Настоящая заявка испрашивает приоритет на основании заявки на патент Китая №201910550238.4 «КРИСТАЛЛИЧЕСКАЯ ФОРМА 1,4-БИС[1,2-БЕНЗИЗОСЕЛЕНАЗОЛА-3(2Н)-ОН]-БУТАНА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ», поданной 24 июня 2019 г. в Национальное Управление Интеллектуальной Собственности Китая, которая полностью включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области получения кристаллической формы и, в частности, к кристаллической форме 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана, способу ее получения и применения.
УРОВЕНЬ ТЕХНИКИ
Структура 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана показана ниже:
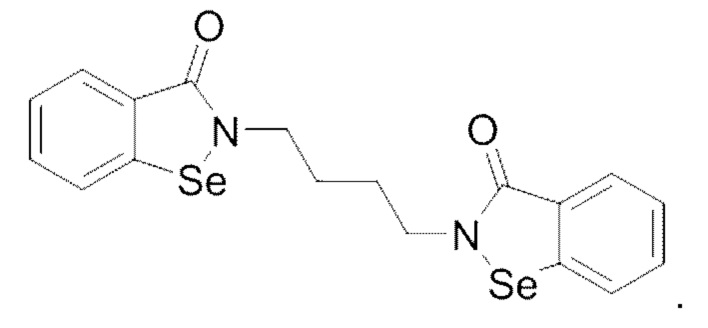
Данное соединение широко применяют, например: 1) при изготовлении лекарственных средств для предварительного лечения различных фиброзных заболеваний, таких как фиброз печени, фиброз легких, фиброз почек, миелофиброз, фиброз кожи, муковисцидоз, подслизистый фиброз полости рта или фиброз миокарда; 2) при изготовлении косметических средств для лечения фиброза кожи; 3) при изготовлении лекарственных средств для профилактики и лечения артрита, таких как лекарственных средств для профилактики и лечения ревматического артрита, ревматоидного артрита и т.д.; 4) при изготовлении лекарственных средств для профилактики и лечения воспалений, таких как лекарственных средств для профилактики и лечения пародонтита, плечелопаточного периартрита или миокардита; 5) при изготовлении лекарственных средств для лечения различных воспалительных заболеваний с фиброзом; 6) при изготовлении лекарственных средств для профилактики и лечения метастазов опухолей.
В ходе исследований и разработок лекарственных средств исследование кристаллических форм играет жизненно важную роль. Твердые формы лекарственного средства могут иметь значительные различия по внешнему виду, растворимости, температуре плавления, скорости растворения, биодоступности и т.п., что влияет на стабильность, биодоступность и эффективность лекарственного средства. Полиморфизм лекарственного средства является важным фактором, влияющим на качество и клиническую эффективность такого лекарственного средства. Следовательно, разработка чистой стабильной кристаллической формы имеет решающее значение и необходима для производства и применения лекарственного средства.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
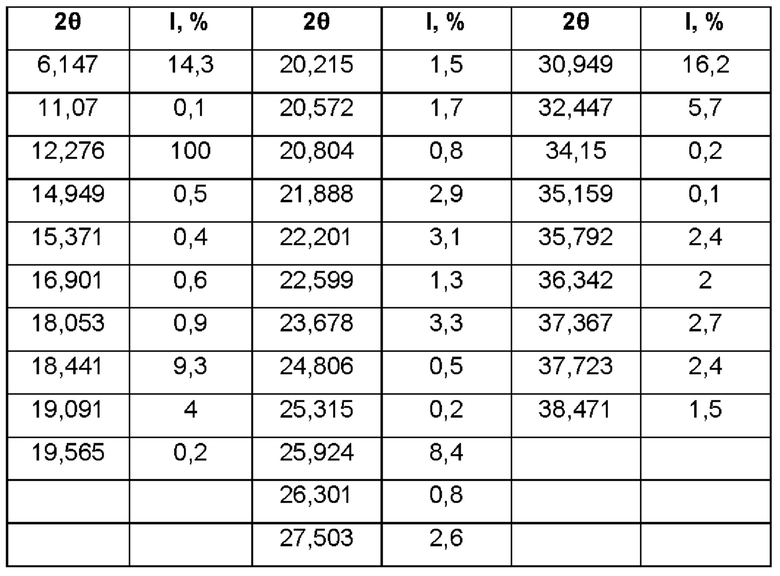
Для решения технических задач, описанных выше, настоящее изобретение, во-первых, обеспечивает кристаллическую форму I 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана, имеющую характеристические пики при углах 2θ, составляющих 6,15±0,20°, 12,28±0,20°, 18,44±0,20°, 25,92±0,20° и 30,95±0,20°, при проведении рентгеновской порошковой дифракции с использованием излучения Cu-Kα.
Согласно одному варианту реализации настоящего изобретения кристаллическая форма I имеет характеристические пики при углах 2θ, составляющих 6,15±0,20°, 12,28±0,20°, 18,44±0,20°, 19,09±0,20°, 22,20±0,20°, 23,68±0,20°, 25,92±0,20°, 30,95±0,20° и 32,45±0,20°, при проведении рентгеновской порошковой дифракции с использованием излучения Cu-Kα.
Согласно одному варианту реализации настоящего изобретения кристаллическая форма I имеет характеристические пики при углах 2θ, составляющих 6,15±0,20°, 12,28±0,20°, 18,44±0,20°, 19,09±0,20°, 21,89±0,20°, 22,20±0,20°, 23,68±0,20°, 25,92±0,20°, 27,50±0,20°, 30,95±0,20°, 32,45±0,20°, 35,79±0,20°, 37,37±0,20° и 37,72±0,20°, при проведении рентгеновской порошковой дифракции с использованием излучения Cu-Kα.
Согласно одному варианту реализации настоящего изобретения кристаллическая форма I имеет следующие характеристические пики при углах 2θ и относительные интенсивности при проведении рентгеновской порошковой дифракции с использованием излучения Cu-Kα:
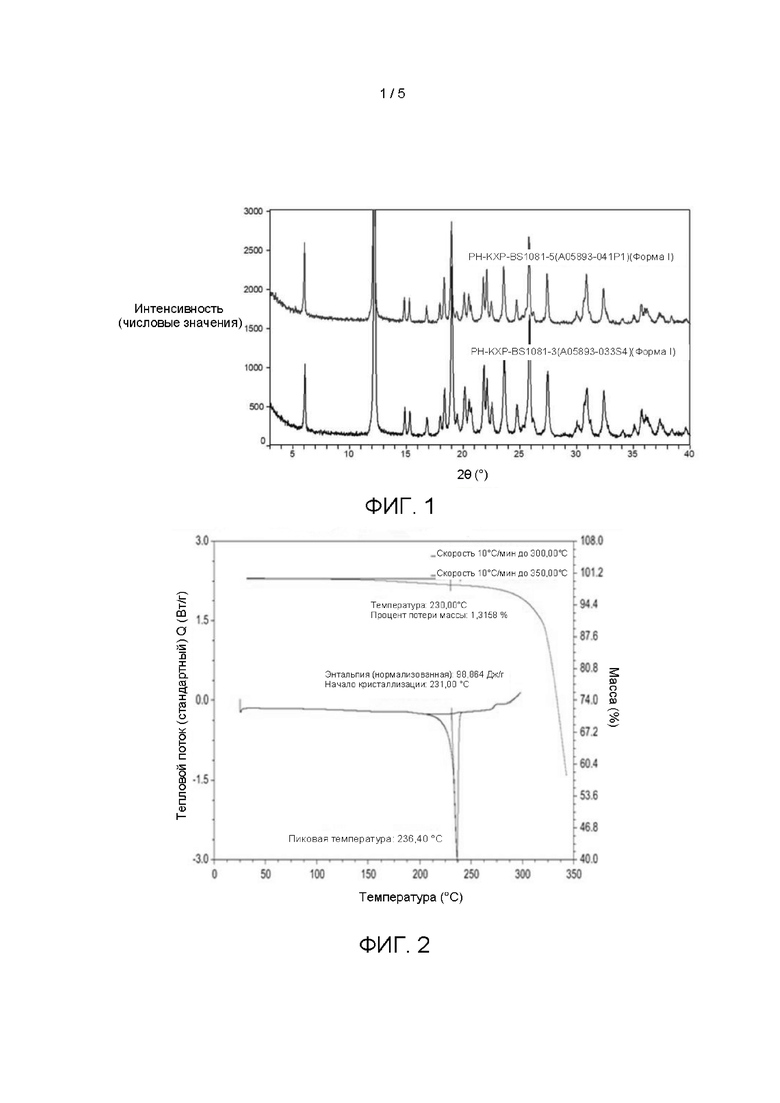
Согласно одному варианту реализации настоящего изобретения дифрактограмма рентгеновской порошковой дифракции кристаллической формы I по существу показана на Фиг. 1.
