Область техники, к которой относится изобретение
Настоящее изобретение относится к биотехнологиям, а именно, к технологиям создания клеточных линий с транслокациями с использованием нуклеаз-генетических редакторов; к медицинским и научным технологиям, а именно, к технологиям моделирования на клетках событий, происходящих при лейкозах, с целью изучения и подбора наиболее эффективных лекарств. Создание клеточных линий с транслокациями позволяет получить удобную модель лейкозов для их изучения на клеточном уровне. Так, можно проводить исследование дифференциальной экспрессии генов на полученных клетках в сравнении с клетками без транслокации или же сравнивать между собой исходно однородные клетки с разными транслокациями. Результаты таких исследований позволят лучше понять особенности лейкозных клеток и выявить потенциальные мишени для терапии. На полученных клетках возможно тестировать различные химиотерапевтические агенты. Для этого проводят обработку клеток с транслокацией и клеток без транслокации различными терапевтическими соединениями и затем ставят цитотоксические тесты для выявления соединений, наиболее эффективно действующих на клетки с транслокацией.
Уровень техники
Получение клеточных линий с транслокациями является важной задачей как для исследовательских целей, так и для практического применения. Изучение клеточных моделей с разными транслокациями позволяет выявить механизмы образования транслокаций и механизмы развития рака, ассоциированного с данными транслокациями. А тестирование лекарств на клеточных моделях с транслокациями позволит лучше подобрать разные лекарства для разных типов рака, в зависимости от конкретной транслокации.
Создание клеток с транслокациями начиналось с применения Cre-Lox систем. В геном клеток в целевые локусы встраивают LoxP-последовательности, и при экспрессии в этих клетках трансгена, кодирующего Cre-рекомбиназу, между LoxP-сайтами происходила рекомбинация, которая приводила к образованию хромосомных транслокаций между целевыми локусами. Примером такого подхода является работа Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse (doi: 10.1093/embo-reports/kvd027). Такой подход является очень времязатратным - требуется отселектировать клетки сначала с LoxP-сайтом в одной хромосоме, потом с LoxP-сайтом в другой, проверить правильность вставки, затем запускать работу Cre-рекомбиназы и потом отбирать клетки с транслокацией. При этом частота возникновения транслокаций в клетках не очень высока, так, в упомянутой работе она не превышала 1 × 10-4, в другой аналогичной работе, Cre-Mediated Site-Specific Translocation between Nonhomologous Mouse Chromosomes (http://www.jstor.org/stable/2368247), частота транслокаций в клетках, содержащих LoxP-сайты в целевых локусах и экспрессирующих Cre, оценивается как 1 на 1,2-2,4 × 10-3.
Технологической революцией в получении клеток с транслокациями стало использование программируемых нуклеаз. Экспрессия в клетке нуклеаз, направленных на два целевых сайта, приводит к возникновению транслокации между этими сайтами в некоторых из клеток. Количество клеток, в которых возникает транслокация, может варьировать, но в среднем составляет от 1 на 300 клеток до 1 на 10000 клеток.
Для получения клеточной культуры, в которой все клетки содержат транслокацию, необходимо отобрать отдельные клетки с транслокацией из смешанной популяции, и получить из них так называемую моноклональную культуру. Чтобы отобрать клетки, содержащие транслокацию, применяют различные подходы. Самый простой - клонирование культуры. Трансфицированные нуклеазами клетки распределяются по одной в лунки планшета. Из единичных клеток вырастают клоны. Когда клоны подрастают, из них выделяют ДНК и проводят ПЦР-анализ на наличие в них искомой транслокации. По такому алгоритму с применением CRISPR/Cas получены линии клеток человека eHAP с перестройкой CD74-ROS1 (Efficient generation and reversion of chromosomal translocations using CRISPR/Cas technology, https://doi.org/10.1186/s12864-016-3084-5). Плазмиды, кодирующие гены гидовых РНК, а также плазмиду, кодирующую Cas9 и плазмиду с устойчивостью к антибиотику бластицидину, трансфицировали вместе в клетки с помощью реагента для трансфекции Turbofectin (Origene). Затем популяцию клеток обогащали трансфицированными клетками за счет обработки антибиотиком бластицидином в течение 2=3 дней. Клетки разводили с помощью серии разведений до концентрации 15 штук в мл, затем распределяли по 50 мкл в лунки 384-луночного планшета и далее ждали, пока из них вырастут клоны для дальнейшего анализа с помощью ПЦР. Всего смогли проанализировать 192 клона, из которых обнаружили 2 положительных. Данный метод показал возможность получения транслокаций, однако отличается довольно большими затратами времени и ресурсов: так, для получения 2 клонов авторы проводили вручную сериальные разведения, следили за нарастающими клонами с помощью специализированного оборудования, которое имеется далеко не во всех лабораториях = робота для работы с клетками Microlab STAR Line robotic system, и провели множество реакций ПЦР (как минимум по одной с каждым из 192-х клонов). Для анализа клеток с помощью ПЦР их лизировали набором реактивов DirectPCR Lysis reagent (PeqLab), предполагающим использование от ≈ 40 000 клеток и более и их лизис в течение ночи.
Другой пример - получение линии мышиных стволовых клеток (mESC) с перестройкой Cdx2-Gsk3α и линии клеток HEK293 c транслокацией BCR-ABL1 (Induction of site-specific chromosomal translocations in embryonic stem cells by CRISPR/Cas9, https://doi.org/10.1038/srep21918). Авторы сначала получили линию клеток mESC, экспрессирующих Cas9, а затем доставляли гены РНК-гидов с помощью лентивирусной трансдукции. Эти клетки сначала распределяли по 5 штук в лунку, и обнаружили с помощью ПЦР-анализа 3 положительные лунки из 33-х. Затем клетки из положительных лунок клонировали уже по 1 клетке. В трех из 16 моноклонах была перестройка. Для клеток HEK293 c транслокацией BCR-ABL1 авторы не указывают число проанализированных клонов, однако отмечают, что этот анализ отличается времязатратностью. Недостатком стратегии является то, что для получения одной и той же транслокации в нескольких разных клеточных линиях требуется предварительно в каждой из них провести интеграцию в геном гена Cas9, а также то, что использование лентивирусной трансдукции будет приводить к постоянной экспрессии системы CRISPR/Cas, которая обладает нецелевой активностью и в полученной линии клеток будет постоянно вносить мутации в нецелевые сайты.
Оба описанных подхода используют клонирование и ПЦР-анализ многочисленнных клонов для обнаружения тех, что содержат транслокацию. Однако был предложен и другой метод, который предполагает возможность отбора (селекции) клеток, в которых произошло событие образования целевой транслокации, что позволяет обойтись без клонирования. Таким подходом является интеграция в точку перестройки селектирующей кассеты. Для этого в клетки помимо кодирующих нуклеазы плазмид вносится плазмида, содержащая участки гомологии к двум перестраивающимся локусам, между которыми заключена селектирующая кассета. Кассета кодирует флуоресцентный белок или белок устойчивости к антибиотику. В этом случае клетки с перестройкой можно отобрать из популяции по флуоресценции или по устойчивости к антибиотику. С помощью такого подхода получили клетки с транслокацией EWSR1-WT1 на основе мезенхимальных стволовых клеток человека hMSC (Generation of chromosomal translocations that lead to conditional fusion protein expression using CRISPR-Cas9 and homology-directed repair, doi: 10.1016/j.ymeth.2017.05.006). Однако этот подход ненадежен, так как во многих случаях отбираются клетки, которые не несут транслокации, а лишь несут кассету, встроившуюся по негомологичному соединению концов разрыва ДНК. Также после получения транслокации необходимо вырезать эту кассету с помощью экспрессии Cre-рекомбиназы. Дополнительной проблемой является сложность диагностики транслокации с помощью ПЦР, так как праймеры, фланкирующие встроившуюся конструкцию и плечи гомологии, располагаются далеко друг от друга, и такая ПЦР требует дополнительной оптимизации для надежной детекции. Использование подобного подхода для получения линий с транслокациями на клетках LCL не было надежным и не дало выигрыш во времени относительно заявляемого способа (данные нашей лаборатории). Так, в попытке моделировать различные транслокации удалось получить только одну линию из 4-х, и на это было затрачено 16 недель.
Техническая проблема, решаемая посредством заявляемого изобретения, заключается в необходимости преодоления недостатков, присущих аналогам за счет разработки способа получения клеточных культур для моделирования и изучения процессов, происходящих в лейкозных клетках с определенной хромосомной транслокацией, и для тестирования лекарств, а также разработка универсального и быстрого способа получения таких линий.
Раскрытие изобретения
Техническим результатом изобретения является возможность получения модельных культур за более короткий промежуток времени (8 недель) из перевиваемых (иммортализованных) клеток с вероятностью воспроизведения не менее 0,95.
Можно выделить следующие преимущества заявляемого способа. Во-первых, надежность способа получения - разработанный способ позволяет оценить вероятность транслокации и, исходя из нее, модифицировать протокол, чтобы максимально быстро получить клеточную линию с целевой перестройкой с вероятностью 95%. Во-вторых, способ требует минимальных затрат рабочего времени и ресурсов лаборатории - при анализе субпопуляций и клонов используется минимально возможное число реакций ПЦР с минимально возможным числом клеток, а процедура субклонирования осуществляется с помощью клеточного сортера, а не методом предельного разведения.
Технический результат достигается способом создания клеточной модели лейкоза для моделирования и изучения процессов, происходящих в лейкозных клетках с определенной хромосомной транслокацией, и для тестирования на них лекарств, включающий забор образца/клеток у пациента, выявление целевых локусов, которые ассоциированы с исследуемой патологией, т.е. определение в геноме локусов перестройки, затем синтезируют по меньшей мере одну плазмидную конструкцию, кодирующую инструменты редактирования генома, РНК-гиды, направленные на целевые локусы (локусы перестройки), и включающие флуоресцентный белок, проводят трансфекцию клеток полученной плазмидой или плазмидами, после трансфекции клетки, экспрессирующие флуоресцентный белок, отбирают с помощью клеточного сортера и распределяют по 20±10 штук в лунки планшета со средой, наращивают субпопуляцию клеток до 20 000±10 000 клеток, затем анализируют полученные субпопуляции на предмет наличия в них клеток с транслокацией с помощью прямой вложенной ПЦР и клонируют субпопуляции с подтвержденной транслокацией на моноклональные линии, среди которых выявляют линии с транслокацией, подтверждают экспрессию слитого гена, при этом для анализа субпопуляций сначала оценивают содержание клеток с транслокацией сначала в группе лунок и ставят одну ПЦР со смешанным материалом, при обнаружении транслокации, проверяют содержимое в индивидуальных лунках. При этом клетки из нескольких лунок объединяют в группы от 2 до 12 лунок, в случае отсутствия транслокации в группе из дальнейшей работы исключают клетки из всех лунок такой группы. Субпопуляцию клеток с подтвержденной перестройкой клонируют с помощью клеточного сортера и наращивают моноклоны до 10000 - 100000 клеток. В качестве инструментов редактирования генома используют нуклеазы, специфически распознающие целевые локусы, а в качестве инструментов редактирования используют системы на основе CRISPR/Cas, нуклеаз-цинковых пальцев, TALE-нуклеаз, эндонуклеаз для встраивания в геном мобильных генетических элементов, а также других нуклеаз, обладающих свойством узнавать и разрезать целевые локусы. В качестве флуоресцентного белка используют зеленый флуоресцентный белок из Aequorea victoria и его гомологи, биливердин-связывающие бактериофитохромы и цианобактерихромы, фикоцианобилин-связывающие цианобактериальные фитохромы, малые дальнекрасные флуоресцентные белки, малые зеленые кислород-независимые флуоресцентные белки, а также флуоресцентно-активируемые метки. При этом плазмидная конструкция для трансфекции содержит ориджин и ген устойчивости к селективному антибиотику и кодирует инструменты редактирования генома, включает участок, содержащий сайт узнавания рестриктаз типа IIS, в который встраивают последовательность, отвечающую за узнавание нуклеазой целевого локуса, а также кодирует гены элементов системы редактирования генома и ген флуоресцентного белка. В качестве клеток используют клетки, способные пройти более 30-40 делений, например, иммортализованные клетки или культуры опухолевых клеток. Трансфекцию клеток осуществляют с помощью химической трансфекции, электропорации, трансдукции с помощью вирусных частиц. Оценку клеток с транслокациями осуществляют методом ПЦР. Для получения моноклонов проводят распределение клеток из популяций, содержащих транслокацию, по одной клетке в лунку.
Для изучения лейкозогенеза (образования гемобластозов) используют клеточную линию, представляющую собой модифицированные иммортализованные лимфобласты линии LCL, содержащие транслокацию MLL-ARHGAP26, полученную заявляемым способом. Для получения клеточной модели используют иммортализованные лимфобласты (линия LCL), определяют локусы перестройки внутри генов MLL-ARHGAP26 и синтезируют генетическую конструкцию px330_gRNA_Cas9 Santaka и плазмиду 53188-PcisI, вставляют в px330_gRNA_Cas9_Santaka ген для второй гидовой РНК из плазмиды 53188-PcisI, по сайтам рестриктаз HindIII и BstAFI, в полученную плазмиду px330_gRNA_Cas9_Santaka_gRNA вносят с помощью рестриктазы BstV21 узнающий участок одной гидовой РНК к MLL, а с помощью рестриктазы PcisI вносят узнающий участок другой гидовой РНК к ARHGAP26, полученной плазмидой px330_gMLL_Santaka_gARH проводят трансфекцию клеток LCL, через 2 суток сортируют клетки по маркеру трансфекции Santaka по 20±10 клеток в лунку 96-луночного планшета, наращивают субпопуляцию клеток до 20 000±10 000 клеток, оценивают содержание клеток с транслокацией с помощью прямой вложенной ПЦР сначала в группе лунок, затем уже в индивидуальных лунках, субпопуляцию клеток с подтвержденной перестройкой клонируют с помощью клеточного сортера и наращивают моноклоны до 10000 - 100000 клеток; затем с помощью прямой вложенной ПЦР определяют наличие транслокации и моноклон клеток с транслокацией используют для дальнейших исследований. Конструкция px330_gRNA_Cas9 Santaka включает элементы системы CRISPR/Cas, вносящие разрывы в локусы в 11-м интроне гена MLL и в 11-м интроне гена ARHGAP26, и содержит ген флуоресцентного белка Santaka, последовательность SV40-промотора, сайт рестриктазы BmtI, полученная на основе коммерческих плазмид px330 и phU6-gRNA, а также набора праймеров с последовательностями SEQ ID № 1 - SEQ ID № 4:
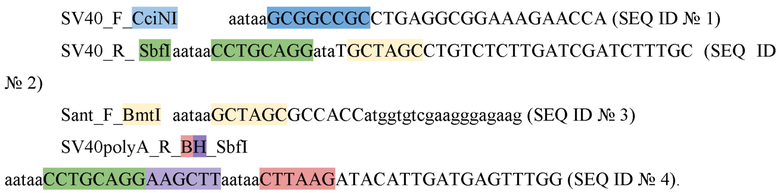
Для синтеза генетической конструкций плазмиды 53188-PcisI используют коммерческую плазмиду phU6-gRNA с олигонуклеотидами, образующими при отжиге друг на друга вставку с сайтами PciSI и набор праймеров с последовательностями SEQ ID № 5 - SEQ ID № 8:
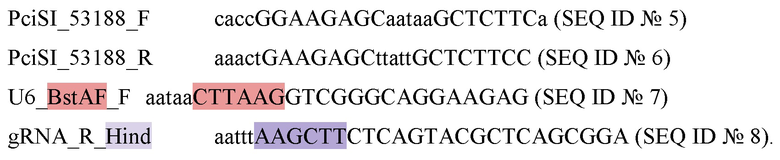
Для проверки наличия транслокации методом вложенной ПЦР используют праймеры с последовательностями SEQ ID № 9 - SEQ ID № 12:
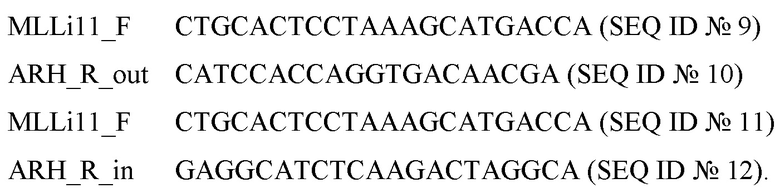
Проверку наличия транслокации проводят методом вложенной ПЦР в два этапа, где на первом этапе ПЦР проводят амплификацию с помощью следующих температурных режимов: 95°C 5 мин, (95°С 20 с, 58°С 1 мин, 72°С 1 мин) × 20, 72°С 5 мин, после прохождения ПЦР 4 мкл реакционной смеси добавляют к 396 мкл деионизированной воды, затем 4 мкл полученного раствора используют в качестве матрицы на втором этапе ПЦР. На втором этапе ПЦР проводят амплификацию с помощью следующих температурных режимов: 95°C 5 мин, (95°С 20°с, 58°С 1 мин, 72°С 40 с) × 30, 72°С 5 мин, результаты анализируют методом электрофореза в агарозном геле.
Краткое описание чертежей
Изобретение поясняется следующим иллюстративным материалом.
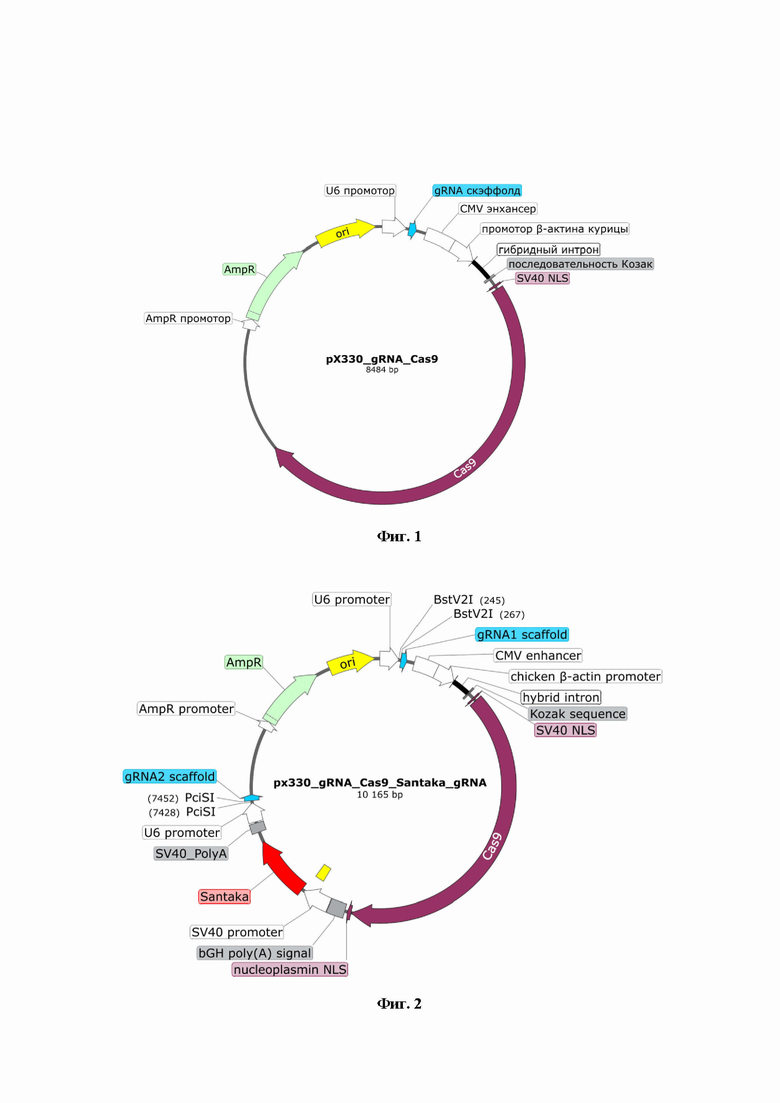
На фиг. 1 показана карта плазмиды pX330_gRNA_dCas9, на основе которой получают плазмидные конструкции, индуцирующие транслокацию. На схеме отмечены: ген нуклеазы Cas9, содержащей SV40 NLS, и необходимые для его экспрессии элементы (CMV-энхансер, промотор бета-актина, гибридный интрон, последовательность Козак), элементы гена гидовой РНК (промотор U6, участок, кодирующий скэффолд гидовой РНК) и другие элементы плазмиды, необходимые при клонировании.
На фиг. 2 показана карта плазмиды, из которой путем клонирования по сайтам BstV2I и PciSI получают плазмиду, индуцирующую перестройку. На схеме отмечены: гены нуклеазы Cas9 и флуоресцентного белка Santaka, содержащей SV40 NLS, и необходимые для его экспрессии элементы (CMV-энхансер, промотор бета-актина, гибридный интрон, последовательность Козак), элементы генов гидовых РНК (промотор U6, участок, кодирующий скэффолд гидовой РНК), и другие элементы плазмиды, необходимые при клонировании.
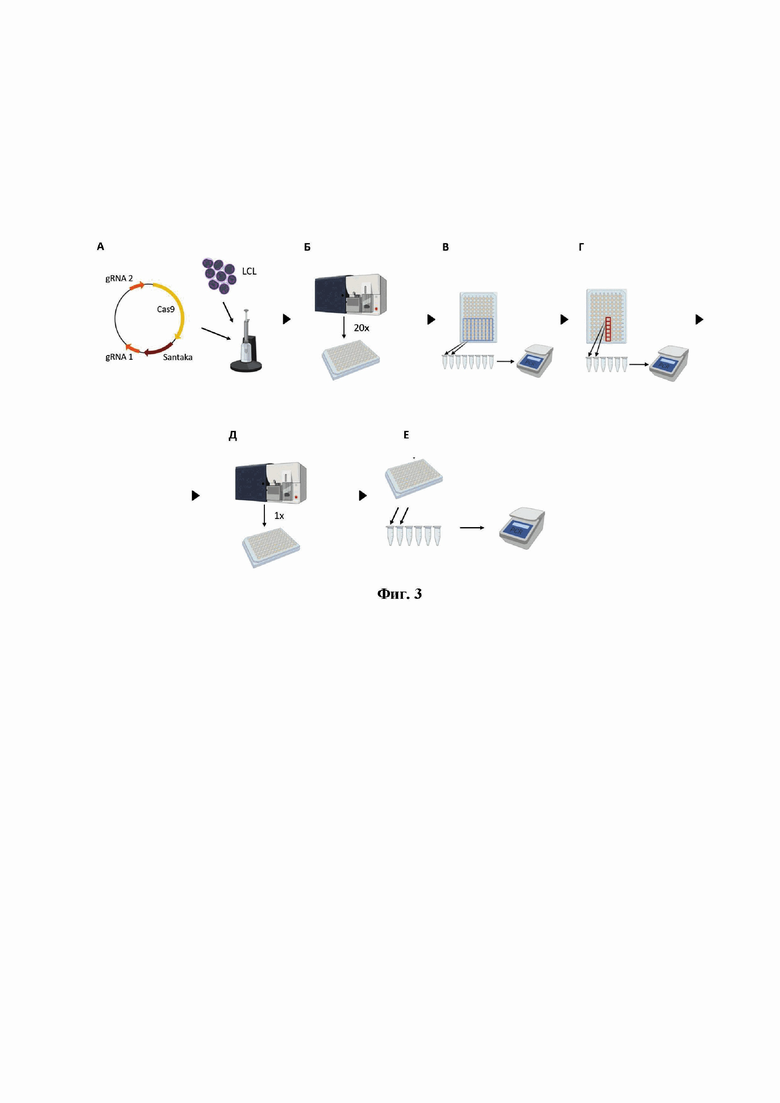
На фиг. 3 представлена схема заявляемого способа получения клеточных моделей, где А. Трансфекция клеток плазмидой для индукции транслокации. Б. Сортинг по флуоресценции Santaka по 20 клеток в лунку планшета. В. Анализ на наличие транслокации групп из 6 лунок с клетками с помощью прямой вложенной ПЦР. Г. Анализ на наличие транслокации индивидуальных лунок групп, которые на предыдущем шаге показали наличие транслокации. Д. Сортинг клеток по одной в лунки планшета для получения моноклонов. Е. Анализ моноклонов на наличие транслокации с помощью прямой ПЦР.
