ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка является международной заявкой, по которой испрашивается приоритет предварительных заявок США № 62/800,996, поданной 4 февраля 2019, № 62/851,433, поданной 22 мая 2019, № 62/894,167, поданной 30 августа 2019, № 62/937,618, поданной 19 ноября 2019 и № 62/962,647, поданной 17 января 2020, содержание каждой из которых включено в настоящий документ в качестве ссылки полностью.
ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к способам лечения или профилактики конкретных симптомов и нарушений, которые ассоциированы с лизосомными болезнями накопления, с использованием соединений хинуклидина формулы (I), необязательно в комбинации с ферментной заместительной терапией. Также включена боль, такая как боль в животе, и . дерматологические нарушения, такие как ангиокератома, у пациентов с болезнью, такой как болезнь Фабри.
УРОВЕНЬ ТЕХНИКИ
Лизосомные болезни накопления
Лизосомные болезни накопления (LSD) представляют собой группу из около 50 редких наследственных заболеваний обмена веществ, вызванных дефектами лизосомальной функции. Как правило, пациенты с LSD накапливают вредные уровни субстрата (т.е. хранимого материала) в лизосомах из-за дефицита или дефекта фермента, ответственного за метаболизм субстрата, или из-за дефицита ферментного активатора, необходимого для правильной ферментативной функции. Большинство LSD вызваны единичным ферментативным дефектом или дефицитом, обычно фермента, вовлеченного в метаболизм жиров или гликопротеинов. Некоторые из наиболее распространенных LSD включают болезнь Гоше, болезнь Фабри и болезнь Ниманна-Пика (тип C). Гоше, Фабри и Ниманн-Пик являются примерами сфинголипидозов. Каждое из этих заболеваний связано с набором симптомов, которые прямо или косвенно вызваны лежащими в основе генетическими дефектами. В результате часто бывает трудно предсказать, какие симптомы или нарушения, связанные с ними, можно эффективно лечить с помощью различных способов лечения. Симптомы, общие для нескольких ЛСД, включают изменения саккадических движений глаз, когнитивную дисфункцию и нарушения походки, такие как атаксия. Эти симптомы особенно распространены при болезни Гоше (например, типа 3) и болезни Неймана-Пика (типа C).
Болезнь Фабри и заболевания кожи
Болезнь Фабри вызвана дефектом в ферменте альфа-галактозидазе, приводящим к накоплению глоботриаосилцерамида (GL-3, также известного как Gb3). GL-3 накапливается в плазме крови, ткани и некоторых органах. Болезнь вызвана X-связанной рецессивной мутацией, такой, что мужчины могут иметь тяжелые симптомы, в то время как у женщин они могут варьироваться от отсутствия симптомов до умеренных или тяжелых. Общим симптомом болезни Фабри является боль всего тела или локализованная боль. Периферическая невропатия, отмеченная осложнениями почек, также распространена, включая хроническую болезнь почек и почечную недостаточность. Накопление сфинголипида в сердечной мышце может вызвать сердечную гипертрофию или рестриктивную кардиомиопатию, а также патологии ритма сердца, такие как тахикардия и брадикардии (включая полную блокаду сердца). Участие кожи включает формирование ангиокератом (маленьких, безболезненных папул), и глазное участие может включать заметмнение роговиц (вихревая кератопатия) и конъюнктивальные и сетчаточные сосудистые патологии и катаракты.
Накопление GL-3 в коже и мягких тканях вызывает самые ранние симптомы болезни Фабри, включая повреждения кожи (например, ангиокератомы), акропарестезию и гипогидроз. См. Lidove, O. et al., Dermatological and Soft-Tissue Manifestations of Fabry Disease: Characteristics and Response to Enzyme Replacement Therapy, Глава 24 в Fabry Disease: Perspectives from 5 years of FOS (Mehta, A. et al. eds., Oxford PharmaGenesis 2006), доступно онлайн через NCBI Bookshelf, National Library of Medicine. Эти симптомы часто начинаются в детстве, за много лет до развития тяжелого повреждения органа, которое является характеристикой более поздних стадий болезни. Кожа включает несколько слоев: эпидермис, дерму и гиподерму, каждый из которых имеют много типов клеток. Болезнь Фабри поражает главным образом эндотелиальные сосуды. Эндотелиальные клетки в поверхностной дерме, чуть ниже бессосудистого эпидермиса, являются главной мишенью заболевания. Эндотелиальные клетки Vasa nervorum в периневрии также затронуты. Кроме того, GL-3 может накопиться в лизосомах сосудистых перицитов, экзокринных клеток железы и кожных фибробластах.
Ангиокератомы являются доброкачественными сосудистыми повреждениями кожи, характеризуемыми пролиферацией расширенных кровеносных сосудов в верхней дерме. Они происходят, когда накопление GL-3 в кожных эндотелиальных клетках приводит к выпуклым сосудам и недостаточности стенки сосуда, сопровождаемой вторичной эктазией. Они являются основными кожными повреждениями, найденными у больных с болезнью Фабри, и могут начать появляться у детей в возрасте 5-15 лет (средний возраст, 13,5 лет). Они найдены у 83% мужчин и 80% женщин с болезнью Фабри. Ангиокератомы растпространяются с возрастом и становятся видимыми на губах и пальцах рук и ног. Они могут быть изолированы или сгруппированы и появиться как маленькие красно-черные папулы с гладкой эпидермальной поверхностью. В то время как болезнь прогрессирует, повреждения растут, достигая диаметра 10 мм, и становятся темно-красными/черными с бородавчатой поверхностью. Исследования электронной микроскопией показывают электроноплотные лизосомальные включения в сосудистые эндотелиальные клетки, сосудистые перициты, экзокринные клетки железы, кожные фибробласты и периневрий.
В дополнение к ангиокератомам, Фабри также ассоциирована с определенной полиневропатией и инфильтрацией потовой железы, часто приводящим к патологиям потения при болезни Фабри. Классические симптомы включают гипогидроз (уменьшенное потоотделение) и ангидроз (отсутствие потоотделения). Эти симптомы могут иметь значительное влияние на качество жизни, вызывая лихорадку и непереносимость нагревания и упражнений. Гипогидроз предрасполагает к акропарестезии, делая больных нетерпимыми к теплу и упражнениям. Уменьшенное слезообразование слезными железами и уменьшенное образование слюны, может быть связано с гипогидрозом у больных с болезнью Фабри.
Гипергидроз намного менее распространен у больных с болезнью Фабри, чем гипогидроз и, кажется, более распространен у женщин, чем мужчины. Вероятно, что гипергидроз является проявлением периферической невропатии, происходящей при болезни Фабри. Когда затронуты кожные и слизистые железы, может быть необходимо ограничить время, которое пациент проводит в теплой среде, или его физическую активность.
У больных с болезнью Фабри, лимфедема, по видимому, связана с накоплением гликолипидов в лимфатических сосудах. В отсутствие терапии лимфедема при болезни Фабри может быть осложнена рожистым воспалением с риском общей инфекции. Тяжелая лимфатическая микроангиопатия, приводящая к лимфедеме, была описана у пациентов с болезнью Фабри. Однако исследование показало, что тяжелые структурные и функциональные изменения в начальных лимфатических сосудах кожи происходят и у мужчин, и у женщин с болезнью Фабри, независимо от того, когда проявляется лимфедема.
Акропарестезия является также одним из самых ранних симптомов болезни Фабри. Она считается результатом ишемии периферических нервов, вторично к патологиям в эндотелиальных клетках периневрия, и характеризуется покалыванием, хроническим ощцщением жара или ноющей болью в руках и ногах. Острые эпизоды выводящей из строя боли, длящейся от нескольких минут до нескольких дней, могут возникать. Они могут произойти спонтанно, но могут также быть вызваны теплом, болезнью, стрессом или упражнением. Усталость, умеренная лихорадка и боль в суставах могут быть связаны с этими кризами острой боли.
Болезнь Фабри и боль
Боль является изнурительным симптомом Фабри относительно способности пациента участвовать в нормальных действиях повседневной жизни. Общим симптомом Фабри является боль всего тела, боль, локализованная в краевых зонах, или желудочно-кишечная боль (например, боль в животе), которая, предположительно, возникает из-за поверждения окончаний периферических нервов и/или из-за накопления жира в капиллярной сосудистой сети, вызывающнго болезненную обструкцию тока крови.
Ферментозаместительная терапия (ERT) является одним из немногих в настоящее время одобренных способов лечения болезни Фабри в Соединенных Штатах полусинтетическим ферментом агалзидазой бета (альфа-галактозидазой). Другой фермент, агалзидаза альфа, одобрен в Европе, но не в Соединенных Штатах. В Европе, низкомолекулярный ингибитор альфа-галактозидазы мгаластат (1-деоксигалактоножиримицин) также одобрен для лечения подмножеств пациентов Фабри. У некоторых пациентов дефект альфа-галактозидазы вызывается неправильным сворачиванием белка, а не ошибкой в аминокислотной последовательности. Для некоторых из таких пациентов, было обнаружено, что мигаластат связымиется с неправильно свернутым белком и заставляет его переориентироваться в надлежащей структуре - но только некоторые виды ошибок неправильного сворачивания поддаются этой коррекции.
Таким образом, ERT остается основным лечением болезни Фабри. ERT не является очень эффективной для облегчения боли, связанной с болезнью Фабри, и обычно необходимо дополнительное лечение анальгетиками, противосудорожными и нестероидными противовоспалительными средствами. Поэтому остается значительная неудовлетворенная потребность в лечении для болезни Фабри, которое также эффективно управляет болью при болезни Фабри.
Описанные здесь хинуклидиновые соединения обладают активностью в качестве ингибиторов фермента глюкозилцерамидсинтазы (GCS). Эти соединения были описаны как обычно используемые при лечении лизосомных болезней накопления, таких как болезнь Фабри, болезнь Гоше и болезнь Ниманна-Пика. См., например, WO 2012/129084 и U.S. 2016/0361301.
В данной области существует реальная потребность в разработке терапевтических средств, эффективных для облегчения или управления болью и дерматологическими расстройствами, связанными с болезнью Фарри.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
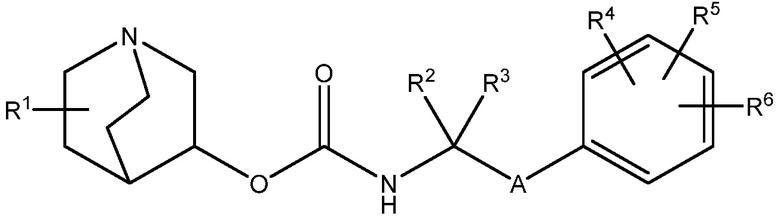
Настоящее изобретение относится к соединению хинуклидина (соединение 1) согласно формуле (I),
(I)
или его фармацевтически приемлемой соли или пролекарству, где:
R1 выбирают из водорода, галогена (например, фтор), циано, нитро, гидрокси, тио, амино, C1-6-алкила (например, метила или этила), C2-6-алкенила, C2-6-алкинил, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанный алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
R2 и R3 независимо выбирают из C1-3-алкила, необязательно замещенного одним или несколькими (например, 1, 2 или 3) галогенами, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) галогенами;
R4, R5 и R6 каждый независимо выбирают из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одним или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси; и
A является 5- или 6-членной арильной или гетеоарильной группой, необязательно замещенной 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6алкокси или C1-6алкила.
В первом аспекте, в настоящей заявке представлен способ лечения или профилактики боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, у субъекта, нуждающегося в этом, где способ включает введение субъекту эффективного количества соединения хинуклидина, как описано в настоящем документе, например, соединения формулы I. В других аспектах, настоящая заявка дополнительно представляет использование соединений хинуклидина, описанных в настоящем документе, для лечения или профилактики боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, и кожные нарушения (например, ангиокератому, акропаретезию, гипогидроз, ангидроз, гипергидроз, лимфедему и акропарестезию). В некоторых вариантах осуществления любых из этих аспектов, субъектом, нудающимся в этом, является субъект, имеющий болезнь Фабри, болезнь Гоше, например, 3 типа или болезнь Ниманна-Пика типа С.
Дополнительные признаки и преимущества соединений, композиций и способов, описанных в настоящем документе, будут очевидны из следующего подробного описания.
ПОДРОБНОЕ ОПИСАНИЕ
Хотя конкретные варианты осуществления настоящего описания теперь будут описаны со ссылкой на примеры получения и схемы, следует понимать, что такие варианты осуществления приведены только в качестве примера и просто иллюстрируют лишь небольшое количество из многих возможных конкретных вариантов осуществления, которые могут представлять заявки принципов настоящего описания. Различные изменения и модификации будут очевидны специалистам в данной области техники, учитывая преимущество настоящего раскрытия, и считаются находящимися в пределах сущности и объема настоящего описания, как дополнительно определено в прилагаемой формуле изобретения.
Определения
Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно понимаются средним специалистом в области, к которой относится это описание. Хотя любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем документе, могут быть использованы на практике или при тестировании настоящего изобретения, далее описаны типовые способы, устройства и материалы. Все технические и патентные публикации, цитируемые в настоящем документе, полностью включены в настоящий документ в качестве ссылки. Ничто в настоящем документе не может быть истолковано как признание того, что изобретение не имеет права датировать такое описание задним числом на основании предшествующего изобретения.
Практика настоящего описания будет использовать, если не указано иное, обычные способы культивирования тканей, иммунологии, молекулярной биологии, микробиологии, клеточной биологии и рекомбинантной ДНК, которые находятся в компетенции специалистов в данной области техники.
Все числовые обозначения, например, pH, температура, время, концентрация, молекулярная масса, включая диапазоны, являются приблизительными, которые варьируются (+) или (-) с шагом 0,1 или 1,0, где это необходимо. Следует понимать, хотя это не всегда явно указано, что всем числовым обозначениям предшествует термин «примерно». Также следует понимать, хотя не всегда явно указано, что реагенты, описанные в настоящем документе, являются просто типовыми, и что их эквиваленты известны в данной области техники.
Используемый в настоящем документе термин «необязательно замещенный» является эквивалентом фразы «незамещенный или замещенный».
Используемая в настоящем документе фраза «в способе лечения или профилактики» (например, во фразе «в способе лечения или профилактики боли») является эквивалентом фразы «при лечении или профилактике» (например, в фразе« при лечении или профилактике боли»).
Как используется в описании и формуле изобретения, формы единственного числа включают множественное число, если контекст явно не диктует иное. Например, термин «клетка» включает множество клеток, включая их смеси. Если специально не указано или не очевидно из контекста, как используется в настоящем документе, термин «или» следует понимать как включающий. Термин «включая» используется в настоящем документе для обозначения и используется взаимозаменяемо с фразой «включая, но не ограничиваясь этим».
Используемый в настоящем документе термин «содержащий» или «содержит» предназначен для обозначения того, что композиции и способы включают перечисленные элементы, но не исключают другие. «Состоящий по существу из», когда используется для определения композиций и способов, означает исключение других элементов, имеющих какое-либо существенное значение для комбинации для заявленной цели. Таким образом, композиция, состоящая по существу из элементов, как определено в настоящем документе, не исключает следовых примесей из способа выделения и очистки и фармацевтически приемлемых носителей, таких как солевой раствор с фосфатным буфером, консерванты и тому подобное. «Состоит из» означает исключение более чем следовых элементов других ингредиентов и существенных стадий способа введения композиций по настоящему изобретению или стадий способа получения композиции или достижения желаемого результата. Варианты осуществления, определенные каждым из этих переходных терминов, входят в объем настоящего изобретения. Использование термина «содержащий» в настоящем документе предназначено для охвата «состоящий по существу из» и «состоящий из».
Термины «субъект», «индивидуум» или «пациент» используются в настоящем документе взаимозаменяемо и относятся к позвоночному, такому как млекопитающее. Млекопитающие включают, но не ограничиваются ими, мышей, крыс, кроликов, обезьян, крупный рогатый скот, овец, свиней, собак, кошек, сельскохозяйственных животных, спортивных животных, домашних животных, лошадей, приматов и людей. В одном варианте осуществления, млекопитающие включают лошадей, собак и кошек. В некоторых вариантах осуществления, млекопитающим является человек, например, человек, страдающий определенным заболеванием или нарушением, таким как болезнь Гоше (например, GD-3) или болезнь Ниманна-Пика типа C.
«Введение» определено в настоящем документе как средство предоставления агента или композиции, содержащей агент, субъекту таким образом, чтобы агент находился внутри тела субъекта. Такое введение может осуществляться любым путем, включая, без ограничений, пероральный, трансдермальный (например, вагинальный, ректальный, перорально-слизистый), путем инъекции (например, подкожный, внутривенный, парентеральный, внутрибрюшинный, в ЦНС) или путем ингаляции (например, пероральный или назальный). Разумеется, фармацевтические препараты выпускаются в формах, подходящих для каждого пути введения.
«Лечить» или «лечение» заболевания обычно включает: (1) ингибирование заболевания, т.е. остановку или уменьшение развития заболевания или его клинических симптомов; и/или (2) облегчение заболевания, т.е. вызывание регресса заболевания или его клинических симптомов.
«Профилактика» или «предотвращение» заболевания обычно включает предотвращение развития клинических симптомов заболевания у пациента, который может быть предрасположен к заболеванию, но еще не испытывает или не проявляет симптомов заболевания.
Термин «страдающий», если он относится к термину «лечение», относится к пациенту или человеку, у которого было диагностировано заболевание. Термин «страдающий», если он относится к термину «профилактика», относится к пациенту или человеку, который предрасположен к заболеванию. Пациента также можно отнести к категории «подверженных риску страдания» заболеванием из-за наличия в анамнезе болезни в его семейном анамнезе, или из-за наличия генетических мутаций, связанных с заболеванием. У пациента, подверженного риску заболевания, еще не развиваются все или некоторые из характерных патологий заболевания.
«Эффективное количество» или «терапевтически эффективное количество» является количеством, достаточным для достижения благоприятных или желаемых результатов. Эффективное количество можно вводить за одно или несколько введений, применений или дозировок. Такая доставка зависит от ряда переменных, включая период времени, в течение которого должна использоваться индивидуальная дозированная форма, биодоступность терапевтического агента и путь введения. Однако понятно, что конкретные уровни доз терапевтических агентов по настоящему изобретению для любого конкретного субъекта зависят от множества факторов, включая, например, активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол и диету субъекта, время введения, скорость выведения, комбинацию лекарственных средств и тяжесть конкретного нарушения, подвергаемого лечению, и форму введения. Лечебные дозировки обычно можно титровать для оптимизации безопасности и эффективности. Как правило, взаимосвязь между дозой и эффектом, полученная при испытаниях in vitro и/или in vivo, первоначально может предоставить полезные рекомендации по правильным дозам для введения пациенту. Обычно желательно вводить такое количество соединения, которое эффективно для достижения уровня в сыворотке, соизмеримого с концентрациями, которые оказались эффективными in vitro. Определение этих параметров находится в компетенции специалиста в данной области техники. Эти соображения, а также эффективные составы и процедуры введения хорошо известны в данной области техники и описаны в стандартных учебниках. В соответствии с этим определением, используемым в настоящем документе, термин «терапевтически эффективным количеством» является количество, достаточное для лечения (например, улучшения) одного или нескольких симптомов, ассоциированных с заболеванием или нарушением, описанным в настоящем документе (например, в любом из Способа 2 и след. или Способа 3 и след.) ex vivo, in vitro или in vivo.
В настоящем документе термин «фармацевтически приемлемый эксципиент» охватывает любые стандартные фармацевтические эксципиенты, включая носители, такие как солевой раствор с фосфатным буфером, воду и эмульсии, такие как эмульсии масло/вода или вода/масло и различные типы смачивающих агентов. Фармацевтические композиции также могут включать стабилизаторы и консерванты. Примеры носителей, стабилизаторов и адъювантов см. Remington’s Pharmaceutical Sciences (20th ed., Mack Publishing Co. 2000).
Используемый в настоящем документе термин «пролекарство» означает фармакологическое производное исходной молекулы лекарственного средства, которое требует биотрансформации, спонтанной или ферментативной, в организме, для высвобождения активного лекарственного средства. Например, пролекарствами являются вариации или производные соединения хинуклидина, описанного в настоящем документе, которые имеют группы, расщепляемые при определенных метаболических условиях, которые, при расщеплении, превращаются в соединение хинуклидина, описанное в настоящем документе, например, соединение формулы I. Такие пролекарства затем фармацевтически активны in vivo, когда они подвергаются сольволизу в физиологических условиях или подвергаются ферментативной деградации. Соединения-пролекарства в настоящем документе могут называться одинарными, двойными, тройными и т.д., в зависимости от количества стадий биопревращения, необходимых для высвобождения активного лекарственного средства в организме, и количества функциональных групп, присутствующих в форме предшественника. Формы пролекарств часто обладают преимуществами растворимости, тканевой совместимости или замедленного высвобождения в организме млекопитающих.
Пролекарства, широко известные в данной области техники, включают хорошо известные производные кислот, такие как, например, сложные эфиры, полученные реакцией кислых соединений с подходящим спиртом, амиды, полученные реакцией кислых соединений с амином и основными группами, которые вступают в реакцию с образованием производного ацилированного основания. Другие производные пролекарства могут быть объединены с другими признаками, описанными в настоящем документе, для повышения биодоступности. Таким образом, специалисты в данной области техники поймут, что некоторые из описанных здесь соединений, имеющие, например, свободные амино- или гидроксигруппы, могут быть превращены в пролекарства. Пролекарства включают соединения, имеющие аминокислотный остаток, или полипептидную цепь из двух или нескольких (например, двух, трех или четырех) аминокислотных остатков, которые ковалентно связаны через пептидные связи со свободными группами амино, гидрокси или карбоновой кислоты описанных здесь соединений. Аминокислотные остатки включают 20 встречающихся в природе аминокислот, обычно обозначаемых трехбуквенными символами, а также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства также включают соединения, имеющие группу карбоната, карбамата, амида или алкилового эфира, ковалентно связанную с любым из вышеуказанных заместителей, описанных в настоящем документе.
Используемый в настоящем документе термин «фармацевтически приемлемая соль» означает фармацевтически приемлемую кислотно-аддитивную соль или фармацевтически приемлемую основно-аддитивную соль описанного соединения, которую можно вводить без каких-либо конечного существенных нежелательных биологических эффектов или любого конечного вредного взаимодействия с любым другим компонентом фармацевтической композиции, в которой она может содержаться.
В настоящем документе, термин «C1-6-алкил» означает насыщенный или разветвленный свободный радикал, состоящий по существу из 1-6 атомов углерода и соответствующего количества атомов водорода. Типовые C1-6-алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил и изобутил. Другие C1-6-алкильные группы будут очевидны специалисту в данной области техники, учитывая преимущество настоящего описания. Термины «C1-3-алкил», «C1-4-алкил» и т.д. имеют эквивалентные значения, т.е., насыщенный линейный или разветвленный свободный радикал, состоящий по существу из 1-3 (или 4) атомов углерода и соответствующего числа атомов водород.
В настоящем документе, термин «C2-6-алкенил» означает ненасыщенный линейный или разветвленный свободный радикал, состоящий, по существу, из 2-6 атомов углерода и соответствующего количества атомов водорода, причем свободный радикал содержит, по меньшей мере, одну двойную связь углерод-углерод. Типовые C2-6-алкенильные группы включают этенил, проп-1-енил, проп-2-енил, изопропенил, бут-1-енил, 2-метилпроп-1-енил и 2-метил-проп-2-енил. Другие C2-6-алкенильные группы будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания.
В настоящем документе, термин «C2-6-алкинил» означает ненасыщенный линейный или разветвленный свободный радикал, состоящий, по существу, из 2-6 атомов углерода и соответствующего количества атомов водорода, причем свободный радикал содержит, по меньшей мере, одну тройную связь углерод-углерод. Типовые C2-6-алкинильные группы включают этинил, проп-1-инил, проп-2-инил, бут-1-инил и 3-метилбут-1-инил. Другие C2-6-алкинильные группы будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания.
В настоящем документе, термин «C1-6-алкилокси» означает насыщенный линейный или разветвленный свободный радикал, состоящий по существу из 1-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода. C1-6-алкилоксигруппа присоединена через атом кислорода. Типовые C1-6-алкилоксигруппы включают метилокси, этилокси, н-пропилокси, изопропилокси, н-бутилокси и изобутилокси. Другие C1-6-алкилоксигруппы будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания. Термины «C1-3-алкилокси», «C1-4-алкилокси» и подобные имеют эквивалентные значения, т.е. насыщенный линейный или разветвленный свободный радикал, состоящий по существу из 1-3 (или 4) атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, где группа присоединена через атом кислорода.
В настоящем документе, термин «C2-6-алкенилокси» означает ненасыщенный линейный или разветвленный свободный радикал, состоящий, по существу, из 2-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, причем свободный радикал содержит, по меньшей мере, одну двойную связь углерод-углерод. C2-6-алкенилоксигруппа присоединена через атом кислорода. Типовой C2-6-алкенилоксигруппой является этенилокси; другие будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания.
В настоящем документе термин «C2-6-алкинилокси» означает ненасыщенный линейный или разветвленный свободный радикал, состоящий, по существу, из 2-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, причем свободный радикал содержит, по меньшей мере, одну тройную связь углерод-углерод. C2-6-алкенилоксигруппа присоединена через атом кислорода. Типовой C2-6-алкенилоксигруппой является этинилокси; другие будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания.
В настоящем документе термин «гетероарил» означает ароматический свободный радикал, содержащий 5 или 6 атомов (т.е. атомы кольца), которые образуют кольцо, где от 1 до 5 атомов кольца являются углеродом, а оставшиеся от 1 до 5 атомов кольца (т.е. гетероатомы кольца) выбраны независимо из группы, состоящей из азота, серы и кислорода. Типовые 5-членные гетероарильные группы включают фурил, тиенил, тиазолил (например, тиазол-2-ил), пиразолил, изотиазолил, оксазолил, изоксазолил, пирролил, триазолил, имидазолил, оксадиазолил и тиадиазолил. Типовые 6-членные гетероарильные группы включают пиридил, пиримидил, пиразинил, пиридазинил, 1,2,4-триазинил, бензоксазолил, бензотиазолил, бензизотиазолил, бензизоксазолил и бензимидазолил. Другие гетероарильные группы будут очевидны специалистам в данной области техники, учитывая преимущество настоящего описания. Обычно гетероарильная группа присоединена к основной структуре через атом углерода. Однако специалисты в данной области поймут, что некоторые другие атомы, например, гетероатомы кольца, могут быть присоединены к основной структуре.
В настоящем документе термин «арил» означает ароматический свободный радикал, содержащий 5 или 6 атомов (т.е. атомов кольца), которые образуют кольцо, где все атомы кольца являются углеродом. Типовой арильной группой является фенильная группа.
В настоящем документе термин «алифатический» означает не ароматическое соединение, содержащее атомы углерода и водорода, например, содержащее от 1 до 9 атомов углерода. Алифатические соединения могут быть линейными или разветвленными, могут содержать одну или несколько кольцевых структур и могут содержать одну или несколько двойных связей углерод-углерод (при условии, что соединение не содержит не насыщенную кольцевую структуру, имеющую ароматический характер). Примеры алифатических соединений включают этан, пропилен, циклобутан и циклогексадиен.
В настоящем документе термины «гало» и «галоген» означают фтор, хлор, бром или йод. Эти термины используются взаимозаменяемо и могут относиться к свободнорадикальной группе галогена или к атому галогена как таковому. Специалисты в данной области техники легко смогут установить идентификацию, с учетом контекста, в котором этот термин используется в настоящем описании.
В настоящем документе термин «циано» означает свободный радикал, имеющий атом углерода, связанный с атомом азота тройной связью. Радикал циано присоединен через атом углерода.
В настоящем документе термин «нитро» означает -NO2 радикал, который присоединен через атом азота.
В настоящем документе термины «гидрокси» и «гидроксил» означают -ОН радикал, который присоединен через атом кислорода. Термин «тио» означает -SH радикал, который присоединен через атом серы.
В настоящем документе термин «амино» означает свободный радикал, содержащий атом азота и 1 или 2 атома водорода. Таким образом, термин «амино» обычно относится к первичным и вторичным аминам. В этом отношении, как используется в настоящем документе, третичный амин представлен общей формулой RR’N-, где R и R’ являются углеродными радикалами, которые могут быть или не быть идентичными. Тем не менее, термин «амино», как правило, может использоваться в настоящем документе для описания первичного, вторичного или третичного амина, и специалисты в данной области техники легко смогут установить идентификацию, с учетом контекста, в которой этот термин используется в настоящем описании.
В настоящем документе термин «оксо» означает кислородный радикал, который присоединен через двойную связь. Если атом, связанный с этим кислородом, представляет собой атом углерода, связью является двойная связь углерод-кислород, которая может обозначаться как -(C=O)- и может называться кетоном.
Перечисление списка химических групп в любом определении переменной в настоящем документе включает определения этой переменной как любой отдельной группы или комбинации перечисленных групп. Перечисление варианта осуществления для переменной или аспекта в настоящем документе включает этот вариант осуществления в качестве любого отдельного варианта осуществления или в комбинации с любыми другими вариантами осуществления или их частями.
Любые композиции или способы, представленные в настоящем документе, могут быть объединены с одной или несколькими другими композициями и способами, представленными в настоящем документе.
В настоящем документе используются следующие сокращения:
Соединения
Настоящее описание относится к соединениям хинуклидина для использования в терапевтических способах, относящихся к лечению или профилактике заболеваний и нарушений, обсуждаемых в настоящем документе. Во всех своих различных аспектах изобретение относится к соединению хинуклидина (Соединение 1) согласно формуле (I),
(I)
или его фармацевтически приемлемой соли или пролекарству, где:
R1 выбирают из водорода, галогена (например, фтора), циано, нитро, гидрокси, тио, амино, C1-6-алкила (например, метила или этила), C2-6-алкенила, C2-6-алкинила, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанный алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
R2 и R3 независимо выбирают из C1-3-алкила, необязательно замещенного одним или несколькими (например, 1, 2 или 3) галогенами, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) галогенами;
R4, R5 и R6 каждый независимо выбирают из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одним или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси; и
A является 5- или 6-членной арильной или гетеоарильной группой (например, фенилом или тиазолилом), необязательно замещенной 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6алкокси или C1-6алкила.
