфин палладия дихлорид (48,9 8 мг, 69,7 8 мкмоль) и карбонат натрия (2 М, 1,05 мл). Реакционную смесь перемешивали при 90°С под защитой азота в течение 15 ч. Реакционную смесь фильтровали через целит, и фильтрат экстрагировали 60 мл этилацетата (20 мл ×3). Органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 3:1) с получением соединения WX04-1.
МС-ЭСИ м/з: 476,1 [М+Н]+.
Этап 2: Синтез соединения WX04-2
К раствору соединения WX04-1 (0,205 г, 431,05 мкмоль) в дихлорметане (5 мл) добавляли м-хлорпероксибензойную кислоту (87,51 мг, 431,05 мкмоль). Реакционную смесь перемешивали при 20°С в течение 15 ч. Реакционную смесь гасили при 20°С 20 мл насыщенного раствора сульфита натрия и затем экстрагировали 40 мл дихлорметана (20 мл ×2). Органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX04-2.
МС-ЭСИ м/з: 492,0 [М+Н]+.
Этап 3: Синтез соединения WX04-3
К раствору соединения WX04-2(213,90 мг, 435,12 мкмоль) и (R)-3-метилморфолина (220,06 мг, 2,18 ммоль) в 1,4-диоксане (5 мл) добавляли диизопропилэтиламин (562,36 мг, 4,35 ммоль). Реакционную смесь перемешивали при 100°С в течение 65 ч. Реакционную смесь экстрагировали 60 мл этилацетата (20 мл ×3), и органическую фазу промывали 60 мл воды (20 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX04-3.
МС-ЭСИ м/з: 529,1 [М+Н]+.
Этап 4: Синтез соединения WX04
К раствору соединения WX04-3 (0,35 г, 662,10 мкмоль) в метаноле (5 мл) добавляли гидроксид натрия (2 М, 993, 14 мкл). Реакционную смесь перемешивали при 60°С в течение 17 ч. Реакционную смесь экстрагировали 40 мл этилацетата (20 мл ×2), и органическую фазу промывали 60 мл воды (20 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX04.
МС-ЭСИ м/з: 375,0 [М+Н]+
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 3,44 (тд, J=12,99, 3,89 Гц, 1 Н) 3,65 (тд, J=11,86, 2,89 Гц, 1 Н) 3,77-3,83 (м, 1 Н) 3,84-3,89 (м, 1 Н) 4,07 (дд, J=11,29, 3,51 Гц, 1 Н) 4,35 (с, 3 Н) 4,54 (уш.д, J=13,80 Гц, 1 Н) 4,90 (уш.д, J=4,02 Гц, 1 Н) 6,78 (д, J=2,01 Гц, 1 Н) 7,16 (уш.с, 1 Н) 7,30-7,35 (м, 1 Н) 7,35-7,38 (м, 2 Н) 7,52-7,57 (м, 2 Н) 7,69 (д, J=7,53 Гц, 1 Н) 8,35 (уш.с, 1 Н)
Пример 5: Соединение WX05
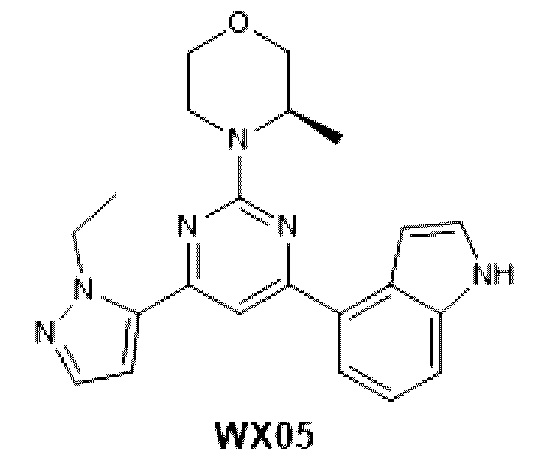
Схема синтеза:
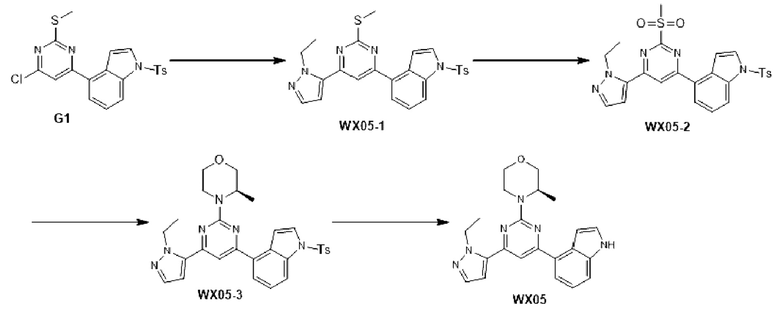
Этап 1: Синтез соединения WX05-1
Кроме использования соответствующих исходных материалов, процедура получения WX05-1 идентична процедуре, использованной для соединения WX04-1 в примере 4 синтеза.
МС-ЭСИ м/з: 490,1 [М+Н]+.
Этап 2: Синтез соединения WX05-2
К раствору соединения WX05-1 (0,193 г, 394,19 мкмоль) в дихлорметане (5 мл) добавляли м-хлорпероксибензойную кислоту (80,03 мг, 394,19 мкмоль). Реакционную смесь перемешивали при 20°С в течение 15 ч. Реакционную смесь гасили добавлением насыщенного раствора сульфита натрия при 20°С и экстрагировали 4 0 мл дихлорметана (20 мл ×2). Органическую фазу промывали 4 5 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX05-2.
МС-ЭСИ м/з: 522,1 [М+Н]+.
Этап 3: Синтез соединения WX05-3
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX05-3 идентична процедуре, использованной для соединения WX04-3 в примере 4 синтеза.
МС-ЭСИ м/з: 543,1 [М+Н]+.
Этап 4: Синтез соединения WX05
Кроме использования соответствующих исходных материалов, процедура получения соединения WX05 идентична процедуре, использованной для соединения WX04 в примере 4 синтеза.
МС-ЭСИ м/з: 389,0 [М+Н]+.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (д, J=6, 78 Гц, 3 Н) 2,29 (с, 3 Н) 3,43 (тд, J=12,86, 3,39 Гц, 1 Н) 3,59-3,69 (м, 1 Н) 3,75-3,81 (м, 1 Н) 3,83-3,88 (м, 1 Н) 4,06 (уш.дд, J=11,04, 3,26 Гц, 1 Н) 4,17 (уш.с, 1 Н) 4,15 (с, 2 Н) 4,56 (уш.д, J=12,05 Гц, 1 Н) 4,92 (уш.д, J=5,02 Гц, 1 Н) 7,17 (уш.с, 1 Н) 7,20 (с, 1 Н) 7,29-7,35 (м, 1 Н) 7,37 (уш.с, 1 Н) 7,40 (с, 1 Н) 7,55 (д, J=8,03 Гц, 1 Н) 7,68 (д, J=7,28 Гц, 1 Н) 8,42 (уш.с, 1 Н).
Пример 6: Соединение WX06
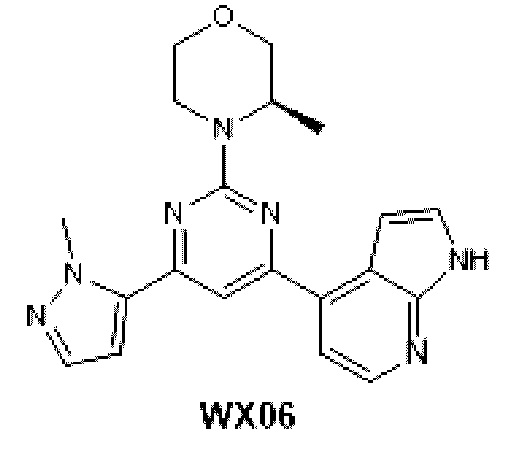
Схема синтеза:
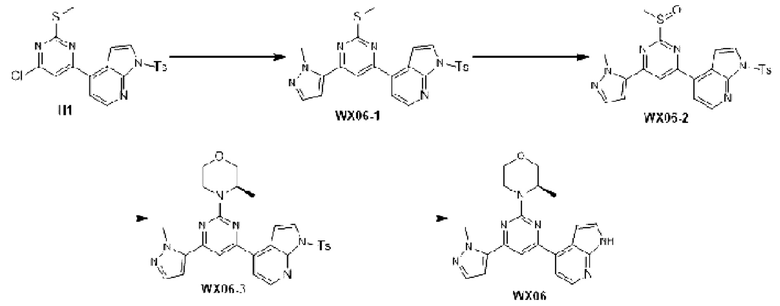
Этап 1: Синтез соединения WX06-1
К раствору соединения H1(200,46 мг, 465,18 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (125,82 мг, 604,73 мкмоль), бистрифенилфосфин палладия дихлорид (16,3 3 мг, 23, 26 мкмоль) и карбонат натрия (2М, 697, 77 мкл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 60 мл этилацетата (20 мл ×3), и органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 3:1) с получением соединения WX06-1.
МС-ЭСИ м/з: 477,0 [М+Н]+.
Этап 2: Синтез соединения WX06-2
К раствору соединения WX06-1 (0,21 г, 440,65 мкмоль) в дихлорметане (5 мл) добавляли м-хлорпероксибензойную кислоту (89,46 мг, 440,65 мкмоль). Реакционную смесь перемешивали при 20°С в течение 15 ч. Реакционную смесь гасили 20 мл насыщенного раствора сульфита натрия при 20°С и затем экстрагировали 40 мл дихлорметана (20 мл ×2). Органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX06-2.
МС-ЭСИ м/з: 493,0 [М+Н]+.
Этап 3: Синтез соединения WX06-3
К раствору соединения WX06-2 (226, 45 мг, 459, 74 мкмоль) и (R)-3-метилморфолина (232,50 мг, 2,30 ммоль) в 1,4-диоксане (5 мл) добавляли диизопропилэтиламин (594,17 мг, 4,60 ммоль). Реакционную смесь перемешивали при 100°С в течение 65 ч. Реакционную смесь экстрагировали 60 мл этилацетата (20 мл ×3), и органическую фазу промывали 30 мл воды (10 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX06-3.
МС-ЭСИ м/з: 530,1 [М+Н]+.
Этап 4: Синтез соединения WX06
К раствору соединения WX06-3 (0,330 г, 623,10 мкмоль) в метаноле (5 мл) добавляли гидроксид натрия (2 М, 934,65 мкл). Реакционную смесь перемешивали при 60°С в течение 17 ч. Реакционную смесь экстрагировали 60 мл этилацетата (20 мл ×3), и органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX06.
МС-ЭСИ м/з: 376,1 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (уш.д, J=6,78 Гц, 3 Н) 3,37-3,53 (м, 1 Н) 3,60-3,71 (м, 1 Н) 3,78-3,84 (м, 1 Н) 3,84-3,91 (м, 1 Н) 4,09 (уш.д, J=9,04 Гц, 1 Н) 4,35 (с, 3 Н) 4,54 (уш.д, J=13,30 Гц, 1 Н) 5,04 (с, 1 Н) 4,88 (уш.д, J=4,52 Гц, 1 Н) 6,75-6,88 (м, 1 Н) 6,81 (с, 1 Н) 6,98-7,13 (м, 1 Н) 7,06 (уш.с, 1 Н) 7,40 (с, 1 Н) 7,50 (уш.с, 1 Н) 7,56 (с, 1 Н) 7,62 (уш.д, J=5,02 Гц, 1 Н) 8,47 (уш.д, J=4,77 Гц, 1 Н) 9,84 (уш.с, 1 Н).
Пример 7: Соединение WX07

Схема синтеза:
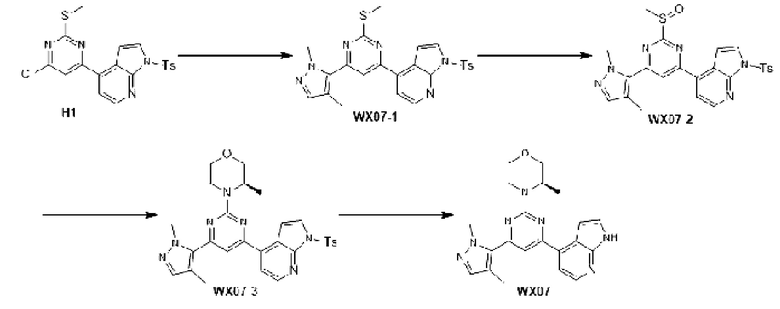
Этап 1: Синтез соединения WX07-1
Кроме использования соответствующих исходных материалов, процедура получения WX07-1 идентична процедуре, использованной для соединения WX06-1 в примере 6 синтеза.
МС-ЭСИ м/з: 491,1 [М+Н]+.
Этап 2: Синтез соединения WX07-2
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX07-2 идентична процедуре, использованной для соединения WX06-2 в примере 6 синтеза.
МС-ЭСИ м/з: 507,1 [М+Н]+.
Этап 3: Синтез соединения WX07-3
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX07-3 идентична процедуре, использованной для соединения WX06-3 в примере 6 синтеза.
МС-ЭСИ м/з: 544,2 [М+Н]+.
Этап 4: Синтез соединения WX07
Кроме использования соответствующих исходных материалов, процедура получения соединения WX07 идентична процедуре, использованной для соединения WX06 в примере 6 синтеза.
МС-ЭСИ м/з: 390,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 2,31 (с, 3 Н) 3,44 (тд, J=12,86, 3,64 Гц, 1 Н) 3,64 (тд, J=11,92, 3,01 Гц, 1 H) 3, 77-3, 82 (м, 1 Н) 3, 84-3, 89 (м, 1 Н) 4,07 (дд, J=11,54, 3,51 Гц, 1 Н) 4,16 (с, 3 Н) 4,55 (уш.д, J=13,30 Гц, 1 Н) 4,90 (уш.д, J=4,77 Гц, 1 Н) 7,06 (уш.с, 1 Н) 7,24 (с, 1 Н) 7,41 (с, 1 Н) 7,48 (уш.с, 1 Н) 7,59-7,66 (м, 1 Н) 8,46 (уш.с, 1 Н) 9,09 (уш.с, 1 Н).
Пример 8: Соединение WX08
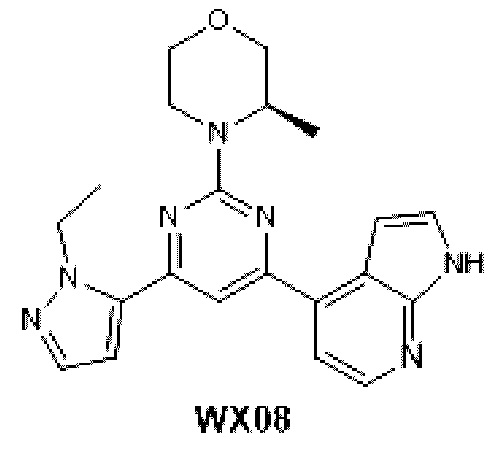
Схема синтеза:

Этап 1: Синтез соединения WX08-1
Кроме использования соответствующих исходных материалов, процедура получения WX08-1 идентична процедуре, использованной для соединения WX06-1 в примере 6 синтеза.
МС-ЭСИ м/з: 491,3 [М+Н]+.
Этап 2: Синтез соединения WX08-2
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX08-2 идентична процедуре, использованной для соединения WX06-2 в примере 6 синтеза.
МС-ЭСИ м/з: 507,1 [М+Н]+.
Этап 3: Синтез соединения WX08-3
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX08-3 идентична процедуре, использованной для соединения WX06-3 в примере 6 синтеза.
МС-ЭСИ м/з: 544,2 [М+Н]+.
Этап 4: Синтез соединения WX08
Кроме использования соответствующих исходных материалов, процедура получения соединения WX08 идентична процедуре, использованной для соединения WX06 в примере 6 синтеза.
МС-ЭСИ м/з: 390,0 [М+Н]+. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,29 (уш.с, 3 Н) 1,39 (уш.с, 3 Н) 3,32 (уш.с, 1 Н) 3,51 (уш.д, J=11,04 Гц, 1 Н) 3,59-3,81 (м, 1 Н) 3,71 (уш.д, J=16,56 Гц, 1 Н) 3,95 (уш.д, J=9,79 Гц, 1 Н) 4,39 (уш.д, J=11,80 Гц, 1 Н) 4,72 (уш.с, 3 Н) 6,66 (уш.с, 1 Н) 6,93 (уш.с, 1 Н) 7,13 (уш.с, 1 Н) 7,32-7,74 (м, 3 Н) 8,35 (уш.с, 1 Н) 9,83 (уш.с, 1 Н).
Пример 9: Соединение WX09
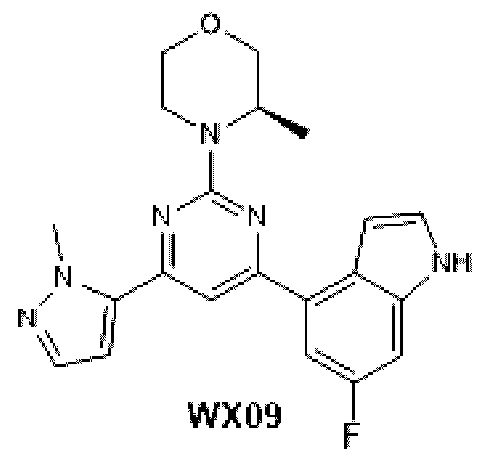
Схема синтеза:
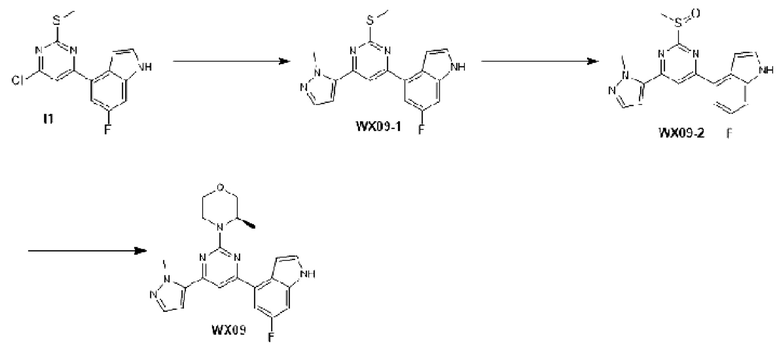
Этап 1: Синтез соединения WX09-1
К раствору соединения II (0,1 г, 340,43 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (92,08 мг, 442,56 мкмоль), бистрифенилфосфин палладия дихлорид (11,95 мг, 17,02 мкмоль) и карбонат натрия (2М, 510,64 мкл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 60 мл этилацетата (20 мл ×3). Органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (петролейный эфир/этилацетат=1:0, 3:1) с получением соединения WX09-1.
МС-ЭСИ м/з: 340,0 [М+Н]+.
Этап 2: Синтез соединения WX09-2
К раствору соединения WX09-1 (102 мг, 300,54 мкмоль) в дихлорметане (5 мл) добавляли м-хлорпероксибензойную кислоту (61,02 мг, 300,54 мкмоль). Реакционную смесь перемешивали при 20°С в течение 15 ч. Реакционную смесь гасили 20 мл насыщенного раствора сульфита натрия при 20°С и затем экстрагировали 40 мл дихлорметана (20 мл ×2). Органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX09-2.
МС-ЭСИ м/з: 356,0[М+Н]+.
Этап 3: Синтез соединения WX09
К раствору соединения WX09-2 (206 мг, 579,65 мкмоль) и (R)-3-метилморфолин (293,15 мг, 2,90 ммоль) в 1,4-диоксане (5 мл) добавляли диизопропилэтиламин (749,14 мг, 5,80 ммоль). Реакционную смесь перемешивали при 100°С в течение 65 ч. Реакционную смесь экстрагировали 60 мл дихлорметана (20 мл×3). Органическую фазу промывали 30 мл воды (10 мл×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX09.
МС-ЭСИ м/з: 393,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (д, J=6,78 Гц, 3 Н) 3,44 (тд, J=12,86, 3,64 Гц, 1 Н) 3,65 (тд, J=11,80, 2,76 Гц, 1 Н) 3,77-3,83 (м, 1 Н) 3,84-3,89 (м, 1 Н) 4,07 (дд, J=11,29, 3,51 Гц, 1 Н) 4,35 (с, 3 Н) 4,53 (уш.д, J=11,80 Гц, 1 Н) 4,88 (уш.д, J=6,53 Гц, 1 Н) 6,79 (д, J=1,76 Гц, 1 Н) 7,08 (уш.с, 1 Н) 7,23 (уш.д, J=8,78 Гц, 1 Н) 7,32-7, 36 (м, 2 Н) 7, 45-7, 50 (м, 1 Н) 7,48 (дд, J=10,67, 2,13 Гц, 1 Н) 7,55 (д, J=2,01 Гц, 1 Н) 8,36 (уш.с, 1 Н).
Пример 10: Соединение WX10
Схема синтеза:
Этап 1: Синтез соединения WX10-1
Кроме использования соответствующих исходных материалов, процедура получения WX10-1 идентична процедуре, использованной для соединения WK09-1 в примере 9 синтеза.
МС-ЭСИ м/з: 353,9 [М+Н]+
Этап 2: Синтез соединения ИХ10-2
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX10-2 идентична процедуре, использованной для соединения НХ09-2 в примере 9 синтеза.
МС-ЭСИ м/з: 3 7 0,0 [М+Н]+.
Этап 3: Синтез соединения ИХ10
Кроме использования соответствующих исходных материалов, процедура получения соединения WX10 идентична процедуре, использованной для соединения WX09 в примере 9 синтеза.
МС-ЭСИ м/з: 407,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (д, J=6,78 Гц, 3 Н) 2,30 (с, 3 Н) 3,43 (тд, J=12,86, 3,64 Гц, 1 Н) 3,61-3,69 (м, 1 Н) 3,76-3,81 (м, 1 Н) 3,83-3,88 (м, 1 Н) 4,06 (дд, J=11,17, 3,39 Гц, 1 Н) 4,15 (с, 3 Н) 4,55 (уш.д, J=12,30 Гц, 1 Н) 4,91 (уш.д, J=4,52 Гц, 1 Н) 7,08 (уш.с, 1 Н) 7,16 (с, 1 Н) 7,23 (дд, J=8,78, 1,51 Гц, 1 Н) 7,34 (т, J=2,76 Гц, 1 Н) 7,40 (с, 1 Н) 7,48 (дд, J=10,54, 2,26 Гц, 1 H) 8,50 (уш.с, 1 Н).
Пример 11: Соединение WX11
Схема синтеза:

