ПЕРЕКРЕСТНАЯ ССЫЛКА
Заявка испрашивает приоритет китайской патентной заявки №202210637542.4, поданной в Национальное управление интеллектуальной собственности Китая 8 июня 2022 года и озаглавленной «Соединение, способ его получения и применение соединения при получении промежуточного соединения для бициклопирона», которая полностью включена в данный документ посредством ссылки.
Область техники
Данная заявка относится к технической области пестицидов, в частности к соединению, способу его получения и применению соединения при приготовлении промежуточного соединения для бициклопирона.
Уровень техники
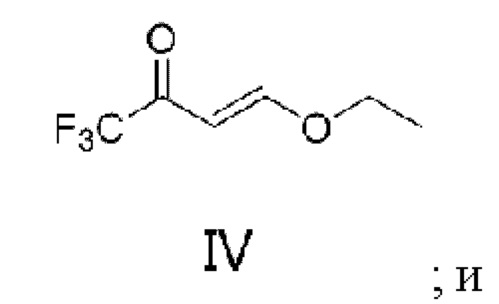
Бициклопирон был впервые выпущен на рынок в 2015 году, и был зарегистрирован и поступил в продажу в США, Канаде, Аргентине, Уругвае, Австралии и других странах. В качестве разновидности ингибиторов 4-гидроксифенилпируватдиоксигеназы (HPPD) бициклопирон разрушает хлорофилл в растениях и может сочетаться с различными гербицидами, такими как мезотрион, изоксафлутол, топрамезон, темботрион и пирасульфатол. Такие отобранные гербициды обладают хорошей активностью в отношении широколиственных сорняков, а также многолетних и однолетних сорняков и могут использоваться для кукурузных полей, пшеничных полей, полей ячменя, полей сахарного тростника и других сельскохозяйственных полей. Бициклопирон имеет следующую структуру:
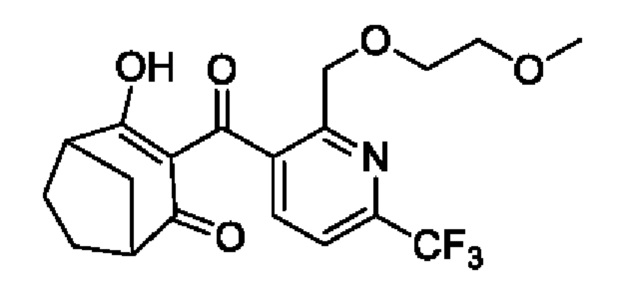
Сообщают, что типичный известный процесс получения бициклопирона (CN 1824662 В) проводят следующим образом:
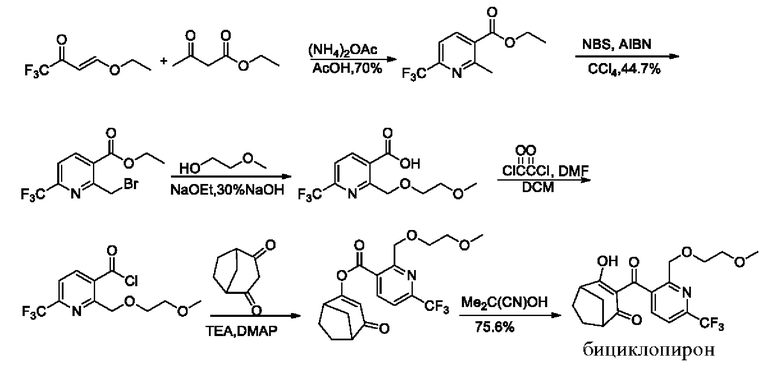
Эксперимент показывает, что на первой стадии затруднена реакция замыкания кольца; на второй стадии селективность замещения бромом в метиле является неудовлетворительной, а выход составляет всего 44,7% (см. CN 1824662 В); и на третьей стадии реакция этерификации склонна к образованию лактонных примесей, и в литературе нет данных о выходе. В вышеуказанном маршруте реакции NBS представляет собой N-бромсукцинимид; AIBN представляет собой азо-бис-изобутиронитрил; ДМФА представляет собой N,N-диметилформамид; DCM представляет собой дихлорметан; TEA представляет собой триэтиламин; и DMAP представляет собой 4-диметиламинопиридин.
В литературе сообщается (A convenient and effective method for synthesizing β-amino-α,β-unsaturated esters and ketones [Удобный и эффективный метод синтеза β-амино β-ненасыщенных сложных эфиров и кетонов], Synthetic Communications, 2004, 34 (5), 909-916, и Synthesis of functionalized pyridinium salts bearing a free amino group [Синтез функционализированных солей пиридиния, имеющих свободную аминогруппу], ARKIVOC, 2014, 2014 (3), 154-169), что этил-(2)-3-аминобут-2-еноат с количественным выходом получают из этилацетоацетата в метаноле с карбаматом аммония, и его далее конденсируют с простым виниловым эфиром с получением этил-2-метил-6-(трифторметил)никотината (см. патентный документ WO 2006059103 A2), аналогично вышеуказанному способу. Маршрут реакции является следующим:
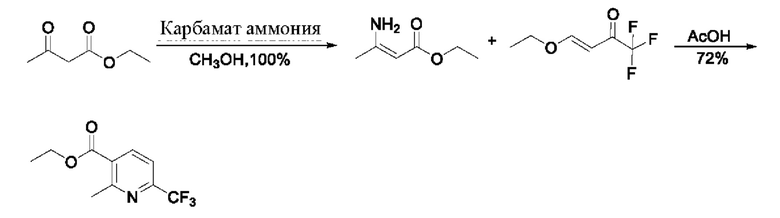
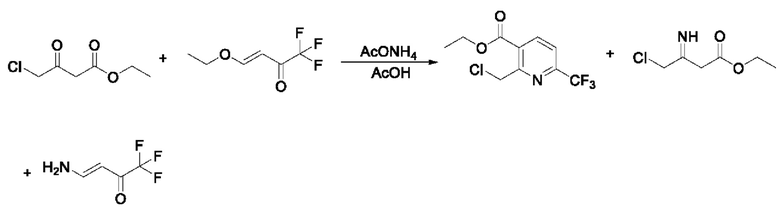
В патентном документе WO 2006059103 A2 сообщают о способе синтеза этил-2-метил-6-(трифторметил)никотината. В настоящее время этот способ широко используется большинством производителей. Эксперимент показывает, что степень превращения в реакции является неполной, отсутствует эффект при удлинении времени реакции, и основными побочными продуктами являются два енамина. Маршрут реакции является следующим:
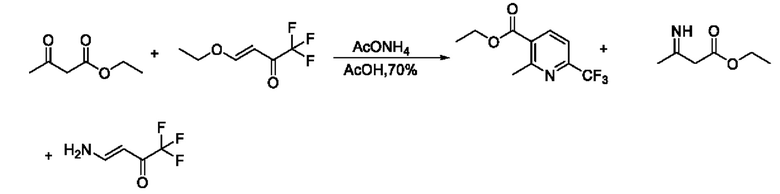
В патентном документе WO 2006059103 A2 сообщают о способе синтеза этил-2-(хлорметил)-6-(трифторметил)никотината из этил-4-хлор-3-оксобутаноата и 4-этокси-1,1,1-трифторбут-3-ен-2-она однореакторным методом в присутствии уксусной кислоты и ацетата аммония. Эксперимент показывает, что продукт является загрязненным, выход его низкий, и его трудно очистить; при повышении температуры или удлинении времени реакции все еще присутствует остаточное промежуточное соединение енамина, и его можно отделить только колоночной хроматографией, а продукт невозможно применять в производстве. Маршрут реакции является следующим:
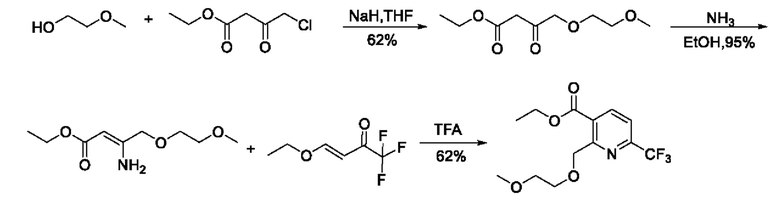
В патентном документе WO 2004078729 A1 от Syngenta сообщают, что этил-4-хлор-3-оксобутаноат и 2-метоксиэтанол соединяют в тетрагидрофуране (ТГФ) под действием NaH для получения продукта с простой эфирной группой; а затем проводят аммонизацию и замыкание кольца для получения этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината. Согласно сообщению, ожидается, что линейный выход составит 36%. В реакционном пути TFA обозначает трифторуксусную кислоту. Есть много проблем в этом пути, например, использование гидрида натрия в реакции на первой стадии. Использование избытка гидрида натрия очень затрудняет разделение и очистку продуктов с двумя или более межмолекулярными макромолекулами, образующимися между этил-4-хлор-3-оксобутаноатом или с его участием; необходимо использовать безводный тетрагидрофуран, и условия реакции являются жесткими, а введение газообразного аммиака на второй стадии является опасной реакцией, хотя и контролируемой. Маршрут реакции является следующим:
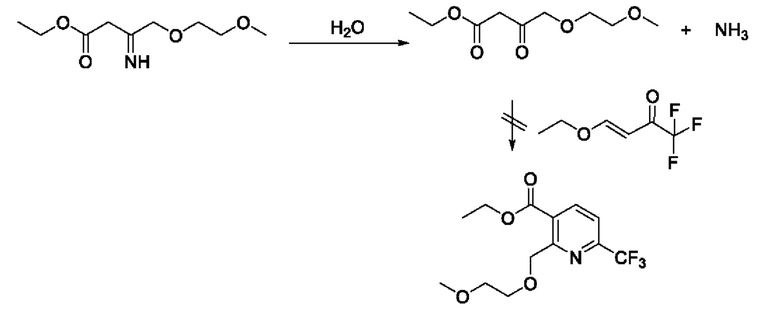
На третьей стадии реакция имеет низкий выход замыкания кольца, основная причина которого заключается в том, что реакции реагента енамина с виниловым эфиром протекают больше в активных функциональных группах и побочных реакциях. По мере протекания реакции полученная вода гидролизует енамин обратно в кетон, который в дальнейшем не может быть превращен в продукт, что приводит к неполной реакции сырьевых материалов. Побочные реакции являются следующими:
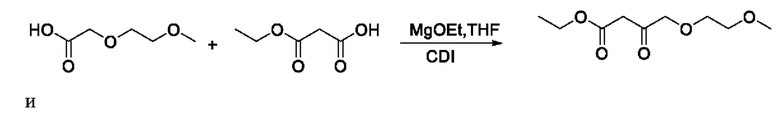
Патентный документ WO 2016102347 A1 описывает способ синтеза с введением боковой цепи. Два исходных материала в этом способе нелегко получить, они имеют относительно высокую стоимость, которая еще больше увеличивается с использованием этоксида магния, N,N-карбонилдиимидазола (CDI) и безводного тетрагидрофурана, они обладаюет плохой атомарной экономичностью и высокой стоимостью очистки и, следовательно, не имеют ценности для промышленного применения. Аналогичным образом, способ патентного документа WO 2005026149 невозможно применять в производстве. Маршруты реакции являются следующими:

В 2015 году в патентном документе ЕР 2821399 А1 от Lonza Ltd сообщили о другом способе синтеза, в котором линейный выход увеличивается в определенной степени; однако исходные материалы трудно синтезировать, стадии являются длительными, и образуется больше отходов, например, фосфорсодержащие сточные воды трудно обрабатывать, необходимо использовать дорогостоящие катализаторы, трифторацетилхлорид нестабилен и тому подобное. Стоимость тоже очень высокая. Маршрут реакции является следующим:
Таким образом, не существует технологического маршрута, который был бы легко доступен в отношении сырья, безопасен и надежен, и пригоден для промышленного масштабирования.
Сущность изобретения
Цель изобретения
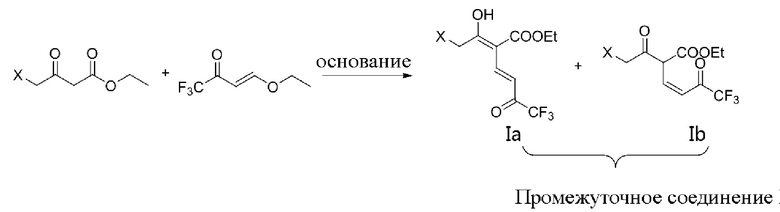
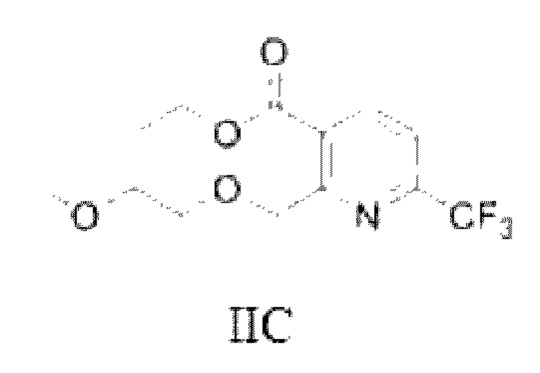
Для преодоления вышеуказанных недостатков, цель данной заявки заключается в том, чтобы предложить соединение, способ его получения и применение соединения при получении промежуточного соединения для бициклопирона. Промежуточное соединение для бициклопирона включает промежуточное соединение I (соединение(я), показанное в формуле (Ia) и/или формуле (Ib)) и промежуточное соединение II (соединение(я), показанное в формуле IIA, формуле IIB и формуле IIC, относящееся к фрагментам никотиновой кислоты).
В данной заявке два фрагмента для получения никотиновой кислоты соединяют сначала под действием основания с образованием промежуточного соединения I, а затем промежуточное соединение I подвергают внутримолекулярному замыканию кольца аммониевой солью, что может значительно увеличить выход промежуточного соединения (II) для бициклопирона, уменьшить побочные реакции и преодолеть недостатки, имеющиеся в уровне техники (например, в способах, описанных в WO 2006059103 A2 и WO 2004078729 A1), заключающиеся в том, что реакция сырьевых материалов часто проходит не полностью ввиду внутримолекулярного замыкания кольца непосредственно аммониевой солью. В данной заявке способ получения промежуточного соединения I под действием основания и последующего проведения реакции замыкания кольца с образованием промежуточного соединения (II) для бициклопирона может уменьшить побочные реакции и может дополнительно увеличивать выход.
Решение
Для достижения цели данной заявки техническое решение, используемое в заявке, является следующим:
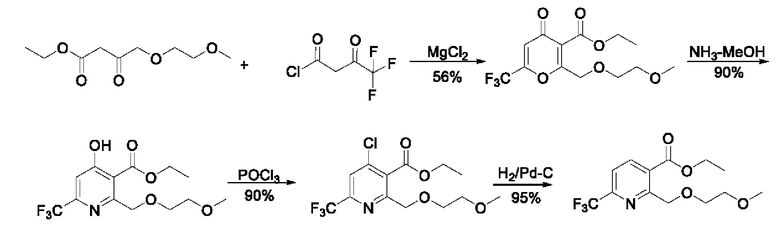
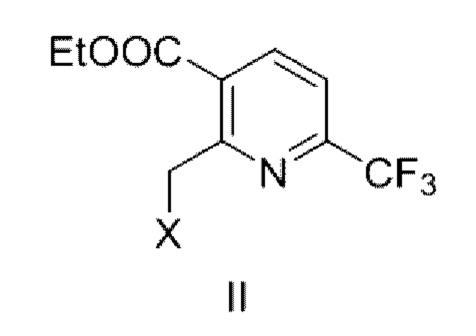
В первом аспекте в данной заявке предложено соединение (называемое промежуточным соединением I), имеющее структурную формулу, показанную в формуле Ia и/или формуле Ib, или его фармацевтически приемлемую соль, или его сольват, или таутомер Ic по отношению к Ib
где X представляет собой -O-R1-O-R2, -Н, -Cl или -Br; и если X представляет собой -O-R1-OR2, R1 выбран из С1-С4 алкиленовых групп, и R2 выбран из С1-С4 алкильных групп.
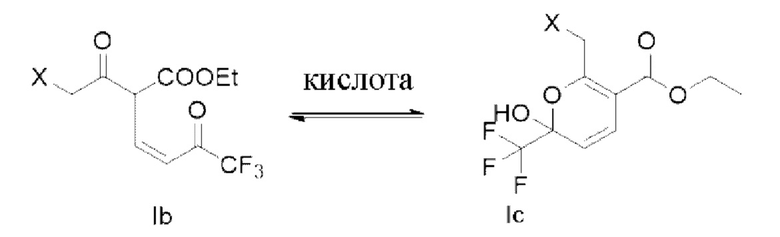
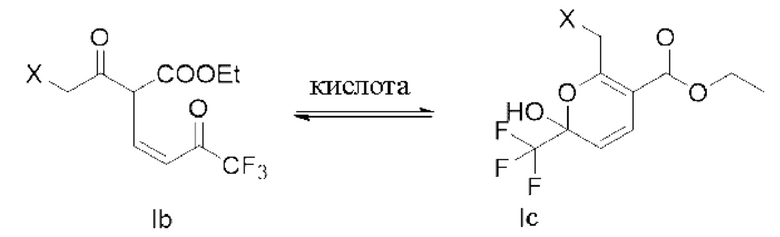
В дальнейших исследованиях обнаружено, что структура формулы lb представляет собой пару кето-рацемических изомеров в основной среде реакции, но эта пара кето-рацемических изомеров существует в форме гемикетальных изомеров (таких как структура, показанная в формуле Iс) после гашения реакции кислотой; и тогда обнаружили, что структура гемикеталя более стабильна, а также в основном присутствует в форме гемикеталя в сухом продукте.
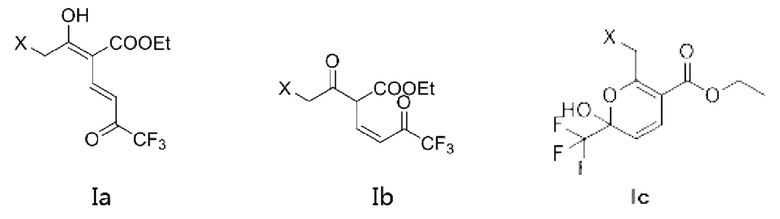
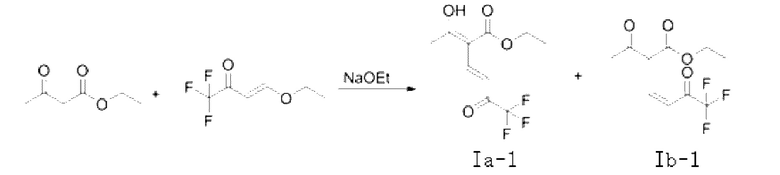
Кроме того, если X является водородом, соединение представляет собой соединение, имеющее структурную формулу, показанную в формуле (Ia-1) или формуле (Ib-1), и таутомер формулы (Ib-1) показан в формуле (Ic-1).
Или X представляет собой -Cl или -Br, возможно, если X представляет собой -Сl, соединение представляет собой соединение, имеющее структурную формулу, показанную в формуле (Ia-2) или формуле (Ib-2), а таутомер формулы (Ib-2) показан в формуле (Iс-2).
