Настоящее изобретение относится к способам лечения или предупреждения конкретных симптомов и нарушений, которые ассоциированы, например, с лизосомными болезнями накопления, с применением хинуклидиновых соединений формулы (I), необязательно в комбинации с ферментозаместительной терапией. Способы обеспечивают улучшение нейронных связей в головном мозге субъекта, увеличение объема ткани головного мозга или предупреждение или задержку потери объема ткани головного мозга у субъекта. Настоящее изобретение также относится к способам осуществления мониторинга прогрессирования или регрессирования неврологического нарушения или оценивания начала проявления неврологического нарушения, ассоциированного с лизосомной болезнью накопления, в которых измеряют объем ткани головного мозга субъекта.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Лизосомные болезни накопления
Лизосомные болезни накопления (LSD) представляют собой группу из приблизительно 50 редких наследственных метаболических заболеваний, вызываемых дефектами лизосомной функции. Как правило, пациенты с LSD накапливают вредные уровни субстрата (т. е. хранящегося материала) в лизосомах вследствие дефицита или дефекта фермента, ответственного за метаболизм субстрата, или вследствие дефицита ферментативного активатора, необходимого для надлежащей ферментативной функции. Большинство LSD вызвано дефектом или дефицитом одного фермента, как правило фермента, участвующего в метаболизме липидов или гликопротеинов. Некоторые из наиболее распространенных LSD предусматривают болезнь Гоше, болезнь Фабри и болезнь Ниманна-Пика (тип С). Болезни Гоше, Фабри и Ниманна-Пика являются примерами сфинголипидозов. Каждое из этих заболеваний ассоциировано с комплексом симптомов, которые непосредственно или опосредованно вызваны лежащими в основе генетическими дефектами. В результате часто бывает трудно предсказать, какие симптомы или нарушения, ассоциированные с каждым заболеванием, можно эффективно подвергать лечению посредством различных способов лечения. Симптомы, которые являются общими для некоторых LSD, предусматривают изменения в саккадических движениях глаз, когнитивную дисфункцию и нарушения походки, например, атаксию. Эти симптомы особенно распространены при болезни Гоше (например, типа 3) и болезни Ниманна-Пика (типа С).
Болезнь Гоше (GD) представляет собой редкую аутосомно-рецессивную лизосомную болезнь накопления. У пациентов с GD имеется мутация в гене GBA1, который кодирует глюкозилцерамидазу (GC), также известную как бета-глюкоцереброзидаза. Этот фермент отвечает за расщепление гликосфинголипидов на их компоненты, например, расщепление глюкозилцерамида (GLC; также известного как глюкоцереброзид) на глюкозу и церамид. Моноциты и макрофаги характеризуются особенно высоким содержанием лизосом, содержащих GLC, и у пациентов с GD эти клетки увеличиваются в размерах и накапливают токсические концентрации GLC. Эти так называемые "клетки Гоше" накапливаются в некоторых органах, в том числе в костях, костном мозге, селезенке, печени, легком и головном мозге. Системно это приводит к спленомегалии, гепатомегалии, анемии, тромбоцитопении, лейкопении, остеопении, остеонекрозу и другим патологическим отклонениям.
Выделяют три подтипа болезни Гоше, которые различаются по возрасту начала, тяжести и наличию неврологических проявлений. Болезнь Гоше типа 1 (GD-1), ненейронопатическая GD, является наиболее распространенной формой с медианным возрастом при постановке диагноза, составляющим 28 лет, и небольшим снижением ожидаемой продолжительности жизни. При GD-1 фермент GC сохраняет некоторую функциональность, и неврологические нарушения отсутствуют. GD типа 2 (GD-2) представляет собой острую нейронопатическую GD с постановкой диагноза в младенчестве, тяжелым неврологическим поражением и смертью, как правило, в течение первых двух лет жизни. Фермент GC у пациента типа 2 характеризуется более тяжелым нарушением функции по сравнению с ферментом при GD-1. GD типа 3 (GD-3) представляет собой хроническую нейронопатическую GD с постановкой диагноза в детстве, с постепенным ухудшением неврологического поражения и ожидаемой продолжительностью жизни, составляющей, как правило, не более 30 лет. Симптомы GD-3 предусматривают отклонения в функционировании селезенки и печени, утомляемость, кровотечение, судорожные припадки и надъядерный паралич взора. Неврологические проявления у пациентов с GD-3 развиваются постепенно с течением заболевания. Одним из наиболее изнурительных признаков является паралич взора, который представляет собой дефект нейронных путей, контролирующих саккадическое движение глаз. На ранних стадиях заболевания наблюдается замедление горизонтальных саккад. Заболевание прогрессирует до полного горизонтального саккадического паралича наряду с различными степенями вертикального саккадического паралича. VOR также может быть нарушен у пациентов с GD-3. Эти признаки заболевания оказывают глубокое влияние на качество жизни пациентов с GD-3 и могут препятствовать получению образования и перспективам трудоустройства.
Существующее лечение GD-1 и GD-3 ограничено ферментозаместительной терапией рекомбинантными ферментами (ERT) с применением имиглюцеразы, велаглюцеразы или талиглюцеразы, а также субстрат-редуцирующей терапией (SRT) с применением миглустата или элиглустата. См., например, Lunawati L. Bennett & Chris Fellner, Pharmacotherapy of Gaucher Disease: Current and Future Options, P&T 43(5): 274-280, 309 (2018). Имиглюцераза, применяемая в ведущей схеме лечения, представляет собой рекомбинантный вариант человеческой GC, полученный из клеток яичника китайского хомячка и вводимый посредством медленной внутривенной инъекции (как правило в течение 1-2 часов) один раз в 1-2 недели. Она доступна с 1998 года в США. Велаглюцераза является еще одним рекомбинантным аналогом человеческой GC, полученным из линии клеток фибросаркомы и одобренным FDA в 2010 году. Талиглюцераза является аналогичной, ее получают с применением генетически модифицированных клеток корня растения моркови, и она одобрена с 2012 года. Все эти виды лечения требуют IV введения в больнице или другом медицинском учреждении, а рекомбинантные ферменты не проникают через гематоэнцефалический барьер и, следовательно, не способны осуществлять лечение неврологических симптомов GD. Таким образом, хотя эти режимы ERT доказали свою эффективность при лечении пациентов с GD-1, у пациентов с GD-3 они эффективны только при лечении симптомов заболевания, отличных от неврологических.
Субстрат-редуцирующая терапия является альтернативным подходом к лечению GD. Целью этой терапии является снижение накопления GLC посредством ингибирования фермента, который отвечает за синтез GLC. Глюкозилцерамидсинтаза (GCS), также известная как UDP-глюкозоцерамидсинтаза, представляет собой фермент, который катализирует начальную стадию гликозилирования церамида с образованием глюкозилцерамида.
Ингибиторы GCS были предложены для лечения различных заболеваний, в том числе болезней накопления гликолипидов и лизосомных болезней накопления, включая болезнь Гоше. См., например, WO 2005/068426 (Actelion Pharm. Ltd.). Миглустат (Завеска) является ингибитором GCS иминоглюкозы. Он представляет собой N-алкилированный иминосахар, который действует как обратимый конкурентный ингибитор GCS, связываясь в активном центре фермента. Хотя он был разработан для лечения нейронопатических форм GD, GD-2 и GD-3, FDA одобрило его только для лечения пациентов с GD-1 от легкой до умеренной степени и только как терапию второй линии (пациенты должны не отвечать критериям прохождения лечения посредством ERT). Хотя миглустат проникает через гематоэнцефалический барьер, в клинических испытаниях было обнаружено, что он был неэффективным в лечении неврологических проявлений GD-3. Элиглустат также является ингибитором GCS, и он является аналогом церемида. Он был одобрен FDA только для лечения системных симптомов у пациентов с GD-1.
Болезнь Ниманна-Пика типа C (NPC) также является лизосомной болезнью накопления. Хотя ее причина совершенно иная, чем у болезни Гоше, в некотором смысле конечный результат аналогичен. NPC вызывается мутациями в генах NPC1 или NPC2. NPC1 представляет собой мембранный белок, который опосредует внутриклеточный транспорт холестерина к постлизосомным местам назначения. В частности, NPC1 действует совместно с NPC2, способствуя выходу холестерина из эндосомного/лизосомного компартмента. Неэтерифицированный холестерин, который был высвобожден из липопротеинов низкой плотности в люмене поздних эндосом/лизосом, переносится с помощью NPC2 в холестерин-связывающий карман NPC1. Примерно 95% пациентов с NPC характеризуются мутациями в NPC1, в то время как большинство остальных характеризуются мутациями в NPC2. Одним из последствий такого нарушенного транспорта холестерина является накопление холестерина и гликосфинголипидов (включая GLC) в клетках печени, селезенки и головного мозга. Одним из характерных признаков NPC, как и GD-3, является прогрессирующее развитие надъядерного паралича взора, в том числе горизонтальных и вертикальных саккадических параличей.
Другая группа заболеваний и нарушений, обычно ассоциированных с саккадическими параличами взора, предусматривает GM2-ганглиозидозы (такие как болезнь Тея-Сакса, болезнь Сандхоффа и вариант AB GM2-ганглиозидоза).
GM2-ганглиозидозы, как и болезнь Гоше, представляют собой лизосомные болезни накопления, характеризующиеся генетическими дефектами в метаболизме гликосфинголипидов. GM2-ганглиозидозы характеризуются дефектами фермента гексозаминидазы А и/или его кофактора, представляющего собой белок-активатор GM2, которые отвечают за распад GM2 до GM3. GM2 и GM3 являются родственными ганглиозидами, которые являются частью одного и того же метаболического пути, в котором глюкозилцерамид разрушается до церамида. Таким образом, GM3 образуется в ходе поэтапного процесса, который начинается с превращения церамида в глюкозилцерамид (через GLC), за которым следует превращение в галактозилглюкозилцерамид, а затем в GM3 (N-ацетил-а-нейраминидилгалактозилглюкозилцерамид), с последующим превращением в GM2 (N-ацетилгалактозил-N-ацетил-а-нейраминидилгалактозилглюкозилцерамид). Таким образом, патологическое накопление GM2, которое является характерным признаком GM2-ганглиозидозов, может быть нормализовано с помощью ингибитора GCS, который ингибирует более раннюю стадию синтеза глюкозилцерамида.
Описанные в данном документе хинуклидиновые соединения характеризуются активностью ингибиторов фермента глюкозилцерамидсинтазы (GCS). Было раскрыто, что эти соединения, как правило, применимы в лечении лизосомных болезней накопления, таких как болезнь Фабри, болезнь Гоше и болезнь Ниманна-Пика. См., например, WO 2012/129084 и патент США № 2016/0361301.
В данной области техники существует реальная потребность в разработке терапевтических средств, эффективных для облегчения или контроля неврологических симптомов, ассоциированных с болезнью Гоше типа 3.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к хинуклидиновому соединению (соединению 1) согласно формуле (I)
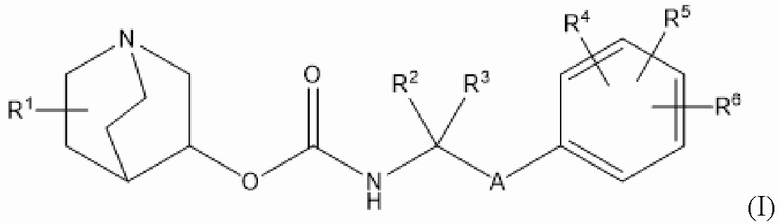
или его фармацевтически приемлемой соли, или пролекарству на его основе, где:
R1 выбран из водорода, галогена (например, фтора), циано, нитро, гидрокси, тио, амино, C1-6-алкила (например, метила или этила), C2-6-алкенила, C2-6-алкинила, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанные алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещены одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио и амино;
R2 и R3 независимо выбраны из C1-3-алкила, необязательно замещенного одним или несколькими (например, 1, 2 или 3) атомами галогена, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) атомами галогена;
каждый из R4, R5 и R6 независимо выбран из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси, и
А представляет собой 5- или 6-членную арильную или гетероарильную группу, необязательно замещенную 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси и C1-6-алкила.
В первом аспекте в настоящей заявке предусмотрен способ лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления, у субъекта, такого как субъект, нуждающийся в этом, при этом способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I). В других аспектах в настоящей заявке дополнительно предусмотрено применение хинуклидиновых соединений, описанных в данном документе, для лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления, и/или для изготовления лекарственного препарата для лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления.
Во втором аспекте в настоящей заявке предусмотрен способ улучшения нейронных связей в головном мозге субъекта, например, субъекта, нуждающегося в этом, при этом способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I). В других аспектах в настоящей заявке дополнительно предусмотрено применение хинуклидиновых соединений, описанных в данном документе, для улучшения нейронных связей в головном мозге субъекта и/или для изготовления лекарственного препарата для улучшения нейронных связей в головном мозге субъекта.
В третьем аспекте в настоящей заявке предусмотрен способ увеличения объема ткани головного мозга или предупреждения или задержки потери объема ткани головного мозга у субъекта, например, у субъекта, нуждающегося в этом, при этом указанный способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I). В других аспектах в настоящей заявке дополнительно предусмотрены хинуклидиновые соединения, описанные в данном документе, для применения в увеличении объема ткани головного мозга или предупреждении или задержке потери объема ткани головного мозга у субъекта, нуждающегося в этом, и/или для изготовления лекарственного препарата для увеличения объема ткани головного мозга или предупреждения или задержки потери объема ткани головного мозга у субъекта, нуждающегося в этом.
В четвертом аспекте в настоящей заявке предусмотрен способ осуществления мониторинга прогрессирования или регрессирования неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта, где субъекта подвергают лечению, которое предусматривает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения формулы (I); при этом указанный способ включает измерение объема ткани головного мозга субъекта в течение определенного периода времени в ходе лечения, например, с применением волюметрической магнитно-резонансной визуализации (vMRI), и оценивание степени любого изменения объема ткани головного мозга за указанный период времени.
В пятом аспекте в настоящей заявке предусмотрен способ оценивания начала проявления неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта с риском развития указанного неврологического нарушения, при этом указанный способ включает: а) измерение объема ткани головного мозга субъекта (например, с применением vMRI) и сравнение с эталонным стандартом для оценки того, меньше ли объем ткани головного мозга, чем эталонный стандарт, и b) если объем ткани головного мозга, идентифицированный на стадии (а), меньше эталонного стандарта, идентифицирование начала проявления указанного неврологического нарушения; при этом способ необязательно дополнительно включает c) инициирование лечения субъекта посредством введения субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения формулы (I), или его фармацевтически приемлемой соли, или пролекарства на его основе.
Дополнительные признаки и преимущества соединений, композиций и способов, раскрытых в данном документе, будут очевидны из следующего подробного описания.
ПОДРОБНОЕ ОПИСАНИЕ
Хотя определенные варианты осуществления настоящего изобретения будут далее описаны со ссылкой на пути получения и схемы, следует понимать, что такие варианты осуществления представлены исключительно в качестве примера и являются лишь иллюстративными, но небольшая часть из множества возможных определенных вариантов осуществления может представлять пути применения принципов настоящего изобретения. Разнообразные изменения и модификации, обеспечивающие преимущества настоящего изобретения, будут очевидны специалистам в данной области техники и считаются находящимися в пределах сути и объема настоящего изобретения, как далее определено в прилагаемой формуле изобретения.
Определения
Если не определено иное, все технические и научные термины, используемые в данном документе, имеют такие же значения, которые обычно понятны специалисту в области техники, к которой принадлежит настоящее изобретение. Хотя при практическом осуществлении или тестировании настоящего изобретения можно применять любые способы и материалы, сходные с описанными в данном документе или эквивалентные им, в данном документе описаны только иллюстративные способы, устройства и материалы. Все технические и патентные публикации, упомянутые в данном документе, включены в данный документ посредством ссылки во всей своей полноте. Ничто в данном документе не следует толковать как признание того, что настоящее изобретение не имеет оснований для противопоставления такому раскрытию как более раннее изобретение.
При осуществлении настоящего изобретения на практике будут применены, если не указано иное, общепринятые методики тканевой культуры, иммунологии, молекулярной биологии, микробиологии, клеточной биологии и рекомбинантной ДНК, которые находятся в пределах компетенции специалистов в данной области техники.
Все числовые обозначения, например, pH, температура, время, концентрация, молекулярная масса, включая диапазоны, являются приближенными значениями, которые варьируются в (+) или (-) сторону с приращением 0,1 или 1,0, если это необходимо. Следует понимать, хотя это не всегда явно указано, что всем числовым обозначениям предшествует термин "приблизительно". Также следует понимать, хотя это не всегда явно указано, что реагенты, описанные в данном документе, являются лишь иллюстративными, и что их эквиваленты известны из уровня техники.
Используемый в данном документе термин "необязательно замещенный" подразумевается как эквивалентный термину "незамещенный или замещенный".
Используемая в данном документе фраза "в способе лечения или предупреждения" (например, во фразе "в способе лечения или предупреждения боли") подразумевается как эквивалентная фразе "в лечении или предупреждении" (например, как во фразе "в лечении или предупреждении боли").
Используемые в настоящем описании и формуле изобретения формы единственного числа включают ссылки на формы множественного числа, если в контексте явно не указано иное. Например, термин "клетка" предусматривает множество клеток, в том числе их смеси. Если специально не указано или не очевидно из контекста, используемый в данном документе термин "или" следует понимать как включающий. Термин "включающий" используется в данном документе для обозначения фразы "включающий без ограничения" и используется взаимозаменяемо с ней.
Подразумевается, что используемые в данном документе термины "содержащий" или "содержит" означают, что композиции и способы предусматривают перечисляемые элементы, но не исключают другие. "Состоящий главным образом из" при применении для определения композиций и способов означает исключение других элементов, имеющих какую-либо существенную значимость для комбинации, для заявляемой цели. Таким образом, композиция, состоящая главным образом из элементов, определенных в данном документе, не будет исключать следовые примеси после осуществления способа выделения и очистки, а также фармацевтически приемлемые носители, такие как фосфатно-солевой буферный раствор, консерванты и т. п. "Состоящий из" означает исключение других ингредиентов на уровне выше следовых элементов и существенных стадий способа для введения композиций в соответствии с настоящим изобретением или стадий способа для получения композиции или достижения намеченного результата. Варианты осуществления, определяемые каждым из этих переходных терминов, входят в объем настоящего изобретения. Подразумевается, что применение термина "содержащий" в данном документе охватывает "состоящий главным образом из" и "состоящий из".
Термины "субъект", "индивидуум" или "пациент" используются в данном документе взаимозаменяемо и относятся к позвоночному, такому как млекопитающее. Млекопитающие включают без ограничения мышей, крыс, кроликов, обезьян, быков, овец, свиней, собак, кошек, сельскохозяйственных животных, животных, используемых в спорте, домашних питомцев, лошадей, приматов и людей. В одном варианте осуществления млекопитающие предусматривают лошадей, собак и кошек. В некоторых вариантах осуществления млекопитающее представляет собой человека, например, человека, страдающего определенным заболеванием или нарушением, таким как болезнь Гоше (например, GD-3) или болезнь Ниманна-Пика типа С.
"Введение" определено в данном документе как способ предоставления средства или композиции, содержащей средство, субъекту таким образом, что это приводит к тому, что средство попадает внутрь организма субъекта. Такое введение может быть осуществлено любым путем, в том числе без ограничения пероральным, трансдермальным (например, через слизистую оболочку влагалища, прямой кишки, ротовой полости), посредством инъекции (например, подкожной, внутривенной, парентеральной, интраперитонеальной, в ЦНС) или посредством ингаляции (например, пероральной или назальной). Фармацевтические препараты, разумеется, представлены формами, подходящими для каждого пути введения.
"Осуществление лечения" или "лечение" заболевания, как правило, предусматривает (1) подавление заболевания, т. е. остановку или снижение развития заболевания или его клинических симптомов, и/или (2) облегчение состояния при заболевании, т. е. обеспечение регрессии заболевания или его клинических симптомов.
Используемые в данном документе термины "осуществление лечения" и "лечение" также относятся либо к устранению когнитивной дисфункции и/или отклонений походки при заболевании, либо к стабилизации таких симптомов. Это связано с тем, что описанные в данном документе заболевания и нарушения являются прогрессирующими нарушениями - в отсутствие лечения состояние пациента будет продолжать ухудшаться. Например, на ранних стадиях течения заболевания пациент может страдать когнитивной дисфункцией и/или отклонениями походки легкой степени, но по мере прогрессирования заболевания у пациентов могут развиться гораздо более тяжелые симптомы. Таким образом, лечение охватывает как замедление такого прогрессирующего ухудшения (например, стабилизацию), так и устранение такого прогрессирующего ухудшения (например, улучшение).
"Осуществление предупреждения" или "предупреждение" заболевания, как правило, предусматривает предотвращение развития клинических симптомов заболевания у пациента, который может быть предрасположен к развитию заболевания, но еще не испытывает симптомов заболевания, или они у него еще не проявляются.
Используемый в данном документе термин "осуществление предупреждения" или "предупреждение" также охватывает предупреждение развития когнитивной дисфункции и/или отклонений походки у пациента с подозрением на наличие заболевания или диагнозом заболевания или нарушения, описанного в данном документе. Поскольку заболевания и нарушения, описанные в данном документе, являются прогрессирующими нарушениями, различные признаки и симптомы могут постепенно проявляться по мере прогрессирования заболевания. Так, например, у пациента может быть диагностирована GD-3 или NPC до начала развития когнитивной дисфункции и/или отклонений походки. У такого пациента описанные в данном документе способы лечения могут быть эффективными в предупреждении развития когнитивной дисфункции и/или отклонений походки.
Термин "паралич" является синонимичным термину "парализованность" и предусматривает любую степень потери двигательной функции одной или нескольких скелетных мышц. Используемый в данном документе термин "паралич", таким образом, охватывает как полный паралич, т. е. полную парализованность, так и частичный паралич, т. е. частичную парализованность. Полный паралич означает, что мышца или группа мышц, например, экстраокулярные мышцы, потеряли способность сокращаться. Таким образом, пораженный глаз или глаза могут быть неспособны двигаться. Частичный паралич может проявляться как торможение движения, замедление движения или другие дефекты движения. Они могут предусматривать потерю диапазона движения. Применительно к саккадам это может предусматривать торможение инициации саккад (например, в ответ на стимулы), изменения частоты саккад, изменения пиковой скорости саккад, изменения амплитуды саккад, изменения латентного периода между саккадами и/или потерю способности удерживать взгляд или перемещать взгляд. В некоторых вариантах осуществления используемый в данном документе термин "паралич" предусматривает офтальмопарез и/или офтальмоплегию. Таким образом, данный термин охватывает как слабость, так и парализованность экстраокулярных мышц. Экстраокулярные мышцы предусматривают любую одну или несколько из верхней прямой, нижней прямой, медиальной прямой, латеральной прямой, нижней косой и верхней косой мышц глаза. Слабость и/или парализованность могут предусматривать одно или несколько горизонтальных движений, вертикальных движений или вращательных движений.
Термин "страдающий", относящийся к термину "лечение", относится к пациенту или индивидууму, у которого было диагностировано заболевание. Термин "страдающий", относящийся к термину "предупреждение", относится к пациенту или индивидууму, который предрасположен к развитию заболевания. Пациент может также называться "характеризующимся риском страдания" заболеванием вследствие анамнеза заболевания в своей семейной родословной или вследствие наличия генетических мутаций, ассоциированных с этим заболеванием. У пациента с риском развития заболевания еще не развились все или некоторые характерные патологические особенности заболевания.
Термин "увеличение", относящийся к способам увеличения объема ткани головного мозга, относится к увеличению объема по меньшей мере одной из отдельных областей ткани головного мозга, предпочтительно множества, и обычно сопровождается увеличением объема ткани головного мозга в целом (т. е. общего объема ткани головного мозга субъекта).
"Эффективное количество" или "терапевтически эффективное количество" представляет собой количество, достаточное для достижения благоприятных или требуемых результатов. Эффективное количество можно вводить за одно или несколько введений, нанесений или дозирований. Такая доставка зависит от ряда переменных, в том числе от периода времени, в течение которого отдельная единица дозирования будет использоваться, биодоступности терапевтического средства и пути введения. Следует понимать, однако, что конкретные уровни доз терапевтических средств по настоящему изобретению для какого-либо конкретного субъекта зависят от ряда факторов, в том числе, например, от активности конкретного применяемого соединения, возраста, веса тела, общего состояния здоровья, пола и рациона субъекта, времени введения, скорости выведения, комбинации лекарственных средств и тяжести конкретного нарушения, подлежащего лечению, а также формы введения. Как правило, лечебные дозировки могут быть подобраны для оптимизации безопасности и эффективности. Как правило, взаимосвязи дозы и эффекта из тестов in vitro и/или in vivo изначально могут служить полезными рекомендациями для введения пациенту надлежащих доз. В целом, желательно вводить количество соединения, которое является эффективным для достижения уровня в сыворотке крови, соизмеримого с концентрациями, которые, как было обнаружено, являются эффективными in vitro. Определение этих параметров находится вполне в пределах квалификации специалистов в данной области техники. Эти соображения, а также эффективные составы и процедуры введения хорошо известны из уровня техники и описаны в стандартных учебниках. Согласно данному определению, используемый в данном документе термин "терапевтически эффективное количество" представляет собой количество, достаточное для лечения (например, улучшения) одного или нескольких симптомов, ассоциированных с заболеванием или нарушением, описанным в данном документе (например, в любом из способа 1 et seq.), ex vivo, in vitro или in vivo.
Используемый в данном документе термин "фармацевтически приемлемое вспомогательное вещество" охватывает любое из стандартных фармацевтических вспомогательных веществ, в том числе носителей, таких как фосфатно-солевой буферный раствор, вода и эмульсии, такие как эмульсия "масло в воде" или "вода в масле", а также разнообразных типов увлажняющих средств. Фармацевтические композиции также могут содержать стабилизаторы и консерванты. Для примеров носителей, стабилизаторов и вспомогательных веществ см. Remington's Pharmaceutical Sciences (20th ed., Mack Publishing Co. 2000).
Используемый в данном документе термин "пролекарство" означает фармакологическое производное молекулы исходного лекарственного средства, которое требует спонтанной либо ферментативной биотрансформации внутри организма для высвобождения активного лекарственного средства. Например, пролекарства представляют собой варианты или производные хинуклидиновых соединений, описанных в данном документе, которые содержат группы, расщепляемые в определенных метаболических условиях, которые при расщеплении становятся хинуклидиновыми соединениями, описанными в данном документе, например, соединением формулы (I). Такие пролекарства становятся фармацевтически активными in vivo тогда, когда они подвергаются сольволизу в физиологических условиях или подвергаются ферментативному разрушению. Соединения, представляющие собой пролекарства, в данном документе могут называться одинарными, двойными, тройными и т. д. в зависимости от количества стадий биотрансформации, требуемых для высвобождения активного лекарственного средства внутри организма, и количества функциональных групп, присутствующих в форме типа предшественника. Формы, представляющие собой пролекарства, часто обеспечивают преимущества в отношении растворимости, тканевой совместимости или замедленного высвобождения в организме млекопитающего.
Пролекарства, широко известные из уровня техники, предусматривают хорошо известные производные кислот, такие как, например, сложные эфиры, полученные посредством осуществления реакции кислотных соединений с подходящим спиртом, амиды, полученные посредством осуществления реакции кислотных соединений с амином, и ацилированные производные оснований, которые образуются посредством осуществления реакции с основными группами. Другие производные, представляющие собой пролекарства, можно комбинировать с другими признаками, раскрытыми в данном документе, для улучшения биодоступности. Таким образом, специалистам в данной области техники будет понятно, что некоторые из раскрытых в данном документе соединений, содержащих, например, свободные амино- или гидроксигруппы, могут быть превращены в пролекарства. Пролекарства предусматривают соединения, содержащие аминокислотный остаток или полипептидную цепь из двух или более (например, двух, трех или четырех) аминокислотных остатков, которые ковалентно соединены посредством пептидных связей со свободными амино-, гидроксигруппами или группами карбоновой кислоты соединений, раскрытых в данном документе. Аминокислотные остатки предусматривают 20 встречающихся в природе аминокислот, обычно обозначаемых трехбуквенными символами, а также предусматривают 4-гидроксипролин, гидроксилизин, десмозин, изодесмозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства также предусматривают соединения, содержащие карбонатный, карбаматный, амидный фрагмент или фрагмент сложного алкилового эфира, ковалентно связанный с любым из вышеупомянутых заместителей, раскрытых в данном документе.
Используемый в данном документе термин "фармацевтически приемлемая соль" означает фармацевтически приемлемую соль присоединения кислоты или фармацевтически приемлемую соль присоединения основания соединения, раскрытого в данном документе, которую можно вводить без какого-либо получаемого значительного нежелательного биологического эффекта(эффектов) или какого-либо получаемого вредного взаимодействия(взаимодействий) с любым другим компонентом фармацевтической композиции, в которой он может содержаться.
Используемый в данном документе термин "C1-6-алкил" означает насыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 1-6 атомов углерода и соответствующего количества атомов водорода. Иллюстративные C1-6-алкильные группы предусматривают метил, этил, н-пропил, изопропил, н-бутил и изобутил. Другие C1-6-алкильные группы будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения. Термины "C1-3-алкил", C1-4-алкил" и т. п. имеют эквивалентные значения, т. e. насыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 1-3 (или 4) атомов углерода и соответствующего количества атомов водорода.
Используемый в данном документе термин "C2-6-алкенил" означает ненасыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 2-6 атомов углерода и соответствующего количества атомов водорода, при этом свободный радикал содержит по меньшей мере одну углерод-углеродную двойную связь. Иллюстративные C2-6-алкенильные группы предусматривают этенил, проп-1-енил, проп-2-енил, изопропенил, бут-1-енил, 2-метилпроп-1-енил и 2-метилпроп-2-енил. Другие C2-6-алкенильные группы будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения.
Используемый в данном документе термин "C2-6-алкинил" означает ненасыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 2-6 атомов углерода и соответствующего количества атомов водорода, при этом свободный радикал содержит по меньшей мере одну углерод-углеродную тройную связь. Иллюстративные C2-6-алкинильные группы предусматривают этинил, проп-1-инил, проп-2-инил, бут-1-инил и 3-метилбут-1-инил. Другие C2-6-алкинильные группы будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения.
Используемый в данном документе термин "C1-6-алкилокси" означает насыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 1-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода. C1-6-алкилоксигруппа присоединяется посредством атома кислорода. Иллюстративные C1-6-алкилоксигруппы предусматривают метилокси, этилокси, н-пропилокси, изопропилокси, н-бутилокси и изобутилокси. Другие C1-6-алкилоксигруппы будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения. Термины "C1-3-алкилокси," "C1-4-алкилокси" и т. п. имеют эквивалентное значение, т. e. насыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 1-3 (или 4) атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, где группа присоединяется посредством атома кислорода.
Используемый в данном документе термин "C2-6-алкенилокси" означает ненасыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 2-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, при этом свободный радикал содержит по меньшей мере одну углерод-углеродную двойную связь. C2-6-алкенилоксигруппа присоединяется посредством атома кислорода. Иллюстративной C2-6-алкенилоксигруппой является этенилокси; другие будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения.
Используемый в данном документе термин "C2-6-алкинилокси" означает ненасыщенный линейный или разветвленный свободный радикал, состоящий главным образом из 2-6 атомов углерода (и соответствующего количества атомов водорода) и атома кислорода, при этом свободный радикал содержит по меньшей мере одну углерод-углеродную тройную связь. C2-6-алкенилоксигруппа присоединяется посредством атома кислорода. Иллюстративной C2-6-алкенилоксигруппой является этинилокси; другие будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения.
Используемый в данном документе термин "гетероарил" означает ароматический свободный радикал с 5 или 6 атомами (т. e. атомами кольца), которые образуют кольцо, где 1-5 атомов кольца представляют собой атомы углерода, а остальные 1-5 атомов кольца (т. e. гетероатомов кольца) независимо выбраны из группы, состоящей из атомов азота, серы и кислорода. Иллюстративные 5-членные гетероарильные группы предусматривают фурил, тиенил, тиазолил (например, тиазол-2-ил), пиразолил, изотиазолил, оксазолил, изоксазолил, пирролил, тиазолил, имидазолил, оксадиазолил и тиадиазолил. Иллюстративные 6-членные гетероарильные группы предусматривают пиридил, пиримидил, пиразинил, пиридазинил, 1,2,4-триазинил, бензоксазолил, бензoтиазолил, бензизотиазолил, бензизоксазолил и бензимидазолил. Другие гетероарильные группы будут вполне очевидными специалистам в данной области техники с учетом преимуществ настоящего изобретения. В целом, гетероарильная группа, как правило, присоединяется к основной структуре посредством атома углерода. Однако, специалистам в данной области техники будет понятно, что некоторые другие атомы, например, гетероатомы кольца, могут быть присоединены к основной структуре.
Используемый в данном документе термин "арил" означает ароматический свободный радикал с 5 или 6 атомами (т. e. атомами кольца), которые образуют кольцо, где все атомы кольца представляют собой атомы углерода. Иллюстративной арильной группой является фенильная группа.
Используемый в данном документе термин "алифатический" означает неароматическое соединение, содержащее атомы углерода и водорода, например, содержащее 1-9 атомов углерода. Алифатические соединения могут быть с прямой цепью или разветвленными, могут содержать одну или несколько кольцевых структур, а также могут содержать одну или несколько углерод-углеродных двойных связей (при условии, что соединение не содержит ненасыщенную кольцевую структуру ароматической природы). Примеры алифатических соединений предусматривают этан, пропилен, циклобутан, циклогексадиен.
Используемые в данном документе термины "галоген" и "галогеновый" означают фтор, хлор, бром или йод. Эти термины используются взаимозаменяемо и могут относиться к группе галогенового свободного радикала или к атому галогена как таковому. Специалисты в данной области техники смогут легко идентифицировать их с учетом контекста, в котором данный термин используется в настоящем изобретении.
Используемый в данном документе термин "циано" означает свободный радикал, содержащий атом углерода, соединенный с атомом азота посредством тройной связи. Цианорадикал присоединяется посредством своего атома углерода.
Используемый в данном документе термин "нитро" означает радикал -NO2, который присоединяется посредством своего атома азота.
Используемые в данном документе термины "гидрокси" и "гидроксил" означают радикал -OH, который присоединяется посредством своего атома кислорода. Термин "тио" означает радикал -SH, который присоединяется посредством своего атома серы.
Используемый в данном документе термин "амино" означает свободный радикал, содержащий атом азота и 1 или 2 атома водорода. В связи с этим термин "амино", как правило, относится к первичным и вторичным аминам. В этом отношении, как используется в данном документе, третичный амин представлен общей формулой RR'N-, где R и R' являются углеродными радикалами, которые могут быть идентичными или неидентичными. Тем не менее, термин "амино", как правило, может быть использован в данном документе для описания первичного, вторичного или третичного амина, и специалисты в данной области техники будут способны легко идентифицировать их с учетом контекста, в котором данный термин используется в настоящем раскрытии.
Используемый в данном документе термин "оксо" означает кислородный радикал, который присоединяется посредством двойной связи. Если атом, связанный с этим атомом кислорода, представляет собой атом углерода, то связь является углерод-кислородной двойной связью, которая может быть обозначена как -(C=O)- и которая может называться кетонной.
Упоминание перечня химических групп в любом определении переменной в данном документе включает определения этой переменной в качестве какой-либо отдельной группы или комбинации перечисленных групп. Упоминание варианта осуществления для переменной или аспекта в данном документе включает этот вариант осуществления в качестве отдельного варианта осуществления или в комбинации с любыми другими вариантами осуществления или их частями.
Любые композиции или способы, предусмотренные в данном документе, можно комбинировать с одним или несколькими из каких-либо других композиций и способов, предусмотренных в данном документе.
В данном документе используются следующие сокращения.
Соединения
Настоящее изобретение относится к хинуклидиновым соединениям для применения в терапевтических способах, связанных с лечением или предупреждением обсуждаемых в данном документе заболеваний и нарушений. Во всех своих различных аспектах настоящее изобретение относится к хинуклидиновому соединению (соединению 1) согласно формуле (I),
или его фармацевтически приемлемой соли, или пролекарству на его основе, где:
R1 выбран из водорода, галогена (например, фтора), циано, нитро, гидрокси, тио, амино, C1-6-алкила (например, метила или этила), C2-6-алкенила, C2-6-алкинила, C1-6-алкилокси, C2-6-алкенилокси и C2-6-алкинилокси, где указанные алкил, алкенил, алкинил, алкилокси, алкенилокси или алкинилокси необязательно замещены одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио или амино;
R2 и R3 независимо выбраны из C1-3-алкила, необязательно замещенного одним или несколькими (например, 1, 2 или 3) атомами галогена, или R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) атомами галогена;
каждый из R4, R5 и R6 независимо выбран из водорода, галогена, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси, и
А представляет собой 5- или 6-членную арильную или гетероарильную группу (например, фенил или триазолил), необязательно замещенную 1, 2 или 3 группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси и C1-6-алкила.