Этап 1: Синтез соединения WX11-1
Кроме использования соответствующих исходных материалов, процедура получения WX11-1 идентична процедуре, использованной для соединения WX09-1 в примере 9 синтеза.
МС-ЭСИ м/з: 354,0 [М+Н]+.
Этап 2: Синтез соединения WX11-2
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX11-2 идентична процедуре, использованной для соединения WX09-2 в примере 9 синтеза.
МС-ЭСИ м/з: 370,0 [М+Н]+.
Этап 3: Синтез соединения WX11
Кроме использования соответствующих исходных материалов, процедура получения соединения WX11 идентична процедуре, использованной для соединения WX09 в примере синтеза 9.
МС-ЭСИ м/з: 407,0 [М+Н]+.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (д, J=6,78 Гц, 3 Н) 1,52 (т, J=7,15 Гц, 3 Н) 3, 40-3, 48 (м, 1 Н) 3, 64-3, 69 (м, 1 Н) 3,77-3,82 (м, 1 Н) 3,83-3,89 (м, 1 Н) 4,04-4,10 (м, 1 Н) 4,53 (уш.д, J=13,05 Гц, 1 Н) 4,73-4,90 (м, 3 Н) 6,77 (д, J=1,76 Гц, 1 Н) 7,08 (уш.с, 1 Н) 7,23 (уш.д, J=8,78 Гц, 1 Н) 7,33 (с, 2 Н) 7,48 (дд, J=10,67, 1,88 Гц, 1 Н) 7,57 (д, J=1,51 Гц, 1 Н) 8,42 (уш.с, 1 Н).
Пример 12: Соединение WX12
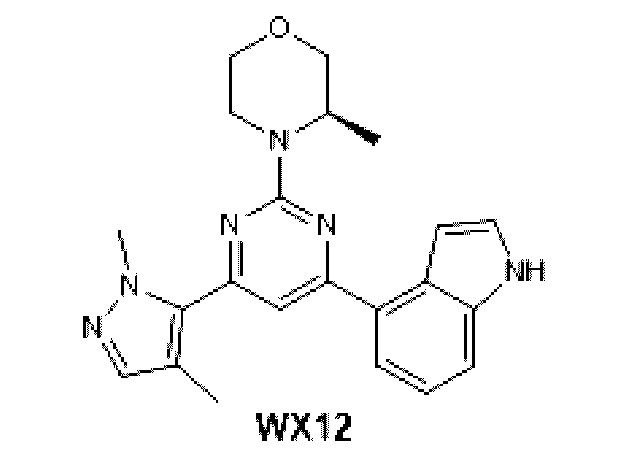
Схема синтеза:
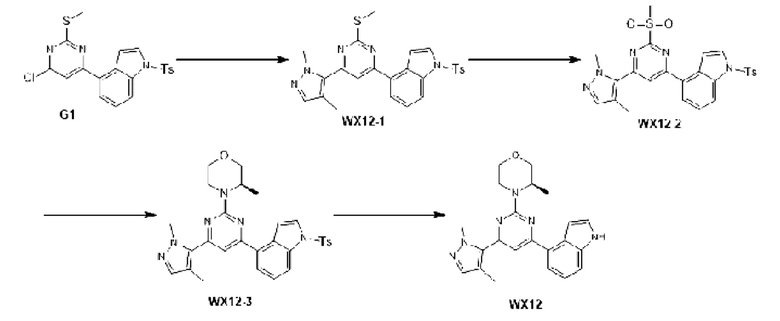
Этап 1: Синтез соединения WX12-1
Кроме использования соответствующих исходных материалов, процедура получения WX12-1 идентична процедуре, использованной для соединения WX04-1 в примере 4 синтеза.
МС-ЭСИ м/з: 490,1 [М+Н]+.
Этап 2: Синтез соединения WX12-2
Кроме использования соответствующих исходных материалов, процедура получением неочищенного WX12-2 желтого цвета идентична процедуре, использованной для соединения WX04-2 в примере 4 синтеза.
МС-ЭСИ м/з: 522,1 [М+Н]+.
Этап 3: Синтез соединения WX12-3
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX12-3 идентична процедуре, использованной для соединения WX04-3 в примере синтеза 4.
МС-ЭСИ м/з: 543,1 [М+Н]+.
Этап 4: Синтез соединения WX12
Кроме использования соответствующих исходных материалов, процедура получения соединения WX12 идентична процедуре, использованной для соединения WX04 в примере 4 синтеза.
МС-ЭСИ м/з: 389,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,41-1,45 (м, 1 Н) 1,43 (д, J=6,78 Гц, 2 Н) 1,53 (т, J=7,15 Гц, 3 Н) 3,45 (тд, J=12,92, 3,51 Гц, 1 Н) 3,66 (тд, J=11,73, 2,64 Гц, 1 Н) 3,77-3,83 (м, 1 Н) 3,84-3,89 (м, 1 Н) 4,07 (уш.дд, J=11,29, 3,01 Гц, 1 Н) 4,55 (уш.д, J=13,55 Гц, 1 Н) 4,74-4,86 (м, 2 Н) 4,88 (уш.д, J=7,03 Гц, 1 Н) 6,76 (д, J=1,51 Гц, 1 Н) 7,16 (уш.с, 1 Н) 7,30-7,35 (м, 1 Н) 7,37 (с, 2 Н) 7,52-7,58 (м, 2 Н) 7,69 (д, J=7,28 Гц, 1 Н) 8,41 (уш.с, 1 Н).
Пример 13: Соединение WX13
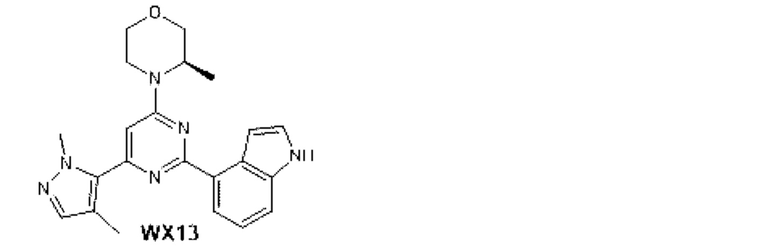
Схема синтеза:
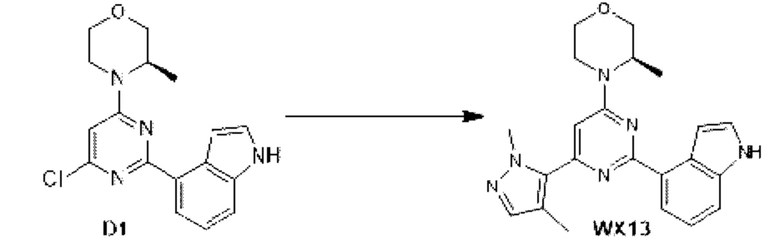
Этап 1: Синтез соединения WX13
К раствору соединения D1 (0,075 г, 228,11 мкмоль) в 1,4-диоксане (5 мл) добавляли 1,4-диметилпиразол-5-пинакол борат (75,99 мг, 342,17 мкмоль), бистрифенилфосфин палладия дихлорид (8,01 мг, 11,41 мкмоль) и карбонат натрия (2 М, 342,17 мкл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 30 мл этилацетата (10 мл ×3), и органическую фазу промывали 30 мл воды (10 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX13.
МС-ЭСИ м/з: 389,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 2,25 (с, 3 Н) 3f43 (тд, J=12,74, 3,89 Гц, 1 Н) 3,69 (тд, J=11,92, 3,01 Гц, 1 Н) 3,80-3,85 (м, 1 Н) 3,87-3,92 (м, 1 Н) 4,12 (дд, J=11,29, 3,51 Гц, 1 Н) 4,15-4,20 (м, 1 Н) 4,16 (с, 2 Н) 4,34 (с, 1 Н) 4,27 (уш.д, J=13,30 Гц, 1 Н) 4,51 (уш.д, J=4,02 Гц, 1 Н) 6,41-6,56 (м, 1 Н) 6,47 (с, 1 Н) 7,29-7, 36 (м, 1 Н) 7,33-7,35 (м, 1 Н) 7,41 (с, 1 Н) 7,50-7,58 (м, 1 Н) 7,52-7,56 (м, 1 Н) 8, 25-8, 37 (м, 2 Н).
Пример 14: Соединение WX14
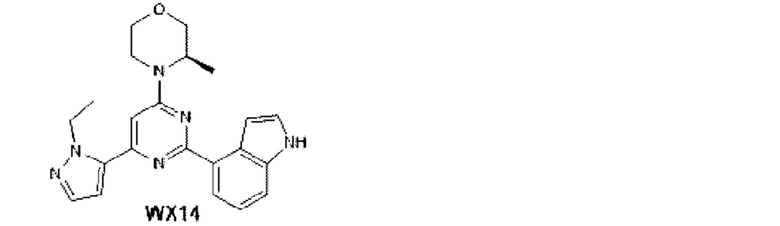
Схема синтеза:
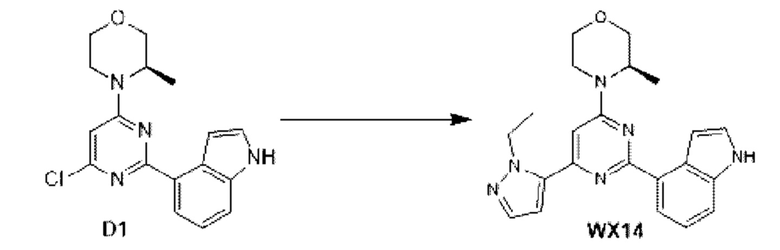
Этап 1: Синтез соединения WX14
К раствору соединения D1 (0,075 г, 228,11 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-этилпиразол-5-пинакол борат (75,99 мг, 342,17 мкмоль), бистрифенилфосфин палладия дихлорид (8,01 мг, 11,41 мкмоль) и карбонат натрия (2 М, 342, 17 мкл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 30 мл этилацетата (10 мл ×3), и органическую фазу промывали 30 мл воды (10 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX14.
МС-ЭСИ м/з: 389, 0 [М+Н]+.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 1,51 (т, J=7,15 Гц, 3 Н) 3,44 (тд, J=12, 74, 3, 89 Гц, 1 Н) 3,68 (тд, J=11,92, 3,01 Гц, 1 Н) 3, 79-3, 86 (м, 1 Н) 3, 87-3, 92 (м, 1 Н) 4,11 (дд, J=11,42, 3,64 Гц, 1 Н) 4,23 (уш.д, J=13,05 Гц, 1 Н) 4,58 (уш.д, J=4,77 Гц, 1 Н) 4,86 (к, J=7,03 Гц, 2 Н) 6,63 (с, 1 Н) 6,65 (д, J=2,01 Гц, 1 Н) 7,30-7,36 (м, 2 Н) 7,50 (уш.с, 1 Н) 7,54 (д, J=8,03 Гц, 1 Н) 7,57 (д, J=1,76 Гц, 1 Н) 8,26 (д, J=7,53 Гц, 1 Н) 8,35 (уш.с, 1 Н).
Пример 15: Соединение WX15
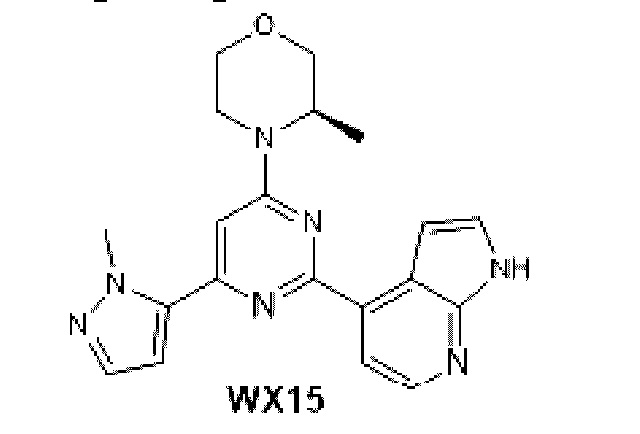
Схема синтеза:
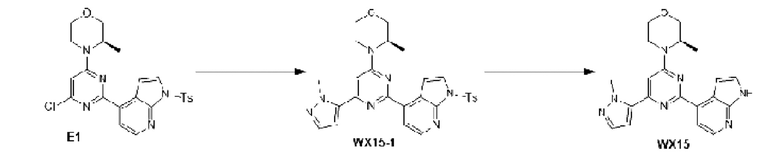
Этап 1: Синтез соединения WX15-1
К раствору соединения Е1 (0,05 г, 103,31 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (25,79 мг, 123,97 мкмоль), бистрифенилфосфин палладия дихлорид (7,25 мг, 10,33 мкмоль) и карбонат натрия (2 М, 154, 97 мкл). Реакционную смесь перемешивали под защитой азота при 90°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 30 мл этилацетата (10 мл ×3), и органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного WX15-1.
МС-ЭСИ м/з: 530,2 [М+Н]+.
Этап 2: Синтез соединения WX15
К раствору соединения WX15-1 (103 мг, 194,48 мкмоль) в метаноле (2 мл) добавляли гидроксид натрия (2 М, 291,72 мкл). Реакционную смесь перемешивали при 60°С в течение 15 ч. Реакционную смесь экстрагировали 60 мл этилацетата (20 мл ×3), и органическую фазу промывали 60 мл воды (20 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения WX15.
МС-ЭСИ м/з: 376,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,44 (д, J=6,78 Гц, 3 Н) 3,45 (тд, J=12,74, 3,89 Гц, 1 Н) 3,68 (тд, J=11,92, 3,01 Гц, 1 Н) 3,80-3,85 (м, 1 Н) 3,88-3,94 (м, 1 Н) 4,13 (дд, J=11,29, 3,51 Гц, 1 Н) 4,22 (уш.д, J=13,05 Гц, 1 Н) 4,38 (с, 3 Н) 4,58 (уш.д, J=4,77 Гц, 1 Н) 6,66-6,71 (м, 2 Н) 7,37 (д, J=1,51 Гц, 1 Н) 7,45-7,50 (м, 1 Н) 7,56 (д, J=2,01 Гц, 1 Н) 8,13 (д, J=5,02 Гц, 1 Н) 8,47 (д, J=5,02 Гц, 1 Н) 9,74 (уш.с, 1 Н).
Пример 16: Соединение 17X16
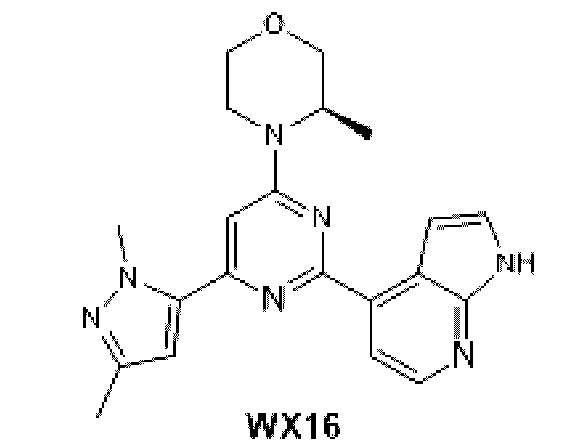
Схема синтеза:
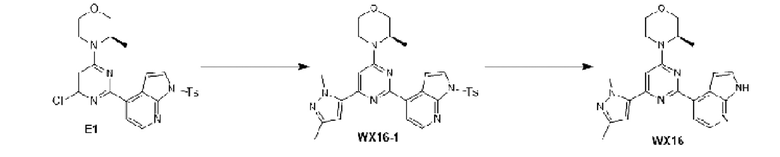
Этап 1: Синтез соединения WX16-1
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX16-1 идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 544, 1 [М+Н]+.
Этап 2: Синтез соединения WX16
Кроме использования соответствующих исходных материалов, процедура получения соединения WX16 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,0 [М+Н]+
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 2,34 (с, 3 Н) 3,44 (тд, J=12,61, 3,89 Гц, 1 Н) 3,67 (тд, J=11,80, 3,01 Гц, 1 Н) 3,78-3,85 (м, 1 Н) 3,87-3,93 (м, 1 Н) 4,12 (дд, J=11, 29, 3, 76 Гц, 1 Н) 4,21 (уш.д, J=12,80 Гц, 1 Н) 4,30 (с, 3 Н) 4,56 (уш.д, J=4,52 Гц, 1 Н) 6,47 (с, 1 Н) 6,66 (с, 1 Н) 7,3
Пример 17: Соединение WX17
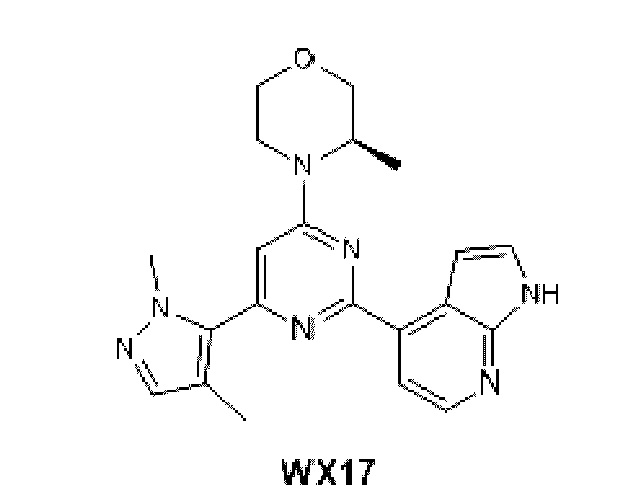
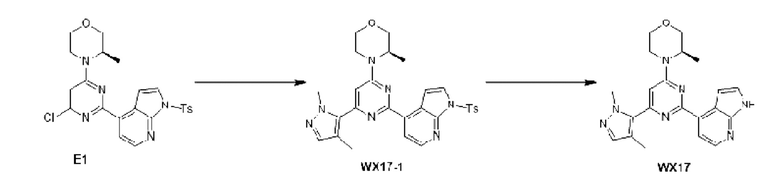
Этап 1: Синтез соединения WX17-1
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX17-1 идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 544,1 [М+Н]+.
Этап 2: Синтез соединения WX17
Кроме использования соответствующих исходных материалов, процедура получения соединения WX17 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,45 (д, J=6,78 Гц, 3 Н) 2,25 (с, 3 Н) 3,45 (тд, J=12, 80, 3, 76 Гц, 1 Н) 3,69 (тд, J=11,92, 2,76 Гц, 1 Н) 3,80-3,86 (м, 1 Н) 3,88-3,93 (м, 1 Н) 4,11-4,17 (м, 4 Н) 4,26 (уш.д, J=12,80 Гц, 1 Н) 4,50 (уш.с, 1 Н) 6,54 (с, 1 Н) 7, 38-7, 44 (м, 2 Н) 7,46 (уш.с, 1 Н) 8,15 (уш.д, J=3,51 Гц, 1 Н) 8,46 (уш.с, 1 Н) 9,67 (уш.с, 1 Н).
Пример 18: Соединение WX18
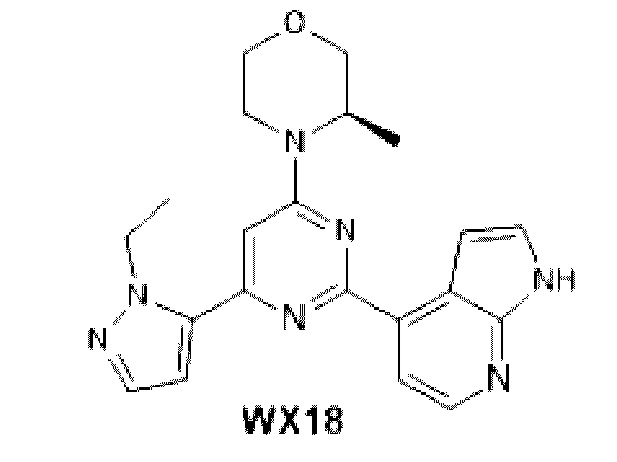
Схема синтеза:
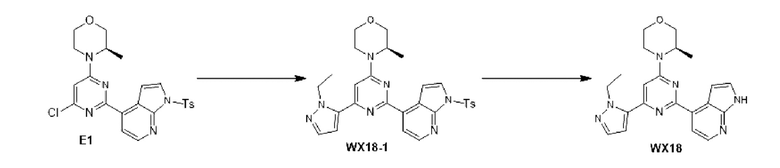
Этап 1: Синтез соединения WX18-1
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX18-1 в виде твердого белого вещества идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 544,2 [М+Н]+.
Этап 2: Синтез соединения WX18
Кроме использования соответствующих исходных материалов, процедура получения соединения WX18 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,46 (уш.д, J=6,53 Гц, 3 Н) 1,51 (уш.т, J=6,90 Гц, 3 Н) 3,42-3,55 (м, 1 Н) 3,68 (уш.т, J=11,67 Гц, 1 Н) 3,75-3,86 (м, 1 Н) 3,89-3, 97 (м, 1 Н) 4,11-4,22 (м, 2 Н) 4,54 (уш.с, 1 Н) 4,69-4,82 (м, 2 Н) 6,70 (с, 1 Н) 6,78 (уш.с, 1 Н) 7,51-7,60 (м, 1 Н) 7, 60-7, 70 (м, 2 Н) 8,38 (уш.д, J=18,32 Гц, 2 Н) 12,42 (уш.с, 1 Н).
Пример 19: Соединение WX19

Схема синтеза:
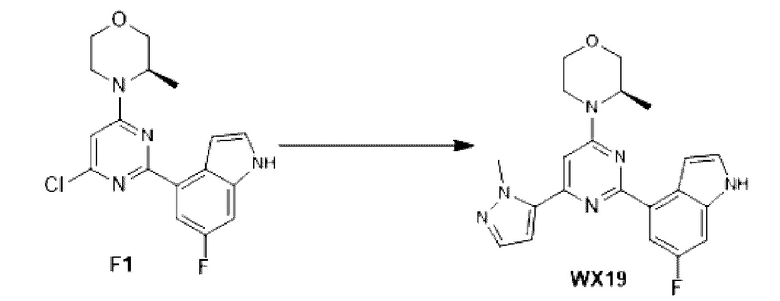
Этап 1: Синтез соединения WX19
К раствору соединения F1 (50 мг, 144,18 мкмоль) в 1,4-диоксане (5 мл) добавляли 1-метилпиразол-5-пинакол борат (36,00 мг, 173,02 мкмоль), бистрифенилфосфин палладия дихлорид (10,12 мг, 14,42 мкмоль) и карбонат натрия (2М, 216,27 мкл). Реакционную смесь перемешивали под защитой азота при 9°С в течение 15 ч. Затем реакционную смесь фильтровали через целит, фильтрат экстрагировали 30 мл этилацетата (10 мл ×3), и органическую фазу промывали 45 мл воды (15 мл ×3) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали с помощью препаративной ЕЭЖХ (нейтральные условия) с получением соединения WX19.
МС-ЭСИ м/з: 393, 0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 3,43 (тд, J=12,61, 3,89 Гц, 1 Н) 3,67 (тд, J=11,80, 2,76 Гц, 1 Н) 3,79-3,84 (м, 1 Н) 3,87-3,92 (м, 1 Н) 4,11 (дд, J=11,54, 3,51 Гц, 1 Н) 4,22 (уш.д, J=13,05 Гц, 1 Н) 4,36 (с, 3 Н) 4,56 (уш.с, 1 Н) 6,64 (с, 1 Н) 6,62-6,65 (м, 1 Н) 6,67 (д, J=l,76 Гц, 1 Н) 7,23 (уш.д, J=7,78 Гц, 1 Н) 7,32 (уш.с, 1 Н) 7,48 (уш.с, 1 Н) 7,55 (д, J=l,76 Гц, 1 Н) 8,03 (дд, J=11,17, 1,88 Гц, 1 Н) 8,34 (уш.с, 1 Н).
Пример 20: Соединение WX20
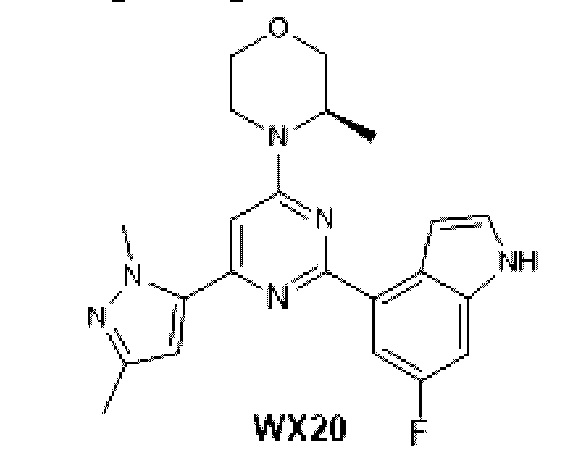
Схема синтеза:
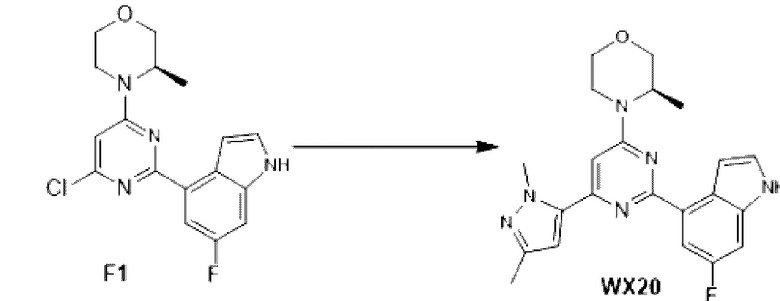
Этап 1: Синтез соединения WX20
Кроме использования соответствующих исходных материалов, процедура получения соединения WX20 идентична процедуре, использованной для соединения WX19 в примере 19 синтеза.
МС-ЭСИ м/з: 407,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,42 (д, J=6,78 Гц, 3 Н) 2,34 (с, 3 Н) 3,42 (тд, J=12,74, 3,64 Гц, 1 Н) 3,66 (тд, J=11,80, 2,76 Гц, 1 Н) 3,78-3,83 (м, 1 Н) 3,86-3,92 (м, 1 Н) 4,11 (уш.дд, J=11,42, 3,39 Гц, 1 Н) 4,21 (уш.д, J=13,30 Гц, 1 Н) 4,28 (с, 3 Н) 4,55 (уш.д, J=5,02 Гц, 1 Н) 6,46 (с, 1 Н) 6,61 (с, 1 Н) 7,21 (уш.д, J=7,53 Гц, 1 Н) 7,31 (уш.с, 1 Н) 7,48 (уш.с, 1 Н) 8,02 (дд, J=11,17, 1,63 Гц, 1 Н) 8,41 (уш.с, 1 Н).
Пример 21: Соединение WX21

