Перекрестная ссылка на родственные заявки
По настоящей заявке испрашивается приоритет для предварительной заявки на патент США № 62/691604, поданной 28 июня 2018, и корейской заявки № 10-2017-0162404, поданной 30 ноября 2017, содержание которых включено в настоящий документ во всей полноте и для всех целей.
Область техники, к которой относится изобретение
В одном аспекте обеспечиваются способы и композиции, включающие PLAG (1-пальмитоил-3-линолеоил-3-ацетилглицерин), для профилактики, лечения, модуляции или ослабления острой лучевой болезни (ОЛБ).
Предшествующий уровень техники
Острая лучевая болезнь (ОЛБ) - это заболевание, которое возникает, когда большая часть человеческого тела подвергается воздействию радиации; это опасное заболевание, которое в конечном итоге приводит к смерти, разрушая иммунную, кроветворную, нервную и/или желудочно-кишечную системы. Ядерный взрыв и ядерная авария могут привести к радиационному облучению, которое может вызвать острую лучевую болезнь.
Основное лечение пациентов, подвергающихся воздействию радиации, заключается в профилактике и лечении инфекции. Клинически адъювантную терапию, такую как применение цитокинов, антибиотиков и переливание крови, проводят на ранней стадии после облучения, а когда длительность нейтропении у пациентов увеличивается, риск вторичной инфекции также увеличивается.
Таким образом, необходимо иметь новые способы лечения острой лучевой болезни.
Изложение сущности изобретения
В одном аспекте обеспечиваются новые способы лечения и профилактики острой лучевой болезни, которые включают PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин).
В другом аспекте предложены новые способы лечения субъекта, который подвергался воздействию ионизирующего излучения (особенно неблагоприятного воздействия, такого как непреднамеренное и/или нетерапевтическое воздействие, и/или воздействие чрезмерного ионизирующего излучения, включая гамма-излучение), которое включает введение субъекту эффективного количества PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина).
В еще одном дополнительном аспекте предлагаются способы лечения и профилактики одного или нескольких субсиндромов острой лучевой болезни, которые включают использование PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина). Субсиндромы ОЛБ включают гемопоэтический, желудочно-кишечный, кожный и/или нейроваскулярный.
В еще одном аспекте предлагаются способы лечения и профилактики гемопоэтического (костномозгового) синдрома острой лучевой болезни, желудочно-кишечного синдрома острой лучевой болезни, кожного синдрома острой лучевой болезни, сердечно-сосудистого синдрома острой лучевой болезни, и/или острой лучевой болезни центральной нервной системы (ЦНС).
В дополнительном аспекте предлагаются способы лечения и профилактики радиационно-индуцированной коагулопатии.
PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) позволяет ослаблять, предотвращать и/или лечить уменьшение иммунных клеток, таких как лейкоциты, нейтрофилы и лимфоциты, вызванное радиацией, и облегчать воспалительные заболевания, такие как оральный мукозит, и дополнительно повышать выживаемость субъектов, подвергшихся воздействию излучения.
PLAG может быть впервые введен индивидууму даже после истечения длительного периода времени с момента воздействия на индивидуума вредного излучения, такого как гамма-излучение. То есть индивидуум может получить терапевтическую пользу, включая увеличение времени выживания, даже после такого отсроченного лечения. См., например, результаты in vivo, изложенные в следующих примерах. Это может быть существенным, поскольку первое лечение индивидуума, подвергающегося радиационному воздействию, часто может быть отсрочено.
Таким образом, в некоторых аспектах PLAG сначала применяют (первая доза для индивидуума) в течение 3, 6, 12, 18, 24, 36, 48, 60 или 72 часов после того, как индивидуум подвергся вредному воздействию излучения (включая воздействие гамма-излучения). В конкретных аспектах PLAG сначала применяют (первая доза для индивидуума) через 3-12, 18, 48 или 72 часа, или между 6 и 18, 24 или 48 часами после того, как индивидуум подвергся вредному воздействию излучения (включая гамма-излучение). В определенных аспектах вредное воздействие излучения может представлять собой воздействие ионизирующего излучения, такого как гамма-излучение, от 1 Гр до 8 Гр или более в течение от 1 секунды до 30, 60 или 120 секунд или более.
В дополнительном аспекте предложена фармацевтическая композиция, содержащая PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) для профилактики или лечения острой лучевой болезни.
Также предлагается фармацевтическая композиция, содержащая PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин), для профилактики или лечения индивидуума, подвергающегося воздействию ионизирующего излучения, в частности индивидуума, подвергающегося воздействию избыточного ионизирующего излучения.
В другом аспекте предложена функциональная оздоровительная пища или пищевая добавка, которая содержит PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) в качестве активного ингредиента для профилактики или ослабления острой лучевой болезни.
В еще одном дополнительном аспекте предлагаются наборы для использования с целью лечения или профилактики острой лучевой болезни, или для лечения или профилактики воздействия чрезмерного ионизирующего излучения.
Подходящие наборы по изобретению могут включать (1) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG); и (2) инструкции по использованию PLAG для лечения или профилактики острой лучевой болезни (ОЛБ) у субъекта или для лечения или профилактики воздействия чрезмерного ионизирующего излучения. Предпочтительно набор будет содержать терапевтически эффективное количество PLAG. Соответствующие инструкции могут быть в письменной форме, в том числе в виде этикетки продукта.
Термины PLAG, EC-18 и 1-пальмитоил-2-линолеоил-3-ацетилглицерин используются в настоящей заявке взаимозаменяемо, и обозначают одно и то же соединение.
В дополнительных аспектах соединение Формулы 1, как изложено ниже, может быть использовано в способах, композициях и наборах по изобретению, в частности, для лечения субъекта, страдающего острой лучевой болезнью или подвергшегося воздействию неблагоприятного ионизирующего излучения.
Другие аспекты раскрыты ниже.
Краткое описание чертежей
Файл патента или заявки содержит как минимум один чертеж, выполненный в цвете. Копии этого патента или публикации патентной заявки с цветными чертежами будут предоставлены Ведомством по запросу и уплате необходимой пошлины.
Фигура 1 представляет схематическую диаграмму дизайна эксперимента, относящуюся к радиационно-индуцированной острой лучевой болезни на животной модели.
На Фигурах 2А-2С показан уровень в крови лейкоцитов (Фигура 2А), нейтрофилов (Фигура 2В) и лимфоцитов (Фигура 2С) после введения PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина) на животной модели острой лучевой болезни.
На Фигуре 3 показана выживаемость при введении PLAG на животных моделях острой лучевой болезни, сопровождающейся оральным мукозитом.
Фигура 4 показывает состояние мукозита после введения PLAG на животной модели острой лучевой болезни.
На Фигуре 5 показана расчетная оценка мукозита у животных, получавших PLAG (EC-18), и не получавших лечения животных на животной модели острой лучевой болезни, сопровождающейся мукозитом полости рта.
На Фигурах 6А-6В показан уровень количества нейтрофилов в крови после введения PLAG на животной модели острой лучевой болезни, сопровождающейся мукозитом полости рта, через 7 дней (Фигура 6А) и через 10 дней (Фигура 6В).
Фигура 7А представляет собой схематическую диаграмму лечения ЕС-18 (30 дней) на животной модели с радиационно-индуцированной острой лучевой болезнью. На Фигуре 7B показана выживаемость мышей (BALB/c, 11 недель), у которых применяли или не применяли EC-18 (250 мг/кг/сутки, перорально), с общим облучением организма (6,5 Гр). Коэффициент выживаемости у мышей, которым вводили ЕС-18, улучшен по сравнению с не получавшими лечения (без ЕС-18) мышами.
Фигура 8 показывает выживаемость мышей (BALB/c, 9 недель), у которых применяли (n = 20) или не применяли (n = 20) EC-18 (250 мг/кг/сутки, перорально), с общим облучением организма (6,5 Гр) согласно схеме на Фигуре 7А. Выживаемость мышей, получавших EC-18, улучшена по сравнению с не получавшими лечения (без EC-18) мышами.
Фигура 9 демонстрирует кривые выживаемости мышей (BALB/c, 9 недель), получавших общее облучение тела (2, 4 и 6 Гр) без лечения EC-18 и с лечением EC-18 (250 мг/кг/сутки, ПО), с общим облучением тела при 6 Гр в соответствии со схемой на Фигуре 7А.
Фигура 10А представляет собой схематическую диаграмму лечения ЕС-18 (17 дней) на животной модели с радиационно-индуцированной острой лучевой болезнью. На Фигуре 10B показана выживаемость мышей (BALB/c, 9 недель), которым вводили или не вводили EC-18 (250 мг/кг/сутки, ПО), с летальным облучением организма (7 Гр). Коэффициент выживаемости мышей, которым вводили ЕС-18, получающих летальное облучение, улучшается примерно до 15 дня по сравнению с не получавшими лечения (без ЕС-18) мышами.
На Фигуре 11 показана выживаемость мышей (BALB/c, 9 недель), которым вводили или не вводили EC-18 (250 мг/кг/сутки, перорально), с летальным облучением всего тела (8 Гр). Показатель выживаемости мышей, которым вводили EC-18, улучшается примерно до 14 дня по сравнению с не получавшими лечения (без ЕС-18) мышами, согласно диаграмме на Фигуре 10A.
Фигура 12А представляет собой схематическую диаграмму лечения ЕС-18 (12 дней) на животной модели с радиационно-индуцированной острой лучевой болезнью. На Фигуре 12B показана выживаемость мышей (BALB/c, 9 недель), у которых применяли или не применяли EC-18 (250 мг/кг/сутки ПО), с летальным облучением всего тела (10 Гр). Показатель выживаемости облученных мышей, которым вводили EC-18, улучшается примерно до 9-го дня по сравнению с не получавшими лечения (без ЕС-18) мышами согласно схеме на Фигуре 12А.
Фигура 13А представляет собой схематическую диаграмму лечения ЕС-18 (17 дней) на животной модели с радиационно-индуцированной острой лучевой болезнью. На Фигуре 13В показана эритема кожи у мышей (BALB/c, 9 недель), у которых применяли или не применяли EC-18 (250 мг/кг/сутки перорально), с летальным облучением всего тела (8 Гр).
Фигура 14А представляет собой схематическую диаграмму лечения ЕС-18 (30 дней) на животной модели с радиационно-индуцированной острой лучевой болезнью. На Фигуре 14В показана эритема кожи у мышей (BALB/c, 9 недель), у которых применяли или не применяли EC-18 (250 мг/кг/сутки перорально), с облучением всего тела (6,5 Гр).
На Фигуре 15 показана эритема кожи у мышей (BALB/c, 11 недель), у которых применяли или не применяли EC-18 (250 мг/кг/сутки перорально), с облучением всего тела (6,5 Гр) в соответствии со схемой на Фигуре 14А.
Фигура 16 иллюстрирует схематический механизм действия EC-18 при радиационно-индуцированной нейтропении (RIN), который показывает, что EC-18 способствует удалению DAMP (молекулярной структуры, ассоциированной с повреждениями) за короткий период времени, ингибируя непрерывное высвобождение нейтрофилов из кровеносных сосудов.
На Фигуре 17А изображена принципиальная схема тестирования влияния EC-18 на антиапоптоз в клетках HaCaT. 5-фторурацил и γ-облучение индуцировали повреждение HaCaT.
Фигура 17B показывает количество клеток в экспериментах, изображенных на Фигуре 17А в соответствии с дозой 5-фторурацила, облучением и лечением PLAG (EC-18).
Фигура 17C показывает частоту раннего апоптоза (%) в экспериментах, приведенных на Фигуре 17А в соответствии с количеством 5-фторурацила, облучением и лечением PLAG (EC-18).
На Фигуре 17D показана частота раннего апоптоза (%) в экспериментах, приведенных на Фигуре 17A, при количестве 5-фторурацила 1 нг/мл, в зависимости от облучения и лечения PLAG (EC-18).
Фигура 17E показывает частоту раннего апоптоза (%) в экспериментах, изображенных на Фигуре 17A, при количестве 5-фторурацила 10 нг/мл в зависимости от облучения и лечения PLAG (EC-18).
Фигура 17F показывает частоту раннего апоптоза (%) в экспериментах, изображенных на Фигуре 17A, при количестве 5-фторурацила 100 нг/мл, в зависимости от облучения и лечения PLAG (EC-18).
Фигура 18А показывает частоту позднего апоптоза (%) в экспериментах, изображенных на Фигуре 17А, в соответствии с количеством 5-фторурацила, облучением и лечением PLAG (EC-18).
Фигура 18B показывает частоту позднего апоптоза (%) в экспериментах, изображенных на Фигуре 17A, при количестве 5-фторурацила 1 нг/мл, в зависимости от облучения и лечения PLAG (EC-18).
Фигура 18С показывает частоту позднего апоптоза (%) в экспериментах, изображенных на Фигуре 17A, при количестве 5-фторурацила 10 нг/мл, в зависимости от облучения и лечения PLAG (EC-18).
Фигура 18B показывает частоту позднего апоптоза (%) в экспериментах, изображенных на Фигуре 17A, при количестве 5-фторурацила 100 нг/мл, в зависимости от облучения и лечения PLAG (EC-18).
Фигура 19А иллюстрирует схематическую диаграмму тестирования влияния EC-18 на внутриклеточную экспрессию АФК (активных форм кислорода) в клетках HaCaT при индуцированном 5-фторурацилом и γ-облучением повреждении HaCaT.
На Фигуре 19B показаны результаты измерения АФК в клетках HaCaT, подвергшихся воздействию 7 Гр γ-излучения после обработки EC-18 после облучения, или без обработки.
На Фигуре 19C показано количество клеток HaCaT против экспрессии АФК, когда клетки подвергали воздействию 7 Гр γ-излучения, после обработки EC-18 после облучения или без обработки (верхняя часть: 6 часов после воздействия, нижняя часть: 24 часа после воздействия).
Фигура 20 иллюстрирует диаграмму для примерного эксперимента по тестированию выживаемости при лечении EC-18 (PLAG) на животной модели острой лучевой болезни, вызванной γ-излучением, 6,5 Гр (мыши, BALB/c, 11 недель).
На Фигуре 21 показаны кривые выживаемости для мышей BALB/c (n = 20 на группу), не подвергавшихся общему облучению тела (контроль), подвергшихся воздействию 6,5 Гр γ-излучения (все тело; без лечения) и получавших EC-18, начиная с Дня 1 (через 24 часа после облучения) или Дня 2 (через 48 часов после облучения).
Фигура 22 показывает массу тела мышей BALB/c, которые не подвергались воздействию 6,5 Гр γ-излучения, и подвергались воздействию 6,5 Гр γ-излучения (всего тела), с лечением EC-18 через 24 или 48 часов после облучения.
Фигура 23 показывает примерную схему эксперимента по тестированию радиационно-индуцированной коагулопатии на мышиной модели.
Фигура 24 изображает диаграмму для примерного эксперимента, включающего три группы (только 5-фторурацил, 5-фторурацил + PLAG 125 мг/кг, 5-фторурацил + PLAG 250 мг/кг) для проверки временной кинетики нейтрофилов при нейтропении, индуцированной 5-фторурацилом (100 мг/кг).
Фигура 25 показывает эксперименты in vitro, сравнивающие эффекты EC-18 и Г-КСФ (гранулоцитарного колониестимулирующего фактора) посредством вестерн-блоттинга.
Фигура 26 является таблицей, иллюстрирующей сравнение EC-18 с Г-КСФ.
На Фигуре 27 представлена диаграмма времени выживания пациентов, подвергшихся облучению, и различные субсиндромы ОЛБ.
На Фигуре 28А показана выживаемость облученных мышей при каждой дозе γ-излучения в течение 30 дней наблюдения. На фигуре 28B показано соотношение дозы облучения (DRR) с использованием пробит-моделей мышей BALB/c, подвергшихся воздействию различных доз γ-излучения.
Фигура 29 показывает нормализованные изменения массы тела облученных мышей, подвергшихся воздействию различных доз γ-излучения.
На Фигурах 30A-B показан терапевтический эффект применения EC-18 при ОЛБ, индуцированной γ-излучением. Фигура 30А показывает выживаемость, а Фигура 30B показывает потерю массы тела облученных мышей с дозой 6,11 Гр γ-излучения. * р <0,05, ** р <0,01, *** р <0,005, для 6,11 Гр против 6,11 Гр + ЕС-18 250 мг/кг.
На Фигурах 31A-F показаны параметры общего анализа крови с подсчетом лейкоцитарной формулы у мышей, подвергшихся воздействию 6,11 Гр γ-излучения. На Фигурах 31A-31F показано количество лейкоцитов (WBC; Фигура 31A), абсолютное количество нейтрофилов (ANC; Фигура 31B), количество моноцитов (Фигура 31C), абсолютное количество лимфоцитов (ALC; Фигура 31D), количество тромбоцитов (PLT; Фигура 31E) и количество эритроцитов (RBC; Фигура 31 F), соответственно. n = 5 мышей на группу.
На Фигурах 32A-J показан терапевтический эффект введения EC-18 в отношении параметров общего анализа крови с лейкоцитарной формулой у мышей, подвергшихся воздействию 6,11 Гр γ-излучения. На Фигурах 32A-32E показана временная зависимость количества лейкоцитов (WBC; Фигура 32A), абсолютного количества нейтрофилов (ANC; Фигура 32B), абсолютного количества лимфоцитов (ALC; Фигура 32C), количества тромбоцитов (PLT; Фигура 32D) и количества эритроцитов (RBC; Фигура 32E) в течение 15 дней, соответственно. На Фигурах 32F-32J показано влияние дозы введения EC-18 на WBC (Фигура 32F), ANC (Фигура 32G), ALC (Фигура 32H), PLT (Фигура 32I) и RВC (Фигура 32J) в день 15, соответственно; ns – незначимые различия; *р<0,05, **р<0,01, ***р <0,005.
Подробное описание изобретения
I. Определения
Сокращения, используемые в настоящей заявке, имеют свое обычное значение в области химии и биологии. Химические структуры и формулы, изложенные в настоящей заявке, конструируются в соответствии со стандартными правилами химической валентности, известными в области химии.
Используемый в настоящей заявке термин «изомеры» относится к соединениям, имеющим одинаковое количество и тип атомов и, следовательно, одинаковую молекулярную массу, но различающимся в отношении структурного расположения или конфигурации атомов.
Используемый в настоящей заявке термин «таутомер» относится к одному из двух или более структурных изомеров, которые существуют в равновесии и которые легко превращаются из одной изомерной формы в другую.
Специалисту в данной области техники будет очевидно, что определенные соединения, раскрытые в данном документе, могут существовать в таутомерных формах, причем все такие таутомерные формы соединений входят в объем изобретения.
Термины единственного числа, используемые в данном документе, означают один или несколько элементов. Например, формы единственного числа предназначены также для включения форм множественного числа, если контекст явно не указывает на иное. Далее будет понятно, что термины «содержать», «включать», «иметь» и т.д. при использовании в данном описании определяют наличие заявленных признаков, областей, целых чисел, этапов, процессов, операций, элементов и/или компонентов, но не исключают наличия или добавления одного или нескольких других признаков, областей, целых чисел, этапов, процессов, операций, элементов, компонентов и/или их комбинаций.
«Фармацевтически приемлемое вспомогательное вещество» и «фармацевтически приемлемый носитель» относятся к веществу, которое способствует введению активного агента и его абсорбции субъектом и может быть включено в композиции по настоящему изобретению, не вызывая значительного неблагоприятного токсикологического воздействия на пациента. Неограничивающие примеры фармацевтически приемлемых вспомогательные вещества включают воду, NaCl, нормальные солевые растворы, раствор Рингера с лактатом, нормальную сахарозу, нормальную глюкозу, связующие вещества, наполнители, дезинтегранты, любриканты, покрытия, подсластители, ароматизаторы, солевые растворы (такие как раствор Рингера), спирты, масла, желатины, углеводы, такие как лактоза, амилоза или крахмал, сложные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон и красители, и тому подобное. Такие препараты могут быть стерилизованы, и при необходимости смешаны со вспомогательными агентами, такими как любриканты, консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, соли для воздействия на осмотическое давление, буферы, красители и/или ароматические вещества и тому подобное, которые не вызывают нежелательных реакций с соединениями по изобретению. Специалист в данной области техники поймет, что другие фармацевтические вспомогательные вещества полезны в настоящем изобретении.
«Лечение» и «лечение» в контексте настоящего описания включают профилактическое лечение. Методы лечения включают введение субъекту терапевтически эффективного количества активного агента. Стадия введения может состоять из одного введения или может включать серию введений. Продолжительность периода лечения зависит от множества факторов, таких как тяжесть состояния, возраст пациента, концентрация активного агента, активность композиций, используемых при лечении, или их комбинации. Также следует понимать, что эффективная дозировка агента, используемого для лечения или профилактики, может увеличиваться или уменьшаться в течение определенного режима лечения или профилактики. Изменения в дозировке могут возникать и проявляться в стандартных диагностических анализах, известных в данной области техники. В некоторых случаях может потребоваться хроническое введение. Например, композиции вводят субъекту в количестве и в течение периода времени, достаточного для лечения пациента. Термин «лечение» и его сочетания могут включать профилактику повреждения, патологии, состояния или заболевания. В некоторых вариантах осуществления лечение является профилактикой. В некоторых вариантах осуществления лечение не включает профилактику.
Термин «предотвращать» относится к уменьшению появления симптомов заболевания у пациента. Как указано выше, профилактика может быть полной (например, без обнаруживаемых симптомов) или частичной, так что наблюдается меньше симптомов, чем при отсутствии лечения.
Термин «модулировать» используется в соответствии с его простым обычным значением и относится к акту изменения или вариации одного или нескольких свойств. «Модуляция» относится к процессу изменения или вариации одного или нескольких свойств. Например, модулятор заболевания уменьшает симптом, причину или характеристику целевого заболевания, такого как ОЛБ и её субсиндромы.
Термины «пациент», «субъект», «пациент, нуждающийся в этом» и «субъект, нуждающийся в этом» в настоящей заявке используются взаимозаменяемо и относятся к живому организму, страдающему или подверженному заболеванию или состоянию, которое можно лечить путем введения фармацевтической композиции, представленной в настоящей заявке. Неограничивающие примеры включают людей, других млекопитающих, крупный рогатый скот, крыс, мышей, собак, обезьян, коз, овец, коров, оленей и других животных, не являющихся млекопитающими. В некоторых вариантах осуществления пациентом или субъектом является человек.
«Эффективное количество» представляет собой количество, достаточное для того, чтобы соединение достигло поставленной цели по сравнению с отсутствием соединения (например, для достижения эффекта, для которого оно вводится, лечения заболевания, снижения активности фермента, увеличения активности фермента, снижения катаболической активности фермента, или уменьшения одного или нескольких симптомов заболевания или состояния). Примером «эффективного количества» является количество, достаточное для содействия лечению, профилактике или уменьшению симптома или симптомов заболевания, которое также можно назвать «терапевтически эффективным количеством». «Уменьшение» симптома или симптомов (и грамматических эквивалентов этой фразы) означает уменьшение тяжести или частоты симптома (симптомов) или устранение симптома (симптомов). «Профилактически эффективное количество» лекарственного средства представляет собой количество лекарственного средства, которое при введении субъекту будет оказывать намеченный профилактический эффект, например, предотвращать или задерживать начало (или повторное возникновение) повреждения, заболевания, патологии или состояния, или снижать вероятность возникновения (или повторного развития) повреждения, заболевания, патологии или состояния, или их симптомов. Полный профилактический эффект не обязательно проявляется при введении одной дозы, и может проявляться только после введения серии доз. Таким образом, профилактически эффективное количество может применяться посредством одного или нескольких введений. «Количество, снижающее активность», как используется в настоящей заявке, относится к количеству антагониста, необходимому для снижения активности фермента по сравнению с отсутствием антагониста. Термин «количество, нарушающее функцию», как он используется в настоящей заявке, относится к количеству антагониста, необходимому для нарушения функции фермента или белка, по сравнению с отсутствием антагониста. Точные количества будут зависеть от цели лечения и будут определены специалистом в данной области техники с использованием известных методов (см., например, Lieberman, Pharmaceutical Dosage Forms (vols. 1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); and Remington: The Science and Practice of Pharmacy, 20th Edition, 2003, Gennaro, Ed., Lippincott, Williams & Wilkins).
Терапевтически эффективное количество PLAG может быть первоначально определено из анализов клеточных культур. Целевые концентрации будут представлять собой такие концентрации активного соединения (соединений), которые способны обеспечить способы, описанные в настоящей заявке, при измерении с использованием способов, описанных в настоящей заявке или известных в данной области техники.
Как хорошо известно в данной области техники, терапевтически эффективные количества для применения у людей также могут быть определены на животных моделях. Например, доза для людей может быть составлена для достижения концентрации, которая, как было установлено, является эффективной для животных. Дозировка у людей может быть отрегулирована путем мониторинга эффективности соединений и корректировки дозировки в сторону повышения или понижения, как описано выше. Регулирование дозы для достижения максимальной эффективности у людей на основе способов, описанных выше, и других способов находится в пределах возможностей обычного специалиста в данной области техники.