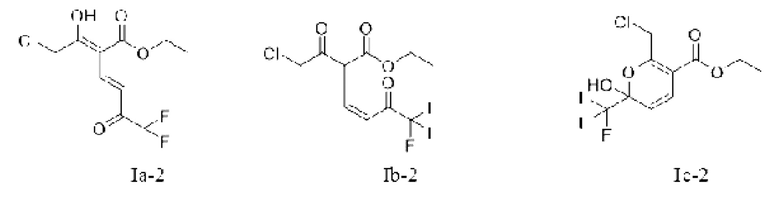
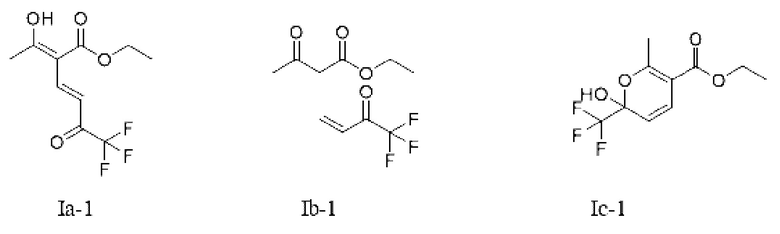
Или X представляет собой -O-R1-O-R, R1 выбран из С1-С4 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Возможно, R1 выбран из C1-С3 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Возможно, R1 выбран из С2-С3 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Предпочтительно, если X представляет собой -O(СН2)2OСН3, соединение представляет собой соединение, имеющее структурную формулу, показанную в формуле Ib-3, или его фармацевтически приемлемую соль, или таутомер, показанный в формуле Iс-3
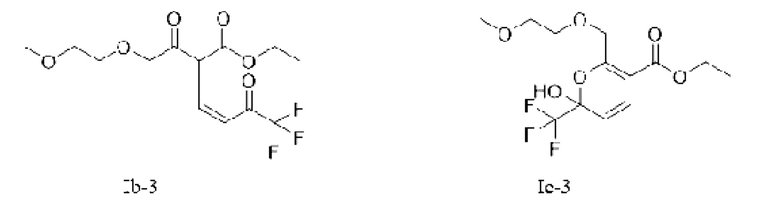
Во втором аспекте в данной заявке предложен способ получения соединения, указанного в первом аспекте, включающий следующую стадию:
в присутствии основания, введение соединения, показанного в формуле III, и/или его енольного таутомера, в реакцию замещения с соединением, показанным в формуле IV, для получения соединения, показанного в формуле Ia и/или Ib и/или Iс,
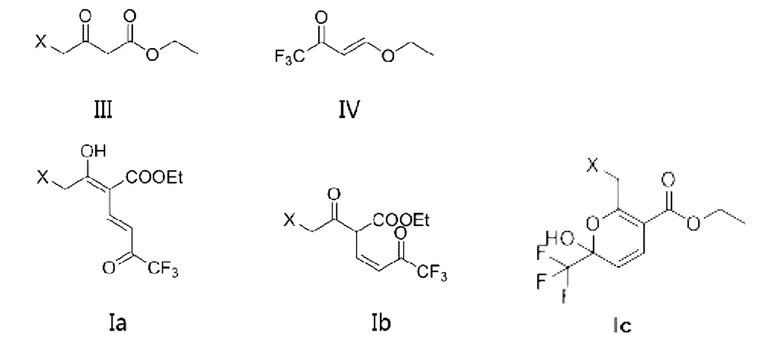
где X представляет собой -O-R1-O-R2, -Н, -Cl или -Br; и если X представляет собой -O-R1-OR2, R1 выбран из С1-С4 алкиленовых групп, и R2 выбран из С1-С4 алкильных групп; и в формулах Ia, Ib, Ic, II и III, X является одним и тем же.
В третьем аспекте в данной заявке предложен способ получения промежуточного соединения для бициклопирона, включающий следующие стадии:
1) в присутствии основания, введение соединение, показанного в формуле III, и/или его енольного таутомера, в реакцию замещения с соединением, показанным в формуле IV,
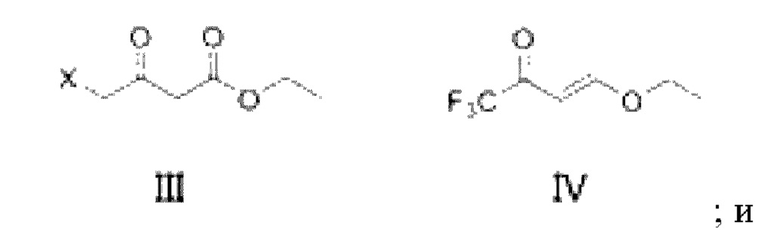
2) в присутствии аммониевой соли и/или аммиака, введение продукта реакции замещения стадии 1) в реакцию замыкания кольца с получением соединения, показанного в формуле II, где в формуле II и формуле III X является одним и тем же,
В способе получения во втором аспекте или третьем аспекте, в реакции замещения, основание представляет собой одно или более, выбранных из группы, состоящей из органического основания, неорганического основания, гидрида натрия или металлического натрия; возможно, органическое основание включает одно или более из алкоксида натрия и алкоксида калия; возможно, органическое основание включает одно или более из метоксида натрия, этоксида натрия, трет-бутоксида калия, метоксида калия, этоксида калия, гексаметилдисилазана натрия и гексаметилдисилазана лития; возможно, неорганическое основание включает одно или более из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия, фосфата натрия, фосфата калия и амида натрия; и возможно, основание представляет собой одно или более, выбранных из группы, состоящей из этоксида натрия, гидроксида натрия и карбоната натрия.
В способе получения во втором или третьем аспекте реакцию замещения проводят в органическом растворителе; возможно, органический растворитель включает один или более из органического спирта, толуола, тетрагидрофурана, диметилсульфоксида (ДМСО), N,N-диметилформамида (ДМФ) и 1,4-диоксана; возможно, органический растворитель включает один или более из метанола, этанола и толуола; и возможно, органический растворитель включает толуол и/или этанол.
В способе получения во втором или третьем аспекте, в реакции замещения, молярное соотношение соединения, показанного в формуле IV, соединения, показанного в формуле III, и/или его енольного таутомера, и основания составляет 1:0,8 - 1,5:0,05 - 1,5, возможно, 1:0,8 - 1,2:0,5 - 1,3, возможно 1:0,9 - 1,1:1 - 1,3, и возможно 1:1:1 - 1,3, предпочтительно 1:1:1-1,2.
В способе получения во втором или третьем аспекте в реакции замещения температура реакции составляет от -15°С до 30°С; и, возможно, 0°С-25°С, предпочтительно 0°С-10°С.
Кроме того, основание добавляют медленно и добавляют по каплям в случае раствора.
Маршрут реакции для промежуточного соединения I (то есть соединения в первом аспекте) данной заявки может быть следующим:
 I
I
Если X - разные заместители, то доля таутомеров (Ia) и (Ib), которые являются енольным соединением Ia и кетосоединением Ib, соответственно, в промежуточном соединении I также может быть разной. Кроме того, (Ib) склонен превращаться в (Iс) путем внутримолекулярного замыкания кольца после гашения кислотой после реакции. Для удобства в описании, в данной заявке Ia и Ib перед гашением реакции представляют собой продукты реакции. Следует отметить, что lb в основном существует в форме Ic после гашения кислотой.
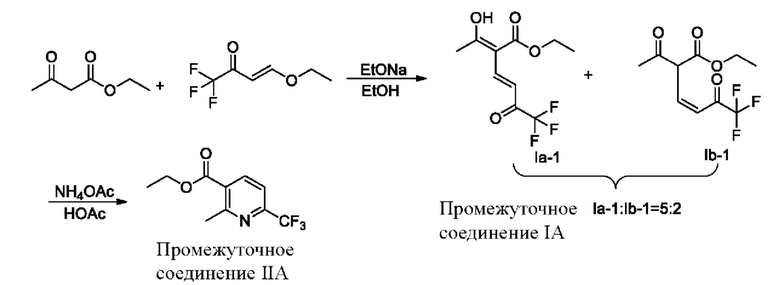
Например, если X представляет собой водород, то полученный продукт реакции включает таутомеры, как показано в формулах (Ia-1) и (Ib-1). Маршрут реакции называется маршрутом А-1. Реакционный маршрут А-1 с этоксидом натрия в качестве основания является следующим:
В реакционном маршруте А-1 ступенчатым способом получают промежуточное соединение I (имеющее структурные формулы (Ia-1) и (Ib-1)), а затем синтезируют промежуточное соединение IIA, которое может значительно уменьшить побочные реакции, улучшить селективность реакции, достичь реакции замыкания кольца в мягких условиях и увеличить выход с 67% до 77,4% (см. пример 1).
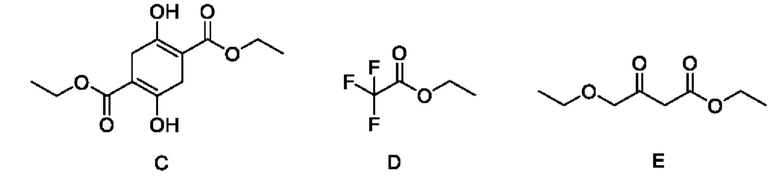
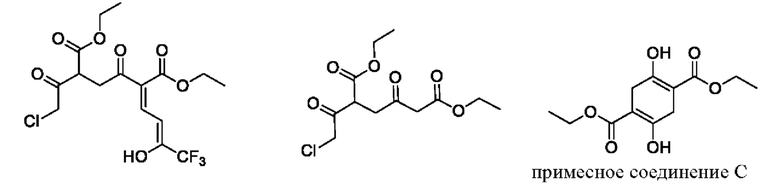
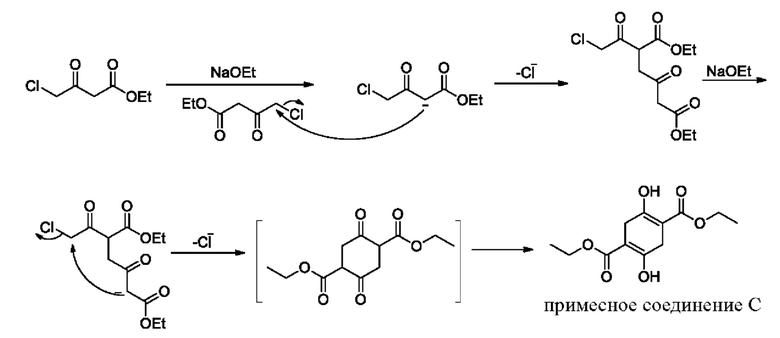
В качестве альтернативы, если X представляет собой -Cl, полученный продукт реакции включает таутомеры, как показано в формулах (Ia-2) и (Ib-2). Полученная специфическая примесь включает по меньшей мере одно из примесного соединения А, примесного соединения В, примесного соединения С и примесного соединения D, причем специфическая примесь включает 2-15% примесного соединения В и 1-5% примесного соединения С. Маршрут реакции называется маршрутом В-1. Маршрут реакции В-1 с этоксидом натрия в качестве основания является следующим:
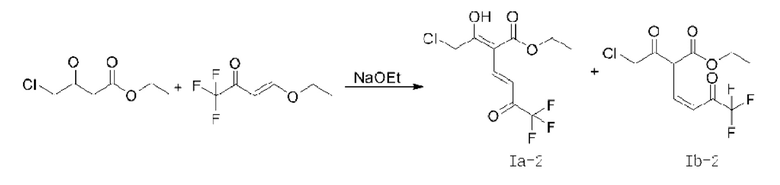
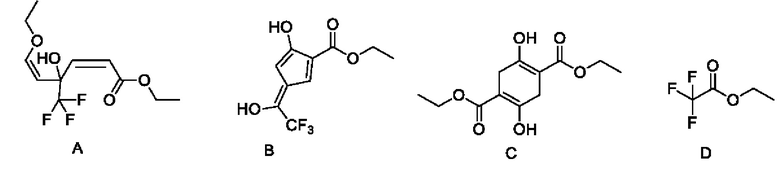
При маршруте реакции В-1, в процессе получения промежуточного соединения I (имеющего структурные формулы (Ia-2) и (Ib-2)) ступенчатым методом, под действием основания, в этил-4-хлорацетоацетате присутствует множество чувствительных групп, так что реакция является очень сложной; и имеется еще четыре основных побочных продукта: примесные соединения А, В, С и D, помимо основного продукта I (пара таутомеров, то есть соединения, имеющие структурную формулу, показанную в формулах (Ia-2) и (Ib-2)). Промежуточное соединение I дополнительно подвергают замыканию кольца для получения промежуточного соединения II с общим выходом 58% (см. пример 2), который также превосходит выход в способе, описанном в WO 2006059103 A2.
Возможно, если IV добавляли по каплям к соединению, показанному в формуле III, и основанию в качестве субстратов, примесь в продукте реакции замещения по меньшей мере включает примесное соединение А, примесное соединение В, примесное соединение С и примесное соединение D.
Возможно, если III и IV используют в качестве субстратов, основание добавляют медленно. Когда контролируют скорость каплепадения основания, продукт реакции замещения по меньшей мере включает примесное соединение В, примесное соединение С и примесное соединение D. Если основание представляет собой раствор основания, то основание добавляют по каплям, и скорость каплепадения равномерно и полностью снижают в течение 1-3 часов, предпочтительно в течение 1,5-2 часов.
В качестве альтернативы, X представляет собой -O-R1-O-R2, R1 выбран из С1-С4 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Возможно, R1 выбран из С1-С3 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Возможно, R1 выбран из С2-С3 алкиленовых групп, и R2 выбран из С1-С2 алкильных групп. Предпочтительно, если X представляет собой -O(СН2)2OСН3, полученный продукт реакции в основном представляет собой соединение, имеющее структурную формулу, показанную в формуле (Ib-3) (которая имеет склонность к превращению в (Ic-3) путем внутримолекулярного замыкания кольца в условиях добавления кислоты); и полученная специфическая примесь включает по меньшей мере одно из примесного соединения С, примесного соединения D и примесного соединения Е. Маршрут реакции называется маршрутом С-1. Маршрут реакции С-1 с этоксидом натрия или карбонатом натрия в качестве основания является следующим:
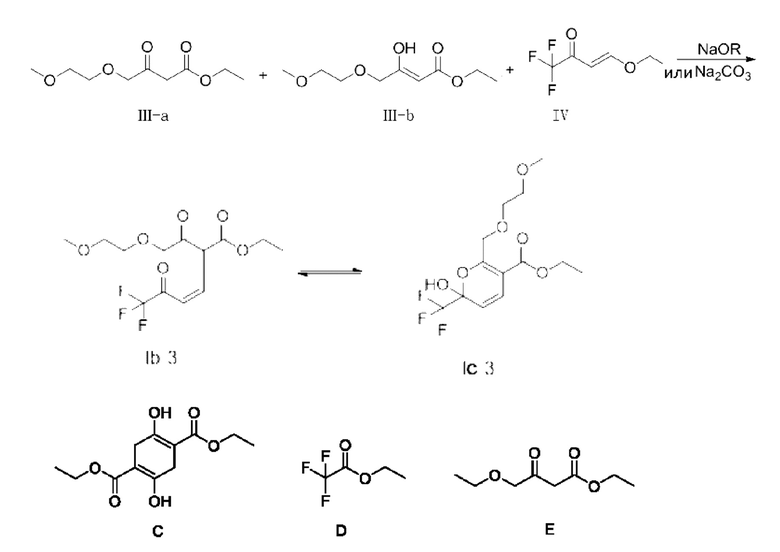
Для промежуточного соединения I (Ia и/или Ib и/или Iс), если X представляет собой -Н, -Cl и -O(СН2)2OСН3, с увеличением заместителя содержание промежуточного соединения I в енольной форме последовательно уменьшается, в то время как содержание промежуточного соединения I в кето-форме, в свою очередь, последовательно увеличивается; после гашения, если X представляет собой -Н и -Сl, изомеризация кетоновой формы до гемикеталя не очевидна; однако, если X представляет собой -O(СН2)2OСН3, кетоновая форма в основном полностью изомеризованав гемикеталь.
В общем случае, промежуточное соединение I (Ia и/или Ib и его таутомер Iс) способствует образованию кинетического продукта при низкой температуре; в то время как в случае высокой температуры оно способствует образованию термодинамического продукта. Авторы данной заявки в результате исследования обнаружили, что промежуточное соединение I, имеющее цис-структуру (кето Ib и его гемикеталь-таутомер Iс), способствует последующей реакции замыкания кольца промежуточного соединения II; в то время как транс-структура (енол Ia) является затрудненной при замыкании кольца и требует более высокой температуры. Например, на маршруте С-1, если промежуточное соединение I (показанное в формуле (Ib-3) и/или формуле (Ic-3)) превращается в транс-Ia-3-структуру, то ввиду стерического препятствия боковой цепи и высокой энергии активации инверсии двойной связи, требуемой для замыкания кольца, замыкание кольца не может быть достигнуто гладко в диапазоне низких температур.
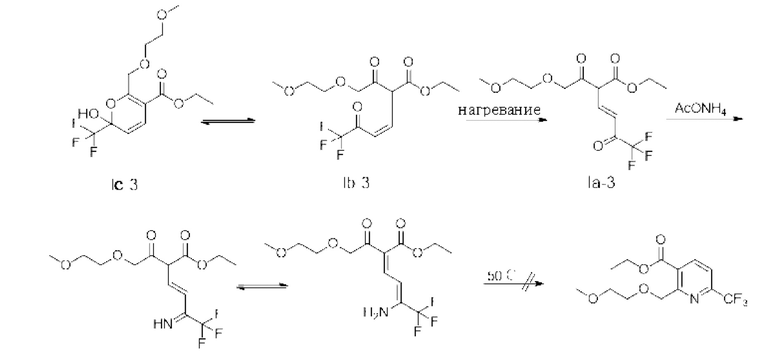
Маршрут реакции С-1 отличается от маршрутов реакции А-1 и В-1 тем, что промежуточное соединение I, полученное ступенчатым методом, обычно содержит только кето-цис-продукт, имеющий структурную формулу (Ib-3); во время реакции кислотного гашения Ib-3 легко изомеризуется в гемикеталь-продукт, имеющий структурную формулу Iс-3 (в дальнейших исследованиях установлено, что Ic-3 все еще способствует последующей реакции после превращения); однако при повышенных температурах или в более кислых условиях также может образовываться небольшое количество енольного транс-продукта Ia-3. Ввиду меньшего количества или даже отсутствия енольного транс-продукта Ia-3, получаемого на маршруте С-1, кето-цис-продукт (Ib-3) получают однореакторным методом, а затем непосредственно используют для последующих реакций без реакции кислотного гашения, что более способствует увеличению выхода реакции. Гемикеталь-цис-продукт (Ic-3), полученный изомеризацией (Ib-3) путем реакции кислотного гашения в ступенчатом способе, также способствует последующей очистке никотиновой кислоты. Поэтому метод маршрута С-1 лучше, чем у маршрутов А-1 и В-1.
В приведенных выше маршрутах реакции А-1, В-1 и С-1 сначала получают промежуточное соединение I (Ia-1 и Ib-1, или Ia-2 и Ib-2, или Ib-3), а затем получают промежуточное соединение II, которое имеет большое значение для снижения образования побочных продуктов, создания крупномасштабного производства и снижения себестоимости продукции; и промежуточное соединение II может быть получено с высокой степенью конверсии и высоким выходом. Среди них маршрут С-1 отличается высоким линейным выходом, небольшим количеством побочных продуктов и больше подходит для промышленного крупномасштабного производства.
Среди трех вышеупомянутых растворов А-1, В-1 и С-1 раствор С-1 является предпочтительным маршрутом ввиду небольшого количества побочных реакций, например, с устранением недостатков маршрута А-1, состоящих в плохой селективности метилгалогенирования, и недостатков маршрута В-1, состоящих в большем количестве побочных реакций этил-4-хлорацетоацетата.
В способе получения в третьем аспекте условие реакции замыкания кольца включает: температуру 0°С-80°С, предпочтительно, 45°С-60°С.
В способе получения в третьем аспекте, в реакции замыкания кольца, аммониевая соль включает один или более из хлорида аммония, карбоната аммония, бикарбоната аммония, нитрата аммония, сульфата аммония, фосфата аммония и ацетата аммония, предпочтительно, аммониевая соль представляет собой ацетат аммония; и аммиак присутствует в форме газообразного аммиака и/или аммиачной воды.
В способе получения в третьем аспекте, в реакции замыкания кольца, молярное отношение соединения, показанного в формуле Ia и/или формуле Ib, к аммониевой соли и/или аммиаку составляет 1:1 - 5, возможно, 1:1 - 2,5, предпочтительно, 1:1,2 - 1,5.
В четвертом аспекте в данной заявке предложена композиция, включающая соединение согласно первому аспекту или продукт, полученный с помощью способа приготовления согласно второму аспекту или третьему аспекту.