Согласно одному варианту реализации настоящего изобретения диаграмма ДСК-ТГА кристаллической формы I по существу показана на Фиг. 2.
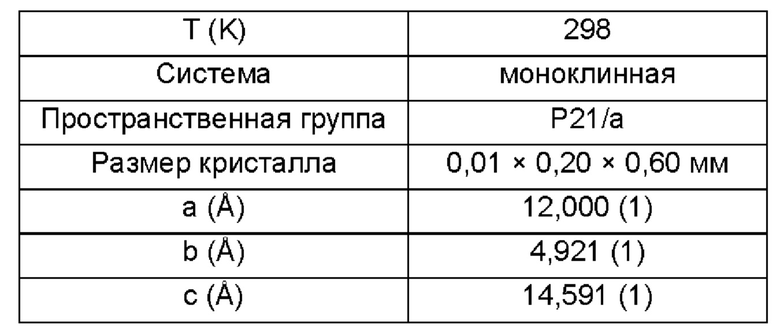
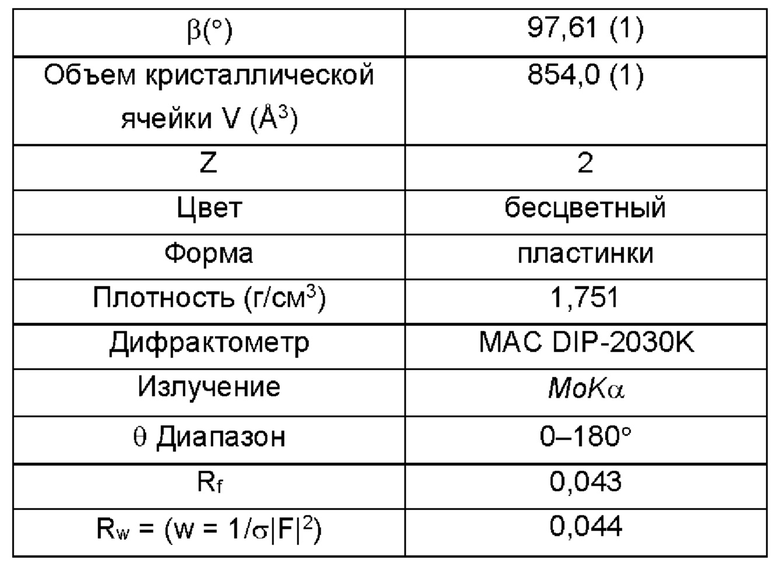
Согласно одному варианту реализации настоящего изобретения кристаллическая форма I представляет собой монокристалл, имеющий следующие монокристаллические свойства:
Настоящее изобретение дополнительно обеспечивает способ получения кристаллической формы I, как она описана выше, включающий:
растворение 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана в растворителе S1 и добавление антирастворителя S2 с получением кристаллической формы I;
где растворитель S1 выбран из ДМСО и смеси, состоящей из ДМСО и 1-10% ацетона или тетрагидрофурана (об./об.);
растворитель S2 выбран из воды, ацетонитрила, МТБЭ, изо про пил ацетата, метанола и этилацетата при условии, что, когда растворителем S1 является ДМСО, растворитель S2 не является этилацетатом.
Согласно одному варианту реализации настоящего изобретения объемное соотношение растворителя S1 и растворителя S2 составляет от 1:0,5 до 1:2, предпочтительно 1:2 или 2:1.
Согласно одному варианту реализации настоящего изобретения процедуру кристаллизации предпочтительно проводят при 30-70°С, и более предпочтительно при 40-60°С.
Настоящее изобретение дополнительно обеспечивает применение кристаллической формы I, как она описана выше, при изготовлении лекарственного средства или косметического средства для ингибирования активности желатиназы-2 и желатиназы-9, или при приготовлении лекарственного средства для лечения повреждающего иммунную систему заболевания или зависящего от функции иммунной регуляции или вызываемого функцией иммунной регуляции релевантного заболевания
Согласно одному варианту реализации настоящего изобретения повреждающее иммунную систему заболевание и зависящее от функции иммунной регуляции или вызываемое функцией иммунной регуляции релевантное заболевание выбраны из инфекции, вирусного гепатита, аллергических заболеваний, аутоиммунных заболеваний и синдрома приобретенного иммунодефицита.
Согласно одному варианту реализации настоящего изобретения лекарственное средство для ингибирования активности желатиназы-2 и желатиназы-9 выбрано из лекарственных средств для профилактики и лечения заболеваний, связанных с фиброзом, для лечения заболеваний, связанных с воспалением, для лечения связанных с фиброзом осложнений с воспалением или для лечения метастазов опухолей.
Согласно предпочтительному варианту реализации настоящего изобретения фиброзное заболевание выбрано из фиброза печени, фиброза легких, фиброза почек, миелофиброза, фиброза кожи, муковисцидоза, подслизистого фиброза полости рта и фиброза миокарда.
Согласно предпочтительному варианту реализации настоящего изобретения заболевание, связанное с воспалением, выбрано из гепатита В, пародонтита, ревматического артрита, ревматоидного артрита, плечелопаточного периартрита и миокардита.
Согласно предпочтительному варианту реализации настоящего изобретения кристаллическую форму I применяют для изготовления лекарственного средства для лечения гепатита В или рака печени.
Согласно одному варианту реализации настоящего изобретения гепатит В представляет собой хронический гепатит В у взрослых с активной репликацией вируса, стойким повышенным уровнем аланинаминотрансферазы (ALT) в сыворотке крови или гистологически активными поражениями в печени, или хроническое инфекционно-компенсированное заболевание печени, вызываемое вирусом гепатита В (HBV) у детей.
Согласно предпочтительному варианту реализации настоящего изобретения рак печени представляет собой ассоциированный с HBV рак печени или, в частности, рак печени с HBV.
Настоящее изобретение также относится к фармацевтической композиции, содержащей кристаллическую форму I, как она описана выше.
Настоящее изобретение также относится к фармацевтической композиции, как она описана выше, для ингибирования активности желатиназы-2 и желатиназы-9 или для лечения вызывающего иммунное повреждение заболевания и зависящего от функции иммунной регуляции или вызываемое функцией иммунной регуляции релевантного заболевания.
ПОЛОЖИТЕЛЬНЫЕ ЭФФЕКТЫ
Настоящее изобретение относится к кристаллической форме I 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана. Кристаллическая форма I имеет хорошую стабильность, она не подвержена полиморфному переходу, пригодна для изготовления фармацевтических композиций и препаратов, их применения, транспортировки и хранения, и полностью обеспечивает безопасность и качество лекарственных средств.
Кроме того, указанная кристаллическая форма отличается высокой растворимостью, подходящей биодоступностью, низкой токсичностью и отличной ингибирующей активностью в отношении опухолей. Авторы настоящего изобретения неожиданно обнаружили, что кристаллическая форма I обладает хорошей ингибирующей активностью в отношении рака печени, особенно рака печени, ассоциированного с HBV. Поскольку 80% пациентов с раком печени в Китае являются носителями HBV, применение кристаллической формы I указанного соединения при лечении рака печени имеет очевидную актуальность.
Наконец, способ получения кристаллической формы I отличается простотой, хорошей воспроизводимостью, высоким выходом и пригодностью для промышленного производства, поэтому он имеет большую прикладную ценность.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 показана дифрактограмма рентгеновской порошковой дифракции (XRD) кристаллической формы I (На фиг.1 показаны результаты для двух партий кристаллической формы I).