Используемый в настоящей заявке термин «терапевтически эффективное количество» или «эффективное количество» относится к количеству терапевтического средства, достаточному для ослабления расстройства, как описано выше. Например, для данного параметра терапевтически эффективное количество будет демонстрировать увеличение или уменьшение по меньшей мере на 5%, 10%, 15%, 20%, 25%, 40%, 50%, 60%, 75%, 80%. 90% или не менее 100%. Терапевтическая эффективность также может быть выражена как «кратное» увеличение или уменьшение. Например, терапевтически эффективное количество может оказывать по меньшей мере 1,2-кратное, 1,5-кратное, 2-кратное, 5-кратное или более влияние, по сравнению с контролем.
Дозировки могут варьировать в зависимости от потребностей пациента и используемого соединения. Доза, вводимая пациенту, в контексте настоящего изобретения должна быть достаточной для осуществления положительного терапевтического ответа у пациента с течением времени. Размер дозы также будет определяться наличием, характером и степенью любых побочных эффектов. Определение правильной дозировки для конкретной ситуации находится в компетенции практикующего врача. Как правило, лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. После этого дозировку увеличивают небольшими приращениями до достижения оптимального эффекта при определенных обстоятельствах. Количества и интервалы дозировки можно регулировать индивидуально, чтобы обеспечить уровни вводимого соединения, эффективные для конкретного клинического показания, подлежащего лечению. Это обеспечит терапевтический режим, соразмерный тяжести патологического состояния человека.
С использованием представленных в настоящей заявке принципов можно спланировать эффективную схему профилактического или терапевтического лечения, которая не вызывает существенной токсичности и в то же время эффективна для лечения клинических симптомов, демонстрируемых конкретным пациентом. Это планирование должно включать тщательный выбор активного соединения с учетом таких факторов, как активность соединения, относительная биодоступность, масса тела пациента, наличие и тяжесть побочных эффектов, предпочтительный способ введения и профиль токсичности выбранного агента.
«Фармацевтически приемлемое вспомогательное вещество» и «фармацевтически приемлемый носитель» относятся к веществу, которое способствует введению активного агента и его абсорбции субъектом и может быть включено в композиции по настоящему изобретению, не вызывая значительного неблагоприятного токсикологического воздействия на пациента. Неограничивающие примеры фармацевтически приемлемых вспомогательных веществ включают воду, NaCl, нормальные солевые растворы, раствор Рингера с лактатом, нормальную сахарозу, нормальную глюкозу, связующие вещества, наполнители, дезинтегранты, любриканты, покрытия, подсластители, ароматизаторы, солевые растворы (такие как раствор Рингера), спирты, масла, желатины, углеводы, такие как лактоза, амилоза или крахмал, сложные эфиры жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон и красители, и тому подобное. Такие препараты могут быть стерилизованы, и при необходимости смешаны со вспомогательными агентами, такими как любриканты, консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, соли для воздействия на осмотическое давление, буферы, красители и/или ароматические вещества и тому подобное, которые не вызывают нежелательных реакций с соединениями по изобретению. Специалист в данной области техники поймет, что другие фармацевтические вспомогательные вещества могут применяться в настоящем изобретении.
Термин «препарат» предназначен для включения композиции активного соединения с инкапсулирующим материалом в качестве носителя, обеспечивающего капсулу, в которой активный компонент с другими носителями или без них окружен носителем, который, таким образом, связан с ним. Точно так же, включены облатки и пастилки. Таблетки, порошки, капсулы, пилюли, облатки и пастилки могут быть использованы в качестве твердых лекарственных форм, подходящих для перорального введения.
Используемый в настоящей заявке термин «введение» означает пероральное введение, введение в виде суппозитория, местное применение, внутривенное, интраперитонеальное, внутримышечное, внутриочаговое, интратекальное, интраназальное или подкожное введение, или имплантацию устройства с медленным высвобождением, например, мини-осмотического насоса, субъекту. Введение любым путем, включая парентеральное и трансмукозальное (например, буккальное, сублингвальное, небное, десневое, назальное, вагинальное, ректальное или трансдермальное), пригодно для препарата. Парентеральное введение включает, например, внутривенное, внутримышечное, внутриартериальное, внутрикожное, подкожное, интраперитонеальное, интравентрикулярное и интракраниальное. Другие способы доставки включают использование липосомальных составов, внутривенное вливание, трансдермальные пластыри и т.д., но не ограничиваются ими.
Композиции, раскрытые в настоящей заявке, могут быть доставлены трансдермально, местным путем, в виде палочек для аппликатора, растворов, суспензий, эмульсий, гелей, кремов, мазей, паст, желе, красок, порошков и аэрозолей. Пероральные препараты включают таблетки, пилюли, порошок, драже, капсулы, жидкости, пастилки, облатки, гели, сиропы, кашицы, суспензии и т.д., пригодные для приема внутрь пациентом. Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Жидкие формы препаратов включают растворы, суспензии и эмульсии, например, растворы в воде или в воде/пропиленгликоле. Композиции по настоящему изобретению могут дополнительно включать компоненты для обеспечения замедленного высвобождения и/или комфорта. Такие компоненты включают высокомолекулярные анионные мукомиметические полимеры, гелеобразующие полисахариды и тонкоизмельченные субстраты лекарственного носителя. Эти компоненты более подробно обсуждаются в патентах США №№ 4911920; 5403841; 5212162; и 4,861,760. Все содержание этих патентов включено в настоящий документ посредством ссылки во всей полноте для всех целей. Композиции, раскрытые в настоящей заявке, также могут быть доставлены в виде микросфер для замедленного высвобождения в организме. Например, микросферы могут быть введены посредством внутрикожной инъекции содержащих лекарственное средство микросфер, которые медленно высвобождаются подкожно (см. Rao, J. Biomater Sci. Polym. Ed. 7: 623-645, 1995); в виде биодеградируемых и инъецируемых гелевых составов (см., например, Gao Pharm. Res. 12: 857-863, 1995) или в качестве микросфер для перорального введения (см., например, Eyles, J. Pharm. Pharmacol. 49: 669-674, 1997). В другом варианте осуществления составы композиций по настоящему изобретению могут быть доставлены с использованием липосом, которые сливаются с клеточной мембраной или способны подвергаться эндоцитозу, то есть с использованием рецепторных лигандов, прикрепленных к липосоме, которые связываются с поверхностными мембранными белковыми рецепторами клетки, что приводит к эндоцитозу. Используя липосомы, особенно когда поверхность липосом несет рецепторные лиганды, специфичные для клеток-мишеней или иным образом предпочтительно направленные на конкретный орган, можно направить доставку композиций по настоящему изобретению в клетки-мишени в in vivo. (См., например, Al-Muhammed, J. Microencapsul. 13:293-306, 1996; Chonn, Curr. Opin. Biotechnol. 6:698-708, 1995; Ostro, Am. J. Hosp. Pharm. 46:1576-1587, 1989). Композиции также могут быть доставлены в виде наночастиц.
Фармацевтические композиции могут включать композиции, в которых соединение PLAG содержится в терапевтически эффективном количестве, то есть в количестве, эффективном для достижения его предполагаемой цели. Фактическое количество, эффективное для конкретного применения, будет зависеть, среди прочего, от состояния, подлежащего лечению. При введении способами для лечения заболевания такие композиции будут содержать количество активного ингредиента, эффективное для достижения необходимого результата, например, модуляции активности молекулы-мишени и/или уменьшения, устранения или замедления прогрессирования симптомов заболевания.
Дозировка и частота (однократные или многократные дозы) применения млекопитающему может варьировать в зависимости от множества факторов, например, от того, страдает ли млекопитающее другим заболеванием, и от способа его введения; роста, возраста, пола, состояния здоровья, массы тела, индекса массы тела и рациона пациента; характера и степени симптомов заболевания, подлежащего лечению, вида сопутствующего лечения, осложнений от заболевания, которое лечат, или других связанных со здоровьем проблем. Другие терапевтические схемы или агенты могут быть использованы в сочетании со способами и соединениями настоящего изобретения. Регулирование и манипулирование установленными дозами (например, частотой и продолжительностью) находятся в пределах компетенции специалистов в данной области техники.
«Болезнь», «расстройство» или «состояние» относятся к самочувствию или состоянию здоровья пациента или субъекта, которые могут получать лечение с помощью соединений или способов, представленных в настоящей заявке. Используемый в настоящей заявке термин «острая лучевая болезнь» или «ОЛБ» относится к заболеванию или расстройству, связанному с лучевой токсичностью или лучевым заболеванием (например, острой лучевой болезнью (ОЛБ), включая гемопоэтический (костномозговой) острый лучевой синдром, желудочно-кишечный острый лучевой синдром, кожный острый лучевой синдром, сердечно-сосудистый острый лучевой синдром и/или острый лучевой синдром центральной нервной системы (ЦНС). Термин «острая лучевая болезнь» или «ОЛБ» также может включать радиационно-индуцированную коагулопатию. В некоторых вариантах осуществления заболевание представляет собой острую лучевую болезнь (ОЛБ). В некоторых вариантах осуществления ОЛБ возникает у субъекта при воздействии излучения примерно 0,1 Гр (или 10 рад) или более, примерно 0,2 Гр (или 20 рад) или более, примерно 0,3 Гр (или 30 рад) или более, примерно 0,4 Гр (или 40 рад) или более, примерно 0,5 Гр (или 50 рад) или более, примерно 0,6 Гр (или 60 рад) или более, примерно 0,7 Гр (или 70 рад) или более, примерно 0,8 Гр (или 80 рад) или более, примерно 0,9 Гр (или 90 рад) или более, примерно 1 Гр (или 100 рад) или более, примерно 2,0 Гр (или 200 рад), примерно 3,0 Гр (или 300 рад) или примерно 4,0 Гр (или 400 рад) или более.
В некоторых вариантах осуществления ОЛБ может возникать у субъекта при воздействии радиации (включая гамма-излучение) примерно от 1 Гр (или 100 рад) до 8 Гр (или 800 рад) в течение любых изменяющихся периодов времени, таких как по меньшей мере 1, 2, 5, 10, 30, 60, 80, 120, 180, 240 или 300 секунд.
В некоторых вариантах осуществления способы и композиции, раскрытые в настоящей заявке, для лечения субъекта, который подвергался воздействию ионизирующего излучения (в частности, подвергался чрезмерному воздействию ионизирующего излучения, включая гамма-излучение), могут включать использование или введение субъекту эффективного количества PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина) подходящим образом, когда субъект подвергается облучению примерно от 1 Гр (или 100 рад) до 8 Гр (или 800 рад) или более для любых изменяющихся периодов времени, таких как по меньшей мере 1, 2, 5, 10, 30, 60, 80, 120, 180, 240 или 300 секунд.
В некоторых вариантах осуществления заболевание или расстройство включает кожный радиационный синдром, такой как повреждения кожи, эритема, измененное ощущение, зуд, отек, образование пузырей, шелушение, язвы, некроз, выпадение волос, онихолиз и тому подобное. В некоторых вариантах осуществления заболевание или расстройство включает уменьшение нейтрофилов или инфекцию, вызванную снижением количества лейкоцитов. В некоторых вариантах осуществления заболевание или расстройство включает кровоизлияние. В некоторых вариантах осуществления заболевание или расстройство включает диарею. В некоторых вариантах осуществления заболевание или расстройство включает дегидратацию или дисбаланс электролитов. В некоторых вариантах осуществления заболевание или расстройство включает судороги и/или кому.
Как обсуждалось, в некоторых вариантах осуществления способы и композиции, раскрытые в настоящей заявке, используются для лечения субъекта, подвергшегося воздействию ионизирующего излучения. В некоторых аспектах облучение может быть непреднамеренным или случайным. В дополнительных аспектах радиационное излучение не будет использоваться в терапевтических целях, например, обычное излучение не будет радиотерапией, которую можно использовать для лечения рака или другой терапии. В дополнительных аспектах по меньшей мере существенная часть (например, вся конечность и/или туловище и/или вся область головы) субъекта может подвергаться воздействию излучения.
Как указано в настоящем документе, комбинация и любое одно из таких непреднамеренных, случайных, нетерапевтических и/или значительных воздействий на часть тела может быть названо «неблагоприятным» облучением.
II. Композиции
Как обсуждалось, один аспект настоящего изобретения обеспечивает терапевтическую фармацевтическую композицию для профилактики или лечения острой лучевой болезни, содержащую PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) (также называемый в настоящей заявке как EC-18) в качестве активного ингредиента.
Как упоминается в настоящей заявке, термин «острая лучевая болезнь» представляет собой острое заболевание, вызванное радиационным воздействием на все тело или на значительную часть тела; острая лучевая болезнь также известна как радиационная токсичность или лучевой синдром. Излучение - это высвобождаемая энергия, когда нестабильное ядро превращается в другое ядро. Когда излучение проходит через тело, энергия излучения поглощается и может вызвать ионизацию в ткани. В это время H2O ионизируется и может трансформировать ДНК субъекта.
Клинические характеристики острой лучевой болезни включают гемопоэтический синдром, желудочно-кишечный синдром, сердечно-сосудистый синдром и нейроваскулярный синдром.
Как описано выше, острая лучевая болезнь может повреждать чувствительные к излучению системы, такие как иммунная, кроветворная и желудочно-кишечная системы, и привести к смерти. Следовательно, наиболее важным фактором, который следует учитывать для профилактического или терапевтического воздействия на острую лучевую болеззнь, является увеличение выживаемости субъектов, подвергшихся облучению. В настоящем изобретении острая лучевая болезнь может представлять собой любой из симптомов гемопоэтического синдрома, желудочно-кишечного синдрома, сердечно-сосудистого синдрома и нейроваскулярного синдрома, но в то же время острая лучевая болезнь не ограничивается вышеуказанными симптомами. Композиция по данному изобретению эффективна в профилактике, лечении или облегчении вышеуказанных симптомов и в конечном счете улучшает выживаемость субъектов, подвергшихся воздействию излучения.
Острая лучевая болезнь обычно прогрессирует с четырьмя клиническими стадиями: продромальная фаза, латентная фаза, манифестная фаза, и выздоровление или смерть. В зависимости от количества поглощенного излучения симптомы могут появиться в течение нескольких часов или недель. Продромальная фаза обычно начинается в течение 48 часов после облучения, но может длиться до 6 дней после облучения, и симптомы могут включать тошноту, рвоту, усталость, расстройство вегетативной нервной системы и потерю сознания. Латентная фаза может длиться от нескольких дней до нескольких недель в зависимости от степени радиационного воздействия, и клинические симптомы могут появляться не полностью или частично. Однако на этой стадии могут возникнуть такие симптомы, как лимфоцитопения, гранулоцитопения и миелогенный дефицит. Симптомы манифестной фазы могут возникать с задержкой в несколько недель. Симптомы манифестной фазы могут включать гемопоэтический синдром, желудочно-кишечный синдром, сердечно-сосудистый синдром и нейроваскулярный синдром, в зависимости от степени радиационного воздействия. Пациенты, подвергающиеся воздействию излучения, могут пройти все четыре этапа в течение нескольких часов и умереть в течение короткого периода времени.
Среди острой лучевой болезни может быть выделен гемопоэтический синдром, вызванный, индуцированный или обусловленный дозой облучения примерно 0,7-10 Гр, желудочно-кишечный синдром, вызванный, индуцированный или обусловленный дозой облучения примерно 10-30 Гр, и сердечно-сосудистый/ нейроваскулярный синдром, вызванный, индуцированный или обусловленный дозой облучения примерно 50 Гр. В примерных вариантах осуществления настоящего изобретения острый лучевой синдром включает синдромы, вызванные, индуцированные или обусловленные дозой облучения примерно от 0,1 до 100 Гр, примерно от 0,1 до 80 Гр, примерно 0,1 до 70 Гр, примерно от 0,1 до 60 Гр, примерно от 0,1 до 50 Гр или примерно 0,7 или 50 Гр, и может быть вызван, индуцирован или обусловлен дозой облучения примерно 0,1 Гр, примерно 0,5 Гр, примерно 0,7 Гр, примерно 1,0 Гр, примерно 2,0 Гр, примерно 3,0 Гр, или примерно 4,0 Гр или более, что может привести к смерти у особо облученного субъекта (Reports of Practical Oncology and Radiotherapy, 2011, 16 (4): 123-130), но не ограничивается этим.
В дополнительных примерных вариантах осуществления острая лучевая болезнь включает синдромы, вызванные, индуцированные или обусловленные дозой облучения примерно от 1 Гр (или 100 рад) до 8 Гр (или 800 рад) в течение любых различных периодов времени, таких как по меньшей мере 1, 2, 5, 10, 30, 60, 80, 120, 180, 240 или 300 секунд.
Используемый в настоящей заявке термин «профилактика» означает любое действие, которое ингибирует или задерживает возникновение, распространение или повторное развитие определенного расстройства или заболевания, такого как острая лучевая болезнь, при введении соединения или композиции, как описано в настоящей заявке. Используемый в настоящей заявке термин «лечение» означает любое действие, которое улучшает или облегчает симптомы определенного расстройства или заболевания, такого как острая лучевая болезнь, после введения соединения или композиции, как описано в настоящей заявке.
В определенных аспектах соединение, вводимое субъекту, имеет следующую Формулу 1:
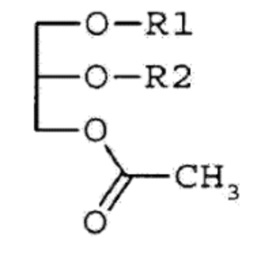 Формула 1,
Формула 1,
где R1 и R2 независимо представляют собой жирнокислотный остаток с 14-20 атомами углерода.
Глицериновые производные Формулы I, приведенной выше, иногда называют в настоящей заявке моноацетилдиацилглицеринами (МДАГ). Остаток жирной кислоты относится к ацильной части, возникающей в результате образования сложноэфирной связи при реакции жирной кислоты и спирта. Неограничивающие примеры R1 и R2, таким образом, включают пальмитоил, олеоил, линолеоил, линоленоил, стеароил, миристоил, арахидоноил и так далее. Предпочтительные комбинации R1 и R2 (R1/ R2) включают олеоил/пальмитоил, пальмитоил/олеоил, пальмитоил/линолеоил, пальмитоил/линоленоил, пальмитоил/арахидоноил, пальмитоил/стеароил, пальмитоил/пальмитоил, олеоил/стеароил, линолеоил/пальмитоил, линолеоил/стеароил, стеароил/линолеоил, стеароил/олеоил, миристоил/линолеоил, миристоил/олеоил и так далее. В оптической активности производные моноацетилдиацилглицерина Формулы 1 могут быть (R)-формой, (S)-формой или рацемической смесью, и могут включать их стереоизомеры. В определенных аспектах в соединениях, где заместители R1 и/или R2 представляют собой остатки ненасыщенной жирной кислоты, одна или несколько двойных связей, которые присутствуют подходящим образом, могут иметь цис-конфигурацию. В других аспектах одна или несколько двойных связей заместителей R1 и/или R2 могут присутствовать в транс-конфигурации. В определенных аспектах такие одна или несколько двойных связей будут присутствовать только в цис-конфигурации.
В определенном предпочтительном аспекте соединение, применяемое у субъекта, представляет собой PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин), имеющий структуру следующей химической Формулы 2 (соединение, которое также иногда обозначают в настоящей заявке как ЕС-18):
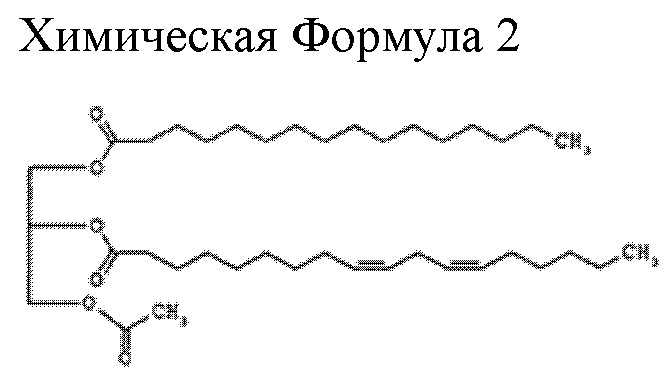
Кроме того, в настоящем изобретении PLAG может включать все вышеупомянутые соединения Химической Формулы 2 и другие очевидные производные из известных в отрасли химических превращений. Например, реакции добавления и замещения для повышения стабильности соединения или для приготовления соединения могут проводиться в диапазоне, не влияющем на фармакологический эффект PLAG, и все эти химические производные включены в объем данного изобретения.
Известно, что PLAG оказывает терапевтическое воздействие на нейтропению, тромбоцитопению и мукозит, вызванные противораковой химиотерапией. Тем не менее, об эффективности PLAG для профилактики или лечения острой лучевой болезни, в том числе о способности к повышению выживаемости людей с острой лучевой болезнью, не сообщается.
В примерном варианте осуществления было обнаружено, что PLAG значительно увеличивает количество лейкоцитов, нейтрофилов и лимфоцитов в крови субъектов с радиационным воздействием (Таблица 1 и Фигура 2) и улучшает мукозит на модели исследования на животных с тяжелым острой лучевой болезнью, где мукозит полости рта был вызван химиолучевой терапией (Фигуры 4 и 5) и повышает число нейтрофилов в крови (Фигура 6), и в результате PLAG значительно увеличивает выживаемость субъектов исследования (Фигура 3). Следовательно, композиция по настоящему изобретению имеет превосходный эффект в качестве терапевтической фармацевтической композиции для профилактики или лечения острой лучевой болезни.
Терапевтическая фармацевтическая композиция по настоящему изобретению может быть применена в течение 1 часа, в течение 2 часов, в течение 3 часов, в течение 4 часов, в течение 5 часов, в течение 6 часов, в течение 7 часов, в течение 8 часов, в течение 9 часов, в течение 10 часов в течение 12 часов, в течение 16 часов, в течение 20 часов, в течение 30 часов, в течение 40 часов или в течение 48 часов после облучения, но не ограничиваясь этим, как обсуждалось выше.
Кроме того, вышеуказанная композиция может быть применена в виде отдельного терапевтического агента, или может быть применена в комбинации с другим лекарственным средством, которое, как известно, обладает эффективностью при лечении острой лучевой болезни. Например, вышеуказанную композицию можно применять с одним или несколькими терапевтическими агентами, включая белки, низкомолекулярные лекарственные средства, нуклеиновые кислоты или тому подобное. Например, композицию можно применять с терапевтическим агентом, включающим гранулоцитарный колониестимулирующий фактор (Г-КСФ), но применение этим не ограничивается. Кроме того, вышеуказанную композицию можно применять вместе с анальгетиками, противоязвенными средствами, антидиарейными средствами, антибиотиками, жаропонижающими средствами, пищевыми добавками и антиоксидантами, которые могут помочь в профилактике или лечении острой лучевой болезни.
Термин «применение» в настоящем изобретении означает введение терапевтической фармацевтической композиции по настоящему изобретению пациенту любым подходящим способом, и путь применения композиции по настоящему изобретению может быть осуществлен различными способами, как перорально, так и не-перорально. Терапевтическая фармацевтическая композиция по настоящему изобретению может быть изготовлена в виде различных композиций в зависимости от способов применения.
Частота применения композиции по настоящему изобретению конкретно не ограничена, но ее можно применять один раз в сутки или несколько раз в сутки в разделенных дозировках.
Терапевтическая фармацевтическая композиция по настоящему изобретению может быть использована в качестве одного лекарственного средства и может быть использована в качестве комбинированного лекарственного средства, содержащего другое лекарственное средство, и может быть приготовлена с использованием фармацевтически приемлемого носителя, наполнителя или разбавителя для получения единичной дозы или контейнера с множеством доз.
Термин «фармацевтическая композиция» в контексте настоящего описания обозначает композицию, приготовленную с целью профилактики или лечения заболеваний, и которая может быть составлена в различных формах в соответствии с обычными способами. Например, она может быть составлена в виде композиций для перорального введения, таких как порошки, гранулы, таблетки, капсулы, суспензии, эмульсии и сиропы, и может быть составлена в форме для наружного применения, в виде суппозиториев и стерилизованных растворов для инъекций.
Кроме того, фармацевтическая композиция по настоящему изобретению может быть изготовлена с дополнительным фармацевтически приемлемым носителем для каждого состава. Используемый в настоящей заявке термин «фармацевтически приемлемый носитель» может относиться к носителю или разбавителю, который не стимулирует организм и не ингибирует биологическую активность и характеристику вводимого соединения. Тип носителя, который можно использовать в настоящем изобретении, конкретно не ограничен, может быть использован любой носитель, обычно применяемый в области промышленности и фармацевтически приемлемый.
Солевой раствор, стерилизованная вода, жидкости для внутривенного введения, буферный солевой раствор, раствор альбумина для инъекций, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол являются неограничивающими примерами используемых носителей. Эти носители могут использоваться отдельно или в комбинации из двух или более из них. Носитель может включать ненатуральный носитель. При необходимости могут быть добавлены и использованы другие обычно применяемые добавки, такие как антиоксидант, буфер и/или бактериостатический агент. Носитель может быть составлен из разбавителя, диспергатора, поверхностно-активного вещества, связующего вещества, любриканта для приготовления инъекционного раствора, такого как водный раствор, суспензия, эмульсия, и пилюль, капсул, гранул или таблеток и тому подобного.
