Область техники
[1] Настоящее изобретение относится к способу получения триазолопиримидинонового производного, проявляющего активность в отношении ингибирования танкиразы.
Уровень техники
[2] Танкираза принадлежит к семейству белков поли(АДФ-рибоза)-полимераз (PARP), которое состоит из 17 членов, имеющих общий каталитический домен PARP. Недавно сообщалось, что на уровни внутриклеточного аксина влияют члены семейства ферментов PARP танкираза-1 и танкираза-2 (также известные как PARP5a и PARP5b, соответственно) (Huang et al., 2009, Nature, 461(7264): 614-620).
[3] Известно, что ингибиторы танкиразы-1 и танкиразы-2 эффективны при различных раковых заболеваниях, таких как солидные виды рака, включая колоректальную карциному, рак толстой кишки, рак желудка, гепатоцеллюлярную карциному, рак молочной железы, медуллобластому, меланому, немелкоклеточный рак легкого, аденокарциному поджелудочной железы и рак предстательной железы. Кроме того, ингибиторы танкиразы-1 и танкиразы-2 обладают терапевтическим потенциалом в отношении заболеваний, отличных от рака, включая остеопороз, остеоартрит, поликистоз почек, легочный фиброз, диабет, шизофрению, сосудистые заболевания, сердечные заболевания, неонкогенные пролиферативные заболевания и нейродегенеративные заболевания, такие как болезнь Альцгеймера.
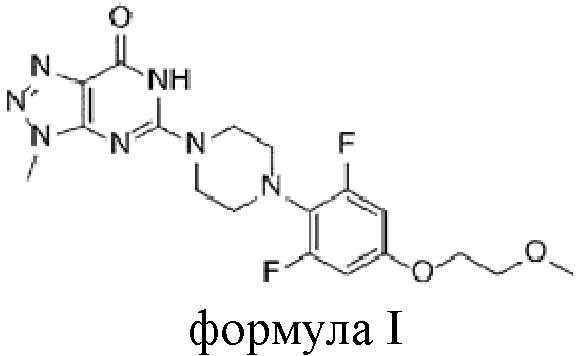
[4] Как отмечалось выше, существует постоянная потребность в новых терапевтических средствах для лечения рака и состояний избыточного роста, и была предпринята попытка разработки новых фармацевтических соединений, способных селективно ингибировать фермент танкиразу. В частности, в качестве селективного ингибитора танкиразы известно триазолопиримидиноновое производное, представленное следующей химической формулой I, которое разрабатывается для лечения колоректального рака у пациентов с мутантными генотипами гомолога вирусного онкогена саркомы крыс Кирстена (KRAS) или у пациентов, не отвечающих на эрбитукс:
[5] [Химическая формула I]
[6] 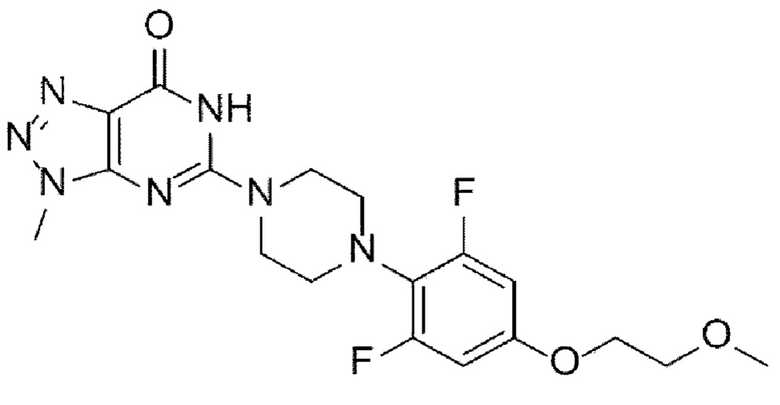
[7] В международной публикации WO 2016/006974 описан способ получения триазолопиримидинонового производного, включая соединение химической формулы I, представленной выше. Однако вышеуказанный способ получения включает реакционный процесс с использованием микроволн и очистки на колонке и не подходит для массового производства, что требует усовершенствования процесса.
[8] Таким образом, существует потребность в разработке нового способа, обеспечивающего возможность получения триазолопиримидинонового производного химической формулы I с высокой чистотой, при одновременном усовершенствовании вышеуказанного неэффективного способа получения.
Сущность изобретения
Техническая проблема
[9] Задача настоящего изобретения заключается в разработке способа получения триазолопиримидинонового производного с высокой чистотой и высоким выходом, со снижением производственных затрат и обеспечением эффективных технологических стадий, пригодных для массового производства. [10] Другая задача настоящего изобретения заключается в обеспечении нового промежуточного соединения, применяемого в вышеуказанном способе получения. Техническое решение
[11] В настоящем изобретении предложен способ получения триазолопиримидинонового производного, представленного следующей химической формулой I:
[12] [Химическая формула I]
[13] 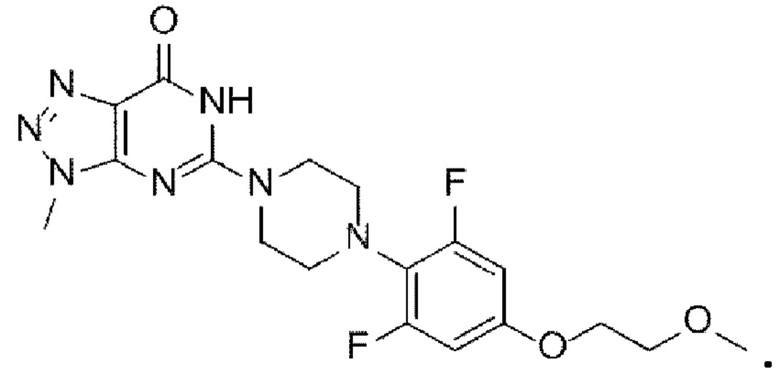
[14] В соответствии с одним из вариантов реализации настоящего изобретения, производное триазолопиримидинона, представленное выше химической формулой I, может быть получено посредством способов синтеза А или В, представленных ниже.
[15] Способ синтеза А
[16] Способ синтеза А способа получения согласно настоящему изобретению включает стадии (А-1) - (А-8), указанные ниже:
[17] (А-1) Стадию 1 получения соединения, представленного следующей химической формулой 2, посредством реакции защиты из соединения, представленного следующей химической формулой 1, или его соли;
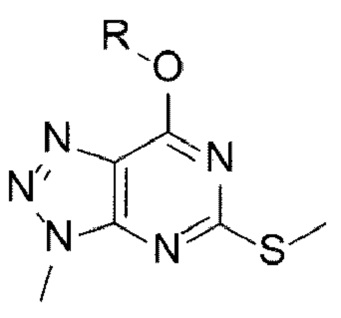
[18] (А-2) Стадию 2 получения соединения, представленного следующей химической формулой 3, посредством реакции окисления из соединения, представленного химической формулой 2;
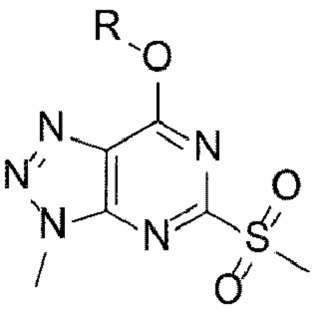
[19] (А-3) Стадию 3 получения соединения, представленного следующей химической формулой 5, посредством реакции аминирования из соединения, представленного следующей химической формулой 4;
[20] (А-4) Стадию 4 получения соединения, представленного следующей химической формулой 6, посредством реакции Дейкина, из соединения, представленного химической формулой 5;
[21] (А-5) Стадию 5 получения соединения, представленного следующей химической формулой 7, посредством реакции алкилирования из соединения, представленного химической формулой 6;
[22] (А-6) Стадию 6 получения соединения, представленного следующей химической формулой 8, или его соли посредством реакции снятия защиты с соединения, представленного химической формулой 7;
[23] (8) Стадию 7 получения соединения, представленного следующей химической формулой 1а, посредством реакции аминирования из соединения, представленного химической формулой 3, и соединения, представленного химической формулой 8, или его соли; и
[24] (А-8) Стадию 8 получения соединения, представленного следующей химической формулой I, посредством реакции снятия защиты с соединения, представленного химической формулой Ia;
[25] [Химическая формула 1]
[26] 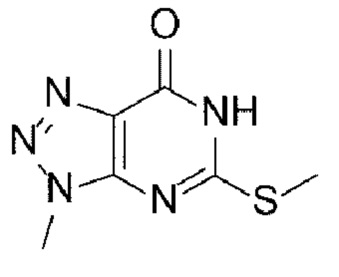
[27] [Химическая формула 2]
[28] 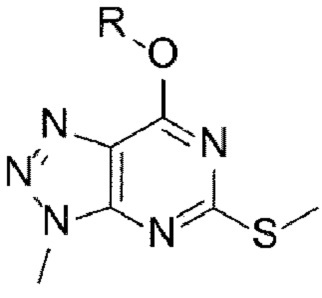
[29] [Химическая формула 3]
[30] 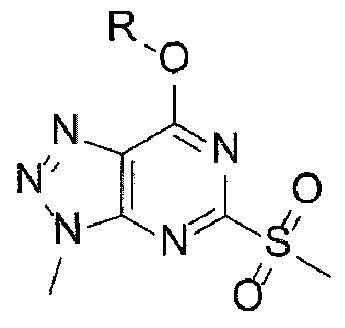
[31] [Химическая формула 4]
[32] 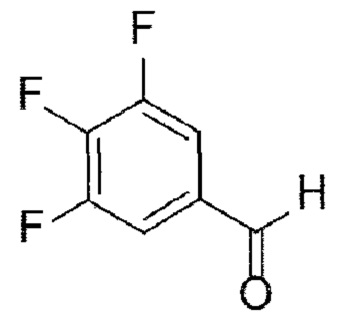
[33] [Химическая формула 5]
[34] 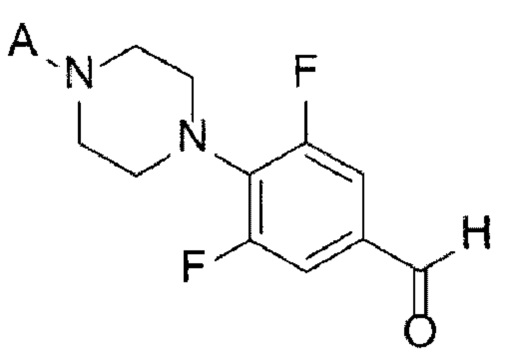
[35] [Химическая формула 6]
[36] 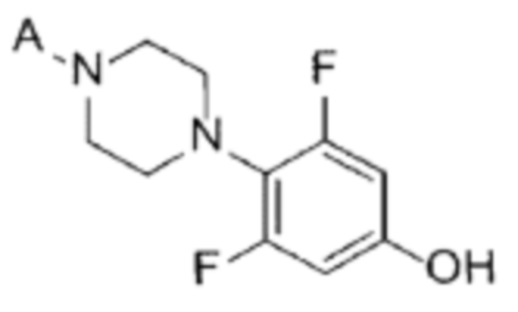
[37] [Химическая формула 7]
[38] 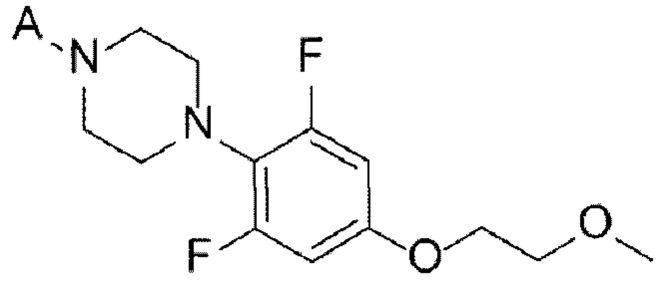
[39] [Химическая формула 8]
[40] 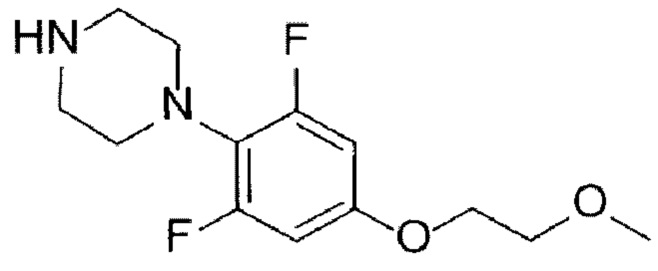
[41] [Химическая формула Ia]
[42] 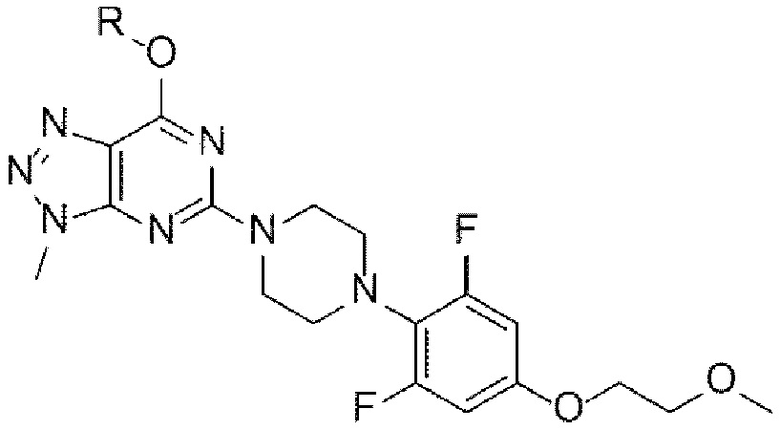
[43] [Химическая формула I]
