Настоящее изобретение относится к области органической химии и фармакологии, к производным пиперидин-4,4-дикарбоновых кислот, а именно способу получения эфиров пиперидин-4,4-дикарбоновых кислот общей формулы

R1, R2=Н, C1-C4-алкил, бензил, арил, либо CH2CH2CO2Alk; R3=Н, С1-С4-алкил, R4=С1-С4-алкил, или их солей. Соединения общей формулы I используются или могут быть использованы как ключевые полупродукты в синтезе фармакологически активных субстанций для лечения таких заболеваний как рак, рассеянный склероз, СПИД, сердечно-сосудистые заболевания и др.
Пиперидиновый цикл составляет основу многих лекарственных препаратов и природных соединений (P. Goel et al, "Recent advancement of piperidine moiety in treatment of cancer- A review", Eur. J. Med. Chem., 2018, 157, 480). TV-незамещенные пиперидины, ввиду наличия реакционноспособной вторичной амино-группы, активно используются как полупродукты в синтезе фармакологически активных соединений (S. Kallstrom et al, "Synthesis of pharmaceutical" active compounds containing a disubstituted piperidine framework", Bioorg. Med. Chem., 2008, 16, 601).
В литературе описаны примеры использования отдельных представителей соединений общей формулы I. Так, в статье (A. Toepfer et al, "Piperidine carboxylic acid derivatives as sialyl Lewis X mimetics", Bioorg. Med. Chem. Lett., 1997, 7, 1311) диэтиловый эфир пиперидин-4,4-дикарбоновой кислоты формулы
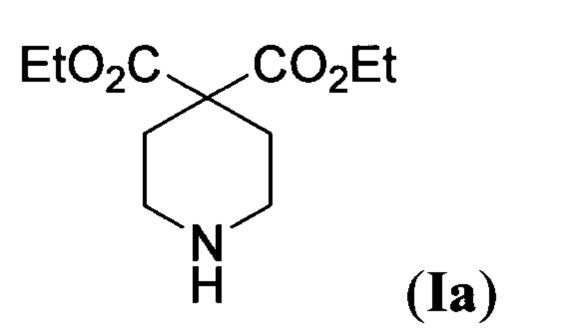
используется в качестве предшественника в синтезе миметиков тетрасахарида Сиалил-Льюис X, который играет жизненно важную роль в процессах распознавания "клетка-клетка". Синтетические миметики тетрасахарида Сиалил-Льюис X рассматриваются в качестве потенциальных противораковых соединений.
В патенте (DE 19537334 А1) соединение Ia используется как предшественник в синтезе аналогов природных лигандов селектинов-трансмембранных гликопротеинов, которые играют важную роль в воспалительных процессах и прогрессировании некоторых видов рака. Производные углеводов, содержащие остаток соединения Ia предлагаются в качестве прототипов препаратов для терапии или профилактики заболеваний, которые связаны с избыточной и опосредованной селектиновыми рецепторами адгезией клеток, например, ревматизма, сердечно-сосудистых заболеваний, таких как реперфузионные повреждения, ишемия или инфаркт.
В патентах (WO 2004083167 Al, JP 2006083133 А и JP 2006083137 А) описано использование соединения Ia в качестве билдинг-блока в синтезе ингибиторов митоген-активируемой протеинкиназы (MEK), предназначенных для лечения рака.
Известно применение соединения Ia качестве реагента для модификации тритерпеноидов, производных бетулиновой кислоты (WO 2015157483 А1). Полученные авторами производные, содержащие остаток соединения Ia проявляют выраженную активность как ингибиторы созревания вируса иммунодефицита человека (ВИЧ).
В заявке (WO 2008152149 А1) соединение Ia используется в качестве предшественника в синтезе агонистов сфингозин-1-фосфатного рецептора 1 (S1P1). Гибридные гетероциклические соединения, содержащие остаток соединения Ia, представляют интерес в качестве прототипов препаратов для лечения отторжения трансплантата, рассеянного склероза, спазма сосудов коронарных артерий, синусовой тахикардии, астмы, рака молочной железы и печени (S.Е. Gardell et al, "Emerging medicinal roles for lysophospholipid signaling", Trends Mol. Med., 2006, 12, 65).
В заявке (WO 2012109108 Al) для создания модуляторов (агонистов и антагонистов) вышеупомянутого сфингозин-1-фосфатного рецептора 1 (S1P1) использован диметиловый эфир пиперидин-4,4-дикарбоновой кислоты формулы
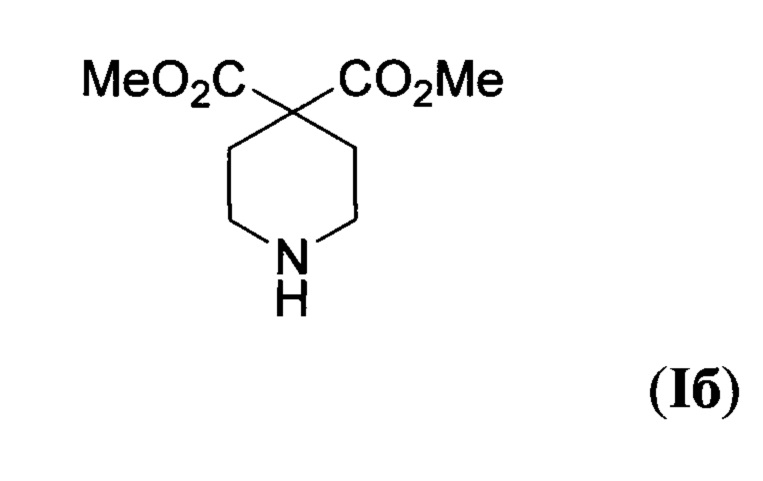
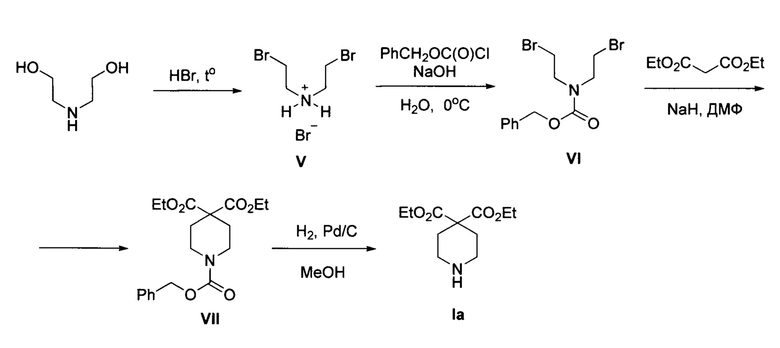