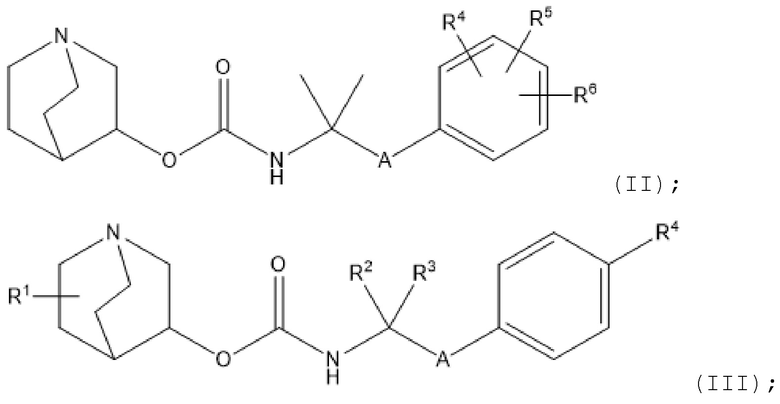
В дополнительных вариантах осуществления любых аспектов настоящего изобретения настоящее изобретение дополнительно относится к следующим соединениям:
1.1 Соединение 1, где R1 выбран из водорода, галогена, циано, нитро, гидрокси, тио, амино, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио и амино;
1.2 Соединение 1, где R1 выбран из водорода, галогена,C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио и амино;
1.3 Соединение 1, где R1 выбран из водорода, галогена,C1-4-алкила и C1-4-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано, нитро, гидрокси, тио и амино;
1.4 Соединение 1, где R1 выбран из водорода, галогена, C1-4-алкила и C1-4-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2, или 3, или 1, или 2) группами, выбранными из циано, нитро, гидрокси, тио и амино;
1.5 Соединение 1, где R1 выбран из водорода, галогена и C1-4-алкила, где указанный алкил необязательно замещен одной или несколькими (например, 1 или 2) группами, выбранными из галогена, гидрокси, тио и амино;
1.6 Соединение 1, где R1 выбран из водорода, фтора, метила и этила, где указанный метил или этил необязательно замещен 1 или 2 группами, выбранными из галогена, гидрокси, тио и амино;
1.7 Соединение 1, где R1 выбран из водорода и метила, где указанный метил необязательно замещен 1 или 2 атомами галогена;
1.8 Соединение 1, где R1 представляет собой водород;
1.9 Соединение 1 или любое из 1.1-1.8, где R1 не присоединен к атому азота хинуклидинового фрагмента;
1.10 Соединение 1 или любое из 1.1-1.9, где каждый из R2 и R3 независимо представляет собой C1-3-алкил, необязательно замещенный одним или несколькими (например, 1, 2 или 3) атомами галогена;
1.11 Соединение 1.10, где каждый из R2 и R3 независимо представляет собой метил или этил, необязательно замещенный 1 или 2 атомами галогена;
1.12 Соединение 1.10, где каждый из R2 и R3 независимо выбран из метила и этила, необязательно замещенных одним или несколькими атомами фтора, например, 1, 2, 3 или 4 атомами фтора;
1.13 Соединение 1.10, где каждый из R2 и R3 независимо представляет собой метил, замещенный 0, 1, 2 или 3 атомами фтора;
1.14 Соединение 1.10, где каждый из R2 и R3 представляет собой метил или трифторметил;
1.15 Соединение 1.10, где каждый из R2 и R3 представляет собой метил;
1.16 Соединение 1 или любое из 1.1-1.9, где R2 и R3 вместе образуют циклопропильную или циклобутильную группу, необязательно замещенную одним или несколькими (например, 1 или 2) атомами галогена;
1.17 Соединение 1.16, где R2 и R3 вместе образуют циклопропильную группу;
1.18 Соединение 1 или любое из 1.1-1.9, где каждый из R2 и R3 представляет собой метил, или R2 и R3 вместе образуют циклопропильную группу;
1.19 Соединение 1 или любое из 1.1-1.9, где каждый из R4, R5 и R6независимо выбран из водорода, галогена, C1-6-алкила и C1-6-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-6-алкилокси;
1.20 Соединение 1 или любое из 1.1-1.9, где каждый из R4, R5 и R6независимо выбран из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, гидрокси, циано и C1-3-алкилокси;
1.21 Соединение 1.19, где каждый из R4, R5 и R6 независимо выбран из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена, циано и C1-3-алкилокси;
1.22 Соединение 1.19, где каждый из R4, R5 и R6 независимо выбран из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси;
1.23 Соединение 1.19, где каждый из R4, R5 и R6 независимо выбран из галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси
1.24 Соединение 1 или любое из 1.19-1.23, где R4 выбран из водорода, галогена, C1-3-алкила и C1-3-алкилокси, где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси;
1.25 Соединение 1.24, где R4 выбран из галогена (например, фтора), C1-3-алкила (например, метила) и C1-3-алкилокси (например, метокси или этокси), где указанный алкил или алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси или этокси);
1.26 Соединение 1.25, где R4 выбран из галогена (например, фтора) и C1-3-алкилокси (например, метокси или этокси), где указанный алкилокси необязательно замещен одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси или этокси);
1.27 Соединение 1.26, где R4 представляет собой фтор или C1-3-алкилокси (например, этокси), необязательно замещенный одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси);
1.28 Соединение 1.26, где R4 представляет собой фтор или этокси, необязательно замещенный одним или несколькими (например, 1, 2 или 3) C1-3-алкилокси (например, метокси);
1.29 Соединение 1 или любое из 1.19-1.28, где R6 представляет собой водород;
1.30 Соединение 1 или любое из 1.19-1.28, где каждый из R5 и R6 представляет собой водород;
1.31 Соединение 1 или любое из 1.19-1.28, где каждый из R5 и R6 представляет собой водород, и R4 представляет собой фтор или C1-3-алкилокси (например, этокси), необязательно замещенный одной или несколькими (например, 1, 2 или 3) группами, выбранными из галогена и C1-3-алкилокси (например, метокси);
1.32 Соединение 1.31, где каждый из R5 и R6 представляет собой водород, и R4 представляет собой фтор или этокси, необязательно замещенный одним или несколькими (например, 1, 2 или 3) C1-3-алкилокси (например, метокси);
1.33 Соединение 1.32, где каждый из R5 и R6 представляет собой водород, и R4 представляет собой фтор или этокси, замещенный метокси (например, 2-метоксиэтокси);
1.34 Соединение 1.32, где R4 представляет собой фтор или 2-метоксиэтокси;
1.35 Соединение 1 или любое из 1.1-1.34, где по меньшей мере один из R4, R5 и R6 не представляет собой водород;
1.36 Соединение 1 или любое из 1.1-1.35, где R6 представляет собой водород, и R4 и R5 расположены в положениях 2, 4 или 6 фенильного кольца, к которому они присоединены (т. е. в орто- или пара-положении по отношению к заместителю А);
1.37 Соединение 1 или любое из 1.1-1.35, где R6 представляет собой водород, и R4 и R5 независимо расположены в положениях 2 и 3 (т. е. смежные орто- и мета-), 3 и 4 (т. е. смежные мета- и пара-) или 3 и 5 (т. е. мета) фенильного кольца, к которому они присоединены (относительно заместителя А);
1.38 Соединение 1 или любое из 1.1-1.35, где R6 представляет собой водород, и R4 и R5 расположены в положениях 3 и 5 (т. е. мета) фенильного кольца, к которому они присоединены (относительно заместителя А);
1.39 Соединение 1 или любое из 1.1-1.35, где R5 и R6 представляют собой водород, и R4 находится в положении 2, 3 или 4 фенильного кольца, к которому он присоединен (например, в орто-, мета- или пара- положении по отношению к заместителю А);
1.40 Соединение 1 или любое из 1.1-1.35, где R5 и R6 представляют собой водород, и R4 находится в положении 2 или 4 фенильного кольца, к которому он присоединен (например, в орто- или пара-положении по отношению к заместителю А);
1.41 Соединение 1 или любое из 1.1-1.35, где R5 и R6 представляют собой водород, и R4 расположен в положении 4 фенильного кольца, к которому он присоединен (например, в пара-положении по отношению к заместителю А);
1.42 Соединение 1 или любое из 1.1-1.35, где ни один из R4, R5 и R6 не представляет собой водород, и каждый из R4, R5 и R6 независимо расположен в положениях 2, 4 или 6 фенильного кольца, к которому они присоединены (т. е. в орто- или пара-положении по отношению к заместителю А);
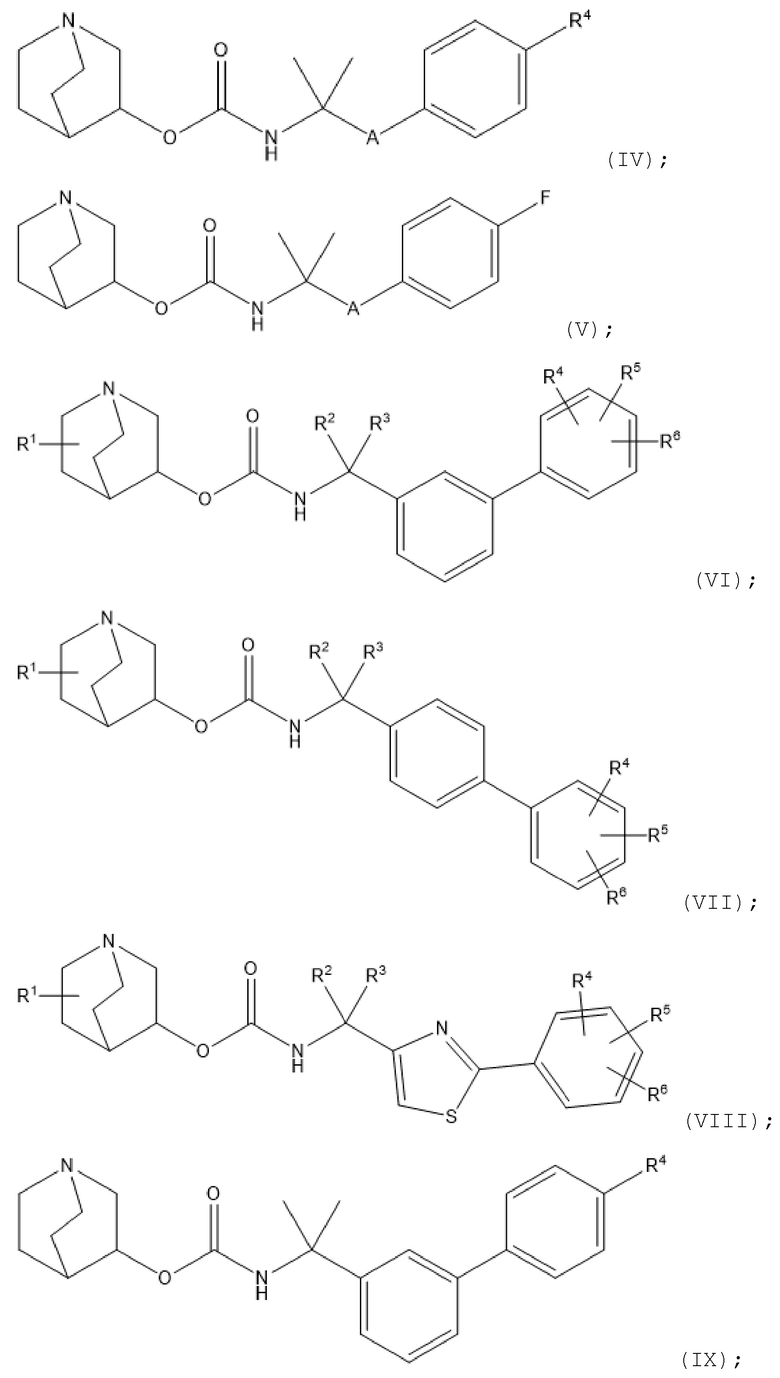
1.43 Соединение 1 или любое из 1.1-1.42, где R4 расположен в положении 4 фенильного кольца, к которому он присоединен (т. е. в пара-положении по отношению к заместителю А);
1.44 Соединение 1 или любое из 1.1-1.43, где А представляет собой 6-членную арильную группу, 5-членную гетероарильную группу (например, содержащую 1, 2 или 3 гетероатома в гетероарильном кольце, независимо выбранных из N, O и S) или 6-членную гетероарильную группу (например, содержащую 1, 2 или 3 атома азота в гетероарильном кольце);
1.45 Соединение 1.44, где A представляет собой 6-членную арильную группу или 5-членную гетероарильную группу (например, содержащую 1, 2 или 3 гетероатома в гетероарильном кольце, независимо выбранных из N, O и S), где необязательно 5-членная гетероарильная группа содержит 1 или 2 гетероатома, выбранных из N и S (например, один N и/или один S);
1.46 Соединение 1.44 или 1.45, где А выбран из группы, состоящей из фенила, фурила, тиенила, тиазолила, пиразолила, изотиазолила, оксазолила, изоксазолила, пирролила, триазолила, имидазолила, оксадиазолила и тиадиазолила;
1.47 Соединение 1.46, где А выбран из группы, состоящей из фенила, тиенила, тиазолила, пирролила и имидазолила;
1.48 Соединение 1.46, где А выбран из группы, состоящей из фенила и тиазолила, например, 2-тиазол-4-ила или 4-тиазол-2-ила;
1.49 Соединение 1 или любое из 1.1-1.48, где А является незамещенным
1.50 Соединение 1 или любое из 1.1-1.48, где А замещен одной или несколькими (например, 1, 2 или 3) группами, независимо выбранными из галогена, гидрокси, тио, амино, нитро, C1-6-алкокси и C1-6-алкила (например, метила);
1.51 Соединение 1.50, где A представляет собой тиазолил, замещенный одним атомом галогена (например, фтором) или C1-6-алкилом (например, метилом);
1.52 Соединение 1.50, где A представляет собой фенил, замещенный 1, 2 или 3 группами, независимо выбранными из галогена (например, фтора) и C1-6-алкила (например, метила);
1.53 Соединение 1.52, где А представляет собой фенил, замещенный 1 или 2 атомами фтора или метильными группами;
1.54 Соединение 1 или любое из 1.1-1.53, где две группы, присоединенные к заместителю А (т. е. фенильное кольцо (-(C6H2R4R5R6)) и группа -C(R2R3)-), расположены в положениях 1,2-, 1,3- или 1,4- друг относительно друга (т. е. в орто-, мета- или пара-положении);
1.55 Соединение 1.54, где две группы, присоединенные к заместителю А, расположены в положении 1,3- друг относительно друга (т. е. в мета-положении);
1.56 Соединение 1.54, где две группы, присоединенные к заместителю А, расположены в положении 1,4- друг относительно друга (т. е. в пара-положении);
1.57 Любое из соединений 1.54-1.56, где заместитель A представляет собой 5-членную гетероарильную группу, и по меньшей мере одна из двух групп, присоединенных к заместителю A (т. е. фенильное кольцо (-(C6H2R4R5R6)) или группа -C(R2R3)-), присоединена к атому углерода гетероарильного кольца, где необязательно обе такие группы присоединены к атомам углерода гетероарильного кольца;
1.58 Соединение 1 или любое из 1.1-1.57, где соединение формулы (I) может быть представлено любой одной или несколькими из следующих подструктур:
1.59 Соединение 1 или любое из 1.1-1.58, где соединение формулы (I) или соединение любой из формул (II)-(XII) характеризуется (S)-конфигурацией;
1.60 Соединение 1 или любое из 1.1-1.58, где соединение формулы (I) или соединение любой из формул (II)-(XII) характеризуется (R)-конфигурацией;
1.61 Соединение 1 или любое из 1.1-1.60, где соединение формулы (I) или соединение любой из формул (II)-(XII) характеризуется энантиомерным избытком (например, (S)-конфигурации), составляющим по меньшей мере 90%, например, по меньшей мере 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99,5% или 99,9%;
1.62 Соединение 1 или любое из 1.1-1.58, где соединение формулы (I) или соединение любой из формул (II)-(XII) является рацемическим (т. е. характеризуется соотношением энантиомеров, составляющим примерно 50:50) или представляет собой смесь энантиомеров с другим соотношением (например, менее чем 50:50 или более чем 50:50);
1.63 Соединение 1 или любое из 1.1-1.62, где соединение формулы (I) выбрано из группы, состоящей из:
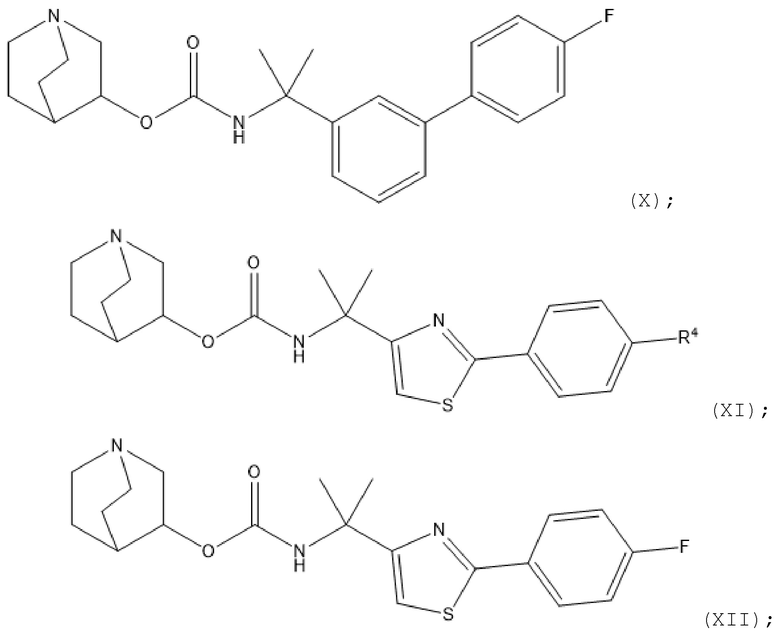
соединения
1.64 Соединение 1 или любое из 1.1-1.63, где соединение выбрано из хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамата, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата и (S)-хинуклидин-3-ил-(2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата;
1.65 Соединение 1 или любое из 1.1-1.63, где соединение представляет собой хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
1.66 Соединение 1 или любое из 1.1-1.63, где соединение представляет собой хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат;
1.67 Соединение 1 или любое из 1.1-1.66, где соединение формулы (I) или любое из (II)-(XII) находится в форме свободного основания;
1.68 Соединение 1 или любое из 1.1-1.66, где соединение формулы (I) или любое из (II)-(XII) находится в форме фармацевтически приемлемой соли;
1.69 Соединение 1.68, где указанная форма соли представляет собой форму соли присоединения кислоты;
1.70 Соединение 1.69, где указанная форма соли присоединения кислоты представляет собой соль, выбранную из гидрохлорида, гидробромида, гидройодида, нитрата, сульфата, бисульфата, фосфата, кислого фосфата, ацетата, лактата, цитрата, кислого цитрата, тартрата, битартрата, сукцината, гидроксисукцината, малата, малеата, фумарата, глюконата, сахарата, бензоата, метансульфоната и памоата;
1.71 Соединение 1.70, где форма соли присоединения кислоты выбрана из гидрохлорида, гидроксисукцината (например, 2-гидроксисукцината) и малата;
1.72 Соединение 1.68, где указанная форма соли представляет собой форму соли присоединения основания;
1.73 Соединение 1 или любое из 1.1-1.72, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в форме малатной соли;
1.74 Соединение 1 или любое из 1.1-1.73, где соединение формулы (I) или любое из (II)-(XII) находится в форме пролекарства, описанного в данном документе;
1.75 Соединение 1 или любое из 1.1-1.74, где соединение формулы (I) или любое из (II)-(XII) находится в форме гидрата, сольвата и/или полиморфа.
Соли
Раскрытые в данном документе соединения, например, любые из соединений 1 или 1.1-1.75, которые являются основными по природе, как правило, способны к образованию широкого разнообразия различных солей с различными неорганическими и/или органическими кислотами. Несмотря на то, что такие соли, как правило, являются фармацевтически приемлемыми для введения животным и людям, часто желательно на практике вначале выделить соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнее обратно в соединение в форме свободного основания путем обработки щелочным реагентом и впоследствии превратить свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты основных соединений можно легко получать с применением общепринятых методик, например, посредством обработки основного соединения по сути эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как, например, метанол или этанол. При аккуратном выпаривании растворителя получают желаемую твердую соль. Раскрытые в данном документе соединения, которые являются положительно заряженными, например, содержащие четвертичный аммоний, также могут образовывать соли с анионным компонентом различных неорганических и/или органических кислот.
Кислоты, которые можно использовать для получения фармацевтически приемлемых солей хинуклидиновых соединений, представляют собой кислоты, которые могут образовывать нетоксичные соли присоединения кислоты, например, соли, содержащие фармакологически приемлемые анионы, такие как хлоридные, бромидные, йодидные, нитратные, сульфатные или бисульфатные, фосфатные или кислые фосфатные, ацетатные, лактатные, цитратные или кислые цитратные, тартратные или битартратные, сукцинатные, малатные, малеатные, фумаратные, глюконатные, сахаратные, бензоатные, метансульфонатные и памоатные [т. е. 1,1'-метиленбис-(2-гидрокси-3-нафтоатные)] соли.
Раскрытые в данном документе соединения, которые являются кислотными по природе, например, соединения, содержащие тиольный фрагмент, как правило, способны к образованию широкого разнообразия различных солей с различными неорганическими и/или органическими основаниями. Несмотря на то, что такие соли, как правило, являются фармацевтически приемлемыми для введения животным и людям, часто желательно на практике вначале выделить соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнее обратно в соединение в форме свободной кислоты путем обработки кислотным реагентом и впоследствии превратить свободную кислоту в фармацевтически приемлемую соль присоединения основания. Эти соли присоединения основания можно легко получать с применением общепринятых методик, например, посредством обработки соответствующих кислотных соединений водным раствором, содержащим требуемые фармакологически приемлемые катионы, и затем выпаривания полученного раствора до сухого состояния, например, при пониженном давлении. В качестве альтернативы, их также можно получать посредством смешивания растворов кислотных соединений в низшем алифатическом спирте вместе с требуемым алкоксидом щелочного металла и затем выпаривания полученного раствора до сухого состояния таким же образом, как описано выше. В любом случае стехиометрические количества реагентов можно использовать для того, чтобы обеспечить завершение реакции и максимально увеличить выходы продукта для желаемой твердой соли.
Основания, которые можно использовать для получения фармацевтически приемлемых солей присоединения основания хинуклидиновых соединений, представляют собой основания, которые могут образовывать нетоксичные соли присоединения основания, например, соли, содержащие фармакологически приемлемые катионы, такие как катионы щелочных металлов (например, калия и натрия), катионы щелочноземельных металлов (например, кальция и магния), соли аммония или другие соли присоединения водорастворимых аминов, таких как N-метилглюкамин (меглумин), соли низшего алканоламмония и другие такие основные формы органических аминов.
В одном варианте осуществления фармацевтически приемлемая соль представляет собой сукцинатную соль. В другом варианте осуществления фармацевтически приемлемая соль представляет собой 2-гидроксисукцинатную соль, например, (S)-2-гидроксисукцинатную соль. В другом варианте осуществления фармацевтически приемлемая соль представляет собой хлористоводородную соль (т. e. соль HCl). В другом варианте осуществления фармацевтически приемлемая соль представляет собой малатную соль.
Пролекарства
Настоящее изобретение дополнительно охватывает пролекарства на основе соединений 1 и 1.1-1.75. Фармацевтически приемлемые пролекарства, раскрытые в данном документе, представляют собой производные хинуклидиновых соединений, которые могут быть превращены in vivo в хинуклидиновые соединения, описанные в данном документе. Пролекарства, которые сами по себе могут обладать некоторой активностью, становятся фармацевтически активными in vivo, когда они подвергаются, например, сольволизу в физиологических условиях или ферментативному разложению. Способы получения пролекарств на основе соединений, описанных в данном документе, будут очевидны специалисту в данной области техники на основании настоящего раскрытия.
В одном варианте осуществления карбаматный фрагмент хинуклидинового соединения является модифицированным. Например, карбаматный фрагмент хинуклидинового соединения может быть модифицирован путем добавления воды и/или одного или двух алифатических спиртов. В этом случае атомы при углерод-кислородной двойной связи карбаматного фрагмента принимают такое расположение, которое можно считать полуацетальной или ацетальной функциональной группой. В одном варианте осуществления карбаматный фрагмент хинуклидинового соединения может быть модифицирован посредством добавления алифатического диола, такого как 1,2-этандиол.
В одном варианте осуществления одна или несколько гидрокси-, тио- или аминогрупп в хинуклидиновом соединении являются модифицированными. Например, одна или несколько гидрокси-, тио- и/или аминогрупп в хинуклидиновом соединении могут быть модифицированы с образованием производных кислот, например, сложных эфиров, сложных тиоэфиров (или сложных тиоловых эфиров) и/или амидов. Производные кислот могут быть образованы, например, посредством осуществления реакции хинуклидинового соединения, которое содержит одну или несколько гидрокси-, тио- или аминогрупп, с ацетилирующим средством. Примеры ацетилирующих средств предусматривают ангидриды, такие как ангидрид уксусной кислоты, хлорангидриды, такие как бензилхлорид, и дикарбонаты, такие как ди-трет-бутилдикарбонат.
Стереохимия
Настоящее изобретение дополнительно охватывает стереоизомеры и смеси стереоизомеров соединений 1 и 1.1-1.75. Стереоизомеры (например, цис- и транс-изомеры) и все оптические изомеры раскрытого в данном документе соединения (например, R- и S-энантиомеры), а также рацемические, диастереомерные и другие смеси таких изомеров находятся в пределах объема настоящего изобретения.
В одном варианте осуществления хинуклидин-3-ильная группа хинуклидинового соединения, определенного в данном документе, характеризуется R-конфигурацией. Следовательно, хинуклидиновое соединение может быть выбрано из группы, состоящей из соединений формул (Ia)-(XIIa):
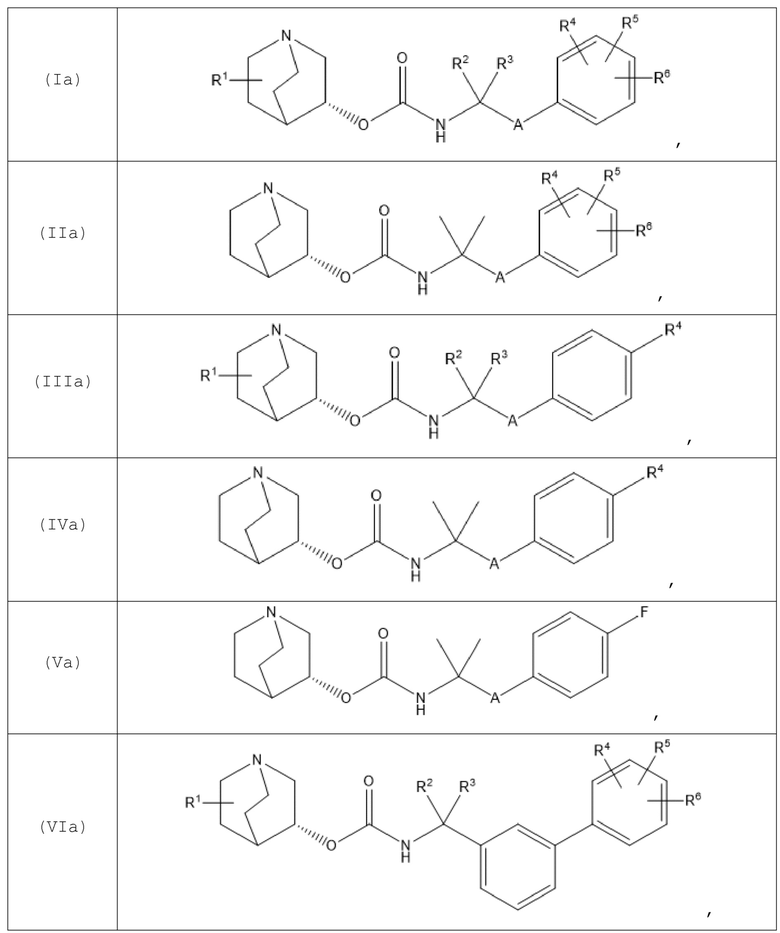
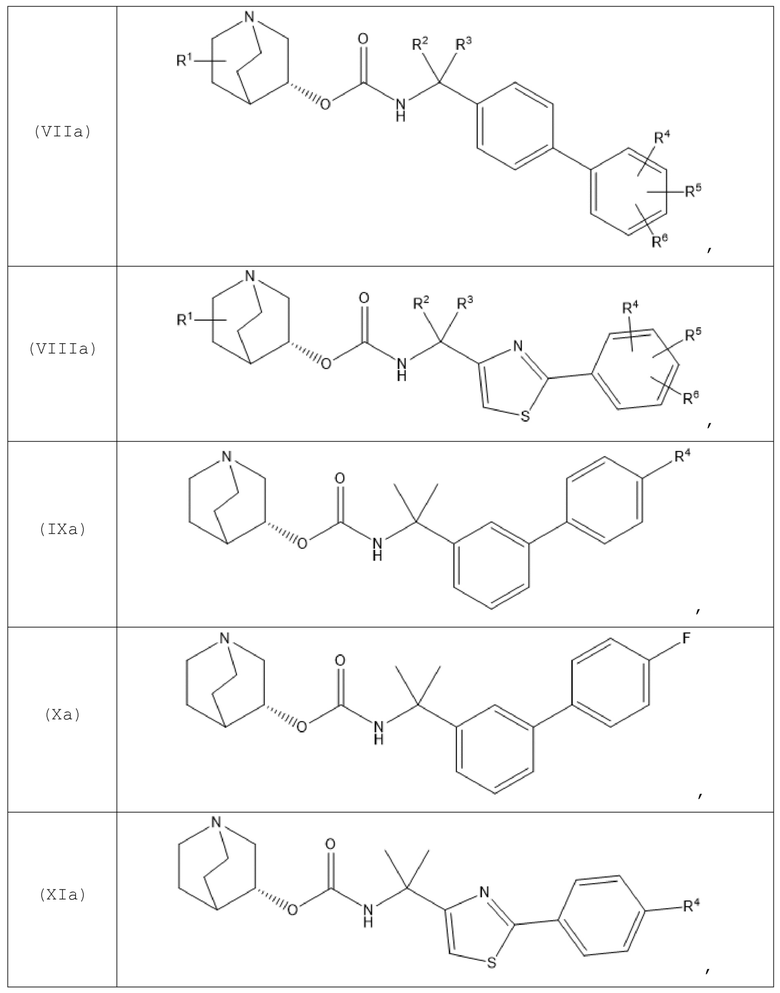
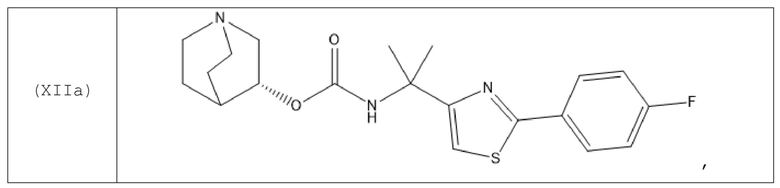
и их фармацевтически приемлемых солей, а также пролекарств на их основе.
В другом варианте осуществления хинуклидин-3-ильная группа хинуклидинового соединения, определенного в данном документе, характеризуется S-конфигурацией. Следовательно, хинуклидиновое соединение может быть выбрано из группы, состоящей из соединений формул (Ib)-(XIIb):
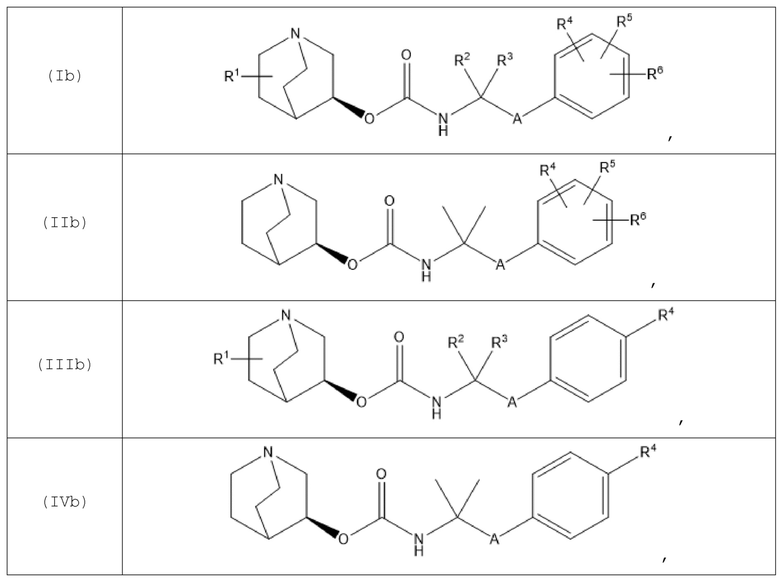
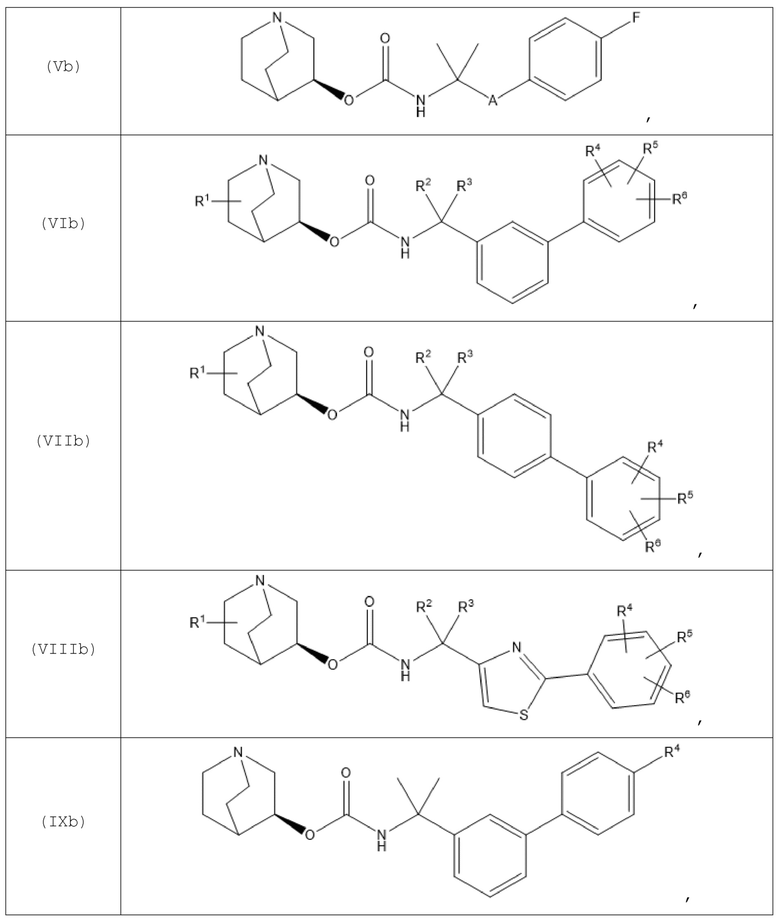
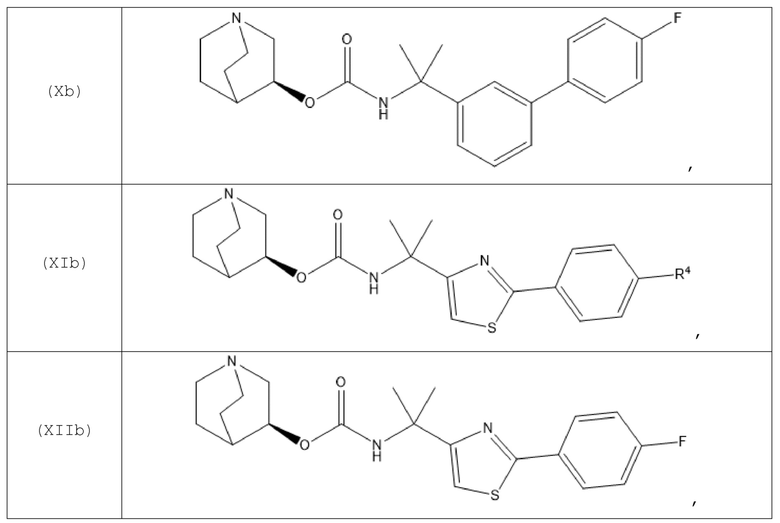
и их фармацевтически приемлемых солей, а также пролекарств на их основе.
В одном варианте осуществления хинуклидиновое соединение представляет собой соединение формулы (Xb), или его фармацевтически приемлемую соль, или пролекарство на его основе. В другом варианте осуществления хинуклидиновое соединение представляет собой соединение формулы (XIIb), или его фармацевтически приемлемую соль, или пролекарство на его основе.
В одном варианте осуществления хинуклидин-3-ильная группа хинуклидинового соединения, определенного в данном документе, существует в смеси изомеров, характеризующихся R- и S-конфигурациями. Например, хинуклидиновое соединение может представлять собой смесь соединений, выбранных из группы, состоящей из соединений формул (Ia) и (Ib), (IIa) и (IIb), (IIIa) и (IIIb), (IVa) и (IVb), (Va) и (Vb), (VIa) и (VIb), (VIIa) и (VIIb), (VIIIa) и (VIIIb), (IXa) и (IXb), (Xa) и (Xb), (XIa) и (XIb) и (XIIa) и (XIIb), а также их фармацевтически приемлемых солей и пролекарств на их основе. В одном варианте осуществления хинуклидиновое соединение представлено в виде рацемической смеси, например, R- и S-изомеры хинуклидин-3-ильной группы присутствуют в приблизительно равных количествах. В другом варианте осуществления хинуклидиновое соединение представлено в виде смеси изомеров, характеризующихся R- и S-конфигурациями, где R- и S-изомеры присутствуют в разных количествах. В одном варианте осуществления S-изомер присутствует в энантиомерном избытке, составляющем по меньшей мере приблизительно 5%, 10%, 25%, 40%, 70%, 80%, 90%, 95%, 97%, 98% или 99%, например, приблизительно 100%. В другом варианте осуществления R-изомер присутствует в энантиомерном избытке, составляющем по меньшей мере приблизительно 5%, 10%, 25%, 40%, 70%, 80%, 90%, 95%, 97%, 98% или 99%, например, приблизительно 100%.
Способы получения энантиомерно обогащенных и/или энантиомерно чистых хинуклидиновых соединений будут очевидны специалисту в данной области техники на основании настоящего раскрытия.
Соединения, раскрытые в данном документе, могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, а также кетоформу и енаминовую форму и их геометрические изомеры и смеси. Таутомеры существуют в виде смесей набора таутомеров в растворе. В твердой форме обычно преобладает один таутомер. Даже если может быть описан один таутомер, все таутомеры находятся в пределах объема настоящего изобретения.
Атропоизомеры также находятся в пределах объема настоящего изобретения. Атропоизомеры относятся к соединениям, которые могут быть разделены на изомеры с ограниченным вращением.
Другие формы
Настоящее изобретение дополнительно охватывает гидраты, сольваты и полиморфы соединения 1 и 1.1-1.75. Фармацевтически приемлемые гидраты, сольваты и полиморфы хинуклидиновых соединений, описанных в данном документе, находятся в пределах объема настоящего изобретения. Хинуклидиновые соединения, описанные в данном документе, могут иметь аморфную форму и/или одну или несколько кристаллических форм.
Изотопно-меченные соединения также находятся в пределах объема настоящего изобретения. Используемый в данном документе термин "изотопно-меченное соединение" относится к раскрытому в данном документе соединению, в том числе к его фармацевтически приемлемым солям и пролекарствам на его основе, каждое из которых описано в данном документе, в котором один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в состав соединений, раскрытых в данном документе, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl соответственно.
Медицинские показания
Хинуклидиновые соединения и содержащие их фармацевтические композиции, описанные в данном документе, применимы в терапии, в частности в терапевтическом лечении неврологических нарушений, в том числе деменции и нарушений походки, например, у пациента, имеющего такое заболевание, как болезнь Гоше. Субъекты, подлежащие лечению согласно способам, описанным в данном документе, предусматривают позвоночных, таких как млекопитающие. В конкретных вариантах осуществления млекопитающим является пациент-человек.
Как обсуждалось выше, одним из характерных признаков болезней накопления гликогена является аномальное накопление различных гликолипидов или гликосфинголипидов в клетках организма. Это накопление является как причиной наблюдаемых симптомов и признаков заболевания, так и диагностическим маркером, свидетельствующим о наличии и/или прогрессировании заболевания. Используемая в данном документе фраза "заметное накопление" в отношении измерения уровня GL-3, GL-1 и других биомаркеров в плазме крови, коже или других мягких тканях означает накопление указанного соединения в концентрации, которая более чем на 25% выше максимальной нормальной концентрации. В некоторых вариантах осуществления "заметное накопление" означает превышение максимальной нормальной концентрации указанного соединения более чем на 50%.
