Перекрестная ссылка на родственные заявки
Данная заявка испрашивает приоритет по патентной заявке на изобретение Китая № CN201510856641.1, поданной 30 ноября 2015 г., которая включена в данный документ в полном объеме посредством ссылки.
Область техники
Данное изобретение относится к области медицины и в частности относится к ряду замещенных 2-(пиридин-2-ил)аминопиримидинов, обладающих активностью ингибирования протеинкиназ, и к способу их получения и их фармацевтическому применению.
Уровень техники
Клеточный цикл является важной частью жизнедеятельности клетки. При нормальном росте клеток осуществление прогрессии клеточного цикла зависит от точной и хорошо организованной регуляции клеточного цикла с помощью регуляторных факторов различных уровней. Ключевым из этих регуляторных факторов является циклин-зависимая киназа (CDK) и ее положительные и отрицательные регуляторы, т.е. циклин и ингибиторы циклин-зависимой киназы (CDI). Комплекс CDK-циклин, образованный циклин-зависимой протеинкиназой и циклином, вовлечен в рост, пролиферацию, состояние покоя или апоптоз клеток. В процессе прогрессии клеточного цикла циклин периодически и постоянно экспрессируется, и разлагается, и связывается с CDK, которые соответственно временно им активируются. Фосфорилирование различных субстратов катализируется активностью CDK для осуществления продвижения и перехода различных фаз клеточного цикла.
На данный момент установлено 13 членов семейства CDK, и они представляют собой CDK1-CDK13, соответственно, среди которых CDK1, CDK2, CDK3, CDK4 и CDK6 вовлечены в регуляцию пролиферации клеток, а CDK7, CDK8, CDK9, CDK11, CDK12 и CDK13 вовлечены в регуляцию транскрипции.
Циклин разделяют на A-L, и различные CDK связываются с различными подтипами циклина. Среди них семейство циклина D (циклин D1, D2, D3) начинает экспрессироваться в фазе G1, связывается и активирует CDK4 и CDK6 с образованием комплекса CDK4/6-циклин D для того, чтобы фосфорилировать ряд субстратов, в том числе белок ретинобластомы (Rb). После фосфорилирования Rb высвобождает белки, которые с ним связываются и ингибируются им, включая главным образом транскрипционный фактор E2F, который активирует и транскрибирует некоторые гены, необходимые для входа в фазу S (MA Ke, Advance in Anti-tumor Effect of CDK4/6 Inhibitors, World Notes on Antibiotics, 2013, 34 (5):197-202). Если равновесие нарушено вследствие различных факторов: или сигнал стимуляции пролиферации клеток увеличен, или сигнал ингибирования пролиферации клеток уменьшен в некоторой степени, пролиферация клеток выйдет из-под контроля, а затем возникнет опухоль. В исследовании установлено, что аномалия пути циклин D-CDK4/6-INK4-Rb встречается в приблизительно 80% случаев рака человека (1. Malumbres M, Barbacid M., To cycle or not to cycle: a critical decision in cancer [J]. Nature Reviews Cancer, 2001, 1 (3): 222; 2. Shapiro GI., Cyclin-dependent kinase pathways as targets for cancer treatment [J]. J Clinical Oncology, 2006, 24 (11):1770). Изменение этого пути ускоряет прогрессию фазы G1, так что ускоряется пролиферация клеток опухоли, и они получают преимущество в выживании. Поэтому вмешательство в этот путь становится терапевтической стратегией, а таким образом CDK4/6 становится одной из потенциальных противоопухолевых мишеней.
Преимущества CDK4/6 в качестве противоопухолевой мишени заключаются в том, что: (1) большинство пролиферирующих клеток зависят от пролиферации CDK2 или CDK4/6, но ингибиторы CDK4/6 не проявляют цитотоксичность, как «неселективные ингибиторы CDK», такую как миелосупрессия и кишечная реакция; и (2) доклинические эксперименты показывают, что если уровень циклина D в клетках повышен, или p16INK4a инактивирован, чувствительность клеток к лекарственному средству может быть увеличена. Поскольку клетки опухоли демонстрируют вышеуказанное явление в отличие от нормальных клеток, направленное действие лекарственных средств в некоторой степени увеличивается.
В дополнение к ингибированию роста опухоли ингибиторы CDK также используются при лечении других расстройств, например сердечно-сосудистых расстройств, в том числе атеросклероза, рестеноза после имплантации сосудистого стента и других сердечно-сосудистых расстройств, вызванных аномальной клеточной пролиферацией; например, при лечении заболеваний, вызванных грибами, простейшими паразитами (такими как Plasmodium falciparum) и инфекциями ДНК- и РНК-содержащими вирусами, включая малярию, СПИД и т.д. Кроме того, в исследованиях дополнительно установлено, что ингибиторы CDK также могут использоваться для лечения аутоиммунных заболеваний (таких как псориаз, ревматоидный артрит, гломерулонефрит и красная волчанка и т.д.) и ингибирования пролиферации воспалительных клеток.
С тех пор, как в WO9811095 был описан ряд соединений 2-пиримидинамина, обладающих активностью ингибирования цитокинов, в данной области техники постепенно появлялось большое количество соединений на основе такой структуры ядра, обладающих ингибирующей активностью относительно CDK4/6, и некоторые стали перспективными лекарственными веществами-кандидатами и даже поступили на клинические исследования III фазы. Например, соединение PD0332991, также известное как палбоциклиб, которое описано в WO2003062236, представлено структурной формулой 1 и разработано компанией Pfizer. PD0332991 имеет IC50, составляющие 11 нмоль/л и 15 нмоль/л для ингибирования CDK4 и CDK6, соответственно; а его IC50 для ингибирования CDK2, CDK1 и CDK5 выше чем 10 мкмоль/л (Fry DW, Harvey PJ, Keller PR, et al., Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts [J]. Molecular Cancer Therapeutics, 2004, 3 (11):1427). Соединение LEE011, которое разрабатывается Novartis (описано в WO2011101409), представлено структурной формулой 2. Соединение LY2835219 (описано в WO2010075074), также известное как бемациклиб, представлено структурной формулой 3; сообщалось, что его IC50 для ингибирования CDK4 и CDK6 составляют 2 нмоль/л и 9,9 нмоль/л, соответственно (Lawrence MG, S.F.Cai, X. Lin et al., Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine [J]. Invest New Drugs, (2014), 32: 825). На данный момент LY2835219 изучается в клиническом исследовании III фазы, проводимом компанией Eli Lilly.
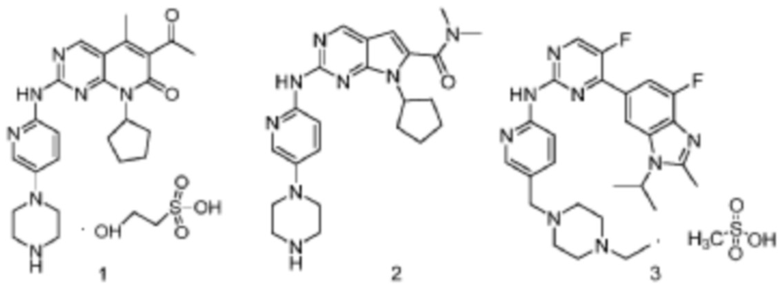
Вследствие появления этих соединений CDK4/6 стала очевидной противоопухолевой мишенью. Заявитель также подавал заявку на патент (№ 201510708487.3, поданная 27 октября 2015 г.) для ряда новых замещенных 2-(пиридин-2-ил)аминопиримидинов, которые демонстрируют активность селективного ингибирования CDK4/6.
Злокачественные опухоли до сих пор являются серьезной угрозой здоровью человека. Следовательно, необходимо и актуально разработать ингибиторы CDK4/6 с более высокой активностью, селективностью и биодоступностью для того, чтобы обеспечить больше клинических вариантов лечения заболеваний, связанных с аномальной пролиферацией клеток, таких как рак.
Сущность изобретения
Принимая во внимание вышеуказанные проблемы, целью данного изобретения является получить новое замещенное соединение 2-(пиридин-2-ил)аминопиримидина. Соединение, предложенное в изобретении, может селективно ингибировать циклин-зависимую киназу CDK4/6 и останавливать клеточный цикл в фазе G1, и таким образом может быть использовано для лечения расстройств, связанных с клеточной пролиферацией.
Для того, чтобы достичь вышеуказанного технического эффекта, в данном изобретении предложены следующие технические решения.
В одном аспекте в данном изобретении предложено соединение структурной формулы I или таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединения структурной формулы I или его таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси,
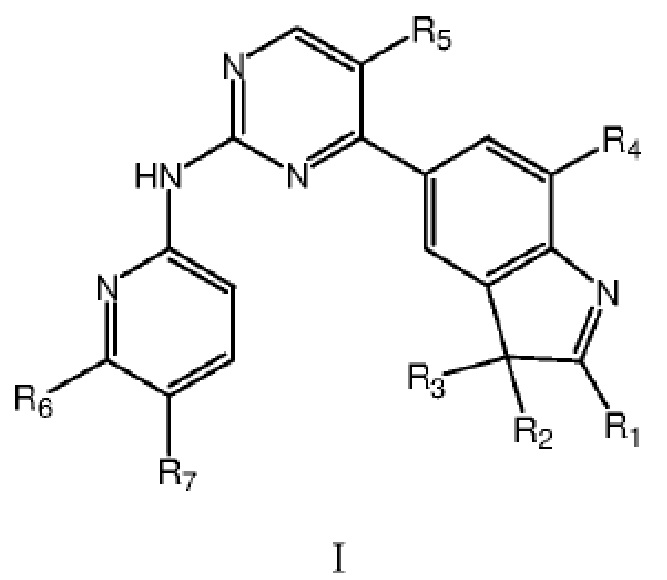
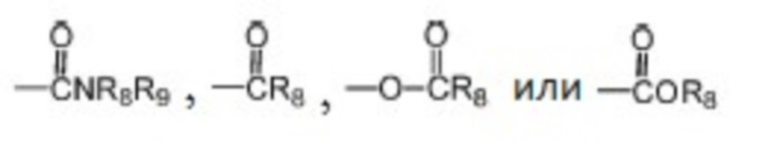
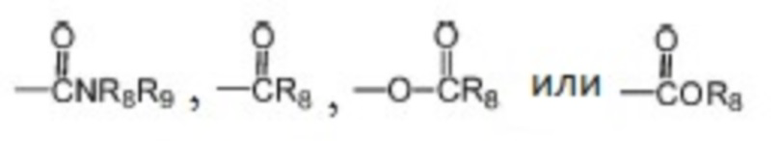
где каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенной C1-C6 углеводородной группы или C1-C6 углеводородной группы, замещенной одним или большим количеством заместителей, выбранных из C1-C6 углеводородной группы, C3-C6 циклоалкана, C1-C6 галогеналкила, C1-C6 алкокси, гидроксила, галогена, циано, -NR8R9,
 ;
;
или любые два из R1, R2 и R3 вместе с атомами C, к которым они соответственно присоединены, образуют насыщенное или ненасыщенное 3-7-членное кольцо;
каждый из R4 и R5 независимо выбран из группы, состоящей из водорода и галогена, и по меньшей мере один из R4 и R5 представляет собой галоген;
R6 выбран из группы, состоящей из атома водорода, C1-C6 алкила, C1-C6 алкокси, гидроксила или галогена;
R7 представляет собой 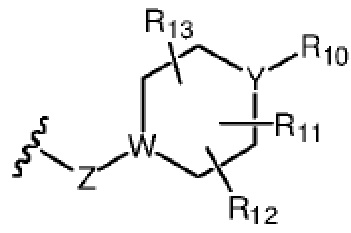 , где Z представляет собой карбонил, O, S, имино, сульфонил или
, где Z представляет собой карбонил, O, S, имино, сульфонил или 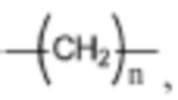 , n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C, N, O или S, но оба W и Y не могут одновременно представлять собой C, и когда Z представляет собой O или S, W представляет собой C; каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 гидроксиалкила, C1-C6 галогеналкила, C1-C6 алкокси, гидроксила, галогена, циано, -NR8R9,
, n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C, N, O или S, но оба W и Y не могут одновременно представлять собой C, и когда Z представляет собой O или S, W представляет собой C; каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 гидроксиалкила, C1-C6 галогеналкила, C1-C6 алкокси, гидроксила, галогена, циано, -NR8R9, 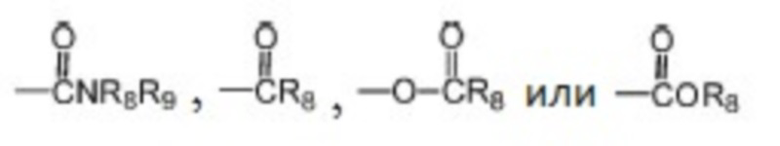 , и если Y=N, R10 не может представлять собой NH2, -NHR8, -NR8R9,
, и если Y=N, R10 не может представлять собой NH2, -NHR8, -NR8R9, 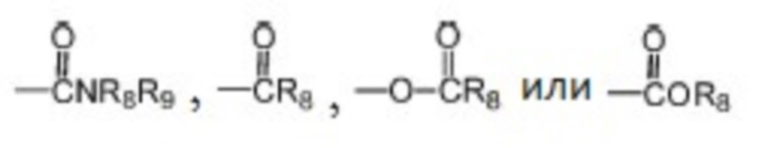 ; или
; или
R6 и R7 вместе с атомами C, к которым они присоединены, образуют 5-7-членный гетероцикл, содержащий один или большее количество атомов, выбранных из N, O или S, и 5-7-членный гетероцикл замещен одним или большим количеством заместителей, выбранных из C1-C6 алкила, C3-C6 циклоалкила, C1-C6 галогеналкила, C1-C6 алкокси, C1-C6 гидроксиалкила, гидроксила, галогена, циано, -NH2, -NHR8, -NR8R9,
 ;
;
где каждый из R8 и R9 независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила и C1-C6 гидроксиалкила.
Каждый из R1, R2 и R3 предпочтительно независимо выбран из атома водорода, незамещенной C1-C6 углеводородной группы или C1-C6 углеводородной группы, замещенной одним или большим количеством заместителей, выбранных из C1-C6 углеводородной группы, C3-C6 циклоалкила, C1-C6 галогеналкила, C1-C6 алкокси, гидроксила или галогена.
Более предпочтительно каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенной C1-C6 углеводородной группы или C1-C6 углеводородной группы, замещенной одним или большим количеством заместителей, выбранных из C1-C6 углеводородной группы, гидроксила или галогена.
Более предпочтительно каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенного линейного или разветвленного C1-C6 алкила, незамещенного линейного или разветвленного C2-C4 алкенила.
Наиболее предпочтительно каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенного линейного или разветвленного C1-C4 алкила.
В качестве другого предпочтительного варианта реализации изобретения R2 и R3 вместе с атомами C, к которым они оба присоединены, образуют насыщенное или ненасыщенное 3-7-членное кольцо.
Более предпочтительно R2 и R3 вместе с атомами C, к которым они оба присоединены, образуют насыщенное 3-7-членное кольцо.
Каждый из R4 и R5 независимо предпочтительно выбран из водорода, фтора или хлора, и по меньшей мере один из R4 и R5 представляет собой фтор или хлор.
Более предпочтительно каждый из R4 и R5 независимо представляет собой водород или фтор, и по меньшей мере один из R4 и R5 представляет собой фтор.
Наиболее предпочтительно R4 представляет собой водород или фтор, и R5 представляет собой фтор.
R6 предпочтительно выбран из атома водорода или C1-C6 алкила.
Z предпочтительно представляет собой карбонильную группу, O или  , n равен целому числу от 0 до 4.
, n равен целому числу от 0 до 4.
Более предпочтительно Z представляет собой , n равен целому числу от 0 до 2, более предпочтительно n= 0 или 1.
Каждый из W и Y предпочтительно независимо выбран из C или N, но W и Y не могут оба одновременно представлять собой C.
Каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 гидроксиалкила, C1-C6 галогеналкила, C1-C6 алкокси, гидроксила или -NR8R9, и если Y=N, R10 не может представлять собой -NR8R9, где каждый из R8 и R9 независимо выбран из атома водорода и C1-C4 алкила.
Более предпочтительно каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила, C1-C6 галогеналкила, C1-C6 алкокси или -NR8R9, где R8 и R9 независимо выбраны из атома водорода и C1-C4 алкила.
Более предпочтительно R7 выбран из заместителей, имеющих следующие структуры:
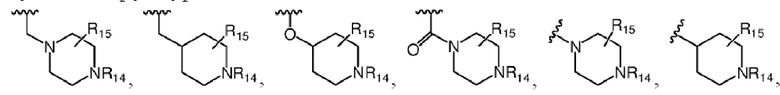
 ,
,
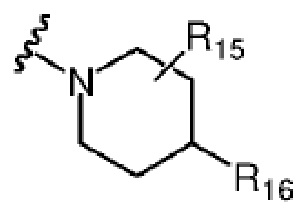
где каждый из R14 и R15 независимо выбран из атома водорода, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 алкокси или гидроксила; R16 выбран из атома водорода, C3-C6 циклоалкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 алкокси, гидроксила или -NR8R9, где R8 и R9 независимо выбраны из атома водорода и C1-C4 алкила.
Более предпочтительно каждый из R14 и R15 независимо выбран из группы, состоящей из атома водорода, C1-C6 алкила, C3-C6 циклоалкила и C1-C6 гидроксиалкила; R16 выбран из атома водорода, C1-C6 алкила, C3-C6 циклоалкила, C1-C6 гидроксиалкила или -NR8R9, где R8 и R9 независимо выбраны из атома водорода и C1-C4 алкила.
В качестве другого предпочтительного варианта реализации изобретения R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий один или большее количество атомов, выбранных из N, O или S.
Более предпочтительно R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий N.
Более предпочтительно R6 и R7 вместе с атомами C, к которым они присоединены, образуют следующую химическую структуру:
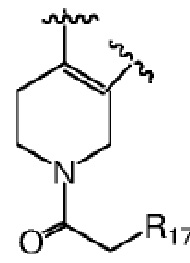 , где R17 выбран из гидроксила или C1-C3 алкокси; дополнительно, R17 предпочтительно представляет собой гидроксил.
, где R17 выбран из гидроксила или C1-C3 алкокси; дополнительно, R17 предпочтительно представляет собой гидроксил.
В качестве предпочтительного варианта реализации в данном изобретении дополнительно предложены соединения структурных формул II, III, IV или V или их соответствующие таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений формул II, III, IV или V или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси,
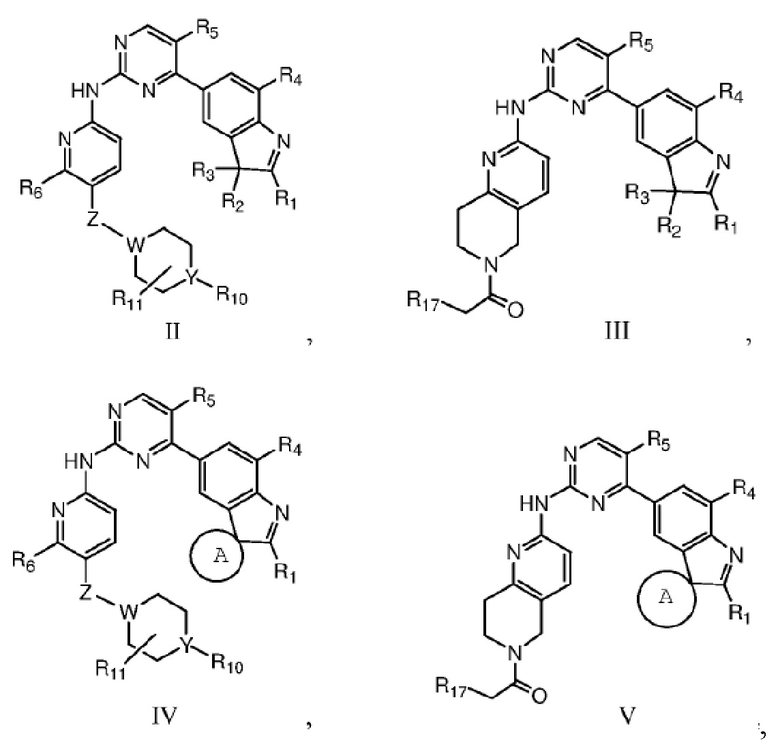
где Z, W, Y, R1, R2, R3, R4, R5, R6, R10, R11 и R17 определены, как указано выше, кольцо A представляет собой насыщенное 3-7-членное кольцо.
Кольцо A предпочтительно представляет собой насыщенное 3-6-членное кольцо.
Более предпочтительно в данном изобретении предложено соединение структурной формулы VIII или таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединения структурной формулы VIII или его таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси,
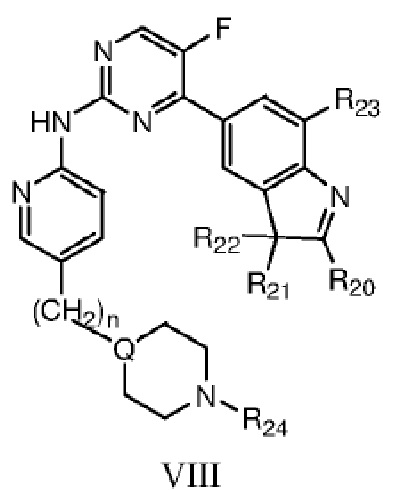
где каждый из R20, R21, R22 независимо выбран из C1-C4 алкила, или R20 представляет собой C1-C4 алкил, а R21 и R22 вместе с атомом C, к которому они присоединены, образуют насыщенное 5-6-членное кольцо; R23 выбран из водорода или фтора; n=0 или 1; R24 выбран из группы, состоящей из водорода, C1-C4 алкила или C1-C4 гидроксиалкила, Q представляет собой C или N.
В качестве более предпочтительного варианта реализации в данном изобретении предложены соединения следующих структур или их таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений указанных структур или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси,
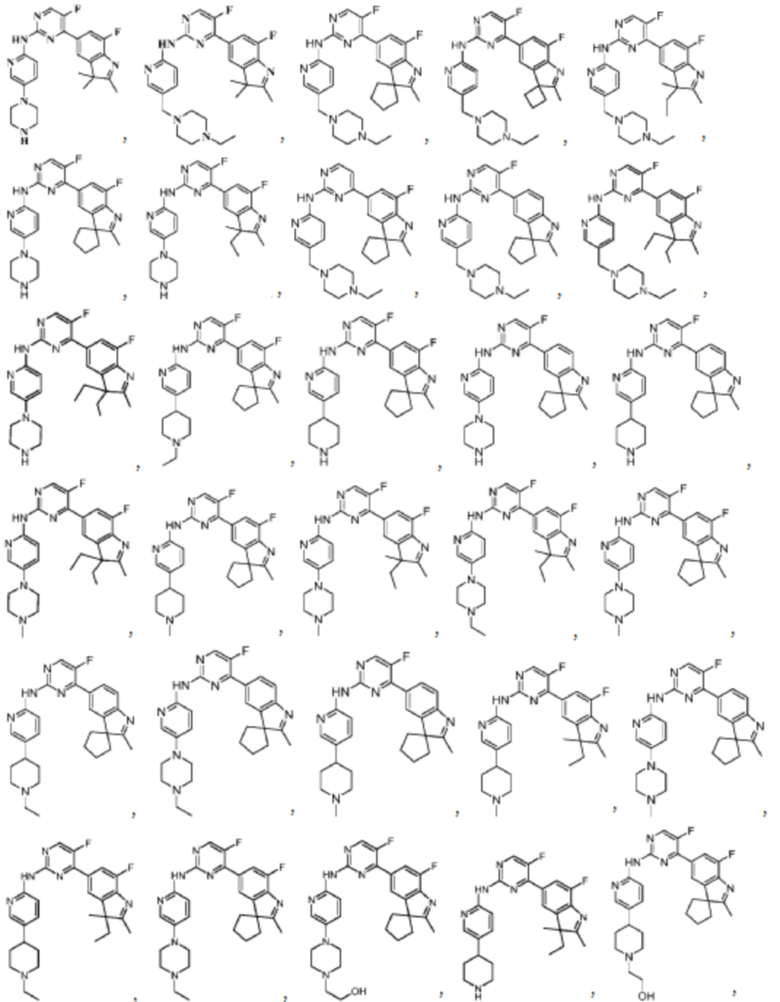
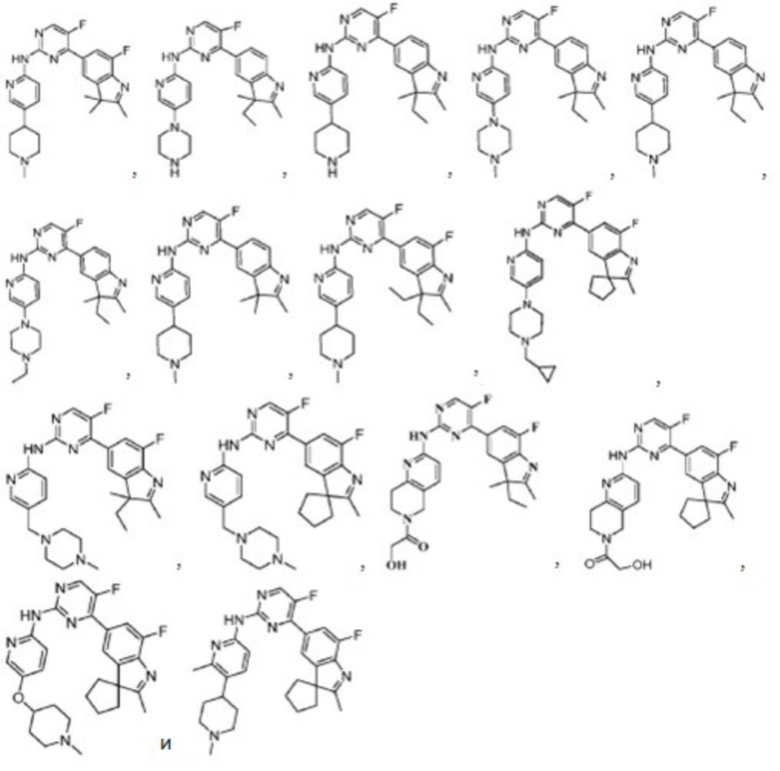
Соединения в соответствии с данным изобретением также включают в себя все вышеупомянутые соединения, которые являются изотопно-мечеными.