Кроме того, фармацевтическая композиция по настоящему изобретению может содержать фармацевтически эффективное количество PLAG. Термин «фармацевтически эффективное количество» в настоящем изобретении означает количество, достаточное для лечения заболевания с разумным соотношением пользы или риска, применимого к медицинскому лечению, и обычно находится в диапазоне от около 0,001 до 5000 мг/кг, предпочтительно примерно от 0,05 до 1000 мг/кг, которое можно вводить один раз в сутки или несколько раз в сутки в разделенных дозировках. Однако для целей настоящего изобретения конкретное терапевтически эффективное количество для конкретного пациента будет зависеть от природы и степени реакции, которая должна быть достигнута, конкретной композиции, включая использование других агентов, возраст, массу тела, пол и диету пациента, время введения, путь введения и пропорцию композиции, продолжительность лечения, лекарства, применяемые или вводимые совместно с конкретной композицией, и аналогичные соединения, хорошо известные в медицинской промышленности.
Как уже говорилось, также обеспечиваются наборы. Например, в этом аспекте соединение PLAG подходящим образом может быть упаковано в подходящие контейнеры, маркированные, например, для использования в качестве терапии для лечения субъекта, страдающего острой лучевой болезнью или ее субсиндромом, или подвергшегося чрезмерному воздействию ионизирующей радиации (например, гамма-излучения). Контейнеры могут включать соединение или композицию PLAG и одно или более из подходящего стабилизатора, молекулы-носителя и/или тому подобного, в зависимости от предполагаемого использования. В других вариантах осуществления набор дополнительно содержит один или несколько терапевтических агентов, которые ослабляют некоторые из симптомов или вторичных инфекций или расстройств, которые могут быть связаны с острой лучевой болезнью или ее субсиндромом, или с воздействием чрезмерного ионизирующего излучения (например, гамма-излучения). Соответственно, упакованные продукты (например, стерильные контейнеры, содержащие одну или несколько композиций, описанных в настоящей заявке, и упакованные для хранения, отгрузки или продажи в концентрированном виде или готовых к употреблению концентрациях) и наборы, включающие соединение PLAG, и инструкции по применению, также входят в объем изобретения. Продукт может включать контейнер (например, флакон, банку, бутылку, пакет или тому подобное), содержащий соединение или композицию PLAG. Кроме того, изделие или набор дополнительно могут включать, например, упаковочные материалы, инструкции по применению, шприцы, устройства для доставки для лечения или мониторинга состояния, для которого требуется профилактика или лечение.
Продукт также может содержать сопроводительную информацию (например, напечатанную этикетку или вкладыш, или другой носитель, описывающий использование продукта (например, аудио- или видеокассету)). Сопроводительная информация может быть связана с контейнером (например, прикреплена к контейнеру) и может описывать способ, которым композиции должны применяться (например, частоту и способ введения), указания и другие виды применения. Композиции могут быть готовы к введению (например, присутствовать в соответствующих единицах дозы) и могут включать один или несколько дополнительных фармацевтически приемлемых адъювантов, носителей или других разбавителей и/или дополнительный терапевтический агент. Альтернативно, композиции, например, могут быть представлены в концентрированной форме с разбавителем и инструкциями для разбавления.
Другим аспектом настоящего изобретения является функциональная пищевая композиция для здоровья, предназначенная для профилактики или ослабления острой лучевой болезни, содержащая PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) в качестве активного ингредиента.
В настоящем изобретении термин «улучшение» означает все действия, которые по меньшей мере уменьшают степень симптома, связанного с состоянием, подвергаемым лечению. При этом пищевая оздоровительная функциональная композиция может быть использована одновременно или отдельно с лекарственным средством для лечения до или после возникновения заболевания для профилактики или облегчения острой лучевой болезни.
PLAG не проявляет заметной токсичности для клеток и оказывает улучшающее действие на острый лучевой синдром, поэтому PLAG можно готовить и принимать в форме пищевой оздоровительной функциональной композиции.
Функциональное питание - это тот же термин, что и питание для специального оздоровительного применения (FoSHU). Это относится к продуктам, которые были обработаны таким образом, что функция биологического контроля оказывается более эффективной в дополнение к пищевой ценности. Пища может быть приготовлена в различных формах, таких как таблетки, капсулы, порошки, гранулы, жидкости, кольца и тому подобное, для достижения полезного влияния в отношении регенерации кожи.
Для этого уровень содержания PLAG, содержащегося в оздоровительной функциональной пище, конкретно не ограничен, но может составлять от 0,01 до 100 масс.%, в частности, от 1 до 80 масс.% в расчете на общую массу оздоровительной функциональной пищи.
Оздоровительная функциональная пищевая композиция по настоящему изобретению может также содержать фармацевтически приемлемый носитель.
Не существует особых ограничений в отношении видов оздоровительных функциональных пищевых продуктов, включая PLAG по настоящему изобретению, и их примеры включают напитки, жевательную резинку, чай, витаминный комплекс, оздоровительные пищевые добавки и тому подобное. К пище могут быть добавлены другие ингредиенты, которые не влияют на эффект облегчения острой лучевой болезни, и их вид не ограничивается конкретно. Например, различные растительные экстракты, диетически приемлемые пищевые добавки или другие натуральные углеводы могут быть добавлены в качестве дополнительного ингредиента.
Пищевую добавку, описанную выше, добавляют для получения оздоровительной функциональной пищи каждого состава, и она может быть подходящим образом выбрана и использована специалистом в соответствующей области техники. Например, могут применяться различные питательные добавки, витамины, минералы (электролиты), синтетические и натуральные ароматизаторы, красители и наполнители, пектиновая кислота и ее соли, альгиновая кислота и ее соли, органические кислоты, защитные коллоидные загустители, регулятор pH, стабилизатор, консервант, глицерин, спирт, газообразующий агент, используемый в газированном напитке, и тому подобное, но их виды не ограничены вышеперечисленным.
Кроме того, описанная выше оздоровительная функциональная пища может содержать дополнительные ингредиенты, которые обычно используются в еде для улучшения запаха, вкуса, внешнего вида и тому подобного. Например, могут быть включены витамины А, С, D, Е, В1, В2, В6, В12, ниацин, биотин, фолат, пантотеновая кислота и тому подобное. Кроме того, пища может включать минералы, такие как цинк (Zn), железо (Fe), кальций (Ca), хром (Cr), магний (Mg), марганец (Mn) и медь (Cu), и тому подобное. Она также может содержать аминокислоты, такие как лизин, триптофан, цистеин, валин и тому подобное.
Кроме того, описанная оздоровительная функциональная пища может включать один или несколько консервантов (таких как сорбат калия, бензоат натрия, салициловая кислота и дегидроацетат натрия), бактерицидные средства (такие как отбеливающий порошок и высокоактивный отбеливающий порошок, гипохлорит натрия), антиоксиданты (бутилгидроксианилид (БГА), бутилгидрокситолуол (БГТ) и т.д.), красители (такие как анилиновые пигменты), цветообразующие агенты (такие как нитрит натрия и ацетаты натрия), отбеливающие вещества (сульфит натрия), приправы (такие как MSG, глутамат натрия), подсластители (такие как дульцин, цикламат, сахарин натрия), ароматизаторы (ванилин, лактоны и т.д.), агенты, вызывающие набухание (квасцы, калия гидро-D-тартрат), обогатитель, эмульгаторы, загустители, инкапсулирующие агенты, гуммиосновы, ингибиторы пенообразования, растворитель, улучшитель и тому подобное. Вышеуказанные добавки выбирают в зависимости от типа пищи и используют в соответствующем количестве.
Оздоровительная функциональная пищевая композиция по настоящему изобретению может быть получена способом, обычно используемым в данной области техники, и может быть приготовлена путем добавления сырья и ингредиентов, которые обычно используются в промышленности. Кроме того, в отличие от обычного медикамента, оздоровительная функциональная пища может иметь преимущество, например, поскольку не может быть никакого побочного эффекта при длительном употреблении, и отмечается лучшая переносимость.
Фармацевтическая композиция может быть приготовлена и введена в широком разнообразии лекарственных форм. Описанные соединения могут быть применены перорально, ректально или посредством инъекций (например, внутривенно, внутримышечно, внутрикожно, подкожно, интрадуоденально или интраперитонеально).
Для приготовления фармацевтических композиций из соединений, описанных в настоящем документе, фармацевтически приемлемые носители могут быть твердыми или жидкими. Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые также могут действовать в качестве разбавителей, ароматизаторов, связующих веществ, консервантов, дезинтегрантов таблеток или инкапсулирующего материала.
В порошках носитель может представлять собой тонкоизмельченное твердое вещество в смеси с тонкоизмельченным активным компонентом. В таблетках активный компонент может быть смешан с носителем, обладающим необходимыми связующими свойствами, в подходящих пропорциях и спрессован до необходимой формы и размера.
Порошки и таблетки предпочтительно содержат от 5% до 70% активного соединения. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, карбоксиметилцеллюлоза натрия, легкоплавкий воск, масло какао и тому подобное. Термин «препарат» предназначен для включения композиции активного соединения с инкапсулирующим материалом в качестве носителя, обеспечивающего капсулу, в которой активный компонент с другими носителями или без них окружен носителем, который, таким образом, связан с ним. Точно так же, включены облатки и пастилки. Таблетки, порошки, капсулы, пилюли, облатки и пастилки могут быть использованы в качестве твердых лекарственных форм, подходящих для перорального введения.
Для приготовления суппозиториев сначала плавят легкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, и активный компонент равномерно диспергируют в нем, например, посредством перемешивания. Расплавленную гомогенную смесь затем разливают в формы удобного размера, дают остыть и тем самым затвердеть.
Жидкие формы препаратов включают растворы, суспензии и эмульсии, например, водные растворы или растворы в воде/пропиленгликоле. Для парентерального введения жидкие препараты могут быть приготовлены в виде раствора в водном растворе полиэтиленгликоля.
Водные растворы, подходящие для перорального применения, могут быть приготовлены путем растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загустителей, при необходимости. Водные суспензии, подходящие для перорального применения, могут быть получены путем диспергирования тонкоизмельченного активного компонента в воде с вязким материалом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, карбоксиметилцеллюлоза натрия и другие хорошо известные суспендирующие агенты.
Также включены твердые формы препаратов, которые предназначены для превращения непосредственно перед применением в жидкие препараты для перорального введения. Такие жидкие формы включают растворы, суспензии и эмульсии. Эти препараты могут содержать в дополнение к активному компоненту красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергаторы, загустители, солюбилизирующие агенты и тому подобное.
Фармацевтический препарат предпочтительно находится в стандартной лекарственной форме. В такой форме препарат разделен на стандартные дозы, содержащие соответствующие количества активного компонента. Стандартная лекарственная форма может представлять собой упакованный препарат, причем упаковка содержит отдельные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, стандартная лекарственная форма может представлять собой капсулу, таблетку, облатку или пастилку, или это может быть соответствующее количество любого из них в упакованной форме.
Количество активного компонента в препарате с единичной дозой может варьировать или регулироваться от 0,1 мг до 10000 мг в зависимости от конкретного применения и содержания активного компонента. Композиция может при необходимости также содержать другие совместимые терапевтические агенты.
Некоторые соединения могут иметь ограниченную растворимость в воде и, следовательно, может потребоваться поверхностно-активное вещество или другой подходящий сорастворитель в композиции. Такие сорастворители включают Полисорбат 20, 60 и 80; Плюроник F-68, F-84 и P-103; циклодекстрин; и полиоксил 35 касторовое масло. Такие сорастворители обычно используют в количестве примерно от 0,01% до 2 масс.%. Вязкость, превышающая вязкость простых водных растворов, может быть необходимой для уменьшения вариабельности при дозировании составов, для уменьшения физического разделения компонентов суспензии или эмульсии состава и/или для улучшения иных свойств состава. Такие агенты, повышающие вязкость, включают, например, поливиниловый спирт, поливинилпирролидон, метилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, хондроитинсульфат и его соли, гиалуроновую кислоту и её соли, и их комбинации. Такие агенты обычно используют в количестве примерно от 0,01 масс.% до 2 масс.%.
Фармацевтические композиции могут дополнительно включать компоненты для обеспечения замедленного высвобождения и/или комфорта. Такие компоненты включают высокомолекулярные анионные мукомиметические полимеры, гелеобразующие полисахариды и тонкоизмельченные субстраты-носители лекарственного средства. Эти компоненты более подробно обсуждаются в патентах США №№ 4911920, 5403841, 5212162 и 4861760. Все содержание этих патентов включено в настоящий документ посредством ссылки во всей полноте для всех целей.
Фармацевтическая композиция может быть предназначена для внутривенного применения. Фармацевтически приемлемый наполнитель может включать буферы для доведения рН до необходимого диапазона для внутривенного применения. Известно много буферов, включая соли неорганических кислот, такие как фосфат, борат и сульфат.
Эффективные дозировки
Фармацевтическая композиция может включать композиции, в которых активный ингредиент содержится в терапевтически эффективном количестве, то есть в количестве, эффективном для достижения его предполагаемой цели. Фактическое количество, эффективное для конкретного применения, будет зависеть, среди прочего, от состояния, подлежащего лечению.
Дозировка и частота (однократные или многократные дозы) вводимых соединений могут варьировать в зависимости от множества факторов, включая путь введения, рост, возраст, пол, состояние здоровья, массу тела, индекс массы тела и рацион реципиента; характер и степень симптомов заболевания, подлежащего лечению; наличие других заболеваний или других проблем со здоровьем; вид сопутствующего лечения; и осложнения от любого заболевания или режима лечения. Другие терапевтические схемы или агенты могут быть использованы в сочетании со способами и соединениями, раскрытыми в настоящей заявке.
Дозировки могут варьировать в зависимости от требований субъекта и используемого соединения. Доза, вводимая субъекту, в контексте фармацевтических композиций, представленных в настоящей заявке, должна быть достаточной для обеспечения полезного терапевтического ответа у субъекта с течением времени. Размер дозы также будет определяться наличием, характером и степенью любых побочных эффектов. Как правило, лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. После этого дозировку увеличивают небольшими приращениями до достижения оптимального эффекта при определенных обстоятельствах.
Количества и интервалы дозировки можно регулировать индивидуально, чтобы обеспечить уровни вводимых соединений, эффективные для конкретного клинического показания, подлежащего лечению. Это обеспечит терапевтический режим, соразмерный тяжести патологического состояния индивидуума.
С использованием представленных в настоящей заявке учений можно спланировать эффективную схему профилактического или терапевтического лечения, которая не вызывает существенной токсичности и все же является полностью эффективной для лечения клинических симптомов, демонстрируемых конкретным пациентом. Это планирование должно включать тщательный выбор активного соединения с учетом таких факторов, как активность соединения, относительная биодоступность, масса тела пациента, наличие и тяжесть неблагоприятных побочных эффектов, предпочтительный способ введения и профиль токсичности выбранного агента.
Токсичность
Соотношение между токсичностью и терапевтическим эффектом для конкретного соединения является его терапевтическим индексом и может быть выражено как соотношение между LD50 (количеством соединения, вызывающим гибель 50% популяции) и ED50 (количеством соединения, обеспечивающим эффект у 50% популяции). Соединения, которые проявляют высокие терапевтические индексы, являются предпочтительными. Данные терапевтического индекса, полученные из анализов клеточных культур и/или исследований на животных, можно использовать при составлении диапазона дозировок для применения у людей. Дозировка таких соединений предпочтительно находится в диапазоне концентраций в плазме, которые включают ED50 с небольшой токсичностью или без нее. Дозировка может варьировать в пределах этого диапазона в зависимости от используемой лекарственной формы и используемого пути введения. См., например, Fingl et al., In: THE PHARMACOLOGICAL BASIS OF THERAPEUTICS, Ch.1, p.l, 1975. Точный состав, способ введения и дозировка могут быть выбраны отдельным врачом с учетом состояния пациента и конкретного метода, в котором используют соединение.
Когда парентеральное применение необходимо или желательно, особенно подходящие смеси для соединений, включенных в фармацевтическую композицию, могут быть инъецируемыми, стерильными растворами, масляными или водными растворами, а также суспензиями, эмульсиями или имплантатами, включая суппозитории. В частности, носители для парентерального введения включают водные растворы декстрозы, физиологический раствор, очищенную воду, этанол, глицерин, пропиленгликоль, арахисовое масло, кунжутное масло, блок-сополимеры полиоксиэтилена, и тому подобное. Ампулы являются удобными стандартными дозами. Фармацевтические добавки, подходящие для использования в фармацевтических композициях, представленных в настоящем документе, могут включать те, которые описаны, например, в Pharmaceutical Sciences (17th Ed., Mack Pub. Co., Easton, PA) и WO 96/05309, при этом идеи обоих этих документов настоящим включены посредством ссылки.
III. Способы лечения
Другим аспектом настоящего изобретения является способ профилактики или лечения острой лучевой болезни, включающий стадию введения PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина) субъекту.
Определение PLAG (также обозначаемого в настоящей заявке как EC-18) и острой лучевой болезни приведено выше.
Поскольку PLAG оказывает профилактическое и терапевтическое воздействие на острую лучевую болезнь, можно предотвратить или лечить острую лучевую болезнь путем введения индивидууму композиции, содержащей PLAG.
PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерин) оказывает превосходное действие в профилактике и лечении острой лучевой болезни за счет увеличения выживаемости субъектов с радиационным облучением. Соответственно, фармацевтическую композицию и пищевую оздоровительную функциональную композицию, содержащую PLAG в качестве активного ингредиента по настоящему изобретению, можно эффективно использовать для профилактики, лечения или облегчения острой лучевой болезни.
IV. Примеры
Хотя предыдущий раздел был описан в некоторых деталях с целью иллюстрации и примера для ясности понимания, для специалистов в данной области техники очевидно, что некоторые незначительные изменения и модификации будут осуществляться на практике в свете вышеизложенного. Следовательно, описание и примеры не должны рассматриваться как ограничивающие объем любого изобретения, изложенного в настоящей заявке.
Все ссылки, цитируемые в настоящей заявке, в том числе заявки на патент и публикации, включены в данное описание посредством ссылки во всей полноте.
Пример 1: Влияние PLAG (1-пальмитоил-2-линолеоил-3-ацетилглицерина) на снижение уровня иммунных клеток в крови на животной модели острой лучевой болезни (ОЛБ).
Мыши BALB/c (9-недельный самец) были получены от Koatech (Pyeongtaek, Республика Корея) и содержались в специфической среде без патогенов (SPF) для создания животной модели острой лучевой болезни. Чтобы оценить влияние PLAG на уменьшение иммунных клеток под действием радиации, для этого исследования были использованы следующие три группы: (1) нормальная контрольная группа (положительный контроль); (2) лучевая терапия - группа с радиационно-индуцированной лейкопенией (RIL) (группа отрицательного контроля), и (3) группа RIL + лечение PLAG (экспериментальная группа).
В частности, мышей в группах RIL и RIL + PLAG подвергали облучению всего тела при 1 Гр (100 рад = 1,06 мин). Для группы лечения RIL + PLAG 50 мг/кг/сутки PLAG (Enzychem Lifesciences Co., Тэджон, Республика Корея) вводили перорально ежедневно в течение 4 последовательных дней после облучения. Группу нормального контроля не подвергали облучению. На 5-й день после облучения отбирали образцы крови, а затем проводили общий анализ крови (CBC) с использованием автоматического гематологического анализатора Mindray BC-5300 (Shenzhen Mindray Bio-medical Electronics, Китай), чтобы подсчитать количество лейкоцитов, нейтрофилов и лимфоцитов в крови (Фигура 1).
В результате в группе RIL + PLAG уровень лейкоцитов, нейтрофилов и лимфоцитов в крови составил 63% (2,18 ± 0,31 против 3,56 ± 0,84), 34% (0,87 ± 0,04 против 1,17 ± 0,35) и 85% ± 0,25 против 2,2 ± 0,42) (Таблица 1 и Фигура 2), увеличившись статистически значимым образом.
Таблица 1
(n=3)
(n=3)
(n=3)
Следовательно, из вышеприведенных результатов видно, что PLAG проявляет эффект индукции повышения уровней различных иммунных клеток, которые были снижены при воздействии радиации на модели исследования ОЛБ.
Пример 2: Влияние PLAG на выживаемость на животной модели острой лучевой болезни (ОЛБ).
Чтобы дополнительно подтвердить влияние PLAG на острую лучевую болезнь, была разработана и испытана животная модель радиационно-индуцированного орального мукозита в качестве животной модели острой лучевой болезни с более тяжелыми условиями, чем в Примере 1 выше. Во-первых, мышей BALB/c (самки в возрасте 8 недель) получали в Koatech (Республика Корея) и содержали в специфической среде без патогенов (SPF) для создания модели радиационно-индуцированного орального мукозита. В этом примере первую группу с индуцированным оральным мукозитом (отрицательный контроль), сравнивали со второй группой, где PLAG вводили перорально животным с индуцированным оральным мукозитом.
В частности, мышей подвергали воздействию гамма-облучения всего тела с дозой 1 Гр, и спустя 0, 7, 10 и 16 дней наносили царапины размером 0,2 см на языке мышей с одной и той же силой и глубиной, используя иглу 18 калибра. На 2-й день после облучения интраперитонеально вводили 5-фторурацил (50 мг/кг/сутки), и применяли PLAG перорально по 250 мг/кг/сутки в течение 18 дней (Таблица 2).
Таблица 2
(0, 2, 10 и 16 дни)
0 или 18 суток
0 или 18 суток
В результате в группе с индуцированным мукозитом полости рта выживаемость составила только 28% (2/7, 72%) на 18-й день, тогда отмечалась 85% (6/7, 15% (Фигура 3)) выживаемость в группе с индуцированным мукозитом полости рта при применении PLAG. Эти результаты свидетельствуют о том, что лечение PLAG значительно повышало выживаемость мышей, несмотря на снижение гемопоэтических клеток от воздействия гамма-излучения и лечения 5-фторурацилом, а также повышенный риск инфекции, вызванной ранами полости рта с царапиной на языке. Это также позволяет предположить, что PLAG проявляет превосходные профилактические и терапевтические эффекты.
Пример 3: Эффекты PLAG в отношении каждого симптома, влияющего на выживаемость на животной модели острой лучевой болезни.
На животной модели, использованной в Примере 2 выше, определяли уровень мукозита полости рта и количество нейтрофилов в крови.
Уровень мукозита полости рта определяли путем подсчета образования язвы и отека языка и частоты атрофии раны, а также расчета показателя мукозита полости рта в общей сложности для 5 наблюдателей с маскированием данных. На 7-й и 10-й день собирали кровь в экспериментальных группах и измеряли количество нейтрофилов в крови путем проведения полного анализа крови (CBC) с использованием автоматического гематологического анализатора Mindray BC-5300 (Shenzhen Mindray Bio-medical Electronics, Китай).
В результате визуально подтвержденные уровни орального мукозита были значительно снижены в группе, получавшей PLAG, по сравнению с группой, не получавшей PLAG, с меньшим образованием язв и отеком языка и меньшей частотой атрофии раны (Фигура 4). Показатель мукозита полости рта также был значительно снижен (Фигура 5). На 7-й и 10-й дни количество нейтрофилов в крови уменьшилось приблизительно на 95% в группе животных с радиационным оральным мукозитом через 10 дней, но количество нейтрофилов было значительно увеличено в группе, которой ежедневно вводили PLAG (Фигура 6).
Пример 4: Улучшение выживаемости
Чтобы оценить влияние PLAG на выживаемость на животных моделях, подвергшихся воздействию радиации, были использованы следующие экспериментальные группы, как показано в Таблице 3, и результаты представлены на Фигуре 7B и Фигурах 8-12. Как обсуждалось в Примерах выше, первую группу не получавших лечение (отрицательный контроль) мышей с индукцией ОЛБ после облучения сравнивали со второй группой получавших лечение мышей с индукцией ОЛБ, получавших PLAG после облучения.
Таблица 3
• γ-облучение: 6,5 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• γ-облучение: 6,5 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• γ-облучение: 2, 4, 6 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения (2 Гр)
2) группа, не получавшая лечения после облучения (4 Гр)
3) группа, не получавшая лечения после облучения (6 Гр)
4) группа, получавшая EC-18 после облучения (6 Гр)
• γ-облучение: 7 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• γ-облучение: 8 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• γ-облучение : 10 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
Пример 5: Лечение эритемы
Чтобы оценить влияние PLAG на восстановление кожного повреждения у животных, подвергшихся воздействию радиации, были использованы следующие экспериментальные группы, как показано в Таблице 4, а результаты приведены на Фигурах 13-15. Как обсуждалось в Примерах выше, первую группу не получавших лечения мышей (отрицательный контроль) после облучения сравнивали со второй группой мышей, получавших лечение PLAG после облучения.
Таблица 4
• γ-облучение: 8 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• Γ-облучение: 6,5 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
• γ-облучение: 6,5 Гр (100 рад =1,06 мин), общее облучение
• EC-18: 250 мг/кг/сутки (перорально)
• Группа: 1) группа, не получавшая лечения после облучения
2) группа, получавшая EC-18 после облучения
Пример 6: Влияние на антиапоптоз клеток HaCaT.
Для оценки воздействия PLAG на клетки HaCaT, подвергшиеся облучению, были использованы следующие экспериментальные группы, и результаты представлены на Фигурах 17B-18D.
• HaCaT: кератиноциты человека
• PLAG: предварительная обработка 100 мкг/мл в течение 1 часа
• 5-фторурацил: 1, 10, 100 нг/мл
• Гамма-излучение: 7 Гр
• проверка апоптоза через 24 часа
• Подсчет клеток после окрашивания трипановым синим
• Апоптоз (аннексин V + 7-амино-актиномицин D)
Пример 7. Влияние на активные формы кислорода (АФК) клеток HaCaT.
