Область техники, к которой относится изобретение
Настоящее изобретение относится к биотехнологии, а именно, к клеточной линии и способу для проверки работоспособности и сравнения активности существующих нуклеаз-генетических редакторов по их эффективности внесения двуцепочечных разрывов в живой клетке.
Уровень техники
Для успешного использования нуклеаз в геномном редактировании требуется предварительно оценивать эффективность внесения двуцепочечных разрывов такими нуклеазами в целевые места генома. Как правило, оценку эффективности редактирования с использованием того или иного геномного редактора (система CRISPR-Cas, TALEN, нуклеазы с доменами цинковых пальцев) проводят по следующей схеме: культуру клеток трансфицируют геном нуклеазы (для системы CRISPR-Cas также геном гидовой РНК), а затем всю популяцию анализируют на предмет наличия клеток, в которых произошла та или иная мутация, так как при репарации двуцепочечных разрывов ДНК могут возникать мутации. Оценив долю мутантных аллелей относительно не мутантных (wt, дикий тип), делают вывод об эффективности редактирования. Среди отредактированных молекул ДНК встречаются как нуклеотидные замены, так и инсерции или делеции, причем различной длины, а также хромосомные перестройки.
На данный момент проверка работоспособности инструментов редактирования генома осуществляется различными способами. Все они направлены на выявление последствий образования двуцепочечных разрывов - мутаций, которые могут возникнуть в точке редактирования.
Известен способ оценки эффективности редактирования генома Next generation sequencing (NGS, секвенирование нового поколения) [Bell C.C. et al., 2014. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing // BMC Genomics, Vol 15, Issue 1, p.1002]. В этом случае ДНК из популяции клеток, в качестве которых используют культивируемые клетки млекопитающих и в которых была активна проверяемая нуклеаза, анализируется путем секвенирования нового поколения на подходящей платформе с целью количественно оценить эффективность внесения мутаций в целевом сайте. NGS (секвенирование нового поколения) для оценки эффективности инструмента редактирования генома обладает следующими недостатками: для секвенирования образцов требуется приготовление библиотеки секвенирования, что занимает несколько часов, а также сложный биоинформатический анализ результатов секвенирования.
Известен способ T7E1-анализа [Mashal R.D. et al., 1995. Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases // Nat. Genet., Vol 9, Issue 2, pp. 177-183] и SURVEYOR [Qiu P. et al., 2004. Mutation detection using Surveyor™ nuclease // BioTechniques. Vol 36 Issue 4, pp. 702-707]. Как в случае T7E1-анализа, так и SURVEYOR, участок ДНК клеток млекопитающих, в которых активна проверяемая нуклеаза, в районе точки редактирования амплифицируют с помощью ПЦР. Затем ПЦР-продукты нагревают для денатурации и медленно охлаждают для ренатурации. В связи с тем, что в ПЦР-смеси присутствуют разные молекулы ДНК одни содержат индели в результате NHEJ, а другие представляют собой неотредактированные молекулы, - после ренатурации формируются гетеродуплексы. Чем эффективнее нуклеаза вносит разрыв, тем больше образуется инделей и больше гетеродуплексных молекул ДНК сформируется после ренатурации. Затем ПЦР-продукты обрабатывают нуклеазами T7E1 или SURVEYOR и продукты разрезания анализируют электрофорезом в агарозном или полиакриламидном геле. По отношению интенсивности полос продуктов разрезания и интактных молекул вычисляют эффективность внесения ДЦР нуклеазой. Однако результат этих способов сильно зависит от типа возникающих мутаций, поскольку они по-разному узнаются нуклеазами T7E1 или SURVEYOR. В конечном счете это делает неточной оценку эффективности, так как многие события редактирования могут остаться незамеченными.
Известен способ RFLP (полиморфизм длин рестрикционных фрагментов) для оценки эффективности редактирования генома [Urnov F.D. et al., 2005. Highly efficient endogenous human gene correction using designed zinc finger nucleases // Nature, Vol 435 Issue 7042, pp. 646-651]. При использовании этого способа РНК-гиды изначально подбирают так, чтобы точка расщепления оказалась внутри сайта узнавания какой-нибудь рестриктазы. Из смешанной популяции клеток млекопитающих, в которых активна проверяемая нуклеаза, после трансфекции кодирующим ее вектором спустя сутки-двое выделяют ДНК и амплифицируют область, включающую точку редактирования. Затем ПЦР-продукт обрабатывают рестриктазой и анализируют электрофоретически. Полосы соответствуют продуктам расщепления и нерасщепленными молекулам, которые несут мутации в сайте узнавания рестриктазой вследствие возникновения инделей. По соотношению интенсивностей полос делают вывод об эффективности внесения разрыва. Однако в данном способе мутация должна попадать на сайт рестрикции, иначе способ будет неприменим. Поэтому данный способ ограничен в том, какие нуклеазы можно проверять с его помощью.