В первом аспекте в настоящем изобретении предусмотрен способ (способ 1) лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления, у субъекта, такого как субъект, нуждающийся в этом, при этом способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75. Также предусмотрено хинуклидиновое соединение, описанное в данном документе, например, соединение согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1 или 1.1-1.75, для применения в способе лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления, у субъекта, нуждающегося в этом, например, для применения в способе 1 или любом из 1.1-1.64. Дополнительно предусмотрено применение хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75 в изготовлении лекарственного препарата для применения в способе лечения или предупреждения когнитивной дисфункции и/или отклонений походки, в том числе атаксии, ассоциированной с лизосомной болезнью накопления, у субъекта, нуждающегося в этом, например, в изготовлении лекарственного препарата для применения в способе 1 или любом из 1.1-1.64.
В конкретных дополнительных вариантах осуществления способа 1 в настоящем изобретении предусмотрены следующие способы.
1.1. Способ 1, где способ включает введение субъекту эффективного количества соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или любых из 1.1-1.75;
1.2. Способ 1, где способ включает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
1.3. Способ 1 или любой из 1.1-1.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1, или любое из 1.1-1.75;
1.4. Способ 1 или любой из 1.1-1.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
1.5. Способ 1.3 или 1.4, где фармацевтическая композиция дополнительно содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, описанное в данном документе;
1.6. Способ 1 или любой из 1.1-1.5, где способ включает введение фармацевтической лекарственной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
1.7. Способ 1.6, где лекарственная форма представляет собой лекарственную форму для перорального введения (например, пилюлю, капсулу, каплету, таблетку, драже, порошок, гранулу, пленку, пастилку или жидкость);
1.8. Способ 1.7, где лекарственная форма представляет собой жевательную таблетку;
1.9. Способ 1.6, где лекарственная форма представляет собой лекарственную форму для парентерального введения (например, где фармацевтическая композиция составлена для инъекции);
1.10. Способ 1.9, где инъекция представляет собой внутривенную, внутримышечную, интратекальную или подкожную инъекцию, необязательно стерильную инъекцию;
1.11. Способ 1.6, где лекарственная форма представляет собой лекарственную форму для местного или ректального применения;
1.12. Способ 1.6, где лекарственная форма представляет собой лекарственную форму для интраназального введения (например, аэрозоль);
1.13. Способ 1 или любой из 1.1-1.12, где способ дополнительно включает одновременное введение второго активного средства, например, второго соединения, способного обеспечивать лечение или предупреждение когнитивной дисфункции и/или отклонений походки у нуждающегося в этом пациента, как описано в данном документе;
1.14. Способ 1.13, где второе активное средство вводят в той же фармацевтической композиции или лекарственной форме, что и хинуклидиновое соединение;
1.15. Способ 1.13 или 1.14, где второе активное средство представляет собой ингибитор GCS (например, миглустат или элиглустат);
1.16. Способ 1 или любой из 1.1-1.15, где субъектом является животное, представляющее собой млекопитающее;
1.17. Способ 1.16, где субъектом является животное, являющееся приматом;
1.18. Способ 1.17, где субъектом является человек;
1.19. Способ 1 или любой из 1.1-1.18, где атаксия представляет собой мозжечковую атаксию;
1.20. Способ 1.19, где атаксия проявляется в виде симптомов, выбранных из нестабильности походки, астении, асинергии, замедленного времени реакции, дисхронометрии, дизартрии, дисфагии, гипотонии, дисметрии, гипометрии, гиперметрии, дисдиадохокинезии, невнятной речи, тремора голоса, атаксического дыхания, постуральной неустойчивости и их комбинации, например, где первичный атаксический дефицит представляет собой неустойчивость походки;
1.21. Способ 1.19 или 1.20, где субъект характеризуется исходной атаксией, соответствующей по меньшей мере 0,5 по Шкале для оценки и определения степени атаксии (SARA) в начале терапии согласно способу, например, исходный показатель согласно SARA составляет по меньшей мере 1, или по меньшей мере 2, или по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5, или по меньшей мере 10, или по меньшей мере 20;
1.22. Способ 1 или любой из 1.1-1.21, где когнитивная дисфункция представляет собой деменцию;
1.23. Способ 1.22, где деменция демонстрирует признаки дефектов скорости визуального поиска, скорости обработки информации при чтении, гибкости мышления и/или исполнительного функционирования, например, что подтверждается прохождением TMT-A за более чем 30 секунд или более чем 45 секунд, или более чем 60 секунд, и/или прохождением TMT-B за более чем 70 секунд, или более чем 90 секунд, или более чем 120 секунд, или более чем 150 секунд, или более чем 180 секунд, и/или где разница между временем прохождения TMT-B и TMT-A составляет более 40 секунд, или более 60 секунд, или более 90 секунд, или более 120 секунд;
1.24. Способ 1 или любой из 1.1-1.23, где у субъекта имеется болезнь Гоше типа 3;
1.25. Способ 1 или любой из 1.1-1.24, где у субъекта имеется болезнь Ниманна-Пика типа C;
1.26. Способ 1 или любой из 1.1-1.24, где у субъекта имеется GM2-ганглиозидоз (например, болезнь Тея-Сакса, болезнь Сандхоффа или вариант AB GM2-ганглиозидоза);
1.27. Способ 1 или любой из 1.1-1.24, где у субъекта диагностирована мутация в гене GBA1;
1.28. Способ 1 или любой из 1.1-1.24, где у субъекта диагностирована мутация в генах NPC1 и/или NPC2;
1.29. Способ 1 или любой из 1.1-1.24, где у субъекта диагностирована мутация в гене HEXA (кодирующем гексозаминидазу A), и/или мутация в гене HEXB (кодирующем гексозаминидазу B), и/или мутация в гене GM2A (кодирующем белок-активатор ганглиозида GM2);
1.30. Способ 1 или любой из 1.1-1.29, где у субъекта диагностирована болезнь Паркинсона;
1.31. Способ 1 или любой из 1.1-1.30, где субъект одновременно подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы (например, имиглюцеразы, велаглюцеразы или талиглюцеразы), где необязательно каждый из таких ферментов представляет собой рекомбинантный фермент;
1.32. Способ 1.31, где субъект одновременно подвергается лечению с применением одной или нескольких из имиглюцеразы, велаглюцеразы (например, велаглюцеразы-альфа) и талиглюцеразы (например, талиглюцеразы-альфа);
1.33. Способ 1.32, где субъект одновременно подвергается лечению с применением имиглюцеразы;
1.34. Способ 1.33, где субъект одновременно подвергается лечению с применением имиглюцеразы в дозировке от 2,5 единиц/кг веса тела до 80 единиц/кг веса тела один раз в 1-3 недели, например, от 40 до 60 единиц/кг веса тела один раз в 2 недели (1 единица имиглюцеразы представляет собой количество фермента, который катализирует гидролиз 1 микромоля синтетического субстрата п-нитрофенил-β-D-глюкопиранозида в минуту при 37°С);
1.35. Способ 1.34, где дозу имиглюцеразы для субъекта при каждом введении (например, один раз в 1-3 недели, например, один раз 2 недели) вводят в виде внутривенной (IV) инфузии в течение периода времени, составляющего 1-3 часа (например, 1-2 часа);
1.36. Способ 1 или любой из 1.1-1.35, где субъекту было введено средство ферментозаместительной терапии (например, имиглюцераза, велаглюцераза и/или талиглюцераза) до начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
1.37. Способ 1.36, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, по меньшей мере 12 месяцев (1 год), или по меньшей мере 18 месяцев, или по меньшей мере 2 лет, или по меньшей мере 3 лет.
1.38. Способ 1.36 или 1.37, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев в стабильной дозе до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
1.39. Способ 1 или любой из 1.1-1.38, где способ дополнительно включает стадию перевода субъекта с ERT-терапии (например, с применением имиглюцеразы, велаглюцеразы или талиглюцеразы) на лечение с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
1.40. Способ 1 или любой из 1.1-1.39, где субъект характеризуется уровнем гемоглобина, составляющим по меньшей мере 11 г/дл в случае женщин и по меньшей мере 12 г/дл в случае мужчин;
1.41. Способ 1 или любой из 1.1-1.40, где количество тромбоцитов у субъекта составляет по меньшей мере 100000/кубический миллиметр;
1.42. Способ 1 или любой из 1.1-1.41, где субъект характеризуется объемом селезенки, составляющим менее чем 10 нормальных объемов (MN), и/или объемом печени, составляющим менее чем 1,5 MN;
1.43. Способ 1 или любой из 1.1-1.42, где у субъекта диагностирована сопутствующая деменция, например, болезнь Альцгеймера или болезнь Паркинсона;
1.44. Способ 1 или любой из 1.1-1.43, где возраст субъекта составляет по меньшей мере 18 лет (например, 18-30 лет) на момент начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
1.45. Способ 1 или любой из 1.1-1.44, где субъект характеризуется концентрацией глюкозилцерамида (GL1), составляющей 4,4-11,1 нг/мл в цереброспинальной жидкости (CSF) и 4,9-8,3 мкг/мл в плазме крови;
1.46. Способ 1 или любой из 1.1-1.45, где субъект характеризуется концентрацией глюкозилсфингозина (лизосомного GL1), составляющей 20,1-67,6 пг/мл в CSF и 8,8-159,0 нг/мл в плазме крови;
1.47. Способ 1 или любой из 1.1-1.46, где субъекту вводят суточную дозу от приблизительно 1 мг до приблизительно 150 мг соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или приблизительно 15 мг, или дозу, выбранную из 2, 5, 15, 25, 50, 100 или 150 мг;
1.48. Способ 1 или любой из 1.1-1.47, где субъектом является взрослый пациент-человек, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
1.49. Способ 1 или любой из 1.1-1.47, где субъектом является пациент детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
1.50. Способ 1 или любой из 1.1-1.49, где способ эффективен для обеспечения снижения по шкале для оценки атаксии SARA на по меньшей мере 0,5, например, снижения показателя согласно SARA на по меньшей мере 1, или на по меньшей мере 2, или на по меньшей мере 3, или на по меньшей мере 5, или на по меньшей мере 10, или где способ эффективен для снижения показателя SARA до значений от 0,00 до 3,00, или от 0,00 до 2,00, или от 0,00 до 1,50, или от 0,00 до 1,00, или от 0,00 до 0,50.
1.51. Способ 1 или любой из 1.1-1.50, где способ эффективен для улучшения когнитивной способности или снижения проявления когнитивных дефицитов, например, как измерено по сокращению времени, необходимого для завершения теста с прокладыванием пути (TMT), TMT-A и/или ТМТ-В, по сокращению разницы между временем прохождения ТМТ-А и временем прохождения ТМТ-В (ТМТ-А - ТМТ-В), например, сокращению на по меньшей мере 10%, или на по меньшей мере 20%, или на по меньшей мере 30%, или на по меньшей мере 40%, или на по меньшей мере 50% (например, где результат прохождения ТМТ-А снижается на 5-20%, и/или результат прохождения ТМТ-В снижается на 25-30%, и/или [ТМТ-А - ТМТ-B] снижается на 25-30%);
1.52. Способ 1 или любой из 1.1-1.51, где способ приводит к снижению концентрации глюкозилцерамида в CSF и/или в плазме крови на по меньшей мере 30% через 6 месяцев лечения, например, на по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70%;
1.53. Способ 1 или любой из 1.1-1.52, где способ приводит к повышению концентрации глюкозилсфингозина в CSF и/или в плазме крови на по меньшей мере 30% через 6 месяцев лечения, например, на по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70%;
1.54. Способ 1 или любой из 1.1-1.53, где способ приводит к статистически или клинически неизменному значению согласно Модифицированному инструменту для оценки тяжести (mSST) неврологического заболевания через 6 месяцев лечения;
1.55. Способ 1 или любой из 1.1-1.54, где способ приводит к усилению кровотока в головном мозге (например, в одной или нескольких лобных, затылочных, теменных или височных долях), например, как показано посредством fMRI-визуализации;
1.56. Способ 1 или любой из 1.1-1.54, где способ приводит к усилению узловых связей в головном мозге (например, между задними и передними отделами головного мозга и/или между затылочно-теменными структурами и лобными, височными и/или лимбическими структурами), например, как показано посредством fMRI-визуализации;
1.57. Способ 1 или любой из 1.1-1.56, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством системного введения, например, парентеральным путем или путем, отличным от парентерального;
1.58. Способ 1.57, где путь введения является пероральным (энтеральным);
1.59. Способ 1.57, где путь введения является парентеральным, например, посредством инъекции, такой как внутривенная инъекция;
1.60. Способ 1 или любой из 1.1-1.59, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством локального введения, например, посредством местного введения;
1.61. Способ 1 или любой из 1.1-1.60, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
1.62. Способ 1.61, где дозировка соединения составляет 15 мг/сутки при пероральном введении;
1.63. Способ 1.62, где дозировка соединения составляет 15 мг/сутки в виде однократной дозы для перорального введения;
1.64. Способ 1 или любой из 1.1-1.63, где субъекту вводят однократную суточную дозу, составляющую 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в виде малатной соли в форме соли присоединения кислоты.
В некоторых вариантах осуществления настоящего изобретения у субъекта диагностировано наличие конкретного заболевания или нарушения и также диагностировано наличие конкретной генетической мутации, например, такой, которая, как известно, вызывает рассматриваемое заболевание или нарушение, хотя часто невозможно доказать, что заболевание или нарушение у конкретного пациента вызвано конкретной мутацией, наличие которой у человека было диагностировано. Используемый таким образом термин "с диагнозом конкретной генетической мутации" означает, что субъект или пациент был подвергнут тестированию, например, посредством секвенирования ДНК или РНК, профилирования белков или других подходящих способов, и у него была обнаружена рассматриваемая мутация. Однако, как обсуждается далее ниже, многие генетические заболевания и нарушения могут иметь несколько генетических причин (например, мутации), и у пациентов может быть несколько мутаций, каждая из которых при некоторых обстоятельствах может быть достаточной, чтобы вызвать заболевание или нарушение, при этом не требуется доказывать, что конкретная мутация вызывает конкретное заболевание или нарушение у конкретного пациента.
Способы согласно способу 1 et seq. могут быть полезными для субъектов, у которых была диагностирована лизосомная болезнь накопления, такая как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа С, но которые еще не испытывают когнитивных и/или атаксических симптомов, ассоциированных с болезненным состоянием. Способы согласно способу 1 et seq. также могут быть полезными для субъектов, которые подвержены риску развития лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, вследствие, например, мутации у субъекта или в семейной родословной субъекта, которая, как известно, вызывает такое заболевание. Таким образом, в некоторых вариантах осуществления способов, описанных в данном документе, субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения, и способ обеспечивает предупреждение или задержку начала проявления и/или развития когнитивных и/или атаксических симптомов заболевания или нарушения у субъекта. В некоторых вариантах осуществления субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения в силу наличия мутации в гене, как описано в данном документе.
Во втором аспекте в настоящем изобретении предусмотрен способ (способ 2) улучшения нейронных связей в головном мозге субъекта, например, субъекта, нуждающегося в этом, при этом способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75. Также предусмотрено хинуклидиновое соединение, описанное в данном документе, например, соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1 или 1.1-1.75 для улучшения нейронных связей в головном мозге субъекта, например, для применения в способе 2 или любом из 2.1-2.67. Дополнительно предусмотрено применение хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75 в изготовлении лекарственного препарата для улучшения нейронных связей в головном мозге субъекта, например, в изготовлении лекарственного препарата для применения в способе 2 или любом из 2.1-2.67.
В конкретных дополнительных вариантах осуществления способа 2 в настоящем изобретении предусмотрены следующие способы.
2.1. Способ 2, где способ включает введение субъекту эффективного количества соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или любых из 1.1-1.75;
2.2. Способ 2, где способ включает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
2.3. Способ 2 или любой из 2.1-2.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1, или любое из 1.1-1.75;
2.4. Способ 2 или любой из 2.1-2.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
2.5. Способ 2.3 или 2.4, где фармацевтическая композиция дополнительно содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, описанное в данном документе;
2.6. Способ 2 или любой из 2.1-2.5, где способ включает введение фармацевтической лекарственной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
2.7. Способ 2.6, где лекарственная форма представляет собой лекарственную форму для перорального введения (например, пилюлю, капсулу, каплету, таблетку, драже, порошок, гранулу, пленку, пастилку или жидкость);
2.8. Способ 2.7, где лекарственная форма представляет собой жевательную таблетку;
2.9. Способ 2.6, где лекарственная форма представляет собой лекарственную форму для парентерального введения (например, где фармацевтическая композиция составлена для инъекции);
2.10. Способ 2.9, где инъекция представляет собой внутривенную, внутримышечную, интратекальную или подкожную инъекцию, необязательно стерильную инъекцию;
2.11. Способ 2.6, где лекарственная форма представляет собой лекарственную форму для местного или ректального применения;
2.12. Способ 2.6, где лекарственная форма представляет собой лекарственную форму для интраназального введения (например, аэрозоль);
2.13. Способ 2 или любой из 2.1-2.12, где способ дополнительно включает одновременное введение второго активного средства, например, второго соединения, способного обеспечивать снижение уровней гликозилцерамида у нуждающегося в этом пациента, как описано в данном документе;
2.14. Способ 2.13, где второе активное средство вводят в той же фармацевтической композиции или лекарственной форме, что и хинуклидиновое соединение;
2.15. Способ 2.13 или 2.14, где второе активное средство представляет собой ингибитор GCS (например, миглустат или элиглустат);
2.16. Способ 2 или любой из 2.1-2.15, где субъектом является животное, представляющее собой млекопитающее;
2.17. Способ 2.16, где субъектом является животное, являющееся приматом;
2.18. Способ 2.17, где субъектом является человек;
2.19. Способ 2 или любой из 2.1-2.18, где у субъекта имеется атаксия, например, симптомы, выбранные из нестабильности походки, астении, асинергии, замедленного времени реакции, дисхронометрии, дизартрии, дисфагии, гипотонии, дисметрии, гипометрии, гиперметрии, дисдиадохокинезии, невнятной речи, тремора голоса, атаксического дыхания, постуральной неустойчивости и их комбинации, например, где первичный атаксический дефицит представляет собой неустойчивость походки;
2.20. Способ 2.19, где субъект характеризуется исходной атаксией, соответствующей по меньшей мере 0,5 по Шкале для оценки и определения степени атаксии (SARA) в начале терапии согласно способу, например, исходный показатель согласно SARA составляет по меньшей мере 1, или по меньшей мере 2, или по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5, или по меньшей мере 10, или по меньшей мере 20;
2.21. Способ 2 или любой из 2.1-2.20, где у субъекта имеется когнитивная дисфункция (например, деменция);
2.22. Способ 2.21, где когнитивная дисфункция представляет собой деменцию;
2.23. Способ 2.22, где деменция демонстрирует признаки дефектов скорости визуального поиска, скорости обработки информации при чтении, гибкости мышления и/или исполнительного функционирования, например, что подтверждается прохождением TMT-A за более чем 30 секунд или более чем 45 секунд, или более чем 60 секунд, и/или прохождением TMT-B за более чем 70 секунд, или более чем 90 секунд, или более чем 120 секунд, или более чем 150 секунд, или более чем 180 секунд, и/или где разница между временем прохождения TMT-B и TMT-A составляет более 40 секунд, или более 60 секунд, или более 90 секунд, или более 120 секунд;
2.24. Способ 2 или любой из 2.1-2.23, где у субъекта имеется болезнь Гоше типа 3;
2.25. Способ 2 или любой из 2.1-2.24, где у субъекта имеется болезнь Ниманна-Пика типа C;
2.26. Способ 2 или любой из 2.1-2.24, где у субъекта имеется GM2-ганглиозидоз (например, болезнь Тея-Сакса, болезнь Сандхоффа или вариант AB GM2-ганглиозидоза);
2.27. Способ 2 или любой из 2.1-2.24, где у субъекта диагностирована мутация в гене GBA1;
2.28. Способ 2 или любой из 2.1-2.24, где у субъекта диагностирована мутация в генах NPC1 и/или NPC2;
2.29. Способ 2 или любой из 2.1-2.24, где у субъекта диагностирована мутация в гене HEXA (кодирующем гексозаминидазу A), и/или мутация в гене HEXB (кодирующем гексозаминидазу B), и/или мутация в гене GM2A (кодирующем белок-активатор ганглиозида GM2);
2.30. Способ 2 или любой из 2.1-2.29, где у субъекта диагностирована болезнь Паркинсона;
2.31. Способ 2 или любой из 2.1-2.30, где субъект одновременно подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы (например, имиглюцеразы, велаглюцеразы или талиглюцеразы), где необязательно каждый из таких ферментов представляет собой рекомбинантный фермент;
2.32. Способ 2.31, где субъект одновременно подвергается лечению с применением одной или нескольких из имиглюцеразы, велаглюцеразы (например, велаглюцеразы-альфа) и талиглюцеразы (например, талиглюцеразы-альфа);
2.33. Способ 2.32, где субъект одновременно подвергается лечению с применением имиглюцеразы;
2.34. Способ 2.33, где субъект одновременно подвергается лечению с применением имиглюцеразы в дозировке от 2,5 единиц/кг веса тела до 80 единиц/кг веса тела один раз в 1-3 недели, например, от 40 до 60 единиц/кг веса тела один раз в 2 недели (1 единица имиглюцеразы представляет собой количество фермента, который катализирует гидролиз 1 микромоля синтетического субстрата п-нитрофенил-β-D-глюкопиранозида в минуту при 37°С);
2.35. Способ 2.34, где дозу имиглюцеразы для субъекта при каждом введении (например, один раз в 1-3 недели, например, один раз 2 недели) вводят в виде внутривенной (IV) инфузии в течение периода времени, составляющего 1-3 часа (например, 1-2 часа);
2.36. Способ 2 или любой из 2.1-2.35, где субъекту было введено средство ферментозаместительной терапии (например, имиглюцераза, велаглюцераза и/или талиглюцераза) до начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
2.37. Способ 2.36, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, по меньшей мере 12 месяцев (1 год), или по меньшей мере 18 месяцев, или по меньшей мере 2 лет, или по меньшей мере 3 лет.
2.38. Способ 2.36 или 2.37, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев в стабильной дозе до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
2.39. Способ 2 или любой из 2.1-2.38, где способ дополнительно включает стадию перевода субъекта с ERT-терапии (например, с применением имиглюцеразы, велаглюцеразы или талиглюцеразы) на лечение с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
2.40. Способ 2 или любой из 2.1-2.39, где субъект характеризуется уровнем гемоглобина, составляющим по меньшей мере 11 г/дл в случае женщин и по меньшей мере 12 г/дл в случае мужчин;
2.41. Способ 2 или любой из 2.1-2.40, где количество тромбоцитов у субъекта составляет по меньшей мере 100000/кубический миллиметр;
2.42. Способ 2 или любой из 2.1-2.41, где субъект характеризуется объемом селезенки, составляющим менее чем 10 нормальных объемов (MN), и/или объемом печени, составляющим менее чем 1,5 MN;
2.43. Способ 2 или любой из 2.1-2.42, где у субъекта диагностирована сопутствующая деменция, например, болезнь Альцгеймера или болезнь Паркинсона;
2.44. Способ 2 или любой из 2.1-2.43, где возраст субъекта составляет по меньшей мере 18 лет (например, 18-30 лет) на момент начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
2.45. Способ 2 или любой из 2.1-2.44, где субъект характеризуется концентрацией глюкозилцерамида (GL1), составляющей 4,4-11,1 нг/мл в цереброспинальной жидкости (CSF) и 4,9-8,3 мкг/мл в плазме крови;
2.46. Способ 2 или любой из 2.1-2.45, где субъект характеризуется концентрацией глюкозилсфингозина (лизосомного GL1), составляющей 20,1-67,6 пг/мл в CSF и 8,8-159,0 нг/мл в плазме крови;
2.47. Способ 2 или любой из 2.1-2.46, где субъекту вводят суточную дозу от приблизительно 1 мг до приблизительно 150 мг соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или приблизительно 15 мг, или дозу, выбранную из 2, 5, 15, 25, 50, 100 или 150 мг;
2.48. Способ 2 или любой из 2.1-2.47, где субъектом является взрослый пациент-человек, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
2.49. Способ 2 или любой из 2.1-2.47, где субъектом является пациент детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
2.50. Способ 2 или любой из 2.1-2.49, где способ эффективен для обеспечения снижения по шкале для оценки атаксии SARA на по меньшей мере 0,5, например, снижения показателя согласно SARA на по меньшей мере 1, или на по меньшей мере 2, или на по меньшей мере 3, или на по меньшей мере 5, или на по меньшей мере 10, или где способ эффективен для снижения показателя SARA до значений от 0,00 до 3,00, или от 0,00 до 2,00, или от 0,00 до 1,50, или от 0,00 до 1,00, или от 0,00 до 0,50.
2.51. Способ 2 или любой из 2.1-2.50, где способ эффективен для улучшения когнитивной способности или снижения проявления когнитивных дефицитов, например, как измерено по сокращению времени, необходимого для завершения теста с прокладыванием пути (TMT), TMT-A и/или ТМТ-В, по сокращению разницы между временем прохождения ТМТ-А и временем прохождения ТМТ-В (ТМТ-А - ТМТ-В), например, сокращению на по меньшей мере 10%, или на по меньшей мере 20%, или на по меньшей мере 30%, или на по меньшей мере 40%, или на по меньшей мере 50% (например, где результат прохождения ТМТ-А снижается на 5-20%, и/или результат прохождения ТМТ-В снижается на 25-30%, и/или [ТМТ-А - ТМТ-B] снижается на 25-30%);
2.52. Способ 2 или любой из 2.1-2.51, где способ приводит к снижению концентрации глюкозилцерамида в CSF и/или в плазме крови на по меньшей мере 30% через 6 месяцев лечения, например, на по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70%;
2.53. Способ 2 или любой из 2.1-2.52, где способ приводит к повышению концентрации глюкозилсфингозина в CSF и/или в плазме крови на по меньшей мере 30% через 6 месяцев лечения, например, на по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70%;
2.54. Способ 2 или любой из 2.1-2.53, где способ приводит к статистически или клинически неизменному значению согласно Модифицированному инструменту для оценки тяжести (mSST) неврологического заболевания через 6 месяцев лечения;
2.55. Способ 2 или любой из 2.1-2.54, где способ приводит к усилению кровотока в головном мозге (например, в одной или нескольких лобных, затылочных, теменных или височных долях), например, как показано посредством fMRI-визуализации;
2.56. Способ 2 или любой из 2.1-2.54, где способ приводит к усилению узловых связей в головном мозге (например, между задними и передними отделами головного мозга и/или между затылочно-теменными структурами и лобными, височными и/или лимбическими структурами, например, как показано посредством fMRI-визуализации);
2.57. Способ 2 или любой из 2.1-2.56, где способ приводит к улучшению связей в областях головного мозга, связанных с исполнительной функцией;
2.58. Способ 2 или любой из 2.1-2.57, где способ приводит к повышению эффективности связей в функциональных сетях в состоянии покоя между пассивным режимом работы и медиальными и фронтальными сетями;
2.59. Способ 2 или любой из 2.1-2.58, где способ приводит к улучшению связей между RSN 1, 2 и 3 (восприятие-зрение, познание-речь-орфография, познание-пространство) и RSN 6, 7 и 8 (сенсомоторная функция, слуховая функция и исполнительный контроль);
2.60. Способ 2 или любой из 2.1-2.59, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством системного введения, например, парентеральным путем или путем, отличным от парентерального;
2.61. Способ 2.60, где путь введения является пероральным (энтеральным);
2.62. Способ 2.60, где путь введения является парентеральным, например, посредством инъекции, такой как внутривенная инъекция;
2.63. Способ 2 или любой из 2.1-2.62, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством локального введения, например, посредством местного введения;
2.64. Способ 2 или любой из 2.1-2.63, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
2.65. Способ 2.64, где дозировка соединения составляет 15 мг/сутки при пероральном введении;
2.66. Способ 2.65, где дозировка соединения составляет 15 мг/сутки в виде однократной дозы для перорального введения;
2.67. Способ 2 или любой из 2.1-2.66, где субъекту вводят однократную суточную дозу, составляющую 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в виде малатной соли в форме соли присоединения кислоты.
Способы согласно способу 2 et seq. могут быть полезными для субъектов, у которых была диагностирована лизосомная болезнь накопления, такая как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа С, но которые еще не испытывают когнитивных и/или атаксических симптомов, ассоциированных с болезненным состоянием. Способы согласно способу 2 et seq. также могут быть полезными для субъектов, которые подвержены риску развития лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, вследствие, например, мутации у субъекта или в семейной родословной субъекта, которая, как известно, вызывает такое заболевание. Таким образом, в некоторых вариантах осуществления способов, описанных в данном документе, субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения, и способ обеспечивает предупреждение или задержку начала проявления и/или развития когнитивных и/или атаксических симптомов заболевания или нарушения у субъекта. В некоторых вариантах осуществления субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения в силу наличия мутации в гене, как описано в данном документе.
В третьем аспекте в настоящем изобретении предусмотрен способ (способ 3) увеличения объема ткани головного мозга или предупреждения задержки потери объема ткани головного мозга у субъекта, например, у субъекта, нуждающегося в этом, при этом указанный способ включает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75. Также предусмотрено хинуклидиновое соединение, описанное в данном документе, например, соединение согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1 или 1.1-1.75 для применения в увеличении объема ткани головного мозга или предупреждении или задержке потери объема ткани головного мозга у субъекта, нуждающегося в этом, например, для применения в способе 3 или любом из 3.1-3.65. Дополнительно предусмотрено применение хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75 в изготовлении лекарственного препарата для увеличения объема ткани головного мозга или предупреждения или задержки потери объема ткани головного мозга у субъекта, нуждающегося в этом, например, в изготовлении лекарственного препарата для применения в способе 3 или любом из 3.1-3.65.
В конкретных дополнительных вариантах осуществления способа 3 в настоящем изобретении предусмотрены следующие способы.
3.1. Способ 3, где способ включает введение субъекту эффективного количества соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или любых из 1.1-1.75;
3.2. Способ 3, где способ включает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
3.3. Способ 3 или любой из 3.1-3.2, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1, или любое из 1.1-1.75;
3.4. Способ 3 или любой из 3.1-3.3, где способ включает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
3.5. Способ 3.3 или 3.4, где фармацевтическая композиция дополнительно содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, описанное в данном документе;
3.6. Способ 3 или любой из 3.1-3.5, где способ включает введение фармацевтической лекарственной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
3.7. Способ 3.6, где лекарственная форма представляет собой лекарственную форму для перорального введения (например, пилюлю, капсулу, каплету, таблетку, драже, порошок, гранулу, пленку, пастилку или жидкость);
3.8. Способ 3.7, где лекарственная форма представляет собой жевательную таблетку;
3.9. Способ 3.6, где лекарственная форма представляет собой лекарственную форму для парентерального введения (например, где фармацевтическая композиция составлена для инъекции);
3.10. Способ 3.9, где инъекция представляет собой внутривенную, внутримышечную, интратекальную или подкожную инъекцию, необязательно стерильную инъекцию;
3.11. Способ 3.6, где лекарственная форма представляет собой лекарственную форму для местного или ректального применения;
3.12. Способ 3.6, где лекарственная форма представляет собой лекарственную форму для интраназального введения (например, аэрозоль);
3.13. Способ 3 или любой из 3.1-3.12, где способ дополнительно включает одновременное введение второго активного средства, например, второго соединения, способного обеспечивать снижение уровней гликозилцерамида у нуждающегося в этом пациента, как описано в данном документе;
3.14. Способ 3.13, где второе активное средство вводят в той же фармацевтической композиции или лекарственной форме, что и хинуклидиновое соединение;
3.15. Способ 3.13 или 3.14, где второе активное средство представляет собой ингибитор GCS (например, миглустат или элиглустат);
3.16. Способ 3 или любой из 3.1-3.15, где субъектом является животное, представляющее собой млекопитающее;
3.17. Способ 3.16, где субъектом является животное, являющееся приматом;
3.18. Способ 3.17, где субъектом является человек;
3.19. Способ 3 или любой из 3.1-3.18, где у субъекта имеется атаксия, например, симптомы, выбранные из нестабильности походки, астении, асинергии, замедленного времени реакции, дисхронометрии, дизартрии, дисфагии, гипотонии, дисметрии, гипометрии, гиперметрии, дисдиадохокинезии, невнятной речи, тремора голоса, атаксического дыхания, постуральной неустойчивости и их комбинации, например, где первичный атаксический дефицит представляет собой неустойчивость походки;
3.20. Способ 3.19, где субъект характеризуется исходной атаксией, соответствующей по меньшей мере 0,5 по Шкале для оценки и определения степени атаксии (SARA) в начале терапии согласно способу, например, исходный показатель согласно SARA составляет по меньшей мере 1, или по меньшей мере 2, или по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5, или по меньшей мере 10, или по меньшей мере 20;
3.21. Способ 3 или любой из 3.1-3.20, где у субъекта имеется когнитивная дисфункция (например, деменция);
3.22. Способ 3.21, где когнитивная дисфункция представляет собой деменцию;
3.23. Способ 3.22, где деменция демонстрирует признаки дефектов скорости визуального поиска, скорости обработки информации при чтении, гибкости мышления и/или исполнительного функционирования, например, что подтверждается прохождением TMT-A за более чем 30 секунд или более чем 45 секунд, или более чем 60 секунд, и/или прохождением TMT-B за более чем 70 секунд, или более чем 90 секунд, или более чем 120 секунд, или более чем 150 секунд, или более чем 180 секунд, и/или где разница между временем прохождения TMT-B и TMT-A составляет более 40 секунд, или более 60 секунд, или более 90 секунд, или более 120 секунд;
3.24. Способ 3 или любой из 3.1-3.23, где у субъекта имеется болезнь Гоше типа 3;
3.25. Способ 3 или любой из 3.1-3.24, где у субъекта имеется болезнь Ниманна-Пика типа C;
3.26. Способ 3 или любой из 3.1-3.24, где у субъекта имеется GM2-ганглиозидоз (например, болезнь Тея-Сакса, болезнь Сандхоффа или вариант AB GM2-ганглиозидоза);
3.27. Способ 3 или любой из 3.1-3.24, где у субъекта диагностирована мутация в гене GBA1;
3.28. Способ 3 или любой из 3.1-3.24, где у субъекта диагностирована мутация в генах NPC1 и/или NPC2;
3.29. Способ 3 или любой из 3.1-3.24, где у субъекта диагностирована мутация в гене HEXA (кодирующем гексозаминидазу A), и/или мутация в гене HEXB (кодирующем гексозаминидазу B), и/или мутация в гене GM2A (кодирующем белок-активатор ганглиозида GM2);
3.30. Способ 3 или любой из 3.1-3.29, где у субъекта диагностирована болезнь Альцгеймера или болезнь Паркинсона;
3.31. Способ 3 или любой из 3.1-3.30, где субъект одновременно подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы (например, имиглюцеразы, велаглюцеразы или талиглюцеразы), где необязательно каждый из таких ферментов представляет собой рекомбинантный фермент;
3.32. Способ 3.31, где субъект одновременно подвергается лечению с применением одной или нескольких из имиглюцеразы, велаглюцеразы (например, велаглюцеразы-альфа) и талиглюцеразы (например, талиглюцеразы-альфа);
3.33. Способ 3.32, где субъект одновременно подвергается лечению с применением имиглюцеразы;
3.34. Способ 3.33, где субъект одновременно подвергается лечению с применением имиглюцеразы в дозировке от 2,5 единиц/кг веса тела до 80 единиц/кг веса тела один раз в 1-3 недели, например, от 40 до 60 единиц/кг веса тела один раз в 2 недели (1 единица имиглюцеразы представляет собой количество фермента, который катализирует гидролиз 1 микромоля синтетического субстрата п-нитрофенил-β-D-глюкопиранозида в минуту при 37°С);
3.35. Способ 3.34, где дозу имиглюцеразы для субъекта при каждом введении (например, один раз в 1-3 недели, например, один раз 2 недели) вводят в виде внутривенной (IV) инфузии в течение периода времени, составляющего 1-3 часа (например, 1-2 часа);
3.36. Способ 3 или любой из 3.1-3.35, где субъекту было введено средство ферментозаместительной терапии (например, имиглюцераза, велаглюцераза и/или талиглюцераза) до начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
3.37. Способ 3.36, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, по меньшей мере 12 месяцев (1 год), или по меньшей мере 18 месяцев, или по меньшей мере 2 лет, или по меньшей мере 3 лет.
3.38. Способ 3.36 или 3.37, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев в стабильной дозе до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
3.39. Способ 3 или любой из 3.1-3.38, где способ дополнительно включает стадию перевода субъекта с ERT-терапии (например, с применением имиглюцеразы, велаглюцеразы или талиглюцеразы) на лечение с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
3.40. Способ 3 или любой из 3.1-3.39, где субъект характеризуется уровнем гемоглобина, составляющим по меньшей мере 11 г/дл в случае женщин и по меньшей мере 12 г/дл в случае мужчин;
3.41. Способ 3 или любой из 3.1-3.40, где количество тромбоцитов у субъекта составляет по меньшей мере 100000/кубический миллиметр;
3.42. Способ 3 или любой из 3.1-3.41, где субъект характеризуется объемом селезенки, составляющим менее чем 10 нормальных объемов (MN), и/или объемом печени, составляющим менее чем 1,5 MN;
3.43. Способ 3 или любой из 3.1-3.42, где у субъекта диагностирована сопутствующая деменция, например, болезнь Альцгеймера или болезнь Паркинсона;
3.44. Способ 3 или любой из 3.1-3.43, где возраст субъекта составляет по меньшей мере 18 лет (например, 18-30 лет) на момент начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
3.45. Способ 3 или любой из 3.1-3.44, где субъект характеризуется концентрацией глюкозилцерамида (GL1), составляющей 4,4-11,1 нг/мл в цереброспинальной жидкости (CSF) и 4,9-8,3 мкг/мл в плазме крови;
3.46. Способ 3 или любой из 3.1-3.45, где субъект характеризуется концентрацией глюкозилсфингозина (лизосомного GL1), составляющей 20,1-67,6 пг/мл в CSF и 8,8-159,0 нг/мл в плазме крови;
3.47. Способ 3 или любой из 3.1-3.46, где субъекту вводят суточную дозу от приблизительно 1 мг до приблизительно 150 мг соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или приблизительно 15 мг, или дозу, выбранную из 2, 5, 15, 25, 50, 100 или 150 мг;
3.48. Способ 3 или любой из 3.1-3.47, где субъектом является взрослый пациент-человек, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
3.49. Способ 3 или любой из 3.1-3.47, где субъектом является пациент детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
3.50. Способ 3 или любой из 3.1-3.49, где способ приводит к увеличению объема ткани головного мозга или предупреждению или задержке потери объема ткани головного мозга в одной или нескольких областях головного мозга, выбранных из правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной области, правого миндалевидного тела, левой области клина и левой лингвальной области, например, по результатам измерений с применением волюметрической магнитно-резонансной визуализации (vMRI).
