ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому типу производного цитидина и применению производных при получении противоопухолевых препаратов.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Злокачественная опухоль является одним из распространенных заболеваний, которые угрожают здоровью человека, и занимает первое место среди всех заболеваний с точки зрения смертности. Что касается противоопухолевых препаратов, доступных для применения в медицинской практике в настоящее время, то их токсичность является значимой проблемой при химиотерапии опухолей. Усиление эффективности лечения опухолей и одновременное снижение токсичности препарата является ключевым предметом исследования существующих в настоящее время противораковых препаратов.
Производные цитидина, обладающие противоопухолевыми эффектами, включают цитарабин и гемцитабин. Для достижения противоракового эффекта цитарабин в организме превращается в активный цитарабина трифосфат. Цитарабина трифосфат предотвращает синтез ДНК и ингибирует рост клеток путем ингибирования ДНК-полимеразы и введения небольшого количества ДНК, и в основном применяется для лечения острого миелобластного лейкоза. Однако цитарабин имеет сравнительно сильные токсические побочные эффекты, которые могут вызывать подавление деятельности костного мозга, снижение уровня лейкоцитов и тромбоцитопению в гемопоэтической системе. В тяжелых случаях могут возникать апластическая анемия или мегалобластная анемия. У пациента с лейкемией или лимфомой при раннем лечении может возникать гиперурикемия, а при тяжелых случаях может возникать нефропатия мочевой кислоты.
Гемцитабин является производным дезоксицитидина и подобен цитарабину по структуре и метаболизму. Гемцитабин в клетке катализируется нуклеозидмонофосфат киназой в активированный дифосфат (dFdCDP) и трифосфат (dFdCTP), и dFdCTP препятствует синтезу ДНК посредством ингибирования ДНК-полимеразы. Цепь ДНК прекращает свое удлинение за счет включения в ДНК, ингибируя тем самым рост опухолевых клеток.
Гемцитабин применим при раке поджелудочной железы (первичное и вторичное лечение), немелкоклеточном раке легкого, раке молочной железы, раке яичника и плоскоклеточном раке головы и шеи. Однако токсичность гемцитабина также относительно велика. Его побочные реакции представляют собой подавление деятельности костного мозга, такое как лейкопения, тромбоцитопения и анемия; желудочно-кишечные реакции, такие как легкая тошнота, рвота и аномальная функция печени; лихорадка, гриппоподобные симптомы, усталость, мукозит и так далее.
После того, как вышеупомянутые производные цитидина попадут в организм человека, опухолевые клетки будут продуцировать ген с множественной лекарственной устойчивостью. Кроме того, аминогруппа на кольце легко ацетилируется, вызывая потерю противораковой активности и других факторов устойчивости. Вышеупомянутые производные цитидина обладают значительными токсическими побочными эффектами и имеют тенденцию вызывать устойчивость к лекарственным средствам.
Чтобы уменьшить токсичность цитарабина и гемцитабина и улучшить или поддержать противоопухолевую эффективность, исследователи модифицировали химическую структуру производных цитидина.
Например, в Synthesis and Biological Activity of a Gemcitabine Phosphoramidate Prodrug (J. Med. Chem 2007, 50, 3743-3746; Weidong Wu) раскрыт вид пролекарства гемцитабина фосфат.
В патенте США №7265096 В2 (заявка №10/701965) раскрыты пролекарства гемцитабина, фармацевтические композиции и их применения. В этой статье описано замещение амина гемцитабина, атома водорода гидроксила и атома водорода гидроксиметила на рибофуранозе. Атом водорода гидроксиметила на рибофуранозе был замещен Н, ацилом, замещенным ацилом, ацилоксикарбонилом, замещенным ацилоксикарбонилом, оксикарбонилом и замещенным оксикарбонилом; атом водорода гидроксила на рибофуранозе был замещен Н, ацилом, замещенным ацилом, ацилоксикарбонилом, замещенным ацилоксикарбонилом, оксикарбонилом и замещенным оксикарбонилом; аминогруппа была замещена -N=C(R10)(R11) или -NHR12, где R12 представлял собой С5-С9 ацил или С5-С9 замещенный ацил. Соединение, полученное в этом патенте, является пролекарством, которое проявляет противоопухолевую активность только после превращения в организме. Кроме того, клинические исследования показали, что пролекарство гемцитабина обладает высокой токсичностью и низкой противоопухолевой активностью, и оно еще не разработано как лекарственное средство.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема, решаемая настоящим изобретением, заключается в обеспечении нового типа производного цитидина и применении таких производных при получении противоопухолевых препаратов.
Техническое решение для достижения цели настоящего изобретения включает: новый тип производного цитидина следующей общей формулы (I):

где R1 представляет собой С1-С10 алкил, С1-С10 замещенный алкил, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, Ph представляет собой фенил; углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, а углеродная цепь или бензольное кольцо замещены независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами.
R2 представляет собой Н, галоген или  , где Х1 представляет собой C1-С10 алкил, C1-С10 замещенный алкил, C1-C10 алкокси, C1-C10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; углеродная цепь указанного замещенного алкокси замещена независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, и углеродная цепь или бензольное кольцо замещены одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами.
, где Х1 представляет собой C1-С10 алкил, C1-С10 замещенный алкил, C1-C10 алкокси, C1-C10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; углеродная цепь указанного замещенного алкокси замещена независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, и углеродная цепь или бензольное кольцо замещены одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами.
R3 представляет собой Н или  , где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо, конденсированное гетероциклическое кольцо, замещенный фенил, который замещен независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами, замещенное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами или замещенное конденсированное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; где указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин или пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин или индол; и Х2 представляет собой -(CH2)n-, где n равно 1, 2, 3, или Х2 представляет собой -О-(CH2)n-, где n равно 0, 1, 2, 3.
, где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо, конденсированное гетероциклическое кольцо, замещенный фенил, который замещен независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами, замещенное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами или замещенное конденсированное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами; где указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин или пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин или индол; и Х2 представляет собой -(CH2)n-, где n равно 1, 2, 3, или Х2 представляет собой -О-(CH2)n-, где n равно 0, 1, 2, 3.
Необязательно, R2 представляет собой Н.
Предпочтительно, R2 не является Н; и R3 не является Н.
Когда R2 не является Н, R2 представляет собой галоген или 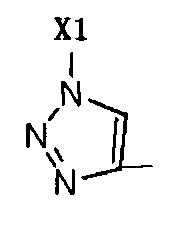 , Х1 представляет собой -(CH2)n-Ph или замещенный -(CH2)n-Ph.
, Х1 представляет собой -(CH2)n-Ph или замещенный -(CH2)n-Ph.
Предпочтительно, R1 представляет собой С1-С4 алкил, С1-С4 замещенный алкил, бензил или замещенный бензил; Х3 R3 представляет собой замещенный имидазол, замещенный независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами, замещенный пиридин, замещенный независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами, или замещенное бензольное кольцо, замещенное независимо одним, или двумя, или тремя атомами галогена, цианогруппами, нитрогруппами, аминогруппами, гидроксильными группами или карбоксильными группами.
Применения вышеуказанных соединений или их солей при получении противоопухолевых препаратов.
Опухоль относится к кровяной опухоли или злокачественной солидной опухоли.
Соль относится к гидрохлориду, фосфату, сульфату, карбонату, нитрату, цитрату, тартрату, малеату, сукцинату, сульфонату, пара-толуолсульфонату, мезилату, бензоату или фумарату.
Фармацевтическая композиция включает: производное цитидина, как показано формулой (I), или его фармацевтически приемлемую соль в качестве активного ингредиента и один или несколько фармацевтических носителей или эксципиентов.
Лекарственная форма вышеупомянутой композиции представляет собой инъекцию или средство для перорального введения, где инъекция относится к инъекции в виде раствора, инъекции в виде суспензии, инъекции в виде эмульсии или стерильному порошку для инъекции; средство для перорального введения относится к таблетке, порошку, грануле, капсуле, пеллету, раствору, суспензии, эмульсии, сиропу или эликсиру.
Настоящее изобретение обладает преимущественными эффектами: для соединения, раскрытого в настоящем изобретении, с помощью экспериментов в отношении эффекта ингибирования роста на ксенотрансплантатах рака толстой кишки НСТ-116 у бестимусных мышей, несущих опухоли, было продемонстрировано, что соединение, раскрытое в настоящем изобретении, обладает высокой противоопухолевой активностью и небольшим влиянием на массу бестимусных мышей, несущих рак толстой кишки человека НСТ-116, что показывает, что токсичность соединения является относительно низкой.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Новое производное цитидина в настоящем изобретении имеет следующую структурную формулу (I):
где, R1 представляет собой C1-С10 алкил, C1-С10 замещенный алкил, -(CH2)n-Ph или замещенный -(CH2)n-Ph; в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, Ph представляет собой фенил; углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро, амино-, гидроксилами или карбоксилами; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10, и его углеродная цепь или бензольное кольцо замещены независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
R2 представляет собой Н, галоген или  , где Х1 представляет собой C1-С10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; углеродная цепь указанного замещенного алкокси замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10 и его углеродная цепь или бензольное кольцо замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
, где Х1 представляет собой C1-С10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; углеродная цепь указанного замещенного алкила замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; углеродная цепь указанного замещенного алкокси замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в указанном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; в указанном замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10 и его углеродная цепь или бензольное кольцо замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
R3 представляет собой Н или  , где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо или конденсированное гетероциклическое кольцо, замещенный фенил, который замещен независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, замещенное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, или замещенное конденсированное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин или пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин или индол; Х2 представляет собой -(СН2)n-, где n равно 1, 2, 3, или Х2 представляет собой -O-(СН2)n-, где n равно 0, 1, 2, 3.
, где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо или конденсированное гетероциклическое кольцо, замещенный фенил, который замещен независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, замещенное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, или замещенное конденсированное гетероциклическое кольцо, которое замещено независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин или пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин или индол; Х2 представляет собой -(СН2)n-, где n равно 1, 2, 3, или Х2 представляет собой -O-(СН2)n-, где n равно 0, 1, 2, 3.
Касательно производного цитидина согласно настоящему изобретению, в Таблице 1 перечислены следующие соединения. Однако производные цитидина настоящего изобретения не ограничены этими соединениями.


При получении соединений приведенной выше таблицы, твердые реагенты, используемые в процессе синтеза, используют непосредственно без дополнительной обработки, жидкие реагенты используют после повторной перегонки и сушки.
(Пример 1)
Производное цитидина в настоящем примере представляет собой 4-N-(н-бутоксикарбонил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 4, код: G2), который может быть получен в три стадии реакции. Схема реакции приведена ниже (в схеме реакции HMDS - гексаметилдисилазан, rt - комнатная температура, a TEA - триэтиламин, далее аббревиатуры те же).