В другом аспекте в данном изобретении дополнительно предложено соединение структурной формулы VI или таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь; или фармацевтически приемлемые соли или сольваты соединения структурной формулы VI или его таутомера, мезомера, рацемата, энантиомера, диастереомера,
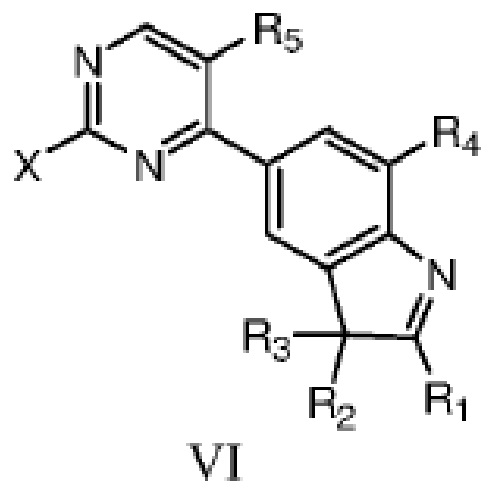
где R1, R2, R3, R4 и R5 определены, как указано выше, X представляет собой уходящую группу или аминогруппу.
X предпочтительно представляет собой галоген или амино, более предпочтительно фтор, бром, хлор или амино.
В другом аспекте в данном изобретении предложен способ получения соединения структурной формулы I, включающий в себя проведение катализируемой палладием реакции кросс-сочетания между соединением формулы VI и соединением формулы VII в растворителе, что дает соединение формулы I,
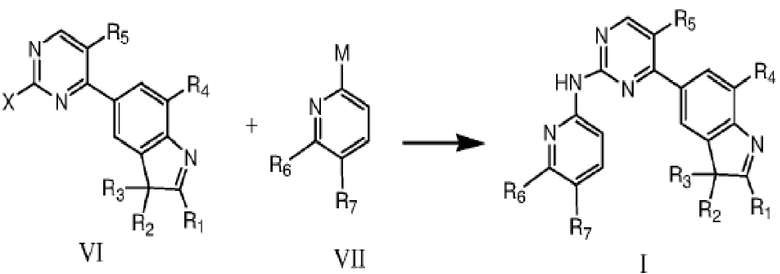
где R1, R2, R3, R4, R5, R6 и R7 определены, как указано выше; каждый из X и M независимо представляют собой уходящую группу или аминогруппу, только один из X и M представляет собой аминогруппу, и один из двух должен представлять собой аминогруппу;
уходящая группа предпочтительно представляет собой галоген;
более предпочтительно уходящая группа представляет собой фтор, бром или хлор.
При этом вышеуказанный способ получения может дополнительно включать в себя удаление защитной группы.
При этом вышеуказанный способ получения может дополнительно включать в себя разделение и/или очистку продуктов, и разделение и/или очистку можно осуществлять с помощью метода, обычно используемого в органическом синтезе, например, пригодной комбинации методов фильтрования, экстракции, промывки, концентрирования, хроматографии и тому подобного.
В другом аспекте в данном изобретении предложено применение соединений структурных формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси; или фармацевтически приемлемых солей или сольватов соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси при производстве готового фармацевтического состава для лечения расстройства связанного с пролиферацией клеток.
Готовый фармацевтический состав предпочтительно содержит фармацевтически приемлемое вспомогательное вещество.
Расстройство, связанное с пролиферацией клеток, предпочтительно относится к раку млекопитающего или человека, более предпочтительно относится к раку человека, в том числе злокачественным солидным опухолям и злокачественным несолидным опухолям, в частности включая, но не ограничиваясь ими, рак молочной железы, рак легких, рак предстательной железы, лейкоз, рак головного мозга, рак желудка и глиому.
Расстройство, связанное с пролиферацией клеток, предпочтительно может также представлять собой СПИД, атеросклероз и рестеноз после имплантации сосудистого стента.
Указанное применение предпочтительно относится к применению соединений структурных формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси; или фармацевтически приемлемых солей или сольватов соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси, в качестве единственного активного ингредиента или в комбинации с другими биологически активными веществами при производстве готового фармацевтического состава для лечения расстройства, связанного с пролиферацией клеток.
Другие биологически активные вещества включают в себя, но не ограничиваясь ими, противораковые агенты, иммуносупрессивные агенты и противовирусные агенты; причем противораковый агент выбран из алкилирующего агента (такого как циклофосфамид, ифосфамид, тиотепа, семустин, мехлоретамина гидрохлорид, бусульфан, хлорамбуцил, мелфалан, нитрокафан, формилмелфалан, кармустин, ломустин, альтретамин, дибромоманнит, темозоломид и тому подобного), антиметаболитных противоопухолевых лекарственных средств (таких как цитарабин, фторурацил, метотрексат, гидроксимочевина, тегафур, меизоиндиго, меркаптопурин и тому подобного), агента, представляющего собой комплекс с платиной (такого как цисплатин, карбоплатин, оксалиплатин и тому подобное), антибиотических противоопухолевых лекарственных средств (актиномицина D, митомицина, доксорубицина, пингиангмицина, эпирубицина, пирарубицина, даунорубицина, блеомицина и тому подобного), противоопухолевых лекарственных средств природного происхождения (гомогаррингтонина и его производных, винкристина и его производных, гидроксикамптотецина и его производных, этопозида и его производных, виндезина и его производных, винбластина и его производных, винорелбина гидротартрата, таксола и его производных, колхицина и его производных, элемена и его производных и тому подобного), гормональных противоопухолевых лекарственных средств (таких как аминоглутетимид, тамоксифен, дексаметазон, дутастерид, флутамид, гонадорелин, лейпролида ацетат, летрозол и тому подобное), ингибиторов VEGFR или EGFR (таких как сунитиниб, сорафениб, иматиниб, гефитиниб, эрлотиниб, вандетаниб, пазопаниб, лапатиниб, канертиниб, афатиниб, мубритиниб, дазатиниб, нератиниб и тому подобное), противоопухолевых лекарственных средств, представляющих собой антитела (таких как трастузумаб, пертузумаб, ритуксимаб, панитумумаб, бевацизумаб, ипилимумаб, офатумумаб, рамуцирумаб и тому подобное), ингибиторов mTOR (таких как эверолимус, сиролимус, зотаролимус и тому подобное) и лекарственных средств для лечения опухоли головного мозга, таких как темозоломид и тому подобное.
В другом аспекте в данном изобретении предложен комбинированный продукт для лечения расстройства, связанного с пролиферацией клеток, причем комбинированный продукт содержит одно или большее количество соединений, выбранных из соединений структурных формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси; или фармацевтически приемлемых солей или сольватов соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси.
Комбинированный продукт предпочтительно дополнительно содержит фармацевтически приемлемые вспомогательные вещества, и/или комбинированный продукт представляет собой набор.
В другом аспекте в данном изобретении дополнительно предложен способ лечения расстройства, связанного с пролиферацией клеток, включающий в себя введения пациенту, нуждающемуся в этом, перорально или неперорально эффективного количества соединений по данному изобретению или вышеупомянутого комбинированного продукта.
Вышеуказанный способ лечения расстройства, связанного с пролиферацией клеток предпочтительно включает в себя введение пациенту перорально или неперорально эффективного количества соединений по данному изобретению и указанных других биологически активных веществ. Указанные другие биологически активные вещества включают в себя, но не ограничиваясь ими, противораковые агенты, иммуносупрессивные агенты и противовирусные агенты; причем противораковый агент выбран из алкилирующего агента (такого как циклофосфамид, ифосфамид, тиотепа, семустин, мехлоретамина гидрохлорид, бусульфан, хлорамбуцил, мелфалан, нитрокафан, формилмелфалан, кармустин, ломустин, альтретамин, дибромоманнит, темозоломид и тому подобное), антиметаболитных противоопухолевых лекарственных средств (таких как цитарабин, фторурацил, метотрексат, гидроксимочевина, тегафур, меизоиндиго, меркаптопурин и тому подобное), агента, представляющего собой комплекс с платиной (такого как цисплатин, карбоплатин и оксалиплатин), антибиотических противоопухолевых лекарственных средств (актиномицина D, митомицина, доксорубицина, пингиангмицина, эпирубицина, пирарубицина, даунорубицина, блеомицина и тому подобного), противоопухолевых лекарственных средств природного происхождения (гомогаррингтонина и его производных, винкристина и его производных, гидроксикамптотецина и его производных, этопозида и его производных, виндезина и его производных, винбластина и его производных, винорелбина гидротартрата, таксола и его производных, колхицина и его производных, элемена и его производных и тому подобного), гормональных противоопухолевых лекарственных средств (таких как аминоглутетимид, тамоксифен, дексаметазон, дутастерид, флутамид, гонадорелин, лейпролида ацетат, летрозол и тому подобное), ингибиторов VEGFR или EGFR (таких как сунитиниб, сорафениб, иматиниб, гефитиниб, эрлотиниб, вандетаниб, пазопаниб, лапатиниб, канертиниб, афатиниб, мубритиниб, дазатиниб, нератиниб и тому подобное), противоопухолевых лекарственных средств, представляющих собой антитела (такие как трастузумаб, пертузумаб, ритуксимаб, панитумумаб, бевацизумаб, ипилимумаб, офатумумаб, рамуцирумаб и тому подобное), ингибиторов mTOR (таких как эверолимус, сиролимус, зотаролимус и тому подобное) и лекарственных средств для лечения опухоли головного мозга, таких как темозоломид и тому подобное.
Пероральный или непероральный путь может представлять собой доставку пациенту перорально или с помощью инъекции, пластыря, спрея и одного или большего количества других известных путей. Эффективное количество может включать в себя количество, эффективное для лечения, уменьшения, смягчения, ослабления, устранения одного или большего количества симптомов патологического состояния, которое пытаются вылечить или в альтернативном варианте пытаются предупредить, или количество, эффективное для дополнительного получения клинически выявляемых полезных изменений в патологическом состоянии или его последствиях.
В другом аспекте в данном изобретении предложено соединение для лечения расстройства, связанного с пролиферацией клеток, или его соответствующий таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси, причем структурная формула соединения представляет собой одну или большее количество структурных формул, выбранных из группы, состоящей из структурных формул I-V и VIII;
Расстройство, связанное с пролиферацией клеток, предпочтительно относится к раку млекопитающего или человека, более предпочтительно относится к раку человека, в том числе злокачественным солидным опухолям и злокачественным несолидным опухолям, в частности включая в себя, но не ограничиваясь ими, рак молочной железы, рак легких, рак предстательной железы, лейкоз, рак головного мозга, глиому и рак желудка; и/или
расстройство, связанное с пролиферацией клеток, представляет собой одно или большее количество заболеваний, выбранных из группы, состоящей из СПИДа, атеросклероза и рестеноза после имплантации сосудистого стента.
При описании данного изобретения, если не указано иное, «C1-C6 алкил» относится к линейному или разветвленному C1-C6 алкилу; «C1-C4 алкил» относится к линейному или разветвленному C1-C4 алкилу, предпочтительно метилу, этилу, пропилу или изопропилу. «C1-C6 алкокси» относится к C1-C6 линейному или разветвленному алкокси, предпочтительно C1-C4 линейному или разветвленному алкокси, более предпочтительно метокси, этокси, пропокси или 2-метилэтокси. «C3-C6 циклоалкил» относится к незамещенному C3-C6 циклоалкилу или C3-C6 циклоалкилу, замещенному C1-C4 алкилом и/или C1-C4 алкокси, предпочтительно незамещенному C3-C6 циклоалкилу или C3-C6 циклоалкилу, замещенному C1-C4 алкилом и/или C1-C4 алкокси, более предпочтительно циклопропилу, циклобутилу, метилциклопропилу, циклопентилу или циклогексилу. «Галоген» относится к брому, хлору или фтору. «C1-C6 галогеналкил» относится к линейному или разветвленному C1-C6 алкилу, замещенному бромом, хлором или фтором, предпочтительно линейному или разветвленному C1-C4 алкилу, замещенному хлором или фтором, более предпочтительно монофторметилу, дифторметилу, трифторметилу, монохлорметилу, дихлорметилу, трихлорметилу, 1-фторэтилу, 1-хлорпропилу, 1-хлорэтилу и 1-хлорпропилу.
Имеющиеся исследования свидетельствуют, что токсичность ингибиторов CDK главным образом связана с ингибированием CDK1 и других протеинкиназ, таких как Pim-1, треонин-/серинкиназа, кодируемые протоонкогеном с таким же названием. Следовательно, ожидается, что как соединения, ингибирующие CDK, они характеризуются более значимым различием между воздействием на CDK4/CDK6 и на CDK1 и другие киназы, т.е. селективным ингибированием CDK4/CDK6. Соединения, предложенные в данном изобретении, лучше или сопоставимы по активности с LY2835219, кандидатом, который на данный момент изучается в клиническом исследовании III фазы, а некоторые соединения демонстрируют лучшую селективность к киназам. Кроме того, предпочтительное соединение (полученное в Примере 17) хорошо абсорбируется при пероральном введении и характеризуется хорошим распределением между кровью и головным мозгом. Вышеприведенные результаты указывают на то, что соединения по данному изобретению являются перспективными для разработки новых лекарственных средств для лечения заболеваний, связанных с пролиферацией клеток, особенно злокачественных опухолей, особенно рака головного мозга.
Подробное описание сущности изобретения
Данное изобретение описано ниже на основании конкретных примеров. Специалисту в данной области техники должно быть понятно, что эти примеры являются только иллюстрацией изобретения и не предназначены для ограничения объема изобретения каким-либо способом.
Соединения формулы VI по данному изобретению являются основными промежуточными веществами для синтеза соединений формулы I, и их подвергают реакции кросс-сочетания, катализируемой палладием, с соединениями формулы VII в растворителе, что дает соединения формулы I.
Соединения формулы VI можно синтезировать с помощью следующей схемы реакций:
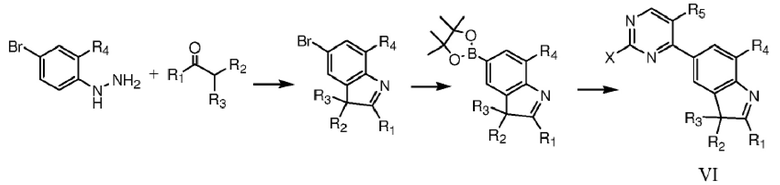
где R1, R2, R3, R4, R5, R6 и R7 определены, как указано выше, X представляет собой уходящую группу или аминогруппу.
Каждый из R1, R2 и R3 предпочтительно независимо выбран из атома водорода, незамещенной C1-C6 углеводородной группы или C1-C6 углеводородной группы, замещенной одним или большим количеством заместителей, выбранных из C1-C6 углеводородной группы, гидроксила или галогена.
Более предпочтительно каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенного линейного или разветвленного C1-C6 алкила, незамещенного линейного или разветвленного C2-C4 алкенила.
Наиболее предпочтительно каждый из R1, R2 и R3 независимо выбран из атома водорода, незамещенного линейного или разветвленного C1-C4 алкила.
Как альтернативный вариант, в качестве другого предпочтительного варианта, R1 определен, как указано выше, а R2 и R3 вместе с атомом C, к которому они присоединены, образуют насыщенное или ненасыщенное 3-7-членное кольцо; более предпочтительно R2 и R3 вместе с атомом C, к которому они присоединены, образует насыщенное 3-7-членное кольцо.
Каждый из R4 и R5 предпочтительно независимо представляет собой водород или фтор, и по меньшей мере один из R4 и R5 представляет собой фтор.
X предпочтительно представляет собой галоген или амино, более предпочтительно фтор, бром, хлор или амино.
R6 предпочтительно представляет собой атом водорода или C1-C4 алкил.
R7 предпочтительно представляет собой заместитель следующей структуры:
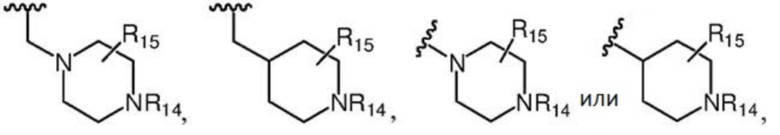
где каждый из R14 и R15 независимо выбран из группы, состоящей из атома водорода, C1-C4 алкила и C1-C4 гидроксиалкила.
Как альтернативный вариант, в качестве другого предпочтительного варианта реализации изобретения R6 и R7 вместе с атомом C, к которому они присоединены, образуют следующую химическую структуру:
, где R17 выбран из гидроксила или C1-C3 алкокси; более предпочтительно гидроксила.
Если не указано иное, все экспериментальные методики в следующих примерах представляют собой общепринятые методики. Если не указано иное, исходные химические вещества, реагенты и тому подобное, используемые в следующих примерах, представляют собой коммерчески доступные продукты.
Сокращения и их значения, встречающиеся в примерах данного изобретения, даны ниже:
ПЭ: петролейный эфир
ЭА: этилацетат
ДХМ: дихлорметан
MeOH: метанол
Pd (dppf)Cl2: [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид
Pd(PPh3)4: тетракис(трифенилфосфин)палладий
Pd2(dba)3: трис(дибензилиденацетон)дипалладий
NaHB(OAc)3: триацетоксиборгидрид натрия
LHMDS: гексаметилдисилазид лития
ДАФИ: флуоресцентный краситель ДАФИ.
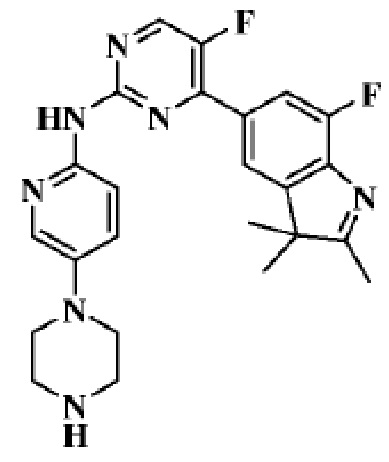
Пример 1
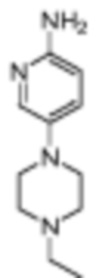
5-Фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
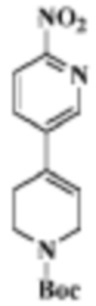
Стадия 1: трет-бутиловый эфир 4-(6-нитропиридин-3-ил)пиперазин-1-карбоновой кислоты
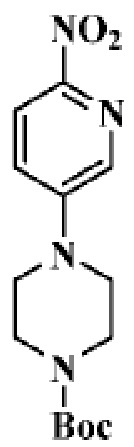
В реакционную колбу помещали 5-бром-2-нитропиридин (5,0 г, 24,63 ммоль), трет-бутиловый эфир пиперазин-1-карбоновой кислоты (5,04 г, 27,09 моль), ацетонитрил (30 мл) и диизопропилэтиламин (4,77 г, 36,94 ммоль). Смеси давали реагировать при нагревании с обратным холодильником в течение 2 ч. Продукт реакции подвергали испарению на роторном испарителе для удаления растворителя и отделяли с помощью колоночной хроматографии (ПЭ/ЭА=1:1 до ДХМ/MeOH=20:1) для получения целевого соединения (3,8 г, желтое твердое вещество).
МС (ЭРИ): рассчит. масса для C14H20N4O4 308,1, найденное m/z 309,1 [M+H]+.
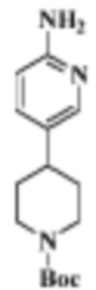
Стадия 2: трет-бутиловый эфир 4-(6-аминопиридин-3-ил)пиперазин-1-карбоновой кислоты
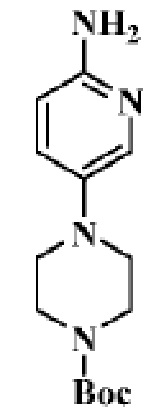
В реакционную колбу помещали трет-бутиловый эфир 4-(6-нитропиридин-3-ил)пиперазин-1-карбоновой кислоты (0,92 г, 3,0 ммоль), полученный на Стадии 1, этилацетат/метанол (10 мл/10 мл) и Pd/C (0,1 г) и продували газообразным водородом. Реакцию осуществляли при комнатной температуре в течение 2 ч. Продукт реакции отфильтровывали и концентрировали, чтобы получить целевое соединение (792 мг, желтоватое твердое вещество).
МС (ЭРИ): рассчит. масса для C14H22N4O2 278,2, найденное m/z 279,2 [M+H]+.
Стадия 3: получение 5-бром-7-фтор-2,3,3-триметил-3H-индола
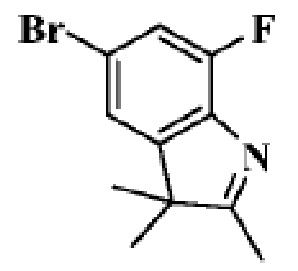
В реакционную колбу помещали (4-бром-2-фторфенил)гидразина гидрохлорид (1,0 г, 4,14 ммоль), уксусную кислоту (10 мл) и 3-метил-2-бутанон (0,32 г, 4,14 ммоль). Смеси давали реагировать при нагревании с обратным холодильником в течение 5 ч. Продукт реакции подвергали испарению на роторном испарителе для удаления растворителя, добавляли 20 мл воды и трижды экстрагировали этилацетатом (по 20 мл каждый раз). Объединенную органическую фазу однократно промывали 25 мл насыщенного водного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, подвергали испарению на роторном испарителе и разделяли с помощью колоночной хроматографии (ДХМ: MeOH=50:1 до 25:1), чтобы получить целевое соединение (420 мг, желтое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 7,23-7,21 (м, 2H), 2,30 (с, 3H), 1,32 (с, 6H).
МС (ЭРИ): m/z 258,0 [M+H]+.
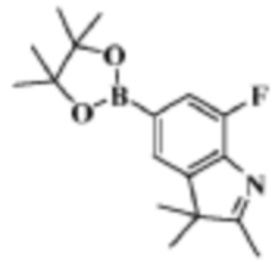
Стадия 4: получение 7-фтор-2,3,3-триметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3H-индола
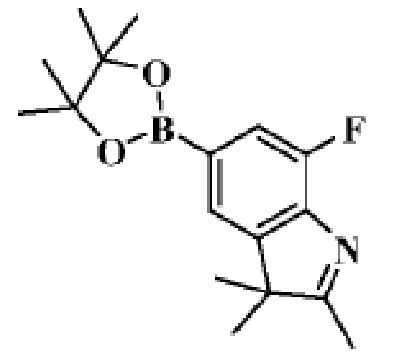
В реакционную колбу помещали 5-бром-7-фтор-2,3,3-триметил-3H-индол (400,0 мг, 1,56 ммоль), полученный на Стадии 3, 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолан) (436,5 мг, 1,71 ммоль), ацетат калия (306,3 мг, 3,12 ммоль), диоксан (10 мл) и Pd(dppf)Cl2 (228,7 мг,0,32 ммоль). Смесь нагревали до 90°C под защитой газообразного азота и давали реагировать в течение ночи. Продукт реакции охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и трижды экстрагировали этилацетатом (по 20 мл каждый раз). Объединяли органические фазы этилацетата, однократно промывали 25 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ: MeOH=50:1-30:1), чтобы получить целевое соединение (306,5 мг, желтое масло).