На Фиг. 2 показана термограмма ДСК-ТГА кристаллической формы I.
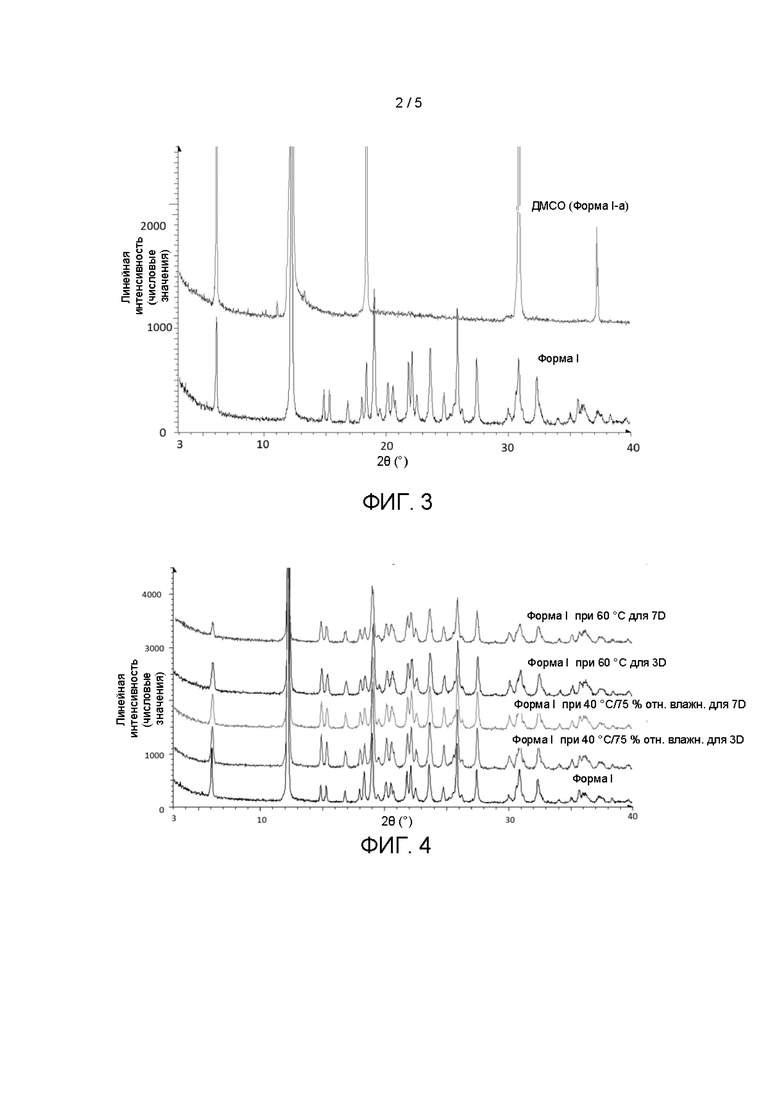
На Фиг. 3 показаны дифрактограммы XRD кристаллической формы l-a и кристаллической формы I.
На Фиг. 4 показаны дифрактограммы XRD кристаллической формы I после выдерживания в различных условиях.
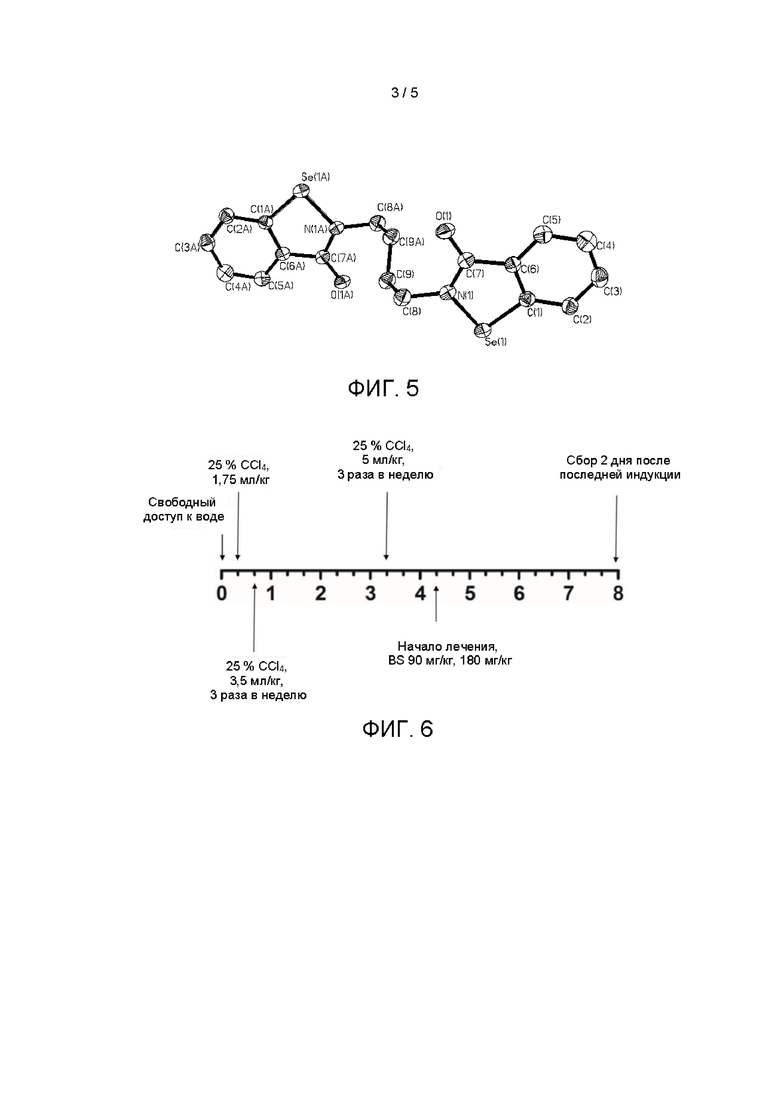
На Фиг. 5 показана монокристаллическая структура кристаллической формы I.
На Фиг. 6 показана схема экспериментальной модели на животных, описанной в испытательном примере 4.
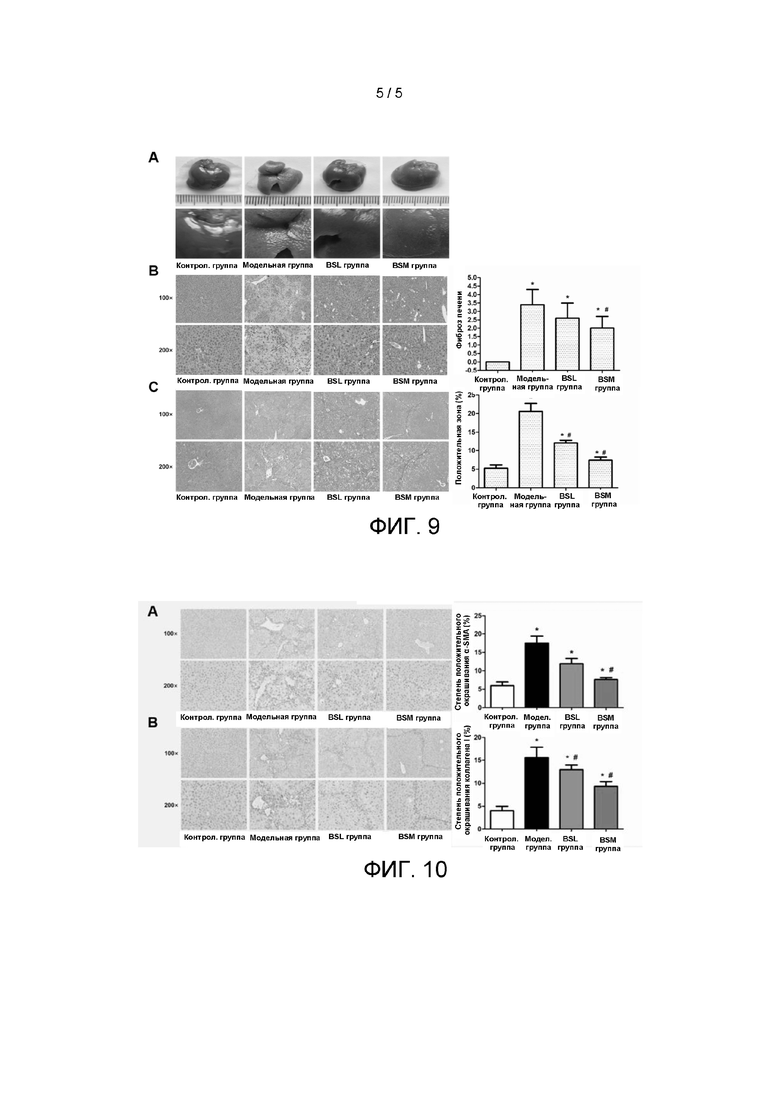
На Фиг. 7 показано ингибирование роста клеток HSC (%) при различных концентрациях кристаллической формы I.