В качестве возможного воплощения, если X представляет собой Н, композиция включает соединение, имеющее структурные формулы (Ia-1) и (Ib-1) с молярным соотношением примерно 5:2 при комнатной температуре (соединения, показанные в формуле Ia-1 и формуле Ib-1, представляют собой пару таутомеров, которые находятся в химическом равновесии в реакционной системе и имеют молярное отношение, связанное с температурой).
В качестве возможного воплощения, если X представляет собой С1, композиция включает соединения, имеющие структурные формулы (Ia-2) и (Ib-2) с молярным соотношением примерно 1:5 при комнатной температуре (соединения, показанные в формуле Ia-2 и формуле Ib-2, представляют собой пару таутомеров, то есть пару продуктов реакции, которые находятся в химическом равновесии в реакционной системе и имеют молярное отношение, связанное с температурой). Возможно, композиция дополнительно включает по меньшей мере одно из примесного соединения А, примесного соединения В, примесного соединения С и примесного соединения D; и возможно, композиция дополнительно включает 2-15% примесного соединения В и 1-5% примесного соединения С.
В качестве возможного воплощения, если X представляет собой -O(СН2)2OСH3, композиция включает соединение, имеющее структурную формулу, показанную в формуле (Ib-3); и возможно, композиция дополнительно включает по меньшей мере одно из примесного соединения С, примесного соединения D и примесного соединения Е.
В пятом аспекте данной заявки предложено применение соединения согласно первому аспекту, продукта, полученного способом получения согласно второму аспекту или третьему аспекту, и композиции согласно четвертому аспекту при получении бициклопирона.
В способе получения во втором или третьем аспекте продукт реакции замещения может быть непосредственно использован для последующих реакций или может быть очищен (например, вакуумнаяой дистилляцией) для последующих реакций.
Предпочтительно, в данной заявке предложен способ получения промежуточного соединения IIC для бициклопирона однореакторным способом (то есть продукт реакции непосредственно используют для последующих реакций), включающий следующие стадии:
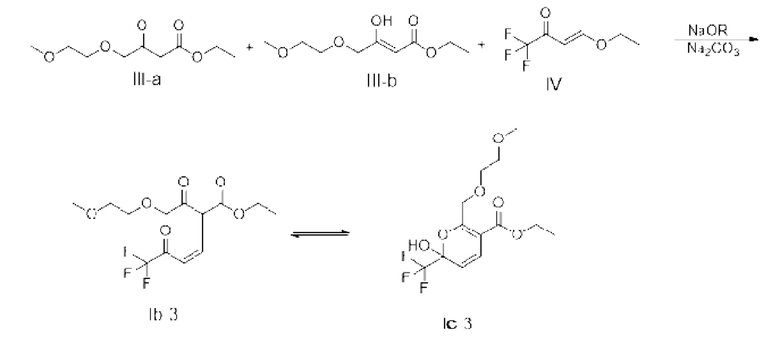
(1) введение материала, содержащего соль 2-метоксиэтанола, в реакцию с этил-4-хлорацетоацетатом для получения реакционного материала, содержащего формулу III-а и/или III-b,

(2) введение реакционного материала стадии (1) в реакцию замещения с соединением, показанным в формуле IV, под действием основания для получения реакционного материала,
(3) добавление аммониевой соли и/или аммиака к реакционному материалу стадии (2) для введения реакционного материала стадии (2) в реакцию замыкания кольца, с получением соединения, показанное в формуле IIC,
При этом соединения, показанные в формуле III-а и формуле III-b, представляют собой пару таутомеров, и их молярное отношение получается в случае достижения равновесия между таутомерами.
Маршрут реакции на этой стадии представляет собой маршрут реакции С-1 однореакторного метода (с основанием, представляющим собой алкоксид натрия или карбонат натрия в качестве примера):
Согласно конкретному воплощению данной заявки, на стадии (1) молярное отношение этил-4-xлорацетоацетата к соли 2-метоксиэтанола составляет 1:1,8 - 2,5. Авторы данной заявки обнаружили в результате исследования, что в случае низкой доли соли 2-метоксиэтанола сырьевые материалы реагируют не полностью; а высокая доля приводит к побочным реакциям, что, в свою очередь, приводит к снижению выхода. Поэтому оптимальным молярным соотношением является 1:2 - 2,3. Отсюда выводят в обратную сторону, что при получении соли 2-метоксиэтанола молярное отношение основания к соединению, показанному в формуле IV, составляет 1 - 3: 1, возможно 2 - 2,5: 1, предпочтительно 2 - 2,3:1.
Кроме того, на стадии (1) материал, содержащий соль 2-метоксиэтанола, представляет собой реакционный материал, полученный реакцией 2-метоксиэтанола под действием основания.
Кроме того, температура реакции для 2-метоксиэтанола и основания составляет 40-180°С, или 80 150°С, или 80-130°С.
Кроме того, основание добавляют медленно и добавляют по каплям в случае раствора.
Кроме того, основание представляет собой одно или более, выбранных из группы, состоящей из органического основания, неорганического основания, гидрида натрия или металлического натрия; органическое основание включает одно или более из алкоксида натрия и алкоксида калия, предпочтительно, органическое основание включает одно или более из метоксида натрия, этоксида натрия, трет-бутоксида калия, метоксида калия и этоксида калия; и неорганическое основание включает одно или более из гидроксида натрия, гидроксида калия и амида натрия; предпочтительно основание представляет собой одно или более, выбранных из группы, состоящей из этоксида натрия, метоксида натрия, метоксида калия и этоксида калия; и выбор правильного основания может повысить селективность маршрута реакции, что увеличит выход продукта.
Кроме того, на стадии (2) температура реакции составляет от -15°С до 30°С, предпочтительно, 0°С - 10°С.
Кроме того, на стадии (3) температура реакции составляет 0°С - 80°С, возможно, 30°С - 80°С и предпочтительно 45°С - 60°С.
Кроме того, на стадии (3) аммониевая соль включает один или более из хлорида аммония, карбоната аммония, бикарбоната аммония, нитрата аммония, сульфата аммония, фосфата аммония и ацетата аммония, предпочтительно, аммониевая соль представляет собой ацетат аммония; и аммиак присутствует в виде газообразного аммиака и/или аммиачной воды.
Кроме того, на стадии (3) молярное отношение соединения, показанного в формуле Ib-3, к соли аммония и/или аммиаку составляет 1:1 - 5, возможно, 1:1 - 2,5 и, предпочтительно, 1:1,2 - 1,5.
Кроме того, на стадии (2) реакционный материал дополнительно включает по меньшей мере одно из примесного соединения С, примесного соединения D и примесного соединения Е,
В воплощении с однореакторным способом реакционного маршрута С-1 реакционный материал стадии (1) непосредственно используют для реакции на стадии (2).
В воплощении с однореакторным способом реакционного маршрута С-1 реакционный материал стадии (2) непосредственно используют для реакции на стадии (3).
В общем случае, промежуточное соединение I способствует образованию кинетического продукта при низкой температуре; в то время как в случае высокой температуры это способствует образованию термодинамического продукта. Эксперимент показывает, что промежуточное соединение I, имеющее цис-структуру, способствует последующей реакции замыкания кольца промежуточного соединения II; в то время как транс-структура (енол) затруднена при замыкании кольца и требует более высокой температуры. Например, если IС имеет транс-структуру, ввиду стерического препятствия боковой цепи и высокой энергии активации инверсии двойной связи, необходимой для замыкания кольца, замыкание кольца не может быть достигнуто гладко в диапазоне низких температур.
Полный маршрут реакции для получения промежуточного соединения II с маршрутами реакции А-1, В-1 и С-1 выглядит следующим образом:
маршрут А-1
маршрут В-1
маршрут С-1
В приведенном выше способе С-1 получения IIC промежуточное соединение Ib-3 может быть получено ступенчатым методом, то есть промежуточные соединения на каждой стадии могут быть очищены вакуумной дистилляцией перед следующей реакцией (в ступенчатом способе кето-цис-Ib-3 легко изомеризуется в гемикеталь Iс-3 в реакции кислотного гашения). Следует отметить, что если температура вакуумной дистилляции слишком высока, кето-цис-Ib-3 или гемикеталь Iс-3 могут быть превращены в енол-транс-1а-3, что приведет к последующему нарушению замыкания кольца. Также может быть использован однореакторный метод, то есть реакционные жидкости, полученные после каждой промежуточной реакции, непосредственно или после простой обработки вводят в следующую реакцию без очистки, преимущество этого заключается в том, что он позволяет избежать образования Ia-3 вследствие ненужной высокой температуры или последующей обработки во время реакции кислотного гашения.
Существует 4 метода получения натриевой соли 2-метоксиэтанола в вышеуказанном маршруте С-1 получения IIC. Первый метод заключается в добавлении 60 масс. % гидрида натрия к толуолу для реакции с 2-метоксиэтанолом; при этом часто используют большое количество или чрезмерное количество гидрида натрия; ввиду включения минерального масла в системе часто остается большое количество непрореагировавшего гидрида натрия; и реакция гашения может выделять большое количество водорода, так что метод не подходит для промышленного производства. Второй метод заключается в добавлении гидроксида натрия к избыточному количеству 2-метоксиэтанола и одновременном кипячении с толуолом для удаления воды из реакции. Этот метод имеет преимущества дешевого сырья, безопасной реакции и возможности массового производства, но имеет недостатки, заключающиеся в том, что воду трудно удалить чисто, а продукт, натриевая соль 2-метоксиэтанола, является темным. Авторы данной заявки обнаружили, что даже следовые количества воды могут влиять на последующие реакции. Третий метод представляет собой метод с использованием металлического натрия, в котором натриевую соль получают реакцией металлического натрия с 2-метоксиэтанолом. Этот метод дешев в отношении сырья, его можно контролировать в производстве и он подходит для непрерывного производства. Ввиду высвобождения большого количества водорода существуют скрытые угрозы для безопасности. Четвертый метод представляет собой способ с обменом алкоксида натрия, то есть спирт с низкой температурой кипения испаряется в реакции метоксида натрия или этоксида натрия с 2-метоксиэтанолом. Метод может быть расширен, но имеет недостаток в высокой стоимости ввиду большого количества используемого алкоксида натрия. Среди вышеуказанных четырех процессов получения натриевой соли 2-метоксиэтанола в качестве основы предпочтителен алкоксид натрия, особенно метоксид натрия и этоксид натрия.
По сравнению со ступенчатым методом, однореакторный метод прост в работе и может выполняться непрерывно. С одной стороны, это может уменьшить потери, вызванные процессом разделения. С другой стороны, соответствующий выход продукта высок, но содержание продукта является низким. Преимущества ступенчатого метода заключаются в том, что побочные продукты, образующиеся на каждой стадии реакции, могут быть удалены путем дистилляции, и их выход довольно низкий; однако содержание продукта является высоким, что способствует последующей кристаллизации никотиновой кислоты.
Однако следует отметить, что промежуточное соединение Ib-3 имеет плохую стабильность, например, стабильность при хранении. Ib-3 легко изомеризуется при подкислении или при высокой температуре (основная часть Ib-3 изомеризуется в Ic-3, а небольшая часть изомеризуется в Ia-3). Ic-3 способствует последующим реакциям, в то время как Ia-3 не может быть использован для последующих реакций. Поэтому, по сравнению со ступенчатым методом, в однореакторном методе Ib-3, полученный в основных условиях, не требует реакции кислотного гашения, и реакционный материал непосредственно подвергается последующим реакциям, которые могут эффективно исключить или уменьшить изомеризацию промежуточного соединения Ib-3 в Ia-3. Учитывая надежность процесса, однореакторный метод превосходит ступенчатый, поэтому в процессе маршрута С-1 в качестве предпочтительного решения использован однореакторный метод.
Следует отметить, что данная заявка не имеет особых ограничений в способе последующей обработки в приведенной выше реакции замещения или реакции замыкания кольца. Способ последующей обработки может быть выполнен со ссылкой на традиционные способы в данной области техники, такие как экстракция, колоночная хроматография, подготовка при высоком давлении и кристаллизация.
Положительные эффекты
(1) В данной заявке два соединения для получения фрагментов никотиновой кислоты соединяют сначала под действием основания с образованием промежуточного соединения I, а затем промежуточное соединение I подвергают внутримолекулярному замыканию кольца аммониевой солью, что может значительно увеличить выход промежуточного соединения (II) для бициклопирона, уменьшить побочные реакции и преодолеть недостатки уровня техники (например, методов, описанных в WO 2006059103 и WO 2004078729 A1), состоящие в том, что реакция сырья часто протекает не полностью ввиду внутримолекулярного замыкания кольца непосредственно аммониевой солью. В данной заявке ступенчатый способ получения промежуточного соединения I путем гидролиза под действием основания и последующего проведения реакции замыкания кольца с получением промежуточного соединения (II) для бициклопирона способствует очистке никотиновой кислоты на последующей стадии; в то время как однореакторный метод может уменьшить побочные реакции и может еще больше увеличить выход.
(2) В данной заявке можно сделать выбор между тремя маршрутами реакции, А-1, В-1 и С-1 в зависимости от сырья, сначала получают промежуточное соединение I (Ia-1 и Ib-1, или Ia-2 и Ib-2, или Ib-3), а затем получают промежуточное соединение II, которое имеет большое значение для снижения образования побочных продуктов, достижения крупномасштабного производства и снижения себестоимости продукции; и промежуточное соединение II может быть получено с высокой степенью конверсии и высоким выходом. Среди них маршрут С-1 отличается высоким линейным выходом, небольшим количеством побочных продуктов и больше подходит для промышленного крупномасштабного производства. Маршрут С-1 является предпочтительным путем ввиду небольшого количества побочных реакций, например, исключая недостатки маршрута А-1, состоящие в плохой селективности метилгалогенирования, и недостатки маршрута В-1, состоящие в большем количестве побочных реакций этил-4-хлорацетоацетата. В двух решениях маршрута С-1 однореакторный метод может избежать риска превращения цис-промежуточного соединения Ib-3 в транс-промежуточное соединение Ia-3 вследствие разделения/очистки в ступенчатом методе. Таким образом, однореакторный метод на маршруте С-1 является предпочтительным решением.
Краткое описание чертежей
Одно или более воплощений в качестве примера описаны чертежами на соответствующих сопроводительных чертежах, и такое примерное описание не является ограничением воплощений. Конкретный термин «примерный», используемый здесь, означает «служащий примером, воплощением или иллюстрацией». Любое воплощение, описанное в данном документе как «примерное», не обязательно должно толковаться как преимущественное по сравнению с другими воплощениями.
Фиг. 1 представляет собой ядерно-магнитный Н-спектр аналитического образца на стадии 1 примера 1 данной заявки;
Фиг. 2 представляет собой ядерно-магнитный С-спектр аналитического образца на стадии 1 примера 1 данной заявки;
Фиг. 3 представляет собой ядерно-магнитный Н-спектр аналитических образцов на стадии 2) стадии 1 примера 2-3 данной заявки;
Фиг. 4 представляет собой ядерно-магнитный С-спектр аналитических образцов на стадии 2) стадии 1 примера 2-3 данной заявки;
Фиг. 5 представляет собой ядерно-магнитный Н-спектр примесного соединения А на стадии 3) стадии 1 примера 2-3 данной заявки;
Фиг. 6 представляет собой ядерно-магнитный С-спектр примесного соединения А на стадии 3) стадии 1 примера 2-3 данной заявки;
Фиг. 7 представляет собой ядерно-магнитный Н-спектр примесного соединения В на стадии 3) стадии 1 примера 2-3 данной заявки;
Фиг. 8 представляет собой ядерно-магнитный С-спектр примесного соединения В на стадии 3) стадии 1 примера 2-3 данной заявки;
Фиг. 9 представляет собой ядерно-магнитный Н-спектр промежуточного соединения IIB на стадии 2 примера 2-3 данной заявки;
Фиг. 10 представляет собой ядерно-магнитный С-спектр промежуточного соединения IIB на стадии 2 примера 2-3 данной заявки;
Фиг. 11 представляет собой ядерно-магнитный Н-спектр промежуточных соединений III-а и III-Ь на стадии 1 примера 3 данной заявки;
Фиг. 12 представляет собой ядерно-магнитный С-спектр промежуточных соединений III-а и III-b на стадии 1 примера 3 данной заявки;
Фиг. 13 представляет собой ядерно-магнитный Н-спектр промежуточного соединения IС на стадии 2 примера 3 данной заявки;
Фиг. 14 представляет собой ядерно-магнитный С-спектр промежуточного соединения IС на стадии 2 примера 3 данной заявки;
Фиг. 15 представляет собой ядерно-магнитный Н-спектр промежуточного соединения IIIС на стадии 4 примера 4-3 данной заявки;
Фиг. 16 представляет собой ядерно-магнитный С-спектр промежуточного соединения IIIС на стадии 4 примера 4-3 данной заявки;
Фиг. 17 представляет собой ядерно-магнитный Н-спектр примесного соединения Е примера 13 данной заявки;
Фиг. 18 представляет собой ядерно-магнитный С-спектр примесного соединения Е примера 13 данной заявки;
Фиг. 19 представляет собой ядерно-магнитный Н-спектр примесного соединения С примера 14 данной заявки; и
Фиг. 20 представляет собой ядерно-магнитный С-спектр примесного соединения С примера 14 данной заявки.
Подробное описание изобретения
Для обеспечения того, чтобы цели, технические решения и преимущества примеров данной заявки были более ясными, технические решения в примерах заявки ниже описаны ясно и полно. Очевидно, что описанные примеры являются лишь частью, а не всеми примерами данной заявки. На основании примеров данной заявки все другие примеры, полученные специалистом в данной области техники без творческих усилий, должны попадать в объем охраны данной заявки.
Кроме того, чтобы лучше проиллюстрировать заявку, следующие конкретные воплощения приведены во многих конкретных деталях. Специалистам в данной области техники понятно, что воплощение данной заявки также может быть реализовано без некоторых конкретных деталей. В некоторых примерах сырье, решения, способы и средства, известные специалистам в данной области техники, не были подробно описаны, чтобы излишне не затемнять аспекты примеров данной заявки.
Во всем описании и формуле изобретения, если прямо не указано иное, термин «включать» или трансформацию этого термина, например, «содержать», нужно понимать как включающий указанные элементы или компоненты, и не исключающий другие элементы или компоненты.
Содержание продуктов в следующих примерах подтверждается жидкостным или газовым хроматографом для облегчения расчета выхода.
В следующих примерах, для того, чтобы промежуточное соединение I соответствовало маршруту реакции и отличалось от него, промежуточное соединение I называют промежуточным соединением IA (соответствующие таутомеры Ia-1, Ib-1 и/или Ic-1), промежуточным соединением IB (соответствующие таутомеры Ia-2, Ib-2 и/или Iс-2) или промежуточным соединением IС (соответствующие структурные формулы Ib-3 и/или Ic-3).
В следующих примерах ГХ-МС относится к газовой хромато-масс-спектрометрии, ЖХ-МС относится к жидкостной хромато-масс-спектрометрии, детектирование ГХ относится к детектированию методом газовой хроматографии, и детектирование ВЭЖХ относится к детектированию методом жидкостной хроматографии.
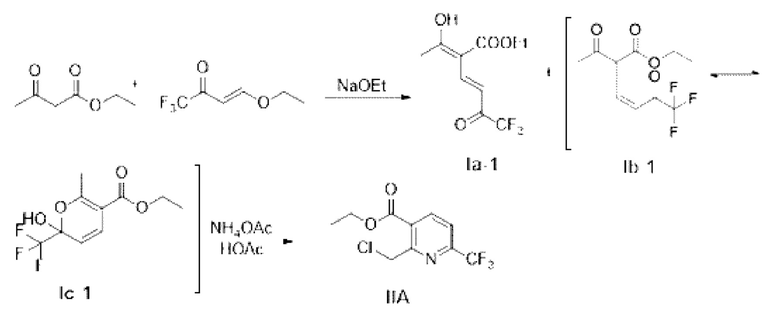
Пример 1
Синтез этил-2-метил-6-(трифторметил)никотината (промежуточное соединение IIA) (один из примеров маршрута А-1)
Стадия 1: синтез промежуточного соединения 1А(енол Ia-1; кето Ib-1)
Этилацетоацетат (6 г, 46,5 ммоль) и 4-этокси-1,1,1-трифторбут-3-ен-2-он (8 г, 47,6 ммоль) добавляли в одногорлую колбу и охлаждали на водяной бане. Этанольный раствор этоксида натрия (56 ммоль) медленно добавляли по каплям до полного исчерпания, перемешивание проводили при 0°С в течение 2 ч, мониторинг проводили методом тонкослойной хроматографии (ТСХ) до окончания реакции. Реакционную жидкость выливали в 50 мл разбавленной соляной кислоты; полученную смесь трижды экстрагировали этилацетатом (60 мл × 3); органические фазы соединяли и промывали насыщенной соленой водой; органическую фазу отделяли и концентрировали с получением 11,96 г коричневой жидкости; и коричневая жидкость была очищена с помощью колоночной хроматографии для получения аналитического образца. Аналитический образец подвергают Н- и С-ядерно-магнитным спектральным анализам, и их результаты показаны на фиг. 1 и фиг. 2.
