ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области техники фармацевтической химии и, в частности, оно относится к индольному соединению, способу получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Атопический дерматит (AD) представляет собой хроническое рецидивирующее воспалительное кожное заболевание. Сообщалось, что многие факторы вовлечены в патофизиологию AD, включая генетические факторы, факторы окружающей среды, изменения в липидном составе эпидермиса, иммунную дисфункцию и микробную дисрегуляцию. В дополнение к кожным симптомам, таким как эритема, папула, экссудация, шелушение, ксероз, бляшки и лихенификация, AD может также вызывать психическую болезнь и повышать риск развития сердечно-сосудистых заболеваний. Кроме того, считается, что он также связан с депрессией и суицидальными тенденциями. С точки зрения эпидемиологических характеристик AD чаще встречается у детей, и большинство симптомов продолжают проявляться во взрослом возрасте. Статистический анализ соответствующих клинических данных в Соединенных Штатах был хорошо изучен, и результаты исследований на популяциях детей и подростков демонстрируют, что только 1% пациентов, которые страдали от данного заболевания до 12 лет, не будут продолжать иметь заболевание после этого.
В недавних исследованиях было показано, что уровень распространенности AD в Китае постепенно повышается, особенно у детей, и уровень распространенности в городских областях выше, чем в сельских областях. В заключение, AD оказывает значительное влияние на качество жизни и состояние психического здоровья пациентов. Постоянное повышение уровня распространенности AD в последние годы, особенно у детей, проживающих в городах, привело к клиническому и социальному запросу на разработку более безопасных и более эффективных лекарственных средств и протоколов для лечения AD.
В настоящее время традиционные лекарственные средства для лечения AD главным образом делятся на три типа: 1) увлажняющие средства, которые улучшают состояние сухой кожи; 2) кортикостероиды, которые характеризуются широким спектром противовоспалительных и иммуносупрессивных эффектов; и 3) ингибиторы кальциневрина, которые облегчают воспаление посредством связывания с иммунофилином с осуществлением ингибирования кальциневрина и в конечном счете ингибирования секреции различных цитокинов. Кроме того, поскольку пациенты с AD часто страдают от инфекций, вызываемых Staphylococcus aureus, они более восприимчивы к грибковым инфекциям, чем здоровые люди, и поэтому можно систематически использовать антибиотики для устранения патогенных микроорганизмов для облегчения состояний, обусловленных AD. Однако приведенные выше традиционные способы лечения имеют свои ограничения: 1) увлажняющее средство главным образом функционирует для замедления испарения кожной влаги, и его терапевтический эффект ограничен; 2) долговременное применение кортикостероида может вызывать системные нежелательные эффекты и ингибировать гипоталамо-гипофизарно-надпочечниковую (HPA) ось, а применение у детей может оказывать нежелательные эффекты в отношении их развития; 3) ингибитор кальциневрина часто вызывает локализованные жжение и зуд; 4) после отмены антибиотика патогенные микроорганизмы склонны к повторной колонизации, а долговременное применение антибиотика может приводить к появлению штаммов, устойчивых к лекарственным средствам.
Поскольку патофизиология AD тесно связана с дисфункцией эпидермального барьера и иммунной дисфункцией, пациенты с AD часто демонстрируют аномальную экспрессию цитокинов. Некоторые исследователи предложили классифицировать AD по разным эндотипам в соответствии с молекулярными механизмами, ассоциированными с различными патологическими фенотипами. В последние годы низкомолекулярные ингибиторы, нацеливающиеся на рецепторы цитокинов, и биологические средства, нацеливающиеся на конкретные цитокины или их рецепторы, стали горячими темами в разработке терапевтических лекарственных средств для лечения AD. Низкомолекулярные ингибиторы фосфодиэстеразы (PDE)-4 и Янус-киназы (JAK), антагонисты рецептора гистамина 4 (H4R), агонисты рецептора ароматических углеводородов (AhR) и моноклональные антитела к рецептору интерлейкина-4 (IL-4) и интерлейкина-13 (IL-13) и т.п. в настоящее время проходят клинические испытания или находятся на стадии клинического одобрения. Среди них дупиксент (дупилумаб, моноклональное антитело к рецептору IL-4), экриза (крисаборол, антагонист фосфодиэстеразы-4 (PDE-4) для местного применения) и сибинко (аброцитиниб, ингибитор JAK1 для перорального применения) были одобрены FDA. В настоящее время лекарственное средство с бенвитимодом в качестве первичного активного ингредиента, служащего в качестве как агониста AhR, так и ингибитора тирозинкиназы T-клеток, был одобрен Национальным управлением медицинскими продуктами Китая, но в настоящее время его применение для лечения псориаза ограничено. Следовательно, с целью решения таких проблем как безопасность и устойчивость к лекарственным средствам, обусловленная долговременным применением лекарственных средств, а также для улучшения эффективности абсорбции лекарственных средств при трансдермальном введении срочно требуется разработать больше низкомолекулярных лекарственных средств из природных источников.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
На основании этого необходимо предоставить класс индольных соединений с хорошим терапевтическим эффектом при атопическом дерматите для решения указанных выше проблем.
В одном аспекте настоящего изобретения предусмотрено индольное соединение, характеризующееся структурой формулы I,
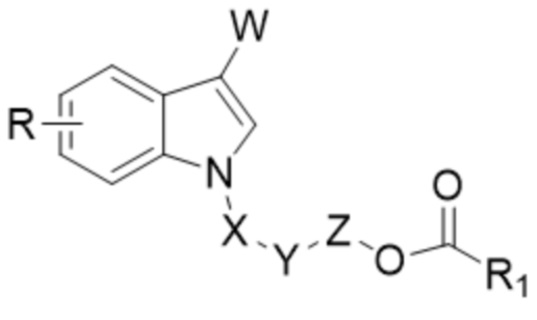
I,
где
W представляет собой COR2 или CR3R4OR5;
X отсутствует или X представляет собой CO или CR3R4;
Y отсутствует или Y представляет собой O;
Z отсутствует или Z представляет собой CR3R4;
R1 выбран из группы, состоящей из незамещенного или замещенного C1-20алкила, незамещенного или замещенного C1-20алкенила, незамещенного или замещенного арила, гетероарила, арилалкила, гетероарилалкила и алкокси;
R выбран из группы, состоящей из H, D, незамещенного или замещенного C1-6алкила, незамещенного или замещенного C1-6алкенила, незамещенного или замещенного арила, гетероарила, галогена, OR5, NR6R7, CO2R7, CONR6R7, OCOR8, NHCOR8, NHSO2R8 или CN;
каждый из R2, R3, R4, R6, R7 и R8 независимо выбран из группы, состоящей из H, D, незамещенного или замещенного C1-6алкила, незамещенного или замещенного C1-6алкенила, незамещенного или замещенного арила и гетероарила; и
R5 представляет собой H, D или COR1.
В одном варианте осуществления соединение характеризуется структурой, показанной в формуле II:
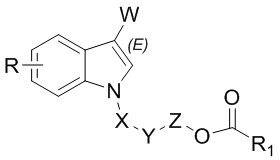
II.
В одном варианте осуществления W представляет собой COR2 или CR3R4OR5;
X отсутствует или X представляет собой CO;
Y отсутствует или Y представляет собой O;
Z отсутствует или Z представляет собой CR3R4;
R1 выбран из группы, состоящей из незамещенного или замещенного C1-20алкила, незамещенного или замещенного арила и алкокси;
R представляет собой H; и
каждый из R2, R3, R4 и R5 представляет собой H.
В одном варианте осуществления R1 выбран из группы, состоящей из незамещенного или замещенного C5-15алкила, ацетоксилзамещенного арила, (2,6-дихлорфенил)аминозамещенного арила и пентаоксапентадеканила.
В одном варианте осуществления W представляет собой COH или CH2OH;
каждый из X, Y и Z представляет собой COOCH2 или CH2;
R1 выбран из C7-С15алкила, ацетоксилзамещенного арила, (2,6-дихлорфенил)аминозамещенного арила и пентаоксапентадеканила; и
R представляет собой H.
В одном варианте осуществления W представляет собой COH;
каждый из X, Y и Z представляет собой CH2;
R1 выбран из группы, состоящей из C7-С15алкила, ацетоксилзамещенного арила, (2,6-дихлорфенил)аминозамещенного арила и пентаоксапентадеканила; и
R представляет собой H.
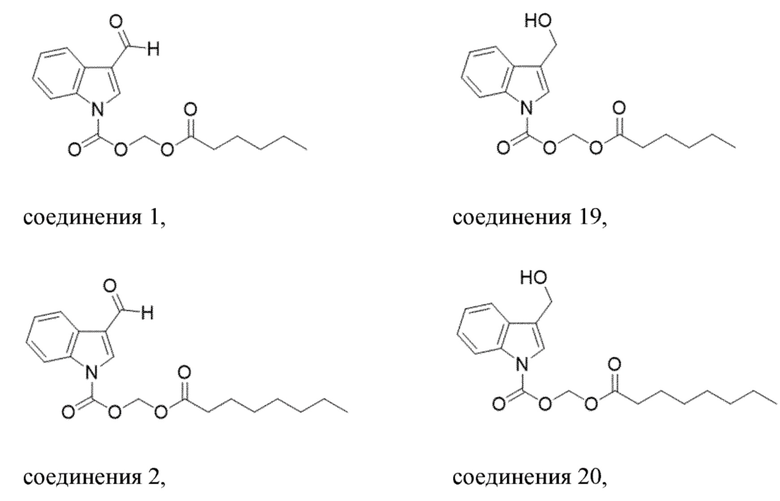
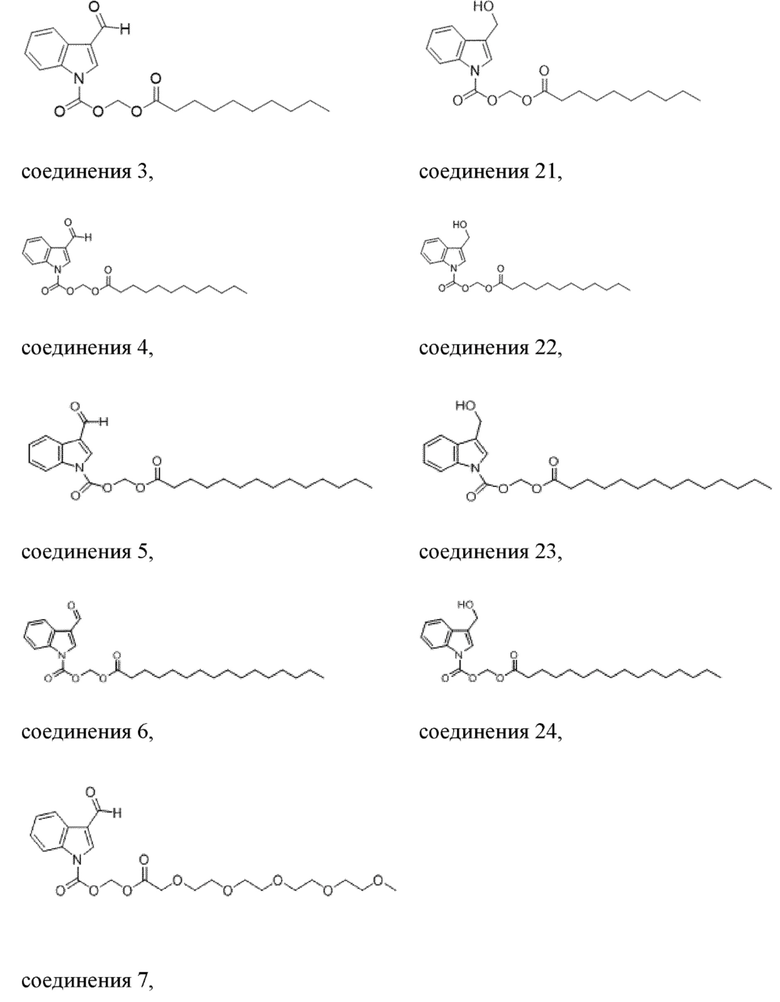
В одном варианте осуществления соединение выбрано из группы, состоящей из
соединения 34 и соединения 35.
Соединение 4 имеет характеристические пики при значениях 2θ, составляющих 4,9±0,2°, 7,3±0,2°, 9,9±0,2°, 14,9±0,2° и 22,0±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 4 имеет характеристические пики при значениях 2θ, составляющих 4,9±0,2°, 7,3±0,2°, 9,9±0,2°, 11,3±0,2°, 11,8±0,2°, 14,9±0,2°, 19,0±0,2°, 19,9±0,2°, 21,6±0,2° и 22,0±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 4 имеет характеристические пики при значениях 2θ, составляющих 4,9±0,2°, 7,3±0,2°, 9,9±0,2°, 10,7±0,2°, 11,3±0,2°, 11,8±0,2°, 13,4±0,2°, 14,6±0,2°, 14,9±0,2°, 18,4±0,2°, 19,0±0,2°, 19,9±0,2°, 21,6±0,2°, 22,0±0,2° и 25,3±0,2, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 8 имеет характеристические пики при значениях 2θ, составляющих 5,2±0,2°, 11,6±0,2°, 12,6±0,2°, 16,0±0,2° и 19,3±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 8 имеет характеристические пики при значениях 2θ, составляющих 5,2±0,2°, 6,3±0,2°, 10,0±0,2°, 11,6±0,2°, 12,6±0,2°, 12,9±0,2°, 14,3±0,2°, 16,0±0,2°, 19,3±0,2° и 21,3±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 8 имеет характеристические пики при значениях 2θ, составляющих 5,2±0,2°, 6,3±0,2°, 10,0±0,2°, 11,6±0,2°, 12,6±0,2°, 12,9±0,2°, 14,3±0,2°, 16,0±0,2°, 19,3±0,2°, 20,4±0,2°, 21,3±0,2°, 23,2±0,2°, 25,2±0,2°, 26,3±0,2° и 27,6±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 9 имеет характеристические пики при значениях 2θ, составляющих 12,3±0,2°, 14,9±0,2°, 19,9±0,2°, 23,4±0,2° и 27,3±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 9 имеет характеристические пики при значениях 2θ, составляющих 10,3±0,2°, 12,3±0,2°, 12,7±0,2°, 14,9±0,2°, 15,6±0,2°, 19,2±0,2°, 19,9±0,2°, 23,4±0,2°, 25,1±0,2° и 27,3±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 9 имеет характеристические пики при значениях 2θ, составляющих 4,9±0,2°, 10,3±0,2°, 12,3±0,2°, 12,7±0,2°, 14,9±0,2°, 15,6±0,2°, 17,3±0,2°, 19,2±0,2°, 19,9±0,2°, 20,3±0,2°, 20,7±0,2°, 23,4±0,2°, 24,8±0,2°, 25,1±0,2° и 27,3±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 22 имеет характеристические пики при значениях 2θ, составляющих 3,4±0,2°, 5,3±0,2°, 6,9±0,2°, 10,2±0,2° и 19,9±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 22 имеет характеристические пики при значениях 2θ, составляющих 3,4±0,2°, 5,3±0,2°, 6,9±0,2°, 9,7±0,2°, 10,2±0,2°, 11,7±0,2°, 14,9±0,2°, 17,7±0,2°, 19,9±0,2° и 20,6±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 22 имеет характеристические пики при значениях 2θ, составляющих 3,4±0,2°, 5,3±0,2°, 6,9±0,2°, 9,7±0,2°, 10,2±0,2°, 11,7±0,2°, 12,5±0,2°, 14,0±0,2°, 14,9±0,2°, 15,4±0,2°, 17,7±0,2°, 19,9±0,2°, 20,6 ± 0,2°, 21,9±0,2° и 23,2±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 23 имеет характеристические пики при значениях 2θ, составляющих 10,6±0,2°, 11,0±0,2°, 18,4±0,2°, 21,2±0,2° и 21,7±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно MC20-875-034A1 имеет характеристические пики при значениях 2θ, составляющих 10,6±0,2°, 11,0±0,2°, 15,2±0,2°, 18,4±0,2°, 19,9±0,2°, 20,6±0,2°, 21,2±0,2°, 21,7±0,2°, 23,6±0,2° и 24,2±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 23 имеет характеристические пики при значениях 2θ, составляющих 5,2±0,2°, 10,2±0,2°, 10,6±0,2°, 11,0±0,2°, 13,0±0,2°, 14,0±0,2°, 15,2±0,2°, 18,4±0,2°, 19,9±0,2°, 20,6±0,2°, 21,2±0,2°, 21,7±0,2°, 22,4±0,2°, 23,6±0,2° и 24,2±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 24 имеет характеристические пики при значениях 2θ, составляющих 4,4±0,2°, 6,6±0,2°, 8,9±0,2°, 21,0±0,2° и 22,6±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 24 имеет характеристические пики при значениях 2θ, составляющих 4,4±0,2°, 6,6±0,2°, 8,9±0,2°, 11,1±0,2°, 12,2±0,2°, 13,4±0,2°, 19,5±0,2°, 20,6±0,2°, 21,0±0,2° и 22,6±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 24 имеет характеристические пики при значениях 2θ, составляющих 4,4±0,2°, 6,6±0,2°, 8,9±0,2°, 10,4±0,2°, 11,1±0,2°, 12,2±0,2°, 13,4±0,2°, 16,1±0,2°, 17,8±0,2°, 19,5±0,2°, 20,6±0,2°, 21,0±0,2°, 22,6±0,2°, 24,8±0,2° и 26,5±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 26 имеет характеристические пики при значениях 2θ, составляющих 3,6±0,2°, 10,5±0,2°, 11,8±0,2°, 13,9±0,2° и 19,7±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 26 имеет характеристические пики при значениях 2θ, составляющих 3,6±0,2°, 7,4±0,2°, 10,5±0,2°, 11,8±0,2°, 13,9±0,2°, 14,9±0,2°, 16,8±0,2°, 19,7±0,2°, 21,2±0,2° и 23,5±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 26 имеет характеристические пики при значениях 2θ, составляющих 3,6±0,2°, 7,4±0,2°, 10,5±0,2°, 11,8±0,2°, 13,9±0,2°, 14,9±0,2°, 16,8±0,2°, 19,7±0,2°, 21,2±0,2°, 21,5±0,2°, 22,0±0,2°, 23,5±0,2° 25,0±0,2° и 26,0±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 27 имеет характеристические пики при значениях 2θ, составляющих 6,5±0,2°, 10,2±0,2°, 13,2±0,2°, 15,0±0,2° и 23,8±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 27 имеет характеристические пики при значениях 2θ, составляющих 6,5±0,2°, 10,2±0,2°, 13,2±0,2°, 15,0±0,2°, 16,3±0,2°, 20,2±0,2°, 20,6±0,2°, 21,4±0,2°, 23,8±0,2° и 27,0±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 27 имеет характеристические пики при значениях 2θ, составляющих 6,5±0,2°, 10,2±0,2°, 13,2±0,2°, 15,0±0,2°, 16,3±0,2°, 19,3±0,2°, 20,2±0,2°, 20,6±0,2°, 21,4±0,2°, 23,8±0,2°, 24,1±0,2°, 26,0±0,2°, 27,0±0,2°, 27,3±0,2° и 30,6±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 35 имеет характеристические пики при значениях 2θ, составляющих 12,4±0,2°, 14,7±0,2°, 15,3±0,2°, 17,3±0,2° и 23,5±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 35 имеет характеристические пики при значениях 2θ, составляющих 10,8±0,2°, 12,4±0,2°, 13,3±0,2°, 14,7±0,2°, 15,3±0,2°, 17,3±0,2°, 21,8±0,2°, 22,7±0,2°, 23,5±0,2° и 24,0±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 35 имеет характеристические пики при значениях 2θ, составляющих 10,8±0,2°, 12,4±0,2°, 13,3±0,2°, 14,7±0,2°, 15,3±0,2°, 17,3±0,2°, 17,8±0,2°, 19,8±0,2°, 21,8±0,2°, 22,7±0,2°, 23,5±0,2°, 24,0±0,2°, 25,6±0,2°, 26,2±0,2° и 27,4±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 41 имеет характеристические пики при значениях 2θ, составляющих 3,1±0,2°, 5,2±0,2°, 6,7±0,2°, 10,2±0,2° и 19,9±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
предпочтительно соединение 41 имеет характеристические пики при значениях 2θ, составляющих 3,1±0,2°, 5,2±0,2°, 6,7±0,2°,9,1±0,2°, 9,8±0,2°, 10,2±0,2°, 11,5±0,2°, 19,9±0,2°, 20,5±0,2° и 21,6±0,2, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
более предпочтительно соединение 41 имеет характеристические пики при значениях 2θ, составляющих 3,1±0,2°, 5,2±0,2°, 6,7±0,2°, 9,1±0,2°, 9,8±0,2°, 10,2±0,2°, 10,5±0,2°, 11,5±0,2°, 14,1±0,2°, 14,8±0,2°, 19,3±0,2°, 19,9±0,2°, 20,5±0,2°, 21,6±0,2° и 23,1±0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения.
Настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей вышеуказанное соединение и фармацевтически приемлемое вспомогательное вещество.
В одном варианте осуществления лекарственная форма фармацевтической композиции представляет собой таблетку, диспергирующее средство, настойку, гель, капсулу, спрей, суппозиторий, гранулу, жидкость для перорального применения или гранулу.
В одном варианте осуществления лекарственная форма фармацевтической композиции представляет собой препарат для местного применения.
Настоящее изобретение дополнительно относится к применению соединения в изготовлении лекарственного препарата для лечения дерматита и/или заболевания иммунной системы.
В одном варианте осуществления лекарственный препарат применяют для лечения атопического дерматита и/или астмы.
По сравнению с предшествующим уровнем техники настоящее изобретение обеспечивает следующие преимущества.
Индольные соединения по настоящему изобретению, структурно оптимизированные на основе соединений-прототипов, а именно природного низкомолекулярного соединения IAId (индол-3-карбоксальдегида) и другой родственной малой молекулы I3C (индол-3-метанола), были проверены на мышиной модели кальципотриол-(MC903)-индуцированных ушных AD-подобных симптомов. Индольные соединения значительно облегчали AD-подобные симптомы в ухе мыши, некоторые из них характеризовались даже лучшими эффектами, чем IAId и I3C, демонстрируя многообещающие медицинские перспективы.
КРАТКОЕ ОПИСАНИЕ
На фигурах 1-10 показаны XRPD-дифрактограммы соединений 4, 22, 23, 24, 8, 26, 9, 27, 41 и 35 в вариантах осуществления 7, 8, 10, 12, 14, 15, 16, 17, 27 и 31 соответственно;
на фигуре 11 показаны схематические диаграммы общих уровней IgE в сыворотке крови мышей в первой серии экспериментов в варианте осуществления 32;
на фигуре 12 показаны схематические диаграммы значений толщины ушей мышей в первой серии экспериментов в варианте осуществления 32;
на фигуре 13 показаны схематические диаграммы потери веса у мышей в первой серии экспериментов в варианте осуществления 32;
на фигуре 14 показаны фотографии ушей иллюстративных мышей после введения различных лекарственных средств в первой серии экспериментов в варианте осуществления 32;
на фигуре 15 показаны профили окрашивания с помощью HE (гематоксилин-эозин) ушных тканей иллюстративных мышей после введения различных лекарственных средств в первой серии экспериментов в варианте осуществления 32;
на фигуре 16 показаны схематические диаграммы общих уровней IgE в сыворотке крови мышей во второй серии экспериментов в варианте осуществления 32;
на фигуре 17 показаны схематические диаграммы толщины ушей мышей во второй серии экспериментов в варианте осуществления 32;
на фигуре 18 показаны схематические диаграммы потери веса у мышей во второй серии экспериментов в варианте осуществления 32;
на фигуре 19 показаны фотографии ушей иллюстративных мышей после введения различных лекарственных средств во второй серии экспериментов в варианте осуществления 32;
на фигуре 20 показаны профили окрашивания с помощью HE ушных тканей иллюстративных мышей после введения различных лекарственных средств во второй серии экспериментов в варианте осуществления 32;
на фигурах 21-23 показаны схематические диаграммы общих уровней IgE в сыворотке крови мышей в каждой группе в варианте осуществления 33;
на фигурах 24-26 показаны схематические диаграммы значений толщины ушей мышей в каждой группе в варианте осуществления 33;
на фигурах 27-29 показаны схематические диаграммы потери веса мышей в каждой группе в варианте осуществления 33; и
на фигуре 30 показаны фотографии ушей иллюстративных мышей в варианте осуществления 33.
ПОДРОБНОЕ ОПИСАНИЕ
Для лучшего понимания настоящего изобретения настоящее изобретение будет полностью описано ниже со ссылкой на соответствующие сопутствующие фигуры. Предпочтительные варианты осуществления показаны на фигурах. Однако настоящее изобретение может быть осуществлено во множестве различных форм и не ограничивается вариантами осуществления, описанными в данном документе. Скорее, эти варианты осуществления предусмотрены с целью сделать раскрытое содержание настоящего изобретения более подробным и полным.
Если не указано иное, все технические и научные термины, применяемые в данном документе, имеют то же значение, которые обычно понятно специалисту в данной области техники в пределах области техники, к которой принадлежит настоящее изобретение. Термины, применяемые в описании настоящего изобретения в данном документе, предназначены только для целей описания вариантов осуществления и не предназначены для ограничения настоящего изобретения. Термин «и/или», применяемый в данном документе, включает любую одну или все комбинации одного или более соответствующих элементов, перечисленных в данном документе.
Фармацевтические композиции, предусмотренные в данном документе, могут быть составлены в любой лекарственной форме, подходящей для местного введения, для достижения местных или системных эффектов, включая эмульсии, растворы, суспензии, кремы, гели, гидрогели, мази, порошки, повязки, крепкие настойки, лосьоны, суспензии, настойки, пасты, пены, пленки, аэрозоли, промывочные жидкости, спреи, суппозитории, перевязочные материалы и кожные пластыри.