300 мг (1 ммоль) 2'-дезокси-2',2'-дифторцитидина гидрохлорида (структурная формула: 1, код: G1), 5 мл (0,023 ммоль) бис(триметилсилил)амина HMDS и 5 мг сульфата аммония в каталитическом количестве растворяли в 5 мл 1,4-диоксана и кипятили с обратным холодильником в течение 2 часов. Химическая структурная формула продукта реакции - 2. После завершения реакции с обратным холодильником реакционную жидкость концентрировали, добавляли толуол и дважды концентрировали досуха. Полученный продукт после концентрирования растворяли в 10 мл дихлорметана.
0,24 мл (3 ммоль) N-метилимидазола и 0,32 мл (3 ммоль) н-бутилхлорформиата добавляли в вышеуказанный раствор дихлорметана для реагирования при перемешивании при комнатной температуре в течение 4 часов. Химическая структурная формула продукта реакции - 3, и реакционную жидкость концентрировали с получением вязкого маслянистого продукта.
Вязкий маслянистый продукт растворяли в смешанном растворе 3 мл триэтиламина и 20 мл метанола и перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли дистилляцией при пониженном давлении. Для очистки сырого продукта проводили хроматографию на силикагеле, а после элюирования дихлорметаном / метанолом (20:1) получали 230 мг G2. Выход трех стадий реакции составил 55,5%.
ЯМР характеристика G2:
1H-ЯМР (MeOD-d4, 400 МГц) δ: 8,30 (d, 1Н, J=7,68 Гц, Н6), 7,34 (d, 1Н, J=7,68 Гц, Н5), 6,28 (t, 1Н, J=7,08 Гц, Н1'), 4,33 (m, 1Н, Н5а'), 4,0 (m, 2Н, O-СН2-СН2-), 3,81 (m, 1Н, Н5b'), 3,79 (m, 1Н, Н4'), 1,68 (m, 2Н, O-СН2-СН2-), 1,45 (m, 2Н, O-СН2-СН2-СН2), 0,98 (t, 3H, J=7,4 Гц, -СН2-СН3).
13С-ЯМР (МеОD-d4, 100 МГц) δ: 164,28, 156,27, 153,50, 144,39, 128,33, 122,72, 95,81, 84,90, 81,71, 74,87, 68,88, 63,69, 59,15, 30,66, 32,40, 18,81, 11,23, 8,06.
Вышеуказанный способ получения может быть адаптирован путем изменения исходных материалов реакции. Помимо н-бутила, R1 может собой другие группы, такими как C1-С10 алкил, C1-С10 замещенный алкил, -(CH2)n-Ph, n равно 0, 1, 2, 3 до 10 или замещенный -(CH2)n-Ph, n равно 0, 1, 2, 3 до 10, Ph представляет собой фенил; где углеродная цепь в замещенном алкиле замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; углеродная цепь или бензольное кольцо в указанном замещенном -(CH2)n-Ph замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
(Пример 2)
Производное цитидина в настоящем примере представляет собой 4-N-(трет-бутилоксикарбонил)-2'-дезокси-2',2,-дифторцитидин (структурная формула: 6, код: G3), который может быть получен в три стадии реакции. Схема реакции приведена ниже.
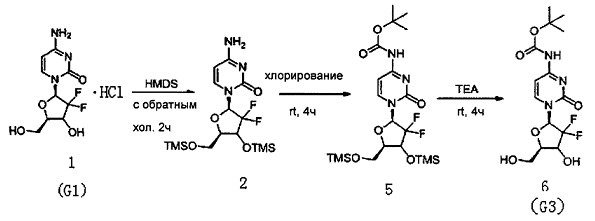
300 мг (1 ммоль) 2'-дезокси-2',2'-дифторцитидина гидрохлорида, 5 мл (0,023 ммоль) HMDS и 5 мг сульфата аммония в каталитическом количестве растворяли в 5 мл 1,4-диоксана и нагревали с обратным холодильником в течение 2 часов. После завершения реакции с обратным холодильником реакционную жидкость концентрировали, добавляли толуол и дважды концентрировали досуха. Полученный продукт после концентрирования растворяли в 10 мл дихлорметана.
0,24 мл (3 ммоль) N-метилимидазола и 416 мг (3 ммоль) ди-трет-бутилдикарбоната добавляли в вышеуказанный раствор дихлорметана для осуществления реакции с перемешиванием при комнатной температуре в течение 4 часов. Химическая структурная формула продукта реакции - 5, и реакционную жидкость концентрировали с получением вязкого маслянистого продукта.
Вязкий маслянистый продукт растворяли в смешанном растворе 3 мл триэтиламина и 20 мл метанола и перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли дистилляцией при пониженном давлении. Для очистки сырого продукта проводили хроматографию на силикагеле и после элюирования дихлорметаном/метанолом (20:1) получали 199 мг G3. Выход трех стадий реакции составил 54%.
ЯМР характеристики G3:
1Н-ЯМР (MeOD-d4, 400 МГц) δ: 8,27 (d, 1Н, J=7,68 Гц, Н6), 7,32 (d, 1Н, J=7,68 Гц, Н5), 6,28 (t, 1Н, J=7,08 Гц, H1'), 4,25 (m, 1Н, Н5а'), 3,93 (m, 2Н, H5b', Н4'), 3,78 (m, 1Н, Н3'), 1,52 (s, 9Н, трет-Bu).
13С-ЯМР (MeOD-d4, 100 МГц) δ: 164,36, 152,17, 144,24, 125,29, 122,71, 120,13, 109,98, 95,78, 82,17, 81,60, 70,45, 69,30, 68,90, 65,57, 55,90, 27,12, 23,57.
(Пример 3)
Производное цитидина в настоящем примере представляет собой 4-N-(карбоксибензил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 8, код: G4), который может быть получен в три стадии реакции. Схема реакции приведена ниже.
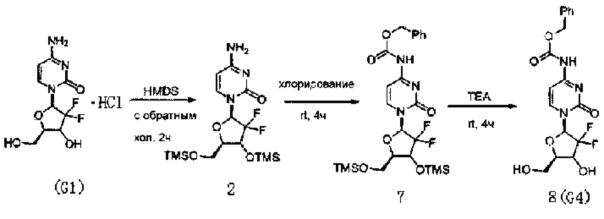
300 мг (1 ммоль) 2'-дезокси-2',2'-дифторцитидина гидрохлорида, 5 мл (0,023 ммоль) HMDS и 5 мг сульфата аммония в каталитическом количестве растворяли в 5 мл 1,4-диоксана и нагревали с обратным холодильником в течение 2 часов. После завершения реакции с обратным холодильником реакционную жидкость концентрировали, добавляли толуол и дважды концентрировали досуха. Полученный продукт после концентрирования растворяли в 10 мл дихлорметана.
0,24 мл (3 ммоль) N-метилимидазола и 340 мг (3 ммоль) карбобензоксихлорида добавляли в вышеуказанный раствор дихлорметана для реагирвоания при перемешивании при комнатной температуре в течение 4 часов. Химическая структурная формула продукта реакции - 7, и реакционную жидкость концентрировали с получением вязкого маслянистого продукта.
Вязкий маслянистый продукт растворяли в смешанном растворе 3 мл триэтиламина и 20 мл метанола и перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли дистилляцией при пониженном давлении. Для очистки сырого продукта проводили хроматографию на силикагеле и после элюирования дихлорметаном/метанолом (20:1) получали 162 мг G4. Выход трех стадий реакции составил 41%.
ЯМР характеристики G4:
1Н-ЯМР (MeOD-d4, 400 МГц) δ: 8,31 (d, 1Н, J=7,64 Гц, Н6), 7,39 (m, 5Н, J=7,68 Гц, Ph), 6,25 (t, 1Н, J=7,12 Гц, Н1'), 5,21 (s, 2H, CH2-Ph), 4,31 (m, 1Н, Н5а'), 3,82 (m, 2Н, H5b, Н4'), 3,79 (m, 1Н, Н3').
13С-ЯМР (MeOD-d4, 100 МГц) δ: 164,22, 156,22, 153,27, 144,48, 135,87, 128,42, 128,10, 125,31, 122,74, 120,16, 95,89, 85,35, 84,91, 81,7, 81,66, 68,87, 67,54, 58,31.
(Пример 4)
Производное цитидина в настоящем примере представляет собой 4-N-(4-нитрокарбоксибензил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 10, код: G5), который может быть получен в три стадии реакции. Схема реакции приведена ниже.

300 мг (1 ммоль) 2'-дезокси-2',2'-дифторцитидина гидрохлорида, 5 мл (0,023 ммоль) HMDS и 5 мг сульфата аммония в каталитическом количестве растворяли в 5 мл 1,4-диоксана и нагревали с обратным холодильником в течение 2 часов. После завершения реакции с обратным холодильником реакционную жидкость концентрировали, добавляли толуол и дважды концентрировали досуха. Полученный продукт после концентрирования растворяли в 10 мл дихлорметана.
0,24 мл (3 ммоль) N-метилимидазола и 430 мг (3 ммоль) 4-нитробензилхлорформиата добавляли в вышеуказанный раствор дихлорметана для реагирования при перемешивании при комнатной температуре в течение 4 часов. Химическая структурная формула продукта реакции - 9, и реакционную жидкость концентрировали с получением вязкого маслянистого продукта.
Вязкий маслянистый продукт растворяли в смешанном растворе 3 мл триэтиламина и 20 мл метанола и перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли дистилляцией при пониженном давлении. Для очистки сырого продукта проводили хроматографию на силикагеле, а после элюирования дихлорметаном/метанолом (20:1) получали 160 мг G5. Выход трех стадий реакции составил 36%.
ЯМР характеристика G5:
1Н-ЯМР (DMSO-d6, 400 МГц) δ: 11,11 (s, 1Н), 8,25 (m, 3H, Ph), 7,68 (d, 2H, J=8,64 Гц, Ph), 7,08 (d, 1H, J=7,44 Гц, H6), 6,29 (d, 1H, J=7,44 Гц, H5), 6,17 (t, 1H, J=7,4 Гц, Н1'), 5,33 (s, 2H, CH2-Ph), 5,29 (t, 1H, J=5,44 Гц), 4,19 (m, 1H, H5a'), 3,66 (m, 1H, H5b'), 3,61 (m, 1H, H4'), 3,29 (m, 1H, H3').
13С-ЯМР (DMSO-d6, 100 МГц) δ: 163,93, 153,47, 147,86, 144,38, 128,97, 126,72, 126,19, 124,26, 123,62, 95,57, 84,82, 83,99, 81,77, 71,91, 69,13, 68,42, 66,06, 62,34, 59,52.
В соответствии с вышеуказанным способом синтеза могут быть синтезированы различные производные с различными замещающими группами, такие как G5-1 и G5-2.
(Пример 5)
Производное цитидина в настоящем примере представляет собой 5-Br-4-N-(н-бутоксикарбонил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 11, код: G6), схема реакции которого приведена ниже.

После того как был получен G2 в соответствии со способом Примера 1, 1 г (2,75 ммоль) G2 растворяли в 150 мл диметилформамида, куда при перемешивании добавляли 500 мг (1,75 ммоль) DBDMH (1,3-дибром-5,5-диметилгидантоин). Полученный бледно-желтый раствор перемешивали при комнатной температуре в течение 1 часа. Согласно ЖХМС, когда реакция была признана завершенной, G2 полностью превратилось в G6. Растворитель сушили на роторном испарителе при пониженном давлении и для концентрирования использовали ацетонитрил. Для очистки сырого продукта проводили хроматографию на силикагеле и получали 263 мг G6 после элюирования дихлорметаном / метанолом (20:1). Общий выход реакции от G1 до G6 составил 51%.
ЯМР характеристика G6:
1Н-ЯМР (MeOD-d4, 400 МГц) δ: 8,62 (s, 1Н, Н5), 6,18 (t, 1Н, J=6,52 Гц, H1'), 4,19 (m, 1Н, Н5а'), 4,15 (m, 2Н, H5b', Н4'), 3,95 (m, 2Н, O-СН2-СН3), 3,17 (m, 1Н, Н3'), 1,36 (m, 2Н, -О-СН2-СН2-СН2-СН3), 1,31 (m, 2Н, -O-СН2-СН2-СН2-СН3), 0,98 (m, 2Н, -О-СН2-СН2-СН2-СН3).
13С-ЯМР (MeOD-d4,100 МГц) δ:143,05, 124,56, 84,59, 81,91, 75,45, 66,17, 58,74, 58,42, 32,96, 30,64, 28,32, 18,84, 12,79, 8,17.
ЭСМС: вычислено для C14H18BrF2N3O6 m/z 442,03 (М+Н)+, найдено 442,02.
(Пример 6)