3.51. Способ 3 или любой из 3.1-3.50, где способ приводит к увеличению объема ткани головного мозга в одной или нескольких областях головного мозга, связанных с исполнительной функцией.
3.52. Способ 3.50 или 3.51, где увеличение объема ткани головного мозга в одной или нескольких областях мозга сопровождается улучшением нейронных связей в одной или нескольких областях головного мозга, например, как показано с применением функциональной магнитно-резонансной визуализации (fMRI).
3.53. Способ 3 или любой из 3.1-3.52, где способ приводит к увеличению объема ткани головного мозга в целом.
3.54. Способ 3 или любой из 3.1-3.53, где увеличение объема ткани головного мозга составляет по меньшей мере 5 мм3 в любой одной или нескольких областях головного мозга, например, по меньшей мере 10 мм3, по меньшей мере 15 мм3, по меньшей мере 20 мм3, по меньшей мере 30 мм3, по меньшей мере 50 мм3, по меньшей мере 70 мм3 или по меньшей мере 90 мм3 в любой одной или нескольких областях головного мозга и/или до 100 мм3 или до 150 мм3 в любой одной или нескольких областях головного мозга.
3.55. Способ 3 или любой из 3.1-3.54, где увеличение объема ткани головного мозга в целом составляет по меньшей мере 5 мм3, например, по меньшей мере 30 мм3, по меньшей мере 60 мм3, по меньшей мере 90 мм3, по меньшей мере 120 мм3, по меньшей мере 150 мм3, по меньшей мере 200 мм3, или по меньшей мере 250 мм3, и/или до 400 мм3, или до 500 мм3 от всего объема головного мозга.
3.56. Способ 3 или любой из 3.1-3.55, где увеличение объема ткани головного мозга составляет по меньшей мере 0,1%, например, от 0,1% до 10,0% по сравнению с исходным объемом ткани головного мозга в любой одной или нескольких областях головного мозга, например, по меньшей мере 0,50%, 0,75%, 1,0%, 2,0% или 5,0% в любой одной или нескольких областях головного мозга.
3.57. Способ 3 или любой из 3.1-3.56, где увеличение объема ткани головного мозга в целом составляет по меньшей мере 0,05%, например, от 0,05% до 0,30% по сравнению с исходным объемом ткани головного мозга в целом, например, по меньшей мере 0,10%, 0,15%, 0,20% или 0,25%.
3.58. Способ 3 или любой из 3.1-3.57, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством системного введения, например, парентеральным путем или путем, отличным от парентерального;
3.59. Способ 3.58, где путь введения является пероральным (энтеральным);
3.60. Способ 3.58, где путь введения является парентеральным, например, посредством инъекции, такой как внутривенная инъекция;
3.61. Способ 3 или любой из 3.1-3.57, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством локального введения, например, посредством местного введения;
3.62. Способ 3 или любой из 3.1-3.61, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил-(2-(4'-фтор-[3,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
3.63. Способ 3.62, где дозировка соединения составляет 15 мг/сутки при пероральном введении;
3.64. Способ 3.63, где дозировка соединения составляет 15 мг/сутки в виде однократной дозы для перорального введения;
3.65. Способ 3 или любой из 3.1-3.61, где субъекту вводят однократную суточную дозу, составляющую 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в виде малатной соли в форме соли присоединения кислоты.
Способы согласно способу 3 et seq. могут быть полезными для субъектов, у которых была диагностирована лизосомная болезнь накопления, такая как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа С, но которые еще не испытывают когнитивных и/или атаксических симптомов, ассоциированных с болезненным состоянием. Способы согласно способу 3 et seq. также могут быть полезными для субъектов, которые подвержены риску развития лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, вследствие, например, мутации у субъекта или в семейной родословной субъекта, которая, как известно, вызывает такое заболевание. Таким образом, в некоторых вариантах осуществления способов, описанных в данном документе, субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения, и способ обеспечивает предупреждение или задержку начала проявления и/или развития когнитивных и/или атаксических симптомов заболевания или нарушения у субъекта. В некоторых вариантах осуществления субъект был диагностирован как характеризующийся риском развития указанного заболевания или нарушения в силу наличия мутации в гене, как описано в данном документе.
Способы согласно способу 3 et seq. направлены на увеличение объема ткани головного мозга, и нет конкретного ограничения в отношении возможных областей головного мозга, которые могут быть преимущественным образом увеличены с точки зрения объема ткани. Соответствующие области головного мозга могут, например, предусматривать правую часть скорлупы, правую постцентральную долю, правое миндалевидное тело, правую лингвальную область, левую область клина, левую лингвальную область, правую верхнюю часть височной доли, правую латеральную часть орбитофронтальной доли, левую околошпорную долю, левую поперечную часть височной доли, правый полюс височной доли, правое прилежащее ядро, левую часть скорлупы, левую часть энторинальной коры, правый бледный шар, правое прилежащее ядро, левый полюс височной доли, правую часть энториальной коры, левое хвостатое ядро, левый полюс лобной доли, правую долю покрышечной части, правую поперечную височную долю, правый гиппокамп, левую парацентральную долю, левую верхнюю часть теменной доли, левую фузиформную долю, левые части STS, правую парацентральную долю, правую медиальную часть височной доли, левую каудальную часть средней лобной доли, правую ростральную часть передней поясной доли, левую часть треугольной доли, левую прецентральную долю, правую часть орбитальной доли, левую часть средней височной доли, левый перешеек поясной доли, правую каудальную часть средней лобной доли, целую правую височную долю, левую нижнюю часть височной доли, правую латеральную часть теменной доли, правую латеральную часть затылочной доли, левую супрамаргинальную долю, целую левую височную долю, правую ростральную часть средней лобной доли, левую ростральную часть средней лобной доли, левую медиальную часть височной доли и левую верхнюю часть лобной доли или любую их комбинацию. Конкретные области головного мозга, которые могут по меньшей мере в некоторых вариантах осуществления демонстрировать наибольшее относительное увеличение объема в результате осуществления способов согласно способу 3 et seq., предусматривают правое прилежащее ядро, левую часть скорлупы, левую часть энторинальной коры, правую часть скорлупы, правую постцентральную долю, левую околошпорную область, правое миндалевидное тело, левую область клина и левую лингвальную область.
Способы мониторинга и оценивания
В четвертом аспекте в настоящем изобретении предусмотрен способ (способ 4) осуществления мониторинга прогрессирования или регрессирования неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта, где субъект подвергается лечению, которое предусматривает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75, при этом указанный способ включает измерение объема ткани головного мозга субъекта в течение определенного периода времени в ходе лечения, например, с применением волюметрической магнитно-резонансной визуализации (vMRI), и оценивание степени любого изменения объема ткани головного мозга за указанный период времени, и необязательно дополнительно включает инициирование или корректирование лечения субъекта посредством введения субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75. Будет понятно, что для оценивания степени любого изменения объема ткани головного мозга в результате лечения измерение объема ткани головного мозга можно проводить, например, в начале вышеуказанного лечения или вскоре после начала лечения (например, в течение от 1 до 14 дней), и либо в конце назначенного периода времени лечения, либо периодически/регулярно (например, один раз в неделю, один раз в месяц, один раз в 2, 3, 4, 6, 9, 12 месяцев и т. д.) в ходе продолжающегося лечения, чтобы оценить и повторно оценить степень любого изменения объема головного мозга в ходе лечения. Накопление сравнительных результатов, относящихся к состоянию субъекта за период времени, в течение которого субъект подвергался лечению, позволяет повысить точность определения состояния субъекта и прогрессирования/регрессирования симптомов заболевания.
В конкретных дополнительных вариантах осуществления способа 4 в настоящем изобретении предусмотрены следующие способы.
4.1. Способ 4, где лечение предусматривает введение субъекту эффективного количества соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или любых из 1.1-1.75;
4.2. Способ 4, где лечение предусматривает введение субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
4.3. Способ 4 или любой из 4.1-4.2, где лечение предусматривает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1, или любое из 1.1-1.75;
4.4. Способ 4 или любой из 4.1-4.2, где лечение предусматривает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
4.5. Способ 4.3 или 4.4, где фармацевтическая композиция дополнительно содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, описанное в данном документе;
4.6. Способ 4 или любой из 4.1-4.5, где лечение предусматривает введение фармацевтической лекарственной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
4.7. Способ 4.6, где лекарственная форма представляет собой лекарственную форму для перорального введения (например, пилюлю, капсулу, каплету, таблетку, драже, порошок, гранулу, пленку, пастилку или жидкость);
4.8. Способ 4.7, где лекарственная форма представляет собой жевательную таблетку;
4.9. Способ 4.6, где лекарственная форма представляет собой лекарственную форму для парентерального введения (например, где фармацевтическая композиция составлена для инъекции);
4.10. Способ 4.9, где инъекция представляет собой внутривенную, внутримышечную, интратекальную или подкожную инъекцию, необязательно стерильную инъекцию;
4.11. Способ 4.6, где лекарственная форма представляет собой лекарственную форму для местного или ректального применения;
4.12. Способ 4.6, где лекарственная форма представляет собой лекарственную форму для интраназального введения (например, аэрозоль);
4.13. Способ 4 или любой из 4.1-4.12, где лечение дополнительно предусматривает одновременное введение второго активного средства, например, второго соединения, способного обеспечивать снижение уровней гликозилцерамида у нуждающегося в этом пациента, как описано в данном документе;
4.14. Способ 4.13, где второе активное средство вводят в той же фармацевтической композиции или лекарственной форме, что и хинуклидиновое соединение;
4.15. Способ 4.13 или 4.14, где второе активное средство представляет собой ингибитор GCS (например, миглустат или элиглустат);
4.16. Способ 4 или любой из 4.1-4.15, где субъектом является животное, представляющее собой млекопитающее;
4.17. Способ 4.16, где субъектом является животное, являющееся приматом;
4.18. Способ 4.17, где субъектом является человек;
4.19. Способ 4 или любой из 4.1-4.18, где у субъекта имеется атаксия, например, симптомы, выбранные из нестабильности походки, астении, асинергии, замедленного времени реакции, дисхронометрии, дизартрии, дисфагии, гипотонии, дисметрии, гипометрии, гиперметрии, дисдиадохокинезии, невнятной речи, тремора голоса, атаксического дыхания, постуральной неустойчивости и их комбинации, например, где первичный атаксический дефицит представляет собой неустойчивость походки;
4.20. Способ 4.19, где субъект характеризуется исходной атаксией, соответствующей по меньшей мере 0,5 по Шкале для оценки и определения степени атаксии (SARA) в начале терапии согласно способу, например, исходный показатель согласно SARA составляет по меньшей мере 1, или по меньшей мере 2, или по меньшей мере 3, или по меньшей мере 4, или по меньшей мере 5, или по меньшей мере 10, или по меньшей мере 20;
4.21. Способ 4 или любой из 4.1-4.20, где у субъекта имеется когнитивная дисфункция (например, деменция);
4.22. Способ 4.21, где когнитивная дисфункция представляет собой деменцию;
4.23. Способ 4.22, где деменция демонстрирует признаки дефектов скорости визуального поиска, скорости обработки информации при чтении, гибкости мышления и/или исполнительного функционирования, например, что подтверждается прохождением TMT-A за более чем 30 секунд или более чем 45 секунд, или более чем 60 секунд, и/или прохождением TMT-B за более чем 70 секунд, или более чем 90 секунд, или более чем 120 секунд, или более чем 150 секунд, или более чем 180 секунд, и/или где разница между временем прохождения TMT-B и TMT-A составляет более 40 секунд, или более 60 секунд, или более 90 секунд, или более 120 секунд;
4.24. Способ 4 или любой из 4.1-4.23, где у субъекта имеется болезнь Гоше типа 3;
4.25. Способ 4 или любой из 4.1-4.24, где у субъекта имеется болезнь Ниманна-Пика типа C;
4.26. Способ 4 или любой из 4.1-4.24, где у субъекта имеется GM2-ганглиозидоз (например, болезнь Тея-Сакса, болезнь Сандхоффа или вариант AB GM2-ганглиозидоза);
4.27. Способ 5 или любой из 4.1-4.24, где у субъекта диагностирована мутация в гене GBA1;
4.28. Способ 5 или любой из 4.1-4.24, где у субъекта диагностирована мутация в генах NPC1 и/или NPC2;
4.29. Способ 5 или любой из 4.1-4.24, где у субъекта диагностирована мутация в гене HEXA (кодирующем гексозаминидазу A), и/или мутация в гене HEXB (кодирующем гексозаминидазу B), и/или мутация в гене GM2A (кодирующем белок-активатор ганглиозида GM2);
4.30. Способ 4 или любой из 4.1-4.29, где у субъекта диагностирована болезнь Альцгеймера или болезнь Паркинсона;
4.31. Способ 4 или любой из 4.1-4.30, где субъект одновременно подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы (например, имиглюцеразы, велаглюцеразы или талиглюцеразы), где необязательно каждый из таких ферментов представляет собой рекомбинантный фермент;
4.32. Способ 4.31, где субъект одновременно подвергается лечению с применением одной или нескольких из имиглюцеразы, велаглюцеразы (например, велаглюцеразы-альфа) и талиглюцеразы (например, талиглюцеразы-альфа);
4.33. Способ 4.32, где субъект одновременно подвергается лечению с применением имиглюцеразы;
4.34. Способ 4.33, где субъект одновременно подвергается лечению с применением имиглюцеразы в дозировке от 2,5 единиц/кг веса тела до 80 единиц/кг веса тела один раз в 1-3 недели, например, от 40 до 60 единиц/кг веса тела один раз в 2 недели (1 единица имиглюцеразы представляет собой количество фермента, который катализирует гидролиз 1 микромоля синтетического субстрата п-нитрофенил-β-D-глюкопиранозида в минуту при 37°С);
4.35. Способ 4.34, где дозу имиглюцеразы для субъекта при каждом введении (например, один раз в 1-3 недели, например, один раз 2 недели) вводят в виде внутривенной (IV) инфузии в течение периода времени, составляющего 1-3 часа (например, 1-2 часа);
4.36. Способ 4 или любой из 4.1-4.35, где субъекту было введено средство ферментозаместительной терапии (например, имиглюцераза, велаглюцераза и/или талиглюцераза) до начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
4.37. Способ 4.36, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, по меньшей мере 12 месяцев (1 год), или по меньшей мере 18 месяцев, или по меньшей мере 2 лет, или по меньшей мере 3 лет;
4.38. Способ 4.36 или 4.37, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев в стабильной дозе до начала терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
4.39. Способ 4 или любой из 4.1-4.38, где способ дополнительно включает стадию перевода субъекта с ERT-терапии (например, с применением имиглюцеразы, велаглюцеразы или талиглюцеразы) на лечение с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
4.40. Способ 4 или любой из 4.1-4.39, где субъект характеризуется уровнем гемоглобина, составляющим по меньшей мере 11 г/дл в случае женщин и по меньшей мере 12 г/дл в случае мужчин;
4.41. Способ 4 или любой из 4.1-4.40, где количество тромбоцитов у субъекта составляет по меньшей мере 100000/кубический миллиметр;
4.42. Способ 4 или любой из 4.1-4.41, где субъект характеризуется объемом селезенки, составляющим менее чем 10 нормальных объемов (MN), и/или объемом печени, составляющим менее чем 1,5 MN;
4.43. Способ 4 или любой из 4.1-4.42, где у субъекта диагностирована сопутствующая деменция, например, болезнь Альцгеймера или болезнь Паркинсона;
4.44. Способ 4 или любой из 4.1-4.43, где возраст субъекта составляет по меньшей мере 18 лет (например, 18-30 лет) на момент начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
4.45. Способ 4 или любой из 4.1-4.44, где субъект характеризуется концентрацией глюкозилцерамида (GL1), составляющей 4,4-11,1 нг/мл в цереброспинальной жидкости (CSF) и 4,9-8,3 мкг/мл в плазме крови;
4.46. Способ 4 или любой из 4.1-4.45, где субъект характеризуется концентрацией глюкозилсфингозина (лизосомного GL1), составляющей 20,1-67,6 пг/мл в CSF и 8,8-159,0 нг/мл в плазме крови;
4.47. Способ 4 или любой из 4.1-4.46, где субъекту вводят суточную дозу от приблизительно 1 мг до приблизительно 150 мг соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или приблизительно 15 мг, или дозу, выбранную из 2, 5, 15, 25, 50, 100 или 150 мг;
4.48. Способ 4 или любой из 4.1-4.47, где субъектом является взрослый пациент-человек, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
4.49. Способ 4 или любой из 4.1-4.47, где субъектом является пациент детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
4.50. Способ 4 или любой из 4.1-4.49, где указанный период времени, в течение которого субъекта подвергают лечению, и в течение которого осуществляют мониторинг объема головного мозга, составляет от 3 месяцев до 24 месяцев, например, от 3 месяцев до 12 месяцев, от 3 месяцев до 6 месяцев, от 6 месяцев до 24 месяцев, от 6 месяцев до 18 месяцев, от 6 месяцев до 12 месяцев, от 12 месяцев до 24 месяцев или от 12 до 18 месяцев;
4.51. Способ 4 или любой из 4.1-4.50, где измерение объема ткани головного мозга субъекта за указанный период времени проводят посредством позитронно-эмиссионной томографии (PET) головного мозга или посредством волюметрической магнитно-резонансной визуализации (vMRI);
4.52. Способ 4.51, где объем ткани головного мозга субъекта измеряют многократно, периодически или регулярно в ходе лечения, например, один раз в неделю, один раз в месяц, один раз в 2, 3, 4, 6, 9, 12 месяцев и т. д.;
4.53. Способ 4 или любой из 4.1-4.52, где если наблюдается уменьшение или отсутствие увеличения объема головного мозга в целом в течение указанного периода времени, то способ дополнительно включает модифицирование лечения посредством повышения дозы соединения формулы (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), вводимого субъекту в ходе лечения, и повторное оценивание степени любого изменения объема ткани головного мозга после дополнительного периода времени в ходе модифицированного лечения с применением повышенной дозы;
4.54. Способ 4 или любой из 4.1-4.53, где если наблюдается уменьшение или отсутствие увеличения объемов в трех или более из следующих областей головного мозга: области правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной области, правого миндалевидного тела, левой области клина и левой лингвальной области, в течение указанного периода времени, то способ дополнительно включает модифицирование лечения посредством повышения дозы соединения формулы (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), вводимого субъекту в ходе лечения, и повторное оценивание степени любого изменения объема ткани головного мозга после дополнительного периода времени в ходе модифицированного лечения с применением повышенной дозы;
4.55. Способ 4 или любой из 4.1-4.54, где лечение приводит к увеличению объема ткани головного мозга или предупреждению или задержке потери объема ткани головного мозга в течение определенного периода времени в одной или нескольких областях головного мозга, выбранных из правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной области, правого миндалевидного тела, левой области клина и левой лингвальной области;
4.56. Способ 4 или любой из 4.1-4.55, где лечение приводит к увеличению объема головного мозга в одной или нескольких областях головного мозга, связанных с исполнительной функцией;
4.57. Способ 4.55 или 4.56, где увеличение объема головного мозга в одной или нескольких областях головного мозга сопровождается улучшением нейронных связей в одной или нескольких областях головного мозга, например, как показано с применением функциональной магнитно-резонансной визуализации (fMRI);
4.58. Способ 4 или любой из 4.1-4.57, где лечение приводит к увеличению объема ткани головного мозга в целом;
4.59. Способ 4 или любой из 4.55-4.58, где увеличение объема ткани головного мозга составляет по меньшей мере 5 мм3 в любой одной или нескольких областях головного мозга, например, по меньшей мере 10 мм3, по меньшей мере 15 мм3, по меньшей мере 20 мм3, по меньшей мере 30 мм3, по меньшей мере 50 мм3, по меньшей мере 70 мм3 или по меньшей мере 90 мм3 в любой одной или нескольких областях головного мозга и/или до 100 мм3 или до 150 мм3 в любой одной или нескольких областях головного мозга.
4.60. Способ 4 или любой из 4.55-4.59, где увеличение объема ткани головного мозга в целом составляет по меньшей мере 5 мм3, например, по меньшей мере 30 мм3, по меньшей мере 60 мм3, по меньшей мере 90 мм3, по меньшей мере 120 мм3, по меньшей мере 150 мм3, по меньшей мере 200 мм3, или по меньшей мере 250 мм3, и/или до 400 мм3, или до 500 мм3 от всего объема головного мозга.
4.61. Способ 4 или любой из 4.55-4.60, где увеличение объема ткани головного мозга составляет по меньшей мере 0,1%, например, от 0,1% до 10,0% по сравнению с исходным объемом ткани головного мозга в любой одной или нескольких областях головного мозга, например, по меньшей мере 0,50%, 0,75%, 1,0%, 2,0% или 5,0% в любой одной или нескольких областях головного мозга.
4.62. Способ 4 или любой из 4.55-4.61, где увеличение объема ткани головного мозга в целом составляет по меньшей мере 0,05%, например, от 0,05% до 0,30% по сравнению с исходным объемом ткани головного мозга в целом, например, по меньшей мере 0,10%, 0,15%, 0,20% или 0,25%.
4.63. Способ 4 или любой из 4.1-4.62, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством системного введения, например, парентеральным путем или путем, отличным от парентерального;
4.64. Способ 4.63, где путь введения является пероральным (энтеральным);
4.65. Способ 4.63, где путь введения является парентеральным, например, посредством инъекции, такой как внутривенная инъекция;
4.66. Способ 4 или любой из 4.1-4.63, где соединение согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любое из соединений 1 или 1.1-1.75), или его фармацевтически приемлемую соль, или пролекарство на его основе вводят посредством локального введения, например, посредством местного введения;
4.67. Способ 4 или любой из 4.1-4.67, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
4.68. Способ 4.63, где дозировка соединения составляет 15 мг/сутки при пероральном введении;
4.69. Способ 4.63, где дозировка соединения составляет 15 мг/сутки в виде однократной дозы для перорального введения;
4.70. Способ 4 или любой из 4.1-4.67, где субъекту вводят однократную суточную дозу, составляющую 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в виде малатной соли в форме соли присоединения кислоты.
Способы согласно способу 4 et seq. в конкретных вариантах осуществления можно осуществлять с применением волюметрической магнитно-резонансной визуализации (vMRI), как описано в данном документе. Аналитические инструменты также можно применять для помощи в оценивании данных vMRI, включая, например, тензорную морфометрию (TBM), как описано в данном документе.
Следует понимать, что в некоторых вариантах осуществления способы 4 et seq. можно в качестве альтернативы рассматривать как способы лечения или предупреждения неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта (например, пациента), нуждающегося в этом, при этом способ включает мониторинг прогрессирования или регрессирования неврологического нарушения, ассоциированного с лизосомной болезнью накопления, где субъекта подвергают лечению, которое предусматривает введение субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75, и где указанный способ включает стадии измерения объема ткани головного мозга субъекта за период времени в ходе лечения, т. е. с применением vMRI, оценивание степени любого изменения объема ткани головного мозга за указанный период времени и корректирование соответствующим образом параметров способа лечения.
В пятом аспекте в настоящем изобретении предусмотрен способ (способ 5) оценивания начала проявления неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта с риском развития указанного неврологического нарушения, при этом указанный способ включает: а) измерение объема ткани головного мозга субъекта (например, с применением vMRI) и сравнение с эталонным стандартом для оценки того, меньше ли объем ткани головного мозга, чем эталонный стандарт, и b) если объем ткани головного мозга, идентифицированный на стадии (а), меньше эталонного стандарта, идентифицирование начала проявления указанного неврологического нарушения; при этом способ необязательно дополнительно включает c) инициирование лечения субъекта посредством введения субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле I или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75. Следует понимать, что посредством измерения объема ткани головного мозга субъекта и сравнения с эталонным стандартом можно определить, достиг ли субъект, у которого имеется лизосомная болезнь накопления и который подвержен риску развития ассоциированного с ней неврологического нарушения, стадии прогрессирования заболевания, при которой лечение, например, с применением соединения формулы (I), может быть особенно полезным.
Эталонный стандарт объема ткани головного мозга (объема ткани головного мозга в целом и/или объемов отдельных областей ткани головного мозга) можно определить на основании данных о здоровом (без деменции) населении (полученных или доступных из опубликованных источников), что позволяет использовать нормативные волюметрические данные с учетом возрастных и гендерных особенностей в качестве эталонного стандарта на основании возраста и пола субъекта. Это позволяет проводить корректное сравнение, посредством которого можно оценить прогрессирование заболевания у субъекта на основании степени, в которой объем ткани головного мозга субъекта меньше эталонного стандарта в областях ткани головного мозга и/или на основании объема ткани головного мозга в целом, из чего у субъекта может быть определено начало неврологического нарушения, ассоциированного с лизосомной болезнью накопления.
В конкретных дополнительных вариантах осуществления способа 5 в настоящем изобретении предусмотрены следующие способы.
5.1. Способ 5, где у субъекта имеется болезнь Гоше типа 3;
5.2. Способ 5, где у субъекта имеется болезнь Ниманна-Пика типа C;
5.3. Способ 5, где у субъекта имеется GM2-ганглиозидоз (например, болезнь Тея-Сакса, болезнь Сандхоффа или вариант AB GM2-ганглиозидоза);
5.4. Способ 5 или любой из 5.1-5.3, где у субъекта диагностирована мутация в гене GBA1;
5.5. Способ 5 или любой из 5.1-5.3, где у субъекта диагностирована мутация в генах NPC1 и/или NPC2;
5.6. Способ 5 или любой из 5.1-5.3, где у субъекта диагностирована мутация в гене HEXA (кодирующем гексозаминидазу A), и/или мутация в гене HEXB (кодирующем гексозаминидазу B), и/или мутация в гене GM2A (кодирующем белок-активатор ганглиозида GM2);
5.7. Способ 5 или любой из 5.1-5.6, где у субъекта диагностирована болезнь Альцгеймера или болезнь Паркинсона;
5.8. Способ 5 или любой из 5.1-5.7, где субъект подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы (например, имиглюцеразы, велаглюцеразы или талиглюцеразы), где необязательно каждый из таких ферментов представляет собой рекомбинантный фермент;
5.9. Способ 5.8, где субъект подвергается лечению с применением одной или нескольких из имиглюцеразы, велаглюцеразы (например, велаглюцеразы-альфа) и талиглюцеразы (например, талиглюцеразы-альфа);
5.10. Способ 5.9, где субъект одновременно подвергается лечению с применением имиглюцеразы;
5.11. Способ 5.10, где субъект подвергается лечению с применением имиглюцеразы в дозировке от 2,5 единиц/кг веса тела до 80 единиц/кг веса тела один раз в 1-3 недели, например, от 40 до 60 единиц/кг веса тела один раз в 2 недели (1 единица имиглюцеразы представляет собой количество фермента, который катализирует гидролиз 1 микромоля синтетического субстрата п-нитрофенил-β-D-глюкопиранозида в минуту при 37°С);
5.12. Способ 5.11, где дозу имиглюцеразы для субъекта при каждом введении (например, один раз в 1-3 недели, например, один раз 2 недели) вводят в виде внутривенной (IV) инфузии в течение периода времени, составляющего 1-3 часа (например, 1-2 часа);
5.13. Способ 5 или любой из 5.1-5.12, где субъекту было введено средство ферментозаместительной терапии (например, имиглюцераза, велаглюцераза и/или талиглюцераза) до начала любого необязательного лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
5.14. Способ 5.13, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев до начала необязательной терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75), например, по меньшей мере 12 месяцев (1 год), или по меньшей мере 18 месяцев, или по меньшей мере 2 лет, или по меньшей мере 3 лет.
5.15. Способ 5.13 или 5.14, где субъекту было введено средство терапии на основе имиглюцеразы в течение по меньшей мере 6 месяцев в стабильной дозе до начала необязательной терапии с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
5.16. Способ 5 или любой из 5.1-5.15, где способ дополнительно включает стадию перевода субъекта с ERT-терапии (например, с применением имиглюцеразы, велаглюцеразы или талиглюцеразы) на необязательное лечение с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
5.17. Способ 5 или любой из 5.1-5.16, где субъект характеризуется уровнем гемоглобина, составляющим по меньшей мере 11 г/дл в случае женщин и по меньшей мере 12 г/дл в случае мужчин;
5.18. Способ 5 или любой из 5.1-5.17, где количество тромбоцитов у субъекта составляет по меньшей мере 100000/кубический миллиметр;
5.19. Способ 5 или любой из 5.1-5.18, где субъект характеризуется объемом селезенки, составляющим менее чем 10 нормальных объемов (MN), и/или объемом печени, составляющим менее чем 1,5 MN;
5.20. Способ 5 или любой из 5.1-5.19, где у субъекта диагностирована сопутствующая деменция, например, болезнь Альцгеймера или болезнь Паркинсона;
5.21. Способ 5 или любой из 5.1-5.20, где возраст субъекта составляет по меньшей мере 18 лет (например, 18-30 лет) на момент начала лечения с применением соединения согласно формуле (I) (или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb) или любого из соединений 1 или 1.1-1.75);
5.22. Способ 5 или любой из 5.1-5.21, где субъект характеризуется концентрацией глюкозилцерамида (GL1), составляющей 4,4-11,1 нг/мл в цереброспинальной жидкости (CSF) и 4,9-8,3 мкг/мл в плазме крови;
5.23. Способ 5 или любой из 5.1-5.22, где субъект характеризуется концентрацией глюкозилсфингозина (лизосомного GL1), составляющей 20,1-67,6 пг/мл в CSF и 8,8-159,0 нг/мл в плазме крови;
5.24. Способ 5 или любой из 5.1-5.23, где субъектом является взрослый пациент-человек, например, в возрасте от 18 до 80 лет, например, от 18 до 60 лет, или от 18 до 40 лет, или от 18 до 30 лет, или от 18 до 25 лет;
5.25. Способ 5 или любой из 5.1-5.23, где субъектом является пациент детского возраста, например, в возрасте от 0 до 18 лет, например, от 1 до 15 лет, или от 1 до 5 лет, или от 5 до 10 лет, или от 10 до 15 лет, или от 10 до 18 лет;
5.26. Способ 5 или любой из 5.1-5.25, где измерение объема ткани головного мозга субъекта проводят посредством позитронно-эмиссионной томографии головного мозга (PET) или посредством волюметрической магнитно-резонансной визуализации (vMRI);
5.27. Способ 5 или любой из 5.1-5.26, где установлено, что субъект характеризуется объемом ткани головного мозга, который меньше эталонного стандарта;
5.28. Способ 5.28, где сравнение с эталонным стандартом указывает на то, что субъект характеризуется меньшим объемом ткани головного мозга в одной или нескольких областях головного мозга, выбранных из правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной области, правого миндалевидного тела, левой области клина и левой лингвальной области;
5.29. Способ 5.27 или 5.28, где сравнение с эталонным стандартом указывает на то, что субъект характеризуется меньшим объемом ткани головного мозга в одной или нескольких областях головного мозга, связанных с исполнительной функцией;
5.30. Способ по любому из абзацев 5.27-5.29, где сравнение с эталонным стандартом указывает на то, что субъект характеризуется меньшим объемом ткани головного мозга в одной или нескольких областях головного мозга, где по результатам оценивания имеет место потеря нейронных связей, например, как показано с применением функциональной магнитно-резонансной визуализации (fMRI);
5.31. Способ по любому из абзацев 5.27-5.30, где сравнение с эталонным стандартом указывает на то, что субъект характеризуется меньшим объемом ткани головного мозга в целом;
5.32. Способ по любому из абзацев 5.27-5.31, где способ дополнительно включает инициирование лечения субъекта посредством введения субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75;
5.33. Способ 5.32, где способ дополнительно включает инициирование лечения субъекта посредством введения субъекту эффективного количества соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или любого из 1.1-1.75;
5.34. Способ 5.32 или 5.33, где способ дополнительно включает инициирование лечения субъекта посредством введения субъекту эффективного количества соединения 1 или любого одного или нескольких из соединений 1.1-1.75;
5.35. Способ 5.32-5.34, где лечение предусматривает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение формулы (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любое из соединений 1, или любое из 1.1-1.75;
5.36. Способ 5.32-5.35, где лечение предусматривает введение субъекту эффективного количества фармацевтической композиции, содержащей соединение 1 или любое одно или несколько из соединений 1.1-1.75;
5.37. Способ 5.35 или 5.36, где фармацевтическая композиция дополнительно содержит по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, описанное в данном документе;
5.38. Способ 5.32-5.37, где способ дополнительно включает инициирование лечения субъекта посредством введения фармацевтической лекарственной формы, содержащей эффективное количество соединения или эффективное количество фармацевтической композиции;
5.39. Способ 5.38, где лекарственная форма представляет собой лекарственную форму для перорального введения (например, пилюлю, капсулу, каплету, таблетку, драже, порошок, гранулу, пленку, пастилку или жидкость);
5.40. Способ 5.39, где лекарственная форма представляет собой жевательную таблетку;
5.41. Способ 5.38, где лекарственная форма представляет собой лекарственную форму для парентерального введения (например, где фармацевтическая композиция составлена для инъекции);
5.42. Способ 5.41, где инъекция представляет собой внутривенную, внутримышечную, интратекальную или подкожную инъекцию, необязательно стерильную инъекцию;
5.43. Способ 5.38, где лекарственная форма представляет собой лекарственную форму для местного или ректального применения;
5.44. Способ 5.38, где лекарственная форма представляет собой лекарственную форму для интраназального введения (например, аэрозоль);
5.45. Способ 5.32-5.44, где лечение дополнительно предусматривает одновременное введение второго активного средства, например, второго соединения, способного обеспечивать снижение уровней гликозилцерамида у нуждающегося в этом пациента, как описано в данном документе;
5.46. Способ 5.45, где второе активное средство вводят в той же фармацевтической композиции или лекарственной форме, что и хинуклидиновое соединение;
5.47. Способ 5.45 или 5.46, где второе активное средство представляет собой ингибитор GCS (например, миглустат или элиглустат);
5.48. Способ по любому из абзацев 5.32-5.47, где соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат или хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат;
5.49. Способ 5.48, где дозировка соединения составляет 15 мг/сутки при пероральном введении;
5.50. Способ 5.49, где дозировка соединения составляет 15 мг/сутки в виде однократной дозы для перорального введения;
5.51. Способ по любому из абзацев 5.32-5.48, где субъекту вводят однократную суточную дозу, составляющую 5 мг, 10 мг, 15 мг или 20 мг соединения, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата, необязательно в виде малатной соли в форме соли присоединения кислоты;
5.52. Способ по любому из абзацев 5.32-5.51, где объем ткани головного мозга субъекта измеряют многократно, периодически или регулярно, например, один раз в неделю, один раз в месяц, один раз в 2, 3, 4, 6, 9, 12 месяцев и т. д. после начала лечения с применением соединения согласно формуле (I) для оценивания изменения объема ткани головного мозга;
5.53. Способ 5 или любой из 5.1-5.52, где субъектом является животное, представляющее собой млекопитающее;
5.54. Способ 5.53, где субъектом является животное, являющееся приматом;
5.55. Способ 5.54, где субъектом является человек.
Следует понимать, что в некоторых вариантах осуществления способы 5 et seq. могут альтернативно рассматриваться как способы лечения или предупреждения неврологического нарушения, ассоциированного с лизосомной болезнью накопления, у субъекта (например, пациента), нуждающегося в этом и подверженного риску развития указанного неврологического нарушения, при этом способ включает оценку начала неврологического нарушения, ассоциированного с лизосомной болезнью накопления, и при этом указанный способ включает следующие стадии: а) измерение объема ткани головного мозга субъекта (например, с применением vMRI) и сравнение с эталонным стандартом для оценки того, меньше ли объем ткани головного мозга, чем эталонный стандарт; б) если объем ткани головного мозга, идентифицированный на стадии (а), меньше эталонного стандарта, идентифицирование начала указанного неврологического нарушения; и c) инициирование лечения субъекта посредством введения субъекту эффективного количества хинуклидинового соединения, описанного в данном документе, например, соединения согласно формуле (I) или любой из (II)-(XII), (Ia)-(XIIa) или (Ib)-(XIIb), или любого из соединений 1 или 1.1-1.75. В вариантах осуществления лечение по пункту с) может быть осуществлено любым из способа 1, способа 2 и способа 3, описанных в данном документе.
Все способы, описанные в данном документе (например, способ 1, способ 2, способ 3, способ 4, способ 5, а также любые варианты осуществления этих способов, описанных выше), могут быть применимы у субъектов, которые соответствуют одному, или нескольким, или всем из следующих критериев:
a) возраст 18 лет или старше;
b) наличие клинического диагноза болезни Гоше, например, наличие диагноза или установленного риска развития болезни Гоше (например, болезни Гоше типа 3). Диагноз может быть установлен с помощью любой оценки, описанной в данном документе, например, в силу наличия мутации в гене, как описано в данном документе;
c) наличие документально подтвержденного дефицита активности кислой бета-глюкозидазы;
d) получение до начала лечения, в течение по меньшей мере 3 лет лечения при помощи ERT, например лечения имиглюцеразой (Церезим), в стабильной ежемесячной дозе в течение по меньшей мере 6 месяцев;
e) уровень гемоглобина ≥11,0 г/дл для женщин и ≥12,0 г/дл для мужчин;
f) количество тромбоцитов ≥100000/мм3;
g) объем селезенки, составляющий <10 нормальных объемов (MN), или общая спленэктомия (при условии, что спленэктомия имела место >3 лет до рандомизации);
h) объем печени <1,5 МН;
i) отсутствие костного криза и отсутствие симптоматического заболевания костей (например, боли в костях, связанной с остеонекрозом и/или патологическими переломами) в течение трех месяцев или одного года до начала лечения;
j) наличие в анамнезе судорог, за исключением миоклонических судорог;
k) болезнь Гоше типа 3, которая характеризуется глазодвигательной апраксией (надъядерным параличом взора), характеризующейся аномалией горизонтальных саккад; а также
l) наличие легкого неврологического поражения или умеренного неврологического поражения, измеренного с применением модифицированного инструмента оценки тяжести (mSST; Davies, et al., 2011), на момент начала лечения.
В варианте осуществления способа 1, описанного в данном документе, или в конкретном подварианте осуществления способа 1, описанного в данном документе, субъект соответствует всем критериям (а)-(l). В варианте осуществления способа 2, описанного в данном документе, или в конкретном подварианте осуществления способа 2, описанного в данном документе, субъект соответствует всем критериям (а)-(l). В варианте осуществления способа 3, описанного в данном документе, или в конкретном подварианте осуществления способа 3, описанного в данном документе, субъект соответствует всем критериям (а)-(l). В варианте осуществления способа 4, описанного в данном документе, или в конкретном подварианте осуществления способа 4, описанного в данном документе, субъект соответствует всем критериям (а)-(l). В варианте осуществления способа 5, описанного в данном документе, или в конкретном подварианте осуществления способа 5, описанного в данном документе, субъект соответствует всем критериям (а)-(l).