В литературе описано несколько способов получения эфиров пиперидин-4,4-дикарбоновых кислот общей формулы I. В статье (A. Toepfer et al, "Piperidine carboxylic acid derivatives as sialyl Lewis X mimetics", Bioorg. Med. Chem. Lett., 1997, 7, 1311) соединение la получено в четыре стадии из коммерчески доступного диэтаноламина. На первой стадии из диэтаноламина получают гидробромид бис(2-бромэтил)амина V, после чего производится постановка защитной бензоилоксикарбонильной группы на атом азота, затем образующийся продукт VI вводится в циклизацию с диэтилмалонатом с получением пиперидина VII. На заключительной стадии производится удаление бензоилоксикарбонильной группы в условиях каталитического гидрирования на палладиевом катализаторе Pd/C. Общий выход продукта Ia из диэтаноламина-30%. Процесс протекает по следующей схеме:
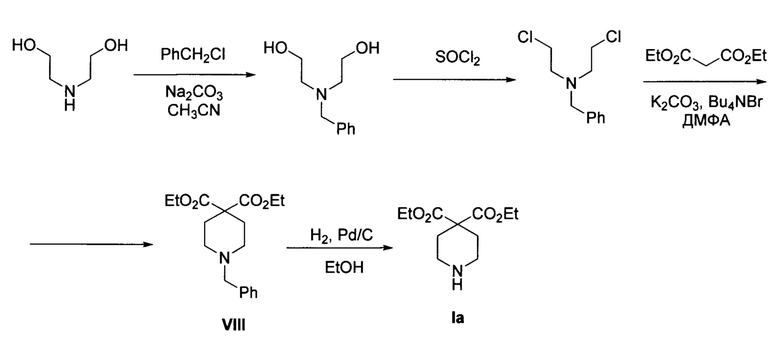
Похожая многостадийная схема использована в заявке (WO 2008152149 А1), однако в качестве ближайшего предшественника Ia здесь используется соответствующее N-бензильное производное VIII как показано ниже. Процесс протекает по следующей схеме:
Главным недостатком вышеупомянутых способов является необходимость использования защитной (бензоилоксикарбонильной или бензильной) группы, постановка и удаление которых на атом азота амино-группы требуют проведения дополнительных ресурсо- и энергозатратных операций и использования дополнительных реагентов, которые не входят в состав продукта. В результате этого снижается выход целевого соединения Ia, увеличивается стоимость его производства и образуется большое количество побочных продуктов, которые необходимо отделять и утилизировать. Кроме того, эти способы не применимы к способу получения эфиров пиперидин-4,4-дикарбоновой кислоты I, содержащих заместители у атомов углерода пиперидинового цикла (R1≠Н и/или R2≠Н и/или R3≠Н) ввиду недоступности необходимых С-замещенных предшественников.
В патенте (CN 101525313) и статье (Skinner et al, "Spiro-1'-benzenesulfonylpiperidine-4',5-barbituric Acid and Related Derivatives of Isonipecotic Acid", J. Am. Chem. Soc, 1955, 77, 2248) описан способ гидробромида пиперидина la (продукт Ia⋅HBr) из диэтаноламина путем его сульфонирования, конденсации бис-сульфоната IX с диэтилмалоновым эфиром в присутствии гидрида натрия с последующим удалением бенсульфамидной группы из продукта X действием бромоводорода в уксусной кислоте. Общий выход соли Ia⋅HBr из диэтаноламина составил 66%. Процесс протекает по следующей схеме:
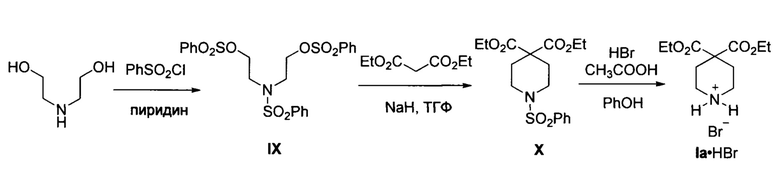
Как и рассмотренные выше способы, этот способ имеет недостаток, связанный с необходимостью использования защитной (фенилсульфонатной группы) на атоме азота амино-группы. Это приводит к необходимости использования дополнительных токсичных реагентов и понижению экологичности процесса ввиду образования побочных продуктов на стадиях постановки и удаления защитных групп, и, как следствие, увеличивает стоимость производства продукта Ia⋅HBr. Кроме того, этот способ не применим к получению других соединений общей формулы I с R1≠Н и/или R2≠Н и/или R3≠Н ввиду отсутствия необходимых С-замещенных производных бис-сульфоната IX. Известен способ получения диметилового эфира пиперидин-4,4-дикарбоновой кислоты (Iб), описанный в заявке (WO 2012109108 А1). Способ заключается в трансформации пиперидин-4-карбоновой кислоты в N-трет(бутоксикарбонил)-замещенное производное XI под действием ди(трет-бутил)дикарбоната в присутствии триэтиламина, с последующей обработкой соединения XI диизопропиламидом лития и метилхлорформиатом. Образующийся продукт XII превращается в целевое соединение Iб, действием трифторуксусной кислоты. Выход продукта Iб-52%. Процесс протекает по следующей схеме:

Этот способ также включает стадии постановки и удаления защитной группы (трет-бутоксикарбонильной) на атоме азота, протекающие с выходами 75% и 90%, соответственно, что заметно снижает общую выход продукта Iб и эффективность синтеза. Практическая значимость этого способа для получения препаративных количеств продукта Iб также ограничена необходимостью поддержания глубоко отрицательных температур (-78°С) на второй стадии процесса. Кроме того, в литературе не известно примеров применения этого способа для получения других соединений общей формулы I с R1≠Н и/или R2≠Н и/или R3≠Н.