Фармацевтические композиции, предусмотренные в данном документе для перорального введения, могут быть предусмотрены в твердых, полутвердых или жидких лекарственных формах для перорального введения.
Применяемое в данном документе пероральное введение также включает трансбуккальное, подъязычное или сублингвальное введение. Подходящие лекарственные формы для перорального применения включают без ограничения таблетки, таблетки быстрого действия, жевательные таблетки, капсулы, пилюли, пластыри, троше, таблетки для рассасывания, пастилки, крахмальные капсулы, гранулы, медицинские жевательные резинки, сыпучие порошки, шипучие или нешипучие порошки или гранулы, аэрозоли для перорального применения, растворы, эмульсии, суспензионные средства, капсулу-имплантат, присыпки, крепкие настойки и сиропы. В дополнение к указанному активному ингредиенту указанная фармацевтическая композиция может содержать одно или более фармацевтически приемлемых вспомогательных веществ, включая без ограничения связующие средства, наполнители, разбавители, разрыхлители, смачивающие средства, смазочные средства, вещества, способствующие скольжению, красящие вещества, средства, препятствующие миграции, подсластители, ароматизирующие средства, эмульгаторы, суспендирующие и диспергирующие средства, консерванты, растворители, неводные жидкости, органические кислоты и источники диоксида углерода.
Если не указано иное, исходные материалы, применяемые в следующих вариантах осуществления, являются коммерчески доступными; если не указано иное, способы, применяемые в следующих вариантах осуществления, являются общепринятыми способами, которые могут быть осуществлены.
Параметры выявления соединений в следующих вариантах осуществления посредством порошковой рентгеновской дифракции (XRPD) показаны следующим образом.
Диапазон сканирования: 3-40°, шаг сканирования: 0,02°, скорость сканирования: 0,1°/стадия, медная мишень, длина волны: 1,54 Å.
Вариант осуществления 1
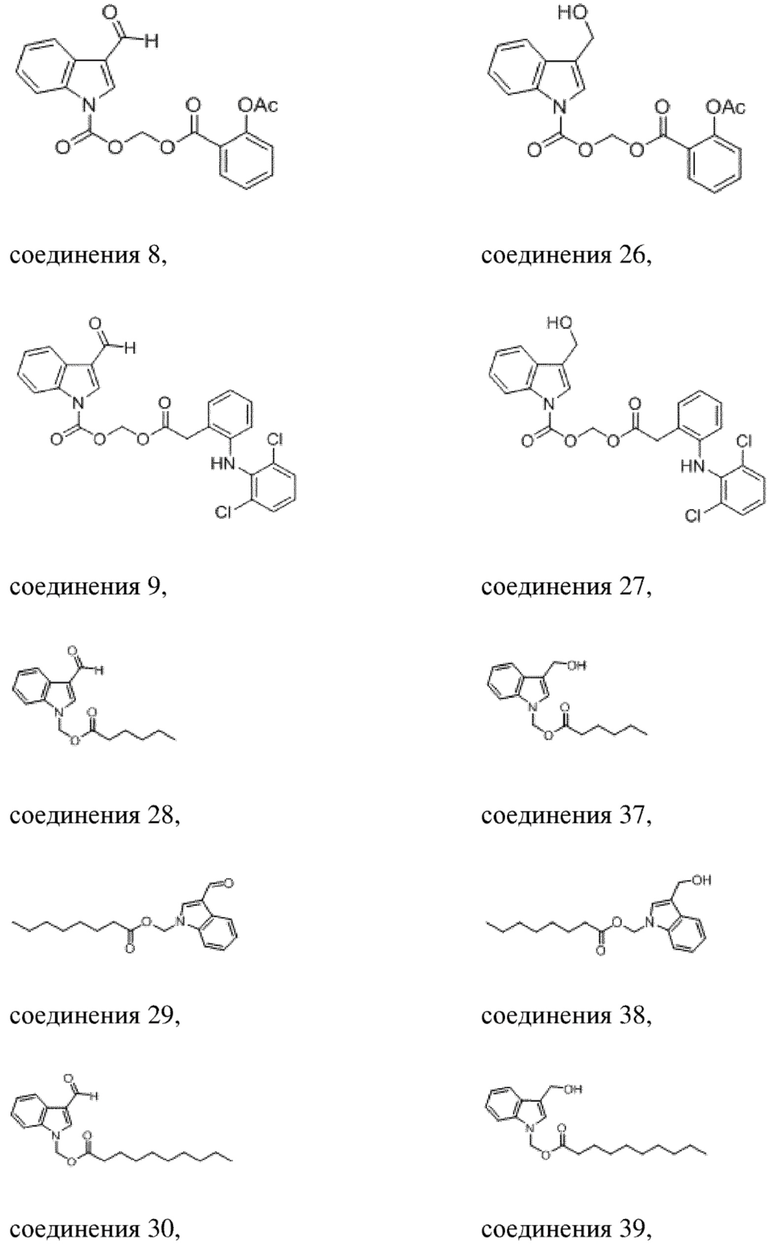
Получение (гексилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 1)
(1) Синтез хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B)
Соединение A Соединение B
3-Индолкарбоксальдегид (соединение A, 5,0 г, 34 ммоль) растворяли в безводном тетрагидрофуране (THF) (60 мл) в защитной атмосфере аргона, а затем после снижения температуры до -78°C в смесь по каплям добавляли раствор бис(триметилсилил)амида лития (LHMDS) в THF (1 M, 51,7 мл, 51,7 ммоль) и обеспечивали осуществление реакции в течение 1 часа при -78°C. Впоследствии в реакционную жидкость по каплям добавляли раствор хлорметилхлорформиата (6,62 г, 51,7 ммоль) в THF (20 мл) и обеспечивали осуществление реакции в течение 2 часов при -78°C. TLC (петролейный эфир/этилацетат = 5:1) демонстрировала, что реакция завершена.
В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (50 мл) с последующим экстрагированием этилацетатом (3×40 мл). Затем органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент: петролейный эфир:этилацетат = от 100:1 до 1:1) с получением желтого твердого вещества, т.е. требуемого соединения, представляющего собой хлорметил-3-формил-1H-индол-1-карбоксилат (2) (3,0 г, выход: 37,5%).
Характеристические данные применительно к соединению B были следующими: 1H ЯМР (400 МГц, DMSO) δ 10,10 (s, 1H), 8,77 (s, 1H), 8,17 (dd, J = 7,9, 3,2 Гц, 2H), 7,55-7,40 (m, 2H), 6,25 (s, 2H).
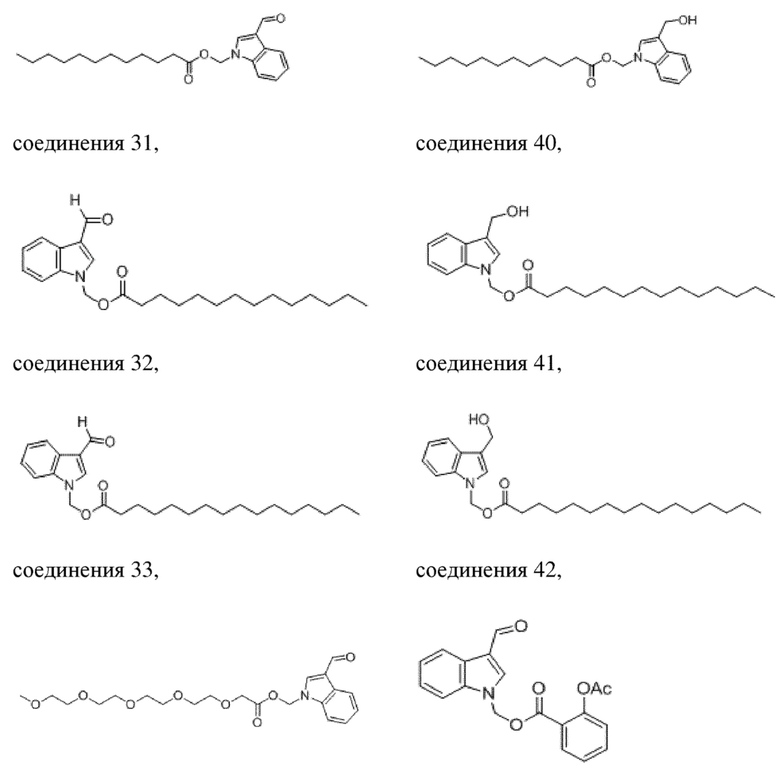
(2) Синтез (гексилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 1)
Соединение B Соединение 1
Йодид натрия (252 мг, 1,69 ммоль) и карбонат калия (1,4 г, 10,11 ммоль) добавляли в раствор хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B) (800 мг, 3,37 ммоль) и гексановой кислоты (391 мг, 3,37 ммоль) в безводном DMF (8,0 мл) при 0°C и обеспечивали осуществление реакции в смеси в течение 18 часов при комнатной температуре. TLC (петролейный эфир:этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент: петролейный эфир:этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (гексилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 1) (570 мг, выход: 53,6%).
Характеристические данные применительно к соединению 1 были следующими: 1H ЯМР (400 МГц, DMS): δ 9,98 (s, 1H), 8,43 (s, 1H), 8,12 (d, J=7,7 Гц, 1H), 7,70 (d, J=8,1 Гц, 1H), 7,34 (dtd, J=14,9, 7,6, 1,1 Гц, 2H), 6,31 (s, 2H), 2,32 (t, J=7,3 Гц, 2H), 1,54-1,40 (m, 2H), 1,23-1,05 (m, 4H), 0,75 (t, J=7,0 Гц, 3H).
Чистота соединения 1, выявленная посредством HPLC, составляла 98,71% при 254 нм и 98,70% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 2
Получение (гексилокси)метил-3-(гидроксиметил)-1H-индолкарбоксилата (соединение 19)
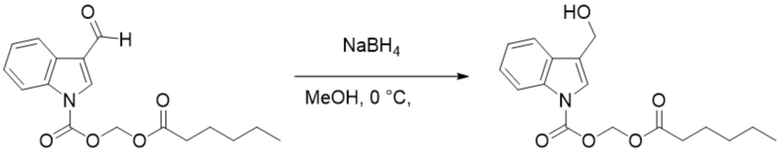
Соединение 1 Соединение 19
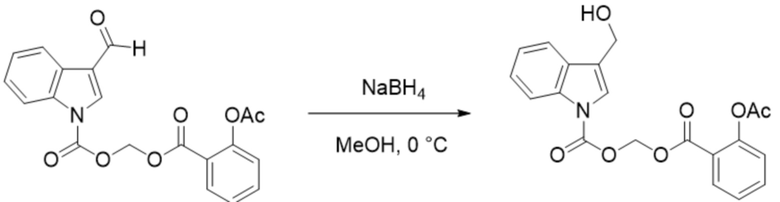
(Гексилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 1) (240 мг, 0,76 ммоль) растворяли в безводном метаноле (8 мл). После снижения температуры до 0°C добавляли борогидрид натрия (28,8 мг, 0,76 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. Выявление посредством TLC (петролейный эфир:этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (гексилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 19) (200 мг, выход: 82,9%).
Характеристические данные применительно к соединению 19 были следующими: 1H ЯМР (400 МГц, DMS): δ 7,61 (d, J=7,8 Гц, 1H), 7,54 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,24-7,16 (m, 1H), 7,13-7,04 (m, 1H), 6,17 (s, 2H), 4,91 (t, J = 5,4 Гц, 1H), 4,62 (d, J = 5,2 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,52-1,41 (m, 2H), 1,30-1,08 (m, 4H), 0,78 (t, J=7,0 Гц, 3H).
Чистота соединения 19, выявленная посредством HPLC, составляла 96,18% при 254 нм и 96,51% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 3
Получение (октилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 2)
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез (октилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 2)
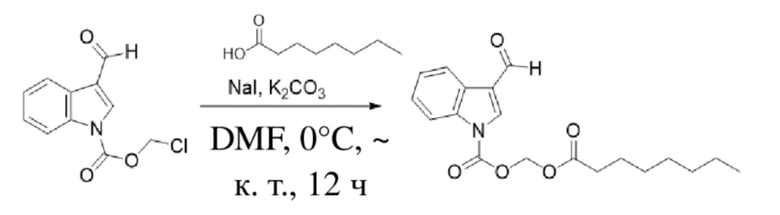
Соединение B Соединение 2
Йодид натрия (504 мг, 3,37 ммоль) и карбонат калия (1,4 г, 10,11 ммоль) добавляли в раствор хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B) (800 мг, 3,37 ммоль) и октановой кислоты (583,2 мг, 4,05 ммоль), растворенных в безводном DMF (8,0 мл), при 0°C и обеспечивали осуществление реакции в смеси в течение 18 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Затем органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (октилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 2) (480 мг, выход: 41,3%).
Характеристические данные применительно к соединению 2 были следующими: 1H ЯМР (400 МГц, DMS) δ 9,97 (s, 1H), 8,42 (s, 1H), 8,11 (d, J=7,7 Гц, 1H), 7,70 (d, J=8,1 Гц, 1H), 7,44-7,26 (m, 2H), 6,30 (s, 2H), 2,32 (t, J=7,2 Гц, 2H), 1,50-1,41 (m, 2H), 1,11 (s, 8H), 0,80 (t, J=7,0 Гц, 3H).
Чистота соединения 2, выявленная посредством HPLC, составляла 99,42% при 254 нм и 98,47% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 4
Получение (октилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 20)
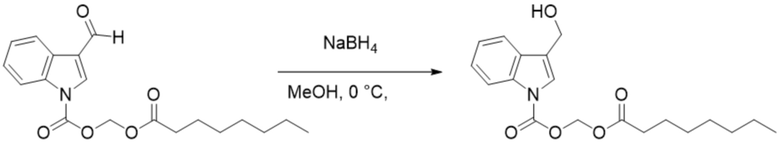
Соединение 2 Соединение 20
(Октилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 2) (240 мг, 0,76 ммоль) растворяли в безводном метаноле (8 мл). После снижения температуры до 0°C добавляли борогидрид натрия (28,8 мг, 0,76 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (октилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 20) (150 мг, выход: 57%).
Характеристические данные применительно к соединению 20 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,8 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,1 Гц, 1H), 7,09 (t, J=7,0 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,62 (d, J=5,4 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,47 (d, J = 6,9 Гц, 2H), 1,22 (d, J=10,1 Гц, 2H), 1,16 (s, 7H), 0,82 (t, J=7,0 Гц, 3H).
Чистота соединения 20, выявленная посредством HPLC, составляла 95,69% при 254 нм и 95,81% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 5
Получение (деканоилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 3)
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез (деканоилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 3)
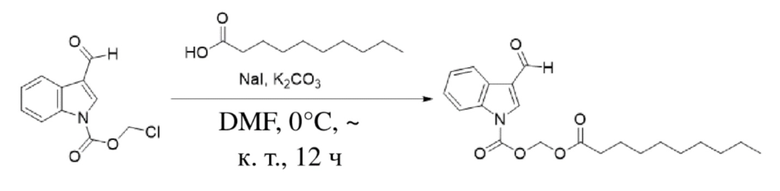
Соединение B. Соединение 3
Йодид натрия (442 мг, 2,95 ммоль) и карбонат калия (1,2 г, 8,85 ммоль) добавляли к раствору хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B) (700 мг, 2,95 ммоль) и декановой кислоты (560 мг, 3,24 ммоль), растворенных в безводном DMF (8,0 мл), при 0°C и обеспечивали осуществление реакции в смеси в течение 18 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (деканоилокси)метил-3-формил-1H-индол-1-карбоксилат (500 мг, выход: 50%).
Характеристические данные применительно к соединению 3 были следующими: 1H ЯМР (400 МГц, DMS) δ 9,97 (s, 1H), 8,42 (s, 1H), 8,11 (d, J=7,8 Гц, 1H), 7,70 (d, J=8,1 Гц, 1H), 7,34 (dt, J=25,0, 7,2 Гц, 2H), 6,30 (s, 2H), 2,31 (t, J=7,2 Гц, 2H), 1,47 (d, J = 6,6 Гц, 2H), 1,23 (d, J=6,0 Гц, 2H), 1,13 (d, J=12,2 Гц, 10H), 0,84 (t, J=7,0 Гц, 3H).
Чистота соединения 3, выявленная посредством HPLC, составляла 98,70% при 254 нм и 98,85% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 6
Получение (деканоилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 21)
Соединение 3 Соединение 21
(Деканоилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 3) (300 мг, 0,803 ммоль) растворяли в безводном метаноле (8 мл). После снижения температуры до 0°C добавляли борогидрид натрия (31 мг, 0,803 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (деканоилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 21) (200 мг, выход: 66,5%).
Характеристические данные применительно к соединению 21 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,8 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,2 Гц, 1H), 7,09 (t, J=7,4 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,2 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,52-1,38 (m, 2H), 1,20 (d, J=31,1 Гц, 12H), 0,85 (t, J=6,9 Гц, 3H).
Чистота соединения 21, выявленная посредством HPLC, составляла 97,36% при 254 нм и 97,56% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 7
Получение (додецилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 4)
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез (додецилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 4)
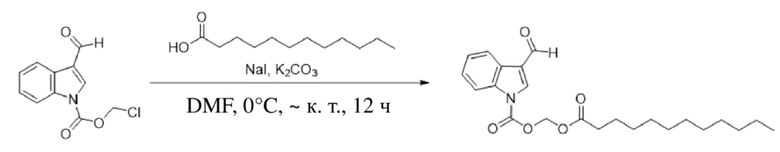
Соединение B Соединение 4
Йодид натрия (504 мг, 3,37 ммоль) и карбонат калия (1,4 г, 10,11 ммоль) добавляли в раствор хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B) (800 мг, 3,36 ммоль) и додекановой кислоты (800 мг, 4,03 ммоль), растворенных в безводном DMF (8,0 мл), при 0°C и обеспечивали осуществление реакции в смеси в течение 18 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде желтого твердого вещества, т.е. требуемого соединения, представляющего собой (додецилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 4) (500 мг, выход: 37%).
Характеристические данные применительно к соединению 4 были следующими: 1H ЯМР (400 МГц, DMS) δ 9,97 (s, 1H), 8,42 (s, 1H), 8,11 (d, J=7,7 Гц, 1H), 7,70 (d, J=8,3 Гц, 1H), 7,34 (dd, J=17,5, 7,4 Гц, 2H), 6,30 (s, 2H), 2,31 (t, J=7,2 Гц, 2H), 1,46 (s, 2H), 1,27-1,05 (m, 17H), 0,85 (t, J=6,9 Гц, 3H).
Чистота соединения 4, выявленная посредством HPLC, составляла 98,06% при 254 нм и 96,38% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
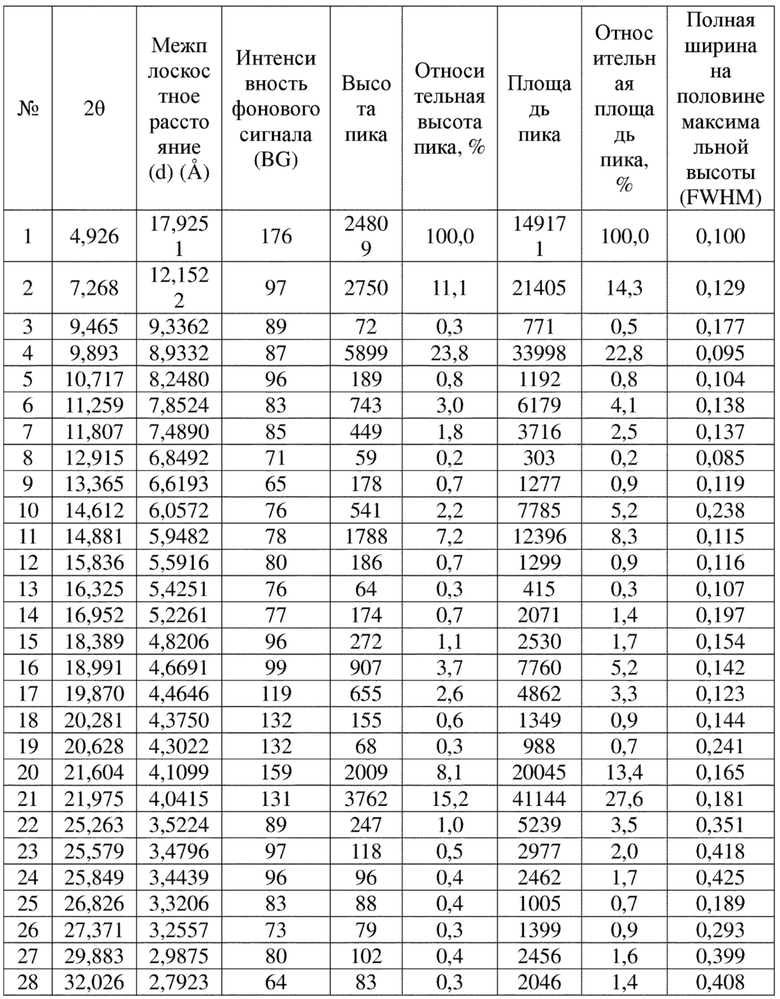
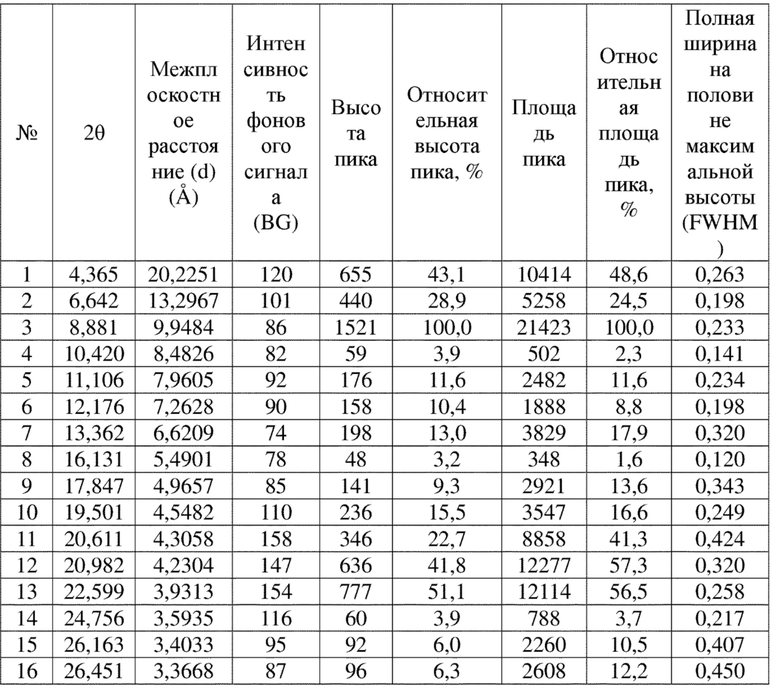
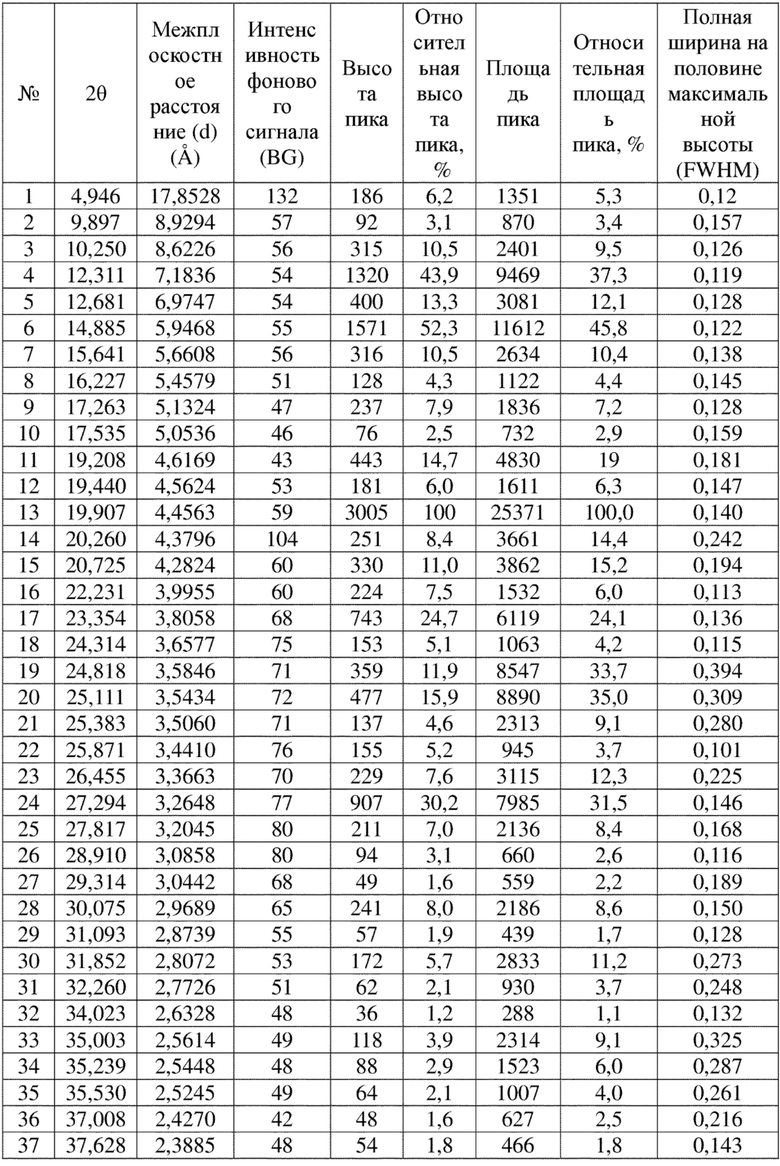
Соединение 4 перекристаллизовывали с получением чистого кристалла I соединения 4. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 1 и на фигуре 1.
