Изобретение относится к масложировой промышленности, в частности к способам количественного и качественного определения содержания монохлорпропандиолов (МХПД) и глицидола и их эфиров в составе липидсодержащих пищевых матриц и может быть использовано для определения 2-монохлорпропандиола, 3-монохлорпропандиола, глицидола в жидких и твердых растительных маслах.
В настоящее время существует множество методик, позволяющих устанавливать количественное содержание монохлорпропандиолов (МХПД), глицидола в масложировой продукции. Основными методами, позволяющими проводить такие исследования являются высокоэффективная жидкостная хроматография с масс-селективным детектором (ВЭЖХ-МСД) и газовая хроматография с масс-селективным детектором (ГХ-МСД). Наиболее доступным для лабораторий, осуществляющих контроль уровня МХПД в продукции, из указанных методов, является косвенный метод ГХ-МСД. Для этого метода применяются ряд методик, отличающихся между собой способами подготовки пробы (щелочной, кислотный, ферментативный гидролиз и т.д.), суть которых заключается в «высвобождении» определяемых соединений (МХПД, глицидол), находящихся в маслах и другой масложировой продукции в виде сложных эфиров с различными жирными кислотами, их дериватизации, выделении при помощи жидкость-жидкостной экстракции, концентрировании и анализе с применением газового хроматографа сопряженного с масс-селективным детектором. Благодаря гидролизу, приводящему к разрушению сложных эфиров, происходит объединение аналитов, что характеризует косвенные методы определения в отличие от прямых, где определяются непосредственно сложные эфиры МХПД и глицидола с различными жирными кислотами. Все методики для ГХ-МСД объединяет то, что для количественного анализа МХПД и глицидола в качестве внутренних стандартов (ВС) применяются пентадейтерированные соединения 3-МХПД, 2-МХПД, глицидола и 3-МБПД, в которых пять атомов водорода замещены на пять атомов дейтерия. Например, при определении МХПД и глицидола по ГОСТ ISO 18363-3: 2020 (медленный кислотный гидролиз) требуется два дейтерированных ВС – эфиры пальмитиновой кислоты 3-МХПД-d5 и глицидола-d5, при определении МХПД и глицидола по ГОСТ ISO 18363-2-2018 (медленный щелочной гидролиз) требуется пять вида ВС – 2-МХПД – d5, 3-МХПД- d5 в свободном виде, а также эфиры жирных кислот 2-МХПД, 3-МХПД и глицидола. Согласно последнему ГОСТ 34900 – 2022 для количественного определении МХПД и глицидола необходимо применять три дейтерированных стандартов: 2-МХПД – d5, 3-МХПД- d5 и 3-МБПД- d5.
Пентадейтерированные ВС, имеют химическую структуру идентичную определяемым веществам и отличаются только молекулярной массой: у дейтерированных стандартов она выше на 5 единиц (Таблица 1).
Таблица 1 - Характеристика аналитов и внутренних стандартов
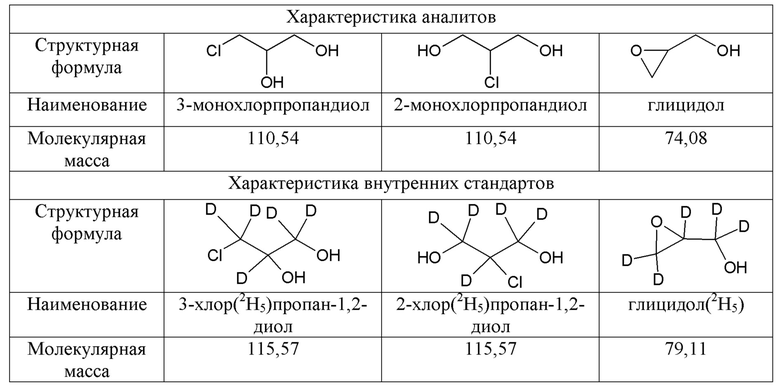
Дейтерированные стандарты обладают рядом достоинств, благодаря которым получили широкое применение в хроматографических методах анализа. Одинаковое структурное строение молекул с определяемыми аналитами, обусловливает их близкие свойства, такие, как время удерживания при хроматографировании, коэффициент распределения при экстракции, фрагментация в источнике ионов масс-спектрометрического детектора, химические свойства и т.д. Все это, безусловно, обеспечивает достаточно высокую точность измерений и воспроизводимость получаемых результатов.
