ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к носителю для твердофазного синтеза, способу его получения и его применению. Носитель предназначен для использования в твердофазном синтезе олигонуклеотидов и принадлежит к области получения функциональных полимерных материалов.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
В качестве одного из эффективных инструментов прецизионной медицины все больше и больше олигонуклеотидных препаратов получают одобрение на продажу. Олигонуклеотидные препараты часто синтезируют с использованием химических методов, и наиболее распространен фосфорамидитный твердофазный синтез, в котором реакционная колонка наполнена носителем твердофазного синтеза, и раствор с растворенными реагирующими веществами быстро протекает через колонку под определенным давлением для совершения реакции.
На ранней стадии развития технологии твердофазного синтеза олигонуклеотидов в число широко используемых носителей твердофазного синтеза входили неорганические частицы, такие как стеклянные микросферы с контролируемым размером пор (CPG) и модифицированный кварц, но они имели очевидные недостатки, такие как низкая загрузка (обычно менее 100 мкмоль/г), которая приводит к низкому выходу одной партии олигонуклеотидов, низкому использованию оборудования и высоким издержкам производства. Чтобы улучшить загрузку носителя, компании Nitto Denko CO. LTD. и Ionis Inc. совместно подали заявку на патент WO 2006029023, которая включала получение органических полимеров в качестве носителей для твердофазного синтеза олигонуклеотидов с использованием стирола, ацетоксистирола и дивинилбензола в качестве мономеров полимеризации, а также изооктана и 2-этил-1-гексанола в качестве порообразующих агентов. Приготовленный носитель имел загрузку до 100–350 мкмоль/г. Для дальнейшего улучшения выхода и чистоты олигонуклеотидов в заявке на патент US 8653152, поданной компанией Nitto Denko CO. LTD, для приготовления носителя для твердофазного синтеза используются стирол, метакрилонитрил, ацетоксистирол и дивинилбензол в качестве мономеров полимеризации, изооктан и 2-этил-1-гексанол в качестве порообразующего агента. Эффективность синтеза повышается за счет добавления метакрилонитрила для подавления эффекта неодинакового набухания носителя для твердофазного синтеза в различных растворителях.
В обоих вышеуказанных полимерных носителях используется дивинилбензол в качестве поперечно-сшивающего агента. При использовании дивинилбензола существуют две основные проблемы, одной из которых является невысокая чистота, а другой – наличие у него трех изомеров с разными свойствами. Это приводит к большой разнице в характеристиках смол, полученных с использованием дивинилбензола разного качества от разных производителей. В настоящее время чистота дивинилбензола в целом по отрасли составляет 40–60%. Помимо собственно дивинилбензола он также содержит определенное количество этилстирола, а в некоторых случаях также содержит небольшое количество диэтилбензола.
Три изомера дивинилбензола демонстрируют различную реакционную способность при сополимеризации с другими мономерами, что значительно влияет на структуру и характеристики сополимера. В исследовании кинетике сополимеризации стирола-дивинилбензола, проведенном He Binglin и др. (Reactive Polymer, 12 (1990), 269-275), было обнаружено, что в процессе сополимеризации п-дивинилбензол и м-дивинилбензол имеют более высокую скорость преобразования, чем стирол. Это приводит к высокой степени поперечного сшивания сополимеров, образуемых на ранней стадии полимеризации, и низкой степени поперечного сшивания сополимеров, образуемых на поздней стадии полимеризации, а это приводит к неоднородной химической структуре в смоле. Из-за неоднородной химической структуры макропористая адсорбционная смола со стирол-дивинилбензолом в качестве скелета имеет ряд проблем во время использования, таких как различная степень фрагментации, долгое достижение адсорбционного равновесия, образование «хвостов» после десорбции, неполная регенерация и т.д.
Диеновые поперечно-сшивающие агенты, такие как 1, 2-бис-(4-винилфенил)этан, у которых две винильные группы находятся не на одном и том же бензольном кольце, имеют реакционную способность, аналогичную стиролу. Sundell и др. (Journal of Polymer Science Part A: Polymer Chemistry, 31 (1993), 2305-2311) получили сильнокислотные смолы с использованием такого рода мономеров в качестве поперечно-сшивающих агентов. Результаты экспериментов показали, что эти смолы обладали более высокой механической прочностью, чем сильнокислотные смолы на основе сополимеров стирол-дивинилбензол. В исследовании синтеза и адсорбционных характеристик 1,2-бис-(п-винилфенил)этановых полимеров были получены сверхсильносшитые полистирольные адсорбционные смолы с 1,2-бис-(п-винилфенил)этаном и дивинилбензолом в качестве сшивающих агентов, соответственно. Результаты экспериментов показали, что у адсорбционных смол с 1,2-бис-(п-винилфенил)этаном в качестве поперечно-сшивающего агента отсутствовало явление образования «хвостов» во время использования, и они легко регенирировались.
Олигонуклеотидные препараты появились при лечении таких заболеваний, как вирусная инфекция, высокий уровень холестерина, редактирование экспрессии генов, ухудшение зрения и слепота, обструкция печеночных вен, благодаря их высокой терапевтической эффективности, низкой лекарственной токсичности, сильной специфичности и широким областям применения. Спрос на олигонуклеотидные препараты растет с каждым годом. В то же время существующие носители для твердофазного синтеза олигонуклеотидов имеют проблемы, такие как неоднородная внутренняя структура и высокое сопротивление переносу масс, что приводит к низкой эффективности синтеза олигонуклеотидов и высоким издержкам производства. Поэтому необходимо разработать носитель для твердофазного синтеза, который обеспечивает возможность крупномасштабного синтеза олигонуклеотидных препаратов при низких затратах и высокой эффективности для удовлетворения рыночного спроса на олигонуклеотидные препараты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Для достижения крупномасштабного производства олигонуклеотидов при низких затратах и преодоления недостатков, таких как неоднородная внутренняя структура и высокое сопротивление переносу масс существующих носителей для твердофазного синтеза нуклеотидов, в настоящем изобретении предложены носитель твердофазного синтеза, способ его получения и его применение.
В настоящем изобретении в качестве поперечно-сшивающих мономеров выбраны диеновые соединения, имеющие две винильные группы, находящиеся не на одном и том же бензольном кольце, которые демонстрируют в основном следующие три эффекта: Во-первых, мономеры такого типа имеют константу сополимеризации, аналогичную стиролу, что приводит к улучшению однородности химической структуры внутри смолы и образованию равномерно распределенных активных участков. Во-вторых, улучшение однородности химической структуры внутри смолы выгодно для образования равномерных поровых каналов внутри смолы, что снижает сопротивление переносу масс. В-третьих, по сравнению с дивинилбензолом поперечно-сшивающие агенты такого типа содержат гибкие молекулярные цепи, которые могут улучшить характеристики набухания смолы в различных органических растворителях.
В настоящем изобретении предложен носитель для твердофазного синтеза, причем носитель имеет полимерный скелет с функциональными группами, которые представлены следующей формулой:
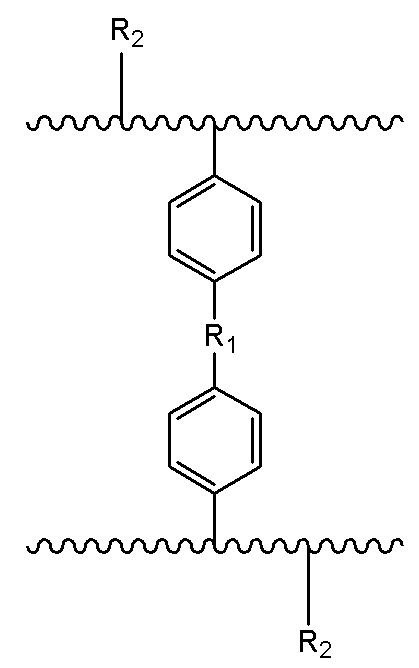
где R1= -(CH2)n-, n – целое число о 0 до 3, или R1= -O-(CH2)m-O-, m – целое число от 1 до 4; и R2= -OH или -NH2.
В некоторых вариантах осуществления носитель для твердофазного синтеза представляет собой сополимер, содержащий в своем скелете повторяющиеся структурные звенья, представленные формулой (I), формулой (II), формулой (III) и формулой (IV):
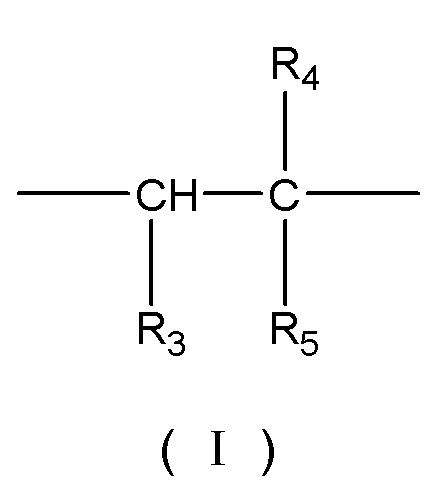
где R3 выбирают из группы, состоящей из -H, -CN и -CH2-CN; R4 представляет собой -H или -CH3; и R5 выбирают из группы, состоящей из -CN, -CH2-CN, -OOCH3 и -CONH2;
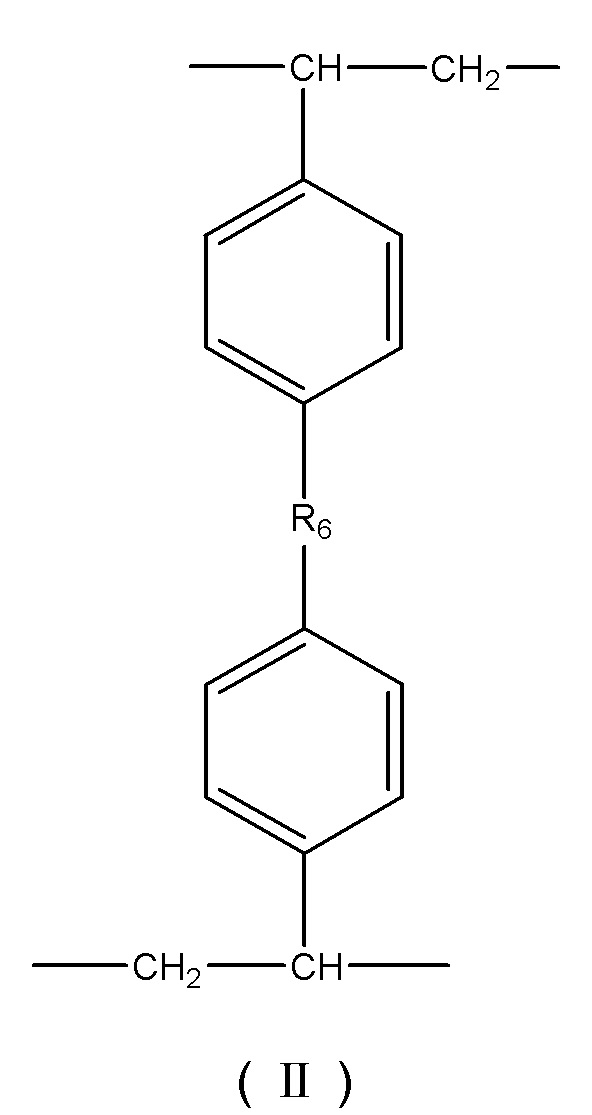
где R6 представляет собой -(CH2)x-, x – целое число от 0 до 3, или R6 представляет собой -O-(CH2)y-O -, y – целое число от 1 до 4;
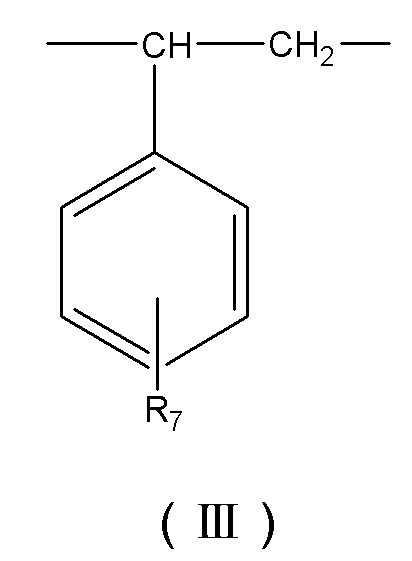
R7 выбирают из группы, состоящей из –H, CH3(CH2)z - или CH3(CH2)ZO-, где z – целое число от 0 до 4, (CH3)2CH-, (CH3)2CH(CH2)-, (CH3)2CH(CH2)2-, (CH3)3C-, CH3CH2CH(CH3)-, CH3CH2C(CH3)2-, и CH3CH2CH2CH(CH3)-;
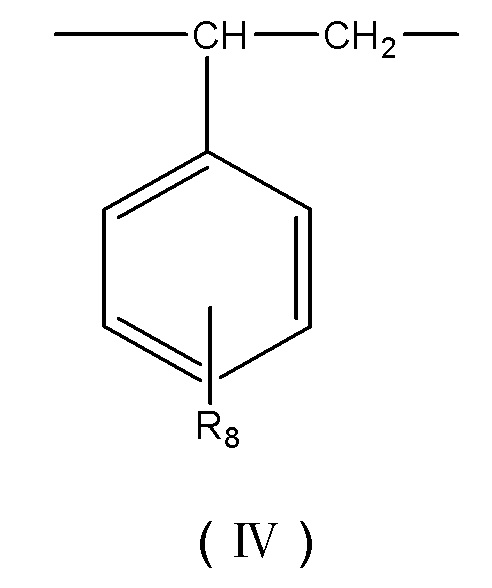
R8 выбирают из группы, состоящей из –OH, -CH2OH, -NH2, -CH2NH2, -CHCOOC-C6H4-OH, -CHCOOCCH2-C6H4-OH, -(CH2)4OOC-C6H4-OH, -(CH2)4OOCCH2-C6H4-OH, -(CH2)4OOCCH2-C6H4-NH2, -CH2COONH-C6H4-NH2, -COO-C6H4-OH и -CH2COO-C6H4-OH.