Таблица 1. Данные XRPD применительно к кристаллу I соединения 4
Вариант осуществления 8
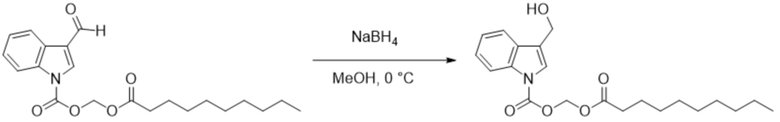
Получение (додецилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 22)
Соединение 4 Соединение 22
(Додецилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 4) (250 мг, 0,76 ммоль) растворяли в безводном метаноле (8 мл). После снижения температуры до 0°C добавляли борогидрид натрия (28,8 мг, 0,76 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде желтого твердого вещества, т.е. требуемого соединения 22, представляющего собой (додецилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (150 мг, выход: 57%).
Характеристические данные применительно к соединению 22 были следующими: 1H ЯМР (400 МГц, DMS°) δ 7,61 (d, J=7,7 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,18 (dd, J=11,2, 4,1 Гц, 1H), 7,09 (t, J=7,0 Гц, 1H), 6,17 (s, 2H), 4,89 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,1 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,48-1,40 (m, 2H), 1,23 (d, J=7,9 Гц, 8H), 1,15 (s, 8H), 0,85 (t, J=6,9 Гц, 3H).
Чистота соединения 22, выявленная посредством HPLC, составляла 98,63% при 254 нм и 98,21% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 22 перекристаллизовывали с получением чистого кристалла I соединения 22. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 2 и на фигуре 2.
Таблица 2. Данные XRPD применительно к кристаллу I соединения 22
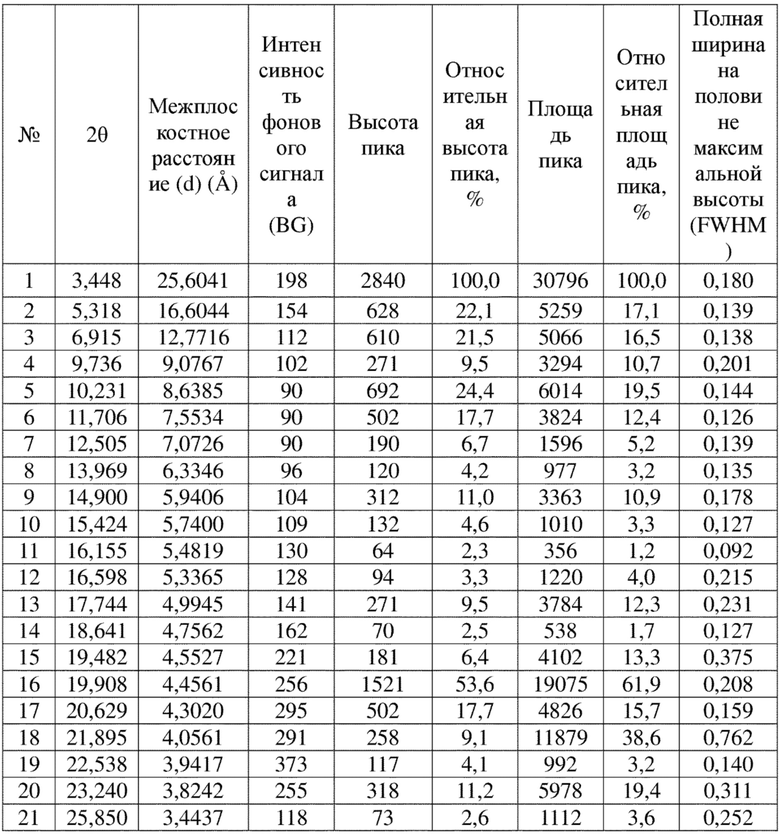
Условия обнаружения: СКАНИРОВАНИЕ: 3,0001/39,9937/0,01948/16 (сек), Cu, I (макс.) = 3038.
ПИК: 19-Pts/параболический фильтр, пороговое значение = 3,0, значение отсечки = 0,1%, BG = 3/1,0, наивысшая точка пика = вершина.
Примечание: интенсивность = количество импульсов, 2T(0) = 0,0 (град.), длина волны для вычисления d-расстояния = 1,54056 Å (Cu/K-альфа1).
Вариант осуществления 9
Получение (тетрадецилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 5)
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез (тетрадецилокси)метил-3-формил-1H-индол-1-карбоксилата
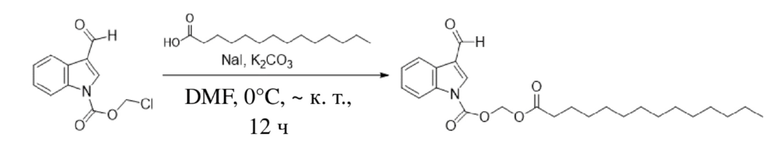
Соединение B Соединение 5
Йодид натрия (442 мг, 2,95 ммоль) и карбонат калия (1,2 г, 8,85 ммоль) добавляли к раствору хлорметил-3-формил-1H-индол-1-карбоксилата (2) (700 мг, 2,95 ммоль) и тетрадекановой кислоты (738 мг, 3,24 ммоль), растворенных в безводном DMF (8,0 мл), при 0°C и обеспечивали осуществление реакции в смеси в течение 18 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (тетрадецилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 5) (500 мг, выход: 39,7%).
Характеристические данные применительно к соединению 5 были следующими: 1H ЯМР (400 МГц, DMS) δ 9,97 (s, 1H), 8,42 (s, 1H), 8,11 (d, J=7,7 Гц, 1H), 7,69 (d, J=8,1 Гц, 1H), 7,46-7,22 (m, 2H), 6,30 (s, 2H), 2,31 (t, J=7,2 Гц, 2H), 1,53-1,37 (m, 2H), 1,27-1,17 (m, 12H), 1,11 (s, 8H), 0,85 (t, J=6,8 Гц, 3H).
Чистота соединения 5, выявленная посредством HPLC, составляла 99,76% при 254 нм и 99,69% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 10
Получение (тетрадецилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 23)
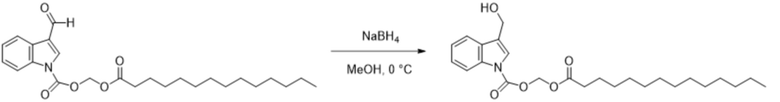
Соединение 5 Соединение 23
(Тетрадеканоилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 5) (250 мг, 0,58 ммоль) растворяли в безводном метаноле/тетрагидрофуране (5 мл/5 мл). После снижения температуры до 0°C добавляли борогидрид натрия (22 мг, 0,58 ммоль) и обеспечивали осуществление реакции в смеси в течение 1 часа при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Затем органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (тетрадецилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 23) (200 мг, выход: 80%).
Характеристические данные применительно к соединению 23 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,60 (dd, J=7,6, 3,7 Гц, 1H), 7,53 (dd, J=8,2, 3,8 Гц, 1H), 7,33 (d, J=3,8 Гц, 1H), 7,19 (t, J=7,5 Гц, 1H), 7,09 (td, J=7,3, 3,5 Гц, 1H), 6,16 (d, J=3,9 Гц, 2H), 4,89 (dd, J=10,0, 5,2 Гц, 1H), 4,68-4,55 (m, 2H), 2,25 (td, J=7,2, 3,8 Гц, 2H), 1,45 (s, 2H), 1,23 (s, 12H), 1,15 (s, 8H), 0,93-0,78 (m, 3H).
Чистота соединения 23, выявленная посредством HPLC, составляла 98,71% при 254 нм и 98,03% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 23 перекристаллизовывали с получением чистого кристалла I соединения 23. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 3 и на фигуре 3.
Таблица 3. Данные XRPD применительно к кристаллу I соединения 23
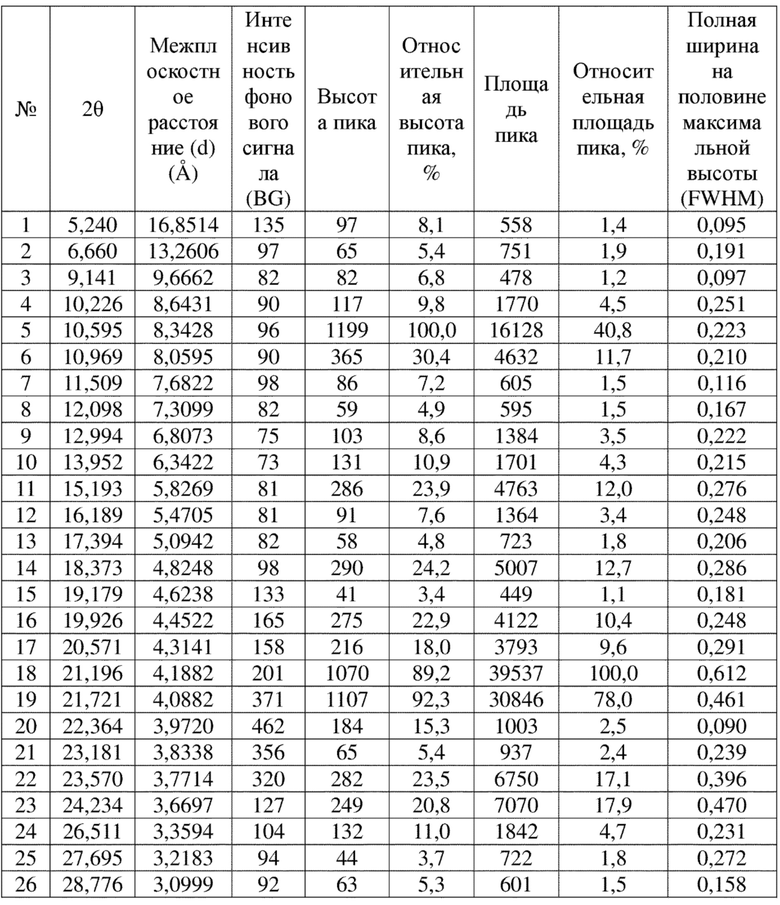
Вариант осуществления 11
Получение (пальмитоилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 6)
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез йодметил-3-формил-1H-индол-1-карбоксилата (соединение C)
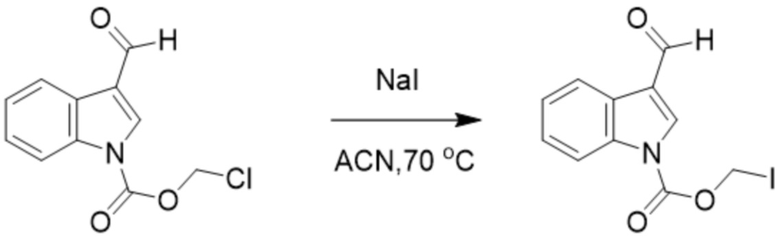
Соединение B Соединение C
Хлорметил-3-формил-1H-индол-1-карбоксилат (соединение B) (2,3 г, 9,66 ммоль) и йодид натрия (4,3 г 29,2 ммоль) растворяли в ацетонитриле (20 мл). После повышения температуры до 70°C обеспечивали осуществление реакции в смеси в течение 4 часов. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Реакционную жидкость фильтровали и фильтрат экстрагировали этилацетатом. Впоследствии жидкость концентрировали с получением йодметил-3-формил-1H-индол-1-карбоксилата (соединение C) (2,4 г, выход: 77%).
(3) Синтез пальмитата серебра (соединение C-2)

Соединение C-1 Соединение C-2
Пальмитиновую кислоту (соединение C-1) (2 г, 7,8 ммоль) растворяли в растворе гидроксида натрия (312 мг/40 мл). После повышения температуры до 80°C в смесь добавляли нитрат серебра (1,32 г, 7,8 ммоль) с осаждением белого твердого вещества. Впоследствии смесь охлаждали до комнатной температуры, фильтровали и осадок на фильтре промывали водой и высушивали с получением пальмитата серебра (соединение C-2) (2,5 г, выход: 89%).
(4) Синтез (пальмитоилокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 6)
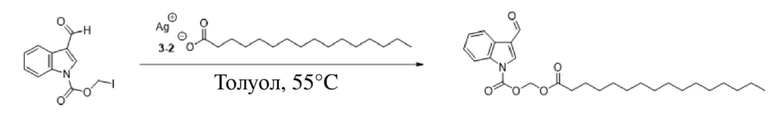
Соединение C Соединение 6
Пальмитат серебра (2,1 г, 5,1 ммоль) добавляли в раствор йодметил-3-формил-1H-индол-1-карбоксилата (2) (1,5 г, 4,6 ммоль), растворенного в безводном метилбензоле (20 мл), при 0°C. После повышения температуры до 55°C обеспечивали осуществление реакции в смеси в течение 4 часов. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. Реакционную жидкость фильтровали и фильтрат экстрагировали этилацетатом (3×50 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (пальмитоилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 6) (800 мг, выход: 38%).
Характеристические данные применительно к соединению 6 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,60 (dd, J=7,6, 3,7 Гц, 1H), 7,53 (dd, J=8,2, 3,8 Гц, 1H), 7,33 (d, J=3,8 Гц, 1H), 7,19 (t, J=7,5 Гц, 1H), 7,09 (td, J=7,3, 3,5 Гц, 1H), 6,16 (d, J=3,9 Гц, 2H), 4,89 (dd, J=10,0, 5,2 Гц, 1H), 4,68-4,55 (m, 2H), 2,25 (td, J=7,2, 3,8 Гц, 2H), 1,45 (s, 2H), 1,23 (s, 12H), 1,15 (s, 8H), 0,93-0,78 (m, 3H).
Чистота соединения 6, выявленная посредством HPLC, составляла 99,03% при 254 нм и 97,54% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 12
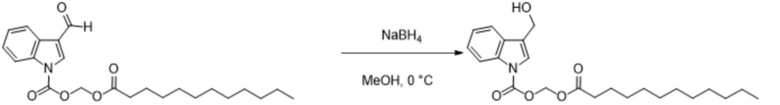
Получение (пальмитоилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 24)
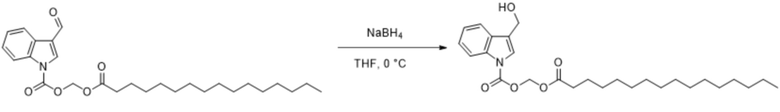
Соединение 6 Соединение 24
(Пальмитоилокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 6) (300 мг, 0,65 ммоль) растворяли в безводном тетрагидрофуране (8 мл). После снижения температуры до 0°C добавляли борогидрид натрия (25,8 мг, 0,65 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (пальмитоилокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 24) (150 мг, выход: 50%).
Характеристические данные применительно к соединению 24 были следующими: 1H ЯМР (400 МГц, DMS) δ 8,05 (d, J=7,8 Гц, 1H), 7,67 (d, J=7,7 Гц, 1H), 7,54 (s, 1H), 7,36 (dd, J=11,3, 4,2 Гц, 1H), 7,29 (t, J=7,5 Гц, 1H), 6,00 (s, 2H), 5,16 (t, J=5,5 Гц, 1H), 4,64 (dd, J=5,5, 1,0 Гц, 2H), 2,41 (t, J=7,2 Гц, 2H), 1,59-1,47 (m, 2H), 1,25 (d, J=16,2 Гц, 16H), 1,14 (s, 8H), 0,85 (t, J=6,8 Гц, 3H).
Чистота соединения 24, выявленная посредством HPLC, составляла 97,66% при 254 нм и 98,28% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 24 перекристаллизовывали с получением чистого кристалла I соединения 24. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 4 и на фигуре 4.
Таблица 4. Данные XRPD применительно к кристаллу I соединения 24
Вариант осуществления 13
Получение эстерифицированного 3-оксо-2,5,8,11,14,17-гексаоксаоктил-3-формил-1H-индол-1-карбоксилата (соединение 7).
(1) Соединение B получали в соответствии со способом из варианта осуществления 1.
(2) Синтез эстерифицированного 3-оксо-2,5,8,11,14,17-гексаоксаоктил-3-формил-1H-индол-1-карбоксилата (соединение 7).
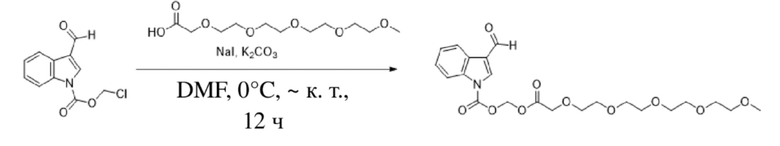
Соединение B Соединение 7
Йодид натрия (1,1 г, 7,5 ммоль) и карбонат калия (3,1 г, 22,5 ммоль) добавляли к раствору хлорметил-3-формил-1H-индол-1-карбоксилата (соединение B) (1,78 г, 7,5 ммоль) и 2,5,8,11,14-пентаоксагексадекан-16-овой кислоты (2,0 г, 7,5 ммоль), растворенных в безводном DMF (8,0 мл), при 0°C и обеспечивали осуществление реакции в смеси в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (3×40 мл). Впоследствии органические фазы объединяли, промывали водой (50 мл) и насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением желтого маслянистого продукта, т.е. требуемого эстерифицированного соединения, представляющего собой 3-оксо-2,5,8,11,14,17-гексаоксаоктил-3-формил-1H-индол-1-карбоксилат (соединение 7) (600 мг, выход: 17,1%).
Характеристические данные применительно к соединению 7 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1H), 8,36-8,25 (m, 1H), 7,95 (s, 1H), 7,54 (d, J=7,4 Гц, 1H), 7,38 (dq, J=7,2, 6,0 Гц, 2H), 6,20 (s, 2H), 4,18 (s, 2H), 3,68 (d, J=5,2 Гц, 2H), 3,66-3,56 (m, 12H), 3,53 (dd, J=5,7, 3,5 Гц, 2H), 3,36 (s, 3H).
Чистота соединения 7, выявленная посредством HPLC, составляла 96, 07% при 254 нм и 95,59% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 14
Получение ((2-ацетоксибензоил)окси)метил-3-формил-1H-индол-1-карбоксилата (соединение 8)
(1) Синтез 2-ацетоксибензоата серебра (R8_Ag+).
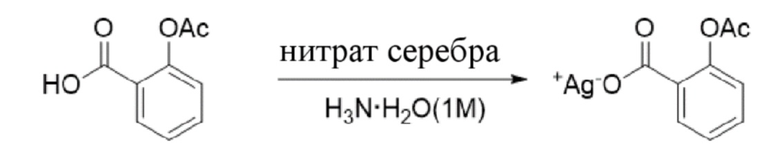
R8 R8_Ag+
2-Ацетоксибензойную кислоту (соединение R8) (2,0 г, 11,1 ммоль) растворяли в 1 M растворе гидроксида аммония (30 мл). После охлаждения до температуры 0°C добавляли водный раствор нитрата серебра (1,89 г, 11,1 ммоль, 10 мл) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Осаждалось большое количество белого твердого вещества, и смесь фильтровали для сбора продукта в виде белого твердого вещества. Осадок на фильтре промывали водой и твердое вещество высушивали с получением 2-ацетоксибензоата серебра (R8_Ag+) (2,5 г, выход: 78,6%).
(2) Синтез ((2-ацетоксибензоил)окси)метил-3-формил-1H-индол-1-карбоксилата
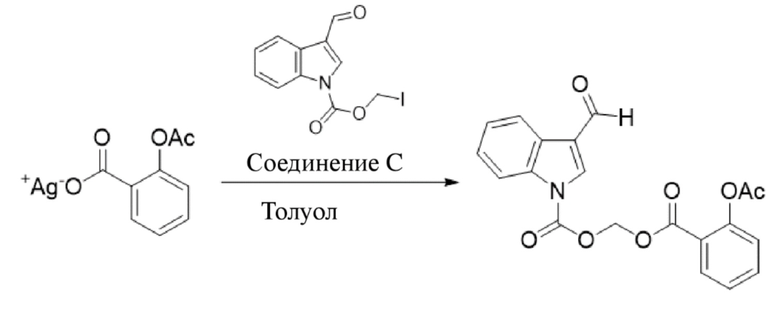
R8_Ag+ Соединение 8
2-Ацетоксибензоат серебра (1,25 г, 4,35 ммоль) и йодметил-3-формил-1H-индол-1-карбоксилат (соединение C) (1,0 г, 3,0 ммоль) добавляли к метилбензолу (10 мл) и обеспечивали осуществление реакции в смеси в течение 4 часов при 40°C. LCMS демонстрировала, что йодметил-3-формил-1H-индол-1-карбоксилат прореагировал полностью. Реакционную жидкость фильтровали и фильтрат экстрагировали этилацетатом (3×20 мл) и водой (40 мл). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде светло-желтого твердого вещества, т.е. требуемого соединения, представляющего собой ((2-ацетоксибензоил)окси)метил-3-формил-1H-индол-1-карбоксилат (соединение 8) (500 мг, выход: 43,7%).
Характеристические данные применительно к соединению 8 были следующими: 1H ЯМР (400 МГц, DMS) δ 10,11 (s, 1H), 8,75 (s, 1H), 8,17 (d, J = 9,1 Гц, 2H), 8,05 (dd, J=7,9, 1,6 Гц, 1H), 7,80-7,70 (m, 1H), 7,47 (ddd, J=24,6, 16,8, 8,2 Гц, 3H), 7,29 (d, J=7,2 Гц, 1H), 6,28 (s, 2H), 2,27 (s, 3H).
Чистота соединения 8, выявленная посредством HPLC, составляла 95,96% при 254 нм и 95,28% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
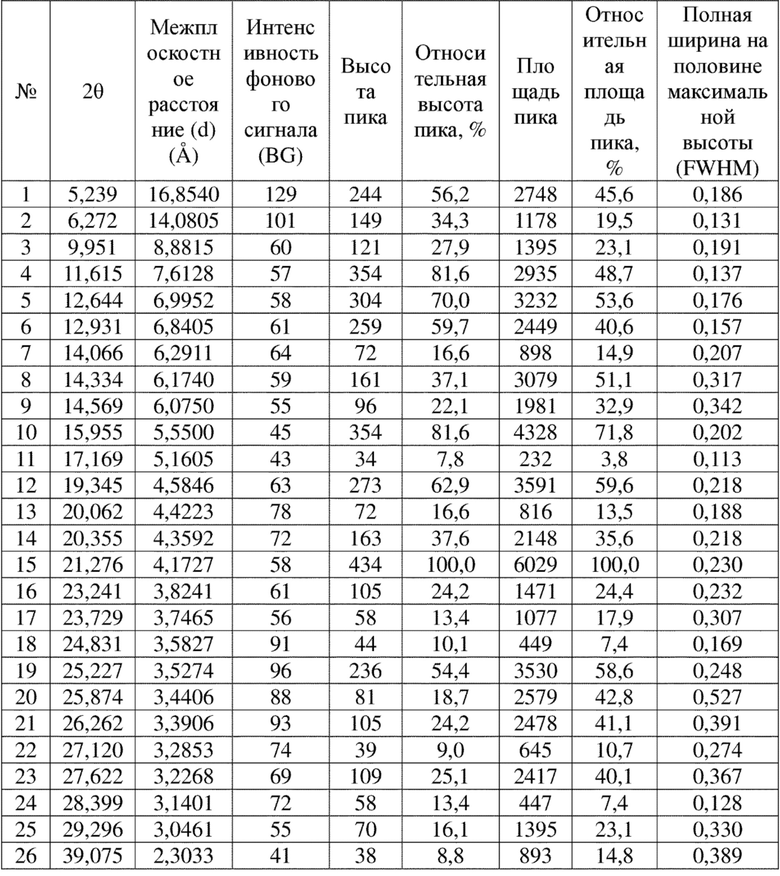
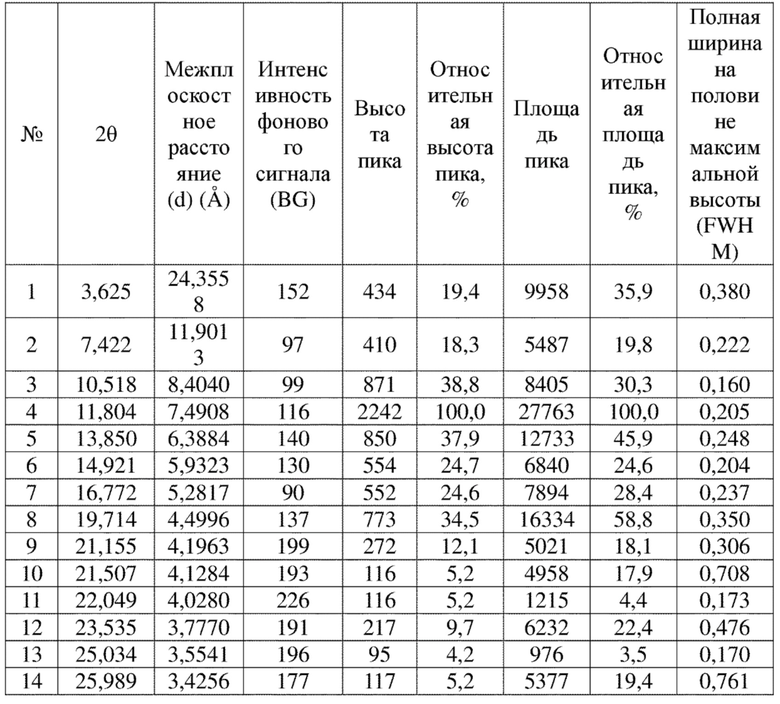
Соединение 8 перекристаллизовывали с получением чистого кристалла I соединения 8. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 5 и на фигуре 5.
Таблица 5. Данные XRPD применительно к кристаллу I соединения 8
Вариант осуществления 15
Получение ((2-ацетоксибензоил)окси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 26)
Соединение 8 Соединение 26
((2-Ацетоксибензоил)окси)метил-3-формил-1H-индол-1-карбоксилат (соединение 8) (170 мг, 0,446 ммоль) растворяли в безводном метаноле/тетрагидрофуране (3 мл/3 мл). После снижения температуры до 0°C добавляли борогидрид натрия (17 мг, 0,446 ммоль) и обеспечивали осуществление реакции в смеси в течение 3 часов при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде светло-желтого твердого вещества, т.е. требуемого соединения, представляющего собой ((2-ацетоксибензоил)окси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 26) (110 мг, выход: 64,7%).
