Перекрестная ссылка на родственные заявки
В настоящей заявке испрашивается приоритет предварительной заявки на патент США №62/884,480, поданной 8 августа 2019 г., содержание которой полностью включено в настоящую заявку посредством ссылки.
Предпосылки создания изобретения
Изоцитратдегидрогеназы (ИДГ) катализируют окислительное декарбоксилирование изоцитрата до 2-оксоглутарата (т.е. α-кетоглутарата). Эти ферменты принадлежат к двум различным подклассам, один из которых использует НАД(+) в качестве акцептора электронов, а другой - НАДФ(+). Было сообщено о пяти изоцитратдегидрогеназах: трех НАД(+)-зависимых изоцитратдегидрогеназах, локализующихся в митохондриальном матриксе, и двух НАДФ(+)-зависимых изоцитратдегидрогеназах, одна из которых является митохондриальной, а другая преимущественно цитозольной. Каждый НАДФ(+)-зависимый изофермент представляет собой гомодимер.
ИДГ1 (изоцитратдегидрогеназа 1 (НАДФ+), цитозольная) также известна как IDH; IDP; IDCD; IDPC или PICD. Белок, кодируемый этим геном, представляет собой НАДФ(+)-зависимую изоцитратдегидрогеназу, обнаруженную в цитоплазме и пероксисомах. Он содержит сигнальную последовательность пероксисомального нацеливания PTS-1. Присутствие этого фермента в пероксисомах предполагает его роль в регенерации NADPH для интрапероксисомального восстановления, такого как превращение 2,4-диеноил-CoAs в 3-еноил-CoAs, а также в пероксисомальных реакциях, в которых расходуется 2-оксоглутарат, а именно альфа-гидроксилирование фитановой кислоты. Цитоплазматический фермент играет важную роль в цитоплазматической продукции NADPH.
Ген ИДГ1 человека кодирует белок из 414 аминокислот. Нуклеотидные и аминокислотные последовательности ИДГ1 человека можно найти как записи GenBank NM_005896.2 и NP_005887.2 соответственно. Нуклеотидные и аминокислотные последовательности для ИДГ1 также описаны, например, в Nekrutenko и соавт., Mol. Biol. Evol. 15: 1674-1684 (1998); Geisbrecht и соавт., J. Biol. Chem. 274: 30527-30533 (1999); Wiemann и соавт., Genome Res. 11: 422-435 (2001); The MGC Project Team, Genome Res. 14: 2121-2127 (2004); Lubec и соавт., представлено (ДЕЦ-2008) в UniProtKB; Kullmann и соавт., представлено (JUN-1996) в базах данных EMBL/GenBank/DDBJ; и Sjoeblom и соавт., Science 314: 268-274 (2006).
Было обнаружено, что мутации ИДГ1, присутствующие в некоторых раковых клетках, приводят к новой способности фермента катализировать NADPH-зависимое восстановление α-кетоглутарата до R(-)-2-гидроксиглутарата (2HG). Считается, что продукция 2HG способствует образованию и прогрессированию рака (Dang, L и соавт., Nature 2009, 462: 739-44).
ИДГ2 (изоцитратдегидрогеназа 2 (НАДФ+), митохондриальная), также известная как IDH; IDP; IDHM; IDPM; ICD-M; или мНАДФ-ИДГ. Белок, кодируемый этим геном, представляет собой НАДФ(+)-зависимую изоцитратдегидрогеназу, обнаруженную в митохондриях. Он играет роль в промежуточном метаболизме и выработке энергии. Этот белок может тесно связываться или взаимодействовать с комплексом пируватдегидрогеназы. Ген человека ИДГ2 кодирует белок из 452 аминокислот. Нуклеотидные и аминокислотные последовательности для ИДГ2 можно найти как записи в GenBank NM_002168.2 и NP_002159.2 соответственно. Нуклеотидная и аминокислотная последовательность для ИДГ2 человека также описаны, например, в публикации Huh и соавт., представленной (NOV-1992) в базах данных EMBL/GenBank/DDBJ; и The MGC Project Team, Genome Res. 14: 2121-2127 (2004).
Немутантный, например, дикий тип ИДГ2 катализирует окислительное декарбоксилирование изоцитрата до α-кетоглутарата (α-KG).
Было обнаружено, что мутации ИДГ2, присутствующие в некоторых раковых клетках, приводят к новой способности фермента катализировать NADPH-зависимое восстановление α-кетоглутарата до R(-)-2-гидроксиглутарата (2HG). 2HG не образуется посредством ИДГ2 дикого типа. Считается, что продукция 2HG способствует образованию и прогрессированию рака (Dang, L и соавт., Nature 2009, 462: 739-44).
Мутации в ИДГ1 или ИДГ2 встречаются в более чем 70% опухолей диффузной глиомы низкой степени злокачественности (LGG). Мутации ИДГ приводят к накоплению 2-HG, который, как полагают, способствует онкогенезу за счет гиперметилирования ДНК, усиления репрессивного метилирования гистонов и ингибирования процессов дифференцировки. Исследования, проведенные с использованием вспомогательного соединения, известного как AGI-5198, которое, как было показано, ингибирует мутантный ИДГ1 (mIDH1), но не мутантный ИДГ2 (mIDH2), продемонстрировали, что ингибирование белков mIDH1 может подавлять рост mIDH1-управляемых глиом в некоторых модельных системах (D. Rohle и соавт. Science 340: 626-630 (2013)).
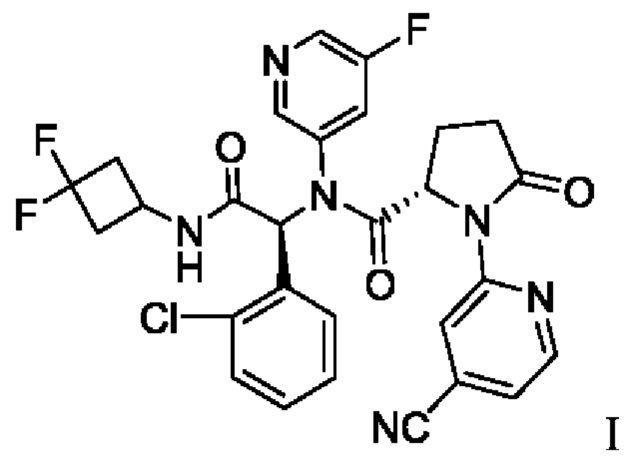
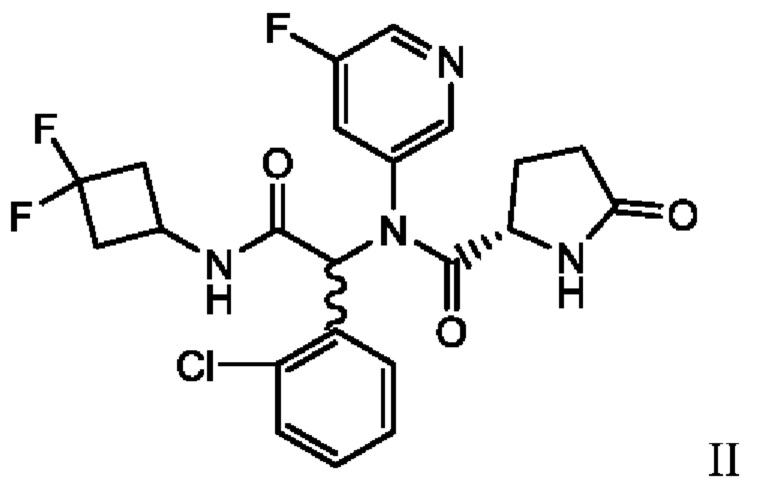
В патенте США №9,474,779 В2 и патенте США №9,968,595 В2, полностью включенных в настоящую заявку посредством ссылки, раскрыто соединение, описанное химическим названием (S)-N-((S)-1-(2-Хлорфенил)-2-(3,3-дифторциклобутиламино)-2-оксоэтил)-1-(4-цианопиридин-2-ил)-N-(5-фторпиридин-3-ил)-5-оксопирролидин-2-карбоксамид, который, как было показано, действует в качестве ингибитора мутантных белков ИДГ1 в биохимических и клеточных анализах. В зависимости от принятого названия это соединение также может обозначено как (2S)-N-{(1S)-1-(2-хлорфенил)-2-[(3,3-дифторциклобутил)амино]-2-оксоэтил}-1-(4-цианопиридин-2-ил)-N-(5-фторпиридин-3-ил)-5-оксопирролидин-2-карбоксамид. Далее, в 2015 году соединению было присвоено следующее МНН: ивосидениб. Структура ивосидениба описана в настоящей заявке как соединение I:
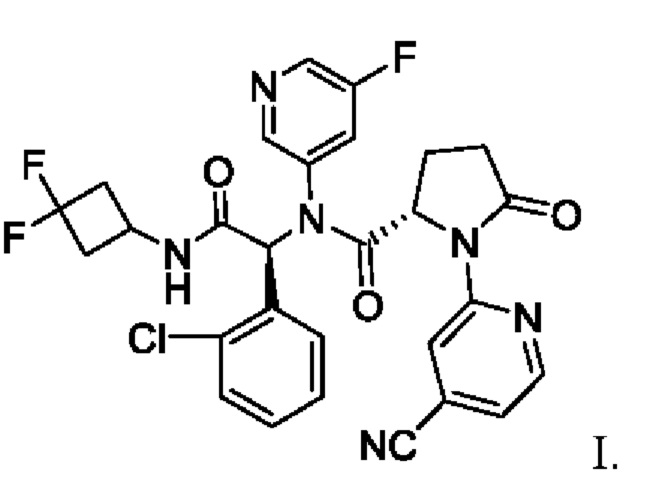
Существует потребность в эффективном и крупномасштабном производственном способе получения соединения I для использования при лечении пациентов с гематологическими злокачественными новообразованиями, такими как острый миелогенный лейкоз (ОМЛ), миелодиспластический синдром (МДС), миелопролиферативные новообразования (МПН), миелодиспластические/миелопролиферативные новообразования, хронический миеломоноцитарный лейкоз (ХММЛ), В-клеточный острый лимфобластный лейкоз (В-ОЛЛ), или лимфома (например, Т-клеточная лимфома), или солидная опухоль, такая как глиома, холангиокарцинома, хондросаркома, рак предстательной железы, рак толстой кишки, меланома или немелкоклеточный рак легкого (НМРЛ), причем каждый из вышеперечисленных характеризуется наличием мутантного аллеля ИДГ1. В некоторых вариантах осуществления гематологическое злокачественное новообразование является рецидивирующим или рефрактерным; в других вариантах осуществления гематологическое злокачественное новообразование диагностировано впервые или пациент не подходит для интенсивной индукционной химиотерапии. В некоторых вариантах осуществления гематологическое злокачественное новообразование представляет собой рецидивирующий или рефрактерный острый миелогенный лейкоз (ОМЛ). В других вариантах осуществления гематологическое злокачественное новообразование представляет собой впервые диагностированный острый миелогенный лейкоз (ОМЛ). В других вариантах осуществления солидная опухоль представляет собой холангиокарциному. В других вариантах осуществления холангиокарциному выбирают из внутрипеченочной холангиокарциномы, околокорневой холангиокарциномы и дистальной холангиокарциномы. В еще других вариантах осуществления у пациента имеется хондросаркома. В других вариантах осуществления гематологическое злокачественное новообразование или солидная опухоль находятся на поздних стадиях.