Производное цитидина в настоящем примере представляет собой 5-Br-4-N-(карбоксибензил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 12, код: G7), схема реакции которого приведена выше (DMF в реакционной смеси представляет собой N,N-диметилформамид и является тем же самым далее).
После того, как G4 был получен согласно способу Примера 3, 1 г (2,75 ммоль) G4 растворяли в 150 мл N,N-диметилформамида, куда при перемешивании добавляли 500 мг (1,75 ммоль) DBDMH. Полученный бледно-желтый раствор перемешивали для реагирования при комнатной температуре в течение 1 часа. Согласно ЖХМС, когда реакция была признана завершенной, G4 полностью превратилось в G7. Растворитель сушили на роторном испарителе при пониженном давлении и для концентрирования использовали ацетонитрил. Для очистки сырого продукта проводили хроматографию на силикагеле и получали 283 мг G7 после элюирования дихлорметаном/метанолом (20:1). Общий выход реакции от G1 до G7 составил 32%.
ЯМР характеристика G7:
1Н-ЯМР (MeOD-d4,400 МГц) δ: 8,60 (s, 1Н, Н5), 7,45 (m, 2Н, Ph), 7,36 (m, 3H, Ph), 6,18 (t, 1Н, J=6,52 Гц, Н1'), 5,24 (s, 2Н, CH2-Ph), 4,33 (m, 1Н, Н5а'), 4,0 (m, 2Н, Н5b', Н4'), 3,81 (m, 1Н, Н3').
13С-ЯМР (MeOD-d4, 100 МГц) δ: 143,23, 135,93, 128,37, 128,27, 125,22, 122,63, 120,05, 85,41, 85,08, 84,76, 81,85, 68,77, 68,53, 68,31, 67,93, 58,76.
ЭСМС: вычислено для C17H16BrF2N3O6 m/z 476,02 (М+Н)+, найдено 477,09,
Аналогично, после того, как аминогруппа замещается согласно способу синтеза Примера 1, позиция 5 может быть замещена Br согласно способу Примера 6 с получением G7-1 и G7-2.
В соответствии с вышеуказанным способом получения могут быть получены производные с другими группами на R2.
(Пример 7)
Производное цитидина в настоящем примере представляет собой 5'-O-[3,5-динитросалицилат]-4-N-(н-бутоксикарбонил)-2'-дезокси-2',2'-дифторцитидин (структурная формула: 14, код: G8).
Схема реакции приведена ниже (в схеме реакции (Вос)2O представляет собой ди-трет-бутилдикарбонат, диоксан представляет собой 1,4-диоксан, DMAP представляет 4-диметиламинопиридин, EDCL представляет собой 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, DCM представляет собой дихлорметан, TFA представляет собой трифторуксусную кислоту, и аббревиатуры далее аналогичны):
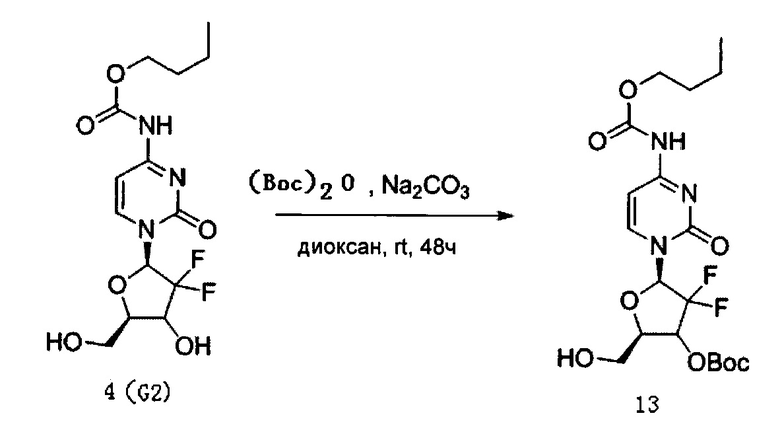

Сначала было получено Соединение 13. 60 мг (0,16 ммоль) G2, полученного в Примере 1, и 106 мг (1 ммоль) карбоната натрия смешивали и добавляли к смешанному раствору 5 мл 1,4-диоксана и воды (4:1 по объему). В раствор добавляли 44 мг (0,2 ммоль) (Вос)2О, перемешивали при 24°C для осуществления реакции. ТСХ проводили в процессе реакции, чтобы определить, полностью ли прореагировал G2. В систему добавляли 2 мл воды для разбавления после завершения реакции и затем дважды экстрагировали этилацетатом, каждый раз по 30 мл. Органическую фазу, полученную экстракцией, последовательно промывали 5 мл воды и 5 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия после завершения промывки и затем концентрировали досуха при пониженном давлении. После концентрирования проводили хроматографию на силикагеле для очистки и элюировали смесью дихлорметан/ацетон/метанол (1:1:0,02) с получением 51 мг Соединения 13. Выход указанной выше реакции составил 76%.
51 мг (0,11 ммоль) полученного Соединения 13, 98 мг (0,42 ммоль) 3,5-динитросалициловой кислоты и 60 мг (0,31 ммоль) EDCL смешивали и добавляли в 15 мл дихлорметана. 2 мг DMAP добавляли в дихлорметан для реагирования при перемешивании при 24°C в течение 24 часов. В ходе реакции проводили тонкослойную хроматографию (ТСХ), чтобы определить, полностью ли прореагировало Соединение 13.
После завершения реакции в вещества, полученные в результате реакции, добавляли 50 мл дихлорметана, а затем для их промывки последовательно добавляли 10 мл воды и 20 мл насыщенного солевого раствора. После завершения промывки для сушки использовали безводный сульфат натрия, концентрировали досуха, в концентрированные вещества добавляли 5 мл трифторуксусной кислоты (TFA) и перемешивали при комнатной температуре в течение 12 часов, а затем концентрировали досуха. После концентрирования для очистки проводили хроматографию на силикагеле, элюировали смесью дихлорметан/метанол (20:1) с получением 18 мг G8. Выход реакции с получением Соединения 14 из Соединения 13 составил 28%.
Путем замещения 3,5-динитросалициловой кислоты другими кислотами может быть получено такое соединение с R3  , где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо или конденсированное гетероциклическое кольцо, а также замещенный фенил, замещенное гетероциклическое кольцо или замещенное конденсированное гетероциклическое кольцо, замещенное независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин и пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин и индол; Х2 представляет собой соединение с С1-С3-(СН2)n- или С0-С3 -O-(СН2)n-.
, где Х3 представляет собой бензольное кольцо, гетероциклическое кольцо или конденсированное гетероциклическое кольцо, а также замещенный фенил, замещенное гетероциклическое кольцо или замещенное конденсированное гетероциклическое кольцо, замещенное независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; указанное гетероциклическое кольцо представляет собой имидазол, пиридин, фуран, тиофен, тиазол, пиримидин, пиперазин и пиперидин; указанное конденсированное гетероциклическое кольцо представляет собой хинолин и индол; Х2 представляет собой соединение с С1-С3-(СН2)n- или С0-С3 -O-(СН2)n-.
(Пример 8)
Производное цитидина в настоящем примере представляет собой 5'-O-[2-(4-нитpo-1H-имидaзoл)aцeтaт]-4-N-(трет-бyтoкcикapбoнил)-2'-дeзoкcи-2',2'-дифторцитидин (структурная формула: 16, код: G9).
Схема реакции указана ниже:


Сначала получали Соединение 15. 60 мг (0,16 ммоль) Соединения 6 (G3), полученного в Примере 2, и 106 мг (1 ммоль) карбоната натрия смешивали и добавляли к смешанному раствору 5 мл 1,4-диоксана и воды (4:1 по объему). 44 мг (0,2 ммоль) (Вос)2O добавляли в раствор, перемешивая при 24°C для осуществления реакции. ТСХ проводили во время реакции, чтобы определить, полностью ли прореагировал G2. 2 мл воды добавляли в систему для разбавления после завершения реакции и затем дважды экстрагировали этилацетатом, каждый раз по 30 мл. Органическую фазу, полученную экстракцией, последовательно промывали 5 мл воды и 5 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия после завершения промывки и затем концентрировали досуха при пониженном давлении. После концентрирования проводили хроматографию на силикагеле для очистки и элюировали смесью дихлорметан/ацетон/этанол (1:1:0,02) с получением 48 мг Соединения 15. Выход указанной выше реакции составил 64%.
51 мг (0,11 ммоль) полученного Соединения 15, 98 мг (0,57 ммоль) 2-(4-нитро-1Н-имидазол)уксусной кислоты и 60 мг (0,31 ммоль) EDCL смешивали и добавляли в 15 мл дихлорметана. 2 мг DMAP добавляли в дихлорметан для осуществления реакции при перемешивании при 24°C в течение 24 часов. ТСХ проводили во время реакции, чтобы определить, полностью ли прореагировало Соединение 15.
После завершения реакции в вещества, полученные в результате реакции, добавляли 50 мл дихлорметана, а затем для их промывки последовательно добавляли 10 мл воды и 20 мл насыщенного солевого раствора. После завершения промывки для сушки использовали безводный сульфат натрия, концентрировали досуха, в концентрированный материал добавляли 5 мл трифторуксусной кислоты (TFA) и перемешивали при комнатной температуре в течение 12 часов, а затем концентрировали досуха. После концентрирования для очистки проводили хроматографию на силикагеле, элюировали смесью дихлорметан/метанол (20:1) с получением 18 мг G9. Выход реакции с получением Соединения 16 из Соединения 15 составил 31%.
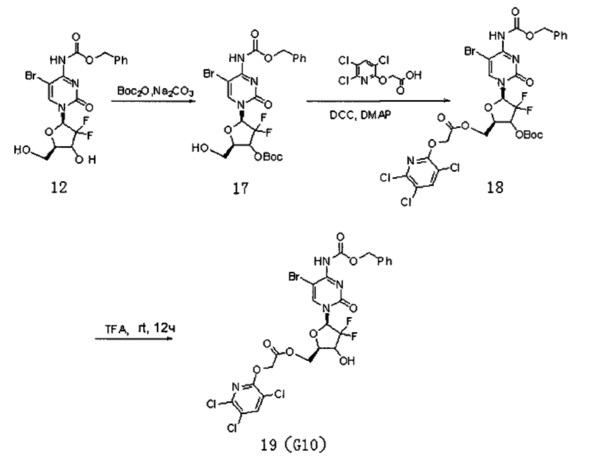
(Пример 9)
Производное цитидина в настоящем примере обозначено как G10, схема реакции является следующей (где DCC представляет собой N,N'-дициклогексилкарбодиимид):
Сначала получали Соединение 22. Схема реакции приведена ниже (где DMF представляет собой N,N-диметилформамид):

