ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет на основании китайской заявки на патент на изобретение №202011445339.4, поданной в Национальное управление интеллектуальной собственности Китая 8 декабря 2020 г. и озаглавленной "СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИ-5-[2-(4-(ТРИФТОРМЕТИЛФЕНИЛ)ЭТИЛАМИНО)]БЕНЗОЙНОЙ КИСЛОТЫ", которая включена в настоящий документ во всей полноте посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
[0002] Настоящее изобретение относится к области способов синтеза лекарственных средств и относится к синтезу лекарственного средства против болезни Альцгеймера, в частности к способу получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты.
УРОВЕНЬ ТЕХНИКИ
[0003] 2-Гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойная кислота представляет собой новый ингибитор некроза клеток, разработанный компанией GNT Pharma Co., Ltd., Корея, который может эффективно лечить неврологические заболевания, такие как болезнь Альцгеймера и синдром Паркинсона, и хорошо зарекомендовал себя в клинической практике.
[0004] В настоящее время опубликовано несколько сообщений о способах синтеза указанного соединения, и применяемые процессы являются сложными, сырье является дорогим, а практическая применимость является недостаточной. By Юлян (Wu Yuliang) et al. сообщали о промышленно масштабируемом синтетическом способе (Wu Yuliang, Lu Xin et al., Synthesis of the Alzheimer's Disease Drag 2-hydroxy-5-[2-(4-(trifluoromethylphenyl)ethylamino)]benzoic acid [J], Chinese Journal of New Drugs, 2012, 21 (16): 1930-1932), который обеспечивали путем конденсации и гидролиза 2-(4-трифторметил)фенэтилметансульфоната (соединение 4) с метил-5-аминосалицилатом (соединение 6) с получением целевого продукта, который применяли в крупносерийном производстве.
[0005] Тем не менее, самая большая проблема, связанная с указанным способом, заключается в том, что 2-(4-трифторметил)фенэтилметансульфонат характеризуется низкой стабильностью, низкой температурой плавления (27-28°С) и сложностью рафинирования и очистки. В частности, примесь мета-изомера 2-(3-трифторметил)фенэтилметансульфоната трудно удаляется, а содержание дизамещенной примеси метил-2-гидрокси-5-[N,N-бис(2-(4-(трифторметил)фенил)этил)амино]бензоата из реакции конденсации является высоким, и его гидролизат попадает в конечный продукт, влияя на качество конечного продукта. Поскольку текущие стандарты контроля качества для АФИ (активных фармацевтических ингредиентов) становятся все выше, возникает много проблем с синтезом с применением 2-(4-трифторметил)фенэтилметансульфоната.
[0006] Что касается проблем, связанных с текущим способом, желательно найти подходящее промежуточное соединение для замены 2-(4-трифторметил)фенэтилметансульфоната для решения указанных выше проблем и получения продуктов высокого качества.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[0007] Для преодоления упомянутых выше недостатков в настоящем изобретении предложен способ получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты с высоким выходом и чистотой.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
[0008] В частности, в настоящем изобретении предложен способ получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты, включающий стадии:
[0009] (1) подвергания соединения I реакции введения защиты с применением защитного реагента с получением соединения II,
[0010] 
[0011] (2) подвергания соединения II реакции конденсации с метил-5-аминосалицилатом с получением соединения III,
[0012] 
[0013] (3) подвергания соединения III реакции гидролиза с получением целевого соединения IV,
[0014] 
[0015] где R представляет собой любой из n-метилбензолсульфонила, n-нитробензолсульфонила, бензолсульфонила, трифторметансульфонила и ацетила, предпочтительно n-метилбензолсульфонила или бензолсульфонила, более предпочтительно n-метилбензолсульфонил.
[0016] В указанном выше способе получения реакцию введения защиты проводят при температуре 0-30°С, предпочтительно 20-25°С.
[0017] В указанном выше способе получения реакцию введения защиты проводят в растворителе, который представляет собой по меньшей мере один из толуола, этилбензола, ксилола, метиленхлорида и хлороформа, предпочтительно толуол.
[0018] Предпочтительно в указанном выше способе получения стадия (1) дополнительно включает стадию рафинирования неочищенного соединения П.
[0019] Более предпочтительно в указанном выше способе получения рафинирование осуществляют путем кристаллизации, которую проводят путем растворения при 40-80°С и выделения путем кристаллизации при 0-40°С, предпочтительно путем растворения при 50-60°С и выделения путем кристаллизации при 20-30°С; растворитель, применяемый для кристаллизации, представляет собой по меньшей мере один из этилацетата, н-гексана, н-гептана, толуола, метиленхлорида, метанола, этанола, изопропанола и воды, предпочтительно н-гептан.
[0020] В указанном выше способе получения реакцию конденсации проводят при температуре 30-100°С, предпочтительно 80-90°С.
[0021] В указанном выше способе получения реакцию конденсации проводят в растворителе, который представляет собой по меньшей мере один из толуола, ацетонитрила, N,N-диметилформамида, диметилсульфоксида и тетрагидрофурана, предпочтительно толуол.
[0022] В указанном выше способе получения реакцию конденсации проводят в присутствии основания, которое представляет собой по меньшей мере одно из триэтиламина, N,N-диизопропилэтиламина, 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) и пиридина, предпочтительно триэтиламин.
[0023] Предпочтительно в указанном выше способе получения стадия (2) дополнительно включает стадию образования соли неочищенного соединения III с применением кислоты или ее водного раствора.
[0024] Более предпочтительно в указанном выше способе получения образование соли проводят с применением неорганической кислоты, которая представляет собой любую из хлористоводородной кислоты, серной кислоты, азотной кислоты и фосфорной кислоты, предпочтительно серную кислоту.