[44] 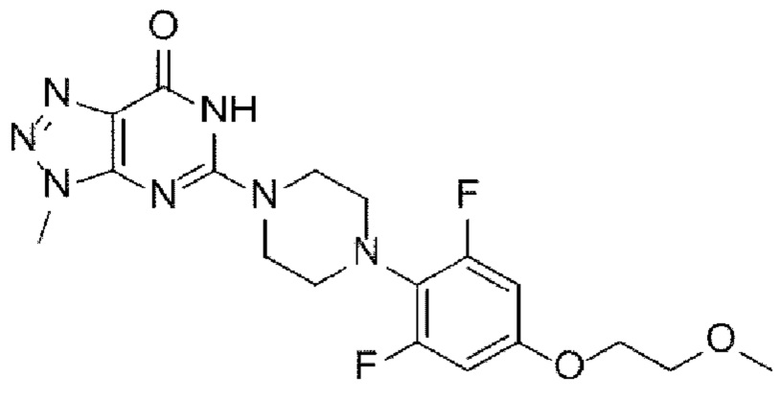
[45] в указанных химических формулах
[46] R представляет собой О-защитную группу; и
[47] А представляет собой N-защитную группу.
[48] Далее будут отдельно описаны стадии (А-1) - (А-8).
[49] Стадия (А-1)
[50] В настоящем изобретении стадия (А-1) представляет собой стадию получения соединения, представленного химической формулой 2, посредством реакции защиты с использованием триазолопиримидинонового производного, представленного химической формулой 1, или его соли в качестве исходного вещества (схема реакции 1):
[51] [Схема реакции 1]
[52] 
[53] в указанной химической формуле R представляет собой O-защитную группу.
[54] В соответствии с одним из вариантов реализации настоящего изобретения, указанная выше реакция приводит к введению защитной группы в триазолопиримидиноновое производное. Например, R может представлять собой C1-С6 алкил, ацетил, бензоил, бензил, п-метоксибензил, метоксиметилацеталь (MOM), тетрагидропиран (ТНР) или простой силиловый эфир. В соответствии с одним из вариантов реализации настоящего изобретения, в указанной выше реакции соединение, представленное химической формулой 1, можно приводить в контакт с алкилгалогенидом с получением соединения, представленного химической формулой 2. Например, R может представлять собой изопропил, и реакцию можно проводить посредством реакции алкилирования с 2-йодпропаном, но не ограничиваясь этим.
[55] В указанной реакции может быть использовано любое основание, обычно используемое для реакций защиты. Например, основание может представлять собой гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия, бикарбонат натрия, карбонат цезия или фторид цезия. В частности, может быть использован фторид цезия (CsF), но основание не ограничивается им.
[56] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций защиты. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид или их смесь. В частности, может быть использован диметилформамид, но растворитель не ограничивается им.
[57] Кроме того, реакцию можно проводить при 30-110°С, и, более конкретно, можно проводить при 60-90°С, но не ограничиваясь этим.
[58] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, изопропаноле, с получением требуемого продукта с высокой степенью чистоты.
[59] Стадия (А-2)
[60] Согласно настоящему изобретению, на стадии (А-2) получают соединение, представленное химической формулой 3, посредством реакции окисления с использованием триазолопиримидинонового производного, представленного химической формулой 2, в качестве исходного вещества (схема реакции 2):
[61] [Схема реакции 2]
[62] 
[63] в указанной химической формуле R является таким, как определено выше.
[64] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции окисления, в которой соединение, представленное химической формулой 2, подвергают взаимодействию с окислителем.
[65] В реакции может быть использован любой окислитель, обычно используемый в реакциях окисления. Например, в качестве окислителя можно использовать пероксид водорода, пероксид бензоила, мета-хлорпероксибензойную кислоту или оксон. В частности, может быть использован оксон, но окислитель не ограничивается им.
[66] В реакции может быть использован любой органический растворитель, обычно используемый в реакциях окисления. Например, растворитель может представлять собой тетрагидрофуран, 1,4-диоксан, ацетон, метанол, этанол, изопропанол, воду или их смесь. В частности, может быть использована смесь тетрагидрофурана и метанола, но растворитель не ограничивается ею.
[67] Кроме того, реакцию можно проводить при 0-70°С, и, более конкретно, можно проводить при 30-50°С, но не ограничиваясь этим.
[68] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, изопропаноле, с получением требуемого продукта с высокой степенью чистоты.
[69] Стадия (А-3)
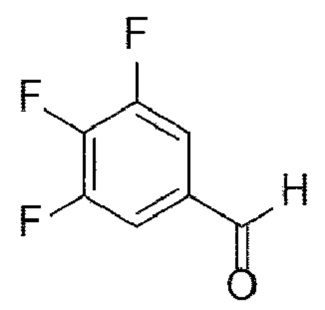
[70] В настоящем изобретении стадия (А-3) представляет собой стадию получения соединения, представленного химической формулой 5, посредством реакции аминирования с использованием производного соединения трифторбензальдегида, представленного химической формулой 4, в качестве исходного вещества (схема реакции 3):
[71] [Схема реакции 3]
[72] 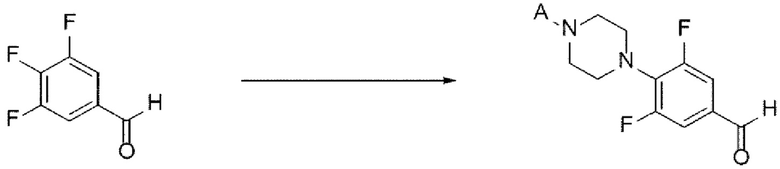
[73] в указанной химической формуле А представляет собой N-защитную группу.
[74] В соответствии с одним из вариантов реализации настоящего изобретения в указанной реакции могут быть использованы трифторбензальдегидные производные и защищенные пиперазиновые производные. Например, А может представлять собой -Boc, -Cbz, -Fmoc, -бензил, п-метоксибензил, тритил или диметокситритил (DMT). В соответствии с одним из вариантов реализации настоящего изобретения реакцию можно осуществлять посредством реакции аминирования, в которой 3,4,5-трифторбензальдегид подвергают взаимодействию с N-Boc-пиперазином, но не ограничиваясь этим.
[75] В указанной реакции может быть использовано любое основание, обычно используемое для реакций аминирования. Например, основание может представлять собой карбонат лития, карбонат натрия, карбонат калия, бикарбонат натрия или карбонат цезия. В частности, может быть использован карбонат лития, но основание не ограничивается им.
[76] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций аминирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид или их смесь. В частности, может быть использован диметилсульфоксид, но растворитель не ограничивается им.
[77] Кроме того, реакцию можно проводить при 80-150°С, и, более конкретно, можно проводить при 110-130°С, но не ограничиваясь этим.
[78] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим.
[79] Стадия (А-4)
[80] В настоящем изобретении стадия (А-4) представляет собой стадию получения соединения, представленного химической формулой 6, посредством реакции Дейкина с использованием производного соединения фенилпиперазина, представленного химической формулой 5, в качестве исходного вещества (схема реакции 4):
[81] [Схема реакции 4]
[82]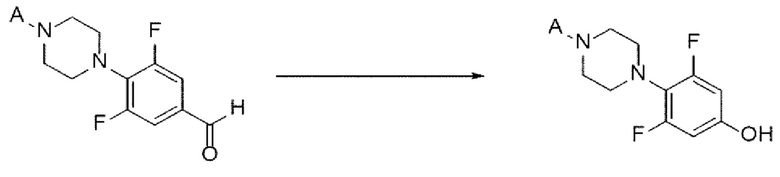
[83] в химической формуле А является таким, как определено выше.
