[1] Настоящая заявка испрашивает приоритет заявки на патент Китая № 201910887908.1, датой подачи которой является 19 сентября 2019 г. Данная заявка на патент Китая включена в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ
[2] Настоящее изобретение относится к способу получения кристаллической формы ингибитора поверхностного антигена вируса гепатита B.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[3] Вирусный гепатит B, сокращенно гепатит B, представляет собой заболевание, обусловленное инфекцией, вызываемой вирусом гепатита B (сокращенно HBV) в организме. Вирус гепатита В относится к семейству hepadnaviridae, главным образом существует в клетках печени и повреждает клетки печени, вызывая воспаление, некроз и фиброз гепатоцитов. Вирусный гепатит В подразделяется на два типа, острый гепатит В и хронический гепатит В. Большинство взрослых с острым гепатитом В могут сами излечиться посредством их собственного иммунного механизма. Однако хронический гепатит В (CHB) стал большой проблемой для глобального здравоохранения и основной причиной хронического заболевания печени, цирроза и гепатоцеллюлярной карциномы (HCC). По оценкам, 2 миллиарда человек во всем мире инфицированы вирусом хронического гепатита В, у более чем 350 миллионов человек развился гепатит В, и около 600000 человек ежегодно умирают от осложнений хронического гепатита В. Китай является регионом с высокой распространенностью гепатита В, где сосредоточено много пациентов с гепатитом В, что представляет серьезную опасность. Согласно данным, в Китае насчитывается приблизительно 93 миллиона человек, инфицированных вирусом гепатита В, и приблизительно у 20 миллионов из них диагностирован хронический гепатит В, у 10-20% из которых может развиться цирроз, и у 1-5% из которых может развиться гепатоцеллюлярная карцинома.
[4] Особенно важным для функционального излечения гепатита В являются клиренс HBsAg (поверхностного антигена гепатита В) и образование поверхностных антител. Количественное определение HBsAg является очень важным биологическим показателем. Снижение уровня HBsAg и сероконверсия HBsAg редко наблюдаются у пациентов с хронической инфекцией и являются конечными точками современных способов терапии.
[5] Поверхностный антигенный белок вируса гепатита В (HBV) играет очень важную роль в процессе проникновения HBV в гепатоциты и имеет большое значение для предупреждения и лечения HBV-инфекции. Поверхностные антигенные белки предусматривают большие (L), средние (M) и малые (S) поверхностные антигенные белки, которые имеют общий C-концевой S-участок. Они экспрессируются с одной и той же открытой рамки считывания и характеризуются разными значениями длины, определяемыми тремя стартовыми кодонами AUG в рамке считывания. Эти три поверхностных антигенных белка содержат домены pre-S1/pre-S2/S, pre-S2/S и S. Поверхностный антигенный белок HBV интегрируется в мембрану эндоплазматического ретикулума (ER), и процесс интеграции инициируется N-концевой сигнальной последовательностью. Они не только составляют основную структуру вирионов, но также образуют сферические и нитевидные субвирусные частицы (SVP, HBsAg), которые агрегируются в ER, ER хозяина и аппарате пре-Гольджи, и SVP содержат большинство поверхностных антигенных белков S. Белок L имеет критическое значение в морфогенезе вируса и взаимодействии нуклеокапсидов, но не является необходимым для образования SVP. Вследствие отсутствия нуклеокапсидов SVP являются неинфекционными. SVP принимают значительное участие в прогрессировании заболевания, особенно в иммунном ответе на вирус гепатита В. В крови инфицированных людей количество SVP в по меньшей мере 10000 раз больше, чем вируса, что блокирует иммунную систему и ослабляет иммунный ответ организма на вирус гепатита В. HBsAg также может подавлять врожденный иммунитет человека, индуцированное полисахаридами (LPS) и IL-2 образование цитокинов, функцию дендритных клеток (DC) и индуцирующую активность LPS в отношении ERK-1/2 и интерферирующей киназы 1/2 N-конца c-Jun в моноцитах. Следует отметить, что прогрессирование цирроза и гепатоцеллюлярной карциномы также в значительной степени связано с устойчивой секрецией HBsAg. Эти выводы свидетельствуют о том, что HBsAg играет важную роль в развитии хронического гепатита.
[6] В настоящее время одобренными и маркированными лекарственными средствами против HBV являются главным образом иммуномодуляторы (интерферон-α и пегинтерферон-α-2α) и противовирусные лекарственные средства (ламивудин, адефовир дипивоксил, энтекавир, телбивудин, тенофовир, клевудин и т. п.). Среди них противовирусные лекарственные средства относятся к лекарственным средствам на основе нуклеотидов, и их механизм действия заключается в ингибировании синтеза ДНК HBV, а не в непосредственном снижении уровня HBsAg. Как и в случае расширенной терапии лекарственные средства на основе нуклеотидов демонстрируют элиминацию HBsAg со скоростью, близкой к результатам наблюдений в естественных условиях.
[7] Существующие клинические терапевтические средства характеризуются низкой эффективностью в снижении уровня HBsAg. Таким образом, разработка низкомолекулярных ингибиторов для перорального применения, которые способны эффективно снижать уровень HBsAg, крайне необходима в современном клиническом медикаментозном лечении.
[8] Компания Roche разработала ингибитор поверхностного антигена под названием RG7834 для лечения гепатита В и сообщила об эффективности данного соединения в модели с сурками против гепатита В: при применении в качестве отдельного лекарственного средства RG7834 способен снижать уровень поверхностных антигенов на 2,57 Log и ДНК HBV на 1,7 Log. По-прежнему существует потребность в новых соединениях, которые способны эффективно снижать уровень поверхностного антигена вируса гепатита B, особенно в лекарственных средствах для предупреждения или лечения хронического гепатита В.
СОДЕРЖАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
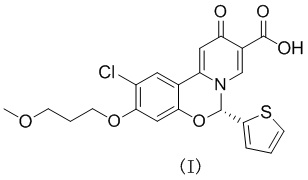
[9] Настоящее изобретение предусматривает способ получения кристаллической формы А соединения формулы (I), где способ включает добавление соединения формулы (I) в любой форме к метанолу, ацетону, этилацетату, ацетонитрилу, смеси ацетонитрил-вода при соотношении 1:1 или смеси ацетон-вода при соотношении 1:1, перемешивание смеси в течение от 1 часа до 72 часов при температуре от 25 °C до 65 °C, затем охлаждение до комнатной температуры, фильтрование и высушивание осадка на фильтре с получением кристаллической формы А; где кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,30 ± 0,20°, 9,30 ± 0,20°, 9,84 ± 0,20°, 18,68 ± 0,20°, 20,16 ± 0,20°, 23,06 ± 0,20°, 24,00 ± 0,20° и 25,38 ± 0,20°;
 .
.
[10] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,30 ± 0,20°, 9,30 ± 0,20°, 9,84 ± 0,20°, 12,84 ± 0,20°, 18,68 ± 0,20°, 20,16 ± 0,20°, 21,26 ± 0,20°, 23,06 ± 0,20°, 24,00 ± 0,20° и 25,38 ± 0,20°.
[11] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,302°, 7,883°, 9,301°, 9,842°, 12,838°, 15,436°, 16,580°, 18,124°, 18,680°, 19,459°, 20,161°, 20,800°, 21,262°, 21,704°, 23,057°, 24,000°, 24,837°, 25,382°, 26,244°, 26,558°, 27,740°, 28,119°, 28,827°, 29,502°, 29,880°, 30,261°, 30,762°, 31,678°, 32,595°, 33,061°, 34,347°, 35,235°, 35,738°, 36,642°, 38,619° и 39,558°.
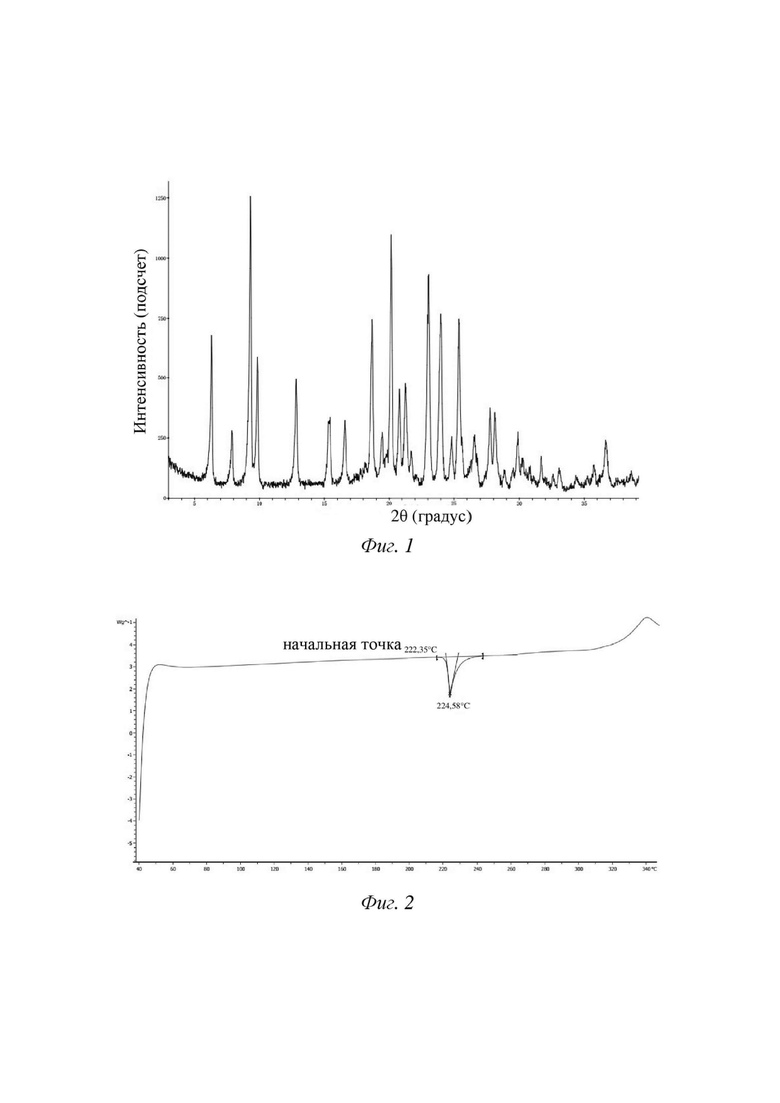
[12] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется дифрактограммой XRPD, показанной на фигуре 1.
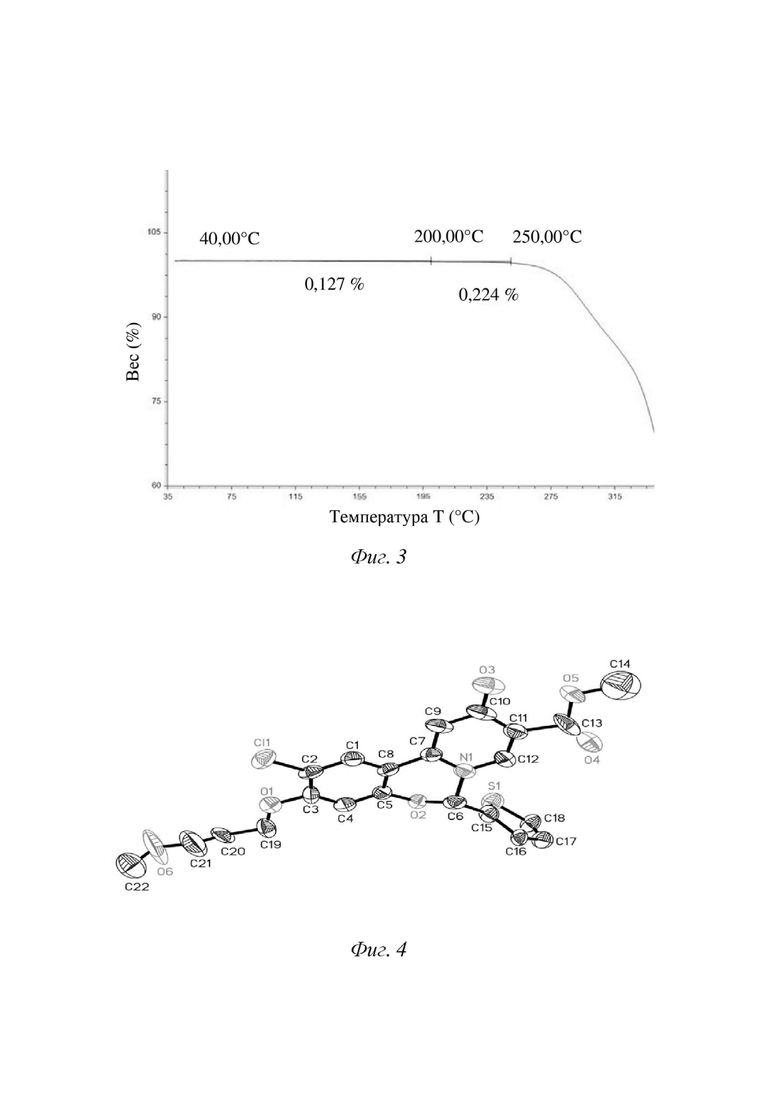
[13] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется профилем дифференциальной сканирующей калориметрии, содержащим эндотермический пик при 224,58oC ± 3oC.
[14] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется термограммой DSC, показанной на фигуре 2.
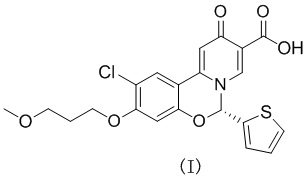
[15] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется кривой термогравиметрического анализа, демонстрирующей потерю веса 0,127% при 200,00oC ± 3oC и потерю веса 0,224% при 250oC ± 3oC.
[16] В некоторых вариантах осуществления настоящего изобретения вышеупомянутая кристаллическая форма А характеризуется термограммой TGA, показанной на фигуре 3.
[17] В некоторых вариантах осуществления настоящего изобретения весовое соотношение вышеупомянутого соединения формулы (I) и растворителя выбрано из 1: 1-30.
[18] Технические эффекты
[19] Соединение по настоящему изобретению характеризуется значительной активностью против вируса гепатита В. Соединение по настоящему изобретению не ингибирует изоферменты цитохрома Р450, что указывает на более низкий риск лекарственного взаимодействия; характеризуется значительной стабильностью в микросомах печени у трех видов, т. е. крысы, человека и мыши, что указывает на то, что соединение легко не метаболизируется; характеризуется лучшим воздействием и биодоступностью и хорошо переносится в исследовании нейротоксичности однократной дозы.
[20] Кристаллическую форму А соединения по настоящему изобретению легко получить, она характеризуется хорошей физической стабильностью и химической стабильностью, а также высокой ценностью для промышленного применения и экономической ценностью.
[21] Определение и описание
[22] Если не указано иное, следующие термины и фразы, используемые в данном документе, имеют следующие значения. Конкретная фраза или термин не должны считаться неопределенными или неясными, если они специально не определены, но должны пониматься в обычном значении. Если в настоящем документе появляется торговое наименование, предполагается, что оно относится к соответствующему продукту или его активному ингредиенту.
[23] Промежуточные соединения по настоящему изобретению могут быть получены посредством различных способов синтеза, хорошо известных специалисту в данной области техники, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные посредством комбинирования с другими способами химического синтеза, и эквивалентные альтернативные варианты осуществления, хорошо известные специалисту в данной области техники, где предпочтительные варианты осуществления включают без ограничения примеры из настоящего раскрытия.
[24] Химические реакции, описанные в конкретных вариантах осуществления настоящего изобретения, завершаются в подходящем растворителе, где растворитель должен быть подходящим для химических изменений по настоящему изобретению и реагентов и материалов, необходимых для этого. Для того, чтобы получить соединения по настоящему изобретению, иногда специалисту в данной области техники необходимо модифицировать или выбрать стадии синтеза или схемы реакций на основе существующих вариантов осуществления.
[25] Настоящее изобретение будет конкретно описано ниже посредством примеров, которые никоим образом не предназначены для ограничения настоящего изобретения.
[26] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными и могут использоваться без дополнительной очистки.
[27] Соединения названы вручную или с помощью программного обеспечения ChemDraw® и доступные соединения названы наименованиями из каталога поставщика.
[28] Способ с применением рентгеновского порошкового дифрактометра (XRPD), используемый в настоящем изобретении
[29] Модель прибора: рентгеновский дифрактометр Dandong Haoyuan DX-2700BH
[30] Способ тестирования: от примерно 10 мг до 20 мг образца используется для выявления посредством XRPD.