Для оценки воздействия PLAG на клетки HaCaT, подвергшиеся облучению, были использованы экспериментальные группы, как в Примере 6, и результаты представлены на Фигурах 19A-19C.
Пример 8: Улучшение выживаемости
Фигура 20 представляет собой примерную диаграмму для тестирования показателей выживаемости при лечении на животной модели острой лучевой болезнью, вызванной γ-излучением 6,5 Гр (мыши, BALB/c, 11 недель). Фигура 21 показывает результаты кривых выживаемости мышей BALB/c, подвергшихся воздействию γ-излучения 6,5 Гр при ежедневном лечении ЕС-18 после облучения. НК представляет не получавший лечения контроль без облучения; 6,5 Гр – выживаемость мышей, подвергнутых γ-излучению 6,5 Гр; 6,5 Гр + PLAG (+ 1d) – выживаемость мышей, подвергнутых γ-излучению 6,5 Гр и получавших лечение PLAG в течение 1 дня; и 6,5 Гр + PLAG (+ 2d) – выживаемость мышей, подвергнутых γ-излучению 6,5 Гр и получавших PLAG в течение 2 дней. Фигура 22 показывает изменения массы тела мышей BALB/c на Фигуре 21.
Могут быть установлены различные тесты для измерения выживаемости после γ-облучения на животных моделях. Например, Фигура 23 изображает примерную диаграмму для примерного эксперимента по тестированию выживаемости на мышиных моделях. Фигура 24 изображает примерную диаграмму для примерного эксперимента, включающего три группы (только 5-FU, 5-FU + PLAG 125 мг/кг, 5-FU + PLAG 250 мг/кг) для проверки временной кинетики нейтрофилов при индуцированной 5-фторурацилом (100 мг/кг) нейтропении.
Пример 9. Эффекты PLAG, измеренные in vitro (вестерн-блоттинг).
Влияние EC-18 по сравнению с Г-КСФ на клетки после γ-излучения измеряли вестерн-блоттингом (Фигура 25). Эксперимент позволяет (i) показать корреляцию между активностью РЭФР (рецептора эпидермального фактора роста) и аномальным ростом и индукцией метастазирования раковых клеток молочной железы в среде TAN; и (ii) подтвердить механизм ингибирующего действия при лечении PLAG в этом состоянии. Фактически было подтверждено, что активность РЭФР увеличивалась в группах, стимулированных нейтрофилами или активированными Г-КСФ нейтрофилами, однако активность РЭФР (фосфорилирование РЭФР) снижалась в группе, получавшей PLAG. Фигура 26 позволяет показать эффекты PLAG по сравнению с Г-КСФ.
Пример 10: Время выживания при различной лучевой болезни.
Фигура 27 представляет диаграмму времени выживания пациентов, подвергшихся облучению, и различные синдромы ОЛБ.
Пример 11: EC-18 для лечения острой лучевой болезни
Мы исследовали эффективность EC-18 для разработки медицинского противодействия острой лучевой болезни (ОЛБ) путем анализа смертности и заболеваемости, вызванной ионизирующим излучением.
Материалы и методы
Животные
Самцов и самок мышей BALB/c, свободных от специфических патогенов (в возрасте 10 недель), получали от Koatech Co. (Pyongtaek, Республика Корея). После получения мышей содержали по 5 особей на клетку в специальном помещении без патогенов и акклиматизировали в течение 1 недели в условиях постоянной температуры и нормальных световых циклов. Все животные получали стандартный рацион для мышей с водой без ограничения. Все экспериментальные процедуры были одобрены Институциональным комитетом по уходу и использованию животных Корейского научно-исследовательского института биологии и биотехнологии, и выполнялись в соответствии с руководящими принципами Национального института здравоохранения по уходу и использованию лабораторных животных и Корейскими национальными законами по гуманному обращению с животными.
Определение летальной дозы (LD) общего γ-облучения тела (TBI)
Облучение всего тела животных проводили с помощью гамма-облучателя (J.L. Shepherd & Associates, Сан-Фернандо, США) с источником 60Co (скорость воздействия 0,833 Гр/мин). Для экспериментов по определению выживаемости после γ-облучения мышей группировали по 20 особей (10 самцов и 10 самок) на экспериментальные группы; 6,0 Гр, 6,2 Гр, 6,4 Гр и 6,5 Гр. Выживаемость и массу тела животных регистрировали ежедневно в течение 30 дней.
Создание мышиной модели острой лучевой болезни (ОЛБ), индуцированной γ-облучением