[84] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить путем добавления окислителя к соединению, представленному химической формулой 5.
[85] В реакции Дейкина может быть использован обычно используемый окислитель. Например, можно использовать пероксид водорода, персульфат аммония, мета-хлорпероксибензойную кислоту (mCPBA) или их смесь. В частности, может быть использована мета-хлорпероксибензойная кислота, но окислитель не ограничивается ею.
[86] Можно использовать обычно используемое основание при гидролизе промежуточного соединения (фенилформиата), полученного после вышеуказанной реакции. Например, основание может представлять собой гидроксид лития, гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия или карбонат кальция. В частности, может быть использован гидроксид натрия, но основание не ограничивается им.
[87] В вышеуказанной реакции может быть использован обычно используемый растворитель. Например, растворитель может представлять собой дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, ацетон, метанол, этанол, изопропанол, воду или их смесь. В частности, может быть использован дихлорметан, но растворитель не ограничивается им.
[88] Кроме того, реакцию можно проводить при температуре от -20 до 30°С, и, более конкретно, можно проводить при температуре от -15 до 15°С, но не ограничиваясь этим.
[89] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. Например, в данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, изопропаноле, с получением требуемого продукта с высокой степенью чистоты.
[90] Стадия (А-5)
[91] В настоящем изобретении стадия (А-5) представляет собой стадию получения соединения, представленного химической формулой 7, посредством реакции алкилирования с использованием производного соединения фенилпиперазина, представленного химической формулой 6, в качестве исходного вещества (схема реакции 5):
[92] [Схема реакции 5]
[93] 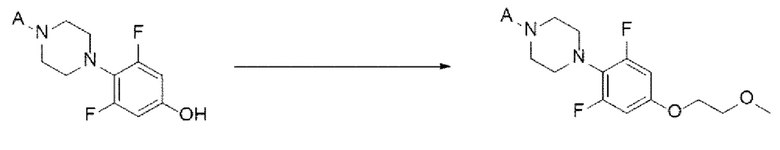
[94] в химической формуле А является таким, как определено выше.
[95] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции алкилирования, в которой соединение, представленное химической формулой 6, подвергают взаимодействию с 1-галоген-2-метоксиэтаном (например, 1-бром-2-метоксиэтаном).
[96] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций алкилирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид или их смесь. В частности, может быть использован ацетонитрил, но растворитель не ограничивается им.
[97] Кроме того, реакцию можно проводить при 50-100°С, и, более конкретно, можно проводить при 75-85°С, но не ограничиваясь этим.
[98] Стадия (А-6)
[99] В настоящем изобретении стадия (А-6) представляет собой стадию получения соединения, представленного химической формулой 8, или его соли посредством реакции снятия защиты с использованием производного соединения фенилпиперазина, представленного химической формулой 7, в качестве исходного вещества (схема реакции 6):
[100] [Схема реакции 6]
[101] 
[102] в химической формуле А является таким, как определено выше.
[103] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции снятия защиты с соединения, представленного химической формулой 7, в кислых условиях.
[104] В указанной реакции может быть использован любой растворитель, обычно используемый для реакций снятия защиты. Например, растворитель может представлять собой метанол, этанол, изопропанол, тетрагидрофуран, ацетонитрил, воду или их смесь. В частности, может быть использован метанол, но растворитель не ограничивается им.
[105] Кроме того, реакцию можно проводить при 0-60°С, и, более конкретно, можно проводить при 30-50°С, но не ограничиваясь этим.
[106] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, дихлорметане, трет-бутилметиловом эфире или их смеси с получением требуемого продукта с высокой степенью чистоты.
[107] Стадия (А-7)
[108] В настоящем изобретении стадия (А-7) представляет собой стадию получения соединения, представленного химической формулой 9, посредством реакции аминирования из соединения, представленного химической формулой 3, и соединения, представленного химической формулой 8, или его соли (схема реакции 7):
[109] [Схема реакции 7]
[110] 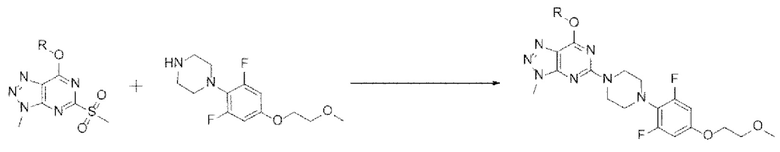
[111] в указанной химической формуле R является таким, как определено выше.
[112] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции аминирования с использованием, например, 7-изопропокси-3-метил-5-(метилсульфонил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина и гидрохлорида 1-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазина.
[113] В указанной реакции может быть использовано любое основание, обычно используемое для реакций аминирования. Например, основание может представлять собой карбонат лития, карбонат натрия, карбонат калия, бикарбонат натрия, карбонат цезия, триэтиламин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). В частности, может быть использован диизопропилэтиламин, но основание не ограничивается им.
[114] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций аминирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид, метанол, этанол, изопропанол или их смесь. В частности, может быть использован этанол, но растворитель не ограничивается им.
[115] Кроме того, реакцию можно проводить при 50-100°С, и, более конкретно, можно проводить при 60-80°С, но не ограничиваясь этим.
[116] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, дихлорметане, диизопропиловом эфире или их смеси с получением требуемого продукта с высокой степенью чистоты.
[117] Стадия (А-8)
[118] В настоящем изобретении стадия (А-8) представляет собой стадию получения соединения, представленного химической формулой I, посредством реакции снятия защиты с соединения, представленного химической формулой 9 (схема реакции 8):
[119] [Схема реакции 8]
[120] 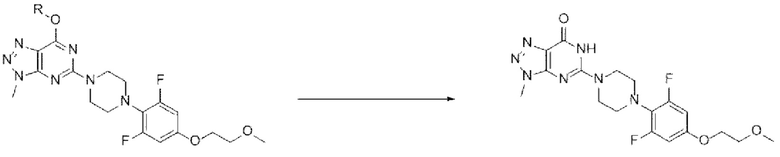
[121] в указанной химической формуле R является таким, как определено выше.
[122] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно, например, проводить посредством реакции снятия защиты с 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидина.
[123] В вышеуказанной реакции в качестве растворителя может быть использована любая кислота, обычно используемая для реакций снятия защиты. В данном случае кислота может представлять собой уксусную кислоту, соляную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту или их смесь. В частности, может быть использована уксусная кислота, серная кислота или их смесь, но кислота не ограничивается ими.
[124] Кроме того, реакцию можно проводить при 30-80°С, и, более конкретно, можно проводить при 40-60°С, но не ограничиваясь этим.
[125] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в ацетоне, воде или их смеси с получением требуемого продукта с высокой степенью чистоты.
[126] Способ синтеза В
[127] Способ синтеза В способа получения согласно настоящему изобретению включает стадии (В-1) - (В-7), указанные ниже:
[128] (В-1) Стадию 1 получения соединения, представленного следующей химической формулой 2, посредством реакции защиты из соединения, представленного следующей химической формулой 1, или его соли;
[129] (В-2) Стадию 2 получения соединения, представленного следующей химической формулой 3, посредством реакции окисления из соединения, представленного химической формулой 2;