Краткое изложение сущности изобретения
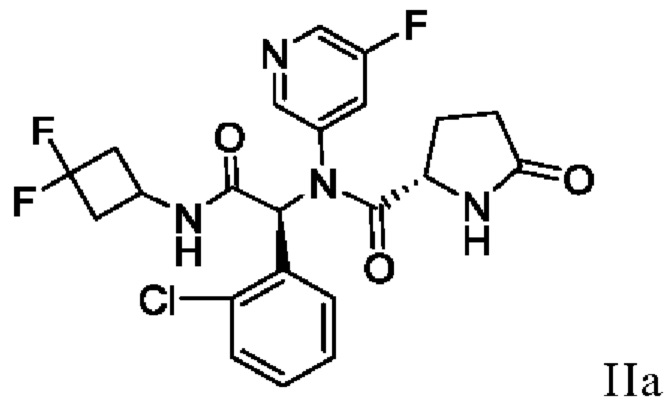
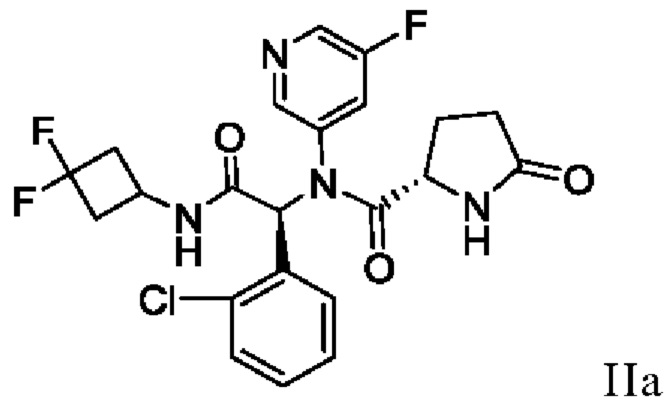
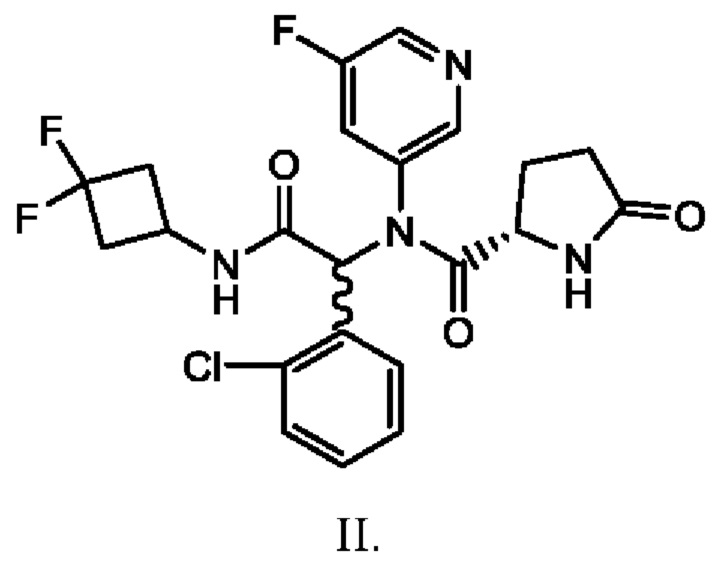
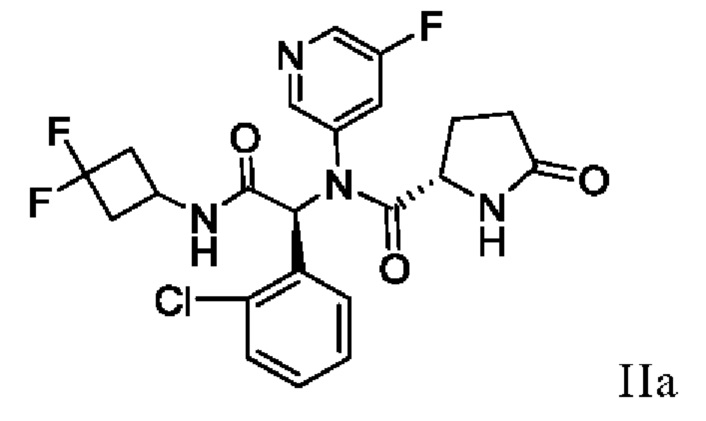
В одном аспекте настоящая заявка относится к способу получения соединения I, включающему в себя выделение кристаллического этанольного сольвата соединения IIa
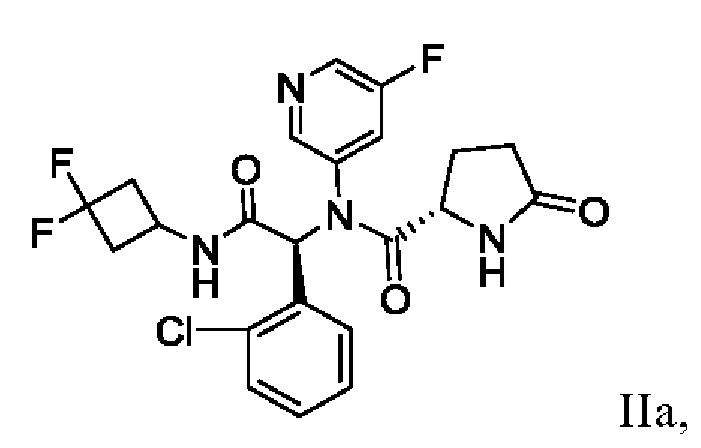
и превращение кристаллического этанольного сольвата соединения IIa в соединение I или его сольват. В некоторых аспектах кристаллический этанольный сольват соединения IIa является по существу химически и диастереомерно чистым. По существу химически и диастереомерно чистый кристаллический этанольный сольват соединения IIa также называют «этанольным сольватом типа А соединения IIa». В некоторых вариантах осуществления соединение I получают в виде сольвата изопропилацетата, который можно превратить в другие сольваты, а также в несольватную или безводную твердую форму.
В другом аспекте настоящая заявка относится к способу получения этанольного сольвата типа А соединения IIa, включающему в себя суспендирование или растворение смеси соединений IIa и IIb в растворе системы растворителей основания и этанола, добавление неполярного растворителя и выделение этанольного сольвата типа А соединения IIa. Используемый в настоящей заявке термин «суспендирование» означает приведение материала в контакт с растворителем или смесью растворителей таким образом, что суспендированный материал растворяется по меньшей мере частично.
В другом аспекте настоящая заявка относится к кристаллическому этанольному сольвату типа А соединения IIa per se. В некоторых вариантах осуществления этанольный сольват типа А является по существу химически чистым. В других вариантах осуществления этанольный сольват типа А является по существу диастереомерно чистым. В других вариантах осуществления этанольный сольват типа А является по существу химически и диастереомерно чистым.
Краткое описание чертежей
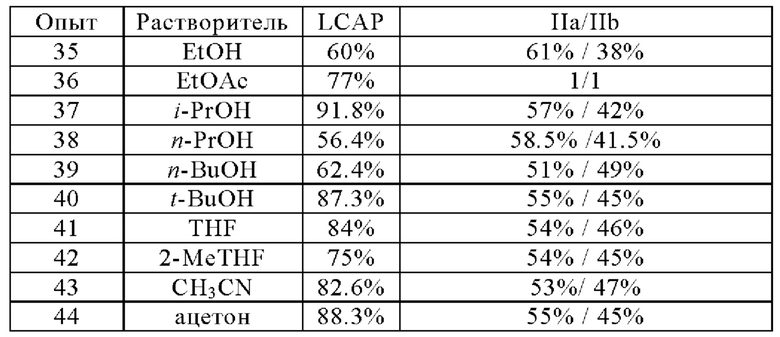
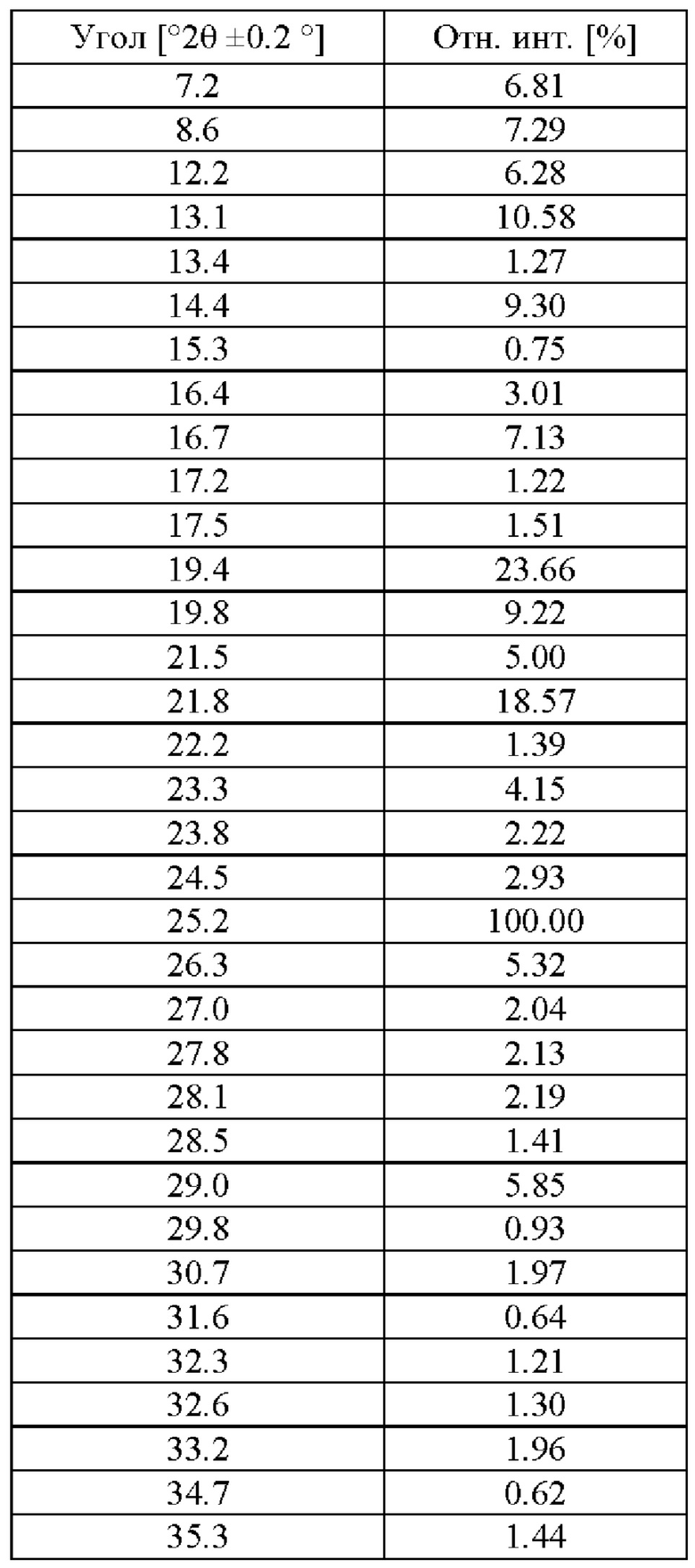
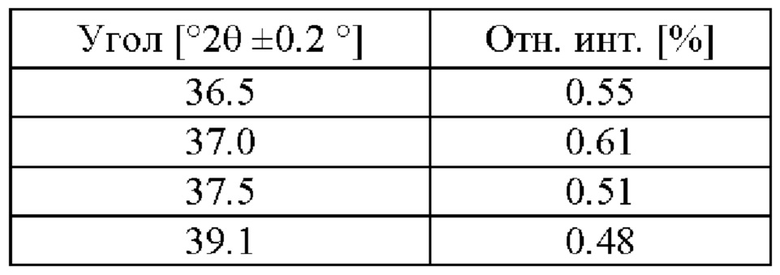
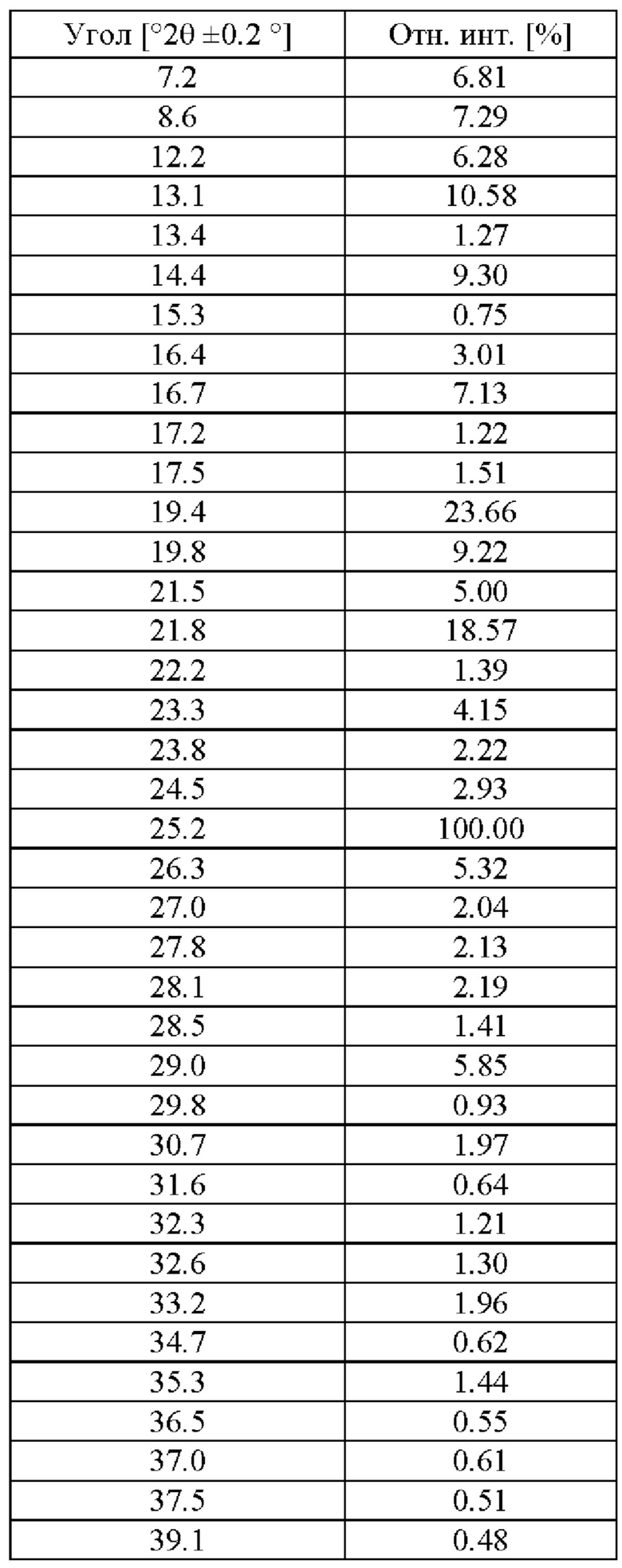
На фигуре 1 представлена рентгеновская порошковая дифрактограмма (РПД) этанольного сольвата типа А соединения IIa.