Характеристические данные применительно к соединению 26 были следующими: 1H ЯМР (400 МГц, DMS) δ 8,09 (d, J=8,4 Гц, 1H), 8,01 (dd, J=7,9, 1,6 Гц, 1H), 7,74 (td, J=7,8, 1,7 Гц, 1H), 7,68 (d, J=7,6 Гц, 1H), 7,58 (s, 1H), 7,44 (dd, J=11,0, 4,3 Гц, 1H), 7,39 (t, J=7,3 Гц, 1H), 7,33-7,23 (m, 2H), 6,22 (s, 2H), 5,16 (t, J=5,5 Гц, 1H), 4,69-4,59 (m, 2H), 2,26 (s, 3H).
Чистота соединения 26, выявленная посредством HPLC, составляла 96,35% при 254 нм и 95,62% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 26 перекристаллизовывали с получением чистого кристалла I соединения 26. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 6 и на фигуре 6.
Таблица 6. Данные XRPD применительно к кристаллу I соединения 26
Вариант осуществления 16
Получение (2-(2-((2,6-дихлорфенил)амино)фенил)ацетокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 9)
(1) Синтез 2-(2-((2,6-дихлорфенил)амино)фенил)ацетата серебра (R9_Ag+).
R9 R9_Ag+
2-(2-((2,6-Дихлорфенил)амино)фенил)уксусную кислоту (соединение R9) (3,0 г, 10,1 ммоль) растворяли в 1 M растворе гидроксида аммония (30 мл). Смесь охлаждали до 0°C с последующим добавлением в смесь водного раствора нитрата серебра (1,7 г, 10,1 ммоль, 10 мл) и затем обеспечивали осуществление реакции в смеси в течение часа при 0°C. Осаждалось большое количество белого твердого вещества, и смесь фильтровали для сбора продукта в виде белого твердого вещества. Осадок на фильтре промывали водой и высушивали с получением 2-(2-((2-,6-дихлорфенил)амино)фенил)ацетата серебра (R9_Ag+) (2,9 г, выход: 71%).
(2) Синтез (2-(2-((2,6-дихлорфенил)амино)фенил)ацетокси)метил-3-формил-1H-индол-1-карбоксилата (соединение 9)
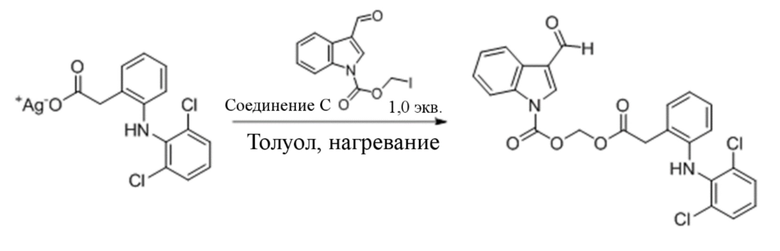
R9_Ag+ Соединение 9
2-(2-((2-,6-Дихлорфенил)амино)фенил)ацетат серебра (R9_Ag+) (2,2 г, 5,5 ммоль) и йодметил-3-формил-1H-индол-1-карбоксилат (соединение C) (1,5 г, 4,6 ммоль) растворяли в метилбензоле и обеспечивали осуществление реакции в смеси в течение 4 часов при 40°C. LCMS демонстрировала, что йодметил-3-формил-1H-индол-1-карбоксилат (соединение C) прореагировал полностью. Реакционную жидкость фильтровали и фильтрат экстрагировали этилацетатом (3×20 мл) и водой (40 мл). Впоследствии органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (2-(2-((2,6-дихлорфенил)амино)фенил)ацетокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 9) (500 мг, выход: 43,7%).
Характеристические данные применительно к соединению 9 были следующими: 1H ЯМР (400 МГц, DMS) δ 10,08 (s, 1H), 8,68 (s, 1H), 8,14 (dd, J=19,1, 7,6 Гц, 2H), 7,45 (t, J=12,0 Гц, 4H), 7,24-7,13 (m, 2H), 7,03 (dd, J=17,5, 10,2 Гц, 2H), 6,82 (d, J=7,4 Гц, 1H), 6,20 (d, J=8,4 Гц, 1H), 6,11 (s, 2H), 3,98 (s, 3H).
Чистота соединения 9, выявленная посредством HPLC, составляла 98,7% при 254 нм и 98,8% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
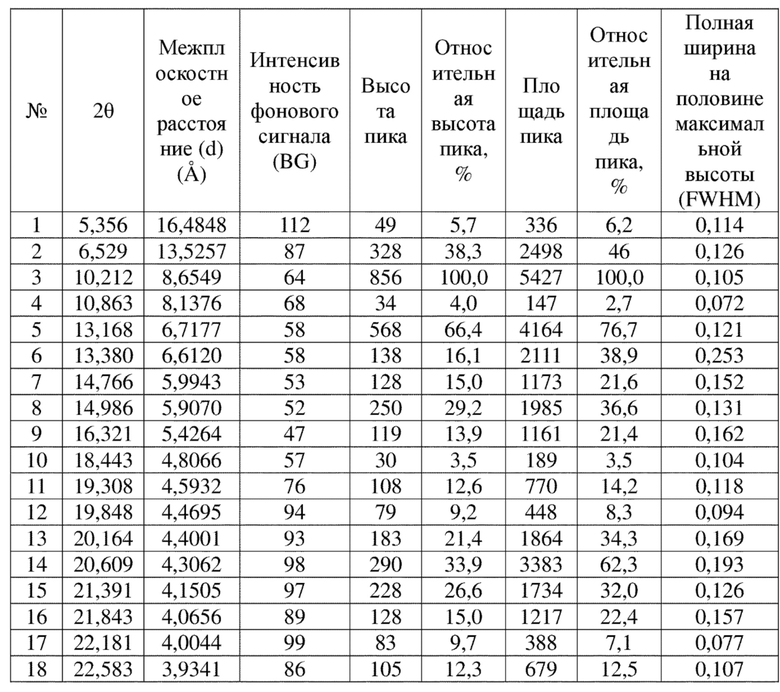
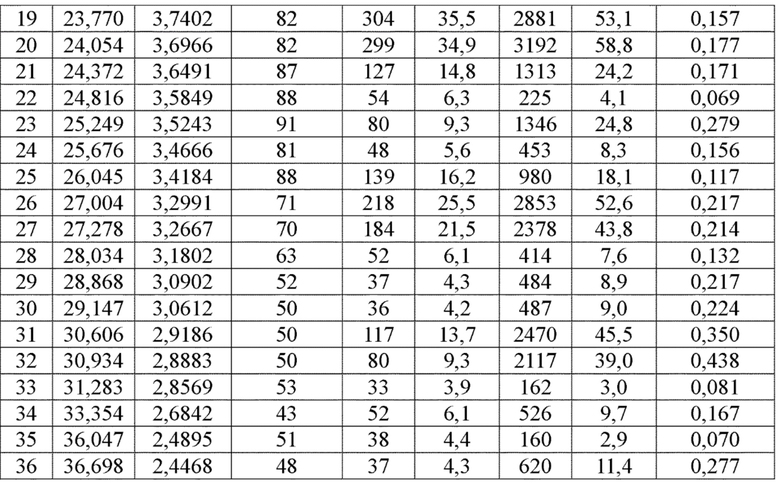
Соединение 9 перекристаллизовывали с получением чистого кристалла I соединения 9. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 7 и на фигуре 7.
Таблица 7. Данные XRPD применительно к кристаллу I соединения 9
Вариант осуществления 17
Получение (2-(2-((2,6-дихлорфенил)амино)фенил)ацетокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилата (соединение 27)
Соединение 9 Соединение 27
(2-(2-((2-,6-Дихлорфенил)амино)фенил)ацетокси)метил-3-формил-1H-индол-1-карбоксилат (соединение 9) (80 мг, 0,16 ммоль) растворяли в безводном тетрагидрофуране (3 мл). После снижения температуры до 0°C добавляли борогидрид натрия (6 мг, 0,16 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при 0°C. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении, объединяли с MC20-875-048 и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 1:1) с получением продукта в виде светло-желтого твердого вещества, т.е. требуемого соединения, представляющего собой (2-(2-((2,6-дихлорфенил)амино)фенил)ацетокси)метил-3-(гидроксиметил)-1H-индол-1-карбоксилат (соединение 27) (110 мг, выход: 52%).
Характеристические данные применительно к соединению 27 были следующими: 1H ЯМР (400 МГц, DMS) δ 8,04 (d, J=7,7 Гц, 1H), 7,67 (d, J=7,7 Гц, 1H), 7,57-7,44 (m, 3H), 7,31 (dt, J=25,2, 6,8 Гц, 2H), 7,19 (t, J=8,1 Гц, 2H), 7,10-6,95 (m, 2H), 6,80 (t, J=7,4 Гц, 1H), 6,25-6,15 (m, 1H), 6,05 (s, 2H), 5,15 (t, J=5,5 Гц, 1H), 4,67-4,56 (m, 2H), 3,95 (s, 2H).
Чистота соединения 27, выявленная посредством HPLC, составляла 95,96% при 254 нм и 95,28% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 27 перекристаллизовывали с получением чистого кристалла I соединения 27. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 8 и на фигуре 8.
Таблица 8. Данные XRPD применительно к кристаллу I соединения 27
Вариант осуществления 18
Получение (3-формил-1H-индол-1-ил)метилгексаноата (соединение 28)
(1) Синтез хлорметилгексаноата.
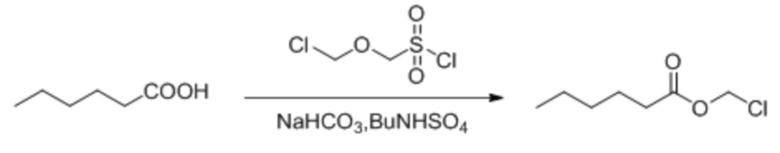
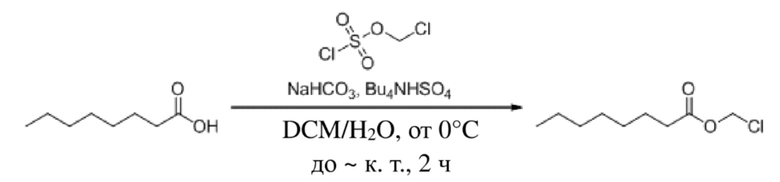
Гексановую кислоту (1,0 г, 8,62 ммоль), бикарбонат натрия (2,89 г, 34,48 ммоль) и BuNHS4 (0,29 г, 0,862 ммоль) растворяли в смешанном растворе дихлорметана и воды (10 мл/10 мл) и смесь перемешивали в течение 10 мин. После охлаждения до температуры 0°C по каплям добавляли (хлорметокси)метансульфонилхлорид (1,71 г, 10,34 ммоль). Затем температуру повышали до комнатной температуры и обеспечивали осуществление реакции в смеси в течение 2 часов. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Реакционную жидкость подвергали разделению жидкостей. Водную фазу экстрагировали дихлорметаном (30 мл × 3) и органическую фазу промывали водой (30 мл × 3). Затем органическую фазу объединяли, промывали насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 5:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой хлорметилгексаноат (400 мг, выход: 28,3%).
(2) Синтез (3-формил-1H-индол-1-ил)метилгексаноата (соединение 28)
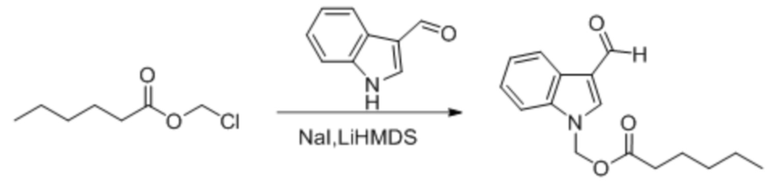
Соединение 28
Йодид натрия (18 мг, 0,122 ммоль) добавляли в раствор 1H-индол-3-карбальдегида (176 мг, 1,22 ммоль), растворенного в тетрагидрофуране. Бис(триметилсилил)амид лития (1,83 мл, 1,83 ммоль) по каплям добавляли в смесь в безводных и бескислородных условиях при -78°C и обеспечивали осуществление реакции в смеси в течение получаса при -78°C. Затем по каплям добавляли раствор хлорметилгексаноата (400 мг, 2,44 ммоль), растворенного в THF, и обеспечивали осуществление реакции в смеси в течение 3 часов при -78°C. LCMC (MC20-874-3-P1B) демонстрировала, что реакция завершена. В реакционную жидкость добавляли водный раствор хлорида аммония (10 мл). Затем для разбавления добавляли воду (50 мл) и реакционную жидкость экстрагировали этилацетатом (30 мл × 3). Впоследствии органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали посредством PRE-TLC (PE: EA = 4:1, Rf = 0,4) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилгексаноат (200 мг, выход: 30,0%).
Характеристические данные применительно к соединению 28 были следующими: 1H ЯМР (400 МГц, DMS): δ 1H ЯМР (400 МГц, CDCl3) δ 10,02 (s, 1H), 8,30 (dd, J=6,9, 1,5 Гц, 1H), 7,91 (s, 1H), 7,58-7,46 (m, 1H), 7,44-7,28 (m, 2H), 6,11 (s, 2H), 2,31 (t, J=7,5 Гц, 2H), 1,64-1,51 (m, 2H), 1,31-1,11 (m, 4H), 0,81 (t, J=7,0 Гц, 3H).
Вариант осуществления 19
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилгексаноата (соединение 37)
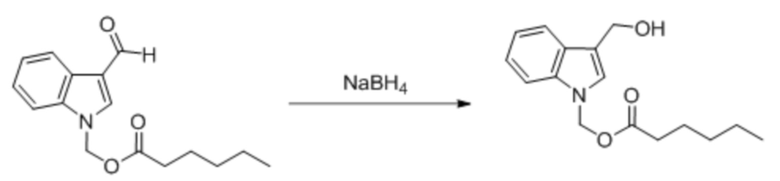
Соединение 28 Соединение 37
(3-Формил-1H-индол-1-ил)метилгексаноат (400 мг, 1,47 ммоль) растворяли в безводном метаноле (6 мл). После снижения температуры до 0°C добавляли борогидрид натрия (56 мг, 1,47 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (15 мл) и жидкость экстрагировали этилацетатом (3×20 мл). Впоследствии органические фазы объединяли, промывали водой (20 мл) и насыщенным солевым раствором (20 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали посредством PRE-TLC (PE: EA = 3:1, Rf = 0,25) с получением светло-желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (3-(гидроксиметил)-1H-индол-1-ил)метилгексаноат (соединение 37) (115 мг, выход: 28,5%).
Характеристические данные применительно к соединению 28 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,8 Гц, 1H), 7,54 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,19 (dd, J=11,2, 4,0 Гц, 1H), 7,10 (t, J=7,4 Гц, 1H), 6,17 (s, 2H), 4,90 (s, 1H), 4,62 (s, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,46 (dd, J=14,6, 7,3 Гц, 2H), 1,23-1,09 (m, 4H), 0,78 (t, J=6,9 Гц, 3H).
Чистота соединения 28, выявленная посредством HPLC, составляла 97,07% при 254 нм и 96,37% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 20
Получение (3-формил-1H-индол-1-ил)метилоктаноата (соединение 29)
(1) Синтез хлорметилоктаноата
Октановую кислоту (2,0 г, 13,868 ммоль), бикарбонат натрия (4,660 г, 55,473 ммоль) и гидросульфат тетрабутиламмония (Bu4NHS°4) (471 мг, 1,387 ммоль) растворяли в дихлорметане и воде (32 мл, 1:1) и смесь перемешивали в течение 5 мин при комнатной температуре. В смесь постепенно добавляли хлорметилсульфохлоридат (2,746 г, 16,642 ммоль) и обеспечивали осуществление реакции в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Смесь экстрагировали дихлорметаном (2×30 мл). Затем органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 80:1 до 100:1) с получением бесцветного маслянистого продукта, т.е. требуемого соединения, представляющего собой хлорметилоктаноат (1,827 г, выход: 68,4%).
Характеристические данные применительно к хлорметилоктаноату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,71 (s, 2H), 2,38 (t, J=7,5 Гц, 2H), 1,73-1,58 (m, 2H), 1,36-1,22 (m, 8H), 0,88 (t, J=6,9 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метилоктаноата (соединение 29)
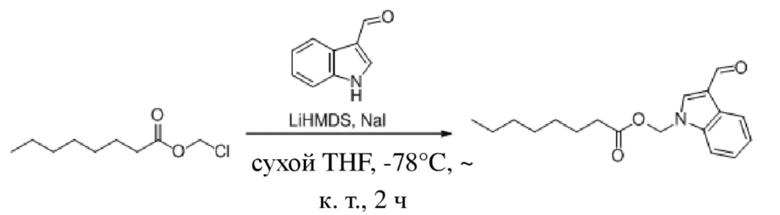
Соединение 29
1H-Индол-3-ацетальдегид (400 мг, 2,755 ммоль) и йодид натрия (83 мг, 0,551 ммоль) растворяли в безводном тетрагидрофуране (6 мл) в защитной атмосфере аргона. После снижения температуры до -78°C в смесь по каплям добавляли бис(триметилсилил)амид лития (1 M, 4,1 мл, 4,132 ммоль) и обеспечивали осуществление реакции в течение получаса при -78°C. Затем в реакционную жидкость по каплям добавляли раствор хлорметилоктаноата (2) (1,061 г, 5,510 ммоль), растворенного в тетрагидрофуране (6 мл), и обеспечивали осуществление реакции в смеси в течение двух часов при -78°C. Затем TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл). Реакционную жидкость экстрагировали этилацетатом (2×50 мл). Затем органические фазы объединяли, промывали водой (100 мл) и насыщенным солевым раствором (100 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = 100:1~70:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилоктаноат (соединение 29) (580 мг, выход: 69,9%).
Характеристические данные применительно к соединению 29 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,03 (s, 1H), 8,31 (dd, J=6,8, 1,5 Гц, 1H), 7,92 (s, 1H), 7,53 (dd, J=7,1, 1,2 Гц, 1H), 7,44-7,31 (m, 2H), 6,12 (s, 2H), 2,32 (t, J=7,5 Гц, 2H), 1,71-1,47 (m, 2H), 1,31-1,08 (m, 8H), 0,84 (t, J=7,0 Гц, 3H).
Чистота соединения 29, выявленная посредством HPLC, составляла 99,06% при 254 нм и 99,06% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 21
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилоктаноата (соединение 38)
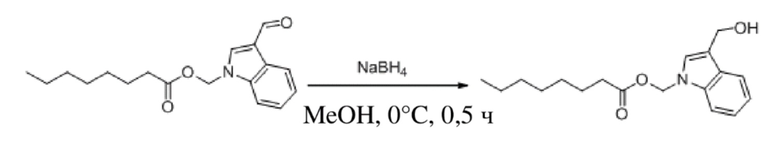
Соединение 29 Соединение 38
(3-Формил-1H-индол-1-ил)метилоктаноат (соединение 29) (250 мг, 0,829 ммоль) растворяли в безводном метаноле (3 мл). После снижения температуры до 0° в смесь добавляли борогидрид натрия (31 мг, 0,829 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (10 мл) и жидкость экстрагировали этилацетатом (2 × 30 мл). Затем органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством препаративной TLC (элюент : петролейный эфир : этилацетат = 3:1) с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой (3-(гидроксиметил)-1H-индол-1-ил)метилгексаноат (соединение 38) (60 мг, выход: 24,0 %), который объединяли с MC20-877-017P и MC20-877-018P с получением 103 мг требуемого соединения, представляющего собой (3-(гидроксиметил)-1H-индол-1-ил)метилгексаноат (соединение 38).
Характеристические данные применительно к соединению 38 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,8 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,1 Гц, 1H), 7,09 (t, J=7,1 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,3 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,51-1,39 (m, 2H), 1,17 (d, J=13,5 Гц, 8H), 0,82 (t, J=7,0 Гц, 3H).
Чистота соединения 38, выявленная посредством HPLC, составляла 93,04% при 254 нм и 93,86% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 22
Получение (3-формил-1H-индол-1-ил)метилдеканоата (соединение 30)
(1) Синтез хлорметилдеканоата
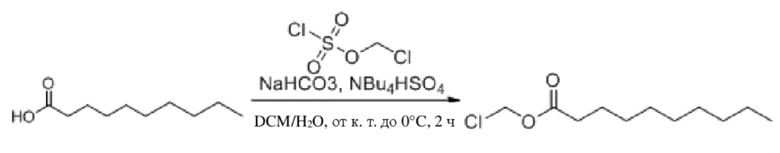
Декановую кислоту (1,0 г, 5,80 ммоль), бикарбонат натрия (1,95 г, 23,20 ммоль) и гидросульфат тетрабутиламмония (197 мг, 0,58 ммоль) растворяли в дихлорметане (10 мл) и воде (10 мл) в защитной атмосфере аргона и смесь перемешивали в течение 5 мин при комнатной температуре. Впоследствии реакционную смесь помещали на баню с ледяной водой при 0°C и в реакционную жидкость по каплям добавляли хлорметилсульфохлоридат (1,15 г, 6,96 ммоль). После добавления материалов температуру повышали до комнатной температуры и продолжали обеспечение реакции в смеси в течение 2 часов. Выявление посредством TLC (петролейный эфир : этилацетат = 20:1) демонстрировало, что реакция завершена. Затем добавляли воду (50 мл) в реакционную жидкость, которую дополнительно выливали в делительную воронку для разделения органических фаз. В водную фазу добавляли дихлорметан для экстракции (3×50 мл). Органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт пропускали через хроматографическую колонку и разделяли и очищали с помощью колонки с силикагелем для быстрого разделения (элюент : петролейный эфир : этилацетат = от 100:1 до 100:5) с получением продукта в виде бесцветной жидкости, т.е. требуемого соединения, представляющего собой хлорметилдеканоат (1,04 г, выход: 81,2%).
Характеристические данные применительно к хлорметилдеканоату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,71 (s, 2H), 2,38 (t, J=7,5 Гц, 2H), 1,69-1,59 (m, 2H), 1,31 (s, 2H), 1,28 (d, J=13,6 Гц, 10H), 0,88 (t, J=6,8 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метилдеканоата (соединение 30)
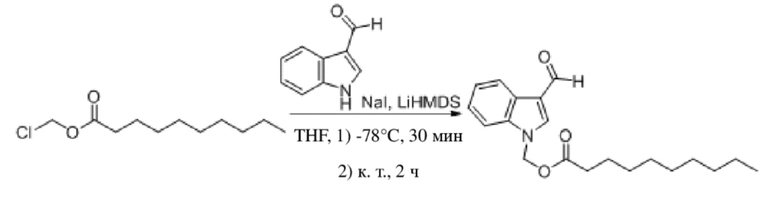
Соединение 30
3-Формил-1H-индол (438 мг, 3,02 ммоль) и йодид натрия (46 мг, 0,30 ммоль) растворяли в безводном тетрагидрофуране (15 мл) в защитной атмосфере аргона при -78°C. При перемешивании смеси добавляли бис(триметилсилил)амид лития (4,6 мл, 4,60 ммоль,1 M) и обеспечивали осуществление реакции в смеси в течение получаса. Впоследствии постепенно добавляли хлорметилдеканоат (1,00 г, 4,56 ммоль). После добавления температуру повышали до комнатной температуры и продолжали обеспечивать реакцию в смеси в течение 2 часов при перемешивании. Выявление посредством LCMS и TLC (петролейный эфир : этилацетат = 15:2) демонстрировало, что реакция в целом завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (50 мл) и реакционную жидкость экстрагировали этилацетатом (2×40 мл). Затем органические фазы объединяли, промывали с помощью насыщенного раствора тиосульфата натрия (100 мл) и насыщенного солевого раствора (50 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли с помощью хроматографической колонки/колонки с силикагелем для быстрого разделения (элюент : петролейный эфир : этилацетат = от 100:1 до 100:15) и очищали посредством препаративной TLC (петролейный эфир : этилацетат = 5:1) с получением бесцветной маслянистой жидкости, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилдеканоат (соединение 30) (660 мг, выход: 66,3%).
Характеристические данные применительно к соединению 30 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1H), 8,31 (dd, J=6,8, 1,5 Гц, 1H), 7,93 (s, 1H), 7,57-7,48 (m, 1H), 7,37 (dtd, J=14,6, 7,2, 1,3 Гц, 2H), 6,12 (s, 2H), 2,32 (t, J=7,5 Гц, 2H), 1,62-1,51 (m, 2H), 1,26 (d, J=6,5 Гц, 2H), 1,19 (d, J=2,6 Гц, 11H), 0,87 (t, J=7,0 Гц, 3H).
Чистота соединения 30, выявленная посредством HPLC, составляла 96,43% при 254 нм и 97,83% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 23
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилдеканоата (соединение 39)
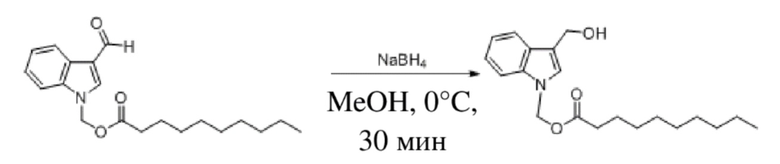
Соединение 30 Соединение 39
(3-Формил-1H-индол-1-ил)метилдеканоат (соединение 30) (450 мг, 1,37 ммоль) растворяли в безводном метаноле (8 мл). После снижения температуры до 0° добавляли борогидрид натрия (52 мг, 1,37 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 5:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли воду (50 мл) и жидкость экстрагировали этилацетатом (2 × 25 мл). Впоследствии органические фазы объединяли, промывали насыщенным солевым раствором (30 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт дважды очищали посредством препаративной TCL (элюент : петролейный эфир : этилацетат = 5:1) и затем получали светло-желтый маслянистый продукт, т.е. требуемое соединение, представляющее собой (3-(гидроксиметил)-1H-индол-1-ил)метилдеканоат (соединение 39) (187 мг, выход: 41,3%).