Для экспериментов по определению выживаемости после дозы LD70/30 6,11 Гр γ-облучения (TBI, 60Co, 0,833 Гр/мин) мышей распределяли по 20 особей (10 самцов и 10 самок) в экспериментальные группы; группу только с γ-облучением (положительный контроль), группу с γ-облучением с ЕС-18 10 мг/кг; группу с γ-облучением с ЕС-18 50 мг/кг, и группу с γ-облучением с ЕС-18 250 мг/кг. EC-18 (Enzychem Lifesciences, Jaecheon, Республика Корея) суспендировали в ФБР и вводили перорально один раз в сутки, начиная с 1 дня после облучения. В группе положительного контроля вводили ФБР. Выживаемость и массу тела животных регистрировали ежедневно в течение 30 дней.
Для временного анализа гемопоэтического синдрома ОЛБ (H-ARS) самок мышей распределяли по 5 особей на экспериментальные группы: группу только с γ-облучением (положительный контроль) и группу с γ-облучением с ЕС-18 250 мг/кг. Для анализа эффекта дозы введения EC-18 при H-ARS мышей распределяли по 8 особей (5 самцов и 3 самки) на экспериментальную группу: группу только с γ-облучением (положительный контроль), группу с γ-облучением с ЕС-18 50 мг/кг, группу с γ-облучением с ЕС-18 100 мг/кг, группу с γ-облучением с ЕС-18 250 мг/кг и группу с γ -облучением с ЕС-18 100 мг/кг. Цельную кровь собирали из орбитальных синусов с использованием капиллярных трубок без ЭДТА (Kimble Chase Life Science and Research Products LLC, Флорида, США) и пробирок для сбора, содержащих K3E-K3EDTA (Greiner Bio-One International, Кремсмюнстер, Австрия). Клетки крови подсчитывали и классифицировали посредством полного анализа крови (CBC) с использованием авто-гематологического анализатора Mindray BC-5000 (Shenzhen Mindray Biomedical Electronics, Guangdong Sheng, Китай). Значения для клеток крови животных регистрировали ежедневно в течение 30 дней.
Статистический анализ
Все данные представлены в виде среднего значения ± стандартное отклонение (SD). Для оценки значимости показателя выживаемости и сравнения длительности нейтропении между контрольной группой и группой, получавшей EC-18, был применен парный логарифмический критерий Мантеля-Кокса для кривой выживаемости. Средняя продолжительность жизни была рассчитана как сумма продолжительности жизни для всех мышей/общее количество мышей. Для сравнения статистических различий более чем в двух группах использовали односторонний тест ANOVA. Все остальные статистические анализы были выполнены с использованием парного t-критерия Стьюдента, и значения p<0,05 считались статистически значимыми.
Результаты
Отношение доза облучения - ответ (DRR) и LDXX/30
Доза облучения является значимым прогностическим фактором смертности и заболеваемости с увеличением дозы. Фигура 28А показывает выживаемость мышей BALB/c, подвергшихся воздействию различных доз γ-облучения 60Co. Доза 6,0; 6,2; 6,4 и 6,5 Гр привела к 60, 80, 100 и 100% смертности, соответственно, после 30 дней наблюдения. Фигура 28B показывает соотношение дозы облучения (DRR) с использованием пробит-моделей. Среднее время выживания умерших для каждой группы дозы облучения составило соответственно 15,30 ± 4,98; 13,69 ± 3,26; 14,15 ± 3,48 и 15,85 ± 4,42 дня (Таблица 5). Тридцатидневную выживаемость рассчитывали при каждой дозе облучения, и отмечали как процент смертности по оси Y. Основываясь на пробит-модели, было определено LD30/30, LD50/30, LD70/30 и LD95/30 с 95% доверительными интервалами для каждой дозы (Таблица 6). Было определено, что LD70/30 составляет 6,11 Гр, и этот параметр использовали для оценки нескольких биологических показателей (например, выживаемости, снижения массы тела и уменьшения гемопоэтических клеток) в последующих исследованиях.
Таблица 5. Смертность мышей BALB/c после γ-облучения в течение 30 дней.
Таблица 6. Расчетная доза облучения у мышей BALB/c после γ-облучения.
Гамма-излучение также вызывало значительное снижение массы тела облученных мышей (Фигура 29). Было трудно найти строгую связь между дозой облучения и потерей массы тела на начальной стадии (0-10 дней), но массы тела групп с более низкой дозой (6,0 и 6,2 Гр) были восстановлены до нормального диапазона на конец периода наблюдения.
Терапевтическое влияние введения EC-18 на выживаемость, среднюю продолжительность жизни и массу тела облученных мышей с LD70/30 при общем облучении.
