ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к конденсированному трициклическому соединению и его фармацевтическому применению. Более конкретно, настоящее изобретение относится к конденсированному трициклическому соединению, или его фармацевтически приемлемой соли, имеющему ингибирующую киназу пируватдегидрогеназы (далее в настоящем документе сокращенно называемую ПДГК) активность, содержащей его фармацевтической композиции, содержащему его терапевтическому или профилактическому средству для лечения диабета (диабета 1 типа, диабета 2 типа и так далее), синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета (диабетической невропатии, диабетической ретинопатии, диабетической нефропатии, катаракты и так далее), сердечной недостаточности (острой сердечной недостаточности, хронической сердечной недостаточности), кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболеваний периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической невропатии или хронического заболевания почек, и тому подобного.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0002]
В тканях для реакций с использованием энергии, таких как биосинтез, активный транспорт, мышечное сокращение и тому подобное, энергию обеспечивает гидролиз аденозинтрифосфата (АТФ). АТФ производится путем окисления метаболического топлива, которое дает много энергии, такого как глюкоза и свободные жирные кислоты. В окислительных тканях, таких как мышцы, АТФ в основном вырабатывается из ацетил-КоА, который входит в цикл лимонной кислоты. Ацетил-КоА образуется путем окисления глюкозы гликолитическим путем или путем β-окисления свободных жирных кислот. Ферментом, который играет ключевую роль в контроле образования ацетил-КоА из глюкозы, является пируватдегидрогеназа (далее в настоящем документе сокращенно называемая ПДГ). ПДГ катализирует восстановление никотинамидадениндинуклеотида (НАД) до НАДН одновременно с окислением пировиноградной кислоты до ацетил-КоА и диоксида углерода (например, непатентные документы 1, 2).
[0003]
ПДГ представляет собой мультиферментный комплекс, состоящий из трех ферментных компонентов (Е1, Е2 и Е3) и нескольких субъединиц, локализованных в митохондриальном матриксе. Е1, Е2 и Е3 отвечают за декарбоксилирование пировиноградной кислоты, образование ацетил-КоА и восстановление НАД до НАДН, соответственно.
С ПДГ связываются ферменты двух классов, обладающие регуляторной функцией. Одним из них является ПДГК, представляющий собой протеинкиназу, обладающую специфичностью в отношении ПДГ. Его роль заключается в инактивации субъединицы E1α комплекса ПДГ путем фосфорилирования. Другим является фосфатаза ПДГ, которая представляет собой специфическую протеинфосфатазу, которая активирует ПДГ посредством дефосфорилирования субъединицы E1α. Доля ПДГ в активном (дефосфорилированном) состоянии определяется балансом активности киназы и активности фосфатазы. Активность киназы регулируется относительной концентрацией метаболических субстратов. Например, киназа активируется увеличением соотношений НАДН/НАД, ацетил-КоА/КоА и АТФ/аденозиндифосфат (АДФ), и ингибируется пировиноградной кислотой (например, непатентный документ 3).
[0004]
В тканях млекопитающих идентифицировано 4 вида изоферментов ПДГК. В частности, ПДГК2 экспрессируется в широком диапазоне тканей, включая печень, скелетные мышцы и жировые ткани, участвующие в метаболизме глюкозы. Кроме того, поскольку ПДГК2 проявляет сравнительно высокую чувствительность к активации увеличением соотношения НАДН/НАД или ацетил-КоА/КоА и ингибированию пировиноградной кислотой, предполагается его участие в кратковременной регуляции метаболизма глюкозы (например, не патентный документ 4).
[0005]
Кроме того, ПДГК1 экспрессируется в больших количествах в сердечной мышце, скелетной мышце, β-клетке поджелудочной железы и тому подобном. Кроме того, поскольку экспрессия ПДГК1 индуцируется путем активации фактора, индуцируемого гипоксией (HIF) 1 в ишемическом состоянии, предполагается ее участие в ишемических и раковых заболеваниях (например, непатентный документ 5).
[0006]
При таких заболеваниях, как инсулинозависимый диабет (1 типа), инсулиннезависимый диабет (2 типа) и тому подобное, стимулируется окисление липидов с одновременным снижением утилизации глюкозы. Это снижение утилизации глюкозы является одним из факторов, вызывающих гипергликемию. При снижении окислительного метаболизма глюкозы при диабете 1 и 2 типа и ожирении активность ПДГ также снижается. Это предполагает участие снижения активности ПДГ в снижении утилизации глюкозы при диабете 1 и 2 типа (например, непатентные документы 6, 7).
Напротив, печеночный глюконеогенез повышается при диабете 1 и 2 типа, что также является одним из факторов, вызывающих гипергликемию. Снижение активности ПДГ приводит к увеличению концентрации пировиноградной кислоты, что, в свою очередь, повышает доступность молочной кислоты в качестве субстрата для печеночного глюконеогенеза. Это предполагает возможное участие снижения активности ПДГ в усилении глюконеогенеза при диабете 1 и 2 типа (например, непатентные документы 8, 9).
Когда ПДГ активируется за счет ингибирования ПДГК, считается, что скорость окисления глюкозы увеличивается. В результате стимулируется утилизация глюкозы в организме и подавляется глюконеогенез в печени, вследствие чего ожидается ослабление гипергликемии при диабете 1 и 2 типа (например, непатентные документы 10, 11, 12).
Другим фактором, способствующим диабету, является нарушение секреции инсулина, которое, как известно, связано со снижением активности ПДГ в β-клетках поджелудочной железы и индукцией ПДГК1, 2 и 4 (например, непатентные документы 13, 14).
Кроме того, известно, что устойчивая гипергликемия вследствие диабета вызывает такие осложнения, как диабетическая невропатия, диабетическая ретинопатия, диабетическая нефропатия и тому подобное. Тиамин и α-липоевая кислота в качестве коферментов способствуют активации ПДГ. Показано, что тиамин и α-липоевая кислота, или производные тиамина и производные α-липоевой кислоты, обладают многообещающим эффектом при лечении диабетических осложнений. Таким образом, ожидается, что активация ПДГ приведет к уменьшению диабетических осложнений (например, не патентные документы 15, 16).
[0007]
В условиях ишемии ограниченное снабжение кислородом приводит к снижению окисления как глюкозы, так и жирных кислот, а также снижению количества АТФ, образующегося в тканях в результате окислительного фосфорилирования. В отсутствие достаточного количества кислорода уровень АТФ поддерживается за счет ускоренного анаэробного гликолиза. В результате увеличивается содержание молочной кислоты и снижается внутриклеточное значение рН. Несмотря на то, что клетки пытаются поддерживать гомеостаз ионов за счет потребления энергии, аномально низкий уровень АТФ и нарушенная осмолярность клеток приводят к гибели клеток. Кроме того, аденозинмонофосфат-активирующая киназа, активируемая в ишемическом состоянии, инактивирует ацетил-КоА-карбоксилазу путем фосфорилирования. Уровни общего малонил-КоА в тканях падают, вследствие этого активность карнитинпальмитоилтрансферазы I повышается, и окисление жирных кислот получает преимущество относительно окисления глюкозы за счет возможности переноса ацил-КоА в митохондрии. Окисление глюкозы способно приводить к большему количеству АТФ на молекулу кислорода, чем окисление жирных кислот. Таким образом, в ишемических условиях, когда в энергетическом метаболизме доминирует окисление глюкозы за счет активации ПДГ, считается, что способность к поддержанию уровня АТФ повышается (например, непатентный документ 17).
Кроме того, поскольку активация ПДГ вызывает окисление пировиноградной кислоты, образующейся в результате гликолиза, и снижение продукции молочной кислоты, считается, что чистая протонная нагрузка в ишемизированных тканях снижается. Следовательно, ожидается, что активация ПДГ за счет ингибирования ПДГК оказывает защитное действие при ишемических заболеваниях, таких как ишемия сердечной мышцы (например, непатентные документы 18, 19).
[0008]
Считается, что лекарственное средство, которое активирует ПДГ за счет ингибирования ПДГК, снижает выработку лактата, поскольку способствует метаболизму пирувата. Следовательно, ожидается, что такое лекарственное средство будет полезно для лечения гиперлактацидемии, такой как митохондриальная болезнь, митохондриальная энцефаломиопатия и сепсис (например, непатентный документ 20).
[0009]
В раковых клетках увеличивается экспрессия ПДГК1 или 2. Кроме того, в раковых клетках продуцирование АТФ за счет окислительного фосфорилирования в митохондриях снижается, а продуцирование АТФ за счет анаэробного гликолиза в цитоплазме возрастает. Ожидается, что активация ПДГ за счет ингибирования ПДГК будет способствовать окислительному фосфорилированию в митохондриях и увеличению продуцирования активного кислорода, что вызовет апоптоз раковых клеток. Следовательно, активация ПДГ путем ингибирования ПДГК полезна для лечения раковых заболеваний (например, непатентный документ 21).
[0010]
Легочная гипертензия характеризуется повышенным артериальным давлением, вызванным частичным сужением легочной артерии вследствие усиленной клеточной пролиферации в ней. Поэтому при легочной гипертензии ожидается, что активация ПДГ в клетках легочной артерии будет способствовать окислительному фосфорилированию в митохондриях, увеличению продуцирования активного кислорода и индукции апоптоза клеток легочной артерии. Следовательно, активация ПДГ путем ингибирования ПДГК считается полезной для лечения легочной гипертензии, например, легочной артериальной гипертензии (например, непатентный документ 22).
[0011]
При болезни Альцгеймера снижается производство энергии и метаболизм глюкозы в головном мозге, а также снижается активность ПДГ. При снижении активности ПДГ снижается продуцирование ацетил-КоА. Ацетил-КоА используется для производства АТФ в системе транспорта электронов через цикл лимонной кислоты. Ацетил-КоА также является исходным материалом для синтеза ацетилхолина, одного из нейротрансмиттеров. Вследствие этого считается, что снижение активности ПДГ в головном мозге при болезни Альцгеймера вызывает гибель нейронов из-за снижения продуцирования АТФ. Более того, считается, что синтез ацетилхолина, который является трансмиттером холинергического нерва, ингибируется, вызывая ухудшение памяти и тому подобное. Ожидается, что активация ПДГ в головном мозге усилит выработку энергии и синтез ацетилхолина при болезни Альцгеймера. Следовательно, активация ПДГ путем ингибирования ПДГК считается полезной для лечения болезни Альцгеймера (например, непатентные документы 23, 24).
[0012]
Сосудистая деменция представляет собой заболевание, которое условно подразделяется на тип с поражением крупных сосудов и тип с поражением мелких сосудов. При типе с поражением крупных сосудов причиной является инфаркт головного мозга, включая реперфузию ишемии, а гибель нейронов индуцируется повышением показателей пировиноградной и молочной кислот за счет снижения внутримозговой активности ПДГ и снижения выработки энергии. При типе с поражением мелких сосудов причиной является поражение белого вещества вследствие церебральной гипоперфузии, что считают причиной когнитивной дисфункции вследствие хронического снижения метаболизма глюкозы. Когда ПДГ в головном мозге активируется при сосудистой деменции, снижение уровня молочной кислоты и тому подобное, а также увеличение выработки энергии, ожидается при типе с поражением крупных сосудов, а повышенный метаболизм глюкозы ожидается при типе с поражением мелких сосудов. Следовательно, активация ПДГ ингибиторами ПДГК считается полезной для лечения сосудистой деменции (например, непатентные документы 28, 29, 30).
[0013]
Показано, что дихлоруксусная кислота, являющаяся лекарственным средством, активирующим ПДГ, обеспечивает многообещающие эффекты при лечении диабета, ишемии миокарда, инфаркта миокарда, стенокардии, сердечной недостаточности, гиперлактацидемии, ишемии головного мозга, церебральной апоплексии, заболеваний периферических артерий, хронической обструктивной болезни легких, ракового заболевания и легочной гипертензии (например, непатентные документы 10, 18, 20, 22, 25, 26, 27).
Было показано, что соединение, обладающее ингибирующим действием на ПДГК, оказывает нейропротекторное действие на ишемически-реперфузионное повреждение сетчатки (непатентный документ 31). Ишемическое повреждение сетчатки связано с такими заболеваниями, как глаукома, диабетическая ретинопатия, ретинопатия недоношенных, окклюзия вен сетчатки и тому подобное.
Также было показано, что у животных с моделью заболевания, имеющих заболевание почек, подобное хронической почечной недостаточности, и сниженную почечную функцию, соединения, обладающие ингибирующим действием на ПДГК, уменьшают тяжесть заболевания (патентный документ 1).
[0014]
Исходя из вышеизложенного, ингибитор ПДГК считается полезным для лечения или профилактики заболеваний, связанных с нарушением утилизации глюкозы, например, диабета (диабета 1 типа, диабета 2 типа и так далее), синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета (диабетической нейропатии, диабетической ретинопатии, диабетической нефропатии, катаракты и так далее). Кроме того, ингибитор ПДГК считается полезным для лечения или профилактики заболеваний, вызванных ограниченным поступлением энергетических субстратов к тканям, например, сердечной недостаточности (острой сердечной недостаточности, хронической сердечной недостаточности), кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболевания периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности. Кроме того, считается, что ингибитор ПДГК полезен для лечения или профилактики митохондриального заболевания, митохондриальной энцефаломиопатии, рака, легочной гипертензии и тому подобного.
[0015]
Таким образом, ингибитор ПДГК считается полезным для лечения или профилактики диабета (диабета 1-го типа, диабета 2-го типа и так далее), синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета (диабетической нейропатии, диабетической ретинопатии, диабетической нефропатии, катаракты и так далее), сердечной недостаточности (острой сердечной недостаточности, хронической сердечной недостаточности), кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболевания периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии или хронической почечной недостаточности.
Список документов
Патентные документы
[0016]
Патентный документ 1: WO 2020/054734
Не патентные документы
[0017]
Непатентный документ 1: Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990 Jun 5; 265(16):8971-4.
Непатентный документ 2: Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990 Nov; 4(14):3224-33.
Непатентный документ 3: Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003 May; 284(5):E855-62.
Непатентный документ 4: Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998 Jan 1; 329 (Pt 1):191-6.
Непатентный документ 5: Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006 Mar; 3(3):177-85.
Непатентный документ 6: Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005 Dec; 115(12):3587-93.
Непатентный документ 7: Caterson ID, Fuller SJ, Randle PJ. Effect of the fatty acid oxidation inhibitor 2-tetradecylglycidic acid on pyruvate dehydrogenase complex activity in starved and alloxan-diabetic rats. Biochem J. 1982 Oct 15; 208(1):53-60.
Непатентный документ 8: Boden G, Chen X, Stein TP. Gluconeogenesis in moderately and severely hyperglycemic patients with type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2001 Jan; 280(1): E23-30.
Непатентный документ 9: Shangraw RE, Fisher DM. Pharmacokinetics and pharmacodynamics of dichloroacetate in patients with cirrhosis. Clin Pharmacol Ther. 1999 Oct; 66(4):380-90.
Непатентный документ 10: Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. NEngl J Med. 1978 Mar 9; 298(10):526-30.
Непатентный документ 11: Mayers RM, Leighton B, Kilgour E. ПДГ kinase inhibitors: a novel therapy for Type II diabetes? Biochem Soc Trans. 2005 Apr; 33(Pt 2):367-70.
Непатентный документ 12: Jeoung NH, Rahimi Y, Wu P, Lee WN, Harris RA. Fasting induces ketoacidosis and hypothermia in ПДГК2/ПДГК4-double-knockout mice. Biochem J.2012 May 1; 443(3):829-39.
Непатентный документ 13: Zhou YP, Berggren PO, Grill V. A fatty acid-induced decrease in pyruvate dehydrogenase activity is an important determinant of beta-cell dysfunction in the obese diabetic db/db mouse. Diabetes. 1996 May; 45(5):580-6.
Непатентный документ 14: Xu J, Han J, Epstein PN, Liu YQ. Regulation of PDK mRNA by high fatty acid and glucose in pancreatic islets. Biochem Biophys Res Commun. 2006 Jun 9; 344(3):827-33.
Непатентный документ 15: Benfotiamine. Monograph. Altern Med Rev. 2006 Sep; 11(3):238-42.
Непатентный документ 16: Vallianou N, Evangelopoulos A, Koutalas P. Alpha-lipoic Acid and diabetic neuropathy. Rev Diabet Stud. 2009 Winter; 6(4):230-6.
Непатентный документ 17: Ussher JR, Lopaschuk GD. The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc Res. 2008 Jul 15; 79(2):259-68.
Непатентный документ 18: Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol. 1988 Jan 1; 61(1):65-70.
Непатентный документ 19: Taniguchi M, Wilson C, Hunter CA, Pehowich DJ, Clanachan AS, Lopaschuk GD. Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. Am J Physiol Heart Circ Physiol. 2001 Apr; 280(4):H1762-9.
Непатентный документ 20: Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003 Jul; 43(7):683-91.
Непатентный документ 21: Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell.2007 Jan; 11(1):37-51.
Непатентный документ 22: McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004 Oct 15; 95(8):830-40.
Непатентный документ 23: Saxena U. Bioenergetics breakdown in Alzheimer's disease: targets for new therapies. Int J Physiol Pathophysiol Pharmacol. 2011; 3(2):133-9.
Непатентный документ 24: Stacpoole PW. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell. 2012 Jun; 11(3):371-7.
Непатентный документ 25: Marangos PJ, Turkel CC, Dziewanwska ZE, Fox AW. Dichloroacetate and cerebral ischaemia therapeutics. Expert Opin Investig Drugs. 1999 Apr; 8(4):373-82.
Непатентный документ 26: Calvert LD, Shelley R, Singh SJ, Greenhaff PL, Bankart J, Morgan MD, et al. Dichloroacetate enhances performance and reduces blood lactate during maximal cycle exercise in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008 May 15; 177(10):1090-4.
Непатентный документ 27: Flavin DF. Non-Hodgkin's Lymphoma Reversal with Dichloroacetate. J Oncol. Hindawi Publishing Corporation Journal of Oncology Volume 2010, Article ID 414726, 4 pages doi:10.1155/2010/414726.
Непатентный документ 28: Froelich L, Goetz ME, Weinmueller M, Youdim MB, Barth N, Dirr A, Gsell W, Jellinger K, Beckmann H, Riederer P. (r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia. J Neural Transm (Vienna). 2004 Mar; 111(3):295-310
Непатентный документ 29: Parnetti L, Reboldi GP, Gallai V. Cerebrospinal fluid pyruvate levels in Alzheimer's disease and vascular dementia. Neurology. 2000 Feb 8; 54(3):735-7.
Непатентный документ 30: Pascual B, Prieto E, Arbizu J, Marti-Climent J, Olier J, Masdeu JC. Brain glucose metabolism in vascular white matter disease with dementia: differentiation from Alzheimer disease. Stroke. 2010 Dec; 41(12):2889-93.
Непатентный документ 31: Sato K, Mochida S, Tomimoto D, Konuma T, Kiyota N, Tsuda S, Shiga Y, Omodaka K, Nakazawa T. A pyruvate dehydrogenase kinase inhibitor prevents retinal cell death and improves energy metabolism in rat retinas after ischemia/reperfusion injury. Experimental eye research 2020 Apr; 193: 107997.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0018]
Настоящее изобретение заключается в следующем.
[1] Соединение формулы [I-a], или его фармацевтически приемлемая соль:
[0019]
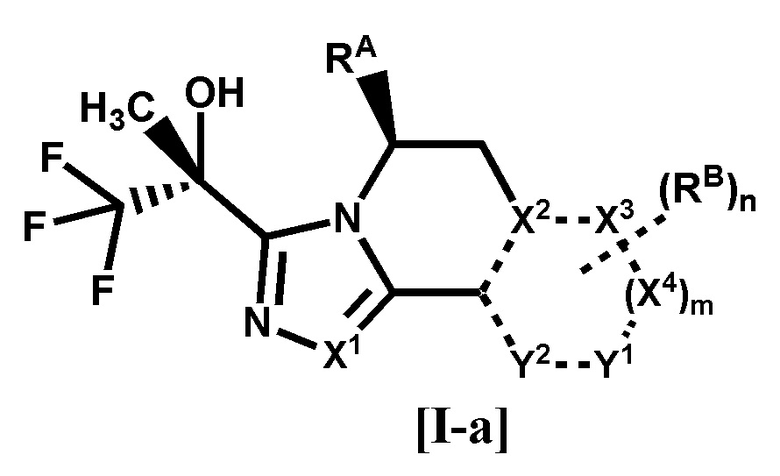
[0020]
где
связь, обозначенная пунктирной линией, представляет собой одинарную связь или двойную связь,
X1, X2, X3 и X4 каждый независимо представляет собой C или N, Y1 и Y2 каждый независимо представляет собой C, N или O (где общее количество N и O для X2, X3, X4, Y1 или Y2 составляет 0-3),
RA представляет собой C1-4 алкил,
RB представляет собой
(1) галоген,
(2) циано,
(3) гидрокси,
(4) оксо,
(5) -COR1 {где R1 представляет собой
(A) водород,
(B) -OH,
(C) -NR2R3 (где R2 и R3 каждый независимо представляет собой водород или C1-4 алкил), или
(D) 4-6-членный насыщенный гетероциклил, имеющий один атом азота (где насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена)},
(6) C1-8 алкил {где C1-8 алкил необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) гидрокси,
(C) фенила, необязательно замещенного галогеном,
(D) пиридила, необязательно замещенного гало-C1-4 алкилом, и
(E) -OR4 (где R4 представляет собой
(a) C1-4 алкил,
(b) фенил, необязательно замещенный галогеном, или
(c) бензил, необязательно замещенный C1-4 алкокси)},
(7) C1-8 алкокси {где C1-8 алкокси необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) циано,
(C) гидрокси,
(D) C1-4 алкокси, необязательно замещенного 1-3 атомами галогена,
(E) C1-4 алкилсульфонила,
(F) C3-6 циклоалкила, необязательно замещенного одним заместителем, выбранным из группы, состоящей из циано и циано-C1-4 алкила,
(G) фенила, необязательно замещенного циано,
(H) -COCy1 (где Cy1 представляет собой 4-6-членный насыщенный гетероциклил, имеющий один атом азота, и насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена), и
(I) 4-6-членного насыщенного гетероциклила, имеющего 1 или 2 гетероатома, независимо выбранных из атома азота, атома кислорода и атома серы (где насыщенный гетероциклил необязательно замещен 1-4 заместителями, независимо выбранными из
(a) C1-4 алкила,
(b) оксо,
(c) C1-4 алкилкарбонила,
(d) бензоила, необязательно замещенного галогеном, и
(e) C1-4 алкилсульфонила,
когда насыщенный гетероциклил замещен двумя C1-4 алкилами, два C1-4 алкила необязательно связаны друг с другом, образуя кольцо с внутренним мостиком вместе с атомами, связанными с ними)},
(8) -Cy2 {где Cy2 представляет собой
(A) C3-6 циклоалкил (где C3-6 циклоалкил необязательно замещен 1 или 2 заместителями, независимо выбранными из
(a) галогена,
(b) C1-4 алкила,
(c) гало-C1-4 алкила, и
(d) фенила, необязательно замещенного галогеном),
(B) фенил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, гало-C1-4 алкила и C1-4 алкокси, или
(C) 4-6-членный насыщенный гетероциклил, имеющий один атом азота или атом кислорода (где насыщенный гетероциклил необязательно замещен одним заместителем, выбранным из группы, состоящей из (a) фенила, необязательно замещенного галогеном, и (b) C1-4 алкилкарбонила)}, или
(9) -OCy3 {где Cy3 представляет собой
(A) 4-6-членный насыщенный гетероциклил, имеющий один атом азота или атом кислорода (где насыщенный гетероциклил необязательно замещен одним заместителем, выбранным из группы, состоящей из (a) бензоила, необязательно замещенного галогеном, и (b) C1-4 алкилкарбонила), или
(B) 6-членный гетероарил, имеющий 1 или 2 атома азота (где гетероарил необязательно замещен 1 или 2 заместителями, независимо выбранными из циано, гало-C1-4 алкила и C3-6 циклоалкила)},
m равно 0 или 1, и
n равно 0, 1 или 2, когда n равно 2, все RB могут быть одинаковыми или разными.
[0021]
[2] Соединение по п. [1], или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-b]:
[0022]
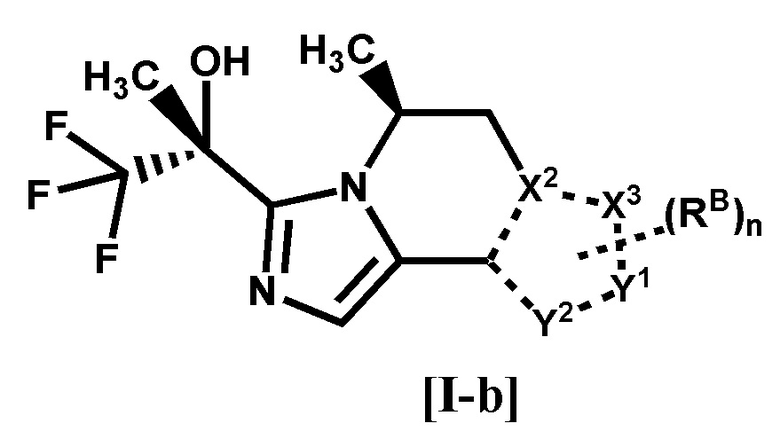
[0023]
где каждый символ является таким, как определено в п. [1], или его фармацевтически приемлемая соль.
[0024]
[3] Соединение по п. [1] или [2], или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-c]:
[0025]
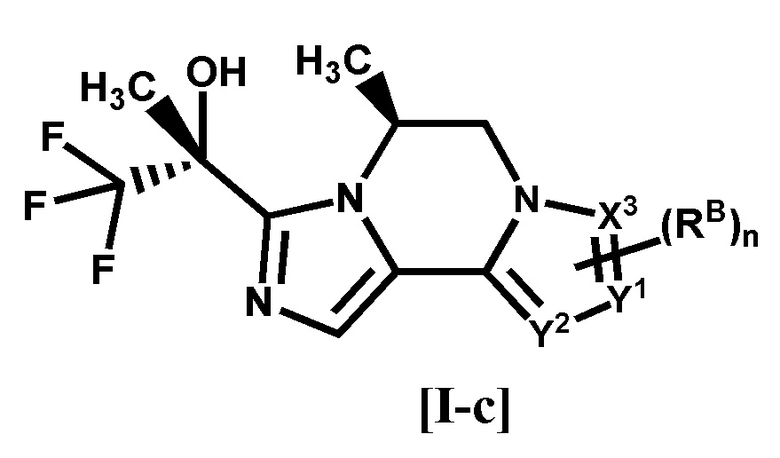
[0026]
где каждый символ является таким, как определено в п. [1], или его фармацевтически приемлемая соль.
[0027]
[4] Соединение по любому из п. [1]-[3], где n равно 1, или его фармацевтически приемлемая соль.
[0028]
[5] Соединение по любому из п. [1]-[4], или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-d]:
[0029]
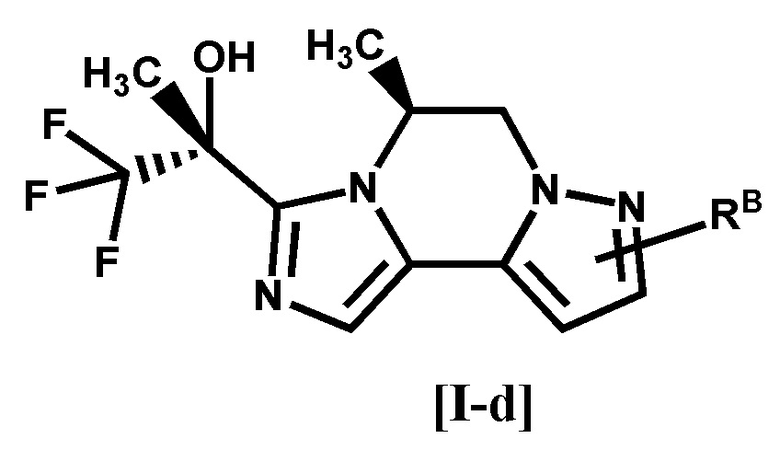
[0030]
где символ является таким, как определено в п. [1], или его фармацевтически приемлемая соль.
[0031]
[6] Соединение по любому из пунктов [1]-[4], или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-e]:
[0032]
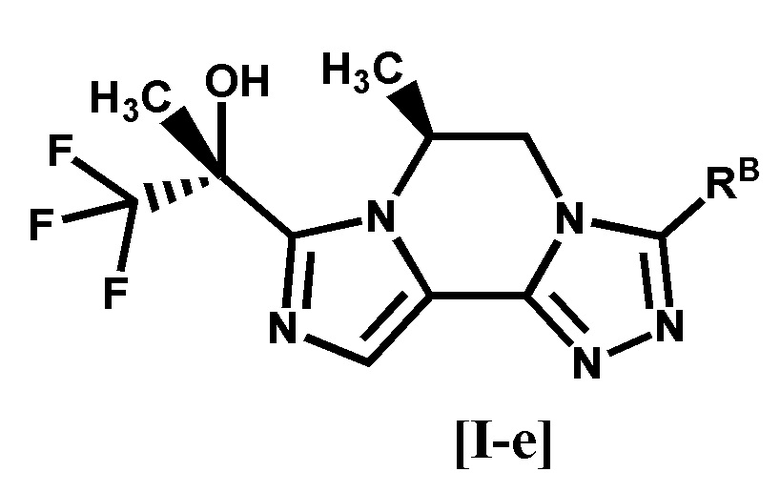
[0033]
где символ является таким, как определено в п. [1], или его фармацевтически приемлемая соль.
[0034]
[7] Соединение по любому из пунктов [1]-[6], или его фармацевтически приемлемая соль, где RB представляет собой
(1) C1-8 алкил {где C1-8 алкил необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) гидрокси,
(C) фенила, необязательно замещенного галогеном,
(D) пиридила, необязательно замещенного гало-C1-4 алкилом, и
(E) -OR4 (где R4 представляет собой
(a) C1-4 алкил,
(b) фенил, необязательно замещенный галогеном, или
(c) бензил, необязательно замещенный C1-4 алкокси)}, или
(2) C1-8 алкокси {где C1-8 алкокси необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) циано,
(C) гидрокси,
(D) C1-4 алкокси, необязательно замещенного 1-3 атомами галогена,
(E) C1-4 алкилсульфонила,
(F) C3-6 циклоалкила, необязательно замещенного одним заместителем, выбранным из группы, состоящей из циано и циано-C1-4 алкила,
(G) фенила, необязательно замещенного циано,
(H) -COCy1 (где Cy1 представляет собой 4-6-членный насыщенный гетероциклил, имеющий один атом азота, и насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена), и
(I) 4-6-членного насыщенного гетероциклила, имеющего 1 или 2 гетероатома, независимо выбранных из атома азота, атома кислорода и атома серы (где насыщенный гетероциклил необязательно замещен 1-4 заместителями, независимо выбранными из
(a) C1-4 алкила,
(b) оксо,
(c) C1-4 алкилкарбонила,
(d) бензоила, необязательно замещенного галогеном, и
(e) C1-4 алкилсульфонила,
когда насыщенный гетероциклил замещен двумя C1-4 алкилами, два C1-4 алкила необязательно связаны друг с другом, образуя кольцо с внутренним мостиком вместе с атомами, связанными с ними)}.
[0035]
[8] Соединение, выбранное из следующих формул:
[0036]
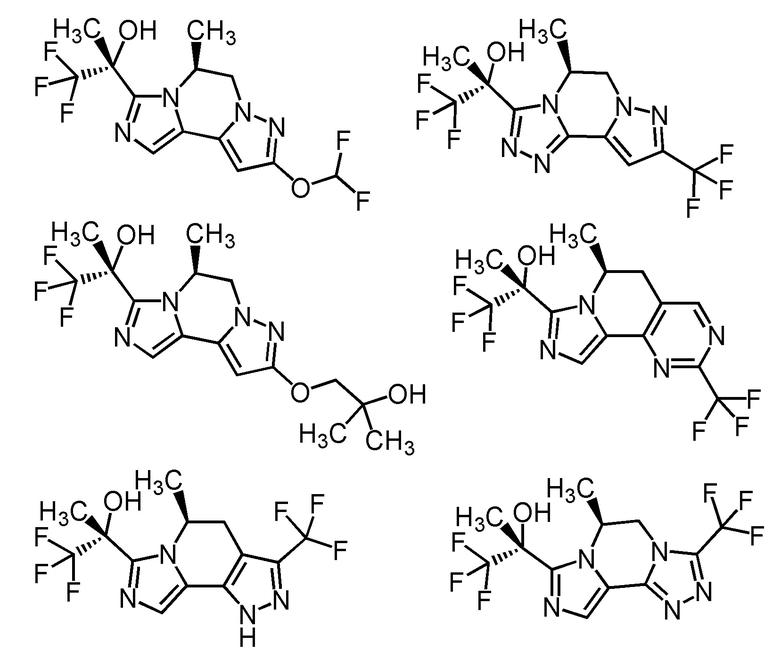 ,
,
[0037]
или его фармацевтически приемлемая соль.
[0038]
[9] Фармацевтическая композиция, содержащая соединение по любому из пунктов [1]-[8], или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
[0039]
[10] Ингибитор ПДГК, включающий соединение по любому из пунктов [1]-[8] или его фармацевтически приемлемую соль.
[0040]
[11] Ингибитор ПДГК2, включающий соединение по любому из пунктов [1]-[8] или его фармацевтически приемлемую соль.
[0041]
[12] Средство для лечения или профилактики диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболеваний периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии или хронической почечной недостаточности, включающее соединение по любому из пунктов [1]-[8] или его фармацевтически приемлемую соль.
[0042]
[13] Средство по п. [12], где диабет представляет собой диабет 1 типа или диабет 2 типа.
[0043]
[14] Средство по п. [12], где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
[0044]
[15] Средство по п. [12], где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
[0045]
[16] Средство по п. [12], где легочная гипертензия представляет собой легочную артериальную гипертензию.
[0046]
[17] Способ ингибирования ПДГК, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пунктов [1]-[8] или его фармацевтически приемлемой соли.
[0047]
[18] Способ лечения или профилактики заболевания, выбранного из группы, состоящей из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности, который включает введение млекопитающему терапевтически эффективного количества соединения по любому из пунктов [1]-[8] или его фармацевтически приемлемой соли.
[0048]
[19] Способ по п. [18], где диабет представляет собой диабет 1 типа или диабет 2 типа.
[0049]
[20] Способ по п. [18], где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
[0050]
[21] Способ по п. [18], где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
[0051]
[22] Способ по п. [18], где легочная гипертензия представляет собой легочную артериальную гипертензию.
[0052]
[23] Применение соединения по любому из пунктов [1]-[8], или его фармацевтически приемлемой соли, в производстве ингибитора ПДГК.
[0053]
[24] Применение соединения по любому из пунктов [1]-[8], или его фармацевтически приемлемой соли, в производстве средства для лечения или профилактики заболевания, выбранного из группы, состоящей из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
[0054]
[25] Применение по п. [24], где диабет представляет собой диабет 1 типа или диабет 2 типа.
[0055]
[26] Применение по п. [24], где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
[0056]
[27] Применение по п. [24], где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
[0057]
[28] Применение по п. [24], где легочная гипертензия представляет собой легочную артериальную гипертензию.
[0058]
[29] Соединение по любому из пунктов [1]-[8], или его фармацевтически приемлемая соль, для применения в лечении или профилактике заболевания, выбранного из группы, состоящей из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
[0059]
[30] Соединение по п. [29], или его фармацевтически приемлемая соль, где диабет представляет собой диабет 1 типа или диабет 2 типа.
[0060]
[31] Соединение по п. [29], или его фармацевтически приемлемая соль, где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
[0061]
[32] Соединение по п. [29], или его фармацевтически приемлемая соль, где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
[0062]
[33] Соединение по п. [29], или его фармацевтически приемлемая соль, где легочная гипертензия представляет собой легочную артериальную гипертензию.
[0063]
[34] Способ ингибирования ПДГК2, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пунктов [1]-[8] или его фармацевтически приемлемой соли.
[0064]
[35] Коммерческая упаковка, содержащая фармацевтическую композицию по п. [9], и связанное с ней письменное сообщение, которое содержит информацию о том, что фармацевтическая композиция может быть использована для лечения или профилактики заболевания, выбранного из группы, состоящей из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
[0065]
[36] Набор, включающий фармацевтическую композицию по п. [9] и связанное с ней письменное сообщение, которое содержит информацию о том, что фармацевтическая композиция может быть использована для лечения или профилактики заболевания, выбранного из группы, состоящей из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0066]
В настоящем документе использованы термины, определения которых приведены ниже.
[0067]
«Галоген» означает фтор, хлор, бром или иод. В качестве «галогена» предпочтительным является фтор или хлор.
[0068]
«C1-4 алкил» означает неразветвленный или разветвленный алкил, имеющий 1-4 атома углерода. Примеры включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. В качестве «C1-4 алкила» предпочтительным является метил.
[0069]
«C1-8 алкил» означает неразветвленный или разветвленный алкил, имеющий 1-8 атомов углерода. Примеры включают метил, этил, пропил, изопропил, 1,1-диметилпропил, 1-этилпропил, 1-метил-1-этилпропил, бутил, изобутил, втор-бутил, трет-бутил, 1-метил-1-пропилбутил, пентил, изопентил, неопентил, гексил, изогексил, 1,1-диметилбутил, 2,2-диметилбутил, 3,3-диметилбутил, 2-этилбутил и тому подобное.
[0070]
«C1-4 алкилкарбонил» означает алкилкарбонил, в котором алкильный фрагмент представляет собой «C1-4 алкил», определение которому дано выше, и включает, например, ацетил, пропаноил, бутаноил, 2-метилпропаноил, пентаноил, 3-метилбутаноил, 2-метилбутаноил и 2,2-диметилпропаноил. В качестве «C1-4 алкилкарбонила» предпочтительным является ацетил.
[0071]
«C1-4 алкилсульфонил» означает алкилсульфонил, в котором алкильный фрагмент представляет собой «C1-4 алкил», определение которому дано выше, и включает, например, метансульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил и трет-бутилсульфонил. В качестве «C1-4 алкилсульфонила» предпочтительным является метансульфонил.