[31] Подробные параметры XRPD являются следующими:
[32] световая трубка: Cu, kα, (λ = 1,54184Ǻ).
[33] Напряжение на световой трубке: 40 кВ, сила тока на световой трубке: 30 мА
[34] Щель расходимости: 1 мм
[35] Щель детектора: 0,3 мм
[36] Антирассеивающая щель: 1 мм
[37] Диапазон сканирования: 3-40 град.
[38] Размер шага: 0,02 град.
[39] Продолжительность шага: 0,5 сек
[40] Способ с применением дифференциального сканирующего калориметра (DSC), используемый в настоящем изобретении
[41] Модель прибора: дифференциальный сканирующий калориметр METTLER TOLEDO DSC1
[42] Способ тестирования: образец (2-6 мг) помещали в покрытый золотом тигель для DSC объемом 30 мкл, выдерживающий высокое давление, для тестирования и нагревали с 40oC до 350oC со скоростью нагревания 10oC/мин.
Способ с применением термогравиметрического анализатора (TGA), используемый в настоящем изобретении
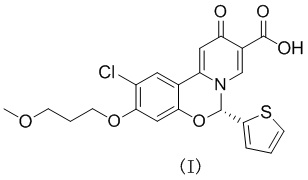
[43] Модель прибора: термогравиметрический анализатор TA TGA550
[44] Способ тестирования: образец (2-10 мг) помещали в алюминиевый тигель, затем помещали в платиновую подвесную корзину для тестирования и нагревали с 40oC до 500oC в условиях атмосферы азота (N2) при скорости потока газа 40 мл/мин и скорости нагревания 10oC/мин.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[45] Фигура 1 представляет собой дифрактограмму XRPD излучения Cu-Kα кристаллической формы А соединения формулы (I).
[46] Фигура 2 представляет собой термограмму DSC кристаллической формы А соединения формулы (I).
[47] Фигура 3 представляет собой термограмму TGA кристаллической формы А соединения формулы (I).
[48] Фигура 4 представляет собой эллипсоидную диаграмму рентгеновской дифракции монокристалла для определения стереохимической структуры соединения формулы (II).
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
[49] Чтобы лучше понять содержание настоящего изобретения для дальнейшего описания используются следующие конкретные примеры, но конкретные варианты осуществления не ограничивают содержание настоящего изобретения.
[50] Пример 1. Получение соединения формулы (I)
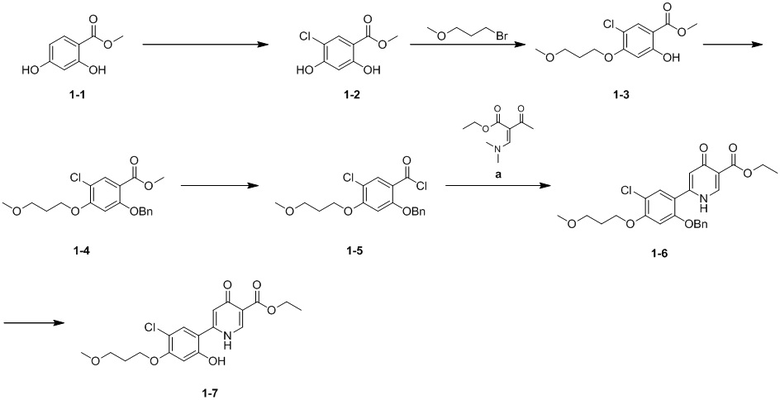
[51] Стадия A. Соединение 1-1 (10,00 г, 59,5 ммоль) растворяли в дихлорметане (500 мл) при 0oC, затем добавляли сульфонилхлорид (10,77 г, 79,77 ммоль, 7,98 мл) и смешанный раствор перемешивали при 35oC в течение 38 часов. Затем раствор выливали в 300 мл насыщенного водного раствора бикарбоната натрия, перемешивали и разделяли. Водную фазу экстрагировали этилацетатом (150 мл × 3) и затем объединенные органические фазы промывали насыщенным солевым раствором (40 мл × 3), высушивали над безводным сульфатом натрия и перегоняли при пониженном давлении с получением белого остатка. Затем белый остаток очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = от 30/1 до 20/1) с получением соединения 1-2.
[52] 1H ЯМР (400 МГц, MeOH-d4) δ 10,75 (s, 1H), 7,74 (s, 1H), 6,54 (s, 1H), 5,98 (s, 1H), 3,85 (s, 3H).
[53] Стадия B. Соединение 1-2 (8,00 г, 39,49 ммоль) и 1-бром-3-метоксипропан (7,25 г, 47,39 ммоль) растворяли в N,N-диметилформамиде (100,00 мл) и охлаждали до 0oC. Затем добавляли карбонат калия (10,92 г, 78,98 ммоль) и смешанный раствор нагревали до 25oC и перемешивали в течение 10 часов. К раствору добавляли этилацетат (300 мл) и воду (50 мл) и полученный раствор перемешивали при 25oC в течение 10 минут. Органическую фазу отделяли и промывали насыщенным солевым раствором (40 мл × 3), затем высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением желтой жидкости. Желтую жидкость очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = от 30/1 до 20/1) с получением соединения 1-3.
[54] 1H ЯМР (400 МГц, CDCl3) δ 10,90 (s, 1H), 7,82 (s, 1H), 6,52 (s, 1H), 4,16 (t, J = 6,0 Гц, 2H), 3,94 (s, 3H), 3,60 (t, J = 6,0 Гц, 2H), 3,38 (s, 3H), 2,13 (t, J = 6,0 Гц, 2H).
[55] Стадия C. К раствору соединения 1-3 (3,64 г, 13,25 ммоль) и бензилхлорида (2,18 г, 17,23 ммоль, 1,98 мл) в N,N-диметилформамиде (10,00 мл) добавляли карбонат калия (4,76 г, 34,45 ммоль) и смешанный раствор перемешивали при 25oC в течение 20 часов. К раствору добавляли этилацетат (150 мл) и воду (30 мл) и полученный раствор перемешивали при 20oC в течение 10 минут. Затем органическую фазу отделяли и промывали водой (30 мл × 2) и насыщенным солевым раствором (30 мл × 2), затем высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 1-4.
[56] Стадия D. Соединение 1-4 (2,00 г, 5,48 ммоль) и моногидрат гидроксида лития (1,38 г, 32,89 ммоль) добавляли к смешанному раствору тетрагидрофурана (20 мл) и воды (10 мл) и затем полученный раствор перемешивали при от 10oC до 20oC в течение 10 часов. Затем раствор промывали смесью этилацетат/петролейный эфир в соотношении 1/1 (5 мл × 3). Значение pH водной фазы доводили до 1-2. Затем раствор экстрагировали дихлорметаном (50 мл × 3) и органические фазы объединяли, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 2-бензилокси-5-хлор-4-(3-метоксилпропан)бензойной кислоты. К раствору 2-бензилокси-5-хлор-4-(3-метоксипропан)бензойной кислоты и муравьиной кислоты (1,00 г, 2,85 ммоль) в дихлорметане (10,00 мл) добавляли тионилхлорид (508,60 мг , 4,28 ммоль, 310,12 мкл) и смешанный раствор перемешивали при 25oC в течение 1 часа. Затем раствор концентрировали при пониженном давлении с получением остатка. Остаток растворяли в толуоле и концентрировали при пониженном давлении с получением остатка и остаток соединения 1-5 хранили в атмосфере азота.
[57] 1H ЯМР (400 МГц, DMSO-d6) δ 12,87-12,22 (m, 1H), 7,74 (s, 1H), 7,53 (br d, J = 7,2 Гц, 2H), 7,40 (t, J = 7,6 Гц, 2H), 7,36-7,30 (m, 1H), 6,94 (s, 1H), 5,27 (s, 2H), 4,20 (s, 2H), 3,50 (s, 2H), 3,26 (s, 3H), 1,98 (t, J = 6,4 Гц, 2H).
[58] Стадия E. Раствор соединения 1-5 (2,94 г, 7,96 ммоль) и соединения а (1,62 г, 8,76 ммоль, 1,10 экв.) в тетрагидрофуране (20 мл) по каплям добавляли (5 мин) к раствору гексаметилдисилазида лития (1 моль/л, 23,88 мл) в тетрагидрофуране (20 мл) при -70oC Затем охлаждающую баню удаляли и обеспечивали продолжение перемешивания смеси в течение 5 минут. К смеси добавляли ацетат аммония (3,23 г, 41,95 ммоль) и уксусную кислоту (67,85 г, 1,13 ммоль) и большую часть тетрагидрофурана удаляли с помощью роторного испарителя при 60oC и остаток нагревали при от 60oC до 65oC в течение 1,5 часа. Реакционную смесь охлаждали и затем к ней добавляли воду (40 мл) и дихлорметан (200 мл). Смесь перемешивали в течение 10 минут и разделяли и органическую фазу промывали водой (10 мл × 3) и водным раствором бикарбоната натрия, высушивали и концентрировали с получением желтого остатка. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1) с получением соединения 1-6.
[59] Стадия F. К раствору соединения 1-6 (3,00 г, 6,36 ммоль) в тетрагидрофуране (20 мл) добавляли палладий на активированном угле (влажный) (500 мг) и раствор перемешивали при 25oC в течение 2 часов в атмосфере водорода (15 фунтов/кв. дюйм). Затем коричневую суспензию фильтровали и фильтрат собирали и концентрировали при пониженном давлении с получением остатка. Затем остаток суспендировали смесью петролейный эфир/этилацетат (4/1) (два раза) и фильтровали с получением соединения 1-7.
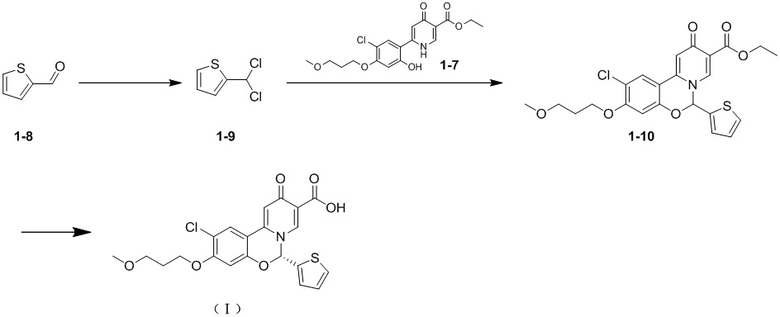
[60] Стадия G. К раствору соединения 1-8 (20,00 г, 178,33 ммоль) в дихлорметане (150,00 мл) добавляли пиридин (2,82 г, 35,67 ммоль) с последующим добавлением пентахлорида фосфора (37,14 г, 178,33 ммоль) при -10oC и полученную смесь подвергали реакции при -10oC в течение 0,5 часа. После завершения реакции в реакционную систему добавляли бикарбонат натрия (44,94 г, 534,99 ммоль). Реакционную смесь перемешивали в течение еще 0,5 часа, затем фильтровали через целит, промывали дихлорметаном (20 мл × 3) и фильтрат концентрировали с получением остатка соединения 1-9.
[61] 1H ЯМР (400 МГц, CDCl3) δ 7,68 (d, J = 3,91 Гц, 1H), 7,35-7,44 (m, 5H), 7,22 (d, J = 3,67 Гц, 1H), 6,91 (s, 1H), 5,34 (s, 2H).
[62] Стадия Н: раствор соединения 1-7 (10,00 г, 26,19 ммоль), карбоната цезия (38,40 г, 117,86 ммоль) и соединения 1-9 (21,88 г, 130,95 ммоль) в диметилсульфоксиде (100,00 мл) перемешивали при 100oC в течение 16 часов. После завершения реакции реакцию гасили с помощью 50 мл воды. Реакционный раствор разбавляли с помощью 150 мл воды и экстрагировали дихлорметаном (100 мл × 3). Затем органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: дихлорметан/этанол = от 100/1 до 8/1) с получением соединения 1-10.
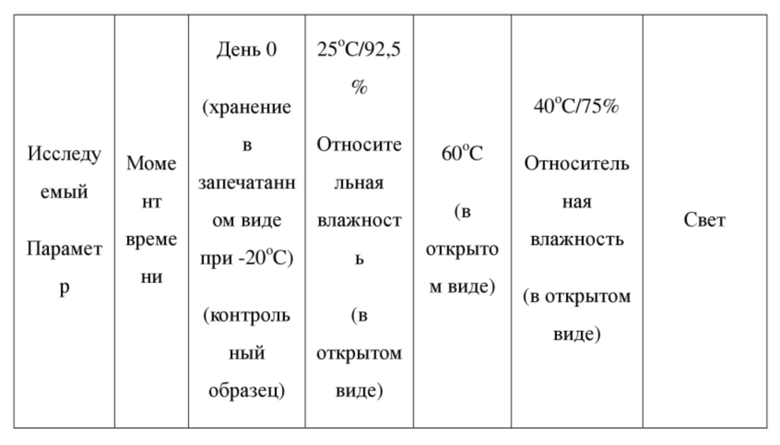
[63] Стадия I. Соединение 1-10 разделяли на колонке для хиральной хроматографии (колонка для разделения: AS (250 мм × 30 мм, 10 мкм); подвижная фаза: [0,1% смесь аммиачная вода-этанол]; градиент элюирования: 40-40%, 4,3 мин; 120 мин) с получением двух конфигурационных изомеров со временем удерживания = 2,516 мин и 5,098 мин соответственно. Затем изомер со временем удерживания = 2,516 мин (55 мг, 116,64 мкмоль) помещали в раствор метанола (10 мл) и добавляли 4 моль/мл гидроксида натрия (145,80 мкл). Реакционный раствор перемешивали при от 25oC до 30oC в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении; остаток растворяли в воде (20 мл) и значение pH доводили до 1-2 с помощью соляной кислоты с концентрацией 2 моль/л; твердое вещество осаждали и затем фильтровали и высушивали с получением соединения формулы (I) (время удерживания = 3,842 мин), значение ee (энантиомерного избытка): 100%. Способ измерения значений ee (энантиомерного избытка): колонка для разделения OD-3S_3_40_3ML: Chiralcel OD-3, I.D. 100 × 4,6 мм, 3 мкм, подвижная фаза: 40% метанол (0,05% диэтиламина) в CO2, скорость потока: 3 мл/мин, длина волны: 220 нм.
[64] Соединение формулы (I): 1H ЯМР (400 МГц, CDCl3) δ 15,40 (s, 1H), 8,32 (s, 1H), 7,62 (s, 1H), 7,41 (dd, J = 1,28, 4,83 Гц, 1H), 6,93-6,98 (m, 2H), 6,91 (s, 1H), 6,88 (s, 1H), 6,62 (s, 1H), 4,09 (t, J = 6,24 Гц, 2H), 3,51 (t, J = 5,87 Гц, 2H), 3,28 (s, 3H), 2,05 (квин., J = 6,08 Гц, 2H).
[65] Пример 2. Получение кристаллической формы А соединения формулы (I)
[66] При температуре, поддерживаемой при 40oC, в реакционную колбу добавляли соединение формулы (I) (1 г) и затем добавляли ацетон (10 мл). Смесь перемешивали при 40oC в течение 24 часов, охлаждали до комнатной температуры и фильтровали, осадок на фильтре промывали ацетоном (10 мл) с получением неочищенного продукта, который высушивали в вакуумном сушильном шкафу (45oC, 16 часов) с получением кристаллической формы А соединения формулы (I).