МС (ЭРИ): m/z 304,1 [M+H]+.
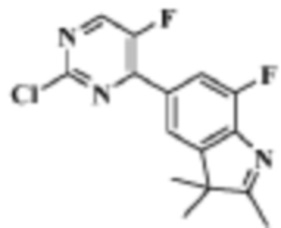
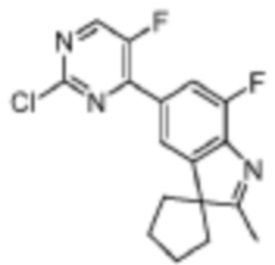
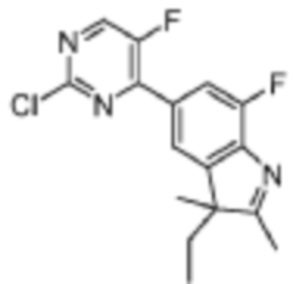
Стадия 5: получение 5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2,3,3-триметил-3H-индола
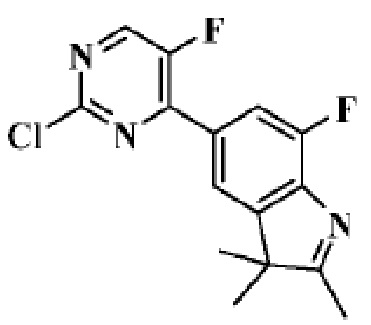
В реакционную колбу для микроволновой обработки помещали 7-фтор-2,3,3-триметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3H-индол (300 мг, 0,99 ммоль), полученный на Стадии 4, 2,4-дихлор-5-фторпиримидин (181,8 мг, 1,08 ммоль), фосфат калия (419,8 мг, 1,98 ммоль), смесь диоксан/вода (4 мл/1 мл) и Pd(PPh3)4 (114,5 мг, 0,09 ммоль). Реакцию под действием микроволнового излучения проводили под защитой газообразного азота при 130°C в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и экстрагировали трижды дихлорметаном (по 15 мл каждый раз). Органические фазы объединяли, однократно промывали 20 мл насыщенного водного раствора хлорида натрия, затем сушили с помощью безводного сульфата натрия, фильтровали, подвергали испарению на роторном испарителе и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ: MeOH=100:1-50:1), чтобы получить целевое соединение (301,2 мг, желтое твердое вещество).
МС (ЭРИ): рассчит. масса для C15H12ClF2N3 307,1, найденное m/z 308,1 [M+H]+.
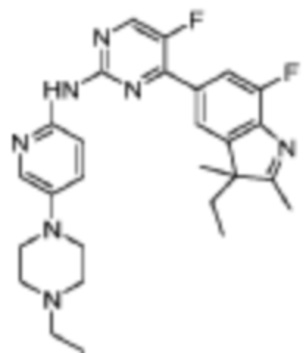
Стадия 6: получение трет-бутилового эфира 4-(6-((5-фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоновой кислоты
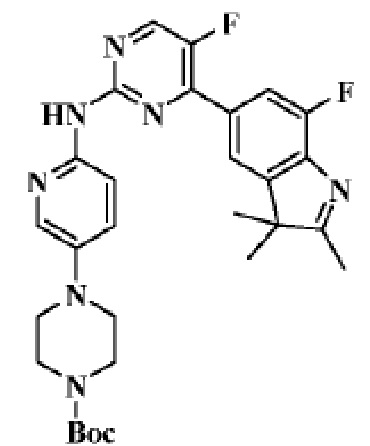
В реакционную колбу помещали 5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2,3,3-триметил-3H-индол (150,0 мг, 0,48 ммоль), полученный на Стадии 5, трет-бутиловый эфир 4-(6-аминопиридин-3-ил)пиперазин-1-карбоновой кислоты (135,8 мг, 0,48 ммоль), полученный на Стадии 2, карбонат цезия (371,6 мг, 0,96 ммоль), диоксан (3 мл), Pd2(dba)3 (44,7 мг, 0,05 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (30,4 мг, 0,05 ммоль). Смесь нагревали до 150°C под защитой газообразного азота, чтобы осуществить реакцию под действием микроволнового излучения в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и трижды экстрагировали дихлорметаном (по 10 мл каждый раз). Органические фазы объединяли, однократно промывали 30 мл насыщенного водного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH=50:1), чтобы получить целевое соединение (53,4 мг, желтое твердое вещество).
МС (ЭРИ): рассчит. масса для C29H33F2N7O2 549,3, найденное m/z 550,3 [M+H]+.
Стадия 7: получение 5-фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)-N-
(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-аминотрифторацетата
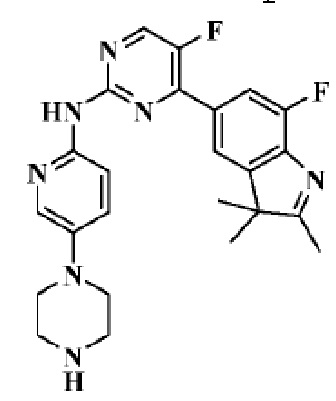
В реакционную колбу помещали трет-бутиловый эфир 4-(6-((5-фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоновой кислоты (30,0 мг, 0,054 ммоль), полученный на Стадии 6, дихлорметан (4 мл) и трифторуксусную кислоту (1 мл), и смесь перемешивали при комнатной температуре в течение 2 ч. Продукт реакции подвергали испарению на роторном испарителе для удаления растворителя, устанавливали pH 8 с помощью насыщенного водного раствора гидрокарбоната натрия и трижды экстрагировали дихлорметаном (по 5 мл каждый раз). Органические фазы объединяли, однократно промывали 10 мл насыщенного водного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, подвергали испарению на роторном испарителе и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH=20:1), чтобы получить целевое соединение (10,5 мг, желтое твердое вещество).
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,75 (ш. с., 1H), 8,65 (д, 1H, J=3,2 Гц), 8,02-7,94 (м, 3H), 7,82 (д, 1H, J=10,8 Гц), 7,43 (д, 1H, J=8,8 Гц), 3,09-3,02 (м, 4H), 2,84-2,83 (м, 4H), 2,31 (с, 3H), 1,34 (с, 6H).
МС (ЭРИ): рассчит. масса для C24H25F2N7 449,50, найденное m/z 450,2 [M+H]+.
Пример 2
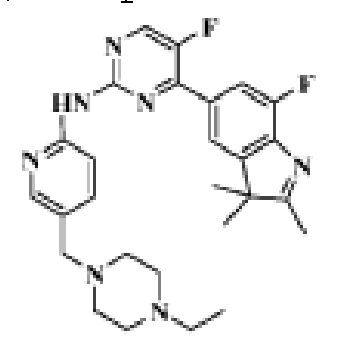
N-(5-((4-Этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)пиримидин-2-амино
Стадия 1: 1-((6-бромпиридин-3-ил)метил)-4-этилпиперазин
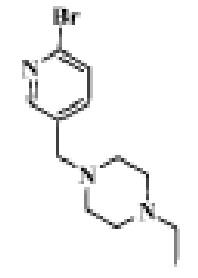
2-Бром-5-формилпиридин (1,5 г, 8,15 ммоль), 1-этилпиперазин (0,93 г, 8,15 ммоль) и дихлорметан (15 мл) помещали в реакционную колбу, а затем порциями добавляли NaHB(OAc)3 (2,58 г, 12,23 ммоль). Реакцию проводили при комнатной температуре в течение ночи. Продукт реакции отфильтровывали, концентрировали и отделяли с помощью колоночной хроматографии (ДХМ/MeOH=100:1 до 10:1), чтобы получить целевой продукт (1,64 г, желтое масло).
МС (ЭРИ): рассчит. масса для C12H18BrN3 285,1, найденное m/z 286,1 [M+H]+.
Стадия 2: 5-((4-этилпиперазин-1-ил)метил)пиридин-2-амино
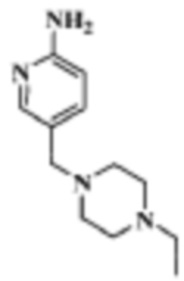
В реакционную колбу помещали 1-((6-бромпиридин-3-ил)метил)-4-этилпиперазин (2,84 г, 10 ммоль), полученный на Стадии 1, 2-(дициклогексилфосфино)бифенил (700 мг, 2 ммоль), Pd2(dba)3 (915 мг, 1 ммоль) и толуол (30 мл), добавляли LHMDS (1 н.) (20 мл, 20 ммоль) под защитой газообразного азота. Смесь нагревали до 80°C, и ей давали реагировать в течение ночи, затем охлаждали до комнатной температуры, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии (ДХМ/MeOH=100:1-10:1), что давало 1,52 г целевого продукта (коричневое твердое вещество).
МС (ЭРИ): рассчит. масса для C13H21N3 220,2, найденное m/z 221,2 [M+H]+.
Стадия 3: получение 5-бром-7-фтор-2,3,3-триметил-3H-индола
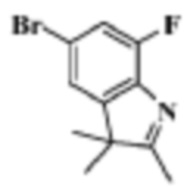
В реакционную колбу помещали (4-бром-2-фторбензол)гидразин (900,0 мг, 3,73 ммоль), уксусную кислоту (5 мл) и 3-метилбутан-2-он (353,3 мг, 4,09 ммоль). Смеси давали реагировать при нагревании с обратным холодильником в течение 5 ч. Продукт реакции подвергали испарению на роторном испарителе для удаления растворителя, добавляли 10 мл воды и трижды экстрагировали этилацетатом (по 20 мл каждый раз). Органические фазы объединяли, однократно промывали 25 мл насыщенного водного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и подвергали испарению на роторном испарителе. Остаток разделяли с помощью колоночной хроматографии на силикагеле (этилацетат:петролейный эфир=1: 50-1: 25), что давало 910 мг целевого соединения (желтое твердое вещество).
МС (ЭРИ): m/z 258,0 [M+H]+.
Стадия 4: получение 7-фтор-2,3,3-триметил-5-(4,4,5,5-тетраметил-
1,3,2-диоксаборолан-2-ил)-3H-индола
В реакционную колбу помещали 5-бром-7-фтор-2,3,3-триметил-3H-индол (1,0 г, 3,91 ммоль), полученный на Стадии 3, 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолан) (1,09 г, 4,29 ммоль), ацетат калия (770 мг, 7,82 ммоль), диоксан (10 мл), Pd(dppf)Cl2 (570 мг, 0,78 ммоль) и нагревали до 90°C под защитой газообразного азота для проведения реакции в течение ночи. Продукт реакции охлаждали до комнатной температуры, фильтровали, разбавляли 10 мл воды и трижды экстрагировали этилацетатом (по 20 мл каждый раз). Органические фазы объединяли, однократно промывали 25 мл насыщенного раствора хлорида натрия, сушили с помощью сульфата натрия, фильтровали, подвергали испарению на роторном испарителе и разделяли с помощью колоночной хроматографии на силикагеле (ЭА: ПЭ=1:100-1:20), что давало 1,02 г целевого соединения (желтое масло).
МС (ЭРИ): m/z 304,2 [M+H]+.
Стадия 5: получение 5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2,3,3-триметил-3H
-индола
В реакционную колбу для микроволновой обработки помещали 7-фтор-2,3,3-триметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3H-индол (1,0 г, 3,30 ммоль), полученный на Стадии 4, 2,4-дихлор-5-фторпиримидин (610 мг, 3,63 ммоль), фосфат калия (1,39 г, 6,60 ммоль), смесь диоксан/вода (8 мл/2 мл) и Pd(PPh3)4 (380 мг, 0,33 ммоль). Реакцию под действием микроволнового излучения проводили под защитой газообразного азота при 130°C в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и трижды экстрагировали дихлорметаном (по 15 мл каждый раз). Органические фазы объединяли, однократно промывали 20 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (ЭА: ПЭ=1:50 до 1:10), что давало целевое соединение (290,0 мг, желтое твердое вещество).
МС (ЭРИ): m/z 308,1 [M+H]+.
Стадия 6: получение N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-
(7-фтор-2,3,3-триметил-3H-индол-5-ил)пиримидин-2-амино
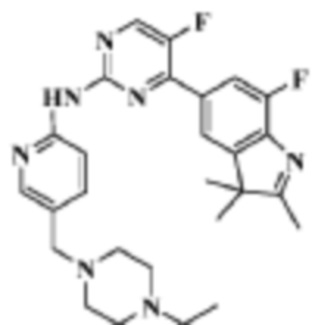
В реакционную колбу помещали 5-(2-хлор-5-фторпиримидин-4-ил)-7-фтор-2,3,3-триметил-3H-индол (290,0 мг, 0,94 ммоль), полученный на Стадии 5, 5-((4-этилпиперазин-1-ил)метил)пиридин-2-амин (228,6 мг, 1,04 ммоль), полученный на Стадии 2, фосфат калия (400,5 мг, 1,88 ммоль), 10 мл диоксана, Pd2(dba)3 (86,4 мг, 0,09 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (109,2 мг, 0,19 ммоль). Реакцию под действием микроволнового излучения проводили под защитой газообразного азота при 150°C в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и трижды экстрагировали дихлорметаном (по 10 мл каждый раз). Органические фазы объединяли, однократно промывали 30 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, подвергали испарению на роторном испарителе для удаления растворителя и разделяли с помощью колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), что давало целевое соединение (140,3 мг, желтое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 8,72 (с,1H), 8,49 (д, 1H, J=3,2 Гц), 8,38 (д, 1H, J=8,4 Гц), 8,31 (с, 1H), 7,92 (с, 1H), 7,89 (с, 1H), 7,73 (д, 1H, J=8,4 Гц), 3,52 (с, 2H), 2,54-2,41 (м, 10H), 2,38 (с, 3H), 1,40 (с, 6H), 1,10 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 492,2 [M+H]+.
Пример 3
N-(5-((4-Этилпиперазин-1-ил)метилпиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
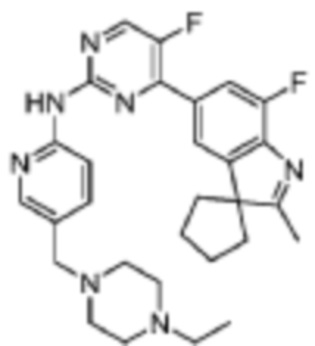
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 9,75 (с, 1H), 8,54 (д, 1H, J=3,2 Гц), 8,38-8,37 (м, 2H), 7,94 (с, 1H), 7,87 (д, 1H, J=10,8 Гц), 7,68 (д, 1H, J=8,4 Гц), 3,49 (с, 2H), 2,99-2,39 (м, 10H), 2,37 (с, 3H), 2,14-2,08 (м, 6H), 1,87-1,84 (м, 2H), 1,06 (т, 3H, J=6,4 Гц).
МС (ЭРИ): m/z 518,3 [M+H]+.
Пример 4
N-(5-((4-Этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклобутан-1,3'-индол]-5'-ил)аминопиримидин-2-амино
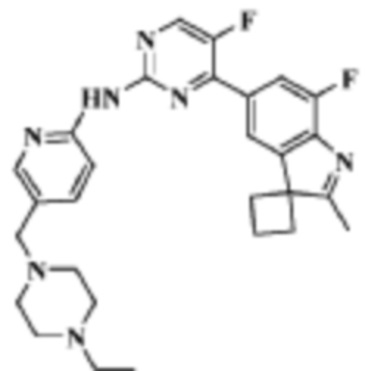
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 8,36-8,32 (м, 2H), 8,23 (с, 1H), 8,04 (с, 1H), 7,78-7,75 (м, 2H), 7,71 (д, 1H, J=8,4 Гц), 5,84-5,83 (м, 1H), 5,63-5,62 (м, 1H), 4,39-4,38 (м, 1H), 3,66-3,64 (м, 1H), 3,51 (с, 2H), 3,06-3,00 (м, 1H), 2,71-2,38 (м, 11H), 1,55 (с, 3H), 1,11 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 504,3 [M+H]+.
Пример 5
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фторпиримидин-2-амино
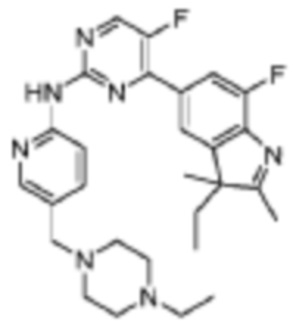
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 8,71 (с, 1H), 8,49 (д, 1H, J=3,6 Гц), 8,38 (д, 1H, J=8,4 Гц), 8,31 (с, 1H), 7,94 (д, 1H, J=11,2 Гц), 7,86 (с, 1H), 7,72 (дд, 1H, J=8,0, 1,2 Гц), 3,52 (с, 2H), 2,53-2,41 (м, 10H), 2,34 (с, 3H), 2,06-1,93 (м, 1H), 1,91-1,84 (м, 1H), 1,39 (с, 3H), 1,09 (т, 3H, J=7,2 Гц), 0,50 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 506,3 [M+H]+.
Пример 6
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
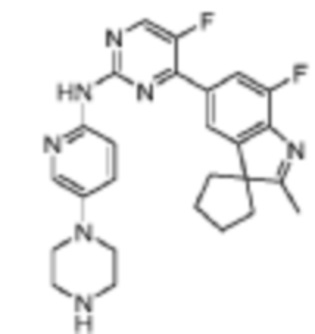
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 1.
1H-ЯМР (400 МГц, CDCl3) δ 8,67 (ш. с., 1H), 8,44 (д, 1H, J=3,6 Гц), 8,29 (д, 1H, J=9,2 Гц), 8,11 (д, 1H, J=2,4 Гц), 7,95 (с, 1H), 7,89 (д, 1H, J=10,8 Гц), 7,36 (дд, 1H, J=9,2, 2,8 Гц), 3,12-3,06 (м, 8H), 2,39 (с, 3H), 2,16-2,10 (м, 6H), 1,89-1,86 (м, 2H).
МС (ЭРИ): m/z 476,2 [M+H]+.
Пример 7
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
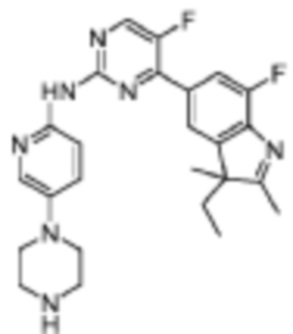
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 1.
1H-ЯМР (400 МГц, CDCl3) δ 9,33 (ш. с., 1H), 8,46 (д, 1H, J=3,6 Гц), 8,28 (д, 1H, J=8,8 Гц), 8,16 (д, 1H, J=2,4 Гц), 7,90 (д, 1H, J=10,8 Гц), 7,82 (с, 1H), 7,35 (дд, 1H, J=8,8 Гц, 2,8 Гц), 3,10-3,03 (м, 8H), 2,31 (с, 3H), 2,03-1,80 (м, 3H), 1,36 (с, 3H), 0,47 (т, 6H, J=7,6 Гц).
МС (ЭРИ): m/z 464,2 [M+H]+.
Пример 8
N-(5-((4-Этилпиперазин-1-ил)метил)пиридин-2-ил)-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
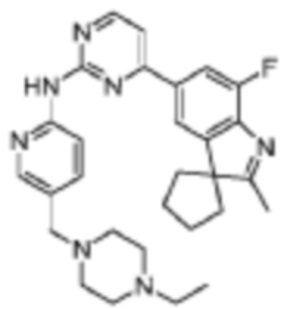
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 8,83 (с, 1H), 8,61 (д, 1H, J=5,2 Гц), 8,49 (д, 1H, J=8,8 Гц), 8,32 (д, 1H, J=1,2 Гц), 7,91 (д, 1H, J=1,2 Гц), 7,78 (д, 1H, J=10,8 Гц), 7,78 (дд, 1H, J=8,4, 1,6 Гц), 7,21 (д, 1H, J=5,2 Гц), 3,52 (с, 2H), 2,54-2,41 (м, 10H), 2,38 (с, 3H), 2,19-2,08 (м, 6H), 1,90-1,87 (м, 2H), 1,10 (т, 3H, J=6,8 Гц).
МС (ЭРИ): m/z 500,3 [M+H]+.
Пример 9
N-(5-((4-Этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
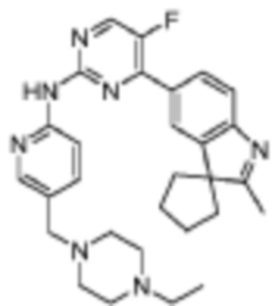
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 8,73 (ш. с., 1H), 8,47 (д, 1H, J=3,6 Гц), 8,43 (д, 1H, J=8,4 Гц), 8,30 (с, 1H), 8,15-8,13 (м, 2H), 7,70-7,64 (м, 2H), 3,52 (с, 2H), 2,53-2,37 (м, 10H), 2,31 (с, 3H), 2,21-2,06 (м, 6H), 1,89-1,86 (м, 2H), 1,10 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 500,3 [M+H]+.
Пример 10
4-(3,3-Диэтил-7-фтор-2-метил-3H-индол-5-ил)-N-(5-((4-этилпиперидин-1-ил)метил)пиридин-2-ил)-5-фторпиримидин-2-амино
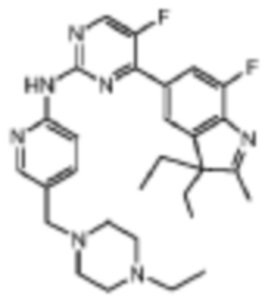
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, CDCl3) δ 9,19 (ш. с., 1H), 8,53 (с, 1H), 8,39-8,34 (м, 2H), 7,95 (д, 1H, J=11,2 Гц), 7,83 (с, 1H), 7,72 (д, 1H, J=8,0 Гц), 3,51 (с, 2H), 2,52-2,41 (м, 10H), 2,31 (с, 3H), 2,06-2,01 (м, 2H), 1,90-1,85 (м, 2H), 1,08 (т, 3H, J=6,8 Гц), 0,46 (т, 6H, J=6,8 Гц).
МС (ЭРИ): m/z 520,3 [M+H]+.
Пример 11
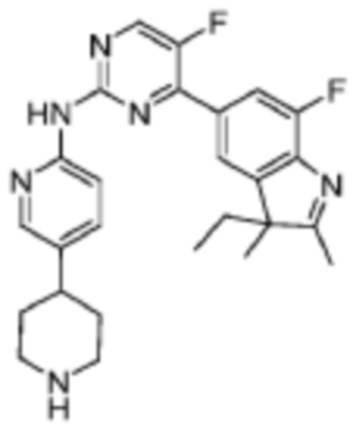
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(пиперидин-1-ил)пиридин-2-ил)пиримидин-2-амино
Стадия 1: трет-бутиловый эфир 6-нитро-3',6'-дигидро-[3,4'-бипиридин]-1'(2'H)-
карбоновой кислоты
В реакционную колбу помещали 5-бром-2-нитропиридин (20,3 г, 0,1 моль), трет-бутиловый эфир 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-
5,6-дигидропиридин-1(2H)-карбоновой кислоты (31 г, 0,1 моль), смесь диоксан/вода (250 мл/30 мл), карбонат цезия (66 г, 0,2 моль) и Pd(dppf)Cl2 (7,33 г, 0,01 моль) и продували газообразным азотом. Смесь нагревали до 85°C в течение 12 ч. Продукт реакции охлаждали до комнатной температуры, концентрировали и разделяли с помощью колоночной хроматографии (ПЭ/ЭА=1:1 до ДХМ/MeOH=20:1) для получения целевого продукта (11 г, желтое твердое вещество).
МС (ЭРИ): рассчит. масса для C15H19N3O4 305,1, найденное m/z 306,1 [M+H]+.
Стадия 2: трет-бутиловый эфир 4-(6-аминопиридин-3-ил)пиперидин-1-карбоновой кислоты
В реакционную колбу помещали трет-бутиловый эфир 6-нитро-3',6'-дигидро-[3,4'-бипиридин]-1'(2'H)-карбоновой кислоты (0,9 г, 3,0 ммоль), полученный на Стадии 1, этилацетат/метанол (10 мл/10 мл) и Pd/C (0,1 г). Туда подавали водород и реакцию осуществляли при комнатной температуре в течение 2 ч. Продукт реакции отфильтровывали и концентрировали, чтобы получить целевой продукт (790 мг, желтоватое твердое вещество).
МС (ЭРИ): рассчит. масса для C15H23N3O2 277,2, найденное m/z 278,2 [M+H]+.
