Область техники, к которой относится изобретение
Настоящее изобретение относится к медицинским технологиям, а именно, к технологиям поиска новых терапевтических препаратов или комбинаций препаратов (химических соединений), улучшающих существующие терапевтические подходы к лечению рака. В настоящее время в химиотерапии онкологических заболеваний используются ингибиторы топоизомераз. Пациенты, прошедшие лечение препаратами данной группы, рискуют в течение нескольких лет получить вторичный лейкоз. Данное заболевание развивается примерно у каждого двадцатого пациента. Развитие заболевания напрямую связано с возникновением определенных хромосомных перестроек (транслокаций). Проблема вторичных лейкозов может быть существенно снижена в случае обнаружения веществ, снижающих вероятность лейкозогенных хромосомных перестроек. Для обнаружения такого действия у различных веществ или комбинаций веществ предложена тест-система, к которой добавляется исследуемый препарат (тестируемое вещество), а затем по числу возникших транслокаций по сравнению с контролем делается вывод о риске возникновения вторичного лейкоза. Тест-система может быть адаптирована под высокопроизводительный скрининг молекул в поисках наиболее эффективной с точки зрения способности снижения числа транслокаций. Вещество или комбинация веществ, у которого(-ых) обнаруживается способность существенно снижать число транслокаций, может применяться (при условии прохождения всех необходимых клинических испытаний) в комбинированной терапии, то есть добавляться к уже существующим химиотерапевтическим препаратам, причем всем, которые могут приводить к возникновению заболеваний, вызванных хромосомными перестройками.
Уровень техники
На сегодня существует проблема вторичных лейкозов. Этот тип лейкоза развивается как побочный эффект химиотерапии с применением препаратов из класса ингибиторов топоизомераз (этопозид, доксирубицин). Данный вид терапии является одним из самых эффективных, и поэтому получил широкое распространение. Причиной вторичного лейкоза становятся определенные межхромосомные перестройки, происходящие при ошибочной репарации двуцепочечных разрывов ДНК, которые возникают вследствие применения ингибиторов топоизомераз. Снижение уровня подобных транслокаций означает снижение вероятности развития вторичных лейкозов. Для поиска веществ, которые способны снижать уровень таких транслокаций, предложена клеточная модель, представляющая культуру клеток, на которой возможно быстро и эффективно моделировать возникновение одной из хромосомных транслокаций, ассоциированных со вторичными лейкозами.
Известно создание культуры клеток, в которых была воспроизведена с помощью системы CRISPR/Cas подобная транслокация AML-ETO (Torres R., Martin М.С., Garcia А., Cigudosa J.C, Ramirez J.С, Rodriguez-Perales S., 2014. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system // Nat. Commun. V. 5. P. 1-8). Полученные клетки изучались на предмет наличия данной транслокации методом FISH и на предмет экспрессии химерного гена на месте транслокации. Авторы работы делают вывод о возможности создания с помощью системы CRISPR/Cas моделей для изучения онкологических заболеваний. Однако данная модель не ориентирована на изучение самого процесса возникновения хромосомной транслокации. Поэтому с ее помощью невозможно испытывать различные химические вещества на предмет снижения вероятности возникновения данной транслокации.
Отдаленными аналогами тест-системы могут быть клеточные линии, в которых в гены AML и ЕТО внесены мутации с применением технологии CRISPR/Cas: www.horizondiscovery.com/human-runx1-20bp-deletion-knockout-cell-line-hzghc004198c004 и www.horizondiscovery.com/human-rmix1t1-knockout-cell-line-hzghc862. Данные клеточные линии могут использоваться для изучения функций генов, но не для изучения процесса возникновения транслокаций.
Изогенные клеточные линии (т.е. имеющих одно происхождение), воспроизводящие ассоциированные с определенными видами рака мутациями (BRAF, KRAS, PIK3CA и EGFR), создают ученые из Италии (https://www.ncbi.nlm.nih.gov/pubmed/19106301) и предлагают стратегию испытаний лекарств с помощью панели этих изогенных мутантных линий https://www.ncbi.nlm.nih.gov/pubmed/20016269. Авторы описывают пример обнаружения с помощью своего метода веществ - потенциальных терапевтических агентов, обладающих терапевтическим действием. При этом из уровня техники не выявлены тест-системы, с помощью которых было бы возможно определять процесс возникновения хромосомной транслокации, ассоциированной со вторичными лейкозами.
Раскрытие изобретения
Технической проблемой, решаемой настоящим изобретением, является создание тест-системы для исследования терапевтического агента (активнодействующего вещества/компонента или комбинации веществ), используемого как компонент комбинированной химиотерапии, на предмет снижения риска возникновения заболеваний, вызванных межхромосомными перестройками, например, вторичного лейкоза, который проявляется как отдаленный побочный эффект при использовании препаратов из класса ингибиторов топоизомераз. Это одни из наиболее широко применяемых химиотерапевтических препаратов при лечении онкологических заболеваний.
Техническим результатом изобретения является выявление терапевтического агента (активнодействующего вещества/компонента/препарата или комбинации), использование которого при химиотерапии может привести к снижению риска возникновения вторичного лейкоза, при этом выявление агента осуществляют посредством детектирования и подсчета лейкозогенных хромосомных транслокаций заявлемой тест-системой после взаимодействия с исследуемым препаратом и сравнения с отрицательным контролем.
Тест-система предполагает поиск в первую очередь универсального терапевтического агента, который мог бы снизить риск заболеваний, вызванных образованием хромосомных транслокаций.
Тест-система представляет из себя культуру генетически модифицированных клеток, в которых запускаются по сигналу извне искомые хромосомные транслокации. Моделируемыми транслокациями могут быть любые перестройки между разными хромосомами или участками одной хромосомы. Для повышения релевантности тест-системы предпочтительнее моделировать транслокации, вероятность возникновения которых необходимо снизить. Например, это могут быть вызывающие вторичный лейкоз транслокации - AML-ETO или другие. После добавления к системе тестируемых веществ и активации системы, искомые транслокации детектируются методом количественной ПЦР с использованием подобранных предварительно праймеров и параметров реакции. Базовый уровень формирования данных транслокаций при активации системы сравнивается с уровнем транслокаций, возникающих при добавлении в момент активации тестируемых веществ. Тест-система позволяет количественно оценивать влияние веществ на частоту транслокаций, а следовательно, достоверно сравнивать вещества между собой по этому параметру. Система адаптируется для процедуры высокопроизводительного скрининга, например, скрининга библиотек химических агентов в поисках наиболее эффективного вещества.
Поставленная проблема решается заявляемой тест-системой для исследования влияния препарата (вещества или комбинации веществ), используемого при химиотерапии, на вероятность возникновения вторичного лейкоза, представляющая собой линию клеток млекопитающего, геном которых содержит генетическую конструкцию, включающую гены систем направленного внесения двуцепочечных разрывов в хромосомы клетки в локусы, между которыми происходит лейкозогенная хромосомная перестройка, а также гены активатора транскрипции ТА и гены селективных маркеров, при этом гены направленного внесения двуцепочечных разрывов, находятся под контролем индуцируемого трансактиватором ТА промотора. При этом в качестве линии клеток млекопитающих используют клетки, скорость деления которых составляет не менее одного деления в 2-3 суток и геном которых изучен настолько, чтобы было возможным подобрать нуклеазы, узнающие локусы, между которыми формируются хромосомные перестройки, например, в качестве линии клеток млекопитающих могут быть использованы лимфобластоидные клетки человека (LCL) RPMI 8866. В качестве генов систем направленного внесения двуцепочечных разрывов используют эндонуклеазу Cas9 и гидовые РНК, при этом в качестве линии клеток млекопитающих, например клетки человека, мыши или любых других млекопитающих, используют клетки, скорость деления которых составляет не менее одного деления в 2-3 суток. В качестве системы, позволяющей вносить направленные двуцепочечные разрывы, используют систему CRISPR/Cas, то есть нуклеаза Cas9 и гидовые РНК, узнающие локусы внутри генов, между которыми возникает хромосомная перестройка, по изменению частоты которой оценивается эффективность вещества или комплекса веществ, в качестве которой выбрана перестройка между генами AML1 и ETO. В качестве системы контроля над экспрессией элементов системы, вносящей специфический разрыв, используют систему Tet-ON, включающую в себя индуцируемый промотор TRE, контролирующий экспрессию гена Cas9, индуцируемый белком трансактиватором, находящимся в комплексе с доксициклином, при этом в качестве селективных маркеров, позволяющих отличить клетки, несущие трансгенные конструкции, используют гены устойчивости к антибиотикам пуромицину и неомицину. Гены системы, позволяющей вносить направленные двуцепочечные разрывы, интегрированы в геном клеток исходной линии в локус, где они будут стабильно экспрессироваться, например, в области первого интрона гена PPP1R12C, расположенного на 19 хромосоме.
Поставленная проблема также решается способом получения заявляемой тест-системы, который заключается в создании генетических конструкций, содержащих гены системы, позволяющей под действием внешнего воздействия вносить направленные двуцепочечные разрывы в нужные локусы, например, генетической конструкции (1), включающей элементы CRISPR/Cas: гены гидовых РНК gRNA_AML1 и gRNA_ETO, ген нуклеазы Cas9, находящийся под индуцируемом промотором TRE, и ген устойчивости к пуромицину, и генетической конструкции (2), включающей ген активатора транскрипции ТА и ген устойчивости к генетицину, с последующими трансфекцией полученных генетических конструкций в клетки исходной клеточной линии и отбором клеток, содержащих генетические конструкции. Способ заключается в выборе моделируемой хромосомной перестройки и участков, внесение разрывов в которые приведет к воспроизведению данной перестройки, затем конструирование системы под узнавание выбранной последовательности и внесение в нее разрыва, а именно конструирование нуклеазы ZFN - подбор аминокислотных последовательностей доменов «цинковых пальцев», распознающих нужную последовательность нуклеотидов ДНК, либо конструирование субъединиц нуклеазы TALEN - подбор доменов белка, распознающих нужную последовательность нуклеотидов ДНК, в случае системы CRISPR-Cas9 подбирается узнающий ДНК-мишень участок гидовой РНК, например, подбор участков в генах AML1 и ЕТО и гидовых РНК, которые их узнают. Конструирование плазмидных векторов, кодирующих отдельные элементы заявляемой тест-системы, например, в случае использования системы CRISPR/Cas - синтез олигонуклеотидов, представляющих из себя участки генов гидовых РНК, отвечающих за специфическое узнавание гидовой РНК разрезаемого локуса; клонирование полученных олигонуклеотидов в экспрессионный вектор, содержащий последовательность гена гидовых РНК, включающую участок для вставки этих олигонуклеотидов методом рестрикционного клонирования, а также промотор для экспрессии гена гидовой РНК эукариотической РНК-полимеразой III, с последующей проверкой работоспособности сконструированных нуклеаз, например методом ((Engineered Nuclease-induced Translocations» (ENIT) (Germini и др., 2017). Создание генетической конструкции (1) проводят амплификацией участков векторов, кодирующих отдельные элементы заявляемой тест-системы, например, в случае использования системы CRISPR/Cas, кодирующих гидовые РНК, содержащие промотор, кодирующую последовательность и терминатор, затем вставка данных последовательностей в плазмиду, содержащую ген Cas9 под индуцируемым промотором TRE и ген устойчивости к пуромицину, а также последовательностей, способствующие встраиванию трансгенов в геном трансфицируемых клеток, при этом в качестве последовательностей, способствующие встраиванию трансгенов в геном трансфицируемых клеток используют так называемые плечи гомологии, представляющие из себя участки, повторяющие последовательности длиной 100-1000 нуклеотидов с каждой стороны от точки разрыва в геноме, куда затем встраивается трансген. Получение генетической конструкции (2), содержащую ген трансактиватора системы Tet-ON, ген устойчивости к неомицину, а также последовательности, способствующие встраиванию трансгенов в геном трансфицируемых клеток (так называемые плечи гомологии). Трансфекцию полученных генетических конструкций в клетки исходной клеточной линии осуществляют посредством электропорации с подобранными заранее параметрами прибора и условиями электропорации. Отбор клеток, содержащих генетические конструкции, осуществляют благодаря наличию в составе генетических конструкций генов - селективных маркеров, при этом отбор клеток производят посредством выращивания клеток на селективных антибиотиках в течение двух недель при предварительно подобранных концентрациях антибиотиков, при которых погибают клетки, не несущие генов устойчивости к данным антибиотикам, но при которых выживают клетки, несущие селективные маркеры.
