Область техники
Настоящее изобретение относится к конъюгированному лекарственному средству на основе трициклического полипептида и его применению, в частности, к соединению, представленному формулой (III), или его фармацевтически приемлемой соли.
Уровень техники
Нектин-4 (белок 4, подобный рецептору полиовирусов, PVRL4) в последние годы является новой появившейся опухолеассоциированной мишенью и принадлежит к семейству белков нектинов, которое включает четыре основных подтипа, т.е. нектины 1-4. Вместе с нектиноподобной молекулой (Necl) нектин-4 представляет собой иммуноглобулиноподобную молекулу клеточной адгезии, играя существенную роль в образовании и поддержании межклеточной адгезии и плотных контактов. Нектины-1, -2 и -3 широко экспрессируются в нормальных тканях человека, а нектин-4 главным образом экспрессируется на высоком уровне в эмбрионах и плаценте, но у взрослых людей уровень его экспрессии значительно снижается. Исследования показывают, что нектин-4 сверхэкспрессируется в различных опухолях, как, например, при раке мочевого пузыря, уротелиальном раке, раке молочной железы, трижды негативном раке молочной железы, раке легкого, раке желудка, раке пищевода и т.д., что делает его потенциальной мишенью для лечения соответствующих форм рака. На сегодняшний момент биологический конъюгат антитело-лекарственное средство энфортумаб ведотин, разработанный для этой мишени, был одобрен для продажи в США в 2019 г. Основным показанием для его применения в качестве единственного зарегистрированного для продажи лекарственного средства для данной мишени является метастатический уротелиальный рак, при этом также проводятся исследования клинической эффективности для нескольких показаний. Таким образом, разработка химиотерапевтических лекарственных средств, нацеленных на нектин-4, имеет обширные перспективы применения.
Сущность изобретения
В настоящем изобретении предусмотрено соединение, представленное формулой (III), или его фармацевтически приемлемая соль,
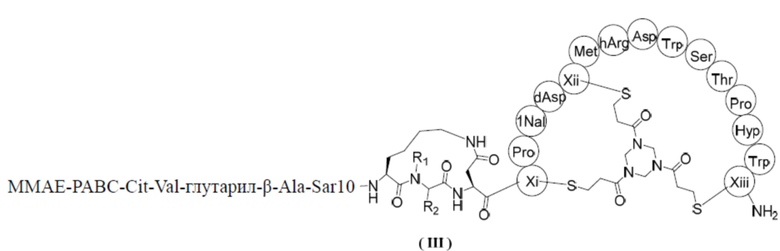 ,
,
где R1 выбран из H и C1-3алкила;
R2 выбран из H, C1-4алкила и 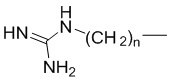 ;
;
n выбран из 1, 2, 3 и 4; и
каждый из Xi, Xii и Xiii независимо выбран из Cys, hCys, βCys и Pen;
ММAE - монометилауристатин Е;
PABC - 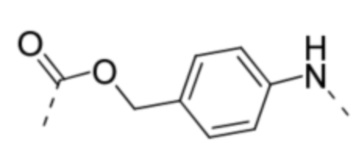 .
.
В некоторых вариантах настоящего изобретения вышеуказанное соединение выбрано из структур, представленных формулами (III-1) и (III-2),
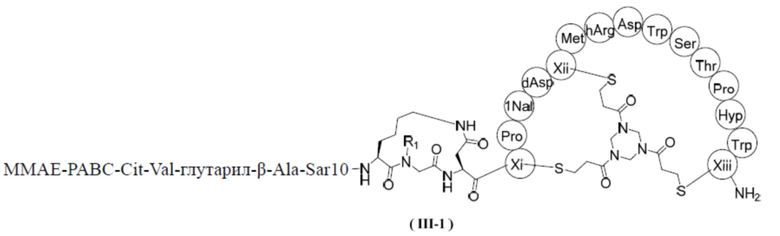 ,
, 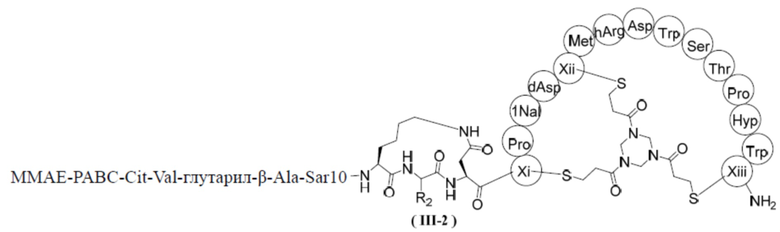 ,
,
где R1, R2, Xi, Xii и Xiii определены в настоящем изобретении.
В настоящем изобретении дополнительно предусмотрено соединение или его фармацевтически приемлемая соль, выбранное из
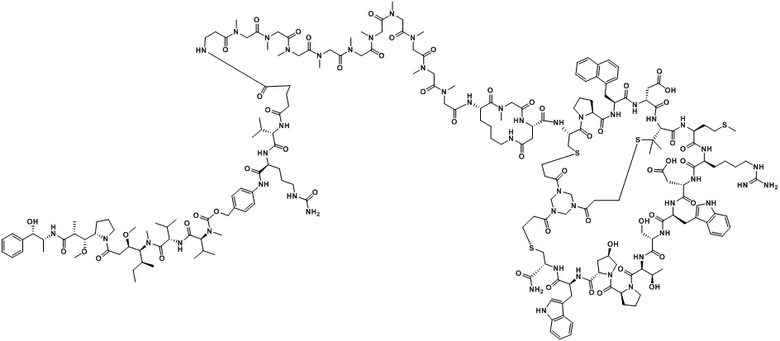 ,
,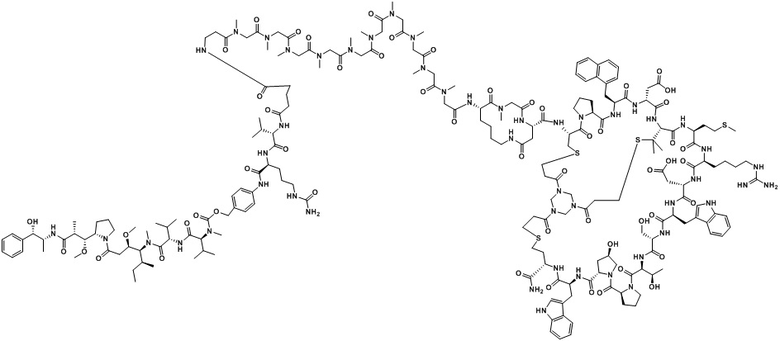 ,
,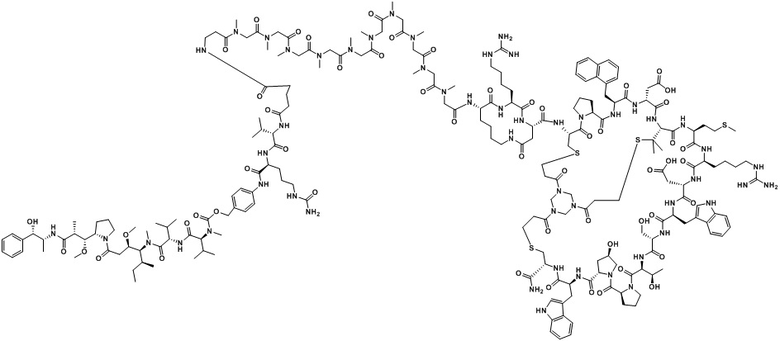 .
.
В настоящем изобретении дополнительно предусмотрена фармацевтическая композиция, содержащая терапевтически или профилактически эффективное количество соединения или его фармацевтически приемлемой соли по настоящему изобретению, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
В настоящем изобретении дополнительно предусмотрено применение соединения или его фармацевтически приемлемой соли по настоящему изобретению, а также фармацевтической композиции по настоящему изобретению, в получении лекарственного средства для лечения солидных опухолей, в которых сверхэкспрессируется нектин-4.
В настоящем изобретении дополнительно предусмотрен способ лечения солидных опухолей, в которых сверхэкспрессируется нектин-4, включающий введение субъекту соединения или его фармацевтически приемлемой соли по настоящему изобретению.
В настоящем изобретении дополнительно предусмотрен способ лечения солидных опухолей, в которых сверхэкспрессируется нектин-4, включающий введение субъекту фармацевтической композиции по настоящему изобретению.
В настоящем изобретении дополнительно предусмотрен продукт, содержащий соединение или его фармацевтически приемлемую соль по настоящему изобретению, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
В настоящем изобретении дополнительно предусмотрен продукт, содержащий фармацевтическую композицию по настоящему изобретению, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
В настоящем изобретении дополнительно предусмотрен следующий способ получения:
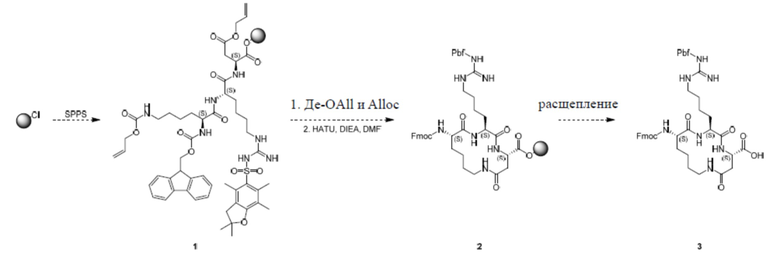
В настоящем изобретении дополнительно предусмотрен следующий способ тестирования.
Способ тестирования 1. Тестирование способности к связыванию соединений по настоящему изобретению с белком нектином-4
1. Цель эксперимента
Протестировать сродство соединения, подлежащего тестированию, к белку-мишени нектину-4 с использованием способа SPR.
2. Материалы и приборы
Biacore 8K (GE Healthcare)
96-луночный планшет (кат. № 650101, Greiner Bio-One)
Чип CM5 (кат. № BR-1005-30, GE Healthcare)
Набор для иммобилизации по аминогруппе (кат. № BR-1000-50, GE Healthcare)
EDC
NHS
1 М этаноламина
10 мМ ацетата натрия, pH 4,5 (кат. № BR-1003-50, GE Healthcare)
DMSO (кат. № D4540, Sigma)
P20 (кат. № BR-1000-54, GE Healthcare)
PBS (кат. № BR-1006-72, GE Healthcare)
Нектин-4 (кат. № 1006-72, GE Healthcare)
3. Протокол эксперимента
В этом эксперименте применяется способ иммобилизации по аминогруппе. Белок-мишень нектин-4 непосредственно иммобилизуют на чипе CM5 с использованием Biacore 8K. Соединение, подлежащее тестированию в качестве аналита, затем разбавляют буферным раствором (10 мМ PBS, pH 7,4, 137 мМ NaCl, 2,7 мМ KCl, 5% DMSO и 0,05% P20) до необходимого градиента концентрации для многоциклового кинетического выявления, в котором каждый цикл состоит из ввода пробы в течение 180 секунд и диссоциации в течение 180 секунд, а затем выполняется следующий цикл, для получения данных кинетического анализа сродства белка-мишени нектина-4 и соединения, подлежащего тестированию. С помощью программного обеспечения для оценки Biacore Insight (версия 2.0.15.12933) окончательные данные подвергают кинетическому анализу соответствия на основе модели 1:1.
4. Методика и процедура эксперимента
1) Получение буферного раствора: 10 мМ PBS, pH 7,4, 137 мМ NaCl, 2,7 мМ KCl, 5% DMSO и 0,05% P20.
2) Активация чипа CM5: чип CM5 активируют с помощью 400 мМ EDC и 100 мМ NHS при скорости потока 10 мкл/мин в течение 420 секунд.
3) Связывание белка-мишени: белок-мишень разбавляют до 10 мкг/мл с помощью 10 мМ ацетата натрия (pH 4,5) и связывают его при скорости потока 10 мкл/мин в течение 284 с. В эксперименте используются каналы №1, №2 и №3 на чипе, и результаты связывания составляют 1639,9 RU, 1747,8 RU и 1702,2 RU соответственно.
4) Блокирование чипа CM5: чип CM5 блокируют с помощью 1 M этаноламина при скорости потока 10 мкл/мин в течение 420 секунд.
5) Приготовление концентраций аналита: соединение, подлежащее тестированию, разбавляют с помощью буферного раствора. Соединение, подлежащее тестированию, разбавляют от 100 нМ до 0,78 нМ с 2-кратным градиентом.
6) Анализ вводимой пробы: каждая концентрация рабочего раствора соединения, подлежащего тестированию, соответствует одному циклу при скорости потока 30 мкл/мин для связывания в течение 180 секунд и диссоциации в течение 180 секунд. Последний цикл представляет собой цикл поправки на 5% растворитель DMSO.