Н- и С-ядерно-магнитные спектральные анализы промежуточного соединения IA (енол Ia-1; кето Ib-1) (фиг. 1 и фиг. 2) являются следующими:
ЖХ-МС: М+1=253, М-1=251
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): (енол) 14,75 (s, 1 Н), 7,82 (d, 1 Н, J=15,0 Гц), 6,89 (d, 1 H, J=15,0 Гц), 4,33 (q,2 H, J=5,0 Гц), 2,33 (s, 3 H), 1,36 (t, 3 H, J=5,0 Гц); (кетон) 6,85 (d, 0,4 Н, J=10,0 Гц), 5,47 (d, 0,4 Н, J=10,0 Гц), 4,16 (q, 1 Н, J=5,0 Гц), 4,02-4,06 (q, 0,2 Н, J=5,0 Гц), 2,35 (s, 1,2 Н), 1,24 (t, 1,2 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), δ (ррm): 186,06, 179,45 (q, JC-F=40,5 Гц), 171,29, 164,44, 162,32, 141,77, 126,02, 121,52, 119,26, 115,79 (q, JC-F=346,5 Гц), 112,91, 107,47, 102,80, 100,05, 93,62 (q, JC-F=19,5 Гц), 61,28, 59,72, 19,74, 18,84, 13,21, 12,92
Данные масс-спектрометрии показывают, что молекулярная масса промежуточного соединения IA равна 252, что эквивалентно молекулярной массе структурной формулы IA (Ia-1, Ib-1). Данные спектра протонного ядерного магнитного резонанса показывают, что δ14,76 ppm в формуле Ia-1 содержит активный водород, имеющий признаки фенольной гидроксильной группы, и образует водородную связь с соседним атомом в пространстве, который является енольным гидроксильным водородом. Константы взаимодействия при δ7,82 ppm и δ6,89 ppm составляют J=15,0 Гц, что доказывает, что эти два атома водорода представляют транс-олефиновые связи; в то время как константы взаимодействия при δ6,84 ppm и δ5,47 ppm в формуле Ib-1 составляют J=10,0 Гц, что доказывает, что эти два атома водорода представляют цис-олефиновые связи. В углеродном спектре δ186,06 ppm и δ179,45 ppm разделены на квартеты, что указывает на то, что они представляют углерод, присоединенный к CF3; 5115,79 ppm - это квартет, и JC-F=346,5 Гц, что указывает на то, что 5115,79 ppm - это углерод в CF3.
Стадия 2: синтез этил-2-метил-6-(трифторметил)никотината (промежуточное соединение IIA)
Коричневую жидкость (11,4 г), приготовленную на стадии 1, растворяли в уксусной кислоте (20 мл); полученный раствор перемешивали при комнатной температуре; ацетат аммония (4,28 г) добавляли для перемешивания в течение примерно 0,5 ч; затем температуру повышали до 50°С для продолжения реакции в течение 1,5 ч; и система стала коричневато-красной. Уксусную кислоту отделяли вакуумным концентрированием при 70°С; остаток трижды экстрагировали 150 мл дихлорметана; органические фазы соединяли и промывали небольшим количеством насыщенного водного раствора бикарбоната натрия; органическую фазу отделяли и концентрировали с получением 10,8 г бурого маслянистого вещества с содержанием 74,1%. Суммарный выход стадий 1 и 2 составляет 77,4%.
В примере 1 промежуточное соединение IA (енол Ia-1; кето Ib-1) получают под действием основания ступенчатым способом, а затем синтезируют промежуточное соединение IIA, что может значительно уменьшить побочные реакции, улучшить селективность реакции, достичь реакции замыкания кольца в мягких условиях и увеличить выход с 67% в уровне техники до 77,4%.
Пример 2
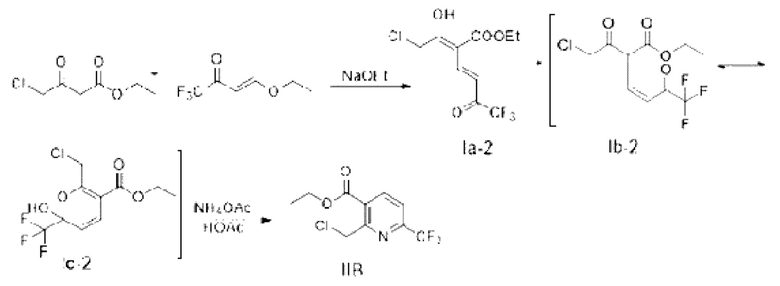
Синтез этил-2-хлорметил-6-(трифторметил)никотината (промежуточное соединение IIВ)
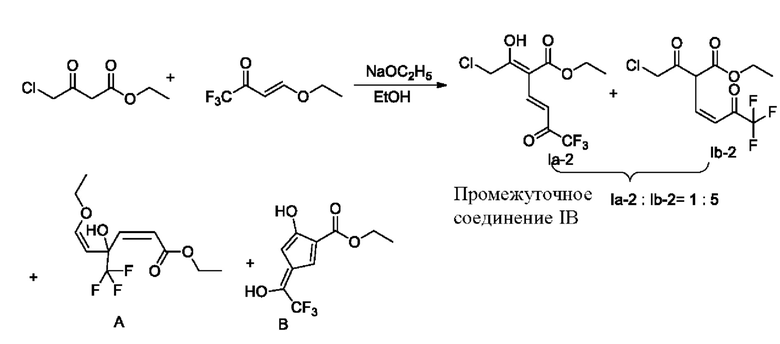
Пример 2-1
Синтез промежуточного соединения IB (енол Ia-2; кето Ib-2)
Безводный этанол (20 г) и этил-4-хлорацетоацетат (3,46 г, 21 ммоль) добавляли в четырехгорлую колбу объемом 250 мл, и при добавляли перемешивании 4-этокси-1,1,1-трифторбут-3-ен-2-он (3,36 г, 20 ммоль). Температуру понижали до -15°С, и медленно добавляли по каплям этоксид натрия (2,04 г, 30 ммоль), растворенный в этаноле, при этом добавление по каплям завершали примерно за 1,0 ч. Температуру поддерживали на уровне -15°С в течение примерно 2,0 ч, и проводили мониторинг с помощью тонкослойной хроматографии (ТСХ) до конца реакции. Реакционную жидкость выливали в 30 мл приготовленной разбавленной соляной кислоты; этанол удаляли ротационным выпариванием; водную фазу трижды экстрагировали этилацетатом (10 мл × 3); органические фазы соединяли, сушили безводным сульфатом магния и подвергали ротационному выпариванию и концентрированию с получением 4,25 г сырого продукта с выходом 54,3%.
Пример 2-2
Синтез промежуточного соединения IB (енол Ia-2; кето Ib-2)
Безводный этанол (35 г), этил-4-хлорацетоацетат (17,0 г, 102 ммоль) и 4-этокси-1,1,1-трифторбут-3-ен-2-он (16,8 г, 100 ммоль) добавляли в четырехгорлую колбу объемом 250 мл. Температуру повышали до 30°С, и медленно добавляли по каплям этоксид натрия (7,0 г, 102,9 ммоль), растворенный в этаноле, при этом добавление по каплям завершали примерно за 1,0 ч. Температуру поддерживали на уровне 30°С в течение примерно 2,0 ч, и реакцию контролировали в середине методом ЖХ и прекращали, когда содержание сырья составляло меньше 1%. Реакционную жидкость выливали в 100 мл разбавленной соляной кислоты; значение рН регулировали до 1-2; водную фазу три раза экстрагировали этилацетатом (100 мл × 3); органические фазы соединяли, сушили безводным сульфатом магния и подвергали ротационному выпариванию и концентрированию с получением 29,0 г сырого продукта с выходом 67,7%.
Пример 2-3
Синтез этил-2-хлорметил-6-(трифторметил)никотината (промежуточное соединение IIВ)
Стадия 1: синтез промежуточного соединения IB (енол Ia-2; кето Ib-2)
1) Безводный этанол (28 г) и этил-4-хлорацетоацетат (17,63 г, 107 ммоль) добавляли в четырехгорлую колбу, и при перемешивании добавляли 4-этокси-1,1,1-трифторбут-3-ен-2-он (17,7 г, 105 ммоль). Температуру понижали до 0°С, и медленно добавляли по каплям этоксид натрия (7,16 г), растворенный в этаноле, при этом добавление по каплям завершали примерно за 2,0 ч. Температуру поддерживали на уровне 0°С в течение примерно 2,0 ч, и реакцию контролировали в середине методом ЖХ и прекращали, когда содержание сырья составляло меньше 1%; реакционную жидкость выливали в 100 мл приготовленного разбавленного раствора соляной кислоты, доводя значение рН до 2-3; и полученную смесь экстрагировали с помощью дихлорметана (150 мл). Водную фазу два раза экстрагировали дихлорметаном (100 мл × 2); и органические фазы объединяли и промывали насыщенной соленой водой один раз для отделения органической фазы. Органическую фазу концентрировали для получения 32,62 г сырого продукта, который представлял собой оранжево-желтую жидкость.
2) Сырой продукт, полученный концентрированием органической фазы на стадии 1), очищали колоночной хроматографией с получением 21,8 г светло-желтого чистого продукта с выходом 72,4%, где соотношение формулы Ia-2 и формулы Ib-2 составляло примерно 1:5. Образец, очищенный колоночной хроматографией, подвергали анализу Н и С ядерно-магнитных спектров, как показано на фиг. 3 и 4.
3) Для концентрированного сырого продукта в 1) были получены примесное соединение А (100 мг, чистота 97%) и примесное соединение В (1,0 г, чистота 95%) путем приготовления жидкой фазы при высоком давлении с ацетонитрилом и водой в качестве подвижных фаз.
Н и С ядерно-магнитные спектральные анализы промежуточного соединения I (енол Ia-2, кето Ib-2) (фиг. 3 и фиг. 4) являются следующими:
ЖХ-МС: M+1=287
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): (соединение Ia-2) 14,54 (s, 1 Н), 7,82 (d, 1 Н, J=15,0 Гц); 7,05 (d, 1 Н, J=15,0 Гц), 4,44 (q, 2 Н, J=5,0 Гц), 4,37 (s, 2 Н), 1,44 (t, 3 Н, J=5,0 Гц); (соединение Ib-2) 6,95 (d, 1,25 Н, J=10,0 Гц), 5,70 (d, 1,25 Н, J=10,0 Гц), 4,93 (d, 1,25 Н, J=10,0 Гц), 4,46 (d, 1,25 Н, J=10,0 Гц), 4,27-4,31 (m, 3,75 Н), 2,01 (s, 2 Н), 1,35 (t, 3,75 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), δ (ррm): 177,20 (d, JC-F=40,5 Гц), 170,05, 160,20, 155,67, 138,49, 135,76, 124,65, 119,35 (q, JC-F=339,0 Гц), 114,76, 114,55, 109,63, 104,12, 100,04, 92,89 (d, JC-F=42,0 Гц), 61,12, 59,62, 46,44, 38,15, 38,05, 12,25, 12,03
В этом примере во время синтеза промежуточного соединения IB (енол Ia-2, кето Ib-2) на стадии 1, под действием основания, в этил-4-хлорацетоацетате присутствуют множество чувствительных групп, так что реакция является очень сложной; и по-прежнему существуют четыре основных примесных соединения - побочных продукты А, В, С и D, помимо промежуточного соединения I в качестве основного продукта (енол Ia-2, кето Ib-2).
Образование примесного соединения А, примесного соединения В, примесного соединения С и примесного соединения D конкурирует с промежуточным соединением IB в качестве основного продукта (енол Ia-2, кето Ib-2), что связано с режимом присоединения основания. В общем, если основание добавляют быстро, вследствие высокой концентрации 4-этокси-1,1,1-трифторбут-3-ен-2-он подвергается кислотному разложению с образованием анионов этилакрилата, а затем выполняют добавление с получением примесного соединения А; тогда как в случае, если основание добавляют медленно, ввиду низкой общей концентрации основания образование А почти не наблюдается. Образование примесного соединения В и примесного соединения С не зависит от способа подачи основания и типа основания. Содержание В составляет примерно 6-12%, и содержание С обычно составляет около 3%. Побочные продукты В и С неизбежно образуются, а также являются основными примесями в этом примере. Возможный механизм образования примесного соединения А является следующим:
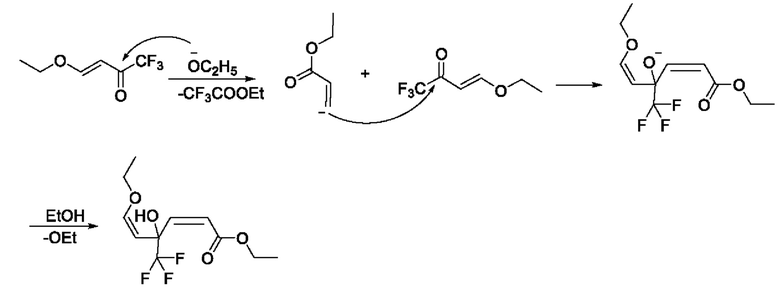
Н и С ядерно-магнитные спектральные анализы примесного соединения А показаны на фиг. 5 и 6.
ЖХ-МС: М+1=269
1Н ЯМР (d6-DMSO, 500 МГц), δ (ррm): 7,05 (d, 1 Н, J=10,0 Гц), 5,48 (d, 1 Н, J=10,0 Гц), 5,03 (d, 1 Н, J=10,0 Гц), 4,41 (d, 1 Н, J=10,0 Гц), 4,27-4,32 (m, 2 Н), 3,56-3,66 (m, 2 Н), 1,35 (t, 3 Н, J=10,0 Гц), 1,24 (t, 3 Н, J=10,0 Гц)
13С ЯМР (d6-DMSO, 150 МГц), δ (ррm): 162,11, 158,09, 126,53, 119,00 (q, JC-F=340,50 Гц), 108,26, 96,79 (t, JC-F=40,5 Гц), 59,50, 57,68, 13,16, 12,26
Данные масс-спектрометрии показывают, что молекулярная масса примесного соединения А равна 268, что эквивалентно молекулярной массе структурной формулы (примесное соединение А). Данные протонного спектра показывают, что молекулярная структура содержит 2 этоксигруппы и 4 олефиновых атома водорода, причем 4 олефиновых атома водорода находятся на разных олефиновых связях. Химический сдвиг δ162,11 ppm в углеродном спектре представляет карбонильный углерод, δ119,00 ppm расщепляется на квартет, и JC-F=340,50 Гц, что указывает на то, что молекула содержит CF3; δ96,79 ppm расщепляется на триплет, a JC-F=40,5 Гц, что указывает на то, что углерод непосредственно присоединен к CF3, где СH2 при δ3,56 - 3,66 ppm расщепляется на две группы, что указывает на то, что структура имеет хиральность и представляет собой пару рацемических изомеров.
Механизм образования примесного соединения В является следующим:
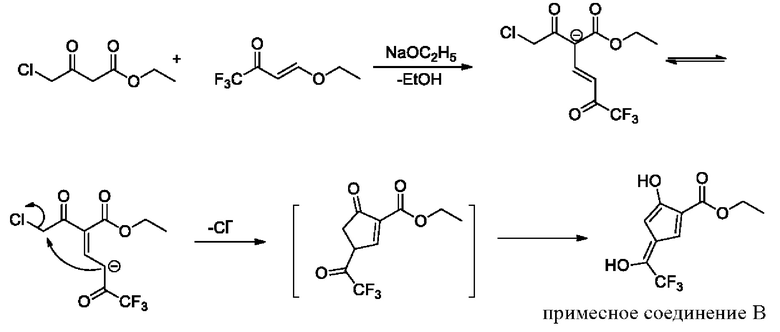
Н и С ядерно-магнитные спектральные анализы примесного соединения В показаны на фиг. 7 и 8.
ЖХ-МС: М+1=251, М-1=249
1Н ЯМР (d6-DMSO, 500 МГц), δ (ррm): 10,56 (brs., 2 Н), 7,35 (d, 1 Н, J=10,0 Гц), 7,09 (d, 1 Н, J=10,0 Гц), 4,41 (q, 2 Н, J=5,0 Гц), 1,36 (t, 3 Н, J=5,0 Гц)
13С ЯМР (d6-DMSO, 150 МГц), δ (ррm): 168,97, 150,16, 145,84, 123,85 (q, JC-F=325,5 Гц), 119,95 (q, JC-F=34,5 Гц), 119,32, 116,63, 116,08 (d, JC-F=6 Гц), 62,40, 14,33.
Данные масс-спектрометрии показывают, что молекулярная масса равна 250, что эквивалентно молекулярной массе структурной формулы (примесное соединение В). Данные протонного спектра показывают, что широкий синглет δ 10,56 ppm содержит 2 активных атома водорода, имеющих признаки фенольной гидроксильной группы, и образует водородные связи с фенольной гидроксильной группой и соседними атомами в пространстве. В другом аспекте δ 168,97 ppm показывает только одну карбонильную группу. Молекула содержит этоксигруппу, которая представляет собой карбонильную группу сложного эфира, указывая на то, что две другие «карбонильные группы» в молекуле существуют в енольной форме, а не в кето-форме.
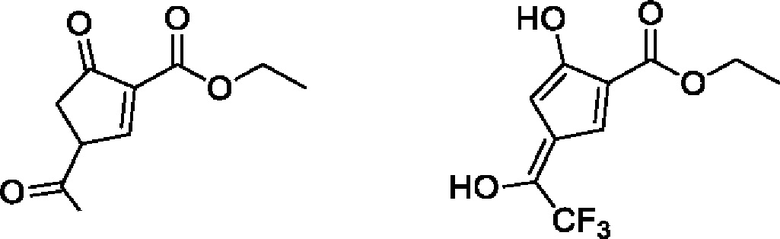
структура кето-формы → структура енольной формы
δ 7,35 ppm и δ 7,09 ppm представляют олефиновый водород, и цвет, проявляющийся под действием ультрафиолета, указывает на то, что молекула должна быть ароматической; в углеродном спектре δ 123,85 ppm расщепляется на квартет, и JC-F=325,5 Гц, что указывает на то, что он содержит группу CF3; и δ 119,95 ppm расщепляется на квартет, и CF3 и JC-F=34,5 Гц, что указывает на то, что углерод присоединен к группе CF3 и не является карбонильным углеродом.
На стадии 1 этого примера также имеется много других возможных побочных продуктов (около 20 видов) с содержанием, в общем, менее 3%, например:
Основная причина заключается в том, что этил-4-хлорацетоацетат содержит множество активных центров, а селективность реакции невысока. Даже если температуру снизить до -15°С, на результаты реакции это мало влияет. Содержание примесного соединения С составляет около 3%. Механизм реакции является следующим:
Масс-спектрометрия показывает, что молекулярная масса примесного соединения С равна 256. Исходя из данных углеродного спектра и протонного спектра, предполагается, что молекулярная структура симметрична. В протонном спектре δ 12,13 ppm является енольным водородом и образует внутримолекулярные водородные связи с соседними группами. δ170,29 ppm доказывает, что существует только один вид карбонильного углерода. Существование этоксигруппы указывает на то, что структура содержит только сложноэфирную группу, отличную от кетонной группы.