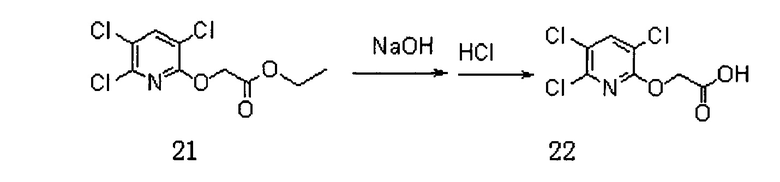
В реакционную колбу добавляли Соединение 20, а именно 3,5,6-трихлор-2-пиридол (5 г, 25,2 ммоль), карбонат калия (7 г, 50,6 ммоль) и DMF (30 мл) и перемешивали в течение 15 мин. После охлаждения до 0°C добавляли капли этилбромацетата (2,78 мл, 25,2 ммоль) для реагирования при комнатной температуре в течение 4 часов. Добавляли воду (15 мл) и экстрагировали дихлорметаном (15 мл×3). Органическую фазу сушили на роторном испарителе и полученное Соединение 21 использовали непосредственно на следующей стадии реакции.
Соединение 21 (5,4 г, 18,8 ммоль) добавляли в 54 мл воды, в которую добавляли гидроксид натрия (0,864 г, 21,6 ммоль), и перемешивали при 80°C в течение 4 часов. Концентрированную соляную кислоту добавляли по каплям для доведения рН до 1. После фильтрования получали 3,3 г (70%) Соединения 22.
Соединение 12, а именно 5-Br-4-N-(бензилоксикарбонил)-2'-дезокси-2',2'-дифторцитидин (2 г, 4,21 ммоль), и карбонат натрия (3,3 г, 31,1 ммоль) смешивали и добавляли в систему 1,4-диоксана и воды (4:1 по объему, 200 мл). Добавляли (Вос)2О (1,8 г, 8,25 ммоль, ди-трет-бутилдикарбонат) и перемешивали при 25°C в течение 48 часов. ТСХ проводили во время реакции. После завершения реакции добавляли 20 мл воды для разбавления, 2×100 мл этилацетата использовали для экстракции два раза. Органическую фазу промывали 50 мл воды и 50 мл насыщенного солевого раствора. Безводный сульфат натрия использовали для сушки и проводили колоночную хроматографию (дихлорметан / ацетон / метанол, 1:1:0,02) для получения Соединения 17 с выходом 690 мг и 29%.
ЭСМС: вычислено для С22H24BrF2N3О8 m/z 576,07 (М+Н)+, найдено 576,16.
Соединение 17 (350 мг, 0,61 ммоль), Соединение 22 (186 мг, 0,73 ммоль) и DCC (250 мг, 1,21 ммоль, дициклогексилкарбодиимид, N,N-дициклогексилкарбодиимид) смешивали и добавляли к 7 мл дихлорметана, куда затем добавляли DMAP (15 мг, 0,12 ммоль, 4-диметиламинопиридин). Реакционную смесь перемешивали в течение ночи при комнатной температуре, проводили ТСХ. После завершения реакции добавляли 5 мл воды для разбавления, для экстракции использовали 2×20 мл дихлорметана. Органическую фазу промывали 50 мл воды и 50 мл насыщенного раствора соли. Для сушки использовали безводный сульфат натрия и проводили колоночную хроматографию (дихлорметан/метанол, 45:1) с получением промежуточного соединения 18 (260 мг, выход: 70%). Промежуточное соединение непосредственно обрабатывали TFA (трифторуксусной кислотой) в дихлорметане с получением продукта G10 (175 мг, выход: 77%).
ЯМР характеристика G10:
1Н-ЯМР (CDCl3, 400 МГц) δ: 7,81 (s, 1Н), 7,75 (s, 1Н), 7,35 (m, 5H), 6,24 (m, 1Н), 5,27 (s, 2H), 5,22 (s, 2H), 5,03 (m, 2H), 4,67 (m, 1H), 4,47 (m, 1H), 4,30 (s, 2H).
13C ЯМР (CDCl3, 100 МГц) δ: 167,80, 162,56, 157,21, 155,57, 143,28, 140,91, 128,90, 128,78, 124,55, 123,44, 118,57,117,47, 72,61, 68,68, 64,94,63,83, 63,37,62,84.
ЭСМС: вычислено для C24H18BrCl3F2N4O8 m/z 712,93 (M+H)+, найдено 712,99.
(Пример 10)
Производное цитидина в настоящем примере обозначено как G11.
Сначала получали Соединение 24. Схема реакции является следующей:
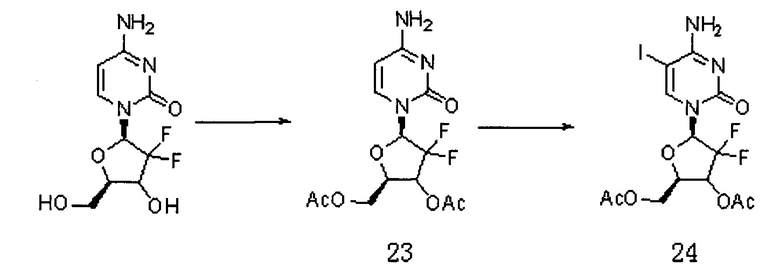
2'-дезокси-2',2'-дифторцитидин (5 г, 16 ммоль) растворяли в уксусной кислоте (56 мл), перемешивали при 35°C в течение 2 часов; добавляли трихлорметан (17 мл) и перемешивали при 0°C в течение 15 мин. Хлороформ/ацетилхлорид (28 мл/33 мл) по каплям добавляли в смешанный раствор, перемешивали при 50°C в течение 24 часов. Растворитель сушили на роторном испарителе. Для выпаривания на роторном испарителе добавляли метанол (35 мл) для получения продукта 23, который использовали непосредственно на следующей стадии.
Простое вещество йод (2,8 г, 11 ммоль), йодистую кислоту (0,83 г, 4,7 ммоль), уксусную кислоту (37,5 мл), четыреххлористый углерод (25,5 мл), воду (25,5 мл) и Соединение 23 добавляли в реакционную колбу, перемешивали при 40°C в течение 24 часов. Растворитель сушили на роторном испарителе. Добавляли дихлорметан и воду. pH доводили до 6-7 и органическую фазу промывали тиосульфатом натрия и водой. Органические фазы объединяли и сушили с безводным сульфатом натрия. Фильтрат после фильтрации сушили на роторном испарителе. Получали 5 г продукта Соединения 24 (выход двух стадий составил 55%).
ЕСМС: вычислено для C13H14F2N3O6 m/z 473,99 (М+Н)+, найдено 474,14.
Схема реакции для получения Соединения G11 такая, как указано ниже:
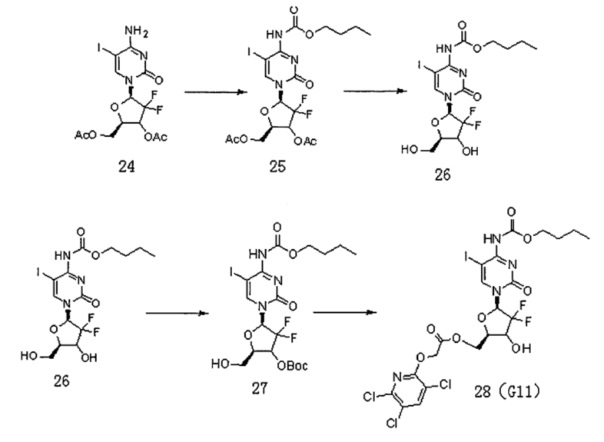
Соединение 24 (2,5 г, 5,3 ммоль) и пиридин (1,24 г, 15,8 ммоль) растворяли в дихлорметане (35 мл), куда по каплям добавляли бутилхлорформиат (2,15 г, 15,8 ммоль) при 0°C и подвергали реакции при 10°C в течение ночи. Растворитель сушили на роторном испарителе. Колоночную хроматографию (дихлорметан / метанол, 60:1) проводили с получением 1,9 г (выход: 63%) Соединения 25.
ЕСМС: вычислено для C18H22F2N3O8 m/z 574,04 (М+Н)+, найдено 574,14.
Соединение 25 (2,5 г, 4,3 ммоль) растворяли в метаноле (40 мл) и перемешивали в течение 5 мин, куда добавляли карбонат калия (2,1 г, 15,2 ммоль) и перемешивали при комнатной температуре в течение ночи. Фильтрат после фильтрации сушили на роторном испарителе и получали 1,5 г (выход 70,4%) Соединения 26.
ЕСМС: вычислено для C14H18F2N3O6 m/z 490,02 (М+Н)+, найдено 490,07.
Соединение 26 (3,9 г, 8 ммоль) и карбонат натрия (4,24 г, 40 ммоль) растворяли в смеси 1,4-диоксана (22,5 мл) и воды (4,5 мл) и перемешивали в течение 10 мин, куда добавляли ди-трет-бутилдикарбонат (2,1 г, 9,6 ммоль), реакция проходила при комнатной температуре в течение по меньшей мере 24 часов. Растворитель сушили на роторном испарителе. Добавляли дихлорметан (70 мл) и воду (100 мл) и для экстракции использовали дихлорметан (70 мл×3). Органическую фазу сушили на роторном испарителе. Проводили колоночную хроматографию (дихлорметан : метанол, 80:1) с получением 2,8 г (выход: 60%) Соединения.27.
ЕСМС: вычислено для C19H26F2N3O8 m/z 590,07 (М+Н)+, найдено 590,02.
Соединение 27 (50 мг, 0,61 ммоль), Соединение 22 (32,4 мг, 0,13 ммоль) и DCC (35 мг, 0,17 ммоль) смешивали и добавляли к 2 мл дихлорметана, куда затем добавляли DMAP (2 мг, 0,016 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Проводили ТСХ. После завершения реакции добавляли 5 мл воды для разбавления, 2×20 мл дихлорметана использовали для экстракции дважды. Органическую фазу промывали 5 мл воды и 5 мл насыщенного раствора соли. Безводный сульфат натрия использовали для сушки и проводили колоночную хроматографию (дихлорметан / метанол, 150:1) с получением промежуточного соединения (50 мг, выход: 71%). Промежуточное соединение непосредственно обрабатывали TFA в дихлорметане с получением G11 (35 мг, выход: 80%).
Характеристики G11 такие как указано ниже:
ЭСМС: вычислено для C21H20C13F2IN4O8 m/z 726,94 (М+Н)+, найдено 727,12.
1Н-ЯМР (MeOD-d4, 400 МГц) δ: 8,06 (s, 1H), 5,49 (s, 1H), 5,07 (m, 3H), 4,51 (s, 1H), 4,48 (m, 1H), 4,21 (s, 5H), 1,69 (m, 3H), 1,44 (m, 3H), 0,97 (m, 3H).
13С-ЯМР (MeOD-d4, 100 МГц) δ: 168,06, 141,08, 140,91, 122,67, 81,73, 66,15, 63,85, 63,54, 59,38, 58,64, 33,27, 30,68, 19,73, 18,89, 15,75, 12,84, 8,78.
(Пример 11)
Производное цитидина в настоящем примере обозначено как G12, схема реакции такая, как указана ниже:

Соединение 27 (300 мг, 0,51 ммоль), 2-(4-нитро-1Н-имидазол)уксусную кислоту (105 мг, 0,61 ммоль) и DCC (210 мг, 1,02 ммоль) смешивали и добавляли к 10 мл дихлорметана, а затем добавляли DMAP (9 мг, 0,073 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, проводили ТСХ. После завершения реакции добавляли 20 мл воды для разбавления, 3×30 мл дихлорметана использовали для экстракции трижды. Органическую фазу промывали 20 мл воды и 20 мл насыщенного раствора соли. Безводный сульфат натрия использовали для сушки и проводили колоночную хроматографию (дихлорметан / метанол, 66:1) с получением 200 мг промежуточного соединения (53%). Промежуточное соединение непосредственно обрабатывали TFA в дихлорметане с получением 70 мг G12 (40%).
(Пример 12)
Производное цитидина в настоящем примере обозначено как G13,
Сначала получали Соединение 35.

В предварительно высушенную реакционную колбу добавляли Соединение 24 (5 г, 0,01 моль), (Ph3)PdCl2 (1,5 г, 2,14 ммоль) и CuI (1,0 г, 5,27 ммоль), куда в атмосфере азота добавляли сухой THF (100 мл), Ме3SiC≡СН (5,2 г, 0,053 моль) и TEA (15 мл), и оставляли для реагирования при комнатной температуре в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (этилацетат/петролейный эфир, 1:1) с получением 2,67 г (выход: 57%) Соединения 30.
Соединение 30 (10 г, 1,1 ммоль) добавляли в МеОН (200 мл), перемешивали до полного растворения и охлаждали при 0°C в течение 15 минут, добавляли K2СО3 (10,92 г, 3,95 ммоль) и перемешивали при 0°C в течение 2 часов. После завершения реакции карбонат калия сначала удаляли фильтрованием, растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 10:1) с получением 5,46 г (выход: 84,27%) Соединения 31.
Соединение 31 (5,46 г, 0,019 моль), CuSO4, 5H2O (475 мг, 1,9 ммоль) и Vc.Na (1,13 г, 5,7 ммоль) добавляли в смесь THF (55 мл) и воды (49 мл) и перемешивали в атмосфере азота при комнатной температуре в течение 10 мин. Затем добавляли  перемешивали в атмосфере азота при комнатной температуре в течение 36 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 15:1) с получением 4,88 г (выход: 61,0%) Соединения 32.
перемешивали в атмосфере азота при комнатной температуре в течение 36 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 15:1) с получением 4,88 г (выход: 61,0%) Соединения 32.
Соединение 32: ЕСМС: вычислено для C18H18F2N6O4 m/z 421,18 (М+Н)+, найдено 421,36.

В приведенной выше формуле реакции 1,4-диоксан представляет собой 1,4-диоксан, HMDS представляет собой гексаметилдисилазан.
Соединение 32 (2,0 г, 4,76 ммоль), HMDS (20 мл) и (NH4)2SO4 (50,3 мг, 0,38 ммоль) добавляли в 1,4-диоксан (20 мл), перемешивали при кипячении с обратным холодильником при 80°C в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Растворитель сушили на роторном испарителе с толуолом (5 мл×2) и затем отсасывали с помощью масляного насоса. Добавляли дихлорметан (40 мл) и пиридин (1,13 г, 14,3 ммоль) и смесь охлаждали до 0°C, затем медленно добавляли нитробензоат (3,08 г, 14,3 ммоль) и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Добавляли метанол (40 мл) и охлаждали при 0°C в течение 15 минут. Затем по каплям добавляли триэтиламин (6,7 мл) и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 40:1) с получением 780 мг (выход трех стадий: 27,3%) Соединения 33.
Соединение 33:
ЕСМС: вычислено для C26H23F2N7O8 m/z 600,22 (М+Н)+, найдено 600,25.

Соединение 33 (780 мг, 1,3 ммоль) и Na2СО3 (690 мг, 6,5 ммоль) добавляли в смесь 1,4-диоксана (20 мл) и воды (5 мл), перемешивали для растворения. (Вос)2О (340,6 мг, 1,56 ммоль) добавляли при комнатной температуре и перемешивали в течение ночи при комнатной температуре. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 80:1) с получением 366 мг (выход: 40,2%) Соединения 34.
Соединение 34:
ЕСМС: вычислено для С31Н31F2N7О10 m/z 700,28 (М+Н)+, найдено 700,08.