В некоторых вариантах осуществления носитель имеет содержание гидроксильной группы и аминогруппы 100–1000 мкмоль/г, предпочтительно 400–700 мкмоль/г.
В некоторых вариантах осуществления носитель имеет размер частиц в диапазоне 35–200 мкм, предпочтительно 50–100 мкм.
В некоторых вариантах осуществления носитель имеет средний диаметр пор 10–200 нм, предпочтительно 40–100 нм.
В настоящем изобретении также предложен способ получения носителя для твердофазного синтеза. Способ получения включает следующие этапы: получение водной фазы и масляной фазы, соответственно; водная фаза содержит воду, диспергирующее средство и неорганическую соль; масляная фаза содержит: поперечно-сшивающий агент, моновинильное соединение, функциональный мономер, модифицированный мономер, порообразующий агент и инициатор, при этом поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер и модифицированный мономер представляют собой мономеры, способные к полимеризации; и добавление масляной фазы в водную фазу, перемешивание и нагревание для проведения реакции, и удаление порообразующего агента по завершении реакции, получение пористой полимерной смолы Пористая полимерная смола способна подвергаться дальнейшей реакции для получения носителя для твердофазного синтеза, содержащего гидроксильную группу и аминогруппу в качестве функциональных групп.
В частности, в некоторых вариантах осуществления поперечно-сшивающий мономер представляет собой диеновый поперечно-сшивающий агент, имеющий две винильные группы не на одном и том же бензольном кольце, который выбирают из группы, состоящей из 4,4'-дивинилбифенила, ди(4-винилфенли)метана, 1,2-ди(4-винилфенил)этана, 1,3-ди(4-винилфенил)пропана, ди(4'-винилфенокси)метана, 1,2-ди(4'-винилфенокси)этана, 1,3-ди(4'-винилфенокси)пропана, 1,4-ди(4'-винилфенокси)бутана и любой их комбинации. Предпочтительно поперечно-сшивающий агент представляет собой 1,2-ди(4-винилфенил)этан.
В некоторых вариантах осуществления моновинильное соединение представляет собой ароматическое моновинильное соединение. Моновинильное соединение представляет собой стирол, незамещенный или замещенный алкилом C1-C5 на своем бензольном кольце, такой как метилстирол, этилстирол, n-пропилстирол, изопропилстирол, n-бутилстирол, изобутилстирол, втор-бутилстирол, трет-бутилстирол, n-амилстирол, изопентилстирол, втор-амилстирол или трет-амилстирол; или стирол, замещенный C1-C5 алкокси, такой как метоксистирол, этоксистирол, пропоксистирол, бутоксистирол или пентоксистирол. Предпочтительно моновинильное соединение представляет собой стирол.
В некоторых вариантах осуществления функциональный мономер имеет двойную связь, способную к свободнорадикальной полимеризации, а также гидроксильную группу, аминогруппу, галогенированную группу или другую группу, способную превращаться в гидроксильную группу и аминогруппу посредством реакции. В процессе синтеза олигонуклеотидов реакционноспособные гидроксильная группа и аминогруппа действуют в качестве активных участков для соединения олигонуклеотидов, включая аминогруппу, аминоалкильную группу, гидроксильную группу, гидроксиалкильную группу и т.д. Предпочтительны первичная аминогруппа, аминометильная группа, гидроксильная группа, гидроксиметильная группа и т.д. В число функциональных мономеров входят, без исключения, гидроксистирол и его производные, такие как 4-гидроксистирол и т.д.; гидроксиалкилстирол и его производные, такие как 4-гидроксиалкилстирол и т.д.; ацилоксистирол и его производные, такие как 4-ацилоксистирол и бензоилоксистирол и т. д.; аминостирол и его производные, такие как 4-аминостирол и т.д.; аминоалкилситрол и его производные, такие как 4-аминометилстирол и т.д.; мономеры галоалкилстирола, такие как 4-(4-бромбутил)стирол и 4-хлорметилстирол и т.д.; мономеры 4-винилфенилэфира, такие как метил-4-винилбензоат и этиловый эфир 4-этенилбензолуксусной кислоты и т.д.
В некоторых вариантах осуществления некоторые из функциональных мономеров содержат гидроксильные защитные группы или аминозащитные группы, которые могут быть непосредственно отрезаны с образованием аминогрупп или гидроксильных групп. Например, ацилоксистирол может быть преобразован посредством щелочного гидролиза или кислотного гидролиза в гидроксильный группы для использования в качестве активных участков для соединения олигонуклеотидов; некоторые из функциональных мономеров необходимо преобразовать посредством реакции функционализации в аминогруппы или гидроксильные группы для использования в качестве активных участков. Например, галоалкилстирол может быть превращен посредством гидролиза в гидроксильную группу или превращен посредством реакции Габриеля в первичные аминогруппы для использования в качестве активных участков для соединения олигонуклеотидов; некоторые из функциональных мономеров должны сшивать соединительные звенья, имеющие аминогруппы или гидроксильные группы для использования в качестве активных участков. Например, галоалкилстирол может реагировать с 4-гидроксибензойной кислотой или 4-гидроксифенилуксусной кислотой с образованием гидроксильных групп, служащих в качестве активных участков для соединения олигонуклеотидов; некоторые из таких функциональных мономеров нуждаются в вышеуказанных многократных реакциях для получения аминогрупп или гидроксильных групп, служащих в качестве активных участков. Например, мономеры 4-винилфенилэфиров должны гидролизироваться, чтобы сначала обнажить карбоксильные группы, а затем реагировать с гидрохиноном или п-фенилендиамином для получения аминогрупп или гидроксильных групп, служащих в качестве активных участков.
В некоторых вариантах осуществления модифицированный мономер имеет двойную связь, способную к свободнорадикальной полимеризации, а также имеет цианогруппу, эфирную группу и амидную группу. В число модифицированных мономеров входят, без ограничения, акрилонитрил, метакрилонитрил, фумаронитрил, 1,4-дициано-2-бутен, метилметакрилат и акриламид.
В некоторых вариантах осуществления инициатор выбирают из группы, состоящей из органических пероксидов и азосоединений. В число инициаторов входят, без исключения, пероксид бензоила, пероксид лауроила, трет-бутилперокси-2-этилгексаноат, 2,2'-азо-бис(-2-метилпропионитрил), 2,2'-азо-бис-(2-метилбутиронитрил) и 2,2'-азо-бис-(2,4-диметил)валеронитрил. На долю инициатора приходится 0,5–5 мас.% в пересчете на общую массу мономеров.
В некоторых вариантах осуществления порообразующий агент представляет собой органический растворитель или поверхностно-активное вещество, которое не полимеризуется и не растворяется или слабо растворяется в воде. Порообразующий агент выбирают из группы, состоящей из ароматических углеводородов, таких как бензол, толуол и этилбензол; алифатических углеводородов, таких как линейные или разветвленные C6–C12 алканы или C6–C12 циклоалканы, такие как гексан, гептан, октан, додекан, изооктан, изододекан и циклогексан; галогенизированные углеводороды, такие как хлороформ и хлорбензол; эфиры, содержащие 4 или более атомов углерода, такие как этилацетат, бутилацетат и дибутилфталат; спирты, такие как линейный или разветвленный C4–C12 алкановый спирт или C4–C12 циклоалкановый спирт, такой как гексанол, циклогексанол, октанол, изооктанол, деканол и додеканол; маслорастворимые поверхностно-активные вещества, такие как сорбитантриолеат, полученный из пчелиного воска полиоксиэтиленсорбит, сорбитантристеарат, полиоксиэтиленсорбитгексастеарат, этиленгликолевый сложный эфир жирной кислоты, пропиленгликольмоностеарат, сорбитансесквиолеат, полиоксиэтиленсорбитолеат, моностеарин, гидроксилированный ланолин, сорбитмоноолеат и пропиленгликольлаурат, и любая их комбинация.
В некоторых вариантах осуществления водная фаза содержит воду, диспергирующее средство и неорганическую соль. Диспергирующее вещество представляет собой водорастворимый полимер, который содержит, без ограничения, одно или более из группы, состоящей из поливинилового спирта, гидроксиэтилцеллюлозы, гидроксиметилцеллюлозы, карбоксиметилцеллюлозы, метилцеллюлозы, этилцеллюлозы, полиакрилата натрия и поливинилпирролидона. Диспергирующее вещество присутствует в водной фазе в количестве 0,1–5 мас.%. Неорганическая соль действует в качестве регулятора плотности водной фазы, при этом снижая растворимость компонентов масляной фазы в водной фазе, так что капли масла более устойчиво диспергируются в водной фазе. В число неорганических солей входят, без ограничения, одна или более солей выбранных из группы, состоящей из хлорида натрия, хлорида калия, хлорида кальция, сульфата натрия, сульфата калия, сульфата кальция и т.д. Неорганическая соль присутствует в водной фазе в количестве 20 мас.% или ниже.
В некоторых вариантах осуществления для уменьшения связывания между смолами и улучшения теплообмена полимеризации, а также для улучшения использования оборудования и эффективности производства, массовое соотношение масляной фазы и водной фазы устанавливают равным 1:3–1:20.