Затем мы исследовали терапевтическое влияние EC-18 на выживаемость облученных мышей с общим облучением LD70/30 (6,11 Гр) в течение 30 дней периода наблюдения. Выживаемость группы положительного контроля составляла 20%, тогда как выживаемость групп, получавших ЕС-18 10, 50 и 250 мг/кг, составляла 20, 40 и 80%, соответственно (Фигура 30А). Более того, EC-18 значительно увеличивал среднюю продолжительность жизни облученных мышей дозозависимым образом (Таблица 7). Соответствующая средняя продолжительность жизни групп, получавших ЕС-18 10, 50 и 250 мг/кг, составила 19,3; 22,3 и 28,2 дня по сравнению с 17,9 днями в группе положительного контроля.
Таблица 7. Влияние дозы введения EC-18 на выживаемость и среднюю продолжительность жизни облученных мышей.
p*
Применение EC-18 также эффективно предотвращало тяжелую потерю массы тела, вызванную γ-облучением (Фигура 30B). В частности, группа, получавшая EC-18 в дозе 250 мг/кг, значительно снижала вызванную γ-излучением потерю массы тела с 18-го дня после облучения до конца эксперимента, по сравнению с группой положительного контроля. Кроме того, количество мышей, испытывающих потерю массы тела на 20% от исходного значения, резко уменьшалось по мере увеличения дозы EC-18 (Таблица 8).
Таблица 8. Дозозависимое влияние введения EC-18 на потерю массы тела у облученных мышей.
Основываясь на наблюдениях в этом исследовании, мы пришли к выводу, что EC-18 обладает терапевтическим потенциалом для улучшения выживаемости и предотвращения потери массы тела при ОЛБ, вызванной γ-излучением.
Терапевтический эффект от введения EC-18 при индуцированном γ-излучением гематологическом синдроме ОЛБ
Единственный сеанс γ-облучения всего тела (6,11 Гр) быстро истощил все виды гемопоэтических клеток, включая абсолютное количество нейтрофилов (ANC), моноцитов, абсолютное количество лимфоцитов (ALC), количество тромбоцитов (PLT) и количество эритроцитов (RBC) в пределах 3 дня после облучения (Фигура 31). Среднее значение первого дня нейтропении составило 2,8 ± 0,45 дня, а продолжительность нейтропенического состояния составила 18,0 ± 1,41 дня. Все особи в облученной группе испытывали тяжелое нейтропеническое состояние (ANC <100 клеток/мкл), а продолжительность тяжелого нейтропенического состояния составила 16±0,00. Среднее значение низшего уровня ANC после γ-облучения составило 0,0±0,00 клеток/мкл, а время восстановления до ANC≥500 или 1000 клеток/мкл составило 27 ± 1,41 дней. Более того, γ-облучение вызывало снижение PLT более чем на 90% в течение 7 дней после облучения, которое начало восстанавливаться через 26 дней после облучения. RBC постепенно снижалось и начало восстанавливаться через 22 дня после облучения.
Затем оценивали терапевтический эффект введения EC-18 при гемопоэтическом синдроме ОЛБ. Введение EC-18 значительно ослабило вызванное γ-облучением истощение количества лейкоцитов (WBC), абсолютного количества нейтрофилов (ANC) и абсолютного количества лимфоцитов (ALC) у облученных мышей (Фигуры 32A-J). Группы мышей BALB/c (n = 5 самок для Фигур 32A-32E и n = 8, 5 самцов, 3 самки для Фигур 32F-32J) подвергали воздействию 6,11 Гр γ-излучения, и вводили перорально различные дозы ЕС-18 один раз в сутки до 15 дня или оставляли без лечения. На Фигурах 32A-32E показана временная зависимость количества лейкоцитов (WBC; Фигура 32A), абсолютного количества нейтрофилов (ANC; Фигура 32B), абсолютного количества лимфоцитов (ALC; Фигура 32C), количества тромбоцитов (PLT; Фигура 32D) и количества эритроцитов (RBC; Фигура 32E) в течение 15 дней, соответственно. На Фигурах 32F-32J показано влияние дозы введения EC-18 на WBC (Фигура 32F), ANC (Фигура 32G), ALC (Фигура 32H), PLT (Фигура 32I) и BRC (Фигура 32J) в день 15, соответственно. ns – незначимые различия; * р <0,05, ** р <0,01, *** р <0,005.