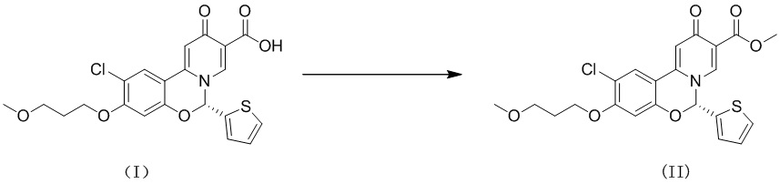
[67] Пример 3. Тест в отношении стабильности кристаллической формы А соединения формулы (I) в виде твердого вещества в условиях высокой температуры и высокой влажности
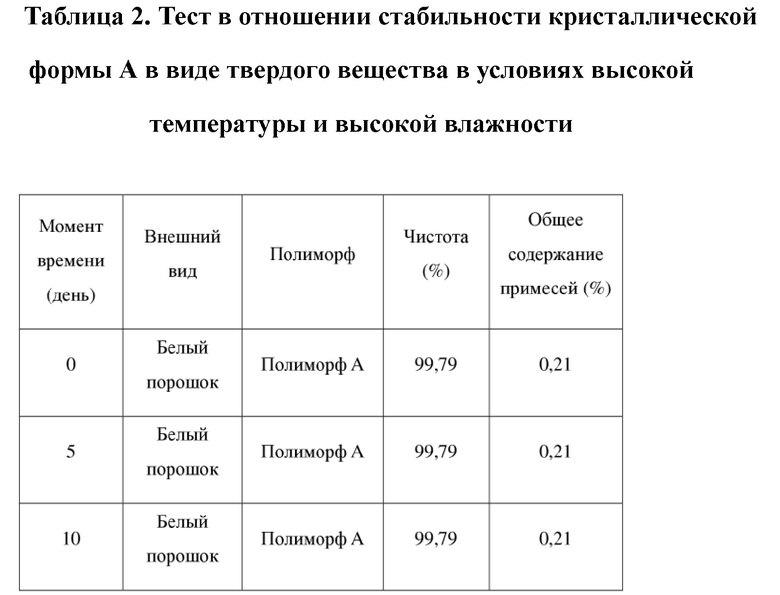
[68] 2 образца кристаллической формы А соединения формулы (I) параллельно взвешивали (вес каждого образца составлял приблизительно 100 мг), помещали на дно стеклянных бутылок для образцов и распределяли в виде тонкого слоя. Бутылки запечатывали алюминиевой фольгой и в алюминиевой фольге делали несколько небольших отверстий, чтобы образцы могли полностью контактировать с окружающим воздухом. Бутылки помещали в камеру с постоянной температурой и влажностью при 40oC/75% влажности. Образцы, помещенные в вышеуказанные условия, отбирали и тестировали в день 5 и день 10. Результаты теста сравнивали с исходными результатами теста в день 0. Результаты теста показаны в таблице 2 ниже.
[69] Экспериментальное заключение: кристаллическая форма А соединения формулы (I) в настоящем изобретении характеризуется хорошей стабильностью и ее легко составлять в лекарственный препарат.
[70] Пример 4. Тест в отношении физической стабильности кристаллической формы А соединения формулы (I) в виде твердого вещества при различных условиях температуры, влажности и освещения
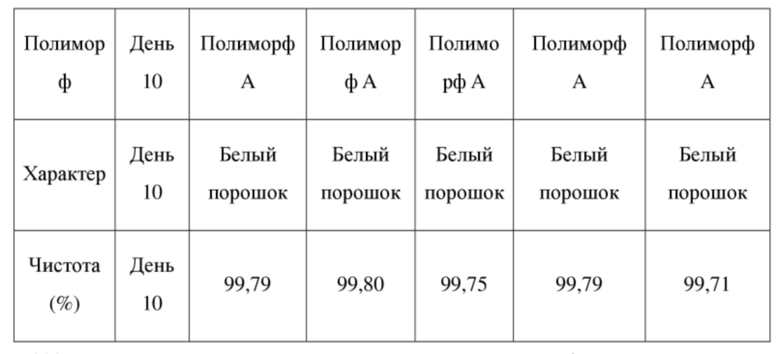
[71] 4 образца кристаллической формы А соединения формулы (I) параллельно взвешивали (вес каждого образца составлял приблизительно 100 мг), помещали на дно стеклянных бутылок для образцов и распределяли в виде тонкого слоя. Бутылки запечатывали алюминиевой фольгой и в алюминиевой фольге делали несколько небольших отверстий, чтобы образцы могли полностью контактировать с окружающим воздухом. Полученные 4 образца помещали в условия относительной влажности 25oC/92,5%, 60oC, 40oC/75% и освещенности соответственно для исследования физической стабильности образцов в день 10. Кроме того, приблизительно 100 мг кристаллической формы А соединения формулы (I) в виде твердого вещества отдельно взвешивали, помещали на дно стеклянной бутылки для образцов, закрывали завинчивающейся крышкой и хранили при -20oC для применения в качестве контрольного образца. В день 10 все образцы извлекали, возвращали в условия комнатной температуры и наблюдали изменения внешнего вида и кристаллические формы образцов выявляли посредством XRPD. Посредством сравнения образца в условиях ускоренного испытания с контрольным образцом определяли физическую стабильность кристаллической формы А соединения формулы (I) в виде твердого вещества. В таблице 3 ниже показаны экспериментальные результаты определения физической стабильности кристаллической формы А в виде твердого вещества.
[72] Таблица 3. Тест в отношении физической стабильности кристаллической формы А в виде твердого вещества в различных условиях температуры, влажности и освещения
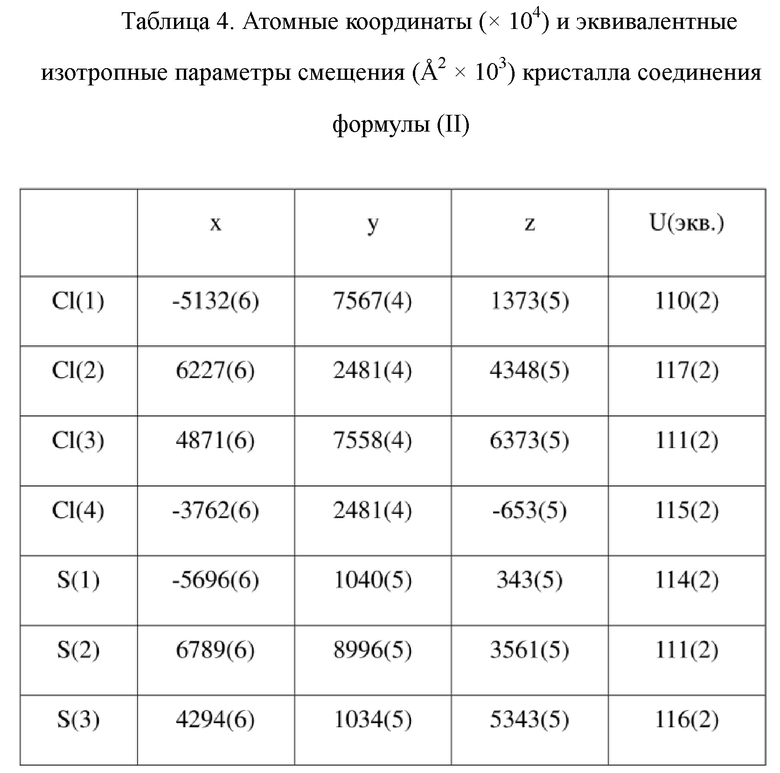
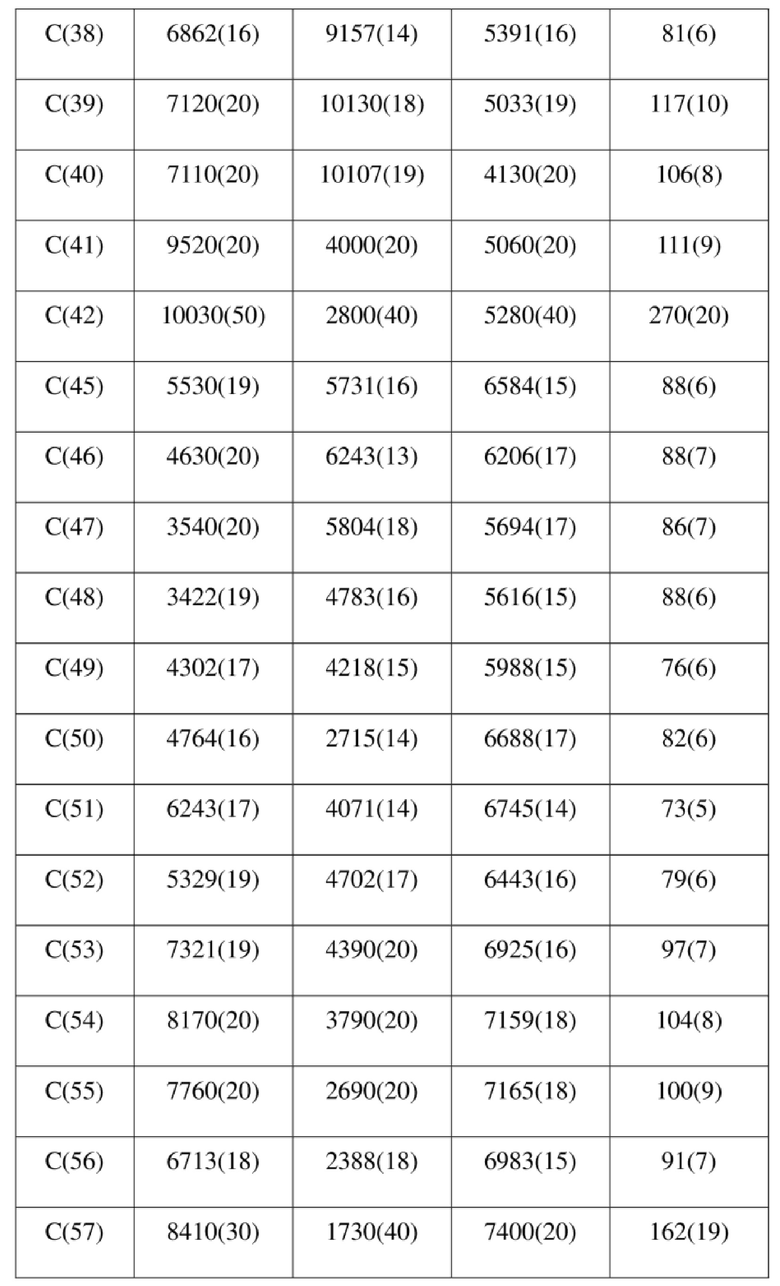
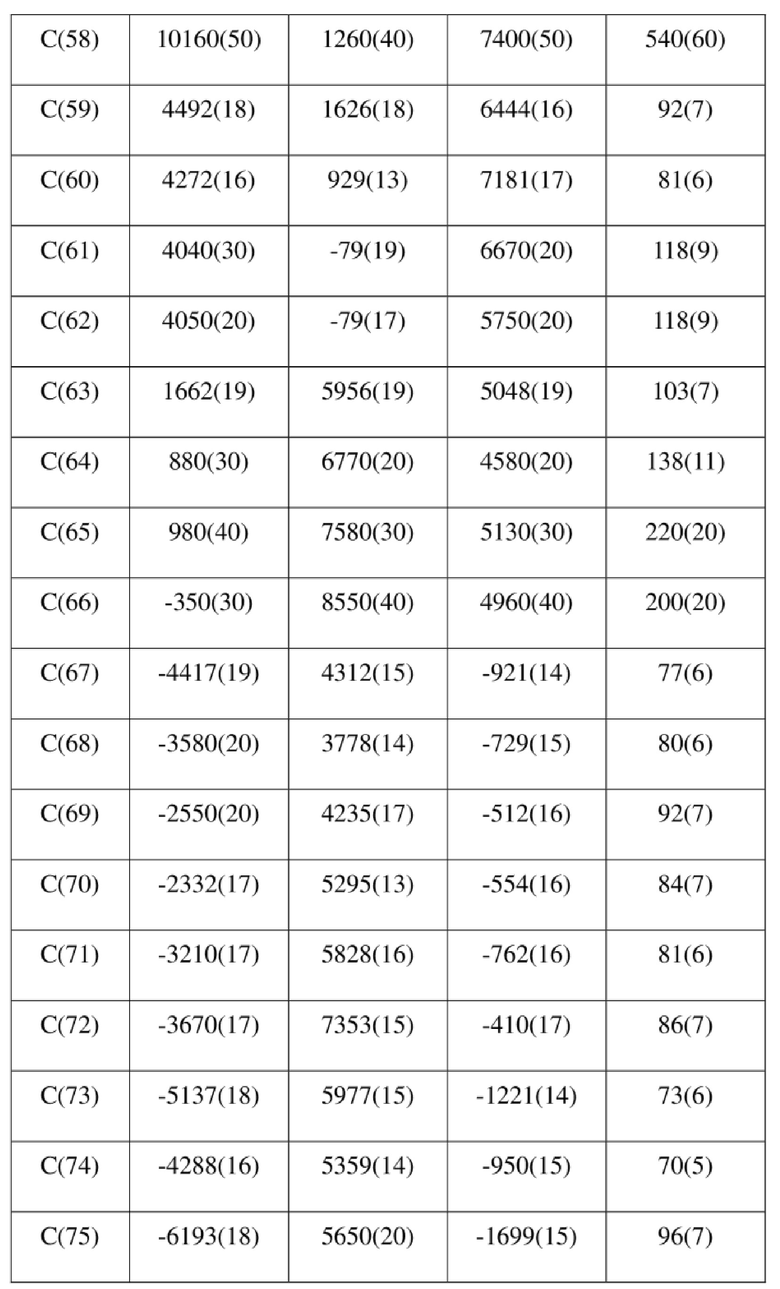
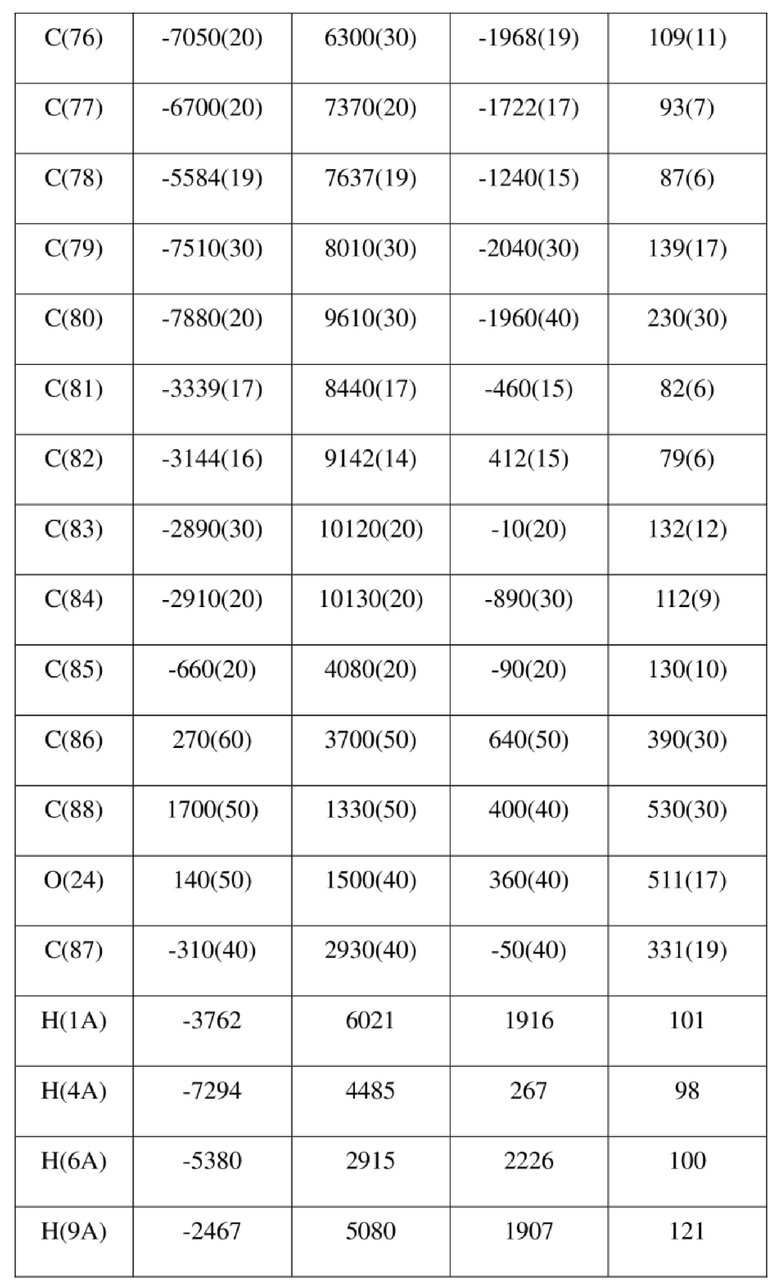
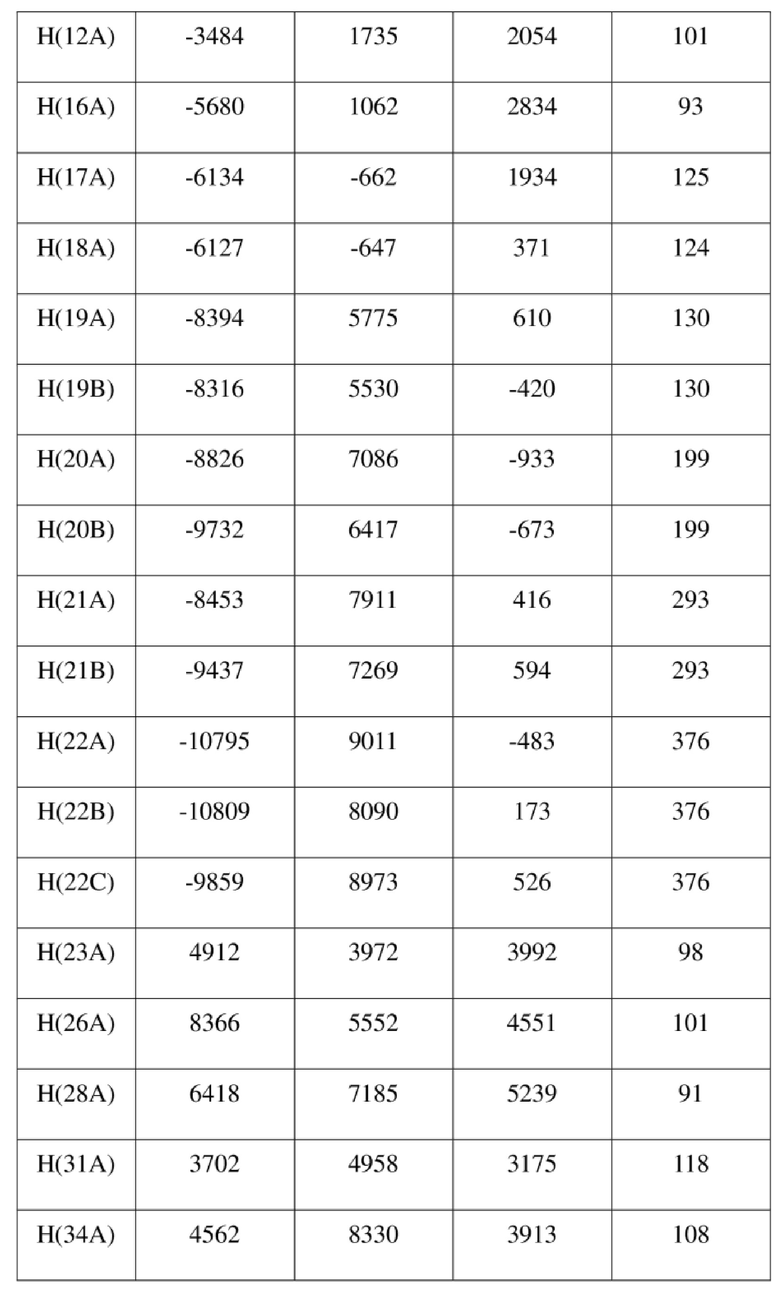
[73] Экспериментальное заключение: Кристаллическая форма А соединения формулы (I) в настоящем изобретении характеризуется хорошей стабильностью и ее легко составлять в лекарственный препарат.