В некоторых вариантах осуществления настоящего изобретения различные компоненты присутствуют в следующих количествах: моновинильное соединение, первоначально содержащееся в масляной фазе, составляет 45–95 мас.% от общей массы мономеров; поперечно-сшивающий мономер, первоначально содержащийся в масляной фазе, составляет 2,9–20 мас.% от общей массы мономеров; функциональные мономеры, первоначально содержащиеся в масляной фазе, составляют 2–20 мас.% от общей массы мономеров; модифицированный мономер, первоначально содержащийся в масляной фазе, составляет 0,1–15 мас.% от общей массы мономеров; и порообразующий агент, первоначально содержащийся в масляной фазе, составляет 15–130 мас.% от общей массы мономеров.
В некоторых наиболее предпочтительных вариантах осуществления настоящего изобретения различные компоненты присутствуют в следующих количествах: моновинильное соединение, первоначально содержащееся в масляной фазе, составляет 62–86 мас.% от общей массы мономеров; поперечно-сшивающий мономер, первоначально содержащийся в масляной фазе, составляет 7–13 мас.% от общей массы мономеров; функциональные мономеры, первоначально содержащиеся в масляной фазе, составляют 5–15 мас.% от общей массы мономеров; модифицированный мономер, первоначально содержащийся в масляной фазе, составляет 2–10 мас.% от общей массы мономеров; и порообразующий агент, первоначально содержащийся в масляной фазе, составляет 30–110 мас.% от общей массы мономеров.
В некоторых вариантах осуществления полимеризацию выполняют при температуре 50–90 °C, предпочтительно 70–85 °C.
В некоторых вариантах осуществления способ получения носителя для твердофазного синтеза включает следующие этапы:
добавление в реактор определенного количества очищенной воды, добавление диспергирующего вещества в количестве 0,1–5 мас.% от водной фазы и неорганическую соль в количестве не более 20 мас.% от водной фазы, и растворение для получения водной фазы;
отвешивание моновинильного соединения, поперечно-сшивающего мономера, функционального мономера, модифицированного мономера, порообразующего агента и инициатора в соответствии с массовым отношением масляной фазы к водной фазе, составляющим 1:3–1:20; при этом моновинильное соединение составляет 45–95 мас.% от общей массы мономеров, поперечно-сшивающий мономер составляет 2,9–20 мас.% от общей массы мономеров, функциональный мономер составляет 2–20 мас.% от общей массы мономеров, модифицированный мономер составляет 0,1–15 мас.% от общей массы мономеров, порообразующий агент составляет 15–130 мас.% от общей массы мономеров, и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы в реактор, перемешивание и нагревание до температуры 50–90 °C для проведения реакции; удаление порообразующего агента по завершении реакции, просеивание и сбор смолы с соответствующим размером частиц и вакуумная сушка для получения полимерной смолы; и
проведение дополнительной реакции со смолой для получения носителя для твердофазного синтеза, имеющего аминогруппу и карбоксильную группу.
В некоторых вариантах осуществления способ получения носителя для твердофазного синтеза включает следующие этапы:
добавление в реактор определенного количества очищенной воды, добавление диспергирующего вещества в количестве 0,1–5 мас.% от водной фазы и неорганическую соль в количестве не более 20 мас.% от водной фазы, и растворение для получения водной фазы;
отвешивание моновинильного соединения, поперечно-сшивающего мономера, функционального мономера, модифицированного мономера, порообразующего агента и инициатора в соответствии с массовым отношением масляной фазы к водной фазе, составляющим 1:3–1:20; при этом моновинильное соединение составляет 62–86 мас.% от общей массы мономеров, поперечно-сшивающий мономер составляет 7–13 мас.% от общей массы мономеров, функциональный мономер составляет 5–15 мас.% от общей массы мономеров, модифицированный мономер составляет 2–10 мас.% от общей массы мономеров, порообразующий агент составляет 30–110 мас.% от общей массы мономеров, и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы в реактор, перемешивание и нагревание до температуры 70–85 °C для проведения реакции; удаление порообразующего агента по завершении реакции, просеивание и сбор смолы с соответствующим размером частиц и вакуумная сушка для получения полимерной смолы; и
проведение дополнительной реакции со смолой для получения носителя для твердофазного синтеза, имеющего аминогруппу и карбоксильную группу.
Вышеупомянутый способ позволяет получать носитель для твердофазного синтеза в соответствии с настоящим изобретением, т.е. пористую смолу, имеющую гидроксильные группы и аминогруппы. В настоящем изобретении содержание аминогруппы или гидроксильной группы, т.е. загрузка, может быть вычислено посредством реакции с реактивом Fmoc-Leu-OH и последующим удалением защитной группы Fmoc. Количество извлеченной Fmoc определяют калориметрическим способом, а затем вычисляют содержание аминогруппы или гидроксильной группы в носителе.
В некоторых вариантах осуществления подробная операция выглядит следующим образом: 1,0 г носителя точно отвешивают и суспендируют в 7 мл раствора ацетонитрила, затем туда добавляют 0,5 г Fmoc-Leu-OH, 0,5 г HBTU и 0,5 мл DIEA и в течение 2 ч перемешивают при комнатной температуре для проведения реакции. По завершении реакции смолу промывают последовательно ацетонитрилом (три раза, по 10 мл каждый раз) и метанолом (три раза, по 10 мл каждый раз), а затем смолу просушивают. Точно отвешивают 0,1000 г смолы, суспендируют в 20%-ном растворе пиперидина/DMF (об./об.) и встряхивают в течение 30 мин при комнатной температуре, фильтруют и собирают фильтрат. Эту смолу промывают DMF и фильтрат собирают. Фильтраты объединяют для получения общего объема, а затем растворяют соответствующее количество раз, чтобы измерить поглощательную способность при 300 нм. Проводят аналогичные реакции удаления Fmoc, используя серию известных концентраций Fmoc-Leu-OH, и измеряют поглощательную способность для построения стандартной кривой. Содержание аминогруппы или гидроксильной группы в носителе вычисляют с помощью стандартной кривой.
Содержимое функциональной группы носителя зависит от процентного содержания функциональных мономеров в перерасчете на общую массу мономеров, и путем регулирования количества функциональных мономеров можно получить ряд носителей с различным содержанием аминогруппы или гидроксильной группы. Содержание функциональной группы носителя определяет количество синтезированных олигонуклеотидов; если содержание функциональной группы слишком мало, выход олигонуклеотидов в одной партии будет уменьшен, а если содержание функциональной группы слишком высокое, это скажется на чистоте олигонуклеотидов. В настоящем изобретении содержание функциональной группы носителя составляет 100–1000 мкмоль/г, предпочтительно 400–700 мкмоль/г.
Размер частиц носителя в настоящем изобретении измеряется с помощью прибора для обработки изображений частиц. Носитель равномерно распределяют на предметном стекле и наблюдают под микроскопом при увеличении, при этом увеличенные изображения частиц носителя фиксируются камерой. Морфологические характеристики и размер частиц носителя анализируются и вычисляются компьютером.
Размер частиц носителя в основном зависит от типа и количества диспергирующего вещества, присутствующего в водной фазе, а также типа и количества порообразующих агентов и скорости перемешивания во время полимеризации суспензии. Размер частиц носителя можно регулировать путем коррекции вышеуказанных условий. Если размер частиц носителя слишком велик, с одной стороны, это приведет к уменьшению удельной площади поверхности носителя и увеличит количество активных участков на единицу площади, что скажется на чистоте олигонуклеотидов; с другой стороны, если размер частиц слишком велик, это замедлит скорость переноса масс и приведет к увеличению загрязняющих примесей во время синтеза олигонуклеотидов. Если размер частиц носителя слишком мал, это приведет к слишком высокому давлению в процессе синтеза и сильно увеличит стоимость оборудования. В настоящем изобретении носитель имеет размер частиц в диапазоне 35–200 мкм, предпочтительно 50–100 мкм.
Средний диаметр частиц носителя измеряют методом интрузии ртути. Образцы массой 0,1500–0,3000 г точно отвесили и поместили в автоматический ртутный порозиметр AutoPore IV 9500 (Micromeritics Instrument Co.). Угол смачивания ртутью установили на 130° и натяжение поверхности установили на 485 дин/см, а затем провели измерение методом интрузии ртути в этих условиях. Средний диаметр пор носителя в основном зависит от типа и количества порообразующих агентов, количества поперечно-сшивающего вещества, температуры и времени реакции и т. д. Средний диаметр пор носителя можно регулировать путем коррекции этих условий. Если средний диаметр пор носителя слишком мал, это затруднит перенос масс и скажется на эффективности синтеза, если средний диаметр пор носителя слишком велик, это приведет к уменьшению удельной площади поверхности носителя и увеличит количество активных участков на единицу площади. Во время синтеза олигонуклеотидов рост нуклеотидов приведет к взаимному влиянию и скажется на чистоте олигонуклеотидов. В настоящем изобретении носитель имеет средний диаметр пор 10–200 нм, предпочтительно 40–100 нм.
По сравнению с известным уровнем техники настоящее изобретение имеет три основных преимущества: Во-первых, в настоящем изобретении в качестве поперечно-сшивающих агентов выбирают мономеры, которые имеют постоянную сополимеризации, аналогичную стиролу, что приводит к улучшению однородности внутренней химической структуры смолы и формированию равномерно распределенных активных участков. Во-вторых, улучшение однородности внутренней химической структуры смолы приводит к образованию равномерных поровых каналов в смоле, тем самым снижая сопротивление переносу масс. В-третьих, по сравнению с дивинилбензолом поперечно-сшивающие агенты такого рода содержат гибкие цепи в своей структуре, которые могут улучшить характеристики набухания смолы в различных органических растворителях.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 1.
На Фиг. 2 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 2.
На Фиг. 3 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 3.
На Фиг. 4 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 4.
На Фиг. 5 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 5.
На Фиг. 6 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 6.
На Фиг. 7 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 7.
На Фиг. 8 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 8.
На Фиг. 9 показано полученное сканирующим электронным микроскопом изображение носителя, приведенного в примере 9.
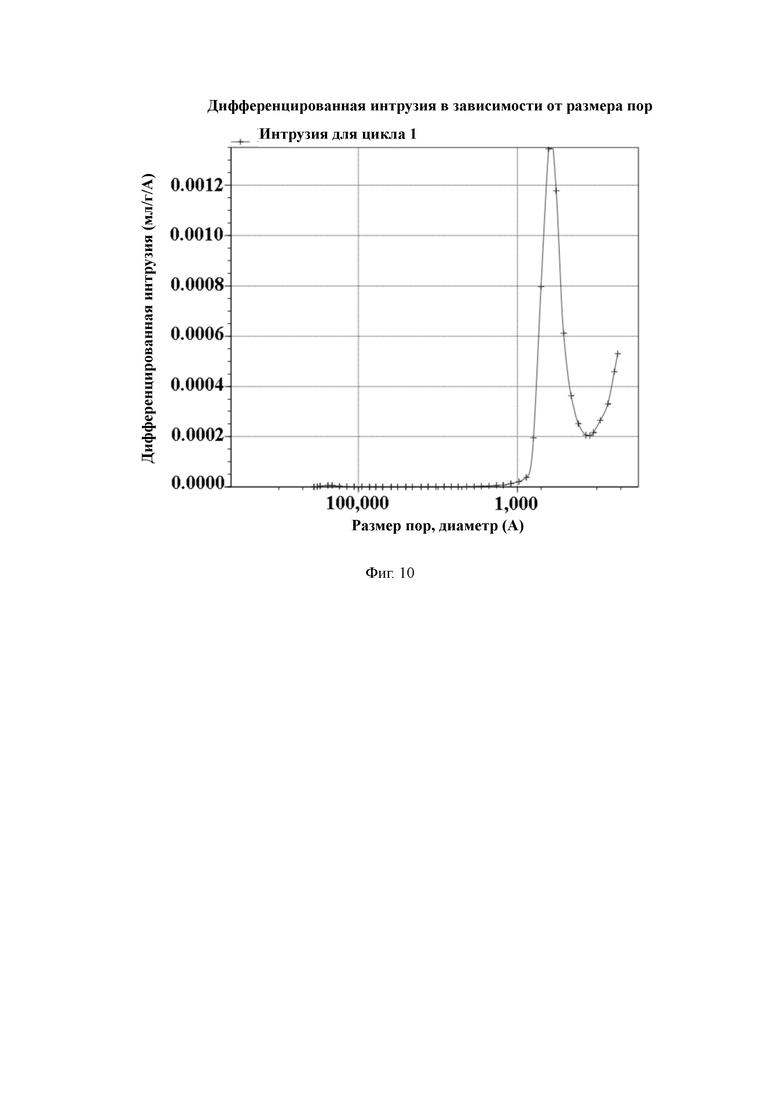
На Фиг. 10 показано распределение пор носителя по размеру в примере 1, измеренное методом интрузии ртути.