В других вариантах осуществления любых аспектов настоящего описания, настоящее описание дополнительно относится к соединениям следующим образом:
1.1 Соединение 1, где R1 выбирают из водорода, галогена, циано, нитро, гидрокси, тио, амино, C1-6-алкила, C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
1.2 Соединение 1, где R1 выбирают из водорода, галогена, C1-6-алкила, C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
1.3 Соединение 1, где R1 выбирают из водорода, галогена, C1-4-алкила, C1-4-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
1.4 Соединение 1, где R1 выбирают из водорода, галогена, C1-4-алкила, C1-4-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3, или 1 или 2) группами, выбранными из циано, нитро, гидрокси, тио или амино;
1.5 Соединение 1, где R1 выбирают из водорода, галогена и C1-4-алкила, где указанный алкил необязательно замещен одной или несколькими (например, 1 или 2) группами, выбранными из галогена, гидрокси, тио или амино;
1.6 Соединение 1, где R1 выбирают из водорода, фтора, метила и этила, где указанный метил или этил необязательно замещен 1 или 2 группами, выбранными из галогена, гидрокси, тио или амино;
1.7 Соединение 1, где R1 выбирают из водорода и метила, где указанный метил необязательно замещен 1 или 2 галогенами;
1.8 Соединение 1, где R1 является водородом;
1.9 Соединение 1, или любое из 1.1-1.8, где R1 не присоединен к атому азота хинуклидиновой группы;
1.10 Соединение 1, или любое из 1.1-1.9, где R2 и R3 каждый независимо является C1-3-алкилом, необязательно замещенным одним или несколькими (например, 1, 2 или 3) галогенами;
1.11 Соединение 1.11, где R2 и R3 каждый независимо является метилом или этилом, необязательно замещенным 1 или 2 галогенами;
1.12 Соединение 1.11, где R2 и R3 каждый независимо выбирают из метила и этила, необязательно замещенного одним или несколькими фторами, например, 1, 2 или 4 фторами;
1.13 Соединение 1.11, где R2 и R3 каждый независимо является метилом, замещенным 0, 1, 2 или 3 фторами;
1.14 Соединение 1.11, где R2 и R3 каждый является метилом или трифторметилом;
1.15 Соединение 1.11, R2 и R3 каждый является метилом;
1.16 Соединение 1, или любое из 1.1-1.9, где R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) галогенами;
1.17 Соединение 1.16, где R2 и R3 вместе образуют циклопропильную группу;
1.18 Соединение 1 или любое из 1.1-1.9, где R2 и R3 каждый является метилом, или R2 и R3 вместе образуют циклопропильную группу;
1.19 Соединение 1, или любое из 1.1-1.9, где R4, R5 и R6 каждый независимо выбирают из водорода, галогена, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси;
1.20 Соединение 1, или любое из 1.1-1.9, где R4, R5 и R6 каждый независимо выбирают из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-3-алкилокси;
1.21 Соединение 1.19, где R4, R5 и R6 каждый независимо выбирают из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано и C1-3-алкилокси;
1.22 Соединение 1.19, где R4, R5 и R6 каждый независимо выбирают из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси;
1.23 Соединение 1.19, где R4, R5 и R6 каждый независимо выбирают из галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси;
1.24 Соединение 1, или любое из 1.19-1.23, R4 выбирают из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси;
1.25 Соединение 1.24, R4 выбирают из галогена (например, фтора), C1-3-алкила (например, метила) и C1-3-алкилокси (например, метокси или этокси), где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси или этокси);
1.26 Соединение 1.26, R4 выбирают из галогена (например, фтора) и C1-3-алкилокси (например, метокси или этокси), где указанный алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси или этокси);
1.27 Соединение 1.26, R4 является фтором или C1-3-алкилокси (например, этокси), необязательно замещенным одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси);
1.28 Соединение 1.26, где R4 является фтором или этокси, необязательно замещенным одним или несколькими (например, 1, 2 или 3) C1-3-алкилокси (например, метокси);
1.29 Соединение 1, или любое из 1.19-1.28, где R6 является водородом;
1.30 Соединение 1, или любое из 1.19-1.28, где R5 и R6 каждый является водородом;
1.31 Соединение 1, или любое из 1.19-1.28, R5 и R6 каждый является водородом и R4 является фтором или C1-3-алкилокси (например, этокси), необязательно замещенным одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси);
1.32 Соединение 1.31, где R5 и R6 каждый является водородом, и R4 является фтором или этокси, необязательно замещенным одним или несколькими (например, 1, 2 или 3) C1-3-алкилокси (например, метокси);
1.33 Соединение 1.32, где R5 и R6 каждый является водородом, и R4 является фтором или этокси, замещенным метокси (например, 2-метоксиэтокси);
1.34 Соединение 1.32, где R4 является фтором или 2-метоксиэтокси;
1.35 Соединение 1, или любое из 1.1-1.34, где, по меньшей мере, один из R4, R5 и R6 не является водородом;
1.36 Соединение 1, или любое из 1.1-1.35, где R6 является водородом, и R4 и R5 расположены в 2, 4 или 6 положениях фенильного кольца, к которому они присоединены (т.е., орто или пара к заместителю А);
1.37 Соединение 1, или любое из 1.1-1.35, где R6 является водородом и R4 и R5 независимо расположены в 2 и 3 (т.е., соседних орто и мета), 3 и 4 (т.е. соседних мета и пара), или 3 и 5 положениях (т.е., мета) фенильного кольца к которому они присоединены (по отношению к заместителю А);
1.38 Соединение 1, или любое из 1.1-1.35, где R6 является водородом и R4 и R5 расположены в 3 и 5 положениях (т.е., мета) фенильного кольца к которому они присоединены (по отношению к заместителю А);
1.39 Соединение 1, или любое из 1.1-1.35, где R5 и R6 являются водородом и R4 расположен в 2, 3 или 4 положении фенильного кольца, к которому он присоединен (например, орто, мета или пара к заместителю А);
1.40 Соединение 1, или любое из 1.1-1.35, где R5 и R6 являются водородом и R4 расположен в 2 или 4 положении фенильного кольца к которому он присоединен (например, орто или пара к заместителю А);
1.41 Соединение 1, или любое из 1.1-1.35, где R5 и R6 являются водородом и R4 расположен в 4 положении фенильного кольца к которому он присоединен (например, пара к заместителю А);
1.42 Соединение 1, или любое из 1.1-1.35, где ни один из R4, R5 и R6 не является водородом, и каждый из R4, R5 и R6 независимо находятся в 2, 4 или 6 положениях фенильного кольца, к которому они присоединены (т.е., орто или пара к заместителю А);
1.43 Соединение 1, или любое из 1.1-1.42, где R4 расположен в 4 положении фенильного кольца, к которому он присоединен (т.е., пара к заместителю А);
1.44 Соединение 1, или любое из 1.1-1.43, где A является 6-членной арильной группой, 5-членной гетероарильной группой (например, содержащей 1, 2 или 3 гетероатома в гетероарильном кольце, выбранном из N, O и S), или 6-членной гетероарильной группой (например, содержащей 1, 2 или 3 атома азота в гетероарильном кольце);
1.45 Соединение 1.44, где A является 6-членной арильной группой или 5-членной гетероарильной группой (например, содержащей 1, 2 или 3 гетероатома в гетероарильном кольце, выбранном из N, O и S), необязательно, где 5-членная гетероарильная группа содержит 1 или 2 гетероатома, выбранных из N и S (например, один N и/или один S);
1.46 Соединение 1.44 или 1.45, где A выбирают из группы, состоящей из фенила, фурила, тиенила, тиазолила, пиразолила, изотиазолила, оксазолила, изоксазолила, пирролила, триазолила, имидазолила, оксадиазолила и тиадиазолила;
1.47 Соединение 1.46, где A выбирают из группы, состоящей из фенила, тиенила, тиазолила, пирролила и имидазолила;
1.48 Соединение 1.46, где A выбирают из группы, состоящей из фенила и тиазолила, например, 2-тиазол-4-ила или 4-тиазол-2-ила;
1.49 Соединение 1, или любое из 1.1-1.48, где A не замещен;
1.50 Соединение 1, или любое из 1.1-1.48, где A замещен одной или несколькими (например, 1, 2 или 3) группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6алкокси и C1-6алкила (например, метила);
1.51 Соединение 1.50, где A является тиазолилом, замещенным одним галогеном (например, фтором), или C1-6алкилом (например, метилом);
1.52 Соединение 1.50, где A является фенилом, замещенным 1, 2 или 3 группами, независимо выбранными из галогена (например, фтора) и C1-6алкила (например, метила);
1.53 Соединение 1.52, где A является фенилом, замещенным 1 или 2 фторами или метильными группами;
1.54 Соединение 1, или любое из 1.1-1.53 где две группы, присоединенные к заместителю А (т.е., фенильное кольцо (-(C6H2R4R5R6)) и -C(R2R3)- группа), находятся в 1,2-, 1,3- или 1,4-отношении друг к другу (т.е., орто, мета или пара);
1.55 Соединение 1.54, где две группы, присоединенные к заместителю А, находятся в 1,3-отношении друг к другу (т.е., мета);
1.56 Соединение 1.54, где две группы, присоединенные к заместителю А находятся в 1,4-отношении друг к другу (т.е., пара);
1.57 Любые из соединений 1.54-1.56, где A заместителем является 5-членная гетероарильная группа и, по меньшей мере, одна из двух групп, присоединенных к заместителю А (т.е., фенильное кольцо (-(C6H2R4R5R6)) и -C(R2R3)-группа), присоединена к атому углерода гетероарильного кольца, необязательно, где оде такие группы присоединены к атомам углерода гетероарильного кольца;
1.58 Соединение 1, или любое из 1.1-1.57, где соединение формулы I может быть представлено одной или несколькими следующими субструктурами:
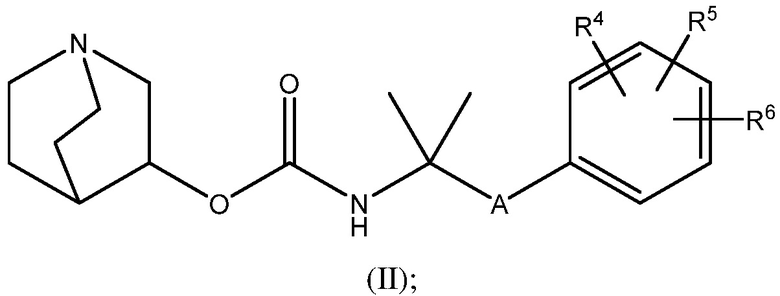
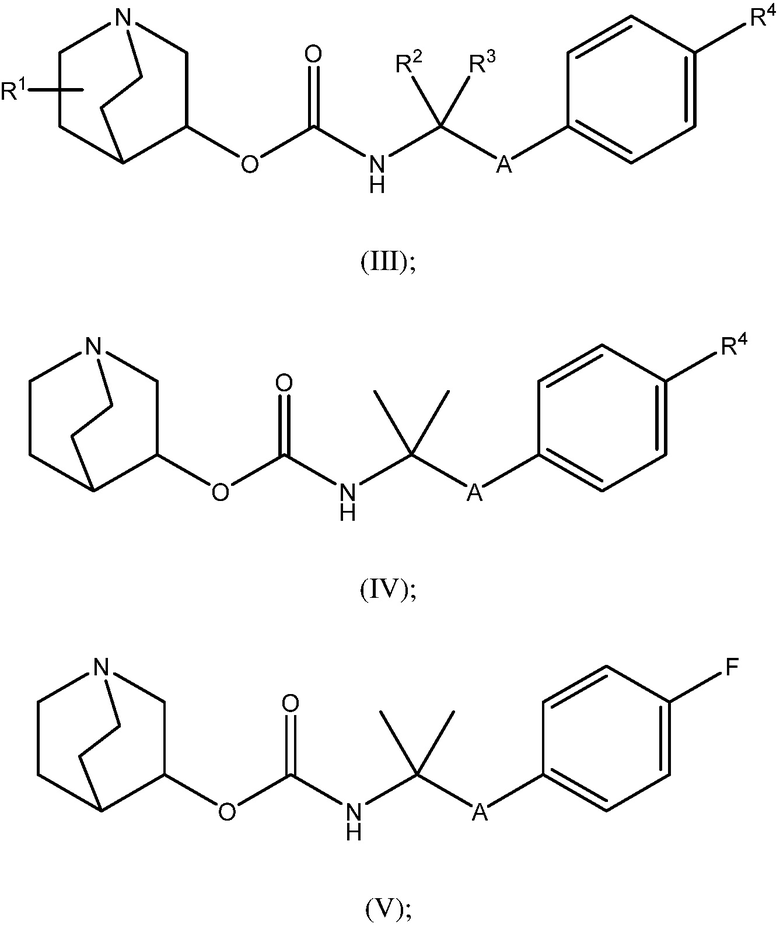
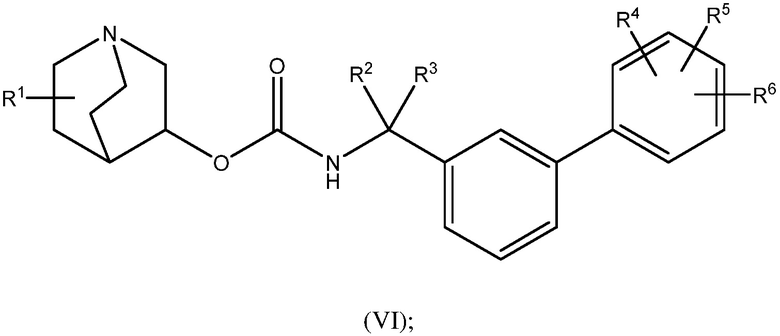
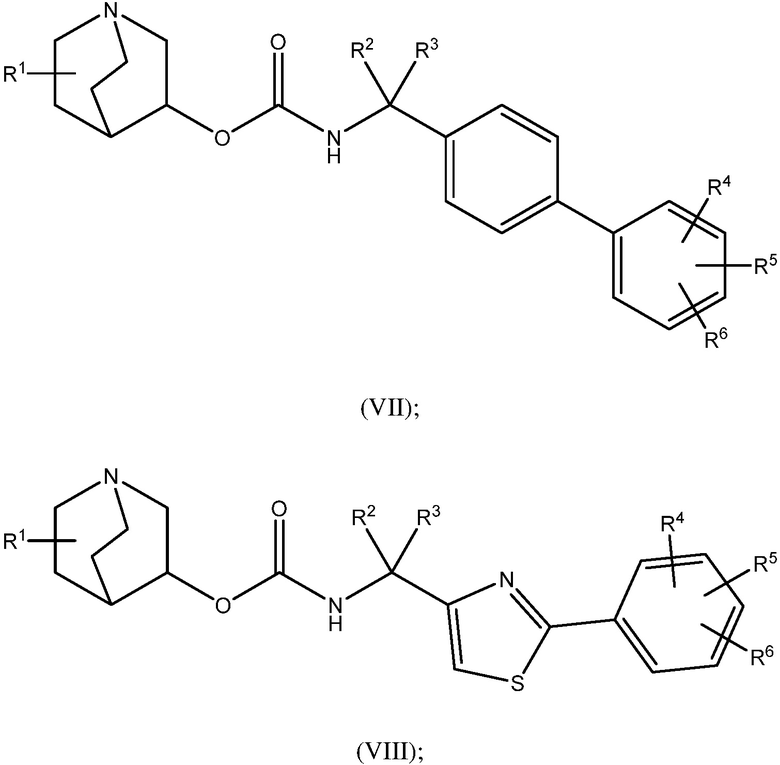
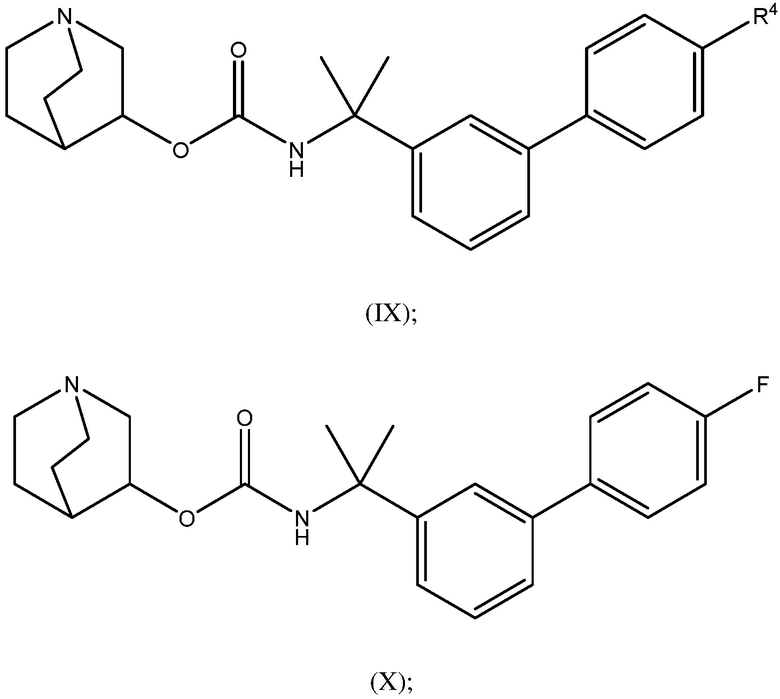
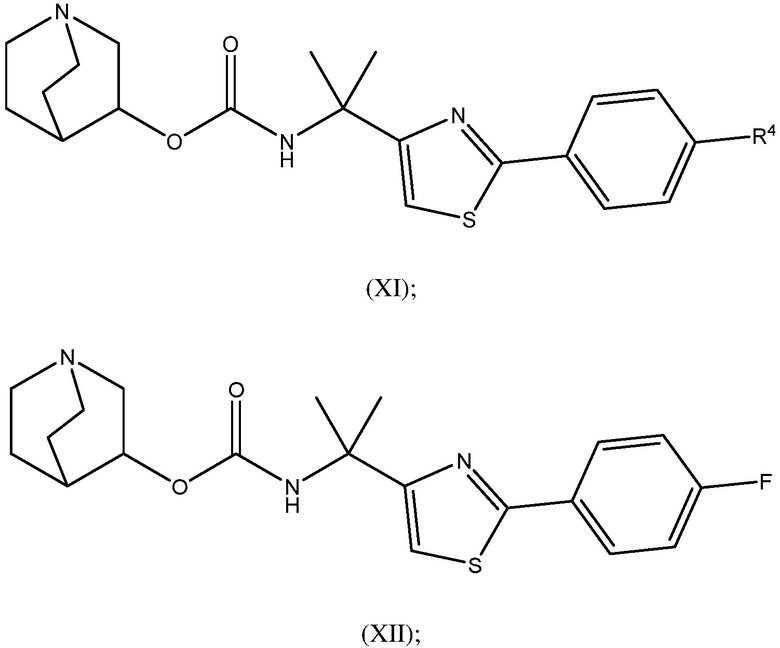
1.59 Соединение 1, или любое из 1.1-1.58, где соединение формулы I или любое из формул II - XII, имеет (S) конфигурацию;
1.60 Соединение 1, или любое из 1.1-1.58, где соединение формулы I или любое из формул II - XII, имеет (R) конфигурацию;
1.61 Соединение 1, или любое из 1.1-1.60, где соединение формулы I или любое из формул II - XII, имеет энантиомерный избыток (например, (S) конфигурации), по меньшей мере, 90%, например, по меньшей мере, 92%, 94%, 95%, 96%, 97%, 98%, 99%, 99,5% или 99.9%;
1.62 Соединение 1, или любое из 1.1-1.58, где соединение формулы I или любое из формул II - XII, является рацемическим (т.е., приблизительно в 50:50 отношении энантиомеров), или является смесью энантиомеров с некоторым другим отношением (например, менее 50:50 или более 50:50);
1.63 Соединение 1, или любое из 1.1-1.62, где соединение формулы I выбирают из группы, состоящей из:
1.64 Соединение 1, или любое из 1.1-1.63, где соединение выбирают из хинуклидин-3-ила (2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамата, (S)-хинуклидин-3-ила (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата и (S)-хинуклидин-3-ила (2-(4’-(2-метоксиэтокси)-[1,1’-бифенил]-4-ил)пропан-2-ил)карбамата;
1.65 Соединение 1, или любое из 1.1-1.63, где соединением является хинуклидин-3-ил (2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
1.66 Соединение 1 или любое из 1.1-1.63, где соединением является хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат, например, (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат;
1.67 Соединение 1, или любое из 1.1-1.66, где соединение формулы I, или любое из II - XII, имеет форму свободного основания;
1.68 Соединение 1, или любое из 1.1-1.66, где соединение формулы I, или любое из II - XII, имеет форму фармацевтически приемлемой соли;
1.69 Соединение 1.68, где указанная солевая форма является формой кислотно-аддитивной соли;
1.70 Соединение 1.69, где указанная форма кислотно-аддитивной соли является солью, выбранной из гидрохлорида, гидробромида, гидроиодида, нитрата, сульфата, бисульфата, фосфата, кислого фосфата, ацетата, лактата, цитрата, кислого цитрата, тартрата, битартрата, сукцината, гидроксисукцинката, малата, малеата, фумарата, глюконата, сахарата, бензоата, метансульфоната и памоата;
1.71 Соединение 1.70, где форму кислотно-аддитивной соли выбирают из гидрохлорида, гидроксисукцината (например, 2-гидроксисукцината) и малата;
1.72 Соединение 1.68, где указанной формой соли является основно-аддитивная форма соли;
1.73 Соединение 1, или любое из 1.1-1.72, где соединением является (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в форме малата;
1.74 Соединение 1, или любое из 1.1-1.73, где соединение формулы I, или любое из II - XII, в форме пролекарства, как описано в настоящем документе;
1.75 Соединение 1, или любое из 1.1-1.74, где соединение формулы I, или любое из II - XII, имеет форму гидрата, сольвата и/или полиморфа.
Соли
Описанные соединения, например, любое из Соединений 1 или 1.1-1.75, которые являются основными по природе, обычно способны образовывать большое количество различных солей с различными неорганическими и/или органическими кислотами. Хотя такие соли обычно фармацевтически приемлемы для введения животным и людям, на практике часто желательно сначала выделить соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение в виде свободного основания путем обработки щелочным реагентом с последующим превращением свободного основания в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений могут быть легко получены с использованием обычных методик, например, путем обработки основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как, например, метанол или этанол. После осторожного выпаривания растворителя получают желаемую твердую соль. Описанные соединения, которые положительно заряжены, например, содержащие четвертичный аммоний, также могут образовывать соли с анионным компонентом различных неорганических и/или органических кислот.
Кислоты, которые можно использовать для получения фармацевтически приемлемых солей соединения хинуклидина, включают такие, которые могут образовывать нетоксичные кислотно-аддитивные соли, например, соли, содержащие фармакологически приемлемые анионы, такие как хлорид, бромид, йодид, нитрат, сульфат или бисульфат, фосфат или кислый фосфат , ацетат, лактат, цитрат или кислый цитрат, тартрат или битартрат, сукцинат, малат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат и памоат [т.е. 1,1'-метиленбис(2-гидрокси-3-нафтоат)].
Описанные соединения, которые являются кислыми по природе, например, соединения, содержащие тиольную группу, обычно способны образовывать большое количество различных солей с различными неорганическими и/или органическими основаниями. Хотя такие соли обычно фармацевтически приемлемы для введения животным и людям, на практике часто бывает желательно сначала выделить соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение свободной кислоты путем обработки кислотным реагентом и затем превратить свободную кислоту в фармацевтически приемлемую основно-аддитивную соль. Эти основно-аддитивные соли могут быть легко получены с использованием обычных методик, например, путем обработки соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и последующим выпариванием полученного раствора досуха, например, при пониженном давлении. Альтернативно, они также могут быть получены путем смешивания низших алканольных растворов кислых соединений и желаемого алкоксида щелочного металла вместе с последующим выпариванием полученного раствора досуха таким же образом, как и раньше. В любом случае можно использовать стехиометрические количества реагентов, чтобы гарантировать полноту реакции и максимальный выход продукта желаемой твердой соли.
Основания, которые могут быть использованы для получения фармацевтически приемлемых основно-аддитивных солей соединения хинуклидина, включают такие, которые могут образовывать нетоксичные основно-аддитивные соли, например, соли, содержащие фармакологически приемлемые катионы, такие как катионы щелочных металлов (например, калия и натрия), катионы щелочноземельных металлов (например, кальция и магния), аммоний или другие водорастворимые амино-аддитивные соли, такие как N-метилглюкамин (меглумин), низший алканоламмоний и другие подобные основания органических аминов.
В одном варианте осуществления, фармацевтически приемлемой солью является сукцинат. В другом варианте осуществления, фармацевтически приемлемой солью является 2-гидроксисукцинат, например, (S)-2-гидроксисукцинат. В другом варианте осуществления, фармацевтически приемлемой солью является гидрохлорид (т.е. соль с HCl). В другом варианте осуществления, фармацевтически приемлемой солью является малат.
Пролекарства
Настоящее описание дополнительно охватывает пролекарства соединений 1 и 1.1-1.75. Фармацевтически приемлемые пролекарства, описанные в настоящем документе, являются производными соединений хинуклидина, которые могут быть превращены in vivo в соединения хинуклидина, описанные в настоящем документе. Пролекарства, которые сами могут обладать некоторой активностью, становятся фармацевтически активными in vivo, когда они подвергаются, например, сольволизу в физиологических условиях или ферментативной деградации. Способы получения пролекарств соединений, как описано в настоящем документе, будут очевидны специалисту в данной области техники на основе настоящего описания.
В одном варианте осуществления, карбаматная группа соединения хинуклидина модифицирована. Например, карбаматная группа соединения хинуклидина может быть модифицирована добавлением воды и/или одного или двух алифатических спиртов. В этом случае двойная связь углерод-кислород карбаматной группы принимает то, что можно было бы рассматривать как полуацетальную или ацетальную функциональную группу. В одном варианте осуществления, карбаматная группа соединения хинуклидина может быть модифицирована добавлением алифатического диола, такого как 1,2-этандиол.
В одном варианте осуществления одна или несколько гидрокси, тио или аминогрупп в соединении хинуклидина модифицированы. Например, одна или несколько гидрокси, тио и/или аминогрупп в соединении хинуклидина могут быть модифицированы с образованием кислых производных, например, сложных эфиров, сложных тиоэфиров (или тиольных эфиров) и/или амидов. Производные кислоты могут быть образованы, например, взаимодействием соединения хинуклидина, которое содержит одну или несколько гидрокси, тио или аминогрупп, с ацетилирующим агентом. Примеры ацетилирующих агентов включают ангидриды, такие как уксусный ангидрид, хлорангидриды, такие как бензилхлорид, и дикарбонаты, такие как ди-трет-бутилдикарбонат.
Стереохимия
Настоящее описание дополнительно охватывает стереоизомеры и смесь стереоизомеров соединений 1 и 1.1-1.75. Стереоизомеры (например, цис- и транс-изомеры) и все оптические изомеры описанного соединения (например, R- и S- энантиомеры), а также рацемические, диастереомерные и другие смеси таких изомеров входят в объем настоящего описания.