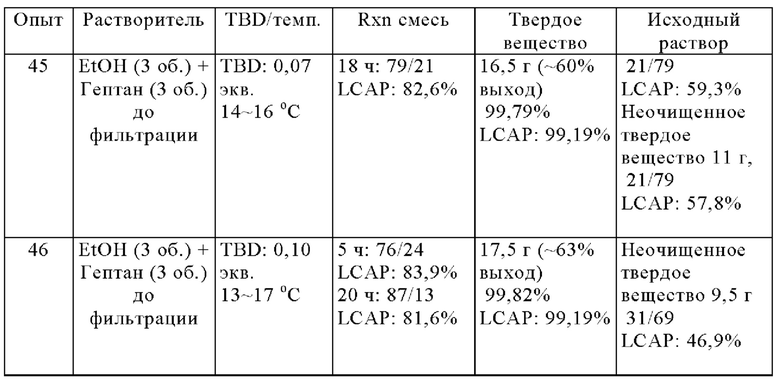
На фигуре 2 представлена дифференциальная сканирующая калориметрия (DCS) и профили термогравиметрического анализа (TGA) этанольного сольвата типа А соединения IIa.
Подробное описание чертежей и различных вариантов осуществления
В одном аспекте настоящая заявка относится к способу стереохимического обогащения смеси соединений формулы II:
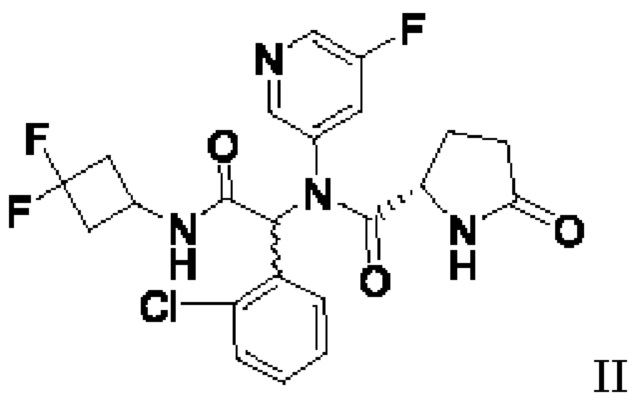
с получением этанольного сольвата соединения IIa:
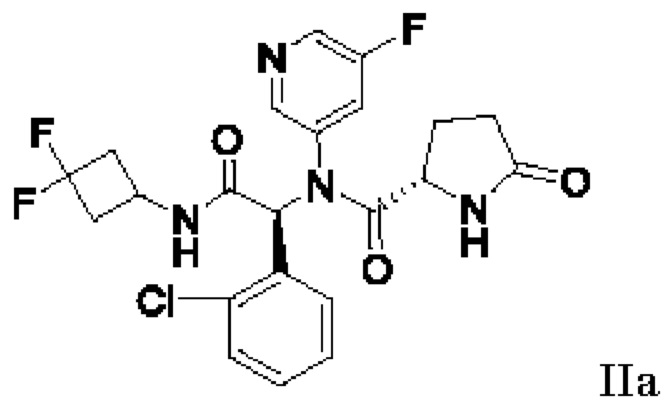
в виде по существу химически и диастереомерно чистого кристаллического материала. Используемый в настоящей заявке термин «по существу химически и диастереомерно чистое» соединение IIa относится к этанольному сольвату соединения IIa, который имеет диастереомерный избыток по меньшей мере приблизительно 90% и химическую чистоту по меньшей мере приблизительно 80%. В некоторых вариантах осуществления по существу диастереомерно чистый кристаллический этанольный сольват соединения IIa имеет диастереомерный избыток по меньшей мере приблизительно 90%, по меньшей мере приблизительно 91%, по меньшей мере приблизительно 92%, по меньшей мере приблизительно 93%, по меньшей мере приблизительно 94%, по меньшей мере приблизительно 95%, по меньшей мере приблизительно 96%, по меньшей мере приблизительно 97%, по меньшей мере приблизительно 98%, по меньшей мере приблизительно 99%, по меньшей мере приблизительно 99,5%, по меньшей мере 99,7%, по меньшей мере 99,8%, по меньшей мере 99,9% (или любой процент между 90% и 100%). Используемый в настоящей заявке в контексте химической чистоты термин «приблизительно» означает ±0,5%. Используемый в настоящей заявке в контексте диастереомерного избытка термин «приблизительно» также означает ±0,5%. Используемый в настоящей заявке диастереомерный избыток (или ди %) рассчитывают по следующей формуле:
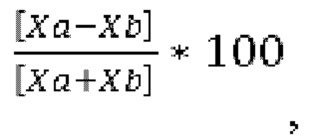
где Ха и Xb представляют собой молярные доли диастереомеров IIa и IIb, где соединение IIb имеет следующую структуру:
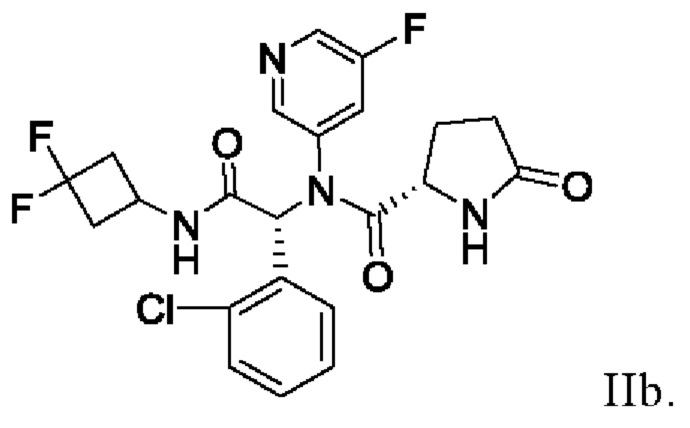
В других вариантах осуществления кристаллический этанольный сольват соединения IIa имеет химическую чистоту по меньшей мере приблизительно 80%, по меньшей мере приблизительно 81%, по меньшей мере приблизительно 82%, по меньшей мере приблизительно 83%, по меньшей мере приблизительно 84%, по меньшей мере приблизительно 85%, по меньшей мере приблизительно 86%, по меньшей мере приблизительно 87%, по меньшей мере приблизительно 88%, по меньшей мере приблизительно 89%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 91%, по меньшей мере приблизительно 92%, по меньшей мере приблизительно 93%, по меньшей мере приблизительно 94%, по меньшей мере приблизительно 95%, по меньшей мере приблизительно 96%, по меньшей мере приблизительно 97%, по меньшей мере приблизительно 98%, по меньшей мере приблизительно 99%, по меньшей мере приблизительно 99.5%, по меньшей мере 99.7%, по меньшей мере 99.8%, по меньшей мере 99,9% по массе (или любой процент между приблизительно 90% до приблизительно 100%) в пересчете на массу соединения IIa (на несольватированной или безводной основе). Следует понимать, что в контексте химической чистоты оставшийся массовый процент обычно включает другие материалы, такие как, например, реакционные примеси, исходные материалы, реагенты, побочные продукты и/или другие технологические примеси, возникающие в результате получения и/или выделения и/или стадий очистки. Например, кристаллический этанольный сольват соединения IIa можно считать по существу химически чистым, если было определено, что он имеет химическую чистоту более чем приблизительно 80% по массе, при измерении стандартными и общепринятыми методами, известными в данной области техники, где оставшиеся менее приблизительно 20% по массе составляют другие материалы, такие как другие реакционные примеси, исходные материалы, реагенты, побочные продукты и/или технологические примеси. Химическую чистоту можно определить методами, известными в данной области техники, например, высокоэффективной жидкостной хроматографией (ВЭЖХ), ЖХ-МС (жидкостная хроматография - масс-спектрометрия), спектроскопией ядерного магнитного резонанса (ЯМР) или инфракрасной спектроскопией. Специалист в данной области легко поймет эти методы и то, как использовать эти дополнительные (или альтернативные) методы для определения или оценки химической чистоты.
Более конкретно, настоящая заявка относится к способу стереохимического обогащения смеси соединений формулы II для получения этанольного сольвата типа А соединения IIa в виде выделенного кристаллического материала с диастереомерным избытком по меньшей мере приблизительно 90%, по меньшей мере приблизительно 91%, по меньшей мере 92%, по меньшей мере 93%, по меньшей мере 94%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, или по меньшей мере 99%.
Используемый в настоящей заявке термин «смесь, содержащая соединения формулы II» и «смесь соединений формулы II» включает в себя смесь соединений IIa и IIb, где IIa и IIb присутствуют в соотношении между 1:99 и 99:1. В некоторых вариантах осуществления смесь соединений формулы II может представлять собой рацемическую смесь. В других вариантах осуществления смеси соединений формулы II соединение IIb может составлять более 50% смеси. В дополнительных вариантах осуществления смеси соединений формулы II соединение IIa может составлять более 50% смеси, но менее 100% смеси.
Было обнаружено, что этанольный сольват соединения IIa может быть селективно кристаллизован из смесей соединений формулы II с получением по существу химически и диастереомерно чистого этанольного сольвата соединения IIa. Селективная кристаллизация позволяет выделить этанольный сольват соединения IIa с диастереомерным избытком. Используемые в настоящей заявке термины «обеспечивать», «получение», «выделенный» и «выделение» относятся к получению и физическому отделению соединения I, или его сольвата, или этанольного сольвата соединения IIa из реакционной смеси. Используемый в настоящей заявке термин «приблизительно» означает ±0,5%. Выделение любого из описанных в настоящей заявке соединений, включая соединение I или его сольват, или этанольный сольват соединения IIa может быть достигнуто с использованием любого метода, известного специалистам в данной области, для отделения твердого материала от жидкости. Примеры методов отделения, пригодных для выделения соединения I или сольвата соединения I или этанольного сольвата соединения IIa включают фильтрацию.
Используемый в настоящей заявке термин «сольват» относится к кристаллической форме молекулы, атома и/или ионов, которая дополнительно содержит одну или несколько молекул растворителя или растворителей, включенных в структуру кристаллической решетки. Молекулы растворителя в сольвате могут находиться в регулярном и/или неупорядоченном расположении. Сольват может содержать либо стехиометрическое, либо нестехиометрическое количество молекул растворителя. Например, сольват с нестехиометрическим количеством молекул растворителя может образоваться в результате частичной потери растворителя из сольвата или может возникнуть непосредственно в процессе кристаллизации. В качестве альтернативы сольваты могут встречаться в виде димеров или олигомеров, содержащих более одной молекулы, или в структуре кристаллической решетки.
Используемые в настоящей заявке термины «несольватный», «несольватированный» или «безводный» относятся к кристаллической форме, которая не имеет одной или нескольких молекул растворителя или растворителей, включенных в структуру кристаллической решетки.
