ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтической химии и медицины и, в частности, к соединениям-ингибиторам FLT3, способам их получения и их применению для лечения заболеваний, связанных с аномальной активностью или уровнем экспрессии FLT3, например, для лечения острого миелоидного лейкоза.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Возникновение и развитие злокачественных опухолей представляет собой сложный процесс. К лекарственным средствам, обычно используемым для их лечения, относятся алкилирующие препараты, антиметаболиты, натуральные препараты и антибиотики, однако все эти препараты обладают низкой эффективностью и серьезными побочными эффектами. Низкомолекулярные ингибиторы киназ в настоящее время активно используются при разработке лекарственных препаратов для лечения онкологических заболеваний. Протеинкиназы являются катализаторами, играющими ключевую роль почти во всех аспектах биологии и биохимии клеток. Эти ферменты генерируют сигнальные молекулы, регулирующие процессы клеточного цикла, пролиферацию, программируемую гибель клеток (апоптоз), цитоскелетную функцию, подвижность, дифференциацию, развитие, транскрипцию и трансляцию. Протеинкиназы играют важную роль в разных процессах, и их точная регуляция имеет критически важное значение, так как нарушения их функций могут приводить к онкологическим заболеваниям, сердечно-сосудистым заболеваниям, воспалительным процессам и заболеваниям нервной системы. Нарушение регуляции, сверхэкспрессия и мутация протеинкиназ являются причинами патогенеза заболеваний человека, что делает эти ферменты перспективными мишенями для лекарственной терапии. Рецепторы факторов роста с активностью протеинтирозинкиназ (PTK) называются рецепторными тирозинкиназами. Рецепторные протеинтирозинкиназы представляют собой класс жестко регулируемых ферментов, и нарушение активации различных членов этого семейства является одним из маркеров онкологических заболеваний. FLT3, также как и KIT, FMS, рецептор тромбоцитарного фактора роста (PDGFR), относится к семейству рецепторных тирозинкиназ и играет важную роль в регуляции кроветворения.
FLT3 (FMS-подобная тирозинкиназа-3) является рецептором цитокинов, экспрессируемым в гемопоэтических стволовых клетках, который регулирует выживание и пролиферацию гемопоэтических стволовых клеток и клеток предшественников, созревание дендритных клеток и поддержание Т-клеточного гомеостаза. Мутации гена FLT3 присутствуют примерно у 30% пациентов с острым миелоидным лейкозом (ОМЛ), и они включают в себя внутреннюю тандемную дупликацию FLT3 (FLT3-ITD) и точечную мутацию домена тирозинкиназы FLT3 (FLT3-TKD). Мутации FLT3-ITD приводят к конститутивной, лиганд-независимой активации тирозинкиназы и, как известно, имеют плохой прогноз выживания пациентов, и они присутствуют примерно у 25% пациентов с ОМЛ, тогда как FLT3-TKD встречаются примерно у 5-10% пациентов. FLT3 может димеризоваться при связывании с лигандом и подвергаться аутофосфорилированию, инициируя тем самым разнообразные внутриклеточные сигнальные программы. Мутации FLT3 могут способствовать пролиферации клеток и ингибировать апоптоз через сигнальные пути MAPK, PI3K/AKT/Mtor и STAT5 в экспериментах in vitro. Высокая частота и плохой прогноз мутаций FLT3 позволяют предположить, что она может стать важной мишенью для терапии ОМЛ.
Таким образом, в данной области техники существует острая необходимость в разработке новых ингибиторов FLT3-киназы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление нового ингибитора FLT3-киназы.
В первом аспекте настоящего изобретения предложено соединение формулы I, либо его фармацевтически приемлемые соли или дейтерированные продукты:

Где, кольцо  представляет собой аза-(5-6-членное гетероароматическое кольцо) и кольцо
представляет собой аза-(5-6-членное гетероароматическое кольцо) и кольцо  представляет собой 5-6-членное ароматическое кольцо или гетероароматическое кольцо, и кольцо
представляет собой 5-6-членное ароматическое кольцо или гетероароматическое кольцо, и кольцо  вместе с кольцом
вместе с кольцом  образуют 9-10-членное гетероароматическое кольцо;
образуют 9-10-членное гетероароматическое кольцо;
Ra выбирают из группы, состоящей из Н, галогена, замещенной или незамещенной C1-C6 алкильной группы, замещенной или незамещенной С2-С6 алкенильной группы, замещенной или незамещенной С2-С6 алкинильной группы, замещенного или незамещенного С3-С8 углеродного кольца (включая насыщенное и частично ненасыщенное кольцо), замещенного или незамещенного 5-9-членного гетероароматического кольца (включая моноциклическое или конденсированное кольцо) содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенного или незамещенного 3-8-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (которое представляет собой насыщенное или частично ненасыщенное гетероциклическое кольцо, предпочтительно замещенную или незамещенную С3-С6 циклоалкильную группу, замещенное или незамещенное 3-6-членное гетероциклическое кольцо, содержащее 1-3 гетероатома, выбранных из атомов кислорода, серы и азота), либо замещенной и незамещенной -[L]m-H группы; где, каждую L независимо выбирают из группы, состоящей из -CH2-, -О-, -NH- или -S-;
m выбирают из группы, состоящей из 1, 2, 3, 4, 5 и 6;
U выбирают из группы, состоящей из химической связи или -О-, -CHR-, карбонильной группы, S, -NH-, -NHC(O)-, -NHS(O)2-, -NHC(O)NH-, -NHC(S)NH-, -COO- и -O-S(O)2-;
Rc выбирают из группы, состоящей из Н, замещенной или незамещенной C1-С6 алкильной группы, замещенного или незамещенного С3-C8 карбоциклического кольца (включая насыщенное и частично ненасыщенное кольцо), замещенного или незамещенного 3-8-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (насыщенное и частично ненасыщенное, включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенной или незамещенной С6-С10 арильной группы, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, либо замещенной и незамещенной -[L]m-H группы; где, L независимо выбирают из группы, состоящей из -CH2-, -О-, -NH- или -S-;
Re выбирают из группы, состоящей из галогена, -NHR, -OR, замещенной или незамещенной C1-С6 алкильной группы, замещенной или незамещенной C1-С6 алкил-NH-группы, замещенной или незамещенной C1-C6 алкоксигруппы, замещенного и незамещенного С3-C8 карбоциклического кольца (включая случаи насыщенного и частично ненасыщенного кольца) или замещенной и незамещенной -[L]m-Н-группы где, L независимо выбирают из группы, состоящей из -СН2-, -О-, -NH- или -S-;
W выбирают из группы, состоящей из Н, -NHR, -OR, галогена, замещенной или незамещенной C1-С6 алкильной группы, замещенной или незамещенной C1-С6 алкоксигруппы, замещенной или незамещенной С2-С6 алкенильной группы, замещенной или незамещенной С2-С6 алкинильной группы, замещенной или незамещенной С3-C8 карбоциклического кольца (включая случаи насыщенного и частично ненасыщенного кольца), замещенной или незамещенной С6-С10 арильной группы, замещенного или незамещенного 4-15-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (насыщенное или частично ненасыщенное кольцо, включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота (включая моноциклическое и конденсированное кольцо), замещенной или незамещенной -C1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы (включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенной или незамещенной С2-С10 ацильной группы, замещенной или незамещенной С2-С10 сложноэфирной группы, замещенной или незамещенной С6-С10 арилоксигруппы и замещенной или незамещенной C1-C6 амидной группы;
Группу W замещают по меньшей мере одной группой со структурой -М-А, при этом М выбирают из группы, состоящей из химической связи или -CHR-, карбонильной группы, S, О, -NH-, -NHC(O)-, -NHS(O)2-, -NHC(O)NH-, -NHC(S)NH-, -COO- и -O-S(O)2-;
А выбирают из группы, состоящей из Н, галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, замещенной или незамещенной C1-C6 алкильной группы, замещенной или незамещенной C1-C6 алкоксигруппы, замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (включая моноциклическое или конденсированное кольцо), замещенной или незамещенной -C1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы, замещенной или незамещенной С2-С10 ацильной группы, замещенной или незамещенной С2-С10 сложноэфирной группы, замещенной или незамещенной С6-С10 арилоксигруппы, замещенной или незамещенной С1-C6 амидной группы, замещенной или незамещенной С1-С4 алкил-S(O)2-группы, замещенной или незамещенной С1-С4 алкил-SO-группы;
В группе А указанное замещение означает замещение одной или несколькими группами, выбранными из группы В, при этому группа В состоит из Н, галогена, =О, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, замещенной или незамещенной С1-C6 алкильной группы, замещенной или незамещенной С1-С6 алкоксигруппы, замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного 3-12-членного (предпочтительно 5-7-членного) гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной или незамещенной -С1-С6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы, замещенной или незамещенной С2-С10 ацильной группы, замещенной или незамещенной С2-С10 сложноэфирной группы, замещенной или незамещенной С6-С10 арилоксигруппы, замещенной или незамещенной С1-C6 амидной группы, замещенной или незамещенной С1-С4 алкил-S(O)2-группы, замещенной или незамещенной С1-С4 алкил-SO-группы; при этом в группе В замещение означает замещение одной или несколькими группами R;
R выбирают из группы, состоящей из Н, галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, замещенной или незамещенной С1-C6 алкильной группы, замещенной или незамещенной С1-C6 алкоксигруппы, замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного 5-7-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной или незамещенной -С1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы, замещенной или незамещенной С2-С10 ацильной группы, замещенной или незамещенной С2-С10 сложноэфирной группы, замещенной или незамещенной С6-С10 арилоксигруппы, замещенной или незамещенной С1-С6 амидной группы, замещенной или незамещенной С1-С4 алкил-S(O)2-группы, замещенной или незамещенной С1-С4 алкил-SO-группы;
Если не указано иное, в приведенных выше формулах замещение означает, что атом водорода в соответствующей группе замещен одним или несколькими заместителями, выбранными из группы, состоящей из дейтерия, трития, галогена, гидроксильной группы, карбоксильной группы, сульфгидрильной группы, бензильной группы, С1-С12 алкоксикарбонильной группы, С1-С6 альдегидной группы, аминогруппы, С1-С6 амидной группы, нитрогруппы, цианогруппы, незамещенной или галогенированной С1-C6 алкильной группы, незамещенной или галогенированной С3-С8 циютоалкильной группы, С2-С10 алкенильной группы, С1-С6 алкоксигруппы, С1-С6 алкиламиногруппы, С6-С10 арильной группы, пяти- или шестичленной гетероарильной группы, пяти- или шестичленной неароматической гетероциклильной группы, -О-(С6-С10-арил)-группы, -О-(пяти- и шестичленной гетероарильной) группы, С1-С12-алкиламинокарбонильной группы, незамещенной или галогенированной С2-С10-ацильной группы, сульфонильной (-SO2-OH) группы, фосфорильной (-РО3-ОН) группы, незамещенной или галогенированной С1-С4-алкил-S(O)2-группы и незамещенной или галогенированной С1-С4-алкил-SO-группы.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой II:

где каждую группу X, Y и Z независимо выбирают из N или CR.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой IIa:

Где кольцо W выбирают из группы, состоящей из замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной или незамещенной -С1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циютоалкильной группы; где в группе W указанное замещение означает замещение одной или несколькими группами, выбранными из группы А.
В другом предпочтительном варианте осуществления изобретения кольцо W выбирают из группы, состоящей из замещенного или незамещенного 4-7-членного гетероциклического кольца, замещенного или незамещенного 5-6-членного гетероароматического кольца, замещенного или незамещенного 9-10-членного гетероароматического кольца с бициклической конденсированной структурой, замещенной и незамещенной фенильной группы и замещенной и незамещенной С3-С6 циклоалкильной группы.
В другом предпочтительном варианте осуществления изобретения кольцо W представляет собой замещенную или незамещенную кольцевую структуру, выбранную из группы, состоящей из фенильной группы, циклопентильной группы, циклогексильной группы, 

В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой III:

Где, кольцо А выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы, замещенной или незамещенной -С1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы; М выбирают из группы, состоящей из химической связи или -О-, -CHR-, карбонильной группы, S, -NH-, -NHC(O)-, -NHS(O)2-, -NHC(O)NH-, -NHC(S)NH-, -COO-, -O-S(O)2-.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой:

где,
Rf выбирают из группы, состоящей из Н, галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, С1-С4 алкил-S(O)2-группы, замещенной или незамещенной С1-С6 алкильной группы, замещенной или незамещенной С1-C6 алкоксигруппы;
t является 0, 1, 2, 3 или 4;
кольцо А выбирают из группы, состоящей из замещенного или незамещенного 5-12-членного насыщенного кольца (включая спироциклическое и мостиковое кольцо), замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы, содержащей по крайней мере один гетероатом, выбранный из N или О в кольце.
В другом предпочтительном варианте осуществления изобретения по крайней мере одно Rf находится в мета-положении участка соединения бензольного кольца и исходного ядра.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой:

где,
Rf выбирают из группы, состоящей из Н, галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, С1-С4 алкил-S(O)2-группы, замещенной или незамещенной С1-С6 алкильной группы, замещенной или незамещенной С1-C6 алкоксигруппы;
t является 0, 1, 2, 3 или 4;
L представляет собой N или СН;
кольцо А выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной или незамещенной С3-С12 циклоалкильной группы, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы.
Где, когда атом азота в  является участком соединения, указанное NH является N (т.е. атом водорода в NH отсутствует для образования участка соединения).
является участком соединения, указанное NH является N (т.е. атом водорода в NH отсутствует для образования участка соединения).
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой IV:
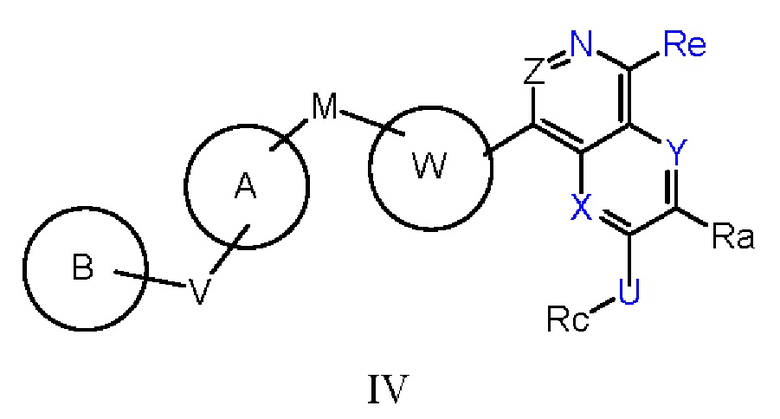
где кольцо А выбирают из группы, состоящей из замещенного или незамещенного 4-7-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы, замещенной или незамещенной -С1-C6 алкилфенильной группы, замещенной или незамещенной С3-С12 циклоалкильной группы;
М выбирают из группы, состоящей из химической связи или -О-, -CHR-, карбонильной группы, S, -NH-, -NHC(O)-, -NHS(O)2-, -NHC(O)NH-, -NHC(S)NH-, -COO-, -O-S(O)2-;
кольцо В выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 13 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного С3-С12 карбоциклического кольца;
V выбирают из группы, состоящей из химической связи или -О-, -CHR-, карбонильной группы, S, -NH-, -NHC(O)-, -NHS(O)2-, -NHC(O)NH-, -NHC(S)NH-, -СОО-, -O-S(O)2-.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру представленную следующей формулой:

где Rg выбирают из группы, состоящей из галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, замещенной или незамещенной С1-C6 алкильной группы, замещенной или незамещенной С1-С6 алкоксигруппы;
Кольцо В выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота (включая моноциклическое, конденсированное, мостиковое или спироциклическое кольцо), замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной и незамещенной С6-С10 арильной группы, замещенного или незамещенного С3-С12 карбоциклического кольца;
u является 0, 1, 2, 3 или 4.
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, представленную следующей формулой:

где Rg выбирают из группы, состоящей из галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, сульфгидрильной группы, альдегидной группы, карбоксильной группы, сульфонильной группы, замещенной или незамещенной С1-C6 алкильной группы, замещенной или незамещенной С1-С6 алкоксигруппы;
u является 0, 1, 2, 3 или 4.
В другом предпочтительном варианте осуществления изобретения кольцо А содержит по крайней мере один заместитель G, при этому группу G выбирают из группы, состоящей из аминогруппы, =O, замещенного или незамещенного 4-7-членного гетероциклического кольца, содержащего 1-3 гетероатома, выбранных из атомов кислорода и азота, замещенной или незамещенной С2-С10 ацильной группы, замещенной и незамещенной С2-С10 сложноэфирной группы и замещенной и незамещенной С1-C6 амидной группы.
Где, когда атом азота в  является участком соединения, указанное NH является N (т.е. атом водорода в NH теряется для образования участка соединения).
является участком соединения, указанное NH является N (т.е. атом водорода в NH теряется для образования участка соединения).
В другом предпочтительном варианте осуществления изобретения соединение формулы I имеет структуру, которую выбирают из группы, состоящей из:

В другом предпочтительном варианте осуществления изобретения W выбирают из группы, состоящей из: 
В другом предпочтительном варианте осуществления изобретения W выбирают из группы, состоящей из: 
В другом предпочтительном варианте осуществления изобретения Rc выбирают из группы, состоящей из:

В другом предпочтительном варианте осуществления изобретения URc выбирают из группы, состоящей из:


В другом предпочтительном варианте осуществления изобретения Re выбирают из группы, состоящей из аминогруппы, замещенной или незамещенной С1-С6 алкил-NH-группы.
Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество одного или нескольких соединений формулы I, их фармацевтически приемлемых солей, рацематов, R-изомеров и S-изомеров, стереоизомеров или таутомеров, как описано в первом аспект изобретения, а также один или несколько фармацевтически приемлемых носителей, вспомогательных веществ, добавок, наполнителей и разбавителей.
В третьем аспекте настоящего изобретения предложено применение соединений формулы I, их рацематов, R-изомеров, S-изомеров или фармацевтически приемлемых солей, как описано в первом аспекте настоящего изобретения, в производстве лекарственного препарата для лечения или профилактики заболеваний, связанных с уровнями аномальных генов или аномальной экспрессией киназ (например, вследствие мутации, удаления или транспозиции соответствующих нуклеотидных последовательностей, либо слияния или сверхэкспрессии указанной киназы), выбранных из группы, состоящей из FLT3, ALK, RET, ROS, AXL, EGFR.
В другом предпочтительном варианте осуществления изобретения заболевание выбирают из группы, состоящей из острого миелоидного лейкоза, нейрофибромы I типа, множественной миеломы, глиобластомы, немелкоклеточного рака легких, рака печени, гепатоцеллюлярной карциномы, рака шейки матки, лимфомы, костных метастазов, гормонорезистентного рака предстательной железы, гормонозависимого рака предстательной железы, аденомы щитовидной железы, медуллярного рака щитовидной железы, мезотелиомы, глиобластомы, метастазов в сфинктере, карциномы из клеток Меркеля, опухоли урогенитального тракта, рака мочевого пузыря, папиллярного рака щитовидной железы, рака молочной железы, саркомы мягких тканей, глиомы, нейроэндокринной опухоли, почечно-клеточного рака, прогрессирующей солидной опухоли, недифференцированной астроцитомы, стромальной опухоли желудочно-кишечного тракта, синдрома Хиппеля-Линдау мелко клеточного рака легких, рака поджелудочной железы, эндокринной опухоли поджелудочной железы, опухоли центральной нервной системы, метастатического рака почки, эндометриоидной карциномы, эндометриоидной аденокарциномы, рака легких, колоректального рака, рака яичников, рабдомиосаркомы, меланомы, ретинобластомы, опухоли центральной и периферической нервной системы, острого лейкоза, хронического лейкоза, холангиокарциномы, бронхиальной карциномы, рака пищевода, рака яичек, рака кожи, рака ротовой полости, нейробластомы, анапластической крупно клеточной лимфомы.
В другом аспекте настоящего изобретения предложен конъюгат образованный соединением по настоящему изобретению и небольшой биомолекулой, либо моноклональным антителом за счет химической связи.
Следует понимать, что в пределах объема настоящего изобретения все технические признаки настоящего изобретения, указанные выше и приведенные далее (в виде примеров), могут комбинироваться друг с другом, образуя новые или предпочтительные технические решения, которые не перечислены здесь из-за ограничений объема.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В ходе обширных и углубленных исследований изобретателями был впервые неожиданно обнаружен класс соединений с ингибирующей активностью в отношении киназ (таких как FLT3, ALK, AXL, EGFR). Настоящее изобретение было совершено на этой основе.
Термины
В настоящем изобретении галоген представляет собой F, Cl, Br или I.
В настоящем изобретении, если не указано иное, используемые здесь термины имеют общепринятые значения, которые известны специалистам в данной области. В настоящем изобретении, если не указано иное, все химические формулы предназначены для охвата всех возможных оптических или геометрических изомеров (R-, S-изомеры или рацематы, или цис- и трансизомеры олефинов и т.д.)
В настоящем изобретении термин «С1-С6 алкильная группа» означает алкильную группу с прямой или разветвленной цепью, содержащую 16 атомов углерода, включая, в том числе, метальную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную, гексильную группы и т.п.; при этом этильная, пропильная, изопропильная, бутильная, изобутильная, втор-бутильная и трет-бутильная группы являются предпочтительными.
В настоящем изобретении термин «С1-С6 алкоксигруппа» означает алкоксигруппу с прямой или разветвленной цепью, содержащую 16 атомов углерода, включая, в том числе, метоксигруппу этоксигруппу, пропоксигруппу изопропоксигруппу бутоксигруппу и другие группы.
В настоящем изобретении термин «С2-С6 алкенильная группа» означает алкенильную группу с прямой или разветвленной цепью, содержащую двойную связь и 2-6 атомов углерода, включая, в том числе, винильную, пропенильную, бутенильную, изобутенильную, пентенильную, гексенильную и другие группы.
В настоящем изобретении термин «С2-С6 алкинильная группа» означает алкинильную группу с прямой или разветвленной цепью, содержащую тройную связь и 2-6 атомов углерода, включая, в том числе, этинильную, пропинильную, бутинильную, изобутинильную, пентинильную, гексинильную и другие группы.
В настоящем изобретении термин «С3-С10 циклоалкильная группа» означает циклоалкильную группу с прямой или разветвленной цепью, содержащую 3-10 атомов углерода в кольце, включая, в том числе, циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклопептильную, циклооктильную, циклодецильную и другие группы. Термины «С3-С8 циклоалкильная группа», «С3-С7 циклоалкильная группа» и «С3-С6 циклоалкильная группа» имеют аналогичные значения.
В настоящем изобретении термин «С3-С10 циклоалкенильная группа» означает цикл о алкенильную группу с 3-10 атомами углерода в кольце, включая, в том числе, циклопропенильную, циклобутенильную, циклопентенильную, циклогексенильную, циклогептенильную, циклооктенильную, циклодециленовую группы. Термин «С3-С7 циклоалкенильная группа» имеет аналогичное значение.
В настоящем изобретении термин «С1-С12 алкоксикарбонильная группа» означает алкоксикарбонильную группу с 1-12 атомами углерода в алкильной цепи, включая, в том числе, метоксикарбонильную, этоксикарбонильную, пропоксикарбонильную, изопропоксикарбонильную, трет-бутоксикарбонильную, бензилоксикарбонильную и другие группы.
В настоящем изобретении термин «С1-С12 алкиламинокарбонильная группа» означает алкиламинокарбонильную группу с 1-12 атомами углерода в алкильной цепи, включая, в том числе, метиламинокарбонильную, этиламинокарбонильную, пропиламинокарбонильную, изопропиламинокарбонильную, трет-бутиламинокарбонильную, бензиламинокарбонильную, диметиламинокарбонильную и другие группы.
В настоящем изобретении термин «С5-С9 фуранозильная группа» означает фуранозильную группу с 5-9 атомами углерода, где 1-положение гликозильной группы связано с основной цепью, включая, в том числе, рибофуранозильную, дезоксирибофуранозильную, галактофуранозильную и другие группы.
В настоящем изобретении термин «С5-С9 пиранозильная группа» означает пиранозильную группу с 5-9 атомами углерода, где 1-положение гликозильной группы связано с основной цепью, включая без ограничений глюкопиранозильную, глюкопирануронильную, рамнопиранозильную, галактопиранозильную, маннопиранозильную и ксилопиранозильную группы.
В настоящем изобретении термины «ароматическое кольцо» или «арильная группа» имеют аналогичное значение, при этом «арильная группа» предпочтительно означает «С6-С12 арильную группу» или «С6-С10 арильную группу». Термин «С6-С12 арильная группа» означает ароматическую циклическую группу с 6-12 атомами углерода без гетероатомов в кольце, например, фенильную, нафтильную и другие группы. Термин «С6-С10 арильная группа» имеет аналогичное значение.
В настоящем изобретении термины «ароматическое гетероциклическое кольцо» или «гетероарильная группа» имеют аналогичное значение и означают гетероароматическую группу, содержащую от одного до нескольких гетероатомов. Указанные здесь гетероатомы включают кислород, серу и азот. К этим группам относятся фурильная, тиенильная, пиридильная, пиразолильная, пирролильная, N-алкилпирролильная, пиримидинильная, пиразинильная, имидазолильная, тетразолильная и другие группы. Гетероарильное кольцо может быть конденсировано с арильным кольцом, гетероциклильным кольцом или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Гетероарильная группа может быть дополнительно замещенной или незамещенной.
В настоящем изобретении термин «3-12-членная гетероциклильная группа» означает насыщенную или ненасыщенную 3-12-членную циклическую группу, содержащую 1-3 гетероатома, выбранных из атомов кислорода, серы и азота в кольце, например, диоксоланильную и другие группы. Термин «3-7-членная гетероциклильная группа» имеет аналогичное значение.
В настоящем изобретении термин «замещенный» означает, что один или несколько атомов водорода в определенной группе замещены определенными заместителями. Определенные заместители являются заместителями, которые описаны выше или представлены в примерах. Если не указано иное, замещенная группа может иметь заместитель, выбранный из определенной группы, в любом замещаемом положении группы, при этом заместитель в каждом положении может быть одинаковым или разным. Циклический заместитель, например гетероциклильная группа, может быть присоединен к другому кольцу, например, к циклоалкильной группе, образуя спиробициклическую систему, например, когда два кольца имеют один общий атом углерода. Специалистам в данной области должно быть понятно, что комбинации заместителей, рассматриваемые в настоящем изобретении, представляют собой стабильные комбинации или те комбинации, которые можно получить путем химического синтеза. Заместителями могут быть (не ограничиваясь перечисленным): С1-8 алкильная группа, С2-8 алкенильная группа, С2-8 алкинильная группа, С3-8 циклоалкильная группа, 3-12-членная гетероциклильная группа, арильная группа, гетероарильная группа, галоген, гидроксильная группа, карбоксильная (-СООН) группа, С1-8 альдегидная группа, С2-10 ацильная группа, С2-10 сложноэфирная группа, С1-С12 алкоксикарбонильная группа, аминогруппа, алкоксигруппа, С1-10 сульфонильная группа и другие группы.
Соединение-ингибитор FLT3
В настоящем изобретении предложено соединение с ингибирующей активностью в отношении FLT3:

Где каждая группа имеет определение, приведенное выше. Предпочтительными соединениями в настоящей заявке являются соединения, выбранные из следующей таблицы 1:




















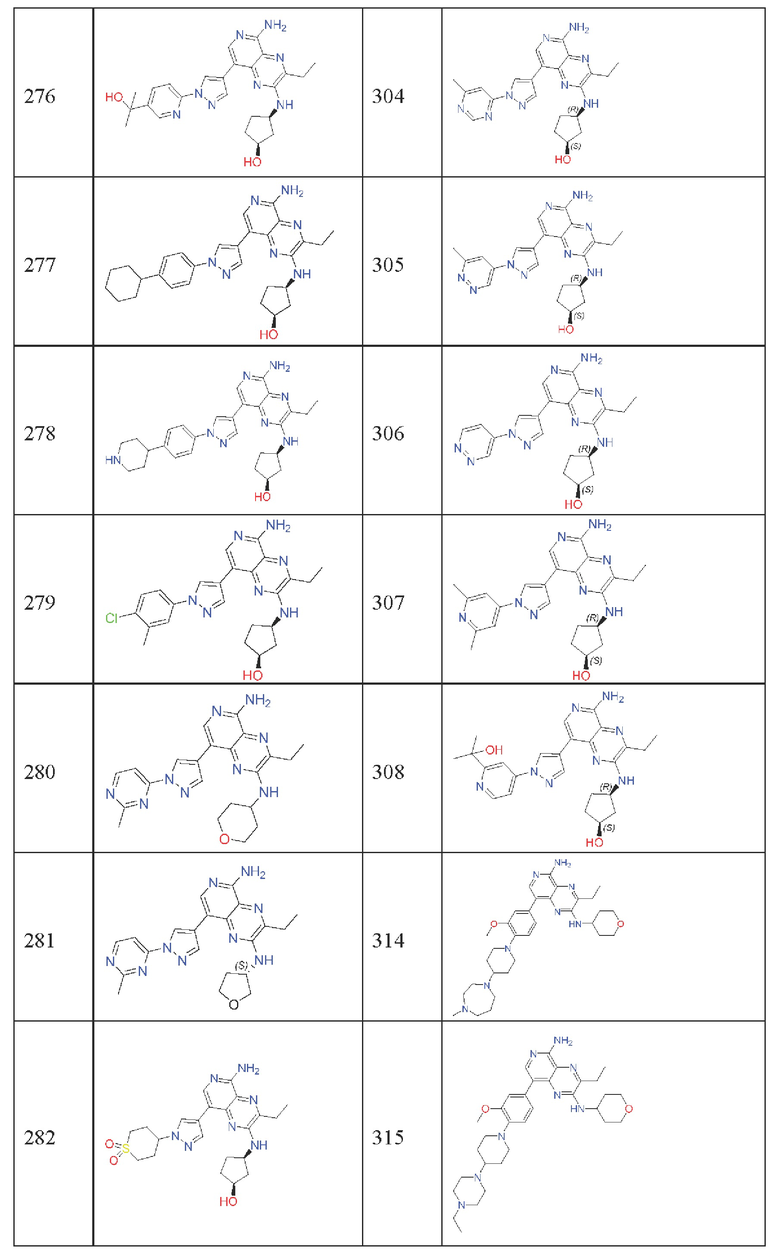




Где соединения 162 и 163 являются  соответственно, но абсолютная конфигурация неизвестна. Где соединения 164 и 165 являются
соответственно, но абсолютная конфигурация неизвестна. Где соединения 164 и 165 являются  соответственно, но абсолютная конфигурация неизвестна. Где соединения 284 и 285 являются
соответственно, но абсолютная конфигурация неизвестна. Где соединения 284 и 285 являются  соответственно, но абсолютная конфигурация неизвестна.
соответственно, но абсолютная конфигурация неизвестна.
Фармацевтические композиции и способы введения
Поскольку соединения по настоящему изобретению имеют высокую ингибирующую активность в отношении киназ, соединения по настоящему изобретению и их разные кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, а также фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве основного действующего вещества, могут применяться для лечения, профилактики и купирования заболеваний, вызванных аномальной активностью или аномальной экспрессией киназ (например, FLT3).
Фармацевтическая композиция по настоящему изобретению содержит безопасное и эффективное количество соединения по настоящему изобретению или его фармакологически приемлемой соли, а также фармакологически приемлемое вспомогательное вещество или носитель. Термин «безопасное и эффективное количество» означает, что количество соединения является достаточным для существенного улучшения состояния, не вызывая при этом серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения по настоящему изобретению на единицу дозирования, более предпочтительно 5-200 мг соединения по настоящему изобретению на единицу дозирования. «Единица дозирования» предпочтительно представляет собой капсулу или таблетку.
Термин «фармацевтически приемлемый носитель» относится к одному или нескольким совместимым твердым, жидким или гелеобразным вспомогательным веществам, пригодным для использования в медицине, при этом они должны быть в должной мере чистыми и малотоксичными. «Совместимость» означает, что каждый компонент композиции можно смешивать с соединениями по настоящему изобретению и друг с другом без существенного снижения эффективности соединений. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (например, натрия карбоксиметилцеллюлозу натрия этилцеллюлозу целлюлозы ацетат и т.д.), желатин, тальк, твердые смазывающие вещества (например, стеариновую кислоту, магния стеарат), кальция сульфат, растительные масла (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (например, пропиленгликоль, глицерин, маннитол, сорбитол и т.д.), эмульгаторы (например, Твин®), смачивающие вещества (например, натрия додецилсульфат), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и др.
Способ введения соединения или фармацевтической композиции по настоящему изобретению не ограничен особым образом, и обычными способами введения являются, в том числе, прием внутрь, внутриопухолевое, ректальное, парентеральное (внутривенное, внутримышечное или подкожное) и местное введение.
Твердые лекарственные формы для приема внутрь включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах действующее вещество смешивают по меньшей мере с одним обычным инертным вспомогательным веществом (или носителем), например, с натрия цитратом или кальция гидрофосфатом, либо смешивают с каким-либо из следующих компонентов: (а) наполнителями или веществами, улучшающими совместимость, например, с крахмалом, лактозой, сахарозой, глюкозой, маннитолом и кремния диоксидом; (b) связующими веществами, например, с гидроксиметилцеллюлозой, альгинатом, желатином, поливинилпирролидоном, сахарозой и аравийской камедью; (с) с увлажняющими веществами, например, с глицерином; (d) с разрыхлителями, например, с агаром, кальция карбонатом, картофельным крахмалом или тапиоковым крахмалом, альгиновой кислотой, некоторыми смешанными силикатами и натрия карбонатом; (е) с веществами, замедляющими высвобождение, например, с парафином, (f) с ускорителями впитывания, например, с четвертичным соединением аммония; (g) со смачивающими веществами, например, с цетиловым спиртом и глицерина моностеаратом; (h) с адсорбирующими веществами, например, с каолином; и (i) со смазывающими веществами, например, с тальком, кальция стеаратом, магния стеаратом, твердым полиэтиленгликолем, натрия лаурилсульфатом, либо их смесью. Лекарственные формы в виде капсул, таблеток и пилюль могут также содержать буферные средства.
Твердые лекарственные формы, например, таблетки, драже, капсулы, пилюли и гранулы, могут быть в оболочке, например, в кишечнорастворимой оболочке, а также содержать другие материалы, применяемые в данной области. Они могут содержать замутнители, при этом высвобождение действующего вещества или соединения в таких композиция может происходить с задержкой в определенной части пищеварительного тракта. Примерами компонентов для включения, которые могут использоваться, являются полимерные материалы и восковые материалы. При необходимости действующее вещество может быть также в микрокапсулированной форме с одним или несколькими из указанных выше вспомогательных веществ.
Жидкие лекарственные формы для приема внутрь включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. Помимо действующего вещества жидкая лекарственная форма может содержать инертные разбавители, обычно применяемые в данной области, например, воду или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, в частности, хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, либо смеси этих веществ.
Кроме этих инертных разбавителей в композициях также могут быть вспомогательные вещества, например, смачивающие, эмульгирующие и суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
Помимо действующего вещества в суспензии также может быть суспендирующее вещество, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и дегидратированный сложный эфир сорбитана, микрокристаллическая целлюлоза, алюминия метоксид и агар, либо их смесь и т.д.
В композициях для парентерального введения могут быть физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки, которые могут быть восстановлены в стерильных растворах или дисперсиях для инъекций. Подходящими водными и неводными носителями, разбавителями, растворителями или вспомогательными веществами являются вода, этанол, полиолы и любые подходящие их смеси.
Лекарственными формами соединений по изобретению для местного применения являются мази, порошки, пластыри, спреи и лекарственные формы для ингаляций. Действующее вещество смешивают в стерильных условиях с физиологически приемлемым вспомогательным веществом и какими-либо консервантами, буферными растворами или распыляющими веществами, которые могут потребоваться при необходимости.
Соединения по настоящему изобретению можно вводить отдельно или в комбинации с другими фармацевтически приемлемыми соединениями. В определенных предпочтительных вариантах осуществления изобретения соединения по настоящему изобретению могут вводиться с другими низкомолекулярными соединениями, образуя PROTAC, либо с другими высокомолекулярными соединениями, например, с моноклональными антителами, образуя ADC.
При применении фармацевтической композиции безопасное и эффективное количество соединения по настоящему изобретению вводят млекопитающему (например, человеку), нуждающемуся в лечении, при этом дозировка при введении представляет собой фармацевтически эффективную дозу. Для людей с массой тела 60 кг суточная доза обычно составляет 1-2000 мг, предпочтительно 50-1000 мг. Конечно, для конкретных доз также следует учитывать такие факторы, как путь введения, состояние здоровья пациента и т.д., находящиеся в компетенции квалифицированного врача.
Настоящее изобретение будет дополнительно проиллюстрировано ниже с ссылкой на конкретные примеры. Следует понимать, что эти примеры используются только для иллюстрации изобретения и не предназначены для ограничения объема изобретения. В экспериментальных методах в следующих примерах, в которых конкретные условия не указаны, данные условия обычно соответствуют общепринятым, либо они соответствуют указанным в инструкциях производителя. Если не указано иное, процентные доли и значения рассчитаны по массе.
Сокращения определены следующим образом:

Исходные материалы могут быть приобретены, либо получены способами, которые известны или раскрыты в данной области техники.
Очистку промежуточных продуктов и соединений проводят обычными методами, используемыми в химических лабораториях, например, методом нормальной или обращенно-фазовой хроматографии, либо путем перекристаллизации. Нормально-фазовую хроматографию проводят на хроматографической колонке, предварительно заполненной силикагелем, или методом препаративной тонкослойной хроматографии. Хроматографические колонки с силикагелем в основном представляют собой стеклянные колонки, либо используют систему высокоэффективной препаративной хроматографии. Подвижную фазу для нормально-фазовой хроматографии выбирают из петролейного эфира/этилацетата, дихлорметана/метанола или других подходящих растворителей и элюирование проводят при заданном соотношении. Обращенно-фазовую препаративную жидкостную хроматографию проводят на колонке С18 с использованием препаративного жидкостного хроматографа или системы высокоэффективной препаративной хроматографии с детектированием при 214 нм и 254 нм, либо с использованием комбинированной системы препаративной жидкостной хроматографии и масс-спектрометрии. Для градиентного элюирования в качестве подвижной фазы используют 0,1% раствор хлористоводородной кислоты в смеси вода/ацетонитрил, смесь вода/ацетонитрил, 0,1% раствор аммония бикарбоната в смеси вода/ацетонитрил, 0,1% раствор муравьиной кислоты в смеси вода/ацетонитрил, 0,1% раствор аммиака в смеси вода/ацетонитрил, 0,1% раствор трифторуксусной кислоты в смеси вода/ацетонитрил или другие подходящие системы растворителей.
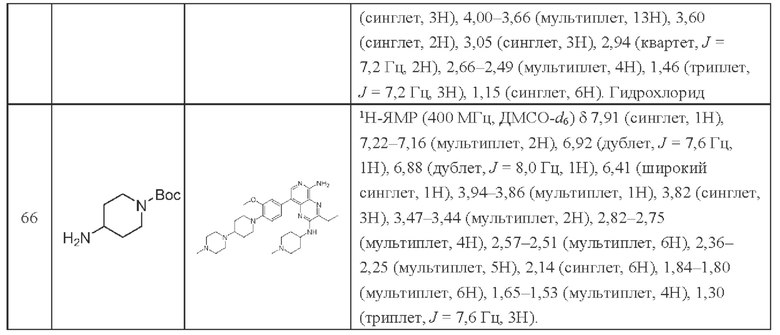
Структуры промежуточных продуктов и соединений устанавливают методами ядерного магнитного резонанса (ЯМР) и масс-спектрометрии (ЖХ-МС). Для ЯМР используют ЯМР-спектрометр Bruker Ascend 400, либо Varian 400, либо ZKNJ BIXI-1 300 МГц, либо Bruker Avance III 400 МГц, либо Bruker AVANCE Neo 400 МГц. Используемыми растворителями являются дейтерированный диметилсульфоксид, дейтерированный хлороформ, дейтерированный метанол или другие дейтерированные растворители. Спектральные данные представлены в следующем виде: химический сдвиг δ (число расщепленных пиков, постоянная спин-спинового взаимодействия J (Гц), число атомов водорода). Тетраметилсилан используют в качестве внутреннего стандарта для химического сдвига, и его химический сдвиг принимается равным нулю (δ, 0 м.д.). Значения нескольких сокращений: s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет), brs (широкий синглет).
Для установления структурных характеристик промежуточных продуктов и соединений использовали следующие репрезентативные методики на основе жидкостной хроматографии/масс-спектрометрии (ЖХ-МС):
Методика I: анализ проводят с использованием системы Agilent LC1260, соединенной с одноквадрупольным масс-спектрометром 6120
Колонка: Waters CORTECS С-18, 2,7 мкм, 4,6×30 мм. Растворитель А: 0,05% водный раствор муравьиной кислоты, растворитель В: 0,05% раствор муравьиной кислоты в ацетонитриле, линейное повышение концентрации ацетонитрила с 5% до 95% в течение одной минуты, выдержка в течение одной минуты, всего 2,5 мин; скорость потока: 1,8 мл/мин; температура колонки: 40°С.
Колонка: XSelect CSH С18, 3,5 мкм, 4,6×50 мм. Растворитель А: 0,05% водный раствор аммиака, растворитель В: 0,05% раствор аммиака в ацетонитриле, повышение концентрации ацетонитрила с 5% до 95% в течение одной минуты, выдержка в течение одной минуты, всего 2,5 мин; скорость потока: 1,8 мл/мин; температура колонки: 40°С.
Методика II: анализ проводят с использованием системы Agilent LC/MSD 1200, соединенной с квадрупольным масс-спектрометром. Колонка: ODS 2000 (50×4,6 мм, 5 мкм) (режим ионизации ИЭР (+) или (-)), температура колонки: 30°С; скорость потока 1,5 мл/мин.
Общие методы: синтез соединения 8
Пример 1: 3-этил-8-(3-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидропиран-4-ил)пиридо[3,4-b]пиразин-2,5-диаминтригидрохлорид (соединение 8)