В одном варианте осуществления, хинуклидин-3-ильная группа соединения хинуклидина, как определено в настоящем документе, имеет R- конфигурацию. Соответственно, соединение хинуклидина может быть выбрано из группы, состоящей из соединений формул (Ia) - (XIIa):
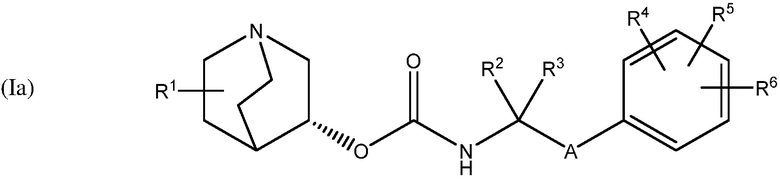
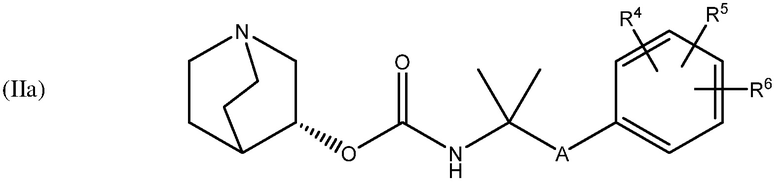
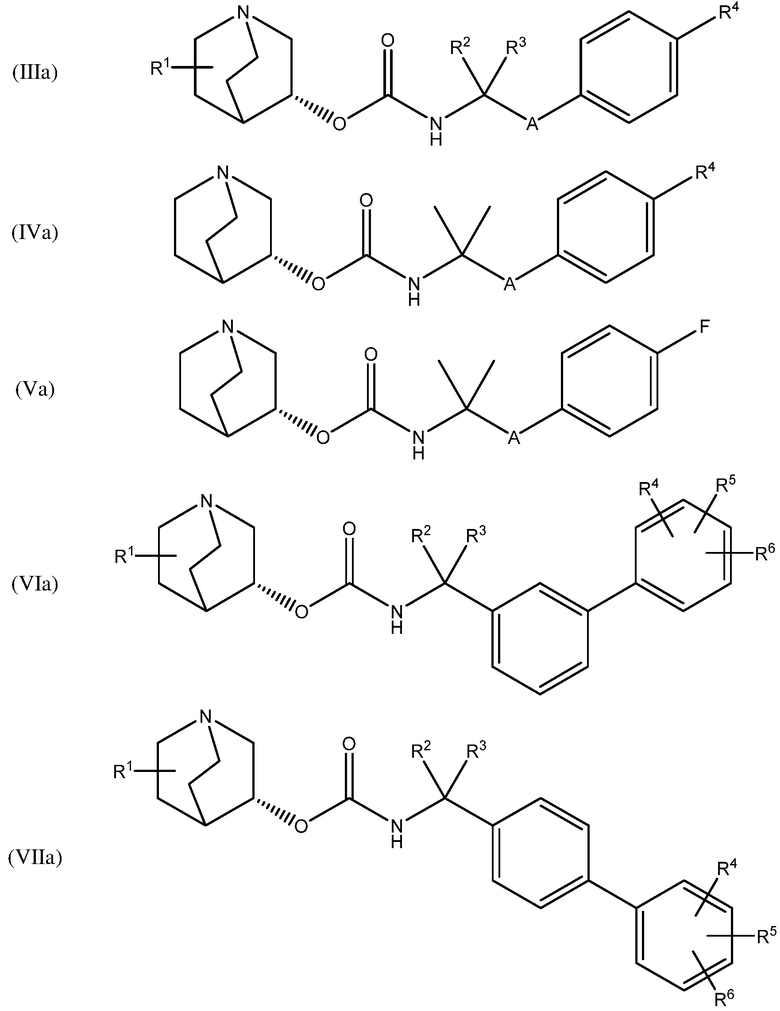
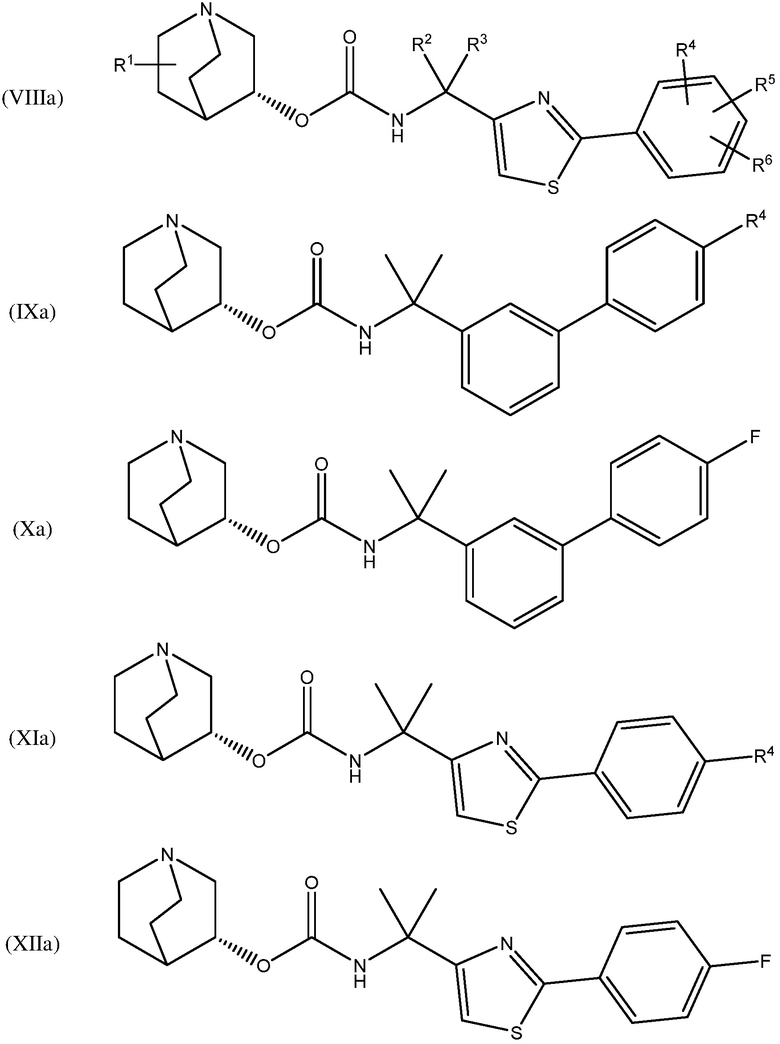
и их фармацевтически приемлемых солей и пролекарств.
В другом варианте осуществления, хинуклидин-3-ильная группа соединения хинуклидина, как определено в настоящем документе, имеет S- конфигурацию. Соответственно, соединение хинуклидина может быть выбрано из группы, состоящей из соединений формул (Ib) - (XIIb):

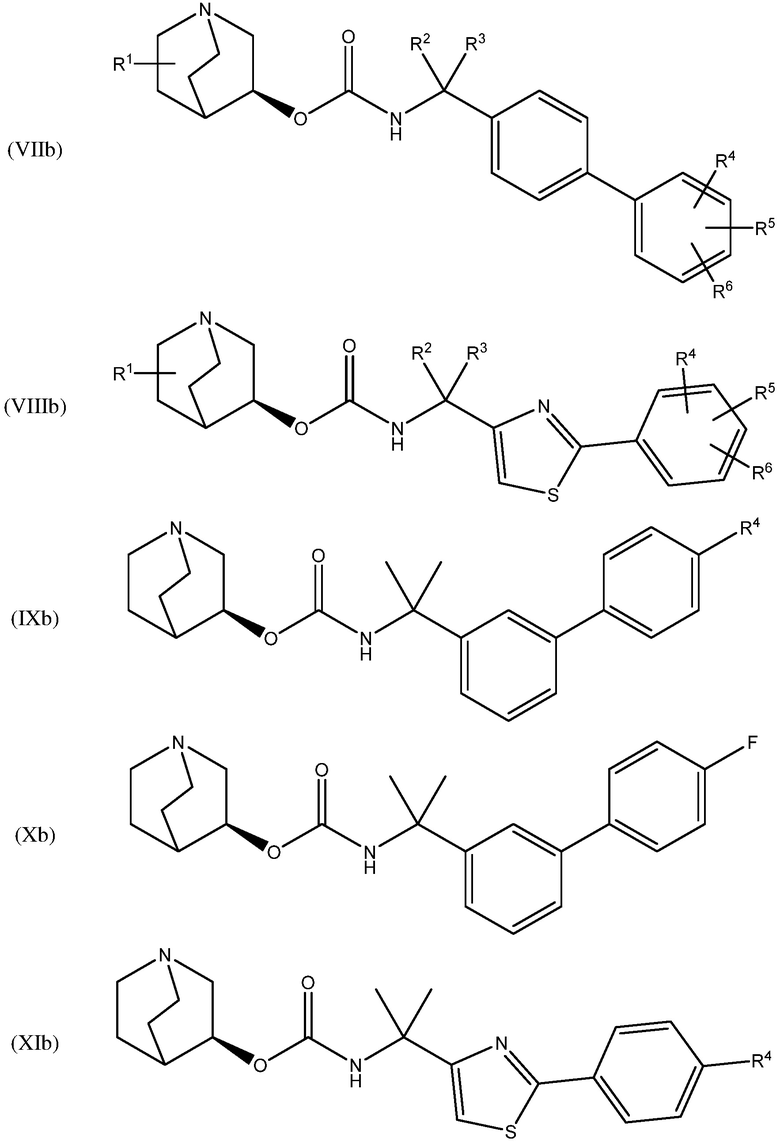
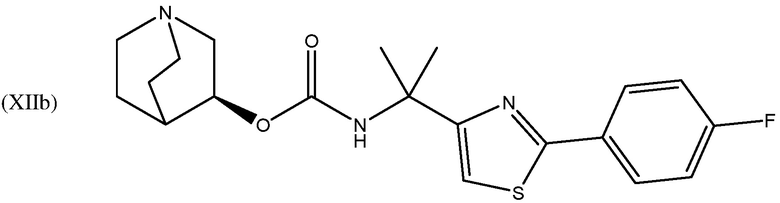
и их фармацевтически приемлемых солей и пролекарств.
В одном варианте осуществления, соединением хинуклидина является соединение формулы (Xb) или его фармацевтически приемлемая соль или пролекарство. В другом варианте осуществления, соединением хинуклидина является соединение формулы (XIIb) или его фармацевтически приемлемая соль или пролекарство.
В одном варианте осуществления, хинуклидин-3-ильная группа соединения хинуклидина, как определено в настоящем документе, существует в смеси изомеров, имеющих R- и S- конфигурации. Например, соединение хинуклидина может быть смесью соединений, выбранных из группы, состоящей из соединений формул (Ia) и (Ib), (IIa) и (IIb), (IIIa) и (IIIb), (IVa) и (IVb), (Va) и (Vb), (VIa) и (VIb), (VIIa) и (VIIb), (VIIIa) и (VIIIb), (IXa) и (IXb), (Xa) и (Xb), (XIa) и (XIb) и (XIIa) и (XIIb) и их фармацевтически приемлемых солей и пролекарства. В одном варианте осуществления, соединение хинуклидина присутствует в виде рацемической смеси, например, R- и S- изомеры хинуклидин-3-ильной группы присутствуют в примерно равных количествах. В другом варианте осуществления, соединение хинуклидина присутствует в виде изомеров, имеющих R- и S- конфигурации, где R- и S- изомеры присутствуют в разных количествах. В одном варианте осуществления, S- изомер присутствует в энантиомерном избытке, по меньшей мере, примерно 5%, 10%, 25%, 40%, 70%, 80%, 90%, 95%, 97%, 98% или 99%, например, примерно 100%. В другом варианте осуществления, R- изомер присутствует в энантиомерном избытке, по меньшей мере, примерно 5%, 10%, 25%, 40%, 70%, 80%, 90%, 95%, 97%, 98% или 99%, например, примерно 100%.
Способы получения энантиообогащенных и/или энантиочувствительных соединений хинуклидина будут очевидны специалисту в данной области техники на основании настоящего описания.
Описанные соединения могут существовать в нескольких таутомерных формах, включая форму енола и имина, форму кето и енамина, геометрические изомеры и их смеси. Таутомеры существуют в виде смесей таутомеров в растворе. В твердой форме обычно преобладает один таутомер. Несмотря на то, что может быть описан один таутомер, все таутомеры входят в объем настоящего описания.
Атропизомеры также входят в объем настоящего описания, Атропизомеры относятся к соединениям, которые могут быть разделены на изомеры с ограниченным вращением.
Другие формы
Настоящее описание также включает гидраты, сольваты и полиморфы соединения 1 и 1.1-1.75. Фармацевтически приемлемые гидраты, сольваты и полиморфы соединений хинуклидина, описанных в настоящем документе, входят в объем настоящего описания. Соединения хинуклидина, описанные в настоящем документе, могут быть в аморфной форме и/или в одной или нескольких кристаллических формах.
Меченые изотопами соединения также входят в объем настоящего описания. В настоящем документе термин «изотопно-меченое соединение» относится к описанному соединению, включая его фармацевтические соли и пролекарства, каждое из которых описано в настоящем документе, в котором один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающегося в природе. Примеры изотопов, которые могут быть включены в описанные соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно.
Медицинские показания
Соединения хинуклидина и фармацевтические композиции, содержащие их, описанные в настоящем документе, применяют в терапии, в частности, при терапевтическом лечении боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, и дерматологических расстройств, таких как ангиокератома, у пациента, имеющего болезнь, такую как болезнь Фабри. Субъекты, подлежащие лечению в соответствии со способами, описанными в настоящем документе, включают позвоночных.
В первом аспекте, настоящее изобретение представляет способ (Способ 2) лечения или профилактики боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, у субъекта, нуждающегося в этом, где способ включает введение субъекту эффективного количества соединения хинуклидина, как описано в настоящем документе, например, соединение формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75. Также представлено соединение хинуклидина, как описано в настоящем документе, например, соединение формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75, для использования в способе лечения или профилактики боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, у нуждающегося в этом субъекта, например, для использования в способе 1, или любом другом из 2.1-2.51. Кроме того, представлено использование соединения хинуклидина, как описано в настоящем документе, например, соединения формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или 1.1-1.75 при производстве лекарственного средства для использования в способе лечения или профилактики боли, включая невропатическую боль, желудочно-кишечную боль (например, боль в животе) и периферическую невропатию, у субъекта, нуждающегося в этом, например, при производстве лекарственного средства для использования в способе 1, или любом из 2.1-2.51.
В других конкретных вариантах способа 1, в настоящем описании представлен:
2.1 Способ 2, где способ включает введение субъекту эффективного количества соединения формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или любого из 1.1-1.75;
2.2 Способ 2, где способ включает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
2.3 Способ 2 или любой из 2.1-2.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение согласно формуле I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из Соединений 1 или любого из 1.1-1.75;
2.4 Способ 2 или любой из 2.1-2.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
2.5 Способ 2.3 или 2.4, где фармацевтическая композиция дополнительно содержит, по меньшей мере, один фармацевтически приемлемый эксципиент, как описано в настоящем документе;
2.6 Способ 2 или любой из 2.1-2.5, где способ включает введение фармацевтической дозированной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
2.7 Способ 2.6, где дозированной формой является пероральная дозированная форма (например, пилюля, капсула, каплет, таблетка, драже, порошок, гранула, пленка, пастилка или жидкость);
2.8 Способ 2.7, где дозированной формой является жевательная таблетка;
2.9 Способ 2.6, где дозированной формой является парентеральная дозированная форма (например, где фармацевтическая композиция составлена для инъекции);
2.10 Способ 2.9, где инъекция является внутривенной, внутримышечной, интратекальной или подкожной, необязательно, стерильной инъекцией;
2.11 Способ 2.6, где дозированной формой является местная или ректальная дозированная форма;
2.12 Способ 2.6, где дозированной формой является интраназальная дозированная форма (например, аэрозоль);
2.13 Способ 2 или любой из 2.1-2.12, где способ дополнительно включает одновременное введение второго активного агента, например, второго соединения, способного лечить или предотвращать боль у пациента, нуждающегося в этом, как описано в настоящем документе;
2.14 Способ 2.13, где второй активный агент вводят в той же фармацевтической композиции или дозированной форме, что и соединение хинуклидина;
2.15 Способ 2.13 или 2.14, где вторым активным агентом является ингибитор галактозидазы (например, ингибитор альфа-галактозидазы, такой как мигаластат);
2.16 Способ 2, или любой из 2.1-2.15, где субъектом является млекопитающее животное;
2.17 Способ 2.16, где субъектом является примат;
2.18 Способ 2.17, где субъектом является человек;
2.19 Способ 2 или любой из 2.1-2.18, где болью является периферическая боль, такая как периферическая невропатия;
2.20 Способ 2 или любой из 2.1-2.19, где болью является желудочно-кишечная боль (например, боль в животе);
2.21 Способ 2, или любой из 2.1-2.20, где болью является боль всего тела;
2.22 Способ 2, или любой из 2.1-2.21, где боль является резистентной или не полностью облегчается лечением аналгетиками;
2.23 Способ 2, или любой из 2.1-2.22, где боль является резистентной или не полностью облегчается нестероидными противовоспалительными агентами;
2.24 Способ 2, или любой из 2.1-2.23, где субъект имеет болезнь Фабри;
2.25 Способ 2.24, где болезнь Фабри не поддается лечению мигаластатом;
2.26 Способ 2, или любой из 2.1-2.25, где субъект имеет тяжелый дефицит или отсутствие активности альфа-галактозидазы (например, <1% от нормальной, например, по данным лейкоцитов в кровотоке);
2.27 Способ 2, или любой из 2.1-2.25, где у субъекта диагностирована мутация в гене GLA (например, гемизиготного самца, гомозиготной самки или гетерозиготной самки), необязательно, где мутацией вляется нонсенс-кодон в GLA гене;
2.28 Способ 2, или любой из 2.1-2.27, где субъект имеет значительное накопление GL-3 в тканях (например, в эндотелиальных клетках капилляров кожи или в плазме);
2.29 Способ 2, или любой из 2.1-2.28, где субъект проходит одновременное лечение ферментозамещающей терапией (ERT), например, с применением агалзидазы альфа или агалзидазы бета;
2.30 Способ 2, или любой из 2.1-2.28, где субъект проходит одновременное лечение ингибитором альфа-галактозидазы (например, мигаластатом);
2.31 Способ 2, или любой из 2.1-2.30, где субъект имеет концентрацию глюкозилцерамида (GL1) в плазме, по меньшей мере, 2 мкг/мл, например, по меньшей мере, 3 мкг/мл или, по меньшей мере, 4 мкг/мл в плазме;
2.32 Способ 2, или любой из 2.1-2.31, где субъект имеет концентрацию глюкозилсфингозина (лизо-GL1) в плазме, по меньшей мере, 65 нг/мл, например, по меньшей мере, 70 нг/мл или, по меньшей мере, 80 нг/мл в плазме;
2.33 Способ 2, или любой из 2.1-2.31, где субъект имеет концентрацию GL3 в плазме, по меньшей мере, 4 мкг/мл, например, по меньшей мере, 6 мкг/мл или, по меньшей мере, 8 мкг/мл в плазме;
2.34 Способ 2 или любой из 2.1-2.33, где субъекту вводят суточную дозу от примерно 1 мг до примерно 150 мг соединения формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или примерно 15 мг, или выбранную из 2, 5, 15, 25, 50, 100, или 150 мг;
2.35 Способ 2, или любой из 2.1-2.34, где субъект является взрослым пациентом-человеком, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
2.36 Способ 2, или любой из 2.1-2.34, где субъектом является пациент-человек детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
2.37 Способ 2, или любой из 2.1-2.36, где способ дает снижение концентрации GL-1 в плазме на, по меньшей мере, 30% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 40%, по меньшей мере, 50%, по меньшей мере, 60%;
2.38 Способ 2, или любой из 2.1-2.37, где способ дает снижение концентрации GL-3 в плазме на, по меньшей мере, 20% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%;
2.39 Способ 2, или любой из 2.1-2.38, где способ дает снижение концентрации GL-3 в плазме на, по меньшей мере, 40% через 26 недель или 52 недели или 104 недели лечения, например, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%;
2.40 Способ 2, или любой из 2.1-2.39, где способ дает снижение концентрации лизо-GL-3 в плазме на, по меньшей мере, 25% через 18 недель или 26 недель или 52 недели, например, по меньшей мере, 35%, по меньшей мере, 45%, по меньшей мере, 55%;
2.41 Способ 2, или любой из 2.1-2.39, где способ дает снижение концентрации GM3 в плазме на, по меньшей мере, 25% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%;
2.42 Способ 2, или любой из 2.1-2.39, где сбособ дает снижение тяжести боли (например, боли в теле и/или желудочно-кишечной боли (например, боли в животе)) через 26 недель или 52 недели или 156 недель;
2.43 Способ 2, или любой из 2.1-2.39, где пособ дает снижение GL-3 уровней в коже (например, эндотелальных клетках капилляров кожи) через 26 недель или 52 недели или 156 недель, например, по давнням степени включений GL-3;
2.44 Способ 2, или любой из 2.1-2.43, где соединение формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75) или его фармацевтически приемлемую соль или пролекарство вводят системным введением, например, патентеральным путем или не парентеральным путем;
2.45 Способ 2.44, где путь введения является пероральным (энтеральным);
2.46 Способ 2.44, где путь введения является парентеральным, например, инъекцией, например, внутривенной инъекцией;
2.47 Способ 2, или любой из 2.1-2.46, где соединение формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемая соль ли пролекарство вводят местным введением, например, местным введением;
2.48 Способ 2, или любой из 2.1-2.49, где соединением является (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил (2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
2.49 Способ 2.48, где доза соединения составляет 15 мг/сутки при пероральном введении;
2.50 Способ 2.49, где доза соединения составляет 15 мг/сутки однократной пероральной дозой;
2.51 Способ 2, или любой из 2.1-2.50, где субъекту вводят однократную суточную дозу 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в форме кислотно-аддитивного малата.
Во втором аспекте, в настоящем изобретении представлен способ (Способ 3) лечения или профилактики дерматологических расстройств, вызванных аккумуляцией GL-3, включая ангиокератому, гипогидроз, ангидроз, гипергидроз, лимфедему и/или акропарестезию, у субъекта, нуждающегося в этом, где способ включает введение субъекту эффективного количества соединения хинуклидина, как описано в настоящем документе, например, соединения формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или 1.1-1.75. Также представлено соединение хинуклидина, как описано в настоящем документе, например, соединение формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75, для применения в способе лечения или профилактики дерматологических расстройств, вызванных аккумуляцией GL-3, включая ангиокератому, гипогидроз, ангидроз, гипергидроз, лимфедему и/или акропарестезию, у субъекта, нуждающегося в этом, например, для применения в способе 3 или любом из 3.1-3.53. Кроме того, представлено применение соединения хинуклидина, как описано в настоящем документе, например, соединения формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или 1.1-1.75, в производстве лекарственного средства для применения в способе лечения или профилактики дерматологических расстройств, вызванных аккумуляцией GL-3, включая ангиокератому, гипогидроз, ангидроз, гипергидроз, лимфедему и/или акропарестезию, у субъекта, нуждающегося в этом, например, производстве лекарственного средства для применения в Способе 4 или любом из 3.1-3.53.
В конкретных дополнительных вариантах осуществления способа 3, в настоящем описании представлен:
3.1 Способ 3, где способ включает введение субъекту эффективного количества соединения формулы I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или любого из 1.1-1.75;
3.2 Способ 3, где способ включает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
3.3 Способ 3 или любой из 3.1-3.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение согласно формуле I или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из Соединений 1 или любого из 1.1-1.75;
3.4 Способ 3 или любой из 3.1-3.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
3.5 Способ 3.3 или 3.4, где фармацевтическая композиция дополнительно содержит, по меньшей мере, один фармацевтически приемлемый эксципиент, как описано в настоящем документе;
3.6 Способ 3 или любой из 3.1-3.5, где способ включает введение фармацевтической дозированной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
3.7 Способ 3.6, где дозированной формой является пероральная дозированная форма (например, пилюля, капсула, каплет, таблетка, драже, порошок, гранула, пленка, пастилка или жидкость);
3.8 Способ 3.7, где дозированной формой является жевательная таблетка;
3.9 Способ 3.6, где дозированной формой является парентеральная дозированная форма (например, где фармацевтическая композиция составлена для инъекции);
3.10 Способ 3.9, где инъекция является внутривенной, внутримышечной, интратекальной или подкожной, необязательно, стерильной инъекцией;
3.11 Способ 3.6, где дозированной формой является местная или ректальная дозированная форма;
3.12 Способ 3.6, где дозированной формой является интраназальная дозированная форма (например, аэрозоль);
3.13 Способ 3 или любой из 3.1-3.12, где способ дополнительно включает одновременное введение второго активного агента, например, второго соединения, способного лечить или предотвращать боль у пациента, нуждающегося в этом, как описано в настоящем документе;
3.14 Способ 3.13, где второй активный агент вводят в той же фармацевтической композиции или дозированной форме, что и соединение хинуклидина;
3.15 Способ 3.13 или 3.14, где вторым активным агентом является ингибитор галактозидазы (например, ингибитор альфа-галактозидазы, такой как мигаластат);
3.16 Способ 3, или любой из 3.1-3.15, где субъектом является млекопитающее животное;
3.17 Способ 3.16, где субъектом является примат;
3.18 Способ 3.17, где субъектом является человек;
3.19 Способ 3 или любой из 3.1-3.18, где дерматологическим расстройством является ангиокератома;
3.20 Способ 3 или любой из 3.1-3.18, где дерматологическим расстройством является гипогидроз и/или ангидроз;
3.21 Способ 3, или любой из 3.1-3.18, где дерматологическим расстройством является лимфедема;
3.22 Способ 3, или любой из 3.1-3.21, где дерматологическим расстройством является акропарестезия;
3.23 Способ 3.22, где боль от акропарестезии является резистентной или не полностью облегчается аналгетиками или нестероидными противовоспалительными агентами;
3.24 Способ 3, или любой из 3.1-3.23, где субъект имеет болезнь Фабри;
3.25 Способ 3.24, где болезнь Фабри не поддается лечению мигаластатом;
3.26 Способ 3, или любой из 3.1-3.25, где субъект имеет тяжелый дефицит или отсутствие активности альфа-галактозидазы (например, <1% от нормальной, например, по данным лейкоцитов в кровотоке);
3.27 Способ 3, или любой из 3.1-3.25, где у субъекта диагностирована мутация в гене GLA (например, гемизиготного самца, гомозиготной самки или гетерозиготной самки), необязательно, где мутацией вляется нонсенс-кодон в GLA гене;
3.28 Способ 3, или любой из 3.1-3.27, где субъект имеет значительное накопление GL-3 в коже и/или в плазме;
3.29 Способ 3.28, где субъект имеет значительное накопление GL-3 в одном или нескольких из эндотелиальных клеток поверхностных сосудов кожи, эндотелиальных клеток глубоких сосудов кожи, клеток гладкой мускулатуры глубоких сосудов кожи и клетках периневрия;
3.30 Способ 3, или любой из 3.1-3.29, где субъект проходит одновременное лечение ферментозамещающей терапией (ERT), например, с применением агалзидазы альфа или агалзидазы бета;
3.31 Способ 3, или любой из 3.1-3.29, где субъект проходит одновременное лечение ингибитором альфа-галактозидазы (например, мигаластатом);
3.32 Способ 3, или любой из 3.1-3.31, где субъект имеет концентрацию глюкозилцерамида (GL1) в плазме, по меньшей мере, 2 мкг/мл, например, по меньшей мере, 3 мкг/мл или, по меньшей мере, 4 мкг/мл в плазме;
3.33 Способ 3, или любой из 3.1-3.32, где субъект имеет концентрацию глюкозилсфингозина (лизо-GL1) в плазме, по меньшей мере, 65 нг/мл, например, по меньшей мере, 70 нг/мл или, по меньшей мере, 80 нг/мл в плазме;
3.34 Способ 3, или любой из 3.1-3.32, где субъект имеет концентрацию GL3 в плазме, по меньшей мере, 4 мкг/мл, например, по меньшей мере, 6 мкг/мл или, по меньшей мере, 8 мкг/мл в плазме;
3.35 Способ 3 или любой из 3.1-3.34, где субъекту вводят суточную дозу от примерно 1 мг до примерно 150 мг соединения формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или примерно 15 мг, или выбранную из 2, 5, 15, 25, 50, 100, или 150 мг;
3.36 Способ 3, или любой из 3.1-3.35, где субъект является взрослым пациентом-человеком, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
3.37 Способ 3, или любой из 3.1-3.35, где субъектом является пациент-человек детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
3.38 Способ 3, или любой из 3.1-3.36, где способ дает снижение концентрации GL-1 в плазме на, по меньшей мере, 30% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 40%, по меньшей мере, 50%, по меньшей мере, 60%;
3.39 Способ 3, или любой из 3.1-3.38, где способ дает снижение концентрации GL-3 в плазме на, по меньшей мере, 20% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%;
3.40 Способ 3, или любой из 3.1-3.39, где способ дает снижение концентрации GL-3 в плазме на, по меньшей мере, 40% через 26 недель или 52 недели или 104 недели лечения, например, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%;
3.41 Способ 3, или любой из 3.1-3.40, где способ дает снижение концентрации лизо-GL-3 в плазме на, по меньшей мере, 25% через 18 недель или 26 недель или 52 недели, например, по меньшей мере, 35%, по меньшей мере, 45%, по меньшей мере, 55%;
3.42 Способ 3, или любой из 3.1-3.40, где способ дает снижение концентрации GM3 в плазме на, по меньшей мере, 25% через 2 недели или 4 недели или 8 недель лечения, например, по меньшей мере, 30%, по меньшей мере, 40%, по меньшей мере, 50%;
3.43 Способ 3, или любой из 3.1-3.40, где пособ дает снижение уровней в коже (например, эндотелальных клетках капилляров кожи) через 26 недель или 52 недели или 156 недель, например, по давнням степени включений GL-3;
3.44 Способ 3.43, где снижение уровней GL-3 в коже (например, включений GL-3) найдено в одном или нескольких из эндотелиальных клеток поверхностных сосудов кожи, эндотелиальных клеток глубоких сосудов кожи, клеток гладкой мускулатуры глубоких сосудов кожи и клетках периневрия; в течение 26 недель или 52 недель или 156 недель;
3.45 Способ 3, или любой из 3.1-3.44, где субъект имеет оценку GL-3 в коже (измеренную по шкале 0-4, как описано в примерах) 2 или более до лечения, и способ дает снижение, по меньшей мере, на один пункт шкалы, например, снижение на один или два пункта; и/или где субъект имеет включения GL-3 в клетках кожи (например, эндотелиальных клеток), показывающие цитоплазматическую объемную долю включений GL-3, по меньшей мере, 0,25 (например, по меньшей мере, 0,27 или 0,3) до лечения, и способ дает снижение цитоплазматической объемной фракции включений GL-3 до менее чем 0,25 (например, менее чем 0,23 или менее чем 0,20 или менее чем 0,19);
3.46 Способ 3 или любой из 3.1-3.45, где соединение формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75) или его фармацевтически приемлемую соль или пролекарство вводят системным введением, например, патентеральным путем или не парентеральным путем;
3.47 Способ 3.46, где путь введения является пероральным (энтеральным);
3.48 Способ 3.46, где путь введения является парентеральным, например, инъекцией, например, внутривенной инъекцией;
3.49 Способ 3, или любой из 3.1-3.48, где соединение формулы I (или любой из II-XII, Ia-XIIa или Ib-XIIb, или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемая соль ли пролекарство вводят местным введением, например, местным введением;
3.50 Способ 3, или любой из 3.1-3.49, где соединением является (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил (2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
3.51 Способ 3.50, где доза соединения составляет 15 мг/сутки при пероральном введении;
3.52 Способ 3.51, где доза соединения составляет 15 мг/сутки однократной пероральной дозой;
3.53 Способ 3, или любой из 3.1-3.52, где субъекту вводят однократную суточную дозу 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в форме кислотно-аддитивного малата.
В некоторых вариантах осуществления настоящего описания у субъекта или субъекта диагностировано конкретное заболевание или нарушение, и также диагностирована конкретная генетическая мутация, например, та, которая, как известно, является причиной рассматриваемого заболевания или нарушения, хотя часто невозможно доказать, что конкретное заболевание или нарушение пациента вызвано конкретной мутацией, которая была диагностирована у человека. В данном случае термин «диагностирована конкретная генетическая мутация» означает, что субъект или пациент были протестированы, например, с помощью ДНК- или РНК-секвенирования, профилирования белков или других подходящих средств, и было обнаружено, что у него имеется рассматриваемая мутация. Однако, как обсуждается ниже, многие генетические заболевания и нарушения могут иметь несколько генетических причин (например, мутаций), и пациенты могут иметь несколько мутаций, каждая из которых может, при некоторых обстоятельствах, быть достаточной, чтобы вызвать заболевание или нарушение, не будучи доказательством того, что конкретная мутация вызывает конкретное заболевание или нарушение у конкретного пациента.