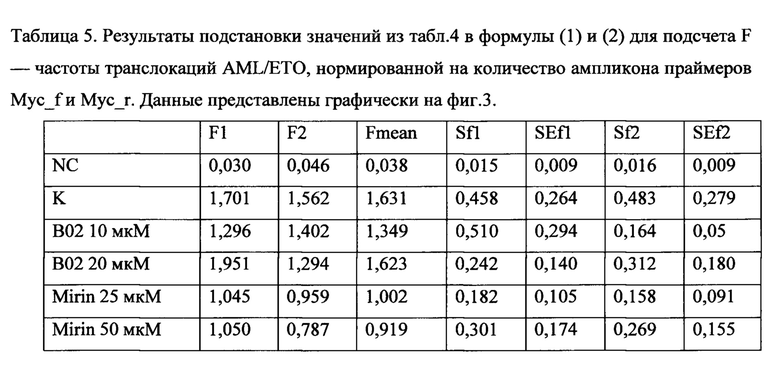
Также поставленная проблема решается способом проведения исследования влияния препарата (вещества или комбинации веществ), используемого при химиотерапии, на вероятность возникновения вторичного лейкоза, с помощью заявляемой тест-системы путем добавления к суспензии, содержащей не менее 2 млн. клеток, исследуемого препарата в концентрациях, не превышающих IC50 для данного препарата, и доксициклина в концентрации, достаточной для запуска экспрессии Cas9; инкубация полученной смеси; проведение количественной ПЦР и анализ результатов; вывод о снижении вероятности возникновения вторичного лейкоза делают по уменьшению количества полученных транслокаций по сравнению с количеством транслокаций в контроле. Предпочтительно концентрация доксициклина составляет от 0,1 до 1 мкг/мл. Образцы для ПЦР готовят любым известным из уровня техники способом или ДНК выделяют с помощью коммерческих наборов для выделения ДНК из клеток согласно инструкции к набору. Предпочтительно количественную ПЦР проводить с использованием предварительно подобранных праймеров ETO_f1-AML_f1 и MYC_f-MYC_r, дающих моноспецифический к участку транслокации продукт амплификации, а именно с последовательностями SEQ ID NO: 3-6. Предпочтительной программой (протоколом) для проведения количественной ПЦР с праймерами с последовательностями SEQ ID NO: 3-6 является: активация полимеразы 95°С 10 мин, денатурация ДНК 95°С 20 сек, отжиг праймеров 60°С 1 мин, стадия элонгации 72°С 30 сек, всего 42 цикла, затем кривая плавления 55-95°С шаг 0.5°С в 5 сек. Подсчет количества транслокаций, проводят следующим образом: полученные значения Ct (значения порогового цикла амплификации образца ДНК с соответствующей парой праймеров) подставляются в формулу (1):
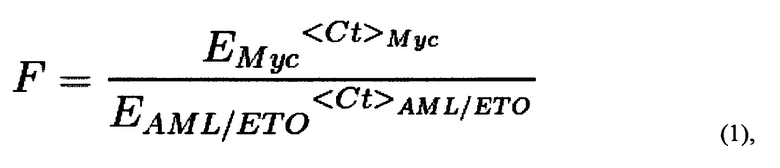
где F - частота транслокаций AML/ETO, нормированная на количество ампликона праймеров Myc_f и Мус_r, EMyc - эффективность амплификации ДНК с пары праймеров Myc_f и Мус_r, EAML/ETO - эффективность амплификации ДНК с пары праймеров ETO_f1 и AML_f1, которая определяется один раз в предварительном эксперименте в условиях, максимально соответствующих условиям дальнейшей работы; <Ct>i - среднее по трем повторностям значение порогового цикла амплификации образца ДНК с соответствующей парой праймеров, где i означает Мус или AML1/ЕТО. Для каждого значения F определяется стандартная ошибка на основании дисперсии CtMyc и CtAML/ETO по формуле (2):
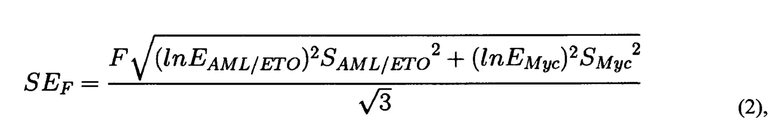
где F - частота транслокации, вычисленная на основании<Ct>Myc и <Ct>AML/ETO, Емус и EAML/ETO - значения эффективности амплификации с праймерами Myc_f и Мус_r и AML_f1/ETO_f1, соответственно. SAML/ETO2 - дисперсия CtAML/ЕТО, SMyc2 - дисперсия Заключение о вероятности возникновения вторичного лейкоза делают по наличию разницы между данными, полученными на обработанных клетках и на контрольных клетках, при этом разницу между данными, полученными на обработанных клетках и на контрольных клетках, делают на основании множественного сравнения экспериментальных выборок с контрольной с помощью непараметрического критерия Даннета.
Краткое описание чертежей
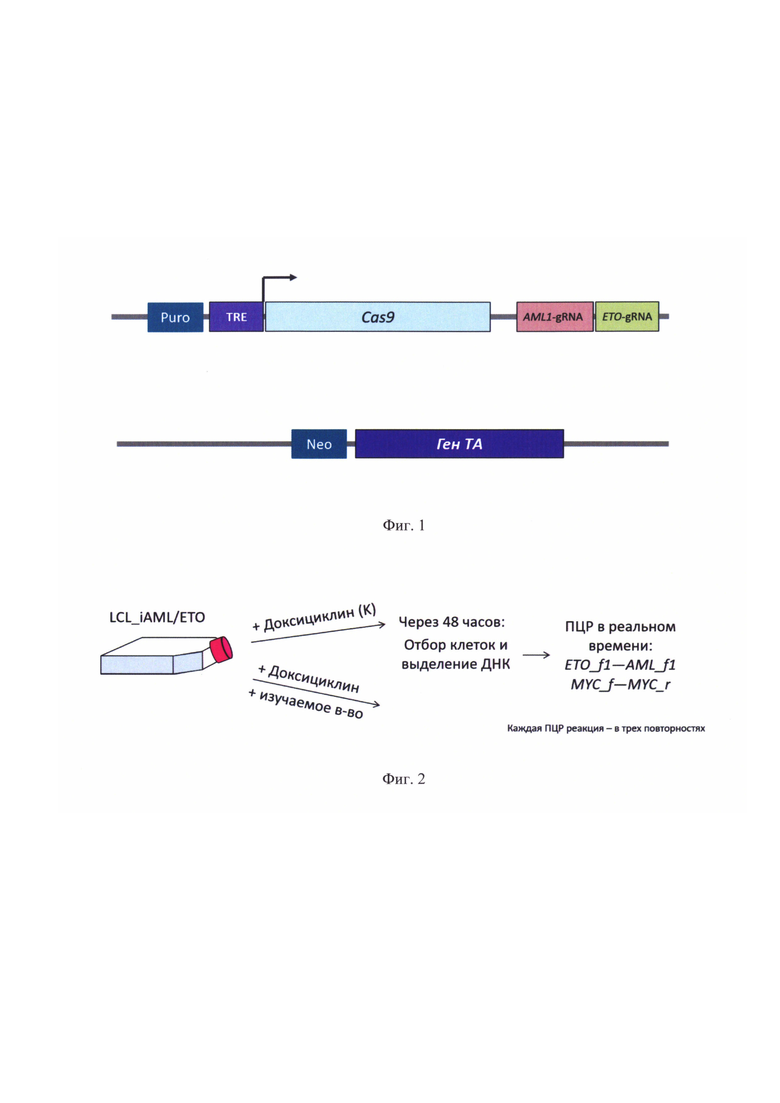
На фиг. 1 представлена схема конструкции, интегрированные в геном клеток описываемой тест-системы. Cas9 - ген нуклеазы Cas9, AML1-gRNA_- ген гидовой РНК, узнающей локус AML1, ETO-gRNA_- ген гидовой РНК, узнающей локус ETO, TRE - промотор гена Cas9, индуцируемый белком транс-активатором, Puro - ген устойчивости к пуромицину, Neo - ген устойчивости к неомицину, ТА - ген, кодирующий белок транс-активатор.
На фиг. 2 показана последовательность действий при анализе изучаемых веществ с помощью тест-системы.
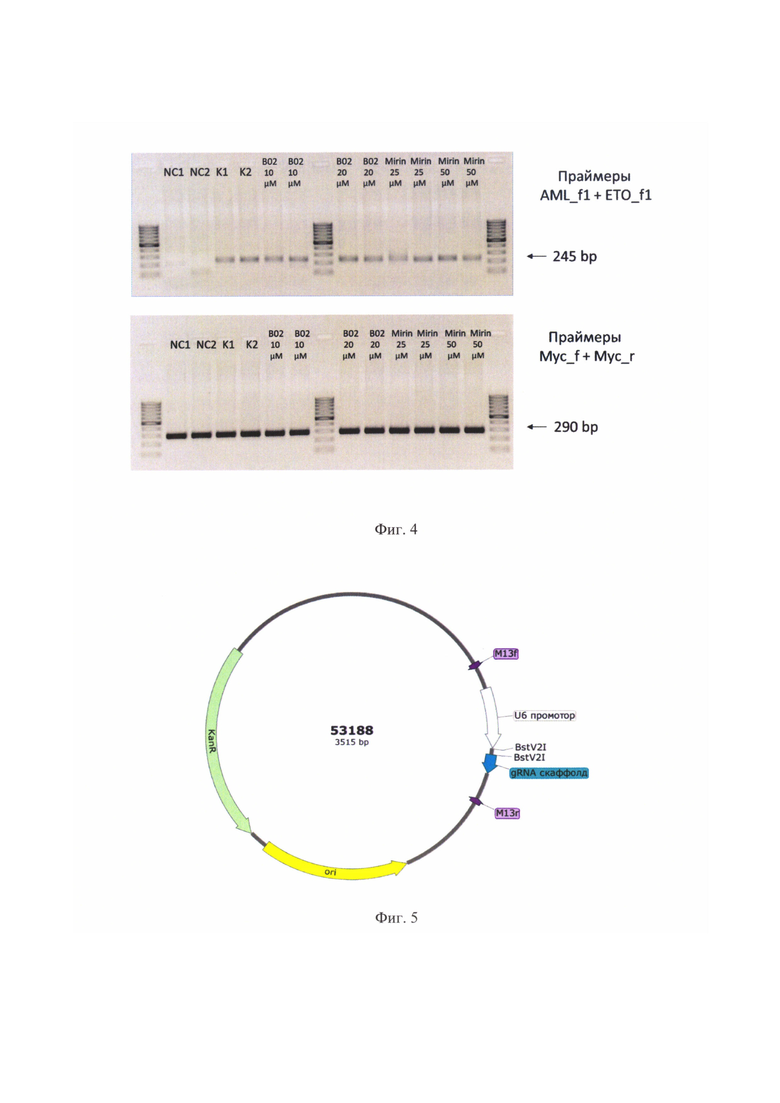
На фиг. 3 продемонстрировано влияние ингибиторов белков репарации RAD51 (В02) и MRE11 (Mirin) на частоту индуцируемых транслокаций AML1-ETO в линии LCL_iAML/ETO. А - Значение F умножено на 10000. Усы - SEF. Круглыми точками отложены значения F для двух повторностей. Каждая точка амплифицировалась с каждой парой праймеров в трех повторностях. Ромбом отмечено среднее значение F для каждой выборки. «*» отмечены достоверные отличия от контроля (р<0.05 по критерию Даннета). Обозначения: NC - негативный контроль: экспрессия Cas9 не активировалась. К - положительный контроль: клетки + доксициклин, но без ингибиторов; В02 10 мкМ, В02 10 мкМ, Mirin 25 мкМ и Mirin 50 мкМ - одновременная активация экспрессии Cas9 и обработка клеток соответствующими ингибиторами в указанных концентрациях. Б - нормировка средних частот F в каждой выборке на среднее значение F в положительном контроле (К). В качестве меры разброса на гистограмме отложена стандартная ошибка (SE) для двух значений F в каждой выборке.