[0025] В указанном выше способе получения реакцию гидролиза проводят при температуре 60-100°С, предпочтительно 80-85°С.
[0026] В указанном выше способе получения реакцию гидролиза проводят в присутствии кислоты, которая представляет собой серную кислоту.
[0027] В указанном выше способе получения реакцию гидролиза проводят с барботированием азота.
ТЕХНИЧЕСКИЕ ЭФФЕКТЫ
[0028] Путем применения конкретных защитных реагентов для защиты гидроксигруппы в соединении I (т.е. 2-(4-трифторметил)фенэтиловом спирте) в сочетании с оптимизацией других параметров процесса способ согласно настоящему изобретению позволяет эффективно удалять примеси мета-изомеров на стадии (1) и дизамещенные примеси на стадии (2), таким образом получая 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с высоким выходом и чистотой, тем самым удовлетворяя требования высоких стандартов качества для АФИ при исследовании препаратов.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
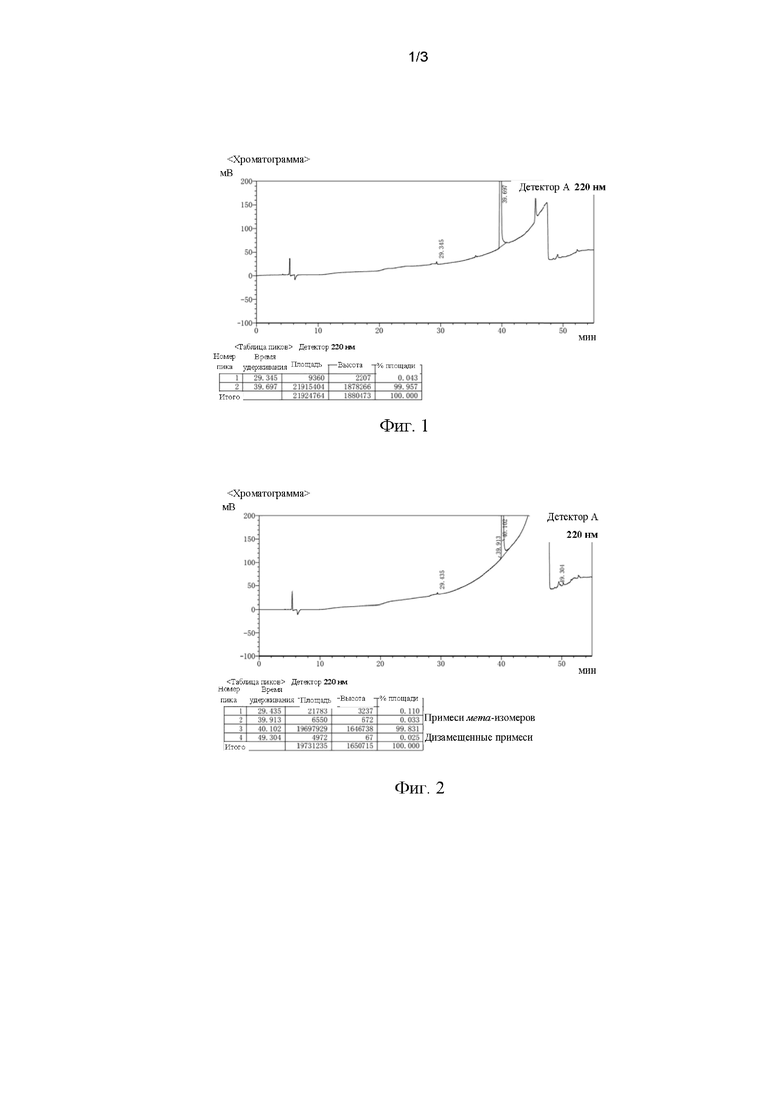
[0029] На фиг. 1 представлена хроматограмма ВЭЖХ целевого продукта, полученного в примере 1.
[0030] На фиг. 2 представлена хроматограмма ВЭЖХ целевого продукта, полученного в примере 2.
[0031] На фиг. 3 представлена хроматограмма ВЭЖХ целевого продукта, полученного в примере 3.
[0032] На фиг. 4 представлена хроматограмма ВЭЖХ целевого продукта, полученного в примере 4.
[0033] На фиг. 5 представлена хроматограмма ВЭЖХ целевого продукта, полученного в примере сравнения.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ
[0034] Объяснение технических отличительных признаков, описанных ниже, основано на репрезентативных вариантах реализации и конкретных примерах настоящего изобретения, но настоящее изобретение не ограничивается указанными вариантами реализации и конкретными примерами.
[0035] Если не указано иное, термин "соединение", применяемый в настоящем описании, включает все стереоизомерные формы, геометрические изомерные формы, таутомерные формы и формы соединения с изотопными метками.
[0036] Если не указано иное, числовой диапазон, представленный в виде "числовое значение А - числовое значение В", применяемый в настоящем описании, относится к диапазону, включающему конечные значения А и В.
[0037] Если не указано иное, числовой диапазон, описанный при помощи модификаторов "выше" или "ниже", применяемый в настоящем описании, относится к числовому диапазону, включающему указанное число.
[0038] Если не указано иное, ссылки в настоящем описании на "некоторые конкретные/предпочтительные варианты реализации", "другие конкретные/предпочтительные варианты реализации", "варианты реализации" и т.п. означают, что конкретные элементы (например, отличительные признаки, структуры, свойства и/или характеристики), описанные в связи с вариантом реализации, включены по меньшей мере в один из вариантов реализации, описанных в настоящем документе, и могут присутствовать или не присутствовать в других вариантах реализации. Кроме того, следует понимать, что элементы можно объединять любым подходящим образом в различных вариантах реализации.