[130] (В-3) Стадию 3 получения соединения, представленного следующей химической формулой 9, посредством реакции аминирования из соединения, представленного следующей химической формулой 4;
[131] (В-4) Стадию 4 получения соединения, представленного следующей химической формулой 10, посредством реакции аминирования из соединения, представленного химической формулой 3, и соединения, представленного химической формулой 9, или его соли;
[132] (В-5) Стадию 5 получения соединения, представленного следующей химической формулой 11, посредством реакции Дейкина, из соединения, представленного химической формулой 10;
[133] (В-6) Стадию 6 получения соединения, представленного следующей химической формулой 1а, посредством реакции алкилирования из соединения, представленного химической формулой 11; и
[134] (В-7) Стадию 7 получения соединения, представленного следующей химической формулой I, посредством реакции снятия защиты с соединения, представленного химической формулой 1а:
[135] [Химическая формула 1]
[136] 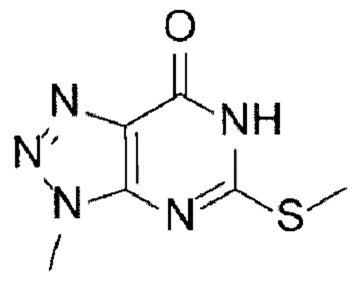
[137] [Химическая формула 2]
[138] 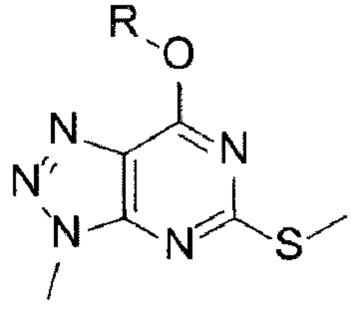
[139] [Химическая формула 3]
[140] 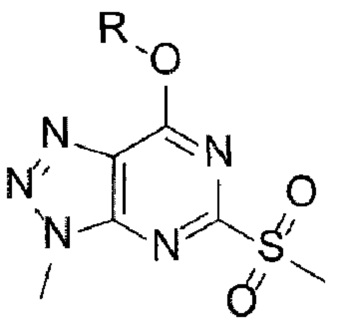
[141] [Химическая формула 4]
[142] 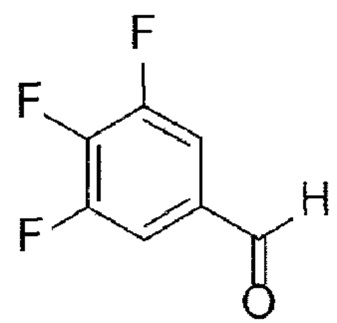
[143] [Химическая формула 9]
[144] 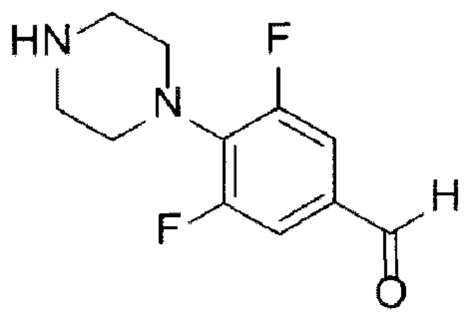
[145] [Химическая формула 10]
[146] 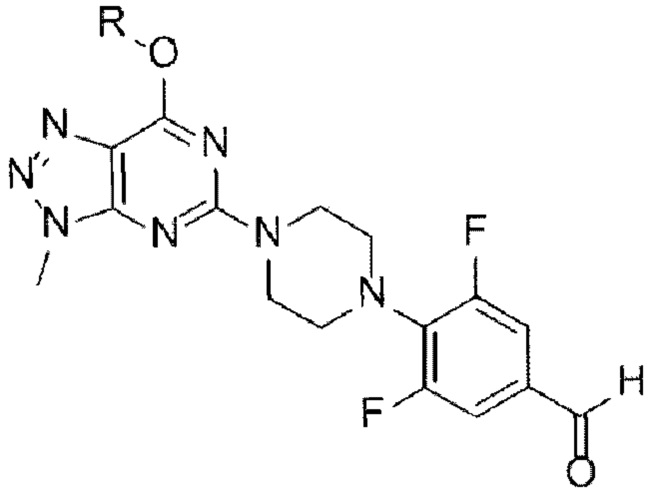
[147] [Химическая формула 11]
[148] 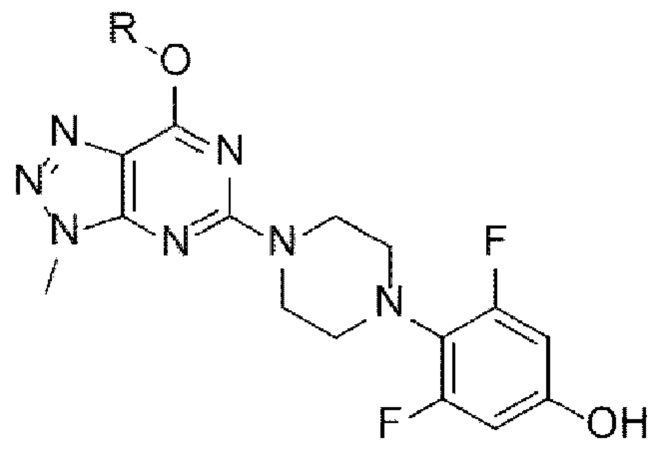
[149] [Химическая формула 1а]
[150] 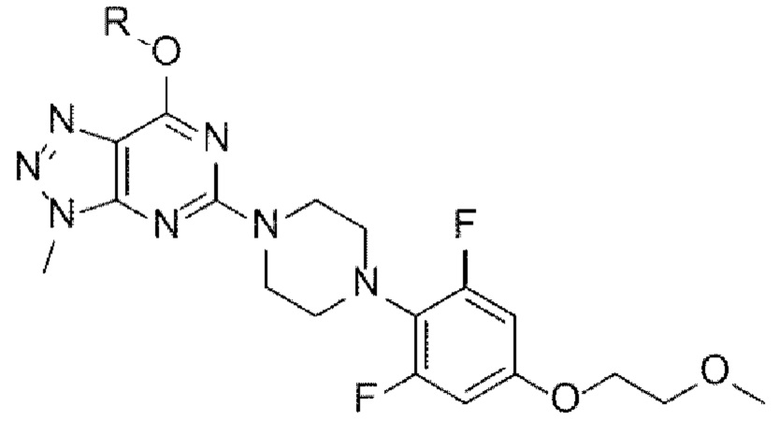
[151] [Химическая формула I]
[152] 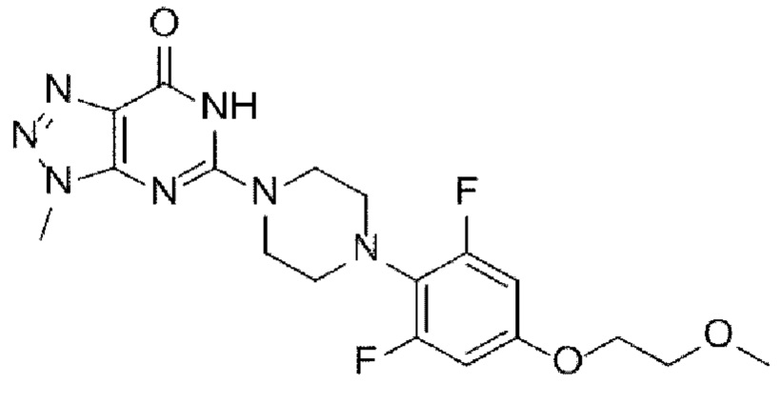
[153] в указанных химических формулах
[154] R представляет собой О-защитную группу.
[155] Далее будут отдельно описаны стадии (В-1) - (В-7). Среди них стадии (В-1), (В-2) и (В-7) являются такими же, как стадии (А-1), (А-2) и (А-8), описанные выше, соответственно, и, таким образом, будут подробно рассмотрены стадии (В-3) - (В-6).
[156] Стадия (В-3)
[157] В настоящем изобретении стадия (В-3) представляет собой стадию получения соединения, представленного химической формулой 8, или его соли посредством реакции аминирования с использованием производного соединения трифторбензальдегида, представленного химической формулой 4, в качестве исходного вещества (схема реакции 9):
[158] [Схема реакции 9]
[159] 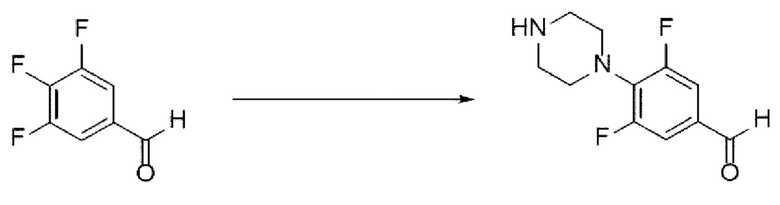
[160] В соответствии с одним из вариантов реализации настоящего изобретения в указанной реакции могут быть использованы трифторбензальдегидные производные и пиперазин.
[161] В указанной реакции может быть использовано любое основание, обычно используемое для реакций аминирования. Например, основание может представлять собой карбонат лития, карбонат натрия, карбонат калия, бикарбонат натрия или карбонат цезия. В частности, может быть использован карбонат калия, но основание не ограничивается им.
[162] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций аминирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, изопропиловый спирт, диметилсульфоксид, диметилацетамид, диметилформамид, этиленгликоль, диэтиленгликоль, диметиловыи эфир, диметоксиэтан или их смесь. В частности, может быть использован диметоксиэтан, но растворитель не ограничивается им.
[163] Кроме того, реакцию можно проводить при 60-150°С, и, более конкретно, можно проводить при 70-90°С, но не ограничиваясь этим.
[164] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим.
[165] Стадия (В-4)
[166] В настоящем изобретении стадия (В-3) представляет собой стадию получения соединения, представленного химической формулой 10, посредством реакции аминирования из соединения, представленного химической формулой 3, и соединения, представленного химической формулой 9, или его соли (схема реакции 10):
[167] [Схема реакции 10]
[168] 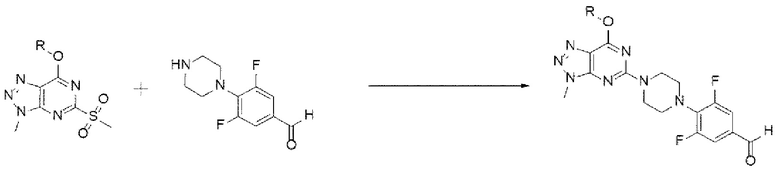
[169] в указанной химической формуле R является таким, как определено выше.
[170] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции аминирования с использованием, например, 7-изопропокси-3-метил-5-(метилсульфонил)-3Н-[1,2,3]триазоло[4,5-d]пиримидина и 3,5-дифтор-4-(пиперазин-1-ил)бензальдегида.
[171] В указанной реакции может быть использовано любое основание, обычно используемое для реакций аминирования. Например, основание может представлять собой карбонат лития, карбонат натрия, карбонат калия, бикарбонат натрия, карбонат цезия, триэтиламин, диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), но не ограничиваясь ими.
[172] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций аминирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид, метанол, этанол, изопропанол или их смесь. В частности, может быть использован диметилацетамид, но растворитель не ограничивается им.
[173] Кроме того, реакцию можно проводить при 50-150°С, и, более конкретно, можно проводить при 80-120°С, но не ограничиваясь этим.
[174] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, диэтиловом эфире, изопропаноле или их смеси с получением требуемого продукта с высокой степенью чистоты.
[175] Стадия (В-5)
[176] В настоящем изобретении стадия (В-5) представляет собой стадию получения соединения, представленного химической формулой 11, посредством реакции Дейкина с использованием соединения, представленного химической формулой 10, в качестве исходного вещества (схема реакции 11):
[177] [Схема реакции 11]
[178] 
[179] в указанной химической формуле R является таким, как определено выше.
[180] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить путем добавления окислителя к соединению, представленному химической формулой 10.
[181] В реакции Дейкина может быть использован обычно используемый окислитель. Например, можно использовать пероксид водорода, персульфат аммония, мета-хлорпероксибензойную кислоту (mCPBA) или их смесь. В частности, может быть использована мета-хлорпероксибензойная кислота, но окислитель не ограничивается ею.
[182] Можно использовать обычно используемое основание при гидролизе промежуточного соединения (фенилформиата), полученного после вышеуказанной реакции. Например, основание может представлять собой гидроксид лития, гидроксид натрия, гидроксид калия, бикарбонат натрия, карбонат натрия или карбонат кальция. В частности, может быть использован гидроксид натрия, но основание не ограничивается им.
[183] В вышеуказанной реакции может быть использован обычно используемый растворитель. Например, растворитель может представлять собой дихлорметан, хлороформ, тетрагидрофуран, 1,4-диоксан, ацетон, метанол, этанол, изопропанол, воду или их смесь. В частности, может быть использован дихлорметан, но растворитель не ограничивается им.
[184] Кроме того, реакцию можно проводить при температуре от -20 до 30°С, и, более конкретно, можно проводить при температуре от -15 до 15°С, но не ограничиваясь этим.
[185] Предложенный способ может дополнительно включать, после вышеуказанной реакции, одну или более стадий разделения или очистки полученного продукта, но не ограничиваясь этим. В данном примере настоящего изобретения продукт вышеуказанной реакции перемешивают в органическом растворителе, изопропаноле, трет-бутилметиловом эфире или их смеси с получением требуемого продукта с высокой степенью чистоты.