В дополнительных вариантах осуществления субъект является взрослым или пациентом детского возраста 12 лет или больше. В вариантах осуществления субъект является взрослым или пациентом детского возраста 12 лет или больше с болезнью Гоше типа 3 (что подтверждается, например, удовлетворением вышеуказанных критериев b) и/или k)), и состояние которого стабилизируется посредством ERT, например имиглюцеразы (Церезим), для системных состояний. Системные состояния могут, например, характеризоваться наличием маркеров системного заболевания, таких как маркеры, ассоциированные с: i) объемом селезенки и печени (например, измеренным с помощью магнитно-резонансной визуализации (MRI)); ii) количеством тромбоцитов и iii) уровнями гемоглобина. В некоторых вариантах осуществления субъект с болезнью Гоше типа 3 получает лечение при помощи ERT (например, с применением имиглюцеразы (Церезим)) в течение по меньшей мере 3 лет и/или достиг следующих терапевтических целей в отношении GD: один, или более, или все из вышеприведенных критериев от e) до i).
В конкретном варианте осуществления в настоящем изобретении представлен (S)-хинуклидин-3-ил(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в однократной пероральной дозе 15 мг/сутки, необязательно в виде малатной соли в форме соли присоединения кислоты, для применения в способах, определенных в данном документе (например, способе 1, способе 2, способе 3, способе 4, способе 5, а также любом из вариантов осуществления указанных способов, описанных выше), у субъекта с болезнью Гоше типа 3 (что подтверждается, например, удовлетворением вышеуказанных критериев b) и/или k)), состояние которого стабилизируется посредством ERT, например имиглюцеразы (Церезим), при системных состояниях (таких как определены выше), где субъектом является взрослый или пациент детского возраста 12 лет или больше.
Положительный эффект в отношении когнитивной дисфункции, и/или нейронных связей, и/или объема ткани головного мозга, и/или регрессии неврологического нарушения, как описано в способе 1, способе 2, способе 3 и способе 4 соответственно, можно понимать как лечение ЦНС-проявлений. Таким образом, в конкретном варианте осуществления в настоящем изобретении представлен (S)-хинуклидин-3-ил(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в однократной пероральной дозе 15 мг/сутки, необязательно в виде малатной соли в форме соли присоединения кислоты, для применения в лечении ЦНС-проявлений у субъекта с болезнью Гоше типа 3 (что подтверждается, например, удовлетворением вышеприведенных критериев b) и/или k)), состояние которого стабилизируется с помощью ERT, например имиглюцеразы (Церезим), при системных состояниях (таких как определены выше), где субъектом является взрослый или пациент детского возраста 12 лет или больше.
В вариантах осуществления каждого из описанных в данном документе способов (например, способ 1, способ 2, способ 3, способ 4, способ 5, а также любой из вариантов осуществления указанных выше способов) подлежащий лечению субъект не подвергается одновременному лечению индуктором CYP3A, например сильным индуктором CYP3A, таким как рифампин, или умеренным индуктором CYP3A, таким как фенобарбитал или эфавиренз. В вариантах осуществления субъект не принимает пищевой добавки, идентифицированной как сильный или умеренный индуктор CYP3A.
В одном варианте осуществления в настоящем изобретении представлен (S)-хинуклидин-3-ил(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в однократной пероральной дозе 15 мг/сутки, необязательно в виде малатной соли в форме соли присоединения кислоты, для применения в лечении ЦНС-проявлений у субъекта с болезнью Гоше типа 3 (что подтверждается, например, удовлетворением вышеприведенных критериев b) и/или k)), состояние которого стабилизируется с помощью ERT, например имиглюцеразы (Церезим), при системных состояниях (таких как определены выше), где субъектом является взрослый или пациент детского возраста 12 лет или больше, и где субъект, подлежащий лечению, не подвергается одновременному лечению индуктором CYP3A, например. сильным индуктором CYP3A, таким как рифампин, или умеренным индуктором CYP3A, таким как фенобарбитал или эфавиренц. В вариантах осуществления субъект не принимает пищевой добавки, идентифицированной как сильный или умеренный индуктор CYP3A.
Все способы, описанные в данном документе (например, способ 1, способ 2, способ 3, способ 4, способ 5, а также любой из вариантов осуществления этих способов, описанных выше), могут быть сочтены неподходящими для определенных групп пациентов, например, тех, кто характеризуется наличием конкретных ранее развившихся состояний, тех, кто в настоящее время подвергается лечению некоторыми лекарственными препаратами или подвергался в прошлом, и тех, у кого в анамнезе было лечение, например хирургическое лечение, как оценено и описано в данном документе. Лечащий врач вправе решать, влияют ли конкретное(конкретные) состояние(состояния) субъекта и/или прием в настоящее время или в прошлом лекарственного(лекарственных) препарата(препаратов) на его пригодность для прохождения способов лечения в соответствии со способами, раскрытыми в данном документе.
В вариантах осуществления субъекты, подлежащие лечению с помощью способов, описанных в данном документе (например, способа 1, способа 2, способа 3, способа 4, способа 5, а также любых вариантов осуществления этих способов, описанных выше), не включают тех, которые соответствуют одному, или нескольким, или всем из следующих критериев:
1) проходил субстрат-редуцирующую терапию или терапию шаперонами при болезни Гоше в течение 6 месяцев до начала лечения;
2) подвергался частичной или общей спленэктомии в течение 3 лет до начала лечения;
3) зависим от переливания крови;
4) характеризуется предшествующим варикозным расширением вен пищевода, или инфарктом печени, или текущими уровнями ферментов печени (аланинаминотрансфераза [ALT] / аспартатаминотрансфераза [AST]) или общего билирубина, более чем в 2 раза превышающими верхний предел нормы, только если у пациента не диагностирован синдром Жильбера;
5) характеризуется клинически значимым заболеванием, отличным от болезни Гоше, в том числе сердечно-сосудистым (врожденный порок сердца, заболевание коронарных артерий, пороки клапанов или левожелудочковая сердечная недостаточность; клинически значимые аритмии или нарушения проводимости), печеночным, желудочно-кишечным, легочным, неврологическим, эндокринным, метаболическим (например, гипокалиемия, гипомагниемия) или психическим заболеванием, другими медицинскими состояниями или серьезными интеркуррентными заболеваниями, которые могут препятствовать участию;
6) характеризуется почечной недостаточностью, определяемой расчетной скоростью клубочковой фильтрации <30 мл/мин/1,73 м2;
7) имеет в анамнезе рак, за исключением базально-клеточной карциномы;
8) характеризуется наличием миоклонических судорог;
9) наличие беременности или лактации;
10) характеризуется кортикальной катарактой более одной четверти окружности хрусталика (кортикальная катаракта 2 степени) или задней субкапсулярной катарактой больше 2 мм (задняя субкапсулярная катаракта 2 степени) в соответствии с классификацией Всемирной организации здравоохранения (ВОЗ);
11) требует применения инвазивной искусственной вентиляции легких;
12) требует применения неинвазивной искусственной вентиляции легких во время бодрствования более 12 часов в день;
13) в настоящее время получает потенциально катарактогенные лекарственные препараты (кортикостероиды, псоралены, используемые в дерматологии с терапией ультрафиолетовым светом [PUVA], типичные нейролептики и лекарственные препараты от глаукомы) или любые лекарственные препараты, которые могут ухудшить зрение пациента с катарактой (например, альфа-адренергические лекарственные препараты от глаукомы);
14) характеризуется введением сильных или умеренных индукторов или ингибиторов CYP3A в течение 15 дней или 5 периодов полувыведения с момента скрининга, в зависимости от того, что дольше, до начала лечения или употреблением грейпфрута, грейпфрутового сока или продуктов, содержащих грейпфрут, в течение 72 часов после начала лечения;
15) запланирована стационарная госпитализация, в том числе плановая операция, во время лечения; а также
16) перенес трансплантацию жизненно важных органов (например, костного мозга или печени).
Фармацевтические композиции
Настоящее изобретение также предусматривает фармацевтические композиции, содержащие по меньшей мере одно хинуклидиновое соединение, описанное в данном документе, и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, например, для применения согласно способам, раскрытым в данном документе. Фармацевтически приемлемое вспомогательное вещество может представлять собой любое такое вспомогательное вещество, известное из уровня техники, включая описанные, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co., (A. R. Gennaro edit. 1985). Фармацевтические композиции на основе соединений, раскрытых в данном документе, можно получить с помощью традиционных способов, известных из уровня техники, включающих, например, смешивание по меньшей мере одного соединения, раскрытого в данном документе, с фармацевтически приемлемым вспомогательным веществом.
Таким образом, в одном аспекте настоящего изобретения предусмотрена фармацевтическая лекарственная форма, содержащая хинуклидиновое соединение, описанное в данном документе, и фармацевтически приемлемое вспомогательное вещество, где лекарственная форма составлена для обеспечения при введении (например, при пероральном введении) количества указанного соединения, достаточного для обеспечения лечения заболевания или нарушения, как предусмотрено в любом из способов, описанных в данном документе (например, способ 1 et seq., способ 2 et seq., способ 3 et seq., способ 4 et seq. или способ 5 et seq.).
Фармацевтическая композиция или лекарственная форма в соответствии с настоящим изобретением может содержать средство и другой носитель, например, соединение или композицию, инертные или активные, такие как детектируемое средство, метка, вспомогательное вещество, разбавитель, связующее, стабилизатор, буферы, соли, липофильные растворители, консервант, вспомогательное вещество или подобное. Носители также предусматривают фармацевтические вспомогательные вещества и добавки, например, белки, пептиды, аминокислоты, липиды и углеводы (например, сахара, в том числе моносахариды, ди-, три-, тетра- и олигосахариды; дериватизированные сахара, такие как альдиты, альдоновые кислоты, этерифицированные сахара и т. п.; а также полисахариды или полимеры сахаров), которые могут присутствовать по отдельности или в комбинации и составляют отдельно или в комбинации 1-99,99% по весу или объему. Иллюстративные белковые вспомогательные вещества предусматривают сывороточный альбумин, такой как человеческий сывороточный альбумин (HSA), рекомбинантный человеческий альбумин (rHA), желатин, казеин и т. п. Типичные аминокислотные компоненты/компоненты-антитела, которые могут также функционировать для обеспечения буферной емкости, предусматривают аланин, глицин, аргинин, бетаин, гистидин, глутаминовую кислоту, аспарагиновую кислоту, цистеин, лизин, лейцин, изолейцин, валин, метионин, фенилаланин, аспартам и т. п. Также предполагается наличие в объеме настоящего изобретения углеводных вспомогательных веществ, примеры которых предусматривают без ограничения моносахариды, такие как фруктоза, мальтоза, галактоза, глюкоза, D-манноза, сорбоза и т. п.; дисахариды, такие как лактоза, сахароза, трегалоза, целлобиоза и т. п.; полисахариды, такие как рафиноза, мелезитоза, мальтодекстрины, декстраны, крахмалы и т. п.; а также альдиты, такие как маннит, ксилит, мальтит, лактит, ксилит, сорбит (глюцит) и миоинозит.
Носители, которые можно применять, предусматривают буфер или средство, регулирующее pH; как правило, буфер представляет собой соль, полученную из органических кислоты или основания. Типичные буферы предусматривают соли органических кислот, такие как соли лимонной кислоты, аскорбиновой кислоты, глюконовой кислоты, угольной кислоты, винной кислоты, янтарной кислоты, уксусной кислоты или фталевой кислоты; трис, гидрохлорид трометамина или фосфатные буферы. Дополнительные носители предусматривают полимерные вспомогательные вещества/добавки, такие как поливинилпирролидоны, фиколлы (полимерный сахар), декстраты (например, циклодекстрины, такие как 2-гидроксипропил-β-циклодекстрин), полиэтиленгликоли, вкусоароматические добавки, противомикробные средства, подсластители, антиоксиданты, антистатические средства, поверхностно-активные вещества (например, полисорбаты, такие как "TWEEN 20" и "TWEEN 80"), липиды (например, фосфолипиды, жирные кислоты), стероиды (например, холестерин) и хелатирующие средства (например, EDTA).
Настоящее раскрытие также предусматривает фармацевтические композиции и наборы, содержащие указанные композиции, которые содержат по меньшей мере одно хинуклидиновое соединение, описанное в данном документе, и по меньшей мере одно дополнительное фармацевтически активное средство. Такие фармацевтические композиции и наборы могут быть приспособлены для обеспечения одновременного, последовательного и/или раздельного введения хинуклидинового соединения и дополнительного активного средства. Например, хинуклидиновое соединение и дополнительное активное средство могут быть составлены в отдельных лекарственных формах, например, в отдельных таблетках, капсулах, лиофилизатах или жидкостях, или они могут быть составлены в одной и той же лекарственной форме, например, в одних и тех же таблетке, капсуле, лиофилизате или жидкости. Если хинуклидиновое соединение и дополнительное активное средство составлены в одной и той же лекарственной форме, то хинуклидиновое соединение и дополнительное активное средство могут присутствовать по сути в смеси, например, в ядре таблетки, или они могут присутствовать по сути в отдельных участках лекарственной формы, например, в отдельных слоях одной и той же таблетки. В одном варианте осуществления фармацевтическая лекарственная форма содержит дополнительное средство, которое способно обеспечивать лечение или предупреждение когнитивной дисфункции и/или отклонений походки, например, у пациента с диагнозом или предрасположенностью к лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, описанных в данном документе.
Согласно дополнительному аспекту настоящее изобретение предусматривает фармацевтическую композицию, содержащую: (i) хинуклидиновое соединение, описанное в данном документе; (ii) дополнительное активное средство и (iii) фармацевтически приемлемое вспомогательное вещество. В одном варианте осуществления дополнительное активное средство представляет собой средство, которое способно обеспечивать лечение или предупреждение когнитивной дисфункции и/или отклонений походки, например, у пациента с диагнозом или предрасположенностью к лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, описанных в данном документе. В одном варианте осуществления дополнительное активное средство способно обеспечивать лечение или предупреждение нарушения походки (например, атаксию) или деменции, например, у пациента с диагнозом или предрасположенностью к лизосомной болезни накопления, такой как болезнь Гоше типа 3 или болезнь Ниманна-Пика типа C, описанных в данном документе.
Раскрытые в данном документе хинуклидиновые соединения и фармацевтические композиции можно применять для животного или человека. Таким образом, раскрытое в данном документе соединение может быть составлено в виде фармацевтической композиции для перорального, трансбуккального, парентерального (например, внутривенного, внутримышечного или подкожного), местного, ректального или интраназального введения или в форме, подходящей для введения с помощью ингаляции или инсуффляции. В конкретных вариантах осуществления хинуклидиновое соединение или фармацевтическая композиция составлены для системного введения, например, посредством пути, отличного от парентерального. В одном варианте осуществления хинуклидиновое соединение или фармацевтическая композиция составлены для перорального введения, например, в твердой форме. Такие способы введения и способы получения соответствующих фармацевтических композиций описаны, например, в Gibaldi's Drug Delivery Systems in Pharmaceutical Care (1st ed., American Society of Health-System Pharmacists 2007).
Фармацевтические композиции могут быть составлены таким образом, чтобы обеспечивать медленное, продленное или контролируемое высвобождение активного ингредиента, содержащегося в них, с применением, например, гидроксипропилметилцеллюлозы в различных соотношениях для обеспечения желаемого профиля высвобождения, других полимерных матриц, липосом и/или микросфер. Фармацевтические композиции также могут необязательно содержать средства, придающие непрозрачность, и могут иметь такой состав, при котором активный(активные) ингредиент(ингредиенты) высвобождаются из них исключительно или предпочтительно в определенном отделе желудочно-кишечного тракта, необязательно с задержкой, например, при использовании энтеросолюбильного покрытия. Примеры капсулирующих композиций включают полимерные вещества и воски. Активный ингредиент также может находиться в микроинкапсулированной форме, при необходимости вместе с одним или несколькими фармацевтически приемлемыми носителями, вспомогательными веществами или разбавителями, хорошо известными из уровня техники (см., например, Remington’s). Соединения, раскрытые в данном документе, могут также быть составлены для стабильной доставки согласно способам, хорошо известным средним специалистам в данной области. Примеры таких составов можно найти в патентах США №№ 3119742, 3492397, 3538214, 4060598 и 4173626.
В твердых лекарственных формах для перорального введения (например, капсулах, таблетках, пилюлях, драже, порошках, гранулах и подобных) активный ингредиент смешан с одним или несколькими фармацевтически приемлемыми носителями, наполнителями или разбавителями, такими как цитрат натрия или фосфат дикальция, и/или чем-либо из следующего: (1) заполнителей или сухих разбавителей, таких как крахмалы, лактоза, сахароза, глюкоза, маннит, микрокристаллическая целлюлоза, фосфат кальция и/или кремниевая кислота; (2) связующих, таких как, например, карбоксиметилцеллюлоза, альгинаты, желатин, прежелатинизированный маисовый крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза, сахароза и/или аравийская камедь; (3) увлажнителей, таких как глицерин; (4) разрыхлителей, таких как агар-агар, карбонат кальция, крахмалгликолят натрия, картофельный или маниоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) замедлителей растворения, таких как парафин; (6) ускорителей всасывания, таких как соединения четвертичного аммония; (7) смачивающих средств, таких как, например, лаурилсульфат натрия, ацетиловый спирт и глицеринмоностеарат; (8) абсорбентов, таких как каолин и бентонитовая глина; (9) смазывающих средств, таких как тальк, диоксид кремния, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси, и (10) красителей. В случае капсул, таблеток и пилюль фармацевтические композиции также могут содержать буферные средства. Твердые композиции подобного типа также можно получать с применением заполнителей в мягких и твердых заполняемых желатиновых капсулах и наполнителей, таких как лактоза или молочные сахара, а также высокомолекулярных полиэтиленгликолей и т. п.
Таблетку можно получать посредством прессования или формования необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно получать с применением связующих (например, желатина или гидроксипропилметилцеллюлозы), смазывающих средств, инертных разбавителей, консервантов, разрыхлителей (например, крахмалгликолята натрия или сшитой карбоксиметилцеллюлозы натрия), поверхностно-активных и/или диспергирующих средств. Формованные таблетки можно получать посредством формования смеси порошкообразного активного ингредиента, увлажненного инертным жидким разбавителем, в подходящем устройстве. Таблетки и другие твердые лекарственные формы, такие как драже, капсулы, пилюли и гранулы, могут необязательно быть делимыми или получаемыми с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные из уровня техники.
Согласно вариантам осуществления фармацевтические композиции вводят перорально в жидкой форме. Жидкие лекарственные формы для перорального введения активного ингредиента предусматривают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие препараты для перорального введения могут быть представлены в виде сухого продукта для разведения водой или другой подходящей средой-носителем перед применением. Вдобавок к активному ингредиенту жидкие лекарственные формы могут содержать инертные разбавители, традиционно применяемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое масло, кукурузное масло, масло зародышей, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, а также их смеси. Вдобавок к инертным разбавителям жидкие фармацевтические композиции могут содержать вспомогательные вещества, такие как смачивающие средства, эмульгирующие и суспендирующие средства, подсластители, вкусоароматические добавки, красители, ароматизирующие средства и консерванты и т. п. Суспензии вдобавок к активному(активным) ингредиенту(ингредиентам) могут содержать суспендирующие средства, такие как без ограничения этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилированного сорбита и сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также их смеси. Приемлемые жидкие препараты можно получить с помощью общепринятых способов с фармацевтически приемлемой(приемлемыми) добавкой(добавками), такой(такими) как суспендирующее вещество (например, сорбитный сироп, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгирующее вещество (например, лецитин или аравийская камедь); безводная среда (например, миндальное масло, масляные сложные эфиры или этиловый спирт); и/или консервант (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота). Активный(активные) ингредиент(ингредиенты) также можно вводить в виде болюса, электуария или пасты.
Для трансбуккального введения композиция может принимать форму таблеток или пастилок, составленных традиционным способом.
Согласно вариантам осуществления фармацевтические композиции вводят отличным от перорального способом, как, например, посредством местного применения, трансдермального применения, инъекции и т. п. В связанных вариантах осуществления фармацевтические композиции вводят парентерально путем инъекции, инфузии или имплантации (например, внутривенно, внутримышечно, внутриартериально, подкожно и т. п.).
Раскрытые в данном документе соединения могут быть составлены для парентерального введения путем инъекции, в том числе с применением традиционных методик катетеризации, или инфузии. Составы для инъекции могут быть представлены в единичной лекарственной форме, например, в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных средах-носителях, и могут содержать средство для получения состава, такое как суспендирующее, стабилизирующее и/или диспергирующее средство, общепризнанное специалистами в данной области. В качестве альтернативы, активный ингредиент может находиться в форме порошка для разведения с помощью подходящей среды, например, стерильной апирогенной воды, перед применением.
Фармацевтические композиции можно вводить непосредственно в центральную нервную систему. Следовательно, согласно некоторым вариантам осуществления композиции вводят непосредственно в центральную нервную систему, чтобы обойти гематоэнцефалический барьер. Согласно некоторым вариантам осуществления композицию можно вводить путем прямой инъекции в спинной мозг. Согласно вариантам осуществления композицию вводят путем интратекальной инъекции. Согласно некоторым вариантам осуществления композицию вводят путем интрацеребровентрикулярной инъекции. Согласно вариантам осуществления композицию вводят в боковой желудочек головного мозга. Согласно вариантам осуществления композицию вводят в оба боковых желудочка головного мозга. Согласно дополнительным вариантам осуществления композицию вводят путем интрагиппокампальной инъекции. Композиции можно вводить одной инъекцией или несколькими инъекциями. Согласно другим вариантам осуществления композицию вводят в более чем одно местоположение (например, в два участка в центральной нервной системе).
Фармацевтические композиции могут находиться в форме стерильных инъекционных растворов. Фармацевтические композиции можно стерилизовать, например, посредством фильтрования через фильтр для удерживания бактерий или посредством включения в их состав стерилизующих средств в форме стерильных твердых композиций, которые можно растворять в стерильной воде или некоторых других стерильных инъекционных средах непосредственно перед применением. Для получения такой композиции активный ингредиент растворяют или суспендируют в приемлемой для парентерального введения жидкой среде-носителе. Иллюстративные среды-носители и растворители включают без ограничения воду, воду с pH, доведенным до приемлемого значения посредством добавления соответствующего количества хлористоводородной кислоты, гидроксида натрия или подходящего буфера, 1,3-бутандиол, раствор Рингера и изотонический раствор хлорида натрия. Фармацевтические композиции также могут содержать один или несколько консервантов, например, метил-, этил- или н-пропил-п-гидроксибензоат. Для улучшения растворимости можно добавлять средство, усиливающее растворение, или солюбилизирующее средство, или растворитель может содержать 10-60% вес/вес пропиленгликоля или подобного.
Фармацевтические композиции могут содержать одно или несколько из фармацевтически приемлемых стерильных изотонических водных или неводных растворов, дисперсий, суспензий или эмульсий или стерильные порошки, которые можно разводить с получением стерильных инъекционных растворов или дисперсий непосредственно перед применением. Такие фармацевтические композиции могут содержать антиоксиданты; буферы; бактериостаты; растворенные вещества, которые делают состав изотоничным крови предполагаемого реципиента; суспендирующие средства; загустители; консерванты и т. п.
Примеры подходящих водных и неводных носителей, которые можно использовать в фармацевтических композициях согласно настоящему изобретению, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т. п.) и их подходящие смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Надлежащую текучесть можно поддерживать, например, путем применения материалов для покрытия, таких как лецитин, путем поддержания необходимого размера частиц в случае дисперсий и путем применения поверхностно-активных веществ. В некоторых вариантах осуществления с целью пролонгирования эффекта активного ингредиента желательно замедлять всасывание соединения после подкожной или внутримышечной инъекции. Этого можно достичь посредством применения жидкой суспензии кристаллического или аморфного материала, характеризующегося слабой растворимостью в воде. Скорость всасывания активного ингредиента, таким образом, зависит от его скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. В качестве альтернативы, задержка всасывания введенного парентерально активного ингредиента достигается посредством растворения или суспендирования соединения в масляной среде-носителе. Кроме того, пролонгированное всасывание инъецируемой фармацевтической формы может быть обусловлено включением средств, которые задерживают всасывание, таких как моностеарат алюминия и желатин.
Композиции для парентерального введения с контролируемым высвобождением могут находиться в форме водных суспензий, микросфер, микрокапсул, магнитных микросфер, масляных растворов, масляных суспензий, эмульсий, или активный ингредиент может быть включен в биосовместимый(биосовместимые) носитель(носители), липосомы, наночастицы, имплантаты или инфузионные устройства. Материалы для применения в получении микросфер и/или микрокапсул включают без ограничения биоразлагаемые/биоразрушаемые полимеры, такие как полиглактин, поли(изобутилцианоакрилат), поли(2-гидроксиэтил-L-глутамин) и поли(молочная кислота). Биосовместимые носители, которые можно применять при составлении состава для парентерального введения с контролируемым высвобождением, включают углеводы, такие как декстраны, белки, такие как альбумин, липопротеины или антитела. Материалы для применения в имплантатах могут быть небиоразлагаемыми, например, полидиметилсилоксан, или биоразлагаемыми, такими как, например, поли(капролактон), поли(молочная кислота), поли(гликолевая кислота) или поли(сложные орто-эфиры).
Для местного введения раскрываемое в данном документе соединение можно составлять в виде мази или крема. Раскрытые в данном документе соединения можно также составлять в ректальные композиции, такие как суппозитории или удерживающие клизмы, например, содержащие общепринятые суппозиторные основы, такие как масло какао или другие глицериды.
Для интраназального введения или введения с помощью ингаляции раскрытые в данном документе соединения можно традиционно доставлять в форме раствора или суспензии из контейнера-пульверизатора, нажимаемого или нагнетаемого пациентом, или в виде распыляемого аэрозоля, подаваемого из находящегося под давлением контейнера или распылителя, с применением подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае находящегося под давлением аэрозоля единицу дозирования можно определить путем обеспечения клапана для доставки отмеренного количества. Находящийся под давлением контейнер или распылитель может содержать раствор или суспензию раскрытого в данном документе соединения. Можно составить капсулы и картриджи (изготовленные, например, из желатина) для применения в ингаляторе или инсуффляторе, содержащие порошковую смесь раскрытого в данном документе соединения и подходящей порошковой основы, такой как лактоза или крахмал.
Как правило, средства и композиции, описанные в данном документе, вводят в эффективном количестве или в количестве, достаточном для лечения или предупреждения когнитивной дисфункции и/или отклонений походки у субъекта, нуждающегося в этом. Как правило, дозу можно отрегулировать в этом диапазоне, исходя из, например, возраста, физического состояния, веса тела, пола, рациона, времени введения и других клинических факторов. Определение эффективного количества находится вполне в пределах возможностей специалистов в данной области.
Будучи описанными в общих чертах в данном документе, следующие неограничивающие примеры и прилагаемые фигуры представлены для дополнительной иллюстрации настоящего изобретения, где:
на ФИГ. 1 представлен график, отображающий изменение среднего объема головного мозга в целом (WBV) после 52 недель лечения с применением соединения формулы (I) для пациентов группы A и изменение WBV для пациента 5, определенные на основании измерений vMRI и анализа TBM в соответствии с примером 5 данного документа.
ПРИМЕРЫ
Общие процедуры химического синтеза
Общая процедура A. Образование карбамата с помощью трифосгена
В суспензию гидрохлорида амина (1 эквивалент) и триэтиламина (3-4 эквивалента) в THF (концентрация ~ 0,2 M) при комнатной температуре добавляли трифосген (0,35 эквивалента). Реакционную смесь перемешивали в течение 10 мин., и добавляли небольшое количество эфира (1-2 мл). Соль триэтиламмония отфильтровывали с получением прозрачного раствора изоцианата в смеси THF/эфир.
К раствору спирта (1,5 эквивалента) в THF (концентрация ~ 0,2 M) при комнатной температуре добавляли NaH [60%, масло] (1,5 эквивалента). Реакционную смесь перемешивали в течение 15 мин., и по каплям добавляли вышеупомянутый раствор (изоцианат в смеси THF/эфир). При стандартной обработке реакцию гасили солевым раствором. Раствор экстрагировали с помощью EtOAc, и органический слой высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали на СombiFlash (картридж с SiO2, CHCl3 и 2 н. NH3 в MeOH) с получением соответствующего карбамата.
Общая процедура B. Алкилирование с помощью церийорганического соединения
Суспензию CeCl3 (4 эквивалента) в THF (концентрация ~ 0,2 M) перемешивали при комнатной температуре в течение 1 ч. Суспензию охлаждали до -78°C, и по каплям добавляли раствор MeLi/эфир [1,6 M] (4 эквивалента). Обеспечивали образование церийорганического комплексного соединения в течение периода 1 ч, и по каплям добавляли раствор нитрила (1 эквивалент) в THF (концентрация 2,0 M). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 ч. Раствор охлаждали до 0°C и гасили водой (~ 1 мл) с последующим добавлением 50% водного раствора гидроксида аммония (~ 3 мл) до образования осадка и осаждения его на дне колбы. Смесь фильтровали через слой целита и концентрировали. Неочищенный материал обрабатывали раствором HCl/диоксан [4,0 M]. Промежуточное соединение гидрохлорид арилпропан-2-амина растирали в эфире и использовали в таком виде для следующей стадии. В качестве альтернативы, неочищенный амин в форме свободного основания очищали на СombiFlash (картридж с SiO2, CHCl3 и 2 н. NH3 в MeOH) с получением соответствующего арилпропиламина.
Общая процедура C. Сочетание по Сузуки
К раствору арилгалогенида (1 эквивалент) в смеси DME/вода [4:1] (концентрация ~ 0,2 M) добавляли бороновую кислоту (2 эквивалента), палладиевый катализатор (0,1-0,25 эквивалента) и карбонат натрия (2 эквивалента). Реакционную смесь подвергали микроволновому облучению в течение 25 мин. при 150°C. После фильтрования через слой целита и концентрирования неочищенный продукт очищали на СombiFlash (картридж с SiO2, CHCl3 и 2 н. NH3 в MeOH) с получением соответствующего аддукта сочетания.
В качестве альтернативы: к раствору арилгалогенида (1 эквивалент) в смеси толуол/вода [20:1] (концентрация ~ 0,2 M) добавляли бороновую кислоту (1,3-2,5 эквивалента), палладиевый катализатор (0,05-0,15 эквивалента), трициклогексилфосфин (0,15-0,45 эквивалента) и фосфат калия (5 эквивалентов). Реакционную смесь подвергали микроволновому облучению в течение 25 мин. при 150°C. После фильтрования через слой целита и концентрирования неочищенный продукт очищали на СombiFlash (картридж с SiO2, CHCl3 и 2 н. NH3 в MeOH) с получением соответствующего аддукта сочетания.
Общая процедура D. Циклопропанирование
В смесь арилнитрила (1 эквивалент) и Ti(Oi-Pr)4 (1,7 эквивалента), перемешиваемую при -70°C, по каплям добавляли EtMgBr [3,0 M в эфире] (1,1 эквивалента). Обеспечивали нагревание реакционной смеси до 25°C, и ее перемешивали в течение 1 ч. В вышеупомянутую смесь по каплям добавляли BF3·Et2O (3 эквивалента) при 25°C. После добавления смесь перемешивали в течение еще 2 ч, а затем гасили водной HCl [2 M]. Затем полученный в результате раствор подщелачивали путем добавления водного NaOH [2 M]. Органический материал экстрагировали этиловым эфиром. Органические слои объединяли, высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали с помощью хроматографии на колонке с силикагелем (элюируя смесью петролейный эфир/EtOAc: 10/1-1/1) с получением соответствующего 1-арилциклопропанамина.
Общая процедура E. Биарильное сочетание с использованием условий по Сузуки
В перемешиваемый раствор арилгалогенидного компонента (1 эквивалент) в смеси диоксан/вода 5:1 (объем/объем) (~ 0,15 M) или N, N-диметилформамиде 5:1 (объем/объем) (~ 0,15 M) добавляли компонент, представляющий собой арилборонат или арилбороновую кислоту (1-1,5 эквивалента), карбонат натрия (2-3 эквивалента) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,05 эквивалента). Смесь нагревали (90°C) в течение ночи, а затем фильтровали через слой целита. Целит ополаскивали этилацетатом, и объединенный фильтрат промывали солевым раствором, высушивали (Na2SO4) и концентрировали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния.
Общая процедура F. Образование карбамата с использованием изоцианата, полученного с помощью смешанного ангидрида/пути перегруппировки Курциуса
В перемешиваемый раствор компонента, представляющего собой карбоновую кислоту (1 эквивалент), в тетрагидрофуране (~ 0,1 M) добавляли триэтиламин (2 эквивалента). Реакционную смесь охлаждали (0°C) и обрабатывали изобутилхлорформиатом (1,5 эквивалента). Через 1 час при 0°C добавляли раствор азида натрия (2 эквивалента) в воде (~ 1 M), и обеспечивали нагревание реакционной смеси до комнатной температуры. После перемешивания в течение ночи реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные экстракты промывали водным раствором бикарбоната натрия и солевым раствором, высушивали (Na2SO4) и концентрировали. Неочищенный ацилазид дополнительно высушивали путем совместного выпаривания с толуолом, а затем поглощали толуолом (~ 0,1 M). Перемешиваемый раствор нагревали с обратным холодильником в течение 2-2,5 часа, охлаждали и обрабатывали спиртовым компонентом (1,25-2 эквивалента). Реакционную смесь нагревали с обратным холодильником в течение ночи, а затем концентрировали. Остаток поглощали этилацетатом или хлороформом, промывали водным раствором карбоната натрия (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния с использованием градиентов растворителей хлороформ/метанол (менее полярные карбаматы) или хлороформ/метанол/аммиак (более полярные карбаматы).
Пример 1. Синтез хинуклидиновых соединений
1-Азабицикло[2.2.2]окт-3-ил-[2-(4'-фторбифенил-3-ил)пропан-2-ил]карбамат ( соединение 1 )
С использованием общей процедуры C, 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)пропан-2-ил]карбамата (600 мг, 1,63 ммоль), 4-фторфенилбороновой кислоты (457 мг, 3,27 ммоль) и ацетата палладия(II) получали указанное в заголовке соединение в виде белого твердого вещества (373 мг, 60%). 1H ЯМР (400 МГц, CDCl3) δ 7,56 (s, 1H), 7,52 (dd, J=5,4, 8,4 Гц, 2H), 7,42-7,38 (m, 3H), 7,12 (m, 2H), 5,18 (5, 1H), 4,62 (s, 1H), 2,66 (m, 6H), 1,72 (s, 6H), 2,01-0,83 (m, 5H) ppm. 13C ЯМР (100 МГц, CDCl3) δ 125,0, 124,0, 123,8, 116,0, 116,0, 71,3, 55,9, 55,5, 47,6, 46,7, 29,6, 25,6, 24,8, 19,8 ppm. Чистота: 98,0% UPLCMS (210 нМ); время удерживания 0,95 минуты; (M+1) 382,9. Анал. расч. для C23H27FN2O2·0,37(CHCl3): C, 65,86; H, 6,47; N, 6,57. Найденное значение: C, 65,85; H, 6,69; N, 6,49.
(S)-Хинуклидин-3-ил-2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-илкарбамат ( соединение 2 )
В перемешиваемый раствор 4-фтортиобензамида (8,94 г, 57,6 ммоль) в этаноле (70 мл) добавляли этил-4-хлорацетоацетат (7,8 мл, 58 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 часов, обрабатывали путем добавления аликвоты этил-4-хлорацетоацетата (1,0 мл, 7,4 ммоль) и нагревали с обратным холодильником еще в течение 3,5 часа. Затем реакционную смесь концентрировали, и остаток разделяли между этилацетатом (200 мл) и водным NaHCO3 (200 мл). Органический слой объединяли с реэкстрактом водного слоя (этилацетат, 1×75 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате масло янтарного цвета очищали с помощью флэш-хроматографии с использованием градиента гексан/этилацетат с получением этил-2-(2-(4-фторфенил)тиазол-4-ил)ацетата в виде легкоплавкого практически бесцветного твердого вещества (13,58 г, 89%).
В перемешиваемый раствор этил-2-(2-(4-фторфенил)тиазол-4-ил)ацетата (6,28 г, 23,7 ммоль) в DMF (50 мл) добавляли гидрид натрия [60% дисперсия в минеральном масле] (2,84 г, 71,0 ммоль). Пенистую смесь перемешивали в течение 15 минут перед охлаждением в ледяной бане и добавлением йодметана (4,4 мл, 71 ммоль). Реакционную смесь перемешивали в течение ночи с обеспечением медленного нагревания охлаждающей бани до комнатной температуры. Затем смесь концентрировали, и остаток разделяли между этилацетатом (80 мл) и водой (200 мл). Органический слой промывали второй порцией воды (1×200 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате масло янтарного цвета очищали с помощью флэш-хроматографии с использованием градиента гексан/этилацетат с получением этил-2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропаноата в виде бесцветного масла (4,57 г, 66%).
В перемешиваемый раствор этил-2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропаноата (4,56 г, 15,5 ммоль) в смеси THF/этанол/вода 1:1:1 (45 мл) добавляли моногидрат гидроксида лития (2,93 г, 69,8 ммоль). Реакционную смесь перемешивали в течение ночи, концентрировали и повторно растворяли в воде (175 мл). Раствор промывали эфиром (1×100 мл), подкисляли путем добавления 1,0 н. HCl (80 мл) и экстрагировали этилацетатом (2×70 мл). Объединенные экстракты высушивали (Na2SO4) и концентрировали с получением 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (4,04 г, 98%). Данный материал применяли на следующей стадии без очистки.