[0039] Если не указано иное, термин "множество", применяемый в настоящем описании, обозначает наличие двух или более элементов, к которым относится указанный термин.
[0040] Далее представлено подробное описание настоящего изобретения.
[0041] В настоящем изобретении предложен способ получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты с высоким выходом и чистотой.
[0042] Способ включает стадии:
[0043] (1) подвергания соединения I реакции введения защиты с применением защитного реагента с получением соединения II,
[0044] 
[0045] (2) подвергания соединения II реакции конденсации с метил-5-аминосалицилатом с получением соединения III,
[0046] 
[0047] (3) подвергания соединения III реакции гидролиза с получением целевого соединения IV, которое представляет собой 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту,
[0048] 
[0049] где R представляет собой любой из n-метилбензолсульфонила, n-нитробензолсульфонила, бензолсульфонила, трифторметансульфонила и ацетила, предпочтительно n-метилбензолсульфонила или бензолсульфонила, более предпочтительно n-метилбензолсульфонил.
[0050] Стадия (1)
[0051] Стадия (1) представляет собой стадию подвергания соединения I (т.е. 2-(4-трифторметил)фенэтилового спирта) реакции введения защиты с применением защитного реагента с получением соединения II (т.е. 2-(4-трифторметил)фенэтилсульфоната или -карбоксилата).
[0052] На стадии (1) выбор защитной группы играет ключевую роль в удалении примесей мета-изомеров на указанной стадии и контролировании дизамещенных примесей на стадии (2).
[0053] В одном варианте реализации настоящего изобретения защитный реагент, применяемый для защиты соединения I на стадии (1), может представлять собой любой из n-метилбензолсульфонилхлорида, трифторметансульфонилхлорида, ацетилхлорида, бензолсульфонилхлорида и n-нитробензолсульфонилхлорида или любой из их соответствующих ангидридов. Соответственно, защитная группа (т.е. группа R) в соединении II может представлять собой любую из n-метилбензолсульфонила, трифторметансульфонила, ацетила, бензолсульфонила и n-нитрофенилсульфонила.
[0054] В предпочтительном варианте реализации настоящего изобретения защитный реагент, применяемый для защиты соединения I на стадии (1), может представлять собой n-метилбензолсульфонилхлорид или бензолсульфонилхлорид.
[0055] В более предпочтительном варианте реализации настоящего изобретения защитный реагент, применяемый для защиты соединения I на стадии (1), может представлять собой n-метилбензолсульфонилхлорид.
[0056] Промежуточный 2-(4-трифторметил)фенэтилметансульфонат, применяемый в уровне техники, характеризуется низкой стабильностью, низкой температурой плавления и сложностью рафинирования и очистки. В частности, примеси изомеров трудно удалять, а содержание дизамещенных примесей в реакции конденсации является высоким, что влияет на качество конечного продукта. Напротив, при применении упомянутых выше защитных реагентов промежуточные соединения, получаемые согласно настоящему изобретению, представляют собой 2-(4-трифторметил)фенэтил-n-метилбензолсульфонат, 2-(4-трифторметил)фенэтилтрифторметансульфонат, 2-(4-трифторметил)фенэтилацетат, 2-(4-трифторметил)фенэтилбензолсульфонат и 2-(4-трифторметил)фенэтил-n-нитробензолсульфонат, соответственно, которые преодолевают упомянутые выше недостатки 2-(4-трифторметил)фенэтилметансульфоната. В результате можно получать конечный продукт 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с высокой чистотой и выходом.
[0057] В одном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) проводят при низкой температуре.
[0058] В предпочтительном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) проводят при 0-30°С.
[0059] В предпочтительном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) проводят при 20-25°С.
[0060] В одном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) можно проводить в растворителе.
[0061] В предпочтительном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) можно проводить в органическом растворителе, который может представлять собой по меньшей мере один из толуола, этилбензола, ксилола, метиленхлорида и хлороформа.
[0062] В предпочтительном варианте реализации настоящего изобретения реакцию введения защиты на стадии (1) можно проводить в толуоле.
[0063] После завершения реакции введения защиты на стадии (1) в систему можно добавлять воду и перемешивать для разделения слоев, а затем отделенную органическую фазу можно концентрировать при пониженном давлении с получением неочищенного соединения II, которое затем рафинируют с получением соединения II высокой чистоты.
[0064] В одном варианте реализации настоящего изобретения стадия (1) может дополнительно включать стадию рафинирования неочищенного соединения II. Как правило, стадию рафинирования можно проводить путем кристаллизации. Выбор растворителя, применяемого для кристаллизации, оказывает большое влияние на удаление мета-изомеров на стадии (1).
[0065] В предпочтительном варианте реализации настоящего изобретения растворитель, применяемый для кристаллизации, может представлять собой по меньшей мере один из этилацетата, н-гексана, н-гептана, толуола, метиленхлорида, метанола, этанола, изопропанола и воды.
[0066] В более предпочтительном варианте реализации настоящего изобретения растворитель, применяемый для кристаллизации, может представлять собой н-гептан.
[0067] Кроме того, при рафинировании неочищенного соединения II путем кристаллизации также можно регулировать температуру кристаллизации для облегчения удаления примесей и осаждения целевого продукта. Если температура кристаллизации является слишком высокой, это влияет на выход, а если температура кристаллизации является слишком низкой, снижается чистота соединения II.
[0068] В одном варианте реализации настоящего изобретения кристаллизацию можно проводить путем растворения при 40-80°С и выделения путем кристаллизации при 0-40°С.
[0069] В предпочтительном варианте реализации настоящего изобретения кристаллизацию можно проводить путем растворения при 50-60°С и выделения путем кристаллизации при 20-30°С.
