Настоящее изобретение относится к кристаллическим формам карбетоцина, способу их получения и их фармацевтическим композициям.
Уровень техники
Карбетоцин (также известный как 1-дезамино-1-монокарба-2-(O-метилтирозин)окситоцин или 1-бутановая кислота-2-(O-метил-L-тирозин)-1-карбаокситоцин) является синтетическим олигопептидным аналогом окситоцина длительного действия с агонистическим действием. Карбетоцин включает следующие замещения по отношению к окситоцину: а) аминогруппы цистеина (положение 1) на атом водорода, б) его дисульфидной связи на тиоэфирную связь и в) гидроксильной группы тирозина (положение 2) на метилоксильную группу. Карбетоцин (PABAL®, DURATOCIN®) в настоящее время одобрен для предотвращения атонии матки после рождения ребенка с помощью кесарева сечения под эпидуральной или спинальной анестезией. Дозы, используемые по этим медицинским показаниям, являются относительно небольшими, например, порядка 100 микрограмм однократно.
В последнее время повысилась потребность в агонистах окситоциновых рецепторов, в особенности, карбетоцине. Например, окситоциновые рецепторы в последнее время были показаны при лечении синдрома Прадера-Вилли (Prader-Willi) (см. WO 2016/044131). Синдром Прадера-Вилли представляет собой генетическое заболевание, отличающееся чрезмерным поглощением пищи, поведением, направленным на поиск пищи, быстрой прибавкой в весе, навязчивым поведением и агрессией у маленьких детей. Как описано в WO 2016/044131, пациенты, которых лечили карбетоцином, показывают статистически значимое улучшение по сравнению с пациентами, которых лечили плацебо, спустя 15 суток при количественных оценках чрезмерного поглощения пищи, обсессивно-компульсивного расстройства, поведения, направленного на поиск пищи, и общего клинического впечатления. Для этого показания к применению необходимо получать относительно большое количество пептида, потому что используемые дозы значительно более высокие чем те, которые используют при лечении атонии матки, например, порядка десятков миллиграмм в сутки, и это лечение более долгосрочное. Было бы желательным для таких показаний получать относительно большое количество карбетоцина высокой чистоты.
Синтез пептидов можно выполнять, используя методики твердофазного синтеза, которые хорошо известны в уровне техники. Жидкофазный синтез является альтернативным способом, который может подходить для небольшого количества пептида. Эта стадия получения пептида известна как «подготовительный процесс» и она приводит к образованию сырого пептидного продукта.
Вслед за синтезом сырого пептида обычно необходимо отделить представляющий интерес пептид от различных пептидных и не пептидных примесей. Эта стадия известна как стадия очистки.
В уровне техники известно множество способов очистки пептидов. Однако способы очистки пептидов обычно включают по меньшей мере одну хроматографическую стадию, например, гель-проникающую хроматографию, хроматографию с гидрофобным взаимодействием, ионообменную хроматографию, электрофорез со свободно движущейся границей, аффинную хроматографию, высокоэффективную жидкостную хроматографию (ВЭЖХ) и т.п. Наиболее применяемой формой ВЭЖХ является обращенно-фазовая ВЭЖХ (также известная как ОФ-ВЭЖХ), в которой пептиды элюируют повышающимся количеством органического растворителя, такого как ацетонитрил, согласно их гидрофобности.
После стадии очистки пептид обычно необходимо отделить от летучих растворителей. Эта стадия известна как стадия выделения. Известные способы отделения пептида от растворителей включают ультрафильтрацию и лиофилизацию.
Лиофилизация (также известная как сублимационная сушка) включает стадию быстрого замораживания содержащего пептид раствора, обычно путем погружения удерживающего раствор контейнера в жидкий азот. Контейнер затем помещают в вакуумную камеру, которая содержит охлаждающий змеевик. Летучие растворители сублимируются в вакууме. В способе сублимации очищенный образец гарантированно поддерживается холодным.
Лиофилизация является технологией, наиболее часто используемой в уровне техники для выделения пептидов из раствора. В принципе, причина состоит в том, что эта технология хорошо известна, воспроизводима и легко выполняется. Дополнительно, стабильность пептидов обычно повышается при низких температурах.
Способы очистки и выделения карбетоцина и родственных пептидов известны в уровне техники.
В CN 104592362 описана стадия очистки жидкостной хроматографией карбетоцина, за которой следует лиофилизация. В большинстве случаев стадия жидкостной хроматографии является ВЭЖХ.
В WO 2015/185584 описана очистка и лиофилизация агонистов окситоцина, отличных от карбетоцина.
В CN 102977192 описан способ очистки карбетоцина путем объединения жидкостной хроматографии и ионообменной хроматографии. После очистки продукт проходит через стадию обессоливания и лиофилизации.
В CN 104744567 описан способ очистки карбетоцина с помощью ионообменной хроматографии, за которой следует лиофилизация.
В CN 101531705 описан способ очистки карбетоцина с использованием обращенно-фазовой ВЭЖХ, за которой следует превращение продукта в ацетатную соль с использованием ионообменного способа. После превращения в соль продукт затем подвергают лиофилизации.
В WO 2009/122285 описан способ очистки аналогов окситоцина, включающий стадию ВЭЖХ, за которой следует стадия лиофилизации. В статье Rudko A D et al. "Crystalline Salts of Oxytocin: X-ray crystallographic data" J. Crystal Growth, vol. 10, no. 3, 1971, pages 260-262 описано определение характеристик кристаллизованных солей окситоцина. В статье Bryn S et al. "Pharmaceutical Solids: A Strategic Approach to Regulatory Considerations" Pharmaceutical Research, vol. 12, no. 7, 1995, pages 945-954 описано определение характеристик фармацевтических твердых веществ.
Из приведенных выше ссылок можно видеть, что в уровне техники существует устойчивая тенденция, направленная на использование лиофилизации в качестве стадии выделения при синтезе карбетоцина и других агонистов окситоциновых рецепторов.
Однако существует несколько проблем, связанных с лиофилизацией, например, необходимо потратить большое количество времени для обработки пептида, и затраты на хладагент и оборудование очень высоки.
Эти проблемы могут быть допустимыми при получении небольшого количества пептида. Однако при массовом получении пептида лиофилизация становится «узким местом» в способе получения. Кроме того, доля от всех затрат на производство, расходуемая на лиофилизацию, возрастает с массой получаемого пептида.
Соответственно, также в уровне техники существует потребность в улучшенном способе выделения карбетоцина с удалением «узкого места» лиофилизации, так чтобы можно было получить большее количество карбетоцина достаточной чистоты, удовлетворяя потребность показаний к применению, таких как лечение синдрома Прадера-Вилли.
Краткое описание изобретения
В одном аспекте настоящее изобретение относится к карбетоцину в кристаллической форме.
В дополнительном аспекте настоящее изобретение относится к способу получения карбетоцина в кристаллической форме, включающему стадию кристаллизации карбетоцина.
В дополнительном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей карбетоцин по настоящему изобретению или карбетоцин, полученный по настоящему изобретению.