На Фиг. 8 показано влияние различных концентраций кристаллической формы I на массу тела (А), биохимические показатели печени (Е, F и G), активность TR (С и D) и уровни TGF (В) в моделях фиброза печени у животных.
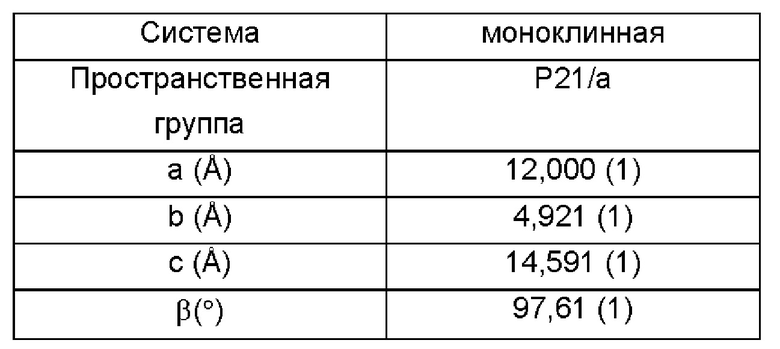
На Фиг. 9 показана шероховатость поверхности печени (А), окрашивание гематоксилином и эозином (НЕ) (В) и окрашивание по Массону (Masson) (С) для различных концентраций кристаллической формы I в моделях фиброза печени у животных.
На Фиг. 10 показана экспрессия (A) α-SMA и (В) Коллагена 1А1 в печени у различных групп мышей в конце эксперимента с использованием модели фиброза печени при различных концентрациях кристаллической формы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая схема настоящего изобретения будет далее подробно проиллюстрирована со ссылкой на следующие конкретные примеры. Следует понимать, что нижеследующие примеры представляют собой просто иллюстративные примеры и объясняют суть настоящего изобретения, и их не следует рассматривать как ограничение объема охраны настоящего изобретения. Все методы, реализованные на основе вышеупомянутого содержания настоящего изобретения, входят в объем охраны настоящего изобретения.
Если не указано иное, исходные материалы и реагенты, используемые в следующих примерах, являются коммерчески доступными продуктами или могут быть получены известными способами.
Рентгеновскую порошковую дифракцию (XRD) проводили с помощью рентгеновского порошкового дифрактометра D8 Advance (Bruker) и рентгеновского порошкового дифрактометра D2 Phaser (Bruker). Платформы были оснащены детектором LynxEye. С помощью рентгеновского порошкового дифрактометра (Bruker) тестировали образцы при углах сканирования 20 от 3° до 40° с шагом 0,02°. Напряжение и ток рентгеновской трубки при тестировании составляли 40 кВ и 40 мА, соответственно.
В качестве термогравиметрического анализатора использовали TGA Q500 или Discovery TGA 55 (ТА, США). Образцы помещали в находящийся в равновесии открытый алюминиевый лоток для образцов, и массу автоматически измеряли в печи ТГА. Образцы нагревали со скоростью 10°С/мин до конечной температуры.
В качестве дифференциального сканирующего калориметра использовали DSC Q200 или Discovery DSC 250 (ТА, США). Образцы точно взвешивали и помещали в лоток для образцов для ДСК с точечным отверстием, и записывали точную массу образцов. Образцы нагревали со скоростью 10°С/мин до конечной температуры.
Пример получения 1. Получение 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана
1.1 Получение 2,2'-диселенибис(бензойной кислоты)
(1) Получение диазония 2-хлорбензоата
4,0 г антраниловой кислоты смешали с 40 мл соляной кислоты в объемном соотношении 1:1. Систему перемешивали на ледяной бане, поддерживая температуру ниже 5°С, затем медленно и по каплям добавили к раствору нитрита натрия (25 г) в воде (20 мл) и выдерживали в течение 2 часов с получением диазония 2-хлорбензоата. Продукт использовали непосредственно на следующей стадии без очистки.
(2) К 120 мл воды добавили 12 г порошка селена и гидроксида натрия, медленно добавили 10 г гидросульфита натрия при перемешивании и выдерживали в течение 2 часов с получением раствора диселенида натрия. Раствор использовали непосредственно на следующей стадии без очистки.
(3) Раствор диазония 2-хлорбензоата, полученный на стадии (1), по каплям добавили к раствору диселенида натрия, полученному на стадии (2), при перемешивании, и смесь непрерывно перемешивали в течение 4 часов, пока полностью не прекратилось выделение азота. Реакционную смесь подкислили соляной кислотой, отфильтровали полученный осадок, промыли его водой и сушили в эксикаторе с получением 2,2'-диселенибис(бензойной кислоты). Температура плавления продукта после перекристаллизации составляла 294°С.
1Н-ЯМР (300 МГц, ДМСО-d6) δ: 7,33-8,04 (м, 4Н, Ph-H), 13,6 (шир., СООН);
ИК (КВr) см-1: (О-Н) 3005, (СО2) 1672, (C-N) 1264, С=С (фенильное кольцо) 1560, 1460, 1417;
MS-FAB (масс-спектрометрия с бомбардировкой ускоренными атомами) (m/z): 201 [1/2М+].
1.2 Получение 2-(хлорселено)бензоилхлорида
2,2'-диселенибис(бензойную кислоту) (40,0 г), 200 мл тионилхлорида и ДМФА перемешивали при кипячении с обратным холодильником в течение 3 часов, и затем избыток тионилхлорида отогнали на роторном испарителе. Остаток перекристаллизовали из н-гексана с получением 2-(хлорселено)бензоилхлорида с температурой плавления 66°С.
1Н-ЯМР (300 МГц, ДМСО-d6) δ: 7,33-8,16 (м, 4Н, Ph-H).
1.3 Получение 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана
3 мл 1,4-бутандиамина растворили в 50 мл ТГФ и добавили триэтиламин на ледяной бане и в атмосфере N2. 2,9 г2-(хлорселено)бензоилхлорида растворили в 60 мл ТГФ и медленно и по каплям добавили раствор 1,4-бутандиамина с получением твердого вещества желтого цвета. После добавления раствор нагрели до комнатной температуры и оставили реагировать в течение 2 часов. Твердое вещество отфильтровали и несколько раз промыли растворителем, этанолом и диэтиловым эфиром. Продукт перекристаллизовали из смеси ДМСО-вода, получив 2 г бледно-желтого твердого вещества с т.пл. 243-248°С.
1Н-ЯМР: (300 МГц, ДМСО-d6) δ: 7,37-8,04 (м, 4Н, Ph-H), 3,75 (с, 2Н, СН2), 1,64 (с, 2Н, СН2);
MS-FAB (m/z): 451 [М]+.