Стадия 1: 1-бензил-4-пиперидон (5,00 г, 26,5 ммоль) и 1-трет-бутоксикарбонилпиперазин (5,41 г, 29,1 ммоль) растворяли в дихлорметане (100 мл). Добавляли уксусную кислоту (2,38 г, 39,7 ммоль), после чего перемешивали в течение 5 ч при комнатной температуре. Затем к полученному раствору порциями добавляли натрия триацетоксиборгидрид (22,4 г, 106 ммоль) и раствор перемешивали в течение ночи при комнатной температуре. Раствор концентрировали и к остатку добавляли воду (100 мл). рН раствора доводили до 10 с помощью 5% раствора натрия гидроксида и трижды экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли и промывали насыщенным солевым раствором (50 мл), высушивали над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали на обращенно-фазовой колонке С18 (при увеличении содержания ацетонитрила в воде с 50% до 90%) с получением 4-(1-бензилпиперидин-4-ил)пиперазин-1-карбоновой кислоты трет-бутилового эфира в виде твердого вещества желтого цвета (5,80 г, выход 61%). Результат МС: 360,4 [М+Н]+.
Стадия 2: трет-бутил-4-(1-бензилпиперидин-4-ил)пиперазин-1-карбоксилат (5,80 г, 16,1 ммоль) растворяли в метаноле (100 мл) и добавляли 10% палладий на углеродном носителе (1,5 г). Смесь перемешивали при 50°С в течение ночи в среде водорода (50 фунт/кв. дюйм). В конце реакции раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении с получением трет-бутил-4-(пиперидин-4-ил)пиперазин-1-формиата в виде твердого вещества белого цвета (4,15 г, выход 96%). 1H-ЯМР (300 МГц, CDCl3): δ 3,44-3,41 (мультиплет, 4Н), 3,17-3,13 (мультиплет, 2Н), 2,63-2,58 (мультиплет, 2Н), 2,55-2,49 (мультиплет, 4Н), 2,41-2,31 (мультиплет, 1Н), 1,82-1,78 (мультиплет, 2Н), 1,46 (синглет, 9Н), 1,43-4,33 (мультиплет, 2Н).
Стадия 3: 1-фтор-2-метокси-4-нитробензол (2,00 г, 11,7 ммоль) и трет-бутил-4-(пиперидин-4-ил)пиперазин-1-карбоксилат (3,46 г, 12,9 ммоль) растворяли в N,N-диметилформамиде (30 мл). Затем добавляли калия карбонат (3,23 г, 23,4 ммоль). Смесь перемешивали при 100°С в течение 2 ч. В конце реакции раствор охлаждали до комнатной температуры, после чего добавляли воду (100 мл). Раствор дважды экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли и дважды промывали насыщенным солевым раствором (10 мл×2), высушивали над безводным натрия сульфатом, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-4-(1-(2-(2-метокси-4-нитрофенил)пиперидин-4-ил)пиперазин-1-карбоксилата в виде твердого вещества желтого цвета (4,90 г, выход 99%). 421,0 [М+1]+.
Стадия 4: трет-бутил-4-(1-(2-(2-метокси-4-нитрофенил)пиперидин-4-ил)пиперазин-1-карбоксилат (4,90 г, 11,7 ммоль) растворяли в метаноле (50 мл) и добавляли 10% палладий на углеродном носителе (0,5 г). Смесь перемешивали при 50°С в течение ночи в среде водорода (из баллона). В конце реакции смесь охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали при пониженном давлении с получением трет-бутил-4-(1-(4-амино-2-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоксилата в виде твердого вещества фиолетового цвета (4,50 г, выход 99%). 1H-ЯМР (400 МГц, CDCl3): δ 6,77 (дублет, J=8,4 Гц, 1H), 6,26-6,23 (мультиплет, 2Н), 3,81 (синглет, 3Н), 3,50-3,38 (мультиплет, 8Н), 2,55-2,54 (мультиплет, 2Н), 2,50-2,47 (мультиплет, 2Н), 2,45-2,37 (мультиплет, 1Н), 1,83-1,78 (мультиплет, 4Н), 1,46 (синглет, 9Н).
Стадия 5: трет-бутил-4-(1-(4-амино-2-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоксилат (3,40 г, 8,72 ммоль) и дийодметан (7,00 г, 26,1 ммоль) растворяли в ацетонитриле (100 мл). Добавляли изоамилнитрит (1,53 г, 13,1 ммоль). Смесь перемешивали при 80°С в течение 4 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан (30 мл), воду (20 мл) и насыщенный раствор натрия карбоната (10 мл). Смесь трижды экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этил ацетат = 2:1 - этилацетат) с получением трет-бутил-4-(4-(4-иод-2-метоксифенил)циклогексил)пиперазин-1-карбоксилата в виде твердого вещества коричневого цвета (1,85 г, выход 42%). 1H-ЯМР (300 МГц, CDCl3) δ 7,21 (дублет дублетов, J=8,1, 1,8 Гц, 1H), 7,09 (дублет, J=1,5 Гц, 1H), 6,65 (дублет, J=8,4 Гц, 1H), 3,84 (синглет, 3Н), 3,52-3,48 (мультиплет, 2Н), 3,46-3,42 (мультиплет, 4Н), 2,56-2,47 (мультиплет, 6Н), 2,42-2,38 (мультиплет, 1Н), 1,87-1,74 (мультиплет, 4Н), 1,46 (синглет, 9Н). Результат МС: 502,5 [М+Н]+.
Стадия 6: трет-бутил-4-(4-(4-йод-2-метоксифенил)циклогексил)пиперазин-1-карбоксилат (1,85 г, 3,69 ммоль) растворяли в N,N-диметилформамиде (100 мл). Добавляли пинаколдиборат (1,13 г, 4,43 ммоль), калия ацетат (1,09 г, 11,1 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (135 мг, 0,17 ммоль). Смесь перемешивали при 100°С в течение 5 ч в защитной среде азота. В конце реакции раствор охлаждали до комнатной температуры и вливали в воду (100 мл). Добавляли насыщенный раствор натрия карбоната (10 мл). Смесь трижды экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат = 1:1 - этилацетат) с получением 4-(1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперидин-4-ил)-1-трет-бутоксикарбонилпиперазина в виде твердого вещества желтого цвета (промежуточный продукт А, 1,30 г, выход 70%). 1Н-ЯМР (300 МГц, CDCl3) δ 7,39 (дублет, J=7,8 Гц, 1H), 7,25 (синглет, 1H), 6,93 (дублет, J=7,8 Гц, 1Н), 3,91 (синглет, 3Н), 3,64-3,60 (мультиплет, 2Н), 3,46-3,43 (мультиплет, 4Н), 2,62-2,55 (мультиплет, 6Н), 2,50-2,41 (мультиплет, 1Н), 1,89-1,76 (мультиплет, 4Н), 1,47 (синглет, 9Н), 1,33 (синглет, 12Н). Результат МС: 502,6 [М+Н]+.
Стадия 7: 5-бром-2-хлорпиридин-3,4-диамин (5,00 г, 22,5 ммоль) и этил-2-оксобутират (3,51 г, 27,0 ммоль) растворяли в этаноле (100 мл) и уксусной кислоте (0,2 мл). Смесь перемешивали при 90°С в течение 36 ч в защитной среде азота. В конце реакции температуру снижали до 50°С. Раствор фильтровали и осадок на фильтре промывали этанолом (10 мл×3). Фильтрат концентрировали при пониженном давлении с получением остатка, который затем растворяли в этаноле (20 мл). Раствор кипятили с обратным холодильником в течение 10 мин, затем охлаждали до 50°С, фильтровали, и осадок на фильтре промывали этанолом (10 мл×2), сушили с получением 8-бром-5-хлор-3-этилпиридо[3,4-b]пиразин-2(1Н)-она в виде твердого вещества желтого цвета (4,1 г, выход 63%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 12,32 (широкий синглет, 1H), 8,50 (синглет, 1H), 2,86 (квартет, J=7,6 Гц, 2Н), 1,25 (триплет, J=7,6 Гц, 3Н).
Стадия 8: 8-бром-5-хлор-3-этилпиридо[3,4-b]пиразин-2(1Н)-он (3,10 г, 10,7 ммоль) суспендировали в дихлорметане (50 мл) и добавляли 5 капель N,N-диметилформамида и оксалилхлорид (5,46 г, 43,0 ммоль). Смесь перемешивали при 40°С в течение ночи в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан (50 мл), воду (30 мл) и насыщенный раствор натрия карбоната (10 мл). Смесь пять раз экстрагировали дихлорметаном (30 мл×5). Органические фазы объединяли, промывали насыщенным солевым раствором, высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении с получением 8-бром-2,5-дихлор-3-этилпиридо[3,4-b]пиразина в виде твердого вещества желтого цвета (2,0 г, выход 61%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ 8,91 (синглет, 1H), 3,20 (квартет, J=7,2 Гц, 2Н), 1,39 (триплет, J=7,2 Гц, 3Н).
Стадия 9: тетрагидропиран-4-амин (1,35 г, 9,77 ммоль) суспендировали в этаноле (20 мл) и добавляли N,N-диизопропилэтиламин (2,52 г, 19,5 ммоль). Раствор перемешивали при комнатной температуре в течение получаса. 8-Бром-2,5-дихлор-3-этилпиридо[3,4-b]пиразин (2,00 г, 6,51 ммоль) суспендировали в этаноле (30 мл) и добавляли N,N-диизопропилэтиламин (1,68 г, 13,0 ммоль). Раствор перемешивали при комнатной температуре в течение получаса. Два раствора объединяли и перемешивали при 70°С в течение ночи. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду (30 мл) и полученный раствор фильтровали. Осадок на фильтре промывали смесью дихлорметан/метанол = 10/1 (5 мл×3) и сушили с получением 8-бром-5-хлор-3-этил-N-(тетрагидропиран-4-ил)пиридо[3,4-b]пиразин-2-амина в виде твердого вещества желтого цвета (1,2 г, выход 50%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,50 (синглет, 1H), 7,81 (дублет, J=7,2 Гц, 1Н), 4,42-4,31 (мультиплет, 1Н), 3,98-3,94 (мультиплет, 2Н), 3,43 (триплет, J=7,5 Гц, 2Н), 2,92 (квартет, J=7,2 Гц, 2Н), 1,98-1,93 (мультиплет, 2Н), 1,80-1,66 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н).
Стадия 10: к смеси 8-бром-5-хлор-3-этил-N-(тетрагидропиран-4-ил)пиридо[3,4-b]пиразин-2-амина (0,960 г, 2,59 ммоль) и 4-(1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил)фенил)пиперидин-4-ил)-1-трет-бутоксикарбонилпиперазина (промежуточный продукт А, 1,30 г, 2,59 ммоль) в N,N-диметилацетамиде (50 мл) добавляли водный раствор (5 мл) натрия карбоната (824 мг, 7,78 ммоль) и [11,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (190 мг, 0,26 ммоль). Смесь перемешивали при 100°С в течение 3 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и вливали в воду (150 мл). Смесь трижды экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (этилацетат дихлорметан/метанол/25% водный раствор аммиака = 30:1:0,1) с получением неочищенного продукта. Неочищенный продукт очищали на обращенно-фазовой хроматографической колонке С18 (при увеличении содержания ацетонитрила в воде с 50% до 80%) с получением трет-бутил-4-(1-(4-(5-хлор-3-этил)-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридил[3,4-b]пиразин-8-ил)-2-метоксифенил)пиперидин-4-илпиперазин-1-карбоксилата в виде твердого вещества желтого цвета (0,60 г, выход 35%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ 8,32 (синглет, 1H), 7,56 (дублет, J=7,2 Гц, 1H), 7,30-7,28 (мультиплет, 2Н), 6,97-6,95 (мультиплет, 1H), 4,19-4,11 (мультиплет, 1H), 3,93-3,89 (мультиплет, 2Н), 3,83 (синглет, 3Н), 3,52-3,49 (мультиплет, 2Н), 3,31-3,30 (мультиплет, 4Н), 3,25-3,22 (мультиплет, 2Н), 2,90 (квартет, J=7,2 Гц, 2Н), 2,61-2,57 (мультиплет, 2Н), 2,50-2,41 (мультиплет, 4Н), 2,38-2,32 (мультиплет, 1H), 1,88-1,81 (мультиплет, 4Н), 1,71-1,55 (мультиплет, 4Н), 1,40 (синглет, 9Н), 1,32 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 666,8 [М+Н]+.
Стадия 11: к смеси трет-бутил 4-(1-(4-(5-хлор-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридинил[3,4-b]пиразин-8-ил)-2-метоксифенил)пиперидин-4-илпиперазин-1-карбоксилата (0,30 г, 0,45 ммоль), (2,4-диметоксифенил)метиламина (113 мг, 0,680 ммоль), 4,5-бис(дифенилфосфин)-9,9-диметилксантена (26 мг, 0,045 ммоль) и натрия трет-бутоксида (130 мг, 1,35 ммоль) в толуоле (10 мл) добавляли трис(дибензилиденацетон)дипалладий (21 мг, 0,024 ммоль). Смесь перемешивали при 110°С в течение 3 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли этилацетат (20 мл) и воду (30 мл). Смесь трижды экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 30:1) с получением неочищенного продукта. Неочищенный продукт очищали на обращенно-фазовой хроматографической колонке С18 (при увеличении содержания ацетонитрила в воде с 40% до 90%) с получением трет-бутил-4-(1-(4-(5-((2,4)-диметоксибензил)амино)-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридинил[3,4-b]пиразин-8-ил)-2-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоновой кислоты в виде твердого вещества желтого цвета (0,13 г, выход 36%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,16 (синглет, 1Н), 7,35-7,33 (мультиплет, 1Н), 7,30-7,27 (мультиплет, 1Н), 7,16 (синглет, 1Н), 6,96-6,94 (мультиплет, 2Н), 6,50-6,49 (мультиплет, 1Н), 6,46-6,43 (мультиплет, 1Н), 4,80-4,78 (мультиплет, 2Н), 4,76-4,74 (мультиплет, 1Н), 4,25-4,16 (мультиплет, 1Н), 4,01-3,98 (мультиплет, 2Н), 3,89-3,88 (мультиплет, 6Н), 3,80 (синглет, 3Н), 3,62-3,59 (мультиплет, 2Н), 3,50-3,45 (мультиплет, 6Н), 2,70 (квартет, J=7,2 Гц, 2Н), 2,64-2,59 (мультиплет, 6Н), 2,53-2,45 (мультиплет, 1Н), 2,08-2,05 (мультиплет, 2Н), 1,89-1,82 (мультиплет, 4Н), 1,56-1,52 (мультиплет, 2Н), 1,47 (синглет, 9Н), 1,41 (триплет, J=7,2 Гц, 3Н). Результат МС: 797,5 [М+Н]+.
Стадия 12: к смеси трет-бутил-4-(1-(4-(5-((2,4-диметоксибензил)амино)амино)-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридинил[3,4-b]пиразин-8-ил)-2-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоновой кислоты (0,13 г, 0,16 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при 30°С в течение 1 ч и концентрировали. К полученному остатку добавляли воду (2 мл) и рН доводили до 10 с помощью 5% раствора натрия гидроксида. Раствор экстрагировали шесть раз (10 мл×6) смесью дихлорметан/метанол = 10/1. Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении с получением 3-этил-8-(3-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридин[3,4-b]пиразин-2,5-диамина в виде твердого вещества желтого цвета (90 мг, выход 100%). Результат МС: 547,6 [М+Н]+.
Стадия 13: к смеси 3-этил-8-(3-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридин[3,4-b]пиразин-2,5-диамина (90 мг, 0,16 ммоль) в дихлорметане (2 мл) и метаноле (1 мл) добавляли 37% водный раствор формальдегида (15 мг, 0,18 ммоль) и 1 каплю уксусной кислоты. После перемешивания при комнатной температуре в течение 20 мин добавляли натрия цианоборгидрид (31 мг, 0,49 ммоль). После перемешивания при комнатной температуре в течение 30 мин добавляли воду (2 мл) и подщелачивали до рН=10 с помощью 5% раствора натрия гидроксида. Смесь экстрагировали шесть раз (10 мл×6) смесью дихлорметан/метанол = 10/1. Органические фазы объединяли и концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали на обращенно-фазовой хроматографической колонке С18 (при увеличении содержания ацетонитрила в воде с 10% до 50%, водная фаза при этом содержала 0,1% хлористоводородной кислоты) с получением 3-этил-8-(3-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридин[3,4-b]пиразин-2,5-диаминтригидрохлорида в виде твердого вещества желтого цвета (65 г, выход 61%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 13,77 (синглет, 1Н), 12,48-12,17 (мультиплет, 2Н), 8,54 (синглет, 2Н), 7,96-7,94 (мультиплет, 2Н), 7,55 (широкий синглет, 1Н), 7,43-7,32 (мультиплет, 2Н), 4,14-4,05 (мультиплет, 1Н). 3,95 (синглет, 3Н), 3,92-3,89 (мультиплет, 2Н), 3,79-3,60 (мультиплет, 12Н), 3,35-3,27 (мультиплет, 1Н), 3,25-3,22 (мультиплет, 2Н), 2,92-2,86 (мультиплет, 5Н), 2,35-2,25 (мультиплет, 4Н), 1,83-1,78 (мультиплет, 2Н), 1,74-1,64 (мультиплет, 2Н), 1,33 (триплет, J=7,2 Гц, 3Н). Результат МС: 561,4 [М+Н]+.
Соединения, указанные в таблицах, получали с использованием того же способа и соответствующих исходных материалов.
Соединения в следующей таблице получали способом синтеза соединения 8, при этом тетрагидропиран-4-амин на стадии 9 заменяли на соответствующие исходные материалы, указанные в таблице:

Соединения в следующей таблице получали способом синтеза соединения 8, при этом тетрагидропиран-4-амин на стадии 9 заменяли на соответствующие исходные материалы, указанные в таблице, а соответствующий продукт на стадии 11 гидролизовали щелочью для снятия ацетильной защитной группы:

Соединения в следующей таблице получали способом синтеза соединения 8, при этом 1-фтор-2-метокси-4-нитробензол на стадии 3 заменяли на соответствующие исходные материалы, указанные в таблице:
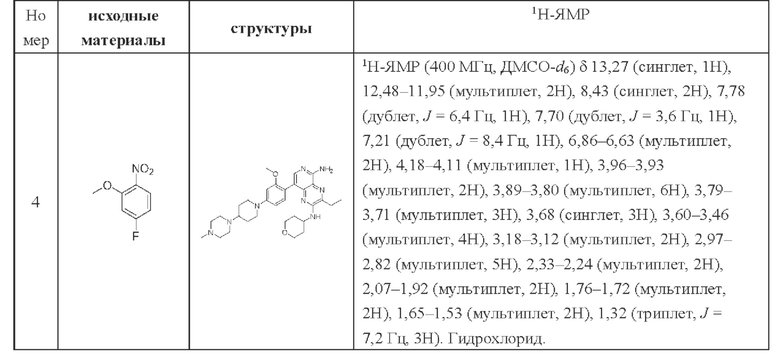


Соединения в следующей таблице получали способом синтеза соединения 8, при этом промежуточный продукт А заменяли на соответствующие исходные материалы, указанные в таблице ниже, для реакции с 8-бром-5-хлор-3-этил-N-(тетрагидропиран-4-ил)пиридо[3,4-b]пиразин-2-амином:

Продукт на стадии 10 способа синтеза соединения 8 представляет собой соединение 12, указанное в следующей таблице:

Соединение 11 в следующей таблице получали путем снятия защиты на стадии 12 в способе синтеза соединения 8, при этом соединение 12 использовали в качестве исходного материала:

Соединения в следующей таблице получали способом синтеза, описанным в примере 11, при этом использовали соответствующие исходные материалы, указанные в таблице:


Соединение в следующей таблице получали способом синтеза соединения 8, при этом (2,4-диметоксифенил)метиламин заменяли на триметилбороксан и на стадии 11 использовали реакцию Сузуки:

Соединения в следующей таблице получали способом синтеза соединения 8, при этом этил-2-оксобутират заменяли на соответствующие исходные материалы, указанные в таблице:


Соединения в следующей таблице получали способом синтеза соединения 8, при этом трет-бутил-4-(пиперидин-4-ил)пиперазин-1-карбоксилат заменяли на соответствующие исходные материалы, указанные в таблице:


Пример 2: промежуточный продукт трет-бутил-4-(1-(4-(5-((2,4-диметоксибензил)амино)-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридино[3,4-b]пиразин-8-ил)-2-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоновую кислоту, полученную на стадии 11 примера 1, также можно синтезировать следующим способом, после чего может быть получено соединение 8 на стадиях 12 и 13 в примере 1:

Стадия 1: смесь 8-бром-5-хлор-3-этил-N-(тетрагидропиран-4-ил)пиридино[3,4-b]пиразин-2-амина (6,00 г, 16,1 ммоль) и 2,4-диметоксибензамина (15 г, 90 ммоль) перемешивали при 145°С в течение ночи. В конце реакции смесь охлаждали до комнатной температуры и очищали на хроматографической колонке с силикагелем (петролейный эфир/этилацетат = 5:1-3:1) с получением 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина в виде твердого вещества желтого цвета (5,5 г, выход 68%). Полученный методом МС результат: 502,5 [М+Н]+.
Стадия 2: к смеси 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина (4,00 г, 7,97 ммоль) и промежуточного продукта А (4,0 г, 7,97 ммоль) в 1,4-диоксане (100 мл) добавляли цезия карбонат (5,20 г, 15,9 ммоль) в воде (15 мл) и Pd(dppf)Cl2 (583 мг, 0,80 ммоль). Эту смесь перемешивали и проводили реакцию при 110°С в течение 3 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении и добавляли этилацетат (50 мл) и воду (50 мл). Раствор трижды экстрагировали этилацетатом (50 мл×3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат = 1:1-0:1 - этилацетат/метанол = 20:1) с получением промежуточного продукта В в виде твердого вещества желтого цвета (3,1 г, выход 49%).
Соединения в следующей таблице получали на стадии 9 в примере 1, при этом тетрагидропиран-4-амин заменяли на соответствующие исходные материалы, указанные в таблице, и подвергали реакции с промежуточным продуктом М согласно способу синтеза соединения 8 в примере 2:


Соединения в следующей таблице получали способом синтеза соединения 8 в примере 1, при этом тетрагидропиран-4-амин на стадии 9 заменяли на соответствующие исходные материалы, указанные в таблице, а затем получали согласно способу, описанному в примере 2:


Соединение 10 в следующей таблице получали способом синтеза, описанным в примере 2, при этом 2,4-диметоксибензамин заменяли на метиламин.

Соединение 23 в следующей таблице получали способом синтеза промежуточного продукта А в примере 1 и способом синтеза в примере 2, при этом 1-фтор-2-метокси-4-нитробензол заменяли на исходный материал, указанный в таблице:


Соединение 31 в следующей таблице получали способом синтеза промежуточного продукта А в примере 1 и способом синтеза в примере 2, при этом 1-фтор-2-метокси-4-нитробензол и трет-бутил-4-(пиперидин-4-ил)пиперазин-1-карбоксилат заменяли на 1-фтор-2-метил-4-нитробензол и трет-бутил-3,9-диазаспиро[5,5]ундекан-3-карбоксилат, соответственно, указанные в таблице.

Соединения в следующей таблице получали способом синтеза промежуточного продукта А в примере 1 и способом синтеза в примере 2, при этом 2,4-(пиперидин-4-ил)пиперазин-1-карбоновой кислоты трет-бутиловый эфир заменяли на соответствующие исходные материалы, указанные в таблице.


Соединения в следующей таблице получали согласно способу синтеза промежуточного продукта А в примере 1 и способу синтеза в примере 2, при этом 1-бензил-4-пиперидон заменяли на исходный материал, указанный в таблице.

Соединения в следующей таблице получали согласно способу, описанному в примере 1, при этом 2-оксобутирата этиловый эфир на стадии 7 заменяли на исходный материал, указанный в таблице, после чего использовали способ, описанный в примере 2


Соединения в следующей таблице получали согласно способу синтеза промежуточного продукта А в примере 1 и способу синтеза в примере 2, при этом 4-(4-(4-иод-2-метоксифенил)циклогексил)пиперазин-1-трет-бутилформиат заменяли на исходный материал, указанный в таблице.

Пример 3: 3-этил-8-(6-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пиридин-3-ил)-N2-тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 21)

Стадия 1: 5-бром-2-фторпиридин (500 мг, 2,84 ммоль) и 1-метил-4-(4-пиперидинил)пиперазин (520 мг, 2,84 ммоль) растворяли в N,N-диметилформамиде (10 мл) и добавляли калия карбонат (784 мг, 5,68 ммоль). Эту смесь перемешивали и проводили реакцию при 110°С в течение 6 ч. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (50 мл). Раствор экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан : метанол = 20:1 - 10:1) с получением твердого вещества белого цвета (800 мг, выход 83%). 1H-ЯМР (400 МГц, CDCl3): δ 8,16 (дублет, J=2,0 Гц, 1Н), 7,49 (дублет дублетов, J=8,8, 2,0 Гц, 1Н), 6,56 (дублет, J=8,8 Гц, 1Н), 4,29-4,26 (мультиплет, 2Н), 2,86-2,79 (мультиплет, 2Н), 2,71-2,55 (мультиплет, 4Н), 2,49-2,42 (мультиплет, 5Н), 2,28 (синглет, 3Н), 1,93-1,85 (мультиплет, 2Н), 1,57-1,47 (мультиплет, 2Н).
Стадия 2: к смеси 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-Ь]пиразин-2,5-диамина (800 мг, 1,59 ммоль) и дивалерилдибора (810 мг, 3,19 ммоль) в N,N-диметилформамиде (5 мл) добавляли калия ацетат (364 мг, 3,19 ммоль) и Pd(dppf)Cl2 (117 мг, 0,16 ммоль). Эту смесь перемешивали и проводили реакцию при 110°С в течение 5 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (30 мл). Раствор трижды экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этил ацетат = 1:1-0:1 - этилацетат/метанол = 10:1) с получением промежуточного продукта С в виде твердого вещества коричневого цвета (230 мг, выход 31%). Полученный методом МС результат: 468,6 [М+Н]+.
Стадия 3: используя способ синтеза, описанный для стадии 2 в примере 2, проводили реакцию 1-(1-(5-бромпиридин-2-ил)пиперидин-4-ил)-4-метилпиперазина (87 мг, 0,26 ммоль) с промежуточным продуктом С (100 мг, 0,21 ммоль) с получением N5-(2,4-диметоксибензил)-3-этил-8-(6-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пиридин-3-ил)-N2-(тетрагидро-2Н)пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина в виде твердого вещества желтого цвета (70 мг, выход 49%). Полученный методом МС результат: 682,6 [М+Н]+.
Стадия 4: используя способ, описанный для стадии 12 в примере 1, проводили реакцию N5-(2,4-диметоксибензил)-3-этил-8-(6-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пиридин-3-ил)-N2-(тетрагидро-2Н)пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина (70 мг, 0,1 ммоль) с получением соединения 21 в виде твердого вещества желтого цвета (70 мг, 0,1 ммоль). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 8,38 (дублет, J=2,0 Гц, 1Н), 7,92-7,88 (мультиплет, 2Н), 7,05 (дублет, J=6,8 Гц, 1Н), 6,85 (дублет, J=8,8 Гц, 1Н), 6,45 (синглет, 2Н), 4,35-4,31 (мультиплет, 2Н), 4,11-4,03 (мультиплет, 1Н), 3,93-3,91 (мультиплет, 2Н), 3,40-3,28 (мультиплет, 4Н), 2,83-2,78 (мультиплет, 4Н), 2,42-2,30 (мультиплет, 7Н), 2,14 (синглет, 3Н), 1,89-1,80 (мультиплет, 4Н), 1,66-1,57 (мультиплет, 2Н), 1,44-1,34 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 532,3[М+Н]+.
Пример 4: 3-этил-8-(4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-3-(метилсульфил)фенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 18)

Стадия 1: коммерчески доступные 1-хлор-2-(метилсульфон)-4-нитробензол (1,20 г, 5,08 ммоль) и 1-метил-4-(4-пиперидинил)пиперазин (931 мг, 5,08 ммоль) растворяли в диметилсульфоксиде (20 мл) и добавляли калия карбонат (1,40 г, 10,2 ммоль). Эту смесь перемешивали и проводили реакцию при 100°С в течение 2 ч. В конце реакции смесь охлаждали до комнатной температуры и добавляли в воду (100 мл). Раствор экстрагировали этилацетатом (20 мл×5). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл×2) и концентрировали при пониженном давлении. Добавляли дихлорметан (20 мл) и раствор вливали в водный раствор (30 мл) концентрированной хлористоводородной кислоты (3 мл) и промывали дихлорметаном (20 мл). Водную фазу подщелачивали 5% раствором натрия гидроксида и экстрагировали дихлорметаном (20 мл×4). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении с получением твердого вещества коричневого цвета (1,70 г, выход 87%). [М+Н]+. 1H-ЯМР (400 МГц, CDCl3) δ 8,95 (дублет, J=2,8 Гц, 1Н), 8,42 (дублет дублетов, J=8,8, 2,8 Гц, 1Н), 7,44 (дублет, J=9,2 Гц, 1Н), 3,55-3,52 (мультиплет, 2Н), 3,32 (синглет, 3Н), 2,87-2,81 (мультиплет, 2Н), 2,73-2,65 (мультиплет, 4Н), 2,58-2,50 (мультиплет, 4Н), 2,43-2,34 (мультиплет, 1Н), 2,30 (синглет, 1Н), 2,08-2,05 (мультиплет, 2Н), 1,82-1,72 (мультиплет, 2Н). Полученный методом МС результат: 383,2.
Стадия 2: используя способ, описанный для стадии 4 в примере 1, в качестве исходного материала использовали 1-метил-4-(1-(2-(метилсульфонил)-4-нитрофенил)пиперидин-4-ил)пиперазин с получением 4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-3-(метилсульфонил)анилина в виде твердого вещества белого цвета (1,50 г, выход 96%). Полученный методом МС результат: 353,5 [М+Н]+.
Стадия 3: 4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-3-(метилсульфонил)анилин (600 мг, 1,70 ммоль) растворяли в водном растворе (5 мл) концентрированной серной кислоты (834 мг, 8,51 ммоль). Температуру раствора доводили до 0°С и к раствору по каплям добавляли водный раствор (0,5 мл) натрия нитрита (117 мг, 8,51 ммоль). Раствор перемешивали при 0-5°С в течение 10 мин, затем к раствору по каплям добавляли водный раствор (2 мл) калия йодида (565 мг, 3,40 ммоль). Раствор перемешивали при 5°С в течение 30 мин, после чего вливали в воду (5 мл). рН раствора доводили до 10 с помощью 5% раствора натрия гидроксида и экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 10:1) на колонке С18 (40-80% водный раствор ацетонитрила) с получением твердого вещества желтого цвета (250 мг, выход 32%). Полученный методом МС результат: 464,1 [М+Н]+.
Стадия 4 и 5: использовали способ, описанный для стадии 3 и стадии 4 в примере 3. Проводили реакцию 1-(1-(4-йод-2-(метилсульфон)фенил)пиперидин-4-ил)-4-метилпиперазина (87 мг, 0,26 ммоль) с промежуточным продуктом С (200 мг, 0,43 ммоль) с получением соединения 18 в виде твердого вещества желтого цвета (60 мг, выход после двух стадий 46%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,46 (дублет, J=1,2 Гц, 1Н), 7,98 (синглет, 1Н), 7,91 (дублет, J=8,4 Гц, 1H), 7,55 (дублет, J=8,4 Гц, 1Н), 7,06 (дублет, J=7,6 Гц, 1Н), 6,62 (синглет, 2Н), 4,28-4,18 (мультиплет, 1Н), 3,84-3,82 (мультиплет, 2Н), 3,40 (синглет, 3Н), 3,38-3,32 (мультиплет, 4Н), 3,26-3,23 (мультиплет, 2Н), 2,84-2,77 (мультиплет, 5Н), 2,73-2,59 (мультиплет, 6Н), 2,37 (синглет, 3Н), 1,93-1,91 (мультиплет, 2Н), 1,82-1,79 (мультиплет, 2Н), 1,64-1,57 (мультиплет, 4Н), 1,31 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 609,4 [М+Н]+.
Пример 5: 2-(5-(5-амино-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридино[3,4-b]пиразин-8-ил)-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пропан-2-ол (соединение 19)

Стадия 1: используя способ, описанный для стадии 1 в примере 4, проводили реакцию 1-(5-бром-2-фторфенил)этанона (1,20 г, 5,53 ммоль) с 1-метил-4-(4-пиперидинил)пиперазином (1,01 г, 5,53 ммоль) с получением твердого вещества желтого цвета (1,40 г, выход 67%). Полученный методом МС результат: 380,1 [М+Н]+.
Стадия 2: к раствору 1-(5-бром-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)этил-1-она (400 мг, 1,05 ммоль) в тетрагидрофуране (10 мл) по каплям при 0°С в защитной среде азота добавляли раствор метилмагния бромида в тетрагидрофуране (3,0 М, 1,05 мл, 3,16 ммоль). Реакционную смесь перемешивали при 0-5°С в течение 10 мин, затем вливали в воду (10 мл) и экстрагировали смесью дихлорметан/метанол (10/1, 10 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный остаток очищали на колонке С18 (водный раствор ацетонитрила 10-60%) с получением твердого вещества белого цвета (170 мг, выход 41%).
Стадии 3 и 4: используя способ, описанный для стадии 3 и стадии 4 в примере 3, проводили реакцию 2-(5-бром-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пропан-2-ола (102 мг, 0,26 ммоль) с промежуточным продуктом С (120 мг, 0,26 ммоль) с получением соединения 19 в виде твердого вещества белого цвета (60 мг, выход после двух стадий 30%). 1H-ЯМР (400 МГц, ДМСО-d6): δ 7,92 (синглет, 1Н), 7,64-7,61 (мультиплет, 2Н), 7,54 (широкий синглет, 1Н), 7,39 (дублет, J=8,0 Гц, 1Н), 7,01 (дублет, J=7,2 Гц, 1Н), 6,52 (синглет, 2Н), 4,20-4,12 (мультиплет, 1Н), 3,90-3,88 (мультиплет, 2Н), 3,30-3,27 (мультиплет, 4Н), 2,97-2,88 (мультиплет, 4Н), 2,81 (квартет, J=7,2 Гц, 2Н), 2,74-2,59 (мультиплет, 7Н), 2,37 (синглет, 3Н), 1,95-1,92 (мультиплет, 2Н), 1,84-1,80 (мультиплет, 2Н), 1,67-1,58 (мультиплет, 2Н), 1,57-1,48 (мультиплет, 8Н), 1,31 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 589,4 [М+Н]+.
Пример 6: 3-этил-8-(3-((4-(4-метилпиперазин-1-ил)пиперидин-1-ил)метил)фенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридо[3,4-b]пиразин-2,5-диамин (соединение 43)

Стадия 1: используя способ, описанный для стадии 2 в примере 2, проводили реакцию 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина (150 мг, 0,3 ммоль) с 3-формилфенилбороновой кислотой (54 мг, 0,36 ммоль) с получением промежуточного продукта В в виде твердого вещества желтого цвета (110 мг, выход 69%).
Стадия 2: к смеси 3-(5-((2,4-диметоксибензил)амино)-3-этил-2-((тетрагидро-2Н-пиран-4 -ил)амино)пиридино[3,4-b]пиразин-8-ил)бензальдегида (110 мг, 0,21 ммоль) и 1-метил-4-(пиперидин-4-ил)пиперазина(57 мг, 0,31 ммоль) в дихлорметане (5 мл) добавляли 1 каплю уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и добавляли натрия триацетоксиборгидрид (133 мг, 0,63 ммоль). После перемешивания при комнатной температуре в течение ночи добавляли воду (5 мл). Смесь экстрагировали дихлорметаном (15 мл×4), органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 30:1-15:1) с получением твердого вещества желтого цвета (100 мг, выход 69%). Полученный методом МС результат: 695,7 [М+Н]+.
Стадия 3: используя способ снятия защиты, описанный для стадии 12 в способе синтеза соединения 8, в качестве исходного материала использовали N5-(2,4-диметоксибензил)-3-этил-8-(3-((4-(4-метилпиперазин-1-ил)пиперидин-1-ил)метил)фенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (80 мг, 0,12 ммоль) с получением соединения 43 в виде твердого вещества белого цвета (40 мг, выход 61%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,01 (синглет, 1Н), 7,76-7,71 (мультиплет, 2Н), 7,42-7,38 (мультиплет, 2Н), 7,16 (дублет, J=7,2 Гц, 2Н), 6,75 (синглет, 2Н), 4,13-4,04 (мультиплет, 1Н), 3,93-3,90 (мультиплет, 3Н), 3,32-3,26 (мультиплет, 5Н), 3,15-3,06 (мультиплет, 3Н), 2,93-2,72 (мультиплет, 10Н), 2,54 (синглет, 3Н), 1,86-1,84 (мультиплет, 4Н), 1,70-1,61 (мультиплет, 4Н), 1,31 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 545,3 [М+Н]+.
Пример 7: 3-этил-8-(3-метокси-4-(4-((4-метилпиперазин-1-ил)метил)пиперидин-1-ил)фенил)-1H-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 45)

Стадия 1: к смеси 1-метил-4-(пиперидин-4-илметил)пиперазина (1,0 г. 5,1 ммоль) и 1-бром-4-хлор-2-метоксибензола (1,1 г, 5,1 ммоль) в 1,4-диоксане (15 мл) добавляли цезия ацетат (5,0 г, 15 ммоль), Pd2(dba)3 (0,50 г, 0,50 ммоль), BINAP (0,60 г, 1,0 ммоль). Эту смесь перемешивали и проводили реакцию при 110°С в течение 3 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (30 мл). Смесь трижды экстрагировали (20 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 4:1) с получением маслянистого вещества желтого цвета (620 мг, выход 35%). (ИЭР) m/z 338,1. [М+Н]+.
Стадия 2: к смеси 1-((1-(4-хлор-2-метоксифенил)пиперидин-4-ил)метил)-4-метилпиперазина (400 мг, 1,2 ммоль) и дивалерилдибора (601 мг, 2,4 ммоль) в 1,4-диоксане (10 мл) добавляли калия ацетат (291 мг, 3,6 ммоль), pd(dba)2 (14 мг.0,020 ммоль) и X-Phos (11 мг, 0,020 ммоль). Полученную смесь перемешивали при 110°С в течение 16 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 4:1) с получением маслянистого вещества желтого цвета (400 мг, выход 75%). (ИЭР) m/z 430,2. [М+Н]+.
Стадия 3: используя способ, описанный для стадии 2 в примере 2, проводили реакцию 1-((1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксоран-2-ил)фенил)пиперидин-4-ил)метил)-4-метилпиперазина (137 мг, 0,31 ммоль) с 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-d]пиразин-2,5-диамином (100 мг, 0,21 ммоль) с получением маслянистого вещества желтого цвета (112 мг, выход 79%). (ИЭР) m/z 725,3. [М+Н]+.
Стадия 4: N5-(2,4-диметоксибензил)-3-этил-8-(3-метокси-4-(4-((4-метилпиперазин-1-ил)метил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (112 мг, 0,16 ммоль) растворяли в метанольном растворе (4 М, 5 мл) хлористоводородной кислоты. Раствор перемешивали при 25°С в течение 16 ч. В конце реакции раствор охлаждали до комнатной температуры и реакцию останавливали насыщенным раствором натрия бикарбоната (10 мл). Смесь трижды экстрагировали (10 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной жидкостной хроматографией с получением формиата соединения 45 в виде твердого вещества желтого цвета (19 мг, выход 20%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,25 (синглет 1Н), 7,92 (синглет, 1Н), 7,24-7,17 (мультиплет, 2Н), 7,00 (дублет, J=7,2 Гц, 1Н), 6,89 (дублет, J=8,0 Гц, 1Н), 6,44 (синглет, 2Н), 4,14-4,10 (мультиплет, 1Н), 3,90 (дублет, J=8,4 Гц, 2Н), 3,81 (синглет, 3Н), 3,24 (синглет, 6Н), 2,83-2,78 (мультиплет, 2Н), 2,54 (дублет, J=12,8 Гц, 3Н), 2,43-2,22 (мультиплет, 5Н), 2,17 (дублет, J=7,2 Гц, 2Н), 2,15 (синглет, 3Н), 1,86 (дублет, J=12,4 Гц, 2Н), 1,77 (дублет, J=12,0 Гц, 2Н), 1,68-1,56 (мультиплет, 3Н), 1,33-1,22 (мультиплет, 5Н). (ПЭР) m/z 575,3. [М+Н]+.
Используя способ, описанный для стадии 1 в примере 7, 1-бром-4-хлор-2-метоксибензол заменяли на исходное вещество, указанное в следующей таблице, с получением соответствующего промежуточного продукта, после чего, используя способ, описанный в примере 7, 1-метил-4-(пиперидин-4-илметил)пиперазин заменяли на 1-метил-4-(4-пиперидинил) с получением соединения, указанного в следующей таблице

Используя способ, описанный для стадии 9 в примере 1, и способ синтеза, описанный для стадии 1 в примере 2, тетрагидропиран-4-амин заменяли на исходные материалы из следующей таблицы с получением соответствующих промежуточных продуктов, после чего, используя способ, описанный в примере 7, 1-метил-4-(пиперидин-4-илметил)пиперазин заменяли на 1-метил-4-(4-пиперидинил) с получением соединений, указанных в следующей таблице:




Используя способ, описанный в примере 7, 1-метил-4-(пиперидин-4-илметил)пиперазин заменяли на исходные материалы, указанные в следующей таблице, с получением следующих соединений:

Используя способ, описанный в примере 7, и способ каталитического гидрирования, описанный для стадии 4 в примере 1, 1-((1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксиборан-2-ил)фенил)пиперидин-4-ил)метил)-4-метилпиперазин заменяли на исходный материал, указанный в таблице ниже, с получением соединения, указанного в таблице:


Используя способ, описанный для стадии 2 в примере 2, и способ снятия защитной группы, описанный для стадии 12 в примере 1, 1-((1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксиборан-2-ил)фенил)пиперидин-4-ил)метил)-4-метилпиперазин заменяли на исходный материал, указанный в таблице ниже, с получением соединения, указанного в таблице:

Используя способ, описанный в примере 7, 1-((1-(2-метокси-4-(4,4,5,5-тетраметил- 1,3,2-диоксоран-2-ил)фенил)пиперидин-4-ил)метил)-4-метилпиперазин заменяли на исходные материалы, указанные в следующей таблице, с получением следующих соединений:



Используя способ, описанный для стадии 9 в примере 1, тетрагидропиран-4-амин заменяли на следующий исходный материал 2 для получения соответствующего промежуточного продукта, после чего, используя способ, описанный в примере 7 со стадии 3 до конца, 1-((1-(2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксоран-2-ил)фенил)пиперидин-4-ил)метил)-4-метилпиперазин заменяли на следующий исходный материал 1 с получением соединения, указанного в следующей таблице:

Пример 8: 3-этил-8-(4-метилпипнрадин-1-ил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 53)

Стадия 1: к смеси 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина (75 мг, 0,15 ммоль) и 1-метилпиперазина (30 мг, 0,30 ммоль) в 1,4-диоксане (5 мл) добавляли калия трет-бутоксид (43 мг, 0,45 ммоль), tBuXPhos Pd G3 (27 мг, 0,030 моль) и tBuXPhos (28 мг, 0,060 ммоль). Смесь перемешивали при 110°С в течение 14 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (10 мл). Смесь трижды экстрагировали (10 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 10:1) с получением маслянистого вещества коричневого цвета (50 мг, выход 64%). (ИЭР) m/z 522,2. [М+1]+.
Стадия 2: используя способ, описанный для стадии 4 в примере 7, проводили реакцию N5-(2,4-диметоксибензил)-3-этил-8-(4-метилпиперазин-1-ил)-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамина (50 мг, 0,096 ммоль) с получением соединения 53 в виде твердого вещества желтого цвета (15 мг, 42 ммоль). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,18 (широкий синглет, 2Н), 7,37 (синглет, 1Н), 7,05 (дублет, J=7,2 Гц, 1Н), 6,16 (широкий синглет, 2Н), 4,25-4,19 (мультиплет, 1Н), 3,98-3,94 (мультиплет, 2Н), 3,41-3,35 (мультиплет, 2Н), 3,22-3,06 (мультиплет, 2Н), 2,79 (дублет, J=7,2 Гц, 2Н), 2,59-2,52 (мультиплет, 4Н), 2,28 (синглет, 3Н), 1,93-1,87 (мультиплет, 2Н), 1,72-1,62 (мультиплет, 2Н), 1,28 (триплет, J=7,2 Гц, 4Н). (ИЭР) m/z 373,1. [М+1]+.
Пример 9: 3-((5-амино-3-этил-8-(1-(1,-метил-[1,4,-бипиперидин]-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 134)
Используя способ, описанный для стадии 2 в примере 6, проводили реакцию исходного материала из следующей таблицы с соединением 206 с получением соединения, указанного в таблице:


Пример 10: используя способ, описанный для стадии 2 в примере 6, проводили реакцию следующих исходных материалов с соединением 58 с получением соединений, указанных в таблице:




Используя способ, описанный для стадии 2 в примере 6, проводили реакцию следующих исходных материалов с соединением 58, после чего использовали способ снятия защитной группы, описанный для стадии 4 в примере 7, с получением соединений, указанных в следующей таблице:

Используя способ, описанный для стадии 2 в примере 6, проводили реакцию следующих исходных материалов с соединением 58, после чего использовали способ снятия защитной группы, описанный для стадии 12 в примере 1, затем использовали способ, описанный для стадии 2 получения соединения 6 для реакции с формальдегидом, с получением соединения, указанного в следующей таблице:

Используя способ, описанный для стадии 2 в примере 6, проводили реакцию параформальдегида с соединением 157 с получением следующего соединения:

Пример 11: 3-этил-8-(1-(1-(пент-3-ил)пиперидин-4-ил)-1Н-пиразол-4-ил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 232)

К раствору соединения 58 (100 мг, 0,20 ммоль) в ацетонитриле (3 мл) добавляли 3-бромпентан (360 мг, 2,4 ммоль) и диизопропилэтиламин (90 мг, 0,70 ммоль). Эту смесь перемешивали и проводили реакцию при 105°С в течение 16 ч в сосуде Шленка. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (10 мл). Смесь трижды экстрагировали (10 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной жидкостной хроматографией с получением формиата соединения 232 в виде твердого вещества желтого цвета (30 мг, выход 23%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,34 (синглет, 1Н), 8,17 (широкий синглет, 4Н), 8,01 (синглет, 1Н), 7,05 (дублет, J=8,0 Гц, 1Н), 6,44 (широкий синглет, 2Н), 4,35-4,26 (мультиплет, 1Н), 4,17-4,09 (мультиплет, 1Н), 4,01-3,94 (мультиплет, 2Н), 3,49-3,43 (мультиплет, 2Н), 2,83-2,82 (мультиплет, 4Н), 2,46 (дублет, J=12,0 Гц, 2Н), 2,30-2,23 (мультиплет, 1Н), 2,06 (дублет, J=8,0 Гц, 2Н), 1,99-1,88 (мультиплет, 4Н), 1,74-1,70 (мультиплет 2Н), 1,52-1,45 (мультиплет, 2Н), 1,37-1,24 (мультиплет, 5Н), 0,91-0,87 (мультиплет, 6Н). (ИЭР) m/z. 493,2 [М+1]+.
Пример 12: 3-этил-8-(5-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пиридин-2-ил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 20)

Стадия 1: к смеси 3-йодпиридина (1,00 г, 4,88 ммоль), 1-метил-4-(4-пиперидинил)пиперазина (893 мг, 4,88 ммоль), Xantphos (282 мг, 0,49 ммоль) и калия трет-бутоксида (1,09 г, 9,76 ммоль) в толуоле (20 мл) добавляли трис(дибензилиденацетон)дипалладий (447 мг, 0,49 ммоль). Эту смесь перемешивали для проведения реакции при 108°С в течение 4 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли метиленхлорид (20 мл) и воду (20 мл). Смесь трижды экстрагировали (20 мл×3) дихлорметаном. Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл), высушивали над безводным натрия сульфатом, фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 10:1 - дихлорметан/метанол/25% водный раствор аммиака = 10:1:0,1) с получением твердого вещества желтого цвета (750 мг, выход 59%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,30 (дублет, J=2,8 Гц, 1Н), 8,07-8,05 (мультиплет, 1Н), 7,20-7,17 (мультиплет, 1Н), 7,15-7,12 (мультиплет, 1Н), 3,77-3,74 (мультиплет, 2Н), 2,81-2,74 (мультиплет, 2Н), 2,65-2,60 (мультиплет, 4Н), 2,51-2,44 (мультиплет, 2Н), 2,41-2,38 (мультиплет, 1Н), 2,31 (синглет, 3Н), 2,19-2,06 (мультиплет, 2Н), 1,98-1,95 (мультиплет, 2Н), 1,72-1,62 (мультиплет, 2Н).
Стадия 2: к смеси 1-метил-4-(1-(пиридин-3-ил)пиперидин-4-ил)пиперазина (750 мг, 2,88 ммоль) в дихлорметане (30 мл) на бане со льдом добавляли порциями NBS (770 мг, 4,33 ммоль). Реакционную смесь перемешивали в течение 2 ч при 5-20°С, после чего вливали в воду (20 мл). Раствор экстрагировали дихлорметаном (20 мл×4). Органические фазы объединяли, промывали насыщенным солевым раствором (5 мл), высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 20:1-10:1) с получением твердого вещества желтого цвета (500 мг, выход 51%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,00 (дублет, J=3,2 Гц, 1Н), 7,28-7,16 (мультиплет, 1Н), 7,07 (дублет дублетов, J=8,8, 3,2 Гц, 1Н), 3,71-3,68 (мультиплет, 2Н), 2,80-2,74 (мультиплет, 2Н), 2,63-2,36 (мультиплет, 9Н), 2,30 (синглет, 3Н), 1,97-1,94 (мультиплет, 2Н), 1,69-1,59 (мультиплет, 2Н). Полученный методом МС результат: 339,1 [М+Н]+.
Стадии 3 и 4: используя способ, описанный для стадии 3 и стадии 4 в примере 3, проводили реакцию 1-(1-(6-бромпиридин-3-ил)пиперидин-4-ил)-4-метилпиперазина (145 мг, 0,43 ммоль) с промежуточным продуктом С (200 мг, 0,43 ммоль) с получением соединения 20 в виде твердого вещества желтого цвета (60 мг, выход после двух стадий 26%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,46 (синглет, 1Н), 8,35-8,31 (мультиплет, 2Н), 7,39-7,36 (мультиплет, 1Н), 7,24-7,22 (мультиплет, 1Н), 6,82 (синглет, 2Н), 4,22-4,14 (мультиплет, 1Н), 3,96-3,94 (мультиплет, 2Н), 3,88-3,85 (мультиплет, 2Н), 3,46-3,40 (мультиплет, 3Н), 3,07-2,73 (мультиплет, 12Н), 2,58 (синглет, 3Н), 1,94-1,91 (мультиплет, 4Н), 1,72-1,58 (мультиплет, 4Н), 1,32 (триплет, J=7,2 Гц, 3Н). Полученный методом МС результат: 532,2 [М+Н]+.
Пример 13: (1S,3R)-3-((5-амино-8-бром-3-этилпиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 151)

Стадии 1 и 2, синтез промежуточного продукта D: используя способы, описанные для стадии 9 в примере 1 и для стадии 1 в примере 2, применяли (1S,3R)-3-аминоциклопентанола гидрохлорид для получения промежуточного продукта D в виде твердого вещества желтого цвета (выход после двух стадий 40%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,01 (синглет, 1Н), 7,31-7,17 (мультиплет, 2Н), 7,07 (дублет, J=8,0 Гц, 1Н), 6,56 (синглет, 1Н), 6,44-6,38 (мультиплет, 1Н), 5,76 (синглет, 1Н), 4,74 (дублет, J=3,6 Гц, 1Н), 4,56 (дублет, J=6,0 Гц, 2Н), 4,52-4,41 (мультиплет, 1Н), 3,83 (синглет, 3Н), 3,72 (синглет, 3Н), 2,83-2,77 (мультиплет, 2Н), 2,13-1,99 (мультиплет, 2Н), 1,83-1,72 (мультиплет, 2Н), 1,68-1,56 (мультиплет, 2Н), 1,30 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z 502,0 [М+Н]+.
Стадия 3: используя способ, описанный для стадии 12 в примере 1, применяли промежуточный продукт D (100 мг, 0,20 ммоль) с получением соединения 151 в виде твердого вещества желтого цвета (56 мг, выход 79%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,14 (широкий синглет, 1Н), 8,00 (синглет, 1Н), 7,43 (дублет, J=6,8 Гц, 1Н), 6,98 (широкий синглет, 2Н), 4,76 (синглет, 1Н), 4,55-4,45 (мультиплет, 1Н), 4,22-4,13 (мультиплет, 1Н), 2,81 (квартет, J=7,2 Гц, 2Н), 2,36-2,27 (мультиплет, 1Н), 2,12-2,01 (мультиплет, 1Н), 1,87-1,73 (мультиплет, 2Н), 1,70-1,58 (мультиплет, 2Н), 1,30 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z 352,1. [М+Н]+.
Пример 14: используя способ, описанный для стадии 2 в примере 2, применяли исходные материалы, указанные в следующей таблице, для реакции с соединением 151 с получением соответствующих соединений:



Пример 15: (1S,3R)-3-((5-амино-3-этил-8-(1-(пиперидин-4-ил)-1Н-пиррол-3-ил)пиридо[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 152) и (1S,3R)-3-((5-амино-3-этил-8-(1-(1-метилпиперидин-4-ил)-1Н-пиррол-3-ил)пиридо[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 153)

Используя способ, описанный в примере 3, из промежуточного продукта D получали промежуточный продукт Е, после чего проводили реакцию с трет-бутиловым эфиром 4-(3-бром-1H-пиррол-1-ил)пиперидин-1-карбоксилата. Проводили снятие защиты с полученного продукта, используя способ, описанный для стадии 12 в примере 1, с получением соединения 152 в виде твердого вещества желтого цвета. Проводили реакцию соединения 152 с формальдегидом, используя способ, описанный для стадии 2 в примере 6, с получением соединения в виде твердого вещества желтого цвета 153.
Соединение 152: 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,68 (синглет, 1Н), 6,96 (дублет, J=7,2 Гц, 1Н), 6,88 (синглет, 1Н), 6,49 (синглет, 2Н), 6,07 (триплет, J=4,0 Гц, 1Н), 5,87 (синглет, 1Н), 4,77 (дублет, J=3,6 Гц, 1Н), 4,31 (дублет, J=6,4 Гц, 1Н), 4,09 (синглет, 1Н), 3,80 (синглет, 1Н), 2,92 (дублет, J=12,0 Гц, 2Н), 2,77-2,72 (мультиплет, 2Н), 2,24 (синглет, 1Н), 2,00-1,90 (мультиплет, 1Н), 1,81-1,47 (мультиплет, 11Н), 1,29 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 422,2 [М+1]+.
Соединение 153: 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,69 (синглет, 1Н), 7,00 (дублет, J=7,2 Гц, 1Н), 6,90 (синглет, 1Н), 6,55 (синглет, 2Н), 6,10 (триплет, J=4,0 Гц, 1Н), 5,90-5,89 (мультиплет, 1Н), 4,79 (дублет, J=3,6 Гц, 1Н), 4,37-4,24 (мультиплет, 1Н), 4,09 (дублет, J=4,0 Гц, 1Н), 3,80 (синглет, 1Н), 2,95 (синглет, 2Н), 2,77-2,72 (мультиплет, 2Н), 2,30 (синглет, 3Н), 2,02-1,57 (мультиплет, 11Н), 1,55-1,49 (мультиплет, 1Н), 1,30 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 436,2 [М+1]+.
Пример 16: используя способ, описанный для стадии 2 в примере 6, применяли исходный материал, указанный в следующей таблице, для реакции с соединением 154 с получением соответствующих соединений:


Пример 17: (1S,3R)-3-((5-амино-3-этил-8-(1-(1,2,2,6,6-пентаметилпиперидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 169)

Стадия 1: к раствору в дихлорметане (30 мл), содержащему 4-гидрокси-1,2,2,6,6-пентаметилпиперидин (3 г, 18 ммоль) при 0°С добавляли триэтиламин (8,9 г, 88 ммоль) и метансульфонилхлорид (2,4 г, 21 ммоль). Реакционную смесь перемешивали при 25°С в течение 6 ч и по окончании реакцию останавливали насыщенным раствором натрия бикарбоната. Раствор дважды экстрагировали дихлорметаном (30 мл×2). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении с получением маслянистого вещества желтого цвета (74,2 г, выход 96%), которое непосредственно использовали в следующей реакции. (ИЭР) m/z 250,2. [М+Н]+.
Стадия 2: к раствору N,N-диметилформамида (20 мл), содержащему 4-пиразола борат (2,3 г, 12 ммоль) добавляли натрия гидрид (0,48 г, 12 ммоль). После перемешивания при 25°С в течение 1 ч добавляли продукт, полученный на стадии 1 (1,2,2,6,6-пентаметилпиперидин-4-ил)мезилат (2,0 г, 8,0 моль). Раствор перемешивали при 110°С в течение 12 ч. Раствор охлаждали до комнатной температуры и экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной жидкостной хроматографией с получением маслянистого вещества желтого цвета (23 мг, 1,3%). (ИЭР) m/z. 348,2 [М+Н]+.
Стадии 3 и 4: используя способ, описанный для стадии 2 в примере 2, а затем способ снятия защитной группы, описанный для стадии 12 в примере 1, проводили реакцию промежуточного продукта D (15 мг, 0,03 ммоль) с продуктом, полученным на стадии 2 (26 мг, 0,075 ммоль), с получением соединения 169 в виде твердого вещества желтого цвета (4,5 мг, выход после двух стадий 28%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,44 (синглет, 1Н), 8,20 (дублет, J=8,0 Гц, 4Н), 8,02 (синглет, 1Н), 7,10 (дублет, J=8,0 Гц, 1Н), 6,38 (синглет, 2Н), 4,54-4,49 (мультиплет, 2Н), 4,25-4,20 (мультиплет, 1Н), 2,81-2,78 (мультиплет, 2Н), 2,33-2,29 (мультиплет, 1Н), 2,25 (синглет, 3Н), 2,09-2,05 (мультиплет, 1Н), 2,00-1,97 (мультиплет, 2Н), 1,89-1,80 (мультиплет, 5Н), 1,71-1,61 (мультиплет, 2Н), 1,32-1,29 (мультиплет, 3Н), 1,16 (синглет, 6Н), 1,11 (синглет, 6Н). (ИЭР) m/z. 493,3 [М+Н]+.
Используя способ, описанный в примере 17, начиная со стадии 3, проводили реакцию исходного материала, указанного в следующей таблице, с промежуточным продуктом D, после чего проводили реакцию с метилйодидом, используя способ, описанный в примере 17, с получением соединения, указанного в таблице:

Используя способ, описанный в примере 17 со стадии 3 до конца, исходные материалы, указанные в следующей таблице, применяли для получения соответствующих соединений:


Используя способ, описанный в примере 17 со стадии 2 до конца, исходные материалы, указанные в следующей таблице, применяли для получения соответствующих соединений: _

Используя способ, описанный в примере 17, 4-гидрокси-1,2,2,6,6-пентаметилпиперидин заменяли на исходные материалы, указанные в следующей таблице, с получением соответствующих соединений:


Пример 18:

Стадия 1: используя способ, описанный для стадии 2 в примере 2, промежуточный продукт D (0,30 г, 0,60 ммоль) применяли в качестве исходного материала для реакции с трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилатом (0,45 г, 1,2 ммоль) с получением твердого вещества желтого цвета (180 мг, выход 45%). (ИЭР) m/z 673,3. [М+Н]+.
Стадия 2: используя способ, описанный для стадии 12 в примере 1, продукт, полученный на стадии 1 (0,18 г, 0,27 ммоль), применяли в качестве исходного материала для получения промежуточного продукта F в виде твердого вещества желтого цвета (80 мг, выход 71%). (ИЭР) m/z 423,3. [М+Н]+.
Используя способ, описанный для стадии 2 в примере 6, исходные материалы, указанные в следующей таблице, использовали для реакции с промежуточным продуктом F, затем проводили снятие защитной группы, используя способ, описанный для стадии 12 в примере 1, после чего проводили реакцию с параформальдегидом, используя способ, описанный для стадии 2 в _примере 6, с получением соединения, указанного в таблице:

Следующие исходные материалы использовали для реакции с промежуточным продуктом F, используя способ, описанный для стадии 2 в примере 6, с получением соединений, указанных в следующей таблице:




Следующие исходные материалы использовали для реакции с промежуточным продуктом F, используя способ, описанный в примере 11, с получением соединений, указанных в таблице:



Следующий исходный материал использовали для реакции с промежуточным продуктом F, используя способ, описанный для стадии 3 в примере 35, с получением соединения, указанного в следующей таблице:

Пример 19: (1S,3R)-3-((5-амино-3-этил-8-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 181)

Раствор промежуточного продукта F (90 мг, 0,21 ммоль) и 2,2-диметилэтиленоксида (46 мг, 0,64 ммоль) в этаноле (10 мл) перемешивали при 80°С в течение 12 ч. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан/метанол = 70%:30%) с получением соединения 181 в виде твердого вещества желтого цвета (65 мг, выход 59%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,44 (синглет, 1Н), 8,20 (синглет, 1Н), 8,02 (синглет, 1Н), 7,11 (дублет, J=7,2 Гц, 1Н), 6,37 (широкий синглет, 2Н), 4,79 (дублет, J=3,6 Гц, 1Н), 4,54-4,44 (мультиплет, 1Н), 4,28-4,20 (мультиплет, 1Н), 4,17-4,05 (мультиплет, 2Н), 3,11-2,99 (мультиплет, 2Н), 2,80 (квартет, J=7,2 Гц, 2Н), 2,38-2,21 (мультиплет, 5Н), 2,12-1,77 (мультиплет, 7Н), 1,75-1,60 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н), 1,11 (синглет, 6Н). (ИЭР) m/z. 495,2 [М+1]+.
Следующие исходные материалы использовали для получения следующего соединения, указанного в таблице, используя способ, описанный в примере 19:


Пример 20: 4-(4-(5-амино-3-этил-2-(((1R,3S)-3-гидроксициклопентил)амино)пиридино[3,4-b]пиразин-8-ил)-1Н-пиразол-1-ил)-N-изопропилпиперазин-1-формамид (соединение 189)

К раствору промежуточного продукта F (80 мг, 0,19 ммоль) и триэтиламина (57 мг, 0,57 ммоль) в тетрагидрофуране (10 мл) добавляли изопропилизоцианат (19 мг, 0,23 ммоль). Смесь перемешивали при 25°С в течение 2 ч и концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан/метанол = 70%:30%) с получением соединения 189 в виде твердого вещества желтого цвета (38 мг, выход 37%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,40 (синглет, 1Н), 8,17 (синглет, 1Н), 8,05 (синглет, 1Н), 7,13 (дублет, J=6,9 Гц, 1Н), 6,47 (широкий синглет, 2Н), 6,24 (дублет, J=7,6 Гц, 1Н), 4,79 (дублет, J=3,6 Гц, 1Н), 4,51-4,42 (мультиплет, 1Н), 4,38-4,28 (мультиплет, 1Н), 4,25-4,18 (мультиплет, 1Н), 4,14-41,06 (мультиплет, 2Н), 3,77 (дублет дублетов, J=13,4, 6,8 Гц, 1Н), 2,89-2,73 (мультиплет, 4Н), 2,27 (дублет дублетов, J=13,4, 6,8 Гц, 1Н), 2,10-1,98 (мультиплет, 3Н), 1,89-1,61 (мультиплет, 7Н), 1,30 (триплет, J=7,2 Гц, 3Н), 1,07 (дублет, J=6,8 Гц, 6Н). (ИЭР) m/z 508,3 [М+1]+.
Пример 21: 1-(4-(4-(5-амино-3-этил-2-(((1R,3S)-3-гидроксициклопентил)амино)пиридино[3,4-b]пиразин-8-ил)-1Н-пиразол-1-ил)пиперадин-1-ил)-2-метилпропил-1-он (соединение 185)

Раствор изомасляной кислоты (25 мг, 0,28 ммоль), EDCI (109 мг, 0,57 ммоль), НОВТ (77 мг, 0,57 ммоль) и диизопропилэтиламина (147 мг, 1,1 ммоль) в N,N-диметилформамиде (5 мл) перемешивали при 0°С в течение получаса, после чего добавляли промежуточный продукт F (80 мг, 0,19 ммоль). Смесь перемешивали при 25°С в течение 2 ч и после реакции добавляли воду (20 мл). Раствор трижды экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан/метанол = 95%:5%) с получением соединения в виде твердого вещества желтого цвета (50 мг, выход 51%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,40 (синглет, 1Н), 8,19 (синглет, 1Н), 8,06 (синглет, 1Н), 7,08 (дублет, J=7,2 Гц, 1Н), 6,37 (широкий синглет, 2Н), 4,79 (дублет, J=3,6 Гц, 1Н), 4,56-4,40 (мультиплет, 3Н), 4,25-4,17 (мультиплет, 1Н), 4,12-4,02 (мультиплет, 1Н), 3,27-3,18 (мультиплет, 1Н), 2,96-2,89 (мультиплет, 1Н), 2,80 (квартет, J=7,2 Гц, 2Н), 2,53-2,52 (мультиплет, 1Н), 2,31-2,22 (мультиплет, 1Н), 2,17-2,00 (мультиплет, 3Н), 1,88-1,63 (мультиплет, 6Н), 1,30 (триплет, J=7,2 Гц, 3Н), 1,06-0,99 (мультиплет, 6Н). (ИЭР) m/z 493,3. [М+1]+.
Пример 22: (1S,3R)-3-((5-амино-3-этил-8-(1-(6-метилпиридин-3-ил)-1Н-пиразол-4-ил)пиридино[3,4-б]пиразин-2-ил)амино)циклопентан-1-ол (соединение 177)

Стадия 1: к раствору 2-метил-5-бромпиридина (2,3 г, 14 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-бромпиразол (1,0 г, 6,8 ммоль), цезия карбонат (6,6 г, 21 ммоль), меди йодид (0,13 г, 0,60 ммоль) и Транс-N,N-диметилциклогексан-1,2-диамин (0,19 г, 1,3 ммоль). Смесь перемешивали при 110°С в течение 12 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (50 мл). Раствор трижды экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (петролейный эфир/этилацетат = 70%:30%) с получением маслянистого вещества желтого цвета (900 мг, выход 29%). (ИЭР) m/z. 238,1 [М+Н]+.
Стадия 2 и 3: использовали способ, описанный для стадии 6 в примере 1, и способ, описанный для стадии 2 в примере 2, соответственно. Получали боратный промежуточный продукт, используя 5-(4-бром-1Н-пиразол-1-ил)-2-метилпиридин в качестве исходного материала, после чего проводили реакцию с соединением 151 с получением соединения 177 в виде твердого вещества желтого цвета (выход после двух стадий 11%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 9,07 (синглет, 1Н), 8,94 (дублет, J=2,8 Гц, 1Н), 8,46 (синглет, 1Н), 8,30 (синглет, 1Н), 8,12-8,09 (мультиплет, 1Н), 7,42 (дублет, J=8,4 Гц, 1Н), 7,23 (дублет, J=6,8 Гц, 1Н), 6,66 (синглет, 2Н), 4,82 (дублет, J=3,6 Гц, 1Н), 4,59-4,51 (мультиплет, 1Н), 4,24-4,18 (мультиплет, 1Н), 2,85-2,79 (мультиплет, 2Н), 2,52 (синглет, 3Н), 2,32-2,27 (мультиплет, 1Н), 2,11-2,03 (мультиплет, 1Н), 1,91-1,76 (мультиплет, 2Н), 1,71-1,64 (мультиплет, 2Н), 1,32 (триплет, J=7,2 Гц, 3Н). ЖХ-МС: Rt=1,051 мин, (ИЭР) m/z. 431,2 [М+1]+.
Использовали способ синтеза, описанный в примере 22. Боратный промежуточный продукт получали из исходных материалов, указанных в следующей таблице, используя способы, описанные для стадий 1 и 2, после чего проводили реакцию с соединением 2 с получением соответствующих соединений, указанных в таблице, согласно способу, описанному для стадии 3:

Боратный промежуточный продукт получали согласно способам, описанным для стадий 1 и 2 в примере 22, а соответствующие соединения, указанные в следующей таблице, получали согласно способам, описанным для стадий 3 и 4 в примере 7:


Боратный промежуточный продукт получали согласно способам, описанным для стадий 1 и 2 в примере 22, а соответствующие соединения, указанные в следующей таблице, получали согласно способам, описанным для стадий 3 и 4 в примере 17



Проводили реакцию соединения 278 с исходным материалом, указанным в следующей таблице, с получением соответствующего соединения согласно способу, описанному для стадии 7 в примере 33:

2-Метил-5-бромпиридин заменяли на исходные материалы, указанные в следующей таблице, с получением соответствующих соединений согласно способу, описанному в примере 22:




Исходные материалы, указанные в следующей таблице, использовали для получения соответствующих соединений согласно способу, описанному в примере 22, начиная со стадии 2.


Используя способ синтеза, описанный для соединения 151 на стадии 1, (1S,3R)-3-аминоциклопентанола гидрохлорид заменяли на (1R,3R)-3-аминоциклопентанол с получением следующего промежуточного продукта 356-А. Затем, используя способ, описанный в примере 22, 2-метил-5-бромпиридин заменяли на исходный материал, указанный в следующей таблице, и соединение 151 заменяли на промежуточный продукт 356-А на стадии 3 с получением соответствующего соединения:

Пример 23: (1S,3R)-3-((5-амино-3-этил-8-(1-(2-метилпиримидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-б]пиразин-2-ил)амино)циклопентан-1-ол (соединение 180)

Стадия 1. натрия гидрид (112 мг, 4,67 ммоль) добавляли к раствору 4-пиразолбората (498 мг, 2,6 ммоль) в тетрагидрофуране (20 мл), затем добавляли 4-хлор-2-метилпиримидин (300 мг, 2,3 ммоль) и раствор перемешивали при 25°С в течение 3 ч. После реакции добавляли воду (30 мл) и раствор экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан/метанол = 9:1) с получением твердого вещества белого цвета (200 мг, выход 28%). (ИЭР) m/z. 287,2. [М+Н]+.
Стадия 2: проводили реакцию продукта, полученного на стадии 1 (81 мг, 0,11 ммоль), с соединением 151 (50 мг, 0,14 ммоль) согласно способу, описанному для стадии 2 в примере 2 с получением соединения 180 в виде твердого вещества желтого цвета (7 мг, выход 12%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,55 (синглет 1Н), 8,74 (дублет, J=5,6 Гц, 1Н), 8,58 (синглет, 1Н), 8,43 (синглет, 1Н), 8,17 (широкий синглет, 1Н), 7,75 (дублет, J=5,6 Гц, 1Н), 7,15 (дублет, J=7,2 Гц, 1Н), 6,58 (широкий синглет, 2Н), 4,62-4,57 (мультиплет, 1Н), 4,31-4,27 (мультиплет, 1Н), 2,84-2,80 (мультиплет, 2Н), 2,64 (синглет, 3Н), 2,40-2,35 (мультиплет, 1Н), 2,29-2,22 (мультиплет, 1Н), 1,99-1,81 (мультиплет, 2Н), 1,79-1,66 (мультиплет, 2Н), 1,33 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 432,2 [М+1]+.
4-Хлор-2-метилпиримидин заменяли на исходные материалы, указанные в следующей таблице, с получением соответствующих соединений согласно способу, описанному в примере 23:


Пример 24: (1S,3R)-3-((5-амино-3-этил-8-(1-(1-(изопропилсульфонил)пиперидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 187)

Стадия 1: трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат (1,0 г, 2,65 ммоль) растворяли в 1,4-диоксане (5 мл), после чего добавляли раствор хлористоводородной кислоты в 1,4-диоксане (4 М, 10 мл). Раствор перемешивали при 25°С в течение 1 ч. После реакции раствор концентрировали при пониженном давлении с получением соединения в виде твердого вещества белого цвета (650 мг, 88%). (ИЭР) m/z 278,2. [М+Н]+.
Стадия 2: к раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)-1Н-пиразол-1-ил)пиперидина (800 мг, 2,9 ммоль) и триэтиламина (876 мг, 8,7 ммоль) в дихлорметане (10 мл) добавляли 2-пропансульфонилхлорид (494 мг, 3,5 ммоль). Эту смесь перемешивали и проводили реакцию при 25°С в течение 16 ч. После реакции добавляли воду (30 мл) и раствор трижды экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении с получением маслянистого вещества желтого цвета (400 мг, выход 95%). (ИЭР) m/z. 384,3. [М+Н]+.
Стадии 3 и 4: продукт, полученный на стадии 2 (73 мг, 0,19 ммоль), использовали в качестве исходного материала для реакции с промежуточным продуктом D (80 мг, 0,16 ммоль) согласно способу, описанному в примере 17, начиная со стадии 3, с получением формиата соединения 187 в виде твердого вещества желтого цвета (30 мг, выход после двух стадий 43%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,43 (синглет, 1Н), 8,19 (синглет, 1Н), 8,14 (синглет, 1Н), 8,07 (синглет, 1Н), 7,12 (дублет, J=7,2 Гц, 1Н), 6,47 (синглет, 2Н), 4,79 (синглет, 1Н), 4,52-4,46 (мультиплет, 1Н), 4,44-4,34 (мультиплет, 1Н), 4,28-4,16 (мультиплет, 1Н), 3,76 (дублет, J=12,0 Гц, 2Н), 3,40 (дублет, J=6,4 Гц, 1Н), 3,13 (триплет, J=12,0 Гц, 2Н), 2,83-2,77 (мультиплет, 2Н), 2,32-2,25 (мультиплет, 1Н), 2,15 (дублет, J=12,0 Гц, 2Н), 2,09-1,99 (мультиплет, 1Н), 1,93-1,76 (мультиплет, 4Н), 1,73-1,61 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н), 1,25 (дублет, J=6,4 Гц, 6Н). (ИЭР) m/z. 529,3 [М+1]+.
Исходный материал, указанный в следующей таблице, использовали для получения соответствующего боратного промежуточного продукта, после чего проводили реакцию боратного промежуточного продукта с соединением 151 с получением соединения, указанного в следующей таблице, согласно способу, описанному в примере 24:

2-Пропансульфонил хлорид заменяли на исходный материал, указанный в следующей таблице, с получением соответствующих соединений согласно способу, описанному в примере 24:


Пример 25: (S)-3-этил-8-(1-(пиперидин-4-ил)-1Н-пиразол-4-ил)-N2-(тетрагидрофуран-3-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 259)

Стадии 1 и 2: получали промежуточный продукт G в виде твердого вещества желтого цвета согласно способу синтеза, описанного для промежуточного продукта D, с использованием (S)-тетрагидрофуран-3-амина (выход после двух стадий 28%). (ИЭР) m/z. 488,0 [М+Н]+.
Стадия 3: промежуточный продукт G (200 мг, 0,41 ммоль) использовали в качестве исходного материала для реакции с трет-бутил-4-(4-(4,4,5,5-тетраметил-1,3,2-диоксоборациклопентан-2)-ил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилатом (186 мг, 0,49 ммоль) согласно способу, описанному для стадии 2 в примере 2 с получением промежуточного продукта Н в виде маслянистого вещества желтого цвета (280 мг, выход 94%). (ИЭР) m/z 659,3. [М+Н]+.
Стадия 4: промежуточный продукт Н (280 мг, 0,42 ммоль) использовали в качестве исходного материала с получением соединения 259 в виде твердого вещества желтого цвета (88 мг, выход 48%) согласно способу, описанному для стадии 12 в примере 1. 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,34 (синглет, 1Н), 8,18 (синглет, 1Н), 8,03 (синглет, 1Н), 7,25 (дублет, J=5,6 Гц, 1Н), 6,39 (синглет, 1Н), 4,71-4,67 (мультиплет, 1Н), 4,21-4,01 (мультиплет, 1Н), 4,04-4,00 (мультиплет, 1Н), 3,94-3,90 (мультиплет, 1Н), 3,82-3,76 (мультиплет, 1Н), 3,74-3,71 (мультиплет, 1Н), 3,06 (дублет, J=12,0 Гц, 2Н), 2,87-2,82 (мультиплет, 2Н), 2,68-2,59 (мультиплет, 2Н), 2,54 (синглет, 1Н), 2,33-2,25 (мультиплет, 1Н), 2,14-2,06 (мультиплет, 1Н), 2,02 (дублет, J=12,0 Гц, 2Н), 1,84-1,75 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 409,2 [М+1]+.
(S)-Тетрагидрофуран-3-амин заменяли на исходный материал 1 в следующей таблице с получением соответствующего промежуточного продукта согласно способу, описанному в примере 25, после чего проводили реакцию промежуточного продукта с исходным материалом 2 с получением соответствующего соединения:

(S)-Тетрагидрофуран-3-амин заменяли на исходный материал, указанный в следующей таблице, с получением промежуточного продукта 261-А согласно способу, описанному в примере 25. После этого проводили реакцию промежуточного продукта 261-А с метилйодидом с получением соединения, указанного в следующей таблице 261 согласно способу, описанному в примере 11.