7) Все результаты подвергают кинетическому анализу соответствия на основе модели 1:1.
ТЕХНИЧЕСКИЙ ЭФФЕКТ
Соединения по настоящему изобретению обладают сильным эффектом связывания с нектином-4 и выраженной активностью противодействия пролиферации опухолевых клеток in vitro; проявляют сильный противоопухолевый эффект в модели подкожной опухоли у мышей in vivo и обладают превосходной метаболической стабильностью и PK-свойствами in vitro.
Краткое описание графических материалов
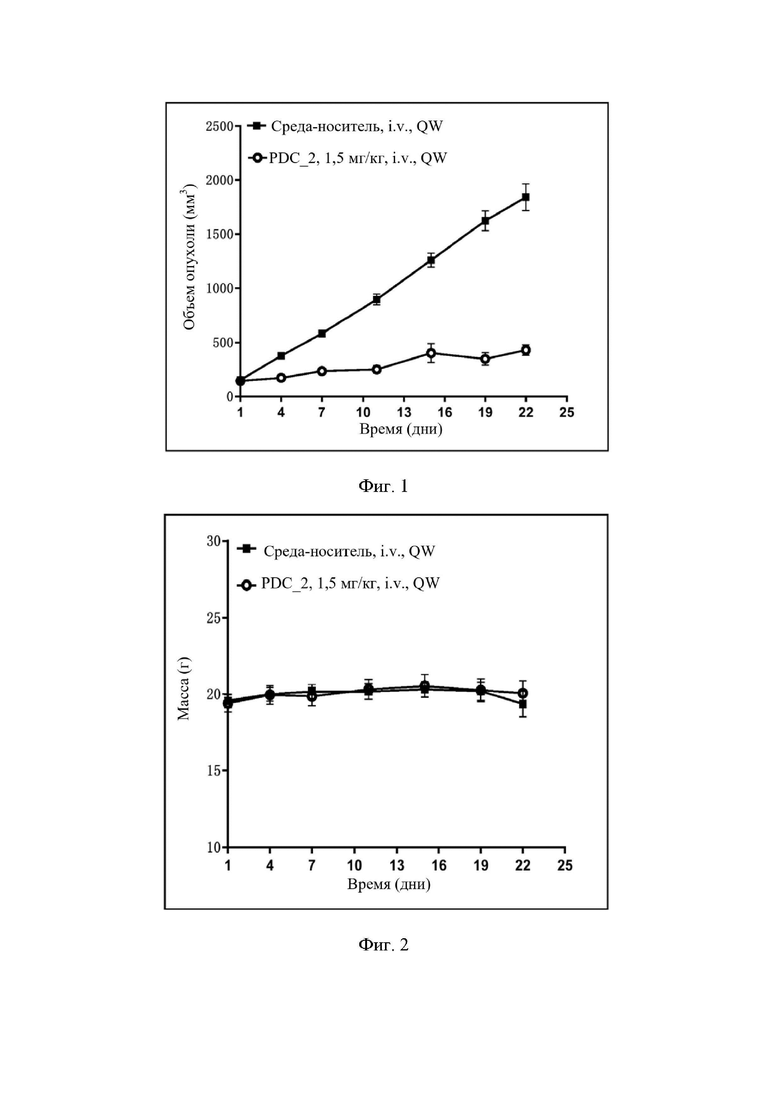
На фиг. 1 показана кривая роста опухоли с соединением по настоящему изобретению в ксенотрансплантатной модели подкожной опухоли из раковых клеток легкого человека NCI-H292.
На фиг. 2 показана кривая изменения массы животных с соединением по настоящему изобретению в ксенотрансплантатной модели подкожной опухоли из раковых клеток легкого человека NCI-H292.
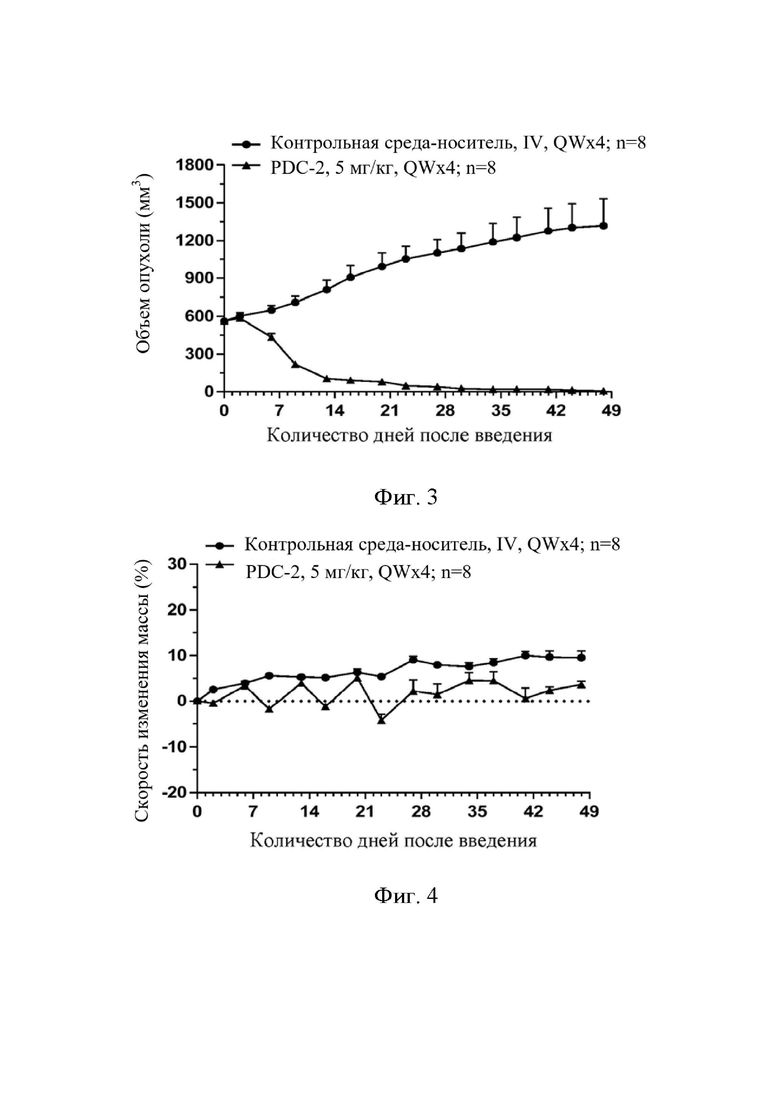
На фиг. 3 показана кривая роста опухоли с соединением по настоящему изобретению, вводимым в группах, когда средний объем опухоли достигает примерно 500-600 мм3, в ксенотрансплантатной модели подкожной опухоли из раковых клеток молочной железы человека MDA-MB-468 на «голых» мышах BALB/c.
На фиг. 4 показана скорость изменения массы животных с соединением по настоящему изобретению, вводимым в группах, когда средний объем опухоли достигает примерно 500-600 мм3, в ксенотрансплантатной модели подкожной опухоли из раковых клеток молочной железы человека MDA-MB-468 на «голых» мышах BALB/c.
Определения и описание
Если не указано иное, предполагается, что следующие термины и фразы, используемые в данном документе, имеют следующие значения. Если не указано иное, конкретный термин или фраза не должны считаться неопределенными или неясными, но должны трактоваться в соответствии с общепринятым значением. Если в данном документе встречаются торговые наименования, то предполагается, что они относятся к их соответствующим товарам или их активным ингредиентам.
Термин «фармацевтически приемлемый», используемый в данном документе, относится к таким соединениям, материалам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения подходят для применения в контакте с тканями людей и животных без излишней токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным отношением польза/риск.
Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, полученной из соединения с помощью конкретных заместителей, раскрытых в настоящем изобретении, и относительно нетоксичной кислоты или основания. Если соединения по настоящему изобретению содержат относительно кислые функциональные группы, соли присоединения основания могут быть получены путем приведения таких соединений в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, органических аминов или магния или подобные соли. Если соединения по настоящему изобретению содержат относительно основные функциональные группы, соли присоединения кислоты могут быть получены путем приведения таких соединений в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Определенные конкретные соединения по настоящему изобретению содержат основные и кислые функциональные группы и, таким образом, могут быть превращены в любую соль присоединения основания или кислоты.
Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы традиционными химическими способами из исходного соединения, содержащего кислотный радикал или основную группу. Как правило, такие соли получают путем осуществления реакции этих соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе или смеси их обоих.
Термин «фармацевтически приемлемое вспомогательное вещество» относится к инертному материалу, который вводится вместе с активным ингредиентом и способствует введению активного ингредиента, включающему без ограничения любое вещество, способствующее скольжению, подсластитель, разбавитель, консервант, краситель/красящее вещество, усилитель вкуса, поверхностно-активное вещество, смачивающее средство, диспергирующее средство, разрыхлитель, суспендирующее средство, стабилизатор, изотоническое средство, растворитель или эмульгатор, одобренные Государственным управлением по контролю качества пищевых продуктов и лекарственных средств для применения у человека или животных (например, домашних животных). Неограничивающие примеры вспомогательного вещества включают карбонат кальция, фосфат кальция, различные сахара и крахмалы, производные целлюлозы, желатин, растительное масло и полиэтиленгликоль.
Термин «фармацевтическая композиция» относится к смеси из одного или более соединений по настоящему изобретению или их солей и фармацевтически приемлемых вспомогательных веществ. Целью фармацевтической композиции является способствование введению соединений по настоящему изобретению в организм.
Фармацевтическая композиция по настоящему изобретению может быть получена путем объединения соединения по настоящему изобретению с подходящими фармацевтически приемлемыми вспомогательными веществами, например, в виде твердых, полутвердых, жидких или газообразных составов, таких как таблетки, пилюли, капсулы, порошки, гранулы, мази, эмульсии, суспензии, суппозитории, инъекционные формы, средства для ингаляции, гели, микросферы, аэрозоли и т.п.
Термин «аминокислота» относится к встречающимся в природе и синтетическим аминокислотам, а также к аналогам аминокислот и миметикам аминокислот, которые функционируют аналогично встречающимся в природе аминокислотам. Встречающиеся в природе аминокислоты представляют собой те аминокислоты, которые кодируются генетическим кодом, а также те аминокислоты, которые были впоследствии модифицированы, как, например, гидроксипролин, гамма-карбоксиглутаминовая кислота и О-фосфосерин. Аналоги аминокислот относятся к соединениям, которые имеют ту же основную химическую структуру, что и встречающиеся в природе аминокислоты (например, альфа-атом углерода, связанный с атомом водорода, карбоксильной группой, аминогруппой и R-группой), таким как гомосерин, норлейцин, метионинсульфоксид и метилметионинсульфоний. Такие аналоги могут иметь модифицированные R-группы (например, норлейцин) или модифицированные пептидные остовы, но сохраняют ту же основную химическую структуру, что и встречающиеся в природе аминокислоты. Миметики аминокислот относятся к химическим соединениям, структура которых отличается от общей химической структуры аминокислот, но которые функционируют аналогично встречающимся в природе аминокислотам.
Последовательности аминокислот по настоящему изобретению содержат стандартные однобуквенные или трехбуквенные коды из двадцати природных аминокислот.
Термин «лечение» включает ингибирование, замедление, остановку или обращение вспять прогрессирования или тяжести существующего симптома или состояния.
Термин «терапевтически эффективное количество» или «эффективное количество» означает количество соединения по настоящему изобретению, обеспечивающее достижение следующих эффектов: (i) лечения или предупреждения конкретного заболевания, состояния или нарушения, (ii) облегчения, уменьшения интенсивности проявлений или устранения одного или более симптомов конкретного заболевания, состояния или нарушения или (iii) предупреждения или задержки начала проявления одного или более симптомов конкретного заболевания, состояния или нарушения, описанного в данном документе. В случае рака терапевтически эффективное количество лекарственного средства может снижать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять и предпочтительно останавливать в определенной степени) инфильтрацию раковых клеток в окружающие органы; ингибировать (т.е. замедлять и предпочтительно останавливать в определенной степени) метастазирование опухоли; ингибировать рост опухоли в определенной степени и/или облегчать один или более симптомов, ассоциированных с раком, в определенной степени. В той степени, в какой лекарственное средство может предотвращать рост существующих раковых клеток и/или уничтожать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим.
Если не указано иное, то предполагается, что термин «изомеры» включает геометрические изомеры, цис-транс-изомеры, стереоизомеры, энантиомеры, оптические изомеры, диастереомеры и таутомеры.