[0070] Стадия (2)
[0071] Стадия (2) представляет собой стадию подвергания соединения II реакции конденсации с метил-5-аминосалицилатом с получением соединения III (т.е. метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата).
[0072] На стадии (2) температура реакции оказывает большое влияние на дизамещенные примеси, и повышение температуры приводит к значительному повышению содержания дизамещенных примесей.
[0073] В одном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить при температуре 30-100°С.
[0074] В предпочтительном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить при температуре 80-90°С.
[0075] В одном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить в растворителе.
[0076] В предпочтительном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить в органическом растворителе, который может представлять собой по меньшей мере один из толуола, ацетонитрила, N,N-диметилформамида, диметилсульфоксида и тетрагидрофурана.
[0077] В более предпочтительном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить в толуоле.
[0078] В одном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить в присутствии основания, которое может представлять собой по меньшей мере одно из триэтиламина, N,N-диизопропилэтиламина, 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) и пиридина.
[0079] В одном варианте реализации настоящего изобретения реакцию конденсации на стадии (2) можно проводить в присутствии триэтиламина.
[0080] После завершения реакции конденсации на стадии (2) в систему можно добавлять воду и перемешивать для разделения слоев, а затем отделенную органическую фазу можно концентрировать при пониженном давлении с получением неочищенного соединения III.
[0081] Поскольку неочищенное соединение III обычно неустойчиво в твердой форме, и его нельзя непосредственно кристаллизовывать, требуется превращать его в форму соли, которая облегчает получение твердого вещества и достижение разделения и очистки путем кристаллизация.
[0082] В одном варианте реализации настоящего изобретения стадия (2) дополнительно включает стадию образования соли неочищенного соединения III с применением кислоты (или ее раствора, предпочтительно ее водного раствора). Как правило, стадию образования соли можно проводить с применением неорганической или органической кислоты. Обычные неорганические кислоты включают (но не ограничиваются ими) хлористоводородную кислоту, серную кислоту, азотную кислоту и фосфорную кислоту. Обычные органические кислоты включают (но не ограничиваются ими) уксусную кислоту, молочную кислоту, лимонную кислоту, яблочную кислоту и винную кислоту.
[0083] В предпочтительном варианте реализации настоящего изобретения кислота, применяемая для образования соли, может представлять собой любую из хлористоводородной кислоты, серной кислоты, азотной кислоты и фосфорной кислоты или их водный раствор любой концентрации.
[0084] В более предпочтительном варианте реализации настоящего изобретения кислота, применяемая для образования соли, может представлять собой серную кислоту или ее водный раствор любой концентрации.
[0085] Соответственно, когда кислота, применяемая для образования соли, представляет собой серную кислоту, соль присоединения кислоты соединения III предпочтительно представляет собой гемисульфат.
[0086] Стадия (3)
[0087] Стадия (3) представляет собой стадию подвергания соединения III (т.е. метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата) или его соли присоединения кислоты (такой как гемисульфат) реакции гидролиза с получением целевого соединения IV (т.е. 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты).
[0088] В одном варианте реализации настоящего изобретения реакцию гидролиза на стадии (3) можно проводить при температуре 60-100°С.
[0089] В предпочтительном варианте реализации настоящего изобретения реакцию гидролиза на стадии (3) можно проводить при температуре 80-85°С.
[0090] В одном варианте реализации настоящего изобретения реакцию гидролиза на стадии (3) можно проводить в присутствии кислоты, которая может представлять собой серную кислоту, или ее водного раствора любой концентрации.
[0091] В одном варианте реализации настоящего изобретения кислота, применямая для гидролиза, может представлять собой серную кислоту.
[0092] Для повышения чистоты получаемого соединения IV можно дополнительно добавлять активированный уголь для обесцвечивания.
[0093] В одном варианте реализации настоящего изобретения реакцию гидролиза на стадии (3) можно проводить с барботированием азота, что, очевидно, может сокращать время реакции и улучшать степень превращения реакции.
[0094] Кроме того, следует отдельно отметить, что, если не указано иное, конечную точку реакции на каждой стадии в способе получения согласно настоящему изобретению определяют путем ВЭЖХ; сырье в реакции обычно реагирует в соответствии со стехиометрическим отношением химической реакции или в избытке; количество растворителя и/или катализатора в реакции можно регулировать в соответствии с количеством сырья в реакции, в частности, можно повышать при большем количестве сырья в реакции и снижать при меньшем количестве сырья в реакции.
[0095] Для лучшего понимания технических решений согласно настоящему изобретению оно дополнительно описано ниже при помощи конкретных примеров, и специалистам в данной области техники понятно, что настоящее изобретение не ограничивается указанными примерами.
[0096] Пример 1
[0097] (1) Получение 2-(4-трифторметил)фенэтил-n-метилбензолсульфоната:
[0098] 
[0099] В 500 мл мерную колбу вносили толуол (230 г) и n-метилбензолсульфонилхлорид (100 г, 0,53 моль), перемешивали до растворения и отставляли в сторону. В 1 л реакционную колбу вносили толуол (140 г), 40 масс. % водный раствор гидроксида натрия (140 г, 1,4 моль) и 2-(4-трифторметил)фенэтиловый спирт (95 г, 0,5 моль) и охлаждали до 0-10°С. Затем по каплям добавляли толуольный раствор n-метилбензолсульфонилхлорида. Затем проводили реакцию при 20-25°С в течение 6 часов и протекание реакции контролировали путем ГХ. После завершения реакции добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении с получением концентрированного неочищенного продукта. Температуру повышали до 50-60°С и по каплям добавляли н-гептан (170 г). Затем смесь охлаждали до 20-25°С, кристаллизовывали, фильтровали с отсасыванием и сушили с получением сухого продукта целевого соединения (163 г) с чистотой по ВЭЖХ 99,6%, содержанием примесей мета-изомеров 0,03% и выходом 95%.