Соединение 34 (250 мг, 0,36 ммоль), Соединение 22 (109 мг, 0,43 ммоль), DCC (148 мг, 0,72 ммоль) и DMAP (5 мг) добавляли к 12 мл дихлорметана и перемешивали в течение ночи при комнатной температуре. После завершения реакции DCC удаляли фильтрованием под вакуумом. Для очистки проводили хроматографию на силикагеле (дихлорметан / метанол, 100:1) с получением 210 мг (выход: 60,0%) Соединения 35.
Соединение 35:
ЕСМС: вычислено для C38H33C13F2N8O12 m/z 937,28 (М+Н)+, найдено 937,28.
Соединение 35 (210 мг, 0,224 ммоль) добавляли в 10 мл системы DCM/TFA 5:1 и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 60:1) с получением 140 мг (выход: 75,0%) G13.
ЕСМС: вычислено для C33H25C13F2N8O10 m/z 837,17 (М+Н)+, найдено 837,27.
1Н-ЯМР (CDCl3, 400 МГц) δ: 8,34 (s, 1H), 8,23 (m, 3H), 7,57 (m, 3H), 7,33 (m, 3H), 7,23 (m, 2H), 6,38 (m, 1H), 5,49 (m, 3H), 5, 30 (s, 3H), 5,13 (m, 2H), 4,73 (m, 1H), 4,33 (m, 3H).

Путем замены  тринитридным соединением, которое коммерчески доступно или может быть легко приготовлено,
тринитридным соединением, которое коммерчески доступно или может быть легко приготовлено,
может быть получено соединение, имеющее такое R2 как 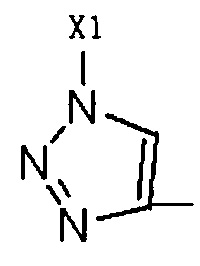 , где Х1 представляет собой C1-C10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкокси замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в -(CH2)n-Ph и замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; и углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
, где Х1 представляет собой C1-C10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-С6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкокси замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в -(CH2)n-Ph и замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; и углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
(Пример 13)
Производное цитидина в настоящем примере обозначено как G14. Сначала было получено Соединение 39.

К 1,4-диоксану (15 мл) добавляли Соединение 32 (1,5 г, 3,57 ммоль), HMDS (15 мл) и (NH4)2SO4 (37,7 мм, 0,285 ммоль) и перемешивали при кипячении с обратным холодильником при 80°C в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Растворитель сушили на роторном испарителе с толуолом (5 мл×2) и затем отсасывали с помощью масляного насоса. Добавляли дихлорметан (30 мл) и пиридин (0,846 г, 0,01 ммоль) и смесь охлаждали до 0°C, куда медленно добавляли карбобензокси хлорид (1,83 г, 0,01 ммоль) и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Добавляли 40 мл метанола и охлаждали при 0°C в течение 15 минут. Затем по каплям добавляли 6 мл триэтиламина и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 40:1) с получением 570 мг (выход: 28,8%) Соединения 37.
ЕСМС: вычислено для C25H24F2N6O6 m/z 555,24 (М+Н)+, найдено 555,04.
Соединение 37 (520 мг, 0,939 ммоль) и Na2CO3 (497,5 мг, 4,69 ммоль) добавляли в смесь из 12 мл 1,4-диоксана и 3 мл воды и перемешивали для растворения. (Вос)2О (245,5 мг, 1,126 ммоль) добавляли при комнатной температуре и перемешивали в течение ночи при комнатной температуре. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 80:1) с получением 278 мг (выход: 45,3%) Соединения 38.
ЕСМС: вычислено для C31H32F2N6O8 m/z 655,28(М+Н)+, найдено 655,08.

Соединение 38 (200 мг, 0,3 ммоль), Соединение 22 (93,6 мг, 0,367 ммоль), DCC (126 мг, 0,612 ммоль) и DMAP (10 мг) добавляли в 10 мл дихлорметана и перемешивали в течение ночи при комнатной температуре. После завершения реакции DCC удаляли фильтрованием под вакуумом. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 125:1) с получением 170 мг (выход: 62,5%) Соединения 39.
ЕСМС: вычислено для C38H34C13F2N7O10 m/z 914,14 (M+Na)+, найдено 914,36.

Соединение 39 (140 мл, 0,157 ммоль) добавляли в 10 мл системы DCM / TFA 5:1 и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан / метанол, 50:1) с получением 95 мг (выход: 76,43%) G14.
ЕСМС: вычислено для С33Н26С13F2N7O8 m/z 814,26 (M+Na)+, найдено 814,61.
1Н-ЯМР (CDCl3, 400 МГц) δ: 8,50 (s, 1H), 8,36 (s, 1H), 7,63 (s, 1H), 7,33 (m, 9Н), 7,23 (m, 2H), 6,38 (m, 1H), 5,49 (m, 2H), 5,30 (s, 4H), 5,13 (m, 1H), 4,80 (m, 1H), 4,50 (m, 2H), 4,30 (m, 1H).
13С-ЯМР (CDCl3, 100 МГц) δ: 168,61 157,65 155,68 146,20 143,22 140,59 128,87 128,02 123,07 117,24 68,19 64,11 54,40 12,69.

Путем замены  тринитридным соединением, которое коммерчески доступно или легко получаемо, может быть получено соединение, имеющее такое R2 как
тринитридным соединением, которое коммерчески доступно или легко получаемо, может быть получено соединение, имеющее такое R2 как  , где Х1 представляет C1-С10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-C6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкоксиде замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в -(CH2)n-Рh и замещенном -(CH2)n-Рh n равно 0, 1, 2, 3 до 10; а углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
, где Х1 представляет C1-С10 алкил, C1-С10 замещенный алкил, C1-С10 алкокси, C1-С10 замещенный алкокси, C1-C6 алкилсульфонил, C1-С6 алкилтиол, -(CH2)n-Ph или замещенный -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкоксиде замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами; в -(CH2)n-Рh и замещенном -(CH2)n-Рh n равно 0, 1, 2, 3 до 10; а углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
(Пример 14)
Производное цитидина в настоящем примере обозначено как G15.
Сначала было получено Соединение 43.

К 1,4-диоксану (13 мл) добавляли Соединение 32 (1,3 г, 3,0 ммоль), HMDS (13 мл) и (NH4)2SO4 (32,7 мм, 0,247 ммоль) и перемешивали при кипячении с обратным холодильником при 80°C в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Растворитель сушили на роторном испарителе с толуолом (5 мл×2) и затем отсасывали с помощью масляного насоса. Добавляли дихлорметан (26 мл) и пиридин (0,733 г, 9,3 ммоль) и смесь охлаждали до 0°C, куда медленно добавляли бутилхлоркарбонат (1,268 г, 9,3 ммоль) и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Добавляли метанол (30 мл) и охлаждали при 0°C в течение 15 минут, а затем добавляли по каплям триэтиламин (4,5 мл) и перемешивали при 10°C в течение ночи. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол: 40:1) с получением 850 мг (выход: 52,8%) Соединения 41.
Соединение 41 (800 мг, 1538 ммоль) и Na2CO3 (815 мг, 7,69 ммоль) добавляли в смесь из 25 мл 1,4-диоксана и 6 мл воды, перемешивая для растворения. (Вос)2О (403 мг, 1,85 ммоль) добавляли при комнатной температуре и перемешивали в течение ночи при комнатной температуре. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 80:1), получая 458 мг (выход: 48,1%) Соединения 42.
ЕСМС: вычислено для C28H34F2N6O8 m/z 637,76 (M+NH4)+, найдено 637,46.

К 20 мл дихлорметана добавляли Соединение 42 (380 мг, 0,613 ммоль), Соединение 22 (187,5 мг, 0,736 ммоль), DCC (252,5 мг, 1226 ммоль) и DMAP (20 мг) и перемешивали в течение ночи при комнатной температуре. После завершения реакции DCC удаляли фильтрованием под вакуумом. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол, 125:1) с получением 333 мг (выход: 63,5%) Соединения 43.
ЕСМС: вычислено для C35H36C13F2N7O10 m/z 858,18 (М+Н)+, найдено 858,28.
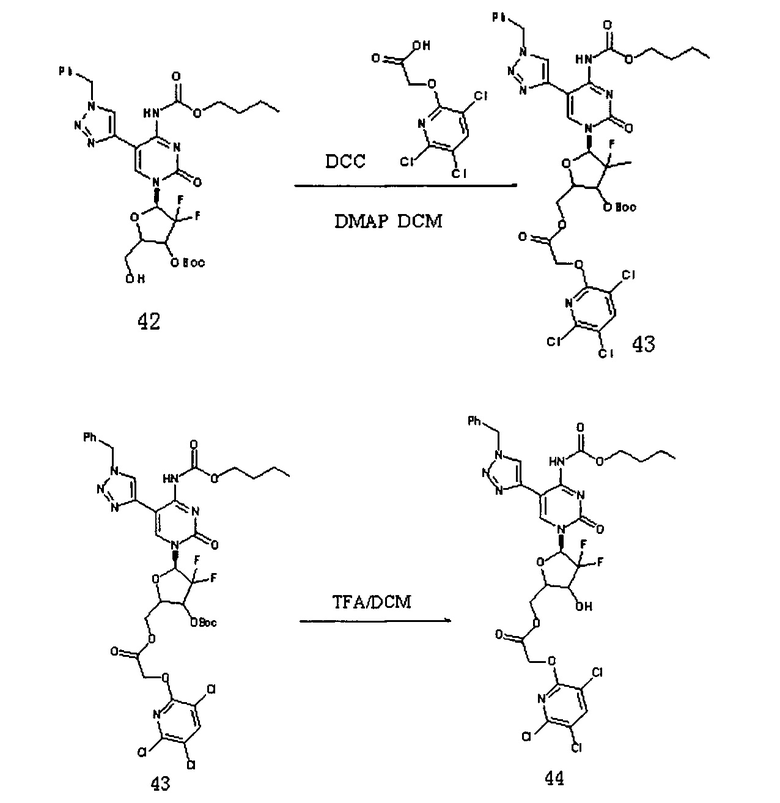
Соединение 43 (310 мг, 0,362 ммоль) добавляли в 12 мл системы DCM / TFA 5:1 и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции растворитель удаляли дистилляцией при пониженном давлении. Для очистки проводили хроматографию на силикагеле (дихлорметан/метанол: 50:1) с получением 235 мг (выход: 85,8%) G15.
ЕСМС: вычислено для C30H28C13F2N7O8 m/z 758,18 (М+Н)+, найдено 758,18.
1НЯМР (CDCl3, 400 МГц) δ: 8,43 (s, 1H), 8,33 (s, 1H), 8,11 (m, 1H), 7,70 (m, 1H), 7,63 (m, 1H), 7,34 (m, 4H), 6,34 (m, 1H), 5,53 (m, 3H), 5,21 (m, 2H), 4,76 (m, 1H), 4,54 (m, 1H), 4,46 (m, 1H), 4,43 (m, 1H), 4,20 (m, 2H), 1,70 (m, 2H), 1,41 (m, 2H), 0,95 (m, 3H).
Путем замещения  тринитридным соединением, которое доступно коммерчески или которое легко получить, может быть получено соединение, имеющее такое R2 как
тринитридным соединением, которое доступно коммерчески или которое легко получить, может быть получено соединение, имеющее такое R2 как 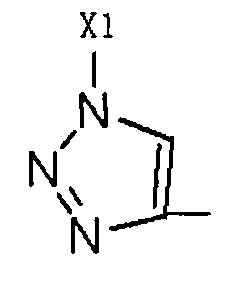 , где Х1 является C1-С10 алкилом, C1-С10 замещенным алкилом, C1-С10 алкокоси, C1-С10 замещенным алкокси, C1-С6 алкилсульфонилом, C1-С6 алкилтиолом, -(CH2)n-Ph или замещенным -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкоксиде замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, в -(CH2)n-Ph и замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; и углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
, где Х1 является C1-С10 алкилом, C1-С10 замещенным алкилом, C1-С10 алкокоси, C1-С10 замещенным алкокси, C1-С6 алкилсульфонилом, C1-С6 алкилтиолом, -(CH2)n-Ph или замещенным -(CH2)n-Ph; где углеродная цепь на замещенном алкиле и замещенном алкоксиде замещена независимо одним, или двумя, или тремя атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами, в -(CH2)n-Ph и замещенном -(CH2)n-Ph n равно 0, 1, 2, 3 до 10; и углеродная цепь или бензольное кольцо в замещенном -(CH2)n-Ph замещены одним, или двумя, или тремя Н, атомами галогена, циано-, нитро-, амино-, гидроксилами или карбоксилами.
(Пример 15)
Производное цитидина в настоящем примере обозначено как G16.