Чертежи, относящиеся к настоящему изобретению
Ниже описаны чертежи, относящиеся к настоящему изобретению:
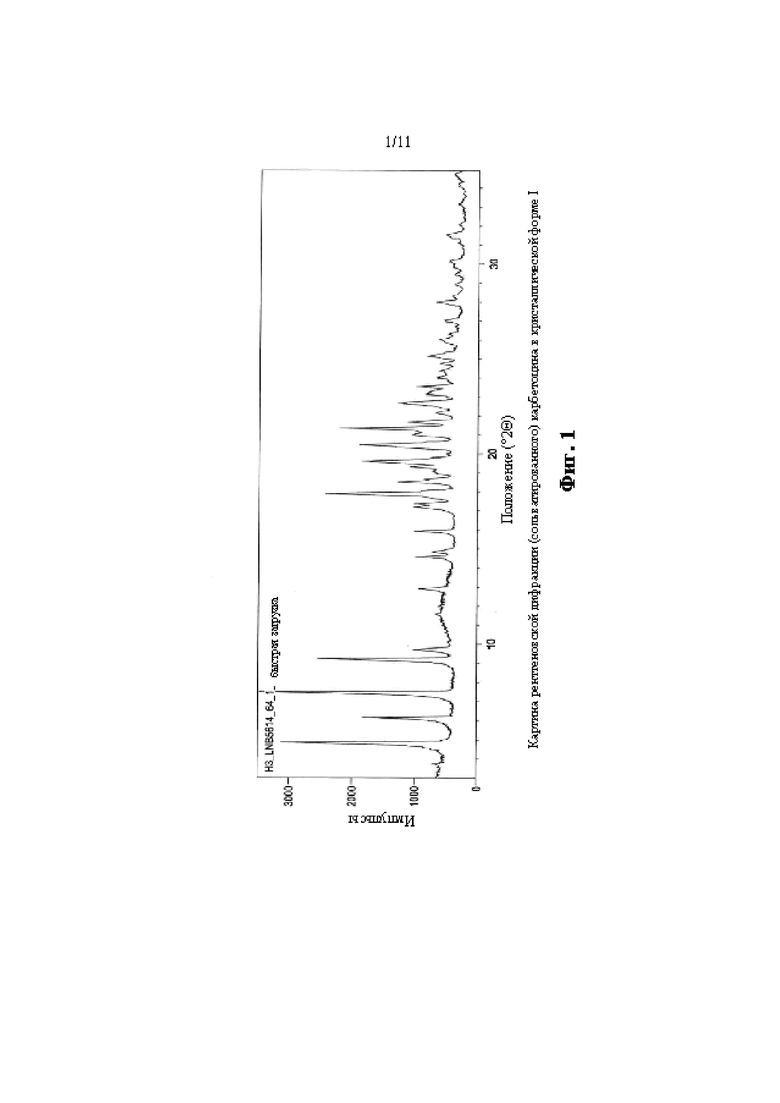
на фиг. 1 показана картина рентгеновской дифракции (Cu) сольватированной кристаллической формы I карбетоцина,
на фиг. 2 показана картина рентгеновской дифракции (Cu) десольватированной кристаллической формы II карбетоцина,
на фиг. 3 показаны хроматограммы ВЭЖХ выделенных твердых веществ из примера 1 (фиг. 3а) и выделенных твердых веществ из примера 4 (фиг. 3b),
на фиг. 4 показаны данные ТГ/ДТА (термогравиметрии/дифференциального термического анализа), относящиеся к сольватированной кристаллической форме I карбетоцина из примера 1 (фиг. 4а) и десольватированной кристаллической форме II карбетоцина из примера 4 (фиг. 4b),
на фиг. 5 показана картина рентгеновской дифракции (Cu-Kα1) кристаллического карбетоцина, полученного в примере 5,
на фиг. 6 показана хроматограмма ВЭЖХ выделенных твердых веществ из примера 5,
на фиг. 7 показаны данные дифференциальной сканирующей калориметрии (ДСК), относящиеся к выделенному кристаллическому карбетоцину из примера 5,
на фиг. 8 показаны данные гравиметрической сорбции пара (ГСП) выделенного кристаллического карбетоцина из примера 5: изменение в графике массы (фиг. 8а) и в графике изотермы (фиг. 8b).
Подробное описание изобретения
Ранее не было известно, что карбетоцин способен образовывать кристаллы. Авторы настоящей заявки неожиданно обнаружили, что можно образовывать три кристаллические формы карбетоцина, как описано в данном документе, две из которых можно обозначить как форма I и форма II. Форма I является сольватированной (например, гидратированной), в то время как форма II является десольватированной. Форма II обладает высокой стабильностью (см. фиг. 4В) и имеет приемлемо низкое содержание этиленгликоля (см. пример 3) и ее можно использовать, например, в качестве лекарственного средства. Под приемлемо низким содержанием этиленгликоля понимают содержание этиленгликоля ниже предела согласно Международной конференции по гармонизации, составляющего 620 ч/млн, определенное с помощью газовой хроматографии. Форму I можно использовать в качестве синтетического промежуточного продукта при получении формы II. Третья кристаллическая форма также описана в данном документе (см. пример 5).
По настоящему изобретению в первом аспекте предоставляют карбетоцин в кристаллической форме. Во втором аспекте предоставляют карбетоцин в сольватированной (например, гидратированной) кристаллической форме. В третьем аспекте предоставляют карбетоцин в десольватированной кристаллической форме.
Под сольватированной понимают, что кристаллическая структура содержит либо упорядоченные, либо неупорядоченные молекулы растворителя. Под неупорядоченными понимают, что положения молекул растворителя или положения атомов в них могут изменяться в пределах кристаллической структуры. Молекулы растворителя могут находиться в жидком или газообразном состоянии при комнатной температуре и атмосферном давлении. Молекулы растворителя могут состоять из молекул только одного типа. Альтернативно, молекулы растворителя могут состоять из двух или более различных типов молекул (одним из которых может в некоторых случаях быть вода). На молекулу карбетоцина может приходиться по меньшей мере 0,1 или более молекул растворителя, например, по меньшей мере 0,2 молекулы растворителя на молекулу карбетоцина, например, по меньшей мере 0,5 молекулы растворителя на молекулу карбетоцина, например, по меньшей мере 1 молекула растворителя на молекулу карбетоцина, например, по меньшей мере 2 молекулы растворителя на молекулу карбетоцина, например, по меньшей мере 5 молекул растворителя на молекулу карбетоцина. Соответственно, карбетоцин в сольватированной кристаллической форме может находиться в форме моно-, ди-, три-, тетра-, пента или гекса- гидрата сольватированной кристаллической формы. Предпочтительно, когда карбетоцин находится в сольватированной кристаллической форме, сольватированная кристаллическая форма является моногидратом или пентагидратом. Соответственно, в одном аспекте карбетоцин находится в кристаллической форме моногидрата или пентагидрата. Такой карбетоцин может содержать либо упорядоченные, либо неупорядоченные молекулы растворителя. Полагают, что число молекул растворителя не влияет на то, являются ли они упорядоченными или неупорядоченными.
Под десольватированной понимают, что кристаллическая структура содержит мало упорядоченных или неупорядоченных молекул растворителя или не содержит их вообще. Может быть не больше 2 молекул растворителя на молекулу карбетоцина, например, не больше 1 молекулы растворителя на молекулу карбетоцина, например, не больше 0,5 молекулы растворителя на молекулу карбетоцина, например, не больше 0,2 молекулы растворителя на молекулу карбетоцина, например, не больше 0,1 молекулы растворителя на молекулу карбетоцина, например, не больше 0,05 молекулы растворителя на молекулу карбетоцина, например, не больше 0,02 молекулы растворителя на молекулу карбетоцина, например, не больше 0,01 молекулы растворителя на молекулу карбетоцина.