[0072]
«Гало-C1-4 алкил» означает неразветвленный или разветвленный алкил, имеющий 1-4 атома углерода и замещенный 1-5 атомами «галогена», определение которому дано выше. Когда алкил замещен несколькими атомами галогена, атомы галогена могут быть одинаковыми или разными. Примеры «гало-C1-4 алкила» включают фторметил, дифторметил, трифторметил, 1-фтор-1-метилэтил, 1,1-дифторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 1,1-дифторпропил, 1,1-дифтор-2-метилпропил и тому подобное. В качестве «гало-C1-4 алкила» предпочтительным является C1-4 алкил, замещенный 1-3 атомами фтора, и более предпочтительным является трифторметил.
[0073]
«Циано-C1-4 алкил» означает «C1-4 алкил», определение которому дано выше, который замещен одной цианогруппой. Примеры включают цианометил, 2-цианоэтил, 1-циано-1-метилэтил, 3-цианопропил, 4-цианобутил и тому подобное.
[0074]
«C1-4 алкокси» означает алкилокси, в котором алкильный фрагмент представляет собой «C1-4 алкил», определение которому дано выше, и включает, например, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутокси. В качестве «C1-4 алкокси» предпочтительным является метокси.
[0075]
«C1-8 алкокси» означает алкокси, в котором алкильный фрагмент представляет собой «C1-8 алкил», определение которому дано выше. Примеры включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, изопентилокси, неопентилокси, 1,2-диметилпропилокси, 1-этилпропилокси, гексилокси, изогексилокси, 1,2,2-триметилпропилокси, 1,1-диметилбутилокси, 2,2-диметилбутилокси, 3,3-диметилбутилокси, 2-этилбутилокси и тому подобное.
[0076]
«C3-6 циклоалкил» означает 3-6-членную моноциклическую углеводородную кольцевую группу и включает, например, циклопропил, циклобутил, циклопентил и циклогексил. В качестве «C3-6 циклоалкила предпочтительным является циклопропил.
[0077]
«4-6-членный насыщенный гетероциклил, имеющий один атом азота» означает 4-6-членную моноциклическую насыщенную гетероциклическую группу, имеющую один атом азота помимо атома углерода. Примеры насыщенного гетероциклила включают азетидинил, пирролидинил и пиперидинил.
[0078]
«4-6-членный насыщенный гетероциклил, имеющий 1 или 2 гетероатома, независимо выбранные из группы, состоящей из атома азота, атома кислорода и атома серы» означает 4-6-членную моноциклическую насыщенную гетероциклическую группу, имеющую, помимо атома углерода, 1 или 2 гетероатома, независимо выбранные из группы, состоящей из атома азота, атома кислорода и атома серы. Примеры насыщенного гетероциклила включают оксетанил, тетрагидрофуранил, тетрагидропиранил, азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тетрагидротиопиранил, изотиазолидинил и тому подобное, и предпочтительными являются оксетанил, тетрагидрофуранил, пиперидинил, тетрагидротиопиранил и изотиазолидинил.
[0079]
«Когда насыщенный гетероциклил замещен двумя C1-4 алкилами, два C1-4 алкила необязательно связаны друг с другом, образуя кольцо с внутренним мостиком вместе с атомами, связанными с ними» означает, например, что насыщенный гетероциклил представляет собой следующую группу:
[0080]
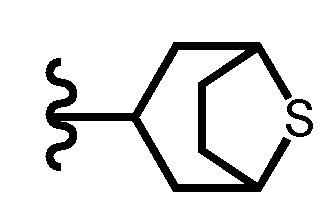 .
.
[0081]
«4-6-членный насыщенный гетероциклил, имеющий один атом азота или атом кислорода» означает 4-6-членную моноциклическую насыщенную гетероциклическую группу, имеющую, помимо атома углерода, один гетероатом, независимо выбранный из группы, состоящей из атома азота и атома кислорода. Примеры насыщенного гетероциклила включают оксетанил, тетрагидрофуранил, тетрагидропиранил, азетидинил, пирролидинил, пиперидинил и тому подобное, и предпочтительными являются оксетанил, тетрагидрофуранил, тетрагидропиранил, азетидинил и пиперидинил.
[0082]
«6-членный гетероарил, имеющий 1 или 2 атома азота» означает 6-членный моноциклический гетероарил, имеющий 1 или 2 атома азота помимо атома углерода. Примеры гетероарила включают пиридил, пиримидинил и пиразинил.
[0083]
Предпочтительный вариант осуществления соединения формулы [I-a] описан ниже.
Одним из предпочтительных вариантов осуществления соединения формулы [I-a] является соединение, представленное формулой [I-a1]:
[0084]
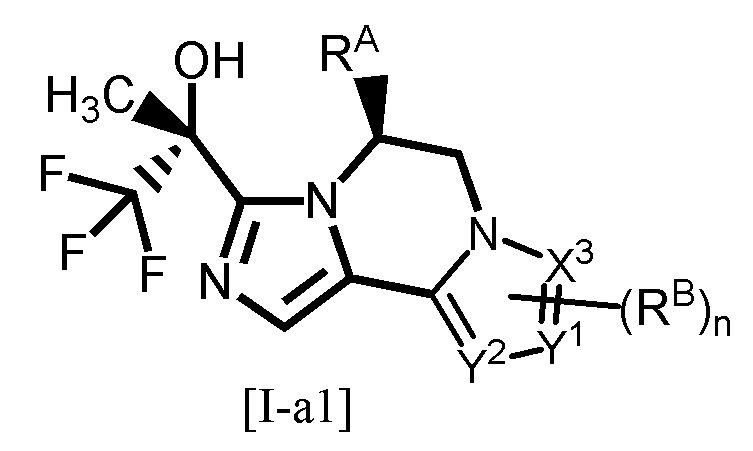
[0085]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0086]
Одним из других предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a2]:
[0087]
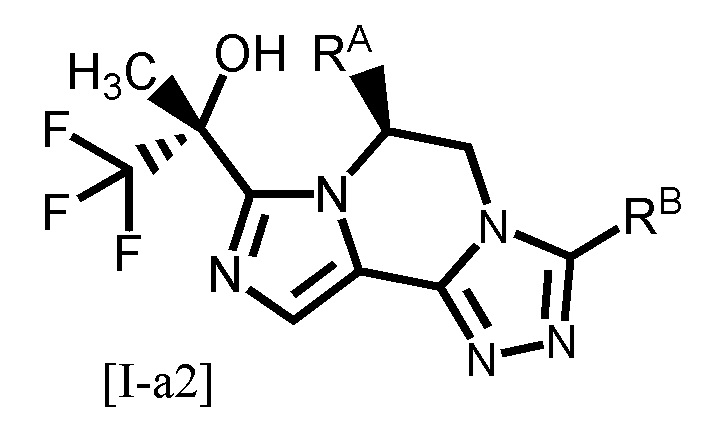
[0088]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0089]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a3]:
[0090]
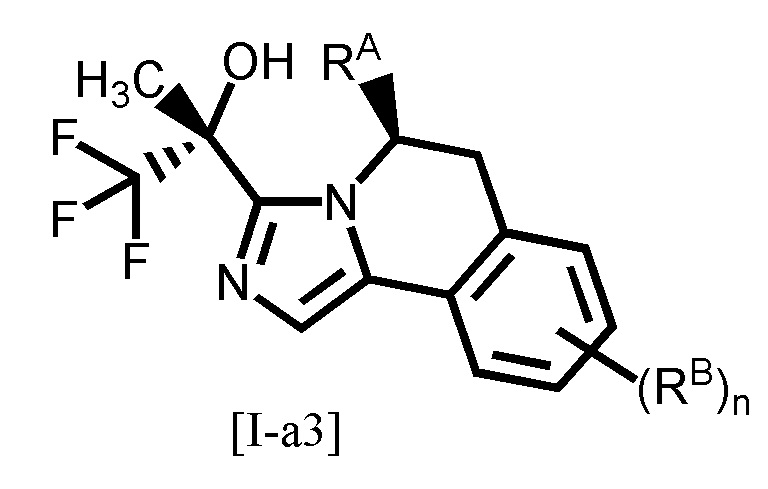 ,
,
[0091]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0092]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a4]:
[0093]
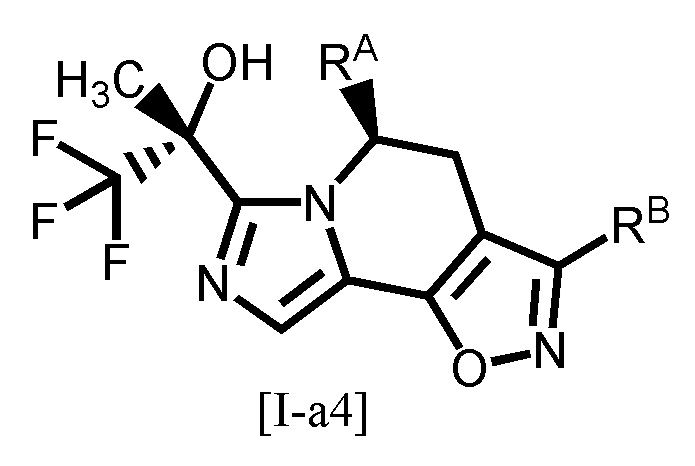 ,
,
[0094]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0095]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a5]:
[0096]
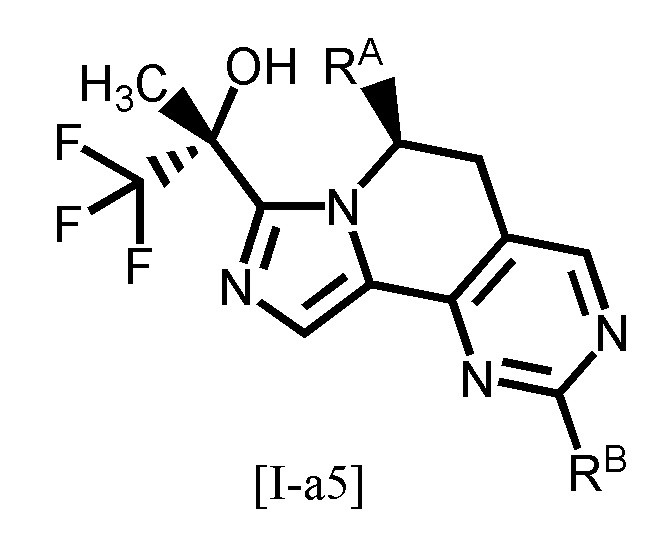 ,
,
[0097]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0098]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a6]:
[0099]
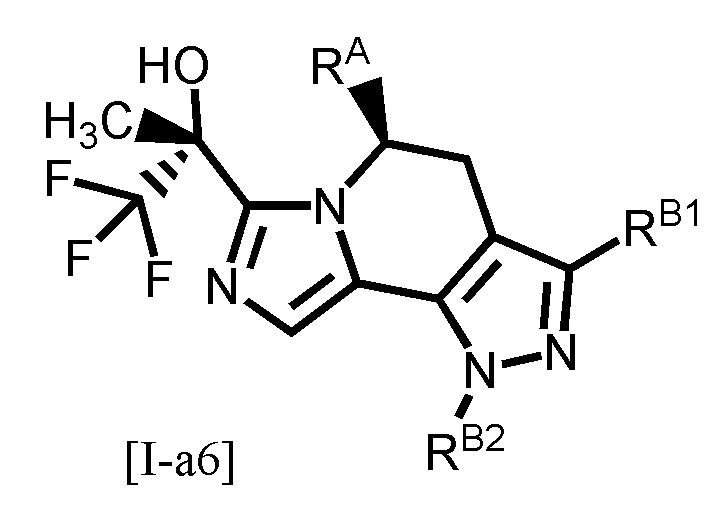 ,
,
[0100]
где RB1 и RB2 каждый независимо является таким, как определено для RB в вышеуказанной формуле [I-a]; и другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0101]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a7]:
[0102]
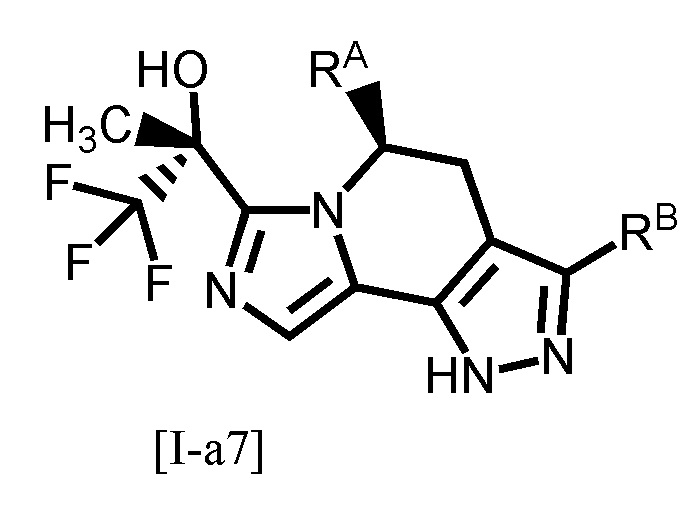 ,
,
[0103]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0104]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a8]:
[0105]
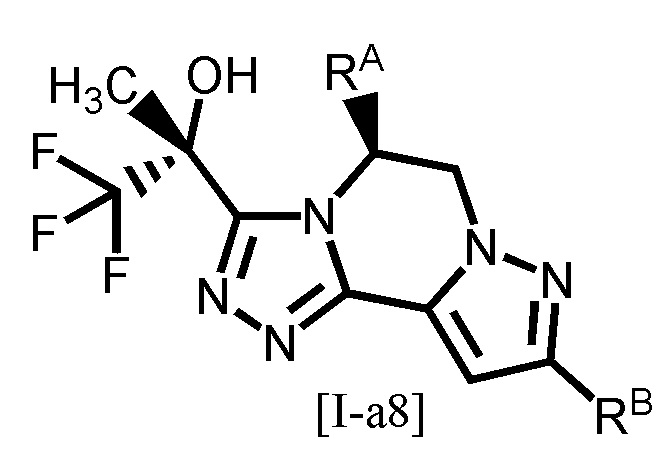 ,
,
[0106]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0107]
Одним из предпочтительных вариантов осуществления формулы [I-a] является соединение, представленное формулой [I-a10]:
[0108]
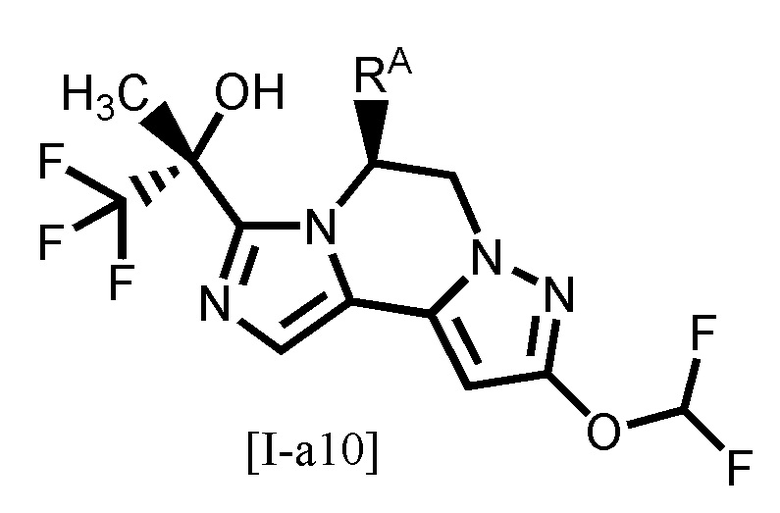 ,
,
[0109]
где символ является таким, как определено в вышеуказанной формуле [I-a].
[0110]
В вышеуказанных формулах [I-a] и [I-a1]-[I-a8] RA предпочтительно представляет собой метил.
В вышеуказанных формулах [I-a], [I-a1] и [I-a3] n предпочтительно равно 1.
[0111]
«Фармацевтически приемлемая соль» может представлять собой любую соль без избыточной токсичности, известную в данной области. В частности, можно упомянуть соли с неорганическими кислотами, соли с органическими кислотами, соли с неорганическими основаниями, соли с органическими основаниями и тому подобное. Различные формы фармацевтически приемлемых солей хорошо известны в данной области и, например, они описаны в следующих справочных документах:
(a) Berge et al., J. Pharm. Sci., 66, p1-19(1977),
(b) Stahl et al., «Handbook of Pharmaceutical Salts: Properties, Selection, and Use» (Wiley-VCH, Weinheim, Germany, 2002),
(c) Paulekuhn et al., J. Med. Chem., 50, p6665-6672 (2007).
Фармацевтически приемлемая соль соединения формулы [I-a] может быть получена путем проведения реакции соединения с неорганической кислотой, органической кислотой, неорганическим основанием или органическим основанием известным способом. Фармацевтически приемлемая соль соединения формулы [I-a] может быть образована из одной половины молекулы, одной молекулы или двух, или более, молекул кислоты или основания на молекулу соединения формулы [I-a].
[0112]
Примеры соли с неорганической кислотой включают соли с фтористоводородной кислотой, соляной кислотой, бромистоводородной кислотой, йодистоводородной кислотой, азотной кислотой, фосфорной кислотой и серной кислотой.
Примеры соли с органической кислотой включают соли с уксусной кислотой, адипиновой кислотой, альгиновой кислотой, 4-аминосалициловой кислотой, ангидрометиленлимонной кислотой, бензойной кислотой, бензолсульфокислотой, эдетатом кальция, камфорной кислотой, камфор-10-сульфокислотой, угольной кислотой, лимонной кислотой, эдетовой кислотой, этан-1,2-дисульфоновой кислотой, додецилсерной кислотой, этансульфоновой кислотой, фумаровой кислотой, глюкогептоновой кислотой, глюконовой кислотой, глюкуроновой кислотой, гликолиларсаниловой кислотой, гексилрезорциловой кислотой, гидроксинафтойной кислотой, 2-гидрокси-1-этансульфоновой кислотой, молочной кислотой, лактобионовой кислотой, яблочной кислотой, малеиновой кислотой, миндальной кислотой, метансульфокислотой, метилсерной кислотой, метилазотной кислотой, метиленбис(салициловой кислотой), галактаровой кислотой, нафталин-2-сульфокислотой, 2-нафтойной кислотой, 1,5-нафталиндисульфоновой кислотой, олеиновой кислотой, щавелевой кислотой, памовой кислотой, пантотеновой кислотой, пектиновой кислотой, пикриновой кислотой, пропионовой кислотой, полигалактуроновой кислотой, салициловой кислотой, стеариновой кислотой, янтарной кислотой, дубильной кислотой, винной кислотой, тейхоевой кислотой, тиоциановой кислотой, трифторуксусной кислотой, п-толуолсульфокислотой, ундекановой кислотой, аспарагиновой кислотой и глутаминовой кислотой.
[0113]
Примеры соли с неорганическим основанием включают соль с литием, натрием, калием, магнием, кальцием, барием, алюминием, цинком, висмутом или аммонием.
Примеры соли с органическим основанием включают соль с ареколином, бетаином, холином, клемизолом, этилендиамином, N-метилглюкамином, N-бензилфенетиламином, трис(гидроксиметил)метиламином, аргинином или лизином.
[0114]
Предпочтительным вариантом осуществления «фармацевтически приемлемой соли» является такая соль, как описано ниже.
Примеры соли с неорганической кислотой включают соли с соляной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой и бромистоводородной кислотой.
Примеры соли с органической кислотой включают соли с щавелевой кислотой, малеиновой кислотой, лимонной кислотой, фумаровой кислотой, молочной кислотой, яблочной кислотой, янтарной кислотой, винной кислотой, уксусной кислотой, трифторуксусной кислотой, бензойной кислотой, глюкуроновой кислотой, олеиновой кислотой, памовой кислотой, метансульфокислотой, бензолсульфокислотой, п-толуолсульфокислотой и 2-гидрокси-1-этансульфокислотой.
Примеры соли с неорганическим основанием включают соли с натрием, калием, кальцием, магнием и цинком.
Примеры соли с органическим основанием включают соли с трис(гидроксиметил)метиламином, N-метилглюкамином и лизином.
[0115]
Соединение формулы [I-a], или его фармацевтически приемлемая соль, может существовать в виде сольвата. Термин «сольват» относится к соединению формулы [I-a], или его фармацевтически приемлемой соли, с которым связана молекула растворителя, а также охватывает гидраты. Такие сольваты предпочтительно представляют собой фармацевтически приемлемые сольваты и включают, например, гидрат, сольват этанола, сольват диметилсульфоксида, и тому подобное, соединения формулы [I-a] или его фармацевтически приемлемой соли.
Конкретные примеры включают полугидрат, моногидрат, дигидрат или моно(этанол)сольват соединения формулы [I-a] или моногидрат гидрохлорида соединения формулы [I-a], дигидрат гидрохлорида того же соединения, и тому подобное. Такие сольваты могут быть получены обычными способами.
[0116]
Соединение формулы [I-a] может существовать в виде стереоизомера, который следует признать цис/транс-изомером. В этом случае соединение формулы [I-a] может существовать в виде цис-изомера, транс-изомера или смеси цис-изомера и транс-изомера.
Соединение формулы [I-a] может существовать в виде таутомера. В этом случае соединение формулы [I-a] может существовать в виде отдельного таутомера или смеси таутомеров.
Соединение формулы [I-a] может содержать один или более асимметричных атомов углерода. В этом случае соединение формулы [I-a] может существовать в виде отдельного энантиомера, отдельного диастереомера, смеси энантиомеров или смеси диастереомеров.
Соединение формулы [I-a] может существовать в виде атропоизомера. В этом случае соединение формулы [I-a] может существовать в виде отдельного атропоизомера или смеси атропоизомеров.
Соединение формулы [I-a] может одновременно иметь несколько структурных характеристик, которые определяют вышеупомянутые изомеры. Кроме того, соединение формулы [I-a] может содержать вышеуказанные изомеры в любом соотношении.
В отсутствие иных указаний, таких как аннотация и тому подобное, формулы, химические структуры и названия соединений, указанные в настоящем описании без указания их стереохимии, охватывают все вышеупомянутые изомеры, которые могут существовать.
[0117]
Диастереомерная смесь может быть разделена на каждый диастереомер обычными способами, такими как хроматография, кристаллизация и тому подобное. Кроме того, каждый диастереомер также может быть получен с использованием стереохимически единственного исходного материала или методом синтеза с использованием стереоселективной реакции.
[0118]
Энантиомерная смесь может быть разделена на каждый отдельный энантиомер методом, хорошо известным в данной области.
Например, смесь энантиомеров может быть подвергнута взаимодействию с практически чистым энантиомером, известным как хиральное вспомогательное вещество, с образованием смеси диастереомеров, которые затем могут быть выделены в диастереомер с повышенным соотношением изомеров или в практически чистый одиночный диастереомер общепринятым методом, таким как фракционная кристаллизация или хроматография. Добавленное хиральное вспомогательное вещество может быть удалено из выделенного диастереомера реакцией расщепления, с получением желаемого энантиомера.
Кроме того, смесь энантиомеров соединения также может быть разделена непосредственно методом хроматографии с использованием хиральной твердой фазы, хорошо известной в данной области. Альтернативно, один из энантиомеров также может быть получен с использованием практически чистого оптически активного исходного материала или стереоселективным синтезом (асимметричная индукция) прохирального промежуточного соединения с использованием хирального вспомогательного вещества или асимметричного катализатора.
[0119]
Абсолютную стерическую конфигурацию можно определять на основании рентгеноструктурного анализа полученного кристаллического продукта или промежуточного продукта. В этом случае при необходимости можно использовать полученный кристаллический продукт или промежуточный продукт, дериватизированный реагентом, имеющим асимметричный центр с известной стерической конфигурацией.
[0120]
Соединение формулы [I-a] может быть помечено изотопом (2H, 3H, 14C, 35S и тому подобным).
[0121]
Соединение формулы [I-a], или его фармацевтически приемлемая соль, предпочтительно является в значительной степени очищенным соединением формулы [I-a], или его фармацевтически приемлемой солью. Также предпочтительно, оно представляет собой соединение формулы [I-a], или его фармацевтически приемлемую соль, очищенное до степени чистоты не менее 80%.
[0122]
Фармацевтическая композиция по настоящему изобретению может быть получена путем соответствующего смешивания подходящего количества соединения формулы [I-a], или его фармацевтически приемлемой соли, с фармацевтически приемлемым носителем по меньшей мере одного вида способом, известным в области изготовления фармацевтических препаратов. Содержание соединения формулы [I-a], или его фармацевтически приемлемой соли, в фармацевтической композиции варьируется в зависимости от лекарственной формы, дозы и тому подобного. Оно составляет, например, от 0,1 до 100 масс% в композиции.
[0123]
Лекарственная форма соединения формулы [I-a], или его фармацевтически приемлемой соли, включает пероральный препарат, такой как таблетка, капсула, гранула, порошок, пастилка, сироп, эмульсия и суспензия, или парентеральный препарат, такой как наружный препарат, суппозиторий, инъекция, глазные капли, назальный препарат и легочный препарат.
[0124]
Примеры «фармацевтически приемлемого носителя» включают различные органические или неорганические вещества-носители, обычно используемые в качестве материалов для изготовления, и включают эксципиент, разрыхлитель, связывающее вещество, флюидизирующее вещество, смазывающее вещество, и тому подобное, для твердых препаратов, и растворитель, солюбилизирующее средство, суспендирующее средство, изотоническое средство, буферное средство, смягчающее средство, и тому подобное, для жидких препаратов, а также основу, эмульгатор, смачивающее средство, стабилизатор, стабилизирующее средство, диспергирующее средство, пластификатор, регулятор pH, усилитель абсорбции, гелеобразующее средство, консервант, наполнитель, растворитель, солюбилизирующие средства, суспендирующее средство, и тому подобное, для полутвердых препаратов. Кроме того, при необходимости можно также использовать добавки, такие как консервант, антиоксидант, краситель, подсластитель и тому подобное.
[0125]
Примеры «эксципиента» включают лактозу, сахарозу, D-маннит, D-сорбит, кукурузный крахмал, декстрин, кристаллическую целлюлозу, кристаллическую целлюлозу, кармеллозу, кармеллозу кальция, карбоксиметилкрахмал натрия, низкозамещенную гидроксипропилцеллюлозу, гуммиарабик и тому подобное.
Примеры «разрыхлителя» включают кармеллозу, кармеллозу кальция, кармеллозу натрия, карбоксиметилкрахмал натрия, кроскармеллозу натрия, кросповидон, низкозамещенную гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, кристаллическую целлюлозу и тому подобное.
Примеры «связывающего вещества» включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон, кристаллическую целлюлозу, сахарозу, декстрин, крахмал, желатин, кармеллозу натрия, гуммиарабик и тому подобное.
Примеры «флюидизирующего вещества» включают легкую безводную кремниевую кислоту, стеарат магния и тому подобное.
Примеры «смазывающего вещества» включают стеарат магния, стеарат кальция, тальк и тому подобное.
Примеры «растворителя» включают очищенную воду, этанол, пропиленгликоль, макрогол, кунжутное масло, кукурузное масло, оливковое масло и тому подобное.
Примеры «солюбилизирующих средств» включают пропиленгликоль, D-маннит, бензилбензоат, этанол, триэтаноламин, карбонат натрия, цитрат натрия и тому подобное.
Примеры «суспендирующего средства» включают хлорид бензалкония, кармеллозу, гидроксипропилцеллюлозу, пропиленгликоль, повидон, метилцеллюлозу, моностеарат глицерина и тому подобное.
Примеры «изотонического средства» включают глюкозу, D-сорбит, хлорид натрия, D-маннит и тому подобное.
Примеры «буферного средства» включают гидрофосфат натрия, ацетат натрия, карбонат натрия, цитрат натрия и тому подобное.
Примеры «смягчающего средства» включают бензиловый спирт и тому подобное.
Примеры «основы» включают воду, животные и растительные масла (оливковое масло, кукурузное масло, арахисовое масло, кунжутное масло, касторовое масло и тому подобное), низшие спирты (этанол, пропанол, пропиленгликоль, 1,3-бутиленгликоль, фенол и тому подобное), высшую жирную кислоту и ее сложный эфир, воски, высший спирт, многоатомный спирт, углеводороды (белый вазелин, жидкий парафин, парафин и тому подобное), гидрофильный вазелин, очищенный ланолин, поглощающую воду мазь, водный ланолин, гидрофильную мазь, крахмал, пуллулан, гуммиарабик, трагакантовую камедь, желатин, декстран, производные целлюлозы (метилцеллюлозу, карбоксиметилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу и тому подобное), синтетический полимер (карбоксивиниловый полимер, полиакрилат натрия, поли(виниловый спирт), поливинилпирролидон и тому подобное), пропиленгликоль, макрогол (макрогол 200-600 и тому подобное), а также сочетание двух или более из них.
Примеры «консерванта» включают этилпараоксибензоат, хлорбутанол, бензиловый спирт, дегидроацетат натрия, сорбиновую кислоту и тому подобное.
Примеры «антиоксиданта» включают сульфит натрия, аскорбиновую кислоту и тому подобное.
Примеры «красителя» включают пищевые красители (например, пищевой краситель красный № 2 или 3, пищевой краситель желтый № 4 или 5 и так далее), β-каротин и тому подобное.
Примеры «подсластителя» включают сахарин натрия, глицирризинат дикалия, аспартам и тому подобное.
[0126]
Фармацевтическую композицию по настоящему изобретению можно вводить перорально или парентерально (местное, ректальное, внутривенное, внутримышечное, подкожное введение и тому подобное) млекопитающим, отличным от человека (таким как мышь, крыса, хомяк, морская свинка, кролик, кошка, собака, свинья, крупный рогатый скот, лошадь, овца, обезьяна и так далее), и человеку. Доза варьируется в зависимости от субъекта, которому вводят препарат, заболевания, симптома, лекарственной формы, пути введения и тому подобного. Например, суточная доза для перорального введения взрослому пациенту, как правило, находится в диапазоне примерно от 0,01 мг до 1 г в расчете на соединение формулы [I-a] в качестве активного ингредиента. Это количество можно вводить в одной или нескольких порциях.
[0127]
Соединение формулы [I-a], или его фармацевтически приемлемая соль, обладает ингибирующим действием на ПДГК и может быть использовано для лечения и/или профилактики различных заболеваний или состояний, которые, как ожидается, будут ослаблены в результате контроля активности ПДГК. Примеры различных заболеваний или состояний, которые, как ожидается, будут ослаблены в результате контроля активности ПДГК, включают такие заболевания, как диабет (диабет 1-го типа, диабет 2-го типа), синдром резистентности к инсулину, метаболический синдром, гипергликемия, гиперлактацидемия, осложнения диабета (диабетическая невропатия, диабетическая ретинопатия, диабетическая нефропатия, катаракта), сердечная недостаточность (острая сердечная недостаточность, хроническая сердечная недостаточность), кардиомиопатия, ишемия миокарда, инфаркт миокарда, стенокардия, дислипидемия, атеросклероз, заболевания периферических артерий, перемежающаяся хромота, хронические обструктивные болезни легких, ишемия головного мозга, мозговая апоплексия, митохондриальная болезнь, митохондриальная энцефаломиопатия, рак, легочная гипертензия (легочная артериальная гипертензия), болезнь Альцгеймера, сосудистая деменция (сосудистая деменция с поражением крупных или мелких сосудов), глаукома, диабетическая ретинопатия, ретинопатия недоношенных, окклюзия вен сетчатки, ишемическая оптическая нейропатия, хроническая почечная недостаточность и тому подобное.
[0128]
Симптомы болезни Альцгеймера включают снижение когнитивной функции, психологические симптомы и поведенческие расстройства, и тому подобное.
[0129]
«Ингибирование ПДГК» означает устранение или ослабление активности ПДГК путем ингибирования ее функции. Например, это означает ингибирование функции ПДГК на основании условий в приведенном ниже экспериментальном примере 1. Для «ингибирования ПДГК» предпочтительно ингибируют человеческую ПДГК. Для «ингибирования ПДГК» предпочтительно «ингибируют ПДГК2».
«Ингибитор ПДГК» означает вещество, которое связывается с ПДГК и ингибирует функцию ПДГК. В качестве «ингибитора ПДГК» предпочтительным является «ингибитор ПДГК человека». В качестве «ингибитора ПДГК» предпочтительным является «ингибитор ПДГК2».
[0130]
В настоящем описании «лечение» также включает улучшение симптомов, предотвращение тяжести, поддержание ремиссии, предотвращение обострения и, кроме того, предотвращение рецидива.
В настоящем описании термин «предотвращение», или «профилактика», означает подавление возникновения симптомов.
[0131]
В настоящем описании представление предпочтительных вариантов осуществления и вариантов соединения, способа, применения и композиции по настоящему изобретению также включает сочетания предпочтительных вариантов осуществления и вариантов при условии, что они могут сочетаться и не противоречат друг другу.
[0132]
Далее описаны способы получения соединения формулы [I-a] или его фармацевтически приемлемой соли. Однако способ получения соединения формулы [I-a], или его фармацевтически приемлемой соли, не ограничивается такими способами получения.
Соединение, полученное на каждой стадии, может быть выделено или очищено при необходимости обычными способами, такими как перегонка, перекристаллизация, колоночная хроматография и тому подобное. В некоторых случаях следующий этап можно проводить без выделения или очистки. Когда реакция, которую необходимо проводить на каждой стадии, представляет собой безводную реакцию, ее предпочтительно проводят в атмосфере инертного газа аргона, азота и тому подобного.
[0133]
[Способ получения 1]
Соединение формулы [I-a1] может быть получено способом получения 1, показанным на следующей схеме.
[0134]
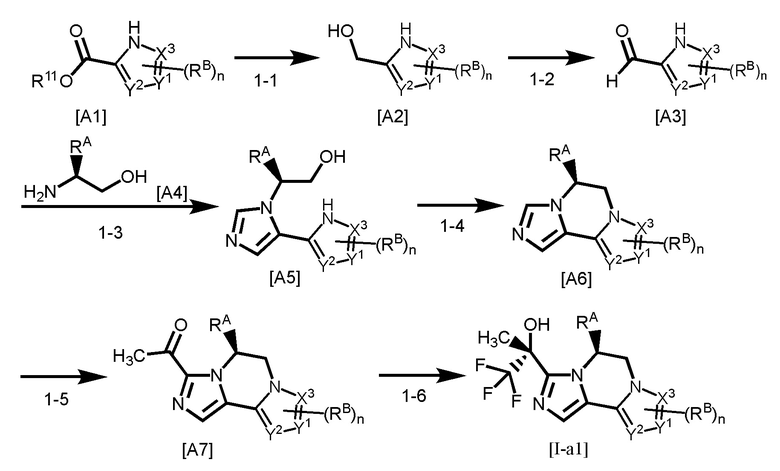
[0135]
где R11 представляет собой C1-4 алкил; и каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0136]
Стадия 1-1
Соединение [A2] может быть получено путем восстановления сложноэфирной группы соединения [A1]. Например, соединение [A2] может быть получено путем проведения реакции соединения [A1] с восстановителем в растворителе при температуре от -40°C до комнатной температуры.
Примеры восстановителя включают алюмогидрид лития, гидрид диизобутилалюминия и боргидрид лития.
Примеры растворителя включают тетрагидрофуран, диэтиловый эфир и циклопентилметиловый эфир.
Соединение [А1] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0137]
Стадия 1-2
Соединение [A3] может быть получено путем окисления гидроксильной группы соединения [A2]. Например, соединение [A3] может быть получено путем проведения реакции соединения [A2] с окислителем в растворителе при температуре от ледяной до комнатной.
Примеры окислителя включают диоксид марганца, периодинан Десса-Мартина и комплекс триоксид серы-пиридин.
Примеры растворителя включают тетрагидрофуран, диметоксиэтан, толуол, диметилсульфоксид, хлороформ и дихлорметан.
[0138]
Стадия 1-3
Соединение [А5] может быть получено путем проведения реакции иминирования соединения [А3] и соединения [А4], и реакции циклизации с использованием п-толуолсульфонилметилизоцианида. Например, реакцию иминирования соединения [А3] и соединения [А4] проводят в растворителе при температуре от комнатной до 60°С. Затем проводят реакцию полученного продукта с п-толуолсульфонилметилизоцианидом в растворителе в присутствии основания при температуре от ледяной до комнатной, с получением соединения [А5].
Примеры растворителя реакции иминирования включают метанол и диметилформамид.
Примеры основания включают карбонат калия.
Примеры растворителя реакции циклизации включают диметоксиэтан.
Соединения [A3] и [A4] могут быть коммерчески доступными продуктами или могут быть получены соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0139]
Стадия 1-4
Соединение [А6] может быть получено путем проведения внутримолекулярной реакции Мицунобу соединения [А5]. Например, соединение [А6] может быть получено путем проведения реакции соединения [А5] с фосфином и диэфиром азодикарбоновой кислоты в растворителе при температуре от комнатной до 100°С.
Примеры фосфина включают триоктилфосфин, трибутилфосфин и трифенилфосфин.
Примеры диэфира азодикарбоновой кислоты включают диизопропилазодикарбоксилат и ди-трет-бутилазодикарбоксилат.
Примеры растворителя включают толуол, тетрагидрофуран и 2-метилтетрагидрофуран.
[0140]
Стадия 1-5
Соединение [A7] может быть получено путем проведения реакции соединения [A6] с N-метокси-N-метилацетамидом. Например, соединение [A7] может быть получено путем проведения реакции соединения [A6] с N-метокси-N-метилацетамидом в растворителе при температуре от -78°C до комнатной температуры в присутствии основания.
Примеры основания включают н-бутиллитий и диизопропиламид лития.
Примеры растворителя включают циклопентилметиловый эфир, тетрагидрофуран и толуол.
[0141]
Стадия 1-6
Соединение [I-a1] может быть получено путем проведения реакции соединения [A7] с (трифторметил)триметилсиланом. Например, соединение [I-a1] может быть получено путем проведения реакции соединения [A7] с (трифторметил)триметилсиланом в растворителе в присутствии добавки при температуре от ледяной до комнатной.
Примеры добавки включают фторид тетра-н-бутиламмония, ацетат лития, карбонат калия и фторид цезия.
Примеры растворителя включают тетрагидрофуран, диметилформамид и диметилацетамид.