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(пиперидин-1-ил)пиридин-2-ил)пиримидин-2-амино
Другие стадии осуществляли в соответствии со стадиями, подобными Стадиям 3-7 Примера 1, чтобы получить целевое соединение этого примера.
1H-ЯМР (400 МГц, CDCl3) δ 8,61 (ш. с., 1H), 8,46 (д, 1H, J=3,6 Гц), 8,34 (д, 1H, J=8,8 Гц), 8,26 (д, 1H, J=1,6 Гц), 7,93 (д, 1H, J=11,2 Гц), 7,64(д, 1H, J=8,0 Гц), 7,86 (с, 1H), 7,61 (дд, 1H, J=8,4 Гц, 2,0 Гц), 3,24-3,21 (м, 2H), 2,77 (т, 1H, J=10,8 Гц), 2,64-2,61 (м, 1H), 2,34 (с, 3H), 2,06-1,91 (м, 4H), 1,89-1,84 (м, 3H), 1,72-1,63 (м, 2H), 1,39 (с, 3H), 0,50 (т, 1H, J=7,2 Гц).
МС (ЭРИ): найденное m/z 463,3 [M+H]+.
Пример 12
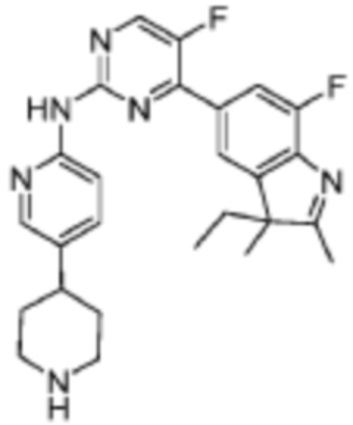
N-(5-(1-Этилпиперидин-4-ил)пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
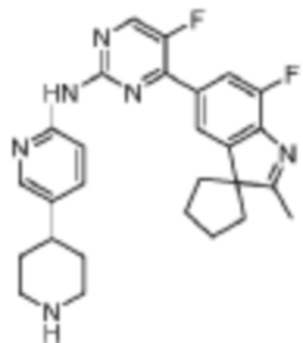
Это промежуточное соединение получали в соответствии со стадией, подобной стадии из Примера 11.
N-(5-(1-Этилпиперидин-4-ил)пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
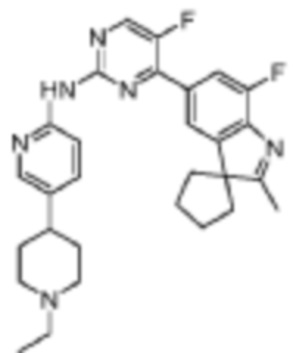
В реакционную колбу помещали 5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино, 50 мг (0,1 ммоль), полученный на вышеуказанной стадии, ацетальдегид, 26 мг (0,6 ммоль) и дихлорметан, 5 мл, и реакцию осуществляли при комнатной температуре в течение 0,5 ч. Затем добавляли триэтилборгидрид натрия, 60 мг (0,28 ммоль), и реакцию осуществляли при комнатной температуре в течение 2 ч. В реакционный раствор добавляли 20 мл насыщенного водного раствора карбоната натрия, а затем трижды экстрагировали дихлорметаном (по 10 мл каждый раз). Органические фазы объединяли, однократно промывали насыщенным раствором хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и разделяли с помощью колоночной хроматографии на силикагеле (ПЭ/ЭА=5:1), что давало целевое соединение (11 мг, выход 22%).
1H-ЯМР (400 МГц, CDCl3) δ 8,45 (д, 1H, J=3,6 Гц), 8,38 (с, 1H), 8,33 (д, 1H, J=8,8 Гц), 8,25 (с, 1H), 7,97 (с, 1H), 7,90 (д, 1H, J=11,2 Гц), 7,62 (дд, 1H, J=8,8, 2,0 Гц), 3,14-3,11 (м, 2H), 2,55-2,46 (м, 3H), 2,40 (с, 3H), 2,18-2,03 (м, 8H), 1,90-1,82 (м, 6H), 1,15 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 503,3 [M+H]+.
Пример 13
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
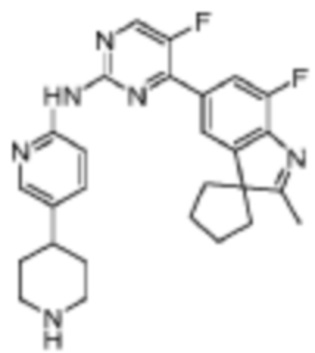
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 11.
1H-ЯМР (400 МГц, CDCl3) δ 9,14 (ш. с., 1H), 8,48 (д, 1H, J=3,2 Гц), 8,34-8,30 (м, 2H), 7,96 (с, 1H), 7,89 (д, 1H, J=10,8 Гц), 7,58 (д, 1H, J=8,4 Гц), 3,22-3,19 (м, 2H), 2,76 (т, 2H, J=11,6 Гц), 2,66-2,60 (м, 1H), 2,38 (с, 3H), 2,16-2,02 (м, 6H), 1,88-1,83 (м, 4H), 1,69-1,61 (м, 2H).
МС (ЭРИ): m/z 475,3 [M+H]+.
Пример 14
5-Фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
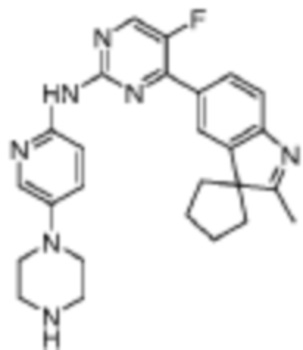
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 11.
1H-ЯМР (400 МГц, CDCl3) δ 9,28 (ш. с., 1H), 8,43 (д, 1H, J=3,6 Гц), 8,33 (д, 1H, J=9,2 Гц), 8,16-8,09 (м, 3H), 7,63 (д, 1H, J=8,0 Гц), 7,32 (дд, 1H, J=9,2 Гц, 2,8 Гц), 3,08-3,06 (м, 4H), 3,03-3,02 (м, 4H), 2,34 (с, 3H), 2,29-2,04 (м, 7H), 1,86-1,83 (м, 2H).
МС (ЭРИ): m/z 458,3 [M+H]+.
Пример 15
5-Фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
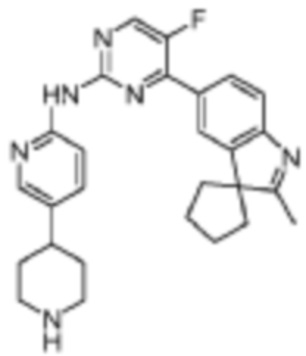
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 11.
1H-ЯМР (400 МГц, CDCl3) δ 8,99 (ш. с., 1H), 8,46 (д, 1H, J=3,6 Гц), 8,39 (д, 1H, J=8,4 Гц), 8,29 (д, 1H, J=1,6 Гц), 8,15-8,11 (м, 2H), 7,65 (д, 1H, J=8,0 Гц), 7,58 (дд, 1H, J=8,8 Гц, 2,0 Гц), 3,22-3,19 (м, 2H), 2,76 (т, 2H, J=10,4 Гц), 2,65-2,59 (м, 1H), 2,36 (с, 3H), 2,18-2,05 (м, 7H), 1,88-1,83 (м, 4H), 1,70-1,60 (м, 2H).
МС (ЭРИ): m/z 457,3 [M+H]+.
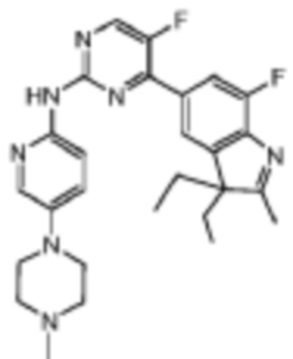
Пример 16
4-(3,3-Диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
Стадия 1: получение 5-бром-3,3-диэтил-7-фтор-2-метил-3H-индола
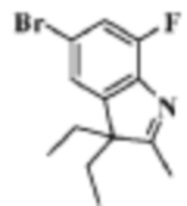
В реакционную колбу помещали 10 мл уксусной кислоты, 5,0 г (20,75 ммоль) (4-бром-2-фторфенил)гидразина гидрохлорида и 2,35 г (20,75 ммоль) 3-этилпентан-2-она, и смеси давали реагировать при нагревании с обратным холодильником в течение 5 ч. Продукт реакции подвергали испарению на роторном испарителе для удаления растворителя, добавляли 50 мл воды и трижды экстрагировали этилацетатом (по 50 мл каждый раз). Органические фазы объединяли, однократно промывали 50 мл раствора хлорида натрия, сушили с помощью сульфата натрия, фильтровали, подвергали испарению на роторном испарителе и разделяли с помощью колоночной хроматографии (ЭА: ПЭ=1:100-1:10), чтобы получить целевое соединение (2,5 г, желтое масло), выход 87,1%.
МС (ЭРИ): m/z 286,1 [M+H]+.
Стадия 2: получение 3,3-диэтил-7-фтор-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3H-индола
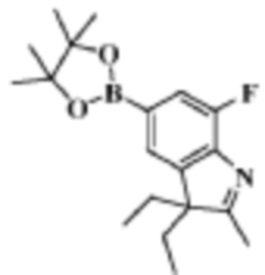
В реакционную колбу помещали 2,0 г (7,07 моль) 5-бром-3,3-диэтил-7-фтор-2-метил-3H-индола, полученного на Стадии 1, 1,97 г (7,77 ммоль) 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолана), 1,38 г (1,41 ммоль) ацетата калия, 10 мл 1,4-диоксана и 1,03 г (1,41 ммоль) Pd(dppf)Cl2 и смесь нагревали до 90°C под защитой газообразного азота для осуществления реакции в течение ночи. Продукт реакции охлаждали до комнатной температуры, фильтровали, разбавляли 10 мл воды и трижды экстрагировали этилацетатом (по 10 мл каждый раз). Органические фазы объединяли, однократно промывали 15 мл раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (ЭА: ПЭ=1:50 до 1:10), что давало целевое соединение (2,0 г, желтое масло), выход 86,96%.
МС (ЭРИ): m/z 332,3 [M+H]+.
Стадия 3: 5-(2-хлор-5-фторпиримидин-4-ил)-3,3-диэтил-7-фтор-2-метил-3H-индол
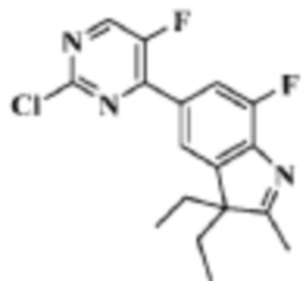
В реакционную колбу помещали 2,0 г (6,05 ммоль) 3,3-диэтил-7-фтор-2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3H-индола, полученного на Стадии 2, 1,10 г (6,65 ммоль) 2,4-дихлор-5-фторпиримидина, 2,56 г (12,1 ммоль) фосфата калия, 20 мл/5 мл диоксана/воды, 0,69 г (0,61 ммоль) Pd (PPh3)4, и смесь нагревали до 120°C под защитой газообразного азота, и давали реагировать в течение 2 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, разбавляли 10 мл воды и трижды экстрагировали 50 мл дихлорметана. Органические фазы объединяли, однократно промывали 20 мл раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и разделяли с помощью колоночной хроматографии на силикагеле (ЭА: ПЭ=1:100-1:10), что давало целевое соединение (1,2 г, желтое твердое вещество), выход 59,4%.
МС (ЭРИ): m/z 336,1 [M+H]+.
Стадия 4: получение трет-бутилового эфира 4-(6-((4-(3,3-диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоновой кислоты
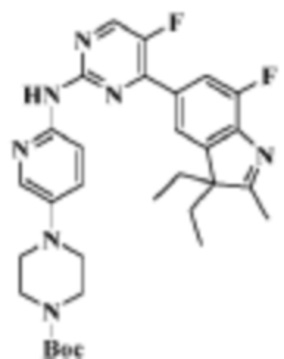
В реакционную колбу помещали 400,0 мг (1,19 ммоль) 5-(2-хлор-5-фторпиримидин-4-ил)-3,3-диэтил-7-фтор-2-метил-3H-индола, полученного на Стадии 3, 331,9 мг (1,19 ммоль) трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилата, полученного в соответствии со Стадиями 1-2 Примера 1, 776,2 мг (2,38 ммоль) карбоната цезия, 10 мл 1,4-диоксана, 109,5 мг (0,12 ммоль) Pd2(dba)3 и 69,0 мг (0,12 ммоль) 4,5-бис(дифенилфосфино)-9,9-диметилксантена и продували азотом. Смеси давали реагировать под действием микроволнового излучения при 130°C в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, разбавляли 10 мл воды, трижды экстрагировали 10 мл дихлорметана. Органические фазы объединяли, однократно промывали 30 мл раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью ТСХ, чтобы получить целевое соединение (253,1 мг, желтое твердое вещество), выход 36,74%.
МС (ЭРИ): m/z 578,3 [M+H]+.
Стадия 5:
4-(3,3-диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
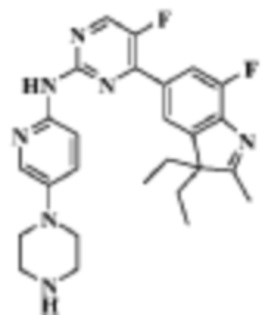
В реакционную колбу помещали трет-бутиловый эфир 4-(6-((4-(3,3-диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоновой кислоты, 250,0 мг (0,43 ммоль), полученный на Стадии 4, дихлорметан, 4 мл и ТФУ, 1 мл. Смесь перемешивали при комнатной температуре в течение 2 ч, а затем удаляли растворитель. Остаток доводили до pH 8 с помощью 5 мл насыщенного раствора гидрокарбоната натрия и трижды экстрагировали дихлорметаном (по 5 мл каждый раз). Органические фазы объединяли, однократно промывали 10 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и разделяли с помощью ТСХ, что давало целевое соединение (201,2 мг, желтое твердое вещество), выход 97,6%.
МС (ЭРИ): m/z 478,3 [M+H]+.
Стадия 6:
4-(3,3-диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
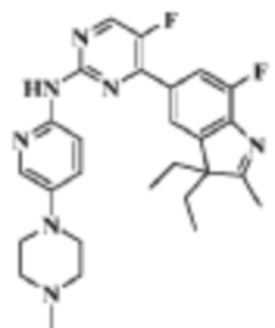
В реакционную колбу помещали 50,0 мг (0,10 ммоль) 4-(3,3-диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино, 30,8 мг (1,0 ммоль) формальдегида, 2 мл 1-этил-(3-диметиламинопропил)карбодиимида, 65,3 мг (0,3 ммоль) триэтилборгидрида натрия, и смеси давали реагировать в течение ночи. Для гашения реакции добавляли 2 мл метанола и продукт реакции трижды экстрагировали дихлорметаном (по 5 мл каждый раз). Органическую фазу однократно промывали 10 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью ТСХ, чтобы получить целевое соединение (20,3 мг, желтое твердое вещество), выход 39,4%.
1H-ЯМР (400 МГц, CDCl3) δ 9,57 (ш. с., 1H), 8,47 (д, 1H, J=3,2 Гц), 8,28 (д, 1H, J=8,8 Гц), 8,18 (д, 1H, J=2,0 Гц), 7,91 (д, 1H, J=11,2 Гц), 7,78 (с, 1H), 7,33 (д, 1H, J=8,8 Гц), 3,16-3,15 (м, 4H), 2,58-2,57 (м, 4H), 2,33 (с, 3H), 2,26 (с, 3H), 2,04-1,95 (м, 2H), 1,85-1,78 (м, 7H), 0,43 (т, 6H, J=7,2 Гц).
МС (ЭРИ): m/z 492,3 [M+H]+.
Пример 17
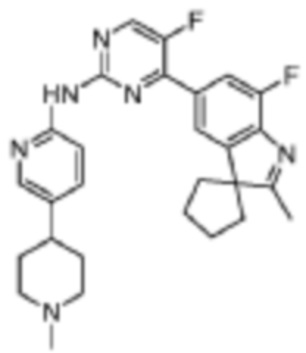
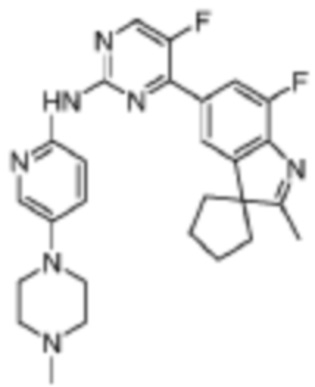
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
Промежуточное соединение 5'-(2-хлор-5-фторпиримидин-4-ил)-7'-фтор-2'-метилспиро[циклопентан-1,3'-индол] получали в соответствии со стадиями, подобными стадиям Примера 1.
Стадия 1: 1'-метил-6-нитро-1',2',3',6'-тетрагидро-3,4'-бипиридин
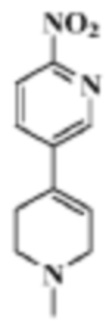
В реакционную колбу помещали 5-бром-2-нитропиридин (20,3 г, 0,1 моль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин (22,3 г, 0,1 моль), смесь диоксан/вода (250 мл/30 мл), карбонат цезия (66 г, 0,2 моль) и Pd(dppf)Cl2 (7,33 г, 0,01 моль). Смесь перемешивали для проведения реакции при 85°C под защитой газообразного азота в течение 12 ч. Продукт реакции охлаждали до комнатной температуры, концентрировали и разделяли с помощью колоночной хроматографии (ПЭ/ЭА=1:1 до ДХМ/MeOH=20:1), что давало целевой продукт (5,7 г, белое твердое вещество).
МС (ЭРИ): рассчит. масса для C11H13N3O2 219,1, найденное m/z 220,1 [M+H]+.
Стадия 2: 5-(1-метилпиперидин-4-ил)пиридин-2-амино
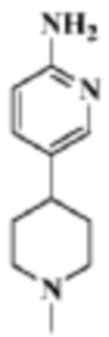
В реакционную колбу помещали 1'-метил-6-нитро-1',2',3',6'-тетрагидро-3,4'-бипиридин (657 мг, 3,0 ммоль), полученный на Стадии 1, этилацетат/метанол (10 мл/10 мл) и Pd/C (0,1 г). В смесь подавали газообразный водород и смесь перемешивали для проведения реакции в течение 2 ч, фильтровали и концентрировали, что давало целевой продукт (550 мг, белое твердое вещество).
МС (ЭРИ): рассчит. масса для C11H17N3 191,1, найденное m/z 192,2 [M+H]+.
Стадия 3:
5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
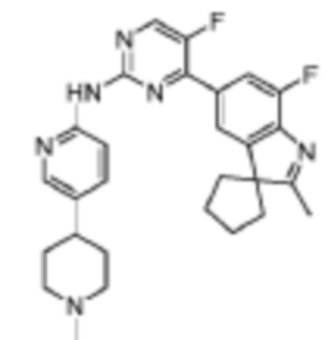
В реакционную колбу помещали промежуточное соединение 5'-(2-хлор-5-фторпиримидин-4-ил)-7'-фтор-2'-метилспиро[циклопентан-1,3'-индол] (150,0 мг, 0,45 ммоль), 5-(1-метилпиперидин-4-ил)пиридин-2-амино (86,6 мг, 0,45 ммоль), полученный на Стадии 2, карбонат цезия (293,2 мг, 0,9 ммоль), диоксан (3 мл), Pd2(dba)3 (44,7 мг, 0,05 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (30,4 мг, 0,05 ммоль). Смесь нагревали до 150°C под защитой газообразного азота для проведения реакции под действием микроволнового излучения в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, фильтровали, добавляли 10 мл воды и трижды экстрагировали дихлорметаном (по 10 мл каждый раз). Объединенную органическую фазу однократно промывали 30 мл насыщенного водного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол=50:1), что давало целевое соединение (51,1 мг, желтое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ 9,38 (ш. с., 1H), 8,49 (д, 1H, J=3,2 Гц), 8,34-8,33 (м, 2H), 7,96 (с, 1H), 7,88 (д, 1H, J=11,2 Гц), 7,58 (дд, 1H, J=8,8 Гц, 1,6 Гц), 3,01-2,98 (м, 2H), 2,52-2,44 (м, 1H), 2,38 (с, 3H), 2,34 (с, 3H), 2,25-2,04 (м, 8H), 1,87-1,76 (м, 6H).
МС (ЭРИ): m/z 489,3 [M+H]+.
Пример 18
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
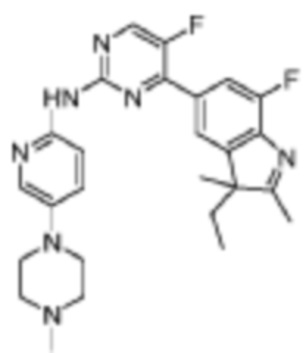
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 16.
1H-ЯМР (400 МГц, CDCl3) δ 9,48 (ш. с., 1H), 8,47 (д, 1H, J=3,2 Гц), 8,28 (д, 1H, J=9,2 Гц), 8,17 (с, 1H), 7,90 (д, 1H, J=10,8 Гц), 7,82 (с, 1H), 7,34 (д, 1H, J=8,4 Гц), 3,17-3,16 (м, 4H), 2,59-2,58 (м, 4H), 2,35 (с, 3H), 2,31 (с, 3H), 2,01-1,96 (м, 1H), 1,87-1,82 (м, 1H), 1,35 (с, 3H), 0,47 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 478,3 [M+H]+.
Пример 19
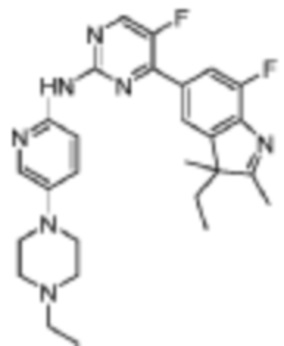
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-5-фторпиримидин-2-амино
Стадия 1: получение 1-этил-4-(6-нитропиридин-3-ил)пиперазина
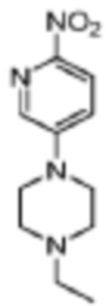
В реакционную колбу помещали 4,00 г (19,7 ммоль) 5-бром-2-нитропиридина, 3,40 г (2,98 ммоль) 1-этилпиперазина, 4,10 г (29,6 ммоль) карбоната калия, 0,4 г (1,2 ммоль) йодида тетрабутиламмония и 40 мл ДМСО и проводили реакцию при 80°C в течение 16 ч. Затем реакционный раствор выливали в ледяную воду и трижды экстрагировали дихлорметаном (по 20 мл каждый раз). Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии (ДХМ/MeOH=100:1-10:1), что давало 3,59 г желтого твердого вещества, выход 56,1%.
МС (ЭРИ): m/z 237,2 [M+H]+.
Стадия 2: получение 5-(4-этилпиперазин-1-ил)пиридин-2-амино
650 мг (2,13 ммоль) 1-этил-4-(6-нитропиридин-3-ил)пиперазина, полученного на Стадии 1, растворяли в 45 мл метанола, добавляли 10 %-ный палладий на угле (250 мг, катализатор), газообразную среду трижды заменяли газообразным водородом и реакцию осуществляли при комнатной температуре в течение 12 ч в атмосфере газообразного водорода под давлением 3 атм. После завершения реакции продукт реакции отфильтровывали с помощью небольшого количества диатомита и осадок на фильтре однократно промывали 20 мл смеси растворителей дихлорметана и метанола (об./об.=10:1). Затем фильтрат собирали и концентрировали при пониженном давлении, что давало 559 мг неочищенного продукта, представляющего собой целевое соединение (прозрачное и вязкое вещество), которое использовали для следующей реакции непосредственно без дополнительной очистки.
МС (ЭРИ): m/z 207,1 [M+H]+.
Стадия 3: получение 5-(2-хлор-5-фторпиримидин-4-ил)-3-этил-7-фтор-2,3-диметил -3H-индола
Это промежуточное соединение получали с помощью той же методики, что и в Стадиях 1-3 Примера 16.
Стадия 4: получение
4-(3-этил-7-фтор-2,3-диметил-3H-индол-5-ил)-N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-5-фторпиримидин-2-амино
В реакционную колбу помещали 321 мг (1 ммоль) 5-(2-хлор-5-фторпиримидин-4-ил)-3-этил-7-фтор-2,3-диметил-3H-индола, полученного на Стадии 3, 206 мг (1 ммоль) 5-(4-этилпиперазин-1-ил)пиридин-2-амино, полученного на Стадии 2, 2 мл 1,4-диоксана, 650 мг (2 ммоль) Cs2CO3, 91 мг (0,1 ммоль) Pd2(dba)3 и 58 мг (0,1 ммоль) дифенилфосфина. Смесь нагревали до 120°C для проведения реакции под действием микроволнового излучения в течение 1 ч. Продукт реакции охлаждали до комнатной температуры, добавляли 10 мл воды, а затем трижды экстрагировали этилацетатом (по 40 мл каждый раз). Органические фазы объединяли, однократно промывали 40 мл насыщенного раствора хлорида натрия, сушили с помощью сульфата натрия, фильтровали, концентрировали при пониженном давлении и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH=10:1), что давало целевое соединение (49 мг, желтое твердое вещество), выход 10 %.