В перемешиваемый и охлажденный (0°C) раствор 2-(2-(4-фторфенил)тиазол-4-ил)-2-метилпропановой кислоты (4,02 г, 15,2 ммоль) в THF (100 мл) добавляли триметиламин (4,2 мл, 30 ммоль), а затем изобутилхлорформиат (3,0 мл, 23 ммоль). Реакционную смесь перемешивали охлажденной в течение еще 1 часа перед добавлением раствора азида натрия (1,98 г, 30,5 ммоль) в воде (20 мл). Реакционную смесь перемешивали в течение ночи с обеспечением медленного нагревания охлаждающей бани до комнатной температуры. Затем смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные экстракты промывали водным NaHCO3 (1×150 мл) и солевым раствором (1×100 мл), высушивали (Na2SO4) и концентрировали. После совместного выпаривания с толуолом (2×50 мл) полученное в результате белое твердое вещество поглощали толуолом (100 мл) и нагревали с обратным холодильником в течение 4 часов. Затем добавляли (S)-3-хинуклидинол (3,87 г, 30,4 ммоль), и нагревание с обратным холодильником продолжали в течение ночи. Реакционную смесь концентрировали, и остаток разделяли между этилацетатом (100 мл) и водным NaHCO3 (150 мл). Органический слой промывали водой (1×150 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате грязно-белое твердое вещество очищали с помощью флэш-хроматографии с использованием градиента хлороформ/метанол/аммиак с получением указанного в заголовке соединения в виде белого твердого вещества (4,34 г, 73%).1H ЯМР (400 МГц, CDCl3) δ 7,96-7,88 (m, 2H), 7,16-7,04 (m, 3H), 5,55 (br s, 1H), 4,69-4,62 (m, 1H), 3,24-3,11 (m, 1H), 3,00-2,50 (m, 5H), 2,01-1,26 (m, 11H) ppm. 13C ЯМР (400 МГц, CDCl3) δ 166,4, 165,1, 163,8 (d, J=250,3 Гц), 162,9, 155,0, 130,1 (d, J=3,3 Гц), 128,4 (d, J= 8,5 Гц), 115,9 (d, J= 22,3 Гц), 112,5, 71,2, 55,7, 54,2, 47,5, 46,5, 28,0, 25,5, 24,7, 19,6 ppm. Чистота: 100% UPLCMS (210 нМ & 254 нМ); время удерживания 0,83 минуты; (M+1) 390.
(S)-Хинуклидин-3-ил-(2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 3 )
С использованием общей процедуры E и исходных веществ для реакции, этил-2-(4-бромфенил)-2-метилпропаноата и 4-(2-метоксиэтокси)фенилбороновой кислоты, получали этил-2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропаноат в виде грязно-белого твердого вещества. В перемешиваемый раствор этого соединения (3,01 г, 8,78 ммоль) в смеси тетрагидрофуран/этанол/вода 1:1:1 (объем/объем/объем) (45 мл) добавляли моногидрат гидроксида лития (1,47 г, 61,4 ммоль). Смесь нагревали с обратным холодильником в течение ночи, а затем концентрировали. Остаток растворяли в воде, обрабатывали 1 н. хлористоводородной кислотой (65 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали с получением 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,75 г, 100%). Осуществляли реакцию этого промежуточного соединения и (S)-хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде бесцветного стекловидного твердого вещества. МГц, DMSO-d6) δ 7,62-7,29 (m, 7H), 7,01 (d, J=8,9 Гц, 2H), 4,47-4,37 (m, 1H), 4,17-4,08 (m, 2H), 3,72-3,62 (m, 2H), 3,32 (s, 3H), 3,09-2,25 (m, 6H), 2,05-1,18 (m, 11H) ppm. 13C ЯМР (100 МГц
1-Азабицикло[2.2.2]окт-3-ил-[2-(бифенил-3-ил)пропан-2-ил]карбамат ( соединение 4 )
С использованием общей процедуры C, 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)пропан-2-ил]карбамата (600 мг, 1,63 ммоль), фенилбороновой кислоты (398 мг, 3,27 ммоль) и ацетата палладия(II) получали указанное в заголовке соединение в виде белого твердого вещества (379 мг, 64%). 1H ЯМР (400 МГц, CDCl3) δ 7,61 (s, 1H), 7,56 (d, J= 7,4 Гц, 2H), 7,50-7,38 (m, 4H), 7,34 (m, 2H), 5,16 (s, 1H), 4,63 (s, 1H), 3,39-2,09 (m, 6H), 1,72 (s, 6H), 2,02-0,73 (m, 5H) ppm. 13C ЯМР (100 МГц, CDCl3) δ 154,8, 147,8, 141,6, 129,0, 129,0, 128,6, 127,5, 125,8, 125,0, 124,0, 71,6, 71,3, 55,9, 55,5, 47,6, 46,8, 31,5, 30,2, 30,0, 29,5, 25,6, 24,8, 19,8 ppm. Чистота: 99% UPLCMS (210 нМ); время удерживания 0,84 минуты; (M+1) 365,0. Анал. расч. для C23H28N2O2·0,29(CHCl3): C, 70,02; H, 7,14; N, 7,01. Найденное значение: C, 70,02; H, 7,37; N, 6,84.
(S)-Хинуклидин-3-ил-2-(бифенил-4-ил)пропан-2-илкарбамат ( соединение 5 )
С использованием общей процедуры B бромбензoнитрил (2,00 г, 11,0 ммоль) превращали в соответствующий 2-(4-бромфенил)пропан-2-амин (1,20 г, 51%) в виде коричневого масла.
С использованием общей процедуры A, 2-(4-бромфенил)пропан-2-амина (1,0 г, 4,7 ммоль) и (S)-хинуклидин-3-ола получали (S)-хинуклидин-3-ил-2-(4-бромфенил)пропан-2-илкарбамат (1,0 г, 58%) в виде коричневого масла.
С использованием общей процедуры C, вышеупомянутого бромида (200 мг, 0,540 ммоль), фенилбороновой кислоты (133 мг, 1,10 ммоль) и [PdCl2(pddf)]CH2Cl2 получали указанное в заголовке соединение в виде белого твердого вещества (70 мг, 35%). 1H ЯМР (500 МГц, CDCl3) δ 7,60-7,53 (m, 4H), 7,47 (d, J=8,5 Гц, 2H), 7,42 (t, J=7,5 Гц, 2H), 7,33 (t, J=7,5 Гц, 1H), 5,26 (br s, 1H), 4,64 (m, 1H), 3,33-3,15 (m, 1H), 3,10-2,45 (m, 5H), 2,40-1,80 (m, 2H), 1,78-1,58 (m, 7H), 1,55-1,33 (m, 2H) ppm. 13C ЯМР (125 МГц, CDCl3) δ 154,5, 146,1, 140,8, 139,5, 128,7, 127,2, 127,1, 127,1, 125,2, 70,9, 55,5, 55,1, 47,4, 46,4, 31,1, 29,5, 25,3, 24,5, 19,5 ppm. Чистота: 100% LCMS (214 нМ & 254 нМ); время удерживания 1,56 минуты; (M+1) 365.
Хинуклидин-3-ил-1-(бифенил-4-ил)циклопропилкарбамат ( соединение 6 )
С использованием общей процедуры D бромбензoнитрил (3,00 г, 16,5 ммоль) превращали в соответствующий 1-(4-бромфенил)циклопропанамин (1,80 г, 51%) в виде желтого твердого вещества.
С использованием общей процедуры A из 1-(4-бромфенил)циклопропанамина (1,0 г, 4,7 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-1-(4-бромфенил)циклопропилкарбамат (1,3 г, 75%) в виде белого полутвердого вещества.
С использованием общей процедуры C, вышеупомянутого карбамата (400 мг, 1,12 ммоль), фенилбороновой кислоты (267 мг, 2,22 ммоль) и [PdCl2(pddf)]CH2Cl2 получали указанное в заголовке соединение в виде вязкого масла (100 мг, 25%). 1H ЯМР (500 МГц, CDCl3) δ 7,47 (d, J= 7,5 Гц, 2H), 7,43 (d, J= 8,0 Гц, 2H), 7,33 (t, J= 7,5 Гц, 2H), 7,26-7,15 (m, 3H), 5,93 (br s, 0,6H), 5,89 (br s, 0,4H), 4,67 (m, 1H), 3,20-3,06 (m, 1H), 2,88-2,42 (m, 5H), 1,98-1,08 (m, 9H) ppm. 13C ЯМР (125 МГц, CDCl3) δ 155,0, 141,0, 139,7, 138,2, 127,7, 126,1, 126,0, 124,8, 124,1, 70,0, 54,5, 46,3, 45,4, 34,1, 24,3, 23,2, 18,3, 17,0 ppm. Чистота: 100% LCMC (214 нМ & 254 нМ); время удерживания 1,52 минуты; (M+1) 363.
(S)-Хинуклидин-3-ил-1-(4'-фторбифенил-4-ил)циклопропилкарбамат ( соединение 7 )
С использованием общей процедуры C, (S)-хинуклидин-3-ил-1-(4-бромфенил)циклопропилкарбамата, 4-F-фенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали указанное в заголовке соединение в виде белого твердого вещества (45%). 1H ЯМР (500 МГц, DMSO-d6) δ 8,06-7,83 (d, 1H), 7,69-7,66 (m, 2H), 7,59-7,55 (m, 2H), 7,29-7,22 (m, 4H), 4,56-4,54 (m, 1H), 3,13-2,32 (m, 6H), 1,91-1,19 (m, 9H) ppm. 13C ЯМР (125 МГц, DMSO-d6) δ 163,2, 161,2, 156,4, 143,7, 136,9, 128,9, 128,8, 126,8, 125,6, 116,2, 116,0, 70,7, 55,8, 47,4, 46,4, 34,8, 25,7, 24,6, 19,6, 18,7, 18,6 ppm. Чистота: > 97% LCMS (214 нМ & 254 нМ); время удерживания 1,96 минуты; (M+1) 381,2.
(S)-1-Азабицикло[2.2.2]окт-3-ил-[1-(2',4'-дифторбифенил-4-ил)циклопропил]карбамат ( соединение 8 )
С использованием общей процедуры C, (S)-хинуклидин-3-ил-1-(4-бромфенил)циклопропилкарбамата (0,446 г, 1,22 ммоль), 2,4-дифторфенилбороновой кислоты (0,386 г, 2,44 ммоль) и Pd(OAc)2 (0,015 г, 0,067 ммоль) получали указанное в заголовке соединение в виде рыжевато-коричневого твердого вещества (0,111 г, 23%). 1H ЯМР (CDCl3) δ 7,43 (dd, J=8,4, 1,6 Гц, 2H), 7,40-7,33 (m, 1H), 7,31 (d, J=7,7 Гц, 2H), 6,99-6,81 (m, 2H), 5,54 (d, J=48,0 Гц, 1H), 4,82-4,65 (m, 1H), 3,30-3,07 (m, 1H), 2,98-2,44 (m, 5H), 1,97 (d, J=32,7 Гц, 1H), 1,83 (d, J=10,3 Гц, 1H), 1,64 (s, 1H), 1,52 (s, 1H), 1,39 (s, 1H), 1,31 (d, J=6,8 Гц, 4H) ppm. 13C ЯМР основной ротамер (CDCl3) δ 162,2 (dd, J=12,8, 249,1 Гц), 159,8 (dd, J=11,8, 251,0 Гц), 156,9, 156,0, 142,6, 133,1, 131,3 (m), 128,9, 125,6, 124,9, 111,5 (dd, J=3,9, 21,2 Гц) 104,4 (dd, J=25,2, 29,4 Гц), 72,1, 71,6, 55,7, 47,4, 46,5, 35,7, 35,3, 25,5, 24,6, 24,4, 19,5, 18,1 ppm. Чистота: LCMS > 99,3% (214 нМ & 254 нМ); время удерживания 0,90 минуты; (M+1) 399,0.
1-Азабицикло[2.2.2]окт-3-ил-[1-(4'-метоксибифенил-4-ил)циклопропил]карбамат ( соединение 9 )
С использованием общей процедуры C, хинуклидин-3-ил-1-(4-бромфенил)циклопропилкарбамата (0,485 г, 1,33 ммоль), 4-метоксифенилбороновой кислоты (0,404 г, 2,66 ммоль) и Pd(OAc)2 (0,016 г, 0,071 ммоль) получали указанное в заголовке соединение в виде серого твердого вещества (0,337 мг, 65%). 1H ЯМР (CDCl3) δ 7,48 (dd, J=8,6, 5,5 Гц, 4H), 7,29 (d, J=7,6 Гц, 2H), 6,96 (d, J=8,8 Гц, 2H), 5,58 (d, J=48,7 Гц, 1H), 4,83-4,63 (m, 1H), 3,84 (s, 3H), 3,20 (dd, J=24,0, 15,5 Гц, 1H), 2,97-2,42 (m, 5H), 1,97 (d, J=30,9 Гц, 1H), 1,81 (s, 1H), 1,75-1,33 (m, 3H), 1,28 (d, J=6,8 Гц, 4H) ppm. 13C ЯМР основной ротамер (CDCl3) δ 159,1, 156,0, 141,4, 139,0, 133,4, 128,0, 126,7, 125,9, 114,2, 71,5, 55,7, 55,3, 47,4, 46,5, 35,3, 25,5, 24,6, 19,6, 17,8 ppm. Чистота: LCMS >97,1% (214 нМ & 254 нМ); время удерживания 0,88 минуты; (M+1) 393,4.
Хинуклидин-3-ил-2-(5-(4-фторфенил)тиофен-3-ил)пропан-2-илкарбамат ( соединение 10 )
В перемешиваемый и охлажденный (0°C) раствор этил-5-бромтиофен-3-карбоксилата (13,30 г, 56,57 ммоль) в THF (100 мл) по каплям добавляли раствор бромида метилмагния в диэтиловом эфире [3,0 M] (55,0 мл, 165 ммоль) в течение 20 минут. Через 2 часа реакционный раствор концентрировали. Остаток поглощали водным NH4Cl (200 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные экстракты высушивали (Na2SO4) и концентрировали. Полученное в результате масло янтарного цвета очищали с помощью флэш-хроматографии с использованием градиента гексан/этилацетат с получением 2-(5-бромтиофен-3-ил)пропан-2-ола в виде масла бледно-янтарного цвета (8,05 г, 64%).
В перемешиваемый раствор 2-(5-бромтиофен-3-ил)пропан-2-ола (8,03 г, 36,3 ммоль) в метиленхлориде (80 мл) добавляли азид натрия (7,08 г, 109 ммоль), а затем трифторуксусную кислоту (8,0 мл, по каплям в течение 5-6 минут). Загустевающую суспензию перемешивали в течение 1,5 часа перед разбавлением водой (350 мл) и экстрагированием этилацетатом (1×200 мл). Органический слой промывали водным NaHCO3 (1×250 мл), высушивали (Na2SO4) и концентрировали с получением неочищенного азидного продукта. В перемешиваемый раствор этого материала в THF (160 мл) добавляли воду (11 мл) с последующим добавлением трифенилфосфина (23,8 г, 90,7 ммоль). Реакционную смесь перемешивали в течение 2 дней перед концентрированием. Полученный в результате остаток растворяли в этилацетате (250 мл) и экстрагировали 1 н. водной HCl (4×75 мл). Объединенные экстракты подщелачивали концентрированным NH4OH и экстрагировали этилацетатом (2×100 мл). Эти экстракты, в свою очередь, высушивали (Na2SO4) и концентрировали. Полученное в результате масло янтарного цвета очищали с помощью флэш-хроматографии с использованием градиента метиленхлорид/метанол/аммиак с получением смеси 2-(5-бромтиофен-3-ил)пропан-2-амина и трифенилфосфиноксида (в соотношении ~ 70/30) в виде вязкого масла янтарного цвета (1,32 г, 17%).
В перемешиваемый раствор 3-хинуклидинола (3,00 г, 23,6 ммоль) в THF (100 мл) добавляли 4-нитрофенилхлорформиат (5,94 г, 29,5). После перемешивания в течение 4 часов осадок отфильтровывали, ополаскивали с помощью THF и высушивали воздухом на фритте в лабораторной вакуумной системе. Осадок на фильтре растворяли в этилацетате (150 мл) и промывали водным NaHCO3 (1×150 мл) и водой (2×150 мл). Органический слой высушивали (Na2SO4) и концентрировали с получением неочищенного продукта, представляющего собой 4-нитрофенилхинуклидин-3-илкарбонат, который использовали на следующей стадии без очистки.
В перемешиваемый раствор 2-(5-бромтиофен-3-ил)пропан-2-амина (0,366 г, 1,66 ммоль) в THF (10 мл) добавляли 4-нитрофенилхинуклидин-3-илкарбонат (0,571 г, 1,95 ммоль) и несколько гранул 4-(диметиламино)пиридина. Смесь нагревали с обратным холодильником в течение ночи, концентрировали и разделяли между этилацетатом (50 мл) и водным NaHCO3 (50 мл). Органический слой снова промывали водным NaHCO3 (1×50 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате грязно-желтое смолистое вещество очищали с помощью флэш-хроматографии с использованием градиента хлороформ/метанол/аммиак с получением хинуклидин-3-ил-(1-(5-бромтиофен-3-ил)циклопропил)карбамата в виде грязно-белого твердого вещества (0,305 г, 49%).
С использованием общей процедуры C, хинуклидин-3-ил-(1-(5-бромтиофен-3-ил)циклопропил)карбамата (0,227 г, 0,742 ммоль), 4-фторфенилбороновой кислоты (0,208 г, 1,49 ммоль), трициклогексилфосфина (0,021 г, 0,075 ммоль), фосфата калия (0,866, 4,08 ммоль) и ацетата палладия (8,0 мг, 36 мкмоль) получали указанное в заголовке соединение в виде серого твердого вещества (0,142 г, 49%). 1H ЯМР (400 МГц, CDCl3) δ 7,60-7,45 (m, 2H), 7,24-7,19 (m, 1H), 7,10-6,97 (m, 3H), 5,23 (br s, 1H), 4,72-4,61 (m, 1H), 3,30-3,04 (m, 1H), 3,03-2,25 (m, 5H), 2,09-1,02 (m, 11H) ppm. 13C ЯМР (400 МГц, CDCl3) δ 162,3 (d, J=247,1 Гц), 154,5, 149,8, 143,6, 130,7, 127,4 (d, J=8,1 Гц), 121,8, 118,9, 115,8 (d, J=21,6 Гц), 70,8, 55,5, 53,4, 47,3, 46,4, 29,0, 25,4, 24,4, 19,4 ppm. Чистота: 95,8% UPLCMS (210 нМ & 254 нМ); время удерживания 0,90 минуты; (M+1) 389.
(S)-Хинуклидин-3-ил-2-(3-(4-фторфенил)изотиазол-5-ил)пропан-2-илкарбамат ( соединение 11 )
В перемешиваемый раствор 2-(3-(4-фторфенил)изотиазол-5-ил)пропан-2-амина (1,21 г, 5,12 ммоль) в толуоле добавляли раствор фосгена в толуоле [~ 1,9 M] (10,8 мл, 20,5 ммоль). Реакционную смесь нагревали с обратным холодильником в течение двух часов, а затем концентрировали. Остаток совместно выпаривали с толуолом (2×15 мл) с получением неочищенного изоцианатного промежуточного соединения в виде золотистого масла. Этот материал поглощали толуолом (10 мл) и обрабатывали (S)-3-хинуклидинолом (0,749 г, 5,89 ммоль). Реакционную смесь нагревали с обратным холодильником в течение ночи и концентрировали. Остаток очищали с помощью флэш-хроматографии с использованием градиента хлороформ/метанол/аммиак с получением указанного в заголовке соединения в виде белого твердого вещества (0,971 г, 49%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,09-8,00 (m, 2H), 7,87 (br s, 1H), 7,75 (s, 1H), 7,35-7,25 (m, 2H), 4,54-4,45 (m, 1H), 3,14-2,92 (m, 1H), 2,87-2,17 (m, 5H), 1,98-0,98 (m, 11H) ppm. 13C ЯМР (400 МГц, DMSO-d6) δ 180,1, 165,6, 162,6 (d, J=246,4 Гц), 154,7, 131,2 (d, J=3,0 Гц), 128,7 (d, J=8,4 Гц), 118,2, 115,7 (d, J=21,8 Гц), 70,6, 55,3, 52,8, 46,9, 45,9, 29,9, 25,2, 24,2, 19,2 ppm. Чистота: 100% UPLCMS (210 нМ & 254 нМ); время удерживания 0,82 минуты; (M+1) 390.
(S)-Хинуклидин-3-ил-2-(4-(4-фторфенил)тиазол-2-ил)пропан-2-илкарбамат ( соединение 12 )
В перемешиваемый раствор этил-3-амино-3-тиоксопропаноата (20,00 г, 135,9 ммоль) в этаноле (120 мл) добавляли 2-бром-4'-фторацетофенон (29,49 г, 135,9 ммоль). Смесь нагревали с обратным холодильником в течение 1 часа, концентрировали и разделяли между этилацетатом (300 мл) и водным NaHCO3 (400 мл). Органический слой объединяли с реэкстрактом водного слоя (этилацетат, 1×100 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате светло-коричневое твердое вещество очищали с помощью флэш-хроматографии с использованием градиента гексан/этилацетат с получением этил-2-(4-(4-фторфенил)тиазол-2-ил)ацетата в виде грязно-белого твердого вещества (29,92 г, 83%).
В перемешиваемый и охлажденный (-78°C) раствор этил-2-(4-(4-фторфенил)тиазол-2-ил)ацетата (10,00 г, 37,69 ммоль) в THF (250 мл) по каплям добавляли раствор трет-бутоксида калия в THF [1,0 M] (136 мл, 136 ммоль) в течение 15 минут с последующим добавлением 18-краун-6 (1,6 мл, 7,5 ммоль). Еще через 30 минут при -78°C по каплям добавляли йодметан (8,5 мл) в течение 5 минут. Реакционную смесь перемешивали охлажденной в течение еще 2 часов перед выливанием в воду (450 мл) и экстрагированием этилацетатом (2×150 мл). Объединенные экстракты промывали солевым раствором (1×200 мл), высушивали (Na2SO4) и концентрировали. Полученное в результате коричневое масло очищали с помощью флэш-хроматографии с использованием градиента гексан/этилацетат с получением этил-2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропаноата в виде масла бледно-янтарного цвета (8,64 г, 78%).
В перемешиваемый раствор этил-2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропаноата (0,900 г, 3,07 ммоль) в смеси THF/этанол/вода 1:1:1 (15 мл) добавляли моногидрат гидроксида лития (0,451 г, 10,7 ммоль). После перемешивания в течение ночи реакционную смесь концентрировали и повторно растворяли в воде (80 мл). Раствор промывали эфиром (1×50 мл), подкисляли путем добавления 1 н. HCl (15 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные экстракты высушивали (Na2SO4) и концентрировали с получением 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропановой кислоты в виде бледно-золотистого твердого вещества (0,808 г, 99%).
В перемешиваемый и охлажденный (0°C) раствор 2-(4-(4-фторфенил)тиазол-2-ил)-2-метилпропановой кислоты (0,784 г, 2,96 ммоль) в THF (25 мл) добавляли триэтиламин (0,82 мл, 5,9 ммоль), а затем изобутилхлорформиат (0,58 мл, 4,4 ммоль). Реакционную смесь перемешивали охлажденной в течение еще 1 часа перед добавлением раствора азида натрия (0,385 г, 5,92 ммоль) в воде (7 мл). Реакционную смесь перемешивали в течение ночи с обеспечением медленного нагревания охлаждающей бани до комнатной температуры. Затем смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные экстракты промывали водным NaHCO3 (1×150 мл) и солевым раствором (1×100 мл), высушивали (Na2SO4) и концентрировали. После совместного выпаривания с толуолом (2×30 мл) полученное в результате грязно-белое твердое вещество поглощали толуолом (25 мл) и нагревали с обратным холодильником в течение 4 часов. Затем добавляли (S)-3-хинуклидинол (0,753 г, 5,92 ммоль), и нагревание с обратным холодильником продолжали в течение 3 часов. Реакционную смесь концентрировали, и остаток очищали с помощью флэш-хроматографии с использованием градиента хлороформ/метанол/аммиак с получением указанного в заголовке соединения в виде белого твердого вещества (0,793 г, 69%). 1H ЯМР (400 МГц, CDCl3) δ 7,90-7,81 (m, 2H), 7,32 (s, 1H), 7,14-7,05 (m, 2H), 5,76 (br s, 1H), 4,72-4,65 (m, 1H), 3,26-3,10 (m, 1H), 3,03-2,37 (m, 5H), 2,05-1,23 (m, 11H) ppm. 13C ЯМР (400 МГц, CDCl3) δ 177,6, 162,6 (d, J=248,4 Гц), 154,8, 153,6, 130,8 (d, J=3,2 Гц), 128,1 (d, J=8,1 Гц), 115,9 (d, J=21,7 Гц), 112,2, 71,6, 55,7, 47,4, 46,5, 29,1, 25,4, 24,7, 19,6 ppm. Чистота: 100% UPLCMS (210 нМ & 254 нМ); время удерживания 0,82 минуты; (M+1) 390.
Хинуклидин-3-ил-(2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 13 )
С использованием общей процедуры F и исходных веществ для реакции, 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты (полученной согласно описанному в примере 3) и хинуклидин-3-ола, получали указанное в заголовке соединение в виде бесцветного стекловидного твердого вещества (23%). Данные ЯМР соответствуют таковым из примера 3. Чистота: 100%, 99,1% (210 & 254 нм) UPLCMS; время удерживания: 0,87 мин.; (M+H+) 439,0.
(S)-Хинуклидин-3-ил-(2-(3'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 14 )
Заменив 4-(2-метоксиэтокси)фенилбороновую кислоту на 3-(2-метоксиэтокси)фенилбороновую кислоту, последовательность реакций, описанную в общих чертах в примере 3, применяли для получения 2-(3'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты. Осуществляли реакцию этого промежуточного соединения и хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде стекловидного бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 7,63-7,31 (m, 6H), 7,24-7,10 (m, 2H), 6,92 (dd, J=8,2, 1,9 Гц, 1H), 4,51-4,34 (m, 1H), 4,21-4,08 (m, 2H), 3,72-3,64 (m, 2H), 3,32 (s, 3H), 3,09-2,26 (m, 5H), 2,04-1,22 (m, 9H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 158,9, 154,6, 147,6, 141,5, 137,6, 129,9, 126,3, 125,2, 118,9, 113,2, 112,5, 70,4, 70,0, 66,9, 58,2, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,2, 19,2 ppm. Чистота: 100%, 100% (210 & 254 нМ) UPLCMS; время удерживания: 0,91 минуты; 15 (M+H+) 439,4.
Хинуклидин-3-ил-(2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат ( соединение 15 )
Заменив этил-2-(4-бромфенил)-2-метилпропаноат на этил-2-(3-бромфенил)-2-метилпропаноат, последовательность реакций, описанную в общих чертах в примере 3, применяли для получения 2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-3-ил)-2-метилпропановой кислоты. Осуществляли реакцию этого промежуточного соединения и хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 7,62-7,20 (m, 7H), 7,03 (d, J=8,7 Гц, 2H), 4,48-4,35 (m, 2H), 4,18-4,08 (m, 2H), 3,72-3,62 (m, 2H), 3,32 (s, 3H), 3,10-2,19 (m, 6H), 2,10-1,10 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 158,0, 154,6, 148,8, 139,5, 133,1, 128,5, 127,7, 123,8, 123,2, 122,7, 114,8, 70,4, 69,9, 67,0, 58,2, 55,3, 54,5, 47,0, 45,9, 29,4, 25,3, 24,2, 19,2 ppm. Чистота: 97,4%, 94,6% (210 & 254 нМ) UPLCMS; время удерживания: 0,88 минуты; (M+H+) 439,3.
Хинуклидин-3-ил-(2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 16 )
В перемешиваемый раствор 4-йодфенола (10,05 г, 45,68 ммоль) в ацетонитриле (100 мл) добавляли карбонат калия (6,95 г, 50,2 ммоль) и 1-хлор-3-метоксипропан (6,4 мл, 57,1 ммоль). Смесь нагревали с обратным холодильником в течение ночи, а затем концентрировали. Остаток поглощали водой и экстрагировали этилацетатом. Объединенные экстракты промывали водным раствором бикарбоната натрия, высушивали (Na2SO4) и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии на диоксиде кремния с использованием элюента гексан/этилацетат с получением 1-йод-4-(3-метоксипропокси)бензола в виде бесцветного масла (4,39 г, 33%). Осуществляли реакцию этого промежуточного соединения и этил-2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата согласно общей процедуре E с получением этил-2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)-2-метилпропаноата. В перемешиваемый раствор этого соединения (0,693 г, 1,94 ммоль) в смеси тетрагидрофуран/этанол/вода 1:1:1 (объем/объем/объем) (10 мл) добавляли моногидрат гидроксида лития (0,326 г, 7,77 ммоль). Смесь нагревали с обратным холодильником в течение ночи, а затем концентрировали. Остаток растворяли в воде, обрабатывали 1 н. хлористоводородной кислотой (10 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали с получением 2-(4'-(3-метоксипропокси)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде восковидного грязно-белого твердого вещества (0,630 г, 99%). Осуществляли реакцию этого промежуточного соединения и хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде стекловидного бесцветного твердого вещества (62%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,61-7,29 (m, 7H), 7,00 (d, J= 8,8 Гц, 2H), 4,47-4,36 (m, 1H), 4,05 (t, J= 6,4 Гц, 2H), 3,48 (t, J=6,3 Гц, 2H), 3,26 (s, 3H), 3,10-2,25 (m, 6H), 2,04-1,74 (m, 4H), 1,65-1,23 (m, 9H) ppm.13C ЯМР (100 МГц, DMSO-d6) δ 158,0, 154,5, 146,7, 137,4, 132,4, 127,5, 125,7, 125,2, 114,8, 69,9, 68,5, 64,6, 57,9, 55,4, 54,2, 46,9, 46,0, 29,4, 29,0, 25,2, 24,1, 19,2 ppm. Чистота: 97,7%, 98,2% (210 & 254 нМ) UPLCMS; время удерживания: 0,96 минуты; (M+H+) 453,5.
Хинуклидин-3-ил-(2-(4'-(гидроксиметил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 17 )
С использованием общей процедуры E и исходных веществ для реакции, этил-2-(4-бромфенил)-2-метилпропаноата и 4-формилфенилбороновой кислоты, получали этил-2-(4'-формил-[1,1'-бифенил]-4-ил)-2-метилпропаноат в виде твердого вещества бледно-янтарного цвета. Осуществляли реакцию этого промежуточного соединения и хинуклидин-3-ола согласно общей процедуре F с получением хинуклидин-3-ил-(2-(4'-формил-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата в виде пенистого желтого твердого вещества. В перемешиваемый раствор этого материала (0,755 г, 1,92 ммоль) в смеси тетрагидрофуран/этанол 2:1 (объем/объем) (15 мл) добавляли борогидрид натрия (0,073 г, 1,93 ммоль). Через 45 минут реакционную смесь разбавляли водой и экстрагировали хлороформом. Объединенные экстракты высушивали (Na2SO4) и концентрировали на диоксиде кремния. С помощью флэш-хроматографии на диоксиде кремния с использованием элюента хлороформ/метанол/аммиак получали указанное в заголовке соединение в виде белого твердого вещества (0,323 г, 43%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,66-7,29 (m, 9H), 5,18 (t, J= 5,7 Гц, 1H), 4,53 (d, J= 5,7 Гц, 2H), 4,46-4,37 (m, 1H), 3,11-2,19 (m, 6H), 2,11-1,10 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 154,7, 147,3, 141,5, 138,4, 137,7, 127,0, 126,2, 126,1, 125,3, 70,0, 62,6, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,2, 19,2 ppm. Чистота: 97,5%, 99,1% (210 & 254 нМ) UPLCMS; время удерживания: 0,73 минуты; (M+H+) 395.
Хинуклидин-3-ил-(2-(4'-(2-гидроксиэтил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 18 )
С использованием общей процедуры E и исходных веществ для реакции, 1-(2-(бензилокси)этил)-4-бромбензола и этил-2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропаноата, получали этил-2-(4'-(2-(бензилокси)этил)-[1,1'-бифенил]-4-ил)-2-метилпропаноат в виде бесцветного смолистого вещества. В перемешиваемый раствор этого соединения (1,34 г, 3,33 ммоль) в смеси тетрагидрофуран/этанол/вода 1:1:1 (объем/объем/объем) (18 мл) добавляли моногидрат гидроксида лития (0,698 г, 16,6 ммоль). После нагревания с обратным холодильником в течение ночи реакционную смесь концентрировали и разделяли между водой и диэтиловым эфиром. Полученную в результате эмульсию повторно экстрагировали 0,2 н. водным раствором гидроксида натрия (5×50 мл). Каждый раз удаляли прозрачную часть водного слоя. Затем объединенные водные слои обрабатывали 1,0 н. хлористоводородной кислотой (80 мл), и полученную в результате суспензию белого твердого вещества экстрагировали этилацетатом. Объединенные органические слои высушивали (Na2SO4) и концентрировали с получением 2-(4'-(2-(бензилокси)этил)-[1,1'-бифенил]-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (1,20 г, 96%). Осуществляли реакцию этого соединения и хинуклидин-3-ола согласно общей процедуре F с получением хинуклидин-3-ил-(2-(4'-(2-бензилоксиэтил)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата. В перемешиваемый раствор этого материала (0,435 г, 0,806 ммоль) в метаноле добавляли 1,0 н. хлористоводородную кислоту (1 мл) и 10% палладий на угле (50% воды; 0,087 г). Смесь циклически обрабатывали вакуумом и продувкой азотом несколько раз с повторным наполнением водородом после последнего вакуумирования. Через 1,25 часа реакционную смесь фильтровали через целит и концентрировали. Остаток поглощали водным раствором карбоната натрия и экстрагировали смесью хлороформ/изопропанол 4:1 (объем/объем). Объединенные экстракты высушивали (Na2SO4) и концентрировали на диоксиде кремния. С помощью флэш-хроматографии на диоксиде кремния с использованием градиента хлороформ/метанол/аммиак получали очищенное указанное в заголовке соединение в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 7,85-7,63 (m, 1H), 7,63-7,19 (m, 8H), 4,78-4,62 (m, 2H), 3,71-2,78 (m, 8H), 2,76 (t, J= 6,8 Гц, 2H), 2,26-1,96 (m, 2H), 1,96-1,40 (m, 9H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 153,8, 146,8, 138,7, 137,9, 137,6, 129,4, 126,3, 126,1, 125,3, 66,2, 62,1, 54,4, 52,8, 45,4, 44,5, 38,6, 29,5, 29,2, 24,0, 19,9, 16,6 ppm. Чистота: 100%, 100% (210 & 254 нМ) UPLCMS; время удерживания: 0,75 минуты; (M+H+) 409.
Хинуклидин-3-ил-(2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)пропан-2-ил)карбамат ( соединение 19 )
В перемешиваемую суспензию 4-метокситиобензамида (9,99 г, 59,7 ммоль) в этаноле (75 мл) добавляли этил-4-хлорацетоацетат (8,1 мл, 60 ммоль). Смесь нагревали с обратным холодильником в течение 4 часов перед охлаждением, добавлением дополнительного количества этил-4-хлорацетоацетата (0,81 мл, 6,0 ммоль) и возвращением к нагреванию с обратным холодильником. Еще после 4 часов нагревания реакционную смесь концентрировали и разделяли между этилацетатом и водным раствором бикарбоната натрия. Органический слой объединяли с дополнительными этилацетатными экстрактами, высушивали (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния с использованием градиента гексан/этилацетат с получением этил-2-(2-(4-метоксифенил)тиазол-4-ил)ацетата в виде масла бледно-янтарного цвета (14,51 г, 87%). В перемешиваемый раствор этого соединения (14,48 г, 52,2 ммоль) в N, N-диметилформамиде (125 мл) порциями добавляли гидрид натрия (60% дисперсия в минеральном масле; 6,27 г, 157 ммоль) в течение 15 минут. Полученную в результате красную суспензию охлаждали (0°C) и обрабатывали, добавляя по каплям йодметан (9,80 мл, 157 ммоль) в течение 10 минут. Охлаждающую баню убирали, и обеспечивали перемешивание реакционной смеси в течение 4 часов перед концентрированием и разделением остатка между этилацетатом и водой. Органический слой еще два раза промывали водой, высушивали (Na2SO4) и концентрировали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния с использованием градиента гексан/этилацетат с получением этил-2-(2-(4-метоксифенил)тиазол-4-ил)-2-метилпропаноата в виде масла бледно-янтарного цвета (14,12 г, 89%). В перемешиваемый раствор этого промежуточного соединения (14,12 г, 46,24 ммоль) в метиленхлориде (250 мл) по каплям добавляли трибромид бора (11,0 мл, 116 ммоль) в течение 5 минут. После перемешивания в течение ночи реакционную смесь гасили путем медленного добавления метанола (~ 20 мл), а затем концентрировали. Остаток поглощали метанолом (250 мл) и концентрированной серной кислотой (7,0 мл). Перемешиваемый раствор нагревали с обратным холодильником в течение 2 часов, концентрировали и разделяли между этилацетатом и водным раствором бикарбоната натрия. Органический слой объединяли со вторым этилацетатным экстрактом водного слоя, высушивали (Na2SO4) и концентрировали с получением метил-2-(2-(4-гидроксифенил)тиазол-4-ил)-2-метилпропаноата в виде белого твердого вещества (12,56 г, 98%). В перемешиваемый раствор 1-бром-3-метоксипропана (1,66 г, 10,8 ммоль) в ацетоне (30 мл) добавляли фенольное промежуточное соединение (2,00 г, 7,21 ммоль) и карбонат калия (1,25 г, 9,04 ммоль). Смесь нагревали в течение ночи с обратным холодильником, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния с использованием градиента гексан/этилацетат с получением метил-2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)-2-метилпропаноата в виде смолистого вещества слабо-янтарного цвета (2,47 г, 98%). В перемешиваемый раствор этого соединения (2,45 г, 7,01 ммоль) в смеси тетрагидрофуран/этанол/вода 1:1:1 (объем/объем/объем) (45 мл) добавляли моногидрат гидроксида лития (1,47 г, 35,0 ммоль). После перемешивания в течение ночи реакционную смесь концентрировали и разделяли между водой и диэтиловым эфиром. Водный слой обрабатывали 1,0 н. хлористоводородной кислотой (40 мл) и экстрагировали этилацетатом. Объединенные экстракты высушивали (Na2SO4) и концентрировали с получением 2-(2-(4-(3-метоксипропокси)фенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,19 г, 40 93%). Осуществляли реакцию этого соединения и хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде твердого вещества мягкой консистенции и слабо-янтарного цвета. 1H ЯМР (400 МГц, DMSO-d6) δ 7,82 (d, J=8,9 Гц, 2H), 7,36 (br s, 1H), 7,24 (br s, 1H), 7,03 (d, J=8,9 Гц, 2H), 4,49-4,41 (m, 1H), 4,07 (t, J=6,4 Гц, 2H), 3,48 (t, J=6,4 Гц, 2H), 3,26 (s, 3H), 3,09-2,26 (m, 6H), 2,02-1,91 (m, 2H), 1,91-1,03 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 165,8, 162,4, 160,0, 154,6, 127,5, 126,1, 114,9, 112,1, 70,1, 68,4, 64,8, 57,9, 55,4, 53,5, 46,9, 45,9, 28,9, 28,3, 25,2, 24,2, 19,2 ppm. Чистота: 100%, 100% (210 & 254 нМ) UPLCMS; время удерживания: 0,87 минуты; (M+H+) 460.