Как уже отмечалось выше, одним из признаков гликогеновых болезней накопления является патологическое накопление различных гликолипидов или гликосфинголипидов в клетках тела. Это накопление является и причиной заметных симптомов и признаками заболевания, а также диагностическим маркером, свидетельствующим о наличии и/или развитии заболевания. В настоящем документе фраза “заметное накопление” в отношении измерения GL-3, GL-1 и других биомаркеров в плазме, коже или других мягких тканях, означает накопление больше чем 25% более максимальной нормальной концентрации указанного соединения. В некоторых вариантах осуществления, “заметное накопление” означает больше чем 50% более максимальной нормальной концентрации указанного соединения.
Способы согласно способу 2 и след. и способу 3 и след. могут быть благоприятны для субъектов, у которых диагностирована лизосомная болезнь накопления, такая как болезнь Фабри, но которые еще не имеют болевые симптомы, ассоциированные с болезненным состоянием, или у которых присутствуют только ранние дерматологические симптомы заболевания. Способы согласно способу 2 и след. и способу 3 и след. также могут быть благоприятны для субъектов, подверженных риску развития лизосомной болезни накопления, такой как болезнь Фабри, из-за, например, мутации у субъекта или семейного анамнеза, которая, как известно, вызывает такое заболевание. Поэтому, в некоторых вариантах осуществления способов, описанных в настоящем документе, субъект диагностирован как имеющий риск развития такого заболевания или нарушения, и способ предотвращает или откладывает наступление и/или развитие болезненных симптомов заболевания или нарушения у субъекта. В некоторых вариантах осуществления, субъект диагностирован как имеющий риск развития указанного заболевания или нарушения через наличие мутации в гене, как описано в настоящем документе.
Фармацевтические композиции
В настоящем описании также представлены фармацевтические композиции, содержащие, по меньшей мере, одно соединение хинуклидина, как описано в настоящем документе, и по меньшей мере, один фармацевтически приемлемый эксципиент, например, для использования в соответствии со способами, описанными в настоящем документе. Фармацевтически приемлемым эксципиентом может быть любой такой эксципиент, известный в данной области техники, включая описанные в, например, Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985). Фармацевтические композиции соединений, описанных в настоящем документе, могут быть получены обычными способами, известными в данной области техники, например, смешиванием, по меньшей мере, одного описанного в настоящем документе соединения с фармацевтически приемлемым эксципиентом.
Таким образом, в одном аспекте настоящее описание представляет фармацевтическую дозированную форму, содержащую соединение хинуклидина, как описано в настоящем документе, и фармацевтически приемлемый эксципиент, где дозированная форма составлена для обеспечения при введении (например, при пероральном введении) количества указанного соединения, достаточного для лечения заболевания или нарушения, описанного в настоящем документе (например, в любом из Способа 2 и след. или Способа 3 и след.).
Фармацевтическая композиция или дозированная форма по изобретению может включать агент и другой носитель, например, соединение или композицию, инертный или активный, такой как поддающийся обнаружению агент, метка, адъювант, разбавитель, связующий агент, стабилизатор, буферы, соли, липофильные растворители, консервант, адъювант или подобные. Носители также включают фармацевтические эксципиенты и добавки, например, белки, пептиды, аминокислоты, жиры и углеводы (например, сахара, включая моносахариды, ди-, три-, тетра- и олигосахариды; дериватизированные сахара, такие как альдиты, альдоновые кислоты, эстерифицированные сахара и подобные; и полисахариды или полимеры сахара), которые могут присутствовать по отдельности или в комбинации, включая отдельно или в комбинации от 1 до 99,99% по массе или по объему. Примеры белковых эксципиентов включают сывороточный альбумин, такой как сывороточный альбумин человека (HSA), рекомбинантный альбумин человека (rHA), желатин, казеин и подобные. Типовые компоненты аминокислоты/антитела, которые также могут функционировать в качестве буферного средства, включают аланин, глицин, аргинин, бетаин, гистидин, глутаминовую кислоту, аспарагиновую кислоту, цистеин, лизин, лейцин, изолейцин, валин, метионин, фенилаланин, аспартам и подобные. Углеводные эксципиенты также входят в объем данного изобретения, их примеры включают, но не ограничиваются ими, моносахариды, такие как фруктоза, мальтоза, галактоза, глюкоза, D-манноза, сорбоза и подобные; дисахариды, такие как лактоза, сахароза, трегалоза, целлобиоза и подобные; полисахариды, такие как раффиноза, мелецитоза, мальтодекстрины, декстраны, крахмалы и подобные; и альдиты, такие как маннит, ксилит, мальтит, лактит, ксилит, сорбит (глюцит) и миоинозит.
Носители, которые можно использовать, включают буфер или агент, регулирующий pH; обычно буфером является соль, полученная из органической кислоты или основания. Типовые буферы включают соли органических кислот, такие как соли лимонной кислоты, аскорбиновой кислоты, глюконовой кислоты, карбоновой кислоты, винной кислоты, янтарной кислоты, уксусной кислоты или фталевой кислоты; Tris, гидрохлорид трометамина или фосфатные буферы. Дополнительные носители включают полимерные эксципиенты/добавки, такие как поливинилпирролидоны, фиколлы (полимерный сахар), декстраты (например, циклодекстрины, такие как 2-гидрокси-пропил-β-циклодекстрин), полиэтиленгликоли, ароматизаторы, антимикробные агенты, подсластители, антиоксиданты, антистатики. поверхностно-активные вещества (например, полисорбаты, такие как «TWEEN 20» и «TWEEN 80»), жиры (например, фосфолипиды, жирные кислоты), стероиды (например, холестерин) и хелатирующие агенты (например, ЭДТК).
В настоящем описании также представлены фармацевтические композиции и наборы, содержащие указанные композиции, которые содержат, по меньшей мере, одно соединение хинуклидина, описанное в настоящем документе, и, по меньшей мере, еще один фармацевтически активный агент. Эти фармацевтические композиции и наборы могут быть адаптированы для одновременного, последовательного и/или раздельного введения соединения хинуклидина и дополнительного активного агента. Например, соединение хинуклидина и дополнительный активный агент могут быть составлены в виде отдельных дозированных форм, например, в отдельных таблетках, капсулах, лиофилизатах или жидкостях, или они могут быть составлены в одной и той же дозированной форме, например, в той же таблетке, капсуле, лиофилизате или жидкости. Если соединение хинуклидина и дополнительный активный агент составлены в одной и той же дозированной форме, соединение хинуклидина и дополнительный активный агент могут присутствовать по существу в смеси, например, в сердцевине таблетки, или они могут присутствовать по существу в отдельных областях дозированной формы, например, в разных слоях одной и той же таблетки. В одном варианте осуществления, фармацевтическая дозированная форма содержит дополнительный агент, который способен лечить или предотвращать надъядерный паралич взора, например, у пациента, имеющего, диагностированного или предрасположенного к лизосомной болезни накопления, такой как Гоше 3 типа или Ниманн-Пик типа С, или боль, например, у пациента, у которого диагностирована лизосомная болезнь накопления, или предрасположенного к ней, например, болезнь Фабри, как описано в настоящем документе.
В дополнительном аспекте, в настоящем описании представлена фармацевтическая композиция, содержащая: (i) соединение хинуклидина, как описано в настоящем документе; (ii) дополнительный активный агент; и (iii) фармацевтически приемлемый эксципиент. В другом варианте осуществления, дополнительным активным агентом является агент, который способен лечить или предотвращать боли или дерматологического нарушения (например, ангиокератомы), например, у пациента, имеющего, диагностированного или предрасположенного к лизосомной болезни накопления, такой как болезнь Фабри, как описано в настоящем документе, например, при пероральном введении субъекту.
Описанные соединения хинуклидина и фармацевтические композиции можно использовать на животных или людях. Таким образом, описанное соединение может быть составлено в виде фармацевтической композиции для перорального, буккального, парентерального (например, внутривенного, внутримышечного или подкожного), местного, ректального или интраназального введения, или в форме, подходящей для введения путем ингаляции или инсуффляции. В конкретных вариантах осуществления, соединение хинуклидина или фармацевтическая композиция составлены для системного введения, например, не парентеральным путем. В одном варианте осуществления, соединение хинуклидина или фармацевтическая композиция составлена для перорального введения, например, в твердой форме. Такие способы введения и способы приготовления соответствующих фармацевтических композиций описаны, например, в Gibaldi’s Drug Delivery Systems in Pharmaceutical Care (1st ed., American Society of Health-System Pharmacists 2007).
Фармацевтические композиции могут быть составлены таким образом, чтобы обеспечивать медленное, пролонгированное или контролируемое высвобождение активного ингредиента с использованием, например, гидроксипропилметилцеллюлозы в различных пропорциях для обеспечения желаемого профиля высвобождения, других полимерных матриц, липосом и/или микросфер. Фармацевтические композиции также могут необязательно содержать замутнители, и могут быть композицией, которая высвобождает активные ингредиенты только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, замедленно, например, с помощью энтеросолюбильного покрытия. Примеры герметизирующих композиций включают полимерные вещества и воски. Активный ингредиент также может быть в микрокапсулированной форме, при необходимости, с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами или разбавителями, хорошо известными в данной области техники (см., например, Remington’s). Описанные соединения могут быть составлены для непрерывной доставки способами, хорошо известными специалистам в данной области техники. Примеры таких составов можно найти в патентах США 3,119,742; 3,492,397; 3,538,214; 4,060,598; и 4,173,626.
В твердых дозированных формах для перорального введения (например, капсулах, таблетках, пилюлях, драже, порошках, гранулах и подобных) активный ингредиент смешан с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами или разбавителями, такими как цитрат натрия или дикальцийфосфат, и/или любой из следующих: (1) наполнители или разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит, микрокристаллическая целлюлоза, фосфат кальция и/или кремниевая кислота; (2) связующие агенты, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, прежелатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза, сахароза и/или аравийская камедь; (3) увлажнители, такие как глицерин; (4) разрыхлители, такие как агар-агар, карбонат кальция, натрия крахмалгликолят, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) агенты, замедляющие растворение, такие как парафин; (6) ускорители абсорбции, такие как соединения четвертичного аммония; (7) смачивающие агенты, такие как, например, лаурилсульфат натрия, ацетиловый спирт и моностеарат глицерина; (8) абсорбенты, такие как каолин и бентонитовая глина; (9) смазывающие вещества, такие как тальк, диоксид кремния, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (10) красители. В случае капсул, таблеток и пилюль, фармацевтические композиции также могут содержать буферные агенты. Твердые композиции подобного типа также могут быть получены с использованием наполнителей в мягких и твердых желатиновых капсулах и эксципиентов, таких как лактоза или молочные сахара, а также высокомолекулярных полиэтиленгликолей и подобных.
Таблетка может быть изготовлена прессованием или формованием, необязательно с одним или несколькими дополнительными ингредиентами. Прессованные таблетки могут быть приготовлены с использованием связующих агентов (например, желатина или гидроксипропилметилцеллюлозы), смазывающих агентов, инертных разбавителей, консервантов, разрыхлителей (например, натрия крахмалгликолята или поперечно-сшитой карбоксиметилцеллюлозы натрия), поверхностно-активных веществ и/или диспергирующих агентов. Формованные таблетки могут быть получены путем формования в подходящей машине смеси порошкообразного активного ингредиента, смоченного инертным жидким разбавителем. Таблетки и другие твердые дозированные формы, такие как драже, капсулы, пилюли и гранулы, необязательно могут иметь насечки или приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в данной области техники.
В вариантах осуществления, фармацевтические композиции вводят перорально в жидкой форме. Жидкие дозированные формы для перорального введения активного ингредиента включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие препараты для перорального введения могут быть представлены в виде сухого продукта для разведения водой или другим подходящим носителем перед использованием. В дополнение к активному ингредиенту, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана и их смеси. В дополнение к инертным разбавителям, жидкие фармацевтические композиции могут включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы, красители, отдушки и консерванты и подобные. Суспензии, в дополнение к активному ингредиенту, могут содержать суспендирующие агенты, такие как, но не ограничиваясь ими, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси. Подходящие жидкие препараты могут быть приготовлены обычными способами с фармацевтически приемлемыми добавками, такими как суспендирующий агент (например, сироп сорбита, метилцеллюлоза или гидрированные пищевые жиры); эмульгатор (например, лецитин или аравийская камедь); не водный носитель (например, миндальное масло, масляные эфиры или этиловый спирт); и/или консервант (например, метил- или пропилпара-гидроксибензоаты или сорбиновая кислота). Активные ингредиенты также можно вводить в виде болюса, электуария или пасты.
Для буккального введения, композиция может принимать форму таблеток или пастилок, приготовленных обычным способом.
В вариантах осуществления, фармацевтические композиции вводят не пероральными средствами, такими как местное нанесение, трансдермальное нанесение, инъекция и подобные. В родственных вариантах осуществления, фармацевтические композиции вводят парентерально путем инъекции, инфузии или имплантации (например, внутривенно, внутримышечно, внутриартериально, подкожно и т.п.).
Описанные соединения могут быть составлены для парентерального введения путем инъекции, в том числе с использованием обычных методов катетеризации или инфузии. Составы для инъекций могут быть представлены в виде стандартной дозированной формы, например, в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать препаратообразующий агент, такой как суспендирующий, стабилизирующий и/или диспергирующий агент, признанный специалистами в данной области техники. Альтернативно, активный ингредиент может быть в форме порошка для восстановления подходящим носителем, например, стерильной апирогенной водой, перед использованием.
Фармацевтические композиции могут быть введены непосредственно в центральную нервную систему. Соответственно, в некоторых вариантах осуществления, композиции вводят непосредственно в центральную нервную систему, чтобы избежать гематоэнцефалического барьера. В некоторых вариантах осуществления, композиция может быть введена через прямую инъекцию в спинной мозг. В вариантах реализации изобретения композиция вводится путем интратекальной инъекции. В некоторых вариантах осуществления, композицию вводят интрацеребровентрикулярной инъекцией. В вариантах осуществления, композицию вводят в церебральный боковой желудочек. В вариантах осуществления, композицию вводят в оба церебральных боковых желудочка. В дополнительных вариантах осуществления, композицию вводят внутригиппокампальной инъекцией. Композиции можно вводить одой инъекцией или несколькими инъекциями. В других вариантах осуществления, композицию вводят более чем в одно место (например, в два участка центральной нервной системы).
Фармацевтические композиции могут быть в форме стерильных инъекций. Фармацевтические композиции могут быть стерилизованы, например, фильтрацией через фильтр, задерживающий бактерии, или введением стерилизующих агентов в виде стерильных твердых композиций, которые могут быть растворены в стерильной воде или какой-либо другой стерильной среде для инъекций непосредственно перед использованием. Для приготовления такой композиции, активный ингредиент растворяют или суспендируют в парентерально приемлемом жидком носителе. Типовые носители и растворители включают, но не ограничиваются ими, воду, воду, доведенную до подходящего pH добавлением соответствующего количества хлористоводородной кислоты, гидроксида натрия или подходящего буфера, 1,3-бутандиол, раствор Рингера и изотонический раствор хлорида натрия. Фармацевтическая композиция также может содержать один или несколько консервантов, например, метил-, этил- или н-пропилпара-гидроксибензоат. Для улучшения растворимости может быть добавлен улучшающий растворение, или солюбилизирующий агент, или растворитель может содержать 10-60% масс. пропиленгликоля или подобного.
Фармацевтические композиции могут содержать один или несколько фармацевтически приемлемых стерильных изотонических водных или не водных растворов, дисперсий, суспензий или эмульсий или стерильных порошков, которые могут быть восстановлены в стерильные растворы или дисперсии для инъекций непосредственно перед применением. Такие фармацевтические композиции могут содержать антиоксиданты; буферы; бактериостатики; растворенные вещества, которые придают составу изотоничность к крови предполагаемого реципиента; суспендирующие агенты; загустители; консерванты; и подобные.
Примеры подходящих водных и не водных носителей, которые могут использоваться в фармацевтических композициях по изобретению, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и подобные) и их подходящие смеси, растительные масла, такие как оливковое масло, и сложные органические эфиры для инъекций, такие как этил олеат. Надлежащую текучесть можно поддерживать, например, путем использования покрывающих материалов, таких как лецитин, путем поддержания необходимого размера частиц в случае дисперсий и путем использования поверхностно-активных веществ. В некоторых вариантах осуществления, чтобы продлить действие активного ингредиента, желательно замедлить абсорбцию соединения при подкожной или внутримышечной инъекции. Это может быть достигнуто за счет использования жидкой суспензии кристаллического или аморфного материала, имеющего плохую растворимость в воде. Скорость абсорбции активного ингредиента затем зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, отсроченная абсорбция парентерально введенного активного ингредиента достигается растворением или суспендированием соединения в масляном носителе. Кроме того, пролонгированная абсорбция фармацевтической формы для инъекций может быть обеспечена за счет включения агентов, замедляющих абсорбцию, таких как моностеарат алюминия и желатин.
Парентеральные композиции с контролируемым высвобождением могут быть в форме водных суспензий, микросфер, микрокапсул, магнитных микросфер, масляных растворов, масляных суспензий, эмульсий, или активный ингредиент может быть включен в биосовместимые носители, липосомы, наночастицы, имплантаты или устройства для инфузии. Материалы для использования в производстве микросфер и/или микрокапсул включают, но не ограничиваются ими, биоразлагаемые/биоразрушаемые полимеры, такие как полиглактин, поли (изобутилцианоакрилат), поли (2-гидроксиэтил-L-глутамин) и поли(молочная кислота). Биосовместимые носители, которые можно использовать при приготовлении парентерального состава с контролируемым высвобождением, включают углеводы, такие как декстраны, белки, такие как альбумин, липопротеины или антитела. Материалы для использования в имплантатах могут быть не биоразлагаемыми, например, полидиметилсилоксан, или биоразлагаемыми, такими как, например, поли(капролактон), поли(молочная кислота), поли(гликолевая кислота) или поли(ортоэфиры).
Для местного применения описанное соединение может быть составлено в виде мази или крема. Описанные соединения также могут быть составлены в ректальных композициях, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Для интраназального введения или введения ингаляцией, описанные соединения могут быть удобно доставлены в форме раствора или суспензии из помпового распылителя, который сжимается или накачивается пациентом, или в виде аэрозольного спрея из находящегося под давлением контейнера или небулайзера с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением, единичная доза может быть определена путем обеспечения клапана для доставки отмеренного количества. Контейнер под давлением или небулайзер может содержать раствор или суспензию описанного соединения. Капсулы и картриджи (сделанные, например, из желатина) для использования в ингаляторе или инсуффляторе могут быть составлены так, чтобы содержать порошковую смесь описанного соединения и подходящей порошковой основы, такой как лактоза или крахмал.
Обычно агенты и композиции, описанные в настоящем документе, вводят в эффективном количестве или количестве, достаточном для лечения или профилактики надъядерного паралич взора у субъекта, нуждающегося в этом. Обычно, доза может быть скорректирована в этом диапазоне на основании, например, возраста, физического состояния, массы тела, пола, диеты, времени введения и других клинических факторов. Определение эффективного количества находится в компетенции специалиста в данной области техники.
Будучи в целом описанными в настоящем документе, следующие не ограничивающие примеры представлены для дополнительной иллюстрации этого изобретения.
ПРИМЕРЫ
Общие методики химического синтеза
Общая методика A: Образование карбамата с трифосгеном
К суспензии гидрохлорида амина (1 эквивалент) и триэтиламина (3-4 эквивалентов) в ТГФ (концентрация ~ 0,2 M) при комнатной температуре добавляют трифосген (0,35 эквивалента). Реакционную смесь перемешивают в течение 10 мин и добавляют небольшое количество простого эфира (1-2 мл). Соль триэтиламмония отфильтровывают с получением прозрачного раствора изоцианата в ТГФ/простом эфире.
К раствору спирта (1,5 эквивалента) в ТГФ (концентрация ~ 0,2 M) при комнатной температуре добавляют NaH [60%, масло] (1,5 эквивалента). Реакционную смесь перемешивают в течение 15 мин и вышеуказанный раствор (изоцианат в ТГФ/простом эфире) добавляют по каплям. Во время стандартной обработки, реакцию гасят насыщенным раствором соли. Раствор экстрагируют EtOAc, и органический слой сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают на combiflash (SiO2 картридж, CHCl3 и 2N NH3 в MeOH) с получением соответствующего карбамата.
Общая методика B: Алкилирование церийорганическим соединением
Суспензию CeCl3 (4 эквивалента) в ТГФ (концентрация ~ 0,2 M) перемешивают при комнатной температуре в течение 1 ч. Суспензию охлаждают до -78°C и MeLi/Простой эфир [1,6M] (4 эквивалента) добавляют по каплям. Церийорганический комплекс образуется в течение 1 ч, и раствор нитрила (1 эквивалент) в ТГФ (концентрация 2.0 M) добавляют по каплям. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 18 ч. Раствор охлаждают до 0°C и гасят водой (~ 1 мл), затем добавляют 50% водный раствор гидроксида аммония (~3 мл) до образования осадка и осаждают на дно колбы. Смесь фильтруют через слой целита и концентрируют. Неочищенный продукт обрабатывают раствором HCl/диоксана [4,0 M]. Промежуточный гидрохлорид арилпропан-2-амина растирают в простом эфире и применяют как есть на следующей стадии. Альтернативно, неочищенное свободное основание амина очищают на combiflash (SiO2 картридж, CHCl3 и 2N NH3 в MeOH) с получением соответствующего арилпропиламина.
Общая методика C: Сочетание Сузуки
К раствору арилгалогенида (1 эквивалент) в смеси ДМЭ/вода [4:1] (концентрация ~ 0,2 M) добавляют бороновую кислоту (2 эквивалента), палладиевый катализатор (0,1-0,25 эквивалента) и карбонат натрия (2 эквивалента). Реакционную смесь облучают микроволнами 25 мин при 150°C. После фильтрации через слой целита и концентрации, неочищенный продукт очищают на combiflash (SiO2 картридж, CHCl3 и 2N NH3 в MeOH) с получением соответствующего продукта сочетания.
Альтернативно: К раствору арилгалогенида (1 эквивалент) в смеси толуола/воды [20:1] (концентрация ~ 0,2 M) добавляют бороновую кислоту (1,3-2,5 эквивалента), палладиевый катализатор (0,05-0,15 эквивалента), трициклогексилфосфин (0,15-0.45 эквивалент) и фосфат калия (5 эквивалентов). Реакционную смесь облучают микроволнами 25 мин при 150°C. После фильтрации через слой целита и концентрации, неочищенный продукт очищают на combiflash (SiO2 картридж, CHCl3 и 2N NH3 в MeOH) с получением соответствующего продукта сочетания.
Общая методика D: Циклопропанирование
К смеси арилнитрила (1 эквивалент) и Ti(Oi-Pr)4 (1,7 эквивалента), перемешиваемой при -70°C, добавляют по каплям EtMgBr [3,0 M в простом эфире] (1,1 эквивалента). Реакционную смесь нагревают до 25°C и перемешивают в течение 1 ч. К вышеуказанной смеси добавляют BF3·Et2O (3 эквивалента) по каплям при 25°C. После добавления, смесь перемешивают в течение еще 2 ч и затем гасят водной HCI [2M]. Полученный раствор затем подщелачивают добавлением водного NaOH [2M]. Органический продукт экстрагируют простым эфиром этила. Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией на силикагеле (элюируя петролейным эфиром/EtOAc: 10/1 до 1/1) с получением соответствующего 1-арилциклопропанамина.
Общая методика E: Биарильное сочетание с применением условий Сузуки
К перемешиваемому раствору арилгалогенидного компонента (1 эквивалент) в 5:1 (об./об.) диоксане/воде (~0,15 M) или 5:1 (об./об.) добавляют N, N-диметилформамид (~0,15 M), арилборонат или компонент арилбороновой кислоты (1-1,5 эквивалента), карбонат натрия (2-3 эквивалента) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (0,05 эквивалента). Смесь нагревают (90°C) в течение ночи и затем фильтруют через слой целита. Целит промывают этилацетатом и объединенный фильтрат промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Остаток очищают флэш-хроматографией над диоксидом кремния.
Общая методика F: Получение карбамата с применением изоцианата, созданного через путь смешанного ангидрида/перестановки Куртиуса
К перемешиваемому раствору компонента карбоновой кислоты (1 эквивалент) в тетрагидрофуране (~0,1 M) добавляют триэтиламин (2 эквивалента). Реакционную смесь охлаждают (0°C) и обрабатывают изобутилхлорформиатом (1,5 эквивалента). Через 1 час при 0°C, добавляют раствор азида натрия (2 эквивалента) в воде (~1 M), и реакционную смесь нагревают до комнатной температуры. После перемешивания в течение ночи, реакционную смесь разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты промывают водным раствором бикарбоната натрия и насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Неочищенный ацилазид дополнительно сушат совместным выпариванием с толуолом и затем помещают в толуол (~0,1 M). Перемешанный раствор кипятят с обратным холодильником в течение 2-2,5 часов, охлаждают и обрабатывают спиртовым компонентом (1,25-2 эквивалента). Реакционную смесь нагревают при кипении с обратным холодильником в течение ночи и затем концентрируют. Остаток помещают либо в этилацетат, либо в хлороформ и промывают водным карбонатом натрия, (Na2SO4) и концентрируют. Неочищенный продукт очищают флэш-хроматографией над диоксидом кремния с применением градиентов растворителей хлороформа/метанола (менее полярных карбаматов) или хлороформа/метанола/аммиака (более полярных карбаматов).
Пример 1: Синтез соединений хинуклидина
1-азабицикло[2,2,2]окт-3-ил [2-(4'-фторбифенил-3-ил)пропан-2-ил]карбамат (Соединение 1)
С применением общей методики C, 1-азабицикло[2,2,2]окт-3-ил [2-(3-бромфенил)пропан-2-ил]карбамат (600 мг, 1,63 ммоль), 4-фторфенилбороновую кислоту (457 мг, 3,27 ммоль) и ацетат палладия (II) дают указанное в заголовке соединение в виде белого твердого вещества (373 мг; 60%). 1H ЯМР (400 MГц, CDCl3) δ 7,56 (с, 1H), 7,52 (дд, J=5,4, 8,4 Гц, 2H), 7,42-7,38 (м,3H), 7,12 (м,2H), 5,18 (5, 1H), 4,62 (с, 1H), 2,66 (м,6H), 1,72 (с, 6H), 2,01-0,83 (м,5H) ч./млн. 13C ЯМР (100 MГц, CDCl3) δ 125,0, 124,0, 123,8, 116,0, 116,0, 71,3, 55,9, 55,5, 47,6, 46,7, 29,6, 25,6, 24,8, 19,8 ч./млн. Чистота: 98,0% УЖХМС (210 нм); время удержания 0,95 мин; (M+1) 382,9. Анал. Рассч. для C23H27FN2O2·0,37 (CHCl3): C, 65,86; H, 6,47; N, 6,57. Найдено: C, 65,85; H, 6,69; N, 6,49.
(S)-хинуклидин-3-ил 2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-илкарбамат (Соединение 2)
К перемешиваемому раствору 4-фтортиобензамида (8,94 г, 57,6 ммоль) в этаноле (70 мл) добавляют этил 4-хлорацетоацетат (7,8 мл, 58 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в течение 4 часов, обрабатывают добавлением аликвоты этил 4-хлорацетоацетата (1,0 мл, 7,4 ммоль) и кипятят с обратным холодильником в течение еще 3,5 часов. Реакционную смесь затем концентрируют, и остаток разделяют между этилацетатом (200 мл) и водным NaHCO3 (200 мл). Органический слой объединяют с обратным экстрактом водного слоя (этилацетат, 1×75 мл), сушат (Na2SO4) и концентрируют. Полученное янтарное масло очищают флэш-хроматографией с применением градиента гексан/этилацетат с получением этил 2-(2-(4-фторфенил)тиазол-4-ил)ацетата в виде низкоплавкого практически бесцветного твердого вещества (13,58 г, 89%).
К перемешиваемому раствору этил 2-(2-(4-фторфенил)тиазол-4-ил)ацетата (6,28 г, 23,7 ммоль) в ДМФ (50 мл) добавляют гидрид натрия [60% дисперсию в минеральном масле] (2,84 г, 71,0 ммоль). Пенистую смесь перемешивают в течение 15 минут, затем охлаждают на ледяной бане и добавляют йодметан (4,4 мл, 71 ммоль). Реакционную смесь перемешивают в течение ночи, позволяя охлаждающей бане медленно нагреваться до комнатной температуры. Смесь затем концентрируют и остаток разделяют между этилацетатом (80 мл) и водой (200 мл). Органический слой промывают второй порцией воды (1×200 мл), сушат (Na2SO4) и концентрируют. Полученное янтарное масло очищают флэш-хроматографией с применением градиента гексан/этилацетат с получением этил 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропаноата в виде бесцветного масла (4,57 г, 66%).
К перемешиваемому раствору этил 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропаноата (4,56 г, 15,5 ммоль) в 1:1:1 ТГФ/этаноле/воде (45 мл) добавляют моногидрат гидроксида лития (2,93 г, 69,8 ммоль). Реакционную смесь перемешивают в течение ночи, концентрируют и повторно растворяют в воде (175 мл). Раствор промывают простым эфиром (1×100 мл), подкисляют добавлением 1,0 N HCl (80 мл) и экстрагируют этилацетатом (2×70 мл). Объединенные экстракты сушат (Na2SO4) и концентрируют с получением 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (4,04 г, 98%). Этот продукт применяют на следующей стадии без очистки.