1H-ЯМР (400 МГц, CDCl3) δ 9,25 (ш. с., 1H), 8,46 (д, 1H, J=3,2 Гц), 8,28 (д, 1H, J=9,2 Гц), 8,17 (д, 1H, J=2,0 Гц), 7,90 (д, 1H, J=11,2 Гц), 7,82 (с, 1H), 7,35 (дд, 1H, J=9,2 Гц, 2,8 Гц), 3,20-3,18 (м, 4H), 2,64-2,61 (м, 4H), 2,51 (к, 2H, J=6,8 Гц), 2,31 (с, 3H), 2,03-1,94 (м, 1H), 1,89-1,82 (м, 1H), 1,36 (с, 3H), 1,13 (т, 3H, J=7,2 Гц), 0,48 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 492,3 [M+H]+.
Пример 20
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 16.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,77 (ш. с., 1H), 8,63 (д, 1H, J=4,0 Гц), 8,02-7,98 (м, 3H), 7,82 (д, 1H, J=11,2 Гц), 7,41 (дд, 1H, J=8,8 Гц, 2,8 Гц), 3,13-3,11 (м, 4H), 2,49-2,46 (м, 4H), 2,33 (с, 3H), 2,26 (с, 3H), 2,23-2,07 (м, 6H), 1,76-1,74 (м, 2H).
МС (ЭРИ): m/z 490,3 [M+H]+.
Пример 21
N-(5-(1-Этилпиперидин-4-ил)пиридин-2-ил)-5-фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
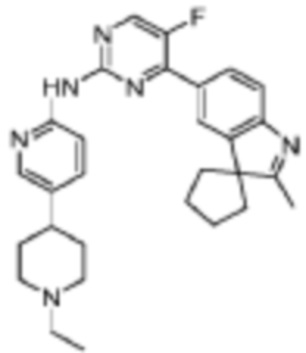
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 12.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,91 (ш. с., 1H), 8,65 (д, 1H, J=4,0 Гц), 8,28-8,14 (м, 3H), 8,04 (д, 1H, J=8,4 Гц), 7,43 (дд, 1H, J=8,4 Гц, 2,0 Гц), 7,60 (д, 1H, J=8,0 Гц), 3,00-2,97 (м, 4H), 2,38-2,33 (м, 2H), 2,30 (с, 3H), 2,08-2,07 (м, 6H), 1,99-1,94 (м, 2H), 1,77-1,64 (м, 6H), 1,02 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 485,3 [M+H]+.
Пример 22
N-(5-(4-Этилпиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
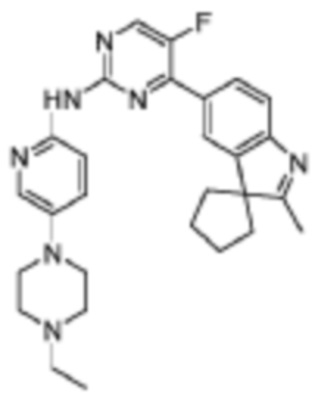
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 19.
1H-ЯМР (400 МГц, CDCl3) δ 8,87 (ш. с., 1H), 8,42 (д, 1H, J=3,6 Гц), 8,33 (д, 1H, J=9,2 Гц), 8,13-8,10 (м, 3H), 7,65 (д, 1H, J=8,0 Гц), 7,35 (дд, 1H, J=8,8 Гц, 2,4 Гц), 3,19-3,18 (м, 4H), 2,64-2,63 (м, 4H), 2,53 (к, 2H, J=6,8 Гц), 2,35 (с, 3H), 2,27-2,06 (м, 6H), 1,88-1,85 (м, 2H), 1,14 (т, 3H, J=6,8 Гц).
МС (ЭРИ): m/z 486,4 [M+H]+.
Пример 23
N-(5-(4-Метилпиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
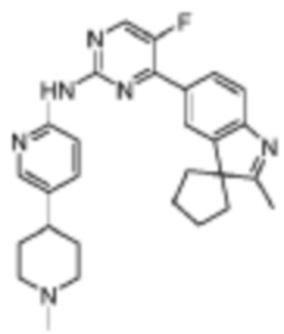
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, CDCl3) δ 9,24 (ш. с., 1H), 8,47 (д, 1H, J=3,2 Гц), 8,38 (д, 1H, J=8,8 Гц), 8,31 (с, 1H), 8,14-8,09 (м, 2H), 7,64 (д, 1H, J=8,4 Гц), 7,57 (дд, 1H, J=8,4 Гц, 2,4 Гц), 3,00-2,98 (м, 2H), 2,49-2,46 (м, 1H), 2,35 (с, 3H), 2,33 (с, 3H), 2,16-1,99 (м, 8H), 1,85-1,76 (м, 6H).
МС (ЭРИ): m/z 471,3 [M+H]+.
Пример 24
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
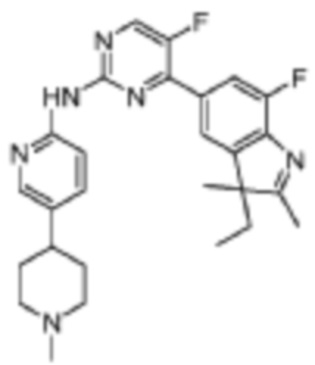
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, CDCl3) δ 9,74 (ш. с., 1H), 8,50 (д, 1H, J=3,6 Гц), 8,35-8,32 (м, 2H), 7,88-7,82 (м, 2H), 7,57 (д, 1H, J=8,8 Гц), 2,97-2,95 (м, 2H), 2,49-2,42 (м, 1H), 2,30 (с, 6H), 2,24-1,93 (м, 3H), 1,88-1,74 (м, 5H), 1,35 (с, 3H), 0,47 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 477,3 [M+H]+.
Пример 25
5-Фтор-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)-4-(2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
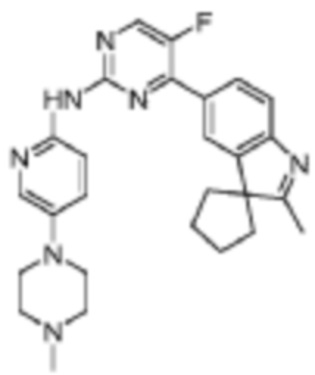
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 16.
1H-ЯМР (400 МГц, CDCl3) δ 9,28 (ш. с., 1H), 8,44 (д, 1H, J=3,6 Гц), 8,33 (д, 1H, J=9,2 Гц), 8,18 (д, 1H, J=2,0 Гц), 8,12-8,09 (м, 2H), 7,64 (д, 1H, J=8,0 Гц), 7,33(дд, 1H, J=9,2 Гц, 2,4 Гц), 3,16-3,14 (м, 4H), 2,59-2,58 (м, 4H), 2,35 (с, 3H), 2,34 (с, 3H), 2,15-2,06 (м, 6H), 1,87-1,83 (м, 2H).
МС (ЭРИ): m/z 472,3 [M+H]+.
Пример 26
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-N-(5-(1-этилпиперидин-4-ил)пиридин-2-ил)-5-фторпиримидин-2-амино
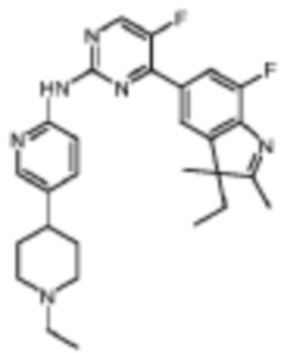
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 12.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,99 (ш. с., 1H), 8,70 (д, 1H, J=3,2 Гц), 8,19 (с, 1H), 8,14 (д, 1H, J=8,8 Гц), 7,92 (с, 1H), 7,85 (д, 1H, J=11,2 Гц), 7,69 (д, 1H, J=8,4 Гц), 3,04-3,02 (м, 2H), 2,42-2,41 (м, 2H), 2,28 (с, 3H), 2,04-1,99 (м, 3H), 1,89-1,84 (м, 1H), 1,78-1,63 (м, 4H), 1,33 (с, 3H), 1,04 (т, 3H, J=6,8 Гц), 0,36 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 491,3 [M+H]+.
Пример 27
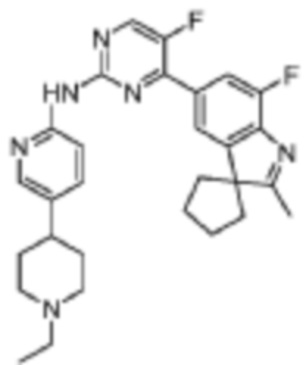
N-(5-(4-Этилпиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
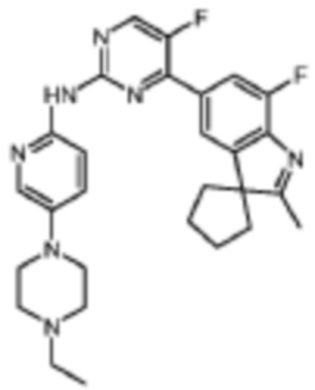
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 19.
1H-ЯМР (400 МГц, CDCl3) δ 8,92 (ш. с., 1H), 8,44 (д, 1H, J=3,6 Гц), 8,29 (д, 1H, J=9,2 Гц), 8,15 (д, 1H, J=2,8 Гц), 7,94 (с, 1H), 7,89 (д, 1H, J=11,2 Гц), 7,36 (дд, 1H, J=9,2 Гц, 2,8 Гц), 3,21-3,19 (м, 4H), 2,65-2,63 (м, 4H), 2,51 (к, 2H, J=7,2 Гц), 2,38 (с, 3H), 2,22-2,07 (м, 6H), 1,89-1,86 (м, 2H), 1,15 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 504,3 [M+H]+.
Пример 28
2-(4-(6-((5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-ил)этанол
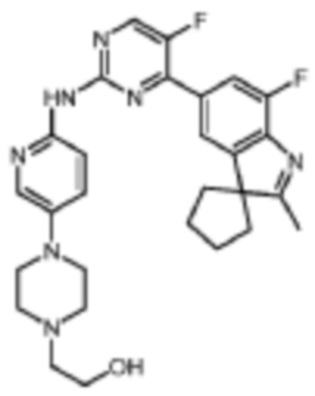
В реакционную колбу помещали 5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино, 20 мг (0,04 ммоль, полученный по той же методике, что и в Примере 6), 16 мг (0,12 ммоль) 2-бромэтанола, 2 мл ДМФА и 17 мг (0,12 ммоль) карбоната цезия. Смесь нагревали до 80°C и проводили реакцию в течение 1 ч, а затем продукт реакции охлаждали до комнатной температуры, добавляли 10 мл воды и трижды экстрагировали этилацетатом (по 40 мл каждый раз). Органические фазы объединяли, однократно промывали 40 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH=10:1), что давало целевое соединение (10 мг, желтое твердое вещество), выход 55 %.
1H-ЯМР (400 МГц, CDCl3) δ 8,52 (ш. с., 1H), 8,43 (д, 1H, J=2,8 Гц), 8,29 (д, 1H, J=9,2 Гц), 8,09 (с, 1H), 7,94 (с, 1H), 7,89 (д, 1H, J=10,8 Гц), 7,36 (д, 1H, J=8,8 Гц), 3,70-3,68 (м, 2H), 3,19-3,18 (м, 4H), 2,71-2,63 (м, 7H), 2,39 (с, 3H), 2,25-2,00 (м, 6H), 1,90-1,87 (м, 2H).
МС (ЭРИ): m/z 520,3 [M+H]+.
Пример 29
4-(3,3-Диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(пиперазин-4-ил)пиридин-2-ил)пиримидин-2-амино
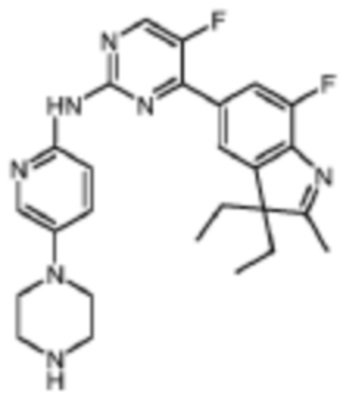
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 1.
1H-ЯМР (400 МГц, CDCl3) δ 8,66 (ш. с., 1H), 8,45 (д, 1H, J=3,6 Гц), 8,29 (д, 1H, J=9,2 Гц), 8,11 (д, 1H, J=2,8 Гц), 7,94 (д, 1H, J=11,2 Гц), 7,81 (с, 1H), 7,37 (дд, 1H, J=8,8 Гц, 2,4 Гц), 3,13-3,12 (м, 4H), 3,08-3,07 (м, 4H), 2,30 (с, 3H), 2,08-1,99 (м, 2H), 1,91-1,82 (м, 3H), 0,46 (т, 6H, J=7,6 Гц).
МС (ЭРИ): m/z 478,3 [M+H]+.
Пример 30
2-(4-(6-((5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пирмидин-2-ил)амино)пиридин-3-ил)пиперидин-1-ил)этанол
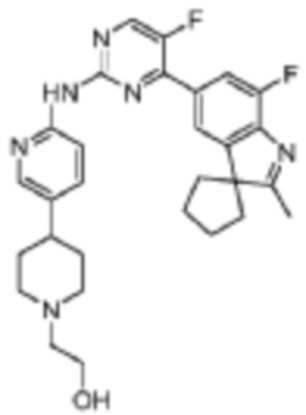
В реакционную колбу помещали 20 мг (0,04 ммоль) 5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино, полученного с помощью тех же методик, что и в Примере 13, 16 мг (0,12 ммоль) 2-бромэтанола, 2 мл ДМФА и 17 мг (0,12 ммоль) карбоната цезия, и смесь нагревали до 80°C, и реакцию проводили в течение 1 ч. Затем продукт реакции охлаждали до комнатной температуры, добавляли 10 мл воды и трижды экстрагировали этилацетатом (по 40 мл каждый раз). Органические фазы объединяли, однократно промывали 40 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, и концентрировали при пониженном давлении, и разделяли с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH=10:1), что давало целевое соединение (10 мг, желтое твердое вещество), выход 55%.
1H-ЯМР (400 МГц, CDCl3) δ 8,63 (ш. с., 1H), 8,46 (д, 1H, J=3,2 Гц), 8,34 (д, 1H, J=8,4 Гц), 8,26 (с, 1H), 7,96 (с, 1H), 7,90 (д, 1H, J=10,8 Гц), 7,60 (д, 1H, J=8,0 Гц), 3,67-3,66 (м, 2H), 3,08-3,06 (м, 2H), 2,60-2,52 (м, 4H), 2,39 (с, 3H), 2,25-2,11 (м, 8H), 1,89-1,83 (м, 4H), 1,80-1,77 (м, 2H).
МС (ЭРИ): m/z 519,3 [M+H]+.
Пример 31
5-Фтор-4-(7-фтор-2,3,3-триметил-3H-индол-5-ил)-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
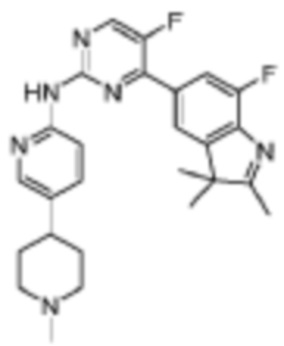
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, метанол-d4) δ 8,57 (ш. с., 1H), 8,37 (д, 1H, J=3,6 Гц), 8,18 (с, 1H), 8,13 (д, 1H, J=8,8 Гц), 7,84 (с, 1H), 7,70 (д, 1H, J=11,2 Гц), 7,51 (д, 1H, J=7,2 Гц), 3,21-3,18 (м, 2H), 2,60-2,54 (м, 4H), 2,50-2,44 (м, 2H), 2,37 (с, 3H), 1,90-1,79 (м, 4H), 1,38 (с, 6H).
МС (ЭРИ): m/z 463,3 [M+H]+.
Пример 32
4-(3-Этил-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
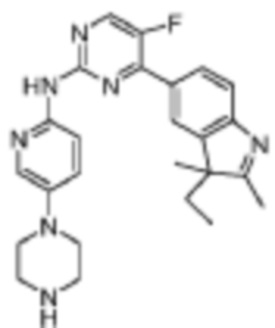
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 6.
1H-ЯМР (400 МГц, CDCl3) δ 9,68 (ш. с., 1H), 8,44 (д, 1H, J=3,6 Гц), 8,32 (д, 1H, J=8,8 Гц), 8,18 (д, 1H, J=2,4 Гц), 8,11 (д, 1H, J=8,0 Гц), 7,98 (с, 1H), 7,64 (д, 1H, J=8,0 Гц), 7,30 (д, 1H, J=2,8 Гц), 3,05-3,04 (м, 4H), 3,00-2,98 (м, 4H), 2,26 (с, 3H), 2,00-1,90 (м, 2H), 1,83-1,74 (м, 1H), 1,32 (с, 3H), 0,45 (т, 1H, J=7,2 Гц).
МС (ЭРИ): m/z 446,3 [M+H]+.
Пример 33
4-(3-Этил-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(пиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
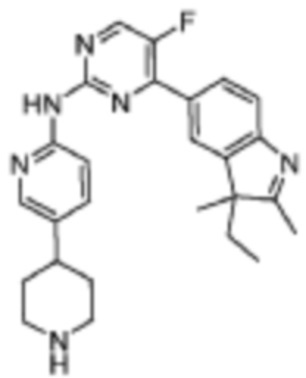
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 13.
1H-ЯМР (400 МГц, метанол-d4) δ 8,33 (д, 1H, J=2,8 Гц), 8,16-8,13 (м, 2H), 7,95-7,93 (м, 2H), 7,48-7,41 (м, 2H), 3,18-3,15 (м, 2H), 2,73 (т, 1H, J=11,6 Гц), 2,59-2,53 (м, 1H), 2,30 (с, 3H), 2,01-1,96 (м, 1H), 1,89-1,83 (м, 1H), 1,79-1,76 (м, 2H), 1,66-1,58 (м, 2H), 1,32 (с, 3H), 0,42 (т, 1H, J=6,8 Гц).
МС (ЭРИ): m/z 445,3 [M+H]+.
Пример 34
4-(3-Этил-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино
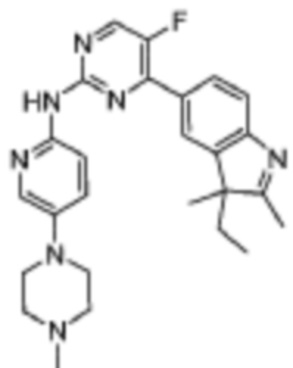
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 16.
1H-ЯМР (400 МГц, метанол-d4) δ 8,42 (д, 1H, J=3,2 Гц), 8,18 (д, 1H, J=8,8 Гц), 8,09-8,01 (м, 3H), 7,57 (д, 1H, J=8,0 Гц), 7,42-7,37 (м, 1H), 3,22-3,15 (м, 4H), 2,61-2,60 (м, 4H), 2,35 (с, 3H), 2,32 (с, 3H), 2,07-2,02 (м, 1H), 1,94-1,89 (м, 1H), 1,38 (с, 3H), 0,43 (т, 1H, J=7,2 Гц).
МС (ЭРИ): m/z 460,3 [M+H]+.
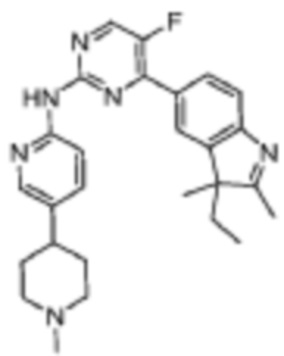
Пример 35
4-(3-Этил-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, CDCl3) δ 8,97 (ш. с., 1H), 8,47 (д, 1H, J=3,6 Гц), 8,39 (д, 1H, J=8,4 Гц), 8,30 (д, 1H, J=2,0 Гц), 8,15 (д, 1H, J=8,4 Гц), 8,04 (с, 1H), 7,68 (д, 1H, J=8,0 Гц), 7,59 (дд, 1H, J=8,8 Гц, 2,4 Гц), 3,01-2,98 (м, 4H), 2,51-2,45 (м, 1H), 2,34 (с, 3H), 2,31 (с, 3H), 2,10-1,97 (м, 3H), 1,85-1,81 (м, 5H), 1,37 (с, 3H), 0,49 (т, 1H, J=7,2 Гц).
МС (ЭРИ): m/z 459,3 [M+H]+.
Пример 36
4-(3-Этил-2,3-диметил-3H-индол-5-ил)-N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-5-фторпиримидин-2-амино
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 19.
1H-ЯМР (400 МГц, CDCl3) δ 8,93 (ш. с., 1H), 8,44 (д, 1H, J=3,6 Гц), 8,33 (д, 1H, J=9,2 Гц), 8,15-8,13 (м, 2H), 8,02 (с, 1H), 7,67 (д, 1H, J=8,0 Гц), 7,35 (д, 1H, J=8,0 Гц), 3,19-3,18 (м, 4H), 2,63-2,62 (м, 4H), 2,52-2,47 (м, 2H), 2,30 (с, 3H), 2,02-1,97 (м, 1H), 1,86-1,80 (м, 1H), 1,36 (с, 3H), 1,14 (т, 3H, J=7,2 Гц), 0,57 (т, 1H, J=7,2 Гц).
МС (ЭРИ): m/z 474,3 [M+H]+.
Пример 37
5-Фтор-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)-4-(2,3,3-триметил-3H-индол-5-ил)пиримидин-2-амино
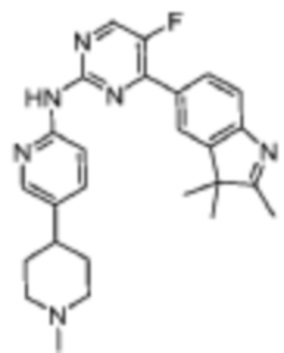
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, CDCl3) δ 9,03 (ш. с., 1H), 8,46 (д, 1H, J=3,2 Гц), 8,38 (д, 1H, J=8,4 Гц), 8,29 (с, 1H), 7,67 (д, 1H, J=8,0 Гц), 7,60 (дд, 1H, J=8,8 Гц, 1,6 Гц), 3,00-2,97 (м, 2H), 2,49-2,44 (м, 1H), 2,34 (с, 3H), 2,33 (с, 3H), 2,10-2,03 (м, 2H), 1,84-1,79 (м, 4H), 1,37 (с, 6H).
МС (ЭРИ): m/z 445,3 [M+H]+.
Пример 38
4-(3,3-Диэтил-7-фтор-2-метил-3H-индол-5-ил)-5-фтор-N-(5-(1-метилпиперидин-4-ил)пиридин-2-ил)пиримидин-2-амино
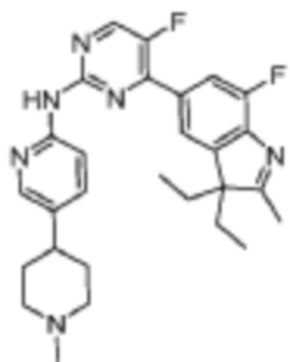
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 17.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,95 (с, 1H), 8,69 (с, 1H), 8,19-8,11 (м, 2H), 7,89-7,84 (м, 2H), 7,67 (д, 1H, J=7,6 Гц), 2,87-2,85 (м, 2H), 2,25 (с, 3H), 2,19 (с, 3H), 2,02-1,92 (м, 6H), 1,90-1,65 (м, 4H), 0,34 (м, 6H).
МС (ЭРИ): m/z 491,3 [M+H]+.
Пример 39
1-(2-((4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фторпиримидин-2-ил)амино)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-гидроксиацетамид
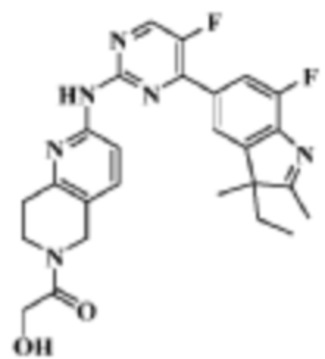
Стадия 1: 2-(2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-ацетоксиацетамид
В реакционную колбу помещали 1,0 г (5,95 ммоль) 2-хлор-5,6,7,8-тетрагидро-1,6-нафтиридина, 1,21 г (11,90 ммоль) триэтиламина и 5 мл дихлорметана, а затем медленно по каплям добавляли 2-хлор-2-ацетоксиацетилхлорид (1,22 г, 8,93 ммоль). Смеси давали реагировать в течение 1 ч при комнатной температуре, и реакцию гасили 5 мл воды. Растворитель удаляли и остаток трижды экстрагировали дихлорметаном (по 15 мл каждый раз). Органические фазы объединяли, однократно промывали 10 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, что давало неочищенный продукт, представляющий собой целевое соединение (1,09 г, желтоватый), выход 68,21%.