Соединение 11 (2 г, 4,52 ммоль) и карбонат натрия (3,4 г) растворяли в смеси 133 мл 1,4-диоксана и 34 мл воды и перемешивали в течение 10 мин. Добавляли (Вос)2О (1,47 г, 6,78 ммоль) и оставляли взаимодействовать при комнатной температуре в течение по меньшей мере 48 часов. Растворитель сушили на роторном испарителе. Добавляли дихлорметан (70 мл) и воду (100 мл) и для экстракции использовали дихлорметан (70 мл×3). Органическую фазу сушили на роторном испарителе. Колоночную хроматографию (дихлорметан:метанол, 80:1) проводили с получением 1,2 г (48,9%) Соединения 45.
ЕСМС: вычислено для C19H26BrF2N3O8 m/z 542,09 (М+Н)+, найдено 542,11

Соединение 45 (355 мг, 0,65 ммоль), защищенное Воc, Соединение 22 (499 мг, 1,95 ммоль) и DCC (401 мг, 1,95 ммоль) смешивали и добавляли в 45 мл дихлорметана, куда затем добавляли DMAP (2 мг, 0,016 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Проводили ТСХ. После завершения реакции добавляли 5 мл воды для разбавления, для экстракции использовали 2×20 мл дихлорметана. Органическую фазу промывали 5 мл воды и 5 мл насыщенного раствора соли. Для сушки использовали безводный сульфат натрия, а затем TFA использовали для непосредственного получения целевого соединения G16 (110 мг, выход двух стадий: 24%).
1Н-ЯМР (MeOD-d4, 400 МГц) δ: 8,05 (s, 2Н, Ar), 6,22 (t, 1Н, J=7,6 Гц, H1'), 5,15 (m, 2Н), 4,67 (m, 1Н, Н5а'), 4,48 (m, 4Н, H5b', Н4', O-СН2-СН3), 1,70 (m, 2Н, -О-СН2-СН2-СН2-СН3), 1,28 (m, 2Н, -О-СН2-СН2-СН2-СН3), 0,97 (m, 2Н, -О-СН2-СН2-СН2-СН3).
13С-ЯМР (MeOD-d4, 100 МГц) δ: 168,12, 155,98, 142,88, 141,03, 124,15, 122,75, 117,32, 82,73, 78,37, 77,97, 63,76, 63,55, 62,67, 41,82, 31,04, 30,63, 29,98, 18,88, 18,84, 12,81, 8,29.
ЕСМС: вычислено для C21H20BrCl3F2N4O8 m/z 681,94 (М+2Н)+, найдено 681,15.
(Пример 16)
Производное цитидина в настоящем примере обозначено как G17.

Соединение 10 (911 мг, 1,75 ммоль) растворяли в 50 мл диметилформамида, куда добавляли дибромгидантоин (500 мг, 1,75 ммоль) для получения слабого желтого раствора и перемешивали при комнатной температуре в течение 1 часа. ЖХМС проводили до завершения реакции. Растворитель сушили при пониженном давлении и проводили колоночную хроматографию (дихлорметан / метанол, 20:1) с получением Соединения 47 (483 мг, общий выход: 52,9%, белое твердое вещество).
1Н-ЯМР (CDCl3, 400 МГц) δ: 8,40 (s, 1Н, Ar), 8,23 (d, J=4 Гц, 2Н, Ar), 7,60 (d, J=4 Гц, 2Н, Ar), 6,15 (m, 1Н, H1'), 5,30 (s, 2Н, Ar-СН2), 4,50 (m, 1Н, Н5а'), 4,48 (m, 4Н, Н5b', Н4', O-СН2-СН3).
Соединение 47 (2,33 г, 5,72 ммоль) и карбонат натрия (4,12 г) растворяли в смеси 133 мл 1,4-диоксана и 34 мл воды и перемешивали в течение 10 мин, куда добавляли ди-трет-бутилдикарбонат (1,72 г, 7,9 ммоль) и оставляли взаимодействовать при комнатной температуре в течение по меньшей мере 48 часов. Растворитель сушили на роторном испарителе. Добавляли дихлорметан (70 мл) и воду (100 мл) и для экстракции использовали дихлорметан (70 мл×3). Органическую фазу сушили на роторном испарителе. Колоночную хроматографию (дихлорметан / метанол 80:1) проводили с получением 1,4 г (49%) Соединения 48.
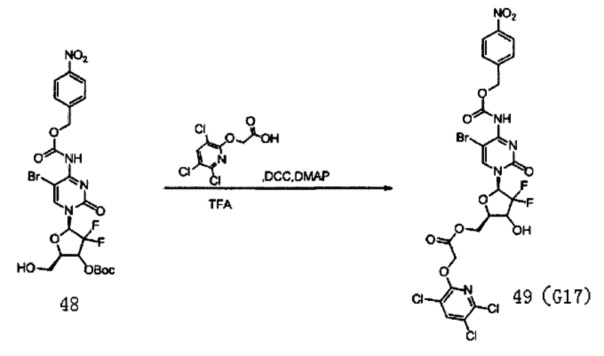
Соединение 48 (540 мг, 0,869 ммоль), защищенное Воc, Соединение 22 (667 мг, 2,60 ммоль) и DCC (537 мг, 2,60 ммоль) смешивали и добавляли в 45 мл дихлорметана, куда затем добавляли DMAP (2 мг, 0,016 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Проводили ТСХ. После завершения реакции добавляли 5 мл воды для разбавления. Для экстракции использовали 2×20 мл дихлорметана. Органическую фазу промывали 5 мл воды и 5 мл насыщенного солевого раствора. Для сушки использовали безводный сульфат натрия, а затем использовали TFA для непосредственного получения целевого соединения G17 (115 мг, выход двух стадий: 17%).
1Н-ЯМР (MeOD-d4, 400 МГц) δ:, 05 (s, 2Н, Ar), 6,22 (t, 1Н, J=7,6 Гц, H1'), 5,15 (m, 2Н), 4,67 (m, 1Н, Н5а'), 4,48 (m, 4Н, H5b', Н4', O-СН2-СН3), 1,70 (m, 2Н, -О-СН2-СН2-СН2-СН3), 1,28 (m, 2Н, -О-СН2-СН2-СН2-СН3), 0,97 (m, 2Н, -O-СН2-СН2-СН2-СН3).
13С-ЯМР (MeOD-d4, 100 МГц) δ: 67,81, 157,47, 155,55, 142,93, 141,00, 128,98, 128,42, 123,99, 117,54, 79,92, 76,89, 76,55, 66,69, 65,80, 63,78, 62,54.
ЭСМС: вычислено для C24H17BrCl3F2N5O10 m/z 760,92 (М+2Н)+, найдено 760,10.
(Пример 17. Гидрохлорид производного цитидина)
Настоящий пример включает получение гидрохлорида соединения Примера 1, а именно 4-N-(н-бутоксикарбонил)-2'-дезокси-2',2'-дифторцитидина.
0,50 г 4-N-(н-бутоксикарбонил)-2'-дезокси-2',2'-дифторцитидина растворяли в 60 мл этилацетата и обрабатывали сухим газом соляной кислоты на ледяной бане. После перемешивания в течение 15 мин растворитель удаляли с получением белого твердого продукта.
Получение гидрохлоридной соли других производных цитидина было аналогично указанному выше.
В дополнение к указанному выше гидрохлориду могут быть также получены фосфат, сульфат, карбонат, нитрат, цитрат, тартрат, малеат, сукцинат, сульфонат, пара-толуолсульфонат, мезилат, бензоат или фумарат производного цитидина.
(Пример 18. Лиофилизированный порошок для инъекции производного цитидина)
Настоящий пример включает получение лиофилизированного порошка для инъекции Соединения G14 Примера 13.
Лиофилизированный порошок для инъекции G14 включает 30 г Соединения G14, 300 г маннита (20% масс/об), 7 г буферного агента дигидрата дигидрофосфата натрия и 4,0 г поверхностно-активного вещества полоксамера 188 (F68).
Дигидрат дигидрофосфата натрия, полоксамер 188 (F68) (CAS No. 9003-11-6) и маннит (20% масс/об), взвешенные в соответствии с вышеуказанной рецептурой, добавляли к 300 г воды для инъекции, предварительно охлажденной до менее 10°C, значение рН раствора доводили до 7,3-7,5, используя раствор NaOH 0,1 моль/л. Затем к вышеуказанному раствору добавляли рецептурное количество G14 и хорошо перемешивали; и значение рН доводили до 7,3±0,2 с использованием 0,1 моль/л раствора NaOH или 0,1 моль/л HCl (в настоящем примере использовали 7,5). В полученный раствор добавляли воду до 2000 г и стерилизовали фильтрованием через 0,22 мкм микропористую мембрану. Фильтрат разливали во флаконы по 2,0 г на флакон. Флаконы частично закупоривали, а затем вымораживали досуха в лиофилизаторе. После завершения сушки флаконы упаковывали в вакууме, закрывали и маркировали с получением 1000 флаконов лиофилизированного порошка для инъекции. Затем температура хранения была от 2 до 8°C.
В дополнение к вышеупомянутому лиофилизированному порошку для инъекции, т.е. стерильному порошку для инъекции, производные цитидина согласно настоящему изобретению могут быть приготовлены для других форм инъекций, таких как инъекция в виде раствора, инъекция в виде суспензии и инъекция в виде эмульсии.
(Пример 19. Пероральная фармацевтическая композиция производного цитидина)
Фармацевтическая композиция производного цитидина в настоящем примере включает активные ингредиенты и необходимые фармацевтические компоненты, при этом фармацевтически активные ингредиенты представляют собой производные цитидина или соответствующие гидрохлориды, полученные в приведенных выше примерах. Масса фармацевтически активных ингредиентов в композиции составляет от 1% до 95% (30% в настоящем примере). Необходимые фармацевтические компоненты представляют собой воду, лактозу, кукурузный крахмал, гидроксипропилметилцеллюлозу (НРМС) и стеарат магния. Лекарственная форма фармацевтической композиции настоящего примера представляет собой таблетку.
Что касается применимой лекарственной формы фармацевтической композиции, в дополнение к вышеуказанной таблетке фармацевтически активный ингредиент может быть приготовлен в виде порошка, гранулы, капсулы, пеллета, раствора, суспензии, эмульсии, сиропа или эликсира для перорального применения или составов замедленного высвобождения и контролируемого высвобождения при пероральном введении или других пероральных фармацевтических композиций, содержащих общеизвестные соответствующие необходимые фармацевтические компоненты (классифицированные как добавки и присадки и т.д., в зависимости от их эффектов). Например, добавки включают маннит, лактозу, крахмал, стеарат магния, соль сахарина, целлюлозу или сульфат магния фармацевтической степени чистоты.
При осуществлении вышеуказанных пероральных лекарственных форм фармацевтические присадки могут быть выбраны в качестве носителей для фармацевтически активных ингредиентов, включая те, которые уже известны в области техники, такие как инертный твердый разбавитель, водный растворитель, липосома, микросфера и/или нетоксичный органический растворитель и так далее. Предпочтительные присадки включают увлажнитель, эмульгатор, буфер рН, человеческий сывороточный альбумин, антиоксидант, консервант, бактериостат, глюкозу, сахарозу, трегалозу, мальтозу, лецитин, глицин, сорбиновую кислоту, пропеновый спирт, полиэтилен, протамин, борную кислоту, хлорид натрия или хлорид калия, минеральное масло, растительное масло и так далее; одна или несколько их комбинаций могут быть выбраны в качестве носителя лекарственного средства.
Фармацевтические композиции согласно настоящему изобретению нацелены на гематологическую опухоль или злокачественные солидные опухоли; в частности, опухоли-мишени включают рак легкого, рак предстательной железы, рак молочной железы, рак толстой кишки, рак желудка, рак поджелудочной железы, рак печени, рак пищевода, опухоли головного мозга, рак яичника, рак матки, рак почки, рак головы и шеи, рак кожи, рак мочевого пузыря, рак вульвы, рак яичка, рак прямой кишки, ворсинчатый рак, герминому, злокачественную лимфому, лейкемию и множественную миелому, и даже более предпочтительные опухоли-мишени могут включать рак поджелудочной железы (первичное и вторичное лечение), немелкоклеточный рак легкого, рак молочной железы, рак яичника и плоскоклеточный рак головы и шеи и рак толстой кишки. Настоящее Изобретение не ограничено ими.
(Пример применения 1. Исследование MTD (максимальной переносимой дозы) однократного внутрибрюшинного введения ряда соединений на мышах ICR)
Этот эксперимент состоял в исследовании токсических эффектов однократного внутрибрюшинного введения на мышах ICR и определении максимальной переносимой дозы (MTD) каждого субъекта. Максимальная переносимая доза (MTD) относится к дозе, которая не может привести к смерти животного, не может привести к тому, что животное перенесет потерю массы более 10% (по сравнению с Сутками 0) или не может вызвать значительных побочных эффектов.
1. Конфигурация исследуемых веществ.
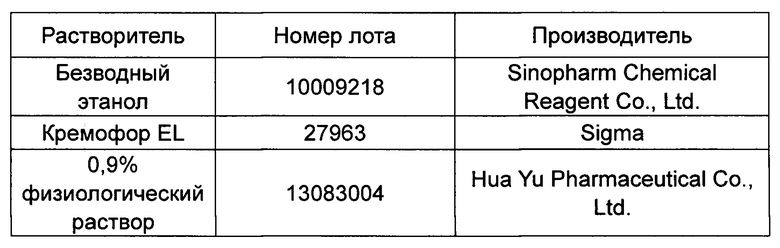
Источник растворителя, используемого для растворения исследуемого вещества, был следующим:

Соответствующее количество исследуемого вещества отбирали в стеклянную пробирку объемом 5 мл и растворяли в этаноле с помощью 5 мм магнитной мешалки для перемешивания. После полного растворения Кремофор EL добавляли при непрерывном перемешивании. Перед использованием добавляли отмеченное количество физиологического раствора и равномерно перемешивали. Объемное соотношение этанола, Кремофора EL и физиологического раствора составляло 5:5:90.
2. Подопытные животные
Разновидности и линии: мыши ICR; уровень: SPF (без специфических патогенов); пол: самки.
Источник: Shanghai Slac Laboratory Animal Co., Ltd.
Сертификационный номер: 0130749.
Масса животных в начале исследования: 18-20 г.
Количество и пол: 41, самки.
Модель питания: шесть на клетку.
Время адаптации: от 5 до 7 суток, условия содержания те же, что и в период эксперимента.
Температура окружающей среды в помещении для животных была 18-26°C, относительная влажность была 30-70%, а свет был включен 12 часов. Период адаптации до испытания для подопытных животных составлял 5-7 суток. Корм для роста и размножения больших и мелких мышей SPF был стерилизован с помощью Со60 и был приобретен у Beijing Kе Ао Xie Li Co., Ltd. Для подопытных животных использовали воду, стерилизованную фильтрацией. Во время эксперимента животные могли свободно есть и пить.
3. Способы исследования
Способ введения: ip (внутрибрюшинно). В случае смерти животных дозу уменьшали до достижения выживаемости животных; дозу увеличивали в случае отсутствия смертей животных; исследование заканчивали, когда животные в большинстве случаев выживали при данной высокой дозе. Наконец, MTD мышей в отношении исследуемых веществ определяли в соответствии с результатами исследований. Животных наблюдали непрерывно в течение 7 суток после однократного введения.
Во время эксперимента все животные находились под тщательным и непрерывным клиническим наблюдением в течение 14 суток, два раза в сутки (10:00, 16:00) после введения. Наблюдению повергались, но не ограничиваясь указанным: кожа, волосы, уши, нос, рот, грудь, живот, внешние гениталии, конечности и ноги, дыхательную систему и систему кровообращения, вегетативные эффекты (такие как слюноотделение), нервная система (например, тремор, судороги, реакция на стресс и ненормальное поведение).
Животных взвешивали перед введением, а затем взвешивали и регистрировали данные в одно и то же время каждый день.
Результаты наблюдения, массу тела животных и выживаемость животных после одной недели введения подробно регистрировали каждый день.
4. Результаты исследования
G3 и G4 могли переноситься при 350 мг/кг, G5 могло переноситься при 300 мг/кг, G6 могло переноситься при 200 мг/кг, G7 могло переноситься при 200 мг/кг, G8 могло переноситься при 300 мг/кг, G10 могло переноситься при 400 мг/кг, G11 могло переноситься при 400 мг/кг, G12 могло переноситься при 400 мг/кг, G13 могло переноситься при 400 мг/кг, G15 могло переноситься при 400 мг/кг, G16 могло переноситься при 400 мг/кг.
(Пример применения 2: Влияние ряда соединений на ингибирование роста опухоли)
В этом примере применения путем наблюдения за образованием и ростом опухоли в местах инокуляции и изменениями массы тела испытуемых животных оценивали ингибирование роста опухоли на ксенотрансплантатах рака толстой кишки НСТ-116 и токсичность одной внутрибрюшинной инъекции соединений от G2 до G17 у бестимусных мышей, несущих опухоль толстой кишки НСТ-116.
1. Цели исследования
Определение эффекта ингибирования роста и токсичности образцов производного цитидина согласно настоящему изобретению на трансплантированной опухоли бестимусных мышей, несущих рак толстой кишки НСТ-116.
2. Приготовление исследуемых веществ
Источник растворителя, используемого для растворения исследуемого вещества, был следующим:
Соответствующее количество исследуемого вещества отбирали в стеклянную 5 мл пробирку и растворяли в этаноле с помощью 5 мм магнитной мешалки для перемешивания. После полного растворения при непрерывном перемешивании добавляли Кремофор EL. Перед использованием добавляли и равномерно перемешивали отмеченное количество физиологического раствора. Объемное соотношение этанола, Кремофора EL и физиологического раствора составляло 5:5:90.
3. Подопытные Животные
Разновидности и линии: Balb/c бестимусные мыши; уровень: SPF; пол: самки.
Источник: Shanghai Sippr-bk Laboratory Animal Co., Ltd.
Количество животных: было заказано 100 животных, и для тестирования были выбраны здоровые особи.
Сертификационный номер для животных: 0123627.
Возраст животных в начале исследования: 7-9 недель
Масса животных в начале исследования: 18-22 г.
Время адаптации к окружению: 5-7 суток.
Мечение животных: номер на хвосте.
В комнате для содержания животных поддерживали температуру 23±2°C, влажность 40%-70%, со сменой света и темноты каждые 12 часов.
Корма (SLAC-M01) были приобретены у Beijing Kе Ао Xie Li Co., Ltd., и для животных, участвующих в исследовании, использовали воду, стерилизованную фильтрованием. В ходе эксперимента животные могли свободно есть и пить.
4. Способы исследования
4.1 Опухолевые клетки: клетки рака толстой кишки НСТ-116, приобретенные у Института Клеточной Биологии Академии Наук Китая. Клетки инкубировали в среде F-12 (содержащей 10% FBS) в CO2-инкубаторе при 37°C и при насыщенной влажности, с объемной долей CO2 5% и воздуха 95%. Перед инокуляцией клетки брали в логарифмической фазе и обрабатывали 0,25% трипсином, один раз промывали PBS и ресуспендировали в PBS для подсчета. Клетки ресуспендировали в бессывороточной среде и доводили примерно до 3×107 клеток/мл.
4.2 Инокуляция и группирование животных. Каждой бестимусной мыши стерильно инокулировали подкожно 0,1 мл клеточной суспензии (3×106 клеток/мышь) в правую сторону задней конечности. Когда опухоль вырастала примерно до 60-150 мм3, для группирования выбирали бестимусных мышей со схожим объемом и желаемой формой опухоли (предпочтительно группировали опухоли одной сферической формы, опухоли, не имеющие неправильной формы или множественные опухоли), по 6 мышей в группе. Группирование было следующим:

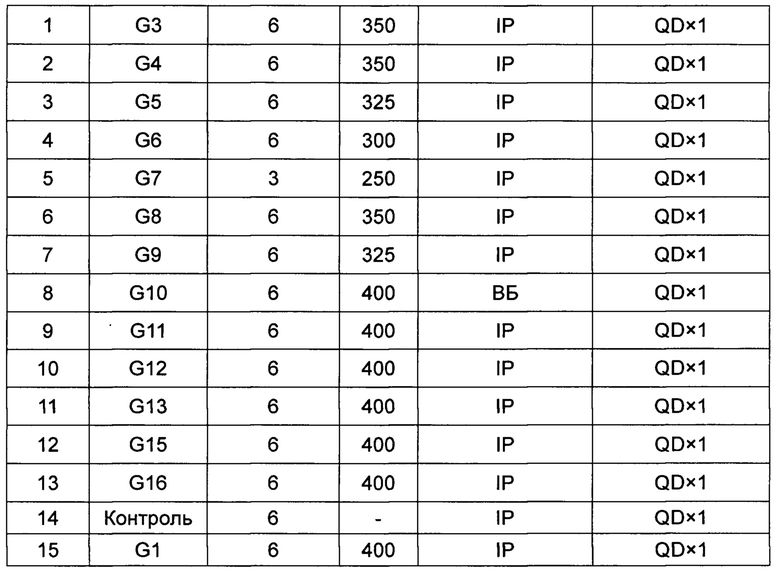
IP: внутрибрюшинная инъекция; QD×1: однократное введение 1 раз в сутки.
Контрольную группу, а именно контрольную группу модели, инъецировали смесью этанола, Кремофора EL и физиологического раствора (5:5:90).
4.3 Введение препаратов животным и наблюдение
В месте инокуляции каждой группы бестимусных мышей наблюдали образование опухоли. Диаметр опухолевого узла (D) измеряли три раза в неделю с помощью линейки с круглыми отверстиями, и объем опухолевого узла (V) рассчитывали по следующей формуле: V=3/4π(D/2)3.
Противоопухолевую активность оценивали по скорости ингибирования роста опухоли TGI (%) и относительной скорости пролиферации опухоли Т/С (%).
Скорость ингибирования роста опухоли TGI (%) рассчитывали как: TGI (%) = (Vконтроль-Vобработка) / Vконтроль) × 100%.
Относительный объем опухоли (RTV) рассчитывали как: RTV=Vt/V0, где V0 - объем опухоли на стадии группирования и введения, a Vt - объем опухоли во время измерения.
Относительную скорость пролиферации Т/С (%) рассчитывали как: Т/С (%) = TRTV/CRTV × 100%.
TRTV: RTV обработанной группы; CRTV: RTV контрольной группы.
Этих мышей взвешивали три раза в неделю.
4.4 Клинические симптомы
Все клинические симптомы каждого животного регистрировали в начале исследования и в ходе исследования. Наблюдения проводили в одно и то же время каждый день.
Животных умерщвляли с помощью CO2, когда потеря массы составляла более 20%, когда они были близки к смерти, или когда объем его опухоли превышал 2800 мм3. Опухоль отделяли и взвешивали, и животное вскрывали и обследовали для оценки и регистрации патологических изменений в органах.
4.5 Данные и статистический анализ
Данные выражали как Среднее ± SEM (стандартная ошибка среднего), если не было специально предусмотрено иное; для оценки данных между двумя группами выполняли непарный Т-тест. Значение Р менее 0,05 принимали как существенное различие.
5. Результаты исследования
(1) Клинические наблюдения и смертность
В группе G4 350 мг/кг 1 животное умерло на Сутки 4 после введения, для 3 животных наблюдали снижение активности, потерю массы тела, снижение температура поверхности тела по сравнению с обычной и другие клинические симптомы. Все животные в группе G8 350 мг/кг умерли на Сутки 4 после введения. Пять животных в группе G6 300 мг/кг умерли на Сутки 4 после введения. В клинических симптомах в контрольной группе модели и других группах, получивших соединения (G3, G5, G7, G9, G10, G11, G12, G13, G15, G16) не наблюдали существенных различий. Смертность каждой группы показана в Таблице 2 (QD*1: однократная внутрибрюшинная инъекция).

(2) Влияние исследуемых соединений на массу тела мышей, несущих рак толстой кишки человека НСТ-116.
Средняя масса каждой группы животных приведена в Таблице 3.


*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель


*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель
Скорости изменения массы групп G3-G9 приведены в Таблице 4-1.

*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель
Скорости изменения массы групп G10-G16 приведены в Таблице 4-2.