Пример 2. Получение кристаллической формы I
1,4-Бис[1,2-бензизоселеназол-3(2Н)-он]-бутан, полученный в примере получения 1, растворили в растворителе и добавили антирастворитель для получения кристаллической формы I. Объемы использованных растворителя и антирастворителя показаны в следующей таблице:
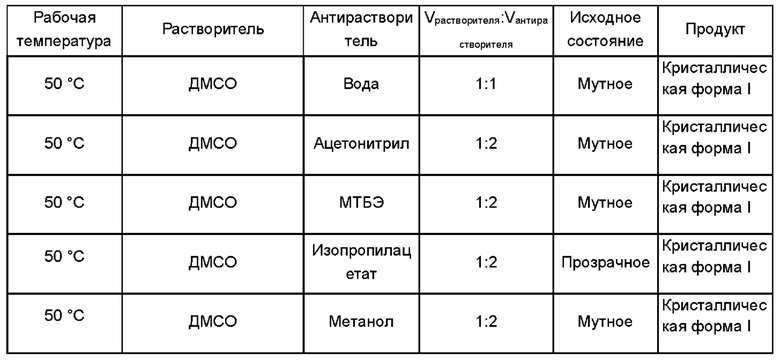
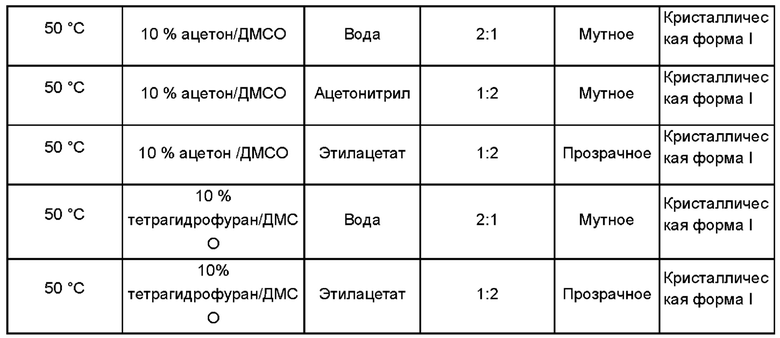
Дифрактограмма XRD кристаллической формы I показана на Фиг. 1. Термограмма ДСК-ТГА показана на Фиг. 2.
Дальнейшие испытания показали, что кристаллическая форма I представляет собой монокристалл, как показано на Фиг. 5, и свойства монокристалла являются следующими:
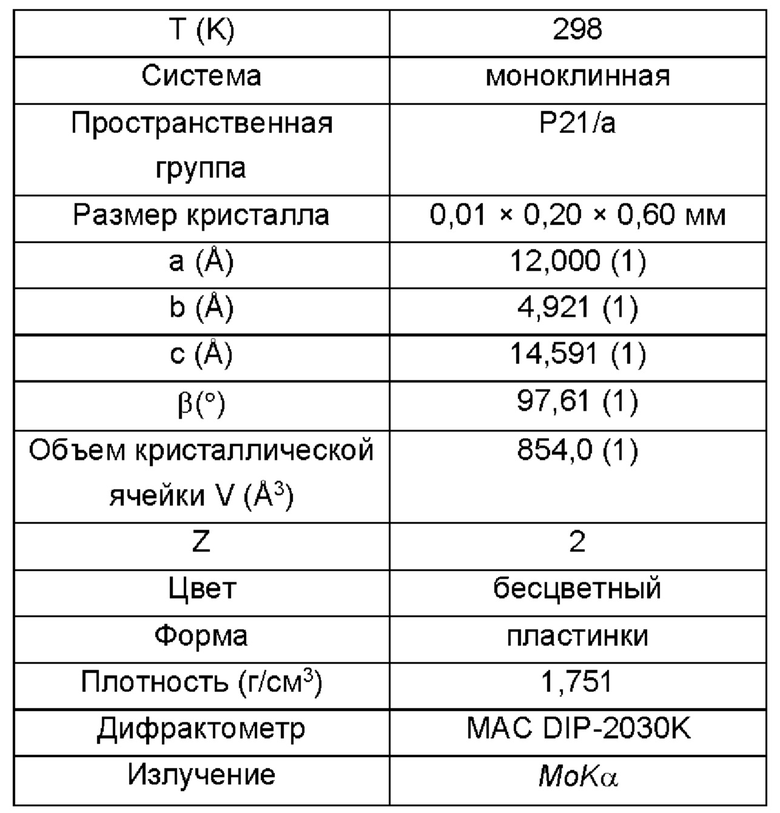

Сравнительный пример 1
1,4-Бис[1,2-бензизоселеназол-3(2Н)-он]-бутан, полученный в примере получения 1, растворили в ДМСО, и растворитель выпарили с получением кристаллической формы I-a, дифрактограмма XRD которой показана на Фиг. 3.
Сравнительный пример 2
1,4-Бис[1,2-бензизоселеназол-3(2Н)-он]-бутан, полученный в примере получения 1, растворили в хорошем растворителе ДМСО и добавили антирастворитель бутанон, этанол или этилацетат (объемное соотношение хороший растворитель:антирастворитель составляло 1:2). Из всех антирастворителей образовалась кристаллическая форма I-a.
Сравнительный пример 3
1,4-Бис[1,2-бензизоселеназол-3(2Н)-он]-бутан, полученный в примере получения 1, растворили в хорошем растворителе (10% ацетон/ДМСО или 10% тетра гидрофуран/ДМ СО) и добавили антирастворитель, этанол (объемное соотношение хорошего растворителя и антирастворителя составляло 1: 2). В обоих хороших растворителях образовалась кристаллическая форма I-a.
Испытательный пример 1. Испытание на стабильность кристаллической формы I
(1) Испытание на стабильность проводили в отношении полученной в примере 1 кристаллической формы I. Процедуры: образцы кристаллической формы I поместили в камеру для испытания на стабильность на 3-7 дней при 40°С/относительной влажности 75% и 60°С. Результаты показаны на Фиг. 4. Как видно из Фиг. 4, дифрактограмма XRD кристаллической формы I не изменилась после хранения кристаллической формы I в различных условиях, что демонстрирует, что кристаллическая форма I стабильна в условиях высокой температуры и высокой влажности и имеет хорошую стабильность.
(2) Кристаллическую форму I-a, полученную в сравнительных примерах 1-3, измельчили. С помощью рентгеновского порошкового дифракционного анализа было определено, что порошок представлял собой кристаллическую форму I. Таким образом, кристаллическая форма I, как было подтверждено, является стабильной кристаллической формой 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана.

Таким образом, кристаллическая форма I имеет хорошую стабильность, не подвержена полиморфному переходу, пригодна для изготовления фармацевтических композиций и препаратов, их применения, транспортировки и хранения, и полностью обеспечивает безопасность и качество лекарственных средств.
Испытательный пример 2. Анализ ингибирующей активности кристаллической формы I в отношении опухоли у мышей с использованием клеток Н22
Кристаллическую форму I вводили в дозе 180 мг/кг: 180 мг кристаллической формы I растворили в 5 мл 5%о раствора КМЦ-Na с получением суспензии, которую вводили в дозе 5 мл/кг.