[00100] (2) Получение гемисульфата метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата:
[00101] 
[00102] В 1 л реакционную колбу вносили 2-(4-трифторметил)фенэтил-n-метилбензолсульфонат (68 г, 0,20 моль), метил-5-аминосалицилат (35 г, 0,21 моль), толуол (200 г) и триэтиламин (24 г, 0,24 моль). Смесь нагревали и выдерживали при 80-90°С, проводили реакцию в течение 14 часов. Реакцию останавливали после отбора образцов и прохождения теста. Затем добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении до полного удаления дистиллята. К смеси добавляли метанол (160 г), перемешивали до растворения, нагревали до 35-40°С и по каплям добавляли 50 масс. % водный раствор серной кислоты (23 г, 0,12 моль). Затем смесь перемешивали в течение 15 минут, медленно охлаждали до 25-30°С, подвергали выделению путем кристаллизации в течение 1 часа, фильтровали с отсасыванием и сушили с получением гемисульфата соединения III (67 г, 0,17 моль) с чистотой по ВЭЖХ 99% и выходом 86,3%.
[00103] (3) Получение 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты:
[00104] 
[00105] В 500 мл реакционную колбу вносили 40 масс. % водный раствор серной кислоты (300 г), ледяную уксусную кислоту (100 г) и гемисульфат соединения III (77 г). Температуру повышали до 80-85°С, при этом в реакционный раствор вводили азот и реакцию барботирования проводили в течение 4 часов. После отбора образцов и прохождения теста добавляли активированный уголь для обесцвечивания и смесь фильтровали. Фильтрат медленно охлаждали до температуры от -5 до 5°С, выдерживали в течение 1 часа, фильтровали с отсасыванием и сушили с получением сухого продукта соединения IV (58,4 г) с чистотой по ВЭЖХ 99,957% и выходом 93%. Не обнаруживали дизамещенные примеси и примеси мета-изомеров (как показано на фиг. 1).
[00106] Пример 2
[00107] (1) Получение 2-(4-трифторметил)фенэтилбензолсульфоната:
[00108] 
[00109] В 500 мл мерную колбу вносили толуол (230 г) и бензолсульфонилхлорид (93 г, 0,53 моль), перемешивали до растворения и отставляли в сторону. В 1 л реакционную колбу вносили толуол (140 г), 40 масс. % водный раствор гидроксида натрия (140 г, 1,4 моль) и 2-(4-трифторметил)фенэтиловый спирт (95 г, 0,5 моль) и охлаждали до 0-10°С.Затем по каплям добавляли толуольный раствор бензолсульфонилхлорида. Затем проводили реакцию при 20-25°С в течение 8 часов и протекание реакции контролировали путем ГХ. После завершения реакции добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении с получением концентрированного неочищенного продукта. Температуру повышали до 50-60°С и по каплям добавляли смешанный растворитель из этилацетата и н-гептана (150 г). Затем смесь охлаждали до 20-25°С, кристаллизовывали, фильтровали с отсасыванием и сушили с получением сухого продукта целевого соединения (145 г, 0,44 моль) с чистотой по ВЭЖХ 98% и выходом 89%.
[00110] (2) Получение гемисульфата метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата:
[00111] 
[00112] В 1 л реакционную колбу вносили 2-(4-трифторметил)фенэтилбензолсульфонат (66 г, 0,20 моль), метил-5-аминосалицилат (35 г, 0,21 моль), толуол (200 г) и 24 г триэтиламина (24 г, 0,24 моль). Смесь нагревали и выдерживали при 90-100°С, проводили реакцию в течение 12 часов. Реакцию останавливали после отбора образцов и прохождения теста. Затем добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении до полного удаления дистиллята. К смеси добавляли метанол (168 г), перемешивали до растворения, нагревали до 35-40°С и по каплям добавляли 50 масс. % водный раствор серной кислоты (23 г, 0,12 моль). Затем смесь перемешивали в течение 15 минут, медленно охлаждали до 25-30°С, подвергали выделению путем кристаллизации в течение 1 часа, фильтровали с отсасыванием и сушили с получением гемисульфата соединения III (62 г) с чистотой по ВЭЖХ 98,5% и выходом 80%.
[00113] (3) Получение 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты:
[00114] В 500 мл реакционную колбу вносили 40 масс. % водный раствор серной кислоты (240 г), ледяную уксусную кислоту (80 г) и гемисульфат соединения III (60 г). Температуру повышали до 80-85°С, при этом в реакционный раствор вводили азот и реакцию барботирования проводили в течение 4 часов. После отбора образцов и прохождения теста добавляли активированный уголь для обесцвечивания и смесь фильтровали. Фильтрат медленно охлаждали до температуры от -5 до 5°С, выдерживали в течение 1 часа, фильтровали с отсасыванием и сушили с получением сухого продукта соединения IV (44,5 г) с чистотой по ВЭЖХ 99,831%, содержанием дизамещенных примесей 0,025%, содержанием примесей мета-изомеров 0,033% (как показано на фиг. 2) и выходом 91%.
[00115] Пример 3
[00116] (1) Получение 2-(4-трифторметил)фенэтилацетата:
[00117] 
[00118] В 500 мл мерную колбу вносили толуол (230 г) и ацетилхлорид (41,6 г, 0,53 моль), перемешивали до растворения и отставляли в сторону. В 1 л реакционную колбу вносили толуол (140 г), 40 масс. % водный раствор гидроксида натрия (140 г, 1,4 моль) и 2-(4-трифторметил)фенэтиловый спирт (95 г, 0,5 моль) и охлаждали до 0-10°С.Затем по каплям добавляли толуольный раствор ацетилхлорида. Затем проводили реакцию при 20-25°С в течение 8 часов и протекание реакции контролировали путем ГХ. После завершения реакции добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении с получением концентрированного неочищенного продукта. Температуру повышали до 50-60°С и по каплям добавляли смешанный растворитель из этилацетата и н-гептана (150 г). Затем смесь охлаждали до 20-25°С, кристаллизовывали, фильтровали с отсасыванием и сушили с получением сухого продукта целевого соединения (93 г, 0,4 моль) с чистотой по ВЭЖХ 98% и выходом 80%.
[00119] (2) Получение гемисульфата метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата:
[00120] 
[00121] В 1 л реакционную колбу вносили 2-(4-трифторметил)фенэтилацетат (46,4 г, 0,20 моль), метил-5-аминосалицилат (35 г, 0,21 моль), толуол (200 г) и 24 г триэтиламина (24 г, 0,24 моль). Смесь нагревали и выдерживали при 90-100°С, проводили реакцию в течение 12 часов. Реакцию останавливали после отбора образцов и прохождения теста. Затем добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении до полного удаления дистиллята. К смеси добавляли метанол (168 г), перемешивали до растворения, нагревали до 35-40°С и по каплям добавляли 50 масс. % водный раствор серной кислоты (23 г, 0,12 моль). Затем смесь перемешивали в течение 15 минут, медленно охлаждали до 25-30°С, подвергали выделению путем кристаллизации в течение 1 часа, фильтровали с отсасыванием и сушили с получением гемисульфата соединения III (60 г, 0,15 моль) с чистотой по ВЭЖХ 98,5% и выходом 77%.
[00122] (3) Получение 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты:
[00123] В 500 мл реакционную колбу вносили 40 масс. % водный раствор серной кислоты (240 г), ледяную уксусную кислоту (80 г) и гемисульфат соединения III (60 г). Температуру повышали до 80-85°С, при этом в реакционный раствор вводили азот и реакцию барботирования проводили в течение 4 часов, После отбора образцов и прохождения теста добавляли активированный уголь для обесцвечивания и смесь фильтровали. Фильтрат медленно охлаждали до температуры от -5 до 5°С, выдерживали в течение 1 часа, фильтровали с отсасыванием и сушили с получением сухого продукта соединения IV (40 г) с чистотой по ВЭЖХ 99,639%, содержанием дизамещенных примесей 0,128%, содержанием примесей мета-изомеров 0,097% (как показано на фиг. 3) и выходом 82%.
[00124] Пример 4
[00125] (1) Получение 2-(4-трифторметил)фенэтилтрифторметансульфоната:
[00126] 
[00127] В 500 мл мерную колбу вносили толуол (230 г) и ангидрид трифторметансульфокислоты (150 г, 0,53 моль), перемешивали до растворения и отставляли в сторону. В 1 л реакционную колбу вносили толуол (140 г), триэтиламин (64 г, 0,63 моль) и 2-(4-трифторметил)фенилэтиловый спирт (95 г, 0,5 моль) и охлаждали до 0-10°С. Затем по каплям добавляли толуольный раствор ангидрида трифторметансульфокислоты. Затем проводили реакцию при 20-25°С в течение 8 часов и протекание реакции контролировали путем ГХ. После завершения реакции добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении с получением концентрированного неочищенного продукта. Температуру повышали до 50-60°С и по каплям добавляли смешанный растворитель из этилацетата и н-гептана (150 г). Затем смесь охлаждали до 20-25°С, кристаллизовывали, фильтровали с отсасыванием и сушили с получением сухого продукта целевого соединения (129 г, 0,4 моль) с чистотой по ВЭЖХ 98,5% и выходом 80%.
[00128] (2) Получение гемисульфата метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата:
[00129] 
[00130] В 1 л реакционную колбу вносили 2-(4-трифторметил)фенэтилтрифторметансульфонат (64,5 г, 0,20 моль), метил-5-аминосалицилат (35 г, 0,21 моль), толуол (200 г) и 24 г триэтиламина (24 г, 0,24 моль). Смесь нагревали и выдерживали при 90-100°С, проводили реакцию в течение 12 часов. Реакцию останавливали после отбора образцов и прохождения теста. Затем добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении до полного удаления дистиллята. К смеси добавляли метанол (168 г), перемешивали до растворения, нагревали до 35-40°С и по каплям добавляли 50 масс. % водный раствор серной кислоты (23 г, 0,12 моль). Затем смесь перемешивали в течение 15 минут, медленно охлаждали до 25-30°С, подвергали выделению путем кристаллизации в течение 1 часа, фильтровали с отсасыванием и сушили с получением гемисульфата соединения III (61 г) с чистотой по ВЭЖХ 98,5% и выходом 78,3%.
[00131] (3) Получение 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты:
[00132] В 500 мл реакционную колбу вносили 40 масс. % водный раствор серной кислоты (240 г), ледяную уксусную кислоту (80 г) и гемисульфат соединения III (60 г). Температуру повышали до 80-85°С, при этом в реакционный раствор вводили азот и реакцию барботирования проводили в течение 4 часов. После отбора образцов и прохождения теста добавляли активированный уголь для обесцвечивания и смесь фильтровали. Фильтрат медленно охлаждали до температуры от -5 до 5°С, выдерживали в течение 1 часа, фильтровали с отсасыванием и сушили с получением сухого продукта соединения IV (40,5 г) с чистотой по ВЭЖХ 99,762%, содержанием дизамещенных примесей 0,023%, содержанием примесей мета-изомеров 0,068% (как показано на фиг. 4) и выходом 83%.