Наиболее близким по характеру химических превращений к предлагаемому изобретению является способ получения диметилового эфира 2-метилпиперидин-4,4-дикарбоновой кислоты формулы
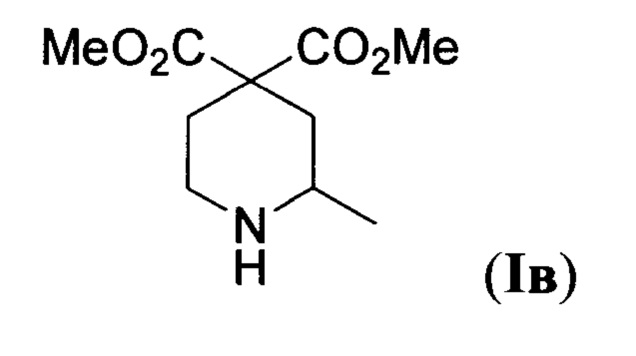
(Iв)-единственного известного С-замещенного представителя соединений общей формулы I (R1=Me, R2, R3=Н, R4=Me)-описанный в статье (S. Kim et al, "Novel radical cyclizations of alkyl azides. A new route to N-Heterocycles", J. Am. Chem. Soc. 1994, 116, 5521). Данный способ заключается в восстановительной циклизации линейного азида XIII под действием системы (Me3Si)3SiH/азобисизобутиронитрил в кипящем бензоле. Процесс протекает по следующей схеме:
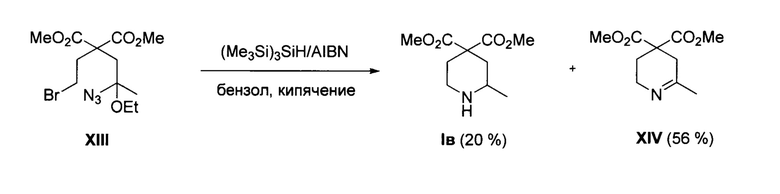
Однако с помощью указанного способа соединение Iв было получено с низким выходом (20%) в качестве побочного продукта, в то время как основным продуктом был соответствующий 2,5-дигидропиридин XIV (выход 56%). Исходное соединение XIII не является коммерчески доступным. Еще одним недостатком описанного метода синтеза соединения Iв является необходимость использования стехиометрических количеств дорогостоящего восстановителя-трис(триметилсилил)силана. Эти обстоятельства делают невозможным практическое применение этого способа для получения продукта Iв. Кроме того, в литературе не известны другие азиды типа XIII, которые могли бы служить предшественниками эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы I отличных от продукта Iв. Как видно из приведенных литературных данных, известные способы получения эфиров пиперидин-4,4-дикарбоновых кислот общей формулы I и их солей имеют общие недостатки, связанные с низкой селективностью и эффективностью, невысокими выходами продуктов, необходимостью использования дополнительных стадий введения и удаления защитных групп, образованием большого числа побочных продуктов. Эти способы не являются универсальными и применимы только для очень узкого круга исходных соединений. Это иллюстрируется наличием в литературе только трех представителей соединений типа I (продукты Ia, Iб и Iв), несмотря на высокую практическую значимость этого класса соединений. Представляется весьма вероятным, что в применениях, описанных в патентах (DE 19537334 Al, WO 2008152149 Al, WO 2004083167 Al, JP 2006083133 A, JP 2006083137 A, WO 2015157483 А1 и WO 2012109108 А1), замена соединений Ia и Iб на их аналоги, содержащие заместители R1≠Н и/или R2≠Н и/или R3≠Н в пиперидиновом цикле, позволит улучшить фармакологический профиль получаемых на основе соединений Ia и Iб фармацевтических субстанций. Технической задачей предлагаемого изобретения является повышение выходов целевых продуктов общей формулы I, а также расширение ассортимента получаемых соединений.
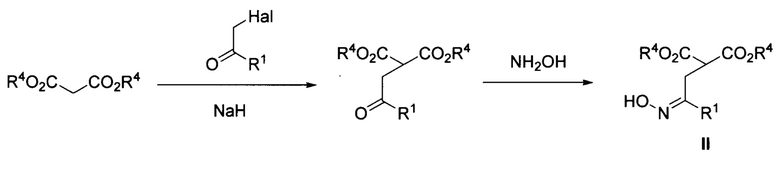
Поставленная задача достигается предложенным способом получения эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы

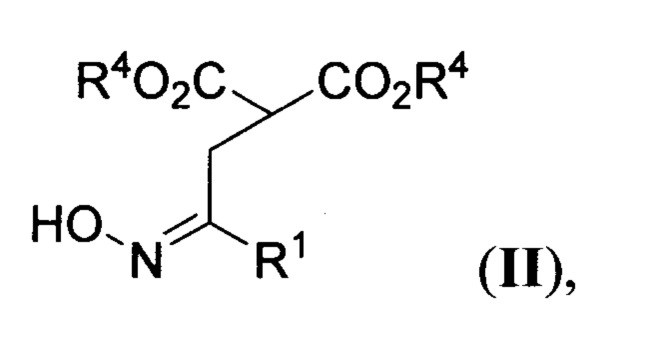
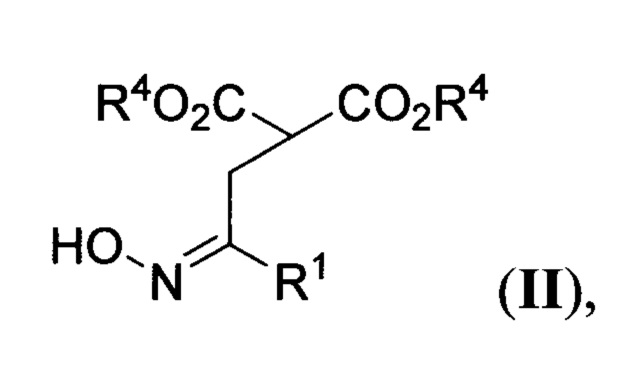
где R1 и R2=Н, С1-С4- алкил, бензил, арил, либо CH2CH2CO2Alk; R3=Н или С1-С4-алкил, R4=С1-С4-алкил, или их солей, заключающимся в том, что соответствующие эфиры 2-(оксиминоалкил)малонатов общей формулы
где R1 и R4 имеют вышеуказанные значения, обрабатывают соответствующими N,N-бис(силокси)енаминами общей формулы
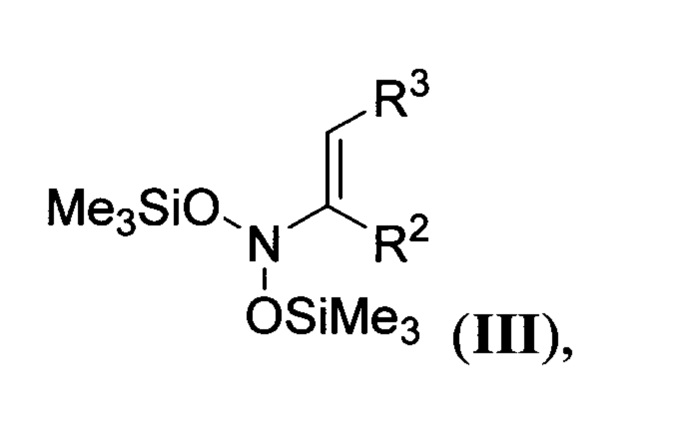
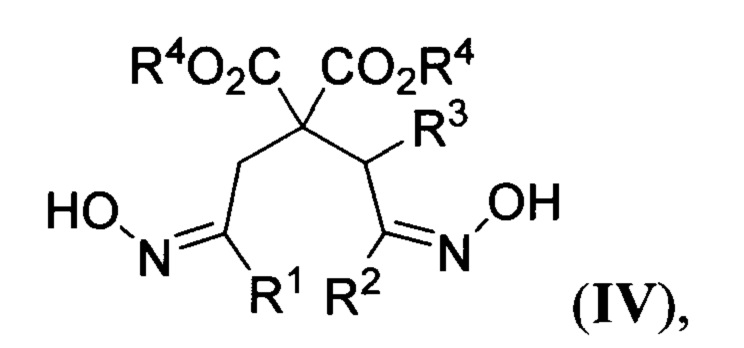
где R2 и R3 имеют вышеуказанные значения, в присутствии азотистого основания при пониженной температуре в среде апротонного растворителя, полученные при этом 2,2-бис(оксиминоалкил)малонаты общей формулы
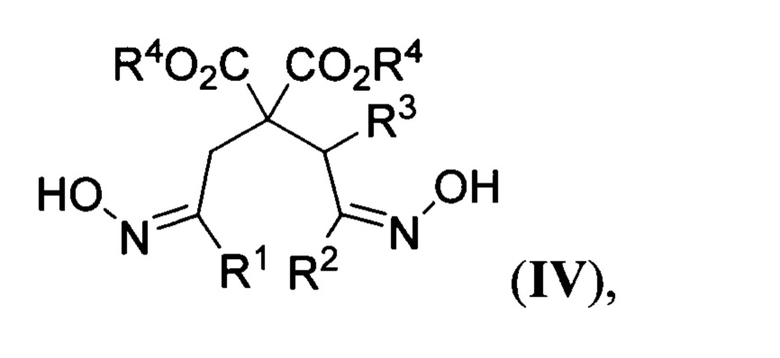
где R1, R2, R3 и R4 имеют вышеуказанные значения, гидрируют водородом при повышенной температуре и повышенном давлении водорода в присутствии металлического катализатора в среде низкомолекулярного спирта. Предложенный новый способ получения соединений общей формулы I заключается в последовательности следующих двух химических стадий: 1) превращения 2-(оксиминоалкил)малонатов II в 2,2-бис(оксиминоалкил)малонаты IV с использованием бис(силокси)енаминов III; 2) восстановительной циклизации 2,2-бис(оксиминоалкил)малонатов IV в эфиры пиперидин-4,4-дикарбоновых кислот I. Процесс протекает по следующей общей схеме:
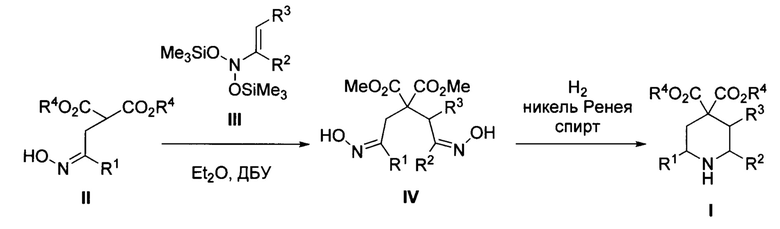