Соединения по настоящему изобретению могут существовать в специфических геометрических или стереоизомерных формах. Настоящее изобретение охватывает все такие соединения, в том числе цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, а также рацемические смеси и другие смеси, как, например, смеси, обогащенные энантиомерами или диастереомерами, все из которых находятся в пределах объема настоящего изобретения. В таких заместителях, как алкильная группа, могут присутствовать дополнительные асимметрические атомы углерода. Все такие изомеры и их смеси включены в объем настоящего изобретения.
Если не указано иное, термин «энантиомеры» или «оптические изомеры» относится к стереоизомерам, которые являются зеркальными отображениями друг друга.
Если не указано иное, термин «цис-транс-изомеры» или «геометрические изомеры» проистекает из неспособности двойной связи или одинарной связи при атоме углерода, образующем кольцо, свободно вращаться.
Если не указано иное, термин «диастереомер» относится к стереоизомерам, в которых молекулы имеют два или более хиральных центра и не являются зеркальными отображениями друг друга.
Если не указано иное, «(+)» означает правовращающий, «(-)» означает левовращающий, а «(±)» означает рацемическую смесь.
Если не указано иное, абсолютная конфигурация стереоцентра представлена с помощью связей в виде клиновидных сплошных линий ( ) и связей в виде клиновидных пунктирных линий (
) и связей в виде клиновидных пунктирных линий ( ), а относительная конфигурация стереоцентра представлена с помощью связей в виде прямых сплошных линий (
), а относительная конфигурация стереоцентра представлена с помощью связей в виде прямых сплошных линий ( ) и связей в виде прямых пунктирных линий (
) и связей в виде прямых пунктирных линий ( ); связи в виде клиновидных сплошных линий () или связи в виде клиновидных пунктирных линий () представлены волнистыми линиями (
); связи в виде клиновидных сплошных линий () или связи в виде клиновидных пунктирных линий () представлены волнистыми линиями ( ), или связи в виде прямых сплошных линий () или связи в виде прямых пунктирных линий () представлены волнистыми линиями ().
), или связи в виде прямых сплошных линий () или связи в виде прямых пунктирных линий () представлены волнистыми линиями ().
Если не указано иное, термин «обогащенный одним изомером», «изомерно обогащенный», «обогащенный одним энантиомером» или «энантиомерно обогащенный» означает, что один изомер или энантиомер присутствует в содержании, которое меньше 100% и больше или равняется 60%, или больше или равняется 70%, или больше или равняется 80%, или больше или равняется 90%, или больше или равняется 95%, или больше или равняется 96%, или больше или равняется 97%, или больше или равняется 98%, или больше или равняется 99%, или больше или равняется 99,5%, или больше или равняется 99,6%, или больше или равняется 99,7%, или больше или равняется 99,8%, или больше или равняется 99,9%.
Если не указано иное, термин «изомерный избыток» или «энантиомерный избыток» относится к разности значений относительного процентного содержания двух изомеров или двух энантиомеров. Например, если один изомер или энантиомер присутствует в содержании 90%, а другой изомер или энантиомер присутствует в содержании 10%, то изомерный или энантиомерный избыток (значение ee) составляет 80%.
Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры могут быть получены посредством хирального синтеза, хиральных реагентов или других традиционных методик. Желаемый энантиомер соединения по настоящему изобретению можно получить путем асимметрического синтеза или дериватизации с использованием хиральных вспомогательных веществ, при этом полученную диастереомерную смесь разделяют и вспомогательную группу отщепляют с получением чистого желаемого энантиомера. В качестве альтернативы, если молекула содержит основную функциональную группу, такую как аминогруппа, или кислую функциональную группу, такую как карбоксильная группа, то образуется диастереомерная соль с соответствующими оптически активными кислотой или основанием с последующим разделением диастереомеров посредством традиционных способов, хорошо известных в данной области техники, с последующим извлечением для получения чистого энантиомера. Кроме того, разделение энантиомеров и диастереомеров обычно осуществляют с помощью хроматографии с использованием хиральной неподвижной фазы, необязательно в комбинации с химической дериватизацией (например, образованием карбаматов из амина).
Соединения по настоящему изобретению могут содержать неприродные доли изотопов атомов по одному или более атомам, составляющим соединения. Например, соединения могут быть помечены радиоактивными изотопами, такими как тритий (3H), йод-125 (125I) или C-14 (14C). Также, например, дейтерированное лекарственное средство может быть образовано путем замещения водорода дейтерием, где связь, образованная между атомом дейтерия и атомом углерода, прочнее, чем связь, образованная между обычным атомом водорода и атомом углерода, и дейтерированное лекарственное средство обладает такими преимуществами, как сниженные токсические побочные эффекты, увеличенная стабильность лекарственного средства, повышенная терапевтическая эффективность и длительный биологический период полужизни, по сравнению с недейтерированным лекарственным средством. Все изотопные варианты соединений по настоящему изобретению, вне зависимости от того, являются ли они радиоактивными или нет, включены в объем настоящего изобретения.
Если в перечисленных присоединяемых группах не указано направление их присоединения, то направление присоединения является произвольным. Например, в 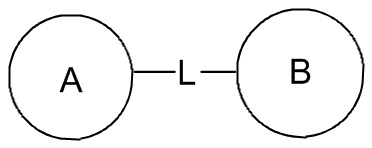 , если присоединяемая группа L представляет собой -M-W-, -M-W- может быть присоединена к кольцу A и кольцу B в том же направлении, что и порядок чтения слева направо, образуя
, если присоединяемая группа L представляет собой -M-W-, -M-W- может быть присоединена к кольцу A и кольцу B в том же направлении, что и порядок чтения слева направо, образуя 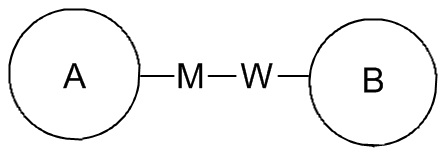 , или она может быть присоединена к кольцу A и кольцу B в направлении, противоположном порядку чтения слева направо, образуя
, или она может быть присоединена к кольцу A и кольцу B в направлении, противоположном порядку чтения слева направо, образуя 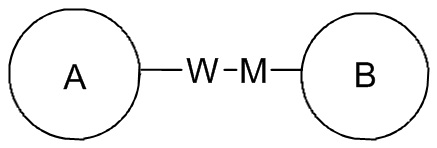 . Комбинации присоединяемых групп, заместителей и/или их вариантов допустимы только в том случае, если такие комбинации приводят к получению стабильных соединений.
. Комбинации присоединяемых групп, заместителей и/или их вариантов допустимы только в том случае, если такие комбинации приводят к получению стабильных соединений.
Если не указано иное, в случае, когда группа имеет один или несколько сайтов присоединения, любые один или более сайтов этой группы могут быть присоединены к другим группам посредством химических связей. Если химические связи присоединяются непозиционно, и в сайтах присоединения присутствуют атомы Н, то количество атомов Н в сайте будет соответствующим образом уменьшаться в зависимости от количества присоединенных химических связей с образованием группы с соответствующей валентностью при присоединении химических связей. Химические связи, присоединяющие сайты к другим группам, могут быть представлены связями в виде прямых сплошных линий ( ), связями в виде прямых пунктирных линий (
), связями в виде прямых пунктирных линий ( ) или волнистыми линиями (
) или волнистыми линиями (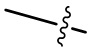 ). Например, связь в виде прямой сплошной линии в -OCH3 представляет присоединение к другой группе посредством атома кислорода в группе; связи в виде прямых пунктирных линий в
). Например, связь в виде прямой сплошной линии в -OCH3 представляет присоединение к другой группе посредством атома кислорода в группе; связи в виде прямых пунктирных линий в 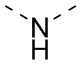 представляют присоединение к другим группам посредством обоих концов атома азота в группе; волнистые линии в
представляют присоединение к другим группам посредством обоих концов атома азота в группе; волнистые линии в 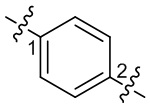 представляют присоединение к другим группам посредством атомов углерода в фенильной группе в положениях 1 и 2.
представляют присоединение к другим группам посредством атомов углерода в фенильной группе в положениях 1 и 2.
Если не указано иное, термин «C1-4алкил» представляет прямую или разветвленную насыщенную углеводородную группу, состоящую из 1-4 атомов углерода. C1-4алкил включает C1-2алкил, C1-3алкил, C2-3алкил и т. п.; который может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры C1-4алкильных групп включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил) и т.п.
Если не указано иное, термин «C1-3алкил» представляет прямую или разветвленную насыщенную углеводородную группу, состоящую из 1-3 атомов углерода. C1-3алкил включает C1-2алкил, C2-3алкил и т.п.; который может быть одновалентным (например, метил), двухвалентным (например, метилен) или поливалентным (например, метин). Примеры C1-3алкильных групп включают без ограничения метил (Me), этил (Et), пропил (включая н-пропил и изопропил) и т.п.
Если не указано иное, аминокислоты Xi, Xii и Xiii в настоящем изобретении присоединены к TATA посредством тиольной группы в остатке. Например, когда Xi представляет собой Pen, то оба из 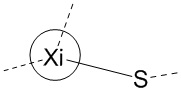 и
и 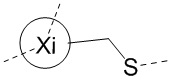 представляют собой
представляют собой 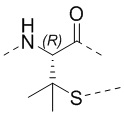 ; а когда Xi представляет собой Cys, то оба из и представляют собой
; а когда Xi представляет собой Cys, то оба из и представляют собой 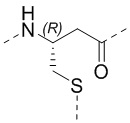 .
.
Структура соединений по настоящему изобретению может быть подтверждена традиционными способами, хорошо известными специалистам в данной области. Если настоящее изобретение относится к абсолютной конфигурации соединений, то абсолютная конфигурация может быть подтверждена традиционными в данной области техники техническими средствами. Например, в случае рентгеновской дифрактометрии монокристаллов (SXRD) данные об интенсивности дифракции культивируемого монокристалла собирают с использованием дифрактометра Bruker D8 Venture, при этом источником света является излучение CuKα, а режим сканирования представляет собой φ/ω, и после сбора соответствующих данных абсолютную конфигурацию можно подтвердить путем дополнительного разрешения кристаллической структуры с использованием прямого способа (Shelxs97).
Соединения по настоящему изобретению могут быть получены различными способами синтеза, хорошо известными специалистам в данной области, включая конкретные варианты осуществления, изложенные ниже, варианты осуществления, образованные комбинацией с другими способами химического синтеза, и их эквиваленты, хорошо известные специалистам в данной области. Предпочтительные варианты осуществления включают без ограничения примеры из настоящего изобретения.
Соединения именуются в соответствии с традиционной в данной области техники номенклатурой или с использованием программного обеспечения ChemDraw®, а для коммерчески доступных соединений используются названия из каталога поставщика.
Растворитель, используемый в настоящем изобретении, коммерчески доступен. Соотношение смешанных растворителей, представленных в настоящем изобретении, является объемным соотношением. Например, 20% MeCN/H2O означает, что объем MeCN в смешанном растворителе составляет 20%.
В настоящем изобретении используются следующие сокращения: экв. - эквивалент; SPPS - твердофазный синтез полипептидов; TFA - трифторуксусная кислота; DIEA - диизопропилэтиламин; DMF - N,N-диметилформамид; HATU - гексафторфосфат 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония; EDC - гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида; NHS - N-гидроксисукцинимид; TIS - триизопропилсилан; DTT - DL-1,4-дитиотреитол; TATA - 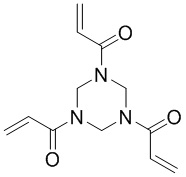 ; ММAE - монометилауристатин Е, имеющий структуру
; ММAE - монометилауристатин Е, имеющий структуру 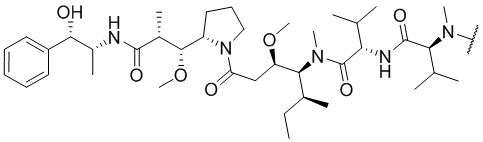 ; PABC -
; PABC - 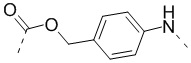 ; Cit - L-цитруллин; Val - L-валин; глутарил -
; Cit - L-цитруллин; Val - L-валин; глутарил - 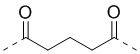 ; β-Ala -
; β-Ala - 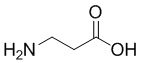 ; Sar -
; Sar - 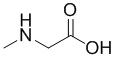 ; Sar10 -
; Sar10 - 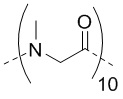 ; Cys - L-цистеин; hCys -
; Cys - L-цистеин; hCys - 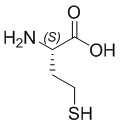 ; βCys -
; βCys - 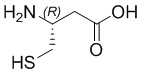 ; Pen -
; Pen - 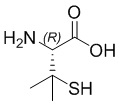 ; N-метил-Dap -
; N-метил-Dap - 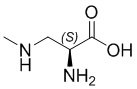 ; 1Nal - 1-нафтилаланин; hArg - L-гомоаргинин; Hyp - L-гидроксипролин; Trp - L-триптофан; Pro - L-пролин; Thr - L-треонин; Ser - L-серин; Asp - L-аспарагиновая кислота; dAsp - D-аспарагиновая кислота; Fmoc - 9-флуоренилметилоксикарбонил; Boc - трет-бутоксикарбонил (Boc); Trt - тритил; Pbf - 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил; и PBS - фосфатный буферный раствор.