Применение EC-18 существенно снижало вызванное γ-облучением уменьшение ANC. Средний первый день нейтропении (ANC <500 клеток/мкл) в контрольной и получавшей EC-18 группах составил 1,8 ± 1,09 и 2,2 ± 1,09 дня (значение двустороннего критерия P = 0,62), соответственно. Хотя EC-18 не защищал облученных мышей от тяжелой нейтропении, он эффективно уменьшал продолжительность тяжелой нейтропении с 13,0 дней до 7,2 ± 1,79 дней. Кроме того, EC-18 значительно увеличил среднее значение наименьшего уровня ANC после γ-облучения с 4,0 ± 5,48 клеток/мкл до 20,0 ± 10,00 клеток/мкл (значение двустороннего критерия P = 0,035). Введение EC-18 ослабляло снижение PLT и RBC, индуцированное облучением всего организма (Фигуры 32D, 32E и 32I, 32J). На основании наблюдений мы пришли к выводу, что EC-18 может обладать терапевтическим потенциалом для ослабления снижения числа клеток крови при ОЛБ, вызванной γ-излучением.
Понятно, что примеры и варианты осуществления, описанные в настоящей заявке, предназначены только для иллюстративных целей и что различные модификации или изменения в их свете будут предложены специалистам в данной области техники и должны быть включены в сущность и сферу применения этой заявки и объем формулы изобретения. Все публикации, патенты и патентные заявки, процитированные здесь, тем самым включены в качестве ссылки во всей полноте для всех целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ ЛЕЙКОПЕНИИ И ТРОМБОЦИТОПЕНИИ | 2015 |
|
RU2702122C2 |
| Способ моделирования острой лучевой болезни в эксперименте | 2023 |
|
RU2811270C1 |
| Способ лечения животных при лучевой болезни | 2024 |
|
RU2836843C1 |
| Способ получения препарата для лечения радиационных поражений организма животных | 2024 |
|
RU2837739C1 |
| Новые пептиды и их аналоги для применения при лечении мукозита полости рта | 2014 |
|
RU2668963C2 |
| КОМПОЗИЦИЯ ДЛЯ ПОДАВЛЕНИЯ РАКОВЫХ ЗАБОЛЕВАНИЙ КРОВИ ИЛИ МЕТАСТАЗИРОВАНИЯ РАКА, СОДЕРЖАЩАЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА МОНОАЦЕТИЛДИГЛИЦЕРИДНОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2632098C2 |
| КОМПОЗИЦИЯ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ЛЕЧЕНИЯ ХРОНИЧЕСКИХ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ЛЕГКИХ, СОДЕРЖАЩАЯ МОНОАЦЕТИЛДИГЛИЦЕРОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2014 |
|
RU2642631C2 |
| СПОСОБ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ОСТРОЙ ЛУЧЕВОЙ БОЛЕЗНИ В ЭКСПЕРИМЕНТЕ | 2014 |
|
RU2551619C1 |
| КОМПОЗИЦИЯ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ЛЕЧЕНИЯ АСТМЫ, СОДЕРЖАЩАЯ МОНОАЦЕТИЛДИАЦИЛГЛИЦЕРИНОВОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2014 |
|
RU2636615C2 |
| Способ экстренной профилактики и лечения острой лучевой болезни (варианты) | 2018 |
|
RU2699040C1 |
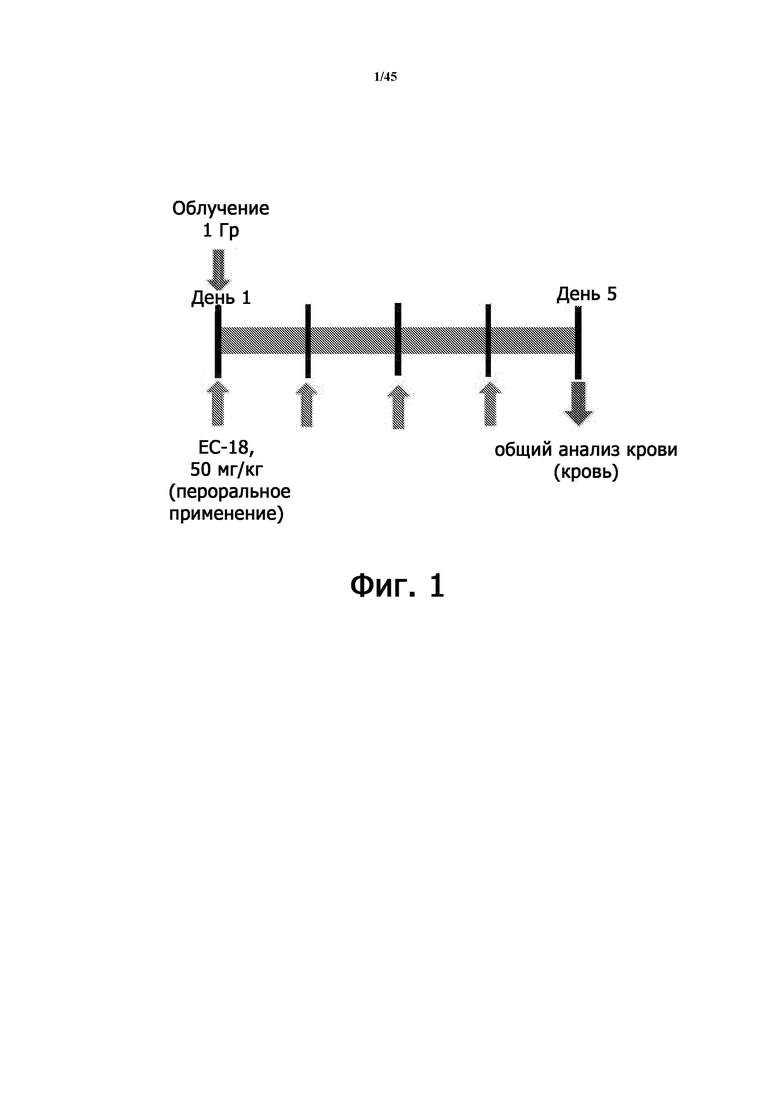
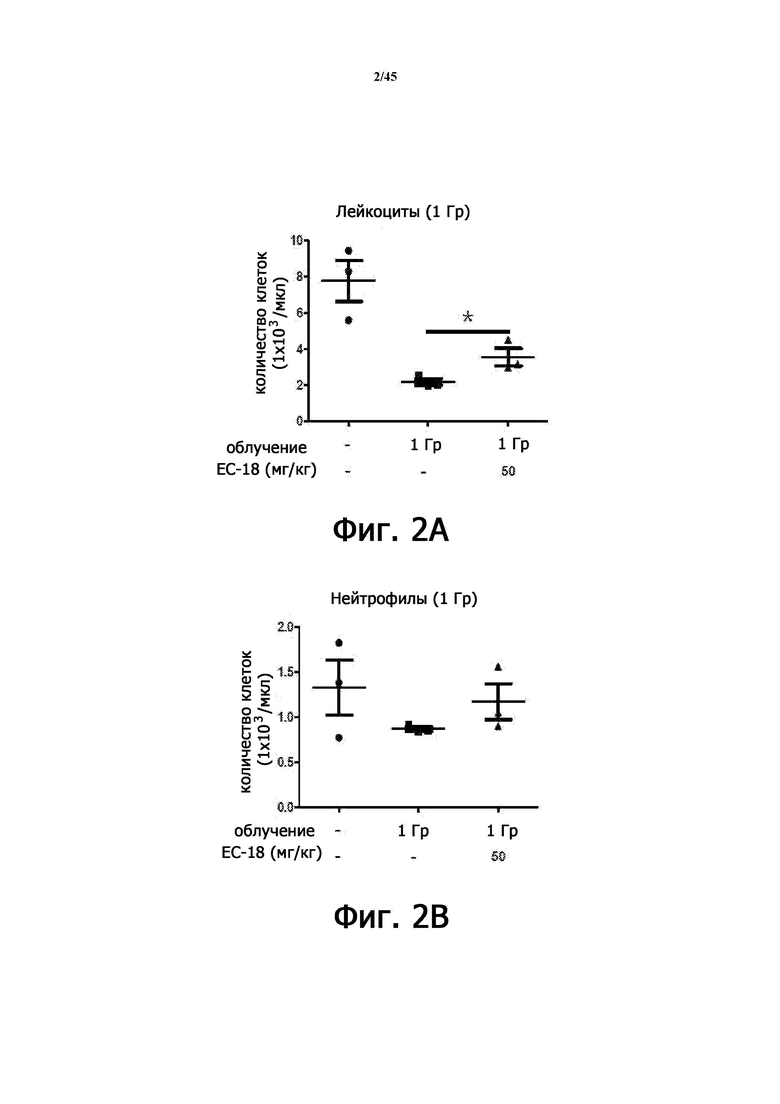
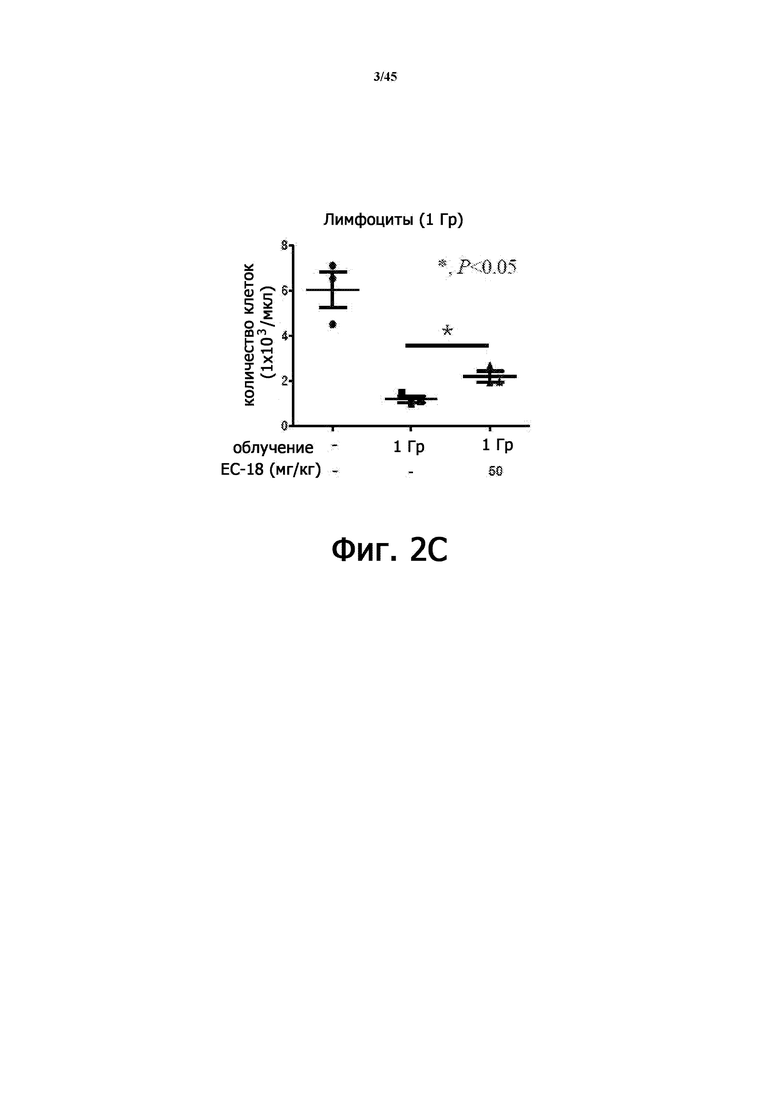

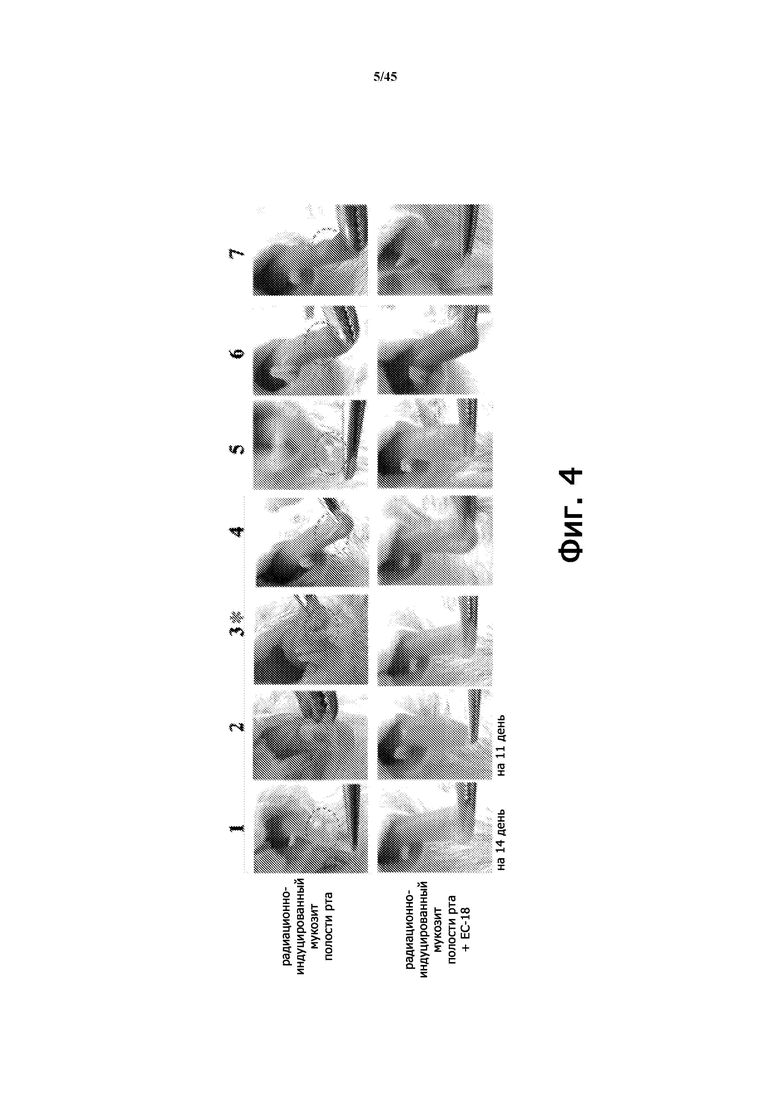
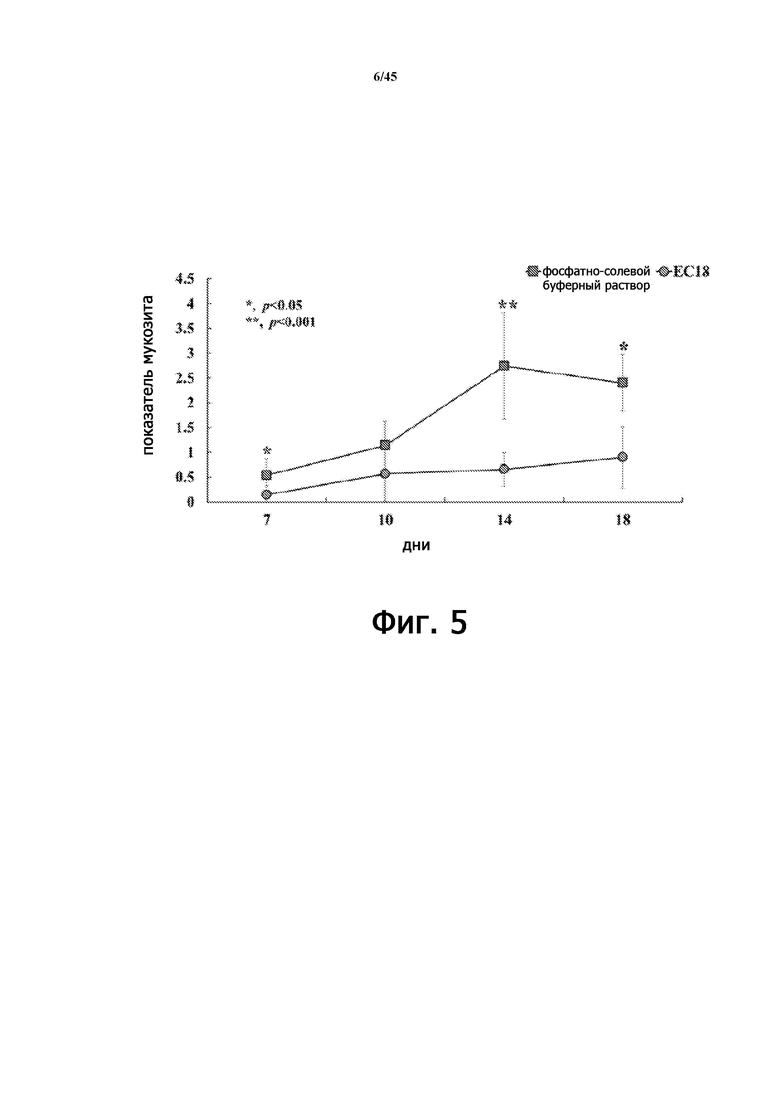
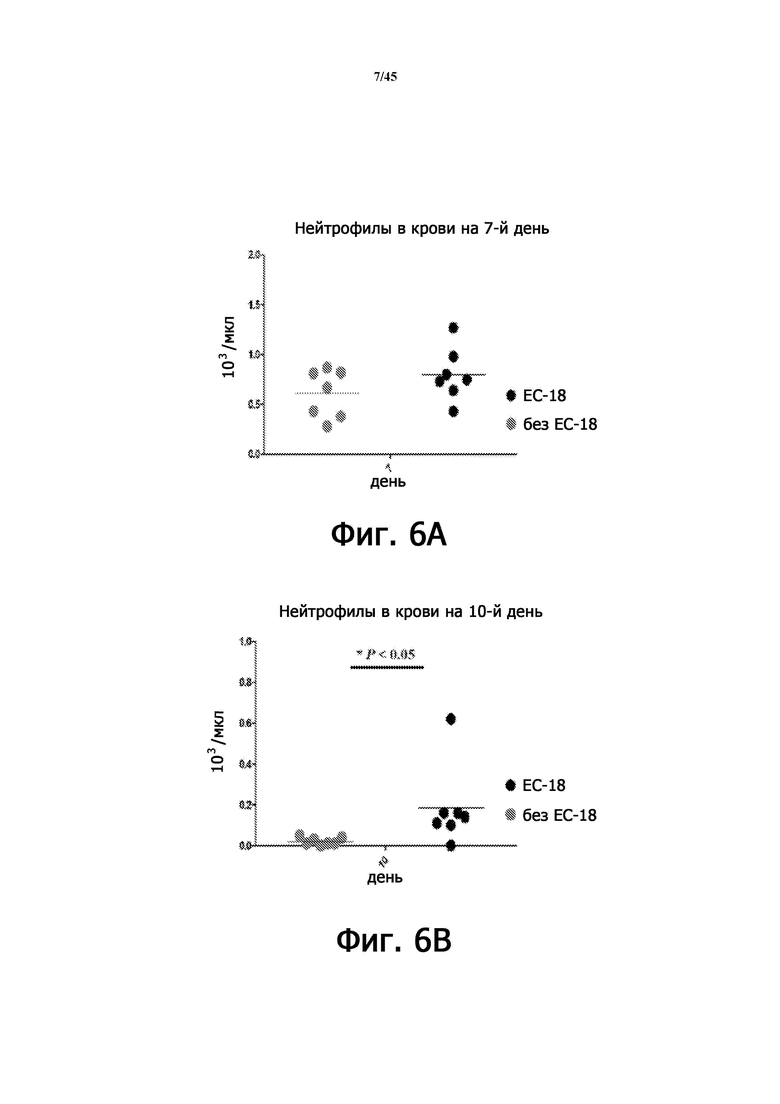
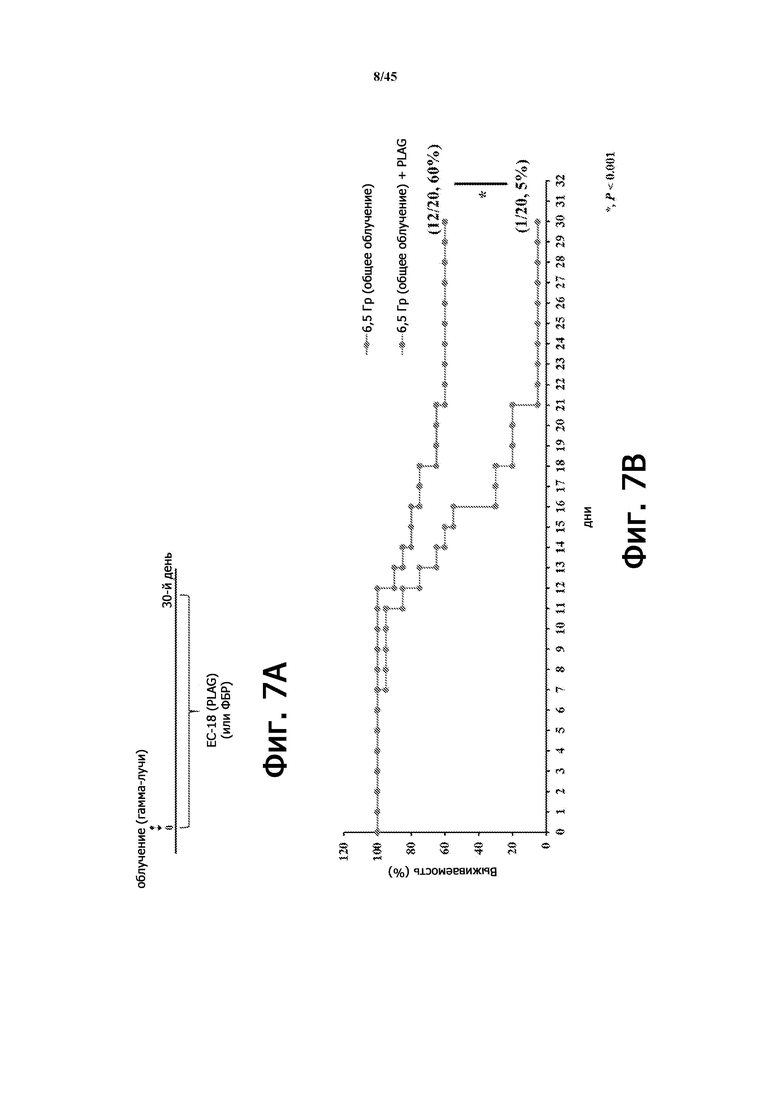
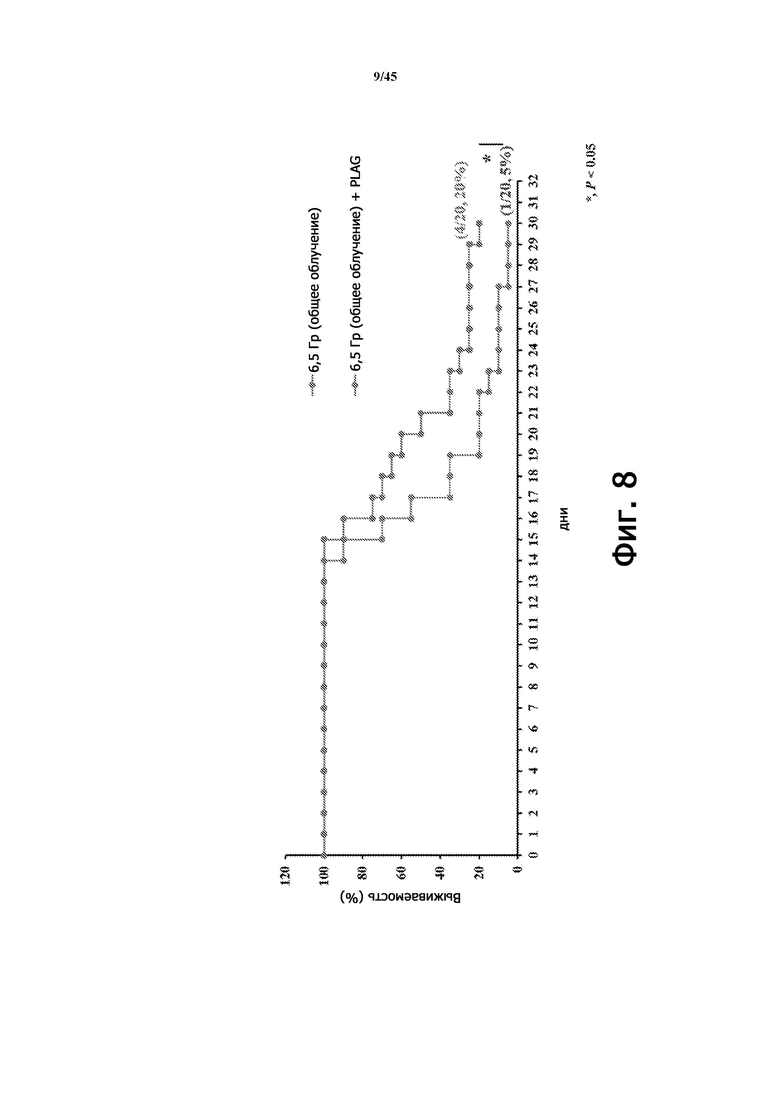
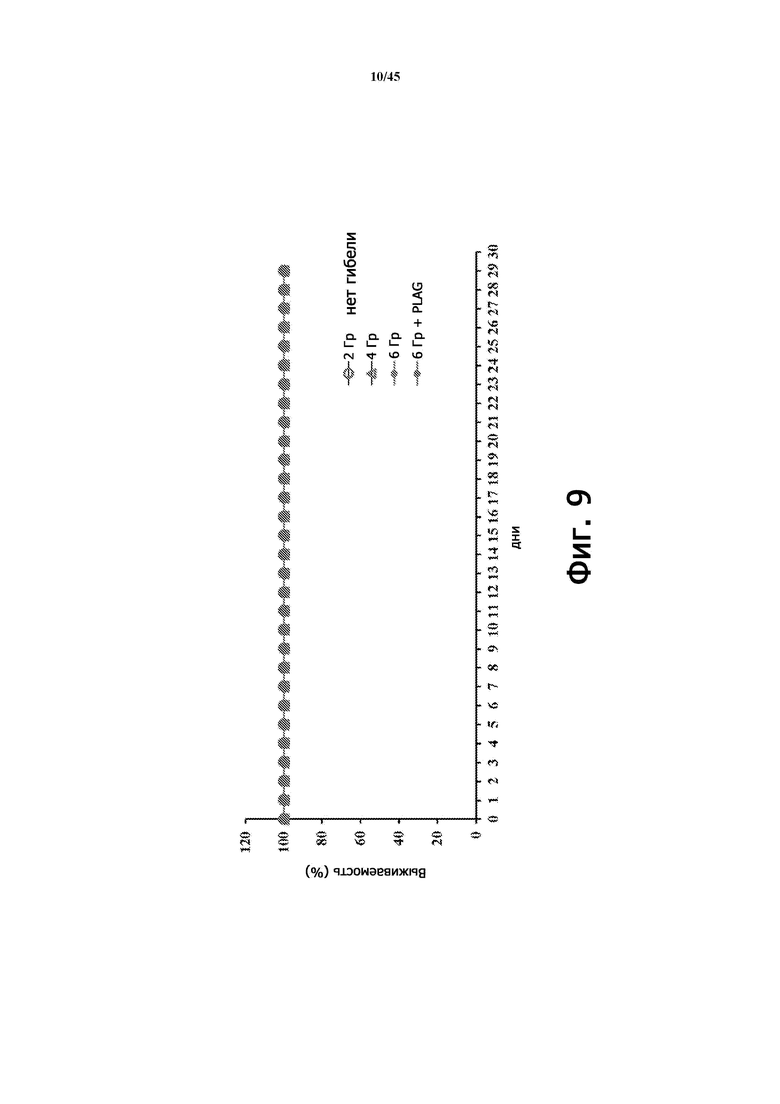
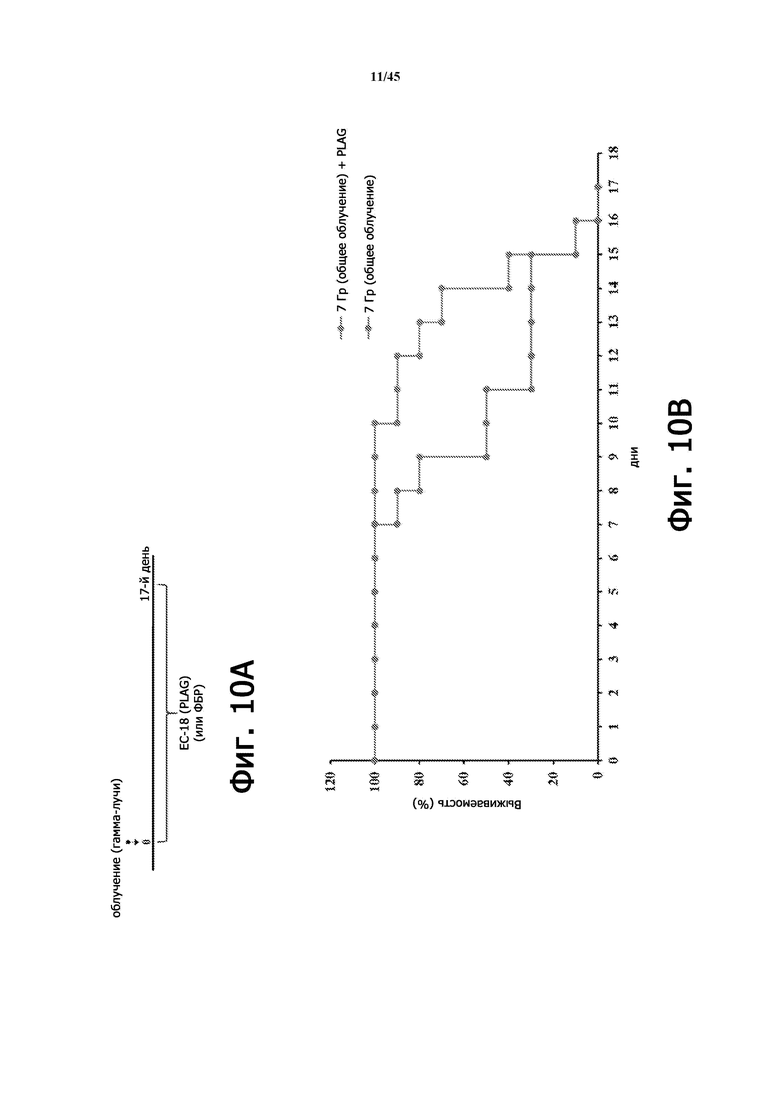
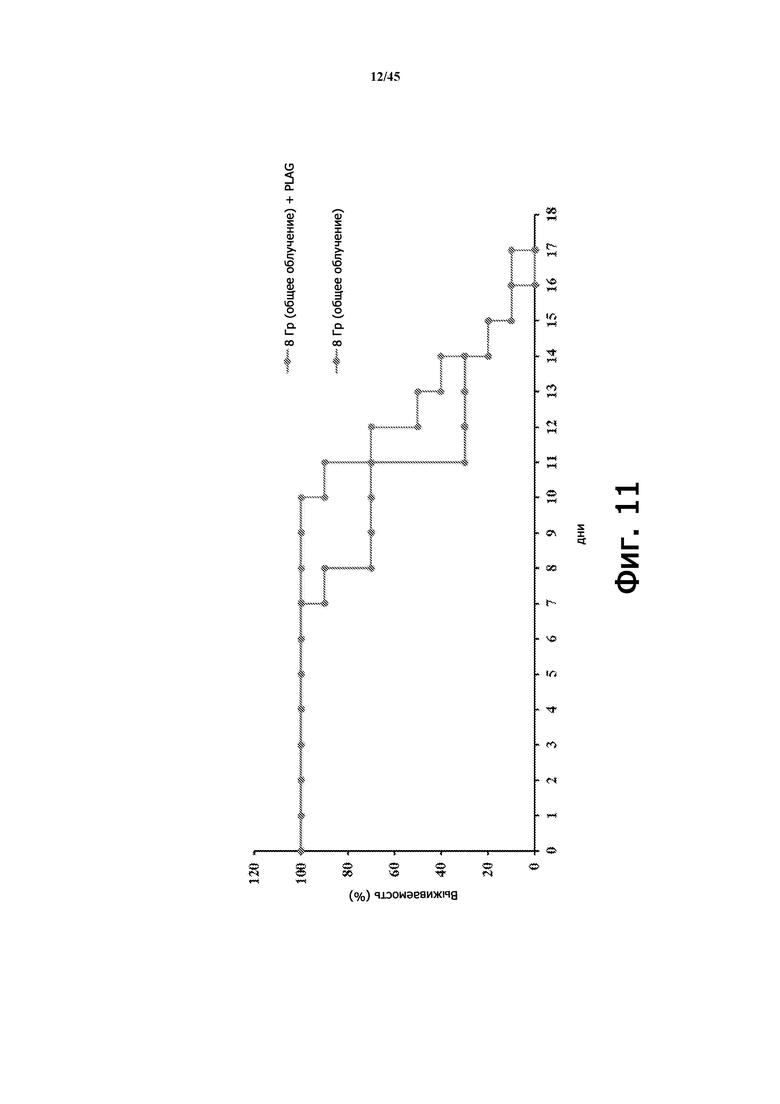
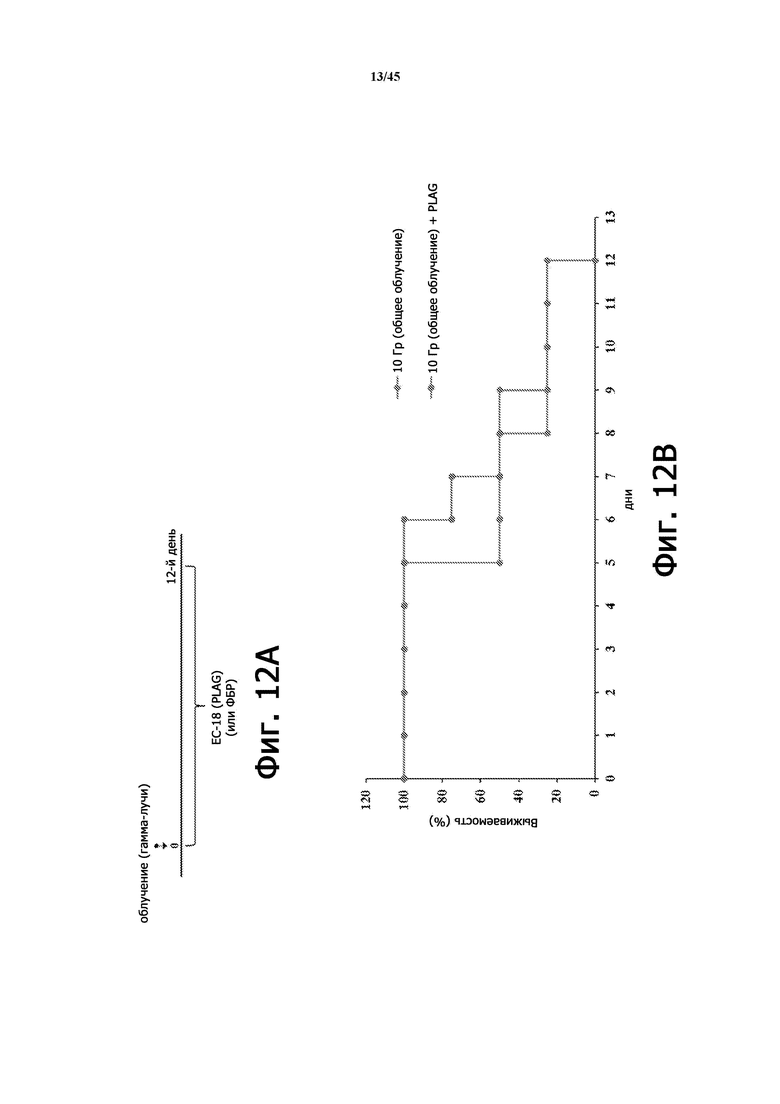
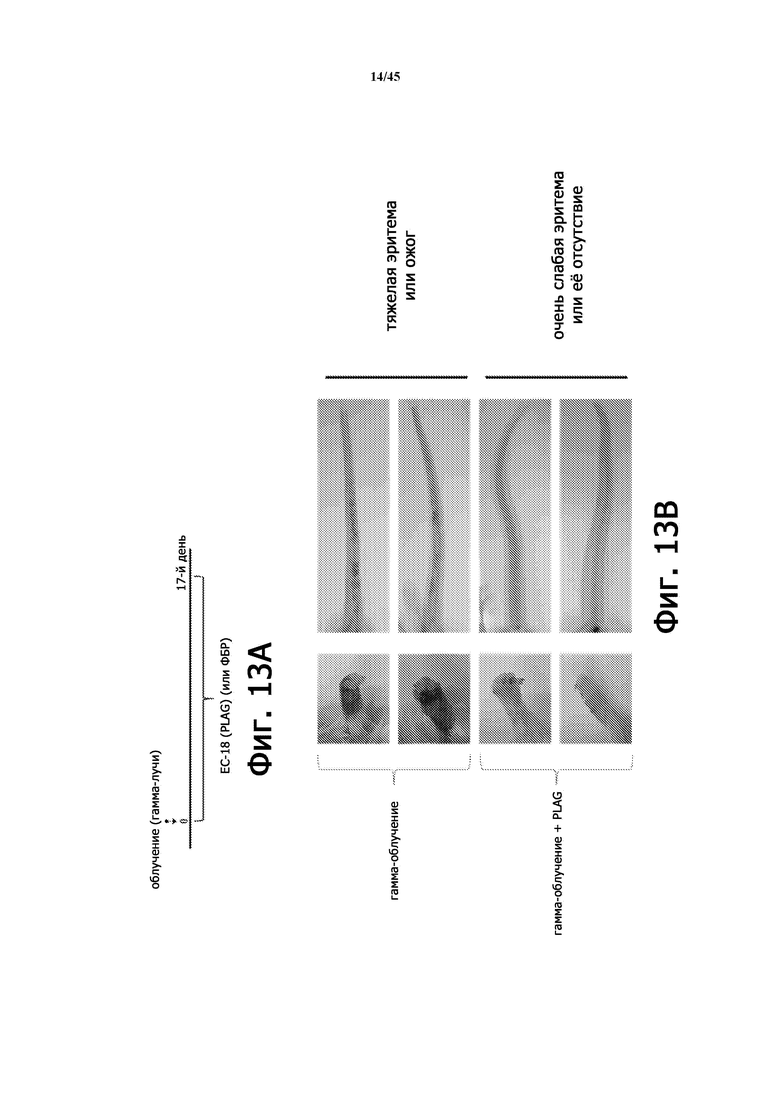


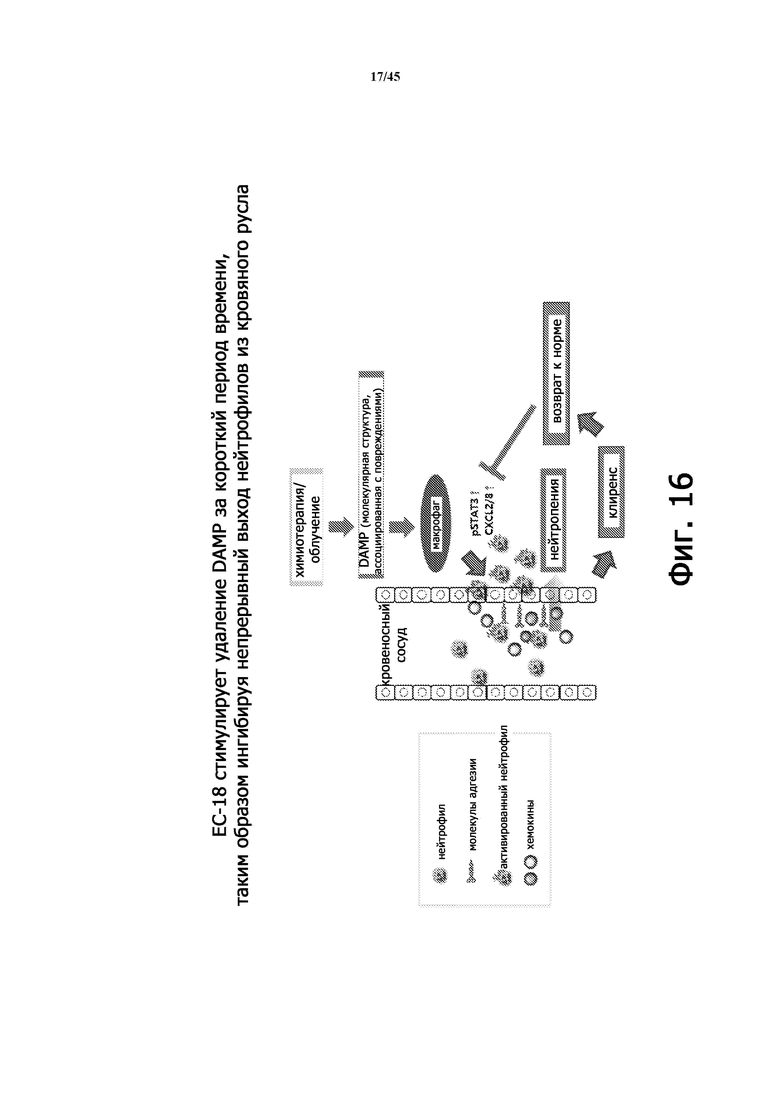

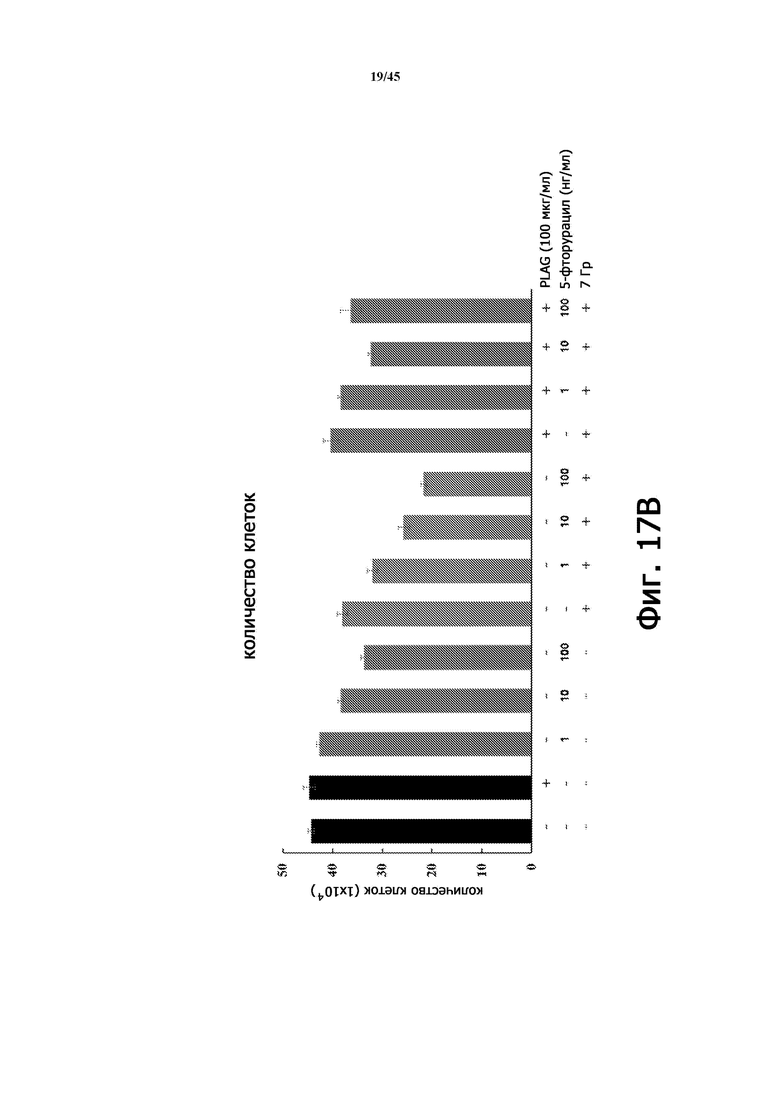
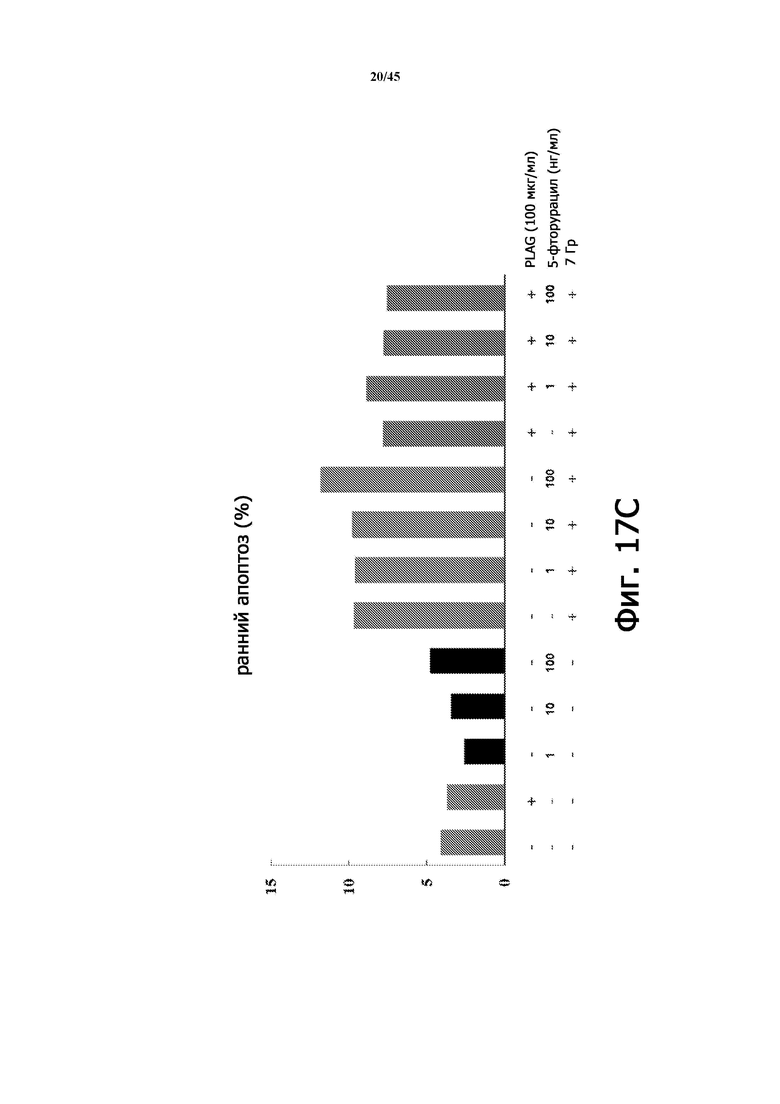
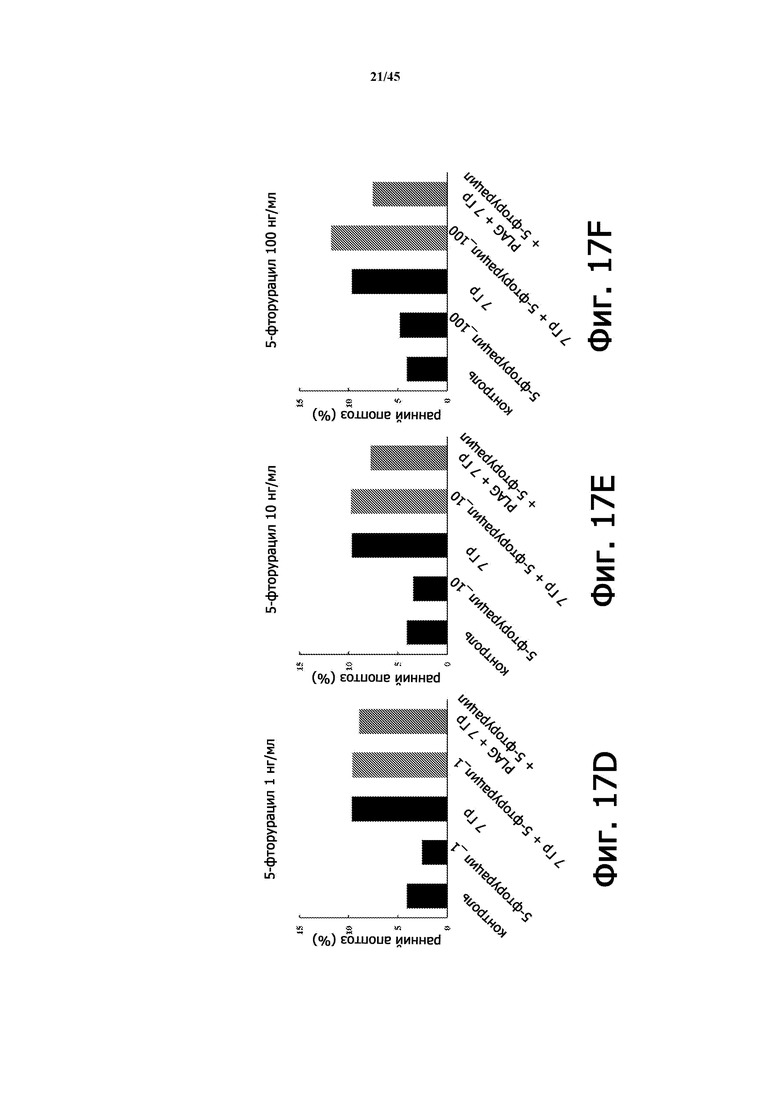
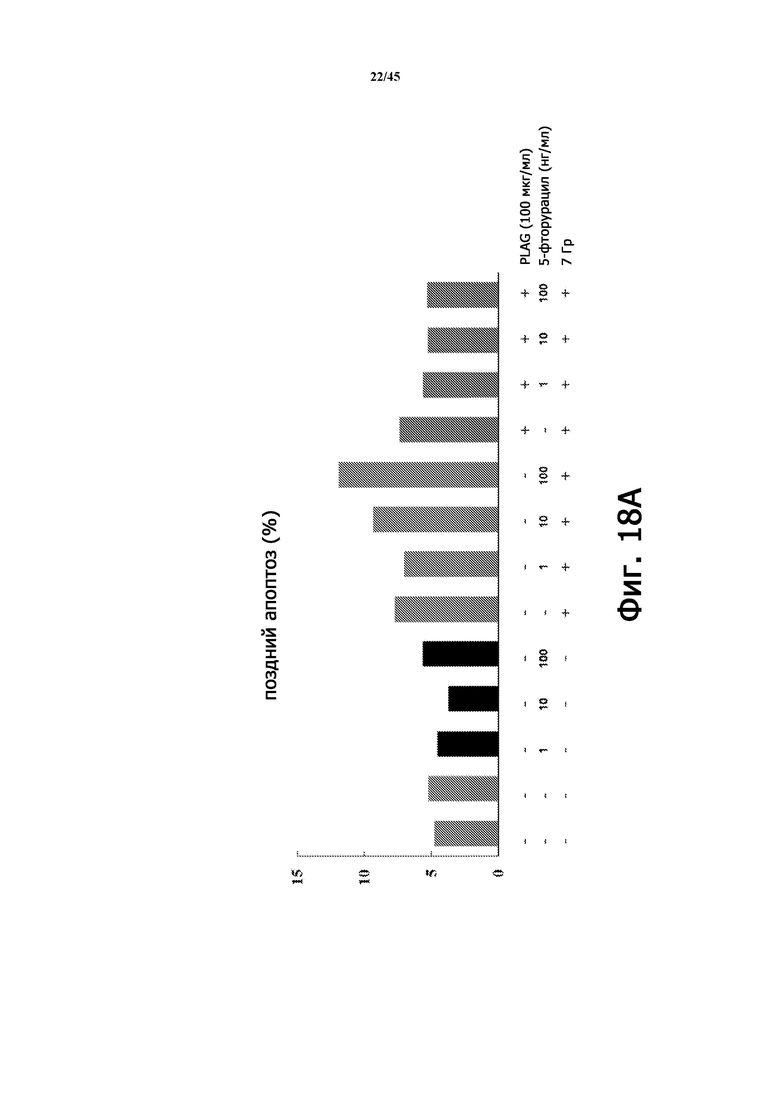
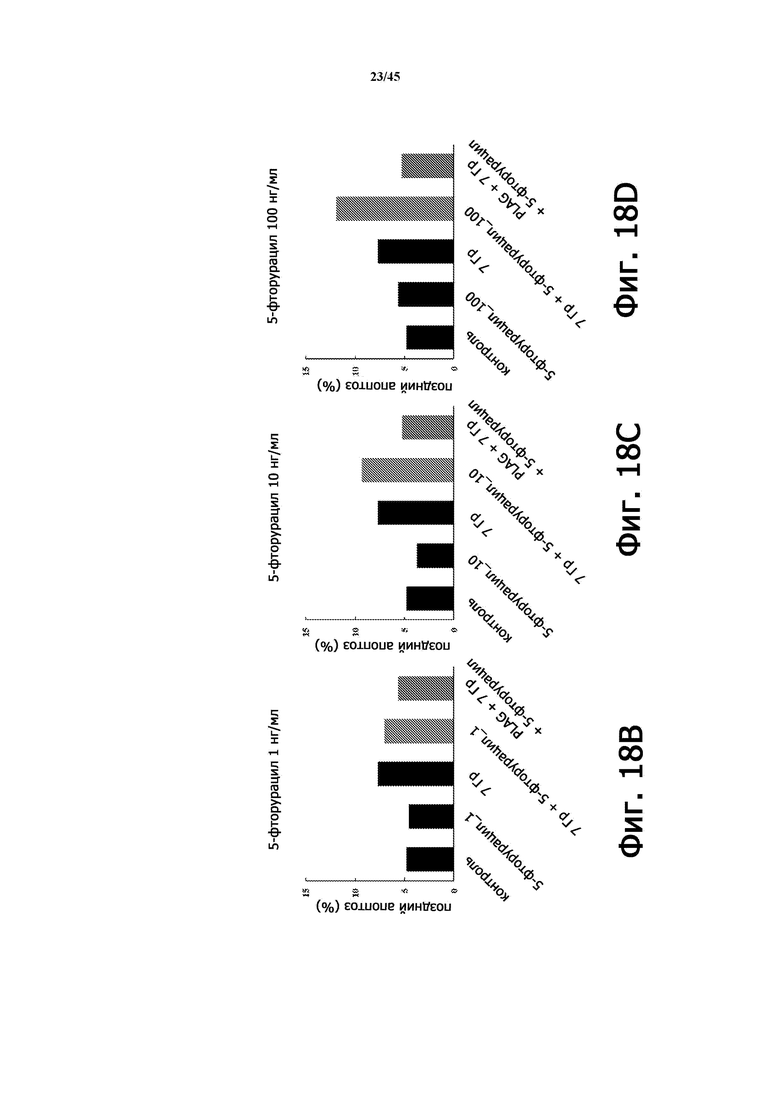
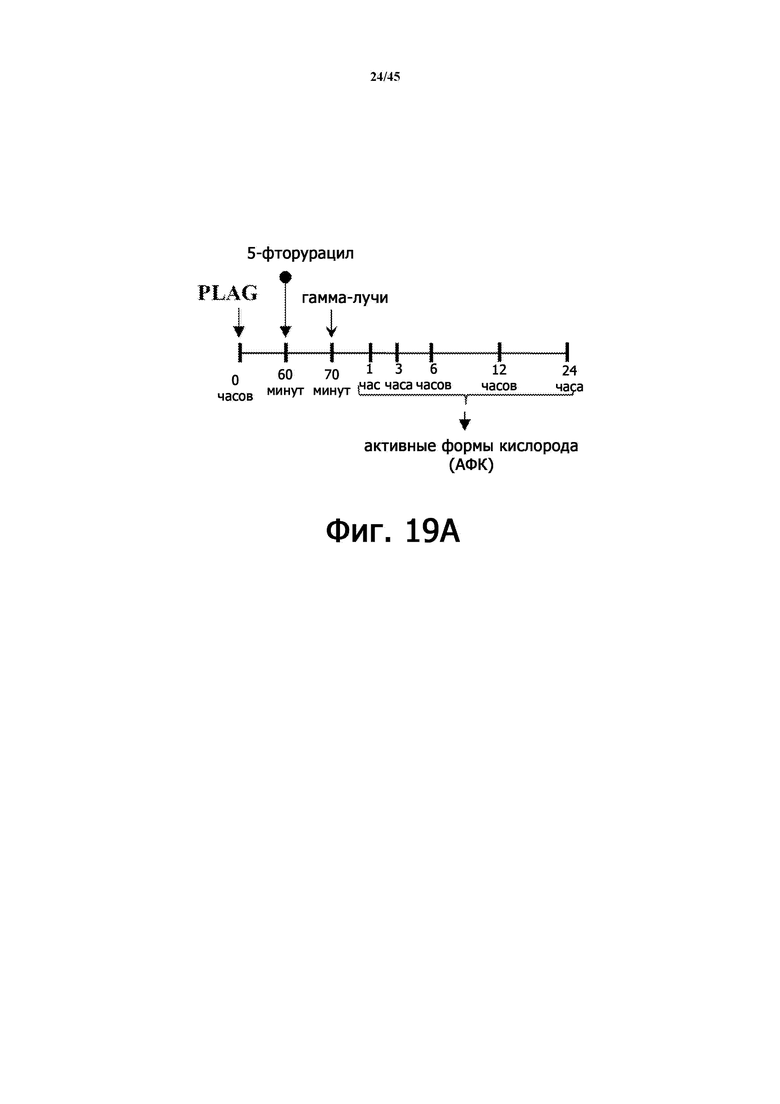
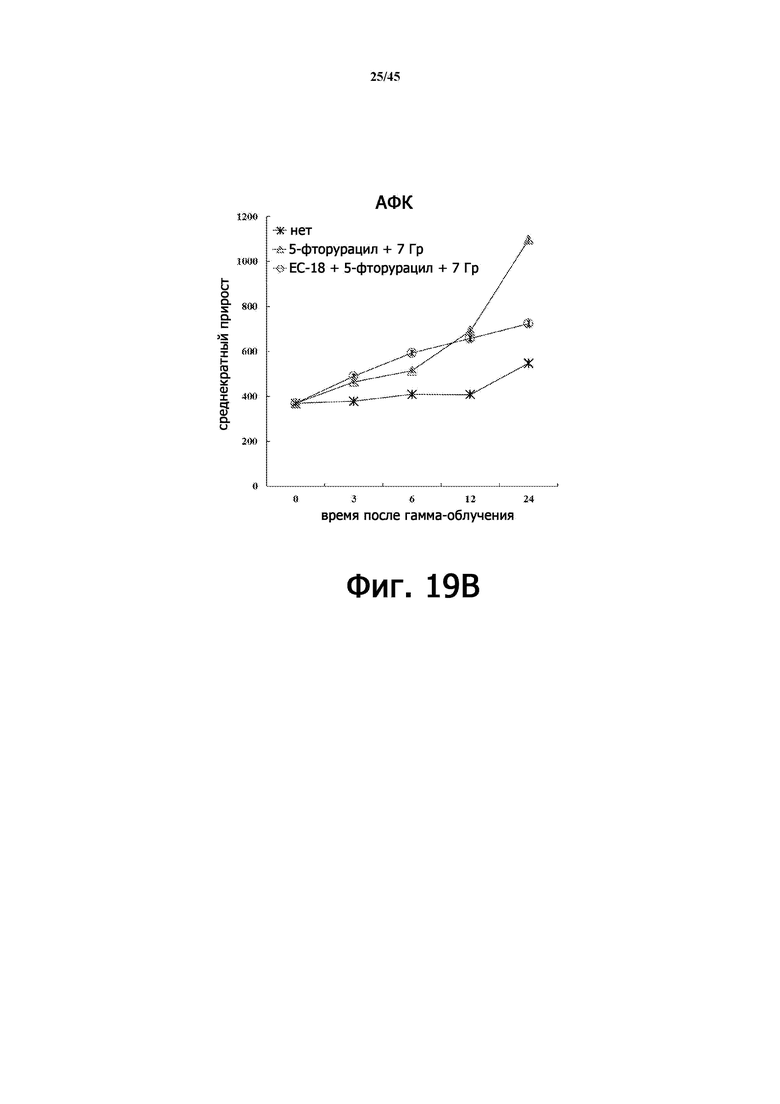
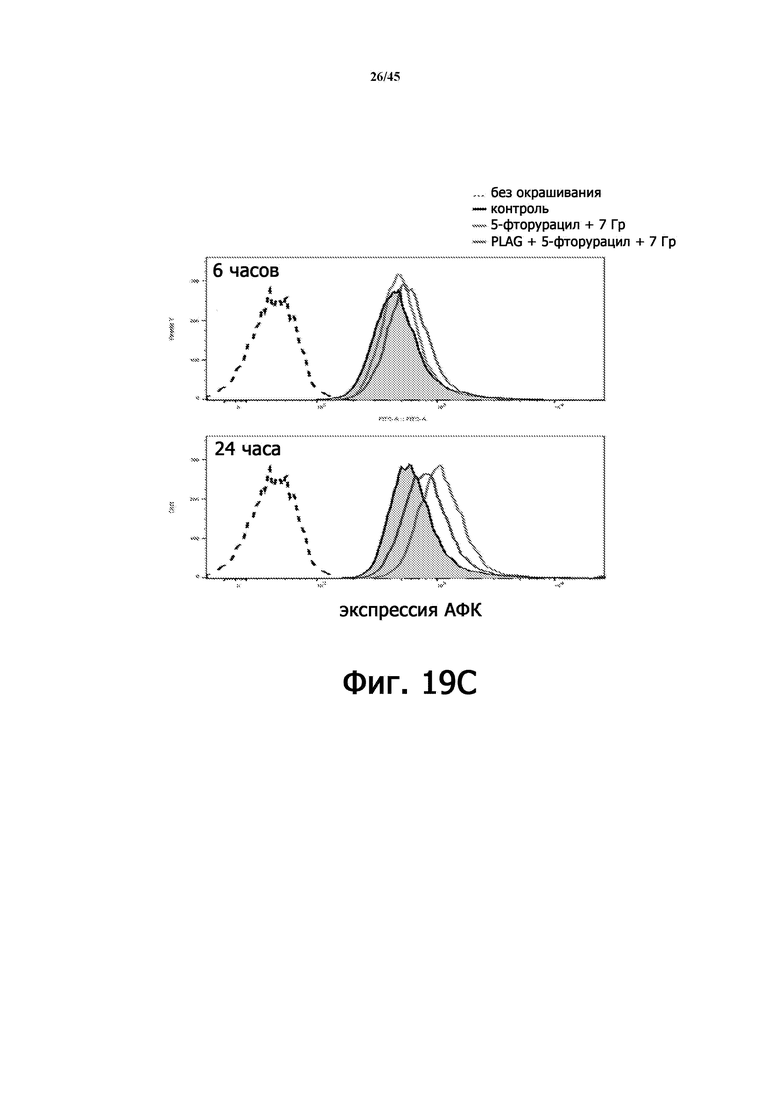
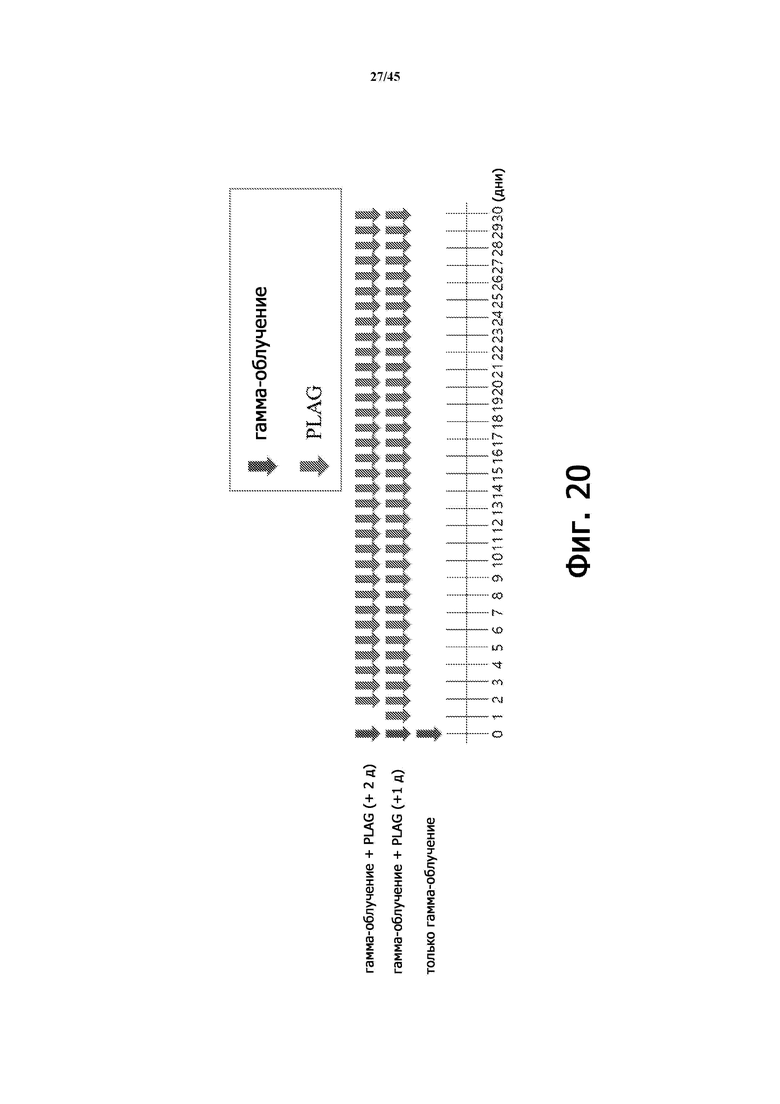
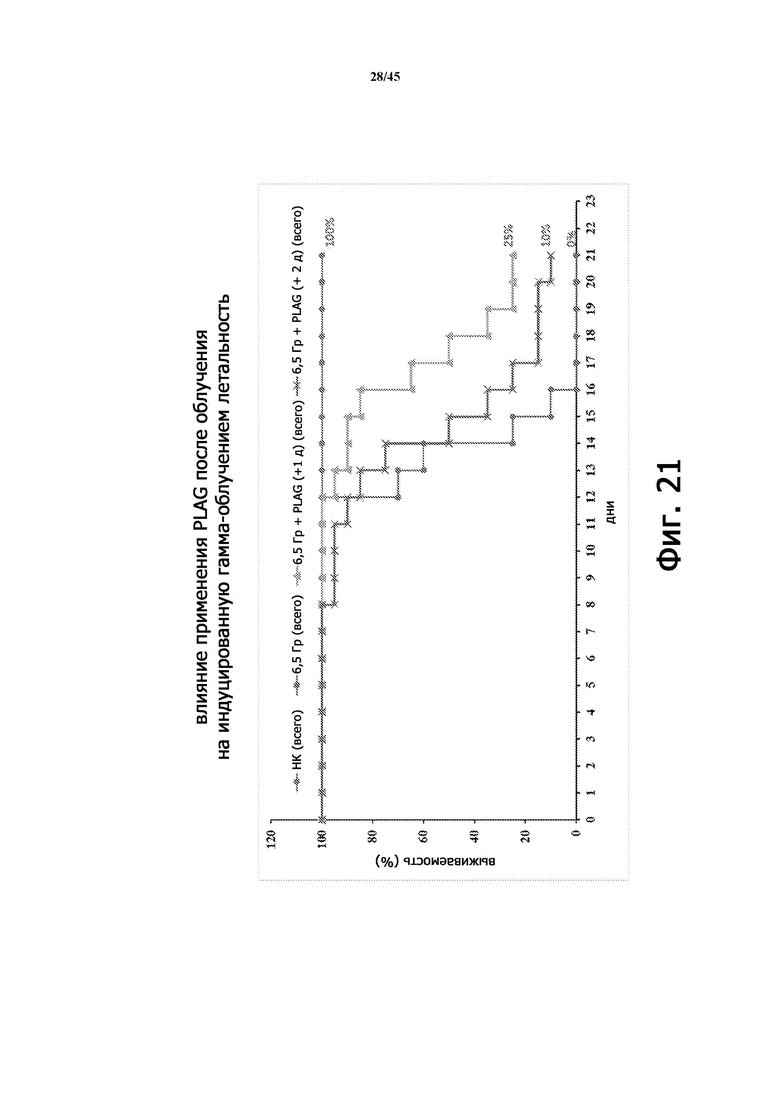
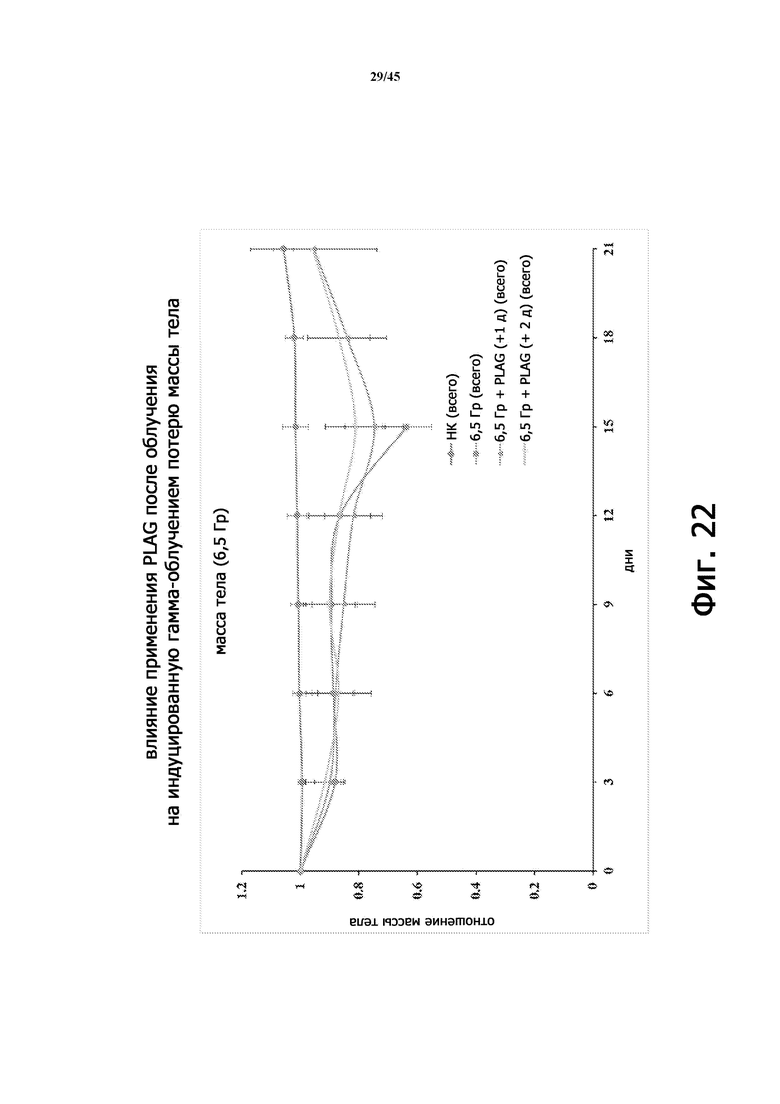
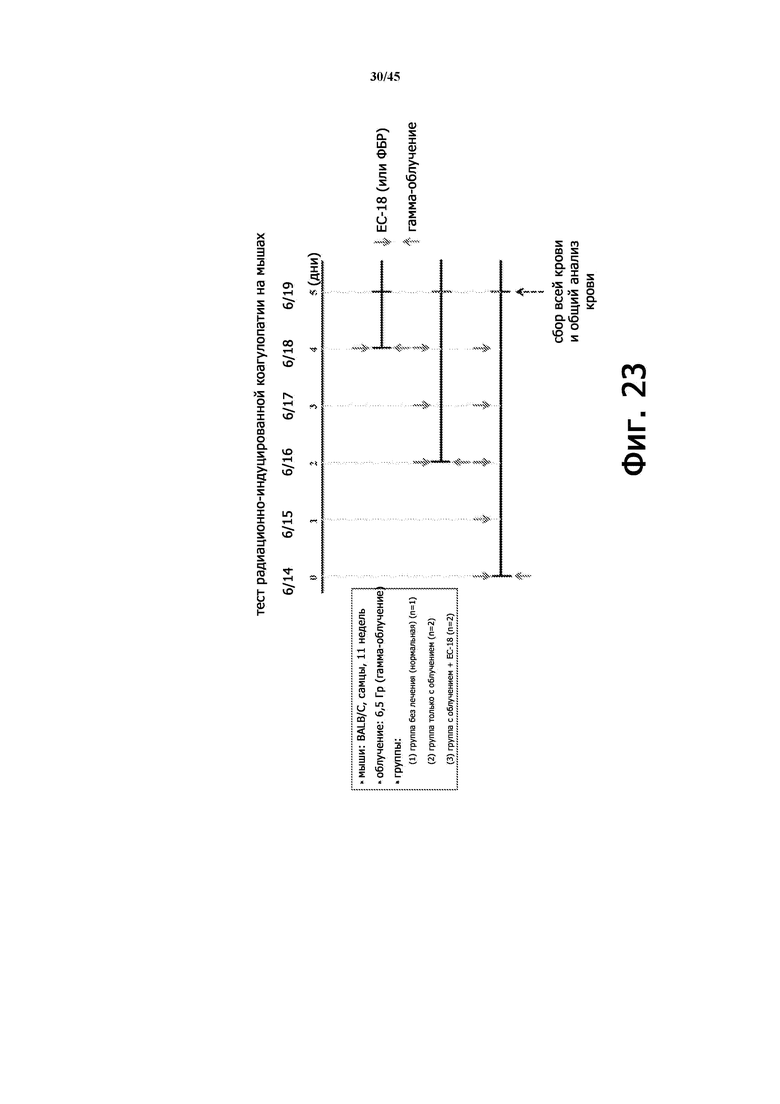


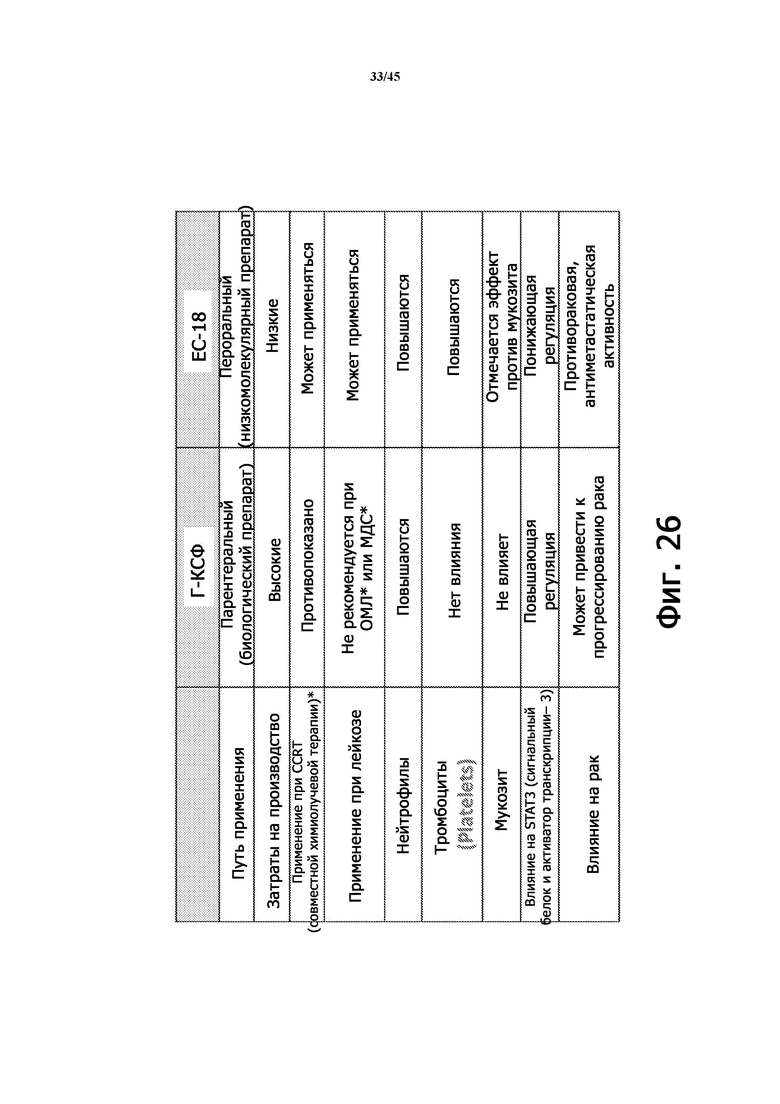
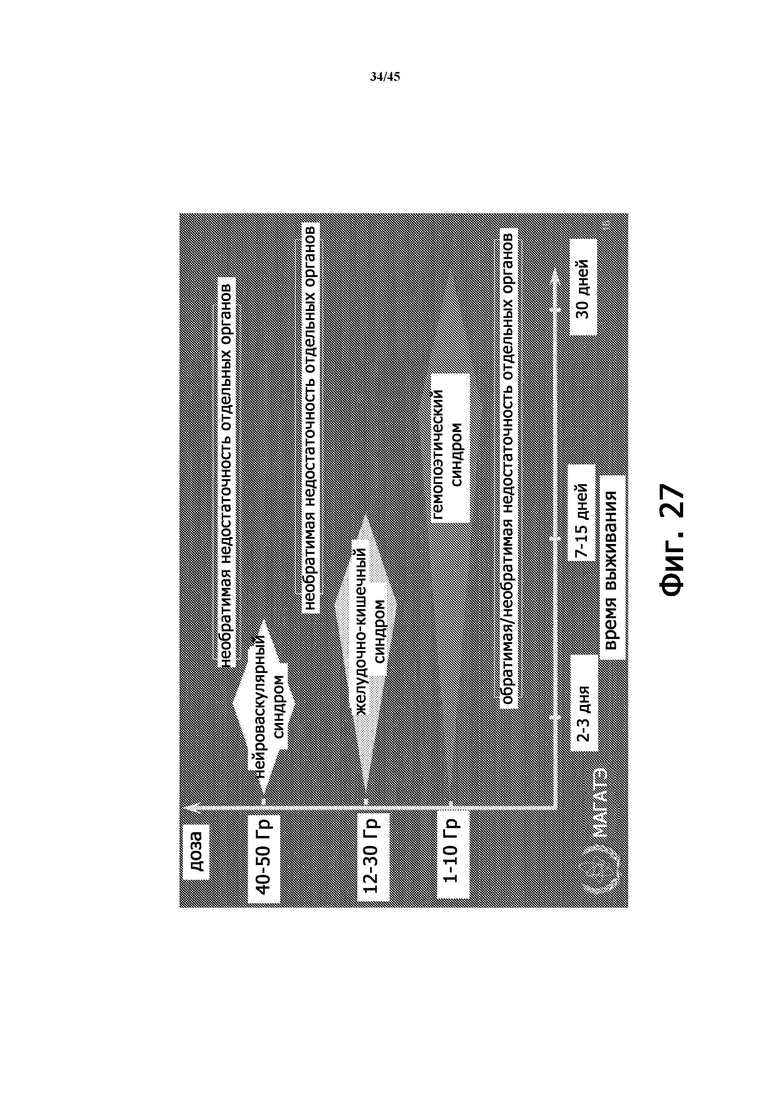
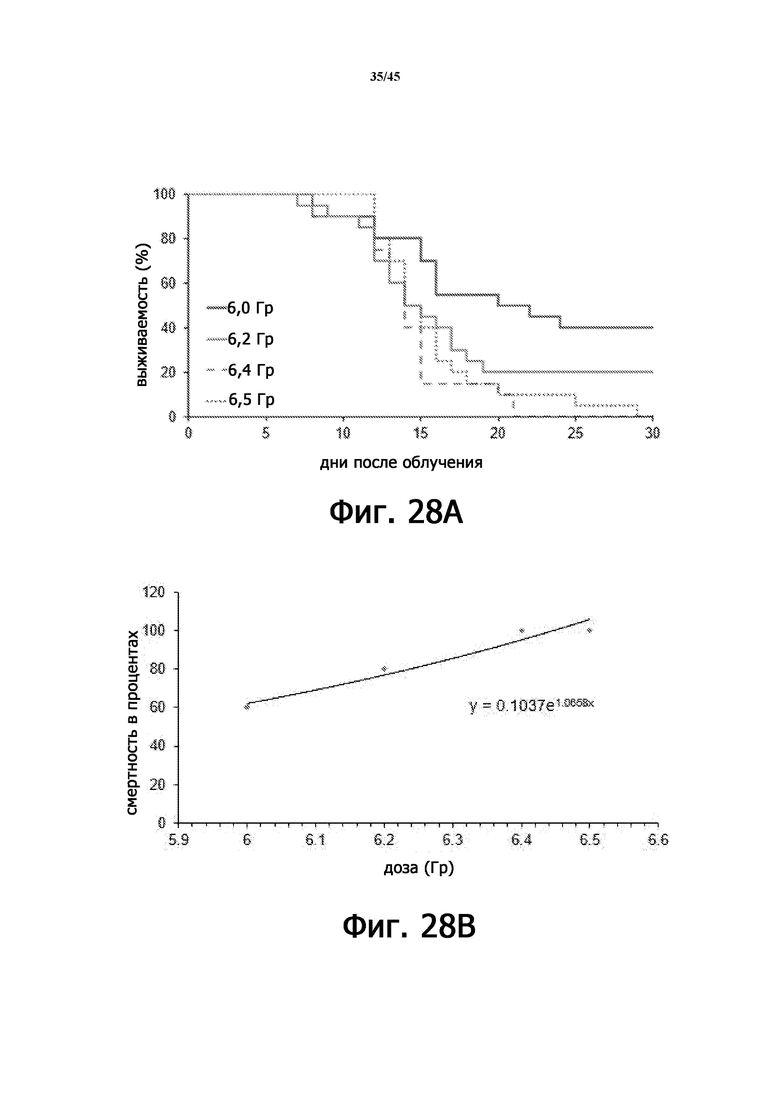
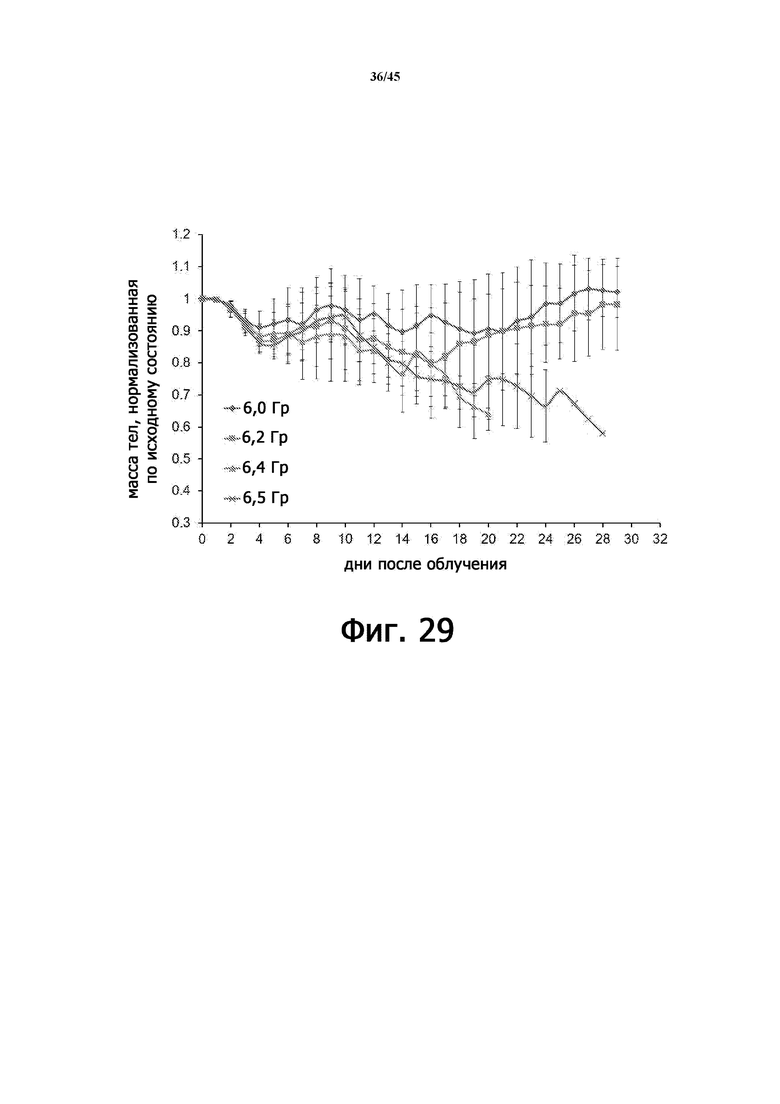
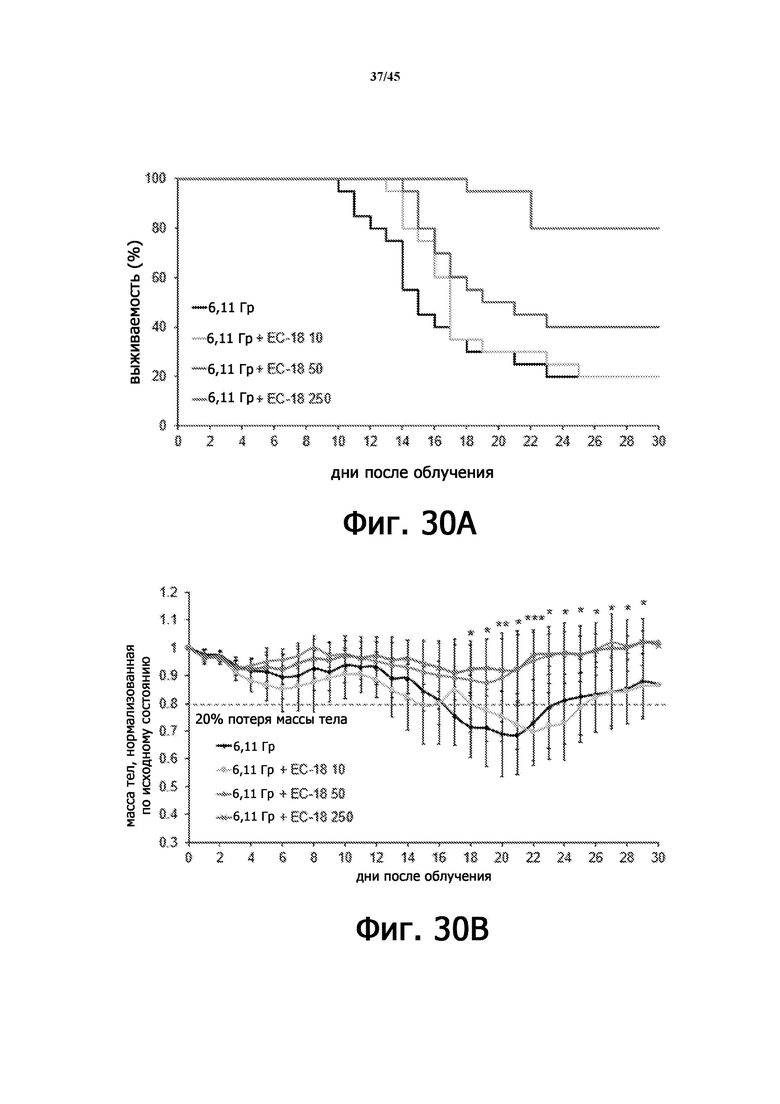
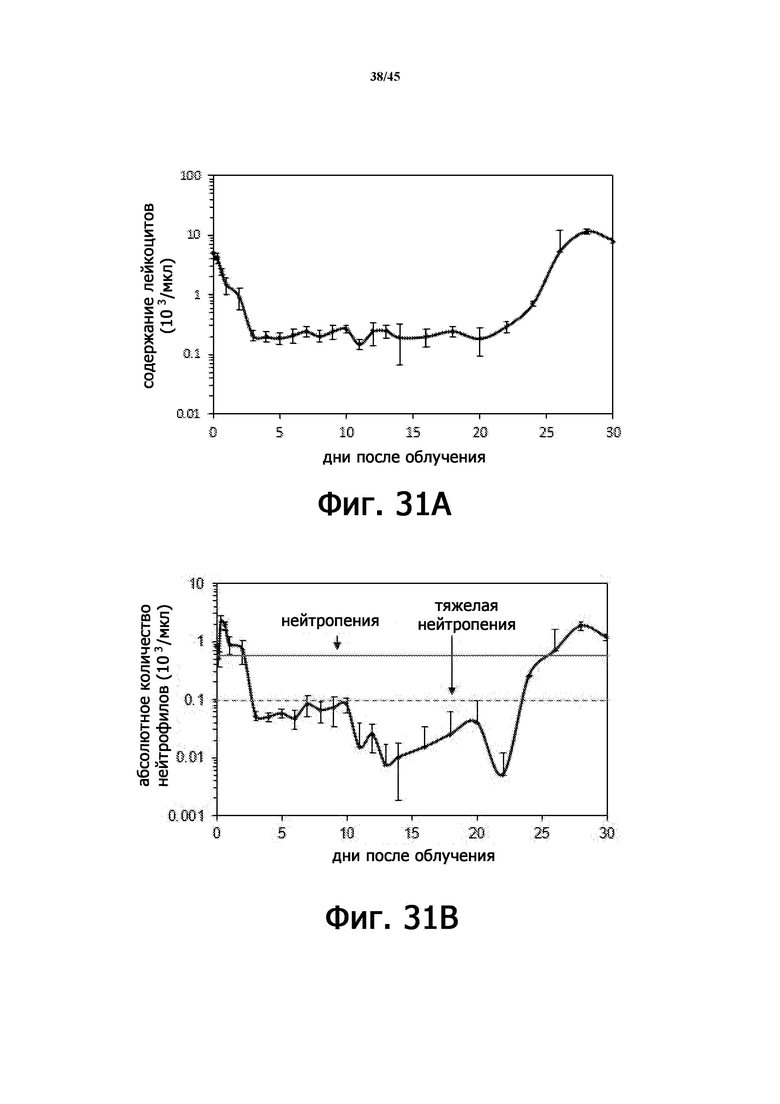
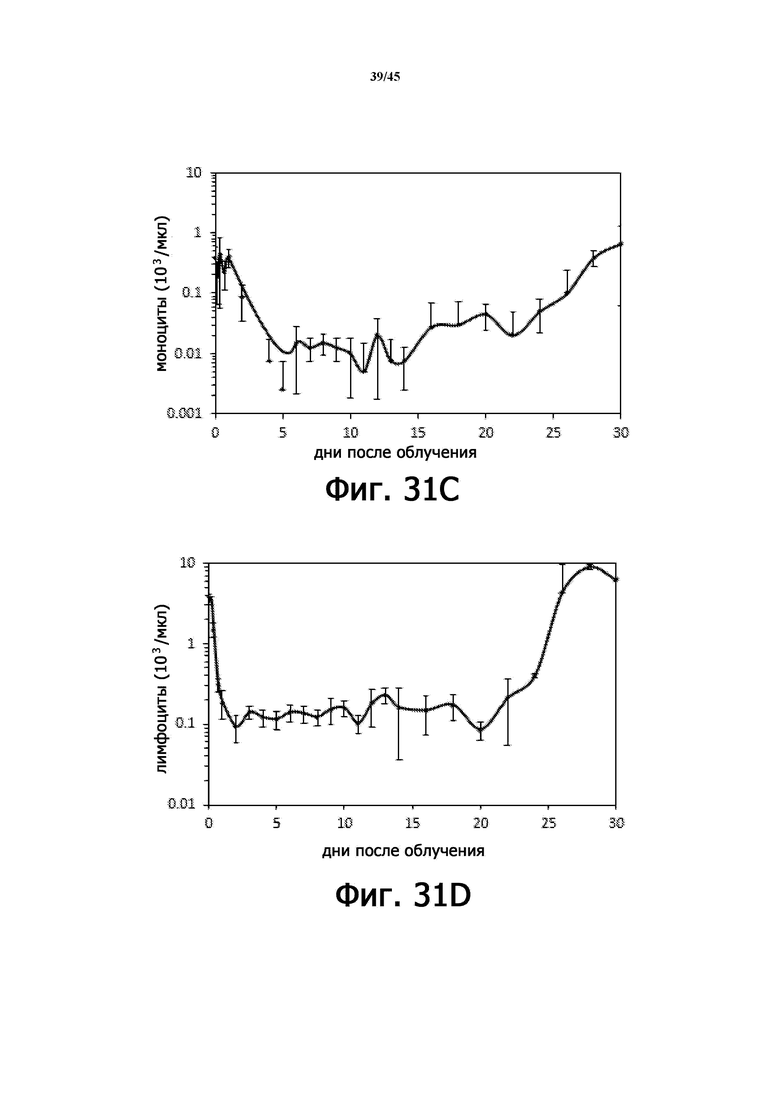
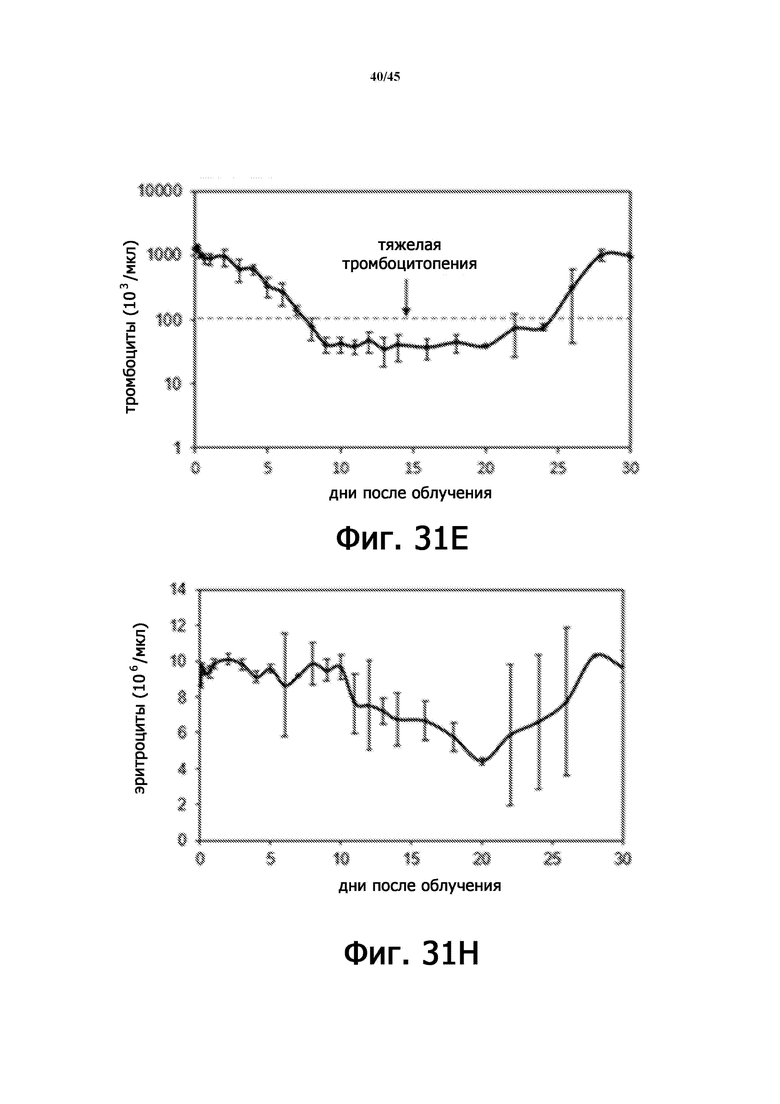
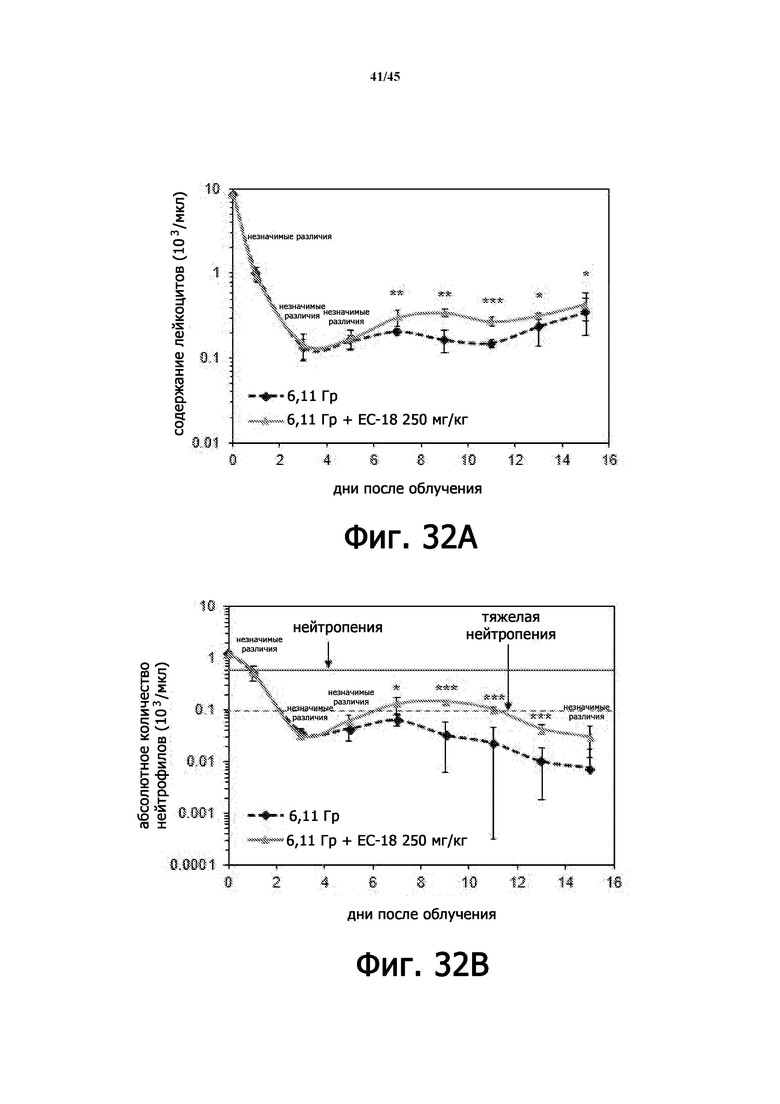
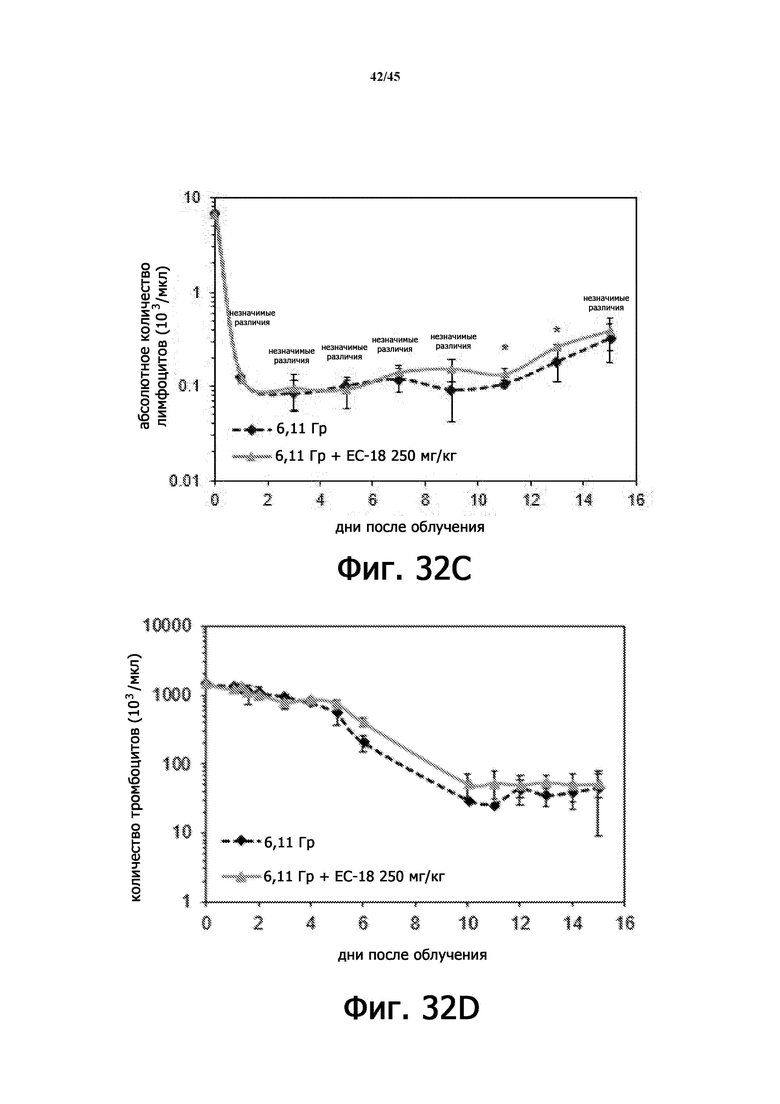
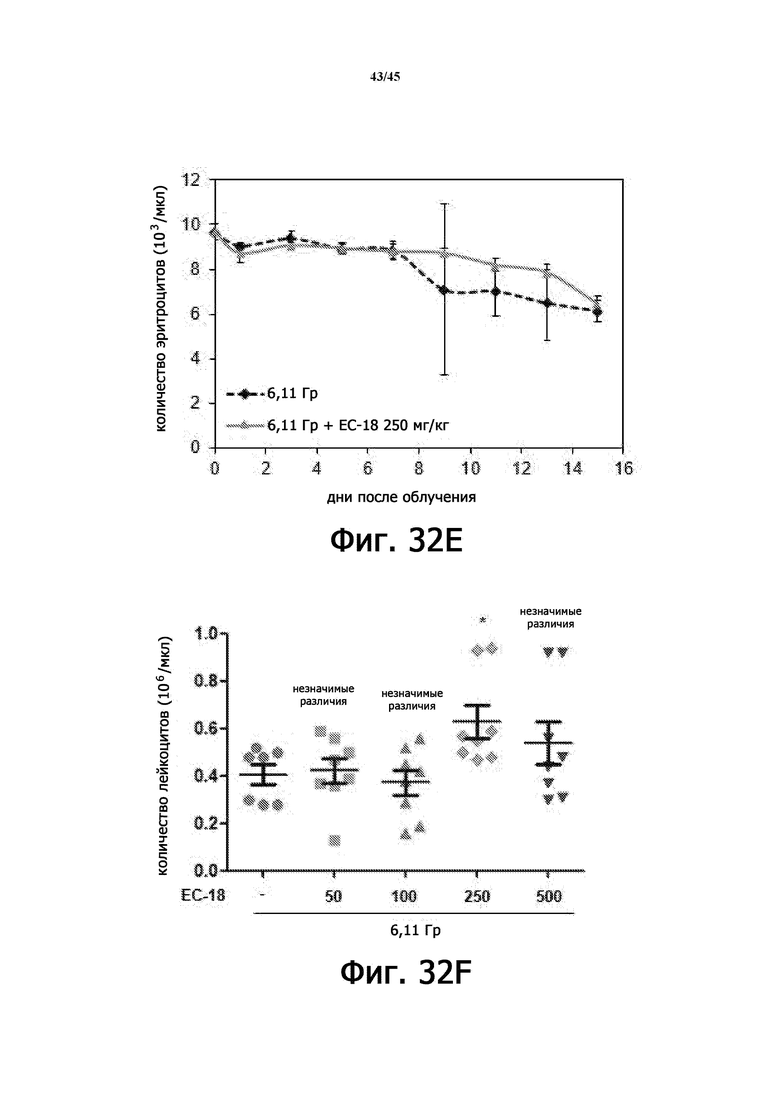
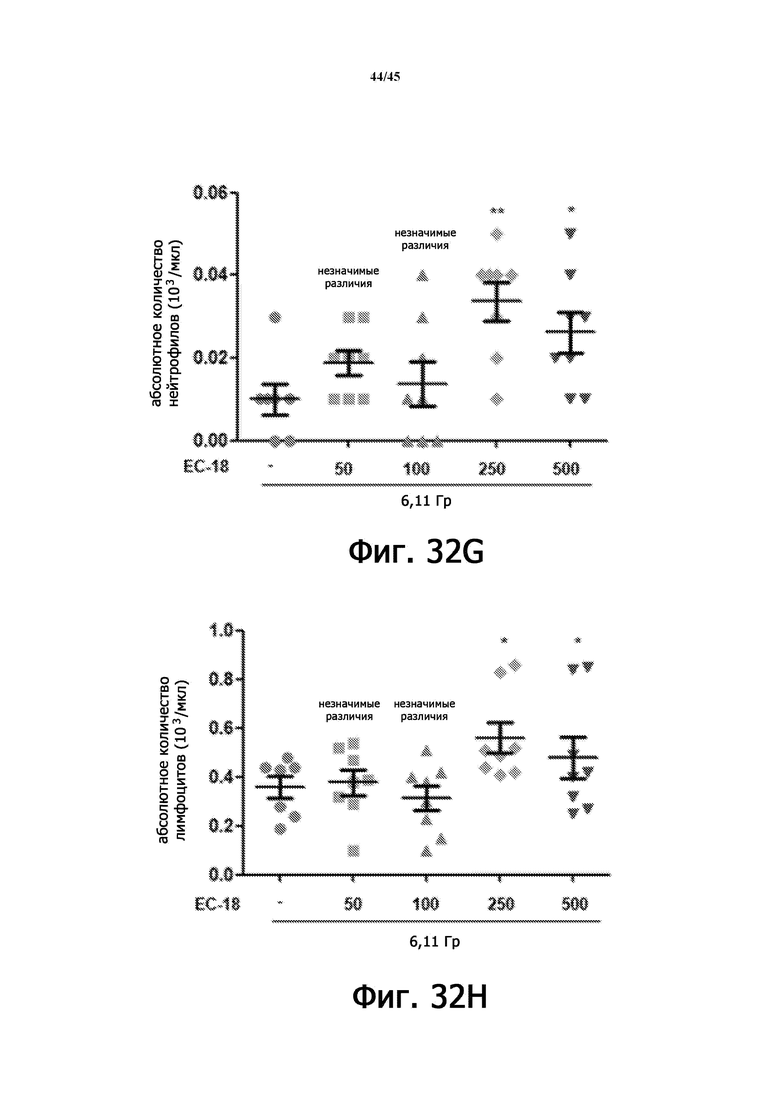
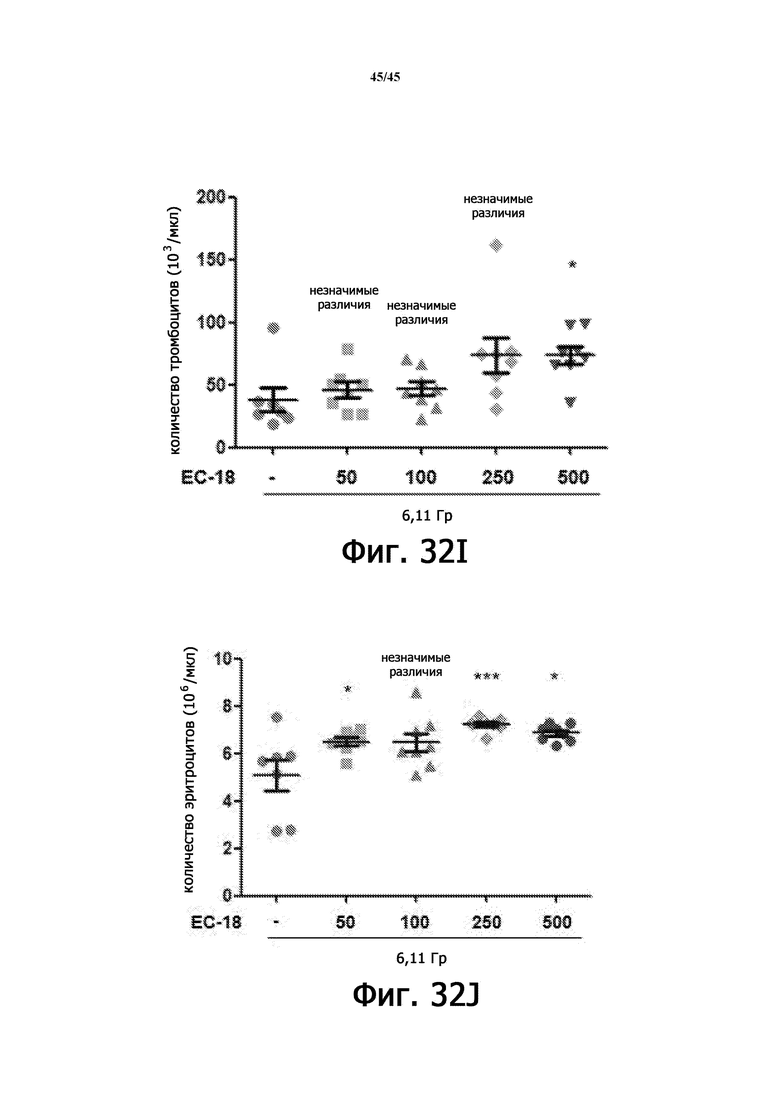