Хинуклидин-3-ил-(2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)пропан-2-ил)карбамат ( соединение 20 )
В перемешиваемый раствор 2-бромэтилметилового эфира (1,88 г, 13,5 ммоль) в ацетоне добавляли метил-2-(2-(4-гидроксифенил)тиазол-4-ил)-2-метилпропаноат (полученный согласно описанному в примере 19, 2,00 г, 7,21 ммоль) и карбонат калия (1,56 г, 11,3 ммоль). После нагревания с обратным холодильником в течение ночи смесь обрабатывали дополнительным количеством 2-бромэтилметилового эфира (1,88 г, 13,5 ммоль) и карбоната калия (1,56 г, 11,3 ммоль). Реакционную смесь нагревали с обратным холодильником в течение второй ночи, фильтровали и концентрировали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния с использованием градиента гексан/этилацетат с получением метил-2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)-2-метилпропаноата в виде белого твердого вещества (2,71 г, 90%). В перемешиваемый раствор этого соединения (2,71 г, 8,08 ммоль) в смеси тетрагидрофуран/этанол/вода 1:1:1 (объем/объем/объем) (50 мл) добавляли моногидрат гидроксида лития (1,70 г, 40,5 ммоль). После перемешивания в течение ночи реакционную смесь концентрировали и разделяли между водой и диэтиловым эфиром. Водный слой обрабатывали 1,0 н. хлористоводородной кислотой (41 мл) и экстрагировали этилацетатом. Объединенные экстракты высушивали (Na2SO4) и концентрировали с получением 2-(2-(4-(2-метоксиэтокси)фенил)тиазол-4-ил)-2-метилпропановой кислоты в виде белого твердого вещества (2,57 г, 99%). Осуществляли реакцию этого соединения и хинуклидин-3-ола согласно общей процедуре F с получением указанного в заголовке соединения в виде твердого вещества бледно-янтарного цвета. 1H ЯМР (400 МГц, DMSO-d6) δ 7,82 (d, J=8,8 Гц, 2H), 7,36 (br s, 1H), 7,24 (br s, 1H), 7,04 (d, J=8,8 Гц, 2H), 4,49-4,41 (m, 1H), 4,19-4,12 (m, 2H), 3,71-3,65 (m, 2H), 3,32 (s, 3H), 3,11-2,87 (m, 1H), 2,86-2,19 (m, 5H), 1,92-1,16 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 165,7, 162,9, 159,9, 154,6, 127,5, 126,2, 114,9, 112,2, 70,3, 70,1, 67,1, 58,2, 55,4, 53,5, 46,9, 45,9, 28,3, 25,2, 24,3, 19,2 ppm. Чистота: 100%, 100% (210 & 254 нМ) UPLCMS; время удерживания: 0,85 минуты; (M+H+) 446.
Хинуклидин-3-ил-2-(5-(4-(2-метоксиэтокси)фенил)пиридин-2-ил)пропан-2-илкарбамат ( соединение 21 )
С использованием общей процедуры E и исходных веществ для реакции, 5-бромпиколинонитрила и 2-(4-(2-метоксиэтокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана, получали 5-(4-(2-метоксиэтокси)фенил)пиколинонитрил. Трихлорид церия (8,05 г, 21,6 ммоль) загружали в колбу и высушивали путем нагревания (170°C) в вакууме в течение 3 часов. Твердое вещество поглощали тетрагидрофураном (20 мл) и энергично перемешивали в течение 30 минут. Суспензию охлаждали до -78°C и обрабатывали, добавляя по каплям 3,0 M раствор метиллития в диэтиловом эфире (7,2 мл, 21,6 ммоль). После добавления реакционную смесь перемешивали при -78°C в течение 1 часа перед добавлением раствора вышеупомянутого арилбората (1,83 г, 7,20 ммоль) в тетрагидрофуране (20 мл). Смесь выдерживали при -78°C в течение 2 часов, а затем обеспечивали ее нагревание до комнатной температуры. В этот момент реакционную смесь гасили путем добавления водного гидроксида аммония (10 мл) и фильтровали через слой целита. Фильтрат экстрагировали этилацетатом, и объединенные экстракты промывали солевым раствором, высушивали (Na2SO4) и концентрировали. Остаток очищали с помощью флэш-хроматографии на диоксиде кремния с использованием этилацетата в качестве элюента с получением 2-(5-(4-(2-метоксиэтокси)фенил)пиридин-2-ил)пропан-2-амина в виде желтого твердого вещества (0,800 г, 39%). В перемешиваемую суспензию этого промежуточного соединения (0,500 г, 1,75 ммоль) в воде (10 мл) и концентрированной хлористоводородной кислоте (0,44 мл) добавляли толуол (10 мл). Смесь охлаждали (0°C) и обрабатывали одновременно в течение 1 часа растворами трифосгена (0,776 г, 2,62 ммоль) в толуоле (10 мл) и бикарбоната натрия (2,2 г, 26 ммоль) в воде (20 мл). После добавлений реакционную смесь перемешивали еще в течение 30 минут перед удалением и высушиванием (Na2SO4) верхнего толуольного слоя. В это же время перемешиваемый раствор хинуклидин-3-ола (0,445 г, 3,64 ммоль) в тетрагидрофуране (10 мл) обрабатывали гидридом натрия (60% дисперсия в минеральном масле; 0,154 г, 3,85 ммоль). Эту смесь перемешивали в течение 5 минут, а затем добавляли в раствор неочищенного изоцианата в толуоле. Реакционную смесь перемешивали в течение 10 минут, гасили путем добавления солевого раствора (5 мл) и экстрагировали этилацетатом. Объединенные экстракты высушивали (Na2SO4) и концентрировали. Остаток очищали с помощью флэш-хроматографии на обращенно-фазовом диоксиде кремния с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (0,100 г, 13%). 1H ЯМР (500 МГц, CDCl3) δ 8,70-8,70 (d, J=2,0 Гц, 1H), 7,83-7,81 (m, 1H), 7,49-7,47 (d, J=9,0 Гц, 2H), 7,45-7,43 (d, J=8,0 Гц, 1H), 7,03-7,01 (d, J=8,5 Гц, 2H), 6,63 (br s, 1H), 4,68-4,66 (m, 1H), 4,16 (t, J=5,0 Гц, 2H), 3,77 (t, J=5,0 Гц, 2H), 3,45 (s, 3H), 3,19-2,70 (m, 6H), 2,15-1,89 (m, 2H), 1,76 (s, 6H), 1,73-1,36 (m, 3H) ppm. 13C ЯМР (125 МГц, CDCl3) δ 162,7, 158,9, 154,9, 145,9, 134,8, 134,3, 130,1, 128,1, 119,2, 115,2, 71,0, 70,8, 67,4, 59,2, 55,9, 55,7, 47,4, 46,5, 46,4, 27,9, 25,4, 24,6, 19,5 ppm. Чистота: >99% (214 & 254 нМ) LCMS; время удерживания: 1,32 минуты; (M+H+) 440,2.
Хинуклидин-3-ил-(2-(4'-(3-цианопропокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 22 )
В перемешиваемый раствор 4-бромфенола (17,1 г, 98,8 ммоль) в ацетонитриле (150 мл) добавляли 1-бромбутилнитрил (12,3 мл, 124 ммоль) и карбонат калия (15,0 г, 109 ммоль). Смесь нагревали с обратным холодильником в течение ночи, охлаждали и концентрировали. Остаток поглощали водой и экстрагировали этилацетатом. Объединенные экстракты высушивали (Na2SO4) и концентрировали, а неочищенный материал очищали с помощью флэш-хроматографии на диоксиде кремния с использованием элюента гексан/этилацетат с получением 4-(4-бромфенокси)бутаннитрила в виде белого твердого вещества (20,8 г, 88%). В перемешиваемый раствор этого продукта в N, N-диметилформамиде (100 мл) добавляли бис(пинаколато)дибор (4,60 г, 18,1 ммоль), ацетат калия (7,41 г, 75,5 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (0,616 г, 1,04 ммоль). Смесь нагревали с обратным холодильником в течение ночи, а затем концентрировали. Остаток поглощали этилацетатом и промывали водой и солевым раствором. Органический слой высушивали (Na2SO4) и концентрировали, а неочищенный продукт очищали с помощью флэш-хроматографии на диоксиде кремния с использованием элюента гексан/этилацетат с получением 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)бутаннитрила в виде белого твердого вещества (3,43 г, 79%). Осуществляли реакцию этого продукта и хинуклидин-3-ил-(2-(4-бромфенил)пропан-2-ил)карбамата (полученного посредством реакции хинуклидин-3-ола и 2-(4-бромфенил)пропан-2-амина с использованием общей процедуры F) согласно общей процедуре E с получением указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 7,67-7,26 (m, 7H), 7,02 (d, J=8,8 Гц, 2H), 4,50-4,33 (m, 1H), 4,08 (t, J=6,0 Гц, 2H), 3,14-2,18 (m, 8H), 2,04 (quin, J=6,7 Гц, 2H), 1,94-1,70 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 157,7, 154,5, 146,8, 137,4, 132,7, 127,6, 125,7, 125,2, 120,2, 114,9, 70,0, 65,8, 55,4, 54,2, 46,9, 45,9, 29,4, 25,3, 24,7, 24,2, 19,2, 13,4 ppm. Чистота: 100%, 98,9% (210 & 254 нМ) UPLCMS; время удерживания: 0,88 минуты; (M+H+) 448,6.
Хинуклидин-3-ил-(2-(4'-(цианометокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамат ( соединение 23 )
С использованием общей процедуры E и исходных веществ для реакции, хинуклидин-3-ил-(2-(4-бромфенил)пропан-2-ил)карбамата (полученного посредством реакции хинуклидин-3-ола и 2-(4-бромфенил)пропан-2-амина с использованием общей процедуры F) и 4-(цианометокси)фенилбороновой кислоты, получали указанное в заголовке соединение в виде твердого вещества бледно-янтарного цвета. 1H ЯМР (400 МГц, DMSO-d6) δ 7,65 (d, J= 8,2 Гц, 2H), 7,60-7,31 (m, 5H), 7,15 (d, J=8,9 Гц, 2H), 5,21 (s, 2H), 4,53-4,30 (m, 1H), 3,18-2,19 (m, 6H), 2,05-1,18 (m, 11H) ppm. 13C ЯМР (100 МГц, DMSO-d6) δ 155,8, 154,6, 147,2, 137,2, 134,4, 127,8, 126,0, 125,3, 116,7, 115,3, 70,0, 55,4, 54,2, 53,5, 46,9, 45,9, 29,4, 25,2, 24,2, 19,2 ppm. Чистота: 100%, 100% (210 & 254 нМ) UPLCMS; время удерживания: 0,85 минуты; (M+H+) 420,3.
Пример 2. Получение ( S )-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата в форме свободного основания
Стадия 1. Диметилирование с помощью метилйодида
3N круглодонная (RB) колба была оснащена термометром, капельной воронкой и системой подвода азота. Колбу продували азотом, и трет-бутоксид калия (MW 112,21, 75,4 ммоль, 8,46 г, 4,0 экв., белый порошок) отвешивали и добавляли в колбу через насыпную воронку с последующим добавлением THF (60 мл). Большая часть трет-бутоксида калия растворялась с получением непрозрачного раствора. Эту смесь охлаждали в ледяной бане до 0-2°C (внутренняя температура). В отдельной колбе исходный сложный эфир (MW 265,3, 18,85 ммоль, 5,0 г, 1,0 экв.) растворяли в THF (18 мл+2 мл на ополаскивание) и переносили в капельную воронку. Этот раствор по каплям добавляли в охлажденную смесь в течение периода 25-30 мин., поддерживая внутреннюю температуру на уровне ниже 5°C в ходе добавления. Реакционную смесь вновь охлаждали до 0-2°C. В отдельной колбе готовили раствор метилйодида (MW 141,94, 47,13 ммоль, 6,7 г, 2,5 экв.) в THF (6 мл) и переносили в капельную воронку. Затем колбу, содержащую раствор метилйодида, ополаскивали с помощью THF (1,5 мл), и этот раствор затем переносили в капельную воронку, уже содержащую прозрачный бесцветный раствор метилйодида в THF. Этот раствор по каплям осторожно добавляли в темно-коричневую реакционную смесь в течение периода 30-40 мин., поддерживая внутреннюю температуру на уровне ниже 10°C все время в ходе добавления. После завершения добавления слегка мутную смесь перемешивали еще в течение 1 ч, причем за это время внутреннюю температуру понижали до 0-5°C. После перемешивания в течение часа при 0-5°C реакционную смесь гасили путем медленного добавления по каплям 5,0 M водной HCl (8 мл) в течение периода 5-7 мин. Внутреннюю температуру в ходе этого добавления поддерживали на уровне ниже 20°C. После добавления добавляли воду (14 мл), и смесь перемешивали в течение 2-3 мин. Перемешивание прекращали, и обеспечивали разделение на два слоя. Затем два слоя переносили в 1N RB колбу емкостью 250 мл, и THF выпаривали in vacuo, насколько это было возможно, с получением двухфазного слоя THF/продукта и воды. Обеспечивали разделение на два слоя. Раствор продукта стадии 1 в THF использовали в следующей реакции.
Стадия 2. Гидролиз сложного этилового эфира с помощью моногидрата LiOH
Неочищенный сложный эфир в THF добавляли в реакционную колбу. Отдельно отвешивали LiOH.H2O (MW 41,96, 75,0 ммоль, 3,15 грамма, 2,2 экв.) в стакан емкостью 100 мл, в который добавляли магнитный мешальник. Добавляли воду (40 мл), и смесь перемешивали до полного растворения твердого вещества с получением прозрачного бесцветного раствора. Затем этот водный раствор добавляли в RB колбу емкостью 250 мл, содержащую раствор сложного эфира в тетрагидрофуране (THF). К горлу колбы прикрепляли конденсатор, и к верхней части конденсатора прикрепляли систему подвода азота. Смесь нагревали с обратным холодильником в течение 16 часов. Через 16 часов нагревание прекращали, и смесь охлаждали до комнатной температуры. THF выпаривали in vacuo с получением коричневого раствора. Аликвоту коричневого водного раствора анализировали с помощью HPLC и LC/MS в отношении завершения гидролиза сложного этилового эфира. Добавляли воду (15 мл), и этот водный основный раствор экстрагировали с помощью TBME (2×40 мл) для удаления сложного трет-бутилового эфира. Водный основный слой охлаждали в ледяной бане до 0-10°C и подкисляли путем добавления по каплям концентрированной HCl до pH ~ 1 при перемешивании. К этому смолистому твердому веществу в водном кислом растворе добавляли TBME (60 мл), и смесь встряхивали, а затем энергично перемешивали для растворения всей кислоты в слое TBME. Два слоя переносили в делительную воронку, и слой TBME отделяли. Бледно-желтый водный кислый раствор повторно экстрагировали с помощью TBME (40 мл), и слой TBME отделяли и объединяли с ранее полученным слоем TBME. Водный кислый слой сливали. Объединенные слои TBME высушивали над безводным Na2SO4, фильтровали и выпаривали in vacuo с удалением TBME и получением неочищенной кислоты в виде оранжевого/темно-желтого масла, которое затвердевало в высоком вакууме с образованием твердого вещества грязно-желтого цвета. Неочищенную кислоту отвешивали и кристаллизовали посредством нагревания ее в смеси гептан/TBME (3:1, 5 мл/г неочищенного вещества) с получением кислоты в виде твердого желтого вещества.
Стадия 3. Образование гидроксамовой кислоты с помощью NH2OH.HCl
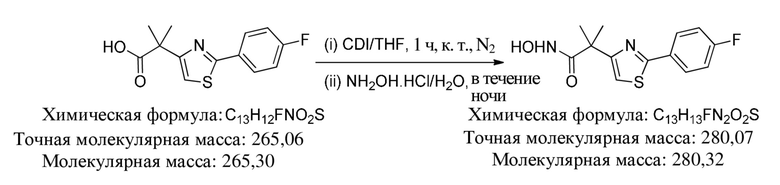
Карбоновую кислоту (MW 265,3, 18,85 ммоль, 5,0 г, 1,0 экв.) отвешивали и переносили в 1N RB колбу емкостью 25 мл в атмосфере азота. Добавляли THF (5,0 мл), и кислота без труда растворялась с получением прозрачного раствора от темно-желтого до коричневого цвета. Раствор охлаждали до 0-2°C (температура бани) в ледяной бане и медленно небольшими порциями добавляли N, N'-карбонилдиимидазол (CDI; MW 162,15, 20,74 ммоль, 3,36 г, 1,1 экв.) в течение периода 10-15 минут. Ледяную баню удаляли, и раствор перемешивали при комнатной температуре в течение 1 ч. После 1-часового перемешивания раствор снова охлаждали в ледяной бане до 0-2°C (температура бани). Медленно небольшими порциями добавляли гидрохлорид гидроксиламина (NH2OH.HCl; MW 69,49, 37,7 ммоль, 2,62 г, 2,0 экв.) в виде твердого вещества в течение периода 3-5 минут, поскольку это добавление было экзотермическим. После завершения добавления в неоднородную смесь по каплям добавляли воду (1,0 мл) в течение периода 2 минут, и реакционную смесь перемешивали при 0-10°C в ледяной бане в течение 5 минут. Охлаждающую баню убирали, и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи, 20-22 ч. Раствор становился прозрачным, как только весь NH2OH.HCl растворялся. Через 20-22 ч аликвоту реакционной смеси анализировали с помощью жидкостной хроматографии высокого давления (HPLC). Затем THF выпаривали in vacuo, и остаток поглощали дихлорметаном (120 мл) и водой (60 мл). Смесь переносили в делительную воронку, где ее встряхивали и обеспечивали разделение на два слоя. Водный слой сливали, а дихлорметановый слой промывали 1 н. гидрохлоридом (HCl; 60 мл). Кислый слой сливали. Дихлорметановый слой высушивали над безводным Na2SO4, фильтровали, и растворитель выпаривали in vacuo с получением неочищенной гидроксамовой кислоты в виде бледно-желтого твердого вещества, которое высушивали в высоком вакууме в течение ночи.
Стадия 3, продолжение. Превращение гидроксамовой кислоты в циклическое промежуточное соединение (не выделяемое)
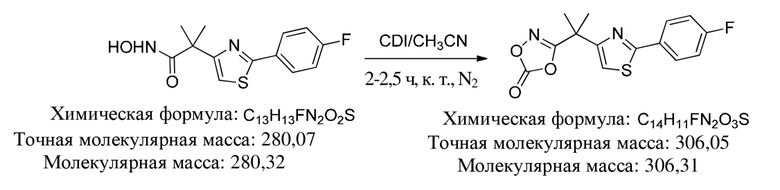
Неочищенную гидроксамовую кислоту (MW 280,32, 5,1 г) переносили в 1N RB колбу емкостью 250 мл с системой подвода азота. Добавляли магнитный мешальник с последующим добавлением ацетонитрила (50 мл). Твердое вещество было нерастворимым в ацетонитриле. Желтую неоднородную смесь перемешивали в течение 2-3 минут в атмосфере азота, и одной порцией добавляли CDI (MW 162,15, 20,74 ммоль, 3,36 г, 1,1 экв.) при комнатной температуре. Экзотермический эффект не наблюдали. Твердое вещество сразу же растворялось, и прозрачный желтый раствор перемешивали при комнатной температуре в течение 2-2,5 ч. Через 2-2,5 ч аликвоту анализировали с помощью HPLC и LC/MS, которые показали превращение гидроксамовой кислоты в желаемое циклическое промежуточное соединение.
Затем ацетонитрил выпаривали in vacuo с получением неочищенного циклического промежуточного соединения в виде красноватого густого масла. Масло поглощали толуолом (60 мл), и красноватую смесь нагревали с обратным холодильником в течение 2 часов, причем за это время циклическое промежуточное соединение перегруппировывалось в изоцианат с выделением CO2 (см. ниже).
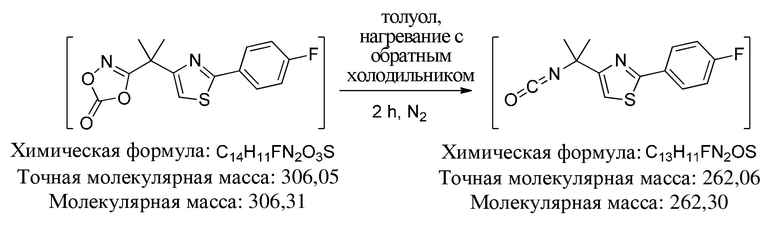
Стадия 3, продолжение. Превращение изоцианата в свободное основание
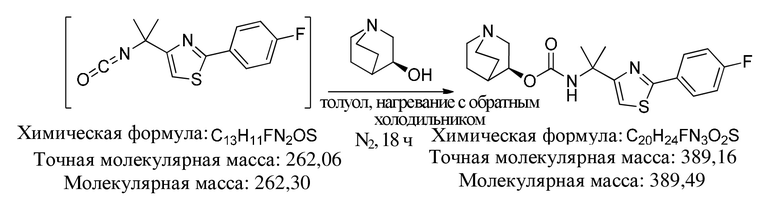
Реакционную смесь охлаждали до 50-60°C, и в смесь одной порцией добавляли (S)-(+)-хинуклидинол (MW 127,18, 28,28 ммоль, 3,6 г, 1,5 экв.) в виде твердого вещества. Смесь повторно нагревали с обратным холодильником в течение 18 ч. Через 18 ч аликвоту анализировали с помощью HPLC и LC/MS, которые показали полное превращение изоцианата в необходимый продукт. Реакционную смесь переносили в делительную воронку, и добавляли толуол (25 мл). Смесь промывали водой (2×40 мл), и водные слои отделяли. Объединенные водные слои повторно экстрагировали толуолом (30 мл), и водный слой сливали. Объединенные толуольные слои экстрагировали с помощью 1 н. HCl (2×60 мл), и толуольный слой (содержащий примесь O-ацила) сливали. Объединенные слои HCl переносили в колбу Эрленмейера емкостью 500 мл, оснащенную магнитным мешальником. Этот перемешиваемый прозрачный желтый/красновато-оранжевый раствор подщелачивали до pH 10-12 путем добавления по каплям 50% вес/вес водного NaOH. Необходимое свободное основание осаждалось из раствора в виде грязно-желтого смолистого твердого вещества, которое могло препятствовать движению магнитного мешальника. В эту смесь добавляли изопропилацетат (100 мл), и смесь энергично перемешивали в течение 5 минут во время перехода смолистого твердого вещества в изопропилацетат. Перемешивание прекращали, и обеспечивали разделение смеси на два слоя. Желтый изопропилацетатный слой отделяли, а основный водный слой повторно экстрагировали изопропилацетатом (30 мл). Основный водный слой сливали, а объединенные изопропилацетатные слои высушивали над безводным Na2SO4, фильтровали в предварительно взвешенную RB колбу, и растворитель выпаривали in vacuo с получением неочищенного свободного основания в виде твердого вещества от бежевого до рыжевато-коричневого цвета, которое высушивали в высоком вакууме в течение ночи.
Стадия 3, продолжение. Перекристаллизация неочищенного свободного основания
Неочищенное свободное основание от бежевого до рыжевато-коричневого цвета взвешивали и перекристаллизовывали из смеси гептан/изопропилацетат (3:1, 9,0 мл растворителя/г неочищенного свободного основания). Соответствующее количество смеси гептан/изопропилацетат добавляли в неочищенное свободное основание вместе с магнитным мешальником, и смесь нагревали с обратным холодильником в течение 10 мин. (изначально свободное основание было частично растворимым, но растворялось с получением прозрачного красновато-оранжевого раствора при нагревании с обратным холодильником). Источник тепла удаляли, и обеспечивали охлаждение смеси до комнатной температуры при перемешивании с образованием белого осадка. После перемешивания при комнатной температуре в течение 3-4 ч осадок отфильтровывали в лабораторной вакуумной системе с использованием воронки Бюхнера, промывали гептаном (20 мл) и высушивали в лабораторной вакуумной системе на воронке Бюхнера в течение ночи. Осадок переносили в чашку-кристаллизатор и высушивали при 55°C в течение ночи в вакуумной печи. 1H ЯМР (400 МГц, CDCl3) δ 8,04-7,83 (m, 2H), 7,20-6,99 (m, 3H), 5,53 (s, 1H), 4,73-4,55 (m, 1H), 3,18 (dd, J=14,5, 8,4 Гц, 1H), 3,05-2,19 (m, 5H), 2,0-1,76 (m, 11H) ppm. 13C ЯМР (100 МГц, CDCl3) δ 166,38, 165,02, 162,54, 162,8-155,0 (d, C-F), 130,06, 128,43, 128,34, 116,01, 115,79, 112,46, 71,18, 55,70, 54,13, 47,42, 46,52, 27,94, 25,41, 24,67, 19,58 ppm.
Пример 3. Получение кристаллических форм ( S )-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбаматных солей
Кристаллические (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбаматные соли могут быть образованы из свободного основания, полученного согласно описанному в примере 23.
Например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в форме свободного основания (приблизительно 50 ммоль) растворяли в IPA (140 мл) при комнатной температуре и фильтровали. Фильтрат добавляли в RB колбу емкостью 1 л, оснащенную мешалкой с верхним приводом и системой подвода/отвода азота. L-яблочную кислоту (приблизительно 50 ммоль) растворяли в IPA (100+30 мл) при комнатной температуре и фильтровали. Фильтрат добавляли в вышеупомянутую 1-литровую колбу. Полученный в результате раствор перемешивали при комнатной температуре (с затравкой или без нее) в атмосфере азота в течение 4-24 часов. На протяжении этого периода времени формировались кристаллы. Продукт собирали путем фильтрования и промывали небольшим количеством IPA (30 мл). Кристаллическое твердое вещество высушивали в вакуумной печи при 55˚C в течение 72 часов с получением необходимой малатной соли.
Кристаллические формы других солей, например, солей присоединения кислоты, образуемых с янтарной кислотой или HCl, могут быть получены аналогичным образом.
Пример 4. Ингибирование GCS in-vitro ( соединение 2 и аналоги )
Ингибирование активности глюкозилцерамидсинтазы можно измерить с помощью одного или нескольких анализов. Первый анализ представляет собой микросомальный анализ, с помощью которого непосредственно измеряют превращение церамида в глюкозилцерамид посредством HPLC. Микросомы являются источником активности глюкозилцерамидсинтазы в микросомальном анализе. Второй анализ представляет собой фенотипический анализ на основании клеток, с помощью которого отслеживается экспрессия нижележащего липида GM3 на клеточной поверхности посредством иммунофлуоресценции, опосредованной антителами. Конкретные протоколы приведены ниже.
Микросомальный анализ активности глюкозилцерамидсинтазы
Ферментный анализ с применением микросом как источника активности глюкозилцерамидсинтазы. Флуоресцентный керамидный субстрат доставляется к мембраносвязанному ферменту в виде комплекса с альбумином. После реакции церамид и глюкозилцерамид разделяют и проводят количественный анализ посредством HPLC с обращенной фазой с обнаружением флуоресценции. Ферментативную активность оценивают с применением субстрата с флуоресцентной меткой и микросом в качестве источника глюкозилцерамидсинтазы. C6-NBD-церамид образует комплекс с альбумином для доставки в микросомы, которые выделяют в соответствии с процедурой, описанной ниже. Конечная концентрация C6-NBD-церамида в исходном растворе составляет 0,5 мМ; конечная концентрация BSA составляет 0,5 мМ. Разделение и количественный анализ субстрата и продукта (глюкозилцерамида) осуществляют посредством HPLC с обращенной фазой с обнаружением флуоресценции.
Получение микросом из клеток А375 меланомы человека
Микросомы выделяют из клеток А375 меланомы человека. С помощью трипсинизации собирают от восьми до десяти миллионов клеток и промывают охлажденным на льду PBS. Клетки ресуспендируют в ледяном буфере для лизиса, содержащем ингибиторы протеазы. Клеточный лизат обрабатывают ультразвуком на льду с помощью ультразвукового зонда. После обработки ультразвуком клеточный лизат отделяют от дебриса центрифугированием при 10000 g в течение 10 минут при 4°C. Супернатант удаляют и очищают дополнительным центрифугированием при 100000 g в течение 1 часа при 4°C. Затем осадок ресуспендируют в буфере для лизиса, разделяют на аликвоты и хранят при -80°С до использования.
Анализ глюкозилцерамидсинтазы
Для определения ингибирования глюкозилцерамидсинтазы субстраты с удвоенной Km (флуоресцентный церамид и UDP-глюкоза, 3 мкМ и 4 мкМ соответственно) и микросомы (разведение 1:50) объединяют 1:1 и инкубируют при комнатной температуре в течение 1 часа в темноте на планшетном шейкере. Реакцию останавливают добавлением 150 мкл 100 мкМ C8-церамида в 50% водн. изопропанол; 10 мкл конечной смеси анализируют с помощью HPLC (с флуоресцентным детектором). Подвижная фаза представляет собой 1% муравьиную кислоту, добавленную к смеси 81% метанола/19% воды со скоростью потока 0,5 мл/мин. Флуоресценцию регистрируют при λex= 470 нм и λem= 530 нм. В этих условиях NBD-C6-GluCer характеризовался временем удерживания приблизительно 1,7 мин., а NBD-C6-Cer элюировался из колонки через приблизительно 2,1 мин. Оба пика отделены друг от друга и исходной линии и автоматически проинтегрированы программным обеспечением HPLC. Процент превращения субстрата в продукт используется в качестве показателя считывания для тестирования ингибитора.
Иммуносорбентный анализ, связанный с флуоресценцией GM3 (FLISA)
Это фенотипический анализ, с помощью которого измеряют экспрессию GM3 в клетках B16 меланомы мыши или клетках C32 меланомы человека после обработки исследуемыми соединениями. Экспрессию GM3 на клеточной поверхности определяют по флуоресценции, опосредованной антителами.
Соединения разбавляют в среде и высевают в 384-луночные планшеты в DMSO. Клетки B16 и C32 анализируют при значении плотности 20000 клеток/мл и 62500 клеток/мл соответственно на лунку. Каждая кривая титрования содержит 10 точек, которые анализируют в двух повторностях при каждом прогоне испытания. Планшеты инкубируют в течение 48 часов при 37°C, 5% CO2, а затем один раз промывают TBS. В каждую лунку добавляют антитело к GM3, а затем планшеты инкубируют еще в течение одного часа при комнатной температуре. Затем планшеты дважды промывают и инкубируют в течение еще одного часа с меченым вторичным антителом. После последней инкубации планшеты дважды промывают, и регистрируют флуоресценцию при λex=D640/20 нм и λem=657 нм на флуоресцентном ридере.
Результаты анализа
Отдельные результаты анализа некоторых иллюстративных соединений в данных анализах представлены в таблице ниже. Результаты микросомальных анализов выражены как «IC50GCS», что представляет собой концентрацию соединения, вызывающую полумаксимальное (50%) ингибирование активности глюкозилцерамидсинтазы. Результаты клеточных анализов выражены как «IC50 GM3 B16» или «IC50 GM3 C32» для анализа B16 и анализа C32 соответственно. Эти значения представляют собой концентрацию соединения, вызывающую полумаксимальное (50%) ингибирование экспрессии GM3 на поверхности клеток.
соединения
Эти сравнительные результаты демонстрируют, что соединения согласно настоящему изобретению характеризуются сравнимой активностью in-vitro, как ингибиторы GCS и, как следствие, ожидается, что они продемонстрируют схожие преимущества in-vivo.
Пример 5. Клиническое исследование соединения 2 на пациентах с GD-3
Было инициировано 156-недельное многокомпонентное открытое многонациональное исследование безопасности, переносимости, фармакокинетики, фармакодинамики и поисковых показателей эффективности соединения 2 в комбинации с имиглюцеразой у взрослых пациентов с болезнью Гоше типа 3, стабилизированной имиглюцеразой (под названием испытание LEAP или LEAP2IT). Соединение 2 вводят перорально в форме малатной соли (L-яблочная кислота) в дозе 15 мг/сутки (измеряется как количество свободного основания) в виде однократной суточной дозы. Оценки конечных точек связаны с безопасностью, биомаркерами CSF, фармакокинетикой/фармакодинамикой, системным заболеванием, нейровизуализацией и неврологической функцией (ЦНС/неврологические проявления).
В исследование были включены пациенты в возрасте 18 лет и старше с клиническим диагнозом GD3 и документально подтвержденным дефицитом активности кислой бета-глюкозидазы, получавшие лечение с помощью ERT в течение по меньшей мере 3 лет и имиглюцеразы (Церезим) в стабильной ежемесячной дозе в течение по меньшей мере 6 месяцев на момент включения в исследование. Пациенты должны достичь следующих терапевтических целей при GD: уровень гемоглобина ≥11,0 г/дл для женщин и ≥12,0 г/дл для мужчин; количество тромбоцитов ≥100000/мм3; объем селезенки составляет <10 нормальных объемов (MN) или тотальная спленэктомия (при условии, что спленэктомия производилась более чем за 3 года до рандомизации); объем печени составляет <1,5 MN; отсутствие костного криза и отсутствие симптоматического заболевания костей, такого как боль в костях, связанная с остеонекрозом и/или патологическими переломами, в течение последнего года. Пациенты должны иметь GD3 с глазодвигательной апраксией (надъядерным параличом взора), характеризующейся аномалией горизонтальных саккад.
(A) 52-недельный промежуточный анализ (N=6)
Промежуточный анализ проводили после того, как 6 пациентов завершили 52 недели одновременного лечения с использованием (1) имиглюцеразы (Церезим от Sanofi Genzyme) по установленной для каждого пациента схеме и (2) соединения 2, вводимого перорально по 15 мг/сутки в разовой дозе. В ходе исследования на примере пациентов оценивали безопасность и переносимость, биомаркеры CSF и плазмы крови (глюкозилцерамид, GL-1; глюкозилсфингозин, лизосомный GL1), фармакокинетику, маркеры системного заболевания (объем селезенки и печени, измеренный с помощью магнитно-резонансной визуализации (MRI), количество тромбоцитов, уровни гемоглобина), признаки интерстициального заболевания легких (компьютерная томография легких (CT) высокого разрешения) и горизонтальное саккадическое движение глаз. Кроме того, в CSF пациентов с GD3 количественно определяли исследовательские биомаркеры: церамид (предшественник GL-1), хитотриозидазу (CHITO), GM3 и GPNMB. Симптомы атаксии измеряли с помощью шкалы SARA, неврологические симптомы измеряли с помощью теста с построением маршрута, а функциональную МРТ использовали для оценки нейронных связей в головном мозге.
На исходном уровне у пяти пациентов было легкое неврологическое поражение, а у одного - умеренное неврологическое поражение, что измерялось с помощью модифицированного инструмента оценки тяжести (mSST; см., например, Davies, et al., J Inherit Metab Dis. (2011) 34(5), стр. 1053-1059).
Анализ концентраций соединения 2 в плазме крови и CSF показал, что соединение 2 эффективно проникает через гематоэнцефалический барьер у всех пациентов. Однако обнаружено, что у пациента 5 концентрации соединения 2 в плазме крови и CSF на неделе 26 ниже приблизительно на 50%, а на неделе 52 концентрации являются неопределяемыми. Считается, что это связано либо с несоблюдением режима лечения, либо с ошибками дозирования, поэтому анализ повторяли без включения данных пациента 5 на неделях 26 и 52. Данные подтверждают вывод о том, что равновесная концентрация соединения 2 достигалась в плазме крови и CSF на неделе 4 или ранее.
Через 52 недели данные также демонстрировали устойчивое значительное улучшение в показателях биомаркеров плазмы крови и CSF для GD-3. У всех шести пациентов с GD3 концентрации GL-1 и лизосомного GL-1 в плазме крови и CSF были следующими.
Таким образом, через 52 недели по сравнению с исходным уровнем концентрации в плазме и CSF изменились следующим образом.
(изменение, %)
Кроме того, в CSF пациентов с GD3 количественно определяли исследовательские биомаркеры: церамид (предшественник GL-1), хитотриозидазу (CHITO; фермент, уровень которого, как известно, повышен у пациентов с GD), GM3 (маркер гликосфинголипидов, уровень которого, как известно, повышен у пациентов с GD) и GPNMB (гликопротеин неметастатического белка B меланомы, который по имеющимся сведениям является биомаркером невропатического GD3). После 52 недель лечения не наблюдалось значительных изменений концентраций церамида, CHITO или GPNMB в CSF. У четырех из шести пациентов наблюдали измеримые концентрации GM3 в CSF на исходном уровне, и у каждого из этих пациентов обнаружили неопределяемое содержание GM3 в CSF через 4 недели, 26 недель и 52 недели.
Кроме того, через 52 недели у 5 из 6 пациентов наблюдали улучшения состояния при атаксии. Степень атаксии на исходном уровне и на протяжении всего исследования оценивали по Шкале оценки и определения степени атаксии (SARA; Schmitz-Hübsch et al. [2006]), с помощью которой оценивают восемь различных признаков мозжечковой атаксии по шкале от 0 до 40. Восемь параметров представляют собой следующее: походку, положение стоя, положение сидя, нарушение речи, управление пальцами, тест «нос-палец», быстрое попеременное движение рук и скольжение «пяткой по голени». Результаты оценки атаксии по SARA для всех шести пациентов представлены в таблице ниже.
Как показано в таблице, пять из шести пациентов характеризовались незначительной атаксией на исходном уровне, при этом средний совокупный балл по шкале SARA составлял 2,8 (SD=1,2). Наиболее частыми дефицитами на исходном уровне были нарушения походки. За исключением пациента 5 из-за низкого уровня воздействия соединения 2 у этого пациента и практически нормального балла в отношении атаксии у пациента (всего 0,5) на исходном уровне, затем у 4 из 5 пациентов наблюдали улучшение в отношении атаксии на неделе 52 (среднее улучшение=-0,9; SD=3,2). Пациент 4 продемонстрировал увеличение балла при оценке атаксии, при этом бал на исходном уровне составлял 3, а на неделе 52-7,5. Следует отметить, что такое явное ухудшение было почти полностью вызвано изменением параметра оценки «положения стоя» (балл положения стоя на исходном уровне и на неделе 26=1; бал на неделе 52=5) и жалобами пациента на боль в левом колене во время проверки. Кроме того, перед обследованием субъект повредил большой палец левой ноги; эта травма считалась устраненной через 11 дней после осмотра. За исключением этого выпадающего эффекта пациента 4, лечение при помощи соединения 2 приводило к значительному снижению среднего балла по SARA к неделе 26, который дополнительно немного улучшился к неделе 52.
Для оценки когнитивной функции у пациентов использовали тест с прокладыванием пути (TMT). ТМТ является одним из наиболее широко используемых нейропсихологических тестов и включен в большинство наборов тестов. TMT - это диагностический инструмент для оценки общего интеллекта и когнитивных дисфункций (Tombaugh et al. [2004]; Cavaco et al. [2013]). В части A TMT (TMT-A) субъектов просят соединить группу чисел в порядке возрастания (путь A). Эта задача представляет собой комбинацию визуального поиска и общей скорости визуальной и двигательной обработки. Часть B (TMT-B) представляет собой последовательность, в которой чередуются цифры и буквы (путь B). Субъекты должны активно «переключаться» между обеими категориями, соединяя их в возрастающем, но чередующемся порядке. Следовательно, считается, что эта задача включает компонент исполнительной функции, поскольку субъект должен активно переключаться между категориями, соединяя символы (MacPherson et al. [2017]).