Процедуры: Выбрали контрольную группу, которой вводили холостой раствор (5%о раствор КМЦ-Na один раз в день через желудочный зонд), и группу, получающую кристаллическую форму I (180 мг/кг один раз в день через желудочный зонд). После 2 дней акклиматизации подготовленных животных случайным образом разделили на 4 группы по 10 животных в каждой. В день О клетки Н22 прививали подкожно в количестве 1×104/мышь в правую подмышечную впадину. Лечение начали через 24 ч после прививки. Ежедневно регистрировали длинный и короткий диаметры опухоли и рассчитывали размер опухоли по формуле: длинный диаметр × короткий диаметр2 × 0,5236. Массу тела измеряли каждые 2 дня. Через 21 день от начала лечения взяли кровь из глазных яблок после анестезии хлоралгидратом. Мышей умерщвляли и опухоли быстро собирали. В конце эксперимента опухоли взвесили для обобщения с соответствующими статистическими данными, приведенными в следующей таблице:
Ингибирование роста опухоли кристаллической формой I у мышей с использованием клеток Н22 (n=10)
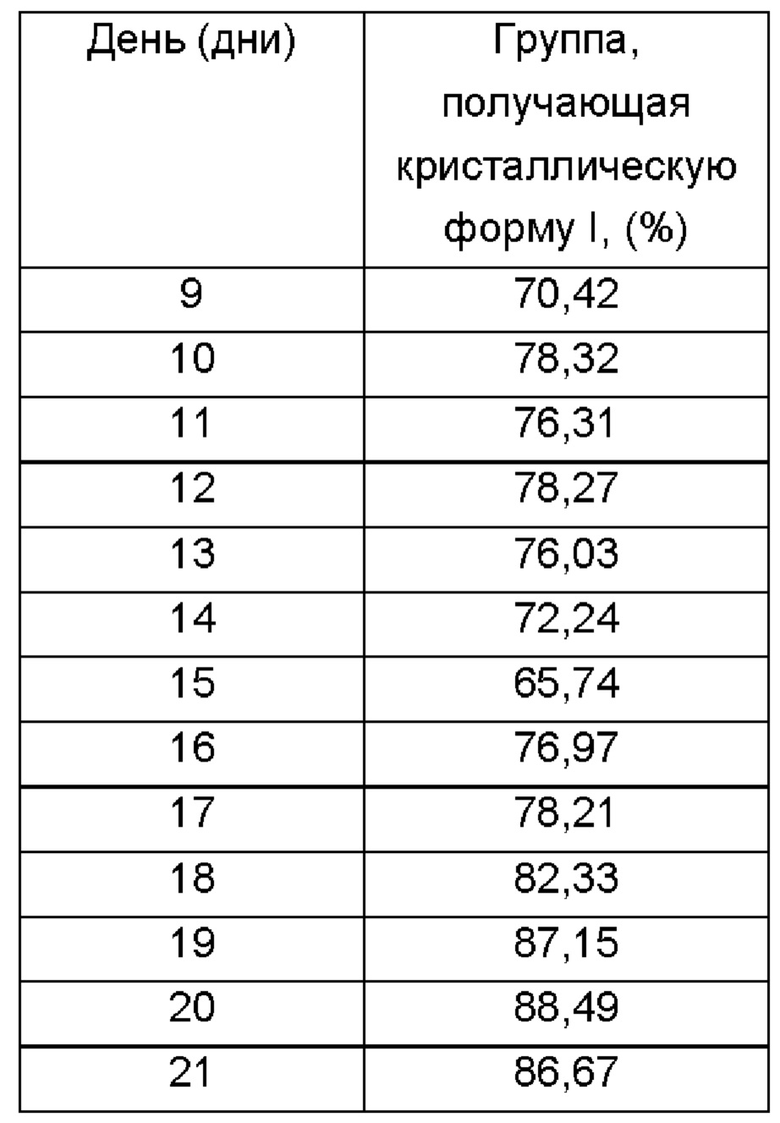
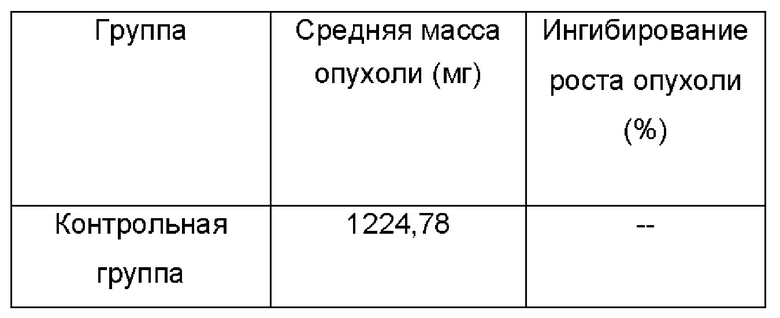
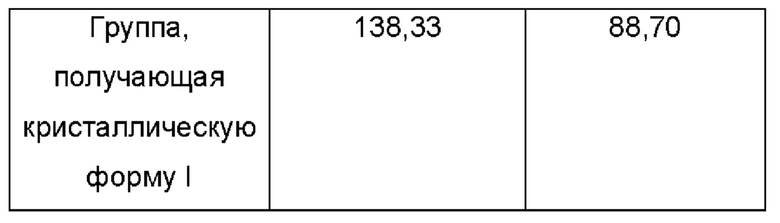
Из приведенных выше результатов видно, что ингибирование роста опухоли кристаллической формой I, полученной согласно настоящему изобретению, составляет до 88,7%. Предполагают, что конкретная кристаллическая форма дополнительно улучшает терапевтическую активность указанного соединения. Предположительно, причина может заключаться в том, что в кристаллической форме I значительно снижено количество примесей, которые неблагоприятны для активности или являются вредными, и кристаллическая форма I также обладает хорошей совместимостью с растворителями при введении, в результате чего облегчается абсорбция препарата.
Испытательный пример 3. Ингибирование HBV кристаллической формой I
Посев клеток: клетки HepAD38 высевали в 24-луночные планшеты с плотностью клеток, составляющей 8×104 на лунку, при этом каждая лунка содержала 500 мкл культуральной среды. Обработка: через 9 ч после посева производили наблюдение за клетками. Когда клетки прилипли к стенке, добавили лекарственные препараты (кристаллическую форму I или ETV (энтекавир)) с получением конечных концентраций 0,5, 1, 5, 20 и 50 мкМ. Клетки культивировали в течение 48 часов, дважды промыли PBS (фосфатно-солевой буферный раствор), добавили вышеуказанные препараты и культивировали еще 48 часов. Через 48 часов супернатант собрали и определяли ДНК HBV, HBsAg и НВеАд в супернатанте. Предпочтительно определяли ДНК HBV.
Анализ ДНК HBV в супернатанте клеточной культуры: согласно набору для экстракции вирусных нуклеиновых кислот от компании TransGen (TransGen's Viral Nucleic Acid Extraction kit), нуклеиновую кислоту экстрагировали и определяли количество вирусной ДНК с помощью способа SYBR green qPCR (Получение флуоресцирующего зеленым цветом комплекса ДНК с цианиновым красителем SYBR green для визуализации ДНК).
Анализ HBsAg и HBeAg в супернатанте клеточной культуры: анализ представлял собой иммунофлуоресцентный метод исследования с временным разрешением (ELISA) в соответствии с наборами для анализа HBsAg и HBeAg от компании РЕ Corporation.
Процедуры определения ДНК HBV, РНК HBV, HBsAg и HBeAg в супернатанте клеточной культуры были следующими:
1) Необходимое количество клеток высевали в 12-луночный планшет. Через 24 ч среду заменяли свежей средой, не содержащей двойных антител или ФБС (FBS). Плазмиды HBV трансфицировали с помощью Lipo2000. Через 6 часов среду заменяли средой, содержащей двойные антитела и 10% FBS, для получения непрерывной культуры.
Через 48 ч отобрали 1 мл супернатанта клеточной культуры для обнаружения HBsAg и HBeAg. Указанные клетки промыли по меньшей мере 5 раз с помощью PBS. Отобрали 1 мл промывочного раствора после последней промывки с помощью PBS и использовали его в качестве базового уровня для определения степени вымывания плазмид. Примечание: при промывании клеток PBS медленно добавляли в чашку вдоль стенки чашки без ресуспендирования клеток. Затем чашку осторожно встряхивали для промывания клеток, и оставшийся PBS удаляли после каждой промывки, чтобы минимизировать количество оставшихся плазмид.
Клетки перенесли в большую чашку для культивирования и культивировали в культуральной среде в течение 3 дней. Для обнаружения ДНК HBV и РНК HBV отобрали 1 мл супернатанта клеточной культуры. Если обнаружение не производили сразу, то образцы хранили при -20°С.
1 мл отобранного супернатанта клеточной культуры и последний промывочный раствор PBS, которые были отобраны через 48 часов, центрифугировали при 5000 об/мин в течение 5 минут. 200 мкл супернатанта после центрифугирования добавили к 5 мкл ДНКазы I (дезоксирибонуклеаза I) + 5 мкл буфера для ДНКазы I, хорошо перемешали и подвергли кратковременному центрифугированию. Супернатант инкубировали в течение 1 ч при 37°С, перемешивали каждые 30 мин и подвергли кратковременному центрифугированию.
Через 1 час добавили 5 мкл ЭДТА и систему инкубировали при 65°С в течение 10 минут, прежде чем реакция с дезоксирибонуклеазой I была остановлена.