МС (ЭРИ): m/z 269,1 [M+H]+.
Стадия 2:
1-(2-((4-(3-этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фторпиримидин-2-ил)амино)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-гидроксиацетамид
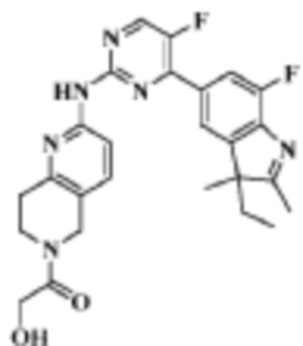
В реакционную колбу помещали 101 мг (0,34 ммоль) 4-(3-этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фторпиримидин-2-амино, полученного по методике, подобной Стадиям 1-3 Примера 1, 85,2 мг (0,32 ммоль) 2-(2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-ацетоксиацетамида, 10 мл диоксана, 65,3 мг (0,68 ммоль) трет-бутоксида натрия, 31,2 мг (0,034 ммоль) Pd2(dba)3, 19,7 мг (0,034 ммоль) 4,5-бис(дифенилфосфино)-9,9-диметилксантена. Смесь нагревали до 120°C и ей давали реагировать под действием микроволнового излучения в течение 1 ч, а затем продукт реакции охлаждали до комнатной температуры, добавляли 50 мл воды и трижды экстрагировали этилацетатом (по 50 мл каждый раз). Органические фазы объединяли, однократно промывали 50 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и разделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол=10:1), что давало целевое соединение (20 мг, желтое твердое вещество), выход 12%.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,98 (с, 1H), 8,70 (д, 1H, J=3,6 Гц), 8,07 (д, 1H, J=8,4 Гц), 7,94 (с, 1H), 7,87 (д, 1H, J=11,2 Гц), 7,64-7,57 (м, 1H), 4,68-4,55 (м, 3H), 4,22-4,21 (м, 2H), 4,21-4,19 (м, 2H), 2,89-2,81 (м, 2H), 2,28 (с, 3H), 2,04-1,84 (м, 2H), 1,33 (с, 3H), 0,37 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 493,2 [M+H]+.
Пример 40
1-(2-((5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3’-индол]-5’-ил)пиримидин-2-ил)амино)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-гидроксиацетамид
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 39.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,07 (ш. с., 1H), 8,69 (д, 1H, J=3,6 Гц), 8,53 (с, 1H), 8,09-8,02 (м, 1H), 7,98 (с, 1H), 7,88 (д, 1H, J=12,4 Гц), 7,62-7,54 (м, 1H), 4,73 (ш. с., 1H), 4,62-4,61 (м, 2H), 4,21-4,19 (м, 2H), 2,89-2,81 (м, 2H), 2,50 (с, 2H), 2,34-2,11 (м, 5H), 1,75-1,72 (м, 2H).
МС (ЭРИ): m/z 505,2 [M+H]+.
Пример 41
N-(5-(4-(Циклопропилметил)пиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3’-индол]-5’-ил)пиримидин-2-амино
Промежуточное соединение 5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N- (5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино получали в соответствии со стадиями, аналогичными стадиям Примера 6.
В реакционную колбу помещали вышеуказанное промежуточное соединение 5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амино (150 мг, 0,32 ммоль), бромметилциклопропан, ацетонитрил (5 мл) и карбонат калия (130,0 мг, 0,96 ммоль), и нагревали до 80°C, и проводили реакцию в течение 4 ч. Продукт реакции охлаждали до комнатной температуры, добавляли 50 мл воды, трижды экстрагировали 10 мл дихлорметана (по 10 мл каждый раз). Органические слои объединяли, однократно промывали 15 мг насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и разделяли с помощью колоночной хроматографии (дихлорметан/метанол=10:1), чтобы получить целевой продукт этого примера (42,1 мг, белое твердое вещество).
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,89 (с, 1H), 8,68 (м, 1H), 8,64-8,01 (м, 3H), 7,87 (д, 1H, J=14,4 Гц), 7,42-7,40 (м, 1H), 3,14-3,13 (м, 5H), 2,60-2,51 (м, 5H), 2,33 (с, 3H), 2,24-2,23 (м, 2H), 2,09-2,02 (м, 6H), 1,99-1,97 (м, 2H), 1,75-1,74 (м, 2H), 0,85-0,83 (м, 2H), 0,48-0,47 (м, 2H).
МС (ЭРИ): m/z 530,3 [M+H]+.
Пример 42
4-(3-Этил-7-фтор-2,3-диметил-3H-индол-5-ил)-5-фтор-N-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амино
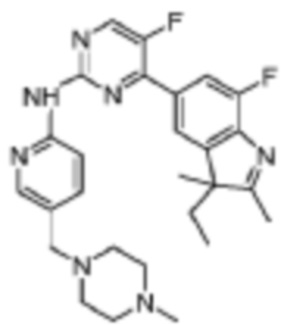
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,06 (ш. с., 1H), 8,72 (д, 1H, J=3,2 Гц), 8,18-8,15 (м, 2H), 7,93 (с, 1H), 7,86 (д, 1H, J=11,6 Гц), 7,67 (д, 1H, J=5,2 Гц), 3,42 (с, 2H), 2,35-2,28 (м, 8H), 2,14 (с, 3H), 2,04-1,98 (м, 1H), 1,89-1,84 (м, 1H), 1,34 (с, 3H), 0,36 (т, 3H, J=7,2 Гц).
МС (ЭРИ): m/z 492,3 [M+H]+.
Пример 43
5-Фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3’-индол]-5’-ил)-N-(5-((4-метилпиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амино
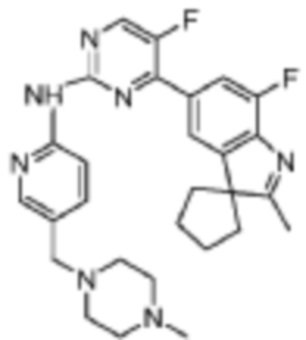
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 2.
1H-ЯМР (400 МГц, ДМСО-d6) δ 10,09 (с, 1H), 8,71 (д, 1H, J=3,2 Гц), 8,18-8,15 (м, 2H), 8,02 (с, 1H), 7,85 (д, 1H, J=11,2 Гц), 7,67 (д, 1H, J=8,4 Гц), 3,43 (с, 2H), 2,50-2,34 (м, 8H), 2,14 (с, 3H), 2,14-2,08 (м, 6H), 1,75-1,74 (м, 2H), 1,75-1,72 (м, 2H).
МС (ЭРИ): m/z 504,2 [M+H]+.
Пример 44
N-(5-(1-Метилпиперидин-4-ил)-(6-метилпиридин)-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино
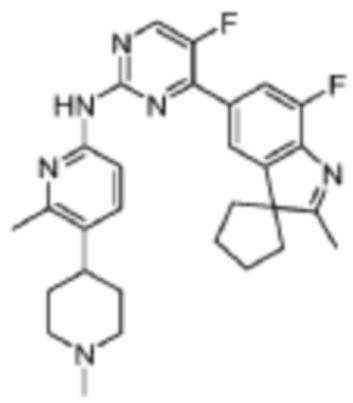
Целевое соединение получали в соответствии со стадиями, подобными стадиям Примера 12.
1HЯМР (400 МГц, ДМСО-d6), δ 9,84 (с, 1H), 8,70 (с, 1H), 8,11 (с, 1H), 7,98-8,01 (д, 2H), 7,81-7,85 (д, 1H), 2,87-2,90 (д, 2H), 2,50-2,51 (м, 1H), 2,20-2,34 (м, 6H), 1,78-1,98 (м, 8H), 1,69-1,75 (м, 6H).
МС (ЭРИ): m/z 503,3 [M+H]+.
Пример 45
N-(5-((1-Метилпиперидин-4-ил)окси)-пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3’-индол]-5’-ил)пиримидин-2-амино
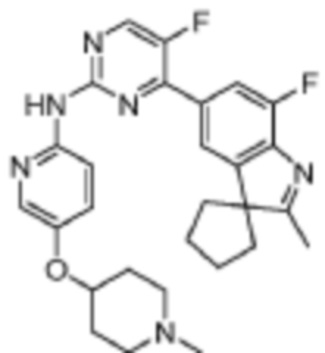
Промежуточное соединение 5'-(2-хлор-5-фторпиримидин-4-ил)-7'-фтор-2'-метилспиро[циклопентан-1,3'-индол] получали в соответствии со стадиями, подобными стадиям Примера 1.
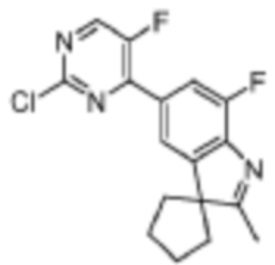
Стадия 1: 1-метилпиперидин-4-ил-4-метилбензолсульфонат
В реакционную колбу помещали 4-гидрокси-1-метилпиперидин (1000 мг, 8,69 ммоль), п-толуолсульфонилхлорид (3310 мг, 17,38 ммоль), дихлорметан (50 мл) и триэтиламин (1 мл) и давали реагировать при комнатной температуре в течение 2 ч. Затем к продукту реакции добавляли 50 мл воды и трижды экстрагировали 30 мл дихлорметана (по 30 мл каждый раз). Органические слои объединяли, однократно промывали 50 мг насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и разделяли с помощью колоночной хроматографии (дихлорметан/метанол=50:1), что давало 1,87 г промежуточного соединения, выход 80,0 % (бледно-желтое твердое вещество).
Стадия 2: 2-бром-5-((1-метилпиперидин-4-ил)окси)пиридин
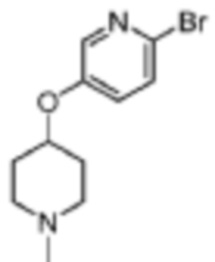
В реакционную колбу помещали промежуточное соединение 1-метилпиперидин-4-ил-4-метилбензолсульфонат (1000 мг, 3,70 ммоль), 2-бром-5-гидроксипиридин (637 мг, 3,70 ммоль) и ДМФА (50 мл), и смесь нагревали до 90°C, и давали реагировать в течение 2 ч. К продукту реакции добавляли 50 мл воды и трижды экстрагировали дихлорметаном (по 30 мл каждый раз). Органические слои объединяли, однократно промывали 50 мг насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и разделяли с помощью колоночной хроматографии (дихлорметан/метанол=40:1), что давало 0,49 г промежуточного соединения (бледно-желтое твердое вещество), выход 50,1 %.
Стадия 3: 5-((1-метилпиперидин-4-ил)окси)-пиридин-2-амин
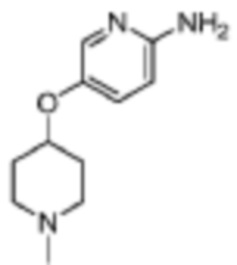
В реакционную колбу помещали промежуточное соединение 2-бром-5-((1-метилпиперидин-4-ил)окси)пиридин (1000 мг, 3,70 ммоль), бис(триметилсилил)амид натрия (618 мг, 3,70 ммоль), тетрагидрофуран (50 мл), 2-(дициклогексилфосфино)бифенил (120 мг, 0,37 ммоль) и трис(дибензилиденинденацетон)дипалладий (338 мг, 0,37 ммоль), и смесь нагревали до 65°C, и проводили реакцию в течение 12 ч. К продукту реакции добавляли 50 мл воды, трижды экстрагировали дихлорметаном (по 30 мл каждый раз). Органические слои объединяли, промывали 50 мл насыщенного раствора хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали и разделяли с помощью колоночной хроматографии (дихлорметан/метанол=10:1), что давало промежуточное соединение (0,37 г, желтое твердое вещество), выход 49,3 %.
Стадия 4:
N-(5-((1-метилпиперидин-4-ил)окси)-пиридин-2-ил)-5-фтор-4-(7'-фтор-2'-метилспиро[циклопентан-1,3'-индол]-5'-ил)пиримидин-2-амино получали по методике, подобной Стадии 6 Примера 1.
1HЯМР (400 МГц, ДМСО-d6), δ 9,69 (с, 1H), 8,63 (с, 1H), 7,93-7,99 (д, 1H), 7,80-7,83 (д, 1H), 7,81-7,85 (д, 1H), 7,12-7,14 (д, 1H), 5,76-5,82 (м, 1H), 5,02-5,13 (м, 2H), 3,79 (с, 2H), 2,56-2,59 (м, 13H), 2,28-2,33 (м, 6H), 1,74-1,99 (м, 2H).
МС (ЭРИ): m/z 505,2 [M+H]+.
Контроль, используемый в следующих примерах экспериментов, с номером LY2835219 получали в соответствии с методикой получения, представленной в WO2010075074, и его структурная формула такая же, как показана на структурной формуле 3 в разделе «Уровень техники».
Экспериментальный пример 1
Измерение активности ингибирования киназы CDK для соединений по данному изобретению
In vitro ингибирующее действие соединений по данному изобретению на киназную активность CDK (CDK1, CDK4 и CDK6) исследовали с помощью следующей методики.
1.1 Информация об оборудовании и наборах
киназная ферментная система CDK1/циклин A2
киназная ферментная система CDK4/циклин E1
киназная ферментная система CDK6/циклин D3
киназная ферментная система CDK9/циклин K
1.2 Приготовление растворов для эксперимента
1.2.1 Приготовление реакционного буферного раствора I для киназ: 5× реакционный буферный раствор A (Promega; V307A-C), поставляемый в наборе, смешивали с и разбавляли H2O Milli Q и 0,1 М ДТТ (дитиотреитол), что давало 4× буферный раствор для киназ, а затем дополнительно добавляли H2O Milli Q, чтобы в итоге приготовить 1× буферный раствор для киназ.
Приготовление реакционного буферного раствора II для киназ: к 1× реакционному буферному раствору для киназ добавляли 0,5% ДМСО (диметилсульфоксид) и хорошо перемешивали.
1.2.2 Приготовление раствора киназы: растворы киназы с концентрациями, необходимыми для каждой реакционной системы, готовили из исходного раствора киназы с концентрацией 100 нг/мкл и 1× реакционного буферного раствора для киназ.
1.2.3 Приготовление раствора исследуемого соединения и контрольного раствора LY2835219:
(1) Приготовление контрольного раствора LY2835219
a. Отбирали 1 мкл исходного раствора стандартного вещества с концентрацией 10 мМ, и добавляли 9 мкл реакционного буферного раствора I для киназ, и хорошо перемешивали; затем добавляли 90 мкл реакционного буферного раствора I для киназ и хорошо перемешивали; затем добавляли 100 мкл реакционного буферного раствора I для киназ и хорошо перемешивали. Конечная концентрация составляла 50 мкМ.
b. 40 мкл реакционного буферного раствора II для киназ добавляли в лунки B2-B10 96-луночного планшета, и 50 мкл вышеуказанного раствора добавляли в B1;
c. Из лунки B1 отбирали 10 мкл раствора, добавляли в B2 и хорошо перемешивали; затем отбирали 10 мкл полученного раствора и добавляли в B3, и разбавление выполняли последовательно до B9, чтобы получить контрольные растворы, которые разбавлены последовательно в 5 раз.
(2) Приготовление раствора исследуемого соединения:
a. Брали растворы исследуемого соединения с определенной концентрацией, соответственно разбавляли реакционным буферным раствором I для киназ, чтобы получить раствор соединения с конечной концентрацией 50 мкМ;
b. 40 мкл реакционного буферного раствора II для киназ добавляли в лунки H2-H10 96-луночного планшета; и 50 мкл вышеуказанного раствора добавляли в H1;
c. Из лунки H1 отбирали 10 мкл раствора, добавляли в H2 и хорошо перемешивали; затем отбирали 10 мкл полученного раствора и добавляли в H3, и разбавление выполняли последовательно до H9, чтобы получить растворы исследуемых соединений, которые разбавлены последовательно в 5 раз.
1.2.4 Приготовление смешанного раствора реакционного субстрата и АТФ
a. Приготовление раствора АТФ:
200 мкл раствора АТФ с концентрацией 0,1 мМ: 2 мкл 10 мМ АТФ добавляли к 198 мкл реакционного буферного раствора I для киназ;
300 мкл раствора АТФ с концентрацией 50 мкМ: 150 мкл реакционного буферного раствора I для киназ добавляли к 150 мкл вышеуказанного раствора АТФ с концентрацией 0,1 мМ;
b. Приготовление 300 мкл раствора реакционного субстрата:
150 мкл исходного раствора реакционного субстрата с концентрацией 1 мкг/мкл добавляли к 150 мкл реакционного буферного раствора I для киназ и хорошо перемешивали;
c. Вышеуказанные растворы a/b смешивали, чтобы получить смешанные растворы, соответственно.
1.3 Процедура эксперимента:
1.3.1: Отбирали 2 мкл растворов соединений с различными концентрациями, добавляли в 384-луночные планшеты и центрифугировали в течение 3 мин;
1.3.2: В каждую лунку добавляли 4 мкл раствора киназы, центрифугировали при 5000 об/мин и 18°C в течение 10 мин и встряхивали на встряхивателе для планшетов в течение 10 мин;
1.3.3: В каждую лунку добавляли 4 мкл смешанного раствора субстрата и АТФ, центрифугировали при 5000 об/мин. и 18°C в течение 10 мин и встряхивали на встряхивателе для планшетов при 37°C в течение 90 мин;
1.3.4: 384-луночный планшет вынимали и давали принять комнатную температуру;
1.3.5: В каждую лунку добавляли 10 мкл реагента ADP-Glo, центрифугировали при 5000 об/мин и 18°C в течение 18 мин и встряхивали на встряхивателе для планшетов при 25°C в течение 40 мин, а затем реакцию останавливали;
1.3.6: В каждую лунку добавляли 20 мкл реагента для определения киназы, центрифугировали при 5000 об/мин и 18°C в течение 10 мин и встряхивали на встряхивателе для планшетов при 25°C в течение 30 мин.; и
1.3.7: Использовали ридер для микропланшетов M1000pro для считывания значений флуоресценции.
1.4 Обработка данных:
Степень ингибирования для каждого соединения при данной концентрации рассчитывали по формуле приведенной ниже, а аппроксимацию кривой осуществляли с помощью программного обеспечения GraphPad Prism 5, чтобы получить величину IC50.

1.5 Результаты исследования:
Ингибирующее действие LY2835219, описанного в WO2010075074, и соединений Примеров 1-43 на CDK1/циклин A2 и CDK6/циклин D3 выражено в виде IC50, и конкретные результаты показаны в Таблице 1.
Таблица 1. Результаты определения ингибирующей активности исследуемых соединений для CDK1/циклин A2 и CDK6/циклин D3 (IC50: нМ)
Ингибирующее действие некоторых иллюстративных соединений по данному изобретению на CDK9/циклин D3, Pim-1 и CDK2/циклин E1 и CDK4/циклин E1 показано в Таблице 2, Таблице 3 и Таблице 4, соответственно.
Таблица 2. Результаты определения ингибирующей активности некоторых исследуемых соединений для CDK9/циклина D3 (IC50: нМ)
Таблица 3. Результаты определения ингибирующей активности некоторых исследуемых соединений для Pim-1 (IC50: нМ)
циклин A2
циклин D3
циклин D3
CDK6
CDK6
CDK6
Таблица 4. Ингибирующее действие части исследуемых соединений на CDK4 и CDK2 (IC50: нМ)
циклин A2
циклин E1
циклин E1
циклин D3
циклин D3
CDK6
CDK6
CDK6
1.6 Заключение по эксперименту:
1) Соединения по данному изобретению обладают значительным ингибирующим действием на CDK6 и CDK4.
2) CDK1/CDK6, CDK9/CKD6 и Pim-1/CDK6 могут отражать селективность соединения к протеинкиназам. Чем больше значение, тем лучше селективность соединения к CDK6, что указывает на то, что токсичность соединения при неселективном ингибировании киназ может быть меньше. Контрольное соединение (LY2835219) демонстрирует CDK1/CDK6=83,83, CDK9/CDK6=1,33 и Pim-1/CDK6=1,03; некоторые из соединений по данному изобретению демонстрируют лучшую селективность, чем LY2835219, в частности, соединение, полученное в Примере 17, показывает более высокую ферментативную активность по отношению к CDK6 и лучшую селективность по сравнению с CDK1, CDK9 и Pim1.
Экспериментальный пример 2
Измерение ингибирующего действия иллюстративных соединений по данному изобретению на пролиферацию клеток рака молочной железы человека MDB-MA-231
2.1 Материалы для эксперимента: клетки рака молочной железы человека MDA-MB-231, приобретенные в Cell Resource Center, Пекинский объединенный медицинский колледж, ДАФИ (5 мг/мл, Beyotime, c1002), 4 %-ый параформальдегид (DINGGUO BIOTECHNOLOGY CO. LTD, AR-0211), 96-луночный планшет с черным прозрачным дном (PE, 6005182), система мультипараметрического клеточного анализа In Cell Analyzer 2200 (GE Healthcare).
2.2 Приготовление растворов для эксперимента:
2.2.1 Приготовление среды для клеток рака молочной железы человека MDA-MB-231: RPIM1640 + 10% ФБС + 1% пенициллина/стрептомицина.
2.2.2 Приготовление растворов исследуемых соединений и растворов стандарта LY2835219:
(1) Приготовление растворов стандарта LY2835219
a. Отбирали 3,6 мкл исходного раствора стандарта с концентрацией 10 мМ, добавляли к 6,4 мкл среды и хорошо перемешивали; затем добавляли 90 мкл среды и хорошо перемешивали; затем добавляли 200 мкл среды и хорошо перемешивали, что давало исходную концентрацию 20 мМ;
b. 200 мкл среды, содержащей 0,2% ДМСО (диметилсульфоксид), добавляли в лунки B2-B10 96-луночного планшета; 300 мкл вышеуказанного раствора добавляли в B1; и
c. отбирали 100 мкл раствора из лунки B1, добавляли в B2 и хорошо перемешивали; затем отбирали 100 мкл полученного раствора и добавляли в B3, и разбавление выполняли последовательно до B9, чтобы получить стандартные растворы, последовательно разбавленные в 3 раза.
(2) Приготовление растворов исследуемых соединений
a. Брали раствор исследуемого соединения с определенной концентрацией и разбавляли средой, что давало раствор соединений с конечной концентрацией 20 мкМ;
b. 200 мкл среды, содержащей 0,2% ДМСО (диметилсульфоксид), добавляли в лунки H2-H10 96-луночного планшета; и 300 мкл вышеуказанного раствора добавляли в H1; и
c. из лунки H1 отбирали 100 мкл раствора, добавляли в H2 и хорошо перемешивали; затем отбирали 100 мкл полученного раствора и добавляли в H3, и разбавление выполняли последовательно до B9, чтобы получить растворы исследуемого соединения, последовательно разбавленные в 3 раза.
2.3 Процедура эксперимента:
2.3.1: Клетки MDA-MB-231 инокулировали в 96-луночные клеточные планшеты с черным прозрачным дном в количестве 4000 клеток/100 мкл/лунка и выращивали в течение ночи при 37°C;
2.3.2: В культуральный планшет с внесенными клетками добавляли вышеуказанные образцы в количестве 100 мкл/лунка, слегка похлопывали, чтобы хорошо перемешать, и инкубировали при 37°C в течение 72 ч;
2.3.3: Фиксация: клеточный планшет вынимали, среду удаляли и добавляли 50 мкл 4%-ного параформальдегида на лунку для фиксации в течение 10 мин;
2.3.4: Добавляли 50 мкл 0,1 M глицина для нейтрализации в течение 10 мин.;
2.3.5: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для двукратного промывания;
2.3.6: Пермеабилизация: добавляли 50 мкл 0,2%-ного тритона X-100 (тритон) на лунку, и пермеабилизацию осуществляли при комнатной температуре в течение 10 мин;
2.3.7: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для двукратного промывания;
2.3.8: Исходный раствор ДАФИ с концентрацией 5 мг/мл разбавляли в соотношении 1:5000 (конечная концентрация 1 мкг/мл), и выполняли окрашивание при комнатной температуре в течение 20 мин;
2.3.9: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для трехкратного промывания; и
2.3.10: Выполняли считывание и анализ с помощью In cell analyzer.