Группа RA соединения [A7] становится стерическим препятствием, и реакция протекает диастереоселективным образом. О стерической конфигурации соединения [I-a1] можно судить по механизму реакции и подтверждать ее рентгеноструктурным анализом.
[0142]
[Способ получения 2]
Соединение формулы [I-a2] может быть получено способом получения 2, показанным на следующей схеме.
[0143]
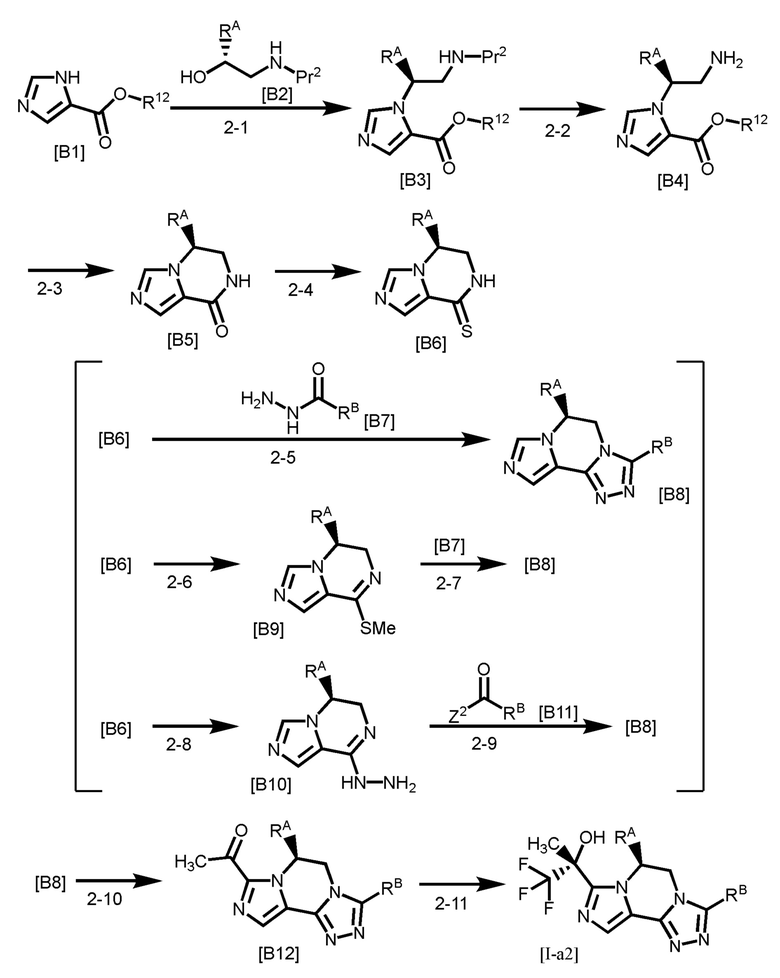
[0144]
где R12 представляет собой C1-4 алкил;
Pr2 представляет собой защитную группу для аминогруппы, такую как трет-бутоксикарбонил и тому подобное;
Z2 представляет собой RBC(O)O- или хлор; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0145]
Стадия 2-1
Соединение [B3] может быть получено путем проведения реакции Мицунобу соединения [B1] и соединения [B2]. Например, соединение [B3] может быть получено путем проведения реакции соединения [B1] с соединением [B2], фосфином и диэфиром азодикарбоновой кислоты в растворителе при температуре от комнатной до 100°C.
Примеры фосфина включают триоктилфосфин, трибутилфосфин и трифенилфосфин.
Примеры диэфира азодикарбоновой кислоты включают диизопропилазодикарбоксилат и ди-трет-бутилазодикарбоксилат.
Примеры растворителя включают толуол и тетрагидрофуран.
Соединение [B1] и соединение [B2] могут быть коммерчески доступными продуктами или могут быть получены соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0146]
Стадия 2-2
Соединение [B4] может быть получено путем снятия защиты с аминогруппы соединения [B3]. Например, когда Pr2 представляет собой трет-бутоксикарбонил, соединение [B4] может быть получено путем обработки соединения [B3] кислотой в растворителе при температуре от ледяной до комнатной. Соединение [B4] может быть получено в виде соли с кислотой, используемой в этой реакции.
Примеры кислоты включают трифторуксусную кислоту и соляную кислоту.
Примеры растворителя включают тетрагидрофуран и этилацетат.
[0147]
Стадия 2-3
Соединение [B5] может быть получено путем лактамизации соединения [B4]. Например, соединение [B5] может быть получено путем проведения реакции соединения [B4] с основанием в растворителе при температуре от ледяной до комнатной.
Примеры основания включают гидрокарбонат натрия.
Примеры растворителя включают метанол и воду.
[0148]
Стадия 2-4
Соединение [B6] может быть получено путем проведения реакции соединения [B5] с сернистым реагентом. Например, соединение [B6] может быть получено путем проведения реакции соединения [B5] с сернистым реагентом в растворителе при температуре от комнатной до 110°C.
Примеры сернистого реагента включают реагент Лавессона (2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид).
Примеры растворителя включают толуол и пиридин.
[0149]
Стадия 2-5
Соединение [B8] может быть получено путем проведения реакции циклизации соединения [B6] и соединения [B7]. Например, соединение [B8] может быть получено путем проведения реакции соединения [B6] с соединением [B7] в растворителе при температуре от 100°C до 200°C. При необходимости можно также использовать микроволновую печь.
Примеры растворителя включают н-бутанол и н-метилпирролидон.
Соединение [B7] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0150]
Стадии 2-6 и 2-7
Соединение [B9] получают путем метилирования соединения [B6], затем соединение [B8] также может быть получено путем проведения реакции соединения [B9] с соединением [B7]. Метилирование соединения [B6] можно выполнять, например, путем проведения реакции соединения [B6] с метилирующим реагентом в растворителе при температуре от ледяной до комнатной. Соединение [B9] также может быть получено в виде соли, такой как йодистоводородная соль или тому подобное.
Примеры метилирующего реагента включают йодистый метил.
Примеры растворителя включают диметилформамид и ацетон.
Реакцию соединения [B9] и соединения [B7] можно проводить, например, методом, аналогичным методу на стадии 2-5.
Соединение [B7] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0151]
Стадии 2-8 и 2-9
Соединение [B10] получают путем проведения реакции соединения [B6] с гидразином, затем соединение [B8] также может быть получено путем проведения реакции соединения [B10] с соединением [B11]. Реакцию соединения [B6] и гидразина можно выполнять, например, путем проведения реакции соединения [B6] с гидразином в растворителе при температуре от комнатной до 80°C.
Примеры растворителя включают этанол и изопропанол.
Реакцию соединения [B10] и соединения [B11] можно выполнять, например, путем проведения реакции соединения [B10] с соединением [B11] в растворителе в присутствии кислоты при температуре от ледяной до комнатной.
Примеры кислоты включают трифторуксусную кислоту.
Примеры растворителя включают хлороформ.
Соединение [B11] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0152]
Стадия 2-10
Соединение [B12] может быть получено путем проведения реакции соединения [B8] с N-метокси-N-метилацетамидом. Например, соединение [B12] может быть получено методом, аналогичным методу на стадии 1-5.
[0153]
Стадия 2-11
Соединение [I-a2] может быть получено путем проведения реакции соединения [B12] с (трифторметил)триметилсиланом. Например, соединение [I-a2] может быть получено методом, аналогичным методу на стадии 1-6.
[0154]
[Способ получения 3]
Соединение формулы [I-a3] может быть получено способом получения 3, показанным на следующей схеме.
[0155]
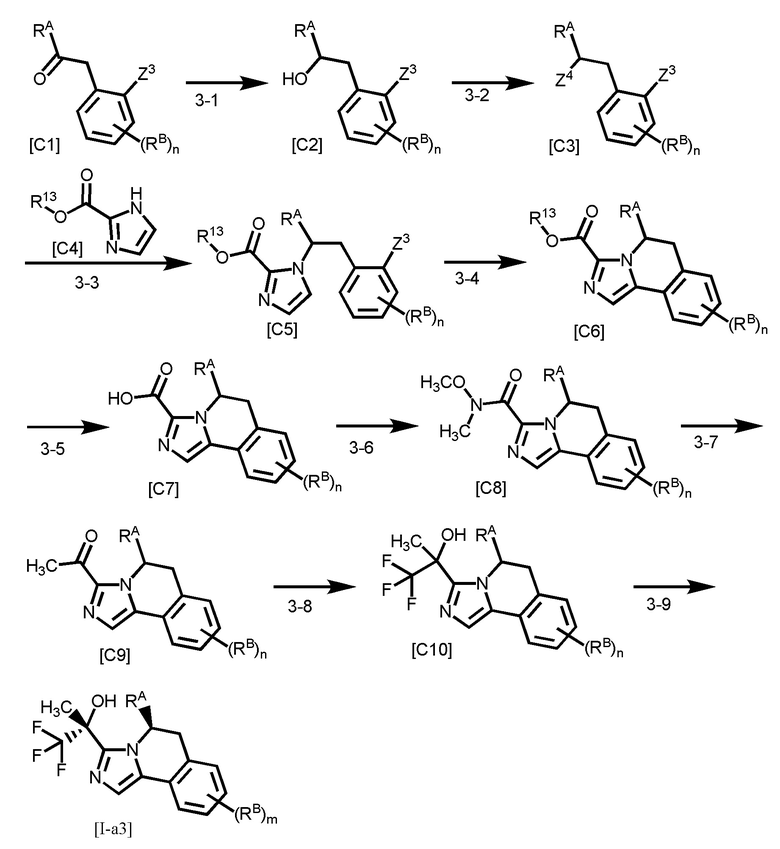
[0156]
где R13 представляет собой C1-4 алкил;
Z3 представляет собой уходящую группу, такую как бром, иод, трифторметансульфонилокси или тому подобное;
Z4 представляет собой уходящую группу, такую как хлор, бром, метансульфонилокси или тому подобное; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0157]
Стадия 3-1
Соединение [C2] может быть получено путем восстановления карбонильной группы соединения [C1]. Например, соединение [C2] может быть получено путем проведения реакции соединения [C1] с восстановителем в растворителе при температуре от ледяной до комнатной.
Примеры восстановителя включают боргидрид натрия.
Примеры растворителя включают тетрагидрофуран и метанол.
Соединение [C1] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0158]
Стадия 3-2
Соединение [С3] может быть получено путем превращения гидроксильной группы соединения [С2] в уходящую группу. Например, когда Z4 представляет собой метансульфонилокси, соединение [C3] может быть получено путем проведения реакции соединения [C2] с метансульфоновым ангидридом в растворителе в присутствии основания при охлаждении льдом.
Примеры основания включают триэтиламин.
Примеры растворителя включают тетрагидрофуран, хлороформ и дихлорметан.
[0159]
Стадия 3-3
Соединение [C5] может быть получено путем проведения реакции соединения [C3] с соединением [C4]. Например, соединение [C5] может быть получено путем проведения реакции соединения [C3] с соединением [C4] в растворителе в присутствии основания при температуре от комнатной до 80°C.
Примеры основания включают карбонат цезия.
Примеры растворителя включают диметилформамид.
Соединение [C4] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0160]
Стадия 3-4
Соединение [C6] может быть получено путем проведения реакции внутримолекулярной циклизации соединения [C5]. Например, соединение [C6] может быть получено путем проведения реакции соединения [C5] в растворителе в присутствии металлического катализатора, лиганда и основания при 120°C.
Примеры металлического катализатора включают ацетат палладия (II).
Примеры лиганда включают ди-1-адамантил-н-бутилфосфин и 2-дициклогексилфосфино-2’,6’-диметоксибифенил.
Примеры основания включают карбонат калия.
Примеры растворителя включают диметилацетамид.
[0161]
Стадия 3-5
Соединение [C7] может быть получено путем гидролиза сложноэфирной группы соединения [C6]. Например, соединение [C7] может быть получено путем обработки соединения [C6] щелочью в растворителе при температуре от ледяной до 60°C.
Примеры щелочи включают гидроксид лития и гидроксид натрия.
Примеры растворителя включают метанол, этанол и воду.
[0162]
Стадия 3-6
Соединение [C8] может быть получено путем проведения реакции амидирования соединения [C7] и N, O-диметилгидроксиламина. Например, соединение [C8] может быть получено путем проведения реакции соединения [C7] с N, O-диметилгидроксиламином в растворителе в присутствии основания и конденсирующего реагента при температуре от ледяной до комнатной.
Примеры основания включают диизопропилэтиламин и триэтиламин.
Примеры конденсирующего реагента включают HATU.
Примеры растворителя включают диметилформамид.
[0163]
Стадия 3-7
Соединение [C9] может быть получено путем проведения реакции соединения [C8] с метилмагнийгалогенидом. Например, соединение [C9] может быть получено путем проведения реакции соединения [C8] с метилмагнийгалогенидом в растворителе при температуре от 0°C до комнатной температуры.
Примеры метилмагнийгалогенида включают метилмагнийбромид.
Примеры растворителя включают тетрагидрофуран и диэтиловый эфир.
[0164]
Стадия 3-8
Соединение [C10] может быть получено путем проведения реакции соединения [C9] с (трифторметил)триметилсиланом. Например, соединение [C10] может быть получено методом, аналогичным методу на стадии 1-6.
[0165]
Стадия 3-9
Соединение [I-a3] может быть получено путем очистки соединения [C10] методом хиральной колоночной хроматографии. Стерическую конфигурацию соединения [I-a3] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0166]
[Способ получения 4]
Соединение формулы [I-a4] может быть получено способом получения 4, показанным на следующей схеме.
[0167]
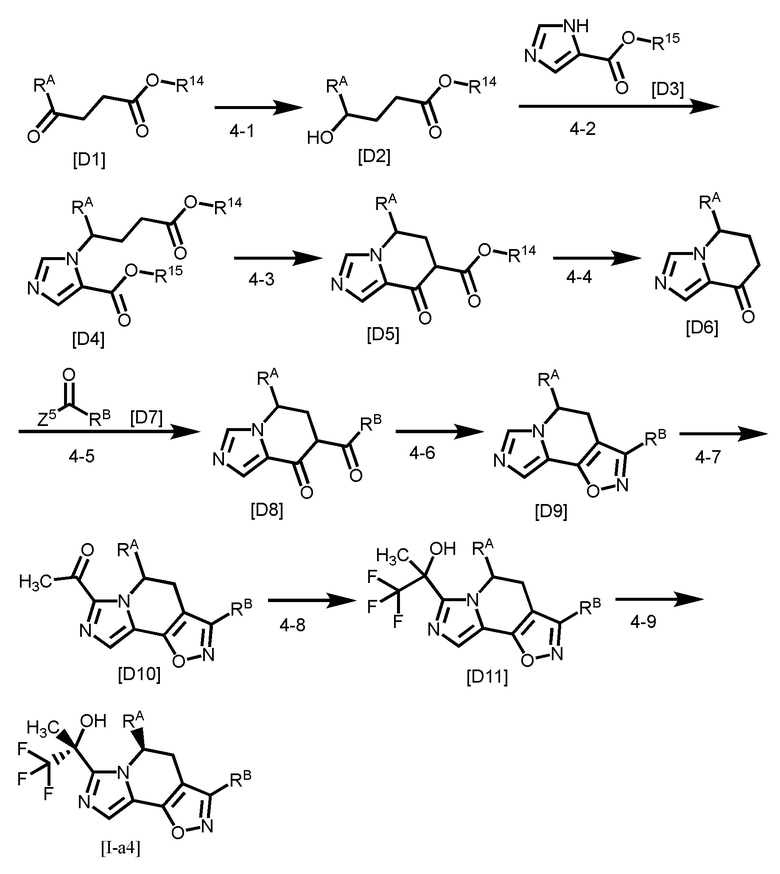
[0168]
где R14 и R15 каждый независимо представляет собой C1-4 алкил;
Z5 представляет собой C1-4 алкокси или хлор; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0169]
Стадия 4-1
Соединение [D2] может быть получено путем восстановления карбонильной группы соединения [D1]. Например, соединение [D2] может быть получено путем проведения реакции соединения [D1] с восстановителем в растворителе при температуре от -78°C до комнатной температуры.
Примеры восстановителя включают боргидрид натрия.
Примеры растворителя включают метанол и тетрагидрофуран.
Соединение [D1] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0170]
Стадия 4-2
Соединение [D4] может быть получено путем проведения реакции Мицунобу соединения [D2] и соединения [D3]. Например, соединение [D4] может быть получено методом, аналогичным методу на стадии 2-1.
Соединение [D3] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0171]
Стадия 4-3
Соединение [D5] может быть получено путем внутримолекулярной конденсации Кляйзена соединения [D4]. Например, соединение [D5] может быть получено путем обработки соединения [D4] основанием в растворителе при температуре от комнатной до 110°C.
Примеры основания включают трет-бутоксид калия.
Примеры растворителя включают толуол и тетрагидрофуран.
[0172]
Стадия 4-4
Соединение [D6] может быть получено путем декарбоксилирования сложноэфирной группы соединения [D5]. Например, соединение [D6] может быть получено путем обработки соединения [D5] кислотой или хлоридом натрия в растворителе при температуре от 100°C до 160°C.
Примеры кислоты включают соляную кислоту.
Примеры растворителя включают воду и диметилсульфоксид.
[0173]
Стадии 4-5 и 4-6
Соединение [D9] может быть получено путем проведения реакции соединения [D6] с соединением [D7], с получением соединения [D8], и проведения реакции циклизации соединения [D8] и гидроксиламина. Соединение [D8] может быть получено, например, путем проведения реакции соединения [D6] с соединением [D7] в растворителе в присутствии основания при температуре от 0°C до 70°C.
Примеры основания включают гидрид натрия и бис(триметилсилил)амид лития.
Примеры растворителя включают тетрагидрофуран.
Соединение [D7] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
Соединение [D9] может быть получено, например, путем проведения реакции соединения [D8] с гидроксиламином в растворителе при температуре от 70°C до 110°C, и при необходимости можно использовать кислоту.
Примеры кислоты включают концентрированную серную кислоту.
Примеры растворителя включают уксусную кислоту и метанол.
[0174]
Стадия 4-7
Соединение [D10] может быть получено путем проведения реакции соединения [D9] с N-метокси-N-метилацетамидом. Например, соединение [D10] может быть получено методом, аналогичным методу на стадии 1-5.
[0175]
Стадия 4-8
Соединение [D11] может быть получено путем проведения реакции соединения [D10] с (трифторметил)триметилсиланом. Например, соединение [D11] может быть получено методом, аналогичным методу на стадии 1-6.
[0176]
Стадия 4-9
Соединение [I-a4] может быть получено путем очистки соединения [D11] методом хиральной колоночной хроматографии. Стерическую конфигурацию соединения [I-a4] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0177]
[Способ получения 4-a]
На Стадии 4-6 способа получения 4 соединение [D12], представленное формулой:
[0178]
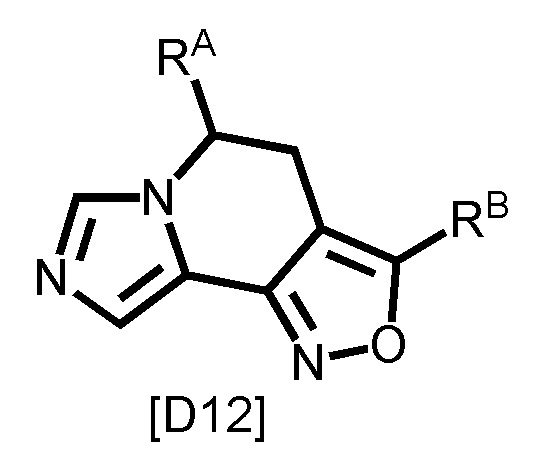
[0179]
может быть получено параллельно с соединением [D9]. Соединение [I-a16], представленное формулой:
[0180]
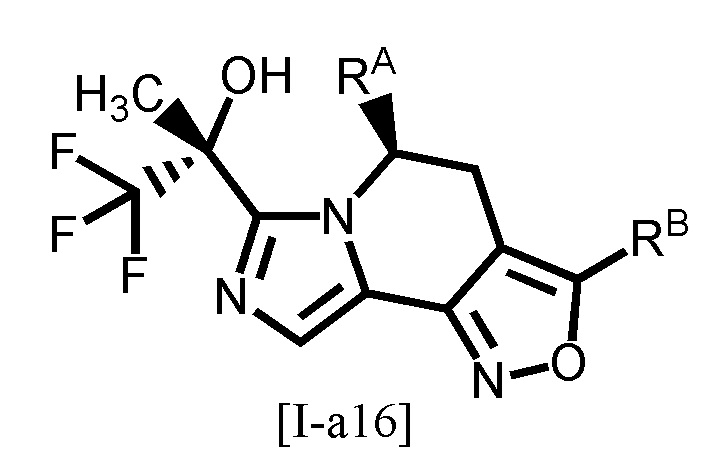
[0181]
может быть получено путем проведения реакций, аналогичных реакциям, проводимым на стадиях от 4-7 до 4-9, и с использованием соединения [D12]. Стерическую конфигурацию соединения [I-a16] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0182]
[Способ получения 5]
Соединение формулы [I-a5] может быть получено способом получения 5, показанным на следующей схеме.
[0183]
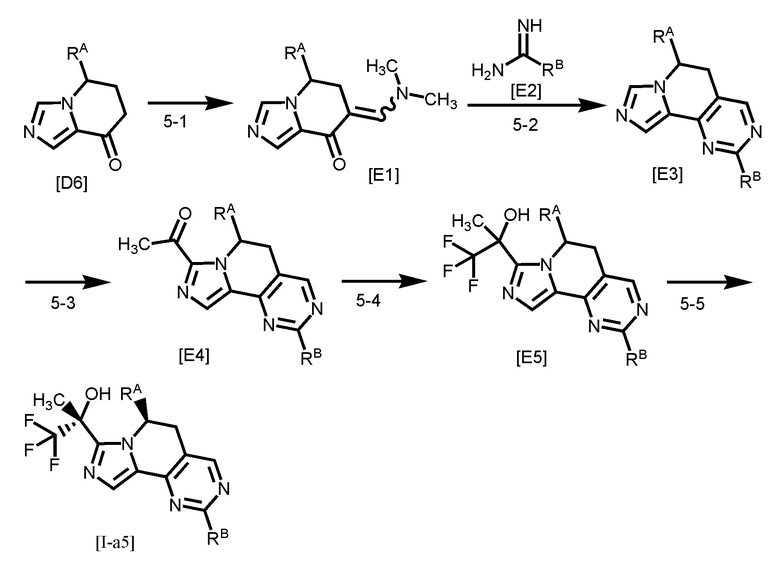
[0184]
где каждый символ является таким, как определено в вышеуказанной формуле [I-a].
[0185]
Стадия 5-1
Соединение [E1] может быть получено путем проведения реакции соединения [D6] с трет-бутоксибис(диметиламино)метаном. Например, соединение [E1] может быть получено путем проведения реакции соединения [D6] с трет-бутоксибис(диметиламино)метаном в растворителе при температуре от комнатной до 110°C.
Примеры растворителя включают диоксан.
[0186]
Стадия 5-2
Соединение [E3] может быть получено путем проведения реакции циклизации соединения [E1] и соединения [E2]. Например, соединение [E3] может быть получено путем проведения реакции соединения [E1] с соединением [E2] в растворителе в присутствии основания при температуре от комнатной до 78°C.
Примеры основания включают этоксид натрия.
Примеры растворителя включают этанол.
Соединение [E2] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0187]
Стадия 5-3
Соединение [E4] может быть получено путем проведения реакции соединения [E3] с N-метокси-N-метилацетамидом. Например, соединение [E4] может быть получено методом, аналогичным методу на стадии 1-5.
[0188]
Стадия 5-4
Соединение [E5] может быть получено путем проведения реакции соединения [E4] с (трифторметил)триметилсиланом. Например, соединение [E5] может быть получено методом, аналогичным методу на стадии 1-6.
[0189]
Стадия 5-5
Соединение [I-a5] может быть получено путем очистки соединения [E5] методом хиральной колоночной хроматографии. Стерическую конфигурацию соединения [I-a5] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0190]
[Способ получения 6]
Соединение формулы [I-a6] может быть получено способом получения 6, показанным на следующей схеме.
[0191]
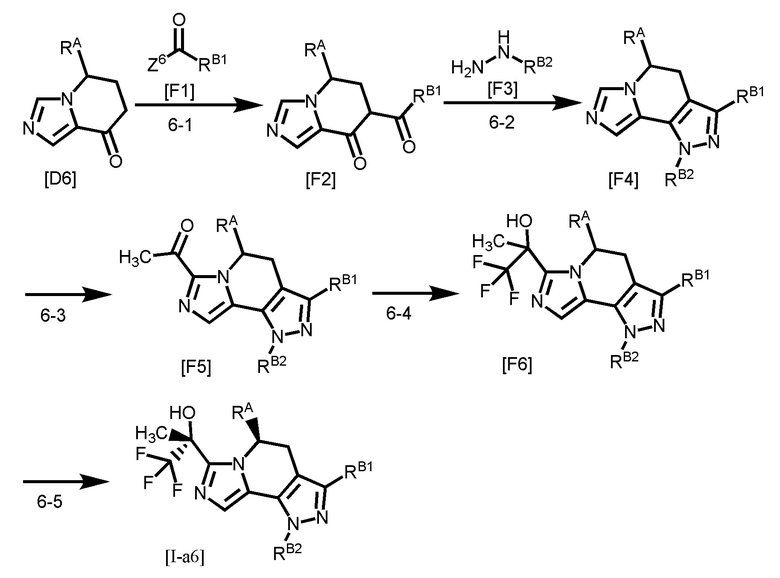
[0192]
где RB1 и RB2 каждый независимо является таким, как определено для RB в вышеуказанной формуле [I-a];
Z6 представляет собой C1-4 алкокси или хлор; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0193]
Стадия 6-1
Соединение [F2] может быть получено путем проведения реакции соединения [D6] с соединением [F1]. Например, соединение [F2] может быть получено методом, аналогичным методу на стадии 4-5.
Соединение [F1] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0194]
Стадия 6-2
Соединение [F4] может быть получено путем проведения реакции циклизации пиразола соединения [F2] и соединения [F3]. Например, соединение [F4] может быть получено путем проведения реакции соединения [F2] с соединением [F3] в растворителе при температуре от комнатной до 100°C.
Примеры растворителя включают уксусную кислоту и этанол.
Соединение [F3] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0195]
Стадия 6-3
Соединение [F5] может быть получено путем проведения реакции соединения [F4] с N-метокси-N-метилацетамидом. Например, соединение [F5] может быть получено методом, аналогичным методу на стадии 1-5.
[0196]
Стадия 6-4
Соединение [F6] может быть получено путем проведения реакции соединения [F5] с (трифторметил)триметилсиланом. Например, соединение [F6] может быть получено методом, аналогичным методу на стадии 1-6.
[0197]
Стадия 6-5
Соединение [I-a6] может быть получено путем очистки соединения [F6] методом хиральной колоночной хроматографии. Стерическую конфигурацию соединения [I-a6] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0198]
[Способ получения 7]
Соединение формулы [I-a7] может быть получено способом получения 7, показанным на следующей схеме.
[0199]
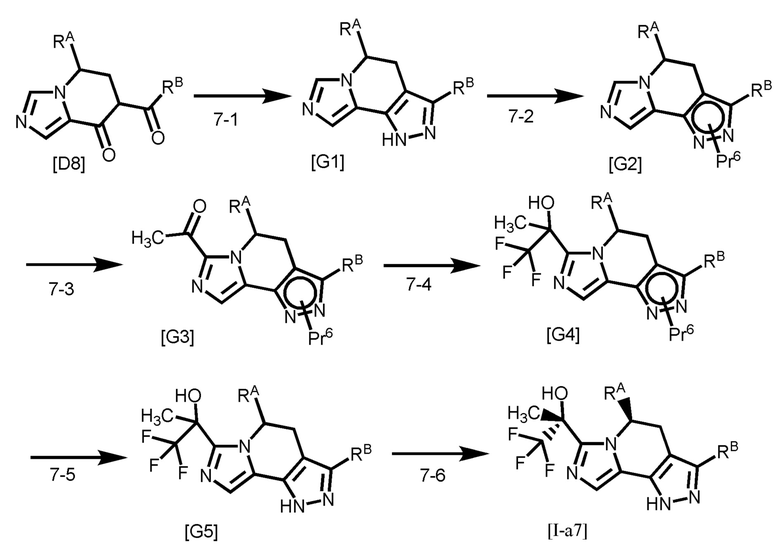
[0200]
где Pr6 представляет собой защитную группу пиразола, такую как п-метоксибензил и тому подобное; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0201]
Стадия 7-1
Соединение [G1] может быть получено путем проведения реакции циклизации пиразола соединения [D8] и гидразина. Например, соединение [G1] может быть получено методом, аналогичным методу на стадии 6-2.
[0202]
Стадия 7-2
Соединение [G2] может быть получено путем введения защитной группы в пиразол соединения [G1]. Например, когда Pr6 представляет собой п-метоксибензильную группу, соединение [G2] может быть получено путем проведения реакции соединения [G1] с п-метоксибензилхлоридом в растворителе в присутствии основания при температуре от ледяной до комнатной. Защитная группа Pr6 может быть связана с любым из двух атомов азота пиразола.
Примеры растворителя включают тетрагидрофуран.
Примеры основания включают гидрид натрия.
[0203]
Стадия 7-3
Соединение [G3] может быть получено путем проведения реакции соединения [G2] с N-метокси-N-метилацетамидом. Например, соединение [G3] может быть получено методом, аналогичным методу на стадии 1-5.
[0204]
Стадия 7-4
Соединение [G4] может быть получено путем проведения реакции соединения [G3] с (трифторметил)триметилсиланом. Например, соединение [G4] может быть получено методом, аналогичным методу на стадии 1-6.
[0205]
Стадия 7-5
Соединение [G5] может быть получено путем снятия защиты с пиразола соединения [G4]. Например, когда Pr6 представляет собой п-метоксибензильную группу, соединение [G5] может быть получено путем обработки соединения [G4] кислотой в растворителе при температуре от 60°C до 80°C.
Примеры кислоты включают трифторуксусную кислоту.
Примеры растворителя включают дихлорметан.
[0206]
Стадия 7-6
Соединение [I-a7] может быть получено путем очистки соединения [G5] методом хиральной колоночной хроматографии. Стерическую конфигурацию соединения [I-a7] можно определять, например, методом рентгеноструктурного анализа кристаллов.
[0207]
[Способ получения 8]
Соединение формулы [I-a8] может быть получено способом получения 8, показанным на следующей схеме.
[0208]
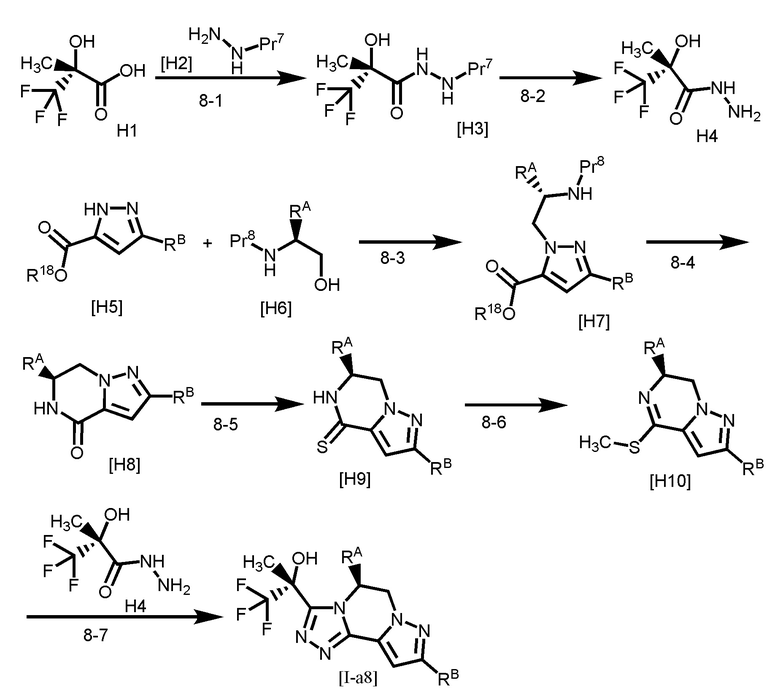
[0209]
где R18 представляет собой C1-4 алкил;
Pr7 и Pr8 каждый независимо представляет собой защитную группу для аминогруппы, такую как трет-бутоксикарбонил и тому подобное; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0210]
Стадия 8-1
Соединение [H3] может быть получено путем проведения реакции (R)-3,3,3-трифтор-2-гидрокси-2-метилпропионовой кислоты (соединения H1) и соединения [H2]. Например, соединение [H3] может быть получено путем проведения реакции соединения H1 с соединением [H2] в растворителе в присутствии конденсирующего реагента при температуре от ледяной до комнатной.
Примеры конденсирующего реагента включают 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид и 1-гидроксибензотриазол моногидрат.
Примеры растворителя включают ацетонитрил и диметилформамид.
Соединение [H2] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0211]
Стадия 8-2
(R)-3,3,3-трифтор-2-гидрокси-2-метилпропангидразин (соединение H4) может быть получен путем снятия защиты с соединения [H3]. Например, когда Pr7 представляет собой трет-бутоксикарбонил, соединение H4 может быть получено путем обработки соединения [H3] кислотой в растворителе при температуре от ледяной до комнатной. Соединение H4 может быть получено в виде соли с кислотой, используемой в этой реакции.
Примеры кислоты включают трифторуксусную кислоту и соляную кислоту.
Примеры растворителя включают тетрагидрофуран, этилацетат и хлороформ.
[0212]
Стадия 8-3
Соединение [H7] может быть получено путем проведения реакции Мицунобу соединения [H5] и соединения [H6]. Например, соединение [H7] может быть получено методом, аналогичным методу на стадии 2-1.
Соединение [H5] и соединение [H6] могут быть коммерчески доступными продуктами или могут быть получены соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0213]
Стадия 8-4
Соединение [H8] может быть получено путем снятия защиты с аминогруппы соединения [H7] и лактамизации. Снятие защиты с аминогруппы можно выполнять, например, методом, аналогичным методу на стадии 2-2. Лактамизацию можно выполнять, например, методом, аналогичным методу на стадии 2-3.
[0214]
Стадия 8-5
Соединение [H9] может быть получено путем проведения реакции соединения [H8] с сернистым реагентом. Например, соединение [H9] может быть получено методом, аналогичным методу на стадии 2-4.
[0215]
Стадия 8-6
Соединение [H10] может быть получено путем метилирования соединения [H9]. Например, соединение [H10] может быть получено методом, аналогичным методу на стадии 2-6.
[0216]
Стадия 8-7
Соединение [I-a8] может быть получено путем проведения реакции циклизации соединения [H10] и соединения H4. Например, соединение [I-a8] может быть получено путем проведения реакции соединения [H10] с соединением H4 в растворителе в присутствии кислоты при температуре от 60°C до 120°C.
Примеры кислоты включают уксусную кислоту.
Примеры растворителя включают изопропанол и воду.
[0217]
[Способ получения 9]
Соединение, имеющее нужную группу RB, может быть получено путем превращения функциональной группы на соответствующей стадии в способах получения 1-8. Например, соединения [I-a10]-[I-a15] могут быть получены путем превращения соединения [I-a9], полученного в любом из способов получения 1-8, способом получения 9, показанным на следующей схеме.
[0218]
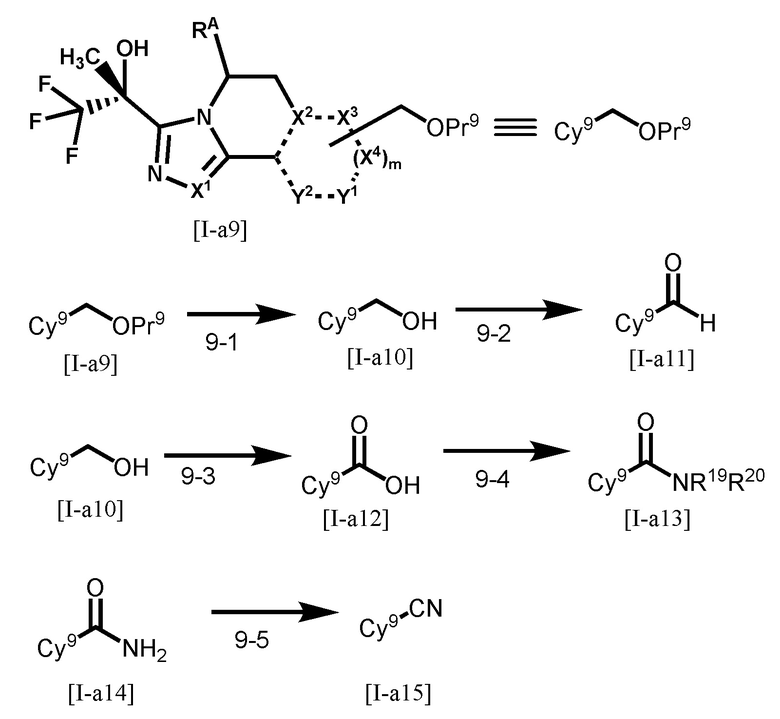
[0219]
где Pr9 представляет собой защитную группу гидроксильной группы, такую как п-метоксибензил и тому подобное;
R19 и R20 каждый независимо представляет собой водород или C1-4 алкил; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0220]
Стадия 9-1
Соединение [I-a10] может быть получено путем снятия защиты с гидроксильной группы соединения [I-a9]. Например, соединение [I-a10] может быть получено путем обработки соединения [I-a9] кислотой в растворителе при комнатной температуре.
Примеры кислоты включают трифторуксусную кислоту.
Примеры растворителя включают дихлорметан.
[0221]
Стадия 9-2
Соединение [I-a11] может быть получено путем окисления гидроксильной группы соединения [I-a10]. Например, соединение [I-a11] может быть получено методом, аналогичным методу на стадии 1-2.
[0222]
Стадия 9-3
Соединение [I-a12] может быть получено путем окисления гидроксильной группы соединения [I-a10]. Например, соединение [I-a12] может быть получено путем проведения реакции соединения [I-a10] с окислителем в растворителе при комнатной температуре.
Примеры окислителя включают перманганат калия.
Примеры растворителя включают ацетон.