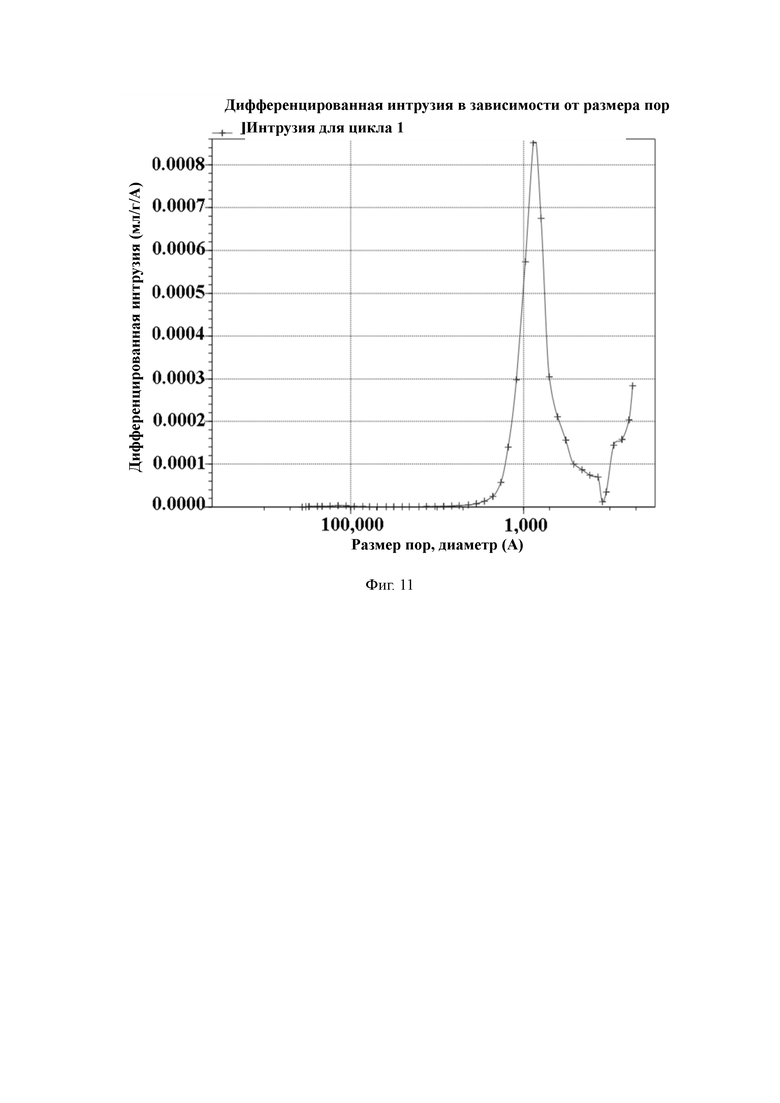
На Фиг. 11 показано распределение пор носителя по размер в примере 2, измеренное методом интрузии ртути.
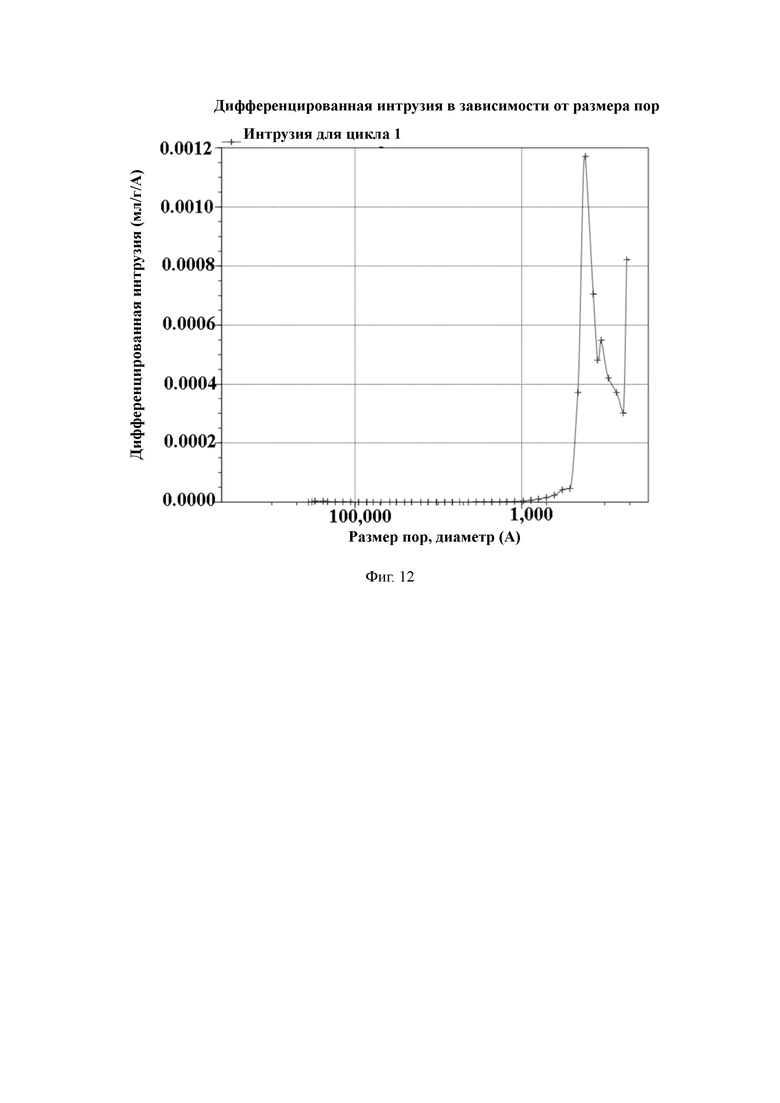
На Фиг. 12 показано распределение пор носителя по размер в примере 3, измеренное методом интрузии ртути.
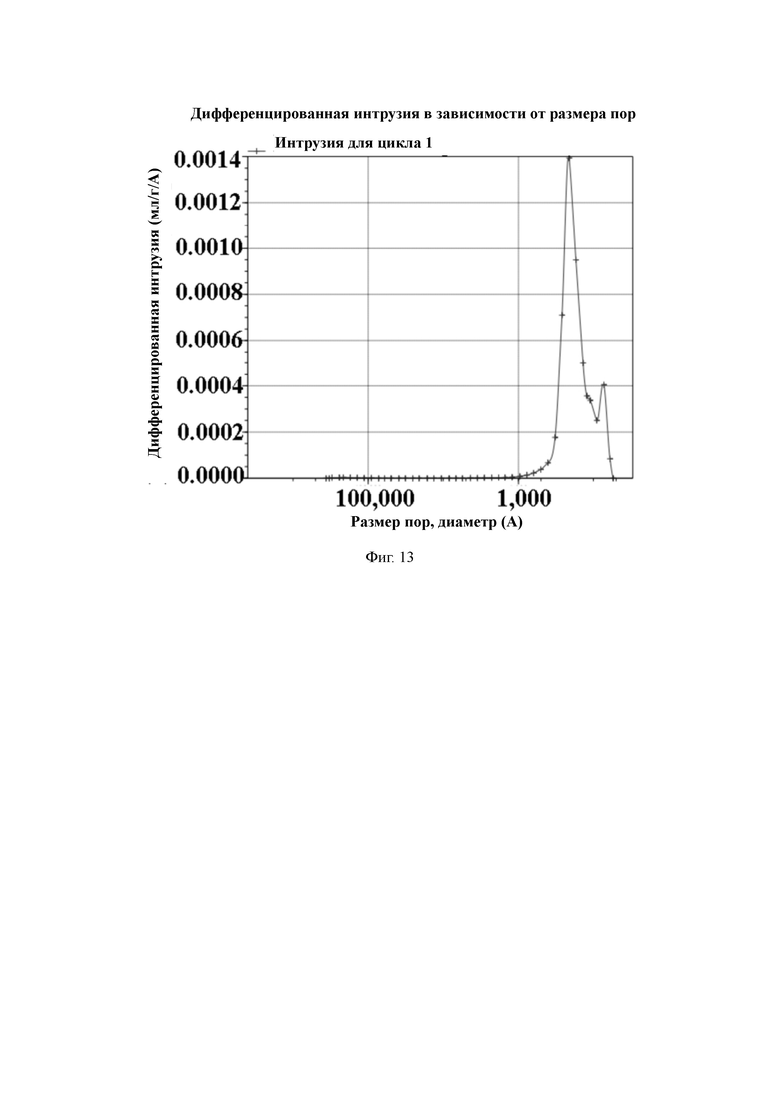
На Фиг. 13 показано распределение пор носителя по размер в примере 4, измеренное методом интрузии ртути.
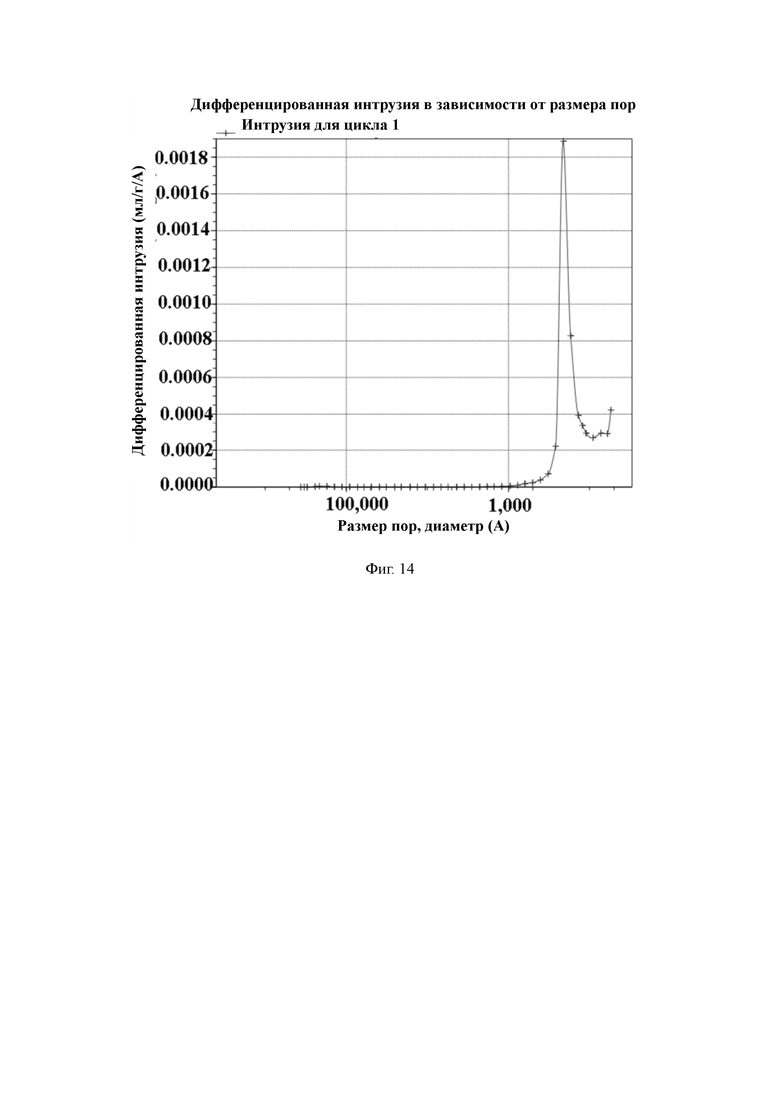
На Фиг. 14 показано распределение пор носителя по размер в примере 5, измеренное методом интрузии ртути.