; 1Nal - 1-нафтилаланин; hArg - L-гомоаргинин; Hyp - L-гидроксипролин; Trp - L-триптофан; Pro - L-пролин; Thr - L-треонин; Ser - L-серин; Asp - L-аспарагиновая кислота; dAsp - D-аспарагиновая кислота; Fmoc - 9-флуоренилметилоксикарбонил; Boc - трет-бутоксикарбонил (Boc); Trt - тритил; Pbf - 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил; и PBS - фосфатный буферный раствор.
Подробное описание изобретения
Нижеследующие примеры подробно описывают настоящее изобретение, но они не предназначены для налагания какого-либо неблагоприятного ограничения на настоящее изобретение. В данном документе было подробно описано настоящее изобретение и также раскрыты его конкретные варианты осуществления. Специалистам в данной области будет очевидно, что в конкретные варианты осуществления можно вносить различные изменения и модификации без отступления от сущности и объема настоящего изобретения.
Пример 1
 Путь синтеза:
Путь синтеза:
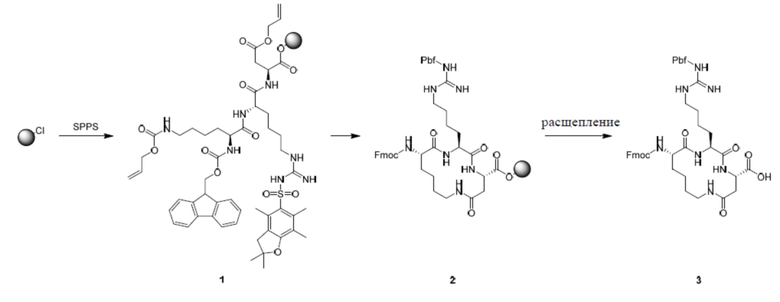
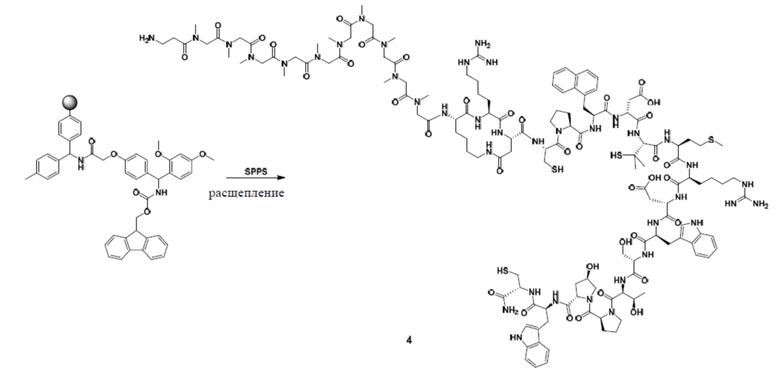
 1. Синтез соединения 3
1. Синтез соединения 3
Синтез полипептида
Полипептид синтезировали с использованием стандартного способа ступенчатого синтеза.
1) Добавляют DCM в контейнер, содержащий CTC-смолу (10,0 ммоль, 10 г, с 1,0 ммоль/г субстрата) и Fmoc-Asp(OAll)-OH (3,95 г, 10,0 ммоль, 1,0 экв).
2) Добавляют DIEA (4,0 экв.) и перемешивают в течение 2 часов.
3) Добавляют MeOH (0,3 мл) и перемешивают в течение 30 мин.
4) Полученную смесь высушивают, а затем прополаскивают три раза с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
5) Добавляют 20% пиперидин/DMF, а затем в системе обеспечивают протекание реакции в течение 30 мин.
6) Полученную смесь высушивают, а затем прополаскивают пять раз с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
7) Добавляют раствор следующей аминокислоты с защитной группой Fmoc на 30 секунд, а затем конденсирующее средство, и в системе обеспечивают протекание реакции в условиях барботирования с помощью N2 в течение примерно 1 часа.
8) Повторяют этапы 4-7 для конденсации следующей аминокислоты до завершения присоединения Fmoc-Lys(Alloc)-OH. Порядок добавления аминокислот и конденсирующих средств, используемых для синтеза соединения 3, показан в таблице 1.
9) Полученную смесь высушивают, а затем прополаскивают три раза с использованием DMF и три раза с использованием DCM, каждый раз барботируя азотом в течение 30 секунд.
10) De-OAll и Alloc: добавляют Pd(PPh3)4 (0,1 экв.) и PhSiH3 (10,0 экв.) в раствор смолы в DCM, и в системе обеспечивают протекание реакции в условиях барботирования с помощью N2 в течение примерно 15 минут, и ее высушивают. Этот этап повторяют три раза.
11) Полученную смесь высушивают, а затем прополаскивают пять раз с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
12) Циклизация: добавляют раствор конденсирующего средства HATU (22,85 экв.) и DIEA (6,0 экв.), и в системе обеспечивают протекание реакции в условиях барботирования с помощью N2 в течение примерно 1 часа.
13) Полученную смесь высушивают, а затем прополаскивают три раза с использованием DMF и три раза с использованием метанола, каждый раз барботируя азотом в течение 30 секунд, и высушивают.
Таблица 1. Порядок добавления
Расщепление и очистка
Добавляют буферный раствор для расщепления (20% HFIP/DCM) в колбу, содержащую полипептид с защищенной боковой цепью, и перемешивают при комнатной температуре в течение 30 минут × 2 раза. Раствор собирают и высушивают в центробежной сушилке, очищают посредством обращенно-фазового процесса (система NH4HCO3) и лиофилизируют с получением промежуточного продукта 3.
Таблица 2. Условия очистки
2. Синтез соли TFA соединения 4
Полипептид синтезировали с использованием стандартного способа ступенчатого синтеза.
1) Добавляют DMF в контейнер, содержащий амидную MBHA-смолу Ринка (0,5 ммоль, 1,56 г, с 0,32 ммоль/г субстрата), и смоле дают набухнуть в течение 2 часов.
2) Полученную смесь высушивают, а затем прополаскивают три раза с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
3) Добавляют 20% пиперидин/DMF, а затем в системе обеспечивают протекание реакции в течение 30 мин.
4) Полученную смесь высушивают, а затем прополаскивают пять раз с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
5) Добавляют раствор аминокислоты с защитной группой Fmoc на 30 секунд, а затем конденсирующее средство, и в системе обеспечивают протекание реакции в условиях барботирования с помощью N2 в течение примерно 1 часа.
6) Повторяют этапы 2-5 для конденсации следующей аминокислоты.
Порядок добавления аминокислот и конденсирующих средств, используемых для синтеза соединения 4, показан в таблице 3.
Таблица 3. Порядок добавления
Расщепление и очистка полипептида
1) Добавляют буферный раствор для расщепления (90% TFA/2,5% TIS/2,5% H2O/5,0% DTT) в колбу, содержащую полипептид с защищенной боковой цепью, и перемешивают при комнатной температуре в течение 2 часов.
2) Полипептид осаждают ледяным изопропиловым эфиром и центрифугируют его с помощью центрифуги (3 мин, 3000 об./мин).
3) Полученный полипептид промывают изопропиловым эфиром еще два раза.
4) Неочищенный полипептид высушивают с получением соли TFA промежуточного продукта 4.
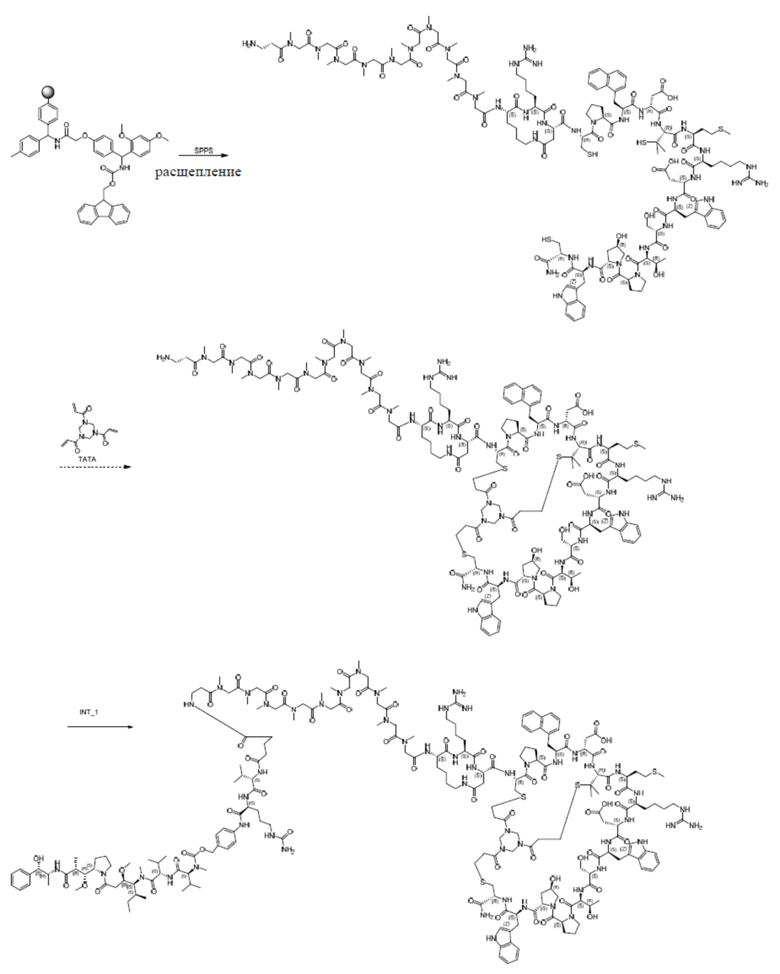
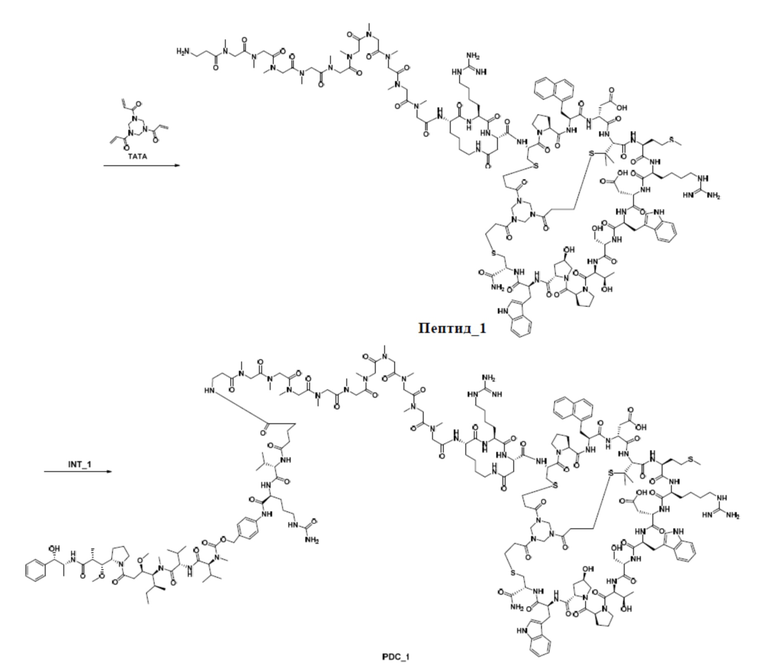
2. Синтез ацетатной соли соединения пептид_1
Неочищенную соль TFA промежуточного продукта 4, описанную выше (2,4 г), растворяют в 50% MeCN/H2O (1 л), и медленно добавляют TATA (0,5 ммоль) к перемешиваемому раствору при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут, а затем pH доводят до 8 с помощью NH4HCO3. Реакционную смесь перемешивают при комнатной температуре в течение дополнительных 12 часов. Когда LC-MS покажет, что реакция завершена, перемешивание прекращают. Смесь очищают посредством обращенно-фазового процесса с получением ацетатной соли соединения пептид_1.