К перемешиваемому и охлажденному (0°С) раствору 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропановой кислоты (4,02 г, 15,2 ммоль) в ТГФ (100 мл) добавляют триметиламин (4,2 мл, 30 ммоль), затем изобутилхлорформиат (3,0 мл, 23 ммоль). Реакционную смесь перемешивают холодной в течение еще 1 часа, затем добавляют раствор азида натрия (1,98 г, 30,5 ммоль) в воде (20 мл). Реакционную смесь перемешивают в течение ночи, позволяя охлаждающей бане медленно нагреваться до комнатной температуры. Смесь затем разбавляют водой (100 мл) и экстрагируют этилацетатом (2×60 мл). Объединенные экстракты промывают водным NaHCO3 (1×150 мл) и насыщенным раствором соли (1×100 мл), сушат (Na2SO4) и концентрируют. После совместного выпаривания с толуолом (2×50 мл), полученное белое твердое вещество помещают в толуол (100 мл) и кипятят с обратным холодильником в течение 4 часов. (S)-3-хинуклидинол (3,87 г, 30,4 ммоль) затем добавляют и кипячение с обратным холодильником продолжают в течение ночи. Реакционную смесь концентрируют, и остаток разделяют между этилацетатом (100 мл) и водным NaHCO3 (150 мл). Органический слой промывают водой (1×150 мл), сушат (Na2SO4) и концентрируют. Полученное беловатое твердое вещество очищают флэш-хроматографией с применением градиента хлороформа/метанола/аммиака с получением указанного в заголовке соединения в виде белого твердого вещества (4,34 г, 73%). 1H ЯМР (400 MГц, CDCl3) δ 7,96-7,88 (м, 2H), 7,16-7,04 (м, 3H), 5,55 (шс, 1H), 4,69-4,62 (м,1H), 3,24-3,11 (м,1H), 3,00-2,50 (м, 5H), 2,01-1,26 (м, 11H) ч./млн. 13C ЯМР (400 MГц, CDCl3) δ 166,4, 165,1, 163,8 (д, J=250,3 Гц), 162,9, 155,0, 130,1 (д, J=3,3 Гц), 128,4 (д, J= 8,5 Гц), 115,9 (д, J= 22,3 Гц), 112,5, 71,2, 55,7, 54,2, 47,5, 46,5, 28,0, 25,5, 24,7, 19,6 ч./млн. Чистота: 100% УЖХМС (210 нм & 254 нм); время удержания 0,83 мин; (M+1) 390.
(S)-хинуклидин-3-ил (2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 3)
С применением общей методики E и введения в реакционную смесь этил 2-(4-бромфенил)-2-метилпропаноата и 4-(2-метоксиэтокси)фенилбороновой кислоты, этил 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропаноат получают в виде беловатого твердого вещества. К перемешиваемому раствору этого соединения (3,01 г, 8,78 ммоль) в 1:1:1 (об./об./об.) тетрагидрофуране/этаноле/воде (45 мл) добавляют моногидрат гидроксида лития (1,47 г, 61,4 ммоль). Смесь нагревают при кипении с обратным холодильником в течение ночи и затем концентрируют. Остаток растворяют в воде, обрабатывают 1N хлористоводородной кислотой (65 мл) и экстрагируют этилацетатом. Объединенные органические слои промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют с получением 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,75 г, 100%). Это промежуточное соединение и (S)-хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде бесцветного, стеклообразного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,62-7,29 (м, 7H), 7,01 (д, J=8,9 Гц, 2H), 4,47-4,37 (м, 1H), 4,17-4,08 (м, 2H), 3,72-3,62 (м, 2H), 3,32 (с, 3H), 3,09-2,25 (м, 6H), 2,05-1,18 (м, 11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 157,9, 154,5, 146,7, 137,4, 132,5, 127,5, 125,7, 125,2, 114,8, 70,4, 70,0, 66,9, 58,2, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,2, 19,2 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,87 мин; (M+H+) 439,5.
1-азабицикло[2,2,2]окт-3-ил [2-(бифенил-3-ил)пропан-2-ил]карбамат (Соединение 4)
С применением общей методики C, 1-азабицикло[2,2,2]окт-3-ил [2-(3-бромфенил)пропан-2-ил]карбамат (600 мг, 1,63 ммоль), фенилбороновая кислота (398 мг, 3,27 ммоль) и ацетат палладия (II) дают указанное в заголовке соединение в виде белого твердого вещества (379 мг, 64%). 1H ЯМР (400 MГц, CDCl3) δ 7,61 (с, 1H), 7,56 (д, J= 7,4 Гц, 2H), 7,50-7,38 (м, 4H), 7,34 (м, 2H), 5,16 (с, 1H), 4,63 (с, 1H), 3,39-2,09 (м, 6H), 1,72 (с, 6H), 2,02-0,73 (м, 5H) ч./млн. 13C ЯМР (100 MГц, CDCl3) δ 154,8, 147,8, 141,6, 129,0, 129,0, 128,6, 127,5, 125,8, 125,0, 124,0, 71,6, 71,3, 55,9, 55,5, 47,6, 46,8, 31,5, 30,2, 30,0, 29,5, 25,6, 24,8, 19,8 ч./млн. Чистота: 99% УЖХМС (210 нм); время удержания 0,84 мин; (M+1) 365,0. Анал. рассч. для C23H28N2O2·0,29 (CHCl3): C, 70,02; H, 7,14; N, 7,01. Найдено: C, 70,02; H, 7,37; N, 6,84.
(S)-хинуклидин-3-ил 2-(бифенил-4-ил)пропан-2-илкарбамат (Соединение 5)
С применением общей методики B, бромбензонитрил (2,00 г, 11,0 ммоль) превращают в соответствующий 2-(4-бромфенил)пропан-2-амин (1,20 г, 51%) в виде коричневого масла.
С применением общей методики A, 2-(4-бромфенил)пропан-2-амин (1,0 г, 4,7 ммоль) и (S)-хинуклидин-3-ол дает (S)-хинуклидин-3-ил 2-(4-бромфенил)пропан-2-илкарбамат (1,0 г, 58%) в виде коричневого масла.
С применением общей методики C, вышеуказанный бромид (200 мг, 0,540 ммоль), фенилбороновая кислота (133 мг, 1,10 ммоль) и [PdCl2(pddf)]CH2Cl2 дают указанное в заголовке соединение в виде белого твердого вещества (70 мг, 35%). 1H ЯМР (500 MГц, CDCl3) δ 7,60-7,53 (м, 4H), 7,47 (д, J=8,5 Гц, 2H), 7,42 (т, J=7,5 Гц, 2H), 7,33 (т, J=7,5 Гц, 1H), 5,26 (шс, 1H), 4,64 (м, 1H), 3,33-3,15 (м, 1H), 3,10-2,45 (м, 5H), 2,40-1,80 (м, 2H), 1,78-1,58 (м, 7H), 1,55-1,33 (м, 2H) ч./млн. 13C ЯМР (125 MГц, CDCl3) δ 154,5, 146,1, 140,8, 139,5, 128,7, 127,2, 127,1, 127,1, 125,2, 70,9, 55,5, 55,1, 47,4, 46,4, 31,1, 29,5, 25,3, 24,5, 19,5 ч./млн. Чистота: 100% ЖХМС (214 нм & 254 нм); время удержания 1,56 мин; (M+1) 365.
Хинуклидин-3-ил 1-(бифенил-4-ил)циклопропилкарбамат (Соединение 6)
С применением общей методики D, бромбензонитрил (3,00 г, 16,5 ммоль) превращают в соответствующий 1-(4-бромфенил)циклопропанамин (1,80 г, 51%) в виде желтого твердого вещества.
С применением общей методики A, 1-(4-бромфенил)циклопропанамин (1,0 г, 4,7 ммоль) и хинуклидин-3-ол дает хинуклидин-3-ил 1-(4-бромфенил)циклопропилкарбамат (1,3 г, 75%) в виде белого полутвердого вещества.
С применением общей методики C, вышеуказанный карбамат (400 мг, 1,12 ммоль), фенилбороновая кислота (267 мг, 2,22 ммоль) и [PdCl2(pddf)]CH2Cl2 дают указанное в заголовке соединение в виде вязкого масла (100 мг, 25%). 1H ЯМР (500 MГц, CDCl3) δ 7,47 (д, J= 7,5 Гц, 2H), 7,43 (д, J= 8,0 Гц, 2H), 7,33 (т, J= 7,5 Гц, 2H), 7,26-7,15 (м, 3H), 5,93 (шс, 0,6H), 5,89 (шс, 0,4H), 4,67 (м, 1H), 3,20-3,06 (м, 1H), 2,88-2,42 (м, 5H), 1,98-1,08 (м, 9H) ч./млн. 13C ЯМР (125 MГц, CDCl3) δ 155,0, 141,0, 139,7, 138,2, 127,7, 126,1, 126,0, 124,8, 124,1, 70,0, 54,5, 46,3, 45,4, 34,1, 24,3, 23,2, 18,3, 17,0 ч./млн. Чистота: 100% ЖХМС (214 нм & 254 нм); время удержания 1,52 мин; (M+1) 363.
(S)-хинуклидин-3-ил 1-(4'-фторбифенил-4-ил)циклопропилкарбамат (Соединение 7)
С применением общей методики C, (S)-хинуклидин-3-ил 1-(4-бромфенил)циклопропил карбамат, 4-F-фенилбороновая кислота и [PdCl2(pddf)]CH2Cl2 дают указанное в заголовке соединение в виде белого твердого вещества (45%). 1H ЯМР (500 MГц, ДМСО-d6) δ 8,06-7,83 (д, 1H), 7,69-7,66 (м, 2H), 7,59-7,55 (м, 2H), 7,29-7,22 (м, 4H), 4,56-4,54 (м, 1H), 3,13-2,32 (м, 6H), 1,91-1,19 (м, 9H) ч./млн. 13C ЯМР (125 MГц, ДМСО-d6) δ 163,2, 161,2, 156,4, 143,7, 136,9, 128,9, 128,8, 126,8, 125,6, 116,2, 116,0, 70,7, 55,8, 47,4, 46,4, 34,8, 25,7, 24,6, 19,6, 18,7, 18,6 ч./млн. Чистота: > 97% ЖХМС (214 нм & 254 нм); время удержания 1,96 мин; (M+1) 381,2.
(S)-1-азабицикло[2,2,2]окт-3-ил [1-(2',4'-дифторбифенил-4-ил)циклопропил]карбамат (Соединение 8)
С применением общей методики C, (S)-хинуклидин-3-ил 1-(4-бромфенил)циклопропилкарбамат (0,446 г, 1,22 ммоль), 2,4-дифторфенилбороновая кислота (0,386 г, 2,44 ммоль) и Pd(OAc)2 (0,015 г, 0,067 ммоль) дают указанное в заголовке соединение в виде рыжевато-коричневого твердого вещества (0,111 г, 23%). 1H ЯМР (CDCl3) δ 7,43 (дд, J=8,4, 1,6 Гц, 2H), 7,40-7,33 (м, 1H), 7,31 (д, J=7,7 Гц, 2H), 6,99-6,81 (м, 2H), 5,54 (д, J=48,0 Гц, 1H), 4,82-4,65 (м, 1H), 3,30-3,07 (м, 1H), 2,98-2,44 (м, 5H), 1,97 (д, J=32,7 Гц, 1H), 1,83 (д, J=10,3 Гц, 1H), 1,64 (с, 1H), 1,52 (с, 1H), 1,39 (с, 1H), 1,31 (д, J=6,8 Гц, 4H) ч./млн. 13C ЯМР основной ротомер (CDCl3) δ 162,2 (дд, J=12,8, 249,1 Гц), 159,8 (дд, J=11,8, 251,0 Гц), 156,9, 156,0, 142,6, 133,1, 131,3 (м), 128,9, 125,6, 124,9, 111,5 (дд, J=3,9, 21,2 Гц) 104,4 (дд, J=25,2, 29,4 Гц), 72,1, 71,6, 55,7, 47,4, 46,5, 35,7, 35,3, 25,5, 24,6, 24,4, 19,5, 18,1 ч./млн. Чистота: ЖХМС >99,3% (214 нм & 254 нм); время удержания 0,90 мин; (M+1) 399,0.
1-азабицикло[2,2,2]окт-3-ил [1-(4'-метоксибифенил-4-ил)циклопропил]карбамат (Соединение 9)
С применением общей методики C, хинуклидин-3-ил 1-(4-бромфенил)циклопропилкарбамат (0,485 г, 1,33 ммоль), 4-метоксифенилбороновая кислота (0,404 г, 2,66 ммоль) и Pd(OAc)2 (0,016 г, 0,071 ммоль) дают указанное в заголовке соединение в виде серого твердого вещества (0,337 мг, 65%). 1H ЯМР (CDCl3) δ 7,48 (дд, J=8,6, 5,5 Гц, 4H), 7,29 (д, J=7,6 Гц, 2H), 6,96 (д, J=8,8 Гц, 2H), 5,58 (д, J=48,7 Гц, 1H), 4,83-4,63 (м, 1H), 3,84 (с, 3H), 3,20 (дд, J=24,0, 15,5 Гц, 1H), 2,97-2,42 (м, 5H), 1,97 (д, J=30,9 Гц, 1H), 1,81 (с, 1H), 1,75-1,33 (м, 3H), 1,28 (д, J=6,8 Гц, 4H) ч./млн. 13C ЯМР основной ротомер (CDCl3) δ 159,1, 156,0, 141,4, 139,0, 133,4, 128,0, 126,7, 125,9, 114,2, 71,5, 55,7, 55,3, 47,4, 46,5, 35,3, 25,5, 24,6, 19,6, 17,8 ч./млн. Чистота: ЖХМС >97,1% (214 нм & 254 нм); время удержания 0,88 мин; (M+1) 393,4.
Хинуклидин-3-ил 2-(5-(4-фторфенил)тиофен-3-ил)пропан-2-илкарбамат (Соединение 10)
К перемешиваемому и охлажденному (0°C) раствору этил 5-бромтиофен-3-карбоксилата (13,30 г, 56,57 ммоль) в ТГФ (100 мл) добавляют раствор бромид метилмагния в диэтиловом эфире [3,0 M] (55,0 мл, 165 ммоль), по каплям в течение 20 минут. Через 2 часа, реакционный раствор концентрируют. Остаток помещают в водный NH4Cl (200 мл) и экстрагируют этилацетатом (2×100 мл). Объединенные экстракты сушат (Na2SO4) и концентрируют. Полученное янтарное масло очищают флэш-хроматографией с применением градиента гексан/этилацетат с получением 2-(5-бромтиофен-3-ил)пропан-2-ола в виде бледно-янтарного масла (8,05 г, 64%).
К перемешиваемому раствору 2-(5-бромтиофен-3-ил)пропан-2-ол (8,03 г, 36,3 ммоль) в метиленхлориде (80 мл) добавляют азид натрия (7,08 г, 109 ммоль) затем трифторуксусную кислоту (8,0 мл; по каплям в течение 5-6 минут). Загущающую суспензию перемешивают в течение 1,5 часа, затем разбавляют водой (350 мл) и экстрагируют этилацетатом (1×200 мл). Органический слой промывают водным NaHCO3 (1×250 мл), сушат (Na2SO4) и концентрируют с получением неочищенного продукта азида. К перемешиваемому раствору этого продукта в ТГФ (160 мл) добавляют воду (11 мл) затем трифенилфосфин (23,8 г, 90,7 ммоль). Реакционную смесь перемешивают в течение 2 дней, затем концентрируют. Полученный остаток растворяют в этилацетате (250 мл) и экстрагируют 1 N водной HCl (4×75 мл). Объединенные экстракты подщелачивают концентрированным NH4OH и экстрагируют этилацетатом (2×100 мл). Эти экстракты, в свою очередь, сушат (Na2SO4) и концентрируют. Полученное янтарное масло очищают флэш-хроматографией с применением градиента метиленхлорид/метанол/аммиак с получением смеси 2-(5-бромтиофен-3-ил)пропан-2-амина и оксида трифенилфосфина (соотношение ~70/30) в виде вязкого янтарного масла (1,32 г, 17%).
К перемешиваемому раствору 3-хинуклидинола (3,00 г, 23,6 ммоль) в ТГФ (100 мл) добавляют 4-нитрофенилхлороформиат (5,94 г, 29,5). После перемешивания в течение 4 часов, осадок отфильтровывают, промывают ТГФ и сушат на воздухе на фритте под лабораторным вакуумом. Фильтровальную лепешку растворяют в этилацетате (150 мл) и промывают водным NaHCO3 (1×150 мл) и водой (2×150 мл). Органический слой сушат (Na2SO4) и концентрируют с получением неочищенного карбоната 4-нитрофенилхинуклидин-3-ила, который применяют на следующей стадии без очистки.
К перемешиваемому раствору 2-(5-бромтиофен-3-ил)пропан-2-амина (0,366 г, 1,66 ммоль) в ТГФ (10 мл) добавляют карбонат 4-нитрофенилхинуклидин-3-ил (0,571 г, 1,95 ммоль) и несколько гранул 4-(диметиламино)пиридина. Смесь кипятят с обратным холодильником в течение ночи, концентрируют и разделяют между этилацетатом (50 мл) и водным NaHCO3 (50 мл). Органический слой снова промывают водным NaHCO3 (1×50 мл), сушат (Na2SO4) и концентрируют. Полученную грязно-желтую камедь очищают флэш-хроматографией с применением градиента хлороформ/метанол/аммиак с получением хинуклидин-3-ил (1-(5-бромтиофен-3-ил)циклопропил)карбамата в виде беловатого твердого вещества (0,305 г, 49%).
С применением общей методики C, хинуклидин-3-ил (1-(5-бромтиофен-3-ил)циклопропил)карбамат (0,227 г, 0,742 ммоль), 4-фторфенилбороновая кислота (0,208 г, 1,49 ммоль), трициклогексилфосфин (0,021 г, 0,075 ммоль), фосфат калия (0,866, 4,08 ммоль) и ацетат палладия (8,0 мг, 36 мкмоль) дают указанное в заголовке соединение в виде серого твердого вещества (0,142 г, 49%). 1H ЯМР (400 MГц, CDCl3) δ 7,60-7,45 (м, 2H), 7,24-7,19 (м, 1H), 7,10-6,97 (м, 3H), 5,23 (шс, 1H), 4,72-4,61 (м, 1H), 3,30-3,04 (м, 1H), 3,03-2,25 (м, 5H), 2,09-1,02 (м, 11H) ч./млн. 13C ЯМР (400 MГц, CDCl3) δ 162,3 (д, J=247,1 Гц), 154,5, 149,8, 143,6, 130,7, 127,4 (д, J=8,1 Гц), 121,8, 118,9, 115,8 (д, J=21,6 Гц), 70,8, 55,5, 53,4, 47,3, 46,4, 29,0, 25,4, 24,4, 19,4 ч./млн. Чистота: 95,8% УЖХМС (210 нм & 254 нм); время удержания 0,90 мин; (M+1) 389.
(S)-хинуклидин-3-ил 2-(3-(4-фторфенил)изотиазол-5-ил)пропан-2-илкарбамат (Соединение 11)
К перемешиваемому раствору 2-(3-(4-фторфенил)изотиазол-5-ил)пропан-2-амина (1,21 г, 5,12 ммоль) в толуоле добавляют раствор фосгена в толуоле [~1,9 M] (10,8 мл, 20,5 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в течение двух часов и затем концентрируют. Остаток совместно выпаривают с толуолом (2×15 мл) с получением неочищенного промежуточного изоцианата в виде золотистого масла. Этот продукт помещают в толуол (10 мл) и обрабатывают (S)-3-хинуклидинолом (0,749 г, 5,89 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в течение ночи и концентрируют. Остаток очищают флэш-хроматографией с применением градиента хлороформ/метанол/аммиак с получением указанного в заголовке соединения в виде белого твердого вещества (0,971 г, 49%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,09-8,00 (м, 2H), 7,87 (шс, 1H), 7,75 (с, 1H), 7,35-7,25 (м, 2H), 4,54-4,45 (м, 1H), 3,14-2,92 (м, 1H), 2,87-2,17 (м, 5H), 1,98-0,98 (м, 11H) ч./млн. 13C ЯМР (400 MГц, ДМСО-d6) δ 180,1, 165,6, 162,6 (д, J=246,4 Гц), 154,7, 131,2 (д, J=3,0 Гц), 128,7 (д, J=8,4 Гц), 118,2, 115,7 (д, J=21,8 Гц), 70,6, 55,3, 52,8, 46,9, 45,9, 29,9, 25,2, 24,2, 19,2 ч./млн. Чистота: 100% УЖХМС (210 нм & 254 нм); время удержания 0,82 мин; (M+1) 390.
(S)-хинуклидин-3-ил 2-(4-(4-фторфенил)тиазол-2-ил)пропан-2-илкарбамат (Соединение 12)
К перемешиваемому раствору этил 3-амино-3-тиоксoпропаноата (20,00 г, 135,9 ммоль) в этаноле (120 мл) добавляют 2-бром-4’-фторацетофенон (29,49 г, 135,9 ммоль). Смесь кипятят с обратным холодильником в течение 1 часа, концентрируют и разделяют между этилацетатом (300 мл) и водным NaHCO3 (400 мл). Органический слой объединяют с обратным экстрактом водного слоя (этилацетат, 1×100 мл), сушат (Na2SO4) и концентрируют. Полученное светло-коричневое твердое вещество очищают флэш-хроматографией с применением градиента гексан/этилацетат с получением этил 2-(4-(4-фторфенил)тиазол-2-ил)ацетата в виде беловатого твердого вещества (29,92 г, 83%).
К перемешиваемому и охлажденному (-78°C) раствору этил 2-(4-(4-фторфенил)тиазол-2-ил)ацетата (10,00 г, 37,69 ммоль) в ТГФ (250 мл) добавляют раствор трет-бутоксида калия в ТГФ [1,0 M] (136 мл, 136 ммоль), по каплям в течение 15 минут, затем 18-краун-6 (1,6 мл, 7,5 ммоль). Через еще 30 минут при -78°C добавляют йодметан (8,5 мл), по каплям в течение 5 минут. Реакционную смесь перемешивают холодной в течение еще 2 часов, затем выливают в воду (450 мл) и экстрагируют этилацетатом (2×150 мл). Объединенные экстракты промывают насыщенным раствором соли (1×200 мл), сушат (Na2SO4) и концентрируют. Полученное коричневое масло очищают флэш-хроматографией с применением градиента гексан/этилацетат с получением этил 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропаноата в виде бледно-янтарного масла (8,64 г, 78%).
К перемешиваемому раствору этил 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропаноата (0,900 г, 3,07 ммоль) в 1:1:1 ТГФ/этаноле/воде (15 мл) добавляют моногидрат гидроксида лития (0,451 г, 10,7 ммоль). После перемешивания в течение ночи, реакционную смесь концентрируют и повторно растворяют в воде (80 мл). Раствор промывают простым эфиром (1×50 мл), подкисляют добавлением 1N HCl (15 мл) и экстрагируют этилацетатом (2×50 мл). Объединенные экстракты сушат (Na2SO4) и концентрируют с получением 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропановой кислоты в виде бледно-золотистого твердого вещества (0,808 г, 99%).
К перемешиваемому и охлажденному (0°C) раствору 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропановой кислоты (0,784 г, 2,96 ммоль) в ТГФ (25 мл) добавляют триэтиламин (0,82 мл, 5,9 ммоль) затем изобутилхлорформиат (0,58 мл, 4,4 ммоль). Реакционную смесь перемешивают холодной в течение еще 1 часа, затем добавляют раствор азида натрия (0,385 г, 5,92 ммоль) в воде (7 мл). Реакционную смесь перемешивают в течение ночи, позволяя охлаждающей бане медленно нагреваться до комнатной температуры. Смесь затем разбавляют водой (100 мл) и экстрагируют этилацетатом (2×60 мл). Объединенные экстракты промывают водным NaHCO3 (1×150 мл) и насыщенным раствором соли (1×100 мл), сушат (Na2SO4) и концентрируют. После совместного выпаривания с толуолом (2×30 мл), полученное беловатое твердое вещество помещают в толуол (25 мл) и кипятят с обратным холодильником в течение 4 часов. (S)-3-хинуклидинол (0,753 г, 5,92 ммоль) затем добавляют, и кипение с обратным холодильником продолжают в течение 3 часов. Реакционную смесь концентрируют, и остаток очищают флэш-хроматографией с применением градиента хлороформ/метанол/аммиак с получением указанного в заголовке соединения в виде белого твердого вещества (0,793 г, 69%). 1H ЯМР (400 MГц, CDCl3) δ 7,90-7,81 (м, 2H), 7,32 (с, 1H), 7,14-7,05 (м, 2H), 5,76 (шс, 1H), 4,72-4,65 (м, 1H), 3,26-3,10 (м, 1H), 3,03-2,37 (м, 5H), 2,05-1,23 (м, 11H) ч./млн. 13C ЯМР (400 MГц, CDCl3) δ 177,6, 162,6 (д, J=248,4 Гц), 154,8, 153,6, 130,8 (д, J=3,2 Гц), 128,1 (д, J=8,1 Гц), 115,9 (д, J=21,7 Гц), 112,2, 71,6, 55,7, 47,4, 46,5, 29,1, 25,4, 24,7, 19,6 ч./млн. Чистота: 100% УЖХМС (210 нм & 254 нм); время удержания 0,82 мин; (M+1) 390.
Хинуклидин-3-ил (2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 13)
С применением общей методики F и введением в реакционную смесь 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты (полученной, как описано в примере 3) и хинуклидин-3-ола, указанное в заголовке соединение получают в виде бесцветного, стеклообразного твердого вещества (23%). Данные ЯМР совпадают с таковым из примера 3. Чистота: 100%, 99,1% (210 & 254 нм) УЖХМС; время удержания: 0,87 мин; (M+H+) 439,0.
(S)-хинуклидин-3-ил (2-(3’-(2-метоксиэтокси)-[1,1’-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 14)
Заменяя 4-(2-метоксиэтокси)фенилбороновую кислоту 3-(2-метоксиэтокси)фенилбороновой кислотой, последовательность реакционной смеси, указанную в примере 3, применяют для получения 2-(3'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты. Это промежуточное соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде стеклообразного, бесцветного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,63-7,31 (м, 6H), 7,24-7,10 (м, 2H), 6,92 (дд, J=8,2, 1,9 Гц, 1H), 4,51-4,34 (м, 1H), 4,21-4,08 (м, 2H), 3,72-3,64 (м, 2H), 3,32 (с, 3H), 3,09-2,26 (м, 5H), 2,04-1,22 (м, 9H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 158,9, 154,6, 147,6, 141,5, 137,6, 129,9, 126,3, 125,2, 118,9, 113,2, 112,5, 70,4, 70,0, 66,9, 58,2, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,2, 19,2 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,91 мин; 15 (M+H+) 439,4.
Хинуклидин-3-ил (2-(4’-(2-метоксиэтокси)-[1,1’-бифенил]-3-ил)пропан-2-ил)карбамат (Соединение 15)
Заменяя этил 2-(4-бромфенил)-2-метилпропаноат этил 2-(3-бромфенил)-2-метилпропаноатом, последовательность реакционной смеси, указанную в примере 3, применяют для получения 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-3-ил)-2-метилпропановой кислоты. Это промежуточное соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,62-7,20 (м, 7H), 7,03 (д, J=8,7 Гц, 2H), 4,48-4,35 (м, 2H), 4,18-4,08 (м, 2H), 3,72-3,62 (м, 2H), 3,32 (с, 3H), 3,10-2,19 (м, 6H), 2,10-1,10 (м, 11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 158,0, 154,6, 148,8, 139,5, 133,1, 128,5, 127,7, 123,8, 123,2, 122,7, 114,8, 70,4, 69,9, 67,0, 58,2, 55,3, 54,5, 47,0, 45,9, 29,4, 25,3, 24,2, 19,2 ч./млн. Чистота: 97,4%, 94,6% (210 & 254 нм) УЖХМС; время удержания: 0,88 мин; (M+H+) 439,3.
Хинуклидин-3-ил (2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 16)
К перемешиваемому раствору 4-йодфенола (10,05 г, 45,68 ммоль) в ацетонтриле (100 мл) добавляют карбонат калия (6,95 г, 50,2 ммоль) и 1-хлор-3-метоксипропан (6,4 мл, 57,1 ммоль). Смесь нагревают при кипении с обратным холодильником в течение ночи и затем концентрируют. Остаток помещают в воду и экстрагируют этилацетатом. Объединенные экстракты промывают водным раствором бикарбоната натрия, сушат (Na2SO4) и концентрируют. Неочищенный продукт очищают флэш-хроматографией над диоксидом кремния с применением элюента гексан/этилацетат с получением 1-йод-4-(3-метоксипропокси)бензола в виде бесцветного масла (4,39 г, 33%). Это промежуточное соединение и этил 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат подвергают взаимодействию согласно общей методике E с получением этил 2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)-2-метилпропаноата. К перемешиваемому раствору этого соединения (0,693 г, 1,94 ммоль) в 1:1:1 (об./об./об.) тетрагидрофуране/этаноле/воде (10 мл) добавляют моногидрат гидроксида лития (0,326 г, 7,77 ммоль). Смесь нагревают при кипении с обратным холодильником в течение ночи и затем концентрируют. Остаток растворяют в воде, обрабатывают 1N хлористоводородной кислотой (10 мл) и экстрагируют этилацетатом. Объединенные органические слои промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют с получением 2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде воскообразного беловатого твердого вещества (0,630 г, 99%). Это промежуточное соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде стеклообразного, бесцветного твердого вещества (62%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,61-7,29 (м, 7H), 7,00 (д, J= 8,8 Гц, 2H), 4,47-4,36 (м, 1H), 4,05 (т, J= 6,4 Гц, 2H), 3,48 (т, J=6,3 Гц, 2H), 3,26 (с, 3H), 3,10-2,25 (м, 6H), 2,04-1,74 (м, 4H), 1,65-1,23 (м, 9H) ч./млн.,13C ЯМР (100 MГц, ДМСО-d6) δ 158,0, 154,5, 146,7, 137,4, 132,4, 127,5, 125,7, 125,2, 114,8, 69,9, 68,5, 64,6, 57,9, 55,4, 54,2, 46,9, 46,0, 29,4, 29,0, 25,2, 24,1, 19,2 ч./млн. Чистота: 97,7%, 98,2% (210 & 254 нм) УЖХМС; время удержания: 0,96 мин; (M+H+) 453,5.