Известен способ TIDE (метод отслеживания инделей через декомпозицию) [Brinkman E.K. et al., 2014. Easy quantitative assessment of genome editing by sequence trace decomposition// Nucleic Acids Res., Vol 42, Issue 22, pii: e168]. Способ отслеживания инделей через декомпозицию (tracking of indeles by decomposition, TIDE) основывается на биоинформатическом анализе результатов сэнгеровского секвенирования ПЦР-продуктов амплификации редактируемого локуса в культуре трансфицированных клеток млекопитающих. Этот способ позволяет предсказывать частоты различных делеций и инсерций, возникающих в редактируемом локусе. Однако способ работает только в случае небольшого разнообразия возникающих мутаций. При большом разнообразии алгоритмы декомпозиции не могут выделить отдельные варианты инсерций и делеций, что не позволяет провести оценку эффективности инструмента редактирования генома.
Известен способ IDAA (анализ ампликонов с инделями) [Yang Z. et al., 2015. Fast and sensitive detection of indels induced by precise gene targeting // Nucleic Acids Res., Vol 43, Issue 9, pii: e59]. Как и описанные ранее способы, IDAA предполагает амплификацию области редактирования с фланкирующих праймеров. В одном из праймеров содержится флуорофор. После амплификации ПЦР-продукты разделяют методом капиллярного электрофореза и детектируют по флуоресценции, оценивая соотношение ПЦР-продуктов с делециями и ПЦР-продуктов нормального размера. Однако для реализации способа необходим прибор для капиллярного электрофореза. Разрешающей способности капиллярного электрофореза бывает недостаточно для различения +1 и -1 инделей с аллелями дикого типа. Нет возможности детектировать замены нуклеотидов и транслокации с участием локуса редактирования.
Известны способы оценки эффективности редактирования генома - pDR/FACS (от названия плазмиды pDR и FACS - fluorescence-activated cell sorting) [Menzorov A.G. et al., 2016. Genome editing using CRISPR/ Cas9 system: A practical guide // Russ. J. Genet.: Appl. Res., Vol 20, Issue 6, pp. 930-944]. Способ pDR/FACS отличается от описанных ранее, так как эффективность редактирования в нем оценивают не по частоте мутаций, возникающих в ходе NHEJ, а по частоте гомологичной рекомбинации - другого пути репарации ДЦР. В этом способе клетки млекопитающих трансфицируют плазмидой, содержащей ген проверяемой нуклеазы, а также плазмидой pDR. Плазмида pDR включает в себя 2 копии кодирующих последовательностей зеленого флуоресцентного белка (GFP), но обе они не функциональны. Первая копия содержит сайт узнавания проверяемой нуклеазы, сдвигающий рамку считывания, а вторая содержит лишь центральную область гена gfp, но с правильной последовательностью. Копии разделены последовательностью длиной почти 4 тыс.п.н., что делает возможным использование одной копии в качестве матрицы для репарации другой копии после внесения в нее разрыва нуклеазой. В случае успешной работы нуклеазы в дефектный ген gfp будет внесен разрыв, и после гомологичной рекомбинации с участием второго гена gfp образуется полноценный ген. Эффективность репарации оценивают с помощью FACS-анализа клеток.
Известен способ оценки эффективности ENIT (индуцированные нуклеазами транслокации) [Germini D. et al., 2017. A one-step PCR-based assay to evaluate efficiency and precision of genomic DNA-editing tools // Mol. Ther. Methods Clin. Dev., Vol 5, Issue 6, pp. 43-50]. Способ ENIT (engineered nuclease-induced translocations) основан на том, что при внесении нескольких разрывов в ДНК образуются хромосомные транслокации. Клетки из культуры клеток млекопитающих трансфицируют двумя плазмидами. Первая плазмида содержит ген нуклеазы, для которой ранее уже была показана высокая эффективность внесения разрывов. Эта нуклеаза направлена на уникальный участок генома (участок ДНК “X”). Вторая плазмида содержит ген проверяемой нуклеазы, направленной на другой уникальный участок генома (участок ДНК “Y”). Если проверяемая нуклеаза эффективно вносит разрывы, то между локусами X и Y возникают хромосомные транслокации, которые могут быть детектированы с помощью ПЦР. Сравнение эффективности гидов может быть проведено по разнице в интенсивности полос на электрофорезе или количественной ПЦР. Однако способ дает только качественную оценку и не обнаруживает инсерции, делеции и точечные мутации.
Способ qEva основан на лигировании двух половинок олигонуклеотидной пробы, подобранной к участку редактирования, и определении количества продуктов лигирования [Dabrowska M. et al., 2018. qEva-CRISPR: a method for quantitative evaluation of CRISPR/Casmediated genome editing in target and off-target sites // Nucleic Acids Res., Vol 46, Issue 17, pii: e101]. Проба подбирается так, что половинки отжигаются на ДНК клеток млекопитающих, в которых активна проверяемая нуклеаза, с двух сторон от точки редактирования, и их концы оказываются рядом. ДНК клеток, трансфицированных для редактирования, смешивают с олигонуклеотидными пробами и проводят реакцию лигирования. Затем количество продуктов лигирования оценивают, амплифицируя их с помощью ПЦР и анализируя на капиллярном электрофорезе. Если происходит мутация, то конец половинки пробы не отжигается на ДНК и лигирования половинок пробы не происходит, что приводит к отсутствию ПЦР-продукта. Количество мутантной ДНК нормируется на общее количество ДНК, для чего в ту же реакцию лигирования добавляют состоящую из двух половинок пробу к интактному участку генома и праймеры для ее амплификации. Однако для реализации способа существует необходимость в длинных олигонуклеотидных пробах, что приводит к увеличению времени на проведение реакции лигирования, и не очень высокая чувствительность (от 5% мутантных клеток в популяции).