Соединения IIa и IIb содержат группу С-Н, которая подвержена эпимеризации и взаимопревращению стереохимической конфигурации хлорфенилзамещенного углерода соединений IIa и IIb. Поскольку соединение IIa истощается из фазы раствора кристаллизационной смеси (из-за селективной кристаллизации соединения IIa), реакция эпимеризации превращает соединение IIb в соединение IIa (и наоборот). Поскольку реакция эпимеризации, вероятно, не является стереоселективной из-за дистального расположения второго хирального центра (за исключением потенциального небольшого эффекта от второго хирального центра или стерического фактора), вероятно, смесь из IIa/IIb сохраняется в фазе раствора в соотношении 50/50. В то же время твердая фаза обогащается соединением IIa (явление, называемое в настоящей заявке стереохимическим обогащением), поскольку было обнаружено, что в некоторых случаях соединение IIa менее растворимо, чем соединение IIb, и поэтому предпочтительно осаждается из раствора. Таким образом, путем сочетания реакции эпимеризации со стадией селективной кристаллизации было обнаружено, что результирующий процесс «динамической кристаллизации», описанный в настоящей заявке стереохимически обогащает исходную смесь диастереомеров (соединения IIa и IIb) соединением IIa (которое затем выделяют), тем самым обеспечивая выход соединения IIa, превышающий теоретически возможное количество, в пересчете на количество соединения IIa в исходной смеси диастереомеров. Например, если 10 граммов смеси соединений формулы II содержат приблизительно по 5 граммов каждого из соединений IIa и IIb, то описанный выше процесс динамической кристаллизации потенциально может позволить выделить соединение IIa в количестве, превышающем 5 граммов.
В некоторых аспектах соединение IIa кристаллизуют из системы растворителей на основе этанола в присутствии основания с получением по существу химически и диастереомерно чистого этанольного сольвата соединения IIa. Используемый в настоящей заявке термин «система этанольных растворителей» представляет собой любую смесь растворителей, которая содержит по меньшей мере приблизительно 30%, по меньшей мере приблизительно 35%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 45%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 55%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 65%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 75%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 85%, по меньшей мере приблизительно 90%, по меньшей мере 92%, по меньшей мере 93%, по меньшей мере 94%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, или по меньшей мере 98% этанола по объему. В некоторых вариантах осуществления система этанольных растворителей содержит 100% этанола. В некоторых вариантах осуществления система этанольных растворителей не содержит толуола, трифторэтанола (TFE), ацетонитрила, N,N-диметилформамида (DMF), метилацетата или МТВЕ или смесей, содержащих любой из них, если только система этанольных растворителей не содержит по меньшей мере 30% (по объему) этанола.
В некоторых вариантах осуществления основание представляет собой первичный, вторичный или третичный амин. Подходящие основания включают в себя 1,5,7-триазабицикло[4.4.0]дец-5-ен («TBD»), 1,5-диазабицикло[4.3.0]нон-5-ен («DBN»), триэтиламин, диизопропиламин, N,N-диизопропилэтиламин, N-метилморфолин, морфолин, N-метилпиперазин, пиридин, бутиламин, дибутиламин или 1,5-диазабицикло(4.3.0)нон-5-ен, или их комбинацию.
Процесс динамической кристаллизации можно проводить при температуре от -5 до 25°С, от -5 до 20°С, от 0 до 25°С, от 5 до 20°С, от 10 до 18°С, от 12 до 18°С, от 12 до 17°С, от 12 до 16°С или от 12 до 15°С.
В некоторых вариантах осуществления процесс динамической кристаллизации осуществляют в присутствии по меньшей мере 0,05 эквивалентов, по меньшей мере 0.10 эквивалентов, по меньшей мере 0,15 эквивалентов, по меньшей мере 0.20 эквивалентов, или по меньшей мере 0,25 эквивалентов основания на эквивалент соединений IIa и IIb. В других вариантах осуществления процесс динамической кристаллизации осуществляют в присутствии приблизительно 0,04 эквивалентов, приблизительно 0,07 эквивалентов, приблизительно 0,10 эквивалентов, приблизительно 0,13 эквивалентов, приблизительно 0,16 эквивалентов, приблизительно 0,19 эквивалентов, приблизительно 0,22 эквивалентов или приблизительно 0,25 эквивалентов основания на эквивалент соединений IIa и IIb. Используемый в настоящей заявке термин «эквивалент соединений IIa и IIb» относится к суммарным эквивалентам двух соединений, присутствующих в смеси соединений формулы П. В других вариантах осуществления динамическую кристаллизацию проводят в присутствии приблизительно от 0,05 до приблизительно 0,10 эквивалентов, приблизительно от 0,10 до приблизительно 0,15 эквивалентов, или приблизительно от 0,15 до приблизительно 0,20 эквивалентов основания на эквивалент соединений IIa и IIb. Используемый в контексте эквивалентов основания термин «приблизительно» означает ±0,02 эквивалентов. В некоторых вариантах осуществления процесса динамической кристаллизации в соответствии с изобретением количество используемого основания является каталитическим. Используемые в настоящей заявке термины «каталитический» и «каталитическое количество» относится к приблизительно от 0,04 эквивалента к приблизительно 0,25 эквивалента основания на эквивалент соединений IIa и IIb. В некоторых вариантах осуществления каталитическое количество основания составляет приблизительно от 0,04 эквивалента до приблизительно 0,07 эквивалента, приблизительно 0,04 эквивалента до приблизительно 0,10 эквивалента, приблизительно 0,04 эквивалента до приблизительно 0,13 эквивалента, приблизительно 0,04 эквивалента до приблизительно 0,16 эквивалента, приблизительно 0,04 эквивалента до приблизительно 0,19 эквивалента, или приблизительно 0.04 эквивалентов до приблизительно 0,22 эквивалента основания на эквивалент соединений IIa и IIb. В других вариантах осуществления каталитическое количество основания составляет приблизительно до 0,04 эквивалента, приблизительно до 0,07 эквивалента, приблизительно до 0,10 эквивалента, приблизительно до 0,13 эквивалентов, приблизительно до 0,16 эквивалента, приблизительно до 0,19 эквивалентов, приблизительно до 0,22 эквивалента, или приблизительно до 0,25 эквивалента основания на эквивалент соединений IIa и IIb.
Оптимальные условия для получения и выделения по существу химически и диастереомерно чистого этанольного сольвата соединения IIa с использованием процесса динамической кристаллизации в соответствии с изобретением обычно зависят от температуры кристаллизации, растворителя, основания и количества основания. Например, в верхней части диапазона температур, описанного выше, процесс разложения становится более распространенным, чем при более низких температурах. Более высокие концентрации основания также могут увеличить разложение продукта. Кроме того, учитывая, что соединения формулы II содержат пептидные связи, нуклеофильность основания также может способствовать разложению соединений. Тем не менее, даже при использовании высоких температур, высоких концентраций основания и/или нуклеофильного основания процесс динамической кристаллизации все же может давать стереохимически обогащенное соединение IIa, хотя и с некоторыми продуктами разложения, которые могут потребовать дополнительной очистки выделенного соединения этанольного сольвата.
В другом аспекте настоящая заявка относится к способу получения по существу химически и диастереомерно чистого кристаллического этанольного сольвата соединения IIa (т.е., этанольного сольвата типа А соединения IIa), включающему в себя кристаллизацию соединения IIa из смеси соединений формулы II из системы этанольных растворителей в присутствии основания. Более конкретно, настоящая заявка относится к способу получения этанольного сольвата типа А соединения IIa, включающему в себя суспендирование или растворение смеси, содержащей соединения формулы II в системе этанольных растворителей, добавление основания и перемешивание полученной суспензии или раствора при температуре ниже 25°С в течение по меньшей мере восьми часов, перед добавлением по каплям неполярного апротонного растворителя (такого, как, например, гексан, циклогексан, или метилциклогексан или гептан) или смеси неполярных апротонных растворителей с последующим отделением этанольного сольвата в виде твердого вещества путем фильтрации. В некоторых вариантах осуществления раствор или суспензию основания и смесь, содержащую соединения формулы II в системе этанольных растворителей перемешивают при температуре от -5 до 25°С, от -5 до 20°С, от 0 до 25°С, от 5 до 20°С, от 10 до 18°С, от 12 до 18°С, от 12 до 17°С, от 12 до 16°С или от 12 до 15°С в течение до 12 часов, до 14 часов, до 16 часов, до 18 часов или до 20 часов до добавления к суспензии или раствору неполярного апротонного растворителя или смеси неполярных апротонных растворителей. В некоторых вариантах осуществления неполярный апротонный растворитель может быть выбран из четыреххлористого углерода, бензола, диэтилового эфира, хлороформа, метил-трет-бутилового эфира (МТВЕ), диоксана или толуола или смеси любых двух или более из вышеперечисленных. В некоторых вариантах осуществления неполярный апротонный растворитель может быть выбран из пентана, циклопентана, н-гептана, н-гексана, циклогексана или метилциклогексана или смеси любых двух или более из вышеперечисленных. В некоторых вариантах осуществления неполярный апротонный растворитель или смесь растворителей добавляют к суспензии или раствору по каплям в течение периода от приблизительно одной минуты до приблизительно двадцати четырех часов. В некоторых вариантах осуществления неполярный апротонный растворитель или смесь растворителей добавляют к суспензии или раствору целиком сразу или двумя или более порциями в течение периода от приблизительно одной минуты до приблизительно двадцати четырех часов.
Подходящим может быть любое основание, которое может эпимеризовать соединение IIa в соединение IIb (или наоборот). Хотя выбор основания может способствовать разложению соединений формулы II (что нежелательно), процесс динамической кристаллизации не обязательно полностью предотвращает разложение. Вместо этого степень разложения соединений следует сопоставлять с выходом выделенного стереохимически обогащенного соединения IIa.
Подходящие основания для реакции эпимеризации включают в себя 1,5,7-триазабицикло[4.4.0]дец-5-ен («TBD»), 1,5-Диазабицикло[4.3.0]нон-5-ен («DBN»), триэтиламин, диизопропиламин, N,N-диизопропилэтиламин, N-метилморфолин, морфолин, N-метилпиперазин, пиридин, бутиламин, дибутиламин или 1,5-диазабицикло(4.3.0)нон-5-ен, или их сочетание.
В некоторых вариантах осуществления процесс динамической кристаллизации осуществляют в присутствии по меньшей мере 0,05 эквивалентов, по меньшей мере 0,10 эквивалентов, по меньшей мере 0,15 эквивалентов, по меньшей мере 0.20 эквивалентов, или по меньшей мере 0,25 эквивалентов основания на эквивалент соединений IIa и IIb. В других вариантах осуществления процесс динамической кристаллизации осуществляют в присутствии приблизительно 0,04 эквивалента, приблизительно 0,07 эквивалента, приблизительно 0,10 эквивалента, приблизительно 0,13 эквивалентов, приблизительно 0,16 эквивалента, приблизительно 0,19 эквивалента, приблизительно 0,22 эквивалента или приблизительно 0,25 эквивалента основания на эквивалент соединений IIa и IIb. В других вариантах осуществления процесс динамической кристаллизации осуществляют в присутствии приблизительно от 0,05 до приблизительно 0,10 эквивалента, приблизительно от 0,10 до приблизительно 0,15 эквивалента, или приблизительно от 0,15 до приблизительно 0,20 эквивалента основания на эквивалент соединений IIa и IIb. В некоторых вариантах осуществления процесса динамической кристаллизации в соответствии с изобретением количество используемого основания является каталитическим.
В некоторых вариантах осуществления этанольный сольват типа А соединения IIa может быть получен посредством добавления смеси соединений формулы II к системе этанольных растворителей, добавления основания и перемешивания смеси при температуре от -5 до 25°С, от -5 до 20°С, от 0 до 25°С, от 5 до 20°С, от 10 до 18°С, от 12 до 18°С, от 12 до 17°С, от 12 до 16°С, или от 12 до 15°С в течение до 12 часов, 14 часов, 16 часов, 18 часов или 20 часов перед добавлением по каплям неполярного растворителя, такого как н-гептан или смесь неполярных растворителей, с последующей стадией выделения.