Фиг. 4. Электрофорез продуктов количественной ПЦР. На гель наносилось по одному образцу для каждой повторности. NC - негативный контроль: клетки без ингибиторов и доксициклина. K - положительный контроль: клетки + доксициклин без ингибиторов; В02 10 мкМ, В02 20 мкМ, Mirin 25 мкМ и Mirin 50 мкМ - одновременная активация экспрессии Cas9 и обработка клеток соответствующими ингибиторами в указанных концентрациях. Дорожка М - маркер молекулярных масс: набор из 10 фрагментов ДНК от 100 п.н. до 1000 п.н. с шагом в 100 п.н. Яркость полос на геле зависит от многих факторов, поэтому не используется для определения исходного количества матрицы.
На фиг. 5 представлена карта плазмиды phU6-gRNA. На карте отмечены элементы: KanR - ген устойчивости к канамицину под контролем бактериального промотора, Ori - ориджин репликации в клетках E. coli, M13f и M13r - праймеры для проверки вставки последовательности гидовой РНК в плазмиду, U6 - промотор РНК полимеразы III человека, gRNA_скаффолд - последовательность константной части гидовой РНК. Отмечены сайты узнавания рестриктазы BstV2I.
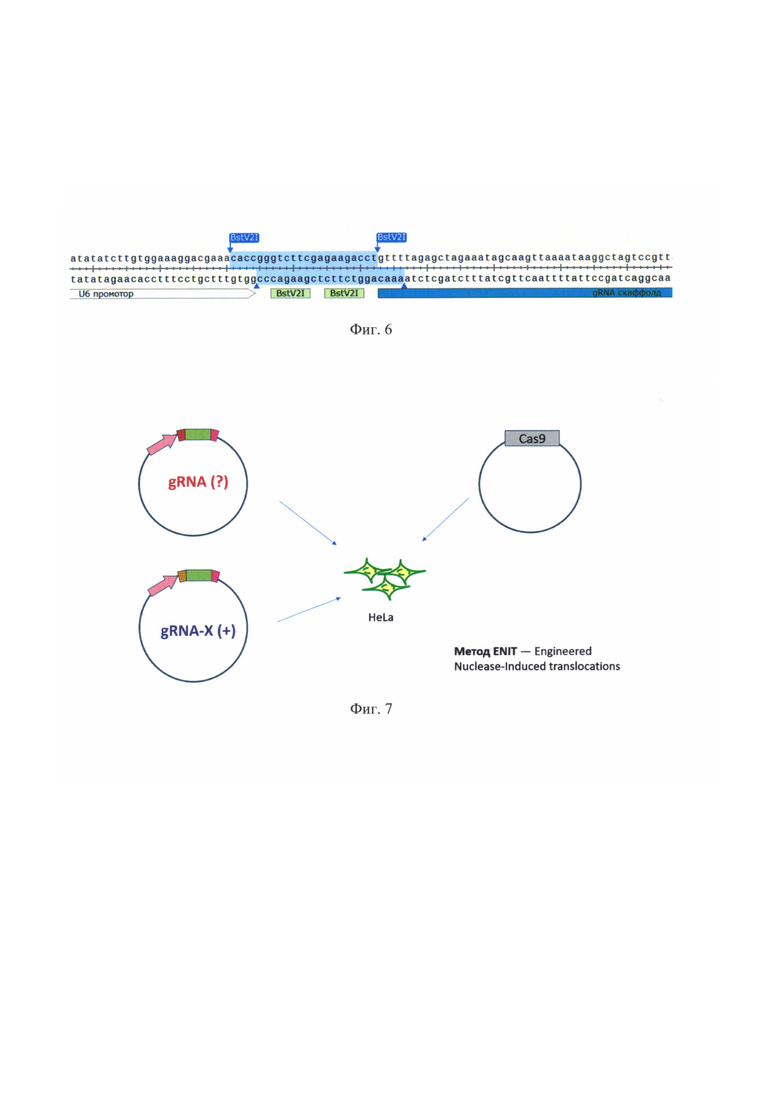
На фиг. 6 показан участок плазмиды phU6-gRNA. Фрагмент, вырезаемый рестриктазой BstV2I, выделен цветом. На участке отмечены сайты узнавания рестриктазы BstV2I. Остальные обозначения как на фиг. 5.
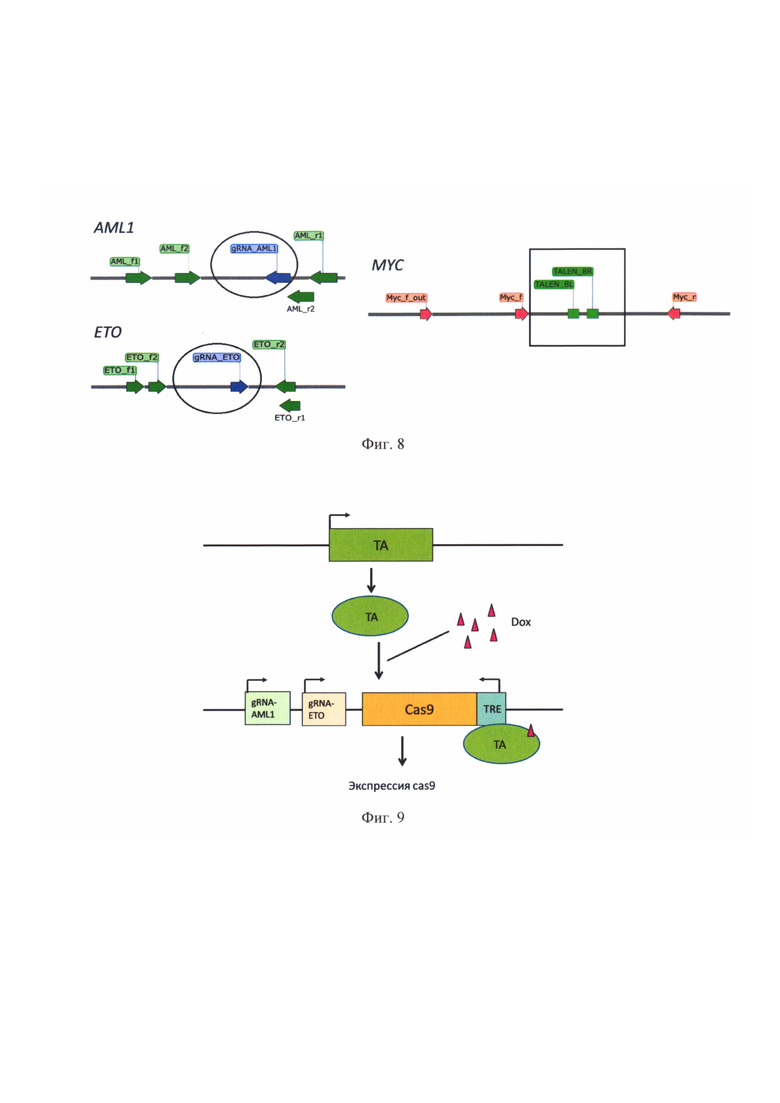
На фиг. 7 представлена схема трансфекции клеток HeLa при проверке работы нуклеаз методом ENIT. Вместо плазмиды с достоверно работающей гидовой РНК («gRNA-X(+)») можно использовать пару плазмид, кодирующих достоверно работающую нуклеазу TALEN. gRNA_(?) - плазмида, содержащая проверяемый РНК-гид.
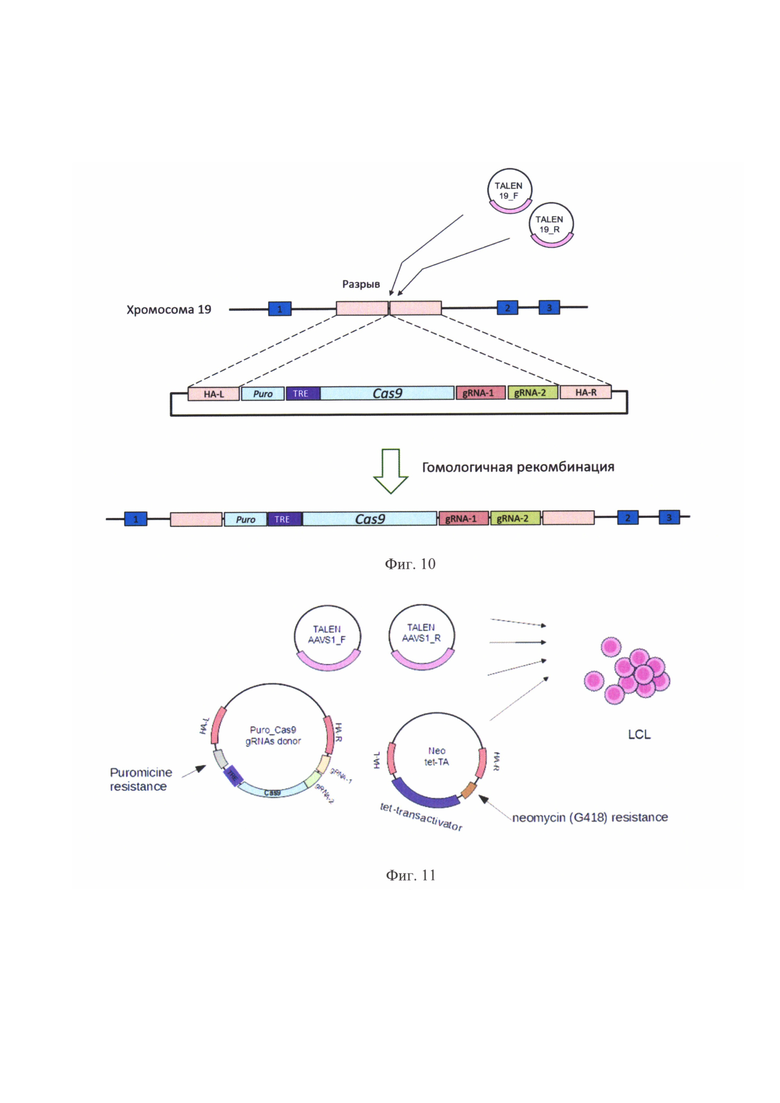
На фиг. 8 показано взаимное расположение праймеров на участках генов AML1, ЕТО и MYC. Овалом отмечены последовательности, узнаваемые гидовыми РНК в генах AML1 и ЕТО, квадратом - субъединицами TALEN в гене MYC.