Все вышеуказанные ранее способы оценки эффективности действия нуклеаз-геномных редакторов дают только непрямую оценку эффективности работы нуклеаз по возникающим мутациям, и не позволяют напрямую оценить частоту внесения двуцепочечного разрыва в клетках. Такие способы дают заведомо заниженную оценку, поскольку не все возникающие в ДНК двуцепочечные разрывы приводят к мутациям - часть из них репарируется без ошибок. В то же время базовой характеристикой геномного редактора является именно способность вносить разрывы в ДНК. Другим общим недостатком способов оценить эффективность действия нуклеаз по результатам репарации разрывов являются большие затраты времени (2-3 дня с момента трансфекции), так как необходимо дополнительное время на осуществление клетками репарации разрывов.
Таким образом, техническая проблема, решаемая посредством заявляемого изобретения, заключается в необходимости преодоления недостатков, присущих аналогам и прототипу за счет разработки способа, позволяющего количественно оценить эффективность внесения двуцепочечного разрыва ДНК различными нуклеазами - геномными редакторами в культуре клеток. Решение такой проблемы актуально в связи с развитием технологий редактирования генома и появлением новых нуклеаз, которые потенциально могут использоваться для редактирования генома, например бактериальные белки Argonaute (Ago).
Раскрытие изобретения
Техническим результатом изобретения является разработка клеточной линии и способа, которые позволяют с помощью микроскопа напрямую визуализировать результаты работы нуклеазы - образование разрывов в ДНК, что возможно уже через 12 часов после трансфекции клеток кодирующими нуклеазу конструкциями. Заявляемый способ является достоверным, наглядным и более быстрым, чем существующие способы проверки эффективности нуклеаз-редакторов.
Технический результат достигается клеточной линией HCT116_dCas9_PCP_sgRNA-TL_53BP1, где sgRNA-TL - гидовая РНК, визуализирующая целевой локус, на который направлена проверяемая нуклеаза, и ген белка-маркера двуцепочечных разрывов ДНК 53BP1, который слит с флуоресцентным белком. Клеточная линия создана на основе клетки млекопитающих, скорость деления которых составляет не менее одного деления в 2-3 суток, в качестве клеток млекопитающих используют клетки человека, или мыши, или кролика. Для визуализации целевого локуса (TL) используют систему для прижизненной визуализации локусов хроматина в клетках, в качестве которой используют систему CRISPR-имаджинга, основанную на связывании каталитически неактивного белка dCas9 с гидовыми РНК, имеющими аптамерные последовательности, связываемые белками, слитыми с флуоресцентным белком. При этом в качестве элементов визуализации целевого локуса используют ген dCas9, ген направляющей sgRNA_TL-8xPP7 и ген визуализирующего слитого белка PCP-sfGFP, в качестве флуоресцентных белков используют sfGFP и tagBFP.
Технический результат также достигается способом получения клеточной линии HCT116_dCas9_PCP_sgRNA-TL_53BP1, заключающийся в том, что предварительно получают 3 плазмидных вектора для интеграции генных кассет в геном клеток, проводят лентивирусную трансдукцию, с последующим отбором трансдуцированных клеток HCT116_dCas9_PCP_sgRNA-TL_53BP1 с помощью клеточного сортинга в флуоресцентных каналах, детектирующих флуоресценцию sfGFP и tagBFP, а также с помощью селектирующего антибиотика пуромицина. При этом плазмидные векторы содержат последовательности, необходимые для упаковки в лентивирусные частицы, а также интегрируемые кассеты: в первом - ген dCas9 и ген устойчивости к селектирующему антибиотику пуромицину, во втором - ген белка PCP, слитого с sfGFP, в третьем - ген визуализирующей целевой локус РНК, и ген белка 53BP1, слитого с tagBFP.
Также технический результат достигается способом оценки эффективности нуклеаз-геномных редакторов с использованием клеточной линии HCT116_dCas9_PCP_sgRNA-TL_53BP1, клетки которой трансфицируют плазмидами, кодирующими проверяемые нуклеазы-редакторы и РНК- или ДНК-гиды, направляющие нуклеазу на целевой локус, и спустя 24-48 часов оценивают с помощью конфокальной микроскопии долю целевых локусов, в которые был внесен разрыв, по колокализации фокусов tagBFP и sfGFP; эффективность нуклеазы-редактора оценивают по доле клеток, в которых сигналы от целевого локуса и сигналы от 53BP1 колокализованы после трансфекции плазмидами, кодирующими необходимые элементы проверяемой системы редактирования генома, по сравнению с нетрансфицированными клетками. При этом трансфекцию проводят методом электопорации, для чего прикрепленные на поверхности культуральной емкости клетки линии HCT116_dCas9_PCP_sgRNA_53BP1 снимают с поверхности с помощью раствора трипсина, суспендируют в буфере для электропорации и электропорируют согласно протоколу, а после трансфекции наносят на стекла для дальнейшей микроскопии. Трансфекцию проводят с использованием нуклеопоратора Neon (ThermoFisher), программы для электропорации клеток HCT116: 2 импульса тока напряжением 1050 В, длительностью 30 мс каждый. Долю целевых локусов, в которые внесен разрыв, оценивают по колокализации фокусов tagBFP и sfGFP в клетках HCT116_dCas9_PCP_sgRNA_53BP1, трансфицированных согласно п.11, а колокализацию оценивают по пересечению сигналов-фокусов tagBFP и sfGFP, при этом подсчет проводят среди 100 сигналов в клетках HCT116_dCas9_PCP_sgRNA_53BP1, трансфицированных методом электопорации и среди 100 сигналов не трансфицированных клеток, после чего значение процента колокализации для контроля вычитают из значения процента колокализации для опыта.