200 мкл нуклеиновой кислоты HBV в супернатанте клеточной культуры сконцентрировали до системы объемом 20 мкл с использованием набора для экстракции нуклеиновых кислот HBV от компании TransGene.
Уровень ДНК HBV в супернатанте и базовый уровень плазмид в PBS после обработки ДНКазой измеряли с помощью теста ПЦР в режиме реального времени. После обратной транскрипции РНК HBV в кДНК провели тест ПЦР в режиме реального времени для определения указанного уровня.
(1) Количественная система ДНК HBV:

Всего 27 мкл (аккуратное перемешивание, кратковременное центрифугирование)

Положение праймера было 386-476 в S-области.
(2) количественная система кДНК после обратной транскрипции РНК HBV:


Всего 27 мкл (аккуратное перемешивание, кратковременное центрифугирование)

Количественная последовательность праймера была создана перед С-доменом/в С-домене (2297-2287).
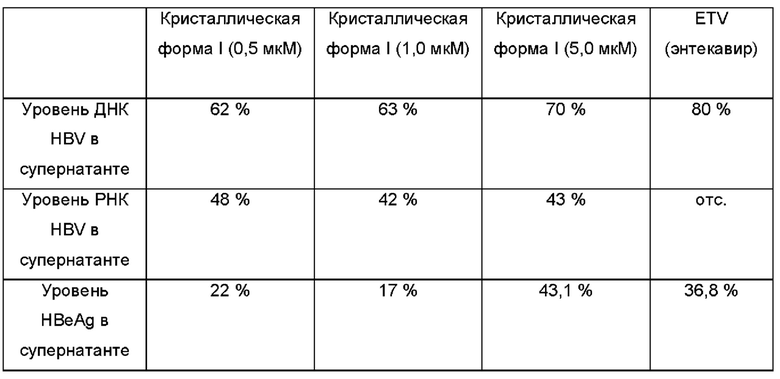
Кристаллическая форма I обладает хорошим ингибирующим действием на ДНК HBV и супернатантный HBeAg. Таким образом, с помощью кристаллической формы I можно более эффективно лечить рак печени, вызванный HBV.
Испытательный пример 4
Исследуемый препарат: кристаллическая форма, полученная в примере 1.
Животные: инбредные мыши-самцы Balb/c, в возрасте шести недель, были приобретены в Пекинском Университете, Научный центр здравоохранения, Департамент лабораторных исследований животных, номер лицензии SCXK (Jing) 2016-0010. Среда для размножения была чистой. Температура в помещении составляла 25±2°С. Мышей разводили в 12-часовом цикле свет/темнота, при этом они получали достаточное количество пищи и воды.
Приготовление раствора для исследования на животных:
Приготовление раствора КМЦ-Na

Смесь перемешивали с высокой скоростью мешалки. КМЦ-Na первоначально находился в виде твердого вещества, напоминающего хлопок, и постепенно растворился после перемешивания с образованием раствора, используемого в качестве растворителя.
Приготовление раствора кристаллической формы I (BS)
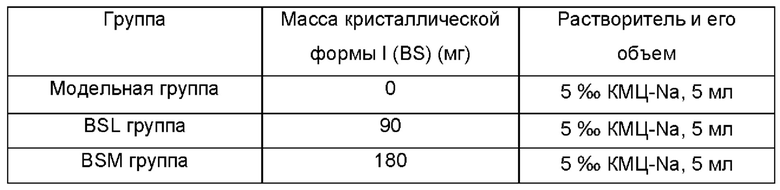
После приготовления раствора или суспензии соединение вводили мышам в дозе 5 мл/кг.
Приготовление 25% раствора ССЦ
Смесь перемешивали с высокой скоростью мешалки и после хорошего перемешивания хранили при комнатной температуре.
Материалы:
BS1801 (Peking University, School of Pharmaceutical Sciences), линия клеток mHSC (Коллекция культур BeNa, Хэбэй, Китай), линия клеток AML12, фетальная бычья сыворотка (Gibco), среда DMEM (М&С), среда F12, TGF-(31 (Novoprotein), смесь культурного фактора клеток (Sigma), дексаметазон (Solarbio), четыреххлористый углерод (Beijing Ouhe Technology Co., Ltd.), оливковое масло (Macklin), сульфородамин В (Sigma), параформальдегид (Yuanye), Трис (Ameresco), натрий-карбоксиметилцеллюлоза (КМЦ-Na) и фиксатор тканей животных.
Процедуры:
1. Культура клеток и анализ жизнеспособности клеток
Жиронакапливающие клетки печени мышей (mHSC) культивировали с использованием 20% FBS в термостате при 37°С, 5% СО2. В эксперименте по пролиферации клеток плотность клеток составляла 5000 клеток на лунку, обработку проводили после индукции с применением TGF-β1 с концентрацией 5 нг/мл в течение 24 часов, а ингибирование пролиферации клеток выявляли с помощью сульфородамина В (SRB).
2. Иммуноблоттинг
Клетки mHSC перенесли в чашки из расчета 106 клеток на чашку и обрабатывали через 12 ч после адгезии. Через 48 ч клетки в супернатанте собрали и объединили с адгезивными клетками. Клетки подвергали лизису на льду в течение 30 минут с использованием лизата RIPA, содержащего ингибитор протеаз, и центрифугировали при 16000 об/мин в течение 15 минут, при этом супернатант сохранили. Концентрацию клеточного лизата определяли с помощью БСА (ВСА) и добавляли определенное количество загрузочного буфера для приготовления образца белка с концентрацией 2,5 мкг/мкл. Содержание a-SMA, коллагена I и коллагена III определяли с помощью ДСН-ПААГ (SDS-PAGE).
3. Исследование на животных
20 мышей-самцов Balb/c в возрасте 6 недель были рандомизированы в 4 группы. После ступенчатой градиентной индукции с использованием 25% CCU в течение 4 недель (три раза в неделю) приступили к лечению группы BSL (90 мг/кг) и группы BSM (180 мг/кг) один раз в день, а также контрольной и модельной группам вводили 5%о раствор КМЦ-Na. После 8 недель индукции мышей умерщвляли, собирали кровь и центрифугировали при 3000 об/мин в течение 15 минут, чтобы получить сыворотку для определения TGF-β1, TrxR1, AST, ALT, ALP, TP и экспрессии (модель фиброза печени у животных показана на Фиг. 6). Часть печени фиксировали 4% параформальдегидом и подвергли иммуногистохимическому окрашиванию гематоксилином и эозином (НЕ) и окрашиванию по Массону (Masson). Определяли экспрессию α-SMA и коллагена I в ткани печени иммуногистохимически. Часть печени консервировали в жидком азоте.
На Фиг. 7 показаны измеренные значения IC50, и из результата на Фиг. 7 видно, что, поскольку TGF β1 играет очень важную патологическую роль в фиброзных поражениях, а клетки HSC состоят на 5% из нормальных клеток печени, повреждение печени обычно приводит к трансформации и активации клеток HSC, при этом TGF β1 является сильным фактором фиброза. Ингибирующий эффект кристаллической формы по настоящему изобретению на клетки HSC является одним из важных факторов противофиброзного эффекта.
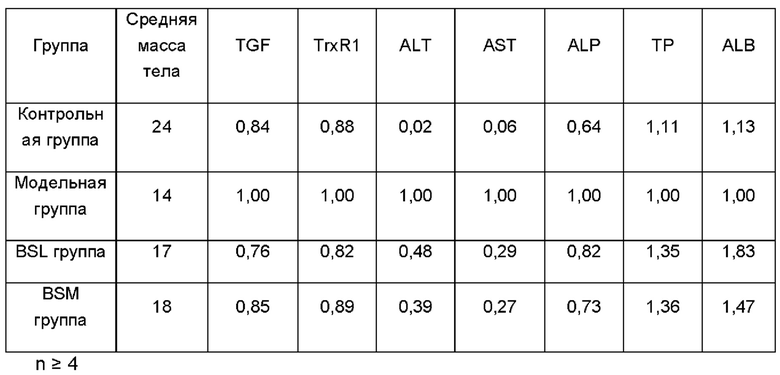
На Фиг. 8 показаны в сравнении различные биохимические показатели и важные белковые маркеры в сыворотке после введения мышам с модельным фиброзом препарата BS (соответствующие результаты показаны в следующей таблице). Как видно из Фиг. 8 и результатов, приведенных в нижеследующей таблице, средняя масса тела мышей в модельной группе и группах лечения была значительно снижена, особенно в модельной группе, по сравнению с таковой в контрольной группе. В конце исследования средняя масса тела в каждой из четырех групп составляла 24,76±4,84 г, 14,08±1,51 г, 17,00±0,68 г и 18,04±2,23 г, соответственно.