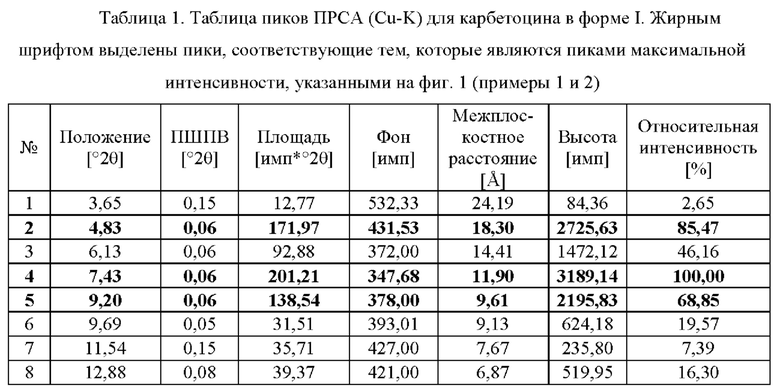
Для того, чтобы определить кристаллическую форму, можно выполнить порошковый рентгеноструктурный анализ (ПРСА). В настоящем изобретении ПРСА выполняли с использованием Cu K излучения (α1 λ=1,54060  , α2=1,54443 , β=1,39225 , отношение α1 : α2=0,5) на PANalytical X'pert pro, как дополнительно подробно описано в примере 1. Карбетоцин в кристаллической форме и/или карбетоцин в сольватированной кристаллической форме может быть охарактеризован пиками порошковой рентгеновской дифракции при примерно 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ (Cu), и/или может быть охарактеризован картиной порошковой рентгеновской дифракции (Cu), по существу такой, как показана на фиг.1, и/или может быть охарактеризован наличием 5 или более, 6 или более, 7 или более, 8 или более, 9 или более, 10 или более, 11 или более, 12 или более, 13 или более, 14 или более, 15 или более или по существу всех пиков порошковой рентгеновской дифракции (Cu), показанных в таблице 1. Соответственно, в одном аспекте карбетоцин в кристаллической форме характеризуется пиками порошковой рентгеновской дифракции при примерно 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ, выполненной с использованием Cu K излучения (α1 λ=1,54060
, α2=1,54443 , β=1,39225 , отношение α1 : α2=0,5) на PANalytical X'pert pro, как дополнительно подробно описано в примере 1. Карбетоцин в кристаллической форме и/или карбетоцин в сольватированной кристаллической форме может быть охарактеризован пиками порошковой рентгеновской дифракции при примерно 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ (Cu), и/или может быть охарактеризован картиной порошковой рентгеновской дифракции (Cu), по существу такой, как показана на фиг.1, и/или может быть охарактеризован наличием 5 или более, 6 или более, 7 или более, 8 или более, 9 или более, 10 или более, 11 или более, 12 или более, 13 или более, 14 или более, 15 или более или по существу всех пиков порошковой рентгеновской дифракции (Cu), показанных в таблице 1. Соответственно, в одном аспекте карбетоцин в кристаллической форме характеризуется пиками порошковой рентгеновской дифракции при примерно 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ, выполненной с использованием Cu K излучения (α1 λ=1,54060 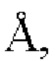 α2=1,54443 , β=1,39225
α2=1,54443 , β=1,39225 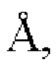 отношение α1 : α2=0,5).
отношение α1 : α2=0,5).
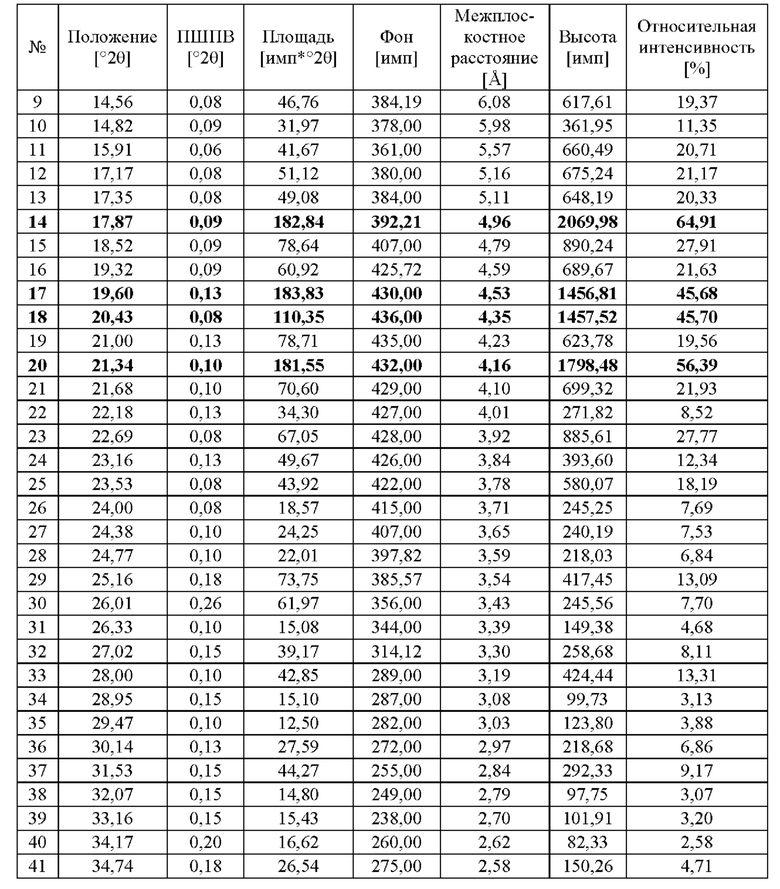
Карбетоцин в кристаллической форме и/или карбетоцин в десольватированной кристаллической форме может быть охарактеризован пиками порошковой рентгеновской дифракции при примерно 4,11, 4,39, 5,60, 7,45, 17,75, 19,16 и 19,45 градусах 2θ (Cu), и/или может быть охарактеризован картиной порошковой рентгеновской дифракции (Cu), по существу такой, как показана на фиг. 2, и/или может быть охарактеризован наличием 5 или более, 6 или более, 7 или более, 8 или более, 9 или более, 10 или более, 11 или более, 12 или более, 13 или более, 14 или более, 15 или более или по существу всех пиков порошковой рентгеновской дифракции (Cu), показанных в таблице 2. Соответственно, в одном аспекте карбетоцин в кристаллической форме характеризуется пиками порошковой рентгеновской дифракции при примерно 4,11, 4,39, 5,60, 7,45, 17,75, 19,16 и 19,454 градусах 2θ, выполненной с использованием Cu K излучения (α1 λ=1,54060  α2=1,54443
α2=1,54443 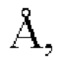 β=1,39225
β=1,39225 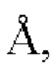 отношение α1 : α2=0,5).
отношение α1 : α2=0,5).