[0223]
Стадия 9-4
Соединение [I-a13] может быть получено путем амидирования соединения [I-a12] и HNR19R20. Например, соединение [I-a13] может быть получено путем проведения реакции соединения [I-a12] с HNR19R20 в растворителе в присутствии основания и конденсирующего реагента при температуре от ледяной до комнатной.
Примеры основания включают диизопропилэтиламин и триэтиламин.
Примеры конденсирующего реагента включают HATU.
Примеры растворителя включают диметилформамид.
Когда каждый из R19 и R20 в HNR19R20 представляет собой водород на данной стадии, может быть получено соединение [I-a14].
[0224]
Стадия 9-5
Соединение [I-a15] может быть получено путем проведения реакции цианирования соединения [I-a14]. Например, соединение [I-a15] может быть получено путем проведения реакции соединения [I-a14] с ангидридом кислоты в растворителе в присутствии основания при температуре от ледяной до комнатной.
Примеры основания включают пиридин.
Примеры ангидрида кислоты включают трифторуксусный ангидрид.
Примеры растворителя включают 1,4-диоксан.
[Способ получения 10]
Соединение формулы [I-a10] может быть получено способом получения 10, показанным на следующей схеме.
[0225]
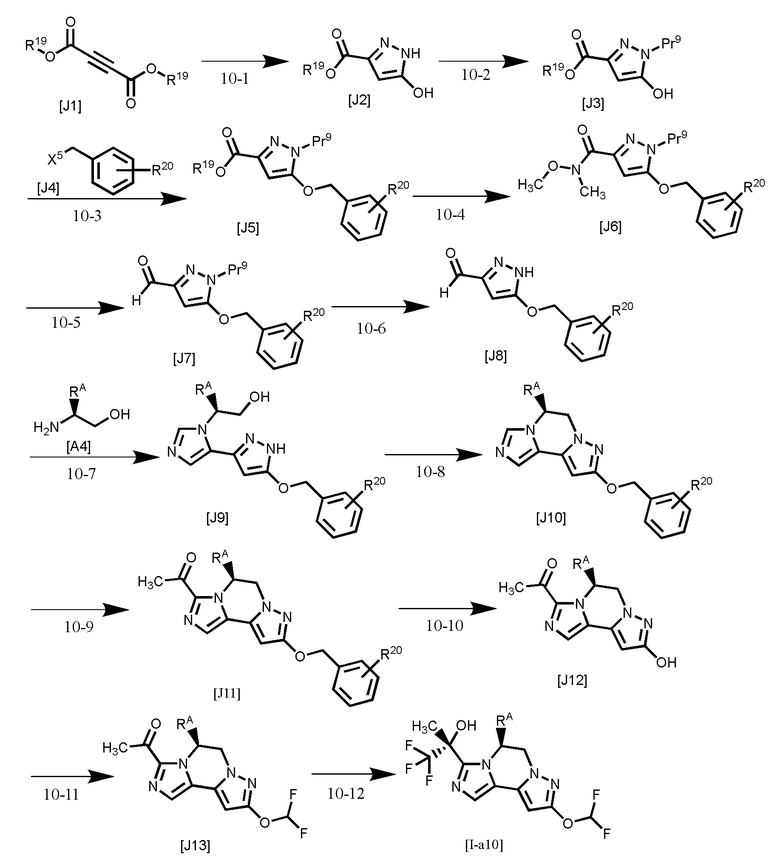
[0226]
где R19 представляет собой C1-4 алкил;
R20 представляет собой водород, галоген, C1-4 алкил или нитро;
Pr9 представляет собой защитную группу пиразола, такую как 2-тетрагидропиранил и тому подобное;
X5 представляет собой уходящую группу, такую как хлор, бром, метансульфонилокси и тому подобное; и
каждый другой символ является таким, как определено в вышеуказанной формуле [I-a].
[0227]
Стадия 10-1
Соединение [J2] может быть получено путем проведения реакции соединения [J1] с гидразином. Например, соединение [J2] может быть получено путем проведения реакции соединения [J1] с гидразином в растворителе при комнатной температуре. При необходимости реакцию можно проводить в присутствии кислоты.
Примеры растворителя включают ацетонитрил, толуол и этанол.
Примеры кислоты включают уксусную кислоту.
Соединение [J1] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0228]
Стадия 10-2
Соединение [J3] может быть получено путем введения защитной группы в пиразол соединения [J2]. Например, когда Pr9 представляет собой 2-тетрагидропиранильную группу, соединение [J3] может быть получено путем проведения реакции соединения [J2] с 3,4-дигидро-2H-пираном в растворителе в присутствии кислоты при комнатной температуре.
Примеры растворителя включают ацетонитрил и N, N-диметилформамид.
Примеры кислоты включают п-толуолсульфонат пиридиния и п-толуолсульфоновую кислоту.
[0229]
Стадия 10-3
Соединение [J5] может быть получено путем проведения реакции соединения [J3] с соединением [J4]. Например, соединение [J5] может быть получено путем проведения реакции соединения [J3] с соединением [J4] в растворителе в присутствии основания при комнатной температуре.
Примеры растворителя включают N-метилпирролидон, N, N-диметилформамид, ацетонитрил, толуол, изопропилацетат, тетрагидрофуран и диметилсульфоксид.
Примеры основания включают карбонат калия, карбонат натрия, карбонат цезия, карбонат лития, трет-бутоксид калия, ацетат калия, фосфат калия, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), N, N-диизопропилэтиламин.
Соединение [J4] может быть коммерчески доступным продуктом или может быть получено соответствующим преобразованием коммерчески доступного продукта способом, хорошо известным специалистам в данной области.
[0230]
Стадия 10-4
Соединение [J6] может быть получено путем гидролиза сложного эфира соединения [J5] и последующего проведения реакции амидирования с N, O-диметилгидроксиламином. Например, соединение [J6] может быть получено путем проведения реакции соединения [J5] со щелочью для гидролиза сложного эфира в растворителе и проведения реакции полученного соединения с N, O-диметилгидроксиламином в присутствии конденсирующего реагента при комнатной температуре.
Примеры щелочи включают гидроксид натрия.
Примеры конденсирующего реагента включают сочетание WSC-HCl и HOBt.
Примеры растворителя включают 1,2-диметоксиэтан.
[0231]
Стадия 10-5
Соединение [J7] может быть получено путем восстановления соединения [J6]. Например, соединение [J7] может быть получено путем проведения реакции соединения [J6] с восстановителем в растворителе при охлаждении льдом.
Примеры восстановителя включают бис(2-метоксиэтокси)алюмогидрид натрия.
Примеры растворителя включают толуол и 1,2-диметоксиэтан.
[0232]
Стадия 10-6
Соединение [J8] может быть получено путем снятия защиты защитной группы пиразола соединения [J7]. Например, когда Pr9 представляет собой 2-тетрагидропиранильную группу, соединение [J8] может быть получено путем обработки соединения [J7] кислотой в растворителе при комнатной температуре.
Примеры кислоты включают соляную кислоту, метансульфоновую кислоту, серную кислоту и фосфорную кислоту.
Примеры растворителя включают 1,2-диметоксиэтан.
[0233]
Стадия 10-7
Соединение [J9] может быть получено путем проведения реакции соединения [J8] с соединением [A4]. Например, соединение [J9] может быть получено методом, аналогичным методу на стадии 1-3.
[0234]
Стадия 10-8
Соединение [J10] может быть получено путем проведения внутримолекулярной реакции Мицунобу соединения [J9]. Например, соединение [J10] может быть получено методом, аналогичным методу на стадии 1-4. Соединение [J10] может быть получено в виде соли с кислотой, такой как соляная кислота и тому подобное.
[0235]
Стадия 10-9
Соединение [J11] может быть получено путем проведения реакции соединения [J10] с N-метокси-N-метилацетамидом. Например, соединение [J11] может быть получено методом, аналогичным методу на стадии 1-5.
[0236]
Стадия 10-10
Соединение [J12] может быть получено путем снятия защиты с соединения [J11]. Например, соединение [J12] может быть получено путем обработки соединения [J11] кислотой при температуре от комнатной до 50°C. Соединение [J12] может быть получено в виде соли с используемой кислотой.
Примеры кислоты включают концентрированную соляную кислоту.
[0237]
Стадия 10-11
Соединение [J13] может быть получено путем проведения реакции соединения [J12] с диэтил(бромдифторметил)фосфонатом. Например, соединение [J13] может быть получено путем проведения реакции соединения [J12] с диэтил(бромдифторметил)фосфонатом в растворителе в присутствии основания при комнатной температуре.
Примеры растворителя включают ацетонитрил, 1,2-диметоксиэтан и тетрагидрофуран.
Примеры основания включают гидроксид калия, гидроксид лития и гидроксид тетрабутиламмония.
[0238]
Стадия 10-12
Соединение [I-a10] может быть получено путем проведения реакции соединения [J13] с (трифторметил)триметилсиланом. Например, соединение [I-a10] может быть получено методом, аналогичным методу на стадии 1-6. Группа RA соединения [J13] становится стерическим препятствием, и реакция протекает диастереоселективным образом. О стерической конфигурации соединения [I-a10] можно судить по механизму реакции и подтверждать ее рентгеноструктурным анализом кристаллов.
ПРИМЕРЫ
[0239]
Способ получения соединения формулы [I-a], или его фармацевтически приемлемой соли, по настоящему изобретению конкретно объяснен с помощью следующих примеров получения. Однако способ получения соединения формулы [I-a], или его фармацевтически приемлемой соли, не ограничен примерами получения.
Если не указано иначе, «%» означает масс%. Если не указано иначе, соотношение в смеси растворителей представляет собой соотношение в смеси по объему.
Используемые в примерах сокращения означают следующее.
ДМСО: диметилсульфоксид
M: моль/л
N: нормальность
HATU: О-(7-азабензотриазол-1-ил)-N, N,N’,N’-тетраметилуроний гексафторфосфат
WSC-HCl: 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид
HOBt-H2O: 1-гидроксибензотриазол моногидрат
Результаты измерения 1H-ЯМР указаны с использованием следующих сокращений.
с: синглет, д: дублет, дд: двойной дублет, дт: двойной триплет, т: триплет, к: квартет, дк: двойной квартет, м: мультиплет, уш.с: уширенный синглет, уш.м: уширенный мультиплет, J: константа взаимодействия, Гц: герц.
1H-ЯМР спектр определяли в CDCl3 или ДМСО-D6 с использованием тетраметилсилана в качестве внутреннего стандарта, и все значения δ приведены в ppm.
[0240]
Пример получения 1
Синтез (R)-2-((S)-9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ола (соединение примера 23)
[0241]
Стадия 1
Этил-3-(бензилокси)-1H-пиразол-5-карбоксилат
[0242]
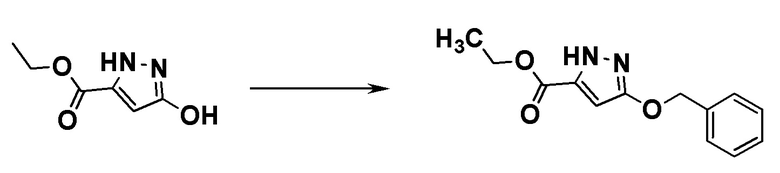
[0243]
Этил-3-гидрокси-1H-пиразол-5-карбоксилат (10 г) смешивали с тетрагидрофураном (100 мл). К смеси добавляли бензиловый спирт (7,99 мл) и трифенилфосфин (18,48 г). При охлаждении льдом по каплям добавляли диизопропил азодикарбоксилат (13,7 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, добавляли этилацетат (70 мл) и гексан (140 мл), и смесь перемешивали при комнатной температуре. Осажденное твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=92:8 до гексан:этилацетат=47:53), с получением указанного в заголовке соединения в виде неочищенного продукта (20 г). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0244]
Стадия 2
(3-(бензилокси)-1H-пиразол-5-ил)метанол
[0245]

[0246]
Неочищенный продукт (19 г) этил-3-(бензилокси)-1H-пиразол-5-карбоксилат, полученный на предыдущей стадии, растворяли в циклопентилметиловом эфире (70 мл). При охлаждении льдом раствор по каплям добавляли к раствору боргидрида лития (4,17 г) в циклопентилметиловом эфире (150 мл). Реакционную смесь перемешивали при 0°C в течение 5 мин. При охлаждении льдом последовательно добавляли по каплям раствор метанола (8 мл) в циклопентилметиловом эфире (50 мл), метанол (8 мл) и метанол (8 мл). Смесь перемешивали при 0°C в течение 10 мин. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и 1Н водный раствор соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от этилацетат:гексан=16:84 до этилацетата, затем этилацетат:метанол=82:18), с получением указанного в заголовке соединения (8,2 г).
1H-ЯМР (400 МГц, CDCl3) 4,66 (с, 2H), 5,19 (с, 2H), 5,64 (с, 1H), 7,26-7,47 (м, 5H)
[0247]
Стадия 3
3-(бензилокси)-1H-пиразол-5-карбальдегид
[0248]
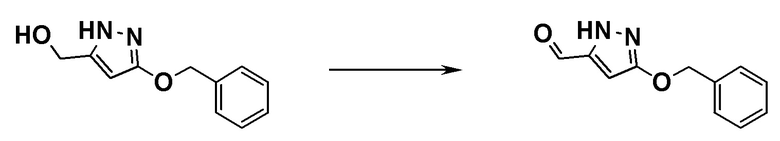
[0249]
(3-(бензилокси)-1H-пиразол-5-ил)метанол (8,2 г), полученный на предыдущей стадии, растворяли в 1,2-диметоксиэтане (164 мл). При комнатной температуре добавляли диоксид марганца (41 г), и смесь перемешивали при 80°C в течение 1,5 час. После остывания смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде неочищенного продукта (5,4 г). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0250]
Стадия 4
(S)-2-(5-(3-(бензилокси)-1H-пиразол-5-ил)-1H-имидазол-1-ил)пропан-1-ол
[0251]
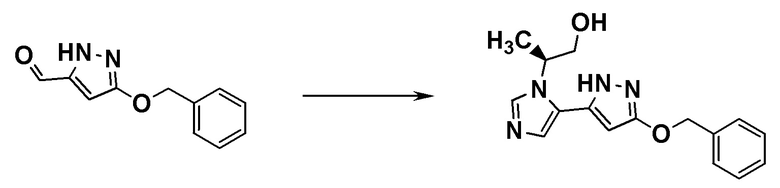
[0252]
Неочищенный продукт (5,4 г) 3-(бензилокси)-1H-пиразол-5-карбальдегид, полученный на предыдущей стадии, смешивали с метанолом (54 мл). (S)-2-аминопропан-1-ол (2 г) добавляли при комнатной температуре, и смесь перемешивали при комнатной температуре в течение ночи. При охлаждении льдом добавляли 1,2-диметоксиэтан (164 мл), п-толуолсульфонилметил изоцианид (7,82 г) и карбонат калия (11,07 г), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от этилацетат:гексан=25:75 до этилацетата, затем этилацетат:метанол=60:40), с получением указанного в заголовке соединения в виде неочищенного продукта (4,1 г).
[0253]
Стадия 5
(S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин
[0254]
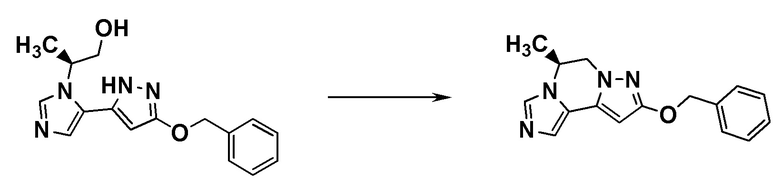
[0255]
Неочищенный продукт (4,1 г) (S)-2-(5-(3-(бензилокси)-1H-пиразол-5-ил)-1H-имидазол-1-ил)пропан-1-ол, полученный на предыдущей стадии, и трифенилфосфин (4,69 г) смешивали с тетрагидрофураном (123 мл). При охлаждении льдом добавляли ди-трет-бутилазодикарбоксилат (4,11 г), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на колонке с силикагелем (от этилацетат:гексан=25:75 до этилацетата, затем этилацетат:метанол=60:40), с получением указанного в заголовке соединения (2,98 г).
1H-ЯМР (400 МГц, CDCl3) 1,64 (д, J=6,70 Гц, 3H), 3,97 (дд, J=12,95, 8,09 Гц, 1H), 4,31 (дд, J=12,95, 4,39 Гц, 1H), 4,54-4,65 (м, 1H), 5,21 (с, 2H), 5,86 (с, 1H), 7,26 (с, 1H), 7,29-7,49 (м, 5H), 7,59 (с, 1H)
[0256]
Стадия 6
(S)-1-(9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он
[0257]
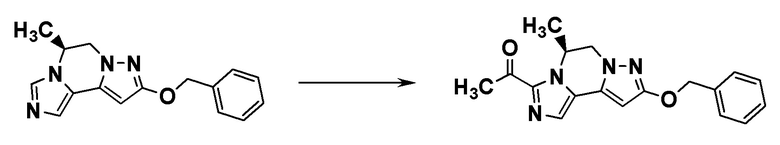
[0258]
(S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин (2,98 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (29,8 мл). Смесь охлаждали до -78°C, и по каплям добавляли 2M раствор смеси диизопропиламид лития/тетрагидрофуран-гептан-этилбензол (13,29 мл). Смесь перемешивали при -78°C в течение 30 мин. К смеси добавляли N-метокси-N-метилацетамид (5,25 мл), и смесь перемешивали при -78°C в течение 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и этилацетат, и выпавшее в осадок твердое вещество собирали фильтрованием. Фильтрат разделяли, органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли смесь этилацетат/гексан=1/1 (10 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, объединяли с твердым веществом, собранным ранее при фильтровании, смешивали с этилацетатом, и концентрировали, с получением указанного в заголовке соединения (2 г).
1H-ЯМР (400 МГц, CDCl3) 1,44 (д, J=6,58 Гц, 3H), 2,69 (с, 3H), 4,19 (дд, J=13,45, 1,20 Гц, 1H), 4,29 (дд, J=13,45, 4,63 Гц, 1H), 5,24 (с, 2H), 5,75-5,84 (м, 1H), 5,98 (с, 1H), 7,32-7,50 (м, 6H)
[0259]
Стадия 7
(R)-2-((S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ол
[0260]
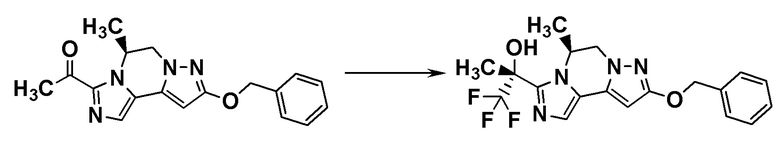
[0261]
(S)-1-(9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он (2 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (29,8 мл). При охлаждении льдом к смеси добавляли фторид цезия (0,195 г) и по каплям добавляли (трифторметил)триметилсилан (1,088 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 мин. При охлаждении льдом добавляли метанол (24,84 мл) и карбонат калия (1,065 г), и смесь перемешивали при комнатной температуре в течение 45 мин. Реакционную смесь концентрировали при пониженном давлении, добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (этилацетат:гексан=12:88 до 4:96), с получением указанного в заголовке соединения (2,2 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,28 (д, J=6,70 Гц, 3H), 1,80 (с, 3H), 4,12-4,24 (м, 2H), 5,15 (с, 2H), 5,27-5,36 (м, 1H), 6,04 (с, 1H), 7,23 (с, 1H), 7,28 (с, 1H), 7,29-7,46 (м, 5H)
[0262]
Стадия 8
(S)-5-метил-3-((R)-1,1,1-трифтор-2-гидроксипропан-2-ил)-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-9-ол
[0263]
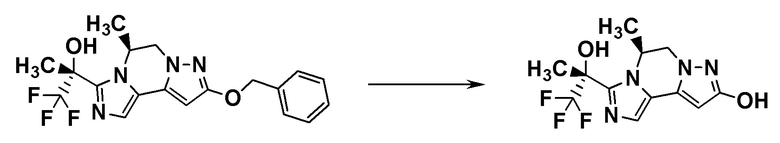
[0264]
(R)-2-((S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ол (2,2 г), полученный на предыдущей стадии, и 10% палладий на угле (0,44 г) смешивали с этанолом (44 г). В атмосфере водорода смесь перемешивали при комнатной температуре в течение 1,5 час. Реакционную смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от этилацетат:гексан=16:84 до этилацетата, затем этилацетат:метанол=80:20). К полученному неочищенному продукту последовательно добавляли этилацетат (6 мл) и гексан (6 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (1,82 г). Стерическую конфигурацию указанного в заголовке соединения определяли рентгеноструктурным анализом кристаллов.
1H-ЯМР (400 МГц, ДМСО-D6) 1,28 (д, J=6,58 Гц, 3H), 1,81 (с, 3H), 4,07-4,11 (м, 2H), 5,23-5,36 (м, 1H), 5,75 (с, 1H), 7,20 (с, 1H), 7,26 (уш.с, 1H), 9,89 (уш.с, 1H)
[0265]
Стадия 9
(R)-2-((S)-9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ол
[0266]
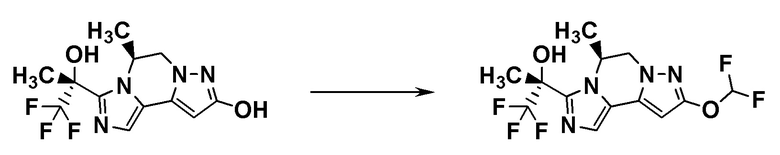
[0267]
(S)-5-метил-3-((R)-1,1,1-трифтор-2-гидроксипропан-2-ил)-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-9-ол (1 г), полученный на предыдущей стадии, карбонат калия (0,914 г) и хлордифторацетат натрия (1,009 г) смешивали с диметилформамидом (6 мл), и смесь перемешивали при 100°C в течение 3 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=88:12 до этилацетата). К полученному неочищенному продукту добавляли смесь этилацетат/гексан=1/2 (1,5 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (0,315 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,27 (д, J=6,70 Гц, 3H), 1,81 (с, 3H), 4,20-4,30 (м, 2H), 5,31-5,40 (м, 1H), 6,31 (с, 1H), 7,27 (т, J=73,29 Гц, 1H), 7,31 (с, 1H), 7,32 (с, 1H)
[0268]
Пример получения 2
Синтез (R)-1,1,1-трифтор-2-((S)-9-(2-гидрокси-2-метилпропокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)пропан-2-ола (соединение примера 52)
[0269]
Стадия 1
Метил-2-(((S)-5-метил-3-((R)-1,1,1-трифтор-2-гидроксипропан-2-ил)-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-9-ил)окси)ацетат
[0270]
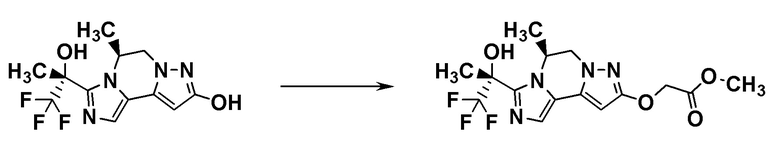
[0271]
(S)-5-метил-3-((R)-1,1,1-трифтор-2-гидроксипропан-2-ил)-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-9-ол (0,5 г) и карбонат калия (0,288 г) смешивали с диметилформамидом (5 мл). При охлаждении льдом добавляли метилбромацетат (0,154 мл), и смесь перемешивали при комнатной температуре в течение 2 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток последовательно очищали хроматографией на силикагеле (от гексан:этилацетат=88:12 до этилацетата) и хроматографией на аминосиликагеле (от этилацетат:гексан=12:88 до этилацетата, затем этилацетат:метанол=90:10), с получением указанного в заголовке соединения (0,411 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,26 (д, J=6,47 Гц, 3H), 1,80 (с, 3H), 3,67 (с, 3H), 4,08-4,21 (м, 2H), 4,78 (с, 2H), 5,24-5,36 (м, 1H), 6,03 (с, 1H), 7,25 (с, 1H), 7,27 (с, 1H)
[0272]
Стадия 2
(R)-1,1,1-трифтор-2-((S)-9-(2-гидрокси-2-метилпропокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)пропан-2-ол
[0273]
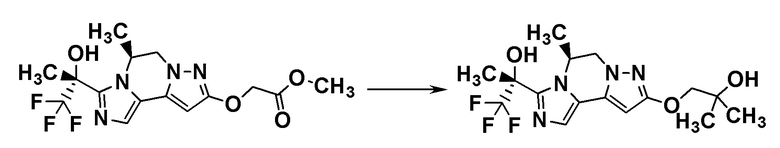
[0274]
Метил-2-(((S)-5-метил-3-((R)-1,1,1-трифтор-2-гидроксипропан-2-ил)-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-9-ил)окси)ацетат (0,411 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (2 мл). При охлаждении льдом добавляли 1,08M раствор смеси метилмагнийбромид/тетрагидрофуран (4 мл), и смесь перемешивали при комнатной температуре в течение 2 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (от гексан:этилацетат=88:12 до этилацетата) и хроматографией на аминосиликагеле (от этилацетат:гексан=24:76 до этилацетата). К полученному неочищенному продукту последовательно добавляли этилацетат (0,7 мл) и гексан (1,4 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (0,251 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,16 (с, 6H), 1,27 (д, J=6,47 Гц, 3H), 1,80 (с, 3H), 3,82 (с, 2H), 4,09-4,19 (м, 2H), 4,57 (с, 1H), 5,25-5,36 (м, 1H), 5,98 (с, 1H), 7,22 (с, 1H), 7,26 (с, 1H).
[0275]
Пример получения 3
Синтез (R)-1,1,1-трифтор-2-((S)-6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин-8-ил)пропан-2-ола (соединение примера 2)
[0276]
Стадия 1
Метил-(S)-1-(1-((трет-бутоксикарбонил)амино)пропан-2-ил)-1H-имидазол-5-карбоксилат
[0277]
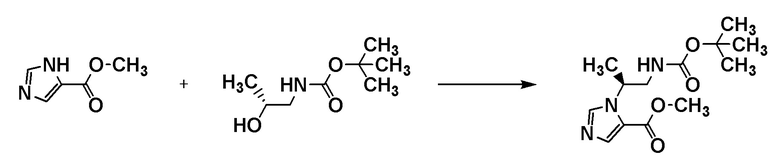
[0278]
Метил-1H-имидазол-5-карбоксилат (250 г), трет-бутил (R)-(2-гидроксипропил)карбамат (521 г) и триоктилфосфин (1505 мл) смешивали с толуолом (1750 мл). Реакционный раствор нагревали до 80°C, и по каплям добавляли 40% раствор смеси диизопропилазодикарбоксилат/толуол (1599 мл). Реакционный раствор перемешивали при 80°C в течение 1 час. Смесь дважды экстрагировали 1Н водным раствором гидросульфата калия. Водный слой промывали смесью этилацетат:гексан=1:1. При охлаждении льдом к водному слою добавляли 1,4 M водный раствор карбоната натрия (1 л) небольшими порциями, и смесь дважды экстрагировали этилацетатом. Добавляли сульфат натрия и силикагель, и смесь перемешивали при комнатной температуре в течение 30 мин. Сульфат натрия и силикагель отфильтровывали, и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат (250 мл), и смесь перемешивали при 80°C в течение 10 мин. Медленно добавляли гексан (1750 мл), и смесь перемешивали, давая возможность охладиться до комнатной температуры. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (432,4 г).
1H-ЯМР (400 МГц, CDCl3) 1,39 (с, 9H), 1,54 (д, J=6,88 Гц, 3H), 3,48 (т, J=6,28 Гц, 2H), 3,85 (с, 3H), 4,63 (уш.с, 1H), 5,17-5,32 (м, 1H), 7,72-7,77 (м, 2H)
[0279]
Стадия 2
Метил(S)-1-(1-аминопропан-2-ил)-1H-имидазол-5-карбоксилат дигидрохлорид
[0280]
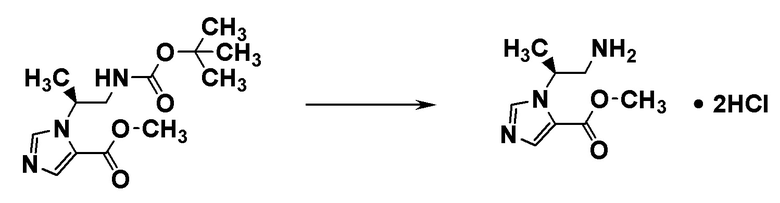
[0281]
Метил(S)-1-(1-((трет-бутоксикарбонил)амино)пропан-2-ил)-1H-имидазол-5-карбоксилат (432,4 г), полученный на предыдущей стадии, смешивали с этилацетатом (865 мл). При охлаждении льдом по каплям добавляли смесь 4Н соляная кислота/этилацетат (1526 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 час. При охлаждении льдом по каплям добавляли смесь 4Н соляная кислота/этилацетат (382 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 час. При охлаждении льдом по каплям добавляли смесь 4Н соляная кислота/этилацетат (38,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 час. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (388,89 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,51 (д, J=6,94 Гц, 3H), 3,20-3,36 (м, 1H), 3,42-3,57 (м, 1H), 3,85 (д, J=4,62 Гц, 3H), 5,29-5,43 (м, 1H), 8,09 (с, 1H), 8,21 (уш.с, 3H), 8,95 (с, 1H)
[0282]
Стадия 3
(S)-5-метил-6,7-дигидроимидазо[1,5-a]пиразин-8(5H)-он
[0283]
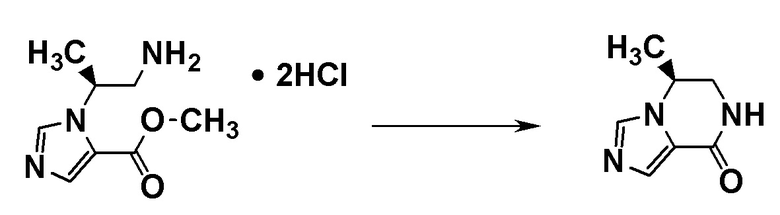
[0284]
Метил(S)-1-(1-аминопропан-2-ил)-1H-имидазол-5-карбоксилат дигидрохлорид (388,89 г), полученный на предыдущей стадии, смешивали с метанолом (1944 мл) и водой (117 мл). К смеси добавляли карбонат натрия (644 г), и смесь перемешивали при 90°C в течение 2 час. При комнатной температуре добавляли тетрагидрофуран (1944 мл), и реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом. К полученному остатку добавляли толуол, и смесь перемешивали при комнатной температуре в течение 1 час. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (278,65 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,46 (д, J=6,28 Гц, 3H), 3,19-3,28 (м, 1H), 3,46-3,58 (м, 1H), 4,37-4,48 (м, 1H), 7,45 (с, 1H), 7,83-7,94 (м, 2H)
[0285]
Стадия 4
(S)-5-метил-6,7-дигидроимидазо[1,5-a]пиразин-8(5H)-тион
[0286]
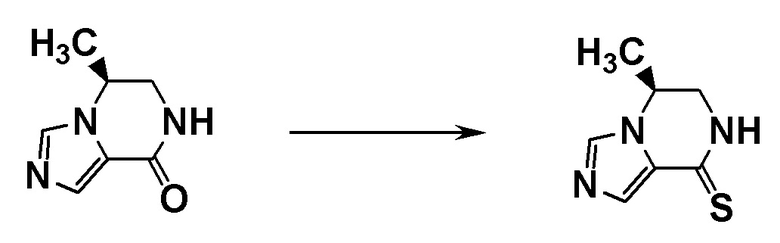
[0287]
(S)-5-метил-6,7-дигидроимидазо[1,5-a]пиразин-8(5H)-он (62,2 г) смешивали с толуолом (600 мл) и пиридином (200 мл). К смеси добавляли реактив Лавессона (50 г), и смесь перемешивали при 110°C в течение ночи. После остывания растворитель декантировали, и смолообразное вещество промывали толуолом. К полученному смолообразному веществу добавляли метанол (187 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (39,9 г). Указанное в заголовке соединение (20,7 г) получали аналогичным способом.
1H-ЯМР (400 МГц, ДМСО-D6) 1,43 (д, J=6,47 Гц, 3H), 3,23-3,31 (м, 1H), 3,58 (дт, J=13,56, 4,10 Гц, 1H), 4,39-4,52 (м, 1H), 7,61 (с, 1H), 7,96 (с, 1H), 10,03 (уш.с, 1H)
[0288]
Стадия 5
(S)-6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин
[0289]
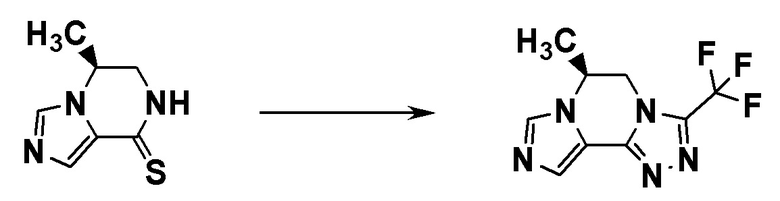
[0290]
(S)-5-метил-6,7-дигидроимидазо[1,5-a]пиразин-8(5H)-тион (3 г), полученный на предыдущей стадии, смешивали с изопропиловым спиртом (30 мл). При комнатной температуре добавляли гидразин гидрат (2,62 мл), и смесь перемешивали при 80°C в течение 3 час. После остывания реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке один раз с этанолом и два раза с толуолом. К полученному остатку добавляли трифторуксусную кислоту (23,68 мл) и трифторуксусный ангидрид (12,65 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и дважды подвергали азеотропной перегонке с толуолом. Полученный остаток очищали хроматографией на колонке с аминосиликагелем (от этилацетат:гексан=75:25 до этилацетата, затем этилацетат:метанол=80:20), с получением указанного в заголовке соединения (3,96 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,57 (д, J=6,58 Гц, 3H), 4,26 (дд, J=13,15, 8,67 Гц, 1H), 4,64 (дд, J=13,15, 4,48 Гц, 1H), 4,74-4,85 (м, 1H), 7,72 (с, 1H), 8,13 (с, 1H)
[0291]
Стадия 6
(S)-1-(6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин-8-ил)этан-1-он
[0292]
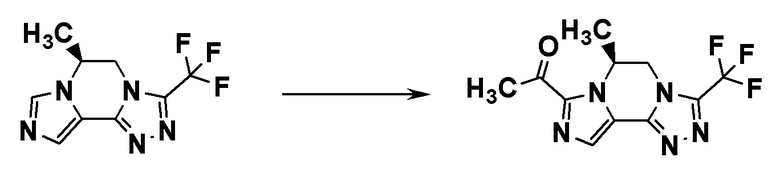
[0293]
(S)-6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин (3,96 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (80 мл). Смесь охлаждали до -78°C и по каплям добавляли 2M раствор смеси диизопропиламид лития/тетрагидрофуран-гептан-этилбензол (24,67 мл). Смесь перемешивали при -78°C в течение 30 мин. К смеси добавляли N-метокси-N-метилацетамид (5,25 мл), и смесь перемешивали при -78°C в течение 35 мин. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата). К полученному неочищенному продукту добавляли толуол (12 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (2,04 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,27 (д, J=6,70 Гц, 3H), 2,63 (с, 3H), 4,49-4,61 (м, 2H), 5,66-5,78 (м, 1H), 7,94 (с, 1H)
[0294]
Стадия 7
(R)-1,1,1-трифтор-2-((S)-6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин-8-ил)пропан-2-ол
[0295]
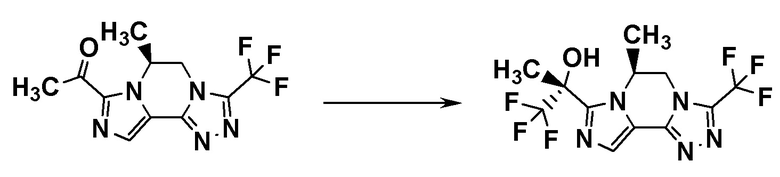
[0296]
(S)-1-(6-метил-3-(трифторметил)-5,6-дигидроимидазо[1,5-a][1,2,4]триазолo[3,4-c]пиразин-8-ил)этан-1-он (2,04 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (40,8 мл). При охлаждении льдом добавляли фторид цезия (0,217 г) и (трифторметил)триметилсилан (2,111 мл), и смесь перемешивали при 0°C в течение 25 мин. При охлаждении льдом добавляли метанол (24,48 мл) и карбонат калия (1,186 г), и смесь перемешивали при комнатной температуре в течение 1 час. К реакционной смеси добавляли силикагель (40 мл), смесь фильтровали через слой силикагеля (20 мл), и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=16:84 до этилацетата). К полученному неочищенному продукту добавляли толуол (20 мл) и этилацетат (4 мл), и смесь перемешивали при 80°C. Выпавшее в осадок твердое вещество собирали фильтрованием. К полученному твердому веществу добавляли этилацетат (6 мл) и гексан (4 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (1,171 г). Стерическую конфигурацию указанного в заголовке соединения определяли рентгеноструктурным анализом кристаллов.
1H-ЯМР (400 МГц, ДМСО-D6) 1,28 (д, J=6,47 Гц, 3H), 1,86 (с, 3H), 4,48 (дд, J=13,64, 3,93 Гц, 1H), 4,55 (д, J=13,64 Гц, 1H), 5,45-5,51 (м, 1H), 7,49 (с, 1H), 7,75 (с, 1H)
[0297]
Пример получения 4
Синтез 1,1,1-трифтор-2-(5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-ил)пропан-2-ола (соединение примера 1)
[0298]
Стадия 1
1-(2-бромфенил)пропан-2-ол
[0299]
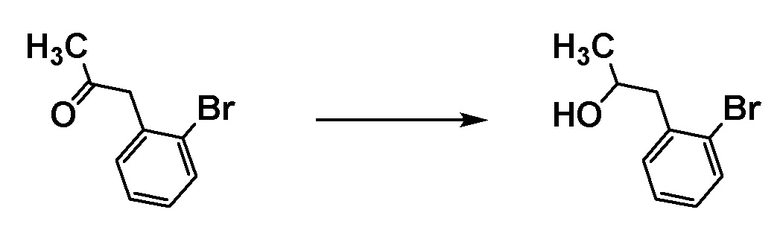
[0300]
1-(2-бромфенил)пропан-2-он (2 г) смешивали с метанолом (10 мл). При охлаждении льдом к смеси добавляли боргидрид натрия (0,533 г), и смесь перемешивали при комнатной температуре в течение 3 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (гексан:этилацетат от 95:5 до 60:40), с получением указанного в заголовке соединения (1,937 г).