Таблица 1. Масса тела и биохимические показатели сыворотки крови мышей для всех групп по окончании исследования
На Фиг. 9 показаны результаты иммуногистохимического окрашивания печени и тканей после проведенных экспериментов над контрольной и модельной группами, а также группой, получающей препарат BS в низкой дозе (BSL), и группой, получающей препарат BS в средней дозе (BSM). Как видно из результатов группы (А), мыши в контрольной группе продемонстрировали гладкую, глянцевую, темно-красную, мягкую поверхность печени, что указывает на нормальное состояние печени. В конце исследования мыши в модельной группе показали снижение активности, потерю массы до 43,2%, матовую, светлую, твердую и зернистую поверхность печени и фиброзное состояние, что свидетельствует об успешном создании модели. Указанные симптомы в печени мышей в группах лечения улучшились, и зернистость на поверхности печени мышей уменьшилась.
Результаты окрашивания с помощью гематоксилина и эозина (НЕ) показывают, что клетки печени демонстрировали сильный отек и сильное воспаление в модельной группе по сравнению с контрольной группой, и большая часть граничных пластин клеток печени была разрушена (>50%); напротив, в группах BSL и BSM симптомы некроза и воспаления гепатоцитов были значительно уменьшены по сравнению с модельной группой.
Результаты окрашивания по Массону показывают, что мыши в модельной группе продемонстрировали мостовидный фиброз между портальными областями, а также образовались фиброзные промежутки; в группах BSL и BSM симптомы постепенно уменьшились по сравнению с модельной группой; контрольная группа продемонстрировала только несколько волокон вокруг портальной области.
На Фиг. 10 показаны экспрессия (A) α-SMA и (В) коллагена 1А1 в печени для различных групп мышей в конце эксперимента на модели фиброза печени при различных концентрациях кристаллической формы I. На Фиг. 10 по результатам окрашивания α-SMA и окрашивания коллагена 1 видно, что уровень экспрессии соответствующих белков в печени мышей в модельной группе был значительно больше по сравнению с контрольной группой, в то время как данные симптомы в группах BSL и BSM были значительно снижены, что явно значительно отличает их от модельной и контрольной групп.
Таким образом, настоящее изобретение обеспечивает кристаллическую форму I 1,4-бис[1,2-бензизоселеназол-3(2Н)-он]-бутана. Кристаллическая форма I имеет хорошую стабильность, пригодна для изготовления фармацевтических композиций и препаратов, их применения, транспортировки и хранения, и полностью обеспечивает безопасность и качество лекарственных средств.
Кроме того, указанная кристаллическая форма отличается высокой растворимостью, подходящей биодоступностью, низкой токсичностью и отличной ингибирующей активностью в отношении опухолей, в частности, отличается общим ингибированием у пациентов с раком печени, имеющих вирус HBV, а также эффектами функционального ингибирования в процессах фиброза печени и фиброза сердца, онкогенеза и роста опухоли. Таким образом, указанная кристаллическая форма выполняет защитную функцию в отношении всего процесса от заражения вирусом HBV до рака печени.
Наконец, способ получения кристаллической формы I, раскрытый в данном документе, отличается простотой, хорошей воспроизводимостью, высоким выходом и пригодностью для промышленного производства, поэтому он имеет большую прикладную ценность.
Примеры согласно настоящему изобретению были описаны выше. Однако настоящее изобретение не ограничивается приведенными выше примерами. Любая модификация, эквивалент, усовершенствование и т.п., сделанные без отклонения от сущности и принципов настоящего изобретения, подпадают под объем охраны настоящего изобретения.
Изобретение относится к способу получения кристаллической формы I 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана, имеющей характеристические пики при углах 2θ, составляющих 6,15 ± 0,20°, 12,28 ± 0,20°, 18,44 ± 0,20°, 19,09 ± 0,20°, 21,89 ± 0,20°, 22,20 ± 0,20°, 23,68 ± 0,20°, 25,92 ± 0,20°, 27,50 ± 0,20°, 30,95 ± 0,20°, 32,45 ± 0,20°, 35,79 ± 0,20°, 37,37 ± 0,20° и 37,72 ± 0,20°, при проведении порошковой рентгеновской дифракции с использованием излучения Cu-Kα. Способ включает растворение 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана в растворителе S1 и добавление антирастворителя S2 с получением кристаллической формы I. Растворитель S1 выбран из ДМСО и смеси, состоящей из ДМСО и 1-10% ацетона или тетрагидрофурана (об./об.); растворитель S2 выбран из воды, ацетонитрила, МТБЭ, изопропилацетата, метанола и этилацетата. При этом, когда растворителем S1 является ДМСО, растворитель S2 не является этилацетатом; объемное соотношение растворителя S1 и растворителя S2 составляет 1:0,5–1:2; и способ осуществляют при температуре 30–70°C. Технический результат - получение кристаллической формы I 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана, имеющей хорошую стабильность, низкую токсичность и ингибирующую активность в отношении рака печени, ассоциированного с HBV. 2 з.п. ф-лы, 10 ил., 1 табл., 9 пр.
1. Способ получения кристаллической формы I 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана, имеющей характеристические пики при углах 2θ, составляющих 6,15 ± 0,20°, 12,28 ± 0,20°, 18,44 ± 0,20°, 19,09 ± 0,20°, 21,89 ± 0,20°, 22,20 ± 0,20°, 23,68 ± 0,20°, 25,92 ± 0,20°, 27,50 ± 0,20°, 30,95 ± 0,20°, 32,45 ± 0,20°, 35,79 ± 0,20°, 37,37 ± 0,20° и 37,72 ± 0,20°, при проведении порошковой рентгеновской дифракции с использованием излучения Cu-Kα, при этом способ включает:
растворение 1,4-бис[1,2-бензизоселеназол-3(2H)-он]-бутана в растворителе S1 и добавление антирастворителя S2 с получением кристаллической формы I;
где растворитель S1 выбран из ДМСО и смеси, состоящей из ДМСО и 1-10% ацетона или тетрагидрофурана (об./об.);
растворитель S2 выбран из воды, ацетонитрила, МТБЭ, изопропилацетата, метанола и этилацетата при условии, что, когда растворителем S1 является ДМСО, растворитель S2 не является этилацетатом;
объемное соотношение растворителя S1 и растворителя S2 составляет 1:0,5–1:2; и
способ осуществляют при температуре 30–70°C.
2. Способ по п. 1, в котором кристаллическая форма I характеризуется дифрактограммой рентгеновской порошковой дифракции (XRD), показанной на Фиг. 1, или эквивалентна кристаллической форме, соответствующей указанной дифрактограмме XRD.
3. Способ по п. 1 или 2, в котором кристаллическая форма I представляет собой монокристалл, имеющий следующие монокристаллические свойства:
| CN 1704409 A, 07.12.2005 | |||
| Jie He; Dongdong Li et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Электрический ртутный выключатель | 1924 |
|
SU3816A1 |
| Способ автоподстройки частоты источника питания резонансной колебательной системы | 1988 |
|
SU1511834A1 |
| CN 101781283 A, 21.07.2010 | |||
| ПРОИЗВОДНЫЕ БИСБЕНЗИЗОСЕЛЕНАЗОЛОНИЛА С ПРОТИВООПУХОЛЕВЫМ, ПРОТИВОВОСПАЛИТЕЛЬНЫМ И АНТИТРОМБОТИЧЕСКИМ ДЕЙСТВИЕМ И ИХ ПРИМЕНЕНИЕ | 2002 |
|
RU2324688C2 |