Этанольный сольват типа А соединения IIa может быть идентифицирован по одной или нескольким из следующих характеристик: по меньшей мере приблизительно 90%, по меньшей мере приблизительно 91%, по меньшей мере 92%, по меньшей мере 93%, по меньшей мере 94%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, или по меньшей мере 99% диастереомерного избытка и рентгеновская порошковая дифрактограмма, по сути такая, как изображена на Фигуре 1, где порошковые рентгеновские дифрактограммы были измерены с использованием порошкового дифрактометра, оснащенного источником рентгеновской трубки Cu. В таблице 1 перечислены углы, °2θ±0.2°, и межплоскостное расстояние d дифракционных пиков, идентифицированных в экспериментальной рентгеновской порошковой дифрактограмме Фиг. 1. Используемый в настоящей заявке в связи с диастереомерным избытком термин «приблизительно» означает ±2,5%.
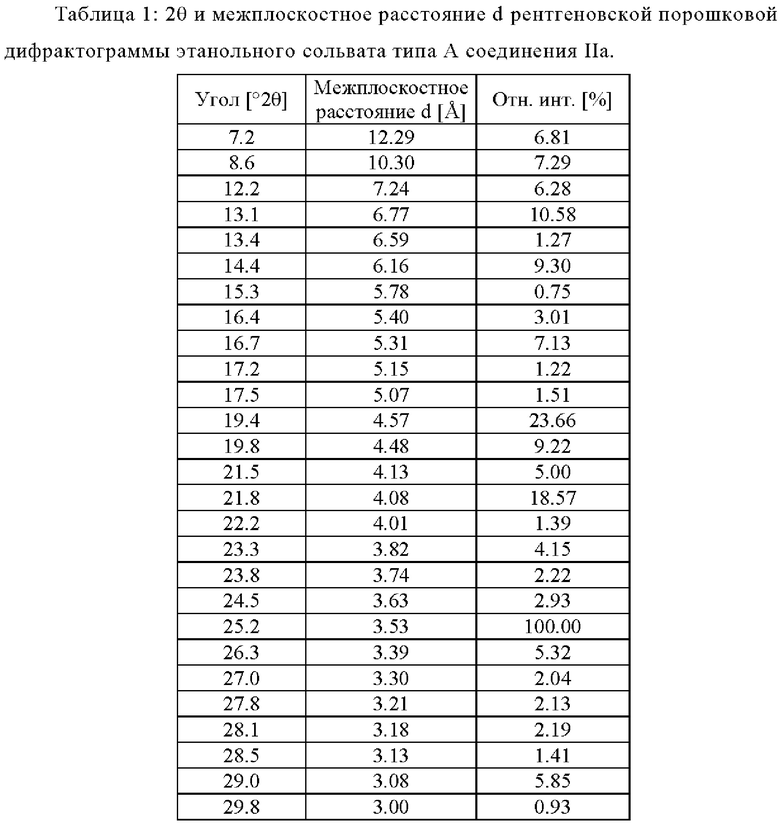
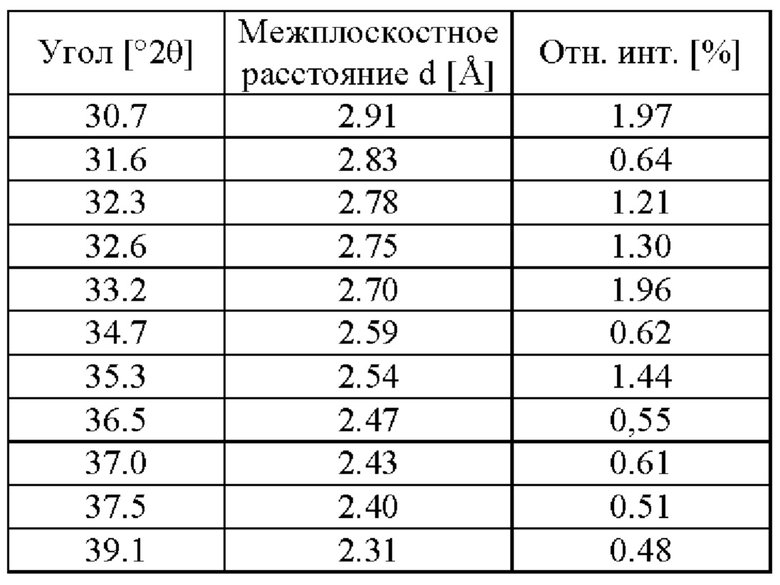
В некоторых вариантах осуществления этанольный сольват типа А соединения IIa может быть охарактеризован рентгеновской порошковой дифрактограммой (РПД), включающей один или несколько из следующих пиков при 2θ: 7.2°±0.2°, 8.6°±0.2°, 12.2°±0.2°, 13.1°±0.2°, 14.4°±0.2°, 16.7°±0.2°, 19.4°±0.2°, 19.8°±0.2°, 21.8°±0.2° и 25.2°±0.2°. В некоторых вариантах осуществления этанольный сольват типа А соединения IIa может быть охарактеризован рентгеновской порошковой дифрактограммой (РПД), включающей один, два, три, четыре, пять или шесть из следующих пиков при 2θ: 7.2°±0.2°, 8.6°±0.2°, 12.2°±0.2°, 13.1°±0.2°, 14.4°±0.2°, 16.7°±0.2°, 19.4°±0.2°, 19.8°±0.2°, 21.8°±0.2° и 25.2°±0.2°. В некоторых вариантах осуществления, этанольный сольват типа А соединения IIa может быть охарактеризован рентгеновской порошковой дифрактограммой (РПД), включающей три или более из следующих пиков при 2θ: 7.2°±0.2°, 8.6°±0.2°, 12.2°±0.2°, 13.1°±0.2°, 14.4°±0.2°, 16.7°±0.2°, 19.4°±0.2°, 19.8°±0.2°, 21.8°±0.2° и 25.2°±0.2°.
Используемый в настоящей заявке термин «рентгеновская порошковая дифрактограмма (РПД) по сути такая, как изображена» на определенной фигуре означает, что в целях сравнения присутствуют по меньшей мере 50% наиболее интенсивных пиков, показанных на фигуре. Кроме того, следует понимать, что в целях сравнения допускается некоторая вариабельность положений пиков 2-тета от показанных, например, ±0,2 градуса, и что эта вариабельность относится к каждому положению пика.
Кристаллический этанольный сольват типа А соединения IIa может быть также охарактеризован с помощью дифференциальной сканирующей калориметрии («ДСК») и термогравиметрического анализа («ТГА»). В одном варианте осуществления кристаллический этанольный сольват типа А соединения IIa показывает термограмму ДСК, включающую в себя три эндотермы при 120.6°С (пиковая температура), 136,4°С (пиковая температура) и 203.4°С (температура начала). Этанольный сольват типа А соединения IIa может быть также охарактеризован потерей массы до 10 мас. % при 150°С или ниже, как определено с помощью ТГА. В некоторых вариантах осуществления этанольный сольват типа А соединения IIa характеризуется потерей массы по меньшей мере 8,0 мас. %.
На фигуре 2 показаны термограммы ДСК и ТГА этанольного сольвата типа А соединения IIa.
В другом аспекте настоящая заявка относится к способу получения соединения I, включающему в себя получение по существу химически и диастереомерно чистого кристаллического этанольного сольвата типа А соединения IIa и превращение его в соединение I.
В некоторых аспектах получение по существу химически и диастереомерно чистого кристаллического этанольного сольвата типа А соединения IIa включает в себя стереохимическое обогащение и селективную кристаллизацию соединения IIa из системы этанольных растворителей с использованием процесса динамической кристаллизации, описанного выше. Любая смесь соединений формулы II может быть отправной точкой для получения по существу химически и диастереомерно чистого кристаллического этанольного сольвата типа А соединения IIa. В некоторых вариантах осуществления по существу химически и диастереомерно чистое соединение IIb может быть отправной точкой для получения по существу химически и диастереомерно чистого кристаллического этанольного сольвата соединения IIa.
Смесь соединений формулы II может быть приготовлена любым способом, известным специалистам в данной области. Исходные материалы для получения смеси соединений формулы II известны в данной области техники или могут быть получены в соответствии со способами, известными специалистам в данной области техники, примерами которых являются способы, описанные в WO 2013107291, WO 2007076034, WO 2006067445, WO 2006067445, Atkinson, J.G. и соавт., J. Amer. Chem. Soc. 90: 498 (1968), Berkessel. А. и соавт., Angew Chemie, Int Ed. 53: 11660 (2014); Angew. Chem. Int. Ed. Sun, X. и соавт., 52: 4440 (2013), Topolovčan, N. и соавт., Eur. J. Org. Chem. 2868 (2015), and Pavlik, J.W. и соавт., J Heterocyclic Chem. 42: 73 (2005), все из которых полностью включены в настоящую заявку посредством ссылки.
Например, смесь соединений формулы II может быть получена в соответствии со схемой 1.
Схема 1:
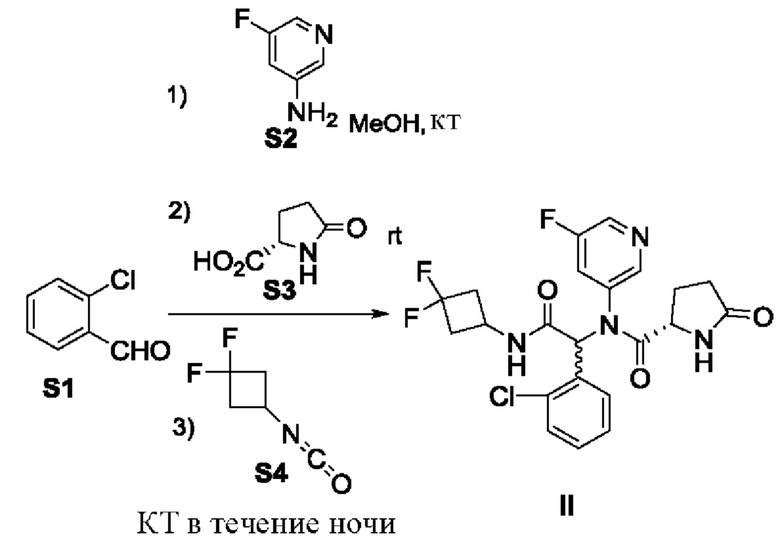
На схеме 1 смесь соединений формулы II может быть синтезирована путем взаимодействия бензальдегида S1 с первичным амином S2 в метаноле с последующим взаимодействием полученного продукта с оксопирролидин-2-карбоновой кислотой S3 и изоцианатоциклобутаном S4 с получением соединений формулы II.
Смесь соединений формулы II может быть подвергнута процессу динамической кристаллизации согласно настоящей заявке с получением по существу химически b диастереомерно чистого кристаллического этанольного сольвата типа А соединения IIa в соответствии со способом, изображенным на схеме 2.
Схема 2:
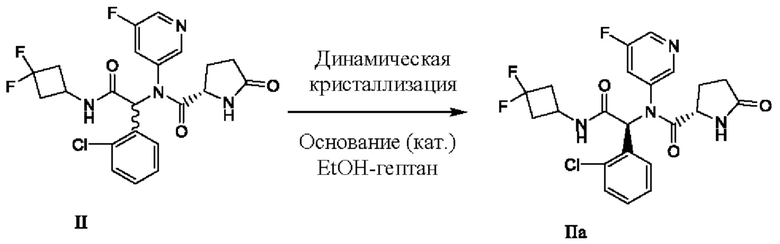
При динамической кристаллизации может быть выделен по существу химически и диастереомерно чистый этанольный сольват типа А соединения IIa с получением диастереомерного избытка по меньшей мере приблизительно 90%, по меньшей мере приблизительно 91%, по меньшей мере 92%, по меньшей мере 93%, по меньшей мере 94%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98% или по меньшей мере 99%.
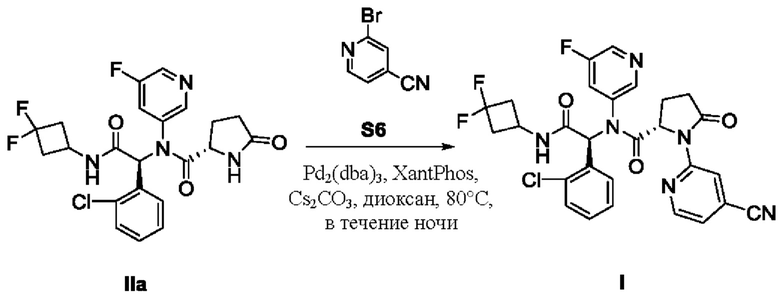
По существу химически и диастереомерно чистый кристаллический этанольный сольват типа А соединения IIa может быть превращен в соединение I в соответствии с любым способом, известным специалистам в данной области. Например, по существу химически и диастереомерно чистый кристаллический этанольный сольват типа А соединения IIa можно превратить в соединение I в соответствии со схемой 3. На схеме 3 реакция Бухвальда соединения IIa с бромизоникотинонитрилом дает соединение I.