Способ оценки основан на использовании культуры генетически модифицированных клеток, в которых визуализирован локус-мишень, на который направляется проверяемая нуклеаза, и которые экспрессируют флуоресцентный белок-репортер двуцепочечных разрывов ДНК. Такие клетки трансфицируют векторами с генами проверяемых нуклеаз или самими нуклеазами и далее с помощью флуоресцентной микроскопии оценивают процент колокализации локусов-мишеней с белком-репортером двуцепочечных разрывов ДНК. Вычисленный процент колокализации служит мерой эффективности работы проверяемой нуклеазы-геномного редактора.
Краткое описание чертежей
Изобретение поясняется следующими чертежами.
На фиг.1 представлена схема создания клеточной линии, иллюстрирующая получение линии HCT116_dCas9_PCP_sgRNA_53BP1: лентивирусную трансфекцию клеток НСТ116 элементами системы CRISPR-имаджинга и 53ВР1-tagBFP. Ниже показана схема эксперимента для количественной оценки эффективности нуклеаз-геномных редакторов. В качестве примера - плазмиды, кодирующие систему CRISPR/Cas или же нуклеазы TALEN или нуклеаза на основе белков-аргонавтов (Ago).
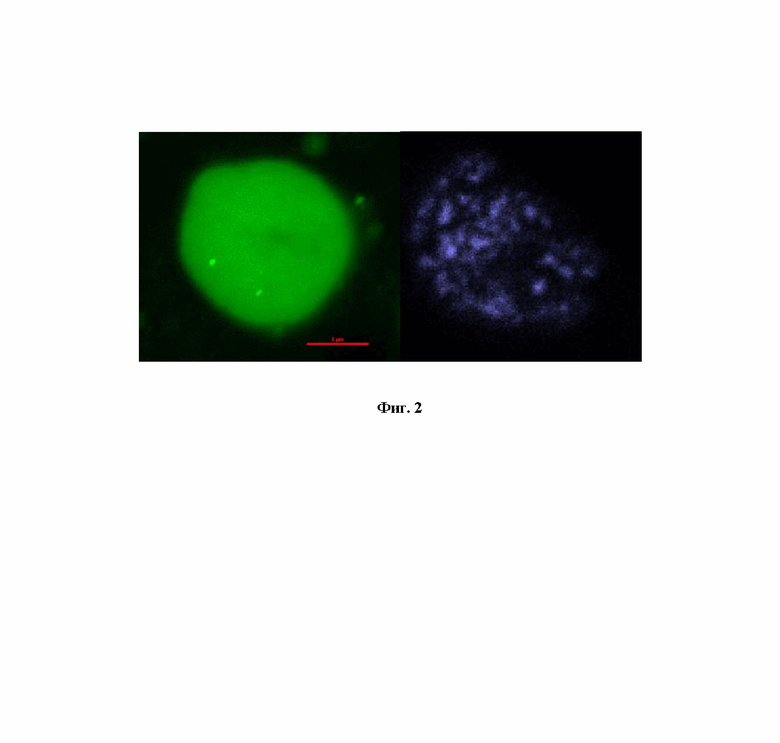
На фиг.2 представлена микроскопия ядер клеток HCT116, экспрессирующих элементы системы CRISPR-Sirius: dCas9, PCP-sfGFP и sgRNA_TL_8xPP7 (слева), и экспрессирующих 53ВР1-tagBFP (справа).
Осуществление изобретения
Ниже представлено более подробное описание заявляемого изобретения. Настоящее изобретение может подвергаться различным изменениям и модификациям, понятным специалисту на основе прочтения данного описания. Такие изменения не ограничивают объем притязаний. Например, могут изменяться тип клеток, используемых для создания описываемой клеточной линии, способ трансфекции клеток при создании описываемой клеточной линии, схемы плазмидных векторов, конкретный белок репарации, который используется для визуализации разрыва, флуоресцентные белки, которые используются в данном способе и т.д.