В качестве азотистого основания на первой стадии используют, например, диазабициклоундецен (ДБУ) либо родственные амидиновые и гуанидиновые основания, в качестве металлического катализатора на второй стадии используют преимущественно никель Ренея, в качестве апротонного растворителя используют, например, диэтиловый эфир, а в качестве низкомолекулярного спирта используют, например, метанол или этанол. Первую стадию процесса-получение бис-оксимов IV-необходимо проводить при пониженной температуре, преимущественно от 0 до -30°С. При нормальной температуре (комнатной) выход продуктов IV заметно снижается из-за осмоления реакционной смеси.
Вторую стадию процесса-каталитическое гидрирование бис-оксимов IV в пиперидины I - необходимо проводить в автоклаве при повышенной температуре, например, при 40-70°С, и давлении водорода от 20 до 60 атм. Снижение температуры процесса до комнатной и давления водорода до атмосферного приводит к понижению выхода продукта из-за неполной конверсии бис-оксимов IV.
При необходимости соединения общей формулы I могут быть переведены в соответствующие аммонийные соли действием любой сильной минеральной кислоты, например, хлороводорода.
Разработанный способ обладает выраженными достоинствами по сравнению с описанными в литературе способами. Во-первых, он позволяет более эффективно получать известные эфиры пиперидин-4,4-дикарбоновых кислот типа I без использования лишних стадий постановки и удаления защитных групп на атом азота амино-группы с высоким выходом, например, до 72% для соединения Ia и до 51% для соединения 1в. Во-вторых, в основе предложенного процесса получения соединений общей формулы I лежит реакция гидрирования на гетерогенном катализаторе, использующая в качестве единственного реагента дешевый, возобновляемый и экологически чистый восстановитель-водород. Каталитические процессы значительно легче реализовать в промышленных масштабах, чем некаталитические. В-третьих, разработанным способом могут быть синтезированы ранее не известные эфиры пиперидин-4,4-дикарбоновых кислот общей формулы I, содержащие различные заместители в пиперидиновом цикле. Это позволяет расширить ассортимент новых замещенных эфиров пиперидин-4,4-дикарбоновых кислот общей формулы I для дальнейшего создания на их основе лекарственных препаратов различного назначения. Исходными веществами для предлагаемого способа получения эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы I являются N,N-бис(силокси)енамины III и 2-(оксиминоалкил)малонаты II, которые могут быть синтезированы из коммерчески доступных реагентов по описанным в литературе методикам в граммовых и килограммовых количествах как показано на схемах ниже.
N,N-Бис(силокси)енамины III получают двойным силилированием алифатических нитросоединений триметилсилилбромидом в присутствии триэтиламина с количественными выходами (A.D. Dilman et al, "Novel Convenient Method for the Synthesis of N,N-Bis(trimethylsilyloxy)enamines" Synthesis 1998, 181 и A.D. Dilman et al, "Synthesis of N,N-bis(silyloxy)enamines with a functionalized double bond" J. Chem. Soc, Perkin Trans. 1 2000, 2926).

2-(Оксиминоалкил)малонаты II получают одним из двух способов, описанных ниже.
Способ 1. Взаимодействие малоновых эфиров с N,N-бис(силокси)енаминами III (А.V. Ustinov et al, "Chemistry of N,N-bis(silyloxy)enamines. 4. Study of the reactions of N,N-bis(silyloxy)enamines with 1,3-diones", Russian Chemical Bulletin, 2002, 51, 1455) по следующей схеме:
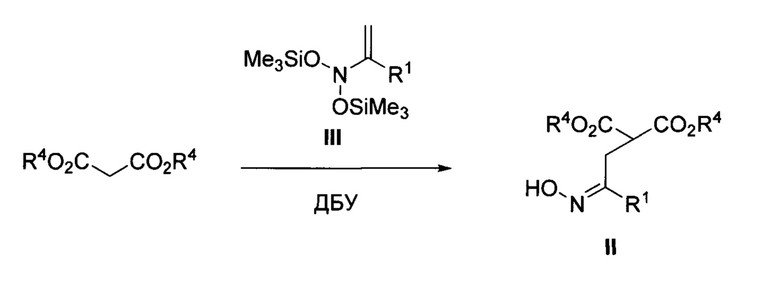
Способ 2. Взаимодействие малоновых эфиров с α-галогенкетонами с последующим оксимированием образующихся кетонов (P.-F. Мао et al, "Cu(OAc)2-Triggered Cascade Reaction of Malonate-Tethered Acyl Oximes with Indoles, Indole-2-alcohols, and Indole-2-carboxamides", Organic Letters, 2019, 21, 3153) по следующей схеме:
Разработанный способ получения эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы I принципиальным образом отличается от известных в литературе тем, что позволяет осуществлять модулярную сборку целевых соединений I, содержащих заместители при углеродных атомах пиперидинового цикла, без использования защитных групп на атоме азота. Универсальность и новизна разработанного метода демонстрируется тем, что большинство полученных и описанных в настоящем патенте производных пиперидин-4,4-дикарбоновой кислоты общей формулы I ранее не были известны.