(S)-Тетрагидрофуран-3-амин заменяли на исходный материал, указанный в следующей таблице, с получением соединения, указанного в таблице, согласно способу, описанному в примере 25.

Исходный материал, указанный в следующей таблице, использовали для реакции с соединением 258 с получением соединения, указанного в следующей таблице, согласно способу, описанному для стадии 2 в примере б.

Исходный материал, указанный в следующей таблице, использовали для получения соединения, указанного в таблице, согласно способу синтеза соединения 250.


Исходные материалы в следующей таблице использовали для реакции с соединением 259 с получением соответствующих соединений, указанных в таблице, согласно способу, описанному для стадии 2 в примере 6.



(S)-Тетрагидрофуран-3-амин заменяли на исходный материал, указанный в следующей таблице, с получением соединения, указанного в таблице, согласно способу синтеза соединения 225.

Пример 26: (1S,3R)-3-((5-амино-3-этил-8-(1-(1-(2-гидроксиэтил)пиперидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 207)

Стадия 1: к раствору промежуточного продукта F (50 мг, 0,10 ммоль) в N,N-диметилформамиде (5 мл) добавляли трет-бутил(2-иодэтокси)диметилсилан (71 мг, 0,25 ммоль) и цезия карбонат (161 мг, 0,50 ммоль) и перемешивали при 45°С в течение 12 ч. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (1 мл). Смесь концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан : метанол = 3:1) с получением соединения в виде твердого вещества желтого цвета (40 мг, выход 41%). (ИЭР) m/z 581,3. [М+Н]+.
Стадия 2: продукт, полученный на стадии 1 (40 мг, 0,070 ммоль), использовали в качестве исходного материала для удаления защитной группы согласно способу, описанному для стадии 4 в примере 7 с получением формиатной соли соединения 207 в виде твердого вещества желтого цвета (5 мг, выход 31%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,42 (синглет, 1Н), 8,18 (синглет, 1Н), 8,15 (широкий синглет, 2Н), 8,05 (синглет, 1Н), 7,12 (дублет, J=7,2 Гц, 1Н), 6,42 (широкий синглет, 2Н), 4,47-4,45 (мультиплет, 1Н), 4,19-4,15 (мультиплет, 2Н), 3,55-3,52 (мультиплет, 2Н), 3,06-3,03 (мультиплет, 2Н), 2,80-2,75 (мультиплет, 2Н), 2,55-2,50 (мультиплет, 4Н), 2,40-2,26 (мультиплет, 2Н), 2,14-1,92 (мультиплет, 6Н), 1,83-1,81 (мультиплет, 2Н), 1,72-1,59 (мультиплет, 2Н), 1,30 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z 467,2 [М+1]+.
Промежуточный продукт F заменяли на исходные материалы, указанные в следующей таблице, с получением соответствующих соединений согласно способу, описанному в примере 26.


Пример 27: (S)-3-этил-8-(1-(пиперидин-4-ил)-1Н-пиразол-4-ил)-1Н-(тетрагидрофуран-3-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 230)

Промежуточный продукт I в виде твердого вещества желтого цвета (350 мг, выход 86%) получали из промежуточного продукта G (588 мг, 1,2 ммоль), используя способ, описанный для стадии 12 в примере 1. (ИЭР) m/z 338,1. [М+Н]+.
Проводили реакцию исходных материалов, указанных в следующей таблице, с промежуточным продуктом I согласно способу, описанному для стадии 2 в примере 2, с получением соответствующих соединений, указанных в таблице,.


Проводили реакцию исходного материала, указанного в следующей таблице, с промежуточным продуктом G согласно способу, описанному в примере 17 со стадии 3 до конца, с получением соответствующего соединения, указанного в таблице

Пример 28: 3-((5-амино-3-этил-8-(1-(1,-этил-[1,4,-бипиперидин]-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклогексан-1-ол (соединение 246)

(S)-Тетрагидрофуран-3-амин заменяли на 3-аминоциклогексанол с получением промежуточного продукта J согласно способу, описанному в примере 25, после чего использовали промежуточный продукт J согласно способу, описанному для стадии 2 в примере 6, с получением формиата соединения 246. 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,47 (синглет, 1Н), 8,22-8,17 (мультиплет, 4Н), 8,02 (синглет, 1Н), 6,87 (дублет, J=7,6 Гц, 1Н), 6,36 (широкий синглет, 2Н), 4,63-4,56 (мультиплет, 1Н), 4,16-4,13 (мультиплет, 1Н), 4,08 (синглет, 1Н), 3,12-2,93 (мультиплет, 6Н), 2,80 (квартет, J=7,2 Гц, 2Н), 2,35-2,27 (мультиплет, 4Н), 2,11-1,41 (мультиплет, 18Н), 1,29 (триплет, J=7,2 Гц, 3Н), 1,08-1,01 (мультиплет, 3Н). (ИЭР) m/z 548,3. [М+1]+.
Пример 29: 3-этил-8-(4-(4-(4-этилпиперазин-1-ил)пиперидин-1-ил)-3-метоксифенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 315)

Стадия 1: к смеси 1-этил-4-(пиперидин-4-илметил)пиперазина (8,0 г, 40 ммоль) и 1-бром-4-хлор-2-метоксибензола (9,6 г, 43 ммоль) в 1,4-диоксане (200 мл) добавляли цезия ацетат (28 г, 87 ммоль), Pd2(dba)3 (4,0 г, 4,3 ммоль), BINAP (5,4 г, 8,7 ммоль). Эту смесь перемешивали и проводили реакцию при 100°С в течение 12 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (100 мл). Раствор трижды экстрагировали (200 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 4:1) с получением маслянистого вещества желтого цвета (7,0 г, выход 52%). (ИЭР) m/z 338,2 [М+Н]+.
Стадия 2: к смеси 1-(1-(4-хлор-2-метоксифенил)пиперидин-4-ил)-4-этилпиперазина (7,0 г, 21 ммоль) и дивалерилдибора (7,9 г, 31 ммоль) в 1,4-диоксане (100 мл) добавляли калия ацетат (5,1 г, 62 ммоль), Pd2(dba)3 (1,2 г, 2,1 ммоль) и X-Phos (2,0 г, 4,1 ммоль). Эту смесь перемешивали для проведения реакции при 100°С в течение 12 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (100 мл). Смесь трижды экстрагировали (300 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 4:1) с получением промежуточного продукта К в виде маслянистого вещества желтого цвета (7,0 г, выход 79%). (ИЭР) m/z 430,3. [М+Н]+.
Стадия 3: к смеси 1,4-диоксан/вода (8 мл, 4/1), содержащей 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (1,4 г, 3,0 ммоль) и промежуточный продукт К (4,0 г, 7,97 ммоль) добавляли цезия карбонат (2,7 г, 8,0 ммоль) и Pd(dppf)Cl2 (0,20 г, 0,20 ммоль). Эту смесь подвергали реакции в СВЧ-печи при 100°С в течение 1 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (50 мл). Смесь трижды экстрагировали (30 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 10:1) с получением твердого вещества желтого цвета (900 мг, выход 42%). (ИЭР) m/z 725,4. [М+Н]+.
Стадия 4: N5-(2,4-диметоксибензил)-3-этил-8-(4-(4-(4-этилпиперазин-1-ил)пиперидин-1-ил)-3-метоксифенил)-1H-(тетрагидро-2Н-пиран-4-ил)пиридин[3,4-b]пиразин-2,5-диамин (900 мг, 1,2 ммоль) в растворе трифторуксусной кислоты/дихлорметана (5 мл, 3/1) перемешивали при 25°С в течение 3 ч. В конце реакции добавляли диизопропилэтиламин (10 мл) для остановки реакции, и смесь концентрировали при пониженном давлении. Остаток очищали препаративной жидкостной хроматографией с получением формиата соединения 315 в виде твердого вещества желтого цвета (255 мг, выход 35%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,14 (широкий синглет, 1Н), 7,91 (синглет, 1Н), 7,25-7,17 (мультиплет, 2Н), 7,09 (дублет, J=8,0 Гц, 1Н), 6,91 (дублет, J=8,0 Гц, 1Н), 6,62 (широкий синглет, 2Н), 4,14-4,10 (мультиплет, 1Н), 3,91 (дублет, J=8,0 Гц, 2Н), 3,82 (синглет, 3Н), 3,48 (дублет, J=12,0 Гц, 3Н), 3,26-3,24 (мультиплет, 2Н), 2,82-2,78 (мультиплет, 2Н), 2,61-2,51 (мультиплет, 6Н), 2,49-2,44 (мультиплет, 6Н), 1,88-1,84 (мультиплет, 4Н), 1,64-1,60 (мультиплет, 4Н), 1,31 (триплет, J=7,2 Гц, 3Н), 1,14 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 575,3 [М+1]+.
1-Этил-4-(пиперидин-4-илметил)пиперазин заменяли на исходный материал, указанный в следующей таблице, с получением соответствующего промежуточного продукта, указанного в таблице, согласно способу, описанному в примере 29. Затем проводили реакцию промежуточного продукта с соединением 2 согласно способу, описанному для стадии 3 в примере 29, с получением соответствующего соединения:

1-Этил-4-(пиперидин-4-илметил)пиперазин заменяли на исходный материал 1, указанный в следующей таблице, согласно способу, описанному в примере 29, а 1-бром-4-хлор-2-метоксибензол заменяли на исходный материал 2, указанный в таблице, для проведения реакции с получением соответствующего соединения.

1-Этил-4-(пиперидин-4-илметил)пиперазин заменяли на исходные материалы в следующей таблице согласно способу, описанному в примере 29 для стадии 1 и 2, с получением соответствующих промежуточных продуктов, после чего проводили реакцию промежуточного продукта с промежуточным продуктом D согласно способу, описанному в примере 29 для стадии 3 и 4, с получением соответствующего соединения.

1-Этил-4-(пиперидин-4-илметил)пиперазин заменяли на исходный материал, указанный в следующей таблице, согласно способу, описанному в примере 29 для стадии 1 и 2, с получением соответствующего промежуточного продукта, указанного в таблице, после чего проводили реакцию промежуточного продукта с соединением 151 согласно способу, описанному в примере 29 для стадии 3, с получением соответствующего соединения.

Использовали исходный материал, указанный в следующей таблице, для получения соответствующего соединения, указанного в таблице, согласно способу, описанному в примере 29 для стадии 3 и 4.

Для реакции с промежуточным продуктом K использовали исходные материалы, указанные в следующей таблице, с получением соответствующих соединений, указанных в таблице, согласно способу, описанному в примере 29.


Пример 30: (1S,3R)-3-((5-амино-3-этилпиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 263)

Соединение 151 (100 мг, 0,28 ммоль) использовали в качестве исходного материала с получением соединения 263 в виде твердого вещества белого цвета (20 мг, 25%) согласно способу, описанному для стадии 4 в примере 1. 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,14 (широкий синглет, 1H), 7,75 (дублет, J=6,0 Гц, 1Н), 7,01 (дублет, J=7,2 Гц, 1Н), 6,59 (дублет, J=6,0 Гц, 1Н), 6,51 (широкий синглет, 2Н), 4,81 (синглет, 1Н), 4,48-4,44 (мультиплет, 1Н), 4,19-4,17 (мультиплет, 1Н). 2,78-2,73 (мультиплет, 2Н), 2,18-2,14 (мультиплет, 1Н), 2,01-1,96 (мультиплет, 1Н), 1,80-1,74 (мультиплет, 2Н), 1,66-1,59 (мультиплет, 2Н), 1,28 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 274,1 [М+1]+.
Пример 31: (1S,3R)-3-((5-амино-3-этил-8-(1-(5-метилпиримидин-4-ил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 269)

Стадия 1: к раствору 4-хлор-5-метилпиримидина (300 мг, 2,3 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиразол (906 мг, 4,6 ммоль) и цезия карбонат (2,3 г, 7,0 ммоль). Смесь перемешивали при 80°С в течение 12 ч в среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (50 мл). Раствор трижды экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении с получением продукта (200 мг, выход 30%). Продукт непосредственно использовали на следующей стадии. (ИЭР) m/z. 287,2 [М+Н]+.
Стадии 2 и 3: проводили реакцию продукта, полученного на стадии 1, с промежуточным продуктом D согласно способу, описанному в примере 29 для стадии 3 и 4, с получением формиата соединения 269 в виде твердого вещества желтого цвета (выход после двух стадий 46%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,61 (синглет, 1Н), 8,90 (синглет, 1Н), 8,77 (синглет, 1Н), 8,56 (синглет, 1Н), 8,38 (синглет, 1Н), 8,14 (широкий синглет, 1Н), 7,37 (дублет, J=7,2 Гц, 1Н), 6,91 (синглет, 2Н), 4,80 (синглет, 1Н), 4,57-4,53 (мультиплет, 1Н), 4,30-4,25 (мультиплет, 1Н), 2,86-2,81 (мультиплет, 2Н), 2,66 (синглет, 3Н), 2,46-2,40 (мультиплет, 2Н), 2,23-2,13 (мультиплет, 1Н), 1,93-1,83 (мультиплет, 2Н), 1,74-1,64 (мультиплет, 2Н), 1,32 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 432,2 [М+1]+.
4-Хлор-5-метилпиримидин заменяли на исходный материал, указанный в следующей таблице, согласно способу, описанному в примере 31, при этом два изомера продукта, полученного на стадии 1, отделяли с получением соответствующих соединений, указанных в таблице.

Использовали исходные материалы, указанные в следующей таблице, для получения соответствующего соединения, указанного в таблице, согласно способу, описанному в примере 6 для стадии 2.

4-Хлор-5-метилпиримидин заменяли на исходные материалы, указанные в следующей таблице, с получением соответствующих соединений, указанных в таблице, согласно способу, описанному в примере 31.

4-Хлор-5-метилпиримидин заменяли на исходные материалы в следующей таблице с получением боратного промежуточного продукта согласно способу, описанному для стадии 1 в примере 31. Затем проводили реакцию промежуточного продукта с соединением 151 согласно способу, описанному для стадии 2 в примере 2 с получением соответствующих соединений, указанных в следующей таблице.



2-Метил-5-бромпиридин заменяли на исходные материалы, указанные в следующей таблице, с получением промежуточных продуктов в виде боратов согласно способу, описанному в примере 2. Затем проводили реакцию боратных промежуточных продуктов с промежуточным продуктом D согласно способу, описанному в примере 31 для стадии 2 и 3, с получением соответствующих соединений, указанных в следующей таблице.



4-Хлор-2-метилпиримидин заменяли на исходный материал, указанный в следующей таблице, с получением промежуточного продукта 335-А согласно способу, описанному для стадии 1 в примере 23. Затем проводили реакцию промежуточного продукта 335-А с промежуточным продуктом D согласно способу, описанному в примере 31 для стадии 2 и 3, с получением соответствующего соединения, указанного в следующей таблице.
4-Хлор-2-метилпиримидин заменяли на исходный материал, указанный в следующей таблице, с получением промежуточного продукта 346-А согласно способу, описанному в примере 23 для стадии 1. Затем промежуточный продукт 346-А использовали для получения соответствующего соединения, указанного в следующей таблице, согласно способу, описанному в примере 7 для стадии 3 и 4.

Пример 32: 8-бром-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 2)
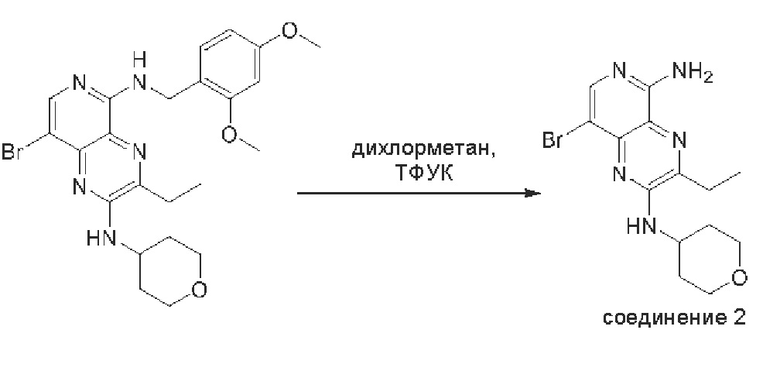
8-Бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (1,0 г, 2,0 ммоль) использовали в качестве исходного материала для получения соединения 2 в виде твердого вещества желтого цвета (650 мг, выход 93%) согласно способу, описанному в примере 1 для стадии 12. (ИЭР) m/z 352,0. [М+Н]+.
Проводили реакцию исходных материалов, указанных в следующей таблице, с соединением 2 согласно способу, описанному в примере 23, с получением соответствующих соединений, указанных в следующей таблице.


Пример 33: 3-этил-8-(3-метокси-4-(4-(8-метил-3,8-диазабицикло[3.2.1]октан-3-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 293) и 8-(4-(4-(3,8-диазабицикло[3.2.1]октан-3-ил)пиперидин-1-ил)-3-метоксифенил)-3-этил-1H-(тетрагидро-2Н-пиран-пиридин-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 319)

Стадии 1 и 2: трет-бутил-3,8-диазабицикло[3,2,1]октан-8-карбоксилат (3,5 г, 0,016 моль) и бензил-4-оксопиперидин-1-карбоксилат (5,7 г, 0,025 моль) использовали в качестве исходных материалов для получения маслянистого вещества желтого цвета (3 г, выход после двух стадий 61%) согласно способам, описанным в примере 1 для стадии 1 и стадии 2. (ИЭР) m/z 296,3. [М+Н]+.
Стадии 3 и 4: трет-бутил-3-(пиперидин-4-ил)-3,8-диазабицикло[3.2.1]октан-8-карбоксилат использовали в качестве исходного материала для получения промежуточного продукта 293-А в виде маслянистого вещества коричневого цвета (выход после двух стадий 66%) согласно способам, описанным в примере 7 для стадии 1 и стадии 2. (ИЭР) m/z 528,3. [М+Н]+.
Стадии 5 и 6: проводили реакцию промежуточного продукта 293-А с 8-бром-N5-(2,4-диметоксибензил)-3-этил-N2-(тетрагидро-2Н-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамином согласно способу синтеза соединения 8, предложенного в примере 2, с получением соединения 319 в виде твердого вещества желтого цвета (выход после двух стадий 35%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,31 (синглет, 2Н), 7,92 (синглет, 1Н), 7,22-7,18 (мультиплет, 2Н), 7,01 (дублет, J=7,2 Гц, 1Н), 6,89 (дублет, J=8,4 Гц, 1Н), 6,45 (синглет, 2Н), 4,17-4,07 (мультиплет, 1Н), 3,92-3,88 (мультиплет, 2Н), 3,81 (синглет, 3Н), 3,77 (синглет, 2Н), 3,41 (дублет, J=11,6 Гц, 3Н), 3,27 (триплет, J=11,2 Гц, 3Н), 2,83-2,74 (мультиплет, 4Н), 2,60-2,54 (мультиплет, 2Н), 2,32-2,29 (мультиплет, 1Н), 1,88-1,75 (мультиплет, 8Н), 1,66-1,54 (мультиплет, 4Н), 1,31 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 573,3 [М+1]+.
Стадия 7: раствор соединения 319 (100 мг, 0,17 ммоль) и формальдегида (8,0 мг, 0,27 ммоль) в метаноле (10 мл) перемешивали при 25°С в течение 10 мин, после чего добавляли натрия боргидрид (20 мг, 0,52 ммоль). Смесь перемешивали при 25°С в течение 30 мин и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 1:1) с получением соединения 293 в виде твердого вещества желтого цвета (40 мг, выход 39%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 7,92 (синглет, 1Н), 7,27-7,12 (мультиплет, 2Н), 7,00 (дублет, J=7,2 Гц, 1Н), 6,88 (дублет, J=8,0 Гц, 1Н), 6,44 (широкий синглет, 2Н), 4,19-4,01 (мультиплет, 1Н), 3,92-3,88 (мультиплет, 2Н), 3,88 (синглет, 3Н), 3,39 (дублет, J=12,0 Гц, 2Н), 3,30-3,22 (мультиплет, 2Н), 3,10 (синглет, 2Н), 2,83-2,78 (мультиплет, 2Н), 2,66-2,54 (мультиплет, 4Н), 2,38 (дублет, J=8,0 Гц, 2Н), 2,22 (дублет, J=12,0 Гц, 4Н), 1,87-1,77 (мультиплет, 6Н), 1,70-1,50 (мультиплет, 6Н), 1,31 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 587,3 [М+1]+.
Пример 34: (1S,3R)-3-((5-амино-3-этил-8-(3-метокси-4-(1-(1-метилпиперидин-4-ил)-1Н-пиразол-4-ил)фенил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 312)

Стадия 1: к смеси тетрагидрофуран/вода (50 мл), содержащей 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)-1H-пиразол-1-ил)пиперидин-1-карбоновой кислоты трет-бутиловый эфир (2,0 г, 5,0 ммоль) и 1-бром-4-хлор-2-метоксибензол (1,8 г, 7,0 ммоль), добавляли цезия карбонат (5,18 г, 15,0 ммоль) и Pd(dppf)Cl2 (0,39 г, 0,50 ммоль). Полученную смесь перемешивали при 70°С в течение 1 ч в защитной среде азота. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (20 мл). Раствор трижды экстрагировали (10 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (петролейный эфир/этилацетат = 70%/30%) с получением маслянистого вещества желтого цвета (1,8 г, выход 83%). (ИЭР) m/z. 392,1 [М+Н]+.
Стадии 2-5: продукт, полученный на стадии 1, подвергали реакции согласно способу, описанному в примере 33 для стадий 4-7, с получением формиата соединения 312 в виде твердого вещества желтого цвета (выход после четырех стадий 1,3%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,18 (дублет, J=4,0 Гц, 3Н), 8,00 (синглет, 1Н), 7,94 (синглет, 1Н), 7,60 (дублет, J=8,0 Гц, 1Н), 7,43 (дублет, J=1,2 Гц, 1Н), 7,31-7,29 (мультиплет, 1Н), 7,05 (дублет, J=7,2 Гц, 1Н), 6,53 (синглет, 2Н), 4,44-4,39 (мультиплет, 1Н), 4,20-4,09 (мультиплет, 2Н), 3,91 (синглет, 3Н), 2,89 (синглет, 2Н), 2,79 (мультиплет, 2Н), 2,24 (синглет, 3Н), 2,19-1,98 (мультиплет, 7Н), 1,96-1,91 (мультиплет, 1Н), 1,84-1,54 (мультиплет, 5Н), 1,31 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 543,3. [М+Н]+.
Пример 35: 3-этил-8-(3-метокси-4-(3-метил-4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 287)