[186] Стадия (В-6)
[187] В настоящем изобретении стадия (В-6) представляет собой стадию получения соединения, представленного химической формулой 1а, посредством реакции алкилирования с использованием соединения, представленного химической формулой 11, в качестве исходного вещества (схема реакции 12):
[188] [Схема реакции 12]
[189] 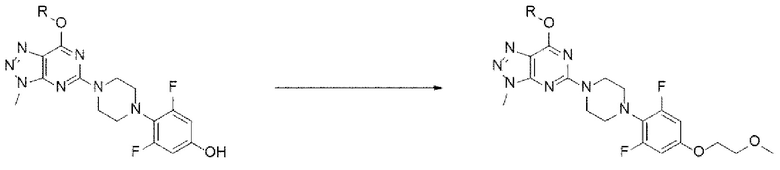
[190] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством реакции алкилирования, в которой соединение, представленное химической формулой 11, подвергают взаимодействию с 1-галоген-2-метоксиэтаном (например, 1-бром-2-метоксиэтаном).
[191] В указанной реакции может быть использован любой органический растворитель, обычно используемый для реакций алкилирования. Например, растворитель может представлять собой ацетонитрил, тетрагидрофуран, 1,4-диоксан, ацетон, диметилсульфоксид, диметилацетамид, диметилформамид или их смесь. В частности, может быть использован диметилформамид, но растворитель не ограничивается им.
[192] Кроме того, реакцию можно проводить при 50-100°С, и, более конкретно, можно проводить при 60-80°С, но не ограничиваясь этим.
[193] В соответствии со способом получения, описанным в международной публикации № WO 2016/006974, соединение, представленное химической формулой I, синтезируют с помощью 11-стадийного процесса. Кроме того, такой способ получения не подходит для массового производства, поскольку в нем используются микроволновые печи или множество стадий очистки на колонках. Однако в соответствии со способом получения согласно настоящему изобретению ни способ синтеза А, ни способ синтеза В не требуют большого количества процессов и включают 8 или 7 стадий. Кроме того, стадии (А-1) - (А-8) способа синтеза А и стадии (В-1) - (В-7) способа синтеза В не проводят с помощью процесса в микроволновом устройстве и очистки на колонке, и они подходят для массового производства, поскольку могут эффективно обеспечивать получение соединений с высоким выходом и высокой чистотой.
[194] Кроме того, соединения, представленные химической формулой 1-11 и химической формулой 1а, полученные и используемые на стадиях (А-1) - (А-8), стадиях (В-1) - (В-7) выше, являются подходящими промежуточными соединениями для получения производных соединений триазолопиримидинона, представленных химической формулой I.
Полезные эффекты
[195] Способ получения согласно настоящему изобретению может обеспечивать разработку эффективных способов и требует меньшего количества процессов по сравнению с обычными способами получения, что позволяет получать производное соединение триазолопиримидинона с высокой чистотой и высоким выходом посредством кристаллизации без необходимости очистки на колонке и проведения реакции в микроволновом устройстве, что существенно снижает стоимость производства, является экономичным и пригодным для массового производства. Наилучший вариант реализации
[196] Далее представлены следующие примеры для улучшения понимания настоящего изобретения. Представленные примеры приведены лишь для облегчения понимания настоящего изобретения и не налагают ограничений на содержание настоящего изобретения.
[197] Пример 1
[198] В примере 1 настоящего изобретения производное соединение триазолопиримидинона, представленное химической формулой I, получали в соответствии со следующей схемой реакции I:
[199] [Схема реакции I]
[200]
[201] 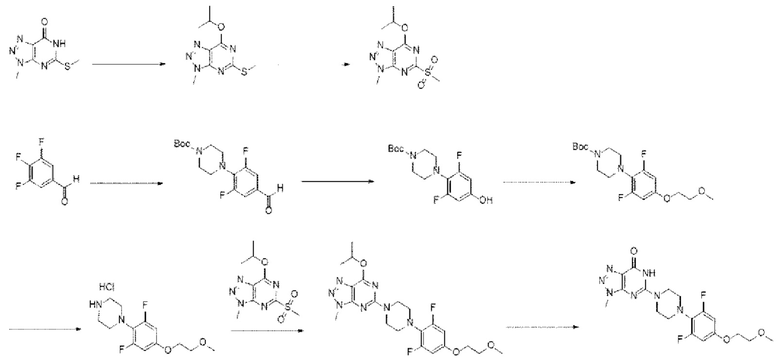
[202] Стадия 1: Получение 7-изопропокси-3-метил-5-(метилтио)-3Н-[1,2,3]триазоло[4,5-d]пиримидина
[203] 3-Метил-5-(метилтио)-3,6-дигидро-7Н-[1,2,3]триазоло[4,5-d]пиримидин-7-он (15 г, 76,1 ммоль) разбавляли в 76 мл диметилформамида. К ним добавляли фторид цезия (46,2 г, 304,2 ммоль) и изопропилйодид (38,1 г, 228,2 ммоль), внутреннюю температуру повышали до 75-80°С и перемешивали реакционную смесь в течение 2 часов. После завершения реакции смесь охлаждали до комнатной температуры, добавляли 300 мл этилацетата и перемешивали смесь в течение 10 минут. Полученные кристаллы фильтровали через целитный фильтр и добавляли к фильтрату 120 мл 5% солевого раствора, и перемешивали в течение 10 минут. Органический слой промывали еще дважды, используя по 120 мл 5% солевого раствора. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К концентрированному остатку добавляли 50 мл изопропанола, нагревали до 40°С и перемешивали в течение 30 минут. Выпавшие в осадок кристаллы перемешивали при 5-10°С в течение 30 минут, фильтровали и сушили при пониженном давлении с получением требуемого продукта (11,3 г, 62%) в виде твердого вещества светло-желтого цвета.
[204] 1Н-ЯМР 400 Гц (ДМСО-d6): 5,62 (м, 1Н), 4,15 (с, 3Н), 2,61 (с, 3Н), 1,44 (д, J=8 Гц, 6Н).
[205] ЖХ-МС (ИЭР, m/z): 240,1 [М+Н+].
[206] Стадия 2: Получение 7-изопропокси-3-метил-5-(метилсульфонил)-3Н-[1,2,3]триаволо[4,5-d]пиримидина
[207] 7-Изопропокси-3-метил-5-(метилтио)-3Н-[1,2,3]триазоло[4,5-d]пиримидин, полученный на стадии 1 (10 г, 41,8 ммоль), разбавляли в 70 мл тетрагидрофурана и перемешивали при охлаждении до внутренней температуры от 5 до 10°С. Оксон (38,5 г, 125,3 ммоль) растворяли в 200 мл очищенной воды, а затем по каплям добавляли к реакционному раствору в течение 30 минут. После завершения капельного добавления повышали внутреннюю температуру до 35-40°С и перемешивали реакционную смесь в течение 2 часов. По завершении реакции реакционный раствор концентрировали при пониженном давлении, добавляли 100 мл дихлорметана и экстрагировали органический слой. Водный слой экстрагировали, используя 100 мл дихлорметана, органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К концентрированному остатку добавляли 50 мл изопропанола, нагревали до 75 Си перемешивали в течение 30 минут. Выпавшие в осадок кристаллы перемешивали при 5-10°С в течение 30 минут, фильтровали и сушили при пониженном давлении с получением требуемого продукта (9,9 г, 87%) в виде твердого вещества почти белого цвета.
[208] 1Н-ЯМР 400 Гц (ДМСО-d6): 5,74 (м, 1Н), 4,30 (с, 3Н), 3,47 (с, 3Н), 1,50 (д, J=6,4 Гц, 6Н).
[209] ЖХ-МС (ИЭР, m/z): 272,0 [М+Н+].
[210] Стадия 3: Получение трет-бутил-4-(2,6-дифтор-4-формилфенил)пиперазин-1-карбоксилата
[211] В диметилсульфоксиде (25 мл) разбавляли 3,4,5-трифторбензальдегид (5 г, 31,2 ммоль), Вос-пиперазин (5,8 г, 31,2 ммоль) и карбонат лития (7,3 г, 94 ммоль) и затем перемешивали смесь в течение 3 часов при внутренней температуре от 115 до 120°С. По завершении реакции реакционный раствор охлаждали до комнатной температуры, разбавляли в смеси 125 мл холодной воды и 50 мл этилацетата и перемешивали в течение 10 минут. Нерастворимый материал удаляли фильтрованием через целит. Отделяли органический слой фильтрата, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Следующую реакцию проводили без очистки остатка.
[212] Стадия 4: Получение трет-бутил-4-(2,6-дифтор-4-гидроксифенил)пиперазин-1-карбоксилата
[213] Трет-бутил-4-(2,6-дифтор-4-формилфенил)пиперазин-1-карбоксилат, полученный на стадии 3, растворяли в 102 мл дихлорметана и перемешивали при охлаждении до внутренней температуры от -15 до -10°С. Медленно, по частям добавляли мета-хлорпероксибензойную кислоту (8,08 г, 46,8 ммоль) и перемешивали в течение 2 часов, поддерживая внутреннюю температуру от -10 до 10°С. По каплям добавляли 2 н. водный раствор гидроксида натрия (78 мл, 156 ммоль) в реакционный раствор, который постепенно доводили до комнатной температуры и перемешивали в течение 2 часов. По завершении реакции водный слой отделяли и доводили до рН 6 с помощью 1 н. водного раствора соляной кислоты при 0-5°С, а затем добавляли 90 мл трет-бутилметилового эфира и перемешивали в течение 10 минут. Органический слой экстрагировали и промывали 30 мл водного раствора бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К остатку добавляли 35 мл изопропанола и перемешивали смесь при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали и сушили при пониженном давлении с получением требуемого продукта (3,9 г, 40%) в виде бледно-желтого твердого вещества.
[214] 1Н-ЯМР 400 Гц (MeOD): 6,33 (д, J=8,2 Гц, 2Н), 4,63 (с, 1Н), 3,52 (с, 4Н), 3,00 (с, 4Н), 1,49 (с, 9Н).
[215] ЖХ-МС (ИЭР, m/z): 315,1 [М+Н+].
[216] Стадия 5: Получение трет-бутил-4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)лилеразин-1-карбоксилата
[217] Трет-бутил-4-(2,6-дифтор-4-гидроксифенил)пиперазин-1-карбоксилат (20 г, 63,6 ммоль), полученный на стадии 4, разбавляли ацетонитрилом. Добавляли карбонат кальция (26,4 г, 190,9 ммоль) и 1-бром-2-метоксиэтан (13,3 г, 95,5 ммоль) и затем доводили реакционную смесь до 75-85°С и перемешивали при кипении с обратным холодильником в течение 8 часов. По завершении реакции реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 140 мл очищенной воды и 280 мл этилацетата и перемешивали при комнатной температуре в течение 15 минут. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Следующую реакцию проводили без очистки остатка.