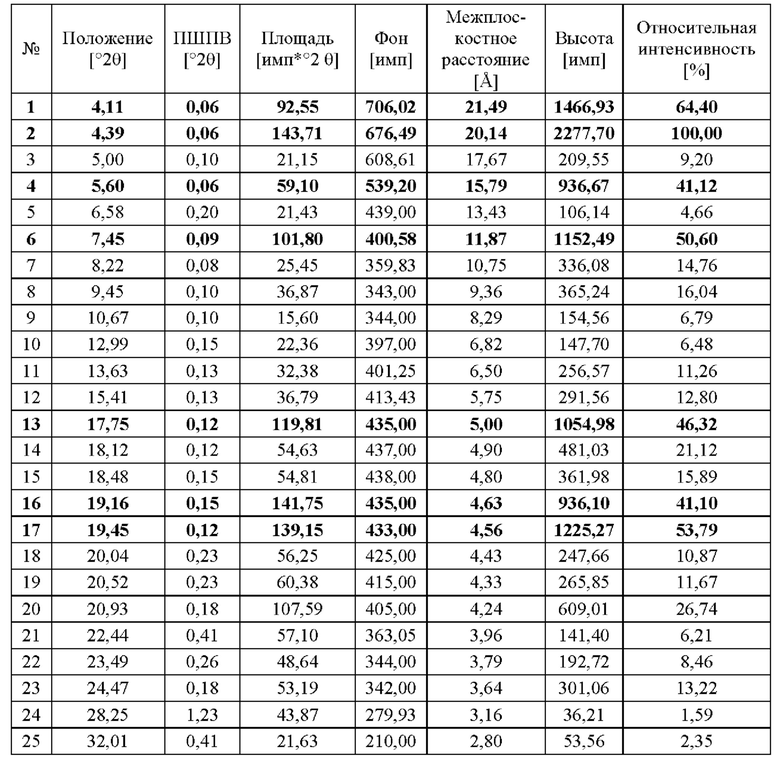
Карбетоцин в кристаллической форме может быть охарактеризован пиками порошковой рентгеновской дифракции при примерно 4,34, 6,43, 8,66, 17,37, 19,03 и 19,39 градусах 2θ (Cu-Kα1), и/или может быть охарактеризован картиной порошковой рентгеновской дифракции (Cu-Kα1), по существу такой, как показана на фиг. 5, и/или может быть охарактеризован наличием 5 или более, 6 или более, 7 или более, 8 или более, 9 или более, 10 или более, 11 или более, 12 или более, 13 или более, 14 или более, 15 или более или по существу всех пиков порошковой рентгеновской дифракции (Cu-Kα1), показанных в таблице 3. Соответственно, в одном аспекте карбетоцин в кристаллической форме характеризуется пиками порошковой рентгеновской дифракции при примерно 4,34, 6,43, 8,66, 17,37, 19,03 и 19,39 градусах 2θ, выполненной с использованием Cu Kα1 излучения (α1 λ=1,54060 ).

(ПШПВ = полная ширина линии на половине высоты)



В таблице 3 показаны данные, полученные из примера 5. Отметим, что источник излучения для значений, представленных в таблице 3, представляет собой источник Cu-Kα1, в то время как источник излучения для значений, представленных в таблицах 1 и 2, представляет собой источник Cu-K. В приведенной выше таблице пиков ПРСА, таблице 3 (фиг. 5, пример 5), тем не менее представлен карбетоцин в другой кристаллической форме или полиморфной модификации по сравнению с примерами 1 и 2 (фиг. 1, таблица 1) и примерами 3 и 4 (фиг. 2, таблица 2).
По настоящему изобретению в дополнительном аспекте предоставляют способ получения карбетоцина в кристаллической форме, включающий стадию кристаллизации карбетоцина.
Под кристаллизацией понимают способ образования кристаллической формы карбетоцина из карбетоцина, растворенного в растворителе. Под кристаллической формой понимают твердое вещество с регулярно повторяющимся внутренним расположением атомов и внешними плоскими гранями. Кристаллические формы можно отличить от аморфных форм на основе порошкового рентгеноструктурного анализа. Кристаллические формы характеризуются описанными в данном документе пиками порошковой рентгеновской дифракции. В аморфных твердых формах картина ПРСА имеет по существу непрерывный вид, то есть без явно выраженных пиков.
Карбетоцин в кристаллической форме можно кристаллизовать из смеси, содержащей карбетоцин и одну или более жидкостей, причем одна или более жидкостей возможно включают одну или более жидкостей из группы, состоящей из воды, водного ацетатного буфера, этиленгликоля, ацетонитрила, этанола, метанола, пропанола, изопропанола, 1,2-пропандиола и диметилформамида, например, смесь этиленгликоля и ацетонитрила, например, смесь этанола, этиленгликоля и ацетонитрила, например, смесь пропанола, этиленгликоля и ацетона, например, смесь изопропанола, этиленгликоля и ацетона, например, смесь диметилформамида, этиленгликоля и ацетонитрила, например, смесь диметилформамида и ацетонитрила, например, смесь диметилформамида и ацетона, например, смесь этанола и ацетонитрила, например, смесь метанола и ацетонитрила, например, смесь 1,2-пропандиола и ацетонитрила, например, смесь 1,2-пропандиола и ацетона. Когда одна или более жидкостей содержит две или более жидкостей, одна из двух или более жидкостей может быть антирастворителем (определенным ниже). Одна или более жидкостей могут содержать этиленгликоль и антирастворитель в отношении от 15:85 до 25:75, например, в отношении от 17,5:82,5 до 22,5:77,5, например, в отношении примерно 20:80, и где добавление антирастворителя изменяет отношение этиленгликоля к ацетонитрилу на отношение от 1:99 до 30:70, например, отношение от 2:98 до 25:75, например, отношение от 3:97 до 20:80, например, отношение от 5:95 до 20:80, например, отношение от 5:95 до 15:85, например, отношение от 7,5:92,5 до 12,5:87,5, например, отношение примерно 10:90, например, отношение от 5:95 до 10:90, например, отношение от 5:95 до 7,5:92,5, например, отношение примерно 6,7:93,3.
Одна или более жидкостей могут включать смесь этиленгликоля и ацетонитрила.
Одна или более жидкостей могут содержать этиленгликоль и ацетонитрил в отношении от 1:99 до 50:50, например, отношении от 2:98 до 40:60, например, отношении от 3:97 до 35:65, например, отношении от 5:95 до 35:65, например, отношении от 8:92 до 30:70, например, отношении от 10:90 до 30:70, например, отношении от 15:85 до 25:75, например, отношении примерно 20:80.
Одна или более жидкостей могут содержать воду. Одна или более жидкостей могут содержать водный ацетатный буфер. В частном аспекте карбетоцин в кристаллической форме кристаллизуют из смеси, содержащей карбетоцин и одну или более жидкостей, содержащих воду и/или водный ацетатный буфер.
В одном из воплощений одна или более жидкостей могут быть водой. Если одна или более жидкостей являются водой, вода может иметь рН примерно от 2 до 6, предпочтительно рН примерно от 3 до 4, более предпочтительно рН примерно 3,5.
В одном из воплощений одна или более жидкостей могут быть водным ацетатным буфером. Водный ацетатный буфер можно образовать из водной смеси уксусной кислоты и ацетатной соли. Ацетатная соль может быть любой солью ацетата с подходящим противоионом. Подходящий противоион может быть, например, ионом щелочного металла, ионом щелочноземельного металла или органическим катионом. Противоион может быть литием, натрием, калием, магнием, кальцием или аммонием. Предпочтительно противоион может быть натрием или калием, наиболее предпочтительно противоион может быть натрием. Предпочтительно водный ацетатный буфер имеет рН примерно от 4 до 7, предпочтительно рН примерно от 5 до 6, наиболее предпочтительно рН составляет примерно 5,5. Предпочтительно водный ацетатный буфер имеет концентрацию от 20 до 30 мМ, наиболее предпочтительно концентрация составляет примерно 25 мМ.
Карбетоцин в кристаллической форме можно получить путем охлаждения смеси, содержащей карбетоцин и одну или более жидкостей, например, от 40°С до 5°С, или путем циклического изменения температуры смеси, содержащей карбетоцин и одну или более жидкостей, например, между 40°С и 5°С. Температуру можно изменять со скоростью от 5°С до 50°С в час, например, 35°С в час. Под циклическим изменением температуры смеси понимают, что температуру необходимо последовательно понижать и затем повышать, или наоборот. Температуру можно понижать и затем повышать, или наоборот, два или более раза, например, три или более раза, например, четыре или более раза, например, пять или более раз, например, десять или более раз. После охлаждения или циклического изменения температуры смеси, содержащей карбетоцин и одну или более жидкостей, температуру можно поддерживать, например, на уровне 5°С, в течение промежутка времени, подходящего для образования кристаллического карбетоцина. Обычно кристаллический карбетоцин может образоваться, и его можно выделить за 6-24 часа, например, за примерно 12 часов или за примерно 18 часов. Охлаждение, циклическое изменение или поддерживание температуры может происходить с перемешиванием или без перемешивания смеси.