1H-ЯМР (400 МГц, CDCl3) 1,27 (д, J=6,01 Гц, 3H), 1,46 (д, J=3,93 Гц, 1H), 2,83 (дд, J=13,52, 7,98 Гц, 1H), 2,96 (дд, J=13,52, 4,74 Гц, 1H), 4,07-4,19 (м, 1H), 7,04-7,13 (м, 1H), 7,21-7,28 (м, 2H), 7,54 (д, J=8,09 Гц, 1H)
[0301]
Стадия 2
1-(2-бромфенил)пропан-2-ил метансульфонат
[0302]
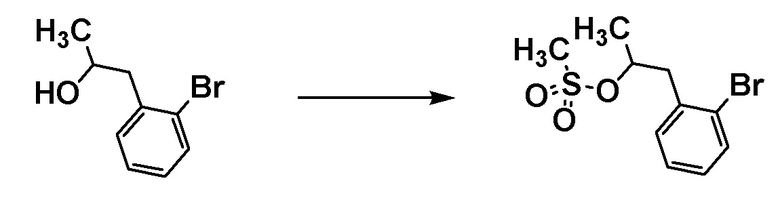
[0303]
Смешивали метансульфоновый ангидрид (2,11 г) и дихлорметан (10 мл). При охлаждении льдом к смеси добавляли 1-(2-бромфенил)пропан-2-ол (1,737 г), полученный на предыдущей стадии, и триэтиламин (3,38 мл), и смесь перемешивали при комнатной температуре в течение 4 час. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле (гексан:этилацетат от 95:5 до 60:40), с получением указанного в заголовке соединения (2,142 г).
1H-ЯМР (400 МГц, CDCl3) 1,51 (д, J=6,24 Гц, 3H), 2,50 (с, 3H), 3,00-3,14 (м, 2H), 4,94-5,06 (м, 1H), 7,08-7,17 (м, 1H), 7,23-7,30 (м, 2H), 7,56 (д, J=7,86 Гц, 1H)
[0304]
Стадия 3
Этил-1-(1-(2-бромфенил)пропан-2-ил)-1H-имидазол-2-карбоксилат
[0305]
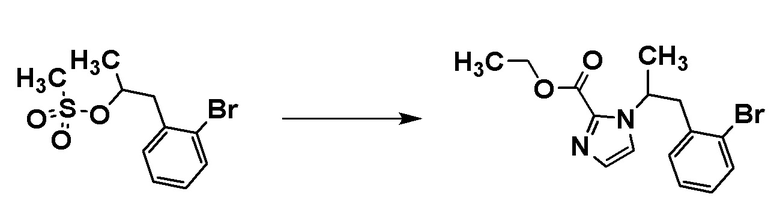
[0306]
Этил-1H-имидазол-2-карбоксилат (3,54 г) и карбонат цезия (3,29 г) смешивали с диметилформамидом (11,85 мл). Реакционный раствор нагревали до 80°C, добавляли 1-(2-бромфенил)пропан-2-ил метансульфонат (1,54 г), полученный на предыдущей стадии, и смесь перемешивали в течение ночи. К реакционной смеси добавляли воду, и смесь дважды экстрагировали этилацетатом. Органический слой последовательно промывали три раза водой и один раз насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (от гексан:этилацетат=88:12 до этилацетата), с получением указанного в заголовке соединения (0,383 г).
1H-ЯМР (400 МГц, CDCl3) 1,33 (т, J=7,17 Гц, 3H), 1,54 (д, J=6,94 Гц, 3H), 3,04 (дд, J=13,64, 7,86 Гц, 1H), 3,27 (дд, J=13,64, 6,24 Гц, 1H), 4,27 (к, J=7,17 Гц, 2H), 5,74-5,86 (м, 1H), 6,87 (дд, J=7,51, 1,73 Гц, 1H), 7,04 (тд, J=7,63, 1,70 Гц, 1H), 7,12 (тд, J=7,46, 1,31 Гц, 1H), 7,17 (с, 1H), 7,50 (дд, J=7,98, 1,27 Гц, 1H)
[0307]
Стадия 4
Этил-5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-карбоксилат
[0308]
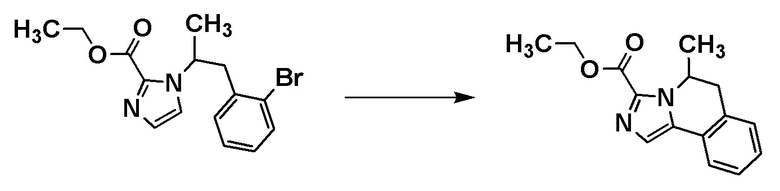
[0309]
Этил-1-(1-(2-бромфенил)пропан-2-ил)-1H-имидазол-2-карбоксилат (383 мг), полученный на предыдущей стадии, ацетат палладия (II) (74,3 мг), карбонат калия (305 мг) и ди-1-адамантил-н-бутилфосфин (178 мг) смешивали с диметилацетамидом (3,7 мл). В атмосфере аргона смесь перемешивали при 120°C в течение 7 час. При охлаждении льдом добавляли воду, и выпавшее в осадок твердое вещество собирали фильтрованием. Фильтрат дважды экстрагировали этилацетатом. Органический слой последовательно промывали два раза водой и один раз насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток и твердое вещество, полученное при фильтровании, очищали хроматографией на силикагеле (от гексан:этилацетат=88:12 до этилацетата), с получением указанного в заголовке соединения (145 мг).
1H-ЯМР (400 МГц, CDCl3) 1,26 (д, J=6,70 Гц, 3H), 1,44 (т, J=7,05 Гц, 3H), 2,84 (д, J=15,95 Гц, 1H), 3,36 (дд, J=15,95, 6,24 Гц, 1H), 4,35-4,50 (м, 2H), 5,59-5,69 (м, 1H), 7,23-7,33 (м, 3H), 7,49 (с, 1H), 7,59 (д, J=6,94 Гц, 1H)
[0310]
Стадия 5
N-метокси-N,5-диметил-5,6-дигидроимидазо[5,1-a]изохинолин-3-карбоксамид
[0311]
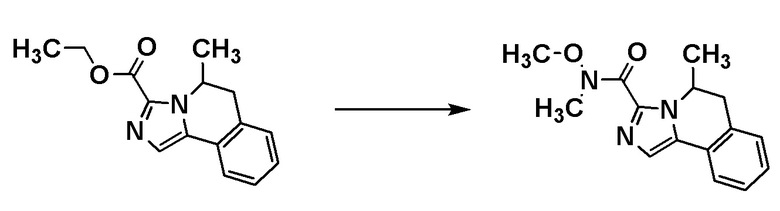
[0312]
Этил-5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-карбоксилат (82,4 мг), полученный на предыдущей стадии, смешивали с этанолом (1,6 мл). При охлаждении льдом добавляли 2Н водный раствор гидроксида натрия, и смесь перемешивали при комнатной температуре в течение 4 час. Реакционную смесь концентрировали и подвергали азеотропной перегонке с толуолом. Полученный остаток смешивали с диметилформамидом. При охлаждении льдом добавляли N, O-диметилгидроксиламин гидрохлорид (94 мг), HATU (183 мг) и диизопропилэтиламин (0,253 мл), и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь дважды экстрагировали этилацетатом. Органический слой промывали последовательно водой и насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (от гексан:этилацетат=88:12 до этилацетата), с получением указанного в заголовке соединения (80 мг).
1H-ЯМР (400 МГц, CDCl3) 1,29 (д, J=6,58 Гц, 3H), 2,82 (дд, J=15,84, 1,49 Гц, 1H), 3,35 (дд, J=15,84, 5,98 Гц, 1H), 3,61 (с, 3H), 3,89 (с, 3H), 5,38-5,50 (м, 1H), 7,11-7,34 (м, 3H), 7,43 (с, 1H), 7,59 (д, J=7,47 Гц, 1H)
[0313]
Стадия 6
1-(5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-ил)этан-1-он
[0314]
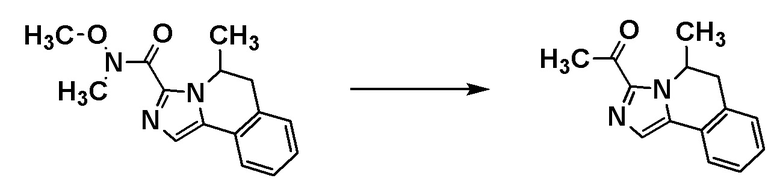
[0315]
N-метокси-N,5-диметил-5,6-дигидроимидазо[5,1-a]изохинолин-3-карбоксамид (80 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (2 мл). При охлаждении льдом к смеси по каплям добавляли 1M раствор смеси метилмагнийбромид/тетрагидрофуран (0,59 мл), и смесь перемешивали в течение 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения (46,3 мг).
1H-ЯМР (400 МГц, CDCl3) 1,24 (д, J=6,70 Гц, 3H), 2,68 (с, 3H), 2,82 (дд, J=15,95, 1,39 Гц, 1H), 3,33 (дд, J=15,95, 6,47 Гц, 1H), 5,61-5,73 (м, 1H), 7,23-7,33 (м, 3H), 7,48 (с, 1H), 7,57-7,64 (м, 1H)
[0316]
Стадия 7
1,1,1-трифтор-2-(5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-ил)пропан-2-ол
[0317]
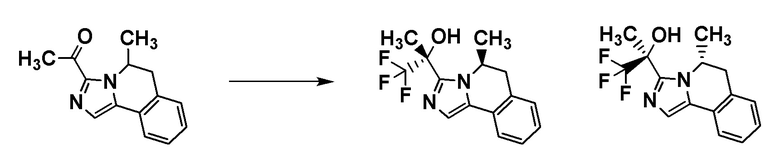
[0318]
1-(5-метил-5,6-дигидроимидазо[5,1-a]изохинолин-3-ил)этан-1-он (46,3 мг), полученный на предыдущей стадии, смешивали с диметилформамидом (1 мл). При охлаждении льдом добавляли фторид цезия (7,42 мг) и (трифторметил)триметилсилан (54,1 мкл), и смесь перемешивали при комнатной температуре в течение 30 мин. При охлаждении льдом добавляли метанол (0,332 мл) и карбонат калия (40,5 мг), и смесь перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой последовательно промывали два раза водой и один раз насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток последовательно очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата) и тонкослойной хроматографией на силикагеле (гексан:этилацетат=2:1), с получением указанного в заголовке соединения (50,8 мг) в виде рацемата.
1H-ЯМР (400 МГц, CDCl3) 1,21 (д, J=6,47 Гц, 3H), 1,95 (с, 3H), 2,77 (дд, J=15,49, 1,62 Гц, 1H), 3,28 (дд, J=15,49, 5,32 Гц, 1H), 3,52 (с, 1H), 5,10-5,20 (м, 1H), 7,17-7,30 (м, 5H), 7,33 (с, 1H), 7,54 (д, J=7,40 Гц, 1H)
[0319]
Пример получения 5
Синтез 2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ола (соединение примера 34) и 2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ола (соединение примера 35)
[0320]
Стадия 1
Метил-4-гидроксипентаноат
[0321]
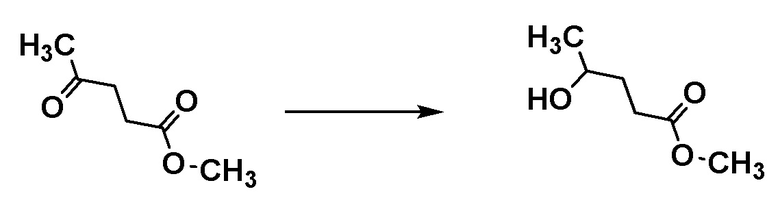
[0322]
Метил-4-оксопентаноат (14 г) смешивали с метанолом (112 мл). Смесь охлаждали до -78°C, добавляли боргидрид натрия (4,07 г), и смесь перемешивали при -50°C в течение 2 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения в виде неочищенного продукта (11,15 г). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0323]
Стадия 2
Метил-1-(5-метокси-5-оксопентан-2-ил)-1H-имидазол-5-карбоксилат
[0324]
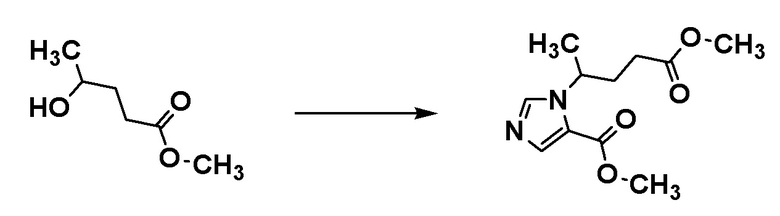
[0325]
Смешивали метил-1H-имидазол-5-карбоксилат (3 г) и толуол (24 мл). К смеси добавляли метил-4-гидроксипентаноат в виде неочищенного продукта (4,85 г), полученный на предыдущей стадии, и триоктилфосфин (10,58 г). Смесь нагревали до 80°C, по каплям добавляли диизопропил азодикарбоксилат (5,77 г), и смесь перемешивали в течение 3 час. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле (от этилацетат:гексан=50:50 до этилацетат, затем этилацетат:метанол=90:10), с получением указанного в заголовке соединения (2,6 г).
1H-ЯМР (400 МГц, CDCl3) 1,52 (д, J=6,94 Гц, 3H), 2,05-2,35 (м, 4H), 3,63 (с, 3H), 3,83 (с, 3H), 5,17-5,30 (м, 1H), 7,70-7,74 (м, 2H)
[0326]
Стадия 3
Метил-5-метил-8-оксо-5,6,7,8-тетрагидроимидазо[1,5-a]пиридин-7-карбоксилат
[0327]
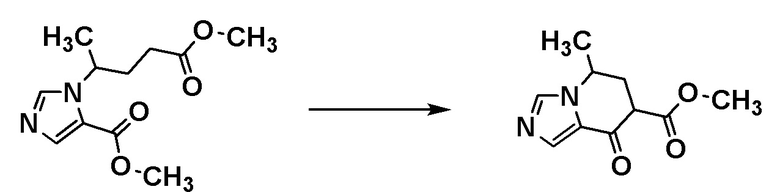
[0328]
Метил-1-(5-метокси-5-оксопентан-2-ил)-1H-имидазол-5-карбоксилат (1,48 г), полученный на предыдущей стадии, смешивали с тетрагидрофураном (45 мл). К смеси добавляли трет-бутоксид калия (0,76 г), и смесь перемешивали при комнатной температуре в течение 3 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде неочищенного продукта (1,04 г). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0329]
Стадия 4
5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он
[0330]
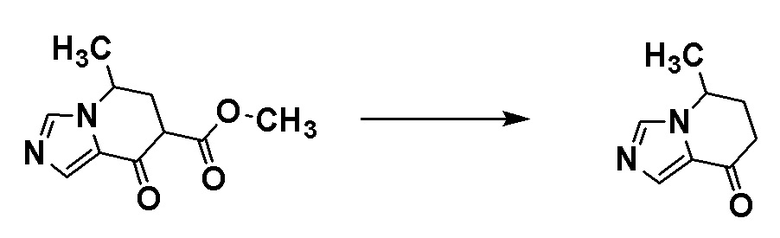
[0331]
Неочищенный продукт (1 г) метил-5-метил-8-оксо-5,6,7,8-тетрагидроимидазо[1,5-a]пиридин-7-карбоксилат, полученный на предыдущей стадии, смешивали с диметилсульфоксидом (6,82 мл). К смеси добавляли хлорид натрия (0,421 г) и воду (1,73 мл), и смесь перемешивали при 160°C в течение 3 час. Реакционную смесь концентрировали при пониженном давлении для выпаривания воды и дважды очищали катионообменной хроматографией на колонке (от метанола до 1Н раствора аммиака в метаноле), с получением указанного в заголовке соединения (0,714 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,54 (д, J=6,70 Гц, 3H), 1,86-2,02 (м, 1H), 2,16-2,28 (м, 1H), 2,52-2,59 (м, 2H), 4,35-4,50 (м, 1H), 7,63 (д, J=0,69 Гц, 1H), 8,05 (с, 1H)
[0332]
Стадия 5
7-(циклопропанкарбонил)-5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он
[0333]
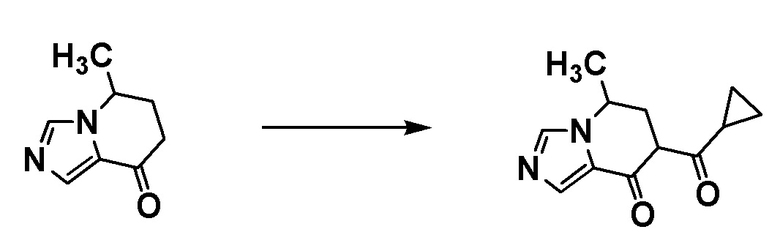
[0334]
5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он (100 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (1,5 мл). При охлаждении льдом к смеси по каплям добавляли раствор смеси 1,1 M бис(триметилсилил)амид лития/н-гексан (0,908 мл), и смесь перемешивали в течение 40 мин. К реакционной смеси добавляли циклопропанкарбонил хлорид (77 мг), и смесь перемешивали при 0°C в течение 5 мин и при комнатной температуре в течение 25 мин. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (от этилацетата до этилацетат:метанол=90:10), с получением указанного в заголовке соединения в виде неочищенного продукта (91,1 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0335]
Стадия 6
Смесь 3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридина и 3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридина
[0336]
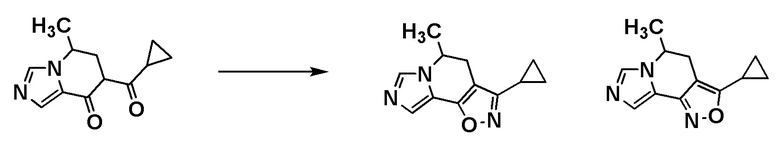
[0337]
Неочищенный продукт (90 мг) 7-(циклопропанкарбонил)-5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он, полученный на предыдущей стадии, смешивали с уксусной кислотой (0,9 мл). К смеси добавляли гидроксиламин гидрохлорид (86 мг), и смесь перемешивали при 90°C в течение 40 мин. После охлаждения до комнатной температуры к реакционной смеси добавляли серную кислоту (81 мг), и смесь перемешивали при 100°C в течение 2 час и при 110°C в течение 4 час. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток последовательно очищали хроматографией на силикагеле (от этилацетата до этилацетат:метанол=90:10) и препаративной тонкослойной хроматографией (этилацетат:метанол=90:10), с получением указанного в заголовке соединения в виде неочищенного продукта (45,2 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0338]
Стадия 7
Смесь 1-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин-7-ил)этан-1-она и 1-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин-7-ил)этан-1-она
[0339]
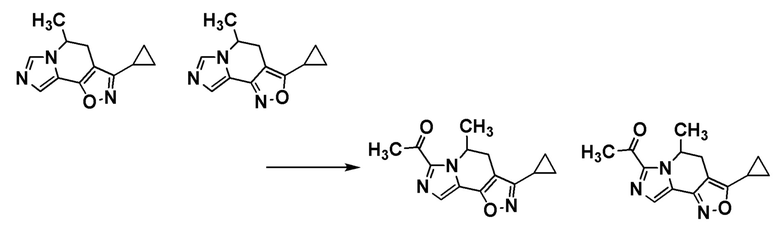
[0340]
Неочищенный продукт (43,2 мг) 3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин и 3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин, полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,864 мл). Смесь охлаждали до -78°C и по каплям добавляли 2M раствор диизопропиламида лития (0,251 мл). Смесь перемешивали при -78°C в течение 40 мин. К смеси добавляли N-метокси-N-метилацетамид (62,1 мг), и смесь перемешивали при -78°C в течение 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (гексан:этилацетат от 75:25 до 50:50), с получением указанного в заголовке соединения в виде неочищенного продукта (18,8 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0341]
Стадия 8
2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ол (соединение примера 34) и 2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ол (соединение примера 35)
[0342]
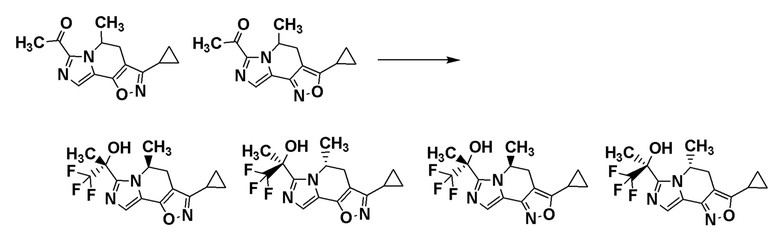
[0343]
Неочищенный продукт (17 мг) 1-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин-7-ил)этан-1-он и 1-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин-7-ил)этан-1-он, полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,34 мл). При охлаждении льдом добавляли фторид цезия (7,42 мг) и (трифторметил)триметилсилан (54,1 мкл), и смесь перемешивали при комнатной температуре в течение 1 час. При охлаждении льдом добавляли метанол (0,332 мл) и карбонат калия (40,5 мг), и смесь перемешивали в течение 1,5 час, давая возможность нагреваться до комнатной температуры. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь разделяли, и органический слой очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением соединения примера 34 (5,2 мг) и соединения примера 35 (8,4 мг), каждого из них в виде рацемата. Структуру изоксазольного фрагмента определяли методом ЯМР.
2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[5,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ол
1H-ЯМР (400 МГц, CDCl3) 1,02-1,04 (м, 4H), 1,30 (д, J=3,29 Гц, 3H), 2,00 (с, 3H), 2,63-2,67 (м, 1H), 3,05-3,11 (м, 1H), 3,33-3,37 (м, 1H), 5,30-5,36 (м, 1H), 7,41 (с, 1H).
2-(3-циклопропил-5-метил-4,5-дигидроимидазо[1,5-a]изоксазолo[3,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ол
1H-ЯМР (400 МГц, CDCl3) 1,06-1,16 (м, 4H), 1,31 (д, J=6,58 Гц, 3H), 1,85 (с, 3H), 2,79 (дд, J=15,55, 1,79 Гц, 1H), 2,96-3,02 (м, 1H), 3,87-4,04 (м, 1H), 5,42-5,44 (м, 1H), 7,60 (с, 1H).
[0344]
Пример получения 6
Синтез 1,1,1-трифтор-2-(6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин-8-ил)пропан-2-ола (соединение примера 6)
[0345]
Стадия 1
7-((диметиламино)метилен)-5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он
[0346]
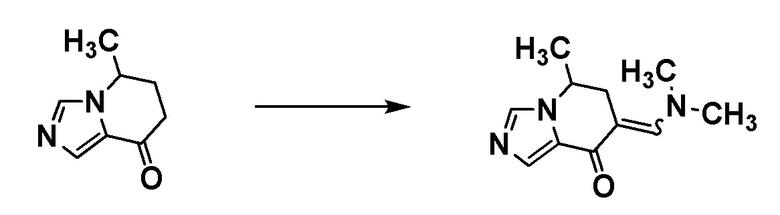
[0347]
5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он (250 мг) смешивали с 1,4-диоксаном (1,25 мл). К смеси добавляли трет-бутоксибис(диметиламино)метан (435 мг), и смесь перемешивали при 110°C в течение 1 час. Реакционную смесь концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде неочищенного продукта. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0348]
Стадия 2
6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин
[0349]
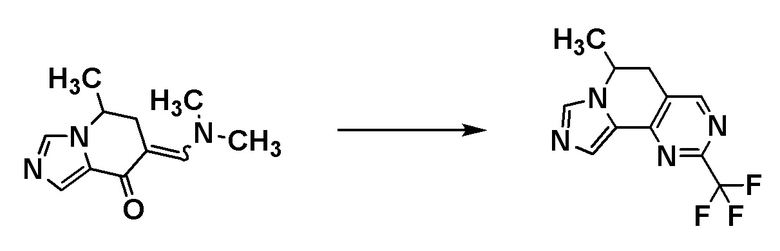
[0350]
Неочищенный продукт 7-((диметиламино)метилен)-5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он, полученный на предыдущей стадии, смешивали с этанолом (0,95 мл). К смеси добавляли трифторацетамидин (142 мг), и смесь перемешивали при комнатной температуре в течение 15 мин. К смеси добавляли этоксид натрия (86 мг), и смесь перемешивали в течение ночи при нагревании с обратным холодильником. Реакционную смесь оставляли охлаждаться до комнатной температуры, добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (гексан:этилацетат от 95:5 до 60:40), с получением указанного в заголовке соединения (172,6 мг).
1H-ЯМР (400 МГц, CDCl3) 1,62 (д, J=6,47 Гц, 3H), 2,94 (дд, J=16,18, 8,32 Гц, 1H), 3,25 (дд, J=16,18, 5,09 Гц, 1H), 4,44-4,58 (м, 1H), 7,76 (с, 1H), 8,00 (с, 1H), 8,64 (с, 1H).
[0351]
Стадия 3
1-(6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин-8-ил)этан-1-он
[0352]
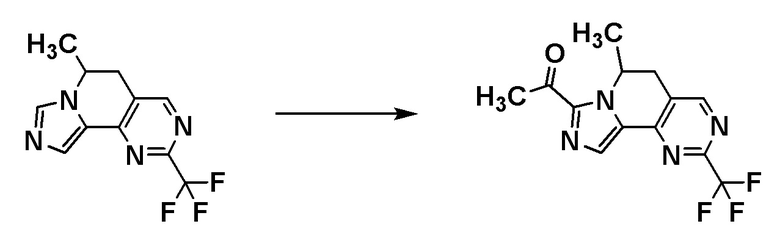
[0353]
6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин (50 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,5 мл). Смесь охлаждали до -78°C и по каплям добавляли 2M раствор диизопропиламида лития (0,118 мл). Смесь перемешивали при -78°C в течение 1 час. К смеси добавляли N-метокси-N-метилацетамид (40,6 мг), и смесь перемешивали при -78°C в течение 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения (17,2 мг).
1H-ЯМР (400 МГц, CDCl3) 1,30 (д, J=6,70 Гц, 3H), 3,00 (дд, J=16,30, 1,16 Гц, 1H), 3,38 (дд, J=16,30, 6,59 Гц, 1H), 5,77-5,86 (м, 1H), 8,04 (с, 1H), 8,74 (с, 1H)
[0354]
Стадия 4
1,1,1-трифтор-2-(6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин-8-ил)пропан-2-ол
[0355]
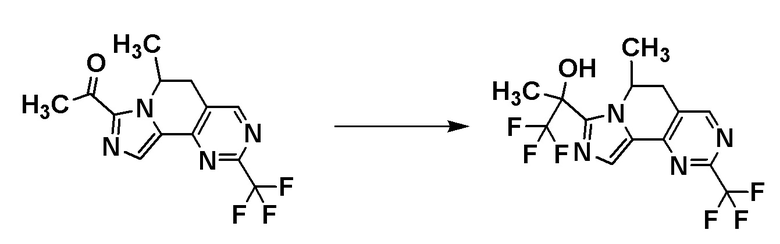
[0356]
1-(6-метил-2-(трифторметил)-5,6-дигидроимидазо[1’,5’:1,2]пиридо[3,4-d]пиримидин-8-ил)этан-1-он (17,2 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,172 мл). При охлаждении льдом к смеси добавляли фторид цезия (1,764 мг) и (трифторметил)триметилсилан (16,51 мг), и смесь перемешивали при комнатной температуре в течение 1 час. При охлаждении льдом к смеси добавляли (трифторметил)триметилсилан (16,51 мг), и смесь перемешивали при комнатной температуре в течение 20 мин. При охлаждении льдом к смеси добавляли (трифторметил)триметилсилан (16,51 мг), и смесь перемешивали при комнатной температуре в течение 20 мин. При охлаждении льдом к реакционной смеси добавляли метанол (0,103 мл) и карбонат калия (9,63 мг), и смесь перемешивали при комнатной температуре в течение 30 мин. К смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой последовательно промывали водой и насыщенным водным раствором хлорида натрия, и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией на силикагеле (гексан:этилацетат=1:1). К полученному неочищенному продукту добавляли смесь этилацетат/гексан=1/1, и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (7,1 мг) в виде рацемата.
1H-ЯМР (400 МГц, ДМСО-D6) 1,20 (д, J=6,47 Гц, 3H), 1,84 (с, 3H), 3,14 (д, J=16,41 Гц, 1H), 3,23 (дд, J=16,41, 5,32 Гц, 1H), 5,34-5,44 (м, 1H), 7,40 (с, 1H), 7,81 (с, 1H), 8,92 (с, 1H)
[0357]
Пример получения 7
Синтез 2-(1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ола (соединение примера 24)
[0358]
Стадия 1
1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин
[0359]
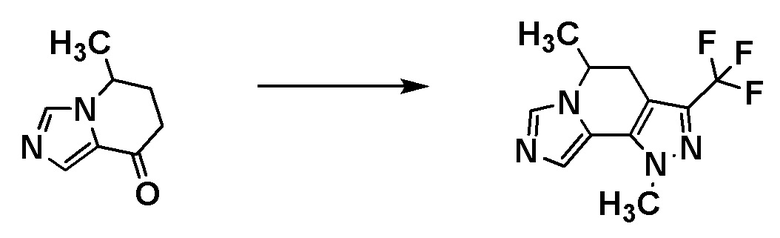
[0360]
5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он (150 мг) смешивали с тетрагидрофураном (1,5 мл). К смеси добавляли этилтрифторацетат (312 мг). При охлаждении льдом к смеси добавляли гидрид натрия (88 мг), и смесь перемешивали при комнатной температуре в течение 10 мин и при 60°C в течение 1 час. После охлаждения до комнатной температуры к реакционной смеси добавляли метилгидразин (138 мг) и этанол (1,5 мл). Реакционную смесь перемешивали при 95°C в течение 3 час. Реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с уксусной кислотой. К полученному остатку добавляли уксусную кислоту (1,5 мл) и метилгидразин (138 мг), и смесь перемешивали при комнатной температуре в течение 1 час. Реакционную смесь концентрировали, добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (этилацетат:метанол от 95:5 до 60:40), с получением указанного в заголовке соединения (170,4 мг). Положение замещения метильной группы в пиразольном фрагменте определяли методом двумерного ЯМР.
1H-ЯМР (400 МГц, ДМСО-D6) 1,44 (д, J=6,47 Гц, 3H), 2,72 (дд, J=15,95, 7,86 Гц, 1H), 3,09 (дд, J=15,95, 5,55 Гц, 1H), 4,01 (с, 3H), 4,41-4,52 (м, 1H), 7,46 (с, 1H), 7,96 (с, 1H)
[0361]
Стадия 2
1-(1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-он
[0362]
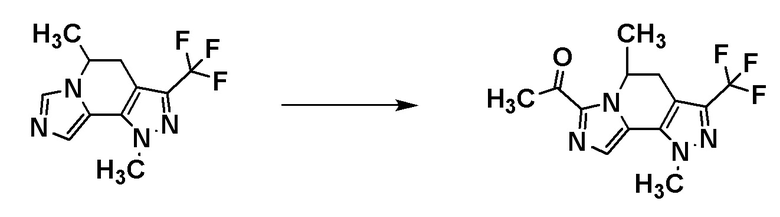
[0363]
1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин (170,4 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (5,112 мл). Смесь охлаждали до -78°C и по каплям добавляли 2M раствор диизопропиламида лития (0,831 мл). Смесь перемешивали при -78°C в течение 30 мин. К смеси добавляли N-метокси-N-метилацетамид (206 мг), и смесь перемешивали при -78°C в течение 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения (150 мг).
[0364]
Стадия 3
2-(1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)-1,1,1-трифторпропан-2-ол
[0365]
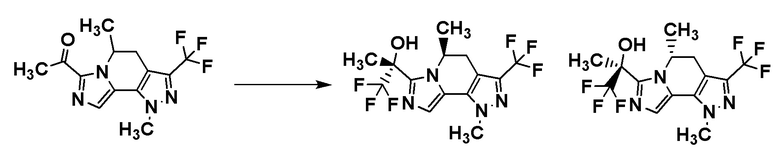
[0366]
1-(1,5-диметил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-он (150 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (1,5 мл). При охлаждении льдом к смеси добавляли фторид цезия (1,764 мг) и (трифторметил)триметилсилан (143 мг), и смесь перемешивали при комнатной температуре в течение 45 мин. При охлаждении льдом к смеси добавляли (трифторметил)триметилсилан (143 мг), и смесь перемешивали при комнатной температуре в течение 15 мин. При охлаждении льдом к реакционной смеси добавляли метанол (0,103 мл) и карбонат калия (9,63 мг), и смесь перемешивали при комнатной температуре в течение 1 час. К смеси добавляли насыщенный водный раствор хлорида аммония, и смесь дважды экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения (127,7 мг) в виде рацемата.
1H-ЯМР (400 МГц, ДМСО-D6) 1,16 (д, J=6,58 Гц, 3H), 1,84 (с, 3H), 2,90 (д, J=16,14 Гц, 1H), 2,97 (дд, J=16,14, 5,38 Гц, 1H), 4,04 (с, 3H), 5,32-5,43 (м, 1H), 7,27 (с, 1H), 7,46 (с, 1H)
[0367]
Пример получения 8
Синтез 1,1,1-трифтор-2-(5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ола (соединение примера 50)
[0368]
Стадия 1
5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин
[0369]
[0370]
5-метил-6,7-дигидроимидазо[1,5-a]пиридин-8(5H)-он (200 мг) смешивали с тетрагидрофураном (8 мл). К смеси добавляли этилтрифторацетат (227 мг). К смеси добавляли гидрид натрия (80 мг), и смесь перемешивали при комнатной температуре в течение 1 час и при 70°C в течение 1,5 час. После охлаждения до комнатной температуры к реакционной смеси добавляли этилтрифторацетат (37,8 мг), и смесь перемешивали при 70°C в течение 30 мин. После охлаждения до комнатной температуры к реакционной смеси добавляли уксусную кислоту (80 мг), и смесь концентрировали при пониженном давлении. К полученному остатку добавляли уксусную кислоту (2 мл), гидразин моногидрат (233 мг) и серную кислоту (261 мг), и смесь перемешивали при 100°C в течение 2 час. Реакционную смесь очищали хроматографией на колонке с силикагелем (от этилацетата до этилацетат:метанол=80:20), с получением указанного в заголовке соединения в виде неочищенного продукта (316,8 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0371]
Стадия 2
Смесь 1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридина и 2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридина
[0372]
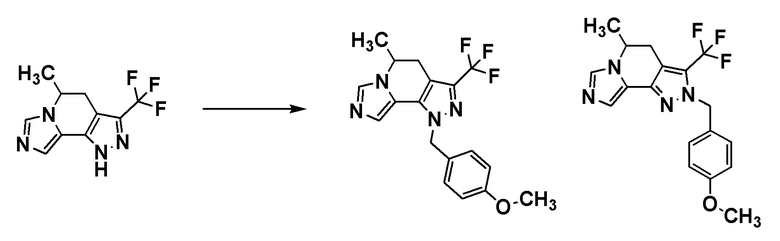
[0373]
Неочищенный продукт (82 мг) 5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин, полученный на предыдущей стадии, смешивали с диметилформамидом (0,82 мл). При охлаждении льдом к смеси добавляли гидрид натрия (16,25 мг), и смесь перемешивали в течение 30 мин. При охлаждении льдом к реакционной смеси добавляли 4-метоксибензилхлорид (58,3 мг), и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=12:88 до этилацетата), с получением указанного в заголовке соединения в виде неочищенного продукта (67,1 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0374]
Стадия 3
Смесь 1-(1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-она и 1-(2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-2H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-она
[0375]
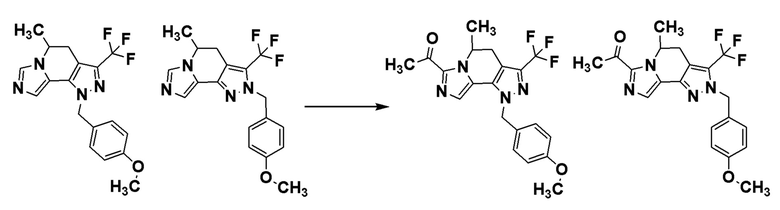
[0376]
Неочищенный продукт (61,1 мг) 1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин и 2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин, полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,611 мл). Смесь охлаждали до -78°C и по каплям добавляли 2M раствор диизопропиламида лития (0,211 мл). Смесь перемешивали при -78°C в течение 45 мин. К смеси добавляли N-метокси-N-метилацетамид (52,2 мг), и смесь перемешивали при -78°C в течение 40 мин. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, смесь оставляли нагреваться до комнатной температуры и разделяли. Органический слой очищали хроматографией на колонке с силикагелем (от гексан:этилацетат=20:80 до этилацетата), с получением указанного в заголовке соединения в виде неочищенного продукта (48 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0377]
Стадия 4
Смесь 1,1,1-трифтор-2-(1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ола и 1,1,1-трифтор-2-(2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-2H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ола
[0378]
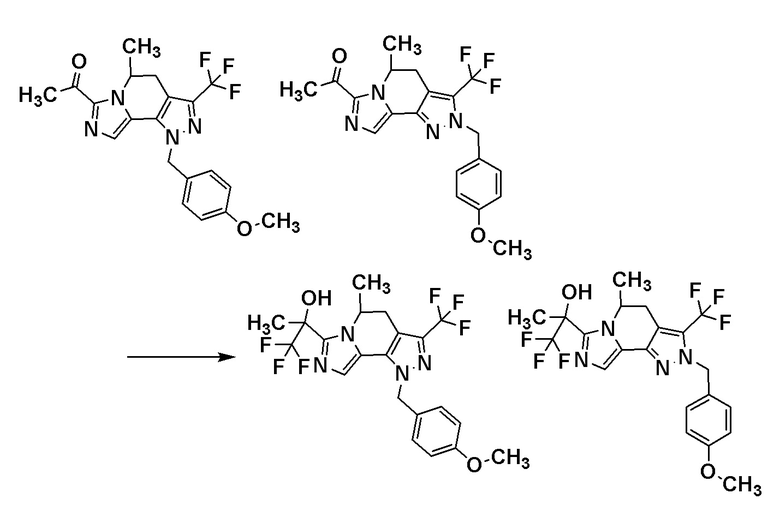
[0379]
Неочищенный продукт (45 мг) 1-(1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-он и 1-(2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-2H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)этан-1-он, полученный на предыдущей стадии, смешивали с тетрагидрофураном (0,9 мл). При охлаждении льдом к смеси добавляли фторид цезия (5,07 мг) и (трифторметил)триметилсилан (47,5 мг), и смесь перемешивали при комнатной температуре в течение 2 час. При охлаждении льдом к реакционной смеси добавляли метанол (0,45 мл) и карбонат калия (77 мг), и смесь перемешивали в течение 2 час. К смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией на силикагеле (гексан:этилацетат=50:50), с получением указанного в заголовке соединения в виде неочищенного продукта (11,8 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0380]
Стадия 5
1,1,1-трифтор-2-(5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ол
[0381]
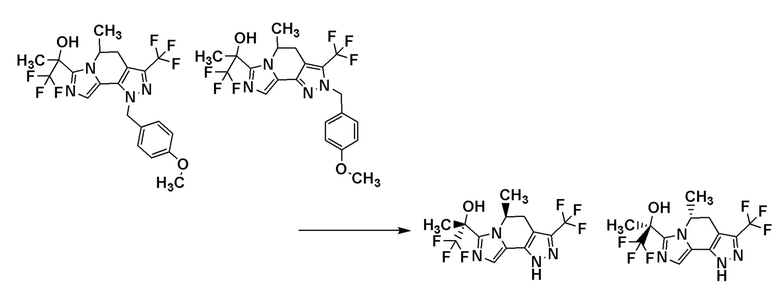
[0382]
Неочищенный продукт (11,5 мг) 1,1,1-трифтор-2-(1-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-1H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ол и 1,1,1-трифтор-2-(2-(4-метоксибензил)-5-метил-3-(трифторметил)-4,5-дигидро-2H-имидазо[1,5-a]пиразолo[3,4-c]пиридин-7-ил)пропан-2-ол, полученный на предыдущей стадии, смешивали с дихлорметаном (0,46 мл). К смеси добавляли трифторуксусную кислоту (0,0467 мл), и смесь перемешивали при комнатной температуре в течение 2 дней и при 60°C в течение 2 час. К смеси добавляли трифторуксусную кислоту (0,3 мл), и смесь перемешивали при 70°C в течение 2 час и при 80°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении, и добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия. Смесь дважды экстрагировали этилацетатом, и органический слой сушили над сульфатом магния. Сульфат магния отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией на силикагеле (гексан:этилацетат=1:2), с получением указанного в заголовке соединения (6,5 мг) в виде рацемата.