Предлагаемый способ позволяет оценивать эффективность внесения разрывов проверяемой нуклеазой-редактором генома по колокализации разрывов с белком репарации 53BP1 в культуре клеток человека. В качестве такой культуры может использоваться иммортализованная прикрепленная клеточная культура HCT116, HeLa, HEK293, DU145 и другие, а также суспензионные клеточные линии Jurkat, HL60, Daudi, Raji, в которые методами генной инженерии добавлены гены элементов системы CRISPR-имаджинга для визуализации локусов генома. В такой культуре визуализируется эндогенный геномный локус, например, субтеломерный локус на 19 хромосоме, имеющий координаты Chr19: 380000-383000 (сборка генома человека hg19), который далее в тексте будет обозначен как TL (target-locus). В этом локусе содержится 45 повторов последовательности AGCAGATGTAGGNGG. Для визуализации этого локуса с помощью опубликованного метода CRISPR-имаджинга, который называется CRISPR-Sirius [Ma H. et al., 2018. CRISPR Sirius: RNA scaffolds for signal amplification in genome imaging // Nat. Methods., Vol 15. Issue 11, pp. 928-931], в клетках должно экспрессироваться три гена: ген каталитически неактивного белка Cas9 из бактерии Streptococcus pyogenes (dCas9, dead Cas9), ген белка PCP, слитого с зеленым флуоресцентным белком superfolderGFP (sfGFP) - PCP-sfGFP, а также ген гидовой РНК для dCas9, узнающая часть которой представлена последовательностью AGCAGATGTAGG, а к константной части добавлено восемь шпилек-аптамеров PP7, последовательности которых опубликованы [Ma H. et al., 2018. CRISPR Sirius: RNA scaffolds for signal amplification in genome imaging // Nat. Methods., Vol 15. Issue 11, pp. 928-931]. Указанные гены доставляются в клетки и интегрируются в геном с помощью лентивирусной трансдукции с использованием самоинактивирующихся лентивирусных векторов второго поколения либо другими способами трансфекции, например, с помощью транспозазы или с помощью других типов трансдукции. В клетках образуется комплекс из dCas9 и гидовой РНК, который связывается с локусом TL. С PP7-повторами в составе гидовой РНК связывается белок PCP-sfGFP. В результате на локусе TL формируется агрегат флуоресцентных белков, который при флуоресцентной микроскопии обнаруживается как флуоресцентный сигнал.
Кроме того, клетки также должны экспрессировать ген белка 53BP1, слитого с флуоресцентным белком, например с белком tagBFP или mTagBFP, shBFP, mKalama1 и другие. Белок 53BP1 связывается с двуцепочечными разрывами в клетках [Roukos V. et al., 2013. Spatial Dynamics of Chromosome Translocations in Living Cells // SCIENCE Vol 341, Issue 6146, pp.660-664; Wikiniyadhanee R. et al., 2020. TRIM29 is required for efficient recruitment of 53BP1 in response to DNA double-strand breaks in vertebrate cells // FEBS Open Bio., Vol 10, Issue 10, pp.2055-2071]. Использование белка 53BP1-tagBFP при флуоресцентной микроскопии, таким образом, позволяет визуализировать двуцепочечные разрывы ДНК в ядре. Ген этого белка доставляется в клетки и интегрируется в геном с помощью лентивирусной трансдукции с использованием самоинактивирующихся лентивирусных векторов второго поколения либо другими способами трансфекции, например, с помощью транспозазы или с помощью других типов трансдукции.
Для оценки эффективности внесения разрывов проверяемая нуклеаза-редактор генома (далее, нуклеаза) направляется на участок ДНК рядом с локусом TL. Для этого в полученные клетки, экспрессирующие PCP, слитый с sfGFP (или другим флуоресцентным белком, отличающимся по спектру от первого - EGFP, YFP, mClover3, fusionRed, RFP и другие), dCas9 и гидовую РНК с PP7-повторами, либо трансфицируется ген нуклеазы и, при необходимости, направляющей молекулы нуклеиновой кислоты, либо трансфцируется сам белок (при необходимости, в комплексе с направляющей молекулой нуклеиновой кислоты). Трансфекцию данной линии клеток генами проверяемой нуклеазы проводят любым доступным способом, например, с помощью реактивов Turbofect Transfection Reagent (Thermo Fisher Scientific), Lipofectamine 3000 (Invitrogen), Effectene (Qiagen), электропорация на приборах Neon Transfection System (Thermo Fisher Scientific) или Gene Pulser (Bio-Rad) и т.д. - по инструкции производителя. Если проверяемая нуклеаза работает, она вносит двуцепочечный разрыв в целевой локус. Спустя 12-24 часа после трансфекции клетки изучаются на флуоресцентном микроскопе в канале флуоресценции sfGFP и в канале флуоресценции tagBFP. В качестве микроскопа можно использовать любой микроскоп, обеспечивающий возможность детектировать флуоресценцию используемых белков и имеющий разрешение от 512×512 пикселей при увеличении от 40-кратного и выше. По изображениям, полученным путем наложения этих каналов, вычисляется доля локусов TL (визуализированных с помощью CRISPR-имаджинга, канал sfGFP) колокализуется с сигналом 53BP1-tagBFP (канал tagBFP). Кроме трансфицированных клеток, также анализируются нетрансфицированные клетки, для вычисления фоновой колокализации. Для расчета параметра эффективности проверяемой нуклеазы, из доли колокализованных локусов в трансфицированных клетках отнимается доля колокализованных локусов в контрольных клетках. То есть расчет проводят по формуле: E = NT - NK, где NT - доля сигналов TL, которые колокализуются с сигналом 53BP1-tagBFP в трансфицированных клетках, NK - доля сигналов TL, которые колокализуются с сигналом 53BP1-tagBFP в нетрансфицированных клетках. Таким образом, значения N находятся в пределах от 0 до 1, а Е - от -1 до +1. Если значение Е>0, то нуклеаза (геномный редактор) эффективна, причем чем выше значение, тем более она эффективна. Если значение Е = 0 или меньше, то нуклеаза не эффективна.