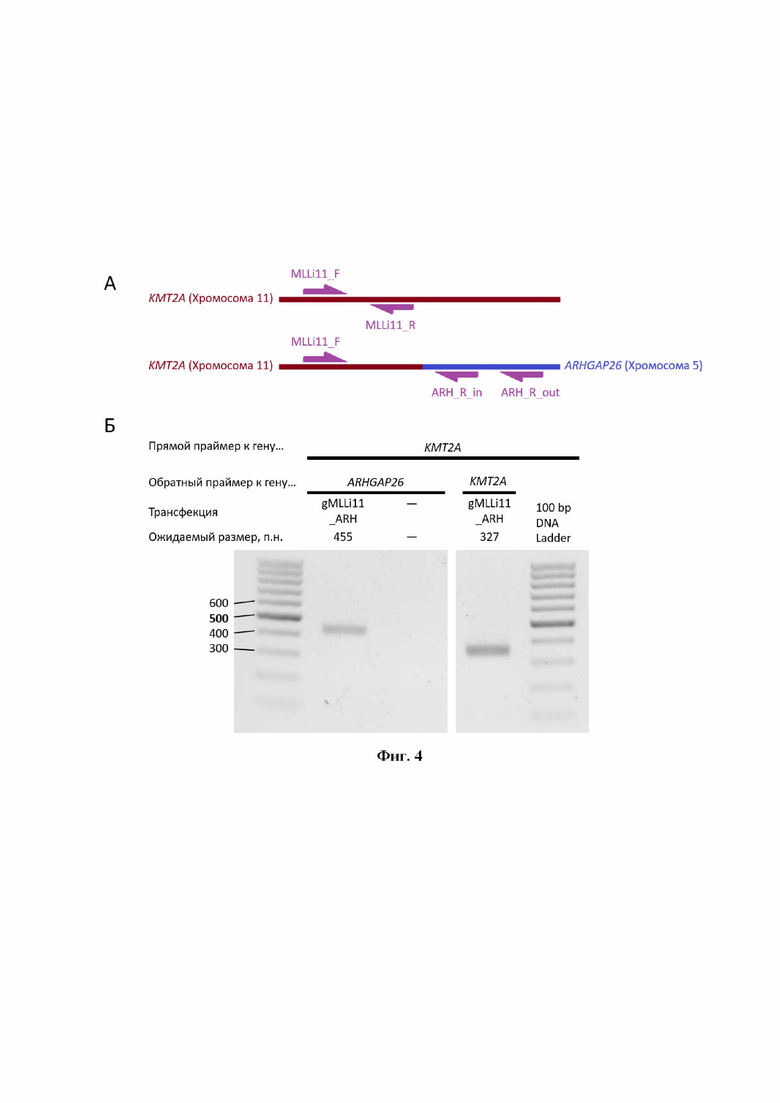
На фиг. 4 представлены: А. схема расположения точек отжига праймеров на интактном гене MLL и на слитом гене MLL-ARHGAP26. Б. электрофореграмма, демонстрирующая результаты ПЦР на транслокацию MLL-ARHGAP26.
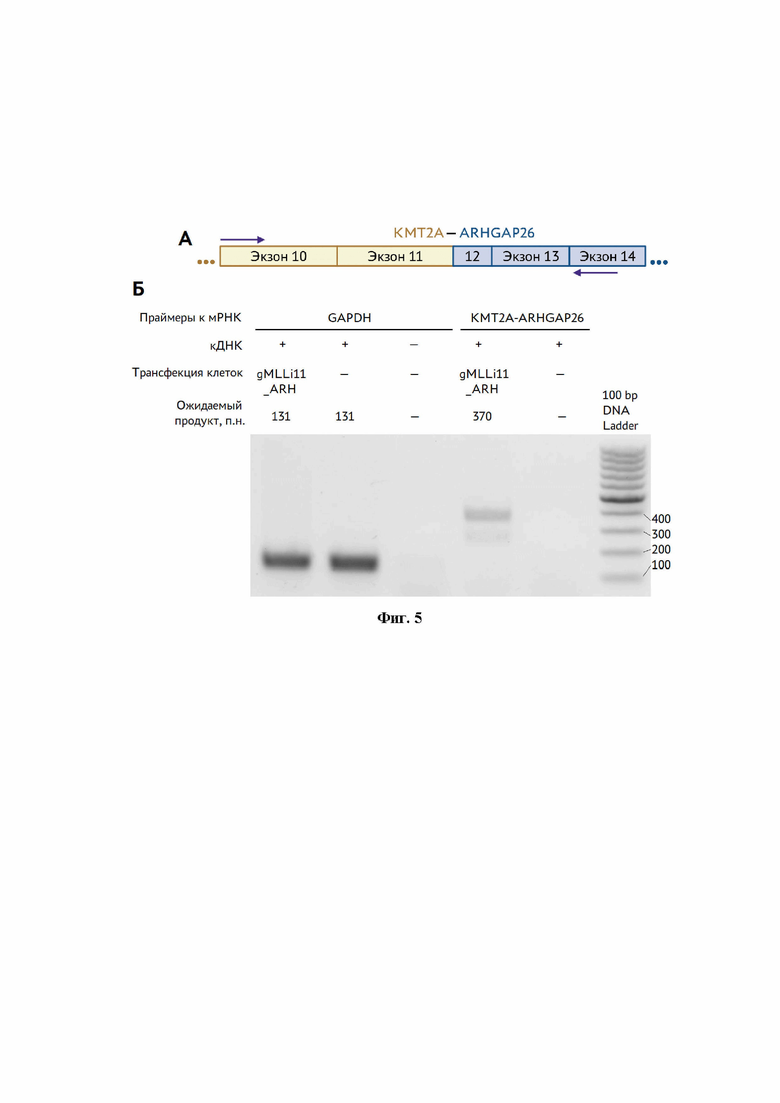
На фиг. 5 представлена электрофореграмма, демонстрирующая результат проверки экспрессии слитого гена. А. Схема расположения праймеров при проверке полученных клеток на экспрессию мРНК слитого гена. Б. Электрофорез продуктов ПЦР с обратной транскрипцией. GAPDH используют в качестве контроля реакции.
Осуществление изобретения
Для моделирования транслокации, ассоциированной с лейкозом, определяют точки, между которыми происходит перестройка у пациентов (PMID: 37019990). Существует множество способов определения локусов перестройки, как с точностью до интрона перестраивающегося гена, так и с точностью до нуклеотида (см. обзор методов: PMID: 33205680). Для моделирования перестройки, как у пациентов, достаточно вносить в каждом из генов-партнеров разрыв в любую точку внутри интрона, в котором произошел разрыв у пациента (кроме сайтов сплайсинга), так как интрон не содержит белок-кодирующей последовательности, а онкогенный эффект перестройки обуславливается химерным белком. Выбор целевого локуса проводят, исходя из данных о точках разрыва, а также возможности внесения разрыва программируемой нуклеазой. Так, при использовании нуклеазы Cas9 из Streptococcus pyogenes для ее работы необходима последовательность GG, следующая через 1 нуклеотид от 20-нуклеотидного участка, узнаваемого РНК-гидом - так называемый РАМ (protospacer-adjacent motif), а ортологи Cas9 из других бактерий используют следующие РАМ’ы: SaCas9 из Staphylococcus aureus - NNGRRT (R=A, G) (PMID: 25830891); CjCas9 из Campylobacter jejuni - N4RYAC (Y=C, T) (PMID: 28220790); St1Cas9 из Streptococcus thermophilus - NNRGAA (PMID: 31900288). Для каждой описанной в научной литературе нуклеазы Cas всегда определен ее РАМ.
Заявляемый способ получения клеточных линий с транслокациями заключается в следующем.
Во-первых, получают плазмидную конструкцию, кодирующую инструменты редактирования генома, направленные на 2 целевых локуса (между которыми происходит перестройка), а также флуоресцентный белок. Плазмиды должны содержать все элементы, необходимые для репликации и селекции. Используются любые способы сборки плазмидных конструкций. Элементы, из которых получают плазмиду (или плазмиды) могут быть получены из других плазмид или синтезированы (проведен синтез гена). Необходимые элементы плазмид и методы сборки описаны в электронной книге Plasmids 101, Addgene. Допустимо разбивать эти компоненты на несколько плазмид, однако это снижает эффективность трансфекции. Также допустимо доставлять инструменты редактирования генома в клетки непосредственно в виде белков или нуклеопротеиновых комплексов любым способом, либо же с помощью трансфекции клеток мРНК, кодирующими эти компоненты.