1H-ЯМР (400 МГц, ДМСО-D6) 1,17 (д, J=6,58 Гц, 3H), 1,84 (с, 3H), 2,87-3,02 (м, 2H), 3,41-3,48 (м, 2H), 5,30-5,45 (м, 1H), 7,23 (с, 1H), 14,06 (с, 1H)
[0383]
Пример получения 9
Синтез (R)-1,1,1-трифтор-2-((S)-5-метил-9-(трифторметил)-5,6-дигидропиразолo[1,5-a][1,2,4]триазолo[3,4-c]пиразин-3-ил)пропан-2-ола (соединение примера 13)
[0384]
Стадия 1
Трет-бутил-(R)-2-(3,3,3-трифтор-2-гидрокси-2-метилпропаноил)гидразин-1-карбоксилат
[0385]
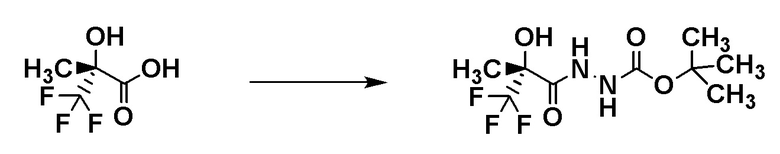
[0386]
Трет-бутоксикарбонилгидразин (1,17 г) смешивали с ацетонитрилом (15 мл). К смеси добавляли (R)-3,3,3-трифтор-2-гидрокси-2-метилпропионовую кислоту (1 г). При охлаждении льдом к смеси добавляли 1-гидроксибензотриазол моногидрат (0,581 г) и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (1,455 г), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, к полученному остатку добавляли этилацетат, и смесь промывали последовательно 0,5Н соляной кислотой и насыщенным водным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия. Сульфат натрия отфильтровывали, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (1,66 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,27-1,42 (м, 9H), 1,46 (с, 3H), 6,99 (с, 1H), 8,31 (с, 0,25H), 8,76 (с, 0,75H), 9,64-9,96 (м, 1H)
[0387]
Стадия 2
(R)-3,3,3-трифтор-2-гидрокси-2-метилпропангидразид
[0388]
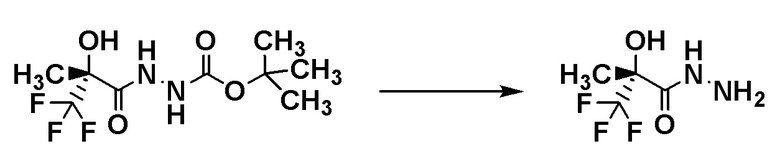
[0389]
Трет-бутил-(R)-2-(3,3,3-трифтор-2-гидрокси-2-метилпропаноил)гидразин-1-карбоксилат (0,6 г), полученный на предыдущей стадии, смешивали с этилацетатом (1,114 мл). При охлаждении льдом к смеси добавляли трифторуксусную кислоту (2,78 мл), и смесь перемешивали при комнатной температуре в течение 5 час. Реакционную смесь концентрировали при пониженном давлении и очищали катионообменной хроматографией на колонке (от метанола до 1Н раствора аммиака в метаноле), с получением указанного в заголовке соединения (155,8 мг).
1H-ЯМР (400 МГц, ДМСО-D6) 1,44 (с, 3H), 4,32 (с, 2H), 6,82 (с, 1H), 9,28 (уш.с, 1H)
[0390]
Стадия 3
Метил-(S)-1-(2-((трет-бутоксикарбонил)амино)пропил)-3-(трифторметил)-1H-пиразол-5-карбоксилат
[0391]
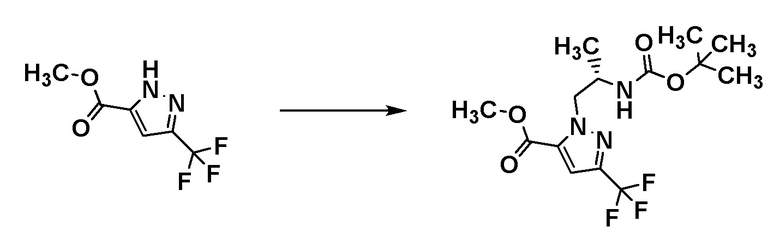
[0392]
Метил-3-(трифторметил)-1H-пиразол-5-карбоксилат (200 мг), N-(трет-бутоксикарбонил)-L-аланинол (271 мг) и трифенилфосфин (405 мг) смешивали с тетрагидрофураном (2 мл). Смесь нагревали до 80°C и по каплям добавляли диизопропилазодикарбоксилат (0,3 мл). Реакционную смесь перемешивали при 80°C в течение 2 час и при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонке с силикагелем (гексан:этилацетат от 92:8 до 44:66), с получением указанного в заголовке соединения (388,9 мг).
1H-ЯМР (400 МГц, CDCl3) 1,18 (д, J=6,70 Гц, 3H), 1,31 (с, 9H), 3,90 (с, 3H), 4,14-4,29 (м, 1H), 4,37-4,52 (м, 1H), 4,55-4,79 (м, 2H), 7,06 (с, 1H)
[0393]
Стадия 4
(S)-6-метил-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин-4(5H)-он
[0394]
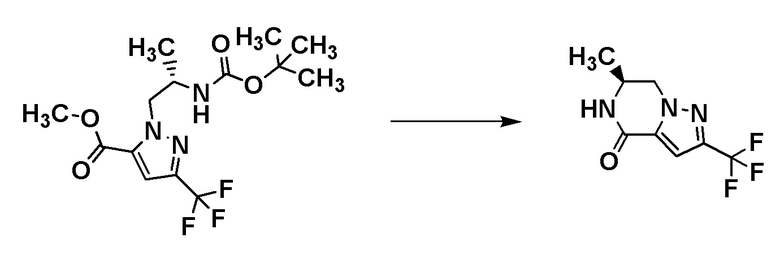
[0395]
Метил-(S)-1-(2-((трет-бутоксикарбонил)амино)пропил)-3-(трифторметил)-1H-пиразол-5-карбоксилат (362 мг), полученный на предыдущей стадии, смешивали с хлороформом (2 мл). При охлаждении льдом к смеси добавляли трифторуксусную кислоту (2 мл), и смесь перемешивали при комнатной температуре в течение 3 час. Реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом. К полученному остатку добавляли метанол (3,62 мл) и карбонат натрия (437 мг), и смесь перемешивали при комнатной температуре в течение 3 час. К реакционной смеси добавляли воду, и смесь перемешивали при комнатной температуре в течение 30 мин. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (193,4 мг).
1H-ЯМР (400 МГц, ДМСО-D6) 1,22 (д, J=6,47 Гц, 3H), 3,97-4,16 (м, 2H), 4,49 (дд, J=12,60, 4,05 Гц, 1H), 7,17 (с, 1H), 8,50 (с, 1H)
[0396]
Стадия 5
(S)-6-метил-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин-4(5H)-тион
[0397]
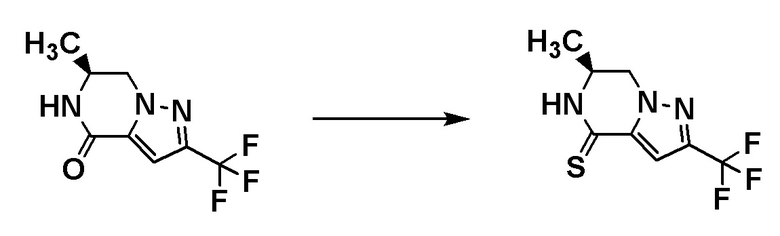
[0398]
(S)-6-метил-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин-4(5H)-он (193,4 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (3,868 мл). К смеси добавляли 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфид (250 мг), и смесь перемешивали при 70°C в течение 7 час. Реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонке с силикагелем (гексан:этилацетат от 92:8 до 44:66), с получением указанного в заголовке соединения (199,8 мг).
1H-ЯМР (400 МГц, CDCl3) 1,47 (д, J=6,47 Гц, 3H), 4,03-4,23 (м, 2H), 4,48 (дд, J=12,25, 3,47 Гц, 1H), 7,29 (с, 1H), 7,62 (уш.с, 1H)
[0399]
Стадия 6
(S)-6-метил-4-(метилтио)-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин гидроиодид
[0400]
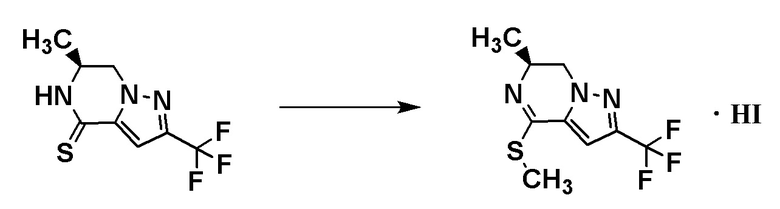
[0401]
(S)-6-метил-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин-4(5H)-тион (80 мг), полученный на предыдущей стадии, смешивали с ацетоном (0,8 мл). При охлаждении льдом к смеси добавляли метилиодид (57,9 мг), и смесь перемешивали при комнатной температуре в течение ночи. При охлаждении льдом к смеси добавляли метилиодид (57,9 мг), и смесь перемешивали при комнатной температуре в течение 7 час. Реакционную смесь концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде неочищенного продукта (128 мг). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
[0402]
Стадия 7
(R)-1,1,1-трифтор-2-((S)-5-метил-9-(трифторметил)-5,6-дигидропиразолo[1,5-a][1,2,4]триазолo[3,4-c]пиразин-3-ил)пропан-2-ол
[0403]
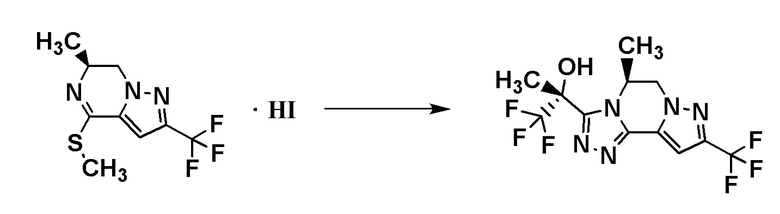
[0404]
Неочищенный продукт (128 мг) (S)-6-метил-4-(метилтио)-2-(трифторметил)-6,7-дигидропиразолo[1,5-a]пиразин гидроиодид, полученный на предыдущей стадии, и (R)-3,3,3-трифтор-2-гидрокси-2-метилпропангидразид (61 мг), полученный на стадии 2, смешивали с изопропанолом (1 мл). К смеси добавляли уксусную кислоту (0,039 мл), и смесь перемешивали при 100°C в течение 1,5 час. Смесь оставляли охлаждаться до комнатной температуры, к реакционной смеси добавляли ацетат натрия (56 мг) и воду (0,256 мл), и смесь перемешивали при 100°C в течение ночи. Реакционную смесь последовательно очищали катионообменной хроматографией на колонке (метанол) и хроматографией на колонке с силикагелем (от гексан:этилацетат=88:12 до этилацетата), с получением указанного в заголовке соединения (32,2 мг).
1H-ЯМР (400 МГц, ДМСО-D6) 1,30 (д, J=6,58 Гц, 3H), 1,91 (с, 3H), 4,62 (д, J=13,75 Гц, 1H), 4,70 (дд, J=13,75, 4,04 Гц, 1H), 5,31-5,42 (м, 1H), 7,47 (с, 1H), 7,71 (с, 1H)
[0405]
Пример получения 10
Синтез (R)-2-((S)-9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ола (соединение примера 23)
[0406]
Стадия 1
Этил-5-гидрокси-1H-пиразол-3-карбоксилат
[0407]
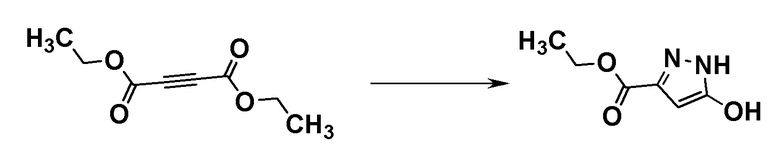
[0408]
Диэтилацетилендикарбоксилат (250 мл) смешивали с ацетонитрилом (797 мл). При охлаждении льдом последовательно по каплям добавляли гидразин моногидрат (80 мл) и уксусную кислоту (17,9 мл), и смесь перемешивали при комнатной температуре в течение 24 час. Реакционную смесь перемешивали при охлаждении льдом в течение 2 час, и выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (156,71 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,26 (т, J=7,05 Гц, 3H), 3,94-4,53 (м, 2H), 5,39-6,26 (м, 1H), 9,47-11,36 (уш.м, 1H), 12,73 (с, 1H)
[0409]
Стадия 2
Этил-5-гидрокси-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксилат
[0410]
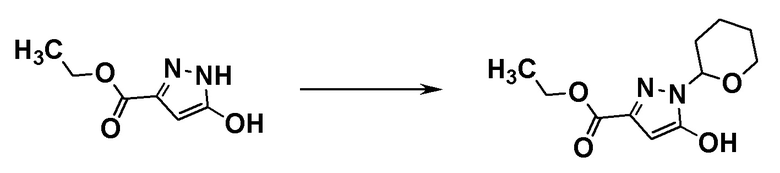
[0411]
Этил-5-гидрокси-1H-пиразол-3-карбоксилат (300 г), полученный на предыдущей стадии, смешивали с ацетонитрилом (720 мл). При комнатной температуре добавляли пиридиний п-толуолсульфонат (24,14 г). К смеси по каплям добавляли 3,4-дигидро-2H-пиран (170 г), и смесь перемешивали при комнатной температуре в течение 2 дней. Реакционную смесь перемешивали при охлаждении льдом в течение 2 час, и выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (410 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,25 (т, J=7,05 Гц, 3H), 1,42-1,77 (м, 4H), 1,88-1,99 (м, 1H), 2,12-2,26 (м, 1H), 3,49-3,61 (м, 1H), 3,85-3,94 (м, 1H), 4,13-4,29 (м, 2H), 5,27-5,35 (м, 1H), 5,73 (с, 1H), 11,63 (с, 1H)
[0412]
Стадия 3
Этил-5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксилат
[0413]
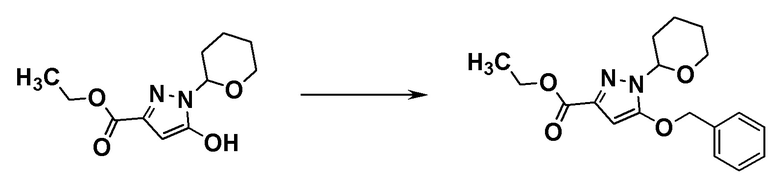
[0414]
Этил-5-гидрокси-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксилат (205 г), полученный на предыдущей стадии, смешивали с N-метилпирролидоном (615 мл). При охлаждении водой по каплям добавляли карбонат калия (142 г), бензилбромид (175 г), и смесь перемешивали при комнатной температуре в течение 2 час. К смеси по каплям добавляли уксусную кислоту (25,6 г), и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду (1,23 л), и смесь экстрагировали изопропилацетатом (2,05 л). Органический слой промывали два раза 10% водным раствором хлорида натрия (1,025 л) и концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с изопропилацетатом (2,05 л). К полученному твердому веществу добавляли изопропилацетат (615 мл), и смесь перемешивали с нагреванием при 55°C для растворения твердого вещества. Раствор оставляли охлаждаться до 40°C и инокулировали затравочные кристаллы. К полученной суспензии по каплям добавляли н-гептан (2,46 л), смесь оставляли охлаждаться до комнатной температуры и перемешивали при комнатной температуре в течение 2 час. Смесь перемешивали при охлаждении льдом в течение 2 час, и выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (235,15 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,26 (т, J=7,05 Гц, 3H), 1,45-1,56 (м, 2H), 1,57-1,80 (м, 2H), 1,89-2,00 (м, 1H), 2,11-2,26 (м, 1H), 3,52-3,62 (м, 1H), 3,84-3,92 (м, 1H), 4,19-4,28 (м, 2H), 5,19-5,31 (м, 2H), 5,35-5,42 (м, 1H), 6,22 (с, 1H), 7,30-7,51 (м, 5H)
На этой стадии кристаллы указанного в заголовке соединения могут быть получены без использования затравочных кристаллов.
[0415]
Стадия 4
5-(бензилокси)-N-метокси-N-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксамид
[0416]
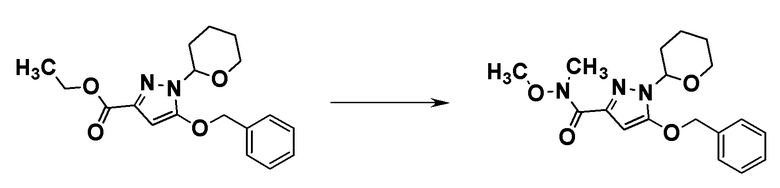
[0417]
Этил-5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксилат (235,15 г), полученный на предыдущей стадии, смешивали с 1,2-диметоксиэтаном (941 мл). К смеси по каплям добавляли 2Н водный раствор гидроксида натрия (428 мл), и смесь перемешивали при комнатной температуре в течение 3,5 час. При охлаждении водой к смеси добавляли N-метокси-N-метиламин гидрохлорид (83 г) и HOBt-H2O (21,8 г), добавляли WSC-HCl (164 г) 4 порциями, и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли 5% водный раствор гидрокарбоната натрия (941 мл), и смесь экстрагировали толуолом (1411 мл). Органический слой промывали 5% водным раствором хлорида натрия (941 мл) и концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с толуолом (2,35 л). К полученному остатку добавляли толуол, с получением раствора указанного в заголовке соединения, содержащего толуол в количестве, в 5 раз превышающем по массе исходный материал.
[0418]
Стадия 5
5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбальдегид
[0419]
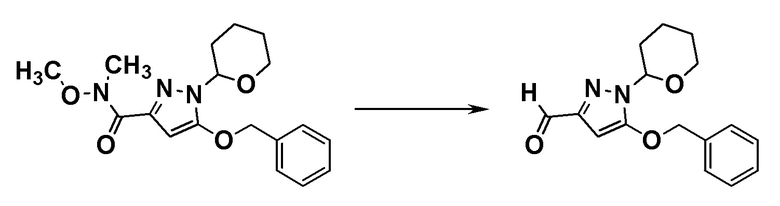
[0420]
При охлаждении льдом к раствору 5-(бензилокси)-N-метокси-N-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбоксамида в толуоле, полученному на предыдущей стадии, по каплям добавляли 70% раствор смеси бис(2-метоксиэтокси)алюмогидрид натрия/толуол (141 мл), и смесь перемешивали в течение 2,5 час. При охлаждении водой к реакционной смеси по каплям добавляли раствор смеси изопропанол (54,8 мл)/толуол (118 мл). Смесь оставляли нагреваться до комнатной температуры, добавляли смесь тартрат калия-натрия (201 г)/вода (941 мл), и смесь перемешивали в течение 1 час. Раствор разделяли, органический слой промывали два раза 5% водным раствором хлорида натрия (941 мл) и концентрировали при пониженном давлении. В полученном остатке растворитель заменяли 1,2-диметоксиэтаном (2,35 л). К полученному остатку добавляли 1,2-диметоксиэтан, с получением раствора указанного в заголовке соединения, содержащего 1,2-диметоксиэтан в количестве, в 13 раз превышающем по массе исходный материал.
[0421]
Стадия 6
5-(бензилокси)-1H-пиразол-3- карбальдегид
[0422]
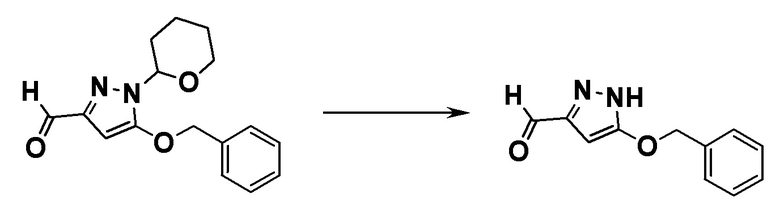
[0423]
При охлаждении льдом к раствору смеси 5-(бензилокси)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-карбальдегид/1,2-диметоксиэтан, полученному на предыдущей стадии, добавляли 1Н соляную кислоту (1423 мл), и смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (132,04 г).
1H-ЯМР (400 МГц, ДМСО-D6) 5,18 (с, 2H), 6,36 (с, 1H), 7,26-7,49 (м, 5H), 9,70 (с, 1H), 13,34 (уш.с, 1H)
[0424]
Стадия 7
(S)-2-(5-(3-(бензилокси)-1H-пиразол-5-ил)-1H-имидазол-1-ил)пропан-1-ол
[0425]
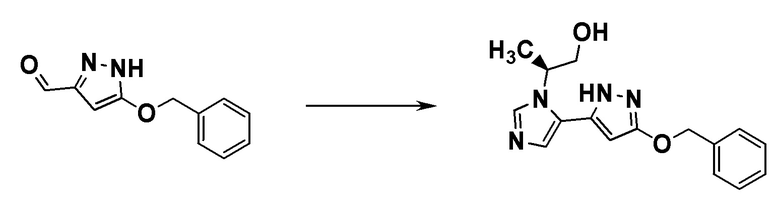
[0426]
5-(бензилокси)-1H-пиразол-3-карбальдегид (261,97 г), полученный на предыдущей стадии, смешивали с диметилформамидом (1040 мл). При комнатной температуре добавляли (S)-2-аминопропан-1-ол (102 г), и смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли карбонат калия (358 г) и добавляли п-толуолсульфонилметилизоцианид (304 г) 3 порциями, и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли трет-бутоксид калия (72,7 г), и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли 2-метилтетрагидрофуран (1572 мл) и KC FLOCK (W-300G, Nippon Paper Industries Co., Ltd., 131 г), и смесь фильтровали с использованием KC FLOCK в качестве адъюванта. К фильтрату добавляли 2-метилтетрагидрофуран (1572 мл), смесь перемешивали при комнатной температуре и фильтровали. Фильтраты объединяли и промывали 3 раза 10% водным раствором хлорида натрия (1048 мл). Первый водный слой экстрагировали 2-метилтетрагидрофураном (2620 мл) и промывали 3 раза 10% водным раствором хлорида натрия (1048 мл). Органические слои объединяли, концентрировали при пониженном давлении и подвергали азеотропной перегонке последовательно с толуолом и 2-метилтетрагидрофураном. К полученному остатку добавляли 2-метилтетрагидрофуран, с получением раствора указанного в заголовке соединения, содержащего 2-метилтетрагидрофуран в количестве, в 4 раза превышающем по массе исходный материал.
[0427]
Стадия 8
(S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин гидрохлорид
[0428]
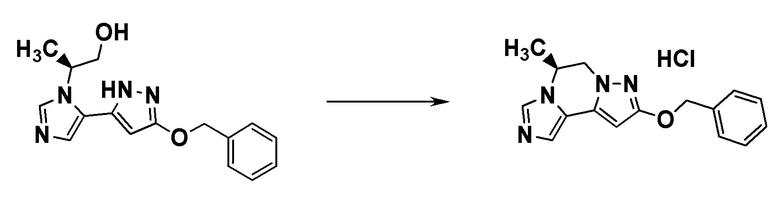
[0429]
Раствор смеси (S)-2-(5-(3-(бензилокси)-1H-пиразол-5-ил)-1H-имидазол-1-ил)пропан-1-ол/2-метилтетрагидрофуран, полученный на предыдущей стадии, смешивали с трифенилфосфином (408 г). При нагревании при 73°C по каплям добавляли 40% раствор смеси диизопропилазодикарбоксилат/толуол (763 мл), и смесь перемешивали в течение 1 час. При нагревании при 60°C по каплям добавляли раствор смеси 2Н соляная кислота/этанол (842 мл), и смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (244,13 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,59 (д, J=6,47 Гц, 3H), 4,05-4,19 (м, 1H), 4,43-4,53 (м, 1H), 4,85-4,99 (м, 1H), 5,19 (с, 2H), 6,23 (с, 1H), 7,26-7,48 (м, 5H), 8,01 (д, J=1,16 Гц, 1H), 9,29 (с, 1H)
[0430]
Стадия 9
(S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин
[0431]
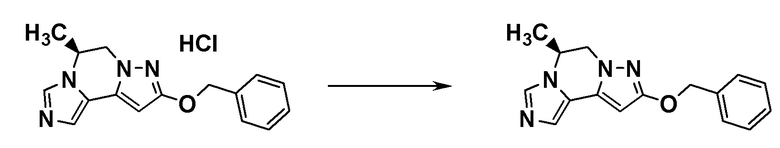
[0432]
(S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин гидрохлорид (224 г), полученный на предыдущей стадии, смешивали с 2-метилтетрагидрофураном (896 мл) и водой (448 мл). К смеси добавляли 4Н водный раствор гидроксида натрия (194 мл), и смесь перемешивали в течение 1 час. Раствор разделяли, органический слой промывали 20% водным раствором хлорида натрия (896 мл) и концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке последовательно с толуолом (1 л) и тетрагидрофураном (1,1 л). К полученному остатку добавляли тетрагидрофуран, с получением раствора указанного в заголовке соединения, содержащего тетрагидрофуран в количестве, в 5,4 раза превышающем по массе исходный материал.
[0433]
Стадия 10
(S)-1-(9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он
[0434]
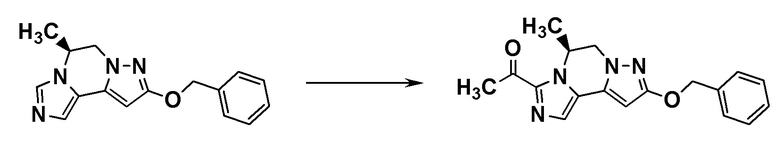
[0435]
С охлаждением при -78°C к раствору (S)-9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин/тетрагидрофурана, полученному на предыдущей стадии, по каплям добавляли раствор смеси 1M диизопропиламид лития/гексан-тетрагидрофуран (634 мл), и смесь перемешивали в течение 1 час. К смеси добавляли N-метокси-N-метилацетамид (173 мл), и смесь перемешивали при -78°C в течение 1,5 час. К смеси по каплям добавляли раствор смеси 1M диизопропиламид лития/гексан-тетрагидрофуран (667 мл), и смесь перемешивали в течение 2,5 час. Реакционную смесь по каплям добавляли к раствору смеси уксусная кислота (405 мл)/этанол (672 мл), охлажденному до 0°C. Выпавшее в осадок твердое вещество собирали фильтрованием. Полученное твердое вещество смешивали со смесью этанол/вода=1/2 (2016 мл), и смесь перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (205,85 г).
1H-ЯМР (400 МГц, CDCl3) 1,44 (д, J=6,58 Гц, 3H), 2,69 (с, 3H), 4,19 (дд, J=13,45, 1,20 Гц, 1H), 4,29 (дд, J=13,45, 4,63 Гц, 1H), 5,24 (с, 2H), 5,75-5,84 (м, 1H), 5,98 (с, 1H), 7,32-7,50 (м, 6H)
[0436]
Стадия 11
(S)-1-(9-гидрокси-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он
[0437]
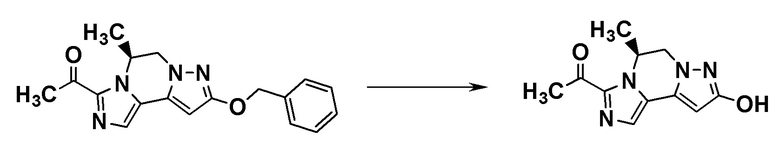
[0438]
(S)-1-(9-(бензилокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он (205,7 г), полученный на предыдущей стадии, смешивали с концентрированной соляной кислотой (563 мл). Смесь перемешивали при 48°C в течение ночи, а затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали толуолом (1,029 л). Полученный водный слой разбавляли этанолом (206 мл) и водой (411 мл), и добавляли 4Н водный раствор гидроксида натрия (1276 мл) и 10% водный раствор цитрата тринатрия (1076 мл). После перемешивания при охлаждении льдом в течение 2 час выпавшее в осадок твердое вещество собирали фильтрованием, с получением указанного в заголовке соединения (144,84 г).
1H-ЯМР (400 МГц, ДМСО-D6) 1,28 (д, J=6,73 Гц, 3H), 2,57 (с, 3H), 4,12 (д, J=13,46 Гц, 1H), 4,22 (дд, J=13,46, 4,49 Гц, 1H), 5,53-5,65 (м, 1H), 5,94 (с, 1H), 7,48 (с, 1H), 10,04 (с, 1H)
[0439]
Стадия 12
(S)-1-(9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он
[0440]
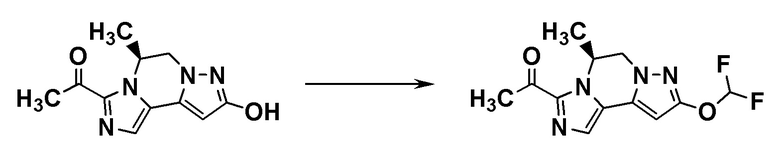
[0441]
(S)-1-(9-гидрокси-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он (1 г), полученный на предыдущей стадии, смешивали с ацетонитрилом (10 мл). К смеси добавляли 8M водный раствор гидроксида калия (168 мл). При охлаждении льдом к смеси по каплям добавляли диэтил(бромдифторметил)фосфонат (1,533 мл), и смесь перемешивали при охлаждении водой в течение 1 час. К реакционной смеси при комнатной температуре добавляли воду (20 мл) и этилацетат (20 мл), и смесь дважды экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонке с силикагелем (гексан:этилацетат от 3:1 до 1:1), с получением указанного в заголовке соединения (561 мг).
1H-ЯМР (400 МГц, ДМСО-D6) 1,27 (д, J=6,70 Гц, 3H), 2,58 (с, 3H), 4,25-4,43 (м, 2H), 5,59-5,69 (м, 1H), 6,50 (с, 1H), 7,30 (т, J=73,06 Гц, 1H), 7,57 (с, 1H)
[0442]
Стадия 13
(R)-2-((S)-9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)-1,1,1-трифторпропан-2-ол
[0443]
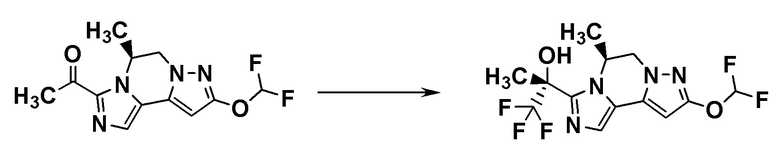
[0444]
(S)-1-(9-(дифторметокси)-5-метил-5,6-дигидроимидазо[1,5-a]пиразолo[5,1-c]пиразин-3-ил)этан-1-он (561 мг), полученный на предыдущей стадии, смешивали с тетрагидрофураном (5,6 мл). К смеси добавляли фторид цезия (0,06 г). При охлаждении льдом по каплям добавляли (трифторметил)триметилсилан (0,587 мл), и смесь перемешивали при комнатной температуре в течение 1,5 час. К реакционной смеси добавляли метанол (3,366 мл) и карбонат калия (0,33 г), и смесь перемешивали при комнатной температуре в течение 1 час. Реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонке с силикагелем (этилацетат:гексан от 1:3 до 1:1), с получением указанного в заголовке соединения (529 мг).
1H-ЯМР (400 МГц, ДМСО-D6) 1,27 (д, J=6,70 Гц, 3H), 1,81 (с, 3H), 4,20-4,30 (м, 2H), 5,31-5,40 (м, 1H), 6,31 (с, 1H), 7,27 (т, J=73,29 Гц, 1H), 7,31 (с, 1H), 7,32 (с, 1H)
[0445]
Соединения примеров 1-103 получали способами, аналогичными вышеописанным способам получения 1-10 и примерам получения 1-10, или иными известными способами по мере необходимости. Структурные формулы и данные о свойствах соединений примеров приведены в следующих далее таблицах. Примечания к таблицам показывают следующее содержание.
[0446]
1 (Примеры 1, 3, 5, 6, 24, 25, 34, 35 и 50)
рацематы
2 (Пример 26)
смесь диастереомеров двух видов
3 (Пример 97)
Стерическая конфигурация циклопропанового фрагмента является транс, смесь диастереомеров двух видов.