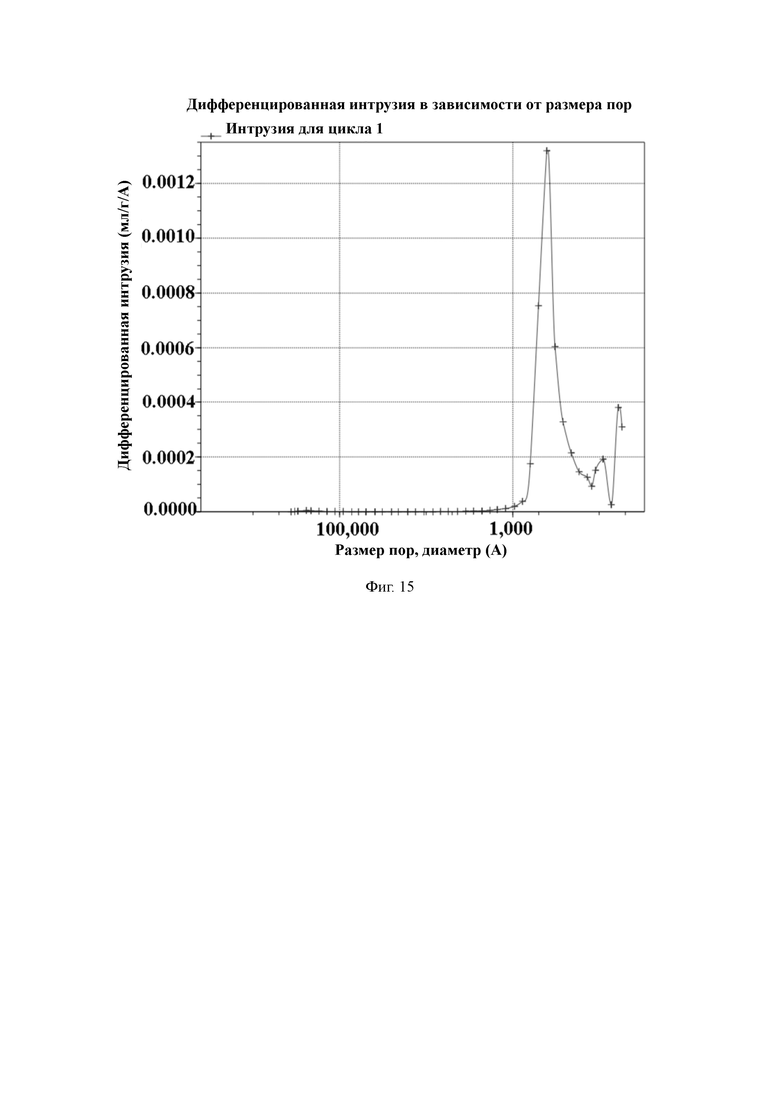
На Фиг. 15 показано распределение пор носителя по размер в примере 6, измеренное методом интрузии ртути.
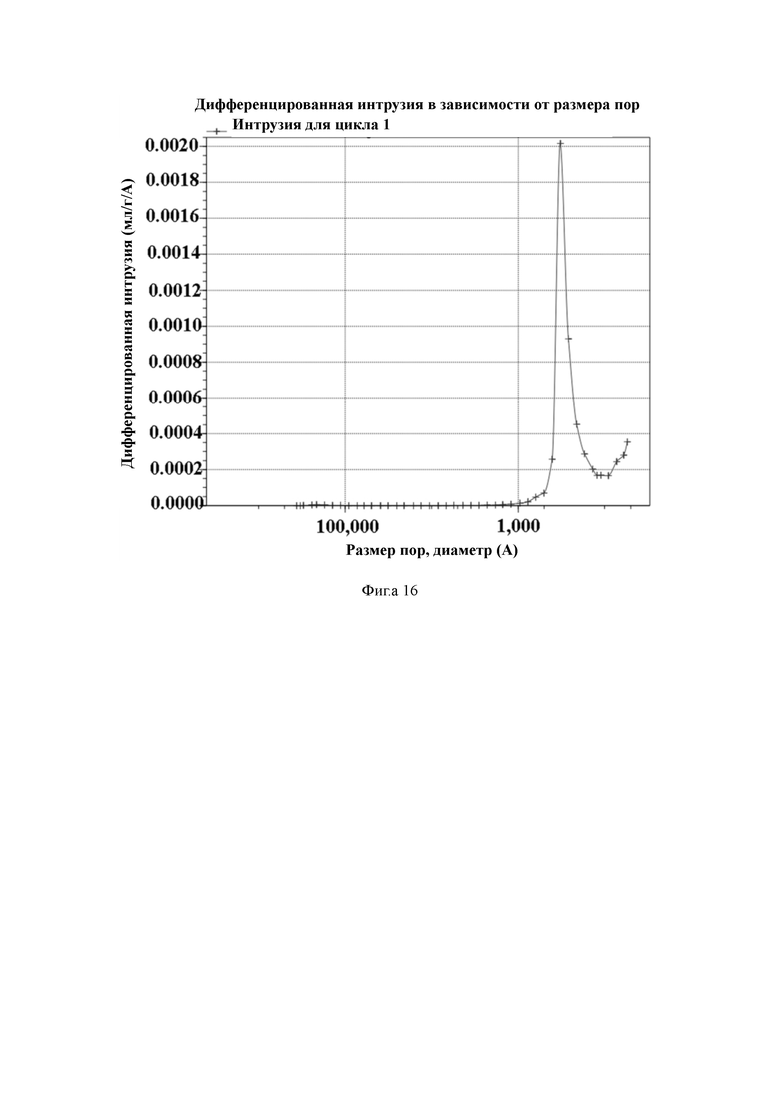
На Фиг. 16 показано распределение пор носителя по размер в примере 7, измеренное методом интрузии ртути.
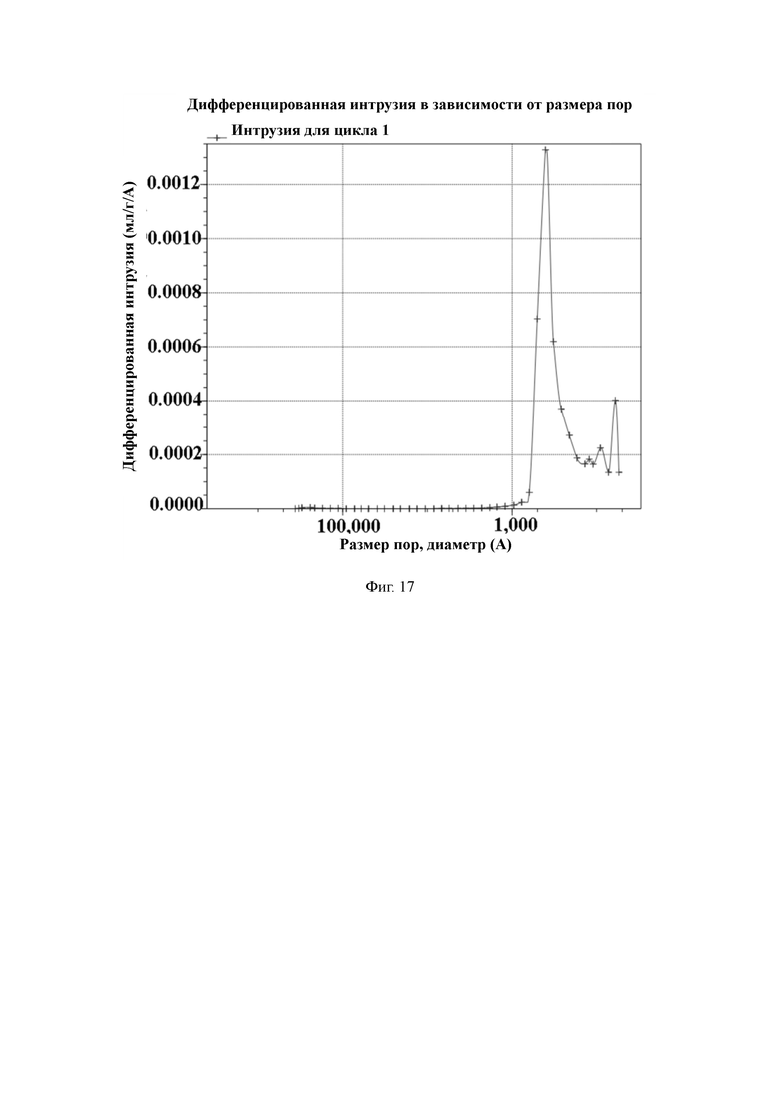
На Фиг. 17 показано распределение пор носителя по размер в примере 8, измеренное методом интрузии ртути.
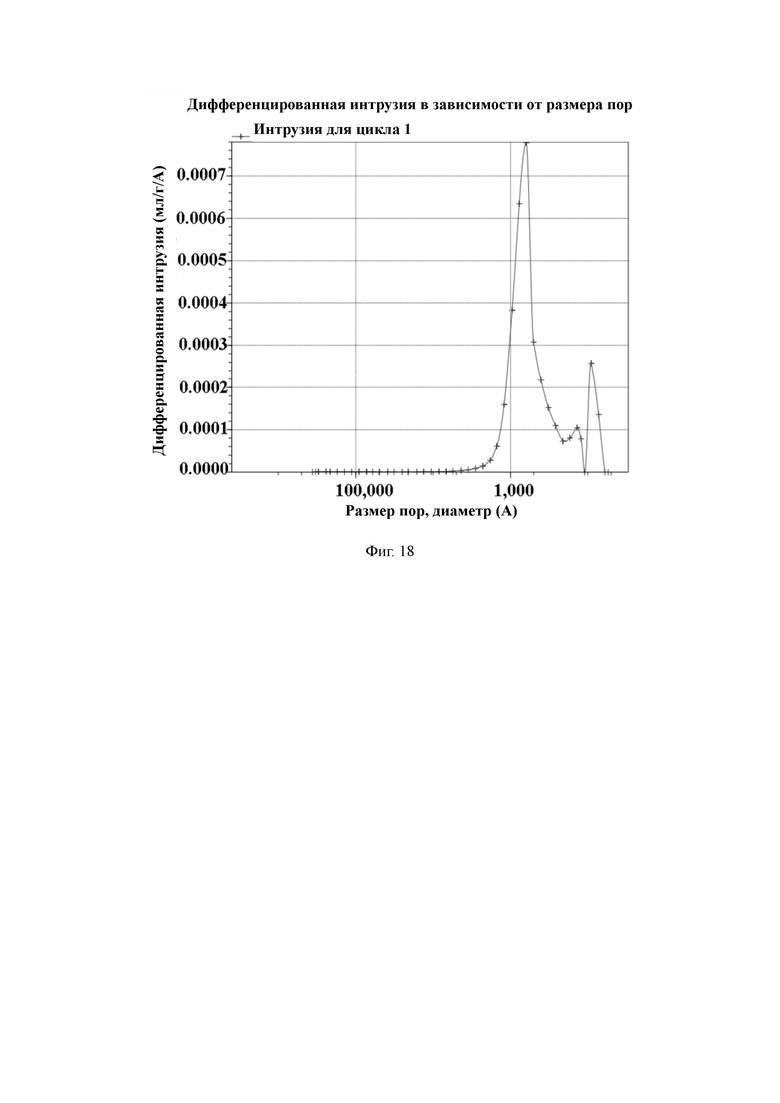
На Фиг. 18 показано распределение пор носителя по размер в примере 9, измеренное методом интрузии ртути.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Настоящая заявка будет проиллюстрирована далее со ссылкой на следующие варианты осуществления. Однако эти варианты осуществления являются лишь иллюстрацией, а не ограничениями настоящего изобретения, подробно изложенного в формуле изобретения.