На стадии 1 примера 2 в качестве субстратов используют этил-4-хлорацетоацетат и 4-этокси-1,1,1-трифторбут-3-ен-2-он. В процессе добавления алкоксида натрия по каплям алкоксид натрия обеспечивает образование анионов этил-4-хлор-3-оксобутаноата, которые далее реагируют с 4-этокси-1,1,1-трифторбутом-3-ен-2-оном с образованием промежуточного соединения IB (енол Ia-2 и кето Ib-2), и промежуточное соединение IB подвергается внутримолекулярной реакции с образованием примесного соединения В; и межмолекулярная реакция этил-4-хлорацетоацетата приводит к образованию примесного соединения С и других полимеров. Влияние алкоксида натрия на 4-этокси-1,1,1-трифторбут-3-ен-2-он заключается в образовании примесного соединения А и примесного соединения D путем кислотного гидролиза. Это является причиной того, что контролируемые в середине реакции сырьевые материалы полностью исчезают, и выход промежуточного соединения IB является низким.
Промежуточное соединение IB, полученное на стадии 1 этого примера, включает цис- и транс-таутомеры в соотношении примерно 1:5, где транс-изомер Н имеет енольную структуру. Ввиду образования внутримолекулярных водородных связей сигнал енольного водорода сдвигается в более низкопольную область до δ 14,54 ppm и J=15,0 Гц, указывая на то, что его структура является транс-структурой. Цис-изомер I фактически содержит хиральность и в общей сложности два олефиновых водорода; и δ 1,34-1,36 в более высоком поле показывает мультиплет вместо простого триплета, указывая на то, что это пара диастереоизомеров.
Стадия 2: синтез этил-2-хлорметил-6-(трифторметил)никотината (промежуточное соединение IIВ)
Уксусную кислоту (130,48 г) и продукт (18,17 г) на стадиях 1 2) добавляли в четырехгорлую колбу; добавляли при перемешивании ацетат аммония (9,44 г); и температура была повышена до 50°С для сохранения тепла в течение 2 часов. Реакцию контролировали методом ЖХ в середине и прекращали, когда содержание сырья было меньше 1%. Растворитель удаляли вакуумной дистилляцией; остаток промывали насыщенным водным раствором бикарбоната натрия до тех пор, пока не прекращалось образование видимых пузырей; и добавляли дихлорметан (200 мл) для экстракции. Водную фазу экстрагировали дихлорметаном (100 мл × 2); и органические фазы объединяли и промывали насыщенной соленой водой (100 мл) один раз. Растворитель удаляли ротационным выпариванием с получением 24,71 г оранжево-желтого сырого продукта. Сырой продукт очищали с помощью колоночной хроматографии с получением 14,3 г бледно-желтого чистого продукта (промежуточного соединения IIВ) с содержанием 95% и выходом 80,1%. Н и С ядерно-магнитные спектральные анализы чистого продукта, полученного с помощью колоночной хроматографии, показаны на фиг. 9 и 10.
Н и С ядерно-магнитные спектральные анализы промежуточного соединения IIB показаны на фиг. 9 и 10.
ЖХ-МС: М+1=268
1ЯМР (CDCl3, 500 МГц), δ (ppm): 8,45 (d, 1 Н, J=10,0 Гц), 7,75 (d, 1 Н, J=10,0 Гц); 5,13 (s, 2 Гц), 4,48 (q, 2 Н, J=5,0 Гц), 1,45 (t, 3 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), δ (ppm): 162,40 (d, JС-F=66 Гц), 155,99, 147,87 (q, JC-F=42 Гц), 139,01, 126,81, 118,89 (q, JC-F=267 Гц), 116,37 (d, JС-F560 Гц), 60,65, 43,00, 12,18.
Пример 3
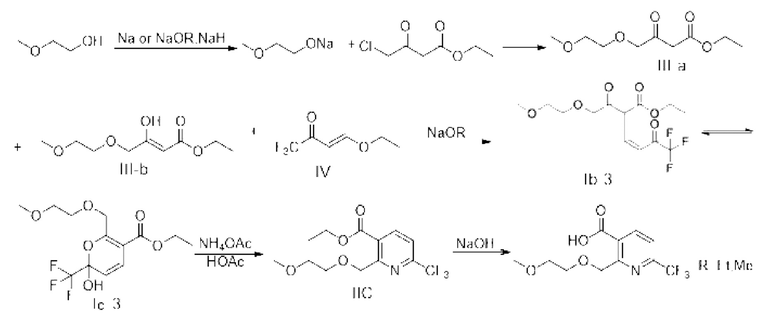
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
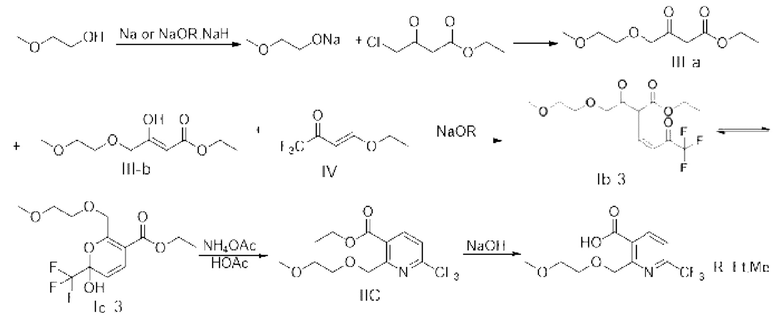
Стадия 1: синтез этил-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-а) и этил-(Z)-3-гидрокси4-(2-метоксиэтокси)-3-оксобутирата (то есть III-b)
В четырехгорлую колбу добавляли тетрагидрофуран (100 мл) и создавали защиту азотом; и добавляли NaH (60%, 10,4 г, 260 ммоль) порциями при перемешивании. Температуру снижали до 10°С, медленно добавляли по каплям 2-метоксиэтанол (21,6 г, 284 ммоль) с последующим образованием пузырьков; и после завершения добавления по каплям выполняли перемешивание в течение 30 мин. Смешанную жидкость этил-4-хлорацетоацетата (10 г, 61 ммоль) и тетрагидрофурана (50 мл) добавляли по каплям в вышеуказанную четырехгорлую колбу; и после завершения добавления по каплям проводили реакцию при комнатной температуре в течение 2 ч. Проводили контроль в середине реакции, чтобы остановить реакцию, когда остаточный этил-4-хлорацетоацетат составлял меньше 1%. Растворитель удаляли при пониженном давлении, и к остатку добавляли 150 мл воды; и значение рН регулировали с помощью 30% НCl до 2-3. Для экстракции добавляли дихлорметан (200 мл), и полученную водную фазу экстрагировали два раза дихлорметаном (100 мл × 2); органические фазы объединяли, промывали один раз насыщенной соленой водой, сушили безводным сульфатом магния и концентрировали с получением 9,15 г оранжево-желтой жидкости с содержанием 73% и выходом 53,6%. Отношение кето-формы (то есть соответствующей структурной формуле III-а) к енольной форме (то есть соответствующей структурной формуле III-b) составляет примерно 9:1.
Н и С ядерно-магнитные спектральные анализы соединений III-а и III-b показаны на фиг. 11 и 12.
ГХ-МС: М=204
1Н ЯМР (CDCl3, 500 МГц), δ (ррm) (III-а): 4,10-4,14 (m, 4 ч), 3,61 (t, 2 Н, J=5,0 Гц), 3,50 (t, 2 Н, J=5,0 Гц), 3,46 (s, 2 Н), 3,31 (s, 3 Н), 1,21 (г, 3 Н, J=5,0 Гц); (III-b): 11,89 (s, 1 Н), 5,24 (s, 1 Н), 4,10-4,14 (m, 2 Н), 4,02 (s, 2 Н), 3,61 (t, 2 Н, J=5,0 Гц), 3,50 (t, 2 Н, J=5,0 Гц), 3,32 (s, 3 Н), 1,21 (t, 3 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), (III-а) δ: 200,85, 166,04, 75,21, 70,87, 70,04, 60,35, 57,99, 44,85, 13,08; (III-b) 5:172.91, 171.64, 87.83, 70.80, 69.65, 68.87, 59.18, 58.07, 13.21.
Стадия 2: Синтез промежуточного соединения 1С
В четырехгорлую колбу добавляли этанол (5,6 г) и 3,06 г сырого продукта (73%, 10,9 ммоль) стадии 1, и при перемешивании добавляли (Е)-4-этокси-1,1,1-трифторбут-3-ен-2-он (17,7 г, 105 ммоль). Температуру понижали до 0°С и медленно добавляли по каплям этоксид натрия (0,72 г, 10,6 ммоль), растворенный в этаноле. После того, как добавление по каплям было завершено, температуру поддерживали примерно 2,0 ч, и реакцию контролировали методом ЖХ до тех пор, пока содержание сырья не становилось меньше 1%. Реакционную жидкость выливали в 20 мл приготовленного раствора соляной кислоты со значением рН около 2-3. Полученную смесь экстрагировали дихлорметаном (20 мл). Водную фазу экстрагировали два раза дихлорметаном (10 мл × 2); и органические фазы объединяли. Объединенную органическую фазу промывали один раз насыщенной соленой водой, чтобы отделить органическую фазу; органическую фазу высушивали безводным сульфатом магния и концентрировали с получением 3,51 г сырого продукта, который представлял собой оранжево-желтую жидкость, и непосредственно вводили в следующую реакцию без очистки. Аналитические образцы были получены путем подготовки жидкой фазы и разделения и подвергнуты Н и С ядерно-магнитному спектральному анализу. Н и С ядерно-магнитные спектральные анализы промежуточного соединения IС показаны на фиг. 13 и 14.
ГХ-МС: М=326
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): 8,19 (d, 1 Н, J=5,0 Гц), 7,61 (d, 1 Н, J=5,0 Гц), 4,94 (s, 2 Н), 3,35 (q, 2 Н, J=5,0 Гц), 3,63 (t, 2 Н, J=5,0 Гц), 3,48 (t, 2 Н, J=5,0 Гц), 3,29 (s, 3 Н), 1,34 (t, 3 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), δ (ррm): 164,43, 158,02, 148,05 (q, JC-F=15,0 Гц), 138,41, 128,71, 120,01 (q, JC-F=327,0 Гц), 118,20, 71,87, 70,74, 69,55, 61,04, 57,95, 13,10
Стадия 3: Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточного соединения IIC)
В четырехгорлую колбу добавляли уксусную кислоту (14,04 г) и сырой продукт (3,51 г) стадии 2; добавляли при перемешивании ацетат аммония (0,94 г, 12,2 ммоль); и температуру повышали до 50°С для сохранения тепла в течение 2 ч. Реакцию контролировали в середине методом ЖХ и прекращали, когда содержание сырья было меньше 1%. Растворитель удаляли при пониженном давлении; остаток промывали насыщенным водным раствором бикарбоната натрия до прекращения образования видимых пузырьков; и для экстракции добавляли дихлорметан (20 мл). Водную фазу экстрагировали дихлорметаном (15 мл × 2); и органические фазы объединяли и промывали один раз насыщенной соленой водой (15 мл). Органическую фазу сушили безводным сульфатом магния, и удаляли растворитель при пониженном давлении с получением 3,75 г сырого продукта, который представлял собой оранжево-желтую жидкость с содержанием 53%. Общий выход стадии 2 и стадии 3 составляет 59,3%. Общий выход стадий 1, 2 и 3 составляет 31,8%.
Пример 4
Пример 4-1:
Синтез этил-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-а) и этил-(2)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-b)
Этоксид натрия (6,8 г, 100 ммоль) добавляли к 2-метоксиэтанолу (8,0 г, 105 ммоль) для перемешивания и нагревания; полученную жидкость нагревали на масляной бане до 40°С и перемешивали в течение 2 ч; этанол удаляли методом вакуумной дистилляции; в это время система представляет собой коричнево-желтое твердое вещество; коричнево-желтое твердое вещество охлаждали до комнатной температуры; и добавляли толуол (40,2 г) для перемешивания и диспергирования с получением толуольного раствора натриевой соли 2-метоксиэтанола.
Температуру контролировали примерно при 25°С, и этил-4-хлорацетоацетат (7,5 г, 45,5 ммоль) добавляли по каплям к толуольному раствору натриевой соли 2-метоксиэтанола; после того, как добавление было завершено, температура была повышена до 40°С; перемешивание проводили для осуществления реакции в течение 6 ч; и выполняли контроль методом ТСХ до тех пор, пока сырьевые материалы полностью не прореагировали. Температуру снижали до уровня ниже 30°С; значение рН реакционной жидкости регулировали раствором соляной кислоты до 4; перемешивание проводили в течение 10 мин; полученную смесь оставляли для отстаивания и разделения жидкости для отделения органической фазы; водную фазу экстрагировали 30 мл толуола; органические фазы объединяли; толуол удаляли вакуумным ротационным выпариванием с получением сырого продукта промежуточных соединений III-а и III-b; и сырой продукт подвергли вакуумному выпариванию с помощью масляного насоса с получением 6,43 г золотисто-желтого продукта с выходом 69,2%.
Пример 4-2:
Синтез этил-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-а) и этил-(Z)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-b)
Этоксид натрия (6,8 г, 100 ммоль) добавляли к 2-метоксиэтанолу (11,4 г, 150 ммоль) для перемешивания и нагревания; полученный продукт нагревали на масляной бане до 100°С и перемешивали в течение 1 ч, а затем продолжали нагревать до 180°С для удаления излишков 2-метоксиэтанола путем выпаривания; в это время система представляла собой коричневато-черное твердое вещество; коричневато-черное твердое вещество охлаждали до комнатной температуры; и добавляли толуол (40,2 г) для перемешивания и диспергирования с получением толуольного раствора натриевой соли 2-метоксиэтанола.
Температуру контролировали примерно при 25°С, и этил-4-хлорацетоацетат (7,5 г, 45,5 ммоль) добавляли по каплям к толуольному раствору натриевой соли 2-метоксиэтанола; после того, как добавление было завершено, температура была повышена до 40°С; перемешивание проводили для осуществления реакции в течение 6 ч; и выполняли контроль методом ТСХ до тех пор, пока сырьевые материалы полностью не прореагировали. Температура была понижена до комнатной; значение рН реакционной жидкости регулировали раствором соляной кислоты до 4; полученную смесь перемешивали и оставляли для отстаивания и разделения жидкости для выделения органической фазы; водную фазу экстрагировали 30 мл толуола; органические фазы объединяли; толуол удаляли вакуумным ротационным выпариванием с получением сырого продукта промежуточных соединений III-а и III-b; сырой продукт подвергали вакуумному выпариванию с помощью масляного насоса для получения 7,23 г золотисто-желтого продукта с выходом 77,8%.
Пример 4-3:
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
Этап 1: синтез этил-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-а) и этил-(Z)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата (то есть III-b)
Этоксид натрия (21,6 г, 317 ммоль) добавляли к 2-метоксиэтанолу (72,6 г, 954 ммоль) для перемешивания и нагревания; полученную жидкость нагревали на масляной бане до 130°С и подвергали вакуумной дистилляции для сбора 67 г фракции; температуру опустили до уровня ниже 100°С; и добавляли толуол (86,6 г) для перемешивания и диспергирования с получением толуольного раствора натриевой соли 2-метоксиэтанола.
Температуру контролировали примерно при 30°С, и этил-4-хлорацетоацетат (18,7 г, 114 ммоль) добавляли по каплям к толуольному раствору натриевой соли 2-метоксиэтанола; после того, как добавление было завершено, температуру поддерживали на уровне 40°С; перемешивание проводили для осуществления реакции в течение 6 ч; и контроль методом ТСХ (петролейный эфир : этилацетат = 6: 1) выполняли до тех пор, пока сырье полностью не вступило в реакцию. Температуру снижали до уровня ниже 30°С; реакционную жидкость выливали в раствор соляной кислоты (112,2 г, 6,8%); полученную жидкость перемешивали в течение 10 мин и оставляли для отстаивания и разделения жидкости; водную фазу экстрагировали два раза толуолом (43,2 г) и оставляли для отстаивания и разделения жидкости; водную фазу снова экстрагировали дихлорметаном (21,6 г); органические фазы объединяли и подвергали вакуумной перегонке (90°С, -0,095 МПа) с получением сырого продукта промежуточных соединений III-а и III-b, который представлял собой коричневое маслянистое вещество массой 22,0 г. Сырой продукт нагревали на масляной бане до 130°С; растворитель испаряли при пониженном давлении (-0,095 МПа) водяным насосом до тех пор, пока не перестал вытекать дистиллят; насос заменили на масляный; масляную баню нагревали до 140°С; и после выпаривания получали 20,42 г золотисто-желтого продукта с выходом 87,6%, который непосредственно использовали в следующей реакции.
При получении сначала натриевой соли 2-метоксиэтанола, а затем получения соединений III-а и III-b на стадии 1 примера 4 выход может достигать 87,6%, что лучше, чем получено прямой реакцией на стадии 1 примера 3 (53,6%).
Стадия 2: Синтез промежуточного соединения IС
Продукт (20,4 г, 100 ммоль), полученный на вышеуказанной стадии 1, и 4-этокси-1,1,1,1-трифторбут-3-ен-2-он (16,8 г, 100 ммоль) добавляли к безводному этанолу (49,0 г); температуру контролировали ниже 10°С; и добавляли по каплям 20 масс. % этанольного раствора этоксида натрия (34 г, 100 ммоль). После завершения добавления по каплям температуру поддерживали на уровне 0°С-10°С в течение 2 ч; и контроль методом ВЭЖХ проводили до тех пор, пока сырье полностью не вступило в реакцию. Реакционную жидкость выливали в смешанный раствор 114,3 г раствора соляной кислоты (приготовленного из 12,2 г 30 масс. % соляной кислоты) и дихлорметана (81,7 г); полученную смесь перемешивали в течение 10 мин и оставляли для отстаивания и разделения жидкости; затем водную фазу экстрагировали дихлорметаном (40,8 г); и органические фазы объединяли и подвергали вакуумной дистилляции (55°С, ниже -0,095 МПа) для получения промежуточного соединения IС, которое представляло собой коричневое маслянистое вещество массой 36,5 г и которое непосредственно использовали для следующей реакции.
Стадия 3: Синтез этил-2-((2-метоксиэтокси) метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
Промежуточное соединение IС (36,5 г) стадии 2 добавляли к уксусной кислоте (130,5 г), добавляли при перемешивании ацетат аммония (9,4 г, 122 ммоль); температуру контролировали на уровне 50°С-60°С для перемешивания в ходе реакции в течение 2 ч; и контроль методом ВЭЖХ проводили до тех пор, пока сырье полностью не вступило в реакцию. В реакционную жидкость добавляли смешанный раствор воды (97,9 г) и дихлорметана (97,9 г); полученное вещество перемешивали в течение 10 мин и оставляли для отстаивания и отделения жидкости; затем водную фазу экстрагировали два раза дихлорметаном (65,2 г); и органические фазы объединяли и подвергали вакуумной дистилляции (60°С, ниже -0,095 МПа) для получения промежуточного соединения IIC, которое представляло собой коричневое маслянистое вещество массой 35,1 г и было непосредственно использовано для следующей реакции.
Стадия 4: Синтез 2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIIC)
Промежуточное соединение ПС (35,1 г, содержание чистого вещества 30,7 г) стадии 3 добавляли к этанолу (30,7 г); добавляли по каплям раствор гидроксида натрия (42,7 г, 28%, 300 ммоль); температуру поддерживали на уровне 50°С-60°С для перемешивания в ходе реакции в течение 1 ч; и контроль методом ГХ выполняли до тех пор, пока сырье полностью не вступило в реакцию. Температура была понижена до комнатной; в реакционную жидкость добавляли воду (92,2 г) и дихлорметан (61,5 г); полученное вещество перемешивали в течение 10 мин и оставляли для отстаивания и разделения жидкости; в водную фазу добавляли дихлорметан (92,2 г); полученную смесь подкисляли 30% соляной кислотой до тех пор, пока значение рН не становилось примерно равным 1,5; перемешивание проводили в течение 10 мин для отделения жидкости; затем водную фазу экстрагировали дихлорметаном (61,5 г); органические фазы объединяли и подвергали вакуумной дистилляции (60°С, ниже -0,095 МПа) с получением 26,5 г промежуточного соединения IIIC, представляющего собой коричневое маслянистое вещество. К полученному сырому продукту добавляли 13,3 г этилацетата и 16,6 г петролейного эфира; перемешивание проводили медленно, и понижали температуру до -5°С для кристаллизации; и проводили фильтрацию на вакуум-фильтре и сушку для получения светло-желтого твердого продукта массой 15,2 г. Выход стадий 2, 3 и 4 составляет 54,5%.