Альтернативно, оказывается, что карбетоцин в кристаллической форме можно получить путем поддерживания смеси, содержащей карбетоцин и воду, или смеси, содержащей карбетоцин и водный ацетатный буфер, при температуре по меньшей мере 15°С, например, 20°С, например, 30°С, например, 40°С, в течение промежутка времени, подходящего для образования кристаллического карбетоцина. Обычно кристаллический карбетоцин образуется за 3-100 суток, чаще за 3-60 суток, наиболее часто за 7-12 суток. В одном альтернативном воплощении карбетоцин в кристаллической форме можно получить, поддерживая смесь, содержащую карбетоцин и воду, или смесь, содержащую карбетоцин и водный ацетатный буфер, при температуре 20°С в течение примерно 3-60 суток. В другом альтернативном воплощении карбетоцин в кристаллической форме можно получить, поддерживая смесь, содержащую карбетоцин и воду, или смесь, содержащую карбетоцин и водный ацетатный буфер, при температуре 40°С в течение примерно 7-12 суток. В этих альтернативных воплощениях можно выполнять все другие описанные ниже стадии, за исключением стадии, связанной с добавлением антирастворителя.
Карбетоцин можно кристаллизовать из смеси, содержащей по меньшей мере один растворитель и по меньшей мере один антирастворитель. Под растворителем понимают жидкость, в которой карбетоцин легко растворяется или является легко растворимым. Растворитель может быть любым растворителем, в котором карбетоцин растворим в количестве, составляющем при стандартных условиях 0,01 мг/мл или более, например, 0,05 мг/мл или более, например, 0,1 мг/мл или более, например, 0,5 мг/мл или более, например, 1 мг/мл или более, например, 5 мг/мл или более, например, 10 мг/мл или более, например, 20 мг/мл или более. Под антирастворителем понимают жидкость, в которой карбетоцин растворяется менее легко относительно растворителя, или в которой карбетоцин является менее растворимым относительно растворителя. Антирастворитель можно выбрать относительно растворителя и он может быть любым растворителем, в котором карбетоцин растворим в количестве, составляющем при стандартных условиях менее 20 мг/мл, например, менее 10 мг/мл, например, менее 5 мг/мл, например, менее 1 мг/мл, например, менее 0,5 мг/мл, например, менее 0,1 мг/мл, например, менее 0,05 мг/мл, например, менее 0,01 мг/мл. Специалисту понятно, что когда карбетоцин растворим в количестве, например, 10 мг/мл или более в растворителе, в антирастворителе он менее растворим, то есть растворяется в количестве менее 10 мг/мл, например, менее 5 мг/мл, например, менее 1 мг/мл, например, менее 0,5 мг/мл, например, менее 0,1 мг/мл, например, менее 0,05 мг/мл, например, менее 0,01 мг/мл. Если не указано другое, термины растворитель и антирастворитель относятся к поведению растворимости карбетоцина при комнатной температуре и атмосферном давлении.
Растворитель может включать одну или более жидкостей из группы, состоящей из воды, водного ацетатного буфера, этиленгликоля, этанола, метанола, пропанола, изопропанола и 1,2-пропандиола. Растворитель может иметь индекс относительной полярности (ИОП), описанный Christian Reichardt (Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Publishers, 3rd ed., 2003), более 0,5, например, более 0,6, например, более 0,7, например, более 0,8, например, более 0,9, например, более 1,0. Растворитель может представлять собой или содержать любое одно или более соединений из воды, водного ацетатного буфера или спирта, например, любое одно или более соединений из воды (ИОП = 1,000), раствора водного ацетатного буфера, этиленгликоля (ИОП = 0,790), этанола (ИОП = 0,654), метанола (ИОП = 0,762), пропанола (ИОП = 0,803), изопропанола (ИОП = 0,787) или 1,2-пропандиола (ИОП = 0,72).
Карбетоцин в кристаллической форме можно кристаллизовать из смеси, содержащей карбетоцин и одну или более жидкостей, причем карбетоцин присутствует в растворителе в концентрации примерно от 1 мг/мл до 200 мг/мл, предпочтительно примерно от 10 мг/мл до 150 мг/мл, наиболее предпочтительно примерно от 20 мг/мл до 100 мг/мл. В одном воплощении растворитель может быть водой. В одном воплощении растворитель может быть водным ацетатным буфером. В одном воплощении растворитель может быть этиленгликолем.
Способ может включать дополнительную стадию добавления антирастворителя в смесь, содержащую карбетоцин и одну или более жидкостей, например, добавления антирастворителя до охлаждения смеси.
Антирастворитель может иметь индекс относительной полярности (ИОП), описанный Christian Reichardt (Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Publishers, 3rd ed., 2003), менее 1, например, менее 0,9, например, менее 0,8, например, менее 0,75, например, менее 0,7, например, менее 0,6, например, менее 0,5. Антирастворитель может представлять собой или содержать любое одно или более соединений из сложного эфира, кетона, нитрила или эфира, например, любое одно или более соединений из ацетонитрила (ИОП = 0,460), этилацетата (ИОП = 0,228), ацетона (ИОП = 0,355) или метилтретбутилового эфира (ИОП = 0,124).
Таким образом, в одном воплощении способа карбетоцин можно кристаллизовать из смеси, содержащей карбетоцин и одну или более жидкостей, содержащих этиленгликоль и ацетонитрил. Карбетоцин может присутствовать в смеси, содержащей карбетоцин и одну или более жидкостей, в концентрации от 10 мг/мл до 150 мг/мл, наиболее предпочтительно примерно 100 мг/мл. Этиленгликоль и ацетонитрил может присутствовать в отношении от 5:95 до 35:65. Карбетоцин в кристаллической форме можно получить путем охлаждения смеси, содержащей карбетоцин и одну или более жидкостей, от 40°С до 5°С со скоростью 35°С в час и поддержания температуры на уровне 5°С в течение промежутка времени, подходящего для образования кристаллического карбетоцина, например, в течение примерно 12 часов или в течение примерно 18 часов.
Способ может включать дополнительную стадию затравливания смеси, содержащей карбетоцин и одну или более жидкостей, кристаллом, например, кристаллом карбетоцина, например, кристаллом сольватированной кристаллической формы I карбетоцина.
Под затравливанием понимают добавление гомогенных или гетерогенных кристаллов, то есть затравочных кристаллов, в смесь для образования зародышей и/или роста дополнительного карбетоцина в кристаллической форме. Под гомогенными кристаллами понимают кристаллический карбетоцин в любой из его форм. Под гетерогенными кристаллами понимают кристаллы из другого материала.
Способ может включать дополнительную стадию индуцирования кристаллизации в смеси, содержащей карбетоцин и одну или более жидкостей. Кристаллизацию можно индуцировать с помощью любых подходящих средств для способствования образованию зародышей и росту кристаллов, например, путем возмущения поверхности смеси, содержащей карбетоцин и одну или более жидкостей, для создания затравочных кристаллов, например, путем отбора пипеткой жидкости с поверхности и внесения пипеткой жидкости на поверхность смеси, содержащей карбетоцин и одну или более жидкостей, или нанесения царапин в месте, где поверхность смеси, содержащей карбетоцин и одну или более жидкостей, соприкасается с поверхностью контейнера, в котором держат смесь.
Способ может включать дополнительную стадию удаления растворителя и возможной сушки карбетоцина в кристаллической форме.