Для реализации заявляемого способа оценки эффективности нуклеаз необходимо было предварительно получить клеточную линию, которая бы экспрессировала гены компонентов системы визуализации целевого локуса, а также ген белка-репортера двуцепочечных разрывов. В качестве технологии визуализации используют способ визуализации, основанный на системе CRISPR/Cas, который называется CRISPR-Sirius [Ma H. et al., 2018. CRISPR Sirius: RNA scaffolds for signal amplification in genome imaging // Nat. Methods., Vol 15. Issue 11, pp. 928-931]. В качестве целевого локуса был выбран субтеломерный локус на 19 хромосоме, имеющий координаты Chr19: 380000-383000 (сборка генома человека hg19), который далее в тексте будет обозначен как TL (target-locus). В этом локусе содержится 45 повторов последовательности AGCAGATGTAGGNGG. Узнающая последовательность РНК-гида, визуализирующего целевой локус TL: AGCAGATGTAGG.
Для осуществления визуализации использовали плазмиду, содержащую указанную выше узнающую последовательность гидовой РНК. Такая плазмида представляет собой трансферный вектор для самоинактивирующихся лентивирусов второго поколения, в котором присутствует узнающая последовательность гидовой РНК AGCAGATGTAGG, следом за которой идет константная часть гидовой РНК для Cas9 из Streptococcus pyogenes к дуплексу repeat-antirepeat которой добавлено восемь пермутированных вариантов PP7-шпильки, которые удовлетворяют консенсусной последовательности SNAGCAGANSATATGGSNTCGCTNS. Дизайн гидовой РНК, содержащей 8 пермутированных повторов PP7, опубликован [Ma H. et al., 2018. CRISPR Sirius: RNA scaffolds for signal amplification in genome imaging // Nat. Methods., Vol 15. Issue 11, pp. 928-931]. Ген гидовой РНК находится под повтором человеческого промотора U6. Рядом с геном гидовой РНК находится ген устойчивости к антибиотику гигромицину под контролем человеческого промотора гена фософглицераткиназы (hPGK). С использованием такого трансферного вектора получают лентивирусные частицы, которые далее будут использованы для трансдукции. Для получения лентивирусных частиц клетки культуры HEK293T трансфицировали описанной трансферной плазмидой, а также плазмидой, кодирующей ген белка оболочки вируса везикулярного стоматита VSV-G, и пакующей плазмидой для лентивирусов второго поколения. Трансфекцию проводили с помощью реактива Turbofect Transfection Reagent (Thermo Fisher Scientific) по инструкции производителя. Спустя 4 дня после трансфекции отбирали среду, в которой культивировались клетки, и профильтровывали ее через 0,22 мкм фильтр. Полученный образец представлял собой суспензию лентивирусных частиц, которые далее использовали для трансдукции. Все работы проводили стерильно в ламинарном боксе в помещении, удовлетворяющем требованиям по работе с лентивирусными векторами (BSL-2).
Кроме того, использовали плазмиду, которая представляет собой трансферный вектор для самоинактивирующихся лентивирусов второго поколения, в котором есть ген каталитически неактивной кодон-оптимизированной для клеток человека Cas9 из бактерии Streptococcus pyogenes, содержащей мутации D10A и H840A (dCas9), к которой через T2A-пептид добавлена последовательность, кодирующая фермент, обеспечивающий устойчивость к антибиотику пуромицину. Этот ген находится под контролем сильного промотора CMV, соединенного с энхансером CMV. С использованием такого трансферного вектора получают лентивирусные частицы, которые далее использованы для трансдукции. Получение лентивирусных частиц описано выше.
Также была использована плазмида, которая представляет собой трансферный вектор для самоинактивирующихся лентивирусов как минимум второго поколения, в котором присутствует ген белка PCP фага PP7, слитого с зеленым флуоресцентным белком superfolder GFP (sfGFP). Этот ген находится под контролем сильного промотора EF-1a. С использованием такого трансферного вектора получают лентивирусные частицы, которые далее были использованы для трансдукции. Получение лентивирусных частиц описано выше.
Описанные три типа лентивирусных частиц последовательно добавляли к культуре клеток человека клеточная линия HCT116, которая обладала следующими свойствами: клетки были прикреплены (адгезированы к поверхности культуральной посуды), клетки были иммортализованы, клетки делились на реже одного раза в 2-3 дня. Культивирование линии проводили при стандартных лабораторных условиях: в среде DMEM c добавлением 10% эмбриональной телячьей сыворотки, в инкубаторе с 5% CO2 в атмосфере при 37°C. Для пересева культуры использовали раствор 0,05% трипсина с ЭДТА (220 мг/л), приготовленный на фосфатном солевом буфере (PBS).