Схема синтеза:
Этап 1: Синтез соединения WX21
Кроме использования соответствующих исходных материалов, процедура получения соединения WX21 идентична процедуре, использованной для соединения WX19 в примере 19 синтеза.
МС-ЭСИ м/з: 407,0 [М+Н]+.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,29 (д, J=6,78 Гц, 3 Н) 2,10 (с, 3 Н) 3,28 (тд, J=12,74, 3,64 Гц, 1 Н) 3,54 (тд, J=11,86, 2,64 Гц, 1 Н) 3,66-3,71 (м, 1 Н) 3,73-3,77 (м, 1 Н) 3,95-4,01 (м, 4 Н) 4,11 (уш.д, J=12,30 Гц, 1 Н) 4,34 (уш.с, 1 Н) 6,34 (с, 1 Н) 7,05-7,16 (м, 2 Н) 7,36 (уш.с, 1 Н) 7,90 (уш.д, J=10,54 Гц, 1 Н) 8,35 (уш.с, 1 Н).
Пример 22: Соединение WX22
Схема синтеза:
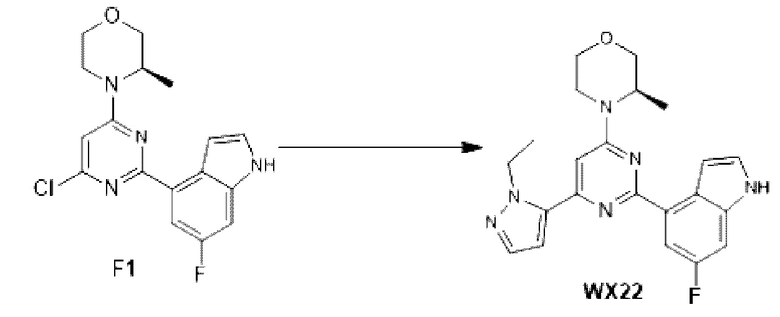
Этап 1: Синтез соединения WX22
Кроме использования соответствующих исходных материалов, процедура получения соединения WX22 идентична процедуре, использованной для соединения WX19 в примере 19 синтеза.
МС-ЭСИ м/з: 407, 0 [М+Н]+.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,43 (д, J=6,78 Гц, 3 Н) 1,52 (т, J=7,15 Гц, 3 Н) 3,43 (тд, J=12,80, 3,76 Гц, 1 Н) 3,67 (тд, J=11,92, 2,76 Гц, 1 Н) 3,78-3,84 (м, 1 Н) 3,87-3,92 (м, 1 Н) 4,11 (дд, J=11,54, 3,51 Гц, 1 Н) 4,21 (уш.д, J=13,05 Гц, 1 Н) 4,56 (уш.д, J=5,02 Гц, 1 Н) 4,83 (к, J=7,03 Гц, 2 Н) 6,62-6,66 (м, 2 Н) 7,22 (дд, J=8, 66, 1, 88 Гц, 1 Н) 7,31 (т, J=2,64 Гц, 1 Н) 7,46 (уш.с, 1 Н) 7,57 (д, J=l,76 Гц, 1 Н) 8,01 (дд, J=11,42, 2,13 Гц, 1 Н) 8,49 (уш.с, 1 Н).
Пример 23: Соединение WX23

Схема синтеза:
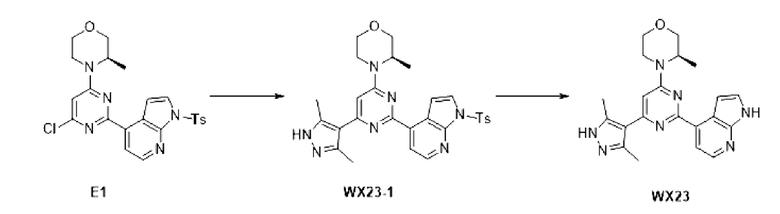
Этап 1: Синтез соединения WX23-1
При комнатной температуре к раствору соединения Е1 (0,16 г, 330,60 мкмоль) в 1,4-диоксане (2,00 мл) добавляли пинаколовый эфир 3,5-диметилпиразол-4-борной кислоты (110,13 мг, 495,90 мкмоль), трис(дибензилацетон) дипалладия (30,27 мг, 33,06 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (38,26 мг, 66,12 мкмоль), фосфат калия (210,53 мг, 991,80 мкмоль) и воду (0,2 мл) и перемешивали при 120°С в микроволновой печи в течение 20 мин. Реакционную систему охлаждали и затем разбавляли этилацетатом (50 мл). Органическую фазу промывали водой (30 мл) и насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=4/1,2/1) с получением соединения WX23-1.
МС м/з: 544,1[М+Н]+.
Этап 2: Синтез соединения WX23
При комнатной температуре к раствору соединения WX23-1 (0,045 г, 82,78 мкмоль) в метаноле (10,00 мл) добавляли раствор гидроксида натрия (2 М, 206,94 мкл) и перемешивали при 30°С в течение 16 ч. Реакционную систему концентрировали при пониженном давлении при 45°С с получением смеси, которую растворяли этилацетатом (30 мл), промывали водой (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (дихлорметан/метанол=100/1,10/1) с получением соединения WX23.
1Н ЯМР (400 МГц, DMSO-d6) 5 ppm 1,30 (уш.д, J=7,03 Гц, 3 Н) 2,43-2,45 (м, 6 Н) 3,37-3,42 (м, 1 Н) 3,51-3,62 (м, 1 Н) 3,71 (уш.д, J=11,54 Гц, 1 Н) 3,82 (уш.д, J=11,54 Гц, 1 Н) 4,03 (уш.д, J=10,04 Гц, 1 Н) 4,21 (уш.д, J=12,55 Гц, 1 Н) 4,60 (уш.с, 1 Н) 6,67 (с, 1 Н) 7,24 (уш.с, 1 Н) 7,58 (уш.с, 1 Н) 8,02 (д, J=4,77 Гц, 1 Н) 8,34 (д, J=5,02 Гц, 1 Н) 11,77 (уш.с, 1 Н) 12,54 (уш.с, 1 Н).
МС м/з: 390,0[М+Н]+.
Пример 24: Соединение WX24
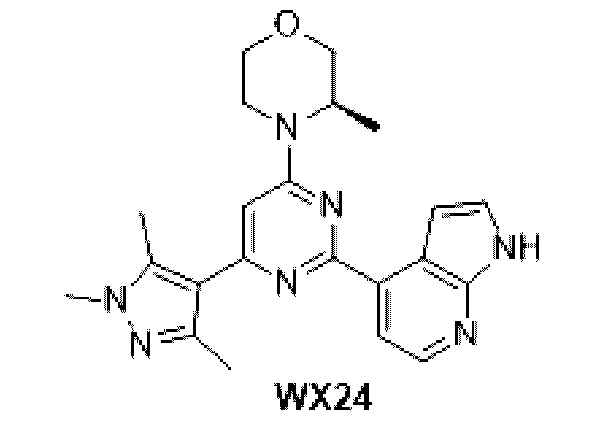
Схема синтеза:

Этап 1: Синтез соединения WX24-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23-1 в примере 23, при этом получали неочищенный продукт, который очищали колоночной хроматографией (петролейный эфир:этилацетат=2/1,1/2) с получением WX24-1.
МС м/з: 558,1[М+Н]+.
Этап 2: Синтез соединения WX24
При комнатной температуре к раствору соединения WX24-1 (0,105 г, 188,28 мкмоль) в метаноле (10,00 мл) добавляли раствор гидроксида натрия (2 М, 941,42 мкл), который перемешивали при 30°С в течение 15 ч. Реакционную систему концентрировали при пониженном давлении при 45°С с получением смеси, которую растворяли в дихлорметане (30 мл), промывали водой (20 мл) и насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (дихлорметан/метанол=100/1,12/1) с получением соединения WX24.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,27 (д, J=6,53 Гц, 3 Н) 2,36 (с, 3 Н) 2,51 (уш.с, 3 Н) 3,30 (уш.с, 1 Н) 3,50-3,57 (м, 1 Н) 3,68 (уш.д, J=8,53 Гц, 1 Н) 3,73 (с, 3 Н) 3,77-3,84 (м, 1 Н) 4,01 (уш.д, J=8,53 Гц, 1 Н) 4,20 (уш.д, J=11,29 Гц, 1 Н) 4,55 (уш.с, 1 Н) 6,63 (с, 1 H) 7,22 (дд, J=3,26, 2,01 Гц, 1 H) 7,56 (т, J=2,89 Гц, 1 Н) 7,99 (д, J=5,02 Гц, 1 Н) 8,32 (д, J=5,02 Гц, 1 Н) 11,75 (уш.с, 1 Н).
МС м/з: 404,0 [М+Н]+.
Пример 25: Соединение WK25
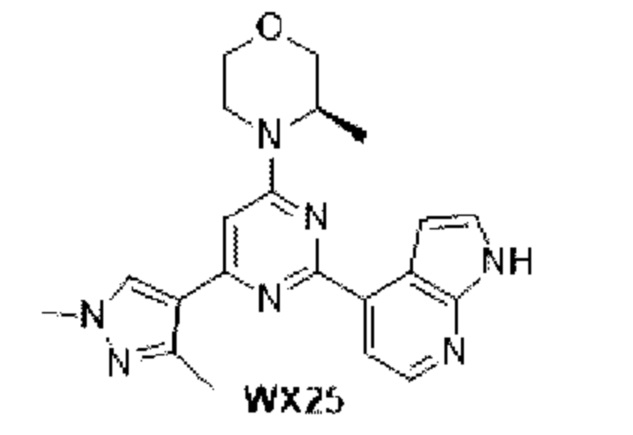
Схема синтеза:

Этап 1: Синтез соединения WX25-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23-1 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (петролейный эфир:этилацетат=2/1,1/2) с получением соединения WX25-1.
МС м/з: 544,1[М+Н]+
Этап 2: Синтез соединения WX25
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=100/1,10/1) с получением соединения WX25.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,27 (д, J=6, 78 Гц, 3 Н) 2,55 (с, 3 Н) 3,20-3,27 (м, 1 Н) 3,49-3,58 (м, 1 Н) 3,68 (дд, (J=11,29, 2,76 Гц, 1 Н) 3, 78-3,85 (м, 4 Н) 4,01 (уш.д, J=8,28 Гц, 1 Н) 4,18 (уш.д, J=13,80 Гц, 1 Н) 4,60 (уш.с, 1 Н) 6,85 (с, 1 Н) 7,23 (дд, J=3,26, 2,01 Гц, 1 Н) 7,57 (т, J=2,89 Гц, 1 Н) 8,02 (д, J=5,02 Гц, 1 Н) 8,32 (д,.J=5,02 Гц, 1 Н) 8,40 (с, 1 Н) 11,75 (уш.с, 1 Н).
МС м/з: 390,0 [М+Н]+.
Пример 26: Соединение WX26
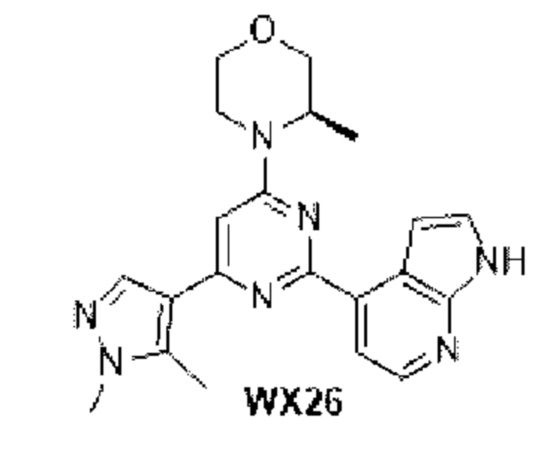
Схема синтеза:
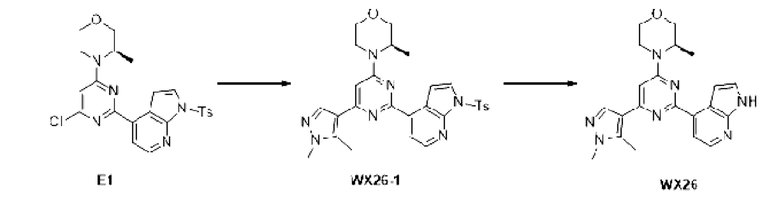
Этап 1: Синтез соединения WX26-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23-1 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (петролейный эфир:этилацетат=2/1,1/2) с получением соединения WX26-1.
МС м/з: 544,2 [М+Н]+.
Этап 2: Синтез соединения WX26
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=100/1,12/1) с получением соединения WX26.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,27 (д, J=6,78 Гц, 3 Н) 2,74 (с, 3 Н) 3,22-3,28 (м, 1 Н) 3,48-3,57 (м, 1 Н) 3,68 (дд, J=11,54, 2,76 Гц, 1 Н) 3,78-3,84 (м, 4 Н) 3,96-4,04 (м, 1 Н) 4,21 (уш.д, J=12,55 Гц, 1 Н) 4,65 (уш.с, 1 Н) 6,92 (с, 1 Н) 7,21 (дд, J=3,26, 2,01 Гц, 1 Н) 7,55-7,59 (м, 1 Н) 7,98 (д, J=5,02 Гц, 1 Н) 8,11 (с, 1 Н) 8,33 (д, J=5,02 Гц, 1 Н) 11,76 (уш.с, 1 Н).
МС м/з: 390,1 [М+Н]+.
Пример 27: Соединение WX27
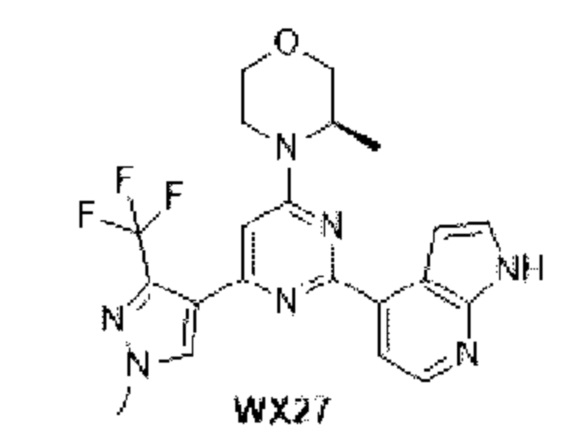
Схема синтеза:
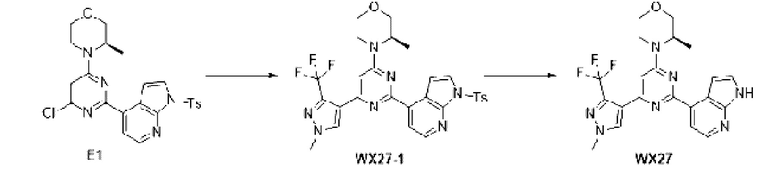
Этап 1: Синтез соединения ИХ.27-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WK23-1 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (петролейный эфир:этилацетат=4/1,2/1) с получением соединения WX27-1.
МС м/з: 598,1 [М+Н]+.
Этап 2: Синтез соединения WX27
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=100/1, 20/1) с получением соединения WX27.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,29 (д, J=6,76 Гц, 3 Н) 3,25-3,30 (м, 1 Н) 3,54 (тд, J=11,84, 3,14 Гц, 1 Н) 3,69 (дд, J=11,52, 2,76 Гц, 1 Н) 3,82 (д, J=11,28 Гц, 1 Н) 3,98-4,06 (м, 4 Н) 4,19 (уш.д, J=11,52 Гц, 1 Н) 4,56 (уш.с, 1 Н) 6,95 (с, 1 Н) 7,22 (дд, J=3,40, 1,88 Гц, 1 Н) 7,55-7,59 (м, 1 Н) 8,07 (д, J=5,28 Гц, 1 Н) 8,32 (д, J=5,04 Гц, 1 Н) 8,67 (с, 1 Н) 11,77 (уш.с, 1 Н).
МС м/з: 444,0 [М+Н]+.
Пример 28: Соединение WX28
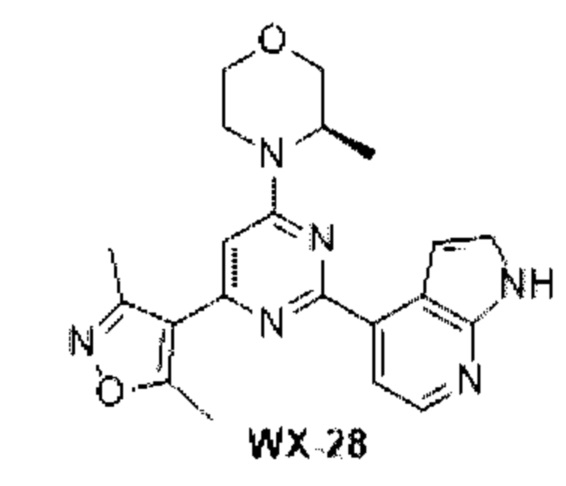
Схема синтеза:
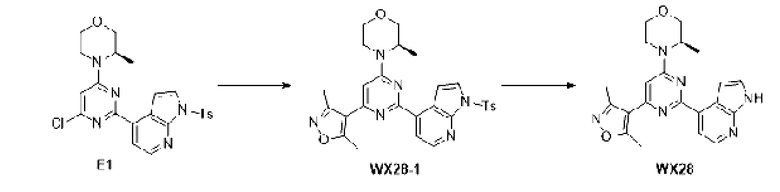
Этап 1: Синтез соединения WX28-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23-1 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (петролейный эфир:этилацетат=2/1,2/1) с получением соединения WX28-1.
МС м/з: 545,1 [М+Н]+
Этап 2: Синтез соединения WX28
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX23 в примере 23, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=100/1,9/1) с получением соединения WX28.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,32 (уш.д, J=6,28 Гц, 3 Н) 2,31-2,47 (м, 3 Н) 2,70 (уш.с, 3 Н) 3,56 (уш.т, J=11,16 Гц, 1 Н) 3,71 (уш.д, J=11,28 Гц, 1 Н) 3, 79-3, 89 (м, 1 Н) 4,04 (уш.д, J=8,76 Гц, 1 Н) 4,27 (уш.д, J=10,56 Гц, 1 Н) 4,64 (уш.с, 1 Н) 6,84 (уш.с, 1 Н) 7,22 (уш.с, 1 Н) 7,60 (уш.с, 1 Н) 8,02 (уш.с, 1 Н) 8,36 (уш.с, 1 Н) 11,82 (уш.с, 1 Н).
МС м/з: 391,0 [М+Н]+.
Пример 29: Соединение WX29
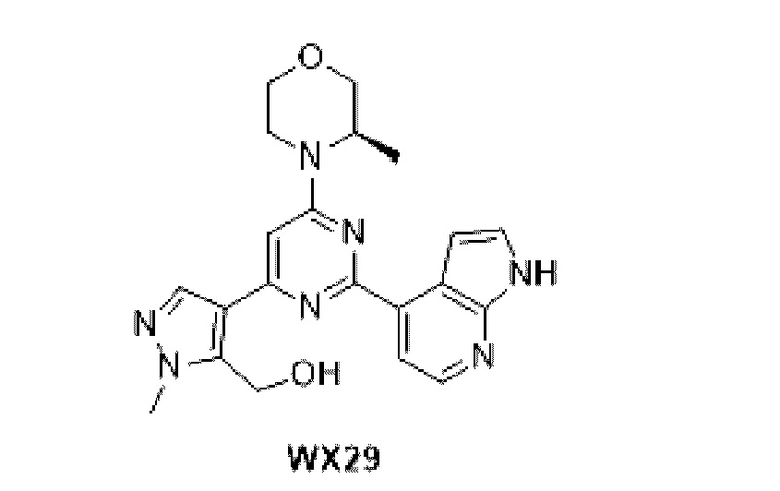
Схема синтеза:

Этап 1: Синтез соединения WX29-2
При комнатной температуре к раствору соединения WX29-1 (0,3 г, 1,59 ммоль) в 1,4-диоксане (8 мл) добавляли биспинакол борат (90,16 мг, 464,91 мкмоль), комплекс [1,1'-бис (дифенилфосфино) ферроцен] палладия дихлорид-дихлорметан (64,81 мг, 79,36 мкмоль) и ацетат калия (467,32 мг, 4,76 ммоль), который перемешивали при 100°С в микроволновой печи в течение 1 ч. Реакционную систему охлаждали и затем разбавляли этилацетатом (30 мл). После фильтрования растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат =10/1,5/1) с получением соединения WK29-2.
МС м/з: 237,0[М+Н]+.
Этап 2: Синтез соединения WX29-3
При комнатной температуре к раствору соединения Е1 (0,15, 309,94 мкмоль) в 1,4-диоксане (2,00 мл) добавляли WX29-2 (87,80 мг, 371,93 мкмоль), трис (дибензилацетон) дипалладия (28,38 мг, 30,99 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (35,87 мг, 61,99 мкмоль), фосфат калия (197,37 мг, 929,82 мкмоль) и воду (0,2 мл), который перемешивали при 120°С в микроволновой печи в течение 20 мин. Реакционную систему охлаждали и затем разбавляли этилацетатом (30 мл). Органическую фазу промывали водой (30 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=2/1,1/2) с получением соединения WX29-3.
МС м/з: 558,1 [М+Н]+.
Этап 3: Синтез соединения WX29-4
При комнатной температуре к раствору соединения WX29-3 (0,12 г, 215,20 мкмоль) в метаноле (8,00 мл) добавляли боргидрид натрия (16,28 мг, 430,40 мкмоль) и перемешивали при 30°С в течение 4 ч. Реакционную систему концентрировали при пониженном давлении при 45°С с получением смеси, которую растворяли этилацетатом (30 мл), промывали водой (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного соединения WX29-4.
МС м/з: 560,1 [М+Н]+.
Этап 4: Синтез соединения WX29
При комнатной температуре к раствору соединения WX29-4 (0,09 г, 150,60 мкмоль) в метаноле (10,00 мл) добавляли раствор гидроксида натрия (2 М, 753,00 мкл) и перемешивали при 30°С в течение 15 ч. Реакционную систему концентрировали при пониженном давлении при 45°С с получением смеси, которую растворяли дихлорметаном (30 мл), промывали насыщенным солевым раствором (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (дихлорметан/метанол=100/1,15/1) с получением соединения WX29.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,30 (д, J=6,52 Гц, 3 Н) 3,26-3,30 (м, 1 Н) 3,51-3,60 (м, 1 Н) 3,71 (уш.д, J=9,04 Гц, 1 Н) 3,79-3,87 (м, 1 Н) 3,93 (с, 3 Н) 4,04 (уш.д, J=8,04 Гц, 1 Н) 4,24 (уш.д, J=12,56 Гц, 1 Н) 4,65 (уш.с, 1 Н) 5,05 (уш.д, J=4,52 Гц, 2 Н) 5,61 (уш.с, 1 Н) 7,04 (с, 1 Н) 7,23 (уш.с, 1 Н) 7,60 (т, J=2,76 Гц, 1 Н) 7,98 (д, J=4,76 Гц, 1 Н) 8,15 (с, 1 Н) 8,36 (д, J=5,04 Гц, 1 Н) 11,79 (уш.с, 1 Н).
МС м/з: 406,0 [М+Н]+.
Пример 30: Соединение WX30
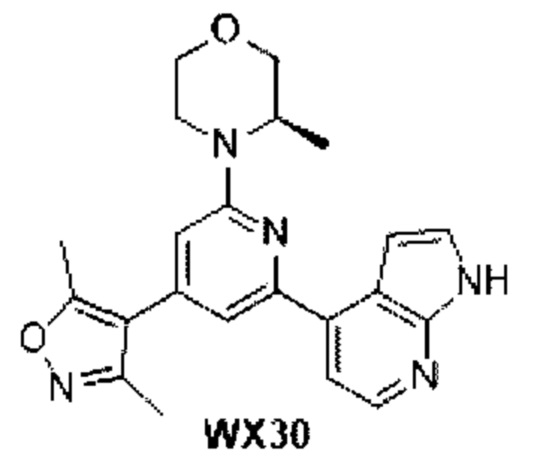
Схема синтеза:
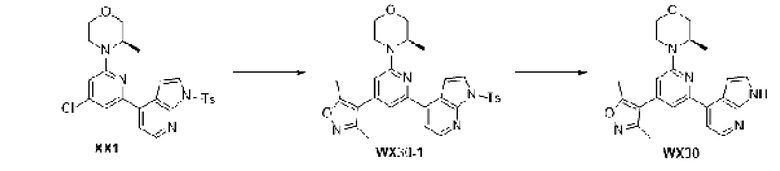
Этап 1: Синтез соединения WX30-1
При комнатной температуре к раствору соединения XX1 (0,1 г, 207,05 мкмоль) в 1,4-диоксане (10,00 мл) добавляли пинаколовый эфир 3,5-диметилизоксазол-4-борной кислоты (55,42 мг, 248,46 мкмоль), трис(дибензилацетон)дипалладий (18,96 мг, 20,70 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (23,96 мг, 41,41 мкмоль), фосфат калия (131,85 мг, 621,14 мкмоль) и воду (0,2 мл) и перемешивали при 120°С в микроволновой печи в течение 20 мин. Реакционную систему охлаждали и затем разбавляли этилацетатом (30 мл). Органическую фазу промывали водой (30 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат=2/1,1/1) с получением соединения WX30-1.
МС м/з: 566,1 [M+Na]+.
Этап 2: Синтез соединения WX30
При комнатной температуре к раствору соединения WX30-1 (0,06 г, 110,37 мкмоль) в метаноле (10,00 мл) добавляли раствор гидроксида натрия (2 М, 551,84 мкл), который перемешивали при 40°С в течение 15 ч. Реакционную систему концентрировали при пониженном давлении при 45°С с получением смеси, которую растворяли этилацетатом (30 мл), промывали водой (20 мл) и сушили над безводным сульфатом натрия. Затем десиккант отфильтровывали, растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (дихлорметан/метанол=40/1,20/1) с получением соединения WX30.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,23 (д, J=6,53 Гц, 3 Н) 2,34 (с, 3 Н) 2,52 (уш.с, 3 Н) 3,20 (тд, J=12,67, 3,76 Гц, 1 Н) 3,50-3,60 (м, 1 Н) 3, 66-3,73 (м, 1 Н) 3,76-3,82 (м, 1 Н) 4,00 (дд, J=11,04, 3,01 Гц, 1 Н) 4,10 (уш.д, J=11,29 Гц, 1 Н) 4,50 (уш.д, J=6,78 Гц, 1 Н) 6,80 (с, 1 Н) 6,93 (дд, J=3,39, 1, 88 Гц, 1 Н) 7,27 (с, 1 Н) 7,55-7,60 (м, 2 Н) 8,30 (д, J=5, 02 Гц, 1 Н) 11,79 (уш.с, 1 Н).
МС м/з: 390,2 [М+Н]+.
Пример 31: Соединение WX31

Схема синтеза:
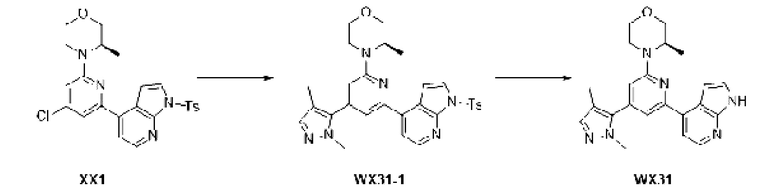
Этап 1: Синтез соединения WX31-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, описанной для соединения WX30-1 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX31-1.
МС м/з: 543,1 [М+Н]+.
Этап 2: Синтез соединения WX31
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX30 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX31.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,24 (д, J=6,53 Гц, 3 Н) 2,08 (с, 3 Н) 3,21 (тд, J=12,61, 3,64 Гц, 1 Н) 3,55 (тд, J=11,73, 2,89 Гц, 1 Н) 3,67-3,73 (м, 1 Н) 3,76-3,81 (м, 1 Н) 3,84 (с, 3 Н) 3,97-4,02 (м, 1 Н) 4,14 (уш.д, J=12,05 Гц, 1 Н) 4,50 (уш.д, J=6, 27 Гц, 1 Н) 6,84 (с, 1 Н) 6,93 (дд, J=3, 39, 1,88 Гц, 1 Н) 7,28 (с, 1 Н) 7,39 (с, 1 Н) 7, 56-7, 60 (м, 2 Н) 8,30 (д, J=5, 02 Гц, 1 Н) 11,80 (уш.с, 1 Н).
МС м/з: 389,2 [М+Н]+.
Припер 32: Соединение WX32
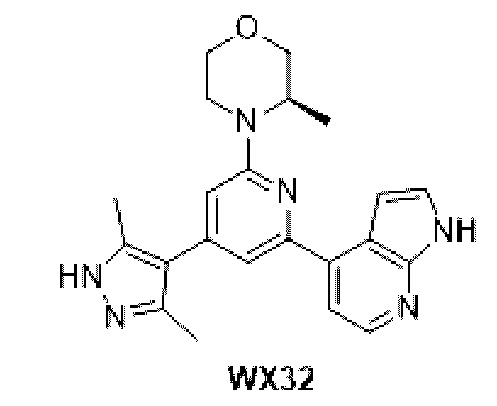
Схема синтеза:

Этап 1: Синтез соединения WX32-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для получения соединения WX30-1 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX32-1.
МС м/з: 543,1 [М+Н]+.
Этап 2: Синтез соединения WX32
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX30 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX32.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,22 (д, J=6, 53 Гц, 3 Н) 2, 27-2, 39 (м, 6 Н) 3,18 (тд, J=12,61, 3,64 Гц, 1 Н) 3,50-3,59 (м, 1 Н) 3, 65-3, 73 (м, 1 Н) 3,75-3,81 (м, 1 Н) 3,95-4,03 (м, 1 Н) 4,07 (уш.д, J=11,54 Гц, 1 Н) 4,47 (уш.д, J=5,27 Гц, 1 Н) 6,68 (с, 1 Н) 6,91 (дд, J=3,39, 1, 88 Гц, 1 Н) 7,21 (с, 1 Н) 7,52-7,59 (м, 2 Н) 8,29 (д, J=5,02 Гц, 1 Н) 11,76 (уш.с, 1 Н) 12,48 (уш.с, 1 Н).
МС м/з: 389,2 [М+Н]+.
Пример 33: Соединение WX33
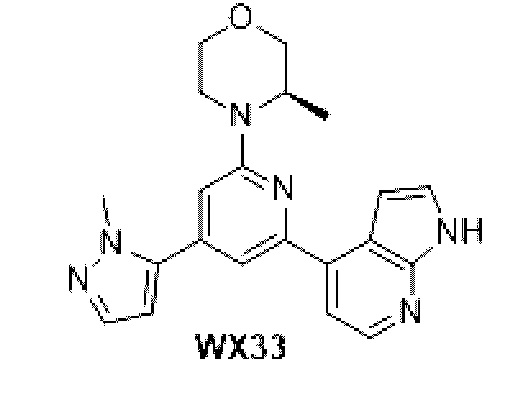
Схема синтеза:
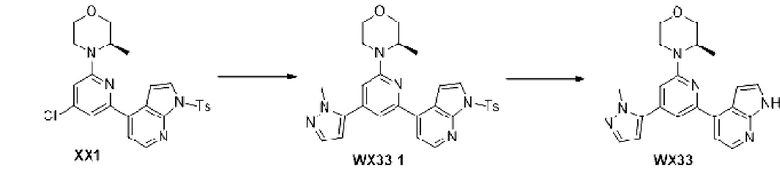
Этап 1: Синтез соединения WX33-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX3Q0-1 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX33-1.
МС м/з: 529,1 [М+Н]+.
Этап 2: Синтез соединения WX33
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX30 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1,10/1) с получением соединения WX33.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,24 (д, J=6, 53 Гц, 3 Н) 3,22 (тд, J=12,61, 3,64 Гц, 1 Н) 3,55 (тд, J=11,73, 2, 64 Гц, 1 Н) 3, 67-3,74 (м, 1 Н) 3,76-3,81 (м, 1 Н) 3,99 (с, 4 Н) 4,11 (уш.д, J=11,54 Гц, 1 Н) 4,55 (уш.д, J=7,03 Гц, 1 Н) 6,67 (д, J=1,76 Гц, 1 Н) 6,89-7,01 (м, 2 Н) 7,40 (с, 1 Н) 7,54 (д, J=1,76 Гц, 1 Н) 7, 56-7,58 (м, 1 Н) 7,60 (д, J=5,02 Гц, 1 Н) 8,31 (д, J=5,02 Гц, 1 Н) 11,79 (уш.с, 1 Н)
МС м/з: 375,2 [М+Н]+.
Пример 34
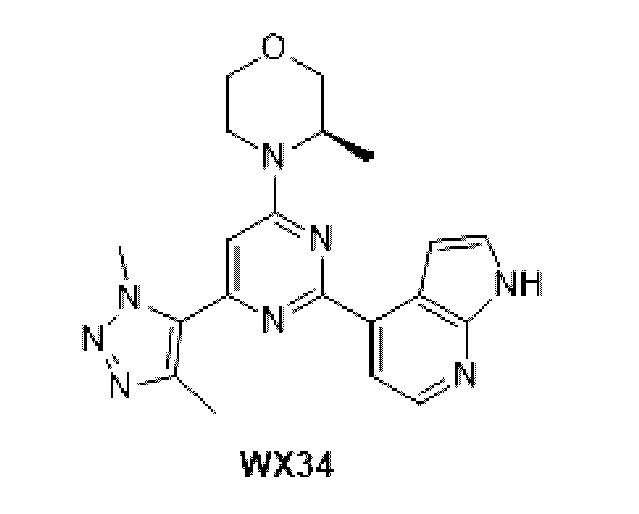
Схема синтеза:
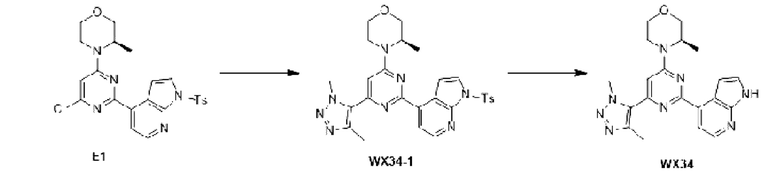
Этап 1: Синтез соединения WX34-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза, причем получали неочищенный продукт, который отделяли с помощью колоночной хроматографии с получением соединения WX34-1.
МС-ЭСИ м/з: 545,4 [М+Н]+.
Этап 2: Синтез соединения 34
Кроме использования соответствующих исходных материалов, процедура получения соединения WX34 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 391,1 [М+Н]+.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,32 (д, J=6,53 Гц, 3 Н) 2,47 (с, 3 Н) 3,37 (уш.д, J=3,51 Гц, 1 Н) 3,56 (тд, J=11,80, 2,76 Гц, 1 Н) 3,71 (дд, J=11, 42, 2, 89 Гц, 1 Н) 3,79-3,89 (м, 1 Н) 4,04 (дд, J=11,17, 3,39 Гц, 1 Н) 4, 22-4, 37 (м, 4 Н) 4,65 (уш.с, 1 Н) 6,99 (с, 1 Н) 7,20 (дд, J=3, 39, 1, 88 Гц, 1 Н) 7,61 (т, J=3,01 Гц, 1 Н) 8,02 (д, J=5,02 Гц, 1 Н) 8,36 (д, J=5,02 Гц, 1 Н) 11,85 (уш.с, 1 Н).
Пример 35

Схема синтеза:
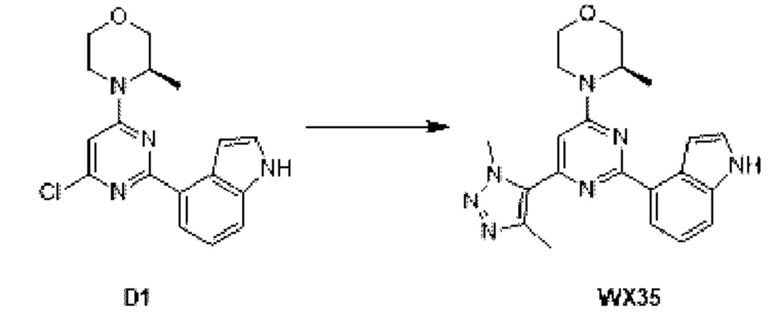
Этап 1: Синтез соединения WX35
Кроме использования соответствующих исходных материалов, процедура получения соединения WX35 идентична процедуре, использованной для соединения WX13 в примере 13 синтеза.
МС-ЭСИ м/з: 390,3 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ ppm 1,28 (с, 3 Н) 2,58 (уш.с, 3 Н) 3,49 (с, 1 Н) 3,73 (с, 1 Н) 3,91 (уш.с, 2 Н) 4,14 (уш.с, 1 Н) 4,29 (с, 1 Н) 4,39 (уш.с, 3 Н) 4,53 (с, 1 Н) 6,52 (с, 1 Н) 7,34 (уш.с, 1 Н) 7,38 (уш.с, 1 Н) 7,48 (уш.с, 1 Н) 7,57 (с, 1 Н) 8,27 (уш.д, J=7,53 Гц, 1 Н) 8,37 (с, 1 Н)
Пример 36
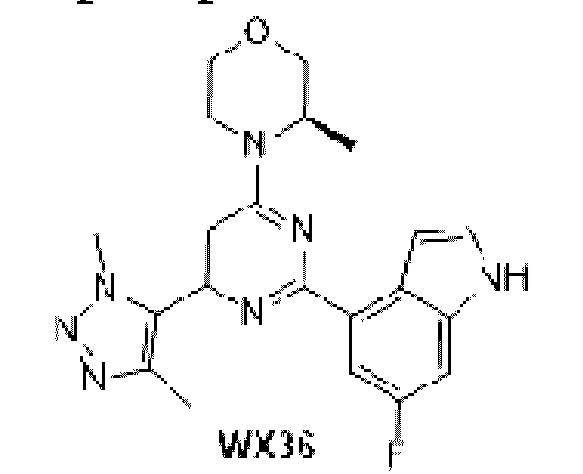
Схема синтеза:
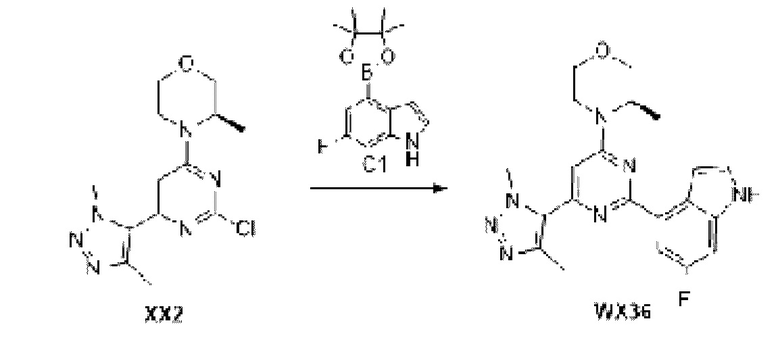
Этап 1: Синтез соединения WX36
Кроме использования соответствующих исходных материалов, процедура получения соединения WX36 идентична процедуре, использованной для соединения WX13 в примере 13 синтеза.
МС-ЭСИ м/з: 408,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,32 (д, J=6, 70 Гц, 3 Н) 2,46 (с, 3 Н) 3,36 (уш.д, J=4,27 Гц, 1 Н) 3, 50-3,62 (м, 1 Н) 3,67-3,74 (м, 1 Н) 3,79-3,86 (м, 1 Н) 4,04 (дд,.J=10,92, 3,14 Гц, 1 Н) 4,25 (с, 3 Н) 4,29 (уш.д, J=11, 29 Гц, 1 Н) 4,63 (уш.с, 1 Н) 6,93 (с, 1 Н) 7,29 (уш.с, 1 Н) 7,36 (дц, J=9,29, 2,01 Гц, 1 Н) 7,48 (т, J=2,64 Гц, 1 Н) 7,90 (дд, J=11,29, 2,51 Гц, 1 Н) 11,36 (уш. с, 1 Н).
Пример 37
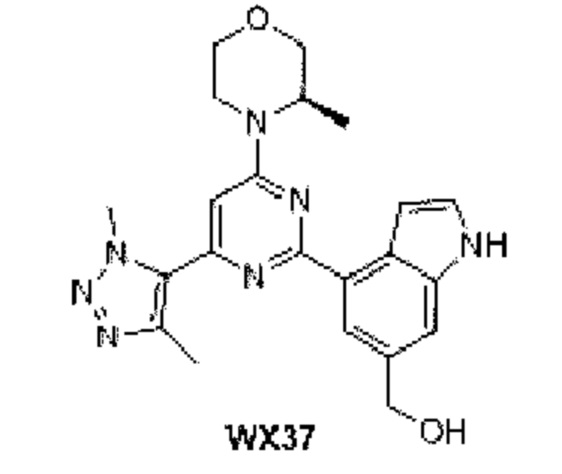
Схема синтеза:
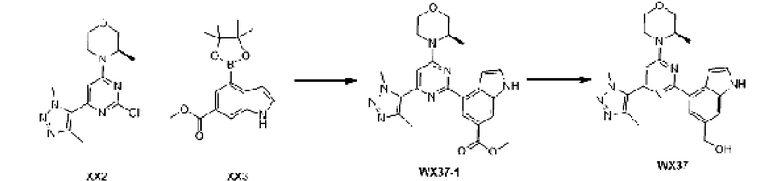
Этап 1: Синтез соединения WX37-1
Кроме использования соответствующий исходных материалов, процедура получения соединения WX37-1 идентична процедуре, использованной для соединения WX13 в примере 13 синтеза.
МС-ЭСИ м/з: 411,3 [М+Н]+.
Этап 2: Синтез соединения WX37
При 0°С к раствору соединения WX37-1 (0,15 г, 335,20 мкмоль) в тетрагидрофуране (10 мл) добавляли алюмогидрид лития (25,44 мг, 670,41 мкмоль), и реакционную смесь перемешивали при 0°С в течение 0,5 ч. При 0°С к реакционной смеси добавляли безводный сульфат натрия, гасили путем добавления воды (1 мл) и фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX37.
МС-ЭСИ м/з: 420,3 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,32 (д, J=6, 78 Гц, 3 Н) 1,34-1,34 (м, 1 Н) 2,46 (с, 3 Н) 3,39 (уш.с, 1 Н) 3,56 (тд, J=11,92, 3,01 Гц, 1 Н) 3,71 (дд, J=11,54, 2,76 Гц, 1 Н) 3,83 (д, J=11,54 Гц, 1 Н) 4,04 (уш.дд, J=11, 42, 3,14 Гц, 1 Н) 4,25 (с, 3 Н) 4,30 (уш.д, J=12,80 Гц, 1 Н) 4,65 (с, 3 Н) 6,88 (с, 1 Н) 7,24 (уш.с, 1 Н) 7,43 (т, J=2, 64 Гц, 1 Н) 7,52 (с, 1 Н) 8,10 (с, 1 Н) 11, 23 (уш. с, 1 Н).
Пример 38
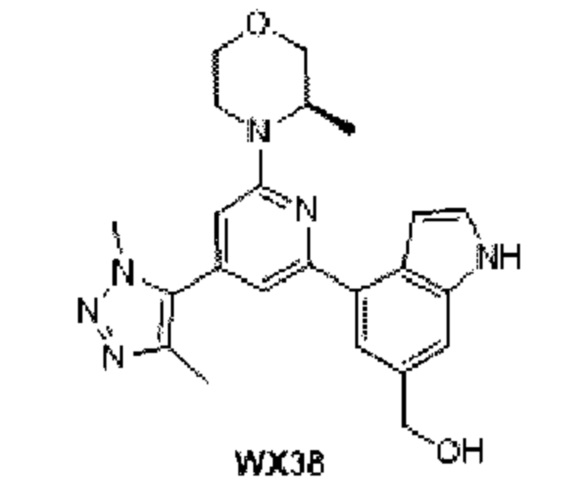
Схема синтеза:
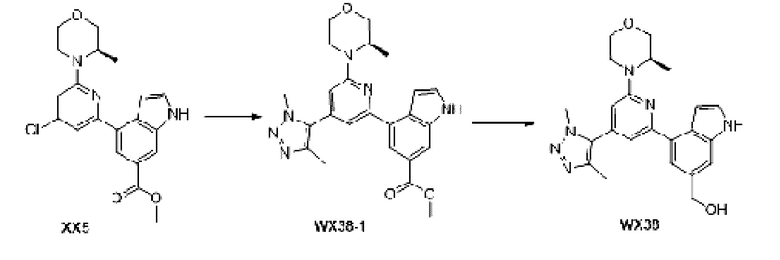
Этап 1: Синтез соединения WX38-1
К раствору соединения ХХ5 (0,28 г, 725, 68 мкмоль) в N,N-диметилформамиде (5 мл) добавляли 1,4-диметил-1Н-1,2,3-триазол (140, 95 мг, 1,45 ммоль), бис (трифенилфосфин) палладия дихлорид (50,94 мг, 72,57 мкмоль) и ацетат тетраметиламмония (115,98 мг, 870,82 мкмоль). Реакционную смесь перемешивали в герметично закрытой пробирке при 140°С при нагревании в течение 4 ч и затем разбавляли этилацетатом (50 мл), промывали водой (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения W38-1.
МС-ЭСИ м/з: 447,3 [М+Н]+.
Этап 2: Синтез соединения WX38
Кроме использования соответствующих исходных материалов, процедура получения соединения WX38 идентична процедуре, использованной для соединения WX37 в примере WX37 синтеза.
МС-ЭСИ м/з: 419,3 [М+Н]+.
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,23 (уш.д, J=6, 53 Гц, 3 Н) 2,32 (с, 3 Н) 3,16-3,23 (м, 1 Н) 3,50-3,60 (м, 1 Н) 3,67-3,73 (м, 1 Н) 3,75-3,81 (м, 1 Н) 4,00 (уш.д, J=8,28 Гц, 1 Н) 4,05 (с, 3 Н) 4,13 (уш.д, J=11,80 Гц, 1 Н) 4,50 (уш.д, J=4,77 Гц, 1 Н) 4,63 (д, J=5,52 Гц, 2 Н) 5,13 (т, J=5, 65 Гц, 1 Н) 6,79 (с, 1 Н) 6,88 (уш.с, 1 Н) 7,16 (с, 1 Н) 7,40 (уш.с, 1 Н) 7,46 (д, J=11,80 Гц, 2 Н) 11, 21 (уш. с, 1 Н)
Пример 39

Схема синтеза:

Этап 1: Синтез соединения WX39-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX39-1 идентична процедуре, использованной для соединения WX38-1 в примере синтеза промежуточного соединения ЖХ38-1.
МС-ЭСИ м/з: 544,4 [М+Н]+.
Этап 2: Синтез соединения WX39
Кроме использования соответствующих исходных материалов, процедура получения соединения WX39 идентична процедуре, использованной для соединения WX15, описанной в примере 15 синтеза.
МС-ЭСИ м/з: 390,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,24 (д, J=6, 78 Гц, 3 Н) 2,33 (с, 3 Н) 3,18-3,26 (м, 1 Н) 3,55 (тд,,J=11,86, 2,64 Гц, 1 Н) 3, 66-3,74 (м, 1 Н) 3,76-3, 82 (м, 1 Н) 4,01 (дд, J=11,42, 3,14 Гц, 1 Н) 4,06 (с, 3 Н) 4,14 (уш.д, J=10, 79 Гц, 1 Н) 4,51 (уш.д, J=7, 03 Гц, 1 Н) 6,92 (с, 1 Н) 6,95 (дд, J=3, 51, 2,01 Гц, 1 Н) 7,36 (с, 1 Н) 7,57-7,61 (м, 2 Н) 8,31 (д, J=5,02 Гц, 1 Н) 11,80 (уш.с, 1 Н)
Пример 40
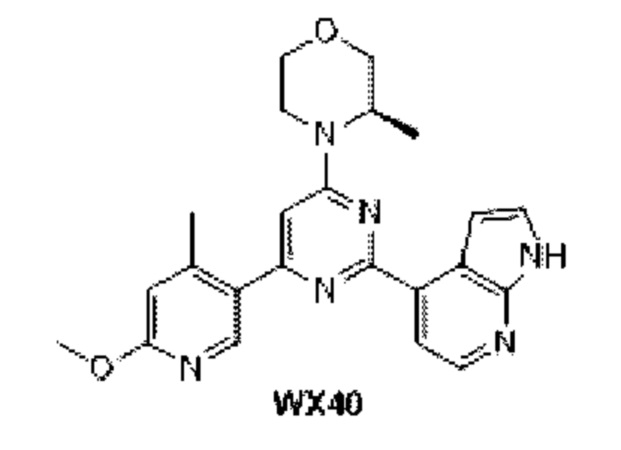
Схема синтеза:
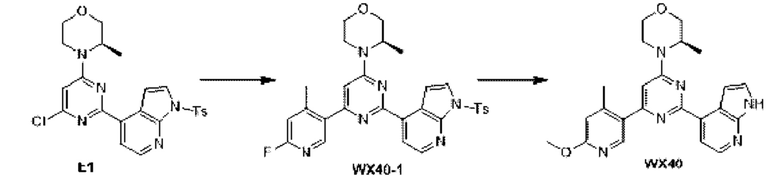
Этап 1: Синтез соединения WX40-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX40-1 идентична процедуре, использованной для соединения WX15-1, описанной в примере синтеза промежуточного соединения WX15-1.
МС-ЭСИ м/з: 559,1 [М+Н]+.
Этап 2: Синтез соединения WX40
К раствору соединения WX40-1 (0,095 г, 170,06 мкмоль) в метаноле (15 мл) добавляли 2 М гидроксид натрия (2М, 1 мл). Реакционную смесь перемешивали при 15-20°С в течение 72 ч, и затем разбавляли водой (30 мл). Водную фазу экстрагировали дихлорметаном (50 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат центрифугировали до сухого состояния с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX40.
МС-ЭСИ м/з: 417,0 [М+Н]+.
1H ЯМР (CDCl3, 400 МГц): δ=9,53 (уш.с, 1Н), 8,44 (д, J=5, 0 Гц, 1Н), 8, 27-8,34 (м, 1Н), 8,14 (д, J=5, 1 Гц, 1Н), 7,37-7,47 (м, 2Н), 6,72 (с, 1Н), 6,55 (с, 1Н), 4,62 (с, 1Н), 4,53 (уш.с, 1Н), 4,26 (уш.д, J=13,3 Гц, 1Н), 3,97-4,02 (м, ЗН), 3,87-3,94 (м, 1Н), 3,79-3,86 (м, 1Н), 3,69 (тд, J=11, 9, 3,1 Гц, 1Н), 3,44 (тд, J=12,8, 3,9 Гц, 1Н), 2,54 (с, 3Н), 1,44 ppm (д, J=6, 9 Гц, 3Н)
Пример 41
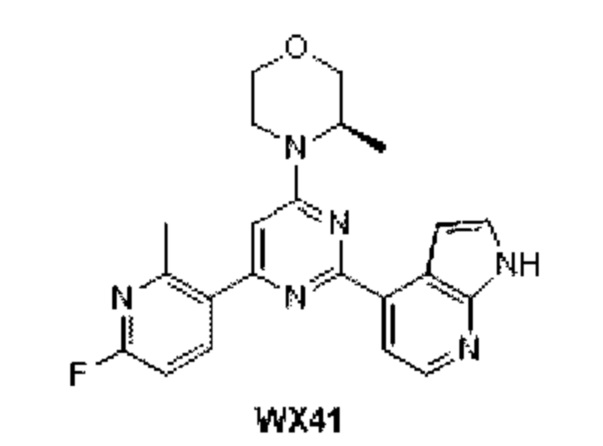
Схема синтеза:
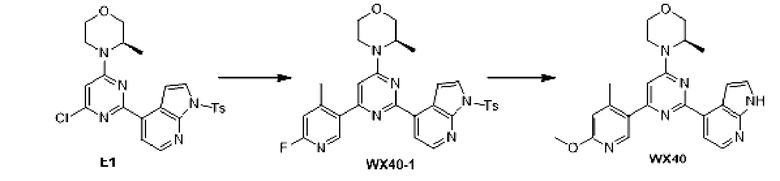
Этап 1: Синтез соединения WX41-1
К раствору соединения Е1 (2 г, 4,13 ммоль) в этаноле (25 мл) добавляли 2 М пидроксид натрия (10,33 мл), и реакционную смесь перемешивали при 15-20°С в течение 14 ч и затем нагревали до 60°С при перемешивании в течение 5 ч. Значение рН реакционной смеси доводили до 6-7 с помощью 2 М хлористоводородной кислоты, разбавляли водой (40 мл) и экстрагировали этилацетатом (60 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (80 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат центрифугировали до сухого состояния с получением неочищенного соединения WX41-1.
МС-ЭСИ м/з: 330,0 [М+Н]+.
Этап 2: Синтез соединения WX41
К раствору соединения WX41-1 (0,06 г, 181,94 мкмоль), 2-фтор-6-метилпиридин-5-борной кислоты (42,28 мг, 272,91 мкмоль) и тетракис(трифенилфосфин)палладия (14,72 мг, 12,74 мкмоль) в 1,4-диоксане (8 мл) добавляли 2 М водный раствор карбоната натрия (2 М, 272,91 мкл), раствор продували азотом три раза. Реакционную смесь перемешивали при нагревании при 95°С в течение 5 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением WX41.
МС-ЭСИ м/з: 405,2 [М+Н]+.
1Н ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=10,24 (уш.с, 1Н), 8,47 (д, J=5,0 Гц, 1Н), 8,15 (д, J=5,3 Гц, 1Н), 7,98 (т, J=8,0 Гц, 1Н), 7,45-7,57 (м, 1Н), 7,36-7,41 (м, 1Н), 6,91 (дд, J=8,3, 3,0 Гц, 1Н), 6,55 (с, 1H), 4,54 (уш.с, 1H), 4,27 (уш.д, J=12,3 Гц, 1H), 4,13 (дд, J=115, 3,5 Гц, 1H), 3,88-3,96 (м, 1H), 3,78-3,86 (м, 1H), 3,69 (тд, J=11, 9, 3,0 Гц, 1H), 3,45 (тд, J=12,8, 3,8 Гц, 1H), 2,71 (с, З3Н), 1,45 ррт (д, J=6, 8 Гц, 3Н)
Пример WX42
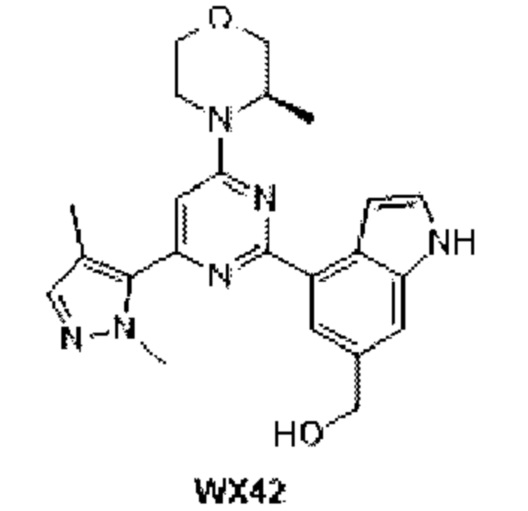
Схема синтеза:
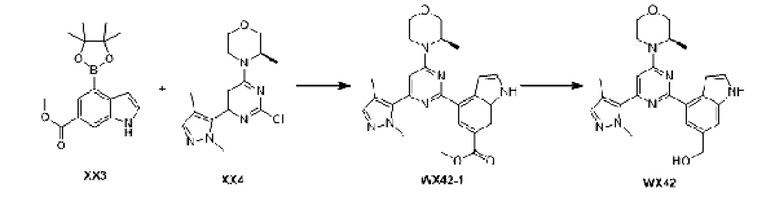
Этап 1: Синтез соединения WX42-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX42-1 идентична процедуре, использованной для соединения WX37-1 в примере синтеза WX37:
К раствору соединения ХХ4 (0,11 г, 357, 40 мкмоль), ХХ3 (161,44 мг, 536,10 мкмоль) и тетракис (трифенилфосфин) палладия (0,03 г, 25,96 мкмоль) в 1, 4-диоксане (8 мл) добавляли 2 М водный раствор карбоната натрия (534, 1 мкл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 100°С в течение 5 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 20-55%) с получением соединения WX42-1.
МС-ЭСИ м/з: 447,1 [М+Н]+.
Этап 2: Синтез соединения WX42
Кроме использования соответствующих исходных материалов, процедура получения соединения WX42 идентична процедуре, использованной для соединения WX37-1 в примере синтеза WX37:
При 0-5°С к раствору соединения WX42-1 (0,13 г, 291,15 мкмоль) в тетрагидрофуране (10 мл) добавляли алюмогидрид лития (0,05 г, 1,32 ммоль). Реакционную смесь перемешивали при 0-5°С в течение 1 ч и затем нагревали до 25°С при перемешивании в течение 2 ч. При 0-5°С к реакционной смеси последовательно медленно добавляли одну каплю воды, две капли 10% гидроксида натрия и три капли воды, затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 50-100%) с получением соединения WX42.
МС-ЭСИ м/з: 419,2 [М+Н]+.
1H ЯМР (CDCl3, 400 МГц): δ=8,44 (уш.с, 1Н), 8,25 (с, 1Н), 7,54 (с, 1Н), 7,49 (уш.с, 1Н), 7,41 (с, 1Н), 7,32 (т, J=2,8 Гц, 1Н), 6,47 (с, 1Н), 4,87 (с, 2Н), 4,48 (уш.д, J=4,5 Гц, 1Н), 4,29 (уш.д, J=13,8 Гц, 1Н), 4,14 (с, 3Н), 4,11 ((м, 1Н), 3,86-3,94 (м, 1Н), 3,79-3,86 (м, 1Н), 3,69 (тд, J=11,9, 3,1 Гц, 1Н), 3,43 (тд, J=12,7, 3,9 Гц, 1Н), 2,24 (с, 3Н), 1,70 (т, J=6,0 Гц, 1Н), 1,43 ррт (д, J=6,8 Гц, 3Н)
Пример 43
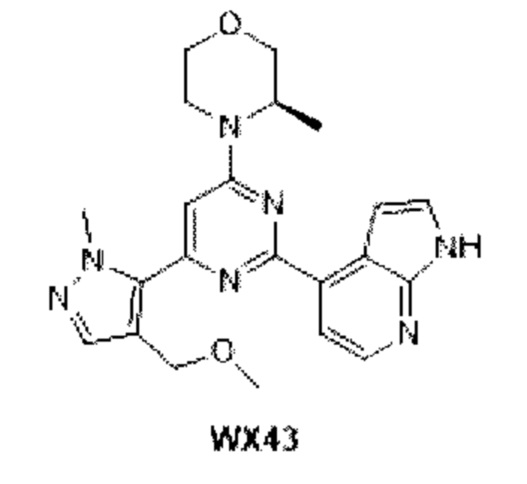
Схема синтеза:
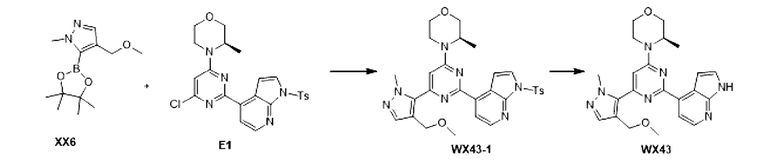
Этап 1: Синтез соединения WX43-1
Кроме использования соответствующих исходных материалов, процедура получения неочищенного WX43-1 идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 574,4 [М+Н]+.
Этап 2: Синтез соединения WX43
Кроме использования соответствующих исходных материалов, процедура получения соединения WX43 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 420, 1 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=9,74 (уш.с, 1Н), 8,46 (уш.с, 1Н), 8,16 (уш.с, 1Н), 7,60 (с, 1Н), 7,48 (д, J=3,3 Гц, 1Н), 7,40 (д, J=3, 0 Гц, 1Н), 7,09 (с, 1Н), 4,53 (уш.с, 1Н), 4,38 (с, 2Н), 4,26 (с, 4Н), 4,13 (дд, J=11,4, 3,6 Гц, 1Н), 3,88-3,95 (м, 1Н), 3,79-3,85 (м, 1Н), 3,65-3,74 (м, 1Н), 3,39-3,49 (м, 4Н), 1,45 ppm (д, J=6, 8 Гц, 3Н).
Пример 44

Схема синтеза:
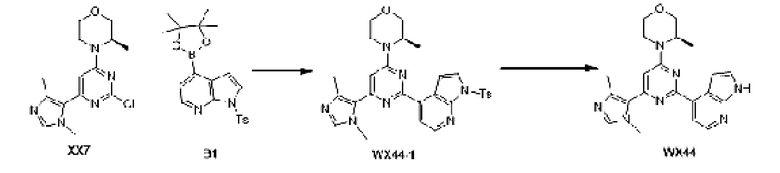
Этап 1: Синтез соединения WX44-1
К раствору соединения ХХ7 (0,05 г, 162, 45 мкмоль), В1 (77,64 мг, 194, 95 мкмоль) и тетракис (трифенилфосфин) палладия (18,77 мг, 16,25 мкмоль) в 1,4-диоксане (5 мл) добавляли 2 М водный раствор карбоната натрия (2 М, 243,68 мкл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 100°С в течение 14 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX44-1.
МС-ЭСИ м/з: 544,4 [М+Н]+.
Этап 2: Синтез соединения WX44
Кроме использования соответствующих исходных материалов, процедура получения соединения WX44 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,3 [М+Н]+.
1Н ЯМР (ХЛОРОФОРМ-d, 400 МГц): δ=9,09 (уш.с, 1Н), 8,43 (д, J=5,0 Гц, 1Н), 8,09 (д, J=5,0 Гц, 1Н), 7,53 (уш.с, 1Н), 7,44 (уш.с, 1Н), 7,35 (д, J=2,3 Гц, 1Н), 6,53 (с, 1Н), 4,51 (уш.д, J=5, 4 Гц, 1Н), 4,23 (уш.д, J=12,6 Гц, 1Н), 4,13 (дд, J=11,5, 3,5 Гц, 1H), 3,97 (с, 3H), 3,89-3,93 (м, 1H), 3,80-3,86 (м, 1H), 3,64-3,70 (м, 1H), 3,44 (тд, J=12,7, 3,9 Гц, 1H), 2,48 (с, 3H), 1,44 ppm (д, J=6,8 Гц, 3H).
Пример 45
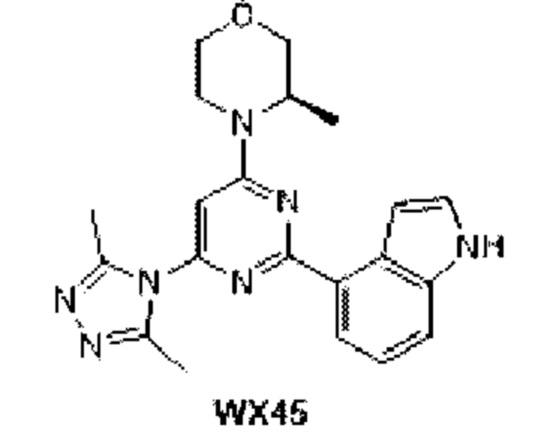
Схема синтеза:
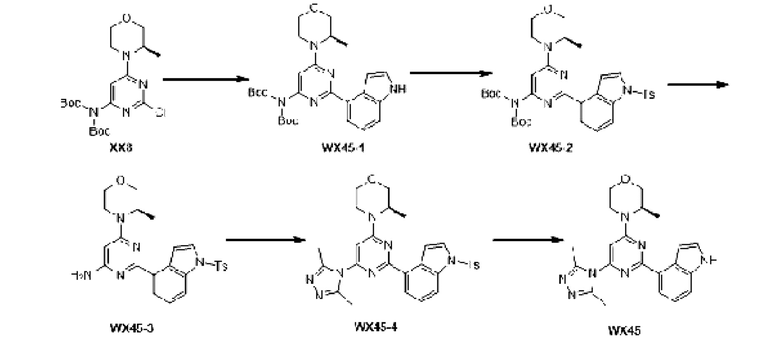
Этап 1: Синтез соединения шх45-1
К раствору соединения ХХ8 (1,6 г, 3,73 ммоль), пинаколовопо эфира индол-4-6орной кислоты (1,18 г, 4,85 ммоль) и бис(трифенилфосфин)палладия дихлорида (183,28 мг, 261,13 мкмоль) в 1,4-диоксане (20 мл) добавляли 2 м водный раствор карбоната натрия (2 М, 5,6 мл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 95°С в течение 16 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX45-1.
МС-ЭСИ м/з: 510,8 [М+H]+.
Этап 2: Синтез соединения WX45-2
При 0-5°С к раствору соединения WX45-1 (1, 25 г, 2,45 ммоль) в N,N-диметилформамиде (10 мл) добавляли гидрид натрия (127,54 мг, 3,19 ммоль, чистота: 60%). Реакционную смесь перемешивали при 0-5°С в течение 10 мин, затем добавляли р-толуолсульфонилхлорид (561,17 мг, 2,94 ммоль). Реакционную смесь перемешивали при 0-5°С в течение 50 мин, гасили путем добавления воды (30 мл) и экстрагировали этилацетатом (40 мл ×3). Органические фазы объединяли и последовательно промывали водой (30 мл ×-2) и насыщенным солевым раствором (50 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного соединения WX45-2.
МС-ЭСИ м/з: 664,5 [М+Н]+.
Этап 3: Синтез соединения WX45-3
К раствору соединения WX45-2 (1,7 г, 2,56 ммоль) в 1,4-диоксане (10 мл) добавляли смесь 4 М хлористоводородной кислоты в 1,4-диоксане (4 М, 5 мл). Реакционную смесь перемешивали при 30°С в течение 2 ч, значение рН доводили до 7-8 с помощью насыщенного раствора бикарбоната натрия, разбавляли водой (30 мл) и экстрагировали этилацетатом (40 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии
(этилацетата/петролейный эфир: 25-75%) с получением соединения WX45-3.
МС-ЭСИ м/з: 464,7 [М+Н]+.
Этап 4: Синтез соединения WX45-4
К раствору соединения WX45-3 (0,3 г, 647,18 мкмоль), 2,5-диметил-1,3,4-оксадиазола (190,47 мг, 1,94 ммоль) в 1-метил-2-пирролидоне (3 мл) добавляли безводную р-толуолсульфоновую кислоту (111,45 мг, 647,18 мкмоль), и реакционную смесь перемешивали при 200°С при воздействии микроволнового излучения в течение 1,5 ч. Реакционную смесь разбавляли водой (20 мл) и экстрагировали дихлорметаном (30 мл ×х4). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. Отфильтрованную органическую фазу концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (метанол/дихлорметан: 0-10%) с получением соединения WX45-4.
МС-ЭСИ м/з: 544,4 [М+Н]+.
Этап 5: Синтез соединения WX45
Кроме использования соответствующих исходных материалов, процедура получения соединения WX45 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,3 [М+Н]+.
1H ЯМР (CDCl3, 400 МГц): δ=8,60 (уш. с, 1Н), 8,26 (дц, J=7,5, 0,8 Гц, 1Н), 7,58 (д, J=8,0 Гц, 1Н), 7,45 (т, J=2,3 Гц, 1Н), 7,36 (т, J=2,8 Гц, 1Н), 7,31 (т, J=7,8 Гц, 1Н), 6,20-6,25 (м, 1Н), 4,49 (уш. с, 1Н), 4,25 (уш. д, J=12,3 Гц, 1Н), 4,13 (дц, J=11,5, 3,8 Гц, 1Н), 3,88-3,96 (м, 1Н), 3,80-3,87 (м, 1Н), 3,68 (тд, J=11,9, 3,1 Гц, 1Н), 3,47 (тд, J=12,8, 3,9 Гц, 1Н), 2,54 (с, 6Н), 1,45 ppm (д, J=6,8 Гц, 3Н).
Пример 46
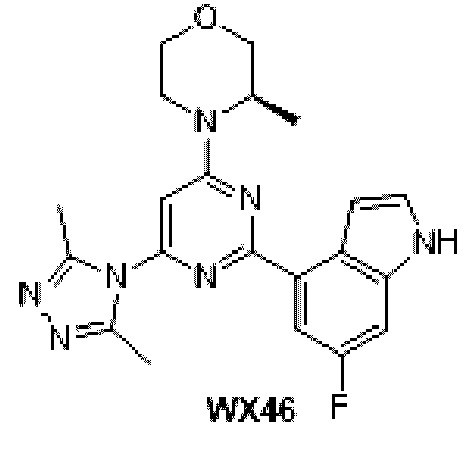
Схема синтеза:
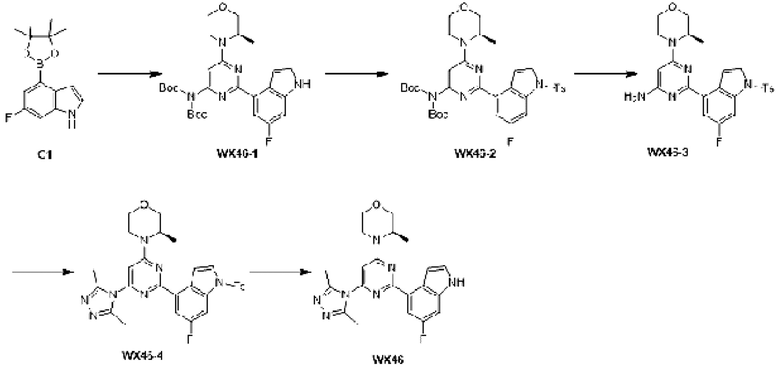
Этап 1: Синтез соединения WX46-1
К раствору соединения С1 (1,3 г, 3,03 ммоль), ХХ8 (830, 95 мг, 3,18 ммоль) и бис(трифенилфосфин)палладия дихлорида (148,92 мг, 212,17 мкмоль) в 1,4-диоксане (20 мл) добавляли 2М водный раствор карбоната натрия (2 М, 4,55 мл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 85°С в течение 16 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX46-1.
МС-ЭСИ м/з: 528,4 [М+Н]+.
Этап 2: Синтез соединения WX46-2
При 0-5°С к раствору соединения WX46-1 (1,35 г, 2,56 ммоль) в N'N-диметилформамиде (10 мл) добавляли гидрид натрия (133,06 мг, 3,33 ммоль, 60% чистоты). Реакционную смесь перемешивали при 0-5°С в течение 10 мин и добавляли р-толуолсульфонилхлорид (585,40 мг, 3,07 ммоль). Реакционную смесь перемешивали при 0-5°С в течение 30 мин, гасили путем добавления воды (30 мл) и экстрагировали этилацетатом (40 мл ×3). Органические фазы объединяли и последовательно промывали водой (30 мл ×2) и насыщенным солевым раствором (50 мл ×2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного соединения WX46-2.
МС-ЭСИ м/з: 682,5 [М+Н]+.
Этап 3: Синтез соединения WX46-3
К раствору соединения WX46-2 (1,75 г, 2,56 ммоль) в 1,4-диоксане (10 мл) добавляли смесь 4М хлористоводородной кислоты в 1,4-диоксане (4 М, 10 мл). Реакционную смесь перемешивали при 30°С в течение 12 ч, значение рН доводили до 7-8 с помощью насыщенного раствора бикарбоната натрия, смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (50 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX46-3.
МС-ЭСИ м/з: 482,3 [М+Н]+.
Этап 4: Синтез соединения WX46-4
Кроме использования соответствующих исходных материалов, процедура получения соединения WX46-4 идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 562,4 [М+Н]+.
Этап 5: Синтез соединения WX46
Кроме использования соответствующих исходных материалов, процедура получения соединения WX46 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 408,2 [М+Н]+.
1Н ЯМР (ХЛОРОФОРМ-d, 400МГц): δ=8,27-8,59 (м, 1Н), 7,93-7,96 (м, 1Н), 7,34 (т, J=2,3 Гц, 1Н), 7,26 (т, J=2,8 Гц, 1Н), 7,20 (уш. д, J=2,4 Гц, 1Н), 6,16-6,22 (м, 1Н), 4,40 (уш. с, 1Н), 4,17 (уш. д, J=10,6 Гц, 1Н), 4,06 (дд, J=11,6, 3,7 Гц, 1Н), 3,79-3,87 (м, 1Н), 3,72-3,78 (м, 1Н), 3,61 (тд, J=11,9, 3,0 Гц, 1Н), 3,39 (тд, J=12, 8, 3,9 Гц, 1Н), 2,46 (с, 6Н), 1,37 ppm (д, J=6, 8 Гц, 3Н).
Пример 47