Группа изобретений относится к применению 1-пальмитоил-2-линолеоил-3-ацетилглицерина (PLAG) для лечения или профилактики острой лучевой болезни у субъекта, также относится к применению 1-пальмитоил-2-линолеоил-3-ацетилглицерина (PLAG) для лечения субъекта, подвергшегося неблагоприятному ионизирующему излучению, также относится к набору для использования при лечении или профилактике острой лучевой болезни у субъекта, включающему: (а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG); (b) инструкции по применению PLAG для лечения или профилактики острой лучевой болезни (ОЛБ) у субъекта, также относится к набору для использования при лечении или профилактике чрезмерного воздействия ионизирующего излучения, включающему: (а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG); (b) инструкции по использованию PLAG для лечения или профилактики чрезмерного воздействия ионизирующего излучения, также относится к набору для использования при лечении или профилактике одного или нескольких субсиндромов острой лучевой болезни, включающему: (а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG); (b) инструкции по применению PLAG для лечения или профилактики одного или нескольких субсиндромов острой лучевой болезни. Группа изобретений обеспечивает индукцию повышения уровней различных иммунных клеток, которые были снижены при воздействии радиации на модели исследования ОЛБ, в значительном повышении выживаемости мышей после облучения, в положительном эффекте PLAG в отношении каждого симптома (снижении уровня мукозита и увеличении количества нейтрофилов в крови), влияющего на выживаемость на животной модели острой лучевой болезни, и на восстановление кожного повреждения у животных, подвергавшихся воздействию радиации. 5 н. и 7 з.п. ф-лы, 8 табл., 11 пр., 66 ил.
1. Применение 1-пальмитоил-2-линолеоил-3-ацетилглицерина (PLAG) для лечения или профилактики острой лучевой болезни у субъекта.
2. Применение 1-пальмитоил-2-линолеоил-3-ацетилглицерина (PLAG) для лечения субъекта, подвергшегося неблагоприятному ионизирующему излучению.
3. Применение по п. 1 или 2, где субъект страдает от гемопоэтического острого лучевого синдрома, радиационной коагулопатии, желудочно-кишечного острого лучевого синдрома, сердечно-сосудистого острого лучевого синдрома, или острого лучевого синдрома центральной нервной системы (ЦНС), или их комбинации или подвержен указанному.
4. Применение по любому из пп. 1-3, где 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG) впервые вводят субъекту в течение 36 часов после радиационного воздействия.
5. Применение по любому из пп. 1-4, где PLAG вводят с гранулоцитарным колониестимулирующим фактором (Г-КСФ).
6. Применение по любому из пп. 1-5, где PLAG вводят с одним или более, выбранными из группы, состоящей из обезболивающих средств, противоязвенных средств, противодиарейных средств, антибиотиков, жаропонижающих средств, пищевых добавок и антиоксидантов.
7. Набор для использования при лечении или профилактике острой лучевой болезни у субъекта, включающий:
(а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG);
(b) инструкции по применению PLAG для лечения или профилактики острой лучевой болезни (ОЛБ) у субъекта.
8. Набор для использования при лечении или профилактике чрезмерного воздействия ионизирующего излучения, включающий:
(а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG);
(b) инструкции по использованию PLAG для лечения или профилактики чрезмерного воздействия ионизирующего излучения.
9. Набор для использования при лечении или профилактике одного или нескольких субсиндромов острой лучевой болезни, включающий:
(а) 1-пальмитоил-2-линолеоил-3-ацетилглицерин (PLAG);
(b) инструкции по применению PLAG для лечения или профилактики одного или нескольких субсиндромов острой лучевой болезни.
10. Набор по любому из пп. 7-9, содержащий терапевтически эффективное количество PLAG.
11. Набор по любому из пп. 7-10, содержащий письменные инструкции по применению PLAG.
12. Набор по любому из пп. 7-11, где инструкции представляют собой этикетку продукта.
| WO 2015176012 A1, 19.11.2015 | |||
| WO 2016109002 A2, 07.07.2016 | |||
| Andrea L | |||
| DiCarlo et al., Medical Countermeasures for Platelet Regeneration after Radiation Exposure / RADIATION RESEARCH, 2011, v.176, pp.e0001-e0015 | |||
| Lars Heslet et al., Acute radiation syndrome (ARS) - treatment of the reduced host defense / International Journal of General Medicine, |