Реакции, используемые в предлагаемом способе получения соединений общей формулы I, в литературе не описаны. Найденный способ каталитического превращения бис-оксимов IV в соединения общей формулы I являются ключевым, новым, неописанным в литературе.
Таким образом, заявляемое изобретение соответствует критерию «новизна» и «изобретательский уровень».
Техническим результатом предлагаемого изобретения является создание эффективного и универсального способа получения эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы I, с высоким выходом для известных соединений, например, до 72% для соединения Ia и до 51% для соединения Iв, а также расширение ассортимента получаемых новых соединений общей формулы I в том числе симметрично- и несимметрично-замещенных, несущих группы R1, R2, R3 в пиперидиновом цикле отличные от водорода. Следует отметить, что предлагаемый способ, в отличие от известных вообще в литературе аналогов, позволяет упростить технологию получения продуктов I за счет: 1) использования каталитического гидрирования вместо некаталитического восстановления на ключевой стадии синтеза; 2) отказа от использования дополнительных стадий постановки и удаления защитных групп на атоме азота пиперидинового кольца.
Изобретение иллюстрируется примерами, не ограничивающими его объем.
Пример 1. Получение диметил пиперидин-4,4-дикарбоксилата (Iб) и его гидрохлорида Iб⋅HCl
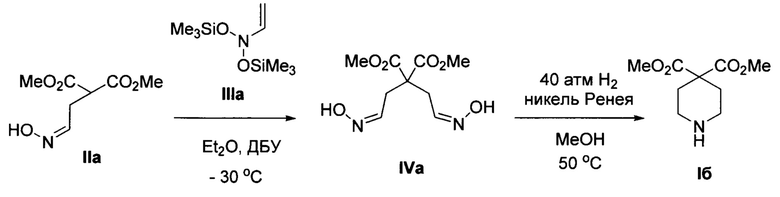
1) К охлажденному до 0°С раствору диметил 2-(2-(гидроксиимино)этил)малоната IIa (2.5 г, 13.2 ммоль) в диэтиловом эфире (120 мл) при интенсивном перемешивании добавляют диазабициклоундецен (2.0 мл, 13.2 ммоль). Смесь перемешивают при этой температуре в течение 15 минут, затем охлаждают до -30°С и по каплям прибавляют 1 М раствор О-(триметилсилил)-N-((триметилсилил)окси)-N-винилгидроксиламина IIIa в CH2Cl2 (14.6 мл, 14.6 ммоль). Реакционную смесь перемешивают при -30°С в течение 2 часов, отогревают до комнатной температуры и добавляют метанол (20 мл) и фторид аммония (2.0 г, 54.1 ммоль). Полученную смесь перемешивают 15 минут и переносят в делительную воронку, содержащую этилацетат (250 мл) и насыщенный водный раствор карбоната натрия (250 мл), и интенсивно встряхивают. Отделенную водную фазу промывают этилацетатом (3 раза по 100 мл), объединенную органическую фазу промывают насыщенным водным раствором хлорида натрия (500 мл) и высушивают от остатков воды выдерживанием над безводным сульфатом натрия. Раствор упаривают при пониженном давлении, получая в остатке сырой диметил 2,2-бис(2-(гидроксиимино)этил)малонат IVa в виде желтоватого масла. Для получения продукта аналитической чистоты его очищают с помощью колоночной хроматографии на силикагеле (элюент петролейный эфир/этилацетат = 2:1). Получают 2.57 г (выход 79%) смеси EIZ-изомеров диметил 2,2-бис(2-(гидроксиимино)этил)малоната IVa. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 9.54 и 9.29 (2 уш. с, 2Н), 7.36, 7.40, 6.81 и 6.76 (т, J=6.1 Гц; т, J=6.1 Гц; т, J=5.4 Гц; т, J=5.5 Гц, 2Н), 3.78, 3.77 и 3.76 (3 с, 6Н), 3.00 и 2.82 (д, J=5.5 Гц и д, J=6.1 Гц, 4Н). 13С ЯМР (75 МГц, CDCl3) δ 170.4, 170.2, 170.0, 147.2, 147.1, 146.9, 146.7, 55.2, 54.8, 54.1, 53.2, 33.8, 33.2, 29.3, 28.9. Масс-спектр высокого разрешения (ESI): m/z 247.0930 (МН+); рассчитано для [C9H15N2O6]+ 47.0925.
2) Раствор диметил 2,2-бис(2-(гидроксиимино)этил)малоната IVa (0.75 г, 3.0 ммоль) в метаноле (10 мл) помещают в стальной автоклав и добавляют суспензию катализатора никеля Ренея (около 500 мг) в метаноле (10 мл). Смесь гидрируют с интенсивным перемешиванием при 50°С и давлении водорода 40 атм до полной конверсии исходного диоксима IVa (3 часа). Полученный раствор отфильтровывают от катализатора и концентрируют при пониженном давлении, получая в остатке сырой диметил пиперидин-4,4-дикарбоксилат Iб в виде желтоватого масла. Для получения продукта аналитической чистоты его очищают с помощью колоночной хроматографии на силикагеле (элюент этилацетат/метанол -2:1). Получают 0.45 г (выход 73%) диметил пиперидин-4,4-дикарбоксилата 16. Общий выход 16 на 2 стадии составляет 58%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 3.73 (с, 6Н), 2.92-2.76 (м, 4Н), 2.11-2.00 (м, 4Н), 1.92-1.76 (уш. с, 1Н). 13С ЯМР (75 МГц, CDCl3) δ 171.8, 53.8, 52.7, 43.4, 31.9. Масс-спектр высокого разрешения (ESI): m/z 202.1078 (МН+); рассчитано для [C9H16NO4]+ 202.1074.
3) К раствору диметил пиперидин-4,4-дикарбоксилата 1б (0.4 г, 2.0 ммоль) в диэтиловом эфире (10 мл) при комнатной температуре добавляют 4 М раствор хлороводорода в диоксане (1.0 мл). Выпавшему осадку гидрохлорида 1б⋅HCl дают отстояться, после чего его отделяют от раствора, промывают диэтиловым эфиром (3×10 мл) и высушивают в вакууме до постоянной массы. Получают 0.43 г (выход 90%) гидрохлорида диметил пиперидин-4,4-дикарбоксилата 1б⋅HCl в виде твердого белого вещества. Тпл=128°С. 1Н ЯМР (300 МГц, D2O) δ 3.82 (с, 6Н), 3.33-3.22 (м, 4Н), 2.44-2.32 (м, 4Н). Элементный анализ: найдено, %: С, 45.32; Н 6.47; N 5.98. Вычислено для C9H16CINO4, %: С, 45.48; Н, 6.79; N,5.89.