Под удалением растворителя понимают удаление части или по существу всех сольватированных молекул из кристаллической структуры карбетоцина, таким образом, чтобы кристаллическая структура включала малое количество или вообще не включала упорядоченных или неупорядоченных молекул растворителя. В предпочтительном воплощении удаление растворителя означает превращение карбетоцина из пентагидратной кристаллической формы в моногидратную кристаллическую форму.
Удаление растворителя из карбетоцина в кристаллической форме можно выполнять путем промывки карбетоцина в кристаллической форме антирастворителем, например, ацетонитрилом, возможно при температуре 20°С или ниже, например, от -30°С до 20°С, например, от -20°С до 20°С, например, от -10°С до 20°С, например, от -5°С до 15°С, например, от 0°С до 10°С, например, примерно 5°С, и затем сушки, например, сушки в вакууме. Сушка может происходить в вакууме в течение промежутка времени, подходящего для осуществления удаления растворителя, например, более 1 часа, например, в течение примерно 24 часов. Предпочтительно карбетоцин в кристаллической форме можно промыть ацетонитрилом при температуре примерно 5°С и высушить в вакууме при температуре примерно 20°С в течение примерно 24 часов для осуществления удаления растворителя.
Удаление растворителя также можно выполнять путем нагрева карбетоцина в кристаллической форме до температуры от по меньшей мере 40°С до не более 190°С или путем воздействия на карбетоцин в кристаллической форме окружающей среды с низкой относительной влажностью, такой как относительная влажность 40% или менее.
Таким образом, в одном воплощении способа карбетоцин можно кристаллизовать из смеси, содержащей карбетоцин и одну или более жидкостей, содержащих этиленгликоль и ацетонитрил. Карбетоцин может присутствовать в смеси, содержащей карбетоцин и одну или более жидкостей, в концентрации от 10 мг/мл до 150 мг/мл, наиболее предпочтительно примерно 100 мг/мл. Этиленгликоль и ацетонитрил могут присутствовать в отношении от 5:95 до 35:65. Можно добавить дополнительный антирастворитель, такой как ацетонитрил. Карбетоцин в кристаллической форме можно получить путем охлаждения смеси, содержащей карбетоцин и одну или более жидкостей, от 40°С до 5°С со скоростью 35°С в час и поддержания температуры на уровне 5°С в течение промежутка времени, подходящего для выделения кристаллического карбетоцина, например, в течение примерно 12 часов или в течение примерно 18 часов. Карбетоцин в кристаллической форме можно промыть ацетонитрилом при температуре примерно 5°С и высушить в вакууме при температуре примерно 20°С в течение примерно 24 часов для осуществления удаления растворителя.
При необходимости, до кристаллизации можно выполнять стадию фильтрации. Стадия фильтрации предпочтительно включает фильтрацию путем центрифугирования. Соответственно, в одном аспекте способ получения карбетоцина в кристаллической форме включает стадии (1) фильтрации, предпочтительно путем центрифугирования, и (2) кристаллизации.
При необходимости, до кристаллизации можно выполнять стадию промывки. Например, карбетоцин, например, сырой карбетоцин, можно суспендировать, например, суспендировать в ацетонитриле, например, суспендировать в ацетонитриле в течение от 2 часов до недели, например, суспендировать в ацетонитриле в течение примерно 18 часов с постоянным перемешиванием. Промывка сырого карбетоцина повышает чистоту до кристаллизации на примерно 1 2% и значительно повышает содержание по результатам анализа от примерно 44% до примерно 70% (в ацетонитриле). Соответственно, в одном аспекте способ получения карбетоцина в кристаллической форме включает стадии (1) промывки карбетоцина, например, сырого карбетоцина, ацетонитрилом, и (2) кристаллизации. В другом аспекте способ получения карбетоцина в кристаллической форме включает стадии (1) промывки карбетоцина, например, сырого карбетоцина, ацетонитрилом, (2) фильтрации, предпочтительно путем центрифугирования, и (3) кристаллизации.
Авторы настоящей заявки преимущественно и неожиданно обнаружили, что можно выделить карбетоцин без какой-либо лиофилизации путем кристаллизации карбетоцина, например, кристаллизации карбетоцина из раствора.
Способ обеспечивает карбетоцин высокой чистоты с приемлемым выходом без потребности в стадии лиофилизации.
Карбетоцин в смеси, содержащей карбетоцин и одну или более жидкостей, может быть по существу чистым карбетоцином или, альтернативно, может быть сырым карбетоцином.
В данном документе термин «сырой», например, в «сыром карбетоцине», означает карбетоцин, который является недостаточно чистым для использования в качестве фармацевтического продукта. Сырой пептид может иметь чистоту менее 95%, например, менее 92,5%, например, от 90% до 93%, например, от 91% до 93%, измеренную с помощью ВЭЖХ с УФ детектированием. Примеси, обнаруженные в сыром пептиде, могут включать одно или более неорганических веществ, остаточный растворитель (например, диметилформамид (ДМФА)), родственные пептиду примеси и остаточные связывающие пептид реагенты.
(Полученный) карбетоцин в кристаллической форме/карбетоцин в сольватированной (например, гидратированной) кристаллической форме/карбетоцин в десольватированной кристаллической форме могут иметь чистоту более или равную 95%.
Сырой карбетоцин можно синтезировать с помощью известных в уровне техники способов, например, способов, аналогичных тем, которые описаны в WO 2009/122285 (международная патентная заявка №. PCT/IB2009/005351) от Ferring B.V.
По изобретению в дополнительном аспекте предоставляют фармацевтическую композицию, содержащую карбетоцин по изобретению, или карбетоцин, полученный согласно способу по изобретению. Фармацевтическую композицию по изобретению можно использовать в качестве лекарственного средства. Фармацевтическую композицию по изобретению можно использовать в лечении неврологического расстройства или репродуктивного расстройства, например, использовать в лечении синдрома Прадера-Вилли (как описано в WO 2016/044131 (международная патентная заявка № PCT/US 2015/04911) от Ferring B.V.), или, например, использовать в лечении или предотвращении атонии матки, например, после влагалищного рождения ребенка, рождения ребенка с помощью кесарева сечения, или использовать в лечении или предотвращении атонии матки у пациентки, которая имеет риск развития послеродового кровотечения, и/или использовать в лечении или предотвращении обильного кровотечения после влагалищного родоразрешения (как описано в WO 2009/122285 (международная патентная заявка № PCT/IB 2009/005351) от Ferring B.V.).
Ниже настоящее изобретение поясняется на примерах. В примерах могут быть описаны предпочтительные воплощения изобретения, но их не рассматривают как ограничивающие каким-либо образом.
Пример 1. Получение сольватированной кристаллической формы I карбетоцина
Стадия 1. Синтез
Сырой карбетоцин с чистотой приблизительно 91% получали с помощью способов синтеза, аналогичных тем, которые описаны в WO 2009/122285 (международная патентная заявка № PCT/IB 2009/005351) от Ferring B.V.
Стадия 2. Получение раствора
60 мг сырого карбетоцина, полученного на стадии 1, растворяли в 0,6 мл смеси 30:70 (об/об) этиленгликоля (первая жидкость): ацетонитрила (вторая жидкость) при 40°С. Сосуд затем затравливали кристаллами (сольватированной) формы I карбетоцина. Понятно, что затравливание не является обязательным, но оно может ускорить кристаллизацию.
Стадия 3. Кристаллизация
Раствор, полученный на стадии 2, нагревали до 40°С и выдерживали при этой температуре в течение 30 минут. Смесь затем фильтровали путем центрифугирования для удаления любых нерастворимых примесей. Смесь затем перемешивали при 40°С в течение тридцати минут, охлаждали до 5°С на протяжении одного часа и затем выдерживали при 5°С в течение ночи с постоянным перемешиванием.
Осажденный материал выделяли.
ПРСА выполняли на PANalytical X'pert pro. Образцы сканировали от 3 до 35° 2θ. Материал аккуратно измельчали для высвобождения любых агломератов и загружали на многолуночный планшет с полимерной пленкой Kapton или Mylar для поддержания образца. Многолуночный планшет затем помещали в дифрактометр и анализировали с использованием Cu K излучения (α1 λ=1,54060 , α2=1,54443 , β=1,39225 , отношение α1 : α2 = 0,5), действуя в трансмиссионном режиме (размер шага 0,0130° 2θ) с использованием настоек генератора 40 кВ/40 мА.
Из раствора кристаллизовался карбетоцин, имеющий картину рентгеновской дифракции, по существу такую, как показана в таблице 1 и на фиг. 1 (форма I).
Твердые вещества анализировали с помощью ТГ/ДТА для для простоты определения от потери массы/тепловых эффектов (фиг. 4а).
Чистота карбетоцина в (сольватированной) кристаллической форме составила 96,2%, вычисленная с помощью УФ-ВЭЖХ (фиг. 3а) согласно способу, изложенному в таблице 4.

Пример 2. Получение сольватированной кристаллической формы I карбетоцина
Стадия 1. Синтез
Сырой карбетоцин с чистотой приблизительно 91% получали с помощью способов синтеза, аналогичных тем, которые описаны в WO 2009/122285 (международная патентная заявка № PCT/IB 2009/005351) от Ferring B.V.
Стадия 2. Получение раствора
60 мг сырого карбетоцина, полученного на стадии 1, растворяли в 0,6 мл смеси 30:70 (об/об) этиленгликоля (первая жидкость): ацетонитрила (вторая жидкость) при 40°С. Сосуд затем затравливали кристаллами (сольватированной) формы I карбетоцина. Понятно, что затравливание не является обязательным, но оно может ускорить кристаллизацию.
Стадия 3. Добавление антирастворителя
Добавляли количество ацетонитрила, достаточное для изменения отношения этиленгликоль: ацетонитрилу до 6,7:93,3 (об/об).
Стадия 4. Кристаллизация
Раствор, полученный на стадии 3, нагревали до 40°С и выдерживали при этой температуре в течение 30 минут. Смесь затем фильтровали путем центрифугирования для удаления любых нерастворимых примесей. Смесь затем перемешивали при 40°С в течение тридцати минут, охлаждали до 5°С на протяжении одного часа и затем выдерживали при 5°С в течение ночи с постоянным перемешиванием.
Осажденный материал выделяли.
ПРСА выполняли так, как описано выше в связи с примером 1.
Из раствора кристаллизовался карбетоцин, имеющий картину рентгеновской дифракции, по существую такую, как показана в таблице 1 и на фиг. 1.
Пример 3. Получение десольватированной кристаллической формы II карбетоцина
Для удаления этиленгликоля, присутствующего в кристаллизованном материале, полученном в примерах 1 и 2, кристаллизованный материал примера 1 или примера 2 подвергали удалению растворителя и сушили.
За промывкой кристаллизованной формы I материала ацетонитрилом при 5°С следовала сушка в вакууме, приводящая к удалению растворителя из сольватированной формы I с получением кристаллов десольватированной формы II, которые имеют картину дифракции по существу такую, как показана в таблице 2 и на фиг. 2. Было обнаружено, что кристаллы формы II имеют уровни содержания этиленгликоля ниже предела согласно Международной конференции по гармонизации, составляющего 620 ч/млн, определенные с помощью газовой хроматографии с параметрами как в таблице 5 ниже.


Пример 4. Получение десольватированной кристаллической формы II карбетоцина К приблизительно 300 мг сырого карбетоцина (чистота приблизительно 91,3%) добавляли 3 мл заранее приготовленной смеси растворителей 30% этиленгликоля: 70% ацетонитрила (об/об) и смесь нагревали до 40°С в течение 30 минут с постоянным перемешиванием.
Спустя 30 минут смесь фильтровали путем центрифугирования для удаления любых нерастворимых примесей. К этой смеси (все еще при 40°С) добавляли 1,5 мл ацетонитрила аликвотами по 0,5 мл. Не наблюдали никакого осаждения при 40°С, даже после полного добавления ацетонитрила.
Смесь затем перемешивали при 40°С в течение 1 часа и охлаждали до 5°С на протяжении 1 часа и затем выдерживали при 5°С в течение 18 часов с постоянным перемешиванием.
Спустя 18 часов выделяли осажденный материал, промывали приблизительно 5 мл ацетонитрила и затем сушили в вакууме при температуре окружающей среды в течение 24 часов.
На следующие сутки твердые вещества анализировали с помощью ВЭЖХ на чистоту и количественный состав (фиг. 3b), ТГ/ДТА для простоты определения потери массы/тепловых эффектов (фиг. 4b), а также с помощью поляризационной микроскопии (ПМ) и ПРСА для определения морфологии и кристаллического содержания.
Анализ ПМ показал, что конечное выделенное твердое вещество содержало смесь агломератов (50-100 мкм), которые легко распадались на очень малые иглоподобные кристаллы (длина <10 мкм).
Данные ТГ/ДТА для кристаллов, образованных с помощью способа примера 4, и приведенные на фиг. 4b, показывают, что существовала общая потеря массы, составляющая приблизительно только 0,8% вплоть до 110°С. Было обнаружено, что этот процесс проходит в две стадии, с первой потерей массы приблизительно 0,5% вплоть до 60°С и второй потерей массы приблизительно 0,3% вплоть до приблизительно 110°С. Эти потери массы соответствуют слабо поверхностно связанным ацетонитрилу и воде и не являются показательными для сольватации самой кристаллической формы. Соответственно, потеря этого растворителя не изменяет степень кристалличности десольватированной кристаллической формы II.
Эти результаты показывают, что десольватированная кристаллическая форма II карбетоцина является высокостабильной.
Пример 5. Получение сольватированной кристаллической формы I карбетоцина
Стадия 1. Синтез
Сырой карбетоцин с чистотой приблизительно 93,5% получали с помощью способов синтеза, аналогичных тем, которые описаны в WO 2009/122285 (международная патентная заявка № PCT/IB 2009/005351) от Ferring B.V.
Стадия 2. Получение раствора
Ацетатный буфер, 25 мМ, рН 5,5, получали из тригидрата ацетата натрия, ледяной уксусной кислоты и сверхчистой воды. 354 мг сырого карбетоцина, полученного на стадии 1, растворяли в 16,6 мл ацетатного буфера. Раствор фильтровали с помощью ПВДФ (поливинилиденфторидного) шприцевого фильтра с размером пор 0,22 мкм и 500 мкл порции раствора аликвотировали в виалы, которые после этого запечатывали. рН раствора карбетоцина составлял 5,3.
Стадия 3. Кристаллизация
Раствор, полученный на стадии 2, нагревали до 40°С и выдерживали в запечатанных виалах при этой температуре. Спустя 3 суток виалы извлекали и стеклянную пипетку Эппендорфа (Eppendorf) использовали для аккуратного отсасывания раствора вверх и внесения обратно в виалы для создания посредством этого некоторого количества зародышей кристаллизации. После пипетирования виалы снова запечатывали и оставляли при 40°С. Спустя 9 суток образовывались частицы с кристаллообразным внешним видом. Осажденный материал выделяли.
ПРСА выполняли на PANalytical X'pert pro с Cu-Kα1 монохроматором (α1 λ=1,54060 ). Образцы сканировали от 2 до 35° 2θ. Материал аккуратно измельчали и намазывали на пластину из Si нулевогофона, которую затем помещали в медленно вращающийся держатель образца в дифрактометре, действующем в трансмиссионном режиме (скорость сканирования 0,01°c, размер шага 0,017° 2θ) с использованием настроек генератора 45 кВ/40 мА. Измерения выполняли с использованием щели с программируемым отклонением угла падения излучения.
Картина рентгеновской дифракции полученных кристаллов карбетоцина представлена в таблице 3 и на фиг. 5. Картина рентгеновской дифракции показывает карбетоцин с другой кристаллической формой или полиморфной модификацией по сравнению с примерами 1 и 2 (фиг. 1, таблица 1) и примерами 3 и 4 (фиг. 2, таблица 2).
Анализ дифференциальной сканирующей калориметрией (ДСК) выполняли на Netzsch DSC 204F1. Несколько миллиграммов кристаллов выделяли из маточного раствора и оставляли сушиться в воздухе в вытяжном шкафу в течение нескольких часов при относительной влажности (ОВ) примерно 20%. Кристаллы аккуратно измельчали в порошковый материал и 1,2 мг этого материала загружали в 25 мкл алюминиевую кювету. На кювету приспосабливали и обжимали крышку, до этого проколотую точечным отверстием (с диаметром 0,25 мм). Образец анализировали от 20 до 250°С, используя скорость нагрева 5 К/мин.
Данные ДСК для кристаллов, образованных с помощью способа примера 5, и показанные на Фиг. 7, показывают, что существует потеря летучего материала в области от 40 до 120°С, что соответствует потере слабо поверхностно связанной воды и сольватированной воды. Эндотермичность плавления с началом при 192°С соответствует плавлению безводного карбетоцина.
Гравиметрическую сорбцию пара (ГСП) выполняли на SMS DVS-1. 1,4 мг кристаллов и порошок добавляли в алюминиевую кювету и подвергали ступенчатым изменениям относительной влажности (ОВ) в течение двух следующих друг за другом циклов: 20-30-40-50-60-70-80-70-60-50-40-30-20-10-0-10-20-30-40-50-60-70-80-90-80-70-60-50-40-30-20-10-0% ОВ в режиме регулирования без обратной связи. Температуру поддерживали на уровне 25°С и использовали чистый азот с расходом 200 мл/мин. Применяемый критерий dm/dt составлял 0,001 масс. %/мин в течение окна 5 минут, с максимальным временем 150 минут для всех стадий, за исключением стадий при ОВ 0%, когда его устанавливали на 6 часов.
Данные ГСП показаны на фиг. 8. График изотермы ГСП показан на фиг. 8b. Плато при примерно 2% (масс./масс.) соответствует моногидрату плюс некоторому количеству слабо связанной поверхностной воды. Второе плато при приблизительно 8% (масс/масс.) соответствует пентагидрату с некоторым количеством слабо связанной поверхностной воды. Пентагидрат существует при ОВ примерно 60% и выше (сорбция) и ОВ от примерно 40 до 90% (десорбция).
Чистота карбетоцина в (сольватированной) кристаллической форме составила 98,7%, вычисленная с помощью УФ-ВЭЖХ согласно способу, указанному в таблице 6.











Изобретение относится к способу получения карбетоцина в сольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ (Cu-K), в котором карбетоцин в кристаллической форме кристаллизуют из смеси, содержащей карбетоцин и одну или более жидкостей, причем одна или более жидкостей представляют собой смесь этиленгликоля и ацетонитрила, путем охлаждения смеси, содержащей карбетоцин и одну или более жидкостей, от 40 до 5°С или путем циклического изменения температуры смеси, содержащей карбетоцин и одну или более жидкостей, от 40 до 5°С. Изобретение относится к способу получения карбетоцина в сольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,34, 6,43, 8,66, 17,37, 19,03 и 19,39 градусах 2θ (Cu-K), в котором карбетоцин в кристаллической форме кристаллизуют из смеси, содержащей карбетоцин и одну или более жидкостей, причем одна или более жидкостей представляет собой водный ацетатный буфер, путем поддержания смеси, содержащей карбетоцин и одну или более жидкостей, при температуре по меньшей мере 15°С в течение 3-100 суток. Также изобретение относится к способу получения карбетоцина в десольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,11, 4,39, 5,60, 7,45, 17,75, 19,16 и 19,45 градусах 2θ (Cu-K), включающему способ по п. 1, который включает дополнительную стадию удаления растворителя и сушки карбетоцина в кристаллической форме. Технический результат – получение карбетоцина в кристаллической форме. 3 н. и 5 з.п. ф-лы, 11 ил., 6 табл., 5 пр.
1. Способ получения карбетоцина в сольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,83, 7,43, 9,20, 17,87, 19,60, 20,43 и 21,34 градусах 2θ (Cu-K), в котором карбетоцин в кристаллической форме кристаллизуют из смеси, содержащей карбетоцин и одну или более жидкостей, причем одна или более жидкостей представляют собой смесь этиленгликоля и ацетонитрила, путем охлаждения смеси, содержащей карбетоцин и одну или более жидкостей, от 40 до 5°С или путем циклического изменения температуры смеси, содержащей карбетоцин и одну или более жидкостей, от 40 до 5°С.
2. Способ получения карбетоцина в сольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,34, 6,43, 8,66, 17,37, 19,03 и 19,39 градусах 2θ (Cu-K), в котором карбетоцин в кристаллической форме кристаллизуют из смеси, содержащей карбетоцин и одну или более жидкостей, причем одна или более жидкостей представляет собой водный ацетатный буфер, путем поддержания смеси, содержащей карбетоцин и одну или более жидкостей, при температуре по меньшей мере 15°С в течение 3-100 суток.
3. Способ по п. 1, в котором одна или более жидкостей содержат этиленгликоль и ацетонитрил в отношении от 1:99 до 50:50.
4. Способ по любому из пп. 1 или 3, включающий дополнительную стадию добавления антирастворителя к смеси, содержащей карбетоцин и одну или более жидкостей, причем антирастворитель, возможно, является ацетонитрилом.
5. Способ по п. 4, в котором одна или более жидкостей содержат этиленгликоль и ацетонитрил в отношении от 15:85 до 25:75 и где добавление ацетонитрила в качестве антирастворителя изменяет отношение этиленгликоля к ацетонитрилу на отношение от 1:99 до 30:70.
6. Способ по любому из пп. 1-5, включающий дополнительную стадию затравливания смеси, содержащей карбетоцин и одну или более жидкостей, кристаллом.
7. Способ получения карбетоцина в десольватированной кристаллической форме, характеризующейся пиками порошковой рентгеновской дифракции при 4,11, 4,39, 5,60, 7,45, 17,75, 19,16 и 19,45 градусах 2θ (Cu-K), включающий способ по п. 1, который включает дополнительную стадию удаления растворителя и сушки карбетоцина в кристаллической форме.
8. Способ по п. 7, в котором удаление растворителя из карбетоцина в кристаллической форме выполняют путем промывки карбетоцина в кристаллической форме антирастворителем, возможно, при температуре 20°С или ниже.
| Колосоуборка | 1923 |
|
SU2009A1 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| CN 101531705 A, 16.09.2009 | |||
| Andrew D | |||
| Rudko; Celia C.H | |||
| Chen; et al.: " Crystalline salts of oxytocin: X-ray crystallographic data", Journal of Crystal Growth, 1971, v.10(3), p.260-262 | |||
| MINO R.CAIRA, Crystalline Polymorphism of Organic Compounds, 1998, p.163-208 | |||
| Sherry L.Morissette et | |||