Схема синтеза:

Этап 1: Синтез соединения WX47-1
К раствору соединения ХХ9 (0,3 г, 857,61 мкмоль), ХХ2-1 (126,74 мг, 1,03 ммоль), карбоната калия (237,05 мг, 1,72 ммоль), трициклогексилфосфина (126,74 мг, 1,03 ммоль) и триметилуксусной кислоты (17,52 мг, 171,52 мкмоль) в N,N-диметилацетамиде (2 мл) добавляли ацетат палладия (19,25 мг, 85,76 мкмоль). Реакционную смесь продували азотом три раза и перемешивали при 130-150°С в течение 18 ч. Реакционную смесь концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX47-1.
МС-ЭСИ м/з: 437,4 [М+Н]+.
Этап 2: Синтез соединения WX47-2
К раствору соединения WX47-1 (, 45 г, 1,03 ммоль) в этаноле (25 мл) добавляли Pd/C (0,1 г, 1,03 ммоль, чистота: 10%). Смесь продували азотом несколько раз и перемешивали в течение 16 ч в атмосфере водорода (15 psi (103 кПа)) при 30°С. Реакционную смесь фильтровали через целит, и фильтрат концентрировали с получением неочищенного соединения WX47-2.
МС-ЭСИ м/з: 317,2 М+Н]+.
Этап 3: Синтез соединения WX47-3
К раствору оксихлорида фосфора (11,75 г, 76,63 ммоль) добавляли WX47-2 (0,33 г, 1,04 ммоль), и реакционный раствор нагревали до 30°С и перемешивали в атмосфере азота в течение 5 ч. Реакционную смесь концентрировали и затем разбавляли дихлорметаном (50 мл). Значение рН органической фазы доводили до 8 с помощью насыщенного раствора бикарбоната натрия, смесь экстрагировали диклорметаном (30 мл ×8). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения WX47-3.
МС-ЭСИ м/з: 335,2 М+Н]+.
Этап 4: Синтез соединения WX47-4
Кроме использования соответствующих исходных материалов, процедура получения соединения WX47-4 идентична процедуре, использованной для соединения WX15-1 в примере 15 синтеза.
МС-ЭСИ м/з: 571,4 М+Н]+.
Этап 5: Синтез соединения WX47
Кроме использования соответствующих исходных материалов, процедура получения соединения WX47 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 417,3 [М+Н]+.
Н ЯМР (CDCl3, 400МГц): δ=9,41 (уш. с, 1Н), 8,45 (д, J=5,0 Гц, 1Н), 8,13 (д, J=5,0 Гц, 1Н), 7,45-7,51 (м, 1Н), 7,39 (дд, J=3,5, 2,0 Гц, 1Н), 6,62 (с, 1Н), 4,52 (уш. с, 1Н), 4,28-4,31 (м, 1Н), 4,25 (tT, J=7,5, 3,8 Гц, 1Н), 4,12-4,17 (м, 1Н), 3,89-3, 95 (м, 1Н), 3, 79-3,88 (м, 1Н), 3,70 (тд, J=11,9, 3,0 Гц, 1Н), 3,47 (тд, J=12,7, 3,8 Гц, 1Н), 2,54 (с, 3Н), 1,46 (д, J=6, 8 Гц, 3Н), 1,35-1,42 (м, 2Н), 1,04-1,14 ppm (м, 2Н).
Пример48
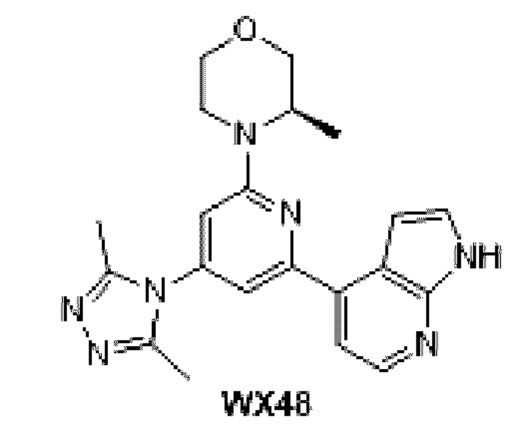
Схема синтеза:
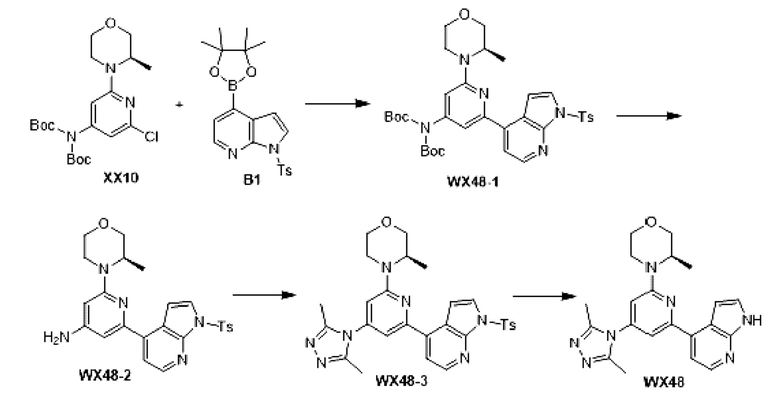
Этап 1: Синтез соединения WX48-1
К раствору соединения ХХ10 (1,6 г, 3,74 ммоль), В1 (2,23 г, 5,61 ммоль) и тетракис(трифенилфосфин)палладия (18,77 мг, 16,25 мкмоль) в 1,4-диоксане (5 мл) добавляли 2М водный раствор карбоната натрия (2 М, 4,67 мл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 100°С в течение 20 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX48-1.
МС-ЭСИ м/з: 664,5 [М+Н]+.
Этап 2: Синтез соединения WX48-2
К раствору соединения WX48-1 (2 г, 3,01 ммоль) в 1,4-диоксане (10 мл) добавляли 4М раствор хлористоводородной кислоты в 1,4-диоксане (4 М, 5 мл). Реакционную смесь перемешивали при 30°С в течение 35 мин, значение рН доводили до 7-8 с помощью насыщенного раствора бикарбоната натрия, разбавляли водой (30 мл) и экстрагировали дихлорметаном (40 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX48-2.
МС-ЭСИ м/з: 464,7 М+Н]+.
Этап 3: Синтез соединения WX48-3
К раствору соединения WX48-2 (0,3 г, 647,18 мкмоль), 2,5-диметил-1,3,4-оксадиазола (1240,00 мг, 2,45 ммоль) в 1-метил-2-пирролидоне (2 мл) добавляли безводную р-толуолсульфоновую кислоту (111,44 мг, 647,18 мкмоль), и реакционную смесь перемешивали при 200°С при воздействии микроволнового излучения в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX48-3.
МС-ЭСИ м/з: 544, 1 М+Н]+.
Этап 4: Синтез соединения WX48
Кроме использования соответствующих исходных материалов, процедура получения соединения WX48 идентична процедуре, использованной для соединения WX15 в примере 15 синтеза.
МС-ЭСИ м/з: 390,3 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400МГц): δ=9,75 (уш. с, 1Н), 6,44 (д, J=5,0 Гц, 1Н), 7,56 (д, J=5,0 Гц, 1Н), 7,42-7,51 (м, 1Н), 7,06 (д, J=1,3 Гц, 1Н), 6,94 (дд, J=3,4, 1,9 Гц, 1Н), 6,38 (д, J=1,3 Гц, 1Н), 4,39 (уш. д, J=6,5 Гц, 1Н), 4,04-4,21 (м, 2Н), 3,87-3,95 (м, 1Н), 3,78-3,86 (м, 1Н), 3,69 (тд, J=12,0, 3,1 Гц, 1Н), 3,39 (тд, J=12,6, 3,9 Гц, 1Н), 2,41 (с, 6Н), 1,39 ppm (д, J=6, 8 Гц, 3Н)
Пример 49
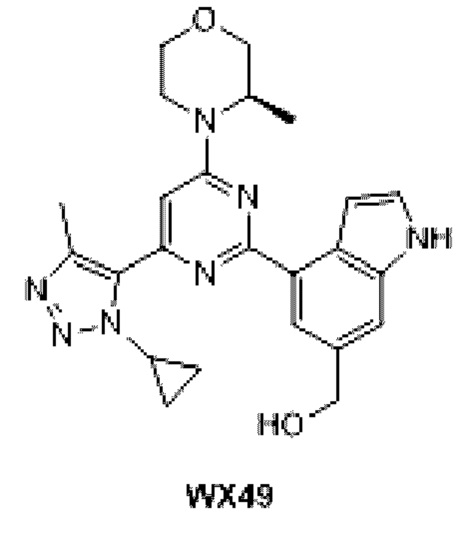
Схема синтеза:
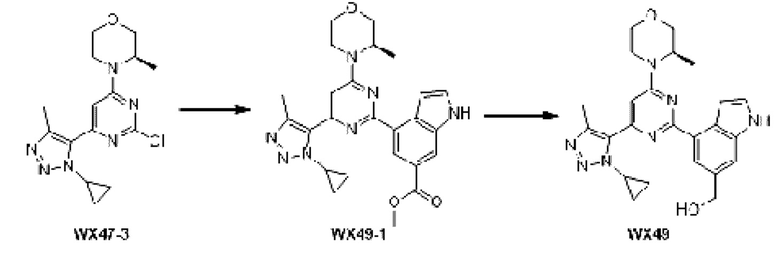
Этап 1: Синтез соединения WX49-1
К раствору соединения WX47-3 (0,1 г, 298, 68 мкмоль), ХХ3 (125,93 мг, 418,16 мкмоль) и тетракис(трифенилфосфин)палладия (24,16 мг, 20,91 мкмоль) в 1,4-диоксане (8 мл) добавляли 2М водный раствор карбоната натрия (2М, 448,02 мкл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 90°С в течение 16 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX49-1.
МС-ЭСИ м/з: 474,4 [М+Н]+.
Этап 2: Синтез соединения WX49
При температуре 0-5°С к раствору соединения WX49-1 (0,06 г, 126,71 мкмоль) в тетрагидрофуране (10 мл) добавляли алюмогидрид лития (0,05 г, 1,32 ммоль). Реакционную смесь нагревали до 30°С при перемешивании в течение 3 ч. При 0-5°С к реакционной смеси последовательно медленно добавляли одну каплю воды, две капли 10% гидроксида натрия и три капли воды, затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии с получением соединения WX49.
МС-ЭСИ м/з: 446,1 [М+Н]+.
1H ЯМР (ХЛОРОФОРМ-d, 400МГц): δ=8,43 (уш. с, 1Н), 8,27 (д, J=1,0 Гц, 1Н), 7,57 (с, 1Н), 7,50 (т, J=2,3 Гц, 1Н), 7,34 (т, J=2,8 Гц, 1Н), 6,54 (с, 1Н), 4,88 (с, 2Н), 4,50 (уш. д, J=4,5 Гц, 1Н), 4,21-4,35 (м, 2Н), 4,13 (дд, J=11,4, 3,6 Гц, 1Н), 3,88-3,93 (м, 1Н), 3,79-3,88 (м, 1Н), 3,64-3,73 (м, 2Н), 3,45 (тд, J=12,8, 4,0 Гц, 1Н), 2,52 (с, 3Н), 1,44 (д, J=6,8 Гц, 3Н), 1, 33-1, 40 (м, 2Н), 1,02-1,11 ppm (м, 2Н).
Пример 50

Схема синтеза:
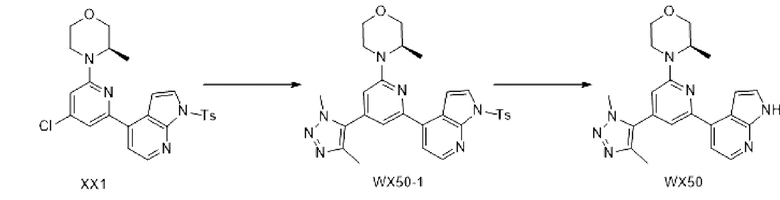
Этап 1: Синтез соединения WX50-1
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX30-1 в примере 30, причем получали неочищенный продукт, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 0-60%) с получением WX50-1.
MS-ESI: 544,4 [М+Н]+
Этап 2: Синтез соединения WX50
Кроме использования соответствующих исходных материалов, процедура идентична процедуре, использованной для соединения WX30 в примере 30, причем получали неочищенный продукт, который очищали колоночной хроматографией (дихлорметан:метанол=30/1, 10/1) с получением соединения WX50.
MS-ESI: 390,1 [М+Н]+
1H ЯМР (400 МГц, DMSO-d6) δ ppm 1,24 (д, J=6, 78 Гц, 3 Н) 2,33 (с, 3 Н) 3,22 (тд, J=12,67, 4,02 Гц, 1 Н) 3,37-3, 42 (м, 1 Н) 3,55 (тд, J=11,86, 2,64 Гц, 1 Н) 3,66-3, 73 (м, 1 Н) 3,76-3,83 (м, 1 Н) 4,01 (дц, J=11,42, 3,14 Гц, 1 Н) 4,06 (с, 3 Н) 4,14 (уш. д, J=10,79 Гц, 1 Н) 4,51 (уш. д, J=7,03 Гц, 1 Н) 6,92 (с, 1 Н) 6,95 (дд, J=3,51, 2,01 Гц, 1 Н) 7,36 (с, 1 Н) 7,57-7,61 (м, 2 Н) 8,31 (д, J=5, 02 Гц, 1 Н) 11,80 (уш. с, 1 Н)
Пример 51
Схема синтеза:

Этап 1: Синтез соединения WX51-1
К раствору соединения D1-2 (4,51 г, 18,18 ммоль), ХХЗ (4,96 г, 16,47 ммоль) и бис (трифенилфосфин) палладия дихлорида (893,11 мг, 1,27 ммоль) в 1, 4-диоксане (60 мл) добавляли 2М водный раствор карбоната натрия (27,27 мл) и продували азотом три раза. Реакционную смесь перемешивали при 110°С при нагревании в течение 15 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 15-35%) с получением WX51-1.
MS-ESI: 386,9[М+Н]+
Этап 2: Синтез соединения WX51-2
При комнатной температуре к раствору соединения WX51-1 (0,21 г, 542,87 мкмоль) в 1,4-диоксане (10 мл) добавляли пинаколовый эфир 3,5-диметилизоксазол-4-борной кислоты (181,65 мг, 814,31 мкмоль), тетракис(трифенилфосфин)палладия (62,73 мг, 54, 29 мкмоль), карбонат натрия (2 М, 814,31 мкл) и перемешивали при 100°С в атмосфере азота в течение 12 ч. Реакционную систему охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл), промывали водой (20 мл) и насыщенным солевым раствором (20 мл), соответственно, сушили над безводным сульфатом натрия и фильтровали. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией (петролейный эфир/этилацетат: 33%-50%) с получением соединения WX51-2.
MS-ESI: 448,3[М+Н]+
Этап 3: Синтез соединения WX51
Кроме использования соответствующих исходных материалов, процедура получения соединения WX50 идентична процедуре, использованной для соединения WX37 в примере синтеза WX37.
МС-ЭСИ м/з: 420,1 [М+Н]+.
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1,30 (д, J=6,78 Гц, 3 Н) 2,48 (с, 3 Н) 2,68 (с, 3 Н) 3,24-3,30 (м, 1 Н) 3,51-3,61 (м, 1 Н) 3,67-3,74 (м, 1 Н) 3,79-3,86 (м, 1 Н) 4,03 (уш. д, J=7,78 Гц, 1 Н) 4,26 (уш. д, J=13,05 Гц, 1 Н) 4, 58-4, 63 (м, 1 Н) 4,65 (д, J=5,77 Гц, 2 Н) 5,16 (т, J=5,77 Гц, 1 Н) 5,76 (с, 1 Н) 6,73 (с, 1 Н) 7,25 (уш. с, 1 Н) 7,41 (т, J=2,64 Гц, 1 Н) 7,50 (с, 1 Н) 8,09 (д, J=l,25 Гц, 1 Н) 11,20 (уш. с, 1 Н)
Пример 52
Схема синтеза:
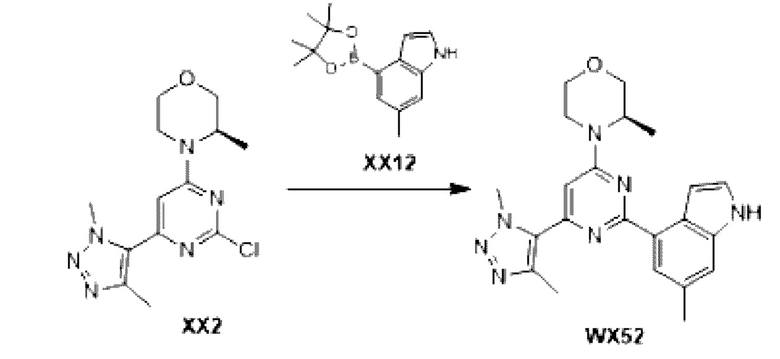
Этап 1: Синтез соединения WX52
Кроме использования соответствующих исходных материалов, процедура получения соединения WX52 идентична процедуре, использованной для соединения WX13 в примере 13 синтеза.
МС-ЭСИ м/з: 404,21 [М+Н]+.
1H ЯМР (400МГц, ХЛОРОФОРМ-d) δ=8,15 (уш. с, 1Н), 7,99 (с, 1Н), 7,30 (т, J=2,3 Гц, 1Н), 7,28 (с, 1Н), 7,20 (уш. с, 1Н), 6,41 (с, 1Н), 4,44 (уш. с, 1Н), 4,28 (с, 3Н), 4,19 (уш. д, J=12, 5 Гц, 1Н), 4,05 (дд, J=3, 6, 11,4 Гц, 1Н), 3,85-3,80 (м, 1Н), 3,77-3,72 (м, 1Н), 3,61 (dт, J=3,0, 11,9 Гц, 1Н), 3,36 (dт, J=4, 0, 12,7 Гц, 1Н), 2,48 (с, 3Н), 2,47 (с, 3Н), 1,36 (д, J=6,8 Гц, 3Н)
Пример 53
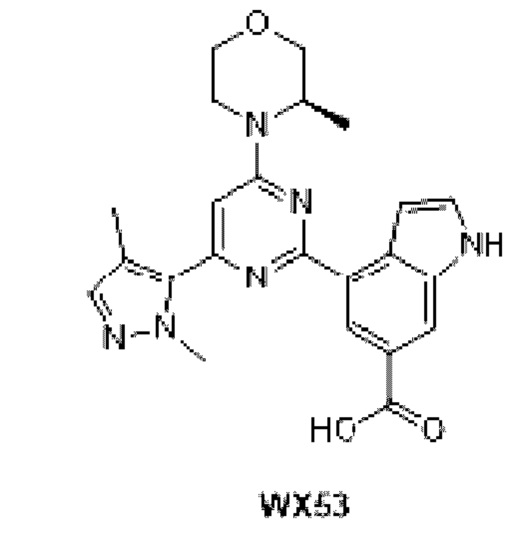
Схема синтеза:
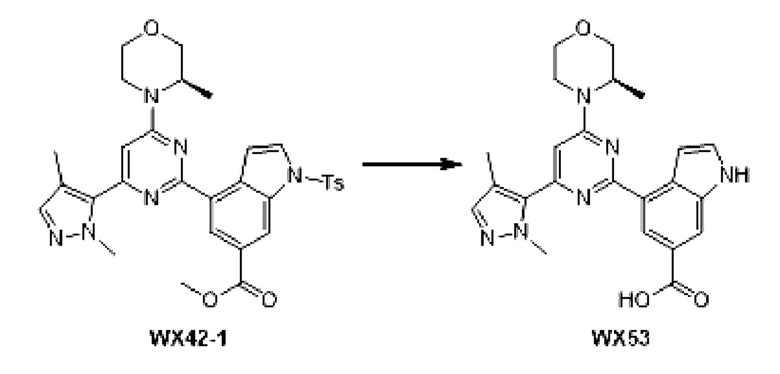
Этап 1: Синтез соединения WX53
К раствору соединения WX42-1 (100 мг, 166,48 мкмоль) в 1,4-диоксане (2 мл) добавляли 2М водный раствор гидроксида натрия (0,25 мл). Реакционную смесь перемешивали при нагревании при 80°С в течение 1,5 ч, затем значение рН доводил до 5 с помощью хлористоводородной кислоты, и затем смесь экстрагировали дихлорметаном и водой. Органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который отделяли с помощью препаративной ВЭЖХ (нейтральные условия) с получением соединения 2.
МС-ЭСИ м/з: 433,31 [М+Н]+.
1H ЯМР (400МГц, DMSO-d6) δ=11,65 (уш. с, 1Н), 8,78 (д, J=l, 5 Гц, 1Н), 8,17 (с, 1Н), 7,69 (т, J=2, 8 Гц, 1Н), 7,41 (с, 1Н), 7,38 (уш. с, 1Н), 6,82 (с, 1Н), 4,60 (уш. с, 1Н), 4,29 (уш. д, J=11,3 Гц, 1Н), 4,05 (уш. с, 1Н), 4,03 (с, 3Н), 3,86-3, 80 (м, 1Н), 3,71 (уш. д, J=9, 4 Гц, 1Н), 3,61-3,53 (м, 1Н), 3,28 (с, 1Н), 2,20 (с, 3Н), 1,32 (д, J=6,6 Гц, 3Н).
Пример 54