TMT-A оценивает в основном скорость восприятия и психомоторных реакций. TMT-B более конкретно оценивает гибкость мышления и способности к переключению. Балл TMT-B минус балл TMT-A используется для устранения расхождения, относящегося к компонентам графомоторного и визуального сканирования TMT-A. Этот производный балл отражает уникальные требования задачи TMT-B.
При изучении нормативных данных для ТМТ-А и ТМТ-В у лиц, проживающих в терапевтической общине, в возрасте 18-89 лет (n=911) средние значения (SD) в возрастной группе 18-24 года (n=155) составляли 22,9 с (6,9) для ТМТ-А и 49 с (12,7) для ТМТ-В (Tombaugh et al. [2004]). Напротив, среднее время, затраченное на прохождение пути A и пути B, для пациентов, принимающих участие в исследовании, составляло 67,8 с (SD=60,3 с) и 193,8 с (SD=197,0) соответственно. На исходном уровне средняя разность во времени, необходимом для прохождения пути B минус путь A, составляла 126,0 с (SD=142,9 с). Это показывает, что пациенты с GD-3 в этом исследовании демонстрировали некоторую степень когнитивной дисфункции на исходном уровне.
На неделе 52 средний период времени, затраченный для прохождения пути А, составлял 56,5 с (SD=55,2 с), а пути В - 122,7 с (SD=91,8 с). У четырех из шести пациентов наблюдалось сокращение времени, затраченного для прохождения пути A, и у шести из шести наблюдалось сокращение времени, затраченного для прохождения пути B. За исключением пациента 5 из-за низкого уровня воздействия соединения 2 у этого пациента, у четырех из пяти пациентов наблюдалось снижение ТМТ-A, и у пяти из пяти пациентов наблюдалось снижение TMT-B.
На неделе 52 у 5 из 6 пациентов наблюдалось сокращение времени (ТМТ-В - ТМТ-А). Отдельные результаты показаны в таблице ниже.
На неделе 52 средняя разность во времени, необходимом для прохождения пути B минус путь A, составляла 66,2 с (SD=54,3). За исключением пациента 5, четыре из пяти пациентов продемонстрировали улучшение в показателе путь B минус путь A на неделе 52 со средним улучшением на -71,4 с (-31,6%) (SD 99,3 с (37,6%)).
Неврологическую функцию дополнительно оценивали с помощью функциональной магнитно-резонансной визуализации (fMRI). Пациент 2 был исключен, поскольку данные fMRI не были собраны на сеансе на неделе 52. Сеансы скрининга fMRI в состоянии покоя проводили при скрининге на исходном уровне, в ходе визитов на неделе 26 и неделе 52. Оценки связи на примере четырех субъектов (пациенты 1, 3, 4 и 5) были введены в анализы второго уровня как «соблюдающая режим» группа. Пациент 5 был исключен из-за вероятного несоблюдения режима лечения исследуемым лекарственным препаратом, как описано выше. Анализы проводили, как описано в другом месте (Smith et al. [2009]).
Было обнаружено, что соблюдающие режим субъекты демонстрируют улучшенную связь между более широко распределенной группой участков головного мозга, чем не соблюдающие режим субъекты, причем наиболее характерным признаком является увеличение силы связи между задним и передним отделами. На анатомическом уровне соблюдающие режим субъекты демонстрируют обширное и сильное укрепление связей между затылочно-теменными структурами и лобными, височными и лимбическими мишенями. Изменения в связях у пациента 5 были более скудными и ограничивались пространственно проксимальными структурами. На функциональном уровне улучшение связи между пассивным режимом работы и медиальными и фронтальными сетями наблюдается у каждого субъекта, кроме пациента 5. Это говорит о том, что сигнал в этих разрозненных сетях становится более когерентным, так что активность головного мозга может более эффективно передаваться между когнитивным резервом (задний отдел) и исполнительными функциями более высокого порядка (передний отдел). Также очевидным является последовательное взаимное картирование сетей состояния покоя (RSN) 2 и 3 («познание-язык-орфография» и «познание-пространство») с RSN 8 и 9 (исполнительная и левая лобно-теменная). У пациента 5 пространственное распределение изменений в связях является гораздо более локальным, отражая прежде всего перекрытие медиально-лобной и лобно-теменной сетей. Обе точки зрения предполагают, что у пациентов, которые полностью соблюдали протокол лечения, развилась большая согласованность между задним и передним отделами головного мозга, так что головной мозг в целом становится податливым к эффективной передаче информации. Там, где это очевидно, измененная связь у пациента 5 проявляется в более узкой группе передних отделов головного мозга и представляет собой менее целостное доказательство терапевтической пользы.
Результаты обобщены в таблице ниже. Пространственный анализ связи между разными анатомическими областями головного мозга выполняется для определения коэффициента корреляции для регрессионной средней интенсивности по вокселям. Результаты показывают, что связь между сетью пассивного режима работы (в состоянии покоя) и сетью исполнительных функций увеличилась у пациентов 1, 3, 4 и 6, но уменьшилась у пациента 5.
(B) 52-недельный промежуточный анализ (N=11)
Дополнительный промежуточный анализ выполняли, когда 11 пациентов достигли 52-недельного рубежа. Результаты этого анализа подтверждают наблюдение, сделанное в предыдущем промежуточном анализе.
Среди 11 пациентов второго промежуточного анализа было 7 мужчин и 4 женщины. 8 пациентов были гомозиготными по мутации p.Leu483Pro (также известной как L444P) в гене бета-галактозидазы, 2 - компаунд-гетерозиготными по вариантам p.Phe523Ile/p.Leu483Pro, и один - компаунд-гетерозиготным по вариантам p.Asp448His/p.Arg502Cys. Девять из 11 пациентов на исходном уровне характеризовались незначительной атаксией, в основном с нарушением походки. Двое считались не имеющими атаксии или имеющими атаксию очень слабой степени. Все 11 пациентов продолжают участие в исследовании. О серьезных нежелательных явлениях не сообщалось, наиболее частыми явлениями были легкая головная боль и боль в спине, при этом было установлено, что они, вероятно, связаны с вмешательством в виде люмбальной пункции для забора CSF.
Концентрации соединения 2 в плазме крови и CSF измеряли, как описано выше. Как было замечено ранее, один пациент (пациент 5) является исключением, демонстрирующим очень низкие уровни соединения 2 в плазме крови и CSF. Данные продолжают подтверждать вывод о том, что равновесная концентрация соединения 2 достигалась в плазме крове и CSF на неделе 4 или ранее.
В следующей таблице пациент 5 исключен из средних значений (N=10).
У одного пациента (пациент 1) также наблюдалось значительное снижение концентраций соединения в плазме крови и CSF, начиная с измерения на неделе 52, и это, вероятно, было результатом совместного лечения с применением индуктора CYP3A4 рифампицина с недели 39 по неделю 51 исследования. Поскольку предполагается, что соединение 2 является субстратом CYP3A, ожидается, что одновременное введение с индукторами CYP3A приведет к снижению системного воздействия.
Через 52 недели данные также демонстрировали устойчивые значительные улучшения с точки зрения биомаркеров плазмы крови и CSF для GD3 за исключением пациента 5. Первоначальные данные о воздействии соединения 2 коррелировали со снижением уровня лизосомного GL1 и GL1 в CSF и плазме крови. Вследствие этого сниженные уровни воздействия соединения 2 соответствовали увеличению уровня как лизосомного GL1, так и GL1, причем как в CSF, так и в плазме крови. На неделе 52 концентрации лизосомного GL1 в CSF у пациента 5 превышали верхний предел количественного определения (ULOQ; >100 пг/мл), поэтому результаты биомаркеров для лизосомного GL1 в CSF были рассчитаны с использованием точных значений ULOQ. Соответственно, у пациента 5 было обнаружено, что уровни лизосомного GL1 и GL1 в CSF увеличились приблизительно на 313% и 37% соответственно на неделе 52 по сравнению с исходным уровнем, а уровни лизосомного GL1 и GL1 в плазме крови увеличились приблизительно на 43% и 14% соответственно на неделе 52 по сравнению с исходным уровнем. Средние результаты для других субъектов исследования были следующими (N=10; пациент 5 исключен).
Таким образом, через 52 недели по сравнению с исходным уровнем концентрации в плазме крови и CSF изменились следующим образом (N=10; пациент 5 исключен).
Результаты оценки атаксии по SARA для пяти новых пациентов представлены в таблице ниже.
Как показано в таблице, за исключением пациента 9, который не страдал атаксией на исходном уровне, у 4 из 4 пациентов наблюдалось улучшение в отношении атаксии на неделе 52. У пациента 7 наблюдалось временное повышение балла при оценке атаксии, которое вернулось к норме на неделе 52.
Чтобы дополнительно продемонстрировать улучшения состояния при атаксии, двух пациентов снимали на видео в следующих временных точках: в ходе скрининга, а также на неделе 26 и неделе 52, при попытке пройти по прямой линии. Сравнение видеодоказательств показывает, что по сравнению с исходным уровнем оба пациента демонстрируют более устойчивую, лучше скоординированную и более быструю походку с меньшим количеством прикосновений к ближайшей стене для опоры и меньшим количеством шагов в сторону.
Сроки ТМТ для новых пациентов показаны в таблице ниже.
Через 52 недели у трех новых пациентов наблюдалось небольшое увеличение времени (TMT-B - TMT-A) неопределенной клинической значимости, наряду с тем, что у двух новых пациентов наблюдалось уменьшение времени (TMT-B - TMT-A), у одного пациента наблюдалось чрезвычайно большое уменьшение (снижение на 92%).
Неврологическую функцию дополнительно оценивали у новых пациентов с использованием функциональной магнитно-резонансной визуализации (fMRI), как описано выше. Результаты fMRI по-прежнему указывают на то, что у пациентов с достаточным воздействием соединения 2 развивается большая согласованность между задним и передним отделами головного мозга, так что головной мозг в целом становится податливым для эффективной передачи информации. Там, где это очевидно, измененная связь у одного пациента с недостаточным воздействием соединения 2 (пациент 5) проявляется в более узком наборе передних отделов головного мозга и представляет собой менее целостное свидетельство терапевтической пользы. Эта улучшенная связь наблюдается в областях, ассоциированных с исполнительной функцией. Функциональная MRI в состоянии покоя демонстрирует улучшенную связь между пассивным режимом работы и медиальными и фронтальными сетями, свидетельствуя о том, что передача сигналов в этих разрозненных сетях становится более когерентной, в результате чего активность головного мозга может более эффективно передаваться между когнитивным резервом (задний отдел) и исполнительными функциями более высокого порядка (передний отдел).
В частности, результаты показывают улучшенную связь между RSN 1, 2 и 3 (восприятие-зрение, познание-речь-орфография и познание-пространство) и RSN 6, 7 и 8 (сенсомоторная функция, слуховая функция и исполнительный контроль). На анатомическом уровне демонстрировалось обширное и сильное укрепление связей между затылочно-теменными структурами и лобными, височными и лимбическими мишенями. Эти данные свидетельствуют об усилении функциональной связи, которая наиболее заметна в сенсорных, моторных и мозжечковых сетях, которые, как считается, нарушены при болезни Гоше.
В таблице ниже для всех пациентов в отношении RSN 3 и 6 (RSN 4 и 8 не включены) обобщены результаты коэффициента корреляции между исходным уровнем и неделей 52 (как указано выше, пациент 2 исключен из-за отсутствия данных).
Обнаружено, что статистический анализ результатов SARA и TMT по сравнению с концентрацией GL-1 в GSF через 52 недели предусматривает хорошую терапевтическую корреляцию. Что касается результатов SARA, в которых более низкие баллы указывают на терапевтическое улучшение, у 8 из 11 пациентов наблюдалась положительная корреляция между снижением GL-1 в CSF (нг/мл) и баллом SARA (от -5 до 5). Для ТМТ, в котором также более короткое (путь В минус путь А) время указывает на терапевтическое улучшение, у 6 из 11 пациентов наблюдалась положительная корреляция между снижением GL-1 в CSF (нг/мл) и временем ТМТ (секунды).
(C) Дополнительный 52-недельный промежуточный анализ (N=9)
Неврологическую функцию дополнительно оценивали с помощью волюметрической магнитно-резонансной визуализации (vMRI). Данные vMRI собирали в ходе сеансов скрининга на исходном уровне и через 52 недели режима лечения для восьми пациентов («Группа A») и дополнительного отдельного пациента (пациент 5). Группа А соответствует одиннадцати пациентам, описанным выше, за исключением пациента 5 и двух других пациентов, у которых отсутствуют поддающиеся анализу данные vMRI. Пациента 5 исключали из-за того, что у него концентрация соединения 2 в плазме крове и CSF была ниже, чем LLOQ через 52 недели, что свидетельствует о несоблюдении режима лечения.
Получали данные vMRI и затем анализировали с использованием анатомической парцелляции с помощью FreeSurfer и цикла анализа на основании тензорной морфометрии (TBM). FreeSurfer представляет собой пакет программного обеспечения с открытым доступом для анализа и визуализации структурных и функциональных данных нейровизуализации, разработанный Лабораторией по вычислительной нейровизуализации. Анализ TBM (также известный как интегрирование по Якоби) заключается в оценке изменений объема, зафиксированных в полях деформации в результате применения метода симметричной регистрации деформируемой области между парой MR-сканов (исходный и последующий) с использованием метода нелинейного симметричного алгоритма регистрации деформаций в логарифмической области с помощью кода Demons и с использованием надежной метрики перекрестной корреляции с обеспечением обратимости преобразования (симметричный процесс). Затем поле деформации анализируется путем вычисления детерминанты его матрицы Якоби, что является мерой локального изменения объема. Интегрирование детерминанта по представляющей интерес области дает оценку скорости изменения объема данной области головного мозга с течением времени.
Общий канал принимает в качестве входных данных исходное (BL) и последующее (FU) изображения и состоит из следующих стадий.
1. Предварительная обработка и переформатирование.
2. Сегментация исходного (BL) и последующего (FU) изображений с использованием FreeSurfer.
3. Регистрация FU и BL со строгим и аффинным преобразованием и переменной разрешающей способностью в серединном пространстве.
4. Симметричная деформируемая нелинейная регистрация между FU и BL.
5. Вычисление согласно изображениям по Якоби.
6. Вычисление изменения объема по вокселям.
7. Интеграция изменения объема по областям.
8. Вывод касательно изменений представляющих интерес областей, указанных для исследования.
Таким образом, TBM представляет собой метод анализа изображений, с помощью которого идентифицируются структурные различия областей по градиентам полей нелинейной деформации, которые выравнивают изображения по общему анатомическому шаблону. TBM представляет собой хорошо известный инструмент для анализа данных vMRI головного мозга, и дополнительное обсуждение применения этого метода в данном контексте, например, представлено в John Ashburner and Karl J Friston., Human Brain Function, Second edition,. Academic Press 2004, Section 1, Chapter 6.3, pages 8 to 13, ISBN: 9780080472959; Moo K Chung., Computational Neuroanatomy, World Scientific 2012, Chapter 3, Pages 49 to 68, ISBN: 9789814472814. doi.org/10.1142/8036; and Thomson et al., Ann N Y Acad Sci. 2007 February; 1097: 183-214. doi:10.1196/annals.1379.017.
Данные vMRI собирали для ткани головного мозга в целом, и анализ позволял количественно оценить изменения объема в отдельных областях ткани головного мозга. Результаты для группы А указывают на то, что объем ткани головного мозга увеличивается во многих отдельных областях головного мозга, что приводит к увеличению объема головного мозга в целом после 52 недель режима лечения, описанного выше. Также было обнаружено, что области головного мозга с увеличенным объемом перекрываются с областями, демонстрирующими увеличенную нейронную связь, как определено с помощью fMRI, обсуждаемой выше. Результаты для пациента 5, напротив, показывают признаки атрофии головного мозга и признаки уменьшения объема головного мозга в целом.
Через 52 недели лечения группа А продемонстрировала увеличение значений среднего объема по меньшей мере в следующих областях головного мозга: области правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной доли, правого миндалевидного тела, левой области клина и левой лингвальной области, а также увеличение среднего значения объема головного мозга в целом. Эти результаты показаны в таблице ниже.
в группе A
Также было обнаружено, что в группе А режим лечения привел к общему увеличению объема ткани головного мозга в целом, тогда как у пациента 5, наоборот, произошло уменьшение объема головного мозга в целом, как видно из Фигуры 1. У пациента 5, который прошел курс лечения безуспешно, наблюдалось довольно сильное уменьшение объема головного мозга в целом, а также уменьшения объема большинства вышеописанных отдельных областей мозга. Эти результаты указывают на то, что режим лечения не только обеспечивает увеличение объема ткани головного мозга в группе А, но также обеспечивает предотвращение и/или задержку возможной потери объема ткани головного мозга в группе А в результате прогрессирования заболевания GD3, как показано у пациента 5.
Пример 6. Фармакокинетика соединения 2 у здоровых добровольцев
Были проведены два клинических исследования фазы 1 для оценки фармакокинетики, фармакодинамики, безопасности и переносимости соединения 2 у здоровых добровольцев в присутствии и в отсутствие пищи. Соединение 2 также известно как венглустат.
Исследование 1
Исследование 1 представляло собой одноцентровое испытание, состоящее из двух частей, с участием здоровых взрослых мужчин-добровольцев. Часть 1 представляла собой двойное слепое, рандомизированное, плацебо-контролируемое исследование последовательно возрастающей однократной дозы соединения 2 в отношении безопасности, переносимости и фармакокинетики. Часть 2 представляла собой открытое, однокогортное, рандомизированное, перекрестное исследование соединения 2 с 2 последовательностями, 2 периодами и 2 видами лечения в отношении PK с использованием пищи с высоким содержанием жира и без нее.
В часть 1 исследования были включены и рандомизированы 55 здоровых мужчин (плацебо, n=14; дозы по 2, 5, 15, 25, 50 и 100 мг, n=6 каждая; доза 150 мг, n=5). В части 2 принимали участие восемь здоровых мужчин.
В части 1 субъекты были рандомизированы для приема 2, 5, 15, 25, 50, 100 или 150 мг соединения 2 (в форме 1-малатной соли, т. е. представлено в форме 1-малатной соли) или соответствующего плацебо утром в первый день после по меньшей мере 10-часового воздержания от пищи. В части 2 субъекты были рандомизированы для перорального приема однократной дозы в 5 мг соединения 2 либо натощак (по меньшей мере за 10 часов до введения и через 4 часа после введения), либо через 30 минут после стандартного завтрака с высоким содержанием жира (~815 ккал). После 7-дневного периода вымывания участников перекрестным образом переводили в другие условия.
В исследовании 1, части 1, отбирали образцы крови для определения концентраций соединения 2 в плазме крове во время введения исследуемого лекарственного средства (0 часов) и через 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24, 48, 72 и 96 часов после введения дозы. Образцы мочи собирали для анализа концентраций соединения 2, начиная с 2 часов до введения исследуемого лекарственного средства и в течение 48 часов после него.
В исследовании 1, части 2, отбирали образцы крови для определения концентраций соединения 2 в плазме крови через 0, 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24 и 48 часов после введения дозы.
В части 1 было обнаружено, что после однократного перорального приема доз соединения 2 от 2 до 150 мг максимальная концентрация (Cmax) в плазме крови достигалась в среднем через 3-5,5 часа, после чего концентрации в плазме крови начинали экспоненциально снижаться со средним геометрическим значением t1/2 28,9 часа. Воздействие увеличивалось почти пропорционально дозе во всем диапазоне доз: 75-кратное увеличение дозы приводило к 97,3-, 89,2- и 85,9-кратному увеличению среднегеометрических значений Cmax, AUClast и AUCinf соответственно. В следующей таблице представлены PK результаты (AUC= площадь под кривой зависимости концентрации от времени, либо до последней поддающейся измерению концентрации, либо экстраполировано до бесконечности; t1/2=конечный период полувыведения; CL/F=кажущийся общий клиренс из плазмы крови; CV=коэффициент вариации; SD=стандартное отклонение; tmax=время до Cmax; Vss/F=кажущийся объем распределения в равновесном состоянии).
(3,00-8,00)
(4,00-8,00)
(2,00-5,00)
(4,00-8,00)
(3,00-6,00)
(2,00-4,00)
(1,00-8,00)
В части 2 было обнаружено, что введение дозы 5 мг с пищей с высоким содержанием жира не влияло на воздействие соединения 2 по сравнению с условиями состояния натощак. Медианное значение tmax составляло 6,00 часов, независимо от состояния сытости или состояния натощак. Соотношения средних геометрических значений для состояния сытости/состояния натощак составляли 0,92 и 0,91 для Cmax и AUClast соответственно. Вариабельность для конкретного субъекта (т. е. в состоянии сытости по сравнению с состоянием натощак) составляла менее половины от общей изменчивости для субъекта.
Исследование 2
Исследование 2 представляло собой одноцентровое, двойное слепое, рандомизированное, плацебо-контролируемое исследование последовательно возрастающей многократно вводимой дозы соединения 2 в отношении безопасности, переносимости, фармакокинетики и фармакодинамики на примере здоровых взрослых добровольцев мужского и женского пола.
В исследование были включены и рандомизированы 36 здоровых взрослых (19 мужчин и 17 женщин) (n=9 в каждой группе). Субъекты были рандомизированы для получения один раз в сутки соединения 2 в дозах 5, 10 или 20 мг (предоставленных как 5 мг капсулы в форме 1-малатной соли) или плацебо на протяжении 14 дней после по меньшей мере 10-часового воздержания от пищи.
Образцы крови отбирали для определения концентраций соединения 2 в плазме крови следующим образом: в день 1 через 0, 0,5, 1, 2, 3, 4, 5, 6, 8, 10, 12 и 16 часов после введения дозы; в дни 2-5, 8, 11 и 13 - в 0 ч; в день 14 через 0,5, 1, 2, 3, 4, 5, 6, 8, 10 и 12 часов после введения дозы; в дни 15-17 через 24, 48 и 72 часа соответственно после введения дозы в день 14. Образцы мочи собирали для анализа концентраций соединения 2 в день 1 (0 часов после введения дозы) и непрерывно в день 14 в период от 0 до 24 часов после введения дозы. Фармакодинамические конечные точки (концентрации GL-1, GL-3 и GM3 в плазме крови) оценивали в дни 1-5, 8, 11, 13 и 14 через 0 часов после введения дозы; и в день 15 через 24 часа после введения дозы в день 14.
Было обнаружено, что у субъектов, получавших 5, 10 или 20 мг соединения 2 один раз в сутки за 14 дней, Cmax в плазме крови наблюдалась в среднем через 2-5 часов после введения дозы в дни 1 и 14. Значения Ctrough достигали плато после дня 5. Воздействие соединения 2 увеличивалось почти пропорционально дозе в диапазоне доз 5-20 мг: такое 4-кратное увеличение дозы приводило к 3,76- и 3,69-кратному увеличению средних геометрических значений Cmax и AUC0-24 соответственно в день 14. PK результаты исследования 2 обобщены в следующей таблице.
После 14 однократных суточных доз соединения 2 его 24-часовая неизмененная фракция, выводимая с мочой (среднее значение fe0-24), находилась в диапазоне от 26,3% до 33,1% без какой-либо очевидной зависимости от дозы. Среднее значение CLR(0-24) изменялось в диапазоне от 1,49 л/ч до 2,07 л/ч, что примерно в 3,18-3,86 раза ниже, чем наблюдаемое значение CL/F в плазме крови.
Уровни GL-1, GL-3 и GM3 в плазме крови участников, получавших плацебо, на протяжении всего времени оставались на исходном уровне, тогда как уровни GL-1 и GM3 в плазме крови участников групп с получением 3 доз соединения 2 снижались по сравнению с исходным уровнем в зависимости от времени и дозы, как показано в следующей таблице (точечные оценки в виде соотношений видов лечения глюкозилцерамидом (GL-1), глоботриаозилцерамидом (GL-3) и ганглиозидом GM3 (GM3) в день 15 в исследовании с многократным введением возрастающей дозы).
Максимально устойчивые эффекты в отношении GL-1 наблюдались в день 11 в группах приема 5 и 10 мг и в день 8 в группе приема 20 мг. Средние расчетные значения снижения GL-1 по сравнению с исходным уровнем в день 15 составляли 41,9%, 69,6% и 74,6% в соответствующих группах приема 5, 10 и 20 мг. Значения GL-1 были ниже нижнего предела количественного определения (LLOQ) на исходном уровне у участника 1, получавшего 5 мг соединения 2, и в день 15 у субъектов 3, 5 и 9 в группах приема 5, 10 и 20 мг соответственно.
Максимальные устойчивые снижения уровня GM3 наблюдалось во всех группах, получавших дозу соединения 2, начиная со дня 13. Средние уровни GM3 в плазме крови в день 15 составляли 42,7%, 49,4% и 57,8% от исходного уровня для групп, получавших дозу в 5, 10 и 20 мг соответственно. Уровень GM3 был ниже LLOQ в день 15 у субъектов 1 и 2 в группах приема доз в 10 и 20 мг соответственно.
Уровень GL-3 в плазме крови также снижался с течением времени во всех группах, получавших дозу соединения 2, но переменные и низкие значения GL-3 на исходном уровне по отношению к LLOQ ограничивали средние расчетные значения снижения GL-3. В группах плацебо с дозами 5, 10 и 20 мг значения GL-3 были ниже LLOQ у субъектов 1, 3, 1 и 6 соответственно на исходном уровне и у субъектов 4, 9, 7 и 9 соответственно в день 15.
Средние предполагаемые снижения уровня GL-1 в плазме крови относительно исходного уровня (CI 90%), связанные с Ctrough соединения 2 в группах введения доз 5, 10 и 20 мг (19,0, 47,5 и 69,9 нг/мл соответственно), составляли 67,0% (54,4-79,7%), 74,4% (63,7-85,2%) и 76,3% (64,8-87,8%) соответственно.
Выводы
В данных исследованиях воздействие соединения 2 у здоровых субъектов (Cmax и AUC) было близко пропорциональным дозе при введении в виде однократных доз в диапазоне от 2 до 150 мг или в виде многократно вводимых ежесуточных доз в диапазоне от 5 до 20 мг в течение 14 дней. По сравнению с приемом в состоянии натощак, прием пищи с высоким содержанием жира не влиял на воздействие у субъектов, получивших однократную дозу в 5 мг. При многократном ежесуточном введении доз от 5 до 20 мг равновесное состояние достигалось в течение 5 дней; при этом ни возраст, ни пол не влияли на накопление. С точки зрения фармакодинамики многократно вводимые ежесуточные дозы соединения 2 обеспечивали снижение концентраций GL-1 и GM3 в плазме крови в зависимости от времени и дозы, что согласуется с опосредованным соединением 2 ингибированием GCS, хотя исходные уровни GL-3 были слишком низкими, чтобы быть полезными как фармакодинамический биомаркер. Зависимое от дозы снижение GL-1 подтверждает предполагаемый механизм действия соединения 2: ингибирование образования GL-1 из церамида с помощью GCS.
Во всех исследованиях профиль безопасности оценивали путем мониторинга нежелательных явлений, возникающих во время лечения (TEAE), в течение 10 дней после приема последней дозы исследуемого лекарственного препарата, включая серьезные нежелательные явления [SAE]), мониторинга ECG, лабораторных показателей и физикального обследования.
Ни в одном из исследований не было случаев наступления смерти, SAE, тяжелых TEAE или TEAE, приводящих к прекращению участия в исследовании.
Ни в одном из исследований не сообщалось о клинически значимых гематологических или биохимических отклонениях. Ни в одном из исследований жизненно важные показатели не указывали на существенные изменения по сравнению с исходным уровнем. Параметры ECG не указывали на значимые изменения в исследованиях с введением однократной возрастающей дозы и влиянием пищи; в исследовании с многократным введением возрастающей дозы никакие параметры ECG не изменялись статистически значимо относительно среднего значения на исходном уровне по сравнению с плацебо у участников, принимающих соединение 2 в любой дозе. Следует понимать, что хотя настоящее изобретение было описано в сочетании с вышеприведенными вариантами осуществления, приведенные выше описание и примеры предназначены для иллюстрации, а не для ограничения объема настоящего изобретения. Другие аспекты, преимущества и модификации в объеме настоящего изобретения будут очевидны специалистам в области, к которой относится настоящее изобретение.
Кроме того, если признаки или аспекты настоящего изобретения описаны с точки зрения групп Маркуша, то специалистам в данной области будет понятно, что настоящее изобретение тем самым также описывается с точки зрения любого отдельного члена или подгруппы членов группы Маркуша.
Все публикации, патентные заявки, патенты и другие литературные источники, упомянутые в данном документе, специально включены посредством ссылки во всей своей полноте в той же степени, как если бы каждый из них был отдельно включен посредством ссылки. В случае противоречий настоящее описание, включая определения, будет иметь преимущественную силу.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ СИМПТОМОВ И НАРУШЕНИЙ, АССОЦИИРОВАННЫХ С ЛИЗОСОМНЫМИ БОЛЕЗНЯМИ НАКОПЛЕНИЯ | 2020 |
|
RU2824599C2 |
| СПОСОБЫ ЛЕЧЕНИЯ СИМПТОМОВ И НАРУШЕНИЙ, АССОЦИИРОВАННЫХ С ЛИЗОСОМНЫМИ БОЛЕЗНЯМИ НАКОПЛЕНИЯ | 2020 |
|
RU2829786C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ | 2015 |
|
RU2711442C2 |
| ИНГИБИТОРЫ ГЛЮКОЗИЛЦЕРАМИД-СИНТАЗЫ | 2012 |
|
RU2645675C2 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2709786C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2014 |
|
RU2673819C2 |
| Амидные соединения, содержащие их фармацевтические композиции и способы их применения | 2017 |
|
RU2759913C2 |
| ПРОЛЕКАРСТВЕННЫЕ МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО ПУТИ СТРЕССА | 2019 |
|
RU2824500C2 |
| ПРОИЗВОДНЫЕ СОБЕТИРОМА | 2017 |
|
RU2776372C2 |
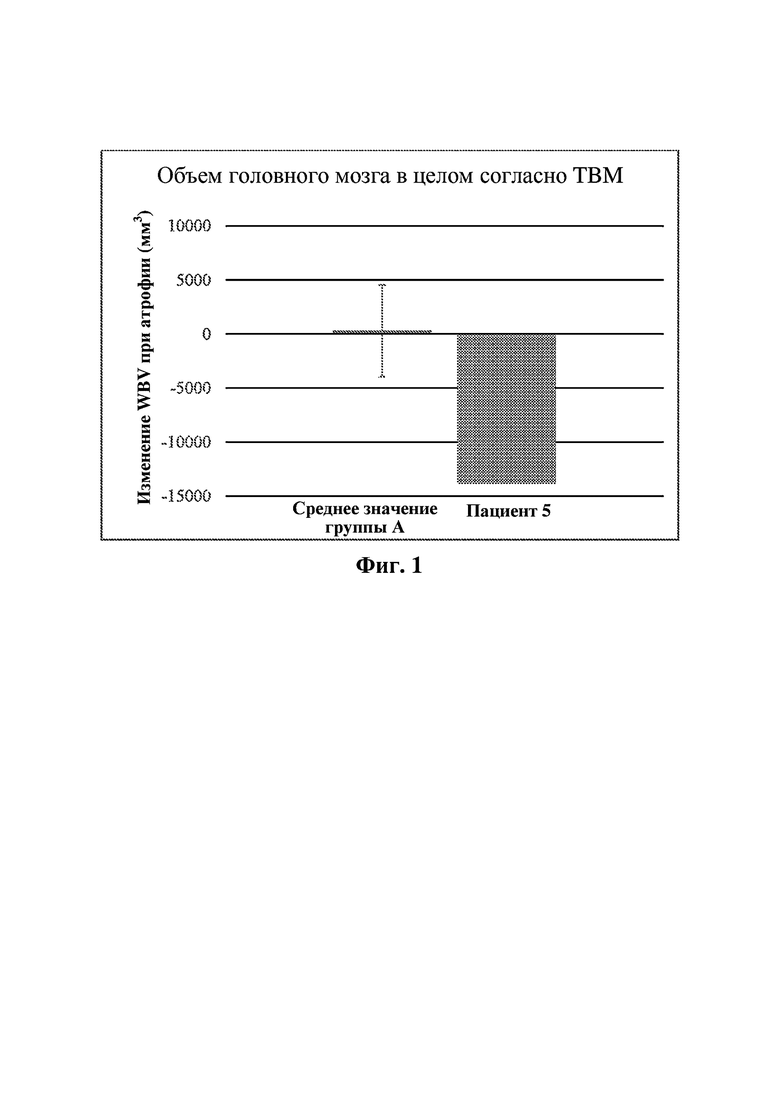
Изобретение относится к химии, фармацевтике и медицине, а именно к способу увеличения объема ткани головного мозга или предупреждения или задержки потери объема ткани головного мозга у субъекта. Согласно предложенному способу субъекту вводят эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, где соединение формулы (I) имеет структуру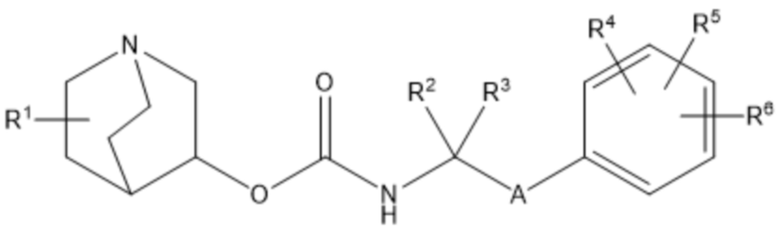 , в которой R1 – водород; R2 и R3 – независимо выбраны из C1-3-алкила; R4, R5 и R6 – независимо выбраны из водорода и галогена; А – тиазолил. Изобретение обеспечивает реализацию назначения. 16 з.п. ф-лы, 1 ил., 19 табл., 6 пр.
, в которой R1 – водород; R2 и R3 – независимо выбраны из C1-3-алкила; R4, R5 и R6 – независимо выбраны из водорода и галогена; А – тиазолил. Изобретение обеспечивает реализацию назначения. 16 з.п. ф-лы, 1 ил., 19 табл., 6 пр.
1. Способ увеличения объема ткани головного мозга или предупреждения или задержки потери объема ткани головного мозга у субъекта, при этом указанный способ включает введение субъекту эффективного количества соединения формулы (I)
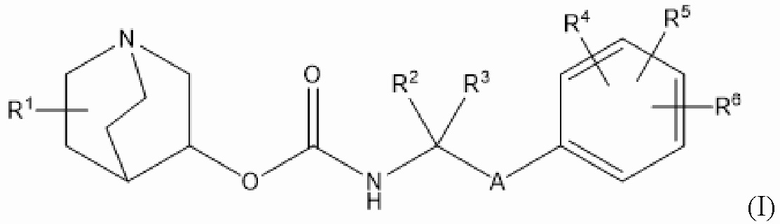
или его фармацевтически приемлемой соли,
где R1 представляет собой водород;
R2 и R3 независимо выбраны из C1-3-алкила;
каждый из R4, R5 и R6 независимо выбран из водорода и галогена; и
А представляет собой тиазолил.
2. Способ по п. 1, где указанное соединение выбрано из хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамата; (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата и (S)-хинуклидин-3-ил-(2-(4'-(2-метоксиэтокси)-[1,1'-бифенил]-4-ил)пропан-2-ил)карбамата, и их фармацевтически приемлемых солей.
3. Способ по п. 1, где указанное соединение представляет собой хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)пропан-2-ил)карбамат.
4. Способ по п. 1, где указанное соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат.
5. Способ по п. 1, где указанное соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамат в форме малатной соли.
6. Способ по любому из пп. 1-5, где способ приводит к увеличению объема ткани головного мозга или предупреждению или задержке потери объема ткани головного мозга в одной или нескольких областях головного мозга, выбранных из правого прилежащего ядра, левой части скорлупы, левой части энторинальной коры, правой части скорлупы, правой постцентральной доли, левой околошпорной области, правого миндалевидного тела, левой области клина и левой лингвальной области.
7. Способ по любому из пп. 1-6, где способ приводит к увеличению объема головного мозга в одной или нескольких областях головного мозга, связанных с исполнительной функцией.
8. Способ по любому из пп. 1-7, где увеличение объема головного мозга в одной или нескольких областях мозга сопровождается улучшением нейронных связей в одной или нескольких областях головного мозга, например, как показано с применением функциональной магнитно-резонансной визуализации (fMRI).
9. Способ по любому из пп. 1-5, где способ приводит к увеличению объема ткани головного мозга в целом.
10. Способ по любому из пп. 1-9, где у субъекта имеется болезнь Гоше типа 3.
11. Способ по любому из пп. 1-10, где указанное соединение или его фармацевтически приемлемую соль вводят путем системного введения, например, посредством пути введения, отличного от парентерального.
12. Способ по п. 11, где указанное соединение или его фармацевтически приемлемую соль вводят перорально.
13. Способ по любому из пп. 1-12, где субъект одновременно подвергается лечению посредством ферментозаместительной терапии (ERT), например, с применением глюкоцереброзидазы.
14. Способ по любому из пп. 1-12, где глюкоцереброзидаза выбрана из имиглюцеразы, велаглюцеразы или талиглюцеразы.
15. Способ по любому из пп. 1-14, где субъекту вводят суточную дозу от 1 мг до 50 мг соединения, например от 5 до 50 мг, или от 10 до 40 мг, или от 10 до 30 мг, или от 10 до 20 мг, или от 20 до 30 мг, или от 30 до 40 мг, или от 40 до 50 мг, или от 5 до 25 мг, или от 20 до 50 мг, или от 5 до 15 мг, или от 15 до 30 мг, или 15 мг.
16. Способ по любому из пп. 1-15, где субъекту вводят однократную суточную дозу (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата в форме малатной соли, составляющую 15 мг в пересчете на количество свободного основания.
17. Способ по любому из пп. 1-16, где субъект является взрослым или пациентом детского возраста 12 лет или больше с болезнью Гоше типа 3, состояние которого стабилизировано посредством ферментозаместительной терапии (ERT) с применением имиглюцеразы для системных состояний, и где субъекту вводят однократную суточную дозу (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)тиазол-4-ил)пропан-2-ил)карбамата в форме малатной соли, составляющую 15 мг в пересчете на количество свободного основания.
| ИНГИБИТОРЫ ГЛЮКОЗИЛЦЕРАМИД-СИНТАЗЫ | 2012 |
|
RU2645675C2 |
| EA 201791993 A1, 29.12.2017 | |||
| John Marshall et al | |||
| CNS-accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease / Molecular Therapy, 2016, V | |||
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| УСТРОЙСТВО ДЛЯ ПОДДЕРЖАНИЯ СИНХРОНИЗМА ВРАЩЕНИЯ АСИНХРОННЫХ ДВИГАТЕЛЕЙ | 1923 |
|
SU1019A1 |
| Sirio Cocozza et al | |||
| Neuroimaging in Fabry disease: current knowledge and future directions / | |||