В качестве инструментов редактирования генома могут выступать любые нуклеазы, специфически распознающие целевые локусы, между которыми получают транслокацию. Выбирают те, что более эффективны, из доступных. Для сравнения эффективности используют методы, описанные в статье PMID: 31876277. В качестве инструментов-редакторов генома выступают системы на основе CRISPR/Cas, нуклеаз-цинковых пальцев, TALE-нуклеаз, эндонуклеаз хоуминга мобильных генетических элементов и другие программируемые нуклеазы или мегануклеазы.
В качестве флуоресцентного белка выступают любые белки-гомологи avGFP (зеленый флуоресцентный белок из Aequorea victoria) - GFP, RFP, CFP, YFP и т.д., а также другие флуоресцентные белки, например, биливердин-связывающие бактериофитохромы и цианобактерихромы, а также фикоцианобилин-связывающие цианобактериальные фитохромы, малые дальнекрасные флуоресцентные белки - smURFP и малые зеленые кислород-независимые флуоресцентные белки, например, на основе флуоресцентных белков из угря (UnaG). Также возможно применение флуоресцентно-активируемых меток (fluorescence-activating and absorption-shifting tag), которые требуют добавления к клеткам флуорогена для флуоресценции. Подойдет любой флуоресцентный белок, обеспечивающий возможность селектировать клетки, несущие плазмидную конструкцию (конструкции), с помощью клеточного сортинга. Чтобы уменьшить время получения нескольких клеточных линий с разными транслокациями, индуцирующие транслокации плазмиды получают на основе универсальной конструкции. Универсальная плазмидная конструкция содержит участки, включающие сайты узнавания рестриктаз типа IIS, для быстрого клонирования в эти места любых последовательностей, отвечающих за узнавание целевого локуса. В случае системы CRISPR/Cas за узнавания целевого локуса отвечают 20 нуклеотидов гена гидовой РНК, кодирующих узнающую мишень часть РНК-гида. Таким образом, на основе одной плазмиды всего лишь в 2 этапа клонирования получают плазмиду, направленную на индукцию одной целевой транслокации.
Во-вторых, проводят трансфекцию клеток для доставки в них элементов системы редактирования генома. В качестве клеток могут выступать любые клетки, которые содержатся в виде культуры и способны пройти более 30-40 делений. Это могут быть иммортализованные клетки или культура опухолевых клеток, например, HeLa, HCT116, LCL, Jurkat, HL60 и т.д. Предпочтительно использовать клетки, близкие по типу к клеткам моделируемой опухоли. Трансфекция клеток осуществляется любым из способов: химическая трансфекция, электропорация, трансдукция с помощью вирусных частиц.
В-третьих, после трансфекции клетки, экспрессирующие флуоресцентный белок, отбирают с помощью клеточного сортера и распределяют по 20±10 штук в лунки планшета со средой. В качестве среды используют среды, обеспечивающие существование этих клеток, их рост и деление. Количество лунок планшета (или нескольких планшетов) рассчитывается, исходя из ожидаемой частоты транслокации в популяции клеток. Данное значение может быть определено в предварительных экспериментах любым способом, позволяющем подсчитать транслокации (ПЦР, секвенирование и т.д.) Для оценки ожидаемой частоты транслокации проводят предварительные эксперименты по трансфекции клеток генетическими конструкциями, индуцирующими транслокации. Спустя 24-72 часа проводят анализ: делают разведения (например, 10000 клеток, 1000 клеток, 100 клеток) и с каждого разведения ставят ПЦР-анализ на наличие транслокации. Предпочтительная методика ПЦР-анализа описана ниже. В случае, если доля клеток с транслокациями ниже 1 на 1000, то целесообразно подбирать другие целевые участки в пределах того же интрона гена, участвующего в транслокации. Например, в случае использования системы CRISPR/Cas синтезируют другие РНК-гиды с целью найти более эффективные. Также можно использовать более эффективные способы трансфекции клеток - различные условия электропорации, химической трансфекции или вирусной трансдукции. Также можно ориентироваться на литературные данные: так, в гематопоэтических клетках человека, трансцифированных рибонуклеопротеиновыми комплексами Cas9 с РНК-гидами, показана частота транслокации примерно 1 на 200 клеток (DOI 10.1182/bloodadvances.2019000450). В итоге количество лунок планшета подбираются так, чтобы хотя бы в несколько лунок попала клетка с транслокацией с вероятностью не менее 0,95. Допустимо также распределять не по 20 клеток в лунку, а другое количество, однако это менее удобно. Число 20 обусловлено тем, что в дальнейшем полученную на основе этих 20 клеток субпопуляцию нужно будет снова клонировать для получения уже моноклонов - популяций, выросших из единичных клеток. И чтобы с вероятностью более 0,95 получить хотя бы один моноклон с транслокацией, нужно вырастить и проверить 60 моноклонов. С учетом того, что не всегда из единичной клетки вырастает популяция (часть клеток погибают после сортинга), берут с запасом - 96 моноклонов (распределяют по лункам одного 96-луночного планшета).
Проверку наличия транслокаций в популяции осуществляют методом ПЦР, как наиболее простым и быстрым среди методов обнаружения транслокаций. Когда проверяют наличие клеток с транслокацией в полученных из 20 клеток субпопуляциях, то для ускорения проверки объединяют в группы по несколько лунок, в диапазоне 2-12 лунок, и ставят одну ПЦР со смешанным материалом каждой группы. Затем группу лунок, в которой обнаружена транслокация, проверяют уже по отдельным лункам. Это уменьшает общее число реакций, так как ориентировочно в одном планшете встречаются лишь единичные лунки с транслокацией.
Чтобы уменьшить затраты времени и материалов, используют так называемую прямую ПЦР, когда ДНК не выделяют, а добавляют в качестве матрицы просто лизат клеток. Допускается выделение ДНК с помощью колонок, шариков и других методов выделения и очистки ДНК из клеток, однако это гораздо более времязатратно и требует большого расхода наборов или реагентов для выделения. Выделение ДНК такими способами требует нескольких сотен тысяч клеток, тогда как для проведения прямой ПЦР берут несколько тысяч клеток, этого размера популяция клеток достигает на неделю быстрее.
Для повышения чувствительности ПЦР (чувствительность ПЦР - насколько мало молекул матрицы необходимо для того, чтобы реакция прошла) используют подход вложенной ПЦР (PMID: 33134100). Этот подход предполагает проведение ПЦР в 2 стадии: на первой стадии проводят реакцию с первой парой праймеров, на второй стадии проводят ПЦР с другой парой праймеров, которые лежат внутри относительно первой пары. Допускается проведение полувложенной ПЦР, когда один из праймеров второй стадии совпадает с одним из праймеров первой стадии.
В-четвертых, проводят распределение клеток из популяций, содержащих транслокацию, по одной клетке в лунку для получения моноклонов. Для этой процедуры используют клеточный сортер, позволяющий распределить по 1 клетке в лунки планшета. Допускается использование других методов клонирования, например метода предельных разведений, но это увеличивает трудозатраты заявляемого способа.
Проверка полученных моноклонов на наличие транслокации осуществляют методом прямой ПЦР. Для подтверждения перестройки ПЦР-продукт секвенируют по Сэнгеру. Для подтверждения экспрессии продукта слитого гена проводят ПЦР с обратной транскрипцией с использованием праймеров, лежащих на экзонах химерной мРНК.
Полученные клеточные линии используют для изучения механизмов развития лейкоза, выявлению новых маркеров для стратификации пациентов и для изучения действия на клетки с данной транслокацией различных препаратов.
Пример
В качестве примера заявляемого способа приводим пример получения клеточной линии, содержащей транслокацию MLL-ARHGAP26. Это одна из транслокаций с участием гена MLL, встречающихся при гемобластозах (раке крови). Для получения данной линии использовали иммортализованные лимфобласты (линия LCL). Была создана генетическая конструкция, содержащая элементы системы CRISPR/Cas, вносящие разрывы в локусы в 11-м интроне гена MLL и в 11-м интроне гена ARHGAP26, и содержащая ген флуоресцентного белка Santaka. Для создания генетической конструкции использовали плазмиду px330_gRNA_Cas9 (фиг. 1).
1) вставили последовательность SV40-промотора по сайтам CciNI и SbfI. При этом добавили сайт рестриктазы  Использовали в качестве матрицы плазмиду phU6-gRNA (#53188 Addgene) и следующие праймеры:
Использовали в качестве матрицы плазмиду phU6-gRNA (#53188 Addgene) и следующие праймеры:
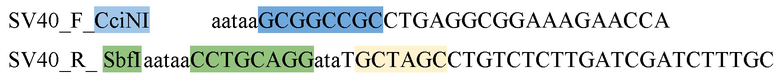
2) наработали последовательность Kozak-Santaka-SV40polyA для ее вставки по сайтам BmtI и SbfI в полученную плазмиду. При этом в праймер со стороны полиА вставили сайты для рестриктаз BstAF I и HindIII. Последовательности праймеров:

Полученную конструкцию назвали px330_gRNA_Cas9_Santaka.
3) вставили в px330_gRNA_Cas9_Santaka ген для второй гидовой РНК из плазмиды 53188-PcisI (она уже содержит сайты не для BstV21, а для PciSI - другой рестриктазы типа IIS) по сайтам рестриктаз HindIII и BstAFI. Для получения плазмиды 53188-PcisI использовали плазмиду phU6-gRNA (#53188 Addgene) и следующие олигонуклеотиды, образующие при отжиге друг на друга вставку с сайтами PciSI:
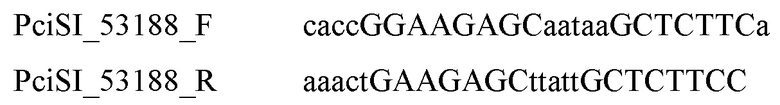
Для вставки в плазмиду px330_gRNA_Cas9_Santaka по сайтам BstAFI и Hind III (W) гена гидовой РНК амплифицировали его с использованием плазмиды 53188-PcisI и следующих праймеров:
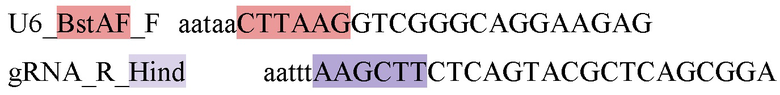
Итоговую конструкцию назвали px330_gRNA_Cas9_Santaka_gRNA (фиг. 2) и использовали для получения плазмиды, индуцирующей целевую перестройку MLL-ARHGAP26. Для этого подбирали последовательности, узнаваемые РНК-гидами, внутри интронов целевых генов, между которыми осуществляется транслокация. Узнаваемые РНК-гидами 20-нулеотидные фрагменты вставляли в плазмиду px330_gRNA_Cas9_Santaka_gRNA в гены гидовых РНК в два этапа с применением рестриктаз BstV21 и PciSI. Для этого клонирования синтезировали одноцепочечные олигонуклеотиды, которые при отжиге друг на друга дают нужную вставку с выступающими липкими концами, соответствующими липким концам обработанной BstV21 или PciSI плазмиды. Правильность сборки плазмиды подтверждали секвенированием по Сэнгеру. Полученную плазмиду назвали px330_gMLL_Santaka_gARH.
Перед получением клеток с транслокацией определяли предел детекции целевых транслокаций. Для этого клетки трансфицировали плазмидой, через 1-2 суток сортировали на наличие флуоресценции и с отобранных клеток поставили по 16 одинаковых реакций прямой вложенной ПЦР с определенным количеством клеток для оценки доли клеток с транслокацией в культуре. Ориентировочно транслокацию можно ожидать в 1 клетке на несколько сотен-тысяч клеток без транслокации. Исходя из этого, ставили по 16 реакций с лизатами 100 клеток в качестве матрицы. Оценив детектируемую долю клеток с транслокациями, разработали протокол, позволяющий получить моноклоны клеток с транслокацией с наименьшим числом сортингов и аналитических ПЦР.
Для получения моноклональных культур с транслокацией MLL-ARHGAP26 проделали следующие шаги (схема на фиг. 3). Провели трансфекцию исходной культуры клеток плазмидой px330_gMLL_Santaka_gARH, индуцирующей целевую транслокацию. Через 2 суток сортировали клетки по маркеру трансфекции Santaka по 20 клеток в лунку 96-луночного планшета. Когда из 20 клеток в лунках вырастали популяции примерно в 20 000 клеток, среди полученных популяций выявляли с помощью прямой вложенной ПЦР лунки, содержащие клетки с транслокацией. Для прямой ПЦР лизаты клеток получали следующим образом: до 10 000 клеток осаждали в ПЦР-пробирках центрифугированием при 1000 g в течение 2 мин. Удаляли супернатант и добавляли 10 мкл лизирующей смеси, содержащей 0,2 мг/мл протеиназы К и по 0,2 мМ каждого дНТФ в 1х ПЦР-буфере. Клетки лизировали 15 мин при 65°С, затем инактивировали протеиназу при 95°С в течение 5 мин. Затем добавляли 10 мкл смеси полимеразы HS TaqPol («Евроген», Россия), праймеров (заказ на синтез выполнен компанией «Евроген», Россия) и дНТФ («Евроген», Россия) в ПЦР-буфере (из набора HS TaqPol, «Евроген», Россия) так, что конечные концентрации соответствовали рекомендуемым производителем полимеразы, и проводили полувложенную ПЦР. При этом объединяли в одну реакцию образцы из 6 лунок.
На первом этапе использовали праймеры:
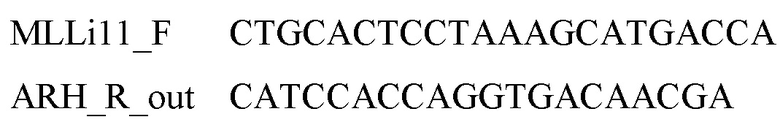
Использовали программу: 95°C 5 мин, (95°С 20 с, 58°С 1 мин, 72°С 1 мин) × 20, 72°С 5 мин. После прохождения ПЦР 4 мкл реакционной смеси добавляли к 396 мкл деионизированной воды. 4 мкл полученного раствора использовали в качестве матрицы для второго раунда ПЦР. На втором этапе ПЦР использовали праймеры:
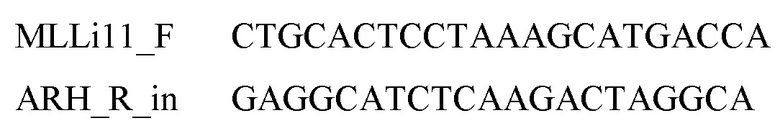
Использовали программу: 95°C 5 мин, (95°С 20 с, 58°С 1 мин, 72°С 40 с) × 30, 72°С 5 мин. Результаты анализировали методом электрофореза в агарозном геле (фиг. 4. - электрофорез продуктов ПЦР).
Если материал из группы лунок давал положительный результат ПЦР, то затем уже лунки этой группы анализировались по-отдельности с помощью такой же ПЦР.
Субпопуляции клеток из положительных лунок (с перестройкой) далее клонировали уже по одной клетке в лунку планшета с помощью клеточного сортера. Подращивали клоны из единичных клеток до примерно 20 тысяч клеток и анализировали эти уже моноклональные линии с помощью прямой ПЦР в одну стадию, используя праймеры MLLi11_F и ARH_R_in, по программе амплификации: 95°C 5 мин, (95°С 20 с, 58°С 20 с мин, 72°С 40 с) × 35, 72°С 5 мин. ПЦР-продукт для дополнительного подтверждения перестройки секвенировали по Сэнгеру.
Клоны, имеющие перестройку, анализировали на предмет экспрессии слитого гена. Для этого из клеток выделяли РНК, проводили обратную транскрипцию и ПЦР с полученной кДНК. Использовали следующие праймеры:
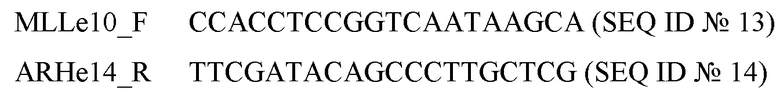
ПЦР проводили с использованием SYBR Green по протоколу: 95°C 2 мин, (95°С 15 с, 60°С 20 с, 72°С 30 с) × 35, 72°С 5 мин. Результаты анализировали на электрофорезе в агарозном геле. Результат проверки экспрессии слитого гена представлена на фиг. 5. Продукт также секвенировали по Сэнгеру для подтверждения результата, также проводили кариотипирование клеток для подтверждения хромосомной транслокации.
Таким образом, за 8 недель из перевиваемых (иммортализованных) клеток была получена клеточная модель для изучения лейкоза с перестройкой MLL-ARHGAP26 на клеточном уровне.
--->
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE ST26SequenceListing PUBLIC "-//WIPO//DTD Sequence Listing
1.3//EN" "ST26SequenceListing_V1_3.dtd">
<ST26SequenceListing dtdVersion="V1_3" fileName="Клеточная модель
MLL.xml" softwareName="WIPO Sequence" softwareVersion="2.3.0"
productionDate="2023-12-29">
<ApplicationIdentification>
<IPOfficeCode>RU</IPOfficeCode>
<ApplicationNumberText></ApplicationNumberText>
<FilingDate></FilingDate>
</ApplicationIdentification>
<ApplicantFileReference>55-2023</ApplicantFileReference>
<ApplicantName languageCode="ru">Федеральное государственное
бюджетное образовательное учреждение высшего образования
"Московский государственный университет имени
М.В.Ломоносова" (МГУ)</ApplicantName>
<ApplicantNameLatin>Federal state budgetary education institution of
higher education "Lomonosov Moscow State University"
(MSU)</ApplicantNameLatin>
<InventionTitle languageCode="ru">СПОСОБ СОЗДАНИЯ КЛЕТОЧНЫХ МОДЕЛЕЙ
ТРАНСЛОКАЦИЙ С ПОМОЩЬЮ ИНСТРУМЕНТОВ РЕДАКТИРОВАНИЯ
ГЕНОМА</InventionTitle>
<SequenceTotalQuantity>14</SequenceTotalQuantity>
<SequenceData sequenceIDNumber="1">
<INSDSeq>
<INSDSeq_length>31</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..31</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q2">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aataagcggccgcctgaggcggaaagaacca</INSDSeq_sequence
>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="2">
<INSDSeq>
<INSDSeq_length>45</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..45</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q4">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aataacctgcaggatatgctagcctgtctcttgatcgatctttgc</IN
SDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="3">
<INSDSeq>
<INSDSeq_length>36</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..36</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q6">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aataagctagcgccaccatggtgtcgaagggagaag</INSDSeq_seq
uence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="4">
<INSDSeq>
<INSDSeq_length>48</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..48</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q8">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aataacctgcaggaagcttaataacttaagatacattgatgagtttgg<
/INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="5">
<INSDSeq>
<INSDSeq_length>25</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..25</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q10">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>caccggaagagcaataagctcttca</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="6">
<INSDSeq>
<INSDSeq_length>25</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..25</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q12">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aaactgaagagcttattgctcttcc</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="7">
<INSDSeq>
<INSDSeq_length>26</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..26</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q14">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aataacttaaggtcgggcaggaagag</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="8">
<INSDSeq>
<INSDSeq_length>29</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..29</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q16">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>aatttaagcttctcagtacgctcagcgga</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="9">
<INSDSeq>
<INSDSeq_length>22</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..22</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q18">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>ctgcactcctaaagcatgacca</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="10">
<INSDSeq>
<INSDSeq_length>20</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..20</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q20">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>catccaccaggtgacaacga</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="11">
<INSDSeq>
<INSDSeq_length>22</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..22</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q22">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>ctgcactcctaaagcatgacca</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="12">
<INSDSeq>
<INSDSeq_length>21</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..21</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q24">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>gaggcatctcaagactaggca</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="13">
<INSDSeq>
<INSDSeq_length>20</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..20</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q26">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>ccacctccggtcaataagca</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="14">
<INSDSeq>
<INSDSeq_length>20</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..20</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>other DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q28">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>synthetic construct</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>ttcgatacagcccttgctcg</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
</ST26SequenceListing>
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕСТ-СИСТЕМА ДЛЯ ПОИСКА ПРЕПАРАТОВ, СНИЖАЮЩИХ РИСК ВОЗНИКНОВЕНИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2720475C2 |
| КЛЕТОЧНАЯ ЛИНИЯ И СПОСОБ ОЦЕНКИ ЭФФЕКТИВНОСТИ ГЕНОМНЫХ РЕДАКТОРОВ | 2022 |
|
RU2831064C2 |
| Рекомбинантные плазмидные ДНК lenti sgRNA(MS2)_zeo_Myc, обеспечивающие активацию экспрессии гена MYC в клетках человека, способ получения клеток человека со стабильно повышенной экспрессией гена MYC и моноклональная линия клеток рака молочной железы человека BT549_Myc со стабильно повышенной экспрессией гена MYC | 2022 |
|
RU2812975C1 |
| Способ получения генно-модифицированных кроликов с нокаутом гена LEPR с помощью системы CRISPR/Cas9 | 2023 |
|
RU2836438C1 |
| Рекомбинантная плазмидная ДНК pSNCA-C-3XFLAG-2XST-donor, обеспечивающая стабильную доксициклин-управляемую экспрессию химерного белка альфа-синуклеина в культурах клеток человека | 2022 |
|
RU2795156C1 |
| Нуклеиновая кислота для аллотопической экспрессии гена MT-ND4 | 2023 |
|
RU2809065C1 |
| МИНИ-БЕЛОК USH2A, НУКЛЕИНОВАЯ КИСЛОТА, КОДИРУЮЩАЯ МИНИБЕЛОК USH2A, И СОДЕРЖАЩИЙ ЕЕ ЭКСПРЕССИОННЫЙ ВЕКТОР ДЛЯ ГЕННОЙ ТЕРАПИИ | 2023 |
|
RU2822884C1 |
| Способ получения мышиной модели для изучения миодистрофии Дюшенна и вариантов ее терапии | 2023 |
|
RU2815936C1 |
| Способ получения рекомбинантного вируса везикулярного стоматита | 2022 |
|
RU2807751C1 |
| Универсальный интеграционный вектор SB7G_HIGH и рекомбинантная плазмида SB7G_HIGH_DEV_57, обеспечивающая синтез и секрецию рекомбинантного человеческого антитела к рецептор-связывающему домену (RBD) коронавируса SARS-CoV-2 в клетках млекопитающих и полученная с использованием универсального вектора SB7G_HIGH, и рекомбинантное моноклональное антитело DEV_K57, обладающее вируснейтрализующей активностью в отношении SARS-CoV-2 | 2023 |
|
RU2829359C1 |
Изобретение относится к области биотехнологии, в частности к способу создания клеточной модели лейкоза для моделирования и изучения процессов, происходящих в лейкозных клетках. Способ направлен на выявление тех единичных клеток в популяции, в которых произошла транслокация, и их отбор. Первичный отбор осуществляют по флуоресценции репортерного белка, и гарантия того, что все необходимые для индукции транслокации элементы (гены Cas9 и РНК-гидов) попали в клетку, обеспечивается тем, что все эти элементы вместе с геном флуоресцентного белка собраны в одну плазмиду. Такая плазмида для индукции новой транслокации создается на основе универсального вектора в два шага клонирования. Для выявления транслокации используют ПЦР, причем без отдельной стадии выделения ДНК (т.н. прямая ПЦР), комбинируя этот подход с вложенной ПЦР, повышающей чувствительность метода. Для надежного получения клеточной линии с транслокацией с вероятностью не менее 0,95 может проводиться предварительная проверка эффективности РНК-гидов и подсчет частоты транслокаций. Основываясь на частоте транслокаций, подбирают оптимальную стратегию клонирования на субпопуляции и стратегию ПЦР-анализа с объединением нескольких образцов в одну реакцию. Клонирование проводят с помощью клеточного сортера. Полученные моноклональные линии характеризуют на предмет наличия транслокации, секвенируют область перестройки, показывают экспрессию продукта слитого гена и могут далее использовать для заявляемых целей. Изобретение позволяет получить моноклональные линии с транслокациями в кратчайший срок и с минимальными затратами времени и ресурсов лаборатории. 20 з.п. ф-лы, 5 ил., 1 пр.
1. Способ создания клеточной модели лейкоза для моделирования и изучения процессов, происходящих в лейкозных клетках, включающий забор образца/клеток у пациента, выявление целевых локусов, которые ассоциированы с исследуемой патологией, затем синтезируют по меньшей мере одну плазмидную конструкцию, кодирующую инструменты редактирования генома, РНК-гиды, направленные на целевые локусы и включающие флуоресцентный белок, проводят трансфекцию клеток полученной плазмидой или плазмидами, после трансфекции клетки, экспрессирующие флуоресцентный белок, отбирают с помощью клеточного сортера и распределяют по 20±10 штук в лунки планшета со средой, наращивают субпопуляцию клеток до 20000±10000 клеток, затем анализируют полученные субпопуляции на предмет наличия в них клеток с транслокацией с помощью прямой вложенной ПЦР и клонируют субпопуляции с подтвержденной транслокацией на моноклональные линии, среди которых выявляют линии с транслокацией, подтверждают экспрессию слитого гена; при этом для анализа субпопуляций сначала оценивают содержание клеток с транслокацией сначала в группе лунок и ставят одну ПЦР со смешанным материалом, при обнаружении транслокации проверяют содержимое в индивидуальных лунках.
2. Способ по п. 1, характеризующийся тем, что клетки из нескольких лунок объединяют в группы от 2 до 12 лунок, в случае отсутствия транслокации в группе из дальнейшей работы исключают клетки из всех лунок такой группы.
3. Способ по п. 1, характеризующийся тем, что субпопуляцию клеток с подтвержденной перестройкой клонируют с помощью клеточного сортера и наращивают моноклоны до 10000-100000 клеток.
4. Способ по п. 1, характеризующийся тем, что в качестве инструментов редактирования генома используют нуклеазы, специфически распознающие целевые локусы.
5. Способ по п. 4, характеризующийся тем, что в качестве инструментов редактирования используют системы на основе CRISPR/Cas, нуклеаз-цинковых пальцев, TALE-нуклеаз, эндонуклеаз для встраивания в геном мобильных генетических элементов, а также других нуклеаз, обладающих свойством узнавать и разрезать целевые локусы.
6. Способ по п. 1, характеризующийся тем, что в качестве флуоресцентного белка используют зеленый флуоресцентный белок из Aequorea victoria и его гомологи, биливердин-связывающие бактериофитохромы и цианобактерихромы, фикоцианобилин-связывающие цианобактериальные фитохромы, малые дальнекрасные флуоресцентные белки, малые зеленые кислород-независимые флуоресцентные белки, а также флуоресцентно-активируемые метки.
7. Способ по п. 1, характеризующийся тем, что плазмидная конструкция содержит ориджин и ген устойчивости к селективному антибиотику и кодирует инструменты редактирования генома, включает участок, содержащий сайт узнавания рестриктаз типа IIS, в который встраивают последовательность, отвечающую за узнавание нуклеазой целевого локуса, а также кодирует гены элементов системы редактирования генома и ген флуоресцентного белка.
8. Способ по п. 1, характеризующийся тем, что в качестве клеток используют клетки, способные пройти более 30-40 делений.
9. Способ по п. 6, характеризующийся тем, что в качестве клеток используют иммортализованные клетки или культуру опухолевых клеток.
10. Способ по п. 1, характеризующийся тем, что трансфекцию клеток осуществляют с помощью химической трансфекции, электропорации, трансдукции с помощью вирусных частиц.
11. Способ по п. 1, характеризующийся тем, что оценку клеток с транслокациями осуществляют методом ПЦР.
12. Способ по п. 1, характеризующийся тем, что для получения моноклонов проводят распределение клеток из популяций, содержащих транслокацию, по одной клетке в лунку.
13. Способ по п. 1, характеризующийся тем, что для изучения лейкозогенеза используют клеточную линию, представляющую собой модифицированные иммортализованные лимфобласты линии LCL, содержащие транслокацию MLL-ARHGAP26.
14. Способ по п. 13, характеризующийся тем, что для получения клеточной модели используют иммортализованные лимфобласты (линия LCL), определяют локусы перестройки внутри генов MLL-ARHGAP26 и синтезируют генетическую конструкцию px330_gRNA_Cas9 Santaka и плазмиду 53188-PcisI, вставляют в px330_gRNA_Cas9_Santaka ген для второй гидовой РНК из плазмиды 53188-PcisI, по сайтам рестриктаз HindIII и BstAFI, в полученную плазмиду px330_gRNA_Cas9_Santaka_gRNA вносят с помощью рестриктазы BstV21 узнающий участок одной гидовой РНК к MLL, а с помощью рестриктазы PcisI вносят узнающий участок другой гидовой РНК к ARHGAP26, полученной плазмидой px330_gMLL_Santaka_gARH проводят трансфекцию клеток LCL, через 2 суток сортируют клетки по маркеру трансфекции Santaka, наращивают субпопуляцию клеток, оценивают содержание клеток с транслокацией с помощью прямой вложенной ПЦР, субпопуляцию клеток с подтвержденной перестройкой клонируют с помощью клеточного сортера и наращивают моноклоны; затем с помощью прямой вложенной ПЦР определяют наличие транслокации и моноклон клеток с транслокацией используют для дальнейших исследований.
15. Способ по п. 14, характеризующийся тем, что конструкция px330_gRNA_Cas9 Santaka включает элементы системы CRISPR/Cas, вносящие разрывы в локусы в 11-м интроне гена MLL и в 11-м интроне гена ARHGAP26, и содержит ген флуоресцентного белка Santaka, последовательность SV40-промотора, сайт рестриктазы BmtI.
16. Способ по п. 15, характеризующийся тем, что для синтеза генетической конструкции px330_gRNA_Cas9 Santaka используют коммерческие плазмиды px330, phU6-gRNA и набор праймеров с последовательностями SEQ ID № 1 - SEQ ID № 4.
17. Способ по п. 14, характеризующийся тем, что для синтеза генетической конструкции плазмиды 53188-PcisI используют плазмиду phU6-gRNA с олигонуклеотидами, образующими при отжиге друг на друга вставку с сайтами PciSI и набор праймеров с последовательностями SEQ ID № 5 - SEQ ID № 8.
18. Способ по п. 14, характеризующийся тем, что для проверки наличия транслокации методом вложенной ПЦР используют праймеры с последовательностями SEQ ID № 9 - SEQ ID № 12.
19. Способ по п. 14, характеризующийся тем, что проверку наличия транслокации проводят методом вложенной ПЦР в два этапа.
20. Способ по п. 19, характеризующийся тем, что на первом этапе ПЦР проводят амплификацию с помощью следующих температурных режимов: 95°C 5 мин, (95°С 20 с, 58°С 1 мин, 72°С 1 мин) × 20, 72°С 5 мин, после прохождения ПЦР 4 мкл реакционной смеси добавляют к 396 мкл деионизированной воды, затем 4 мкл полученного раствора используют в качестве матрицы на втором этапе ПЦР.
21. Способ по п. 19, характеризующийся тем, что на втором этапе ПЦР проводят амплификацию с помощью следующих температурных режимов: 95°C 5 мин, (95°С 20 с, 58°С 1 мин, 72°С 40 с) × 30, 72°С 5 мин, результаты анализируют методом электрофореза в агарозном геле.
| ТЕСТ-СИСТЕМА ДЛЯ ПОИСКА ПРЕПАРАТОВ, СНИЖАЮЩИХ РИСК ВОЗНИКНОВЕНИЯ ВТОРИЧНЫХ ЛЕЙКОЗОВ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2720475C2 |
| CN 111599003 A, 28.08.2020 | |||
| CN 104726494 B, 23.10.2018 | |||
| SHMAKOVA SH.A | |||
| et al., Cell models with inducible oncogenic translocations allow to evaluate the potential of drugs to favor secondary translocations, Cancer Communications (London), 2023, Volume 43(1), pp | |||
| Способ приготовления кирпичей для футеровки печей, служащих для получения сернистого натрия из серно-натриевой соли | 1921 |
|
SU154A1 |
| LOMOV N.A | |||
| et al., Direct ENIT: An easy and | |||