Схема синтеза:
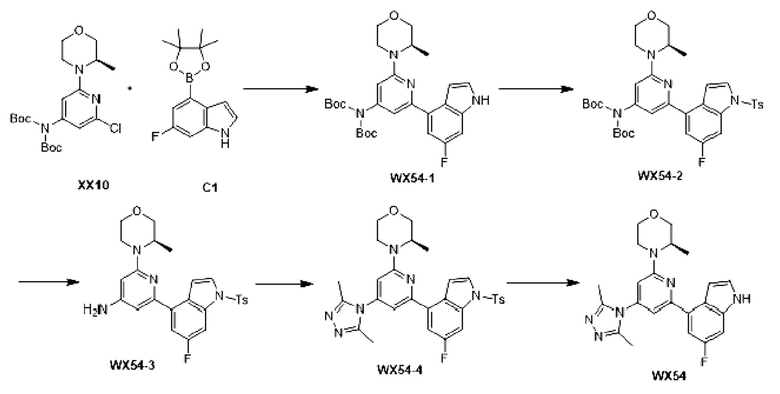
Этап 1: Синтез соединения WX54-1
К раствору соединения ХХ10 (0,35 г, 817,91 мкмоль), С1 (256,27 мг, 981,49 мкмоль) и тетракис (трифенилфосфин)палладия ((66,16 мг, 57,25 мкмоль) в 1,4-диоксане (8 мл)) добавляли 2М водный раствор карбоната натрия (2М, 1,02 мл) и продували азотом три раза. Реакционную смесь перемешивали при нагревании при 90°С в течение 36 ч и затем фильтровали. Раствор концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 15-45%) с получением соединения WX54-1.
МС-ЭСИ м/з: 527,4 [М+Н]+.
Этап 2: Синтез соединения WX54-2
При 0-5°С к раствору соединения WX54-1 (0,43 г, 816,56 ммоль) в N'N-диметилформамиде (10 мл) добавляли гидрид натрия (37,56 мг, 939,05 ммоль, чистота: 60%). Реакционную смесь перемешивали при 0-5°С в течение 15 мин и добавляли р-толуолсульфонилхлорид (186,81 мг, 979,87 мкмоль). Реакционную смесь перемешивали при 0-5°С в течение 2 ч, гасили путем добавления воды (40 мл) при 0-5°С и экстрагировали этилацетатом (50 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (80 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением WX54-2.
МС-ЭСИ м/з: 681,5 [М+Н]+.
Этап 3: Синтез соединения WX54-3
К раствору соединения WX54-2 (0,56 г, 822,58 ммоль) в 1,4-диоксане (10 мл) добавляли 4М раствор хлористоводородной кислоты в 1,4-диоксане (4М, 4,67 мл). Реакционную смесь перемешивали при 30°С в течение 36 ч, значение рН доводили до 7-8 с помощью насыщенного раствора бикарбоната натрия, разбавляли водой (30 мл), и смесь экстрагировали дихлорметаном (40 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (этилацетата/петролейный эфир: 25-50%) с получением соединения WX54-3.
МС-ЭСИ м/з: 481,3[М+Н]+
Этап 4: Синтез соединения WX54-4
К раствору соединения WX54-3 (0,12 г, 249,71 мкмоль), 2,5-диметил-1,3,4-оксадиазола (97,99 мг, 998,85 мкмоль) в 1-метил-2-пирролидоне (2 мл) добавляли безводную р-толуолсульфоновую кислоту (43,00 мг, 249,71 мкмоль) и перемешивали при 220°С при воздействии микроволнового излучения в течение 1,5 ч. Реакционную смесь концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (метанол/дихлорметан: 2-8%) с получением соединения WX54-4.
МС-ЭСИ м/з: 561,4[М+Н]+
Этап 5: Синтез соединения WX54
К раствору соединения WX54-4 (0,05 г, 89,18 мкмоль) в 1,4-диоксане (10 мл) добавляли 2М гидроксид натрия (2 М, 3 мл). Реакционную смесь перемешивали при 80°С в течение 12 ч, значение рН доводили до 7 с помощью 1М хлористоводородной кислоты, смесь разбавляли водой (15 мл) и экстрагировали дихлорметаном (25 мл ×3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. Отфильтрованную органическую фазу концентрировали с получением неочищенного продукта, который отделяли с помощью колоночной хроматографии (метанол/дихлорметан: 10:1) с получением соединения WX54.
МС-ЭСИ м/з: 407,2 [М+Н]+
1Н ЯМР (ХЛОРОФОРМ-d, 400МГц): δ=8,50 (уш. с, 1Н), 7,32 (дд, J=10,5, 2,3 Гц, 1Н), 7,25 (т, J=2,8 Гц, 1Н), 7,12 (дд, J=8,9, 1,6 Гц, 1Н), 6,88 (с, 1Н), 6,85 (уш. с, 1Н), 6,26 (с, 1Н), 4,30 (уш. д, J=6,0 Гц, 1Н), 3,97-4,08 (м, 2Н), 3,72-3,83 (м, 2Н), 3,60 (тд, J=11,9, 3,1 Гц, 1Н), 3,29 (тд, J=12,7, 3,9 Гц, 1Н), 2,34 (с, 6Н), 1,30 ppm (д, J=6,8 Гц, 3Н).
Пример 55

Схема синтеза:
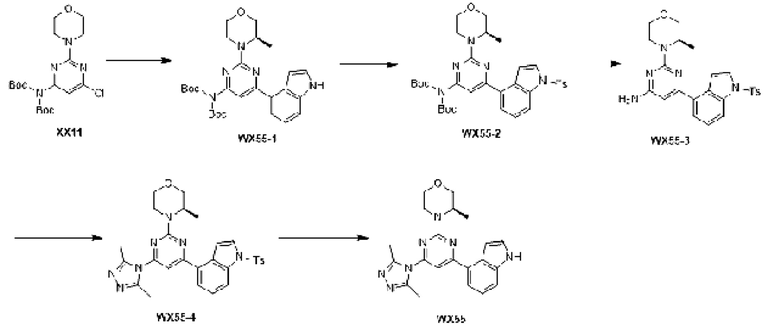
Этап 1: Синтез соединения WX55-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX55-1 идентична процедуре, использованной для соединения WX54-1 в примере 54 синтеза.
МС-ЭСИ м/з: 510,4 [М+Н]+
Этап 2: Синтез соединения WX55-2
Кроме использования соответствующих исходных материалов, процедура получения соединения WX55-2 идентична процедуре, использованной для соединения WX54-2 в примере 54 синтеза.
МС-ЭСИ м/з: 664,5 [М+Н]+
Этап 3: Синтез соединения WX55-3
Кроме использования соответствующих исходных материалов, процедура получения соединения WX55-3 идентична процедуре, использованной для соединения WX54-3 в примере 54 синтеза.
МС-ЭСИ м/з: 464,3 [М+Н]+
Этап 4: Синтез соединения WX55-4
Кроме использования соответствующих исходных материалов, процедура получения соединения WX55-4 идентична процедуре, использованной для соединения WX54-3 в примере 54 синтеза.
МС-ЭСИ м/з: 544,4 [М+Н]+
Этап 5: Синтез соединения WX55
Кроме использования соответствующих исходных материалов, процедура получения соединения WX55 идентична процедуре, использованной для соединения WX54 в примере 54 синтеза.
МС-ЭСИ м/з: 390,2 [М+Н]+
1H ЯМР (ХЛОРОФОРМ-d, 400МГц): δ=8,80 (уш. с, 1Н), 7,68 (д, J=7,5 Гц, 1Н), 7,62 (д, J=8, 1 Гц, 1Н), 7,41-7,45 (м, 1Н), 7,34 (т, J=7,8 Гц, 1Н), 7,13 (уш. с, 1Н), 6,95 (с, 1Н), 4,85 (уш. с, 1Н), 4,54 (уш. д, J=12,8 Гц, 1Н), 4,08 (дд, J=11,4, 3,1 Гц, 1Н), 3,83-3,92 (м, 1Н), 3,74-3, 83 (м, 1Н), 3,64 (тд, J=11,9, 2,6 Гц, 1Н), 3,45 (тд, J=12, 9, 3,4 Гц, 1Н), 2,58 (с, 6Н), 1,44 ppm (д, J=6, 8 Гц, 3Н)
Пример 56
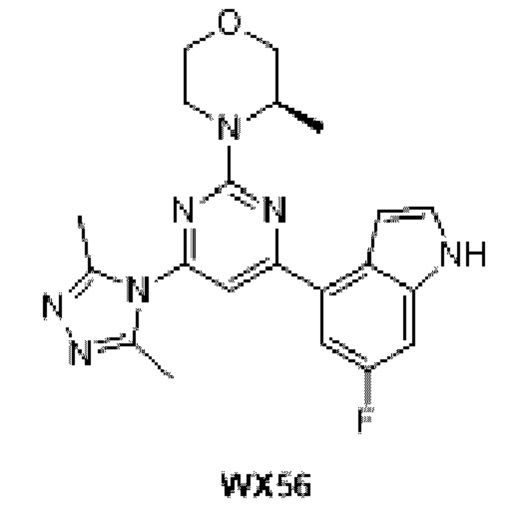
Схема синтеза:
Этап 1: Синтез соединения WX56-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX56-1 идентична процедуре, использованной для соединения WX54-1 в примере 54 синтеза.
МС-ЭСИ м/з: 528,4 [М+Н]+
Этап 2: Синтез соединения WX56-2
Кроме использования соответствующих исходных материалов, процедура получения соединения WX56-2 идентична процедуре, использованной для соединения WX54-2 в примере 54 синтеза.
МС-ЭСИ м/з: 682,5 [М+Н]+
Этап 3: Синтез соединения WX56-3
Кроме использования соответствующих исходных материалов, процедура получения соединения WX56-3 идентична процедуре, использованной для соединения WX54-3 в примере 54 синтеза.
МС-ЭСИ м/з: 482,6 [М+Н]+
Этап 4: Синтез соединения WX56-4
Кроме использования соответствующих исходных материалов, процедура получения соединения WX56-4 идентична процедуре, использованной для соединения ИХ54-3 в примере 54 синтеза.
МС-ЭСИ м/з: 562,3 [М+Н]+
Этап 5: Синтез соединения WX56
Кроме использования соответствующих исходных материалов, процедура получения соединения WX56 идентична процедуре, использованной для соединения WX54 в примере 54 синтеза.
МС-ЭСИ м/з: 407,9 [М+Н]+
1H ЯМР (400МГц, ХЛОРОФОРМ-d) δ=8,47 (уш. с, 1Н), 7,48 (уш. д, J=10,6 Гц, 1Н), 7,39 (уш. с, 1Н), 7,31 (с, 1Н), 7,03 (уш. с, 1Н), 6,92 (с, 1Н), 4,84 (уш. с, 1Н), 4,51 (уш. с, 1Н), 4,08 (уш. д, J=10,4 Гц, 1Н), 3, 90-3, 83 (м, 1Н), 3,81-3,75 (м, 1Н), 3,64 (уш. т, J=11,6 Гц, 1Н), 3, 50-3,39 (м, 1Н), 2,58 (с, бН), 1,44 (уш. д, J=6,5 Гц, 3Н).
Пример 57
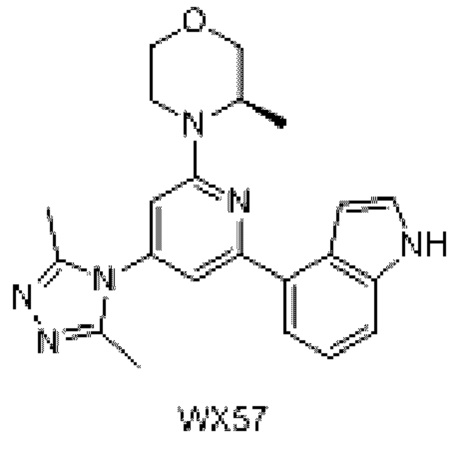
Схема синтеза:
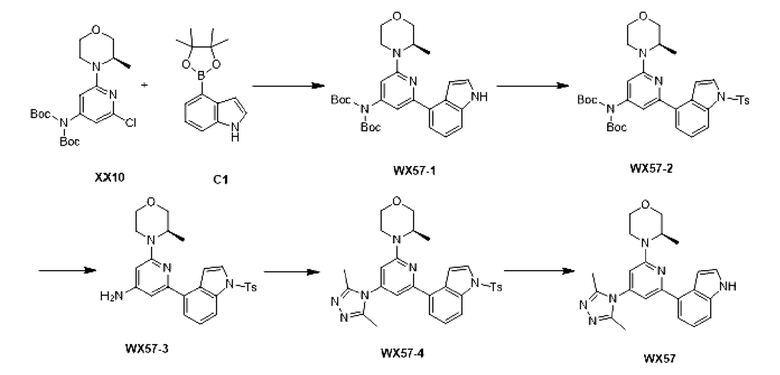
Этап 1: Синтез соединения WX57-1
Кроме использования соответствующих исходных материалов, процедура получения соединения WX57-1 идентична процедуре, использованной для соединения WX54-1 в примере 54 синтеза.
МС-ЭСИ м/з: 509, 4 [М+Н]+.
Этап 2: Синтез соединения WX57-2
Кроме использования соответствующих исходных материалов, процедура получения соединения WX57-2 идентична процедуре, использованной для соединения WX54-2 в примере 54 синтеза.
МС-ЭСИ м/з: 663,5 [М+Н]+.
Этап 3: Синтез соединения WX57-3
Кроме использования соответствующих исходных материалов, процедура получения соединения WX57-3 идентична процедуре, использованной для соединения WX54-3 в примере 54 синтеза.
МС-ЭСИ м/з: 463,3 [М+Н]+
Этап 4: Синтез соединения WX57-4
Кроме использования соответствующих исходных материалов, процедура получения соединения WX57-4 идентична процедуре, использованной для соединения WX54-4 в примере 54 синтеза.
МС-ЭСИ м/з: 542,9 [М+Н]+
Этап 5: Синтез соединения WX57
Кроме использования соответствующих исходных материалов, процедура получения соединения WX57 идентична процедуре, использованной для соединения WX54 в примере 54 синтеза.
МС-ЭСИ м/з: 389,0 [М+Н]+
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1,38 (д, J=6,78 Гц, 3 Н) 2,41 (с, 6 Н) 3,37 (тд, J=12,67, 4,02 Гц, 1 Н) 3,68 (тд, J=11,86, 3,14 Гц, 1 H) 3,80-3,89 (м, 2 Н) 4,05-4,15 (м, 2 Н) 4,39 (уш. д, J=6,78 Гц, 1 Н) 6,28 (д, J=l,25 Гц, 1 Н) 6,98 (д, J=1,00 Гц, 1 Н) 7,01 (уш. с, 1 Н) 7,28-7,33 (м, 1 Н) 7,36 (т, J=2,89 Гц, 1 Н) 7,52 (д, J=8,28 Гц, 1 Н) 7,58 (д, J=7,28 Гц, 1 Н) 8,69 (уш. с, 1 Н)
Экспериментальный пример 1: In vitro оценка
Значения IC50 определяли для оценки ингибиторной активности тестируемых соединений в отношении человеческой киназы ATR.
ATR/ATRIP (h) инкубировали в буфере для анализа, содержащем 50 нМ GST-cMyc-p53 и Mg/АТФ (в требуемой концентрации). Реакцию инициировали добавлением смеси Mg/АТФ. После инкубации в течение 30 мин при комнатной температуре добавляли стоп-раствор, содержащий EDTA, для прекращения реакции. Наконец, добавляли буфер для детектирования, содержащий d2-меченное моноклональное анти-GST антитело и антифосфо-Ser15-фосфорилированное р53 антитело, меченное европием. Затем считывали планшет в режиме гомогенной флуоресценции с временным разрешением.
Сигнал флуоресценции (HTRF) определяли по формуле: HTRF=10000 × (Em665 нм/Em620 нм).
Данные IC50 анализировали с помощью программного обеспечения XLFit версии 5.3 (ID Business Solutions). Нелинейный регрессионный анализ использовали для подгонки сигмоидной кривой зависимости доза-эффект (изменяемый наклон). Результаты экспериментов представлены в таблице 1.
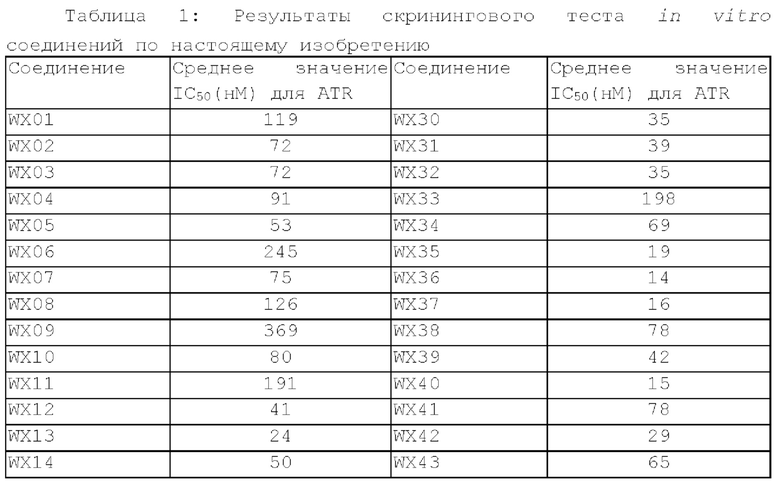
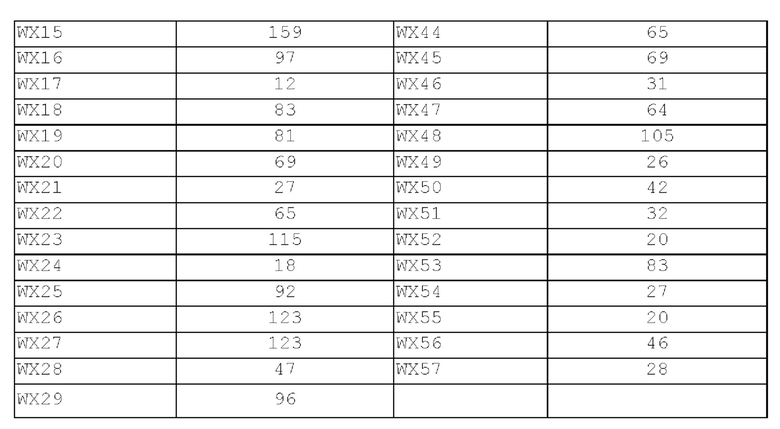
Заключение: Соединения по настоящему изобретению имеют хорошую ингибирующую активность в отношении ATR.
Экспериментальный пример 2: In vitro тест жизнеспособности клеток
В этом эксперименте исследовали ингибирующее действие соединений на пролиферацию клеток путем тестирования влияния на жизнеспособность клеток in vitro в линии опухолевых клеток LoVo.
Тест на жизнеспособность клеток по интенсивности люминесценции CellTiter-Glo
Описанные ниже протоколы выполняли в соответствии с инструкциями, прилагаемыми к набору Promega (Promega-G7573), для тестирования жизнеспособности клеток по интенсивности люминесценции CellTiter-Glo.
(1) Разморозить буфер CellTiter-Glo и дать ему нагреться до комнатной температуры.
(2) Дать субстрату CellTiter-Glo нагреться до комнатной температуры.
(3) Добавить буфер CellTiter-Glo во флакон с субстратом CellTiter-Glo для растворения субстрата с получением рабочего раствора CellTiter-Glo.
(4) Медленно перемешать до полного растворения.
(5) Вынуть чашки с культурой клеток и оставить их на 30 мин для доведения до комнатной температуры.
(6) Добавить 50 мкл (что эквивалентно половине объема среды для культивирования клеток в каждой лунке) рабочего раствора CellTiter-Glo в каждую лунку, обернуть планшет с клетками алюминиевой фольгой для защиты от света.
(7) Встряхивать культуральный планшет на орбитальном шейкере в течение 2 минут для индукции лизиса клеток.
(8) Оставить чашки для культивирования при комнатной температуре на 10 мин для стабилизации сигнала люминесценции.
(9) Выполнить обнаружение сигнала люминесценции на планшетном ридере SpectraMax i3x компании Molecular Devices.
Анализ данных
Для расчета уровня ингибирования, обеспечиваемого тестируемым соединением, использовали следующую формулу (уровень ингибирования, IR): IR(%)=(1-(RLU соединения - RLU пустой контроль)/(RLU контроль с носителем - RLU пустой контроль)) * 100%.
Уровень ингибирования, обеспечиваемый различными концентрациями соединений, вычисляли в Excel, и для построения кривой ингибирования и вычисления релевантных параметров, включая минимальную степень ингибирования, максимальную степень ингибирования и IC50, использовали программное обеспечение GraphPad Prism.
Результаты экспериментов приведены в таблице 2.
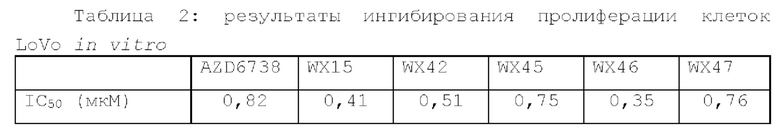
Заключение: соединения по настоящему изобретению обладают хорошим ингибирующим действием на опухолевые клетки LoVo с мутациями в сигнальном пути ATM.
Экспериментальный пример 3: Исследование фармакокинетических свойств In vivo.
Образцы для тестирования: На основе результатов, полученных в описанных выше экспериментах, для дальнейших исследований отбирали некоторые из высокоактивных соединений с репрезентативными структурами.
Экспериментальный метод: Целью этого исследования было определение фармакокинетических параметров соединений и измерение биодоступности при пероральном введении самкам мышей Balb/c Nude.
В эксперименте использовали 6 самок мышей Balb/c Nude. 3 мышам выполняли внутривенное введение в дозе 1 мг/кг, и образцы плазмы собирали через 0 ч (до введения) и через 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения, остальным 3 мышам введение осуществляли через желудочный зонд в дозе 10 мг/кг или 25 мг/кг, и образцы плазмы собирали через 0 ч (до введения дозы) и через 0,5, 1, 2, 3, 4, 6, 8, 24 часа после введения. Затем собранные образцы тестировали с помощью анализа ЖХ/МС/МС и собирали данные. Собранные данные использовали для вычисления соответствующих фармакокинетических параметров с помощью программного обеспечения Phoenix WinNonlin 6.2.1.
Результаты экспериментов представлены в Таблице 3.1 и Таблице 3.2.
3.1 Результаты внутривенного введения
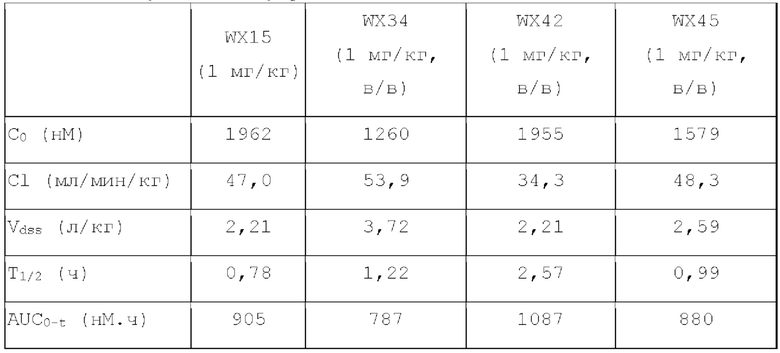
3.2 Результаты перорального введения
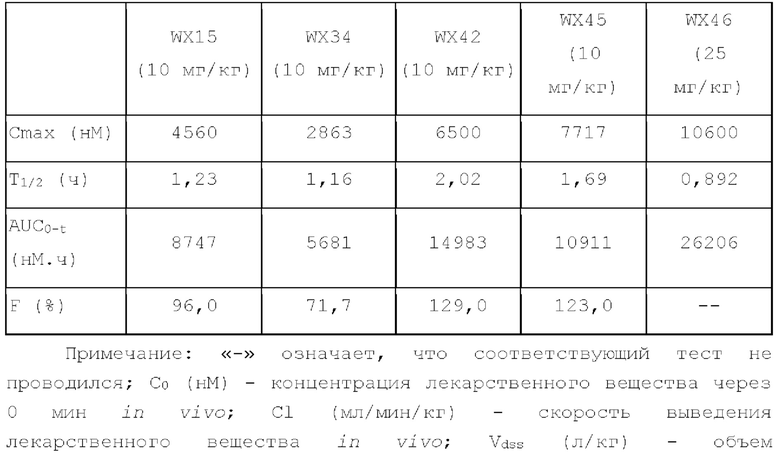