Трансдукцию осуществляли следующим образом: сначала добавляли лентивирусы с dCas9, потом лентивирусы с PCP-sfGFP и затем лентивирусы с гидовой РНК к локусу TL и PP7-повторами (sgRNA_TL-8xPP7). После каждой трансдукции проводили этап селекции трансдуцированных клеток на третий день после трансдукции. В случае трансдукции вирусами с dCas9 клетки пересевали на среду с пуромицином в концентрации 0,2 мкг/мл. Параллельно на такую же среду засевали контрольные (нетрансдуцированные) клетки. Селекцию трансдуцированной культуры считали завершенной, когда в контрольной культуре на среде с антибиотиком не остается живых клеток, а в трансдуцированных клетках видны колонии живых клеток. В случае трансдукции вирусами с sgRNA_TL-8xPP7 клетки пересевали на среду с гигромицином в концентрации 150 мкг/мл. Параллельно на такую же среду засевали контрольные (нетрансдуцированные) клетки. Селекцию трансдуцированной культуры считали завершенной, когда в контрольной культуре на среде с антибиотиком не остается живых клеток, а в трансдуцированных клетках видны колонии живых клеток. В случае трансдукции вирусами с PCP-sfGFP GFP-позитивные клетки отбирали с помощью клеточного сортера FACSAria SORP (BD). Полученная клеточная культура, которая экспрессирует гены dCas9, PCP-sfGFP и sgRNA_TL-8xPP7, далее называется HCT116_dCas9_PCP_sgRNA.
Для визуализации двуцепочечных разрывов в клетках полученной клеточной культуры использовали белок 53BP1, слитый с флуоресцентным белком tagBFP (53BP1-tagBFP). Для этого плазмиду, представляющую собой трансферный вектор для самоинактивирующихся лентивирусов второго поколения, в котором закодирован ген белка 53BP1-tagBFP под контролем сильного промотора CMV, соединенного с энхансером CMV. С использованием такого трансферного вектора получают лентивирусные частицы, которые далее используют для трансдукции культуры HCT116_dCas9_PCP_sgRNA. Получение лентивирусных частиц описано выше. Спустя два дня после трансдукции tagBFP-позитивные клетки отбирали с помощью клеточного сортера FACSAria SORP (BD). Полученная клеточная культура, которая экспрессирует гены dCas9, PCP-sfGFP, sgRNA_TL-8xPP7 и 53BP1-tagBFP будет далее называться HCT116_dCas9_PCP_sgRNA_53BP1.
Проверку эффективности исследуемой нуклеазы-геномного редактора (далее - нуклеазы) проводили следующим образом. В клетки HCT116_dCas9_PCP_sgRNA_53BP1 трансфицировали плазмиду с геном проверяемой нуклеазы и (при использовании нуклеаз, требующих направляющей молекулы нуклеиновой кислоты, например системы CRISPR-Cas) также плазмиду с геном направляющей РНК к визуализируемому локусу. Трансфекцию проводили путем электропорации с использованием прибора Neon Transfection system (Thermo Fisher Scientific) по инструкции производителя по следующим параметрам: напряжение - 1050 В, число импульсов тока - 2, длительность каждого импульса - 30 мс. Для трансфекции использовали 3-5 мкг каждой плазмиды и 1 млн клеток и 100 мкл носы из комплекта прибора. Трансфицированые клетки засевали на 35 мм культуральные чашки со стеклянным дном и спустя 12 часов анализировали на флуоресцентном микроскопе Nikon Eclipse Ti с конфокальным модулем A1, объективом CFI Apochromat TIRF 60x Oil, детектором «A1-DU4», лазерами Sapphire 488 для возбуждения флуоресценции sfGFP и Cobolt 06-MLD 405 nm для возбуждения флуоресценции tagBFP, а также системой для прижизненной микроскопии клеток. При микроскопировании клетки находятся в CO2 -камере и на подогреваемом столик при температуре 37°C. Управление микроскопом осуществляли с помощью программного обеспечения NIS Elements. Разрешение снимков - не менее 512x512 пикселей. Для анализа необходимо проанализировать не менее 100 клеток, содержащих сигналы sfGFP - визуализированные целевые локусы TL. Для каждого визуализированного локуса TL определяли, есть ли колокализация с сигналом tagBFP. На основании анализа получали значение доли сигналов TL в трансфицированных клетках HCT116_dCas9_PCP_sgRNA_53BP1, которые колокализуются с сигналом 53BP1-tagBFP - NT = 0,45. Кроме трансфицированных клеток HCT116_dCas9_PCP_sgRNA_53BP1, на микроскопе также изучали нетрансфицированные клетки, для которых также определяли долю сигналов TL, которые колокализуются с сигналом 53BP1-tagBFP - NK = 0,04. Значение эффективности нуклеазы определяли по формуле E = NT - NK = 0,45 - 0,04 = 0,41. Так как данное значение Е > 0, то нуклеаза считается эффективной.

Изобретение относится к области биотехнологии, в частности к клеточной линии, в которой клетки линии содержат следующие гены: ген белка-маркера двуцепочечных разрывов ДНК 53BP1, который слит с флуоресцентным белком, ген каталитически неактивного белка Cas9 (dCas9), ген белка PCP, слитого с зеленым флуоресцентным белком, ген гидовой РНК для dCas9, который связывается с локусом TL, узнающая часть которой представлена последовательностью AGCAGATGTAGG, а к константной части добавлено восемь шпилек-аптамеров PP7, а также к способу ее получения. Изобретение эффективно для оценки эффективности нуклеаз-геномных редакторов. 3 н. и 10 з.п. ф-лы, 2 ил.