Таблица 4. Условия очистки
Пептид_2 и пептид_3 синтезировали в соответствии с синтезом пептид_1, и их структуры показаны в таблице 5.
Таблица 5. Структурные последовательности соединений пептид_1~3
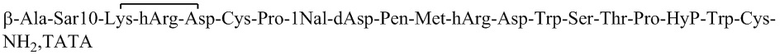
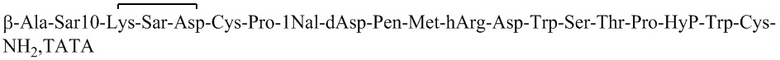
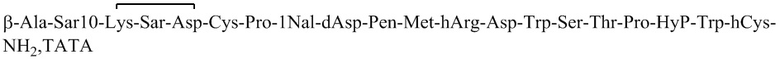
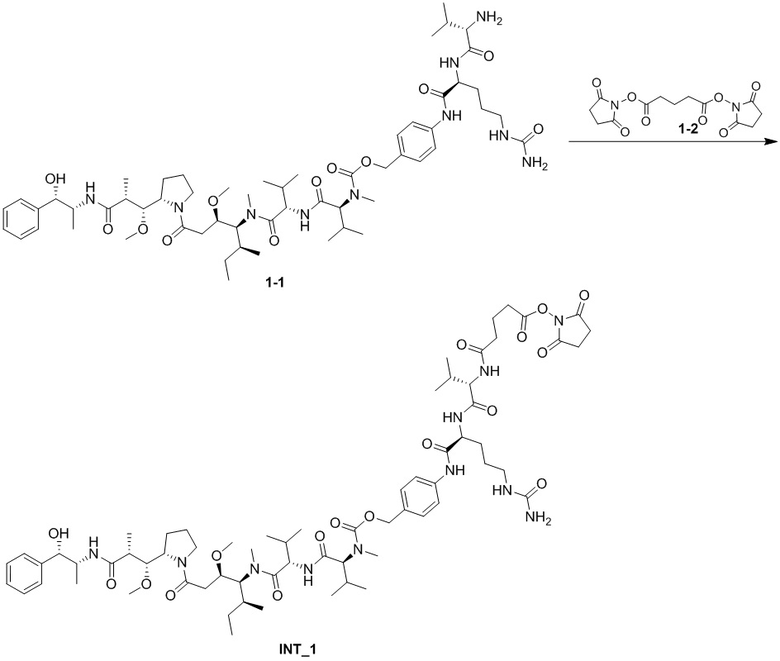
4. Синтез соли TFA соединения INT_1
Соединение 1-1 (200,0 мг, 178,0 мкмоль) растворяют в DMF (5 мл), и DIEA (31,0 мкл, 178,0 мкмоль) добавляют при 0°С и перемешивают в течение 10 мин. Соединение 1-2 (290,4 мг, 890,1 мкмоль) растворяют в DMF (5 мл) в другой реакционной колбе и перемешивают при 0°С в течение 10 мин. Затем по каплям добавляют реакционный раствор соединения 1-1 при 0°С к перемешиваемому реакционному раствору соединения 1-2 при 0°С и перемешивают при 0°C в течение 30 мин. Реакционный раствор фильтруют для удаления нерастворимых остатков. Фильтрат непосредственно очищают посредством обращенно-фазового процесса (подвижная фаза: A: H2O, содержащая 0,075% TFA, B: CH3CN, градиент: 10%-40% (B), 42 мин) с получением соли TFA соединения INT_1.
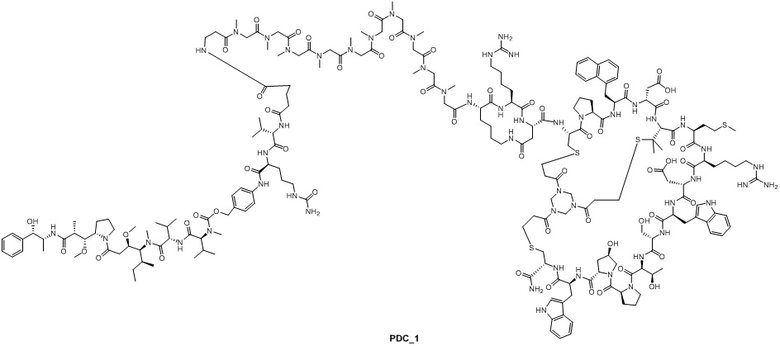
5. Синтез ацетатной соли соединения PDC_1
Ацетатную соль соединения пептид_1 (34,0 мг, 10,1 мкмоль) растворяют в DMF (0,3 мл), а затем добавляют DIEA (7,00 мкл, 40,4 мкмоль) и перемешивают при комнатной температуре в течение 10 минут. Затем соль TFA соединения INT_1 (13,4 мг, 10,1 мкмоль) растворяют в DMF (0,2 мл), по каплям добавляют в вышеуказанный реакционный раствор, а затем перемешивают при комнатной температуре в течение 2 часов. Реакционный раствор фильтруют для удаления нерастворимых остатков. Фильтрат непосредственно очищают посредством обращенно-фазового процесса (подвижная фаза: A: H2O, содержащая 0,075% TFA, B: CH3CN, градиент: 10%-40% (B), 42 мин), лиофилизируют, а затем превращают в ацетатную соль путем обработки с получением ацетатной соли соединения PDC_1. MS m/z: 1533,4 (M+3H+)/3.
Пример 2
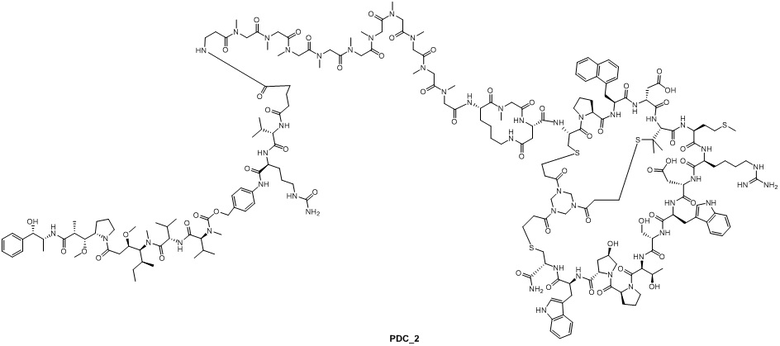
Путь синтеза 1:
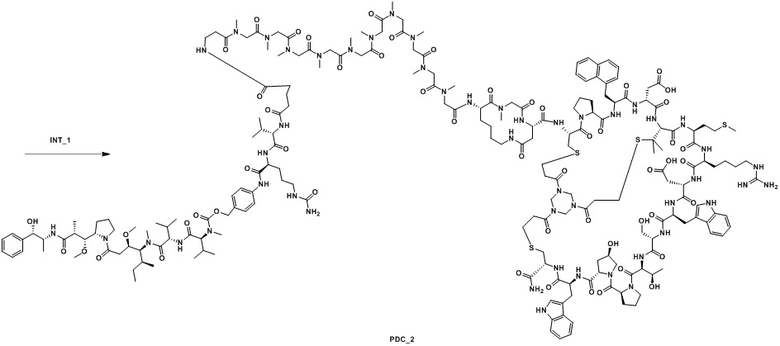
Ацетатную соль PDC_2 получают в соответствии с путем синтеза ацетатной соли PDC_1 (заменяя пептид_1 на пептид_2 в реакции). MS m/z: 1500,5 (M+3H+)/3.
Путь синтеза 2:
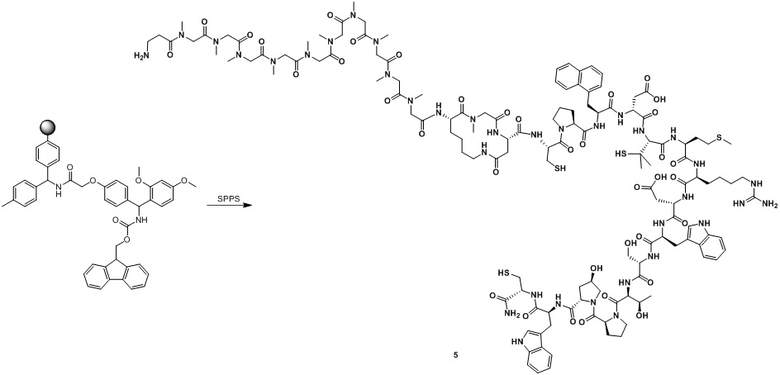
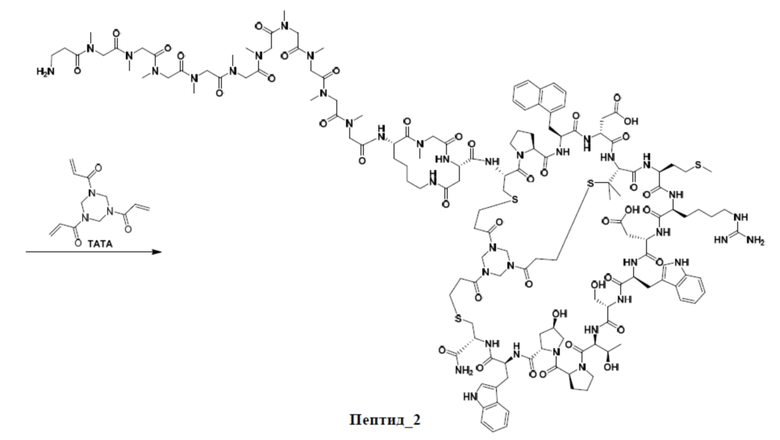
Этап 1. Синтез промежуточного продукта 5
Полипептид синтезировали с использованием стандартного способа ступенчатого синтеза.
1) Добавляют DMF в контейнер, содержащий амидную MBHA-смолу Ринка (5,0 ммоль, 8,33 г, с 0,60 ммоль/г субстрата), и смоле дают набухнуть в течение 2 часов.
2) Полученную смесь высушивают, а затем прополаскивают три раза с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
3) Добавляют 20% пиперидин/DMF, а затем в системе обеспечивают протекание реакции в течение 30 мин.
4) Полученную смесь высушивают, а затем прополаскивают пять раз с использованием DMF, каждый раз барботируя азотом в течение 30 секунд.
5) Добавляют раствор аминокислоты с защитной группой Fmoc на 30 секунд, а затем конденсирующее средство, и в системе обеспечивают протекание реакции в условиях барботирования с помощью N2 в течение примерно 1 часа.
6) Повторяют этапы 2-5 для конденсации следующей аминокислоты.
Порядок добавления аминокислот и конденсирующих средств, используемых для синтеза промежуточного соединения 5, показан в таблице 6.
Таблица 6. Порядок добавления
7) Добавляют буферный раствор для расщепления (90% TFA/2,5% TIS/2,5% H2O/5,0% DTT) в колбу, содержащую полипептид с защищенной боковой цепью, и перемешивают при комнатной температуре в течение 2 часов.
8) Полипептид осаждают ледяным изопропиловым эфиром и центрифугируют его с помощью центрифуги (3 мин, 3000 об./мин).
9) Полученный полипептид промывают изопропиловым эфиром еще два раза.
10) Неочищенный полипептид высушивают с получением неочищенного промежуточного продукта 5.
Этап 2. Синтез ацетатной соли соединения пептид_2
Неочищенный промежуточный продукт 5 (15,0 г) растворяют в 50% MeCN/H2O (5 л), и медленно добавляют TATA (7,5 ммоль) к перемешиваемому раствору при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут, а затем pH доводят до 8 с помощью NH4HCO3. Реакционную смесь перемешивают при комнатной температуре в течение дополнительных 12 часов. Когда LC-MS покажет, что реакция завершена, перемешивание прекращают. Смесь очищают посредством обращенно-фазового процесса (первая очистка: подвижная фаза: A: H2O, содержащая 0,075% TFA, B: CH3CN, градиент: 10%-40% фазы B, 42 мин, время удерживания: 29 мин; вторая очистка: подвижная фаза: A: H2O, содержащая 0,5% AcOH, B: CH3CN, градиент: 20%-40%, 40 мин, время удерживания: 21 мин; третья очистка: подвижная фаза: A: H2O, содержащая 0,5% AcOH, B: CH3CN, градиент: 16%-36% (B); 40 мин, время удерживания: 26 мин) с получением ацетатной соли промежуточного продукта пептид_2.