Пример 2. Получение диэтил пиперидин-4,4-дикарбоксилата (Ia)

Аналогично примеру 1, с тем отличием, что стадия 2 проводилась при 40°С и давлении водорода 20 атм в этаноле, из диэтил 2-(2-(гидроксиимино)этил)малоната Пб и 0-(триметилсилил)-N-((триметилсилил)окси)-N-винилгидроксиламина IIIa получено соединение 1а с выходом 72%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 6.26-5.88 (уш., 1Н), 4.18 (кв, J=7.1 Гц, 4Н), 3.04 (т, J=5.5 Гц, 4Н), 2.22 (т, J=5.5 Гц, 4Н), 1.22 (т, J=7.1 Гц, 6Н). 13С ЯМР (75 МГц, CDCl3) δ 170.22, 61.90, 52.37, 41.94, 29.24, 14.08. Масс-спектр высокого разрешения (ESI): m/z 230.1392 (МН+); рассчитано для [C11H20NO4]+ 230.1387.
Пример 3. Получение диметил 2-метилпиперидин-4,4-дикарбоксилата и его гидрохлорида 1в⋅HCl

Аналогично примеру 1, с тем отличием, что стадия 1 проводилась при 0°С, из диметил 2-(2-(гидроксиимино)этил)малоната II и N-(проп-1-ен-2-ил)-O-(триметилсилил)-N-((триметилсилил)окси)гидрокси-амина IIIб получено соединение Iв с выходом 51%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 5.47-4.79 (уш., 1 Н), 3.80 (с, 3Н), 3.75 (с, 3Н), 3.40-3.23 (м, 1Н), 3.17-2.99 (м, 1Н), 2.90 (тд, J=13.1, 2.5 Гц, 1H), 2.46 (д, J=14.1 Гц, 2Н), 2.24-2.07 (м, 1Н), 1.89 (дд, J=14.1, 12.0 Гц, 1Н), 1.39 (д, J=6.4 Гц, 3Н). Масс-спектр высокого разрешения (ESI): m/z 216.1234 (МН+); рассчитано для [C10H18NO4]+ 216.1230.
Аналогично примеру 1 из соединения Iв был получен гидрохлорид Iв⋅HCl с выходом 95%. Твердое белое вещество. 1Н ЯМР (300 МГц, CDCl3) δ 3.84 (с, 3Н), 3.77 (с, 3Н), 3.56-3.46 (м, 1H), 3.39-3.28 (м, 1H), 3.10 (тд, J=13.7, 3.0 Гц, 1Н), 2.72-2.56 (м, 2Н), 2.18-2.04 (м, 1Н), 1.95 (дд, J=14.9, 12.6 Гц, 1Н), 1.37 (д, J=6.5 Гц, 3Н). Элементный анализ: найдено, %: С, 47.72; Н, 7.21; N, 5.56. Вычислено для C10H18ClNO4, %: С, 47.85; Н, 7.02; N, 5.44.
Пример 4. Получение диметил цис-2,6-диметилпиперидин-4,4-дикарбоксилата (Iг)
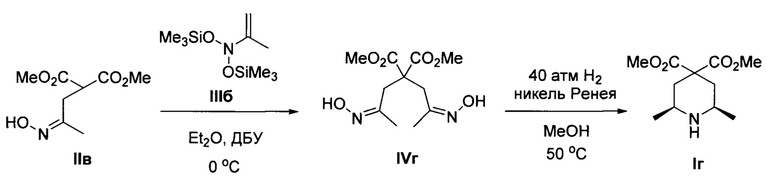
Аналогично примеру 1, с тем отличием, что стадия 1 проводилась при 0°С, из диметил 2-(2-(гидроксиимино)пропил)малоната IIв и N-(проп-1-ен-2-ил)-O-(триметилсилил)-N-((триметилсилил)окси)гидрокси-амина IIIб получено соединение Iг с выходом 38%. Желтоватое масло. 1Н ЯМР (300 МГц, CDCl3) δ 4.12-3.83 (уш. с, 1Н), 3.71 (с, 3Н), 3.67 (с, 3Н), 2.90-2.71 (м, 2Н), 2.28 (д, J =13.0 Гц, 2Н), 1.55-1.38 (дд, J=13.0, 12.5 Гц, 2Н), 1.12 (д, J=6.2 Гц, 6Н). 13С ЯМР (76 МГц, CDCl3) δ 171.8, 171.2, 54.4, 52.8, 52.7, 48.8, 37.8, 21.9. Масс-спектр высокого разрешения (ESI): m/z 230.1390 (МН+); рассчитано для [C11H20NO4]+ 230.1387.
Пример 5. Получение диметил 2-фенилпиперидин-4,4-дикарбоксилата (Iд)

Аналогично примеру 1 из диметил 2-(2-(гидроксиимино)-2-фенилэтил)малоната IIг и O-(триметилсилил)-N-((триметилсилил)окси)-N-винилгидроксиламина IIIa получено соединение Iд с выходом 46%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 7.45-7.27 (м, 5Н), 3.82 (с, 3Н), 3.76 (дд, J=11.7, 2.5 Гц, 1Н), 3.70 (с, 3Н), 3.18 (ддд, J=12.4, 4.5, 2.5 Гц, 1Н), 2.87 (тд, J=12.4, 2.5 Гц, 1Н), 2.49 (дт, J=13.3, 2.5 Гц, 1Н), 2.38 (д кв, J=13.4, 2.4 Гц, 1Н), 1.99 (тд, J=13.4, 4.5 Гц, 1Н), 1.95-1.85 (уш. м, 1Н), 1.86 (дд, J=13.3, 11.7 Гц, 1Н). 13С ЯМР (76 МГц, CDCl3) δ 172.1, 171.4, 143.8, 128.7, 127.7, 126.8, 58.2, 54.6, 52.9, 52.8, 44.0, 39.2, 30.9. Масс-спектр высокого разрешения (ESI): m/z 278.1383 (МН+); рассчитано для [C15H20NO4]+ 278.1387.