Прототипом изобретения является ГОСТ 34900-2022 "Жиры и масла животные и растительные. Определение содержания 2-монохлорпропандиола и эфиров жирных кислот 2-монохлорпропандиола, 3-монохлорпропандиола и эфиров жирных кислот 3-монохлорпропандиола и глицидиловых эфиров жирных кислот с применением ферментативного гидролиза", предусматривающий подготовку эталонных образцов определяемых соединений, путем смешивания внутренних стандартов с растворителем, дериватезации фенилборной кислотой, извлечения гексаном, концентрирования, с последующим анализом эталонных проб путем газовой хроматографии с масс-детектором, построение на основе полученных данных калибровочной зависимости и уравнения регрессии для каждого определяемого компонента относительно внутреннего стандарта, подготовка исследуемого образца, путем смешивания образца масла с растворителем, ферментативного гидролиза смеси раствором липазы Candida rugosa, с последующим нагреванием смеси при 80°С в течение 10 минут, с последующим ее охлаждением проточной водой до температуры 20-25°С и очисткой от свободных жирных кислот гексаном, дериватизацией 2,5% раствором фенилборной кислоты, извлечения фенилборпроизводных определяемых компонентов гексаном, концентрирования в токе азота, анализ исследуемого образца на газовом хроматографе с масс-селективным детектором, расчет содержания каждого определяемого компонента в исследуемом образце осуществляют по предварительно составленному уравнению, описывающему калибровочную прямую, построенную в координатах отношения значения площадей пиков эталонных определяемых компонентов к значениям площадей пиков внутреннего стандарта и отношения массы внесенных эталонных определяемых компонентов к массе внесенного внутреннего стандарта:
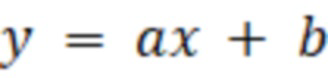 , (1)
, (1)
где 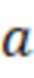 - тангенс угла наклона калибровочной кривой;
- тангенс угла наклона калибровочной кривой;
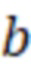 - точка пересечения калибровочной кривой с осью Y,
- точка пересечения калибровочной кривой с осью Y,
при этом количественное содержание определяемых компонентов в образце масла рассчитывали по формуле:
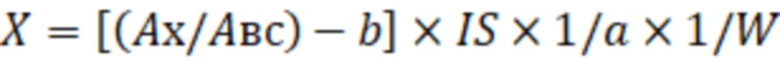 ,
,
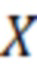 - концентрация определяемого соединения в исследуемом образце, мг/кг;
- концентрация определяемого соединения в исследуемом образце, мг/кг;
Ах – площадь пика фенилборпроизводного количественного иона определяемого соединения;
Авс - площадь пика иона внутреннего стандарта;
- тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y;
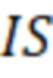 - количество введённого в исследуемый образец внутреннего стандарта, мкг;
- количество введённого в исследуемый образец внутреннего стандарта, мкг;
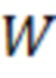 - масса исследуемого образца, г.
- масса исследуемого образца, г.
Главным недостатком таких стандартов является их высокая стоимость, низкая доступность на российском рынке, а также ограниченные сроки годности. К сожалению, до настоящего времени отечественные производители реактивов не освоили технологию производства изотопно-меченных стандартов. Кроме того, дейтерированные стандарты имеют близкие значения времени элюирования с определяемыми аналитами, что приводит к частичному наложению пиков.
Задачей изобретения является усовершенствование способа определения МХПД и глицидола в масложировой продукции.
Техническим результатом изобретения является расширение арсенала способов определения МХПД и глицидола, обеспечивающих возможность одновременного определения в растительных маслах: 3-МХПД, 2-МХПД и глицидола при сохранении высокой точности и реализация данного способа.