Этап 3. Синтез ацетатной соли PDC_2
Ацетатную соль промежуточного продукта пептид_2 (369 мг) и INT_1 (150 мг) растворяют в DMF (6,00 мл), и добавляют DIEA (58,0 мг). Смесь перемешивают при комнатной температуре в течение 5 часов. Когда LC-MS покажет, что реакция завершена, реакцию прекращают. Реакционный раствор фильтруют для удаления нерастворимых остатков. Фильтрат непосредственно очищают посредством обращенно-фазового процесса (подвижная фаза: A: H2O, содержащая 0,075% TFA, B: CH3CN, градиент: 10%-40% (B), 42 мин), лиофилизируют, а затем превращают в соль AcOH путем обработки с получением ацетатной соли PDC_2. MS m/z: 1500,3 (M+3H+)/3.
Пример 3
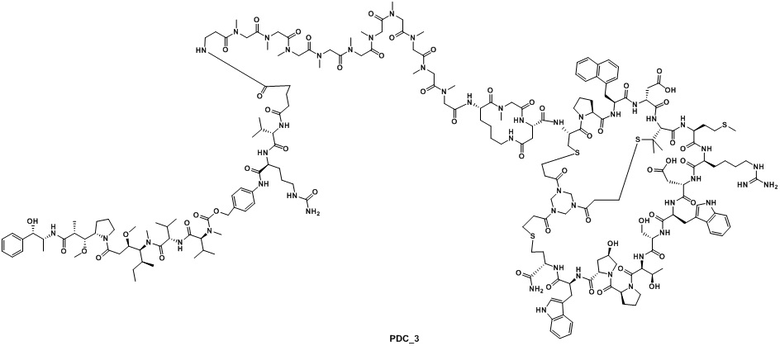
Ацетатную соль PDC_3 получают в соответствии с синтезом ацетатной соли PDC_1 (заменяя пептид_1 на пептид_3 в реакции). MS m/z: 1504,8 (M+3H+)/3.
Данные биологического анализа
Тестовый пример 1. Тестирование способности к связыванию соединений по настоящему изобретению с белком нектином-4
1. Цель эксперимента
Определить сродство соединения, подлежащего тестированию, к белку-мишени нектину-4 с использованием способа SPR.
2. Материалы и приборы
Biacore 8K (GE Healthcare)
96-луночный планшет (кат. № 650101, Greiner Bio-One)
Чип CM5 (кат. № BR-1005-30, GE Healthcare)
Набор для иммобилизации по аминогруппе (кат. № BR-1000-50, GE Healthcare)
EDC
NHS
1 М этаноламина
10 мМ ацетата натрия, pH 4,5 (кат. № BR-1003-50, GE Healthcare)
DMSO (кат. № D4540, Sigma)
P20 (кат. № BR-1000-54, GE Healthcare)
PBS (кат. № BR-1006-72, GE Healthcare)
Нектин-4 (кат. № 1006-72, GE Healthcare)
3. Протокол эксперимента
В этом эксперименте применяется способ иммобилизации по аминогруппе. Белок-мишень нектин-4 непосредственно иммобилизуют на чипе CM5 с использованием Biacore 8K. Соединение, подлежащее тестированию в качестве аналита, затем разбавляют буферным раствором (10 мМ PBS, pH 7,4, 137 мМ NaCl, 2,7 мМ KCl, 5% DMSO и 0,05% P20) до необходимого градиента концентрации для многоциклового кинетического выявления, в котором каждый цикл состоит из ввода пробы в течение 180 секунд и диссоциации в течение 180 секунд, а затем выполняется следующий цикл, для получения данных кинетического анализа сродства белка-мишени нектина-4 и соединения, подлежащего тестированию. С помощью программного обеспечения для оценки Biacore Insight (версия 2.0.15.12933) окончательные данные подвергают кинетическому анализу соответствия на основе модели 1:1.
4. Методика и процедура эксперимента
1) Получение буферного раствора: 10 мМ PBS, pH 7,4, 137 мМ NaCl, 2,7 мМ KCl, 5% DMSO и 0,05% P20.
2) Активация чипа CM5: чип CM5 активируют с помощью 400 мМ EDC и 100 мМ NHS при скорости потока 10 мкл/мин в течение 420 секунд.
3) Связывание белка-мишени: белок-мишень разбавляют до 10 мкг/мл с помощью 10 мМ ацетата натрия (pH 4,5) и связывают его при скорости потока 10 мкл/мин в течение 284 с. В эксперименте используются каналы №1, №2 и №3 на чипе, и результаты связывания составляют 1639,9 RU, 1747,8 RU и 1702,2 RU соответственно.
4) Блокирование чипа CM5: чип CM5 блокируют с помощью 1 M этаноламина при скорости потока 10 мкл/мин в течение 420 секунд.
5) Приготовление концентраций аналита: соединение, подлежащее тестированию, разбавляют с помощью буферного раствора. Соединение, подлежащее тестированию, разбавляют от 100 нМ до 0,78 нМ с 2-кратным градиентом.
6) Анализ вводимой пробы: каждая концентрация рабочего раствора соединения, подлежащего тестированию, соответствует одному циклу при скорости потока 30 мкл/мин для связывания в течение 180 секунд и диссоциации в течение 180 секунд. Последний цикл представляет собой цикл поправки на 5% растворитель DMSO.
7) Все результаты подвергают кинетическому анализу соответствия на основе модели 1:1.
5. Результаты эксперимента
Экспериментальные данные о пяти эффективных концентрациях отбирают для кинетического анализа соответствия на основе модели 1:1 с помощью программного обеспечения для оценки Biacore Insight (версия 2.0.15.12933). Результаты показаны в таблице 7.
Таблица 7. Результаты исследования связывания соединений по настоящему изобретению с белком нектином-4 человека методом SPR
a Среднее значение по двум тестам.
Заключение: соединения по настоящему изобретению обладают сильным эффектом связывания с нектином-4.
Тестовый пример 2. Антипролиферативная активность соединений по настоящему изобретению in vitro в отношении клеток NCI-H292 и MDA-MB-468
1. Цель эксперимента
Исследовать ингибирующий эффект соединений по настоящему изобретению в отношении пролиферации клеток путем обнаружения эффекта соединений по настоящему изобретению в отношении клеточной активности in vitro в линиях опухолевых клеток MDA-MB-468 и NCI-H292.
2. План эксперимента
Культивирование клеток
Линии опухолевых клеток культивируют в инкубаторе при 37°C без CO2 и в инкубаторе при 37°C с 5% CO2 в соответствии с условиями культивирования, показанными соответственно в таблице 8. Клетки периодически пассируют, и для посева отбирают клетки в логарифмической фазе роста.
Таблица 8. Линии клеток и способы их культивирования
H292
MB-468
Посев клеток
Клетки в логарифмической фазе роста собирают и центрифугируют при 1000 об./мин в течение 3 мин при комнатной температуре. Надосадочную жидкость удаляют, и клетки ресуспендируют в 5 мл культуральной среды. Затем отбирают пипеткой 20 мкл клеточной суспензии и смешивают с трипановым синим в соотношении 1:1 для окрашивания в течение 3 мин с целью определения жизнеспособности клеток и подсчета жизнеспособных клеток. Плотность клеток доводят до 3000 клеток/лунка. Затем в каждую лунку культурального планшета добавляют 90 мкл клеточной суспензии, а в лунки холостого контроля добавляют бесклеточную культуральную среду. Культуральный планшет инкубируют в течение ночи в инкубаторе при 37°C без CO2 и со 100% относительной влажностью и в инкубаторе при 37°C с 5% CO2 и 100% относительной влажностью соответственно.
Подготовка планшета с исходным раствором соединения
Подготовка планшета с 400X исходным раствором соединения. Соединение, подлежащее тестированию, подвергают градиентному разведению с помощью DMSO от наиболее высокой до наиболее низкой концентрации.
Таблица 9. Концентрации разведения в планшете с 400X исходным раствором
Получение 10X рабочего раствора соединения и обработка клеток соединением
Получение 10X рабочего раствора соединения. 78 мкл среды для культивирования клеток добавляют в 96-луночный планшет с V-образным дном, и 2 мкл раствора соединения каждой концентрации отбирают пипеткой из планшета с 400X исходным раствором соединения в среду для культивирования клеток в 96-луночном планшете. Затем 2 мкл DMSO добавляют к контролю в виде среды-носителя и холостому контролю. После добавления соединения или DMSO смесь равномерно продувают с помощью многоканального пипеточного дозатора. Добавление лекарственного средства: в планшет для культуры клеток добавляют 10 мкл 10X рабочего раствора соединения. Затем 10 мкл смеси среды для культивирования клеток и DMSO добавляют к контролю в виде среды-носителя и холостому контролю. 96-луночный планшет для культуры клеток помещают обратно в инкубатор на 72 часа.
Люминесцентный анализ жизнеспособности клеток CellTiter-Glo
Обнаружение выполняется в соответствии с инструкциями набора для люминесцентного анализа жизнеспособности клеток Promega CellTiter-Glo (Promega-G7573). Люминесцентный сигнал обнаруживается с помощью многорежимного планшет-ридера EnVision® (EnVision 2104-10).
Анализ данных
Степень ингибирования (IR) у соединения, подлежащего тестированию, рассчитывают согласно следующей формуле: IR (%) = (1- (RLU соединения - RLU холостого контроля) / (RLU контроля в виде среды-носителя - RLU холостого контроля) * 100%. Значения степени ингибирования у соединений в различных концентрациях рассчитывают в Excel. Затем с помощью программного обеспечения GraphPad Prism 6.02 на графике откладывают кривые ингибирования и рассчитывают соответствующие параметры, включая минимальную степень ингибирования, максимальную степень ингибирования и IC50.
Результаты эксперимента показаны в таблице 10.
Таблица 10. Результаты анализа ингибирования пролиферации опухолевых клеток соединением по настоящему изобретению
Разница в IC50 между двумя прогонами находится в пределах 2-кратной.
Заключение: соединение по настоящему изобретению оказывает значительный ингибирующий эффект в отношении пролиферации клеток, культивируемых in vitro, в линиях опухолевых клеток MDA-MB-468 и NCI-H292.
Тестовый пример 3. Эффективность соединений по настоящему изобретению in vivo в ксенотрансплантатной модели подкожной опухоли из раковых клеток легкого человека NCI-H292
1. Цель эксперимента. Исследовать эффективность соединений по настоящему изобретению in vivo в ксенотрансплантатной мышиной модели подкожной опухоли из раковых клеток легкого человека NCI-H292.
2. План эксперимента
Культивирование клеток. Раковые клетки легкого человека NCI-H292 (ATCC, Манассас, Вирджиния, кат. №CRL-1848) культивируют в виде монослоев in vitro в культуральной среде RPMI 1640, дополненной 10% фетальной телячьей сывороткой, в инкубаторе при 37°C с 5% CO2. Клетки стандартным образом подвергают расщеплению дважды в неделю с помощью панкреатического фермента/EDTA для пассирования. Когда насыщенность клетками достигает 80-90% и их количество соответствует требованиям, клетки собирают, подсчитывают и инокулируют.
Животные. «Голые» мыши BALB/c, самки, в возрасте 6-8 недель, массой 17-21 грамм.
Инокуляция опухоли. Инокулируют 0,2 мл (1×107 NCI-H292 подкожно в правую часть спины каждой мыши.
Исследование эффективности. Когда средний объем опухоли достигает приблизительно 100-200 мм3, соединение вводят внутривенно группам по 6 животных на группу. Доза составляет 1,5 мг/кг, а частота введения составляет QW×3.
Наблюдения. Подготовка и любые модификации данного протокола эксперимента будут реализованы только после оценки и одобрения комитетом по этическому обращению с экспериментальными животными (IACUC) WuXi AppTec Shanghai. Использование и благополучие лабораторных животных будет соответствовать правилам Международной ассоциации по оценке и аккредитации ухода за лабораторными животными (AAALAC). Выполняется ежедневный контроль состояния здоровья и смертности животных в качестве стандартного обследования, включающего наблюдение за ростом опухоли и эффектами лекарственного лечения в отношении повседневного характера поведения животных, такого как поведенческая активность, потребление пищи и воды, изменения массы (масса измеряется два раза в неделю), признаки внешнего вида или другие аномальные состояния. Смертность животных и побочные эффекты внутри группы регистрируются, исходя из количества животных в каждой группе.
Прекращение эксперимента. Животных следует подвергнуть эвтаназии, если состояние их здоровья продолжает ухудшаться, если объем опухоли превышает 2000 мм3, или если у них имеется тяжелое заболевание или боль.
Анализ данных. Для сравнения между двумя группами используется T-критерий. Для сравнения между тремя или более группами используется однофакторный ANOVA. Если F-значения существенно различаются, после анализа ANOVA следует провести множественные сравнения. Анализ всех данных проводится с использованием SPSS 17.0. p < 0,05 указывает на значимое различие.