Схема 3:
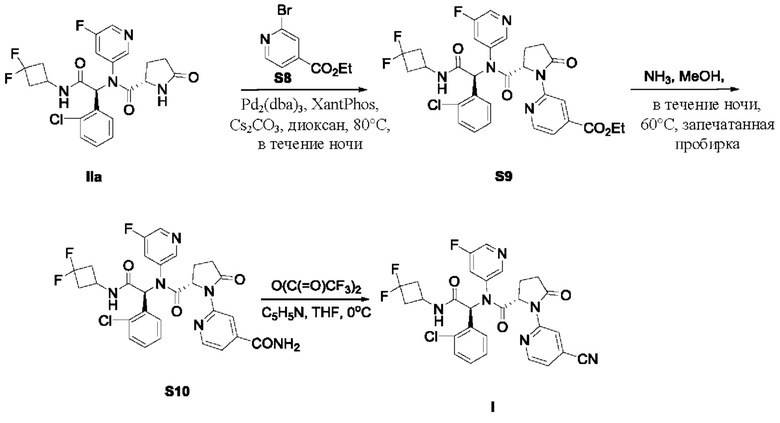
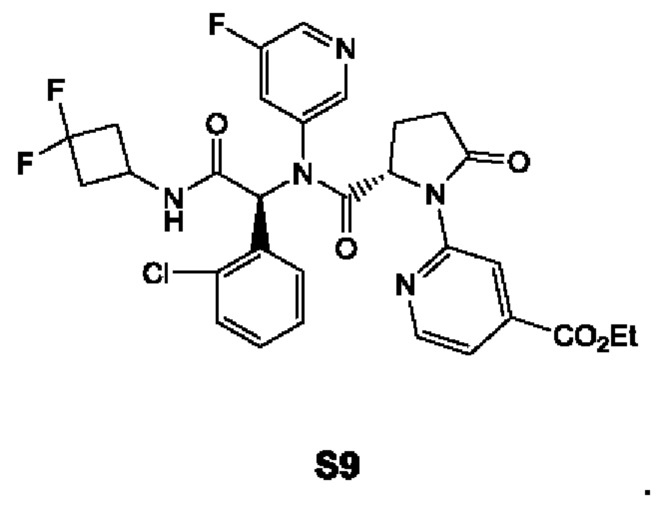
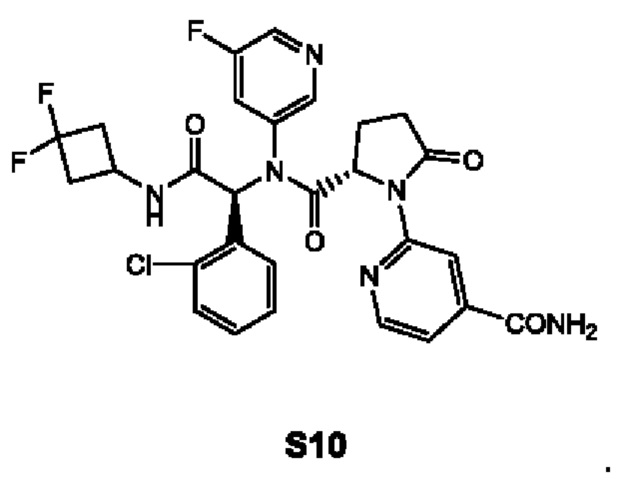
В качестве альтернативы, по существу химически и диастереомерно чистый кристаллический этанольный сольват типа А соединения IIa может быть превращен в соединение I в соответствии со схемой 4. На схеме 4 соединение I синтезируют реакцией соединения IIa с этил-2-бромизоникотинатом с получением промежуточного соединения S9. Восстановление промежуточного соединения S9 в присутствии NH3 дает амид S10. Затем амид S10 подвергают взаимодействию с трифторуксусным ангидридом (TFAA) в присутствии пиридина и подходящего растворителя с получением соединения I. Подходящие растворители включают в себя ТГФ.
Схема 4:
Экспериментальная часть
Нижеследующие примеры приведены в настоящей заявке в иллюстративных целях и никоим образом не ограничивают любой из пунктов формулы изобретения.
Сокращения:
TBD: 1,5,7-триазабицикло[4.4.0]дец-5-ен.
DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен
DBN: 1,5-Диазабицикло[4.3.0]нон-5-ен.
EtOAc: этилацетат.
IP Ас: изопропилацетат.
EtOH: этанол.
I-PrOH: изопропиловый спирт.
n-BuOH: н-бутанол.
t-BuOH: m-бутанол.
DCM: дихлорметан.
THF: тетрагидрофуран.
2-MeTHF: 2-метилтетрагидрофуран.
DMF: диметилформамид.
DMSO: диметилсульфоксид.
КТ (или кт): комнатная температура
Используемый в настоящей заявке термин «LCAP», который означает «процент площади жидкостной хроматографии», означает процент площади пика интересующего соединения по отношению к общей площади пиков.
Эксперименты ВЭЖХ проводили с использованием Agilent 1100 с детектором DAD с Phenomenex Cellulos-4, 250×4.6 мм, колонка 5 мкм и гексан/этанол в качестве подвижной фазы.
1H ЯМР: 1Н раствор ЯМР собирали на ЯМР-спектрометре Bruker 400 МГц ЯМР с использованием ДМСО-d6.
Термический анализ ДСК выполняли на ТА Q2000 DSC (от ТА Instruments). Образцы готовили в гофрированных алюминиевых ванночках и испытывали от КТ до желаемой температуры при скорости нагрева 10°С/мин с использованием N2 в качестве продувочного газа.
Термический анализ ТГА проводили на ТА Q5000 TGA (также от ТА Instruments). Образцы были приготовлены в гофрированных алюминиевых ванночках и испытаны от КТ до желаемой температуры при скорости нагрева 10°С/мин с модуляцией 60 с, используя N2 в качестве продувочного газа.
Образцы рентгеновской порошковой дифракции освещали излучением Cu Kα1 (1.540598 Å) и Cu Kα2 (1.544426 Å) при соотношении Kα1/Kα2, равном 0,50 и данные РПД собирали при от 3 до 40° 2θ.
Динамическая кристаллизация смеси, содержащей соединения IIa и IIb
Смесь 53:47 соединений формулы II (100 г, 89% А по данным ВЭЖХ, 185.0 ммоль) в этаноле (300 мл) объединяли при 15°С с 1,5,7-триазабицикло[4.4.0]дец-5-еном (2.0 г, 14,4 ммоль) затем перемешивали при 14-17°С в течение 20 ч. К реакционной смеси по каплям добавляли н-гептан (300 мл). После дальнейшего перемешивания при 14-17°С в течение 1 ч. твердые вещества собирали фильтрацией. Осадок на фильтре промывали смесью EtOH/н-гептан (200 мл, 1v/1v) ниже 25°С и сушили в вакууме при 50°С с получением соединения IIa в виде белого твердого вещества, этанольный сольват (58,5 г, чистота А 99,4%, 65% выход) при соотношении соединений IIa к IIb >99,9:0.1.
1H ЯМР (400 МГц, DMSO-d6) δ 9.0-8.9 (bm, 2Н), 8.41 (d, J=2.6 Гц, 1H), 8.02 (bs, 1Н), 7.75 (s, 1H), 7.41 (dd, J=8.1, 1.3 Гц, 1H), 7.21 (td, J=7.7, 1.7 Гц, 1H), 7.11 (td, J=7.6, 1.3 Гц, 1H), 6.89 (dd, J=7.7, 1.6 Гц, 1H), 6.38 (bs, 1H), 4.14 (p, J=6.5 Гц, 1H), 4.00 (bs, 1H), 3.05 до 2.84 (m, 2 H), 2.7 до 2.3 (m, 2H), 2.10 (ddd, J=16.8, 9.5, 7.6 Гц, 1H), 2.03 к 1.92 (m, 1H), 1.90 к 1.70 (m, 2H). Этанол δ (4.34, t, J=5.1 Гц, 1H), 3.43 (dq, J=6.9, 4.9 Гц, 2H), 1.04 (t, J=7.0 Гц, 3Н). ЖХ MC для соединения IIa ЭРИ(+) обнаружено [М+Н]+ 481.
Синтез соединения I в соответствии со схемой 2
В одном варианте осуществления по существу химически и диастереомерно чистый кристаллический этанольный сольват соединения IIa превращают в соединение I. Такое превращение включает в себя перемешивание соединения IIa (0.20 ммоль), 2-бромизоникотинонитрила (0,30 ммоль), Cs2CO3 (129 мг, 0.39 ммоль), Pd2(dba)3 (18 мг, 0,02 ммоль) и Xant-Phos (9.4 мг, 0,02 ммоль) в 1,4-диоксане (10 мл) в атмосфере N2 при 80°С в течение ночи. После фильтрации фильтрат концентрируют в вакууме, а остаток очищают стандартным методом с получением соединения I.
Синтез соединения I в соответствии со схемой 3
В качестве альтернативы, в соответствии со схемой 3, соединение IIa и этил-2-бромизоникотинат (соединение S8) объединяют в растворителе для проведения реакции Бухвальда с получением соединения S9. Этиловый эфир соединения S9 может быть восстановлен с образованием амида S10, который может подвергнуться дальнейшей реакции конденсации для введения нитрильной функциональной группы в соединение I.
В частности, смесь по существу химически и диастереомерно чистого кристаллического этанольного сольвата соединения IIa (0,20 ммоль), 2-бромизоникотината (0,30 ммоль), Cs2CO3 (129 мг, 0.39 ммоль), Pd2(dba)3 (18 мг, 0,02 ммоль) и Xant-Phos (9,4 мг, 0,02 ммоль) в 1,4-диоксане (10 мл) перемешивают в атмосфере N2 при 80°С в течение ночи. После фильтрации фильтрат концентрируют в вакууме и остаток очищают стандартным методом с получением соединения S9. Смесь соединения S9 (0,3 ммоль), NH3 (0,4-1 ммоль) и МеОН перемешивают в запечатанной пробирке в течение ночи при 60°С. После фильтрации фильтрат концентрируют в вакууме и остаток очищают стандартным методом с получением соединения S10. Смесь соединения S10 (0,3 ммоль), пиридин и TFAA перемешивают в ТГФ при 0°С в течение ночи. Реакционную смесь концентрируют в вакууме и остаток очищают стандартным методом с получением соединения I.
Химия эпимеризации
Оптимальные условия для динамической кристаллизации обычно включают в себя быструю реакцию эпимеризации с минимальным разложением соединений. Для соединения, восприимчивого к эпимеризации, оптимальные условия для проведения быстрой эпимеризации с минимальным разложением обычно зависят от температуры реакции, основания, концентрации основания и растворителя. Были проведены многочисленные эксперименты, чтобы показать, что процесс динамической кристаллизации в соответствии с настоящей заявкой повысит эффективность крупномасштабного производственного способа получения Соединения I.
Когда по существу химически и диастереомерно чистое соединение IIa объединяли с TBD (0,1 экв) в 95% EtOH при комнатной температуре, было обнаружено, что соотношение соединения IIa к IIb составляет 87,5%/12,5% через 2 ч. (опыт 1). Также наблюдали некоторое разложение, о чем свидетельствуют дополнительные пики на спектрограмме ВЭЖХ. Через 24 ч. соотношение по-прежнему составляло 87,1%/12,9%. Это показало, что реакция эпимеризации была довольно быстрой (хотя и неполной) с каталитическим количеством TBD при комнатной температуре.
Было обнаружено, что реакция эпимеризации протекает медленнее с DBN (0,1 экв) в ЕЮН, и только 2,8% соединения IIb наблюдали через 2 ч. при комнатной температуре (опыт 2). Тем не менее, было обнаружено, что соотношение IIa к IIb составляет 86,3%/13,7% через 24 ч. Пики разложения также наблюдали с помощью ВЭЖХ.
Когда соединение IIa объединяли с TBD (0,1 экв) в смеси EtOAc/н-гептан (1/1), только 4,2% соединения IIb наблюдали через 4 ч. при комнатной температуре, а количество IIb не увеличивалось через 24 ч. (опыт 3).