Общий выход этого примера составляет 47,8%. Выход на стадии 2 и стадии 3 примера 4 является высоким, что значительно лучше, чем общий выход в примере 3 (выход реакции получения ПС в примере 3 не слишком высок, и дальнейшее приготовление IIIC не проводили). Промежуточное соединение IIIC, полученное на стадии 4 примера 4, может быть использовано для синтеза бициклопирона.
Н и С ядерно-магнитные спектральные анализы промежуточного соединения IIIC показаны на фиг. 15 и 16.
ЖХ-МС: М-1=278
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): 10,464 (s, 1 Н), 8,395 (d, 1 Н, J=8,0 Гц), 7,716 (d, 1 Н, J=8,0 Гц), 5,074 (s, 2 Н), 3,776-3,795 (m, 2 Н), 3,611-3,629 (m, 2 Н), 3,377 (s, 3 Н);
13С ЯМР (CDCl3, 150 МГц), δ (ррm): 167,298, 157,784, 148,538 (q, JC-F=42,3 Гц), 139,641, 128,100, 119,921 (q, JC-F=327,3 Гц), 118,574, 71,932, 70,582, 69,383, 57,384.
Пример 5
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC) (однореакторный метод)
В четырехгорлую колбу добавляли 2-метоксиэтанол (19,1 г, 251 ммоль) и толуол (82,7 г); и добавляли при перемешивании металлический натрий (5,3 г, 230 ммоль); постепенно повышали температуру; и реакцию проводили при 80°С до прекращения образования частиц металлического натрия. Температуру снижали и контролировали на уровне ниже 40°С; добавляли по каплям этил-4-хлорацетоацетат (16,5 г, 101 ммоль); температуру поддерживали на уровне 45°С для проведения реакции в течение примерно 3 ч, а затем снижали. Была взята проба для определения методом ГХ, с получением степени конверсии 94,6%; и реакционную жидкость непосредственно использовали для следующей реакции.
Вышеуказанную реакционную жидкость охлаждали до температуры ниже 10°С; в систему добавляли смешанную жидкость 4-этокси-1,1,1-трифторбут-3-ен-2-она (16,8 г, 100 ммоль) и толуола (14,0 г); и после 2 часов реакции был выполнен контроль методом ГХ, показавший отсутствие остаточных сырьевых материалов. Разбавленную соляную кислоту добавляли при перемешивании для подкисления; полученное вещество было оставлен для отстаивания и разделения жидкости; и было обнаружено, что толуольная фаза имеет содержание 87,1%. Реакционную жидкость непосредственно использовали для следующей реакции.
К вышеуказанному толуольному раствору при перемешивании добавляли ацетат аммония (9,3 г, 121 ммоль); температуру повышали до 50°С для проведения реакции в течение 2 ч; и был проведен контроль ВЭЖХ в середине реакции, и реакция была завершена примерно через 3 часа. Полученное вещество промывали водой (50,0 г) для отделения жидкости; было взвешено 25,7 г толуольной фазы, и было обнаружено, что содержание составляет 87,2%. Общий выход промежуточного соединения IIC в три стадии этого примера составляет 72,9%.
Пример 6
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC) (однореакторный метод)
В четырехгорлую колбу добавляли 2-метоксиэтанол (19,1 г, 251 ммоль) и толуол (82,7 г); и добавляли при перемешивании металлический натрий (5,1 г, 222 ммоль); температуру постепенно повышали до 80°С; и реакцию проводили до прекращения образования частиц металлического натрия. Температуру снижали до уровня ниже 40°С; добавляли по каплям этил-4-хлорацетоацетат (16,5 г, 101 ммоль); температуру поддерживали на уровне 40°С для проведения реакции в течение примерно 6 часов; реакцию проводили при 65°С в течение 2 ч; и потом температуру снижали. Была взята проба для контроля методом ГХ, с получением степени конверсии 88,8%; и реакционную жидкость непосредственно использовали для следующей реакции.
Температуру системы охлаждали до уровня ниже 10°С; в реакционную жидкость по каплям добавляли смешанную жидкость из 4-этокси-1,1,1-трифторбут-3-ен-2-она (16,8 г, 100 ммоль) и толуола (14,0 г); и после 2 часов реакции был выполнен контроль методом ГХ, показавший отсутствие остаточных сырьевых материалов. Разбавленную соляную кислоту добавляли для подкисления при перемешивании; полученное вещество было оставлено для отстаивания и разделения жидкости; и было обнаружено, что толуольная фаза имеет содержание 75,3%. Реакционную жидкость непосредственно использовали для следующей реакции.
В вышеуказанный толуольный раствор вводили газообразный аммиак в течение 15 мин при перемешивании; добавляли ацетат аммония (15,0 г); температуру повышали до 50°С для осуществления реакции в течение 2 ч; был проведен контроль методом ВЭЖХ в середине реакции, и реакция была завершена примерно через 3 ч. Полученное вещество промывали водой (50,0 г) для разделения жидкости; было взвешено 20,6 г толуольной фазы, и было обнаружено, что содержание в ней продукта составляет 86,2%. Выход промежуточного соединения ПС за три стадии этого примера составляет 57,2%, и выход реакции в этом примере немного низок. Авторы данной заявки предполагают, что причиной этого может быть использование газообразного аммиака при приготовлении IIC.
Пример 7
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC) (однореакторный метод)
В четырехгорлую колбу добавляли 2-метоксиэтанол (114,4 г, 1,5 моль), гидроксид натрия (48,3 г, 1,21 моль) и толуол (250,3 г); эту четырехгорлую колбу погружали в масляную баню при высокой температуре 140°С-150°С для кипячения с обратным холодильником; воду, образующуюся в результате реакции, удаляли с помощью водоотделителя до тех пор, пока в водоотделителе не оставалось видимых капель воды; четырехгорлую колбу охлаждали на водяной бане с ледяной водой примерно до 30°С; и добавляли по каплям этил-4-хлорацетоацетат (90,61 г, 0,55 моль); реакцию проводили при 40°С в течение ночи; была взята проба для контроля методом ГХ, с получением степени конверсии 95,9%; и реакционную жидкость непосредственно использовали для следующей реакции.
Часть реакционной жидкости отобрали, и обеспечивали содержание натриевой соли 2-метоксиэтанола (0,1 моль); температуру регулировали на уровне ниже 10°С; по каплям добавляли толуольный раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (содержащий 16,8 г 4-этокси-1,1,1-трифторбут-3-ен-2-она и 16,89 г толуола); через 2 часа реакции проводили контроль методом ГХ до отсутствия остаточных сырьевых материалов; и добавляли разбавленную соляную кислоту для подкисления при перемешивании; полученное вещество было оставлено для отстаивания и разделения жидкости; обнаружено содержание толуольной фазы 82,7%; и реакционную жидкость непосредственно использовали для следующей реакции.
Ацетат аммония (9,25 г, 0,12 моль, разделенный на три равные части, с добавлением одной порции каждые 20 мин) добавляли к вышеуказанному толуольному раствору порциями при перемешивании; температуру повышали до 50°С для проведения реакции в течение 2 ч; выполняли контроль методом ВЭЖХ в середине реакции; после завершения реакции добавляли воду (80 г) для промывки и разделения жидкости; было взвешено 17,4 г толуольной фазы, и было обнаружено, что содержание в ней продукта составляет 75,7%. Выход промежуточного соединения ПС в три стадии этого примера составляет 42,9%, и выход реакции этого примера немного низок. Авторы данной заявки предполагают, что на него могут влиять остаточные следовые количества воды при получении соли 2-метоксиэтанола.
Пример 8
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
В четырехгорлую круглодонную колбу объемом 250 мл добавляли смесь (приготовленную в соответствии со стадией 1 примера 4, 20,42 г, 100 ммоль) этанола (50 г), этил-4-(2-метоксиэтокси)-3-оксобутирата и этил-(2)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата. Четырехгорлую колбу помещали в холодную ловушку при температуре 0°С.Начинали перемешивание; после снижения температуры реакционной жидкости до 5±3°С добавляли по каплям 20% этанольный раствор этоксида натрия (34,04 г, 100 ммоль) с помощью капельной воронки постоянного давления; и добавление по каплям было завершено примерно через 30 минут. После завершения добавления по каплям температуру поддерживали для протекания реакции в течение 0,5 ч. Затем по каплям добавляли раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (18,28 г, 109 ммоль) в толуоле (90 мл); и добавление по каплям было завершено примерно через 20 минут. После того, как капельное добавление было завершено, температуру поддерживали для протекания реакции в течение 1 ч. Деионизированную воду (100 г) добавляли в 30% раствор соляной кислоты (12,40 г) для перемешивания до однородности. Реакционную жидкость непосредственно выливали в кислую воду и экстрагировали дихлорметаном (80 г) для выделения органической фазы; водную фазу экстрагировали дихлорметаном (20 г); и органические фазы объединяли; дихлорметан был извлечен при пониженном давлении; и остаток подвергали вакуумной дистилляции при 55°С для получения промежуточного соединения IС (32,63 г).
В четырехгорлую круглодонную колбу объемом 250 мл, нагретую до 50°С на масляной бане, добавляли уксусную кислоту (130 г), ацетат аммония (9,37 г, 122 ммоль) и полученное промежуточное соединение IС (32,63 г); и температуру поддерживали для протекания реакции в течение 2 ч. Температура повышали до 65°С; проводили вакуумную дистилляцию при 2 мм рт. ст.до тех пор, пока не завершалась перегонка фракции; и в четырехгорлую колбу добавляли деионизированную воду (100 г) и дихлорметан (100 г) для экстракции с отделением органической фазы. Водную фазу экстрагировали дихлорметаном (30 г × 2); органические фазы объединяли; растворитель удаляли при пониженном давлении; и остаток нагревали до 60°С и подвергали вакуумной дистилляции при давлении, меньшем или равном 0,095 МПа, с получением желто-черного маслянистого вещества массой 27,2 г с содержанием 89,2%. Выход промежуточного соединения IIC в две стадии этого примера составляет 79,0%.
Пример 9
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
В четырехгорлую круглодонную колбу объемом 250 мл добавляли смесь (приготовленную в соответствии со стадией 1 примера 4, 10,21 г, 50 ммоль) толуола (51,05 г), этил-4-(2-метоксиэтокси)-3-оксобутирата и этил-(2)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата с перемешиванием при постоянной температуре 25°С; и добавляли твердый этоксид натрия (4,08 г, 60 ммоль) для проведения реакции в течение 0,5 ч. Реакционную жидкость охлаждали до 5°С±3°С; по каплям добавляли смешанный раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (9,14 г, 54 ммоль) и толуола (10,21 г); и добавление по каплям было завершено примерно через 20 минут. После того, как добавление по каплям было завершено, температуру поддерживали для проведения реакции в течение 1 ч. Реакцию контролировали с помощью пластины для ТСХ, и реакцию прекратили после того, как сырьевые материалы полностью вступили в реакцию. К 30% раствору соляной кислоты (7,9 г) добавляли деионизированную воду (51,05 г), и кислую воду непосредственно вливали в реакционную жидкость с перемешиванием в течение 10 мин. Проводили жидкостную сепарацию с получением толуольного раствора IС.
Толуольный раствор промежуточного соединения IIC переносили в четырехгорлую круглодонную колбу объемом 250 мл и добавляли уксусную кислоту (3,00 г) и ацетат аммония (4,68 г, 61 ммоль). Эту четырехгорлую колбу помещали в емкость с масляной баней и нагревали до 50°С; и температуру поддерживали для проведения реакции в течение 2 ч. Реакцию контролировали с помощью пластины с лунками для ТСХ; и после того, как сырье полностью вступило в реакцию, реакцию прекратили. Реакционную жидкость переносили в разделительную воронку для отстаивания и разделения жидкости. Была отобрана верхняя органическая фаза и подвергнута вакуумной дистилляции с помощью водоструйного насоса до тех пор, пока не прекратилась отгонка фракции; и затем перегонку прекратили. Получено 14,2 г продукта замыкания кольца с содержанием 88,6%. Выход промежуточного соединения ПС в данном примере составляет 81,9%.
Пример 10
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
В четырехгорлую круглодонную колбу объемом 250 мл добавляли смесь (приготовленную в соответствии со стадией 1 примера 4, 10,21 г, 50 ммоль) толуола (51,05 г), этил-4-(2-метоксиэтокси)-3-оксобутирата и этил-(2)-3-гидрокси-4-(2-метоксиэтокси)-3-оксобутирата с перемешиванием при постоянной температуре 25°С; и добавляли карбонат натрия (6,36 г, 60 ммоль) для проведения реакции в течение 0,5 ч. Реакционную жидкость охлаждали до 5°С±3°С; добавляли по каплям смешанный раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (9,14 г, 54 ммоль) и толуола (10,21 г); и добавление по каплям было завершено примерно через 20 минут. После того, как добавление по каплям было завершено, температуру поддерживали для проведения реакции в течение 1 ч. Реакцию контролировали с помощью пластины для ТСХ; и после того, как сырье полностью вступило в реакцию, реакцию прекратили. Деионизированную воду (51,05 г) добавляли к 30% раствору соляной кислоты (7,9 г), и эту кислую воду непосредственно вливали в реакционную жидкость с перемешиванием в течение 10 мин. Проводили жидкостную сепарацию с получением толуольного раствора IС.
Толуольный раствор IС переносили в четырехгорлую круглодонную колбу объемом 250 мл и добавляли уксусную кислоту (3,00 г) и ацетат аммония (4,68 г, 61 ммоль). Эту четырехгорлую колбу помещали в емкость с масляной баней и нагревали до 50°С, и температуру поддерживали для проведения реакции в течение 2 ч. Реакцию контролировали с помощью пластины для ТСХ, и после того, как сырье полностью вступило в реакцию, реакцию прекратили. Реакционную жидкость переносили в разделительную воронку для отстаивания и разделения жидкости. Была отобрана верхняя органическая фаза и подвергнута вакуумной дистилляции с помощью водоструйного насоса до тех пор, пока не прекратилась отгонка фракции; и затем перегонку прекратили. Получено 13,9 г продукта замыкания кольца с содержанием 90,7%. Выход промежуточного соединения IIC в данном примере составляет 82,1%.
Пример 11 (однореакторный метод)
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
Этоксид натрия (8,46 г, 124,3 ммоль) добавляли к 2-метоксиэтанолу (28,4 г, 373,2 ммоль) для перемешивания и нагревания; полученное вещество нагревали на масляной бане до 130°С и подвергали вакуумной дистилляции с получением 12 г фракции; температуру опустили ниже 100°С и добавляли толуол (51,1 г) при перемешивании и диспергировании с получением толуольного раствора натриевой соли 2-метоксиэтанола.
Температуру контролировали на уровне примерно при 30°С, и к толуольному раствору натриевой соли 2-метоксиэтанола добавляли по каплям этил-4-хлорацетоацетат (8,89 г, 54 ммоль); после того, как добавление было завершено, температуру поддерживали на уровне 40°С; перемешивание проводили для проведения реакции в течение 6 ч; и выполняли контроль с помощью ТСХ (петролейный эфир (ПЭ):этилацетат (ЭА) = 6:1) до тех пор, пока сырье полностью не вступило в реакцию.
Систему охлаждали для охлаждения реакционной жидкости до 5°С±3°С; добавляли по каплям смешанный раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (9,14 г, 54 ммоль) и толуола (10,21 г); и добавление по каплям было завершено примерно через 20 минут. После того, как добавление по каплям было завершено, температуру поддерживали для проведения реакции в течение 1 ч. Реакцию контролировали с помощью пластины для ТСХ; после того, как сырье полностью вступило в реакцию, добавляли уксусную кислоту (4,20 г) и ацетат аммония (4,68 г, 61 ммоль). Четырехгорлую колбу помещали в емкость масляной бани и нагревали до 50°С и поддерживали температуру для проведения реакции в течение 2 ч. Реакцию контролировали с помощью пластины для ТСХ; и после того, как сырье полностью вступило в реакцию, реакцию прекратили. Реакционную жидкость переносили в разделительную воронку для отстаивания и разделения жидкости. Была отобрана верхняя органическая фаза и подвергнута вакуумной дистилляции с помощью водоструйного насоса до тех пор, пока не прекратилась отгонка фракции; и затем перегонку прекратили. Получено 15,1 г продукта замыкания кольца с содержанием 88,7%. Выход промежуточного соединения IIC в этом примере составляет 80,7%.
Пример 12 (однореакторный метод)
Синтез этил-2-((2-метоксиэтокси)метил)-6-(трифторметил)никотината (промежуточное соединение IIC)
Метоксид натрия (6,71 г, 124,3 ммоль) добавляли к 2-метоксиэтанолу (28,4 г, 373,2 ммоль) для перемешивания и нагревания; полученный продукт нагревали на масляной бане до 130°С и подвергали вакуумной дистилляции с получением 6,6 г фракции; температуру опустили ниже 100°С; и добавляли толуол (51,1 г) при перемешивании и диспергировании с получением толуольного раствора натриевой соли 2-метоксиэтанола.
Температуру контролировали примерно при 30°С, и этил-4-хлорацетоацетат (8,89 г, 54 ммоль) добавляли по каплям к толуольному раствору натриевой соли 2-метоксиэтанола; после того, как добавление было завершено, температуру поддерживали на уровне 40°С; перемешивание проводили для реакции в течение 6 ч; и выполняли контроль методом ТСХ (ПЭ: ЭА=6: 1) до тех пор, пока сырье полностью не вступило в реакцию.
Систему охлаждали для охлаждения реакционной жидкости до 5°С±3°С; добавляли по каплям смешанный раствор 4-этокси-1,1,1-трифторбут-3-ен-2-она (9,14 г, 54 ммоль) и толуола (10,21 г); и добавление по каплям было завершено примерно через 20 минут. После того, как добавление по каплям было завершено, температуру поддерживали для проведения реакции в течение 1 ч. Реакцию контролировали с помощью пластины для ТСХ; и после того, как сырье полностью вступило в реакцию, добавляли уксусную кислоту (4,20 г) и ацетат аммония (4,68 г, 61 ммоль). Четырехгорлую колбу помещали в емкость масляной бани и нагревали до 50°С; и поддерживали температуру для проведения реакции в течение 2 ч. Реакцию контролировали с помощью пластины для ТСХ; и после того, как сырье полностью вступило в реакцию, реакцию прекратили. Реакционную жидкость переносили в разделительную воронку для отстаивания и разделения жидкости. Была отобрана верхняя органическая фаза и подвергнута вакуумной дистилляции с помощью водоструйного насоса до тех пор, пока не прекратилась отгонка фракции; а затем перегонку прекратили. Получено 14,8 г продукта замыкания кольца с содержанием 92,5%. Выход промежуточного соединения IIC в этом примере составляет 82,5%.
Пример 13
Синтез этил-4-этокси-3-оксобутирата и этил-(Z)-4-этокси-3-гидроксибут-2-еноата (примесное соединение Е, пара кето- и енольных изомеров)
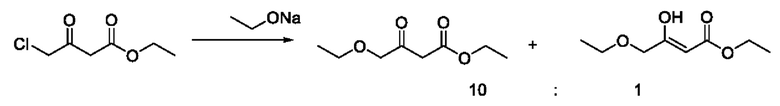
В четырехгорлую колбу объемом 250 мл добавляли этанол (33,6 г) и этил-4-хлорацетоацетат (16,8 г, 100 ммоль), и при перемешивании добавляли 20% этанольный раствор этоксида натрия (68,01 г, 200 ммоль). Температуру повышали до 50°С и поддерживали в течение 2 ч; проводили детектирование методом ГХ до тех пор, пока содержание сырья не стало меньше или равно 1%; а затем реакцию остановили. Реакционную жидкость вливали в приготовленный раствор соляной кислоты (150 мл) со значением рН 2-3. Полученное вещество было обработано дихлорметаном (150 мл); водную фазу экстрагировали два раза дихлорметаном (100 мл × 2); и органические фазы объединяли. Органическую фазу один раз промывали насыщенным водным раствором соли. Органическую фазу отделяли, сушили безводным сульфатом магния и концентрировали с получением сырого продукта, который представлял собой светло-желтую жидкость. Неочищенный продукт очищали с помощью колоночной хроматографии для получения аналитического образца, содержание в котором было больше или равно 97%, согласно показаниям ГХ. Структура была подтверждена стандартными методами ГХ-МС и ЯМР. Метод ЯМР показывает, что отношение кето-структуры к енольной структуре составляет 10:1. Приготовление примесного соединения Е предназначено для характеристики примесного соединения Е в маршруте реакции согласно данной заявке.