Составление лекарственного средства
Частота составления лекарственного средства. Перед первым введением составляют однородный исходный раствор, распределяют его на порции и подвергают криоконсервации в холодильнике при -80°С.
Объем введения. Объем введения корректируют в соответствии с массой животного (объем введения = 10 мкл/г)
Таблица 11. Подробный протокол составления соединения
Отвешивают 5 г сахарозы и добавляют 50 мл стерильной воды с составлением 10% раствора сахарозы.
Отвешивают 193,94 мг L-гистидина, добавляют 10% сахарозы, перемешивают, доводят pH до 7 и разбавляют до 50 мл с составлением однородного раствора.
Результаты эксперимента. См. фиг. 1 и фиг. 2.
Заключение по результатам эксперимента. Соединение по настоящему изобретению демонстрирует значительный эффект ингибирования роста опухоли в ксенотрансплантатной модели подкожной опухоли из раковых клеток легкого человека NCI-H292.
Тестовый пример 4. Исследование фармакодинамических характеристик соединений по настоящему изобретению in vivo в ксенотрансплантатной модели подкожной опухоли из раковых клеток молочной железы человека MDA-MB-468 на «голых» мышах BALB/c
Цель эксперимента. оценить эффективность соединений по настоящему изобретению in vivo в ксенотрансплантатной модели подкожной опухоли из раковых клеток молочной железы человека MDA-MB-468.
Культивирование клеток. Раковые клетки молочной железы человека MDA-MB-468 (ATCC, Манассас, Вирджиния, кат. № HTB-132) культивируют в виде монослоев in vitro в культуральной среде L-15, дополненной 10% фетальной телячьей сывороткой, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина, в инкубаторе при 37°C с 0% CO2. Клетки стандартным образом подвергают расщеплению дважды в неделю с помощью панкреатического фермента/EDTA для пассирования. Когда насыщенность клетками достигает 80-90% и их количество соответствует требованиям, клетки собирают, подсчитывают и инокулируют.
Животные. «Голые» мыши BALB/c, самки, в возрасте 6-8 недель, массой 18-22 грамма, предоставлены компанией Beijing Vital River Laboratory Animal Technology Co., Ltd.
Инокуляция опухоли. Инокулируют 0,2 мл (1×107) клеток MDA-MB-468 (с матригелем, в объемном соотношении 1:1) подкожно в правую часть спины каждой мыши.
Объем введения. Объем введения корректируют в соответствии с массой животного (объем введения = 10 мкл/г)
Составление лекарственного средства. Соединение, подлежащее тестированию, составляют в виде 1,5 мг/мл однородного раствора, используя 25 мМ L-гистидина (pH = 7) в 10% сахарозе в качестве среды-носителя, и хранят в холодильнике при -80°C. В день введения однородный раствор разбавляют до соответствующей концентрации для IV введения (внутривенной инъекции) группе.
Группировка. Когда средний объем опухоли достигает приблизительно 500-600 мм3, соединение вводят группам. Доза составляет 5 мг/кг, а частота введения составляет QW×4.
Результаты эксперимента показаны на фиг. 3 и фиг. 4.
Заключение по результатам эксперимента. В ксенотрансплантатной модели подкожной опухоли из раковых клеток молочной железы человека MDA-MB-468 соединение по настоящему изобретению в группах большого объема по-прежнему демонстрирует значительный эффект ингибирования роста опухоли, корреляцию с уровнем дозы, отсутствие неблагоприятных реакций, среднюю потерю массы, не достигающую 5% или больше, и удовлетворительную безопасность.
Тестовый пример 5. Фармакокинетический анализ соединений по настоящему изобретению в плазме крови крыс
А. Цель эксперимента
Тестирование фармакокинетических характеристик соединений по настоящему изобретению у крыс SD.
B. Процедуры эксперимента
Фармакокинетические характеристики соединения после внутривенной инъекции тестируют у крыс согласно стандартному протоколу. Соединение, подлежащее тестированию, составляют в виде прозрачного раствора, используя 25 мМ L-гистидина (pH = 7) в 10% сахарозе в качестве среды-носителя. Двум крысам дают однократную внутривенную инъекцию 3 мг/кг соединения, подлежащего тестированию. В моменты времени 0,083, 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения собирают цельную кровь для получения плазмы крови. Концентрацию соединения по настоящему изобретению и концентрацию его потенциального метаболита ММАЕ анализируют методом LC-MS/MS, и фармакокинетические параметры рассчитывают с использованием программного обеспечения Phoenix WinNonlin.
С. Результаты эксперимента
Результаты эксперимента показаны в таблице 12.
Таблица 12. Результаты фармакокинетического тестирования у крыс
Заключение: соединение по настоящему изобретению характеризуется коротким периодом полужизни и быстрым клиренсом в крови крыс, и воздействие метаболита MMAE составляет всего приблизительно 1/40 от воздействия соединения по настоящему изобретению, что указывает на его удовлетворительную безопасность.
Тестовый пример 6. Фармакокинетический анализ в плазме крови яванских макаков
А. Цель эксперимента
Тестирование фармакокинетических характеристик соединений по настоящему изобретению у яванских макаков.
B. Процедуры эксперимента
Фармакокинетические характеристики соединения после внутривенной инъекции тестируют у яванских макаков согласно стандартному протоколу. Соединение, подлежащее тестированию, составляют в виде прозрачного раствора, используя 25 мМ L-гистидина (pH = 7) в 10% сахарозе в качестве среды-носителя. Яванским макакам дают однократную внутривенную инъекцию 1 мг/кг соединения, подлежащего тестированию. В моменты времени 0,083, 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения собирают цельную кровь для получения плазмы крови. Концентрацию соединения, подлежащего тестированию, и концентрацию его потенциального метаболита ММАЕ анализируют методом LC-MS/MS, и фармакокинетические параметры рассчитывают с использованием программного обеспечения Phoenix WinNonlin.
С. Результаты эксперимента
Результаты эксперимента показаны в таблице 13.
Таблица 13. Результаты фармакокинетического тестирования у яванских макаков
Заключение: соединение по настоящему изобретению характеризуется коротким периодом полужизни и быстрым клиренсом в крови яванского макака, высвобождением MMAE в кровь ниже предела обнаружения и удовлетворительной безопасностью.
Тестовый пример 7. Метаболическая стабильность соединений по настоящему изобретению в гепатоцитах мышей, крыс, яванских макаков и человека
А. Цель эксперимента
Исследовать метаболическую стабильность соединений по настоящему изобретению в гепатоцитах мышей, крыс, яванских макаков и человека.
B. Процедуры эксперимента
Инкубацию выполняют в 96-луночных планшетах с использованием способа наружной экстракции. Готовят соответственно несколько 96-луночных планшетов для осаждения образцов, обозначаемых T0, T15, T30, T60, T90, T0-MC, T90-MC, и холостую матрицу. Заранее отбирают культуральную среду для восстановления и культуральную среду для инкубации и помещают их на водяную баню при 37°C для предварительного нагревания. Замороженные гепатоциты мышей CD-1, крыс SD, яванских макаков и человека извлекают из резервуара с жидким азотом, восстанавливают и разбавляют до 0,51×106 клеток/мл с помощью культуральной среды для инкубации. Затем 198 мкл суспензии гепатоцитов (0,5×106 клеток/мл) добавляют в предварительно нагретые инкубационные планшеты, и 198 мкл культуральной среды для инкубации, не содержащей гепатоцитов, добавляют в инкубационные планшеты T0-MC и T90-MC в качестве контрольных групп с культуральной средой. Все инкубационные планшеты предварительно инкубируют в инкубаторе при 37°С в течение 10 минут. Затем добавляют по 2 мкл рабочих растворов тестируемого образца и контрольного соединения и равномерно перемешивают. Инкубационные планшеты помещают в шейкер внутри инкубатора для инкубации. Готовят по три повторности для каждого момента времени. Условия инкубации представляют собой 37°С, влажность насыщения и 5% CO2. В тест-системе конечная концентрация тестируемого образца составляет 1 мкМ, конечная концентрация контрольного образца составляет 3 мкМ, конечная концентрация гепатоцитов составляет 0,5×106 клеток/мл, а общая конечная концентрация органического растворителя составляет 1,0%, при этом конечная концентрация DMSO составляет 0,1%.
По окончании инкубации в соответствующий момент времени извлекают инкубационные планшеты, из них удаляют 25 мкл смеси соединения с клетками и смеси контрольного соединения с клетками и соответственно добавляют в планшеты для образцов, содержащие 125 мкл останавливающего буферного раствора. В планшет с холостым образцом непосредственно добавляют 25 мкл культуральной среды для инкубации, не содержащей гепатоцитов. Все планшеты с образцами запечатывают пленкой, встряхивают при 600 об./мин на встряхивателе для планшетов в течение 10 минут, а затем центрифугируют при 3220×g в течение 20 минут. Надосадочную жидкость тестируемого образца и контрольного соединения разбавляют очищенной водой в соотношении 1:3. Все образцы равномерно перемешивают и затем анализируют методом LC-MS/MS.
Концентрацию соединения, подлежащего тестированию, и концентрацию контрольного соединения в образце определяют полуколичественным методом жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS/MS) без стандартных кривых и образца для контроля качества. Соотношение площади пика аналита и площади пика внутреннего стандарта используется для выражения концентрации в образце. Значения времени удерживания, получение хроматограмм и интегрирование хроматограмм аналита и внутреннего стандарта обрабатываются программным обеспечением Analyst (Sciex, Фреймингем, Массачусетс, США).
С. Результаты эксперимента
Результаты эксперимента показаны в таблице 14.
Таблица 14. Результаты исследования метаболической стабильности соединения по настоящему изобретению в гепатоцитах мышей CD-1, крыс SD, яванских макаков и человека
(мл/мин/кг)
Заключение: соединение по настоящему изобретению обладает превосходной метаболической стабильностью в микросомах печени из 4 видов.
Тестовый пример 8. Метаболическая стабильность соединений по настоящему изобретению во фракции S9 почек мышей, крыс, яванских макаков и человека
А. Цель эксперимента
Исследовать метаболическую стабильность соединений по настоящему изобретению во фракции S9 почек мышей, крыс, яванских макаков и человека.
B. Процедуры эксперимента
Материалы. Фракцию S9 почек мышей CD-1, крыс SD, яванских макаков и человека приобретали у BioIVT или XenoTech LLC и хранили в холодильнике при -80°C.
Процедура эксперимента. Готовят восемь 96-луночных планшетов, обозначаемых T0, T5, T15, T30, T45, T60, «холостой» и NCF60. Первые шесть инкубационных планшетов соответствовали моментам времени реакции 0, 5, 15, 30, 45 и 60 минут соответственно. В планшет «холостой» не добавляют тестируемый образец или контрольное соединение.
В планшеты T0, T5, T15, T30, T45, T60 добавляют по 2 мкл рабочего раствора тестируемого образца (10 мМ раствора соединения, подлежащего тестированию, в DMSO, разбавленного 100% ацетонитрилом до 100 мкМ) и 100 мкл рабочего раствора S9 (концентрация почечного белка S9 составляет 1,0 мг/мл), а в планшет «холостой» добавляют только рабочий раствор S9. Вышеуказанные инкубационные планшеты затем помещают на водяную баню при 37°C для предварительной инкубации в течение приблизительно 10 минут.
По окончании предварительной инкубации в каждую лунку с образцом в каждом планшете, за исключением Т0, добавляют 98 мкл рабочего раствора вспомогательного фермента для инициирования реакции. После инкубации в течение соответствующего времени (т.е. 5, 15, 30, 45 и 60 минут) в каждую лунку с образцом добавляют 600 мкл останавливающего буферного раствора (раствора ацетонитрила, содержащего 100 нг/мл толбутамида и 100 нг/мл лабеталола) для прекращения реакции. Подготовка планшета T0. Сначала в планшет Т0 добавляют 600 мкл останавливающего буферного раствора, а затем 98 мкл рабочего раствора вспомогательного фермента. Все планшеты с образцами равномерно встряхивают и центрифугируют при 3220×g в течение 20 минут. Затем 100 мкл надосадочной жидкости из каждой лунки удаляют и разбавляют в 300 мкл чистой воды для анализа методом жидкостной хроматографии с тандемной масс-спектрометрией.
С. Результаты эксперимента
Результаты эксперимента показаны в таблице 15.
Таблица 15. Результаты исследования метаболической стабильности соединения по настоящему изобретению во фракции S9 почек мышей CD-1, крыс SD, яванских макаков и человека
(мкл/мин/мг)
Заключение: соединение по настоящему изобретению обладает превосходной метаболической стабильностью во фракции S9 почек из 4 видов.