Пример 6. Получение диметил 3-метилпиперидин-4,4-дикарбоксилата (Iе)
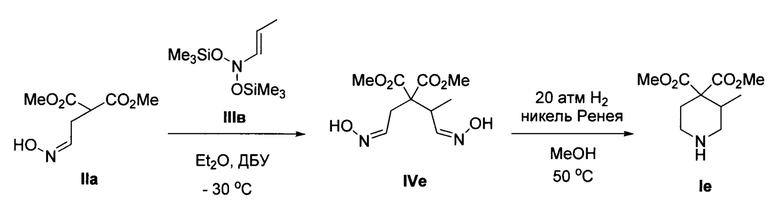
Аналогично примеру 1, с тем отличием, что стадия 2 проводилась при давлении водорода 20 атм, из диметил 2-(2-(гидроксиимино)этил)малоната IIa и N-(проп-1-ен-1-ил)-O-(триметилсилил)-N-((триметилсилил)окси)гидроксиамина IIIв получено соединение Iе с выходом 35%. Желтоватое масло. 1Н ЯМР (300 МГц, CDCl3) δ 5.9-5.7 (уш. с, 1Н), 3.76 (с, 3Н), 3.75 (с, 3Н), 3.14 (дд, J=13.2, 3.8 Гц, 1Н), 3.05 (м, 1Н), 2.89-2.74 (м, 1Н), 2.82 (дд, J=13.2, 7.4 Гц, 1Н), 2.50 (м, 1Н), 2.29 (ддд, J=14.3, 7.9, 3.7 Гц, 1Н), 2.09 (ддд, J=14.3, 7.9, 3.7 Гц, 1Н), 1.07 (д, J=7.1 Гц, 3Н). Масс-спектр высокого разрешения (ESI): m/z 216.1228 (МН+); рассчитано для [C10H18NO4]+ 216.1230.
Пример 7. Получение диметил 2-(3-метокси-3-оксопропил)пиперидин-4,4-дикарбоксилата (Iж)
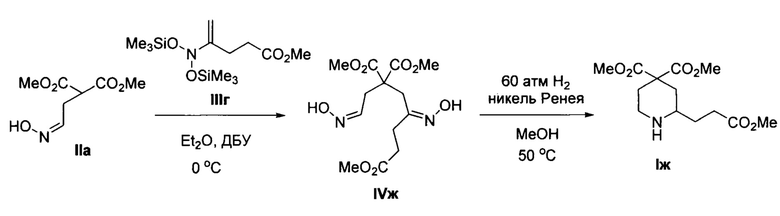
Аналогично примеру 1, с тем отличием, что стадия 1 проводилась при 0°С, а стадия 2 при давлении водорода 60 атм, из диметил 2-(2-(гидроксиимино)этил)малоната IIa и метил 4-(бис((триметилсилил)окси)амино)пент-4-еноата IIIг получено соединение Iж с выходом 21%. Желтоватое масло. 1Н ЯМР (300 МГц, CDCl3) δ 3.76 (с, 3Н), 3.71 (с, 3Н), 3.67 (с, 3Н), 3.11-2.99 (м, 1Н), 2.69 (тд, J=13.0, 2.6 Гц, 1H), 2.58 (м, 1Н), 2.48-2.24 (м, 4Н), 1.81-1.43 (2 м, 4Н), 1.41-1.22 (уш. м, 1Н). Масс-спектр высокого разрешения (ESI): m/z 288.1431 (МН+); рассчитано для [C13H22NO6]+ 288.1442.
Пример 8. Получение диметил 2-бензилпиперидин-4,4-дикарбоксилата (Iз)

Аналогично примеру 1 с тем отличием, что стадия 2 проводилась при температуре 70°С, из диметил 2-(2-(гидроксиимино)этил)малоната IIa и N-(3-фенилпроп-1-ен-2-ил)-O-(триметилсилил)-N-((триметилсилил)окси)гидроксиламин IIIд получено соединение Iз с выходом 46%. Желтоватое масло. 1Н ЯМР (300 МГц, CDCl3) δ 7.37-7.20 (м, 5Н), 3.77 (с, 3Н), 3.73 (с, 3Н), 3.06-2.94 (м, 1Н), 2.88-2.76 (м, 1Н), 2.69-2.56 (м, 2Н), 2.46-2.29 (м, 2Н), 2.20-2.03 (м, 1Н), 1.87 (тд, J=13.1, 4.5 Гц, 1Н), 1.61 (дд, J=13.2, 11.2 Гц, 1Н). Масс-спектр высокого разрешения (ESI): m/z 292.1544 (МН+); рассчитано для [C16H22NO4]+ 292.1543.
Пример 9. Получение диметил 2-(3-метоксифенил)пиперидин-4,4-дикарбоксилата (Iи)
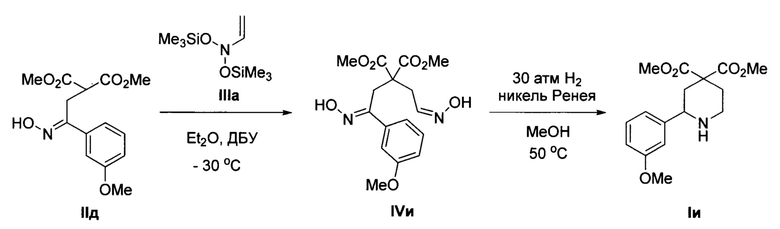
Аналогично примеру 1 с тем отличием, что стадия 2 проводилась при давлении водорода 30 атм, из диметил 2-(2-(гидроксимино)-2-(3-метоксифенил)этил)малоната IIд и O-(триметилсилил)-N-((триметилсилил)окси)-N-винилгидроксиламина IIIa получено соединение Iи с выходом 44%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 7.37-7.29 (м, 2Н), 7.27 (т, J=6.3 Гц, 1Н), 7.00 (дд, J=7.4, 2.0 Гц, 1Н), 3.85 (с, 3Н), 3.82 (с, 3Н), 3.79 (дд, J=11.0, 1.9 Гц, 1Н), 3.71 (с, 3Н), 3.15 (м, 1Н), 2.85 (м, 1Н), 2.50 (дд, J=13.5, 1.9 Гц, 1Н), 2.38 (м, 1H), 2.21-2.10 (уш., 1Н), 2.00 (м, 1Н), 1.87 (дд, J=13.5, 11.0 Гц, 1Н). Масс-спектр высокого разрешения (ESI): m/z 308.1499 (МН+); рассчитано для [C66H22NO5]+ 308.1493.