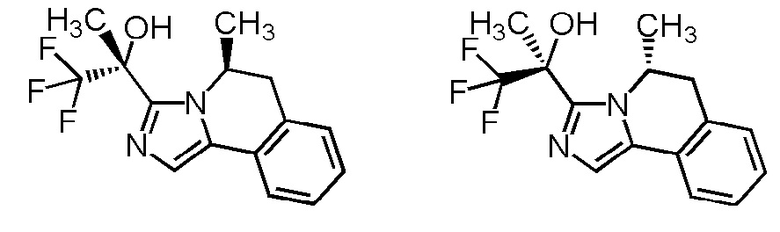
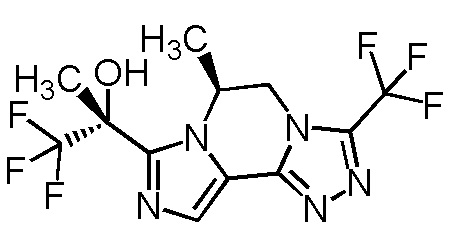
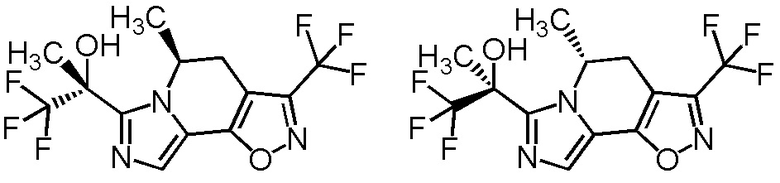
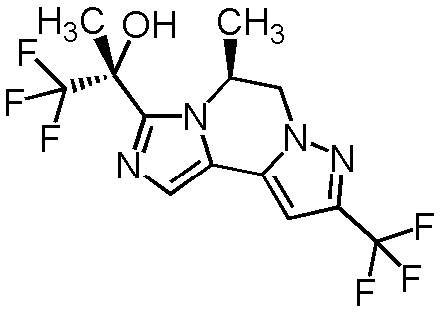
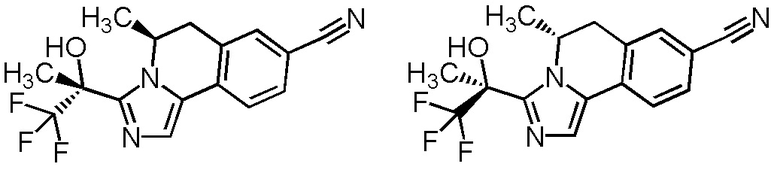
[0448]
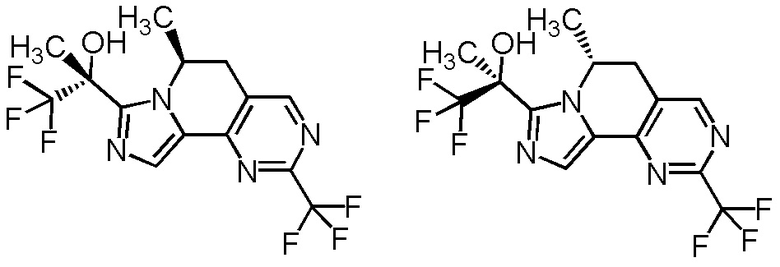
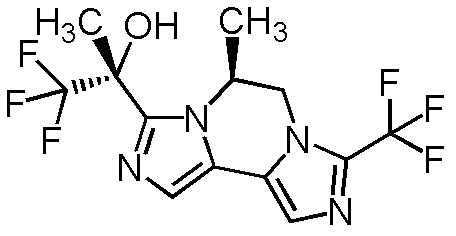
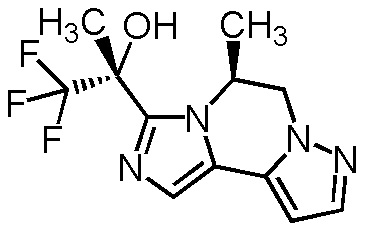
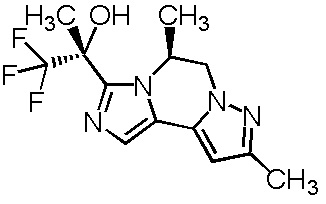
[0449]
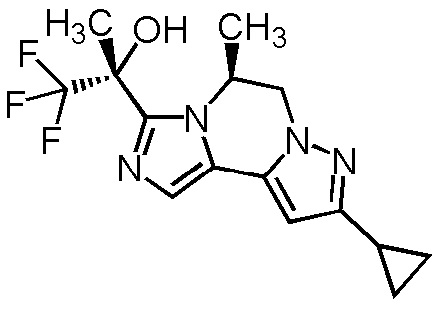
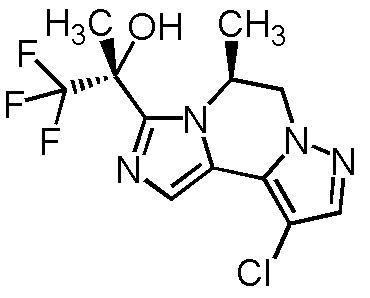
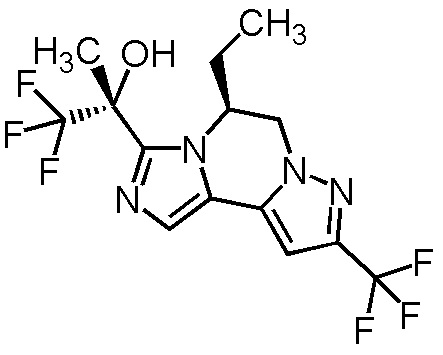
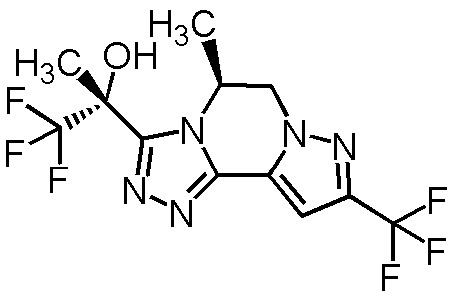
[0450]
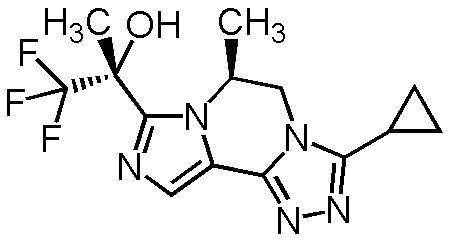
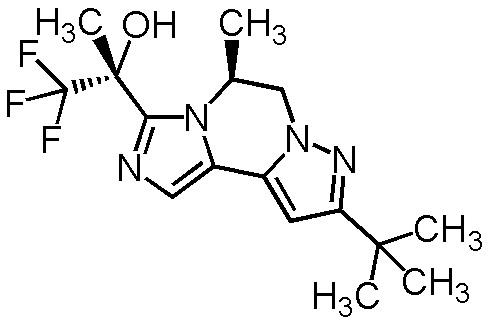
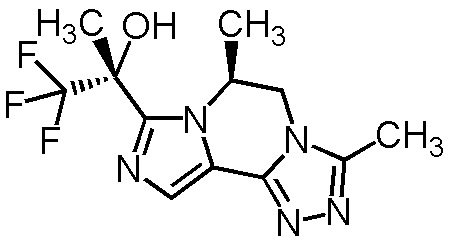
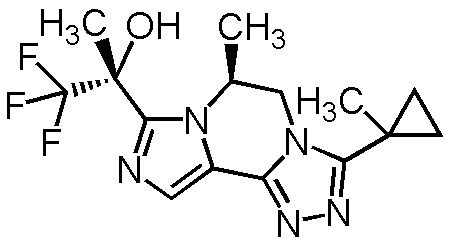
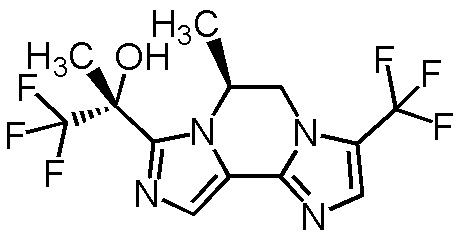
[0451]
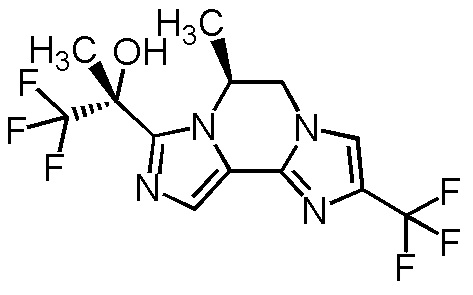
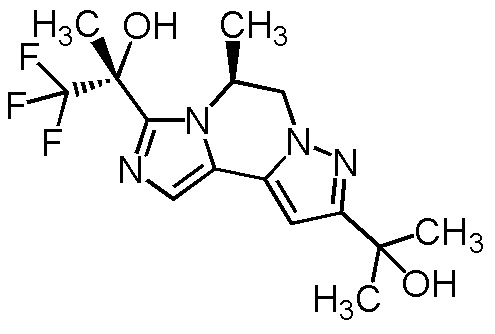
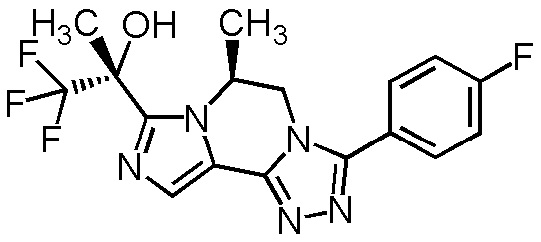
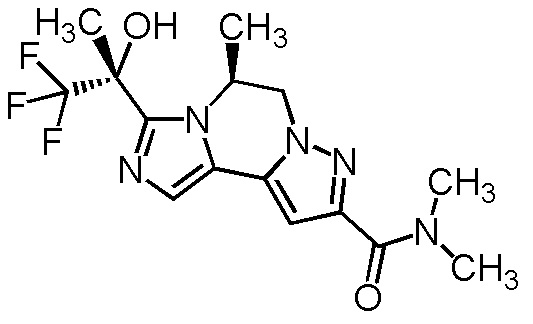
[0452]
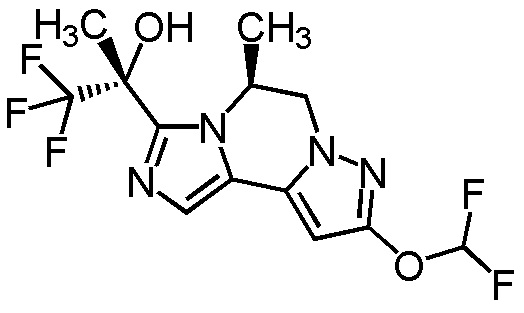
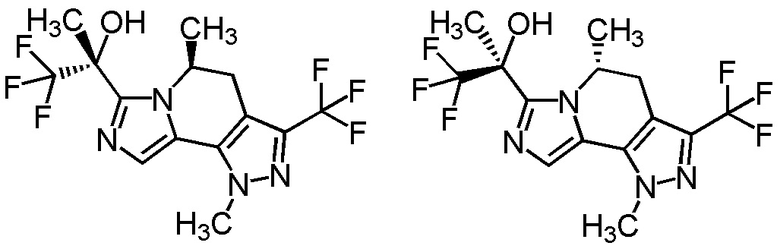
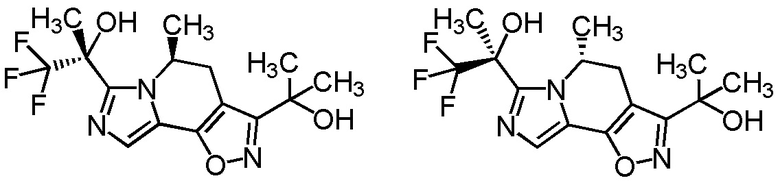
[0453]
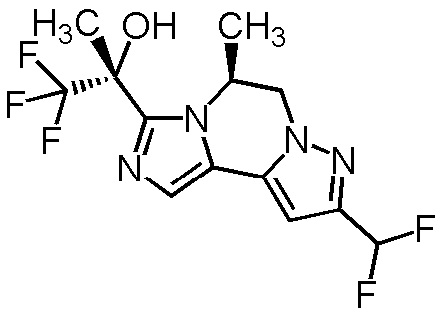
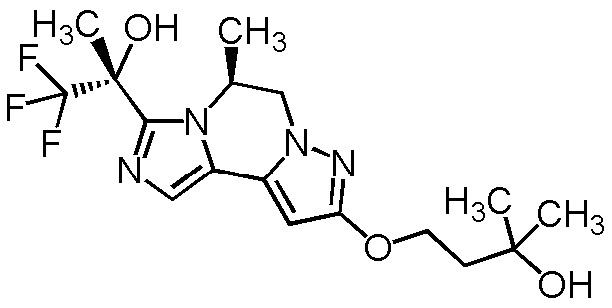
[0454]
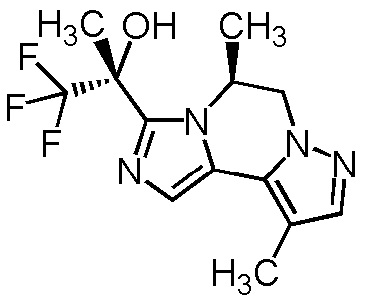
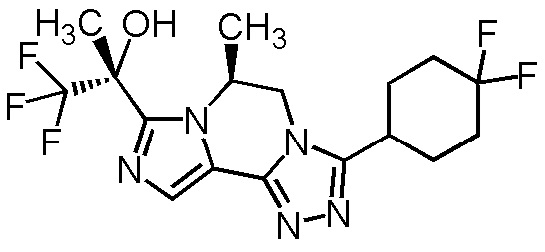
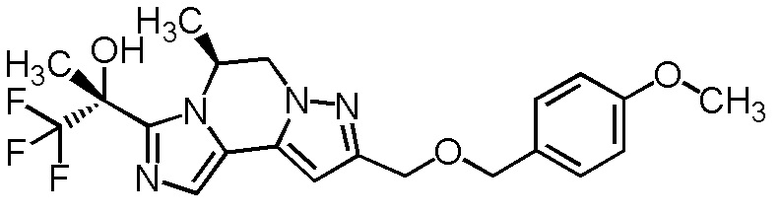
[0455]
(Mx2-1)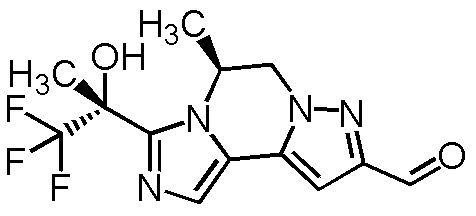
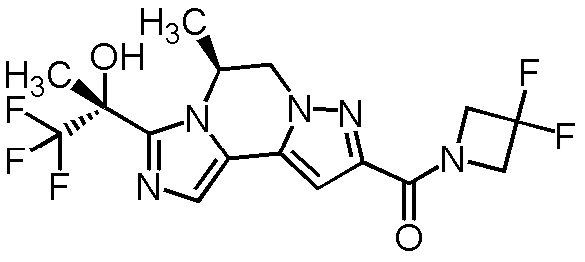
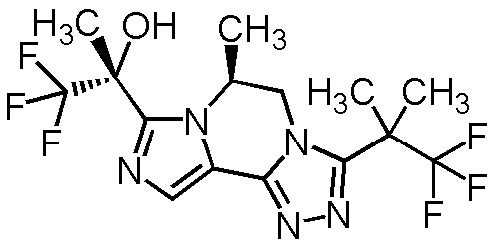
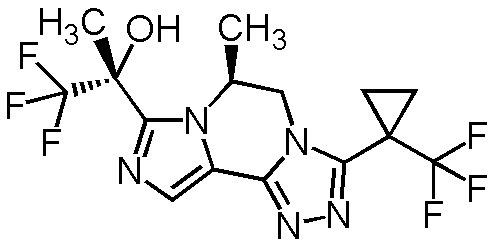
[0456]
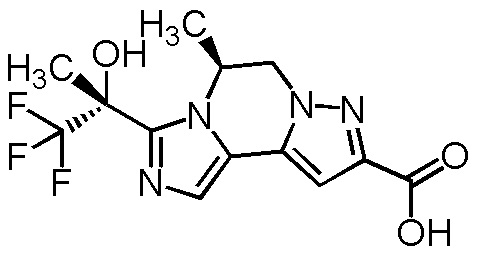
(Mx2-1)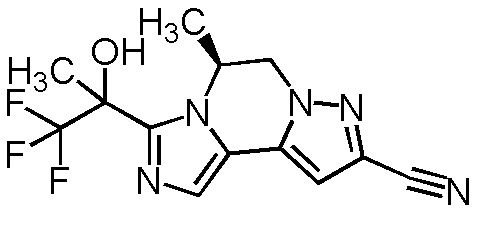
(Mx2-1)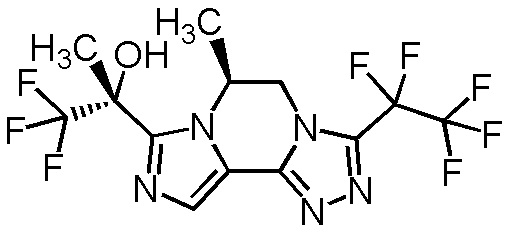
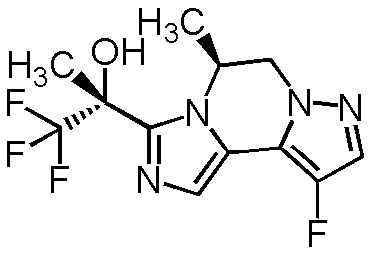
(M-1 +46)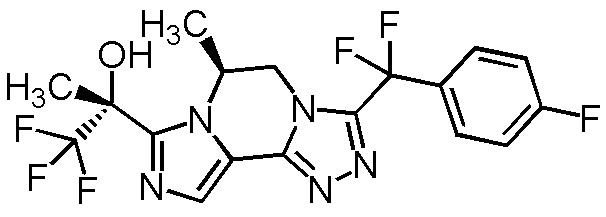
[0457]
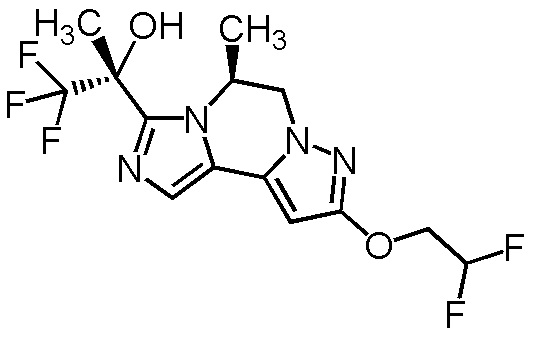
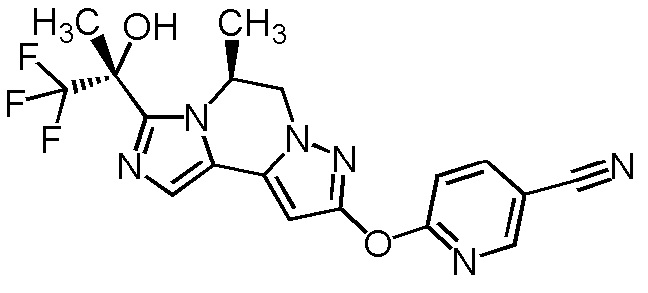
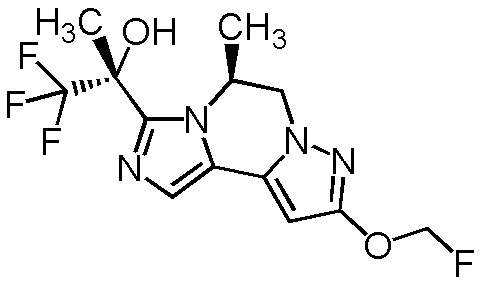
(Mx2-1)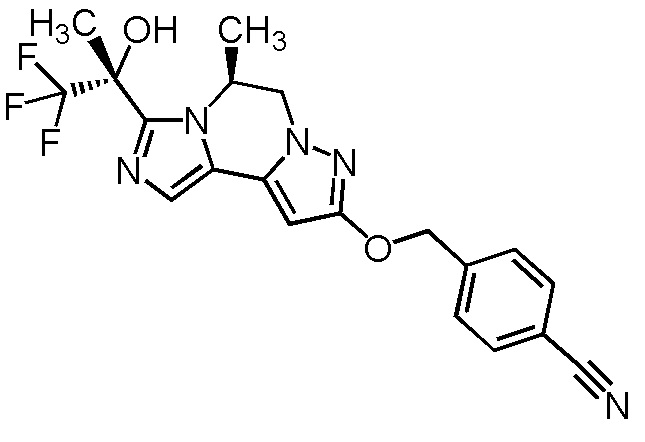
[0458]
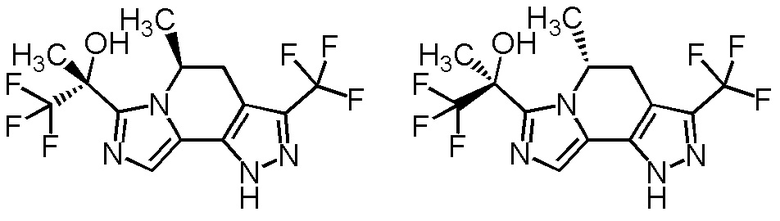
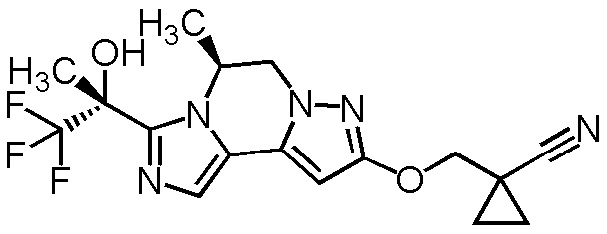
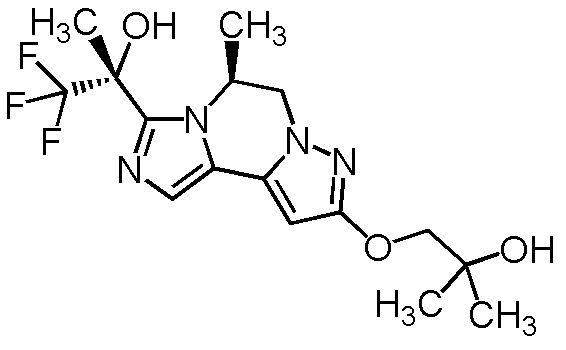
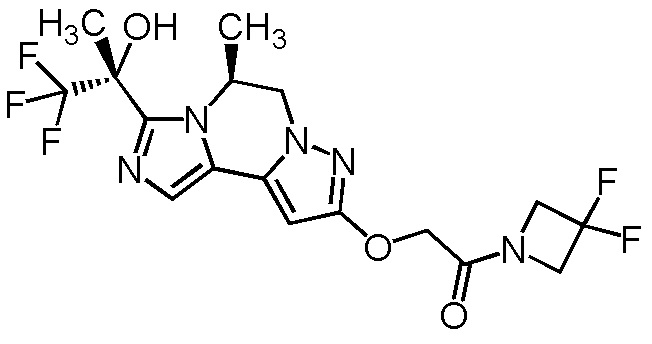
[0459]
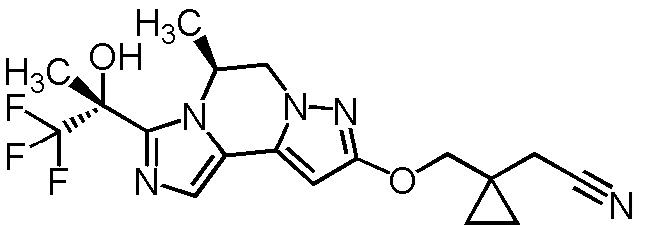
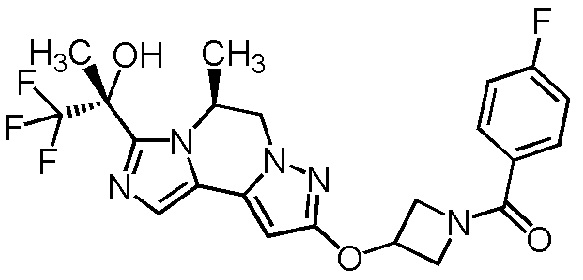
[0460]
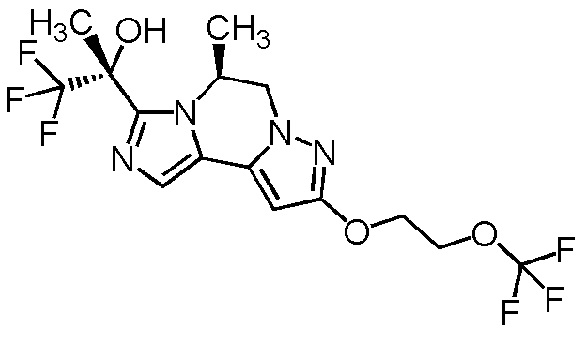
(M-1 +46)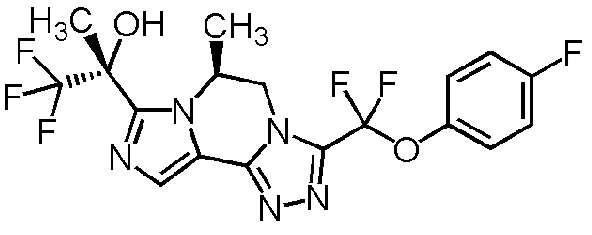
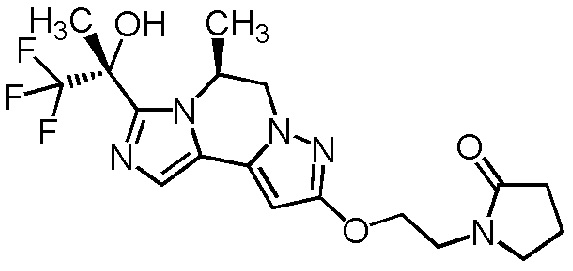
[0461]
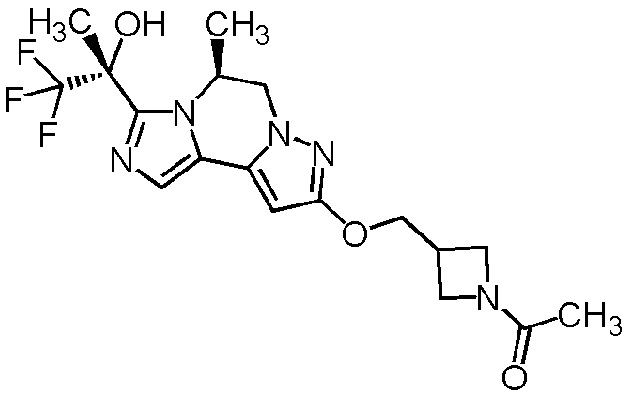
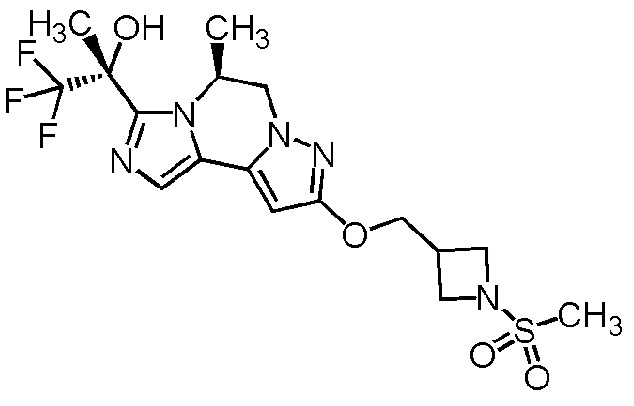
[0462]
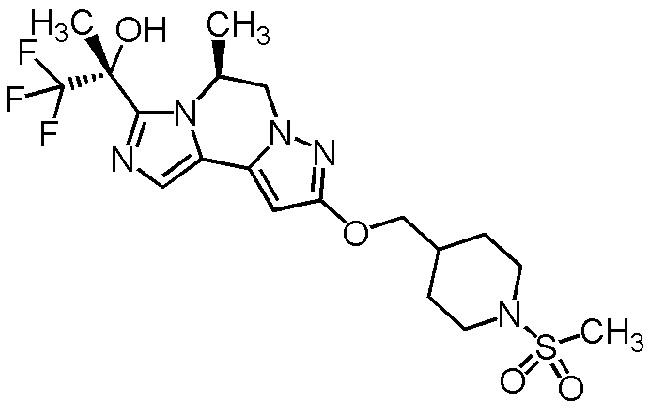
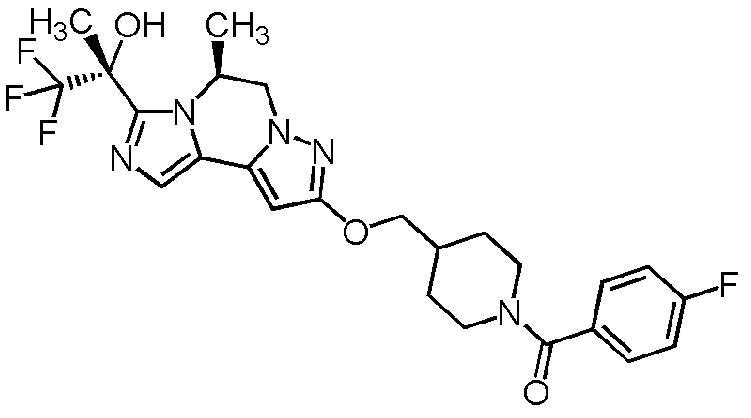
[0463]
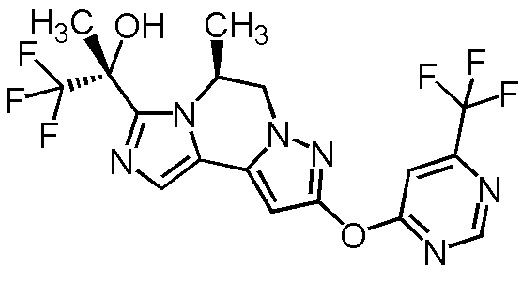
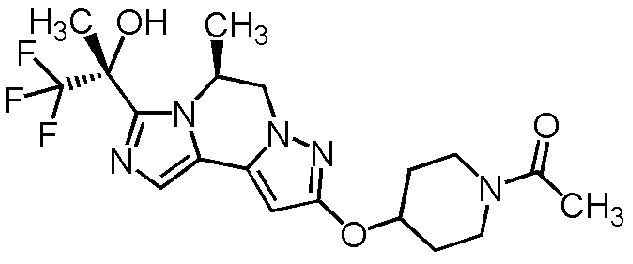
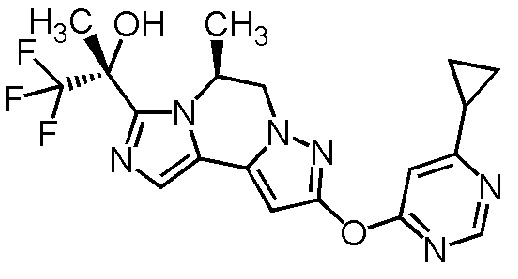
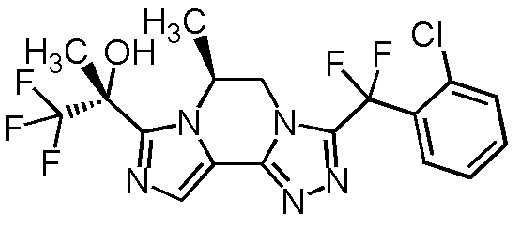
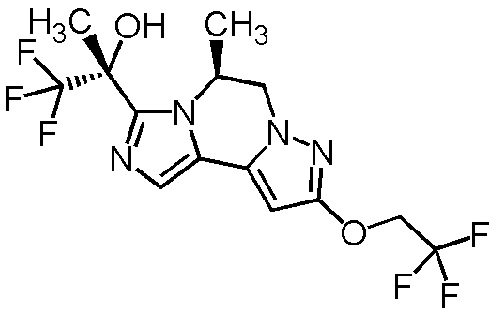
[0464]
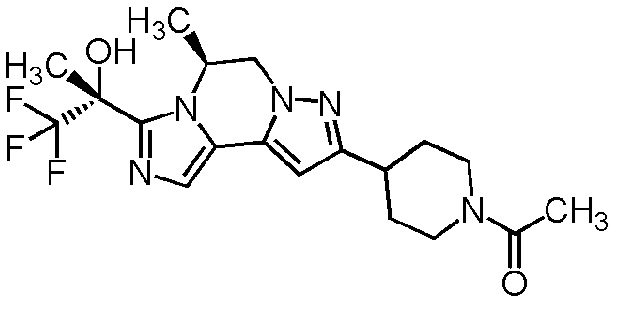
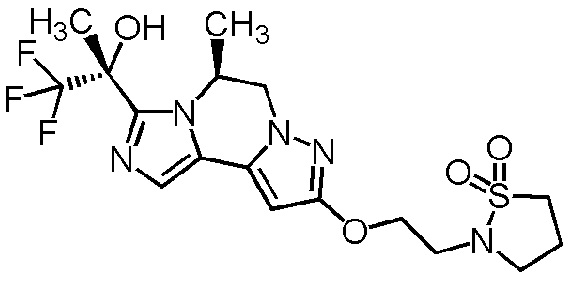
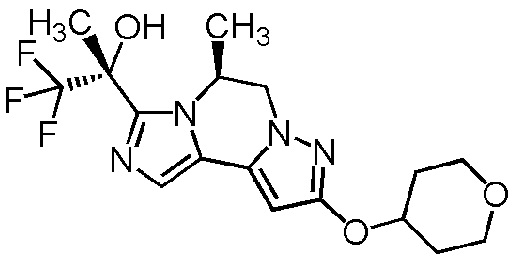
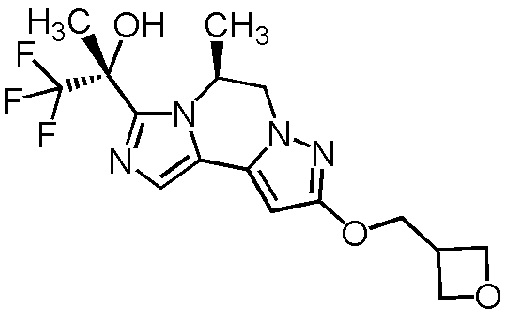
[0465]
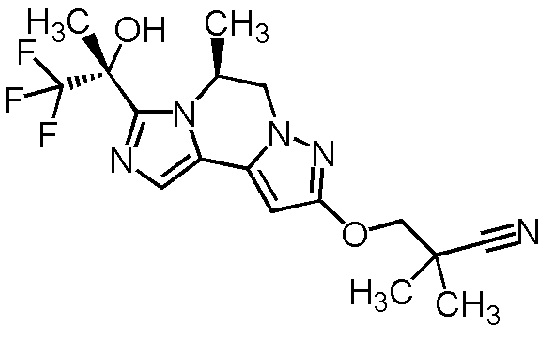
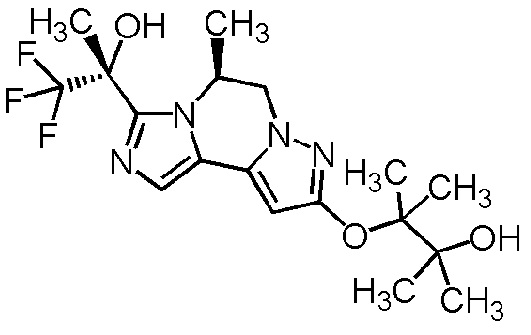
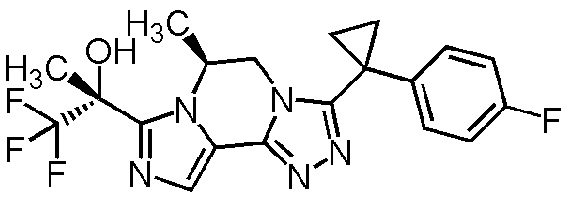
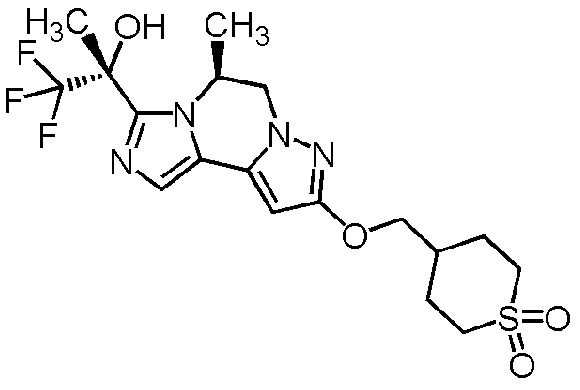
[0466]
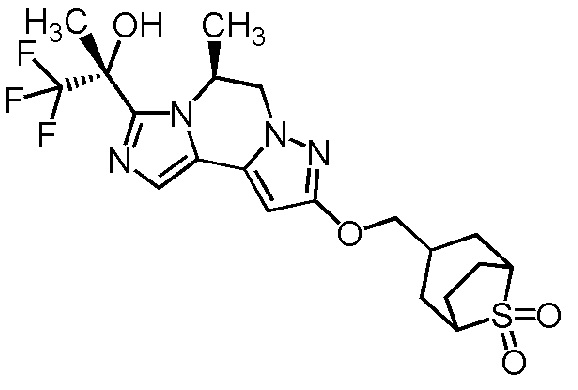
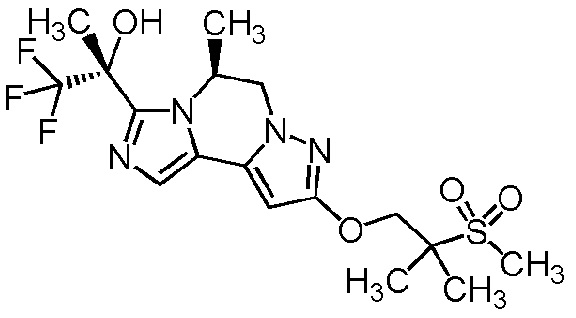
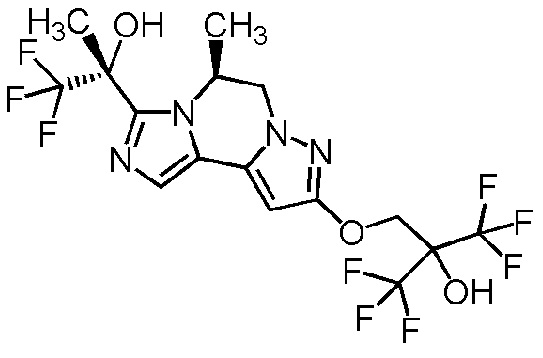
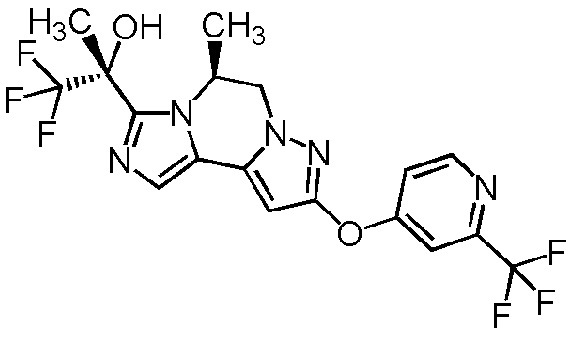
[0467]
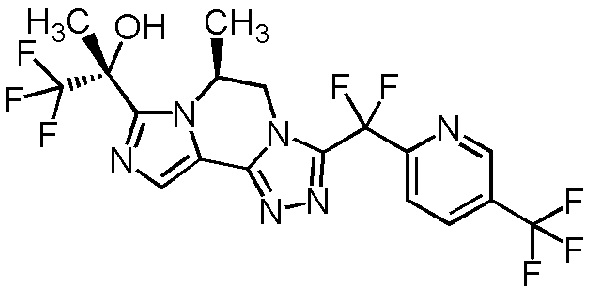
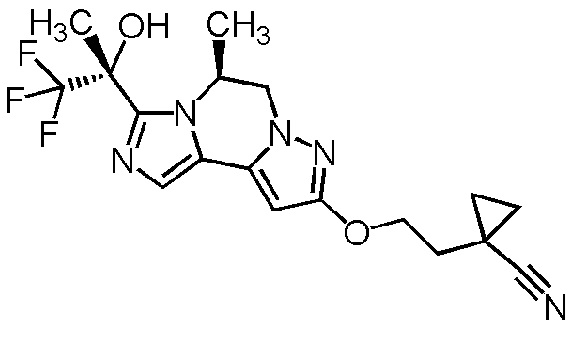
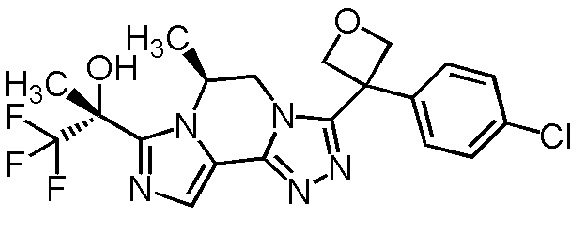
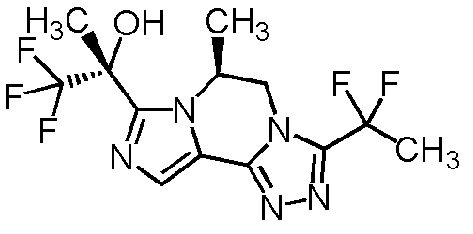
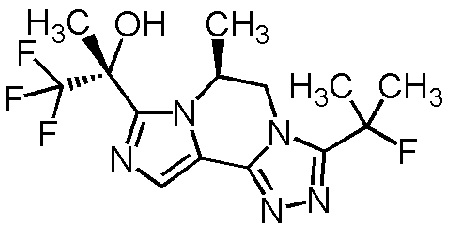
[0468]
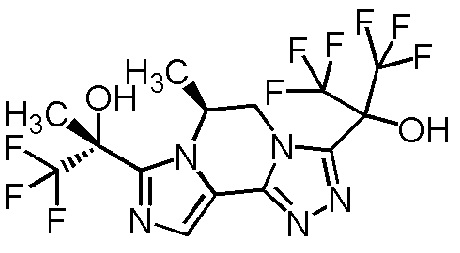
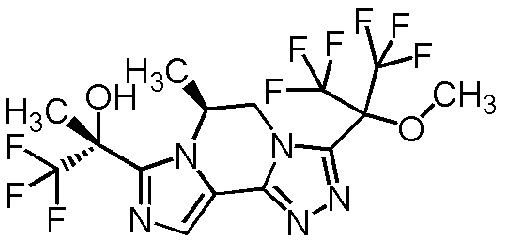
[0469]
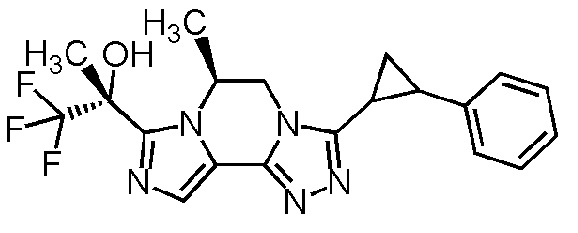
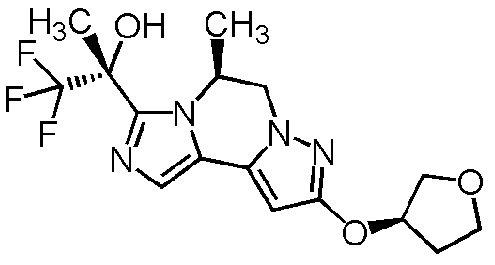
[0470]
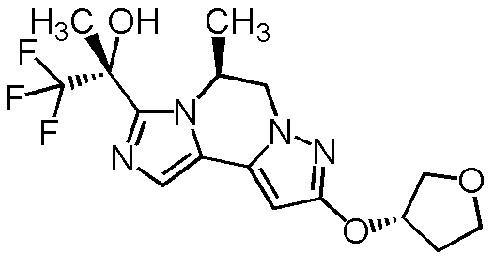
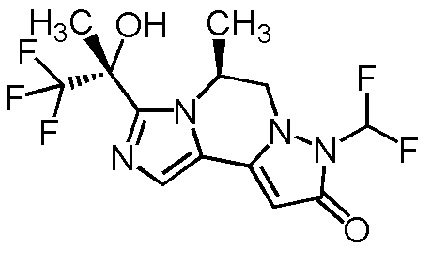
[0471]
Экспериментальный пример 1
Ингибирующее действие на активность ПДГК2
В случае ПДГК2 человека (чПДГК2, регистрационный номер справочной базы данных NCBI NM_002611.4) модифицированную кДНК чПДГК2, где последовательность FLAG-метки добавляли к N-концу клона кДНК чПДГК2 (pReceiver-M01/PDK2-GeneCopoeia) в качестве основы, получали полимеразной цепной реакцией (ПЦР) и лигировали в сайт NdeI/EcoRI вектора pET-17b (Merck KGaA, каталожный номер 69663-3). Рекомбинантную конструкцию вводили трансформацией в Escherichia coli DH5α. Рекомбинантные клоны идентифицировали, ДНК плазмиды выделяли и проводили анализ последовательности ДНК. Один клон, имеющий ожидаемую нуклеотидную последовательность, выбирали для экспериментов по экспрессии.