Однако, когда соединение IIa объединяли с TBD в смеси EtOAc/н-гептан (3/1), соотношение IIa к IIb составляло 85,1%/14,9% через 4 ч. при комнатной температуре (опыт 4) и соотношение не сильно изменилось через 24 ч. Наблюдали меньшее разложение продуктов (по сравнению с приведенной выше реакцией EtOH).
В другом эксперименте, опыт 5, 20 г соединения IIa объединяли с TBD (0,1 экв) в EtOAc (200 мл, 10V) при 40°С в течение 5 ч. Было обнаружено, что соотношение IIa к IIb составляет 63,0%/37,0%, и разложение не было зафиксировано. Затем реакционную смесь охлаждали до комнатной температуры и промывали водным раствором HCl, а затем рассолом. Органический слой сушили безводным Na2SO4, и концентрировали досуха с получением 20 г смеси из IIa/IIb в виде белого твердого вещества с наблюдаемым соотношением 63,6%/36,4%. Этот материал использовали для дальнейших исследований эпимеризации.
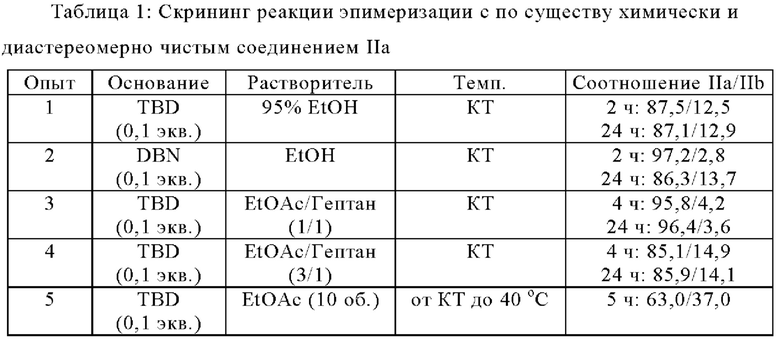
Смесь, содержащую соединения IIa и IIb в соответствующем соотношении 15/85 (и с 83,7% LCAP), эпимеризовали в различных растворителях и в присутствии 0,1 экв. TBD, DBU или DBN в EtOAc или IPAc (опыты 6-10), соотношение IIa/IIb увеличилось до ~1/1 без дальнейшего увеличения даже через 24 ч. Наблюдали значительные степени разложения со снижением химической чистоты до 42~68% (от 83,7%). См. опыты 6-10 в таблице 2.
По сравнению с DBU и DBN эпимеризация была быстрее с TBD в IPAc, при этом соотношение IIa/IIb увеличилось до ~1/1 за ~2 ч. (опыт 8). Поэтому TBD был выбран в качестве основания для дальнейшего исследования.
В смеси EtOAc/EtOH (3/1 об./об.) с 0,1 экв. TBD соотношение IIa/IIb увеличилось до ~1/1 за несколько часов при 10°С (опыт 11). Это соотношение больше не увеличивалось через 20 ч. Тем не менее, по сравнению с EtOAc или IPAc в качестве растворителя, разложение в смеси EtOAc/EtOH (3/1 об./об.) значительно не увеличилось в течение ночи при 10°С (с сохранением LCAP ~74% в течение всего времени).
Эпимеризацию доводили до 20 г с использованием условий опыта 12. Через 3 ч. при 10°С реакционную смесь (IIa/IIb ~ 49/51) фильтровали, чтобы получить 4 г соединения IIa с хиральной чистотой 99,8% и чистотой 98,2% LCAP (опыт 12).
При перемешивании смеси IIa и IIb (с соотношением IIa/IIb ~ 49/51) в EtOH с 0,1 экв. TBD при 15°С в течение 26 ч., соотношение IIa/IIb в твердом веществе составляло ~75/25 с LCAP по-прежнему ~77% (опыт 13).
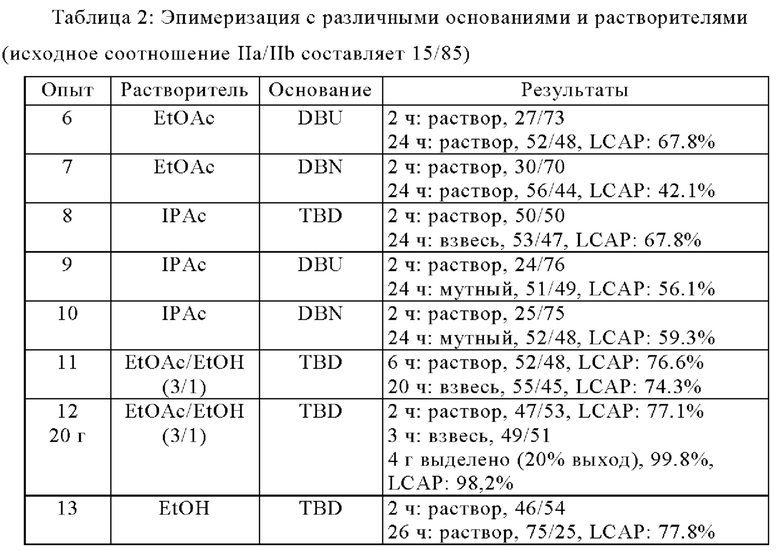
Условия реакции: Неочищенную IIa/IIb (15/85, LCAP: 83.7%) (опыт 6-10: 100 мг, опыт 11 и 13: 1 г, опыт 12: 20 г,) перемешивали с 0.1 экв. оснований в растворителях (2 об.) при КТ (за исключением опытов 11 и 12: 10°С, опыт 13: 15°С).
Оптимизация температуры эпимеризации в EtOH (исходное соотношение IIa/IIb составляет 15/85)
Температуру реакции дополнительно оптимизировали для реакции эпимеризации в EtOH. При использовании 0,1 экв. TBD эпимеризация была слишком медленной при -5°С и 5°С в EtOH. Соотношение IIa/IIb увеличилось до ~50/50 с 15/85 даже через ~24 ч (опыты 14-16). Только 5,5 г твердого вещества было получено из партии 20 г.
Соотношение IIa/IIb увеличилось до 70/30 и 68/32 через 24 ч. при 10°С и соответственно 12°С (опыты 17 и 18). При 15°С (опыты 19 и 20) в реакционной смеси наблюдали соотношение IIa/IIb в 82/18 с 15/85 через 24 ч. После фильтрации и сушки получают 9,5 г твердого вещества из партии 20 г. Хиральная чистота составляла 99,9% и 98,4% LCAP. При 20°С через 24 ч. разложение было более сильным с чистотой ~55% LCAP для реакционной смеси, хотя соотношение соединение IIa/соединение IIb увеличилось до 90/10 (опыт 21). Эти эксперименты показали, что ~15°С является подходящей температурой для реакции эпимеризации в EtOH с 0,1 экв. TBD, и в этих условиях может быть получен по существу химически и диастереомерно чистый кристаллический этанольный сольват соединения IIa с выходом ~32% для 2-й порции.
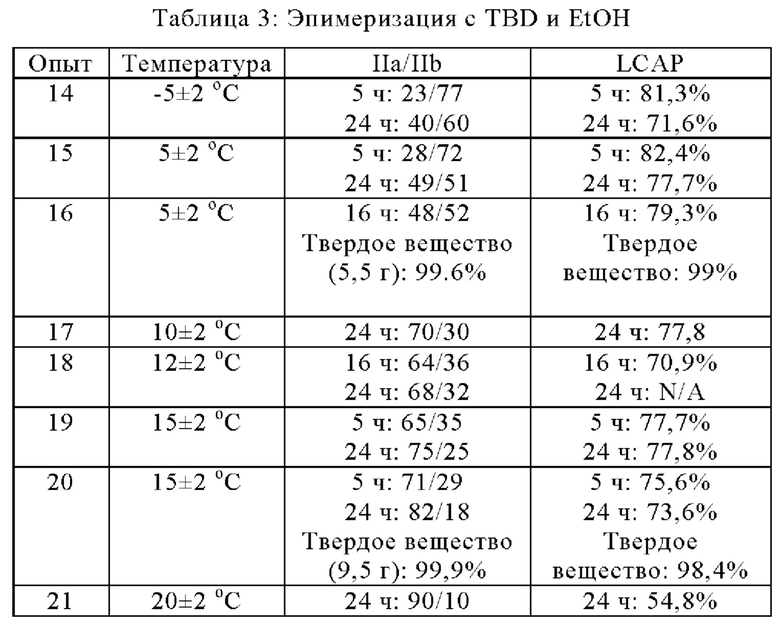
Условия реакции: неочищенную IIa/IIb (15/85, LCAP: 83,7%; 2 г, за исключением 16, 17 и 19: 20 г,) перемешивали с 0,1 экв. TBD в EtOH (2 об.) при различных температурах.
Скрининг различных растворителей для реакции эпимеризации:
Различные растворители подвергали скринингу для эпимеризации неочищенной смеси IIa/IIb, полученной из исходного раствора 1-й порции соединения IIa. Неочищенную смесь (LCAP: 83%, хиральная IIa/IIb: 17% / 83%) перемешивали в этих растворителях в присутствии TBD (0,1 экв) при 15°С в течение 24 ч. С EtOH для реакционной смеси было достигнуто высокое соотношение IIa/IIb в 90%/10% (опыт 22). Тем не менее, разложение IIa было тяжелым, чистота реакционной смеси, определенная по данным ВЭЖХ, составляла всего ~37%. В EtOAc в качестве растворителя наблюдали лишь незначительное разложение (чистота 78% по данным ВЭЖХ), а соотношение IIa/IIb составляло 53% / 47% (опыт 23). Аналогичные результаты были получены с использованием IPA в качестве растворителя, а именно: чистота 78% по данным ВЭЖХ и соотношение 49%/51% IIa/IIb (опыт 24).
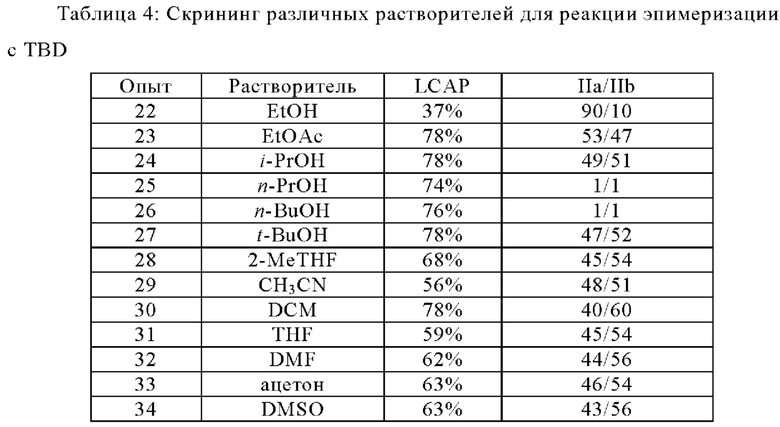
Условия реакции: IIa/IIb (LCAP: 83%, хиральная IIa/IIb: 17%/83%) (0,5 г, 1 экв) и TBD (15 мг, 0.1 экв) перемешивали в различных растворителях (2 об.) при 15°С в течение 24 ч.
i-PrOH в качестве растворителя для динамической кристаллизации (т.е. кристаллизации и эпимеризации)
Различные растворители (EtOH, EtOAc, i-PrOH, n-PrOH, n-BuOH, t-BuOH, THF, 2-MeTHF, CH3CN и ацетон) подвергали скринингу, начиная с практически химически и диастереомерно чистого соединения IIa и TBD (0,1 экв) при 15°С в течение 2 ч. с целью оптимизации реакции эпимеризации с минимальным химическим разложением. Данные в нижеследующей таблице собраны для различных растворителей. i-PrOH обеспечивает хорошую эпимеризацию с минимальным химическим разложением через 2 часа, при котором достигается соотношение IIa/IIb в 57%/42% с химической чистотой 91,8% (см. опыт 37).
Использование TBD в EtOH/н-гептан для эпимеризации
В опытах 45 и 46 смесь соединений IIa и IIb растворяли в этаноле в присутствии TBD (0,07 или 0,10 экв). При перемешивании реакционной смеси в течение определенного времени к раствору добавляли н-гептан перед фильтрацией твердого вещества. Кристаллический этанольный сольват соединения IIa собирали в виде твердого вещества с хиральной чистотой 99+% и 99+% LCAP.