Хинуклидин-3-ил (2-(4'-(гидроксиметил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 17)
С применением общей методики E и вводя в реакционную смесь этил 2-(4-бромфенил)-2-метилпропаноат и 4-формилфенилбороновую кислоту, этил 2-(4'-формил-[1,1'-бифенил]-4-ил)-2-метилпропаноат получают в виде бледно-янтарного твердого вещества. Это промежуточное соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением хинуклидин-3-ил (2-(4'-формил-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата в виде пенистого желтого твердого вещества. К перемешиваемому раствору этого продукта (0,755 г, 1,92 ммоль) в 2:1 (об./об.) тетрагидрофуране/этаноле (15 мл) добавляют боргидрид натрия (0,073 г, 1,93 ммоль). Через 45 минут, реакционную смесь разбавляют водой и экстрагируют хлороформом. Объединенные экстракты сушат (Na2SO4) и концентрируют на диоксиде кремния. Флэш-хроматография на диоксиде кремния с применением элюента хлороформ/метанол/аммиак дает указанное в заголовке соединения в виде белого твердого вещества (0,323 г, 43%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,66-7,29 (м,9H), 5,18 (т, J= 5,7 Гц, 1H), 4,53 (д, J= 5,7 Гц, 2H), 4,46-4,37 (м,1H), 3,11-2,19 (м,6H), 2,11-1,10 (м,11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 154,7, 147,3, 141,5, 138,4, 137,7, 127,0, 126,2, 126,1, 125,3, 70,0, 62,6, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,2, 19,2 ч./млн. Чистота: 97,5%, 99,1% (210 & 254 нм) УЖХМС; время удержания: 0,73 мин; (M+H+) 395.
Хинуклидин-3-ил (2-(4'-(2-гидроксиэтил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 18)
С применением общей методики E и вводя в реакционную смесь 1-(2-(бензилокси)этил)-4-бромбензол и этил 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноат, этил 2-(4'-(2-(бензилокси)этил)-[1,1'-бифенил]-4-ил)-2-метилпропаноат получают в виде бесцветной камеди. К перемешиваемому раствору этого соединения (1,34 г, 3,33 ммоль) в 1:1:1 (об./об./об.) тетрагидрофурана/этанола/воды (18 мл) добавляют моногидрат гидроксида лития (0,698 г, 16,6 ммоль). После нагревания при температуре кипения с обратным холодильником в течение ночи, реакционную смесь концентрируют и разделяют между водой и диэтиловым эфиром. Полученную эмульсию повторно экстрагируют 0,2 N водным раствором гидроксида натрия (5×50 мл). Прозрачную часть водного слоя удаляют каждый раз. Объединенные водные слои затем обрабатывают 1,0 N хлористоводородной кислотой (80 мл) и полученную суспензию белого твердого вещества экстрагируют этилацетатом. Объединенные органические слои сушат (Na2SO4) и концентрируют с получением 2-(4'-(2-(бензилокси)этил)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (1,20 г, 96%). Это соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением хинуклидин-3-ил (2-(4'-(2-бензилоксиэтил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата. К перемешиваемому раствору этого продукта (0,435 г, 0,806 ммоль) в метаноле добавляют 1,0 N хлористоводородной кислоты (1 мл) и 10% палладия на угле (50% воды; 0,087 г). Смесь циклируют между вакуумом и продувкой азотом несколько раз, повторно заполняют водородом после последнего вакуумирования. Через 1,25 часа реакционную смесь фильтруют через Целит и концентрируют. Остаток помещают в водный раствор карбоната натрия и экстрагируют 4:1 (об./об.) хлороформом/изопропанолом. Объединенные экстракты сушат (Na2SO4) и концентрируют в диоксид кремния. Флэш-хроматография над диоксидом кремния с применением градиента хлороформ/метанол/аммиак дает очищенное указанное в заголовке соединение в виде бесцветного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,85-7,63 (м,1H), 7,63-7,19 (м,8H), 4,78-4,62 (м,2H), 3,71-2,78 (м,8H), 2,76 (т, J= 6,8 Гц, 2H), 2,26-1,96 (м,2H), 1,96-1,40 (м,9H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 153,8, 146,8, 138,7, 137,9, 137,6, 129,4, 126,3, 126,1, 125,3, 66,2, 62,1, 54,4, 52,8, 45,4, 44,5, 38,6, 29,5, 29,2, 24,0, 19,9, 16,6 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,75 мин; (M+H+) 409.
Хинуклидин-3-ил (2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)пропан-2-ил)карбамат (Соединение 19)
К перемешиваемой суспензии 4-метокситиобензамида (9,99 г, 59,7 ммоль) в этаноле (75 мл) добавляют этил 4-хлорацетоацетат (8,1 мл, 60 ммоль). Смесь нагревают при кипении с обратным холодильником в течение 4 часов, затем охлаждают, добавляют еще этил 4-хлорацетоацетат (0,81 мл, 6,0 ммоль) и возвращают к кипению с обратным холодильником. Через еще 4 часа нагревания, реакционную смесь концентрируют и разделяют между этилацетатом и водным раствором бикарбоната натрия. Органический слой объединяют с дополнительными экстрактами этилацетата, сушат (Na2SO4) и концентрируют. Неочищенный продукт очищают флэш-хроматографией над диоксидом кремния с применением градиента гексан/этилацетат с получением этил 2-(2-(4-метоксифенил)тиазол-4-ил)ацетат в виде бледно-янтарного масла (14,51 г, 87%). К перемешиваемому раствору этого соединения (14,48 г, 52,2 ммоль) в N, N-диметилформамиде (125 мл) добавляют гидрид натрия (60% дисперсию в минеральном масле; 6,27 г, 157 ммоль), порциями, в течение более 15 минут. Полученную красную суспензию охлаждают (0°C) и обрабатывают, по каплям, в течение 10 минут, йодметаном (9,80 мл, 157 ммоль). Охлаждающую баню удаляют, и реакционную смесь перемешивают 4 часа, затем концентрируют и разделяют остаток между этилацетатом и водой. Органический слой дважды промывают водой, сушат (Na2SO4) и концентрируют. Остаток очищают флэш-хроматографией над диоксидом кремния с применением градиента гексан/этилацетат с получением этил 2-(2-(4-метоксифенил)тиазол-4-ил)-2-метилпропаноата в виде бледно-янтарного масла (14,12 г, 89%). К перемешиваемому раствору этого промежуточного соединения (14,12 г, 46,24 ммоль) в метиленхлориде (250 мл) добавляют трибромид бора (11,0 мл, 116 ммоль), по каплям в течение 5 минут. После перемешивания в течение ночи, реакцию гасят медленным добавлением метанола (~20 мл) и затем концентрируют. Остаток помещают в метанол (250 мл) и концентрированную серную кислоту (7,0 мл). Перемешанный раствор нагревают при кипении с обратным холодильником в течение 2 часов, концентрируют и разделяют между этилацетатом и водным раствором бикарбоната натрия. Органический слой объединяют со вторым экстрактом этилацетата водного слоя, сушат (Na2SO4) и концентрируют с получением метил 2-(2-(4-гидроксифенил)тиазол-4-ил)-2-метилпропаноата в виде белого твердого вещества (12,56 г, 98%). К перемешиваемому раствору 1-бром-3-метоксипропана (1,66 г, 10,8 ммоль) в ацетоне (30 мл) добавляют промежуточный фенол (2,00 г, 7,21 ммоль) и карбонат калия (1,25 г, 9,04 ммоль). Смесь нагревают в течение ночи при кипении с обратным холодильником, фильтруют и концентрируют. Остаток очищают флэш-хроматографией над диоксидом кремния с применением градиента гексан/этилацетат с получением метил 2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)-2-метилпропаноата в виде блеклой янтарной камеди (2,47 г, 98%). К перемешиваемому раствору этого соединения (2,45 г, 7,01 ммоль) в 1:1:1 (об./об./об.) тетрагидрофурана/этанола/воды (45 мл) добавляют моногидрат гидроксида лития (1,47 г, 35,0 ммоль). После перемешивания в течение ночи, реакционную смесь концентрируют и разделяют между водой и диэтиловым эфиром. Водный слой обрабатывают 1,0 N хлористоводородной кислотой (40 мл) и экстрагируют этилацетатом. Объединенные экстракты сушат (Na2SO4) и концентрируют с получением 2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,19 г, 40 93%). Это соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде мягкого блеклого янтарного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,82 (д, J=8,9 Гц, 2H), 7,36 (шс, 1H), 7,24 (шс, 1H), 7,03 (д, J=8,9 Гц, 2H), 4,49-4,41 (м,1H), 4,07 (т, J=6,4 Гц, 2H), 3,48 (т, J=6,4 Гц, 2H), 3,26 (с, 3H), 3,09-2,26 (м,6H), 2,02-1,91 (м,2H), 1,91-1,03 (м,11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 165,8, 162,4, 160,0, 154,6, 127,5, 126,1, 114,9, 112,1, 70,1, 68,4, 64,8, 57,9, 55,4, 53,5, 46,9, 45,9, 28,9, 28,3, 25,2, 24,2, 19,2 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,87 мин; (M+H+) 460.
Хинуклидин-3-ил (2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)пропан-2-ил)карбамат (Соединение 20)
К перемешиваемому раствору 2-бромэтилметилового эфира (1,88 г, 13,5 ммоль) в ацетоне добавляют метил 2-(2-(4-гидроксифенил)тиазол-4-ил)-2-метилпропаноат (полученный, как описано в примере 19, 2,00 г, 7,21 ммоль) и карбонат калия (1,56 г, 11,3 ммоль). После нагревания при температуре кипения с обратным холодильником в течение ночи, смесь обрабатывают дополнительным 2-бромэтилметиловым эфиром (1,88 г, 13,5 ммоль) и карбоната калия (1,56 г, 11,3 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в течение второй ночи, фильтруют и концентрируют. Остаток очищают флэш-хроматографией над диоксидом кремния с применением градиента гексан/этилацетат с получением метил 2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)-2-метилпропаноата в виде белого твердого вещества (2,71 г, 90%). К перемешиваемому раствору этого соединения (2,71 г, 8,08 ммоль) в 1:1:1 (об./об./об.) тетрагидрофуране/этаноле/воде (50 мл) добавляют моногидрат гидроксида лития (1,70 г, 40,5 ммоль). После перемешивания в течение ночи, реакционную смесь концентрируют и разделяют между водой и диэтиловым эфиром. Водный слой обрабатывают 1,0 N хлористоводородной кислотой (41 мл) и экстрагируют этилацетатом. Объединенные экстракты сушат (Na2SO4) и концентрируют с получением 2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,57 г, 99%). Это соединение и хинуклидин-3-ол подвергают взаимодействию согласно общей методике F с получением указанного в заголовке соединения в виде бледно-янтарного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,82 (д, J=8,8 Гц, 2H), 7,36 (шс, 1H), 7,24 (шс, 1H), 7,04 (д, J=8,8 Гц, 2H), 4,49-4,41 (м,1H), 4,19-4,12 (м,2H), 3,71-3,65 (м,2H), 3,32 (с, 3H), 3,11-2,87 (м,1H), 2,86-2,19 (м,5H), 1,92-1,16 (м,11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 165,7, 162,9, 159,9, 154,6, 127,5, 126,2, 114,9, 112,2, 70,3, 70,1, 67,1, 58,2, 55,4, 53,5, 46,9, 45,9, 28,3, 25,2, 24,3, 19,2 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,85 мин; (M+H+) 446.
Хинуклидин-3-ил 2-(5-(4-(2-метоксиэтокси)фенил)пиридин-2-ил)пропан-2-илкарбамат (Соединение 21)
С применением общей методики E и вводя в реакционную смесь 5-бромпиколинонитрил и 2-(4-(2-метоксиэтокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан, получают 5-(4-(2-метоксиэтокси)фенил)пиколинонитрил. Трихлорид церия (8,05 г, 21,6 ммоль) загружают в колбу и сушат нагреванием (170°C) под вакуумом в течение 3 часов. Твердое вещество помещают в тетрагидрофуран (20 мл) и энергично перемешивают в течение 30 минут. Суспензию охлаждают до -78°C и обрабатывают, по каплям, 3,0 M раствором метиллития в диэтиловом эфире (7,2 мл, 21,6 ммоль). После добавления, реакционную смесь перемешивают при -78°C в течение 1 часа, затем добавляют раствор вышеуказанного арилбората (1,83 г, 7,20 ммоль) в тетрагидрофуране (20 мл). Смесь выдерживают при -78°C в течение 2 часов и затем нагревают до комнатной температуры. В это время, реакцию гасят добавлением водного гидроксида аммония (10 мл) и фильтруют через слой целита. Фильтрат экстрагируют этилацетатом, и объединенные экстракты промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Остаток очищают флэш-хроматографией над диоксидом кремния с применением элюента этилацетата с получением 2-(5-(4-(2-метоксиэтокси)фенил)пиридин-2-ил)пропан-2-амина в виде желтого твердого вещества (0,800 г, 39%). К перемешиваемой суспензии этого промежуточного соединения (0,500 г, 1,75 ммоль) в воде (10 мл) и концентрированной хлористоводородной кислоты (0,44 мл) добавляют толуол (10 мл). Смесь охлаждают (0°C) и обрабатывают, одновременно, в течение 1 часа, растворами трифосгена (0,776 г, 2,62 ммоль) в толуоле (10 мл) и бикарбоната натрия (2,2 г, 26 ммоль) в воде (20 мл). После добавлений, реакционную смесь перемешивают в течение еще 30 минут, затем верхний слой толуола удаляют и сушат (Na2SO4). В это время перемешиваемый раствор хинуклидин-3-ола (0,445 г, 3,64 ммоль) в тетрагидрофуране (10 мл) обрабатывают гидридом натрия (60% дисперсию в минеральном масле; 0,154 г, 3,85 ммоль). Эту смесь перемешивают в течение 5 минут и затем добавляют к раствору неочищенного изоцианата в толуоле. Реакционную смесь перемешивают в течение 10 минут, гасят добавлением насыщенного раствора соли (5 мл) и экстрагируют этилацетатом. Объединенные экстракты сушат (Na2SO4) и концентрируют. Остаток очищают флэш-хроматографией над диосидом кремния с обращенной фазой с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (0,100 г, 13%). 1H ЯМР (500 MГц, CDCl3) δ 8,70-8,70 (д, J=2,0 Гц, 1H), 7,83-7,81 (м,1H), 7,49-7,47 (д, J=9,0 Гц, 2H), 7,45-7,43 (д, J=8,0 Гц, 1H), 7,03-7,01 (д, J=8,5 Гц, 2H), 6,63 (шс, 1H), 4,68-4,66 (м,1H), 4,16 (т, J=5,0 Гц, 2H), 3,77 (т, J=5,0 Гц, 2H), 3,45 (с, 3H), 3,19-2,70 (м,6H), 2,15-1,89 (м,2H), 1,76 (с, 6H), 1,73-1,36 (м,3H) ч./млн. 13C ЯМР (125 MГц, CDCl3) δ 162,7, 158,9, 154,9, 145,9, 134,8, 134,3, 130,1, 128,1, 119,2, 115,2, 71,0, 70,8, 67,4, 59,2, 55,9, 55,7, 47,4, 46,5, 46,4, 27,9, 25,4, 24,6, 19,5 ч./млн. Чистота: >99% (214 & 254 нм) ЖХМС; время удержания: 1,32 мин; (M+H+) 440,2.
Хинуклидин-3-ил (2-(4'-(3-цианопропокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 22)
К перемешиваемому раствору 4-бромфенола (17,1 г, 98,8 ммоль) в ацетонитриле (150 мл) добавляют 1-бромбутилнитрил (12,3 мл, 124 ммоль) и карбонат калия (15,0 г, 109 ммоль). Смесь нагревают до кипения с обратным холодильником в течение ночи, охлаждают и концентрируют. Остаток помещают в воду и экстрагируют этилацетатом. Объединенные экстракты сушат (Na2SO4) и концентрируют, и неочищенный продукт очищают флэш-хроматографией над диоксидом кремния с применением элюента гексан/этилацетат с получением 4-(4-бромфенокси)бутаннитрила в виде белого твердого вещества (20,8 г, 88%). К перемешиваемому раствору этого продукта в N, N-диметилформамиде (100 мл), добавляют бис(пинаколато)диборон (4,60 г, 18,1 ммоль), ацетат калия (7,41 г, 75,5 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) с дихлометаном (0,616 г, 1,04 ммоль). Смесь нагревают до кипения с обратным холодильником в течение ночи и затем концентрируют. Остаток помещают в этилацетат и промывают водой и насыщенным раствором соли. Органический слой сушат (Na2SO4) и концентрируют, и неочищенный продукт очищают флэш-хроматографией над диоксидом кремния с применением элюента гексан/этилацетат с получением 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бутаннитрила в виде белого твердого вещества (3,43 г, 79%). Этот продукт и хинуклидин-3-ил (2-(4-бромфенил)пропан-2-ил)карбамат (полученный взаимодействием хинуклидин-3-ола и 2-(4-бромфенил)пропан-2-амина с применением общей методики F) подвергают взаимодействию согласно общей методике E с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,67-7,26 (м,7H), 7,02 (д, J=8,8 Гц, 2H), 4,50-4,33 (м,1H), 4,08 (т, J=6,0 Гц, 2H), 3,14-2,18 (м,8H), 2,04 (квин, J=6,7 Гц, 2H), 1,94-1,70 (м,11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 157,7, 154,5, 146,8, 137,4, 132,7, 127,6, 125,7, 125,2, 120,2, 114,9, 70,0, 65,8, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,7, 24,2, 19,2, 13,4 ч./млн. Чистота: 100%, 98,9% (210 & 254 нм) УЖХМС; время удержания: 0,88 мин; (M+H+) 448,6.
Хинуклидин-3-ил (2-(4'-(цианометокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат (Соединение 23)
С применением общей методики E и вводя в реакционную смесь хинуклидин-3-ил (2-(4-бромфенил)пропан-2-ил)карбамат (полученный взаимодействием хинуклидин-3-ола и 2-(4-бромфенил)пропан-2-амина с применением общей методики F) и 4-(цианометокси)фенилбороновую кислоту, указанное в заголовке соединение получают в виде бледно-янтарного твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,65 (д, J= 8,2 Гц, 2H), 7,60-7,31 (м,5H), 7,15 (д, J=8,9 Гц, 2H), 5,21 (с, 2H), 4,53-4,30 (м,1H), 3,18-2,19 (м,6H), 2,05-1,18 (м,11H) ч./млн. 13C ЯМР (100 MГц, ДМСО-d6) δ 155,8, 154,6, 147,2, 137,2, 134,4, 127,8, 126,0, 125,3, 116,7, 115,3, 70,0, 55,4, 54,2, 53,5, 46,9, 45,9, 29,4, 25,2, 24,2, 19,2 ч./млн. Чистота: 100%, 100% (210 & 254 нм) УЖХМС; время удержания: 0,85 мин; (M+H+) 420,3.
Пример 2: Получение свободного основания (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата
Стадия 1: Диметилирование метилйодидом
3N КД колбу оборудуют термометром, капельной воронкой и входом для азота. Колбу промывают азотом, и трет-бутоксид калия (ММ 112,21, 75,4 ммоль, 8,46 г, 4,0 экв., белый порошок) взвешивают и добавляют в колбу через порошковую воронку, затем добавляют ТГФ (60 мл). Большая часть трет-бутоксида калия растворяется с получением мутного раствора. Эту смесь охлаждают на бане лед-вода до 0-2°C (внутренняя температура). В отдельной колбе, исходный сложный эфир (ММ 265,3, 18,85 ммоль, 5,0 г, 1,0 экв.) растворяют в ТГФ (18 мл+2 мл в качестве промывки) и переносят в капельную воронку. Этот раствор добавляют по каплям в охлажденную смесь в течение 25-30 мин, сохраняя внутреннюю температуру ниже 5°C во время добавления. Реакционную смесь охлаждают обратно до 0-2°C. В отдельной колбе, получают раствор метилйодида (ММ 141,94, 47,13 ммоль, 6,7 г, 2,5 экв.) в ТГФ (6 мл) и переносят в капельную воронку. Колбу, содержащую раствор метилйодида, затем промывают ТГФ (1,5 мл), затем переносят в капельную воронку, уже содержащую прозрачный бесцветный раствор метилйодид в ТГФ. Этот раствор осторожно добавляют по каплям в темно-коричневую реакционную смесь в течение 30-40 мин, сохраняя внутреннюю температуру ниже 10°C все время в течение добавления. После завершения добавления, слегка мутную смесь перемешивают в течение еще 1 ч, в течение которого внутренняя температура падает до 0-5°C. После перемешивания в течение часа при 0-5°C, реакционную смесь гасят медленным по каплям добавлением 5,0M водной HCl (8 мл) в течение 5-7 мин. Внутреннюю температуру сохраняют ниже 20°C во время этого добавления. После добавления, добавляют воду (14 мл) и смесь перемешивают в течение 2-3 мин. Перемешивание останавливают, и два слоя разделяют. Затем два слоя переносят в 250 мл 1N КД колбу, и ТГФ выпаривают в вакууме насколько возможно с получением двухфазного слоя ТГФ/продукта и воды. Два слоя разделяют. ТГФ раствор продукта со стадии 1 применяют в следующей реакции.
Стадия 2: Гидролиз этилового эфира с моногидратом LiOH
Неочищенный сложный эфир в ТГФ добавляют в реакционную колбу. Отдельно, LiOH.H2O (ММ 41,96, 75,0 ммоль, 3,15 граммов, 2,2 экв.) взвешивают в 100 мл стакане, куда добавляют мешалку. Добавляют воду (40 мл), и смесь перемешивают до тех пор, пока все твердые вещества не растворятся с получением прозрачного бесцветного раствора. Этот водный раствор затем добавляют в 250 мл КД колбу, содержащую раствор сложного эфира в тетрагидрофуране (ТГФ). Конденсатор присоединяют к горлу колбы, и вход для азота присоединяют к верхней части конденсатора. Смесь нагревают при кипении с обратным холодильником в течение 16 часов. Через 16 часов, нагревание останавливают, и смесь охлаждают до комнатной температуры. ТГФ выпаривают в вакууме с получением коричневого раствора. Аликвоту коричневого водного раствора анализируют ВЭЖХ и ЖХ/МС для полного гидролиза этилового эфира. Добавляют воду (15 мл), и этот водный щелочной раствор экстрагируют ТБМЭ (2×40 мл) для удаления трет-бутиловогт эфира. Водный щелочной слой охлаждают на бане лед-вода до 0-10°C и подкисляют добавлением по каплям концентрированной HCl до pH ~ 1 при перемешивании. К этому липкому твердому веществу в водном кислот растворе добавляют ТБМЭ (60 мл), и смесь встряхивают и затем энергично перемешивают для растворения всей кислоты в слое ТБМЭ. Два слоя переносят в делительную воронку, и слой ТБМЭ отделяют. Бледно-желтый водный кислый раствор повторно экстрагируют ТБМЭ (40 мл), и слой ТБМЭ отделяют и объединяют с предыдущим слоем ТБМЭ. Водный кислый слой отбрасывают. Объединенные слои ТБМЭ сушат над безводным Na2SO4, фильтруют и выпаривают в вакууме для удаления ТБМЭ и получения неочищенной кислоты в виде оранжевого/темно-желтого масла, которое затвердевает в высоком вакууме до грязно-желтого твердого вещества. Неочищенную кислоту взвешивают и кристаллизуют нагреванием в гептане/ТБМЭ (3:1, 5 мл/г неочищенного продукта) с получением кислоты в виде желтого твердого вещества.
Стадия 3: Образование гидроксаминовой кислоты с NH2OH.HCl
Карбоновую кислоту (ММ 265,3, 18,85 ммоль, 5,0 г, 1,0 экв.) взвешивают и переносят в 25 мл 1N КД колбу под азотом. ТГФ (5,0 мл) добавляют, и кислоту легко растворяют с получением прозрачного темно-желтого/коричневого раствора. Раствор охлаждают до 0-2°C (температура бани) в ледяной бане и медленно добавляют N, N’-карбонилдиимидазол (КДИ; ММ 162,15, 20,74 ммоль, 3,36 г, 1,1 экв.) небольшими порциями в течение 10-15 минут. Ледяную баню удаляют, и раствор перемешивают при комнатной температуре в течение 1 ч. Через 1 ч перемешивания, раствор снова охлаждают в бане лед-вода до 0-2°C (температура бани). Медленно добавляют гидрохлорид гидроксиламина (NH2OH.HCl; ММ 69,49, 37,7 ммоль, 2,62 г, 2,0 экв.) небольшими порциями в виде твердого вещества в течение 3-5 минут, так как это добавление является экзотермическим. После завершения добавления, воду (1,0 мл) добавляют в гетерогенную смесь по каплям в течение 2 минут, и реакционную смесь перемешивают при 0-10°C в бане лед-вода в течение 5 минут. Охлаждающую баню удаляют, и реакционную смесь перемешивают под азотом при комнатной температуре в течение ночи в течение 20-22 ч. Раствор становится прозрачным по мере растворения всего NH2OH.HCl. Через 20-22 ч, аликвоту реакционной смеси анализируют жидкостной хроматографией высокого давления (ЖХВД). ТГФ затем выпаривают в вакууме, и остаток помещают в дихлорметан (120 мл) и воду (60 мл). Смесь переносят в делительную воронку, где ее встряхивают, и два слоя разделяют. Водный слой отбрасывают, и слой дихлорметана промывают 1N гидрохлоридом (HCl; 60 мл). Кислый слой отбрасывают. Слой дихлорметана сушат над безводным Na2SO4, фильтруют, и растворитель выпаривают в вакууме с получением неочищенной гидроксаминовой кислоты в виде бледно-желтого твердого вещества, которое сушат в высоком вакууме в течение ночи.
Стадия 3 (продолжение): Превращение гидроксаминовой кислоты в циклическое промежуточное соединение (не выделяют)
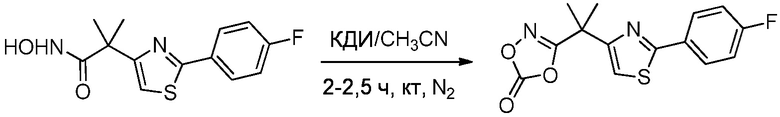
Неочищенную гидроксаминовую кислоту (ММ 280,32, 5,1 г) переносят в 250 мл 1N КД колбу с азотным входом. Добавляют мешалку, затем добавляют ацетонитрил (50 мл). Твердое вещество нерастворимо в ацетонитриле. Желтую гетерогенную смесь перемешивают в течение 2-3 минут под азотом, и КДИ (ММ 162,15, 20,74 ммоль, 3,36 г, 1,1 экв.) добавляют одной порцией при комнатной температуре. Экзотерм не наблюдается. Твердое вещества сразу же растворяют, и прозрачный желтый раствор перемешивают при комнатной температуре в течение 2-2,5 ч. Через 2-2,5 ч, аликвоту анализируют ВЭЖХ и ЖХ/МС, которые показали превращение гидроксаминовой кислоты в желаемое циклическое промежуточное соединение.
Затем ацетонитрил выпаривают в вакууме с получением неочищенного циклического промежуточного соединения в виде красноватого густого масла. Масло помещают в толуол (60 мл), и красноватую смесь нагревают до кипения с обратным холодильником в течение 2 часов, в течение которых циклическое промежуточное соединение выделяет CO2 и перестраивается в изоцианат (см. ниже.
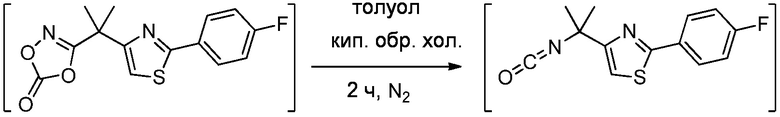
Стадия 3 (продолжение): Превращение изоцианата в свободное основание
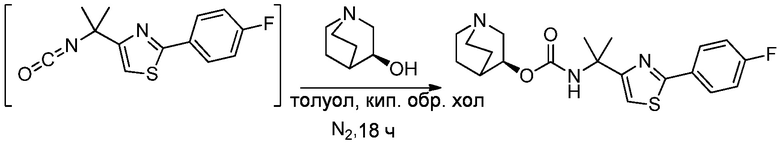
Реакционную смесь охлаждают до 50-60°C и (S)-(+)-хинуклидинол (ММ 127,18, 28,28 ммоль, 3,6 г, 1,5 экв.) добавляют к смеси в виде твердого вещества одной порцией. Смесь повторно нагревают до кипения с обратным холодильником в течение 18 ч. Через 18 ч, аликвоту анализируют ВЭЖХ и ЖХ/МС, которые показывают полное превращение изоцианата в желаемый продукт. Реакционную смесь переносят в делительную воронку, и добавляют толуол (25 мл). Смесь промывают водой (2×40 мл), и водные слои отделяют. Объединенные водные слои повторно экстрагируют толуолом (30 мл), и водный слой отбрасывают. Объединенные толуольные слои экстрагируют 1N HCl (2×60 мл), и толуольный слой (содержащий O-ацильную примесь) отбрасывают. Объединенные HCl слои переносят в 500 мл колбу Эрленмейера, оборудованную мешалкой. Перемешиваемый желтый/красновато-оранжевый раствор подщелачивают до pH 10-12 добавлением по каплям 50% масс./масс. водного NaOH. Желаемое свободное основание выпадает в осадок в виде грязно-желтого липкого твердого вещества, которое удерживает лопатку мешалки. К этой смеси добавляют изопропилацетат (100 мл), и смесь энергично перемешивают в течение 5 минут, когда липкое твердое вещество превращается в изопропилацетат. Перемешивание останавливают, и два слоя разделяют. Желтый слой изопропилацетата отделяют, и щелочной слой повторно экстрагируют изопропилацетатом (30 мл). Щелочной водный слой отбрасывают, и объединенные слои изопропилацетата сушат над безводным Na2SO4, фильтруют в предварительно взвешенную КД колбу, и растворитель выпаривают в вакууме с получением неочищенного свободного основания в виде бежевого/рыжевато-коричневого твердого вещества, которое сушат в высоком вакууме в течение ночи.