Технический результат достигается тем, что способ определения качественного и количественного состава монохлорпропандиолов (МХПД), глицидола в растительном масле, включает подготовку эталонных образцов определяемых соединений, с последующим анализом эталонных проб путем газовой хроматографии с масс-детектором, построение на основе полученных данных калибровочной зависимости и уравнения регрессии для каждого определяемого компонента, подготовка исследуемого образца, анализ исследуемого образца, расчет содержания определяемых соединений в исследуемом образце осуществляют по предварительно составленному уравнению, описывающему калибровочную прямую, построенную в координатах отношения значения площадей пиков эталонных монохлорпропандиолов (МХПД), глицидола определяемых компонентов к значениям площадей пиков внутреннего стандарта и отношения массы внесенных эталонных определяемых компонентов к массе внесенного внутреннего стандарта:
, (1)
где a - тангенс угла наклона калибровочной кривой;
b - точка пересечения калибровочной кривой с осью Y, количественное содержание определяемых компонентов в образце масла рассчитывают по формуле:
X=[(Aх/Aвс)-b]·IS·1/a·1/W, (2)
где X - концентрация определяемого соединения в исследуемом образце, мг/кг;
Ах – площадь пика фенилборпроизводного количественного иона определяемого соединения;
Авс – площадь пика иона внутреннего стандарта;
- тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y;
IS – количество введённого в исследуемый образец внутреннего стандарта, мкг;
W – масса исследуемого образца, г,
при этом подготовку эталонных образцов и исследуемого образца осуществляют путем смешивания эталонных образцов и исследуемого образца соответственно с растворителем и внутренним стандартом, в качестве которого используют 1,3-бутандиол концентрацией 5,0 мкг/мл, последующего ферментного гидролиза образцов раствором липазы Candida rugosa, их нагреванием при температуре 80оС в течение 10 минут, очистки от свободных жирных кислот гексаном, дериватизации 2,5% раствором фенилборной кислоты, извлечения фенилборпроизводных определяемых компонентов гексаном, концентрирования в токе азота при температуре 400, анализа на газовом хроматографе и регистрацией компонентов масс-спектрометрическим детектором в режиме SIM (Selected Ion Monitoring) в том числе для ионов фенилборпроизводного 1,3-бутандиола.
Проведенные исследования подтверждают возможность применения 1,3-бутандиола концентрацией 5,0 мкг/мл в качестве внутреннего стандарта при анализе МХПД и глицидола, и как следствие расширение линейки внутренних стандартов на ряду с изотопномеченными при анализе МХПД и глицидола. Данное вещество по химической структуре, как и определяемые аналиты, относится к классу органических соединений – гликоли (диолы). Это означает что механизмы процессов растворения, дериватизации и экстракции у 1,3-бутандиола будут аналогичны определяемым веществам. Таким образом, применение 1,3-бутандиола в качестве внутреннего стандарта при анализе МХПД и глицидола расширяет спектр соединений пригодных для анализа МХПД и глицидола при сохранении высокой точности и воспроизводимости результатов анализа, что значительно облегчает задачу выбора методического пути решения поставленной аналитической задачи. Также при применении 1,3-бутандиола в качестве внутреннего стандарта отсутствуют зоны перекрывания его пика с пиками определяемых аналитов.
Реализация способа.
Готовят растворы стандартов 3-МХПД, 2-МХПД концентрацией 1,035 мкг/мл (раствор II) и 10,35 мкг/мл (раствор I) в метаноле; растворы глицидола концентрацией 2,37 мкг/мл (раствор I) и 23,70 мкг/мл (раствор II) Для построения калибровочного графика готовят 8 градуировочных растворов путем внесения в пробирку по 10, 20, 50, 100 мкл растворов II и по 20, 35, 50, 80 мкл растворов I. Также в каждую пробирку добавляют по 20 мкл альтернативного внутреннего стандарта 1,3-бутандиола, концентрацией 5,0 мкг/мл (После ряда экспериментов с различными соединениями был предложен 1,3-бутандиол производства компании Shanghai Macklin Biochemical Technology Co., Ltd., Китай).
К полученным смесям приливают по 3,0 мл 30%-го раствора бромида натрия (рН=5,2), перемешивают вихревым движением содержимое пробирок в течение 10 секунд и помещают пробирки в водяную баню на 10 минут при температуре 80оС, для преобразования глицидола в 3-МБПД.
После нагревания содержимое пробирок охлаждают водой до температуры 23-25оС. Далее в каждую пробирку добавляют по 100 мкл 2,5% раствора фенилборной кислоты, в системе дистиллированная вода/ацетон в соотношении 1/19, для дериватизации и перемешивают содержимое пробирок с помощью вихревого смесителя в течение 10 с.