Пример 1
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 108,8 г стирола, 14 г 1,2-ди(п-винилфенил)этана, 12,2 г п-хлорметилстирола, 5 г. фумаронитрила, 6 г сорбитолтриолеата, 40 г изооктанола, 20 г изододекана и 2,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 80 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы с содержанием хлора 565 мкмоль/г.
50 г полимерной пористой смолы и 500 мл N,N-диметилформамида добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем добавляют 30 г фталимида калия и увеличивают температуру до 95 °C для проведения реакции в течение 16 часов. По завершении реакции охлаждают до комнатной температуры, дважды промывают N,N-диметилформамидом, промывают очищенной водой до нейтрального состояния и трижды промывают абсолютным этанолом, а затем смолу отфильтровывают и сушат. 200 г абсолютного этанола и 50 г гидразингидрата добавляют в реактор и нагревают до температуры 75 ℃ для проведения реакции в течение 16 часов. После этого трижды промывают раствором этанол/очищенная вода в объемном соотношении 50:50, промывают очищенной водой до нейтрального состояния, трижды промывают абсолютным этанолом, а затем смолу отфильтровывают и сушат. 200 г абсолютного этанола и 50 г концентрированной хлористо-водородной кислоты добавляют в реактор и нагревают до температуры 60 °C для проведения реакции в течение 6 часов. После этого охлаждают до комнатной температуры, промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием аминогрупп 554 мкмоль/г и средним диаметром пор 54 нм, измеренным методом интрузии ртути.
Пример 2
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 77 г метилстирола, 28 г ди(п-винилфенил)метана, 14 г п-хлорметилстирола, 21 г 1,4-дициано-2-бутена, 15 г сорбитмоноолеата, 60 г изооктанола, 30 г дибутилфталата и 1 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 70 °C для выполнения полимеризации в течение 8 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы с содержанием хлора 646 мкмоль/г.
50 г полимерной пористой смолы и 500 мл N,N-диметилформамида добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем добавляют 35 г фталимида калия и увеличивают температуру до 95 °C для проведения реакции в течение 16 часов. По завершении реакции охлаждают до комнатной температуры, дважды промывают N,N-диметилформамидом, промывают очищенной водой до нейтрального состояния и трижды промывают абсолютным этанолом, а затем смолу отфильтровывают и сушат. 200 г абсолютного этанола и 50 г гидразингидрата добавляют в реактор и нагревают до температуры 75 ℃ для проведения реакции в течение 16 часов. После этого трижды промывают раствором этанол/очищенная вода в объемном соотношении 50:50, промывают очищенной водой до нейтрального состояния, трижды промывают абсолютным этанолом, а затем смолу отфильтровывают и сушат. 200 г абсолютного этанола и 50 г концентрированной хлористо-водородной кислоты добавляют в реактор и нагревают до температуры 60 °C для проведения реакции в течение 6 часов. После этого охлаждают до комнатной температуры, промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием аминогрупп 635 мкмоль/г и средним диаметром пор 145 нм, измеренным методом интрузии ртути.
Пример 3
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 123 г этилстирола, 8 г 1,3-ди(п-винилфенил)пропана, 8,8 г 4-(4-бромбутил)стирола, 0,2 г акрилонитрила, 3 г полиоксиэтиленсорбитолеата, 18 г изододекана, 5 г дибутилфталата и 2 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 65 °C для выполнения полимеризации в течение 10 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы с содержанием брома 257 мкмоль/г.
50 г полимерной пористой смолы и 600 мл N,N-диметилформамида добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем добавляют 5,4 г 4-гидроксибензойной кислоты, 5,4 г обезвоженного карбоната калия и 0,3 г йодида калия и нагревают до температуры 75 °C для проведения реакции в течение 6 часов. По завершении реакции охлаждают до комнатной температуры, промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильных групп 250 мкмоль/г и средним диаметром пор 23 нм, измеренным методом интрузии ртути.
Пример 4
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 112 г стирола, 9 г 1,4-ди(4‘-винилфенокси)бутана, 9 г п-хлорметилстирола, 10 г акриламида, 1 г этиленгликолевого сложного эфира жирной кислоты, 30 г толуола, 60 г дибутилфталата и 4,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 60 °C для выполнения полимеризации в течение 7 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы с содержанием хлора 418 мкмоль/г.
50 г полимерной пористой смолы и 300 мл абсолютного спирта добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. После этого в химическом стакане отвешивают 30 г гидроокиси натрия, растворяют в 300 мл деионизированной воды и затем медленно добавляют в реактор. Нагревают до температуры 65 °C для проведения реакции в течение 6 ч. После этого охлаждают до комнатной температуры, промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильных групп 410 мкмоль/г и средним диаметром пор 38 нм, измеренным методом интрузии ртути.
Пример 5
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 83 г стирола, 23 г 1,2-ди(п-винилфенил)этана, 18 г N-(4-винилфенил)ацетамида, 14 г фумаронитрила, 2 г сорбитансесквиолеата, 10 г додеканола, 10 г циклогексана и 2 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 55 °C для выполнения полимеризации в течение 10 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл абсолютного спирта добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. После этого в химическом стакане отвешивают 30 г гидроокиси натрия, растворяют в 300 мл деионизированной воды и затем медленно добавляют в реактор. Нагревают до температуры 65 °C для проведения реакции в течение 6 ч. После этого охлаждают до комнатной температуры, промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием аминогрупп 822 мкмоль/г и средним диаметром пор 34 нм, измеренным методом интрузии ртути.
Пример 6
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 107,7 г стирола, 12,8 г 1,2-ди(п-винилфенил)этана, 12,5 г 4-ацетоксистирола, 7 г фумаронитрила, 10 г сорбитантриолеата, 42 г изооктанола, 21 г изододекана и 2,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 78 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации смолу промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл ацетонитрила добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем медленно добавляют 7,5 мл гидразингидрата и проводят реакцию в течение 3 ч при комнатной температуре. После этого промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 538 мкмоль/г и средним диаметром пор 58 нм, измеренным методом интрузии ртути.
Пример 7
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 128 г стирола, 8 г 1,3-ди(п-винилфенил)пропана, 3 г 4-ацетоксистирола, 1 г фумаронитрила, 0,4 г полиоксиэтиленсорбитгексастеарата, 35 г дибутилфталата, 12 толуола и 3,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 70 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл ацетонитрила добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем медленно добавляют 7,5 мл гидразингидрата и проводят реакцию в течение 3 ч при комнатной температуре. После этого промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 125 мкмоль/г и средним диаметром пор 46 нм, измеренным методом интрузии ртути.
Пример 8
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 83 г стирола, 17 г 1,4-ди(4'-винилфенил)бутана, 27 г бензоилоксистирола, 13 г фумаронитрила, 103 г изооктанола, 51 г изододекана и 3 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 80 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации смолу промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл ацетонитрила добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем медленно добавляют 7,5 мл гидразингидрата и проводят реакцию в течение 3 ч при комнатной температуре. После этого промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 843 мкмоль/г и средним диаметром пор 42 нм, измеренным методом интрузии ртути.
Пример 9
2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 85 г стирола, 24 г 1,2-ди(п-винилфенил)этана, 18 г метил-4-винилбензоата, 13 г фумаронитрила, 4 г пропиленгликольлаурата, 40 г дибутилфталата, 40 г изододекана и 2,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 70 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации смолу промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл ацетонитрила добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем добавляют 8,8 г гидрохинона, 23 г HBTU и 13 мл DIEA и проводят реакцию в течение 2 ч при комнатной температуре. После этого промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 724 мкмоль/г и средним диаметром пор 124 нм, измеренным методом интрузии ртути.
Сравнительный пример 1
2 л очищенной воды, 20 г поливинилового спирта и 60 г хлорида натрия добавляют в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворяют для получения водяной фазы. Отвешивают 111 г стирола, 11 г дивинилбензола (80 мас.%), 13 г 4-ацетоксистирола, 5 г фумаронитрила, 8 г сорбитмоноолеата, 40 г изооктанола, 20 г изододекана и 2,5 г пероксида бензоила и хорошо перемешивают для получения масляной фазы. Масляную фазу добавляют в реактор, перемешивают и нагревают до температуры 78 °C для выполнения полимеризации в течение 6 часов. По завершении полимеризации смолу промывают горячей водой. Удаляют порообразующие агенты путем экстракции орошением этанолом. Смолу с размером частиц 50–100 мкм отсеивают и собирают, а затем сушат в вакууме для получения полимерной пористой смолы.
50 г полимерной пористой смолы и 300 мл ацетонитрила добавляют в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивают. Затем добавляют 7,5 мл гидразингидрата и проводят реакцию в течение 3 ч при комнатной температуре. После этого промывают водой до нейтрального состояния, и затем сушат в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 550 мкмоль/г и средним диаметром пор 64 нм, измеренным методом интрузии ртути.
Пример 10
Свойства набухания носителей твердофазного синтеза, полученных в примерах 1–9, измеряют в ацетонитриле и толуоле, соответственно. Способ следующий: отвешивают около 1,5 г образца и помещают в измерительную пробирку с притертой пробкой. В соответствующей пропорции добавляют толуол или ацетонитрил. Затем смолы и растворитель перемешивают стеклянной палочкой до полного набухания и плотно закрывают пробкой. 2–3 часа спустя смолы и медленно перемешивают стеклянной палочкой, чтобы удалить пузырьки воздуха и равномерно распределить смолу без образования комков. Палочку для перемешивания вынимают, и измерительную пробирку помещают на стол с резиновым ковриком и подвергают вибрации для компактного уплотнения смолы. После выдерживания в течение 24 часов объем записывают и вычисляют степень набухания.
Результаты показаны в таблице 1:
Пример 11
Носители для твердофазного синтеза, полученные в примерах 1, 2, 6 и 9 и сравнительном примере 1, и носители для твердофазного синтеза NittoPhase HL выбрали для синтеза фрагментов олигонуклеотидов и оценили характеристики смол. Чтобы лучше показать преимущества настоящего изобретения, объем (г) загрузки носителей = объему (мл) колонки для синтеза/степень набухания носителя в толуоле (мл/г), а объем промывки во время синтеза олигонуклеотидов ограничен объемом одной колонки для синтеза.