Характеристические данные применительно к соединению 39 были следующими: 1H ЯМР (400 МГц, DMS°): δ 7,61 (d, J=7,8 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,23-7,14 (m, 1H), 7,09 (t, J=7,3 Гц, 1H), 6,17 (s, 2H), 4,90 (s, 1H), 4,62 (d, J=2,8 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,51-1,38 (m, 2H), 1,23 (d, J=7,3 Гц, 2H), 1,16 (s, 10H), 0,85 (t, J=6,9 Гц, 3H).
Чистота соединения 39, выявленная посредством HPLC, составляла 94,60% при 254 нм и 93,91% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 24
Получение (3-формил-1H-индол-1-ил)метилдодеканоата (соединение 31)
(1) Синтез хлорметилдодеканоата
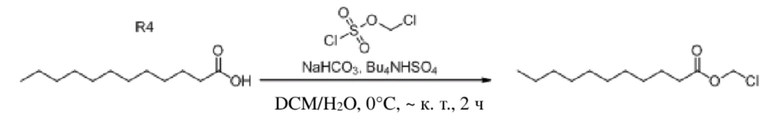
Додекановую кислоту (3,0 г, 14,976 ммоль), бикарбонат натрия (5,032 г, 59,904 ммоль) и гидросульфат тетрабутиламмония (508 мг, 1,498 ммоль) растворяли в дихлорметане и воде (40 мл, 1:1) и смесь перемешивали в течение 5 мин при комнатной температуре. Впоследствии постепенно добавляли хлорметилсульфохлоридат (2,965 г, 17,971 ммоль) и обеспечивали осуществление реакции в смеси в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Для осуществления экстракции применяли дихлорметан (2 × 40 мл). Органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 80:1) с получением бесцветного маслянистого продукта, т.е. требуемого соединения, представляющего собой хлорметилдодеканоат (3,490 г, выход: 93,7%).
Характеристические данные применительно к хлорметилдодеканоату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,71 (s, 2H), 2,38 (t, J=7,5 Гц, 2H), 1,69-1,59 (m, 2H), 1,31 (s, 2H), 1,28 (d, J=13,6 Гц, 10H), 0,88 (t, J=6,8 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метилдодеканоата (соединение 31)
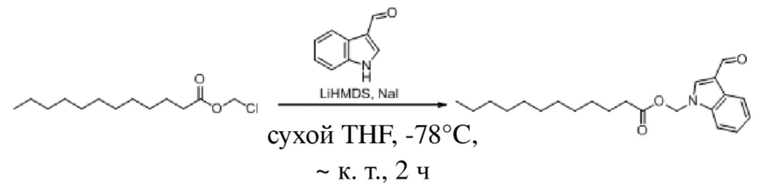
Соединение 31
1H-Индол-3-ацетальдегид (600 мг, 4,133 ммоль) и йодид натрия (62 мг, 0,413 ммоль) растворяли в безводном тетрагидрофуране (9 мл) в защитной атмосфере аргона. После снижения температуры до -78°C в реакционную жидкость по каплям добавляли раствор бис(триметилсилил)амида лития (1 M, 6,2 мл, 6,200 ммоль) в тетрагидрофуране (10 мл) и обеспечивали осуществление реакции в жидкости в течение получаса при -78°C. Впоследствии в реакционную жидкость по каплям добавляли раствор хлорметилдодеканоата (2,0 г, 8,267 ммоль) в тетрагидрофуране (10 мл) и обеспечивали осуществление реакции в жидкости в течение 2 часов при -78°C. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл) и реакционную жидкость экстрагировали этилацетатом (2 × 30 мл). Впоследствии органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = 100:1~70:1) с получением продукта в виде светло-желтого твердого вещества, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилдодеканоат (соединение 31) (665 мг, выход: 45,0%).
Характеристические данные применительно к соединению 31 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,05 (s, 1H), 8,44-8,24 (m, 1H), 7,92 (s, 1H), 7,53 (d, J=7,4 Гц, 1H), 7,45-7,29 (m, 2H), 6,12 (s, 2H), 2,32 (t, J=7,5 Гц, 2H), 1,57 (s, 2H), 1,21 (d, J=16,2 Гц, 16H), 0,88 (t, J=6,9 Гц, 3H).
Чистота соединения 31, выявленная посредством HPLC, составляла 99,17% при 254 нм и 97,86% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 25
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилдодеканоата (соединение 40)
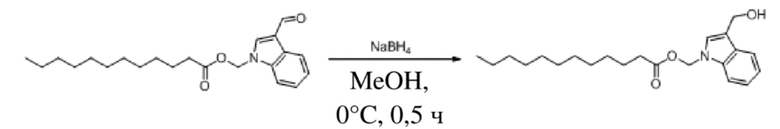
Соединение 31 Соединение 40
(3-Формил-1H-индол-1-ил)метилдодеканоат (соединение 31) (250 мг, 0,695 ммоль) растворяли в безводном метаноле (5 мл). После снижения температуры до 0°C добавляли борогидрид натрия (26 мг, 0,695 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (10 мл) и жидкость экстрагировали этилацетатом (2 × 20 мл). Впоследствии органические фазы объединяли, промывали водой (40 мл) и насыщенным солевым раствором (40 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством препаративной TCL (элюент : петролейный эфир : этилацетат = 3:1) и затем получали продукт в виде светло-желтого твердого вещества, т.е. требуемое соединение, представляющее собой (3-(гидроксиметил)-1H-индол-1-ил)метилдодеканоат (соединение 40) (140 мг, выход: 56,0%).
Характеристические данные применительно к соединению 40 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,8 Гц, 1H), 7,53 (d, J=8,2 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,2 Гц, 1H), 7,09 (t, J=7,2 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,3 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,45 (s, 2H), 1,19 (d, J=26,6 Гц, 16H), 0,85 (t, J=6,8 Гц, 3H).
Чистота соединения 40, выявленная посредством HPLC, составляла 98,23% при 254 нм и 97,03% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 26
Получение (3-формил-1H-индол-1-ил)метилтетрадеканоата (соединение 32)
(1) Синтез хлорметилтетрадеканоата
Тетрадекановую кислоту (3,0 г, 13,136 ммоль), бикарбонат натрия (4,414 г, 52,544 ммоль) и гидросульфат тетрабутиламмония (446 мг, 1,313 ммоль) растворяли в дихлорметане и воде (40 мл, 1:1) и смесь перемешивали в течение 5 мин при комнатной температуре. Затем в реакционную жидкость постепенно добавляли хлорметилсульфохлоридат (2,6 г, 15,764 ммоль) при 0°C. После добавления обеспечивали осуществление реакции в реакционной жидкости в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. Для осуществления экстракции применяли дихлорметан (2×40 мл). Органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 80:1) с получением продукта в виде полупрозрачного твердого вещества, представляющего собой требуемое соединение, т.е. хлорметилтетрадеканоат (3,316 г, выход: 91,2%).
Характеристические данные применительно к хлорметилтетрадеканоату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,70 (s, 2H), 2,38 (t, J=7,5 Гц, 2H), 1,78-1,51 (m, 2H), 1,37-1,23 (m, 20H), 0,88 (t, J=6,8 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метилтетрадеканоата (соединение 32)
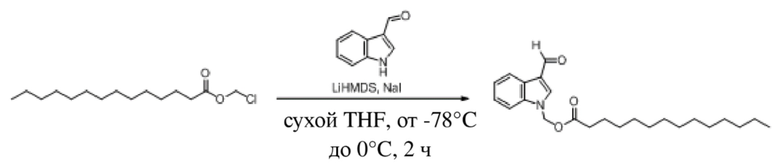
Соединение 32
1H-Индол-3-ацетальдегид (400 мг, 2,755 ммоль) и йодид натрия (41 мг, 0,275 ммоль) растворяли в безводном тетрагидрофуране (8 мл) под защитой атмосферы аргона. После снижения температуры до -78°C в реакционную жидкость по каплям добавляли раствор бис(триметилсилил)амида лития (1 M, 4,1 мл, 4,132 ммоль) в тетрагидрофуране и обеспечивали осуществление реакции в жидкости в течение 1 часа при -78°C. Затем в реакционную жидкость по каплям добавляли раствор хлорметилтетрадеканоата (1,525 г, 5,510 ммоль) в тетрагидрофуране (8 мл) и обеспечивали осуществление реакции в жидкости в течение 2 часов при -78°C. TLC (петролейный эфир : этилацетат = 3:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл) и реакционную жидкость экстрагировали этилацетатом (2×20 мл). Затем органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = 100:1~70:1) с получением продукта в виде светло-желтого твердого вещества, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилтетрадеканоат (соединение 32) (827 мг, выход: 77,8%).
Характеристические данные применительно к соединению 32 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1H), 8,32 (d, J=7,0 Гц, 1H), 7,92 (s, 1H), 7,53 (d, J=7,6 Гц, 1H), 7,44-7,32 (m, 2H), 6,12 (s, 2H), 2,32 (t, J=7,5 Гц, 2H), 1,59 (s, 2H), 1,22 (d, J=22,3 Гц, 20H), 0,88 (t, J=6,8 Гц, 3H).
Чистота соединения 32, выявленная посредством HPLC, составляла 98,67% при 254 нм и 99,35% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 27
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилтетрадеканоата (соединение 41)
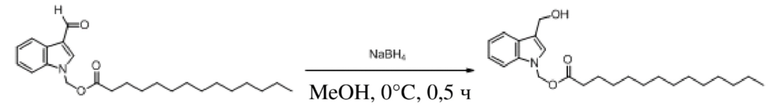
Соединение 32 Соединение 41
(3-Формил-1H-индол-1-ил)метилтетрадеканоат (соединение 32) (300 мг, 0,778 ммоль) растворяли в безводном метаноле (6 мл). После снижения температуры до 0°C добавляли борогидрид натрия (29,4 мг, 0,778 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (10 мл) и жидкость экстрагировали этилацетатом (2 × 30 мл). Впоследствии органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством препаративной TCL (элюент : петролейный эфир : этилацетат = 2:1) и затем получали продукт в виде светло-желтого твердого вещества, т.е. требуемое соединение, представляющее собой (3-(гидроксиметил)-1H-индол-1-ил)метилтетрадеканоат (соединение 41) (130 мг, выход: 43,1%).
Характеристические данные применительно к соединению 41 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,9 Гц, 1H), 7,53 (d, J=8,1 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,5 Гц, 1H), 7,09 (t, J=7,5 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,1 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,45 (s, 2H), 1,19 (d, J=32,7 Гц, 20H), 0,85 (t, J=6,8 Гц, 3H).
Чистота соединения 41, выявленная посредством HPLC, составляла 97,17% при 254 нм и 96,91% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 41 перекристаллизовывали с получением чистого кристалла I соединения 41. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 9 и на фигуре 9.
Таблица 9. Данные XRPD применительно к кристаллу I соединения 41
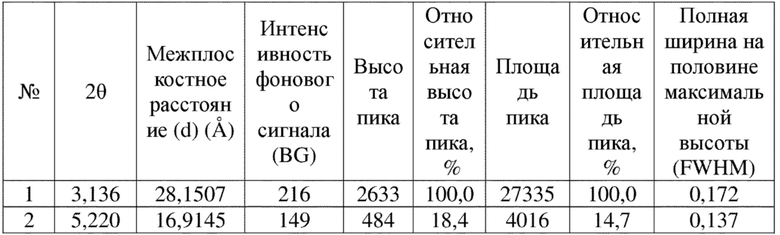
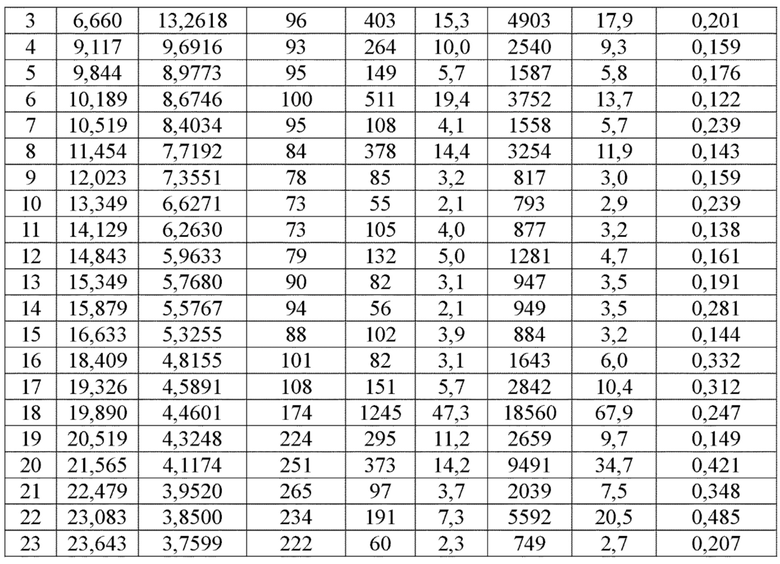
Вариант осуществления 28
Получение (3-формил-1H-индол-1-ил)метилпальмитата (соединение 33)
(1) Синтез хлорметилпальмитата.
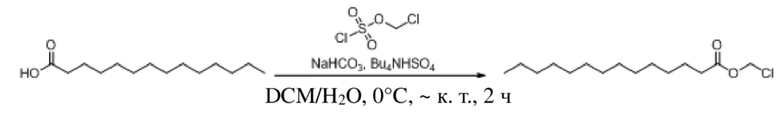
Пальмитиновую кислоту (4,0 г, 15,599 ммоль), бикарбонат натрия (5,241 г, 62,396 ммоль) и гидросульфат тетрабутиламмония (530 мг, 1,560 ммоль) растворяли в дихлорметане и воде (60 мл, 1:1), смесь перемешивали в течение 5 мин при комнатной температуре. Затем постепенно добавляли хлорметилсульфохлоридат (3,080 г, 18,719 ммоль) при 0°C и обеспечивали осуществление реакции в смеси в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Реакционную жидкость экстрагировали дихлорметаном (2×50 мл). Впоследствии органические фазы объединяли, промывали водой (100 мл) и насыщенным солевым раствором (100 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 80:1) с получением продукта в виде полупрозрачного твердого вещества, т.е. требуемого соединения, представляющего собой хлорметилпальмитат (3,727 г, выход: 78,4%).
Характеристические данные применительно к хлорметилпальмитату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,70 (s, 2H), 2,38 (t, J=7,5 Гц, 2H), 1,72-1,54 (m, 2H), 1,33-1,23 (m, 24H), 0,88 (t, J=6,8 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метилпальмитата (соединение 33)
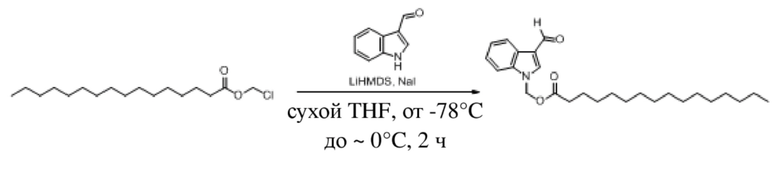
Соединение 33
1H-индол-3-ацетальдегид (400 мг, 2,755 ммоль) и йодид натрия (41 мг, 0,275 ммоль) растворяли в безводном тетрагидрофуране (8 мл) под защитой атмосферы аргона. После снижения температуры до -78°C в реакционную жидкость по каплям добавляли раствор бис(триметилсилил)амида лития (1 M, 4,1 мл, 4,132 ммоль) в тетрагидрофуране и обеспечивали осуществление реакции в жидкости в течение 1 часа при -78°C. Впоследствии в реакционную жидкость по каплям добавляли раствор хлорметилпальмитата (2) (1,680 г, 5,511 ммоль) в тетрагидрофуране (8 мл) и обеспечивали осуществление реакции в жидкости в течение 2 часов при -78°C. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл) и реакционную жидкость экстрагировали этилацетатом (2×40 мл). Затем органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = 100:1~80:1) с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метилпальмитат (соединение 33) (697 мг, выход: 61,2%).
Характеристические данные применительно к соединению 33 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1H), 8,32 (d, J=6,8 Гц, 1H), 7,92 (s, 1H), 7,53 (d, J=7,5 Гц, 1H), 7,37 (td, J=13,8, 6,0 Гц, 2H), 6,12 (s, 2H), 2,32 (t, J=7,5 Гц, 2H), 1,58 (s, 8H), 1,22 (d, J=24,3 Гц, 24H), 0,88 (t, J=6,8 Гц, 3H).
Чистота соединения 31, выявленная посредством HPLC, составляла 97,69% при 254 нм и 98,46% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 29
Получение (3-(гидроксиметил)-1H-индол-1-ил)метилпальмитата (соединение 42)
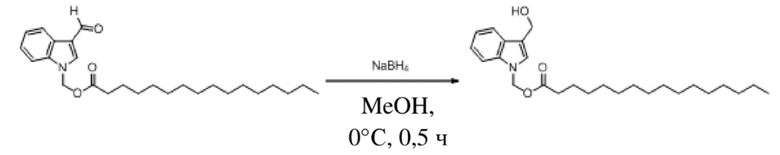
Соединение 33 Соединение 42
(3-Формил-1H-индол-1-ил)метилпальмитат (соединение 33) (300 мг, 0,725 ммоль) растворяли в безводном метаноле (10 мл) и тетрагидрофуране (5 мл). После снижения температуры до 0°C в смесь добавляли борогидрид натрия (27 мг, 0,725 ммоль) и обеспечивали осуществление реакции в смеси в течение получаса при 0°C. Выявление посредством TLC (петролейный эфир : этилацетат = 3:1) демонстрировало, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (10 мл) и жидкость экстрагировали этилацетатом (2 × 30 мл). Затем органические фазы объединяли, промывали водой (60 мл) и насыщенным солевым раствором (60 мл), высушивали с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством препаративной TCL (элюент : петролейный эфир : этилацетат = 2:1) и затем получали белый твердый продукт, т.е. требуемое соединение, (3-(гидроксиметил)-1H-индол-1-ил)метилпальмитат (соединение 42) (250 мг, выход: 83,0%).
Характеристические данные применительно к соединению 42 были следующими: 1H ЯМР (400 МГц, DMS) δ 7,61 (d, J=7,9 Гц, 1H), 7,53 (d, J=8,3 Гц, 1H), 7,34 (s, 1H), 7,19 (t, J=7,1 Гц, 1H), 7,09 (t, J=7,0 Гц, 1H), 6,17 (s, 2H), 4,90 (t, J=5,4 Гц, 1H), 4,61 (d, J=5,1 Гц, 2H), 2,26 (t, J=7,3 Гц, 2H), 1,45 (s, 2H), 1,19 (d, J=33,6 Гц, 24H), 0,85 (t, J=6,8 Гц, 3H).
Чистота соединения 42, выявленная посредством HPLC, составляла 97,87% при 254 нм и 98,43% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 30
Получение (3-формил-1H-индол-1-ил)метил-2,5,8,11,14-пентаоксагексадекан-16-оата (соединение 34)
(1) Синтез хлорметил-2,5,8,11,14-пентаоксагексадекан-16-оата
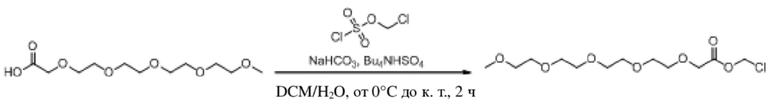
2,5,8,11,14-Пентаоксагексадекан-16-овую кислоту (1,6 г, 6,008 ммоль), бикарбонат натрия (2,019 г, 24,032 ммоль) и гидросульфат тетрабутиламмония (204 мг, 0,600 ммоль) растворяли в дихлорметане и воде (50 мл, 1:1) и смесь перемешивали в течение 5 мин при комнатной температуре. Затем хлорметилсульфохлоридат (1,189 г, 7,210 ммоль) постепенно добавляли в реакционную жидкость при 0°C и обеспечивали осуществление реакции в реакционной жидкости в течение 2 часов при комнатной температуре. Для осуществления экстракции применяли дихлорметан (2×50 мл). Органические фазы объединяли, промывали водой (100 мл) и насыщенным солевым раствором (100 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении с получением желтого маслянистого продукта, т.е. требуемого соединения, представляющего собой хлорметил-2,5,8,11,14-пентаоксагексадекан-16-оат (1,2 г, неочищенный продукт).
Характеристические данные применительно к хлорметил-2,5,8,11,14-пентаоксагексадекан-16-оату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 5,76 (s, 2H), 4,25 (s, 2H), 3,78-3,74 (m, 2H), 3,72-3,65 (m, 12H), 3,55 (dd, J=5,6, 3,6 Гц, 2H), 3,39 (d, J=2,8 Гц, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метил-2,5,8,11,14-пентаоксагексадекан-16-оата (соединение 34)
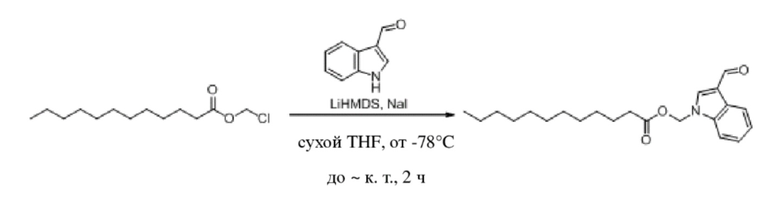
Соединение 34
1H-Индол-3-ацетальдегид (503 мг, 3,812 ммоль), йодид натрия (52 мг, 0,347 ммоль), хлорметил-2,5,8,11,14-пентаоксагексадекан-16-оат (1,2 г, 3,812 ммоль) и триэтиламин (1,052 мг, 10,398 ммоль) растворяли в N,N-диметилформамиде (20 мл) и обеспечивали осуществление реакции в смеси в течение 12 часов при 30°C. После добавления 4-диметиламинопиридина (42 мг, 0,347 ммоль) обеспечивали осуществление реакции в смеси в течение 12 часов при 30°C. LCMS продемонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл) и реакционную жидкость экстрагировали этилацетатом (2×60 мл). Затем органические фазы объединяли, промывали водой (120 мл) и насыщенным солевым раствором (120 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали посредством препаративной HPLC с получением продукта в виде желтого твердого вещества, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метил-2,5,8,11,14-пентаоксагексадекан-16-оат (соединение 34) (320 мг, выход: 21,8%).
Характеристические данные применительно к соединению 34 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1H), 8,31 (d, J=6,9 Гц, 1H), 7,95 (s, 1H), 7,54 (d, J=7,4 Гц, 1H), 7,43-7,32 (m, 2H), 6,20 (s, 2H), 4,18 (s, 2H), 3,70-3,51 (m, 16H), 3,36 (s, 3H).
Чистота соединения 34, выявленная посредством HPLC, составляла 99,32% при 254 нм и 99,38% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Вариант осуществления 31
Получение (3-формил-1H-индол-1-ил)метил-2-ацетоксибензоата (соединение 35)
(1) Синтез 2,14-хлорметил-2-ацетоксибензоата (2)
2-Ацетоксибензойную кислоту (3,0 г, 15,599 ммоль), бикарбонат натрия (5,595 г, 66,604 ммоль) и гидросульфат тетрабутиламмония (565 мг, 1,665 ммоль) растворяли в дихлорметане и воде (40 мл, 1:1) и смесь перемешивали в течение 5 мин при комнатной температуре. Затем хлорметилсульфонилхлорид (3,297 г, 19,982 ммоль) постепенно добавляли в реакционную жидкость при 0°C и обеспечивали осуществление реакции в реакционной жидкости в течение 2 часов при комнатной температуре. TLC (петролейный эфир : этилацетат = 5:1) демонстрировала, что реакция завершена. Для осуществления экстракции применяли дихлорметан (2×40 мл). Органические фазы объединяли, промывали водой (80 мл) и насыщенным солевым раствором (80 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали с применением хроматографической колонки (элюент : петролейный эфир : этилацетат = от 100:1 до 80:1) с получением бесцветного маслянистого продукта, т.е. требуемого соединения, представляющего собой хлорметил-2-ацетоксибензоат (3,0 г, выход: 78,8%).
Характеристические данные применительно к хлорметил-2-ацетоксибензоату были следующими: 1H ЯМР (400 МГц, CDCl3) δ 8,06 (dd, J=7,9, 1,6 Гц, 1H), 7,62 (td, J=7,9, 1,6 Гц, 1H), 7,41-7,29 (m, 1H), 7,22-7,07 (m, 1H), 5,90 (s, 2H), 2,38 (s, 3H).
(2) Синтез (3-формил-1H-индол-1-ил)метил-2-ацетоксибензоата (соединение 35)
Соединение 35
1H-Индол-3-ацетальдегид (800 мг, 5,511 ммоль), йодид натрия (83 мг, 0,551 ммоль), 4-диметиламинопиридин (67 мг, 0,551 ммоль), хлорметил-2-ацетоксибензоат (2) (2,520 г, 11,022 ммоль) и триэтиламин (1,673 мг, 16,533 ммоль) растворяли в N,N-диметилформамиде (32 мл) и обеспечивали осуществление реакции в смеси в течение 24 часов при 30°C. TLC (петролейный эфир/этилацетат = 1,5: 1) демонстрировала, что реакция завершена. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония (20 мл) и реакционную жидкость экстрагировали этилацетатом (2×50 мл). Затем органические фазы объединяли, промывали водой (100 мл) и насыщенным солевым раствором (100 мл), высушивали с помощью безводного сульфата натрия, фильтровали, концентрировали при пониженном давлении и очищали посредством препаративной HPLC с получением продукта в виде белого твердого вещества, т.е. требуемого соединения, представляющего собой (3-формил-1H-индол-1-ил)метил-2-ацетоксибензоат (соединение 35) (233 мг, выход: 12,4%).