Далее в каждую пробирку вносят по 3 мл н-гексана, закрывают винтовой крышкой и встряхивают пробирку на высокоскоростном смесителе в течение 10 мин. После окончания дериватизации фенилборные производные диолов распределяются в верхнем гексановом слое. Для предупреждения попадания в пробы водной фазы содержимое пробирок центрифугируют при 3000—3500 об/мин в течение 1 мин. Гексановый слой, содержащий фенилборные производные стандартов, переносят при помощи пипетки Пастера в новую пробирку, содержащую небольшое количество высаливающего агента, в качестве которого использовали сульфат натрия. После выпаривания гексана до объема пробы 200-300 мкл в потоке азота при 40°С, перемешивают содержимое вихревым движением в течение 10 с и переносят пробу в виалу для газовой хроматографии для последующего анализа методом ГХ-МС.
Для анализа проб используют хромато-масс-спектрометр компании «Хроматэк» - «Кристалл 5000.2» с масс-селективным детектором, оборудованный кварцевой капиллярной колонкой длиной 30 м, диаметром 0,25 мм, с неподвижной фазой толщиной 0,25 мкм (ZB-5plus).
Установленные параметры:
Газовый хроматограф:
- газ-носитель: гелий, постоянный поток 1,2 мл/мин;
- объем пробы: 1,5 мкл;
- режим ввода пробы: без деления потока, время продувки: 1,5 мин;
- температура инжектора: 250°C;
- программирование температуры термостата ГХ (общее время 21 мин):
- начальная температура - 60°С;
- изотермический режим - 1 мин;
- программируемый нагрев - до температуры 170°С со скоростью 10°С/мин;
- изотермический режим - 3 мин;
- программируемый нагрев - до температуры 300°С со скоростью 100°С/мин;
- изотермический режим - 5 мин;
Масс-спектрометр:
- метод ионизации: электронный удар, положительный режим;
- температура интерфейса: 300°C;
- температура ионного источника: 230°C;
- время сканирования: 0,05-0,10.
Регистрацию компонентов осуществляют масс-спектрометрическим детектором в режиме SIM в том числе для ионов фенилборпроизводного 1,3-бутандиола.
Контроль ионов осуществляют согласно данным, приведенным в таблице 2.
Таблица 2 – Регистрируемые ионы
Временной диапазон регистрации определяемых веществ составлял от 8 до 14 мин.
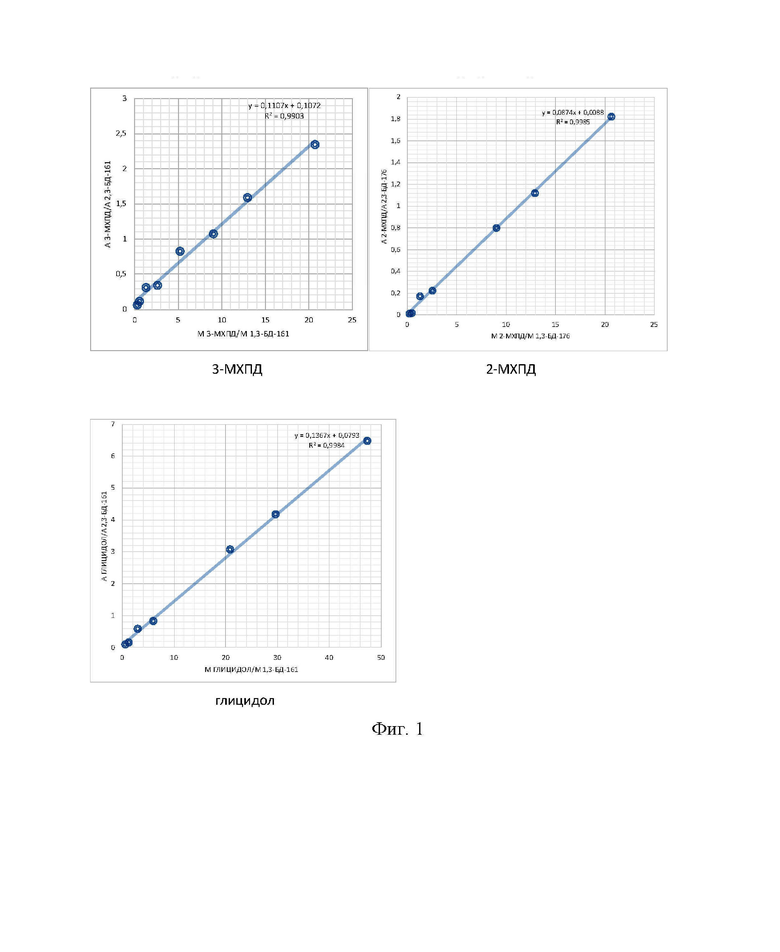
По полученным данным строят калибровочные графики (фиг.1) путем нанесения по оси Y отношения значения площадей пиков фенилборных производных 3-МХПД (m/z 147), 2-МХПД (m/z 196) и 3-МБПД (m/z 147) к значениям площадей пика фенилборпроизводного внутреннего стандарта (Таблица 3), а по оси Х - отношения массы внесенных 3-МХПД, 2-МХПД, 3-МБПД к массе внесенного ВС.
Расчет площади пика осуществляли в соответствии с методикой расчёта площади пика в газовой хроматографии, описанной в пособии ЦАРЕВ Н.И., ЦАРЕВ В.И., КАТРАКОВ И.Б. «Практическая газовая хроматография: Учебно-методическое пособие для студентов химического факультета по спецкурсу «Газохроматографические методы анализа», Барнаул, Изд-во Алт. ун-та, 2000, стр.101-105).
Линейную регрессию рассчитывали по формуле:
, (1)
где - тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y.
Количественное содержание 3-МХПД в образце масла рассчитывали по формуле:
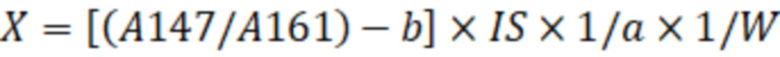 ,
,
где - концентрация 3-МХПД исследуемого образца, мг/кг;
А147 – площадь пика фенилборпроизводного 3-МХПД по m/z 147;
А161 – площадь пика фенилборпроизводного 1,3-бутандиола по m/z 161;
- тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y;
– количество введённого в исследуемый образец внутреннего стандарта, мкг;
– масса исследуемого образца, г.
Количественное содержание 2-МХПД в образце масла рассчитывали по формуле:
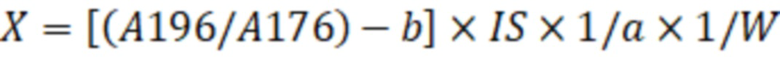 ,
,
где - концентрация 3-МХПД исследуемого образца, мг/кг;
А196 – площадь пика фенилборпроизводного 3-МХПД по m/z 196;
А176 – площадь пика фенилборпроизводного 1,3-бутандиола по m/z 176;
- тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y;
IS - количество введённого в исследуемый образец внутреннего стандарта, мкг;
- масса исследуемого образца, г.
Количественное содержание глицидола в образце масла рассчитывали по формуле:
,
где - концентрация 3-МХПД исследуемого образца, мг/кг;
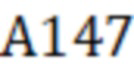 – площадь пика фенилборпроизводного 3-МБПД по m/z 147;
– площадь пика фенилборпроизводного 3-МБПД по m/z 147;
А161 – площадь пика фенилборпроизводного 1,3-бутандиола по m/z 161;
- тангенс угла наклона калибровочной кривой;
- точка пересечения калибровочной кривой с осью Y;
– количество введённого в исследуемый образец внутреннего стандарта, мкг;
– масса исследуемого образца, г.
Таким образом, совокупность заявляемых признаков позволяет обеспечить возможность одновременного определения с высокой точностью, присутствующих в рафинированных дезодорированных растительных маслах определяемых соединений: 3-МХПД, 2-МХПД, глицидола с применением доступного альтернативного внутреннего стандарта 1,3-бутандиола.
Навеску рафинированного дезодорированного пальмового масла в количестве 100 мг помещали в стеклянную пробирку, добавляли 20 мкл раствора внутреннего стандарта - 1,3-бутандиол концентрацией 5,0 мкг/мл и растворяют в 0,5 см3 изооктана. К смеси приливали 3,0 см3 30%-го раствора бромида натрия, содержащего липазу Candida rugosa, закрывали завинчивающейся крышкой и встряхивали пробирку на вихревом смесителе при комнатной температуре в течение 30 минут на 1800 об/мин. Далее пробирку центрифугировали при 3000 об/мин в течение 10 секунд и помещали в водяную баню с температурой 80оС на 10 минут для преобразования глицидола в 3-бромпроизводное. После нагревания охлаждали пробирку в токе холодной воды до температуры 20-25°С. Далее очищали образец от образовавшихся в результате гидролиза свободных жирных кислот путем добавления в пробирку 3 см3 гексана, закрывали крышкой и взбалтывали в течение 10 секунд. После разделения фаз переносили нижний водный слой в новую пробирку при помощи пипетки Пастера и дополнительно очищали раствор, приливая 3 см3 гексана, закрывали крышкой и взбалтывали в течение 10 секунд. После разделения фаз удаляли верхний гексановый слой пипеткой Пастера. В очищенный от жирных кислот раствор, содержащий исследуемые вещества в свободной форме добавляли 100 мкл 2,5% раствор фенилборной кислоты в вода/ацетон (1/19). Перемешивали содержимое пробирке вихревым движением 10 секунд. Далее добавляли 3 см3 гексана и закрывали завинчивающейся крышкой. Встряхивали пробирку в течение 10 минут при температуре 23-25оС. После дериватизации, фенилборпроизводные определяемых компонентов распределяются в верхнем гексановом слое. Если в верхнем слое происходит гелеобразование необходимо центрифугировать пробирку при 3000-3500 об/мин в течение 1-10 минут. Верхний слой, содержащий производные ФБК при помощи пипетки Пастера переносили в новую пробирку, содержащую небольшое количество сульфата натрия и упаривали в токе азота при температуре 40оС до объема приблизительно 300 мкл. Полученную пробу анализировали на газовом хроматографе с масс-селективным детектором при условиях, указанных выше.
Согласно предложенному способу определения качественного и количественного состава монохлорпропандиолов и глицидола был проведен анализ дезодорированных и рафинированных пальмового, подсолнечного и рапсового масел.
В таблице 3 представлены данные по количественному содержанию 3-МХПД, 2-МХПД и глицидола в исследуемых образцах пальмового, подсолнечного и рапсового масел.
Таблица 3 - Количественное содержание определяемых соединений
Из данных представленных в таблице 2, видно, что -заявляемый способ позволяет осуществить возможность одновременного определения с высокой точностью определяемых соединений в рафинированных дезодорированных растительных маслах с применением в качестве внутреннего стандарта 1,3-бутандиола.
Исследования выполнялись с использованием оборудования ЦКП "Исследовательский центр пищевых и химических технологий" КубГТУ (CKP_3111), развитие которого поддерживается Минобрнауки РФ (Соглашение № 075-15-2021-679).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения качественного и количественного содержания монохлорпропандиолов, глицидола в растительных маслах | 2024 |
|
RU2841492C1 |
| СПОСОБ ПОДГОТОВКИ ПРОБ ПИЩЕВОЙ ПРОДУКЦИИ К КОЛИЧЕСТВЕННОМУ ОПРЕДЕЛЕНИЮ СОДЕРЖАНИЯ 3-МОНОХЛОРПРОПАНДИОЛА (3-МХПД) И ГЛИЦИДОЛА МЕТОДОМ ГАЗОЖИДКОСТНОЙ ХРОМАТОГРАФИИ С МАСС-СПЕКТРОМЕТРИЧЕСКИМ ДЕТЕКТИРОВАНИЕМ (ГХ-МС) | 2022 |
|
RU2789504C1 |
| Способ количественного определения антиконвульсантов в плазме крови больных эпилепсией | 2021 |
|
RU2771430C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
| Способ определения содержания свободного глицерина, моно- и диглицеридов | 2021 |
|
RU2780469C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| Способ измерения массовой концентрации метиловых эфиров жирных кислот в биологических средах методом газожидкостной хроматографии | 2020 |
|
RU2758932C1 |
| Способ количественного определения окадаиковой кислоты в морепродуктах | 2017 |
|
RU2666247C1 |
| Способ определения аминогликозидных антибиотиков методом обращенно-фазной высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786839C1 |
| Способ количественного определения ликарбазепина в плазме крови | 2017 |
|
RU2660364C1 |
Изобретение относится к масложировой промышленности. Раскрыт способ определения качественного и количественного состава монохлорпропандиолов, глицидола в растительном масле, включающий подготовку эталонных образцов определяемых соединений, с последующим анализом эталонных проб путем газовой хроматографии с масс-детектором, построение на основе полученных данных калибровочной зависимости и уравнения регрессии для каждого определяемого компонента, подготовка исследуемого образца, анализ исследуемого образца, причем подготовку эталонных образцов и исследуемого образца осуществляют путем смешивания эталонных образцов и исследуемого образца соответственно с растворителем и внутренним стандартом, в качестве которого используют 1,3-бутандиол концентрацией 5,0 мкг/мл, с последующими дериватизацией 2,5% раствором фенилборной кислоты, извлечением фенилборпроизводных определяемых компонентов гексаном, концентрированием в токе азота при температуре 40°С, и анализом на газовом хроматографе и регистрацией компонентов масс-спектрометрическим детектором в режиме SIM (Selected Ion Monitoring) в том числе для ионов фенилборпроизводного 1,3-бутандиола. Изобретение обеспечивает расширение арсенала способов определения МХПД и глицидола, обеспечивающих возможность одновременного определения в растительных маслах: 3-МХПД, 2-МХПД и глицидола при сохранении высокой точности. 1 ил., 3 табл.