[00133] Пример сравнения
[00134] (1) Получение 2-(4-трифторметил)фенэтилметилсульфоната:
[00135] 
[00136] В 500 мл мерную колбу вносили толуол (230 г) и метилсульфонилхлорид (60 г, 0,53 моль), перемешивали до растворения и отставляли в сторону. В 1 л реакционную колбу вносили толуол (140 г), 40 масс. % водный раствор гидроксида натрия (140 г, 1,4 моль) и 2-(4-трифторметил)фенэтиловый спирт (95 г, 0,5 моль) и охлаждали до 0-10°С. Затем по каплям добавляли толуольный раствор метилсульфонилхлорида. Затем проводили реакцию при 20-25°С в течение 10 часов и протекание реакции контролировали путем ГХ. После завершения реакции добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении, а затем добавляли н-гексан (200 г). Затем смесь охлаждали до температуры от -10 до 0°С, кристаллизовывали, фильтровали с отсасыванием и сушили в вакууме при 0-10°С с получением сухого продукта целевого соединения (116 г, 0,43 моль) с чистотой по ВЭЖХ 97%, содержанием мета-изомеров 0,3% и выходом 86%.
[00137] (2) Получение гемисульфата метил-2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензоата:
[00138] 
[00139] В 1 л реакционную колбу вносили 2-(4-трифторметил)фенэтилметилсульфонат (53,6 г, 0,20 моль), метил-5-аминосалицилат (35 г, 0,21 моль), толуол (200 г) и триэтиламин (24 г, 0,24 моль). Смесь нагревали и выдерживали при 80-90°С, проводили реакцию в течение 18 часов. Реакцию останавливали после отбора образцов и прохождения теста. Затем добавляли питьевую воду и смесь перемешивали для разделения слоев. Органическую фазу концентрировали при пониженном давлении до полного удаления дистиллята. К смеси добавляли метанол (160 г), перемешивали до растворения, нагревали до 35-40°С и по каплям добавляли 50 масс. % водный раствор серной кислоты (23 г, 0,12 моль). Затем смесь перемешивали в течение 15 минут, медленно охлаждали до 25-30°С, подвергали выделению путем кристаллизации в течение 1 часа, фильтровали с отсасыванием и сушили с получением гемисульфата соединения III (57 г) с чистотой по ВЭЖХ 97,3%, содержанием мета-изомеров 0,24%, содержанием дизамещенных примесей 1,8% и выходом 73%.
[00140] (3) Получение 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты:
[00141] В 500 мл реакционную колбу вносили 40% водный раствор серной кислоты (300 г), ледяную уксусную кислоту (100 г) и соединение III (77 г). Температуру повышали до 80-95°С и смесь выдерживали в течение 24 часов. После отбора образцов и прохождения теста добавляли активированный уголь для обесцвечивания и смесь фильтровали. Фильтрат медленно охлаждали до температуры от -5 до 5°С, выдерживали в течение 1 часа, фильтровали с отсасыванием и сушили с получением сухого продукта соединения IV (52 г) с чистотой по ВЭЖХ 99,388%, содержанием дизамещенных примесей 0,285%, содержанием примесей мета-изомеров 0,155% (как показано на фиг. 5) и выходом 81%.
[00142] В приведенном выше примере 1 при применении n-метилбензолсульфонилхлорида в качестве защитного реагента (т.е. при применении n-метилбензолсульфонила в качестве защитной группы) в конечном продукте не обнаруживали дизамещенные примеси и примеси мета-изомеров и, таким образом, получали 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с очень высокой чистотой 99,957% и высоким выходом 93%.
[00143] В приведенном выше примере 2 при применении бензолсульфонилхлорида в качестве защитного реагента (т.е. при применении бензолсульфонила в качестве защитной группы) в конечном продукте обнаруживали дизамещенные примеси и примеси мета-изомеров с содержанием менее 0,04% и, таким образом, получали 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с высокой чистотой 99,831% и выходом 91%.
[00144] В приведенном выше примере 3 при применении ацетилхлорида в качестве защитного реагента (т.е. при применении ацетила в качестве защитной группы) в конечном продукте обнаруживали дизамещенные примеси и примеси мета-изомеров с содержанием менее 0,13% и, таким образом, получали 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с высокой чистотой 99,639% и выходом 82%.
[00145] В приведенном выше примере 4 при применении ангидрида трифторметансульфокислоты в качестве защитного реагента (т.е. при применении трифторметансульфонила в качестве защитной группы) в конечном продукте обнаруживали дизамещенные примеси и примеси мета-изомеров с содержанием менее 0,07% и, таким образом, получали 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойную кислоту с высокой чистотой 99,762% и выходом 83%.
[00146] Тем не менее, в примере сравнения из-за применения метилсульфонилхлорида в качестве защитного реагента образовывался промежуточный 2-(4-трифторметил)фенэтилметилсульфонат с низкой стабильностью, что приводило к получению конечного продукта с содержанием дизамещенных примесей 0,285%, содержанием примесей мета-изомеров 0,155%, чистотой только 99,388% и выходом только 81%.
[00147] Как видно из приведенного выше сравнения, за счет применения конкретных защитных реагентов в настоящем изобретении эффективно контролируют содержание примесей изомеров и дизамещенных примесей в конечном продукте 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоте, значительно повышают качество продукта, а также значительно повышают выход реакции.