[218] Стадия 6: Получение гидрохлорида 1-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазина
[219] В реактор добавляли 240 мл метанола и охлаждали до внутренней температуры ниже 5°С, а затем медленно добавляли ацетилхлорид (31,3 мл, 439 ммоль) и перемешивали в течение 15 минут. Трет-бутил-4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-карбоксилат, полученный на стадии 5, растворяли в 64 мл дихлорметана, а затем медленно добавляли в реактор, и повышали внутреннюю температуру до 40-45°С, и перемешивали в течение 2 часов. По завершении реакции реакционный раствор концентрировали при пониженном давлении, а к остатку добавляли 60 мл метанола и растворяли путем перемешивания при комнатной температуре. Внутреннюю температуру понижали до 15-20°С и по каплям добавляли 180 мл трет-бутилметилового эфира, и перемешивали в течение 30 минут. Выпавшие в осадок кристаллы отфильтровывали и сушили при пониженном давлении с получением требуемого продукта (14,2 г, 72%) в виде твердого вещества почти белого цвета.
[220] 1Н-ЯМР 400 Гц (ДМСО-d6): 9,25 (шс, 2Н), 6,85-6,64 (м, 2Н), 4,12-4,02 (м, 2Н), 3,65-3,58 (м, 2Н), 3,28 (с, 3Н), 3,25-3,18 (м, 4Н), 3,17-3,09 (м, 4Н).
[221] ЖХ-МС (ИЭР, m/z): 273,0 [М+Н+]. (Свободная форма)
[222] Стадия 7: Получение 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидина
[223] 7-Изопропокси-3-метил-5-(метилсульфонил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин, полученный на стадии 2 (10 г, 36,9 ммоль), разбавляли 146 мл этанола, затем добавляли диизопропилэтиламин (19,3 мл, 110,5 ммоль) и гидрохлорид 1-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазина (13,1 г, 42,4 ммоль), полученный на стадии 6, повышали температуру до 60-80°С и кипятили реакционную смесь с обратным холодильником, и перемешивали в течение 3 часов. По завершении реакции реакционный раствор концентрировали при пониженном давлении, к остатку добавляли 150 мл дихлорметана и 53 мл воды и разделяли слои. Водный слой дважды экстрагировали, используя по 50 мл дихлорметана. Органические слои объединяли, промывали 50 мл 1 н. водного раствора соляной кислоты и 50 мл 5% солевого раствора, сушили над безводным сульфатом натрия и фильтровали. К фильтрату добавляли 2 г активированного угля и перемешивали в течение 30 минут. Фильтрат фильтровали через целит и концентрировали при пониженном давлении. К концентрированному остатку добавляли 30 мл дихлорметана для растворения, затем по каплям добавляли 200 мл диизопропилового эфира и перемешивали при 5-10°С в течение 1 часа. Выпавшие в осадок кристаллы отфильтровывали и сушили при пониженном давлении с получением требуемого продукта (15,1 г, 89%) в виде твердого вещества почти белого цвета.
[224] 1Н-ЯМР 400 Гц (ДМСО-d6): 6,77-6,69 (м, 2Н), 5,55 (м, 1Н), 4,09-4,06 (м, 2Н), 3,99 (с, 3Н), 3,93 (т, J=4,8 Гц, 4Н), 3,64-3,62 (м, 2Н), 3,28 (с, 3Н), 3,09 (т, J=4,8 Гц, 4Н), 1,42 (д, J=6,4 Гц, 6Н).
[225] ЖХ-МС (ИЭР, m/z): 464,1 [М+Н+].
[226] Стадия 8: Получение 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-3-метил-3,6-дигидро-7Н-[1,2,3]триазоло[4,5-d]пиримидин-7-она
[227] К 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d] пиримидину (15 г, 32,4 ммоль), полученному на стадии 7, добавляли 92 мл уксусной кислоты и 16.1 мл серной кислоты, нагревали до внутренней температуры от 45 до 50°С и перемешивали в течение 3 часов. По завершении реакции реакционный раствор охлаждали до комнатной температуры и по каплям добавляли 230 мл очищенной воды в течение 30 минут при перемешивании. После завершения капельного добавления понижали внутреннюю температуру до 5-10°С и перемешивали полученную смесь в течение 1 часа. Выпавшие в осадок кристаллы отифильтровывали и разбавляли в 150 мл ацетона, затем повышали внутреннюю температуру до 40-45°С и перемешивали смесь в течение 1 часа. Реакционный раствор постепенно охлаждали до 5-10°С и добавляли 150 мл очищенной воды. Выпавшие в осадок кристаллы перемешивали в течение 30 минут, фильтровали и сушили при пониженном давлении с получением требуемого продукта (11,5 г, 84%) в виде твердого вещества почти белого цвета.
[228] 1Н-ЯМР 400 Гц (DMCO-d6): 11,29 (шс, 1Н), 6,74 (д, J=11.2 Гц, 2Н), 4,08 (т, J=4,4 Гц, 2Н), 3,92 (с, 3Н), 3,82-3,76 (м, 4Н), 3,62 (т, J=4,4 Гц, 2Н), 3,29 (с, 3Н), 3,12-3,06 (м, 4Н).
[229] ЖХ-МС (ИЭР, m/z): 422,1 [М+Н+].
[230] Пример 2
[231] В примере 2 настоящего изобретения производное соединение триазолопиримидинона, представленное химической формулой I, получали в соответствии со следующей схемой реакции II:
[232] [Схема реакции II]
[233]
[234] 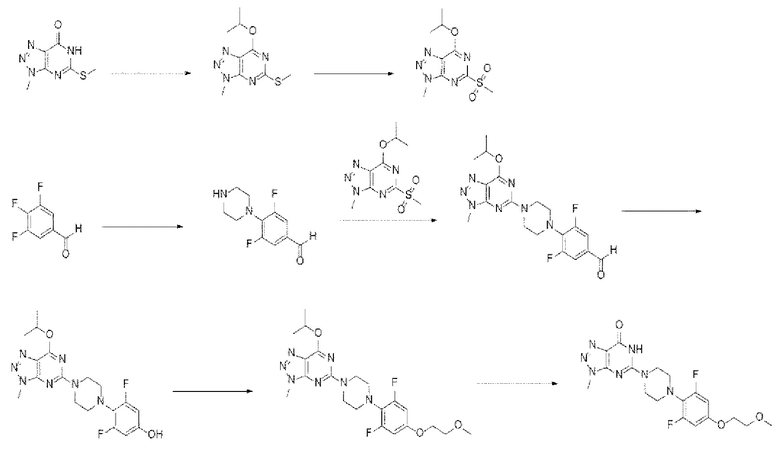
[235] Стадия l: Получение 3,5-дифтор-4-(пиперазин-1-ил)бензальдегида
[236] В диметоксиэтане (50 мл) разбавляли 3,4,5-трифторбензальдегид (4 г, 24,99 ммоль), пиперазин (6,46 г, 75 ммоль) и карбонат калия (6,91 г, 50 ммоль) и затем перемешивали смесь при внутренней температуре от 75 до 80°С в течение 20 часов. По завершении реакции реакционный раствор охлаждали до 0-5°С и доводили до рН 2 путем добавления 2 н. водного раствора соляной кислоты. Водный слой дважды промывали, используя по 50 мл дихлорметана, и затем доводили до нейтрального состояния 1 н. водным раствором гидроксида натрия. К водному слою добавляли 100 мл дихлорметана, затем органический слой экстрагировали, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением требуемого продукта (4,65 г, 82%) в виде желтого твердого вещества.
[237] 1Н-ЯМР 400 Гц (DMCO-d6): 9,80 (с, 1Н), 7,56-7,54 (д, J=8,2 Гц, 2Н), 3,19 (с, 4Н), 2,79 (с, 4Н).
[238] ЖХ-МС (ИЭР, m/z): 227,1 [М+Н+].
[239] Стадия 2: Получение 3,5-дифтор-4-(4-(7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидин-5-ил)пиперазин-1-ил)бензальдегида
[240] 7-Изопропокси-3-метил-5-(метилсульфонил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин, полученный на стадии 2 примера 1 (1 г, 3,69 ммоль) и 3,5-дифтор-4-(пиперазин-1-ил)бензальдегид (0,959 г, 4,24 ммоль), полученный на стадии 1 примера 2, разбавляли в 15 мл диметилацетамида, нагревали до 110-120°С и перемешивали в течение 2 часов. По завершении реакции реакционный раствор охлаждали до комнатной температуры и разбавляли 75 мл этилацетата и 30 мл воды, и отделяли органический слой, и промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К концентрированному остатку добавляли 5 мл изопропанола и перемешивали в течение 30 минут при комнатной температуре. Выпавшие в осадок кристаллы отфильтровывали, промывали изопропанолом и сушили при пониженном давлении с получением требуемого продукта (1,25 г, 81%) в виде бледно-желтого твердого вещества.
[241] 1Н-ЯМР 400 Гц (CDCl3): 9,81 (с, 1Н), 7,44-7,36 (м, 2Н), 5,65-5,55 (м, 1Н), 4,07 (с, 3Н), 4,05-4,03 (т, J=5,0 Гц, 4Н), 3,43 (с, 4Н), 1,50-1,48 (д, J=6,2 Гц, 6Н).
[242] ЖХ-МС (ИЭР, m/z): 418,0 [М+Н+].
[243] Стадия 3: Получение 3,5-дифтор-4-(4-(7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидин-5-ил)пиперазин-1-ил)фенола
[244] 3,5-Дифтор-4-(4-(7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидин-5-ил)пиперазин-1-ил)бензальдегид, полученный на стадии 2 (1 г, 2,4 ммоль), разбавляли в 8 мл дихлорметана, затем медленно, по частям добавляли мета-хлорпероксибензойную кислоту (1076 мг, 4,8 ммоль) при температуре от -10 до 0°С и перемешивали в течение 2 часов, поддерживая внутреннюю температуру от -10 до 10°С. По каплям добавляли 2 н. водный раствор гидроксида натрия (9,6 мл, 19,2 ммоль) в реакционный раствор, который постепенно доводили до комнатной температуры и перемешивали в течение 2 часов. По завершении реакции водный слой отделяли и доводили до рН 6 с помощью 1 н. водного раствора соляной кислоты при 0-5°С, а затем добавляли 50 мл трет-бутилметилового эфира и перемешивали в течение 10 минут. Органический слой экстрагировали и промывали 10 мл водного раствора бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К остатку добавляли 3 мл изопропанола и перемешивали смесь при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали и сушили при пониженном давлении с получением требуемого продукта (388 мг, 40%) в виде бледно-желтого твердого вещества.