[74] Пример 5. Рентгеноструктурный анализ монокристаллов
[75] Культивировать монокристалл соединения формулы (I) непросто, и для подтверждения его абсолютной конфигурации соединение формулы (I) метилируется с помощью триметилсилилдиазометана с получением соединения формулы (II) и затем культивируется монокристалл соединения формулы (II).
[76] Способ получения был следующим:
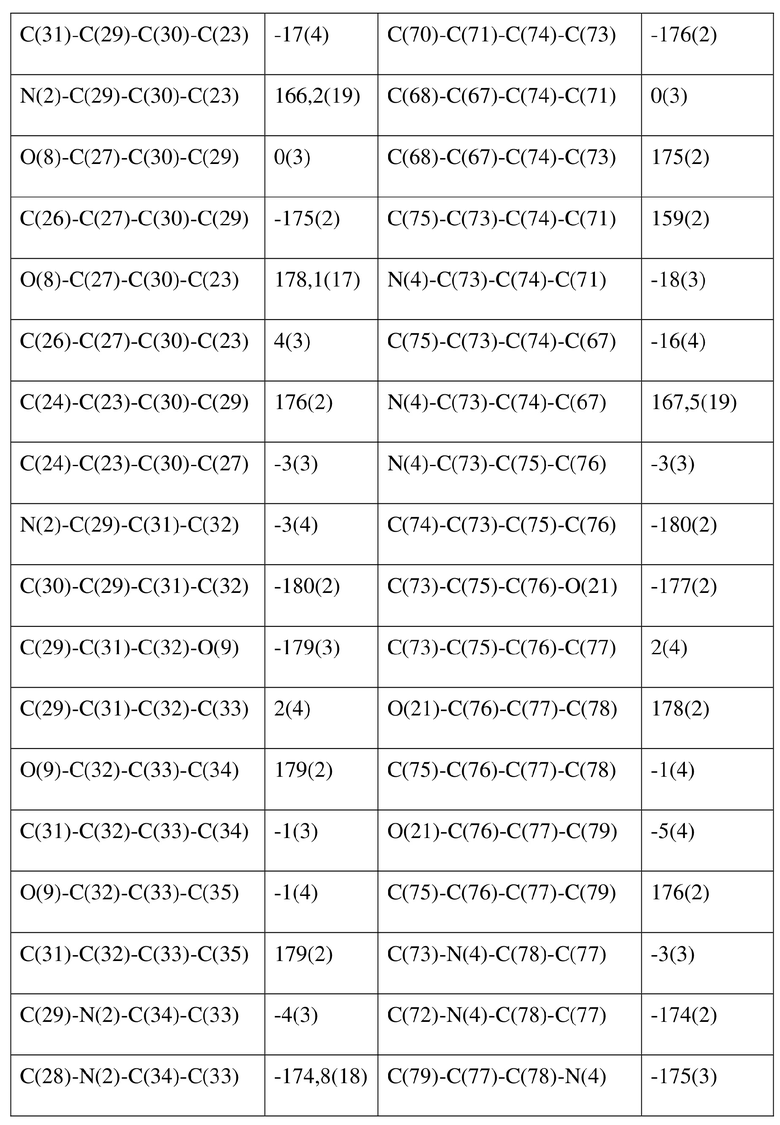
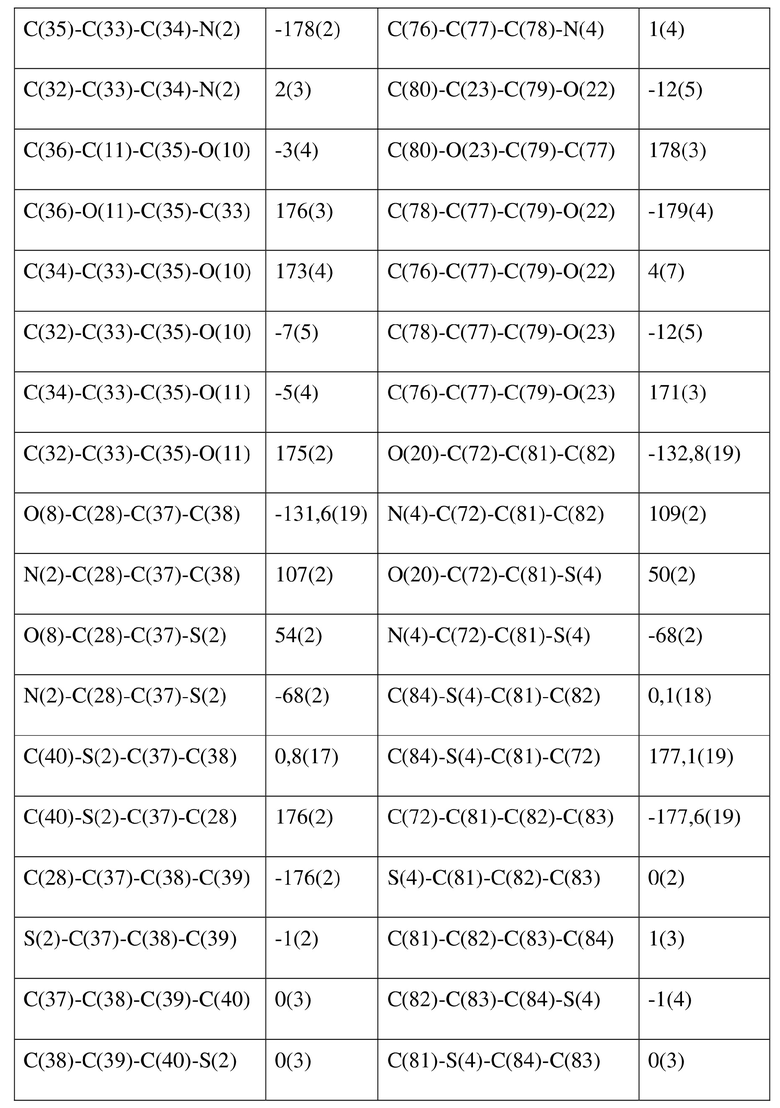
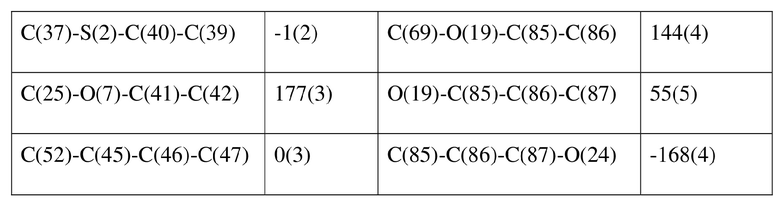
[77] соединение формулы (I) (5 г, 11,16 ммоль) добавляли к смешанному раствору метанола (15 мл) и тетрагидрофурана (45 мл) и добавляли одну порцию триметилсилилдиазометана (2 моль/л, 8,37 мл) при комнатной температуре. Затем полученный раствор перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор концентрировали и затем добавляли 20 мл трет-бутилметилового эфира и полученный раствор перемешивали, фильтровали и высушивали с получением соединения формулы (II). Соединение формулы (II) растворяли в метаноле и полученный раствор культивировали при комнатной температуре в течение 10 дней с применением способа выпаривания растворителя с получением монокристалла соединения формулы (II) с размером кристалла для дифракции, составляющим 0,06 × 0,08 × 0,16 мм. Кристалл относится к триклинной системе с пространственной группой P1, параметры кристаллической ячейки: a = 13,0669(13), b = 13,4585(12), c = 14,4699(14)Å, α = 90,348(4), β = 109,374(4), γ = 96,367(4), объем кристаллической ячейки V = 2383,3(4)Å3, количество асимметричных звеньев в кристаллической ячейке Z = 1.
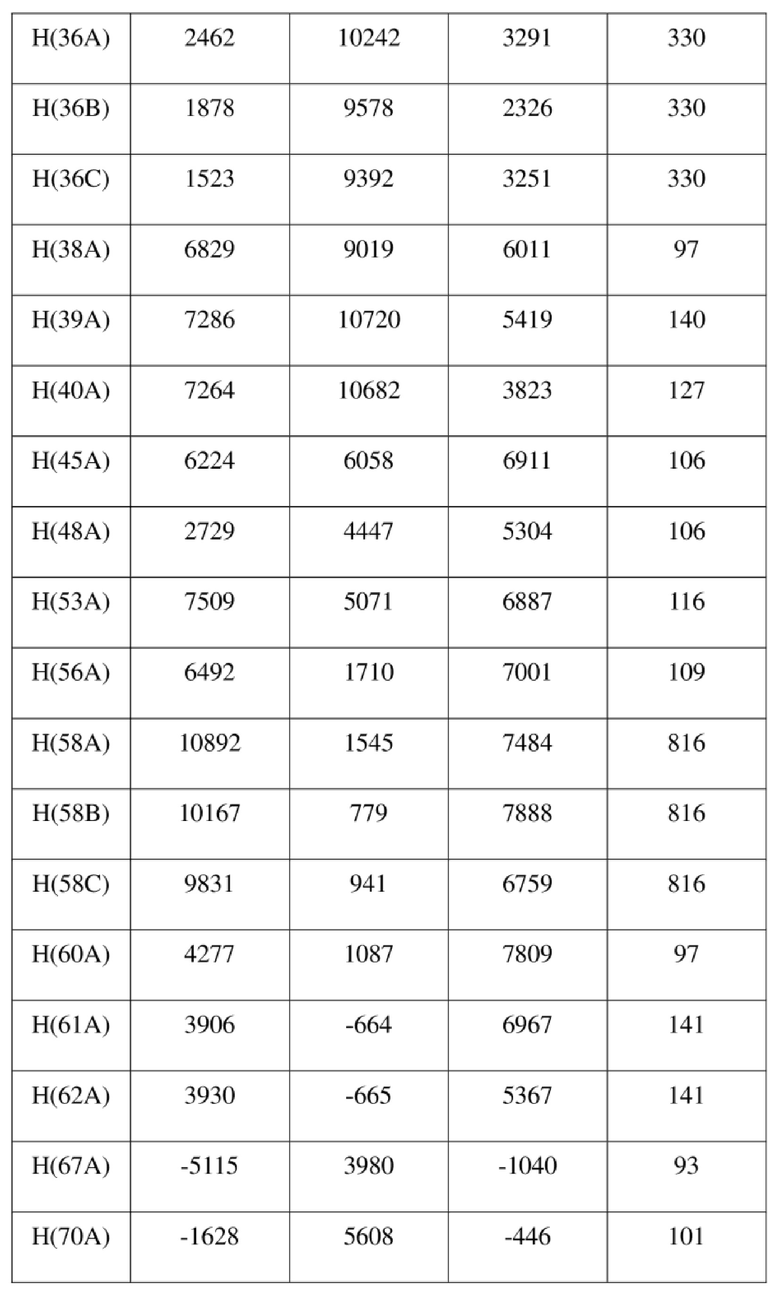
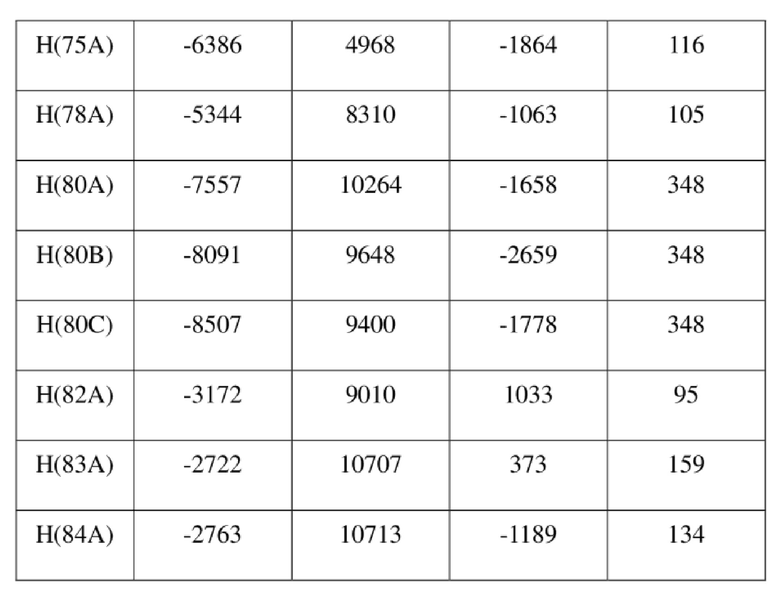
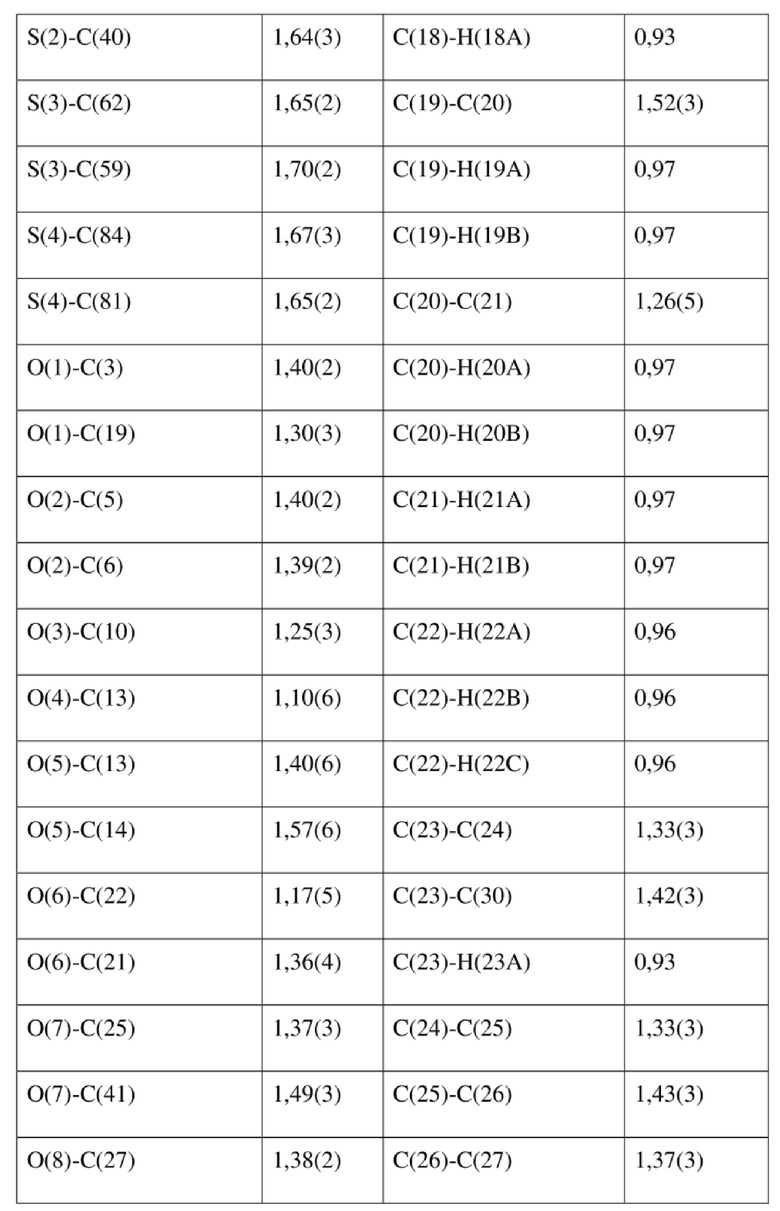
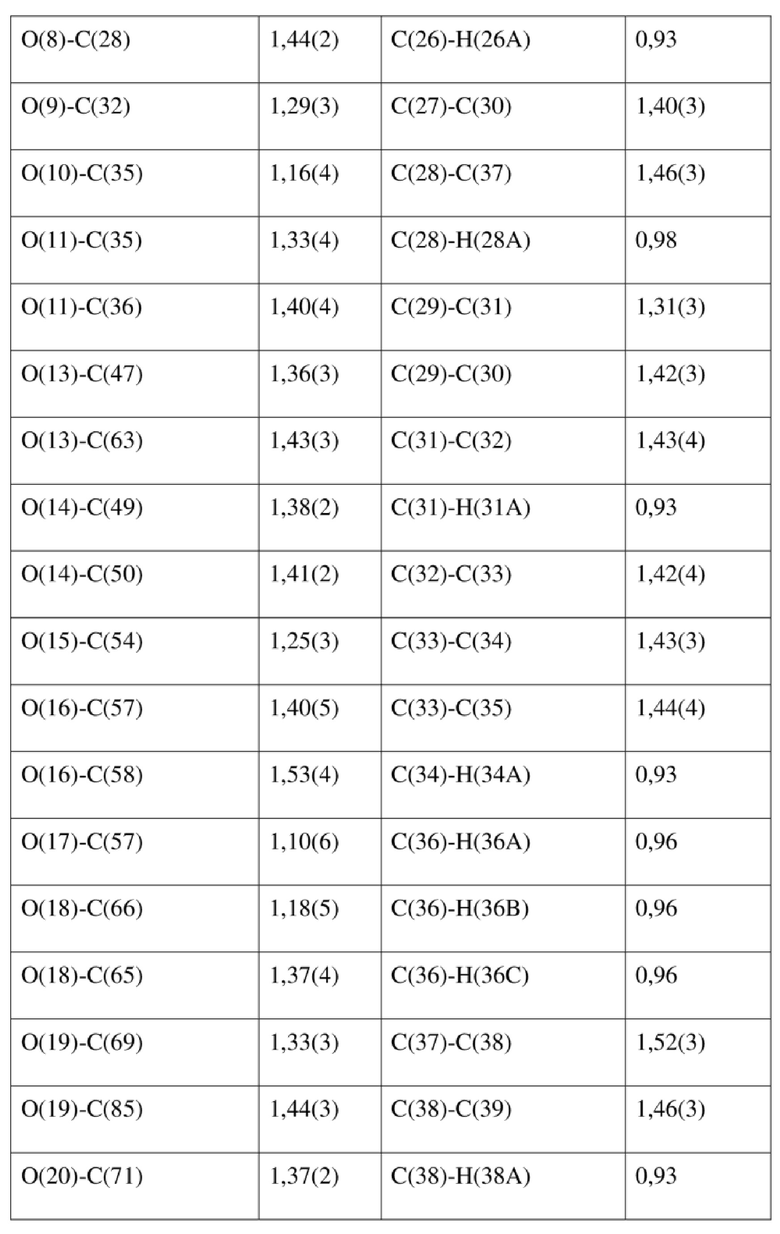
[78] Данные интенсивности дифракции собирали с помощью венчурного дифрактометра Bruker D8 с излучением CuKα в качестве источника света и сканированием φ/ω в качестве режима сканирования, где общее количество собранных точек дифракции составляло 40999, количество независимых точек дифракции составляло 15341, и количество наблюдаемых точек (I/сигма ≥ 2) составляло 5471.