Стадия 1: 1-метилпиперазин (1,5 г, 15 ммоль) и N-трет-бутоксикарбонил-3-метил-4-пиперидон (3,2 г, 15 ммоль) использовали в качестве исходных материалов для получения промежуточного продукта 287-А в виде твердого вещества белого цвета (2,5 г, выход 53%) согласно способу, описанному в примере 6 для стадии 2. (ИЭР) m/z. 298,3 [М+1]+.
Стадия 2: промежуточный продукт 287-А (2,5 г, 8,0 ммоль) использовали в качестве исходного материала для получения промежуточного продукта 287-В в виде твердого вещества белого цвета (1,7 г, выход 87%) согласно способу, описанному для стадии 4 в примере 7. (ИЭР) m/z. 198,3 [М+1]+.
Стадия 3: к смеси промежуточного продукта 287-В (300 мг, 1,5 ммоль) и 1-бром-4-хлор-2-метоксибензола (336 мг, 1,5 ммоль) в растворе 1,4-диоксана(10 мл) добавляли калия трет-бутанол (438 мг, 4,6 ммоль), tBuXPhos Pd G3 (120 мг, 0,15 ммоль) и tBuXPhos (129 мг, 0,30 ммоль). Полученную смесь перемешивали при 100°С в течение 1 ч в атмосфере азота под воздействием СВЧ-излучения. В конце реакции смесь охлаждали до комнатной температуры и добавляли воду (30 мл). Раствор трижды экстрагировали (30 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол = 70%:30%) с получением промежуточного продукта 287-С в виде маслянистого вещества желтого цвета (280 мг, выход 54%). (ИЭР) m/z 338,2. [М+1]+.
Стадии 4 и 5: проводили реакцию промежуточного продукта 287-С согласно способу, описанному в примере 7 для стадии 2, с получением промежуточного продукта 287-D, затем проводили реакцию промежуточного продукта 287-D с соединением 2 с получением формиата соединения 287 в виде твердого вещества желтого цвета согласно способу, описанному для стадии 2 в примере 2 (выход после двух стадий 37%). 1Н NR (400 МГц, ДМСО-d6) δ 8,14 (широкий синглет, 1Н), 7,91 (синглет, 1Н), 7,22-7,20 (мультиплет, 2Н), 7,05 (дублет, J=7,2 Гц, 1Н), 6,88-6,85 (мультиплет, 1Н), 6,55 (синглет, 2Н), 4,14-4,10 (мультиплет, 1Н), 3,92-3,89 (мультиплет, 2Н), 3,82 (синглет, 3Н), 3,41-3,37 (мультиплет, 5Н), 3,30-3,24 (мультиплет, 5Н), 2,84-2,78 (мультиплет, 3Н), 2,54-2,52 (мультиплет, 2Н), 2,51 (синглет, 3Н), 2,17 (синглет, 3Н), 1,88-1,84 (мультиплет, 3Н), 1,63-1,57 (мультиплет, 3Н), 1,31 (триплет, J=7,2 Гц,3Н), 1,09(дублет, J=6,8 Гц,3Н). (ИЭР) m/z. 575,3 [М+1]+.
Пример 36: (1S,3R)-3-((5-амино-3-этил-8-(3-метокси-4-(8-метил-3,8-диазабицикло[3.2.1]октил-3-ил)фенил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 286)

Стадия 1: трет-бутил-3,8-диазабицикло[3,2,1]октан-8-формиат (1,1 г, 5,2 ммоль) использовали для получения промежуточного продукта 286-А в виде маслянистого вещества желтого цвета (0,97 г, выход 50%) согласно способу, описанному в примере 7 для стадии 1. (ИЭР) m/z. 353,3 [М+1]+.
Стадия 2: промежуточный продукт 286-А (970 мг, 2,7 ммоль) использовали в качестве исходного материала для получения промежуточного продукта 287-В в виде твердого вещества белого цвета (450 мг, выход 65%) согласно способу, описанному в примере 7 для стадии 4. (ИЭР) m/z. 253,1 [М+1]+.
Стадия 3: промежуточный продукт 286-В (450 мг, 1,8 ммоль) использовали в качестве исходного материала с получением промежуточного продукта 286-С в виде твердого вещества белого цвета (370 мг, выход 74%) согласно способу, описанному в примере 33 для стадии 7. (ИЭР) m/z 267,1. [М+Н]+.
Стадии 4 и 5: промежуточный продукт 286-С использовали для получения промежуточного продукта 286-D согласно способу, описанному для стадии 2 в примере 7, после чего проводили реакцию промежуточного продукта 286-D с соединением 151 согласно способу, описанному в примере 2 для стадии 2, с получением соединения 286 в виде твердого вещества желтого цвета (выход после двух стадий 2,8%). 1Н-ЯМР (400 МГц, ДМСО-d6)) δ 7,90 (синглет, 1Н), 7,32 (дублет, J=1,6 Гц, 1Н), 7,19-7,16 (мультиплет, 1Н), 7,00 (дублет, J=7,2 Гц, 1Н), 6,81 (дублет, J=8,4 Гц, 1Н), 6,41 (синглет, 2Н), 4,75 (дублет, J=3,6 Гц, 1Н), 4,42-4,37 (мультиплет, 1Н), 4,13 (дублет, J=4,4 Гц, 1Н), 3,81 (синглет, 3Н), 3,21-3,06 (мультиплет, 4Н), 2,85-2,72 (мультиплет, 4Н), 2,21 (синглет, 3Н), 2,16-2,11 (мультиплет, 1Н), 1,96-1,81 (мультиплет, 5Н), 1,77-1,54 (мультиплет, 4Н), 1,30 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 504,3. [М+Н]+.
Пример 37: 3-этил-8-(3-метокси-4-(4-(3-метил-3,8-диазабицикло[3.2.1]октил-8-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2H-пиран-4-ил)пиридино[3,4-b]пиразин-2,5-диамин (соединение 327)

Стадии 1-3: 1,4-диокса-8-азаноло[4,5]декан использовали для получения промежуточного продукта 327-А в виде твердого вещества желтого цвета (выход после трех стадий 20%) согласно способу, описанному в примере 7 для стадий 1-3. (ИЭР) m/z 671,3 [М+1]+.
Стадия 4: раствор промежуточного продукта 327-А (1,0 г, 1,5 ммоль) в смеси трифторуксусной кислоты/дихлорметана (1 мл/10 мл) перемешивали при 25°С в течение 1,5 ч. После реакции раствор концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (1% раствор аммиака в метаноле/дихлорметан = 30%/70%) с получением промежуточного продукта 327-В в виде твердого вещества желтого цвета (750 мг, выход 92%). (ИЭР) m/z. [М+Н]+ 521,4.
Стадия 5: к раствору промежуточного продукта 327-В (600 мг, 1,2 ммоль) в 6 н. растворе хлористоводородной кислоты (6 мл) добавляли уксусную кислоту (3 мл), после чего раствор перемешивали при 70°С в течение 3 ч. После реакции раствор концентрировали при пониженном давлении и добавляли раствор аммиака в метаноле (20 мл) для подщелачивания, после чего раствор снова концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (1% раствор аммиака в метаноле/дихлорметан = 30%/70%) с получением промежуточного продукта L в виде твердого вещества желтого цвета (350 мг, выход 32%). (ИЭР) m/z. [М+Н]+ 477,2.
Стадия 6: к смеси промежуточного продукта L (150 мг, 0,30 ммоль) и 3-метил-3,8-диазабицикло[3,2,1]октана (79 мг, 0,60 ммоль) в 1,2 дихлорэтане (20 мл) добавляли натрия триацетоксиборгидрид (200 мг, 0,90 ммоль). Раствор перемешивали при 80°С в течение 16 ч. В конце реакции смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали препаративной жидкостной хроматографией с получением формиата соединения 327 в виде твердого вещества желтого цвета (17 мг, выход 9%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,20 (широкий синглет, 1,8Н), 7,92 (синглет, 1Н), 7,26-7,17 (мультиплет, 2Н), 7,01 (дублет, J=7,2 Гц, 1Н), 6,89 (дублет, J=8,0 Гц, 1Н), 6,45 (широкий синглет, 2Н), 4,14-4,10 (мультиплет, 1Н), 3,91 (дублет, J=8,4 Гц, 2Н), 3,82 (синглет, 3Н), 3,48 (синглет, 4Н), 3,25-3,20 (мультиплет, 2Н), 2,80 (триплет, J=7,2 Гц, 2Н), 2,64-2,52 (мультиплет, 4Н), 2,48-2,41 (мультиплет, 4Н), 2,22 (дублет, J=12,0 Гц, 2Н), 1,96 (дублет, J=12,0 Гц, 2Н), 1,86 (дублет, J=12,4 Гц, 2Н), 1,75-1,57 (мультиплет, 6Н), 1,50-1,39 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 587,4 [М+Н]+.
Исходные материалы, указанные в следующей таблице, использовали для реакции с промежуточным продуктом L согласно способу восстановительного аминирования, описанному в примере 37 для стадии 6, с получением соответствующих соединений, указанных в таблице.


Пример 38: 3-этил-8-(3-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N2-(тетрагидро-2Н-1 шран-4-ил)-1,6-нафтиридин-2,5-диамин (соединение 113)

Синтез промежуточного продукта М: 1-метил-4-(4-пиперидинил)пиперазин (5,0 г, 30 ммоль) использовали для получения промежуточного продукта М в виде твердого вещества желтого цвета согласно способу, описанному в примере 7 для стадий 1 и 2 (выход после двух стадий 24%). 1H ЯМР (400 Мгц, CDCl3) δ 7,39 (дублет, J=8,0 Гц, 1Н), 7,25 (синглет, 1Н), 6,92 (дублет, J=8,0 Гц, 1Н), 3,90 (синглет, 3Н), 2,51-2,34 (мультиплет, 13Н), 2,34 (синглет, 3Н), 1,89-1,76 (мультиплет, 4Н), 1,33 (синглет, 12Н). (ИЭР) m/z. [М+1]+ 416,2.
Стадия 1: к раствору 4-амино-2-хлорпиридин-3-формальдегида (200 мг, 1,3 ммоль) в N,N-диметилформамиде (5 мл) добавляли NBS (260 мг, 1,5 ммоль). Реакционную смесь перемешивали при 25°С в течение 5 ч и затем вливали в воду (10 мл). Твердое вещество осаждали, фильтровали и высушивали с получением твердого промежуточного продукта 113-А белого цвета (215 мг, выход 72%). 1Н-ЯМР (400 МГц, CDCl3) δ 10,39 (широкий синглет, 1Н), 9,24 (синглет, 1Н), 8,24 (синглет, 1Н), 5,73 (синглет, 1Н). (ИЭР) m/z 234,9 [М+Н]+
Стадия 2: к раствору этилбутирата (440 мг, 3,8 ммоль) в тетрагидрофуране (10 мл) при -78°С добавляли раствор FDA (1,9 мл, 3,8 ммоль, 2 моль/л). Реакционную смесь перемешивали при -78°С в течение 1 ч, после чего добавляли промежуточный продукта 113-А (0,21 г, 0,90 ммоль). Реакционную смесь перемешивали при 25°С в течение 14 ч и добавляли к насыщенному раствору аммония хлорида (10 мл). Раствор трижды экстрагировали (10 мл×3) этилацетатом. Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (петролейный эфир/этилацетат = 10:1) с получением промежуточного продукта 113-В в виде твердого вещества белого цвета (88 мг, выход 40%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 11,51 (широкий синглет, 1Н), 8,50 (синглет, 1Н), 7,85 (синглет, 1Н), 2,59 (дублет дублетов, J=14,8, 7,2 Гц, 2Н), 1,19 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z 286,9 [М+Н]+
Стадия 3: промежуточный продукт 113-В (88 мг, 0,31 ммоль) использовали в качестве исходного материала для получения промежуточного продукта 113-С в виде твердого вещества светло-желтого цвета согласно способу, описанному для стадии 1 в примере 2 (80 мг, выход 62%). (ИЭР) m/z 418,0 [М+Н]+.
Стадия 4: промежуточный продукт 113-С (80 мг, 0,19 ммоль) использовали в качестве исходного материала с получением промежуточного продукта 113-D в виде твердого вещества коричневого цвета согласно способу, описанному для стадии 8 в примере 1 (90 мг, неочищенный продукт непосредственно использовали для следующей реакции).
Стадия 5: смесь промежуточного продукта 113-D (90 мг) и тетрагидропиран-4-амина (412 мг, 4,1 ммоль) перемешивали при 145°С в течение 1 ч. В конце реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (петролейный эфир/этилацетат = 1:1) с получением промежуточного продукта 113-Е в виде твердого вещества коричневого цвета (60 мг, выход 89%). (ИЭР) m/z 353,0 [М+Н]+.
Стадия 6: промежуточный продукт 113-Е (60 мг, 0,17 ммоль) и промежуточный продукт М (142 мг, 0,34 ммоль) использовали в качестве исходных материалов для получения формиата соединения 113 в виде твердого вещества желтого цвета согласно способу, описанному для стадии 2 в примере 2 (23 мг, выход 23%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,20 (широкий синглет, 3Н), 8,00 (синглет, 1Н), 7,81 (синглет, 1Н), 7,23-7,13 (мультиплет, 2Н), 6,88 (дублет, J=8,0 Гц, 1Н), 6,75 (синглет, 2Н), 6,45 (дублет, J=7,2 Гц, 1Н), 4,19-4,11 (мультиплет, 1Н), 3,92-3,84 (мультиплет, 2Н), 3,84-3,77 (мультиплет, 3Н), 3,45 (дублет, J=11,2 Гц, 2Н), 3,23 (триплет, J=11,2 Гц, 2Н), 2,62-2,54 (мультиплет, 6Н), 2,53-2,51 (мультиплет, 1Н), 2,48-2,20 (мультиплет, 2Н), 2,22 (синглет, 3Н), 1,90-1,76 (мультиплет, 4Н), 1,65-1,48 (мультиплет, 4Н), 1,25 (дублет дублетов, J=12,0, 4,8 Гц, 3Н). (ИЭР) m/z. 560,3 [М+1]+.
Исходный материал, указанный в следующей таблице, использовали для реакции с промежуточным продуктом М с получением соответствующего соединения, указанного в таблице, согласно способу синтеза, описанному в примере 38 для стадии 6.


Пример 39: 3-этил-8-(3-метоксифенил)-N2-(тетрагидро-2Н-пиран-4-ил)пиразино[2,3-d]пиридазин-2,5-диамин (соединение 116)

Стадия 1: к раствору этанола (40 мл) и насыщенного аммония хлорида (40 мл), содержащему 3,6-дихлор-5-нитропиридин-4-амин (4,4 г, 21 ммоль), добавляли железный порошок (12 г, 0,21 моль). Реакционную смесь перемешивали при 25°С в течение 18 ч, фильтровали, и осадок на фильтре промывали этанолом (40 мл). Фильтрат выпаривали для удаления этанола и остаток трижды экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, высушивали над безводным натрия сульфатом, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (дихлорметан:метанол = 20:1) с получением промежуточного продукта 116-А в виде твердого вещества желтого цвета (1,8 г, выход 43%). (ИЭР) m/z 178,9 [М+Н]+
Стадии 2-4: согласно способу, описанному в примере 1 для стадий 7-9, используя промежуточный продукт 116-А, получали промежуточный продукт 116-D в виде твердого вещества белого цвета (выход после трех стадий 2,2%). (ИЭР) m/z 312,1 [М+Н]+
Стадия 5: согласно способу, описанному в примере 8 для стадии 1, используя промежуточный продукт 116-D (66 мг, 0,21 ммоль), получали промежуточный продукт 116-Е в виде твердого вещества желтого цвета (60 мг, выход 61%). (ИЭР) m/z 441,1 [М+Н]+
Стадия 6: согласно способу, описанному в примере 17 для стадии 1, используя промежуточный продукт 116-Е (50 мг, 0,12 ммоль) для реакции с ангидридом трифторметансульфоновой кислоты (40 мг, 0,14 ммоль), получали промежуточный продукт 116-Е в виде твердого вещества желтого цвета (50 мг, выход 65%). (ИЭР) m/z 573,0 [М+Н]+
Стадии 7 и 8: проводили реакцию промежуточного продукта 116-Е (70 мг, 0,12 ммоль) с (3-метоксифенил)борной кислотой (37 мг, 0,24 ммоль) согласно способу, описанному в примере 17 для стадии 1, с получением формиата соединения 116 в виде твердого вещества белого цвета (5 мг, выход после двух стадий 11%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,23 (синглет, 1Н), 7,73-7,71 (мультиплет, 2Н), 7,41 (дублет, J=7,2 Гц, 1Н), 7,38-7,34 (мультиплет, 1Н), 7,00-6,97 (мультиплет, 1Н), 6,70 (синглет, 2Н), 4,16-4,12 (мультиплет, 1Н), 3,96-3,92 (мультиплет, 2Н), 3,82 (синглет, 3Н), 3,42 (синглет, 2Н), 2,90-2,84 (мультиплет, 2Н), 1,89 (дублет, J=12,0 Гц, 2Н), 1,67-1,61 (мультиплет, 2Н), 1,35-1,31 (мультиплет, 3Н). (ИЭР) m/z. 381,1 [М+1]+.
Метоксифенилбороновую кислоту заменяли на промежуточный продукт М, полученный на стадии 7, с получением соединения, указанного в следующей таблице, согласно методу синтеза, описанному в примере 39.

Пример 40: (1S,3R)-3-((5-амино-3-этил-8-(3-метокси-4-(2-металпиримидин-4-ил)фенил)пиридо[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 288)

Стадия 1: проводили реакцию 4-хлор-2-метилпиримидина (1,0 г, 7,8 ммоль) с (4-хлор-2-метоксифенил)борной кислотой (1,7 г, 9,3 ммоль) согласно способу, описанному для стадии 2 в примере 2, с получением промежуточного продукта 288-А в виде твердого вещества коричневого цвета (1,6 г, выход 87%).
Стадии 2 и 3: промежуточный продукт 288-А использовали для получения промежуточного продукта согласно способу, описанному для стадии 2 в примере 7, после чего проводили реакцию промежуточного продукта с соединением 151 согласно способу, описанному для стадии 2 в примере 2, с получением формиата соединения 288 в виде твердого вещества желтого цвета (выход после двух стадий составил около 59%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,67 (дублет, J=5,6 Гц, 1Н), 8,14 (широкий синглет, 1Н), 8,07 (синглет, 1Н), 8,01 (дублет, J=8,4 Гц, 1Н), 7,87 (дублет, J=5,6 Гц, 1Н), 7,58-7,54 (мультиплет, 1Н), 7,51-7,47 (мультиплет, 1Н), 7,16 (дублет, J=7,2 Гц, 1Н), 6,81 (широкий синглет, 2Н), 4,87-4,65 (мультиплет, 1Н), 4,46-4,38 (мультиплет, 1Н), 4,17-4,09 (мультиплет, 1Н), 3,94 (синглет, 3Н), 2,81 (квартет, J=7,2 Гц, 2Н), 2,67 (синглет, 3Н), 2,20-2,11 (мультиплет, 1Н), 1,97-1,88 (мультиплет, 1Н), 1,82-1,58 (мультиплет, 4Н), 1,32 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z 472,2. [М+1]+.
Проводили реакцию исходного материала, указанного в следующей таблице, согласно способу, описанному в примере 40 для стадий 1 и 2, с получением промежуточного продукта в виде боратного эфира, после чего проводили реакцию промежуточного продукта в виде боратного эфира с промежуточным продуктом D согласно способу синтеза соединения 8 со стадии 2 до конца с получением соответствующего соединения, указанного в таблице


Пример 41: цис- и транс-4-(4-(5-амино-3-этил-2-(((1R,3S)-3-гидроксициклопентил)амино)пиридино[3,4-b]пиразин-8-ил)-1Н-пиразол-1-ил)-1-метилциююгексил-1-ол (соединения 284 и 285)
Согласно следующему способу цис- и транс-изомеры исходных материалов подвергали реакции с получением соединения 284 и соединения 285, соответственно

Стадия 1: к раствору цис- или транс-4-(4-йод-1Н-пиразол-1-ил)-1-метилциклогексан-1-ола в тетрагидрофуране добавляли при 0°С изопропилмагний хлорид (около 5 эквивалентов). Эту смесь перемешивали при 0°С в атмосфере азота в течение 1 ч, после чего добавляли 2-метокси-4,4,5,5-тетраметил-1,3,2-диоксапентаборан (около 5,5 эквивалентов). Смесь перемешивали при 0°С в течение 1 ч в атмосфере азота, затем нагревали до 25°С и перемешивали в течение 3 ч. После реакции смесь концентрировали при пониженном давлении и остаток очищали на хроматографической колонке с силикагелем (петролейный эфир: этил ацетат = 3:7 - этилацетат) с получением промежуточного продукта N в виде бесцветного маслянистого вещества (выход около 72%). (ИЭР) m/z. 307,2. [М+Н]+.
Стадии 2 и 3: проводили реакцию промежуточного продукта N с промежуточным продуктом D согласно способу, описанному в примере 31 для стадий 2 и 3, с получением соединения 284 (выход после двух стадий 30%) и соединения 285 (выход после двух стадий 6,5%), оба соединения в виде твердого вещества желтого цвета. Соединение 284: 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,45 (синглет, 1Н), 8,17-8,13 (мультиплет, 2Н), 8,01 (синглет, 1Н), 7,28 (дублет, 3=6,8 Гц, 1Н), 6,82 (синглет, 2Н), 4,92-4,65 (мультиплет, 1Н), 4,54-4,46 (мультиплет, 1Н), 4,26-4,07 (мультиплет, 3Н), 2,82 (квартет, 3=7,2 Гц, 2Н), 2,34-2,27 (мультиплет, 1Н), 2,12-2,03 (мультиплет, 3Н), 1,88-1,80 (мультиплет, 4Н), 1,71-1,63 (мультиплет, 4Н), 1,53-1,46 (мультиплет, 2Н), 1,31 (триплет, 3=7,2 Гц, 3Н), 1,16 (синглет, 3Н). (ИЭР) m/z 452,3. [М+1]+. Соединение 285: 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,43 (синглет, 1Н), 8,19 (синглет, 2Н), 8,02 (синглет, 1Н), 7,10 (дублет, 3=7,2 Гц, 1Н), 6,38 (широкий синглет, 2Н), 4,50-4,47 (мультиплет, 1Н), 4,28-4,11 (мультиплет, 2Н), 2,80 (квартет, J=7,2 Гц, 2Н), 2,35-2,25 (мультиплет, 1Н), 2,05-2,00 (мультиплет, 3Н), 2,00-1,52 (мультиплет, 11Н), 1,30 (триплет, J=7,2 Гц, 3Н), 1,19 (синглет, 3Н). (ИЭР) m/z. 452,3 [М+1]+.
Пример 42: (1S,3R)-3-((5-амино-3-этил-8-(1-(4-((1-метилпиперидин-4-ил)окси)фенил)-1Н-пиразол-4-ил)пиридино[3,4-b]пиразин-2-ил)амино)циклопентан-1-ол (соединение 292)

Стадия 1: проводили реакцию 4-(4-йодфенокси)-1-метилпиперидина (800 мг, 2,5 ммоль) с 4-бром-1H-пиразолом (389 мг, 2,6 ммоль) согласно способу, описанному в примере 22 для стадии 1, с получением промежуточного продукта 292-А в виде твердого вещества желтого цвета (410 мг, 48%). (ИЭР) m/z 336,1 [М+Н]+.
Стадия 2: к раствору промежуточного продукта 292-А (350 мг, 1,04 ммоль) в тетрагидрофуране (10 мл) по каплям добавляли раствор н-бутила (0,5 мл, 2,4 н.) при -78°С в атмосфере азота. Раствор перемешивали в течение 20 мин, после чего добавляли 2-метокси-4,4,5,5-тетраметил-1,3,2-диокспентаборан (328 мг, 2,08 ммоль) в тетрагидрофуране (5 мл). Смесь перемешивали при -78°С в течение 1 ч в атмосфере азота, затем температуру доводили до 0°С и перемешивали в течение 1 ч. В конце реакции добавляли метанол (2 мл) для остановки реакции, и раствор концентрировали при пониженном давлении. Остаток очищали на хроматографической колонке с силикагелем (дихлорметан : метанол = 9:1) с получением промежуточного продукта 292-В в виде маслянистого вещества желтого цвета (150 мг, выход 86%). (ИЭР) m/z. 302,2. [М+Н]+.
Стадии 3 и 4: проводили реакцию промежуточного продукта 292-В (114 мг, 0,398 ммоль) с промежуточным продуктом D (100 мг, 0,199 ммоль) согласно способу, описанному в примере 31 для стадии 2 и 3, с получением формиата соединения 292 в виде твердого вещества желтого цвета (38 мг, выход после двух стадий 3,9%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,77-7,70 (мультиплет, 2Н), 7,22-7,15 (мультиплет, 4Н), 6,91 (дублет, J=8,8 Гц, 2Н), 6,52 (дублет, J=1,6 Гц, 1Н), 4,72-4,68 (мультиплет, 1Н), 4,60-4,54 (мультиплет, 1Н), 4,09 (синглет, 1Н), 4,00-3,96 (мультиплет, 1Н), 3,34-3,20 (мультиплет, 4Н), 2,79 (синглет, 3Н), 2,73-2,66 (мультиплет, 2Н), 2,14-1,85 (мультиплет, 5Н), 1,76-1,53 (мультиплет, 5H), 1,48-1,38 (мультиплет, 1Н), 1,25 (триплет, J=7,2 Гц, 3Н). (ИЭР) m/z. 529,3. [М+Н]+.
Пример 43: (S,Е)-N-(1-((2-((4-(5-амино-3-этил-2-((тетрагидро-2Н-пиран-4-ил)амино)пиридино[3,4b]пиразин-8-ил)-2-метоксифенил)амино)этил)амино)-1-оксопропан-2-ил)-4-(диметиламино)-N-метилбутил-2-енамид (соединение 358)

Стадия 1: проводили реакцию 4-бром-2-метоксианилина (8,0 г, 39 ммоль) с трет-бутил(2-оксиэтил)карбаматом (6,6 г, 41 ммоль) согласно способу, описанному для стадии 2 в примере 6, с получением промежуточного продукта 358-А в виде твердого вещества желтого цвета (4,5 г, 30%). (ИЭР) m/z 346,1 [М+Н]+.
Стадии проводили реакцию промежуточного продукта 358-А согласно способу, описанному в примере 7 для стадий 2-4, с получением промежуточного продукта 358-В в виде твердого вещества желтого цвета (выход после трех стадий 23%). (ИЭР) m/z. 438,2 [М+Н]+.
Стадия 5: к раствору (Е)-4-(диметиламино)бутил-2-еновой кислоты (500 мг, 3,9 ммоль) и (2S)-2-(метиламино)пропионата (678 мг, 4,3 ммоль) в дихлорметане (10 мл) добавляли HATU (1,9 г, 5,0 ммоль) и триэтиламин (0,77 г, 7,7 ммоль). Смесь перемешивали при 25°С в течение 4 ч, затем концентрировали при пониженном давлении. Полученный остаток очищали на хроматографической колонке с силикагелем (метанол (содержащий 1% аммиака)/дихлорметан = от 0% до 100% за 20 мин) с получением промежуточного продукта 358-С в виде твердого вещества желтого цвета (980 мг, выход 84%). (ИЭР) m/z. 271,1. [М+Н]+.
Стадия 6: проводили реакцию промежуточного продукта 358-С (500 мг, 1,9 ммоль) согласно способу, описанному для стадии 1 в примере 24, с получением промежуточного продукта 358-D в виде твердого вещества желтого цвета (350 мг, выход 88%). (ИЭР) m/z. 215,1. [М+Н]+.
Стадия 7: проводили реакцию промежуточного продукта 358-D (62 мг, 0,29 ммоль) с промежуточным продуктом 358-В (120 мг, 0,27 ммоль) согласно способу, описанному для стадии 1 в данном примере, с получением соединения 358 в виде твердого вещества желтого цвета (18,5 мг, выход 11%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 8,17 (синглет, 2Н), 7,98 (синглет, 1Н), 7,88 (синглет, 1Н), 7,15 (дублет, J=6,0 Гц, 2Н), 6,98 (дублет, J=7,2 Гц, 1Н), 6,67-6,59 (мультиплет, 2Н), 6,52 (синглет, 1Н), 6,42 (синглет, 2Н), 5,04-5,00 (мультиплет, 2Н), 4,22-4,11 (мультиплет, 1Н), 3,96-3,87 (мультиплет, 2Н), 3,81 (синглет, 3Н), 3,31-3,28 (мультиплет, 3Н), 3,16-3,12 (мультиплет, 2Н), 3,10-3,00 (мультиплет, 2Н), 2,90 (синглет, 2Н), 2,80-2,75 (мультиплет, 3Н), 2,16-2,10 (мультиплет, 6Н), 1,87-1,84 (мультиплет, 2Н), 1,63-1,60 (мультиплет, 2Н), 1,31 (триплет, J=7,2 Гц, 1Н), 1,23 (дублет, J=7,2 Гц, 1Н). (ИЭР) m/z. 634,3. [М+Н]+.
Пример биологического анализа 1: эксперимент для оценки ингибирующей активности в отношении FLT3-киназы in vitro:
Реактивы и расходные материалы

Оборудование
Центрифуга (производитель: Eppendorf, модель: 5430), считыватель для микропланшетов (производитель: Perkin Elmer, модель: Caliper EZ Reader II), Echo 550 (производитель: Labcyte, модель: Echo 550)
Методика эксперимента
1. Процесс киназной реакции
(1) Готовили 1х киназный буфер.
(2) Приготовление последовательных разведений соединения: выполняли последовательные 3-кратные разведения исходного раствора (концентрация испытуемого соединения составляла 1 мкМ) до 10 концентраций и проводили их анализ в отдельных лунках. Соединения последовательно разбавляли до растворов 10 разных концентраций (конечная концентрация представляла собой 100-кратное разведение) в 384-луночных планшетах. В каждую лунку 384-луночного планшета переносили по 250 нл раствора при помощи Echo550. 100% ДМСО (250 нл) добавляли в лунки отрицательного контроля и лунки положительного контроля соответственно.
(3) Киназный раствор (конечная концентрация представляла собой 2,5-кратное разведение) готовили с 1х киназным буфером.
(4) 10 мкл киназного раствора (конечная концентрация представляла собой 2,5-кратное разведение) добавляли в лунки с соединением и лунки с положительным контролем соответственно; 1х киназный буфер (10 мкл) добавляли в лунки отрицательного контроля.
(5) Раствор центрифугировали при 1000 об/мин в течение 30 с, встряхивали, перемешивали и инкубировали при комнатной температуре в течение 10 мин.
(6) Готовили смешанный раствор АТФ и киназного субстрата 2 (при соотношении в конечной концентрации 25/15) с 1х киназным буфером.
(7) 15 мкл смешанного раствора АТФ и киназного субстрата 2 (при соотношении в конечной концентрации 25/15) добавляли для инициирования реакции.
(8) 384-луночный планшет центрифугировали при 1000 об/мин в течение 30 с, встряхивали и перемешивали, после чего инкубировали при комнатной температуре в течение 30 мин.
(9) Для остановки киназной реакции добавляли останавливающий раствор (30 мкл), раствор центрифугировали при 1000 об/мин в течение 30 с, встряхивали и перемешивали.
(10) Показатель преобразования считывали с помощью Caliper EZ Reader II.
2. Анализ данных
Формула для расчета

Где: Преобразование(%)_исп - показатель преобразования испытуемого образца; Преобразование(%)_мин: лунка отрицательного контроля.
Пример биологического анализа 2: анализ ингибирования пролиферации клеток линии MV4-11 in vitro:
Экспериментальные материалы:
Линии клеток MV4-11 были приобретены в банке клеток Китайской академии наук, среда IMDM (Gibco, № по каталогу 12440), фетальная бычья сыворотка (Gibco, № по каталогу 10099141С), пенициллин-стрептомициновые антибиотики (Gibco, № по каталогу 15140122), ДМСО (Sigma, № по каталогу D2650), набор Celltiter-Glo (CTG) (Promega, № по каталогу G7573), 384-луночный прозрачный планшет для культивирования клеток с плоским дном (Corning, № по каталогу 3764), цифровой дозатор D300e (Tecan, № по каталогу D300e), считыватель микропланшетов SpectraMax i3x (MOLECULAR DEVICES, № по каталогу i3x)
Методика эксперимента:
Культура клеток:
Клетки MV4-11 культивировали в питательной среде IMDM с 10% фетальной бычьей сыворотки, 1% пенициллин-стрептомициновых антибиотиков, при этом плотность клеток поддерживали на уровне менее 1,0×106 клеток/мл для обеспечения их постоянного нахождения в логарифмической фазе роста. Жизнеспособность клеток была выше 95%.
Приготовление испытуемых растворов соединений:
Проводили 3-кратные разведения испытуемых соединений от 10 мкМ в ДМСО до 9 концентраций. 60 нл разбавленного соединения добавляли в планшет для культивирования клеток и устанавливали три дубликата.
Клетки, обработанные соединением:
Приготовленную суспензию клеток MV4-11 вносили в 384-луночный планшет путем добавления 20 мкл клеточной суспензии в каждую лунку, т.е. каждая лунка содержала 2000 клеток MV4-11, при этом конечная концентрация ДМСО составляла 0,3%. Планшет с клеточной культурой помещали в инкубатор при 37°С с 5% углерода диоксида и инкубировали в течение 72 ч.
В каждую лунку добавляли по 20 мкл реактива Promega CellTiter-Glo, и планшет инкубировали при комнатной температуре в течение 10 мин для стабилизации люминесцентных сигналов. Для считывания сигналов использовали считыватель микропланшетов SpectraMax i3x.
Анализ данных
Исходные данные преобразовывали в показатель ингибирования, при этом значение IC50 (концентрация полумаксимального ингибирования) соединения определяли путем аппроксимации нелинейной четырехпараметрической кривой.
В следующей таблице приведены результаты, полученные в примере биологического анализа 1 и в примере биологического анализа 2. Результаты показали, что образец соединения по настоящему изобретению оказывает сильное ингибирующее действие на FLT3-киназу и обладает высокой ингибирующей активностью в отношении пролиферации клеток MV4-11.
А: IC50≤10 нМ;
В: 10 нМ<IC50≤100 нМ;
С: 100нМ<IC50≤500 нМ;
D: >500 нМ
Результаты анализа активности некоторых соединений представлены в следующей таблице.






Все литературные источники, упомянутые в настоящем изобретении, включены в настоящий документ в виде ссылок, как если бы все фрагменты из литературных источников были включены в виде отдельных ссылок. Кроме того, следует понимать, что после изучения вышеприведенного описания специалистами в данной области техники могут быть внесены многочисленные изменения и модификации, и такие эквиваленты также попадают в объем, определяемый формулой изобретения, прилагаемой к настоящей заявке.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ НА ОСНОВЕ КЕТОАМИДА | 2021 |
|
RU2819346C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО АМИДА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ОБЛАДАЮЩАЯ АНТИАЦЕТИЛХОЛИНЭСТЕРОДНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2010794C1 |
| ПРОИЗВОДНЫЕ ИНДОЛОНА В ВИДЕ РАЦЕМАТА ИЛИ ОПТИЧЕСКИ АКТИВНОГО ИЗОМЕРА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ АДДИТИВНЫЕ СОЛИ КИСЛОТЫ И КЕТОПРОИЗВОДНЫЕ ИНДОЛОНА | 1991 |
|
RU2068414C1 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАНИЛ-ЦИАНОГУАНИДИНА | 1990 |
|
RU2057129C1 |
Группа изобретений относится к органической химии и включает соединения формул II, IV, а также индивидуальные соединения, указанные в формуле изобретения, фармацевтическую композицию на их основе и применение. В формуле II X и Y представляют собой N, а Z представляет собой CR; R представляет собой H; Ra выбирают из группы, состоящей из H, замещенной или незамещенной C1-C6 алкильной группы; U представляет собой химическую связь или -NH-; и когда U представляет собой химическую связь, то Rc представляет собой пирролидин; Rc выбирают из группы, состоящей из замещенной или незамещенной C1-C6 алкильной группы, замещенного или незамещенного C3-C8 карбоциклического кольца, замещенного или незамещенного 3-8-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода, серы и азота; Re выбирают из группы, состоящей из -NHR; при этом R выбирают из группы, состоящей из H; W представляет собой замещенный или незамещенный фенил, и замещенное или незамещенное 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из атомов серы и азота; помимо общего заместителя группу W далее замещают по меньшей мере одной группой со структурой -M-A, где M выбирают из группы, состоящей из химической связи или -CHR-; остальные заместители являются такими, как указано в формуле изобретения. В формуле IV заместители являются такими, как указано в формуле изобретения. Технический результат - производные пиридино[3,4-b]пиразина, применяемые для лечения заболеваний, связанных с аномальными уровнями генов или аномальной экспрессией киназ, выбранных из группы, состоящей из FLT3, ALK и AXL. 5 н. и 11 з.п. ф-лы, 2 табл., 360 пр.


1. Соединение, представленное следующей формулой II, либо его фармацевтически приемлемые соли:

II
где X и Y представляют собой N, а Z представляет собой CR; при этом R выбирают из группы, состоящей из H;
Ra выбирают из группы, состоящей из H, замещенной или незамещенной C1-C6 алкильной группы;
U представляет собой химическую связь или -NH-; и когда U представляет собой химическую связь, то Rc представляет собой пирролидин;
Rc выбирают из группы, состоящей из замещенной или незамещенной C1-C6 алкильной группы, замещенного или незамещенного C3-C8 карбоциклического кольца, замещенного или незамещенного 3-8-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода, серы и азота;
Re выбирают из группы, состоящей из -NHR; при этом R выбирают из группы, состоящей из H;
W представляет собой замещенный или незамещенный фенил, и замещенное или незамещенное 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из атомов серы и азота;
помимо общего заместителя группу W далее замещают по меньшей мере одной группой со структурой -M-A, где M выбирают из группы, состоящей из химической связи или -CHR-;
А выбирают из группы, состоящей из замещенной или незамещенной C1-C6 алкильной группы, замещенной или незамещенной C1-C6 алкоксигруппы, замещенной или незамещенной C6 арильной группы, замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-2 гетероатома, выбранных из атомов азота, замещенной или незамещенной C3-C12 циклоалкильной группы;
в группе A замещение означает замещение одной или несколькими группами, выбранными из группы B, где группа B состоит из H, галогена, =O, цианогруппы, гидроксильной группы, альдегидной группы, карбоксильной группы, замещенной или незамещенной C1-C6 алкильной группы, замещенной или незамещенной C1-C6 алкоксигруппы, замещенного или незамещенного 3-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-2 гетероатома, выбранных из атомов и азота, замещенной или незамещенной C3-C12 циклоалкильной группы, замещенной или незамещенной C2-C10 сложноэфирной группы, замещенной или незамещенной C1-C6 амидной группы, замещенной или незамещенной C1-C4 алкил-S(O)2-группы, при этом в группе B замещение означает замещение одной или несколькими группами R; при этом R выбирают из группы, состоящей из H, галогена, цианогруппы, гидроксильной группы, замещенной или незамещенной C1-C6 алкильной группы, замещенной или незамещенной C1-C6 алкоксигруппы, замещенной или незамещенной C3 циклоалкильной группы, замещенной или незамещенной C2-C10 сложноэфирной группы;
если не указано иное, в приведенных выше формулах замещение означает, что атом водорода в соответствующей группе замещен одним или несколькими заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидроксильной группы, аминогруппы, C1-C6 амидной группы, цианогруппы, незамещенной или галогенированной C1-C6 алкильной группы, C1-C6 алкоксигруппы, C1-C6 алкиламиногруппы, C1-C12-алкиламинокарбонильной группы, незамещенной или галогенированной C2-C10-ацильной группы, незамещенной или галогенированной C1-C4-алкил-S(O)2-группы.
2. Соединение по п. 1, либо его фармацевтически приемлемые соли, где соединение имеет структуру, представленную в формуле IIa:

IIa
где кольцо W выбирают из группы, состоящей из замещенной или незамещенной фенильной группы, замещенного или незамещенного пиридина, и замещенного или незамещенного пиразолила; при этом в группе W указанная замена означает замещение одной или несколькими группами, выбранными из группы А.
3. Соединение по п. 1, либо его фармацевтически приемлемые соли, где W представляет собой замещенный или незамещенный фенил.
4. Соединение по п. 1, либо его фармацевтически приемлемые соли, где W представляет собой замещенную или незамещенную кольцевую структуру, выбранную из группы, состоящей из 
5. Соединение по п. 1, либо его фармацевтически приемлемые соли, где соединение имеет структуру, представленную в формуле III:

III
где кольцо A выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-2 гетероатома, выбранных из атомов азота, замещенной или незамещенной C6 арильной группы и замещенной или незамещенной C3-C12 циклоалкильной группы;
M выбирают из группы, состоящей из химической связи или -CHR-.
6. Соединение по п. 5, либо его фармацевтически приемлемые соли, где соединение имеет структуру, представленную в следующей формуле:

где
Rf выбирают из группы, состоящей из H, галогена, цианогруппы, аминогруппы, нитрогруппы, гидроксильной группы, C1-C4 алкил-S(O)2-группы, замещенной или незамещенной C1-C6 алкильной группы и замещенной или незамещенной C1-C6 алкоксигруппы;
t является 0, 1, 2, 3 или 4;
кольцо A выбирают из группы, состоящей из замещенного или незамещенного 5-12-членного насыщенного кольца; при этом кольцо A содержит по крайней мере один гетероатом, выбранный из N или O.
7. Соединение по п. 5, либо его фармацевтически приемлемые соли, где соединение имеет структуру, представленную в следующей формуле:

где
Rf выбирают из группы, состоящей из H;
t является 0, 1, 2, 3 или 4;
L представляет собой N;
кольцо A выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенной или незамещенной C3-C12 циклоалкильной группы, замещенного или незамещенного 5-12-членного гетероароматического кольца, содержащего 1-2 гетероатома, выбранных из атомов азота и замещенной или незамещенной C6 арильной группы.
8. Соединение, либо его фармацевтически приемлемые соли по п. 5, где соединение имеет структуру, представленную в следующей формуле IV:

IV
где кольцо A выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенной или незамещенной C6 арильной группы;
M выбирают из группы, состоящей из химической связи или -CHR-;
кольцо B выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота;
V является химической связью.
9. Соединение по п. 5, либо его фармацевтически приемлемые соли, где кольцо A содержит по крайней мере один заместитель G, при этому группу G выбирают из группы, состоящей из =O, замещенного или незамещенного 4-7-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота, замещенной или незамещенной C2-C10 сложноэфирной группы и замещенной или незамещенной C1-C6 амидной группы.
10. Соединение по п. 1, либо его фармацевтически приемлемые соли, где соединение выбирают из следующих соединений:


























































































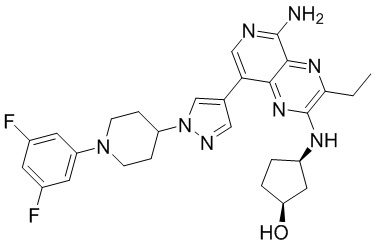





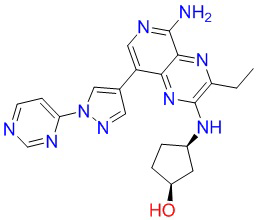















































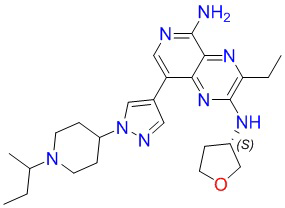











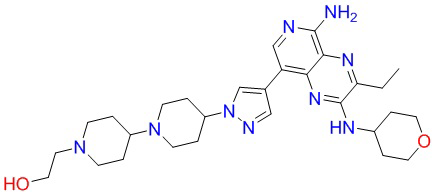






































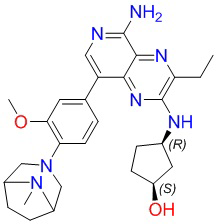




















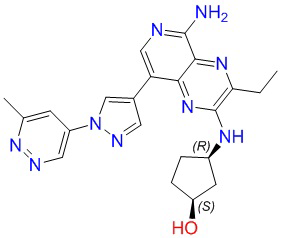





















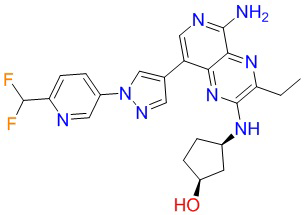




























11. Соединение формулы IV, либо его фармацевтически приемлемые соли,
IV
где X и Y представляют собой N, а Z представляет собой CR; при этом R выбирают из группы, состоящей из H;
Ra выбирают из группы, состоящей из H, замещенной или незамещенной C1-C6 алкильной группы;
U представляет собой -NH-;
Rc выбирают из группы, состоящей из замещенной или незамещенной C1-C6 алкильной группы, замещенного или незамещенного C3-C8 карбоциклического кольца, незамещенного 3-8-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода и азота;
Re выбирают из группы, состоящей из -NHR; при этом R выбирают из группы, состоящей из H;
W представляет собой замещенный фенил, и замещенное или незамещенное 5-6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома, выбранных из группы, состоящей из атомов серы и азота;
где кольцо A выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода, серы и азота, замещенной или незамещенной C6 арильной группы;
M выбирают из группы, состоящей из химической связи или -CHR-;
кольцо B выбирают из группы, состоящей из замещенного или незамещенного 4-12-членного гетероциклического кольца, содержащего 1-2 гетероатома, выбранных из атомов кислорода, серы и азота;
V выбирают из группы, состоящей из -CH2-;
если не указано иное, в приведенных выше формулах замещение означает, что атом водорода в соответствующей группе замещен одним или несколькими заместителями, выбранными из группы, состоящей из дейтерия, галогена, гидроксильной группы, аминогруппы, C1-C6 амидной группы, цианогруппы, незамещенной или галогенированной C1-C6 алкильной группы, C1-C6 алкоксигруппы, C1-C6 алкиламиногруппы, C1-C12-алкиламинокарбонильной группы, галогенированной C2-C10-ацильной группы, незамещенной C1-C4-алкил-S(O)2-группы.
12. Соединение, выбранное из группы, состоящей из:











13. Фармацевтическая композиция для лечения заболеваний, связанных с аномальными уровнями генов или аномальной экспрессией киназ, выбранных из группы, состоящей из FLT3, ALK и AXL, содержащая терапевтически эффективное количество одного или нескольких соединений по любому из пп. 1-12, либо их фармацевтически приемлемых солей, а также один или несколько фармацевтически приемлемых носителей, вспомогательных веществ, добавок, наполнителей и разбавителей.
14. Применение соединения по любому из пп. 1-12, или фармацевтически приемлемых солей в производстве лекарственных препаратов для лечения заболеваний, связанных с уровнями аномальных генов или аномальной экспрессией киназ, выбранных из группы, состоящей из FLT3, ALK and AXL.
15. Применение по п. 14, где заболевание выбирают из группы, состоящей из рака легких, острого лейкоза, хронического лейкоза.
16. Применение по п. 14, где заболевание выбрано из группы, состоящей из острого миелолейкоза, немелкоклеточного рака легкого, мелкоклеточного рака легкого.
| WO 2019062803 A1, 04.04.2019 | |||
| CN110944994 A, 31.03.2020 | |||
| CN 106459042 A, 22.02.2017 | |||
| CN104822687 A, 05.08.2015 | |||
| CN 1918158 A, 21.02.2007 | |||
| EA 201791781 A1, 28.02.2018. |