10 г носителя для твердофазного синтеза отвешивают в реактор и оставляют набухать в 50 мл ацетонитрила в течение 10 минут, а затем туда добавляют 3,0 г о DMT-dT-3'-янтарной кислоты, 1,5 г HBTU и 1,3 мл DIEA для проведения реакции при комнатной температуре в течение 12 ч. По завершении реакции 5 раз промывают ацетонитрилом. Затем добавляют кэп А (состоящий из 20 мл ацетонитрила, 7,5 мл пиридина и 5,0 мл N-метилимидазола) и кэп В (состоящий из 10 мл ацетонитрила и 4 мл уксусного ангидрида) для проведения реакции при комнатной температуре в течение 30 минут. По завершении реакции смолу промывают 5 раз ацетонитрилом и сушат в вакууме для получения носителя, загруженного DMT-dT. Загруженные группы DMT удаляют с помощью раствора p-толуолсульфоновой кислоты/ацетонитрила. Содержание групп DMT, загруженных в носитель, определяют с помощью спектрометрии при длине волн 412 нм, и результаты показаны в таблице 2.
Носители, загруженные DMT-dT, взвешивают, и заполняют ими колонку для синтеза (32 мм). Колонку для синтеза устанавливают на AKTA OligoPilot 100, чтобы синтезировать олигонуклеотид длиной в 20 оснований, имеющий последовательность d[ACGTACGTACGTACGTACGT]. Процесс синтаза: 1. смолу подвергают набуханию в дихлорметане; 2. удаляют группы DMT 10%-м DCA/DCM; 3. промывают обезвоженным ацетонитрилом; 4. добавляют мономер фосфорамидита и активирующий реагент для проведения конденсации; 5. промывают обезвоженным ацетонитрилом; 6. добавляют окислительный агент для проведения окисления; 7. промывают обезвоженным ацетонитрилом; 8. добавляют кэпирующий реагент для проведения кэпирования конца; 9. промывают обезвоженным ацетонитрилом; и 10. повторяют этап 2, чтобы начать следующий цикл.
По завершении синтеза носители извлекают для сушки. Затем их помещают в склянку, и туда добавляют надлежащее количество концентрированной аммиачной воды для проведения реакции при температуре 55 °C в течение 16 ч, чтобы олигонуклеотиды отщепились от носителей при одновременном удалении защитных групп на основаниях. Носитель и олигонуклеотиды отделяют друг от друга посредством фильтрации, и фильтрат высушивают для получения необработанного порошка олигонуклеотидов. Измеряют чистоту олигонуклеотидов с помощью ВЭЖХ и вычисляют выход олигонуклеотидов. Результаты показаны в таблице 2.
Как можно увидеть из таблицы 2, использование для носителя твердофазного синтеза олигонуклеотидов, согласно настоящему изобретению, может улучшить выход и чистоту олигонуклеотидов и, таким образом, помогает уменьшить издержки производства нуклеотидов.
Вышеприведенные примеры и технические решения являются лишь предпочтительными вариантами осуществления настоящего изобретения. Следует отметить, что для специалистов в данной области техники любые изменения или модификации, вытекающие из сущности настоящего изобретения, остаются в пределах объема охраны настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Пористая смола, используемая для твердофазного синтеза и способ ее получения | 2021 |
|
RU2815371C2 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ, АКТИВИРУЮЩИЕ РНКазу Н | 2017 |
|
RU2740501C2 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2708237C2 |
| КОМПОЗИЦИЯ ДЛЯ ИММОБИЛИЗАЦИИ БИОЛОГИЧЕСКИХ МАКРОМОЛЕКУЛ В ГИДРОГЕЛЯХ, СПОСОБ ПРИГОТОВЛЕНИЯ КОМПОЗИЦИИ, БИОЧИП, СПОСОБ ПРОВЕДЕНИЯ ПЦР НА БИОЧИПЕ | 2001 |
|
RU2206575C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛИРОВАННЫХ ИОНООБМЕННЫХСМОЛ | 1978 |
|
SU826959A3 |
| ГИДРОФИЛЬНЫЕ ПОЛИМЕРНЫЕ ЧАСТИЦЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2643034C2 |
| ВОДНЫЕ ПЕРВИЧНЫЕ ДИСПЕРСИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2662226C1 |
| СЕЛЕКТИВНОЕ ОТДЕЛЕНИЕ НИТРОЗОСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2012 |
|
RU2622410C2 |
| Блок-сополимерная композиция и способ ее получения | 2020 |
|
RU2767539C1 |
| ВОДНЫЕ ПЕРВИЧНЫЕ ДИСПЕРСИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2669697C1 |





Изобретение относится к области получения функциональных полимерных материалов, а именно, представляет собой носитель для использования в твердофазном синтезе олигонуклеотидов. Носитель для твердофазного синтеза описывается формулой: 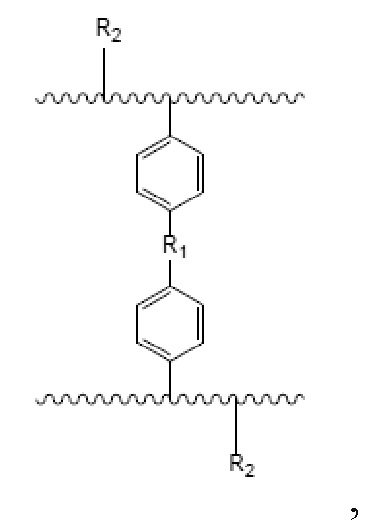
где R1= -(CH2)n-, n – целое число о 0 до 3, или R1= -O-(CH2)m-O-, m – целое число от 1 до 4; и R2= -OH или -NH2; при этом получение носителя для твердофазного синтеза включает следующие этапы: вначале получают водную и масляную фазы; водная фаза содержит воду, диспергирующее средство и неорганическую соль; масляная фаза содержит: поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер, модифицированный мономер; порообразующий агент и инициатор. Затем масляную фазу добавляют к водной при перемешивании и нагревании для проведения реакции полимеризации, после чего, удаляют порообразующий агент. В результате получают пористую полимерную смолу, которая способна подвергаться дальнейшей реакции для получения носителя твердофазного синтеза, содержащего гидроксильную группу и аминогруппу в качестве функциональных групп. При этом дальнейшая реакция включает: получение носителя для твердофазного синтеза, содержащего аминогруппу в качестве функциональной группы посредством реакции Габриеля. Полученный носитель имеет содержание гидроксильных групп или аминогрупп от 100 до 1000 мкмоль/г; размер частиц в диапазоне 35-200 мкм и средний диаметр пор 10-200 нм. Получение олигонуклеотидов с использованием предложенного в изобретении носителя для твердофазного синтеза позволяет увеличить выход и чистоту олигонуклеотидов. 3 н. и 11 з.п. ф-лы, 18 ил., 2 табл., 11 пр.
1. Носитель для твердофазного синтеза, который имеет полимерный скелет с функциональными группами, которые представлены следующей формулой:
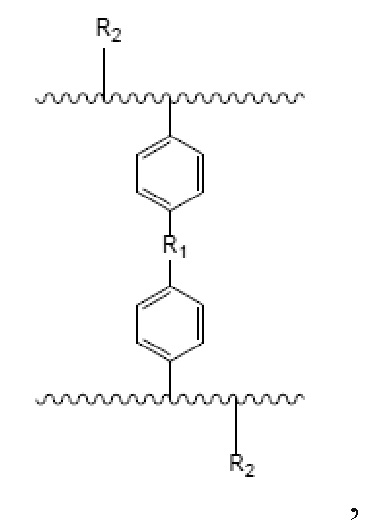
где R1= -(CH2)n-, n – целое число о 0 до 3, или R1= -O-(CH2)m-O-, m – целое число от 1 до 4; и R2= -OH или –NH2;
при этом получение носителя для твердофазного синтеза включает следующие этапы:
A. получение водной фазы и масляной фазы, соответственно; водная фаза содержит воду, диспергирующее средство и неорганическую соль; масляная фаза содержит: поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер, модифицированный мономер; порообразующий агент и инициатор,
при этом поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер и модифицированный мономер представляют собой мономеры, способные к полимеризации;
при этом диспергирующее вещество представляет собой поливиниловый спирт; неорганическая соль представляет собой хлорид натрия;
поперечно-сшивающий мономер выбирают из группы, состоящей из 4,4'- дивинилбифенила, ди(4-винилфенил)метана, 1,2-ди(4-винилфенил)этана, 1,3-ди(4- винилфенил)пропана, 1,4-ди(4'-винилфенокси)бутана;
моновинильное соединение выбирают из группы, состоящей из стирола, метилстирола, этилстирола;
функциональный мономер выбирают из группы, состоящей из п-хлорметилстирола, 4-(4-бромбутил)стирола, N-(4-винилфенил)ацетамида, 4-ацетоксистирола, бензоилоксистирола, метил-4-винилбензоата;
модифицированный мономер выбирают из группы, состоящей из фумаронитрила, 1,4-дициано-2-бутена, акрилонитрила, акриламида;
порообразующий агент выбирают из группы, состоящей из сорбитолтриолеата, изооктанола, изододекана, сорбитмоноолеата, дибутилфталата, полиоксиэтиленсорбитолеата, этиленгликолевого сложного эфира жирной кислоты, толуола, сорбитансесквиолеата, додеканола, циклогексана, полиоксиэтиленсорбитгексастеарата, пропиленгликольлаурата;
инициатор выбирают из группы, состоящей из пероксида бензоила, пероксида лауроила, трет-бутилперокси-2-этилгексаноата, 2,2'-азо-бис(-2-метилпропионитрила), 2,2'-азо-бис-(2-метилбутиронитрила) и 2,2'-азо-бис-(2,4-диметил)валеронитрила;
B. добавление масляной фазы к водной фазе, перемешивание и нагревание для проведения реакции полимеризации, и удаление порообразующего агента по завершении реакции, получение пористой полимерной смолы;
пористая полимерная смола способна подвергаться дальнейшей реакции для получения носителя твердофазного синтеза, содержащего гидроксильную группу и аминогруппу в качестве функциональных групп;
при этом дальнейшая реакция включает:
получение носителя для твердофазного синтеза, содержащего аминогруппу в качестве функциональной группы посредством реакции Габриеля пористой полимерной смолы, которую получают полимеризацией мономеров 4-хлорметилстирола и 4-(4-бромбутил)стирола;
получение носителя для твердофазного синтеза, содержащего аминогруппу в качестве функциональной группы посредством гидролиза пористой полимерной смолы, которую получают полимеризацией мономера N-(4-винилфенил)ацетамида; или
получение носителя для твердофазного синтеза, содержащего гидроксильную группу в качестве функциональной группы посредством гидролиза пористой полимерной смолы, которую получают полимеризацией 4-ацетоксистирола, бензоилоксистирола и мономеров метил-4-винилбензоата;
при этом носитель имеет содержание гидроксильной группы или аминогруппы 100–1000 мкмоль/г;
носитель имеет размер частиц в диапазоне 35–200 мкм; носитель имеет средний диаметр пор 10–200 нм.
2. Носитель для твердофазного синтеза по п. 1, который имеет содержание гидроксильной группы или аминогруппы 400–700 мкмоль/г.
3. Носитель для твердофазного синтеза по п. 1, который имеет размер частиц в диапазоне 50–100 мкм.
4. Носитель для твердофазного синтеза по п. 1, который имеет средний диаметр пор 40–100 нм.