[79] Кристаллическую структуру анализировали посредством прямого способа (Shelxs97) с определением 124 позиций атомов, отличных от атомов водорода. Использовали метод наименьших квадратов для корректировки структурных параметров и идентификации атомных разновидностей. Метод геометрического расчета и разностный метод Фурье использовали для определения всех положений атомов водорода. После уточнения R1 = 0,1357, wR2 = 0,4252 (w = 1/α|F|2), S = 1,817. Окончательная стехиометрическая формула представляла собой 4(C22H20ClNO6S); расчетная молекулярная масса одной молекулы составляла 461,90, и расчетная плотность кристаллов составляла 1,287 г/см3.
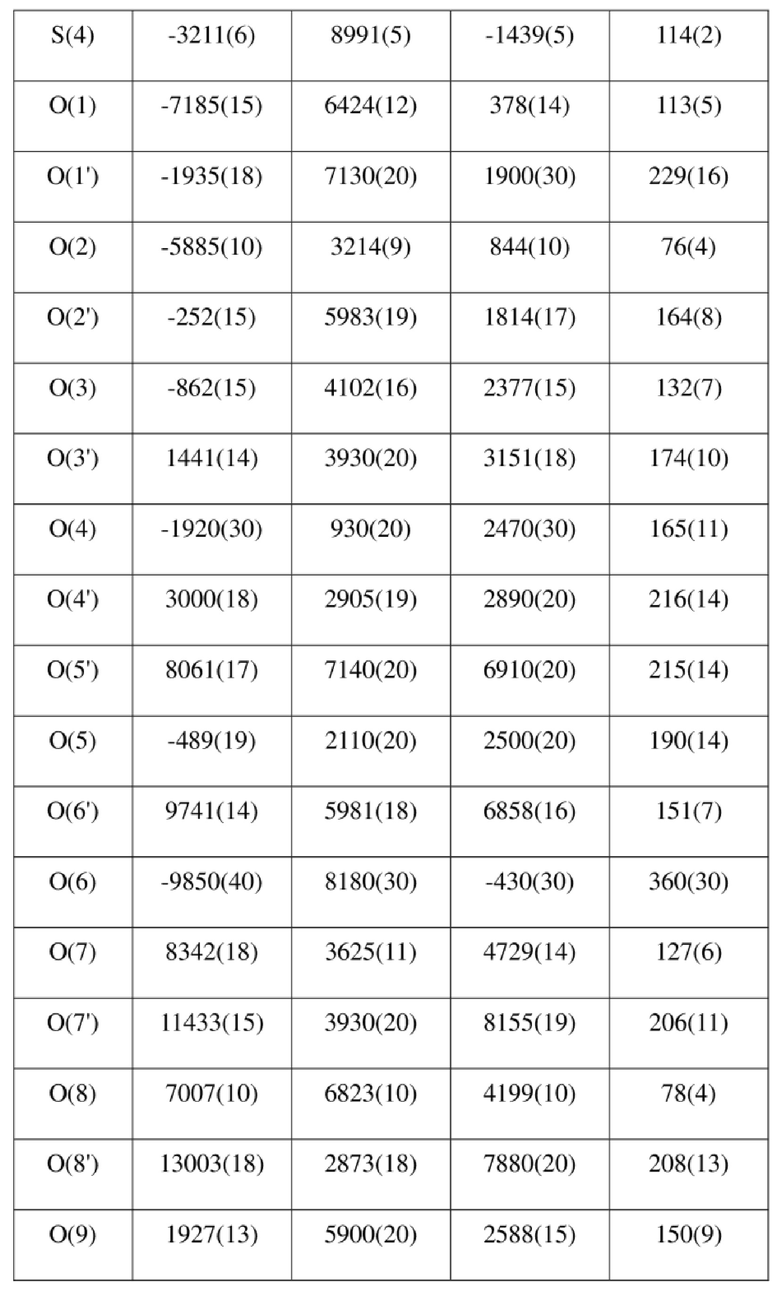
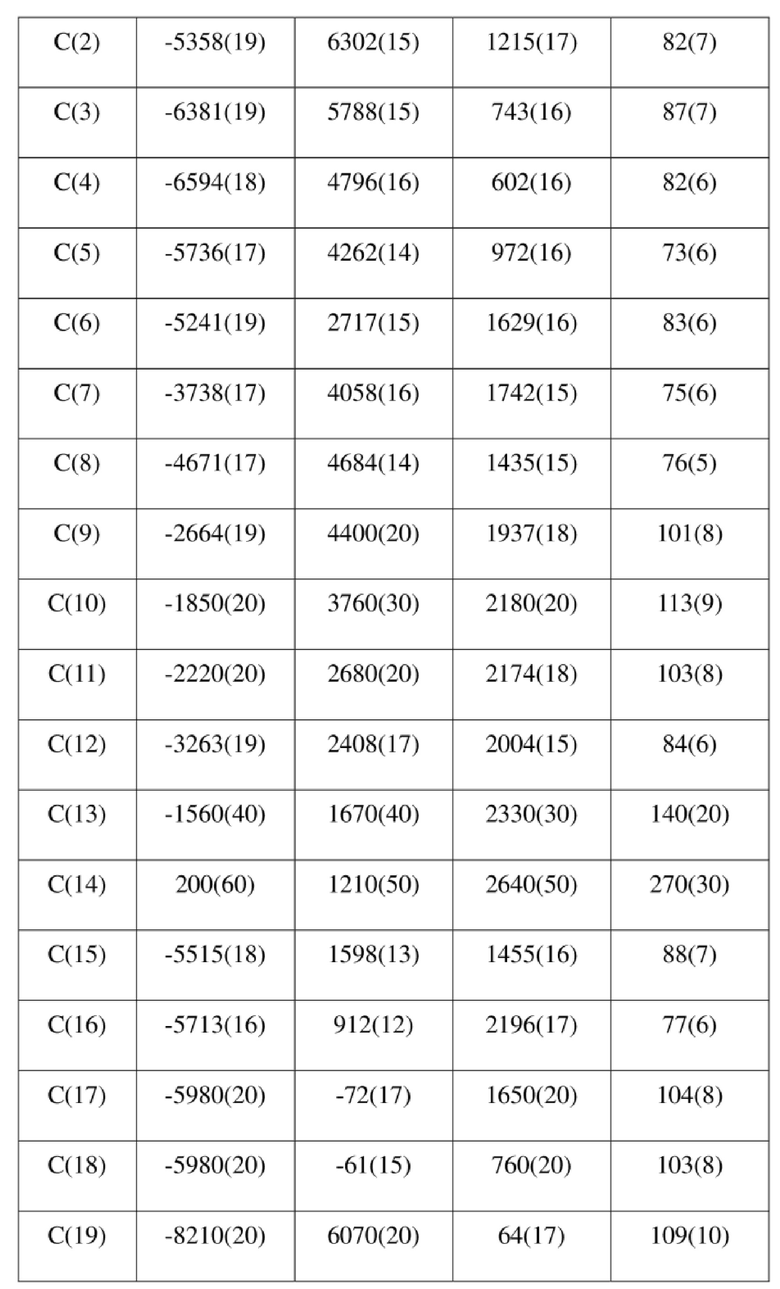
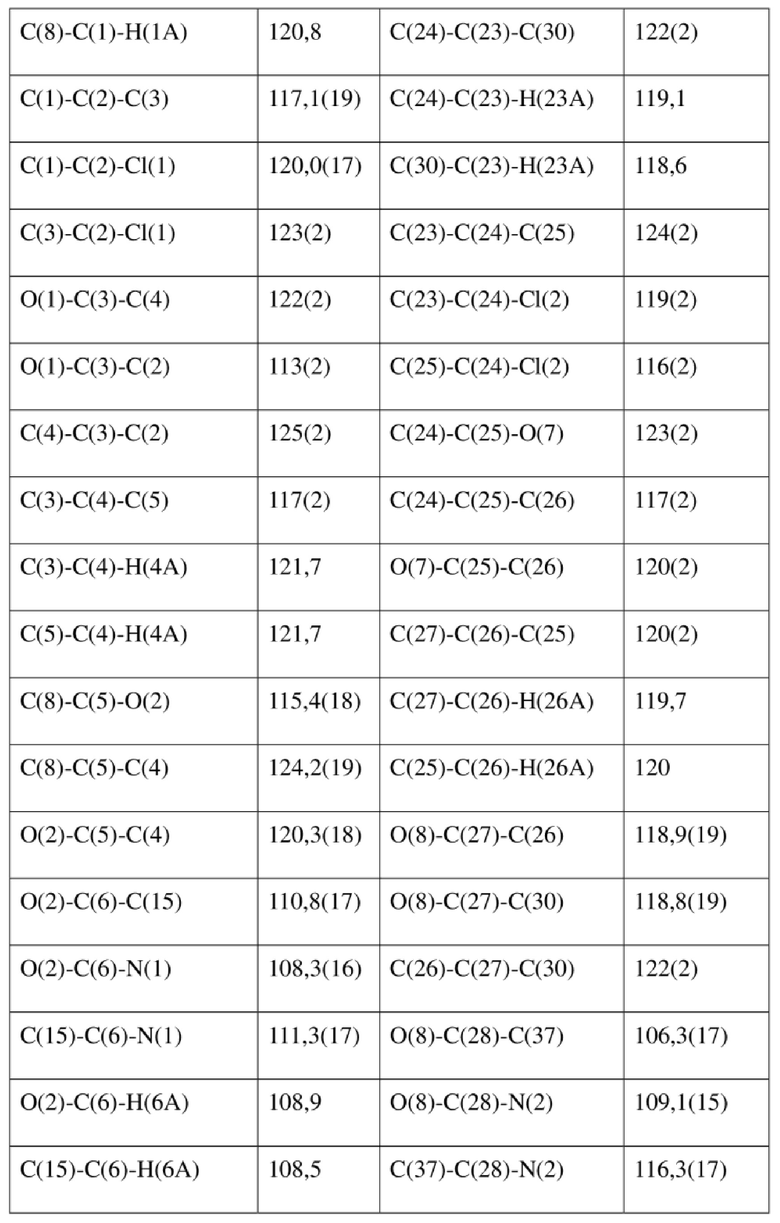
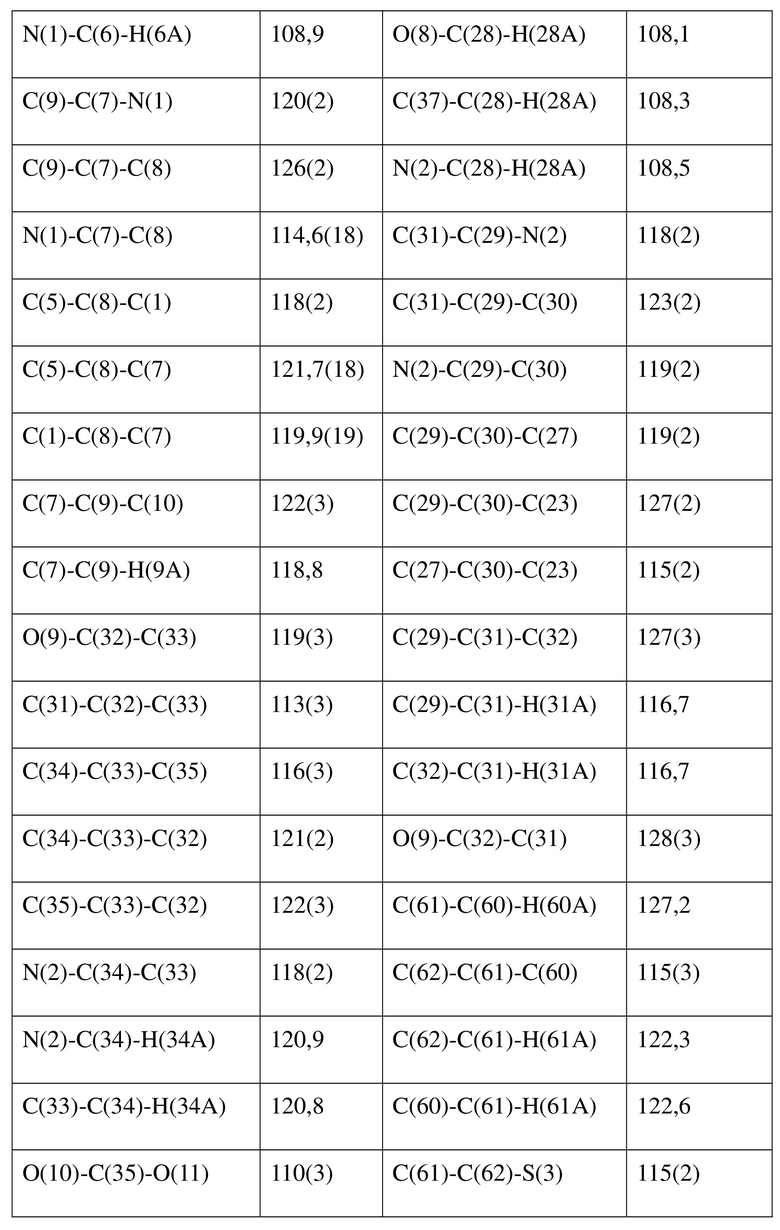
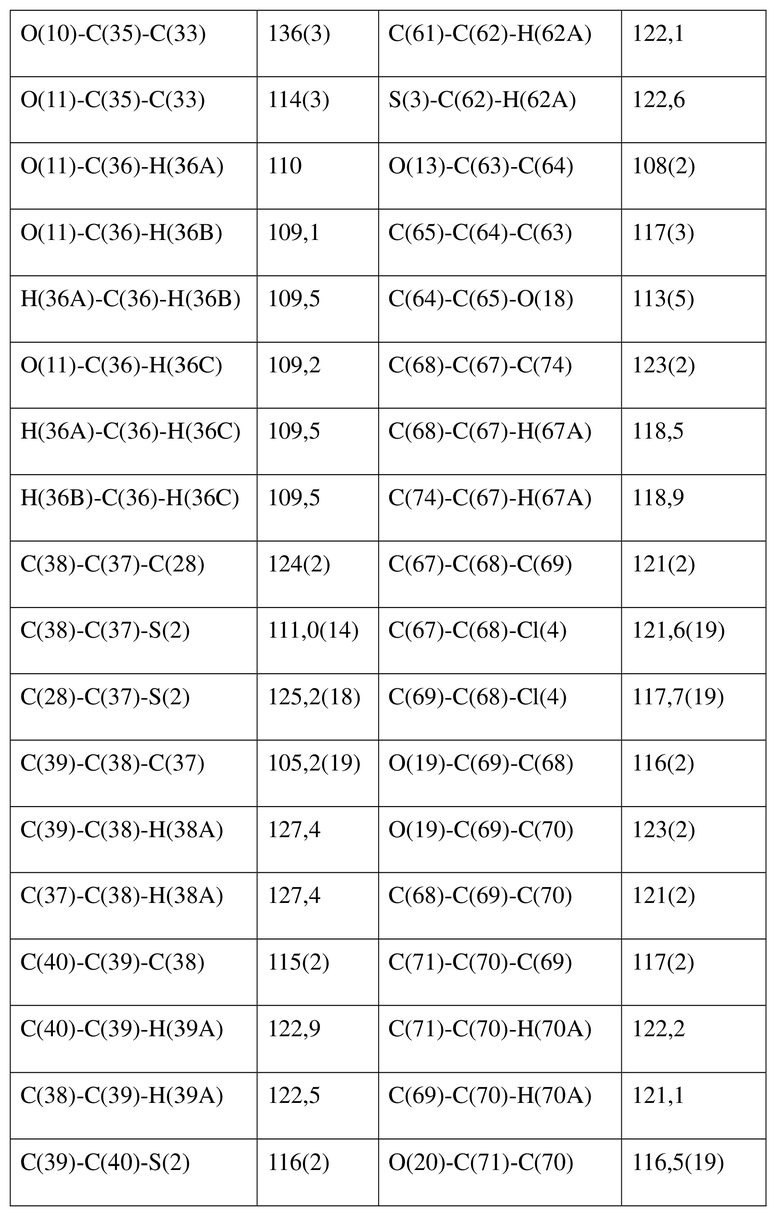
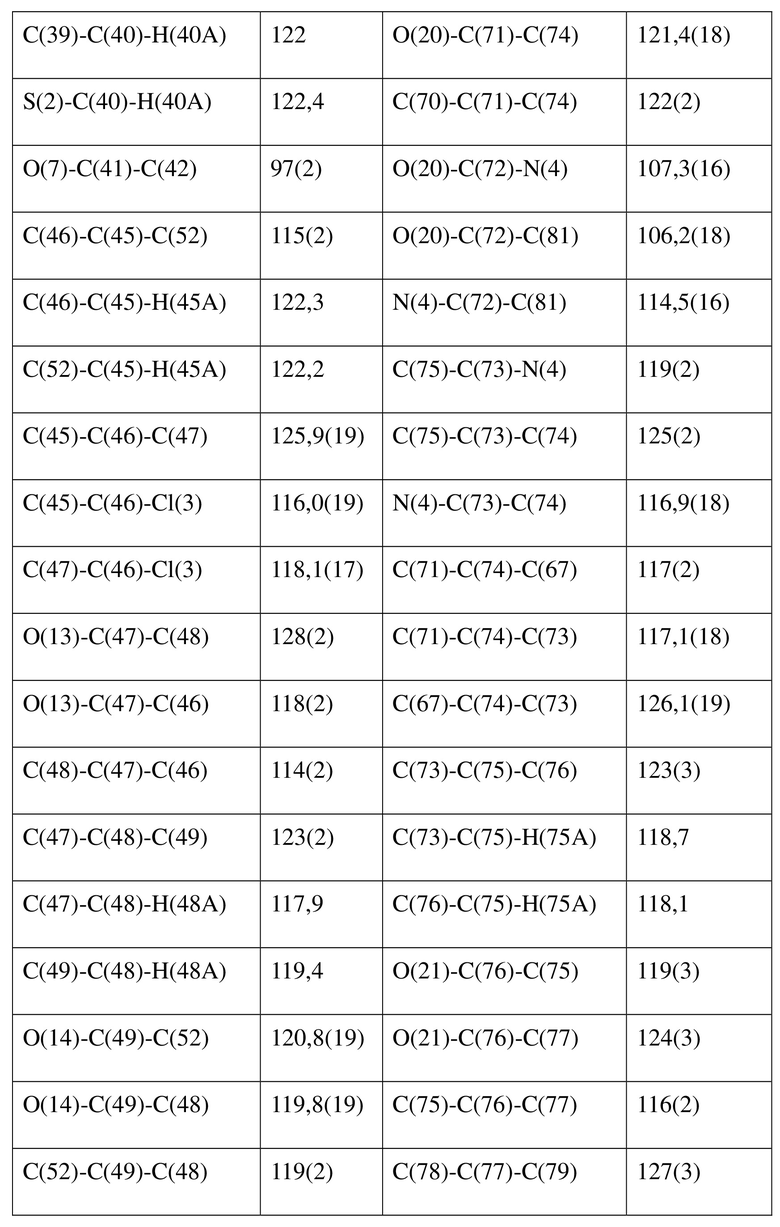
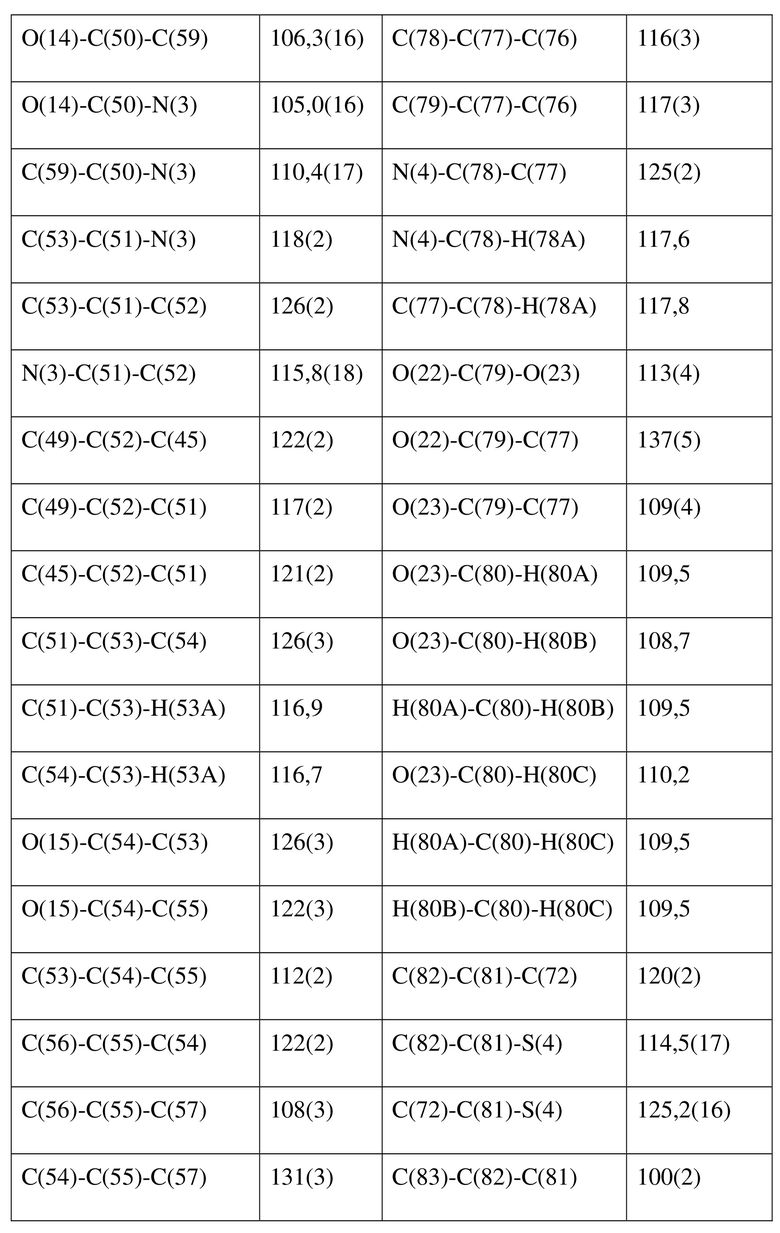
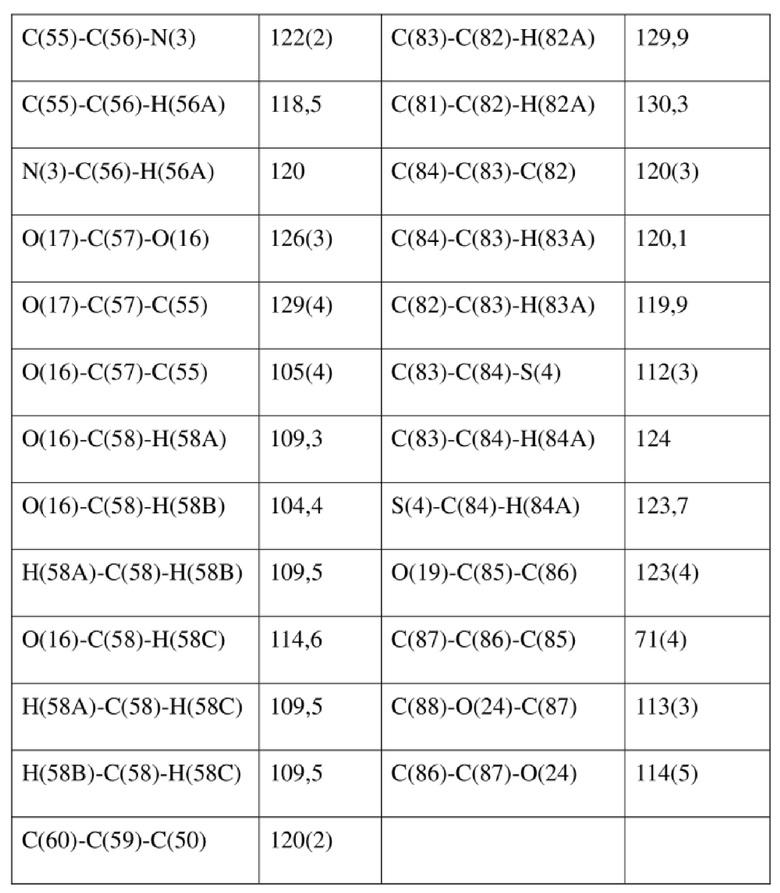
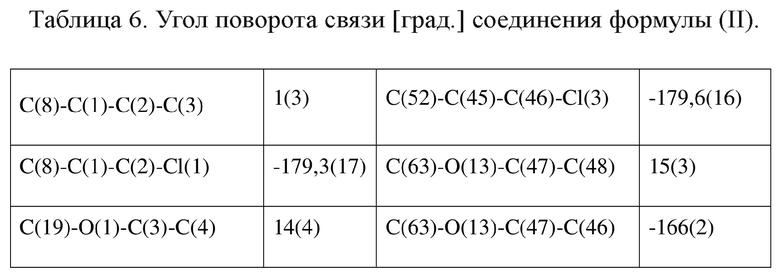
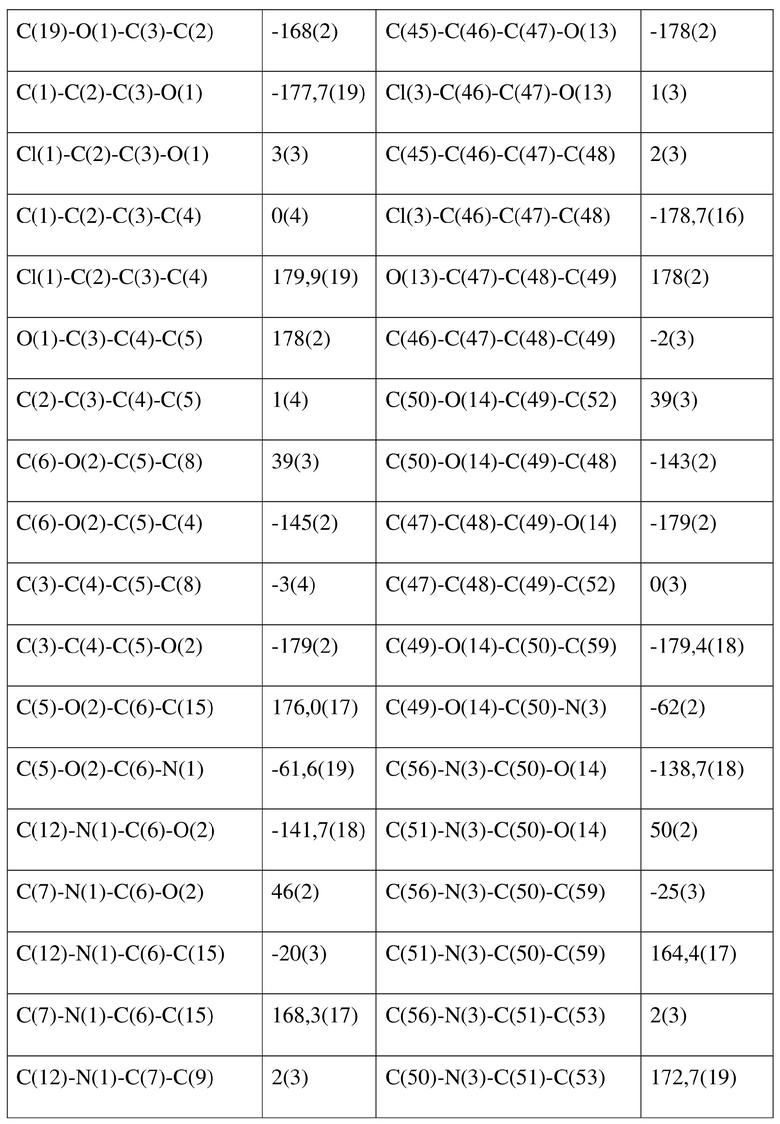
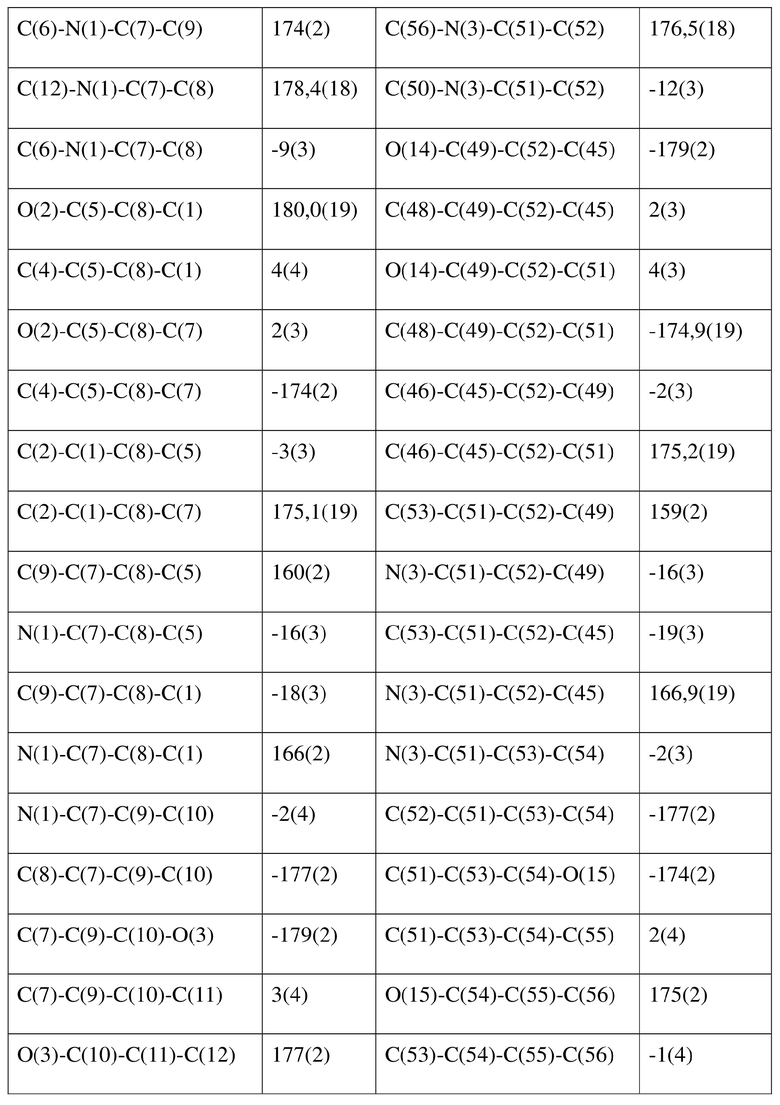
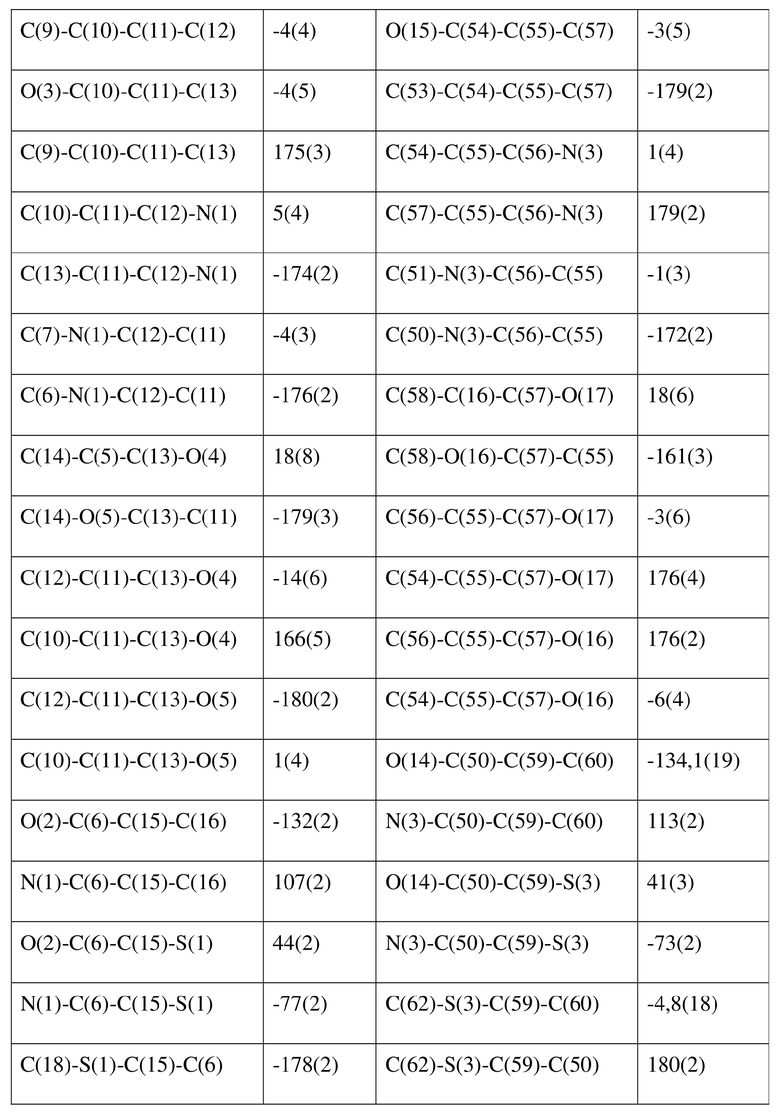
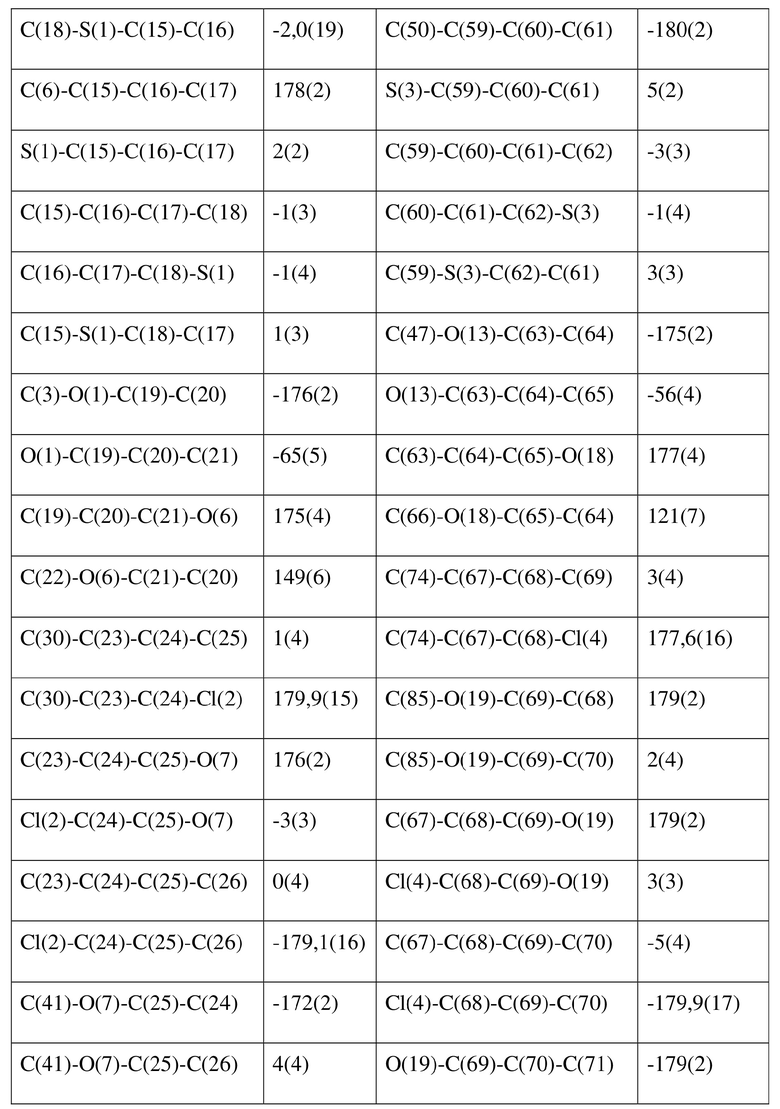
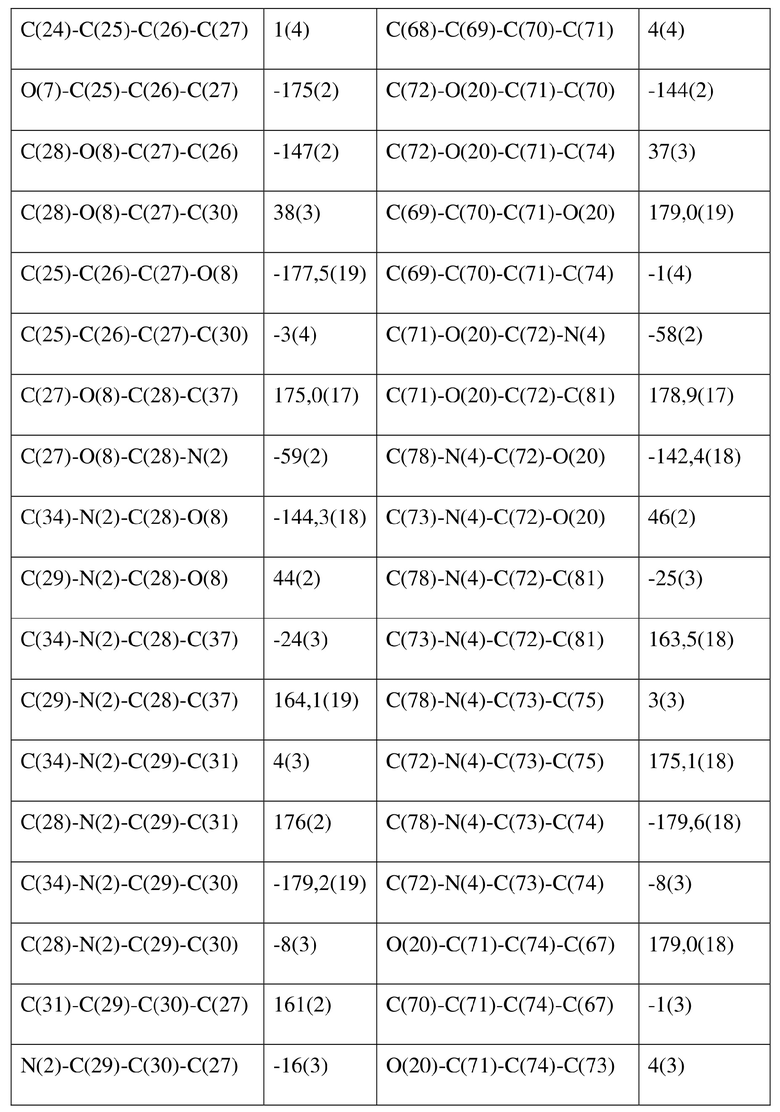
[80] Реакцию проводили в нейтральной среде в мягких условиях без инверсии при хиральном атоме углерода. Таким образом, абсолютная конфигурация соединения формулы (I) соответствовала абсолютной конфигурации соединения формулы (II). Из данных, полученных в случае монокристалла соединения формулы (II), можно определить абсолютную конфигурацию соединения формулы (I). Эллипсоидная диаграмма мономолекулярной стереохимической структуры соединения формулы (II) показана на фигуре 4. См. таблицу 4 для атомных координат (× 104) и эквивалентных изотропных параметров смещения (Å2 × 103) кристалла соединения формулы (II). См. таблицу 5 для длины связи (Å) и угла связи [град.] соединения формулы (II). См. таблицу 6 для угла поворота связи [град.] соединения формулы (II).

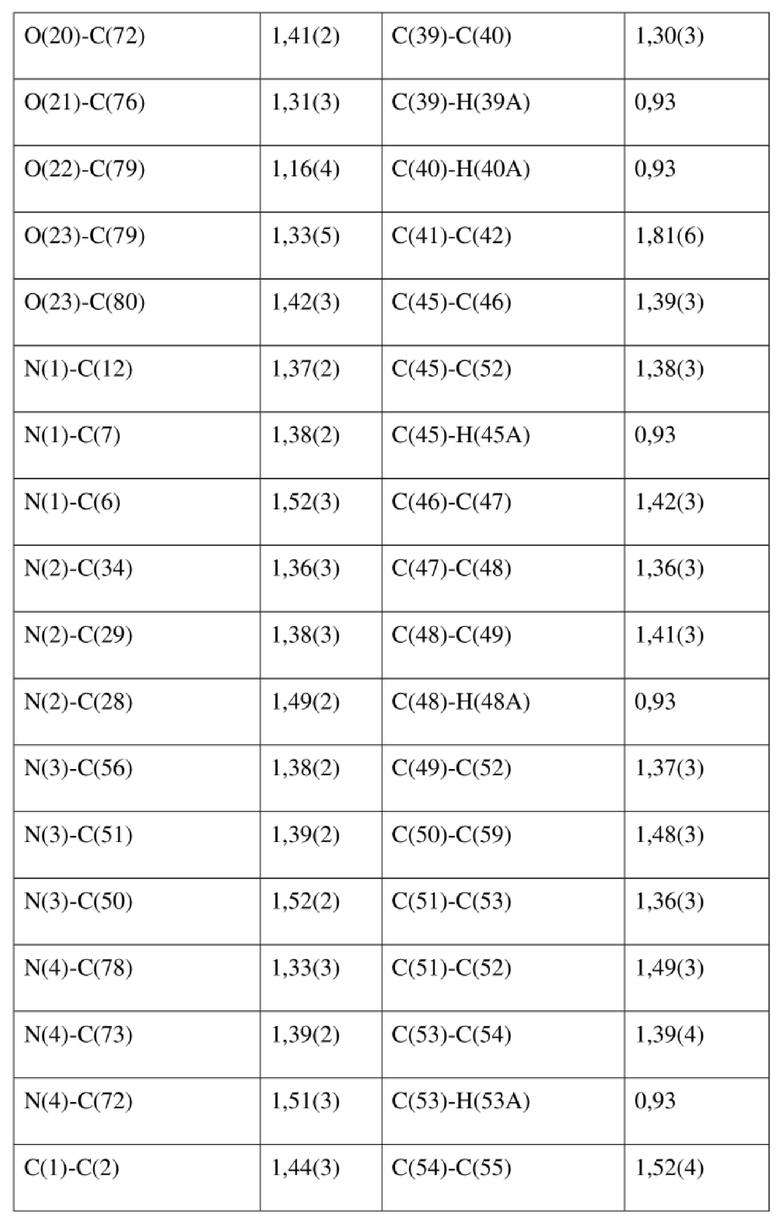
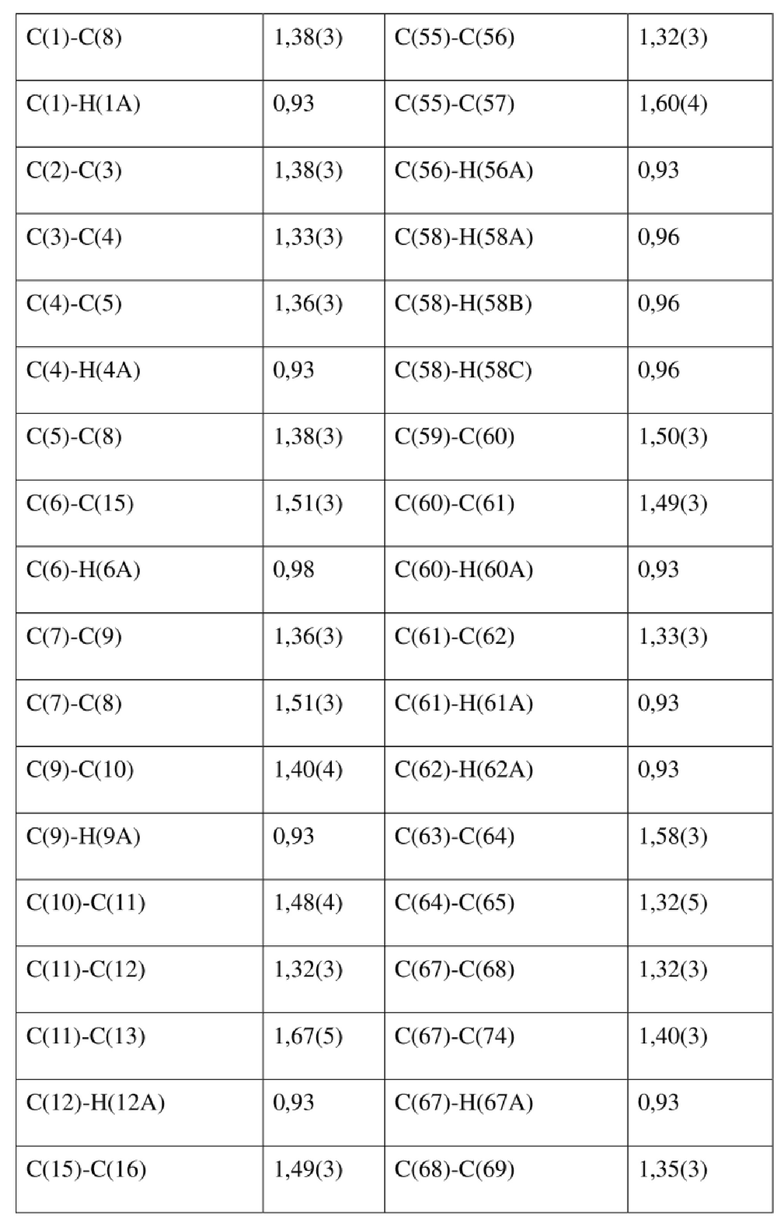
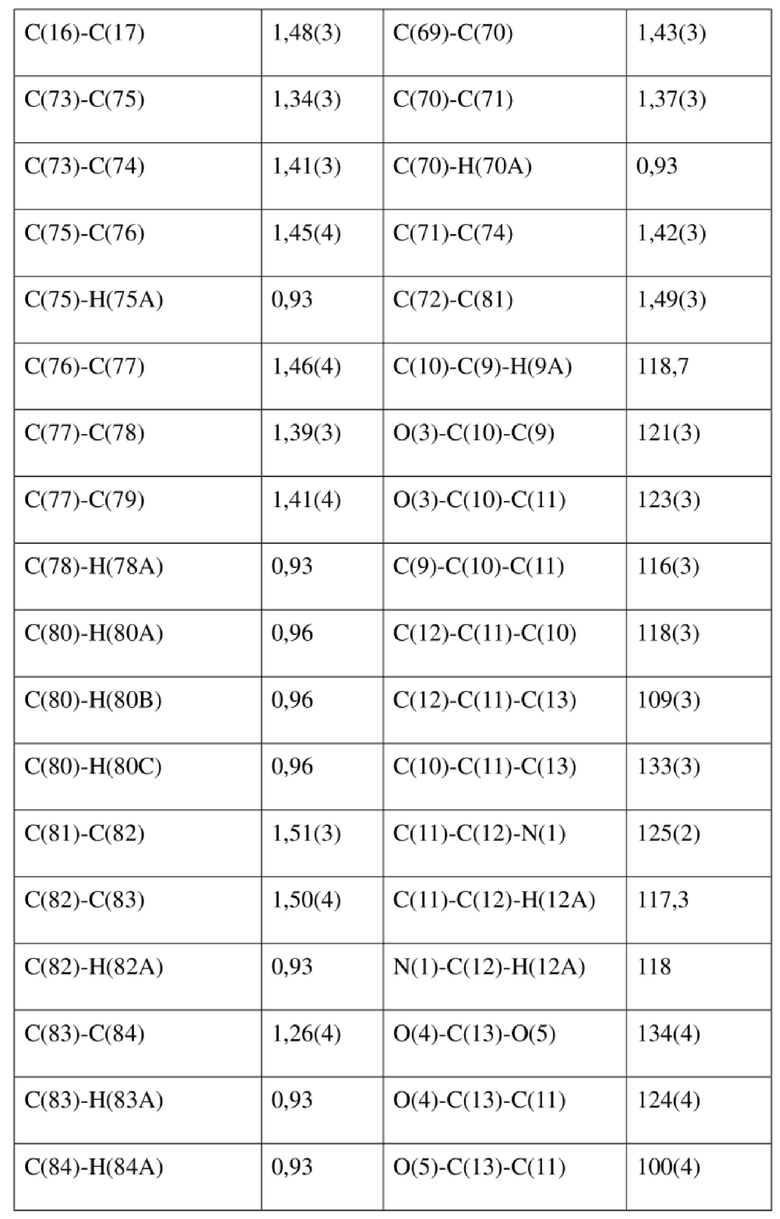
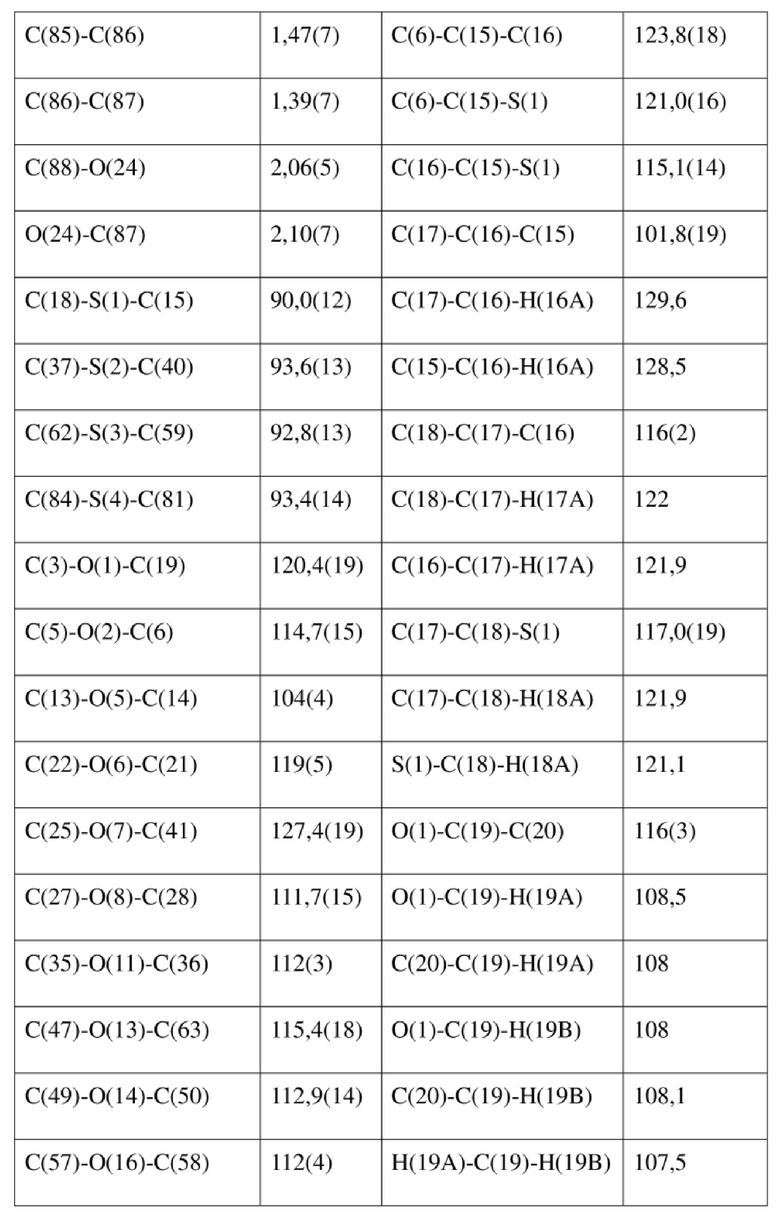

[81] Экспериментальный пример 1: тест соединения формулы (I) в отношении активности HBV in vitro
[82] Материалы эксперимента.
[83] 1. Линии клеток: клетки HepG2.2.15
[84] Среда для культивирования клеток HepG2.2.15, DMEM/F12, Invitrogen-11330032; 10% сыворотка крови, Invitrogen-10099141; 100 единиц/мл пенициллина и 100 мкг/мл стрептомицина, Hyclone-SV30010; 1% заменимых аминокислот, Invitrogen-11140050; 2 мМ L-ГЛУТАМИНА, Invitrogen-25030081; 300 мкг/мл генетицина, Invitrogen-10131027
[85] 2. Реагенты:
[86] панкреатин (Invitrogen-25300062);
[87] DPBS (Corning-21031CVR);
[88] диметилсульфоксид (Sigma-D2650-100ML)
[89] набор для высокоэффективной очистки ДНК (QIAamp 96 DNA Blood Kit, Qiagen-51162)
[90] универсальный реагент faststart на основе зонда для количественного определения (FastStart Universal Probe Master, Roche-04914058001)
[91] набор для количественного выявления поверхностного антигена вируса гепатита B (Autobio, CL 0310)
[92] 3. Расходные материалы и инструменты:
[93] 96-луночный планшет для культивирования клеток (Corning-3599)
[94] CO2-инкубатор (HERA-CELL-240)
[95] оптически прозрачная герметизирующая пленка (ABI-4311971)
[96] 96-луночный планшет для количественной ПЦР (Applied Biosystems-4306737)
[97] прибор для количественной ПЦР с флуоресцентным выявлением (Applied Biosystems-7500 real time PCR system)
[98] Способ проведения эксперимента:
[99] 1. Клетки HepG2.2.15 (4×104 клеток/лунка) высевали в 96-луночный планшет и инкубировали в течение ночи при 37oC, 5% CO2.
[100] 2. В день 2 соединение разбавляли до в общей сложности 8 концентраций с 3-кратным градиентным разбавлением. Соединения при различных концентрациях добавляли в лунки с культурой в двух повторностях. Конечная концентрация диметилсульфоксида в культуральной среде составляла 0,5%. 10 мкМ ETV использовали в качестве контроля со 100% подавлением; 0,5% диметилсульфоксид использовали в качестве контроля с 0% подавлением.
[101] 3. В день 5 культуральную среду заменяли свежей культуральной средой, содержащей соединение.
[102] 4. В день 8 культуральные среды в лунках с культурой собирали и часть образцов отбирали для измерения содержания антигена S вируса гепатита В посредством ELISA; часть образцов отбирали для выделения ДНК с применением набора для высокоэффективной очистки ДНК (Qiagen-51162).
[103] 5. Получение реакционного раствора для ПЦР показано в таблице 7.

[104] Последовательность прямого праймера: GTGTCTGCGGCGTTTTATCA
[105] Последовательность обратного праймера: GACAAACGGGCAACATACCTT
[106] Последовательность зонда: 5'+ FAM + CCTCTKCATCCTGCTGCTATGCCTCATC + TAMRA -3'
[107] 6.1. 15 мкл реакционного смешанного раствора добавляли в каждую лунку 96-луночного планшета для ПЦР и затем в каждую лунку добавляли 10 мкл образца ДНК или стандартов, представляющих собой ДНК HBV.
[108] 6.2. Условия реакции ПЦР включают нагревание при 95oC в течение 10 минут, последующую денатурацию при 95oC в течение 15 секунд и удлинение при 60oC в течение 1 минуты, всего 40 циклов.
[109] 6.3. Определение содержания антигена S вируса гепатита В посредством ELISA включает соответственно добавление 50 мкл образцов и стандартов в реакционный планшет и затем добавление 50 мкл конъюгата фермента в каждую лунку; встряхивание полученного раствора для перемешивания до однородного состояния и инкубацию при 37oC в течение 60 минут; промывание планшета промывочным раствором 5 раз, добавление в каждую лунку 50 мкл люминесцентного субстрата, перемешивание до однородного состояния и осуществление реакции в полученном растворе в темноте при комнатной температуре в течение 10 минут, и, наконец, выявление интенсивности хемилюминесценции с помощью микропланшет-ридера.
[110] 6.4. Анализ данных.
[111] Расчет процента подавления: % подавл. = (1-значение для образца/значение для контроля с диметилсульфоксидом) × 100.
[112] Расчет EC50: Значение 50% ингибирующей концентрации (EC50) соединения в отношении HBV рассчитывали с применением программного обеспечения GraphPad Prism.
[113] Экспериментальные результаты показаны в таблице 8.
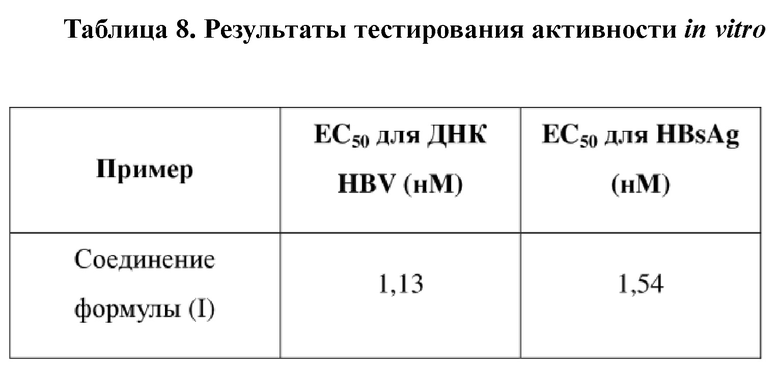
[114] Экспериментальное заключение: соединение формулы (I) способно эффективно подавлять ДНК HBV и поверхностный антиген вируса гепатита B (HBsAg).
Изобретение относится к способу получения кристаллической формы А соединения формулы (I), которая характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,30±0,20°, 9,30±0,20°, 9,84±0,20°, 18,68±0,20°, 20,16±0,20°, 23,06±0,20°, 24,00±0,20° и 25,38±0,20°. Способ включает добавление соединения формулы (I) в любой форме к метанолу, ацетону, этилацетату, ацетонитрилу, смеси ацетонитрил-вода при соотношении 1:1 или смеси ацетон-вода при соотношении 1:1, перемешивание смеси в течение от 1 до 72 ч при температуре от 25 до 65°C. Затем охлаждают смесь до комнатной температуры, фильтруют и высушивают осадок на фильтре с получением кристаллической формы А. Весовое соотношение соединения формулы (I) и растворителя выбрано из 1:1-30. Технический результат – получение кристаллической формы А соединения формулы (I), характеризующейся хорошей физической стабильностью и химической стабильностью. 8 з.п. ф-лы, 4 ил., 8 табл., 6 пр.
1. Способ получения кристаллической формы А соединения формулы (I)