Способ определения качественного и количественного состава монохлорпропандиолов, глицидола в растительном масле, включающий подготовку эталонных образцов определяемых соединений, с последующим анализом эталонных проб путем газовой хроматографии с масс-детектором, построение на основе полученных данных калибровочной зависимости и уравнения регрессии для каждого определяемого компонента, подготовка исследуемого образца, анализ исследуемого образца, расчет содержания определяемых соединений в исследуемом образце осуществляют по предварительно составленному уравнению, описывающему калибровочную прямую, построенную в координатах отношения значения площадей пиков эталонных определяемых компонентов к значениям площадей пиков внутреннего стандарта и отношения массы внесенных эталонных определяемых компонентов к массе внесенного внутреннего стандарта:
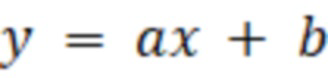 (1)
(1)
где a – тангенс угла наклона калибровочной кривой;
b – точка пересечения калибровочной кривой с осью Y, количественное содержание определяемых компонентов в образце масла рассчитывают по формуле:
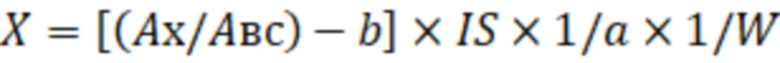 (2)
(2)
где X – концентрация определяемого соединения в исследуемом образце, мг/кг;
AX – площадь пика фенилборпроизводного количественного иона определяемого соединения;
ABC – площадь пика иона внутреннего стандарта;
a – тангенс угла наклона калибровочной кривой;
b – точка пересечения калибровочной кривой с осью Y;
IS – количество введённого в исследуемый образец внутреннего стандарта, мкг;
W – масса исследуемого образца, г,
отличающийся тем, что подготовку эталонных образцов и исследуемого образца осуществляют путем смешивания эталонных образцов и исследуемого образца соответственно с растворителем и внутренним стандартом, в качестве которого используют 1,3-бутандиол концентрацией 5,0 мкг/мл, с последующими дериватизацией 2,5% раствором фенилборной кислоты, извлечением фенилборпроизводных определяемых компонентов гексаном, концентрированием в токе азота при температуре 40°С, и анализом на газовом хроматографе и регистрацией компонентов масс-спектрометрическим детектором в режиме SIM (Selected Ion Monitoring) в том числе для ионов фенилборпроизводного 1,3-бутандиола.
| EP 2874501 B1, 20.07.2016 | |||
| СПОСОБ ПОДГОТОВКИ ПРОБ ПИЩЕВОЙ ПРОДУКЦИИ К КОЛИЧЕСТВЕННОМУ ОПРЕДЕЛЕНИЮ СОДЕРЖАНИЯ 3-МОНОХЛОРПРОПАНДИОЛА (3-МХПД) И ГЛИЦИДОЛА МЕТОДОМ ГАЗОЖИДКОСТНОЙ ХРОМАТОГРАФИИ С МАСС-СПЕКТРОМЕТРИЧЕСКИМ ДЕТЕКТИРОВАНИЕМ (ГХ-МС) | 2022 |
|
RU2789504C1 |
| MACMAHON S | |||
| et al | |||
| Analysis of Processing Contaminants in Edible Oils | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Agric | |||
| Food Chem., 2013, V | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Атмосферическая машина | 1926 |
|
SU4737A1 |
| Ленточный транспортер для подачи ткани под лапку швейной машины | 1932 |
|
SU34900A1 |
| Жиры и | |||