[245] 1Н-ЯМР 400 Гц (DMCO-d6): 6,44-6,42 (д, J=11,0 Гц, 2Н), 5,57-5,51 (м, 1Н), 3,99 (с, 3Н), 3,92 (с, 3Н), 1,42-1,41 (д, J=6,2 Гц, 6Н).
[246] ЖХ-МС (ИЭР, m/z): 406,0 [М+Н+].
[247] Стадия 4: Получение 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидина
[248] 3,5-Дифтор-4-(4-(7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидин-5-ил)пиперазин-1-ил)фенол, полученный на стадии 3 (1 г, 2,5 ммоль), разбавляли 10 мл диметилформамида, затем добавляли карбонат калия (1,02 г, 7,4 ммоль) и перемешивали при 65-70°С в течение 7 часов. По завершении реакции добавляли 200 мл очищенной воды и 550 мл этилацетата и перемешивали при комнатной температуре в течение 15 минут. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К концентрированному остатку по каплям добавляли дихлорметан и диизопропиловый эфир для осаждения кристаллов. Выпавшие в осадок кристаллы отфильтровывали и сушили при пониженном давлении с получением требуемого продукта (780 мг, 68%) в виде твердого вещества почти белого цвета.
[249] 1Н-ЯМР 400 Гц (ДМСО-d6): 6,77-6,69 (м, 2Н), 5,55 (м, 1Н), 4,09-4,06 (м, 2Н), 3,99 (с, 3Н), 3,93 (т, J=4,8 Гц, 4Н), 3,64-3,62 (м, 2Н), 3,28 (с, 3Н), 3,09 (т, J=4,8 Гц, 4Н), 1,42 (д, J=6,4 Гц, 6Н).
[250] ЖХ-МС (ИЭР, m/z): 464,1 [М+Н+].
[251] Стадия 5: Получение 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-3-метил-3,6-дигидро-7Н-[1,2,3]триазоло[4,5-d]пиримидин-7-она
[252] К 5-(4-(2,6-дифтор-4-(2-метоксиэтокси)фенил)пиперазин-1-ил)-7-изопропокси-3-метил-3Н-[1,2,3]триазоло[4,5-d]пиримидину (750 мг, 1,62 ммоль), полученному на стадии 4, добавляли 4,6 мл уксусной кислоты и 0,81 мл серной кислоты, нагревали до внутренней температуры от 45 до 50°С и перемешивали в течение 3 часов. По завершении реакции охлаждали реакционный раствор до комнатной температуры и по каплям добавляли 11 мл очищенной воды в течение 30 минут при перемешивании. После завершения капельного добавления понижали внутреннюю температуру до 5-10°С и перемешивали полученную смесь в течение 1 часа. Выпавшие в осадок кристаллы отфильтровывали и разбавляли в 7,5 мл ацетона, затем внутреннюю температуру повышали до 40-45°С и перемешивали смесь в течение 1 часа. Реакционный раствор постепенно охлаждали до 5-10°С и добавляли 7,5 мл очищенной воды. Выпавшие в осадок кристаллы перемешивали в течение 30 минут, фильтровали и сушили при пониженном давлении с получением требуемого продукта (575 мг, 84%) в виде твердого вещества почти белого цвета.
[253] 1Н-ЯМР 400 Гц (ДМСО-d6): 11,29 (шс, 1Н), 6,74 (д, J=11,2 Гц, 2Н), 4,08 (т, J=4,4 Гц, 2Н), 3,92 (с, 3Н), 3,82-3,76 (м, 4Н), 3,62 (т, J=4,4 Гц, 2Н), 3,29 (с, 3Н), 3,12-3,06 (м, 4Н).
[254] ЖХ-МС (ИЭР, m/z): 422,1 [М+Н+].
[255]
[256] Несмотря на то, что выше подробно описаны некоторые аспекты настоящего изобретения, для специалиста в данной области техники будет очевидно, что представленные конкретные описания являются лишь предпочтительными вариантами реализации и не предназначены для ограничения объема настоящего изобретения. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРИМИДИНА, ПОДАВЛЯЮЩЕЕ РОСТ РАКОВОЙ КЛЕТКИ, И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2792849C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРРОЛОПИРИДИНОВОГО ПРОИЗВОДНОГО | 2021 |
|
RU2830589C1 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| ПИРИМИДИН- И АЗОТСОДЕРЖАЩЕЕ БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2827867C1 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | 2015 |
|
RU2658912C1 |
Настоящее изобретение относится к технологии получения триазолопиримидинонового производного, проявляющего активность в отношении ингибирования танкиразы. Способы получения соединения формулы I характеризуются этапами, состоящими из восьми и семи стадий. Технический результат – предоставление эффективных методов для получения целевого продукта с высокой чистотой и высоким выходом. 2 н. и 8 з.п. ф-лы, 2 пр.
1. Способ получения триазолопиримидинонового производного, включающий:
(А-1) Стадию 1 получения соединения, представленного следующей химической формулой 2, посредством реакции защиты из соединения, представленного следующей химической формулой 1, или его соли;
(А-2) Стадию 2 получения соединения, представленного следующей химической формулой 3, посредством реакции окисления из соединения, представленного химической формулой 2;
(А-3) Стадию 3 получения соединения, представленного следующей химической формулой 5, посредством реакции аминирования из соединения, представленного следующей химической формулой 4;
(А-4) Стадию 4 получения соединения, представленного следующей химической формулой 6, посредством реакции Дейкина из соединения, представленного химической формулой 5;
(А-5) Стадию 5 получения соединения, представленного следующей химической формулой 7, посредством реакции алкилирования из соединения, представленного химической формулой 6;
(А-6) Стадию 6 получения соединения, представленного следующей химической формулой 8, или его соли посредством реакции снятия защиты с соединения, представленного химической формулой 7;
(А-7) Стадию 7 получения соединения, представленного следующей химической формулой Ia, посредством реакции аминирования из соединения, представленного химической формулой 3, и соединения, представленного химической формулой 8, или его соли; и
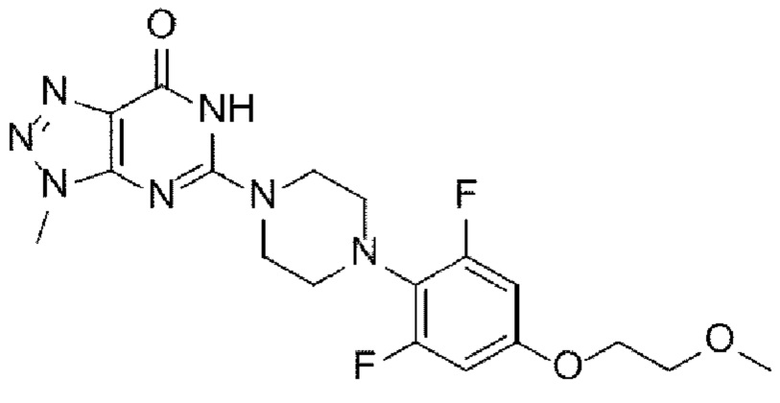
(А-8) Стадию 8 получения соединения, представленного следующей химической формулой I, посредством реакции снятия защиты с соединения, представленного химической формулой Ia:
[Химическая формула 1]
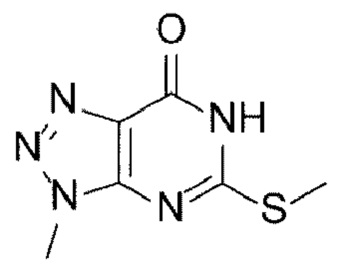
[Химическая формула 2]
[Химическая формула 3]
[Химическая формула 4]
[Химическая формула 5]
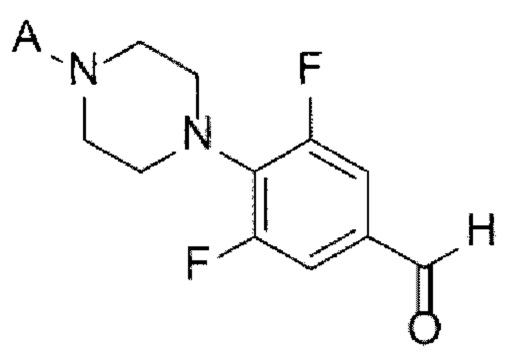
[Химическая формула 6]
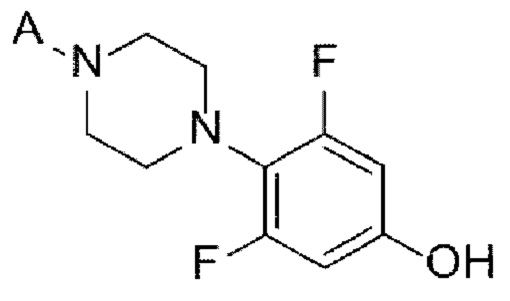
[Химическая формула 7]
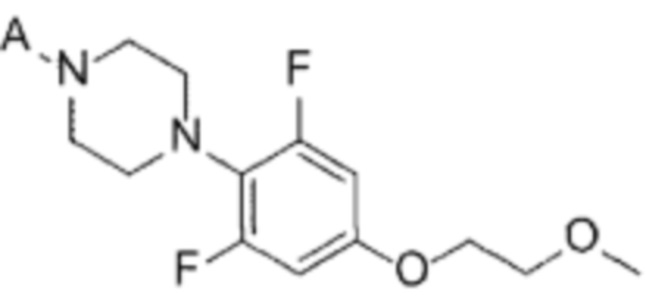
[Химическая формула 8]
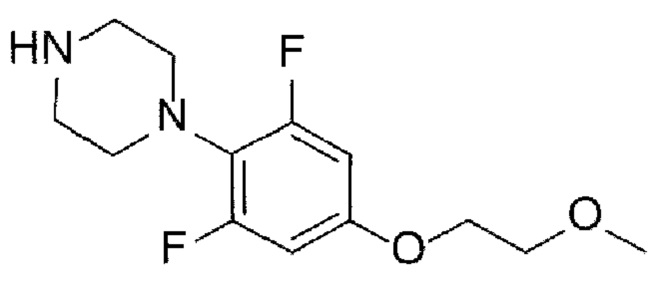
[Химическая формула Ia]
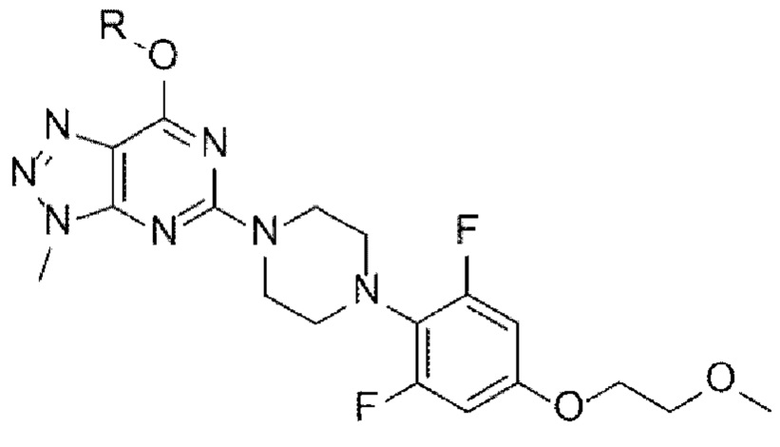
[Химическая формула I]
в указанных химических формулах
R представляет собой O-защитную группу; и
А представляет собой N-защитную группу.
2. Способ по п. 1, отличающийся тем, что R представляет собой C1-С6 алкил, ацетил, бензоил, бензил, п-метоксибензил, метоксиметилацеталь (MOM), тетрагидропиран (ТНР) или простой силиловый эфир.
3. Способ по п. 1, отличающийся тем, что А представляет собой -Boc, -Cbz, -Fmoc, -бензил, п-метоксибензил, тритил или диметокситритил (DMT).
4. Способ по п. 3, отличающийся тем, что А представляет собой -Boc.
5. Способ по п. 1, отличающийся тем, что стадия (А-2) включает взаимодействие по меньшей мере с одним окислителем, выбранным из группы, состоящей из пероксида водорода, пероксида бензоила, мета-хлорпероксибензойной кислоты и оксона.
6. Способ по п. 5, отличающийся тем, что окислитель представляет собой оксон.
7. Способ по п. 1, отличающийся тем, что стадия (А-4) включает взаимодействие по меньшей мере с одним окислителем, выбранным из группы, состоящей из пероксида водорода, персульфата аммония и мета-хлорпероксибензойной кислоты (mCPBA).
8. Способ по п. 7, отличающийся тем, что окислитель представляет собой мета-хлорпероксибензойную кислоту (mCPBA).
9. Способ получения триазолопиримидинонового производного, включающий:
(В-1) Стадию 1 получения соединения, представленного следующей химической формулой 2, посредством реакции защиты из соединения, представленного следующей химической формулой 1, или его соли;
(В-2) Стадию 2 получения соединения, представленного следующей химической формулой 3, посредством реакции окисления из соединения, представленного химической формулой 2;
(В-3) Стадию 3 получения соединения, представленного следующей химической формулой 9, посредством реакции аминирования из соединения, представленного следующей химической формулой 4;
(В-4) Стадию 4 получения соединения, представленного следующей химической формулой 10, посредством реакции аминирования из соединения, представленного химической формулой 9;
(В-5) Стадию 5 получения соединения, представленного следующей химической формулой 11, посредством реакции Дейкина из соединения, представленного химической формулой 10;
(В-6) Стадию 6 получения соединения, представленного следующей химической формулой Ia, посредством реакции алкилирования из соединения, представленного химической формулой 11; и
(В-7) Стадию 7 получения соединения, представленного следующей химической формулой I, посредством реакции снятия защиты с соединения, представленного химической формулой Ia:
[Химическая формула 1]
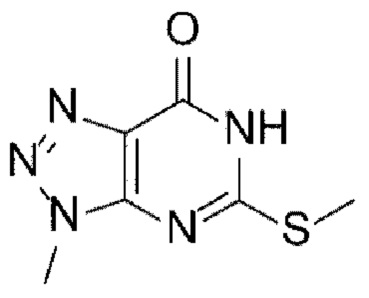
[Химическая формула 2]
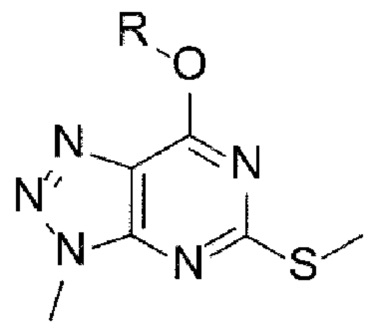
[Химическая формула 3]
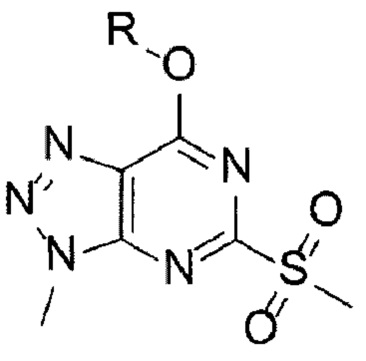
[Химическая формула 4]
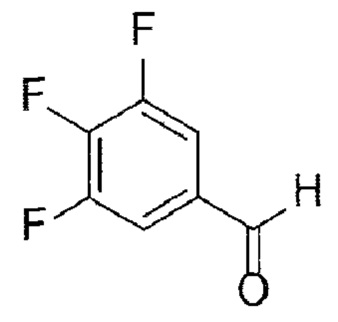
[Химическая формула 9]
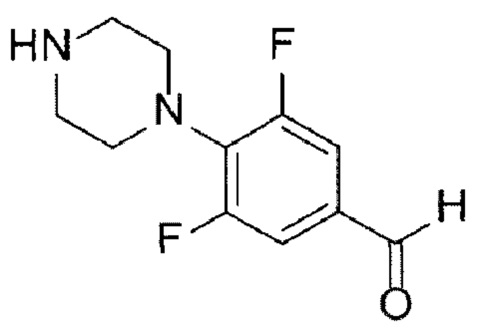
[Химическая формула 10]
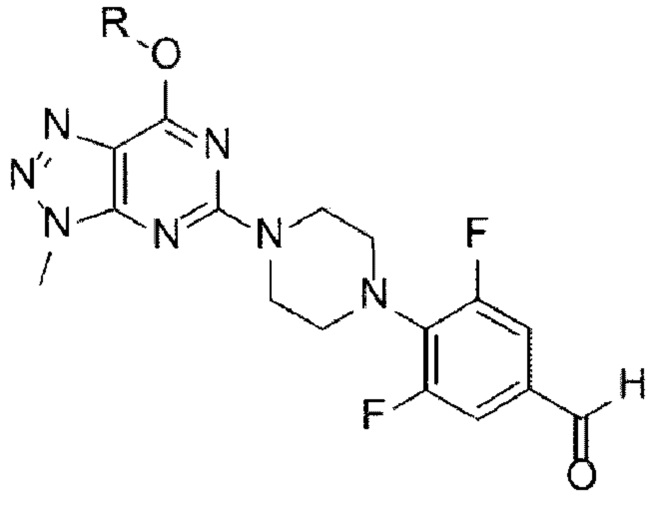
[Химическая формула 11]
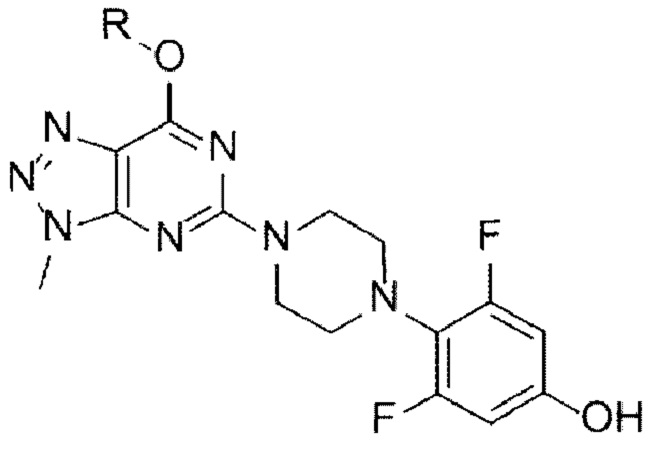
[Химическая формула Ia]
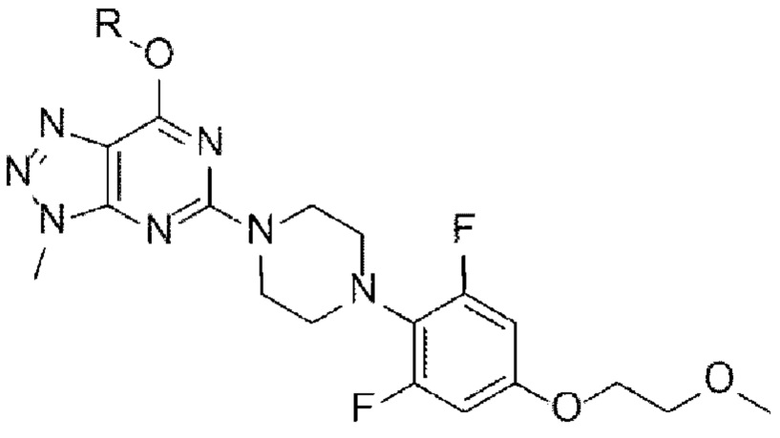
[Химическая формула I]
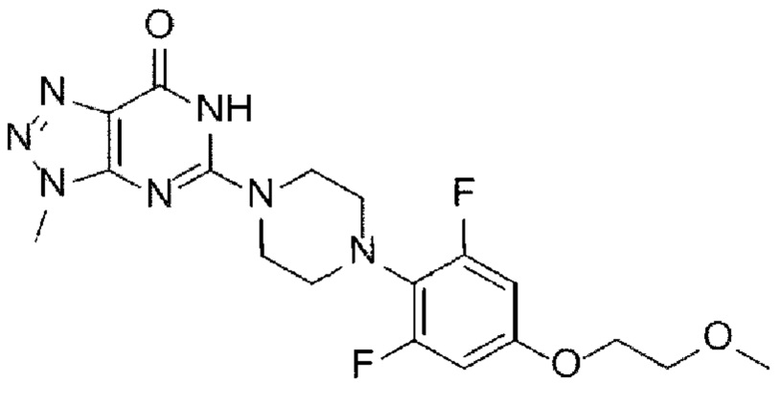
в указанных химических формулах
R представляет собой O-защитную группу.
10. Способ по п. 9, отличающийся тем, что R представляет собой C1-C6 алкил, ацетил, бензоил, бензил, п-метоксибензил, метоксиметилацеталь (MOM), тетрагидропиран (ТГП) или простой силиловый эфир.
| WO 2016006974 A2, 14.01.2016 | |||
| EP 3166945 B1, 09.10.2019 | |||
| WO 2013182580 A1, 12.12.2013 | |||
| RU 2014151009 A, 10.08.2016. |