Хотя в настоящей заявке было описано несколько вариантов осуществления, объем данного раскрытия определяется прилагаемой формулой изобретения, а также конкретными вариантами осуществления, которые были включены сюда в качестве примеров. Содержание всех ссылок (включая ссылки на литературные источники, выданные патенты, опубликованные патентные заявки и одновременно находящиеся на рассмотрении патентные заявки), процитированных в настоящей заявке, тем самым прямо включено в настоящую заявку во всей своей полноте посредством ссылки. Если не указано иное, всем техническим и научным терминам, используемым в данном изобретении, придают значение, обычно известное специалистам в данной области техники.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ МОДИФИКАЦИЯ | 2014 |
|
RU2666368C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 1-(АЦИЛОКСИ)-АЛКИЛКАРБАМАТНЫХ КОНЪЮГАТОВ ЛЕКАРСТВЕННЫХ СРЕДСТВ НАПРОКСЕНА И ПРЕГАБАЛИНА | 2019 |
|
RU2799809C2 |
| ВИНОРЕЛБИНА МОНОТАРТРАТ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730521C2 |
| Соединения имидазолонилхинолина и их терапевтическое применение | 2020 |
|
RU2822479C2 |
| ПО СУЩЕСТВУ, КРИСТАЛЛИЧЕСКАЯ ФОРМА МЕЛАГАТРАНА | 2000 |
|
RU2243973C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО НЕБИВОЛОЛА | 2006 |
|
RU2392277C2 |
| БЕЗВОДНЫЙ КРИСТАЛЛИЧЕСКИЙ АЗИДОЦИТОЗИНПОЛУСУЛЬФАТ | 2003 |
|
RU2334756C2 |
| ИНГИБИТОРЫ АМИД-ГИДРОЛАЗЫ ЖИРНЫХ КИСЛОТ | 2011 |
|
RU2569061C2 |
| ПОЛИМОРФНЫЕ И ПСЕВДОПОЛИМОРФНЫЕ ФОРМЫ ФАРМАЦЕВТИЧЕСКОГО СОЕДИНЕНИЯ | 2010 |
|
RU2575173C2 |
| Полиморфные и псевдополиморфные формы фармацевтического соединения | 2010 |
|
RU2727509C2 |
Изобретение относится к способу получения соединения I (ивосидениба), используемого при лечении пациентов с гематологическими злокачественными новообразованиями. Предлагаемый способ включает выделение кристаллического этанольного сольвата соединения IIa и превращение этанольного сольвата соединения IIa в соединение I или его сольват. Предлагаемый способ позволяет эффективно синтезировать ивосидениб с использованием диастереомерно чистого кристаллического этанольного сольвата соединения IIa. Изобретение относится также к кристаллическому этанольному сольвату соединения IIa и способу его получения. 3 н. и 26 з.п. ф-лы, 2 ил., 4 табл., 46 пр.
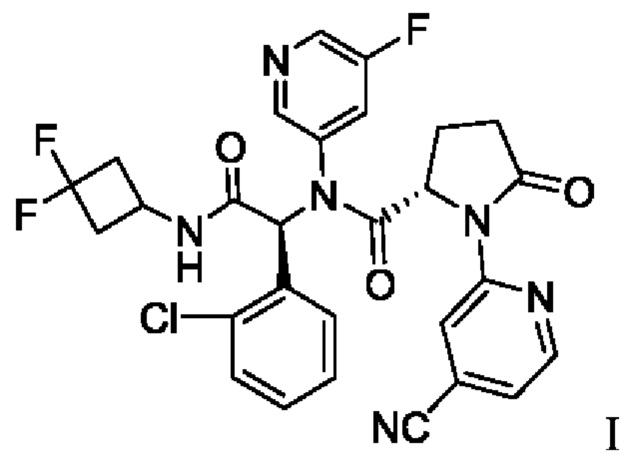
1. Способ получения соединения I
 ,
,
включающий в себя выделение кристаллического этанольного сольвата соединения IIa
и превращение этанольного сольвата соединения IIa в соединение I или его сольват.
2. Способ по п. 1, где выделение кристаллического этанольного сольвата соединения IIa включает в себя кристаллизацию соединения IIa из смеси соединений формулы II
3. Способ по п. 1, где превращение кристаллического этанольного сольвата соединения IIa включает в себя взаимодействие соединения IIa с 2-гало-4-цианопиридином с получением соединения I.
4. Способ по п. 2, где кристаллизация кристаллического этанольного сольвата соединения IIa включает в себя суспендирование или растворение диастереомерной смеси соединений формулы II в системе этанольных растворителей, добавление основания и перемешивание смеси при температуре от 0 до 25°С и добавление по меньшей мере одного неполярного растворителя.
5. Способ по п. 4, где основание выбирают из группы, включающей в себя 1,5,7-триазабицикло[4.4.0]дец-5-ен («TBD»), 1,5-диазабицикло[4.3.0]нон-5-ен («DBN»), триэтиламин, диизопропиламин, N,N-диизопропилэтиламин, N-метилморфолин, морфолин, N-метилпиперазин, пиридин, бутиламин, дибутиламин и 1,5-диазабицикло[4.3.0]нон-5-ен или их смесь.
6. Способ по п. 2, где смесь соединений формулы II изготавливают путем взаимодействия 2-хлорбензальдегида с 5-фторпиридин-3-амином в метаноле с последующим взаимодействием полученного продукта с (S)-5-оксопирролидин-2-карбоновой кислотой и 1,1-дифтор-3-изоцианатоциклобутаном с получением смеси соединений формулы II.
7. Способ по п. 1 включающий в себя взаимодействие кристаллического этанольного сольвата соединения IIa с 2-бромизоникотинонитрилом с получением соединения I.
8. Способ по п. 1, включающий в себя взаимодействие кристаллического этанольного сольвата соединения IIa с этил-2-бромизоникотинатом с получением промежуточного соединения S9
9. Способ по п. 8, включающий в себя восстановление промежуточного соединения S9 в присутствии NH3 с получением амида S10
10. Способ по п. 9, включающий в себя смешивание S10 с трифторуксусным ангидридом (TFAA) в присутствии пиридина с получением соединения I.
11. Способ по п. 1 или 2, где кристаллический этанольный сольват характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей в себя один или более из следующих пиков при 2θ: 7.2±0.2°, 8.6±0.2°, 12.2±0.2°, 13.1±0.2°, 14.4±0.2°, 16.7±0.2°, 19.4±0.2°, 19.8±0.2°, 21.8±0.2° и 25.2±0.2°.
12. Способ по п. 1 или 2, где кристаллический этанольный сольват характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей в себя три или более из следующих пиков при 2θ: 7.2±0.2°, 8.6±0.2°, 12.2±0.2°, 13.1±0.2°, 14.4±0.2°, 16.7±0.2°, 19.4±0.2°, 19.8±0.2°, 21.8±0.2° и 25.2±0.2°.
13. Способ по п. 1 или 2, где кристаллический этанольный сольват соединения IIa имеет диастереомерную чистоту по меньшей мере 90% и химическую чистоту по меньшей мере 80%.
14. Способ по п. 1 или 2, где кристаллический этанольный сольват соединения IIa имеет диастереомерную чистоту приблизительно от 90% до приблизительно 99%.
15. Способ по п. 1 или 2, где кристаллический этанольный сольват соединения IIa имеет химическую чистоту приблизительно от 80% до приблизительно 99%.
16. Способ получения кристаллического этанольного сольвата соединения IIa
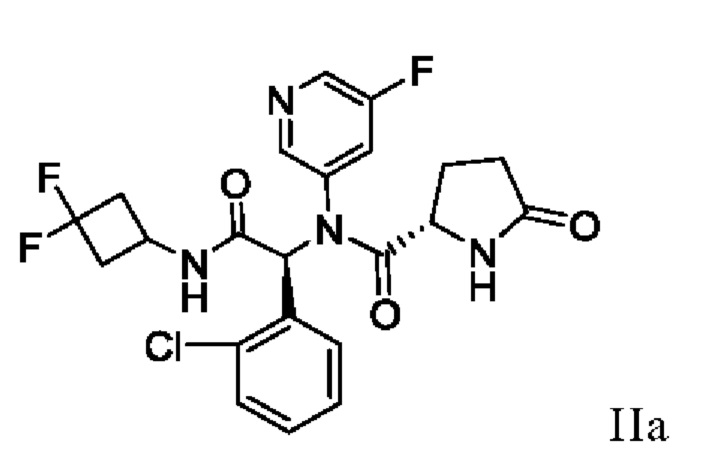 ,
,
включающий в себя суспендирование или растворение смеси соединений формулы II
и основания в системе этанольных растворителей, добавление по меньшей мере одного неполярного растворителя и выделение кристаллического этанольного сольвата соединения IIa.
17. Способ по п. 16, где основание выбирают из группы, включающей в себя 1,5,7-триазабицикло[4.4.0]дец-5-ен («TBD»), 1,5-диазабицикло[4.3.0]нон-5-ен («DBN»), триэтиламин, диизопропиламин, N,N-диизопропилэтиламин, N-метилморфолин, морфолин, N-метилпиперазин, пиридин, бутиламин, дибутиламин и 1,5-диазабицикло[4.3.0]нон-5-ен или их смесь.
18. Способ по п. 16 или 17, где кристаллический этанольный сольват соединения IIa имеет диастереомерную чистоту по меньшей мере 90% и химическую чистоту по меньшей мере 80%.
19. Способ по любому из пп. 16-18, где кристаллический этанольный сольват соединения IIa имеет диастереомерную чистоту приблизительно от 90% до приблизительно 99%.
20. Способ по любому из пп. 16-19, где кристаллический этанольный сольват соединения IIa характеризуется рентгеновской порошковой дифрактограммой (РПД), где присутствует по меньшей мере 50% наиболее интенсивных пиков, показанных в таблице ниже:
21. Способ по любому из пп. 16-19, где кристаллический этанольный сольват соединения IIa характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей в себя один или более из следующих пиков при 2θ: 7.2±0.2°, 8.6±0.2°, 12.2±0.2°, 13.1±0.2°, 14.4±0.2°, 16.7±0.2°, 19.4±0.2°, 19.8±0.2°, 21.8±0.2° и 25.2±0.2°.
22. Способ по любому из пп. 16-19, где кристаллический этанольный сольват соединения IIa характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей в себя три или более из следующих пиков при 2θ: 7.2±0.2°, 8.6±0.2°, 12.2±0.2°, 13.1±0.2°, 14.4±0.2°, 16.7±0.2°, 19.4±0.2°, 19.8±0.2°, 21.8±0.2° и 25.2±0.2°.
23. Способ по любому из пп. 16-22, где кристаллический этанольный сольват соединения IIa имеет химическую чистоту приблизительно от 80% до приблизительно 99%.
24. Кристаллический этанольный сольват соединения IIa
 ,
,
характеризующийся рентгеновской порошковой дифрактограммой (РПД), включающей в себя пять или более из следующих пиков при 2θ: 7.2±0.2°, 8.6±0.2°, 12.2±0.2°, 13.1±0.2°, 14.4±0.2°, 16.7±0.2°, 19.4±0.2°, 19.8±0.2°, 21.8±0.2° и 25.2±0.2°.
25. Кристаллический этанольный сольват по п. 24, где кристаллический этанольный сольват соединения IIa характеризуется рентгеновской порошковой дифрактограммой (РПД), где присутствует по меньшей мере 50% наиболее интенсивных пиков, показанных в таблице ниже:
26. Кристаллический этанольный сольват по п. 24, имеющий диастереомерную чистоту приблизительно от 90% до приблизительно 99%.
27. Кристаллический этанольный сольват по п. 24, имеющий диастереомерную чистоту по меньшей мере приблизительно 99%.
28. Кристаллический этанольный сольват по п. 24, имеющий диастереомерную чистоту по меньшей мере 90% и химическую чистоту по меньшей мере 80%.
29. Кристаллический этанольный сольват по п. 24, имеющий химическую чистоту приблизительно от 80% до приблизительно 99%.
| J | |||
| POPOVICI-MULLER ET AL, Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers, ACS MEDICINAL CHEMISTRY LETTERS, 2018, vol | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ТКАЦКИЙ СТАНОК | 1920 |
|
SU300A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| ПРОИЗВОДНОЕ ИЗОКСАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА МУТАНТНОЙ ИЗОЦИТРАТДЕГИДРОГЕНАЗЫ 1 | 2015 |
|
RU2692782C2 |