1. Клеточная линия HCT116_dCas9_PCP_sgRNA-TL_53BP1 для оценки эффективности нуклеаз-геномных редакторов, в которой клетки линии содержат следующие гены: ген белка-маркера двуцепочечных разрывов ДНК 53BP1, который слит с флуоресцентным белком, ген каталитически неактивного белка Cas9 (dCas9), ген белка PCP, слитого с зеленым флуоресцентным белком, ген гидовой РНК для dCas9, который связывается с локусом TL, узнающая часть которой представлена последовательностью AGCAGATGTAGG, а к константной части добавлено восемь шпилек-аптамеров PP7.
2. Клеточная линия по п. 1, характеризующаяся тем, что для визуализации целевого локуса (TL) используют систему для прижизненной визуализации локусов хроматина в клетках.
3. Клеточная линия по п. 1, характеризующаяся тем, что для прижизненной визуализации локусов хроматина в клетках используют систему CRISPR-имаджинга, основанную на связывании каталитически неактивного белка dCas9 с гидовыми РНК, имеющими аптамерные последовательности, связываемые белками, слитыми с флуоресцентным белком.
4. Клеточная линия по п. 1, характеризующаяся тем, что в качестве элементов визуализации целевого локуса используют ген dCas9, ген направляющей sgRNA_TL-8xPP7 и ген визуализирующего слитого белка PCP-sfGFP.
5. Клеточная линия по п. 1, характеризующаяся тем, что в качестве флуоресцентных белков используют sfGFP и tagBFP.
6. Способ получения клеточной линии по п. 1, характеризующийся тем, что предварительно получают 3 плазмидных вектора для интеграции генных кассет в геном клеток, проводят лентивирусную трансдукцию, с последующим отбором трансдуцированных клеток HCT116_dCas9_PCP_sgRNA-TL_53BP1 с помощью клеточного сортинга в флуоресцентных каналах, детектирующих флуоресценцию белков, слитых с геном белка PCP и геном белка 53BP1, а также с помощью селектирующего антибиотика, устойчивость к которому кодируется в генетических конструкциях, при этом плазмидные векторы содержат интегрируемые кассеты: в первом - ген dCas9 и ген устойчивости к селектирующему антибиотику пуромицину, во втором - ген белка PCP, слитого с зеленым флуоресцентным белком, в третьем - ген визуализирующей целевой локус РНК, и ген белка 53BP1, слитого с флуоресцентным белком.
7. Способ по п. 6, характеризующийся тем, что в качестве клеток используют клетки человека, скорость деления которых составляет не менее одного деления в 2-3 суток.
8. Способ по п. 6, характеризующийся тем, что ген белка PCP слит с зеленым флуоресцентным белком sfGFP, ген белка 53BP1 слит с флуоресцентным белком tagBFP.
9. Способ оценки эффективности нуклеаз-геномных редакторов, характеризующийся тем, что используют клеточную линию HCT116_dCas9_PCP_sgRNA-TL_53BP1 по п.1, клетки которой трансфицируют плазмидами, кодирующими проверяемые нуклеазы-редакторы и РНК- или ДНК-гиды, направляющие нуклеазу на целевой локус, и спустя 24−48 часов оценивают с помощью конфокальной микроскопии долю целевых локусов, в которые был внесен разрыв, по колокализации фокусов используемых флуоресцентных белков, слитых с белками PCP и 53BP1; эффективность нуклеазы-редактора оценивают по доле клеток, в которых сигналы от целевого локуса и сигналы от 53BP1 колокализованы после трансфекции плазмидами, кодирующими необходимые элементы проверяемой системы редактирования генома, по сравнению с нетрансфицированными клетками.
10. Способ по п.9, характеризующийся тем, что трансфекцию проводят методом электопорации, для чего прикрепленные на поверхности культуральной емкости клетки линии HCT116_dCas9_PCP_sgRNA_53BP1 снимают с поверхности с помощью раствора трипсина, суспендируют в буфере для электропорации и электропорируют согласно протоколу, а после трансфекции наносят на стекла для дальнейшей микроскопии.
11. Способ по п.10, характеризующийся тем, что используют нуклеопоратор Neon (ThermoFisher), программу для электропорации клеток HСТ116: 2 импульса тока напряжением 1050 В, длительностью 30 мс каждый.
12. Способ по п.10, характеризующийся тем, что долю целевых локусов, в которые внесен разрыв, оценивают по колокализации фокусов флуоресцентных белков, слитых с белками PCP и 53BP1 в трансфицированных клетках HCT116_dCas9_PCP_sgRNA_53BP1, а колокализацию оценивают по пересечению сигналов-фокусов флуоресцентных белков.
13. Способ по п.12, характеризующийся тем, что подсчет проводят среди 100 сигналов в клетках HCT116_dCas9_PCP_sgRNA_53BP1, полученной согласно п.6, и среди 100 сигналов нетрансфицированных клеток, после чего значение процента колокализации нетрансфицированных клеток вычитают из значения процента колокализации для опыта.
| MAGDALENA DABROWSKA et al., qEva-CRISPR: a method for quantitative evaluation of CRISPR/Cas-mediated genome editing in target and off-target sites, Nucleic Acids Res, 2018 Sep 28;46(17):e101, doi: 10.1093/nar/gky505 | |||
| DIEGO GERMINI et al., A One-Step PCR-Based Assay to Evaluate the Efficiency and Precision of Genomic DNA-Editing Tools, Mol Ther |