5. Способ получения носителя для твердофазного синтеза по п. 1, включающий следующие этапы:
A. получение водной фазы и масляной фазы, соответственно; водная фаза содержит воду, диспергирующее средство и неорганическую соль; масляная фаза содержит: поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер, модифицированный мономер; порообразующий агент и инициатор, при этом поперечно-сшивающий мономер, моновинильное соединение, функциональный мономер и модифицированный мономер представляют собой мономеры, способные к полимеризации;
при этом диспергирующее вещество представляет собой поливиниловый спирт; неорганическая соль представляет собой хлорид натрия;
поперечно-сшивающий мономер выбирают из группы, состоящей из 4,4'- дивинилбифенила, ди(4-винилфенил)метана, 1,2-ди(4-винилфенил)этана, 1,3-ди(4- винилфенил)пропана, 1,4-ди(4'-винилфенокси)бутана;
моновинильное соединение выбирают из группы, состоящей из стирола, метилстирола, этилстирола;
функциональный мономер выбирают из группы, состоящей из п-хлорметилстирола, 4- (4-бромбутил)стирола, N-(4-винилфенил)ацетамида, 4-ацетоксистирола, бензоилоксистирола, метил-4-винилбензоата;
модифицированный мономер выбирают из группы, состоящей из фумаронитрила, 1,4- дициано-2-бутена, акрилонитрила, акриламида;
порообразующий агент выбирают из группы, состоящей из сорбитолтриолеата, изооктанола, изододекана, сорбитмоноолеата, дибутилфталата, полиоксиэтиленсорбитолеата, этиленгликолевого сложного эфира жирной кислоты, толуола, сорбитансесквиолеата, додеканола, циклогексана,
полиоксиэтиленсорбитгексастеарата, пропиленгликольлаурата;
инициатор выбирают из группы, состоящей из пероксида бензоила, пероксида лауроила, трет-бутилперокси-2-этилгексаноата, 2,2'-азо-бис(-2-метилпропионитрила), 2,2'-азо-бис-(2-метилбутиронитрила) и 2,2'-азо-бис-(2,4-диметил)валеронитрила;
B. добавление масляной фазы к водной фазе, перемешивание и нагревание для проведения реакции полимеризации, и удаление порообразующего агента по завершении реакции, получение пористой полимерной смолы;
пористая полимерная смола способна подвергаться дальнейшей реакции для получения носителя твердофазного синтеза, содержащего гидроксильную группу и аминогруппу в качестве функциональных групп;
при этом дальнейшая реакция включает:
получение носителя для твердофазного синтеза, содержащего аминогруппу в качестве функциональной группы посредством реакции Габриеля пористой полимерной смолы, которую получают полимеризацией мономеров 4-хлорметилстирола и 4-(4-бромбутил)стирола;
получение носителя для твердофазного синтеза, содержащего аминогруппу в качестве функциональной группы посредством гидролиза пористой полимерной смолы, которую получают полимеризацией мономера N-(4-винилфенил)ацетамида; или
получение носителя для твердофазного синтеза, содержащего гидроксильную группу в качестве функциональной группы посредством гидролиза пористой полимерной смолы, которую получают полимеризацией 4-ацетоксистирола, бензоилоксистирола и мономеров метил-4-винилбензоата эфира;
при этом носитель имеет содержание гидроксильной группы или аминогруппы 100–1000 мкмоль/г;
носитель имеет размер частиц в диапазоне 35–200 мкм; носитель имеет средний диаметр пор 10–200 нм.
6. Способ по п. 5, в котором:
диспергирующее вещество присутствует в количестве 0,1–5 мас.% в водной фазе, а неорганическая соль присутствует в количестве 20 мас.% или ниже в водной фазе; массовое отношение масляной фазы к водной фазе составляет 1:3–1:20;
моновинильное соединение в масляной фазе составляет 45–95 мас.% от общей массы мономеров;
поперечно-сшивающий мономер в масляной фазе составляет 2,9–20 мас.% от общей массы мономеров;
функциональные мономеры в масляной фазе составляют 2–20 мас.% от общей массы мономеров;
модифицированный мономер в масляной фазе составляет 0,1–15 мас.% от общей массы мономеров; и
порообразующий агент в масляной фазе составляет 15–130 мас.% от общей массы мономеров.
7. Способ по п. 6, в котором:
моновинильное соединение в масляной фазе составляет 62–86 мас.% от общей массы мономеров;
поперечно-сшивающий мономер в масляной фазе составляет 7–13 мас.% от общей массы мономеров;
функциональные мономеры в масляной фазе составляют 5–15 мас.% от общей массы мономеров;
модифицированный мономер в масляной фазе составляет 2–10 мас.% от общей массы мономеров; и
порообразующий агент в масляной фазе составляет 30–110 мас.% от общей массы мономеров.
8. Способ по п. 5, в котором полимеризацию проводят при температуре 50–90 °C.
9. Способ по п. 8, в котором полимеризацию проводят при температуре 70–85 °C.
10. Способ по любому из пп. 5-9, включающий следующие этапы:
добавление в реактор определенного количества очищенной воды, добавление диспергирующего вещества в количестве 0,1–5 мас.% от водной фазы и неорганическую соль в количестве не более 20 мас.% от водной фазы, и растворение для получения водной фазы;
отвешивание моновинильного соединения, поперечно-сшивающего мономера,
функционального мономера, модифицированного мономера, порообразующего агента и инициатора в соответствии с массовым отношением масляной фазы к водной фазе, составляющим 1:3–1:20; при этом моновинильное соединение составляет 45–95 мас.% от общей массы мономеров, поперечно-сшивающий мономер составляет 2,9–20 мас.% от общей массы мономеров, функциональный мономер составляет 2–20 мас.% от общей массы мономеров, модифицированный мономер составляет 0,1–15 мас.% от общей массы мономеров, порообразующий агент составляет 15–130 мас.% от общей массы мономеров, и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы в реактор, перемешивание и нагревание до температуры 50–90 °C для проведения реакции; удаление порообразующего агента по завершении реакции, просеивание и сбор смолы с соответствующим размером частиц и вакуумная сушка для получения полимерной смолы; и
проведение дополнительной реакции со смолой для получения носителя для твердофазного синтеза, имеющего аминогруппу и гидроксильную группу.
11. Способ по п. 10, включающий следующие этапы:
добавление в реактор определенного количества очищенной воды, добавление диспергирующего вещества в количестве 0,1–5 мас.% от водной фазы и неорганическую соль в количестве не более 20 мас.% от водной фазы, и растворение для получения водной фазы;
отвешивание моновинильного соединения, поперечно-сшивающего мономера, функционального мономера, модифицированного мономера, порообразующего агента и инициатора в соответствии с массовым отношением масляной фазы к водной фазе, составляющим 1:3–1:20; при этом моновинильное соединение составляет 62–86мас.% от общей массы мономеров, поперечно-сшивающий мономер составляет 7–13 мас.% от общей массы мономеров, функциональный мономер составляет 5–15 мас.% от общей массы мономеров, модифицированный мономер составляет 2–10 мас.% от общей массы мономеров, порообразующий агент составляет 30–110 мас.% от общей массы мономеров, и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы в реактор, перемешивание и нагревание до температуры 70–85 °C для проведения реакции; удаление порообразующего агента по завершении реакции, просеивание и сбор смолы с соответствующим размером частиц и вакуумная сушка для получения полимерной смолы; и
проведение дополнительной реакции со смолой для получения носителя для твердофазного синтеза, имеющего аминогруппу и гидроксильную группу.
12. Способ по любому из пп. 5-11, включающий следующие этапы:
добавление 2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворение для получения водяной фазы;
отвешивание 108,8 г стирола, 14 г 1,2-ди(п-винилфенил)этана, 12,2 г п-хлорметилстирола, 5 г. фумаронитрила, 6 г сорбитолтриолеата, 40 г изооктанола, 20 г изододекана и 2,5 г пероксида бензоила и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы, перемешивание и нагревание до 80 °C для проведения полимеризации в течение 6 ч; по завершении полимеризации промывка горячей водой, удаление порообразующего агента путем экстракции орошением этанолом, отсеивание и сбор смолы с размером частиц 50–100 мкм и сушка в вакууме для получения полимерной пористой смолы с содержанием хлора 565 мкмоль/г;
добавление 50 г полимерной пористой смолы и 500 мл N,N-диметилформамида в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивание; затем добавление 30 г фталата калия и нагревание до температуры до 95 °C для проведения реакции в течение 16 часов; по завершении реакции охлаждение до комнатной температуры, промывка дважды N,N-диметилформамидом, промывка очищенной водой до нейтрального состояния, промывка трижды абсолютным этанолом и отфильтровывание и сушка смолы; добавление 200 г абсолютного этанола и 50 г гидразингидрата в реактор, повышение температуры до 75 °C и проведение реакции в течение 16 часов; после этого промывка три раза раствором этанол/очищенная вода в объемном соотношении 50:50, промывка очищенной водой до нейтрального состояния, промывка три раза абсолютным этанолом и отфильтровывание и сушка; добавление 200 г абсолютного этанола и 50 г концентрированной хлористо-водородной кислоты в реактор, нагревание до температуры до 60 °C и проведение реакции в течение 6 ч, после этого охлаждение до комнатной температуры, промывка водой до нейтрального состояния и затем сушка в вакууме для получения носителя для твердофазоного синтеза, имеющего содержание аминогруппы 554 мкмоль/г и средним диаметром пор 54 нм, измеренным методом интрузии ртути.
13. Способ по любому из пп. 5-11, включающий следующие этапы:
добавление 2 л очищенной воды, 20 г поливинилового спирта и 30 г хлорида натрия в 3-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и растворение для получения водяной фазы;
отвешивание 107,7 г стирола, 12,8 г 1,2-ди(п-винилфенил)этана, 12,5 г 4-ацетоксистирола, 7 г фумаронитрила, 10 г сорбитолтриолеата, 42 г изооктанола, 21 г изододекана и 2,5 г пероксида бензоила и тщательное перемешивание для получения масляной фазы;
добавление масляной фазы, перемешивание и нагревание до 78 °C для проведения полимеризации в течение 6 ч; по завершении полимеризации промывка горячей водой, удаление порообразующего агента путем экстракции орошением этанолом, отсеивание и сбор смолы с размером частиц 50–100 мкм и сушка в вакууме для получения полимерной пористой смолы;
добавление 50 г полимерной пористой смолы и 300 мл ацетонитрила в 1-литровый реактор, оборудованный конденсатором, мешалкой и термометром, и перемешивание; затем медленное добавление 7,5 мл гидразингидрата и проведение реакции в течение 3 ч при комнатной температуре; промывка водой до нейтрального состояния и затем сушка в вакууме для получения носителя для твердофазного синтеза с содержанием гидроксильной группы 538 мкмоль/г и средним диаметром пор 58 нм, измеренным методом интрузии ртути.
14. Использование носителя для твердофазного синтеза по п. 1, полученного посредством способа по любому из пп. 5-13, в твердофазном синтезе олигонуклеотидов.
| US 8592542 B2, 26.11.2013 | |||
| Sundell M.J | |||
| et al | |||
| Crosslinked polystyrene with improved mechanical properties // Journal of Polymer Science Part A: Polymer Chemistry | |||
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| Способ очистки нефти и нефтяных продуктов и уничтожения их флюоресценции | 1921 |
|
SU31A1 |
| Способ получения средней яри медянки | 1923 |
|
SU2305A1 |
| Lu J., Shouting W., Binglin H | |||
| The mechanism of copolymerization of vinyl/divinyl monomers | |||
| VII the role of pendant double bonds in network formation during | |||