Тестовый пример 9. Анализ стабильности соединений по настоящему изобретению в плазме крови различных видов
А. Цель эксперимента
Протестировать стабильность соединений по настоящему изобретению в плазме крови крыс SD, яванских макаков и человека.
B. Процедуры эксперимента
По 2 мкл рабочего раствора соединения, подлежащего тестированию (100 мкМ), добавляют в соответствующие инкубационные планшеты, включая инкубационные планшеты T0, T10, T30, T60, T120 и T240. Для каждого образца готовят три параллельные лунки. Затем добавляют 98 мкл холостой плазмы крови крыс SD, яванских макаков и человека в соответствующие инкубационные планшеты, в которые был добавлен рабочий раствор. Все образцы инкубируют на водяной бане при 37°С. Конечная инкубационная концентрация соединения, подлежащего тестированию, составляет 2 мкМ. По окончании каждого момента времени инкубации извлекают соответствующий инкубационный планшет, добавляют останавливающий буферный раствор, осаждают и центрифугируют в течение 20 минут. Затем 150 мкл надосадочной жидкости анализируют методом LC-MS/MS. Концентрацию соединения, подлежащего тестированию, в образце определяют полуколичественным методом жидкостной хроматографии с тандемной масс-спектрометрией (LC-MS/MS).
С. Результаты эксперимента
Результаты эксперимента показаны в таблице 16.
Таблица 16. Результаты тестирования стабильности соединения по настоящему изобретению в плазме крови крыс SD, яванских макаков и человека
Заключение: соединение по настоящему изобретению обладает превосходной метаболической стабильностью, при этом период полужизни составляет более 578,1 мин у всех трех видов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ПОЛИПЕПТИДА | 2022 |
|
RU2830694C2 |
| РАДИОФАРМАЦЕВТИЧЕСКИЕ СРЕДСТВА, РАДИОВИЗУАЛИЗИРУЮЩИЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2804297C2 |
| ПОЛИЛИГАНДНЫЕ ЛЕКАРСТВЕННЫЕ КОНЪЮГАТЫ И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2722449C2 |
| Лиганды PSMA для визуализации и эндорадиотерапии | 2018 |
|
RU2807076C2 |
| БИЛИГАНДНЫЙ КОНЪЮГАТ ЛЕКАРСТВЕННОГО СРЕДСТВА И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820346C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2018 |
|
RU2798085C2 |
| ГОМОДЕТНЫЕ ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ИНТЕГРИН α4β7 | 2018 |
|
RU2773443C2 |
| ХИМЕРНЫЕ АНАЛОГИ СОМАТОСТАТИНА-ДОФАМИНА | 2002 |
|
RU2277539C2 |
| ПРОЛЕКАРСТВО, СОДЕРЖАЩЕЕ САМОРАСЩЕПЛЯЕМЫЙ ЛИНКЕР | 2014 |
|
RU2676324C2 |
| БЕНЗО-N-ГИДРОКСИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2017 |
|
RU2737433C2 |
Изобретение относится к конъюгированному лекарственному средству на основе трициклического полипептида, в частности к соединению, представленному формулой (III), или его фармацевтически приемлемой соли, и его применению в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4. 9 н. и 1 з.п. ф-лы, 4 ил., 16 таб., 9 пр.
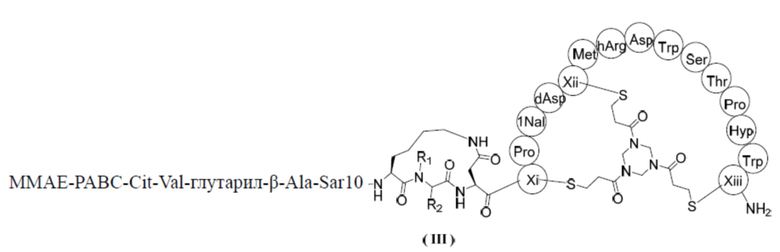
1. Соединение, представленное формулой (III), или его фармацевтически приемлемая соль
ММАЕ-РАВС-Cit-Val-глутарил-β-Ala-Sar10 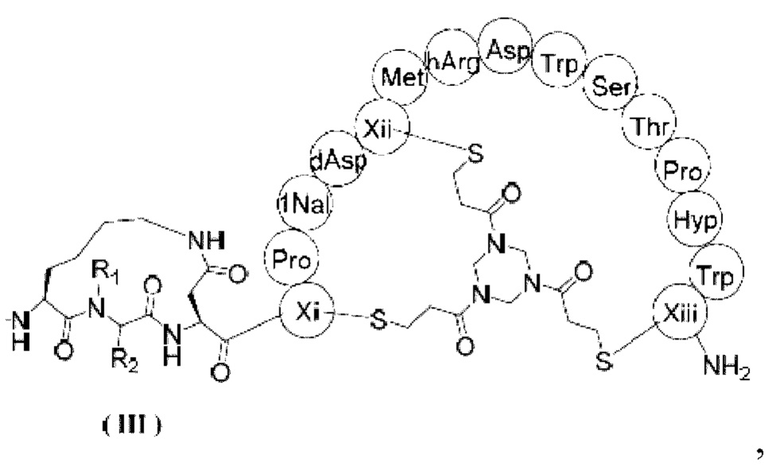
где R1 выбран из Н и С1-3залкила;
R2 выбран из Н, С1-4алкила и 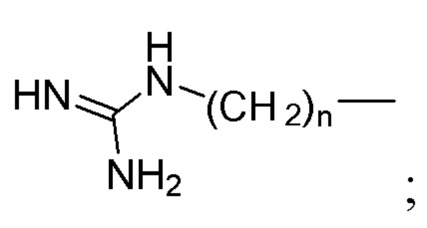
n выбран из 1, 2, 3 и 4; и
каждый из Xi, Xii и Xiii независимо выбран из Cys, hCys, βCys и Pen;
ММАЕ - монометилауристатин Е;
РАВС -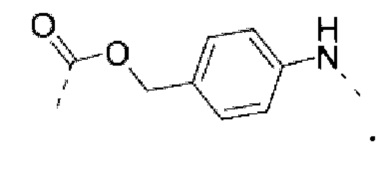
2. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из структур, представленных формулами (III-1) и (III-2),
ММАЕ-РАВС-Cit-Val-глутарил-β-Ala-Sar10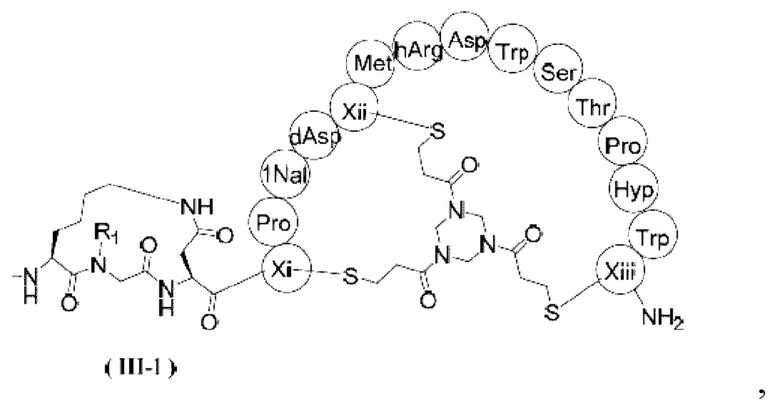
ММАЕ-РАВС-Cit-Val-глутарил-β-Ala-Sar10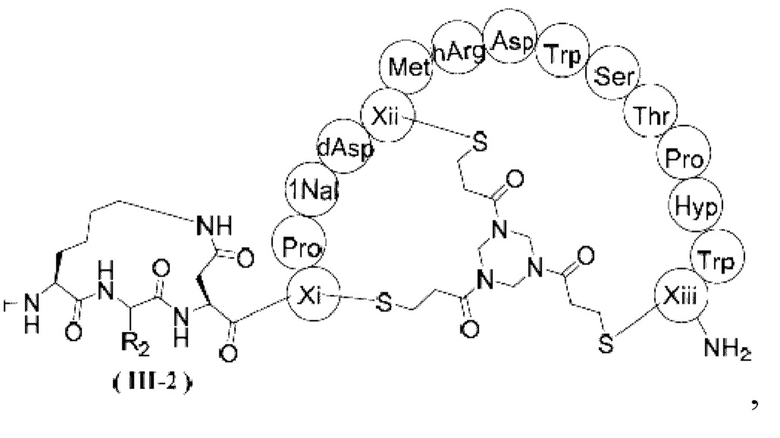
где R1, R2, Xi, Xii и Xiii определены в п. 1.
3. Соединение или его фармацевтически приемлемая соль, выбранное из
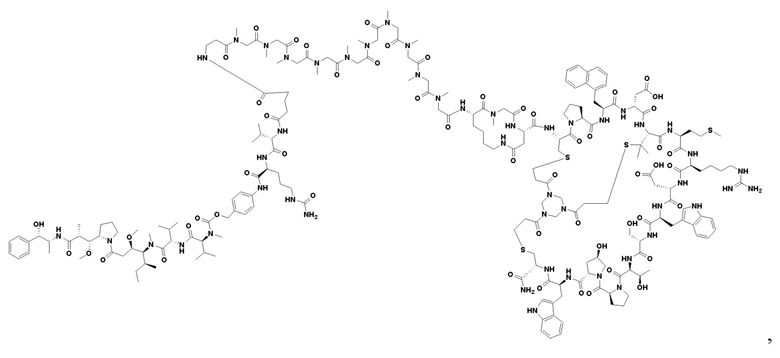
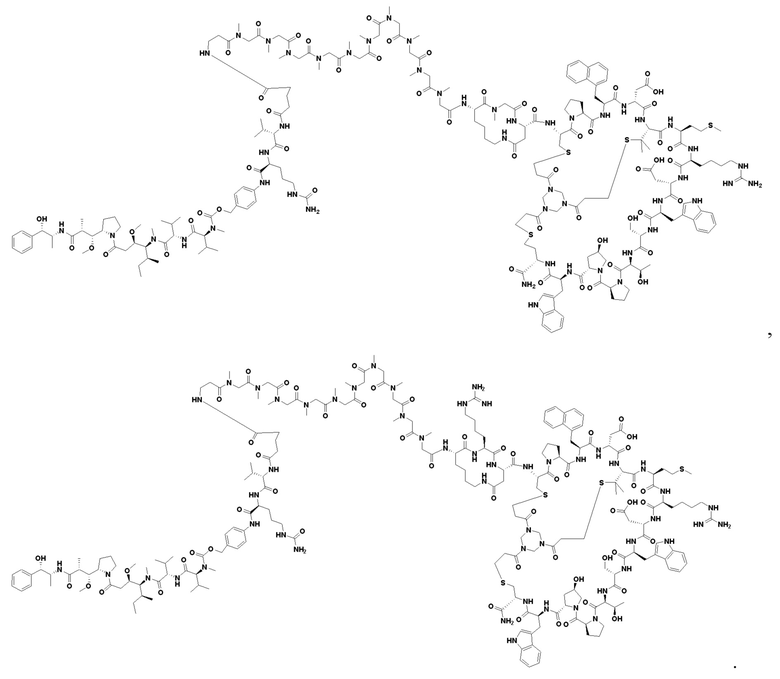
4. Фармацевтическая композиция, содержащая терапевтически или профилактически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-3, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
5. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-3 в получении лекарственного средства для лечения солидных опухолей, в которых сверхэкспрессируется нектин-4.
6. Применение фармацевтической композиции по п. 4 в получении лекарственного средства для лечения солидных опухолей, в которых сверхэкспрессируется нектин-4.
7. Способ лечения солидных опухолей, в которых сверхэкспрессируется нектин-4, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-3.
8. Способ лечения солидных опухолей, в которых сверхэкспрессируется нектин-4, включающий введение субъекту фармацевтической композиции по п. 4.
9. Продукт, содержащий соединение или его фармацевтически приемлемую соль по любому из пп. 1-3, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
10. Продукт, содержащий фармацевтическую композицию по п. 4, для применения в лечении солидных опухолей, в которых сверхэкспрессируется нектин-4.
| WO 2019193328 A1, 10.10.2019 | |||
| WO 2019243833 A1, 26.12.2019 | |||
| RU 2017118326 A, 29.11.2018 | |||
| WO 2020120984 A1, 18.06.2020 | |||
| WO 2020225577 A1, 12.11.2020 | |||
| WO 2021019243 A1, 04.02.2021 | |||
| WO 2021028686 A1, 18.02.2021 | |||
| WO 2019122860 A1, 27.06.2019. |