Заключение: соединения по настоящему изобретению демонстрируют хорошую абсорбцию и экспозицию при пероральном введении и подходят для перорального введения.
Экспериментальный пример 4: Исследование эффективности in vivo в модели колоректального рака LoVo CDX
Цель:
LoVo - это линия опухолевых клеток колоректальной аденокарциномы с мутацией MRE11A (MRE11A - ключевой компонент пути передачи сигнала ATM, восстанавливающий двухцепочечный разрыв ДНК), которая чувствительна к ингибитору ATR. В этом эксперименте для проверки ингибирующего действия ингибитора ATR при монотерапии опухоли с дефектом сигнального пути ATM использовали модель LoVo CDX колоректального рака.
Процедуры:
1. Лабораторное животное
Вид: мышь
Штамм: мыши BALB/c nude
Поставщик: Beijing Vital River Laboratory Animal Technology Co., Ltd.
Недели и вес: 6-8 недель, вес 18-22 г
Пол: самки
2. Клеточная культура
Клетки LoVo колоректального рака человека (ЕСАСС, CatLog: 87060101), монослойная культура in vitro, условия культивирования: среда F-12 Хэма с 10% фетальной бычьей сывороткой, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 2 мМ глутамина, 37°С, 5% СО2 культура. Общий пассаж с трипсин-EDTA перевариванием выполняли два раза в неделю. Когда насыщение клеток составляло 80%-90%, клетки собирали, подсчитывали и инокулировали. 0,1 мл (10×106) клеток LoVo инокулировали подкожно в правый бок каждой мыши Nude. Когда средний объем опухоли достиг 173 мм3, животных разбивали на группы и начинали введение.
3. Приготовление и доза исследуемых веществ.
1) Соединение WX15
Брали навеску 25,51 мг WX15 и растворяли в 0,500 мл DMSO, к этому раствору добавляли 2,000 мл пропиленгликоля и 2,500 мл деионизованной воды, встряхивали до гомогенности, доводили рН до 6,0, получая прозрачный раствор. Способы приготовления соединения WX42, соединения WX45, соединения WX46 аналогичны способу приготовления соединения WX15.
Дозировка: Все тестируемые соединения вводили перорально в дозе 25 мг/кг два раза в сутки с интервалом 8 часов в сутки.
4. Измерение опухолей и экспериментальные индикаторы
Для измерения диаметра опухоли, которое выполняли два раза в неделю, использовали штангенциркуль. Объем опухоли вычисляли по следующей формуле: V=0,5a×b2, а и b представляют собой длинный и короткий диаметры опухоли, соответственно.
Противоопухолевую эффективность соединения оценивали по TGI(%) или относительной скорости пролиферации опухоли Т/С(%).
Относительная скорость пролиферации опухоли Т/С(%)=TRTV/CRTV × 100% (TRTV: среднее значение RTV для группы лечения; CRTV: среднее значение RTV для группы отрицательного контроля). Относительный объем опухоли (относительный объем опухоли, RTV) рассчитывали на основе результатов измерения опухоли по следующей формуле: RTV=Vt/V0, где V0 - объем опухоли, измеренный в момент разбивки по группам введения (т.е. DO), Vt - объем опухоли в точках измерения, и для TRTV и CRTV использовали данные, полученные в один и тот же день.
TGI(%) отражает степень подавления роста опухоли. TGI(%)=[1-(средний объем опухоли в конце введения в группе лечения - средний объем опухоли в начале введения в этой группе лечения)/(средний объем опухоли в конце лечения в контрольной группе с растворителем - средний объем опухоли в начале лечения в контрольной группе с растворителем)]×100%.
В конце эксперимента вес опухолей взвешивали и рассчитывали процентное отношение Т/С. Твес и Свес представляют собой веса опухоли в группе введения и контрольной группе с носителем, соответственно.
5. Результаты эксперимента.
В этом эксперименте оценивали эффективность соединений в модели ксенотрансплантата колоректального рака человека относительно контрольной группы с растворителем в качестве эталона. Объемы опухолей в каждой группе в разные моменты времени показаны на Фиг. 1. На день 17 введения Т/С и TGI в группе WX42 (25 мг/кг) составляли 27,8% и 90,7%, соответственно, по сравнению с контрольной группой с носителем; Т/С и TGI в группе WX45 (25 мг/кг) составили 32,3% и 79,9%, соответственно, по сравнению с контрольной группой с носителем; Т/С и TGI в группе WX46 (25 мг/кг) составили 43,8% и 79,9%, соответственно, по сравнению с контрольной группой с носителем; Т/С и TGI в группе WX15 (25 мг/кг) составили 46,7% и 66,8%, соответственно, по сравнению с контрольной группой с носителем.
6. Заключение
В этом эксперименте соединения по настоящему изобретению показали ингибирующее действие на рост опухоли у мышей в модели опухоли с подкожным ксенотрансплантатом клеток LoVo колоректального рака человека.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНПИРРОЛИДИН-2-ОНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2771314C1 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| Производное индола, используемое в качестве ингибитора CRTH2 | 2017 |
|
RU2756270C2 |
| АЛЬФА- И БЕТА-НЕНАСЫЩЕННОЕ АМИДНОЕ СОЕДИНЕНИЕ - ПРОИЗВОДНОЕ БЕНЗОТРИАЗОЛА, ПРИМЕНЯЕМОЕ В КАЧЕСТВЕ ИНГИБИТОРА TGF-βRI | 2017 |
|
RU2737737C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ПРИМЕНЕНИЕ МОНОЦИКЛИЧЕСКОГО β-ЛАКТАМНОГО СОЕДИНЕНИЯ В ФАРМАЦИИ | 2019 |
|
RU2785715C1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |

Изобретение относится к соединению формулы (I) или его таутомеру или фармацевтически приемлемой соли. В формуле (I): n равно 1, 2, 3 или 4; структурную единицу выбирают из группы, состоящей из
выбирают из группы, состоящей из  , кольцо А выбирают из группы, состоящей из 5-6-членного гетероарила; каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, C1-6 алкила, С1-6 алкокси и С3-6 циклоалкила, причем C1-6 алкил, C1-6 алкокси и С3-6 циклоалкил необязательно замещены 1, 2 или 3 R; каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, СООН и C1-3 алкила, причем C1-3 алкил необязательно замещен 1, 2 или 3 R; каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН, C1-3 алкила и C1-3 алкокси, причем C1-3 алкил и C1-3 алкокси необязательно замещены 1, 2 или 3 R'; каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I и ОН; 5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбираемых из группы, состоящей из -NH-, -О- и N; и Z1 представляет собой N, Z2 и Z3 представляют собой СН; Z2 представляет собой N, Z1 и Z3 представляют собой СН; Z3 представляет собой N, Z1 и Z2 представляют собой СН; Z1 и Z2 представляют собой N, Z3 представляет собой СН; Z2 и Z3 представляют собой N, Z1 представляет собой СН; или Z1 и Z3 представляют собой N, Z2 представляет собой СН. Также предложено применение указанного соединения для производства лекарственного средства. Предложенное соединение может применяться в качестве ингибитора ATR. 3 н. и 12 з.п. ф-лы, 1 ил., 4 табл., 61 пр.
, кольцо А выбирают из группы, состоящей из 5-6-членного гетероарила; каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, C1-6 алкила, С1-6 алкокси и С3-6 циклоалкила, причем C1-6 алкил, C1-6 алкокси и С3-6 циклоалкил необязательно замещены 1, 2 или 3 R; каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, СООН и C1-3 алкила, причем C1-3 алкил необязательно замещен 1, 2 или 3 R; каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН, C1-3 алкила и C1-3 алкокси, причем C1-3 алкил и C1-3 алкокси необязательно замещены 1, 2 или 3 R'; каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I и ОН; 5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбираемых из группы, состоящей из -NH-, -О- и N; и Z1 представляет собой N, Z2 и Z3 представляют собой СН; Z2 представляет собой N, Z1 и Z3 представляют собой СН; Z3 представляет собой N, Z1 и Z2 представляют собой СН; Z1 и Z2 представляют собой N, Z3 представляет собой СН; Z2 и Z3 представляют собой N, Z1 представляет собой СН; или Z1 и Z3 представляют собой N, Z2 представляет собой СН. Также предложено применение указанного соединения для производства лекарственного средства. Предложенное соединение может применяться в качестве ингибитора ATR. 3 н. и 12 з.п. ф-лы, 1 ил., 4 табл., 61 пр.
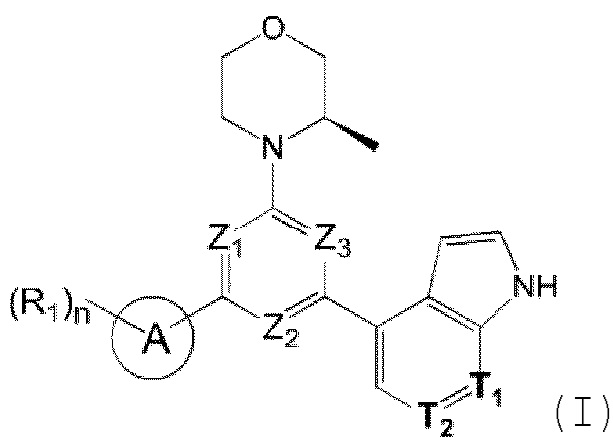
1. Соединение формулы (I) или его таутомер или фармацевтически приемлемая соль
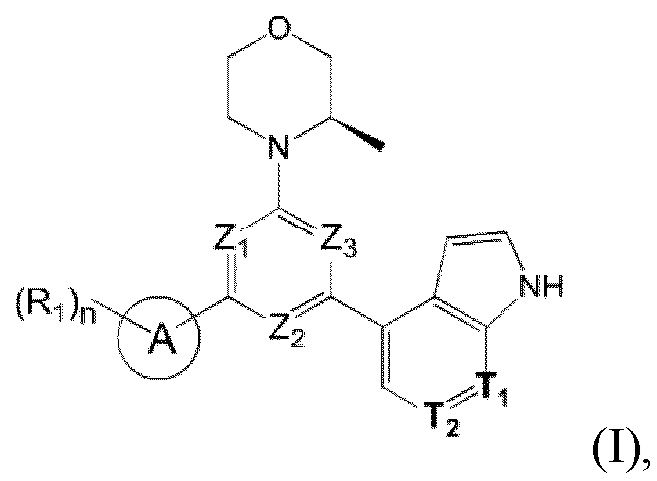
где n равно 1, 2, 3 или 4;
структурную единицу выбирают из группы, состоящей из
выбирают из группы, состоящей из 
кольцо А выбирают из группы, состоящей из 5-6-членного гетероарила;
каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, C1-6 алкила, С1-6 алкокси и С3-6 циклоалкила, причем C1-6 алкил, C1-6 алкокси и С3-6 циклоалкил необязательно замещены 1, 2 или 3 R;
каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, СООН и C1-3 алкила, причем C1-3 алкил необязательно замещен 1, 2 или 3 R;
каждый R независимо выбирают из группы, состоящей из F, Cl, Br, I, ОН, C1-3 алкила и C1-3 алкокси, причем C1-3 алкил и C1-3 алкокси необязательно замещены 1, 2 или 3 R';
каждый R' независимо выбирают из группы, состоящей из F, Cl, Br, I и ОН;
5-6-членный гетероарил содержит 1, 2, 3 или 4 гетероатома или гетерорадикала, независимо выбираемых из группы, состоящей из -NH-, -О- и N; и
Z1 представляет собой N, Z2 и Z3 представляют собой СН;
Z2 представляет собой N, Z1 и Z3 представляют собой СН;
Z3 представляет собой N, Z1 и Z2 представляют собой СН;
Z1 и Z2 представляют собой N, Z3 представляет собой СН;
Z2 и Z3 представляют собой N, Z1 представляет собой СН; или
Z1 и Z3 представляют собой N, Z2 представляет собой СН.
2. Соединение по п. 1 или его таутомер или фармацевтически приемлемая соль, где каждый R независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, ОН, СН3, Et и -O-СН3.
3. Соединение по п. 1 или 2 или его таутомер или фармацевтически приемлемая соль, где каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, C1-3 алкила, C1-3 алкокси и циклопропила, причем C1-3 алкил, C1-3 алкокси и циклопропил необязательно замещены 1, 2 или 3 R.
4. Соединение по п. 3 или его таутомер или фармацевтически приемлемая соль, где каждый R1 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, СН3, CH2F, CHF2, CF3, Et, -CH2OH, -О-СН3,  и
и  .
.
5. Соединение по п. 1 или 2 или его таутомер или фармацевтически приемлемая соль, где каждый R2 независимо выбирают из группы, состоящей из Н, F, Cl, Br, I, СООН, СН3, Et и -СН2-ОН.
6. Соединение по п. 1 или 2 или его таутомер или фармацевтически приемлемая соль, где кольцо А выбирают из группы, состоящей из пиразолила, изоксазолила, оксазолила, имидазолила, 1,2,3-триазолила, 1,2,4-триазолила и пиридила.
7. Соединение по п. 6 или его таутомер или фармацевтически приемлемая соль, где кольцо А выбирают из группы, состоящей из 
8. Соединение по п. 1 или 2 или его таутомер или фармацевтически приемлемая соль, где структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
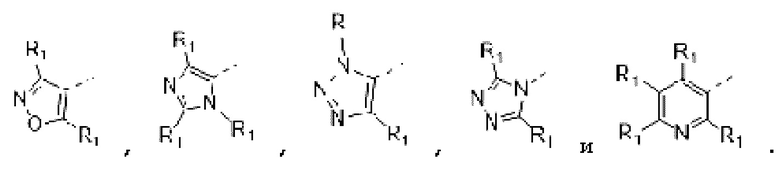
9. Соединение по п. 8 или его таутомер или фармацевтически приемлемая соль, где структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
10. Соединение по п. 1 или 2 или его таутомер или фармацевтически приемлемая соль, где структурную единицу 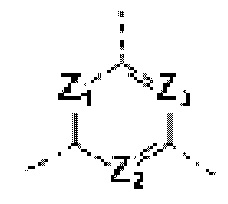 выбирают из группы, состоящей из
выбирают из группы, состоящей из 
11. Соединение по любому из пп. 1-8 или его таутомер или фармацевтически приемлемая соль, причем соединение выбирают из:
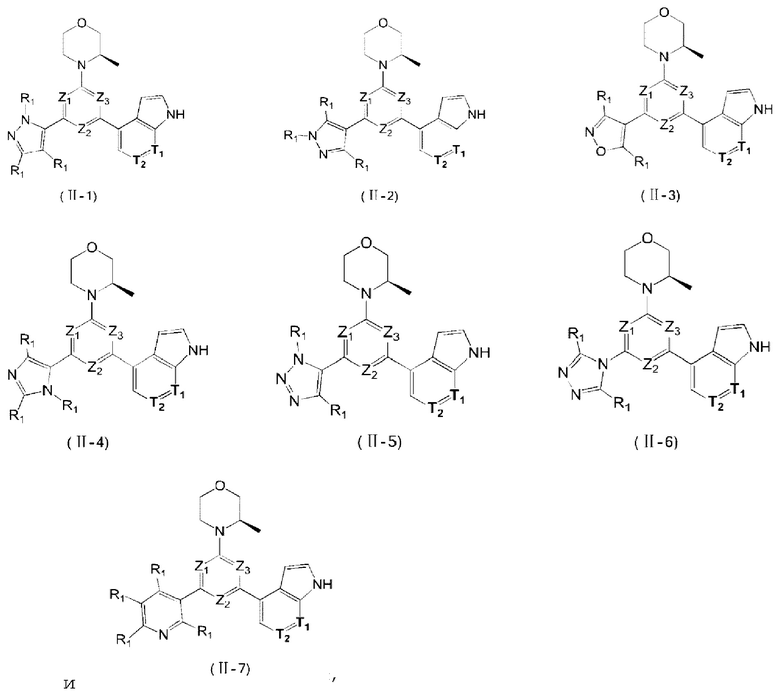
где структурную единицу  выбирают из группы, состоящей из
выбирают из группы, состоящей из 
Z1, Z2 и Z3 являются такими, как определены в п. 1;
R1 является таким, как определен в любом из пп. 1, 2, 3 или 4;
R2 является таким, как определен в п. 1 или 5.
12. Соединение по п. 11 или его таутомер или фармацевтически приемлемая соль, причем соединение выбирают из:
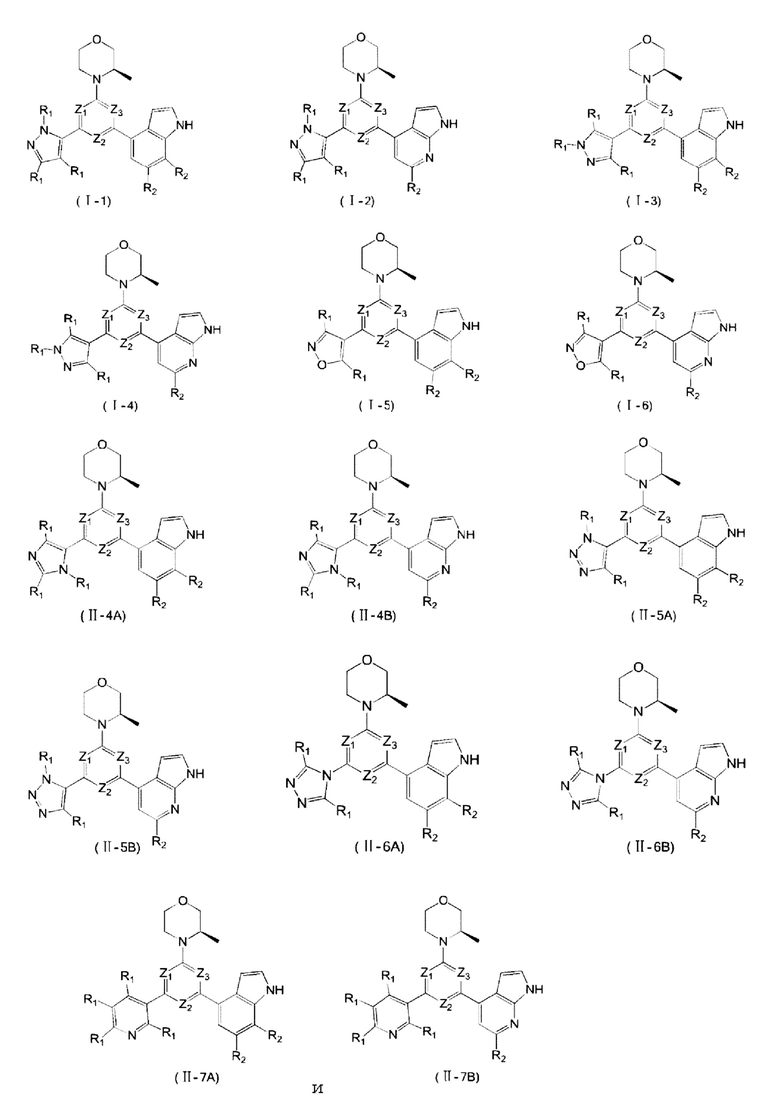
где Z1, Z2 и Z3 являются такими, как определены в п. 1;
R1 является таким как определен в любом из пп. 1, 2, 3 или 4;
R2 является таким как определен в п. 1 или 5.
13. Соединение или его таутомер или фармацевтически приемлемая соль, выбранное из:
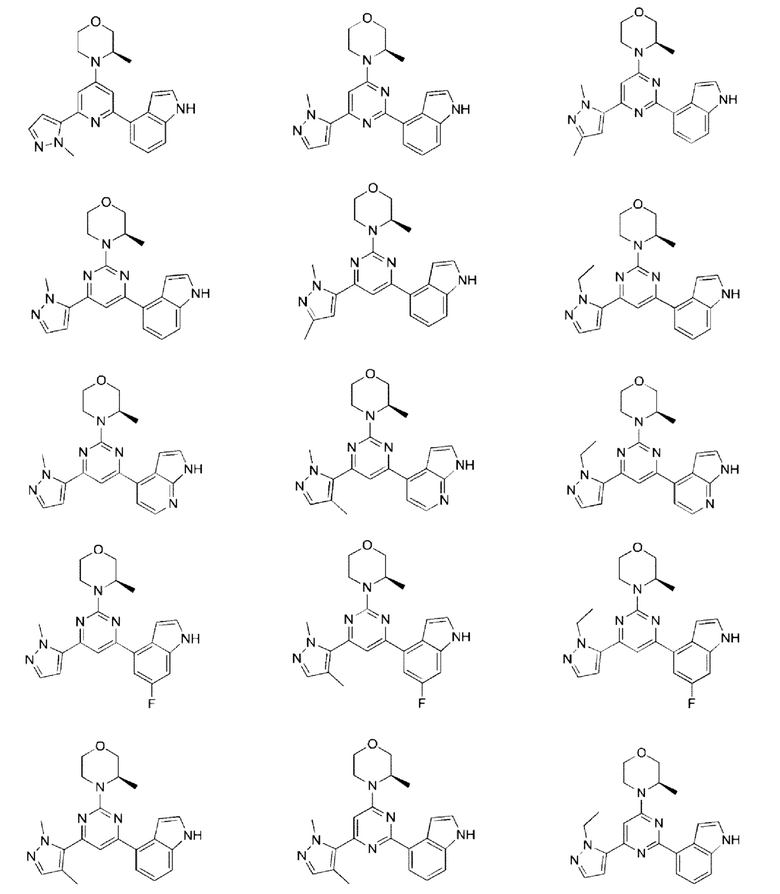

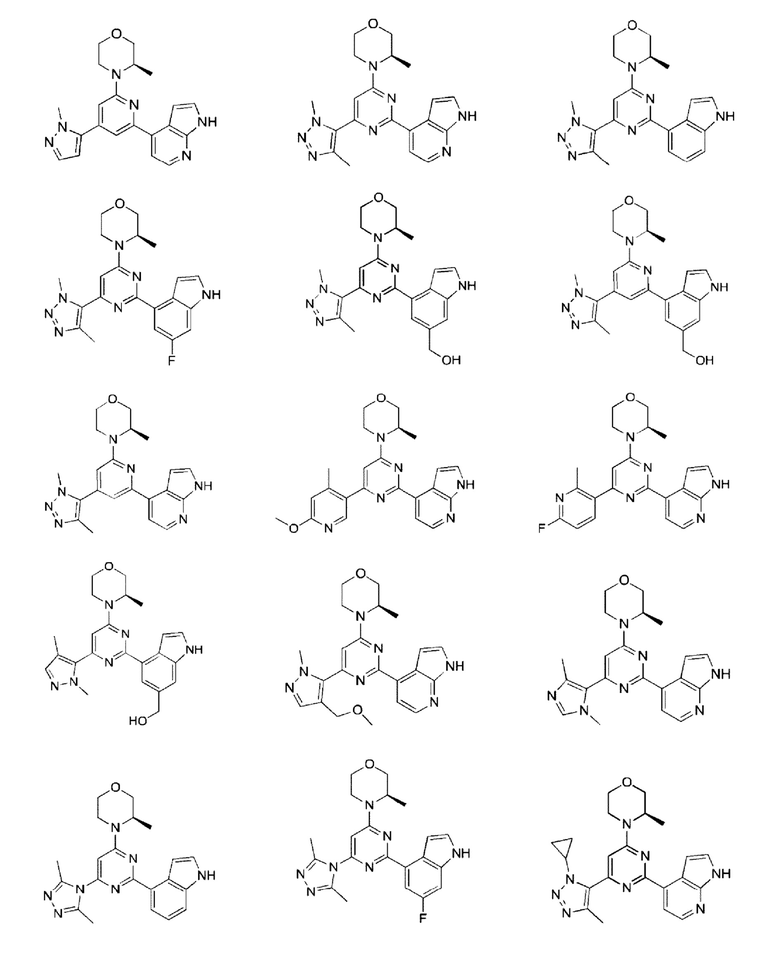

14. Применение соединения по любому из пп. 1-13 или его таутомера или фармацевтически приемлемой соли для производства лекарственного средства для лечения ATR-ассоциированного заболевания.
15. Применение по п. 14, где ATR-ассоциированное заболевание представляет собой солидную опухоль или гематологическую опухоль.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ТРИАЗИНОВЫЕ, ПИРИМИДИНОВЫЕ И ПИРИДИНОВЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ И ДИАГНОСТИЧЕСКИХ ПРОБ | 2009 |
|
RU2537945C2 |
| ТРИАЗИНОВЫЕ, ПИРИМИДИНОВЫЕ И ПИРИДИНОВЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ И ДИАГНОСТИЧЕСКИХ ПРОБ | 2009 |
|
RU2537945C2 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Приспособление для стягивания тормозных тяг | 1930 |
|
SU22087A1 |