[00148] Хотя настоящее изобретение описано выше при помощи конкретных вариантов реализации, объем защиты настоящего изобретения не ограничивается указанными вариантами реализации. Все изменения или эквивалентные замены, которые не выходят за рамки концепции настоящего изобретения, включены в объем защиты настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2-ЦИАН-3-ГИДРОКСИ-N-(ФЕНИЛ) БУТ-2-ЕНАМИДОВ | 2004 |
|
RU2330838C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ТЕРИФЛУНОМИДА | 2016 |
|
RU2722316C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ГИДРОКСИ-3-[4-[[[2-(2-МЕТИЛ-1Н-ИНДОЛ-3-ИЛ)ЭТИЛ]АМИНО]МЕТИЛ]ФЕНИЛ]-2Е-2-ПРОПЕНАМИДА И ИСХОДНЫХ МАТЕРИАЛОВ ДЛЯ НЕГО | 2007 |
|
RU2448090C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗ | 2008 |
|
RU2445309C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОНИКОТИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ VEGF-РЕЦЕПТОРА ТИРОЗИНКИНАЗЫ | 2001 |
|
RU2296124C2 |
| 1,3,4-ОКСАДИАЗОЛ-2-ОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ PPAR-ДЕЛЬТА И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2375358C2 |
| СПОСОБ ДЛЯ ПОЛУЧЕНИЯ (5S)-4-[5-(3,5-ДИХЛОРФЕНИЛ)-5-(ТРИФТОРМЕТИЛ)-4H-ИЗОКСАЗОЛ-3-ИЛ]-2-МЕТИЛ-БЕНЗОЙНОЙ КИСЛОТЫ | 2019 |
|
RU2809762C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ NPY | 2004 |
|
RU2321586C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОКСЕТИНА | 1997 |
|
RU2173679C2 |


Настоящее изобретение относится к области технологии лекарственных средств, к синтезу лекарственного средства против болезни Альцгеймера, конкретно к способу получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты. Способ включает такие стадии, как введение защиты соединения I с получением соединения II, дальнейшую конденсацию соединения II с метил-5-аминосалицилатом с получением соединения III и его последующий гидролиз с получением желаемого соединения IV, где R представляет собой любой из n-метилбензолсульфонила, n-нитробензолсульфонила, бензолсульфонила, трифторметансульфонила и ацетила. Техническим результатом изобретения является повышение качества и выхода целевого продукта. 12 з.п. ф-лы, 5 ил., 5 пр.




1. Способ получения 2-гидрокси-5-[2-(4-(трифторметилфенил)этиламино)]бензойной кислоты, включающий стадии:
1) подвергания соединения I реакции введения защиты с применением защитного реагента с получением соединения II,

2) подвергания соединения II реакции конденсации с метил-5-аминосалицилатом с получением соединения III,

3) подвергания соединения III реакции гидролиза с получением целевого соединения IV,

где R представляет собой любой из n-метилбензолсульфонила, n-нитробензолсульфонила, бензолсульфонила, трифторметансульфонила и ацетила, предпочтительно n-метилбензолсульфонила или бензолсульфонила, более предпочтительно n-метилбензолсульфонил.
2. Способ по п. 1, отличающийся тем, что
указанную реакцию введения защиты проводят при температуре 0-30°С, предпочтительно 20-25°С.
3. Способ по п. 1, отличающийся тем, что
указанную реакцию введения защиты проводят в растворителе, который представляет собой по меньшей мере один из толуола, этилбензола, ксилола, метиленхлорида и хлороформа, предпочтительно толуол.
4. Способ по п. 1, отличающийся тем, что
стадия 1) дополнительно включает стадию рафинирования неочищенного соединения II.
5. Способ по п. 4, отличающийся тем, что
указанное рафинирование осуществляют путем кристаллизации, которую проводят путем растворения при 40-80°С и выделения путем кристаллизации при 0-40°С, предпочтительно путем растворения при 50-60°С и выделения путем кристаллизации при 20-30°С;
указанный растворитель, применяемый для кристаллизации, представляет собой по меньшей мере один из этилацетата, н-гексана, н-гептана, толуола, метиленхлорида, метанола, этанола, изопропанола и воды, предпочтительно н-гептан.
6. Способ по п. 1, отличающийся тем, что
указанную реакцию конденсации проводят при температуре 30-100°С, предпочтительно 80-90°С.
7. Способ по п. 1, отличающийся тем, что
указанную реакцию конденсации проводят в растворителе, который представляет собой по меньшей мере один из толуола, ацетонитрила, N,N-диметилформамида, диметилсульфоксида и тетрагидрофурана, предпочтительно толуол.
8. Способ по п. 1, отличающийся тем, что
указанную реакцию конденсации проводят в присутствии основания, которое представляет собой по меньшей мере одно из триэтиламина, N,N-диизопропилэтиламина, 1,8-диазабицикло[5.4.0]ундец-7-ена и пиридина, предпочтительно триэтиламин.
9. Способ по п. 1, отличающийся тем, что
стадия 2) дополнительно включает стадию образования соли неочищенного соединения III с применением кислоты или ее водного раствора.
10. Способ по п. 9, отличающийся тем, что
указанное образование соли проводят с применением неорганической кислоты, которая представляет собой любую из хлористоводородной кислоты, серной кислоты, азотной кислоты и фосфорной кислоты, предпочтительно серную кислоту.
11. Способ по п. 1, отличающийся тем, что
указанную реакцию гидролиза проводят при температуре 60-100°С, предпочтительно 80-85°С.
12. Способ по п. 1, отличающийся тем, что
указанную реакцию гидролиза проводят в присутствии кислоты, которая представляет собой серную кислоту.
13. Способ по п. 1, отличающийся тем, что
указанную реакцию гидролиза проводят с барботированием азота.
| WU, Yuliang et al | |||
| Chinese Jornal of New Drugs, v | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| Русская печь | 1925 |
|
SU1930A1 |
| СПОСОБ ПЕРЕДЕЛА РУДНОГО СЫРЬЯ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2002 |
|
RU2215050C1 |
| KR 20110007741 A, 25.01.2011. | |||