 ,
,
где способ включает добавление соединения формулы (I) в любой форме к метанолу, ацетону, этилацетату, ацетонитрилу, смеси ацетонитрил-вода при соотношении 1:1 или смеси ацетон-вода при соотношении 1:1, перемешивание смеси в течение от 1 до 72 ч при температуре от 25 до 65°C, затем охлаждение до комнатной температуры, фильтрование и высушивание осадка на фильтре с получением кристаллической формы А;
где кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,30 ± 0,20°, 9,30 ± 0,20°, 9,84 ± 0,20°, 18,68 ± 0,20°, 20,16 ± 0,20°, 23,06 ± 0,20°, 24,00 ± 0,20° и 25,38 ± 0,20°.
2. Способ получения по п. 1, где кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,30 ± 0,20°, 9,30 ± 0,20°, 9,84 ± 0,20°, 12,84 ± 0,20°, 18,68 ± 0,20°, 20,16 ± 0,20°, 21,26 ± 0,20°, 23,06 ± 0,20°, 24,00 ± 0,20° и 25,38 ± 0,20°.
3. Способ получения по п. 2, где кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 6,302°, 7,883°, 9,301°, 9,842°, 12,838°, 15,436°, 16,580°, 18,124°, 18,680°, 19,459°, 20,161°, 20,800°, 21,262°, 21,704°, 23,057°, 24,000°, 24,837°, 25,382°, 26,244°, 26,558°, 27,740°, 28,119°, 28,827°, 29,502°, 29,880°, 30,261°, 30,762°, 31,678°, 32,595°, 33,061°, 34,347°, 35,235°, 35,738°, 36,642°, 38,619° и 39,558°.
4. Способ получения по п. 3, где кристаллическая форма характеризуется дифрактограммой XRPD, показанной на фиг. 1.
5. Способ получения по любому из пп. 1-4, где кристаллическая форма характеризуется профилем дифференциальной сканирующей калориметрии, содержащим эндотермический пик при 224,58 ± 3°C.
6. Способ получения по п. 5, где кристаллическая форма характеризуется термограммой DSC, показанной на фиг. 2.
7. Способ получения по любому из пп. 1-4, где кристаллическая форма характеризуется кривой термогравиметрического анализа, демонстрирующей потерю веса 0,127% при 200,00 ± 3°C и потерю веса 0,224% при 250 ± 3°C.
8. Способ получения по п. 1, где кристаллическая форма характеризуется термограммой TGA, показанной на фиг. 3.
9. Способ получения по п. 1, где весовое соотношение соединения формулы (I) и растворителя выбрано из 1:1-30.
| WO 2018161960 A1, 13.09.2018 | |||
| MINO R.CAIRA, Crystalline Polymorphism of Organic Compounds, 1998, p.163-208 | |||
| Sherry L.Morissette et al.: "High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids", ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, pp.275-300 | |||
| WO 2019169539 A1, 12.09.2019 | |||
| WO 2018214875 A1, |