Характеристические данные применительно к соединению 35 были следующими: 1H ЯМР (400 МГц, CDCl3) δ 10,07 (s, 1H), 8,33 (d, J=7,3 Гц, 1H), 8,13-7,86 (m, 2H), 7,58 (ddd, J=7,8, 4,6, 1,7 Гц, 2H), 7,40 (ddd, J=15,1, 13,9, 6,9 Гц, 2H), 7,29 (t, J=8,1 Гц, 1H), 7,09 (d, J=8,1 Гц, 1H), 6,33 (s, 2H), 2,14 (s, 3H).
Чистота соединения 35, выявленная посредством HPLC, составляла 99,7% при 254 нм и 99,63% при 214 нм, как измерено в соответствии со способом нормализации площади пика.
Соединение 35 перекристаллизовывали с получением чистого кристалла I соединения 35. Чистое соединение выявляли посредством порошковой рентгеновской дифракции (XRPD), и результаты показаны в следующей таблице 10 и на фигуре 10.
Таблица 10. Данные XRPD применительно к кристаллу I соединения 35
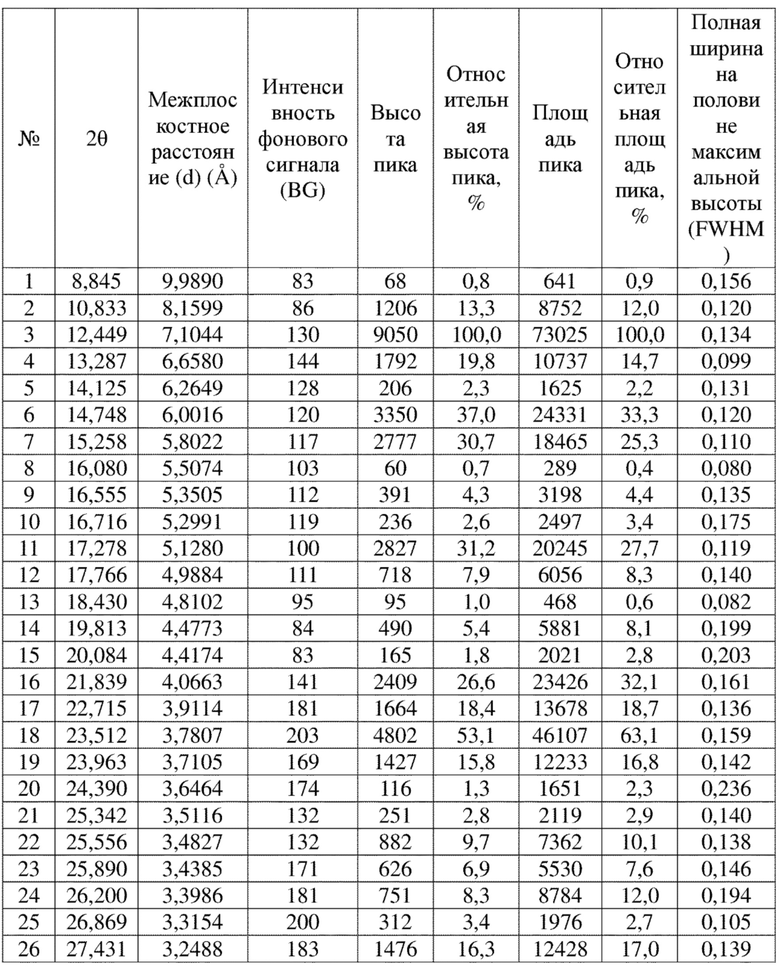
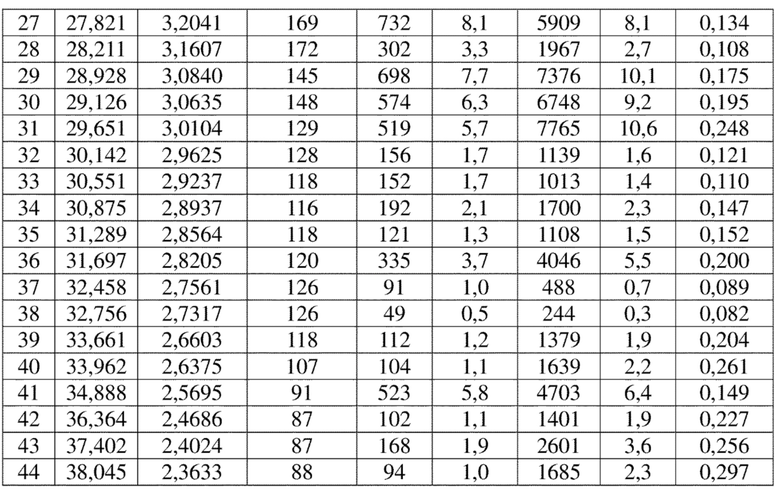
Вариант осуществления 32
Соединения, полученные в вариантах осуществления, приведенных выше, подвергали тестированию в отношении активности.
1. Способ.
Мышиную модель с ушными AD-подобными симптомами, индуцированными кальципотриолом (MC903), применяли для изучения этих соединений в терминах лечебной активности при AD.
Ссылаясь на патент с номером публикации CN 110368386 A, получали мышиную модель, индуцированную MC903 (кальципотриен), где оба уха животных, относящихся к мышиной модели BALB/c, обрабатывали с помощью MC903, MC903 + индол-3-альдегид (IAId) или MC903 + соединение, полученное в вариантах осуществления, в течение 11 последовательных дней соответственно, и общие уровни IgE в сыворотке крови, значения толщины ушей и потерю веса относительно веса в начале эксперимента тестировали в конце эксперимента. Вследствие большого количества лекарственных средств, подлежащих тестированию, терапевтические эффекты лекарственных средств оценивали в двух сериях.
2. Результаты
Данные, показанные в результатах, представляли собой средние значения ± SD из пяти независимо повторенных испытаний. Анализ значимости проводили с применением критерия Манна-Уитни в отношении отличий соответствующих параметров мышей от группы обработки (MC903 + этанол (Et°H)), которой вводили только MC903, где * означало, что P < 0,05, ** означало, что P < 0,01; *** означало, что P < 0,001.
2.1. Первая серия экспериментов
В данной серии экспериментов дозировка MC903, лекарственного средства для конструирования моделей, составляла 6,9 нмоль/мышь каждый день, и экспериментальные результаты для соединений 1, 2, 3, 6, 7, 19, 20, 28, 29, 30, 34, 37, 38 и 39 в данной серии показаны на фигурах 11-13.
На фигуре 11 показаны общие уровни IgE в сыворотке крови мышей после введения мышам различных лекарственных средств. На фигуре 12 показаны значения толщины ушей мышей после введения мышам различных лекарственных средств. На фигуре 13 показана потеря веса мышей относительно веса в начале экспериментов после введения мышам различных лекарственных средств. На фигуре 14 показаны фотографии со значениями толщины ушей иллюстративных мышей после введения мышам различных лекарственных средств. На фигуре 15 показаны изображения окрашивания гематоксилином и эозином (HE) ушных тканей иллюстративных мышей после введения мышам различных лекарственных средств. Группа EtOH представляла собой группу отрицательного контроля, которой вводили только этанол. Группа MC903 + Et°H представляла собой модельную контрольную группу, которой вводили кальципотриен и этанол. Остальные группы представляли собой группы, которым вводили MC903 для конструирования модели, а затем вводили различные лекарственные средства. Группа индол-3-альдегида (IAId) представляла собой группу, которой вводили IAId, группа индол-3-метанола (I3C) представляла собой группу, которой вводили I3C. Группы других соединений представляли собой группы, которым вводили соответствующие соединения.
На фигурах можно видеть, что общие уровни IgE в сыворотке крови, значения толщины ушей и потеря веса относительно веса в начале эксперимента у мышей в модельной контрольной группе (MC903+ Et°H) значительно отличались по сравнению с группой отрицательного контроля (EtoH) (P < 0,001), что указывает на то, что моделирование было успешным. Как в группе IAId, так и в группе I3C общие уровни IgE в сыворотке крови и значения толщины ушей мышей были в значительной степени сниженными (P < 0,05 или P < 0,01) и все соединения оказывали положительные эффекты в отношении общих уровней IgE в сыворотке крови, значений толщины ушей и потери веса мышей. Конкретно, с точки зрения снижения общих уровней IgE в сыворотке крови мышей, соединения 1, 2, 29, 30, 34, 37, 38 и 39 характеризовались статистически значимой разницей по сравнению с таковыми в модельной контрольной группе, и в частности соединения 30 и 38 демонстрировали превосходные эффекты в отношении снижения общего уровня IgE в сыворотке крови мышей. С точки зрения снижения значений толщины ушей мышей соединения 1, 19, 20, 29, 30, 34 и 39 характеризовались статистически значимой разницей по сравнению с таковыми в модельной контрольной группе, и в частности соединения 29 и 30 демонстрировали превосходные эффекты в отношении снижения значений толщины ушей мышей. В сочетании с воспалением ушей и результатами окрашивания тканей мышей с помощью HE было обнаружено, что соединения 29, 30 и 38 характеризовались значительными терапевтическими эффектами при AD-подобных симптомах в ухе мышей.
2.2. Вторая серия экспериментов
Было установлено, что в первой серии экспериментов на статус выживания мышей оказывало большое влияние лекарственное средство для конструирования модели, т.е. MC903, что проявлялось в значительной потере веса (фигура 13). Следовательно, во второй серии тестов лекарственных средств дозировку MC903 снижали до 5 нмоль/мышь каждый день и период тестирования сокращали до 9 дней. Наконец, статус выживания мышей был значительно улучшен (фигура 18), что соответствует ожиданиям.
Экспериментальные результаты для соединений 30, 38 4, 5, 9, 21, 22, 23, 24, 26, 27, 31, 32, 33, 35, 40, 41, 42, 8 и 29 в данной серии экспериментов показаны на фигурах 16-20.
На фигуре 16 показаны общие уровни IgE в сыворотке крови мышей после введения мышам различных лекарственных средств. На фигуре 17 показаны значения толщины ушей мышей после введения мышам различных лекарственных средств. На фигуре 18 показана потеря веса мышей относительно веса в начале экспериментов после введения мышам различных лекарственных средств. На фигуре 19 показаны фотографии со значениями толщины ушей иллюстративных мышей после введения мышам различных лекарственных средств. На фигуре 20 показаны изображения окрашивания гематоксилином и эозином (HE) ушных тканей иллюстративных мышей после введения мышам различных лекарственных средств. Группа EtoH представляла собой группу отрицательного контроля, которой вводили только этанол. Группа MC903 + EtoH представляла собой модельную контрольную группу, которой вводили кальципотриен и этанол. Остальные группы представляли собой группы, которым вводили MC903 для конструирования модели, а затем вводили различные лекарственные средства. Группа индол-3-альдегида (IAId) представляла собой группу, которой вводили IAId. Группа индол-3-метанола (I3C) представляла собой группу, которой вводили IAId. Группы других соединений представляли собой группы, которым вводили соответствующие соединения.
На фигурах можно видеть, что общий уровень IgE в сыворотке крови, значения толщины ушей и потеря веса относительно веса в начале эксперимента у мышей в модельной контрольной группе (MC903+ EtoH) значительно отличались по сравнению с группой отрицательного контроля (EtoH) (P < 0,01 или P < 0,001), что указывает на то, что моделирование было успешным. С точки зрения снижения общих уровней IgE в сыворотке крови мышей соединения 30, 38, 4, 9, 22, 23, 24, 26, 27, 33, 35, 41 и 8 характеризовались статистически значимой разницей по сравнению с таковыми в модельной контрольной группе, и в частности соединения 30, 4, 9, 22 и 35 демонстрировали превосходные эффекты в отношении снижения общих уровней IgE в сыворотке крови мышей. С точки зрения снижения значений толщины ушей мышей соединения 30, 38, 4, 9, 27, 8 и 29 характеризовались статистически значимой разницей по сравнению с таковыми в модельной контрольной группе, и в частности соединение 9 демонстрировало превосходные эффекты в отношении снижения значений толщины ушей мышей. В сочетании с воспалением ушей и результатами окрашивания тканей мышей с помощью HE, а также с учетом параметров многочисленных экспериментов и тестов, лекарственные средства, которые характеризуются терапевтическими эффектами при AD-подобных симптомах в ухе мышей, согласно результатам скрининга представляли собой соединения 4, 9, 29, 30, 38, 22 и 35.
Вариант осуществления 33
Оптимальная эффективная концентрация при лечении AD-подобных симптомов
1. Способ.
Соединения, подвергнутые скринингу, как указано выше, характеризующиеся терапевтическим эффектом при AD-подобных симптомах, отбирали для дополнительного определения оптимальной эффективной концентрации.
Ссылаясь на способ с животной моделью в варианте осуществления 32, дозировки лекарственных средств для каждой группы определяли в соответствии с таблицей ниже, и оптимальную эффективную концентрацию при лечении AD-подобных симптомов исследовали посредством сравнения различий в значениях толщины ушей, общих уровнях IgE в сыворотке крови и потере веса мышей с таковыми в группе обработки, которой вводили только MC903.
Таблица 11. Группы и дозировки для введения
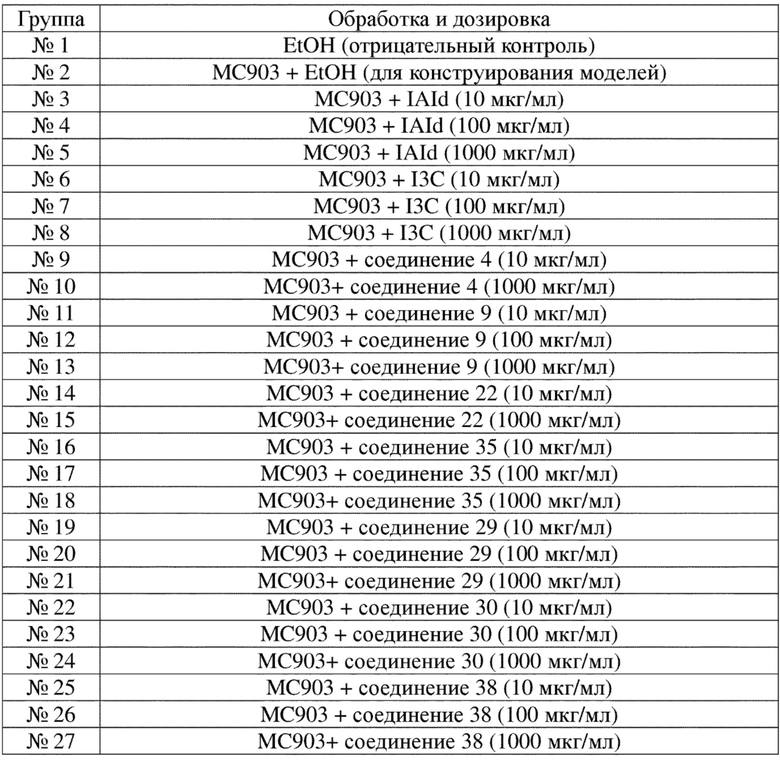
Примечание: указанное выше введение проводили путем местного нанесения.
2. Результаты.
Результаты показаны на фигурах 21-30, где на фигурах 21-23 показаны общие уровни в сыворотке крови мышей в каждой группе, на фигурах 24-26 показаны значения толщины ушей мышей в каждой группе, на фигурах 27-29 показана потеря веса мышей в каждой группе и на фигуре 30 показаны фотографии ушей иллюстративных мышей.
Посредством сравнения различий в значениях толщины ушей, общих уровнях IgE в сыворотке крови и потере веса мышей в группах обработки с таковыми в группе обработки, которой вводили только MC903, было обнаружено, что соединение 9 характеризовалось значительным терапевтическим эффектом при AD-подобных симптомах в ухе мыши при различных концентрациях. Соединения 4, 9, 29, 30, 38, 22 и 35 и т. д. характеризовались хорошим ингибирующим эффектом при систематическом воспалении, проявляющимся как сниженные общие уровни IgE в сыворотке крови и улучшение при AD-подобных симптомах в ухе мыши, и в частности соединения 4, 30 и 38 характеризовались превосходным ингибирующим эффектом при систематическом воспалении.
Вариант осуществления 34
В данном варианте осуществления предусмотрено получение крема для местного применения.
1. Рецептура субстанции
Стеариновая кислота 120 г
Глицерилмоностеарат 35 г
Жидкий парафин 60 г
Альболен 10 г
Ланум 50 г
Триэтаноламин 4 г
Дистиллированная вода добавление до 100. 0 г
2. Получение
2.1. Получение субстанции.
Стеариновую кислоту, глицерилмоностеарат, жидкий парафин, альболен и ланум брали в качестве масляной фазы, помещали в чашку для выпаривания и нагревали до приблизительно 80°C на водяной бане для смешивания и плавления. В колбу добавляли триэтаноламин и дистиллированную воду и нагревали до приблизительно 80°C на водяной бане. Водную фазу постепенно выливали в масляную фазу при изотермической температуре и смесь постоянно перемешивали на водяной бане, пока она не стала полутвердой и не приобрела молочный цвет, затем ее перемешивали при комнатной температуре, пока она практически не конденсировалась, с получением субстанции.
2.2. Получение крема
Активный ингредиент (каждое из соединений) в предварительно определенном количестве добавляли к субстанции, указанной выше. В качестве альтернативы, каждое из соединений растворяли и затем добавляли к субстанции и перемешивали. При этом каждый из ингредиентов/соединений добавляли при перемешивании и равномерно диспергировали в субстанции с получением кремов с различными концентрациями.
Технические признаки приведенных выше вариантов осуществления можно объединять в произвольном порядке. Чтобы сделать описание кратким, описаны не все возможные комбинации технических признаков в приведенных выше вариантах осуществления. Однако до тех пор, пока между этими техническими признаками в комбинациях нет противоречия, следует считать, что эти комбинации входят в объем настоящего описания.