2.4 Обработка данных:
Степень ингибирования для каждого соединения при данной концентрации рассчитывали по формуле, приведенной ниже, а аппроксимацию кривой осуществляли с помощью программного обеспечения GraphPad Prism 5, чтобы получить величину IC50.

2.5 Результаты определения:
Результаты определения цитологической активности LY2835219, описанного в WO2010075074, и соединений Примеров 12 и 17 выражены в виде IC50, а конкретные результаты показаны в Таблице 5.
Таблица 5. Ингибирующая активность иллюстративных соединений по данному изобретению на пролиферацию клеток рака молочной железы человека MDB-MA-231 (IC50: нМ)
2.6 Заключение по эксперименту:
Соединения Примеров 12 и 17 обладают значительной ингибирующей активностью на пролиферацию клеточной линии MDA-MB-231, и иллюстративные соединения по данному изобретению обладают более сильным ингибирующим действием на пролиферацию по сравнению с контрольным соединением LY2835219.
Экспериментальный пример 3
Определение фармакокинетики иллюстративных соединений по данному изобретению у крыс
3.1 Краткое изложение эксперимента
Крыс линии SD использовали в качестве подопытных животных, и определяли концентрации лекарственных средств в плазме крыс в различные моменты времени после внутривенного введения и внутрижелудочного введения иллюстративных соединений с помощью ЖХ/МС/МС для того, чтобы изучить фармакокинетическое поведение соединений по данному изобретению у крыс и оценить их фармакокинетические характеристики.
3.2 Порядок проведения эксперимента
3.2.1 Исследуемые лекарственные вещества:
Соединение, полученное в Примере 17 по данному изобретению.
Контрольное лекарственное вещество LY2835219, полученное нами.
3.2.2 Подопытные животные:
12 здоровых взрослых крыс линии SD, самцов, в возрасте 6-8 недель, массой 200-250 г, приобретенных у Suzhou ZhaoYan New Drug Research Center Co., Ltd., лицензия на разведение животных №: SCXK (Su) 2013-0003
3.2.3 Подготовка исследуемых лекарственных веществ
Внутрижелудочное введение: взвешивали необходимое количество образца и добавляли 0,1 % гидроксиэтилцеллюлозы/0,5 % твина 80 так, чтобы при конечном объеме получить раствор с концентрацией 1 мг/мл.
Внутривенное введение: взвешивали необходимое количество образца и добавляли 10 % N-метил-2-пирролидона и 90 % 18%-ного сульфобутил-β-циклодекстрина так, чтобы при конечном объеме получить раствор с концентрацией 0,4 мг/мл для внутривенного введения.
3.2.4 Введение исследуемых лекарственных веществ
Внутривенное введение: каждое исследуемое соединение вводили внутривенно 3-м самцам крыс линии SD, которых не кормили в течение ночи, в дозе 2 мг/кг и в объеме введения 1 мл/кг.
Внутрижелудочное введение: каждое исследуемое соединение вводили внутрижелудочно 3-м самцам крыс линии SD, которых не кормили в течение ночи, в дозе 5 мг/кг и в объеме введения 5 мл/кг.
3.3 Проведение эксперимента
До введения и через 0,0833, 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после введения отбирали кровь через канюлю в сонной артерии. Цельную кровь предохраняли от свертывания с помощью ЭДТА-K2 и центрифугировали, надосадочную жидкость удаляли, а остаток замораживали при -20°C до анализа образцов. Образцы плазмы анализировали с помощью ЖХ-МС/МС, предварительную обработку образцов выполняли с использованием метода осаждения белков, линейный диапазон при анализе образцов составлял 1-2000 нг/мл, и нижний предел количественного определения составлял 1 нг/мл.
3.4 Полученные фармакокинетические данные
Фармакокинетические параметры соединений по данному изобретению показаны в Таблице 6 и Таблице 7.
Таблица 6. ФК параметры соединения 17 по данному изобретению у крыс после однократного внутривенного введения (среднее ± СО)
Таблица 7. ФК параметры соединения 17 по данному изобретению у крыс после однократного внутрижелудочного введения (среднее ± СО)
3.5 Заключение по эксперименту: иллюстративное соединение по данному изобретению (полученное в Примере 17) обладает более высокой биодоступностью у крыс по сравнению с соединением LY2835219 и хорошо всасывается при пероральном введении.
Экспериментальный пример 4
Определение фармакокинетики иллюстративных соединений по данному изобретению у мышей
4.1 Краткое изложение эксперимента
Мышей линии ICR использовали в качестве подопытных животных, и определяли концентрации лекарственных веществ в плазме мышей в различные моменты времени после внутрижелудочного введения и внутривенного введения иллюстративного соединения по данному изобретению с помощью ЖХ/МС/МС для того, чтобы изучить фармакокинетическое поведение соединений по данному изобретению у мышей и оценить их фармакокинетические характеристики.
4.2 Порядок проведения эксперимента
4.2.1 Исследуемые лекарственные вещества:
Соединение, полученное в Примере 17 по данному изобретению.
Контрольное лекарственное вещество LY2835219, полученное нами.
4.2.2 Подопытные животные:
12 здоровых взрослых мышей линии ICR, самцов, в возрасте 6-8 недель, массой 20-25 г, приобретенных у Suzhou ZhaoYan New Drug Research Center Co., Ltd., лицензия на разведение животных № SCXK (Su) 2013-0003.
4.2.3 Подготовка исследуемых лекарственных веществ
Взвешивали необходимое количество образца и добавляли 0,1% гидроксиэтилцеллюлозы/0,5% твина 80 так, чтобы при конечном объеме получить раствор с концентрацией 0,5 мг/мл для внутрижелудочного введения.
Взвешивали необходимое количество образца и добавляли 10% N-метил-2-пирролидона и 90% 18%-ного сульфобутил-β-циклодекстрина так, чтобы при конечном объеме получить раствор с концентрацией 0,2 мг/мл для внутривенного введения.
4.2.4 Введение исследуемых лекарственных веществ
Каждое исследуемое соединение вводили внутрижелудочно 3-м самцам мышей линии ICR, которых не кормили в течение ночи, в дозе 5 мг/кг и в объеме введения 10 мл/кг.
Каждое исследуемое соединение вводили внутривенно 3-м самцам мышей линии ICR, которых не кормили в течение ночи, в дозе 2 мг/кг и в объеме введения 10 мл/кг.
4.3 Проведение эксперимента
До введения и через 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после введения отбирали кровь у группы, которой введение производили внутрижелудочно, через канюлю в сонной артерии. Цельную кровь предохраняли от свертывания с помощью ЭДТА-K2 и центрифугировали, надосадочную жидкость удаляли, а остаток замораживали при -20°C до анализа образцов. До введения и через 0,083, 0,25, 0,5, 1, 2, 4, 8, 12 и 24 часа после введения отбирали кровь у группы, которой проводили внутривенное введение, через канюлю в сонной артерии. Образцы плазмы обрабатывали так же, как и образцы плазмы группы, которой проводили внутрижелудочное введение. Образцы плазмы анализировали с помощью ЖХ-МС/МС, предварительную обработку образцов выполняли с использованием метода осаждения белков, линейный диапазон при анализе образцов составлял 1-2000 нг/мл, и нижний предел количественного определения составлял 1 нг/мл.
4.4 Полученные фармакокинетические данные: см. Таблицу 8 и Таблицу 9.
Таблица 8. ФК параметры соединения 17 по данному изобретению у мышей после однократного внутривенного введения (среднее ± СО)
Таблица 9. ФК параметры соединения 17 по данному изобретению у мышей после однократного внутрижелудочного введения (среднее ± СО)
4.5 Заключение по эксперименту: иллюстративное соединение 17 по данному изобретению обладает более высокой биодоступностью и более длительным периодом полувыведения у мышей по сравнению с соединением LY2835219 и хорошо всасывается при пероральном введении.
Экспериментальный пример 5
Определение уровней воздействия в плазме и головном мозге иллюстративного соединения 17 по данному изобретению
5.1 Краткое изложение эксперимента
Мышей линии CD-1 использовали в качестве подопытных животных, и определяли концентрации лекарственных веществ в плазме и ткани головного мозга мышей в различные моменты времени после однократного внутрижелудочного введения иллюстративного соединения по данному изобретению с помощью ЖХ/МС/МС для того, чтобы изучить уровень в плазме и уровень воздействия в головном мозге соединений по данному изобретению у мышей.
5.2 Порядок проведения эксперимента
5.2.1 Исследуемые лекарственные вещества:
Соединение, полученное в Примере 17 по данному изобретению.
Контрольное лекарственное вещество LY2835219, полученное нами.
5.2.2 Подопытные животные:
24 здоровые взрослые мыши линии CD-1, самцы, в возрасте 6-8 недель с массой тела 20-25 г, приобретенные у Shanghai sippr BK Laboratory Animal Co., Ltd., лицензия на разведение животных № SCXK (Shanghai) 2013-0016.
5.2.3 Подготовка исследуемых лекарственных веществ
Взвешивали необходимое количество образца и добавляли 0,1 % гидроксиэтилцеллюлозы/0,5 % твина 80 так, чтобы при конечном объеме получить раствор с концентрацией 1,0 мг/мл.
5.2.4 Введение исследуемых лекарственных веществ
Каждое исследуемое соединение вводили внутрижелудочно 12 самцам мышей линии CD-1, которых не кормили в течение ночи, в дозе 10 мг/кг и в объеме введения 10 мл/кг.
5.3 Проведение эксперимента
LY2835219: до введения и через 0,25, 1,5 и 6 часов после введения отбирали кровь через канюлю в сонной артерии, и три мыши умерщвляли в то же время. Извлекали весь головной мозг, разминали и замораживали в жидком азоте. Через 10 часов после введения умерщвляли оставшихся животных, собирали цельную кровь при помощи пункции сердца, и извлекали весь головной мозг, разминали и замораживали в жидком азоте.
Пример 17: до введения и через 2, 4 и 24 часа после введения отбирали кровь через канюлю в сонной артерии, и три мыши умерщвляли в то же время. Извлекали весь головной мозг, разминали и замораживали в жидком азоте. Через 48 часов после введения умерщвляли оставшихся животных, собирали цельную кровь при помощи пункции сердца, и извлекали весь головной мозг, разминали и замораживали в жидком азоте.
Обработка образцов цельной крови: собранную цельную кровь предохраняли от коагуляции с помощью ЭДТА-K2 и центрифугировали, удаляли надосадочную жидкость и остаток замораживали и хранили при -20°C до анализа образцов с помощью ЖХ-МС/МС.
Пробоподготовка гомогената головного мозга: гомогенат головного мозга диспергировали в растворе ФСБ (pH=7,4): MeOH (об.:об., 2: 1) в объеме, который в пять раз превышает объем гомогената головного мозга. Отбирали 100 мкл раствора и в нем осаждали белок с помощью 600 мкл IS. Смесь центрифугировали при 13000 об/мин и 20-25°C в течение 15 минут. 50 мкл надосадочной жидкости смешивали со 150 мкл воды, содержащей 0,3 % FA, и центрифугировали при 4°C. 5 мкл образца анализировали с помощью ЖХ-МС/МС.
Линейный диапазон при анализе образцов составлял 1-2000 нг/мл, а нижний предел количественного определения составлял 1 нг/мл.
5.4 Результаты измерений воздействия в крови и головном мозге показаны в Таблице 10.
Таблица 10. Средняя степень воздействия исследуемого соединения в плазме и головном мозге у мышей линии CD-1
5.5 Заключение по эксперименту: иллюстративное соединение по данному изобретению (полученное в Примере 17) обладает лучшим распределением между кровью и головным мозгом, более высоким соотношением AUC0-последн. (головной мозг/плазма) и более высоким соотношением Cmax (головной мозг/плазма) по сравнению с соединением LY2835219, и Tmax в головном мозге соответствует Tmax в плазме, что указывает на то, что лекарственное вещество обладает подобным ФК поведением в головном мозге и в плазме. Предполагается, что соединения по данному изобретению могут проходить через гематоэнцефалический барьер, чтобы ингибировать рост опухолей головного мозга (рак головного мозга) и лечить рак головного мозга.
Экспериментальный пример 6
Определение ингибирующего действия иллюстративного соединения 17 по данному изобретению на пролиферацию клеточной линии U87 MG.
6.1 Материалы для эксперимента: клеточная линия глиомы человека U87 MG приобретена в клеточном фонде Китайской академии наук (Cell Bank of Chinese Academy of Sciences), Шанхай, ДАФИ (5 мг/мл, Beyotime, c1002), 4 % параформальдегид (DINGGUO BIOTECHNOLOGY CO. LTD, AR-0211), 96-луночный планшет с черным прозрачным дном (PE, 6005182), система мультипараметрического клеточного анализа In Cell Analyzer 2200 (GE Healthcare).
6.2 Приготовление растворов для эксперимента:
6.2.1 Приготовление среды для U87 MG: RPIM1640 + 10% ФБС + 1% пенициллина/стрептомицина.
6.2.2 Приготовление растворов исследуемого соединения и растворов стандарта LY2835219:
(1) Приготовление растворов стандарта LY2835219
a. отбирали 3,6 мкл исходного раствора стандарта с концентрацией 10 мМ, добавляли к 6,4 мкл среды и хорошо перемешивали; затем добавляли 90 мкл среды и хорошо перемешивали; затем добавляли 200 мкл среды и хорошо перемешивали, что давало исходную концентрацию 20 мкМ;
b. 200 мкл среды, содержащей 0,2% ДМСО (диметилсульфоксид), добавляли в лунки B2-B10 96-луночного планшета; 300 мкл вышеуказанного раствора добавляли в B1;
c. отбирали 100 мкл раствора из лунки B1, добавляли в B2 и хорошо перемешивали; затем отбирали 100 мкл полученного раствора и добавляли в B3, и разбавление выполняли последовательно до B9, чтобы получить стандартные растворы, последовательно разбавленные в 3 раза.
(2) Приготовление растворов исследуемого соединения:
a. брали раствор исследуемого соединения с определенной концентрацией и разбавляли средой, что давало раствор соединения с конечной концентрацией 20 мкМ;
b. 200 мкл среды, содержащей 0,2% ДМСО (диметилсульфоксид), добавляли в лунки H2-H10 96-луночного планшета; и 300 мкл вышеуказанного раствора добавляли в H1;
c. из лунки H1 отбирали 100 мкл раствора, добавляли в H2 и хорошо перемешивали; затем отбирали 100 мкл полученного раствора и добавляли в H3, и разбавление выполняли последовательно до H9, чтобы получить растворы исследуемого соединения, последовательно разбавленные в 3 раза.
6.3 Процедура эксперимента:
6.3.1: Клетки U87 MG инокулировали в 96-луночные планшеты с черным прозрачным дном в количестве 4000 клеток/100 мкл/лунка и выращивали в течение ночи при 37°C;
6.3.2: В культуральный планшет с внесенными клетками добавляли вышеуказанные образцы в количестве 100 мкл/лунка, слегка похлопывали, чтобы хорошо перемешать, и инкубировали при 37°C в течение 72 ч;
6.3.3: Фиксация: клеточный планшет вынимали, среду удаляли и добавляли 50 мкл 4%-ного параформальдегида на лунку для фиксации в течение 10 мин.;
6.3.4: Добавляли 50 мкл 0,1 M глицина для нейтрализации в течение 10 мин.;
6.3.5: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для двукратного промывания;
6.3.6: Пермеабилизация: добавляли 50 мкл 0,2%-ного тритона X-100 (тритон) на лунку, и пермеабилизацию осуществляли при комнатной температуре в течение 10 мин;
6.3.7: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для двукратного промывания;
6.3.8: Исходный раствор ДАФИ с концентрацией 5 мг/мл разбавляли в соотношении 1:5000 (конечная концентрация 1 мкг/мл), и выполняли окрашивание при комнатной температуре в течение 20 мин;
6.3.9: Использовали 1× ФСБ (фосфатный буферный раствор, pH 7,2) для трехкратного промывания; и
6.3.10: Выполняли считывание и анализ с помощью In cell analyzer.
6.4 Обработка данных:
Степень ингибирования каждого соединения при данной концентрации рассчитывали по следующей формуле, а аппроксимацию кривой осуществляли с помощью программного обеспечения GraphPad Prism 5, чтобы получить величину IC50.
6.5 Результаты определения:
Результаты определения цитологической активности LY2835219, описанного в WO2010075074, и соединения Примера 17 были выражены в виде IC50, а конкретные результаты показаны в Таблице 11.
Таблица 11. Ингибирующая активность иллюстративного соединения по данному изобретению на пролиферацию клеточной линии U87MG (IC50: нМ)
6.6 Заключение по эксперименту:
Соединение Примера 17 обладает значительной ингибирующей активностью на пролиферацию клеточной линии U87MG, и иллюстративное соединение по данному изобретению обладают более сильным ингибирующим действием на пролиферацию по сравнению с контрольным соединением LY2835219.
Экспериментальный пример 7
Фармакодинамическое исследование соединения 17 по данному изобретению и комбинации соединения 17 по данному изобретению и темозоломида в модели ортотопической ксенотрансплантации клеток U87-luc в головной мозг
7.1 Краткое изложение эксперимента
В качестве подопытных животных использовали взрослых самок голых мышей линии BALB/c. Для изучения влияния иллюстративного соединения 17 по данному изобретению на медианную выживаемость самок голых мышей линии BALB/c после внутрижелудочного введения использовали модель ортотопической ксенотрансплантации клеток U87-luc в головной мозг.
7.2 Порядок проведения эксперимента
7.2.1 Исследуемые лекарственные вещества:
Соединение, полученное в Примере 17 по данному изобретению.
Контрольное лекарственное вещество LY2835219, полученное нами.
Темозоломид был приобретен в selleck.
7.2.2 Подопытные животные:
Здоровые взрослые самки голых мышей линии BALB/c, 8 мышей/группа, в возрасте 6-8 недель, массой 18-22 г, приобретенные в Shanghai sippr BK Laboratory Animal Co., Ltd., лицензия на разведение животных № 2008001658261; 2008001658263.
7.2.3 Подготовка исследуемых соединений
Взвешивали необходимое количество соединения 17 и добавляли 0,1% гидроксиэтилцеллюлозы/0,5% твина 80 в качестве наполнителя до 0,3125 мг/мл;
Взвешивали необходимое количество образца темозоломида и добавляли 0,1% КМЦ-Na + 0,25% твина 80 в качестве наполнителя до 0,3 мг/мл.
7.3 Модель ортотопической ксенотрансплантации в головной мозг
Взрослым самкам голых мышей линии BALB/c давали анестезию с помощью внутрибрюшинной инъекции пентобарбитала натрия в дозе 80 мг/кг. Для облегчения боли животным подкожно вводили бупренорфин в моменты времени 30 минут до операции и 6 часов после операции в дозе 0,1 мг/кг. За животными наблюдали после анестезии, пока все животные не проснулись.
Анестезированных животных должным образом фиксировали, стерилизовали кожу головы животных 70%-ным этанолом и выполняли разрез длиной приблизительно 10 мм справа от срединной линии от лба до линии ушей. Вводили 3×105 клеток U87-luc (3 мкл, смесь ФСБ и Matrigel в соотношении 4:1) в правую лобную долю, которая находится на расстоянии 2 мм от правой части брегмы и на расстоянии 1 мм от передней части венечного шва у каждого животного. Разрез зашивали хирургической нитью № 6 и стерилизовали поливинилпирролидоном. Поддерживали температуру животных до восстановления жизнедеятельности. Через 6 дней после трансплантации клеток опухоли животных с опухолями группировали с использованием стратифицированной рандомизации на основании величин интенсивности сигнала флуоресценции, и среднее значение биолюминесценции достигало 2,812E+07 фотонов/с, когда введение выполняли по группам. Различным группам животных вводили различные дозы в течение в общем 35 дней.
7.4 Медианная выживаемость (дни) для соединения 17 по данному изобретению и комбинации соединения 17 по данному изобретению и темозоломида в модели ортотопической ксенотрансплантации клеток U87-luc в головной мозг
Таблица 12. Медианная выживаемость, обусловленная соединением 17 и комбинацией соединения 17 с темозоломидом
a. Период времени выживания;
b. Р-значения, при сравнении каждой группы с группой «Наполнитель;.
c. Р-значения, при сравнении каждой группы с группой, получавшей разовую дозу темозоломида.
7.5 Заключение по эксперименту: иллюстративное соединение 17 по данному изобретению может значительно увеличивать медианную выживаемость животных в модели ортотопической ксенотрансплантации клеток U87-luc в головной мозг зависимым от дозировки способом. В исследовании комбинированного лекарственного препарата с темозоломидом комбинированный лекарственный препарат дополнительно увеличивает медианную выживаемость животных по сравнению с темозоломидом, используемым отдельно.
Таким образом, в данном изобретении предложен ряд новых соединений, обладающих селективной ингибирующей активностью по отношению к киназам CDK4/6, которая выше чем или сопоставима с активностью LY2835219, лекарственного вещества-кандидата, которое изучается в данный момент в клиническом исследовании III фазы, при этом некоторые соединения демонстрируют лучшую селективность. Кроме того, предпочтительное соединение хорошо всасывается при пероральном введении, и демонстрирует хорошее преодоление гематоэнцефалического барьера, и обладает значительным фармакологическим действием в модели ортотопической ксенотрансплантации клеток U87-luc в головной мозг, свидетельствуя о том, что соединения по данному изобретению перспективны для разработки новых лекарственных средств для лечения заболеваний, связанных с пролиферацией клеток, в частности злокачественных опухолей, особенно различных типов рака головного мозга, и дают новые возможности врачам и пациентам.
Наборы
В данном изобретении также предложен набор, содержащий соединения структурных формул I-V и VIII или их соответствующие таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений структурных формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси.
Кроме того, набор может дополнительно содержать инструкцию по применению.
Фармацевтические композиции
Данное изобретение также относится к комбинированному продукту для лечения расстройства, связанного с пролиферацией клеток, причем комбинированный продукт содержит фармацевтически приемлемый носитель и соединения структурных формул I-V и VIII или их соответствующие таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси. Соединения или их соответствующие таутомер, мезомер, рацемат, энантиомер, диастереомер, дейтерированное соединение, пролекарство или их смесь; или фармацевтически приемлемые соли или сольваты соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси могут находиться в фармацевтической композиции в эффективном количестве или терапевтически эффективном количестве.
При использовании в данном документе, «эффективное количество» относится к количеству, которое является функциональным и активным по отношению к людям и/или животным и приемлемым по отношению к людям и/или животным.
При использовании в данном документе, «фармацевтически приемлемые» ингредиенты представляют собой пригодные для применения у людей и/или животных (таких как млекопитающие или птицы) без нежелательных побочных эффектов (таких как токсичность, раздражение и аллергия), т.е. вещество с разумным соотношением польза/риск. «Фармацевтически приемлемый носитель» означает носитель для введения и может включать в себя различные вспомогательные вещества и разбавители и тому подобное. Такие носители могут включать в себя, но не ограничиваясь ими, воду, изотонический раствор хлорида натрия, липосомы, липиды, белки, конъюгаты белок-антитело, пептиды, целлюлозу, наногель, буферный раствор, глюкозу, глицерин, этанол и их комбинации. Выбор носителя в общем следует осуществлять таким образом, чтобы он был совместим со способом введения, что хорошо известно специалисту в данной области техники.
Эффективное количество по данному изобретению может варьироваться в зависимости от способа введения и тяжести заболевания, подвергаемого лечению. Предпочтительное эффективное количество может определить специалист в данной области техники на основании различных факторов (например, с помощью клинического исследования). Такие факторы включают в себя, но не ограничиваясь ими: фармакокинетические параметры активного ингредиента, такие как биодоступность, метаболизм, период полувыведения и т.д.; тяжесть заболевания пациента, которого подвергают лечению, массу тела пациента, иммунологический статус пациента, путь введения и т.д.
Способ лечения
Данное изобретение также относится к способу лечения расстройства, связанного с пролиферацией клеток, включающему в себя введение пациенту перорально или неперорально эффективного количества соединений структурных формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси; или фармацевтически приемлемых солей или сольватов соединений формул I-V и VIII или их соответствующих таутомера, мезомера, рацемата, энантиомера, диастереомера, дейтерированного соединения, пролекарства или их смеси, или вышеуказанной фармацевтической композиции.
Пероральный или непероральный пути могут представлять собой введение в желудочно-кишечный тракт, назальное введение, интратрахеальное введение, внутрилегочное введение, введение через вены или эпидермис неповрежденных участков, внутрикожное введение, подкожное введение, внутрисердечное введение, внутримышечное введение, внутрикостное введение, внутрибрюшинное введение, эпидуральное введение, трансбуккальное введение, сублингвальное введение, введение через глаз, ректальное введение, вагинальное введение, уретральное введение, введение через наружный слуховой канал или другие пути. Предпочтительные способы введения включают в себя пероральное введение, введение через дыхательные пути, инъекцию, трансдермальное введение, введение через слизистые или введение в полости.