Пример 10. Получение диметил 2-(3-метоксифенил)пиперидин-4,4-дикарбоксилата (Iи)
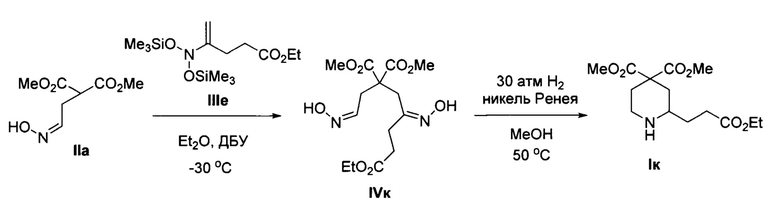
Аналогично примеру 1 с тем отличием, что стадия 2 проводилась при давлении водорода 30 атм из диметил 2-(2-(гидроксиимино)этил)малоната IIa и метил 4-(бис((триметилсилил)окси)амино)пент-4-еноата IIIе получено соединение Iк с выходом 25%. Бесцветное масло. 1Н ЯМР (300 МГц, CDCl3) δ 4.98-4.54 (уш., 1Н), 4.20 (кв, J=7.2 Гц, 2Н), 3.73 (с, 3Н), 3.69 (с, 3Н), 3.12-3.00 (м, 1Н), 2.67 (м, 1H), 2.58 (м, 1Н), 2.48-2.19 (м, 4Н), 1.80-1.44 (2 м, 4Н), 1.33 (т, J=7.2 Гц, 3Н). Масс-спектр высокого разрешения (ESI): m/z 302.1597 (МН+); рассчитано для [C14H24NO6]+ 302.1599.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения замещенных 3-арилпирролов | 2024 |
|
RU2831117C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БИЦИКЛО [3.1.0]ГЕКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЭТОЙ ЦЕЛИ | 2004 |
|
RU2388747C2 |
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| N1/N2-ЛАКТАМНЫЕ ИНГИБИТОРЫ АЦЕТИЛ-КоА-КАРБОКСИЛАЗ | 2011 |
|
RU2540337C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ, ПРОТИВОВИРУСНЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2452735C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА | 2004 |
|
RU2359971C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРАЗИНА В КАЧЕСТВЕ ИНГИБИТОРОВ CSF-1R КИНАЗЫ | 2013 |
|
RU2642777C2 |
| ПРОИЗВОДНОЕ ПИРРОЛИДИН-3-ИЛУКСУСНОЙ КИСЛОТЫ | 2012 |
|
RU2615135C2 |
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
Изобретение относится к способу получения эфиров пиперидин-4,4-дикарбоновых кислот общей формулы (I), R1, R2=Н, C1-C4-алкил, бензил, арил, CH2CH2CO2Alk; R3=Н, С1-С4-алкил, R4=С1-С4-алкил, или их солей, заключающийся в том, что соответствующие эфиры 2-(оксиминоалкил)малонатов общей формулы (II), где R1 и R4 имеют вышеуказанные значения, обрабатывают соответствующими N,N-бис(силокси)енаминами общей формулы (III), где R2 и R3 имеют вышеуказанные значения, в присутствии азотистого основания при пониженной температуре в апротонном растворителе, и полученные при этом 2,2-бис(оксиминоалкил)малонаты общей формулы (IV), где R1, R2, R3 и R4 имеют вышеуказанные значения, гидрируют водородом при повышенной температуре и повышенном давлении водорода в присутствии металлического катализатора в среде низкомолекулярного спирта с последующим выделением целевого продукта в свободном виде или при необходимости в виде соли. Технический результат заключается в создании эффективного и универсального способа получения соединений общей формулы I, которые применимы как ключевые полупродукты в синтезе фармакологически активных субстанций для лечения таких заболеваний как рак, рассеянный склероз, СПИД, сердечно-сосудистые заболевания и др., при этом способ не требует использования дополнительных стадий постановки и удаления защитных групп на атоме азота пиперидинового кольца. 6 з.п. ф-лы, 10 пр.
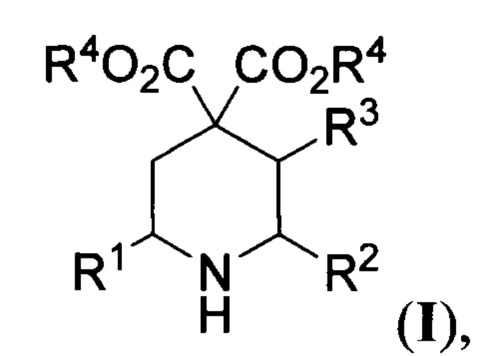

1. Способ получения эфиров пиперидин-4,4-дикарбоновой кислоты общей формулы

R1, R2=Н, C1-C4 - алкил, бензил, арил, либо CH2CH2CO2Alk; R3=Н, С1-С4 - алкил, R4=С1-С4 - алкил, или их солей, заключающийся в том, что соответствующие эфиры 2-(оксиминоалкил)малонатов общей формулы

где R1 и R4 имеют вышеуказанные значения, обрабатывают соответствующими N,N-бис(силокси)енаминами общей формулы

где R2 и R3 имеют вышеуказанные значения, в присутствии азотистого основания при пониженной температуре в апротонном растворителе и полученные при этом 2,2-бис(оксиминоалкил)малонаты общей формулы
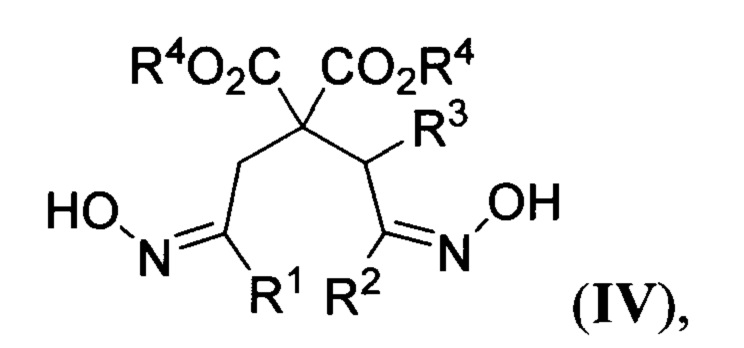
где R1, R2, R3 и R4 имеют вышеуказанные значения, гидрируют водородом при повышенной температуре и повышенном давлении водорода в присутствии металлического катализатора в среде низкомолекулярного спирта с последующим выделением целевого продукта в свободном виде или при необходимости в виде соли.
2. Способ по п. 1, отличающийся тем, что в качестве азотистого основания используют диазабициклоундецен.
3. Способ по п. 1, отличающийся тем, что в качестве апротонного растворителя используют диэтиловый эфир.
4. Способ по п. 1, отличающийся тем, что в качестве металлического катализатора используют никель Ренея.
5. Способ по п. 1, отличающийся тем, что процесс получения соединений общей формулы IV проводят при температуре 0 - (-30)°С.
6. Способ по п. 1, отличающийся тем, что процесс гидрирования соединений общей формулы IV проводят при температуре 40-70°С.
7. Способ по п. 1, отличающийся тем, что процесс гидрирования соединений общей формулы IV проводят при давлении водорода 20 до 60 атм.
| WO 2012109108 A1, 16.08.2012 | |||
| СПОСОБ СКРУТКИ КОНЦА ПРОВОДА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1990 |
|
RU2014653C1 |
| Alexander Toepfer и др.: "PIPERIDINE CARBOXYLIC ACID DERIVATIVES AS SIALYL LEWIS X MIMETICS", 1997, 7(10), с.1311-1316 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |