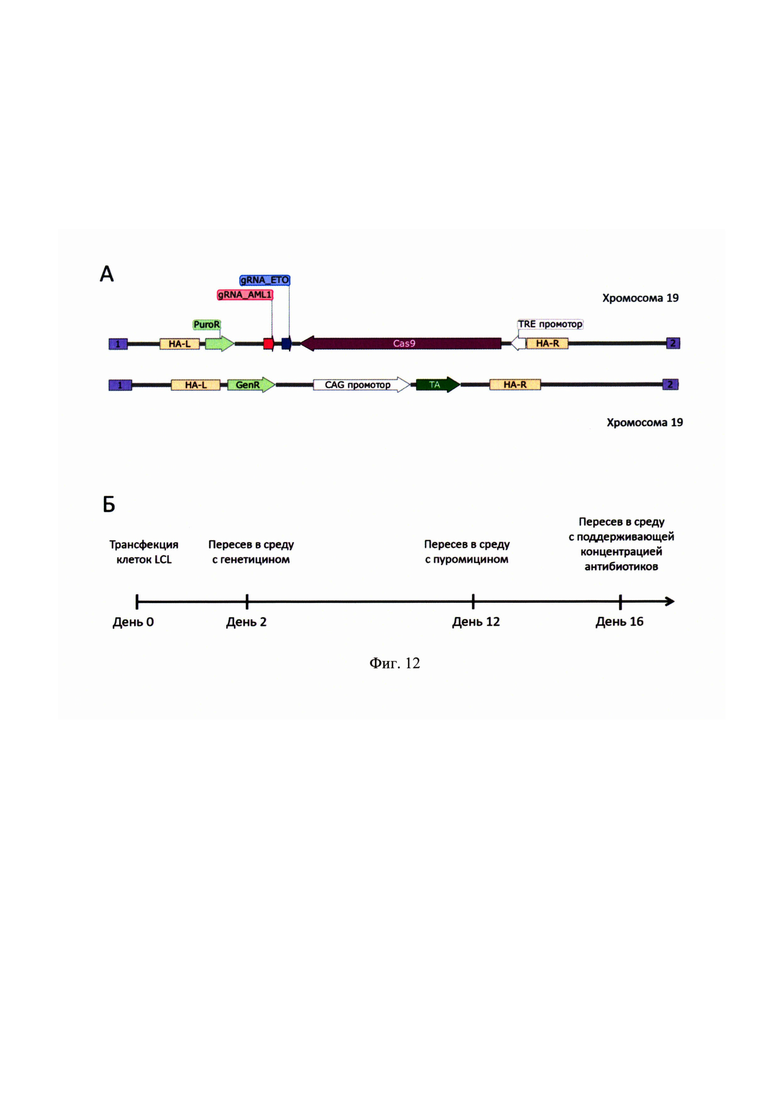
На фиг. 9 представлен механизм активации экспрессии Cas9. Активатор транскрипции (ТА) экспрессируется конститутивно. ТА связывается с добавляемым к клеткам доксициклином (Dox) и активирует экспрессию Cas9. TRE - промотор, содержащий сайты посадки ТА.
На фиг. 10 представлена схема интеграции кассеты в геном по механизму гомологичной рекомбинации.
На фиг. 11 показана схема трансфекции клеток при получении линии LCL_iAML/ETO.
На фиг. 12 отображено получение линии LCL_iAML/ETO. (А) - карта первого интрона гена PPP1R12C, с интегрированными кассетами генов. Цифрами 1 и 2 обозначены экзоны этого гена. HA-L и HA-R - плечи гомологии, использовавшиеся для интеграции кассет генов из плазмид. PuroR и GenR - гены устойчивости к пуромицину и генетицину, соответственно. TRE - индуцируемый промотор. CAG - конститутивный промотор. U6 - промотор для РНК-полимеразы III. ТА - ген активатора транскрипции M2rtTA. (Б) - временная шкала селекции клеток на антибиотиках. Селектирующие концентрации: генетицин - 800 мкг/мл, пуромицин - 0,3 мкг/мл. Поддерживающая концентрация - 300 мкг/мл генетицина и 0,1 мкг/мл пуромицина. Последовательная селекция трансфицированных клеток на генетицине и пуромицине позволяет отобрать клетки, у которых в одну хромосому встроены гены гидовых РНК gRNA_AML1 и gRNA_ETO, ген нуклеазы Cas9 и ген устойчивости к пуромицину, а в другую гомологичную хромосому встроены гены активатора транскрипции ТА и устойчивости к генетицину.
Осуществление изобретения
Заявляемая тест-система для определения частоты образования транслокаций представляет из себя культуру клеток млекопитающих с модифицированным геномом. Выбор исходной культуры клеток, на основе которой сделана тест-система, не является критичным для осуществления изобретения, но чем более близка исходная культура к клеткам, заболевание которых моделируется, тем более релевантными к реальному заболеванию будут полученные на этой культуре результаты.
В геном клеток интегрированы конструкции, представляющие из себя элементы нуклеаз, которые позволяют вносить точные разрывы в хромосомы клетки в локусы, между которыми происходит моделируемая перестройка, например в гены AML1 и ЕТО-транслокации между этими локусами характерны для вторичного лейкоза. В качестве таких нуклеаз в нашем примере используется нуклеаза Cas9 системы CRISPR/Cas, которая направляется в нужные локусы гидовыми РНК. Такими нуклеазами могут быть любые нуклеазы, вносящие направленный разрыв в геном клеток (например, ZFN, TALEN, Cpf1 и другие).
Чтобы нуклеазы вносили точный разрыв в необходимый локус, предварительно подбирается несколько вариантов нуклеаз, в случае системы CRISPR/Cas подбираются вариабельные участки этих РНК-гидов, отвечающих за узнавание. Работа РНК-гидов проверяется различными методами, например, мы проверяли наши гидовые РНК методом ((Engineered Nuclease-induced Translocations» (ENIT). Он предполагает котрансфекцию клеток (например, HeLa) плазмидами, кодирующими нуклеазу Cas9 и гидовые РНК. В случае, если нуклеаза эффективно вносит разрывы, то при репарации с некоторой вероятностью образуются хромосомные перестройки между точками разрывов. Спустя 48 часов после трансфекции с ДНК клеток ставится ПЦР на предмет наличия ожидаемых транслокаций, которые происходят между разрезаемыми локусами (Germini D., Saada Y.B., Tsfasman Т., Osina K., Robin С, Lomov K, Rubtsov М., Sjakste N., Lipinski M., Vassetzky Y., 2017. A one-step PCR-based assay to evaluate efficiency and precision of genomic DNA-editing tools // Mol. Ther. - Methods Clin. Dev. V. 5. № June. P. 43-50). Последовательности вариабельных участков используемых гидов в табл. 1.
Заявляемая тест-система отличается тем, что запускается по сигналу извне. Для этого ключевой элемент системы - нуклеаза, вносящая разрыв в нужные локусы, - кодируется геном под контролем индуцируемого промотора. Например, может быть использована система Тет-ON. В этом случае для индукции экспрессии гена, находящегося под индуцируемым промотором, необходим белок транс-активатор, причем этот активатор работает только в присутствии доксициклина или других химических аналогов тетрациклина, которые способны связываться с белком транс-активатором системы Тет-ON. Поэтому ген транс-активатора также встраивается в геном клеток. Взаимное расположение генов относительно друг друга, а также точка в геноме, куда интегрируются эти конструкции, не важны, принципиальное значение имеет размещение гена Cas9 под контроль индуцируемого промотора.
В результате добавления в среду к клеткам доксициклина происходит активация экспрессии нуклеазы Cas9, и она вносит в необходимых локусах разрывы, которые приводят к образованию транслокаций. Эти транслокации надежно детектируются методом ПЦР через 48 часов после добавления доксициклина. Также не так важно, гены какой именно нуклеазы будут закодированы в геноме, важно, чтобы они делали по сигналу двуцепочечные разрывы в требуемых локусах. Например, за основу культуры LCL_iAML/ETO, взята линия лимфобластоидных клеток (LCL) RPMI 8866 (из коллекции клеточных культур Health Protection Agency (HPA), Великобритания, поставляется Sigma-Aldrich, США), в геном которой были интегрированы элементы системы CRISPR/Cas и системы Tet-ON: гены гидовых РНК gRNA_AML1 и gRNA_ETO, ген нуклеазы Cas9 и ген устойчивости к пуромицину, ген активатора транскрипции ТА и ген устойчивости к генетицину. Для изготовления тест-системы может быть взята и другая линия иммортализированных клеток млекопитающих, чей геном расшифрован и опубликована его последовательность, чтобы была возможность подобрать нуклеазы под конкретные локусы в геноме. Выбор используемой культуры клеток определяется тем, насколько близки данные клетки к клеткам крови человека, заболевания в которых моделируются. Чем ближе - тем выше вероятность, что полученные на тест-системе результаты будут релевантны по отношению к настоящему заболеванию. Другим фактором является удобство работы с данными клетками - скорость деления клеток в культуре должна быть не слишком медленной - не менее одного деления в 2 суток.
Интеграция необходимых элементов (фиг. 1) была осуществлена в участок в области первого интрона гена PPP1R12C, расположенного на 19 хромосоме. Интеграция может быть осуществлена в любые другие участки генома, в которых трансген будет стабильно экспрессироваться (так называемые Safe harbours (Sadelain М., Papapetrou Е.Р., Bushman F.D., 2011. Safe harbours for the integration of new DNA in the human genome // Nat. Rev. Cancer. V. 12. №1. P. 51-58). В одну хромосому интегрирована конструкция, содержащая ген нуклеазы Cas9 под промотором TRE (индуцируемый тетрациклином элемент), ген гидовой РНК, узнающей локус AML1 и ген гидовой РНК, узнающей локус ЕТО (таблица 1), а также ген селекивного маркера (элемента, позволяющего отобрать клетки, несущие трансгенную конструкцию), например, ген устойчивости к селективному антибиотику. В другую гомологичную хромосому 19 интегрирован ген белка-активатора и другой селективный маркер, например, ген устойчивости к антибиотику неомицину. Гены устойчивости к антибиотикам могут быть другими, с помощью которых можно осуществлять отбор клеток, несущих конструкции. Могут быть использованы также другие методы селекции клеток, несущих конструкцию, например, добавление гена флуоресцентного белка и последующий сортинг клеток. Исследования показывают, что уровень экспрессии гена белка-активатора постоянен, а экспрессия гена Cas9 запускается в ответ на добавление в среду к клеткам доксициклина в концентрации 1 мкг/мл и спустя сутки уровень экспрессии гена Cas9 выходит на плато. Транслокации между локусами AML1 и ЕТО при этом надежно детектируются спустя 48 часов после добавления доксициклина.
Для работы с клеточной культурой - тест-системой и для измерения уровня (частоты) образования транслокаций применяется следующая последовательность действий. В качестве примера взята тест-система - клеточная линия LCL_iAML/ETO. Для тест-системы, созданной на основе другой исходной клеточной линии, протокол будет отличаться в методах культивирования этой линии, и будет соответствовать протоколу культивирования исходной линии. Подробно протокол создания клеточной линии LCL_iAML/ETO приведен ниже.
Клеточная линия LCL_iAML/ETO содержится согласно общепринятым методикам ведения суспензионных клеточных культур. Клетки культивируются в инкубаторе (например, «Binder», Германия) при 37°С во влажной атмосфере с содержанием 5% СО2. Работа с культурами ведется с соблюдением техник асептики. Клетки LCL_культивируются в среде RPMI-1640 с добавлением эмбриональной телячьей сыворотки «FBS certified)) («Gibco», США) до 10% объема, а также антибиотиков пенициллин в конечной концентрации 50 ед/мл («ПанЭко», Россия) и стрептомицин в конечной концентрации 50 мкг/мл («ПанЭко», Россия). Пассаж культуры LCL_проводится по достижению плотности 0,8-1 млн клеток/мл путем замены части объема культуры свежей полной средой.
Хранения клеток возможно в замороженном состоянии при -80°С или, что предпочтительнее, в хранилищах клеток в жидком азоте. Для работы следует разморозить ампулу с клетками при комнатной температуре. Затем перенести клетки в 10 мл среды, суспендировать и затем сменить эту среду на свежую, чтобы избавиться от DMSO в среде. Замораживание клеток производится в среде с добавлением 5-7% DMSO в специальных контейнерах для заморозки, например «Мг.Frosty® Cryo 1 С Freezing Containers». Все описанные процедуры культивирования клеток соответствуют общепринятым протоколам и рекомендациям по культивированию суспензионных клеток эукариот.

Для изучения влияния различных веществ на частоту образования транслокаций производится следующая последовательность действий. Линия клеток LCL_iAML/ETO (генетически модифицированные клетки со встроенными элементами систем CRISPR/Cas и TetON) размораживается, и клетки наращиваются до необходимого количества от 2 млн на экспериментальную точку (одна повторность эксперимента в определенных условиях, влияние которых изучается) при концентрации суспензии 0,3-0,8 млн клеток/мл. Общее количество клеток определяется удобством экспериментатора - чем меньше клеток, тем быстрее они нарастают до необходимого количества. С другой стороны, из меньшего количества клеток сложнее выделить геномную ДНК. Оптимальным является 3 млн клеток на экспериментальную точку. Перед началом эксперимента клетки разводятся до 0,3-0,4 млн клеток/мл. Затем клетки распределяются по 5 мл по лункам шестилуночного планшета: по одной лунке на каждую экспериментальную точку. Минимум одна из точек должна быть для контроля, остальные - для изучаемых веществ. В лунки добавляется доксициклин до концентрации 1 мкг/мл, а также (кроме лунки с контролем) изучаемые вещества в исследуемых концентрациях (можно в разных экспериментах брать одно и то же вещество, но в разных концентрациях). Концентрации подбираются исходя из влияния веществ на выживаемость клеток, что может быть взято из литературных данных или проведен соответствующий тест на выживаемость (например, МТТ или ХТТ тесты). В экспериментах были использованы концентрации, которые меньше, чем те, что приводят к гибели хотя бы половины из обработанных клеток. Осадок клеток ресуспендируется в необходимом объеме среды - 1 мл на 0,5 млн клеток или ином, согласно протоколу эксперимента), и суспензия перемещается в новый матрас для культивирования. Спустя 48 часов из клеток выделяется геномная ДНК, например, с помощью коммерческих наборов для выделения ДНК из клеток и согласно инструкции к набору. Затем с этой ДНК ставится количественная ПЦР, с ДНК каждой экспериментальной точки по 6 реакций: 3 реакции с предварительно подобранной парой праймеров ЕТО f1-AML_f1 и 3 реакции с парой MYC_f-MYC_r (последовательности праймеров в таблице 2). Праймеры подбираются исходя из того, какую транслокацию необходимо детектировать. Оптимальным является выбор праймеров с помощью сервиса Primer-blast (ncbi.nlm.nih.gov/tools/primer-blast). Расстояние до точки разрыва - 30-100 нуклеотидов, чтобы использовать праймеры для количественной ПЦР. Лучше не допускать больше 2 GC в последних 5 нуклеотидах на 3'-конце праймера, всего длина 18-30 нуклеотидов. Программа количественной ПЦР: активация полимеразы 95°C - 10 мин, денатурация ДНК 95°C - 20 сек, отжиг праймеров 60°C - 1 мин, стадия элонгации 72°C - 30 сек, всего 42 цикла, затем кривая плавления 55-95°С шаг 0.5°С в 5 сек. Общая схема действий представлена на фиг. 2.

Результаты ПЦР анализируются следующим образом. Полученные значения Ct (значения порогового цикла амплификации образца ДНК с соответствующей парой праймеров) подставляются в формулу (1):
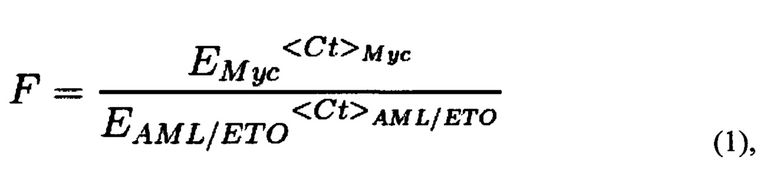
где F - частота транслокаций AML/ETO, нормированная на количество ампликона праймеров Myc_f и Мус_r. Емус - эффективность амплификации ДНК с пары праймеров Myc_f и Мус_r. EAML/ETO - эффективность амплификации ДНК с пары праймеров ETO_f1 и AML_f1 (были определены согласно принятым методикам проведения количественной ПЦР; в качестве матрицы была взята геномная ДНК, и приготовлены разные разведения, затем поставлены реакции количественной ПЦР с соответствующими парами праймеров и строился график зависимости полученных значений Ct> от десятичного логарифма концентрации кДНК, значения эффективности определялись по формуле E=10∧(-1/tg а), где а - угол наклона прямой на графике; значения в таблице 3). Эффективность праймеров определяется один раз в предварительном эксперименте в условиях, максимально соответствующих условиям дальнейшей работы: конкретный прибор, реагенты, протоколы выделения ДНК. В случае изменения этих условий, эффективность определяется заново. <Ct>i - среднее по трем повторностям значение порогового цикла амплификации образца ДНК с соответствующей парой праймеров.
Для каждого значения F определяется стандартная ошибка на основании дисперсии CtMyc и CtAML/ETO по формуле (2):
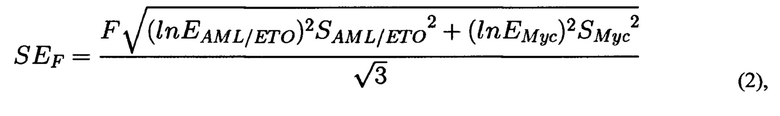
где F - частота транслокации, вычисленная на основании <Ct>Myc и<Ct>AML/ETO, Емус и EAML/ETO - значения эффективности амплификации с праймерами Myc_f и Мус_r и AML_f1/ETO_f1, соответственно. SAML/ETO2 - дисперсия CtAML/ETO, SMyc2 - дисперсия Множественное сравнение экспериментальных выборок с контрольной проводится с помощью непараметрического критерия Даннета (С. Гланц, 1999. Медико-биологическая статистика. Пер. с англ. М.: Практика. Р. 1-459). Использование этого критерия оправдано, когда не известно, распределены ли значения нормально, а также проводится множественное попарное сравнение нескольких выборок с одной контрольной. Эти рассуждения применимы в случае 2-3 экспериментальных точек (повторностей) для обработки каждым из изучаемых веществ. В случае большого числа экспериментальных точек могут применяться другие инструменты статистической обработки результатов. Формула для расчета критерия Даннета (3):
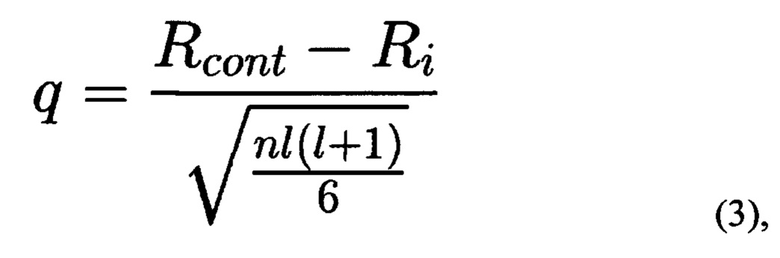
где Rcont и Ri - суммарные ранги контрольной и опытной выборок, соответственно, n - число элементов в каждой выборке, 1 - число сравниваемых выборок. Ранги определяются на основании значений F для каждого тестируемого вещества (или контроля). Полученное значение q сравнивается с табличным (С. Гланц, 1999. Медико-биологическая статистика. Пер. с англ. М.: Практика. Р. 1-459) для определения статистической значимости.
Для демонстрации того, что культура LCL_iAML/ETO может служить моделью ранних событий при возникновении лейкозогенных транслокаций, было исследовано влияние ингибирования белков репарации ДЦР на частоту транслокаций. Относительная частота транслокаций определялась с помощью количественной ПЦР. Для осуществления такой ПЦР в ходе предварительных экспериментов была подобрана пара праймеров (ETO_f1 и AML_f1), которая дает моноспецифический продукт при амплификации с геномной ДНК в одну стадию ПЦР.
Было протестировано два ингибитора - Мирин (Mirin) и В02. Мирин (Mirin, Sigma-Aldrich, США) является ингибитором экзонуклеазной активности белка MRE11 (Dupre и др., 2008). Этот белок инициирует укорочение концов при гомологичной рекомбинации, а также сигнализирует о возникновении ДЦР путем активации киназы ATM (Stracker Т.Н., Petrini J.H.J., 2011. The MRE11 complex: starting from the ends // Nat. Rev. Mol. Cell Biol. V. 12. №2. P. 90-103). Кроме того, MRE11 принимает участие и в негомологичном соединении концов, причем как в классическом, так и в альтернативном пути (Zha S., Boboila С, Alt F.W., 2009. Mrell: Roles in DNA repair beyond homologous recombination // Nat. Struct. Mol. Biol. T. 16. №8. C. 798-800). Второй используемый ингибитор - B02 (Sigma-Aldrich, США) - нарушает процесс гомологичной рекомбинации, препятствуя сборке филамента RAD51 на одноцепочечной ДНК {Huang F., Mazina О.М., Zentner I. J., Cocklin S., Mazin A. V., 2012. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase // J. Med. Chem. V. 55. №7. P. 3011-3020). Используемые в работе концентрации ингибиторов приводили к возникновению физиологического эффекта в культурах клеток в описанных исследованиях (Dupre A., Boyer-Chatenet L., Sattler R.M., Modi А.Р., Lee J.-H., Nicolette M.L., Kopelovich L., Jasin M., Baer R., Paull T.T., Gautier J., 2008. A forward chemical genetic screen reveals an inhibitor of the Mrell-Rad50-Nbsl complex. // Nat. Chem. Biol. V. 4. №2. P. 119-125; Huang F., Mazina O.M., Zentner I.J., Cocklin S., Mazin A. V., 2012. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase // J. Med. Chem. V. 55. №7. P. 3011-3020).
Указанные ингибиторы добавлялись в концентрации 25 мкМ или 50 мкМ для мирина и 10 мкМ или 20 мкМ для В02 к культуре LCL_iAML/ETO (по 2 млн на каждую экспериментальную точку) параллельно с ее активацией доксициклином 1 мкг/мл. Спустя 48 часов из клеток выделялась геномная ДНК с помощью специальных коммерческих наборов, например «diaGene для выделения ДНК из культур клеток» («Диаэм», Россия), и ставилась количественная ПЦР на транслокации (фиг. 2). Отметим, что инкубация клеток с ингибиторами на протяжении указанного времени не приводила к гибели клеток, что проверялось окрашиванием трипановым синим. Эксперимент был проведен в двух биологических повторностях.
Для проведения количественной ПЦР использовался набор реактивов «Maxima SYBR Green qPCR Master mix» («Thermo Fisher Scientific)), США), позволяющий проводить реакцию с использованием «горячего старта». Объем одной реакции - 25 мкл. Концентрация праймеров составляла 250 нМ. Последовательности праймеров приведены в таблице 2. Параметры циклов ПЦР указаны выше. Для работы использовался прибор «CFX96» («Bio-Rad Laboratories», США). Анализ результатов количественной ПЦР проводился с помощью программы «Bio-Rad CFX Manager» версии 3.1 («Bio-Rad Laboratories», США). Пороговое значение флуоресценции и значение порогового цикла (Ct) определялось автоматически. По окончании циклов ПЦР прибор снимал кривую плавления для подтверждения специфичности амплификации.
Для дополнительного подтверждения специфичности амплификации ПЦР-продукты амплификации в каждой повторности были проанализированы с помощью электрофореза (фиг. 3). Во всех случаях, кроме негативного контроля (клетки без ингибиторов и доксициклина) размер ПЦР-продуктов соответствовал ожидаемым. В негативном контроле обнаруживаются продукты неспецифической амплификации, поэтому частота «транслокаций» в этом случае несколько больше нуля (фиг. 3).
В результате были получены данные Ct (таблица 4), которые подставлялись в формулу (1). Значения эффективностей для праймеров (EAML/ETO) брали из табл 3. Стандартная ошибка вычислялась на основании дисперсии CtMyc и CtAML/ETO по формуле (2). Полученные данные (табл. 5) наносились на график. Множественное сравнение экспериментальных выборок с контрольной проводилось с помощью непараметрического критерия Даннета (3).
Таблица 4. Данные Ct из результатов обработки клеточной линии ингибиторами одновременно с индукцией Cas9. NC - отрицательный контроль, К - индукция транслокаций без ингибиторов, В10 и В20 - кообработка ингибитором В02 в концентрациях 10 и 20 мкМ соответственно, М25 и М50 - кообработка ингибитором Мирин в концентрациях 25 и 50 мкМ, соответственно.
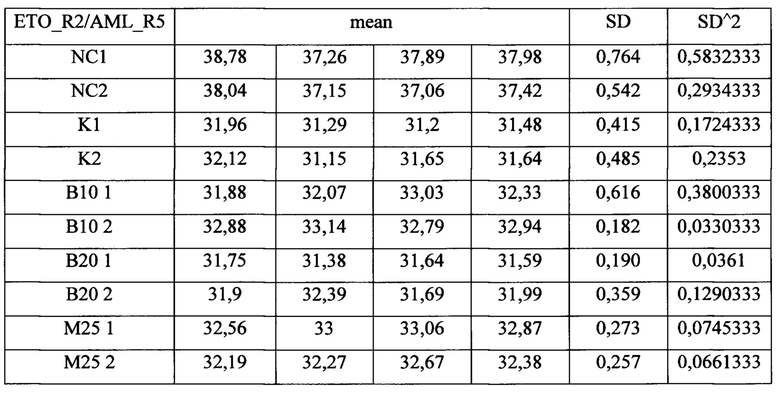
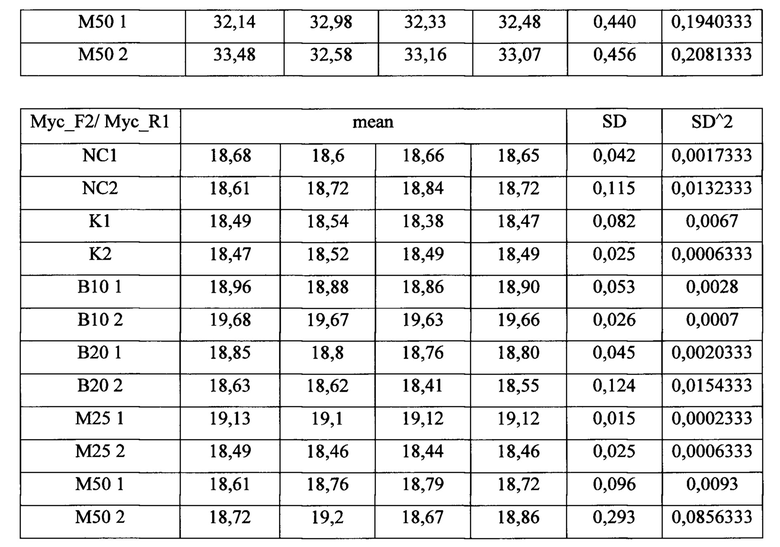
Измеряемая частота транслокаций в данном эксперименте не является абсолютной, поскольку мы не знаем доли клеток в популяции, которые несут хромосомную перестройку. Фактически, мы измеряем долю молекул транслокации по отношению к ДНК гена MYC, по которому проводилась нормировка. Тем не менее, этого вполне достаточно для сравнения частот транслокации в различных экспериментальных условиях.
Можно видеть, что ингибирование Rad51 с помощью В02 не снижает частоту транслокаций (фиг. 3). Это согласуется с предположением о том, что ошибочная репарация ДЦР, приводящая к возникновению перестроек, осуществляется путем негомологичного соединения концов. В то же время, при ингибировании MRE11 с помощью Mirin наблюдается снижение частоты транслокаций на 39% и 44% для 25 мкМ и 50 мкМ ингибитора, соответственно. Для определения статистической достоверности различий был использован непараметрический критерий Даннета (С. Гланц, 1999. Медико-биологическая статистика. Пер. с англ. М.: Практика. Р. 1-459). Этот критерий позволяет проводить попарное сравнение нескольких выборок с одной контрольной. Использование именно непараметрической версии критерия объясняется малым объемом выборки и отсутствием уверенности в том, что случайная величина F распределена нормально. Значение частот транслокаций были заменены рангами, как того требует используемый критерий. При этом для составления выборки использовались именно биологические повторности - по два значения F для каждой выборки.
Таким образом, как показали проведенные эксперименты:
- заявляемая тест-система позволяет изучать влияние различных веществ на частоту образования перестроек между локусами AML и ЕТО, а значит, их влияние на вероятность возникновения вторичных лейкозов.
- в заявляемой тест-системе Mirin в концентрации 25 мкМ и 50 мкМ на 39% и 44% снижает частоту образования транслокаций между AML и ЕТО. Эта транслокация приводит ко вторичным лейкозам, и поэтому имеет смысл провести следующий этап испытаний данного вещества на предмет снижения частоты лейкозогенных перестроек при использовании химиотерапевтических препаратов из класса ингибиторов топоизомераз.
Разница в уровне транслокаций напрямую связана с разницей в уровне риска возникновения вторичных лейкозов.
Протокол создания клеточной линии LCL_iAML/ETO.
Для выполнения этой задачи сначала были подобраны гидовые РНК (гиды) и собраны плазмиды на основе плазмиды phU6-gRNA_(номер в Addgene #53188). Каждая такая плазмида содержала ген одной из гидовых РНК. Эти плазмиды затем использовали для проверки работоспособности гидов. Потом участок, содержащий ген гидовой РНК (вместе с промотором и терминатором), амплифицировали с помощью ПЦР, чтобы затем вставить его в плазмиду Puro_Cas9 donor (номер #58409 в Addgene). Сборка осуществлялась методом Gibson Assembly. Полученная плазмида Puro_Cas9+gRNA_AML1_ETO использовалась при трансфекции клеток.
Подбор гидов
В качестве гидов использовали последовательности узнающих участков гидовых РНК, узнающих последовательности внутри пятого интрона гена AML1 и второго интрона гена ЕТО. Последовательности, узнаваемые гидовыми РНК, располагаются в указанных интронах вблизи кластеров точек разрыва. В качестве исходного материала для выбора узнающих участков гидовых РНК в онлайн-сервис crispr.mit.edu загружали последовательность размером 250 нуклеотидов, включавшая в себя максимальное число точек разрыва, обнаруженных у пациентов (Smith et al., 2014). Для каждого локуса были отобраны по три гидовые РНК, имеющих температуру отжига около 60°С с учетом добавленных нуклеотидов САСС, необходимых для дальнейшего встраивания в плазмиду phU6-gRNA, и начинающихся с G, в противном случае добавляли G на 5'-конец.
Подбор праймеров
Вместе с гидами подбирались и праймеры для ПЦР, с помощью сервиса Primer-blast (ncbi.nlm.nih.gov/tools/primer-blast). Расстояние до точки разрыва выбирали 30-100 нуклеотидов, чтобы использовать праймеры впоследствии и для ПЦР в реальном времени. В отобранных праймерах старались допускать не больше 2 GC в последних 5 нуклеотидах на 3'-конце праймера, всего длина 18-30 нуклеотидов. Синтез праймеров осуществлен на заказ в компании ООО «ДНК-синтез» (Москва, Россия) или в Eurofins Genomics (Германия).
Сборка плазмид с генами гидовых РНК
Сборка экспрессионных векторов, несущих гены гидовых РНК, осуществляли на основе плазмиды phU6-gRNA_(номер #53188, «Addgene», США). Этот вектор содержит промотор гена мяРНК U6, последовательность конститутивной части гидовой РНК (скаффолд), и небольшой спейсер между ними (фиг. 5). Спейсер содержит сайты рестрикции для эндонуклеазы BstV21, вырезающей этот фрагмент из плазмиды. На его место встраивали олигонуклеотид, представляющий из себя последовательность узнающего участка гидовой РНК (фиг. 6). Таким образом плазмида phU6-gRNA_использовалась для создания целого семейства плазмид, несущих гены разных гидовых РНК.
Получение олигонуклеотидов. Последовательность узнающего участка гидовой РНК далее называется «прямым» гидом, а комплементарная ей - «обратным». К «прямому» гиду добавлялся на 5'-конец САСС, к обратному - АААС. Заказывали получившиеся олигонуклеотиды сразу 5'-фосфорелированными.
Отжиг олигонуклеотидов. Олигонуклеотиды разводили до 10 мМ, брали 31 пмоль прямого и столько же соответствующего ему обратного (3,1 мкл), смешивали, нагревали до 95°С и инкубировали 5 мин, затем давали остыть 1 час до комнатной температуры.
Рестрикция плазмиды phU6-gRNA. К 1 мкг плазмиды phU6-gRNA_добавляли 10 единиц активности рестриктазы BstV2I («СибЭнзим», Россия) в поставляемом с ферментом буфере. Рестрикцию проводили при 55°С в течение двух часов. Дефосфорилировали плазмиду с помощью 1 единицы активности щелочной фосфатазы «FastAP» («Thermo Fisher Scientific)), США) 1 час при 37°С, затем инактивировали фермент 15 мин при 75°С. Проводили очистку с помощью набора для очистки ПЦР-продукта NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGEL, Германия) или по протоколу производителя, элюцию проводили в 40 мкл воды, получали концентрацию около 20 нг/мкл.
Лигирование и трансформация бактерий. Реакцию лигирования проводили в 15 мкл: 2 мкл (5 пмоль) олигонуклеотидов, 4 мкл плазмиды (100 нг), 0,5 мкл лигазы Т4 ДНК-лигазы («Thermo Fisher Scientific», США), 1,5 мкл соответствующего лигазного буфера, 7 мкл воды. Негативный контроль - аналогичная реакция, но вместо олигонуклеотидов брали 2 мкл воды. Положительный контроль: 33 нг чистой плазмиды, 0,5 мкл лиг. буфера, 3 мкл воды. Лигирование проводили 20 минут при комнатной температуре.
Затем проводили трансформацию: 3-5 мкл лигазной смеси добавляли к 50 мкл компетентных клеток, подвергали клетки тепловому шоку 40 с при 42°С. Охлаждали 2 мин на льду, добавляли 750 мкл среды SOC, которая представляет собой среду SOB с добавлением глюкозы до 20 мМ. Инкубировали 1 час при 37°С в термомиксере. По 200 мкл размазывали на чашку с агаризованной средой LB (1% w/v триптона, 0,5% w/v дрожжевого экстракта, 170 мМ NaCl, 1,5% агара) с добавлением в качестве селектирующего антибиотика канамицина («Панэко», Россия) 25 мкг/мл. Колонии бактерий выращивались в течение ночи в инкубаторе при 37°С.
Скрининг клонов. На следующий день после трансформации оценивали число колоний в опыте и контролях. Если в опыте выросло колоний значительно больше, чем в негативном контроле, то ставили ПЦР-скрининг: скалывали по несколько колоний в 200 мкл воды, брали этой суспензии 1 мкл и замешивали ПЦР с использованием обратного праймера М13 г и прямого олигонуклеотида (тот, который с добавлением САСС) в качестве прямого праймера. Результаты ПЦР визуализировали на электрофорезе. Так определялись колонии, несущие плазмиду с необходимой вставкой. Из таких колоний затем наращивали ночную культуру (в 5-10 мл среды LB с соответствующим антибиотиком) и выделяли плазмиду с помощью набора «NucleoSpin® Plasmid» (MACHEREY-NAGEL, Германия) или «GeneJET Plasmid Miniprep Kit» («Thermo Fisher Scientific», США) по протоколу производителя. Плазмиду затем секвенировали на предмет корректности вставки, используя праймер М13 г. Метод ENIT для проверки гидовых РНК
Метод «Engineered Nuclease-induced Translocations» (ENIT) предполагает котрансфекцию клеток (например, HeLa) плазмидами, кодирующими нуклеазы и гидовые РНК (в случае проверки работы элементов системы CRISPR/Cas) (фиг. 7). В случае, если нуклеазы эффективно вносят разрывы, то при репарации с некоторой вероятностью образуются хромосомные перестройки между точками разрывов. Спустя 48 часов после трансфекции с ДНК клеток ставится ПЦР на предмет наличия ожидаемых транслокаций, которые происходят между разрезаемыми локусами.
Трансфекция HeLa
За день до трансфекции засеивали в шестилуночный планшет клетки культуры HeLa на 30% конфлюентности. На следующий день трансфицировали клетки: меняли клеткам среду на OptiMEM, затем смешивали плазмиды в равном соотношении, 3-4 мкг суммарно из расчета на одну лунку в 300 мкл среды«Орй-МЕМ» («Gibco», США). Затем добавляли реагент «TurboFect» («Thermo Fisher Scientific)), США) согласно инструкции производителя - 6-8 мкл. Оставляли на 15-20 мин на столе. Затем весь объем добавляли по каплям к клеткам. На следующий день меняли среду на свежую полную DMEM.
Direct-Nested PCR
Транслокации лучше всего детектируются, если в методе ENIT использовать стратегию вложенной (nested) ПЦР. Также вложенная ПЦР менее требовательна к подготовке образцов, поэтому возможно использование «прямой» ПЦР (Direct PCR) без выделения ДНК с помощью специальных наборов.
Спустя 48 часов после трансфекции клетки снимали с чашки с помощью раствора 0,05% трипсина - 0,53 мМ ЭДТА («ПанЭко», Россия) - 300 мкл на лунку, затем добавляли 2,7 мл PBS.
На одну реакцию ПЦР брали 5-100 тысяч клеток. Осаждали 3 мин 5000 rpm на настольной центрифуге. К осадку клеток добавляли 2Х премикс для ПЦР (буфер, нуклеотиды, ионы магния - согласно инструкции производителя к ДНК-полимеразе) и протеиназу К до 0,1 мг/мл так, чтобы получилось по 10 мкл на каждую предстоящую реакцию. Распределяли по 10 мкл в ПЦР-пробирки. Ставили в амплификатор, программа: 10 мин 60° и 3 мин 95°.
После в пробирки добавляли HotStart Taq-полимеразу, праймеры для первой стадии вложенной ПЦР и воду до 20 мкл. Ставился ПЦР1 с внешними праймерами с программой: 95°С 5 мин, 20×(95°С 20 сек, 60°С 1 мин, 72°С 45 сек), 72°С 5 мин.
Затем 1 мкл из реакции разбавлялся в 100 мкл воды и 1 мкл этого разбавленного раствора использовался для ПЦР2 (в 20 мкл) с внутренними праймерами: 95°С 5 мин, 25×(95°С 20 сек, 60°С 1 мин, 72°С 30 сек), 72°С 5 мин. Результаты ПЦР2 анализировались с помощью электрофореза, длины полос сравнивались с теоретически ожидаемыми. Последовательности праймеров даны в таблице 6, а их взаимное расположение на фиг. 8.

Вставка генов гидовых РНК в плазмиду Puro_Cas9 donor
Метод Gibson Assembly. Метод позволяет вставлять в одной реакции сразу несколько фрагментов, причем в любое место плазмиды, где возможно внести разрыв. Для встраивания 2 фрагментов, в качестве места встраивания выбрали сайт узнавания рестриктазой Sail. Использовался набор «Gibson Assembly Master Mix» (NEB, Великобритания), выполняемый протокол основан на протоколе производителя набора.
Подбор праймеров для Gibson Assembly
Праймеры со «свешивающимися» 5'-концами подбирались согласно протоколу Gibson Assembly от NEB, исходя из последовательности плазмиды вокруг точки, куда вставляются фрагменты, в нашем случае вокруг сайта узнавания рестриктазой Sail.
Наработка фрагментов для вставки
В плазмиду Puro_Cas9 donor вставляли гены гидовых РНК, наработанных с помощью ПЦР и с использованием праймеров со свешивающимися 5'-концами, в качестве матрицы использовались проверенные плазмиды phU6-gRNA со вставкой узнающего участка гРНК. Сначала ставили оптимизацию условий ПЦР: ставили градиент по температурам отжига праймеров. В качестве точки отсчета брали температуру отжига, рассчитанную в онлайн-сервисах «NEB Tm Calculator» или «Thermo Fisher Scientific Tm Calculator)). Оценивали качество ПЦР и концентрацию фрагментов по яркости полос на электрофорезе с помощью программы ImageJ. Затем ставили препаративный ПЦР в 50 мкл. Очистили ПЦР-продукт специальным набором для очистки ПЦР NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGEL, Германия), элюировали с колонки в 12 мкл воды (концентрация получалась около 300 нг/мкл).
Подготовка вектора
Ставили рестрикцию 1-2 мкг плазмиды Puro_Cas9 donor (номер #58409, «Addgene», США) рестриктазой SalIl на 3 часа при 37°С. Очищали продукт рестрикции с помощью набора для очистки ПЦР-продукта NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGEL, Германия), элюировали в 10-20 мкл воды.
Gibson Assembly
Каждую реакцию ставили в 4-8 мкл. Ставили контроли: с положительным контролем из набора и отрицательным контролем (вектор без вставок). Предварительно готовили смесь фрагментов для вставки, содержащую по 10 нг каждого фрагмента. В каждую реакцию брали плазмиды 110 нг, 1 мкл олигонуклеотидов (или воды для контроля) и воды до 4 мкл, добавляли 4 мкл Gibson Assembly Master Mix. Помещали в амплификатор на 30 мин на 50°С, затем оставляли на 4°С.
Брали по 2 мкл этой смеси к 50 мкл компетентных клеток, затем проводили трансформацию по обычному протоколу. Высевали клетки на агаровые чашки с ампициллином в качестве селективного антибиотика.
Скрининг колоний
После трансформации на чашках вырастало от нескольких десятков колоний в положительном контроле; в опыте вырастало на порядок больше колоний, чем в негативном контроле. В этом случае ставили ПЦР-скрининг колоний - скалывали штук 20 колоний в 200 мкл воды, брали 1 мкл этой суспензии и замешивали ПЦР с праймерами PuroCas9_F_Sal и PuroCas9_R_Sal, 30 циклов, 60°С отжиг. На электрофорезе выявляются полоски: 200 п. н. - если вставки нет, 500 п.н. - если вставился только один фрагмент, 800 п.н. - если вставились оба фрагмента. В последнем случае такую колонию скалывали, наращивали ночную культуру, выделяли плазмиду с помощью набора «NucleoSpin® Plasmid» и отправляли на секвенирование. Для секвенирования использовали те же праймеры: PuroCas9_F_Sal и PuroCas9_R_Sal. Если секвенирование показывало корректность вставки, то наращивали большой объем культуры (150 мл) с этой плазмид ой и выделяли плазмиду с помощью набора «NucleoBond® Xtra Midi» (MACHEREY-NAGEL, Германия) для дальнейшей трансфекции плазмидой клеток.
Получение линии LCL_iAML/ETO
Чтобы двуцепочечные разрывы не вносились в ДНК еще до начала экспериментов, ген Cas9 находится под индуцируемым промотором системы Tet-On, которая позволяет регулировать экспрессию гена. Система основана на бактериальной системе регуляции экспрессии Tet-оперона, отвечающего за устойчивость бактерии к антибиотикам тетрациклинового ряда. Система состоит двух компонентов: промотора перед регулируемым геном и белка транс-активатора. В состав промотора входит минимальный CMV-промотор и несколько операторных последовательностей (TRE - tetracycline response element), а транс-активатор содержит домен, связывающий эти операторные последовательности только в присутствии тетрациклина или его аналогов, например доксициклина. Также в состав транс-активатора входит сильный трансактивирующий домен вирусного белка VP 16, который запускает сборку инициаторного комплекса РНК-полимеразы II. Принцип работы системы - на фиг. 9
Для интеграции элементов системы CRISPR/Cas и системы Tet-On в геном клеток собрали все элементы в две кассеты в составе двух плазмид. В состав одной кассеты входили ген Cas9 под промотором TRE, гены двух гидовых РНК и ген устойчивости к селективному антибиотику пуромицину. В состав второй кассеты входил ген транс-активатора под конститутивным промотором и ген устойчивости к антибиотику неомицину (генитицину). Чтобы повысить вероятность встраивания этих кассет в геном, мы выбрали стратегию интеграции трансгенов по механизму гомологичной рекомбинации. Встраиваемые кассеты находились в составе плазмидных векторов и были окружены плечами гомологии к определенному участку генома. В качестве такого места в геноме использовался локус AAVS1, который находится внутри первого интрона гена PPP1R12C на 19 хромосоме. Назвали этот локус так, потому что в него in vitro интегрировался вирус AAV, хотя in vivo это и не является местом предпочтительной интеграции этого вируса. Так или иначе, было показано, что трансгены, встроенные в этот локус, демонстрируют стабильную экспрессию, отчего его часто используют, как место для интеграции в геном трансгенов («Safe harbour») (Sadelain, 2011). Одновременно с плазмидами, несущими интегрируемые кассеты, LCL_трансфицировались плазмидами, кодирующими субъединицы TALEN-нуклеазы, которая вносит разрыв в этот локус. Выбор TALEN-нуклеаз вместо более простой системы CRISPR/Cas в данном случае обуславливался тем, что гены гидовых РНК к генам AML1 и ЕТО, находящиеся в состав интегрируемых кассет, находятся под конститутивным промотором hU6 (промотор мяРНК для РНК-полимеразы III). И появление в клетке плазмид, конститутивно экспрессирующих Cas9, привело бы к преждевременным разрывам в локусах AML1 и ЕТО и к нежелательным хромосомным перестройкам еще на стадии получения линии клеток. Схема интеграции кассеты в геном по механизму гомологичной рекомбинации отражена на фиг. 10. Выбор стратегии создания линии также был обусловлен тем, что уже существовали плазмидные векторы, содержащие почти все необходимые элементы системы. Нам оставалось лишь встроить в вектор Puro_Cas9 donor гены гидовых РНК, которые мы подобрали, вставили в плазмиды phU6-gRNA_и проверили методом ENIT. Получение итогового вектора Puro_Cas9+gRNA_AML1_ETO описано в разделе «Вставка генов гидовых РНК в плазмиду Puro_Cas9 donor». Для получения нашей клеточной модели мы использовали культуру LCL, поскольку она является удобной в работе линией лейкоцитов человека. Итак, мы трансфицировали клетки этой культуры одновременно четырьмя плазмидами: Puro_Cas9+gRNA_AML1_ETO, AAVS1-M2rtTA (#60843 «Addgene», США) и двумя плазмидами, содержащими гены субъединиц нуклеазы TALEN, которая вносит разрыв по сайту AAVS1: TALENL (#59025, «Addgene», США) и TALENR (#59026, «Addgene», США) (фиг. 11). Трансфекцию всех плазмид осуществляли путем электропорации по протоколу «Jurkat» с помощью прибора «Gene Pulser Xcell» («Bio-Rad Laboratories», США). Для электропорации использовалось по 2,5 мкг плазмид TALEN_L и TALEN_R, и по 2,3 мкг плазмид AAVS1-M2rtTA и Cas9+gRNA_AML1_ЕТО. После попадания в клетку плазмид начинается транзиентная экспрессия, и начинает нарабатываться TALEN-нуклеаза, которая вносит разрыв в локусы AAVS1 на обеих гомологичных хромосомах. В результате репарации по механизму гомологичной рекомбинации в место разрывов будут встраиваться кассеты, окруженные плечами гомологии. В ряде случаев в одну гомологичную хромосому клетки встроится кассета из плазмиды Puro_Cas9+gRNA_AML1_ETO, содержащая гены гидовых РНК gRNA_AML1 и gRNA_ETO, ген нуклеазы Cas9 и ген устойчивости к пуромицину, а в другую гомологичную хромосому встроится кассета из плазмиды AAVS1-M2rtTA, содержащая ген активатора транскрипции ТА и ген устойчивости к генетицину. Селекция трансфицированных клеток на генетицине и пуромицине позволяет отобрать клетки, содержащие в геноме обе конструкции (фиг. 12), для краткости полученную линию назвали линией LCL_iAML/ETO.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования "Московский государственный университет имени М.В.Ломоносова" (МГУ)
<120> ТЕСТ-СИСТЕМА ДЛЯ ПОИСКА ПРЕПАРАТОВ, СНИЖАЮЩИХ РИСК ВОЗНИКНОВЕНИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ
<160> SEQ ID NО:1
<210> 1
<211> 20
<212> AML
<213> Artificial sequence
<400> 1
1 GACTCCCCCA TGTACCCCTA
<160> SEQ ID NО:2
<210> 2
<211> 20
<212> ETO
<213> Artificial sequence
<400> 2
1 GATGTAAGAG GAAGCAGCTT
<160> SEQ ID NО:3
<210> 3
<211> 20
<212> AML_f1
<213> Artificial sequence
<400> 3
1 TGGGGAAGCT CACCAGATAG
<160> SEQ ID NО:4
<210> 4
<211> 20
<212> ETO_f1
<213> Artificial sequence
<400> 4
1 TTGGGACACC TAGGAGTGGT
<160> SEQ ID NО:5
<210> 5
<211> 24
<212> Myc_f
<213> Artificial sequence
<400> 5
1 CCAGTAACTC CTCTTTCTTC GGAC
<160> SEQ ID NО:6
<210> 6
<211> 22
<212> Myc_r
<213> Artificial sequence
<400> 6
1 CGCTATGCTG GATTTTGCTG CA
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОЗДАНИЯ КЛЕТОЧНЫХ МОДЕЛЕЙ ТРАНСЛОКАЦИЙ С ПОМОЩЬЮ ИНСТРУМЕНТОВ РЕДАКТИРОВАНИЯ ГЕНОМА | 2023 |
|
RU2827872C1 |
| КЛЕТОЧНАЯ ЛИНИЯ И СПОСОБ ОЦЕНКИ ЭФФЕКТИВНОСТИ ГЕНОМНЫХ РЕДАКТОРОВ | 2022 |
|
RU2831064C2 |
| Способ получения генно-модифицированных кроликов с нокаутом гена LEPR с помощью системы CRISPR/Cas9 | 2023 |
|
RU2836438C1 |
| Способ диагностики острых лейкозов и наборы для его выполнения | 2021 |
|
RU2806591C2 |
| Способ конструирования минигенов млекопитающих и рекомбинантная плазмида pgC1HDR, кодирующая миниген ингибитора С1 эстеразы человека, предназначенная для получения гуманизированных по гену Serping1 мышей | 2022 |
|
RU2805177C1 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ НАЦЕЛЕННОЙ ГЕНЕТИЧЕСКОЙ МОДИФИКАЦИИ С ИСПОЛЬЗОВАНИЕМ ПАРНЫХ ГИДОВЫХ РНК | 2015 |
|
RU2734770C2 |
| КОМПОНЕНТЫ СИСТЕМЫ CRISPR-CAS, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ | 2013 |
|
RU2701662C2 |
| Способ выявления структурных перестроек генома опухолевых клеток (химерных генов), определяющих выбор терапии и прогноз при острых лейкозах у детей, с использованием ОТ-ПЦР и последующей гибридизацией с олигонуклеотидным биологическим микрочипом (биочипом) | 2016 |
|
RU2639513C1 |
| СПОСОБ СОЗДАНИЯ НОВОГО ГЕНА В ОРГАНИЗМЕ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2832668C1 |
| Набор для определения копийности вставки интересующей конструкции в AAVS1 локус генома человека | 2021 |
|
RU2786396C1 |
Изобретение относится к биотехнологии, а именно к технологиям поиска новых терапевтических препаратов или комбинаций препаратов (химических соединений), улучшающих существующие терапевтические подходы к лечению рака. Предложена тест-система, к которой добавляется исследуемый препарат (тестируемое вещество), а затем по числу возникших транслокаций по сравнению с контролем делается вывод о риске возникновения вторичного лейкоза. Предложена тест-система, позволяющая изучать влияние различных веществ на частоту образования перестроек между локусами AML и ЕТО, а значит их влияние на вероятность возникновения вторичных лейкозов. 3 н. и 15 з.п. ф-лы, 12 ил., 6 табл., 1 пр.
1. Тест-система для исследования влияния препарата, используемого при химиотерапии, на вероятность возникновения вторичного лейкоза, представляющая собой лимфобластоидные клетки человека (LCL) RPMI 8866, геном которых содержит генетическую конструкцию, включающую гены систем направленного внесения двуцепочечных разрывов в хромосомы клетки в локусы, в качестве которых используют эндонуклеазу Cas9 и гидовые РНК, между которыми происходит лейкозогенная хромосомная перестройка, а также гены активатора транскрипции ТА и гены селективных маркеров, при этом гены направленного внесения двуцепочечных разрывов находятся под контролем индуцируемого трансактиватором ТА промотора, а в качестве локуса, между которым происходит лейкозогенная хромосомная перестройка, используют перестройку между генами АМL1 и ЕТО.
2. Тест-система по п. 1, характеризующаяся тем, что в качестве линии клеток млекопитающих используют клетки, скорость деления которых составляет не менее одного деления в 2-3 суток и геном которых изучен настолько, чтобы было возможным подобрать нуклеазы, узнающие локусы, между которыми формируются хромосомные перестройки.
3. Тест-система по п. 1, характеризующаяся тем, что в качестве селективных маркеров используют гены устойчивости к антибиотикам пуромицину и неомицину.
4. Тест-система по п. 1, характеризующаяся тем, что гены системы направленного внесения двуцепочечных разрывов интегрированы в геном клеток исходной линии в локус, обеспечивающий их стабильную экспрессию.
5. Тест-система по п. 1, характеризующаяся тем, что гены системы направленного внесения двуцепочечных разрывов интегрированы в области первого интрона гена PPP1R12C, расположенного на 19 хромосоме.
6. Способ получения тест-системы по п. 1, характеризующийся тем, что производят создание генетической конструкции (1), включающей гены гидовых РНК, ген нуклеазы Cas9, находящийся под индуцируемом промотором TRE, и ген устойчивости к пуромицину; и генетической конструкции (2), включающей ген активатора транскрипции ТА и ген устойчивости к генетицину, затем полученные генетические конструкции трансфицируют в клетки исходной клеточной линии с последующим отбором клеток, содержащих генетические конструкции.
7. Способ по п. 6, характеризующийся тем, что используют гидовые РНК gRNA _AML1 и gRNA_ETO.
8. Способ по п. 6, характеризующийся тем, что создание генетической конструкции (1) проводят амплификацией участков векторов, кодирующих гидовые РНК, и последовательностей, способствующих их встраиванию в геном трансфицируемых клеток, встраиванием полученных участков в плазмиду, содержащую ген Cas9 под индуцируемым промотором TRE и ген устойчивости к пуромицину.
9. Способ по п. 8, характеризующийся тем, что в качестве последовательностей, способствующих встраиванию, используют участки, повторяющие последовательности длиной от 100 до 1000 нуклеотидов с каждой стороны от точки разрыва в геноме.
10. Способ по п. 6, характеризующийся тем, что трансфекцию полученных генетических конструкций в клетки исходной клеточной линии осуществляют посредством электропорации.
11. Способ по п. 6, характеризующийся тем, что отбор клеток производят посредством выращивания клеток в средах с селективными антибиотиками в концентрациях, при которых погибают клетки, не несущие генов устойчивости к данным антибиотикам, но при которых выживают клетки, несущие селективные маркеры.
12. Способ проведения исследования влияния препарата, используемого при химиотерапии, на вероятность возникновения вторичного лейкоза, с помощью тест-системы по п. 1, характеризующийся тем, что к суспензии, содержащей не менее 2 млн. клеток тест-системы, добавляют исследуемый препарат в концентрации, не превышающей IC50 для данного препарата, и доксициклин в концентрации, достаточной для запуска экспрессии Cas9, проводят инкубацию полученной смеси с последующим проведением количественной ПЦР и анализом полученных результатов и вывод о снижении вероятности возникновения вторичного лейкоза делают по уменьшению количества полученных транслокаций по сравнению с количеством транслокаций в контрольном образце.
13. Способ по п. 12, характеризующийся тем, что концентрация доксициклина составляет от 0,1 до 1 мкг/мл.
14. Способ по п. 12, характеризующийся тем, что количественную ПЦР проводят с использованием праймеров ETO_f1-AML_f1 и MYC_f-MYC_r.
15. Способ по п. 14, характеризующийся тем, что используют праймеры с последовательностями SEQ ID NO: 3-6.
16. Способ по п. 14, характеризующийся тем, что количественную ПЦР проводят по программе: активация полимеразы: 95°С, 10 мин; денатурация ДНК: 95°С, 20 сек, отжиг праймеров: 60°С, 1 мин; стадия элонгации: 72°С, 30 сек, всего 42 цикла, затем кривая плавления: 55-95°С, шаг 0.5°С в 5 сек.
17. Способ по п. 14, характеризующийся тем, что анализ результатов ПЦР проводят путем подсчета количества транслокаций, полученные значения порогового цикла амплификации образца ДНК с соответствующей парой праймеров (Ct) подставляют в формулу (1):
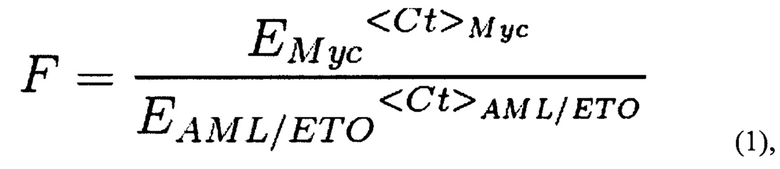
где F - частота транслокаций AML/ETO, нормированная на количество ампликона праймеров Myc_f и Мус_r, Емус - эффективность амплификации ДНК с пары праймеров Myc_f и Myc_r, Eaml/eto - эффективность амплификации ДНК с пары праймеров ETO_f1 и AML_f1, которая определяется один раз в предварительном эксперименте в условиях, максимально соответствующих условиям дальнейшей работы; <Ct>i - среднее по трем повторностям значение порогового цикла амплификации образца ДНК с соответствующей парой праймеров, где i означает Мус или AML1/ETO.
18. Способ по п. 14, характеризующийся тем, что сравнение данных, полученных исследуемых образцах и на контрольных, делают на основании множественного сравнения экспериментальных выборок с контрольной с помощью непараметрического критерия Даннета.
| ДИАГНОСТИКУМ И ТЕСТ-СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ АКТИВНОСТИ АНТИРАБИЧЕСКИХ СЫВОРОТОК И ПРЕПАРАТА ГЕТЕРОЛОГИЧНОГО АНТИРАБИЧЕСКОГО ИММУНОГЛОБУЛИНА IN VITRO МЕТОДОМ ДОТ-ИММУНОАНАЛИЗА | 2008 |
|
RU2360252C1 |
| WO 2003070972 A2, 28.08.2003 | |||
| E Kostareli et al, AML1/ETO and POU4F1 synergy drives B-lymphoid gene expression typical of t(8;21) acute myeloid leukemia, Leukemia (2012), 26, 8.11.2011, p | |||
| ЦЕНТРОБЕЖНАЯ ЗЕРНОСУШИЛКА | 1919 |
|
SU1106A1 |