Стадия 3 (продолжение): Перекристаллизация неочищенного свободного основания
Бежевое/рыжевато-коричневое твердое вещество взвешивают и перекристаллизуют из гептана/изопропилацетата (3:1, 9,0 мл растворителя/г неочищенного свободного основания). Подходящее количество гептана/изопропилацетата добавляют к неочищенному свободному основанию вместе с мешалкой, и смесь нагревают до кипения с обратным холодильником в течение 10 мин (свободное основание изначально было частично растворимым, но растворилось с получением прозрачного красновато-оранжевого раствора при нагревании при кипении с обратным холодильником). Источник тепла удаляют, и смесь охлаждают до комнатной температуры при перемешивании, когда образуется белый осадок. После перемешивания при комнатной температуре в течение 3-4 ч, осадок отфильтровывают через вакуумный шланг с применением воронки Бюхнера, промывают гептаном (20 мл) и сушат под шлангом с вакуумом на воронке Бюхнера в течение ночи. Осадок переносят в кристаллизатор и сушат при 55°C в течение ночи в вакуумной печи. 1H ЯМР (400 MГц, CDCl3) δ 8,04-7,83 (м,2H), 7,20-6,99 (м,3H), 5,53 (с, 1H), 4,73-4,55 (м,1H), 3,18 (дд, J=14,5, 8,4 Гц, 1H), 3,05-2,19 (м,5H), 2,0-1,76 (м,11H) ч./млн. 13C ЯМР (100 MГц, CDCl3) δ 166,38, 165,02, 162,54, 162,8-155,0 (д, C-F), 130,06, 128,43, 128,34, 116,01, 115,79, 112,46, 71,18, 55,70, 54,13, 47,42, 46,52, 27,94, 25,41, 24,67, 19,58 ч./млн.
Пример 3: Получение кристаллических форм солей (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата
Кристаллические соли (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата могут быть образованы из свободного основания, полученного как описано в Примере 23.
Например, свободное основание (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата (примерно 50 ммоль) растворяют в ИПС (140 мл) при комнатной температуре и фильтруют. Фильтрат добавляют в 1 л к.д. колбы, которую оборудуют верхней мешалкой и входом/выходом азота. L-яблочную кислоту (примерно 50 ммоль) растворяют в ИПС (100+30 мл) при комнатной температуре и фильтруют. Фильтрат добавляют в вышеуказанную 1-литровую колбу. Полученный раствор перемешивают при комнатной температуре (с или без высевания) под азотом в течение 4-24 часов. Во время этого периода времени образуются кристаллы. Продукт собирают фильтрацией и промывают небольшим количеством ИПС (30 мл). Кристаллическое твердое вещество сушат в вакуумной печи при 55°C в течение 72 часов с получением желаемого малата.
Кристаллические формы других солей, например, кислотно-аддитивных солей с янтарной кислотой или HCl, могут быть получены по аналогичной методике.
Пример 4: In vitro GCS ингибирование (Соединение 2 и аналоги)
Ингибирование активности глюкозилцерамидсинтазы может быть измерено с применением одного или нескольких анализов. Первым анализом является микросомальный анализ, в котором прямо измеряется превращение церамида в глюкозилцерамид с применением ВЭЖХ. Микросомы являются источником активности глюкозилцерамидсинтазы в микросомальном анализе. Вторым анализом является клеточный, фенотипический анализ, который отслеживает экспрессию на поверхности клеток нисходящего жира GM3 с применением опосредованной антителом иммунофлуоресценции. Конкретные протоколы представлены ниже.
Микросомальный анализ активности глюкозилцерамидсинтазы:
Ферментный анализ с применением микросом в качестве источника активности глюкозилцерамидсинтазы. Флуоресцентный церамидный субстрат доставляют к мембраносвязанному ферменту в виде комплекса с альбумином. После реакции, церамид и глюкозилцерамид разделяют и количественно оценивают с помощью ВЭЖХ с обращенной фазой с флуоресцентным определением. Ферментативную активность оценивают с использованием флуоресцентно меченого субстрата и микросом в качестве источника глюкозилцерамидсинтазы. C6-NBD-церамид образует комплекс с альбумином для доставки в микросомы, которые выделяют в соответствии с процедурой, описанной ниже. Конечная концентрация C6-NBD-церамида в исходном растворе составляет 0,5 мМ; Конечная плотность АБС составляет 0,5 мм. Разделение и количественное определение субстрата и продукта (глюкозилцерамида) проводят с применением ВЭЖХ с обращенной фазой с флуоресцентным определением.
Получение микросом из A375 клеток меланомы человека;
Микросомы выделяют из A375 клеток меланомы человека. От восьми до десяти миллионов клеток собирают трипсинизацией и промывают ледяным ФРФБ. Клетки ресуспендируют в ледяном лизисном буфере, содержащем ингибиторы протеазы. Лизат клеток обрабатывают ультразвуком на льду с помощью зондового соникатора. После обработки ультразвуком, клеточный лизат отделяют от дебриса центрифугированием при 10000 g в течение 10 минут при 4°C. Супернатант удаляют и очищают дополнительным центрифугированием при 100000 g в течение 1 часа при 4°C. Затем осадок в пробирке ресуспендируют в лизисном буфере, аликвотируют и хранят при -80°C до использования.
Анализ глюкозилцерамидсинтазы
Для определения ингибирования глюкозилцерамидсинтазы, субстраты при 2x от их Km (флуоресцентный церамид и UDP-глюкоза, 3 мкМ и 4 мкМ, соответственно) и микросомы (1:50 разведение) объединяют 1:1 и инкубируют при комнатной температуре в течение 1 часа в темноте на планшетном шейкере. Реакционную смесь останавливают добавлением 150 мкл 100 мкМ C8-церамида в 50% водн. изопропаноле; 10 мкл конечной смеси анализируют на ВЭЖХ (с датчиком флуоресценции). Подвижной фазой является 1% муравьиная кислота, добавленная в 81% метанол/19% воду со скоростью потока 0,5 мл/мин. Флуоресценцию определяют с λex= 470 нм и λem= 530 нм. В этих условиях, NBD-C6-GluCer имеет время удержания примерно 1,7 мин, и NBD-C6-Cer элюируется из колонки через примерно 2,1 мин. Оба пика отделяют друг от друга и базовой линией, и интегрируют автоматически с применением программы ВЭЖХ. Долю превращения субстрата в продукт применяют в качестве данных для тестирования ингибитора.
GM3 флуоресцентно-связанный ферментный анализ (FLISA):
Это фенотипический анализ, который измеряет экспрессию GM3 в клетках B16 меланомы мыши или C32 меланомы человека после обработки тестируемыми соединениями. Экспрессия GM3 на клеточной поверхности определяется по флуоресценции, опосредованной антителом.
Соединения разводят в середе и высевают в 384-луночные планшеты в ДМСО. Клетки B16 и C32 анализируют при плотности 20000 клеток/мл и 62500 клеток/мл, соответственно, на лунку. Каждая кривая фильтрации содержит 10 точек, которые анализируют дважды при каждом прогоне теста. Планшеты инкубируют в течение 48 часов при 37°C, 5% CO2 и затем промывают один раз TBS. Анти-GM3 антитело добавляют в каждую лунку, и затем планшеты инкубируют в течение еще одного часа при комнатной температуре. Планшеты затем дважды промывают и инкубируют в течение еще часа с меченым вторичным антителом. После конечной инкубации, планшеты промывают дважды, и флуоресценцию при λex =D640/20 нм и λem =657 нм определяют на флуоресцентном ридере.
Результаты анализа
Результаты индивидуальных анализов некоторых типовых соединений в этих анализах представлены в таблице ниже. Результаты микросомальных анализов показывают как «GCS IC50», которая является концентрацией соединения, вызывающей 50% ингибирование активности глюкозилцерамидсинтазы. Результаты клеточных анализов показывают как «GM3 B16 IC50» или «GM3 C32 IC50» для анализа B16 и анализа C32, соответственно. Эти значения представляют собой концентрацию соединения, вызывающую 50% ингибирование экспрессии GM3 на поверхности клетки.
Эти сравнительные результаты демонстрируют, что соединения согласно настоящему описанию обладают сопоставимой активностью in vitro в качестве ингибиторов GCS и, как результат, ожидается, что они продемонстрируют аналогичные преимущества in vivo.
Пример 5: Клиническое исследование Соединения 2 у пациентов с GD-3
Начинают 156-недельное, открытое, мультинациональное, в несколько частей исследование безопасности, переносимости, фармакокинетики, фармакодинамики и поисковых показателе эффективности Соединения 2 в комбинации с имиглюцеразой у взрослых пациентов с болезнью Гоше 3 типа, стабилизированной имиглюцеразой.
Пациенты в возрасте 18 лет и старше с клиническим диагнозом GD3 и документально подтвержденным дефицитом активности кислой бета-глюкозидазы, получавшие лечение ERT в течение, по меньшей мере, 3 лет и имиглюцеразу (Церезим) в стабильной ежемесячной дозе в течение, по меньше мере, 6 месяцев до включения, были включены в исследование. Пациенты должны достичь следующих терапевтических целей GD: уровень гемоглобина ≥11,0 г/дл для женщин и ≥12,0 г/дл для мужчин; количество тромбоцитов ≥100000/мм3; объем селезенки <10 кратно нормальному (MN) или тотальная спленэктомия (при условии, что спленэктомия произошла >3 лет до рандомизации); объем печени <1,5 MN; и отсутствие криза костей, и отсутствие симптоматических заболеваний костей, таких как боль в костях, связанная с остеонекрозом и/или патологическими переломами в течение последнего года. Пациенты должны иметь GD3 с глазодвигательными нарушениями (надъядерный паралич взора), характеризующимися аномалией горизонтального скачкообразного движения.
(A) промежуточный анализ на 26 неделе
Промежуточный анализ был проведен, когда 5 пациентов завершили 26 недель одновременного лечения (1) имиглюцеразой (Cerezyme от Sanofi Genzyme) в соответствии с установленной схемой для каждого пациента и (2) соединением 2, вводимым перорально в дозе 15 мг/сутки в разовой дозе. В ходе исследования пациентов оценивают на безопасность и переносимость, CSF и биомаркеры плазмы (глюкозилцерамид, GL-1; глюкозилсфингозин, лизо-GL1), фармакокинетику, маркеры системного заболевания (объем селезенки и печени, измеренный с помощью магнитно-резонансной томографии (МРТ), количество тромбоцитов, уровень гемоглобина), признаки интерстициального заболевания легких (компьютерная томография легких высокого разрешения (КТ)) и горизонтальное скачкообразное движение глаз.
Исходно у четырех пациентов было неврологическое поражение легкой степени, и один имел умеренное неврологическое поражение, как было измерено с помощью модифицированного инструмента оценки тяжести (mSST; Davies, et al., 2011). У всех пациентов были обнаружены признаки интерстициального заболевания легких на основании КТ грудной клетки. У одного пациента была анемия, о чем свидетельствует уровень гемоглобина в плазме 10,6 г/дл.
Все пациенты не сообщили о серьезных или длительных нежелательных явлениях, вызванных лечением. Наиболее частыми событиями были головная боль и боль в спине, они считались легкими или умеренными и временными по продолжительности (вероятно, связанными с люмбальной пункцией, выполненной для тестирования CSF).
Было обнаружено, что соединение 2 эффективно преодолевает гематоэнцефалический барьер у всех пациентов, как показано в таблице ниже:
Более высокая вариабельность CSF концентрации соединения 2, наблюдаемая на 26 неделе, объясняется уменьшением воздействия на одного из пациентов (Пациент 5) на 12-26 неделе по неизвестным причинам. Пациент 1 был исключен из определения CSF на 26 неделе из-за ошибки при сборе образца.
Было обнаружено значительное улучшение биомаркеров в плазме и CSF для GD-3 через 26 недель. Исходно, средняя (± СО) концентрация GL-1 в CSF составляло 7,1 ± 2,8 нг/мл (диапазон 4,4-11,1 нг/мл), в то время как средняя (± СО) концентрация лизо-GL-1 в CSF составила 39,3 ± 22,9 пг/мл (диапазон 20,1-67,6 пг/мл). Для сравнения: концентрация GL-1 в CSF здорового человека составляет 4,5-5,9 нг/мл, а концентрация лизо-GL-1 в CSF составляет менее 5,0 пг/мл. Через 4 недели и через 26 недель было найдено следующее индивидуальное снижение биомаркеров CSF (показано как доля снижения от исходной концентрации CSF):
Тяжесть интерстициального заболевания легких характеризуют долей объема легкого, пораженного ILD, по данным КТ высокого разрешения в четырех областях легких (дуга аорты, киль, нижняя зона L3, нижняя зона L4). Пациентов ранжируют как имеющих тяжелое ILD (поражение 51-100% объема легких), умеренное ILD (поражение 26-50% объема легких), легкое ILD (поражение 1-25% объема легких) или нормальное состояние (0% объема легких, показывающего ILD). Все пациенты имели исходное ILD, и у 4 из 5 пациентов наблюдался регресс ILD через 26 недель лечения (пациент 5 показал незначительное прогрессирование ILD):
У всех пациентов системного ухудшения не выявлено. Два пациента показали уменьшение объема селезенки на 10% и более. Клинически значимых изменений уровня гемоглобина не было. В среднем количество тромбоцитов выросло на 17% от исходного уровня до 26 недель, и три пациента с самым низким исходным уровнем тромбоцитов показали индивидуальное увеличение через 26 недель 23-42%. Индивидуальные данные пациентов по количеству тромбоцитов показаны в таблице ниже (показано как 109 тромбоцитов/л):
Количественную оценку горизонтальных и вертикальных скачкообразных движений глаз (HSEM и VSEM, соответственно) проводят для всех 5 пациентов. У пяти пациентов, средняя пиковая скорость (PV) горизонтальных 15° быстрых скачкообразных движений вправо составляет 50,8 °/с (±8,1 °/с) на исходном уровне, и 47,5 °/мин (±12,6 °/с) на 26 неделе; и средняя PV горизонтальных 15° быстрых скачкообразных движений влево составляет 44,7 °/с (±17,9 °/с) на исходном уровне и 32,3 °/с (±15,9 °/с) на 26 неделе. Более медленная скорость подразумевает более значительную степень неврологического ухудшения. Средняя PV горизонтальных 30° быстрых скачкообразных движений вправо составляет 77,7 °/с (±16,4 °/с) на исходном уровне и 68,1 °/мин (±24,7 °/с) на 26 неделе; и средняя PV горизонтальных 30° быстрых скачкообразных движений влево составляет 58,7 °/с (±21,5 °/с) на исходном уровне и 49,9 °/с (±8,5 °/с) на 26 неделе. Нормальный диапазон для 15° и 30° для горизонтальных быстрых скачкообразных движений ранее был описан как >200 °/с и >400 °/с (Bremova-Ertl et al, 2018). Кратко, никаких клинически значимых изменений HSEM не наблюдалось в течение периода лечения 26 недель. Как и для HSEM, измерения VSEM были стабильными между исходным уровнем и 26 неделей.
Четыре поисковых биомаркера были количественно определены в плазме, сыворотке и/или CSF пациентов с GD3 на 4 неделе и 26 неделе: хитотриозидазу (CHITO; фермент, который, как известно, повышен у пациентов с GD) измеряют в CSF и сыворотке; GM3 (уровень гликосфинголипидного маркера, о котором известно, что он повышен у пациентов с GD) измеряют в CSF и плазме; гликопротеиновый не метастатический белок меланомы B (GPNMB; по сообщениям, биомаркер невропатической GD3) измеряют CSF. Результаты показаны в таблице ниже как доля изменения от базового уровня на 4 неделе и 26 неделе для каждого параметра:
(B) Промежуточный анализ на 52 неделе
Второй промежуточный анализ проводят, когда первые 6 пациентов достигают 52 недели лечения, как описано выше в разделе (A). Этот анализ включает пациентов 1-5, как описано в разделе (A), а также нового пациента 6. Все шесть пациентов имели гомозиготный фенотип Гоше L444P (1448T/C).
На 52 неделе все пациенты остаются включенными в исследование. Всего было зарегистрировано 30 нежелательных явлений, возникших в результате лечения у шести пациентов, все из которых имели легкую или среднюю степень тяжести, и ни одно из них не считается связанным с лечением соединением 2 или имиглюцеразой. В основном это были головная боль и боль в спине, вероятно, связанная с люмбальной пункцией.
Анализ концентраций соединения 2 в плазме и CSF показывает в значительной степени сопоставимые значения со значениями, полученными на 26 неделе. Однако пациент 5 имеет примерно на 50% более низкую концентрацию соединения 2 в плазме и CSF на 26 неделе и не определяемую концентрацию на 52 неделе. Считается, что это связано либо с не соблюдением требований, либо с ошибками дозирования, поэтому анализ повторяют без данных пациента 5 для недель 26 и 52. Эти данные подтверждают вывод о том, что устойчивая концентрация соединения 2 достигается в плазме и CSF на или до 4 недели:
На 52 неделе, данные также показали устойчивые значительные улучшения биомаркеров в плазме и CSF для GD-3. Результаты аналогичны тем, которые получены через 26 недель. Для всех шести GD3 пациентов, концентрации GL-1 и lyso-GL-1 в плазме и CSF были следующие:
Таким образом, на 52 неделе, по сравнению с исходным уровнем, концентрации в плазме и CSF изменились следующим образом:
Кроме того, поисковые биомаркеры количественно определяют в CSF пациентов с GD3: церамид (предшественник GL-1), хитотриозидаза (CHITO), GM3 и GPNMB. Через 52 недели лечения, существенных изменений CSF концентраций церамида, CHITO или GPNMB не наблюдалось. Четыре из шести пациентов имели измеримые концентрации GM3 в CSF на исходном уровне, и у каждого из этих пациентов был неопределяемый GM3 в CSF через 4 недели, 26 недель и 52 недели.
Через 52 недели проводят количественную оценку горизонтальных и вертикальных саккадических движений глаз у всех шести пациенток аналогичным образом, как описано в разделе (A). Однако было установлено, что методология, используемая для учета шума (например, вызванного морганием или движениями головы), могла внести систематическую ошибку в результаты. Поэтому способ учета шума был изменен, и был разработан набор критериев контроля для оценки достоверности наборов данных, полученных устройством для считывания движений глаз. При переоценке данных саккадических движений глаз за 26 недель и при оценке данных за 52 недели было обнаружено, что уровень шума был слишком высоким, чтобы можно было сделать какие-либо выводы из данных.
Кроме того, через 52 недели 5 из 6 пациентов показали улучшение атаксии. Степень атаксии на исходном уровне и на протяжении всего исследования оценивают по шкале оценки и ранжированию атаксии (SARA; Schmitz-Hübsch et al. [2006]), которая оценивает восемь отдельных признаков мозжечковой атаксии по шкале от 0 до 40. Восемь признаков: походка, фаза опоры, сидение, нарушение речи, слежение за пальцем, тест «нос-палец», быстрое попеременное движение рук и пяточно-коленная проба. Результаты оценки атаксии по SARA для всех шести пациентов представлены в таблице ниже:
Как показано в таблице, пять из шести пациентов имеют легкую атаксию на исходном уровне, при этом средний суммарный балл по SARA составляет 2,8 (СО=1,2). Наиболее частым дефицитом на исходном уровне являются нарушения походки. Исключая пациента 5 из-за низкого уровня воздействия соединения 2 на этого пациента и по существу нормальной исходной оценки атаксии у пациента (только 0,5), 4 из 5 пациентов демонстрируют улучшение атаксии на 52 неделе (среднее улучшение = -0,9; СО=3,2). Пациент 4 демонстрирует увеличение оценки атаксии, при этом оценка на исходном уровне была 3, а на 52 неделе, 7,5. Следует отметить, что это очевидное ухудшение почти полностью вызвано изменением параметра оценки «фазы опоры» (оценка фазы опоры на исходном уровне и 26 неделе=1; оценка на 52 неделе=5) и что пациент жалуется на боль в левом колене во время исследования. Кроме того, перед исследованием субъект повредил большой палец левой ноги; через 11 дней после исследования эта травма была разрешена. За исключением этого резко выделяющегося эффекта пациента 4, лечение Соединением 2 привело к значительному снижению средней оценки по SARA на 26 неделе, которая была далее немного улучшена на 52 неделе.
Тест построения маршрута (TMT) используют для оценки когнитивных функций у пациентов. TMT является одним из наиболее широко используемых нейропсихологических тестов и входит в состав большинства панелей тестов. TMT является диагностическим инструментом для оценки общего интеллекта и когнитивных дисфункций (Tombaugh et al. [2004]; Cavaco et al. [2013]). В части A TMT, субъектов просят соединить группу чисел в порядке возрастания. Эта задача представляет собой комбинацию визуального поиска и общей визуальной и моторной скорости обработки. Часть B представляет собой последовательность, в которой чередуются цифры и буквы. Субъекты должны активно переключаться между обеими категориями, соединяя их в возрастающем, но чередующемся порядке. Следовательно, считается, что эта задача включает компонент исполнительной функции, поскольку субъект должен активно переключаться между категориями при соединении символов (MacPherson et al. [2017]).
TMT-A оценивает в основном скорость восприятия и психомоторную скорость. TMT-B более конкретно оценивает умственную гибкость и способность к переключению. Оценку TMT B минус TMT А используют для устранения вариабельности, относящейся к компонентам графомоторного и визуального сканирования TMT A. Полученная оценка отражает уникальные требования к задаче TMT B.
В исследовании нормативных данных для TMT A и TMT B у проживающих вне дома престарелых индивидуумов в возрасте 18-89 лет (n=911), средние (SD) значения в группе 18-24 лет (n=155) составляли 22,9 с (6,9) для TMT A и 49 с (12,7) для TMT B (Tombauch et al. [2004]). Наоборот, среднее время, затраченное для завершения испытания A и испытания B для пациентов этого исследования составляло 67,8 с (SD=60,3 с) и 193,8 с (SD=197,0), соответственно. На исходном уровне, средняя разница по времени, затраченном для завершения испытания B минус испытание A составляла 126,0 с (SD=142,9 с). Это показывает, что пациенты с GD-3 в этом исследовании демонстрируют некоторую степень когнитивной дисфункции на исходном уровне.
На 52 неделе среднее время, затраченное для завершения испытания A, составило 56,5 с (SD=55,2 с), а для испытания B - 122,7 с (SD=91,8 с). Четыре из шести пациентов продемонстрировали сокращение времени, затраченного для завершения испытания А, и шесть из шести продемонстрировали сокращение времени, затраченного для завершения испытания B. За исключением Пациента 5 из-за низкого уровня воздействия Соединения 2 у этого пациента, четыре из пяти пациентов продемонстрировали снижение TMT А, и пять из пяти пациентов продемонстрировали уменьшение TMT-B.
На 52 неделе у 5 из 6 пациентов наблюдают сокращение времени (TMT B - TMT A). Индивидуальные результаты показаны в таблице ниже.
На 52 неделе, средняя разница во времени, затраченном для завершения испытания B минус испытание A, составила 66,2 с (SD=54,3). За исключением пациента 5, четыре из пяти пациентов продемонстрировали улучшение в испытании B минус испытание A на 52 неделе, со средним улучшением -71,4 с (-31,6%) (SD 99,3 с (37,6%)).
Неврологическую функцию дополнительно оценивают с помощью функциональной магнитно-резонансной томографии (фМРТ). Пациент 2 был исключен, поскольку на сеансе на 52 неделе данные фМРТ не собирали. Сеансы фМРТ в состоянии покоя проводят в посещения на исходном уровне, при скрининге, 26 неделе и 52 неделе. Оценки связности от четырех субъектов (пациенты 1, 3, 4 и 5) вводят в анализ второго уровня как «соответствующая» группа. Пациент 5 был изолирован из-за вероятного несоблюдения режима приема исследуемого препарата, описанного выше. Анализы проводят, как описано в другом месте (Smith et al. [2009]).
Было обнаружено, что соответствующие субъекты демонстрируют повышенную связь между более широко распределенным набором областей мозга, чем субъекты, не соответствующие требованиям, с увеличением силы между задними и передними аспектами как наиболее заметной особенности. На анатомическом уровне соответствующие пациенты демонстрируют широко распространенное и устойчивое усиление связей между затылочно-теменными структурами и лобными, височными и лимбическими мишенями. Изменения связности у пациента 5 были скромнее и ограничены пространственно проксимальными структурами. На функциональном уровне, улучшенная связность между режимом по умолчанию и медиальными фронтальными сетями наблюдается у всех субъектов, кроме пациента 5. Это говорит о том, что сигнал в этих разрозненных сетях становится более связным, так что активность мозга может более эффективно передаваться между когнитивным резервом (задним) и исполнительными функциями высшего порядка (передними). Также очевидно согласованное реципрокное картирование сетей в состоянии покоя (RSN) 2 и 3 («когнитивные функции-язык-ортография» и «когнитивные функции-пространство») в RSN 8 и 9 (исполнительное и левое лобно-теменное). Пространственное распределение изменений связности гораздо более важно для пациента 5, в первую очередь отражая перекрытие медиально-лобной и лобно-теменной сетей. Обе точки зрения предполагают, что пациент, полностью соблюдающий протокол лечения, развил большую согласованность между задними и передними частями мозга, так что весь мозг стал поддающимся эффективной передаче информации. Там, где это очевидно, измененная связность для пациента 5 проявляется в более узком наборе передних отделов мозга и представляет собой менее целостное свидетельство терапевтического эффекта.
Результаты представлены в таблице ниже. Пространственный анализ связи между различными анатомическими областями мозга выполняется для определения коэффициента корреляции для регрессивной средней интенсивности вокселя за один проход. Результаты показывают, что связь между сетью в режиме по умолчанию (в состоянии покоя) и сетью исполнительных функций увеличилась у пациентов 1, 3, 4 и 6, но уменьшилась у пациента 5.
Дополнительно обнаружено, что два пациента показывают снижение объема селезенки на 52 неделе, и средняя концентрация тромбоцитов повышается на 9,3% в среднем (диапазон от -8,2% до +45,3%), все пациенты сохраняют терапевтическую цель более чем 120×109/л количество тромбоцитов. Повышение средней концентрации тромбоцитов в первую очередь управляется повышениями в 3 из 6 пациентов. Клинически значимые изменения уровня гемоглобина отсутствуют.
Пример 6: Фармакокинетика Соединения 2 у здоровых добровольцев-людей
Два клинических исследования в фазе 1 были проведены для оценки фармакокинетики, фармакодинамики, безопасности и переносимости Соединения 2 на здоровых добровольцах-людях в присутствии и отсутствии пищи. Соединение 2 также известно как венглюстат.
Исследование 1
Исследование 1 представляет собой одноцентровое исследование из 2 частей на здоровых взрослых мужчинах-добровольцах. Часть 1 представляет собой двойное слепое рандомизированное плацебо-контролируемое последовательное восходящее исследование однократной дозы Соединения 2 на безопасность, переносимость и ФК. Часть 2 представляет собой открытое, однокогортное, рандомизированное, перекрестное исследование с 2 последовательностями, 2 периодами и 2 видами лечения Соединением 2 для определения ФК с пищей с высоким содержанием жиров и без нее.
В 1 часть исследования включают и рандомизируют 55 здоровых мужчин (плацебо, n=14; дозы 2, 5, 15, 25, 50 и 100 мг, n=6 каждый; доза 150 мг, n=5). Во 2 части приняли участие восемь здоровых мужчин.
В 1 части субъектов произвольно распределяют для получения 2, 5, 15, 25, 50, 100 или 150 мг Соединения 2 (форма L-яблочной соли) или соответствующего плацебо утром первого дня после, по меньшей мере, 10-часового голодания. Во 2 части субъектов произвольно распределяют для получения однократной пероральной дозы 5 мг Соединения 2 либо во время голодания (по меньшей мере, 10 часов до и 4 часов после приема), либо через 30 минут после стандартизированного завтрака с высоким содержанием жиров (~815 ккал). После 7-дневного периода отмывки участников переводят в другое состояние.
В исследовании 1, часть 1, кровь собирают для определения концентраций в плазме Соединения 2 во время введения исследуемого лекарственного средства (0 час) и через 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24, 48, 72 и 96 часов после введения дозы. Образцы мочи собирают для анализа концентраций Соединения 2, начиная с 2 часов до введения исследуемого лекарственного средства и через 48 часов после этого.
В исследовании 1, часть 2, кровь берут для определения концентраций Соединения 2 в плазме через 0, 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24 и 48 часов после введения дозы.
В 1 части было обнаружено, что после однократного перорального дозирования 2-150 мг доз Соединения 2, максимальная концентрация в плазме (Cmax) возникает в медианное время 3-5,5 часов, затем концентрации в плазме начинают экспоненциально снижаться, при средней геометрической t1/2 28,9 часов. Воздействие увеличивается пропорционально дозе во всем диапазоне доз: 75-кратное повышение дозы дает 97,3-, 89,2- и 85,9-кратное повышение значений средней геометрической Cmax, ППКlast и ППКinf, соответственно. Результаты ФК показаны в следующей таблице (ППК= площадь под кривой время-концентрация, либо для последней измеримой концентрации, либо экстраполированная на бесконечность; t1/2=конечный период полувыведения; CL/F=очевидный общий клиренс из плазмы; CV=коэффициент вариаций; СО=стандартное отклонение; tmax=время до Cmax; Vss/F=очевидный объем распределения в состоянии покоя):
Из 2 части было обнаружено, что введение 5 мг дозы при питании с высоким содержанием жиров не оказывает влияния на действие Соединения 2 по сравнению с состоянием голодания. Медианное tmax составляет 6,00 часов независимо от питания или голодания. Средние геометрические отношения при питании/голодании составляют 0,92 и 0,91 для Cmax и ППКlast, соответственно. Внутрисубъектная вариабельность (т.е., питание/голодание) отмечена при менее половины общей субъектной вариабельности.
Исследование 2
Исследование 2 представляет собой одноцентровое, двойное слепое, рандомизированное, плацебо-контролируемое, последовательное восходящее с повторными дозами исследование безопасности, переносимости, ФК и фармакодинамики Соединения 2 у здоровых взрослых добровольцев мужского и женского пола.
В исследование включены и произвольно распределены 36 здоровых взрослых людей (19 мужчин и 17 женщин) (n=9 в каждой группе). Субъекты произвольно распределены для получения один раз в сутки Соединения 2 в дозе 5, 10 или 20 мг (в виде капсул по 5 мг в форме L-яблочной соли) или плацебо в течение 14 дней после, по меньшей мере, 10 часов голодания.
Кровь собирают для определения концентраций в плазме Соединения 2 следующим образом: День 1 в 0, 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12 и 16 часов после дозирования; в дни 2-5, 8, 11 и 13, в 0 ч; в день 14, в 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12 часов после дозирования; в дни 15-17, в 24, 48 и 72 часов, соответственно, после дозирования на 14 день. Образцы мочи собирают для анализа концентраций Соединения 2 в день 1 (0 часов после дозирования) и далее в день 14 через 0-24 часов после дозирования. Фармакодинамические конечные точки (концентрации GL-1, GL-3 и GM3 в плазме) оценивают в дни 1-5, 8, 11, 13 и 14, в 0 часов после дозирования; и в день 15 через 24 часа после дозирования на 14 день.