*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель
Согласно вышеприведенным данным, для соединения с точки зрения эффекта ингибирования роста трансплантированной опухоли у бестимусных мышей, несущих рак толстой кишки НСТ-116, массы животных в группе G4 350 мг/кг значительно снижались на Сутки 4 после введения (p менее 0,05), со средней скоростью потери массы, составлявшей 10,91±3,45%. После этого, у этих животных наблюдался устойчивый рост массы тела. Их масса значительно увеличивалась в период с Суток 18 по Сутки 20 по сравнению с контрольной группой модели (p менее 0,05). Масса животных в группе G5 325 мг/кг значительно снижалась на Сутки 4 сутки после введения, при этом скорость потери массы составляла менее 10%. После этого у этих животных наблюдался устойчивый рост массы тела. Их масса значительно увеличивалась в период с Суток 13 по Сутки 20 по сравнению с контрольной группой модели (p менее 0,05~0,01). Масса животных в группе G7 250 мг/кг значительно снижалась на Сутки 4 и Сутки 6 сутки после введения (p менее 0,05), при этом скорость потери массы составляла 12,28±4,78% и 4,39±3,6%, соответственно. После этого у этих животных наблюдался устойчивый рост массы тела. В значениях массы тела между другими обработанными группами и контрольными группами модели не было существенной разницы.
(3) Влияние исследуемых соединений на объем опухоли у мышей, несущих рак толстой кишки человека НСТ-116
Удельный объем опухоли в каждой группе приведен в Таблице 5.

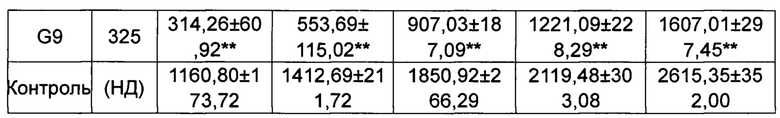
*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель


*p менее 0,05, **p менее 0,01 по сравнению с группой, получавшей носитель
В соответствии с вышеуказанными данными по объему опухоли производные цитидина согласно настоящему изобретению оказывают значительный ингибирующий эффект на опухоль.
(4) Влияние исследуемых соединений на уровень ингибирования роста (TGI%) мышей, несущих рак толстой кишки человека НСТ-116
Влияние исследуемых соединений на TGI% мышей, несущих рак толстой кишки человека НСТ-116, показано в Таблице 6 ниже:


Значение TGI % группы G3 350 мг/кг достигло максимального значения 58,10% на Сутки 8 и составило около 46,82% на Сутки 22. TGI % группы G4 350 мг/кг хорошо контролировались и достигали максимального значения 92,58% на Сутки 11 и все еще оставались на 70% выше на Сутки 22. TGI % группы G5 325 мг/кг хорошо контролировались и достигали максимального значения 94,46% на Сутки 11 и все еще оставались на 70% выше на Сутки 24. TGI % группы G9 325 мг/кг достигало максимального значения 80,77% на Сутки 4 и составляло около 40% на Сутки 22. TG 1% группы G7 250 мг/кг достигало максимального значения 82,62% на Сутки 8 и составляло 44,07% на Сутки 22.
(5) Относительный объем опухоли (RTV) мышей, несущих рак толстой кишки человека НСТ-116, которым вводили исследуемые соединения
Относительные объемы опухолей (RTV) мышей, несущих рак толстой кишки человека НСТ-116, которым вводили соединения G3-G9, показаны в Таблице 7-1:

Относительный объем опухоли в группе G4 350 мг/кг значительно снижался в период с Суток 4 по Сутки 18 по сравнению с контрольной группой модели (p менее 0,05-0,01). Относительный объем опухоли в группе G5 325 мг/кг значительно снижался в период с Суток 4 по Сутки 18 по сравнению с контрольной группой модели (p менее 0,05-0,01). Относительный объем опухоли в группе G7 250 мг/кг значительно снижался в период с Суток 4 по Сутки 15 по сравнению с контрольной группой модели (p менее 0,05-0,01). Относительный объем опухоли в группе G9 325 мг/кг значительно снижался только на Сутки 4 по сравнению с контрольной группой модели (p менее 0,05). Относительный объем опухоли в других обработанных группах по сравнению с контрольными группами модели существенной не различался.
Относительные объемы опухолей (RTV) мышей, несущих рак толстой кишки человека НСТ-116, которым вводили G10-G16, показаны в Таблице 7-2:


(6) Относительная скорость пролиферации опухоли (Т/С %) у мышей, несущих рак толстой кишки человека НСТ-116, которым вводили исследуемые соединения
Влияние исследуемых соединений на Т/С % мышей, несущих рак толстой кишки человека НСТ-116, показано в Таблице 8 ниже:

Т/С % в группе G4 350 мг/кг достигло минимального значения 17,62% на Сутки 13 и составило 75,38% на Сутки 22. Т/С % в группе G5 325 мг/кг достигло минимального значения 10,01% на Сутки 11 и составило 68,77% на Сутки 22. Т/С % в группе G7 250 мг/кг достигло минимального значения 17,67% на Сутки 8 и составило 58,62% на Сутки 22.
В исследовании в отношении влияния ряда соединений на эффект ингибирования роста трансплантированной опухоли бестимусных мышей, несущих раком толстой кишки НСТ-116, Соединения G4, G5 и G7 показали хорошее TGI % в отношении трансплантированной опухоли бестимусных мышей, несущих рак толстой кишки НСТ-116, с хорошим эффектом ингибирования опухоли в период с Суток 8 по Сутки 13 после однократного внутрибрюшинного введения, где G5 достигал минимального Т/С % 10,01% на Сутки 11, оказывали меньшее влияние на потерю массы тела и имели среднюю скорость потери массы менее 10%.
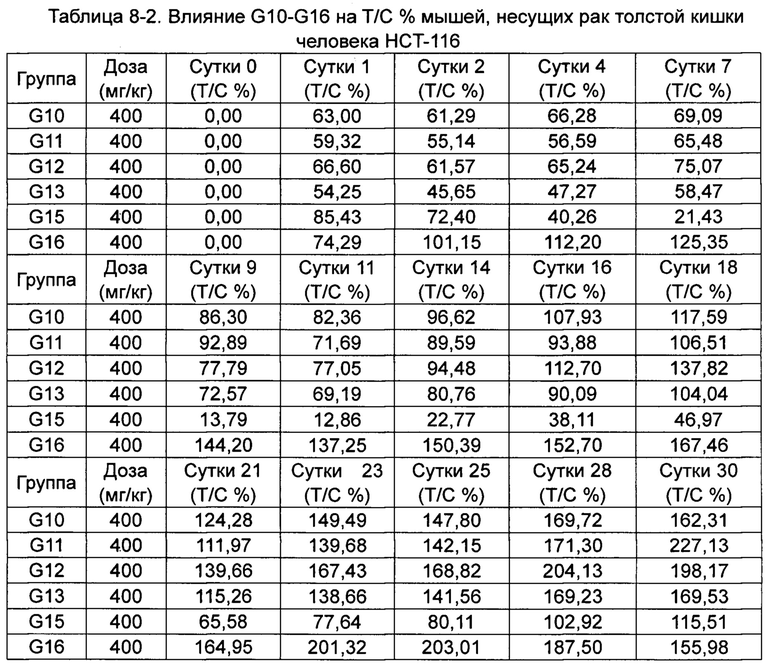
На основании результатов приведенных выше исследований было подтверждено, что новый тип производного цитидина согласно настоящему изобретению оказывает превосходные эффекты в качестве противоопухолевого агента.
Вышеприведенные примеры хорошо иллюстрируют настоящее изобретение, технический персонал в данной области понимает, что могут быть проведены различные модификации, и эти модификации входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ДИМЕРЫ ПРОИЗВОДНЫХ ЦИТИДИНА И ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2687252C2 |
| ПРОИЗВОДНЫЕ 6-АМИНОХИНАЗОЛИНА ИЛИ 3-ЦИАНОХИНОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ EGFR ИЛИ HER-2 | 2010 |
|
RU2536102C2 |
| Регулятор на основе азотсодержащего гетероциклического производного, способ его получения и его применение | 2020 |
|
RU2829553C2 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АЗОТИСТЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2473543C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ОРЕКСИНА | 2015 |
|
RU2669701C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОИНДОЛОНА | 2009 |
|
RU2468022C2 |
| Соединение в качестве ингибитора циклинзависимой киназы 9 и его применение | 2020 |
|
RU2819762C1 |
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
Изобретение относится к производному цитидина, применимому в онкологии, формулы (I):

где R1 является C1-С10 алкилом, -(CH2)n-Ph, где n равно 1, Ph представляет собой фенил, необязательно замещенный нитрогруппой или атомом галогена;
R2 представляет собой  , где X1 представляет собой -(CH2)n-Ph, в котором n равно 1;
, где X1 представляет собой -(CH2)n-Ph, в котором n равно 1;
R3 представляет собой Н или 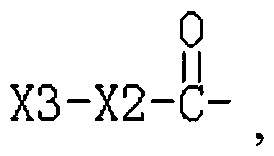 где Х3 представляет собой замещенное гетероциклическое кольцо, которое замещено тремя атомами галогена; где указанное гетероциклическое кольцо представляет собой пиридин; и Х2 представляет собой -O-(СН2)n-, где n равно 1. Предложено новое производное цитидина со сниженной токсичностью и фармацевтические композиции на его основе, эффективные для лечения злокачественной солидной опухоли. 3 н. и 2 з.п. ф-лы, 17 табл., 19 пр.
где Х3 представляет собой замещенное гетероциклическое кольцо, которое замещено тремя атомами галогена; где указанное гетероциклическое кольцо представляет собой пиридин; и Х2 представляет собой -O-(СН2)n-, где n равно 1. Предложено новое производное цитидина со сниженной токсичностью и фармацевтические композиции на его основе, эффективные для лечения злокачественной солидной опухоли. 3 н. и 2 з.п. ф-лы, 17 табл., 19 пр.
1. Новое производное цитидина, имеющее следующую общую формулу (I):

где R1 является C1-С10 алкилом, -(CH2)n-Ph, где n равно 1, Ph представляет собой фенил, необязательно замещенный нитрогруппой или атомом галогена;
R2 представляет собой 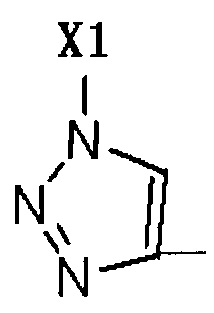 , где X1 представляет собой -(CH2)n-Ph, в котором n равно 1;
, где X1 представляет собой -(CH2)n-Ph, в котором n равно 1;
R3 представляет собой Н или 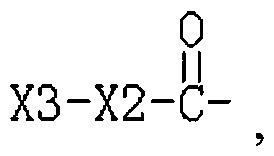 где Х3 представляет собой замещенное гетероциклическое кольцо, которое замещено тремя атомами галогена; где указанное гетероциклическое кольцо представляет собой пиридин; и Х2 представляет собой -O-(СН2)n-, где n равно 1.
где Х3 представляет собой замещенное гетероциклическое кольцо, которое замещено тремя атомами галогена; где указанное гетероциклическое кольцо представляет собой пиридин; и Х2 представляет собой -O-(СН2)n-, где n равно 1.
2. Новое производное цитидина по п. 1, отличающееся тем, что R1 представляет собой С1-С4алкил, бензил или замещенный бензил.
3. Применение производного цитидина по п. 1 для получения противоопухолевых препаратов для лечения злокачественной солидной опухоли.
4. Фармацевтическая композиция для лечения злокачественной солидной опухоли, включающая производное, как показано на общей формуле (I) по п. 1, в качестве активного ингредиента и один или несколько фармацевтических носителей или эксципиентов.
5. Фармацевтическая композиция по п. 4, отличающаяся тем, что лекарственная форма композиции представляет собой инъекцию или средство для перорального введения, где инъекция относится к инъекции в виде раствора, инъекции в виде суспензии, инъекции в виде эмульсии или стерильному порошку для инъекции; средство для перорального введения относится к таблетке, порошку, грануле, капсуле, пеллету, раствору, суспензии, эмульсии, сиропу или эликсиру.
| US 20140134160 A1, 15.05.2014 | |||
| WO 1991015498 A1, 17.10.1991 | |||
| WO 2004041203 A2, 21.05.2004 | |||
| Предохранительное приспособление у передней части кузова трамвайного вагона или иной повозки | 1928 |
|
SU11868A1 |