[0472]
Для экспрессии активности чПДГК2 клетки Escherichia coli штамма BL21(DE3) (Merck KGaA, каталожный номер 69450-4) трансформировали вектором pET17b, содержащим модифицированную кДНК чПДГК2. Клетки Escherichia coli выращивали до оптической плотности 0,6 (600 нмоль/л) при 30°C. Экспрессию белка индуцировали добавлением 500 мкмоль/л изопропил-β-тиогалактопиранозида. Клетки Escherichia coli культивировали при 20°C в течение 17-18 час и собирали центрифугированием. Собранные клетки Escherichia coli ресуспендировали в суспензионном буфере (20 ммоль/л HEPES-NaOH, 500 ммоль/л хлорид натрия, 1% этиленгликоль, 0,1% плюроник (зарегистрированная торговая марка) F-68 (pH 8,0), полный, без ЭДТА (Roche) (pH 8,0)) и разрушали микрофлюидизатором M-110H (MIZUHO INDUSTRIAL CO., LTD.). Осадок удаляли центрифугированием, и супернатант добавляли к DDDDK-меченому гелю для очистки белков (MEDICAL & BIOLOGICAL LABORATORIES CO., LTD., Code No. 3329). DDDDK-меченый гель для очистки белков промывали промывающим буфером (20 ммоль/л HEPES-NaOH, 500 ммоль/л хлорид натрия, 1% этиленгликоль, 0,1% плюроник F-68 (pH 8,0)), и связанный белок элюировали элюирующим буфером 1 (20 ммоль/л HEPES-NaOH, 100 мкг/мл пептид (аминокислотная последовательность DYKDDDDK) (SEQ ID NO: 1), 500 ммоль/л хлорид натрия, 1% этиленгликоль, 0,1% плюроник F-68 (pH 8,0)). Элюированные фракции, содержащие FLAG-меченый белок, объединяли, концентрировали методом ультрафильтрации, наносили на колонку для гель-фильтрации (HiLoad 26/60 Superdex 200 (GE Healthcare Life Scienceс, Code No. 17-1070-01)) и элюировали элюирующим буфером 2 (20 ммоль/л HEPES-NaOH, 150 ммоль/л хлорид натрия, 0,5 ммоль/л этилендиаминтетрауксусная кислота (ЭДТА), 1% этиленгликоль, 0,1% плюроник F-68 (pH 8,0)). Элюированные фракции объединяли и хранили при -80°C.
[0473]
ПДГ (комплекс ПДГ из свиного сердца, Sigma P7032) и 0,5 мкг/мл чПДГК2 смешивали в буфере для анализа (50 ммоль/л 3-морфолинопропансульфоновая кислота (pH 7,0), 20 ммоль/л гидрофосфат дикалия, 60 ммоль/л хлорид калия, 2 ммоль/л хлорид магния, 0,4 ммоль/л ЭДТА, 0,2% полоксамер, 2 ммоль/л дитиотреитол) так, что конечная концентрация ПДГ составляла 0,025 Ед/мл, и смесь инкубировали при 4°C в течение ночи, с получением раствора комплекса ПДГ/чПДГК2.
ПДГ смешивали с буфером для анализа так, что конечная концентрация составляла 0,025 Ед/мл, и смесь инкубировали при 4°C в течение ночи, с получением раствора ПДГ.
Тестируемые соединения разбавляли ДМСО. Для определения ингибирующего действия тестируемого соединения на активность ПДГК в растворе комплекса ПДГ/чПДГК2 раствор комплекса ПДГ/чПДГК2 (20 мкл), тестируемое соединение (1,5 мкл) и 1,06 мкмоль/л АТФ (разбавленные буфером для анализа) (8,5 мкл) добавляли в лунки 384-луночного микропланшета (Greiner Bio-он 781801) и проводили реакцию ПДГК при комнатной температуре в течение 45 мин (лунка с тестируемым соединением). В контрольные лунки вместо тестируемого соединения добавляли ДМСО (1,5 мкл). Кроме того, вместо тестируемого соединения в лунки с пустой пробой добавляли ДМСО (1,5 мкл), и вместо раствора комплекса ПДГ/чПДГК2 добавляли раствор ПДГ. Для определения ингибирующего действия тестируемого соединения на активность ПДГК, имеющуюся в растворе ПДГ, тестируемое соединение и раствор ПДГ вместо раствора комплекса ПДГ/чПДГК2 добавляли в лунку пустая проба+тестируемое соединение.
[0474]
Затем добавляли 10 мкл субстратов (5 ммоль/л пируват натрия, 5 ммоль/л кофермент A, 12 ммоль/л НАД, 5 ммоль/л тиаминпирофосфат, разбавленные буфером для анализа). Смесь инкубировали при комнатной температуре в течение 90 мин и определяли остаточную активность ПДГ.
[0475]
Поглощение при 340 нм в каждой лунке измеряли с использованием микропланшетного ридера, определяя НАДH, продуцируемый в реакции ПДГ. Активность ПДГ для каждой лунки рассчитывали на основании изменений в поглощении до и после реакции ПДГ. Активность ПДГ в образце, обработанном тестируемым соединением, рассчитывали по формуле {активность ПДГ в лунке с тестируемым соединением - (активность ПДГ в лунке с пустой пробой+тестируемым соединением - активность ПДГ в лунке с пустой пробой)}. Степень ингибирования чПДГК2 (%) для тестируемого соединения рассчитывали по формуле [{(активность ПДГ в образце, обработанном тестируемым соединением - активность ПДГ в контрольной лунке)/активность ПДГ в лунке с пустой пробой - активность ПДГ в контрольной лунке)} × 100]. Значение IC50 рассчитывали методом логистической регрессии на основании концентрации тестируемого соединения и степени ингибирования чПДГК2 (%).
[0476]
Результаты приведены в следующих далее таблицах. Когда значение IC50 невозможно было рассчитать, приведена степень ингибирования при самой низкой или самой высокой концентрации тестируемого соединения в анализе. Например, соединение примера 12 демонстрировало степень ингибирования чПДГК2 35% при концентрации 0,1 мкМ.
[0477]
[0478]
[0479]
[0480]
[0481]
В качестве примеров препаратов по настоящему изобретению могут быть упомянуты следующие препараты. Однако настоящее изобретение не ограничено данными примерами препаратов.
[0482]
Пример препарата 1: Получение капсулы
1) соединение примера 1-30 мг
2) кристаллическая целлюлоза - 10 мг
3) лактоза - 19 мг
4) стеарат магния - 1 мг
1), 2), 3) и 4) смешивают и вносят в желатиновую капсулу.
[0483]
Пример препарата 2: Получение таблетки
1) соединение примера 1-10 г
2) лактоза - 50 г
3) кукурузный крахмал - 15 г
4) кармеллоза кальция - 44 г
5) стеарат магния - 1 г
Общее количество 1), 2), 3) и 30 г 4) замешивают с водой, сушат в вакууме и просеивают. Просеянный порошок смешивают с 14 г 4) и 1 г 5), и смесь прессуют таблеточным прессом. Таким образом получают 1000 таблеток, каждая из которых содержит 10 мг соединения примера 1 на каждую таблетку.
[0484]
Пример препарата 3: Получение инъекционного препарата
1) соединение примера 1-5 мг
2) D-маннит - 5 г
3) дистиллированная вода - 100 мл
1) и 2) растворяют в 3), и раствор вносят в контейнер для инъекции, запечатывают и стерилизуют.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0485]
Поскольку соединение формулы [I-a], или его фармацевтически приемлемая соль, обладает ингибирующим действием на ПДГК, оно может быть использовано в качестве активного ингредиента лекарственного препарата для лечения или профилактики диабета (диабета 1 типа, диабета 2 типа и так далее), синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета (диабетической нейропатии, диабетической ретинопатии, диабетической нефропатии, катаракты и так далее), сердечной недостаточности (острой сердечной недостаточности, хронической сердечной недостаточности), кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболеваний периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции (сосудистой деменции с поражением крупных сосудов или мелких сосудов), глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии или хронической почечной недостаточности.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> JAPAN TOBACCO INC.
<120> КОНДЕНСИРОВАННОЕ ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО
ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ
<130> 093110
<150> JP 2020-036931
<151> 2020-03-04
<150> JP 2021-001452
<151> 2021-01-07
<160> 1
<170> PatentIn version 3.5
<210> 1
<211> 8
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> FLAG-метка
<400> 1
Asp Tyr Lys Asp Asp Asp Asp Lys
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
| МОДУЛЯТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2797323C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2702732C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2005 |
|
RU2409582C2 |
| СЛОЖНЫЕ АРИЛАЛКИЛОВЫЕ ЭФИРЫ 4-АМИНО-6-(ЗАМЕЩЕННЫЙ ФЕНИЛ)ПИКОЛИНАТОВ И 6-АМИНО-2-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-4-ПИРИМИДИНКАРБОКСИЛАТОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2012 |
|
RU2566760C2 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| ТРИАЗОЛОПИРИДИНОВОЕ СОЕДИНЕНИЕ И ЕГО ДЕЙСТВИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОЛИЛГИДРОКСИЛАЗЫ И ИНДУКТОРА ВЫРАБОТКИ ЭРИТРОПОЭТИНА | 2010 |
|
RU2538963C2 |
| АНТАГОНИСТ CASR | 2004 |
|
RU2315036C2 |
Группа изобретений относится к области органической химии и фармацевтики и включает трициклическое соединение формулы [I-a], его фармацевтически приемлемые соли, индивидуальные соединения и их фармацевтически приемлемые соли, указанные в формуле изобретения, фармацевтическую композицию, содержащую такие соединения, ингибитор ПДГК, ингибитор ПДГК2, средство для лечения заболевания, связанного с ингибирующим действием на активность ПДГК, способ ингибирования ПДГК, способ лечения заболевания, связанного с ингибирующим действием на активность ПДГК, и применения соединений. В формуле [I-a] связь, обозначенная пунктирной линией, представляет собой одинарную связь или двойную связь, при условии, что кольцо, образованное пунктирными линиями, является ароматическим кольцом, X1, X2, X3 и X4 каждый независимо представляет собой CH или N, Y1 и Y2 каждый независимо представляет собой CH, N или O (где общее количество N и O для X2, X3, X4, Y1 или Y2 составляет 0-3), RA представляет собой C1-4 алкил, RB представляет собой (1) галоген, (2) циано, (3) гидрокси, (4) оксо, (5) -COR1, (6) C1-8 алкил, (7) C1-8 алкокси, (8) -Cy2, (9) -OCy3, m равно 0 или 1, n равно 0, 1 или 2, когда n равно 2, все RB могут быть одинаковыми или разными, остальные заместители являются такими, как представлено в формуле изобретения. Конденсированные трициклические производные формулы [I-a] обладают ингибирующим действием на ПДГК. 15 н. и 23 з.п. ф-лы, 28 табл., 107 пр.
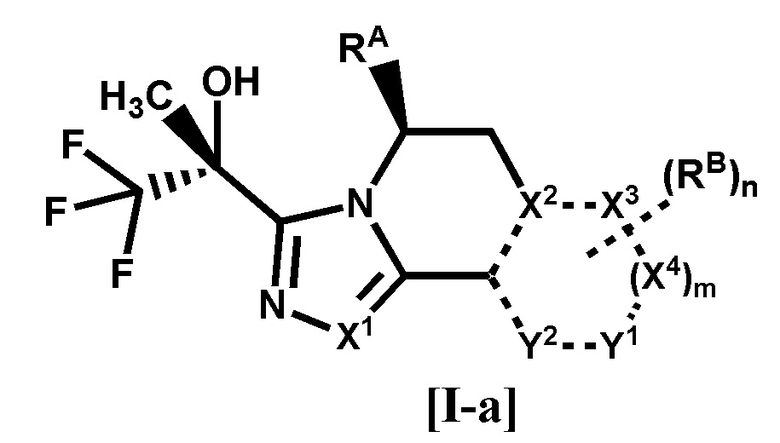
1. Соединение формулы [I-a], или его фармацевтически приемлемая соль:
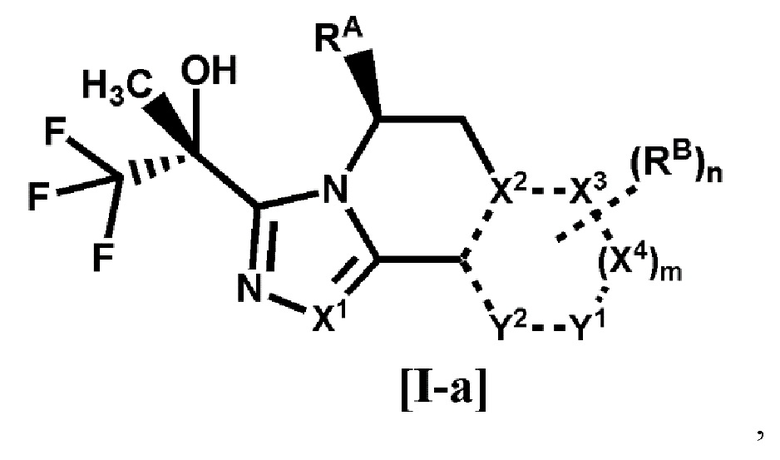
где
связь, обозначенная пунктирной линией, представляет собой одинарную связь или двойную связь, при условии, что кольцо, образованное пунктирными линиями, является ароматическим кольцом,
X1, X2, X3 и X4 каждый независимо представляет собой CH или N, Y1 и Y2 каждый независимо представляет собой CH, N или O (где общее количество N и O для X2, X3, X4, Y1 или Y2 составляет 0-3),
RA представляет собой C1-4 алкил,
RB представляет собой
(1) галоген,
(2) циано,
(3) гидрокси,
(4) оксо,
(5) -COR1 {где R1 представляет собой
(A) водород,
(B) -OH,
(C) -NR2R3 (где R2 и R3 каждый независимо представляет собой водород или C1-4 алкил), или
(D) 4-6-членный насыщенный гетероциклил, имеющий один атом азота (где насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена)},
(6) C1-8 алкил {где C1-8 алкил необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) гидрокси,
(C) фенила, необязательно замещенного галогеном,
(D) пиридила, необязательно замещенного гало-C1-4 алкилом, и
(E) -OR4 (где R4 представляет собой
(a) C1-4 алкил,
(b) фенил, необязательно замещенный галогеном, или
(c) бензил, необязательно замещенный C1-4 алкокси)},
(7) C1-8 алкокси {где C1-8 алкокси необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) циано,
(C) гидрокси,
(D) C1-4 алкокси, необязательно замещенного 1-3 атомами галогена,
(E) C1-4 алкилсульфонила,
(F) C3-6 циклоалкила, необязательно замещенного одним заместителем, выбранным из группы, состоящей из циано и циано-C1-4 алкила,
(G) фенила, необязательно замещенного циано,
(H) -COCy1 (где Cy1 представляет собой 4-6-членный насыщенный гетероциклил, имеющий один атом азота, и насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена), и
(I) 4-6-членного насыщенного гетероциклила, имеющего 1 или 2 гетероатома, независимо выбранных из атома азота, атома кислорода и атома серы (где насыщенный гетероциклил необязательно замещен 1-4 заместителями, независимо выбранными из
(a) C1-4 алкила,
(b) оксо,
(c) C1-4 алкилкарбонила,
(d) бензоила, необязательно замещенного галогеном, и
(e) C1-4 алкилсульфонила,
когда насыщенный гетероциклил замещен двумя C1-4 алкилами, два C1-4 алкила необязательно связаны друг с другом, образуя кольцо с внутренним мостиком вместе с атомами, связанными с ними)},
(8) -Cy2 {где Cy2 представляет собой
(A) C3-6 циклоалкил (где C3-6 циклоалкил необязательно замещен 1 или 2 заместителями, независимо выбранными из
(a) галогена,
(b) C1-4 алкила,
(c) гало-C1-4 алкила, и
(d) фенила, необязательно замещенного галогеном),
(B) фенил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, гало-C1-4 алкила и C1-4 алкокси, или
(C) 4-6-членный насыщенный гетероциклил, имеющий один атом азота или атом кислорода (где насыщенный гетероциклил необязательно замещен одним заместителем, выбранным из группы, состоящей из (a) фенила, необязательно замещенного галогеном, и (b) C1-4 алкилкарбонила)}, или
(9) -OCy3 {где Cy3 представляет собой
(A) 4-6-членный насыщенный гетероциклил, имеющий один атом азота или атом кислорода (где насыщенный гетероциклил необязательно замещен одним заместителем, выбранным из группы, состоящей из (a) бензоила, необязательно замещенного галогеном, и (b) C1-4 алкилкарбонила), или
(B) 6-членный гетероарил, имеющий 1 или 2 атома азота (где гетероарил необязательно замещен 1 или 2 заместителями, независимо выбранными из циано, гало-C1-4 алкила и C3-6 циклоалкила)},
m равно 0 или 1, и
n равно 0, 1 или 2, когда n равно 2, все RB могут быть одинаковыми или разными.
2. Соединение по п. 1, или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-b]:
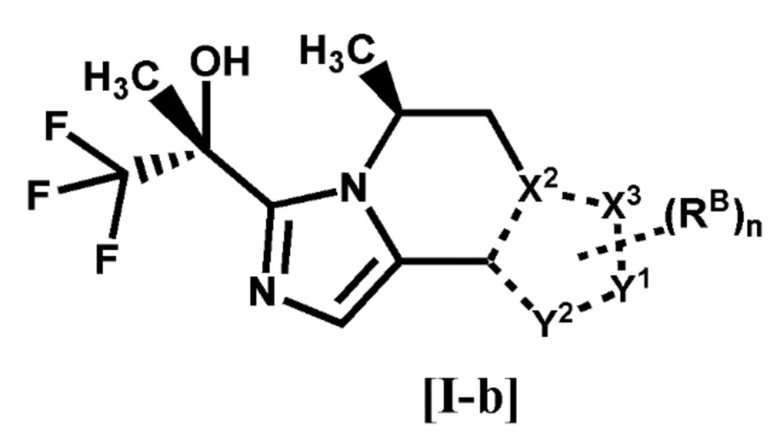 ,
,
где каждый символ является таким, как определено в п. 1, или его фармацевтически приемлемая соль.
3. Соединение по п. 1 или 2, или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-c]:
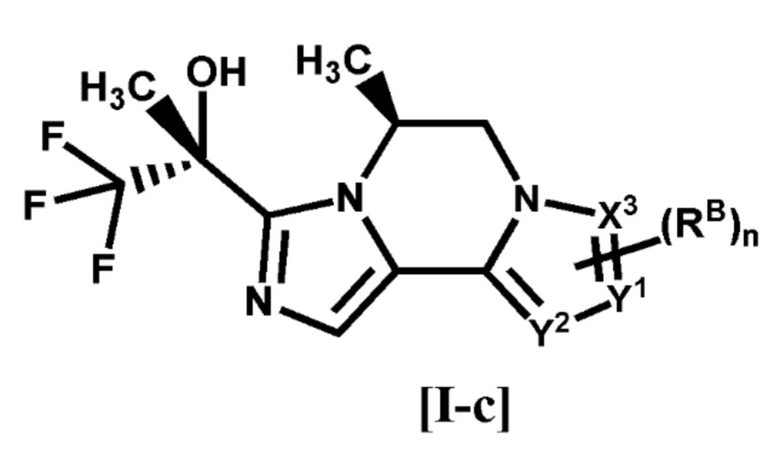 ,
,
где каждый символ является таким, как определено в п. 1, или его фармацевтически приемлемая соль.
4. Соединение по любому из пп. 1-3, где n равно 1, или его фармацевтически приемлемая соль.
5. Соединение по любому из пп. 1-4, или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-d]:
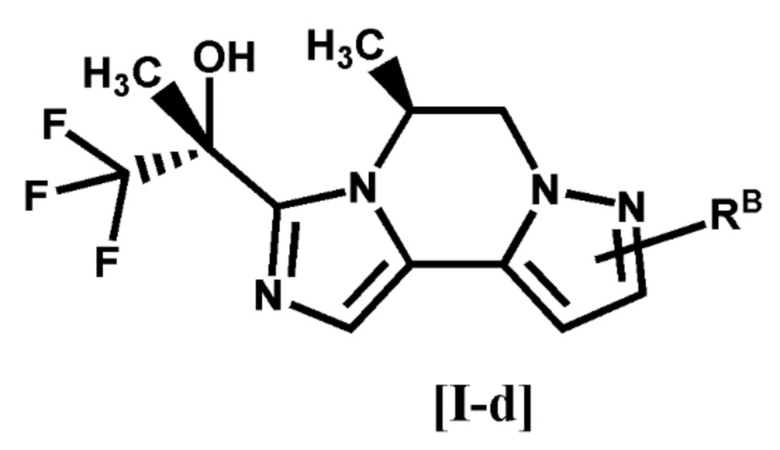 ,
,
где символ является таким, как определено в п. 1, или его фармацевтически приемлемая соль.
6. Соединение по любому из пп. 1-4, или его фармацевтически приемлемая соль, которое представляет собой соединение формулы [I-e]:
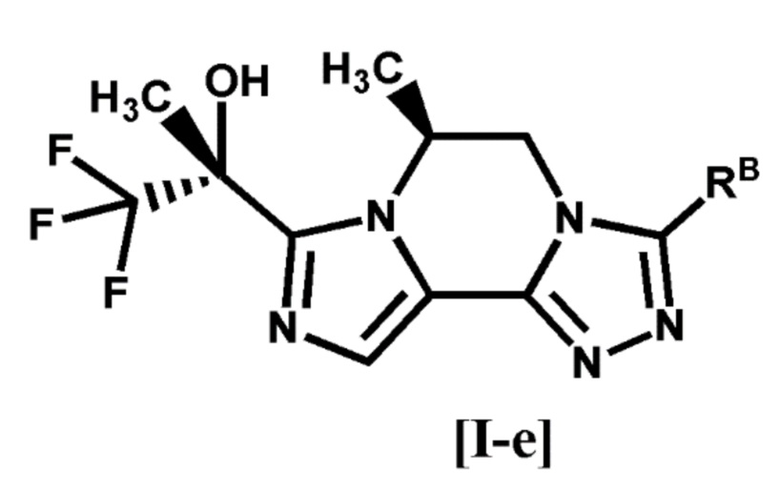 ,
,
где символ является таким, как определено в п. 1, или его фармацевтически приемлемая соль.
7. Соединение по любому из пп. 1-6, или его фармацевтически приемлемая соль, где RB представляет собой
(1) C1-8 алкил {где C1-8 алкил необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) гидрокси,
(C) фенила, необязательно замещенного галогеном,
(D) пиридила, необязательно замещенного гало-C1-4 алкилом, и
(E) -OR4 (где R4 представляет собой
(a) C1-4 алкил,
(b) фенил, необязательно замещенный галогеном, или
(c) бензил, необязательно замещенный C1-4 алкокси)}, или
(2) C1-8 алкокси {где C1-8 алкокси необязательно замещен 1-8 заместителями, независимо выбранными из группы, состоящей из
(A) галогена,
(B) циано,
(C) гидрокси,
(D) C1-4 алкокси, необязательно замещенного 1-3 атомами галогена,
(E) C1-4 алкилсульфонила,
(F) C3-6 циклоалкила, необязательно замещенного одним заместителем, выбранным из группы, состоящей из циано и циано-C1-4 алкила,
(G) фенила, необязательно замещенного циано,
(H) -COCy1 (где Cy1 представляет собой 4-6-членный насыщенный гетероциклил, имеющий один атом азота, и насыщенный гетероциклил необязательно замещен 1 или 2 атомами галогена), и
(I) 4-6-членного насыщенного гетероциклила, имеющего 1 или 2 гетероатома, независимо выбранных из атома азота, атома кислорода и атома серы (где насыщенный гетероциклил необязательно замещен 1-4 заместителями, независимо выбранными из
(a) C1-4 алкила,
(b) оксо,
(c) C1-4 алкилкарбонила,
(d) бензоила, необязательно замещенного галогеном, и
(e) C1-4 алкилсульфонила,
когда насыщенный гетероциклил замещен двумя C1-4 алкилами, два C1-4 алкила необязательно связаны друг с другом, образуя кольцо с внутренним мостиком вместе с атомами, связанными с ними)}.
8. Соединение формулы:
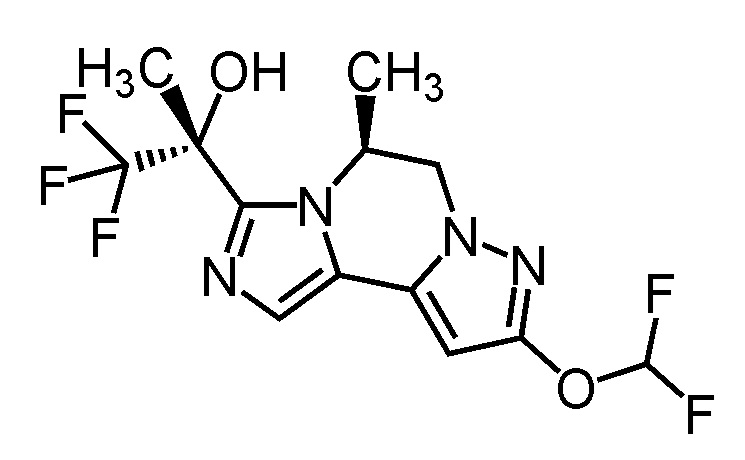
или его фармацевтически приемлемая соль.
9. Соединение формулы:
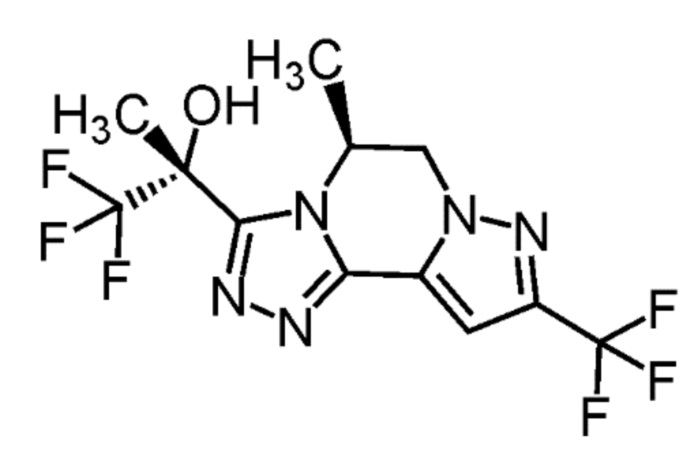
или его фармацевтически приемлемая соль.
10. Соединение формулы:
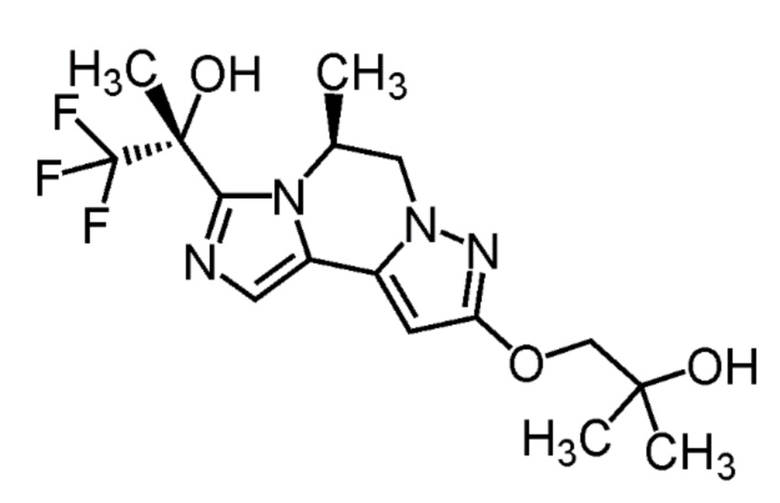
или его фармацевтически приемлемая соль.
11. Соединение формулы:
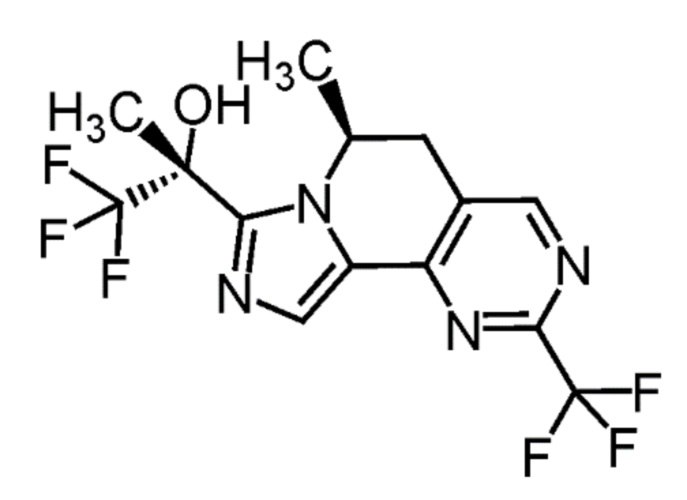
или его фармацевтически приемлемая соль.
12. Соединение формулы:
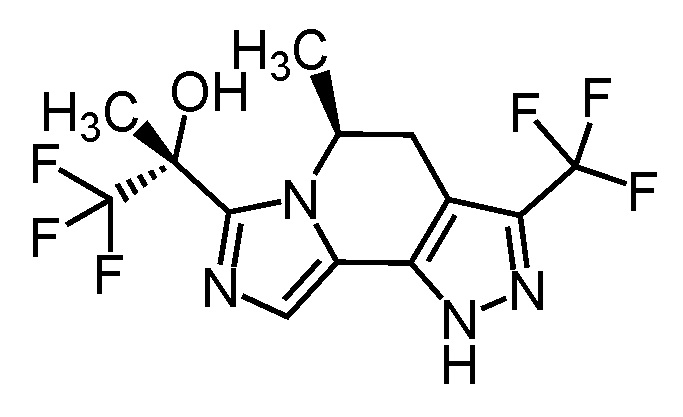
или его фармацевтически приемлемая соль.
13. Соединение формулы:
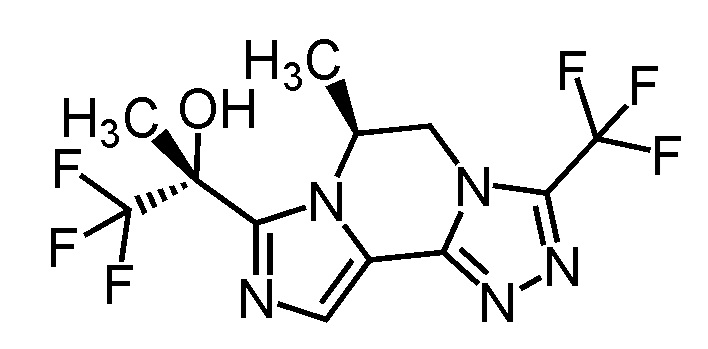
или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция для ингибирования ПДГК2, содержащая эффективное количество соединения по любому из пп. 1-13, или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
15. Ингибитор ПДГК, представляющий собой соединение по любому из пп. 1-13 или его фармацевтически приемлемую соль.
16. Ингибитор ПДГК2, представляющий собой соединение по любому из пп. 1-13 или его фармацевтически приемлемую соль.
17. Средство для лечения заболевания, связанного с ингибирующим действием на активность ПДГК, выбранного из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнений диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, заболеваний периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии или хронической почечной недостаточности, представляющее собой соединение по любому из пп. 1-13 или его фармацевтически приемлемую соль.
18. Средство по п. 17, где диабет представляет собой диабет 1 типа или диабет 2 типа.
19. Средство по п. 17, где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
20. Средство по п. 17, где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
21. Средство по п. 17, где легочная гипертензия представляет собой легочную артериальную гипертензию.
22. Способ ингибирования ПДГК, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли.
23. Способ лечения заболевания, связанного с ингибирующим действием на активность ПДГК, выбранного из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности, который включает введение млекопитающему терапевтически эффективного количества соединения по любому из пп. 1-13 или его фармацевтически приемлемой соли.
24. Способ по п. 23, где диабет представляет собой диабет 1 типа или диабет 2 типа.
25. Способ по п. 23, где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
26. Способ по п. 23, где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
27. Способ по п. 23, где легочная гипертензия представляет собой легочную артериальную гипертензию.
28. Применение соединения по любому из пп. 1-13, или его фармацевтически приемлемой соли, в производстве ингибитора ПДГК.
29. Применение соединения по любому из пп. 1-13, или его фармацевтически приемлемой соли, в производстве средства для лечения заболевания, связанного с ингибирующим действием на активность ПДГК, выбранного из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
30. Применение по п. 29, где диабет представляет собой диабет 1 типа или диабет 2 типа.
31. Применение по п. 29, где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
32. Применение по п. 29, где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
33. Применение по п. 29, где легочная гипертензия представляет собой легочную артериальную гипертензию.
34. Соединение по любому из пп. 1-13, или его фармацевтически приемлемая соль, для использования в лечении заболевания, связанного с ингибирующим действием на активность PDHK, выбранного из диабета, синдрома резистентности к инсулину, метаболического синдрома, гипергликемии, гиперлактацидемии, осложнения диабета, сердечной недостаточности, кардиомиопатии, ишемии миокарда, инфаркта миокарда, стенокардии, дислипидемии, атеросклероза, болезни периферических артерий, перемежающейся хромоты, хронической обструктивной болезни легких, ишемии головного мозга, церебральной апоплексии, митохондриальной болезни, митохондриальной энцефаломиопатии, рака, легочной гипертензии, болезни Альцгеймера, сосудистой деменции, глаукомы, диабетической ретинопатии, ретинопатии недоношенных, окклюзии вен сетчатки, ишемической оптической нейропатии и хронической почечной недостаточности.
35. Соединение по п. 34, или его фармацевтически приемлемая соль, где диабет представляет собой диабет 1 типа или диабет 2 типа.
36. Соединение по п. 34, или его фармацевтически приемлемая соль, где сосудистая деменция представляет собой тип сосудистой деменции с поражением крупных сосудов или тип сосудистой деменции с поражением мелких сосудов.
37. Соединение по п. 34, или его фармацевтически приемлемая соль, где сердечная недостаточность представляет собой острую сердечную недостаточность или хроническую сердечную недостаточность.
38. Соединение по п. 34, или его фармацевтически приемлемая соль, где легочная гипертензия представляет собой легочную артериальную гипертензию.
| WO 2019151274 A1, 08.08.2019 | |||
| WO 2017020981 A1, 09.02.2017 | |||
| WO 2004089380 A2, 21.10.2004 | |||
| WO 2005033102 A2, 14.04.2005 | |||
| KR 100808551 B1, 03.03.2008 | |||
| US 20140296315 A1, 02.10.2014 | |||
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ГИДРОКСИ-2-МЕТИЛ-3,3,3-ТРИФТОРПРОПАНАМИДА, ПОВЫШАЮЩИЕ АКТИВНОСТЬ ПИРУВАТДЕГИДРОГЕНАЗЫ | 2000 |
|
RU2255085C2 |
| EA 200100792 A1, 31.10.2002. | |||