Спектральные анализы Н и С ядерно-магнитного резонанса примесного соединения Е показаны на фиг. 17 и 18. ГХ-МС: М=174
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): (кетон) 4,20 (q, 2 Н, J=5,0 Гц), 4,11 (s, 2 Н), 3,57 (q, 2 Н, J=5,0 Гц), 3,52 (s, 2 Н), 3,63 (t, 2 Н, J=5,0 Гц), 1,28 (t, 3 Н, J=5,0 Гц), 1,24 (t, 3 Н, J=5,0 Гц)
13С ЯМР (CDCl2, 150 МГц), δ (ррm): (кетон) 202,19, 167,06, 75,52, 67,28, 61,34, 45,95, 14,95, 14,07; (Енол) 174,35, 172,67, 88,59, 69,29, 66,95, 60,16, 15,06, 14,21.
Пример 14
Синтез этил-2,5-дигидроксициклогексан-1,4-диен-1,4-дикарбоксилата (примесное соединение С)
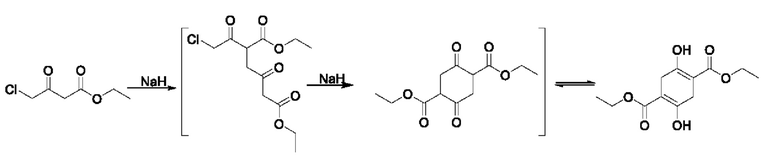
В четырехгорлую колбу объемом 250 мл добавляли тетрагидрофуран (40,0 г) и поддерживали ниже температуру 15°С; добавляли гидрид натрия (2,4 г, 60%, 60 ммоль) порциями при перемешивании в течение 10 мин; затем полученный продукт охлаждали до 5°С - 10°С; медленно добавляли по каплям раствор этил-4-хлорацетоацетата (10 г, растворенный в 30 мл тетрагидрофурана, 61 ммоль); четырехгорлую колбу ставили на ледяную водяную баню для охлаждения с последующим постоянным образованием пузырьков. После того, как добавление по каплям было завершено, реакционная жидкость была светло-желтовато-коричневой. Температуру постепенно повышали до 25°С-30°С, и реакционная жидкость постепенно становилась коричнево-желтой и прозрачной. Растворитель удаляли вакуумным ротационным испарением; добавляли воду (150 мл); дихлорметан (150 мл) добавляли при перемешивании; и значение рН регулировали до 2-3. Проводили экстракцию и сепарацию жидкости; водную фазу экстрагировали два раза дихлорметаном (100 мл × 2); и органические фазы объединяли. Органическую фазу один раз промывали насыщенным водным раствором соли. Органическую фазу отделяли, сушили безводным сульфатом магния и концентрировали для получения сырого продукта. Сырой продукт подвергали колоночной хроматографии с получением 2,72 г продукта с выходом 35,4%. Продукт очищали путем перекристаллизации и очистки с получением аналитического образца, который представлял собой светло-желтые кристаллы.
Спектральные анализы Н и С ядерно-магнитного резонанса примесного соединения С показаны на фиг. 19 и 20.
ЖХ-МС: М-1=255, М+1=257
1Н ЯМР (CDCl3, 500 МГц), δ (ррm): 12,13 (s, 2 Н), 4,18 (t, 4 Н, J=5,0 Гц), 3,11 (s, 4 Н), 1,25 (t, 6 Н, J=5,0 Гц)
13С ЯМР (CDCl3, 150 МГц), δ (ррm): 170,29, 167,43, 92,23, 59,71, 27,51, 13,21.
В заключение следует отметить, что приведенные выше примеры предназначены только для иллюстрации технических решений данной заявки, но не для ограничения технических решений данной заявки. В то время как данная заявка подробно описана в связи с представленными выше примерами, специалист в данной области техники должен понимать, что технические решения, приведенные в предыдущих примерах, могут быть изменены, или часть технических характеристик в них заменена эквивалентами; однако эти модификации или замены не приводят к отклонению сущности соответствующих технических решений от замысла и объема охраны технических решений различных примеров данной заявки.
Промышленная применимость
В данной заявке описаны соединение, способ его получения и применение соединения при получении промежуточного соединения для бициклопирона, где промежуточное соединение II для бициклопирона может быть получено с высоким выходом через промежуточное соединение I (имеющее структурную формулу, показанную в формуле Ia и/или формуле Ib), или его фармацевтически приемлемую соль, его сольват и таутомер Ib (соединение, показанное в формуле Iс). В данной заявке способ получения промежуточного соединения I под действием основания и последующего проведения реакции замыкания кольца с образованием промежуточного соединения (II) для бициклопирона может уменьшить побочные реакции и может дополнительно увеличить выход.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ АРИЛКОНДЕНСИРОВАННЫХ АЗАПОЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2282619C9 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В ПОЛУЧЕНИИ C5aR АНТАГОНИСТОВ | 2015 |
|
RU2712233C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-ЭНАНТИОМЕРОВ | 1994 |
|
RU2114103C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛОПИРИМИДИНОНОВОГО ПРОИЗВОДНОГО | 2021 |
|
RU2836183C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ГАЛОГЕН-4,5-ДИГИДРО-1Н-ПИРАЗОЛОВ | 2003 |
|
RU2326877C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ 7-ЗАМЕЩЕННОГО ПИРРОЛОТРИАЗИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2745548C1 |