При этом пероральное введение включает в себя проглатывание, сублингвальное введение и тому подобное. Способ введения через дыхательные пути включает в себя ингаляцию, такую как ингаляция с помощью ультразвукового распылителя, ингаляция с помощью кислородного аэрозольного распылителя, ингаляция с помощью распылителя ручного действия и тому подобное. Инъекционный способ введения включает в себя артериальную инъекцию, внутривенную инъекцию, внутримышечную инъекцию, внутрисердечную инъекцию, внутрикожную инъекцию и тому подобное. Способы трансдермального введения включают в себя ионтофорез, электропорацию и тому подобное. Способ введения через слизистые включает в себя введение через слизистую оболочку носа, введение через слизистую оболочку ротовой полости, введение через слизистую оболочку глаза, введение через слизистую оболочку прямой кишки, введение через слизистую оболочку матки и введение через слизистую оболочку влагалища. Способ введения в полость включает в себя ректальное введение, вагинальное введение, уретральное введение, назальное введение и введение через наружный слуховой канал.
Все упоминания, приведенные в данном изобретении (в том числе патентные документы или документы, не являющиеся патентами), включены в данный документ посредством ссылки, как если бы каждый был отдельно включен посредством ссылки.
Хотя изобретение было в известной мере описано, можно сделать несомненно пригодные изменения различных условий, не отклоняясь от сущности и объема изобретения. Понятно, что изобретение не ограничено описанием вариантов реализации изобретения, а определяется объемом формулы изобретения, которая включает в себя эквиваленты каждого описанного элемента.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 4-АМИНО-3-ГАЛОГЕН-6(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ И 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2658825C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| АЗЕТИДИНИЛДИАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИН-ЛИПАЗЫ | 2010 |
|
RU2569298C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДОЛ-5-ОЛА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2674249C2 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2667788C2 |
| АЗЕТИДИНИЛДИАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИН-ЛИПАЗЫ | 2010 |
|
RU2549547C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2788916C2 |
Изобретение относится к соединениям формулы I, их дейтерированным производным и фармацевтически приемлемым солям, где в формуле I R1 представляет собой атом водорода или C1-C6 алкил; R2 и R3 представляют собой C1-C6 алкил или R2 и R3 вместе с атомом C, к которому они присоединены, образуют C3-C6 циклоалкил; R4 и R5 независимо выбраны из водорода и галогена, и по меньшей мере один из R4 и R5 представляет собой галоген; R6 выбран из атома водорода или C1-C6 алкила; R7 представляет собой 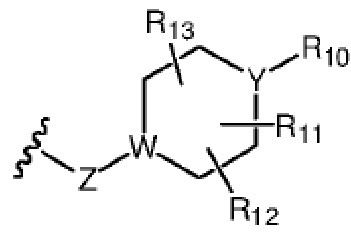 , Z представляет собой O или
, Z представляет собой O или 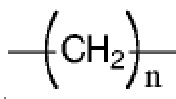 , и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C; R10, R11, R12 и R13 независимо выбраны из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила или циклопропилметила, или R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий один атом N, который замещен
, и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C; R10, R11, R12 и R13 независимо выбраны из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила или циклопропилметила, или R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий один атом N, который замещен  ; R8 представляет собой C1-C6 гидроксиалкил. Также изобретение относится к промежуточным соединениям формулы VI, способу получения и применению соединений формулы I, фармацевтической композиции, набору и способам лечения. Технический результат – соединения формулы I, обладающие ингибирующей активностью в отношении CDK 4/6. 9 н. и 20 з.п. ф-лы, 12 табл., 52 пр.
; R8 представляет собой C1-C6 гидроксиалкил. Также изобретение относится к промежуточным соединениям формулы VI, способу получения и применению соединений формулы I, фармацевтической композиции, набору и способам лечения. Технический результат – соединения формулы I, обладающие ингибирующей активностью в отношении CDK 4/6. 9 н. и 20 з.п. ф-лы, 12 табл., 52 пр.
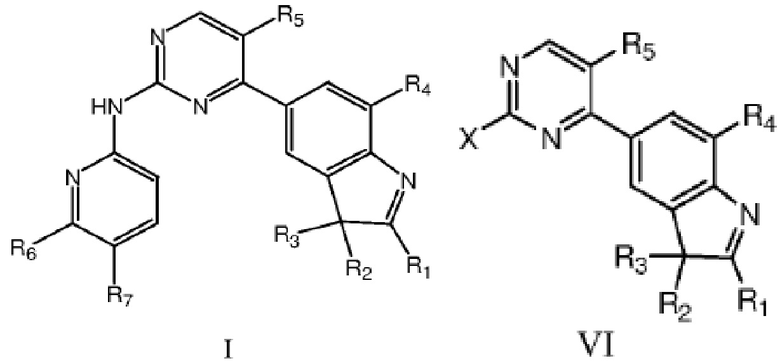
1. Соединение структурной формулы I,
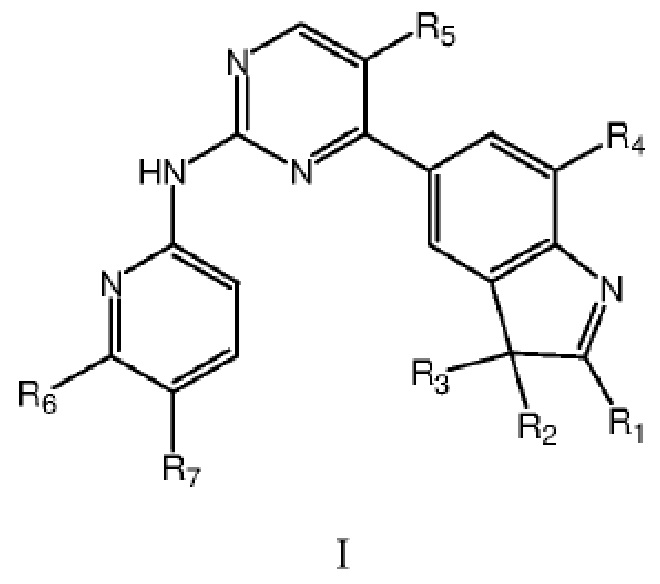
где R1 представляет собой атом водорода или C1-C6 алкил;
каждый из R2 и R3 независимо представляет собой C1-C6 алкил или R2 и R3 вместе с атомом C, к которому они оба присоединены, образуют C3-C6 циклоалкил;
каждый из R4 и R5 независимо выбран из группы, состоящей из водорода и галогена, и по меньшей мере один из R4 и R5 представляет собой галоген;
R6 выбран из атома водорода или C1-C6 алкила;
R7 представляет собой 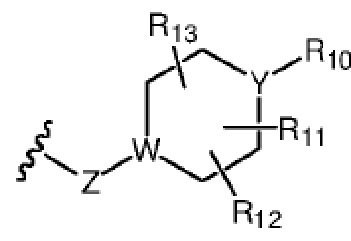 , где Z представляет собой O или
, где Z представляет собой O или 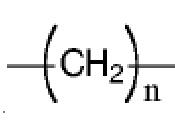 , и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C;
, и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C;
каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила или циклопропилметила; или
R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий один атом N, который замещен  ;
;
где R8 представляет собой C1-C6 гидроксиалкил;
или его дейтерированное соединение, или фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 выбран из атома водорода, незамещенного линейного или разветвленного C1-C4 алкила, а R2 и R3, каждый независимо представляет собой незамещенный линейный или разветвленный C1-C4 алкил.
3. Соединение по п.1 или 2, где R2 и R3 вместе с атомом C, к которому они оба присоединены, образуют C3-C6 циклоалкил.
4. Соединение по любому из пп. 1-3, где R4 и R5, каждый независимо выбран из водорода, фтора или хлора, и по меньшей мере один из R4 и R5 представляет собой фтор или хлор.
5. Соединение по п.4, где R4 и R5, каждый независимо представляет собой водород или фтор, и по меньшей мере один из R4 и R5 представляет собой фтор.
6. Соединение по п.5, где R4 представляет собой водород или фтор, а R5 представляет собой фтор.
7. Соединение по любому из пп. 1-6, где Z представляет собой  , n равно целому числу от 0 до 2.
, n равно целому числу от 0 до 2.
8. Соединение по п.7, где n= 0 или 1.
9. Соединение по любому из пп. 1-6, где R7 выбран из заместителей следующих структур:
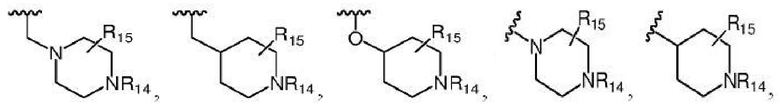
и 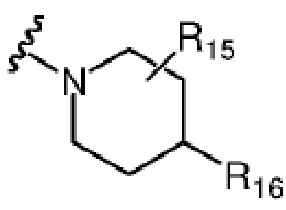 ,
,
где R14 и R15, каждый независимо выбран из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила, циклопропилметила; R16 выбран из атома водорода, C1-C6 гидроксиалкила.
10. Соединение по любому из пп. 1-6, где R6 и R7 вместе с атомами C, к которым они присоединены, образуют следующую химическую структуру:
 , где R17 выбран из гидроксила.
, где R17 выбран из гидроксила.
11. Соединение по п. 1, где соединение представлено структурной формулой II, III, IV или V:
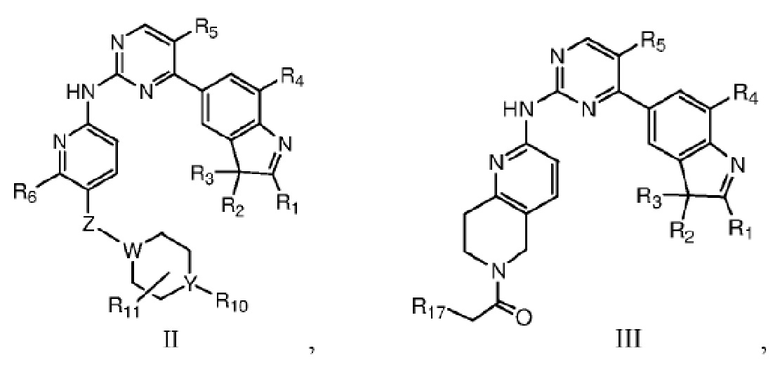
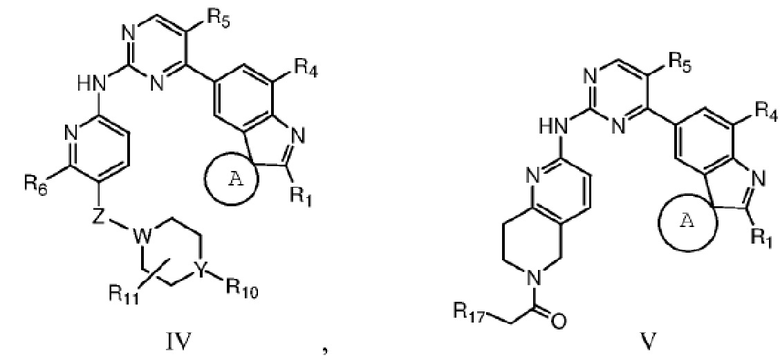 ,
,
где R1, R2 и R3 такие, как определено в любом из пп. 1-3, R4 и R5 такие, как определено в пп. 1, 4, 5 или 6; R6 такой, как определено в п. 1; R10 и R11 такие, как определено в п. 1; R17 такой, как определено в п. 10; Z, W и Y такие, как определено в пп. 1, 7 или 8;
кольцо A представляет собой насыщенное 3-6-членное кольцо.
12. Соединение по п. 1, где соединение представлено структурной формулой VIII:
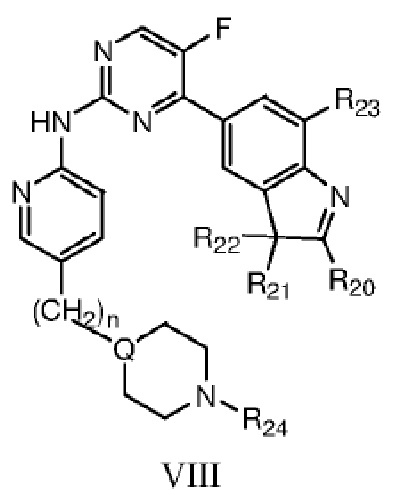
где R20, R21, R22, каждый независимо выбран из C1-C4 алкила, или R20 представляет собой C1-C4 алкил, а R21 и R22 вместе с атомом C, к которому они присоединены, образуют насыщенное 5-6-членное кольцо; R23 выбран из водорода или фтора; n=0 или 1; R24 выбран из водорода, C1-C4 алкила или C1-C4 гидроксиалкила, и Q представляет собой C или N.
13. Соединение по п. 1, где соединение представлено одной из следующих структур:
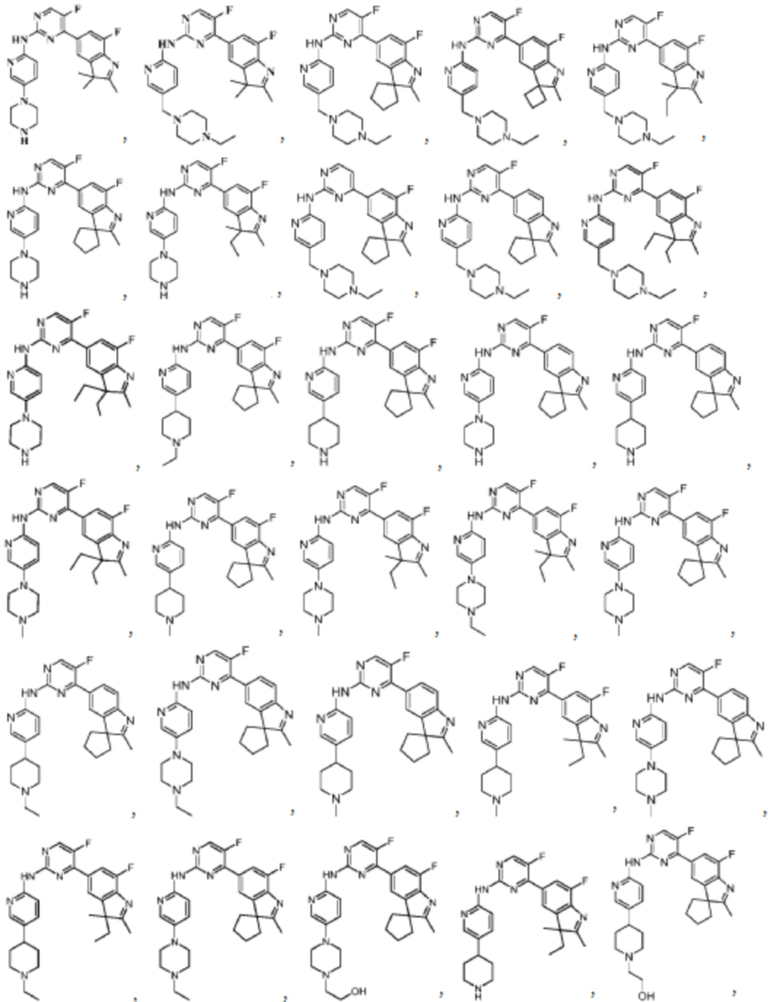
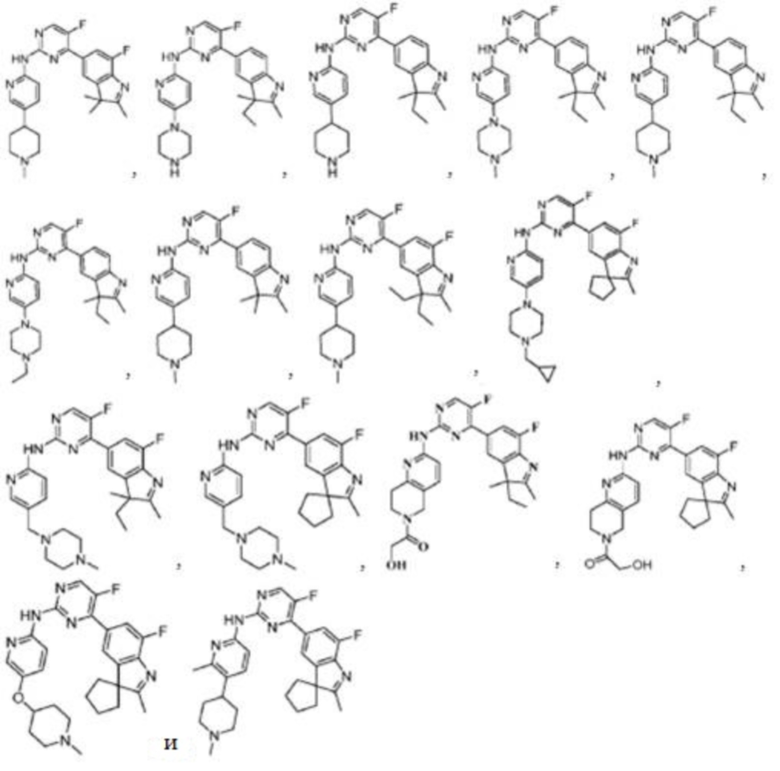 .
.
14. Соединение структурной формулы VI:
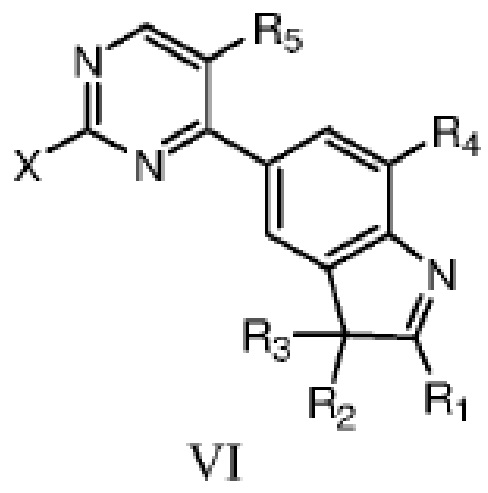
где R1 представляет собой атом водорода или C1-C6 алкил, каждый из R2 и R3 независимо представляет собой C1-C6 алкил или R2 и R3 вместе с атомом C, к которому они оба присоединены, образуют C3-C6 циклоалкил;
каждый из R4 и R5 независимо выбран из группы, состоящей из водорода и галогена, и по меньшей мере один из R4 и R5 представляет собой галоген;
X представляет собой галоген или амино;
или его дейтерированное соединение, или фармацевтически приемлемая соль.
15. Соединение по п. 14, где Х представляет собой фтор, бром, хлор или амино.
16. Способ получения соединения структурной формулы I, или его дейтерированного соединения, или фармацевтически приемлемой соли, который включает в себя проведение катализируемой палладием реакции кросс-сочетания между соединением формулы VI и соединением формулы VII в растворителе, что дает соединение формулы I,
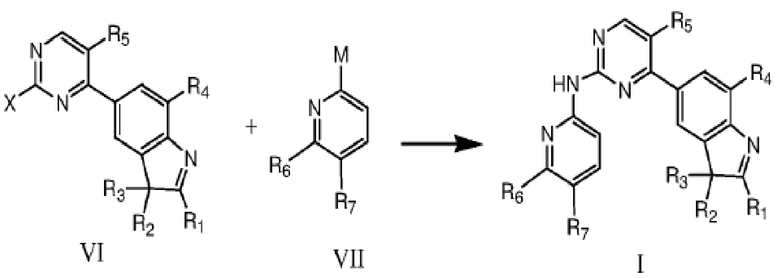
где R1 представляет собой атом водорода или C1-C6 алкил;
каждый из R2 и R3 независимо представляет собой C1-C6 алкил или R2 и R3 вместе с атомом C, к которому они оба присоединены, образуют C3-C6 циклоалкил;
каждый из R4 и R5 независимо выбран из группы, состоящей из водорода и галогена, и по меньшей мере один из R4 и R5 представляет собой галоген;
R6 выбран из атома водорода или C1-C6 алкила;
R7 представляет собой 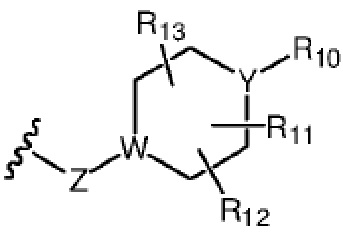 , где Z представляет собой O или
, где Z представляет собой O или  , и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C;
, и n равен целому числу от 0 до 4; каждый из W и Y независимо представляет собой C или N, но оба W и Y не могут одновременно представлять собой C, и если Z представляет собой O, W представляет собой C;
каждый из R10, R11, R12 и R13 независимо выбран из атома водорода, C1-C6 алкила, C1-C6 гидроксиалкила или циклопропилметила; или
R6 и R7 вместе с атомами C, к которым они присоединены, образуют 6-членный гетероцикл, содержащий один атом N, который замещен  ;
;
где R8 представляет собой C1-C6 гидроксиалкил;
Х и М каждый независимо представляет собой галоген или амино, и только один из Х и М представляет собой амино, и один из двух должен представлять собой амино.
17. Способ по п. 16, где галоген представляет собой фтор, бром или хлор.
18. Применение соединения 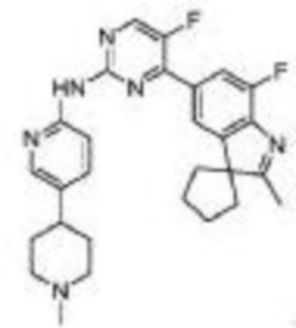 , или его дейтерированного соединения, или фармацевтически приемлемой соли при производстве лекарственного средства, обладающего ингибирующей активностью в отношении CDK 4/6, для лечения рака молочной железы или глиомы.
, или его дейтерированного соединения, или фармацевтически приемлемой соли при производстве лекарственного средства, обладающего ингибирующей активностью в отношении CDK 4/6, для лечения рака молочной железы или глиомы.
19. Применение по п. 18, где лекарственное средство содержит фармацевтически приемлемое вспомогательное вещество.
20. Применение по п. 18 или 19, где соединение вводят субъекту, нуждающемуся в этом, в качестве единственного активного ингредиента или в комбинации с темозоломидом.
21. Применение соединения 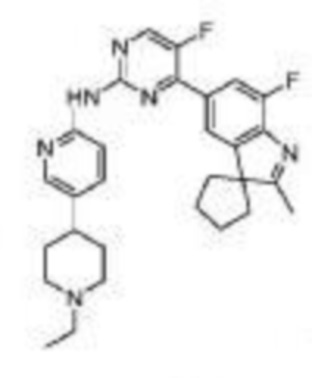 , или его дейтерированного соединения, или фармацевтически приемлемой соли при производстве лекарственного средства, обладающего ингибирующей активностью в отношении CDK 4/6, для лечения рака молочной железы.
, или его дейтерированного соединения, или фармацевтически приемлемой соли при производстве лекарственного средства, обладающего ингибирующей активностью в отношении CDK 4/6, для лечения рака молочной железы.
22. Применение по п. 21, где лекарственное средство содержит фармацевтически приемлемое вспомогательное вещество.
23. Применение по п. 21 или 22, где соединение вводят субъекту, нуждающемуся в этом, в качестве единственного активного ингредиента или в комбинации с темозоломидом.
24. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении CDK 4/6, для лечения рака молочной железы или глиомы, содержащая эффективное количество соединения по любому из пп. 1-13 и фармацевтически приемлемые вспомогательные вещества.
25. Набор, содержащий фармацевтическую композицию по п. 24 и инструкцию по применению.
26. Способ лечения рака молочной железы или глиомы, включающий в себя введение субъекту, нуждающемуся в этом, перорально или парентерально эффективного количества соединения , или его дейтерированного соединения, или фармацевтически приемлемой соли.
27. Способ по п. 26, где соединение вводят субъекту, нуждающемуся в этом, в качестве единственного активного ингредиента или в комбинации с темозоломидом.
28. Способ лечения рака молочной железы, включающий в себя введение субъекту, нуждающемуся в этом, перорально или парентерально эффективного количества соединения, или его дейтерированного соединения, или фармацевтически приемлемой соли.
29. Способ по п. 28, где соединение вводят субъекту, нуждающемуся в этом, в качестве единственного активного ингредиента или в комбинации с темозоломидом.
| ЕА 201170872 А1, 31.12.2011 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| CN 0102007124 A, 06.04.2011 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ИНГИБИТОРЫ JNK | 2009 |
|
RU2504545C2 |