Приведенные выше варианты осуществления отражают лишь несколько примеров настоящего изобретения, и они описаны более конкретно и подробно, но их не следует рассматривать как ограничения объема настоящего изобретения. Следует отметить, что специалисты в данной области техники могут внести ряд модификаций и улучшений без отклонения от идеи настоящего изобретения, которые находятся в пределах объема охраны настоящего изобретения. Таким образом, объем охраны настоящего патента будет определяться прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ИНГИБИТОРА ПОВЕРХНОСТНОГО АНТИГЕНА ВИРУСА ГЕПАТИТА В | 2020 |
|
RU2823673C1 |
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| ПРОИЗВОДНОЕ ПИРИДОНА, ИМЕЮЩЕЕ ТЕТРАГИДРОПИРАНИЛМЕТИЛЬНУЮ ГРУППУ | 2015 |
|
RU2707953C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИАЗАБЕНЗОФЛУОРАНТРЕНОВЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2762189C2 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МИМЕТИКА SMAC, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ ИНГИБИТОРА IAP, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2819398C2 |
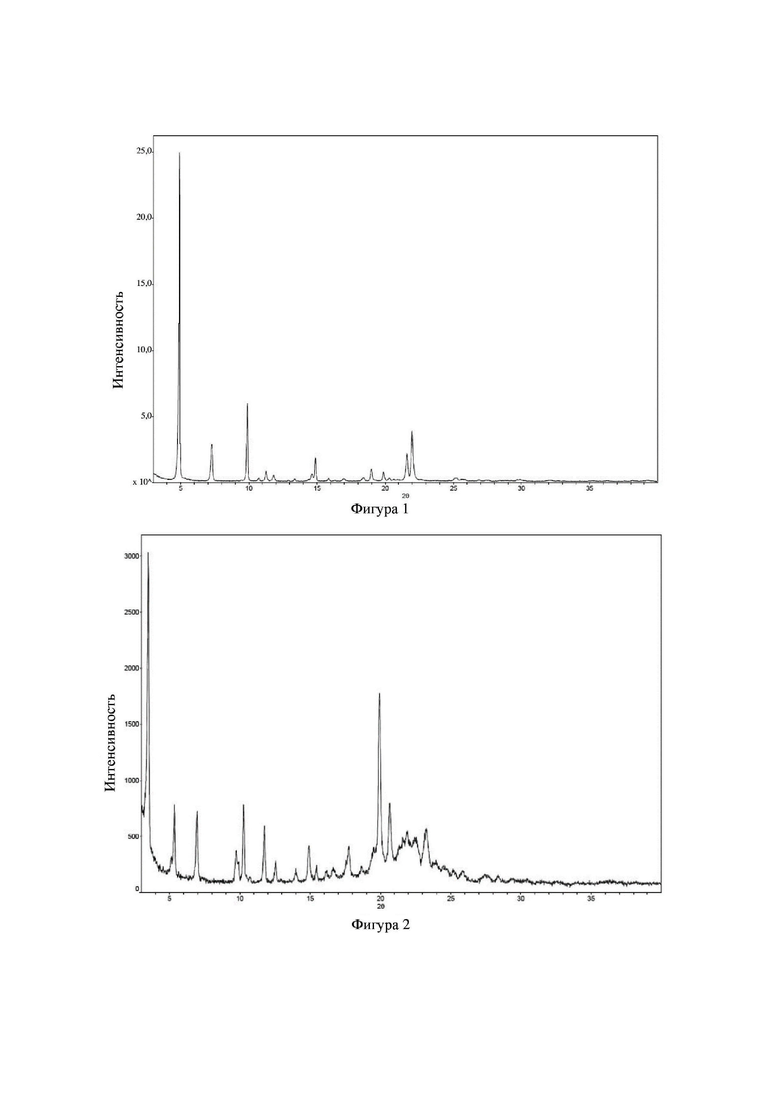
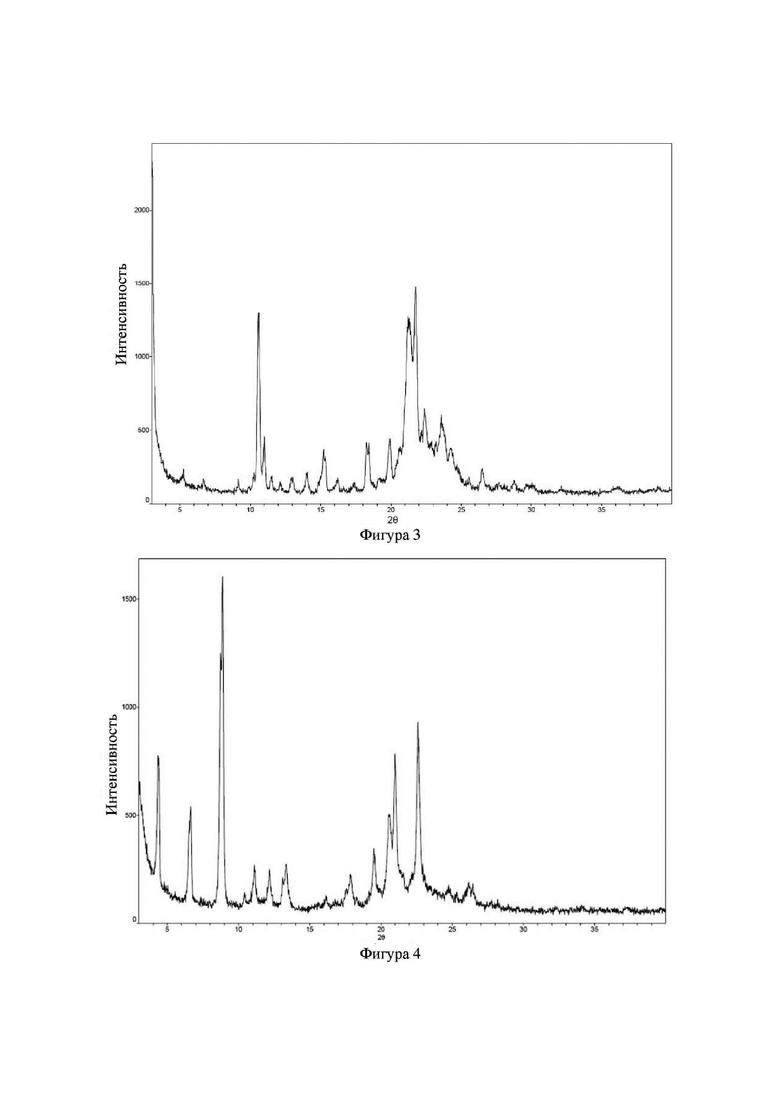
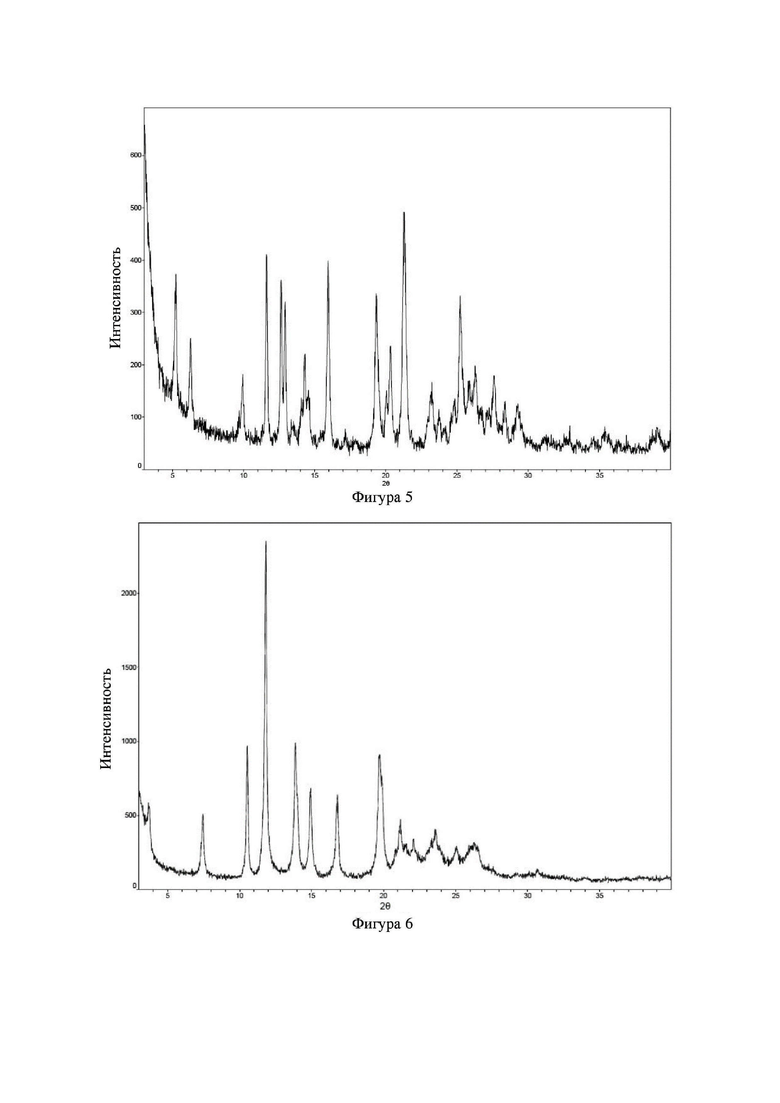
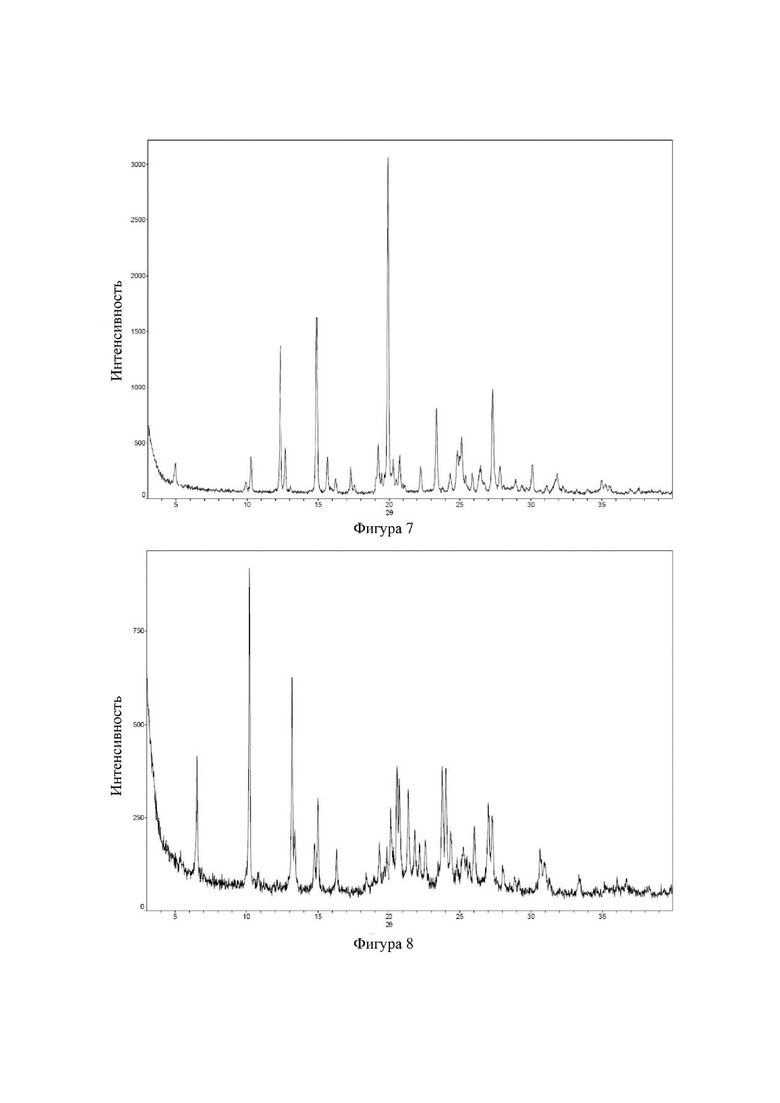
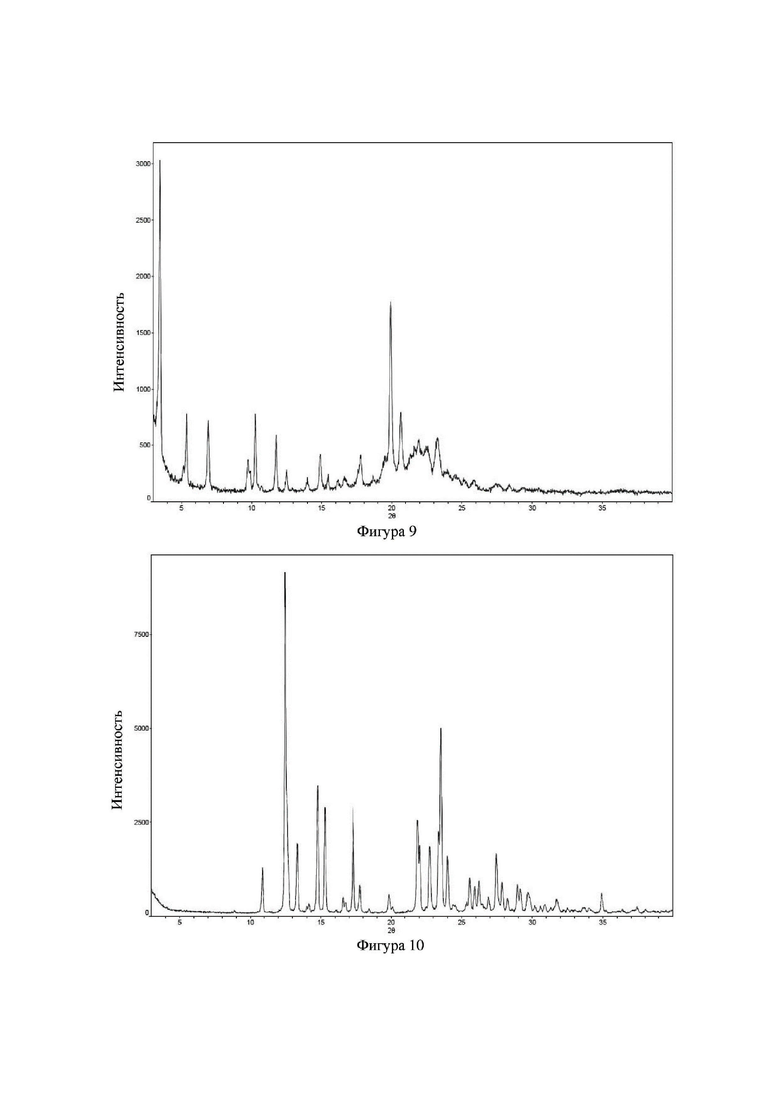
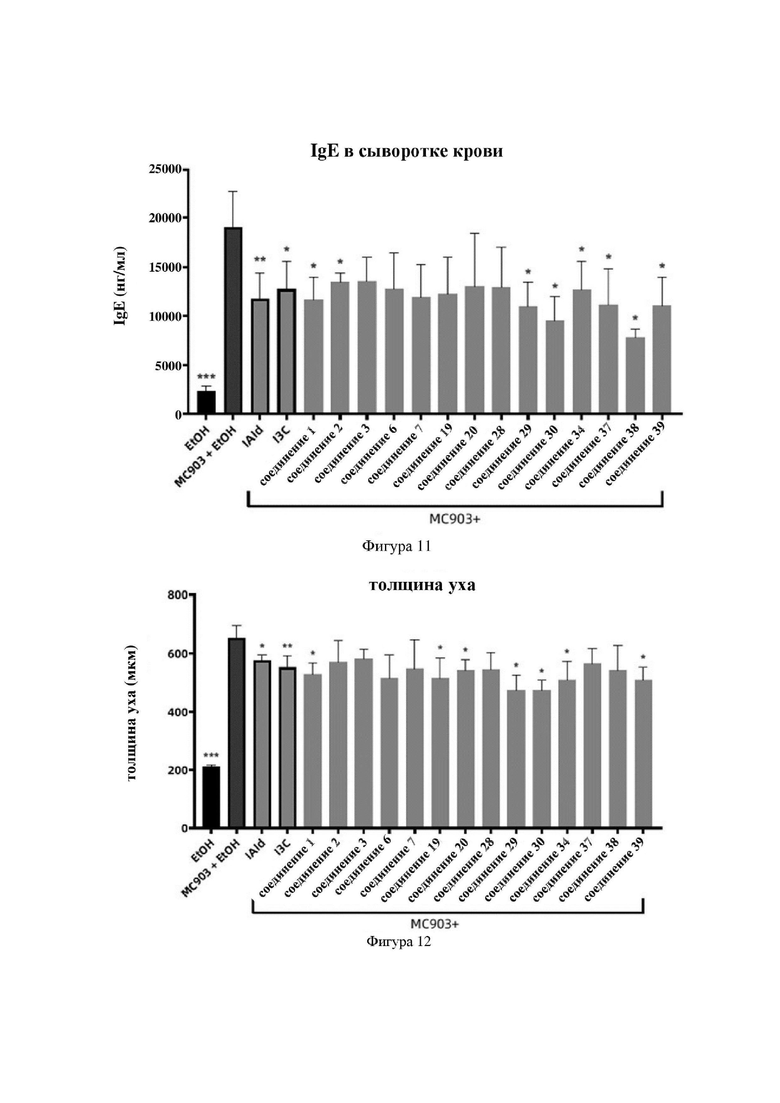
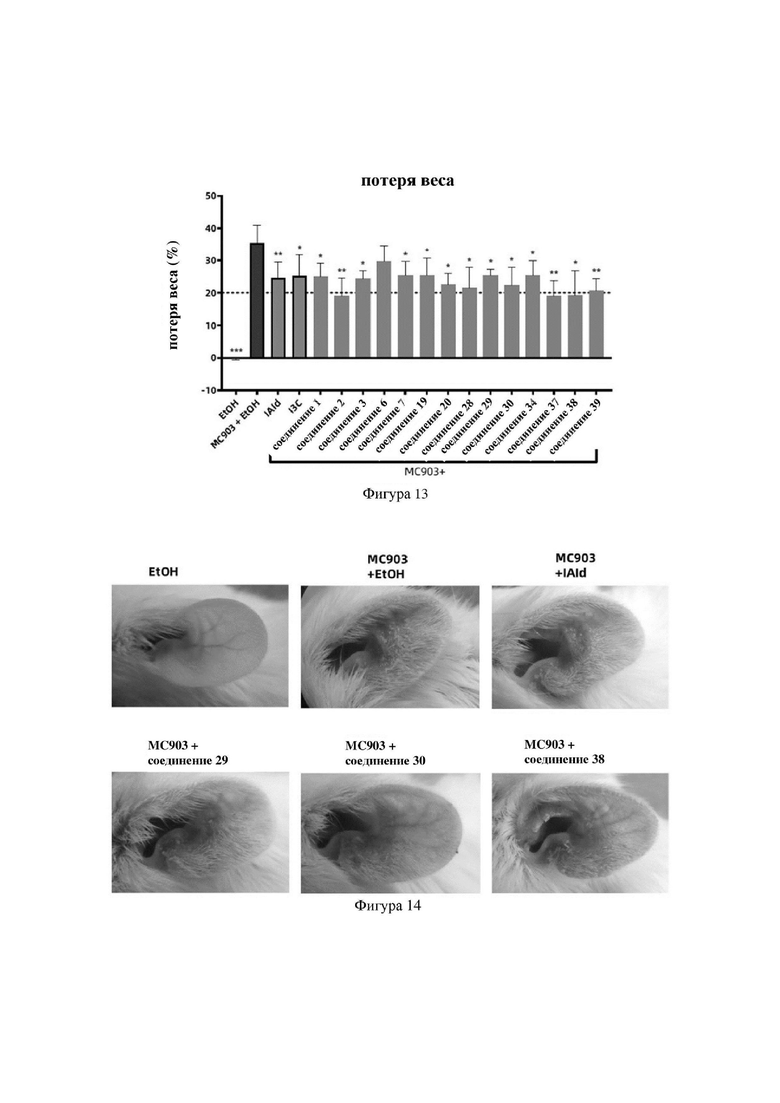
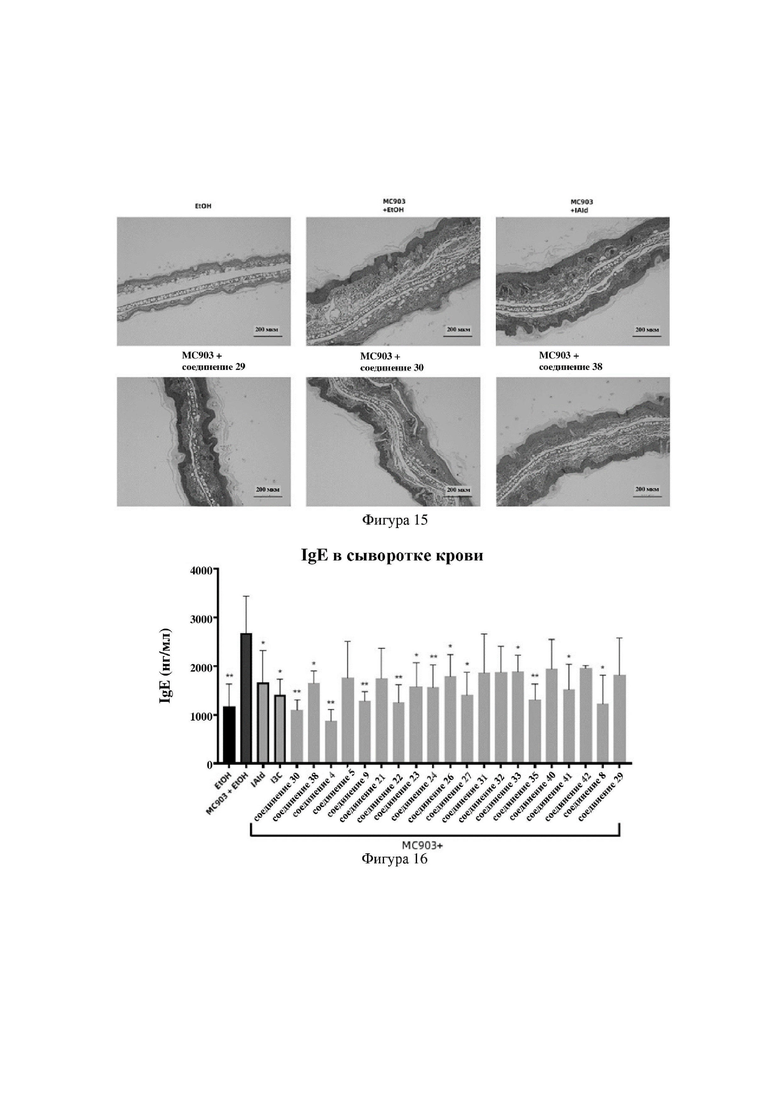
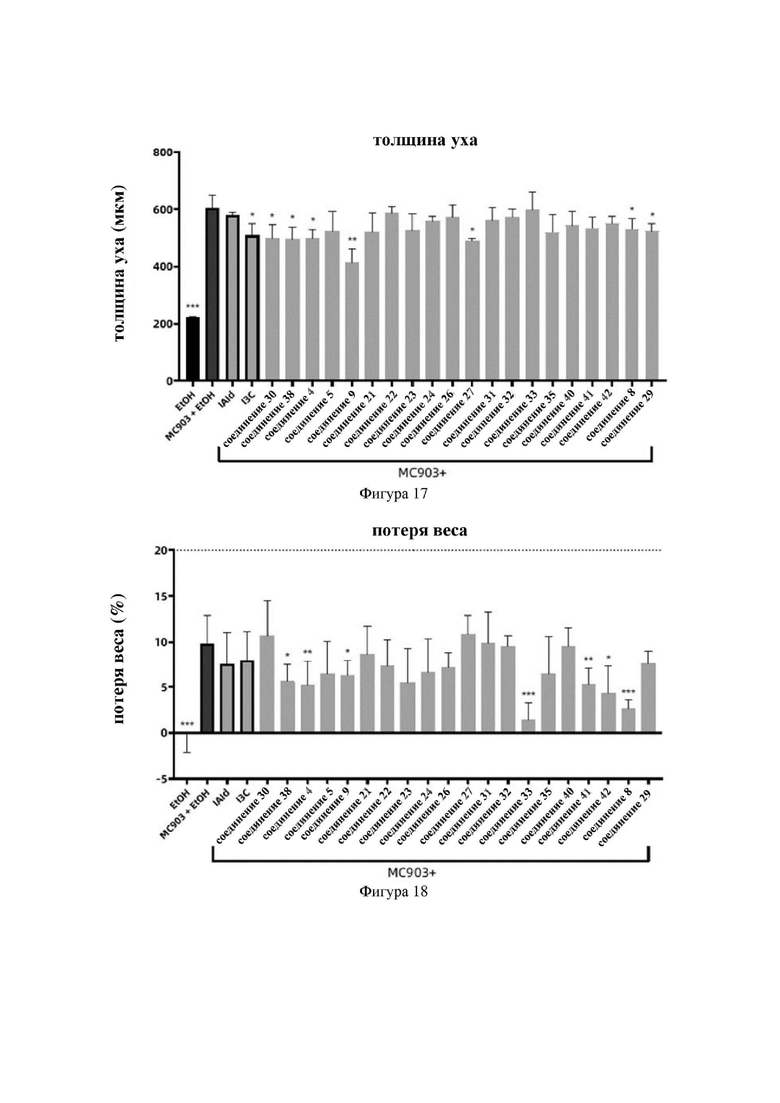
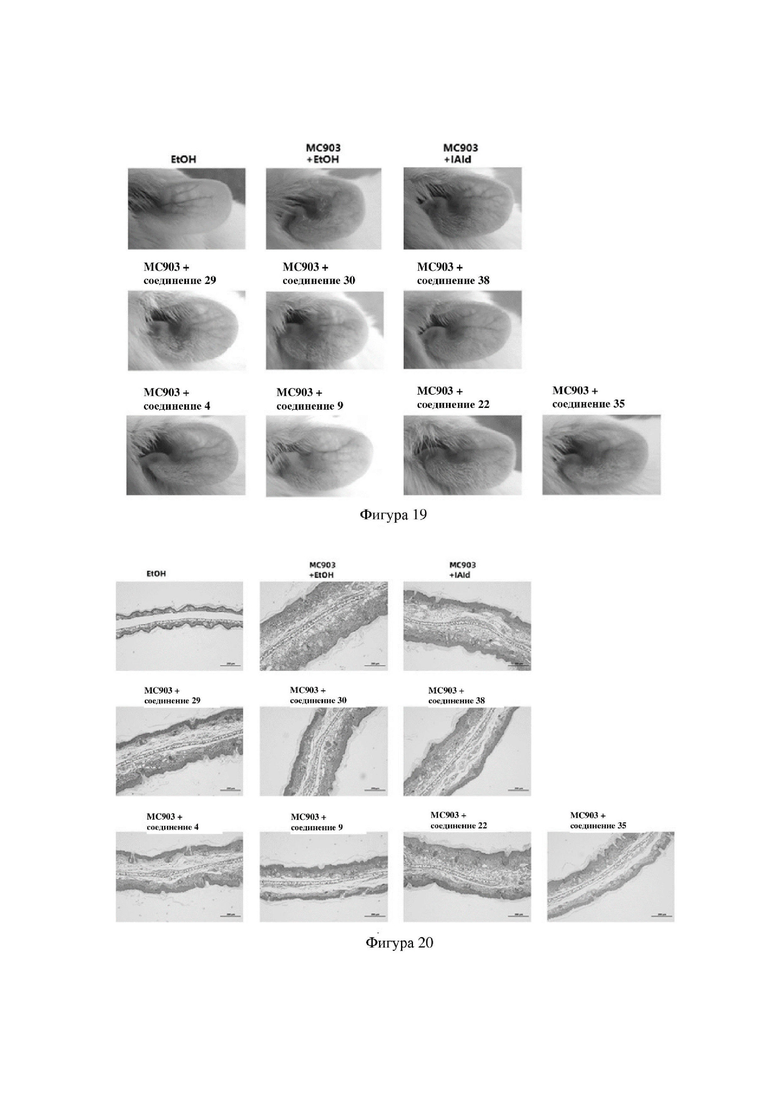
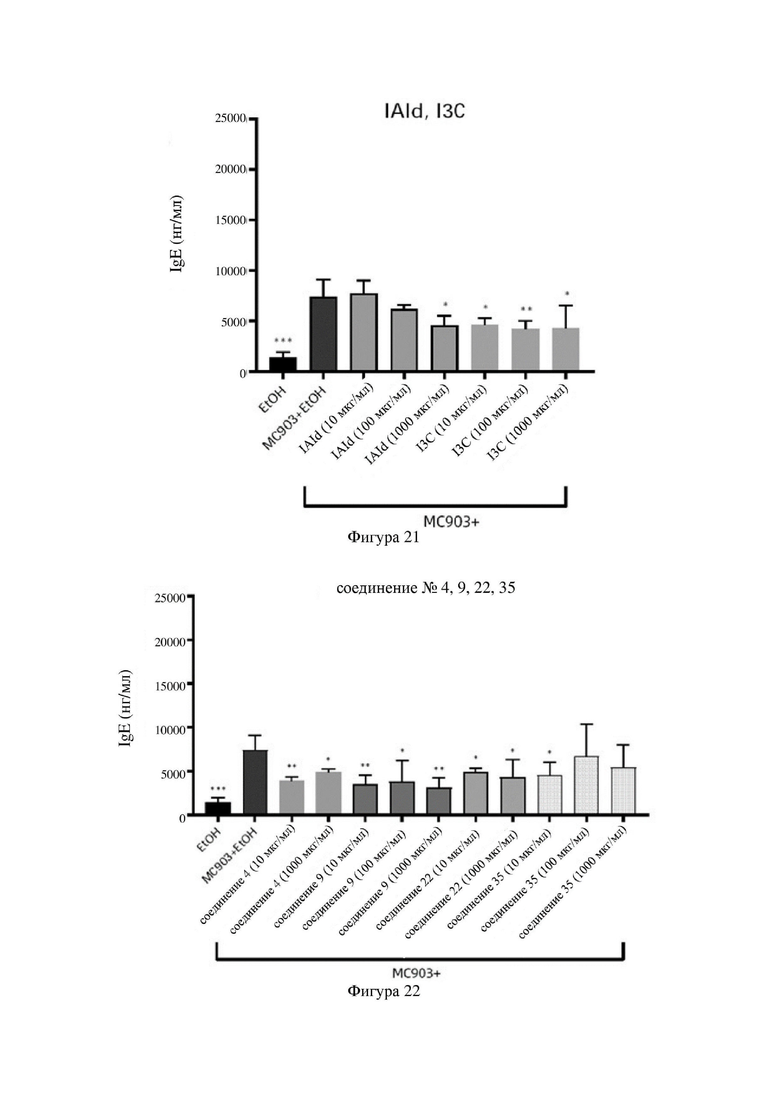
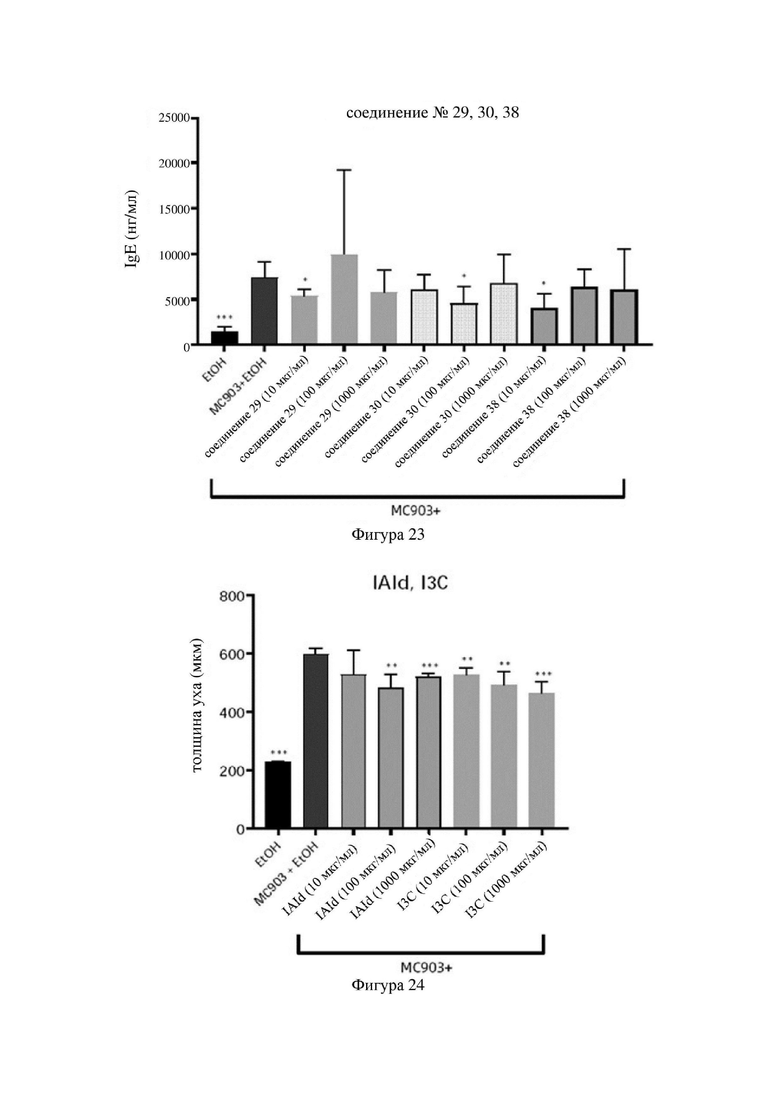
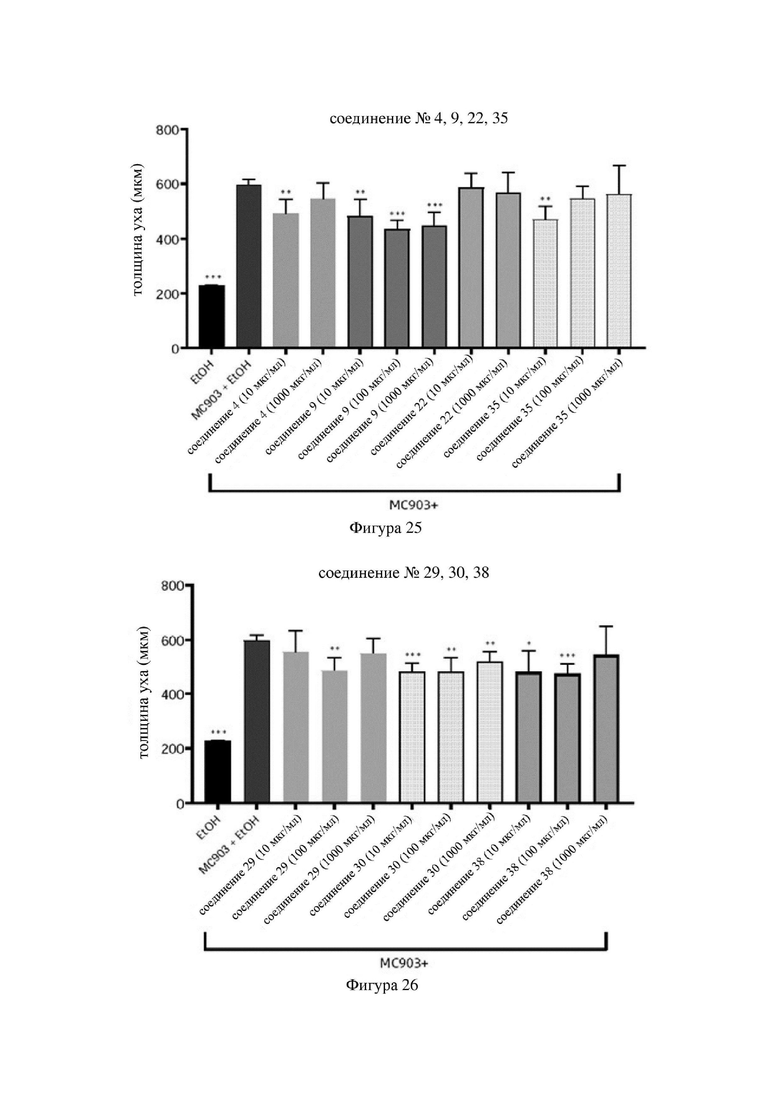
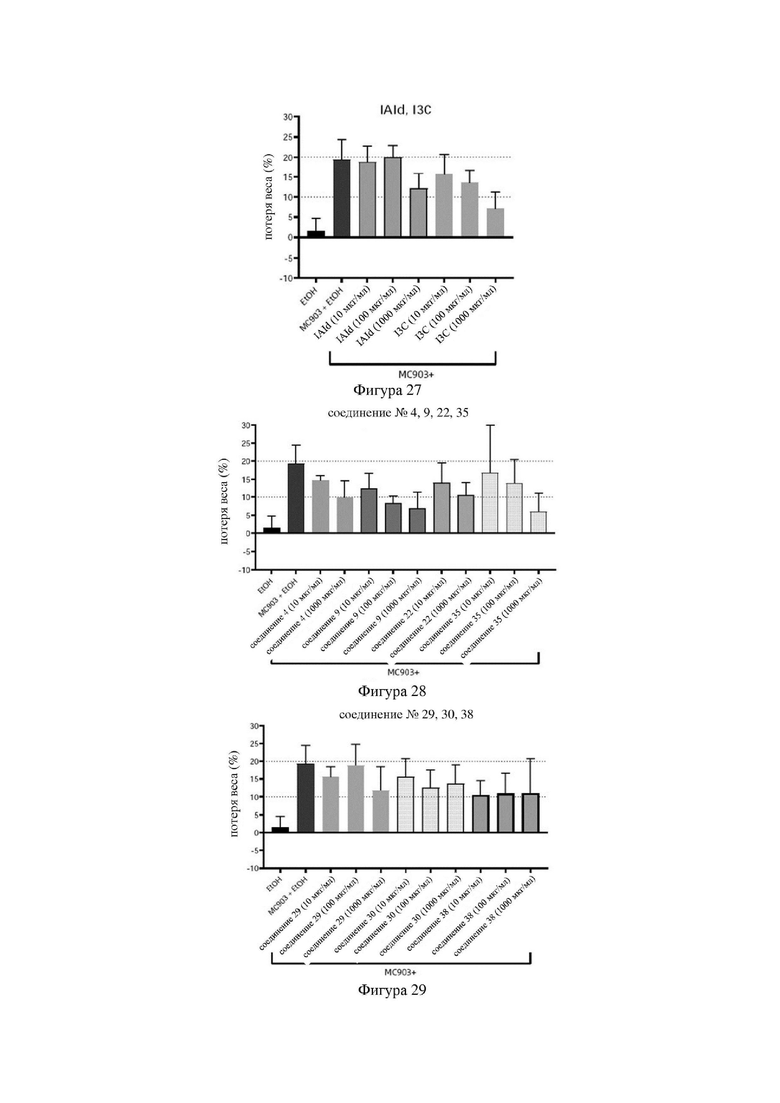
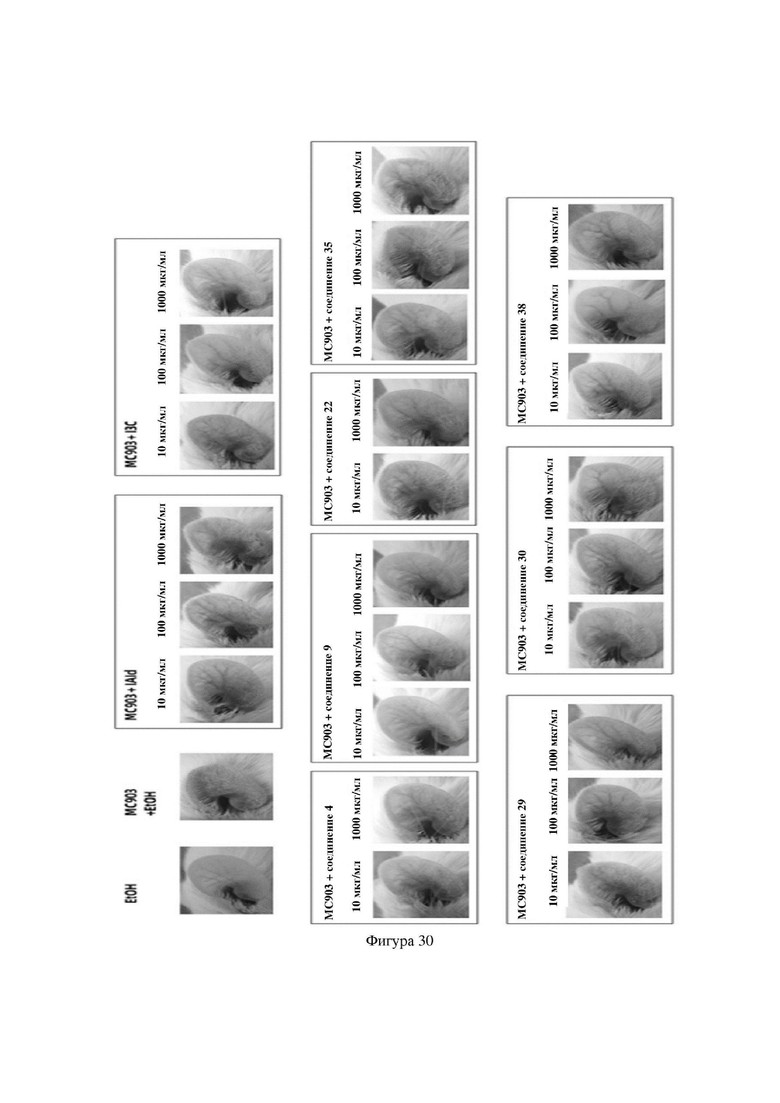
Изобретение относится к соединению формулы I, где W представляет собой COR2 или CR3R4OR5; X отсутствует или X представляет собой CO или CR3R4; Y отсутствует или Y представляет собой O; Z отсутствует или Z представляет собой CR3R4; R1 выбран из группы, состоящей из незамещенного C5-20алкила, С6арила, замещенного 1-2 заместителями, выбранными из ацетокси и хлора, и фрагмента (а); R представляет собой H; каждый из R2, R3 и R4 представляет собой H; и R5 представляет собой H. Также изобретение относится к соединению, где соединение выбрано из группы соединений, представленных в формуле, фармацевтической композиции, содержащей соединение формулы I или соединение, выбранное из группы соединений, и применению соединения формулы I или соединения, выбранного из группы соединений, в изготовлении лекарственного препарата для лечения атопического дерматита. 4 н. и 8 з.п. ф-лы, 30 ил., 11 табл., 34 пр.
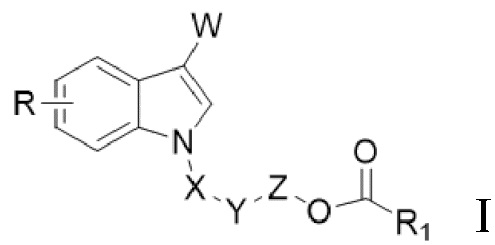
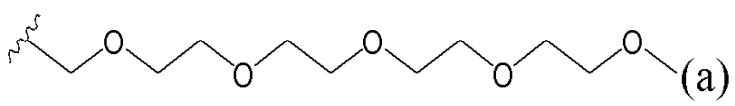
1. Соединение, характеризующееся структурой формулы I
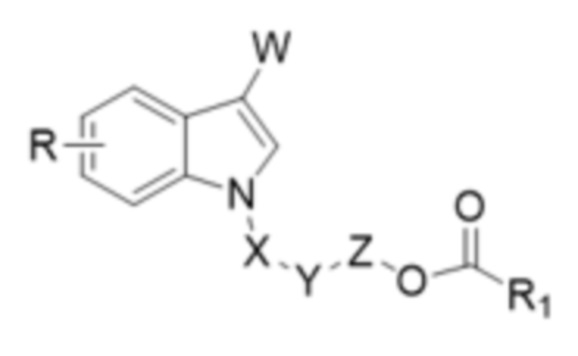
I,
где W представляет собой COR2 или CR3R4OR5;
X отсутствует или X представляет собой CO или CR3R4;
Y отсутствует или Y представляет собой O;
Z отсутствует или Z представляет собой CR3R4;
R1 выбран из группы, состоящей из незамещенного C5-20алкила, С6арила, замещенного 1-2 заместителями, выбранными из ацетокси и хлора, и 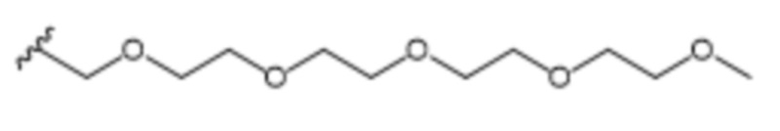 ;
;
R представляет собой H;
каждый из R2, R3 и R4 представляет собой H; и
R5 представляет собой H.
2. Соединение по п. 1, где соединение характеризуется структурой, показанной в формуле II
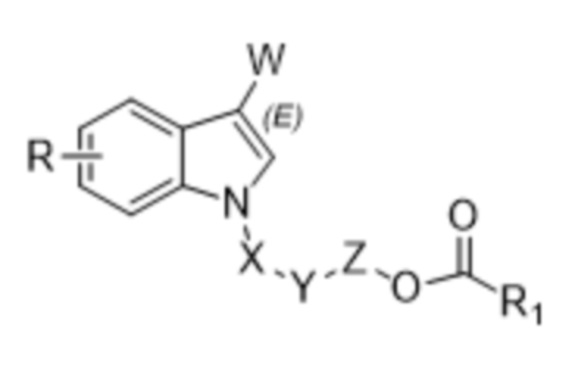
II.
3. Соединение по п. 1 или 2, где
X отсутствует или X представляет собой CO.
4. Соединение по п. 3, где R1 выбран из группы, состоящей из незамещенного C5-15алкила, ацетоксилзамещенного С6арила, (2,6-дихлорфенил)аминозамещенного С6арила и 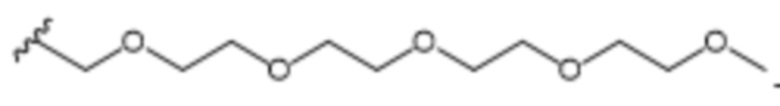