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
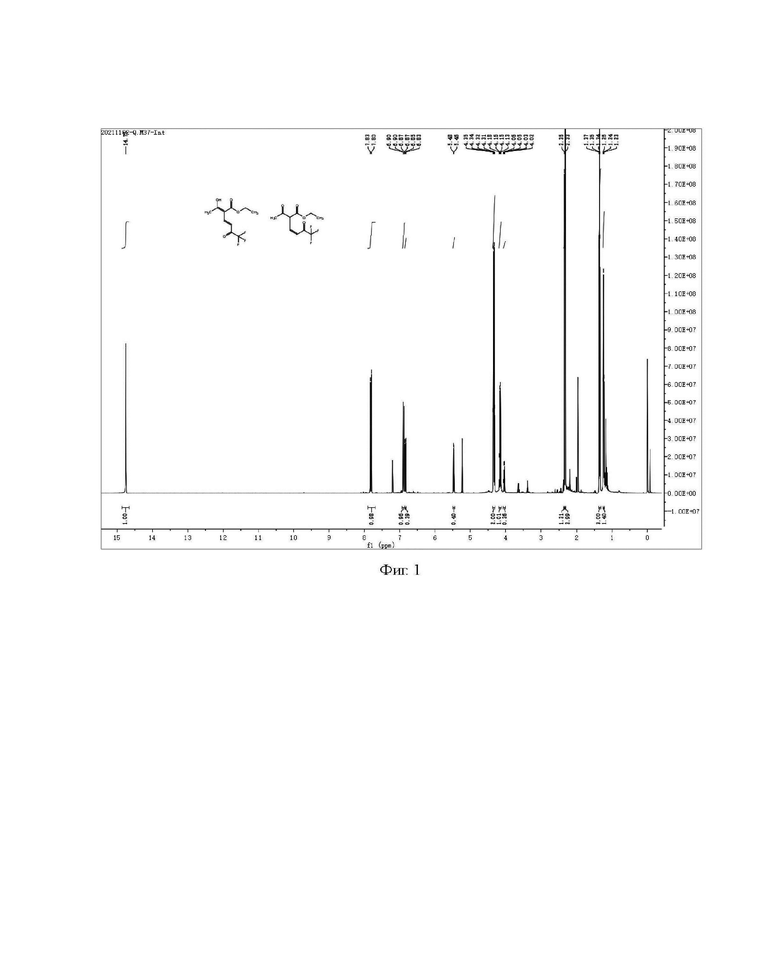
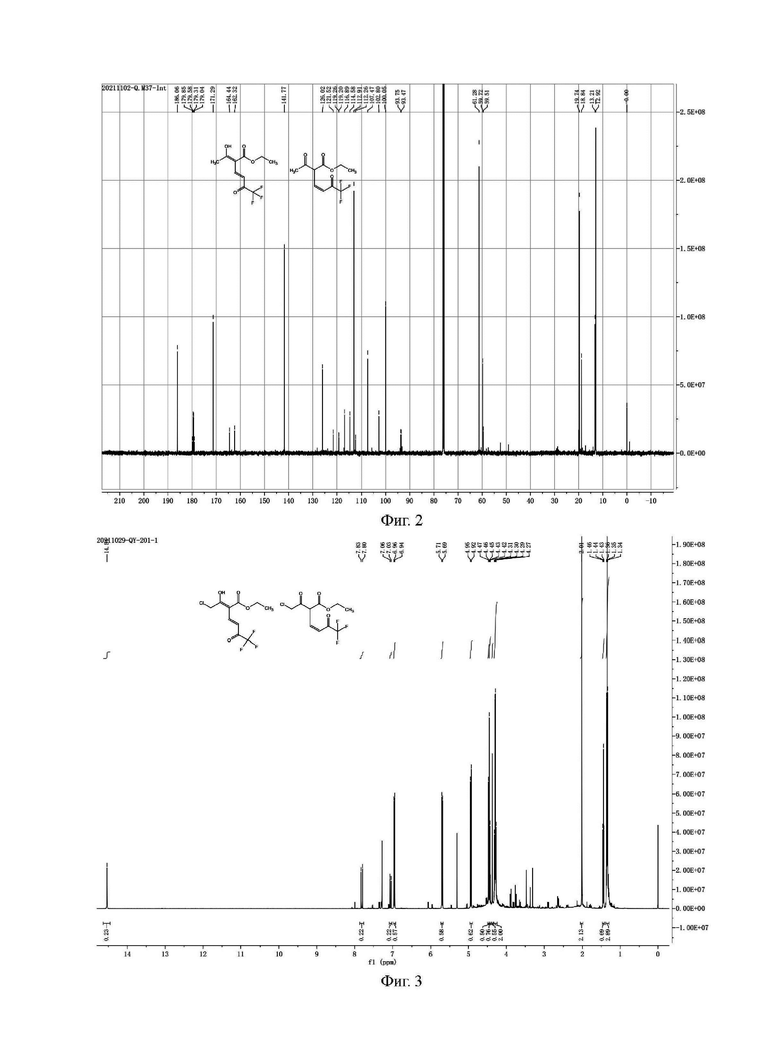
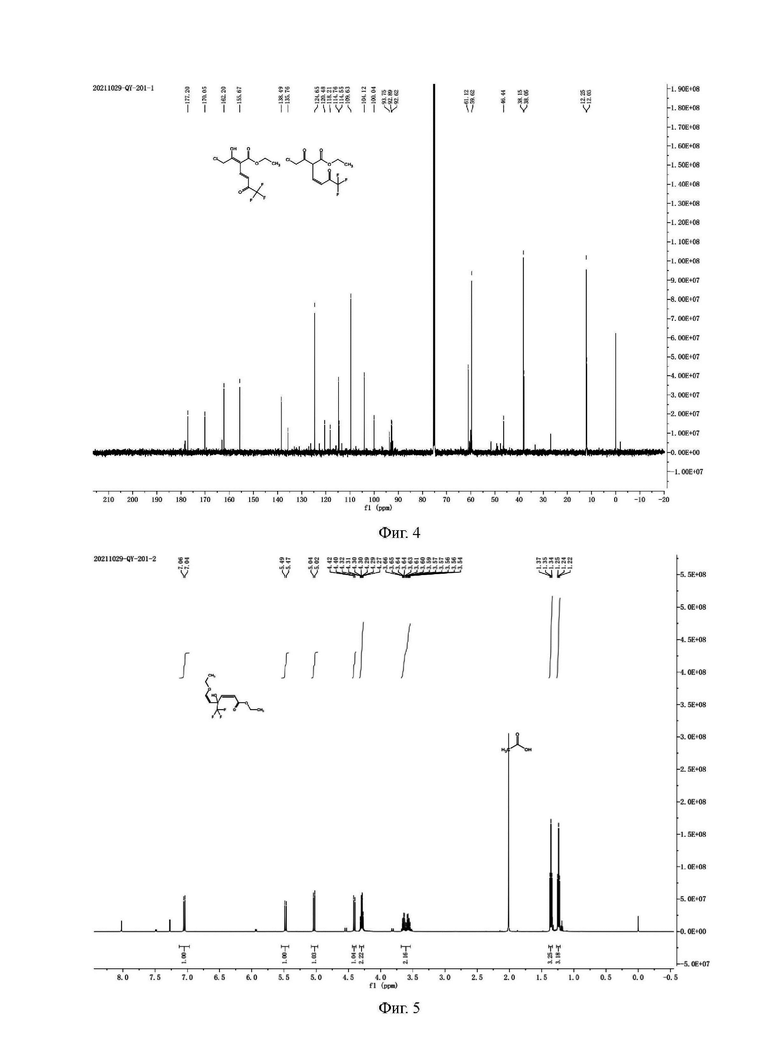
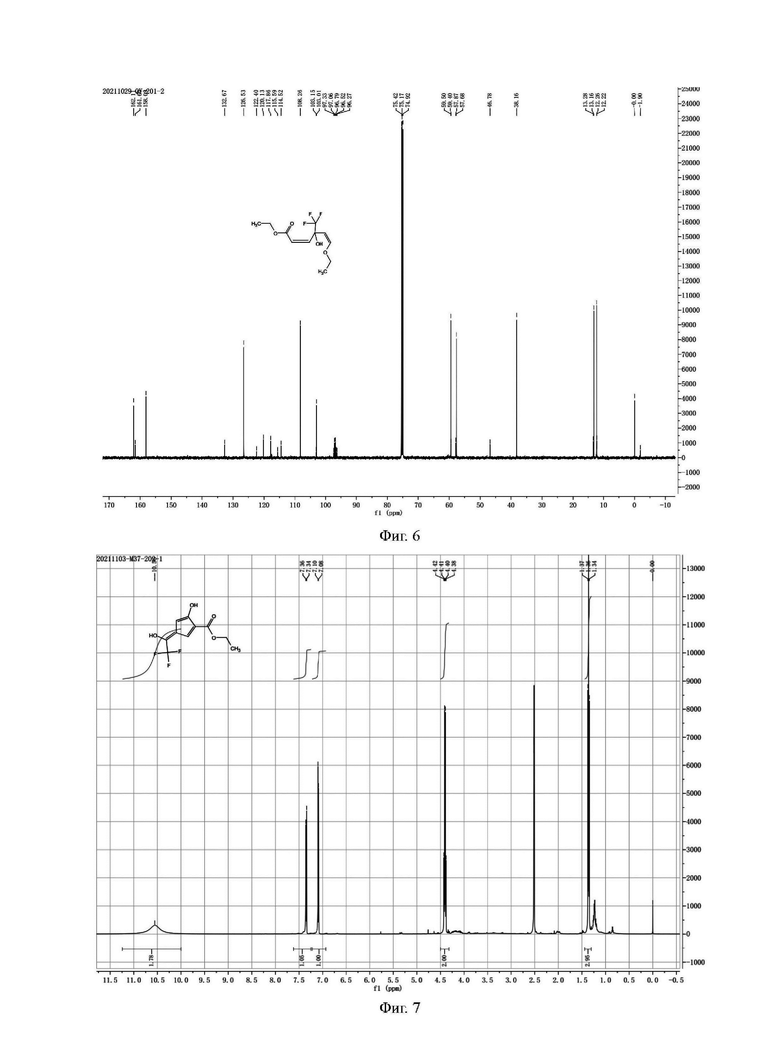
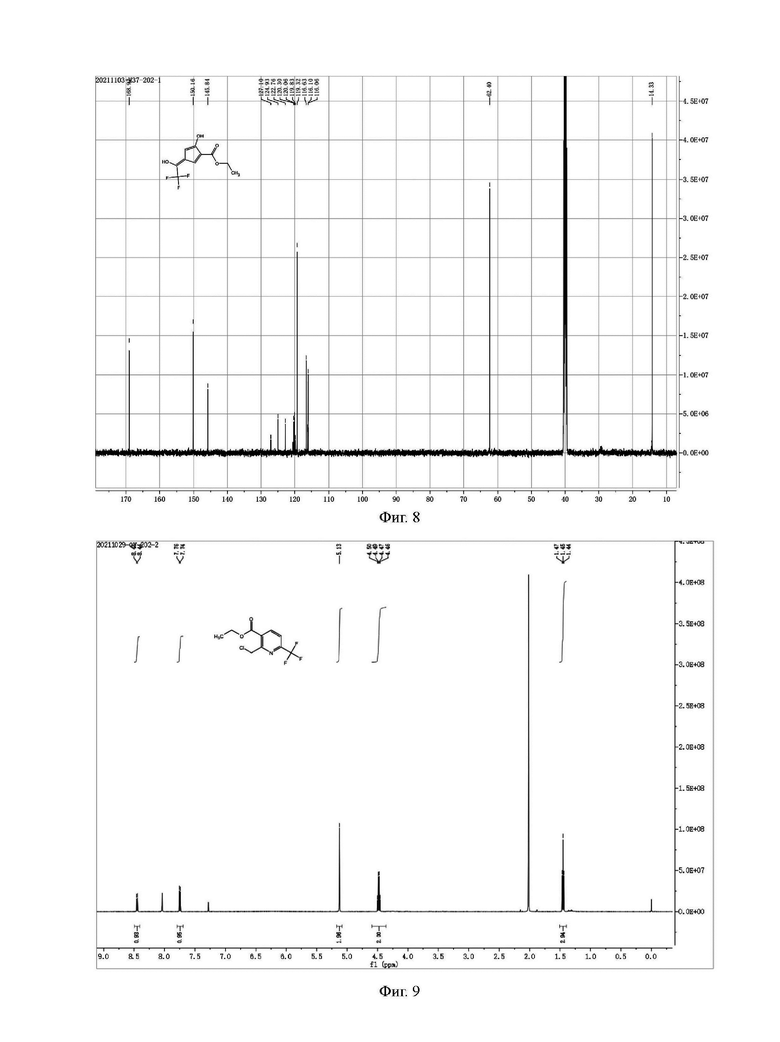
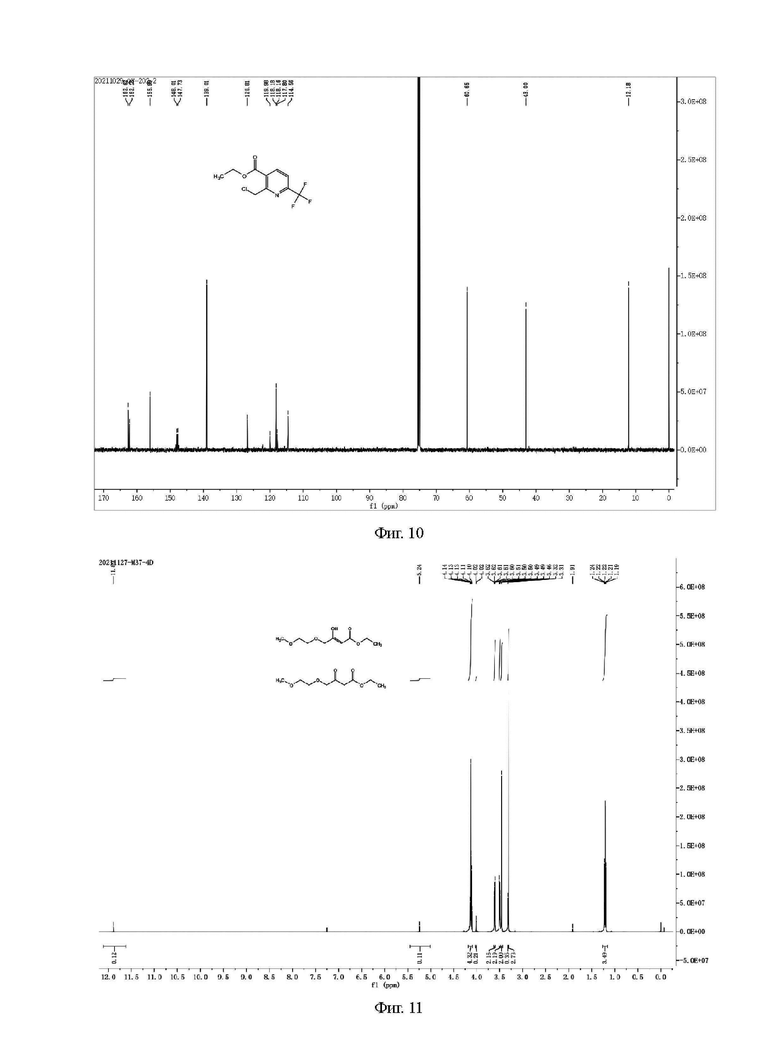
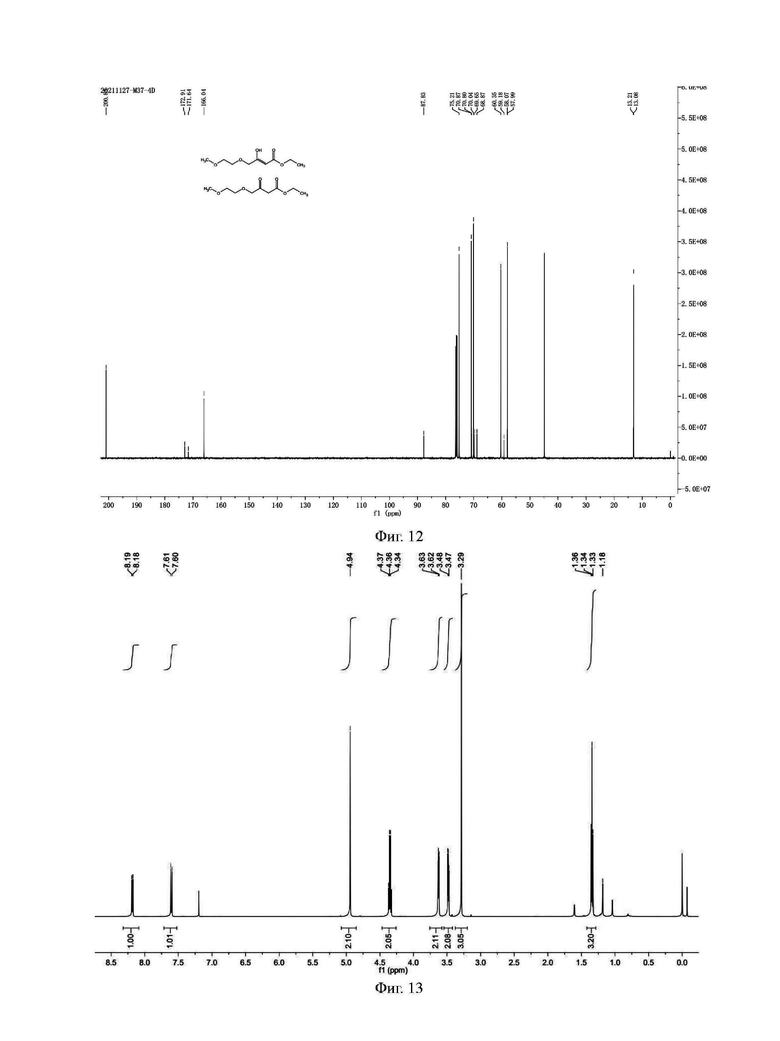
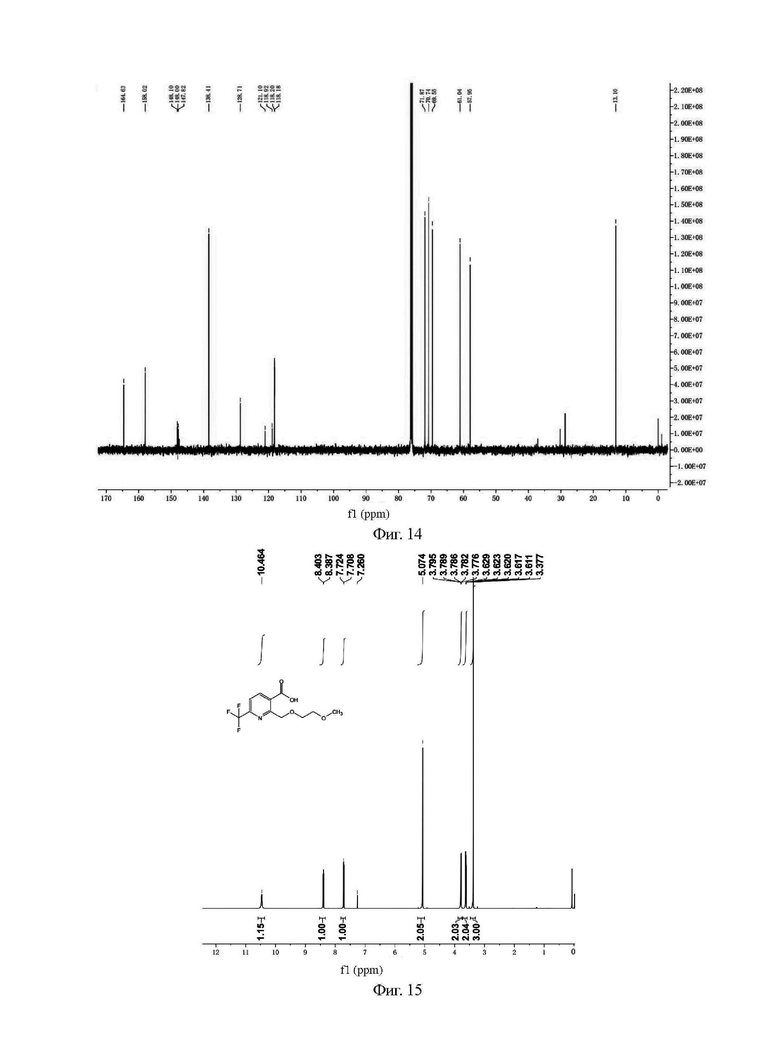
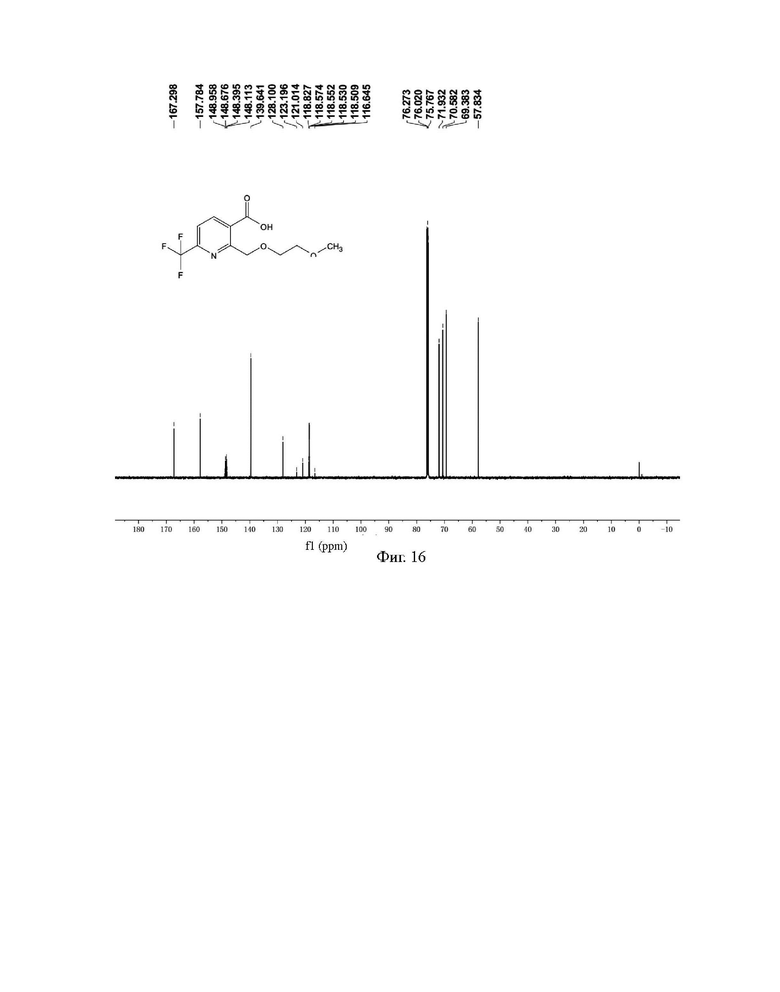
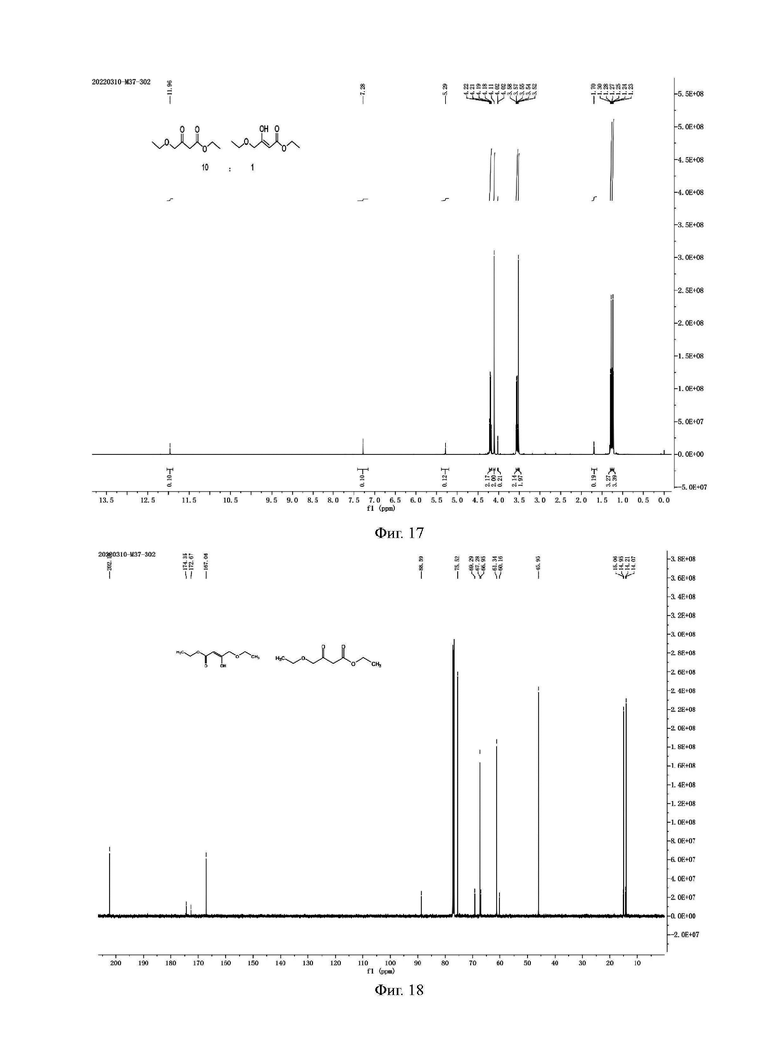
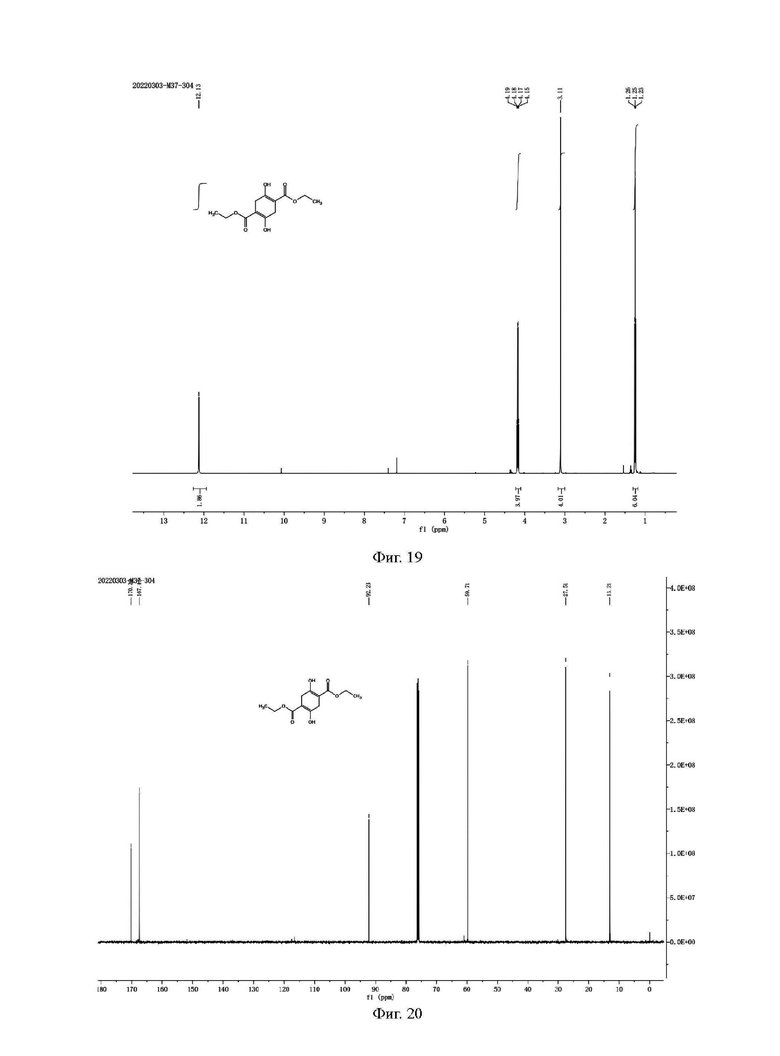
Изобретение относится к соединению, имеющему структуру, показанную в формуле Ib-3, или таутомеру, показанному в формуле Ic-3, которые могут найти применение при получении промежуточного соединения в синтезе бициклопирона. Изобретение относится также к способу получения соединения, имеющего структуру, показанную в формуле Ib-3, или таутомера, показанного в формуле Ic-3, композиции, содержащей указанные соединения и/или реакционный материал, приготовленный указанным способом, и их применению при получении бициклопирона. Использование предлагаемых соединений позволяет повысить выход промежуточного соединения в синтезе бициклопирона и снизить количество примесных соединений. 4 н. и 1 з.п. ф-лы, 20 ил., 14 пр.
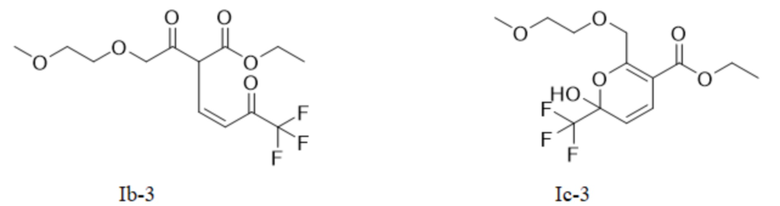
1. Соединение, имеющее структуру, показанную в формуле Ib-3, или таутомер, показанный в формуле Ic-3
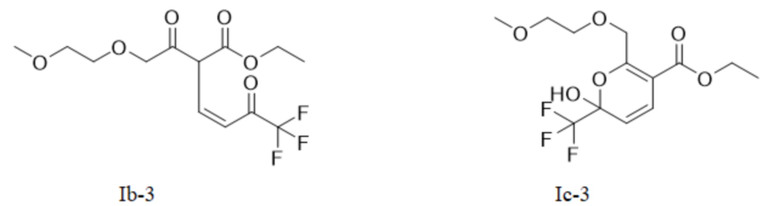
2. Способ получения соединения, соответствующего п. 1, включающий следующую стадию:
в присутствии основания введение соединения, показанного в формуле III-a, и/или его енольного таутомера III-b в реакцию замещения с соединением, показанным в формуле IV, для получения соединения, показанного в формуле Ib-3 и/или Ic-3
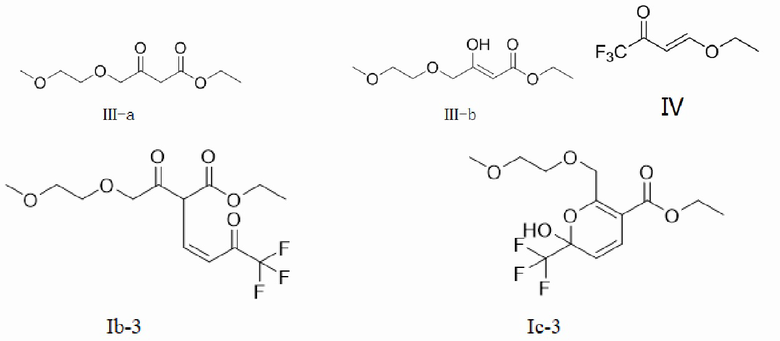
3. Способ получения по п. 2, в котором в реакции замещения основание представляет собой одно или более, выбранных из группы, состоящей из органического основания, неорганического основания, гидрида натрия или металлического натрия, причем органическое основание содержит одно или более из алкоксида натрия и алкоксида калия; органическое основание включает одно или более из метоксида натрия, этоксида натрия, трет-бутоксида калия, метоксида калия, этоксида калия, гексаметилдисилазана натрия и гексаметилдисилазана лития; неорганическое основание включает одно или более из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия, фосфата натрия, фосфата калия и амида натрия; возможно, основание представляет собой одно или более, выбранных из группы, состоящей из метоксида натрия, этоксида натрия, гидроксида натрия и карбоната натрия; и/или
реакцию замещения проводят в органическом растворителе и органический растворитель включает один или более из органического спирта, толуола, тетрагидрофурана, диметилсульфоксида, N,N-диметилформамида и 1,4-диоксана; возможно, органический растворитель включает один или более из метанола, этанола и толуола; и/или
температура реакции замещения составляет от -15°C до 30°C; возможно, температура реакции замещения составляет 0-25°C или 0-10°C; и/или
молярное соотношение соединения, показанного в формуле IV, соединения, показанного в формуле III-a, и/или его енольного таутомера и основания составляет 1:0,8-1,5:0,05-1,5, возможно 1:0,8-1,2:0,5-1,3, возможно 1:0,9-1,1:1-1,3, или 1:1:1-1,3, или 1:1:1-1,2.
4. Композиция для получения бициклопирона, содержащая соединение по п. 1 и/или реакционный материал, приготовленный способом по п. 2 или 3, причем композиция дополнительно содержит по меньшей мере одно из примесного соединения C, соединения D и соединения E:
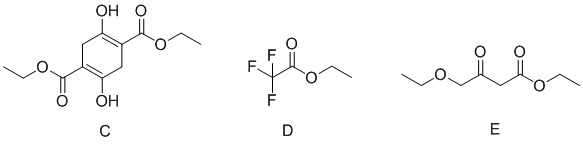
5. Применение соединения по п. 1, или продукта, полученного способом по п. 2 или 3, или композиции по п. 4 при получении бициклопирона.
| N | |||
| ZANATTA ET AL., Reactions of 1,1,1-Trifluoro[chloro]-4-ethoxybut-3-en-2-ones with 1,3-Dicarbonyl Compounds: Synthesis of 5-Acetyl[carboxyethyl]-1,1,1-trifluoro[chloro]hept-3-ene-2,6-diones and their Cyclic Derivatives Phenol, Pyridines, and Azetone, SYNTHESIS, 1999, No | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Приспособление для сбрасывания тресты с мяльной машины | 1924 |
|
SU765A1 |
| Воздухозаборник гондолы с акустической панелью | 2020 |
|
RU2803661C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНЫ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 2001 |
|
RU2326866C2 |
| WO | |||