Было обнаружено, что у субъектов, получающих 5, 10, или 20 мг Соединения 2 один раз в сутки в течение 14 дней, Cmax в плазме возникает в медианное время 2-5 часов после дозирования в дни 1 и 14. Значения Ctrough достигают плато на 5 день 5. Воздействие Соединения 2 повышается близко пропорционально дозе в интервале дозирования 5-20 мг: это 4-кратное повышение дозы дает 3,76- и 3,69-кратное повышение значений средней геометрической Cmax и ППК0-24 на 14 день, соответственно. ФК результаты из исследования 2 суммированы в следующей таблице:
Через 14 доз один раз в сутки Соединения 2, 24-часовая неизмененная фракция выделения с мочой (средняя fe0-24) варьировалась между 26,3% и 33,1% без очевидного соотнесения с дозой. Средняя CLR(0-24) варьируется от 1,49 л/ч до 2,07 л/ч, приблизительно 3,18-3,86-кратно ниже, чем наблюдаемые CL/F в плазме.
GL-1, GL-3 и GM3 в плазме у получателей плацебо остается аналогичной исходному уровню, в то время уровни GL-1 и GM3 в плазме снижаются от исходного уровня в зависимости от времени и дозы в группе 3 дозы Соединения 2, как показано в следующей таблице (точечная оценка соотношений при лечении глюкозилцерамида (GL-1), глоботриаозилцерамида (GL-3) и ганглиозида GM3 (GM3) на 15 день в исследовании с повторным возрастанием дозы):
Максимальное устойчивое действие на GL-1 наблюдают на 11 день в группах 5 и 10 мг и на 8 день в группе 20 мг. Среднее рассчитанное снижение GL-1 по сравнению с исходным уровнем на 15-й день составляет 41,9%, 69,6% и 74,6% в соответствующих группах 5, 10 и 20 мг. Значения GL-1 были ниже нижнего предела количественной оценки (LLOQ) на исходном уровне у 1 реципиента 5 мг соединения 2 и на день 15 у 3, 5 и 9 субъектов в группах 5, 10 и 20 мг доз, соответственно.
Максимальное устойчивое снижение GM3 наблюдают во всех группах дозировки Соединения 2, начиная со дня 13. Средние уровни GM3 в плазме на день 15 составляют 42,7%, 49,4% и 57,8% от исходного уровня для групп 5, 10 и 20 мг доз, соответственно. GM3 был ниже LLOQ на 15 день у 1 и 2 субъектов в группах 10 и 20 мг дозами, соответственно.
GL-3 в плазме также уменьшается со временем во всех группах дозирования Соединения 2, но вариабельные и низкие исходные значения GL-3 по сравнению с LLOQ ограниченными средними расчетными снижениями GL-3. В группах плацебо, 5, 10 и 20 мг доз значения GL-3 были ниже LLOQ у 1, 3, 1 и 6 субъектов, соответственно, на исходном уровне, и у 4, 9, 7 и 9 субъектов, соответственно, на день 15.
Среднее оценочное снижение GL-1 в плазме от исходного уровня (90% CI), связанные с Соединением 2 Ctrough в группах 5, 10 и 20 мг дозы (19,0, 47,5 и 69,9 нг/мл, соответственно) составляет 67,0% (54,4-79,7%), 74,4% (63,7-85,2%) и 76,3% (64,8-87,8%), соответственно.
Заключения
В этих исследованиях, действие Соединения 2 на здоровых субъектов (Cmax и ППК) было близко к дозозависимому при введении однократных доз в диапазоне от 2 до 150 мг или в виде повторных, однократных доз в диапазоне от 5 до 20 мг в течение 14 дней. По сравнению с голоданием, пища с высоким содержанием жиров не влияла на действие у субъектов, получивших однократную дозу 5 мг. При повторных дозах от 5 до 20 мг один раз в сутки устойчивое состояние достигалось в течение 5 дней; на накопление не повлияли ни возраст, ни пол. Фармакодинамически, повторные ежедневные дозы Соединения 2 снижали GL-1 и GM3 в плазме в зависимости от времени и дозы, что согласуется с опосредованным Соединением 2 ингибированием GCS, хотя исходные уровни GL-3 были слишком низкими, чтобы быть полезными в качестве фармакодинамического биомаркера. Дозозависимое снижение GL-1 подтвердило предполагаемый механизм действия Соединения 2: ингибирование образования GL-1 из церамида через GCS.
Во всех исследованиях, профиль безопасности оценивают путем мониторинга нежелательных явлений, возникающих в результате лечения (TEAE), в течение 10 дней после последней дозы исследуемого препарата, включая серьезные нежелательные явления [SAE]), мониторинг ЭКГ, лабораторные показатели и физические обследования.
Ни в одном из исследований не было летальных исходов, SAE, тяжелых TEAE или TEAE, приведших к прекращению исследования.
Ни в одном из исследований не сообщалось о клинически значимых гематологических или биохимических аномалиях. Жизненно важные показатели не показали существенных изменений по сравнению с исходным уровнем ни в одном из исследований. Параметры ЭКГ не показали значимых изменений в исследованиях однократной возрастающей дозы и влияния пищи; в исследовании с множественными возрастающими дозами параметры ЭКГ не изменились статистически значимо по сравнению со средним исходным уровнем по сравнению с плацебо у реципиентов Соединения 2 при любой дозе. Следует понимать, что хотя изобретение было описано в связи с приведенными выше вариантами осуществления, приведенное выше описание и примеры предназначены для иллюстрации, а не для ограничения объема изобретения. Другие аспекты, преимущества и модификации в пределах объема изобретения будут очевидны специалистам в области, к которой относится изобретение.
Пример 7: Клиническое исследование Соединения 2 у пациентов с болезнью Фабри
Способ
Трехлетнее открытое исследование Соединения 2 у молодых пациентов с классической болезнью Фабри было проведено с целью оценки долгосрочной безопасности, фармакодинамики и поисковых показателей эффективности Соединения 2 у взрослых мужчин-пациентов с болезнью Фабри. Одиннадцать субъектов были включены в исследование, и семь субъектов завершили все аспекты исследования. Все субъекты были мужчинами с диагностированной классической болезнью Фабри, подтвержденной генотипом и остаточной активностью альфа-галактозидазы ниже уровней обнаружения (9 из 11 имели нонсенс-мутацию в гене GLA). Все субъекты имели уровни лизо-GL3 в плазме, по меньшей мере, 65 нг/мл и не получали ранее специфического лечения болезни Фабри. Средний возраст субъектов составлял 24 года (диапазон 19-37 лет).
Пациентам вводят суточную пероральную дозу 15 мг Соединения 2. Клиренс отложений GL-3 в коже контролируют путем взятия биопсий через 12, 26, 52 и 156 недель, которые оценивают полуколичественно с помощью световой микроскопии (фокусирующейся на эндотелиальных клетках капилляров кожи). Каждый образец был независимо оценен 3 патологами на наличие включений GL-3 по четырехбалльной шкале и ранжированы как 0 (нет/след), 1 (легкие), 2 (умеренные) или 3 (тяжелые) в соответствии с Eng et al., N. Engl. J. Med. 345:9-16 (2001). Единая оценка для каждого пациента на момент времени получена путем взятия оценок, выставленных большинством из трех патологов. Если не удалось получить оценку большинства, используют среднюю оценку (что приводит к некоторым дробным оценкам). Образцы плазмы также были проанализированы на GL-3, лизо-GL-3, GL-1 и GM3 на исходном уровне и неделях 12, 26, 52 и 156. Оценки боли и абдоминальных симптомов анализируют на исходном уровне и неделях 12, 26, 52, 104 и 156 с использованием протокола подсчета оценок SF-36.
Пациентов оценивают с помощью анкеты Short Form-36 (SF-36) во время многочисленных посещений от исходного уровня до недели 156. Это анкета из 36 пунктов, используемая для измерения 8 различных аспектов здоровья (жизнеспособность, физическое функционирование, телесная боль, общее восприятие здоровья, физическое ролевое функционирование, эмоциональное ролевое функционирование, социальное ролевое функционирование и психическое здоровье). Оценка по каждому из восьми аспектов варьируется от 0 (максимальная инвалидность) до 100 (нет инвалидности), и, таким образом, более высокие оценки указывают на хорошее состояние здоровья. Кроме того, желудочно-кишечные симптомы, включая боль в животе, вздутие живота и движения кишечника, оценивают с использованием модифицированной версии системы оценки тяжести воспаления кишечника. Конкретные вопросы, задаваемые в рамках этих оценок, включают: (1) страдает ли пациент от боли в животе в течение последних десяти дней, (2) какова была тяжесть боли в животе за последние десять дней, используя оценку по шкале от 0 (отсутствие боли) до 100 (очень сильная боль) и (3) сколько дней в течение последних десяти дней пациент испытывал боль в животе.
Результаты
На 156 неделе у пяти пациентов наблюдают снижение показателя GL-3 кожи на 1 балл, у двух пациентов было полное удаление включений GL-3, у одного пациента не было изменений и у одного пациента не было образцов. В течение 156-недельного исследования, средний уровень GL-1 в плазме снизился на 69%, средний уровень GM3 в плазме снизился на 60%, средний уровень GL-3 в плазме снизился на 77%, а средний уровень lyso-GL3 в плазме снизился на 52%. GL-1 и GM3 в плазме показали очень быстрое снижение в течение первых 2-4 недель лечения. Все четыре показателя показали устойчивое сохранение сниженной плазменной нагрузки, которая в значительной степени стабилизировалась к 52 неделе.
Данные по плазме и моче сведены в таблице ниже:
(n=8)
(n=7)
(n=7)
(n=7)
Эти результаты демонстрируют, что Соединение 2, вводимое в дозе 15 мкг/сутки, последовательно снижает уровни GL-1, лизо-GL-1 и GM3 в организме в целом прогрессивным образом.
Кроме того, для сравнения были проанализированы данные 3 фазы ранее завершенного плацебо-контролируемого исследования агалсидазы бета (Fabrazyme) для сравнения (см. Eng et al., N. Eng. J. Med., 345:9 (2001). Для сравнения с агалсидазой бета, историческая контрольная группа включает пациентов, леченных агалсидазой бета на 3 фазе испытания, и изменение GL-3 в плазме сравнивают в нескольких временных точках до трех лет. Критерии включения и исходные характеристики были схожими между двумя исследованиями. При усиления сравнения, пациентов, получавших Соединение 2, сопоставляют с пациентами 3 фазы исследования на основе оценок предрасположенности с использованием исходных переменных возраста, GL-3 в плазме, пола, UPCR (<500 мг/г к с 500-1000 мг/г к с >1000 мг/г). г) и eGFR (<80 к ≥ 80 мл/мин/1,73 м2). 11 пациентов, получавших Соединение 2, сопоставляют с 19 пациентами для сравнения с плацебо и с 28 пациентами для сравнения с агалсидазой бета. Все пациенты во всех трех группах были мужчинами и имели повышенный уровень GL-3 в плазме, UPCR <500 мг/г и eGFR ≥80 мл/ мин/1,73 м2. Средний возраст во всех трех группах был аналогичным. Сравнение показывает, что лечение Соединением 2 в течение 26 недель привело к значительному снижению GL-3 в плазме по сравнению с плацебо, -3,62 мкг/мл по сравнению с -1,06 мкг/мл (P <0,0001). Хотя лечение Соединением 2 дает аналогичное снижение GL-3 в плазме на 52 неделе по сравнению с агалсидазой бета, на 104 неделе и на 156 неделе, снижение GL-3 в плазме при лечении Соединением было значительно больше (p=0,0351 на 104 неделе; p=0,0081 на 156 неделе). Уровни GL-3 в плазме через 156 недели составляют 1,90 мкг/мл для пациентов, леченных Соединением 2, по сравнению с 4,44 мкг/мл для пациентов, леченных агалсидазой бета.
Подробные результаты оценки включения GL-3 в кожу показаны в таблицах ниже (оценка 0 означает отсутствие включений GL-3):
В дополнение к подсчету включений GL-3 в коже с помощью световой микроскопии, долю объема цитоплазмы эндотелиальных клеток, занятая включениями GL-3, оценивают с использованием точечного подсчета электронных микроскопических изображений маскированным ридером. Изображения, по меньшей мере, 50 капилляров поверхностных эндотелиальных клеток получают с помощью электронной микроскопии при 7500х увеличении. Двухфакторные t-тесты используют для оценки различий между исходными значениями и значениями после лечения в каждый момент времени. Результаты представлены в таблице ниже:
Эти результаты демонстрируют, что Соединение 2, вводимое в дозе 15 мг/сутки, последовательно снижает уровни включений GL-3 в коже в целом прогрессивным образом. Результаты в целом были более выраженными для эндотелия поверхностных сосудов по сравнению с эндотелиальными клетками глубоких сосудов и другими тканями кожи.
Семь из девяти пациентов улучшили общие показатели боли в теле на 26 неделе (SF-36), в то время как три из шести пациентов улучшили общие показатели боли в теле на 156 неделе (SF-36). Среди пациентов с желудочно-кишечными болями на исходном уровне, тяжесть боли (боли в животе) снизилась у четырех из пяти на 26 неделе и у четырех из четырех на 156 неделе. Количество дней с желудочно-кишечными болями уменьшилось у пяти из пяти пациентов на 26 неделе и у трех из четырех пациентов на 156 неделе.
Подробные результаты измерения боли в животе показаны в таблицах ниже.
Эти результаты демонстрируют, что Соединение 2, вводимое в дозе 15 мг/сутки, последовательно уменьшает боль в животе и дискомфорт в теле в целом прогрессивным образом.
Кроме того, если признаки или аспекты изобретения описаны в терминах групп Маркуша, специалисты в данной области техники поймут, что изобретение таким же образом описано в терминах любого отдельного члена или подгруппы членов группы Маркуша.
Все публикации, заявки на патенты, патенты и другие ссылки, упомянутые в настоящем документе, прямо включены в качестве ссылки во всей своей полноте в той же степени, как если бы каждая была включена в качестве ссылки по отдельности. В случае противоречия преимущественную силу имеет настоящее описание, включая определения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ СИМПТОМОВ И НАРУШЕНИЙ, АССОЦИИРОВАННЫХ С ЛИЗОСОМНЫМИ БОЛЕЗНЯМИ НАКОПЛЕНИЯ | 2020 |
|
RU2824599C2 |
| СПОСОБЫ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ СИМПТОМОВ, АССОЦИИРОВАННЫХ С ЛИЗОСОМНЫМИ БОЛЕЗНЯМИ НАКОПЛЕНИЯ | 2021 |
|
RU2839414C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| ИНГИБИТОРЫ ГЛЮКОЗИЛЦЕРАМИД-СИНТАЗЫ | 2012 |
|
RU2645675C2 |
| АДЕНОЗИНОВОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2824528C2 |
| ТЕТРАЗАМЕЩЕННЫЕ АЛКЕНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2733741C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ПОЛИМОРФНАЯ ФОРМА ДИАРИЛЬНОГО МАКРОЦИКЛА | 2016 |
|
RU2765181C2 |
| ПРОЛЕКАРСТВЕННЫЕ МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО ПУТИ СТРЕССА | 2019 |
|
RU2824500C2 |
| ПРОИЗВОДНЫЕ АМИДА В КАЧЕСТВЕ БЛОКАТОРОВ Nav1.7 И Nav1.8 | 2018 |
|
RU2768149C2 |
Изобретение относится к области фармацевтической промышленности, а именно к способу лечения или профилактики боли. Способ лечения или профилактики боли у субъекта-человека, нуждающегося в этом, где человек имеет болезнь Фарби и где боль выбрана из невропатической боли, желудочно-кишечной боли и периферической невропатии, включающий введение субъекту эффективного количества соединения формулы (I)
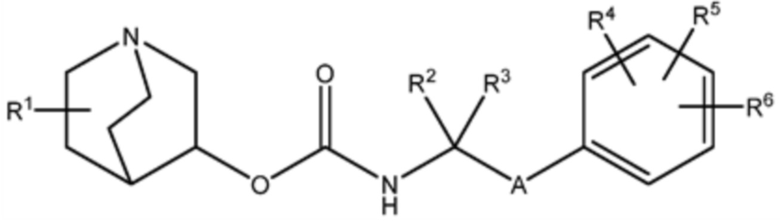
или его фармацевтически приемлемой соли, или пролекарства, где R1 выбирают из водорода, галогена, циано, нитро, гидрокси, тио, амино, C1-6-алкила, C2-6-алкенила, C2-6-алкинил, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанный алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещен одной или несколькими группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино; R2 и R3 независимо выбирают из C1-3-алкила, необязательно замещенного одним или несколькими галогенами, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими галогенами; R4, R5 и R6, каждый, независимо выбирают из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одним или несколькими группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси; и A является 5- или 6-членной арильной или гетеоарильной группой, необязательно замещенной 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси или C1-6-алкила. Использование изобретения обеспечивает лечение или профилактику боли, включая невропатическую боль, желудочно-кишечную боль и периферическую невропатию, у субъекта-человека, нуждающегося в этом, где человек имеет болезнь Фабри. 14 з.п. ф-лы, 19 табл., 7 пр.
1. Способ лечения или профилактики боли у субъекта-человека, нуждающегося в этом, где человек имеет болезнь Фабри и где боль выбрана из невропатической боли, желудочно-кишечной боли и периферической невропатии,
включающий введение субъекту эффективного количества соединения формулы (I)
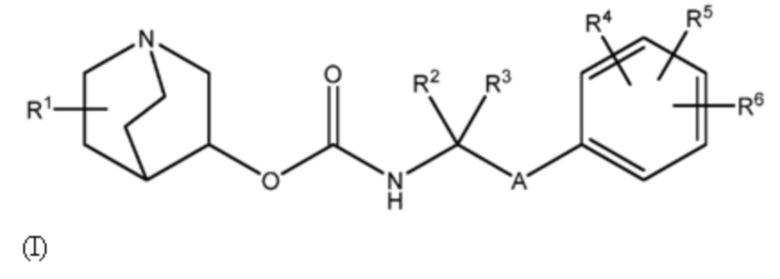
или его фармацевтически приемлемой соли, или пролекарства, где
R1 выбирают из водорода, галогена, циано, нитро, гидрокси, тио, амино, C1-6-алкила, C2-6-алкенила, C2-6-алкинила, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанный алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещен одной или несколькими группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
R2 и R3 независимо выбирают из C1-3-алкила, необязательно замещенного одним или несколькими галогенами, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими галогенами;
R4, R5 и R6, каждый, независимо выбирают из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одним или более группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси; и
A является 5- или 6-членной арильной или гетеоарильной группой, необязательно замещенной 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси или C1-6-алкила.
2. Способ по п. 1, где
i) R1 выбирают из водорода, фтора, метила и этила, где указанный метил или этил необязательно замещен 1 или 2 группами, выбранными из галогена, гидрокси, тио или амино;
ii) R2 и R3, каждый, независимо выбирают из метильной и этильной групп, необязательно замещенных одним или несколькими атомами фтора;
iii) R4 выбирают из галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одним или более группами, выбранными из галогена и C1-3-алкилокси;
iv) R5 и R6, каждый, является водородом; и/или
v) А представляет собой фенил, необязательно замещенный 1,2 или 3 группами независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси и C1-6-алкила или 5-членной гетероарильной группы, которая содержит 1 или 2 гетероатома, выбранных из N и S.
3. Способ по п. 1 или 2, где R4 является фтором или 2-метоксиэтокси и R5 и R6 являются водородом и/или
где R4 расположен в 4 положении фенильного кольца, к которому он присоединен.
4. Способ по любому из пп. 1-3, где A является фенилом, необязательно замещенным 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси и C1-6-алкила, и где две группы, присоединенные к заместителю А, расположены в 1, 3 или 1, 4 положении друг к другу.
5. Способ по любому из пп. 1-3, где A является 5-членной гетеоарильной группой, которая содержит 1 или 2 гетероатома, выбранных из N и S, и где две группы, присоединенные к заместителю А, расположены в 1,3-положении друг к другу.
6. Способ по любому из пп. 1-5, где указанное соединение представлено следующей структурой формулы (II), (III), (IV), (V), (VI), (VII), (VIII), (IX) или (XI)
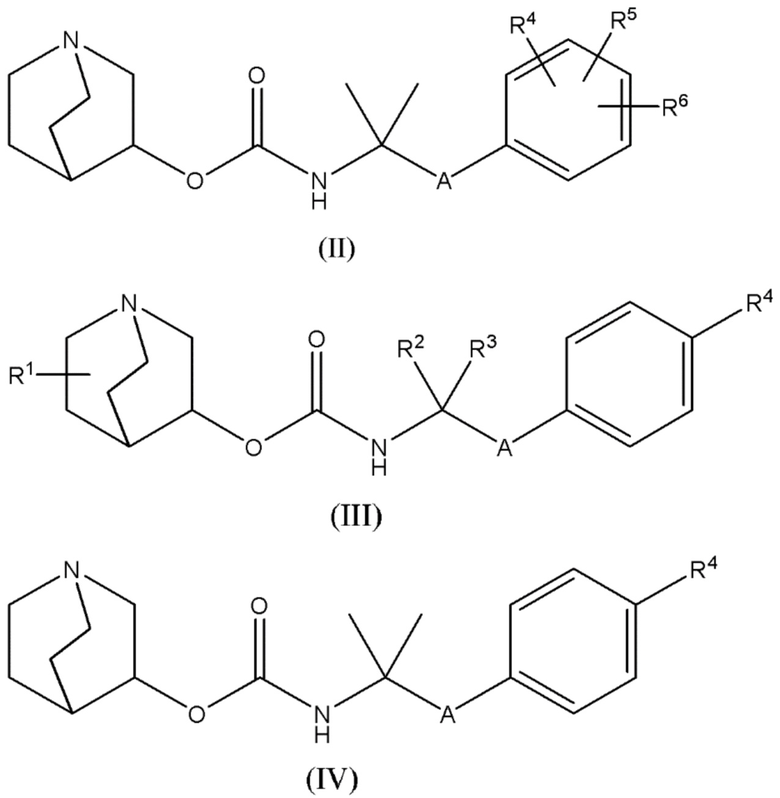
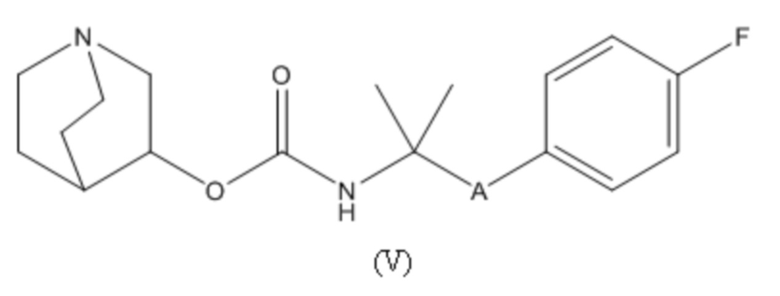
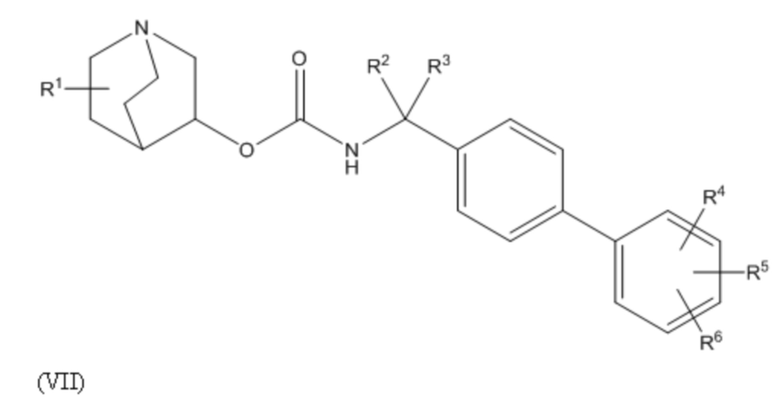
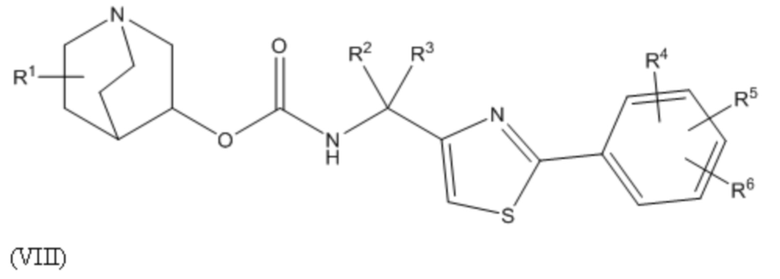
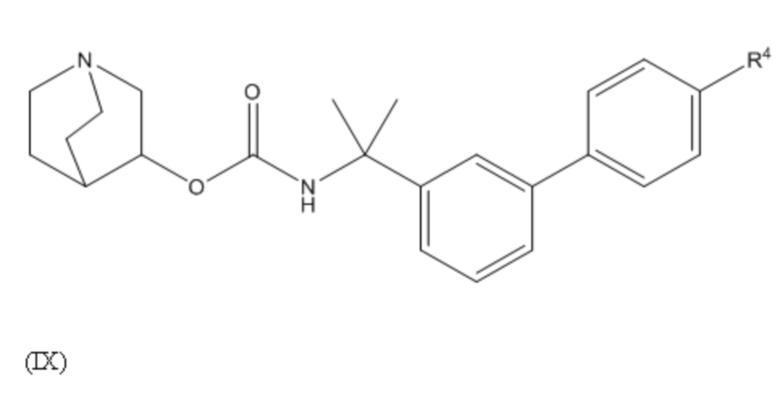
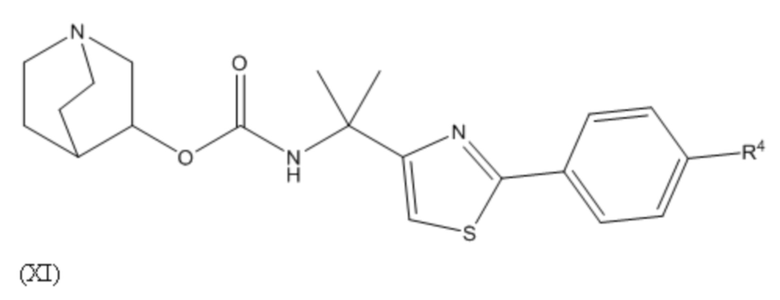
или его фармацевтически приемлемая соль или пролекарство.
7. Способ по п. 6, где указанным соединением является соединение формулы (IX) или (XI)
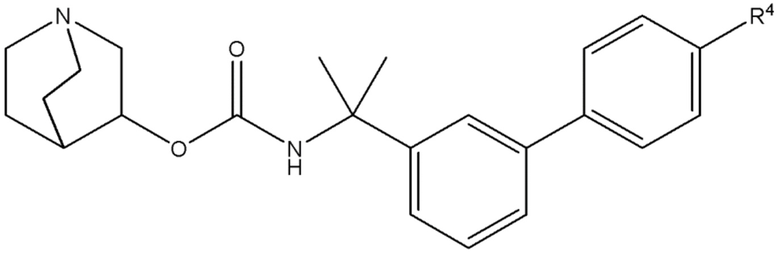
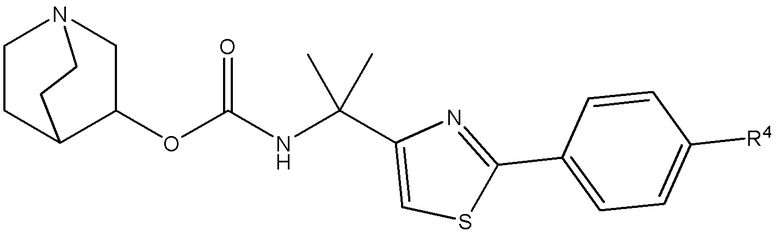
или его фармацевтически приемлемая соль, или пролекарство и
где R4 является фтором.
8. Способ по п. 1, где указанное соединение выбирают из: хинуклидин-3-ил(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамата; (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата; (S)-хинуклидин-3-ил (2-(4’-(2-метоксиэтокси)-[1,1’-бифенил]-4-ил)пропан-2-ил)карбамата; и их фармацевтически приемлемых солей и пролекарств; и (S)-хинуклидин-3-ил (2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата в форме малатной соли.
9. Способ по любому из пп. 1-8, где боль является желудочно-кишечной болью или невропатической болью.
10. Способ по любому из пп. 1-9, где способ дополнительно включает одновременное введение второго активного агента, например второго соединения, способного лечить или предотвращать боль у пациента, нуждающегося в этом.
11. Способ по любому из пп. 1-10, где субъект имеет заметное накопление GL-3 в тканях.
12. Способ по любому из пп. 1-11, где способ приводит к уменьшению тяжести боли в течение 26 недель, или 52 недель, или 156 недель.
13. Способ по любому из пп. 1-12, где субъект проходит одновременное лечение ферментозамещающей терапией (ERT) с применением агалзидазы альфа или агалзидазы бета или с применением глюкоцереброзидазы или ингибитора альфа-галактозидазы.
14. Способ по любому из пп. 1-13, где указанное соединение и/или его фармацевтически приемлемую соль или пролекарство вводят системно.
15. Способ по любому из пп. 1-14, где субъекту вводят суточную дозу от 1 до 150 мг указанного соединения.
| US 2018036295 A1, 08.02.2018 | |||
| US 2016207933 A1, 21.07.2016 | |||
| A | |||
| METHA et al | |||
| Fabry disease: a review of current management strategies | |||
| QJM, 2010, 9, p | |||
| ПРИБОР ДЛЯ ОТОПЛЕНИЯ НЕФТЬЮ | 1923 |
|
SU641A1 |
| ФИРСОВ К.В | |||
| и др | |||
| Неврологические проявления при болезни Фабри | |||
| Журнал неврологии и психиатрии им | |||
| С.С | |||
| Корсакова, 2016, 116(9), с.98-105 | |||
| R | |||
| MESSIKH et al | |||
| Botulinum toxin in disabling | |||