5. Соединение по п. 1 или 2, где
X, Y и Z вместе образуют COOCH2 или CH2; и
R1 выбран из группы, состоящей из C7-C15алкила, ацетоксилзамещенного С6арила, (2,6-дихлорфенил)аминозамещенного С6арила и
6. Соединение по п. 5, где
W представляет собой COH; и
X, Y и Z вместе образуют CH2.
7. Соединение, где соединение выбрано из группы, состоящей из
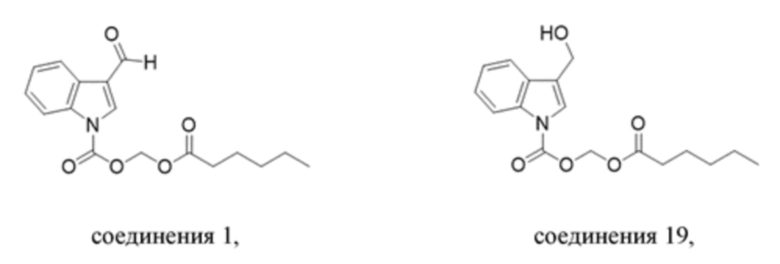
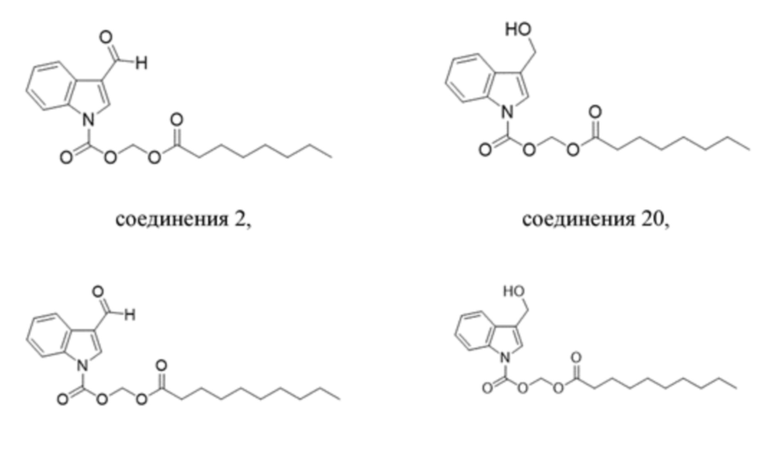
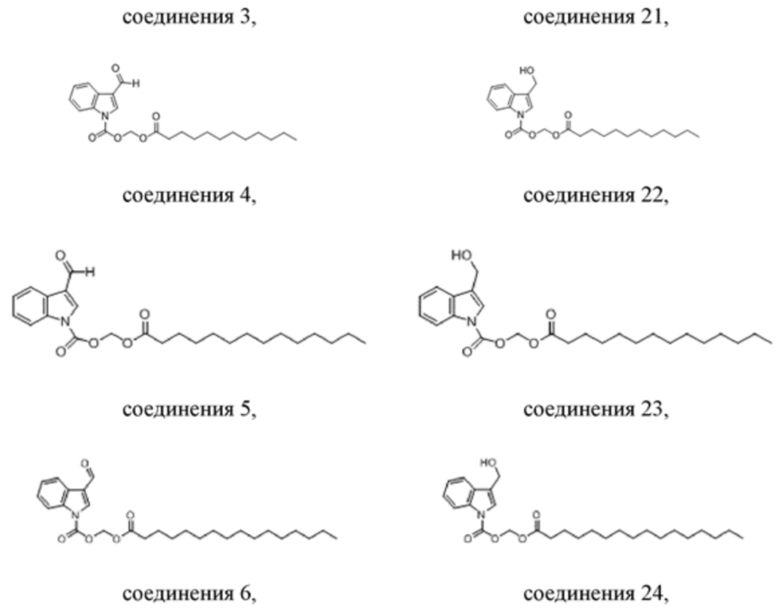
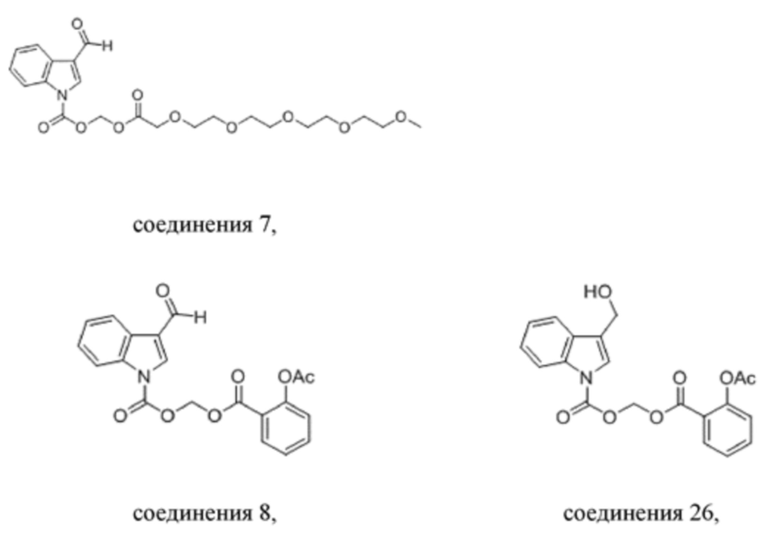
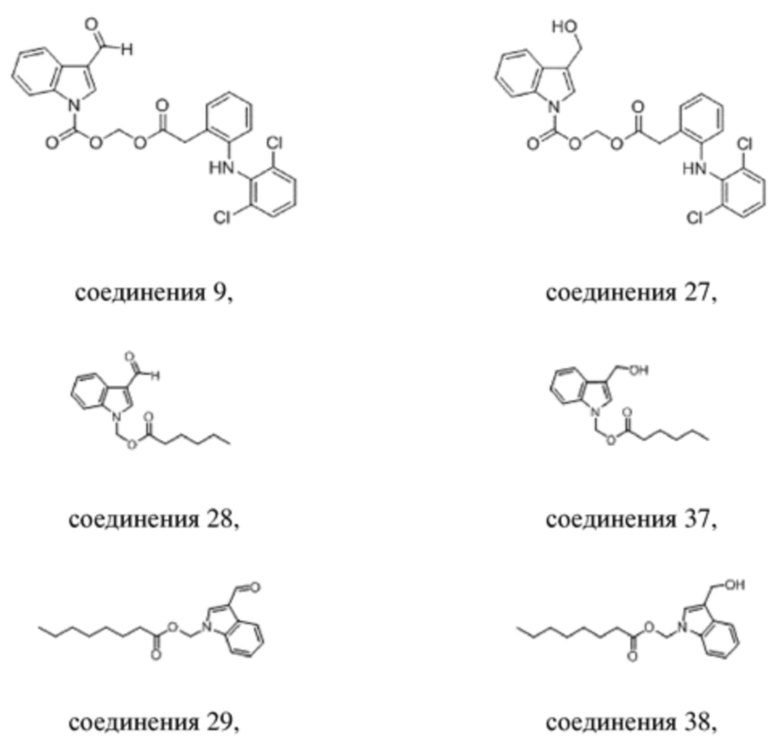
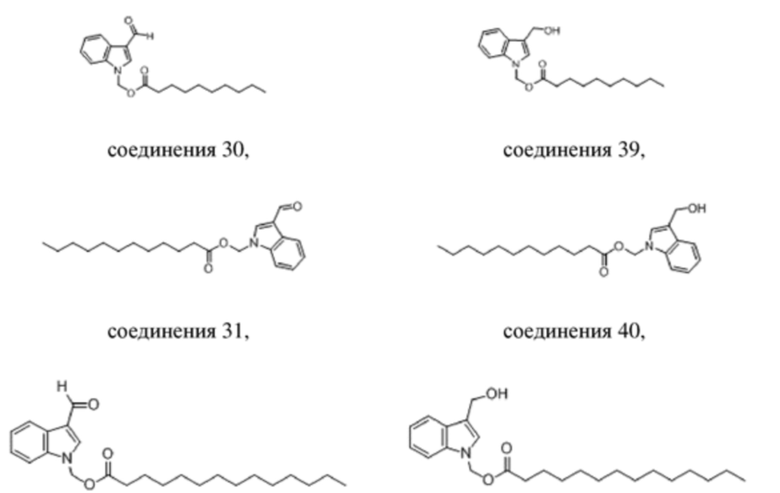
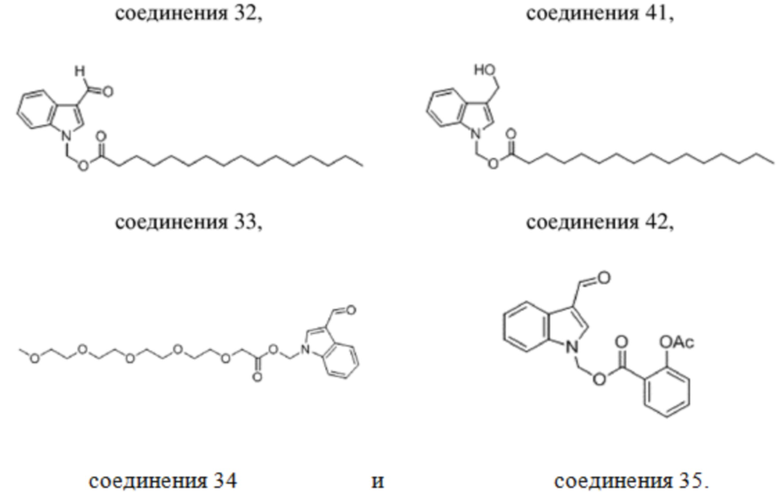
8. Соединение по п. 7, где
соединение 4 имеет характеристические пики при значениях 2θ, составляющих 4,9 ± 0,2°, 7,3 ± 0,2°, 9,9 ± 0,2°, 14,9 ± 0,2° и 22,0 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 8 имеет характеристические пики при значениях 2θ, составляющих 5,2 ± 0,2°, 11,6 ± 0,2°, 12,6 ± 0,2°, 16,0 ± 0,2° и 19,3 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 9 имеет характеристические пики при значениях 2θ, составляющих 12,3 ± 0,2°, 14,9 ± 0,2°, 19,9 ± 0,2°, 23,4 ± 0,2° и 27,3 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 22 имеет характеристические пики при значениях 2θ, составляющих 3,4 ± 0,2°, 5,3 ± 0,2°, 6,9 ± 0,2°, 10,2 ± 0,2° и 19,9 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 23 имеет характеристические пики при значениях 2θ, составляющих 10,6 ± 0,2°, 11,0 ± 0,2°, 18,4 ± 0,2°, 21,2 ± 0,2° и 21,7 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 24 имеет характеристические пики при значениях 2θ, составляющих 4,4 ± 0,2°, 6,6 ± 0,2°, 8,9 ± 0,2°, 21,0 ± 0,2° и 22,6 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 26 имеет характеристические пики при значениях 2θ, составляющих 3,6 ± 0,2°, 10,5 ± 0,2°, 11,8 ± 0,2°, 13,9 ± 0,2° и 19,7 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 27 имеет характеристические пики при значениях 2θ, составляющих 6,5 ± 0,2°, 10,2 ± 0,2°, 13,2 ± 0,2°, 15,0 ± 0,2° и 23,8 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения;
соединение 35 имеет характеристические пики при значениях 2θ, составляющих 12,4 ± 0,2°, 14,7 ± 0,2°, 15,3 ± 0,2°, 17,3 ± 0,2° и 23,5 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения; и
соединение 41 имеет характеристические пики при значениях 2θ, составляющих 3,1 ± 0,2°, 5,2 ± 0,2°, 6,7 ± 0,2°, 10,2 ± 0,2° и 19,9 ± 0,2°, на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения.
9. Фармацевтическая композиция для лечения атопического дерматита, содержащая соединение по любому из пп. 1-8 и фармацевтически приемлемое вспомогательное вещество.
10. Фармацевтическая композиция по п. 9, где лекарственная форма фармацевтической композиции представляет собой таблетку, диспергирующее средство, настойку, гель, капсулу, спрей, суппозиторий, гранулу, жидкость для перорального применения или гранулу.
11. Фармацевтическая композиция по п. 9, где лекарственная форма фармацевтической композиции представляет собой препарат для местного применения.
12. Применение соединения по любому из пп. 1-8 в изготовлении лекарственного препарата для лечения атопического дерматита.
| CN 106008346 B, 13.08.2019 | |||
| WO 2006105796 A1, 12.10.2006 | |||
| WO 2011079102 A1, 30.06.2011 | |||
| Механическая застежка для обуви | 1930 |
|
SU22634A1 |
| US 20190284149 A1, 19.09.2019 | |||
| WO 2006069196 A1, 29.06.2006 | |||
| US 2018110757 A1, 26.04.2018 | |||
| Протез бедра с управляемым коленным шарниром | 1985 |
|
SU1351599A1 |
| WO 2009108551 A2, 03.09.2009 | |||
| VO, Q.V | |||
| et al | |||
| In Silico Study of the Radical Scavenging Activities of Natural | |||

